Область изобретения
Данное изобретение относится к новым твердым формам некоторых лекарственных соединений, к фармацевтическим композициям, содержащим их, к способам их получения и применению новых твердых форм и композиций, содержащих их, в лечении.
Предшествующий уровень техники
При приготовлении лекарственных композиций в виде препарата важно, чтобы лекарственное средство находилось в форме, в которой с ним можно удобно обращаться и его обрабатывать. Это важно не только с точки зрения достижения рентабельного производственного процесса, но также с точки зрения последующего изготовления фармацевтических композиций (например, пероральных лекарственных форм, таких как таблетка), содержащих активное соединение.
Кроме того, при изготовлении пероральных лекарственных композиций, важно, чтобы обеспечивался надежный и воспроизводимый профиль концентрации лекарственного вещества в плазме после введения пациенту. Это особенно важно в изготовлении композиций, содержащих антитромботические агенты.
Химическая стабильность, стабильность твердого состояния и "срок годности" активных ингредиентов также являются очень важными факторами. Лекарственное средство и композиции, содержащие его, должны обладать способностью эффективно храниться в течение длительных периодов времени без демонстрации значительного изменения физико-химических характеристик активного компонента (например, его химического строения, плотности, гигроскопичности и растворимости).
Кроме того, также важна возможность обеспечить лекарственное средство в форме, которая является химически чистой настолько, насколько это возможно.
Аморфные вещества могут создавать трудности в этом отношении. Например, такие вещества обычно трудны в обращении и для приготовления в виде препарата, обеспечивают ненадежную растворимость, и часто обнаруживают нестабильность и химическую неоднородность.
Специалисту понятно, что, если лекарственное средство можно легко получать в стабильной кристаллической форме, вышеуказанные проблемы могут быть решены.
Таким образом, в изготовлении рентабельных и фармацевтически приемлемых лекарственных композиций важно, когда это возможно, получить лекарственное средство в по существу кристаллической и стабильной форме.
Однако следует заметить, что такая цель не всегда достижима. Действительно, обычно невозможно предвидеть, исходя только из молекулярной структуры, какой будет характер кристаллизации соединения, либо как такового, либо в форме соли. Это можно определять только опытным путем.
В международной патентной заявке WO 02/44145 раскрыт ряд соединений, которые, как было обнаружено, полезны в качестве ингибиторов тромбина или пролекарств ингибиторов тромбина, где ингибиторы тромбина имеют общую формулу I
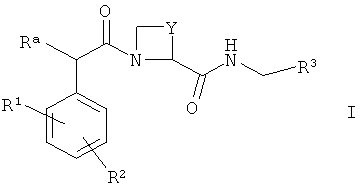
(где Ra, R1, R2, Y и R3 имеют значения, приведенные в описании WO 02/44145), и их фармацевтически приемлемые производные (включая пролекарства).
В WO 02/44145 также конкретно раскрыты соединения Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe), (упоминаемый ниже в данном описании изобретения как Соединение А), где Aze представляет собой (S)-азетидин-2-карбоксилат и Pab представляет собой пара-амидинобензиламино, и Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH) (упоминаемый ниже в данном описании изобретения как Соединение В).

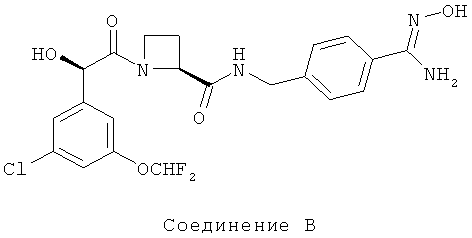
Способ синтеза Соединений А и В описан в Примерах в WO 02/44145, но Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) и Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH) в кристаллической форме не раскрыты.
Соединения А и В метаболизируются после перорального и/или парентерального введения в соответствующее свободное амидиновое соединение (Соединение С), которое, как было обнаружено, является ингибитором тромбина (смотри WO 02/44145, релевантное описание которой включено в данное описание изобретения посредством ссылки).
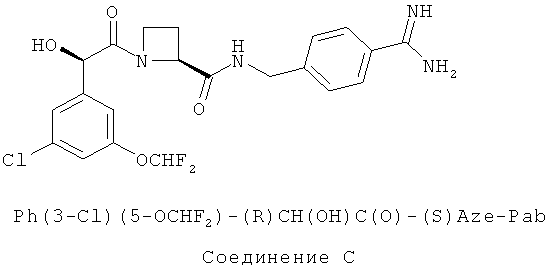
В международной патентной заявке WO 03/101957 предложены кристаллические фармацевтически приемлемые соли присоединения кислот, таких как этансульфоновая кислота, н-пропансульфоновая кислота, н-бутансульфоновая кислота и соли бензолсульфоновой кислоты соединений, таких как Соединение А.
Однако сохраняется необходимость найти кристаллические формы таких активных соединений, которые находятся не в форме солей. Такие кристаллические формы свободного основания позволяют получать композиции без условий в отношении противоиона (который, например, вносит "нетерапевтическую" массу в конечную композицию). Например, использование свободного основания Соединения А вместо соли бензолсульфоновой кислоты позволяет понизить массу приблизительно на 30%, с соответствующим меньшим размером таблетки.
Описание изобретения
Авторы изобретения обнаружили, что Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) и Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH) можно получать в одной или более чем одной несолевых формах, которые являются по существу кристаллическими по своей природе.
Таким образом, согласно первому аспекту изобретения предложены соединения Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) и Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH) в по существу кристаллических, несолевых формах (ниже упоминаемых как "соединения по изобретению" или "твердые или кристаллические формы по изобретению").
Хотя авторы изобретения обнаружили, что возможно получить Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) и Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH) в формах, которые являются кристаллическими более чем на 80% под "по существу кристаллической" авторы изобретения подразумевают более чем на 10% (например, более 20%), в частности более 30%, и более конкретно более 40% кристалличность. Степень (%) кристалличности может определить специалист, используя дифракцию рентгеновских лучей на порошке (XRPD). Можно использовать другие способы, такие как ЯМР твердого тела (ядерно-магнитно-резонансная) спектроскопия, FT-IR (инфракрасная спектроскопия с Фурье-преобразованием), Рамановская спектроскопия, дифференциальная сканирующая калориметрия (DSC) и микрокалориметрию.
Соединения по изобретению могут находиться в несольватированной форме (такой как ангидрат) или в форме сольвата, и все такие формы включены как часть изобретения. Сольваты Соединения А или В включают алкоголяты, такие как изопропанольный сольват (например, соединение по изобретению, которое находится в форме изопропанольного сольвата, содержащего 1/3 молекулы изопропанола на молекулу соединения по изобретению).
Кроме того, как часть изобретения также включены любые таутомеры соединений по изобретению.
Соединения по изобретению могут иметь улучшенные свойства, например стабильность, например по сравнению с Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) и Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH), полученными, как описано в WO 02/44145. Соединения по изобретению имеют другие свойства по сравнению с их аморфными формами свободного основания и/или кристаллическими солевыми формами. Например, другая растворимость и/или скорость растворения (в различных растворителях, например водных системах) может быть полезной в композициях, содержащих соединения по изобретению (такие как, например, кристаллическая форма Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe), описанная в данном описании изобретения).
Согласно дополнительному аспекту изобретения, таким образом в данной заявке предложена стабильная кристаллическая форма Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) и стабильная кристаллическая форма Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH).
Термин "стабильность", как определено в данном описании изобретения, включает химическую стабильность и/или стабильность твердого состояния.
Под "химической стабильностью" авторы изобретения подразумевают, что соответствующие соединения можно хранить в отдельной форме, или в форме композиции, в которой они представлены в смеси с фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами (например, в пероральной лекарственной форме, такой как таблетка, капсула и так далее), при нормальных условиях хранения, с ограниченной степенью химической деструкции или разложения.
Под "стабильностью твердого тела", авторы изобретения подразумевают, что соответствующие соединения можно хранить в отдельной твердой форме, или в форме твердой композиции, в которой они представлены в смеси с фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами (например, в пероральной лекарственной форме, такой как таблетка, капсула и так далее), при нормальных условиях хранения, с незначительной степенью фазовых превращений в твердом состоянии (например, кристаллизации, перекристаллизации, твердофазного перехода, гидратации, дегидратации, сольватации или десольватации).
Примеры "нормальных условий хранения" включают температуру от минус 80°С до плюс 50°С (в частности, 0°С-40°С и более конкретно комнатную температуру, такую как 15°С-30°С), давление 0,1-2 бар (0,1-2×105 Па) (в частности, атмосферное давление), относительную влажность от 5 до 95% (в частности, от 10 до 75%), и/или воздействие УФ-излучения/видимого света до 460 люкс, в течение длительных периодов (например, больше или равных шести месяцам). Соединения по изобретению при таких условиях могут обнаруживать соответствующую химическую деструкцию/разложение или полиморфную трансформацию менее 15%, более конкретно менее 10%, и особенно менее 5%. Специалистам очевидно, что вышеуказанные верхние и нижние пределы для температуры, давления и относительной влажности представляют собой крайние значения нормальных условий хранения, и что некоторые комбинации таких крайних значений не будут иметь место во время нормального хранения (например, температура 50°С и давление 0,1 бар (0,1×105 Па)).
Соединения по изобретению можно предпочтительно получать путем кристаллизации Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) или Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH), как описано в данном описании изобретения.
Согласно дополнительному аспекту изобретения, предложен способ получения соединения по изобретению, который включает кристаллизацию Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) или Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH) из перенасыщенного раствора соответствующего соединения в подходящем растворителе. В таком способе важно оставить перенасыщенный раствор перемешиваться в течение достаточного периода времени. Длительность этого промежутка времени зависит от перенасыщения так, что сильно перенасыщенные растворы могут кристаллизоваться в течение суток или двух, тогда как для менее перенасыщенных растворов может требоваться большее время (например, неделя или более).
Было обнаружено, что в среде, не содержащей затравочных кристаллов, выпаривание растворителя не приводит к кристаллизации, то есть важно поддерживать постоянный уровень перенасыщения (например, при использовании закрытого сосуда).
Представляется важным подходящее смешивание, например посредством перемешивания, возможно потому, что оно создает сайты для первичного, а также вторичного зарождения кристаллов, таким образом ускоряя процесс кристаллизации. Если возможно, добавление затравочных кристаллов (кристаллизуемой формы) к перенасыщенному раствору ускорит процесс кристаллизации в связи с тем, что время первичного зарождения кристаллов в таком случае сократится. Таким образом, в дополнительном способе по изобретению предложено получение соединения по изобретению, которое включает кристаллизацию Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) или Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH) из (перенасыщенного) раствора или суспензии соединения и подходящего растворителя с использованием затравки соответствующего соединения для инициации и/или облегчения кристаллизации. Подходящие растворители включают спирты (такие как этанол и изопропанол), этилацетат, изопропилацетат, водные системы и подходящие их смеси (например, вода/этанол, изопропанол/этилацетат и изопропилацетат/этанол/гептан). При необходимости также можно применять антирастворители. Конкретный способ по изобретению включает применение трех систем растворителей, которые способствуют агрегации кристаллов, то есть применение сильного растворителя, умеренного растворителя и антирастворителя, таких как сильный растворитель этанол, умеренный растворитель изопропилацетат и антирастворитель гептан.
Таким образом можно сокращать период кристаллизации от нескольких суток до примерно одних суток с получением выхода примерно 90%. Увеличенное количество затравочных кристаллов (например, вплоть до 4% масс./масс.) и время добавления антирастворителя может снижать время кристаллизации посредством такого образования центров кристаллизации.
Согласно изобретению кристаллическое Соединение А или В может быть получено путем кристаллизации из перенасыщенного раствора соответствующего аморфного соединения в подходящем растворителе. Перенасыщенный раствор можно получить после растворения соединения в подходящем растворителе и упаривания с получением интересующего (аморфного) соединения, с последующим получением подходящего перенасыщенного раствора. Более предпочтительно перенасыщенный раствор может быть получен из растворов, которые экстрагировали непосредственно из реакционных растворов, в которых интересующие соединения были образованы; или, особенно предпочтительно, непосредственно из реакционных растворов, внутри которых интересующее соединение было образовано.
Дополнительная информация о способах изобретения и продуктах, получаемых посредством этого, описана в Примерах данного описания изобретения.
Кристаллическое вещество - ангидрат можно получать, как описано в данном описании изобретения, посредством кристаллизации Соединения А или В из одного или более подходящих растворителей или их смесей. Ангидрат может быть получен посредством кристаллизации из системы растворителя, которая является по существу не содержащей воду (которая может быть высушена, и/или которую можно сушить в процессе кристаллизации). Однако кристаллическое безводное вещество (например, Соединение А) можно также получать из воды или смесей вода/этанол.
Соединения по изобретению, которые представляют собой ангидраты, обычно содержат не более 2%, в частности 1%, более конкретно 0,5% и более конкретно 0,2% (масс./масс.) воды, причем эта вода либо связана (кристаллизационная вода или иная), либо не связана.
Кристаллический изопропанольный сольват можно получить, как описано в данном описании изобретения путем кристаллизации (например, Соединения В) из системы растворителя, содержащей изопропанол, или комбинации изопропанола и одного или более других подходящих растворителей.
Соединения по изобретению можно выделять, используя методы, которые хорошо известны специалистам в данной области техники, например путем декантации, фильтрации или центрифугирования.
Соединения по изобретению можно сушить, используя стандартные методы. Специалисту очевидно, что температура сушки и продолжительность сушки могут влиять на характеристики твердого состояния соединений, которые находятся в форме сольватов (например, при повышенных температурах и/или пониженном давлении может происходить десольватация).
Если соединения по изобретению кристаллизуются, или перекристаллизуются, как описано в данном описании изобретения, то полученное соединение находится в форме, которая имеет улучшенную химическую стабильность и/или стабильность твердого состояния, упоминаемую здесь.
Кристаллические формы соединений по изобретению можно легко охарактеризовать, используя методы дифракции рентгеновских лучей на порошке (XRPD), например, как описано ниже. Также можно использовать стандартные методы DSC, TGA.
Фармацевтические препараты и медицинские применения
Согласно изобретению кристаллические формы по изобретению можно вводить перорально, внутривенно, подкожно, трансбуккально, ректально, дермально, назально, трахеально, бронхиально, любым другим парентеральным путем, или посредством ингаляции, в форме фармацевтического препарата, содержащего соединение по изобретению в фармацевтически приемлемой лекарственной форме. Однако авторы изобретения предпочитают, чтобы соединение по изобретению представляло собой форму, которая пригодна для перорального введения.
В зависимости от расстройства и пациента, подлежащего лечению, а также пути введения, кристаллические формы по изобретению можно вводить в различных дозах (смотри ниже).
Кристаллические формы по изобретению можно дополнительно обрабатывать перед приготовлением в виде подходящей фармацевтической композиции, например, их можно размалывать или измельчать на более мелкие частицы.
Согласно дополнительному аспекту изобретения предложена фармацевтическая композиция, содержащая кристаллическую форму по изобретению в смеси с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Количество кристаллического соединения по изобретению, которое используют в такой композиции, зависит от состояния и пациента, подлежащего лечению, а также кристаллических(ой) форм(ы), которые(ую) используют, но может быть определено без изобретательского подхода.
Согласно еще одному аспекту изобретения, предложена фармацевтическая композиция, содержащая соединение по изобретению в композиции с немедленным высвобождением, как описано в международной патентной заявке WO 03/101423, релевантная часть описания которой включена в данное описание изобретения посредством ссылки.
Термин фармацевтическая композиция с "немедленным высвобождением" включает любую композицию, в которой скорость высвобождения лекарственного средства из композиции и/или абсорбции лекарственного средства ни заметно, ни намеренно, не замедлена галеновыми манипуляциями. В настоящем случае немедленное высвобождение может быть обеспечено с помощью подходящего фармацевтически приемлемого разбавителя или носителя, причем разбавитель или носитель не пролонгируют в значительной степени, скорость высвобождения лекарственного средства и/или абсорбцию. Таким образом, термин исключает композиции, которые адаптированы для обеспечения "модифицированного", "контролируемого", "замедленного", "продолжительного", "пролонгированного" или "отсроченного" высвобождения лекарственного средства.
В данном контексте термин "высвобождение" включает предоставление (или презентацию) лекарственного средства из композиции в желудочно-кишечный тракт, к тканям организма и/или в системный кровоток. Для высвобождения в желудочно-кишечном тракте (натощак) высвобождение происходит при таких значениях рН, как рН=1-3, особенно при значении, равном или примерно равном рН=1. В одном аспекте изобретения композиция, как описано в данном описании изобретения, с Соединением А или В в кристаллической форме (ниже "Соединение А или В") высвобождает лекарственное средство в интервале значений рН. В другом аспекте изобретения композиция, как описано в данном описании изобретения, с Соединением А или В (в кристаллической форме) высвобождает лекарственное средство в интервале значений рН, таком как рН=1-3, в частности, при рН, равном или примерно равном рН=1. Таким образом, композиции по изобретению могут высвобождать по меньшей мере 70% (в частности 80%) активного ингредиента в течение 4 часов, например в течение 3 часов, в частности 2 часов, более конкретно в течение 1,5 часов, и особенно в течение одного часа (например, в течение 30 минут) после либо перорального, либо парентерального введения.
Композиции по изобретению можно готовить в виде препарата согласно ряду известных методик, например, как описано в М.Е.Aulton "Pharmaceutics: The Science of Dosage Form Design" (1988) (Churchill Livingstone), релевантные части описания данного документа включены в данную заявку посредством ссылки.
Композиции по изобретению могут являться, или их можно адаптировать стандартными методами, чтобы они являлись пригодными для перорального введения, например в форме таблетки с немедленным высвобождением, капсулы с немедленным высвобождением или в виде жидкой лекарственной формы, содержащей активный ингредиент. Такие типы композиций хорошо известны специалистам и могут быть получены методами, известными в данной области техники.
Подходящие разбавители/носители (которые также можно называть "наполнителями") для использования в пероральных композициях по изобретению, например в композициях в форме таблеток с немедленным высвобождением, включают одноосновный фосфат кальция, двухосновный фосфат кальция (включая дигидрат двухосновного фосфата кальция и ангидрат двухосновного фосфата кальция), трехосновный фосфат кальция, лактозу, микрокристаллическую целлюлозу, силикатированную микрокристаллическую целлюлозу, маннит, сорбит, крахмал (например, кукурузный, картофельный или рисовый), глюкозу, лактат кальция, карбонат кальция и тому подобное. Конкретные разбавители/носители включают двухосновный фосфат кальция и микрокристаллическую целлюлозу, которые можно использовать самостоятельно или в комбинации с другим разбавителем/носителем, таким как маннит.
Композиция по изобретению в форме таблетки с немедленным высвобождением может содержать один или более эксципиентов для улучшения физических и/или химических свойств конечной композиции, и/или для облегчения способа изготовления. Такие эксципиенты являются обычными в изготовлении композиций с немедленным высвобождением для пероральной доставки лекарственного средства и включают один или более чем один из следующего: один или более чем один смазывающий агент (такой как стеарат магния, стеариновая кислота, стеарат кальция, стеариловый спирт или, в частности, стеарилфумарат натрия); скользящее вещество (например, тальк или коллоидный оксид кремния); один или более чем один наполнитель (такой как поливинилпирролидон, микрокристаллическая целлюлоза, полиэтиленгликоль (PEG), полиэтиленоксид, гидроксипропилметилцеллюлоза (НРМС) с низкой молекулярной массой, метилцеллюлоза (МС) с низкой молекулярной массой, гидроксипропилцеллюлоза (НРС) с низкой молекулярной массой, гидроксиэтилцеллюлоза (НЕС) с низкой молекулярной массой, крахмал (такой как кукурузный, картофельный или рисовый) или натрий-карбоксиметилцеллюлоза с низкой молекулярной массой; (конкретными связующими веществами являются поливинилпирролидон или НРМС с низкой молекулярной массой); один или более чем один агент, регулирующий рН (такой как органическая кислота (например, лимонная кислота) или соль щелочного металла (например, натрия), оксид магния, щелочной или щелочноземельный металл (например, сульфат, метасульфат, пропионат или сорбат натрия, кальция или калия); один или более чем один разрыхлитель (например, натрия крахмала гликолят, сшитый поливинилпирролидон, сшитая натрий-карбоксиметилцеллюлоза, крахмал (такой как кукурузный, картофельный или рисовый) или альгинат); краситель, вкусоароматизатор, агент, изменяющий тоничность, покрывающее вещество или консервант.
Очевидно, что некоторые из вышеуказанных эксципиентов, которые могут присутствовать в конечной пероральной композиции с немедленным высвобождением (например, таблетке) по изобретению, могут обладать более чем одной из вышеуказанных функций.
В дополнительном аспекте изобретения жидкая композиция по изобретению адаптирована, чтобы быть пригодной для перорального введения.
Подходящие жидкие композиции, которые следует вводить перорально, включают такие, в которых Соединение А или В присутствует вместе с водным носителем, таким как вода. Композиция по настоящему изобретению, содержащая водный носитель, также может содержать один или более чем один эксципиент, такой как противомикробный консервант; модификатор тоничности (например, хлорид натрия, маннит или глюкоза); агент, корректирующий рН (например, обычная неорганическая кислота или основание, включая соляную кислоту или гидроксид натрия); агенты, регулирующие рН (то есть буфер; например винная кислота, уксусная кислота или лимонная кислота); поверхностно-активное вещество (например, додецилсульфат натрия (SDS) или Solutol™); солюбилизатор, который служит для содействия солюбилизации активного ингредиента (например, этанол, полиэтиленгликоль или гидроксипропил-β-циклодекстрин или поливинилхлорид (PVP)); или антиоксидант.
Жидкие пероральные композиции могут находиться в форме суспензий активного ингредиента вместе с водным растворителем или, более конкретно, водными растворами (то есть растворы активного соединения, включающие воду в качестве растворителя). В данном контексте термин "водный раствор" включает композиции, в которых по меньшей мере 99% активного ингредиента находится в растворе примерно при 5°С и атмосферном давлении, и термин "суспензия" означает, что более 1% активного ингредиента не находится в растворе при таких условиях. Типичные дисперсионные агенты для суспензий представляют собой гидроксипропилметилцеллюлозу, АОТ (диоктилсульфосукцинат натрия), PVP (поливинилпирролидон) и SDS (додецилсульфат натрия). Могут быть возможны другие альтернативы.
В другом аспекте настоящего изобретения предлагается жидкая пероральная композиция, содержащая Соединения А или В, воду и по меньшей мере один дополнительный агент. Дополнительные агенты включают:
1) полиэтиленгликоль (PEG) и возможно также этанол, и/или винную кислоту, и/или лимонную кислоту, и/или соляную кислоту; или
2) хлорид натрия (который растворяют в композиции), и возможно также этанол; или
3) соляную кислоту и/или гидроксид натрия для доведения рН до подходящего значения (в частности, в диапазоне 3-8); или
4) DMA (диметилацетамид) и возможно также триглицерид со средней длиной цепи (такой как миглиол); или
5) β-циклодекстрин (такой как гидроксипропил-β-циклодекстрин);
6) модификатор тоничности, такой как хлорид натрия и/или маннит.
В дополнительном аспекте настоящего изобретения предложен пероральный раствор, содержащий Соединение А или В, воду и по меньшей мере один дополнительный агент, как перечислено выше в (1)-(6).
В другом аспекте изобретения предлагается водная композиция Соединения А или В, содержащая солюбилизирующий агент, такой как полиэтиленгликоль, β-циклодекстрин (такой как гидроксипропил-β-циклодекстрин), сорбит или этанол.
В дополнительном аспекте настоящего изобретения предлагается композиция в виде перорального раствора, содержащая Соединения А или В и этанол. Такая композиция может дополнительно содержать триглицерид со средней длиной цепи (такой как миглиол).
В еще одном дополнительном аспекте настоящего изобретения предлагается композиция в виде перорального раствора, содержащая соединение формулы (I) и DMA. Такая композиция может дополнительно содержать триглицерид со средней длиной цепи (такой как миглиол).
В дополнительном аспекте изобретения композиция по изобретению адаптирована, чтобы быть пригодной для парентерального введения. Термин "парентеральный" включает любой способ введения, который не включает пероральное введение в желудочно-кишечный тракт, и включает введение подкожно, внутривенно, внутриартериально, трансдермально, интраназально, внутрибуккально, внутрикожно, внутримышечно, внутрь липомы, внутрибрюшинно, ректально, сублингвально, местно, посредством ингаляции, или любым другим парентеральным путем.
Подходящие композиции по изобретению, которые предназначены для парентерального введения, включают композиции, в которых Соединение А или В присутствует вместе с водным носителем, таким как вода.
Композиция по настоящему изобретению, содержащая водный носитель, может дополнительно содержать один или более чем один эксципиент, такой как противомикробный консервант; модификатор тоничности (например, хлорид натрия, маннит или глюкоза); рН-корректирующий агент (например, обычная неорганичная кислота или основание, включая соляную кислоту или гидроксид натрия); агенты, регулирующий рН (то есть буфер; например винная кислота, уксусная кислота или лимонная кислота); поверхностно-активное вещество (например, додецилсульфат натрия (SDS) или Solutol™); солюбилизатор, который служит для содействия солюбилизации активного ингредиента (например, этанол, полиэтиленгликоль или гидроксипропил-β-циклодекстрин или поливинилхлорид (PVP)); или антиоксидант.
Парентеральные композиции могут находиться в форме суспензий активного ингредиента вместе с водным растворителем или, более конкретно, водными растворами (то есть растворы активного соединения, включающие воду в качестве растворителя). В данном контексте термин "водный раствор" включает композиции, в которых по меньшей мере 99% активного ингредиента находится в растворе при приблизительно 5°С и атмосферном давлении, и термин "суспензия" означает, что более 1% активного ингредиента не находится в растворе при таких условиях. Типичные дисперсионные агенты для суспензий представляют собой гидроксипропилметилцеллюлозу, АОТ, PVP и SDS, но возможны и другие варианты.
Число эксципиентов, используемых в пероральных и парентеральных композициях по изобретению, зависит от многих факторов, таких как природа и количество присутствующего активного ингредиента и количество включенного разбавителя/носителя (водного растворителя или иного).
В другом аспекте настоящего изобретения предложена парентеральная композиция, содержащая Соединение А или В, воду и по меньшей мере один дополнительный агент. Дополнительные агенты включают:
1) полиэтиленгликоль (PEG) и также возможно этанол, и/или винную кислоту, и/или соляную кислоту; или
2) хлорид натрия (который будет растворен в композиции), и возможно также этанол; или
3) соляную кислоту и/или гидроксид натрия для доведения рН до подходящего значения (в частности, в диапазоне 3,5-8); или
4) DMA (диметилацетамид) и возможно также триглицерид со средней длиной цепи (такой как миглиол); или
5) β-циклодекстрин (такой как гидроксипропил-β-циклодекстрин);
6) модификатор тоничности, такой как хлорид натрия и/или маннит.
В дополнительном аспекте настоящего изобретения предлагается инъецируемый раствор, содержащий Соединение А или В, воду и по меньшей мере один дополнительный агент, как перечислено выше в (1)-(6).
В другом аспекте изобретения предлагается водная композиция Соединения А или В, содержащая солюбилизирующий агент, такой как полиэтиленгликоль, β-циклодекстрин (такой как гидроксипропил-β-циклодекстрин), сорбит или этанол.
В дополнительном аспекте настоящего изобретения предлагается парентеральная композиция, содержащая Соединение А или В и этанол. Такая композиция может дополнительно содержать триглицерид со средней длиной цепи (такой как миглиол).
В еще одном дополнительном аспекте настоящего изобретения предлагается парентеральная композиция, содержащая соединение формулы (I) и DMA. Кроме того, данная композиция может содержать триглицерид со средней длиной цепи (такой как миглиол).
В еще одном дополнительном аспекте настоящего изобретения предлагается твердая композиция, содержащая микрокристаллическую целлюлозу и поливинилпирролидон (PVP); или содержащая микрокристаллическую целлюлозу и натрия крахмала гликолят.
Композиции по изобретению, например парентеральные композиции, содержащие соли, можно получать путем добавления разбавителя/носителя к подходящей предварительно приготовленной соли.
Композиции, содержащие активный ингредиент, можно также представить в твердой форме, пригодной для применения в получении композиции по изобретению (например, раствора, такого как водный раствор, например, для парентерального введения) ex tempore. Такие композиции могут находиться в форме твердого вещества, содержащего активный ингредиент, возможно в присутствии одного или более дополнительных эксципиентов, как определено выше в данном описании изобретения, и возможно вплоть до 10% (масс./масс.) разбавителя и/или носителя, как определено выше в данном описании, эти композиции упоминаются выше в данном описании изобретения как "твердые композиции по изобретению".
Твердые композиции по изобретению можно получать путем удаления разбавителя/носителя (например, растворителя) из композиции по изобретению или из концентрированной композиции по изобретению, которые могут, например, находиться в форме раствора, например водного раствора.
В другом аспекте настоящего изобретения предлагается перорально вводимая композиция с немедленным высвобождением, содержащая Соединение А или В, носитель (такой как микрокристаллическая целлюлоза), разрыхлитель (такой как натрия крахмала гликолят), связующее вещество (такое как поливинилпирролидон) и смазывающее вещество (такое как натрия стеарилфумарат). Такая композиция может также содержать дополнительный носитель (или наполнитель), такой как маннит.
Композиции по изобретению, которые находятся в форме таблеток с немедленным высвобождением, можно получать путем объединения активного ингредиента с разбавителем/носителем, используя стандартные методы, и применяя стандартное оборудование, известное специалисту, включающие влажное или сухое гранулирование, прямое прессование/уплотнение, сушку, измельчение, смешивание, таблетирование и нанесение покрытия, а также комбинации таких методов, например, как описано ниже. В одном аспекте изобретения соли присоединения кислоты соединений формулы (I) в кристаллической форме готовят в таблетках.
Таким образом, предложен способ для создания твердой композиции, пригодной для применения в получении препарата по изобретению (например, раствора, такого как водный раствор) ex tempore, включающий удаление разбавителя/носителя (например, растворителя) из композиции по изобретению, или концентрированной композиции по изобретению.
Растворитель можно удалять различными способами, известными специалистам в данной области техники, например выпариванием (при пониженном давлении или иным способом), лиофильной сушкой или любым способом удаления (высушивания) растворителя, который удаляет растворитель (такой как вода) с сохранением целостности активного ингредиента. Примером сушки является сублимационная сушка.
Таким образом, согласно дополнительному аспекту изобретения предлагается высушенная сублимацией (лиофилизированная) твердая композиция по изобретению.
Специалистам ясно, что при получении твердых композиций по изобретению можно добавлять подходящие дополнительные эксципиенты на подходящей стадии до удаления разбавителя/носителя. Например, в случае водных растворов, рН можно регулировать и/или корректировать, как описано выше в данном описании изобретения. Кроме того, можно добавлять подходящий дополнительный эксципиент с целью облегчения образования твердой композиции по изобретению в процессе удаления разбавителя/носителя (например, маннит, сахарозу, глюкозу, маннозу или трегалозу).
Твердая композиция Соединения А или В, таким образом, включает композицию, в которой содержание растворителя (например, воды), отличного от кристаллизационного растворителя, составляет не более 10%, например, менее 2% несвязанного растворителя, такого как вода.
Композиции по изобретению можно стерилизовать, например, посредством стерильной фильтрации или автоклавирования, и/или заполнять ими первичные упаковки, такие как флаконы, картриджи и предварительно заполняемые шприцы. Такие технологические операции также могут иметь место перед сушкой с образованием твердой композиции по изобретению.
Перед введением высушенную твердую композицию можно восстанавливать и/или разбавлять, например, в воде, физиологическом растворе, растворе глюкозы или любом другом подходящем растворе.
Количество разбавителя/носителя в пероральной (например, таблетка с немедленным высвобождением) композиции по изобретению зависит от многих факторов, таких как природа и количество используемого активного ингредиента и природа и количества любых других компонентов (например, дополнительных эксципиентов), присутствующих в композиции, но обычно составляет вплоть до 40% (масс./масс.), в частности вплоть до 30%, более конкретно вплоть до 20%, и в частности вплоть до 10% (масс./масс.) конечной композиции. Количество дополнительных эксципиентов в такой пероральной композиции по изобретению также зависит от таких факторов, как природа и количество используемого активного ингредиента, а также природы и количеств любых других компонентов (например, разбавителей/носителей и/или других дополнительных эксципиентов), которые присутствуют в композиции, но для смазывающих и скользящих веществ составляет обычно вплоть до 5% (масс./масс.), и для связующих веществ и разрыхлителей составляет обычно вплоть до 10% (масс./масс.) конечной композиции.
Композиции по изобретению вводят пациентам-млекопитающим (включая человека), и согласно дополнительному аспекту изобретения, таким образом, предлагается композиция по изобретению для применения в качестве лекарственного средства.
Согласно дополнительному аспекту изобретения предлагается фармацевтическая композиция, включающая соединение по изобретению в композиции с модифицированным высвобождением, как описано в международной патентной заявке WO 03/101424, релевантная часть описания которой включена в данную заявку посредством ссылки.
Специалистам очевидно, что термин фармацевтическая композиция "с модифицированным высвобождением" включает любую композицию/препарат, в которой(ом) начало и/или скорость высвобождения лекарственного средства изменены посредством галеновых манипуляций, и, таким образом, включает определение, предложенное в United States Pharmacopeia (USP XXII) на страницах xliii и xliv вступления/вводной части, релевантная часть данного документа включена в данную заявку посредством ссылки.
В данном случае, модифицированное высвобождение может быть обеспечено при помощи подходящего фармацевтически приемлемого носителя, и/или других средств, где носитель или средства (как это целесообразно) вызывают изменение начала и/или скорости высвобождения активного ингредиента. Таким образом, термин, как очевидно специалистами в данной области техники, включает композиции, которые адаптированы (например, как описано в данном описании изобретения) для обеспечения "замедленного", "пролонгированного" или "длительного" высвобождения лекарственного средства (когда лекарственное средство высвобождается со значительно замедленной скоростью с получением терапевтического ответа в течение требуемого периода времени, возможно, включая предоставление начального количества лекарственного средства, сделанного доступным в пределах заданного времени после введения для вызывания начального желаемого терапевтического отклика); композиции, которые обеспечивают "отсроченное" высвобождение лекарственного средства (когда высвобождение лекарственного средства отложено до достижения определенной области желудочно-кишечного тракта, после чего высвобождение лекарственного средства может быть либо пульсирующим, либо дополнительно модифицированным, как указано выше); а также так называемые композиции "повторного действия" (в которых одна доза лекарственного средства высвобождается либо немедленно, либо через некоторое время после введения, и следующие дозы высвобождаются позже).
Авторы изобретения отдают предпочтение композициям по изобретению, обеспечивающим отсроченное высвобождения или, более конкретно, замедленное (то есть пролонгированное или длительное) высвобождение лекарственного средства в течение периода времени. Более конкретные композиции по изобретению могут быть адаптированы (например, как описано в данном описании изобретения) для обеспечения достаточной дозы лекарственного средства в течение интервала времени между введением доз (независимо от количества доз в единицу времени) с получением желательного терапевтического эффекта. Высвобождение может быть равномерным и/или постоянным в течение длительного периода времени, или иным.
Композиции по изобретению могут, например, находиться в форме одного или более из следующего, все из которых хорошо известны специалистам в данной области техники.
А. Покрытые оболочкой гранулы, таблетки или капсулы, которые могут быть предназначены для высвобождения по меньшей мере некоторого количества лекарственного средства, когда интересующая композиция достигнет определенного участок желудочно-кишечного тракта. Такие таблетки могут, например, быть снабжены некоторой разновидностью покрытия, устойчивого в желудочной среде, такого как слой кишечно-растворимого покрытия, обеспечивая высвобождение по меньшей мере части лекарственного средства, присутствующего в композиции, в конкретной части желудочно-кишечного тракта, такой как участки кишечника.
Б. Сложные единицы или многочастичные системы, которые могут находиться в форме микрочастиц, микросфер или гранул, содержащих лекарственное средство (причем сложные единицы/многочастичные системы могут обеспечивать постепенное перемещение композиции, содержащей лекарственное средство, из желудка в двенадцатиперстную кишку и далее через тонкий и толстый кишечник с высвобождением лекарственного средства с заданной скоростью).
В. Композиции, содержащие дисперсии или твердые растворы активного соединения в матрице, которые могут находиться в форме воска, камеди или жира, или, в частности, в форме полимера, где высвобождение лекарственного средства происходит путем постепенного разрушения поверхности таблетки и/или диффузии.
Г. Системы, которые содержат биоадгезивный слой, который может обеспечивать пролонгированное удерживание композиции по изобретению в определенном участке желудочно-кишечного тракта (например, желудке). Они включают плавающие или погружающиеся системы (то есть системы низкой и высокой плотности соответственно), а также так называемые системы "с расширением объема".
Д. Так называемые "висячие" устройства, в которых лекарственное средство присоединено к ионообменной смоле, которая обеспечивает постепенное высвобождение лекарственного средства под воздействием других ионов, присутствующих в желудочно-кишечном тракте, например кислотной среды желудка.
Е. Устройства, в которых скорость высвобождения лекарственного средства контролируют с помощью его химического потенциала (например, осмотический насос).
Ж. Системы, в которых лекарственное средство высвобождается посредством диффузии через мембраны, включающие многослойные системы.
З. Устройства, которые действуют в соответствии с внешним сигналом с высвобождением небольшого количества лекарственного средства.
И. Активные, самопрограммируемые системы, которые могут содержать чувствительный элемент, который реагирует на конкретную биологическую среду изменением доставки лекарственного средства.
К. Силастиковые депо с контролируемым высвобождением, которые высвобождают лекарственное средство в зависимости от диффузии воды и/или желудочно-кишечных жидкостей в устройство через входное/выходное отверстие, что приводит к растворению и последующему высвобождению лекарственного средства.
Вышеуказанные принципы подробно обсуждаются в ссылках на известный уровень техники, включая Pharmaceutisch Weekblad Scientific Edition, 6, 57 (1984); Medical Applications of Controlled Release, Vol II, eds. Langer and Wise (1984) Bocaraton, Florida, на страницах 1-34; Industrial Aspects of Pharmaceuticals, ed. Sandel, Swedish Pharmaceutical Press (1993) на страницах 93-104; и страницах 191-211 "Pharmaceutics: The Science of Dosage Form Design", ред. M.Е.Aulton (1988) (Churchill Livingstone); а также ссылках, указанных в вышеупомянутых документах, содержание всех этих документов включено в данное описание избретения посредством ссылки.
В дополнительном аспекте изобретения предлагается композиция с модифицированным высвобождением, которая содержит гелеобразующую матрицу. Матрица, в частности, содержит гидроксипропилметилцеллюлозу (НРМС), йота-каррагинан, натрия додецилсульфат (SDS) и/или ксантановую камедь. Более конкретно матрица содержит гидроксипропилметилцеллюлозу (НРМС), йота-каррагинан и/или РЕО (полиэтиленоксид). НРМС может представлять собой одну или смесь двух или более НРМС с разными вязкостями или молекулярными массами (как описано ниже).
В изобретении также предлагается композиция с модифицированным высвобождением, содержащая одну или более НРМС и один или более чем один дополнительный компонент, выбранный из группы, содержащей: йота-каррагинан, микрокристаллическую целлюлозу, смазывающее вещество (такое как натрия стеарилфумарат) или маннит.
Кроме того, в дополнительном аспекте, в изобретении предлагается композиция с модифицированным высвобождением, содержащая ксантановую камедь; или содержащая йота-каррагинан и РЕО (как описано ниже).
Подходящие композиции с модифицированным высвобождением можно таким образом получать согласно стандартным фармацевтическим методам, как описано в данном описании изобретения или в вышеупомянутых документах, и/или хорошо известными методами.
В частности, в композициях по изобретению активный ингредиент предложен вместе с фармацевтически приемлемым носителем. Конкретная композиция по изобретению представлена в форме активного ингредиента в полимерной матрице.
В этом отношении, в частности, предложены композиции по изобретению для перорального введения в форме так называемой "набухающей" системы с модифицированным высвобождением, или системы с модифицированным высвобождением "гелеобразующая матрица", в которых активный ингредиент представлен вместе с полимером, который набухает в водной среде (то есть "гидрофильный гелеобразующий компонент"). Следует понимать, что термин "водная среда" в данном контексте включает воду и жидкости, которые представляют собой жидкости, или которые близки к жидкостям, присутствующим в желудочно-кишечном тракте млекопитающего. Такие полимерные системы обычно содержат гидрофильные макромолекулярные структуры, которые в сухой форме могут находиться в стеклообразном, или по меньшей мере частично кристаллическом состоянии, и которые набухают при соприкосновении с водной средой.
Таким образом, модифицированное высвобождение лекарственного средства осуществляют посредством одного или более из следующих процессов: транспорт растворителя в полимерную матрицу, набухание полимера, диффузия лекарственного средства через набухший полимер и/или разрушение полимера, один или более из которых может служить для медленного высвобождения лекарственного средства из полимерной матрицы в водную среду.
Таким образом, пригодные полимерные вещества (действующие в качестве носителей), которые можно применять в качестве гидрофильного гелеобразующего компонента композиции с модифицированного высвобождения «гелеобразующая матрица», включают полимеры с молекулярной массой примерно 5000 г/моль, и которые или:
а) являются по меньшей мере умеренно растворимыми в водной среде; или
б) набухают при помещении в контакт с водной средой
(как описано выше в данном описании изобретения), тем самым облегчая высвобождение лекарственного средства из носителя.
Таким образом подходящие полимеры для гелеобразующей матрицы, которые могут быть синтетическими или натуральными, включают полисахариды, такие как мальтодекстрин, ксантан, склероглюкан, декстран, крахмал, альгинаты, пуллулан, гиалуроновую кислоту, хитин, хитозан и подобные; другие натуральные полимеры, такие как протеины (альбумин, желатин и так далее), поли-L-лизин; полиакрилат натрия; поли(гидроксиалкилметакрилаты) (например, поли(гидроксиэтилметакрилат)); карбоксиполиметилен (например, Carbopol™); карбомер; поливинилпирролидон; камеди, такие как гуаровая камедь, аравийская камедь, камедь карайи, камедь гхатти, камедь плодов рожкового дерева, камедь тамаринда, геллановая камедь, камедь трагаканта, агар, пектин, глютен и тому подобное; поливиниловый спирт; этиленвиниловый спирт; полиэтиленоксид (РЕО); и эфиры целлюлозы, такие как гидроксиметилцеллюлоза (НМС), гидроксиэтилцеллюлоза (НЕС), гидроксипропилцеллюлоза (НРС), метилцеллюлоза (МС), этилцеллюлоза (ЕС), карбоксиэтилцеллюлоза (СЕС), этилгидроксиэтилцеллюлоза (ЕНЕС), карбоксиметилгидроксиэтилцеллюлоза (СМНЕС), гидроксипропилметилцеллюлоза (НРМС), гидроксипропилэтилцеллюлоза (НРЕС) и натрий-карбоксиметилцеллюлоза (Na CMC); а также сополимеры и/или (простые) смеси любых вышеуказанных полимеров. Некоторые из вышеуказанных полимеров могут быть дополнительно сшиты стандартными методами.
Авторы изобретения предпочитают, чтобы для композиций по изобретению в форме гелеобразующих матричных систем основной используемый набухающий полимер представлял собой НРС, мальтодекстрин, склероглюкан или карбоксиполиметилен, более конкретно РЕО или ксантан, и особенно НРМС, а также сополимеры и/или (простые) смеси любых таких полимеров. Альтернативой является йота-каррагинан.
Если используют РЕО, ксантан и НРМС в (то есть в качестве по меньшей мере одного из полимеров) гидрофильном гелеобразующем компоненте, то предпочтительные молекулярные массы (то есть средние молекулярные массы, которые определяют стандартными методами, такими как осмометрия, гель-фильтрационная хроматография с рефракционным детектором (где молекулярную массу определяют при помощи стандартных калибровочных кривых), рассеяние света и/или методы ультрацентрифугирования), для таких полимеров находятся в диапазоне от 5000 г/моль вплоть до 200000000 г/моль, например, вплоть до 100000000 г/моль, в частности вплоть до 25000000 г/моль, и более конкретно вплоть до 20000000 г/моль. Можно использовать смеси РЕО, ксантана и НРМС-полимеров с разными молекулярными массами в данных диапазонах.
Подходящие НРМС-полимеры также включают те, которые образуют 2% масс./масс. растворы полимера в воде с вязкостями, при измерении стандартными методами, такими как описаны в общем в United States Pharmacopeia XXIV (USP XXIV/NF19) на странице 2002 и след., а также, конкретно, на страницах 843 и 844 (релевантная часть описания изобретения этого документа включена в данную заявку посредством ссылки), от 3 до 150000 спз (3-150000 мПа·с) (при 20°С), например от 10 до 120000 сП (10-120000 мПа·с), в частности от 30 до 50000 сП (30-50000 мПа·с) и более конкретно от 50 до 15000 сП (50-15000 мПа·с). Можно использовать смеси полимеров НРМС с разными вязкостями в этих диапазонах для того, чтобы, например, получать смеси НРМС, которые дают растворы, как указано выше, со "средними" вязкостями (то есть вязкости для смеси) в вышеуказанных предпочтительных диапазонах. Аналогично можно использовать смеси НРМС-полимеров (с вязкостями и/или "средними" вязкостями в этих диапазонах) с другими вышеуказанными полимерами. Подходящие НРМС-полимеры включают полимеры, соответствующие стандартным типам замещения 2208, 2906, 2910 и 1828 согласно United States Pharmacopeia (смотри USP XXIV/NF19 относительно дополнительных сведений). Таким образом, подходящие НРМС-полимеры включают полимеры, реализуемые под торговой маркой METHOCEL™ (Dow Chemical Corporation) или торговой маркой METOLOSE™ (Shin-Etsu).
Подходящие ксантановые полимеры включают те, которые обеспечивают 1% масс./масс. растворы полимера в воде с вязкостями, при измерении стандартными способами, такими, как описано в общем в United States Pharmacopeia XXIV (USP XXIV/NF19) на странице 2002 и далее, а также, конкретно, на страницах 2537 и 2538 (релевантная часть данного документа включена в данное описание изобретения посредством ссылки), от 60 до 2000 сП (60-2000 мПа·с) (при 24°С), например от 600 до 1800 сП (600-1800 мПа·с) и, в частности, от 1200 до 1600 сП (1200-1600 мПа·с). Можно использовать смеси ксантановых полимеров с разными вязкостями в данных диапазонах с целью, например, получения ксантановых смесей, которые дают растворы, как указано выше, со "средними" вязкостями (то есть вязкостью смеси) в указанных выше предпочтительных диапазонах. Аналогично можно использовать смеси ксантановых полимеров (с вязкостями и/или "средними" вязкостями в данных диапазонах) с другими вышеуказанными полимерами. Подходящие ксантановые полимеры включают полимеры, реализуемые под торговыми марками XANTURAL™ и KELTROL™ (CPKelco), и SATIAXANE™ (Degussa, Texturant Systems).
Выбор полимера определяется природой активного ингредиента/лекарственного средства, которое используют в композиции по изобретению, а также требуемой скоростью высвобождения. В частности, специалисту очевидно, например в случае НРМС, что более высокая молекулярная масса, в общем случае обеспечивает более низкую скорость высвобождения лекарственного средства из композиции. Кроме того, в случае НРМС разные степени замещения метоксигрупп и гидроксипропоксигрупп вызывают изменения скорости высвобождения лекарственного средства из композиции. В этом отношении и как указано выше, может быть желательно представить композиции по изобретению в форме гелеобразующих матричных систем, в которых полимерный носитель получен из смеси двух или более полимеров, например с разными молекулярными массами, например, как описанные выше в данном описании изобретения, с целью получения конкретного требуемого или желательного профиля высвобождения.
Авторы изобретения также обнаружили, что скорость высвобождения лекарственного средства из композиций по изобретению в форме гелеобразующих матричных систем можно также контролировать путем регулирования соотношения лекарственное средство: полимер и соотношения площадь поверхности: объем в отдельных композициях (например, таблетках), содержащих лекарственное средство и систему полимерных носителей.
Композиции по изобретению, либо в форме гелеобразующей матричной системы, либо в иной форме, могут содержать один или более чем один дополнительный эксципиент (в дополнение к системе полимерных носителей) для дополнительной модификации высвобождения лекарственного средства, для улучшения физических и/или химических свойств конечной композиции, и/или для облегчения способа изготовления. Такие эксципиенты являются стандартными в изготовлении композиций с модифицированным высвобождением.
Например, композиции по изобретению могут содержать один или более чем один из следующих разбавителей: фосфат кальция (однозамещенный фосфат кальция, двухзамещенный фосфат кальция и трикальцийфосфат), лактозу, микрокристаллическую целлюлозу, маннит, сорбит, диоксид титана, силикат алюминия и подобные. Конкретные разбавители включают микрокристаллическую целлюлозу, а также маннит.
Композиции по изобретению могут содержать одно или более из следующих смазывающих веществ: стеарат магния, стеарилфумарат натрия и тому подобное.
Композиции по изобретению могут содержать скользящее вещество, такое как коллоидный диоксид кремния.
Композиции по изобретению могут содержать одно или более из следующих связующих веществ: поливинилпирролидон, лактозу, маннит, микрокристаллическую целлюлозу, полиэтиленгликоль (PEG), HPMC с низкой молекулярной массой, МС с низкой молекулярной массой, НРС с низкой молекулярной массой и тому подобное. Конкретные связующие вещества включают микрокристаллическую целлюлозу.
Композиции по изобретению могут содержать один или более чем один из следующих агентов, регулирующих рН: органические кислоты (например, лимонную кислоту и тому подобное) или их соли щелочных металлов (например, натрия), фармацевтически приемлемые соли (например, соли натрия, магния или кальция) неорганических кислот (таких как угольная кислота или фосфорная кислота), оксиды магния, а также сульфаты, метабисульфаты, пропионаты и сорбаты щелочных и щелочноземельных металлов (например, натрия, кальция, калия и подобных).
Другие дополнительные эксципиенты могут включать красители, вкусоароматизаторы, солюбилизирующие агенты (такие как SDS), покрывающие вещества, консерванты и так далее.
Можно использовать комбинации вышеупомянутых дополнительных эксципиентов.
Очевидно, что некоторые из указанных выше дополнительных эксципиентов, которые могут присутствовать в конечной композиции по изобретению, могут иметь более чем одну вышеупомянутую функцию. Кроме того, дополнительные эксципиенты, указанные выше, могут также действовать как часть гидрофильного гелеобразующего компонента в гелеобразующей матричной системе.
Общее количество дополнительных эксципиентов (не включая, в случае гелеобразующих матричных систем, основной(ые) полимерный(ые) носитель(носители)), которые могут присутствовать в композиции по изобретению, зависит от природы композиции, также от природы и количества других компонентов данной композиции, и может составлять вплоть до 85%, например от 0,1 до 75%, например от 0,2 до 65%, в частности от 0,3 до 55%, более конкретно от 0,5 до 45% и особенно 1-40%, например от 2 до 35% масс./масс. В любом случае выбор и количество эксципиента(ов) может быть определено специалистом обычным путем (то есть без обращения к изобретательскому вкладу).
В гелеобразующих матричных системах количество полимера в системе должно быть достаточным, для обеспечения дозы лекарственного средства в интервале между приемами лекарственного средства, достаточной для получения требуемого терапевтического эффекта. Таким образом, для гелеобразующей матричной системы авторы изобретения предпочитают такую, где требуется по меньшей мере 2 часа (в частности, по меньшей мере 4 часа, главным образом по меньшей мере 6 часов) для высвобождения 80% (главным образом 60%) от начального содержания лекарственного средства в композиции у пациента после введения в условиях тестирования, описанных ниже, и в частности за период от 8 до 24 часов. Более конкретно, по меньшей мере 80% от начального содержания лекарственного средства в композиции высвобождается за период времени приблизительно от 8 до 24 часов. Подходящие количества полимера, который может быть включен, которые будут зависеть inter alia от активного ингредиента, используемого в композиции, любые эксципиенты, которые могут присутствовать, и природа используемого полимера, находятся в диапазоне 5-99,5%, например 10-95%, в частности 30-80% масс./масс. В любом случае, выбор и количество полимера может быть определено специалистом обычным способом.
В другой конкретном препарате авторы изобретения предпочитают, чтобы соединения по изобретению были приготовлены в виде препарата вместе с гелеобразующей матричной композицией, содержащей йота-каррагинан и один или более нейтральных гелеобразующих полимеров.
Йота-каррагинан присутствует, в частности, в таком конкретном препарате в количестве более 15% по массе. Конкретные категории йота-каррагинана включают йота-каррагинан фармацевтической категории (например, имеющийся у FMC Biopolymer), который имеет вязкость не менее 5 сантипуаз (сП) (5 мПа·с), в частности в диапазоне 5-10 сП (5-10 мПа·с) (для 1,5% раствора, нагретого до 82°С, после чего измеряют вязкость при 75°С с помощью вискозиметра Brookfield LV, оснащенного валом №1, вращающимся со скоростью 30 об/мин), и йота-каррагинан технической категории (например, имеющийся у Fluka Biochemica), который, в частности, имеет вязкость не менее 14 мПа·с, для 0,3% водного раствора, нагретого до 20°С, после чего измеряют вязкость, используя вискозиметр типа Haake с падающим шариком, используемый вместе с термостатом Lauda C3 и Hakke Mess-System III, и используя шарики из нержавеющей стали, покрытые золотом, с плотностью 7,8 г/см3.
Нейтральный гелеобразующий полимер может представлять собой один нейтральный полимер, или смесь более чем одного нейтральных полимеров, обладающий(их) гелеобразующими свойствами и растворимостью, по существу не зависящей от рН. Нейтральный гелеобразующий полимер присутствует, в частности, в композиции в количестве более 10%, но, в частности, более 20% по массе.
Подходящие нейтральные гелеобразующие полимеры включают полиэтиленоксид (РЕО), производные и члены группы РЕО (например, полиэтиленгликоль (PEG)), особенно существующие в природе в твердом состоянии, с подходящей молекулярной массой или вязкостью. При использовании в качестве единственного нейтрального гелеобразующего полимера, РЕО, в частности, имеет MW (молекулярную массу), больше или равную 4 миллионам (4М), соответствующую диапазону вязкости водного раствора 1650-5500 мПа·с (или 1650-5500 сП; измеренную для 1% водного раствора при 25°С, используя вискозиметр Brookfield RVF, с валом №2, при 2 об/мин). Другие примеры подходящих РЕО включают РЕО с MW приблизительно 5 миллионов (5М), соответствующей диапазону вязкости водного раствора 5500-7500 мПа·с, или РЕО с MW приблизительно 8 миллионов (8М), соответствующей диапазону вязкости водного раствора 10000-15000 мПа·с. Этот диапазон охватывает величину обычной вязкости раствора (в сП), измеренную при 25°С, приведенную для данного полимера в USP 24/NF 19, выпуск 2000, стр.2285-2286. Если PEG используют в качестве единственного нейтрального гелеобразующего полимера, он, в частности, имеет высокую молекулярную массу, например MW приблизительно 20000, соответствующую диапазону вязкости 2700-3500 мПа·с (или 2700-3500 сП), измеренной с использованием 50%-ного водного раствора (масс./масс.) при 20°С, с применением капиллярного вискозиметра (Ubbelohde или эквивалентный). [Ref: European Pharmacopoeia 3rd Ed., 2000, Supplement, стр.908-909].
Другие подходящие нейтральные гелеобразующие полимеры включают производные целлюлозы, такие как гидроксипропилметилцеллюлоза (НРМС) или гидроксиэтилцеллюлоза (НЕС) с подходящими высокими значениями вязкости (например, "НРМС 50 сП", "НРМС 10000 сП", "НРМС 15000 сП", "НЕС тип НН" или "НЕС тип Н"). При использовании в качестве единственного нейтрального полимера гидроксипропилметилцеллюлозные полимеры, такие как "НРМС 10000 сП" и "НРМС 15000 сП", имеют, соответственно, кажущиеся вязкости 7500-14000 мПа·с (или 7500-14000 сП) и 11250-21000 мПа·с (или 11250-21000 сП) при измерении при 20°С с 2%-ным (масс./масс.) водным раствором, в расчете на сухое вещество, с использованием капиллярного вискозиметра (Ubbelohde или эквивалентного). Один тип гидроксиэтилцеллюлозного полимера, например "Natrosol 250 Pharma, тип НН" от Hercules Incorporated (Аквалон), обычно демонстрирует вязкость на Brookfield примерно 20000 мПа·с, с использованием устройства Brookfield Synchro-Lectric Model LVF, в условиях концентрации раствора 1%, вал №4, скорость вала 30 об/мин, коэффициент 200, 25°С (смотри Natrosol Physical and Chemical Properties booklet, 33.007-E6 (1993), стр.21).
Конкретные композиции, которые можно упомянуть, включают те, в которых соединения по изобретению приготовлены вместе с йота-каррагинаном и НРМС (10000 сП (10000 мПа·с)) в отношении 50:50 (масс.%), или вместе с йота-каррагинаном и НРМС (50 сП (50 мПа·с)) & НРМС (10000 сП (10000 мПа·с)) в отношении 35:60:5 (масс.%), или вместе с йота-каррагинаном и РЕО 4М в отношении 50:50 (масс.%). Конкретные дополнительные эксципиенты в таких композициях включают смазывающие вещества, такие как натрия стеарилфумарат.
В одном аспекте изобретения предложена неинъецируемая композиция по изобретению, содержащая Соединение А или В; НРМС и смазывающее вещество (такое как натрия стеарилфумарат). В дополнительном аспекте композиция может содержать смесь 2 или более НРМС с разными вязкостями (например, 10000 сП (10000 мПа·с) и 50 сП (50 мПа·с)). Кроме того, композиция может дополнительно содержать солюбилизирующий агент [такой как натрия додецилсульфат (SDS), натрия лаурилсульфат или полиокси-40 гидрогенизированное касторовое масло].
Подходящие количества активного ингредиента в композициях по изобретению, либо в форме гелеобразующих матричных систем или в другой форме, зависят от многих факторов, таких как природа этого ингредиента (свободное основание/соль и так далее), дозы, которая требуется, и природы, и количеств других компонентов композиции. Однако они могут находиться в диапазоне 0,5-80%, например 1-75%, таком как 3-70%, в частности 5-65%, более конкретно 10-60% и главным образом 15-55% масс./масс. В любом случае количество активного ингредиента, подлежащего включению, может быть определено специалистом обычным образом.
Обычная суточная доза Соединения А или В находится в диапазоне 0,001-100 мг свободного основания на кг массы тела, независимо от количества индивидуальных доз, которые вводят в течение этих суток. Конкретная суточная доза находится в диапазоне 20-600 мг.
Композиции по изобретению, такие как описанные выше в данном описании изобретения, могут быть изготовлены в соответствии с хорошо известными способами, такими как описанные в документах, указанных выше в данном описании изобретения. Композиции по изобретению, которые находятся в форме гелеобразующих матричных систем, можно получать стандартными методами и применяя стандартное оборудование, известное специалистам, включая влажное или сухое гранулирование, прямое прессование/сжатие, сушку, измельчение, смешивание, таблетирование и покрытие, а также комбинации этих способов, например, как описано ниже.
Хотя композиции по изобретению, в частности, адаптированы для перорального введения, их применение не ограничено этим способом введения. Парентеральные композиции с модифицированным высвобождением по изобретению, которые могут включать системы, хорошо известные специалистам в данной области техники, например системы на основе полоксамеров, биоразлагаемых микросфер, липосом, суспензий в маслах и/или эмульсий, могут быть получены стандартными методами, например, как описано в Leung et al. "Controlled Drug Delivery: Fundamentals and Applications" (Drugs and the Pharmaceutical Sciences; vol.29), 2nd edition, eds. Robinson and Lee, Dekker (1987) at Chapter 10, страница 433, описание данного документа включено в данную заявку посредством ссылки.
Композиции по изобретению можно принимать один или более раз в сутки (в частности, один, но не более чем два раза в сутки), независимо от количества отдельных единиц (препаратов/композиций), которые вводят как часть одной "дозы".
Композиции по изобретению вводят млекопитающим пациентам (включая человека) и, согласно дополнительному аспекту изобретения, таким образом, предложен препарат по изобретению для применения в качестве лекарственного средства.
Соединения по изобретению являются полезными, поскольку они метаболизируются в организме после введения с образованием соединений, которые обладают фармакологической активностью. Вследствие этого они показаны в качестве фармацевтических средств, и, в частности, в качестве пролекарств фармакологически активных соединений.
В частности, соединения по изобретению метаболизируются в организме с образованием эффективных ингибиторов тромбина, например, как показано в исследованиях, описанных в WO 02/44145. Ожидается, что соединения по изобретению будут полезными в лечении состояний, где требуется или желательно ингибирование тромбина, включая описанные в WO 02/44145, описание данного документа включено в данную заявку посредством ссылки. В Примере 46 WO 02/44145 раскрыто, что соединения из Примера 3 WO 02/44145 (то есть Соединение С, раскрытое в данном описании изобретения) исследовали в Тесте А WO 02/44145, и было обнаружено, что оно демонстрирует значения IC50TT менее 0,02 мкМ. В Примере 47 из WO 02/44145 раскрыто, что соединения из Примера 3 в WO 02/44145 (то есть Соединение С, раскрытое в данном описании изобретения) исследовали в Тесте D в WO 02/44145, и было обнаружено, что оно демонстрирует значение IC50 APTT менее 1 мкМ. Соединение С образуется посредством метаболизма (in vivo) соединений по настоящему изобретению.
Подходящие дозы соединения по изобретению в терапевтическом и/или профилактическом лечении млекопитающих пациентов, особенно человека, находятся в диапазоне 2-600 мг (в частности в диапазоне 20-500 мг) в сутки при пероральном введении, и 1-100 мг в сутки при парентеральном введении, и/или 0,003-60 мг/кг, в частности 0,03-15 мг/кг массы тела в сутки при пероральном введении, и 0,0015-30 мг/кг, в частности 0,015-7,5 мг/кг массы тела при парентеральном введении.
Согласно дополнительному аспекту изобретения предложен способ лечения состояния, где требуется или желательно ингибирование тромбина, включающий введение терапевтически эффективного количества соединения по изобретению пациенту, нуждающемуся в таком лечении. Во избежание сомнений, в "лечение" авторы изобретения включают терапевтическое лечение, а также профилактику, состояния (такого как сердечно-сосудистые расстройства, включая венозный тромбоз, легочную эмболию, артериальный тромбоз, системную эмболию и мерцательную аритмию). Также предложен способ введения соединения С путем введения соединения по изобретению, и способ применения соединения по изобретению для введения соединения С.
Также предложено соединение по изобретению для применения в качестве лекарственного средства, в частности для применения в лечении сердечно-сосудистого расстройства, такого как венозный тромбоз, легочная эмболия, артериальный тромбоз, системная эмболия или мерцательная аритмия.
Преимуществами соединений по изобретению является то, что они находятся в форме, которая обеспечивает упрощенное обращение. Кроме того, преимуществом соединений по изобретению является то, что они могут быть получены в формах, которые имеют улучшенную химическую стабильность и твердотельную стабильность твердого тела (включая более низкую гигроскопичность). Таким образом, соединения могут оставаться стабильными при хранении в течение длительных периодов времени.
Также преимуществом соединений по изобретению может являться то, что они могут кристаллизоваться с хорошими выходами, с более высокой чистотой, более удобно и/или дешевле, чем формы Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) или Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH), полученные ранее.
Согласно дополнительному аспекту изобретения предложено соединение, получаемое согласно любым способам или Примерам, описанным в данном описании изобретения.
Примеры
Данное изобретение проиллюстрировано, но ни в коей мере не ограничено, следующими Примерами и ссылкой на прилагаемые Фигуры.
Могут быть использованы следующие аббревиатуры:
Aze = (S)-азетидин-2-карбоксилат (если не указано иное)
Boc = трет-бутоксикарбонил
СВА = пара-цианобензиламин
Pab = пара-амидинобензиламино
EDC = 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид
TBTU = [N,N,N',N'-тетраметил-O-(бензотриазол-1-ил)урония тетрафторборат]
SMB = хроматографическая система с имитацией подвижного слоя
Общие методики
Анализ дифракции рентгеновских лучей на порошке (XRPD) выполняли на образцах, полученных согласно стандартным методам, например, описанным в Giacovazzo, С. et al. (1995), Fundamentals of Crystallography, Oxford University Press; Jenkins, R. and Snyder, R.L. (1996), Introduction to X-Ray Powder Diffractometry, John Wiley & Sons, New York; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; или Klug, H.P. & Alexander, L.E. (1974), X-ray Diffraction Procedures, John Wiley and Sons, New York. Рентгеновский анализ выполняли, используя дифрактометр Siemens D5000 и/или Philips X'Pert PRO. Некоторые образцы получали также путем перемешивания осадка с образованием суспензии и затем, с использованием пастеровской пипетки, отбирая приблизительно 0,2 мл суспензии и помещая ее на силиконовый держатель с нулевым фоном. Данную суспензию затем анализировали стандартным образом, и влажную, и затем также после сушки. Образцы, полученные данным образом, более предрасположены к "предпочтительным ориентационным эффектам", чем образцы, полученные с использованием сухого образца стандартными способами.
Дифференциальную сканирующую калориметрию (DSC) выполняли, используя прибор Mettler DSC820 или Perkin Elmer DSC7, согласно стандартным методам, например описанным в Hohne, G.W.H. et al. (1996), Differential Scanning Calorimetry, Springer, Berlin.
Термогравиметрический анализ (TGA) выполняли, используя прибор Mettler Toledo TGA850 или Perkin Elmer TGA7.
Формы, полученные согласно Примерам ниже, демонстрировали "по существу" аналогичные картины дифракции XRPD, и/или термограммы DSC и/или TGA, как и другие Примеры, раскрытые ниже, когда, как это было ясно из соответствующих картин/термограмм (с учетом ошибки эксперимента), образовалась та же самая кристаллическая форма. Таким образом, начальные температуры DSC могут варьироваться в диапазоне ±5°С (например, ±2°С), и значения расстояний XRPD могут варьироваться в диапазоне ±2 в последнем десятичном разряде. Следует заметить, что некоторые образцы XRPD демонстрировали такие сильные предпочтительные ориентационные эффекты, что интенсивность некоторых пиков была полностью уменьшена, в то время как другие были значительно усилены. Дифрактограммы, показанные на Фигурах, представляют собой тип, демонстрирующий меньшие предпочтительные эффекты ориентации.
Получение исходных веществ
Аморфное Соединение А и аморфное Соединение В могут быть получены, как описано, например способами, описанными в WO 02/44145 (релевантные разделы которой включены в данную заявку посредством ссылки). Соответствующие исходные вещества и промежуточные продукты могут быть получены, например, как описано в релевантных разделах WO/054168 и WO 06/090153 (релевантные разделы которых также включены в данную заявку посредством ссылки).
Аморфные Соединения А могут быть получены согласно следующей общей схеме 1
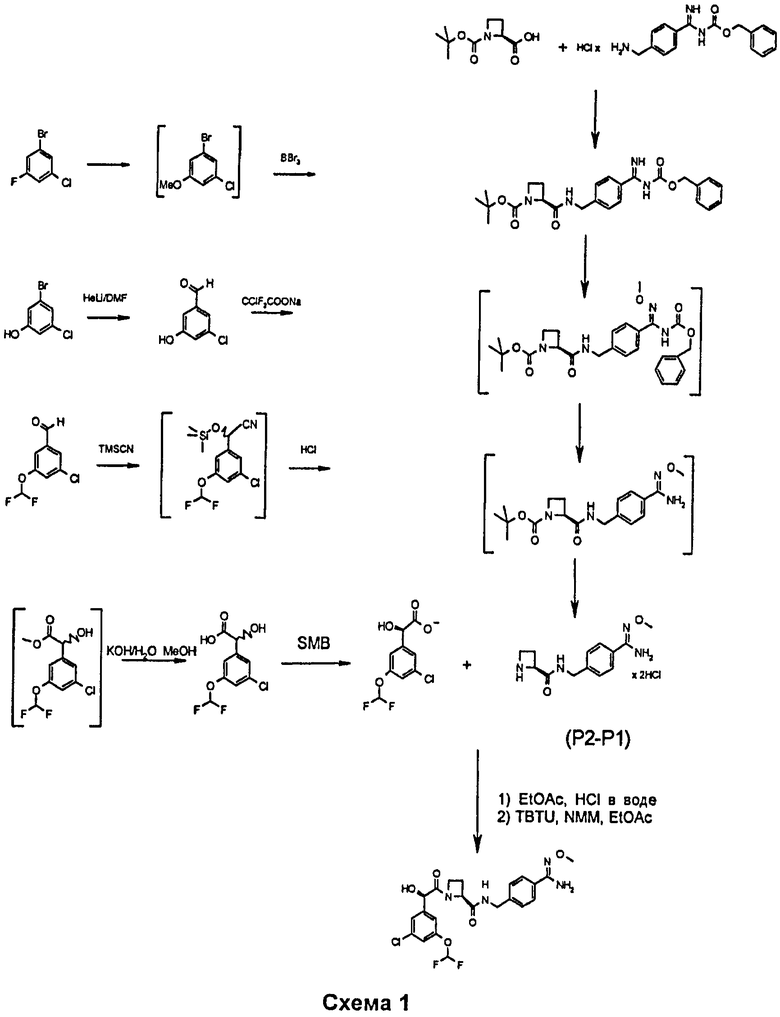
Последнее сочетание также выполняли в растворителе дихлорметане приблизительно при 22°С, используя DIPEA и TBTU (замещенная миндальная кислота: 1,08 кг (4,16 моль); (Р2-Р1): 1,40 кг (4,16 моль); дихлорметан: 16,5 л; DIPEA: 2,1 л (12,27 моль); TBTU: 1,37 кг (4,16 моль)). Обработку выполняли путем добавления EtOAc (22 л), концентрирования в вакууме и экстракции, используя воду (17 л + 17,5 л + EtOAc 16 л) и промывая полунасыщенным раствором карбоната натрия (8,5 л). Неочищенное вещество очищали посредством хроматографии, используя колонку, заполненную SiO2 (кондиционированную элюентом дихлорметан/метанол: 20/1), и элюировали элюентом дихлорметан/метанол: 20/1. После концентрирования в вакууме объединенных фракций продукта остаток растворяли в метаноле, фильтровали, концентрировали и сушили.
Аморфное Соединение В может быть получено путем сочетания 3-хлор-5-дифторметоксиминдальной кислоты (пролинамидная соль - смотри структурную формулу ниже) с Aze-CBA гидрохлоридной солью (которую можно получить путем удаления защиты Boc-Aze-CBA - смотри структурную формулу ниже).

3-хлор-5-дифторметоксиминдальная кислота (пролинамидная соль)

Сочетание выполняли следующим образом.
Пролинамидную соль 3-хлор-5-дифторметоксиминдальной кислоты (5,699 кг, 15,54 моль) добавляли к EtOAc (28,5 л) при температуре ниже 7°С, и затем добавляли HCl (3,6%, 28 л), поддерживая температуру ниже 5°С. После перемешивания в течение 35 минут слои отделяли, и водный слой экстрагировали, используя EtOAc (27 л). Оба органических слоя объединяли, фильтровали через MgSO4 (2,1 кг) и промывали EtOAc (16 л). К высушенному органическому слою добавляли Aze-CBA.HCl (3,91 кг, 15,54 моль) при температуре ниже 22°С; DIPEA: 8 л (46,62 моль) и TBTU: 7,49 кг (23,31 моль), и смесь перемешивали в течение приблизительно 17 часов приблизительно при 22°С.
Смесь промывали водой (2×29 л), 2×Na2CO3 (27 л + 29,5 л), 2×HCl (3,6%; 29 л + 28,6 л), 2×NaCl (12%; 28,2 л + 28,5 л), затем во время экстракции NaCl (23,1%: 2 л). Промывание повторяли с NaHCO3 (7,4%; 2 л) и NaCl (12%; 28 л), затем во время экстракции с NaCl (23,1%; 3 л). К органическому объему (37 л) добавляли EtOAc (18 л), и 20 л отгоняли. Добавляли метилциклогексан (35 л), смесь охлаждали до 3°С, фильтровали через приблизительно 16,5 часов, и осадок на фильтре промывали смесью EtOAc/метилциклогексан (1/1; 6 л каждого) с получением 5,147 кг твердого продукта.
Затем получали аморфное Соединение В путем превращения цианогруппы объединенного продукта 3-хлор-5-дифторметоксиминдальной кислоты-Aze-CBA в требуемую гидроксил-амидиновую функциональную группу путем использования NH2OH·HCl и основания Хюнига (или DIPEA или триэтиламина) в этаноле (при температуре ниже 2°С).
В частности, это взаимодействие выполняли следующим образом.
Гидроксиламина гидрохлорид (1,49 кг, 21,53 моль) добавляли к смеси объединенного продукта 3-хлор-5-дифторметоксиминдальной кислоты-Aze-CBA (3,229 кг, 7,18 моль) в этаноле (21 л) приблизительно при 2°С. Добавляли DIPEA (3,7 л, 21,62 моль), и смесь перемешивали в течение приблизительно 46 часов примерно при 2°С.
Обработку выполняли путем добавления ацетона (1,32 л, 17,95 моль) при температуре ниже 5°С и EtOAc (61 л) и воды (61 л). Отделенный органический слой экстрагировали водой (25 л) и раствором NaCl (23,1%, 5 л), используемым во время экстракции. Растворитель (30 л) отгоняли, и затем добавляли EtOAc (11 л). Растворитель снова отгоняли и добавляли МеОН (5 л). Окончательная перегонка давала 3,378 кг твердого продукта.
Продукт очищали посредством смешивания с изопропанолом, нагревания с обратным холодильником в течение примерно 4 часов, охлаждения и фильтрации. Наконец, твердое вещество суспендировали в метаноле и упаривали, затем использовали этилацетатный растворитель и упаривали.
Краткое описание графических материалов
Кристаллы, полученные, как описано в данном описании изобретения, анализировали с помощью XRPD, и результаты приведены в таблицы ниже (RI представляет собой относительную интенсивность) и показаны на соответствующих Фигурах.
На Фиг.1 показана XRPD-дифрактограмма кристаллического ангидрата Соединения А.
В Таблице 1 непосредственно ниже показаны наиболее значительные пики в XRPD-дифрактограмме кристаллического ангидрата Соединения А.
Ряд слабых и очень слабых пиков были исключен. Значения RI в скобках показывают, насколько пик подвергается воздействию предпочтительных ориентационных эффектов. Вследствие предпочтительных ориентационных эффектов некоторые слабые исключенные пики могут становиться более значительными.
На Фиг.2 показана XRPD-дифрактограмма кристаллического ангидрата Соединения В.
В Таблице 2 непосредственно внизу показаны наиболее значительные пики в XRPD-дифрактограмме кристаллического ангидрата Соединения В.
Ряд слабых и очень слабых пиков был исключен.
На Фиг.3 показана XRPD-дифрактограмма кристаллического 1/3-изопропанолата Соединения В.
В Таблице 3 непосредственно внизу показаны наиболее значительные пики в XRPD-дифрактограмме кристаллического изопропанолата Соединения В.
Ряд слабых и очень слабых пиков был исключен.
Сокращения
vs = очень сильный; s = сильный; m = средний; w = слабый; vw = очень слабый; широкий = наложение нескольких пиков.
Конкретные аспекты изобретения включают
1) Соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe) ангидрат в кристаллической форме с картиной дифракции рентгеновских лучей на порошке, характеризующейся пиками со значениями d 5,2 Ǻ, 4,79 Ǻ, 4,23 Ǻ и 3,98 Ǻ.
2) Соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH) ангидрат в кристаллической форме с картиной дифракции рентгеновских лучей на порошке, характеризующейся пиками со значениями d 4,41 Ǻ, 3,94 Ǻ и 3,59 Ǻ.
3) Соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)·1/3 изопропанолат в кристаллической форме с картиной дифракции рентгеновских лучей на порошке, характеризующейся пиками со значениями d 11,7 Ǻ, 6,4 Ǻ, 4,1 Ǻ и 3,97 Ǻ.
На Фиг.4 показан профиль высвобождения 150 мг и 200 мг гелеобразующих матричных композиций для Примера 26.
Пример 1: Кристаллизация Соединения В
Аморфное Соединение В (31,21 мг) добавляли к смеси этанол/вода 20:80 масс./масс. (3,06 г), и образец встряхивали в течение двух часов. Когда твердое вещество почти полностью растворялось, добавляли еще 10 мг аморфного Соединения В, и полученную суспензию оставляли встряхиваться в течение 5 суток. Затем насыщенный раствор декантировали, и твердый образец помещали в холодильник на два часа до его исследования с помощью XRPD. Образец теперь являлся кристаллическим, и полученная дифрактограмма показана на Фиг.2. Затем образец анализировали с помощью TGA и DSC. Потеря массы при 25-110°С составляла 0,13% и термограмма DSC имела один эндотермический пик при начальной температуре 170°С и энтальпии 106 Дж/г.
Пример 2: Кристаллизация Соединения В
Аморфное Соединение В растворяли в изопропаноле и перемешивали в течение 2 часов в закрытом сосуде. В течение этого времени происходила кристаллизация. Образец анализировали с помощью XRPD и микроскопии в поляризованном свете и обнаружили, что он является только частично кристаллическим. Следовательно, его оставляли суспендироваться в течение еще двух суток, также в закрытом сосуде. Затем образец отбирали на стеклянный фильтр (пористость 4) и анализировали с помощью XRPD. Теперь он имел высокую кристалличность, и полученная XRPD-дифрактограмма показана на Фиг.3. Анализ с помощью DSC показал одну эндотерму с пиком при 95°С, начальным значением при 82°С и энтальпией 127 Дж/г. TGA Анализ показал потерю массы 8% вплоть до 105°С. Затем образец сушили в течение 2 суток при 22°С в вакууме и снова анализировали. Теперь DSC показала эндометрический пик при 97°С (начальное значение = 84°С) и энтальпии 112 Дж/г. Потеря массы в TGA при температуре ниже 105°С теперь составляла 6,8%.
Пример 3: Кристаллизация Соединения А
2,5 мл изопропанола смешивали и перемешивали с аморфным Соединением А (75 мг) в закрытом сосуде. Через 17 часов оно кристаллизовалось. Добавляли небольшое количество изопропанола и перемешивали с образованием суспензии, которую затем фильтровали на стеклянном фильтре с пористостью 4. Полученное твердое вещество сушили в вакууме при 22°С, и образование кристаллов подтверждали с помощью микроскопии в поляризованном свете (посредством визуального просмотра кристаллов между скрещенными поляризаторами).
Пример 4: Кристаллизация Соединения В
Аморфное Соединение В (0,5 г) смешивали с приблизительно 5 мл изопропанола в закрытом сосуде. Через несколько часов образовался плотный осадок. Добавили пару миллилитров изопропанола, чтобы перемешивание было более плавным. Осаждение проверяли с помощью микроскопии в поляризованном свете и, как для Примера 2 выше, было обнаружено, что кристаллы были смешаны с аморфным гелем. Смесь перемешивали в течение 2 суток (также в закрытом сосуде) и затем анализировали с помощью микроскопии в поляризованном свете, чтобы показать, что аморфные области пропали, и теперь образец был высоко кристаллическим. Образец отбирали на стеклянный фильтр с пористостью 4 и сушили в вакууме при 22°С и затем анализировали. Термограмма DSC содержала один эндометрический пик при 95°С (начальная = 86°С) с энтальпией 64 Дж/г, и потеря массы в TGA при температуре ниже 105°С составляла 7,1%. Более низкая энтальпия, полученная в этом Примере, по сравнению с Примером 1 выше, является результатом фоновых эффектов.
Пример 5: Кристаллизация Соединения А
Приблизительно 10 мл аморфного Соединения А в этилацетате упаривали до геля. К нему добавляли 2 мл изопропанола плюс несколько затравок Соединения А (из Примера 3), и смесь оставляли перемешиваться в течение ночи в закрытом сосуде. На следующее утро она кристаллизовалась, что было подтверждено с помощью микроскопии в поляризованном свете (посредством визуального просмотра кристаллов между скрещенными поляризаторами).
Пример 6: Крупномасштабная кристаллизация Соединения А
1 л аморфного Соединения А в этилацетате упаривали до густого желтоватого геля. К нему добавляли приблизительно 30 мл изопропанола, и затем образец перемешивали вплоть до его растворения. Затем перемешивание продолжали в закрытом сосуде, и через одни сутки наблюдалось небольшое образование осадка. Так как развитие было медленным, добавляли некоторое количество затравки Соединения А (из Примера 5), и затем образец оставляли перемешиваться также в закрытом сосуде. Через трое суток смесь кристаллизовалась в плотный твердый ком. Добавляли приблизительно 50 мл + 50 мл изопропанола, чтобы твердое вещество можно было снова диспергировать, и фильтровать через стеклянный фильтр с пористостью 3. Затем образец переносили с фильтра в большую стеклянную чашку, в которой его сушили в вакууме при 50°С в течение 1 суток (вещество вынимали, и комки время от времени разрушали вручную). Образец был кристаллическим и имел XRPD-дифрактограмму, очень похожую на дифрактограмму на Фиг.1 (смотри Пример 20). Термограмма DSC содержала один эндометрический пик при 112°С (начальная = 106°С) с энтальпией плавления 89 Дж/г, и потеря массы в TGA при температуре ниже 120°С составляла 0,1%.
Пример 7: Растворимость Соединения А в этилацетате
Избыток Соединения А из Примера 3 выше суспендировали в этилацетате в течение одних суток. Затем часть насыщенного раствора отбирали через фильтр 0,5 микрометра в предварительно взвешенную колбу Эрленмейера. Колбу Эрленмейера взвешивали и затем открывали и упаривали на воздухе, и затем сушили в вакууме при 40°С до постоянной массы. Определяли массу раствора, массу сухого вещества, массу растворителя и объем растворителя (используя плотность этилацетата при 22°С = 0,90 г/мл).
Растворимость (в виде мг Соединения А на мл этилацетата) = 36 мг/мл.
Растворимость в виде масс.% = 4% масс./масс.
Пример 8: Растворимость Соединения В в изопропаноле
Избыток Соединения В из Примера 2 выше суспендировали в изопропаноле в течение одних суток в закрытом сосуде. Затем часть насыщенного раствора отбирали через фильтр 0,5 микрометра в предварительно взвешенную колбу Эрленмейера. Колбу Эрленмейера взвешивали и затем открывали и упаривали на воздухе, и затем сушили в вакууме при 70°С вплоть до постоянной массы. Определяли массу раствора, массу сухого вещества, массу растворителя и объем растворителя (используя плотность изопропанола при 22°С = 0,79 г/мл).
Растворимость (в виде мг Соединения В на мл изопропанола) = 3 мг/мл.
Растворимость в виде % массы = 0,4% масс./масс.
Пример 9: Растворимость ангидрата Соединения А в изопропаноле
Избыток Соединения А из Примера 6 выше суспендировали в изопропаноле в течение одних суток в закрытом сосуде. Затем часть насыщенного раствора отбирали через фильтр 0,5 микрометра в предварительно взвешенную колбу Эрленмейера. Колбу Эрленмейера взвешивали и затем открывали и упаривали на воздухе, и затем сушили в вакууме при 70°С до постоянной массы. Определяли массу раствора, массу сухого вещества, массу растворителя и объем растворителя (используя плотность изопропанола при 22°С = 0,79 г/мл).
Растворимость (в виде мг Соединения А на мл изопропанола) = 15 мг/мл.
Растворимость как масс.% = 2% масс./масс.
Пример 10: Кристаллизация Соединения А в изопропаноле
Аморфное Соединение А (100,1 мг) взвешивали в пробирке для исследования и затем добавляли перемешивающий магнит и 1,00 мл изопропанола. Пробирку закрывали герметично закупоривающей крышкой и затем помещали на магнитный смеситель при 22°С. Твердое вещество растворялось через несколько минут перемешивания, и затем образец оставляли перемешиваться. Через 17,5 часов пробирку проверяли, и было обнаружено, что большое количество твердого вещества выпало в осадок. Для того чтобы получить более полное осаждение, образец перемешивали еще 3 суток, и затем образец суспензии отбирали и капали на силиконовый держатель с нулевым фоном. Образец оставляли сушиться и затем анализировали с помощью XRPD и получали дифрактограмму, аналогичную дифрактограмме на Фиг.1 (смотри Пример 20), но с предпочтительными ориентационными эффектами.
Пример 11: Кристаллизация Соединения А в изопропаноле
Аморфное Соединение А (80,1 мг) взвешивали в пробирке для исследования и затем добавляли перемешивающий магнит и 1,00 мл изопропанола. Пробирку закрывали герметично закупоривающей крышкой и затем помещали на перемешивающий магнит при 22°С. Твердое вещество растворялось через несколько минут перемешивания, и затем образец оставляли перемешиваться. Через 17,5 часов пробирку проверяли, и было обнаружено, что большое количество твердого вещества выпало в осадок. Для того чтобы получить более полное осаждение, образец перемешивали еще 3 суток, и затем образец суспензии отбирали и капали на силиконовый держатель с нулевым фоном. Образец оставляли сушиться и затем анализировали с помощью XRPD и получали дифрактограмму, аналогичную дифрактограмме на Фиг.1 (смотри Пример 20), но с предпочтительными ориентационными эффектами.
Пример 12: Кристаллизация Соединения А в изопропаноле
Аморфное Соединение А (59,8 мг) взвешивали в пробирке для исследования и затем добавляли перемешивающую перемешивающий магнит и 1,00 мл изопропанола. Пробирку закрывали герметично закупоривающей крышкой и затем помещали на магнитный смеситель при 22°С. Твердое вещество растворялось через несколько минут перемешивания, и затем образец оставляли перемешиваться. Через 17,5 часов пробирку проверяли, и было обнаружено, что большое количество твердого вещества выпало в осадок. Для того чтобы получить более полное осаждение, образец перемешивали еще 3 суток и затем образец суспензии отбирали и капали на силиконовый держатель с нулевым фоном. Образец оставляли сушиться и затем анализировали с помощью XRPD и получали дифрактограмму, аналогичную дифрактограмме на Фиг.1 (смотри Пример 20), но с предпочтительными ориентационными эффектами.
Пример 13: Кристаллизация Соединения А в изопропаноле
Аморфное Соединение А (40,9 мг) взвешивали в пробирке для исследования и затем добавляли перемешивающий магнит и 1,00 мл изопропанола. Пробирку закрывали герметично закупоривающей крышкой и затем помещали на магнитный смеситель при 22°С. Твердое вещество растворялось через несколько минут перемешивания, и затем образец оставляли перемешиваться. Через 17,5 часов пробирку проверяли, но твердое вещество не выпадало в осадок. Образец перемешивали в течение еще 3 суток и затем было замечено незначительное осаждение. Через еще 3 суток образец содержал большое количество твердого вещества. Теперь образец суспензии отбирали и капали на силиконовый держатель с нулевым фоном. Образец оставляли сушиться и затем анализировали с помощью XRPD и получали дифрактограмму, аналогичную дифрактограмме на Фиг.1 (смотри Пример 20), но с предпочтительными ориентационными эффектами.
Пример 14: Кристаллизация Соединения А в изопропаноле
Аморфное Соединение А (19,6 мг) взвешивали в пробирке для исследования и затем добавляли перемешивающий магнит и 1,00 мл изопропанола. Пробирку закрывали герметично закупоривающей крышкой и затем помещали на магнитный смеситель при 22°С. Твердое вещество растворялось через несколько минут перемешивания, и затем образец оставляли перемешиваться. В результате низкого перенасыщения через 1,5 месяца образец все еще не выпадал в осадок.
Пример 15: Дополнительные анализы образцов из экспериментов 10-13
Через 7,5 суток перемешивания все образцы в вышеупомянутых Примерах 10-13 собрали в один образец на фильтре с пористостью 1,2 микрометра. Образец сушили в течение 1 часа в вакуумной печи при 50°С и затем наносили мазком на силиконовый держатель с нулевым фоном и анализировали с помощью XRPD. Дифрактограмма рентгеновских лучей была очень похожа на дифрактограмму на Фиг.1 (смотри Пример 20). Часть высушенного образца затем анализировали с помощью TGA. Было показано отсутствие значительных потерь массы при температуре ниже 200°С. Затем образец анализировали с помощью DSC, используя сканирование 10°С/мин, и обнаружили эндотерму плавления с корректированной начальной температурой плавления 109°С.
Пример 16: Кристаллизация Соединения А в этилацетате
Аморфное Соединение А (160,5 мг) взвешивали в пробирке для исследования и затем добавляли перемешивающий магнит и 1,00 мл этилацетата. Пробирку закрывали герметично закупоривающей крышкой и затем помещали на магнитный смеситель при 22°С. Твердое вещество растворялось через несколько минут перемешивания, и затем образец оставляли перемешиваться. Через 17,5 часов пробирку проверяли, и было обнаружено, что незначительное количество твердого вещества выпало в осадок. Для того чтобы получить более полное осаждение, образец перемешивали еще 3 суток, и затем образец суспензии для XRPD отбирали и капали на силиконовый держатель с нулевым фоном. После сушки выполняли анализ XRPD, который давал дифрактограмму, аналогичную изображенной на Фиг.1 (смотри Пример 20), но с предпочтительными ориентационными эффектами.
Пример 17: Кристаллизация Соединения А в этилацетате
Аморфное Соединение А (140,8 мг) взвешивали в пробирке для исследования, и затем добавляли перемешивающий магнит и 1,00 мл этилацетата. Пробирку закрывали герметично закупоривающей крышкой и затем помещали на магнитный смеситель при 22°С. Твердое вещество растворялось через несколько минут перемешивания, и затем образец оставляли перемешиваться. Через 17,5 часов пробирку проверяли, и было обнаружено, что незначительное количество твердого вещества выпало в осадок. Для того чтобы получить более полное осаждение, образец перемешивали в течение еще 3 суток, и затем образец суспензии для XRPD отбирали и капали на силиконовый держатель с нулевым фоном. После сушки выполняли анализ XRPD, который давал дифрактограмму, аналогичную изображенной на Фиг.1 (смотри Пример 20), но с предпочтительными ориентационными эффектами.
Пример 18: Кристаллизация Соединения А в этилацетате
Аморфное Соединение А (120,3 мг) взвешивали в пробирке для исследования и затем добавляли перемешивающий магнит и 1,00 мл этилацетата. Пробирку закрывали герметично закупоривающей крышкой и затем помещали на магнитный смеситель при 22°С. Твердое вещество растворялось через несколько минут перемешивания, и затем образец оставляли перемешиваться. Через 17,5 часов пробирку проверяли, и было обнаружено, что незначительное количество твердого вещества выпало в осадок. Для того чтобы получить более полное осаждение, образец перемешивали в течение еще 3 суток, и затем образец суспензии для XRPD отбирали и капали на силиконовый держатель с нулевым фоном. После сушки выполняли анализ XRPD, который давал дифрактограмму, аналогичную изображенной на Фиг.1 (смотри Пример 20), но с предпочтительными ориентационными эффектами.
Пример 19: Кристаллизация Соединения А в этилацетате
Аморфное Соединение А (99,5 мг) взвешивали в пробирке для исследования и затем добавляли перемешивающий магнит и 1,00 мл этилацетата. Пробирку закрывали герметично закупоривающей крышкой и затем помещали на магнитный смеситель при 22°С. Твердое вещество растворялось через несколько минут перемешивания, и затем образец оставляли перемешиваться. Через 17,5 часов пробирку проверяли, но твердое вещество не выпадало в осадок. Образец перемешивали еще в течение 3 суток, и затем было замечено незначительное осаждение. Еще через 3 суток образец содержал большое количество твердого вещества. Теперь образец суспензии отбирали и капали на силиконовый держатель с нулевым фоном. Образец оставляли сушиться и затем анализировали с помощью XRPD, которая давала дифрактограмму, аналогичную изображенной на Фиг.1 (смотри Пример 20), но с предпочтительными ориентационными эффектами.
Пример 20: Кристаллизация Соединения А в этилацетате
Аморфное Соединение А (80,4 мг) взвешивали в пробирке для исследования и затем добавляли перемешивающий магнит и 1,00 мл этилацетата. Пробирку закрывали герметично закупоривающей крышкой и затем помещали на магнитный смеситель при 22°С. Твердое вещество растворялось через несколько минут перемешивания, и затем образец оставляли перемешиваться. Через 17,5 часов пробирку проверяли, но твердое вещество не выпадало в осадок. Через 7,5 суток начал появляться небольшой осадок. Однако количество было слишком маленьким для анализа. Через примерно 40 суток образец осаждался более полно. Твердое вещество собирали на фильтре с пористостью 1,2 микрометров. Образец сушили в течение 1 часа в вакуумной печи при 50°С и затем наносили мазком на силиконовый держатель с нулевым фоном и анализировали с помощью XRPD. Полученная дифрактограмма рентгеновских лучей показана на Фиг.1.
Пример 21: Анализ образцов из экпериментов 16-19
Через 7,5 суток перемешивания все образцы в вышеуказанных экспериментах 16-19 собирали в один образец на фильтре с пористостью 1,2 микрометров. Образец сушили в течение 1 часа в вакуумной печи при 50°С и затем наносили на силиконовый держатель с нулевым фоном и анализировали с помощью XRPD. Дифрактограмма рентгеновских лучей была очень похожей на дифрактограмму на Фиг.1 (смотри Пример 20). Часть высушенного образца затем анализировали с помощью TGA. Было показано отсутствие значительных потерь массы ниже 200°С. Затем образец анализировали с помощью DSC, используя сканирование 10°С/мин, и была обнаружена эндотерма плавления со скорректированным значением начала плавления 109°С.
Пример 22: Растворимость Соединения А в этаноле
Избыток Соединения А из вышеуказанного Примера 6 суспендировали в этаноле в течение одних суток в закрытом сосуде. Затем часть насыщенного раствора отбирали через фильтр 0,5 микрометра в предварительно взвешенную колбу Эрленмейера. Колбу Эрленмейера взвешивали и затем открывали и упаривали на воздухе, и затем сушили в вакууме при 40-50°С вплоть до постоянной массы. Определяли массу раствора, массу сухого вещества, массу растворителя и объем растворителя (используя плотность этанола при 22°С = 0,79 г/мл).
Растворимость (в виде мг Соединения А на мл этанола) = 87 мг/мл.
Растворимость в виде % массы = 10% масс./масс.
Пример 23: Растворимость Соединения А в метаноле
Избыток Соединения А из вышеуказанного Примера 6 суспендировали в метаноле в течение одних суток в закрытом сосуде. Затем часть насыщенного раствора отбирали через фильтр 0,5 микрометра в предварительно взвешенную колбу Эрленмейера. Колбу Эрленмейера взвешивали и затем открывали и упаривали на воздухе, и затем сушили в вакууме при 40-50°С вплоть до постоянной массы. Определяли массу раствора, массу сухого вещества, массу растворителя и объем растворителя (используя плотность метанола при 22°С = 0,79 г/мл).
Растворимость (в виде мг Соединения А на мл метанола) = 655 мг/мл.
Растворимость в виде % массы = 45% масс./масс.
Пример 24: Растворимость Соединения А в ацетоне
Избыток Соединения А из вышеуказанного Примера 6 суспендировали в ацетоне в течение одних суток в закрытом сосуде. Затем часть насыщенного раствора отбирали через 0,5 микрометровый фильтр в предварительно взвешенную колбу Эрленмейера. Колбу Эрленмейера взвешивали и затем открывали и упаривали на воздухе, и затем сушили в вакууме при 70°С вплоть до постоянной массы. Определяли массу раствора, массу сухого вещества, массу растворителя и объем растворителя (используя плотность ацетона при 22°С = 0,82 г/мл).
Растворимость (в виде мг Соединения А на мл ацетона) = 398 мг/мл.
Растворимость в виде масс.% = 33% масс./масс.
Пример 25: Гранульная композиция с кишечнорастворимым покрытием
Кристаллический ангидрат соединения А (223 г) суспендировали в 750 г 2,5% (масс./масс.) водном растворе НРМС (6 сП (6 мПа·с)) путем смешивания в смесителе Сильверсона в течение более 4 часов.
Ядра из микрокристаллической целлюлозы (МСС) (170 г; Celphere 305, Asahi Casei) использовали в качестве исходного материала, и распылительное нанесение слоев выполняли в устройстве Wurster с псевдоожиженным слоем с выходом 370 г гранул.
Этилцеллюлозу (16,7 г) и гидроксипропилцеллюлозу (15,6 г) растворяли в 450 г 99,5% EtOH.
Затем 360 г гранул покрывали раствором ЕС/НРС/EtOH с выходом 389 г покрытых оболочкой гранул.
В заключение такие гранулы (350 г) покрывали водной дисперсией Eudragit L30D (Degussa), соответствующей 56 г сухого вещества, и с выходом 401 г гранул с энтеросолюбильным покрытием.
Профиль высвобождения гранульной композиции изучали, используя стандартные методы (LC использовали для определения высвобожденного вещества) при рН 1 (0,1 М HCl) и рН 6,8 (фосфатный буфер). Результаты показывают, что при рН 1 из композиции в течение 24 часов практически не высвобождается вещество активного ингредиента (приблизительно 1-2%). Однако при рН 6,8 приблизительно 21% активного вещества высвобождается в течение 3 часов; 39% в течение 6 часов; 49% в течение 8 часов; 79% в течение 16 часов; 88% в течение 20 часов и 99% в течение 32 часов.
Пример 26: Гелеобразующая матричная композиция
Получение гранулирующего раствора
Гранулирующую жидкость получали путем растворения гидроксипропилцеллюлозы в этаноле при перемешивании.
Гранулирование
Гидроксипропилметилцеллюлозу и кристаллический ангидрат Соединения А смешивали затем в миксере Mini Diosna с емкостью 1 л. Во время смешивания добавляли гранулирующую жидкость в течение 4-6 минут. При необходимости добавляют больше этанола.
Сушка и измельчение
Влажную массу сушили в сушильном шкафу и высушенную массу измельчали, используя Quadro Comill с отверстиями 1,57 мм.
Итоговое смешивание и таблетирование
Затем гранулят смешивали с 2%-ным натрия стеарилфумаратом в смесителе Turbula. Гранулят прессовали в Excenterpress, используя пуансоны 9 мм. Масса таблетки 273 мг, твердость таблетки >60 Н.
Конечная композиция
Профиль высвобождения 150 мг и 200 мг гелеобразующих матричных композиций изучали, используя стандартные методы (LC использовали для определения высвобожденного вещества) при рН 1 (0,1 М HCl) и рН 6,8 (фосфатный буфер). Результаты показаны на Фиг.4 для Примера 26.
Пример 27: Кристаллизация соединения А в смеси изопропилацетат/этанол/гептан
Раствор изопропилацетата (165,27 г), содержащий Соединение А (40 г), фильтровали через стеклокерамический фильтр при температуре окружающей среды. Фильтр промывали 36 мл этанола, и два раствора сливали в 0,5 л реактор, оборудованный механической мешалкой, и нагревали до 30°С. Примерно через 20 минут добавляли затравки кристаллического ангидрата Соединения А (4% масс./масс. в пересчете на 100% продукта) к перемешанному раствору, чтобы инициировать кристаллизацию. Через еще 15 минут в течение 2 ч добавляли 120 мл гептана. Суспензию перемешивали в течение примерно 14 ч при 30°С и затем осуществляли второе добавление гептана (160 мл) в течение 2 ч. Суспензию охлаждали до 20°С во время этого добавления (время охлаждения 2 ч). После еще 10 ч твердое вещество выделяли фильтрацией. Осадок на фильтре промывали два раза смесью 25 мл изопропилацетата, 6 мл этанола и 49 мл гептана. Наконец, его сушили в течение ночи под вакуумом при 40°С (выход 38,2 г). XRPD соответствовала ссылке на ангидрат Соединения А, раскрытый в данном описании изобретения.
Затравочные кристаллы, используемые выше, получали из аналогичных более ранних экспериментов, используя более низкое количество затравок Соединения А (например, затравок, полученных из более раннего подобного эксперимента, выполненного в течение длительного времени). Затравки Соединения А для аналогичных более ранних экспериментов получают из аналогичных экспериментов, включая описанные в данном описании изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ | 2003 |
|
RU2352323C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ И ЕЕ ПРИМЕНЕНИЕ | 2003 |
|
RU2474416C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2323006C2 |
| ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ С НЕМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ | 2003 |
|
RU2351314C2 |
| ПО СУЩЕСТВУ, КРИСТАЛЛИЧЕСКАЯ ФОРМА МЕЛАГАТРАНА | 2000 |
|
RU2243973C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ МИНДАЛЬНОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТРОМБИНА | 2001 |
|
RU2300521C2 |
| ЧАСТИЦЫ АНТАГОНИСТА CRTH2 | 2008 |
|
RU2468012C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ МИНДАЛЬНОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТРОМБИНА | 2002 |
|
RU2341516C2 |
| ТВЁРДЫЕ ФОРМЫ ИНГИБИТОРА КИНАЗЫ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА | 2013 |
|
RU2673077C2 |
| Твёрдые формы цефтолозана | 2014 |
|
RU2703457C2 |

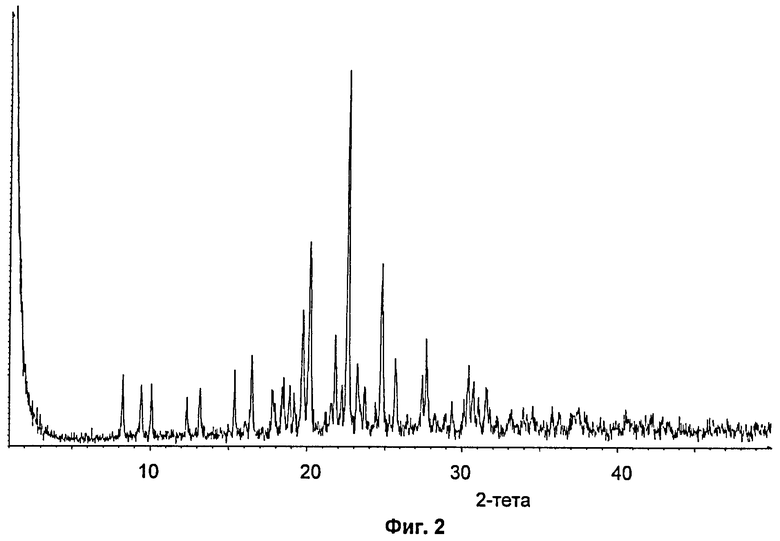

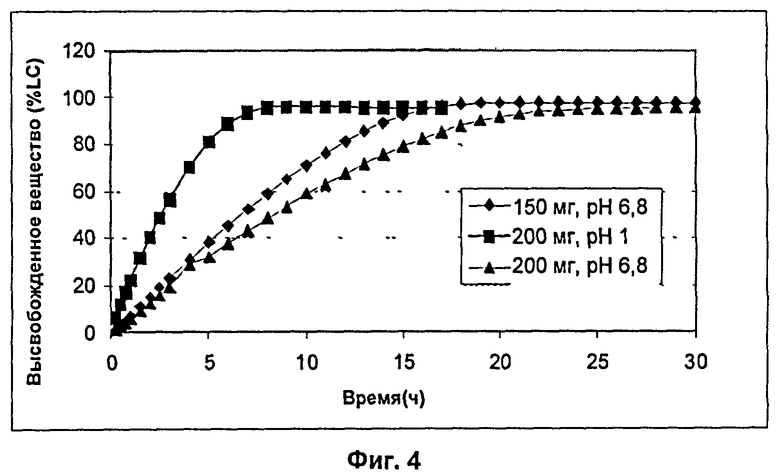
Изобретение относится к кристаллической ангидратной форме (2S)-1-{(2R)-2-[3-хлор-5-(дифторметокси)фенил]-2-гидроксиацетил}-N-[4-(N'-метоксикарбамимидоил)бензил]азетидин-2-карбоксамида формулы:
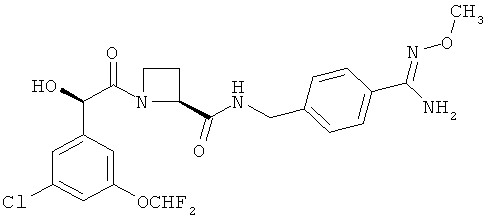
имеющей картину дифракции рентгеновских лучей на порошке, характеризующуюся пиками со значениями d 15,2 Å, 10,8 Å, 6,8 Å, 6,0 Å, 5,2 Å, 4,79 Å, 4,23 Å и 3,98 Å, которое может быть использовано в фармацевтической композиции в качестве ингибитора тромбина. 2 н. и 4 з.п. ф-лы, 27 пр., 4 ил.
1. Соединение (2S)-1-{(2R)-2-[3-хлор-5-(дифторметокси)фенил]-2-гидроксиацетил}-N-[4-(N'-метоксикарбамимидоил)бензил]азетидин-2-карбоксамид
в, по существу, кристаллической ангидратной форме, имеющей картину дифракции рентгеновских лучей на порошке, характеризующуюся пиками со значениями d 5,2 Å, 4,79 Å, 4,23 Å и 3,98 Å.
2. Соединение по п.1, имеющее картину дифракции рентгеновских лучей на порошке, характеризующуюся пиками со значениями d 15,2 Å, 10,8 Å, 6,8 Å, 6,0 Å, 5,2 Å, 4,79 Å, 4,23 Å и 3,98 Å.
3. Соединение по п.2, отличающееся кривой дифференциальной сканирующей калориметрии при скорости нагрева 10°С/мин в закрытой чашке с микроотверстиями в потоке азота, показывающей эндотерму с экстраполированной начальной температурой примерно 109°С и максимальной температурой примерно 115°С.
4. Соединение по п.1, где кристалличность составляет более 80%.
5. Соединение по любому из пп.1-3, которое метаболизируется после перорального и/или парентерального введения, для применения в качестве ингибитора тромбина.
6. Фармацевтическая композиция в качестве ингибитора тромбина, содержащая соединение по любому из пп.1-3, которое метаболизируется после перорального и/или парентерального введения, в смеси с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| ПО СУЩЕСТВУ, КРИСТАЛЛИЧЕСКАЯ ФОРМА МЕЛАГАТРАНА | 2000 |
|
RU2243973C2 |

