Настоящее изобретение относится к некоторым производным гидантоина, полезным в ингибировании металлопротеиназ, к способам их получения, к содержащим их фармацевтическим композициям и к их применению в терапии.
Соединения по данному изобретению представляют собой ингибиторы одного или более ферментов металлопротеиназ. Металлопротеиназы представляют собой суперсемейство протеиназ (ферментов), известное число которых резко увеличилось в последние годы. На основании структурных и функциональных особенностей эти ферменты были классифицированы на семейства и подсемейства, как описано в N. М Hooper (1994) FEBS Letters 354:1-6. Примеры металлопротеиназ включают в себя семейство матриксных металлопротеиназ (матриксины, ММР), таких как коллагеназы (ММР1, ММР8, ММР13, ММР18), желатиназы (ММР2, ММР9), стромелизины (ММР3, ММР10, ММР11), матрилизины (ММР7, ММР26), металлоэластаза (ММР12), энамелизин (ММР19), мембранные типы МТ-ММР (ММР14, ММР15, ММР16, ММР17, ММР24, ММР25) и другие (ММР20, ММР21, ММР22, MMP23a/b, MMP28); семейство ADAM (дезинтегрин, металлопротеиназа, также известная как репролизин или адамализин, или MDC), которое в настоящее время включает в себя 32 известных ADAM с секретазной и шеддазной активностью, таких как ТМР (фактор некроза опухоли)-превращающий фермент (ADAM 17), и 18 известных ADAMTS (дезинтегрин металлопротеиназа тромбоспондин), включая аггреканазы (ADAMTS4, ADAMTS5); семейство астацинов, которое включает в себя такие ферменты как протеиназа, участвующая в процессинге проколлагена (РСР); и другие металлопротеиназы, такие как семейство эндотелин-превращающего фермента и семейство ангиотензин-превращающего фермента.
Считают, что металлопротеиназы играют важную роль в развитии многих физиологических процессов, которые вовлекают ремоделирование ткани, такое как эмбриональное развитие, остеогенез и ремоделирование матки во время менструации. Эта точка зрения основана на способности металлопротеиназ расщеплять широкий спектр матриксных субстратов, таких как коллаген, протеогликан и фибронектин. Считают также, что металлопротеиназы важны в процессинге, или секреции, биологически важных клеточных медиаторов, таких как фактор некроза опухоли (TNF); и в посттрансляционном протеолитическом процессинге, или шеддинге, биологически важных мембранных белков, таких как низкоаффинный IgE-рецептор CD23 (для более полного перечня смотри N.М.Hooper et al., (1997) Biochem J. 321:265-279).
Металлопротеиназы ассоциированы со многими болезненными состояниями. Ингибирование активности одной или более металлопротеиназ может быть полезным при этих болезненных состояниях, например при различных воспалительных и аллергических заболеваниях, таких как воспаление сустава (в частности, ревматоидный артрит, остеоартрит и подагра), воспаление желудочно-кишечного тракта (особенно энтерит, язвенный колит и гастрит), воспаление кожи (в частности, псориаз, экзема, дерматит); при опухолевом метастазировании или инвазии; при заболевании, ассоциированном с преобладанием деградации внеклеточного матрикса над синтезом, таком как остеоартрит; при резорбтивном заболевании кости (таком как остеопороз или болезнь Педжета); при заболеваниях, ассоциированных с аномальным ангиогенезом; при усиленном ремоделировании коллагена, ассоциированного с диабетом, периодонтальным заболеванием (таким как периодонтит), изъязвлением роговицы, изъязвлением кожи, послеоперационными состояниями (такими как анастамоз ободочной кишки) и заживлением кожных ран; при демиелинизирующих заболеваниях центральной и периферической нервной системы (таких как рассеянный склероз); при болезни Альцгеймера; при ремоделировании внеклеточного матрикса, наблюдающейся при сердечно-сосудистых заболеваниях, таком как рестеноз и атеросклероз; и при хронических обструктивных заболеваниях легких, ХОЗЛ.
Известен целый ряд ингибиторов металлопротеиназ; различные классы соединений могут проявлять различные степени эффективности и селективности ингибирования различных металлопротеиназ. Авторы настоящего изобретения открыли новый класс соединений, которые являются ингибиторами металлопротеиназ и представляют особый интерес при ингибировании коллагеназы-3 (также известной как ММР-13). Соединения по данному изобретению имеют превосходную активность и/или фармакокинетические свойства.
Коллагеназу-3 (ММР13) первоначально клонировали из кДНК-библиотеки, происходящей из опухоли молочной железы [J.M.P. Freije et al. (1994) Journal of Biological Chemistry 269(24): 16766-16773]. ПЦР-РНК-анализ РНК из широкого спектра тканей показал, что экспрессия коллагеназы-3 (ММР13) ограничена карциномами молочной железы, поскольку она не была обнаружена в фиброаденомах молочной железы, в нормальной или находящейся в покое молочной железе, плаценте, печени, яичнике, матке, предстательной железе или околоушной слюнной железе или в клеточных линиях рака молочной железы (T47-D, MCF-7 and ZR75-1). После этого наблюдения коллагеназа-3 (ММР13) была обнаружена в трансформированных эпидермальных кератиноцитах [N. Johansson et al., (1997) Cell Growth Differ. 8(2):243-250], плоскоклеточных карциномах [N. Johansson et al., (1997) Am. J. Pathol. 151(2):499-508] и эпидермальных опухолях [К. Airola et al., (1997) J. Invest. Dermatol. 109(2):225-231]. На основании этих результатов предположили, что колагеназа-3 (ММР13) секретируется трансформированными эпителиальными клетками и может быть вовлечена в деградацию внеклеточного матрикса и клеточно-матриксное взаимодействие, ассоциированное с метастазированием, что наблюдается при инвазивных раковых поражениях молочной железы и злокачественном эпителиальном росте при кожном канцерогенезе.
Из последних опубликованных данных вытекает, что коллагеназа-3 (ММР13) играет роль в метаболизме других соединительных тканей. Например, в соответствии с субстратной специфичностью коллагеназы-3 (ММР13) и предпочтением к деградации коллагена II типа [Р.G.Mitchell et al., (1996) J. Clin. Invest. 97(3):761-768; V. Knauper et al., (1996) The Biochemical Journal 271:1544-1550] предположили, что коллагеназа-3 (ММР13) играет роль при первичной оссификации и скелетном ремоделировании [M.Stahle-Backdahl et al., (1997) Lab. Invest. 76(5):717-728; N. Johansson et al., (1997) Dev. Dyn. 208(3):387-397] при деструктивных заболеваниях суставов, таких как ревматоидный и остеоартрит [D. Wernicke et al., (1996) J. Rheumatol. 23:590-595; P.G.Mitchell et al., (1996) J. Clin. Invest. 97(3):761-768; O. Lindy et al., (1997) Arthritis Rheum 40(8): 1391-1399], и при асептическом разрыхлении замещений тазобедренных суставов [S. Imai et al., (1998) J. Bone Joint Surg. Br. 80(4):701-710]. Коллагеназа-3 (MMP13) также вовлечена в хронический периодонтит взрослых, поскольку она локализована в эпителии хронически воспаленной слизистой оболочки ткани десны человека [V.J.Uitto et al., (1998) Am.J.Pathol 152(6):1489-1499], и в ремоделирование коллагенового матрикса в хронических ранах [М.Vaalamo et al., (1997) J.Invest. Dermatol. 109(1):96-101].
Соединения, которые ингибируют действие металлопротеиназ, в частности коллагеназы-3 (MMP13) и ММР12, описаны в WO 00/12478, WO 00/75108, WO 01/62742 и WO 02/074767. В число этих описанных ингибиторов включены арилоксипиперидинсульфонилметил-замещенные соединения гидантоина, в которых арильное кольцо замещено рядом возможных заместителей, включая среди прочих трифторметокси. Не описано, что трифторметокси-заместитель в таких соединениях может быть дополнительно замещен.
Замещенные алкокси- или арилоксипиперидинсульфонилметил-замещенные соединения гидантоина в качестве ингибиторов матриксных металлопротеиназ охвачены в общем описании WO 02/074767. Среди многочисленных возможных заместителей алкоксигруппы указан галоген. Одно из описанных соединений представляет собой (5S)-5-метил-5-[({4-[4-(трифторметокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион (Соединение Х для сравнения).
Авторы настоящего изобретения обнаружили, что замещенные арилоксипиперидинсульфонилметил-замещенные соединения гидантоина, в которых заместитель представляет собой С2-4алкоксигруппу, которая сама замещена двумя или более группами фтора, представляют собой особенно эффективные ингибиторы металлопротеиназ, особенно коллагеназы-3 (MMP13), и имеют желаемые профили активности.
Согласно настоящему изобретению предложено соединение формулы (I)
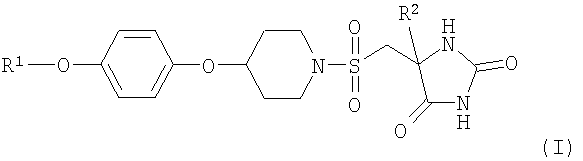 ,
,
где
R1 представляет собой (С2-4)алкил и замещен двумя или более группами фтора, и
R2 представляет собой метил или этил;
или его фармацевтически приемлемая соль.
В данном описании термин (С2-4)алкил включает в себя алкильные группы с неразветвленной и разветвленной цепью, такие как этил, пропил, изопропил, бутил, изобутил и трет-бутил и тому подобное. Ссылки на индивидуальные алкильные группы, такие как этил, пропил и бутил, характерны для варианта с неразветвленной цепью.
Подходящая фармацевтически приемлемая соль соединения формулы (I), например соль присоединения кислоты соединения формулы I, которая является достаточно основной, например соль присоединения кислоты с неорганической или органической кислотой, такой как соляная, бромистоводородная, серная, фосфорная, трифторуксусная, лимонная, малеиновая, винная, фумаровая, гемифумаровая, янтарная, гемиянтарная, миндальная, метансульфоновая, диметансульфоновая, этан-1,2-сульфоновая, бензолсульфоновая, салициловая или 4-толуолсульфоновая кислота, или, например, соль соединения формулы (I), которая является достаточно кислой, например соль щелочного или щелочноземельного металла, такая как кальциевая или натриевая соль, или аммонийная соль, или соль с органическим основанием, таким как метиламин, диметиламин, триметиламин, пиперидин, морфолин или трис-(2-гидроксиэтил)амин.
В тех случаях, когда соединения по данному изобретению содержат один или более асимметрично замещенных атомов углерода, изобретение включает все стереоизомеры, включая энантиомеры и диастереомеры, и смеси, включая их рацемические смеси. Также включены таутомеры и их смеси.
Другие значения R1 и R2 приведены ниже. Такие значения могут быть использованы, когда целесообразно, с любым из определений, пунктов формулы или воплощений, определенных выше или ниже.
R1 представляет собой (С2-4)алкил и замещен двумя или более группами фтора.
R1 представляет собой (С2-4)алкил и замещен двумя-шестью группами фтора.
R1 представляет собой (С2-4)алкил и замещен двумя-пятью группами фтора.
R1 представляет собой этил, пропил или бутил и замещен двумя или более группами фтора.
R1 представляет собой этил или пропил и замещен двумя или более группами фтора.
R1 представляет собой этил, пропил или бутил и замещен двумя-шестью группами фтора.
R1 представляет собой этил, пропил или бутил и замещен двумя-семью группами фтора.
R1 представляет собой этил или пропил и замещен двумя-шестью группами фтора.
R1 представляет собой этил или пропил и замещен двумя-пятью группами фтора.
R1 представляет собой CF3CH2-, CF2HCF2-, CF3CF2-, CF3CH2CH2-, CF2HCF2CH2- или CF3CF2CH2-.
R2 представляет собой метил или этил.
R2 представляет собой метил.
R2 представляет собой этил.
Конкретные новые соединения по изобретению включают, например, соединение формулы (I) или его фармацевтически приемлемые соли, где:
(а) R1 представляет собой (С2-4)алкил и замещен двумя или более группами фтора; а R2 представляет собой метил.
(б) R1 представляет собой (С2-4)алкил и замещен двумя-шестью группами фтора; а R2 представляет собой метил или этил.
(в) R1 представляет собой этил, пропил или бутил и замещен двумя или более группами фтора; а R2 представляет собой метил или этил.
(г) R1 представляет собой этил, пропил или бутил и замещен двумя-шестью группами фтора; а R2 представляет собой метил или этил.
(д) R1 представляет собой этил или пропил и замещен двумя-пятью группами фтора; а R2 представляет собой метил или этил.
(е) R1 представляет собой CF3CH2-, CF2HCF2-, CF3CF2-, CF3CH2CH2-, CF2HCF2CH2- или CF3CF2CH2; а R2 представляет собой метил или этил.
Особенно предпочтительное соединение по изобретению представляет собой, например:
(5S)-5-метил-5-[({4-[4-(2,2,2-трифторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион;
(5S)-5-этил-5-[({4-[4-(2,2,2-трифторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион;
5S-метил-5-[({4-[4-(1,1,2,2-тетрафторэтокси)фенокси]пиперидин-ил}сульфонил)метил]имидазолидин-2,4-дион;
5S-этил-5-[({4-[4-(1,1,2,2-тетрафторэтокси)фенокси]пиперидин-ил}сульфонил)метил]имидазолидин-2,4-дион;
(5S)-5-метил-5-[({4-[4-(пентафторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион;
(5S)-5-этил-5-[({4-[4-(пентафторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион;
5S-метил-5-[({4-[3,3,3-трифторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион;
5S-этил-5-[({4-[3,3,3-трифторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион;
(5S)-5-метил-5-[({4-[4-(2,2,3,3-тетрафторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион;
(5S)-5-этил-5-[({4-[4-(2,2,3,3-тетрафторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион;
(5S)-5-метил-5-[({4-[4-(2,2,3,3,3-пентафторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион; и
(5S)-5-этил-5-[({4-[4-(2,2,3,3,3-пентафторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион.
Рацематы могут быть разделены на индивидуальные энантиомеры с использованием известных процессов (ср. Advanced Organic Chemistry: 3rd Edition: author J March, p.104-107). Подходящая методика включает в себя образование диастереомерных производных путем взаимодействия рацемического вещества с хиральным вспомогательным веществом с последующим разделением, например, с помощью хроматографии, диастереомеров, а затем гидролиз вспомогательных веществ.
Не желая ограничиваться первоначальными определениями, авторы изобретения считают, что в настоящей заявке активный энантиомер имеет S-стереохимию. Это утверждение основано на сравнении с родственными соединениями, для которых была определена абсолютная конфигурация. Соответственно, S-структура представлена в формулах, приведенных в примерах ниже. Однако следует принимать во внимание, что рацемат любого соединения по изобретению может быть разделен на отдельные энантиомеры способом, описанном выше, и более активный энантиомер затем может быть идентифицирован с помощью подходящего анализа без необходимости определения абсолютных конфигураций.
Соединения формулы I или их фармацевтически приемлемые соли могут быть изготовлены любым известным способом, подходящим для получения химически родственных соединений. Подходящие способы проиллюстрированы, например, способами, раскрытыми в WO 02/074767. Такие способы, когда они используются для получения нового соединения формулы I, предложены в качестве дополнительного признака изобретения и проиллюстрированы нижеследующими типичными вариантами способа, в которых, если не указано иначе, R1 и R2 принимают любое из определенных выше значений. Необходимые исходные вещества могут быть получены стандартными методами органической химии. Получение таких исходных веществ описано в сочетании с нижеследующими типичными вариантами способа и в сопутствующих примерах. Альтернативно, необходимые исходные вещества могут быть получены способами, аналогичными проиллюстрированным способам, которые находятся в компетенции среднего специалиста по органической химии.
Соединение формулы 1 или его фармацевтически приемлемая соль могут быть получены путем взаимодействия феноксипиперидина формулы II с сульфонилхлоридом формулы III
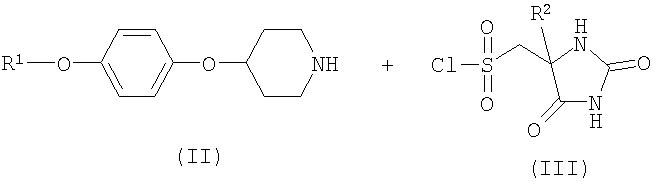
где R1 и R2 такие, как определено выше, и где любая функциональная группа при необходимости защищена, и:
(1) удаления любых защитных групп; и
(2) возможного образования фармацевтически приемлемой соли.
Реакцию предпочтительно осуществляют в подходящем растворителе, возможно в присутствии основания, в течение 1-24 часов при температуре, варьирующей от температуры окружающей среды до температуры дефлегмации. Предпочтительно, такие растворители как пиридин, диметилформамид, тетрагидрофуран, ацетонитрил или дихлорметан используют с основаниями, такими как триэтиламин, N-метилморфолин, пиридин или карбонаты щелочных металлов, при температуре окружающей среды в течение 2-18 часов реакции или до окончания реакции, что определяют хроматографическими или спектроскопическими способами. Реакции сульфонилхлорида формулы III с различными первичными и вторичными аминами ранее описаны в литературе, а варианты условий будут очевидны специалистам в данной области.
Синтез сульфонилхлоридов формулы III описан в литературе и может быть осуществлен, например, из цистеина или гомоцистеина (Mosher J.:J. Org. Chem 23, 1257 (1958). Сульфонилхлориды формулы III также удобно получать в соответствии с Griffith, О.: J. Biol. Chem., 1983, 258, 3, 1591.
Соединения формулы (II) могут быть получены согласно Bioorg Med Chem 2003, 11 (3), 367 и Tet Lett 2002, 43 (12), 2157 с использованием подходящего фторалкоксифенола и трет-бутил-4-гидрокси-1-пиперидин-карбоксилата.
Следует принимать во внимание, что получение соединений формулы (I) может включать на разных стадиях добавление и удаление одной или более защитных групп. Защита и снятие защиты функциональной группы описано в 'Protective Groups in Organic Chemistry', edited by J.W.F. McOmie, Plenum Press (1973) и 'Protective Groups in Organic Synthesis', 2nd edition, T.W. Greene and P.G.M. Wuts, Wiley-lnterscience (1991).
Соединения по данному изобретению представляют собой ингибиторы металлопротеиназ, в частности они представляют собой ингибиторы коллагеназы-3 (ММР13) и, следовательно, показаны для лечения заболеваний или состояний, опосредованных ферментами-металлопротеиназами, включая артрит (такой как остеоартрит), рак, атеросклероз и хронические обструктивные заболевания легких (ХОЗЛ), как обсуждалось выше. В частности, соединения по изобретению показаны для лечения заболеваний или состояний, опосредованных коллагеназой-3 (ММР13). Особым преимуществом ингибиторов коллагеназы-3 по изобретению является то, что они проявляют улучшенную селективность относительно других металлопротеиназ.
Поэтому согласно другому аспекту в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определенно выше, для применения в терапии организма человека или животного.
В данном изобретении также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для применения в терапии.
Следует принимать во внимание, что "терапия" также включает "профилактику", если не указано иначе. Термины "терапевтический" и "терапевтически" следует понимать соответственно.
В еще одном аспекте в настоящем изобретении предложен способ лечения опосредованного металлопротеиназами болезненного состояния, включающий введение теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Следует принимать во внимание, что вводимая доза будет варьировать в зависимости от используемого соединения, способа введения, желаемого лечения и указанного расстройства. Типично, принимают суточную дозу от 0,1 до 75 мг/кг массы тела (и предпочтительно от 0,1 до 30 мг/кг массы тела). При необходимости эту суточную дозу можно давать в виде разделенных доз, при этом количество полученного соединения и путь введения зависят от массы, возраста и пола пациента, которого лечат, и от конкретного болезненного состояния, которое лечат согласно принципам, известным в данной области.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы сами по себе, но обычно будут вводиться в форме фармацевтической композиции вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Поэтому в настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Фармацевтические композиции по изобретению можно вводить стандартным способом для болезненного состояния, требующего лечения, например пероральным, местным, парентеральным, внутрисуставным, трансбуккальным, назальным, вагинальным или ректальным способом введения или путем ингаляции. Для этих целей соединения по данному изобретению могут быть приготовлены известными в данной области способами в форме, например, таблеток, капсул, водных или масляных растворов, суспензий, эмульсий, кремов, мазей, гелей, назальных спреев, суппозиториев, мелкодисперсных порошков или аэрозолей для ингаляции и стерильных водных или масляных растворов, или суспензий, или стерильных эмульсий для парентерального применения (включая внутривенное, внутримышечное или инфузию).
В дополнение к соединениям по настоящему изобретению фармацевтическая композиция по изобретению может также содержать или совместно вводиться (одновременно или последовательно) вместе с одним или более фармакологическими агентами, эффективными в лечении одного или более болезненных состояний, упомянутых выше. Типично, стандартные лекарственные формы будут содержать приблизительно от 1 мг до 500 мг соединения по изобретению.
Активность и селективность соединений по изобретению может быть определена с использованием подходящего теста на ингибирование фермента, как описано в WO 00/12478, WO 00/75108 и WO 01/62742. Ингибирующая активность коллагеназы-3 (ММР13) может быть оценена, например, с использованием методики, изложенной ниже:
Рекомбинантная человеческая проММР13 (коллагеназа-3) может быть экспрессирована и очищена, как описано Knauper и др. [V. Knauper et al., (1996) The Biochemical Journal 271:1544-1550 (1996)]. Очищенный фермент может быть использован для контроля ингибиторов активности следующим образом: очищенную проММР13 активируют с использованием 1 мМ аминофенилртутной кислоты (АРМА) 20 часов при 21°С; активированную ММР13 (11,25 нг на анализ) инкубируют в течение 4-5 часов при 35°С в буфере для анализа (0,1 М Трис-HCl, pH 7,5, содержащий 0,1 М NaCl, 20 мМ CaCl2, 0,02 мМ ZnCl2 и 0,05% (м/о) Brij 35 с использованием синтетического субстрата 7-метоксикумарин-4-ил)-ацетил. Pro. Leu. Gly. Leu. N-3-(2,4-динитрофенил)-L-2,3-диаминопропионил.Ala.Arg.NH2 в присутствии или в отсутствие ингибиторов. Активность определяют путем измерения флуоресценции при λех 328 нм и λem 393 нм. Путем измерения активности ряда концентраций можно построить кривую связывания, из которой можно определить IC50, причем этот показатель означает концентрацию ингибитора, при которой ферментная активность снижена на 50%.
Следует принимать во внимание, что фармакологические свойства соединений по изобретению будут варьировать в соответствии с их структурой, но в общем случае соединения по изобретению демонстрируют ингибиторную активность в отношении коллагеназы-3, как определено вышеприведенным анализом при концентрациях IC50 в пределах от 0,01 до 20 нМ. В нижеследующей таблице приведены показатели IC50 для типичной выборки соединений по изобретению, а также для соединения Х для сравнения, раскрытого в WO 02/074767, при тестировании в вышеприведенном анализе.
Соединение формулы I может быть использовано в комбинации с другими лекарственными средствами и способами лечения, используемыми в лечении болезненных состояний, при которых было бы полезным ингибирование металлопротеиназ, в частности коллагеназы-3 (ММР13). Например, соединение формулы I можно использовать в комбинации с лекарственными средствами и способами лечения, используемыми в лечении ревматоидного артрита, астмы, рака, воспалительного заболевания кишечника, рассеянного склероза, СПИДа, септического шока, застойной сердечной недостаточности, ишемической болезни сердца, псориаза и других болезненных состояний, упомянутых выше в данном описании.
Например, в силу своей способности ингибировать металлопротеиназы соединение формулы I является полезным в лечении некоторых воспалительных и невоспалительных заболеваний, которые в настоящее время лечат ингибирующими циклооксигеназу нестероидными противовоспалительными средствами (NSAID), такими как индометацин, кеторолак, ацетилсалициловая кислота, ибупрофен, сулиндак, толметин и пироксикам. Совместное введение соединения формулы I по настоящему изобретению вместе с NSAID может приводить к снижению количества последнего агента, необходимого для обеспечения терапевтического эффекта. Посредством этого снижают вероятность нежелательных побочных эффектов NSAID, таких как воздействия на желудочно-кишечный тракт. Таким образом, согласно другому признаку данного изобретения предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль в сочетании или в смеси с ингибирующим циклооксигеназу нестероидным противовоспалительным агентом и фармацевтически приемлемым разбавителем или носителем.
Соединение формулы I также может быть использовано вместе с противовоспалительными агентами, такими как ингибитор фермента 5-липоксигеназы.
Соединение формулы I также может быть использовано в лечении состояний, таких как ревматоидный артрит, в комбинации с противоартритными агентами, такими как золото, метотрексат, стероиды и пенициллинамин, и при состояниях, таких как остеоартрит, в комбинации со стероидами.
Соединение формулы I также можно вводить при дегенеративных заболеваниях, например при остеоартрите, вместе с хондропротективными, антидегенеративными и/или репаративными агентами, такими как диацереин, препаратами гиалуроновой кислоты, такими как гиалан, румалон, артепарон, солями хондроитинсульфата и глюкозамина, такими как антрил.
Соединение формулы I можно использовать в лечении астмы в комбинации с противоастматическими агентами, такими как стероиды, бронходилататоры и антагонисты лейкотриенов.
В частности, для лечения воспалительных заболеваний - ревматоидного артрита, остеоартрита, псориаза, воспалительного заболевания кишечника, хронического обструктивного заболевания легких, астмы и аллергического ринита - соединение по настоящему изобретению можно комбинировать с такими агентами как ингибиторы TNF-α, такие как моноклональные антитела против TNF (такие как Ремикейд (Ramicade), CDP-870 и D2E7) и иммуноглобулиновые молекулы к рецептору TNF (такие как per. как Enbrel), неселективные ингибиторы СОХ(циклооксигеназа)-1/СОХ-2 (такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин), ингибиторы СОХ-2 (такие как мелоксикам, целекоксиб, рофекоксиб, валдекоксиб и эторикоксиб), низкая доза метотрекстата, лефунамида, циклезонида, гидроксихлорохина, d-пеницилламина, ауранофина или парентерального или перорального золота.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы 1 вместе с ингибитором биосинтеза лейкотриенов, ингибитором 5-липоксигеназы (5-LO) или антагонистом белка, активирующего 5-липоксигеназу (FLAP), таким как зилейтон, АВТ-761; фенлейтон; тепоксалин; Abbott-79175; Abbott-85761; N-(5-замещенные)-тиофен-2-алкилсульфонамиды; 2,6-ди-трет-бутилфенолгидразоны; метокситетрагидропираны, такие как Zeneca ZD-2138; соединение SB-210661; пиридинил-замещенные 2-цианонафталиновые соединения, такие как L-739010; 2-цианохинолиновые соединения, такие как L-746530; соединения индола и хинолина, такие как МК-591, МК-886 и BAY×1005.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с антагонистом рецепторов лейкотриенов LTB4, LTC4, LTD4 и LTE4, выбранного из группы, состоящей из фенотиазин-3-онов, таких как L-651392; амидино-соединений, таких как CGS-25019C; бензоксаламинов, таких как онтазоласт; бензолкарбоксимидамидов, таких как BIIL 284/260; и соединений, таких как зафирлукаст, аблукаст, монтелукаст, пранлукаст, верлукаст (МК-679), RG-12525, Ro-245913, иралукаст (CGP 45715А) и BAY×7195.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с ингибитором PDE4, включая ингибиторы изоформы PDE4D.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с антигистаминными антагонистами Н1-рецепторов, такими как цетиризин, лоратадин, деслоратадин, фексофенадин, астемизол, азеластин и хлорфенирамин.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с гастропротективным антагонистом Н2-рецептора.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с сосудосуживающим симпатомиметическим агентом - агонистом α1- и α2-адренорецепторов, таким как пропилгекседрин, фенилэфрин, фенилпропаноламин, псевдоэфедрин, нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид и этилнорэпинефрина гидрохлорид.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с антихолинергическими агентами, такими как ипратропия бромид; тиотропия бромид; окситропия бромид; пирензепин и телензепин.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с агонистами β1-β4-адренорецепторов, такими как метапротеренол, изопротеренол, изопреналин, альбутерол, сальбутамол, формотерол, салметерол, тербуталин, орципреналин, битолтерола мезилат и пирбутерол; или метилксантанинами, включая теофиллин и аминофиллин; кромогликатом натрия; или антагонистом мускариновых рецепторов (М1, М2 и М3).
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с миметиком инсулиноподобного фактора роста I типа (IGF-1).
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с ингаляционным глюкокортикоидом со сниженными системными побочными эффектами, таким как преднизон, преднизолон, флунизолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат и мометазона фуроат.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с другими модуляторами функции хемокиновых рецепторов, такими как CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10
и CCR11 (для семейства С-С); CXCR1, CXCR3, CXCR4 и CXCR5 (для семейства С-Х-С) и CX3CR-1 для семейства С-Х3-С.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с противовирусными агентами, такими как вирацепт, AZT, ацикловир и фамцикловир, и антисептическими соединениями, такими как валант (valant).
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с сердечно-сосудистыми агентами, такими как блокаторы кальциевых каналов, агентами, снижающими уровень липидов, такими как статины, фибраты, бета-блокаторы, ингибиторы АПФ, антагонисты рецепторов ангиотензина-2 и ингибиторы агрегации тромбоцитов.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы I вместе с агентами ЦНС, такими как антидепрессанты (такие как сертралин), противопаркинсонические лекарственные средства (такие как депренил, L-допа, Requip, Mirapex, ингибиторы МАОВ, такие как селегин и разагилин, ингибиторы соmР, такие как Tasmar, ингибиторы А-2, ингибиторы обратного захвата дофамина, антагонисты NMDA, агонисты никотина, агонисты дофамина и ингибиторы нейронной синтазы оксида азота) и лекарственные средства, используемые при болезни Альцгеймера, такие как донепезил, такрин, ингибиторы СОХ-2, пропентофиллин или метрифонат.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы 1 вместе с (1) ингибиторами триптазы; (2) антагонистами тромбоцит-активирующего фактора (PAF); (3) ингибиторами интерлейкин-превращающего фермента (ICE); (4) ингибиторами IMPDH; (5) ингибиторами молекул адгезии, включая антагонисты VLA-4; (6) ингибиторами катепсинов, например катепсина В, катепсина К, катепсина L; (7) ингибиторами МАР-киназы; (8) ингибиторами глюкозо-6-фосфат-дегидрогеназы; (9) антагонистами кинин-B1- и B2-рецепторов; (10) противоподагрическими агентами, например колхицином; (11) ингибиторами ксантиноксидазы, например аллопуринолом; (12) урикозурическими агентами, например пробенецидом, сульфинпиразоном и бензбромароном; (13) средствами, усиливающими секрецию гормона роста; (14) модуляторами трансформирующего фактора роста (TGFβ); (15) модуляторами тромбоцитарного фактора роста (PDGF); (16) модуляторами фактора роста фибробластов, например основным фактором роста фибробластов (bFGF); (17) модуляторами гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF); (18) капсаициновым кремом; (19) антагонистами NK1- и NK3-рецепторов тахикинина, выбранных из группы, состоящей из NKP-608C; SB-233412 (талнетант) и D-4418; (20) ингибиторами эластазы, выбранными из группы, состоящей из UT-77 и ZD-0892; (21) ингибиторами TNFα-превращающего фермента (ТАСЕ); (22) ингибиторами индуцибельной синтазы оксида азота (iNOS) или (23) хемоаттрактантной рецептор-гомологичной молекулы, экспрессирующейся на клетках ТН2 (антагонисты CRTH2).
Соединение формулы 1 также можно использовать в комбинации с агентами, используемыми при остеопорозе, такими как ролоксифен, дролоксифен, лазофоксифен или фозомакс, и иммуносупрессивными агентами, такими как FK-506, рапамицин, циклоспорин, азатиоприн и метотрексат.
Соединение формулы 1 также можно использовать в комбинации с уже существующими терапевтическими агентами для лечения остеоартрита. Подходящие агенты для применения в комбинации включают в себя стандартные нестероидные противовоспалительные агенты (в дальнейшем NSAID), такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин, ингибиторы СОХ-2, такие как целекоксиб, валдекоксиб, рофекоксиб и эторикоксиб, анальгетики и внутрисуставные лекарственные средства, такие как кортикостероиды, и гиалуроновые кислоты, такие как гиалган и синвиск, и антагонисты Р2Х7-рецепторов.
Соединение формулы 1 также можно использовать в комбинации с уже существующими терапевтическими агентами для лечения рака. Подходящие агенты для применения в комбинации включают в себя:
(1) антипролиферативные/антинеопластические лекарственные средства и их комбинации, используемые в медицинской онкологии, такие как алкилирующие агенты (например цис-платин, карбоплатин, циклофосфамид, азотистый иприт, мелфалан, хлорамбуцил, бусульфан и нитрозомочевины); антиметаболиты (например антифолаты, такие как фторпиримидины, например 5-фторурацил и тегафур, ралтитрексед, метотрексат, цитозина арабинозид, гидроксимочевина, гемцитабин и паклитаксел (Taxol®)); противоопухолевые антибиотики (например антрациклины, такие как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митамицин-С, дактиномицин и митрамицин); антимитотические агенты (например алкалоиды барвинка, такие как винкристин, винбластин, виндезин и винорелбин, и таксоиды, такие как таксол и таксотер) и ингибиторы топоизомеразы (например эпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан и камптотецин);
(2) цитостатические агенты, такие как антиэстрогены (например тамоксифен, торемифен, ралоксифен, дролоксифен и идоксифен), понижающие регуляторы рецепторов эстрогенов (например фулвестрант), антиандрогены (например бикалутамид, флутамид, нилутамид и ципротерона ацетат), антагонисты LHRH или агонисты LHRH (например гозерелин, лейпрорелин и бусерелин), прогестогены (например мегестрола ацетат), ингибиторы ароматазы (например анастрозол, летрозол, воразол и эксеместан) и ингибиторы 5α-редуктазы, такие как финастерид;
(3) агенты, которые ингибируют инвазию раковых клеток (например ингибиторы металлопротеиназ, такие как маримастат, и ингибиторы функции рецептора активатора плазминогена урокиназного типа);
(4) модуляторы функции фактора роста, например такие ингибиторы включают в себя антитела к факторам роста, антитела к рецепторам факторов роста (например анти-erbb2 антитело трастузумаб [Herceptin™] и анти-erbb1 антитело цетуксимаб [С225]), ингибиторы фарнезилтрансферазы, ингибиторы тирозинкиназы и ингибиторы серин/треонинкиназы, например ингибиторы семейства эпидермальных факторов роста (EGFR) (например ингибиторы тирозинкиназы семейства EGFR, такие как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, ZD1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) и 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)хиназолин-4-амин (CI 1033)), например ингибиторы семейства тромбоцитарных факторов роста и, например, ингибиторы семейства факторов роста гепатоцитов;
(5) антиангиогенные агенты, ингибирующие эффекты фактора роста эндотелия сосудов (например антитело к фактору роста эндотелия сосудов бевацизумаб [Avastin™], соединения, описанные в международных патентных заявках WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354, и соединения с другими механизмами действия (например линомид, ингибиторы функции интегрина-αvβ3 и ангиостатин);
(6) сосудоповреждающие агенты, такие как комбретастатин А4 и соединения, описанные в международных патентных заявках WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 и WO 02/08213;
(7) антисмысловые терапии, например, направленные на перечисленные выше мишени, такие как ISIS 2503, анти-ras антисмысловая терапия;
(8) генотерапевтические подходы, включая, например, подходы с заменой аберрантных генов, таких как аберрантный р53 или аберрантный BRCA1, или BRCA2, подходы GDEPT (ген-направленная ферментная пролекарственная терапия), такие как применение цитозиндезаминазы, тимидинкиназы или бактериального фермента нитроредуктазы, и подходы с повышением толерантности пациента к химиотерапии или радиотерапии, такие как генная терапия множественной лекарственной устойчивости; и
(9) иммунотерапевтические подходы, включая, например, подходы ех-vivo и in-vivo для повышения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, такими как интерлейкин-2, интерлейкин-4 или гранулоцитарно-макрофагальный колониестимулирующий фактор, подходы для снижения Т-клеточной анэргии, подходы с использованием трансфицированных иммунных клеток, таких как цитокин-трансфицированные дендритные клетки, подходы с использованием цитокин-трансфицированных опухолевых клеточных линий и подходы с использованием антиидиотипических антител.
При приготовлении в виде фиксированной дозы в таких комбинированных продуктах используют соединение формулы I в дозовом диапазоне, описанном в данной заявке, и другой фармацевтически активный агент в утвержденном дозовом диапазоне. Последовательное использование предполагается, когда комбинированный препарат является нецелесообразным.
Хотя соединение формулы I в первую очередь полезно в качестве терапевтического агента для применения у теплокровных животных (включая человека), оно также полезно всякий раз, когда требуется ингибирование эффектов металлопротеиназы. Таким образом, оно полезно в качестве фармакологического стандарта для применения в разработке новых биологических тестов и в поиске новых фармакологических агентов.
Далее изобретение проиллюстрировано нижеследующими неограничивающими примерами.
Соответствующие исходные вещества имеются в продаже или могут быть получены любым подходящим способом, как описано в литературе или известно квалифицированному химику, или описано в этих примерах. Кроме того, нижеследующая таблица отражает детали промежуточных соединений и соответствующие регистрационные номера в Chemical Abstracts.
Пример 1
(5S)-5-Метил-5-[({4-[4-(2,2,2-трифторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион
К раствору 4-[4-(2,2,2-трифторэтокси)фенокси]пиперидина гидрохлорида (0,3 г) и диизопропилэтиламина (0,37 мл) в дихлорметане (100 мл) добавляли [(4S)-4-метил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорид (0,261 г). Полученный раствор перемешивали при температуре окружающей среды в течение 18 часов.
Реакционный раствор предварительно адсорбировали непосредственно на силикагель и очищали с помощью хроматографии на колонке с силикагелем, элюируемой этилацетатом. Полученное вещество растирали в порошок, фильтровали и промывали диэтиловым эфиром с получением (5S)-5-метил-5-[({4-[4-(2,2,2-трифторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]-имидазолидин-2,4-диона [0,33 г].
ЯМР-спектр: (ДMCOd6) 1.15 (s, 3Н), 1.6 (s, 2Н), 1.8 (m, 2H), 3.1 (m, 2H), 3.2-3.6 (m, 4H), 4.4 (m, 1H), 4.6-4.7 (m, 2H), 6.9 (m, 4H), 8.9 (s, 1H) 10.7 (широкий, 1Н). Масс-спектр: М-Н- 464.
Соответствующее исходное вещество синтезировали следующим образом:
К раствору 4-бензилоксифенола (10 г), трет-бутил-4-гидрокси-1-пиперидинкарбоксилата (11 г) и трифенилфосфина (19,7 г) в дихлорметане (300 мл) добавляли раствор диизопропилдиазодикарбоксилата (14,8 мл) в дихлорметане (15 мл) по каплям в течение 15 минут. Реакционную смесь нагревали до температуры дефлегмации в течение 4 часов.
Растворитель удаляли. Остаток, перемешанный со смесью 20% этилацетата/изогексан (250 мл) и трифенилфосфиноксидом, отфильтровывали. Фильтрат упаривали и повторно растворяли в дихлорметане (100 мл) и предварительно адсорбировали на силикагеле. Очистку осуществляли с использованием слоя силикагеля с помощью градиентной элюции смесью 2-20% этилацетат/изогексан. Выделенное вещество растирали со смесью 10% диэтилового эфира/изогексан (100 мл) с получением трет-бутил-4-[4-(бензилокси)фенокси]пиперидин-1-карбоксилата (12,2 г). ЯМР-спектр: (ДMCOd6) 1.4 (s, 9Н), 1.5 (m, 2Н), 1.8 (m, 2Н), 3.1 (m, 2H), 3.6 (m, 2H), 4.4 (m, 1H), 5.0 (s, 2H), 6.9 (m, 4H), 7.3-7.5 (m, 5H). Масс-спектр: М-Н- 284.
К 10% палладия на углероде (0,75 г) в токе аргона добавляли раствор трет-бутил-4-[4-(бензилокси)фенокси]пиперидин-1-карбоксилата (7,5 г) в этаноле (250 мл). Сосуд продували аргоном три раза перед введением в систему водорода через баллон. Реакционную смесь энергично перемешивали при температуре окружающей среды в течение 3 часов. Водород удаляли из системы и продували три раза аргоном перед фильтрацией через слой целита. Слой тщательно промывали. Фильтрат и промывки объединяли и упаривали, твердое вещество растирали со смесью 20% диэтилового эфира/изогексан с получением трет-бутил-4-гидрокси-1-пиперидинкарбоксилата (5,7 г). ЯМР-спектр: (CDCl3) 1.5 (s, 9Н), 1.7 (m, 2H), 1.9 (m, 2H), 3.3 (m, 2H), 3.7 (m, 2H), 4.3 (m, 1H), 4.8 (s, 1H), 6.7 (m,4H).
К суспензии трет-бутил-4-гидрокси-1-пиперидинкарбоксилата (4 г) и свежеизмельченного карбоната калия (4,2 г) в ацетоне (200 мл) добавляли чистый 2,2,2-трифторэтилнонафторбутан-сульфонат (6,7 г) и оставляли перемешиваться в течение 3 часов при температуре окружающей среды. Через 4 часа осуществляли дальнейшее добавление нонафлата (3,3 г) и температуру увеличивали до температуры дефлегмации в течение 18 часов.
Карбонат калия отфильтровывали, остаток упаривали и растворяли в этилацетате (200 мл), промывали водой (100 мл) и насыщенным рассолом (100 мл), сушили над сульфатом магния и упаривали для получения грубого белого твердого вещества, которое очищали с помощью хроматографии на силикагеле, элюируя смесью 20% этилацетата/изогексан, с получением трет-бутил-4-[4-(2,2,2-трифторэтокси)фенокси]пиперидин-1-карбоксилата (3 г). ЯМР-спектр: (CDCl3) 1.45 (s, 9Н), 1.7 (m, 2H), 1.9 (m, 2H), 3.3 (m, 2H), 3.7 (m, 2H), 4.3 (m, 3Н), 6.85 (m, 4H).
К трет-бутил-4-[4-(2,2,2-трифторэтокси)фенокси]пиперидин-1-карбоксилату (3 г) добавляли 4 молярный хлористый водород в 1,4-диоксане (50 мл), перемешивали при температуре окружающей среды в течение 1 часа. Растворитель удаляли, и полученное твердое вещество растирали в порошок и дважды промывали небольшим количеством диэтилового эфира с получением 4-[4-(2,2,2-трифторэтокси)фенокси]пиперидина в виде гидрохлоридной соли (2,4 г). ЯМР-спектр: (ДMCOd6) 1.7 (m, 2H), 2.0 (m, 2H), 3.0 (m, 2H), 3.2 (m, 2H), 4.5 (m, 1H), 4.6 (m, 2H), 6.9 (m, 4H), 8.8 (широкий, 2H). Масс-спектр: М-Н- 276.
Пример 2
(5S)-5-Этил-5-[({4-[4-(2,2,2-трифторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион
[(4S)-4-Этил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорид (WO 2004/024698) (869 мг) добавляли к перемешиваемому раствору 4-[4-(2,2,2-трифторэтокси)фенокси]пиперидина гидрохлорида (750 мг) и триэтиламина (1,68 мл) в дихлорметане (50 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов. Растворители удаляли в вакууме и остаток перемешивали в воде в течение 2 часов. Полученное твердое вещество отфильтровывали, промывали водой, затем эфиром и сушили с получением указанного в заголовке соединения (1,13 г); ЯМР-спектр: (ДMCOd6) 0.78 (t, 3Н), 1.25 (m, 2H), 1.66 (m, 4H), 1.94 (m, 2H), 3.11 (m, 2H), 3.49 (d, 1H), 3.58 (m, 1H), 4.41 (m, 1H), 4.64 (q, 2H), 6.97 (m, 4H), 8.5 (s, 1H), 10.71 (s, 1 Н); Масс-спектр: М-Н- 478.
Пример 3
5S-Метил-5-[({4-[4-(1,1,2,2-тетрафторэтокси)фенокси]пиперидин-ил}сульфонил)метил]имидазолидин-2,4-дион
Диизопропилэтиламин (0,35 мл) и (45-метил-2,5-диоксоимидазолидин-4-ил)метансульфонилхлорид (248 мг) добавляли к суспензии 4-[4-(1,1,2,2-тетрафторэтокси)фенокси]пиперидина (300 мг) в дихлорметане (50 мл). Смесь перемешивали при температуре окружающей среды в течение 18 часов. Смесь затем предварительно адсорбировали на силикагель при пониженном давлении и очищали с помощью хроматографии на колонке с силикагелем, элюируя этилацетатом. Выделенный продукт затем перекристаллизовывали из этанола (5 мл) и фильтровали. Твердое вещество затем перемешивали в диэтиловом эфире, фильтровали и сушили под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (245 мг); ЯМР-спектр: (ДMCOd6) 1.34 (s, 3H), 1.66-1.78 (m, 2Н), 1.92-2.04 (m, 2H), 3.08-3.20 (m, 2H), 3.31-3.42 (m, 2H), 3.35 (d, 1H), 3.52 (d, 1H), 4.50-4.60 (m, 1H), 6.76 (tt, 1H), 7.06 (d, 2H), 7.20 (d, 2H), 7.98 (s, 1H), 10.72 (s. 1H); Масс-спектр: М-Н- 482.
4-[4-(1,1,2,2-Тетрафторэтокси)фенокси]пиперидин получали следующим образом:
Диизопропилазодикарбоксилат (2,25 мл) добавляли к раствору 4-(1,1,2,2-тетрафторэтокси)фенола (2,0 г), трет-бутил-4-гидроксипиперидин-1-карбоксилата (2,3 г) и трифенилфосфина (3,5 г) в дихлорметане (30 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 18 часов и затем упаривали при пониженном давлении. Полученную смесь очищали с помощью хроматографии на колонке с силикагелем, элюируя градиентом 0-15% этилацетата в гексане, с получением трет-бутил-4-[4-(1,1,2,2-тетрафторэтокси)фенокси]пиперидин-1-карбоксилата в виде светло-зеленого масла (3,3 г); ЯМР-спектр: (ДMCOd6) 1.39 (s, 9H), 1.43-1.57 (m, 2H), 1.82-1.95 (m, 2H), 3.09-3.24 (m, 2H), 3.56-3.70 (m, 2H), 4.47-4.59 (m, 1H), 6.73 (tt, 1H), 7.03 (d,2H), 7.16 (d,2H).
4 M HCl в диоксане (30 мл) добавляли к трет-бутил-4-[4-(1,1,2,2-тетрафторэтокси)фенокси]пиперидин-1-карбоксилату (3,3 г). Смесь перемешивали при температуре окружающей среды в течение 30 минут. Затем смесь концентрировали при пониженном давлении и растирали с диэтиловым эфиром. Полученный осадок фильтровали, промывали диэтиловым эфиром и сушили под вакуумом с получением 4-[4-(1,1,2,2-тетрафторэтокси)фенокси]-пиперидина в виде гидрохлоридной соли (2,76 г); ЯМР-спектр: (ДMCOd6) 1.75-1.91 (m, 2H), 2.02-2.17 (m, 2H), 2.95-3.12 (m, 2H), 3.13-3.29 (m, 2H), 4.57-4.69 (m, 1H), 6.75 (tt, 1H), 7.06 (d, 2H), 7.19 (d, 2H), 8.95 (bs, 2H); Масс-спектр: M+H+ 294.
Пример 4
5S-Этил-5-[({4-[4-(1,1,2,2-тетрафторэтокси)фенокси]пиперидин-ил}сульфонил)метил]имидазолидин-2,4-дион
Диизопропилэтиламин (0,35 мл) и (4S-этил-2,5-диоксоимидазолидин-4-ил)метансульфонилхлорид (262 мг) добавляли к суспензии 4-[4-(1,1,2,2-тетрафторэтокси)фенокси]пиперидина (300 мг) в дихлорметане (50 мл). Смесь перемешивали при температуре окружающей среды в течение 18 часов. Затем смесь предварительно адсорбировали на силикагель при пониженном давлении и очищали с помощью хроматографии на колонке с силикагелем, элюируя этилацетатом. Выделенный продукт затем перекристаллизовывали из этанола (5 мл) и фильтровали. Твердое вещество затем перемешивали в диэтиловом эфире, фильтровали и сушили под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (250 мг); ЯМР-спектр: (ДMCOd6) 0.78 (t, 3H), 1.60-1.79 (m, 2H), 1.65 (q, 2H), 1.90-2.09 (m, 2H), 3.09-3.20 (m, 2H), 3.31-3.42 (m, 2H), 3.35 (d, 1H), 3.50 (d, 1H), 4.50-4.60 (m, 1H), 6.76 (tt, 1H), 7.06 (d, 2H), 7.20 (d, 2H), 7.95 (s, 1H), 10.74 (s, 1H); Масс-спектр: М-Н- 496.
Пример 5
(5S)-5-Метил-5-[({4-[4-(пентафторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион
К раствору 4-[4-(пентафторэтокси)фенокси]пиперидина гидрохлорида (0,15 г) и диизопропилэтиламина (0,19 мл) в дихлорметане (100 мл) добавляли [(4S)-4-метил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорид (0,12 г). Реакционную смесь перемешивали при температуре окружающей среды в течение 18 часов.
Реакционный раствор подвергали немедленной очистке с использованием колоночной хроматографии, элюируя градиентом 1-5% метанола в дихлорметане. Полученное вещество растирали с небольшим объемом 50% этанола/диэтилового эфира. Полученное твердое вещество промывали диэтиловым эфиром, фильтровали и сушили при пониженном давлении с получением указанного в заголовке соединения (0,18 г); ЯМР-спектр: (ДMCOd6) 1.0 (m, 3H), 1.35 (s, 3H), 1.7 (m, 2H), 1.9 (m, 2H), 3.1 (m, 2H), 3.3-3.5 (m, 4H), 4.5 (m, 1H), 7.05 (m, 2H), 7.25 (m, 2H) 8.0 (s, 1H), 10.7 (s, 1H); Масс-спектр: М-Н- 500.
4-[4-(Пентафторэтокси)фенокси]пиперидина гидрохлорид, используемый в качестве исходного вещества, получали следующим образом:
К раствору 4-(1,1,2,2,2-пентафторэтокси)фенола (6,5 г), трет-бутил-4-гидрокси-1-пиперидинкарбоксилата (6,3 г), трифенилфосфина (11,2 г) в дихлорметане (400 мл) добавляли чистый диизопропилдиазодикарбоксилат (5,6 мл) по каплям в течение 5 минут. Затем реакционную смесь нагревали до температуры дефлегмации в течение 18 часов.
Реакционный раствор предварительно адсорбировали на силикагеле и очищали с использованием колоночной хроматографии, элюируя смесью этилацетата и изогексана (1:4), с получением трет-бутил-4-[4-(пентафторэтокси)фенокси]пиперидин-1-карбоксилата (3,9 г). ЯМР-спектр: (CDCl3) 1.2 (s, 9H), 1.5 (m, 2H), 1.8 (m, 2H), 3.1 (m, 2H), 3.6 (m, 2H), 4.6 (m, 1H), 7.05 (m, 2H), 7.25 (m, 2H); Масс-спектр: M-tBu- 354.
Трет-бутил-4-[4-(пентафторэтокси)фенокси]пиперидин-1-карбоксилат (3,8 г) размешивали в 4,0 M HCl в 1,4-диоксане (50 мл) в течение 1 часа. Растворитель удаляли, полученное твердое вещество растирали с диэтиловым эфиром (50 мл), отфильтровывали и промывали диэтиловым эфиром (2×50 мл) с получением 4-[4-(пентафторэтокси)фенокси]пиперидина гидрохлорида в виде белого твердого вещества (2,9 г). ЯМР-спектр: (CDCl3) 2.1 (m, 2H), 2.3 (m, 2H), 3.3 (m, 4H), 4.6.(s, 1H), 6.9(m, 2H), 7.2 (m, 2H), 9.8 (широкий, 1H); Масс-спектр: М-Н- 312.
Пример 6
(5S)-5-Этил-5-[({4-[4-(пентафторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион
К раствору 4-[4-(пентафторэтокси)фенокси]пиперидина гидрохлорида (0,17 г) и диизопропилэтиламина (0,19 мл) в дихлорметане (100 мл) добавляли [(4S)-4-этил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорид (0,13). Реакционную смесь перемешивали при температуре окружающей среды в течение 18 часов.
Реакционный раствор подвергали немедленной очистке с использованием колоночной хроматографии, элюируя градиентом 1-5% метанола в дихлорметане. Полученное вещество растирали с небольшим объемом смеси 50% этанола/диэтиловый эфир. Полученное твердое вещество промывали диэтиловым эфиром, фильтровали и сушили под вакуумом с получением указанного в заголовке соединения (0,17 г). ЯМР-спектр: (ДMCOd6) 0.8 (m, 3Н), 1.2 (s, 2Н), 1.7 (m, 2H), 1.8 (m, 2H), 2.0 (m, 2H), 3.1 (m, 2H), 3.3-3.5 (m, 4H), 4,6 (m, 1H), 7,1 (m, 2H), 7,25 (m, 2H), 7,9 (s, 1H), 10,7 (s, 1H). Масс-спектр: М-Н- 514.
Пример 7
5S-Метил-5-[({4-[3,3,3-трифторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион
Диизопропилэтиламин (0,37 мл) и (4S-метил-2,5-диоксоимидазолидин-4-ил)метансульфонилхлорид (117 мг) добавляли к суспензии 4-[4-(3,3,3-трифторпропокси)фенокси]пиперидина гидрохлорида (140 мг) в метиленхлориде (20 мл). Смесь перемешивали при температуре окружающей среды в течение 18 часов. Реакция не завершалась, поэтому добавляли (4S-метил-2,5-диоксоимидазолидин-4-ил)метансульфонилхлорид (50 мг). Смесь перемешивали при температуре окружающей среды в течение 4 часов. Затем смесь концентрировали при пониженном давлении и очищали с помощью хроматографии на колонке с силикагелем, элюируя градиентом 0-100% метанола в дихлорметане, с получением указанного в заголовке соединения в виде белого твердого вещества (90 мг). ЯМР-спектр: (ДMCOd6) 1.34 (s, 3Н), 1.63-1.75 (m, 2H), 1.87-1.98 (m, 2H), 2.65-2.82 (m, 2H), 3.06-3.19 (m, 2H), 3.30-3.41 (m, 2H), 3.34 (а, 1H), 3.51 (d, 1H), 4.15 (t, 2H), 4.35-4.45 (m, 1H), 6.87-6.98 (m, 4H), 7.98 (s, 1H), 10.71 (s, 1H); Масс-спектр: М-Н- 478.
Исходное вещество 4-[4-(3,3,3-трифторпропокси)фенокси]пиперидин получали следующим образом:
Диизопропилазодикарбоксилат (2,36 мл) добавляли к раствору 4-(бензилокси)фенола (2,0 г), трет-бутил-4-гидроксипиперидин-1-карбоксилата (2,41 г) и трифенилфосфина (3,67 г) в дихлорметане (30 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 18 часов и затем упаривали при пониженном давлении. Эту полученную смесь очищали с помощью хроматографии на колонке с силикагелем, элюируя градиентом 0-20% этилацетата в гексане, с получением трет-бутил-4-[4-(бензилокси)фенокси]пиперидин-1-карбоксилата в виде светло-оранжевого масла (3,25 г). ЯМР-спектр: (CDCl3) 1.47 (s, 9H), 1.65-1.77 (m, 2H), 1.83-1.93 (m, 2H), 3.24-3.34 (m, 2H), 3.65-3.76 (m, 2H), 4.27-4.35 (m, 1H), 5.01 (s, 2H), 6.80-6.93 (m, 4H), 7.28-7.45 (m, 5H); Масс-спектр: (M-tBuOCO)+H+ 284.
10% палладия на углероде (0,75 г, 50% об./об.) добавляли к раствору трет-бутил-4-[4-(бензилокси)фенокси]пиперидин-1-карбоксилата (1,5 г) в этаноле (100 мл). Смесь удаляли и дважды продували водородом, и затем перемешивали в атмосфере водорода в течение 2 часов. Смесь фильтровали через целит и фильтровальный слой промывали этанолом. Фильтрат концентрировали при пониженном давлении с получением трет-бутил-4-(гидроксифенокси)пиперидин-1-капроксилата в виде коричневого твердого вещества (1,24 г); ЯМР-спектр: (CDCl3) 1.47 (s, 9H), 1.65-1.76 (m, 2H), 1.82-1.93 (m, 2H), 3.23-3.33 (m, 2H), 3.65-3.76 (m, 2H), 4.25-4.34 (m, 1H), 5.07 (s, 1H), 6.70-6.85 (m, 4H).
Диизопропилазодикарбоксилат (0,97 мл) добавляли к раствору трет-бутил-4-(гидроксифенокси)пиперидин-1-карбоксилата (1,2 г), 3,3,3-трифтор-1-пропанола (0,56 г) и трифенилфосфина (1,5 г) в дихлорметане (15 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 18 часов и затем концентрировали при пониженном давлении. Эту полученную смесь очищали с помощью хроматографии на колонке с силикагелем, элюируя градиентом 0-15% этилацетата в гексане, с получением трет-бутил-4-[4-(3,3,3-трифторпропокси)фенокси]пиперидин-1-карбоксилата в виде белого твердого вещества (0,6 г); ЯМР-спектр: (CDCl3) 1.47 (s, 9H), 1.66-1.77 (m, 2H), 1.83-1.94 (m, 2H), 2.52-2.66 (m, 2H), 3.24-3.34 (m, 2H), 3.65-3.75 (m, 2H), 4.11 (t, 2H), 4.27-4.37 (m, 1H), 6.78-6.89 (m, 4H); Масс-спектр: M+H+ 390 and (M-tBuOCO)+H+ 290.
4 M HCl в диоксане (10 мл) добавляли к трет-бутил-4-[4-(3,3,3-трифторпропокси)фенокси]пиперидин-1-карбоксилату (550 мг). Смесь перемешивали при температуре окружающей среды в течение 10 минут. Затем смесь концентрировали при пониженном давлении и растирали с диэтиловым эфиром, и перемешивали в течение 10 минут. Полученный осадок фильтровали и сушили под вакуумом с получением 4-[4-(3,3,3-трифторпропокси)фенокси]пиперидина в виде гидрохлоридной соли (286 мг); ЯМР-спектр: (ДMCOd6) 1.76-1.88 (m, 2Н), 2.01-2.12 (m, 2H), 2.66-2.82 (m, 2H), 2.97-3.10 (m, 2H), 3.14-3.27 (m, 2H), 4.15 (t, 2H), 4.46-4.56 (m, 1H), 6.87-6.98 (m, 4H), 9.03 (bs, 2H); Масс-спектр: M+H+ 290.
Пример 8
5S-Этил-5-[({4-[3,3,3-трифторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион
Диизопропилэтиламин (0,37 мл, 2,12 ммоль) и (4S-этил-2,5-диоксоимидазолидин-4-ил)метансульфонилхлорид (124 мг, 0,52 ммоль) добавляли к суспензии 4-[4-(3,3,3-трифторпропокси)фенокси]пиперидина гидрохлорида (140 мг) в дихлорметане (20 мл). Смесь перемешивали при температуре окружающей среды в течение 18 часов. Реакция не завершалась, поэтому добавляли (4S-этил-2,5-диоксоимидазолидин-4-ил)метансульфонилхлорид (50 мг). Смесь перемешивали при температуре окружающей среды в течение 4 часов. Затем смесь концентрировали при пониженном давлении и очищали с помощью хроматографии на колонке с силикагелем, элюируя градиентом 0-100% этилацетата в гексане, с получением указанного в заголовке соединения в виде белого твердого вещества (90 мг); ЯМР-спектр: (ДMCOd6) 0.78 (t, 3Н), 1.60-1.65 (m, 2H), 1.65 (q, 2H), 1.88-1.98 (m, 2H), 2.66-2.81 (m, 2H), 3.06-3.16 (m, 2H), 3.30-3.40 (m, 2H), 3.33 (d, 1H), 3.49 (d, 1H), 4.15 (t, 2H), 4.36-4.45 (m, 1H), 6.87-6.97 (m, 4H), 7.95 (s, 1H), 10.73 (s, 1H); Масс-спектр: М-Н- 492.
Пример 9
(5S)-5-Метил-5-[({4-[4-(2,2,3,3-тетрафторпропокси)фенокси]-пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион
[(4S)-4-Метил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорид (0,453 г) добавляли к раствору 4-{4-(2,2,3,3-тетрафторпропокси)фенокси]-пиперидина гидрохлорида (0,69 г) в дихлорметане (25 мл) и триэтиламине (1,7 мл) при температуре окружающей среды. Перемешивали в течение 16 часов и упаривали досуха. Остаток очищали с помощью колоночной хроматографии (с использованием λ 230 нм в качестве детектирующей длины волны), элюируя 0-10% метанола и дихлорметаном. Получали твердый продукт, который сушили под вакуумом при 50°С с получением указанного в заголовке соединения (0,24 г). ЯМР-спектр: (ДMCOd6) δ 10.7 (s, 1H), 8.0 (s, 1H), 6.95 (m, 4H), 6.6 (tt, 1H), 4.5 (m, 2H), 4.4 (m, 1H), 3.5 (d, 1H), 3.3 (m, 3H), 3.1 (m, 2H), 1.9 (m, 2H), 1.7 (m, 2H), 1.35 (s, 3H). Масс-спектр: М-Н- 495.89.
4-{4-(2,2,3,3-Тетрафторпропокси)фенокси]пиперидина гидрохлорид, используемый в качестве исходного вещества, получали следующим образом:
2,2,3,3-Тетрафторпропанол (2,64 г) добавляли к суспензии гидрида натрия (1,08 г) в сухом эфире (50 мл) при 0°С в атмосфере аргона. Перемешивали при 0°С в течение 15 минут. Медленно добавляли перфтор-1-бутансульфонилфторид (12,08 г). Перемешивали при температуре дефлегмации в течение 3 часов, охлаждали и осторожно гасили H2O. Дважды экстрагировали эфиром. Объединенные экстракты промывали насыщенным рассолом, сушили над MgSO4, фильтровали и упаривали с получением 2,2,3,3-тетрафторпропил-1,1,2,2,3,3,4,4,4-нонафторбутан-1-сульфоната в виде масла. Выход 5,47 г. ЯМР-спектр: (CDCl3) δ 5.9 (tt, 1H), 4.75 (t, 2H).
2,2,3,3-Тетрафторпропил-1,1,2,2,3,3,4,4,4-нонафторбутан-1-сульфонат (4,84 г) растворяли в ацетоне (50 мл). Добавляли трет-бутил-4-(4-гидроксифенокси)пиперидин-1-карбоксилат (1,71 г) и карбонат калия (2,42 г) и перемешивали при температуре окружающей среды в течение 16 часов. Отфильтровывали нерастворимое вещество и фильтрат упаривали досуха с получением масла. Очищали с помощью колоночной хроматографии с использованием смеси 0-25% этилацетата/изогексан в качестве элюента. Получили 2,66 г трет-бутил-4-[4-(2,2,3,3-тетрафторпропокси)фенокси]-пиперидин-1-карбоксилата в виде масла. ЯМР-спектр: (CDCl3) δ 6.85 (m, 4H), 6.0 (tt, 1H), 4.6 (m, 2H), 4.26 (m, 1H), 3.7 (m, 2H), 3.3 (m, 2H), 1.85 (m, 2H), 1.7 (m, 2H), 1.45 (s, 9H).
Трет-бутил-4-[4-(2,2,3,3-тетрафторпропокси)фенокси]пиперидин-1-карбоксилат (2,66 г) растворяли в 1,4-диоксане (25 мл) и добавляли 4 M HCl в 1,4-диоксане (9,75 мл). Перемешивали при температуре окружающей среды в течение 16 часов. Реакционную смесь упаривали досуха с получением белого твердого вещества. Твердое вещество растирали с эфиром, выделяли и сушили под вакуумом при 50°С. Получили 4-{4-(2,2,3,3-тетрафторпропокси)фенокси]пиперидина гидрохлорид в виде твердого вещества, 1,38 г. ЯМР-спектр: (ДMCOd6) δ 9.0 (br, 1H), 7.0 (m, 4H), 6.65 (tt, 1H), 4.52 (m, 2H), 4.4 (m, 1H), 3.2 (m, 2H), 3.05 (m, 2H), 2.1 (m,2H), 1.8 (m, 2H). Масс-спектр: M+H+ 308.
Пример 10
(5S)-5-Этил-5-[({4-[4-(2,2,3,3-тетрафторпропокси)фенокси]-пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион
Методику, аналогичную описанной в примере 9, использовали для получения указанного в заголовке соединения, используя [(4S)-4-этил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорид по той же схеме. Получили 136 мг. ЯМР-спектр: (ДMCOd6) δ 10.8 (s, 1H), 7.95 (s, 1H), 6.95 (m, 4H), 6.7 (tt, 1H), 4.5 (m, 2H), 4.4 (m, 1H), 3.5 (d, 1H), 3.3 (m, 3H), 3.1 (m, 2H), 1.9 (m, 2H), 1.65 (m, 4H), 0.8 (t, 3H). Масс спектр: М-Н- 510.
Пример 11
(5S)-5-Метил-5-[({4-[4-(2,2,3,3,3-пентафторпропокси)фенокси]-пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион
[(4S)-4-Метил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорид (0,188 г) добавляли к раствору 4-{4-(2,2,3,3,3-пентафторпропокси)фенокси]-пиперидина гидрохлорида (0,30 г) в дихлорметане (10 мл) и триэтиламине (0,70 мл) при температуре окружающей среды. Перемешивали в течение 16 часов и упаривали досуха. Остаток очищали с помощью препаративной ВЭЖХ (с использованием λ 230 нм в качестве детектирующей длины волны), элюируя 0-95% ацетонитрила, H2O+0,2% трифторуксусной кислоты. Получили твердый продукт, который сушили под вакуумом при 50°С с получением указанного в заголовке соединения (0,076 г). ЯМР-спектр: (ДMCOd6) δ 10.7 (s, 1H), 8.0 (s, 1H), 7.0 (m, 4H), 4.75 (t, 2H), 4.45 (m, 1H), 3.5 (d, 1H), 3.3 (m, 3H), 3.1 (m, 2H), 1.95 (m, 2H), 1.7 (m, 2H), 1.35 (s, 3H). Масс-спектр: М-Н- 513.
4-{4-(2,2,3,3,3-Пентафторпропокси)фенокси]пиперидина гидрохлорид, используемый в качестве исходного вещества, получали следующим образом:
2,2,3,3,3-Пентафторпропанол (3,0 г) добавляли к суспензии гидрида натрия (1,08 г) в безводном эфире (50 мл) при 0°С в атмосфере аргона. Перемешивали при 0°С в течение 15 минут. Медленно добавляли перфтор-1-бутансульфонилфторид (12,08 г). Перемешивали при температуре дефлегмации в течение 3 часов, охлаждали и осторожно гасили H2O. Дважды экстрагировали эфиром. Объединенные экстракты промывали насыщенным рассолом, сушили над MgSO4, фильтровали и упаривали с получением 2,2,3,3,3-пентафторпропил-1,1,2,2,3,3,4,4,4-нонафторбутан-1-сульфоната в виде масла. Получили 7,9 г. ЯМР: (CDCl3) δ 4.9 (m, 2H).
2,2,3,3,3-Пентафторпропил-1,1,2,2,3,3,4,4,4-нонафторбутан-1-сульфонат (6,7 г) растворяли в ацетоне (75 мл). Добавляли трет-бутил-4-(4-гидроксифенокси)пиперидин-1-карбоксилат (2,34 г) и карбонат калия (3,31 г). Перемешивали при температуре окружающей среды в течение 16 часов. Отфильтровывали нерастворимое вещество и упаривали фильтрат досуха с получением масла. Очищали с помощью колоночной хроматографии, используя 0-20% этилацетата/изогексан в качестве элюента. Получили трет-бутил-4-[4-(2,2,3,3,3-пентафторпропокси)фенокси]пиперидин-1-карбоксилат, 0,81 г в виде масла. ЯМР-спектр: (CDCl3) δ 6.35 (m, 4H), 3.8 (m, 3H), 3.2 (m, 2H), 2.8 (m, 2H), 1.3 (m, 2H), 1.2 (m, 2H), 0.9 (s, 9H).
Трет-бутил-4-[4-(2,2,3,3,3-пентафторпропокси)фенокси]пиперидин-1-карбоксилат (0,81 г) растворяли в 4 M HCl в 1,4-диоксане (10 мл). Перемешивали при температуре окружающей среды в течение 16 часов. Реакционную смесь упаривали досуха с получением белого твердого вещества. Твердое вещество растирали с эфиром, выделяли и сушили под вакуумом при 50°С. Получили 4-{4-(2,2,3,3,3-пентафторпропокси)фенокси]пиперидина гидрохлорид в виде твердого вещества, 0,76 г. ЯМР-спектр: (ДMCOd6) δ 9.0 (br, 1Н), 7.0 (m, 4H), 4.7 (m, 2H), 4.5 (m, 1H), 3.2 (m, 2H), 3.0 (m, 2H), 2.05 (m, 2H), 1.8 (m, 2H). Масс спектр: M+H+ 326.
Пример 12
(5S)-5-Этил-5-[({4-[4-(2,2,3,3,3-пентафторпропокси)фенокси]-пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дион
Методику, аналогичную описанной в примере 11, использовали для получения указанного в заголовке соединения, используя [(4S)-4-этил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорида по той же схеме. Получили 0,85 г. ЯМР-спектр: (ДMCOd6) δ 10.8 (s, 1H), 7.95 (s, 1H), 6.95 (m, 4H), 6.7 (tt, 1H), 4.5 (m, 2H), 4.4 (m, 1H), 3.5 (d, 1H), 3.3 (m, 3H), 3.1 (m, 2H), 1.9 (m, 2H), 1.65 (m, 4H), 0.8 (t, 3H).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ГИДАНТОИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОБСТРУКТИВНЫХ ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2005 |
|
RU2386629C2 |
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2288228C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2378269C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2376301C2 |
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2293730C2 |
| N-ГИДРОКСИФОРМАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2004 |
|
RU2351595C2 |
| ПИПЕРИДИН- И ПИПЕРАЗИНЗАМЕЩЕННЫЕ N-ГИДРОКСИФОРМАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2001 |
|
RU2283306C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛИДИН-2,4-ДИОНА, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2002 |
|
RU2285695C2 |
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2293729C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2131425C1 |
Изобретение относится к новым соединениям формулы (I):
,
где
R1 представляет собой (С2-4)алкил и замещен двумя или более группами фтора; и
R2 представляет собой метил или этил;
или к их фармацевтически приемлемым солям. Изобретение также относится к способу получения соединений формулы (I), к фармацевтической композиции, к применению соединений, а также к способу ингибирования активности коллагеназы-3. Технический результат - получение новых биологически активных соединений, обладающих ингибирующей активностью в отношении коллагеназы-3. 8 н. и 5 з.п. ф-лы.
1. Соединение формулы (I):
,
где R1 представляет собой (С2-4)алкил и замещен двумя или более группами фтора; и
R2 представляет собой метил или этил;
или его фармацевтически приемлемая соль.
2. Соединение формулы (I) по п.1, где R1 представляет собой этил, пропил или бутил и замещен двумя или более группами фтора.
3. Соединение формулы (I) по п.1 или 2, где R1 представляет собой этил, пропил или бутил и замещен двумя-шестью группами фтора.
4. Соединение формулы (I) по п.1, где R1 представляет собой CF3CH2-, CF2HCF2-, CF3CF2-, CF3CH2CH2-, CF2HCF2CH2- или CF3CF2CH2-.
5. Соединение формулы (I) по п.1, выбранное из:
(5S)-5-метил-5-[({4-[4-(2,2,2-трифторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дио-на;
(5S)-5-этил-5-[({4-[4-(2,2,2-трифторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дио-на;
5S-метил-5-[({4-[4-(1,1,2,2-тетрафторэтокси)фенокси]пиперидин-ил}сульфонил)метил]имидазолидин-2,4-
диона;
5S-этил-5-[({4-[4-(1,1,2,2-тетрафторэтокси)фенокси]пиперидин-ил}сульфонил)метил]имидазолидин-2,4-
диона;
(5S)-5-метил-5-[({4-[4-(пентафторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дио-на;
(5S)-5-этил-5-[({4-[4-(пентафторэтокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-дио-на;
5S-метил-5-[({4-[3,3,3-трифторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-диона;
5S-этил-5-[({4-[3,3,3-трифторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-диона;
(5S)-5-метил-5-[({4-[4-(2,2,3,3-тетрафторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-диона;
(5S)-5-этил-5-[({4-[4-(2,2,3,3-тетрафторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]имидазолидин-2,4-диона;
(5S)-5-метил-5-[({4-[4-(2,2,3,3,3-пентафторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]
имидазолидин-2,4-диона; и
(5S)-5-этил-5-[({4-[4-(2,2,3,3,3-пентафторпропокси)фенокси]пиперидин-1-ил}сульфонил)метил]
имидазолидин-2,4-диона.
6. Способ получения соединения формулы (I) по любому из пп.1-5 или его фармацевтически приемлемой соли, включающий взаимодействие феноксипиперидина формулы (II) с сульфонилхлоридом формулы (III):
где R1 и R2 такие, как определено в п.1, и где любая функциональная группа защищена, если необходимо, и
(1) удаление любых защитных групп и
(2) возможно образование фармацевтически приемлемой соли.
7. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении коллагеназы-3, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как определено в любом из пп.1-5, вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем.
8. Соединение формулы (I) или его фармацевтически приемлемая соль, как определено в любом из пп.1-5, обладающие ингибирующей активностью в отношении коллагеназы-3.
9. Применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено в любом из пп.1-5, для изготовления лекарственного средства для применения в лечении болезненного состояния, опосредованного одним или более ферментами металлопротеиназами.
10. Применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено в любом из пп.1-5, для изготовления лекарственного средства для применения в лечении болезненного состояния, опосредованного коллагеназой-3.
11. Применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено в любом из пп.1-5, для изготовления лекарственного средства для применения в лечении остеоартрита.
12. Способ ингибирования активности коллагеназы-3, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено в любом из пп.1-5.
13. Применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено в любом из пп.1-5, для изготовления лекарственного средства для применения в лечении ревматоидного артрита.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| RU 2002112462 A, 20.08.2004. | |||

