Настоящее изобретение относится к некоторым N-гидроксиформамидным производным, полезным при ингибировании металлопротеиназ, способам их получения, фармацевтическим композициям, содержащим их, и их применению в терапии.
Соединения по этому изобретению являются ингибиторами одного или более чем одного фермента металлопротеиназы. Металлопротеиназы представляют собой суперсемейство протеиназ (ферментов), известное число которых за последние годы значительно увеличилось. Исходя из структурного и функционального анализа эти ферменты классифицированы на семейства и подсемейства, как описано в N.M. Hooper (1994) FEBS Letters 354:1-6. Примеры металлопротеиназ включают матриксные металлопротеиназы (ММР), такие как коллагеназы (ММР1, ММР8, ММР13), желатиназы (ММР2, ММР9), стромелизины (ММРЗ, ММР10, ММР11), матрилизин (ММР7), металлоэластазу (ММР12), энамелизин (ММР19), МТ-ММР (ММР14, ММР15, ММР16, ММР17); репролизин или адамализин, или семейство MDC, которое включает секретазы и шеддазы, такие как ФНО-конвертирующие ферменты (ADAM10 и ТАСЕ); астациновое семейство, которое включает такие ферменты, как протеиназа, катализирующая процессинг проколлагена (procollagen processing proteinase, РСР); и другие металлопротеиназы, такие как аггреканаза, семейство эндотелин-конвертирующих ферментов и семейство ангиотензин-конвертирующих ферментов.
Полагают, что металлопротеиназы имеют важное значение при плеторе при болезненных физиологических процессах, в которые вовлечено ремоделирование тканей, такое как эмбриональное развитие, формирование костей и маточное ремоделирование во время менструации. Это обосновано способностью металлопротеиназ расщеплять широкий ряд матриксных субстратов, таких как коллаген, протеогликан и фибронектин. Также металлопротеиназы, как полагают, являются важными при процессинге, или секреции, биологически важных клеточных медиаторов, таких как фактор некроза опухоли (ФНО), и пост-трансляционном протеолитическом процессинге, или шеддинге, биологически важных мембранных белков, таких как IgE рецептор CD23 с низкой аффинностью (более полный перечень смотри в N.M. Hooper et al., (1997) Biochem J. 321:265-279).
Металлопротеиназы ассоциированы со многими болезненными состояниями. Ингибирование активности одной или более чем одной металлопротеиназы может быть очень полезным при этих болезненных состояниях, например при различных воспалительных и аллергических заболеваниях, таких как воспаление суставов (в частности, ревматоидный артрит, остеоартрит и подагра), воспаление желудочно-кишечного тракта (в частности, воспалительное заболевание кишечника, неспецифический язвенный колит и гастрит), воспаление кожи (в частности псориаз, экзема, дерматит); при метастазировании или инвазии опухолей; при заболевании, ассоциированном с неконтролируемым разрушением внеклеточного матрикса, таком как остеоартрит; при заболевании, характеризующемся резорбцией кости (таком как остеопороз или болезнь Педжета); при заболеваниях, ассоциированных с аберрантным ангиогенезом; при усиленном коллагеновом ремоделировании, ассоциированном с диабетом, заболеванием периодонта (таким как гингивит), образованием язвы роговицы, образованием кожных язв, послеоперационными состояниями (такими как ободочный анастомоз) и заживлением кожных ран; при демиелинизирующих заболеваниях центральной и периферической нервной системы (таких как рассеянный склероз); болезни Альцгеймера; ремоделировании внеклеточного матрикса, наблюдаемом при сердечно-сосудистых заболеваниях, таких как рестеноз и атеросклероз, и хронических обструктивных болезнях легких, ХОБЛ (в которые вовлечена ММР12).
Известен ряд ингибиторов металлопротеиназ; разные классы соединений могут иметь разные степени эффективности и селективности в отношении ингибирования различных металлопротеиназ. Авторы настоящего изобретения обнаружили новый класс соединений, которые являются ингибиторами металлопротеиназ и, в частности, представляют особый интерес в отношении ингибирования коллагеназы 3 (также известной как ММР-13). Соединения по этому изобретению обладают благоприятными воздействием и/или фармакокинетическими свойствами.
Первоначально коллагеназа 3 (ММР13) была клонирована из библиотеки кДНК, полученной из опухоли груди [J. М. Р. Freije et al. (1994) Journal of Biological Chemistry 269(24):16766-16773]. ПЦР-РНК анализ РНК широкого ряда тканей показал, что экспрессия коллагеназы 3 (ММР13) была ограничена карциномами груди, так как она не была обнаружена в фиброаденомах груди, нормальной или покоящейся молочной железе, плаценте, печени, яичнике, матке, предстательной железе или околоушной железе или в клеточных линиях рака груди (T47-D, MCF-7 и ZR75-1). После этого коллагеназа 3 (ММР13) была обнаружена в трансформированных эпидермальных кератиноцитах [N. Johansson et al., (1997) Cell Growth Differ. 8(2):242-250], плоскоклеточных карциномах [N. Johansson et al., (1997) Am. J. Pathol. 151 (2):499-508] и эпидермальных опухолях [К. Airola et al., (1997) J. Invest. Dermatol. 109(2):225-231]. Эти результаты подтверждают, что коллагеназа 3 (ММР13) секретируется трансформированными эпителиальными клетками и может быть вовлечена в деградацию внеклеточного матрикса и взаимодействие "клетка-матрикс", ассоциированное с метастазированием, как это особенно наблюдается при инвазивных очагах рака груди и при злокачественной эпителиальной опухоли при кожных карциномах.
В недавно опубликованных данных подразумевается, что коллагеназа 3 (ММР13) играет роль при обновлении других соединительных тканей. Например, в соответствии со специфичностью субстрата коллагеназы 3 (ММР13) и предпочтительностью для распада коллагена типа II [Р.G.Mitchell et al., (1996) J.Clin. Invest. 97(3):761-768; V.Knauper et al., (1996) The Biochemical Journal 271:1544-1550], коллагеназа 3 (ММР13), как предположили, играет роль во время первичной оссификации и ремоделировании скелета [М.Stahle-Backdahl et al., (1997) Lab. Invest. 76(5):717-728; N.Johansson et al., (1997) Dev. Dyn. 208(3): 387-397], при деструктивных заболеваниях суставов, таких как ревматоидный и остеоартрит [D.Wernicke et al., (1996) J.Rheumatol. 23:590-595; P.G.Mitchell et al., (1996) J. Clin. Invest. 97(3):761-768: O.Lindy et al., (1997) Arthritis Rheum 40(8):1391-1399] и во время асептического расшатывания эндопротезов тазобедренного сустава [S.Imai et al., (1998) J.Bone Joint Surg. Br. 80(4):701-7101. Коллагеназа 3 (ММР13) также вовлечена в хронические периодонтиты у взрослых, так как она локализована в эпителии хронически воспаленной слизистой оболочки десневой ткани человека [V.J.Uitto et al., (1998) Am. J.Pathol 152(6): 1489-1499] и при ремоделировании коллагенового матрикса в хронических ранах [М.Vaalamo et al., (1997) J.Invest. Dermatoi. 109(1):96-101].
Соединения, которые ингибируют действие металлопротеиназ, в частности коллагеназы 3 (ММР13), описаны в WO 00/12478, WO 00/75108 и WO 01/62742. В число этих ингибиторов, о которых сообщалось, включены арил/гетероарилпиперазинсульфонилметилзамещенные N-гидроксиформамидные соединения, в которых арильное кольцо замещено рядом возможных заместителей, включая среди прочего алкокси и арилокси. Нигде не раскрыто, что алкокси- или арилоксизаместитель в таких соединениях сам дополнительно может быть замещен.
Замещенные алкокси- или арилокси-арил/гетероарилпиперазинсульфонилметилзамещенные N-гидроксиформамидные соединения как ингибиторы матриксных металлопротеиназ описаны в общем в описании WO 99/38843. Среди множества перечисленных возможных заместителей для алкоксигруппы присутствует галоген. Однако такое алкоксизамещенное соединение не раскрыто, и действительно, единственным конкретно раскрытым N-гидроксиформамидным соединением является N-{1S-[4-(4-хлорфенил)пиперазин-1-сульфонилметил]-2-метилпропил}-N-гидроксиформамид.
Авторы настоящего изобретения обнаружили, что замещенные арил- или гетероарил-пиперазинсульфонилметилзамещенные N-гидроксиформамидные соединения, в которых заместителем является алкоксигруппа, которая сама замещена одной или более чем одной группой фторо, являются особенно полезными ингибиторами металлопротеиназ, в частности коллагеназы 3 (ММР13), и обладают желаемыми профилями активности.
В первом аспекте настоящего изобретения предложено соединение формулы (I)
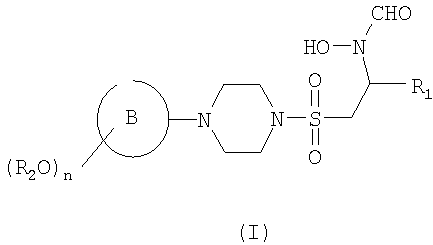
или его фармацевтически приемлемые соль, пролекарство или сольват,
где кольцо В представляет собой моноциклическое арильное кольцо, имеющее шесть кольцевых атомов, или моноциклическое гетероарильное кольцо, имеющее вплоть до шести кольцевых атомов и содержащее один или более чем один кольцевой гетероатом, где каждый указанный гетероатом представляет собой азот;
R2 представляет собой группу, выбранную из С1-6алкила или арила, где указанная группа замещена одной или более чем одной группой фторо;
n равен 1, 2 или 3 и
R1 представляет собой возможно замещенную группу, выбранную из С1-6алкила, С5-7циклоалкила, гетероциклоалкила, арила, гетероарила, С1-6алкил-арила, С1-6алкил-гетероарила, C1-6алкил-циклоалкила или С1-6алкил-гетероциклоалкила.
Термин "арил", как он использован здесь, означает ароматический карбоциклический радикал с одним или двумя кольцами, имеющими вплоть до десяти кольцевых атомов, такой как фенил или нафтил. В тех случаях, когда подразумевается ароматический карбоциклический радикал с одним кольцом, его обозначают как "моноциклическое арильное кольцо". В тех случаях, когда подразумевается, что арильное кольцо имеет шесть кольцевых атомов, это указывают конкретно.
"Гетероарил" относится к ароматическим кольцевым системам, имеющим вплоть до десяти атомов, в частности вплоть до шести кольцевых атомов, и содержащим один или более кольцевых гетероатомов, которые могут быть одинаковыми или разными, выбранных из N, О и S. Примеры включают пирролил, фуранил, тиофенил, имидазолил, тиазолил, пиридинил, пиримидинил и пиразинил. Гетероатомы азота будут замещенными по необходимости; также они могут быть в виде N-оксидов. Атомы серы могут быть в виде S, S(O) или S(O)2. В тех случаях, когда подразумевается гетероароматическая система с одним кольцом, ее обозначают как "моноциклическое гетероарильное кольцо" и в тех случаях, когда подразумевается, что максимальное количество кольцевых атомов гетероарильного кольца составляет менее десяти, это указывают конкретно. В тех случаях, когда подразумевается, что кольцевым гетероатомом является один из N, S или О, в частности, или, что гетероарильное кольцо содержит более чем один гетероатом, в определенном сочетании, например, где гетероатомы являются одинаковыми, это указывают конкретно.
Термин "галоген" включает фтор, хлор, бром и йод, в частности фтор.
Если не оговорено особо, термин "С1-6алкил", когда его используют отдельно или в комбинации, относится к алкильной группировке с неразветвленной или разветвленной цепью, имеющей от одного до шести атомов углерода, включая метил, этил, пропил, изопропил, бутил, трет-бутил, пентил, гексил и тому подобное. Соответственно, под "С1-4алкилом" следует понимать обозначение алкильной группировки с неразветвленной или разветвленной цепью, имеющей от одного до четырех атомов углерода.
Термин "циклоалкил" относится к насыщенной алициклической группировке, имеющей пять, шесть или семь атомов углерода, и включает, например, циклопентил и циклогексил. Гетероциклоалкильное кольцо относится к насыщенному пяти-, шести- или семичленному кольцу, содержащему один или более кольцевых гетероатомов, которые могут быть одинаковыми или разными, выбранных из N, О и S, и включает, например, пиперидинил, пирролидинил, тетрагидрофуранил, тетрагидропиранил.
Термин "возможно замещенный" используют здесь для указания на возможное замещение группой или группами, предусмотренное в любом подходящем доступном положении.
Соответственно, кольцо В представляет собой моноциклическое арильное кольцо, имеющее шесть кольцевых атомов, такое как фенил, или моноциклическое гетероарильное кольцо, имеющее вплоть до шести кольцевых атомов и содержащее от одного до четырех кольцевых атомов азота, такое как пиридинил или пиримидинил, триазинил или тетразинил.
В тех случаях, когда кольцо В представляет собой гетероарильное кольцо, предпочтительным является шестичленное кольцо, содержащее от одного до четырех кольцевых атомов азота, более предпочтительно шестичленное кольцо, содержащее один или два кольцевых атома азота, такое как пиридинил или пиримидинил.
В одном из предпочтительных воплощений кольцо В представляет собой фенильное кольцо.
В другом предпочтительном воплощении кольцо В представляет собой шестичленное гетероарильное кольцо, содержащее один или два кольцевых атома азота. Одним из предпочтительных значений для кольца В является пиридинил, в частности 2-пиридинил. Особо предпочтительным значением для кольца В является пиримидинил, в частности 2-пиримидинил.
R2 может представлять собой арильную группу, имеющую вплоть до десяти кольцевых атомов, в частности моноциклическую арильную группу, имеющую шесть кольцевых атомов (такую как фенил), замещенную одной или более чем одной группой фторо, но предпочтительно представляет собой С1-6алкильную, в частности С1-4алкильную, группу (такую как метил и особенно этил), замещенную одной или более чем одной группой фторо.
Предпочтительно R2 замещен группами фторо в количестве от одной до пяти, в частности тремя или четырьмя группами фторо.
В одном из предпочтительных воплощений R2 представляет собой С1-6алкил, в частности С1-4алкил, замещенный тремя или четырьмя группами фторо.
Одним из предпочтительных значений для R2 является CF2CHF2.
В другом особо предпочтительном воплощении R2 представляет собой CH2CF3.
Подходящее значение n равно 1 или 2 и предпочтительно 1. Предпочтительно заместитель R2O- по кольцу В находится в пара-положении относительно места присоединения к кольцу.
R1 представляет собой подходящую возможно замещенную группу, выбранную из C1-4-алкила (такого как метил или этил), арила, имеющего шесть кольцевых атомов (такого как фенил), 5-6-членного гетероциклоалкильного кольца, содержащего один или два кольцевых гетероатома, которые могут быть одинаковыми или разными, выбранных из N, О и S (такого как пиперидинил или тетрагидропиранил), или С1-4алкил-гетероарила, где гетероарил имеет вплоть до шести кольцевых атомов и содержит один или два кольцевых гетероатома, выбранных из N, О и S (такого как алкилпиримидинил или алкилпиридинил).
Предпочтительно R1 представляет собой возможно замещенное 5-6-членное гетероциклоалкильное кольцо, содержащее один или два кольцевых гетероатома, которые могут быть одинаковыми или разными, выбранных из N, О и S, или С1-4алкил-гетероарильную группу, имеющую вплоть до шести кольцевых атомов и содержащую один или более гетероатомов, которые могут быть одинаковыми или разными, выбранных из N, О и S, возможно замещенную по гетероарильному кольцу.
В одном из предпочтительных воплощений R1 является незамещенным.
В одном из предпочтительных воплощений R1 представляет собой тетрагидропиранильную группу, в частности 4-тетрагидропиранил.
В другом предпочтительном воплощении R1 представляет собой С2-3алкил-пиримидинильную группу, возможно замещенную по пиримидинильному кольцу.
Одним из предпочтительных значений для R1 является 2-пиримидинил-СН2СН2-. Другим особо предпочтительным значением для R1 является 2-пиримидинил-СН2СН2СН2-.
Подходящие возможные заместители для R1 включают одну или более чем одну группу, независимо выбранную из NO2, CF3, CN, галогена, С1-4алкила, карбокси(С1-4)алкила, циклоалкила, -OR4, -SR4, С1-4алкила, замещенного -OR4, SR4 (и его окисленными аналогами), NR4, N-Y-R4, или С1-4алкил-Y-NR4, где R4 представляет собой водород, С1-6алкил, арил, гетероарил или С1-6алкил-арил, каждый из которых независимо возможно замещен галогеном, NO2, CN, CF3, С1-6алкилом, -S-C1-6алкил, -SO-C1-6алкил, -SO2-С1-6алкил или С1-6алкокси, и Y выбран из -SO2- и -СО-.
Когда R1 в соединениях формулы (I) является замещенным, то предпочтительно он замещен одним или двумя заместителями, которые могут быть одинаковыми или разными, выбранными из С1-4алкила, галогена, CF3 и CN. Предпочтительным заместителем является галоген, в частности фтор. Предпочтительно, если R1 является замещенным, то он является монозамещенным. Одним из предпочтительных значений для R1 в соединениях формулы (I), где R1 замещен, является 5-F-2-пиримидинил-СН2СН2-.
Следует понимать, что количество и природа заместителей по кольцам, образованным R1 и/или R2, в соединениях по изобретению будут выбраны таким образом, чтобы избежать стерически нежелательных комбинаций.
В одной из предпочтительных групп соединений по изобретению R2 представляет собой С1-6алкил, замещенный группами фторо в количестве от одной до пяти; n равен 1; кольцо В представляет собой фенил, пиридинил или пиримидинил и R1 представляет собой возможно замещенное 5-6-членное гетероциклоалкильное кольцо, содержащее один или два кольцевых гетероатома, которые могут быть одинаковыми или разными, выбранных из N, О и S, или С1-4алкил-гетероарильную группу, имеющую вплоть до шести кольцевых атомов и содержащую один или более гетероатомов, которые могут быть одинаковыми или разными, выбранных из N, О и S, возможно замещенную по гетероарильному кольцу.
Особо предпочтительными соединениями по изобретению в этой группе являются соединения, в которых R1 не замещен или замещен галогеном, в частности фтором.
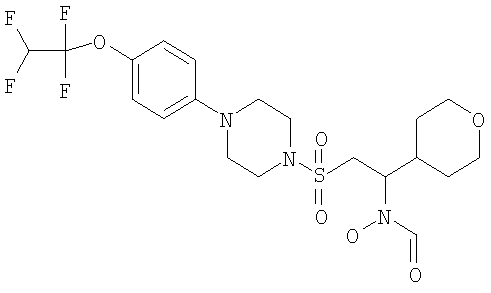
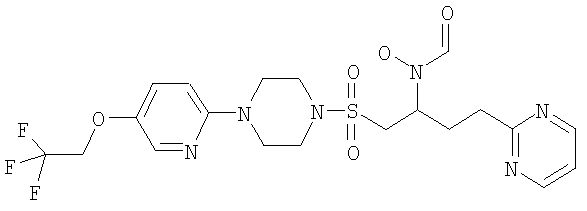
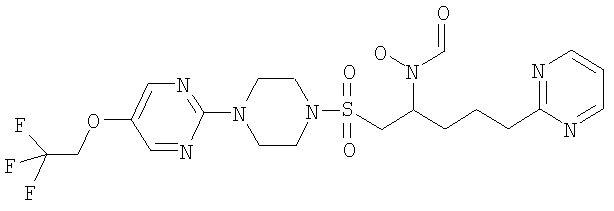
Конкретные соединения включают
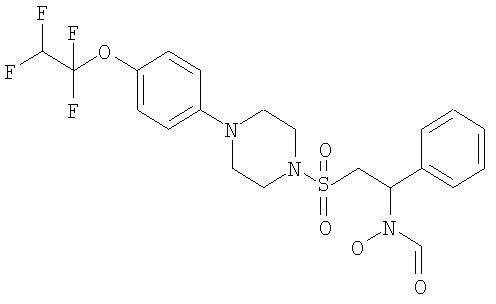
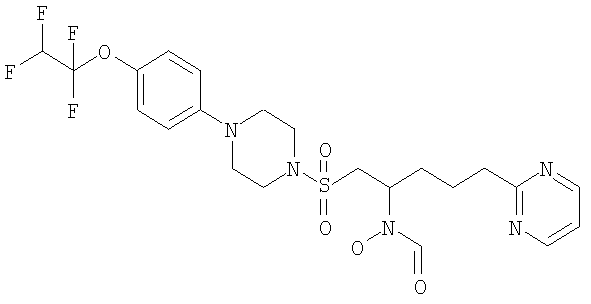
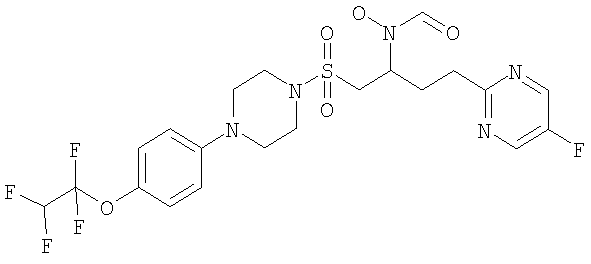
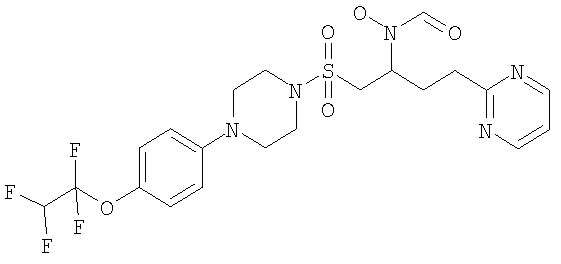

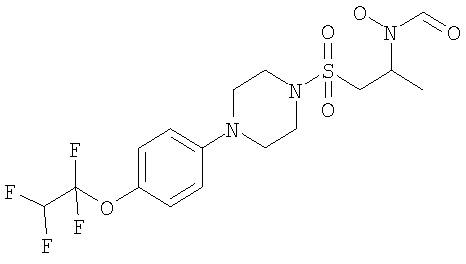
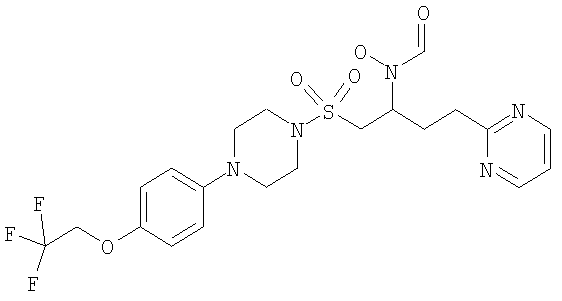
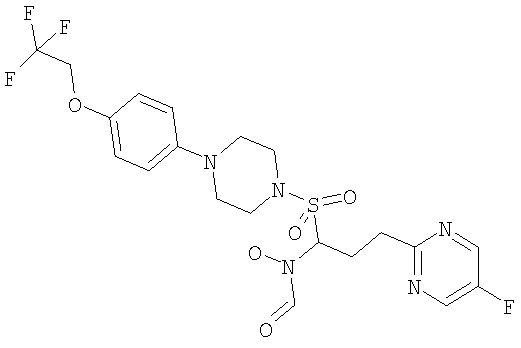
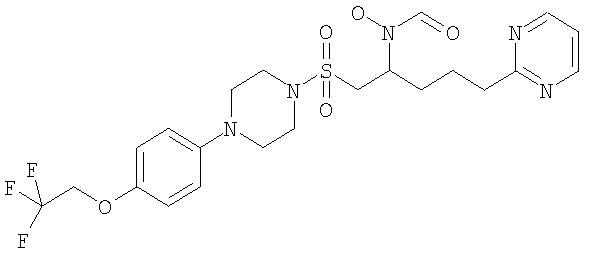
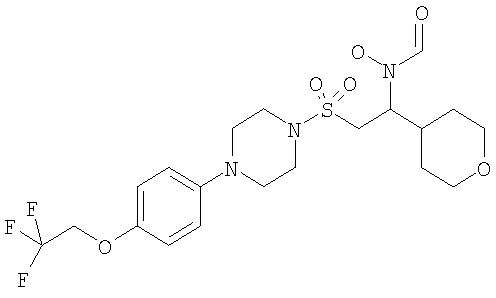
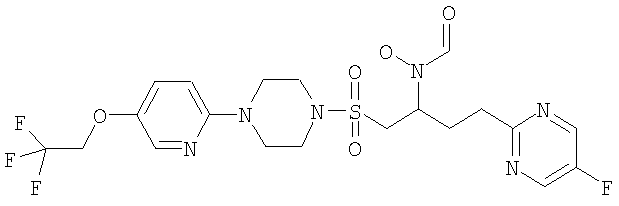
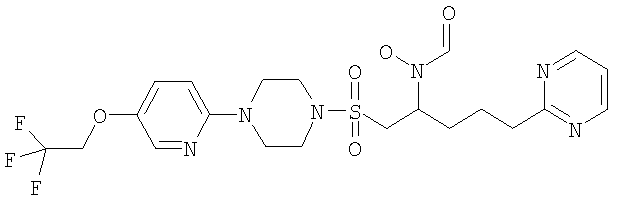
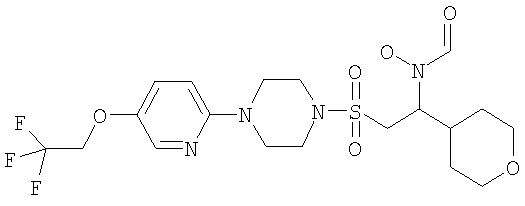
В тех случаях, когда соединения по изобретению содержат один или более чем один асимметрически замещенный атом углерода, изобретение включает все стереоизомеры, в том числе энантиомеры и диастереомеры и смеси, включая их рацемические смеси. Также включены таутомеры и их смеси.
Рацематы могут быть разделены на индивидуальные энантиомеры с использованием известных методик (см. Advanced Organic Chemistry: 3rd Edition: author J March, p.104-107). Подходящая методика включает образование диастереомерных производных путем взаимодействия рацемического вещества со вспомогательным хиральным веществом с последующим разделением диастереомеров, например, хроматографией, и затем отщеплением этих вспомогательных веществ.
Не желая ограничиваться первоначальными определениями, полагают, что в настоящем случае активный энантиомер имеет S-стереохимию. Это основано на сравнении с родственными соединениями, для которых была подтверждена эта абсолютная конфигурация. Соответственно, S-структура указана в формулах, приведенных ниже в примерах. Однако следует понимать, что рацемат любого соединения по изобретению может быть разделен на индивидуальные энантиомеры способом, изложенным выше, и более активный энантиомер может быть затем идентифицирован подходящим методом анализа без необходимости определения абсолютных конфигураций.
Соединения по изобретению могут быть предложены в виде фармацевтически приемлемых солей. Подходящие фармацевтически приемлемые соли включают основные соли, такие как соль щелочного металла, например натрия, соль щелочноземельного металла, например кальция или магния, соль органического амина, например триэтиламина, морфолина, N-метилпиперидина, N-этилпиперидина, прокаина, дибензиламина, N,N-дибензилэтиламина, или аминокислот, например лизина. В другом аспекте, в тех случаях, когда соединение является достаточно основным, подходящие соли включают соли присоединения кислоты, такие как метансульфонат, фумарат, гидрохлорид, гидробромид, цитрат, малеат, и соли, образованные с фосфорной и серной кислотой.
Подходящие пролекарства соединений формулы (I) представляют собой соединения, которые гидролизуются in vivo с образованием соединений формулы (I). Они могут быть получены традиционными способами.
В настоящем изобретении также предложен способ получения соединения формулы (I), как оно определено выше, или его фармацевтически приемлемых солей, пролекарств или сольватов, включающий превращение подходящего гидроксиаминосоединения формулы (IV)
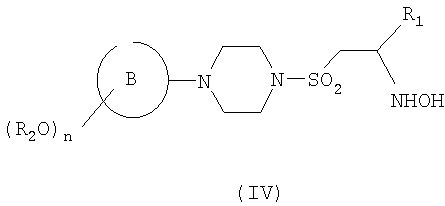
где R2, n, кольцо В и R1 являются такими, как определено в формуле (I), в соединение формулы (I) путем формилирования с подходящим смешанным ангидридом и затем, если необходимо превращение полученного соединения в другое соединение по изобретению и/или образование фармацевтически приемлемых соли, пролекарства или сольвата данного соединения.
Процесс формилирования можно подходящим образом проводить путем взаимодействия соединения формулы (IV) со смешанным ангидридом, полученным в результате взаимодействия муравьиной кислоты и уксусного ангидрида. Взаимодействие легко осуществляют в присутствии органической кислоты, такой как муравьиная кислота. Взаимодействие предпочтительно проводят в подходящем инертном растворителе или разбавителе, таком как дихлорметан (ДХМ) или тетрагидрофуран, и при температуре в интервале, например, от 0 до 50°С.
Соединения формулы (IV) могут быть получены из соответствующего алкена формулы (III)
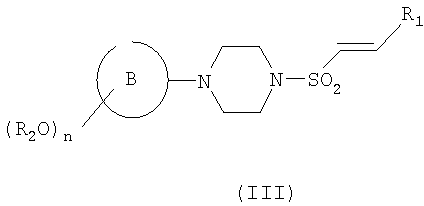
где R2, n, В и R1 являются такими, как определено в формуле (I), который сам может быть получен из соответствующего соединения формулы (II)
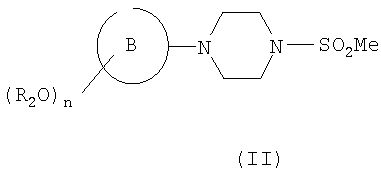
где R2, n и кольцо В являются такими, как определено в формуле (I), путем взаимодействия с подходящим соединением формулы R1CHO (где R1 является таким, как определено для формулы (I)) или путем взаимодействия с подходящим сложным эфиром с образованием кетона, с последующими восстановлением до соответствующего спирта и дегидратацией. Следует понимать, что соединение формулы (III) может быть в виде Е- или Z-изомера, или в виде их смеси. Структура, представленная в формуле (III), не подразумевает ограничения какой-либо конкретной геометрической изомерии вокруг двойной связи.
Соединения формул (III) и (IV) могут быть получены с использованием известных методик способами, аналогичными способам, описанным в упомянутых выше WO 00/12478, WO 00/75108 и WO 01/62742. Примеры способов получения некоторых из этих соединений даны ниже в примерах.
Соединения формулы (II), (III) и (IV) являются новыми и составляют дополнительный аспект изобретения. Конкретные соединения формулы (II) включают
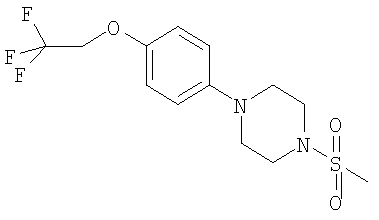
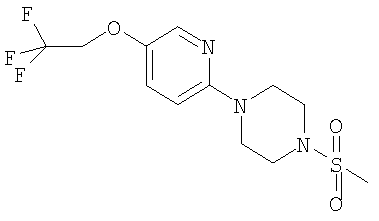
Примеры способов получения этих соединений приведены ниже в примерах.
Соединения формулы (I) могут быть превращены в другие соединения формулы (I) с использованием стандартных методик, общепринятых в данной области техники.
Следует понимать, что получение соединений формулы (I) может включать на различных стадиях присоединение и удаление одной или более чем одной защитной группы. Введение защитных групп и снятие защиты с функциональных групп описаны в "Protective Groups in Organic Chemistry", edited by J.W.F. McOmie, Plenum Press (1973), и "Protective Groups in Organic Synthesis", 2nd edition, T.W. Greene and P.G.M. Wuts, Wiley-lnterscience (1991).
Соединения по изобретению являются ингибиторами металлопротеиназ, в частности они являются ингибиторами коллагеназы 3 (ММР13) и поэтому показаны при лечении заболеваний или состояний, опосредованных ферментами металлопротеиназами, включая артрит (такой как остеоартрит), атеросклероз и хронические обструктивные болезни легких (ХОБЛ), как рассмотрено выше. В частности, соединения по изобретению показаны при лечении заболеваний или состояний, опосредованных коллагеназой 3 (ММР13). Конкретное преимущество ингибиторов коллагеназы 3 по изобретению заключается в том, что они проявляют улучшенную селективность по сравнению с другими металлопротеиназами.
Таким образом, в дополнительном аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемые соль, пролекарство или сольват, как они определены выше, для применения в терапии человека или животного.
Также в изобретении предложено применение соединения формулы (I) или его фармацевтически приемлемых соли, пролекарства или сольвата, как они определены выше, в производстве лекарства для применения в терапии.
Следует понимать, что "терапия" также включает "профилактику", если не указано иного. Термины "терапевтический" и "терапевтически" следует понимать соответствующим образом.
В еще одном дополнительном аспекте настоящего изобретения предложен способ лечения болезненного состояния, опосредованного металлопротеиназой, включающий введение теплокровному животному терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемых соли, пролекарства или сольвата.
Следует понимать, что вводимая дозировка будет варьировать в зависимости от используемого соединения, способа введения, требуемого лечения и указанного расстройства. Обычно принимают суточную дозу от 0,5 до 75 мг/кг массы тела (и предпочтительно от 0,5 до 30 мг/кг массы тела). Эту суточную дозу можно давать в разделенных дозах, если необходимо, при этом точное количество принимаемого соединения и путь введения зависят от массы, возраста и пола пациента, которого лечат, и от конкретного болезненного состояния, которое лечат, в соответствии с принципами, известными в данной области.
Соединения формулы (I) и их фармацевтически приемлемые соли, пролекарство и сольваты могут быть использованы сами по себе, но обычно их будут вводить в форме фармацевтической композиции вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Таким образом, в настоящем изобретении также предложена фармацевтическая композиция, включающая соединение формулы (I) или его фармацевтически приемлемые соль, пролекарство или сольват вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Фармацевтические композиции по изобретению можно вводить способом, общепринятым для болезненного состояния, которое требуется лечить, например пероральным, местным, парентеральным, буккальным, назальным, вагинальным или ректальным введением или путем ингаляции. Для этих целей соединения по этому изобретению могут быть приготовлены с помощью известных в данной области средств в форме, например, таблеток, капсул, водных или масляных растворов, суспензий, эмульсий, кремов, мазей, гелей, назальных спреев, суппозиториев, тонкоизмельченных порошков или аэрозолей для ингаляции, а для парентерального применения (в том числе внутривенного, внутримышечного введения или инфузии) в форме стерильных водных или масляных растворов или суспензий либо стерильных эмульсий.
В дополнение к соединениям по настоящему изобретению фармацевтическая композиция по изобретению может также содержать один или более чем один фармакологический агент, показанный при лечении одного или более чем одного болезненного состояния, указанного выше, либо она может быть введена совместно с таким(и) агентом(ами) (одновременно или последовательно). Обычно стандартные лекарственные формы будут содержать приблизительно от 1 до 500 мг соединения по изобретению.
Активность и селективность соединений по изобретению могут быть определены с использованием соответствующего теста на ингибирование фермента, как описано в WO 00/12478, WO 00/75108 и WO 01/62742. Ингибиторную активность в отношении коллагеназы 3 (ММР13) можно оценить, например, с использованием методики, изложенной ниже.
Рекомбинантная человеческая proMMP13 может быть экспрессирована и очищена, как описано Knauper et al. [V. Knauper et al., (1996) The Biochemical Journal 271:1544-1550 (1996)]. Очищенный фермент может быть использован для контроля активности ингибиторов следующим образом: очищенную proMMP13 активируют с использованием 1 мМ аминофенилртутной кислоты (amino phenyl mercuric acid, АРМА), 20 часов при 21°С; активированную ММР13 (11,25 нг на анализ) инкубируют в течение 4-5 часов при 35°С в буфере для тестирования (0,1 М Трис-HCl, рН 7,5, содержащем 0,1 М NaCl, 20 мМ CaCl2, 0,02 мМ ZnCl и 0,05% (мас./об.) Brij 35 с использованием синтетического субстрата 7-метоксикумарин-4-ил)ацетил. Pro. Leu.Giy.Leu. N-3-(2,4-динитрофенил)-L-2,3-диаминопропионил.Ala.Arg.NH2 в присутствии или в отсутствие ингибиторов. Активность определяют путем измерения флуоресцентности при λех 328 нм и λem 393 нм. Измеряя активность в диапазоне концентраций можно получить кривую, из которой может быть определена ИК50, представляющая собой ингибиторную концентрацию, при которой активность фермента уменьшается на 50%.
Следует понимать, что фармакологические свойства соединений по изобретению будут варьировать в зависимости от их структуры, но, в общем, как определено анализом, описанным выше, соединения по изобретению демонстрируют ингибиторную активность в отношении коллагеназы 3 с концентрациями ИК50 в интервале от 0,01 до 1000 нМ. В следующей таблице приведены значения ИК50 для репрезентативной выборки соединений по изобретению, которые были протестированы в описанном выше анализе.
Соединение формулы I можно применять в комбинации с другими лекарственными средствами и терапиями, применяемыми при лечении болезненных состояний, при которых полезно ингибирование металлопротеиназ, в частности коллагеназы 3 (ММР13). Например, соединение формулы I можно применять в комбинации с лекарственными средствами и терапиями, применяемыми при лечении ревматоидного артрита, астмы, воспалительного заболевания кишечника, рассеянного склероза, СПИДа, септического шока, застойной сердечной недостаточности, ишемической болезни сердца, псориаза и других болезненных состояний, упомянутых выше в этом описании.
Например, благодаря своей способности ингибировать металлопротеиназы соединение формулы I полезно при лечении некоторых воспалительных и невоспалительных заболеваний, которые в настоящее время лечат нестероидным противовоспалительным лекарственным средством (NSAID), ингибирующим циклооксигеназу, таким как индометацин, кеторолак, ацетилсалициловая кислота, ибупрофен, сулиндак, толметин и пироксикам. Совместное введение соединения формулы 1 по настоящему изобретению с NSAID может привести к снижению количества последнего агента, необходимого для достижения терапевтического эффекта. В связи с этим уменьшается вероятность неблагоприятных побочных эффектов от NSAlD, таких как влияние на желудочно-кишечный тракт. Таким образом, в соответствии с дополнительным аспектом изобретения предложена фармацевтическая композиция, которая включает соединение формулы I или его фармацевтически приемлемую соль в сочетании или смеси с нестероидным противовоспалительным агентом, ингибирующим циклооксигеназу, и фармацевтически приемлемый разбавитель или носитель.
Соединение формулы I также можно применять с противовоспалительными агентами, такими как ингибитор фермента 5-липоксигеназы.
Соединение формулы I также можно применять при лечении таких состояний, как ревматоидный артрит, в комбинации с противоартритными агентами, такими как золото, метотрексат, стероиды и пенициллинамин, и таких состояний, как остеоартрит, в комбинации со стероидами.
Соединение формулы I также можно вводить при деградативных заболеваниях, например, остеоартрите, с хондропротекторными, антидеградативными и/или репаративными агентами, такими как Diacerhein, препараты гиалуроновой кислоты, такие как гиалган, румалон, артепарон и глюкозаминовые соли, такие как антрил.
Соединение формулы I можно применять при лечении астмы в комбинации с противоастматическими агентами, такими как стероиды, бронходилататоры и антагонисты лейкотриенов.
В частности, для лечения воспалительных заболеваний, ревматоидного артрита, остеоартрита, псориаза, воспалительного заболевания кишечника, хронической обструктивной болезни легких, астмы и аллергического ринита соединение по настоящему изобретению можно объединять с такими агентами, как ингибиторы ФНО-α, такие как анти-ФНО моноклональные антитела (такие как Ремикейд, CDP-870 и D.sub2.E.sub7.) и ФНО-рецепторные иммуноглобулиновые молекулы (такие как Enbrel.reg.), неселективные ингибиторы циклооксигеназы-1 (СОХ-1)/циклооксигеназы-2 (СОХ-2) (такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин), ингибиторы СОХ-2 (такие как мелоксикам, целекоксиб, рофекоксиб, валдекоксиб или эторикоксиб), метотрексат в низких дозах, лефуномид; циклесонид; гидроксихлорохин, d-пеницилламин, ауранофин или парентеральное или пероральное золото.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с ингибитором биосинтеза лейкотриенов, ингибитором 5-липоксигеназы (5-LO) или антагонистом белка, активирующего 5-липоксигеназу (FLAP), таким как зилеутон; АВТ-761; фенлеутон; тепоксалин; Abbott-79175; Abbott-85761; N-(5-замещенные)-тиофен-2-алкилсульфонамиды; 2,6-ди-трет-бутилфенолгидразоны; метокситетрагидропираны, такие как Zeneca ZD-2138; соединение SB-210661; пиридинил-замещенные 2-цианонафталиновые соединения, такие как L-739010; 2-цианохинолиновые соединения, такие как L-746530; индольные и хинолиновые соединения, такие как МК-591, МК-886 и BAY x 1005.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с антагонистами рецепторов лейкотриенов LTB4, LTC4, LTD4 и LTF4, выбранными из группы, состоящей из фенотиазин-3-онов, таких как L-651392; амидиносоединений, таких как CGS-25019с; бензоксаламинов, таких как онтазоласт; бензолкарбоксимидамидов, таких как BIIL 284/260, и таких соединений, как зафирлукаст, аблукаст, монтелукаст, пранлукаст, верлукаст (МК-679), RG-12525, Ro-254913, иралукаст (CGP 45715А) и BAY × 7195.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с ингибитором PDE4, включая ингибиторы изоформы PDE4D.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с антагонистами антигистаминовых рецепторов H1, такими как цетиризин, лоратадин, деслоратадин, фексофенадин, астемизол, азеластин и хлорфенирамин.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы 1 вместе с гастропротекторным антагонистом рецепторов Н2.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с агонистом α1- и α2-адреноцепторов симпатомиметическим агентом, таким как пропилгекседрин, фенилэфрин, фенилпропаноламин, псевдоэфедрин, гидрохлорид нафазолина, гидрохлорид оксиметазолина, гидрохлорид тетрагидрозолина, гидрохлорид ксилометазолина и гидрохлорид этилнорэпинефрина.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с антихолинергическими агентами, такими как ипратропия бромид; тиотропия бромид; окситропия бромид; пирензепин и телензепин.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с агонистами β1-β4-адренорецепторов, такими как метапротеренол, изопротеренол, изопреналин, альбутерол, сальбутамол, формотерол, сальметерол, тербуталин, оксипреналин, мезилат битолтерола и пирбутерол, или метилксантанинами, в том числе теофиллином и аминофиллином; кромогликатом натрия, или антагонистом мускариновых рецепторов (М1, М2 и М3).
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с миметиком инсулиноподобного фактора роста типа I (IGF-I).
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с ингаляторным глюкокортикоидом с пониженными системными побочными эффектами, таким как преднизон, преднизолон, флунизолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат и мометазона фуроат.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с другими модуляторами функции хемокиновых рецепторов, такими как CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для С-С семейства); CXCR1, CXCR3, CXR4 и CXCR5 (для С-Х-С семейства) и CX3CR1 для С-Х3-С семейства.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с противовирусными агентами, такими как Вирацепт, AZT, ацикловир и фамцикловир, и антисептическими соединениями, такими как Валант.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с сердечно-сосудистыми агентами, такими как блокаторы кальциевых каналов, агентами, снижающими уровень липидов, такими как статины, фибраты, бета-блокаторы, ингибиторы ангиотензин-превращающего фермента (Асе-ингибиторы), антагонисты рецепторов ангиотензина-2 и ингибиторы агрегации тромбоцитов.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с агентами для ЦНС, такими как антидепрессанты (такие как сертралин), антипаркинсонические лекарственные средства (такие как депренил, L-дофа, Реквип, Мирапекс, ингибиторы МАОВ, такие как селегин и расагилин, ингибиторы comP, такие как Тасмар, ингибиторы А-2, ингибиторы обратного захвата дофамина, ингибиторы NMDA, агонисты никотина, агонисты дофамина и ингибиторы нейрональной синтазы оксида азота) и лекарственные средства для лечения болезни Альцгеймера, такие как донепезил, такрин, ингибиторы СОХ-2, пропентофиллин или метрифонат.
Кроме того, настоящее изобретение дополнительно относится к комбинации соединения формулы I вместе с (1) ингибиторами триптазы; (2) антагонистами фактора активации тромбоцитов (PAF); (3) ингибиторами интерлейкин-превращающего фермента (ICE); (4) ингибиторами IMPDH; (5) ингибиторами молекул адгезии, в том числе антагонистами VLA-4; (6) катепсинами; (7) ингибиторами МАР-киназы; (8) ингибиторами глюкозо-6-фосфат-дегидрогеназы; (9) антагонистами рецепторов кинин-В1 и -B2; (10) средствами против подагры, например колхинином; (11) ингибиторами ксантин-оксидазы, например аллопуринолом; (12) средствами, способствующими выведению мочевой кислоты, например пробенецидом, сульфинпиразоном и бензбромароном; (13) стимуляторами секреции гормона роста; (14) трансформирующим фактором роста (TGFβ); (15) тромбоцитарным фактором роста (PDGF); (16) фактором роста фибробластов, например основным фактором роста фибробластов (bFGF); (17) гранулоцитарно-макрофагеальным колониестимулирующим фактором (GM-CSF); (18) капсаициновым кремом; (19) антагонистами рецепторов тахикинина NK1 и NK3, выбранными из группы, состоящей из NKP-608C, SB-233412 (талнетант) и D-4418; (20) ингибиторами эластазы, выбранными из группы, состоящей из UT-77 и ZD-0892; (21) ингибиторами ФНО-конвертирующего фермента (ТАСЕ); (22) ингибиторами индуцибельной синтазы оксида азота (ингибиторами iNOS) или (23) рецептор-гомологичной молекулой для хемоаттрактанта, экспрессированной на ТН2-клетках (антагонисты CRTH2).
Соединение формулы I также можно применять в комбинации с агентами против остеопороза, такими как ролоксифен, дролоксифен, лазофоксифен или фосомакс, и иммунодепрессантами, такими как FK-506, рапамицин, циклоспорин, азатиоприн и метотрексат.
Соединение формулы I также можно применять в комбинации с существующими терапевтическими агентами для лечения остеоартрита. Подходящие агенты, применяемые в комбинации, включают стандартные нестероидные противовоспалительные агенты (далее NSAID), такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин, ингибиторы СОХ-2, такие как целекоксиб, валдекоксиб, рофекоксиб или эторикоксиб, анальгетики и внутрисуставные терапевтические агенты, такие как кортикостероиды и гиалуроновые кислоты, такие как гиалган и синвиск, и антагонисты рецепторов Р2Х7.
Соединение формулы I также можно применять в комбинации с существующими терапевтическими агентами для лечения рака. Подходящие агенты, применяемые в комбинации, включают:
(1) антипролиферативные/противоопухолевые лекарственные средства и их комбинации, применяемые в медицинской онкологии, такие как алкилирующие агенты (например, цис-платина, карбоплатин, циклофосфамид, азотная горчица, мелфалан, хлорамбуцил, бусульфан и нитрозомочевины); антиметаболиты (например, антифолаты, такие как фторпиримидины, подобные 5-фторурацилу и тегафуру, ралтитрексед, метотрексат, цитозинарабинозид, гидроксимочевина, гемцитабин и паклитаксел (Taxol®); противоопухолевые антибиотики (например, антрациклины, подобные адриамицину, блеомицину, доксорубицину, дауномицину, эпирубицину, идарубицину, митомицину-С, дактиномицину и митрамицину); антимитотические агенты (например, винкаалкалоиды, подобные винкристину, винбластину, виндезину и винорелбину, и таксоиды, подобные таксолу и таксотеру) и ингибиторы топоизомеразы (например, эпиподофиллотоксины, подобные этопозиду и тенипозиду, амсакрин, топотекан и камптотецин);
(2) цитостатические агенты, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен и йодоксифен), негативные регуляторы эстрогеновых рецепторов (например, фулвестрант), антиандрогены (например, бикалутамид, флутамид, нилутамид и ацетат кипротерона), антагонисты LHRH или агонисты LHRH (например, гозерелин, лейпрорелин и бусерелин), прогестогены (например, мегестрола ацетат), ингибиторы ароматазы (например, анастрозол, летрозол, воразол и эксеместан) и ингибиторы 5α-редуктазы, такие как финастерид;
(3) агенты, которые ингибируют инвазию раковых клеток (например, ингибиторы металлопротеиназы, подобные маримастату, и ингибиторы функции урокиназных рецепторов активаторов плазминогена);
(4) ингибиторы функции фактора роста, например, такие ингибиторы включают антитела фактора роста, антитела к рецепторам фактора роста (например, анти-erbb2 антитело трастузумаб [Herceptin™] и анти-erbb1 антитело цетуксимаб [С225]), ингибиторы фарнезилтрансферазы, ингибиторы тирозинкиназы и ингибиторы серин/треонинкиназы, например, ингибиторы семейства фактора роста эпидермиса (например ингибиторы тирозинкиназы семейства EGFR, такие как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, AZD1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) и 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)хиназолин-4-амин (CI 1033)), например ингибиторы семейства тромбоцитарных факторов роста, и, например, ингибиторы семейства факторов роста гепатоцитов;
(5) антиангиогенные агенты, такие как агенты, которые ингибируют эффекты сосудистого эндотелиального фактора роста (например, антитело против сосудистого эндотелиального фактора роста бевацизумаб [Avastin™], соединения, такие как соединения, раскрытые в международных заявках на патент WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354), и соединения с другими механизмами действия (например, линомид, ингибиторы функции αvβ3 интегрина и ангиостатин);
(6) сосудоповреждающие агенты, такие как Комбретастатин А4, и соединения, раскрытые в международных заявках на патент WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 и WO 02/08213;
(7) антисмысловые терапии, например такие, которые направлены на мишени, перечисленные выше, такие как ISIS 2503, антисмысловая anti-ras;
(8) подходы генной терапии, включая, например, подходы для замещения аберрантных генов, таких как аберрантный р53, или аберрантный BRCA1 или BRCA2, подходы GDEPT (ген-направленный фермент пролекарственной терапии), такие как подходы, использующие цитозиндеаминазу, тимидинкиназу или бактериальный фермент нитроредуктазу, и подходы увеличения толерантности пациента к химиотерапии или радиотерапии, такие как терапия гена множественной лекарственной устойчивости, и
(9) иммуннотерапевтические подходы, включая, например, ex vivo и in vivo подходы для увеличения иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4, или гранулоцитарно-макрофагеальным колониестимулирующим фактором, подходы для уменьшения анергии Т-клеток, подходы, использующие трансфицированные иммунные клетки, такие как цитокин-трансфицированные дендритные клетки, подходы, использующие цитокин-трансфицированные опухолевые клеточные линии, и подходы, использующие анти-идиотипические антитела.
Если фармацевтический препарат представлен в виде фиксированной дозы, то в таких комбинированных продуктах используют соединение формулы I в диапазоне дозировки, описанном в этом документе, и другой фармацевтически-активный агент в соответствующем ему диапазоне доз. Последовательное применение предусматривается в тех случаях, когда комбинированный фармацевтический препарат является неподходящим.
Хотя соединение формулы I главным образом полезно в качестве терапевтического агента для применения у теплокровных животных (включая человека), также оно является полезным всегда, когда требуется ингибирование эффектов металлопротеиназы. Таким образом, соединение является полезным в качестве фармакологического стандарта для применения при разработке новых биологических тестов и при поиске новых фармакологических агентов.
Далее изобретение проиллюстрировано следующими неограничивающими примерами.
Подходящие исходные вещества имеются в продаже или они могут быть получены любым подходящим способом, описанным в литературе, или известным специалисту-химику, или описанным в примерах, приведенных в данном описании. В дополнение, в следующей таблице представлена подробная информация о промежуточных соединениях и их соответствующие регистрационные номера в Chemical Abstracts.
Спектры ядерного магнитного резонанса (ЯМР-спектры), указанные в примерах, измеряли при комнатной температуре на спектрометре BRUKER DPX при напряженности поля 400 МГц, если не указано иного. Спектры содержат ссылку на внутренний дейтериевый стандарт. Масс-спектры (МС) измеряли на спектрометре Micromass MZD (электрораспыление).
Использованы следующие сокращения:
ДХМ - дихлорметан
ТГФ - тетрагидрофуран
LiHMDS - гексаметилдисилазид лития
ДМСО -диметилсульфоксид
ТФУ - трифторуксусная кислота
ПРИМЕР 1
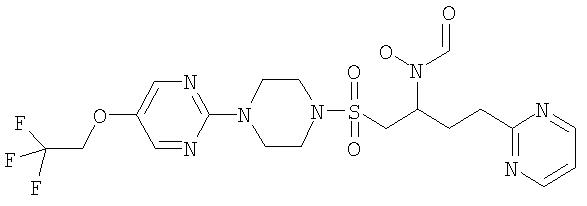
Гидрокси{(1S)-4-пиримидин-2-ил-1-[({4-[5-(2,2,2-трифторэтокси)пиримидин-2-ил]пиперазин-1-ил}сульфонил)метил]бутил}формамид

К муравьиной кислоте (114 мл, 3,03 моль) при 0°С добавляли уксусный ангидрид (28,6 мл, 0,303 моль) и смесь перемешивали при комнатной температуре (КТ) в течение 10 минут. Затем реакционную смесь снова охлаждали до 0°С и добавляли к раствору 2-(4-{[2-(гидроксиамино)-5-пиримидин-2-илпентил]сульфонил}пиперазин-1-ил)-5-(2,2,2-трифторэтокси)пиримидина (30,6 г, 60,5 ммоль) и муравьиной кислоты (114 мл, 3,03 моль) в ТГФ (600 мл). Реакционную смесь доводили до комнатной температуры и перемешивали в течение одного часа. Затем в вакууме удаляли летучие вещества и остаток подвергали азеотропной перегонке с толуолом (2×300 мл). Затем остаток растворяли в метаноле (300 мл) и нагревали до 40°С в течение одного часа. Затем раствор охлаждали до комнатной температуры и концентрировали в вакууме. После этого остаток очищали флэш-хроматографией (силикагель, 10% МеОН в EtOAc) с образованием рацемического соединения в виде бледно-оранжевой пены (22,94 г, 43 ммоль, 71%).
Рацемическую смесь разделяли хиральной ВЭЖХ с использованием условий, указанных ниже:
Колонка 20 мкм Chiralpak AD, Merck 100 мм,
Элюент MeCN/МеОН 90/10 (7 мин, изократическое элюирование),
MeCN/MeOH 90/10 (ступенчатое), MeCN/МеОН 85/15 (10 мин, изократическое), MeCN/МеОН 85/15 (градиентное, 1 мин), MeCN/EtOH 85/15 (изократическое, 37 мин).
Скорость потока 120 мл/мин.
Индивидуальные энантиомеры могут быть получены в кристаллической форме с использованием следующей методики.
40 г указанного в заголовке соединения перемешивали с этанолом (50 мл) при комнатной температуре в течение 30 минут. Растворитель удаляли в вакууме. Образовавшееся твердое вещество перемешивали в ацетоне (20 мл) при комнатной температуре в течение 24 часов. Растворитель удаляли в потоке аргона, а затем в вакууме.
1H ЯМР (ДМСО-D6, 373К): 9.39 (br s, 1H), 8.67 (d, 2H), 8.32 (s, 2H), 8.15 (br s, 1H),7.28(t, 1H), 4.70 (q, 2H), 4.39 (br s,1H), 3.79 (m, 4H), 3.47 (dd, 1H), 3.29 (m, 4H), 3.17 (dd, 1H), 2.91 (m, 2H), 1.75 (m, 4H).
MS (ESI): 534,01 (MH+).
Т.пл. 129-133°С.
Исходное вещество получали следующим образом.
К перемешиваемой суспензии 5-йод-2-[4-(метилсульфонил)пиперазин-1-ил]пиримидина (25,0 г, 67,9 ммоль), бензилового спирта (125 мл), 1,10-фенантролина (2,45 г, 20 мол.%) и карбоната цезия добавляли йодид меди(I) (12,9 г, 67,9 ммоль) и реакционную смесь нагревали до 110°С в течение 90 минут, а затем охлаждали до комнатной температуры. Затем добавляли ДХМ (250 мл) и нерастворимые вещества отфильтровывали через набивку из целита. Осадок промывали ДХМ (250 мл) и ДХМ фильтраты промывали водой. Затем водную фазу подвергали обратной экстракции дополнительным количеством ДХМ (500 мл), объединенные ДХМ экстракты промывали рассолом (500 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме с получением темно-коричневого густого осадка. Этот густой осадок затем очищали флэш-хроматографией (силикагель, 50% EtOAc/гексаны), получая 5-(бензилокси)-2-[4-(метилсульфонил)пиперазин-1-ил]пиримидин в виде беловатого твердого вещества (14,2 г, 40,7 ммоль, 60%).
1H ЯМР (CDCl3): 8.20 (s, 2H), 7.49 (m, 5H), 5.05 (s, 2H), 3.88 (m, 4H), 3.30 (m, 4H), 2.79 (s, 3Н).
MS (ESI): 349,08 (MH+).
5-(Бензилокси)-2-[4-(метилсульфонил)пиперазин-1 -ил]пиримидин (57,9 г, 0,17 моль) растворяли в ТФУ (600 мл) и реакционную смесь кипятили с обратным холодильником при перемешивании в течение 7 часов, затем охлаждали до комнатной температуры. Затем в вакууме удаляли ТФУ и остаток подвергали азеотропной перегонке с толуолом (2×300 мл). Образовавшееся твердое вещество растирали с ДХМ, отфильтровывали, промывали эфиром и сушили, получая 2-[4-(метилсульфонил)пиперазин-1-ил]пиримидин-5-ол в виде бледно-желтого твердого вещества (54,4 г, 0,15 моль, 88%, соль ТФУ).
1H ЯМР (ДМСО-D6): 8.02 (s, 2H), 3.68 (m, 4H), 3.12 (m, 4H), 2.86 (s, 3Н).
К перемешиваемой суспензии 2-[4-(метилсульфонил)пиперазин-1-ил]пиримидин-5-ола (53,5 г, 0,145 моль), К2СО3 (100,1 г, 0,725 моль) в ацетоне (1 л) добавляли 2,2,2-трифторэтил-нонафторбутансульфонат (78 г, 0,203 моль) и реакционную смесь нагревали до 60°С в течение 6 часов, а потом охлаждали до комнатной температуры. Затем реакционную смесь фильтровали и фильтрат упаривали до сухого состояния. Остаток распределяли между ДХМ (500 мл) и водой (500 мл), экстрагировали ДХМ (500 мл), объединенные органические экстракты промывали рассолом (500 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме, получая 2-[4-(метилсульфонил)пиперазин-1-ил]-5-(2,2,2-трифторэтокси)пиримидин в виде беловатого твердого вещества (45,3 г, 0,133 моль, 92%).
1H ЯМР (CDCl3): 8.15 (s, 2H), 4.32 (m, 2H), 3.90 (m, 4H), 3.30 (m, 4H), 2.78 (s, 3Н).
MS (ESI): 341,08 (МН+).
Способ 1
К перемешиваемой суспензии 2-[4-(метилсульфонил)пиперазин-1-ил]-5-(2,2,2-трифторэтокси)пиримидина (8,05 г, 23,6 ммоль) в ТГФ (175 мл) по каплям при -78°С добавляли LHMDS (47,2 мл, 47,2 ммоль) и реакционную смесь перемешивали в течение 15 минут. Затем при -78°С добавляли раствор этил-4-пиримидин-2-илбутаноата (5,5 г, 28,3 ммоль) в ТГФ (50 мл), нагревали до -20°С и перемешивали в течение 2 часов. Затем реакцию гасили добавлением насыщенного раствора NH4Cl (250 мл), дважды экстрагировали EtOAc (2×250 мл), объединенные органические экстракты промывали рассолом (250 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме с образованием желтого твердого вещества. Это вещество затем очищали флэш-хроматографией (силикагель, 50% EtOAc/гексаны), получая 5-пиримидин-2-ил-1-({4-[5-(2,2,2-трифторэтокси)пиримидин-2-ил]пиперазин-1-ил}сульфонил)пентан-2-он в виде беловатого твердого вещества (9,47 г, 19,4 ммоль, 82%).
1H ЯМР (CDCl3): 8.66 (d, 2H), 8.16 (s, 2H), 7.12 (t, 1H), 4.30 (m, 2H), 4.00 (s, 2H), 3.82 (m, 4H), 3.34 (m, 4H), 2.98 (t, 2H), 2.85 (t, 2H), 2.16 (m, 2H).
MS (ESI): 489,02 (МН+).
К перемешиваемому раствору 5-пиримидин-2-ил-1-({4-[5-(2,2,2-трифторэтокси)пиримидин-2-ил]пиперазин-1 -ил}сульфонил)пентан-2-она (9,47 г, 19,4 ммоль) в смеси ДХМ/МеОН (100 мл/100 мл) порциями добавляли NaBH4 (807 мг, 21,3 ммоль) и реакционную смесь перемешивали при комнатной температуре. Затем реакцию гасили добавлением насыщенного раствора NH4Cl (250 мл) и органические вещества удаляли в вакууме. Водный остаток затем экстрагировали EtOAc (2×250 мл), объединенные органические экстракты промывали рассолом (250 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме, получая 5-пиримидин-2-ил-1-({4-[5-(2,2,2-трифторэтокси)пиримидин-2-ил]пиперазин-1-ил}сульфонил)пентан-2-ол в виде белого твердого вещества (9,10 г, 18,6 ммоль, 96%).
MS (ESI): 491,13(MH+).
К перемешиваемому раствору 5-пиримидин-2-ил-1-({4-[5-(2,2,2-трифторэтокси)пиримидин-2-ил]пиперазин-1-ил}сульфонил)пентан-2-ола (9,10 г, 18,6 ммоль), триэтиламина (13 мл, 93,0 ммоль) в ДХМ (200 мл) при 0°С добавляли метансульфонилхлорид (2,16 мл, 27,9 ммоль). Реакционную смесь перемешивали при 0°С в течение 15 минут, нагревали до комнатной температуры и перемешивали в течение 16 часов. Затем реакционную смесь промывали водой (200 мл) и водную часть подвергали обратной экстракции ДХМ (200 мл). Объединенные ДХМ экстракты промывали рассолом (250 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме, получая 2-(4-{[(1Е)-5-пиримидин-2-илпент-1-ен-1-ил]сульфонил}пиперазин-1-ил)-5-(2,2,2-трифторэтокси)пиримидин в виде оранжевого твердого вещества (8,79 г, 18,6 ммоль, 100%).
MS (ESl): 472,49 (МН+).
К перемешиваемому раствору 2-(4-{[(1Е)-5-пиримидин-2-илпент-1-ен-1-ил]сульфонил}пиперазин-1-ил)-5-(2,2,2-трифторэтокси)пиримидина (8,79 г, 18,6 ммоль) в ТГФ (90 мл) добавляли 50%-ный водный раствор гидроксиламина (18 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавляли насыщенный раствор NH4Cl (200 мл), а затем дважды экстрагировали EtOAc (2×250 мл), объединенные органические экстракты промывали рассолом (250 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме, получая 2-(4-{[2-(гидроксиамино)-5-пиримидин-2-илпентил]сульфонил}пиперазин-1 -ил)-5-(2,2,2-трифторэтокси)пиримидин в виде бледно-желтого твердого вещества (8,96 г, 17,7 ммоль, 95%).
MS (ESl): 506,05 (MH+).
Способ 2
К перемешиваемой суспензии 2-[4-(метилсульфонил)пиперазин-1-ил]-5-(2,2,2-трифторэтокси)пиримидина (850 мг, 2,50 ммоль) в ТГФ (25 мл) по каплям при -78°С добавляли LHMDS (5,5 мл, 5,5 ммоль) и реакционную смесь перемешивали в течение 15 минут. Затем добавляли диэтилхлорфосфат (0,4 мл, 2,75 ммоль) и перемешивали в течение 15 минут. Затем этот раствор обрабатывали раствором 4-пиримидин-2-илбутаналя (413 мг, 2,75 ммоль) в ТГФ (5 мл) по каплям, оставляли нагреваться до -20°С и перемешивали в течение 1 часа. Затем реакцию гасили добавлением насыщенного раствора NH4Cl (100 мл), дважды экстрагировали EtOAc (2×100 мл), объединенные органические экстракты промывали рассолом (100 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме с образованием желтого масла. Это масло затем очищали флэш-хроматографией (силикагель, 50% EtOAc/гексаны), получая 2-(4-{[(1Е)-5-пиримидин-2-илпент-1-ен-1-ил]сульфонил}пиперазин-1-ил)-5-(2,2,2-трифторэтокси)пиримидин в виде желтого твердого вещества (1,13 г, 2,39 ммоль, 96%).
MS (ESl): 472,49 (MH+).
Затем это вещество превращали в 2-(4-{[2-(гидроксиамино)-5-пиримидин-2-илпентил]сульфонил}пиперазин-1-ил)-5-(2,2,2-трифторэтокси)пиримидин и затем в гидрокси{(1S)-4-пиримидин-2-ил-1 -[({4-[5-(2,2,2-трифторэтокси)пиримидин-2-ил]пиперазин-1-ил}сульфонил)метил]бутил}формамид посредством той же методики, как описано в способе 1.
ПРИМЕР 2
Также были получены следующие соединения.

ПРИМЕР 3
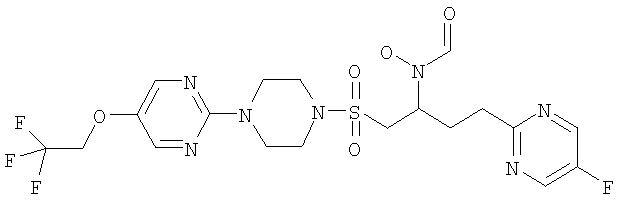
Гидрокси{(1S)-3-пиримидин-2-ил-1-[({4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазин-1-ил}сульфонил)метил]пропил}формамид

К муравьиной кислоте (54 мл, 1,4 моль) при 8°С добавляли уксусный ангидрид (11 мл, 100 ммоль) и смесь перемешивали при КТ в течение 10 минут. Затем смешанный ангидрид снова охлаждали до 8°С и добавляли к предварительно охлажденному до 0°С раствору 2-[3-(гидроксиамино)-4-({4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазин-1-ил}сульфонил)бутил]пиримидина (16,32 г, 33,3 ммоль) в ДХМ (170 мл) и муравьиной кислоте (65 мл, 1,7 моль). Реакционную смесь доводили до комнатной температуры и перемешивали в течение одного часа. Затем в вакууме удаляли летучие вещества и остаток подвергали азеотропной перегонке с толуолом (2×50 мл). Затем остаток растворяли в смеси МеОН/ДХМ (1:1, 250 мл) и перемешивали в течение ночи при комнатной температуре. Затем раствор концентрировали в вакууме и распределяли между ДХМ (250 мл) и насыщенным NaHCO3 (250 мл). После этого ДХМ слой фильтровали через силикагель (20 г), промывая смесью 5% МеОН/ДХМ, и летучие вещества удаляли в вакууме, получая указанное в заголовке рацемическое соединение в виде бледно-желтой пены (15,68 г, 302 ммоль, 91%).
Рацемическую смесь (86,5 г) разделяли на энантиомеры посредством хиральной ВЭЖХ с использованием следующих условий:
Колонка 20 мкм Chiralpak AD, Merck 100 мм
Элюент MeCN/MeOH 90/10 (17 мин, изократическое элюирование), MeCN/MeOH 90/10 (ступенчатое), MeCN/EtOH 90/10 (8 мин, изократическое), MeCN/EtOH 90/10 (градиентное, 1 мин), MeCN/EtOH 85/15 (изократическое, 39 мин).
Скорость потока 120 мл/мин
Концентрировали в вакууме до пены. Подвергали кристаллизации из горячего этанола (430 мл), фильтровали и промывали этанолом и эфиром. Сушили, получая указанное в заголовке соединение в виде белого кристаллического твердого вещества (28 г, 54 ммоль).
1H ЯМР (ДМСО, 373К): 9.41 (s, 1Н), 8.66 (d, 2Н), 8.07 (s, 1H), 7.99 (d, 1H), 7.38 (dd, 1H), 7.26 (t, 1H), 6.83 (d, 1H), 4.61 (q, 2H), 4.45 (В, 1H), 3.51 (t, 4H), 3.47 (d, 1H), 3.27 (t, 4H), 3.24 (d, 1H), 2.91 (t, 2H), 2.17 (m, 2H).
MS (ESI): 519(MH+);
Т.пл. 149-151°С.
Исходное вещество получали следующим образом.
Интенсивно перемешиваемую суспензию 1-(5-бромпиридин-2-ил)пиперазина (номер в CAS 73406-97-0, 116 г, 479 ммоль), 1,10-фенантролина (17,3 г, 96 моль), карбоната цезия (312 г, 960 ммоль) и йодида меди(I) (91 г, 480 ммоль) в бензиловом спирте (960 мл) перемешивали при 120°С в атмосфере инертного газа в течение 24 часов, добавляя каждый час дополнительные аликвоты йодида меди(I) (5×91 г).
Охлаждали до 40°С и разбавляли ДХМ (1 л), перемешивая при комнатной температуре в течение 30 минут. Фильтровали через целит, хорошо промывая ДХМ (500 мл). Фракции промывали NaOH (2 M, 300 мл), объединяли и экстрагировали HCl (2 M, 5×1 л). Объединенные кислотные экстракты промывали ДХМ (500 мл), охлаждали до 0°С и экстрагировали ДХМ (1 л), медленно подщелачивая NaOH (~46%-ным по массе) до рН 10. Водный слой дополнительно экстрагировали ДХМ (2×500 мл) и летучие вещества удаляли в вакууме, получая 1-[5-(бензилокси)пиридин-2-ил]пиперазин в виде черной жидкости (104 г, 278 ммоль @ 72 мас.%, 58%).
1H ЯМР (CDCl3): 8.0 (d, 1Н), 7.2 (dd, 1H), 6.3 (d, 1Н), 5.0 (s, 2Н), 3.50 (s, 8Н), 1.48 (s, 9H), 3.4 (В, 5Н), 3.0 (В, 4Н);.
MS (ESI): 270 (MH+)
Перемешиваемый раствор 1-[5-(бензилокси)пиридин-2-ил]пиперазина (104 г, 278 ммоль) в CH2Cl2 (1,1 л) при 0°С последовательно обрабатывали триэтиламином (94 мл, 672 ммоль) и метансульфонилхлоридом (31 мл, 400 ммоль). Реакционную смесь доводили до комнатной температуры и перемешивали в течение 3 часов. Затем реакционную смесь разбавляли ДХМ (3 л) и промывали водой (1 л), HCl (0,5 М, 2×800 мл) и насыщенным NaHCO3 (800 мл), подвергая обратной экстракции ДХМ (500 мл). Затем объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали в вакууме, получая 1 -[5-(бензилокси)пиридин-2-ил]-4-(метилсульфонил)пиперазин в виде темной жидкости (120 г, 278 ммоль @ 81 мас.%, 100%).
1H ЯМР (CDCl3): 8.0 (d, 1H), 7.35 (m, 5H), 7.2 (dd, 1H), 6.65 (d, 1H), 5.05 (s, 2H), 3.55 (t, 4Н), 3.3 (t, 4H), 2.8 (s, 3Н).
MS (ESl): 348 (МН+).
1-[5-(Бензилокси)пиридин-2-ил]-4-(метилсульфонил)пиперазин (120 г, 278 ммоль) растворяли в ТФУ (1,3 л) и реакционную смесь кипятили с обратным холодильником при перемешивании в течение 3 часов, затем охлаждали до комнатной температуры. Затем ТФУ удаляли в вакууме и остаток подвергали азеотропной перегонке с толуолом (2×300 мл). Образовавшую жидкость разбавляли ДХМ (100 мл) и медленно нейтрализовали насыщенным NaHCO3 (700 мл) до рН 8. Суспензию фильтровали, промывали водой, минимальным количеством ДХМ и эфиром и сушили, получая 6-[4-(метилсульфонил)пиперазин-1-ил]пиридин-3-ол в виде бежевого твердого вещества (69 г, 270 моль, 97%).
1H ЯМР (ДМСО-D6): 7.7 (d, 1Н), 7.1 (dd, 1Н), 6.75 (d, 1H), 3.45 (t, 4H), 3.2 (t, 4H), 2.85 (s, 3Н).
MS (ESl): 257 (MH+).
К перемешиваемой суспензии 6-[4-(метилсульфонил)пиперазин-1-ил]пиридин-3-ола (69 г, 270 ммоль), К2СО3 (112 г, 810 ммоль) в ацетоне (1,8 л) добавляли 2,2,2-трифторэтил-нонафторбутансульфонат и/или 2,2,2-трифторэтил-трифторметансульфонат (суммарное количество 324 ммоль) и реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Затем реакционную смесь фильтровали и фильтрат упаривали до сухого состояния. Остаток распределяли ДХМ (2,5 л, 500 мл) и водой (1,5 л, 300 мл), экстрагировали ДХМ (500 мл), сушили (MgSO4) и фильтровали. Концентрировали в вакууме, разбавляя EtOH до небольшого объема, фильтровали и сушили, получая 1-(метилсульфонил)-4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазин в виде беловатого твердого вещества (62 г, 183 ммоль, 68%).
1H ЯМР (CDCl3): 8.0 (d, 1H), 7.25 (dd, 1H), 6.65 (d, 1H), 4.3 (q, 2H), 3.6 (t, 4H), 3.35 (t, 4H), 2.8 (s, 3Н).
MS (ESI): 340 (МН+).
К перемешиваемой суспензии 1-(метилсульфонил)-4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазина (13,3 г, 39,2 ммоль) в ТГФ (200 мл) по каплям при -70°С добавляли LHMDS (75 мл, 75 ммоль) и реакционную смесь перемешивали в течение 20 минут. Затем при -70°С добавляли раствор этил-3-пиримидин-2-илпропаноата (9,2 г, 51 ммоль) в ТГФ (55 мл), нагревали до -20°С и перемешивали в течение 2 часов. Затем реакцию гасили добавлением насыщенного раствора NH4CI (250 мл), дважды экстрагировали EtOAc (3×250 мл), объединенные органические экстракты промывали рассолом (250 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме с образованием желтого твердого вещества. Это твердое вещество перемешивали в течение 20 минут в смеси 20% изогексан/эфир (100 мл), фильтровали, промывали изогексаном и сушили, получая 4-пиримидин-2-ил-1-({4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазин-1 -ил}сульфонил)бутан-2-он в виде беловатого твердого вещества (15,2 г, 32,2 ммоль, 82%).
1H ЯМР (CDCl3): 8.6 (d, 2H), 7.95 (d, 1H), 7.2 (dd, 1H), 7.1 (t, 1H), 6.6 (d, 1Н), 4.30 (q, 2H), 4.15 (s, 2H), 3.55 (t, 4Н), 3.4 (t, 4Н), 3.35 (t, 2H), 3,3 (t, 2H).
MS (ESl): 472 (МН+).
К перемешиваемому раствору 4-пиримидин-2-ил-1-({4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазин-1-ил}сульфонил)бутан-2-она (15 г, 31,6 ммоль) в смеси 10% МеОН/ДХМ (300 мл) порциями добавляли NaBH4 (0,52 г, 15,8 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 45 минут. Затем реакцию гасили добавлением насыщенного раствора NH4Cl (100 мл), разбавляли водой (150 мл) и экстрагировали ДХМ (3×200 мл), объединенные органические экстракты сушили (рассол, MgSO4), фильтровали и концентрировали в вакууме. Растирали с эфиром, фильтровали и сушили, получая 4-пиримидин-2-ил-1-({4-[5-(2,2,2-трифторэтокси)пиримидин-2-ил]пиперазин-1-ил}сульфонил)бутан-2-ол в виде кремового твердого вещества (13,8 г, 29,0 ммоль, 92%).
MS (ESl): 476 (МН+).
К перемешиваемому раствору 4-пиримидин-2-ил-1-({4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазин-1 -ил}сульфонил)бутан-2-ола (13,7 г, 28,8 ммоль) в ДХМ (250 мл) при 0°С добавляли метансульфонилхлорид (2,68 мл, 34,6 ммоль). Реакционную смесь перемешивали при 0°С в течение 20 минут, после чего по каплям добавляли триэтиламин (18,1 мл, 129 ммоль). Нагревали до комнатной температуры и перемешивали в течение 16 часов. Затем реакционную смесь разбавляли ДХМ (1 л), промывали водой (150 мл) и сушили (рассол, MgSO4), фильтровали и концентрировали в вакууме. Затем остаток очищали флэш-хроматографией (диоксид кремния, 0-5% МеОН в ДХМ), получая 2-[(3Е)-4-({4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазин-1-ил}сульфонил)бут-3-ен-1-ил]пиримидин в виде желтого твердого вещества (11,9 г, 18,6 ммоль, 90%).
MS (ESl): 458 (МН+).
К перемешиваемому раствору 2-[(3Е)-4-({4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазин-1-ил}сульфонил)бут-3-ен-1-ил]пиримидина (10,9 г, 23,7 ммоль) в ТГФ (200 мл) добавляли 50%-ный водный раствор гидроксиламина (11 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавляли воду (100 мл), затем экстрагировали EtOAc (3×100 мл) и сушили (рассол, MgSO4), фильтровали и концентрировали в вакууме, получая 2-[3-(гидроксиамино)-4-({4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазин-1-ил}сульфонил)бутил]пиримидин в виде бледно-желтого твердого вещества (11,1 г, 22,6 ммоль, 96%).
MS (ESl): 491 (MH+).
Альтернативно исходное вещество получали следующим образом.
К перемешиваемой суспензии 1-(метилсульфонил)-4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазина (23 г, 67,8 ммоль, полученного, как описано выше) в ТГФ (450 мл) по каплям при -65°С добавляли раствор LiHMDS в ТГФ (149 мл, 1,0 М раствор, 149 ммоль). Перемешивали в течение 30 минут. Добавляли диэтилхлорфосфат (11,3 мл, 78 ммоль) и перемешивали в течение 1 часа. Раствор обрабатывали по каплям раствором 3-(2-пиридинил)пропионового альдегида (12 г, 88,1 ммоль) в ТГФ (290 мл), затем оставляли нагреваться до 0°С в течение 3 часов, после чего гасили раствором гидроксиламина (41 мл, 50%-ный водный раствор, 680 ммоль). Реакционную смесь перемешивали в течение 16 часов при КТ. Реакционную смесь промывали насыщенным NH4Cl (250 мл), подвергая обратной экстракции этилацетатом (250 мл). Затем объединенные органические экстракты сушили (рассол и MgSO4), фильтровали и концентрировали в вакууме. Затем остаток растирали с эфиром в течение 1 часа, фильтровали и сушили, получая 2-[3-(гидроксиамино)-4-({4-[5-(2,2,2-трифторэтокси)пиридин-2-ил]пиперазин-1-ил}сульфонил)бутил]пиримидин (31,5 г, 64,3 ммоль, 95%).
1H ЯМР (CDCl3): 8.65 (d, 2Н), 8.0 (d, 1Н), 7.25 (dd, 1H), 7.15 (t, 1H), 6.65 (d, 1H), 4.3 (q, 2Н), 3.55 (m, 6H), 3.4 (t, 4H), 3.2 (t, 2H), 2.9 (d, 1H), 2.25 (m, 1H), 2.1 (m,1H);
MS (ESl): 491 (MH+).
Пример 4
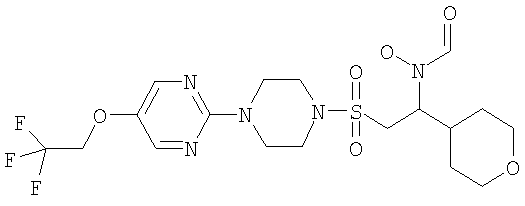
Гидрокси[(1S)-2-({4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1-ил}сульфонил)-1-(тетрагидро-2H-пиран-4-ил)этил]формамид
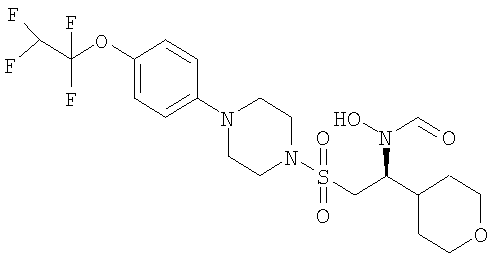
К охлажденному во льду раствору 1-{[(2S)-2-(гидроксиамино)-2-(тетрагидро-2H-пиран-4-ил)этил]сульфонил}-4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазина (52,9 г, 0,1 моль) в смешанной системе растворителей ТГФ/муравьиная кислота (1 л /20 мл) добавляли предварительно приготовленную смесь муравьиной кислоты (19 мл) и уксусного ангидрида (65 мл). Смесь перемешивали при комнатной температуре в течение ночи. Затем растворители выпаривали до небольшого объема и остаток распределяли между дихлорметаном (500 мл) и насыщенным раствором гидрокарбоната натрия. Органический слой отделяли, сушили (MgSO4), фильтровали и концентрировали до масла. Затем его перемешивали в метаноле (500 мл) в течение ночи, а потом концентрировали, получая моноформилированный продукт в виде белого твердого вещества. Это твердое вещество содержало незначительное количество примесей, поэтому его перемешивали в диэтиловом эфире в течение 4 часов, после чего отфильтровывали и сушили, получая гидрокси[(1S)-2-({4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1-ил}сульфонил)-1-(тетрагидро-2H-пиран-4-ил)этил]формамид (51,41 г, 92%).
Гидрокси[(1S)-2-({4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1 -ил}сульфонил)-1-(тетрагидро-2H-пиран-4-ил)этил]формамид (51,4 г) растворяли в горячем метаноле (80 мл) и затем оставляли медленно охлаждаться до комнатной температуры в течение ночи. Белое кристаллическое вещество отфильтровывали и сушили. Затем это твердое вещество перемешивали в изопропиловом спирте (190 мл) в течение 24 часов, после чего отфильтровывали и сушили при 50°С в течение ночи. Кристаллическое вещество промывали диэтиловым эфиром и снова сушили в течение 2 суток.
1H ЯМР (ДМСО-D6): 9.95 и 9.60 (1Н, s), 8.30 и 8.00 (1Н, s), 7.15 (2Н, d), 7.05 (2Н, d), 6.75 (1Н, tt), 4.45 и 3.85 (1Н, t), 3.85 (2Н, m), 3.40 (2Н, m), 3.25 (10Н, m), 1.75 (2Н, m), 1.50 (1Н, m), 1.25 (2Н, m).
MS (ES): 514 (MH+).
Т.пл. 175-176°C.
Исходное вещество получали следующим образом.
1-Бром-4-тетрафторэтоксибензол (номер в CAS 68835-05-9, 12 г, 0.044 М) растворяли в толуоле (250 мл) в атмосфере аргона. Добавляли N-boc-пиперазин (номер в CAS 57260-71-6, 9,79 г, 0,053 М), трет-бутилат натрия (5,93 г, 0,062 М), BINAP (96 мг) и дипалладийтридибензилиденацетон (96 мг). Перемешивали при 80°С в течение 4 часов, охлаждали и отфильтровывали нерастворимое вещество (промывая толуолом). Фильтрат упаривали до сухого состояния, получая неочищенный трет-бутил-4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1-карбоксилат. Выход 15,36 г (92%).
1H ЯМР (CDCl3): δ 7.10 (D, 2H), 6.90 (D, 2H), 5.90 (TT, 1H), 3.60 (M, 4H), 3.15 (M, 4H), 1.50 (S,9H).
MS (ES): 323,0 (МН-трет-бутил).
Неочищенный трет-бутил-4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1-карбоксилат (15,30 г, 0,04 М) растворяли в ДХМ (150 мл) и добавляли ТФУ (30 мл). Смесь перемешивали при комнатной температуре в течение ночи, упаривали до сухого состояния и подвергали азеотропной перегонке с толуолом. Остаток распределяли между этилацетатом и насыщенным раствором бикарбоната натрия и органическую фазу собирали, сушили над MgSO4, фильтровали и упаривали до сухого состояния, получая 1-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин в виде твердого вещества (10,97 г, 98%).
1H ЯМР (CDCl3): δ 7.15 (d, 2H), 6.90 (d, 2H), 5.90 (t, 1H), 3.35 (m, 8H).
MS (ES): 279,0.
1-[4-(1,1,2,2-Тетрафторэтокси)фенил]пиперазин (10,95 г, 0,04 М) растворяли в ДХМ (500 мл) и добавляли триэтиламин (18,5 мл, 0,13 ммоль). Смесь охлаждали до 0°С и добавляли метансульфонилхлорид (7,4 мл, 0,048 моль). Оставляли достигать температуры окружающей среды и перемешивали в течение ночи. Реакционную смесь промывали водой и органическую фазу собирали, сушили над MgSO4, фильтровали и упаривали до сухого состояния. Оставшееся твердое вещество подвергали кристаллизации из этанола, получая 1-(метилсульфонил)-4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин в виде белого твердого вещества. Выход 12,3 г (78,5%).
1H ЯМР (CDCl3): δ 7.15 (d, 2H), 6.95 (d, 2Н), 5.9 (tt, 1H), 3.35 (m, 4H), 3.3 (m, 4H), 2.8 (s, 3Н).
MS (ES): 357,26 (MH+).
1-(Метилсульфонил)-4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин (2,85 г, 0,008 моль) растворяли в безводном ТГФ (200 мл) и охлаждали до -10°С в атмосфере аргона. При охлаждении до -30°С добавляли 1,0 М раствор бис(триметилсилил)амида лития в ТГФ (17,6 мл, 0,0176 моль) и к смеси добавляли раствор метилтетрагидро-2Н-пиран-4-карбоксилата (номер в CAS 110238-91-0) в ТГФ (2 мл). Смесь оставляли достигать комнатной температуры и перемешивали в течение 2 часов. Реакцию гасили насыщенным раствором NH4Cl и смесь разбавляли Н2О и этилацетатом. Органическую фазу собирали, сушили над MgSO4, фильтровали и упаривали до сухого состояния, получая неочищенный 2-({4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1-ил}сульфонил)-1-(тетрагидро-2Н-пиран-4-ил)этанон (3,64 г, 97%).
1H ЯМР (CDCl3): δ 7.15 (d, 2H), 6.95 (d, 2H), 5.90 (tt, 1H), 4.05 (s, 2H), 4.00 (m, 2H), 3.50 (m, 6H), 3.25 (m, 4H), 2.95 (m, 1H), 1.85 (m, 2H), 1.70 (m, 2H).
MS (ES): 469,08 (МН+).
Неочищенный 2-({4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1 -ил}сульфонил)-1-(тетрагидро-2Н-пиран-4-ил)этанон (3,60 г, 0,008 М) растворяли в ДХМ (120 мл) и метаноле (40 мл) при температуре окружающей среды и добавляли борогидрид натрия (334 мг, 0,0088 моль). Перемешивали в течение 2 часов, добавляли H2O (250 мл) и экстрагировали ДХМ. Органическую фазу собирали, сушили над MgSO4, фильтровали и упаривали до сухого состояния, получая 2-({4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1-ил}сульфонил)-1-(тетрагидро-2Н-пиран-4-ил)этанол (3,6 г, 95%).
1H ЯМР (CDCl3): δ 7.15 (d, 2H), 6.90 (d, 2H), 5.90 (tt, 1H), 4.00 (m, 2H), 4.00 (m, 1H), 3.45 (m, 4H), 3.40 (m, 2H), 3.25 (m, 4H), 3.10 (m, 2H), 3.05 (m, 1H), 1.75 (m,2H), 1.50 (m,3H).
MS (ES): 471,08 (MH+).
2-({4-[4-(1,1,2,2-Тетрафторэтокси)фенил]пиперазин-1-ил}сульфонил)-1-(тетрагидро-2Н-пиран-4-ил)этанол (3,6 г, 0,008 М) растворяли в ДХМ (100 мл) и добавляли триэтиламин (5,58 мл, 0,04 моль). Смесь охлаждали до 0°С и в течение ночи при комнатной температуре и перемешивании добавляли метансульфонилхлорид (0,94 мл, 0,012 М). Добавляли воду и органическую фазу отделяли, сушили над MgSO4, фильтровали и упаривали до сухого состояния, получая 1-[4-(1,1,2,2-тетрафторэтокси)фенил]-4-{[(E)-2-(тетрагидро-2H-пиран-4-ил)винил]сульфонил}пиперазин. Выход 3,15 г (86,6%).
1H ЯМР (CDCl3): δ 7.1 (d, 2Н), 6.9 (d, 2Н), 6.75 (dd, 1H), 6.1 (d, 1H), 5.85 (tt, 1H), 4.0 (m, 2Н), 3.4 (m, 2H), 3.25 (m, 8H), 2.5 (m, 1H), 1.7 (m, 2H), 1.55 (m, 2H).
MS (ES): 452,88 (MH+).
1 -[4-(1,1,2,2-Тетрафторэтокси)фенил]-4-{[(E)-2-(тетрагидро-2Н-пиран-4-ил)винил]сульфонил}пиперазин (3,13 г, 0,007 моль) растворяли в ТГФ (50 мл) и добавляли 50%-ный гидроксиламин в Н2О (12 мл). Перемешивали при температуре окружающей среды в течение ночи, гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом. Органическую фазу сушили над MgSO4, фильтровали и упаривали до сухого состояния, получая рацемический 1-{[2-(гидроксиамино)-2-(тетрагидро-2H-пиран-4-ил)этил]сульфонил}-4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин. Его разделяли на энантиомеры с использованием AD Chiralpak хиральной препаративной ВЭЖХ колонки и элюируя смесью 20% метанол/ацетонитрил. Второе соединение, выделенное из колонки, представляло собой требуемый энантиомер, 1-{[(2S)-2-(гидроксиамино)-2-(тетрагидро-2H-пиран-4-ил)этил]сульфонил}-4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин.
1H ЯМР (CDCl3): δ 7.2 (d, 2H), 6.9 (d, 2H), 5.9 (tt, 1H), 3.85 (m, 2H), 3.5-3.1 (m, 11H), 3.05 (m, 2H), 1.951.8 (dd, 2H), 1.6 (d, 2H), 1.35 (m, 2H).
MS (ES): 485,92 (MH+).
ПРИМЕР 5
Гидрокси[1-фенил-2-({4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1-ил}сульфонил)этил]формамид
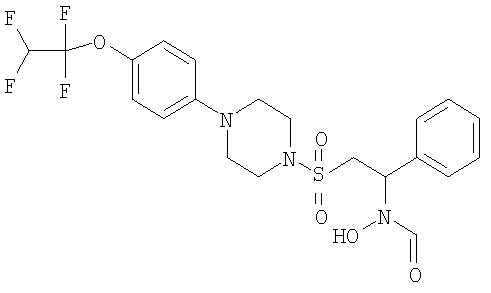
Это соединение получали с использованием способа, приведенного в примере 4.
1H ЯМР (CDCl3): 8.45 и 8.2 (d, 1H), 7.4 (m, 5H), 7.15 (d, 2H), 6.85 (d, 2H), 5.9 (tt, 1H), 5.5 (d, 1H), 3.4 (br s, 4Н), 3.3 (s, 2H), 3.15 (br, 4H).
Промежуточный 1-[(-2-фенилвинил)сульфонил]-4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин получали, как описано ниже.
1-[4-(1,1,2,2-Тетрафторэтокси)фенил]пиперазин (1,39 г, 0,005 моль) растворяли в ДХМ (250 мл) и добавляли триэтиламин (2,1 мл, 0,015 моль). Смесь охлаждали до 0°С и добавляли стиролсульфонилхлорид (номер в CAS 52147-97-4, 1,11 г, 0,0055 моль). Оставляли достигать температуры окружающей среды и перемешивали в течение ночи. Промывали Н2О и отделяли органическую фазу. Сушили над MgSO4, фильтровали и упаривали до сухого состояния с получением масла, которое очищали флэш-колоночной хроматографией (Merck 9385 диоксид кремния), элюируя смесью 80% изо-гексан/этилацетат и получая 1-[(2-фенилвинил)сульфонил]-4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин в виде желтого твердого вещества. Выход 650 мг (25%).
1H ЯМР (CDCl3): δ 7.5 (m, 3Н), 7.4 (m, 3Н), 7.15 (d, 2H), 6.9 (d, 2H), 6.7 (d, 1H), 5.85 (tt, 1H), 3.4 (m, 4H), 3.25 (m, 4H).
MS (ES): 445,27 (MH+).
ПРИМЕР 6
Гидрокси{4-пиримидин-2-ил-1 -[({4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1-ил}сульфонил)метил]бутил}формамид

Это соединение получали с использованием способа, приведенного в примере 4.
MS (ES): 550,03 (MH+).
Промежуточный 2-[5-({4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1-ил}сульфонил)пент-4-ен-1-ил]пиримидин получали, как описано ниже.
1-(Метилсульфонил)-4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин (356 мг, 0,001 моль) растворяли в безводном ТГФ (100 мл) и охлаждали до -10°С в атмосфере аргона. Добавляли 1,0 М раствор бис-(триметилсилил)амида лития в ТГФ (2,2 мл, 0,0022 моль) и перемешивали при -10°С в течение 30 минут, а затем в течение еще 30 минут при перемешивании и -10°С добавляли диэтилхлорфосфат (0,15 мл, 0,001 моль). Добавляли раствор 2-пиримидинил-4-масляного альдегида в безводном ТГФ (5 мл), смесь перемешивали при -10°С в течение 60 минут и, пока смесь оставалась холодной, гасили реакцию насыщенным раствором NH4Cl. Затем разбавляли Н2О и этилацетатом, органическую фазу собирали, сушили над MgSO4, фильтровали и упаривали до сухого состояния с образованием масла. Очистка флэш-колоночной хроматографией (Merck 9385 диоксид кремния) с элюированием этилацетатом дала 2-[5-({4-[4-(1,1,2,2-тетрафторэтокси)фенил]пиперазин-1-ил}сульфонил)пент-4-ен-1-ил]пиримидин. Выход 230 мг (47%).
1H ЯМР (CDCl3): 8.8 (d, 2Н), 7.2 (s, 1H), 7.1 (d, 2Н), 6.85 (d, 2H), 6.2 (d, 2H), 5.8 (tt, 1H), 4.05 (br, 1H), 3.25 (br, 8H), 3.05 (m, 2H), 2.3 (m, 1H), 2.05 (m, 2H), 1.4 (m, 2H).
MS (ESI): 489 (MH+).
ПРИМЕР 7
Также были синтезированы следующие соединения.
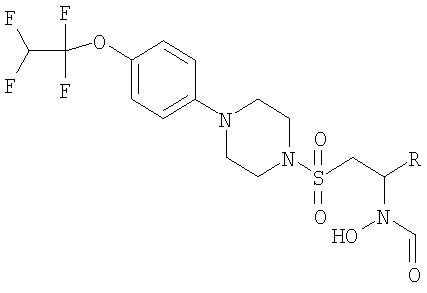
// знантиомер выделяли с помощью AD chiralpak препаративной ВЭЖХ колонки, элюируя смесью 20% метанол/ацетонитрил.
ПРИМЕР 8
Следующие соединения получали, как описано в предыдущих примерах.

// выделяли на Chiralpak AD колонке, элюируя смесью: 15% МеОН, MeCN,
/// выделяли на стадии гидроксиламина на Chiralpak AD колонке, элюируя смесью: 20% МеОН, MeCN.
Исходное вещество для этих синтезов получали следующим образом.
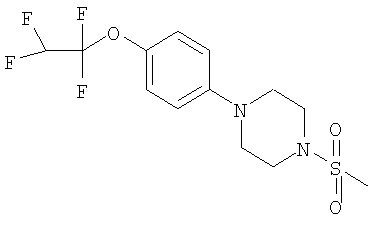
1-(Метилсульфонил)-4-[4-(2,2,2-трифторэтокси)фенил]пиперазин
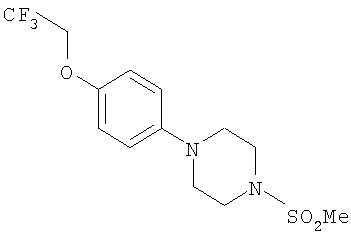
К раствору 4-бромфенола (9,57 г, 55 ммоль) в ацетоне (200 мл) добавляли карбонат калия (22,89 г, 166 ммоль) и 2,2,2-трифторэтил-трифторметансульфонат (16,0 г, 69 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем фильтровали и концентрировали при 300 мбар (30 кПа) и 30°С для удаления ацетона. Получили 1-бром-4-(2,2,2-трифторэтокси)бензол в виде воскообразного твердого вещества (выход >100%, так как присутствует некоторое количество ацетона).
1H ЯМР (ДМСО-D6), δ: 7.50 (2Н, d), 7.05 (2Н, d), 4.75 (2Н, q).
1-Бром-4-(2,2,2-трифторэтокси)бензол (14,5 г, 57 ммоль) растворяли в толуоле (250 мл) в атмосфере аргона. Добавляли трет-бутилпиперазин-1-карбоксилат (12,7 г, 68 ммоль), трет-бутилат натрия (7,6 г, 79,5 ммоль), рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (200 мг, 0,32 ммоль) и трис(дибензилиденацетон)дипалладий(0) (200 мг, 0,2 ммоль) и реакционную смесь нагревали до 80°С в течение 4 часов. Затем смесь охлаждали и фильтровали через целит, получая неочищенный трет-бутил-4-[4-(2,2,2-трифторэтокси)фенил]пиперазин-1-карбоксилат (32,47 г).
1H ЯМР (400 МГц, CDCl3): δ 6.90 (4Н, s), 4.30 (2Н, q), 3.60 (4Н, m), 3.05 (4H,m), 1.45(9H,s).
m/z (ES) 305 (MH+-tBu).
Неочищенный трет-бутил-4-[4-(2,2,2-трифторэтокси)фенил]пиперазин-1-карбоксилат (32,47 г, приблизительно 57 ммоль) растворяли в ДХМ (300 мл) и добавляли ТФУ (69 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем упаривали до сухого состояния, подвергая азеотропной перегонке с толуолом. Остаток распределяли между ДХМ и насыщенным раствором бикарбоната натрия. Органическую фазу отделяли, сушили (MgSO4), фильтровали и концентрировали, получая 1-[4-(2,2,2-трифторэтокси)фенил]пиперазин в виде твердого вещества (13,48 г, 91%).
1H ЯМР (400 МГц, CDCl3): δ 6.90 (4Н, s), 4.30 (2Н, q), 3.30 (8Н, m).
MS (ES) 261 MH+.
1-[4-(2,2,2-Трифторэтокси)фенил]пиперазин (13,48 г, 50 ммоль) растворяли в ДХМ (500 мл) и охлаждали до 0°С. Добавляли триэтиламин (29 мл, 0,2 моль), а затем по каплям метансульфонилхлорид (4,2 мл, 55 ммоль). Затем реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи, после чего гасили добавлением воды. Слои разделяли и органическую фазу сушили (MgSO4), фильтровали и концентрировали. Остаток перекристаллизовывали из горячего этанола, получая чистый 1-(метилсульфонил)-4-[4-(2,2,2-трифторэтокси)фенил]пиперазин (3,3 г, 18%).
1H ЯМР (CDCl3): δ 6.90 (4Н, s), 4.30 (2Н, q), 3.40 (4Н, m), 3.20 (4Н, m), 2.85 (3Н, s).
m/z (ES) 339 МН+.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ГИДАНТОИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2388758C2 |
| СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ КИНАЗЫ EGFR, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2020 |
|
RU2832925C2 |
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2288228C2 |
| ПИПЕРИДИН- И ПИПЕРАЗИНЗАМЕЩЕННЫЕ N-ГИДРОКСИФОРМАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2001 |
|
RU2283306C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2376301C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2697712C2 |
| НОВЫЕ ТИЕНОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2605403C2 |
| ПРОИЗВОДНЫЕ АРИЛАМИДОВ В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2011 |
|
RU2535671C1 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2760554C1 |
| НОВЫЕ ГЕТЕРОЦИКЛЫ | 2009 |
|
RU2572616C2 |
Описывается N-гидроксиформамидное соединение формулы (I) или его фармацевтически приемлемая соль, где кольцо В представляет собой фенил, пиридинил или пиримидинил; R2 представляет собой группу, выбранную из С1-6алкила, фенила или нафтила, где указанная группа замещена одной или более чем одной группой фторо, n равен 1, 2 или 3 и R1 представляет собой тетрагидропиранил, 2-пиримидинил-СН2СН2-, 2-пиримидинил-СН2СН2СН2-, 5F-2-пиримидинил-СН2СН2-, С1-6алкил или фенил. Соединения являются ингибиторами металлопротеиназ. 1 н. и 5 з.п. ф-лы.

1. N-Гидроксиформамидное соединение формулы (I)
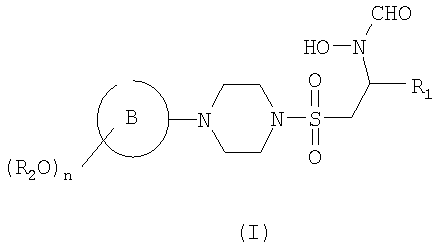
или его фармацевтически приемлемая соль,
где кольцо В представляет собой фенил, пиридинил или пиримидинил;
R2 представляет собой группу, выбранную из С1-6алкила, фенила или нафтила, где указанная группа замещена одной или более чем одной группой фторо;
n равен 1, 2 или 3 и
R1 представляет собой тетрагидропиранил, 2-пиримидинил-СН2СН2-, 2-пиримидинил-СН2СН2СН2-, 5F-2-пиримидинил-СН2СН2-, С1-6алкил или фенил.
2. Соединение по п.1, где R2 представляет собой С1-6алкильную группу, замещенную группами фторо в количестве от одной до пяти.
3. Соединение по п.1, где R2 замещен тремя или четырьмя группами фторо.
4. Соединение по п.3, где R2 представляет собой группу -CF2CHCF2.
5. Соединение по п.3, где R2 представляет собой группу -CH2CF3.
6. Соединение по любому из пп.1-5, где n равен 1.
| WO 00/75108 А1, 14.12.2000 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| WO 00/12478 A1, 09.03.2000 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

