Настоящее изобретение относится к новым производным гидантоина, способам их получения, фармацевтическим композициям, содержащим их, и их применению в терапии.
Металлопротеиназы представляют собой надсемейство протеиназ (ферментов), число которых в последние годы резко возросло. По структурным и функциональным соображениям эти ферменты разделены на семейства и подсемейства, как описано в N.М.Hooper (1994) FEBS Letters 354: 1-6. Примерами металлопротеиназ являются матриксные металлопротеиназы (MMPs), такие как коллагеназы (ММР1, ММР8, ММР13), желатиназы (ММР2, ММР9), стромелизины (ММР3, ММР10, ММР11), матрилизин (ММР7), металлоэластаза (ММР12), энамелизин (ММР19), МТ-ММР (ММР14, ММР15, ММР16, ММР17); репролизин или адамализин или семейство MDC, которое включает в себя секретазы и шеддазы, такие как TNF-конвертирующие ферменты (ADAM10 и ТАСЕ); семейство астацинов, которое включает в себя такие ферменты, как протеиназа процессинга проколлагена (РСР); а также другие металлопротеиназы, такие как аггриканаза, семейство эндотелин-конвертирующих ферментов и семейство ангиотензин-конвертирующих ферментов.
Считается, что металлопротеиназы имеют важное значение при многих физиологических болезненных процессах, в которые вовлечено ремоделирование тканей, такое как развитие эмбриона, костеобразование и маточное ремоделирование во время менструации. В основе этого лежит способность металлопротеиназ расщеплять целый ряд матриксных субстратов, таких как коллаген, протеогликан и фибронектин. Считается также, что металлопротеиназы играют важную роль в процессинге, или секреции, биологически важных клеточных медиаторов, таких как фактор некроза опухоли (TNF), и в посттрансляционном протеолитическом процессинге, или шеддинге, биологически значимых мембранных белков, таких как низкоаффинный рецептор к IgE, CD23 (более полный перечень смотри в N.М.Hooper et al., (1997) Biochem. J. 321: 265-279).
Металлопротеиназы ассоциированы со многими заболеваниями или состояниями. Ингибирование активности одной или более металлопротеиназ может быть полезно при этих заболеваниях или состояниях, например при различных воспалительных и аллергических заболеваниях, таких как воспаление сустава (особенно ревматоидный артрит, остеоартрит и подагра), воспаление желудочно-кишечного тракта (особенно воспалительное заболевание кишечника, неспецифический язвенный колит и гастрит), воспаление кожи (особенно псориаз, экзема, дерматит); при метастазировании или инвазии опухоли; при заболевании, связанном с неконтролируемым разрушением внеклеточного матрикса, таком как остеоартрит; при заболевании, связанном с резорбцией кости (таком как остеопороз и болезнь Педжета); при заболеваниях, связанных с аберрантным ангиогенезом; при усиленном ремоделировании коллагена, ассоциированном с диабетом, заболеванием периодонта (таким как гингивит), изъязвлением роговицы, изъязвлением кожи, послеоперационными состояниями (такими как кишечный анастомоз) и заживлением кожных ран; при заболеваниях, связанных с демиелинизацией центральной и периферической нервных систем (таких как рассеянный склероз); при болезни Альцгеймера; при ремоделировании внеклеточного матрикса, которое наблюдается при сердечно-сосудистых заболеваниях, таких как рестеноз и атеросклероз; при астме; при рините; и при хронических обструктивных болезнях легких (COPD).
ММР12, известная также как макрофагальная эластаза или металлоэластаза, первоначально была клонирована в мыши Shapiro et al. [1992, Journal of Biological Chemistry 267: 4664] и в человеке той же группой исследователей в 1995 году. ММР-12 преимущественно экспрессируется в активированных макрофагах, и, как было показано, секретируется из альвеолярных макрофагов курящих людей [Shapiro et al., 1993, Journal of Biological Chemistry 268: 23824], а также в пенистых клетках в атеросклеротических повреждениях [Matsumoto et al., 1998, Am J Pathol 153:109]. Мышиная модель COPD основана на провокации мышей сигаретным дымом в течение шести месяцев, по две сигареты в сутки, шесть суток в неделю. После такой обработки у мышей дикого типа развивалась легочная эмфизема. Когда в данной модели тестировали мышей, нокаутированных по ММР12, значительная эмфизема у них не развивалась, что четко указывает на то, что ММР12 является ключевым ферментом в патогенезе COPD. Роль ММР, таких как ММР12, при COPD (эмфиземе и бронхите) обсуждалась в Anderson and Shinagawa, 1999, Current Opinion in Anti-inflammatory and Immunomodulatory Investigational Drugs 1(1): 29-38. Недавно было обнаружено, что курение усиливает инфильтрацию макрофагов и экспрессию ММР-12 макрофагального происхождения в бляшках Кангавари сонной артерии человека [Matetzky S., Fishbein MC et al., Circulation 102(18), 36-39 Suppl. S, Oct 31, 2000].
MMP9 (желатиназа В; коллагеназа типа IV 92 кДа; желатиназа 92 кДа) представляет собой секретируемый белок, который впервые был очищен, а затем клонирован и секвенирован в 1989 году [S.M.Wilhelm et al. (1989) J. Biol. Chem. 264(29): 17213-17221; опечатки опубликованы в J. Biol. Chem. (1990) 265(36): 22570]. Недавно опубликованный обзор по MMP9 [Т.Н.Vu & Z.Werb (1998) (In: Matrix Metalloproteases, 1998. Edited by W.C. Parks & R.P.Mecnam. pp 115-148. Academic Press. ISBN 0-12-545090-7] является отличным источником подробной информации и ссылок по данной протеазе. Из указанного обзора Т.Н. Vu & Z.Werb (1998) вытекает следующее.
Экспрессия MMP9 обычно ограничена несколькими типами клеток, в том числе трофобластами, остеокластами, нейтрофилами и макрофагами. Однако эта экспрессия может быть индуцирована в этих клетках и в клетках других типов несколькими медиаторами, включая воздействие на эти клетки факторов роста или цитокинов. Они представляют собой те же самые медиаторы, которые часто вовлечены в инициирование воспалительной реакции. Как и другие секретируемые ММР, MMP9 высвобождается в виде неактивного профермента, который затем расщепляется с образованием ферментативно активного фермента. Протеазы, необходимые для такой активации in vivo, неизвестны. Баланс активной MMP9 и неактивного фермента дополнительно регулируется in vivo взаимодействием с природным белком TIMP-1 (тканевый ингибитор металлопротеаз-1). TIMP-1 связывается с С-концевым участком ММР9, что приводит к ингибированию каталитического домена ММР9. Сочетание баланса индуцированной экспрессии ProMMP9, расщепления Pro- до активной ММР9 и присутствия TIMP-1 определяет количество каталитически активной ММР9, присутствующей в сайте локализации. Протеолитически активная ММР9 атакует субстраты, которые включают в себя желатин, эластин и природные коллагены типа IV и типа V; она не обладает активностью в отношении нативного коллагена типа I, протеогликанов или ламининов.
Возрастает масса сведений о роли ММР9 в различных физиологических и патологических процессах. Физиологические роли включают в себя инвазию эмбриональных трофобластов через эпителий матки на ранних стадиях эмбриональной имплантации; определенное участие в росте и развитии костей; и миграцию воспалительных клеток из сосудистой сети в ткани.
Высвобождение ММР-9, измеренное с применением иммуноферментного анализа, было значительно более высоким в жидкостях и в AM супернатантах от не подвергавшихся лечению астматиков по сравнению с другими популяциями [Am. J. Resp. Cell & Mol. Biol., Nov 1997, 17(5): 583-591]. Повышенную экспрессию ММР9 наблюдали также при некоторых других патологических состояниях, и эти наблюдения свидетельствуют о том, что ММР9 вовлечена в такие болезненные процессы как COPD, артрит, метастазирование опухолей, болезнь Альцгеймера, рассеянный склероз и перфорация бляшек при атеросклерозе, приводящая к острым коронарным состояниям, таким как инфаркт миокарда.
Известно множество ингибиторов металлопротеиназ (см., например, обзор по ингибиторам ММР Beckett R.P. and Whittaker M., 1998, Exp. Opin. Ther. Patents, 8(3): 259-282; и Whittaker M. et al., 1999, Chemical Reviews 99(9): 2735-2776).
В WO 02/074767 раскрыты производные гидантоина формулы
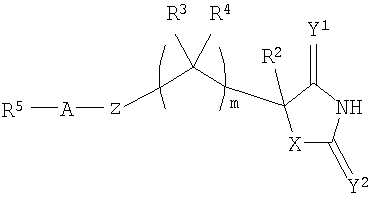 ,
,
которые полезны в качестве ингибиторов ММР, в частности в качестве эффективных ингибиторов ММР12.
В данной работе авторы описывают дополнительную группу производных гидантоина, которые являются ингибиторами металлопротеиназ и, в частности, представляют интерес в ингибировании ММР, таких как ММР12 и ММР9. Соединения по данному изобретению обладают полезной эффективностью, селективностью и/или фармакокинетическими свойствами. Соединения по данному изобретению входят в общий объем WO 02/074767, но конкретно не указаны в ней.
Согласно настоящему изобретению предложено соединение формулы (I)
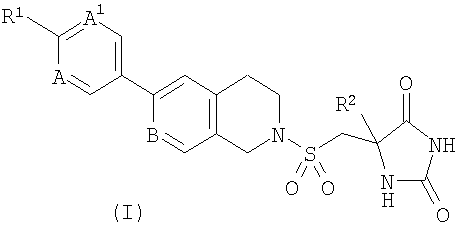 ,
,
где R1 представляет собой Н, галоген, CF3 или CH2CN;
R2 представляет собой C1-3алкил; и
каждый из А, А1 и В независимо представляет собой СН или N;
и его фармацевтически приемлемые соли.
Соединения формулы (I) могут существовать в энантиомерных формах. Следует понимать, что все энантиомеры, диастереомеры, рацематы и их смеси входят в объем данного изобретения.
Соединения формулы (I) могут существовать в таутомерных формах. Все возможные таутомерные формы и их смеси включены в объем данного изобретения.
В одном воплощении R1 представляет собой хлоро.
В одном воплощении R1 представляет собой CF3.
В одном воплощении R2 представляет собой метил или этил. В одном воплощении
R2 представляет собой метил.
В одном воплощении каждый из А и А1 представляет собой N. В другом воплощении А представляет собой N, и А1 представляет собой СН. В другом воплощении каждый из А и А1 представляют собой СН.
В одном воплощении В представляет собой N. В другом воплощении В представляет собой СН.
В одном воплощении R1 представляет собой CF3; R2 представляет собой метил или этил; каждый из А и А1 представляет собой N; и В представляет собой СН.
В одном воплощении R1 представляет собой CF3; R2 представляет собой метил или этил; каждый из А и А1 представляет собой N; и В представляет собой N.
В одном воплощении R1 представляет собой хлоро; R2 представляет собой метил или этил; А представляет собой N, и А1 представляет собой СН; и В представляет собой N.
В одном воплощении R1 представляет собой хлор; R2 представляет собой метил или этил; каждый из А, А1, В представляет собой СН.
Если не указано иное, термин "С1-3алкил" в данном описании означает прямую или разветвленную алкильную группу, содержащую от 1 до 3 атомов углерода. Примерами таких групп являются метил, этил, н-пропил и изопропил.
Если не указано иное, термин "галоген" в данном описании означает фтор, хлор, бром или йод.
Примеры соединений по изобретению включают:
(5S)-5-метил-5-({[6-[2-(трифторметил)пиримидин-5-ил]-3,4-дигидроизохинолин-2(1H)-ил]сульфонил}метил)имидазолидин-2,4-дион;
(5S)-5-({[6-(4-хлорфенил)-3,4-дигидроизохинолин-2(1H)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион;
{4-[2-({[(4S)-4-метил-2,5-диоксоимидазолидин-4-ил]метил}сульфонил)-1,2,3,4-тетрагидроизохинолин-6-ил]фенил}ацетонитрил;
(5S)-5-метил-5-{[(6-пиридин-3-ил-3,4-дигидроизохинолин-2(1Н)-ил)сульфонил]метил}имидазолидин-2,4-дион;
(5S)-5-({[6-(4-хлорфенил)3,4-дигидро-2,7-нафтиридин-2(1H)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион;
и их фармацевтически приемлемые соли.
Каждое приведенное в примере соединение представляет частный и независимый аспект данного изобретения.
Соединения формулы (I) могут находиться в энантиомерных формах. Вследствие этого, все энантиомеры, рацематы и их смеси включены в объем настоящего изобретения. Разные оптические изомеры могут быть выделены посредством разделения рацемической смеси соединений с использованием общепринятых методов, например фракционной кристаллизации или ВЭЖХ (высокоэффективной жидкостной хроматографии). Кроме того, оптические изомеры могут быть получены посредством асимметрического синтеза или посредством синтеза из оптически активных исходных веществ.
Когда у соединений по изобретению существуют оптические изомеры, авторы изобретения раскрывают все отдельные оптически активные формы и их комбинации отдельными конкретными воплощениями данного изобретения так же, как и их соответствующие рацематы.
Предпочтительно соединения формулы (I) обладают (5S)-стереохимией, как показано ниже:

Когда у соединений по изобретению существуют таутомеры, авторы изобретения раскрывают все отдельные таутомерные формы и их сочетания как отдельные конкретные воплощения изобретения.
Настоящее изобретение включает соединение формулы (I) в форме солей. Приемлемые соли включают соли, образованные органическими или неорганическими кислотами, или органическими или неорганическими основаниями. Обычно такие соли будут фармацевтически приемлемыми солями, хотя фармацевтически неприемлемые соли могут быть полезны в получении и очистке отдельных соединений. Такие соли включают соли присоединения кислот, такие как хлористоводородная, бромистоводородная соль, цитрат, тозилат и малеат, и соли, образованные фосфорной и серной кислотой. В другом аспекте приемлемые соли являются основными солями, такими как соль щелочного металла, например натрия или калия, соль щелочноземельного металла, например кальция или магния, или соль органического амина, например триэтиламина.
Соли соединения формулы (I) могут быть образованы посредством взаимодействия свободного основания или другой его соли с одним или более эквивалентами подходящей кислоты или основания.
Соединения формулы (1) являются полезными, поскольку они обладают фармакологической активностью у животных и, таким образом, являются потенциально полезными в качестве лекарственных средств. В частности, соединения по изобретению являются ингибиторами металлопротеиназы, и, соответственно, могут применяться в лечении заболеваний и состояний, опосредованных ММР12 и/или ММР9, таких как астма, ринит, хроническое обструктивное заболевание легких (COPD), артрит (например, ревматоидный артрит и остеоартрит), атеросклероз и рестеноз, рак, инвазия и метастазы, заболевания, включающие в себя деструкцию тканей, расшатывание эндопротезов тазобедренных суставов, заболевание периодонта, фиброзное заболевание, инфаркт и сердечное заболевание, фиброз печени и почек, эндометриоз, заболевания, относящиеся к ослаблению внеклеточного матрикса, сердечная недостаточность, аневризмы аорты, заболевания, относящиеся к ЦНС, такие как болезнь Альцгеймера и рассеянный склероз (MS), и гематологические заболевания.
В общем случае соединения по изобретению являются эффективными ингибиторами ММР9 и ММР12. Соединения по настоящему изобретению также показывают хорошую селективность с точки зрения относительного отсутствия ингибирования разных других ММР, например ММР14.
Таким образом, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль, как определено выше, для применения в терапии.
В другом аспекте изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, для изготовления лекарственного средства для применения в терапии.
В другом аспекте изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, для изготовления лекарственного средства для применения в лечении заболеваний и состояний, при которых полезно ингибирование ММР12 и/или ММР9.
В другом аспекте изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для применения в лечении воспалительного заболевания.
В другом аспекте изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для применения в лечении обструктивных заболевания дыхательных путей, например астмы или COPD.
В другом аспекте изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, в изготовлении лекарственного средства для применения в лечении ревматоидного артрита, остеоартрита, атеросклероза, рака или рассеянного склероза.
В контексте данной заявки, термин "терапия" также включает "профилактику", если не указано иное. Термины "терапевтический" и "терапевтически" должны истолковываться соответственно.
Как ожидается, профилактика наиболее уместна в лечении субъектов, у которых уже имел место эпизод заболевания или состояния, о котором идет речь или которые, как предполагается, имеют повышенный риск такого заболевания или состояния. Субъекты с повышенным риском развития конкретного заболевания или состояния, как правило, включают тех, у кого имеется семейная история данного заболевания или состояния, или тех, у кого в результате генетического тестирования или скрининга обнаружена особенная чувствительность к развитию заболевания или состояния.
Кроме того, в изобретении предложен способ лечения заболевания или состояния, для которых полезно ингибирование ММР12 и/или ММР9, который включает введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как указано выше.
В изобретении также предложен способ лечения обструктивного заболевания дыхательных путей, например астмы или COPD, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как указано выше.
Для указанных выше терапевтических применений вводимые дозы будут, несомненно, меняться в зависимости от используемого соединения, пути введения, требуемого лечения и заболевания, которое лечат. Суточная доза соединения формулы (I)/соли (активного ингредиента) может находиться в интервале от 0,001 мг/кг до 75 мг/кг, в частности от 0,5 мг/кг до 30 мг/кг. Эта суточная доза при необходимости может быть введена в разделенных дозах. Обычно стандартная лекарственная доза будет содержать примерно от 1 мг до 500 мг соединения по данному изобретению.
Соединения формулы (I) или их фармацевтически приемлемые соли могут применяться сами по себе, но в основном будут вводиться в виде фармацевтической композиции, в которой соединение формулы (I)/соль (активный компонент) находится вместе с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем. В зависимости от способа введения, фармацевтическая композиция будет предпочтительно содержать от 0,05 до 99 мас.% (процентов по массе), более предпочтительно от 0,10 до 70 мас.% активного компонента и от 1 до 99,95 мас.%, более предпочтительно от 39 до 99,9 мас.% фармацевтически приемлемого вспомогательного вещества, разбавителя или носителя, причем все проценты по массе рассчитываются исходя из общей композиции. Общепринятые метода выбора и изготовления подходящих фармацевтических композиций описаны, например, в "Pharmaceuticals - The Science of dosage Form Designs", M.E.Aulton, Churchill Livingstone, 1998.
Таким образом, в настоящем изобретении также предлагается фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, как указано выше, вместе с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Кроме того, в изобретении предложен способ изготовления фармацевтической композиции по изобретению, которая содержит смесь соединения формулы (I) или его фармацевтически приемлемой соли, как указано выше, с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Фармацевтические композиции по данному изобретению могут быть введены обычным для данного заболевания или состояния способом, который желателен для лечения, например, при помощи перорального, местного, парентерального, трансбуккального, назального, вагинального или ректального введения или ингаляции. Для этих целей соединения по данному изобретению могут быть приготовлены в виде препарата способами, известными в данной области, в форме, например, таблеток, капсул, водных или масляных растворов, суспензий, эмульсий, кремов, мазей, гелей, назальных спреев, суппозиториев, тонкоизмельченных порошков или аэрозолей для ингаляции, и стерильных водных или масляных растворов или суспензий или стерильных эмульсий для парентерального применения (включая внутривенное, внутримышечное или инфузию).
Кроме соединений по настоящему изобретению фармацевтическая композиция по данному изобретению также может содержать или быть введена вместе (одновременно или последовательно) с одним или более фармацевтическими компонентами, полезными в лечении одного или более заболеваний или состояний, упомянутых выше, таких как продукт Symbicort (товарный знак).
Кроме того, в изобретении также предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, который включает:
а) взаимодействие соединения формулы (II)
 ,
,
где R2 такой, как указан в формуле (I), и L1 представляет собой уходящую группу с соединением формулы (III) (или его солью)
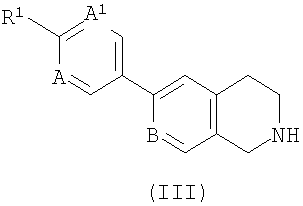
где R1, А, А1 и В такие, как указано в формуле (I); или
б) взаимодействие соединения формулы (V)
 ,
,
где R2 и В такие, как указано в формуле (I), и LG представляет собой уходящую группу; с производным бороновой кислоты формулы (XII)
 ,
,
где R1, А и А1 такие, как указано в формуле (I); или
в) взаимодействие соединения формулы (IX)
 ,
,
где R1, R2, А, А1 и В такие, как указано в формуле (I); с карбонатом аммония и цианидом калия;
и возможно после этого получение его фармацевтически приемлемой соли.
В описанном выше процессе (а), подходящие уходящие группы L1 включают галогено, в частности хлоро или трифторметилсульфонат. Взаимодействие предпочтительно осуществляют в подходящем растворителе, возможно в присутствии добавленного основания в течение подходящего периода времени, обычно от 0,5 до 16 ч, в интервале температур от комнатной до температуры кипения. Используют обычные растворители, такие как N,N-диметилформамид, пиридин, тетрагидрофуран, ацетонитрил, N-метилпирролидин или дихлорметан. Добавленное основание, в случае его применения, может представлять собой органическое основание, такое как триэтиламин, N,N-диизопропилэтиламин, N-метилморфолин или пиридин, или неорганическое основание, такое как карбонат щелочного металла. Взаимодействие обычно проводят при температуре окружающей среды в течение от 0,5 до 16 ч или до завершения реакции, определенного хроматографическими и спектроскопическими методами. Реакции сульфонилгалогенидов с различными первичными и вторичными аминами, хорошо известны в литературе, и различия в условиях очевидны для специалиста в данной области.
Сульфонилхлориды формулы (II), где L1 представляет хлор и R2 представляет Me, раскрыты в WO 02/074767 и ссылках, приведенных в данном описании. Соответствующие соединения, где R2 представляет собой С1-3алкил, могут быть получены с использованием аналогичных методов.
Подходящие способы получения соединений формулы (I) описаны ретросинтетическом методе на Схеме 1.
Схема 1
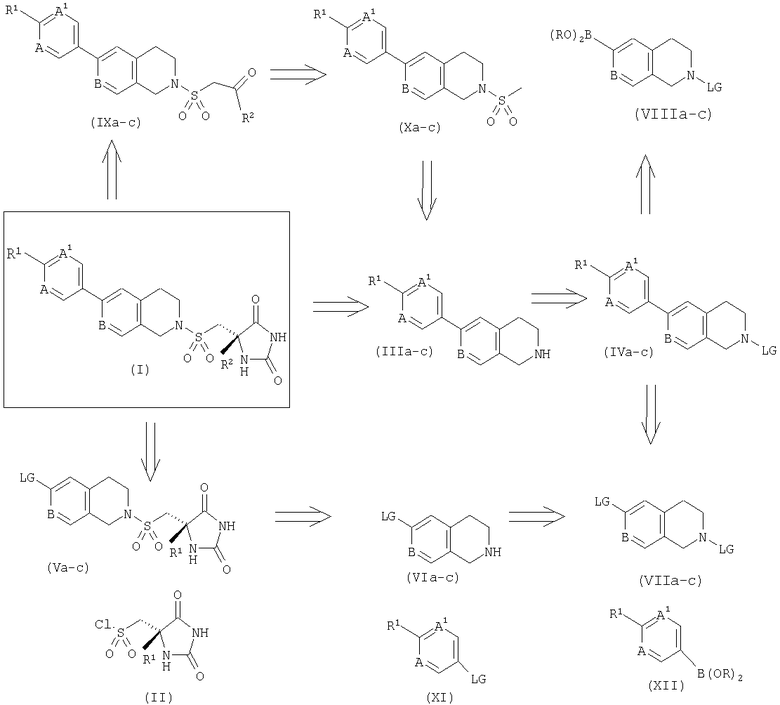
На Схеме 1 защитные группы (PG) могут быть либо карбаматами (например, трет-бутоксикарбамат), либо амидами (например, трифторацетил) или алкилом (например, трет-бутил или бензил). Уходящие группы (LG) могут быть либо хлоридом, бромидом, иодидом или трифторметилсульфонатом. В катализируемом палладием сочетании Сузуки можно использовать либо бороновые кислоты, либо пинаколборонаты. Промежуточное соединение (IVa-c) можно получить посредством стандартных реакций сочетания Сузуки (Chem. Rev. 1995, 95, 2457), или другого способа, между электрофильным (VIIa-c) и бороновым реагентом (XII), между электрофилом (XI) и бороновым реагентом (VIIIa-в). Последний может быть получен из (VIIa-c), применяя стандартные условия Мияура (J. Org. Chem. 1995, 60, 7508-7510). В результате снятия защитных групп (IVa-c) либо хлористым водородом в метаноле (PG = трет-бутоксикарбонил), либо кипящим 1-хлорэтилхлорформиатом/кипящим метанолом (PG = трет-бутил или бензил) (Synlett. 1993, 195-196) получают амин (IIIa-c) в виде хлористоводородной соли. Свободное основание можно получить посредством обработки (IIIa-c) основанием и экстракции органическим растворителем, например этилацетатом или толуолом. В результате взаимодействия (IIIa-c) либо в виде соли, либо в виде основания в подходящем растворителе (например, ацетонитриле, тетрагидрофуране, N-метилпирролидине или N,N-диметилформамиде) с сульфонилхлоридом (II) в присутствии третичного амина (например, триэтиламина, пиридина или N,N-диизопропилэтиламина) в течение от 0,5 до 16 ч получают соединение формулы (I).
Другой способ получения соединений формулы (I) из промежуточного соединения (IIIa-c) через метансульфонамид (Ха-с) и кетон (IXa-c) был описан ранее (WO 02/074767). Описывая кратко, в результате взаимодействия (IIIa-c) с метансульфонилхлоридом и третичным амином (например, триэтиламином, пиридином или N,N-диизопропилэтиламином) в подходящем растворителе (например, дихлорметане или тетрагидрофуране) получают метансульфонамид (Ха-с), который, в свою очередь, может быть превращен в кетон (IXa-c), при использовании стандартных методов. В результате нагревания кетона (IXa-c) с карбонатом аммония и цианидом калия в 50%-м водном этаноле в закрытом сосуде при 80-90°С в течение 1-5 часов получают рацемический гидантоин, который может быть разделен посредством хиральной хроматографии (например, на OD-H со 100%-ным этанолом).
Согласно третьему способу с промежуточного соединения (VIIa-c) снимают защиту, как это описано выше, с получением амина (VIa-c) в виде хлористоводородной соли. Свободное основание может быть выделено путем обработки основанием и экстракции органическим растворителем, например этилацетатом или толуолом. В результате взаимодействия (VIa-c) либо в виде соли, либо в виде основания в приемлемом растворителе (например, ацетонитриле, тетрагидрофуране, N-метилпирролидине или N,N-диметилформамиде) с сульфонилхлоридом (II) в присутствии третичного амина (например, триэтиламина, пиридина или N,N-диизопропилэтиламина) в течение 0,5-16 часов получают хиральный сульфонамид (Va-c). Последний может быть связан с бороновым реагентом (XII) с использованием стандартных условий Сузуки, в результате чего получают соединения формулы (I).
Промежуточные соединения (VIIa-b) легко получить, используя следующие методы.
1,2,3,4-тетрагидроизохинолиновое промежуточное соединение (VIIa)
Методы синтеза 1,2,3,4-тетрагидроизохинолинов хорошо известны в литературе. Классическим способом является реакция Померанца-Фрича, то есть реакция бензальдегидов с диацеталь-защищенным аминоацетальдегидом (Org. React. 1951, 6, 191) с получением изохинолинового ядра, из которого после каталитического восстановления получают 1,2,3,4-тетрагидроизохинолины. Другим способом является реакция Бишлера-Напиральского (Org. React. 1951, 6, 74), реакция карбамата 2-фенилэтанаминов с фосфорилхлоридом в кипящем толуоле или ксилоле. Восстановление получающегося циклического бензамида алюмогидридом лития в тетрагидрофуране (J. Med. Chem. 1987, 30(12), 2208-2216) или дибораном в тетрагидрофуране (J. Med. Chem. 1980, 23(5), 506-511) приводит к образованию 1,2,3,4-тетрагидроизохинолина. Вариантом реакции Бишлера-Напиральского является синтез Пиктета-Шпенглера (Org. React. 1951, 6, 151). В этой реакции амиды, карбаматы или сульфонамиды 2-фенилэтанаминов нагревают с параформальдегидом и сильными протонными кислотами (например, трифторуксусной кислотой, серной кислотой) или кислотами Льюиса в растворителе (например, дихлорметане, толуоле, муравьиной кислоте) с получением 1,2,3,4-тетрагидроизохинолина за одну стадию (Tetrahedron 2002, 58(8), 1471-1478).
Схема 2

Реактивы:
a) (CF3CO)2O, Et3N; +4°C. b) (НСНО)n, H2SO4, HOAc; RT. c) NaBH4, EtOH; RT или NH3 (конц), EtOH, нагрев
d)(t-BuOCO)2O, Et3N, DCM, RT.
Предпочтительно 1,2,3,4-тетрагидроизохинолиновое промежуточное соединение (VIIa) синтезируют способом А, показанным на Схеме 2. Этот способ представляет собой реакцию по типу Фриделя-Крафтса, то есть реакцию N-[2-(3-бромфенил)этил]-2,2,2-трифторацетамида с формальдегидом и серной кислотой в уксусной кислоте (Tetrahedron 1996, 37(31), 5453-5456), в результате которой получается смесь 6-бром- и 8-бромизомера в отношении 3 к 1. В результате замены трифторацетамидной группы группой ВОС получают (VIIa). На этой стадии региоизомеры разделить сложно.
1,2,3,4-тетрагидро-2,7-нафтиридиновое промежуточное соединение (VIIb)
В отличие от 1,2,3,4-тетрагидроизохинолинов, в литературе известно лишь несколько примеров методов синтеза 1,2,3,4-тетрагидро-2,7-нафтиридинов. Одним важным способом получения 1,2,3,4-тетрагидро-2,7-нафтиридина является региоселективное каталитическое восстановление 2,7-нафтиридина (Eur. J. Med. Chem. Ther. 1996, 31(11), 875-888). Синтез 2,7-нафтиридина и некоторых его производных описан в литературе. Один из классических способов включает несколько этапов и начинается с кислотно-катализируемой конденсации малононитрила с диэтил-1,3-ацетондикарбоксилатом (J. Chem. Soc. 1960, 3513-3515; см. также J. Heterocycl. Chem. 1970, 7, 419-421). Слегка отличный способ для получения 2,7-нафтиридина включает окисление 4-формил-2,7-нафтиридина с получением 2,7-нафтиридин-4-карбоновую кислоту, с последующим декарбоксилированием (Synthesis 1973, 46-47). Полностью отличный метод основан на внутримолекулярной реакции Дильса-Альдера N-(этоксикарбонил)-N-(бут-3-инил)аминометилпиразина и приводит к получению смеси 1,2,3,4-тетрагидро-2,7-нафтиридина и 5,6,7,8-тетрагидро-1,7-нафтиридина после гидролиза карбаматной группы (WO 02/064574).
Схема 3
Способ В

Реактивы:
a) LiCH3NCH2CH2N(CH3), THF, -70°C, b) n-BuLi в гексанах, -70°С, затем I2, c) TMS-ацетилен, PdCl2(PPh3)2, CuL Et3N THF 60°C d) 7M NH3, EtOH, 80°C. e) H2, PtO2, HOAc. f) 48% HBr (водн.), 120°C. g) (BOC)2O, Et3N, H2O, THF. h) Tf2O, PhMe, 30% K2PO4.
Предпочтительно промежуточное 1,2,3,4-тетрагидро-2,7-нафтиридиновое соединение (VIIb) может быть синтезировано, как указано на Схемах 3 и 4. Согласно способу В, имеющийся в продаже 6-метоксиникотинальдегид обрабатывают последовательно литиевой солью N,N,N'-триметилэтилендиамина, затем н-BuLi в гексане, и в заключение йодом с получением 4-йод-6-метоксиникотинальдегида (Tetrahedron Lett. 1993, 34(39), 6173-6176). Йодсодержащее соединение связывают с триметилсилилацетиленом в обычных условиях Сонагашира-Хагихара (Synthesis 1980, 627-630) и полученный 6-метокси-4-[(триметилсилил)этинил]никотинальдегид конденсируется с гидроксидом аммония в этаноле с образованием 3-метокси-2,7-нафтиридина (Synthesis 1999, 2, 306-311). Региоселективное каталитическое восстановление (Eur. J. Med. Chem. Ther. 1996, 31(11), 875-888) приводит к получению 6-метокси-1,2,3,4-тетрагидро-2,7-нафтиридина. В результате деметилирования, защиты атома N ВОС-ангидридом и заключительной обработки образующегося трет-бутил-6-гидрокси-3,4-дигидро-2,7-нафтиридин-2(1Н)-карбоксилата ангидридом трифторметилсульфоновой кислоты в двухфазной системе получают (VIIb).
Схема 4
Способ С

Реактивы:
а) n-BuLi, THF, -70°С затем DMF, -70°С до RT. b) t-BuNH, DCM, 3Ǻ mol. aievce. c) Li-TMP, -20°C, затем DMP, -20 до -10°C. d) NaBH3CN, MeOH, HOAo:RT. e) 48% HBr (водн.), обр. холод.; смешали с К2СО3 (водн.) f) Tf2O, пиридин +4°С.
Согласно способу С имеющийся в продаже 5-бром-2-метокси-4-метилпиридин в безводном тетрагидрофуране металлируют н-BuLi и затем обрабатывают N,N-диметилформамидом с получением 6-метокси-4-метилникотинальдегида. Его превращали в трет-бутилимин с использованием трет-бутиламина в дихлорметане. Металлизация лития 2,2,6,6-тетраметилпиперидидом (Li-TMP) (J. Org. Chem. Ther. 1993, 58, 2463-2467) и добавление N,N-диметилформамида приводит к получению иминоацетальдегида, который восстанавливают цианоборогидридом натрия в метаноле с получением 2-трет-бутил-6-метокси-1,2,3,4-тетрагидро-2,7-нафтиридина. Отщепление метильной группы кипящей 48%-ной бромистоводородной кислотой и обработка ангидридом трифторметилсульфоновой кислоты в присутствии основания приводит к образованию (VIIb), защищенного в виде трет-бутиламина.
Специалисту в данной области понятно, что в способах по настоящему изобретению некоторые потенциально реакционно-способные функциональные группы, например гидроксильные или аминогруппы, исходных реагентах или промежуточных соединениях могут нуждаться в защите подходящими защитными группами. Таким образом, получение соединений по изобретению может включать, на различных стадиях, добавление или удаление одной или более защитных групп.
Подходящие защитные группы и подробные описания процессов добавления или удаления защитных групп приведены в 'Protective Groups in Organic Chemistry', под редакцией J.W.F. McOmie, Plenum Press (1973) и 'Protective Groups in Organic Synthesis', 3rd edition, T.W.Greene and P.G.M.Wuts, Wiley-Interscience (1999).
Соединения по данному изобретению и промежуточные соединения, кроме того, могут быть выделены из их реакционных смесей и, при необходимости, затем очищены с использованием стандартных методик.
Ниже настоящее изобретение пояснено ссылкой на следующие иллюстративные примеры.
Общие методы
1Н-ЯМР и 13С-ЯМР спектры регистрировали на приборе Varian Inova 400 МГц или Varian Mercury-VX 300 МГц. Центральные пики хлороформа-d (δн 7.27 м.д.), диметилсульфоксида-d6 (δн 2.5 м.д.), ацетонитрила-d3 (δн 1.95 м.д.) или метанола-d4 (δн 3.31 м.д.) использовали в качестве внутренних стандартов. Колоночную хроматографию проводили с использованием силикагеля (0,04-0,063 мм, Merck) при незначительно повышенном давлении (0,2-0,4 бар (0,2-0,4×105 Па)), прилагаемом к колонке. Колонка Kromasil KR-100-5-C18 (250×20 мм, Akzo Nobel) и смеси ацетонитрил/вода с 0,1% TFA (трифторуксусная кислота) при величине потока 10 мл/мин применялись для препаративной ВЭЖХ (высокоэффективной жидкостной хроматографии). Если не указано иное, исходные вещества имелись в продаже. Все растворители и приобретенные реагенты были лабораторного качества и их использовали такими, как они были получены. Органические фазы из экстракций сушили над безводным сульфатом натрия, если не указано иное. Органические фазы или растворы концентрировали посредством упаривания на роторном испарителе. Выходы продукта не оптимизировали.
Для LC-MS (сочетание жидкостной хроматографии и масс-спектрометрии) анализа использовали следующий метод.
Прибор Agilent 1100; колонка Waters Symmetry 2,1×30 мм; масс-спектрометрия APCI (химическая ионизация при атмосферном давлении); скорость потока 0,7 мл/мин; длина волны 254 или 220 нм; растворитель А: вода + 0,1% TFA (трифторуксусная кислота); растворитель Б: ацетонитрил + 0,1% TFA; Градиент 15-95%/Б 2,7 мин, 95% Б 0,3 мин.
Следующие методы использовали для LC-MS анализа (газовая хроматография и масс-спектрометрия).
Прибор Hewlett Packard 5890 Series II; Колонка Agilent HP-5 (30 м × 0,32 мм ID); масс-селективный детектор Hewlett Packard 5971 Series; Давление 55 кПа Не; программа печи 100°С (3 мин), 25°С/мин.
Сокращения:
рид
Пример 1 (5S)-5-Метил-5-({[6-[2-(трифторметил)пиримидин-5-ил]-3,4-дигидроизохинолин-2(1H)-ил]сульфонил}метил)имидазолидин-2,4-дион

[(4S)-4-Метил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорид (0,0295 г, 0,13 ммоль) в сухом TFA (0,6 мл) добавляли по каплям к перемешиваемому раствору 6-[2-(трифторметил)пиримидин-5-ил]-1,2,3,4-тетрагидроизохинолина (0,039 г, 0,14 ммоль), DIPEA (0,034 мл, 0,2 ммоль) и сухой TFA (0,6 мл) при температуре ледяной бани. После окончания добавления раствор перемешивали при комнатной температуре в течение 2 ч и затем переносили в водный рассол и дважды экстрагировали EtOAc. Объединенные органические фазы промывали рассолом, сушили, фильтровали и центрифугировали с получением неочищенного продукта. В результате очистки препаративной ВЭЖХ получали 0,050 г (76%) указанного в заголовке соединения в виде белого твердого вещества.
LC-MS m/z 470 (M+1)
1H-ЯМР (CD3CN) δ 9.19 (s, 2H), 8.51 (br s, 1H), 7.62 (s, 1H), 7.61 (dd, 1H), 7.36 (d, 1H), 6.33 (br s, 1H), 4.51 (s, 2H), 3.57 (t, 2H), 3.52 (d, 1H), 3.42 (d, 1H), 3.04 (t, 2H) и 1.48 (s, 3H) м.д. (миллионные доли)
Исходные вещества получали следующим образом.
6-[2-(Трифторметил)пиримидин-5-ил]-1,2,3,4-тетрагидроизохинолин
трет-Бутил-6-[2-(трифторметил)пиримидин-5-ил]-3,4-дигидроизохинолин-2(1H)-карбоксилат (0,051 г, 0,13 ммоль) перемешивали в TFA (1 мл) и DCM (1 мл) при комнатной температуре в течение ночи, затем дважды концентрировали, второй раз с добавлением толуола (5 мл), с получением трифторацетатной соли.
LC-MS m/z 280 (M+1);
1H-ЯМР (CD3CN) δ 9.25 (s, 2H), 7.73 (m, 2H), 7.44 (d, 1H), 4.45 (s, 2H), 3.56 (t, 2H) и 3.24 (t, 2H) м.д.
Неочищенный продукт переносили в 1 М раствор карбоната натрия (10 мл) и экстрагировали дважды EtOAc. Объединенные органические фазы промывали рассолом, сушили, фильтровали и концентрировали с получением 0,039 г (100%) указанного в заголовке соединения в виде белого твердого вещества.
2-(Трифторметил)пиримидин-5-ила трифторметансульфонат
Ангидрид трифторметилсульфоновой кислоты (13,9 г, 85 ммоль) в сухом DCM (70 мл) медленно добавляли к ледяному раствору 2-(трифторметил)пиримидин-5-ола (13,9 г, 85 ммоль) (US 4558039), DIPEA (16 мл, 93 ммоль) и сухом DCM (260 мл) с такой скоростью, чтобы поддерживать температуру между 4°С и 6°С. После того как добавление было закончено, раствор перемешивали в течение 2,5 ч при 4°С и затем оставляли нагреваться до комнатной температуры. Добавляли воду (50 мл) и 1 М фосфорную кислоту (4,5 мл), и фазы промывали и разделяли. Органическую фазу последовательно промывали водой и насыщенным бикарбонатом натрия, сушили, фильтровали и осторожно концентрировали путем упаривания на роторном испарителе (давление 300-400 мбар (3×104-4×104 Па)). Темно-красное масло очищали при помощи колоночной хроматографии со смесью EtOAc-гептаны (от 1:8 до 1:4) в качестве элюента с получением 22,5 г (90%) указанного в заголовке соединения в виде бесцветного масла, кристаллизующегося на холоде. Альтернативно, продукт может быть очищен перегонкой, точка кипения 75-77°С/1×103 Па).
1H-ЯМР (CDCl3) δ 8.9 (s, 2H) м.д.
трет-Бутил-6-[2-(трифторметил)пиримидин-5-ил]-3,4-дигидроизохинолин-2(1H)-карбоксилат
Смесь (0,1 г, 0,28 ммоль) 1:4 трет-бутил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата и трет-бутил-8-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин2(1Н)-карбоксилата, 2-(трифторметил)пиримидин-5-ил трифторметансульфоната (0,83 г, 0,28 ммоль), PdCl2×dppf (0,0048 г), 2 М карбоната натрия (1,1 мл), толуола (4 мл) и этанола (1 мл) продували сухим аргоном в течение 10 минут, затем нагревали в закрытом сосуде при 81°С в течение 6 ч. Черный раствор фильтровали через стекловату, переносили в водный рассол и дважды промывали EtOAc. Объединенные органические фазы сушили, фильтровали и концентрировали с кремнеземом (5 г). В результате колоночной хроматографии со смесью EtOAc-гептаны (от 1:8 до 1:5) получали 0,051 г (48%) указанного в заголовке соединения в виде белого твердого вещества.
LC-MS m/z 380 (M+1);
1H-ЯМР (CDCl3) δ 9.06 (s, 2H), 7.44 (dd, 1H), 7.38 (br s, 1H), 7.3 (d, 1H), 4.66 (s, 2H), 3.71 (t, 2H), 2.95 (t, 2H) и 1.51 (s, 9H) м.д.
трет-Бутил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Смесь (0,49 г, 1,6 ммоль) 3:1 трет-бутил-6-бром-3,4-дигидроизохинолин-2(1H)-карбоксилата и трет-бутил-8-бром-3,4-дигидроизохинолин-2(1Н)-карбоксилата, бис(пинаколят)диборана (0,45 г, 1,8 ммоль), PdCl2×dppf (0,039 г, 0,048 ммоль), ацетата калия (0,48 г, 4,8 ммоль) и DMF (8 мл) нагревали при 81°С в течение ночи. Растворитель выпаривали, остаток переносили в водный рассол и промывали дважды EtOAc. Органическую фазу сушили, фильтровали и концентрировали. В результате колоночной хроматографии со смесью EtOAc-гептаны (от 1:10 до 1:4) получали 0,24 г смеси 4:1 указанного в заголовке соединения и трет-бутил-8-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-карбоксилата.
1H-ЯМР (CDCl3) δ 7.62 (d, 1H), 7.6 (s, 1H), 7.13 (d, 1H), 4.59 (s, 2H), 3.64 (t, 2H), 2.85 (t, 2H), 1.5 (s, 9H) и 1.35 (s, 12H) м.д. (6-изомер).
1H-ЯМР (CDCl3) δ 7.69 (d, 1H), 7.24-7.14 (m, 2H), 4.88 (s, 2H), 3.64 (t, 2H), 2.85 (t, 2H), 1.5 (s, 9H) и 1.35 (s, 12H) м.д. (8-изомер).
трет-Бутил-6-бром-3,4-дигидроизохинолин-2(1Н)-карбоксилат
6-Бром-2-(трифторацетил)-1,2,3,4-тетрагидроизохинолин получали в две стадии из [2-(3-бромфенил)этил]амина (4 г, 20 ммоль), следуя методике Стоккера (Tetrahedron. Lett. 1996, 37(31), 5453-5456). В результате колоночной хроматографии со смесью EtOAc-гептаны (от 1:10 до 1:6) получали 2,3 г (7,5 ммоль) смеси 3:1 6-бром-2-(трифторацетил)-1,2,3,4-тетрагидроизохинолина и 8-бром-2-(трифторацетил)-1,2,3,4-тетрагидроизохинолина.
1H-ЯМР (CDCl3) δ 7.62 (d, 1H), 7.6 (s, 1H), 7.13 (d, 1H), 4.59 (s, 2H), 3.64 (t, 2H), 2.85 (t, 2H) и 1.5 (s, 9H) и 1.35 (s, 12H) м.д. (6-изомер).
1H-ЯМР (CDCl3) δ 7.69 (d, 1H), 7.24-7.14 (m, 2H), 4.88 (s, 2H), 3.64 (t, 2H), 2.85 (t, 2H) и 1.5 (s, 9H) и 1.35 (s, 12H) м.д. (8-изомер).
Вышеуказанное вещество перемешивали с абсолютным EtOH (100 мл) и 25%-ным гидроксидом аммония (10 мл) при 60°С в течение 4 ч. Затем добавляли еще 15 мл 25%-го гидроксида аммония и перемешивание продолжали при комнатной температуре в течение ночи. Летучие вещества выпаривали до получения неочищенного амина в виде белого твердого вещества.
LC-MS m/z 212,214 (M+1).
Добавляли сухой тетрагидрофуран (50 мл) и DIPEA (1,3 мл, 7,5 ммоль), затем ВОС-ангидрид (1,8 г, 8,2 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Летучие вещества выпаривали и остаток переносили в воду. Значение рН доводили до 2 с помощью 1 М фосфорной кислоты и продукт экстрагировали дважды EtOAc. Объединенные органические фазы промывали рассолом, который немного подщелачивали насыщенным бикарбонатом натрия, сушили, фильтровали и концентрировали. Неочищенный продукт очищали при помощи колоночной хроматографии со смесью EtOAc-гептаны (от 1:50 до 1:20) с получением 2,24 г (96%) смеси 3:1 указанного в заголовке соединения и трет-бутил-8-бром-3,4-дигидроизохинолин-2(1Н)-карбоксилата.
LC-MS m/z 256, 258 (М-56).
1H-ЯМР (CDCl3) δ 7.31 (dd, 1H), 7.3 (br s, 1H), 6.98 (d, 1H), 4.52 (s, 2H), 3.63 (t, 2H), 2.81 (t, 2H) и 1.5 (s, 9H) м.д. (6-изомер).
1H-ЯМР (CDCl3) δ 7.42 (dd, 1H), 7.12-7.01 (m, 2H), 4.55 (s, 2H), 3.64 (t, 2H), 2.84 (t, 2H) и 1.5 (s, 9H) м.д. (8-изомер).
Пример 2 (5S)-5-({[6-(4-Хлорфенил)-3,4-дигидроизохинолин-2(1Н)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион
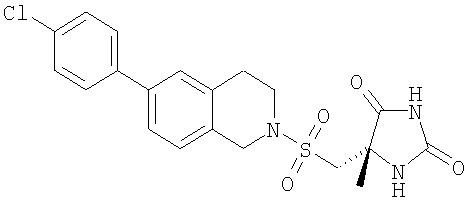
(5S)-5-{[(6-Бром-3,4-дигидроизохинолин-2(1Н)-ил)сульфонил]метил}-5-метилимидазолидин-2,4-дион (0,016 г, 0,04 ммоль), 4-хлорфенилбороновую кислоту (0,0072 г, 0,045 ммоль), PdCl2×dppf (0,003 г), 2 М карбонат натрия (0,15 мл), толуол (0,8 мл) и этанол (0,2 мл) перемешивали в закрытом сосуде при 95°С в течение 17 ч. Растворитель выпаривали и остаток переносили в воду. Раствор подкисляли 10%-ной НОАс до рН 6, а затем дважды экстрагировали EtOAc. Объединенные органические фазы промывали насыщенным рассолом бикарбоната натрия, сушили, фильтровали и концентрировали с получением неочищенного продукта.
LC-MS m/z 434 (M+1).
В результате очистки посредством препаративной ВЭЖХ получали 0,008 г (46%) указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (CD3CN) δ 8.53 (br s, 1H), 7.62 (m, 2H), 7.46 (m, 4H), 7.23 (d, 1H), 6.34 (br s, 1H), 4.45 (s, 2H), 3.53 (m, 2H), 3.49 (d, 1H), 3.39 (d, 1H), 2.99 (m, 2H) и 1.46 (s, 3H) м.д.
Соединения Примеров 3 и 4 получали, используя общий метод Примера 2.
Пример 3 {4-[2-({[(4S)-4-Метил-2,5-диоксоимидазолидин-4-ил]метил}сульфонил)-1,2,3,4-тетрагидроизохинолин-6-ил]фенил}ацетонитрил

Белое твердое вещество.
LC-MS m/z 439 (M+1).
1H-ЯМР (CD3CN) δ 8.61 (br s, 1H), 7.65 (m, 2H), 7.48 (m, 2H), 7.43 (m, 2H), 7.23 (d, 1H), 6.38 (br s, 1H), 4.46 (s, 2H), 3.87 (s, 2H), 3.53 (m, 2H), 3.5 (d, 1H), 3.4 (d, 1H), 3.0 (m, 2H) и 1.46 (s, 3H) м.д.
Пример 4 (5S)-5-Метил-5-{[(6-пиридин-3-ил-3,4-дигидроизохинолин-2(1Н)-ил)сульфонил]метил}имидазолидин-2,4-дион
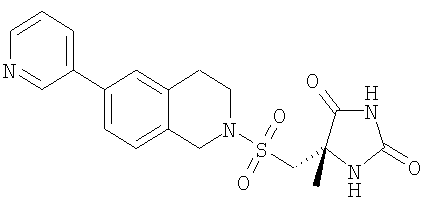
Белый порошок.
LC-MS m/z 401 (M+1).
1H-ЯМР (CD3CN) δ 8.98 (br s, 1H), 8.71 (m, 1H), 8.54 (d, 2H), 7.89 (m, 1H), 7.56 (m, 2H), 7.34 (m, 1H), 6.34 (br s, 1H), 4.49 (s, 2H), 3.55 (m, 2H), 3.52 (d, 1H), 3.41 (d, 1H), 3.03 (m, 2H) и 1.47 (s, 3H) м.д.
Исходное вещество было получено следующим образом.
(5S)-5-({[6-Бром-3,4-дигидроизохинолин-2(1H)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион
Смесь (0,44 г, 1,4 ммоль) 3:1 6-бром-2-(трифторацетил)-1,2,3,4-тетрагидроизохинолина и 8-бром-2-(трифторацетил)-1,2,3,4-тетрагидроизохинолина (полученного согласно Tetrahedron Lett. 1996, 37(31), 5453-5456) перемешивали при комнатной температуре в этаноле (10 мл), содержащем несколько капель 25%-го гидроксида аммония. Через 2,5 ч раствор концентрировали, растворяли в сухом TFA (1 мл) в атмосфере аргона и охлаждали на бане со льдом. Добавляли DIPEA (0,41 мл, 2,4 ммоль), затем раствор [(4S)-4-метил-2,5-диоксиимидазолидин-4-ил]метансульфонилхлорида (0,27 г, 1,2 ммоль) и сухой тетрагидрофуран (1,0 мл). Смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали. Неочищенный продукт переносили в воду и экстрагировали дважды EtOAc. Объединенные органические фазы промывали рассолом, сушили, фильтровали и концентрировали с получением 0,55 г смеси (5S)-5-({[6-бром-3,4-дигидроизохинолин-2(1H)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-диона и (5S)-5-({[8-бром-3,4-дигидроизохинолин-2(1Н)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-диона. Региоизомеры разделяли посредством препаративной ВЭЖХ.
(5S)-5-({[8-Бром-3,4-дигидроизохинолин-2(1H)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион (элюировали первым)
Выход: 0,13 г белого твердого вещества.
LC-MS m/z 402/404 (М+1), 419/421 (М+18);
1H-ЯМР (CD3CN) δ 8.48 (br s, 1H), 7.48 (m, 1H), 7.21 (m, 1H), 7.14 (m, 1H), 6.31 (br s, 1H), 4.36 (s, 2H), 3.48 (m, 4H), 2.95 (m, 2H) и 1.46 (s, 3Н) м.д.
(5S)-5-({[6-Бром-3,4-дигидроизохинолин-2(1Н)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион (элюировали вторым)
Выход: 0,25 г белого твердого вещества.
LC-MS m/z 402/404 (М+1), 419/421 (М+18);
1H-ЯМР (CD3CN) δ 8.47 (br s, 1H), 7.38 (m, 1H), 7.36 (m, 1H), 7.08 (m, 1H), 6.29 (br s, 1H), 4.36 (s, 2H), 3.48 (m, 2H), 3.47 (d, 1H), 3.37 (d, 1H), 2.92 (m, 2H) и 1.45 (s, 3H) м.д.
Пример 5 (5S)-5-({[6-(4-Хлорфенил)-3,4-дигидро-2,7-нафтиридин-2(1Н)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-дион
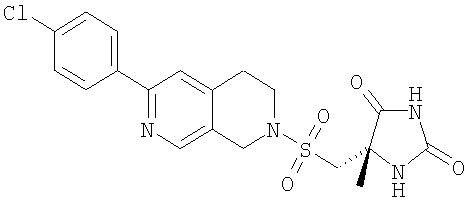
[(4S)-4-Метил-2,5-диоксоимидазолидин-4-ил]метансульфонилхлорид (0,086 г, 0,38 ммоль) в безводном NMP (0,5 мл) добавляли по каплям к перемешиваемому раствору 6-(4-хлорфенил)-1,2,3,4-тетрагидро-2,7-нафтиридина (0,046 г, 0,19 ммоль), DIPEA (0,066 мл, 0,38 ммоль) и безводного NMP (1,5 мл) при комнатной температуре. После завершения добавления раствор перемешивали при комнатной температуре в течение 1,5 ч, затем разбавляли водой (1 мл) и очищали при помощи препаративной ВЭЖХ с получением 0,0070 г (8%) указанного в заголовке соединения в виде белого твердого вещества.
LC-MS m/z 435, 436 (M+1);
1H-ЯМР (DMSO-d6) δ 10.8 (s, 1Н), 8.49 (s, 1Н), 8.1 (d, 2H), 8.06 (s, 1H), 7.84 (s, 1Н), 7.54 (d, 2H), 4.45 (s, 2H), 3.61 (d, 1Н), 3.48 (d, 1Н), 3.47 (t, 2H), 2.98 (t, 2H) и 1.34 (s, 3H) м.д.
Исходные вещества получали следующим образом.
6-(4-Хлорфенил)-1,2,3,4-тетрагидро-2,7-нафтиридин
трет-Бутил-6-{[(трифторметил)сульфонил]окси}-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилат (0,69 г, 1,8 ммоль), 4-хлорфенилбороновая кислота (0,39 г, 2,5 ммоль), PdCl3×dppf (0,05 г), насыщенный карбонат натрия (2 мл), EtOH (4 мл) и толуол (4 мл) перемешивали при 80°С в течение 6 ч. Раствор охлаждали до комнатной температуры, переносили в воду (10 мл) и экстрагировали EtOAc (25 мл). Объединенные органические фазы промывали рассолом, сушили, фильтровали и концентрировали. В результате очистки посредством колоночной хроматографии со смесью EtOAc-гептаны (1:1) в качестве элюента получали 0,065 г (10%) трет-бутил-6-(4-хлорфенил)-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилата.
LC-MS m/z 345 (M+1).
Это вещество растворяли в МеОН (2 мл) и медленно добавляли ацетилхлорид (0,2 мл). После перемешивания при 40°С в течение ночи раствор концентрировали, остаток переносили в 1 М гидроксид натрия (10 мл) и экстрагировали смесью EtOAc-эфир (1:1) (4×30 мл). Объединенные органические фазы сушили, фильтровали и концентрировали с получением 0,046 г (100%) неочищенного соединения, указанного в заголовке.
LC-MS m/z 245 (M+1).
трет-Бутил-6-{[(трифторметил)сульфонил]окси}-3,4-дигидро-2,7-нафтиридин-2(1H)-карбоксилат
Неочищенный 3-метокси-2,7-нафтиридин (полученный из 4,4 ммоль 6-метокси-4-[(триметилсилил)этинил]никотинальдегида) гидрировали (давление 30 фунт/кв. дюйм (206,85 кПа) при комнатной температуре над PtO2 (приблизительно 0,1 г) в НОАс (25 мл) в течение 2,5 ч. Раствор фильтровали через подушку целит и чистый фильтрат концентрировали при помощи лиофилизации с получением неочищенного 6-метокси-1,2,3,4-тетрагидро-2,7-нафтиридина в виде уксуснокислой соли.
LC-MS m/z 165 (M+1).
Это вещество кипятили с обратным холодильником в 48%-ной бромистоводородной кислоте в течение 10 ч. Летучие вещества выпаривали и остаток сушили в вакууме при 45°С с получением приблизительно 0,70 г неочищенного 5,6,7,8-тетрогидро-2,7-нафтиридин-3-ола гидробромида.
LC-MS m/z 151 (M+1).
Это вещество (приблизительно 4,8 ммоль) растворяли в воде (13 мл) и обрабатывали TFA (33 мл), Et3N (0,85 мл, 6 ммоль) и ВОС-ангидридом (1,6 г, 7,3 ммоль) при комнатной температуре. После перемешивания при той же температуре в течение 6 ч раствор концентрировали до одной третьей от его первоначального объема, и остаток переносили в воду и экстрагировали EtOAc три раза. Объединенные органические фазы сушили, фильтровали и концентрировали с получением 0,80 г (выход 67% неочищенного вещества) трет-бутил-6-гидрокси-3,4-дигидро-2,7-нафтиридин-2(1Н)-карбоксилата в виде белого твердого вещества.
LC-MS m/z 251 (M+1), 195 (М-55).
Это вещество (приблизительно 5,4 ммоль) растворяли в двухфазной системе: толуол (20 мл) и 30%-ный водный трехзамещенный ортофосфат калия (20 мл) и обрабатывали ангидридом трифторметилсульфоновой кислоты (1,6 мл, 6,8 ммоль) при 4°С [Org. Lett. 2002, 4(26), 4717-4718]. Баню со льдом удаляли и перемешивание продолжали в течение 2 ч при комнатной температуре, после чего две фазы разделялись. Водную фазу один раз промывали толуолом. Объединенные органические фазы промывали рассолом, сушили и концентрировали. В результате очистки колоночной хроматографией со смесью EtOAc-гептаны (2:1) в качестве элюента получали 0,45 г (выход 17%) указанного в заголовке продукта.
LC-MS m/z 383 (M+1), 283 (М-99).
3-Метокси-2,7-нафтиридин
К перемешиваемому раствору N,N,N'-триметилэтилендиамина (1,9 мл, 15 ммоль) в безводном TFA (65 мл) в атмосфере аргона при -70°С медленно добавляли 1,6 М н-BuLi
в гексанах (9 мл, 14 ммоль). После перемешивания при -70°С в течение 15 мин по каплям добавляли 6-метоксиникотинальдегид (1,3 г, 9,8 ммоль). После того как добавление было закончено, перемешивание продолжали при -70°С в течение еще 15 мин. Затем по каплям добавляли 1,6 М н-BuLi в гексанах (10 мл, 16 ммоль) и перемешивание продолжали при -45°С в течение 4 ч. Раствор охлаждали до -70°С и затем по каплям добавляли раствор йода (3 г, 12 ммоль) и безводный TFA (25 мл). После завершения добавления перемешивание продолжали при -70°С в течение 30 мин и затем при комнатной температуре в течение 3 ч. Неочищенный продукт перемещали в эфир (40 мл) и последовательно промывали насыщенным раствором хлорида аммония (2×40 мл) и 5%-ным тиосульфатом натрия (2×20 мл). Органическую фазу сушили, фильтровали и концентрировали.
В результате очистки колоночной хроматографией со смесью EtOAc-гептаны (1:1) в качестве элюента получали 0,41 г (15%-ный выход) 4-иод-6-метоксиникотинальдегида.
LC-MS m/z 264 (M+1);
1H-ЯМР (CDCl3) δ 9.95 (s, 1H), 8.53 (s, 1H), 7.32 (s, 1H) и 3.98 (s, 3H) м.д.
4-Йод-6-метоксиникотинальдегид (0,41 г, 1,6 ммоль), триметилсилилацетилен (0,35 мл, 2,8 ммоль), PdCl2(PPh3)2 (каталитическое количество), Cul (каталитическое количество), TEA (2 мл) и TFA (10 мл) перемешивали при 60°С в течение 2 ч. Летучие вещества выпаривали, остаток переносили в воду и экстрагировали эфиром. Органическую фазу сушили, фильтровали и концентрировали. В результате очистки колоночной хроматографией со смесью EtOAc-гептаны (1:3) в качестве элюента получали 0,25 г (выход 68%) 6-метокси-4-[(триметилсилил)этинил]никотинальдегида.
LC-MS m/z 234 (M+1);
1H-ЯМР (CDCl3) δ 10.4 (s, 1H), 8.73 (s, 1H), 6.84 (s, 1H), 4.03 (s, 3H) и 0.3 (s, 9H) м.д.
6-Метокси-4-[(триметилсилил)этинил]никотинальдегид (0,25 г, 1,1 ммоль) и 7 М аммиак в метаноле (5 мл) перемешивали в закрытом сосуде при 80°С в течение ночи. Раствор концентрировали, переносили в насыщенный раствор карбоната натрия и экстрагировали эфиром. Органическую фазу сушили, фильтровали и концентрировали с получением 0,25 г указанного в заголовке продукта.
LC-MS m/z 160 (M);
1H-ЯМР (CDCl3) δ 9.41 (s, 1H), 9.27 (s, 1H), 8.47 (d, 1H), 7.64 (d, 1H), 7.03 (s, 1Н) и 4.12 (s, 3Н) м.д.
Фармакологический пример
Отдельные ферментные анализы
ММР12
Рекомбинантный каталитический домен ММР12 человека может быть экспрессирован и очищен, как описано в Parkar A.A. et al., (2000), Protein Expression and Purification, 20, 152. Очищенный фермент можно использовать для проверки ингибиторов активности следующим образом: ММР12 (конечная концентрация 50 нг/мл) инкубируют в течение 60 мин при комнатной температуре с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (10 мкМ) в аналитическом буфере (0,1 М "Tris-HCl" (товарный знак) буфер, рН 7,3, содержащий 0,1 М NaCl, 20 мМ CaCl2, 0,020 мМ ZnCl и 0,05% (мас./об.) детергент "Brij 35" (товарный знак)) в присутствии (10 концентраций) или отсутствии ингибиторов. Активность определяется путем измерения флюоресценции при λех 320 нм (длина волны возбуждения) и λem 405 нм (длина волны эмиссии). Процент ингибирования вычисляют следующим образом.
% Ингибирования равен [Флюоресценцияплюс ингибитор - Флюоресценцияфон] делить на [Флюоресценцияминус ингибитор - Флюоресценцияфон].
ММР8
Очищенную про-ММР8 приобретают у Calbiochem. Фермент (10 мкг/мл) активируют пара-аминофенилртути ацетатом (АРМА) с концентрацией 1 мМ в течение 2,5 ч, 35°С. Активированный фермент можно использовать для проверки ингибиторов активности следующим образом: ММР8 (конечная концентрация 200 нг/мл) инкубируют в течение 90 мин при 35°С (80% H2O) с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (12,5 мкМ) в аналитическом буфере (0,1 М "Tris-HCl" (товарный знак) буфер, рН 7,5, содержащий 0,1 М NaCl, 30 мМ CaCl2, 0,04 мМ ZnCl и 0,05% (мас./об.) детергент "Brij 35" (товарный знак)) в присутствии (10 концентраций) или отсутствии ингибиторов. Активность определяли путем измерения флюоресценции при λех 320 нм и λem 405 нм. Процент ингибирования вычисляли следующим образом.
% Ингибирования равен [Флюоресценцияплюс ингибитор - Флюоресценцияфон] деленное на [Флюоресценцияминус ингибитор - Флюоресценцияфон].
ММР9
Рекомбинантный каталитический домен ММР9 человека может быть экспрессирован и затем очищен при помощи Zn-хелатной колоночной хроматографии с последующей гидроксаматной аффинной колоночной хроматографией. Фермент можно использовать для изучения ингибиторов активности следующим образом: ММР9 (конечная концентрация 5 нг/мл) инкубируют в течение 30 мин при комнатной температуре с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (5 мкМ) в аналитическом буфере (0,1 М "Tris-HCl" (товарный знак) буфер, рН 7,3, содержащий 0,1 М NaCl, 20 мМ CaCl2, 0,02 мМ ZnCl и 0,05% (мас./об.) детергент "Brij 35" (товарный знак)) в присутствии (10 концентраций) или отсутствии ингибиторов. Активность определяют путем измерения флюоресценции при λех 320 нм и λem 405 нм. Процент ингибирования вычисляют следующим образом.
% Ингибирования равен [Флюоресценцияплюс ингибитор - Флюоресценцияфон] делить на [Флюоресценцияминус ингибитор - Флюоресценцияфон].
ММР14
Рекомбинантный каталитический домен ММР14 человека может быть экспрессирован и очищен как описано в Parkar A.A. et al., (2000), Protein Expression and Purification, 20, 152. Очищенный фермент можно использовать для проверки ингибиторов активности следующим образом: ММР14 (конечная концентрация 10 нг/мл) инкубируют в течение 60 мин при комнатной температуре с синтетическим субстратом Mca-Pro-Cha-Gly-Nva-His-Ala-Dpa-NH2 (10 мкМ) в аналитическом буфере (0,1 М "Tris-HCl" (товарный знак) буфер, рН 7,5, содержащий 0,1 М NaCl, 20 мМ CaCl2, 0,02 мМ ZnCl и 0,05% (мас./об.) детергент "Brij 35" (товарный знак)) в присутствии (5 концентраций) или отсутствии ингибиторов. Активность определяют путем измерения флюоресценции при λех 320 нм и λem 405 нм. Процент ингибирования вычисляют следующим образом.
% Ингибирования равен [Флюоресценцияплюс ингибитор - Флюоресценцияфон] делить на [Флюоресценцияминус ингибитор - Флюоресценцияфон].
Протокол тестирования по сравнению с другими матриксными протеиназами, включая ММР9, с использованием экспрессированных и очищенных про-ММР описан, например, в С.Graham Knight et al., (1992) FEBS Lett., 296(3), 263-266.
MMP19
Рекомбинантный каталитический домен MMP19 человека может быть экспрессирован и очищен, как описано в Parkar A.A. et al., (2000), Protein Expression and Purification, 20:152. Очищенный фермент можно использовать для проверки ингибиторов активности следующим образом: MMP19 (конечная концентрация 40 нг/мл) инкубируют в течение 120 мин при 35°С с синтетическим субстратом Mca-Pro-Leu-Ala-Nva- Dpa-Ala-Arg-NH2 (5 мкМ) в аналитическом буфере (0,1 М "Tris-HCl" (товарный знак) буфер, рН 7,3, содержащий 0,1 М NaCl, 20 мМ CaCl2, 0,02 мМ ZnCl и 0,05% (мас./об.) детергент "Brij 35" (товарный знак)) в присутствии (5 концентраций) или отсутствии ингибиторов. Активность определяют путем измерения флюоресценции при λех 320 нм и λem 405 нм. Процент ингибирования вычисляют следующим образом.
% Ингибирования равен [Флюоресценцияплюс ингибитор - Флюоресценцияфон] делить на [Флюоресценцияминус ингибитор - Флюоресценцияфон].
В следующей таблице указаны данные по репрезентативной выборке соединений по настоящему изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2376301C2 |
| НОВЫЕ ГИДАНТОИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОБСТРУКТИВНЫХ ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2005 |
|
RU2386629C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PRMT5 | 2018 |
|
RU2797822C2 |
| ПРОИЗВОДНЫЕ ГИДАНТОИНА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ММР | 2007 |
|
RU2463301C2 |
| ИНГИБИТОРЫ МЕТАЛЛОПРОТЕИНАЗ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2002 |
|
RU2288228C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2838028C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2525116C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ FGFR4, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СОСТАВЫ, СОДЕРЖАЩИЕ ИХ, И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2827699C1 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО[2,7]-НАФТИРИДИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2243218C1 |
Настоящее изобретение относится к новым производным гидантоина формулы (I) или его фармацевтически приемлемая соль, где R1 представляет собой Н, галоген, CF3 или CH2CN; R2 представляет собой C1-3алкил; и каждый из А, А1 и В независимо представляет собой СН или N. Также изобретение относится к способу получения соединения формулы I, к фармацевтической композиции на основе соединения формулы I и его применению в изготовлении лекарственного средства. Технический результат: получены новые производные гидантоина, обладающие активностью ингибитора металлопротеиназ. 3 н. и 7 з.п. ф-лы, 1 табл.

1. Соединение формулы (I) или его фармацевтически приемлемая соль
,
где R1 представляет собой Н, галоген, CF3 или CH2CN;
R2 представляет собой С1-3алкил; и каждый из А, А1 и В независимо представляет собой СН или N.
2. Соединение по п.1, где R1 представляет собой хлоро.
3. Соединение по п.1, где R1 представляет собой CF3.
4. Соединение по п.1, где R2 представляет собой метил или этил.
5. Соединение по п.1, где каждый из А и А1 представляет собой N.
6. Соединение по п.1, выбранное из группы, состоящей из:
(5S)-5-метил-5-({[6-[2-(трифторметил)пиримидин-5-ил]-3,4-дигидроизохинолин-2(1Н)-ил]сульфонил}метил)имидазолидин-2,4-диона;
(5S)-5-({[6-(4-хлорфенил)-3,4-дигидроизохинолин-2(1Н)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-диона;
{4-[2-({[(4S)-4-метил-2,5-диоксоимидазолидин-4-ил]метил}сульфонил)-1,2,3,4-тетрагидроизохинолин-6-ил]фенил}ацетонитрила;
(5S)-5-метил-5-{[(6-пиридин-3-ил-3,4-дигидроизохинолин-2(1Н)-ил)сульфонил]метил}имидазолидин-2,4-диона;
(5S)-5-({[6-(4-хлорфенил)-3,4-дигидро-2,7-нафтиридин-2(1Н)-ил]сульфонил}метил)-5-метилимидазолидин-2,4-диона;
и его фармацевтически приемлемые соли.
7. Способ получения соединения формулы (I), как определено в п.1, или его фармацевтически приемлемой соли, включающий:
взаимодействие соединения формулы (II)
,
где R2 такой, как определено в формуле (I), и L1 представляет собой уходящую группу, с соединением формулы (III) (или его солью)
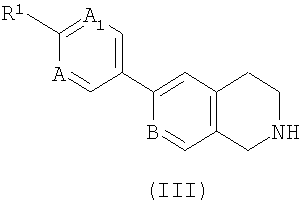 ,
,
где R1, А, А1 и В такие, как определено в формуле (I);
и возможно после этого получение его фармацевтически приемлемой соли.
8. Фармацевтическая композиция, обладающая активностью ингибитора металлопротеиназ, содержащая соединение формулы (I) или его фармацевтически приемлемую соль по любому из пп.1-6 вместе с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
9. Применение соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-6 в изготовлении лекарственного средства для применения в лечении обструктивного заболевания дыхательных путей.
10. Применение по п.9, где обструктивное заболевание дыхательных путей представляет собой астму или хроническое обструктивное заболевание легких.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| ЭЛЕМЕНТ НЕРЕГУЛЯРНОЙ НАСАДКИ ДЛЯ ТЕПЛОМАССООБМЕННЫХ АППАРАТОВ | 1993 |
|
RU2074767C1 |
| СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ НЕРВНЫХ КЛЕТОК, СПОСОБ ПРОФИЛАКТИКИ | 2001 |
|
RU2230060C2 |

