Изобретение относится к газохроматографическому анализу различных химических соединений и может быть использовано в медицине, биологии, экологии и допинговом контроле.
Известны способы определения различных химических соединений методом газовой или жидкостной хроматографии в сочетании с масс-спектрометрией, например [1-3].
Общим недостатком таких технических решений является невысокая степень воспроизводимости при получении хроматографических спектров, что в свою очередь препятствует однозначной идентификации химических соединений и их фрагментов в произвольных комбинациях.
Известен также способ осуществления масс-спектрального анализа многокомпонентной системы, заключающийся в регистрации масс-спектров образца, деконвалюции масс-спектров и ассоциированных с ними хроматограмм одного вещества и распознавании сравнением со справочным спектром вещества, сравнении интенсивностей пиков и времени удерживания распознанных веществ с предварительно определенными параметрами искомого вещества с подтверждением распознавания [4].
Недостатками указанного способа являются высокий порог чувствительности определения, заключающийся в том, что он не позволяет определять исследуемые вещества при низких концентрациях, и вытекающая из этого высокая вероятность получения недостоверных результатов анализа при таком двухстадийном распознавании.
Наиболее близким по своей технической сущности и достигаемому результату к заявляемому техническому решению является способ идентификации неизвестных веществ методом газовой хроматографии в сочетании с масс-спектрометрией. По указанному способу регистрируют хроматограмму как функцию времени удерживания и регистрируют масс-спектр в период времени, соответствующего выходу вещества из колонки, и сравнивают с масс-спектрами известных веществ, находящимися в базе данных, далее определяют индекс удерживания, сравнивают его с таковыми из базы данных и идентифицируют вещество по двум параметрам - масс-спектру и индексу удерживания [5].
Недостатком указанного способа несмотря на привлекательность использования индекса удерживания является высокая вероятность получения недостоверных результатов анализа, поскольку индексы удерживания изначально привязаны к конкретной колонке с определенными параметрами и зачастую либо не воспроизводятся, либо воспроизводятся на другом оборудовании с искажением, что может привести к недостоверной интерпретации результатов анализа.
Техническим результатом, на достижение которого направлено создание данного изобретения, является обеспечение возможности однозначной идентификации химических соединений и их фрагментов в произвольных комбинациях, повышение оперативности и точности определения.
Поставленный технический результат достигается тем, что в ходе анализа готовят две аликвоты пробы исследуемого образца - нативную и дериватизированную, каждую из них пропускают через колонку по меньшей мере в двух режимах кондиционирования параметров изменением градиента температуры и дополнительно каждую из указанных аликвот пропускают через колонку с разделением потока газа-носителя при тех же режимах кондиционирования, далее регистрируют сигналы детектора на хроматограммах, выбирают на них пики со значениями асимметрии на 0,1; 0,5 и 0,6 высоты пика от основания не более 1,05, как наиболее соответствующие биномиальному распределению плотности вероятности и недеформированные влиянием фоновых компонентов, и идентифицируют определяемые вещества по отобранным пикам сопоставлением с эталонными аналитическими характеристиками определяемых веществ.
Следует отметить, что заявляемый способ также может быть осуществлен с использованием аналитической системы ВЭЖХ-МС/МС, поскольку принципиальными положениями заявляемого технического решения являются разделение пробы на различные аликвоты, пропускание через колонку в различных режимах кондиционирования параметров, отбор пиков, наиболее соответствующих биномиальному распределению, и последующая идентификация определяемых веществ сопоставлением отобранных пиков с таковыми у эталонных веществ.
Основным источником недостоверной идентификации является искаженная форма хроматографического пика как результат наличия высоких концентраций фоновых веществ, коэлюирующихся с искомыми компонентами и вызывающих эффект муара - наложение друг на друга и маскировку пиков.
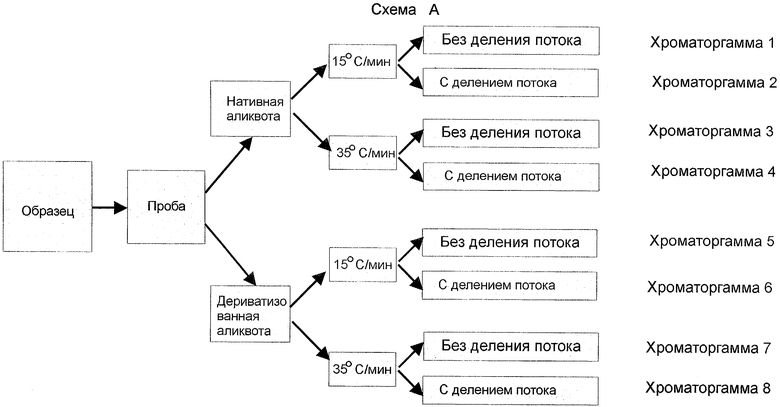
Для устранения фактора влияния фоновых веществ необходимо хроматографические пики «развести» между собой, что должно обеспечить получение неискаженных хроматографических пиков. Для достижения указанной цели анализ выполняют по восьми процедурам: нативные и дериватизированные экстракты анализируют по двум хроматографическим программам: быстрой и плавной, без деления потока и с делением потока.
Следует отметить, что регистрация всех восьми хроматограмм является предельным случаем и на практике не исключены варианты, как это будет показано на конкретных примерах, когда достаточно получение меньшего числа хроматограмм.
В качестве биологической жидкости используют кровь, мочу, слюну, водные экстракты дезинтегрированных органов и тканей и т.п.
В качестве аналитической системы ГХ-МС могут быть использованы например хромато-масс-спектрометр с квадрупольным анализатором Agilent-5973, соединенным с газовым хроматографом модели Agilent-6890.
В качестве вспомогательного оборудования могут быть использованы:
- автоматический шейкер фирмы Glas-Col®, США - для ЖЖЭ;
- центрифуга марки Rotina 46R фирмы Hettich, (ФРГ) для получения контрастной поверхности раздела между органической и водной фазами;
- вакуумный концентратор фирмы Barnstead Inc.(CLUA) для упаривания органического экстракта;
- колонки HP-5MS и VF-5MS для хроматографического разделения.
Разумеется для реализации заявленного изобретения могут быть использованы и другие приборы и устройства с характеристиками, аналогичными вышеуказанным.
В качестве газа-носителя может быть использован гелий, азот или водород.
В качестве внутренних стандартов (ISTD) используют дифениламин или др. подходящее соединение.
Ионизацию осуществляют методом электронного удара в вакууме. Детектирование определяемых веществ проводят в режиме регистрации полного сканирования или по выбранным ионам.
Обработку данных проводят с применением программного обеспечения Chemstation G1701DA.
Изобретение может быть осуществлено следующим образом.
Предварительно, для создания библиотеки (базы данных) готовят растворы веществ-эталонов в органических растворителях. Снимают и регистрируют хроматографические и масс-спектрометрические характеристики веществ-эталонов (детектируют не менее трех характеристических ионов каждого эталонного вещества, определяют время удержания, молекулярную массу, прекурсор-ионы, характеристичные ионы, нижний предел обнаружения).
Готовят раствор внутреннего стандарта, далее проводят пробоподготовку, при которой в образец исследуемой биологической жидкости вводят раствор внутреннего стандарта, доводят pH образца до 9,0 твердым буфером, проводят жидкостно-жидкостную экстракцию смесью органических растворителей различной полярности, органический слой выпаривают досуха в токе азота, перерастворяют в этилацетате и далее разделяют пробу на два аналита, один из которых дериватизируют (обрабатывают пентафторпропионовым ангидридом). Таким образом получают две пробы - нативную и дериватизированную. Пробы далее вводят в систему ГХ-МС. Снимают и регистрируют хроматографические и масс-спектрометрические характеристики обеих проб по четырем процедурам: быстрая программа, плавная программа, без деления потока, с делением потока. Далее анализируют хроматограммы визуально и отбирают на них пики определяемого вещества, соответствующие условиям симметрии по их высоте. Указанные пики далее сравнивают с эталонными и по результатам сравнения идентифицируют анализируемое вещество по наличию и концентрации его в пробе.
Для лучшего понимания изобретение может быть проиллюстрировано, но не исчерпано следующими примерами его конкретного осуществления.
Пример 1.
Определение морфина. Подготовка эталонного образца и анализ пробы.
А. Под колонку HP-5MS.
В качестве внутреннего стандарта используют дифениламин, который добавляют к анализируемым пробам до концентрации 2 мг/дм3. К 1 мл исследуемой эталонной жидкости добавляют внутренний стандарт до концентрации 2 мг/л и экстрагируют 1 мл смеси гексан/диэтиловый эфир 1:1. Экстракт упаривают, добавляют 100 мкл этилацетата и 1 мкл пробы вводят в хроматограф.
Анализ проводят на хромато-масс-спектрометрической системе Agilent 6890/5973N с масс-селективным детектором. Температура узла ввода пробы - 280°C, аналитического интерфейса 240°C. Разделение проводят на кварцевой капиллярной колонке HP-5MS длиной 30 м, внутренним диаметром 0,25 мм, толщина пленки НФ 0,25 мкм. Температурная программа: 700°C (1 мин), 20°C/мин, 280°C. Скорость потока газа-носителя 0,62 мл/мин, средняя линейная скорость газа-носителя 29 см/сек. Объем вводимой пробы 1 мкл. Регистрацию сигнала проводят по полному ионному току (SCAN) в диапазоне масс m/z 29-550 а.е.м. Количественный анализ проводят по выбранным ионам.
Быстрая ГХ программа: 100°C, 1 мин выдержки, (35°C/мин); 300°C (15 мин).
Время удерживания внутреннего стандарта составляло 4,52 мин.
Плавная ГХ программа: 50°C; 0,5 мин выдержки; 99°C/мин; (100°C, 1 мин) (15°C/мин); 280°C (35 мин).
Время удерживания внутреннего стандарта составляло 9,27 мин.
Б. Подготовка пробы и анализ исследуемой мочи.
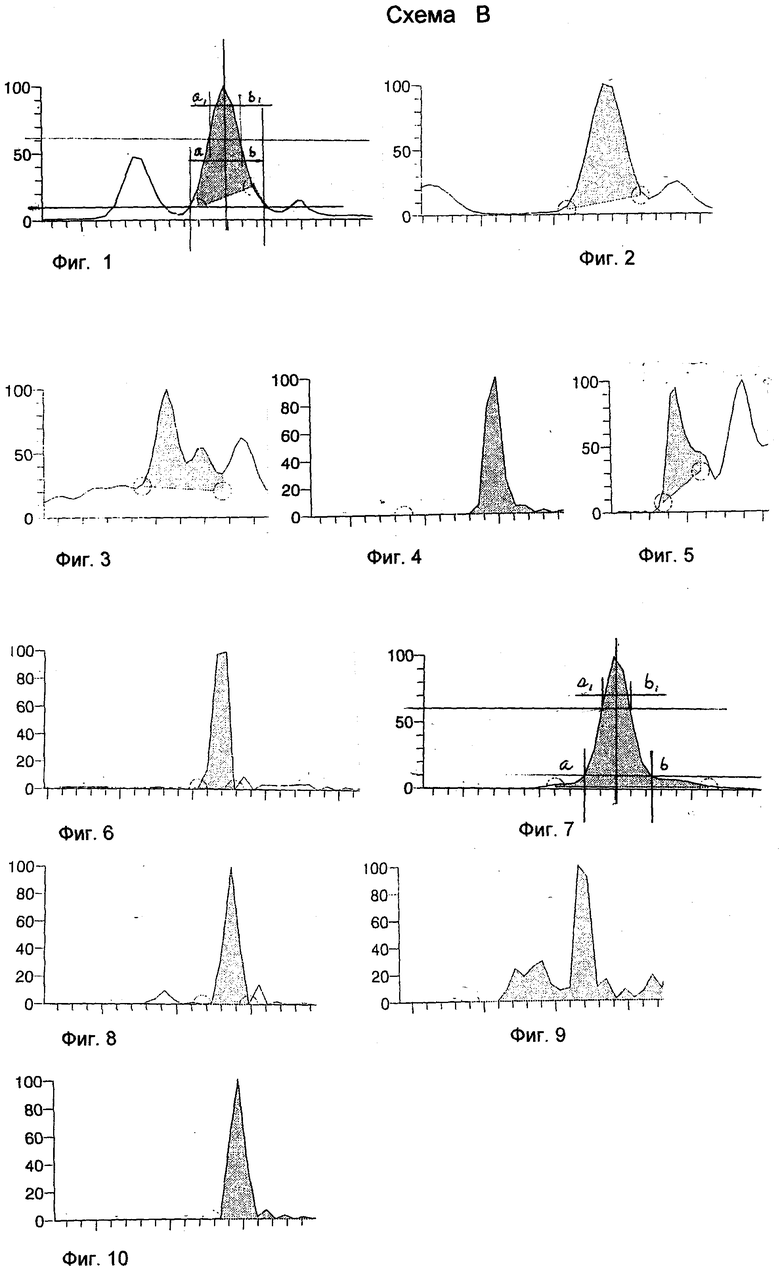
К образцу мочи (5 мл) добавляют 5 мкл раствора внутреннего стандарта, содержащего дифениламин (100 мкг/мл), 0,1 г твердого буфера, 5,0 г сульфата аммония и 5 мл диэтилового эфира, перемешивают в течение 2 минут, далее центрифугируют при 1000 об/мин в течение 5 мин, органический слой отделяют, упаривают досуха в токе азота при 40°C и перерастворяют сухой остаток в 50 мкл этилацетата, разделяют на две аликвоты, одну из которых дериватизируют пентафторпропионовым ангидридом, а вторую оставляют нативной. Снимают и регистрируют хроматографические и масс-спектрометрические характеристики пробы (детектируют не менее трех характеристических ионов каждого эталонного вещества, определяют время удержания, молекулярную массу, прекурсор-ионы, характеристичные ионы, нижний предел обнаружения): отбирают по 5 мкл раствора и вводят в систему ГХ-МС с электрораспылительной ионизацией при атмосферном давлении в режиме регистрации положительных ионов. Анализ ведут как в части «А» и при тех же параметрах процесса за исключением того, что анализ ведут по 8-ми процедурам: быстрая, медленная, с делением и без деления потока. На Схеме A представлен ход проведения анализа. На Схеме B представлены результаты анализа образца биологической жидкости на наличие морфина, где Фиг.1 и 2 - хроматограммы вещества-эталона (морфин); Фиг.3-10 - хроматограммы анализируемых проб по вышеописанным режимам анализа. При анализе хроматограмм 3-10 выбрана таковая на Фиг.7 как наиболее отвечающая условиям симметрии (a=b и a1=b1 на 0,1 и 0,6 высоты пика). Таким образом, анализ пика на Фиг.7 позволяет получить достоверные сведения о наличии искомого вещества, его концентрации и т.п. характеристиках (предел обнаружения 20 нг/мл).
Пример 2.
Определение клофелина.
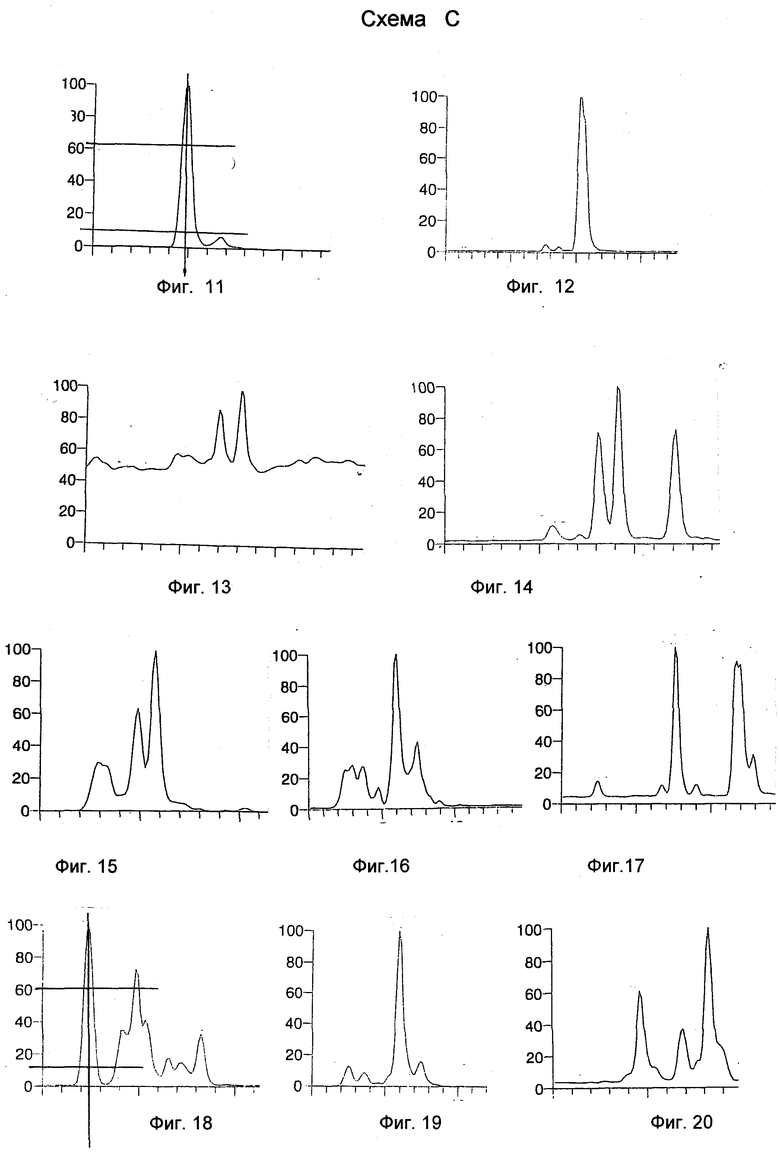
Анализ проводят в режимах, описанных в Примере 1. Результаты анализа представлены на Схеме C. На Схеме C представлены результаты анализа образца биологической жидкости на наличие клофелина, где Фиг.11 и 12 - хроматограммы вещества-эталона (клофелин); Фиг.13-20 - хроматограммы анализируемых проб по вышеописанным режимам анализа. При анализе хроматограмм 13-20 выбрана таковая на Фиг.18 как наиболее отвечающая условиям симметрии (a=b и a1=b1 на 0,1 и 0,6 высоты пика). Таким образом, анализ пика на Фиг.18 позволяет получить достоверные сведения о наличии искомого вещества, его концентрации и т.п. характеристиках (предел обнаружения 50 нг/мл).
Пример 3.
Определение подлинности алкоголя.
Анализ проводят по методике «A» Примера 1. Результаты анализа представлены на Схеме D. На Схеме D представлены результаты анализа образца алкоголя на наличие изопропанола, где Фиг.21 - хроматограмма вещества-эталона; Фиг.22-26 - хроматограммы анализируемых проб по вышеописанным режимам анализа. При анализе хроматограмм 22-25 выбрана таковая на Фиг.24 как наиболее отвечающая условиям симметрии (a=b и a1=b1 на 0,1 и 0,6 высоты пика). Таким образом, анализ пика на Фиг.24 позволяет получить достоверные сведения о наличии искомого вещества, его концентрации и т.п. характеристиках. Следует отметить, что на Фиг.25 (процесс ведут в «плавной» программе с делением потока 1/14) проявились дополнительные признаки фальсификата - наличие бензола и пропанола-1.
Как видно из описания и приведенных примеров осуществления способа, заявляемое техническое решение обеспечивает возможность однозначной идентификации химических соединений и их фрагментов в произвольных комбинациях, повышает оперативность и точность определения.
Источники информации
1. RU 2022265 C1, М.кл. G01N 30/86, публ. 1994 г.
2. RU 2165618 C1, М.кл. G01N 30/46, публ. 2001 г.
3. RU 2321850 C1, М.кл. G01N 30/72, публ. 2008 г.
4. EP 1846757 A2, М.кл. G01N 30/86, публ. 2007 г.
5. WO 2004/104571, М.кл. G01N 30/86, публ. 2004 г. - прототип.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ВЫЯВЛЕНИЯ НЕИЗВЕСТНЫХ ВЕЩЕСТВ В БИОЛОГИЧЕСКИХ ЖИДКОСТЯХ ПАЦИЕНТОВ, ПРИНИМАВШИХ НАРКОТИЧЕСКИЕ ИЛИ ПСИХОАКТИВНЫЕ ВЕЩЕСТВА | 2009 |
|
RU2419788C2 |
| Способ идентификации наркотических и психоактивных веществ в сложных биологических матрицах организма человека | 2019 |
|
RU2705932C1 |
| Способ идентификации этилглюкуронида в сухих пятнах крови | 2020 |
|
RU2740269C1 |
| СПОСОБ ВЫЯВЛЕНИЯ И ОПРЕДЕЛЕНИЯ ПРОИСХОЖДЕНИЯ НЕИЗВЕСТНЫХ ВЕЩЕСТВ В СПИРТНЫХ НАПИТКАХ | 2009 |
|
RU2392616C1 |
| Способ идентификации этилглюкуронида в крови | 2020 |
|
RU2750408C1 |
| Способ идентификации наркотических и психоактивных веществ в биосубстрате человека | 2019 |
|
RU2723907C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ДОПИНГА У ЛОШАДЕЙ | 2011 |
|
RU2489719C2 |
| СПОСОБ РАСПОЗНАВАНИЯ И КЛАССИФИКАЦИИ 3-ОКСОСТЕРОИДОВ И ИХ МЕТАБОЛИТОВ ПРИ ДОПИНГОВОМ КОНТРОЛЕ СПОРТСМЕНОВ | 2010 |
|
RU2452967C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЭНДОГЕННЫХ СТЕРОИДОВ В ПЛАЗМЕ КРОВИ ЧЕЛОВЕКА | 2010 |
|
RU2451292C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КСЕНОБИОТИКОВ В МОЧЕ ЧЕЛОВЕКА ПРИ ДОПИНГОВОМ КОНТРОЛЕ | 2008 |
|
RU2390773C2 |

Изобретение относится к газохроматографическому анализу различных химических соединений и может быть использовано в медицине, биологии, экологии и допинговом контроле. Предложен способ идентификации наркотических и психоактивных веществ в биологических жидкостях, при котором готовят две аликвоты пробы исследуемого образца - нативную и дериватизированную. При этом каждую из двух аликвот пробы пропускают через колонку по меньшей мере в двух режимах кондиционирования параметров изменением градиента температуры. Причем дополнительно каждую из указанных аликвот пропускают через колонку с разделением потока при тех же режимах кондиционирования. Далее регистрируют сигналы детектора на хроматограммах, выбирают на них пики со значениями асимметрии на 0,1; 0,5 и 0,6 высоты пика от основания ≤1,05, как наиболее соответствующие биномиальному распределению плотности вероятности и недеформированные влиянием фоновых компонентов. Затем идентифицируют определяемые вещества по отобранным пикам сопоставлением с эталонными аналитическими характеристиками определяемых веществ. Техническим результатом изобретения является повышение оперативности и точности определения, а также возможность однозначной идентификации химических соединений и их фрагментов в произвольных комбинациях. 26 ил.
Способ идентификации наркотических и психоактивных веществ в биологических жидкостях, при котором готовят пробу исследуемого образца, пропускают ее через хроматографическую колонку с неподвижной жидкой фазой и регистрируют сигналы детектора в виде профиля пиков анализируемых веществ на хроматограмме с последующим определением принадлежности каждого пика анализируемому веществу сравнением с эталонными аналитическими характеристиками вещества, отличающийся тем, что готовят две аликвоты пробы исследуемого образца - нативную и дериватизированную и каждую из них пропускают через колонку по меньшей мере в двух режимах кондиционирования параметров изменением градиента температуры, и дополнительно каждую из указанных аликвот пропускают через колонку с разделением потока при тех же режимах кондиционирования, далее регистрируют сигналы детектора на хроматограммах, выбирают на них пики со значениями асимметрии на 0,1; 0,5 и 0,6 высоты пика от основания ≤1,05, как наиболее соответствующие биномиальному распределению плотности вероятности и недеформированные влиянием фоновых компонентов и идентифицируют определяемые вещества по отобранным пикам сопоставлением с эталонными аналитическими характеристиками определяемых веществ.
| WO 2004104571 A1, 02.12.2004 | |||
| WO 2006082042 A2, 01.02.2006 | |||
| Broad Spectrum Drug Identification Directly from Urine, Using Liquid Chromatography-Tandem Mass Spectrometry (Clinical Chemistry 45, №8, Robert L | |||
| et al, 1224-1233), 1999 | |||
| СПОСОБ МАСС-СПЕКТРОМЕТРИЧЕСКОГО АНАЛИЗА РАЗЛИЧНЫХ ХИМИЧЕСКИХ СОЕДИНЕНИЙ | 2005 |
|
RU2321850C2 |
| Fast gas chromatographfic/mass spectrometric determination of diuretics and masking | |||

