Данное изобретение касается способа получения производных (3-оксо-2,3-дигидро-1H-изоиндол-1-ил)-ацетилгуанидина через производные 3-гидрокси-2,3-дигидро-1H-изоиндол-1-она или производные сложных эфиров 3-(2-карбамоил-фенил)-акриловой кислоты в качестве промежуточных соединений, способа разделения рацемата, а также промежуточных продуктов способа согласно изобретению.
Производные (3-оксо-2,3-дигидро-1H-изоиндол-1-ил)-ацетилгуанидина формулы I

являются ингибиторами NHE1 и описаны в PCT/EP 03/05279. Однако описанные там синтезы приводят к рацемическим смесям региоизомеров, что требует дорогостоящих способов разделения и уменьшает выход желаемых соединений. До сих пор доступ к энантиомерам был возможен только через дорогостоящее хроматографическое разделение на хиральных носителях. Однако пропускная способность веществ при хроматографическом разделении ограничена.
Поэтому представляло большой интерес разработать региоселективный способ получения производных (3-оксо-2,3-дигидро-1H-изоиндол-1-ил)-ацетилгуанидина, а также способ получения энантиомеров. Улучшенное, региоселективное получение рацемических производных (3-оксо-2,3-дигидро-1H-изоиндол-1-ил)-ацетилгуанидина достигается двумя независимыми путями, представленными на схеме 1 и схеме 3. Разделение рацематов достигается путем кристаллизации в виде солей 2,3-O-ацилированных D- или L-винных кислот, как представлено на схеме 5. Посредством щадящей катализируемой основанием рацемизации нежелательных в данном случае энантиомеров возможно значительное превращение рацемата в желаемый энантиомер. Названные способы делают возможным простое получение энантиомерно обогащенных или энантиомерно чистых производных (3-оксо-2,3-дигидро-1H-изоиндол-1-ил)-ацетилгуанидина. С помощью новых способов в настоящее время возможно простое получение больших количеств соединений формулы I в промышленном масштабе.
Таким образом, данное соединение касается способа получения соединений формулы I,

причем
R1 и R2
независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 C-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 C-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 C-атомами,
а также их солей,
отличающегося тем, что, как представлено на схеме 1
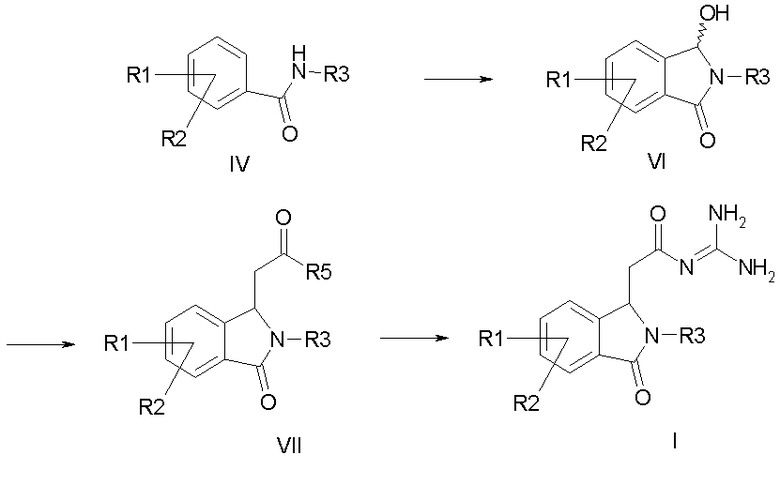
Схема 1
a) формилируют амид формулы IV и затем циклизуют его с получением соединения формулы VI,
b) соединение формулы VI подвергают превращению с алкоксикарбонилметилентрифенилфосфораном, с 1-алкокси-1-триметилсилоксиэтиленом или с триалкилфосфоноацетатом с получением соединения формулы VII, и
c) соединение формулы VII подвергают превращению с гуанидином с получением соединения формулы I,
причем в соединениях формул IV, VI и VII
R1-R3 имеют значения как в формуле I и
R5 обозначает алкокси с 1, 2, 3 или 4 C-атомами;
а также его солей.
Объектом изобретения является также способ получения соединений формулы I,
причем
R1 и R2
независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 C-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 C-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 C-атомами;
а также их солей,
отличающийся тем, что, как представлено на схеме 2

Схема 2
a) соединение формулы II подвергают превращению с амином формулы III с получением амида формулы IV,
b) амид формулы IV формилируют в орто-положение к амидной функции с получением формил-амида формулы V,
c) формил-амид формулы V циклизуют с получением соединения формулы VI,
d) соединение формулы VI подвергают превращению с алкоксикарбонилметилентрифенилфосфораном, с 1-алкокси-1-триметилсилоксиэтиленом или с триалкилфосфоноацетатом с получением соединения формулы VII и
e) соединение формулы VII подвергают превращению с гуанидином с получением соединения формулы I,
причем в соединениях формул II, III, IV, V, VI и VII
R1-R3 имеют значения как в формуле I,
R5 обозначает алкокси с 1, 2, 3 или 4 C-атомами и
X обозначает Cl, Br, OH или алкокси с 1, 2, 3 или 4 C-атомами;
а также его солей.
Соединение формулы II обычным образом подвергают превращению в инертном растворителе, например простом эфире, углеводороде или галогенированном углеводороде, например дихлорметане, при температуре между -30°C и температурой кипения растворителя, предпочтительно при комнатной температуре (КТ), с амином формулы III, в случае необходимости в присутствии активатора, с получением амида формулы IV.
Орто-формилирование можно проводить, например, тем, что используют соединение алкилметалла, например соединение алкиллития, предпочтительно трет.-BuLi, с комплексными лигандами, предпочтительно TMEDA, в инертном растворителе как простой эфир или углеводород, например ТГФ, при температуре между -100°C и 0°C, предпочтительно между -80°C и -50°C. Затем добавляют амид формулы IV и депротонируют в течение интервала времени между 10 минутами и 10 часами, предпочтительно между 10 минутами и 60 минутами, при температуре между -100°C и 0°C, предпочтительно между -80°C и -50°C. Затем добавляют формилирующий реагент, предпочтительно ДМФ, и при температуре между -100°C и 40°C, предпочтительно между -80°C и комнатной температурой, осуществляют реакцию с анионом. Предпочтительно раствор после добавления ДМФ оставляют при КТ в течение периода от 10 минут до 3 часов, например, в течение 30 минут. При этом образующийся промежуточный амид формулы V в целом циклизуется непосредственно с получением изоиндолона формулы VI.
Изоиндолон формулы VI подвергают превращению с (C1-C4)-алкоксикарбонилметилен-трифенилфосфораном в инертном растворителе, таком как простой эфир, углеводород или галогенированный углеводород, например толуол, при температуре между 0°C и температурой кипения растворителя, предпочтительно между 20°C и температурой кипения растворителя, или с три-(C1-C4)-алкилфосфонацетатом в присутствии основания, например гидрида натрия, в инертном растворителе, как простой эфир, углеводород или галогенированный углеводород, например 1,2-диметоксиэтан, при температуре между 0°C и температурой кипения растворителя, предпочтительно между 20°C и температурой кипения растворителя. Альтернативно изоиндолон формулы VI подвергают превращению с 1-(C1-C4)-алкокси-1-триметилсилоксиэтиленом в присутствии кислоты Льюиса, например хлорида титана(IV) или триметилсилилтрифлата, в инертном растворителе, как простой эфир, углеводород или галогенированный углеводород, например дихлорметан, при температуре между -80°C и температурой кипения растворителя, предпочтительно при температуре между -80°C и 20°C (Synth. Commun. 1987, 17, 1).
Сложный эфир формулы VII в целом известными способами может быть превращен с помощью гуанидина до ацилгуанидина формулы I. Превращение осуществляют предпочтительно известными специалистам способами в протонном или апротонном, полярном, но инертном органическом растворителе. При этом при превращении, например, сложного метилового эфира (формула VII; R5=OCH3) с гуанидином пригодны растворители, как метанол, изопропанол или ТГФ при температуре от 20°C вплоть до температуры кипения этого растворителя. При большинстве превращений соединений формулы VII с бессолевым гуанидином используют, например апротонные инертные растворители, например простые эфиры, как ТГФ, диметоксиэтан или диоксан. Но также при необходимости основания, например NaOH, в качестве растворителя при превращении соединений формулы VII с гуанидином может быть использована вода. При превращении соединений формулы VII с солями гуанидина, как, например гидрохлоридом гуанидина, обычно осуществляют превращение в присутствии основания, например калий-трет.-бутоксида, метилата натрия или этилата натрия в инертном растворителе, как диметилформамид, НМП (NMP), 2-пропанол при температуре между 20°C и температурой кипения растворителя.
Помимо эфиров карбоновых кислот формулы VII для превращения с гуанидином могут использоваться также другие активированные производные кислот, например хлорангидриды, тиоэфиры или ангидриды карбоновых кислот. Активирование карбоновых кислот можно осуществлять также, например, с помощью ДЦГКДИ (DCC). Активированные производные кислот могут получаться известным специалисту способом непосредственно из лежащих в основе сложных эфиров карбоновых кислот формулы VII или из соответствующих карбоновых кислот, которые могут быть получены из сложных эфиров посредством типичных методов гидролиза. Ряд пригодных способов получения активированных производных карбоновых кислот имеются в сведениях из литературных источников: J. March, Advanced Organic Chemistry, Third Edition (John Wiley & Sons, 1985, S.350).
Стадии способа, приведенные на схемах 1 и 2, независимо друг от друга могут осуществляться непрерывно или периодически. Обновление реакционной смеси может происходить после каждой стадии способа. Обновление и, при желании, очистка продукта происходит общепринятыми способами, такими как экстракция, pH-разделение, хроматография или кристаллизация и обычная сушка.
Исходные соединения формул II и III являются коммерчески доступными или могут быть получены известными специалисту способами, описанными в литературе или аналогичными им.
Объектом изобретения также является способ получения соединений формулы I
причем
R1 и R2
независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 C-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 C-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 C-атомами,
а также их солей,
отличающийся тем, что, как представлено на схеме 3

Схема 3
a) амин формулы IX подвергают превращению через соль диазония с алкиловым эфиром акриловой кислоты с получением производного коричной кислоты формулы XI,
b) соединение формулы XI подвергают превращению с амином формулы III и гуанидином с получением ацилгуанидина формулы I,
причем в соединениях формул III, IX и XI
R1-R3 имеют значения как в формуле I, и
R6 обозначает алкокси с 1, 2, 3 или 4 C-атомами;
а также его солей.
Данное соединение касается также способа получения соединений формулы I,
причем
R1 и R2
независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 C-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 C-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 C-атомами,
а также их солей,
отличающегося тем, что, как представлено на схеме 4

Схема 4
a) нитросоединение формулы VIII подвергают превращению с получением амина формулы IX,
b) амин формулы IX подвергают превращению с получением соли диазония формулы X,
c) соль диазония формулы X подвергают превращению с алкиловым эфиром акриловой кислоты с получением производного коричной кислоты формулы XI,
d) соединение формулы XI подвергают превращению с получением амида формулы XII и
e) соединение формулы XII подвергают превращению с получением ацилгуанидина формулы I, или посредством превращения соединения формулы XII в присутствии основания с получением производного изоиндолона формулы XIII и затем посредством превращения с гуанидином при активировании с получением ацилгуанидина формулы I (вариант A), или
после образования производного изоиндолона формулы XIII в присутствии основания из соединения формулы XII посредством превращения соединения формулы XIII в сложный эфир формулы XIV и затем посредством превращения с гуанидином с получением ацилгуанидина формулы I (вариант B), или
посредством превращения соединения формулы XII в присутствии сильного основания с получением сложного эфира формулы XIV и затем посредством превращения с гуанидином с получением ацилгуанидина формулы I (вариант C), или
посредством прямого превращения соединения формулы XII с гуанидином в присутствии основания при одновременно следующем гуанилировании и циклизации с получением изоиндолона формулы I (вариант D),
причем в соединениях формул VIII, IX, X, XI, XII, XIII и XIV
R1-R3 имеют значения как в формуле I и
R6 и R7 независимо друг от друга обозначают алкокси с 1, 2, 3 или 4 C-атомами,
а также касается их солей.
Нитросоединение формулы VIII известными способами (как, например, описано в публикации „Houben-Weyl, Methoden der organischen Chemie“, Band XI/1, Stickstoffverbindungen II, Georg Thieme Verlag Stuttgart, 1957, S.360ff) может быть восстановлено до анилина формулы IX. Предпочтительным является каталитическое гидрирование, например с Pd/C, например с 5% Pd/C или 10% Pd/C, в растворителе, как, например, спирт, предпочтительно этанол, в атмосфере водорода при давлении от 1 бар до 200 бар, предпочтительно при давлении 1 бар - 10 бар.
Последующее диазотирование анилина формулы IX происходит в инертном растворителе, предпочтительно этаноле, в присутствии кислоты, анион которой не сам замещает ион диазония, как, например, HBF4 или HPF6, предпочтительно HBF4, или, например, H2SO4 и в присутствии нитрита, предпочтительно NaNO2, при температуре между -30°C и температурой кипения растворителя, предпочтительно между 0°C до 30°C.
Соль диазония формулы X предпочтительно непосредственно подвергают превращению с (C1-C4)-алкиловым эфиром акриловой кислоты, предпочтительно этиловым эфиром акриловой кислоты, в присутствии палладиевого катализатора, предпочтительно Pd(OAc)2, при температуре между 0°C и температурой кипения растворителя, предпочтительно между 45°C до 55°C, до производного коричной кислоты формулы XI.
Остаток бензойной кислоты соединения формулы XI известными специалисту способами может быть переведен в амид формулы XII, предпочтительно через хлорангидрид или с помощью ДЦГКДИ (DCC). Эта реакция также может быть проведена таким образом, что амид формулы XII в реакционной смеси непосредственно циклизуют до сложного эфира формулы XIV, так что реакцию от соединения формулы XI до сложного эфира формулы XIV проводят в одну стадию. Это может происходить или при основных условиях реакции амидообразования, или циклизации может способствовать добавка основания, например, триэтиламина, основания Хюнига (Hünig-Base) или трет.-бутилат калия. Другая альтернатива состоит в том, что соединение формулы XI непосредственно превращается в соединение формулы I, тем что последовательно друг за другом проводят амидообразование, циклизацию и гуанидирование в том же реакционном сосуде, причем реакция может осуществляться без выделения промежуточных соединений.
Для следующего превращения соединения формулы XII до ацилгуанидина формулы I имеется 4 варианта:
Вариант A: Превращение амида формулы XII происходит предпочтительно с водным раствором щелочи, предпочтительно водным раствором NaOH, в растворителе как спирт, предпочтительно метанол или этанол, при температуре между -30°C и температурой кипения растворителя, предпочтительно при КТ. Имеет место как омыление сложноэфирной группы, так и циклизация с образованием производного изоиндолона формулы XIII. Соединение формулы XIII активируют общеизвестным способом (и как изображено на схеме 1) до ацилирования, например через хлорангидрид или с помощью ДЦГКДИ (DCC), и получают ацилгуанидин формулы I.
Вариант B: Синтез карбоновой кислоты формулы XIII происходит как в альтернативном варианте A. Затем стандартным способом для получения сложного эфира, предпочтительно с SOCl2 в спирте, таком как метанол или этанол, получают, например, сложный метиловый или этиловый эфир формулы XIV. Последующее превращение сложного эфира формулы XIV происходит как показано на схеме 1 с образованием ацилгуанидина формулы I.
Вариант C: Превращение амида формулы XII происходит в растворе сильного основания, предпочтительно метилата или трет.-бутилата в спирте, таком как метанол или этанол, и получают сложный метиловый или этиловый эфир формулы XIV. Превращение сложного эфира формулы XIV до ацилгуанидина формулы I происходит как показано на схеме 1.
Вариант D: Амид формулы XII подвергают превращению при обычных условиях до ацилирования гуанидина. В качестве растворителя используют инертный растворитель, такой как простой эфир, углеводород или галогенированный углеводород, предпочтительно ДМФ. Обычно сначала подвергают превращению соль гуанидиния с сильным основанием, предпочтительно KOtBu, причем высвобождается свободный гуанидин. Смесь добавляют к раствору соединения формулы XII в растворителе, таком как спирт, простой эфир, углеводород или галогенированный углеводород, например ДМФ, НМП (NMP) или 2-пропанол. При этом происходит одновременно гуанилирование и циклизация до изоиндолона формулы I. Вариант состоит в том, что последовательно друг за другом с каталитическим количеством сильного основания, например трет.-бутилата калия или метилата натрия, или этилата натрия в растворителе, например ДМФ, НМП (NMP) или 2-пропаноле, соединение формулы XI циклизуют до соединения формулы XIV и затем in situ подвергают превращению до соединения формулы I.
Предпочтительным является вариант D, при котором проводят превращение производного бензойной кислоты формулы XI темплатным способом (несколько стадий в одном сосуде) вплоть до ацилгуанидина формулы I.
Стадии способа, приведенные на схеме 2, могут осуществляться непрерывно или периодически. Обновление реакционной смеси может происходить после каждой стадии способа. Обновление и, при желании, очистка продукта происходит общепринятыми способами, такими как экстракция, pH-разделение, хроматография или кристаллизация и обычная сушка.
Исходные соединения формул III и VIII являются коммерчески доступными или могут быть получены известными специалисту способами, описанными в литературе или аналогичными им.
Объектом изобретения также являются соединения формулы XII
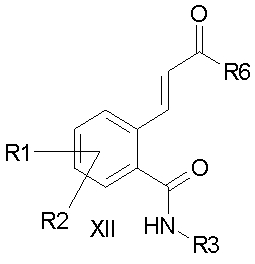
причем
R1 и R2
независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 C-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 C-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 C-атомами,
R6 обозначает алкокси с 1, 2, 3 или 4 C-атомами,
а также их соли.
Заявлено также применение соединений формулы XII в качестве промежуточных продуктов синтеза.
Соединения формулы I в энантиомерно обогащенной или энантиомерно чистой форме предпочтительно могут быть получены новым способом разделения рацемата, который также является объектом данного изобретения. Для этого рацематы соединений формулы I в виде солей 2,3-O-ацилированной D- или L-винной кислоты используют для кристаллизации, причем энантиомеры обогащаются в кристаллизате или в маточнике. Затем опять высвобождают свободные основания из солей.
Таким образом, данное изобретение касается способа выделения соединений формулы Ia и Ib

причем
R1 и R2
независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 C-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 C-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 C-атомами,
а также их солей,
отличающегося тем, что, как представлено на схеме 5

Схема 5
a) соединение формулы I подвергают превращению с получением солей 2,3-O-ацилированной D- или L-винной кислоты и посредством кристаллизации обе соли формул XVa и XVb получают раздельно,
b) свободные основания формул Ia или Ib высвобождают из обеих солей формул XVa или XVb,
причем в соединениях формул I, XVa и XVb
R1-R3 имеют значения как в формулах Ia и Ib и
R* обозначает
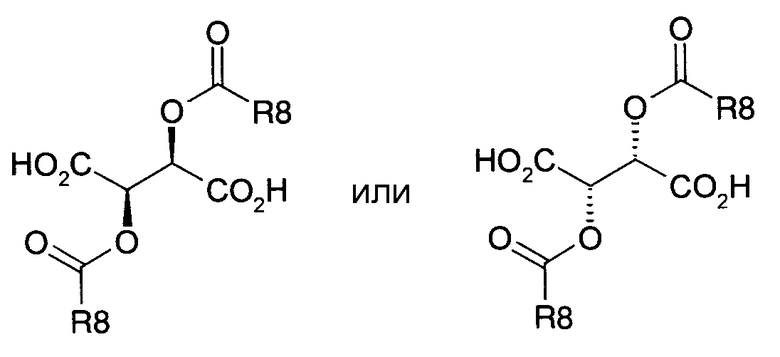
R8 обозначает алкил с 1, 2, 3, 4, 5 или 6 C-атомами или фенил, незамещенный или замещенный 1, 2 или 3 заместителями из группы F, Cl, Br, I, алкил с 1, 2, 3 или 4 C-атомами или алкокси с 1, 2, 3 или 4 C-атомами.
Изобретение также распространяется на вышеописанный способ, при котором нежелательные энантиомеры формулы Ia или Ib вновь рацемизируют.
Рацемат соединения формулы I с производным винной кислоты R*, например, как O,O'-дибензоил-D-винная кислота, O,O'-дибензоил-L-винная кислота, O,O'-ди(4-метилбензоил)-L-винная кислота, O,O'-ди(4-метилбензоил)-D-винная кислота, O,O'-ди(4-метоксибензоил)-L-винная кислота или O,O'-ди(4-метоксибензоил)-D-винная кислота, предпочтительно с O,O'-дибензоил-L-винной кислотой или O,O'-дибензоил-D-винной кислотой, подвергают кристаллизации в пригодном растворителе, например в простом эфире, например диэтиловом эфире, диизопропиловом эфире, диметоксиэтане, тетрагидрофуране или диоксане, в галогенированном углеводороде, например дихлорметане, трихлорметане, тетрахлорметане, 1,2-дихлорэтане или трихлорэтилене, в спирте, например метаноле, этаноле, н-пропаноле, 2-пропаноле, бутаноле, в сложном эфире, например этилацетате или бутилацетате, в воде, или в смеси растворителей, предпочтительно в 2-пропаноле, диметоксиэтане или этилацетате, при температуре между -10°C и температурой кипения растворителя, предпочтительно при 0°C до 40°C. Вариант способа состоит в том, что смеси из двух или нескольких 2,3-O-ацилированных D- или L-винных кислот этих конфигураций, имеющие различные ацильные группы, могут использоваться для разделения.
Образование солей из соединений формулы I и производного винной кислоты R* может происходить при использовании эквивалентных количеств, т.е. могут быть использованы 0,5 моль производного винной кислоты R*, имеющего две карбоксильные группы, на моль соединения формулы I. Соединение формулы I также может подвергаться кристаллизации с менее чем 0,5 мольных эквивалентов 2,3-O-ацилированной D- или L-винной кислоты, например с 0,25 моль до 0,5 моль производного винной кислоты R* на моль соединения формулы I, в особенности с 0,25 моль до 0,3 моль производного винной кислоты R* на моль соединения формулы I. Желаемый энантиомер затем выкристаллизовывается в форме соли формулы XVa или XVb, а нежелательный энантиомер находится в маточнике большей частью в форме энантиомера формулы Ib или Ia и не содержится в форме соли формул XVa или XVb. Чистота энантиомеров солей формул XVa и XVb может быть увеличена посредством повторной кристаллизации или путем перемешивания первичных кристаллов со свежим растворителем при повышенной температуре и последующем охлаждении.
После разделения обеих солей формул XVa и XVb или отделения соли формулы XVa или XVb от нежелательных энантиомеров Ib или Ia, энантиомерно обогащенные соединения формул Ia и Ib затем обычно высвобождают из солей путем добавления вспомогательного основания, например амина, как, например, триэтиламин, неорганического основания, как NaHCO3, Na2CO3 или их водных растворов. При этом обычно используют пригодный растворитель, как, например простой эфир, например, диэтиловый эфир, диизопропиловый эфир, диметоксиэтан, тетрагидрофуран или диоксан, галогенированный углеводород, например, дихлорметан, трихлорметан, тетрахлорметан, 1,2-дихлорэтан или трихлорэтилен, спирт, например метанол, этанол, н-пропанол, 2-пропанол или бутанол, сложный эфир, например этилацетат или бутилацетат, или вода, или смесь растворителей, предпочтительно этилацетат, 2-пропанол, дихлорметан или вода или их смеси, причем реакционная смесь может иметь одну или несколько фаз, при температуре между -10°C и температурой кипения растворителя, предпочтительно при 10°C до 40°C. Это может происходить, например, таким образом, что соль растворяют в водном растворе NaHCO3 и затем с органическим растворителем, например этилацетатом, экстрагируют энантиомер формулы Ia или Ib.
В каждом случае, нежелательный энантиомер Ia или Ib опять может быть переведен в рацемат формулы I посредством способа рацемизации, и таким образом использован для новой стадии разделения рацемата. При этом нежелательные энантиомеры предпочтительно в растворителе, как спирт, например 2-пропанол, при температуре между -10°C и температурой кипения растворителя, предпочтительно при 0°C до 40°C, обрабатывают малым количеством основания, например KOH, нейтрализуют реакционную смесь и после водно-экстракционной обработки отделяют рацемат. Этот способ, путем пригодного выбора количества основания и температуры реакции, может быть проведен так, что практически происходит исключительно образование рацемата, и не наблюдается никаких химических изменений вещества.
Объектом данного изобретения далее являются соединения формул XVa и XVb

причем
R1 и R2
независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 C-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 C-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 C-атомами,
R* обозначает

R8 обозначает алкил с 1, 2, 3, 4, 5 или 6 C-атомами или фенил, незамещенный или замещенный 1, 2 или 3 заместителями из группы F, Cl, Br, I, алкил с 1, 2, 3 или 4 C-атомами или алкокси с 1, 2, 3 или 4 C-атомами.
Если вышеописанные соединения, например соединения формул I, Ia, Ib, VII, XIII, XIV, XVa или XVb, содержат один или несколько центров асимметрии, то независимо друг от друга они могут иметь как S-, так и R-конфигурацию, если не оговорено другое. Соединения могут существовать в виде оптических изомеров, в виде диастереомеров, в виде рацемата или в виде смеси таковых, если это точно не обозначено. У соединений с двойной связью может быть как E- так и Z-конфигурация, если не оговорено другое. Данное изобретение включает таутомерные формы вышеописанных соединений, например соединений формул I, Ia, Ib, XVa и XVb.
Алкильные остатки могут быть неразветвленными или разветвленными. Это также имеет место, если они имеют заместители или выступают в качестве заместителей других остатков, например во фторалкильных остатках или алкоксильных остатках. Примерами алкильных остатков являются метил, этил, н-пропил, изопропил (=1-метилэтил), н-бутил, изобутил (=2-метилпропил), втор.-бутил (=1-метилпропил), трет.-бутил (=1,1-диметилэтил), н-пентил, изопентил, трет.-пентил, неопентил и гексил. Предпочтительными алкильными остатками являются метил, этил, н-пропил и изопропил, особенно предпочтительны метил или этил. В алкильных остатках один или несколько, например, 1, 2, 3, 4 или 5 атомов водорода могут быть замещены атомами фтора. Примерами таких фторалкильных остатков являются трифторметил, 2,2,2-трифторэтил и пентафторэтил, предпочтительно трифторметил или 2,2,2-трифторэтил. Замещенные алкильные остатки могут быть замещены в любых положениях.
Примерами циклоалкильных остатков являются циклопропил, циклобутил, циклопентил, циклогексил или циклогептил.
Фенильные остатки могут быть незамещенными или однократно, или многократно, например однократно, двукратно или трехкратно замещенными одинаковыми или разными остатками. Если фенильный остаток замещен, предпочтительно он имеет один или два одинаковых или разных заместителя. В монозамещенных фенильных остатках заместитель может находиться в 2-положении, 3-положении или 4-положении. Двукратно замещенный фенил может быть замещен в 2,3-положении, 2,4-положении, 2,5-положении, 2,6-положении, 3,4-положении или 3,5-положении. В трехкратно замещенных фенильных остатках заместители могут находиться в 2,3,4-положении, 2,3,5-положении, 2,4,5-положении, 2,4,6-положении, 2,3,6-положении или 3,4,5-положении.
Вышеописанные соединения, например соединения формул I, Ia и Ib, могут использоваться в способе согласно изобретению в форме их солей и/или в форме солей выделяться. Соли могут быть получены обычными методами, например путем превращения с кислотами или основаниями в растворителе, или посредством анионного или катионного обмена из других солей. В качестве солей кислотного присоединения, например, соединений формул I, Ia и Ib при этом имеют в виду, например, галогениды, в особенности гидрохлориды, гидробромиды, лактаты, сульфаты, цитраты, тартраты, ацетаты, фосфаты, метилсульфонаты, бензолсульфонаты, п-толуолсульфонаты, адипинаты, фумараты, глюконаты, глутаматы, глицеринфосфаты, малеинаты, бензоаты, оксалаты и памоаты, а также трифторацетаты. В случае получения биологически активных веществ предпочтительны физиологически и фармацевтически совместимые соли. В качестве примера можно было бы назвать соли соединений формул I, Ia и Ib с фумаровой кислотой, в особенности соли, содержащие один моль фумаровой кислоты на моль соединения формулы I, Ia или Ib, которые, таким образом, являются гидрогенфумаратами или полуфумаратами. Благодаря полезным свойствам, таким как кристалличность, стабильность, особенно малая гигроскопичность, малая склонность к рацемизации и хорошая растворимость, в особенности отличается, например, (S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин гидрогенфумарат гидрат формулы XVI, который также во всех своих таутомерных формах является объектом данного изобретения.

Если соединения содержат кислотную группу, они могут образовывать соли с основаниями, например соли щелочных металлов, предпочтительно натриевые или калиевые соли или соли аммония, например соли с аммиаком или органическими аминами или аминокислотами. Соединения, содержащие основную группу и кислотную группу, также могут существовать в виде амфотерного иона.
Одна форма выполнения данного изобретения касается соединений, в которых R1 и R2 оба не являются водородом, в особенности соединений, в которых R1 обозначает водород и R2 обозначает фтор, хлор или трифторметил, в особенности трифторметил. В соединениях, в которых R1 обозначает водород, заместитель R2 предпочтительно находится в параположении бензольного кольца относительно C=O-группы в системе изоиндолона.
Группа Alk предпочтительно обозначает алкил с 1, 2 или 3 C-атомами, в особенности с 1 или 2 C-атомами, особенно с 1 C-атомом. R4 предпочтительно обозначает трифторметил или циклоалкил с 3, 5 или 6 C-атомами, в особенности 3 C-атомами, особенно предпочтительно трифторметил. Одна форма выполнения данного изобретения касается соединений, в которых R3 обозначает трифторметил или 2,2,2-трифторэтил, в особенности 2,2,2-трифторэтил.
Специальная форма выполнения данного изобретения касается получения N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидина и его энантиомерных форм и их солей.
X предпочтительно обозначает хлор или метокси, в особенности хлор. R5 предпочтительно обозначает метокси или этокси, в особенности этокси. R6 предпочтительно обозначает метокси или этокси, в особенности этокси. R7 предпочтительно обозначает метокси или этокси, в особенности этокси.
В одной форме выполнения данного изобретения R8 обозначает фенил, незамещенный или замещенный 1, 2 или 3 заместителями из группы F, Cl, алкил с 1, 2, 3 или 4 C-атомами или алкокси с 1, 2, 3 или 4 C-атомами, в особенности незамещенный фенил.
Соединения формул I, Ia, Ib, XVa и XVb и их фармацевтически совместимые соли являются замещенными ацилгуанидинами и ингибируют клеточный натрий/протонный обмен (Na+/H+-обменник, NHE), в особенности подтип NHE-1.
На основе свойств ингибирования NHE соединения формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимые соли пригодны для профилактики и лечения заболеваний, вызываемых активацией NHE или активированным NHE, а также вторичных заболеваний, вызываемых повреждениями, обусловленными NHE.
Соединения формул I, Ia, Ib, XVa и XVb также могут использоваться для лечения и профилактики заболеваний, причем NHE ингибируется лишь частично, например, путем использования сниженной дозировки.
Так как ингибиторы NHE преобладающим образом оказывают влияние посредством клеточной регуляции pH, они благоприятным образом могут комбинироваться с другими соединениями, также регулирующими внутриклеточное значение pH, причем имеются в виду ингибиторы ферментных групп карбоангидраз, ингибиторы систем, транспортирующих бикарбонатные ионы, такие как ингибиторы натрий-бикарбонатного котранспортера (NBC) или зависимого от натрия хлорид-бикарбонатного обмена (NCBE), а также с ингибиторами NHE с ингибирующим действием на другие подтипы NHE, в качестве компонентов для комбинации, так как с их помощью могут быть усилены или модулированы существенные фармакологические эффекты, регулирующие pH, описанных здесь ингибиторов NHE.
Применение соединений формул I, Ia, Ib, XVa, XVb или XVI касается профилактики и лечения острых и хронических заболеваний в ветеринарной медицине и медицине человека. Таким образом, ингибиторы NHE согласно изобретению пригодны для лечения заболеваний, вызванных ишемией и реперфузией.
Описанные здесь соединения вследствие своих фармакологических свойств пригодны в качестве антиаритмических лекарственных средств. Благодаря своим кардиозащитным компонентам ингибиторы NHE исключительно пригодны для профилактики инфаркта и лечения инфаркта, а также для лечения стенокардии, причем они также превентивно ингибируют или сильно сокращают патофизиологические процессы при возникновении вызванных ишемией нарушений, в особенности при возникновении вызванных ишемией сердечных аритмий. Благодаря их защитному действию против патологических гипоксических и ишемических ситуаций, соединения формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимые соли, используемые согласно изобретению, вследствие ингибирования клеточного механизма Na+/H+-обмена могут быть использованы в качестве лекарственных средств для лечения всех острых или хронических нарушений, вызванных ишемией, или первичных или вторичных индуцированных ею заболеваний.
Это касается также их применения в качестве лекарственных средств при хирургических вмешательствах. Так, соединения могут быть использованы при трансплантации органов, причем соединения могут быть использованы как для защиты органов донора перед и во время изъятия, для защиты изъятых органов, например, при лечении или их хранении в физиологическом растворе, так и при введении в организм реципиента.
Соединения согласно изобретению также являются ценными лекарственными средствами с защитным действием при проведении ангиопластических операций, например, на сердце, а также на периферических органах и сосудах.
Далее, соединения согласно изобретению могут быть использованы при операциях шунтирования, например при шунтировании коронарных сосудов и при коронарном артериальном шунтировании (Coronary Artery Bypass Graft) (CABG).
В соответствии со своим действием в отношении вызванных ишемией нарушений соединения согласно изобретению формулы I также могут быть использованы для реанимации после остановки сердца.
Соединения согласно изобретению представляют интерес для лекарственных средств против угрожающих жизни аритмий. Они останавливают мерцание желудочков и восстанавливают физиологический синусоидальный ритм сердца.
Так как ингибиторы NHE-1 не только эффективно защищают ткани и органы человека, в особенности сердце, от повреждений, вызываемых ишемией и реперфузией, но и от цитотоксического действия лекарственных средств, в особенности используемых при лечении рака и аутоиммунных заболеваний, пригодно комбинированное применение с ингибиторами NHE, которые могут ингибировать цитотоксическое, в особенности кардиотоксическое побочное действие названных соединений. Путем уменьшения цитотоксического эффекта, в особенности кардиотоксичности, вследствие дополнительного назначения лекарства с ингибиторами NHE-1, кроме того, доза цитотоксического терапевтического средства может быть увеличена и/или предписание врача с такими лекарствами продлено. Терапевтическая польза такой цитотоксической терапии может быть существенно увеличена посредством комбинации с ингибиторами NHE.
Кроме того, ингибиторы NHE1 могут быть использованы при повреждающем сердце перепроизводстве гормонов щитовидной железы, тиреотоксикозе, или при внешнем введении гормонов щитовидной железы. Поэтому соединения формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимые соли пригодны для улучшения лечения кардиотоксическими лекарственными средствами.
В соответствии со своим защитным действием против вызванных ишемией повреждений соединения согласно изобретению также пригодны в качестве лекарственных средств для лечения ишемии нервной системы, в особенности центральной нервной системы, причем они, например, пригодны для лечения апоплексического удара или отека головного мозга.
Ингибиторы NHE пригодны также для лечения и профилактики заболеваний и нарушений, вызванных повышенной возбудимостью центральной нервной системы, в особенности для лечения эпилептических заболеваний, клонических и тонических спазмов, вызванных центральной нервной системой, психических депрессивных состояний, состояний страха и психозов. При этом описанные ингибиторы NHE могут быть использованы по отдельности или в комбинации с другими антиэпилептически действующими веществами или антипсихотическими биологически активными веществами, или ингибиторами карбоангидраз, например, с ацетазоламидом, а также с другими ингибиторами NHE или натриево-зависимыми хлорид-бикарбонатными ионообменниками (NCBE).
В дополнение к этому ингибиторы NHE также пригодны для лечения форм шока таких, как, например, аллергический, кардиогенный, гиповолемический и бактериальный шок.
Соединения формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимые соли также могут использоваться для профилактики и лечения тромбозных заболеваний, так как они в качестве ингибиторов NHE сами могут ингибировать агрегацию тромбоцитов. Кроме этого они могут ингибировать или предотвращать высвобождение медиаторов воспаления и коагуляции, в избытке присутствующих после ишемии и реперфузии, в особенности фактор Виллебранда и тромбогенные селективные протеины. Тем самым может быть уменьшено или исключено патогенное действие существенных тромбогенных факторов. Поэтому ингибиторы NHE согласно данному изобретению можно комбинировать с другими антикоагуляционными и/или тромболитическими биологически активными веществами, как, например, плазминогенные активаторы рекомбинантных или естественных тканей, стрептокиназы, урокиназы, ацетилсалициловая кислота, антагонисты тромбина, антагонисты фактора Xa, фибринолитически действующие лекарственные средства, антагонисты рецептора тромбоксана, ингибиторы фосфодиэстеразы, антагонисты фактора-VIIa, клопидогрел (Clopidogrel), тиколопидин и т.д. Комбинированное использование данных ингибиторов NHE с ингибиторами NCBE и/или с ингибиторами карбоангидразы, например, с ацетазоламидом, особенно предпочтительно.
Кроме этого, ингибиторы NHE характеризуются сильным ингибирующим действием на пролиферацию клеток, например клеточную пролиферацию фибробластов и пролиферацию клеток гладких мышц сосудов. Поэтому речь идет о соединениях формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимых солях в качестве ценных терапевтических средств для заболеваний, в случае которых пролиферация является первичной или вторичной причиной, и поэтому они могут использоваться в качестве средств против атеросклероза, средств против хронической почечной недостаточности, онкологических заболеваний.
Было показано, что с помощью ингибиторов NHE ингибируется миграция клеток. Поэтому речь идет о соединениях формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимых солях в качестве ценных терапевтических средств для заболеваний, у которых миграция клеток является первичной или вторичной причиной, как, например, онкологические заболевания с явно выраженной склонностью к метастазированию.
Ингибиторы NHE далее характеризуются замедлением или предотвращением фиброзных заболеваний. Таким образом, они пригодны в качестве отличных средств для лечения фиброзов сердца, а также фиброза легких, фиброза печени, фиброза почек и других фиброзных заболеваний. Таким образом, они могут использоваться для лечения гипертрофии и гиперплазии органов, например сердца и простаты. Поэтому они пригодны для профилактики и лечения сердечной недостаточности (конгестивная сердечная недостаточность, =CHF), а также для лечения и профилактики гиперплазии простаты или гипертрофии простаты.
Так как NHE у эссенциальных гипертоников является значительно повышенным, соединения формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимые соли пригодны для профилактики и лечения повышенного кровяного давления и лечения сердечно-сосудистых заболеваний. При этом они могут использоваться отдельно или совместно с пригодными компонентами для комбинаций и препаративных готовых форм для лечения повышенного кровяного давления и сердечно-сосудистых заболеваний. Так, например, одно или несколько тиазидо-подобно действующих мочегонных средств, петлевых диуретиков, антагонистов альдостерона и псевдоальдостерона, таких как гидрохлоротиазид, индапамид, политиазид, фуросемид, пиретанид, торасемид, буметанид, амилорид, триамтерен, спиронолактон или эплерон, могут быть скомбинированы с соединениями согласно изобретению. Далее, ингибиторы NHE данного изобретения могут использоваться в комбинации с антагонистами кальция, такими как верапамил, дилтиазем, амлодипин или нифедипин, а также с ингибиторами ACE, как, например, рамиприл, эналаприл, лисиноприл, фосиноприл или каптоприл. Другими благоприятными компонентами для комбинаций также являются β-блокаторы, такие как метопролол, албутерол и т.д., антагонисты рецептора ангиотензина и субтипов его рецепторов, такие как лосартан, ирбесартан, валсартан, омапатрилат, гемопатрилат, антагонисты эндотелина, ингибиторы ренина, агонисты рецепторов аденозина, ингибиторы и активаторы калиевых каналов, такие как глибенкламид, глимепирид, диазоксид, кромакалим, миноксидил и их производные, активаторы митохондриального чувствительного к ATP (АТФ) калиевого канала, ингибиторы Kv1.5 и т.д.
Было показано, что ингибиторы NHE-1 характеризуются существенным противовоспалительным действием и поэтому могут быть использованы в качестве противовоспалительных средств. При этом обращает на себя внимание ингибирование высвобождения медиаторов воспаления. Таким образом, соединения могут использоваться по отдельности или в комбинации с противовоспалительным средством для профилактики или лечения хронических и острых воспалительных заболеваний. В качестве компонентов для комбинаций предпочтительно используют стероидные и нестероидные противовоспалительные средства. Далее соединения согласно изобретению могут использоваться для профилактики или лечения заболеваний, вызванных простейшими, как малярия и куриный кокцидиоз.
Кроме того, было обнаружено, что ингибиторы NHE оказывают благоприятное влияние на сывороточные липопротеины. Общепризнано, что для возникновения атеросклеротических изменений сосудов, в особенности коронарных сердечных заболеваний, высокое значение жиров в крови, так называемая гиперлипидемия, представляет существенный фактор риска. Поэтому для профилактики и регрессии атеросклеротических изменений снижение повышенного содержания сывороточных липопротеинов имеет чрезвычайное значение. Помимо сокращения общего сывороточного холестерина особенное значение имеет снижение доли специфической атерогенной липидной фракции этого общего холестерина, в особенности липопротеинов низкой плотности (low density Lipoprotein) (LDL) и липопротеинов очень низкой плотности (very low density Lipoprotein) (VLDL), так как эти фракции липидов представляют атерогенный фактор риска. Напротив, липопротеинам высокой плотности приписывают защитную функцию против коронарных заболеваний сердца. Соответственно этому гиполипидемические средства должны быть в состоянии понижать не только общий холестерин, но и в особенности VLDL и LDL-фракции сывороточного холестерина. Теперь было найдено, что ингибиторы NHE-1 характеризуются ценными терапевтически используемыми свойствами в отношении влияния на уровень сывороточных липидов. Так, они существенно снижают повышенные концентрации LDL и VLDL в сыворотке до уровня, который может наблюдаться, например, при усиленном диетическом приеме не содержащей холестерина и жиры пищи или при патологических изменениях обмена веществ, например генетически обусловленной гиперлипидемии. Поэтому они могут использоваться для профилактики и регрессии атеросклеротических изменений, в которых они исключают причинный фактор риска. Сюда же относят не только первичные гиперлипидемии, но и известные вторичные гиперлипидемии, которые имеют место, например, при диабете. Кроме того, ингибиторы NHE приводят к явному сокращению инфарктов, вызванных аномалиями обмена веществ, и в особенности к существенному уменьшению величины индуцированного инфаркта и его степени тяжести.
Поэтому соединения формул I, Ia, Ib, XVa, XVb и XVI согласно изобретению предпочтительно находят применение для получения медикамента для лечения гиперхолистеринемии; для получения медикамента для предупреждения атерогенеза; для получения медикамента для профилактики и лечения атеросклероза, для получения медикамента для профилактики и лечения заболеваний, вызванных повышенным уровнем холестерина, для получения медикамента для профилактики и лечения заболеваний, вызванных эндотелиальной дисфункцией; для получения медикамента для профилактики и лечения гипертонии, индуцированной атеросклерозом, для получения медикамента для профилактики и лечения тромбозов, индуцированных атеросклерозом, для получения медикамента для профилактики и лечения ишемических нарушений и постишемических реперфузионных нарушений, индуцированных гиперхолистеринемией и эндотелиальной дисфункцией, для получения медикамента для профилактики и лечения сердечной гипертрофии и кардиомиопатии и застойной сердечной недостаточности (CHF), индуцированных гиперхолистеринемией и эндотелиальной дисфункцией, для получения медикамента для профилактики и лечения коронарных спазмов сосудов и миокардиального инфаркта, индуцированных гиперхолистеринемией и эндотелиальной дисфункцией, для получения медикамента для лечения названных болезней в комбинации с веществами, снижающими кровяное давление, предпочтительно с ингибиторами ангиотензин-превращающего фермента (ACE) и антагонистов рецепторов ангиотензина. Комбинация ингибитора NHE с биологически активными веществами, снижающими уровень жира в крови, предпочтительно с ингибитором HMG-CoA-редуктазы (например, ловастатином или правастатином), причем последний приводит к гиполипидемическому действию и вследствие этого повышает гиполипидемические свойства ингибитора NHE, представляет собой благоприятную комбинацию с усиленным действием и сокращенным использованием биологически активных веществ.
Таким образом, ингибиторы NHE ведут к эффективной защите против повреждений эндотелия различного генеза. С этой защитой сосудов против синдрома эндотелиальной дисфункции соединения формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимые соли являются ценными лекарственными средствами для профилактики и лечения спазмов коронарных сосудов, заболеваний периферических сосудов, в особенности интермиттирующей хромоты, атерогенеза и атеросклероза, левожелудочковой гипертрофии и дилатационной кардиомиопатии, и заболеваний на основе тромбоза.
Кроме того, было показано, что ингибиторы NHE пригодны для лечения неинсулинозависимого диабета (NIDDM), причем резистентность к инсулину подавляется. При этом усилению антидиабетического эффекта и качеству действия соединений согласно изобретению может способствовать их комбинирование с бигуанидом, как метформин, с антидиабетической сульфонилмочевиной, как глибурид, глимепирид, толбутамид, и т.д., ингибитором глюкозидазы, агонистом PPAR, как росиглитазон, пиоглитазон и т.д., с инсулиновыми препаратами различных форм применения, с ингибитором DB4, с сенсибилизатором инсулина или с меглитинидом.
Помимо острых антидиабетических эффектов, ингибиторы NHE противодействуют возникновению поздних диабетических осложнений и поэтому могут использоваться в качестве лекарственных средств для профилактики и лечения поздних диабетических нарушений, таких как диабетическая нефропатия, диабетическая невропатия, диабетическая ретинопатия, диабетическая кардиомиопатия и другие заболевания, возникающие как следствие диабета. При этом они могут предпочтительно комбинироваться с антидиабетическими лекарственными средствами, описанными выше при лечении NIDDM. Комбинация с благоприятными формами приема инсулина при этом может иметь особое значение.
Ингибиторы NHE, помимо защитного действия против острых ишемических ситуаций и последующих также остро отягощенных реперфузионных ситуаций, обнаруживают также прямое терапевтически пригодное для использования действие против заболеваний и нарушений всего организма млекопитающих, которые тесно связаны с симптомами хронически протекающего процесса старения и которые также являются независимыми от острых состояний недостаточного кровоснабжения и также могут встречаться при нормальных, неишемических условиях. Для этих патологических, возникающих после продолжительного старения возрастных проявлений, таких как недомогания, хилость и смерть, которые в последнее время лечат с помощью ингибиторов NHE, речь идет о заболеваниях и нарушениях, в значительной степени обусловленных возрастными изменениями жизненно необходимых органов и их функций, приобретающих в стареющем организме возрастающее значение.
К заболеваниям, связанным с функциональными возрастными нарушениями, с обусловленными возрастом симптомами износа органов, относятся, например, недостаточная чувствительность и способность кровеносных сосудов к реакции по отношению к реакциям контракции и релаксации. Это возрастное ослабление способности к реакции сосудов на сжимающее и релаксирующее возбуждение, которое является существенным процессом сердечно-сосудистой системы и тем самым жизни и здоровья, может быть в значительной степени ликвидировано или сокращено с помощью ингибиторов NHE. Важной функцией и мерой для поддерживания способности сосудов к реакции является блокада или ретардация возрастной прогрессирующей эндотелиальной дисфункции, которая может быть значительно сокращена посредством ингибиторов NHE. Таким образом, ингибиторы NHE исключительно пригодны для лечения и профилактики возрастной прогрессирующей эндотелиальной дисфункции, в особенности интермиттирующей хромоты.
Примером показателя, характеризующего следующий процесс старения, является ослабление сокращаемости сердца и ослабление адаптации сердца к требуемой нагнетаемой мощности сердца. Эта пониженная сердечная деятельность как следствие процесса старения в большинстве случаев связана с дисфункцией сердца, вызванной, кроме прочего, отложением соединительной ткани в ткани сердца. Это отложение соединительной ткани характеризуется увеличением веса сердца, увеличением сердца и ограниченной сердечной функцией. Неожиданно было обнаружено, что подобное старение органа сердца могло быть ингибировано почти полностью. Тем самым ингибиторы NHE исключительно пригодны для лечения и профилактики сердечной недостаточности, застойной сердечной недостаточности (CHF).
Путем подавления пролиферации могут вылечиваться не только уже наступившие онкологические заболевания, но и с помощью ингибиторов NHE сокращаться или сильно замедляться множественные возрастные формы возникновения рака. Особенно достойно внимания заключение о том, что сокращаются или сильно замедляются возникающие возрастные заболевания всех органов, а не только парализуются определенные формы рака. Таким образом, ингибиторы NHE пригодны для лечения и, в особенности, для профилактики обусловленных возрастом форм рака.
С помощью ингибиторов NHE констатируют существенное запаздывание наступления возрастных заболеваний всех исследованных органов, включая сердце, сосуды, печень и т.д., а также значительное запаздывание старческого рака. Более того, это также может неожиданно приводить к продлению жизни в той мере, которая до сих пор не могла быть достигнута никакими другими группами медикаментов или какими-либо природными материалами. Это единственное в своем роде действие ингибиторов NHE также дает возможность, помимо исключительного использования биологически активных веществ для человека и животных, комбинировать эти ингибиторы NHE с другими геронтологически используемыми принципами действия, мероприятиями, веществами и природными материалами, в основе которых лежит другой механизм действия. Такого рода классами биологически активных веществ, используемых в геронтологической терапии, являются, в особенности, витамины и вещества с антиокислительным действием. Так как существует корреляция между энергетической (калорийной) нагрузкой или приемом пищи и процессом старения, может существовать комбинация диетических мероприятий, например, с регулированием аппетита. Таким же образом может быть упомянута комбинация с понижающими кровяное давление медикаментами, как, например, ингибиторы ACE, антагонисты рецепторов ангиотензина, мочегонные средства, антагонисты Ca+2 и т.д., или с медикаментами, нормализирующими обмен веществ, как вещества, понижающие холестерин.
Таким образом, ингибиторы NHE исключительно пригодны для профилактики возрастных изменений тканей и для продления жизни при сохранении высокого качества жизни.
Соединения согласно изобретению являются эффективными ингибиторами клеточного антипорта натрий/протон (Na/H-ионообменник), который при многочисленных заболеваниях (эссенциальная гипертония, атеросклероз, диабет и т.д.) также в таких клетках является повышенным, измерения являются легко доступными, как, например, в эритроцитах, тромбоцитах или лейкоцитах. Поэтому используемые согласно изобретению соединения пригодны в качестве исключительных и простых научных инструментов, например при их применении в качестве диагностикумов для определения и распознавания определенных форм гипертонии, а также атеросклероза, диабета и поздних диабетических осложнений, пролиферативных заболеваний и т.д.
Далее, в медицине человека, ветеринарии или для защиты растений используют лекарственные средства, содержащие вместе с фармацевтически приемлемыми носителями и добавками эффективное количество одного или нескольких соединений формул Xva, XVb и XVI и/или их фармацевтически совместимых солей по отдельности или в комбинации с другими фармакологическими биологически активными веществами или лекарственными средствами. Лекарственные средства, содержащие соединения формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимые соли, можно принимать, например, орально, па-рентерально, внутривенно, ректально, чрескожно или путем ингаляции, причем предпочтительное применение зависит от соответствующей формы проявления болезни. При этом соединения формул I, Ia, Ib, XVa, XVb и XVI могут использоваться по отдельности или вместе с галеновыми вспомогательными веществами, как в ветеринарной медицине, так и в медицине человека. Лекарственные средства содержат биологически активные вещества формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимые соли, в общем, в количестве от 0,01 мг до 1 г на разовую дозу.
Какие вспомогательные вещества пригодны для желаемых лекарственных готовых форм, известно специалисту благодаря его специальным знаниям. Помимо растворителей, гелеобразователей, основ суппозиториев, вспомогательных веществ для таблеток и других носителей биологически активных веществ, могут быть использованы, например, антиоксиданты, диспергаторы, эмульгаторы, антивспениватели, вещества, улучшающие вкус, консервирующие средства, агенты растворения или красители.
Для оральной формы введения активные соединения смешивают с пригодными для этого добавками, такими как носители, стабилизаторы или инертные разбавители, и с помощью общепринятых методов доводят до пригодных форм применения, таких как таблетки, драже, разъемные капсулы, водные, спиртовые или масляные растворы. В качестве инертных носителей могут быть использованы, например, гуммиарабик, магнезия, карбонат магния, фосфат калия, молочный сахар, глюкоза или крахмал, в особенности кукурузный крахмал. При этом композиция может существовать в виде как сухого, так и влажного гранулята. В качестве масляных носителей или в качестве растворителей используют, например, растительные или животные масла, например подсолнечное масло или рыбий жир.
Для подкожного, внутримышечного или внутривенного введения употребляют активные соединения, по желанию с общепринятыми для этого веществами как агенты растворения, эмульгаторы или другие вспомогательные вещества в растворе, суспензии или эмульсии. В качестве растворителей имеют в виду, например, воду, физиологический раствор поваренной соли или спирты, например этанол, пропанол, глицерин, наряду с этим также растворы сахаров, таких как растворы глюкозы или маннита, или также смесь из различных названных растворителей.
В качестве фармацевтических препаративных готовых форм для приема в форме аэрозолей или спреев пригодны, например, растворы, суспензии или эмульсии биологически активных веществ формул I, Ia, Ib, XVa, XVb и XVI и/или их фармацевтически совместимых солей в фармацевтически безвредных растворителях, в особенности этаноле или воде, или смеси таких растворителей. Препаративная готовая форма может при необходимости содержать и другие фармацевтические вспомогательные вещества, такие как тензиды, эмульгаторы и стабилизаторы, а также пропеллент. Одна такая готовая форма обычно содержит биологически активное вещество в концентрации приблизительно от 0,1 до 10, в особенности приблизительно от 0,3 до 3 вес.-%.
Дозировка принимаемого биологически активного вещества формул I, Ia, Ib, XVa, XVb и XVI и частота приема зависят от эффективности и времени воздействия используемых соединений; кроме того, также влияют вид и интенсивность болезни, подвергаемой лечению, а также пол, возраст, вес и индивидуальная предрасположенность млекопитающего, подвергаемого лечению.
В среднем дневная доза соединения формул I, Ia, Ib, XVa, XVb и XVI и/или его фармацевтически совместимых солей при весе пациента около 75 кг составляет, по меньшей мере, 0,001 мг/кг, например, 0,01 мг/кг, вплоть до максимально 10 мг/кг, например, 1 мг/кг веса тела. В острых ситуациях болезни, например, непосредственно после перенесения инфаркта миокарда, могут быть необходимы еще более высокие и, прежде всего, более частые дозировки, например, вплоть до 4 разовых доз в день. В особенности при внутривенном введении, например, у пациентов с инфарктом в отделении интенсивной терапии, может быть необходима доза вплоть до 700 мг в день, и соединения согласно изобретению могут вводиться путем инъекций.
Лист сокращений:
Времена удерживания (rt), приведенные далее, относятся к результатам ВЭЖХ со следующими параметрами:
Метод A:
Стационарная фаза: Waters Symmetry C8 (5 мк) 3,9 (150 мм
Подвижная фаза: изократный CH3CN/0,1% водная CF3CO2H 35:65; λ=220 нм; 1 мл/мин.
Метод B:
Стационарная фаза: Waters Symmetry C8 (5 мк) 3,9 (150 мм
Подвижная фаза: изократный CH3CN/0,1% водная CF3CO2H 40:60; λ=230 нм; 1 мл/мин.
Метод C:
Стационарная фаза: Waters Symmetry C8 (5 мк) 3,9 (150 мм
Подвижная фаза: изократный CH3CN/0,1% водная CF3CO2H 50:50; λ=220 нм; 1 мл/мин.
Пример 1
a) N-(2,2,2-трифтор-этил)-4-трифторметил-бензамид

5,0 г (24 ммоль) 4-трифторметил-бензоилхлорида и 5,0 мл (36 ммоль) триэтиламина растворяют в 50 мл CH2Cl2 и при КТ медленно прикапывают 2,4 г (24 ммоль) 2,2,2-трифтор-этиламина. 4 ч перемешивают при КТ, затем летучие компоненты отгоняют под вакуумом. В остаток добавляют 100 мл ТБМЭ (MTB) и промывают сначала 30 мл насыщенного водного раствора Na2CO3, а затем 30 мл насыщенного водного раствора NaHSO4. Сушат над MgSO4 и получают 6,1 г (94%) бесцветной смолы, которая кристаллизуется при выдерживании; т.пл: 117°C.
b) (R,S)-3-гидрокси-2-(2,2,2-трифтор-этил)-5-трифторметил-2,3-дигидро-изоиндол-1-он

0,37 мл (2,4 ммоль) TMEDA и 1,4 мл (2,3 ммоль) 1,5 M раствора t-BuLi в н-пентане растворяют при -75°C в 2 мл ТГФ (безводном) и при -75°C прикапывают раствор 0,30 г (1,1 ммоль) N-(2,2,2-трифтор-этил)-4-трифторметил-бензамида в 2 мл ТГФ. 3 ч перемешивают при -75°C, затем прикапывают 0,43 мл (5,5 ммоль) ДМФ и в течение 30 минут нагревают до КТ. Реакционную смесь выливают в 100 мл насыщенного водного раствора NaHCO3 и 3 раза экстрагируют с ЭА (по 30 мл). Сушат над MgSO4 и растворитель отгоняют под вакуумом. Хроматография на кизельгеле с ДИП (DIP) дает 80 мг (R,S)-3-гидрокси-2-(2,2,2-трифтор-этил)-5-трифторметил-2,3-дигидро-изоиндол-1-она помимо 110 мг смеси с исходным продуктом. Эту смесь вновь разделяют с помощью ВЭЖХ с обратной фазой (условия смотри ниже) и далее получают 40 мг (R,S)-3-гидрокси-2-(2,2,2-трифтор-этил)-5-трифторметил-2,3-дигидро-изоиндол-1-она; Общий выход продукта 30%.
ВЭЖХ: Градиент, время пробега 20 мин
Растворитель: 0,1% водная CF3CO2H, ацетонитрил (Chromasolv); Поток: 30 мл/мин
Колонка: Waters Xterra™ MS C18 5 мкМ, 30 (100 мм
Градиент:
0-2,5 мин 10% ацетонитрил
3,0 мин 25% ацетонитрил
14,0 мин 75% ацетонитрил
15,0 мин 95% ацетонитрил
17,5 мин 10% ацетонитрил
c) этиловый эфир (RS)-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-уксусной кислоты

В токе аргона этиловый эфир (диэтокси-фосфорил)-уксусной кислоты (135 мг, 0,6 ммоль) растворяют в безводном диметоксиэтане (10 мл). К этому раствору при КТ добавляют 17,6 мг NaH (60%-ный в масле) и перемешивают 10 мин при КТ. После этого добавляют раствор (RS)-3-гидрокси-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-изоиндол-1-она 120 мг (0,04 ммоль) в безводном диметоксиэтане (5 мл) и затем 2 ч перемешивают с обратным потоком. Реакционный раствор охлаждают, затем его выливают в 50 мл 5%-ного раствора гидрокарбоната натрия, 2 раза экстрагируют этилацетатом (по 20 мл), органическую фазу сушат над MgSO4, выпаривают под вакуумом и остаток очищают посредством хроматографии на кизельгеле с ДИП (DIP) в качестве элюента. Получают 90 мг (61%) этиловый эфир (RS)-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-уксусной кислоты в виде бесцветного масла, которое кристаллизуется из гептана в виде бежевого твердого вещества.
Rf(DIP)=0,31
ЯМР-спектр идентичен продукту, полученному в примере 4.
d) (R,S)-N-{2-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-H-изоиндол-1-ил]-ацетил}-гуанидин

Этиловый эфир (RS)-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-уксусной кислоты может быть подвергнут превращению с гуанидином как описано в примере 2g).
Пример 2
a) 2-нитро-4-трифторметил-бензойная кислота
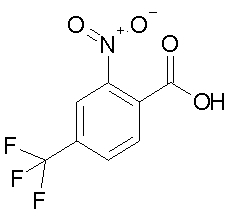
11,97 г 4-трифторметил-бензойной кислоты (63 ммоль) медленно порциями добавляют к 48 мл HNO3 (100%) при КТ. Затем смесь в течение 1 ч нагревают с обратным потоком, затем охлаждают до КТ и выливают на 600 г льда. 1 ч смесь перемешивают, затем осадок отфильтровывают и промывают 1 л воды. Фильтрат 300 мл CH2Cl2 экстрагируют, органическую фазу объединяют с осадком и сушат над Na2SO4. Растворитель отгоняют под вакуумом и осадок перекристаллизовывают тем, что растворяют в 1 л ДИП (DIP) при 68°C, добавляют 2 л н-гептана при этой температуре и в заключение медленно охлаждают раствор до КТ. Закристаллизованный продукт промывают 1 л гептана, сушат под вакуумом и получают 7,1 г (48%), т.пл. 136°C-138°C.
b) 2-амино-4-трифторметил-бензойная кислота

250 г 2-нитро-4-трифторметил-бензойной кислоты (1,06 моль) растворяют в 1 л EtOH и добавляют 7,5 г Pd/C (5%). Смесь гидрируют при давлении водорода 1-2,5 бар. Во время приема водорода температура повышается в интервале от 10°C до 104°C. Через 2 ч прием водорода заканчивается. Затем катализатор отфильтровывают, растворитель отгоняют под вакуумом и получают 215 г (99%) бледно-желтого твердого вещества, т.пл. 174-176°C.
c) 2-((E)-2-этоксикарбонил-винил)-4-трифторметил-бензойная кислота
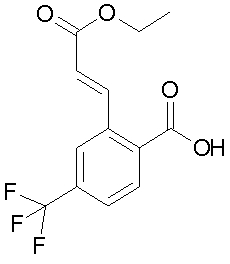
520 мг NaNO2 (7,6 ммоль) растворяют в 2 мл воды и прикапывают к раствору 1,3 г 2-амино-4-трифторметил-бензойной кислоты (6,5 ммоль) в 2,6 мл 48% водного раствора HBF4 и 30 мл этанола при 0°C. 10 минут перемешивают при 0°C, затем нагревают до КТ. Далее добавляют 0,3 мл 48% водного раствора HBF4, затем 30 мл этанола, 0,9 г этилового эфира акриловой кислоты (9,0 ммоль) и 26,9 мг Pd(OAc)2 (0,12 ммоль). Затем смесь перемешивают 1 ч при 50-60°C. Потом растворитель отгоняют под вакуумом, остаток смешивают с 25 мл ЭА и промывают сначала 25 мл 1N водного раствора HCl, затем 25 мл насыщенного водного раствора NaCl. Органическую фазу сушат над Na2SO4, и растворитель отгоняют под вакуумом. Остаток суспендируют в 25 мл гептана и отфильтровывают выпавший продукт. Выход 1,3 г (69%) светло-коричневатого твердого вещества. Аналитическую пробу очищают кристаллизацией из гептана/этилацетата.
ЯМР-спектр идентичен продукту, полученному в примере 3a.
d) этиловый эфир (E)-3-[2-(2,2,2-трифтор-этилкарбамоил)-5-трифторметил-фенил]-акриловой кислоты

1,3 г 2-(2-этоксикарбонил-винил)-4-трифторметил бензойной кислоты (4,5 ммоль) и 453 мг 2,2,2-трифтор-этиламин (4,5 ммоль) растворяют в 5 мл ДМФ и добавляют 0,93 г ДЦГКДИ (DCC). Смесь перемешивают в течение 4 ч при КТ. Побочный продукт мочевину отделяют фильтрацией, и затем растворитель отгоняют под вакуумом. Осадок перекристаллизовывают из DIP и получают 1,6 г (96%) белых кристаллов.
ЯМР-спектр идентичен продукту, полученному в примере 3b.
e) (RS)-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-уксусная кислота

2,2 г этилового эфира (E)-3-[2-(2,2,2-трифтор-этилкарбамоил)-5-трифторметил-фенил]-акриловой кислоты (5,9 ммоль) растворяют в 10 мл метанола и 1,5 мл 5 M водного раствора NaOH (7,5 ммоль). Смесь 18 ч перемешивают при КТ и затем устанавливают pH 7 водным раствором HCl. Растворители отгоняют под вакуумом, и остаток суспендируют в 10 мл воды. В этой суспензии устанавливают pH 2 посредством 2N водного раствора HCl и 3 раза экстрагируют ЭА (по 10 мл). Сушат над Na2SO4 и растворитель отгоняют под вакуумом. Остаток подвергают кристаллизации с системой диэтиловый эфир/ДИП(DIP), т.пл.: 202-204°C.
Выход 1,8 г (89%).
1H-ЯМР (400 МГц, CDCl3): (=3,07 (дд, J1=17 Гц, J2=6 Гц, 1 H), 3,23 (дд, J1=17 Гц, J2=5 Гц, 1 H), 4,27 (м, 1 H), 4,58 (м, 1 H), 5,08 (т, J=5 Гц, 1 H), 7,91 (д, J=8 Гц, 1 H), 7,96 (д, J=8 Гц, 1 H), 8,12 (с, 1 H), 12,50 (ш.с., 1 H) ч./млн.
Элементарный анализ сжиганием: C13H9F6NO3 (341,2): расч. C 45,76, H 2,66, N4,10; получ. C 45,71, H 2,43, N 4,11.
f) этиловый эфир (RS)-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-уксусной кислоты
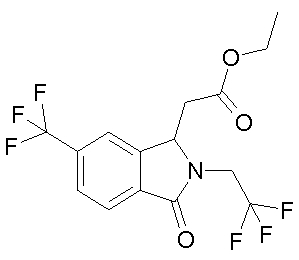
2,6 мл SOCl2 (35 ммоль) растворяют в 20 мл этанола и добавляют 3,4 г (R,S)-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-уксусной кислоты (10 ммоль) при -10°C 18 ч перемешивают при КТ и затем летучие компоненты отгоняют под вакуумом. Остаток подвергают хроматографии на кизельгеле с системой элюентов HEP/EE 3:1. Выход 3,0 г (81%) бесцветного масла, которое кристаллизуется из гептана в виде бежевого твердого вещества.
ЯМР-спектр идентичен продукту, полученному в примере 4.
g) (RS)-N-{2-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-H-изоиндол-1-ил]-ацетил}-гуанидин
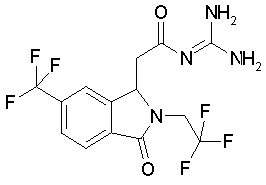
Гуанидин-гидрохлорид (11,5 г, 120 ммоль) растворяют в НМП (NMP) (45 мл) и добавляют KOtBu (11,2 г, 100 ммоль) при перемешивании, 1,5 часа перемешивают при КТ и смесь фильтруют.Фильтрат прикапывают к раствору этилового эфира (RS)-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-уксусной кислоты (7,38 г, 20 ммоль) в НМП (NMP) (12 мл) при перемешивании при КТ и оставляют перемешиваться в течение 60 мин при КТ. Затем добавляют ледяную воду (270 мл), устанавливают pH 7 с помощью 2N HCl, добавляют этилацетат (60 мл) и затем путем добавления раствора NaHCO3 устанавливают значение pH на уровне 8-8,5. Смесь сильно перемешивают в течение 1 часа при КТ, отсасывают образующийся осадок и промывают водой. Получают 7,06 г (83%) (R,S)-N-{2-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-H-изоиндол-1-ил]-ацетил}-гуанидина, соединение включения с 0,5 эквивалентами этилацетата, в виде светло-желтых кристаллов, т.пл. 160-161°C при медленном нагревании, выделение этилацетата около 90°C.
Rf (этилацетат/метанол)=0,45
1H-ЯМР (400 МГц, CDCl3): (=2,54 (дд, J1=8 Гц, J2=16 Гц, 1 H), 3,09 (дд, J1=4 Гц, J2=16 Гц, 1 H), 4,25 (м, 1 H), 4,64 (м, 1 H), 5,18 (м, 1 H), 6,65 (ш.с., 2 H), 7,75 (ш.с., 2 H), 7,88 (д, J=8 Гц, 1 H), 7,95 (д, J=8 Гц, 1 H), 8,02 (с, 1 H) ч./млн.
C14H12F6N4O2 (Ѕ C4H8O2 (426,33): расч. C 45,08, H 3,78, N 13,14; найд. C 45,07, H 3,79, N 13,01.
Пример 3
a) 2-((E)-2-этоксикарбонилвинил)-4-трифторметил-бензойная кислота (Вариант примера 2c)
К 339 г 2-амино-4-трифторметил-бензойной кислоты (1,65 моль) в 6,8 л EtOH (безводного) добавляют при КТ 658 мл 48-50% водного раствора HBF4. При этом температура повышается от 21°C до 26°C. Затем смесь охлаждают до 0°C и прикапывают раствор 125 г NaNO2 в 500 мл воды в течение 17 минут между 0°C и 5°C. При этом из первоначально бледно-желтого раствор сначала превращался в оранжево-красную суспензию, а затем в светло-желтую суспензию. Ход реакции отслеживают с помощью ВЭЖХ (метод B; время удерживания 2-амино-4-трифторметил-бензойной кислоты=6,4 мин; побочного продукта соли 2-карбокси-5-трифторметил-бензолдиазония=1,1 мин). В течение 30 минут заканчивают превращение соли 2-карбокси-5-трифторметил-бензолдиазония до>99%. Затем к смеси добавляют 231 г этилового эфира акриловой кислоты (2,31 моль), 11,1 г Pd(OAc)2 (49 ммоль) и 6,8 л этанола (безводного) и реакционную смесь нагревают до 49-51°C. При этом наблюдают равномерное, увеличивающееся с повышением температуры выделение азота. Превращение происходит с помощью ВЭЖХ (метод B; время удерживания 2-((E)-2-этоксикарбонил-винил)-4-трифторметил-бензойной кислоты = 16,4 мин). Через 45 мин степень превращения составляет более 99%. Затем смесь охлаждают до КТ и растворитель отгоняют под вакуумом. Остаток смешивают с 3 л ЭА и отфильтровывают. Затем фильтрат промывают сначала 3 раза водным раствором HCl (по 2,1 л), затем 1 л насыщенного водного раствора NaCl. Сушат над Na2SO4, растворитель отгоняют под вакуумом и получают 449 г светло-коричневого твердого вещества. При включении примеси (4-трифторметил-бензойная кислота; 6,3%) и остатков растворителя (ЭА; 4%) выход составляет 83%. Аналитическую пробу очищают посредством кристаллизации из смеси гептан/этилацетат.Т.пл.: 132-133°C
1H-ЯМР (400 МГц, CDCl3): (=1,36 (т, J=7 Гц, 3 H), 4,31 (кв, J=7 Гц, 2 H), 6,41 (д, J=16 Гц, 1 H), 7,72 (д, J=8 Гц, 1 H), 7,86 (с, 1 H), 8,21 (д, J=8 Гц, 1 H), 8,51 (д, J=16 Гц, 1 H), 8,5-9,5 (ш.с., 1 H) ч./млн.
Элементарный анализ сжиганием: C13H11F3O4 (288,23): расч. C 54,17, H 3,85; найд. C 54,24, H 3,74.
b) этиловый эфир (E)-3-[2-(2,2,2-трифторэтилкарбамоил)-5-трифторметил-фенил]-акриловой кислоты

315 г оксалилхлорида (2,48 моль) при температуре между 15 и 18°C в течение 24 мин. прибавляют к смеси из 650 г 2-((E)-2-этоксикарбонил-винил)-4-трифторметил-бензойной кислоты (2,25 моль), 33 мл ДМФ и 7,8 л CH2Cl2. Во время добавления наблюдают газовыделение. 1 ч перемешивают при КТ, затем охлаждают до 5°C и добавляют 285 г Et3N (2,81 моль) при температуре между 5°C и 10°C в течение 27 мин. Перемешивают 10 мин при 5°C, затем прибавляют 279 г 2,2,2-трифтор-этиламин (2,81 моль) при температуре между 9°C и 20°C в течение 27 мин. 10 мин перемешивают при КТ, при этом выпадает тонкий осадок, и для улучшения перемешиваемости смеси дополнительно добавляют 1 л CH2Cl2. Реакцию отслеживают с помощью ВЭЖХ (метод C; время удерживания: 2-((E)-2-этоксикарбонил-винил)-4-трифторметил-бензойная кислота = 5,9 мин; время удерживания: этиловый эфир (E)-3-[2-(2,2,2-трифторэтилкарбамоил)-5-трифторметил-фенил]-акриловой кислоты = 13,2 мин). После последующих 50 мин перемешивания при КТ превращение закончено. Затем летучие компоненты отгоняют под вакуумом, остаток смешивают с 12 л ЭА и промывают 3 раза водой (по 2,5 л), затем 2 раза насыщенным водным раствором NaHCO3 (по 2,5 л) и, наконец, 1,5 л насыщенного водного раствора NaCl. Сушат над MgSO4, растворитель отгоняют под вакуумом и получают 802 г этилового эфира (E)-3-[2-(2,2,2-трифторэтилкарбамоил)-5-трифторметил-фенил]-акриловой кислоты в виде коричневого твердого вещества. Этот необработанный продукт объединяют с сырьем из другого осадка (177 г) и растворяют в 3 л ЭА при 60-70°C, и при этой температуре смешивают с 14 л HEP порциями по 1 л. Затем смесь нагревают до 80°C и 1,5 ч перемешивают при этой температуре. Затем эту смесь добавляют к 5,6 л нагретого до 70°C HEP, и затем смесь при перемешивании в течение 5 ч охлаждают до КТ. Затем продукт отфильтровывают, промывают 3 л HEP, сушат на воздухе и получают 689 г этилового эфира (E)-3-[2-(2,2,2-трифторэтилкарбамоил)-5-трифторметил-фенил]-акриловой кислоты (67%) в виде светло-коричневого твердого вещества. Т.пл.: 161,5-162°C.
1H-ЯМР (400 МГц, CDCl3): (=1,33 (т, J=7 Гц, 3 H), 4,05 (м, 2 H), 4,26 (кв, J=7 Гц, 2 H), 6,19 (ш.с., 1 H), 6,46 (д, J=16 Гц, 1 H), 7,63 (д, J=8 Гц, 1 H), 7,68 (д, J=8 Гц, 1 H), 7,87 (с, 1 H), 7,90 (д, J=16 Гц, 1 H) ч./млн.
Элементарный анализ сжиганием: C15H13F6NO3 (369,27): расч. C 48,79, H 3,55, N 3,79; найд. C 48,93, H 3,51, N 3,92.
c) (R,S)-N-{2-[3-оксо-2-(2,2,2-трифтор-этил)-6-трифторметил-2,3-дигидро-H-изоиндол-1-ил]-ацетил}-гуанидин

386 г этилового эфира (E)-3-[2-(2,2,2-трифторэтилкарбамоил)-5-трифторметил-фенил]-акриловой кислоты (1,05 моль) суспендируют в 600 мл ДМФ и при температуре между 5°C и 15°C порционно медленно добавляют 4,7 г KOtBu (42 ммоль). Циклизацию с образованием изоиндолона отслеживают с помощью ТСХ (HEP/EE=2:1; этиловый эфир акриловой кислоты: Rf=0,32; изоиндолон: Rf=0,41). Через час превращение закончено. В промежутке времени 587 г KOtBu суспендируют в 2,2 л ДМФ и добавляют 600 г гуанидиний-хлорида при температуре между 20°C и 25°C. Смесь перемешивают 1 ч при 25°C и затем отфильтровывают KCl. Фильтрат с высвобожденным гуанидином затем прибавляют к реакционной смеси, содержащей изоиндолон и 2 ч перемешивают при КТ. Превращение с образованием ацилгуанидина отслеживают с помощью ВЭЖХ (метод B; длина волны 230 нм и 254 нм; изоиндолон: время удерживания = 15,1 мин; ацилгуанидин: время удерживания = 2,9 мин). Затем реакционную смесь выливают в 14 л ледяной воды, с помощью водного раствора HCl устанавливают pH 8,5-9,0 и 4 раза экстрагируют ЭА (по 3 л). Затем промывают 3 раза по 3 л насыщенным водным раствором NaCl, сушат над Na2SO4 и растворитель отгоняют под вакуумом. Получают 329 г (82%) коричневого твердого вещества. Продукт объединяют с 3 другими осадками из таких же способов получения; общее количество 842 г. Эти 842 г (2,2 моль) настаивают в 2 л ЭА и 5 л Et2O 2 часа при 30°C. Затем отфильтровывают твердое вещество, промывают 2 раза по 2 л Et2O и сушат под вакуумом. Получают 693 г (82% регенерации) почти белого твердого вещества. Соединение выкристаллизовывается из 2-пропанола в качестве соединения включения с 0,5 эквивалентами 2-пропанола.
ЯМР-спектр идентичен S-энантиомеру, полученному в примере 5b.
Пример 4
Этиловый эфир (RS)-(2-(2,2,2-трифторэтил)-3-оксо-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил)-уксусной кислоты посредством реакции в одном сосуде, исходя из 2-((E)-2-этоксикарбонил-винил)-4-трифторметил-бензойной кислоты
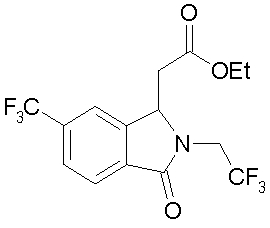
К суспензии 2-((E)-2-этоксикарбонил-винил)-4-трифторметил-бензойной кислоты (2,9 г, 10,1 ммоль) в толуоле (30 мл) добавляют при комнатной температуре SOCl2 (1,98 г, 27,2 ммоль). Перемешивают 5 мин при комнатной температуре и затем нагревают в течение 30 мин до 105°C (температура бани). Примерно при 70°C начинается газовыделение. Перемешивают 3 ч при 105°C, затем охлаждают до комнатной температуры и отсасывают через слой кизельгура (2,5 (0,5 см), промывают толуолом и фильтрат выпаривают под вакуумом. Получают хлорангидрид в форме красно-коричневого масла (3,34 г). 2,2,2-трифторэтиламин (1,2 г, 12,1 ммоль) и триэтиламин (2,58 г, 25,3 ммоль) растворяют в дихлорметане (15 мл) при 5°C и при охлаждении льдом прикапывают хлорангидрид, растворенный в дихлорметане (20 мл), с такой скоростью, что температура сохраняется между 5°C и 10°C. Затем баню со льдом удаляют, и избыток трифторэтиламина и часть дихлорметана отгоняют под легким вакуумом. Затем смесь 10 ч нагревают до кипения с обратным холодильником. После охлаждения смесь разбавляют дихлорметаном (50 мл) и дважды встряхивают с водным раствором 2N HCl (по 50 мл), объединенные органические фазы промывают водой (100 мл), сушат над Na2SO4 и выпаривают под вакуумом. Получают этиловый эфир (RS)-(2-(2,2,2-трифторэтил)-3-оксо-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил)-уксусной кислоты (3,51 г, 94%) в виде темно-коричневого масла, которое очищают кристаллизацией из н-гептана. Т.пл.: 54,5-55,5°C.
1H-ЯМР (400 МГц, CDCl3): (=1,15 (т, J=7 Гц, 3 H), 2,85 (дд, J1=6 Гц, J2=16 Гц, 1 H), 3,01 (дд, J1=5 Гц, J2=16 Гц, 1 H), 3,83 (м, 1 H), 4,12 (кв, J=7 Гц, 2 H), 4,73 (м, 1 H), 5,17 (т, J=6 Гц, 1 H), 7,80 (м, 2 H), 8,01 (д, J=8 Гц, 1 H) ч./млн.
Элементарный анализ сжиганием: C15H13F6NO3 (369,27): расч. C 48,79, H 3,55, N 3,79; найд. C 48,54, H 3,49, N 3,79.
Пример 5
a) (S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин, соль O,Oґ-дибензоил-L-винной кислоты

(RS)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин (соединение включения с этилацетатом, содержание 87,06% в соответствии с ЯМР, 44 г, 100 ммоль) и O,Oґ-дибензоил-L-винную кислоту (11,2 г, 31 ммоль) используют в виде твердых веществ и при перемешивании прикапывают 2-пропанол (500 мл). При этом твердые вещества растворяются сначала полностью, затем выпадает белый осадок. Через 30 мин смесь нагревают до 70°C. При этом опять возникает почти прозрачный раствор. Его в течение 4 ч охлаждают до комнатной температуры и затем при этой температуре перемешивают в течение ночи. После этого перемешивают еще 4 ч при 10°C и затем отсасывают. Осадок дважды промывают 2-пропанолом (по 100 мл) и сушат на воздухе. Получают 28,05 г (S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин, соль O,Oґ-дибензоил-L-винной кислоты (74% выход в расчете на (S)-энантиомер), чистота энантиомера 82% в соответствии с ВЭЖХ (Chiracel OD/21, 250 (4,6 мм, н-гептан/этанол/метанол 50:5:2, 1 мл/мин, 30°C) в виде бесцветных кристаллов. Берут 20 г (14,6 ммоль) этих кристаллов и прикапывают 2-пропанол (400 мл). Смесь при перемешивании нагревают до 80°C и затем оставляют медленно охлаждаться до комнатной температуры. Еще 2 часа перемешивают при этой температуре и затем отсасывают, осадок дважды промывают 2-пропанолом (по 50 мл) и сушат на воздухе. Получают 16,3 г (100% выход в расчете на (S)-энантиомер) (S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин, соль O,Oґ-дибензоил-L-винной кислоты в виде бесцветных кристаллов, т.пл.: 192-193°C, чистота энантиомера>97% в соответствии с ВЭЖХ (условия см. выше).
Элементарный анализ сжиганием: C14H12F6N4O2 (Ѕ C18H14O8 (561,43): расч. C 49,21, H 3,41, N 9,98; найд. C 49,17, H 3,30, N 9,97.
b) (S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин

(S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин, соль O,Oґ-дибензоил-L-винной кислоты (113 мг, 0,20 ммоль) растворяют в смеси из воды (1 мл) и этилацетата (5 мл) и добавляют раствор NaHCO3 (50 мг) в воде (7,5 мл). Смесь перемешивают 16 ч при комнатной температуре и затем трижды экстрагируют этилацетатом (по 5 мл). Объединенные органические фазы один раз встряхивают с раствором NaHCO3 (50 мг) в воде (20 мл) и затем с чистой водой (20 мл), сушат Na2SO4 и выпаривают в вакууме. Получают 75 мг (97%) (S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидина. Продукт кристаллизуется из 2-пропанола в виде соединения включения с 0,5 эквивалентами 2-пропанола, т.пл.: 80-82°C. Вещество также может быть перекристаллизовано из этилацетата и кристаллизовано с 0,5 эквивалентами этилацетата, т.пл.: 121,5-122°C.
Чистота энантиомера >97% в соответствии с ВЭЖХ (Chiracel OD/21, 250 (4,6 мм, н-гептан/2-пропанол 4:1, 1 мл/мин, 30°C).
1H-ЯМР (400 МГц, CDCl3): (=2,54 (дд, J1=8 Гц, J2=16 Гц, 1 H), 3,09 (дд, J1=4 Гц, J2=16 Гц, 1 H), 4,25 (м, 1 H), 4,64 (м, 1 H), 5,18 (м, 1 H), 6,65 (ш.с., 2 H), 7,75 (ш.с., 2 H), 7,88 (д, J=8 Гц, 1 H), 7,95 (д, J=8 Гц, 1 H), 8,02 (с, 1 H) ч./млн.
Элементарный анализ сжиганием (Соединение включения с 0,5 эквивалентами 2-пропанола): C14H12F6N4O2 (Ѕ C3H8O (412,32): расч. C 45,15, H 3,91, N 13,59; найд. C 45,23, H 4,27, N 13,10.
Пример 6
(RS)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин посредством рацемирования (R)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидина

(R)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин (соединение включения с 0,5 эквивалентами 2-пропанола (M=412,3), 43 г, 104 ммоль, полученный путем концентрирования маточного раствора, который образуется при осаждении (S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин, соли O,Oґ-дибензоил-L-винной кислоты в примере 5a), и обработки с NaHCO3 как описано в примере 5b) растворяют в 2-пропаноле (1,8 л) и при комнатной температуре при перемешивании добавляют раствор KOH (85%, 660 мг, 10 ммоль) в 2-пропаноле (400 мл). Смесь перемешивают 24 ч при комнатной температуре и затем подкисляют ледяной уксусной кислотой (720 мг, 1,5 мл), выпаривают под вакуумом при макс.температуре в бане 40°C и остаток распределяют между водой (500 мл) и этилацетатом (400 мл). Водную фазу дважды встряхивают с этилацетатом (по 300 мл). Объединенные органические фазы встряхивают с раствором NaHCO3 (10 г) в воде (500 мл) и затем еще раз с чистой водой. Органическую фазу сушат с Na2SO4 и выпаривают под вакуумом при макс.температуре бани 40°C. Получают 39,8 г (RS)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидина, содержание 87% в соответствии с ЯМР, соединение включения с 0,5 эквивалентами этилацетата, в виде светло-желтых кристаллов, выход 87%. Т.пл.: 164-166°C при медленном нагревании, удаление этилацетата ок. 100°C.
Соотношение энантиомеров 49:51 в соответствии с ВЭЖХ (Chiracel OD/21, 250 (4,6 мм, н-гептан/2-пропанол 4:1, 1 мл/мин, 30°C).
Пример 7
(S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин гидрофумарат гидрат

(S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин (соединение включения с 0,5 эквивалентами 2-пропанола, (M=412,3), 110 г, 266 ммоль) растворяют в диметоксиэтане (2 л) и смешивают с раствором фумаровой кислоты (0,5 M в смеси диметоксиэтан/вода 9:1, 512 мл) и образованный прозрачный раствор выпаривают под вакуумом. Остаток смешивают с дихлорметаном (2 л), и смесь вновь выпаривают под вакуумом. Остаток суспендируют в воде (1,5 л), отсасывают при комнатной температуре и в течение ночи сушат на воздухе при комнатной температуре. Получают 125,9 г (95%) (S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил]-ацетил}-гуанидин гидрогенфумарат гидрата в виде бесцветных кристаллов. Т.пл. 210°C.
Определение ингибирования NHE
Концентрацию ингибирования IC50 для подавления NHE-1 определяли следующим образом.
IC50 для подавления NHE-1 определяли анализом FLIPR посредством измерения роста pHi в трансфицированных линиях клеток, которые экспрессируют NHE-1 человека.
Анализ проводили в приборе FLIPR (флуометрический планшетный ридер) с 96-луночным микротитрационным планшетом (с черными стенками) с прозрачным дном. Трансфицированные линии клеток, которые экспрессируют различные подтипы NHE (родительская линия LAP-1 как результат мутагенеза и последующей селекции не характеризуется никакой эндогенной NHE активностью), высевали в предыдущий день с плотностью ~25000 клеток/ на лунку.
Питательная среда трансфицированных клеток (Iscove+10% зародышевой телячьей сыворотки) дополнительно содержит G418 в качестве селекционного антибиотика, чтобы обеспечивать присутствие трансфицированных последовательностей.
Непосредственный анализ начинают с удаления питательной среды и добавления 100 мкл/на лунку нагружаемого буфера (5 мкМ BCECF-AM [2',7'-бис-(карбоксиэтил)-5-(и-6)-карбоксифлуоресцеин, сложный ацетоксиметиловый эфир] в 20 мМ NH4Cl, 115 мМ холинхлорида, 1 мМ MgCl2, 1 мМ CaCl2, 5 мМ KCl, 20 мМ HEPES, 5 мМ глюкозы; pH 7,4 [установлен KOH]). Затем клетки в течение 20 минут инкубируют при 37°C. Эта инкубация приводит к нагружению клеток флуоресцирующим красителем, интенсивность флуоресценции которого зависит от pHi, и NH4Cl, что приводит к слабой щелочности клеток.
Не флуоресцирующий краситель предварительной стадии BCECF-AM в виде сложного эфира является мембранопроницаемым. Внутриклеточно с помощью эстеразы высвобождается непосредственный краситель BCECF, который является не мембранопроницаемым.
После 20-минутной инкубации удаляют нагрузочный буфер, содержащий NH4Cl и свободный BCECF-AM, путем трехкратной промывки в промывателе клеток (Tecan Columbus), каждый раз используя по 400 мкл промывочного буфера (133,8 мМ холинхлорида, 4,7 мМ KCl, 1,25 мМ MgCl2, 1,25 мМ CaCl2, 0,97 мМ K2HPO4, 0,23 мМ KH2PO4, 5 мМ HEPES, 5 мМ Glucose; pH 7,4 [установлен KOH]). Остающийся в лунках остаточный объем составляет 90 мкл (возможно 50-125 мкл). Эта стадия промывки удаляет свободный BCECF-AM и как следствие ведет к удалению внешних ионов NH4+до внутриклеточного подкисления (~ pHi 6,3-6,4).
Так как равновесие внутриклеточных ионов NH4+с NH3 и H+нарушается вследствие удаления внеклеточных ионов NH4+и посредством последующих, мгновенно протекающих переходов NH3 через клеточную мембрану, процесс промывки ведет к тому, что внутриклеточные H+остаются, что является причиной внутриклеточного подкисления. В конечном итоге это может привести к смерти клеток, если они удерживаются достаточно долго. В этом положении было важно, что промывочный буфер не содержал натрия (<1 мМ), так как внеклеточные ионы натрия приводили бы в мгновенному росту pHi посредством активности клонированных изоформ NHE.
Также было важно, что все используемые буферы (нагрузочный буфер, промывочный буфер, восстанавливающий буфер) не содержали никаких ионов HCO3-, так как присутствие бикарбоната привело бы к активации нарушающей, зависимой от бикарбоната системы регуляции pHi, которая содержится в родительской линии клеток LAP-1.
Микротитрационный планшет с подкисленными клетками затем (через 20 минут после подкисления) переносили к FLIPR. В FLIPR внутриклеточный флуоресцентный краситель возбуждали светом с длиной волны 488 нм, который создавали аргонным лазером, и параметры измерения (мощность лазера, выдержка и диафрагма (Blende) CCD-камеры, встроенной в FLIPR) выбирали таким образом, что средний сигнал флуоресценции на лунку имел значение между 30000 и 35000 относительных единиц флуоресценции.
Непосредственное измерение в FLIPR начинают с того, что управляемая программа каждые две секунды делает съемку CCD-камерой. Через десять секунд возбуждали рост внутриклеточного pH's путем добавки 90 мкл восстанавливающего буфера (133,8 мМ NaCl, 4,7 мМ KCl, 1,25 мМ MgCl2, 1,25 мМ CaCl2, 0,97 мМ K2HPO4, 0,23 мМ KH2PO4, 10 мМ HEPES, 5 мМ глюкозы; pH 7,4 [установлен NaOH]) с помощью 96-луночного микротитрационного планшета, встроенного в FLIPR.
В качестве положительного контроля (100% активность NHE) служат лунки, в которые был добавлен чистый восстанавливающий буфер, отрицательного контроля (0% активность NHE) - содержащие промывочный буфер. Во все другие лунки был добавлен восстанавливающий буфер с двукратно концентрированными тестируемыми веществами. Измерения в FLIPR заканчивали через 60 точек измерения (две минуты).
Исходные данные экспортировали в программу ActivityBase. С помощью этой программы сначала рассчитывали активность NHE для каждой тестируемой концентрации вещества и из этого рассчитывали значение IC50 для веществ. Так как ход повышения pHi не был линейным в течение всего эксперимента, а в конце снижался вследствие уменьшающейся активности NHE при повышенных значениях pHi, для оценки измерения было важно выбирать ту часть (кривой), в которой прирост флуоресценции положительного контроля был линеен.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ N-((3-ОКСО-2,3-ДИГИДРО-1H-ИЗОИНДОЛ-1-ИЛ)АЦЕТИЛ)ГУАНИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ NHE-1 ДЛЯ ЛЕЧЕНИЯ ИНФАРКТА И СТЕНОКАРДИИ | 2003 |
|
RU2318809C2 |
| ПЕНТАФТОРСУЛЬФАНИЛ-БЕНЗОИЛГУАНИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ ИЛИ ДИАГНОСТИЧЕСКИХ СРЕДСТВ, А ТАКЖЕ СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2004 |
|
RU2380358C2 |
| ГЕТЕРОЦИКЛИЛОКСИ-БЕНЗОИЛГУАНИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2160732C2 |
| ФТОРСОДЕРЖАЩИЕ БЕНЗОИЛГУАНИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1996 |
|
RU2159230C2 |
| 4-АМИНО-1-ПИПЕРИДИЛБЕНЗОИЛГУАНИДИНЫ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2140412C1 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
| Способ получения производных 1-полифторалкил-1,4-бензодиазепина | 1971 |
|
SU437295A1 |
| ГЕТЕРОЦИКЛИЛ-БЕНЗОИЛ-ГУАНИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2152390C1 |
| ПРОИЗВОДНЫЕ 4-СУЛЬФОНИЛГУАНИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2159232C2 |
Изобретение относится к способу получения соединений формулы I

где R1, R2 независимо друг от друга обозначают Н, F, Cl, OCF3, 2,2,2-трифторэтокси, -CF3, 2,2,2-трифторэтил, С1-С4алкил; R3 означает Alk-R4, -CF3; Alk означает С1-С4 алкил; R4 означает Н, -CF3, С3-С7 циклоалкил; а также их фармацевтически совместимых солей, отличающийся тем, что как определено на схеме 1:
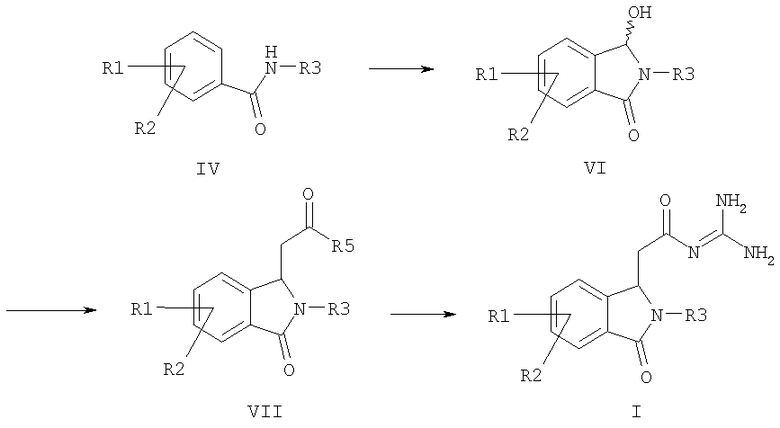
а) формилируют амид формулы IV в орто-положение к амидной группе и затем, без выделения полученного промежуточного соединения, его циклизуют с получением соединения формулы VI, b) соединение формулы VI подвергают превращению с алкоксикарбонилметилентрифенилфосфораном, с 1-алкокси-1-триметилсилоксиэтиленом или с триалкилфосфоноацетатом с получением соединения формулы VII, и с) соединение формулы VII подвергают превращению с гуанидином с получением соединения формулы I, где в соединениях формул IV-VII R1-R3 имеют значения как в формуле I; R5 означает -ОС1-С4алкокси. Описаны промежуточные соединения, способ рацемического разделения. 8 н. и 7 з.п. ф-лы.
1. Способ получения соединений формулы I
причем R1 и R2 независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 С-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 С-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 С-атомами;
а также их фармацевтически совместимых солей,
отличающийся тем, что, как определено на схеме 1
а) формилируют амид формулы IV в ортоположение к амидной группе и затем без выделения полученного промежуточного соединения его циклизуют с получением соединения формулы VI,
b) соединение формулы VI подвергают превращению с алкоксикарбонилметилентрифенилфосфораном, с 1-алкокси-1-триметилсилоксиэтиленом или с триалкилфосфоноацетатом с получением соединения формулы VII и
c) соединение формулы VII подвергают превращению с гуанидином с получением соединения формулы I, причем в соединениях формул IV, VI и VII R1-R3 имеют значения, как в формуле I, и R5 обозначает алкокси с 1, 2, 3 или 4 С-атомами.
2. Способ получения соединений формулы I по п.1, причем R1 и R2 независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 С-атомами;
R3 обозначает Alk-R4 или трифторметил; Alk обозначает алкил с 1, 2, 3 или 4 С-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 С-атомами, а также их фармацевтически совместимых солей, отличающийся тем, что, как определено на схеме 2

a) соединение формулы II подвергают превращению с амином формулы III с получением амида формулы IV,
b) амид формулы IV формилируют в ортоположение к амидной группе с получением формил-амида формулы V, который без выделения подвергают последующей стадии с),
c) формил-амид формулы V циклизуют с получением соединения формулы VI,
d) соединение формулы VI подвергают превращению с алкоксикарбонилметилентрифенилфосфораном, с 1-алкокси-1-триметилсилоксиэтиленом или с триалкилфосфоноацетатом с получением соединения формулы VII и
e) соединение формулы VII подвергают превращению с гуанидином с получением соединения формулы I, причем в соединениях формул II, III, IV, V, VI и VII R1-R3 имеют значения, как в формуле I,
R5 обозначает алкокси с 1, 2, 3 или 4 С-атомами и
Х обозначает Cl, Br, ОН или алкокси с 1, 2, 3 или 4 С-атомами.
3. Способ по п.1 или 2, при котором стадии способа независимо друг от друга проводят непрерывно или периодически.
4. Способ по п.1 или 2, причем соединение формулы I представляет собой N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]-ацетил}-гуанидин, а также его фармацевтически совместимые соли.
5. Способ получения соединений формулы I
причем R1 и R2 независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 С-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 С-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 С-атомами, а также их фармацевтически совместимых солей,
отличающийся тем, что, как это определено на схеме 3

a) амин формулы IX подвергают диазотированию с получением соответствующей соли диазония, которую без выделения подвергают взаимодействию с С1-4 алкиловым эфиром акриловой кислоты с получением производного коричной кислоты формулы XI,
b) соединение формулы XI подвергают превращению с амином формулы III и гуанидином с получением ацилгуанидина формулы I, причем в соединениях формул III, IX и XI
R1-R3 имеют значения, как в формуле I, и
R6 обозначает алкокси с 1, 2, 3 или 4 С-атомами.
6. Способ получения соединений формулы I по п.5, причем R1 и R2 независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2, 2, 2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 С-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 С-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 С-атомами, а также их фармацевтически совместимых солей, отличающийся тем, что, как определено на схеме 4

a) нитросоединение формулы VIII подвергают каталитическому восстановлению до амина формулы IX,
b) амин формулы IX подвергают диазотированию с получением соли диазония формулы X,
c) соль диазония формулы Х без ее выделения подвергают превращению с С1-4 алкиловым эфиром акриловой кислоты с получением производного коричной кислоты формулы XI,
d) соединение формулы XI подвергают амидированию амином формулы III H2NR3 с получением амида формулы XII и
e) соединение формулы XII подвергают превращению с получением ацилгуанидина формулы I, или путем циклизации соединения формулы XII в присутствии сильного основания в производное изоиндолона формулы XIII, которое активируют для получения соответствующего хлорангидрида и затем превращают с гуанидином в изоиндол формулы I (вариант А), или
путем циклизации соединения формулы XII в присутствии сильного основания в производное изоиндола формулы XIII, которое затем превращают в сложный эфир формулы XIV и затем путем взаимодействия с гуанидином превращают в изоиндол формулы I (вариант В), или
путем превращения соединения формулы XII в присутствии сильного основания и спирта, такого, как метанол или этанол, в сложный эфир формулы XIV и затем путем превращения с гуанидином получают изоиндолон формулы I (вариант С), или
путем циклизации соединения формулы XII в присутствии сильного основания при одновременном гуанилировании получают изоиндол формулы I (вариант D),
причем в соединениях формул VIII, IX, X, XI, XII, XIII и XIV
R1-R3 имеют значения, как в формуле I, и
R6 и R7 независимо друг от друга обозначают алкокси с 1, 2, 3 или 4 С-атомами.
7. Способ по п.6, причем на стадии способа е) используют вариант D.
8. Способ по п.6 или 7, при котором стадии способа d) и е) проводят по способу «в одном сосуде».
9. Способ по п.5 или 6, при котором стадии способа независимо друг от друга проводят непрерывно или периодически.
10. Способ по п.5 или 6, причем соединение формулы I представляет собой N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]-ацетил}-гуанидин, а также его фармацевтически совместимые соли.
11. Соединения формулы XII

причем R1 и R2 независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1, 2, 3 или 4 С-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 С-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 С-атомами,
R6 обозначает алкокси с 1, 2, 3 или 4 С-атомами.
12. Соединения формулы XII по п.11 для применения в качестве промежуточного соединения в способе по п.6.
13. Способ выделения соединения формулы Ib
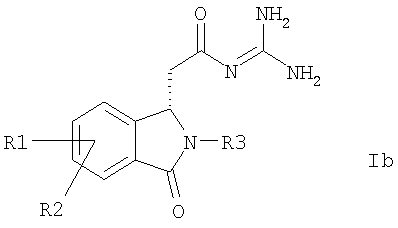
где R1 и R2 независимо друг от друга обозначают водород, F, Cl, трифторметокси, 2,2,2-трифторэтокси, трифторметил, 2,2,2-трифторэтил или алкил с 1,2,3 или 4 С-атомами;
R3 обозначает Alk-R4 или трифторметил;
Alk обозначает алкил с 1, 2, 3 или 4 С-атомами;
R4 обозначает водород, трифторметил или циклоалкил с 3, 4, 5, 6 или 7 С-атомами, а также их солей, отличающийся тем, что
а) рацемат соединения формулы I
подвергают превращению с O,O'-дибензоил-L-винной кислотой формулы R*

с получением соответствующей соли 2,3-O-ацилированной L-винной кислоты формулы XVb


где R8 означает фенил незамещенный или замещенный 1, 2 или 3 заместителями из группы F, Cl, Br, I, алкил с 1, 2, 3 или 4 С-атомами или алкокси с 1, 2, 3 или 4 С-атомами,
и посредством кристаллизации в простом или сложном эфире, галогенированном углеводороде, алифатическом спирте, в воде или смеси растворителей выделяют соль формулы XVb и
b) путем добавления вспомогательного основания и простого или сложного эфира, галогенированного углеводорода, алифатического спирта, или воды, или смеси растворителей выделяют свободное основание формулы Ib, причем в соединениях формул I, XVb R1-R3 имеют значения, как в формуле Ib.
14. Способ по п.13, причем соединение формулы Ib представляет собой (S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]-ацетил}-гуанидин.
15. Соединение формулы XVb
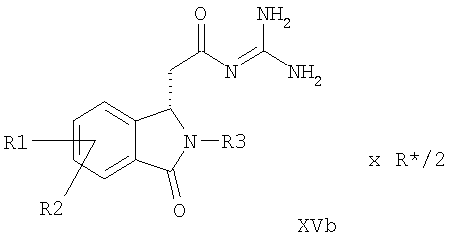
где R1 и R2 независимо друг от друга означают Н, CF3, 2,2,2-трифторэтил;
R3 означает Alk-R4, где Alk означает С1-4алкил; R4 означает CF3; R* означает

где R8 означает фенил.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| 4-НИТРО-3-ТРИФТОРМЕТИЛ-ПЕРФТОРНОНАНОИЛАНИЛИД (ФЛУСТАТ), СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2001 |
|
RU2186057C1 |

