Настоящее изобретение относится к новым соединениям изоиндолона формулы I
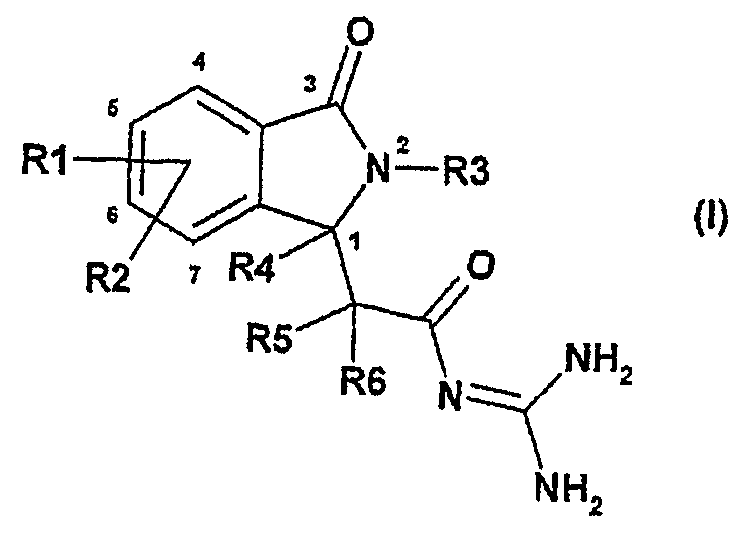
Предложенные соединения являются подходящими в качестве противоаритмических лекарственных средств с кардиозащитным компонентом для профилактики инфаркта и лечения инфаркта и для лечения стенокардии. Они также ингибируют превентивным способом патофизиологические процессы, связанные с развитием нарушений, вызванных ишемией, в частности возникновение вызванной ишемией сердечной аритмии и сердечной недостаточности.
Данное изобретение относится к соединениям формулы I, в которой:
R1 и R2 независимо друг от друга представляют собой водород, алкил, имеющий 1, 2, 3 или 4 атома углерода, алкенил, имеющий 2, 3, 4, 5 или 6 атомов углерода, алкинил, имеющий 2, 3, 4, 5 или 6 атомов углерода, арил, гетероарил, F, Cl, Br, I, NO2, NH2, алкиламино, имеющий 1, 2, 3 или 4 атома углерода, NRaRb, алкилкарбониламино, имеющий 1, 2, 3 или 4 атома углерода, ОН, алкокси, имеющий 1, 2, 3 или 4 атома углерода, S(O)nR7, CO2H, алкоксикарбонил, имеющий 1, 2, 3 или 4 атома углерода, алкилкарбонил, имеющий 1, 2, 3 или 4 атома углерода, CONH2, CONRaRb, CN, полифторалкил, имеющий 1, 2, 3 или 4 атома углерода, полифторалкокси, имеющий 1,2 или 3 атома углерода, или SO3H;
R1 и R2, сами по себе, необязательно замещены линейной или разветвленной алкильной группой, имеющей 1, 2, 3 или 4 атома углерода;
n равно нулю, 1 или 2
R3 представляет собой водород, арил, гетероарил, группу типа Alk-R8 или циклоалкил, имеющий 3, 4, 5, 6, 7 или 8 атомов углерода,
где циклоалкил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из F, Cl, Br или I,
Alk представляет собой алкил, имеющий 1, 2, 3, 4 или 5 атомов углерода в линейной или разветвленной цепи,
R8 представляет собой водород, циклоалкил, имеющий 3, 4, 5, 6, 7 или 8 атомов углерода, полифторалкил, имеющий 1, 2, 3 или 4 атома углерода, арил, гетероарил, ОН, алкокси, имеющий 1, 2, 3 или 4 атома углерода, CO2H, CONH2, CONRaRb, NH2, алкиламино, имеющий 1, 2, 3 или 4 атома углерода или NRaRb;
R4, R5 и R6 независимо друг от друга представляют собой водород или линейный или разветвленный алкил, имеющий 1, 2, 3 или 4 атома углерода;
R7 представляет собой линейный или разветвленный алкил, имеющий 1, 2, 3 или 4 атома углерода;
Ra и Rb независимо друг от друга представляют собой линейный или разветвленный алкил, имеющий 1, 2, 3 или 4 атома углерода, или альтернативно Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членный гетероцикл, необязательно содержащий другой гетероатом, выбранный из O, S или N;
и их рацемическим смесям, энантиомерам и диастереоизомерам и их смесям, их таутомерам и их фармацевтически приемлемым солям.
Предпочтение отдано соединениям формулы I, в которых значения являются следующими:
R1 и R2 независимо друг от друга представляют собой водород, алкил, имеющий 1, 2, 3 или 4 атома углерода, F, Cl, Br, I, NH2, алкиламино, имеющий 1, 2, 3 или 4 атома углерода, NRaRb, алкилкарбониламино, имеющий 1, 2, 3 или 4 атома углерода, ОН, алкокси, имеющий 1, 2, 3 или 4 атома углерода, CO2H, алкоксикарбонил, имеющий 1, 2, 3 или 4 атома углерода, полифторалкил, имеющий 1, 2, 3 или 4 атома углерода, полифторалкокси, имеющий 1,2 или 3 атома углерода, или SO3H,
R1 и R2, сами по себе, необязательно замещены линейной или разветвленной алкильной группой, имеющей 1, 2, 3 или 4 атома углерода;
R3 представляет собой группу типа Alk-R8 или циклоалкил, имеющий 3, 4, 5, 6, 7 или 8 атомов углерода,
где циклоалкил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из F, Cl или Br,
Alk представляет собой алкил, имеющий 1, 2, 3, 4 или 5 атомов углерода в линейной или разветвленной цепи,
R8 представляет собой водород, циклоалкил, имеющий 3, 4, 5, 6, 7 или 8 атомов углерода, полифторалкил, имеющий 1, 2, 3 или 4 атома углерода, арил или гетероарил;
R4, R5 и R6 независимо друг от друга представляют собой водород или линейный или разветвленный алкил, имеющий 1, 2, 3 или 4 атома углерода;
Ra и Rb независимо друг от друга представляют собой линейный или разветвленный алкил, имеющий 1, 2, 3 или 4 атома углерода, или Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членный гетероцикл, необязательно содержащий другой гетероатом, выбранный из O, S и N,
и их рацемическим смесям, энантиомерам и диастереоизомерам и их смесям, их таутомерам и их фармацевтически приемлемым солям.
Особое предпочтение отдано соединениям формулы I, в которых значения являются следующими;
R1 и R2 независимо друг от друга представляют собой водород, алкил, имеющий 1, 2, 3 или 4 атома углерода, F, Cl, Br, I, OH, алкокси, имеющий 1, 2, 3 или 4 атома углерода, полифторалкил, имеющий 1, 2, 3 или 4 атома углерода, или полифторалкокси, имеющий 1,2 или 3 атома углерода, R1 и R2, сами по себе, необязательно замещены линейным или разветвленным алкилом, имеющим 1, 2, 3 или 4 атома углерода;
R3 представляет собой группу типа Alk-R8 или циклоалкил, имеющий 3, 4, 5, 6, 7 или 8 атомов углерода,
где циклоалкил необязательно замещен одним или более заместителями, выбранными из группы, состоящей из F или Cl,
Alk представляет собой алкил, имеющий 1, 2, 3, 4 или 5 атомов углерода в линейной или разветвленной цепи,
R8 представляет собой водород, циклоалкил, имеющий 3, 4, 5, 6, 7 или 8 атомов углерода, или полифторалкил, имеющий 1, 2, 3 или 4 атома углерода;
R4, R5 и R6 независимо друг от друга представляют собой водород или линейный или разветвленный алкил, имеющий 1, 2, 3 или 4 атома углерода;
и их рацемическим смесям, энантиомерам и диастереоизомерам и их смесям, их таутомерам и их фармацевтически приемлемым солям.
В одном варианте соединения формулы I являются такими, как определено выше, и R3 представляют собой атом водорода, арильную или гетероарильную группу или цепь типа Alk-R8, где Alk представляет собой цепь, состоящую из 1-5 атомов углерода в линейной или разветвленной цепи, и R8 представляет собой атом водорода, (С3-С8)циклоалкильную группу, (С1-С4)полифторалкильную группу, арильную группу, гетероарильную группу, гидроксильную группу, (С1-С4)алкоксигруппу, карбоксильную группу, карбоксамидную группу, аминогруппу, (С1-С4)алкиламиногруппу или группу NRaRb.
В одном варианте соединения формулы I являются такими, как определено выше, и R4 представляет собой атом водорода. В другом варианте R5 представляет собой атом водорода.
Отдельное предпочтение отдано соединениям формулы I, отличающимся тем, что они выбраны из группы, включающей:
N-[2-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)-2-метилпропионил]гуанидин,
N-[2-(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)-2-метилпропионил]гуанидин,
N-[(3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-7-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4-амино-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-амино-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-амино-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(7-амино-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4-гидрокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-гидрокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-гидрокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(7-гидрокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4,7-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4,5-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6,7-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4-карбокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-карбокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-карбокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(7-карбокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-1-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
и их рацемические смеси, энантиомеры, диастереоизомеры и их смеси, их таутомеры и их фармацевтически приемлемые соли.
Другое предпочтение отдано соединениям формулы I, отличающимся тем, что они выбраны из группы, включающей:
N-[(2-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-этил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[2-(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин,
N-[(2-изопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[2-(2-бутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин,
N-[(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[2-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин,
N-[(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-бензил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-4-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-5-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-6-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-5-изопропокси-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-хлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-хлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(7-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4,7-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-циклопропилметил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-5-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-6-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(4,4,4-трифторбутил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(4,4,4-трифторбутил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
[1-(2-гуанидино-1-метил-2-оксоэтил)-3-оксо-1,3-дигидроизоиндол-2-ил]уксусная кислота,
N-{2-[3-оксо-2-(2-пирролидин-1-илэтил)-2,3-дигидро-1Н-изоиндол-1-ил]пропионил}гуанидин,
N-[2-(2-гидроксиэтил)-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин,
N-{2-[6-метансульфонил-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
N-[2-(2-циклопропилметил-6-метансульфонил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидиний,
N-{2-[5,6-дифтор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
N-{2-[5,6-дихлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
N-[2-(5,6-дихлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[2-(5,6-дихлор-2-циклопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-{2-[5,6-дихлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
N-{2-[5,6-дифтор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
и их рацемические смеси, энантиомеры и диастереоизомеры, их таутомеры и их фармацевтически приемлемые соли.
Еще одно другое предпочтение отдано соединениям формулы I, отличающимся тем, что они выбраны из группы, включающей:
(R)-N-{2-[6-метансульфонил-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[6-метансульфонил-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-[2-(2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-{2-[5,6-дифтор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[5,6-дифтор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[5,6-дихлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[5,6-дихлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-[2-(5,6-дихлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(5,6-дихлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-[2-(5,6-дихлор-2-циклопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(5,6-дихлор-2-циклопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-{2-[5,6-дихлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[5,6-дихлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-[2-(6-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(6-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-[2-(5-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(5-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-{2-[3-оксо-6-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[3-оксо-6-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[3-оксо-5-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[3-оксо-5-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[6-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[6-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[5-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[5-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-[2-(6-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(6-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-[2-(5-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(5-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
и их фармацевтически приемлемые соли и таутомеры.
Если соединения изобретения содержат один или более асимметрических центров, они независимо друг от друга могут иметь S- и R-конфигурацию. Соединения могут быть в форме оптических изомеров, диастереоизомеров, рацематов или их смесей в любом соотношении.
Настоящее изобретение включает все таутомерные формы соединений формулы I.
Алкильные радикалы могут представлять собой прямую или разветвленную цепь. Указанное относится и к тем случаям, если они имеют заместители или встречаются в качестве заместителей других радикалов, например в алкиламино, алкилкарбониламино, алкокси, алкоксикарбонильном, алкилкарбонильном, полифторалкильном или полифторалкоксирадикалах. Примеры алкильных радикалов включают метил, этил, н-пропил, изопропил (=1-метилэтил), н-бутил, изобутил (=2-метилпропил), втор-бутил (=1-метилпропил), трет-бутил (=1,1-диметилэтил) или пентил. Предпочтительные алкильные радикалы представляют собой метил, этил, н-пропил, изопропил, трет-бутил и изобутил. Один или более, например 1, 2, 3, 4, 5, 6, 7, 8 или 9 атомов водорода в алкильных радикалах могут быть замещены атомами фтора с образованием полифторалкильных радикалов. Примеры таких радикалов включают дифторметил, трифторметил, пентафторэтил, 2,2,2-трифторэтил; 3,3,3-трифторпропил; 3,3,3-трифторбутил, 4,4,4-трифторбутил. Полифторалкоксирадикалы представляют собой алкоксирадикалы, содержащие 1-3 атома углерода, замещенные 1, 2, 3, 4, 5, 6 или 7 атомами фтора, в частности трифторметокси.
Примеры циклоалкильных радикалов включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил. Один или более, например 1 или 2 атома водорода в циклоалкильных радикалах могут быть замещены атомами фтора, хлора, брома или иода, в частности атомами фтора. Замещенные циклоалкильные радикалы могут быть замещены в любых положениях.
Алкенильные радикалы содержат 2, 3, 4, 5 или 6 атомов углерода и 1,2 или 3 сопряженных или несопряженных двойных связей в прямой или разветвленной цепи. Алкинильные радикалы содержат 2, 3, 4, 5 или 6 атомов углерода и 1,2 или 3 сопряженных или несопряженных тройных связей в прямой или разветвленной цепи.
Арильные радикалы выбраны из фенила, 1-нафтила, 2-нафтила и инденила. Замещенные арильные радикалы могут быть замещены в любых положениях.
Гетероарильные радикалы представляют собой моноциклические или бициклические ароматические 3, 4, 5, 6, 7, 8, 9 или 10-членные циклические соединения, в которых 1, 2, 3 или 4 кольцевых атомов представляют собой атомы кислорода, атомы серы или атомы азота, например 1,2 или 3 атома азота, 1 или 2 атома кислорода, 1 или 2 атома серы или комбинацию различных гетероатомов. Гетероарильные радикалы могут быть присоединены через любое положение, например положение 1, положение 2, положение 3, положение 4, положение 5, положение 6, положение 7 или положение 8. Примеры гетероарилов представляют собой фурил, тиенил, пирролил, имидазолил, пиразолил, триазолил, тетразолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиридинил, пиразинил, пиримидинил, пиридазинил, индолил, индазолил, хинолил, изохинолил, фталазинил, хиноксалинил, хиназолинил и циннолинил, в частности тиазолил, тиенил, пирролил, пиридазинил, пиридинил, пиримидинил, фурил, имидазолил, изоксазолил, оксазолил, пиразинил, тетразолил и триазолил. Замещенные гетероарильные радикалы могут быть замещены в любых положениях.
Соединения формулы I ингибируют клеточный антипорт натрий-протон (Na+/H+-обмен, NHE), в частности они ингибируют подтип NHE1. Вследствие ингибирующих свойств в отношении NHE, соединения формулы I и/или их фармацевтически приемлемые соли являются подходящими для профилактики и лечения заболеваний, вызванных активацией NHE или активированным NHE, и заболеваний, вызванных вторично нарушением, связанным с NHE.
Поскольку ингибиторы NHE предпочтительно действуют через их влияние на регуляцию клеточного рН, они могут быть обычно выгодно объединены с другими соединениями, которые регулируют внутриклеточный рН, при этом подходящие для комбинации партнеры представляют собой ингибиторы ферментной группы карбонат-дегидратазы, ингибиторы систем, переносящих бикарбонат-ионы, таких как котранспортер бикарбоната натрия (NBC) или натрийзависимого обмена хлорид-бикарбонат (NCBE), и ингибиторы NHE с ингибирующим действием в отношении других подтипов NHE, потому что через них можно усилить или модулировать фармакологически релевантные эффекты регулирования рН представленных в данном описании ингибиторов NHE.
Применение соединений изобретения относится к профилактике и лечению острых и хронических заболеваний в ветеринарии и медицине, в частности в медицине.
Таким образом, ингибиторы NHE изобретения являются подходящими для лечения заболеваний, вызванных ишемией и реперфузией.
Представленные в данном описании соединения являются подходящими в качестве противоаритмических лекарственных средств вследствие их фармакологических свойств.
Благодаря их кардиозащитному компоненту ингибиторы NHE формулы I и/или их фармацевтически приемлемые соли являются в значительной степени подходящими для профилактики инфаркта и лечения инфаркта и для лечения стенокардии, при этом в указанных случаях они также превентивно ингибируют или в значительной степени ослабляют патофизиологические процессы, связанные с развитием нарушений, вызванных ишемией, в частности возникновение вызванной ишемией сердечной аритмии. Благодаря их защитному действию против патологических гипоксических и ишемических состояний соединения формулы I и/или их фармацевтически приемлемые соли, используемые в соответствии с изобретением, вследствие ингибирования механизма Na+/H+ клеточного обмена, могут быть использованы в качестве лекарственных средств для лечения всех вызванных ишемией острых или хронических нарушений или заболеваний, вызванных первично или вторично.
Данное изобретение также относится к применению указанных соединений в качестве лекарственных средств при хирургических вмешательствах. Поэтому соединения могут быть использованы во время трансплантации органов; при этом соединения могут быть использованы для защиты органов донора перед удалением и во время удаления, для защиты удаленных органов, например во время обработки или их хранения в физиологических растворах и во время переноса в организм реципиента.
Соединения изобретения являются также ценными лекарственными средствами с защитным действием при проведении ангиопластических хирургических операций, например на сердце, а также на периферических органах и сосудах.
Выявлено, что соединения изобретения являются исключительно эффективными лекарственными средствами против аритмии, опасной для жизни. Прекращается фибрилляция желудочков сердца, и восстанавливается физиологический синусовый ритм сердца.
Поскольку ингибиторы NHE1 в тканях и органах человека, в частности сердце, эффективно защищают не только от повреждений, вызванных ишемией и реперфузией, но также и от цитотоксического действия лекарственных препаратов, подобных тем, которые используются, в частности, в терапии рака и терапии аутоиммунных заболеваний, совместное введение с соединениями формулы I и/или их фармацевтически приемлемыми солями является подходящим для ингибирования цитотоксических, в частности кардиотоксических побочных эффектов указанных соединений. Уменьшение цитотоксического действия, в частности кардиотоксичности, в результате комбинированной лекарственной терапии, включающей использование ингибиторов NHE1, дает дополнительную возможность увеличения дозы цитотоксических терапевтических средств и/или пролонгирования лекарственной терапии такими лекарственными средствами. Терапевтическая выгода указанной цитотоксической терапии может быть заметно увеличена за счет комбинации с ингибиторами NHE.
Кроме этого, ингибиторы NHE1 формулы I изобретения и/или их фармацевтически приемлемые соли могут быть использованы в том случае, когда имеется избыточное продуцирование гормонов щитовидной железы, поражающее сердце, тиреотоксикоз или внешний приток гормонов щитовидной железы. Соединения формулы I и/или их фармацевтически приемлемые соли являются, поэтому подходящими для улучшения качества терапии кардиотоксическими лекарственными препаратами.
В соответствии с их защитным действием против повреждения, вызванного ишемией, соединения изобретения являются также подходящими в качестве лекарственных средств для лечения ишемии нервной системы, в частности центральной нервной системы, при этом они являются подходящими, например, для лечения удара или отека мозга.
Соединения формулы I и/или их фармацевтически приемлемые соли являются также подходящими для терапии и профилактики заболеваний и расстройств, вызванных повышенной возбудимостью центральной нервной системы, в частности для лечения эпилептических расстройств, клонических и тонических судорог, состояний психологической депрессии, тревожных расстройств и психоза. В таких случаях раскрытые в данном описании ингибиторы NHE можно использовать в отдельности или в комбинации с другими веществами с антиэпилептической активностью или с противопсихотическими активными ингредиентами или с ингибиторами карбонат-дегидратазы, например с ацетазоламидом, и с другими ингибиторами NHE или натрийзависимого обмена хлорид-бикарбонат (NCBE).
Соединения формулы I в соответствии с изобретением и/или их фармацевтически приемлемые соли дополнительно являются также подходящими для лечения различных типов шока, такого как, например, аллергический, кардиогенный, гиповолемический и бактериальный шок.
Соединения формулы I и/или их фармацевтически приемлемые соли могут быть также использованы для профилактики и лечения тромботических расстройств, потому что они в качестве ингибиторов способны сами ингибировать агрегацию тромбоцитов. Они, кроме того, способны ингибировать или предотвращать встречающееся после ишемии и реперфузии избыточное высвобождение медиаторов воспаления и коагуляции, в частности фактора Виллебранда и белков тромбообразующего селектина. Таким образом можно уменьшить и устранить патогенное действие значимых тромбообразующих факторов. Ингибиторы NHE настоящего изобретения могут быть поэтому объединены с другими антикоагулянтными и/или тромболитическими активными ингредиентами, такими как, например, рекомбинантный или природный активатор плазминогена ткани, стрептокиназа, урокиназа, ацетилсалициловая кислота, антагонисты тромбина, антагонисты фактора Ха, лекарственные средства с фибринолитической активностью, антагонисты тромбоксанового рецептора, ингибиторы фосфодиэстеразы, антагонисты фактора VIIa, клопидогрел, тиклопидин и т.д. В частности, полезным является совместное применение ингибиторов NHE настоящего изобретения с ингибиторами NCBE и/или с ингибиторами карбонат-дегидратазы, такими как, например, ацетазоламид.
Соединения формулы I и/или их фармацевтически приемлемые соли, используемые в соответствии с изобретением, кроме того отличаются сильным ингибирующим действием на пролиферацию клеток, например, пролиферацию фибробласта и пролиферацию клеток гладкой сосудистой мышцы. Соединения формулы I и/или их фармацевтически приемлемые соли являются поэтому подходящими в качестве ценных лекарственных средств при заболеваниях, в которых пролиферация клеток представляет собой первичную или вторичную причину и, следовательно, могут быть использованы в качестве противоатеросклеротических средств, средств для лечения хронической почечной недостаточности и злокачественных образований.
Было показано, что с помощью ингибиторов NHE подавляется миграция клеток. Соединения формулы I и/или их фармацевтически приемлемые соли являются, поэтому подходящими в качестве ценных лекарственных средств при заболеваниях, в которых миграция клеток представляет собой первичную или вторичную причину, таких как, например, злокачественные образования с явной склонностью к метастазу.
Соединения формулы I и/или их фармацевтически приемлемые соли дополнительно отличаются способностью замедлять или предотвращать фиброзные расстройства. Поэтому они являются подходящими в качестве превосходных средств для лечения фиброза сердца и фиброза легких, фиброза печени, фиброза почек и других фиброзных расстройств. Следовательно, они могут быть использованы для лечения гипертрофии и гиперплазии органов, например сердца и простаты. Поэтому они являются подходящими для профилактики и лечения сердечной недостаточности (застойной сердечной недостаточности=CHF) и для лечения и профилактики гиперплазии простаты или гипертрофии простаты.
Поскольку при гипертензии наблюдается значительное усиление NHE, соединения формулы I и/или их фармацевтически приемлемые соли являются подходящими для профилактики и лечения высокого кровяного давления и сердечно-сосудистых расстройств. В данных случаях они могут быть использованы в отдельности или в подходящей комбинации со средствами и композициями, предназначенными для лечения высокого кровяного давления и сердечно-сосудистых расстройств. Таким образом, они могут быть объединены, например, с одним или несколькими диуретиками с тиазидоподобными действием, диуретиками, угнетающими реабсорбцию натрия и хлоридов не только в проксимальных и дистальных канальцах, но и в петле Генле, альдостероном и антагонистами псевдоальдостерона, такими как гидрохлортиазид, индапамид, политиазид, фуроземид, пиретанид, тораземид, буметанид, амилорид, триамтерен, спиронолактон или эплерон. Ингибиторы NHE настоящего изобретения могут быть, кроме того, использованы в комбинации с блокаторами кальциевого канала, такими как верапамил, дилтиазем, амлодипин или нифедипин, и с ингибиторами АСЕ, такими как, например, рамиприл, эналаприл, лизиноприл, фозиноприл или каптоприл. Дополнительными выгодными партнерами для комбинации являются также бета-блокаторы, такие как метопролол, альбутерол и т.д., антагонисты ангиотензинового рецептора и принадлежащих рецептору подтипов, такие как лозартан, ирбезартан, валзартан; омапатрилат, гемопатрилат, антагонисты эндотелина, ингибиторы ренина, агонисты аденозинового рецептора, ингибиторы и активаторы калиевых каналов, такие как глибенкламид, глимепирид, диазоксид, кромакалим, миноксидил и их производные, активаторы калиевого канала, восприимчивые к митохондральному АТФ (канал митоК (АТФ)), ингибиторы Kv 1.5 и т.д.
Выявлено, что ингибиторы NHE1 формулы I и/или их фармацевтически приемлемые соли оказывают значительное противовоспалительное действие и поэтому могут быть использованы в качестве противовоспалительных лекарственных средств. Ингибирование высвобождения медиаторов воспаления в связи с этим заслуживает особого внимания. Следовательно, соединения могут быть использованы в отдельности или в комбинации с противовоспалительным лекарственным средством для профилактики или лечения хронических и острых воспалительных расстройств. Особо полезными для использования в комбинации являются стероидные и нестероидные противовоспалительные лекарственные средства. Соединения изобретения могут быть также использованы для лечения расстройств, вызванных простейшими, малярии и кокцидиоза у домашней птицы.
Дополнительно было найдено, что соединения формулы I и/или их фармацевтически приемлемые соли показывают благоприятное воздействие на сывороточные липопротеины. Общепризнанно, что слишком высокие уровни жира в крови, называемые гиперлипопротеинемией, представляют собой существенный фактор риска развития атеросклеротических сосудистых поражений, в частности коронарной болезни сердца. Поэтому снижение повышенного уровня сывороточных липопротеинов имеет исключительную важность для профилактики и регрессии атеросклеротических поражений. Кроме снижения общего содержания сывороточного холестерина, в частности, важным является снижение доли в общем содержании холестерина специфических атерогенных липидных фракций, в частности липопротеинов низкой плотности (LDL) и липопротеинов очень низкой плотности (VLDL), потому что указанные липидные фракции представляют собой атерогенный фактор риска. И наоборот, защитная функция против коронарной болезни сердца приписывается липопротеинам высокой плотности. Соответственно, гиполипидемические лекарственные средства должны быть способны снижать не только общее содержание холестерина, но в частности содержание фракций VLDL и LDL в сывороточном холестерине. В настоящее время найдено, что ингибиторы NHE1 показывают ценные терапевтически полезные свойства, влияющие на уровни сывороточных липидов. Поэтому они значительно снижают повышенные концентрации сывороточных LDL и VLDL, которые наблюдаются, например, вследствие повышенного потребления богатых холестерином и липидами продуктов или в случаях патологических метаболических изменений, например генетически родственных гиперлипидемий. Следовательно, они могут быть использованы для профилактики и регрессии атеросклеротических поражений устранением этиологического фактора риска. В данном случае включены не только первичные гиперлипидемии, но также некоторые вторичные гиперлипидемии, встречающиеся, например, вместе с диабетом. Кроме этого, соединения формулы I и/или их фармацевтически приемлемые соли приводят к заметному уменьшению инфарктов, вызванных метаболическими расстройствами, и, в частности, к значительному уменьшению размера вызванного инфаркта и его тяжести. Поэтому указанные соединения полезно использовать для получения лекарственного средства для лечения гиперхолестеринемии; для получения лекарственного средства для профилактики атерогенеза; для получения лекарственного средства для профилактики и лечения атеросклероза, для получения лекарственного средства для профилактики и лечения заболеваний, вызванных повышенными уровнями холестерина, для получения лекарственного средства для профилактики и лечения заболеваний, вызванных эндотелиальной дисфункцией, для получения лекарственного средства для профилактики и лечения вызванной атеросклерозом гипертензии, для получения лекарственного средства для профилактики и лечения вызванных атеросклерозом тромбозов, для получения лекарственного средства для профилактики и лечения вызванного гиперхолестеринемией и вызванного эндотелиальной дисфункцией ишемического поражения и постишемической реперфузии, для получения лекарственного средства для профилактики и лечения вызванных гиперхолестеринемией и вызванных эндотелиальной дисфункцией сердечных гипертрофий и кардиомиопатий и застойной сердечной недостаточности (CHF), для получения лекарственного средства для профилактики и лечения вызванных гиперхолестеринемией и вызванных эндотелиальной дисфункцией сердечных коронарных вазоспазмов и инфарктов миокарда, для получения лекарственного средства для лечения указанных расстройств в комбинациях с гипотензивными средствами, предпочтительно с ингибиторами ангиотензин-конвертирующего фермента (ACE) и антагонистами ангиотензинового рецептора. Доказано, что комбинация ингибитора NHE формулы I и/или его фармацевтически приемлемых солей с активным ингредиентом, понижающим уровни жира в крови, предпочтительно с ингибитором HMG-CoA редуктазы (например, ловастатином или правастатином), последний из которых оказывает почти гиполипидемическое действие и таким образом усиливает гиполипидемические свойства ингибитора NHE формулы I и/или его фармацевтически приемлемых солей, представляет собой полезную комбинацию с усиленным действием и пониженным использованием активного ингредиента.
Таким образом, соединения формулы I и/или их фармацевтически приемлемые соли приводят к эффективной защите против эндотелиального повреждения различного происхождения. Указанная защита сосудов от синдрома эндотелиальной дисфункции означает, что соединения формулы I и/или их фармацевтически приемлемые соли являются ценными лекарственными средствами для профилактики и лечения вазоспазмов коронарных сосудов, периферических сосудистых заболеваний, в частности перемежающейся хромоты, атерогенеза и атеросклероза, гипертрофии левого желудочка сердца и застойной кардиомиопатии и тромботических заболеваний.
Кроме того, было найдено, что соединения формулы I и/или их фармацевтически приемлемые соли являются подходящими при лечении инсулинонезависимого диабета (NIDDM) с ограниченной инсулинорезистентностью. В этой связи может быть полезно повышение антидиабетической активности и качества действия соединений изобретения при объединении их с бигуанидом, таким как метформин, с антидиабетической сульфонилмочевиной, такой как глибурид, глимепирид, толбутамид и т.д., с ингибитором глюкозидазы, с агонистом PPAR, таким как розиглитазон, пиоглитазон и т.д., с инсулиновым продуктом, имеющим различные формы для введения, с ингибитором DB4, с инсулиносенсибилизатором или с меглитинидом.
Кроме неотложных антидиабетических действий соединения формулы I и/или их фармацевтически приемлемые соли противодействуют развитию осложнений диабета и поэтому могут быть использованы в качестве лекарственных средств для профилактики и лечения застойных поражений в результате диабета, таких как диабетическая нефропатия, диабетическая ретинопатия, диабетическая кардиомиопатия, и других расстройств, являющихся следствием диабета. В данной связи, они могут быть с пользой объединены с антидиабетическими лекарственными средствами, указанными выше для лечения NIDDM. В данной связи должна быть, в частности, важна комбинация с оказывающей благоприятное действие дозированной формой инсулина.
Ингибиторы NHE формулы I данного изобретения и/или их фармацевтически приемлемые соли, кроме защитных действий от острых ишемических состояний и от последующей подверженности в равной степени острой реперфузии, показывают также прямые терапевтически применимые действия против заболеваний и расстройств всего организма млекопитающего, которые связаны с проявлениями хронически прогрессирующего процесса старения и которые возникают независимо от состояний острой гипоперфузии и при нормальных неишемических состояниях. Указанные патологические, обусловленные возрастом проявления, индуцируемые в течение длительного периода старения, такие как болезнь, инвалидность и смерть, которые теперь могут быть подвергнуты лечению ингибиторами NHE, представляют собой заболевания и расстройства, которые вызываются по существу связанными с возрастом изменениями жизненных органов и их функции и становятся все более значимыми при старении организма.
Расстройства, связанные с функциональными нарушениями, обусловленными возрастом или с проявлениями изнашивания органов, обусловленного возрастом, представляют собой, например, неадекватную реакцию и реактивность кровеносных сосудов на реакции сокращения и расслабления. Обусловленное возрастом снижение реактивности сосудов на сокращающие и расслабляющие раздражители, которая является важным процессом сердечно-сосудистой системы и, следовательно, для жизни и здоровья, может быть в значительной степени устранено или уменьшено ингибиторами NHE. Одна важная функция и мера поддержания реактивности сосудов представляет собой блокаду или задержку обусловленного возрастом прогрессирования эндотелиальной дисфункции, которое может быть в значительной степени устранено ингибиторами NHE. Соединения формулы I и/или их фармацевтически приемлемые соли являются поэтому весьма подходящими для лечения и профилактики обусловленного возрастом прогрессирования эндотелиальной дисфункции, в частности перемежающейся хромоты.
Примером другой переменной, характеризующей процесс старения, является снижение сократительной способности сердца и снижение адаптации сердца к требуемому минутному сердечному выбросу. Пониженная активность сердца вследствие процесса старения в большинстве случаев связана с дисфункцией сердца, которая вызвана, среди прочего, отложением соединительной ткани в ткани миокарда. Указанное отложение соединительной ткани характеризуется увеличением массы сердца, расширением желудочков сердца и ограниченной сердечной функцией. Неожиданно было обнаружено, что можно почти полностью ингибировать также старение сердечного органа. Поэтому соединения формулы I и/или их фармацевтически приемлемые соли являются в значительной степени подходящими для лечения и профилактики сердечной недостаточности, застойной сердечной недостаточности (CHF).
Хотя в предшествующих патентах и заявках на патент предлагалось лечение различных форм уже появившегося рака, авторами неожиданно было обнаружено, что можно не только лечить уже появившийся рак ингибированием пролиферации клеток, но также предотвратить и значительно задержать заболеваемость раком, обусловленную возрастом, благодаря использованию ингибиторов NHE. Особенно заслуживающее внимания обнаружение состоит в том, что встречающиеся в результате старения нарушения всех органов, и не только некоторые разновидности рака, подавляются или появляются с весьма значительной задержкой. Соединения формулы I и/или их фармацевтически приемлемые соли поэтому являются в значительной степени подходящими для лечения и, в частности, для профилактики обусловленных возрастом разновидностей рака.
В настоящее время авторами найдено, что имеется не только в значительной степени сдвинутая во времени и сверх обычных статистических данных задержка в возникновении обусловленных возрастом нарушений всех исследованных органов, включая сердце, сосуды, печень и т.д., но и весьма значительная задержка в возникновении рака у пожилых людей. И наоборот, неожиданно было также обнаружено увеличение продолжительности жизни до такой степени, которая не была достижима до сих пор никакие другие группы лекарственных средств или природными продуктами. Уникальное действие ингибиторов NHE, кроме использования активных ингредиентов в отдельности для людей и животных, также делает возможным объединение указанных ингибиторов NHE с другими активными началами лекарственных средств, мерами, веществами и природными продуктами, которые применяются в геронтологии и которые основаны на различном механизме действия. Такие классы активных ингредиентов, используемых в геронтологической терапии, представляют собой, в частности, витамины и вещества с антиоксидантной активностью. Поскольку существует корреляция между калорийной нагрузкой или потреблением пищи и процессом старения, может иметь место комбинация с диетическими мерами, например средствами, подавляющими аппетит. Подобно можно рассмотреть комбинацию с гипотензивными лекарственными препаратами, например ингибиторами АСЕ, антагонистами ангиотензинового рецептора, диуретиками, антагонистами Са2+ и т.д. или с лекарственными средствами, нормализующими обмен веществ, такими как средства, снижающие уровень холестерина. Соединения формулы I и/или их фармацевтически приемлемые соли являются поэтому в значительной степени подходящими для профилактики обусловленных возрастом изменений тканей и для увеличения продолжительности жизни при одновременном сохранении высокого качества жизни.
Соединения изобретения являются эффективными ингибиторами клеточного антипорта натрий-протон (Na/H обмен), который при большом числе расстройств (эссенциальная гипертензия, атеросклероз, диабет и т.д.) также усиливается в клетках, которые легко поддаются оценкам в таких как, например, эритроциты, тромбоциты или лейкоциты. Соединения в соответствии с изобретением, следовательно, являются подходящими в качестве замечательных и простых научных средств, например при их использовании в качестве диагностических средств для определения и различения разных типов гипертензии, а также атеросклероза, диабета и застойных осложнений в результате диабета, пролиферативных расстройств и т.д.
Настоящее изобретение также относится к способам синтеза производных изоиндолона формулы I
Кроме того, соединения формулы I могут быть в форме таутомеров, рацемических смесей, энантиомеров и диастереоизомеров. Они также образуют часть изобретения.
Соединения формулы I, в которых R4 и R6 представляют собой водород, могут быть получены из фталимидов формулы (I) в соответствии со следующей общей схемой синтеза:
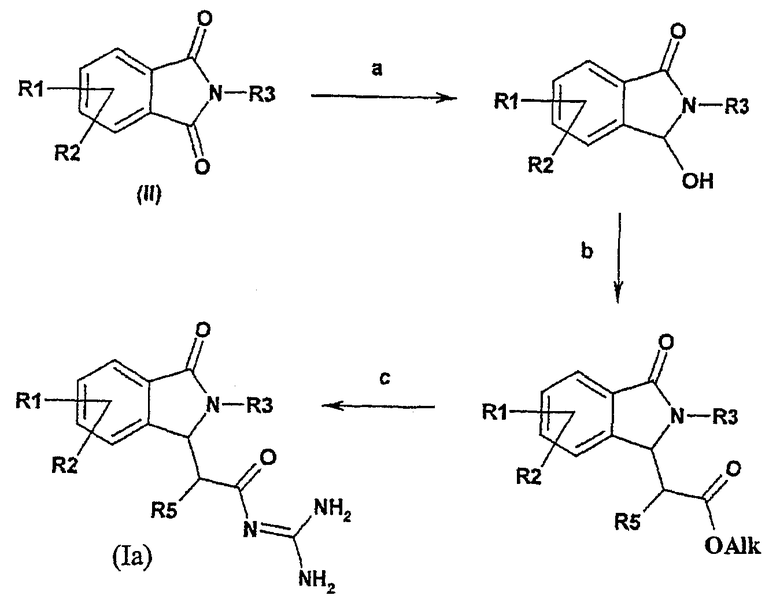
Общая схема синтеза является следующей:
а) комплексный гидрид подвергают взаимодействию с фталимидом (формулы II) в алифатическом спирте,
b) затем полученный продукт подвергают взаимодействию с алкоксикарбонилметилентрифенилфосфораном в толуоле или с триалкилфосфоноацетатом и основанием,
с) полученный продукт подвергают взаимодействию с гуанидинийхлоридом и основанием или с гуанидином, например, в спирте, имеющем 1, 2, 3 или 4 атома углерода.
Реакцию восстановления а) предпочтительно осуществляют с использованием гидрида, такого как боргидрид калия или боргидрид натрия в алифатическом спирте, имеющем 1, 2, 3 или 4 атома углерода, предпочтительно в метаноле, или в тетрагидрофуране, при температуре между 0°С и температурой кипения реакционной смеси.
Реакцию b) обычно осуществляют в присутствии подходящего алкоксикарбонилметилентрифенилфосфорана в растворителе, таком как толуол, при температуре между 20°С и температурой кипения реакционной смеси или в присутствии подходящего триалкилфосфоноацетата и основания, такого как гидрид натрия, в растворителе, таком как 1,2-диметоксиэтан, при температуре между 0°С и температурой кипения реакционной смеси.
Реакцию с) обычно осуществляют в присутствии гидрохлорида гуанидиния и основания, такого как трет-бутоксид калия, в инертном растворителе, таком как диметилформамид, при температуре между 20°С и температурой кипения реакционной смеси или в присутствии гуанидина в растворителе, таком как спирт, имеющий 1, 2, 3 или 4 атома углерода, предпочтительно в изопропаноле, при температуре между 20°С и температурой кипения реакционной смеси.
Альтернативно некоторые соединения формулы I, в которой R4 и R6 представляют собой водород, могут быть получены из альдегидов формулы (III) в соответствии с общей схемой синтеза:
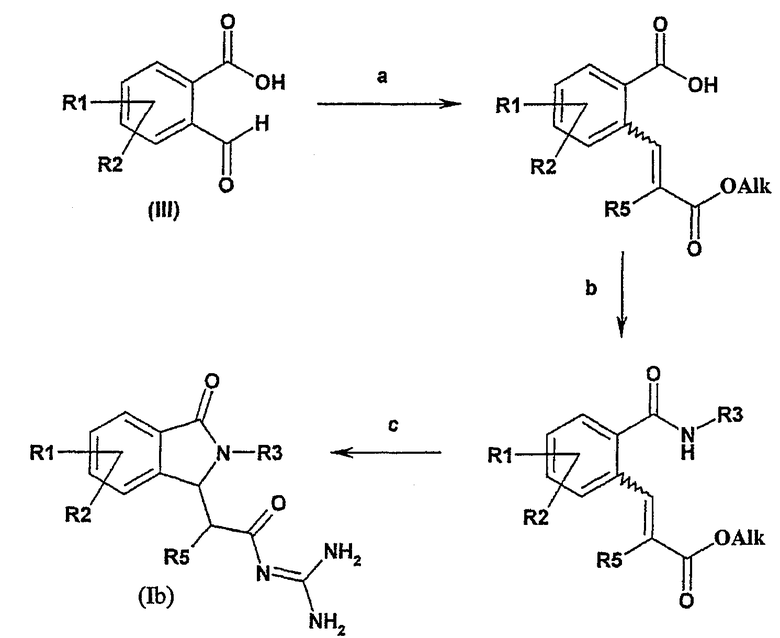
Общая схема синтеза является следующей:
а) соединение формулы III подвергают взаимодействию с алкоксикарбонилметилентрифенилфосфораном в толуоле или с триалкилфосфоноацетатом и основанием,
b) полученный продукт подвергают взаимодействию с амином формулы R3NH2 (R3 имеет такое же значение, как в формуле I) и карбодиимидом,
с) полученный продукт подвергают взаимодействию с гуанидинийхлоридом и основанием, или с гуанидином, например, в спирте, имеющем 1, 2, 3 или 4 атома углерода.
Реакцию а) обычно осуществляют в присутствии подходящего алкоксикарбонилметилентрифенилфосфорана в инертном растворителе, таком как толуол, при температуре между 20°С и температурой кипения реакционной смеси или в присутствии подходящего триалкилфосфоноацетата и основания, такого как гидрид натрия, в растворителе, таком как 1,2-диметоксиэтан, при температуре между 0°С и температурой кипения реакционной смеси.
Реакцию b) осуществляют в присутствии подходящего амина R3NH2. Процесс обычно осуществляют в присутствии реагента сочетания, используемого в химии пептидов, такого как карбодиимид (например, N,N'-дициклогексилкарбодиимид) или N,N'-карбонилдиимидазол в инертном растворителе, таком как простой эфир (например тетрагидрофуран или диоксан), амид (например, диметилформамид) или хлорированный инертный растворитель (например, метиленхлорид, 1,2-дихлорэтан или хлороформ), при температуре между 0°С и температурой кипения реакционной смеси.
Реакцию с) обычно осуществляют в присутствии гидрохлорида гуанидиния и основания, такого как трет-бутоксид калия, в инертном растворителе, таком как диметилформамид, при температуре между 20°С и температурой кипения реакционной смеси или в присутствии гуанидина в растворителе, таком как спирт, имеющий 1, 2, 3 или 4 атома углерода, предпочтительно в изопропаноле, при температуре между 20°С и температурой кипения реакционной смеси.
Соединение формулы I, в котором R4 представляет собой алкильную группу и R6 представляет собой водород, может быть получено из фталимидов формулы (II) в соответствии со следующей общей схемой синтеза:
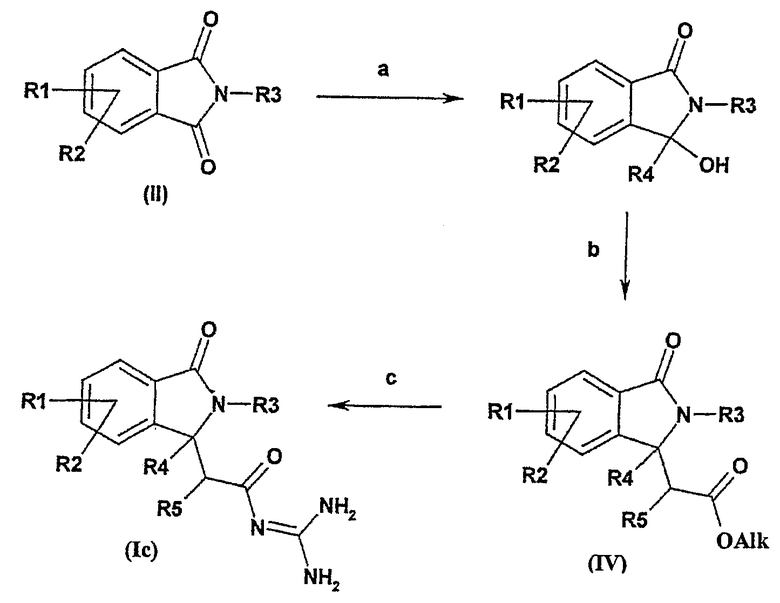
Общая схема синтеза является следующей:
а) фталимид (формулы II) подвергают взаимодействию с галогенидом алкилмагния или с алкиллитиевым реагентом, например в простом эфире,
b) полученный продукт затем подвергают взаимодействию с алкоксикарбонилметилентрифенилфосфораном в толуоле или с 1-этокси-1-триметилсилоксиэтиленом и кислотой Льюиса,
с) полученный продукт подвергают взаимодействию с гуанидинийхлоридом и основанием или с гуанидином, например, в спирте, имеющем 1, 2, 3 или 4 атома углерода.
Реакцию а) предпочтительно осуществляют с использованием галогенида алкилмагния или алкиллитиевого реагента в растворителе, таком как простой эфир, предпочтительно тетрагидрофуран, при температуре между 0°С и температурой кипения реакционной смеси.
Реакцию b) можно осуществлять в присутствии подходящего алкоксикарбонилметилентрифенилфосфорана в растворителе, таком как толуол, при температуре между 20°С и температурой кипения реакционной смеси, или в присутствии 1-этокси-1-триметилсилоксиэтилена и кислоты Льюиса, такой как хлорид титана (IV) или триметилсилилтрифлат, в инертном растворителе, таком как дихлорметан, при температуре между -78°С и 20°С. Получение производных, таких как 1-этокси-1-триметилсилоксиэтен, описано в: Synth. Commun. 1987, 17, 1.
Реакцию с) обычно осуществляют в присутствии гидрохлорида гуанидиния и основания, такого как трет-бутоксид калия, в инертном растворителе, таком как диметилформамид, при температуре между 20°С и температурой кипения реакционной смеси, или в присутствии гуанидина в растворителе, таком как спирт, имеющий 1, 2, 3 или 4 атома углерода, предпочтительно в изопропаноле, при температуре между 20°С и температурой кипения реакционной смеси.
Соединения формулы I, в которых R6 представляет собой алкильную группу, могут быть получены из сложных эфиров формулы (IV) в соответствии со следующей общей схемой синтеза:
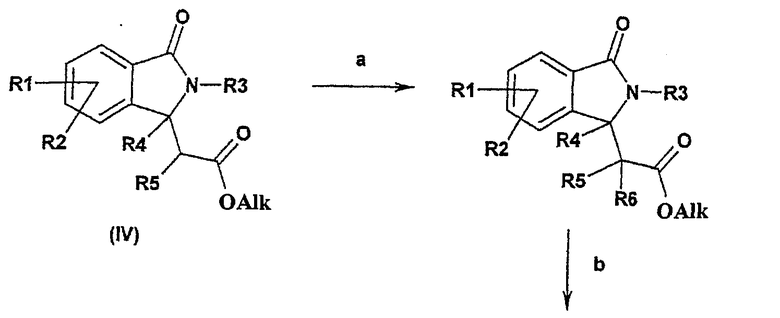
Общая схема синтеза является следующей:
а) соединение формулы IV подвергают взаимодействию в присутствии диизопропиламида лития с R6-Hal, где Hal означает F, Cl, Br или I,
b) полученный продукт подвергают взаимодействию с гуанидинийхлоридом и основанием или с гуанидином, например в спирте, имеющем 1, 2, 3 или 4 атома углерода.
Реакцию а) можно осуществлять в присутствии диизопропиламида лития в инертном растворителе, таком как простой эфир (предпочтительно тетрагидрофуран), и в присутствии подходящего алкилгалогенида R6-Hal при температуре между -78°С и 0°С.
Реакцию b) обычно осуществляют в присутствии гидрохлорида гуанидиния и основания, такого как трет-бутоксид калия, в инертном растворителе, таком как диметилформамид при температуре между 20°С и температурой кипения реакционной смеси, или в присутствии гуанидина в растворителе, таком как спирт, имеющий 1, 2, 3 или 4 атома углерода, предпочтительно в изопропаноле, при температуре между 20°С и температурой кипения реакционной смеси.
Если соединения формулы (II) являются коммерчески недоступными, они могут быть получены, например (путь а) из соответствующих ангидридов формулы (V) в присутствии подходящего амина R3NH2 и кислоты, такой как паратолуолсульфоновая кислота, в растворителе, таком как толуол, при температуре между 20°С и температурой кипения реакционной смеси; или (путь b) по методу Габриэля с использованием в качестве исходных продуктов фталимидов калия формулы (VI) в присутствии соответствующего алкилгалогенида формулы R3Hal и в растворителе, таком как диметилформамид, при температуре между 0°С и температурой кипения реакционной смеси, с применением или адаптацией методики, раскрытой в: Tetrahedron, 1998, 54, 14437.
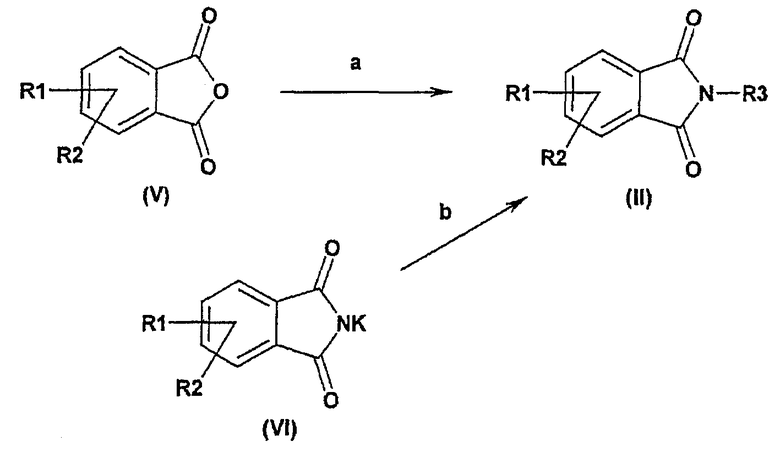
Если соединения формулы (V) являются коммерчески недоступными, они могут быть получены, например, из соответствующих фталевых кислот в ангидриде уксусной кислоты при температуре между 20°С и температурой кипения реакционной смеси.
Если необходимо, для аминной, спиртовой или кислотной функциональной группы используют защитную группу и такие методики удаления защитной группы, которые описаны T.W. Greene, Protective Groups in Organic Synthesis, J.Wiley-Interscience Publication (1991).
Соединения формулы I могут быть необязательно превращены в аддитивные соли неорганической или органической кислоты взаимодействием такой кислоты в растворителе, например, в органическом растворителе, таком как спирт, кетон, простой эфир или хлорированный растворитель. Данные соли также образуют часть изобретения. Примеры фармацевтически приемлемых солей, которые могут быть приведены, включают следующие соли: бензолсульфонат, гидробромид, гидрохлорид, ацетат, цитрат, этансульфонат, фумарат, глюконат, иодат, малеат, изетионат, метансульфонат, метиленбис(β-оксинафтоат), нитрат, оксалат, памоат, фосфат, салицилат, сукцинат, сульфат, тартрат, теофиллинацетат и п-толуолсульфонат.
Если соединения содержат кислотную группу, они способны образовывать соли с основаниями, например, соли щелочного металла, предпочтительно натриевые или калиевые соли или соли аммония, например, соли с аммиаком или органическими аминами или аминокислотами. Они могут также присутствовать в виде цвиттериона.
Список сокращений:
Изобретение иллюстрируют следующие примеры.
Пример 1:
а) N-[2-(2-Метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
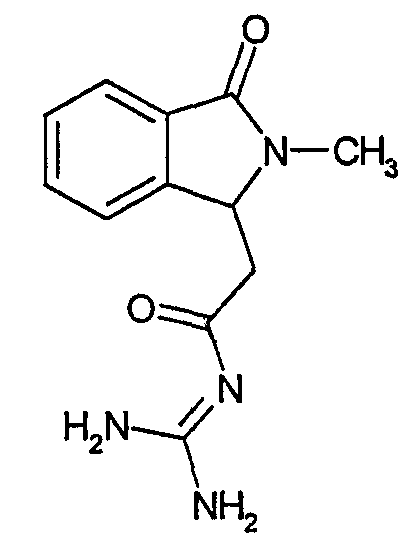
К суспензии 5,2 г трет-бутоксида калия в 100 мл диметилформамида добавляют 4,3 г гуанидинийхлорида. Реакционную смесь перемешивают в инертной атмосфере при температуре примерно 20°С в течение 1 часа и затем добавляют раствор 2 г этил(2-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в 20 мл диметилформамида. Реакционную смесь перемешивают при температуре примерно 20°С в течение 16 часов, затем добавляют 100 мл воды. Доводят рН до 8 добавлением 50 мл 1 н. хлористоводородной кислоты и смесь концентрируют при пониженном давлении (0,6 кПа) при температуре примерно 30°С. Полученный после выпаривания остаток переносят в воду и затем фильтруют. В результате получают 0,35 г N-[(2-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде не совсем белого твердого вещества, плавящегося при 214°С. Масс-спектр Е1: m/e 246 (M+), m/e 159 (основной пик), m/e 146.
b) Этил(2-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
11,5 г этоксикарбонилметилентрифенилфосфорана добавляют к суспензии 4,5 г 3-гидрокси-2-метил-2,3-дигидроизоиндол-1-она в 110 мл толуола. Реакционную смесь кипятят с обратным холодильником при перемешивании в течение 16 часов и затем охлаждают до температуры примерно 20°С. Затем смесь концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Оставшееся масло переносят в 50 мл диэтилового эфира. Образовавшийся остаток отфильтровывают и затем промывают дважды 10 мл диэтилового эфира. Фильтрат концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С с получением окрашенного в оранжевый цвет масла, которое очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 20-45 мкм) при последовательном элюировании смесями циклогексан/этилацетат (70/30, 65/35, 60/40 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют при пониженном давлении (2 кПа) при температуре примерно 30°С. В результате получают 4,1 г этил(2-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде желтого масла. (Rf=0,25, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
с) 3-Гидрокси-2-метил-2,3-дигидроизоиндол-1-он
3,4 г Боргидрида калия медленно добавляют к суспензии 10 г N-метилфталимида в 220 мл метанола в инертной атмосфере. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов и затем по каплям добавляют 200 мл дистиллированной воды. Затем частично (около 120 мл) выпаривают растворитель при пониженном давлении (2 кПа) и температуре около 35°С и остаток разбавляют 400 мл дистиллированной воды. Смесь экстрагируют 400 мл этилацетата. Органическую фазу сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 30°С. В результате получают 4,5 г 3-гидрокси-2-метил-2,3-дигидроизоиндол-1-она в виде белого порошка, плавящегося при 130°С.
Пример 2
а) N-[2-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
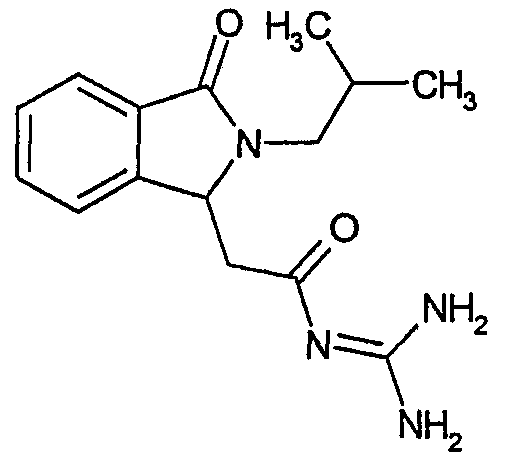
N-[(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 5 г трет-бутоксида калия, 5,2 г гуанидинийхлорида и 2,5 г этил(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 24-х часов и затем фильтруют. Фильтрат переносят в 150 мл воды и 200 мл этилацетата. После разделения фаз осаждением отделяют органическую фазу и водную фазу экстрагируют дважды 200 мл этилацетата. Органические экстракты объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха при пониженном давлении (0,6 кПа) и температуре примерно 45°С. Остаток после выпаривания переносят в диэтиловый эфир, образовавшийся осадок отфильтровывают и затем несколько раз промывают диэтиловым эфиром. Твердое вещество сушат при пониженном давлении (10 Па) и температуре примерно 45°С. В результате получают 1,5 г N-[(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде белого твердого вещества, плавящегося при 250°С. Масс-спектр EI: m/e 288 (M+), m/e 201 (основной пик).
b) Этил(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
К суспензии 1,6 г 60% гидрида натрия в 60 мл 1,2-диметоксиэтана в инертной атмосфере добавляют по каплям 7,7 мл триэтилфосфоноацетата при поддержании температуры ниже 10°С и охлаждают до 0°С при перемешивании. Реакционной смеси дают возможность нагреться до температуры примерно 20°С и затем перемешивают в течение 45 минут. Затем добавляют 5,3 г 3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она, смесь кипятят с обратным холодильником в течение 3,5 часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь обрабатывают 40 мл дистиллированной воды и затем 100 мл диэтилового эфира. После разделения фаз осаждением водную фазу экстрагируют дважды 100 мл диэтилового эфира. Органические экстракты объединяют, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 18°С с получением бледно-желтого масла, которое очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при последовательном элюировании смесями циклогексан/этилацетат (60/40 и затем 50/50 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 30°С. В результате получают 6,3 г этил(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде бледно-желтого масла. (Rf=0,56, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему).
с) 3-Гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он
3-Гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 6,5 г N-изобутилфталимида в 60 мл метанола и 1,7 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям 50 мл дистиллированной воды. Метанол частично выпаривают при пониженном давлении (2 кПа) и температуре примерно 35°С и остаток экстрагируют три раза 60 мл дихлорметана. Органические экстракты объединяют, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 25°С с получением бледно-желтого масла, которое очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 40-63 мкм) при элюировании смесью циклогексан/этилацетат (60/40 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) при температуре около 40°С. В результате получают 5,8 г 3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде белого твердого вещества, плавящегося при 82°С.
d) N-изобутилфталимид
Раствор 3,2 мл изобутиламина в 3 мл толуола при перемешивании добавляют к суспензии 5,2 г ангидрида фталевой кислоты в 50 мл толуола. Реакционную смесь нагревают при температуре примерно 60°С в течение 1 часа и затем при температуре примерно 100°С в течение 2-х часов. Затем в реактор устанавливают аппаратуру Дина-Старка, реакционную смесь нагревают при температуре примерно 130°С в течение 2-х часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в 50 мл насыщенного раствора бикарбоната натрия и экстрагируют дважды 75 мл дихлорметана. Органические экстракты объединяют, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) при температуре примерно 20°С. В результате получают 6,5 г N-изобутилфталимида в виде белого твердого вещества, плавящегося при 92°С.
Пример 3
а) (-)-N-[2-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
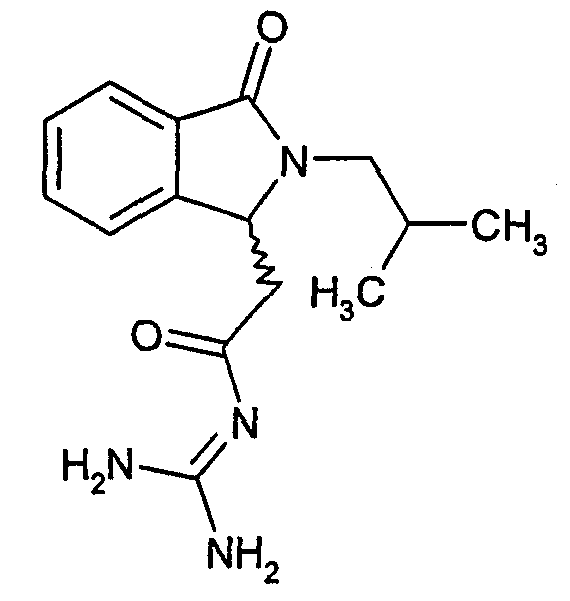
(-)-N-[(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 2,6 г трет-бутоксида калия, 2,6 г гуанидинийхлорида и 1,25 г этил(-)-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 40 часов и затем фильтруют. Фильтрат переносят в 80 мл воды и 120 мл этилацетата. После разделения фаз осаждением отделяют органическую фазу и водную фазу экстрагируют дважды 120 мл этилацетата. Органические экстракты объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха при пониженном давлении (0,6 кПа) и температуре примерно 40°С. Остаток после выпаривания переносят в 30 мл диэтилового эфира, образовавшийся осадок отфильтровывают и затем три раза промывают 5 мл диэтилового эфира. Твердое вещество сушат при пониженном давлении (10 Па) и температуре примерно 45°С. В результате получают 0,75 г (-)-N-[(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде не совсем белого твердого вещества, плавящегося при 264°С (αD 20=-10,2° ± 0,6° в метаноле при 0,5%). Масс-спектр EI: m/e 288 (M+), m/e 245, m/e 201, m/e 132.
b) Этил-(-)-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат и этил-(+)-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил-(-)-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат и этил-(+)-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают разделением 3,0 г этил(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата ВЭЖХ хроматографией на колонке с хиральной стационарной фазой WHELK-01SS с размером частиц 10 мкм при последовательном элюировании смесями гептан/изопропанол (90/10 по объему) и затем гептан/этанол (90/10 и затем 50/50 по объему). Фракции, содержащие первый энантиомер, объединяют и концентрируют при пониженном давлении (1 кПа) и температуре примерно 40°С. Остаток сушат при пониженном давлении (3 кПа) и температуре примерно 40°С. В результате получают 1,3 г этил-(-)-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде вязкого масла, окрашенного в бледный коричневато-желтый цвет (αD 20=-16,2°±0,6° в ДМСО при 0,5%). Фракции, содержащие второй энантиомер, объединяют и концентрируют при пониженном давлении (1 кПа) и температуре примерно 40°С. Остаток сушат при пониженном давлении (3 кПа) и температуре примерно 40°С. В результате получают 1,0 г этил-(+)-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде вязкого бледно-желтого масла (αD 20=-15,1°±0,7° в ДМСО при 0,5%). Этил(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат описан в примере 2.
Пример 4
(+)-N-[2-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
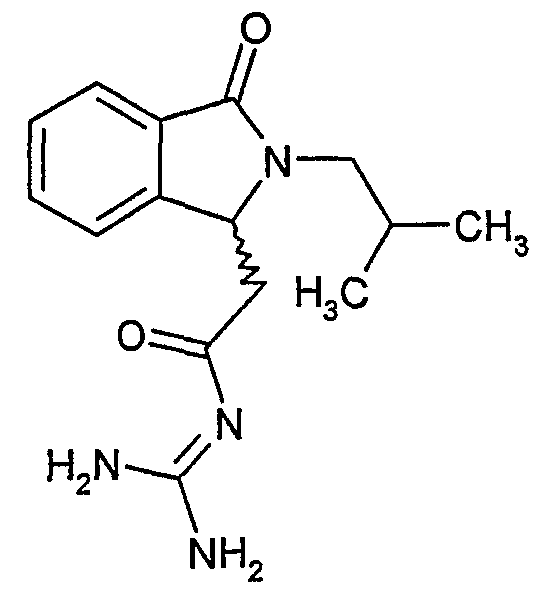
(+)-N-[(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 2,0 г трет-бутоксида калия, 2,1 г гуанидинийхлорида и 1,0 г этил-(+)-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 40 часов и затем фильтруют. Фильтрат переносят в 70 мл воды и 100 мл этилацетата. После разделения фаз осаждением отделяют органическую фазу и водную фазу экстрагируют дважды 100 мл этилацетата. Органические экстракты объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха при пониженном давлении (0,6 кПа) и температуре примерно 40°С. Остаток от выпаривания переносят в 30 мл диэтилового эфира, образовавшийся осадок отфильтровывают и затем промывают три раза 5 мл диэтилового эфира. Твердое вещество сушат при пониженном давлении (10 Па) и температуре примерно 45°С. В результате получают 0,56 г (+)-N-[(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде оранжево-желтого твердого вещества, плавящегося при 264°С (αD 20=+13,9±0,6° в метаноле при 0,5%). Этил-(+)-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат описан в примере 3. Масс-спектр DCI: m/e 289 (M+H)+.
Пример 5
а) N-[2-(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
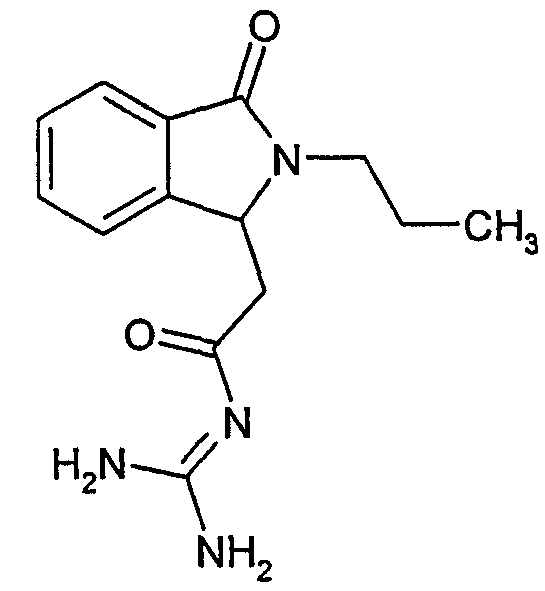
N-[(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 3,9 г трет-бутоксида калия, 3,3 г гуанидинийхлорида и 1,8 г этил(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 1 часа и затем добавляют 60 мл воды. Водную фазу экстрагируют 3 раза 50 мл этилацетата и затем концентрируют досуха при пониженном давлении (0,6 кПа) и температуре примерно 45°С. Остаток переносят в воду, растирают и фильтруют. Твердое вещество переносят в метанол и затем растворитель выпаривают досуха при пониженном давлении (0,6 кПа) и температуре примерно 45°С. В результате получают 0,6 г N-[(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде бледно-желтого пушистого твердого вещества, плавящегося при 229°С. Масс-спектр EI: m/e 274 (M+), m/e 187, m/e 86 (основной пик).
b) Этил(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,8 г 60% гидрида натрия в 20 мл 1,2-диметоксиэтана, 4,0 мл триэтилфосфоноацетата и 1,9 г 3-гидрокси-2-пропил-2,3-дигидроизоиндол-1-она. Неочищенный продукт очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при элюировании смесью циклогексан/этилацетат (50/50 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 30°С. В результате получают 1,9 г этил(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде желтого масла. (Rf=0,7, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (30/70 по объему).
с) 3-Гидрокси-2-пропил-2,3-дигидроизоиндол-1-он
3-Гидрокси-2-пропил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 1,5 г N-пропилфталимида в 25 мл метанола и 0,48 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям дистиллированную воду. Затем частично выпаривают метанол при пониженном давлении (2 кПа) и температуре примерно 35°С и остаток охлаждают до 0°С. Полученный осадок отфильтровывают и затем промывают холодной водой. Твердое вещество переносят в дихлорметан и затем растворитель выпаривают досуха при пониженном давлении (2 кПа) и температуре примерно 35°С. В результате получают 1,0 г 3-гидрокси-2-пропил-2,3-дигидроизоиндол-1-она в виде окрашенного в бежевый цвет порошка. (Rf=0,6, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (30/70 по объему)).
Пример 6
а) N-[2-(2-этил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
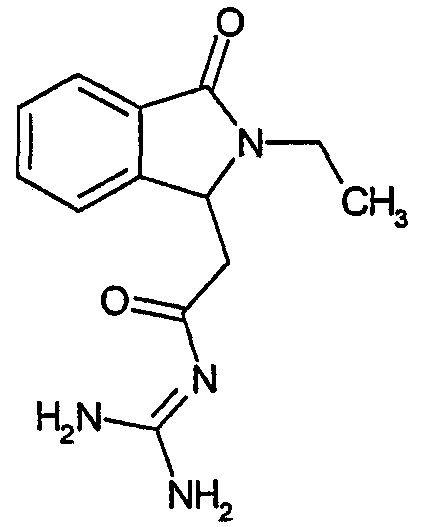
N-[(2-этил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 4,3 г трет-бутоксида калия, 3,7 г гуанидинийхлорида и 1,9 г этил(2-этил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов и затем добавляют 60 мл воды. Водную фазу экстрагируют 3 раза 50 мл этилацетата и затем концентрируют досуха при пониженном давлении (0,6 кПа) и температуре примерно 45°С. Остаток переносят в воду, растирают и фильтруют. Твердое вещество переносят в метанол и затем растворитель выпаривают досуха при пониженном давлении (0,6 кПа) и температуре примерно 45°С. В результате получают 0,56 г N-[(2-этил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде не совсем белого твердого вещества, плавящегося при 223°С. Масс-спектр EI: m/e 260 (M+), m/e 173, m/e 160, m/e 132.
b) Этил(2-этил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(2-этил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,6 г 60% гидрида натрия в 20 мл 1,2-диметоксиэтана, 3,2 мл триэтилфосфоноацетата и 1,8 г 3-гидрокси-2-этил-2,3-дигидроизоиндол-1-она. Неочищенный продукт очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при элюировании смесью циклогексан/этилацетат (50/50 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 30°С. В результате получают 2,0 г этил(2-этил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде бледно-желтого масла. (Rf=0,7, тонкослойная хроматография на силикагеле, элюент: смесь дихлорметан/метанол (90/10 по объему)).
с) 3-Гидрокси-2-этил-2,3-дигидроизоиндол-1-он
3-Гидрокси-2-этил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 4,0 г N-этилфталимида в 20 мл метанола и 1,2 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов, затем охлаждают до температуры примерно 0°С и по каплям добавляют дистиллированную воду. Полученный осадок отфильтровывают и затем промывают холодной водой. Затем из фильтрата частично выпаривают метанол при пониженном давлении (2 кПа) и температуре примерно 35°С и остаток охлаждают до 0°С. Полученный таким образом второй остаток отфильтровывают и затем промывают холодной водой. Две фракции твердого вещества сушат при пониженном давлении (2 кПа) и температуре примерно 35°С. В результате получают 1,9 г 3-гидрокси-2-этил-2,3-дигидроизоиндол-1-она в виде хлопьевидного белого порошка. (Rf=0,5, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (30/70 по объему)).
Пример 7
а) N-[2-(2-изопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
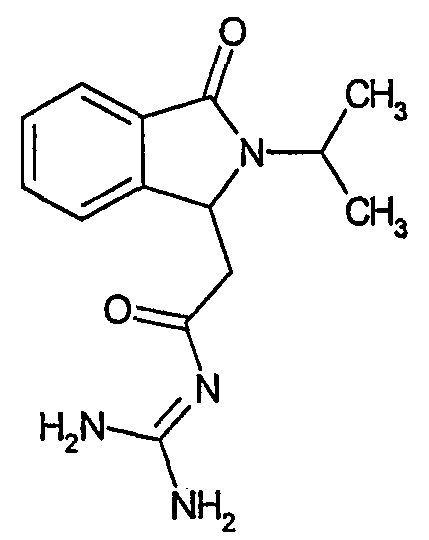
N-[(2-изопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 1,5 г трет-бутоксида калия, 1,3 г гуанидинийхлорида и 0,7 г этил(2-изопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов и затем добавляют 30 мл воды. Водную фазу экстрагируют 3 раза 50 мл этилацетата и затем концентрируют досуха при пониженном давлении (0,6 кПа) и температуре примерно 45°С. Остаток переносят в воду, растирают и фильтруют. Твердое вещество переносят в смесь дихлорметан/метанол (90/10 по объему), фильтруют и фильтрат очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при элюировании смесью дихлорметан/метанол (90/10 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 0,05 г N-[(2-изопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде белого твердого вещества. Масс-спектр EI: m/e 274 (M+), m/e 187, m/e 132. Инфракрасный спектр (KBr): 3412; 1974; 1667; 1603; 1531; 1367 и 698 см-1.
b) Этил(2-изопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(2-изопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,63 г 60% гидрида натрия в 20 мл 1,2-диметоксиэтана, 3,1 мл триэтилфосфоноацетата и 2,0 г 3-гидрокси-2-изопропил-2,3-дигидроизоиндол-1-она. Неочищенный продукт очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при элюировании смесью циклогексан/этилацетат (60/40 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 30°С. В результате получают 0,84 г этил(2-изопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде бледно-желтого масла. (Rf=0,7, тонкослойная хроматография на силикагеле, элюент: смесь дихлорметанол/метанол (90/10 по объему)).
с) 3-Гидрокси-2-изопропил-2,3-дигидроизоиндол-1-он
3-Гидрокси-2-изопропил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 4,0 г N-изопропилфталимида в 20 мл метанола и 1,1 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям дистиллированную воду. Затем растворитель выпаривают досуха при пониженном давлении (2 кПа) и температуре примерно 35°С. В результате получают 6,1 г 3-гидрокси-2-изопропил-2,3-дигидроизоиндол-1-она в виде белого воска. (Rf=0,65, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (30/70 по объему)).
Пример 8
а) N-[2-(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
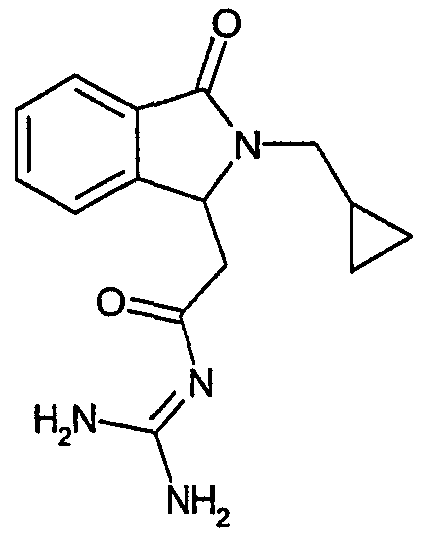
N-[(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 2,9 г трет-бутоксида калия, 2,5 г гуанидинийхлорида и 1,5 г этил(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов и затем добавляют 20 мл воды. Водную фазу экстрагируют 3 раза 100 мл этилацетата. Органические экстракты объединяют и затем концентрируют досуха при пониженном давлении (0,6 кПа) и температуре примерно 35°С. Остаток переносят в воду, растирают, отфильтровывают и затем сушат в эксикаторе. В результате получают 1,2 г N-[(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде не совсем белого порошка, плавящегося при 229°С. Масс-спектр: DCI: m/e 287 (M+H)+.
b) Этил(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,61 г 60% гидрида натрия в 20 мл 1,2-диметоксиэтана, 3,0 мл триэтилфосфоноацетата и 1,55 г 3-гидрокси-2-циклопропилметил-2,3-дигидроизоиндол-1-она. Неочищенный продукт очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при элюировании смесью циклогексан/этилацетат (70/30 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 30°С. В результате получают 1,45 г этил(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде бесцветного масла (Rf=0,52, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат 50/50 по объему)).
с) 3-Гидрокси-2-циклопропилметил-2,3-дигидроизоиндол-1-он
3-Гидрокси-2-циклопропилметил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 4,3 г N-циклопропилметилфталимида в 40 мл метанола и 1,2 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям дистиллированную воду. Полученный остаток отфильтровывают, полученное твердое вещество затем переносят в дихлорметан и концентрируют досуха при пониженном давлении (2 кПа) и температуре около 40°С. В результате получают 4,1 г 3-гидрокси-2-циклопропилметил-2,3-дигидроизоиндол-1-она в виде белого порошка (Rf=0,38, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
d) N-циклопропилметилфталимид
N-циклопропилметилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 4 г ангидрида фталевой кислоты, 2,3 мл циклопропилметиламина и каталитического количества паратолуолсульфоновой кислоты в 40 мл толуола. Реакционную смесь нагревают при температуре примерно 140°С в течение 2-х часов, затем охлаждают до температуры примерно 20°С и перемешивают в течение 16 часов. Реакционную смесь концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в дихлорметан и дважды промывают насыщенным водным раствором бикарбоната натрия. После осаждения фаз отделяют органическую фазу, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 4,3 г N-циклопропилметилфталимида в виде пушистого белого твердого вещества (Rf=0,46, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (40/60 по объему)).
Пример 9
а) N-[2-(2-бензил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
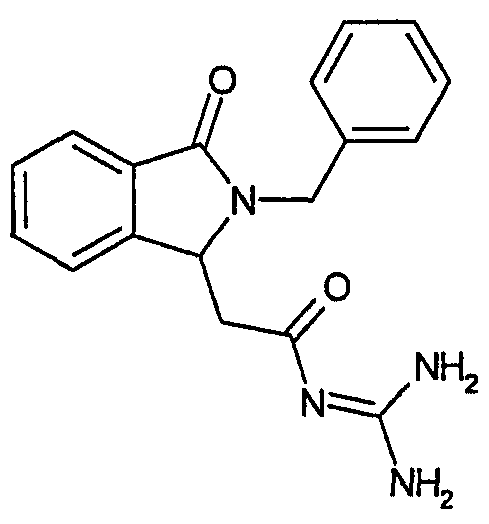
0,22 г натрия добавляют к 20 мл абсолютного этанола в инертной атмосфере. После полного исчезновения натрия добавляют 0,94 г гуанидинийхлорида. Реакционную смесь перемешивают в инертной атмосфере при температуре примерно 20°С в течение 1 часа и затем добавляют раствор 2 г этил(2-бензил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в 5 мл абсолютного этанола. Реакционную смесь перемешивают при температуре примерно 20°С в течение 18 часов и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре около 40°С. Остаток переносят в смесь 10 мл воды и 30 мл диэтилового эфира и затем перемешивают при температуре примерно 0°С в течение 15 минут. Полученный осадок отфильтровывают, промывают дважды 10 мл охлажденной льдом воды и затем сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 1,2 г N-[(2-бензил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде белого порошка, плавящегося при 222-225°С. Инфракрасный спектр (KBr) 3488; 3410; 3344; 1662; 1621; 1521; 1382 и 704 см-1.
b) Этил(2-бензил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(2-бензил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 3,2 г 60% гидрида натрия в 200 мл 1,2-диметоксиэтана, 16,4 мл триэтилфосфоноацетата и 9,5 г 3-гидрокси-2-бензил-2,3-дигидроизоиндол-1-она. Смесь кипятят с обратным холодильником в течение 18 часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь обрабатывают 100 мл воды и затем 100 мл этилацетата. После разделения фаз осаждением водную фазу экстрагируют дважды 100 мл этилацетата. Органические экстракты объединяют, промывают 50 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при последовательном элюировании смесями циклогексан/этилацетат (75/25 и затем 67/33 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 8,7 г этил(2-бензил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде вязкого желтого масла. (Rf=0,35, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
с) 3-Гидрокси-2-бензил-2,3-дигидроизоиндол-1-он
3-Гидрокси-2-бензил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 10,2 г N-бензилфталимида в 100 мл метанола и 2,6 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям 50 мл дистиллированной воды. Затем метанол частично выпаривают при повышенном давлении (2 кПа) и температуре примерно 40°С и дополнительно добавляют 50 мл дистиллированной воды. Водную фазу экстрагируют 3 раза 50 мл этилацетата. Органические экстракты объединяют, промывают 50 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 9,5 г 3-гидрокси-2-бензил-2,3-дигидроизоиндол-1-она в виде белого порошка. (Rf=0,20, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
d) N-бензилфталимид
N-бензилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 10 г ангидрида фталевой кислоты, 7,3 мл бензиламина и каталитического количества паратолуолсульфоновой кислоты в 100 мл толуола. Реакционную смесь нагревают при температуре примерно 140°С в течение 3-х часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь переносят в 100 мл насыщенного водного раствора бикарбоната натрия и водную фазу экстрагируют дважды 100 мл этилацетата. Органические экстракты объединяют, промывают 50 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 10,3 г N-бензилфталимида в виде белого порошка. (Rf=0,44, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
Пример 10
а) N-[2-(6-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
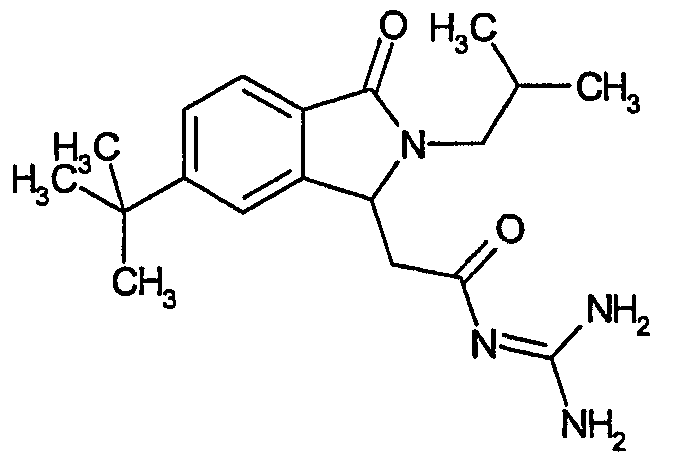
N-[(6-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 9, с использованием в качестве исходных продуктов 20 мл абсолютного этанола, 0,37 г натрия, 1,56 г гуанидинийхлорида и 3,53 г этил(6-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в 10 млабсолютного этанола. Реакционную смесь перемешивают при температуре примерно 20°С в течение 16 часов и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в смесь 15 мл воды и 45 мл диэтилового эфира и затем перемешивают при температуре примерно 0°С в течение 2-х часов. Полученный осадок отфильтровывают, дважды промывают 10 мл охлажденной льдом воды и затем сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 2,5 г N-[(6-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде коричневого порошка, плавящегося при 214-215°С. Масс-спектр: DCI: m/e 345 (M+H)+.
b) Этил(5-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат и этил(6-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(5-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат и этил(6-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 2,9 г 60% гидрида натрия в 200 мл 1,2-диметоксиэтана, 15,2 мл триэтилфосфоноацетата и 9,6 г смеси 5-трет-бутил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она и 6-трет-бутил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она. Смесь кипятят с обратным холодильником в течение 18 часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь обрабатывают 110 мл воды и затем 100 мл этилацетата. После разделения фаз осаждением водную фазу экстрагируют дважды 100 мл этилацетата. Органические экстракты объединяют, промывают 100 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток разделяют ВЭЖХ хроматографией на колонке с хиральной стационарной фазой WHELK-01SS с размером частиц 10 мкм при последовательном элюировании смесями гептан/изопропанол (90/10 и затем 50/50 по объему). Фракции, содержащие первый региоизомер, объединяют и концентрируют при пониженном давлении (1 кПа) и температуре примерно 40°С. Остаток сушат при пониженном давлении (3 кПа) и температуре примерно 40°С. В результате получают 1,81 г этил(5-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде вязкого серого масла (Rf=0,43, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)). Фракции, содержащие второй региоизомер, объединяют и концентрируют при пониженном давлении (1 кПа) и температуре примерно 40°С. Остаток сушат при пониженном давлении (3 кПа) и температуре примерно 40°С. В результате получают 3,53 г этил(6-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде вязкого серого масла. (Rf=0,38, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
с) 5-Трет-бутил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он и 6-трет-бутил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он
5-Трет-бутил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он и 6-трет-бутил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 10,4 г 4-трет-бутил-N-изобутилфталимида в 60 мл метанола и 2,3 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 19 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям 50 мл дистиллированной воды. Затем метанол частично выпаривают при пониженном давлении (2 кПа) и температуре примерно 40°С и дополнительно добавляют 50 мл дистиллированной воды. Водную фазу дважды экстрагируют 100 мл этилацетата. Органические экстракты объединяют, промывают 50 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 9,9 г смеси 5-трет-бутил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она и 6-трет-бутил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде желтой пены. (Rf=0,26 и 0,30, пределы не определены, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
d) 4-Трет-бутил-N-изобутилфталимид
4-Трет-бутил-N-изобутилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 10 г ангидрида 4-трет-бутилфталевой кислоты и 4,9 мл изобутиламида в 100 мл толуола. Реакционную смесь нагревают при температуре примерно 75°С в течение 10 минут, затем добавляют каталитическое количество паратолуолсульфоновой кислоты и смесь нагревают при температуре примерно 140°С в течение 3-х часов. После охлаждения до температуры примерно 60°С, реакционную смесь концентрируют досуха при пониженном давлении (2 кПа). Остаток переносят в смесь 50 мл воды и 30 мл насыщенного водного раствора бикарбоната натрия и водную фазу экстрагируют дважды 200 мл дихлорметана. Органические экстракты объединяют, промывают 50 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 10,4 г 4-трет-бутил-N-изобутилфталимида в виде вязкого желтого масла. (Rf=0,75, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
Пример 11
N-[2-(5-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
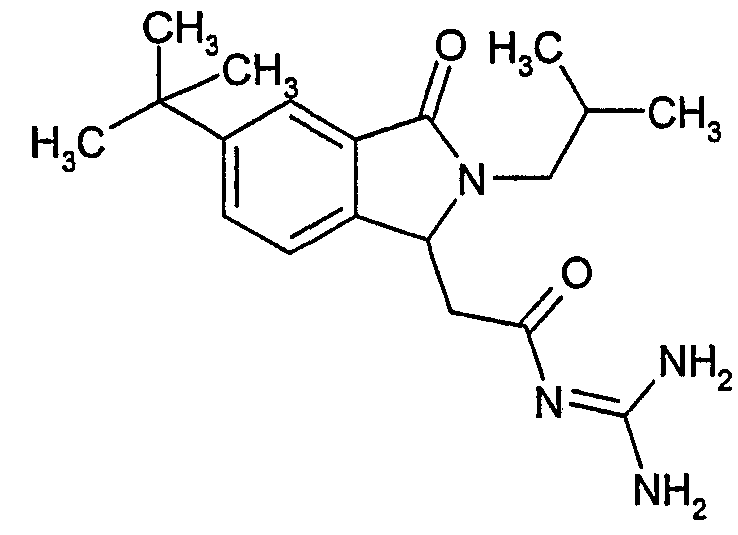
N-[(5-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 9, с использованием в качестве исходных продуктов 10 мл абсолютного этанола, 0,19 г натрия, 0,80 г гуанидинийхлорида и 1,81 г этил(5-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в 10 мл абсолютного этанола. Реакционную смесь перемешивают при температуре примерно 20°С в течение 18 часов и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в смесь 10 мл воды и 30 мл диэтилового эфира и затем перемешивают при температуре примерно 0°С в течение 1 часа. Полученный осадок отфильтровывают, промывают дважды 10 мл охлажденной льдом воды и затем сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 1,1 г N-[(5-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде белого порошка, плавящегося при 262-263°С. Этил(5-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат описан в примере 10. (Анализ: С19Н28N4O2, вычислено, % С: 66,25, Н: 8,19, N:16,27, O: 9,29; найдено, % С: 66,13, Н: 8,50, N:16,12).
Пример 12
а) N-[2-(5-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(6-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
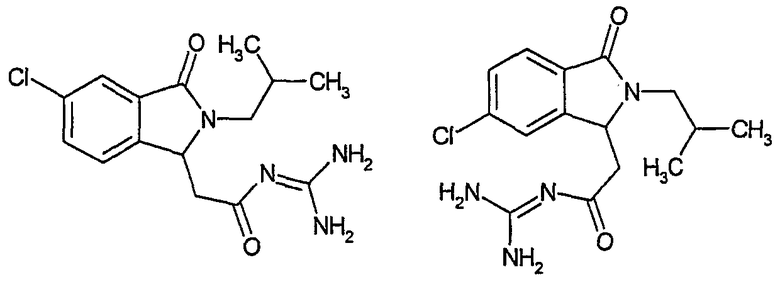
N-[(5-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(6-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 9, с использованием в качестве исходных продуктов 20 мл абсолютного этанола, 0,35 г натрия, 1,46 г гуанидинийхлорида и 3,1 г этил(5-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата и этил(6-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в 15 мл абсолютного этанола. Реакционную смесь перемешивают при температуре примерно 20°С в течение 18 часов и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в смесь 15 мл воды и 45 мл диэтилового эфира и затем перемешивают при температуре примерно 0°С в течение 4-х часов. Полученный осадок отфильтровывают, промывают дважды 20 мл охлажденной льдом воды и затем сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 1 г N-[(5-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина и N-[(6-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде белого порошка. (Анализ: C15H19ClN4O2, вычислено, % C: 55,81, Н: 5,93, Cl: 10,98, N: 17,36, O: 9,91; найдено, % С: 55,67, Н: 6,18, Cl: 11,32, N:17,05). Масс-спектр: DCI: m/e 323 (M+H)+.
b) Этил(5-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат и этил(6-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(5-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат и этил(6-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 4 г 60% гидрида натрия в 200 мл 1,2-диметоксиэтана, 20,3 мл триэтилфосфоноацетата и 12 г 5-хлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она и 6-хлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она. Смесь кипятят с обратным холодильником в течение 18 часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь обрабатывают 200 мл воды и затем 100 мл этилацетата. После разделения фаз осаждением водную фазу экстрагируют дважды 200 мл этилацетата. Органические экстракты объединяют, промывают 100 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 32-63 мкм) при последовательном элюировании смесями циклогексан/этилацетат (90/10 и затем 80/20 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 3,1 г этил(5-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата и этил(6-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде вязкого желтого масла. (Rf=0,34, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
с) 5-Хлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он и 6-хлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он
5-Хлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он и 6-хлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 12 г 4-хлор-N-изобутилфталимида 100 мл метанола и 3 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям 100 мл дистиллированной воды. Затем частично выпаривают метанол при пониженном давлении (2 кПа) и температуре примерно 40°С. Водную фазу экстрагируют дважды 150 мл этилацетата. Органические экстракты объединяют, промывают 50 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 12 г 5-хлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она и 6-хлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде белого порошка. (Rf=0,15, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
d) 4-Хлор-N-изобутилфталимид
4-Хлор-N-изобутилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 10 г ангидрида 4-хлорфталевой кислоты, 5,4 мл изобутиламина и каталитического количества паратолуолсульфоновой кислоты в 100 мл толуола. Реакционную смесь нагревают при температуре примерно 140°С в течение 16 часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь переносят в 100 мл насыщенного водного раствора бикарбоната натрия и водную фазу экстрагируют дважды 100 мл этилацетата. Органические экстракты объединяют, промывают 50 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 12 г 4-хлор-N-изобутилфталимида в виде белого порошка. (Rf=0,85, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
Пример 13
а) N-[2-(5-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(6-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
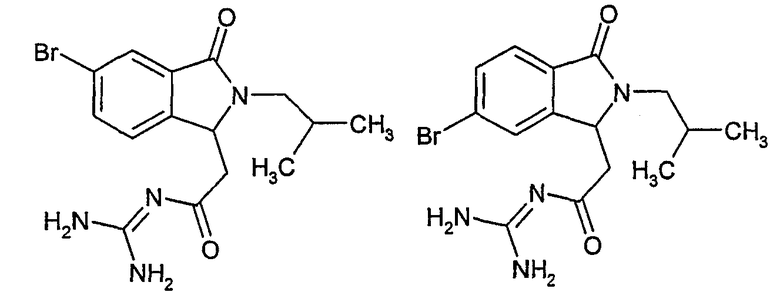
N-[(5-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(6-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 9, с использованием в качестве исходных продуктов 30 мл абсолютного этанола, 0,36 г натрия, 1,5 г гуанидинийхлорида и 3,7 г этил(5-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата и этил(6-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в 20 мл абсолютного этанола. Реакционную смесь перемешивают при температуре примерно 20°С в течение 16 часов и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в смесь 100 мл воды и 200 мл этилацетата. Органическую фазу промывают 100 мл воды и затем 150 мл насыщенного раствора хлорида натрия. Указанную органическую фазу сушат над сульфатом магния и концентрируют при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают белое твердое вещество, которое затем снова переносят в смесь 15 мл воды и 50 мл диэтилового эфира и перемешивают в течение 1,5 часов при температуре примерно 20°С. Полученное таким образом белое твердое вещество отфильтровывают, промывают дважды 50 мл диэтилового эфира и сушат в эксикаторе при пониженном давлении (2 Па) и температуре примерно 35°С. В результате получают 0,92 г N-[(5-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина и N-[(6-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде белого порошка. (Анализ: C15H19BrN4O2, вычислено, % С: 49,06, Н: 5,21, Br: 21,76, N: 15,26, O:8,71; найдено, % С: 48,97, Н: 5,34, Cl: 14,72, N: 21,52). Масс-спектр: EI: m/e 366 (M+), m/e 279, m/e 86 (основной пик).
b) Этил(5-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат и этил(6-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(5-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат и этил(6-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 5,0 г 60% гидрида натрия в 250 мл 1,2-диметоксиэтана, 24,8 мл триэтилфосфоноацетата и 23,7 г 5-бром-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она и 6-бром-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она. Смесь кипятят с обратным холодильником в течение 4-х часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь обрабатывают 300 мл воды и затем смесь экстрагируют 3 раза 250 мл диэтилового эфира. Органические экстракты объединяют, промывают 300 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток очищают хроматографией под давлением аргона (80 кПа) на колонке с силикагелем (размер частиц 32-63 мкм) при последовательном элюировании смесями циклогексан/этилацетат (95/5 по объему), циклогексан/метанол (90/10) и циклогексан/этилацетат (80,20). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре около 40°С. В результате получают 3,72 г этил(5-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата и этил(6-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде бесцветного масла. (Rf=0,70, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
с) 5-Бром-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он и 6-бром-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он
5-Бром-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он и 6-бром-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 23,6 г 4-бром-N-изобутилфталимида в 200 мл метанола и 4,5 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 16 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям 175 мл дистиллированной воды. Затем частично выпаривают метанол при пониженном давлении (2 кПа) и температуре примерно 40°С. Водную фазу экстрагируют 3 раза 200 мл этилацетата. Органические экстракты объединяют, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 23,7 г 5-бром-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она и 6-бром-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде белого порошка. (Rf=0,79, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
d) 4-Бром-N-изобутилфталимид
4-Бром-N-изобутилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 20 г ангидрида 4-бромфталевой кислоты, 9,2 мл изобутиламина и каталитического количества паратолуолсульфоновой кислоты в 200 мл толуола. Реакционную смесь нагревают при температуре примерно 140°С в течение 6 часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С и затем остаток переносят в 350 мл этилацетата и 300 мл насыщенного раствора бикарбоната натрия. После осаждения отделяют водную фазу и затем экстрагируют дважды 350 мл этилацетата. Объединенные органические экстракты промывают 300 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 23,6 г 4-бром-N-изобутилфталимида в виде белого порошка. (Rf=0,91, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
Пример 14
N-[2-(2-изобутил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
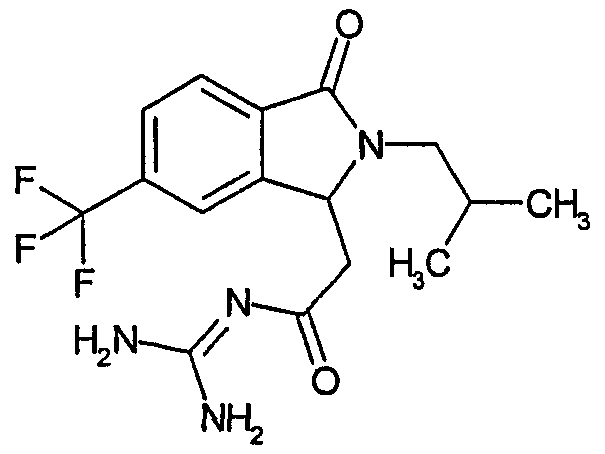
N-[2-(2-изобутил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 9, с использованием в качестве исходных продуктов 10 мл абсолютного этанола, 0,19 г натрия, 0,78 г гуанидинийхлорида и 1,86 г этил(5-трифторметил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в 10 мл абсолютного этанола. Реакционную смесь перемешивают при температуре примерно 20°С в течение 16 часов и затем снова концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Затем остаток переносят в смесь 5 мл воды и 15 мл диэтилового эфира и затем снова концентрируют досуха при таких же условиях. Остаток переносят в 100 мл этилацетата, органическую фазу промывают дважды 60 мл водного 1Н раствора гидроксида натрия и затем дважды 60 мл 4Н раствора хлористоводородной кислоты. Кислые водные экстракты объединяют, обрабатывают водным 30% раствором гидроксида натрия до рН 14 и затем экстрагируют 3 раза 50 мл этилацетата. Органические экстракты объединяют, промывают 100 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Полученное таким образом желтое твердое вещество переносят в 20 мл диэтилового эфира, перемешивают при температуре примерно 20°С в течение 1 часа и затем отфильтровывают. Твердое вещество промывают дважды 20 мл диэтилового эфира и затем сушат в эксикаторе при пониженном давлении (2 Па) и температуре примерно 35°С. В результате получают 0,34 г N-[(5-трифторметил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде желтого порошка, плавящегося при 224°С. (Анализ: С16H19F3N4O2, вычислено, % С: 53,93, Н: 5,37, F: 15,99, N: 15,72, О: 8,98%; найдено, % С: 53,95, Н: 5,15, F: 15,00, N: 15,59).
Пример 15
а) N-[2-(2-изобутил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
N-[2-(2-изобутил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 9, с использованием в качестве исходных продуктов 10 мл абсолютного метанола, 0,13 г натрия, 0,52 г гуанидинийхлорида и 1,25 г этил(6-трифторметил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в 10 мл абсолютного этанола. Реакционную смесь перемешивают при температуре примерно 20°С в течение 16 часов и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Затем остаток переносят в смесь 5 мл воды и 15 мл диэтилового эфира и затем снова концентрируют досуха при таких же условиях. Остаток переносят в 100 мл этилацетата и органическую фазу промывают дважды 50 мл водного 1 н. раствора гидроксида натрия и затем 30 мл водного 4N раствора хлористоводородной кислоты. Кислую водную фазу обрабатывают водным 30% раствором гидроксида натрия до рН 14 и затем экстрагируют 3 раза 50 мл этилацетата. Органические экстракты объединяют и промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Полученное таким образом белое твердое вещество переносят в 20 мл диэтилового эфира, перемешивают при температуре примерно 20°С и затем фильтруют. Твердое вещество сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 0,099 г N-[2-(2-изобутил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде бледно-желтого твердого вещества, плавящегося при 154°С. Масс-спектр: EI: m/e 356 (M+), m/e 269, m/e 200, m/e 86 (основной пик).
b) Этил(5-трифторметил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат и этил(6-трифторметил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(5-трифторметил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат и этил(6-трифторметил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 1,32 г 60% гидрида натрия в 120 мл 1,2-диметоксиэтана, 6,57 мл триэтилфосфоноацетата и 12,07 г 5-трифторметил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она и 6-трифторметил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она. Смесь кипятят с обратным холодильником в течение 24-х часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь затем обрабатывают 150 мл воды и затем смесь экстрагируют 3 раза 250 мл диэтилового эфира. Органические экстракты объединяют, промывают 150 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Смесь 2,3 г остатка впрыскивают на колонку диаметром 8 см, содержащую 1,2 кг хиральной стационарной фазы WHELK 01SS. Осуществляют элюирование подвижной фазой, состоящей из смеси дихлорметана, этанола и гептана, взятых в соотношении 14,5/0,5/85 об./об. В идентичных условиях проводят два дополнительных впрыска 2,7 г и 3 г. Фракции, содержащие первый региоизомер, объединяют и концентрируют досуха при пониженном давлении (1 кПа) и температуре примерно 40°С. В результате получают 1,86 г этил(5-трифторметил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде белого твердого вещества (Rf=0,64, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)). Фракции, содержащие второй региоизомер, объединяют и концентрируют досуха при пониженном давлении (1 кПа) и температуре примерно 40°С с получением 2,79 г белого твердого вещества. Образовавшийся продукт повторно очищают хроматографией под давлением аргона (80 кПа) на колонке с силикагелем (размер частиц 32-63 мкм) при элюировании смесью циклогексан/этилацетат (90/10 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 1,25 г этил(6-трифторметил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде бесцветного вязкого масла. (Rf=0,46, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
с) 5-Трифторметил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он и 6-трифторметил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он
5-Трифторметил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он и 6-трифторметил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 14,9 г 4-трифторметил-N-изобутилфталимида в 300 мл метанола и 2,96 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 16 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям 100 мл дистиллированной воды. Затем частично выпаривают метанол при пониженном давлении (2 кПа) и температуре примерно 40°С. Водную фазу экстрагируют 3 раза 200 мл этилацетата. Органические экстракты объединяют, промывают 300 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 12,07 г 5-трифторметил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она и 6-трифторметил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде белого порошка. (Rf=0,64, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
d) 4-Трифторметил-N-изобутилфталимид
4-Трифторметил-N-изобутилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 14,8 г ангидрида 4-трифторметилфталевой кислоты, 7,2 мл изобутиламина и каталитического количества паратолуолсульфоновой кислоты в 160 мл толуола. Реакционную смесь нагревают при температуре примерно 140°С в течение 5 часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С и остаток переносят в 250 мл этилацетата и 200 мл насыщенного водного раствора бикарбоната натрия. После осаждения отделяют водную фазу и затем экстрагируют дважды 250 мл этилацетата. Органические экстракты объединяют, промывают насыщенном раствором соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 14,9 г 4-трифторметил-N-изобутилфталимида в виде желтого порошка. (Rf=0,59, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
Ангидрид 4-трифторметилфталевой кислоты может быть получен адаптацией или применением методики, описанной Cavalleri et al., J. Med. Chem., 13(1), 148-149, (1970).
Пример 16
а) N-[2-(2-изобутил-5-изопропокси-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
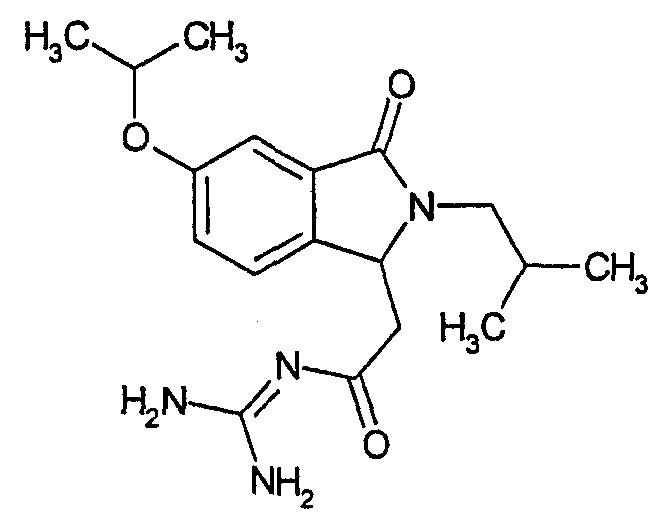
N-[2-(2-изобутил-5-изопропокси-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 9, с использованием в качестве исходных продуктов 20 мл абсолютного этанола, 0,086 г натрия, 0,36 г гуанидинийхлорида и 0,42 г этил(5-изопропилокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 18 часов и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Затем остаток переносят в 50 мл этилацетата и органическую фазу промывают дважды 30 мл водного 1 н. раствора гидроксида натрия и затем дважды 50 мл водного 1 н. раствора хлористоводородной кислоты. Кислую водную фазу обрабатывают водным 30% раствором гидроксида натрия до рН 14 и затем экстрагируют 3 раза 30 мл этилацетата. Органические экстракты объединяют, промывают 50 мл насыщенного водного раствора соли, сушат над сульфатом магния и концентрируют при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток растирают в диэтиловом эфире, затем фильтруют и сушат в эксикаторе при пониженном давлении (2 Па) и температуре примерно 40°С в течение 2-х часов. В результате получают 0,0175 г N-[2-(2-изобутил-5-изопропокси-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде белого порошка, плавящегося при 208°С. 1H ЯМР (300 МГц, d6-(CD3)2SO, δ в м.д.): 0,75 (д, J=6,5 Гц: 3H); 0,90 (д, J=6,5 Гц: 3H); 1,30 (мт: 6H); 2,01 (мт: 1H); 2,40 (дд, J=15 и 6,5 Гц: 1H); 2,65 (дд, J=15 и 6,5 Гц: 1H); 3,00 (дд, J=13,5 и 5,5 Гц: 1H); 3,53 (дд, J=13,5 и 10 Гц: 1H); 4,68 (мт: 1H); 4,96 (широкий т, J=6,5 Гц: 1H); от 6,40 до 7,10 (широкий мультиплет: 2H); 6,98 (дд, J=8,5 и 2 Гц: 1H); 7,09 (д, J=2 Гц: 1H); 7,54 (д, J=8,5 Гц: 1H); от 7,60 до 8,20 (широкий мультиплет: 2H).
b) Этил(5-изопропилокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(5-изопропилокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,149 г 60% гидрида натрия в 20 мл 1,2-диметоксиэтана, 1,23 мл триэтилфосфоноацетата и 1,08 г 5-изопропилокси-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она. Смесь кипятят с обратным холодильником в течение 5 часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь обрабатывают 100 мл воды и затем смесь экстрагируют 3 раза 100 мл диэтилового эфира. Органические экстракты объединяют, промывают 100 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток очищают хроматографией под давлением аргона (80 кПа) на колонке с силикагелем (размер частиц 32-63 мкм) при элюировании смесью циклогексан/этилацетат (90/10 по объему) и затем смесью циклогексан/этилацетат (80/20 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 0,42 г этил(5-изопропилокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде бесцветного масла. (Rf=0,57, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
с) 5-Изопропилокси-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он
5-Изопропилокси-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 1,16 г N-изобутил-4-изопропилоксифталимида в 20 мл метанола и 0,24 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 17 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям 30 мл дистиллированной воды. Затем частично выпаривают метанол при пониженном давлении (2 кПа) и температуре примерно 40°С и добавляют 50 мл воды. Водную фазу экстрагируют 3 раза 70 мл этилацетата. Органические экстракты объединяют, промывают 100 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 1,08 г 5-изопропилокси-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде вязкого бесцветного масла. (Rf=0,50, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
d) N-изобутил-4-изопропилоксифталимид
Смесь 1 г 4-гидрокси-N-изобутилфталимида, 0,85 мл 2-бромпропана и 1,38 г карбоната калия в 5 мл диметилформамида перемешивают при температуре примерно 60°С в течение 17 часов. Затем реакционную смесь охлаждают до температуры примерно 20°С и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в 70 мл воды и затем экстрагируют 3 раза 75 мл этилацетата. Органические фазы объединяют, промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 1,16 г N-изобутил-4-изопропилоксифталимида в виде белого твердого вещества. (Rf=0,50, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
е) 4-Гидрокси-N-изобутилфталимид
4-Гидрокси-N-изобутилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 7,26 г ангидрида 4-ацетоксифталевой кислоты, 7,35 мл изобутиламина и каталитического количества паратолуолсульфоновой кислоты в 75 мл толуола. Реакционную смесь нагревают при температуре примерно 140°С в течение 4-х часов, затем охлаждают до температуры примерно 40°С и концентрируют досуха при пониженном давлении (2 кПа). Остаток переносят в 175 мл этилацетата и 150 мл насыщенного раствора бикарбоната натрия. После осаждения отделяют водную фазу и затем экстрагируют дважды 150 мл этилацетата. Органические экстракты объединяют, промывают 200 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в 100 мл циклогексана, перемешивают при температуре примерно 20°С в течение 1,5 часов и затем фильтруют. Твердое вещество сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 4,0 г 4-гидрокси-N-изобутилфталимида в виде белого твердого вещества. (Rf=0,25, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (75/25 по объему)).
Ангидрид 4-ацетоксифталевой кислоты может быть получен адаптацией или применением методики, описанной J. Sah, J. Med. Chem., 42 (16), 3014-3017, (1999) и N.J. Hinde, J.Chem.Soc., Perkin Trans. 2, 5, 1249-125, (1998).
Пример 17
а) N-[2-(7-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
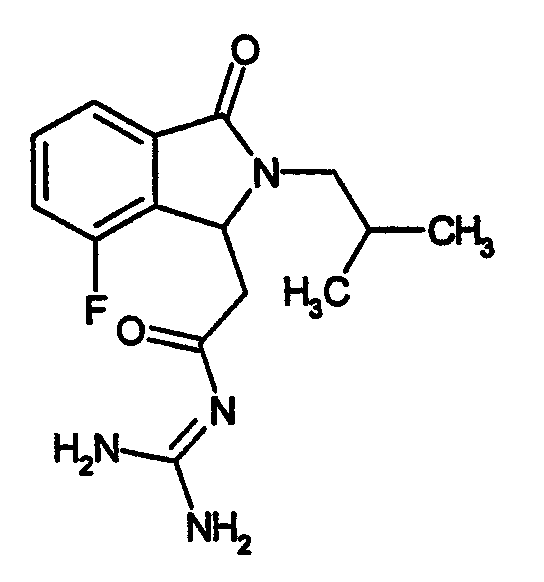
N-[(7-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 1,29 г трет-бутоксида калия, 1,32 г гуанидинийхлорида и 0,67 г этил(7-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов и затем фильтруют. Фильтрат переносят в 40 мл воды и 60 мл этилацетата. После разделения фаз осаждением отделяют органическую фазу и водную фазу экстрагируют дважды 60 мл этилацетата. Органические экстракты объединяют, сушат над сульфатом натрия, фильтруют и концентрируют досуха при пониженном давлении (0,6 кПа) и температуре примерно 55°С. Остаток после выпаривания переносят в диэтиловый эфир, снова концентрируют досуха в таких же условиях, затем переносят в воду и снова концентрируют досуха в таких же условиях. Остаток очищают хроматографией под давлением аргона (50 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при элюировании смесью дихлорметан/метанол (90/10 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 20°С. Остаток переносят в простой диизопропиловый эфир, растирают, фильтруют и затем сушат. В результате получают 0,15 г N-[(7-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде бледно-желтого твердого вещества, плавящегося при 205°С. 1Н ЯМР (300 МГц, d6-(CD3)2SO, δ м.д.): 0,74 (д, J=6,5 Гц: 3Н); 0,89 (д, J=6,5 Гц: 3Н); 2,06 (мт: 1Н); 2,38 (дд, J=15 и 7,5 Гц: 1Н); 2,90 (дд, J=15 и 4,5 Гц: 1Н); 3,04 (дд, J=13,5 и 4,5 Гц: 1Н); 3,56 (дд, J=13,5 и 10 Гц: 1Н); 5,23 (дд, J=7,5 и 4,5 Гц: 1Н); от 6,20 до 7,00 (широкий мультиплет: 2Н); от 7,35 до 7,60 (мт: 3Н); от 7,50 до 8,20 (широкий мультиплет: 2Н).
b) Этил(7-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(7-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,2 г 60% гидрида натрия в 15 мл 1,2-диметоксиэтана, 1,1 мл триэтилфосфоноацетата и 0,8 г 4-фтор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она. Неочищенный продукт очищают хроматографией под давлением аргона (50 кПа) на колонке с силикагелем (размер частиц 40-63 мкм) при элюировании смесью циклогексан/этилацетат (80/20 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 0,44 г этил(7-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде желтого масла. (Rf=0,59, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
с) 4-Фтор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он
4-Фтор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 3,9 г 3-фтор-N-изобутилфталимида в 20 мл метанола и 0,95 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 19 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям дистиллированную воду. Полученный осадок отфильтровывают, промывают холодной водой и затем сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 3,5 г 4-фтор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде липкого белого порошка. (Rf=0,36, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (60/40 по объему)).
d) 3-Фтор-N-изобутилфталимид
3-Фтор-N-изобутилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 3,4 г ангидрида 3-фторфталевой кислоты, 2,0 мл изобутиламина и каталитического количества паратолуолсульфоновой кислоты в 20 мл толуола. Реакционную смесь нагревают при температуре примерно 140°С в течение 3-х часов. После охлаждения до температуры примерно 20°С реакционную смесь концентрируют досуха при пониженном давлении (2 кПа). Остаток переносят в дихлорметан и промывают насыщенным водным раствором бикарбоната натрия. После осаждения отделяют органическую фазу, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 4,0 г 3-фтор-N-изобутилфталимида в виде не совсем белого порошка. (Rf=0,73, тонкослойная хроматография на силикагеле, элюент: дихлорметан).
Пример 18
а) N-[2-(5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
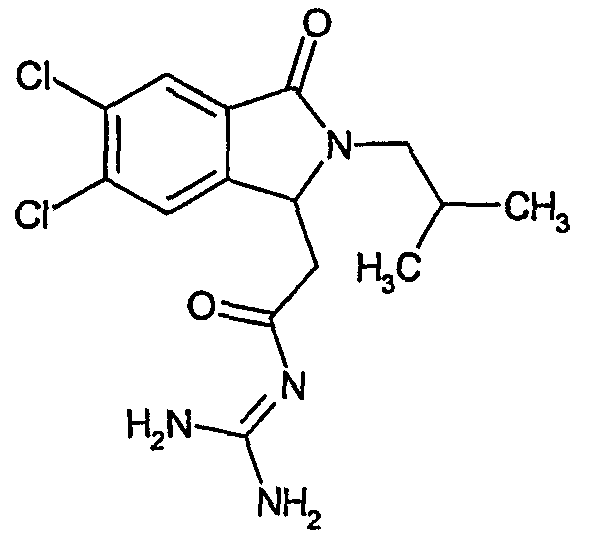
К 4,0 мл абсолютного этанола в инертной атмосфере добавляют 0,04 г натрия. После полного исчезновения натрия добавляют 0,16 г гуанидинийхлорида. Реакционную смесь перемешивают в инертной атмосфере при температуре примерно 20°С в течение 45 минут и затем фильтруют. Фильтрат концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток разбавляют 10 мл тетрагидрофурана и затем добавляют полученный ниже (на стадии b) раствор 5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетилхлорида в 5 мл тетрагидрофурана. Реакционную смесь перемешивают при температуре примерно 20°С в течение 16 часов и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в 50 мл этилацетата и затем промывают дважды 50 мл 1 н. раствора хлористоводородной кислоты. Водные экстракты объединяют, рН доводят до 14 добавлением 30% раствора гидроксида натрия и смесь экстрагируют 3 раза 50 мл этилацетата. Органические экстракты объединяют, промывают 100 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в этилацетат и затем концентрируют досуха при таких же условиях. Остаток переносят в 20 мл диэтилового эфира, растирают и затем фильтруют. Твердое вещество сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 35°С. В результате получают 0,04 г N-[(5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде не совсем белого порошка, плавящегося при 154°С. Масс-спектр: DCI: m/e 357 (M+H)+.
b) 5,6-Дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетилхлорид
К раствору 0,36 г (5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)уксусной кислоты в 10 мл дихлорметана в инертной атмосфере добавляют по каплям 0,72 г оксалилхлорида. Реакционную смесь перемешивают в инертной атмосфере при температуре примерно 20°С в течение 3-х часов, затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают неочищенный 5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетилхлорид в виде вязкого зеленого масла, которое непосредственно используют выше (на стадии а).
с) 5,6-Дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)уксусная кислота
К 5 мл абсолютного этанола в инертной атмосфере добавляют 0,09 г натрия. После полного исчезновения натрия добавляют 0,04 г гуанидинийхлорида. Реакционную смесь перемешивают в инертной атмосфере при температуре примерно 20°С в течение 1,5 часов и затем добавляют раствор 0,93 г этил(5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в 10 мл абсолютного этанола. Реакционную смесь перемешивают при температуре примерно 20°С в течение 18 часов и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в смесь 5 мл воды и 15 мл диэтилового эфира и затем перемешивают при температуре 20°С в течение 1 часа. Смесь концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в смесь дихлорметана и этилацетата и затем фильтруют. Фильтрат очищают хроматографией под давлением аргона (80 кПа) на колонке с силикагелем (размер частиц 32-63 мкм) при элюировании смесью дихлорметан/метанол (95/5 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в диэтиловый эфир, растирают и затем фильтруют. Твердое вещество сушат в эксикаторе при пониженном давлении (2 Па) и температуре примерно 35°С, затем снова растворяют в 70 мл этилацетата и обрабатывают 50 мл водного 1 н. раствора гидроксида натрия. После разделения фаз осаждением органическую фазу промывают 50 мл водного 1 н. раствора гидроксида натрия. Водные экстракты объединяют и затем обрабатывают водным 5 н. раствором хлористоводородной кислоты до рН 1. После трехкратной экстракции 50 мл этилацетата органические экстракты объединяют, промывают 100 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 0,36 г (5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)уксусной кислоты в виде желтого твердого вещества. (Rf=0,33, тонкослойная хроматография на силикагеле, элюент: дихлорметан/метанол (90/10 по объему)).
d) Этил(5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,75 г 60% гидрида натрия в 45 мл 1,2-диметоксиэтана, 3,71 мл триэтилфосфоноацетата и 3,42 г 5,6-дихлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она. Смесь кипятят с обратным холодильником в течение 5,5 часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь обрабатывают 75 мл воды и затем 100 мл диэтилового эфира. После разделения фаз осаждением водную фазу экстрагируют дважды 100 мл диэтилового эфира. Органические экстракты объединяют, промывают 100 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток очищают хроматографией под давлением аргона (80 кПа) на колонке с силикагелем (размер частиц 32-63 мкм) при последовательном элюировании смесями циклогексан/этилацетат (90/10 и затем 80/20 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 0,93 г этил(5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде белого твердого вещества. (Rf=0,74, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
е) 5,6-Дихлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он
5,6-Дихлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 3,78 г 4,5-дихлор-N-изобутилфталимида в 75 мл метанола и 0,75 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 16 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям дистиллированную воду. Затем частично выпаривают метанол при пониженном давлении (2 кПа) и температуре примерно 40°С и затем добавляют 100 мл этилацетата. После разделения фаз осаждением водную фазу экстрагируют дважды 75 мл этилацетата. Органические экстракты объединяют, промывают 150 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 3,42 г 5,6-дихлор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде не совсем белого твердого вещества. (Rf=0,69, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
f) 4,5-Дихлор-N-изобутилфталимид
4,5-Дихлор-N-изобутилфталимид может быть получен, следуя методике примера 2, с использованием в качестве исходных продуктов 10 г ангидрида 4,5-дихлорфталевой кислоты и 4,6 мл изобутиламина в 100 мл толуола. Реакционную смесь нагревают при температуре примерно 140°С в течение 10 минут, затем добавляют каталитическое количество паратолуолсульфоновой кислоты и смесь нагревают при температуре примерно 140°С в течение 4-х часов. После охлаждения до температуры примерно 40°С реакционную смесь концентрируют досуха при пониженном давлении (2 кПа). Остаток переносят в 250 мл насыщенного водного раствора бикарбоната натрия и смесь экстрагируют 3 раза 200 мл этилацетата. Органические экстракты объединяют, промывают 200 мл насыщенного раствора соли, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 2,8 г 4,5-дихлор-N-изобутилфталимида в виде окрашенного в бежевый цвет твердого вещества. (Rf=0,80, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
Пример 19
а) N-[2-(4,7-Дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
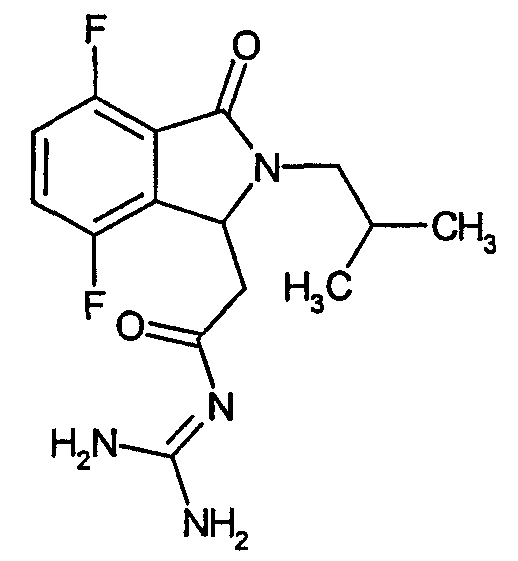
N-[(4,7-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 2,68 г трет-бутоксида калия, 2,74 г гуанидинийхлорида и 1,49 г этил(4,7-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов и затем фильтруют. Фильтрат переносят в 80 мл воды и 120 мл этилацетата. После разделения фаз осаждением отделяют органическую фазу и водную фазу экстрагируют дважды 120 мл этилацетата. Органические экстракты объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха при пониженном давлении (0,6 кПа) и температуре примерно 40°С. Остаток от выпаривания очищают хроматографией под давлением аргона (50 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при последовательном элюировании смесями дихлорметан/метанол (95/5 и затем 90/10 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 35°С. Остаток переносят в диэтиловый эфир, растирают, фильтруют и затем сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 0,16 г N-[(4,7-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде бледно-желтого твердого вещества, плавящегося при 212°С. Масс-спектр: DCI: m/e 325 (M+H)+, m/e 263 (основной пик).
b) Этил(4,7-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(4,7-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,6 г 60% гидрида натрия в 35 мл 1,2-диметоксиэтана, 3,1 мл триэтилфосфоноацетата и 2,5 г 4,7-дифтор-3-гидрокси-2-изобутил-2,3-дигидро-изоиндол-1-она. Неочищенный продукт очищают хроматографией под давлением аргона (60 кПа) на колонке с силикагелем (размер частиц 40-63 мкм) при последовательном элюировании смесями циклогексан/этилацетат (80/20 и затем 70/30 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 1,49 г чистого этил(4,7-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде темно-желтого масла, которое непосредственно используют на следующей стадии. (Rf=0,27, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (70/30 по объему)).
с) 4,7-Дифтор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он
4,7-Дифтор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 6,41 г 3,6-дифтор-N-изобутилфталимида в 70 мл метанола и 1,44 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 3-х часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям 45 мл дистиллированной воды. Затем частично выпаривают метанол при пониженном давлении (2 кПа) и температуре примерно 30°С и затем добавляют 80 мл дихлорметана. После разделения фаз осаждением водную фазу экстрагируют дважды 80 мл дихлорметана. Органические экстракты объединяют, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 30°С. Остаток сушат в эксикаторе при пониженном давлении (2 кПа) и температуре примерно 20°С. В результате получают 5,68 г 4,7-дифтор-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде желтого твердого вещества, плавящегося при 129,5°С. (Rf=0,48, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
d) 3,6-Дифтор-N-изобутилфталимид
3,6-Дифтор-N-изобутилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 5,0 г ангидрида 3,6-дифторфталевой кислоты, 2,7 мл изобутиламина и каталитического количества паратолуолсульфоновой кислоты в 50 мл толуола. Реакционную смесь кипятят в течение 3-х часов. После охлаждения до температуры примерно 20°С реакционную смесь фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа). Остаток переносят в 50 мл насыщенного водного раствора бикарбоната натрия и промывают 3 раза 80 мл дихлорметана. Органические экстракты объединяют, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 6,41 г 3,6-дифтор-N-изобутилфталимида в виде бледно-желтого твердого вещества. (Rf=0,74, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
Пример 20
a) N-[2-(2-изобутил-4-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
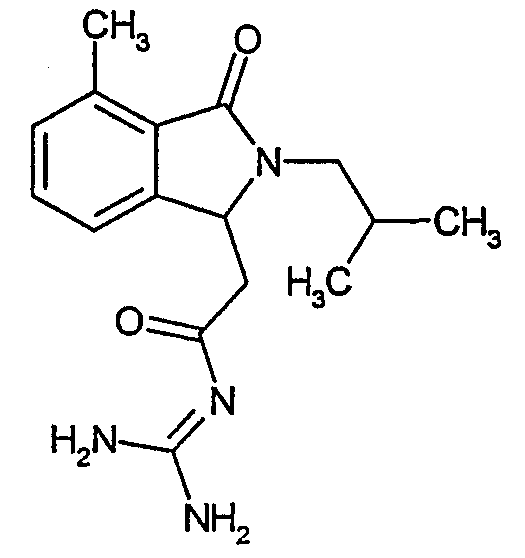
N-[2-(2-изобутил-4-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 0,68 г трет-бутоксида калия, 0,58 г гуанидинийхлорида и 0,36 г этил(4-метил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 20 часов и затем добавляют 20 мл воды. Водную фазу экстрагируют 3 раза 100 мл этилацетата. Органические экстракты объединяют и затем концентрируют досуха при пониженном давлении (0,5 кПа) и температуре примерно 40°С. Остаток переносят в дихлорметан и затем снова концентрируют при таких же условиях. Остаток переносят в воду, растирают и фильтруют и затем переносят в дихлорметан, растирают и фильтруют. Затем твердое вещество сушат в эксикаторе. В результате получают 0,21 г N-[2-(2-изобутил-4-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде белого порошка, плавящегося при 270-272°С (разложение). 1H NMR (300 МГц, d6-(CD3)2SO, δ м.д.): 0,76 (д, J=7 Гц: 3H); 0,91 (д, J=7 Гц: 3H); 2,03 (мт: 1H); 2,42 (дд, J=15,5 и 6,5 Гц: 1H); 2,60 (дд, J=15,5 и 6,5 Гц: 1H); 2,62 (с: 3H); 3,00 (дд, J=14 и 5,5 Гц: 1H); 3,56 (дд, J=14 и 10 Гц: 1H); 4,98 (т, J=6,5 Гц: 1H); от 6,40 до 7,10 (широкий мультиплет: 2H); 7,21 (широкий д, J=7,5 Гц: 1H); 7,36 (широкий д, J=7,5 Гц: 1H); 7,42 (т, J=7,5 Гц: 1H); от 7,50 до 8,30 (широкий мультиплет: 2H).
b) Этил(4-метил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(4-метил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,23 г 60% гидрида натрия в 15 мл 1,2-диметоксиэтана, 1,1 мл триэтилфосфоноацетата и 0,63 г 7-метил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она. Неочищенный продукт очищают хроматографией под давлением аргона (50 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при элюировании смесью циклогексан/этилацетат (70/30 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 0,36 г этил(4-метил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде бесцветного масла.
с) 4-Метил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он и 7-метил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он
4-Метил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он и 7-метил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 5,4 г N-изобутил-3-метилфталимида в 20 мл метанола и 1,4 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 65 часов, затем охлаждают до температуры примерно 0°С и добавляют по каплям 10 мл дистиллированной воды. Смесь концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С, остаток последовательно переносят в диэтиловый эфир и затем в дихлорметан. Полученный осадок отфильтровывают и затем фильтрат концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток очищают хроматографией под давлением аргона (50 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при элюировании смесью циклогексан/этилацетат (70/30 по объему). Фракции, содержащие каждый ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Фракции, содержащие смесь двух ожидаемых продуктов, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток снова очищают хроматографией под давлением аргона (50 кПа) на колонке с силикагелем (размер частиц 15-40 мкм) при элюировании смесью циклогексан/этилацетат (80/20 по объему). Фракции, содержащие каждый ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 0,74 г 7-метил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде белого порошка. (Rf=0,54, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)) и 0,89 г 4-метил-3-гидрокси-2-изобутил-2,3-дигидроизоиндол-1-она в виде пушистого белого твердого вещества. (Rf=0,74, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)).
d) N-изобутил-3-метилфталимид
N-изобутил-3-метилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 5,0 г ангидрида 3-метилфталевой кислоты, 3,0 мл изобутиламина и каталитического количества паратолуолсульфоновой кислоты в 50 мл толуола. Реакционную смесь нагревают при температуре примерно 140°С в течение 2,5 часов и затем перемешивают при температуре примерно 20°С в течение 16 часов. Реакционную смесь концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в дихлорметан и промывают дважды насыщенным водным раствором бикарбоната натрия. После осаждения отделяют органическую фазу, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. В результате получают 6,0 г N-изобутил-3-метилфталимида в виде масла, окрашенного в карамельный цвет, Rf=0,76 (тонкослойная хроматография на силикагеле, элюент: дихлорметан).
Пример 21
а) N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
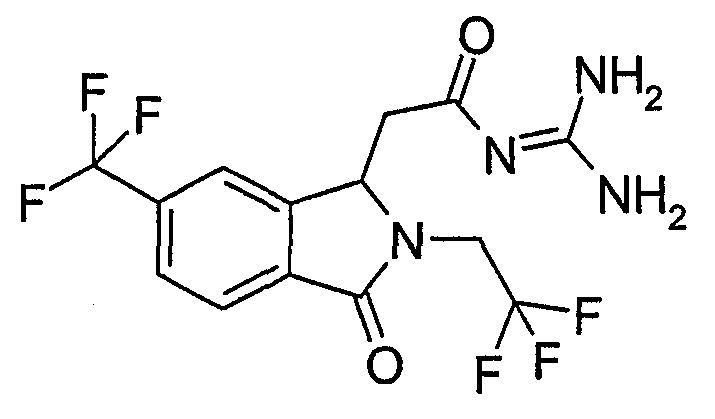
1,24 г гидрохлорида гуанидиния и 1,21 г KOtBu суспендируют с использованием 30 мл ДМФА (безводного) и перемешивают в течение 30 минут при комнатной температуре. Затем добавляют раствор 0,8 г этилового эфира [3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]уксусной кислоты в 5 мл ДМФА (безводного) и смесь перемешивают в течение 17 ч при комнатной температуре. Затем смесь вливают в 100 мл воды и доводят рН до рН 8 с использованием водного раствора HCl. Водный слой экстрагируют три раза с использованием каждый раз 100 мл ЕА. Органический слой сушат над MgSO4 и в вакууме выпаривают растворитель. В результате хроматографии на силикагеле с использованием смеси ЕА/МеОН 3:1 получают 0,56 г аморфного твердого вещества. (Rf (смесь ЕА/МеОН 3:1)=0,45, МС (ES+): 383 (М+1)+).
b) Этиловый эфир [3-оксо-2-(2,2,2-трифторметил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]уксусной кислоты и этиловый эфир [3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]уксусной кислоты
0,84 г NaH суспендируют с использованием 60 мл ДМФА (безводного) и при температуре между 10 и 25°С добавляют 4,5 мл этилового эфира (диэтоксифосфорил)уксусной кислоты. Смесь перемешивают при комнатной температуре в течение 30 минут и затем добавляют раствор 4,2 г 3-гидрокси-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидроизоиндол-1-она и 3-гидрокси-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидроизоиндол-1-она в 20 мл ДМФА (безводного). Реакционную смесь кипятят с обратным холодильником в течение 1 часа, затем охлаждают до комнатной температуры. Затем добавляют 100 мл ЕА и смесь промывают дважды с использованием 200 мл полунасыщенного водного раствора NaHCO3. Органический слой сушат над MgSO4 и в вакууме выпаривают растворитель. Хроматографию осуществляют на колонке Merck Lichrospher RP18, 10 мкм, 50 × 250 мм. Условия хроматографии:
скорость потока 150 мл/минута
элюент А: вода +2% ТФУК
элюент В: ацетонитрил
00 минута: 90% А, 10% В
04 минута: 90% А, 10% В
24 минута: 25% А, 75% В
25 минута: 5% А, 95% В
30 минута: 5% А, 95% В
31 минута: 90% А, 10% В
35 минута: 90% А, 10% В
В результате получают 3,3 г смеси региоизомерных этилового эфира [3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]уксусной кислоты и этилового эфира [3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]уксусной кислоты. В результате осуществления хроматографии на силикагеле с использованием DIP получают 0,8 г этилового эфира [3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]уксусной кислоты в виде бесцветного масла. (Rf (DIP)=0,37, МС (ES+): 370 (М+1)+) и 0,63 г этилового эфира [3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]уксусной кислоты в виде бесцветного масла. (Rf (DIP)=0,30, МС (ES+): 370 (М+1)+).
c) 3-Гидрокси-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидроизоиндол-1-он и 3-гидрокси-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидроизоиндол-1-он
5,5 г 2-(2,2,2-трифторэтил)-5-трифторметилизоиндол-1,3-диона растворяют с использованием 150 мл МеОН (безводного) и при комнатной температуре добавляют 1,0 г КВН4. Смесь выдерживают при комнатной температуре в течение 16 ч, охлаждают до 0°С и вливают в 100 мл воды. В вакууме удаляют метанол. Водный слой экстрагируют три раза с использованием каждый раз 100 мл CH2Cl2. Органический слой сушат над MgSO4 и в вакууме удаляют растворитель с получением 4,2 г вязкого масла. (Rf (DIP)=0,37, МС (DCI): 300 (М+1)+).
d) 2-(2,2,2-Трифторэтил)-5-трифторметилизоиндол-1,3-дион
5,0 г 5-трифторметилизобензофуран-1,3-диона растворяют с использованием 50 мл толуола (безводного) и 4,6 г 2,2,2-трифторэтиламина. Смесь выдерживают при комнатной температуре в течение 16-ти часов. Затем добавляют 50 мг толуол-4-сульфоновой кислоты и смесь кипятят с обратным холодильником в течение 5-ти часов. Добавляют 200 мл ЕА и смесь промывают дважды с использованием 50 мл 10% водного раствора Na2CO3. Органический слой сушат над MgSO4 и в вакууме удаляют растворитель с получением 5,5 г вязкого масла. (Rf (DIP)=0,57; МС (EI): 298 (М+1)+).
е) 5-Трифторметилизобензофуран-1,3-дион
25,0 г 4-трифторметилфталевой кислоты растворяют в 50 мл НОАс и добавляют 15,1 мл ангидрида уксусной кислоты. Смесь кипятят с обратным холодильником в течение 6 часов. Затем в вакууме удаляют растворитель. Остаток обрабатывают три раза толуолом, с использованием каждый раз 50 мл толуола и затем в вакууме удаляют растворитель. Выход бесцветного масла составляет 23,1 г (Rf (CH2Cl2)=0,60).
Пример 22
N-[2-(3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
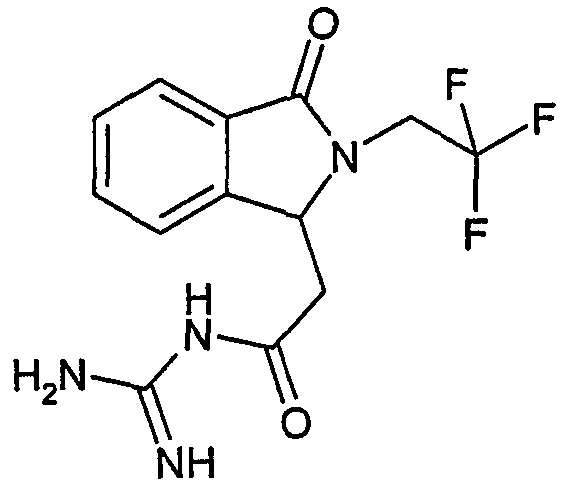
N-[(3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 21. В результате получают аморфный белый порошок, Rf=0,10 (тонкослойная хроматография на силикагеле, элюент: смесь этилацетат/метанол (5:1 по объему). Масса-спектр: ES+: m/e 315.
Пример 23
N-[2-(5-хлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[2-(6-хлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
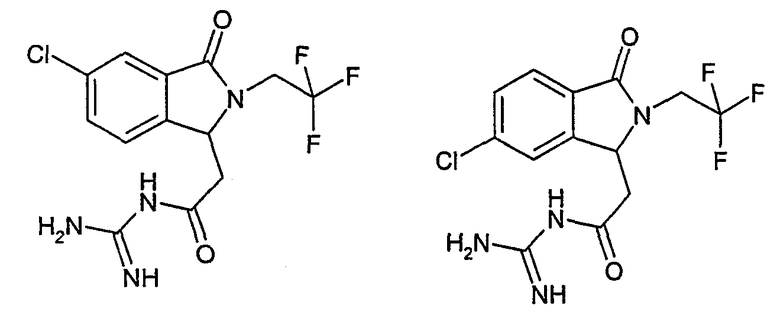
N-[(5-хлор-3-оксо-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(6-хлор-3-оксо-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают в виде смеси двух региоизомеров, следуя методике примера 21. В результате получают аморфный белый порошок, Rf=0,20 (тонкослойная хроматография на силикагеле, элюент: смесь этилацетат/метанол (5:1 по объему). Масса-спектр: ES+: m/e 349.
Пример 24
N-[2-(6-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[2-(5-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
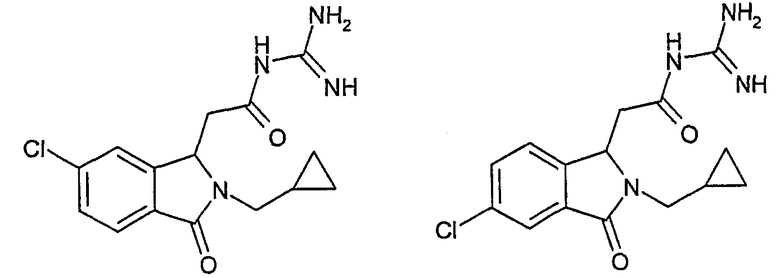
N-[(5-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(6-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают в виде смеси двух региоизомеров, следуя методике примера 8, Rf=0,09 (тонкослойная хроматография на силикагеле, элюент: смесь этилацетат/метанол (5:1 по объему). Масса-спектр: ES+: m/e 321.
Пример 25
N-[2-(2-циклопропилметил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[2-(2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
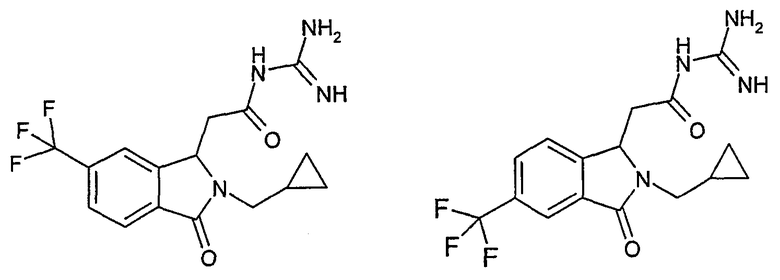
N-[(2-циклопропилметил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают в виде смеси двух региоизомеров, следуя методике примера 8, Rf=0,11 (тонкослойная хроматография на силикагеле, элюент: смесь этилацетат/метанол (5:1 по объему)). Масса-спектр: ES+: m/e 355.
Пример 26
а) N-[2-(5-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[2-(6-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
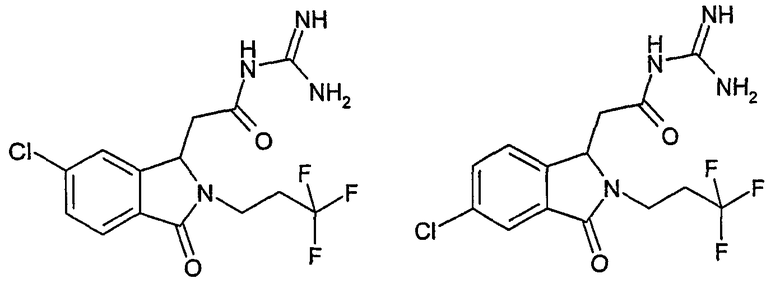
N-[(5-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(6-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают в виде смеси двух региоизомеров, следуя методике примера 1, с использованием в качестве исходных продуктов 4-хлор-N-(3,3,3-трифторпропил)фталимида, Rf=0,12 (тонкослойная хроматография на силикагеле, элюент: смесь этилацетат/метанол (5:1 по объему). Масса-спектр: ES+: m/e 363.
b) 4-Хлор-N-(3,3,3-трифторпропил)фталимид
Смесь 5,0 г 4-хлор-N-(3,3,3-трифторпропил)фталамовой кислоты и 30 мг паратолуолсульфоновой кислоты в 100 мл кипятят с обратным холодильником при перемешивании в течение 7 часов. Затем реакционную смесь концентрируют досуха при пониженном давлении. В результате получают 4,2 г 4-хлор-N-(3,3,3-трифторпропил)фталимида, Rf=0,5 (тонкослойная хроматография на силикагеле, элюент: простой диизопропиловый эфир). Масса-спектр: DCI: m/e 278.
с) 4-Хлор-N-(3,3,3-трифторпропил)фталамовая кислота
К смеси 7,5 г 4-хлорфталимида и 4,6 г карбоната калия в 40 мл диметилформамида при температуре примерно 110°С и перемешивании добавляют по каплям 3,6 г 1,1,1-трифтор-3-иодпропана. После перемешивания в течение 11 часов при температуре примерно 120°С реакционную смесь охлаждают до температуры примерно 20°С и затем вливают в 200 мл воды. Смесь подкисляют разбавленным водным раствором хлористоводородной кислоты до рН около 3 и затем экстрагируют 3 раза 100 мл этилацетата. Органическую фазу сушат над сульфатом магния и затем концентрируют досуха. В результате получают 5,0 г 4-хлор-N-(3,3,3-трифторпропил)фталамовой кислоты, Rf=0,12 (тонкослойная хроматография на силикагеле, элюент: этилацетат).
d) 4-Хлорфталимид
Раствор 10,0 г ангидрида 4-хлорфталевой кислоты в 24,6 г формамида нагревают при температуре примерно 120°С при перемешивании в течение 3-х часов, затем охлаждают до температуры примерно 20°С и вливают в 100 мл воды. После перемешивания в течение 30 минут смесь фильтруют и затем осадок сушат в вакууме при температуре примерно 60°С. В результате получают 10,4 г 4-хлорфталимида в виде белого твердого вещества, плавящегося при 171°С. Rf=0,07 (тонкослойная хроматография на силикагеле, элюент: дихлорметан).
Пример 27
а) N-[2-(3-оксо-5-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[2-(3-оксо-6-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
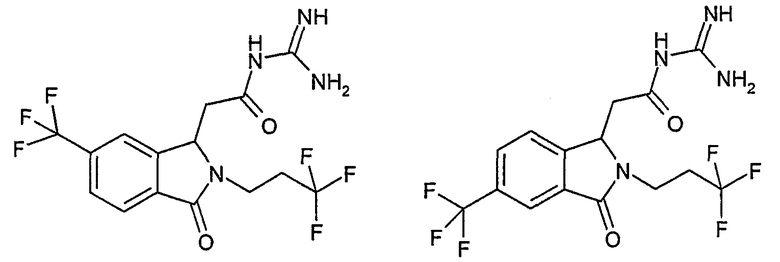
N-[(3-оксо-5-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(3-оксо-6-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают в виде смеси двух региоизомеров, следуя методике примера 1, с использованием в качестве исходных продуктов 4-трифторметил-N-(3,3,3-трифторпропил)фталимида, Rf=0,12 (тонкослойная хроматография на силикагеле, элюент: смесь этилацетат/метанол (5:1 по объему)). Масса-спектр: ES+: m/e 397.
b) 4-Трифторметил-N-(3,3,3-трифторпропил)фталимид получают методикой, подобной методике получения 4-хлор-N-(3,3,3-трифторпропил)фталимида, описанной в примере 26, с использованием в качестве исходных продуктов ангидрида 4-трифторметилфталевой кислоты, Rf=0,12 (тонкослойная хроматография на силикагеле, элюент: этилацетат).
Ангидрид 4-трифторметилфталевой кислоты может быть получен переделкой или применением методики, описанной Cavalleri et al., J. Med. Chem., 13(1), 148-149, (1970).
Пример 28
N-[2-(5-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[2-(6-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
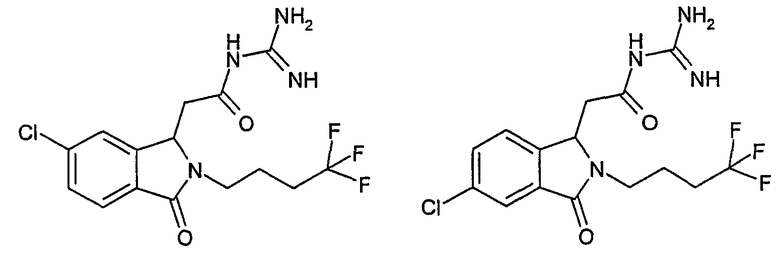
N-[(5-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(6-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают в виде смеси двух региоизомеров, следуя методике примера 8, Rf=0,11 (тонкослойная хроматография на силикагеле, элюент: смесь этилацетат/метанол (5:1 по объему)). Масса-спектр: ES+: m/e 377.
Пример 29
N-[2-(3-оксо-2-(4,4,4-трифторбутил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[2-(3-оксо-2-(4,4,4-трифторбутил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
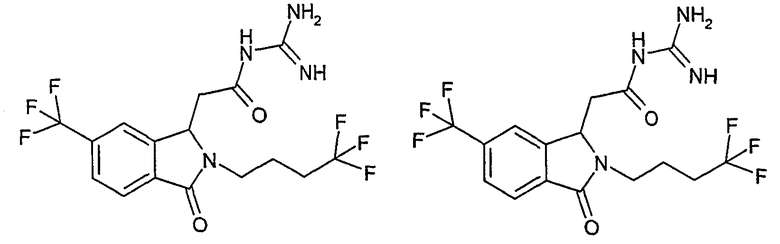
N-[(3-оксо-2-(4,4,4-трифторбутил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и N-[(3-оксо-2-(4,4,4-трифторбутил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают в виде смеси двух региоизомеров, следуя методике примера 27, с использованием в качестве исходных продуктов 4-трифторметил-N-(4,4,4-трифторбутил)фталимида, Rf=0,12 (тонкослойная хроматография на силикагеле, элюент: смесь этилацетат/метанол (5:1 по объему)). Масса-спектр: ES+: m/e 411.
Пример 30
а) N-[2-(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин
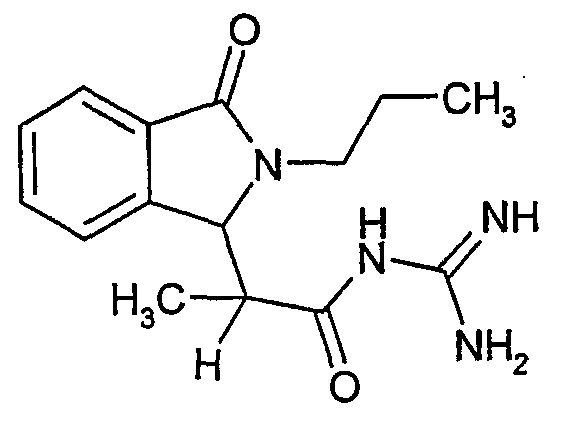
3,75 г (39,2 ммоль) гуанидинийхлорида растворяют в 20 мл диметилформамида и добавляют 3,96 г (35,3 ммоль) трет-бутоксида калия. Смесь перемешивают в течение 45 минут при температуре примерно 20°С и затем добавляют по каплям раствор 1,08 г (3,92 ммоль) этил-2-метил-3-(2-пропилкарбамоилфенил)акрилата в 15 мл диметилформамида. После перемешивания в течение 16 часов при температуре примерно 20°С выпаривают растворитель и остаток растворяют в водном 2 н. растворе хлористоводородной кислоты. Сразу после экстракции дихлорметаном водную фазу подщелачивают гидроксидом калия до рН 12 и отфильтровывают полученный таким образом осадок*. Осадок переносят в водный 2 н. раствор хлористоводородной кислоты, отфильтровывают и затем сушат вымораживанием; таким образом выделяют около 400 мг одного из двух полученных диастереоизомеров (диастереоизомер А) в форме, обогащенной гидрохлоридом, при соотношении примерно 6:1 (MC (ES): M+H: 289,1).
*Первый полученный фильтрат подвергают двойной экстракции дихлорметаном. Объединенные органические экстракты сушат над сульфатом натрия и затем концентрируют. Остаток переносят в водный 2 н. раствор хлористоводородной кислоты, фильтруют и затем сушат вымораживанием. Таким образом выделяют 77 мг второго диастереоизомера (диастереоизомер В) в форме, обогащенной гидрохлоридом, при соотношении 10:1 (MC (ES): M+H: 289,1).
Диастереоизомер А разделяют на два энантиомера ВЭЖХ хроматографией на колонке с хиральной стационарной фазой (Chiralpak AD 250×4,6) при элюировании смесью ацетонитрил/изопропанол/н-гептан (50/3/4 по объему), содержащей 0,3% диэтиламина; скорость потока: 1 мл/мин. В результате получают энантиомер А1: 3,106 мин и энантиомер А2: 3,522 мин.
Оба энантиомера превращают в соответствующие трифторацетаты растворением свободных оснований в водной разбавленной трифторуксусной кислоте, затем осуществляют сушку вымораживанием; энантиомер А1 в виде трифторацетата (αD 20=-73° в метаноле при 0,1%), энантиомер А2 в виде трифторацетата (αD 20=+56° в метаноле при 0,1%).
b) Этил-2-метил-3-(2-пропилкарбамоилфенил)акрилат
Раствор 0,69 мл (5,0 ммоль) триэтиламина и 1,64 г (5,0 ммоль) О-[(циано(этоксикарбонил)метилен)амино]-1,1,3,3-тетраметилуронийтетрафторбората («TOTU») в 6 мл диметилформамида добавляют при 0°С к раствору 1,17 г (5,0 ммоль) 2-(2-этоксикарбонилпропен-1-ил)бензойной кислоты в 10 мл диметилформамида. После перемешивания в течение 30 минут при 0°С и затем в течение 30 минут при температуре примерно 20°С указанный раствор вводят по каплям во второй раствор, состоящий из 296 мг (5,0 ммоль) н-пропиламина и 0,69 мл (5,0 ммоль) триэтиламина в 10 мл диметилформамида, и смесь перемешивают в течение еще 6 часов при температуре примерно 20°С. После выдерживания раствора в течение ночи в вакууме удаляют растворитель и остаток растворяют в этилацетате. После двукратной промывки раствором бикарбоната натрия и последующей однократной промывки раствором хлорида натрия органическую фазу сушат над сульфатом натрия и затем концентрируют. В результате получают 1,10 г этил-2-метил-3-(2-пропилкарбамоилфенил)акрилата в виде желтоватого масла, которое может взаимодействовать на следующей стадии без дальнейшей очистки.
с) 2-(2-Этоксикарбонилпропен-1-ил)бензойная кислота
Смесь 5,0 г (33,3 ммоль) 2-формилбензойной кислоты и 14,5 г (40,0 ммоль) этил-2-(трифенилфосфанилиден)пропионата в 100 мл диметилформамида перемешивают при температуре примерно 20°С в течение 1 часа. После удаления растворителя остаток растворяют в дихлорметане и затем экстрагируют дважды раствором бикарбоната натрия. Водные фазы объединяют, промывают дихлорметаном и затем подкисляют водным 6 н. раствором хлористоводородной кислоты до рН между 1 и 2. После двукратной экстракции дихлорметаном органические фазы объединяют, сушат над сульфатом магния и затем концентрируют. В результате получают 2-(2-этоксикарбонилпропен-1-ил)бензойную кислоту в виде желтого масла, которое непосредственно используют на следующей стадии без дальнейшей очистки.
Пример 31
а) N-[2-(2-бутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин
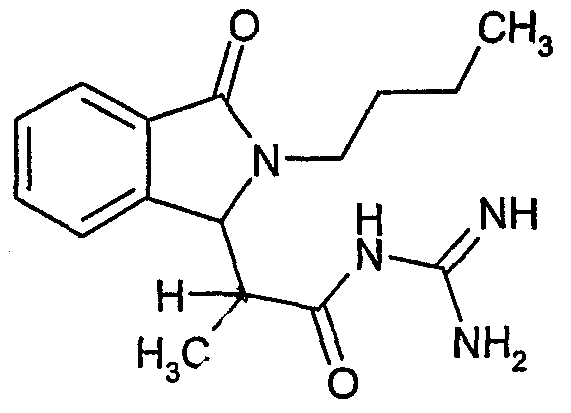
N-[2-(2-бутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин получают, следуя методике примера 30, с использованием в качестве исходных продуктов 3,63 г (38,0 ммоль) гуанидинийхлорида в 20 мл диметилформамида и 3,84 г (34,2 ммоль) трет-бутоксида калия. Смесь перемешивают в течение 45 минут при температуре примерно 20°С, затем добавляют по каплям раствор 1,10 г (3,80 ммоль) этил(2-бутилкарбамоилфенил)-2-метилакрилата в 15 мл диметилформамида и смесь перемешивают в течение 3-х часов при температуре примерно 20°С. После выдерживания раствора в течение ночи растворитель удаляют и остаток растворяют в водном 2 н. растворе хлористоводородной кислоты. После однократной экстракции дихлорметаном органическую фазу концентрируют и остаток затем переносят в водный 2 н. раствор хлористоводородной кислоты, фильтруют и затем сушат вымораживанием. В результате получают 1,07 г N-[2-(2-бутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидина в виде смеси диастереоизомеров; рН водной фазы доводят до значения 12 гидроксидом калия и отфильтровывают полученный таким образом осадок*. Полученное твердое вещество переносят в водный 2 н. раствор хлористоводородной кислоты и после фильтрования и сушки вымораживанием выделяют около 106 мг одного их двух полученных диастереоизомеров (диастереоизомер А) в форме, обогащенной гидрохлоридом, при соотношении 5:1. (MC (ES): M+H: 303,1).
*Первый полученный фильтрат подвергают двойной экстракции дихлорметаном. Объединенные органические экстракты сушат над сульфатом натрия и затем концентрируют. Остаток переносят в водный 2 н. раствор хлористоводородной кислоты, фильтруют и затем сушат вымораживанием; таким образом выделяют 38 мг второго диастереоизомера (диастереоизомер В) в форме, обогащенной гидрохлоридом, при соотношении более 10:1 (MC (ES): M+H: 303,1).
Диастереоизомер А разделяют на два энантиомера ВЭЖХ хроматографией на колонке с хиральной стационарной фазой (Chiralpak AD 250×4,6) при элюировании смесью ацетонитрил/изопропанол/н-гептан (50/3/4 по объему), содержащей 0,3% диэтиламина; скорость потока: 1 мл/мин. В результате получают энантиомер А1: 3,195 мин, энантиомер А2: 3,714 мин.
Оба энантиомера превращают в соответствующие трифторацетаты растворением свободных оснований в водной разбавленной трифторуксусной кислоте, затем осуществляют сушку вымораживанием; энантиомер А1 в виде трифторацетата (αD 20=-32° в метаноле при 0,2%), энантиомер А2 в виде трифторацетата (αD 20=+42° в метаноле при 0,2%).
b) Этил-3-(2-бутилкарбамоилфенил)-2-метилакрилат
Этил-3-(2-бутилкарбамоилфенил)-2-метилакрилат получают, следуя методике примера 30, с использованием в качестве исходных продуктов 1,17 г (5,0 ммоль) 2-(2-этоксикарбонилпропен-1-ил)бензойной кислоты в 10 мл диметилформамида, раствора 0,69 мл (5,0 ммоль) триэтиламина, 1,64 г (5,0 ммоль) О-[(циано(этоксикарбонил)метилен)амино]-1,1,3,3-тетраметилуронийтетрафторбората («TOTU») в 6 мл диметилформамида и второго раствора, состоящего из 366 мг (5,0 ммоль) н-бутиламина и 0,69 мл (5,0 ммоль) триэтилфамина в 10 мл диметилформамида. В результате получают 1,13 г этил-3-(2-бутилкарбамоилфенил)-2-метилакрилата в виде желтоватого масла, которое может взаимодействовать на следующей стадии без дальнейшей очистки.
Пример 32
а) N-[2-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин
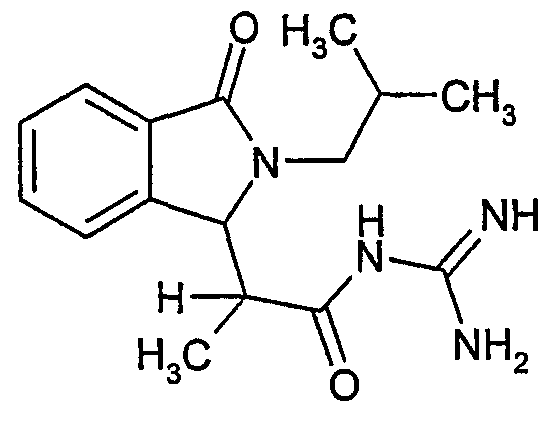
N-[2-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин получают, следуя методике примера 30, с использованием в качестве исходных продуктов 3,73 г (39,0 ммоль) гуанидинийхлорида в 20 мл диметилформамида и 3,94 г (35,1 ммоль) трет-бутоксида калия. Смесь перемешивают в течение 45 минут при температуре примерно 20°С, затем добавляют по каплям раствор 1,13 г (3,90 ммоль) этил-3-(2-изобутилкарбамоилфенил)-2-метилакрилата в 15 мл диметилформамида и смесь перемешивают в течение 3-х часов при температуре примерно 20°С. После стояния раствора в течение ночи растворитель удаляют и остаток растворяют в водном 2 н. растворе хлористоводородной кислоты. После однократной экстракции дихлорметаном органическую фазу концентрируют, остаток затем переносят в 2 н. раствор хлористоводородной кислоты, фильтруют и затем сушат вымораживанием. В результате получают 862 мг N-[2-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидина в виде смеси диастереизомеров; рН водной фазы доводят до значения 12 гидроксидом калия и полученный таким образом осадок отфильтровывают*. Полученное таким образом твердое вещество переносят в водный 2 н. раствор хлористоводородной кислоты и после фильтрования и сушки вымораживанием выделяют около 255 мг одного из двух полученных диастереоизомеров (диастереоизомер А) в форме, обогащенной гидрохлоридом, при соотношении 9:1 (MS (ES): M+H: 303,1.
*Первый полученный фильтрат подвергают двойной экстракции дихлорметаном. Объединенные органические экстракты сушат над сульфатом натрия и затем концентрируют. Остаток переносят в водный 2 н. раствор хлористоводородной кислоты, фильтруют и затем сушат вымораживанием; таким образом выделяют 78 мг второго диастереоизомера (диастереоизомер В) в форме, обогащенной гидрохлоридом, при соотношении более 10:1 (MC (ES): M+H: 303,1).
Диастереоизомер А разделяют на два энантиомера ВЭЖХ хроматографией на колонке с хиральной стационарной фазой (Chiralpak AD 250×4,6) при элюировании смесью ацетонитрил/изопропанол/н-гептан (50/3/4 по объему), содержащей 0,3% диэтиламина; скорость потока: 1 мл/мин. В результате получают энантиомер А1: 3,895 мин, энантиомер А2: 4,437 мин.
Оба энантиомера превращают в соответствующие трифторацетаты растворением свободных оснований в водной разбавленной трифторуксусной кислоте, затем осуществляют сушку вымораживанием; энантиомер А1 в виде трифторацетата (αD 20=-43° в метаноле при 0,2%), энантиомер А2 в виде трифторацетата (αD 20=+57° в метаноле при 0,2%).
b) Этил-3-(2-изобутилкарбамоилфенил)-2-метилакрилат
Этил-3-(2-изобутилкарбамоилфенил)-2-метилакрилат получают, следуя методике примера 30, с использованием в качестве исходных продуктов 1,17 г (5,0 ммоль) 2-(2-этоксикарбонилпропен-1-ил)бензойной кислоты в 10 мл диметилформамида, раствора 0,69 мл (5,0 ммоль) триэтиламина, 1,64 г (5,0 ммоль) О-[(циано(этоксикарбонил)метилен)амино]-1,1,3,3-тетраметилуронийтетрафторбората («TOTU») в 6 мл диметилформамида и второго раствора, состоящего из 366 мг (5,0 ммоль) изобутиламина, 0,69 мл (5,0 ммоль) триэтиламина в 10 мл диметилформамида. В результате получают 1,15 г этил-3-(2-изобутилкарбамоилфенил)-2-метилакрилата в виде желтоватого масла, которое может взаимодействовать на следующей стадии без дальнейшей очистки.
Пример 33
а) N-[2-(2-гидроксиэтил)-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин
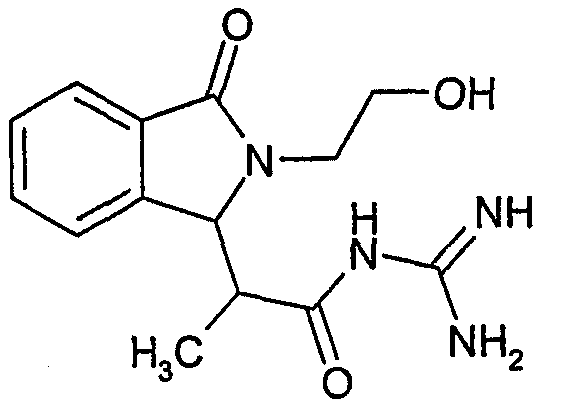
N-[2-(2-гидроксиэтил)-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин получают, следуя методике примера 30, с использованием в качестве исходных продуктов 3,75 г (39,2 ммоль) гуанидинийхлорида в 20 мл диметилформамида и 3,96 г (35,3 ммоль) трет-бутоксида калия. Смесь перемешивают в течение 45 минут при температуре примерно 20°С, затем добавляют по каплям раствор 1,09 г (3,92 ммоль) этил-3-[2-(2-гидроксиэтилкарбамоил)фенил]-2-метилакрилата в 15 мл диметилформамида и смесь перемешивают в течение 3-х часов при температуре примерно 20°С. После выдерживания раствора в течение ночи удаляют растворитель и остаток растворяют в водном 2 н. растворе хлористоводородной кислоты. После однократной экстракции дихлорметаном рН водной фазы доводят до значения 12 гидроксидом калия и полученный таким образом остаток отфильтровывают*. Полученное таким образом твердое вещество переносят в водный 2 н. раствор хлористоводородной кислоты и после фильтрования и сушки вымораживанием выделяют около 207 мг одного из двух полученных диастереоизомеров (диастереоизомер А) в форме, обогащенной гидрохлоридом, при соотношении 10:1 (MS (ES): M+H: 291,1).
*Первый полученный фильтрат подвергают двойной экстракции дихлорметаном. Объединенные органические экстракты сушат над сульфатом натрия и затем концентрируют. Остаток переносят в водный 2 н. раствор хлористоводородной кислоты, фильтруют и затем сушат вымораживанием; таким образом выделяют 50 мг смеси двух диастереоизомеров (диастереоизомеры А и В) в виде гидрохлорида при соотношении 1:1 (MC (ES): M+H: 291,1).
b) Этил-3-[2-(2-гидроксиэтилкарбамоил)фенил]-2-метилакрилат
Этил-3-[2-(2-гидроксиэтилкарбамоил)фенил]-2-метилакрилат получают, следуя методике примера 30, с использованием в качестве исходных продуктов 1,17 г (5,0 ммоль) 2-(2-этоксикарбонилпропен-1-ил)бензойной кислоты в 10 мл диметилформамида, раствора 0,69 мл (5,0 ммоль) триэтиламина и 1,64 г (5,0 ммоль) О-[(циано(этоксикарбонил)метилен)амино]-1,1,3,3-тетраметилуронийтетрафторбората («TOTU») в 6 мл диметилформамида и второго раствора, состоящего из 306 мг (5,0 ммоль) этаноламина и 0,69 мл (5,0 ммоль) триэтиламина в 10 мл диметилформамида. В результате получают 1,14 г этил-3-[2-(2-гидроксиэтилкарбамоил)фенил]-2-метакрилата в виде желтоватого масла, которое может взаимодействовать на следующей стадии без дальнейшей очистки.
Пример 34
а) N-[2-(2-изобутил-5-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
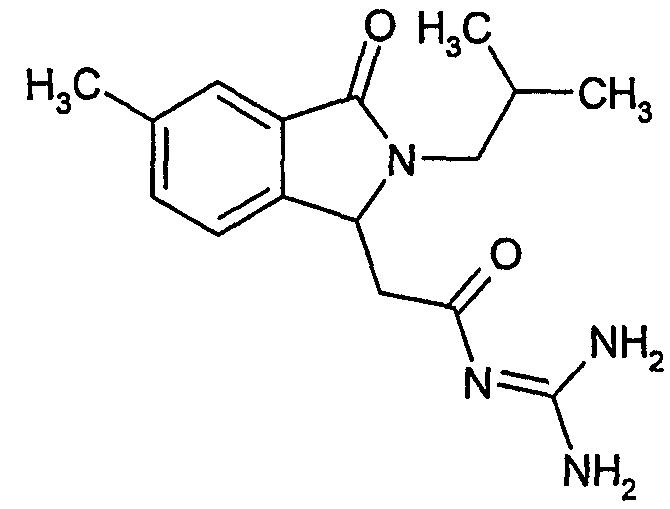
N-[(2-изобутил-5-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 1,75 г трет-бутоксида калия, 1,78 г гуанидинийхлорида и 0,9 г этил(2-изобутил-5-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 40 часов и затем вливают в 150 мл воды и экстрагируют 3 раза 150 мл этилацетата. Органические экстракты объединяют, промывают 3 раза 50 мл воды, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2,7 кПа) и температуре примерно 40°С. Остаток переносят в диэтиловый эфир и затем фильтруют с получением 0,66 г N-[(2-изобутил-5-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде белого твердого вещества, плавящегося при 266°С. (Анализ: С16Н22N4O2; вычислено, % С: 63,56, Н: 7,33, N: 18,53, O: 10,58; найдено, % С: 63,57, Н: 7,48, N: 18,50).
b) Этил(2-изобутил-5-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(2-изобутил-5-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,19 г 75% гидрида натрия в 25 мл 1,2-диметоксиэтана, 1,2 мл триэтилфосфоноацетата и 0,85 г 3-гидрокси-2-изобутил-6-метил-2,3-дигидроизоиндол-1-она. Неочищенный продукт очищают хроматографией на колонке с силикагелем (размер частиц 15-45 мкм) при элюировании смесью дихлорметан/метанол (99/1 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2,7 кПа) и температуре примерно 40°С. В результате получают 1 г этил(2-изобутил-5-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде бесцветного масла, которое непосредственно используют на следующей стадии.
Пример 35
а) N-[(2-изобутил-6-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
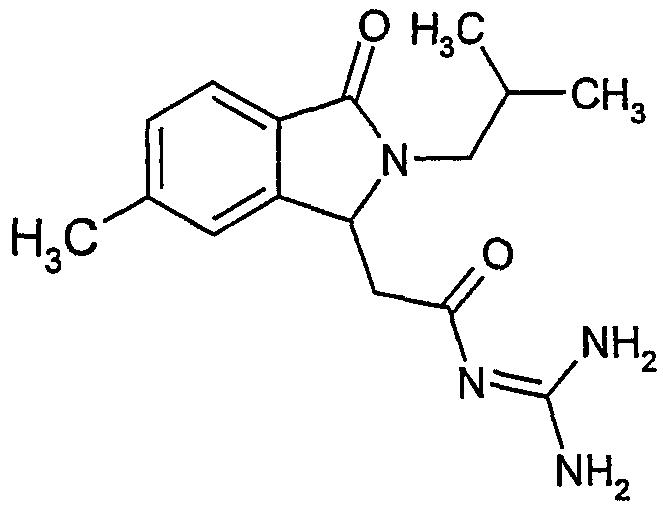
N-[(2-изобутил-6-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин получают, следуя методике примера 1, с использованием в качестве исходных продуктов 2,52 г трет-бутоксида калия, 2,58 г гуанидинийхлорида и 1,3 г этил(2-изобутил-6-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата. Реакционную смесь перемешивают при температуре примерно 20°С в течение 40 часов, затем вливают в 150 мл воды и экстрагируют 3 раза 150 мл этилацетата. Органические экстракты объединяют, промывают дважды 50 мл воды, сушат над сульфатом магния, фильтруют и затем концентрируют досуха при пониженном давлении (2,7 кПа) и температуре примерно 40°С. Остаток переносят в диэтиловый эфир и затем фильтруют с получением 0,81 г N-[(2-изобутил-6-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидина в виде белого твердого вещества, плавящегося при 260°С. (Rf=0,28, тонкослойная хроматография на силикагеле, элюент: смесь дихлорметан/метанол (90/10 по объему)). (Анализ: С16Н22N4O2; вычислено, % С: 63,56, Н: 7,33, N: 18,53, O: 10,58; найдено, % С: 63,40, Н: 7,33, N: 18,37).
b) Этил(2-изобутил-6-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат
Этил(2-изобутил-6-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетат получают, следуя методике примера 2, с использованием в качестве исходных продуктов 0,24 г 75% гидрида натрия в 30 мл 1,2-диметоксиэтана, 1,5 мл триэтилфосфоноацетата и 1,1 г 3-гидрокси-2-изобутил-5-метил-2,3-дигидроизоиндол-1-она. Неочищенный продукт очищают хроматографией на колонке с силикагелем (размер частиц 15-45 мкм) при элюировании смесью циклогексан/этилацетат (50/50 по объему). Фракции, содержащие ожидаемый продукт, объединяют и концентрируют досуха при пониженном давлении (2,7 кПа) и температуре примерно 40°С. В результате получают 1,4 г этил(2-изобутил-6-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетата в виде бесцветного масла, которое непосредственно используют на следующей стадии.
с) 3-Гидрокси-2-изобутил-5-метил-2,3-дигидроизоиндол-1-он и 3-гидрокси-2-изобутил-6-метил-2,3-дигидроизоиндол-1-он
3-Гидрокси-2-изобутил-5-метил-2,3-дигидроизоиндол-1-он и 3-гидрокси-2-изобутил-6-метил-2,3-дигидроизоиндол-1-он получают, следуя методике примера 1, с использованием в качестве исходных продуктов 6,7 г N-изобутил-4-метилфталимида в 65 мл метанола и 1,7 г боргидрида калия. Реакционную смесь перемешивают при температуре примерно 20°С в течение 16 часов, затем добавляют 0,3 г боргидрида калия и реакционную смесь перемешивают при температуре примерно 20°С в течение двух часов. Затем смесь охлаждают до температуры примерно 0°С и добавляют по каплям 40 мл дистиллированной воды. Полученный остаток отфильтровывают и затем фильтрат концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 25°С. Остаток очищают хроматографией под давлением аргона (50 кПа) на колонке с силикагелем (размер частиц 40-63 мкм) при последовательном элюировании смесями циклогексан/этилацетат (70/30; 60/40 по объему). Фракции, содержащие смесь двух ожидаемых продуктов, объединяют и концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 25°С. В результате получают 2,27 г смеси 3-гидрокси-2-изобутил-5-метил-2,3-дигидроизоиндол-1-она и 3-гидрокси-2-изобутил-6-метил-2,3-дигидроизоиндол-1-она в виде белого твердого вещества (Rf=0,54, тонкослойная хроматография на силикагеле, элюент: смесь циклогексан/этилацетат (50/50 по объему)). Вторая порция исходных продуктов, в которой используют 3,5 г N-изобутил-4-метилфталимида, таким же путем дает 2,2 г смеси 3-гидрокси-2-изобутил-5-метил-2,3-дигидроизоиндол-1-она и 3-гидрокси-2-изобутил-6-метил-2,3-дигидроизоиндол-1-она в виде белого твердого вещества. Две порции объединяют и затем разделяют ВЭЖХ хроматографией на системе, содержащей две последовательно расположенные колонки диаметром 60 мм, содержащие соответственно 700 г и 475 г хиральной стационарной фазы CHIRALPAK AS с размером частиц 20 мкм, при элюировании смесью гептан/изопропанол (90/10 по объему) со скоростью потока 90 мл/мин. Первый и второй элюированные изомеры соответствуют первой паре региоизомеров. Третий и четвертый элюированные изомеры соответствуют второй паре региоизомеров. В указанных условиях осуществляют три впрыска 1 г, 1,6 г и 1,7 г соответственно. После концентрирования фракций при пониженном давлении (1 кПа) и температуре примерно 40°С получают следующие соединения: 0,77 г (+)-3-гидрокси-2-изобутил-5-метил-2,3-дигидроизоиндол-1-она в виде белого твердого вещества (αD 20=+17,3°±0,8° в метаноле при концентрации 0,5%), 1,09 г (+)-3-гидрокси-2-изобутил-6-метил-2,3-дигидроизоиндол-1-она в виде белого твердого вещества (αD 20=+23,2°±0,7° в метаноле при концентрации 0,5%), 1,17 г (-)-3-гидрокси-2-изобутил-6-метил-2,3-дигидроизоиндол-1-она в виде белого твердого вещества (αD 20=-20,1°±0,8° в метаноле при концентрации 0,5%) и 0,85 г (-)-3-гидрокси-2-изобутил-5-метил-2,3-дигидроизоиндол-1-она в виде белого твердого вещества (αD 20=-15,6°±0,6° в дихлорметане при концентрации 0,5%).
d) N-изобутил-4-метилфталимид
N-изобутил-4-метилфталимид получают, следуя методике примера 2, с использованием в качестве исходных продуктов 6,0 г ангидрида 4-метилфталевой кислоты, 3,7 мл изобутиламина и каталитического количества паратолуолсульфоновой кислоты в 60 мл толуола. Реакционную смесь нагревают при температуре примерно 140°С в течение трех часов и затем охлаждают до температуры примерно 20°С. Реакционную смесь концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 40°С. Остаток переносят в 50 мл насыщенного водного раствора бикарбоната натрия и затем смесь экстрагируют дважды 75 мл дихлорметана. Органические экстракты объединяют, сушат над сульфатом натрия, фильтруют и затем концентрируют досуха при пониженном давлении (2 кПа) и температуре примерно 30°С с получением 6,7 г N-изобутил-4-метилфталимида в виде белого твердого вещества, плавящегося при 102°С.
Пример 36
а) (R)-N-{2-[6-метансульфонил-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин и
(S)-N-{2-[6-метансульфонил-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
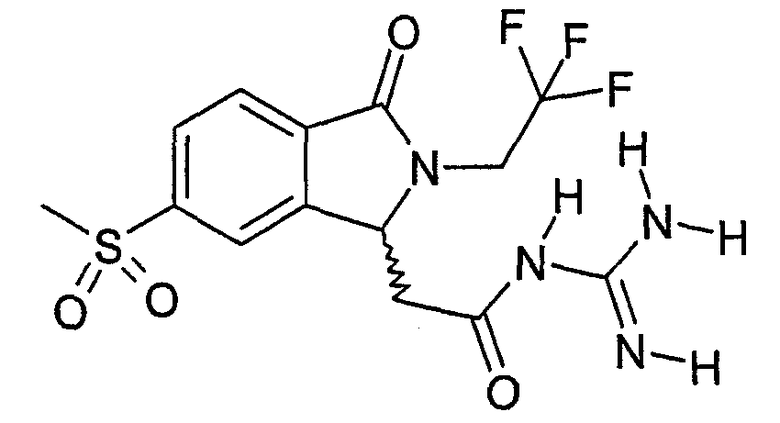
1,6 г KOtBu растворяют с использованием 22 мл ДМФА (безводного). Полученный раствор добавляют к раствору, приготовленному из 1,5 г гидрохлорида гуанидина с использованием 15 мл ДМФА (безводного). Смесь перемешивают в течение 30 минут при комнатной температуре. Затем добавляют раствор, приготовленный из 1,1 г этилового эфира [6-метансульфонил-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]уксусной кислоты с использованием 15 мл ДМФА (безводного). Реакционную смесь перемешивают при комнатной температуре в течение 22-х часов. Затем смесь переносят в 500 мл полунасыщенного водного раствора NaHCO3 и экстрагируют дважды с использованием каждый раз 200 мл этилацетата. ЕА слой сушат над Na2SO4 и в вакууме удаляют растворитель. В результате хроматографии на силикагеле с использованием смеси ЕА/МеОН 3:1 получают 0,28 г аморфного твердого вещества. (Rf (ЕА/МеОН 3:1)=0,15, MC (ES+):393 (M+1)+).
b) Этиловый эфир [6-метансульфонил-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]уксусной кислоты
0,56 г 60% суспензии NaH в минеральном масле суспендируют в 20 мл DME. Затем при комнатной температуре добавляют по каплям 2,8 мл этилового эфира (диэтоксифосфорил)уксусной кислоты и смесь перемешивают в течение 1 часа при указанной температуре. Затем добавляют раствор, приготовленный из 3-гидрокси-5-метансульфонил-2-(2,2,2-трифторэтил)-2,3-дигидроизоиндол-1-она с использованием 30 мл ДМЕ, и смесь нагревают до температуры образования флегмы. Смесь кипятят с обратным холодильником в течение 4-х часов и затем дают возможность охлаждаться до комнатной температуры. Добавляют 300 мл ЕА и смесь промывают 3 раза с использованием каждый раз 100 мл насыщенного водного раствора NaHCO3. Затем водный слой экстрагируют дважды с использованием каждый раз 100 мл ЕА. Объединенные ЕА слои сушат над Na2SO4 и в вакууме удаляют растворитель. В результате хроматографии на силикагеле с использованием МТВ получают 1,1 г бесцветного масла. (Rf (МТВ)=0,40, MC (DCI):380 (M+1)+).
b) 3-Гидрокси-5-метансульфонил-2-(2,2,2-трифторэтил)-2,3-дигидроизоиндол-1-он
3,4 г 5-метансульфонил-2-(2,2,2-трифторэтил)изоиндол-1,3-диона растворяют с использованием 240 мл МеОН. Затем маленькими порциями при комнатной температуре добавляют 0,63 г КВН4. Реакционную смесь перемешивают в течение 20 часов при комнатной температуре и затем при 0°С вливают в 1л воды. Затем водный слой экстрагируют три раза с использованием каждый раз 300 мл CH2Cl2. Затем водный слой экстрагируют четыре раза с использованием каждый раз 300 мл ЕА. Каждый органический слой сушат отдельно над Na2SO4. В вакууме удаляют растворители. ЕА слой дает 1,4 г чистого продукта в виде бледно-желтого масла. CH2Cl2 слой подвергают хроматографии на силикагеле с использованием смеси ЕА/НЕР 1:2, в результате которой получают еще 1,5 г продукта. (Rf (ЕА/НЕР)=0,035, MC (DCI):310 (M+1)+).
c) 5-Метансульфонил-2-(2,2,2-трифторэтил)изоиндол-1,3-дион
4,0 г 5-метилсульфанил-2-(2,2,2-трифторэтил)изоиндол-1,3-диона растворяют с использованием 100 мл CH2Cl2 и маленькими порциями при комнатной температуре добавляют 7,2 г 3-хлорбензолкарбопероксокислоты. Смесь перемешивают при комнатной температуре в течение 12 часов и выдерживают при указанной температуре в течение еще 60 часов. Затем добавляют 400 мл CH2Cl2, промывают дважды с использованием 150 мл насыщенного водного раствора Na2SO3 и наконец промывают три раза с использованием полунасыщенного водного раствора Na2CO3. Органический слой сушат над Na2SO4. В вакууме удаляют растворители с получением 3,4 г аморфного твердого вещества. (Rf (ЕА)=0,13, MC (DCI): 308 (M+1)+).
d) 5-Метилсульфанил-2-(2,2,2-трифторэтил)изоиндол-1,3-дион
4,0 г 5-хлор-2-(2,2,2-трифторэтил)изоиндол-1,3-диона (см. пример 23), 4,1 г К2СО3 и 1,1 г метантиолата натрия суспендируют с использованием 50 мл ДМФА (безводного). Смесь перемешивают при 80°С в течение 9 часов и после охлаждения разбавляют 400 мл полунасыщенного водного раствора NaHCO3 и экстрагируют четыре раза с использованием каждый раз 200 мл этилацетата. Органический слой сушат над Na2SO4. В вакууме удаляют растворители с получением 4,1 г аморфного твердого вещества. (Rf (ЕА/НЕР 1:2)=0,43, MC (DCI): 276 (M+1)+).
Указанные в заголовке соединения примеров 36а и 36b получают хроматографией 270 мг примера 36 на колонке размером 250×20 мм с хиральной стационарной фазой Chiralpak AD-H, имеющей размер частиц 10 мкм, с использованием смеси ACN/HEP/i-PrOH 50:6:3, при скорости потока 6 мл/минута.
Пример 36а
Выход аморфного твердого вещества: 12 мг.
Аналитическая ВЭЖХ на Chiralpak AD-H/33, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:6:3, при скорости потока 1 мл/минута: RT=3,772 минут.
Пример 36b
Выход аморфного твердого вещества: 11 мг.
Аналитическая ВЭЖХ на Chiralpak AD-H/33, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:6:3, при скорости потока 1 мл/минута: RT=5,601 минут.
Пример 37
N-[2-(2-цикопропилметил-6-метансульфонил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидинийацетат
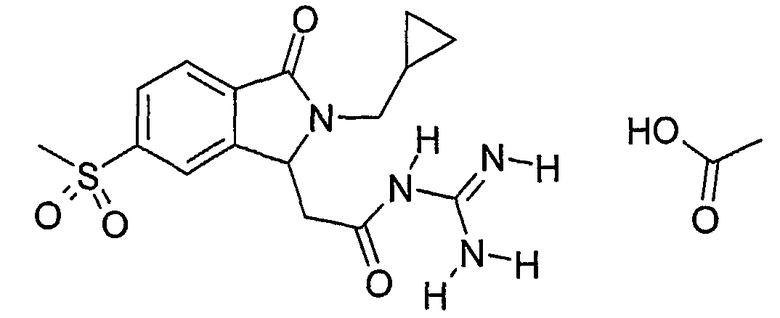
Указанное в заголовке соединение примера 37 синтезируют, следуя методике примера 36, с использованием в качестве исходного продукта 5-хлор-2-циклопропилметилизоиндол-1,3-диона (см. пример 24).
Rf (EA/5% HOAc)=0,047; MC (ES+): 365 (M+1)+).
Пример 38
а) (R)-N-[2-(2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и
(S)-N-[2-(2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
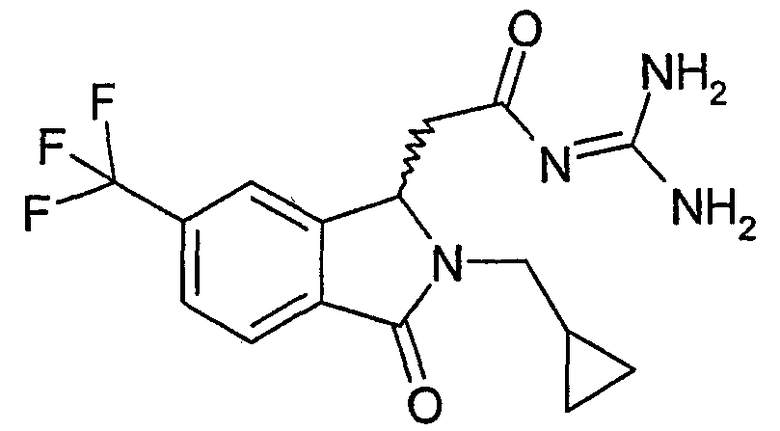
0,3 г (2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)уксусной кислоты растворяют с использованием 5 мл NMP (безводного). Добавляют 0,3 г CDI и смесь перемешивают в течение 17 часов при комнатной температуре с получением промежуточного продукта, представляющего собой 2-циклопропилметил-3-(2-имидазол-1-ил-2-оксоэтил)-5-трифторметил-2,3-дигидро-изоиндол-1-он. Тем временем, суспендируют 0,55 г гидрохлорида гуанидина и 0,54 г KOtBu с использованием 10 мл NMP (безводного) и смесь перемешивают в течение 30 минут при комнатной температуре. Затем образовавшийся раствор добавляют к вышеуказанному имидазолиду и реакционную смесь выдерживают в течение 15 часов при комнатной температуре. Добавляют 100 мл ЕА и промывают три раза с использованием каждый раз 100 мл полунасыщенного водного раствора NaHCO3. Органический слой сушат над MgSO4 и в вакууме удаляют растворитель. В результате хроматографии на силикагеле с использованием смеси ЕА/МеОН 3:1 получают 0,21 г аморфного твердого вещества. (Rf (EA/МеОН 5:1)=0,25; MC (ES+): 355 (M+1)+).
b) (2-Циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)уксусная кислота
1,0 г этилового эфира (2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)уксусной кислоты растворяют с использованием 10 мл этанола и добавляют 3,5 мл 1 н. водного раствора NaOH. Смесь перемешивают в течение 3-х часов при комнатной температуре и затем в вакууме удаляют растворитель. После этого добавляют 30 мл воды и рН раствора доводят до рН 2 с использованием водного раствора HCl. Затем раствор экстрагируют три раза с использованием каждый раз 50 мл ЕА. Органический слой сушат над MgSO4 и в вакууме удаляют растворитель с получением 0,35 г аморфного твердого вещества. (Rf (EA/МеОН 5:1)=0,43; MC (ES+): 314 (M+1)+).
с) Смесь этилового эфира (2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)уксусной кислоты и этилового эфира (2-циклопропилметил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)уксусной кислоты синтезируют, следуя методике примера 8.
Для разделения региоизомеров осуществляют хроматографию 10,5 г смеси 9 раз на колонке Merck Lichrospher, RP18, 10 мкм, 50×250 мм. Условия хроматографии:
скорость потока: 150 мл/минута
элюент А: вода +0,2% ТФУК
элюент В: ацетонитрил
00 минута: 65% А, 35% В
38 минута: 65% А, 35% В
40 минута: 10% А, 90% В
45 минута: 10% А, 90% В
46 минута: 65% А, 35% В
50 минута: 65% А, 35% В
Выход:
0,84 г этилового эфира (2-циклопропилметил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)уксусной кислоты;
Rf (DIP)=0,24
и
1,0 г этилового эфира (2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)уксусной кислоты;
Rf (DIP)=0,30.
Указанные в заголовке соединения примеров 38а и 38b получают хроматографией 207 мг соединения примера 38 на Chiralcel OJ, 250×50 мм, 20 мкм с использованием смеси НЕР/EtOH/MeOH 25:1:1+0,1% DEA при скорости потока 100 мл/минута.
Пример 38а
Выход аморфного твердого вещества: 30 мг.
Аналитическая ВЭЖХ на Chiralcel OJ/16, 250×4,6 с использованием смеси HEP/EtOH/MeOH 25:1:1+0,1% DEA при скорости потока 1,0 мл/минута: RT=8,763 минут.
Пример 38b
Выход аморфного твердого вещества: 25 мг.
Аналитическая ВЭЖХ на Chiralcel OJ/16, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=10,598 минут.
Пример 39
N-[2-(2-циклопропилметил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
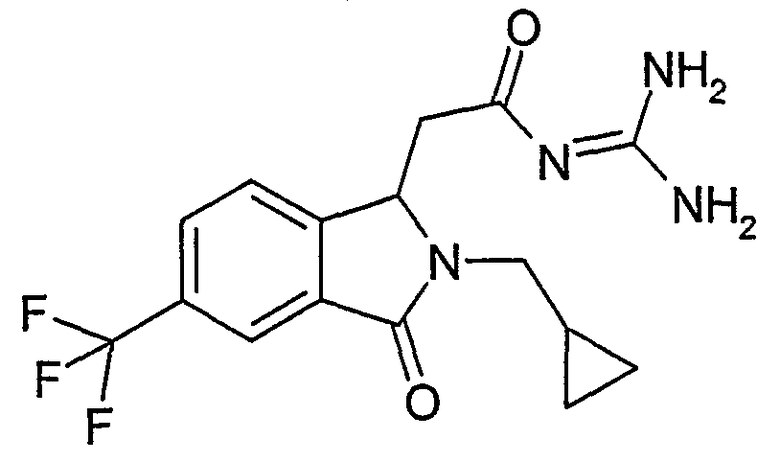
Указанное в заголовке соединение примера 39 синтезируют, следуя методике примера 38, с использованием в качестве исходного продукта этилового эфира (2-циклопропилметил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)уксусной кислоты.
Rf (EA/МеОН 3:1)=0,21; MC (ES+): 355 (M+1)+).
Пример 40
(R)-N-{2-[5,6-дифтор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин и
(S)-N-{2-[5,6-дифтор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
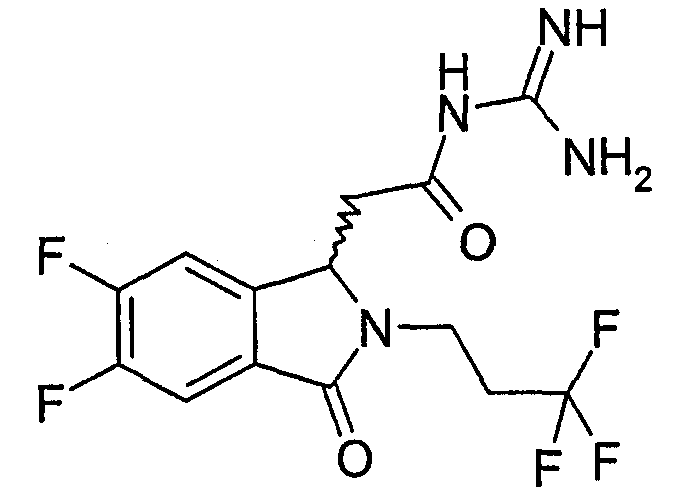
Указанное в заголовке соединение примера 40 синтезируют, следуя методике примера 27.
Rf (EA/МеОН 3:1)=0,33; MC (ES+): 365 (M+1)+).
Указанные в заголовке соединения примеров 40а и 40b получают хроматографией 207 мг примера 40 на Chiralpak AD-H, 250×20 мм, 10 мкм с использованием смеси ACN/HEP/i-PrOH 50:3:6+0,1% DEA при скорости потока 19 мл/минута.
Пример 40а
Выход аморфного твердого вещества: 30 мг.
Аналитическая ВЭЖХ на Chiralpak AD-H/33, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:3:6+0,1% DEA при скорости потока 1,0 мл/минута: RT=4,708 минут.
Пример 40b
Выход аморфного твердого вещества: 30 мг.
Аналитическая ВЭЖХ на Chiralpak AD-H/33, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:3:6+0,1% DEA при скорости потока 1,0 мл/минута: RT=9,719 минут.
Пример 41
(R)-N-{2-[5,6-дихлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин и
(S)-N-{2-[5,6-дихлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
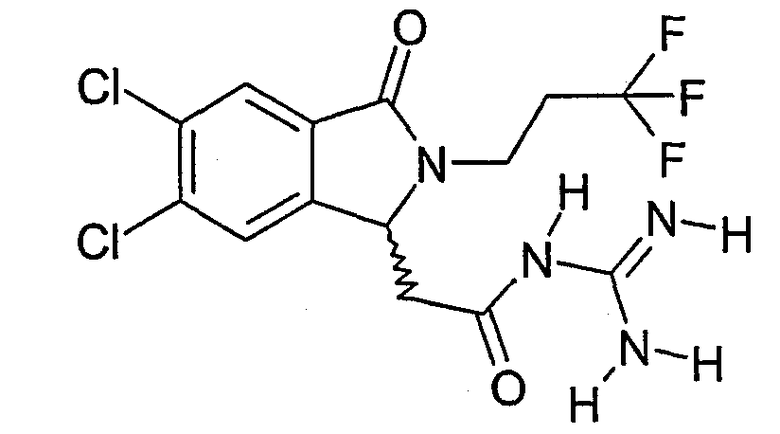
Указанные в заголовке соединения примера 41 синтезируют, следуя методике примера 27. Свободное основание превращают в ацетат во время хроматографии на силикагеле с использованием смеси ЕА/5% НОАс.
Пример 41а
N-{2-[5,6-дихлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидинийацетат
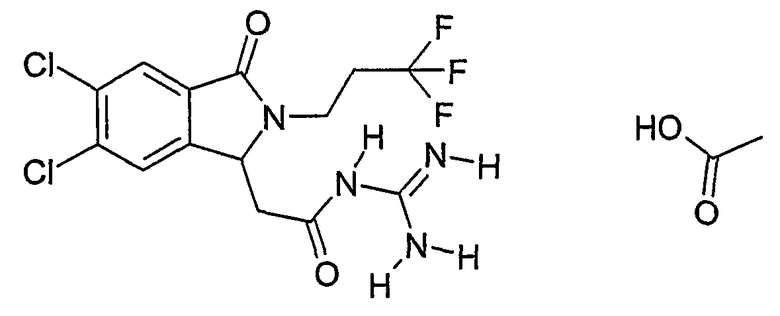
Rf (EA/5% НОАс)=0,16; MC (ES+): 397 (M+1)+).
Указанные в заголовке соединения примера 41b и 41с получают хроматографией 530 мг примера 41а на Chiralcel OJ, 250×50 мм, 20 мкм с использованием смеси НЕР/EtOH/MeOH 10:1:1+0,1% DEA при скорости потока 100 мл/минута.
Пример 41b
Выход аморфного свободного основания: 165 мг.
Аналитическая ВЭЖХ на Chiralcel OJ/37, 250×4,6 мм с использованием смеси HEP/EtOH/MeOH 20:1:1+0,1% DEA при скорости потока 1,0 мл/минута: RT=11,838 минут.
Пример 41с
Выход аморфного свободного основания: 122 мг.
Аналитическая ВЭЖХ на Chiralcel OJ/37, 250×4,6 мм с использованием смеси HEP/EtOH/MeOH 20:1:1+0,1% DEA при скорости потока 1,0 мл/минута: RT=16,029 минут.
Указанные в заголовке соединения примеров 42-45 синтезируют, следуя методике примера 8.
Пример 42
(R)-N-[2-(5,6-дихлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и
(S)-N-[2-(5,6-дихлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
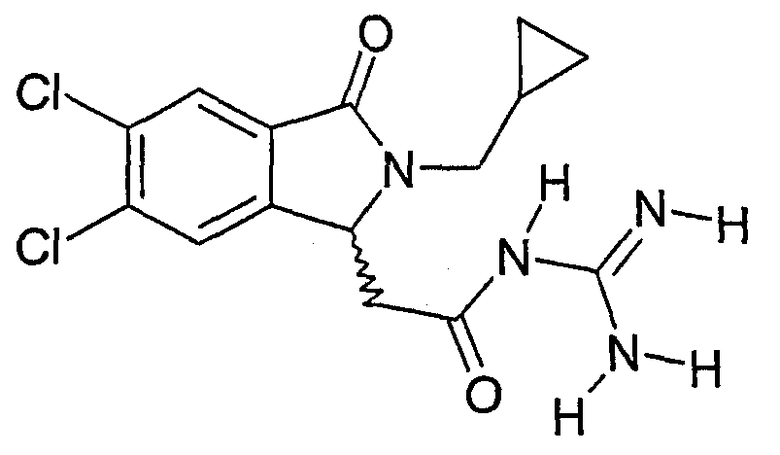
Rf (EA/MeOH 5:1)=0,082; MC (ES+): 355 (M+1)+).
Указанные в заголовке соединения примера 42а и 42b получают хроматографией 830 мг соединения примера 42 на колонке Chiralcel OJ, 250×50 мм, 20 мкм с использованием смеси НЕР/EtOH/MeOH 25:1:1+0,1% DEA при скорости потока 100 мл/минута.
Пример 42a
Выход аморфного твердого вещества: 113 мг.
Аналитическая ВЭЖХ на Chiralcel OJ/16, 250×4,6 с использованием смеси HEP/EtOH/MeOH 25:1:1+0,1% DEA при скорости потока 1,0 мл/минута: RT=11,691 минут.
Пример 42b
Выход аморфного твердого вещества: 199 мг.
Аналитическая ВЭЖХ на Chiralcel OJ/16, 250×4,6 с использованием смеси HEP/EtOH/MeOH 25:1:1+0,1% DEA при скорости потока 1,0 мл/минута: RT=14,032 минут.
Пример 43
(R)-N-[2-(5,6-дихлор-2-циклопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и
(S)-N-[2-(5,6-дихлор-2-циклопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
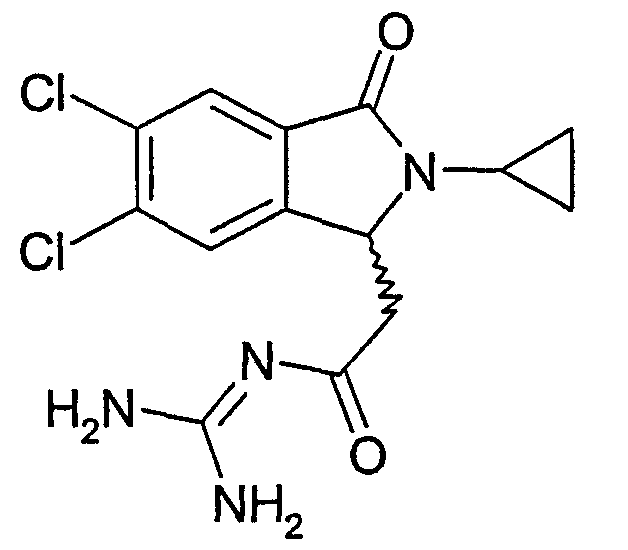
Rf (EA/MeOH 2:1)=0,13; MC (ES+): 341 (M+1)+).
Указанные в заголовке соединения примера 43а и 43b получают хроматографией 110 мг соединения примера 43 на колонке Chiralpak AD-H, 250×20 мм, 10 мкм с использованием смеси НЕР/EtOH/MeOH 1:1:1+0,1% DEA при скорости потока 10-15 мл/минута.
Пример 43a
Выход аморфного твердого вещества: 60 мг.
Аналитическая ВЭЖХ на Chiralpak AD-H/31, 250×4,6, с использованием смеси HEP/EtOH/MeOH 1:1:1+0,1% DEA при скорости потока 1,0 мл/минута: RT=6,420 минут.
Пример 43b
Выход аморфного твердого вещества: 41 мг.
Аналитическая ВЭЖХ на Chiralpak AD-H/31, 250×4,6 с использованием смеси HEP/EtOH/MeOH 1:1:1+0,1% DEA при скорости потока 1,0 мл/минута: RT=15,401 минут.
Пример 44
(R)-N-{2-[5,6-дихлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин и
(S)-N-{2-[5,6-дихлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
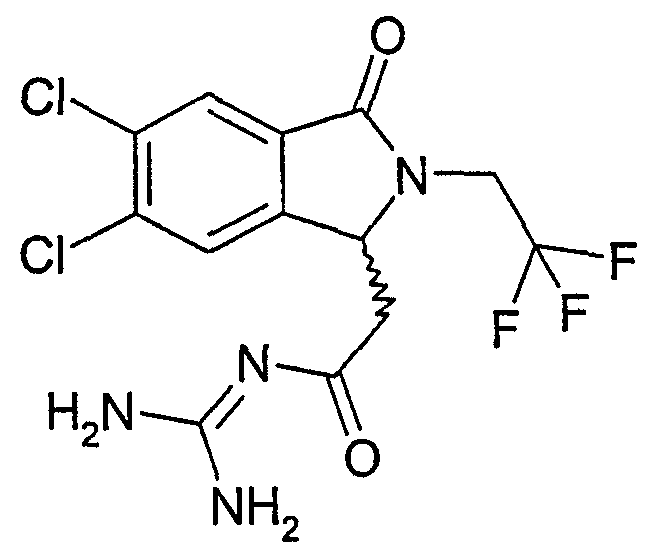
Свободное основание превращают в ацетат во время хроматографии на силикагеле с использованием смеси ЕА/5% НОАс.
Пример 44а
N-{2-[5,6-дихлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидинийацетат
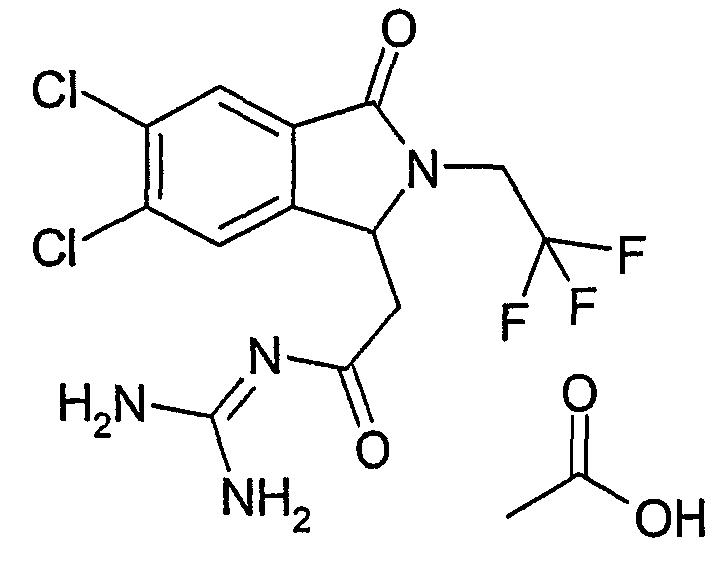
Rf (EA/5% НОАс)=0,27; MC (ES+): 383 (M+1)+).
Указанные в заголовке соединения примера 44b и 44с получают хроматографией 100 мг соединения примера 44а на Chiralpak AD-H, 250×20 мм, 10 мкм с использованием смеси НЕР/EtOH/MeOH 2:1:1+0,1% DEA при скорости потока 14 мл/минута.
Пример 44b
Выход аморфного свободного основания: 25 мг.
Аналитическая ВЭЖХ на Chiralpak AD-H/31, 250×4,6 мм с использованием смеси HEP/EtOH/MeOH 20:1:1+0,1% DEA при скорости потока 1,0 мл/минута: RT=5,027 минут.
Пример 44с
Выход аморфного свободного основания: 26 мг.
Аналитическая ВЭЖХ на Chiralpak AD-H/31, 250×4,6 мм с использованием смеси HEP/EtOH/MeOH 20:1:1+0,1% DEA при скорости потока 1,0 мл/минута: RT=9,012 минут.
Пример 45
N-{2-[5,6-дифтор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
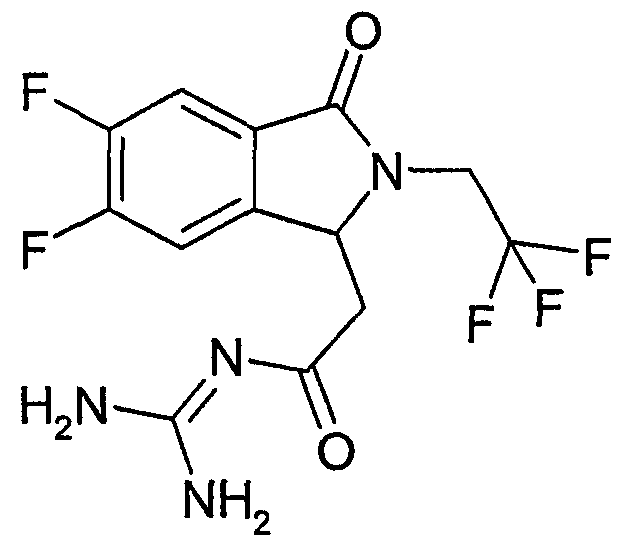
Rf (EA/МеОН 3:1)=0,21; MC (ES+): 351 (M+1)+).
Пример 46
(R)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин и
(S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
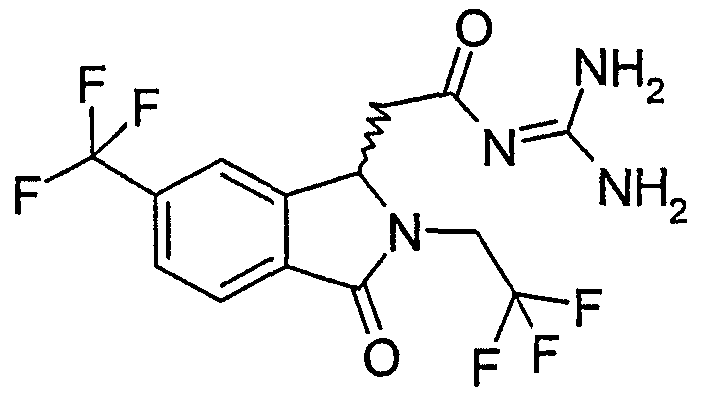
Указанные в заголовке соединения примера 46а и 46b получают хроматографией 0,56 г соединения примера 21 на Chiralcel OD, 250×50 мм, 20 мкм с использованием смеси НЕР/EtOH/MeOH 50:5:2 при скорости потока 100 мл/минута, разделяя два энантиомера.
Пример 46а
Выход аморфного твердого вещества: 120 мг.
Аналитическая ВЭЖХ на Chiralcel OD/20, 250×4,6 с использованием смеси HEP/EtOH/MeOH 50:5:2 при скорости потока 1,0 мл/минута: RT=9,620 минут.
Пример 46b
Выход аморфного твердого вещества: 190 мг.
Аналитическая ВЭЖХ на Chiralcel OD/20, 250×4,6 с использованием смеси HEP/EtOH/MeOH 50:5:2 при скорости потока 1,0 мл/минута: RT=11,899 минут.
Пример 47
(R)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин и
(S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
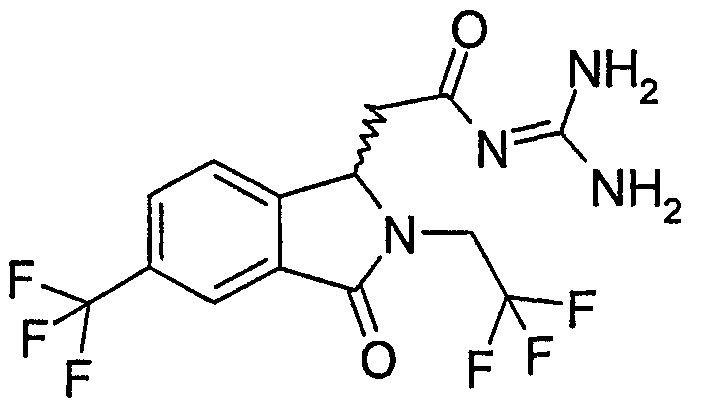
Указанные в заголовке соединения синтезируют, следуя методике примера 21, с использованием в качестве исходного продукта этилового эфира [3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]уксусной кислоты (пример 21). Rf (EA/МеОН 3:1)=0,40; MC (ES+): 383 (M+1)+).
Энантиомеры разделяли хроматографией на Chiralpak AD-H, 250×20 мм, 10 мкм с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 3-6 мл/минута.
Пример 47а
Выход аморфного твердого вещества: 24 мг.
Аналитическая ВЭЖХ на Chiralcel OD/20, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=4,236 минут.
Пример 47b
Выход аморфного твердого вещества: 31 мг.
Аналитическая ВЭЖХ на Chiralcel OD/20, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=6,296 минут.
Пример 48
N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидинийбифумарат
1,5 г указанного в заголовке соединения примера 46b и 0,45 г фумаровой кислоты растворяют вместе с использованием смеси ацетон/ACN 1:1+1 мл воды и перемешивают в течение 15 минут при комнатной температуре. В вакууме удаляют растворители, остаток суспендируют с использованием 50 мл CH2Cl2 и отфильтровывают продукт. Продукт сушат в вакууме с получением 1,65 г белых кристаллов, т.пл. 202°С.
Указанные в заголовке соединения примера 49 и 50 получают хроматографией 600 мг соединения примера 26 на Chiralpak AD, 250×50 мм, 20 мкм с использованием смеси ACN/HEP/i-PrOH 50:4:3+0,3% DEA при скорости потока 100 мл/минута.
Пример 49
(R)-N-[2-(6-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и
(S)-N-[2-(6-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
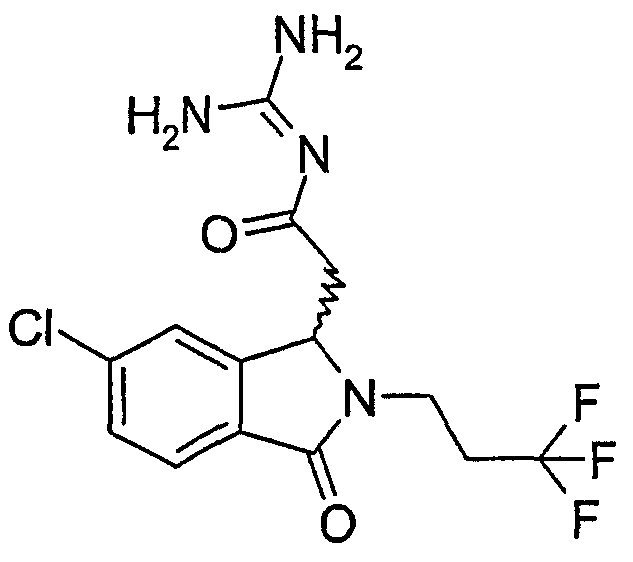
Пример 49а
Выход аморфного твердого вещества: 90 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:3:4+0,3% DEA при скорости потока 1,0 мл/минута: RT=6,092 минут.
Пример 49b
Выход аморфного твердого вещества: 110 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:3:4+0,3% DEA при скорости потока 1,0 мл/минута: RT=11,876 минут.
Пример 50
(R)-N-[2-(5-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин и
(S)-N-[2-(5-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин
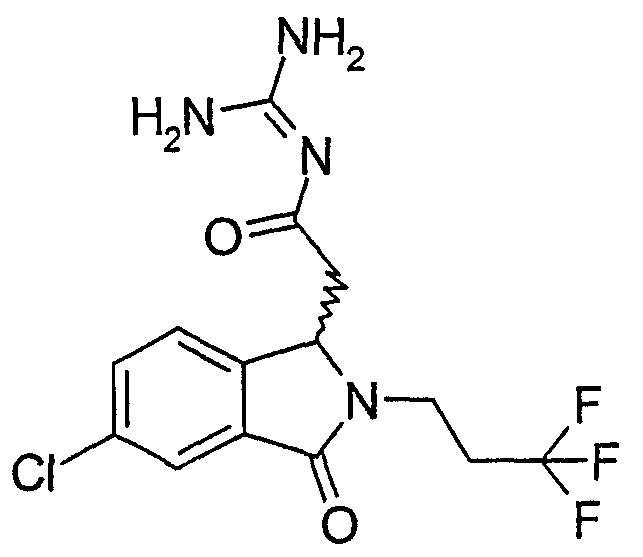
Пример 50а
Выход аморфного твердого вещества: 35 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:3:4+0,3% DEA при скорости потока 1,0 мл/минута: RT=5,663 минут.
Пример 50b
Выход аморфного твердого вещества: 51 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:3:4+0,3% DEA при скорости потока 1,0 мл/минута: RT=23,673 минут.
Указанные в заголовке соединения примеров 51 и 52 получают хроматографией 280 мг примера 27 на Chiralpac AD, 250×50 мм, 20 мкм с использованием смеси ACN/HEP/i-PrOH 50:4:3+0,3% DEA при скорости потока 50 мл/минута.
Пример 51
(R)-N-{2-[3-оксо-6-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин и
(S)-N-{2-[3-оксо-6-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
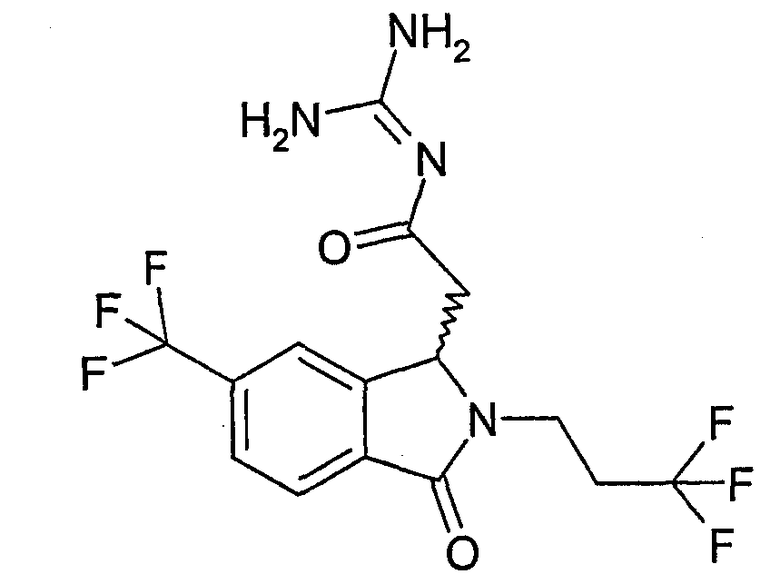
Пример 51а
Выход аморфного твердого вещества: 29 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=4,037 минут.
Пример 51b
Выход аморфного твердого вещества: 27 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=5,083 минут.
Пример 52
(R)-N-{2-[3-оксо-5-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин и
(S)-N-{2-[3-оксо-5-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
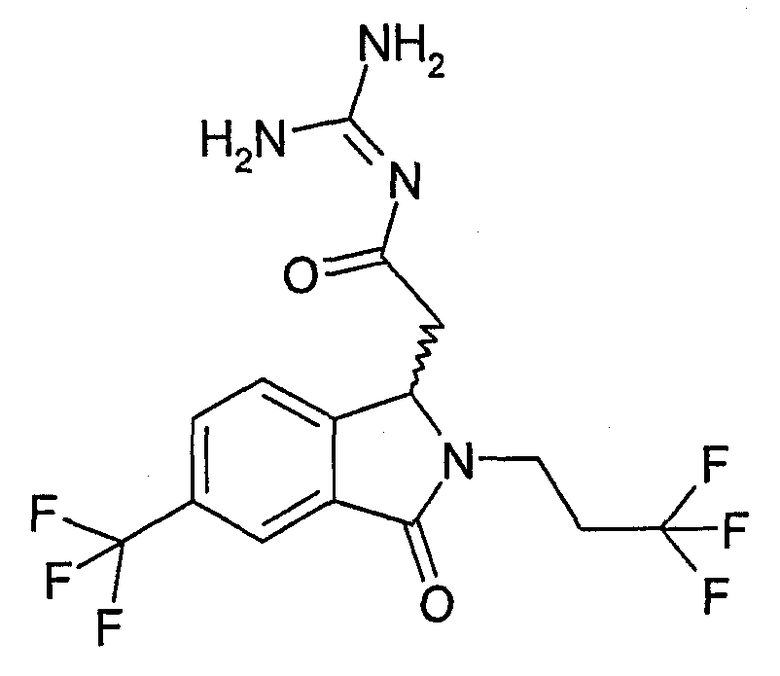
Пример 52а
Выход аморфного твердого вещества: 15 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=4,497 минут.
Пример 52b
Выход аморфного твердого вещества: 68 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=8,228 минут.
Указанные в заголовке соединения примеров 53 и 54 получают хроматографией 300 мг соединения примера 28 на Chiralpac AD, 250×50 мм, 20 мкм с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 100 мл/минута.
Пример 53
(R)-N-{2-[6-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин и
(S)-N-{2-[6-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
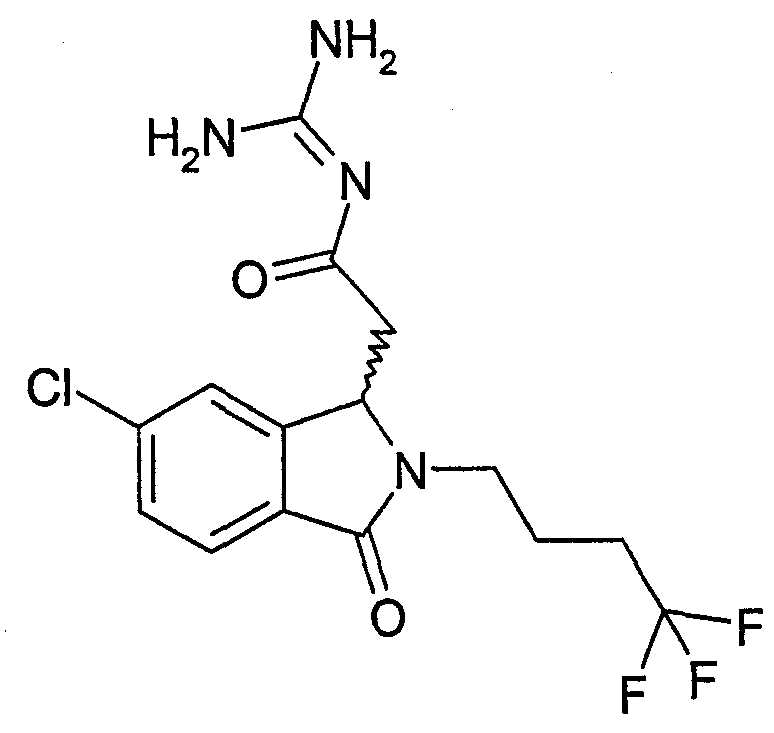
Пример 53а
Выход аморфного твердого вещества: 76 мг
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:4:3+0,3% DEA при скорости потока 1,0 мл/минута: RT=5,761 минут.
Пример 53b
Выход аморфного твердого вещества: 34 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:4:3+0,3% DEA при скорости потока 1,0 мл/минута: RT=7,079 минут.
Пример 54
(R)-N-{2-[5-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин и
(S)-N-{2-[5-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин
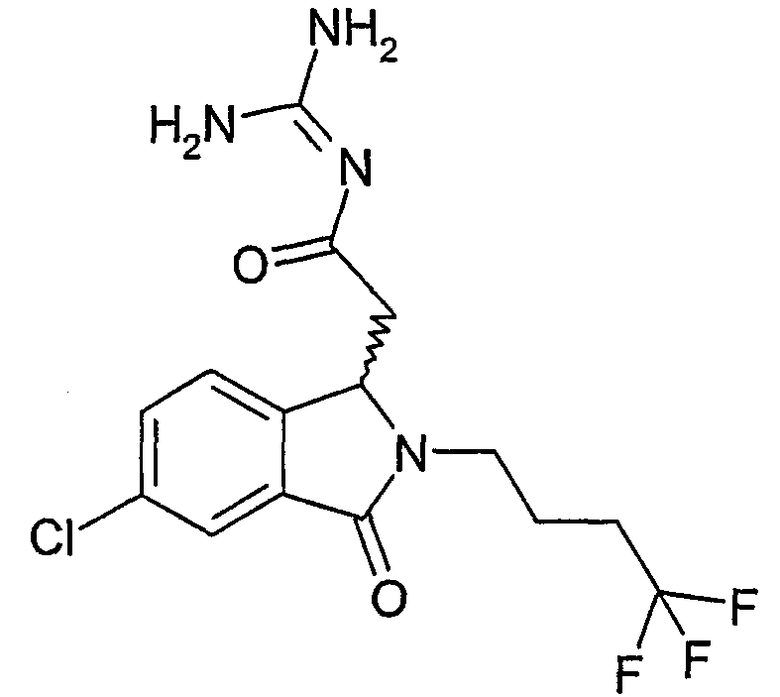
Пример 54а
Выход аморфного твердого вещества: 34 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:4:3+0,3% DEA при скорости потока 1,0 мл/минута: RT=5,421 минут.
Пример 54b
Выход аморфного твердого вещества: 72 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:4:3+0,3% DEA при скорости потока 1,0 мл/минута: RT=9,865 минут.
Указанные в заголовке соединения примеров 55 и 56 получают хроматографией 330 мг соединения примера 24 на Chiralpac AD, 250×50 мм, 20 мкм с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 100 мл/минута.
Пример 55
(R)-N-[2-[6-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил]ацетил]гуанидин и
(S)-N-[2-[6-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил]ацетил]гуанидин
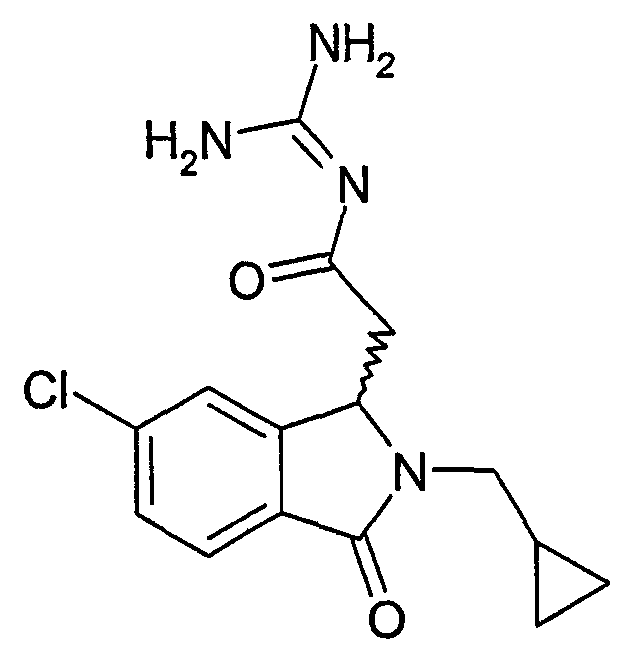
Пример 55а
Выход аморфного твердого вещества: 16 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=7,330 минут.
Пример 55b
Выход аморфного твердого вещества: 46 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=11,908 минут.
Пример 56
(R)-N-[2-[5-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил]ацетил]гуанидин и
(S)-N-[2-[5-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил]ацетил]гуанидин
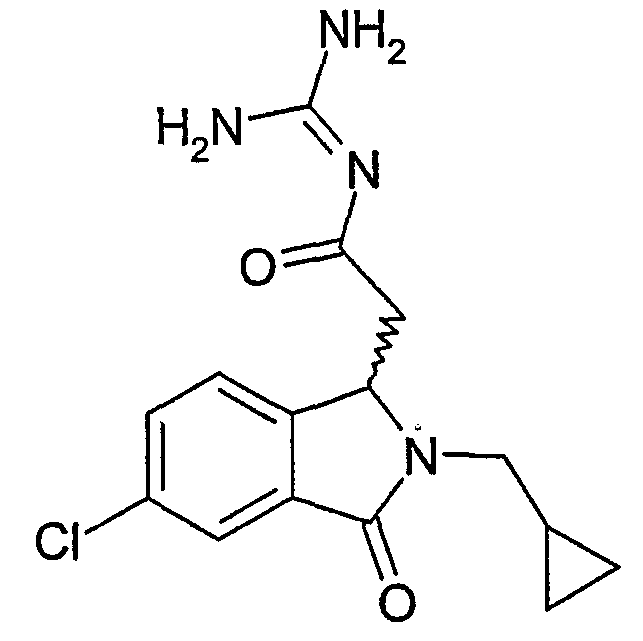
Пример 56а
Выход аморфного твердого вещества: 32 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=6,821 минут.
Пример 56b
Выход аморфного твердого вещества: 47 мг.
Аналитическая ВЭЖХ на Chiralpac AD/H, 250×4,6 с использованием смеси ACN/HEP/i-PrOH 50:5:2+0,3% DEA при скорости потока 1,0 мл/минута: RT=27,738 минут.
Способы ингибирования NHE
Ингибирующие активности в отношении NHE (значения IC50) соединений в соответствии с изобретением определяли испытанием на FLIPR.
Испытание проводят на FLIPR (планшетридер для считывания флуоресцирующего изображения), снабженном 96-луночными планшетами для микротитрования с прозрачным дном и темными стенками. За день до испытания высевают линии трансфицированных клеток при плотности ˜25000 клеток/лунка, экспрессирующих различные подтипы NHE (линия родительских клеток LAP-1 не показывает эндогенную NHE активность в результате мутагенеза и последующей селекции).
Питательная среда для трансфицированных клеток (Iscove +10% фетальная телячья сыворотка) содержит также в качестве выбранного антибиотика G418 для обеспечения присутствия трансфицированных последовательностей.
Действительное испытание начинается удалением питательной среды и добавлением 100 мкл загрузочного буфера на лунку (5 мкМ BCECF-AM) [сложный 2',7'-бис(2-карбоксиэтил)-5(6)-карбоксифлуоресцеинацетоксиметиловый эфир] в 20 мМ NH4Cl, 115 мМ холинхлорида, 1мМ CaCl2, 5 мМ KCl, 20 мМ HEPES и 5 мМ глюкозы; рН 7,4 (доведенный до указанного значения КОН). Затем клетки выращивают в течение 20 минут при 37°С. Указанная инкубация приводит к загрузке в клетки флуоресцирующего красителя, интенсивность флуоресценции которого зависит от pHi и от NH4Cl, который приводит к слабому подщелачиванию клеток.
Предшественник BCECF-AM, нефлуоресцирующий краситель, представляет собой сложный эфир, способный пересекать мембрану. Действительный краситель, который не способен пересекать мембрану, высвобождается внутри клеток эстеразами.
После 20-минутной инкубации загрузочный буфер, содержащий NH4Cl и свободный BCECF-AM, удаляют трехразовой промывкой в устройстве для промывки клеток (Tecan Columbus), при этом каждую промывку осуществляют с использованием 400 мкл промывочного буфера (133,8 мМ холинхлорида, 4 мМ KCl, 1,25 мМ MgCl2, 1,25 мМ CaCl2, 0,97 мМ K2HPO4, 0,23 мМ KH2PO4, 5 мМ HEPES и 5 мМ глюкозы; рН 7,4 (доведенный до указанного значения КОН). Оставшийся в клетках остаточный объем равен 90 мкл (возможно между 50 и 125 мкл). На стадии промывки удаляется свободный BCECF-AM, что приводит к внутриклеточному подкислению (pHi равен 6,3-6,4) вследствие удаления внеклеточных ионов аммония.
Когда равновесие между внутриклеточным аммонием и водным раствором аммиака и протонами нарушается за счет удаления внеклеточного аммония и последующего немедленного перехода водного раствора аммиака через клеточную мембрану, процесс промывки приводит к удерживанию внутриклеточных протонов, которое является причиной внутриклеточного подкисления. Такое подкисление может привести, в конечном счете, к гибели клеток, если оно продолжается достаточно долго. Поэтому в данном случае важно, чтобы промывочный буфер не содержал натрий (<1 мМ), иначе ионы внеклеточного натрия будут приводить к немедленному повышению pHi вследствие активности клонированных изоформ NHE. Важно также, чтобы все используемые буферы (загрузочный буфер, промывочный буфер и регенерационный буфер) не содержали HCO3-ионы, в противном случае присутствие бикарбоната будет приводить к активации бикарбонатзависимых систем, содержащихся в линии родительских клеток LAP-1, которые нарушают регулирование pHi.
Затем планшет для микротитрования, содержащий подкисленные клетки, переносят (через период времени после подкисления до 20 минут) на FLIPR. На FLIPR внутриклеточный флуоресцирующий краситель активируют светом с длиной волны 488 нм, который генерируется аргоновым лазером, и выбирают параметры для измерения (мощность лазера, время облучения и диафрагму CDD камеры, вмонтированной в FLIPR) таким образом, чтобы среднее значение флуоресцирующего сигнала на лунку находилось между 30000 и 35000 относительными единицами флуоресценции.
Действительное измерение на FLIPR начинается с фотографирования, осуществляемого CDD камерой каждые две секунды. Через 10 секунд инициируют повышение внутриклеточного рН добавлением 90 мкл регенерационного буфера (133,8 мМ NaCl, 4,7 мМ KCl, 1,25 мМ MgCl2, 1,25 мМ CaCl2, 0,97 мМ K2HPO4, 0,23 мМ KH2PO4, 10 мМ HEPES и 5 мМ глюкозы; рН 7,4 (доведенный NaOH)) с использованием пипетки, приспособленной для 96 лунок, включенного в FLIPR. Некоторые лунки, в которые добавляют чистый регенерационный буфер, служат в качестве положительных контролей (100% активность в отношении NHE). Отрицательные контроли (0% активность в отношении NHE) содержат промывочный буфер. Во все другие лунки добавляют регенерационный буфер с удвоенной концентрацией испытуемого вещества. Измерение на FLIPR заканчивается после 60 измерений (две минуты).
Экспериментальные данные дают возможность вычислить активности испытуемого вещества в отношении NHE при любой его концентрации и из полученных данных определить значения IC50. Для подтипа NHE1 получают следующие результаты.
(как гидрохлорид)
(как трифторацетат)
(как гидрохлорид)
(как трифторацетат)
(как гидрохлорид)
(как трифторацетат)
Изобретение также относится к применению производных изоиндолона формулы I и/или их фармацевтически приемлемых солей для получения лекарственных средств и фармацевтических композиций в качестве ингибиторов NHE. Изобретение включает лекарственное средство, предназначенное для лечения людей, применения в ветеринарии или для защиты растений, которое содержит эффективное количество соединения формулы I и/или его фармацевтически приемлемых солей вместе с фармацевтически приемлемыми носителями и добавками в отдельности или в комбинации с другими фармацевтически ингредиентами или лекарственными средствами.
Фармацевтические композиции в соответствии с изобретением состоят из соединения формулы I и/или его фармацевтически приемлемой соли в чистой форме или в форме композиции, в которой оно (они) объединено(ы) с любым другими фармацевтически совместимым продуктом, который может быть инертным или физиологически активным. Лекарственные средства в соответствии с изобретением могут быть введены, например, перорально, парентерально, внутривенно, ректально, чрескожно, местно или ингаляцией. Лекарственные средства обычно содержат активные ингредиенты формулы I и/или их фармацевтически приемлемые соли в количестве от 0,001 мг до 1 г на дозированную единицу.
Подходящие для желательной фармацевтической композиции эксципиенты знакомы специалистам в данной области на основе знания специалиста. Кроме растворителей, гелеобразователей, основ для суппозиториев, эксципиентов для таблеток и других носителей активного ингредиента, возможно использование, например, антиоксидантов, диспергирующих веществ, эмульгаторов, противовспенивателей, корригентов, консервантов, солюбилизаторов или красителей.
Активные соединения, предназначенные для фармацевтической композиции для перорального введения, смешивают с добавками, подходящими для данной цели, такими как носители, стабилизаторы или инертные разбавители, и превращают традиционными методами в подходящие дозированные формы, такие как таблетки, таблетки с покрытием, твердые желатиновые капсулы, водные, спиртовые или масляные растворы. Примеры возможных для использования инертных носителей включают гуммиарабик, магнезию, карбонат магния, фосфат калия, лактозу, глюкозу или крахмал, в частности кукурузный крахмал. Более того, возможно использование для получения как сухих гранул, так и влажных гранул. Примерами подходящих масляных носителей или растворителей являются растительные масла или животные жиры, такие как подсолнечное масло или масло печени рыб.
В качестве твердой композиции для перорального введения могут быть использованы таблетки, пилюли, порошки (желатиновые капсулы или крахмальные капсулы) или гранулы. В данных композициях активный ингредиент согласно изобретению смешивают с одним или более инертными разбавителями, такими как крахмал, целлюлоза, сахароза, лактоза или диоксид кремния, в токе аргона. Данные композиции могут также содержать иные вещества, чем разбавители, например одно или более лубрикантов, таких как стеарат магния или тальк, краситель, покрытие (драже) или лак.
В качестве жидких композиций для перорального введения могут быть использованы фармацевтически приемлемые растворы, суспензии, эмульсии, сиропы и эликсиры, содержащие инертные разбавители, такие как вода, этанол, глицерин, растительные масла или жидкий парафин. Данные композиции могут содержать иные вещества, чем разбавители, например, увлажняющие продукты, подсластители, загустители, корригенты или стабилизаторы.
Стерильные композиции для парентерального введения могут предпочтительно представлять собой водные или неводные растворы, суспензии или эмульсии. Возможные для использования растворители или наполнители (носители) включают воду, пропиленгликоль, полиэтиленгликоль, растительные масла, в частности оливковое масло, инъецируемые органические сложные эфиры, например этилолеат, или другие подходящие органические растворители. Данные композиции могут также содержать адъюванты, в частности увлажняющие средства, тонизирующие средства, эмульгаторы, диспергирующие вещества и стабилизаторы. Стерилизация может быть осуществлена несколькими способами, например асептической фильтрацией, включением стерилизующих средств в композицию, облучением или нагревом. Они могут быть также приготовлены в форме стерильных твердых композиций, которые могут быть растворены во время использования в стерильной воде или любой другой стерильной среде для инъекций.
Композиции для ректального введения представляют собой суппозитории или ректальные капсулы, которые, кроме активного ингредиента, содержат эксципиенты, такие как масло какао, полусинтетические глицериды или полиэтиленгликоли.
Композиции для местного введения могут представлять собой, например, кремы, лосьоны, глазные капли, средства для полоскания рта, капли для носа или аэрозоли.
Для подкожного, внутримышечного или внутривенного введения используемые активные соединения превращают, если требуется, вместе с веществами, обычными для этой цели, такими как солюбилизаторы, эмульгаторы или другие эксципиенты, в раствор, суспензию или эмульсию. Примерами подходящих растворителей являются: вода, физиологический раствор или спирты, например этанол, пропанол, глицерин, а также растворы сахаров, такие как растворы глюкозы или маннита, или, кроме того, смесь различных указанных растворителей.
Подходящими в качестве фармацевтической композиции для введения в форме аэрозолей или распыляемых растворов являются, например, растворы, суспензии или эмульсии активного ингредиента формулы I и/или его фармацевтически приемлемых солей в фармацевтически приемлемом растворителе, в частности таком как этанол, или вода, или в смеси указанных растворителей. В случае необходимости композиция может также содержать иные фармацевтические эксципиенты, такие как поверхностно-активные вещества, эмульгаторы и стабилизаторы и газ-вытеснитель. Такой препарат содержит, например, активный ингредиент с концентрацией от около 1 до 10, в частности от около 0,3 до 3 мас.%.
Доза вводимого активного ингредиента формулы I и частота введения зависят от требуемого эффекта, эффективности и продолжительности действия используемых соединений и дополнительно также от природы и тяжести подвергаемого лечению расстройства и от пола, возраста, массы тела и от индивидуальной восприимчивости млекопитающего, подвергаемого лечению. Обычно лечащий врач определяет соответствующую дозу как функцию возраста и массы тела больного и всех других факторов, специфических для подвергаемого лечению индивидуума.
Суточная доза соединения формулы I и/или его фармацевтически приемлемых солей для больного весом 75 кг составляет в среднем, по меньшей мере, 0,001 мг/кг, предпочтительно 1 мг/кг и максимально 1000 мг/кг, предпочтительно 100 мг/кг массы тела. Для острых периодов расстройства, например, сразу же после перенесенного инфаркта миокарда, могут быть также необходимы более высокие и, в частности, более частые дозы, например до 4-х разовых доз в день. Может быть необходимо введение вплоть до 2000 мг в день, в частности при внутривенном введении, например больному инфарктом в отделении интенсивной терапии, и соединения изобретения могут быть введены вливанием.
Следующие примеры иллюстрируют композиции в соответствии с изобретением.
ПРИМЕР А
Гелевые капсулы, содержащие 50 мг дозу активного продукта, имеющие указанный ниже состав, получают в соответствии с общепринятой методикой:
ПРИМЕР В
Таблетки, содержащие 50 мг дозу активного ингредиента, имеющие указанный ниже состав, получают в соответствии с общепринятой методикой:
ПРИМЕР С
Получают инъецируемый раствор, содержащий 10 мг активного ингредиента, имеющий указанный ниже состав:
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ (3-ОКСО-2,3-ДИГИДРО-1Н-ИЗОИНДОЛ-1-ИЛ)-АЦЕТИЛГУАНИДИНА | 2004 |
|
RU2397159C2 |
| ПРОИЗВОДНЫЕ 2-ИМИНОПИРРОЛИДИНА | 2002 |
|
RU2270192C2 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2797295C1 |
| ПРОИЗВОДНЫЕ ГИДРОКСИБЕНЗАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Hsp90 | 2006 |
|
RU2458919C2 |
| ТИАЗОЛБЕНЗОГЕТЕРОЦИКЛЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ПОЛУЧЕНИЯ ТИАЗОЛБЕНЗОГЕТЕРОЦИКЛОВ | 1998 |
|
RU2198889C2 |
| ИЗОИНДОЛИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ РАКА | 2009 |
|
RU2527952C2 |
| НОВЫЕ ИЗОИНДОЛИНОВЫЕ ИЛИ ИЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2689305C2 |
| ПРОИЗВОДНЫЕ 3,4-ДИГИДРО-2Н-ИЗОХИНОЛИН-1-ОНА И 2,3-ДИГИДРОИЗОИНДОЛ-1-OHA, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА АЛЬДОСТЕРОНСИНТАЗЫ | 2014 |
|
RU2695524C2 |
| ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА У МЛЕКОПИТАЮЩЕГО | 2000 |
|
RU2263109C2 |
| НОВЫЕ СОЕДИНЕНИЯ 3,4-ДИГИДРО-2Н-ИЗОХИНОЛИН-1-ОНА И 2,3-ДИГИДРОИЗОИНДОЛ-1-ОНА | 2014 |
|
RU2693698C2 |
Описываются соединения формулы (I), в которой R1 и R2 обозначают Н, алкил, F, Cl, Br, алкокси, S(O)nR7, полифторалкил, полифторалкокси; R3 обозначает Alk-R8, циклоалкил; R8 обозначает Н, циклоалкил, полифторалкил, фенил или ОН; R4, R5 и R6 обозначают Н или алкил; R7 обозначает алкил, а также способ их получения, фармацевтическая композиция, обладающая ингибирующей активностью в отношении Na+/K+ клеточного обмена. Заявляемые соединения являются подходящими в качестве противоаритмических лекарственных средств с кардиозащитным компонентом для профилактики инфаркта и лечения инфаркта и лечения стенокардии. Они также ингибируют превентивным образом патофизиологические процессы, связанные с развитием нарушений, вызванных ишемией, в частности возникновение вызванной ишемией сердечной аритмии и сердечной недостаточности. 8 н. и 9 з.п. ф-лы.
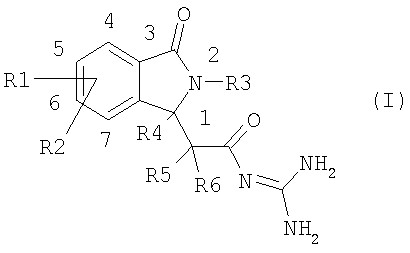
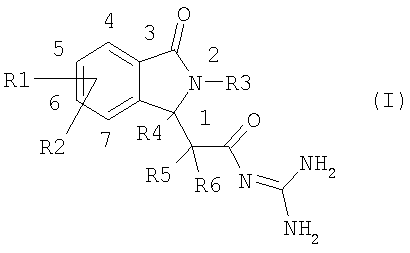
в которой R1 и R2 независимо друг от друга представляют собой водород, алкил, имеющий 1, 2, 3 или 4 атома углерода, F, Cl, Br, алкокси, имеющий 1, 2, 3 или 4 атома углерода, S(O)nR7, полифторалкил, имеющий 1, 2, 3 или 4 атома углерода, или полифторалкокси, имеющий 1, 2 или 3 атома углерода;
n равно 2;
R3 представляет собой группу типа Alk-R8 или циклоалкил, имеющий 3, 4, 5, 6, 7 или 8 атомов углерода;
Alk представляет собой алкил, имеющий 1, 2, 3, 4 или 5 атомов углерода в линейной или разветвленной цепи;
R8 представляет собой водород, циклоалкил, имеющий 3, 4, 5, 6, 7 или 8 атомов углерода, полифторалкил, имеющий 1, 2, 3 или 4 атома углерода, фенил или ОН;
R4, R5 и R6 независимо друг от друга представляют собой водород или линейный или разветвленный алкил, имеющий 1, 2, 3 или 4 атома углерода;
R7 представляет собой линейный или разветвленный алкил, имеющий 1, 2, 3 или 4 атома углерода;
и их рацемические смеси, энантиомеры и диастереоизомеры и их смеси, их таутомеры и их фармацевтически приемлемые соли.
R1 и R2 независимо друг от друга представляют собой водород, алкил, имеющий 1, 2, 3 или 4 атома углерода, F, Cl, Br, алкокси, имеющий 1, 2, 3 или 4 атома углерода, полифторалкил, имеющий 1, 2, 3 или 4 атома углерода, или полифторалкокси, имеющий 1, 2 или 3 атома углерода,
R3 представляет собой группу типа Alk-R8 или циклоалкил, имеющий 3, 4, 5, 6, 7 или 8 атомов углерода,
Alk представляет собой алкил, имеющий 1, 2, 3, 4 или 5 атомов углерода в линейной или разветвленной цепи,
R8 представляет собой водород, циклоалкил, имеющий 3, 4, 5, 6, 7 или 8 атомов углерода, или полифторалкил, имеющий 1, 2, 3 или 4 атома углерода;
R4, R5 и R6 независимо друг от друга представляют водород или линейный или разветвленный алкил, имеющий 1, 2, 3 или 4 атома углерода;
и их рацемические смеси, энантиомеры и диастереоизомеры и их смеси, их таутомеры и их фармацевтически приемлемые соли.
N-[2-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)-2-метилпропионил]гуанидин,
N-[2-(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)-2-метилпропионил]гуанидин,
N-[(3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-7-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4-амино-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-амино-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-амино-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(7-амино-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4-гидрокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-гидрокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-гидрокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(7-гидрокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4,7-дихлор-2-изобутил-3-оксо-2,3-дигидро-1H-изоиндол-1-ил)ацетил]гуанидин,
N-[(4-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил] гуанидин,
N-[(6-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4,5-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6,7-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4-карбокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-карбокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-карбокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(7-карбокси-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-1-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
и их рацемические смеси, энантиомеры и диастереоизомеры и их смеси, их таутомеры и их фармацевтически приемлемые соли.
N-[(2-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-этил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[2-(3-оксо-2-пропил-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин,
N-[(2-изопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[2-(2-бутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин,
N-[(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[2-(2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин,
N-[(2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-бензил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-4-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-5-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-6-метил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-трет-бутил-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-5-изопропокси-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-хлор-2-изобутил-3-оксо-2,3-дигидро-1H-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-хлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1H-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-хлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-хлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5,6-дихлор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(7-фтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(4,7-дифтор-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(5-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(6-бром-2-изобутил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-изобутил-3-оксо-6-трифторметил-2,3-дигидро-1H-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-циклопропилметил-3-оксо-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-5-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-6-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(4,4,4-трифторбутил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[(3-оксо-2-(4,4,4-трифторбутил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[1-(2-гуанидино-1-метил-2-оксоэтил)-3-оксо-1,3-дигидроизоиндол-2-ил]уксусная кислота,
N-{2-[3-оксо-2-(2-пирролидин-1-илэтил)-2,3-дигидро-1H-изоиндол-1-ил]пропионил}гуанидин,
N-[2-(2-гидроксиэтил)-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)пропионил]гуанидин,
N-{2-[6-метансульфонил-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
N-[2-(2-циклопропилметил-6-метансульфонил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-{2-[5,6-дифтор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
N-{2-[5,6-дихлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
N-[2-(5,6-дихлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-[2-(5,6-дихлор-2-циклопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
N-{2-[5,6-дихлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
N-{2-[5,6-дифтор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
и их рацемические смеси, энантиомеры и диастереоизомеры, их таутомеры и их фармацевтически приемлемые соли.
(R)-N-{2-[6-метансульфонил-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[6-метансульфонил-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-[2-(2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(2-циклопропилметил-3-оксо-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-{2-[5,6-дифтор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[5,6-дифтор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[5,6-дихлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[5,6-дихлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-[2-(5,6-дихлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(5,6-дихлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-[2-(5,6-дихлор-2-циклопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(5,6-дихлор-2-циклопропил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-{2-[5,6-дихлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[5,6-дихлор-3-оксо-2-(2,2,2-трифторэтил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-6-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[3-оксо-2-(2,2,2-трифторэтил)-5-трифторметил-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-[2-(6-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(6-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-[2-(5-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(5-хлор-3-оксо-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-{2-[3-оксо-6-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[3-оксо-6-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[3-оксо-5-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[3-оксо-5-трифторметил-2-(3,3,3-трифторпропил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[6-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[6-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-{2-[5-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(S)-N-{2-[5-хлор-3-оксо-2-(4,4,4-трифторбутил)-2,3-дигидро-1Н-изоиндол-1-ил]ацетил}гуанидин,
(R)-N-[2-(6-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(6-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(R)-N-[2-(5-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин,
(S)-N-[2-(5-хлор-2-циклопропилметил-3-оксо-2,3-дигидро-1Н-изоиндол-1-ил)ацетил]гуанидин;
и их фармацевтически приемлемые соли и таутомеры.
a) комплексный гидрид подвергают взаимодействию с фталимидом (формулы II, R1, R2 и R3 имеют такие же значения, как определено в пп.1-5)
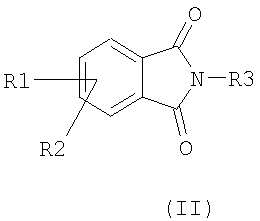
b) полученный продукт затем подвергают взаимодействию с алкоксикарбонилметилентрифенилфосфораном в толуоле, или с триалкилфосфоноацетатом и основанием;
c) полученный продукт подвергают взаимодействию с гуанидинийхлоридом и основанием или с гуанидином, например, в спирте, имеющем 1, 2, 3 или 4 атома углерода.
а) соединение формулы III (R1 и R2 имеют такие же значения, как определено в пп.1-5) подвергают взаимодействию с алкоксикарбонилметилентрифенилфосфораном в толуоле, или с триалкилфосфоноацетатом и основанием
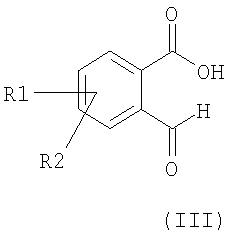
b) полученный продукт подвергают взаимодействию с амином формулы R3NH2 (R3 имеет такие же значения, как определено в любом из пп.1-5) и карбодиимидом;
c) полученный продукт подвергают взаимодействию с гуанидинийхлоридом и основанием, или с гуанидином, например, в спирте, имеющем 1, 2, 3 или 4 атома углерода.
a) фталимид (формулы II, как определено в п.14) подвергают взаимодействию с алкилмагнийгалогенидом или алкиллитиевым реагентом в простом эфире;
b) полученный продукт затем подвергают взаимодействию с алкоксикарбонилметилентрифенилфосфораном в толуоле или с 1-этокси-!-триметилсилоксиэтиленом и кислотой Льюиса;
c) полученный продукт подвергают взаимодействию с гуанидинийхлоридом и основанием, или с гуанидином, например в спирте, имеющем 1, 2, 3 или 4 атома углерода.
а) соединение формулы IV (R1-R5 имеют такие же значения, как определено в пп.1-5) подвергают взаимодействию в присутствии литийдиизопропиламида с R6-Hal, где Hal представляет собой F, Cl, Br или I
b) полученный продукт подвергают взаимодействию с гуанидинийхлоридом и основанием, или с гуанидином, например в спирте, имеющем 1, 2, 3 или 4 атома углерода.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Kawamoto T at al, Potent and selective inhibition of the human Na+/H+ exchanger isoform NHE1 by a novel aminoguanidine derivative T-162559, European journal of pharmacology, 2001, vol.420, №1, p.1-8 | |||
| RU 2141476 C1, 20.11.1999. | |||

