Настоящее изобретение относится к фенилэтиламинопроизводным, их фармацевтически приемлемым солям, фармацевтическим композициям, содержащим такие производные, и к их применению в качестве модуляторов натриевых и/или кальциевых каналов.
Фенилэтиламинопроизводные, являясь объектом настоящего изобретения, действуют как активные модуляторы кальциевых и/или натриевых каналов и, следовательно, применимы для предупреждения, ослабления и лечения широкого круга патологий, включающих, но без ограничения, неврологические, психические, сердечно-сосудистые, воспалительные, офтальмологические, мочеполовые и желудочно-кишечные заболевания, где вышеупомянутые механизмы описаны как играющие патологическую роль.
Соединения по настоящему изобретению по существу не обладают никаким ингибирующим действием в отношении МАО (моноаминоксидазы) или проявляют значительно уменьшенное ингибирующее действие на МАО в дозах, которые являются терапевтически эффективными для предупреждения, ослабления и/или лечения вышеуказанных заболеваний.
УРОВЕНЬ ТЕХНИКИ
Уровень техники (химия)
Патентная заявка WO 90/14334 описывает монозамещенные N-фенилалкил-альфа-аминокарбоксамидные производные общей формулы
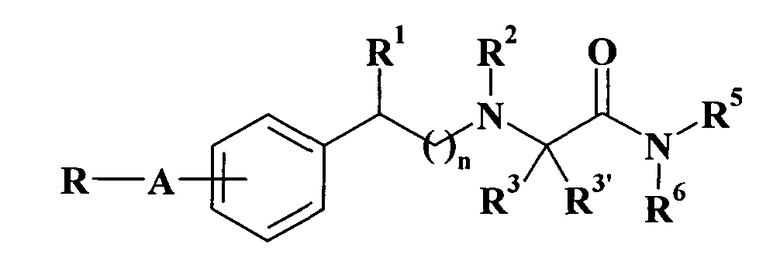
где
R представляет собой (C1-C8)алкил, (C3-C8)циклоалкил, фурил, тиенил, пиридил или фенильное кольцо, необязательно замещенное 1-4 заместителями, независимо выбранными из галогена, (C1-C6)алкила, (C1-C6)алкокси и трифторметила; A представляет собой -(CH2)m-, -(CH2)p-X-(CH2)q-группу, в которой m является целым числом от 1 до 4, один из р и q равен нулю, а другой равен нулю или представляет собой целое число от 1 до 4, Х представляет собой -O-, -S- или -NR4-группу, в которой R4 представляет собой водород или (C1-C4)алкил; n равно 0 или 1; каждый из R1 и R2 независимо представляет собой водород или (C1-C4)алкил; R3 представляет собой водород, (C1-C4)алкил, необязательно замещенный гидроксилом или фенилом, необязательно замещенным, как указано выше; R3' представляет собой водород или R3 и R3' вместе образуют (C3-C6)циклоалкильное кольцо; каждый из R5 и R6 независимо представляет собой водород или (C1-C6)алкил; и их применение в качестве противоэпилептических, антипаркинсонических, нейропротекторных, антидепрессивных, противоспазматических и/или снотворных средств.
Соединение 2-[2-[4-(3-хлорбензилокси)фенил]этиламино]ацетамид и его гидрохлоридная соль и их получение конкретно описаны в вышеупомянутой патентной заявке (см. также P. Pevarello et al., J. Med. Chem., 1998, 41, 579-590).
В WO 90/14334 упомянуты, но не охарактеризованы соединения (S)-2-[2-[4-бензилоксифенил]этиламино]ацетамид, (S)-2-[2-[4-(2-хлорбензилокси)фенил]этиламино]ацетамид, 2-[2-(4-бензилфенил)этиламино]ацетамид и 2-[2-(4-бензиламинофенил)этиламино]ацетамид.
В патентной заявке WO 04/089353 описаны способ и комбинированная терапия для лечения болезни Паркинсона с помощью сафинамида ((S)-(+)-2-[4-(3-фторбензилокси)бензиламино]пропанамида), производного сафинамида или ингибитора MAO-B вместе с антипаркинсоническими средствами. Соединение 2-[2-[4-(3-хлорбензилокси)фенил]этиламино]ацетамид приводится в качестве примера в этой заявке.
Вышеупомянутое соединение также получено и описано в качестве противосудорожного средства (Pevarello P., Bonsignori A., Dostert P., Heidempergher F., Pinciroli V., Colombo M., McArthur R.A., Salvati P., Post C., Fariello R.G., Varasi M.J. Med. Chem. (1998) 41: 579-590).
Патентная заявка WO 99/35125 описывает альфа-аминоамидные производные общей формулы
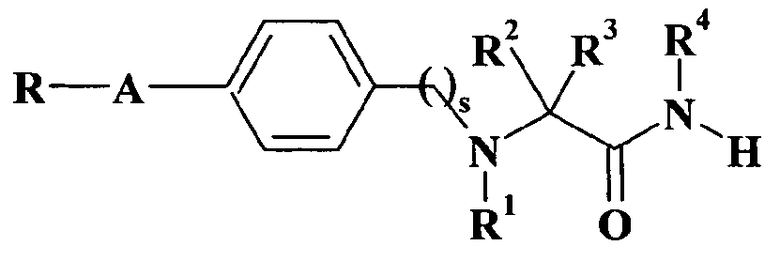
где R представляет собой фурил, тиенил, пиридил или фенильное кольцо; A представляет собой -(CH2)m-, -(CH2)n-X- или -(CH2)v-O- группу, в которой m представляет собой целое число от 1 до 4, n равно нулю или целому числу от 1 до 4, X представляет собой -S- или -NH- и v равно нулю или целому числу от 1 до 5; s равно 1 или 2; R1 представляет собой водород или (C1-C4)алкил; один из R2 и R3 представляет собой водород, а другой представляет собой водород или (C1-C4)алкил, необязательно замещенный гидрокси или фенилом; или R2 и R3 вместе образуют (C3-C6)циклоалкильное кольцо; или R2 и R3, оба, представляют собой метил; R4 представляет собой водород или (C1-C4)алкил; и их применение в качестве анальгетических средств.
В вышеуказанной патентной заявке упоминается соединение 2-[2-[4-(3-хлорбензилокси)фенил]этиламино]пропанамид.
Патентная заявка WO 03/091219 описывает 5-(бензилокси)-2-(йодфенил)этиламинопроизводные (см. формулу XII), которые используют как промежуточные соединения при получении изохинолинов в качестве ингибиторов моноаминоксидазы В, применяемых против болезни Альцгеймера и сенильной деменции:
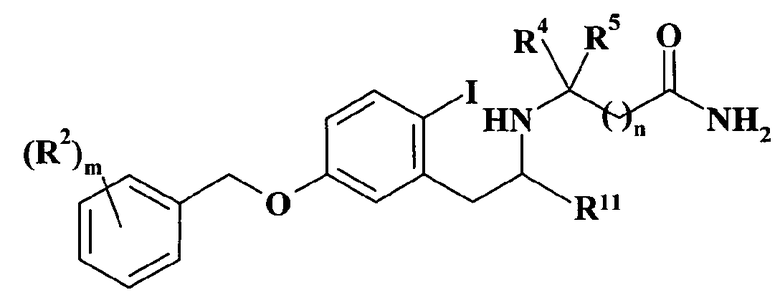
где, в частности, m равно 1, 2 или 3; R2 выбран из галогена, галоген(C1-C6)алкила, циано, (C1-C6)алкокси или галоген-(C1-C6)алкокси; R11 представляет собой водород; n равно 0, 1 или 2; R4 и R5 независимо выбраны из водорода, (C1-C6)алкила, -(CH2)р-OR8, -(CH2)р-SR8 или бензила, где p равно 1 или 2 и R8 представляет собой водород или (C1-C6)алкил.
В WO 99/26614 раскрываются замещенные 2-(бензиламино)ацетамиды и их применение для лечения нарушений, реагирующих на блокаду каналов ионов натрия, включающего предотвращение или облегчение невропатической боли.
WO 03/037865 относится к соединениям, применяемым в лечении рака, общей формулы
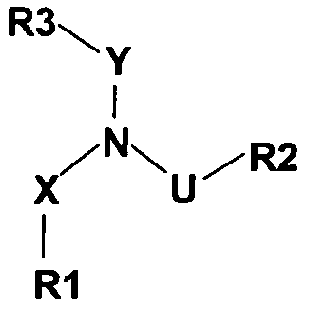
в которой обозначения R1, R2, R3, X, U и Y могут принимать широкий ряд значений. Хотя некоторые комбинации указанных широких родовых значений могли бы включать фенетиламинопроизводные, ни одно из соединений, описанных в настоящей заявке, в действительности не раскрывается в WO 03/037865.
Патент США № 5366982 (WO 92/01675) относится к соединениям, обладающим свойствами селективного антагониста лейкотриена B4 (LTB4), охватываемым общей формулой
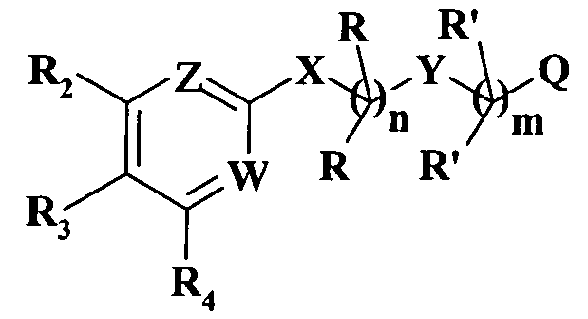
в которой обозначения R, R', R2, R3, R4, X, Y, Z, W, n, m и Q могут принимать широкий ряд значений. Несмотря на то, что некоторые комбинации указанных родовых значений могли бы охватывать также фенетиламинопроизводные, в действительности ни одно из соединений, описанных в настоящей заявке, не раскрывается в патенте США № 5366982.
В WO 98/35957 раскрываются производные ацетамида, активные в качестве антагонистов нейропептидного Y рецептора, в частности, применяемые для лечения ожирения, общей формулы
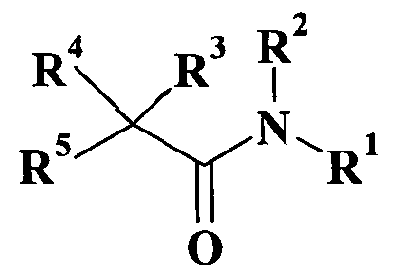
в которой обозначения R1, R2, R3, R4 и R5 могут принимать широкий ряд значений. В действительности ни одно из соединений, описанных в настоящей заявке, не раскрывается в WO 98/35957.
В EP 1588704A описаны альфа-аминоамидные производные, включающие (S)-(+)-2-[4-(2-фторбензилокси)бензиламино]пропанамид, т.е. ралфинамид, для применения в лечении синдрома усталых ног.
WO 2005/018627 раскрывает альфа-аминоамидные производные, включая ралфинамид, для применения в качестве терапевтических противовоспалительных средств.
Уровень техники (биология)
Кальциевые каналы представляют собой мембранные, состоящие из многих субъединиц белки, которые делают возможным регулируемый вход кальциевых ионов в клетки из внеклеточной жидкости. Как правило, кальциевые каналы являются потенциалзависимыми и называются потенциалзависимыми кальциевыми каналами (VGCC). Каналы VGCC находят по всей нервной системе млекопитающих, где они регулируют внутриклеточные уровни ионов кальция, которые важны для жизнеспособности и функций клеток. Внутриклеточные концентрации ионов кальция тесно связаны с рядом жизненных процессов у животных, таких как высвобождение нейротрансмиттеров, мышечные сокращения, двигательная активность и секреция гормонов. Все «чувствительные к раздражению» клетки у животных, такие как нейроны центральной нервной системы (ЦНС), периферические нервные клетки и мышечные клетки, включая клетки скелетных мышц, сердечных мышц и клетки гладких мышц вен и артерий, имеют потенциалзависимые кальциевые каналы.
Кальциевые каналы представляют собой большое семейство с множеством подтипов, отличающихся генетически, физиологически и фармакологически. На основании биофизических свойств кальциевых токов, зарегистрированных от отдельных нейронов, описаны два суперсемейства: высокопороговые (с высоким High Voltage Activated (HVA) и низкопороговые Low Voltage Activated (LVA) кальциевые каналы. Кальциевые токи, которые называют токами L-типа, P-типа, Q-типа, N-типа, R-типа, представляют собой HVA, а токи T-типа представляют собой LVA. Конкретно, термин «L-тип» был первоначально применен к каналам с высокой проводимостью одиночных каналов и длительным временем открытия, и термин «T-тип» применяют к каналам с очень низкой проводимостью одиночных каналов и переходным временем открытия. Дальнейшее исследование многообразия функциональных кальциевых каналов выявило канал «N-типа», экспрессированный в нейронах, и канал «P-типа», который представляет собой доминирующий тип, экспрессированный в мозжечковых нейронах Пуркинье и фармакологически устойчивый к известным блокаторам кальциевых каналов L-типа и N-типа. Исходя из молекулярного родства, десять отличающихся подтипов кальциевых токов выявлены, клонированы, экспрессированы и сгруппированы в три семейства: семейство Cav1 (Cav 1.1, 1.2, 1.3, 1.4) функционально относится к Ca току L-типа; семейство Cav2 (Cav 2.1, 2.2, 2.3) функционально относится к токам P/Q, N, R-типа, и семейство Cav3 (Cav 3.1, 3.2, 3.3) функционально относится к току T-типа.
Считается, что кальциевые каналы имеют отношение к некоторым болезненным состояниям. Полагают, что ряд соединений, применимых для лечения различных сердечно-сосудистых заболеваний у млекопитающих, включая человека, оказывают полезное действие путем модуляции функций потенциалзависимых кальциевых каналов, присутствующих в сердечных и/или сосудистых гладких мышцах. Соединения с активностью, направленной против кальциевых каналов, также непосредственно связаны с лечением боли. Конкретно кальциевые каналы N-типа (Cav2.2), ответственные за регуляцию высвобождения нейротрансмиттера, играют значительную роль в ноцицептивной трансмиссии, как полагают, вследствие их распределения в тканях и на основании результатов нескольких фармакологических исследований. Активированные кальциевые каналы N-типа были обнаружены в ипсилатеральном спинном роге на моделях невропатической боли при повреждениях (Cizkova D., Marsala J., Lukacova N., Marsala M., Jergova S., Orendacova J., Yaksh T.L. Exp. Brain Res. (2002) 147: 456-463). Специфические блокаторы кальциевых каналов N-типа, как показано, являются эффективными средствами для уменьшения болевых реакций на моделях невропатической боли (Mattews E.A., Dickenson A.H. Pain (2001) 92: 235-246), в фазе II формалинового теста (Diaz A., Dickenson A.H. Pain (1997) 69: 93-100) и при гипералгезии, вызванной воспалением коленного сустава (Nebe J., Vanegas H., Schaible H.G. Exp. Brain Res. (1998) 120: 61-69). Показано, что мыши-мутанты, не имеющие кальциевых каналов N-типа, обладают пониженной реакцией на постоянную боль, что видно по уменьшению болевой реакции во время фазы II формалинового теста (Kim C., Jun K., Lee T., Kim S.S., Mcenery M.W., Chin H., Kim H.L., Park J.M., Kim D.K., Jung S.J., Kim J., Shin H.S. Mol. Cell Neurosci. (2001) 18: 235-245; Hatakeyama S., Wakamori M., Ino M., Miyamoto N., Takahashi E., Yoshinaga T., Sawada K., Imoto K., Tanaka I., Yoshizawa T., Nishizawa Y., Mori Y., Nidome T., Shoji S. Neuroreport (2001) 12: 2423-2427), а также на невропатическую боль, оцениваемую по уменьшению механической аллодинии и термической гипералгезии на модели лигирования спинального нерва. Интересно, что упомянутые мыши показали также пониженные уровни тревожности по сравнению с мышами немутантного типа (Saegusa H., Kurihara T., Zong S., Kazuno A., Matsuda Y., Nonaka T., Han W., Toriyama H., Tanabe T., EMBO J. (2001) 20: 2349-2356). Участие кальциевых каналов N-типа в боли дополнительно подтверждено в клинике зиконотидом, пептидом, полученным из яда морского моллюска Conus Magnus. Ограничение в терапевтическом использовании указанного пептида состоит в том, что его приходится вводить человеку интратекально (Bowersox S.S. и Luther R. Toxicon (1998) 36: 1651-1658).
Натриевые каналы играют важную роль в нейронной сети, быстро передавая электрические импульсы по всем клеткам и клеточным сетям, координируя таким образом высшие психические процессы, начиная от локомоторных до познавательных. Указанные каналы представляют собой большие трансмембранные белки, которые способны переключать различные состояния, чтобы обеспечить селективную проницаемость для ионов натрия. Для этого процесса требуется действие потенциала для деполяризации мембраны, и поэтому такие каналы являются потенциалзависимыми. В последние несколько лет разработано намного лучшее объяснение взаимодействия натриевых каналов и лекарственных средств.
Потенциалзависимые натриевые каналы первоначально систематизировали по их чувствительности к тетродотоксину от низкой наномолярной (тетродотоксин-чувствительные TTXs) до высокой микромолярной (тетродотоксин-устойчивые, TTXr). Пока 10 различных альфа-субъединиц натриевых каналов идентифицированы и систематизированы как Nav1.1 и до Nav1.9. Субъединицы Nav1.1-Nav1.4, Nav1.6 и Navl.7 представляют собой TTXs, тогда как Nav.1.5, Nav.1.8 и Nav.1,9 представляют собой TTXr с различными степенями чувствительности. Nav1.1-Nav1.3 и Nav1.6 экспрессированы главным образом в ЦНС, тогда как Nav1.4 и Nav1.5 экспрессированы в основном в мышцах (скелетных и сердечных соответственно), а Nav1.8 и Nav1.9 экспрессированы преимущественно в небольших DRGs.
Стало понятно, что ряд лекарственных средств, включая местные анестетики, антиаритмические средства I класса и противосудорожные средства с неизвестным механизмом действия, на самом деле действуют путем модуляции проводимости натриевых каналов. Блокаторы нейрональных натриевых каналов нашли применение для лечения эпилепсии (фенитоин и карбамазепин), биполярных расстройств (ламотригин), предотвращения нейродегенерации и для ослабления невропатической боли. Различные противоэпилептические лекарственные средства, которые стабилизируют нейрональную возбудимость, эффективны при невропатической боли (габапентин, карбамазепин).
Кроме того, наблюдается также увеличение экспрессии или активности натриевых каналов на некоторых моделях воспалительной боли, заставляя предположить роль натриевых каналов в воспалительной боли.
Все вместе эти данные указывают, что соединения, приводящие к блокаде натриевых и/или кальциевых каналов, обладают высоким терапевтическим потенциалом для предупреждения, ослабления и лечения широкого спектра патологий, включающих неврологические, психиатрические, сердечно-сосудистые, воспалительные, офтальмологические, мочеполовые и желудочно-кишечные заболевания, где вышеупомянутые механизмы описаны как играющие патологическую роль.
Имеется множество статей и патентов, в которых описаны модуляторы или антагонисты натриевых и/или кальциевых каналов для лечения или модуляции плеторы заболеваний, например, их применение в качестве местных анестетиков, антиаритмических и противорвотных средств, антиманиакальных антидепрессантов, средств для лечения однополюсной депрессии, сердечно-сосудистых заболеваний, энуреза, диареи, воспаления, эпилепсии, нейродегенеративных состояний, гибели нервных клеток, невропатической боли, мигрени, острой гипералгезии и воспаления, заболевания почек, аллергии, астмы, бронхоспазма, дисменореи, спазма пищевода, глаукомы, нарушений мочевыводящих путей, нарушений моторики желудочно-кишечного тракта, преждевременных родов, ожирения. Неисчерпывающий список таких статей и патентов/патентных заявок, описывающих блокаторы натриевых и/или кальциевых каналов и виды их применения, включает ссылки, представленные ниже.
C. Alzheimer в Adv. Exp. Med. Biol. 2002, 513, 161-181, описывает натриевые и/или кальциевые каналы в качестве мишеней нейропротективных веществ.
Vanegas e Schaible (Pain 2000, 85, 9-18) обсуждают действие антагонистов кальциевых каналов по спинальным механизмам боли, гипералгезии и аллодинии.
Патент США № 5051403 относится к способу уменьшения нейронального повреждения, связанного с ишемическим состоянием, таким как удар, путем введения связывающего/ингибирующего омега-конотоксин пептида, где пептид характеризуется специфическим ингибированием потенциалзависимых токов кальциевых каналов избирательно в нейрональных тканях.
Патент США № 5587454 относится к композициям и способам для получения обезболивания конкретно в лечении боли и невропатической боли.
Патент США № 5863952 относится к антагонистам кальциевых каналов для лечения ишемического инсульта.
Патент США № 6011035 относится к блокаторам кальциевых каналов, применимым для лечения таких заболеваний, как удар и боль.
Патент США № 6117841 относится к блокаторам кальциевых каналов и их применению в лечении инсульта, церебральной ишемии, боли, травмы головы или эпилепсии.
Патент США № 6362174 относится к блокаторам кальциевых каналов N-типа в лечении инсульта, церебральной ишемии, боли, эпилепсии и травмы головы.
Патент США № 6380198 относится к применению блокатора кальциевых каналов флунаризина для местного лечения глаукомы.
Патенты США № 6420383 и № 6472530 относятся к новым блокаторам кальциевых каналов, применимым для лечения и профилактики ряда заболеваний, таких как гиперчувствительность, аллергия, астма, бронхоспазм, дисменорея, спазм пищевода, глаукома, преждевременные роды, заболевания мочевыводящих путей, нарушения моторики желудочно-кишечного тракта и сердечно-сосудистые заболевания.
Патент США № 6458781 относится к соединениям, которые действуют, блокируя кальциевые каналы, и их применению для лечения инсульта, церебральной ишемии, боли, травмы головы или эпилепсии.
Патент США № 6521647 относится к применению блокаторов кальциевых каналов в лечении заболеваний почек у животных, главным образом хронической почечной недостаточности.
WO 97/10210 относится к трициклическим гетероциклическим производным и их применению в терапии, конкретно в качестве антагонистов кальциевых каналов, например для лечения ишемии, конкретно ишемического инсульта.
WO 03/018561 относится к соединениям хинолина в качестве антагонистов кальциевых каналов N-типа и способам применения таких соединений для лечения или профилактики боли или ноцицепции.
WO 03/057219 относится к блокаторам кальциевых каналов, применимым в качестве средств для лечения или модуляции нарушений центральной нервной системы, таких как невропатическая боль, воспалительная боль, боль, связанная с воспалением или эпилепсия.
В WO 99/14199 описаны замещенные 1,2,3,4,5,6-гексагидро-2,6-метано-3-бензазоцин-10-олы в качестве сильных блокаторов кальциевых каналов, применимых для лечения нескольких заболеваний, таких как удар, нейродегенеративные расстройства, болезнь Альцгеймера, болезнь Паркинсона и сердечно-сосудистые заболевания.
WO 01/74779 раскрывает новые аминопиридиновые блокаторы натриевых каналов и их применение в качестве противосудорожных средств, местных анестетиков, в качестве антиаритмических средств для лечения или предотвращения нейродегенеративных состояний, таких как боковой амиотрофический склероз (ALS), для лечения или предотвращения как острой, так и хронической боли и для лечения или предотвращения диабетической нейропатии.
В WO 04/087125 описаны производные аминокислот в качестве ингибиторов натриевых каналов млекопитающих, применимых для лечения хронической и острой боли, звона в ушах, заболеваний кишечника, дисфункции мочевого пузыря и демиелинизирующих заболеваний.
Моноаминоксидаза (MAO) представляет собой фермент во внешней митохондриальной мембране нейрональных и не нейрональных клеток. Существуют две изоформы MAO: MAO-A и MAO-B. Ферменты MAO ответственны за окислительное деаминирование эндогенных и ксенобиотических аминов и имеют различные предпочтительные субстраты, специфичность ингибиторов и распределение в тканях. Для MAO-A предпочтительными субстратами являются серотонин, норадреналин и адреналин, а клоргилин представляет собой селективный ингибитор MAO-A; тогда как для MAO-B в качестве субстрата предпочтителен β-фенилэтиламин, и она почти селективно ингибируется селегилином. Допамин, тирамин и триптамин окисляются как MAO-A, так и MAO-B, конкретно в мозге человека допамин деаминирован ферментом MAO-B на 80%.
Ингибирование MAO позволяет эндогенным и экзогенным субстратам накапливаться и, возможно, при почти полном ингибировании (>90%) изменять динамику нормальных моноаминных медиаторов. MAO регулирует концентрации в мозгу наиболее важных нейромедиаторов, таких как норадреналин, серотонин и допамин, которые связаны с эмоциями, тревожностью и движением.
Таким образом, полагают, что MAO тесно связана с различными психиатрическими и неврологическими заболеваниями, такими как депрессия, тревожные расстройства и болезнь Паркинсона (PD).
Ингибиторы MAO-A используют главным образом в психиатрии для лечения большой, рефракторной и атипичной депрессии как следствия их способности увеличивать пониженные уровни серотонина и норадреналина. Позже ингибиторы MAO-A стали использовать для лечения пациентов с тревожными расстройствами, такими как социальная фобия, расстройства панического типа, посттравматические стрессовые расстройства и обсессивно-компульсивные расстройства.
Ингибиторы MAO-B используют в основном в неврологии для лечения PD.
Имеются также последние данные и интерес к роли MAO-B в других патологических состояниях, таких как болезнь Альцгеймера (AD). До настоящего времени нет сообщений, свидетельствующих о вовлечении MAO-B в метаболизм сомедиаторов, таких как колецистокинин, вещество Р, соматостатин и нейротензин, которые участвуют в модуляции болевой чувствительности. По этой причине не имеется разумного научного объяснения для применения ингибиторов MAO-B в болевых синдромах.
Сообщают о негативных реакциях на лекарственные средства во время клинической практики с ингибиторами MAO. Первое поколение неселективных и необратимых ингибиторов MAO, таких как транилципромин и фенелзин, имеет серьезные побочные эффекты, включающие гепатотоксичность, ортостатическую гипотензию и наиболее важно гипертонический криз, который имеет место после глотания пищи, содержащей тирамин (Cooper A.J. Tyramine and irreversible monoamine oxidase inhibitors in clinical pratice.- Br. J. Psych. Suppl. 1989: 38-45).
Когда используют упомянутые неселективные и необратимые ингибиторы MAO, необходимо соблюдать строгую диету с пониженным содержанием тирамина. Прессорная чувствительность в отношении тирамина нормализуется в течение 4 недель после прекращения транилципроминовой терапии и более 11 недель после прекращения терапии фенелзином.
Селегилин, почти избирательный и необратимый ингибитор MAO-B, особенно при использовании в комбинации с леводопой, может вызывать анорексию/тошноту, сухость во рту, дискинезию и ортостатическую гипотензию у пациентов с PD, причем последняя является наиболее проблематичной (Volz H.P. and Gleiter C.H.- Monoamine oxidase inhibitors. A perspective on their use in the elderly.- Drugs Aging 13 (1998), pp. 341-355).
При монотерапии у пациентов, получавших селегилин, чаще имели место анорексия/тошнота, скелетно-мышечные поражения и сердечная аритмия, по сравнению с больными, получавшими плацебо. Кроме указанных негативных эффектов, отмечались увеличенные показатели повышенных уровней AST и ALT в сыворотке крови. Наиболее часто описываемыми отрицательными эффектами моклобемида, селективного и обратимого ингибитора MAO-A, являются нарушения сна, повышенная тревожность, дисфория и головная боль.
Комбинация селективных ингибиторов обратного захвата серотонина (SSRIs) и моклобемида обладает высокой эффективностью в случаях не поддающейся лечению депрессии, но идут споры о том, возникают ли в результате такой комбинации токсические побочные эффекты, такие как серотонергический синдром (Baumann P.- Pharmacokinetic-pharmacodynamic relationship of the selective serotonin reuptake inhibitors. Clin. Pharmacokinet. 31 (1996), pp. 444-469). Из-за сердечной аритмии и повышенных уровней ферментов следует регулярно проверять электрокардиограмму и данные лабораторных анализов.
Множество видов физиологических изменений, которые происходят с возрастом, влияют на фармакодинамику и фармакокинетику ингибиторов MAO. Действительно, фармакокинетические переменные заметно отличаются у пожилых пациентов от переменных у молодых пациентов. Указанные переменные, включающие всасывание, распределение, метаболизм и экскрецию, приходится принимать во внимание, чтобы избежать или свести к минимуму некоторые негативные неблагоприятные эффекты взаимодействий лекарственное средство - лекарственное средство. Пожилые больные обычно более чувствительны к побочным эффектам, включая неблагоприятные реакции на лекарства, чем молодые. У пожилых пациентов чаще, чем у молодых, может происходить гипертонический криз, так как сердечно-сосудистая система пожилых людей уже нарушена с возрастом.
Применение симпатомиметических лекарственных средств в комбинации с ингибиторами MAO может также повышать кровяное давление. Кроме того, по сравнению с плацебо при применении фенелзина значительно чаще имеет место дремота, тремор, дискинезия, диарея, затрудненное мочеиспускание, ортостатические эффекты и неблагоприятные дерматологические эффекты. Интересно отметить, что во время лечения моклобемидом, как сообщают, у пожилых пациентов головная боль имеет место чаще, чем у более молодых пациентов (Volz H.P. и Gleiter C.H.- Monoamine oxidase inhibitors. A perspective on their use in the elderly. Drugs Aging 13 (1998), pp. 341-355).
Ингибиторы MAO иногда прописывают при депрессии. Из-за потенциального риска суицида, неблагоприятные побочные реакции и токсичность, обусловленная передозировкой, являются важными факторами, рассматриваемыми при выборе антидепрессанта. Кроме того, когда ингибиторы MAO используют в больших дозах, неблагоприятные сердечно-сосудистые эффекты, по-видимому, значительно возрастают, и, так как селективность MAO теряется при таких высоких дозах, тирамин может вызывать потенциально опасные гипертензивные реакции. Сильная передозировка ингибиторами MAO вызывает ажитацию, галлюцинации, гиперпирексию, гиперрефлексию и судороги. Аномальное кровяное давление также является признаком токсичности, и может потребоваться промывание желудка и поддержание пневмокардиальной функции. Передозировка традиционных неселективных и необратимых ингибиторов MAO является достаточно опасной и иногда смертельной (Yamada and Richelson, 1996. Pharmacology of antidepressants in the elderly. In: David J.R., Snyder L., editors. Handbook of pharmacology of aging. Boca Raton: CRC Press 1996).
В лечении заболеваний, в которых механизм(ы) натриевых и кальциевых каналов играет(ют) патологическую роль, и, конкретно, в лечении болевых синдромов (или невропатического, или воспалительного типа) ингибирование MAO ферментов бесполезно (невыгодно). Наиболее активные в клинике антиноцицептивные лекарственные средства не обладают способностью ингибировать MAO. Наоборот, побочные эффекты, ингибирующие MAO, могут налагать по меньшей мере два типа негативных ограничений.
1) Диетические: употребление пищи с высоким содержанием тирамина может вызвать серьезное, даже угрожающее жизни, повышение системного кровяного давления (так называемый «сырный эффект»).
2) Фармакологические: боль часто лечат комбинацией лекарственных средств, таких как опиоидные производные и трициклические антидепрессанты. Такая комбинация в сочетании с ингибиторами MAO опасна, так как она может вызвать серотонинергический синдром (возбуждение, тремор, галлюцинации, гипертермию и аритмию).
Следовательно, устранение или значительное уменьшение ингибирующей MAO активности в лекарственных средствах, действующих в качестве модуляторов натриевых и/или кальциевых каналов, применимых для предупреждения, облегчения и излечения широкого спектра патологий, в которых указанный(е) механизм(ы) играет(ют) патологическую роль, включающих неврологические, психические, сердечно-сосудистые, воспалительные, офтальмологические, мочеполовые и желудочно-кишечные заболевания, является неожиданным и существенным улучшением для терапии по сравнению с соединениями, имеющими аналогичную эффективность, но обладающими вышеупомянутыми побочными эффектами. Указанное улучшение является особенно желательным для лекарственных средств, действующих в качестве модуляторов натриевых и/или кальциевых каналов, применимых, в частности, для лечения болевых синдромов.
Принимая во внимание эти данные по ингибиторам MAO, конкретно отсутствие каких-либо доказательств о роли MAO-B в патологических поражениях, подобных боли, мигрени, сердечно-сосудистым, воспалительным, мочеполовым и желудочно-кишечным заболеваниям, можно допустить, что ингибирование MAO-B не должно быть существенным признаком для соединений, указанных для вышеупомянутых патологий, не допускающих побочных эффектов во время хронического и/или длительного лечения.
Предпочтительное решение описанной выше проблемы состояло бы в создании лекарственных средств, которые «избирательно действуют как модуляторы натриевых и/или кальциевых каналов» или применимы для «избирательного лечения» повреждений, расстройств или заболеваний, в которых механизм(ы) натриевых и/или кальциевых каналов играет(ют) патологическую роль. Это выражение подразумевает лекарственные средства, которые при введении пациенту, нуждающемуся в лечении, в количествах, эффективных для лечения вышеуказанных болезней, где вышеупомянутые механизм(ы) играет(ют) патологическую роль, не проявляют какой-либо ингибирующей активности в отношении MAO или проявляют значительно сниженную ингибирующую активность в отношении MAO, таким образом, в результате не допускают побочных эффектов, обусловленных накоплением эндогенных и экзогенных моноаминных медиаторов.
Основная задача настоящего изобретения состоит в применении фенилэтиламинопроизводных для изготовления лекарственных средств, активных в качестве модуляторов натриевых и/или кальциевых каналов, для лечения патологий, в которых вышеуказанные механизм(ы) играет(ют) патологическую роль, причем указанные лекарственные средства по существу не обладают ингибирующей активностью в отношении MAO или обладают значительно сниженной ингибирующей активностью в отношении MAO и, следовательно, возможности нежелательных побочных эффектов уменьшены. Указанное применение обеспечивает повышенный селективный ресурс для предупреждения, ослабления и/или излечения вышеуказанных патологических поражений.
Описание изобретения
Авторы настоящего изобретения обнаружили новый класс фенилэтиламинопроизводных, высокоэффективных в качестве модуляторов натриевых и/или кальциевых каналов, по существу не обладающих ингибирующей активностью в отношении MAO или обладающих значительно сниженной ингибирующей активностью в отношении MAO и, следовательно, имеющих потенциально сниженные побочные эффекты, для предупреждения, ослабления и излечения широкого спектра патологий, включающих, но без ограничения, нейрогенные, психиатрические, сердечно-сосудистые, воспалительные, офтальмологические, мочеполовые и желудочно-кишечные заболевания, в которых, как сообщается, вышеупомянутые механизмы играют патологическую роль.
В настоящем описании и формуле изобретения выражение «модулятор(ы) натриевых и/или кальциевых каналов» обозначает соединения, способные блокировать натриевые и/или кальциевые токи потенциалзависимым способом.
Следовательно, задача настоящего изобретения состоит в обеспечении соединения общей формулы I
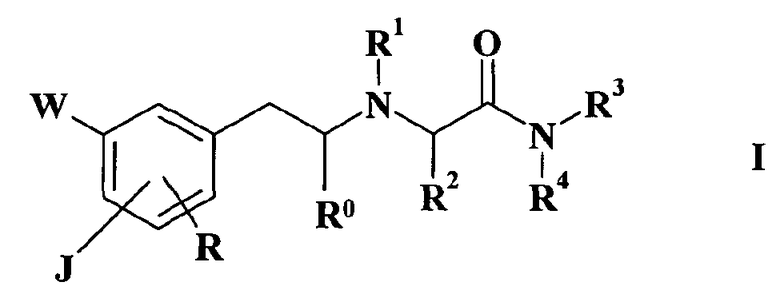
где
(a)
J представляет собой группу A-[(CH2)n-O]r- в пара-положении по отношению к этиламиновому фрагменту,
в которой
n равно нулю или 1, и
r равно 1;
A представляет собой трифторметил; или фенил, необязательно замещенный галогеном;
W представляет собой (C1-C4)алкокси;
R представляет собой водород;
R0 представляет собой водород или (C1-C2)алкил;
R1 представляет собой водород; (C1-C4)алкил, необязательно замещенный гидроксигруппой; циклопропилметил; 2-пропин-1-ил; бензил, необязательно замещенный одной или двумя (C1-C2)алкоксигруппами в бензольном кольце; тиазолил; 5-6-членный насыщенный гетероциклил, содержащий атом азота, необязательно замещенный (C1-C2)алкильной группой; или гетероциклилметил, в котором гетероциклическая группа представляет собой 5-6-членный гетероциклил, содержащий 1-3 гетероатома, выбранных из азота, кислорода и серы, необязательно замещенный одной или двумя группами, выбранными из (C1-C2)алкила, гидроксиметила и (C1-C2)алкокси;
R2 представляет собой водород; (C1-C4)алкил или фенил;
R3 представляет собой водород или (C1-C4)алкил; и
R4 представляет собой водород; (C1-C4)алкил, необязательно замещенный группой, выбранной из амино, (C1-C4)алкиламино, ди(C1-C4)алкиламино, имидазолила и пирролидинила, причем имидазолильная и пирролидинильная группы необязательно замещены (C1-C2)алкильной группой; или бензил; или
R3 и R4 вместе с соседним атомом азота образуют пирролидинильное, морфолинильное или пиперазинильное кольцо, необязательно замещенное (C1-C2)алкильной группой;
или
(b)
J представляет собой водород;
W представляет собой группу A-[(CH2)n-O]r-,
в которой
n равно нулю, 1 или 2; и
r равно нулю или 1;
A представляет собой (C1-C4)алкил, трифторметил; циклопропил; циклопентил; фенил, необязательно замещенный группой, выбранной из галогена, метила, метокси, трифторметила, ацетиламино и диметиламинометила; тиенил, необязательно замещенный хлором; фуранил; изоксазолил, необязательно замещенный одной или двумя метильными группами; пиперидинил; морфолинил; пиридинил или пиримидинил, причем пиридинильное и пиримидинильное кольца необязательно замещены одной или двумя метоксигруппами;
R представляет собой водород или фтор;
R0 представляет собой водород или (C1-C2)алкил;
R1 представляет собой циклопропилметил, фуранилметил, тетрагидрофуранил или тетрагидрофуранилметил;
R2 представляет собой водород или (C1-C4)алкил;
R3 представляет собой водород или (C1-C4)алкил; и
R4 представляет собой водород, (C1-C4)алкил, необязательно замещенный группой, выбранной из (C1-C2)алкокси, амино, (C1-C4)алкиламино и ди(C1-C4)алкиламино; или гетероциклил, где гетероциклил выбран из изоксазолила, пиразолила, имидазолила, тиазолила и 1,3,4-тиадиазолилила и может быть необязательно замещен (C1-C2)алкильной группой; или
R3 и R4 вместе с соседним атомом азота образуют пирролидиновое кольцо;
при условии, что когда A представляет собой (C1-C4)алкил, трифторметил, циклопропил или циклопентил, тогда r равен 1;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера, либо в виде их смеси, и его фармацевтически приемлемые соли.
Tермин «(C1-C4)алкильная» или «(C1-C4) алкильная» группа в других заместителях (например, в терминах «алкокси», «моно- и диалкиламино»), используемый в настоящем описании и формуле изобретения, когда не указано иное, обозначает линейный или разветвленный алкильный радикал или группу; примеры указанных радикалов или групп включают соответственно метил, этил, пропил, изопропил, бутил, изобутил и трет-бутил или метокси, этокси, пропокси, изопропокси, бутокси, изобутокси и трет-бутокси.
Tермин «галоген», когда не указано иное, обозначает радикал атома галогена, такого как фтор, хлор, бром и йод.
Термин «гетероцикл» и «гетероциклил», когда в настоящем описании не указано иное, означает полностью ненасыщенный, частично ненасыщенный или насыщенный моноциклический 5- или 6-членный гетероцикл, содержащий от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы. Примеры моноциклического 5- или 6-членного полностью ненасыщенного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы, включают, например, пиррол, фуран, тиофен, пиразол, имидазол, изоксазол, оксазол, изотиазол, тиазол, 1,2,3- и 1,3,4-тиадиазол, пиридин, пиран, пиридазин, пиримидин, пиразин и триазин.
Примеры моноциклического 5- или 6-членного частично ненасыщенного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы, включают, например, пирролин, пиразолин, имидазолин, оксазолин, изоксазолидин и тиазолин.
Примеры моноциклического 5- или 6-членного насыщенного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы, включают, например, пирролидин, пиразолидин, имидазолидин, оксазолидин, изоксазолидин, пиперидин, пиперазин, тетрагидрофуран, тетрагидропиран, морфолин и тиоморфолин.
В тех случаях, когда соединения по настоящему изобретению содержат по меньшей мере один асимметрический атом, они могут существовать в виде индивидуальных энантиомеров или диастереоизомеров или их смеси; объем настоящего изобретения включает все возможные индивидуальные энантиомеры или диастереоизомеры указанных соединений и их смеси, например рацемические смеси.
Примерами фармацевтически приемлемых солей соединений формулы I являются их соли с органическими и неорганическими кислотами, такими как хлористоводородная, бромистоводородная, иодистоводородная, азотная, серная, фосфорная, уксусная, пропионовая, винная, фумаровая, лимонная, бензойная, янтарная, коричная, миндальная, салициловая, гликолевая, молочная, щавелевая, яблочная, малеиновая, малоновая, фумаровая, винная, п-толуолсульфоновая, метансульфоновая, глутаровая кислота и им подобные.
Соединения формулы I действуют как модуляторы кальциевых и/или натриевых каналов и, следовательно, применимы для предупреждения, ослабления и излечения широкого спектра патологий, включающих (но не ограниченных перечисленными ниже) неврологические, психиатрические, сердечно-сосудистые, воспалительные, офтальмологические, урологические и желудочно-кишечные заболевания, в которых вышеупомянутые механизмы описаны, как играющие главную роль.
Предпочтительная группа соединений формулы I по настоящему изобретению включает соединение группы (a), определенной выше, где
J представляет собой группу A-[(CH2)n-O]r- в пара-положении по отношению к этиламиновому фрагменту, где
n представляет собой 1; и
r представляет собой 1;
A представляет собой фенил или фенил, необязательно замещенный фтором или хлором;
W представляет собой метоксигруппу;
R представляет собой водород;
R0 представляет собой водород;
R1 представляет собой водород; (C1-C4)алкил; циклопропилметил; бензил или гетероциклилметил, где гетероциклическая группа выбрана из фуранила, тетрагидрофуранила и пиридинила, необязательно замещенного метоксигруппой;
R2 представляет собой водород; (C1-C4)алкил или фенил;
R3 представляет собой водород или (C1-C4)алкил; и
R4 представляет собой водород; (C1-C4)алкил, необязательно замещенный группой, выбранной из амино, диметиламино и пирролидинила, где пирролидинил необязательно замещен метильной группой; или
R3 и R4 вместе с соседним атомом азота образуют пирролидинильное или морфолинильное кольцо;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера, либо в виде их смеси, и его
фармацевтически приемлемые соли.
Дополнительная группа предпочтительных соединений формулы I по настоящему изобретению включает соединение группы (b), определенной выше, в котором
J представляет собой водород;
W представляет собой группу A-[(CH2)n-O]r-, в которой
n равно нулю, 1 или 2;
r равно нулю или 1;
A представляет собой (C1-C4)алкил; трифторметил; циклопропил; циклопентил; фенил, необязательно замещенный группой, выбранной из фтора, хлора, метила, метокси, трифторметила, ацетиламино и диметиламинометила; тиенил, необязательно замещенный хлором; фуранил; изоксазолил, необязательно замещенный одной или двумя метильными группами; пиперидинил; морфолинил; пиридинил или пиримидинил, причем пиридинильная и пиримидинильная группа необязательно замещены одной или двумя метоксигруппами;
R представляет собой водород или фтор;
R0 представляет собой водород;
R1 представляет собой циклопропилметил; фуранилметил; тетрагидрофуранил или тетрагидрофуранилметил;
R2 представляет собой водород или метил;
R3 представляет собой водород или (C1-C4)алкил; и
R4 представляет собой водород; (C1-C4)алкил, необязательно замещенный группой, выбранной из метокси, амино, метиламино и диметиламино; изоксазолил, необязательно замещенный метильной группой; пиразолил; имидазолил; тиазолил; или 1,3,4-тиадиазолил;
или
R3 и R4 вместе с соседним атомом азота образуют пирролидинильное кольцо;
при условии, что когда А представляет собой (C1-C4)алкил, трифторметил, циклопропил или циклопентил, тогда
r равно 1;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера, либо в виде их смеси, и его фармацевтически приемлемые соли.
Более предпочтительная группа соединений формулы I, охватываемая группой (b), определенной выше, включает соединение, в котором
J представляет собой водород;
W представляет собой группу A-[(CH2)]n-O]r-, в которой
n равно 1 или 2,
r равно 1;
A представляет собой (C1-C4)алкил; трифторметил; циклопропил; циклопентил; фенил, необязательно замещенный группой, выбранной из фтора, хлора, метила, метокси и трифторметила; тиенил, необязательно замещенный хлором; изоксазолил, необязательно замещенный одной или двумя метильными группами; пиридинил; пиперидинил или морфолинил;
R представляет собой водород или фтор;
R0 представляет собой водород;
R1 представляет собой циклопропилметил; фуранилметил; тетрагидрофуранил или тетрагидрофуранилметил;
R2 представляет собой водород;
R3 представляет собой водород или (C1-C4)алкил; и
R4 представляет собой водород; (C1-C4)алкил, необязательно замещенный группой, выбранной из метокси, амино, метиламино и диметиламино; изоксазолил, необязательно замещенный метильной группой; пиразолил; имидазолил; тиазолил или 1,3,4-тиадиазолил; или
R3 и R4 вместе с соседним атомом азота образуют пирролидинильное кольцо;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера, либо в виде их смеси, и его фармацевтически приемлемые соли.
В этой группе более предпочтительных соединений формулы I группы (b), определенной выше в настоящем описании, наиболее предпочтительная группа соединений включает соединение, в котором
J представляет собой водород;
W представляет собой группу A-[(CH2)n-O]r-, в которой
n равно 1;
r равно 1;
A представляет собой (C1-C4)алкил;
R представляет собой водород;
R0 представляет собой водород;
R1 представляет собой фуранилметил или тетрагидрофуранилметил;
R2 представляет собой водород;
R3 представляет собой водород или (C1-C4)алкил; и
R4 представляет собой (C1-C4)алкил;
в определенных случаях или в виде индивидуального изомера
или диастереоизомера, или их смеси и его фармацевтически приемлемые соли.
Еще более предпочтительная группа соединений формулы I по настоящему изобретению, охватываемая группой (b), определенной выше, включает соединение, в котором
J представляет собой водород;
W представляет собой группу A-[(CH2)n-O]r-, в которой
n равно нулю;
r равно 1;
A представляет собой циклопентил или фенил, необязательно замещенный фтором;
R представляет собой водород;
R1 представляет собой фуранилметил;
R2 представляет собой водород;
R3 представляет собой водород или (C1-C4)алкил; и
R4 представляет собой водород или (C1-C4)алкил;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера, либо в виде их смеси, и его фармацевтически приемлемые соли.
Еще более предпочтительная группа соединений формулы I по настоящему изобретению, охватываемая группой (b), определенной выше, включает соединение, в котором
J представляет собой водород;
W представляет собой группу A-[(CH2)n-O]r-, в которой
n равно нулю;
r равно нулю;
A представляет собой фенил, необязательно замещенный
группой, выбранной из фтора, метокси, ацетиламино и диметиламинометила; тиенил; фуранил; изоксазолил, необязательно замещенный одной или двумя метильными группами; пиперидинил; пиридинил или пиримидинил, причем пиридинильная и пиримидинильная группы, необязательно замещены одной или двумя метоксигруппами;
R представляет собой водород;
R0 представляет собой водород;
R1 представляет собой фуранилметил или тетрагидрофуранилметил;
R2 представляет собой водород;
R3 представляет собой водород или (C1-C4)алкил; и
R4 представляет собой водород или (C1-C4)алкил;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера, либо в виде их смеси, и его фармацевтически приемлемые соли.
Наиболее предпочтительно, если соединение формулы I по настоящему изобретению выбрано из группы, состоящей из
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N-метилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]изобутиламино]-N-метилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](тетрагидрофуран-3-илметил)амино]-N-метилацетамида;
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N,N-диметилацетамида;
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметилпропионамида;
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметил-2-фенилацетамида;
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-1-(морфолин-4-ил)-2-фенилэтанона;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-1-(пирролидин-1-ил)этанона;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-(2-амино-2-метилпропил)ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-(2-диметиламиноэтил)ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-[2-(1-метилпирролидин-2-ил)этил]ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-этилацетамида;
2-[[2-[4-(бензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-метилацетамида;
2-[[2-(3-бензилоксифенил)этил](циклопропилметил)амино]-N-метилацетамида;
2-[[2-(3-бензилоксифенил)этил](фуран-2-илметил)амино]-N- метилацетамида;
2-[[2-[3-(2-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида;
2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]ацетамида;
2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(2-диметиламиноэтил)ацетамида;
2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(2-амино-2-метилпропил)ацетамида;
2-[[2-(3-бензилоксифенил)этил](циклопропилметил)амино]-N-метилпропионамида;
2-[[2-[3-метокси-4-(2,2,2-трифторэтокси)фенил]этил](циклопропилметил)амино]-N-метилацетамида;
2-[[2-(3'-фторбифенил-3-ил)этил](фуран-2-илметил)амино]-N-метилацетамида;
2-[[2-(3-бензилоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида;
2-[[2-(3-бутоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида;
2-[[2-(3-бутоксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида;
2-[[2-[4-фтор-3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида;
2-[[2-[3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида;
2-[[2-[3-(3,5-диметилизоксазол-4-ил)фенил]этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида;
2-[[2-[3-(пиперидин-1-ил)фенил]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-метилацетамида;
(S)-2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N-метил-4-метилвалерамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](фуран-3-илметил)амино]-N-метилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-этилацетамида и
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](6-метоксипиридин-3-илметил)амино]-N-метилацетамида;
в определенных случаях либо в виде индивидуального энантиомера, либо в виде их смеси, и его фармацевтически приемлемые соли, предпочтительно его соль с хлористоводородной кислотой.
Соединения формулы I, объект настоящего изобретения, получают по способу синтеза, который включает
a) реакцию соединения формулы II
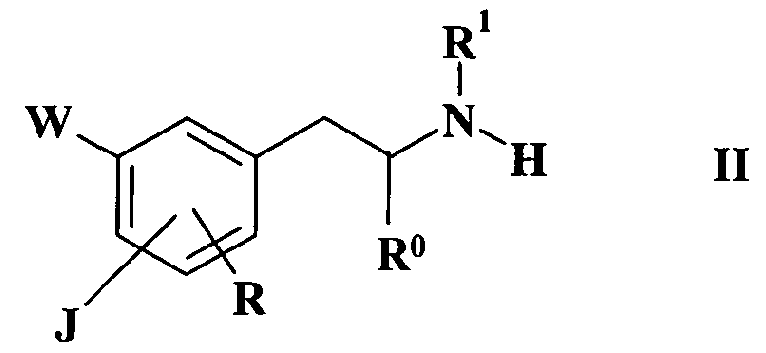
где
J, W, R, R0 и R1 имеют такие же значения, как определено
выше в формуле I,
с соединением формулы III
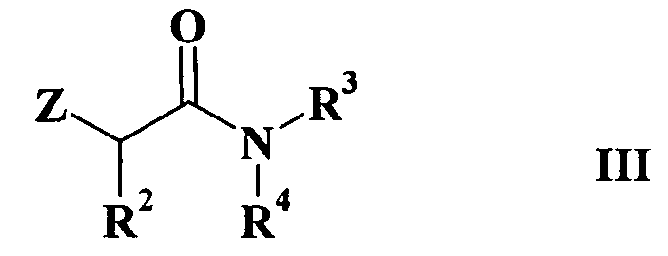
где
R2, R3 и R4 имеют такие же значения, как определено выше в формуле I и Z представляет собой атом галогена или подходящую уходящую группу, такую, например, как метансульфонилокси, пара-толуолсульфонилокси или трифторметансульфонатная группы, или альтернативно проводят указанную реакцию между соединением формулы II и соединением формулы IV
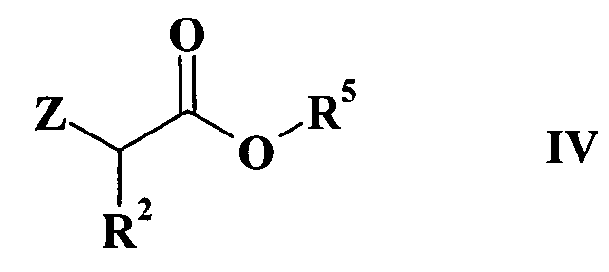
где
R2 и Z имеют значения, определенные выше и R5 представляет собой (C1-C4)aлкильную группу, и получают соединение формулы V
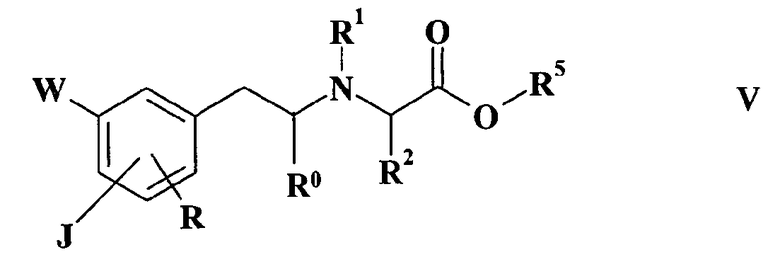
где
J, W, R, R0, R1, R2 и R5 имеют такие же значения, как определено выше, которое далее реагирует с амином формулы HNR3R4, где R3 и R4 такие, как определено выше, давая соединения по изобретению.
Реакцию амидирования, которая делает возможным введение заместителя -NR3R4, проводят по общепринятой методике амидирования, в которой сложный эфир превращают в соответствующий амид взаимодействием с выбранным амином. Согласно варианту осуществления изобретения, имеющему практическое значение, амидирование проводят в присутствии триметилалюминия.
Соединение формулы I, в котором J, W, R, R0, R2, R3 и R4 имеют такие же значения, как указано выше, и R1 имеет такие же значения, как и выше, кроме водорода, может быть также получено реакцией соединения формулы VI
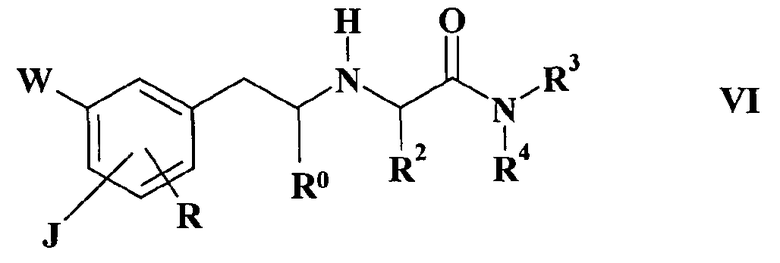
где
J, W, R, R0, R2, R3 и R4 имеют такие же значения, как и в вышеупомянутой формуле I, с соединением R1-Z, в котором R1 имеет значения, указанные выше, кроме водорода, а Z представляет собой атом галогена или подходящую уходящую группу, например, метансульфонилокси, пара-толуолсульфонилокси или трифторметансульфонилокси, в присутствии основания, или с карбонильным соединением формулы R6R7CO в присутствии восстановителя, где R6 представляет собой водород; (C1-C3)алкил, необязательно замещенный гидроксигруппой; циклопропил; этинил; фенил, необязательно замещенный одной или двумя (C1-C2)алкоксигруппами; 5- или 6-членный гетероциклил, содержащий от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы, где гетероциклил необязательно замещен одной или двумя группами, выбранными из (C1-C2)алкила, гидроксиметила и (C1-C2)алкокси; R7 представляет собой водород; или R6 и R7 вместе с соседней карбонильной группой представляет собой (C3-C4)алифатический кетон или 5-6-членный насыщенный гетероциклический кетон, содержащий атом азота или атом кислорода, необязательно замещенный (C1-C2)алкильной группой, например 1-метилпиперидин-4-он или дигидрофуран-3(2H)-он.
Соединение по изобретению может быть превращено в еще одно соединение по изобретению.
Например, соединение формулы I, в котором J представляет собой бензилоксирадикал, может быть превращено в соответствующее гидроксипроизводное каталитическим гидрированием и затем введено в реакцию с соответствующим реагентом для замещения первоначальной бензильной группы различными группами, например трифторметилбензильной, фенилэтильной, трифторэтильной, циклопентильной, циклопропилметильной и гетероциклилметильной группой, как определено выше. Если требуется, соединение по изобретению может быть превращено в фармацевтически приемлемую соль, и/или, если требуется, соль можно превратить в свободное соединение, и/или, если требуется, смесь энантиомеров или диастереоизомеров соединения по изобретению может быть разделена на соответствующие индивидуальные изомеры.
Соединения формул II, III, IV и VI коммерчески доступны, или их получают из коммерчески доступных соединений известными способами.
Согласно варианту осуществления изобретения, имеющему практическое значение, получение соединения формулы II осуществляют взаимодействием соединения формулы VII
где
J, W и R имеют значения, такие как определено в формуле I, с нитроалканом формулы R0-CH2-NO2, где R0 имеет такие же значения, как определено в формуле I, что приводит к образованию соединения формулы VIII
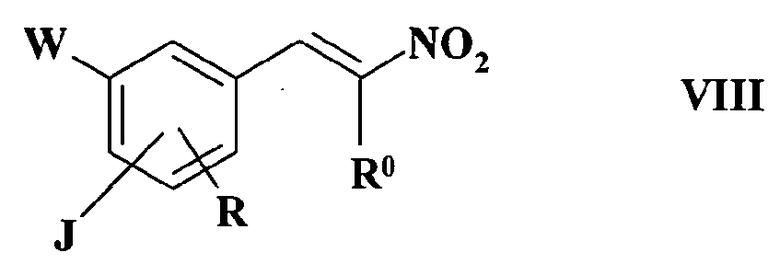
где
J, W, R и R0 имеют такие же значения, как и в формуле I, которое восстанавливают восстановителем, таким как LiAlH4, или каталитическим восстановлением, используя Pt/H2 или Pd/H2, и получают соединение формулы II, в котором R1 представляет собой водород.
Когда требуется, соединение формулы II, где R1 имеет такие же значения, как указано выше, кроме водорода, соединение формулы II, где R1 представляет собой водород, подвергают взаимодействию с соединением формулы R1Z, где R1 имеет такие же значения, как определено выше, кроме водорода, в присутствии основания, или с карбонильным соединением формулы R6R7CO, где R6 и R7 имеют такие значения, как определено выше, в присутствии восстановителя.
Реакцию соединения формулы II с соединением формулы III, которая дает соединение по изобретению, проводят по известным методикам.
Согласно предпочтительному варианту осуществления изобретения указанную реакцию проводят в присутствии основания и более предпочтительно указанное основание выбирают из K2CO3, триэтиламина или диизопропилэтиламина.
Когда получают соединение формулы I, где R1 представляет собой водород (т.e. соединение формулы VI), введение радикала R1, который не является водородом, определенного выше, проводят обычными способами получения вторичных и третичных аминов, такими как алкилирование или восстановительное аминирование.
Согласно предпочтительному варианту осуществления изобретения указанную реакцию алкилирования проводят в присутствии основания и более предпочтительно указанное основание выбирают из K2CO3, триэтиламина или диизопропилэтиламина.
Согласно еще одному предпочтительному варианту осуществления изобретения указанное восстановительное аминирование с соединением R6R7CO, где R6 и R7 имеют такие же значения, как определено выше, проводят в присутствии восстановителя, выбранного из NaBH4, NaBH3CN и цианоборгидрида (полистирилметил)триметиламмония.
В качестве альтернативного способа соединения формулы I получают способом синтеза, который включает реакцию соединения формулы IХ
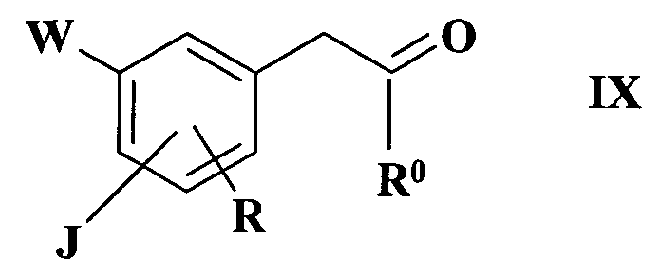
где
J, W, R и R0 имеют такие же значения, как определено в формуле I, или соединения формулы X
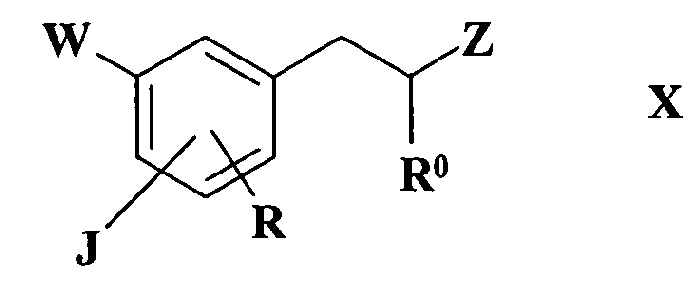
где
J, W, R и R0 имеют такие же значения, как определено в формуле I, и Z такой, как определено выше; с соединением формулы XI
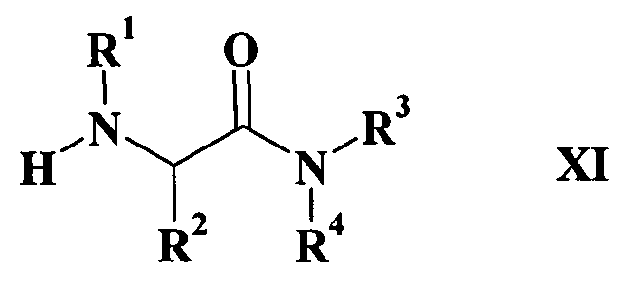
где
R1, R2, R3 и R4 имеют такие значения, как определено в формуле I, в присутствии восстановителя в случае реакции IX с XI или в присутствии основания в случае реакции соединения X с соединением XI.
Реакция соединения формулы IX с соединением формулы XI, которая дает соединение по изобретению, представляет собой восстановительное аминирование, а реакция соединения формулы X с соединением формулы XI представляет собой реакцию алкилирования: указанные реакции осуществляют по общепринятым методикам.
Предпочтительные восстановители, используемые в реакции соединения формулы IX с соединением формулы XI, выбирают из NaBH4, NaBH3CN и цианоборгидрида (полистирилметил)триметиламмония.
Согласно предпочтительному варианту осуществления изобретения реакцию соединения формулы X с соединением формулы XI проводят в присутствии основания и более предпочтительно указанное основание выбирают из K2CO3, триэтиламина или диизопропилэтиламина.
При получении соединения формулы I и исходных веществ и/или промежуточных соединений, описанных в настоящем документе, может быть полезной защита некоторых групп, которые чувствительны к условиям реакции.
Оценка полезности необязательной защиты, а также выбор подходящего защитного агента в соответствии с реакцией, осуществляемой при получении соединения по изобретению, и функциональной группы, которую следует защищать, общеизвестны специалистам в данной области.
Удаление необязательных защитных групп проводят по общепринятым методикам. Для ссылки обзорного характера на применение защитных групп в органической химии см. Theodora W. Greene и Peter G.M. Wuts "Protective groups in organic synthesis", John Wiley & Sons, Inc., II Ed., 1991.
Получение солей соединений формулы I осуществляют известными способами.
Для получения индивидуальных энантиомеров или диастереоизомеров (если таковые имеются) соединения формулы I указанное соединение может быть получено стерически контролируемым синтезом или путем применения реагентов, обладающих соответствующей хиральностью или выделением нужного изомера из энантиомерной или диастереомерной смеси по обычным методикам.
Например, индивидуальные оптически активные энантиомеры можно получать из их рацематов хиральной хроматографией или превращением их в смесь диастереоизомерных производных, разделением диастереоизомерных производных и восстановлением соответствующих энантиомеров.
Диастереоизомеры могут быть выделены из их смесей общепринятыми способами, основанными на их различных физико-химических свойствах, такими как хроматография, перегонка или фракционная кристаллизация.
Фармакология
Соединения по изобретению можно использовать для изготовления лекарственного средства, активного в качестве модулятора кальциевых и/или натриевых каналов против заболеваний, вызванных дисфункциями потенциалзависимых кальциевых и/или натриевых каналов.
Активность соединений, представителей настоящего изобретения, сравнивают с активностью внутреннего стандарта «ралфинамида» (S)-(+)-2-[4-(2-фторбензилокси)бензиламино]пропанамида (используемого авторами изобретения) и/или «сафинамида» (S)-(+)-2-[4-(3-фторбензилокси)бензиламино]пропанамида.
Такие соединения являются потенциалзависимыми блокаторами кальциевых и/или натриевых каналов, действующими в низком микромолярном интервале, как показано блокадой притока ионов кальция и/или натрия (флуоресцентный анализ) и потенциалзависимой блокадой токов (методы пэтч-кламп). Модулирующую активность фенилэтиламинопроизводных в отношении кальциевых каналов N-типа и L-типа определяют по анализам притока ионов кальция, основанным на флуоресценции (таблица 1 для N-типа и таблица 2 для L-типа) и методами пэтч-кламп в конститутивных и/или Cav 2.2 трансфецированных клеточных линиях (таблица 4).
Активность фениламинопроизводных как модуляторов натриевых каналов измеряют анализом притока ионов натрия, основанным на флуоресценции (таблица 3), методом пэтч-кламп в конститутивных и/или Nav 1.3 трансфецированных линиях клеток (таблица 5) и в кортикальных нейронах (таблица 6).
Активность вышеупомянутых соединений в отношении MAO-B измеряют, используя радиоферментный анализ (таблица 7).
Аналгетическую активность in vivo вышеуказанных соединений оценивают на «модели полного адъюванта Фрейнда на крысах» и на «модели невропатической боли Беннета на крысах» (таблица 8).
Противосудорожную активность определяют с помощью "теста максимального электрошока" на мышах (таблица 9).
Антиманиакальную активность определяют на модели «локомоторной гиперактивности мышей, вызванной амфетамином и хлордиазепоксидом» (фиг.1).
Активность против шизофрении и против привыкания (аддиктивности) оценивают с помощью «теста когнитивного нарушения при шизофрении» (таблица 10) и «тест поведенческой сенситизации, индуцированной кокаином» на крысах.
Тесты «острое раздражение мочевого пузыря крыс уксусной кислотой» и «умеренное раздражение мочевого пузыря крыс циклофосфамидом» используют в качестве моделей урологических заболеваний.
Активность против мигрени определяют с помощью «теста на мигрень» у крыс.
Такие вещества демонстрируют также «применение и частота-зависимость», т.e. усиление блокировки во время высокочастотной стимуляции, когда имеется большое накопление каналов в неактивированном состоянии, такое как в неврональных патологических состояниях. Функционально, зависимая от применения блокировка приводит к депрессии неврональной активности при высокочастотном возбуждении и пониженной блокирующей активности при нормальной доле работающих нейронов, заставляя предположить, что соединения по настоящему изобретению, возможно, селективно подавляют аномальную активность кальциевых и/или натриевых каналов, оставляя не затронутой физиологическую активность, уменьшая таким образом успокаивающее действие на ЦНС (W.A. Catterall, Trends Pharmacol. Sci. (1987) 8: 57-65).
Соединения по изобретению активны in vivo, когда их вводят перорально или внутрибрюшинно в количестве от 0,1 до 100 мг/кг на различных моделях животных, описанных далее в настоящем документе.
Принимая во внимание описанные выше механизмы действия, соединения по настоящему изобретению можно применять для предупреждения или лечения невропатической боли. Синдромы невропатической боли включают, но без ограничения, диабетическую невропатию; ишиаз; неспецифическую боль в нижнем отделе спины; боль при рассеянном склерозе; фибромиалгию; невропатию, вызванную ВИЧ; невралгию, такую как постгерпетическая невралгия и тригеминальная невралгия, мортоновская метатарзальная невралгия, каузалгия; и боль, возникающую в результате физической травмы, ампутации, фантома конечности, рака, токсинов или хронических воспалительных состояний; центральную боль, такую как боль, наблюдаемую при таламических синдромах, смешанные центральные и периферические формы боли, такие как комплексный регионарный болевой синдром (CRPS), называемый также симпатической рефлекторной дистрофией.
Соединения по изобретению применимы также для лечения хронической боли. Хроническая боль включает, но без ограничения, хроническую боль, вызванную воспалением или связанным с воспалением состоянием, остеоартрит, ревматоидный артрит, сильное ранение или травму, боль в верхнем отделе спины или боль в нижнем отделе спины (возникающую в результате систематического, регионарного или первичного заболевания позвоночника, такого как радикулопатия), боль в костях (обусловленную остеоартритом, остеопорозом, метастазом кости или другими причинами), тазовую боль, боль, связанную с повреждением спинного мозга, сердечную боль в груди, не сердечную боль в груди, центральную боль после удара, миофасциальную боль, менискоцитарную боль, боль при раке, болезнь Фабри, боль при СПИД, гериатрическую боль или боль, вызванную головной болью, боль в височно-нижнечелюстном суставе, подагру, фиброз или компрессионные синдромы верхней апертуры грудной клетки, в особенности ревматоидный артрит и остеоартрит.
Соединения по изобретению применимы также для лечения острой боли (вызванной сильным ранением, заболеванием, повреждениями при спортивной медицине, кистевым туннельным синдромом, ожогами, растяжениями и деформациями скелетных мышц, растяжением мышечных сухожилий, шейно-плечевыми болевыми синдромами, диспепсией, язвой желудка, язвой двенадцатиперстной кишки, дисменореей, эндометриозом или хирургической операцией (такой, как операция на открытом сердце или операция шунтирования), послеоперационной боли, боли при камнях в почках, боли в желчном пузыре, боли при родах или зубной боли.
Соединения по изобретению применимы также для лечения различных типов головной боли, таких как мигрень, головная боль тензионного типа, трансформированная мигрень или развивающаяся головная боль, сильная приступообразная головная боль с периодическими рецидивами, а также вторичные головные боли, такие как головные боли в результате инфекций, метаболических расстройств или других системных заболеваний, и других типов острой головной боли, пароксизмальной гемикрании и т.п., возникающих в результате ухудшения вышеупомянутых первичных и вторичных типов головной боли.
Соединения по изобретению применимы также для лечения неврологических заболеваний, таких как эпилепсия, включающая простые парциальные припадки, сложные парциальные припадки, вторично генерализованные припадки, дополнительно включающие отсутствие припадков, миоклонические припадки, клонические припадки, тонические припадки, тонические клонические припадки и атонические припадки. Соединения по изобретению применимы также для лечения нейродегенеративных расстройств различного происхождения, таких как болезнь Альцгеймера, и другие состояния деменции, такие как деменция с тельцами Леви, лобно-височная деменция и таупатия; боковой амиотрофический склероз, болезнь Паркинсона и другие паркинсониальные синдромы, другие типы спиномозжечковой дегенерации и невропатия Шарко-Mари-Тута.
Соединения по изобретению применимы также для лечения когнитивных нарушений и психических заболеваний. Психические заболевания включают, но без ограничения, большую депрессию, психическую депрессию, манию, биполярное расстройство (такое как биполярное расстройство типа I, биполярное расстройство типа II), циклотимическое расстройство, быстрые циклы, ультрадианные циклы, манию, гипоманию, шизофрению, шизофреноформные расстройства, шизоаффективные расстройства, расстройство личности, нарушения внимания с или без гиперактивного поведения, бредовые расстройства, кратковременные психические расстройства, долевые психические расстройства, психические расстройства, обусловленные общим медицинским состоянием, психические расстройства, вызванные веществами, или психическое расстройство, не оговоренное иным образом, тревожные расстройства, такие как генерализованное тревожное расстройство, расстройства панического типа, посттравматический стресс, волевые расстройства, фобические расстройства, диссоциативные состояния и, кроме того, расстройства при курении, наркомании и алкоголизме. В частности, биполярные расстройства, психоз, тревожные расстройства и аддиктивность.
Соединения по изобретению применимы также для лечения таких болезней, как головокружение, звон в ушах, мышечный спазм, мышечный склероз, и других заболеваний, включающих, но без ограничения, сердечно-сосудистые заболевания (такие как аритмия сердца, инфаркт сердца или стенокардия, повышенное кровяное давление, ишемическая болезнь сердца, ишемия головного мозга), эндокринные нарушения (такие как акромегалия или несахарный диабет), заболевания, в которых патофизиология нарушения затрагивает избыточную или гиперсекреторную или иным образом несоответствующую клеточную секрецию эндогенного вещества (такого как катехоламин, гормон или фактор роста).
Соединения по изобретению применимы также для избирательного лечения заболеваний печени, таких как воспалительные заболевания печени, например, хронический вирусный гепатит В, хронический вирусный гепатит С, алкогольное поражение печени, билиарный первичный цирроз печени, аутоиммунный гепатит, неалкогольный стеатогепатит и отторжение трансплантата печени.
Соединения по изобретению ингибируют воспалительные процессы, влияющие на все системы организма. Следовательно, они применимы в лечении воспалительных процессов скелетно-мышечной системы, примеры которых перечислены ниже, но этот список целевых заболеваний не является исчерпывающим: артритические нарушения, такие как анкилозирующий спондилит, затылочный артрит, фибромиалгия, подагра, ювенильный ревматоидный артрит, пояснично-крестцовый артрит, остеоартрит, остеопороз, псориазный артрит, ревматизм; заболевания, влияющие на кожу и родственные ткани: экзема, псориаз, дерматит и воспалительные состояния, такие как солнечные ожоги; заболевания дыхательной системы: астма, аллергический ринит и синдром дыхательной недостаточности, заболевания легких, связанные с воспалением, такие как астма и бронхит; хроническое обструктивное заболевание легких; расстройства иммунной и эндокринной систем: узелковый периартериит, тиреоидит, апластическая анемия, склеродома, астенический бульбарный паралич, рассеянный склероз и другие демиелинизирующие расстройства, энцефаломиелит, саркоидоз, нефритный синдром, синдром Бечета, полимиозит, гингивит.
Соединения по изобретению применимы также для лечения заболеваний желудочно-кишечного тракта (GI), таких как воспалительные заболевания кишечника, включающие, но без ограничения, язвенный колит, болезнь Крона, илеит, проктит, глютеновую болезнь, энтеропатию, микроскопический или коллагенозный колит, эозинофильный гастроэнтерит или резервуарный илеит, возникающий после проктоколэктомии и постилеонатальный анастомоз и синдром раздраженного кишечника, включающий любые заболевания, связанные с болью в животе и/или желудочно-кишечным дискомфортом, таким как пилороспазм, нервная диспепсия, спастическая толстая кишка, спастический колит, спастический кишечник, невроз кишечника, функциональный колит, спастический колит, послабляющий колит и функциональная диспепсия; но также для лечения атрофического гастрита, гастрита varialoforme, язвенного колита, пептической язвы, пиреза и других повреждений GI тракта, например, бактериями Helicobacter pylori, гастроэзофагеальной рефлюксной болезни, пареза желудка, такого как диабетический парез желудка; и других функциональных заболеваний кишечника, таких как неязвенная диспепсия (NUD); рвоты, диареи и внутреннего воспаления.
Соединения по изобретению применимы также для лечения заболеваний мочеполового тракта, таких как повышенная деятельность мочевого пузыря, простатит (хронический бактериальный и хронический небактериальный простатит), простадиния, интерстициальный цистит, непроизвольное мочеиспускание и доброкачественная гиперплазия простаты, аннекситы, тазовое воспаление, бартолинит и вагинит. В частности, повышенная деятельность мочевого пузыря и непроизвольное мочеиспускание.
Соединения по изобретению применимы также для лечения офтальмологических заболеваний, таких как ретинит, ретинопатии, увеит и сильное повреждение тканей глаза, дегенерация желтого пятна или глаукома, конъюнктивит.
Следует понимать, что соединения по изобретению можно эффективно использовать в сочетании с одним или несколькими другими терапевтическими средствами. Примеры подходящих средств для дополнительной терапии включают модулятор серотониновых рецепторов, включающий агонист 5HT1B/1D, такой как триптан (например, суматриптан или наратриптан); агонист аденозиновых A1 рецепторов; антагонист аденозиновых A2 рецепторов; антагонист пуринергических P2X рецепторов, EP лиганд; модулятор NMDA, такой как антагонист глицина; модулятор AMPA; антагонист вещества P (например, антагонист NKl); каннабиноид; агонист никотиновых рецепторов; агонист адренергических рецепторов альфа-1 или -2; ацетаминофен или фенацетин; ингибитор 5-липоксигеназы; антагонист лейкотриеновых рецепторов; DMARD (например, метотрексат); габапентин и родственные соединения; агонисты L-допа и/или допамина; ингибитор катехол-О-метилтрансферазы; трициклический антидепрессант (например, амитриптилин); стабилизирующие нейроны антиэпилептические лекарственные средства; ингибитор моноаминергического захвата (например, венлафаксин); ингибитор матричной металлопротеиназы; ингибитор синтазы оксида азота (NOS), такой как iNOS или ингибитор nNOS; акцептор свободных радикалов; ингибитор агрегации альфа-синуклеина; ингибитор холинэстеразы, средство, снижающее уровень холестерина; модулятор альфа-секретазы; модулятор бета-секретазы; ингибитор агрегации бета-амилоидов; ингибитор высвобождения или действия фактора альфа некроза опухолей; терапия антителами, такая как терапия моноклональными антителами; противовирусное средство, такое как ингибитор нуклеозидов (например, ламивудин) или модулятор иммунной системы (например, интерферон); опиоидный анальгетик, такой как морфин; антагонист ваниллоидных рецепторов; анальгетик, такой как ингибитор циклооксигеназы-1 и/или циклооксигеназы-2; местный анестетик, такой как лидокаин и производные; стимулятор, включающий кофеин; антагонист H2 (например, ранитидин); ингибитор протонного насоса (например, омепразол); антацид (например, гидроксид алюминия или магния; ветрогонное средство (например, семетикон); деконгестант (например, фенилэфрин, фенилпропаноламин, псевдоэфедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгекседрин или лево-дезоксиэфедрин, нафтазолин, ксилометазолин, пропилгекседрин или лево-дезоксиэфедрин); противокашлевое средство (например, кодеин, гидрокодон, карбифен, карбетапентан или декстрометорфан); диуретик или седативный или неседативный антигистамин; другие блокаторы кальциевых или натриевых каналов. Следует понимать, что настоящее изобретение охватывает применение соединения формулы (I) или его фармацевтически приемлемой соли в сочетании с одним или несколькими терапевтическими средствами.
Соединения по настоящему изобретению применимы для лекарственных средств, предназначенных для людей, и для ветеринарии. Следует понимать, что используемый здесь термин «лечение» во всех случаях, когда конкретно не определено иное, включает профилактику, уменьшение и лечение патологического заболевания, конкретно, он включает как лечение установленных симптомов, так и профилактическое лечение. Соединения по настоящему изобретению для их терапевтического или профилактического применения при вышеупомянутых патологиях предпочтительно используют в качестве активных ингредиентов в фармацевтической композиции.
Поэтому дополнительным объектом настоящего изобретения являются фармацевтические композиции, содержащие терапевтически эффективное количество соединения по изобретению или его соль в смеси с фармацевтически приемлемым носителем.
Соответственно, когда выражение «терапевтически эффективный» относится к «количеству», «дозе» или «дозировке» соединений по настоящему изобретению, подразумевается «количество», «доза» или «дозировка» любых соединений, достаточное(ая) для применения как для лечения установленных симптомов, так и для предупреждения вышеуказанных патологических заболеваний.
Фармацевтические композиции по настоящему изобретению можно вводить в виде множества лекарственных форм с быстрым и модифицированным высвобождением, например перорально в виде таблеток, пастилок, капсул, таблеток с покрытием из сахара или пленки, жидких растворов, эмульсий или суспензий; ректально в виде суппозиториев; парентерально, например, в виде препаратов для внутримышечных инъекций и/или препаратов замедленного всасывания; внутривенных инъекций и инфузий; местно и трансдермально в виде пластыря, геля и крема.
Подходящие фармацевтически приемлемые терапевтически инертные органические и/или неорганические вещества-носители, применимые для получения таких композиций, включают, например, воду, желатин, аравийскую камедь, лактозу, крахмал, целлюлозу, стеарат магния, тальк, растительные масла, циклодекстрины, полиалкиленгликоли и т.п.
Композиция, содержащая фенилэтиламинопроизводные формулы I, такая как определено выше, может быть стерилизована и может содержать дополнительно хорошо известные компоненты, такие как, например, консерванты, стабилизаторы, увлажняющие вещества и эмульгаторы, например парафиновое масло, моноолеат маннида, соли для регулирования осмотического давления, буферы и т.п.
Например, твердые формы для перорального введения вместе с активным ингредиентом могут содержать разбавители, например лактозу, декстрозу, сахарозу, целлюлозу, кукурузный или картофельный крахмал; вещества, способствующие скольжению, например диоксид кремния, тальк, стеариновую кислоту, стеарат магния или кальция и/или полиэтиленгликоли; связывающие вещества, такие как крахмалы, акациевые камеди, желатин, метилцеллюлоза, карбоксиметилцеллюлоза или поливинилпирролидон; разрыхлители, например крахмал, альгиновая кислота, альгинаты или натрий-крахмалгликолят; смеси, выделяющие газ; красящие и подслащивающие вещества; увлажняющие агенты, такие как лецитин, полисорбаты, лаурилсульфаты; и вообще нетоксичные и фармакологически неактивные вещества, используемые в фармацевтических рецептурах. Указанные фармацевтические препараты можно изготавливать известными способами, например смешиванием, гранулированием, таблетированием, способами нанесения сахарного или пленочного покрытия.
Приготовление фармацевтических композиций по настоящему изобретению можно осуществлять по общепринятым методикам.
Препараты для перорального введения включают препараты с замедленным высвобождением, которые можно получать общепринятыми способами, например путем нанесения энтеросолюбильного покрытия на таблетки и гранулы.
Жидкая дисперсия для перорального введения может быть приготовлена, например, в виде сиропов, эмульсий и суспензий. В качестве носителя сиропы могут содержать, например, сахарозу, или сахарозу с глицерином, и/или маннит, и/или сорбит.
Суспензии и эмульсии могут содержать в качестве носителя, например, природную камедь, агар, альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензии или растворы для внутримышечных инъекций могут содержать вместе с активным соединением фармацевтически приемлемый носитель, например стерильную воду, оливковое масло, этилолеат, гликоли, например, пропиленгликоль и, если требуется, подходящее количество гидрохлорида лидокаина.
Растворы для внутривенных инъекций или инфузии могут содержать в качестве носителя, например, стерильную воду, или предпочтительно, чтобы они были в виде стерильных водных изотонических физиологических растворов.
Суппозитории могут содержать вместе с активным ингредиентом фармацевтически приемлемый носитель, например кокосовое масло, полиэтиленгликоль, поверхностно-активные вещества сложные эфиры жирных кислот и полиоксиэтиленсорбитана или лецитин.
Фармацевтические композиции, содержащие фенилэтиламинопроизводные формулы I, как определено выше, содержат на единицу лекарственной формы, например капсулу, таблетку, порошок для инъекций, чайную ложку, суппозиторий и т.п., от около 0,1 до около 500 мг одного или нескольких активных ингредиентов, наиболее предпочтительно от 1 до 10 мг.
Специалисты могут легко определить оптимальные терапевтически эффективные дозы для введения, которые могут варьироваться в основном в зависимости от силы препарата, типа введения и развития состояния или заболевания, которое подвергается лечению. Кроме того, факторы, связанные с конкретным субъектом, которого лечат, включающие возраст, массу тела, режим питания и время введения, вызывают необходимость регулирования дозы до терапевтически эффективного уровня.
Понятно, что хотя здесь изобретение описывается в отношении предпочтительных вариантов осуществления изобретения, специалисты в данной области техники знают, что можно реализовать и другие варианты осуществления изобретения, не выходя за пределы сущности изобретения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
1Н ЯМР спектры регистрируют в растворе CDCl3 или ДМСО-d6 на спектрометре Varian Gemini (200 MГц). Химические сдвиги определяют как δ при использовании CDCl3 или ДМСО-d6 и D2O в качестве внутреннего стандарта. ВЭЖХ/МС анализы регистрируют на приборе Gilson, используя колонку X-Terra RP 18 column (5 мкм, 4,6×50 мм), соединенную с УФ-детектором (220 нм) и масс-спектрометром Finnigan Aqa (электроспрей, ионизация, область положительных ионов). Условия, используемые для анализов: поток: 1,2 мл/мин; температура колонки: 50°С; градиент A/B элюирования (элюент A: 0,1% муравьиная кислота в воде; элюент B: 0,1% муравьиная кислота в ацетонитриле): 5-95% B от 0 до 8,0 минут, 95% B от 8,0 до 9,5 минут.
Для лучшей иллюстрации изобретения приводятся нижеследующие примеры.
ПРИМЕРЫ
Пример 1. Гидрохлорид 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N-метилацетамида
Вышеупомянутое соединение синтезируют по схеме 1.
Схема 1
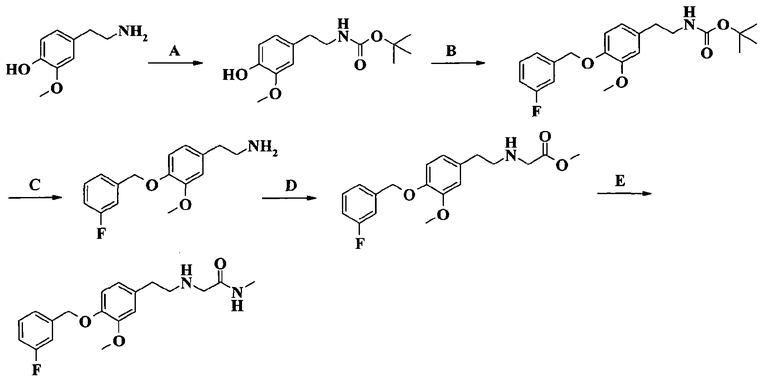
Стадия A. трет-Бутиловый эфир [2-(4-гидрокси-3-метоксифенил)этил]карбаминовой кислоты
(Boc)2O (54 г, 0,24 моль), растворенный в 100 мл диоксана, прибавляют при 0°С к раствору, содержащему 39 г (0,23 моль) 2-(4-гидрокси-3-метоксифенил)этиламина в 230 мл 1М раствора гидроксида натрия и 390 мл диоксана. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Диоксан удаляют и к остатку прибавляют водный KHSO4 до достижения рН 6. Экстрация этилацетатом приводит к образованию масла, которое растирают в гексане и получают 54,7 г (выход 88%) белого твердого вещества.
1H ЯМР (CDCl3): 7,26 (с, 1H); 6,88-6,64 (м, 2H); 5,51 (с, 1H); 4,54 (ушир.с, 1H); 3,80 (с, 3H); 3,41-3,27 (м, 2H); 2,72 (т, 2H, J=7,25 Гц); 1,44 (с, 9H).
Стадия B. трет-Бутиловый эфир [2-(4-(3-фторбензилокси)-3-метоксифенил)этил]карбаминовой кислоты
1-Хлорметил-3-фторбензол (34,6 г, 0,23 моль) в 50 мл сухого диметилформамида прибавляют к суспензии 55,9 г (0,209 моль) трет-бутилового эфира [2-(4-гидрокси-3-метоксифенил)этил]карбаминовой кислоты, 43 г K2CO3 и 3,4 г иодида калия в 400 мл сухого диметилформамида. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Удаляют растворитель, к остатку прибавляют воду и продукт экстрагируют этилацетатом. Полученное неочищенное масло растирают с диэтиловым эфиром. Твердое вещество отфильтровывают и получают 58,2 г (выход 74%) продукта, указанного в заголовке.
1H ЯМР (CDCl3): 7,40-6,60 (м, 7H); 5,10 (с, 2H); 4,50-4,60 (ушир.с, 1H); 3,90 (с, 3H); 3,30-3,40 (м, 2H); 3,75 (т, 2H, J=7,2 Гц); 1,44 (с, 9H).
Стадия C. Гидрохлорид 2-[4-(3-фторбензилокси)-3-метоксифенил]этиламина
трет-Бутиловый эфир [2-(4-(3-фторбензилокси)-3-метоксифенил)этил]карбаминовой кислоты (58,2 г, 0,155 моль) растворяют в 300 мл этилацетата. Прибавляют 150 мл безводного 2М раствора хлористоводородной кислоты в этилацетате и смесь перемешивают при комнатной температуре в течение ночи. Твердое вещество отфильтровывают и промывают этилацетатом и диэтиловым эфиром и получают 44,2 г (выход 91%) белого твердого вещества.
1H ЯМР (D2O): 7,31-7,17 (м, 1H); 7,11-6,80 (м, 5H); 6,69-6,61 (м, 1H); 5,02 (с, 2H); 3,69 (с, 3H); 3,05 (т, 2H, J=6,85 Гц); 2,74 (т, 2H, J=6,85 Гц).
Стадия D. Гидрохлорид метилового эфира [2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]уксусной кислоты
2-[4-(3-Фторбензилокси)-3-метоксифенил]этиламин (1 г, 3,2 ммол), K2CO3 (833 мг, 6 ммол), 50 мг иодида калия и 0,27 мл (2,9 ммол) метилового эфира бромуксусной кислоты растворяют в 10 мл диметилформамида и смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют, к остатку прибавляют воду и остаток экстрагируют этилацетатом. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:0:0→100:2,5:0,25 градиент об.:об.:об.). Полученный продукт растворяют в безводной хлористоводородной кислоте в этилацетате. Растворитель удаляют и остаток растирают в диэтиловом эфире. Получают 482 мг (выход 39%) указанного в заголовке соединения в виде коричневого твердого вещества.
1H-ЯМР (CDCl3): 7,39-7,28 (м, 2H); 7,22-6,92 (м, 2H); 7,04-6,92 (м, 1H); 6,86-6,74 (м, 3H); 5,10 (с, 2H); 3,88 (с, 3H); 3,87-3,80 (м, 2H); 3,78 (с, 3H); 3,41-3,19 (м, 4H).
Стадия E. Гидрохлорид 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N-метилацетамида
Метиловый эфир [2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]уксусной кислоты (900 мг, 2,34 ммол) растворяют в 10 мл сухого толуола и прибавляют 5 мл (10 ммол) 2M раствора метиламина в тетрагидрофуране при 0°С, затем 5 мл (10 ммол) 2 M раствора триметилалюминия в гептане. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Раствор охлаждают до 0°С и выливают в метанол. Растворитель удаляют и неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:0:0→100:4:0,4 градиент об.:об.:об.). Продукт растворяют в безводной хлористоводородной кислоте в этилацетате и твердое вещество отфильтровывают. Выделяют 590 мг (выход 66%) указанного в заголовке соединения в виде гигроскопического твердого вещества.
1H-ЯМР (диметилсульфоксид-d6): 9,06 (м, 2H); 8,46 (ушир.м, 1H); 7,49-6,67 (м, 7H); 5,07 (с, 2H); 3,76 (с, 3H); 3,73-3,63 (ушир.м, 2H); 3,23-3,04 (м, 2H); 2,94-2,80 (ушир.м, 2H); 2,65 (д, 3H, J=4,36 Гц).
ЖХ-МС: MH+=347,4.
Пример 2. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]изобутиламино]-N-метилацетамида
Вышеуказанное соединение синтезируют согласно схеме 2.
Схема 2
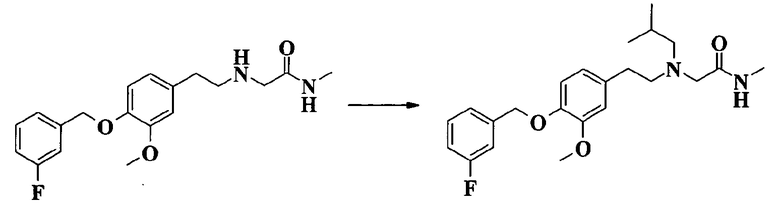
2-[2-[4-(3-Фторбензилокси)-3-метоксифенил]этиламино]-N-метилацетамид (90 мг, 0,235 ммол) и 2-метилпропионовый альдегид (19 мг, 0,263 ммол) растворяют в 6 мл смеси дихлорметан/уксусная кислота (8:2, об.:об.) и 1,5 мл метанола. Прибавляют 100 мг (0,425 ммол) цианоборгидрида (полистирилметил)триметиламмония (загрузка: 4,25 ммол/г) и смесь перемешивают при комнатной температуре в течение ночи. Смолу отфильтровывают и растворитель удаляют. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:0:0→100:2:0,2 градиент об.:об.:об.). Продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате, растворитель удаляют и остаток растирают в диэтиловом эфире. Выделяют 80 мг (выход 77%) указанного в заголовке соединения в виде гигроскопического твердого вещества.
1H-ЯМР (диметилсульфоксид-d6): 9,42 (ушир.м, 1H); 8,73 (ушир.м, 1H); 7,49-6,72 (м, 7H); 5,07 (с, 2H); 4,14-3,87 (м, 2H); 3,77 (с, 3H); 3,42-3,24 (м, 2H); 3,10-2,86 (м, 4H); 2,69-2,65 (м, 3H); 2,16-1,92 (м, 1H); 0,95 (д, 6H). ЖХ-МС: MH+=403.
Примеры 3-13. Соединения примеров 3-13 получают аналогично по методике, описанной в схеме 2.
Пример 3. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](2-пропин-1-ил)амино]-N-метилацетамида
ЖХ-МС: MH+=385.
Пример 4. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](3-метилизоксазол-5-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=442.
Пример 5. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](3-метоксиизоксазол-5-илметил)амино]-N-метилацетамида.
Пример 6. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](1-метилимидазол-5-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=441.
Пример 7. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](тетрагидрофуран-3-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=431.
Пример 8. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-метилацетамида
ЖХ-МС: MH+=401.
Пример 9. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=427.
Пример 10. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](фуран-3-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=427.
Пример 11. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](5-гидроксиметилфуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=457.
Пример 12. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]-[(1,2,3-тиадиазол-4-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=445.
Пример 13. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](1,3-диметилпиразол-5-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=455.
Примеры 14-15. Соединения примеров 14-15 получают аналогично по методике, описанной в схеме 2, но без образования соли с хлористоводородной кислотой.
Пример 14. 2-[[2-[4-(3-Фторбензилокси)-3-метоксифенил]этил](6-метоксипиридин-3-илметил)амино]-N-метилацетамид
ЖХ-МС: MH+=468.
Пример 15. 2-[[2-[4-(3-Фторбензилокси)-3-метоксифенил]этил](3,5-диметоксибензил)амино]-N-метилацетамид
ЖХ-МС: MH+=497.
Примеры 16-25. Соединения примеров 16-25 получают аналогично по методике, описанной в схеме 2 примера 2, исходя из 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметилацетамида, вместо 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N-метилацетамида.
Пример 16. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]метиламино]-N,N-диметилацетамида
ЖХ-МС: MH+=375.
Пример 17. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N,N-диметилацетамида
ЖХ-МС: MH+=415.
Пример 18. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]изопропиламино]-N,N-диметилацетамида
ЖХ-МС: MH+=403.
Пример 19. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](1-метилпиперидин-4-ил)амино]-N,N-диметилацетамида
ЖХ-МС: MH+=458.
Пример 20. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N,N-диметилацетамида
ЖХ-МС: MH+=451.
Пример 21. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]этиламино]-N,N-диметилацетамида
ЖХ-МС: MH+=389.
Пример 22. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](фуран-2-илметил)амино]-N,N-диметилацетамида
ЖХ-МС: MH+=441.
Пример 23. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](тиен-2-илметил)амино]-N,N-диметилацетамида
ЖХ-МС: MH+=457.
Пример 24. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](тиазол-2-илметил)амино]-N,N-диметилацетамида
ЖХ-МС: MH+=458.
Пример 25. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](пиридин-3-илметил)амино]-N,N-диметилацетамида
ЖХ-МС: MH+=452.
Примеры 26-27. Соединения примеров 26-27 получают аналогично по методике, описанной в схеме 2, исходя из 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметилацетамида вместо 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N-метилацетамида, но без образования соли с хлористоводородной кислотой.
Пример 26. 2-[[2-[4-(3-Фторбензилокси)-3-метоксифенил]этил](2-гидроксиэтил)амино]-N,N-диметилацетамид
ЖХ-МС: MH+=405.
Пример 27. 2-[[2-[4-(3-Фторбензилокси)-3-метоксифенил]этил](тиазол-2-ил)амино]-N,N-диметилацетамид.
Пример 28. Гидрохлорид 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметилацетамида.
Вышеупомянутое соединение синтезируют по схеме 3
Схема 3
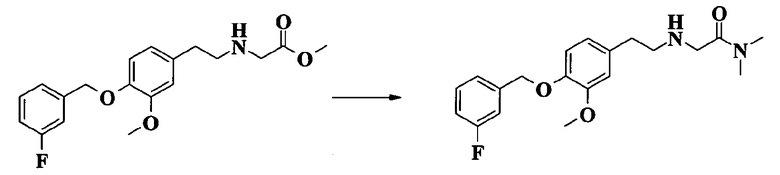
Метиловый эфир 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]уксусной кислоты (100 мг, 0,26 ммол) растворяют в 5 мл сухого толуола. Прибавляют 0,4 мл (0,8 ммол) 2M раствора диметиламина в тетрагидрофуране при 0°С, затем 0,4 мл (0,8 ммол) 2M раствора триметилалюминия в гептане. Реакционную смесь перемешивают в течение 4 часов при комнатной температуре. Раствор охлаждают до 0°С и выливают в метанол. Растворитель удаляют и неочищенный остаток очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:5:0,5 об.:об.:об.). Продукт растворяют в смеси этилацетат/хлористоводородная кислота. Растворитель удаляют и остаток растирают в диэтиловом эфире. Выделяют 70 мг (выход 68%), указанного в заголовке соединения в виде гигроскопического твердого вещества.
1H-ЯМР (CDCl3): 9,56 (ушир.с, 1H); 7,38-7,27 (м, 2H); 7,21-7,10 (м, 2H); 7,04-6,86 (м, 2H); 6,79-6,76 (м, 2H); 5,10 (с, 2H); 3,93 (ушир.т, 2H); 3,89 (с, 3H); 3,41-3,15 (м, 4H); 2,94 (с, 6H).
ЖХ-МС: MH+=361.
Пример 29. Гидрохлорид 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-1-(пирролидин-1-ил)этанона
Указанное соединение получают, как описано на схеме 3, используя пирролидин в диметилформамиде вместо N,N-диметиламин, для получения нужного соединения в виде белого твердого вещества (выход 48%).
1H-ЯМР (CDCl3): 9,57 (ушир.с, 1H); 7,38-7,27 (м, 2H); 7,21-7,10 (м, 2H); 7,04-6,76 (м, 4H); 5,10 (с, 2H); 3,89 (с, 3H); 3,84 (ушир. т, 2H); 3,50-3,14 (м, 8H); 2,07-1,80 (м, 4H).
ЖХ-МС: MH+=387.
Пример 30. Гидрохлорид 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметилпропионамида
Вышеупомянутое соединение синтезируют по схеме 4.
Схема 4
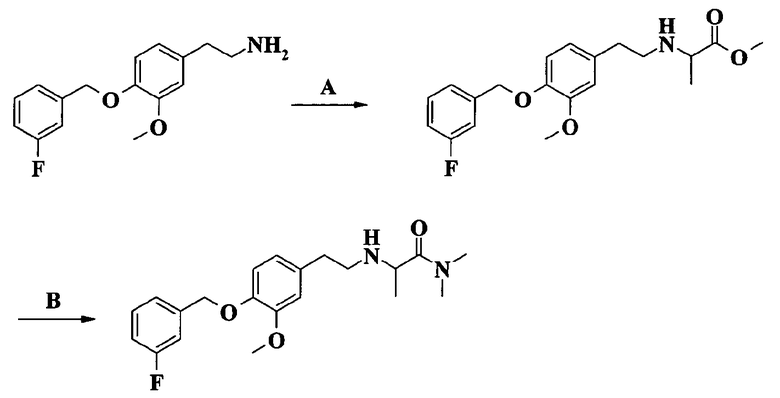
Стадия A. Гидрохлорид метилового эфира 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]пропионовой кислоты
Раствор 0,75 г (2,4 ммол) 2-[4-(3-фторбензилокси)-3-метоксифенил]этиламина, 0,88 мл (5,05 ммол) диизопропилэтиламина и 0,294 мл (2,64 ммол) метилового эфира 2-бромпропионовой кислоты в 10 мл сухого тетрагидрофурана выдерживают при 75°С в течение 48 часов. Реакционную смесь выливают в воду и продукт экстрагируют этилацетатом. Растворитель удаляют и неочищенный остаток очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:0:0→100:2:0,2 градиент). Продукт растворяют в смеси этилацетат/хлористоводородная кислота. Растворитель удаляют и остаток растирают в диэтиловом эфире. Выделяют 300 мг (выход 31%) белого твердого вещества.
1H-ЯМР (D2O): 7,35-7,18 (м, 1H); 7,16-6,80 (м, 5H); 6,75-6,62 (м, 1H); 5,05 (с, 2H); 4,06-3,88 (м, 1H); 3,77-3,64 (м, 6H); 3,18 (ушир.т, 2H), 2,83 (ушир.т, 2H); 1,43-1,34 (м, 3H).
Стадия B. Гидрохлорид 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметилпропионамида
Метиловый эфир 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]пропионовой кислоты (125 мг, 0,31 ммол) растворяют в 5 мл сухого толуола. Прибавляют 0,785 мл (1,57 ммол) 2M раствора диметиламина в тетрагидрофуране при 0°С, затем 0,47 мл (0,94 ммол) 2M раствора триметилалюминия в гептане. Реакционную смесь перемешивают в течение 5 часов при комнатной температуре. Раствор охлаждают до 0°С и выливают в метанол. Растворитель удаляют и неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:5:0,5). Продукт растворяют в смеси этилацетат/хлористоводородная кислота. Растворитель удаляют и твердое вещество отфильтровывают. Выделяют 94 мг (выход 74%) указанного в заголовке соединения в виде гигроскопического твердого вещества.
1H-ЯМР (CDCl3): 8,01 (ушир.с, 1H); 7,38-7,09 (м, 3H); 7,03-6,72 (м, 4H); 5,08 (с, 2H); 4,49-4,30 (м, 1H); 3,86 (с, 3H); 3,42-3,07 (м, 4H); 2,98 (д, 6H, J=7,51 Гц); 1,69-1,60 (м, 3H).
Пример 31. (S)-2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N-метил-4-метилвалерамид
ЖХ-МС: MH+=403.
Указанное соединение получают аналогично по методике, описанной в схеме 4.
Пример 32. Гидрохлорид 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-2-фенил-N,N-диметилацетамида
Вышеуказанное соединение получают по схеме 5.
Схема 5
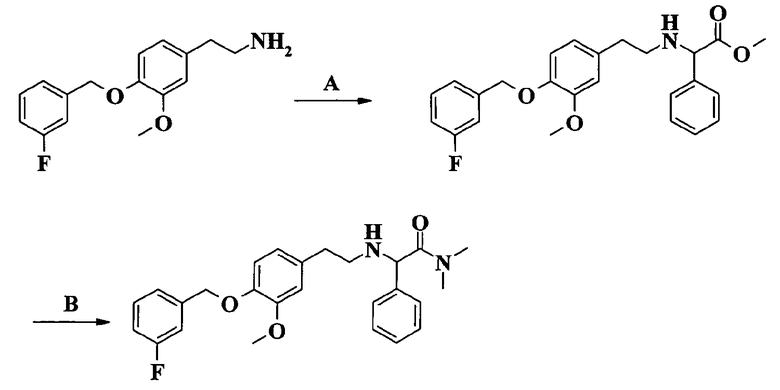
Стадия A. Метиловый эфир 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-2-фенилуксусной кислоты
Раствор 0,75 г (2,4 ммол) 2-[4-(3-фторбензилокси)-3-метоксифенил]этиламина, 0,88 мл (5,05 ммол) диизопропилэтиламина и 0,416 мл (2,64 ммол) метилового эфира 2-бром-2-фенилуксусной кислоты в 10 мл сухого тетрагидрофурана выдерживают при 75°С в течение 48 часов. Реакционную смесь выливают в воду и продукт экстрагируют этилацетатом. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:0:0→100:2:0,2 градиент об.:об.:об.). Получают 600 мг (выход 50%) указанного в заголовке соединения в виде желтого масла.
1H-ЯМР (D2O): 7,45-7,16 (м, 6H); 7,10-6,84 (м, 3H); 6,81-6,70 (м, 2H); 6,57 (дд, 1H, J=8,37 и 2,16 Гц); 4,99 (с, 2H); 4,93 (с, 1H); 3,63 (д, 6H, J=2,38 Гц); 3,13-2,68 (м, 4H).
Стадия B. 2-[2-[4-(3-Фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметил-2-фенилацетамид
Указанное соединение синтезируют по методике, описанной на схеме 6 (стадия В), используя 115 мг (0,27 ммол) метилового эфира 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-2-фенилуксусной кислоты, 1,06 мл (2,1 ммол) 2M раствора диметиламина в тетрагидрофуране и 0,53 мл (1,06 ммол) 2M раствора триметилалюминия в гептане. Выделяют 66 мг (выход 52%) указанного в заголовке соединения выделяют в виде белого твердого вещества.
1H-ЯМР (CDCl3): 8,52 (ушир.с, 1H); 7,47-7,10 (м, 9H); 7,03-6,92 (м, 2H); 6,81-6,62 (м, 3H); 5,42 (ушир.с, 1H); 5,10 (с, 2H); 3,58 (с, 3H); 3,24-2,99 (м, 4H); 2,91 (д, 6H).
Пример 33-35. Упомянутые соединения получают по методике, описанной в схеме 5, используя соответствующий амин на стадии В.
Пример 33. Гидрохлорид 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-1-(морфолин-4-ил)-2-фенилэтанона
(выход 51%).
1H-ЯМР (CDCl3): 8,59 (ушир.с, 1H); 7,48-7,26 (м, 6H); 7,21-7,10 (м, 2H); 7,04-6,92 (м, 2H); 6,79-6,63 (м, 3H); 5,50 (ушир.с, 1H); 5,09 (с, 2H); 3,85 (с, 3H); 3,76-3,33 (м, 6H); 3,23-2,91 (м, 6H).
Пример 34. Гидрохлорид 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-1-(пирролидин-1-ил)-2-фенилэтанона
ЖХ-МС: MH+=463.
Пример 35. Гидрохлорид 2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-1-(4-метилпиперазин-1-ил)-2-фенилэтанона
ЖХ-МС: MH+=492.
Пример 36. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]ацетамида
Указанное соединение синтезируют по схеме 6
Схема 6
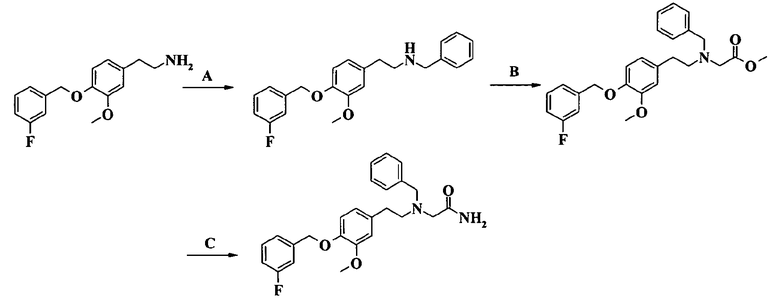
Стадия A. [2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламин
Смесь 4,4 г (16 ммол) 2-[4-(3-фторбензилокси)-3-метоксифенил]этиламина, 1,72 г (16 ммол) бензальдегида, 100 мл этанола и 30 г молекулярных сит 4Å кипятят с обратным холодильником в течение ночи. Реакционную смесь охлаждают до комнатной температуры, прибавляют 50 мг PtO2 и смесь гидрируют при 15 фунт/кв.дюйм в течение 5 часов. Катализатор отфильтровывают и растворитель удаляют при пониженном давлении. Неочищенный продукт реакции очищают флэш-хроматографией (дихлорметан/метанол/NH3 85:15:1,5, об.:об.:об.) и 2,72 г (выход 46%) указанного в заголовке соединения выделяют в виде желтого масла.
1H-ЯМР (CDCl3): 10,12 (ушир.с, 1H); 7,60-7,26 (м, 8H); 7,19-7,09 (м, 2H); 7,03-6,91 (м, 1H); 6,77-6,59 (м, 3H); 5,08 (с, 2H); 4,01 (ушир.т, 2H); 3,18-2,88 (м, 4H).
Стадия B. Метиловый эфир 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]уксусной кислоты
[2-[4-(3-Фторбензилокси)-3-метоксифенил]этил]бензиламин (1,7 г, 4,65 ммол), 1,74 мл (10 ммол) диизопропилэтиламина и 0,5 мл (5,11 ммол) метилового эфира 2-бромуксусной кислоты растворяют в 20 мл ацетонитрила и реакционную смесь перемешивают при 70°С в течение ночи. Растворитель удаляют при пониженном давлении, к остатку прибавляют воду и продукт экстрагируют этилацетатом. Неочищенный продукт очищают флэш-хроматографией (гексан/этилацетат 100:0→80:20 градиент об.:об.) и выделяют 1,94 г (выход 95%) указанного в заголовке соединения в виде желтого масла.
1H-ЯМР (CDCl3): 7,72-7,65 (м, 2H); 7,47-7,28 (м, 5H); 7,21-7,10 (м, 2H); 7,04-6,93 (м, 1H); 6,85-6,67 (м, 3H); 5,11 (с, 2H); 4,68-4,30 (м, 2H); 3,89 (с, 3H); 3,72 (с, 3H); 3,68-3,15 (м, 6H).
Стадия C. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]ацетамида
Метиловый эфир 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]уксусной кислоты (80 мг, 0,18 ммол) растворяют в 3 мл диоксана и 2 мл NH3 (30%). Раствор нагревают микроволновым облучением при 100°С в течение 8 часов. Растворитель удаляют и неочищенный остаток очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:0:0→95:5:0,5 градиент об.:об.:об.). Продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате. Растворитель удаляют и остаток растирают в диэтиловом эфире. Выделяют 30 мг (выход 36%) указанного в заголовке соединения в выделяют в виде желтого твердого вещества.
1H-ЯМР (диметилсульфоксид-d6): 10,00 (ушир.с, 1H); 7,95, 7,69 (2 ушир.с, 2H); 7,63-6,67 (м, 12H); 5,06 (с, 2H); 4,42 (ушир.с, 2H); 3,86 (ушир.с, 2H); 3,75 (с, 3H); 3,38-3,12 (ушир.с, 2H); 3,10-2,87 (ушир.с, 2H).
ЖХ-МС: MH+=423.
Примеры 37-45. Указанные соединения получают по методике, описанной на схеме 6 (стадия С), используя соответствующий амин.
Пример 37. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-этилацетамида
ЖХ-МС: MH+=451.
Пример 38. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-изопропилацетамид
ЖХ-МС: MH+=465.
Пример 39. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-этил-N-метилацетамида
ЖХ-МС: MH+=465.
Пример 40. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-1-(пирролидин-1-ил)этанона
ЖХ-МС: MH+=477.
Пример 41. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-бензилацетамида
ЖХ-МС: MH+=513.
Пример 42. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-(2-амино-2-метилпропил)ацетамида
ЖХ-МС: MH+=494.
Пример 43. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-(2-диметиламиноэтил)ацетамида
ЖХ-МС: MH+=494.
Пример 44. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-[2-(1-метилпирролидин-2-ил)этил]ацетамида
ЖХ-МС: MH+=534.
Пример 45. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-(3-имидазол-1-илпропил)ацетамида
ЖХ-МС: MH+=531.
Пример 46. 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-метилацетамид
Упомянутое соединение получают аналогично по методике, описанной на схеме 6 (стадия C), используя соответствующий амин, но не превращают в соль действием хлористоводородной кислоты.
ЖХ-МС: MH+=437,4.
Пример 47. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-этилацетамида
Вышеупомянутое соединение синтезируют по схеме 7
Схема 7
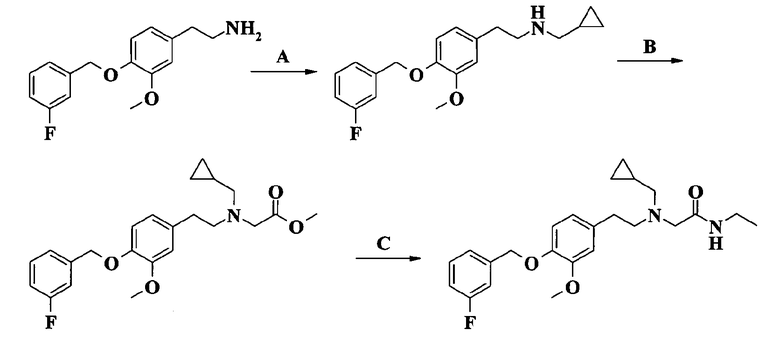
Стадия A. [2-[4-(3-Фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амин
Суспензию 0,66 г (2,1 ммол) 2-[4-(3-фторбензилокси)-3-метоксифенил]этиламина, 0,151 г (2,1 ммол) циклопропанкарбальдегида, 0,3 мл триэтиламина и 3 г молекулярных сит в 6 мл этанола перемешивают при кипячении с обратным холодильником в течение 3 часов. Смесь охлаждают до 0°С и порциями прибавляют 0,2 г (5 ммол) NaBH4. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Прибавляют 3 мл водного хлорида аммония, растворитель удаляют при пониженном давлении, остаток экстрагируют этилацетатом. Неочищенный остаток очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:0:0→95:5:0,5 градиентградиент об.:об.:об.) и получают 0,3 г требуемого соединения в виде масла (выход 43%).
1H-ЯМР (CDCl3): 9,80 (ушир.м, 2H); 7,37-6,67 (м, 7H); 5,09 (с, 2H); 3,86 (с, 3H); 3,22 (ушир.с, 4H); 2,92-2,80 (м, 2H); 0,93-0,78 (м, 1H); 0,75-0,63 (м, 2H); 0,49-0,38 (м, 2H).
Стадия B. Метиловый эфир 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]уксусной кислоты
[2-[4-(3-Фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амин, (0,271 г, 0,82 ммол), триэтиламин (0,140 мл, 1 ммол) и 0,155 г (0,89 ммол) метилового эфира бромуксусной кислоты растворяют в 5 мл ацетонитрила и проводят реакцию при 70°С в течение ночи. Растворитель удаляют в вакууме, к остатку прибавляют воду и продукт экстрагируют этилацетатом. Неочищенный продукт очищают флэш-хроматографией (гексан/этилацетат 100:0→80:20 градиент об.:об.) и выделяют 0,32 г (выход 97%) указанного в заголовке соединения в виде желтого масла.
1H-ЯМР (CDCl3): 7,38-7,28 (м, 1H); 7,22-7,10 (м, 2H); 7,04-6,91 (м, 1H); 6,80-6,62 (м, 3H); 5,11 (с, 2H); 3,88 (с, 3H); 3,70 (с, 3H); 3,59-3,50 (м, 2H); 3,00-2,53 (м, 6H); 0,96-0,78 (м, 1H); 0,58-0,46 (м, 2H); 0,18-0,07 (ушир.кв, 2H).
Стадия C. 2-[[2-[4-(3-Фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-этилацетамид
Метиловый эфир 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]уксусной кислоты (105 мг, 0,26 ммол) растворяют в 5 мл сухого толуола. Прибавляют 0,5 мл 2M (1 ммол) раствора этиламина в тетрагидрофуране прибавляют при 0°C, затем 0,4 мл (0,8 ммол) 2M раствора триметилалюминия в гептане. Реакционную смесь перемешивают в течение 4 часов при комнатной температуре. Раствор охлаждают до 0°С и выливают в метанол. Растворитель удаляют, неочищенный продукт очищают флэш-хроматографией (этилацетат/гексан 0:100→85:15 градиент об.:об.) и получают 52 мг (выход 48%) указанного в заголовке соединения.
ЖХ-МС: MH+=415.
1H-ЯМР (CDCl3): 8,85 (ушир.с, 1H); 7,39-6,64 (м, 7H); 5,09 (с, 2H); 4,19 (м, 2H); 3,86 (с, 3H); 3,61-3,42 (м, 2H); 3,42-3,09 (м, 6H); 1,36-1,17 (м, 1H); 1,22 (т, 3H, J=7,3 Гц); 0,81-0,65 (м, 2H); 0,53-0,41 (м, 2H).
Пример 48. Гидрохлорид 2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-изопропилацетамида.
Указанное соединение получают по методике, описанной в настоящем описании выше, используя изопропиламин вместо этиламина. Выделяют 63 мг требуемого соединения (выход 52%) в виде гигроскопичного твердого вещества.
ЖХ-МС: MH+=429.
Пример 49. 2-[[2-[4-(Бензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-метилацетамид
Вышеупомянутое соединение синтезируют по схеме 8
Схема 8
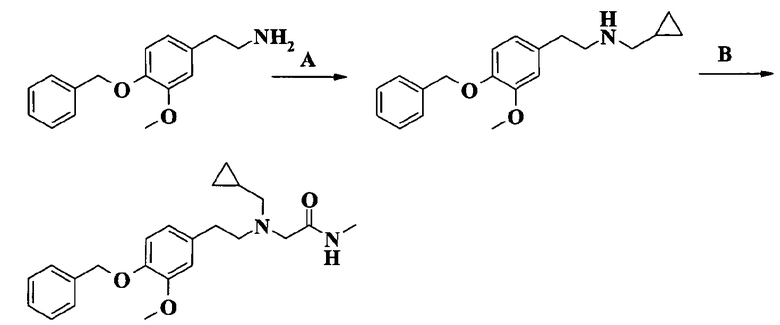
Стадия A. [2-(4-Бензилокси-3-метоксифенил)этил](циклопропилметил)амин
Суспензию 1,5 г (5,1 ммол) 2-(4-бензилокси-3-метоксифенил)этиламина, 0,365 г (5,1 ммол) циклопропанкарбоксальдегида, 0,7 мл триэтиламина и 8 г молекулярных сит в 15 мл этанола перемешивают при кипячении с обратным холодильником в течение 3 часов. Смесь охлаждают до 0°С и прибавляют порциями 0,19 г (5 ммол) NaBH4. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Прибавляют 3 мл водного хлорида аммония, растворитель удаляют в вакууме и остаток экстрагируют этилацетатом. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:0:0→95:5:0,5 градиент об.:об.:об.) и получают 0,850 г (выход 53%) требуемого соединения в виде масла.
ЖХ-МС: MH+=312.
Стадия B. 2-[[2-[4-(Бензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-метилацетамид
Смесь 0,5 г (1,6 ммол) [2-(4-бензилокси-3-метоксифенил)этил] (циклопропилметил)амина, 0,27 мл (1,92 ммол) триэтиламина, 0,207 г (1,92 ммол) 2-хлор-N-метилацетамида в 4 мл диметилформамида нагревают микроволновым облучением до 120°С в течение 2 часов. Растворитель удаляют в вакууме и неочищенный продукт очищают флэш-хроматографией (этилацетат/гексан 0:10→9:1 градиент). Выделяют 0,52 г (выход 84%) указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (CDCl3): 7,49-6,62 (м, 8H); 5,14 (с, 2H); 3,88 (с, 3H); 3,14 (с, 2H); 2,83-2,58 (м, 4H); 2,52 (д, 3H, J=5,58 Гц); 2,43 (д, 2H, J=6,62 Гц); 0,91-0,69 (м, 1H); 0,57-0,44 (м, 2H); 0,1-0,05 (м, 2H).
ЖХ-МС: MH+=383.
Пример 50. Гидрохлорид 2-[[2-(3-бензилоксифенил)этил](циклопропилметил)амино]-N-метилацетамида
Вышеуказанное соединение синтезируют по схеме 9
Схема 9
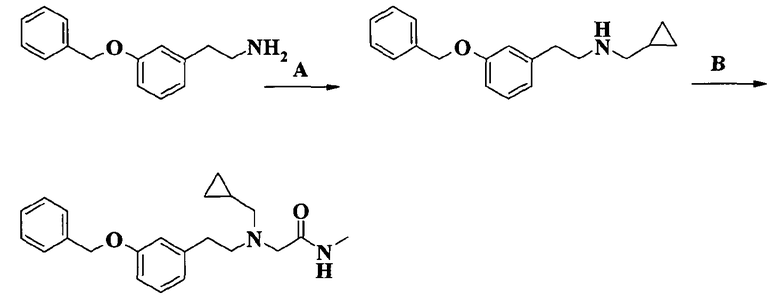
Стадия A. [2-(3-Бензилоксифенил)этил](циклопропилметил)амин
Суспензию 0,264 г (1 ммол) 2-(3-бензилоксифенил)этиламина, 70 мг (1 ммол) циклопропанкарбоксальдегида и 5 г молекулярных сит в 4 мл этанола перемешивают при кипячении с обратным холодильником в течение 3 часов. Смесь охлаждают до 0°С и прибавляют порциями 37,8 мг (1 ммол) NaBH4. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Прибавляют 3 мл водного раствора хлорида аммония, растворитель удаляют в вакууме и остаток экстрагируют этилацетатом. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:0:0→95:5:0,5 градиент об.:об.:об.) и получают 0,24 г (85% выход) требуемого соединения в виде желтого масла.
1H-ЯМР (CDCl3): 7,48-6,78 (м, 9H); 5,06 (с, 2H); 3,00-2,71 (м, 4H); 2,50 (д, 2H); 1,03-0,81 (м, 1H); 0,55-0,41 (м, 2H); 0,16-0,06 (м, 2H).
Стадия B. Гидрохлорид 2-[[2-(3-бензилоксифенил)этил](циклопропилметил)амино]-N-метилацетамида
Смесь 0,24 г (0,85 ммол) [2-(3-бензилоксифенил)этил] (циклопропилметил)амина, 0,14 мл (1,00 ммол) триэтиламина, 0,11 г (1,02 ммол) 2-хлор-N-метилацетамида в 3 мл диметилформамида подвергают микроволновому нагреву до 120°С в течение 2 часов. Растворитель удаляют и твердый остаток очищают флэш-хроматографией (дихлорметан/метанол/NН3 100:0:0→95:5:0,5 градиент). Полученный продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате. Растворитель удаляют в вакууме и остаток растирают в диэтиловом эфире. Выделяют 0,24 г (выход 80%) указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (CDCl3): 7,47-6,68 (м, 9H); 5,06 (с, 2H); 3,15 (с, 2H); 2,86-2,62 (м, 4H); 2,56 (д, 3H); 2,43 (д, 2H); 0,92-0,67 (м, 1H); 0,59-0,44 (м, 2H); 0,16-0,04 (м, 2H).
ЖХ-МС: MH+=353.
Пример 51: Гидрохлорид 2-[[2-(3-бензилоксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида
Вышеуказанное соединение синтезируют по схеме 10
Схема 10
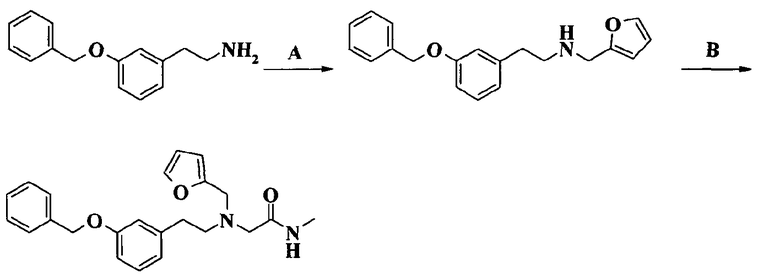
Стадия A. [2-(3-Бензилоксифенил)этил](фуран-2-илметил)амин
Суспензию 30,2 г (133 ммол) 2-(3-бензилоксифенил)этиламина, 11,0 мл (133 ммол) фуран-2-карбоксальдегида и 60 г молекулярных сит 4Е в 300 мл этанола кипятят с обратным холодильником в течение 3 часов. Смесь охлаждают до 0°С и прибавляют порциями 10,8 г (286 ммол) NaBH4. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Прибавляют 60 мл водного раствора хлорида аммония. Растворитель удаляют в вакууме, остаток экстрагируют этилацетатом. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:1:0,1 об.:об.:об.) и выделяют 22,4 г (выход 55%) указанного в заголовке соединения в виде желтого масла.
1H-ЯМР (CDCl3): 10,1 (ушир., 1H); 7,4-6,3 (м, 12H); 5 (с, 2H); 4,2 (т, 2H, J=4,9 Гц); 3,2-3,0 (м, 4H).
Стадия B. Гидрохлорид 2-[[2-(3-бензилоксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида
Раствор 3,0 г (9,8 ммол) [2-(3-бензилоксифенил)этил](фуран-2-илметил)амина, 15,0 г (10,7 ммол) 2-хлор-N-метилацетамида и 1,87 мл (10,7 ммол) диизопропилэтиламина в 50 мл ацетонитрила перемешивают при кипячении с обратным холодильником в течение 24 часов. Растворитель удаляют при пониженном давлении, неочищенную реакционную смесь очищают флэш-хроматографией (этилацетат/гексан 1:1 об.:об.). Выделенный продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате. Растворитель удаляют в вакууме, остаток растирают в диэтиловом эфире и выделяют 2,66 г (выход 65%) указанного в заголовке соединения в виде гигроскопического твердого вещества.
1H-ЯМР (CDCl3): 8,8 (ушир., 1H); 7,5-7,2 (м, 7Н); 6,9-6,8 (м, 4Н); 6,5 (м, 1H); 5,0 (с, 2H); 4,5-4,3 (м, 2H); 4,0-3,8 (м, 2H); 3,2 (м, 4H); 3-2,8 (м, 3H).
ЖХ-МС: MH+=379.
Пример 52: Гидрохлорид 2-[[2-[3-(2-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамид
Вышеуказанное соединение синтезируют по схеме 11
Схема 11
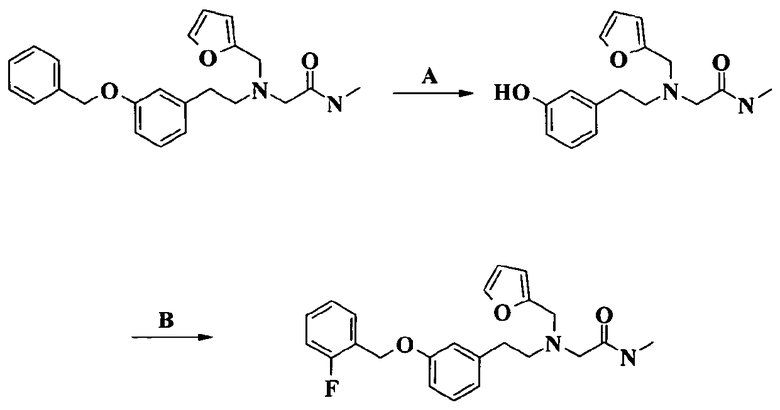
Стадия A. 2-[[2-(3-Гидроксифенил)этил](фуран-2-илметил)амино]-N-метилацетамид
К раствору 4,12 г (10,9 ммол) гидрохлорида 2-[[2-(3-бензилоксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида в 100 мл метанола прибавляют 400 мг Pd/C (10%). Гидрирование проводят при 30 фунт/кв.дюйм в течение 90 минут при комнатной температуре. Катализатор отфильтровывают, растворитель удаляют и неочищенный продукт очищают флэш-хроматографией (этилацетат/гексан 1:1+триэтиламин). Выделяют 2,1 г (выход 67%) указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (CDCl3): 7,37 (д, 1H, J=2,1); 7,20 (т, 1H, J=7,2); 6,75-6,67 (м, 4H); 6,33-6,31 (м, 1H); 6,20 (м, 1H); 5,72 (ушир., 1H); 3,72 (с, 2H); 3,14 (с, 2H); 2,74 (м, 4H); 2,56 (д, 3H, J=4,1).
ЖХ-МС: MH+=289.
Стадия B. Гидрохлорид 2-[[2-[3-(2-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
Раствор 60 мг (0,21 ммол) 2-[[2-(3-гидроксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида, 36 мг (0,25 ммол) 1-хлорметил-2-фторбензола, 44 мг K2CO3 (0,32 ммол) и 3 мг иодида калия в 4 мл диметилформамида кипятят с обратным холодильником в течение ночи. Растворитель удаляют в вакууме и неочищенный продукт очищают препаративной ВЭЖХ. Выделенный продукт растворяют в смеси этилацетат/хлористоводородная кислота. Растворитель удаляют в вакууме и остаток растирают в диэтиловом эфире. Выделяют 65 мг (выход 72%) указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (CDCl3): 12,67 (ушир., 1H); 8,79 (м, 1H); 7,55-7,05 (м, 5H); 6,88 (м, 4H); 6,49 (м, 1H); 5,11 (с, 2H); 4,45 (м, 2H); 3,72 (м, 2H); 3,24 (м, 4H); 2,88 (д, 3H, J=4,56 Гц).
ЖХ-МС: MH+=397,3.
Примеры 53-68. Указанные соединения получают по методике, описанной на схеме 11, используя соответствующие реагенты.
Гидрохлорид 2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида.
Пример 54. Гидрохлорид 2-[[2-[3-(2-хлорбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=413.
Пример 55. Гидрохлорид 2-[[2-[3-(3-хлорбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=413.
Пример 56. Гидрохлорид 2-[[2-[3-(3-метилбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=393.
Пример 57. Гидрохлорид 2-[[2-[3-(4-метилбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=393.
Пример 58. Гидрохлорид 2-[[2-[3-(3-трифторметилбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=447.
Пример 59. Гидрохлорид 2-[[2-[3-(3-фторфенокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=383.
Пример 60. Гидрохлорид 2-[[2-[3-(2-фенилэтокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=393.
Пример 61. Гидрохлорид 2-[[2-(3-циклопропилметоксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=343.
Пример 62. Гидрохлорид 2-[[2-[3-(2-пиперидин-1-илэтокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=340.
Пример 63. Гидрохлорид 2-[[2-[3-(2-морфолин-4-ил)этокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=402.
Пример 64. Гидрохлорид 2-[[2-[3-(2,2,2-трифторэтокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=371.
Пример 65. Гидрохлорид 2-[[2-[3-(3,5-диметилизоксазол-4-илметокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=398.
Пример 66. Гидрохлорид 2-[[2-[3-(5-хлортиен-2-илметокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=419.
Пример 67. Гидрохлорид 2-[[2-[3-(пиридин-2-илметокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=380.
Примеры 68-69. Указанные соединения получают аналогично по методике, описанной на схеме 11, используя соответствующие реагенты, но не превращают их в соль действием хлористоводородной кислоты.
Пример 68. 2-[[2-[3-(4-Трифторметилбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамид
ЖХ-МС: MH+=447.
Пример 69. 2-[[2-(3-Циклопентилоксифенил)этил](фуран-2-илметил)амино]-N-метилацетамид
ЖХ-МС: MH+=357.
Пример 70. 2-[[2-[3-(3-Фторбензилокси)фенил]этил](фуран-2-илметил)амино]ацетамид
Вышеуказанное соединение синтезируют по схеме 12
Схема 12
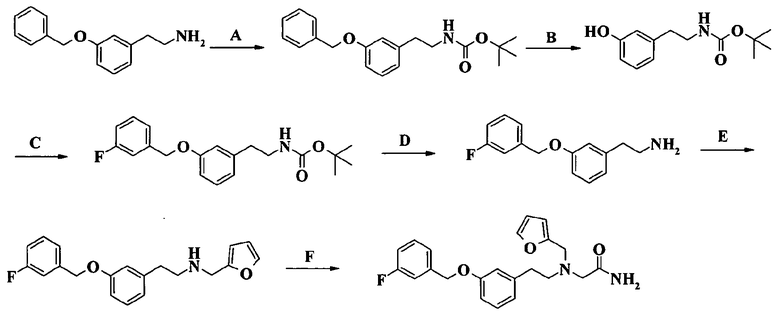
Стадия A. трет-Бутиловый эфир [2-(3-бензилоксифенил)этил]карбаминовой кислоты
К суспензии 5,27 г гидрохлорида 2-(3-бензилоксифенил)этиламина (20 ммол) в 20 мл дихлорметана и 2,78 мл триэтиламина (20 ммол) прибавляют 4,8 г (Boc)2О (22 ммол) в 10 мл дихлорметана. Реакционную смесь перемешивают в течение 1 часа при комнатной температуре. После упаривания растворителя к остатку прибавляют водный раствор, содержащий 5% лимонной кислоты, и продукт экстрагируют этилацетатом. Выделяют указанный в заголовке продукт с количественным выходом в виде бесцветного масла.
1H-ЯМР (CDCl3): 7,45-6,78 (м, 9H); 5,05 (с, 2H); 4,54 (ушир.с, 1H); 3,48-3,28 (м, 2H); 2,77 (т, 2H); 1,44 (с, 9H).
Стадия B. трет-Бутиловый эфир [2-(3-гидроксифенил)этил]карбаминовой кислоты
К раствору 13 г (0,039 моль) трет-бутилового эфира [2-(3-бензилоксифенил)этил]карбаминовой кислоты в 100 мл этанола прибавляют 1 г Pd/C 10%. Смесь гидрируют при 40 фунт/кв.дюйм в течение ночи. Катализатор отфильтровывают и промывают этанолом. Растворитель удаляют в вакууме и получают 9,4 г указанного в заголовке соединения в виде бесцветного масла с количественным выходом.
1H-ЯМР (CDCl3): 7,22-7,12 (м, 1H); 6,78-6,66 (м, 3H); 4,56 (ушир.с, 1H); 3,42-3,30 (м, 2H); 2,74 (т, 2H); 1,44 (с, 9H).
Стадия C. трет-Бутиловый эфир [2-[3-(3-фторбензилокси)фенил]этил]карбаминовой кислоты
1-Хлорметил-3-фторбензол (2,87 г, 19,8 ммол) в 5 мл сухого диметилформамида прибавляют к суспензии 4,66 г (19,6 ммол) трет-бутилового эфира [2-(3-гидроксифенил)этил]карбаминовой кислоты, 4 г K2CO3 и 0,3 г иодида калия в 50 мл сухого диметилформамида. Реакционную смесь сначала перемешивают при комнатной температуре в течение ночи, затем нагревают до 50°С в течение 6 часов. После упаривания растворителя к остатку прибавляют воду и продукт экстрагируют этилацетатом. Получают 7 г неочищенного масла. Очистка флэш-хроматографией с использованием смеси этилацетат/гексан (1:9→2:8 градиент) дает 5,9 г (выход 86%) указанного в заголовке продукта в виде бесцветного масла.
1H-ЯМР (CDCl3): 7,40-6,68 (м, 8H); 5,05 (с, 2H); 4,53 (ушир.с, 1H); 3,44-3,30 (м, 2H); 2,77 (т, 2H); 1,44 (с, 9H).
Стадия D. 2-[3-(3-Фторбензилокси)фенил]этиламин
Раствор 10,36 г (30 ммол) трет-бутилового эфира [2-[3-(3-фторбензилокси)фенил]этил]карбаминовой кислоты в 100 мл дихлорметана и 15 мл трифторуксусной кислоты перемешивают при комнатной температуре в течение ночи. Растворитель удаляют, прибавляют 5% раствор K2CO3 в воде, продукт экстрагируют этилацетатом и получают указанное в заголовке соединение с количественным выходом в виде липкого масла.
1H-ЯМР (диметилсульфоксид-d6): 8,04 (ушир.с, 3H); 7,49-6,72 (м, 8H); 5,09 (с, 2H); 3,08-2,75 (м, 4H).
Стадия E. [2-[3-(3-Фторбензилокси)фенил]этил](фуран-2-илметил)амин
К раствору 2,45 г (10 ммол) 2-[3-(3-фторбензилокси)фенил]этиламина в 50 мл сухого этанола прибавляют 1,44 г (15 ммол) фуран-2-карбоксальдегида и 7,5 г молекулярных сит 3Å. Реакционную смесь кипятят с обратным холодильником в течение 3 часов. Молекулярные сита отфильтровывают и раствор охлаждают до 5°С. Прибавляют 0,57 г (15 ммол) NaBH4 в атмосфере N2 и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют, к остатку прибавляют 5% раствор NaHCO3 и продукт экстрагируют этилацетатом. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:0:0→100:2:0,2, об.:об.:об.), получая 2,2 г (выход 68%)масла.
1H-ЯМР (CDCl3): 7,44-6,12 (м, 11H); 5,04 (с, 2H); 3,79 (с, 2H); 2,96-2,73 (м, 4H).
Стадия F. 2-[[2-[3-(3-Фторбензилокси)фенил]этил](фуран-2-илметил)амино]ацетамид
Раствор 1,8 г (5,53 ммол) [2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амина, 0,57 г (6,08 ммол) 2-хлорацетамида и 0,92 мл (6,62 ммол) триэтиламина в 5 мл сухого диметилформамида нагревают при 120°С в течение 2 часов микроволновым облучением. Растворитель удаляют в вакууме, прибавляют воду и продукт экстрагируют этилацетатом. Неочищенную реакционную смесь очищают флэш-хроматографией (дихлорметан/метанол 95:5 об.:об.). Выделяют 2,1 г (выход 99%) желтого масла.
1H-ЯМР (DMSO-d6): 7,84-6,48 (м, 11H); 5,08 (с, 2H); 4,48 (с, 2H); 3,87 (с, 2H); 3,33-2,87 (м, 4H).
Пример 71. Дигидрохлорид 2-[[2-[3-(3-Фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(2-диметиламиноэтил)ацетамида
Вышеуказанное соединение синтезируют по схеме 13
Схема 13
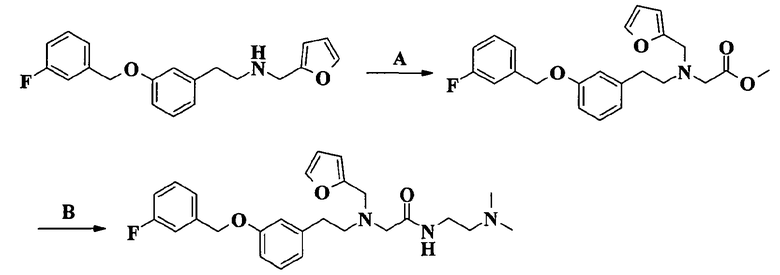
Стадия A. Метиловый эфир 2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]уксусной кислоты
К раствору 0,9 г (2,76 ммол) [2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амина и 0,39 г (3,05 ммол) диизопропилэтиламина в 15 мл ацетонитрила прибавляют 0,46 г (3,05 ммол) метилового эфира 2-бромуксусной кислоты. Растворитель удаляют, к остатку прибавляют воду и продукт экстрагируют этилацетатом. Очистка флэш-хроматографией (этилацетат/гексан 1:9→2:8 градиент об.:об.) дает 0,9 г (выход 82%) прозрачного масла.
1H-ЯМР (CDCl3): 7,40-7,11 (м, 5H); 7,06-6,94 (м, 1H); 6,83-6,74 (м, 3H); 6,33-6,29 (м, 1H); 6,20 (д, 1H, J=3,34 Гц); 5,04 (с, 2H); 3,90 (с, 2H); 3,70 (с, 3H); 3,40 (с, 2H); 2,92-2,72 (м, 4H).
Стадия B. Дигидрохлорид 2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(2-диметиламиноэтил)ацетамида
Метиловый эфир 2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]уксусной кислоты (100 мг, 0,25 ммол) растворяют в 5 мл сухого толуола. Прибавляют 66 мг (0,75 ммол) N,N-диметилэтан-1,2-диамина при 0°С, затем 0,4 мл (0,8 ммол) 2 M триэтилалюминия в гептане. Реакционную смесь нагревают до 60°С в течение ночи. Раствор охлаждают до 0°С и выливают в метанол. Растворитель удаляют в вакууме и неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:2:0,2, об.:об.:об.). Продукт растворяют в смеси этилацетат/хлористоводородная кислота и полученное твердое вещество отфильтровывают. Выделяют 80 мг (выход 65%) указанного в заголовке соединения в виде гигроскопического твердого вещества.
1H-ЯМР (D2О): 7,48-6,29 (м, 11H); 4,95 (с, 2H); 4,33 (с, 2H); 3,88 (с, 2H); 3,48-3,34 (м, 2H); 3,32-3,17 (м, 2H); 3,15-3,04 (м, 4H); 2,97-2,77 (м, 2H); 2,72 (с, 6H).
Пример 72. Дигидрохлорид 2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(2-амино-2-метилпропил)ацетамида
Метиловый эфир 2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]уксусной кислоты (100 мг, 0,25 ммол) и 1 мл 2-метилпропан-1,2-диамина нагревают при 120°С в течение 3 часов микроволновым облучением. Реакционную смесь охлаждают до комнатной температуры, прибавляют воду и продукт экстрагируют этилацетатом. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол 95:5 об.:об.). Продукт растворяют в смеси этилацетат/хлористоводородная кислота. Растворитель удаляют и полученную соль растирают в диэтиловом эфире. Выделяют 95 мг (выход 72%) указанного в заголовке соединения в виде гигроскопического твердого вещества.
1H-ЯМР (CDCl3): 10,95 (ушир.с, 1H); 9,13 (ушир.с, 1H); 8,46 (ушир.с, 3H); 7,47-6,32 (м, 11H); 4,99 (с, 2H); 4,89-4,45 (м, 2H); 4,45-4,09 (ушир.с, 2H); 3,87-3,00 (м, 6H); 1,52 (с, 6H).
ЖХ-МС: MH+=454.
Пример 73-76. Указанные соединения получают по методике, описанной на схеме 13, используя соответствующие амины.
Пример 73. Гидрохлорид 2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(2-метоксиэтил)ацетамида
ЖХ-МС: MH+=441.
Пример 74. Гидрохлорид 2-[[2-[3-(3- фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(1,3,4-тиадиазол-2-ил)ацетамида
ЖХ-МС: MH+=467.
Пример 75. Гидрохлорид 2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(3-метилизоксазол-5-ил)ацетамида
ЖХ-МС: MH+=464.
Пример 76. Гидрохлорид 2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(1H-пиразол-3-ил)ацетамида
ЖХ-МС: MH+=449.
Примеры 77-78. Указанные соединения получают по методике, описанной на схеме 13, используя соответствующий амин, но не превращают их в соли действием хлористоводородной кислоты.
Пример 77. 2-[[2-[3-(3-Фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(1H-имидазол-2-ил)ацетамид
ЖХ-МС: MH+=449.
Пример 78. 2-[[2-[3-(3-Фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-тиазол-2-илацетамид
ЖХ-МС: MH+=466.
Пример 79. Гидрохлорид 2-[[2-(3-бензилоксифенил)этил](циклопропилметил)амино]-N-метилпропионамид
Вышеупомянутое соединение синтезируют по схеме 14
Схема 14
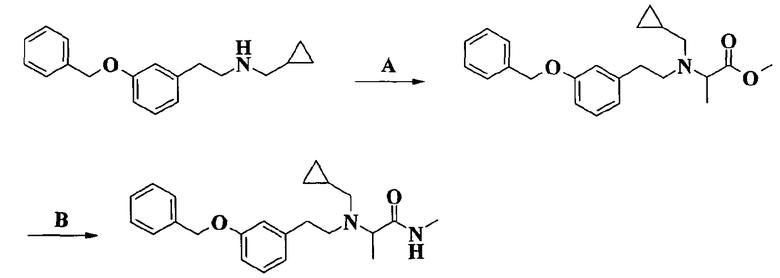
Стадия A. Метиловый эфир 2-[[2-(3-бензилоксифенил)этил](циклопропилметил)амино]пропионовой кислоты
Раствор 562 мг (2 ммол) [2-(3-бензилоксифенил)этил](циклопропилметил)амина, 0,3 мл (2,2 ммол) триэтиламина и 367 мг (2,2 ммол) метилового эфира 2-бромпропионовой кислоты в 20 мл ацетонитрила кипятят с обратным холодильником в течение 24 часов. Растворитель удаляют, прибавляют воду и продукт экстрагируют этилацетатом. Выделяют 730 мг указанного в заголовке соединения с количественным выходом в виде бесцветного масла.
1H-ЯМР (CDCl3): 7,48-6,75 (м, 9H); 5,05 (с, 2H); 3,74 (кв, 1H); 3,67 (с, 3H); 3,00-2,37 (м, 6H); 1,24 (д, 3H); 0,96-0,74 (м, 1H); 0,60-0,40 (м, 2H); 0,22-0,04 (м, 2H).
ЖХ-МС: MH+=368.
Стадия B. Гидрохлорид 2-[[2-(3-бензилоксифенил)этил](циклопропилметил)амино]-N-метилпропионамида
Метиловый эфир 2-[[2-(3-бензилоксифенил)этил](циклопропилметил)амино]пропионовой кислоты (730 мг, 2 ммол) растворяют в 10 мл сухого толуола. Прибавляют 4 мл (8 ммол) 2М раствора метиламина в тетрагидрофуране, затем 4 мл (8 ммол) 2M триметилалюминия в гептане. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Раствор охлаждают до 0°С и выливают в метанол. Растворитель удаляют в вакууме и неочищенную реакционную смесь очищают флэш-хроматографией (дихлорметан/метанол 100:5 об.:об.). Продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате и образовавшуюся соль отфильтровывают. Выделяют 500 мг (выход 62%) указанного в заголовке соединения в виде гигроскопического твердого вещества.
1H-ЯМР (CDCl3): 7,48-6,69 (м, 9H); 5,06 (с, 2H); 3,56 (кв, 1H); 2,80-2,14 (м, 6H); 2,47 (д, 3H); 1,17 (д, 3H); 0,89-0,65 (1H); 0,63-0,37 (м, 2H); 0,21-0,02 (м, 2H).
ЖХ-МС: MH+=366.
Пример 80. Гидрохлорид 2-[[2-[3-метокси-4-(2,2,2-трифторэтокси)фенил]этил](циклопропилметил)амино]-N-метилацетамида
Вышеупомянутое соединение синтезируют по схеме 15
Схема 15
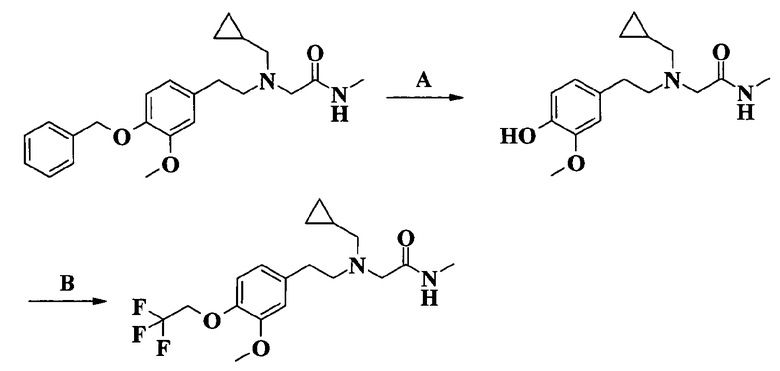
Стадия A. 2-[[2-(4-Гидрокси-3-
метоксифенил)этил](циклопропилметил)амино]-N-метилацетамид
Смесь 0,49 г (1,28 ммол) 2-[[2-(4-бензилокси-3-метоксифенил)этил](циклопропилметил)амино]-N-метилацетамида и 50 мг Pd/C 10% в 10 мл этанола гидрируют при 40 фунт/кв.дюйм в течение 2 часов. Катализатор отфильтровывают и растворитель удаляют. Выделяют 0,366 г (выход 98%) желтого масла.
ЖХ-МС: MH+=293.
Стадия B. Гидрохлорид 2-[[2-(3-метокси-4-(2,2,2-трифторэтокси)фенил)этил](циклопропилметил)амино]-N-метилацетамида
Смесь 90 мг (0,3 ммол) 2-[[2-(4-гидрокси-3-метоксифенил)этил](циклопропилметил)амино]-N-метилацетамида, 107 мг (0,51 ммол) 1,1,1-трифтор-2-йодэтана, 71 мг (0,51 ммол) K2CO3 и 5 мг иодида калия в 5 мл диметилформамида нагревают при 120°С в течение ночи. Смесь фильтруют через целит и растворитель удаляют в вакууме. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол 100:0→100:1,5 градиент, об.:об.). Продукт растворяют в безводной хлористоводородной кислоте в этилацетате. Растворитель удаляют в вакууме и продукт растирают в диэтиловом эфире. Выделяют 30 мг (выход 27%) указанного в заголовке соединения в виде белого гигроскопического твердого вещества.
1H-ЯМР (CDCl3): 11,49 (ушир.с, 1H); 8,48 (ушир.с, 1H); 6,92-6,70 (м, 3H); 4,35 (кв, 2H, JHF=8,89 Гц); 4,21 (д, 2H); 3,84 (с, 3H); 3,66-3,10 (м, 6H); 2,85 (д, 3H, J=4,62 Гц); 1,37-1,17 (м, 1H); 0,81-0,69 (м, 2H); 0,53-0,42 (м, 2H).
ЖХ-МС: MH+=374.
Пример 81. Гидрохлорид 2-[[2-(3'-фторбифенил-3-
ил)этил](фуран-2-илметил)амино]-N-метилацетамида
Вышеупомянутое соединение синтезируют по схеме 16
Схема 16
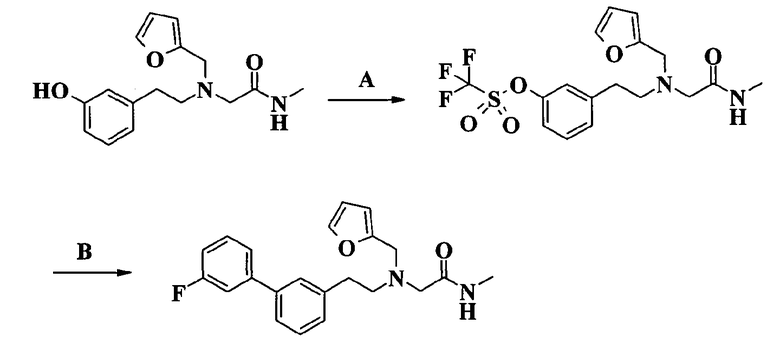
Стадия A. 2-[[2-(3-Трифторметилсульфонилоксифенил)этил](фуран-2-илметил)амино]-N-метилацетамид
N-Фенил-бис(трифторметансульфонимид) (1,36 г, 3,8 ммол) в 10 мл ацетонитрила прибавляют в атмосфере N2 к смеси 1 г (3,5 ммол) 2-[[2-(3-гидроксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида и 960 мг (7 ммол) K2CO3 в 30 мл смеси ацетонитрил/дихлорметан (2:1). Раствор перемешивают при комнатной температуре в течение ночи. Растворитель удаляют в вакууме, прибавляют воду и продукт экстрагируют этилацетатом. Необработанную реакционную смесь очищают флэш-хроматографией (гексан/этилацетат/диметилформамид 1:2:0,2). Выделяют 1,3 г (выход 90%) указанного в заголовке соединения в виде желтого масла.
ЖХ-МС: MH+=421.
Стадия B. Гидрохлорид 2-[[2-(3'-фторбифенил-3-ил)этил](фуран-2-илметил)амино]-N-метилацетамида
Смесь 100 мг (0,24 ммол) 2-[N-(3-
трифторметилсульфонилоксифенил)этил](фуран-2-илметил)амино)-N-метилацетамида, 48 мг (0,34 ммол) 3-фторфенилбороновой кислоты, 46 мг (0,34 ммол) K2CO3 и 10 мг Pd(PPh3)4 в 2 мл этанола нагревают при 110°С микроволновым облучением в течение 15 минут. Смесь фильтруют через целит и растворитель удаляют в вакууме. Неочищенный продукт очищают препаративной ВЭЖХ. Продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате. Растворитель удаляют в вакууме и продукт растирают в диэтиловом эфире. Выделяют 34 мг (выход 36%) указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (CDCl3): 8,79 (ушир., 1H); 7,56-6,81 (м, 10H); 6,51-6,48 (м, 1H); 4,61-4,35 (м, 2H); 3,77 (ушир.с, 2H); 3,40-3,25 (м, 4H); 2,89 (д, 3H, J=4,6 Гц).
ЖХ-МС: MH+=367.
Пример 82-89. Указанные соединения получают методике, описанной на схеме 16, используя соответствующий реагент бороновой кислоты.
Пример 82. Гидрохлорид 2-[[2-[3-(тиен-3-ил)фенил)]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=355.
Пример 83. Гидрохлорид 2-[[2-(3'-метоксибифенил-3-ил)этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=379.
Пример 84. Гидрохлорид 2-[[2-(3'-ацетиламинобифенил-3-ил)этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=406.
Пример 85. Гидрохлорид 2-[[2-(2'-диметиламинометилбифенил-3-
ил)этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=406.
Пример 86. Гидрохлорид 2-[[2-[3-(пиридин-3-ил)фенил)]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=350.
Пример 87. Гидрохлорид 2-[[2-[3-(6-метоксипиридин-3-ил)фенил)]этил](фуран-2-илметил)амино]-N-метилацетамид
ЖХ-МС: MH+=380.
Пример 88. Гидрохлорид 2-[[2-[3-(2,4-диметоксипиримидин-5-ил)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=411.
Пример 89. Гидрохлорид 2-[[2-[3-(фуран-3-ил)фенил)]этил](фуран-2-илметил)амино]-N-метилацетамида
ЖХ-МС: MH+=339.
Пример 90. 2-[[2-[3-(3,5-Диметилизоксазол-4-ил)фенил]этил](фуран-2-илметил)амино]-N-метилацетамид
Указанное соединение получают по методике, описанной схемой 16, в которой используют соответствующий реагент бороновой кислоты, но не превращают полученное соединение в соль действием хлористоводородной кислоты.
ЖХ-МС: MH+=368.
Пример 91. Гидрохлорид 2-[[2-[3-(пиперидин-1-ил)фенил)]этил](фуран-2-илметил)амино]-N-метилацетамида
Смесь 100 мг (0,24 ммол) 2-[2-[(3-трифторметилсульфонилоксифенил)этил](фуран-2-илметил)амино)-N-метилацетамида, 41 мг (0,48 ммол) пиперидина, 28 мг (0,29 ммол) трет-бутоксида натрия, 10 мг Pd(CH3COO)2 и 10 мг N-фенил-2-(ди-
трет-бутилфосфинил)индола в 2 мл толуола нагревают при 100°С микроволновым облучением в течение 15 минут. Реакционную смесь фильтруют через целит и растворитель удаляют в вакууме. Неочищенную реакционную смесь очищают препаративной ВЭЖХ. Продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате, растворитель удаляют при пониженном давлении, продукт растирают в диэтиловом эфире.
Выделяют 51 мг (выход 55%) указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (CDCl3): 8,66 (м, 1H); 8,10 (ушир.с, 1H); 7,72-7,66 (м, 2H); 7,51-7,33 (м, 2H); 6,86-6,84 (м, 1H); 6,50-6,47 (м, 1H); 4,55-4,52 (м, 2H); 3,86-3,65 (м, 4H); 3,34 (ушир.с, 6H); 2,83 (д, 3H, J=4,9 Гц); 2,72-2,65 (м, 2H); 2,09-1,92 (м, 5H).
ЖХ-МС: MH+=356.
Пример 92. Гидрохлорид 2-[[2-(3-бензилоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида
Вышеуказанное соединение синтезируют по схеме 17.
Схема 17
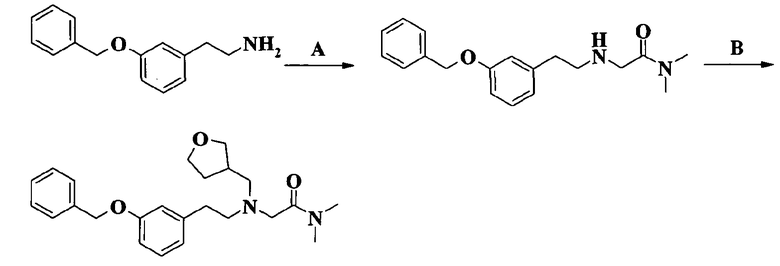
Стадия A. 2-[2-(3-Бензилоксифенил)этиламино]-N,N-диметилацетамид
Смесь 4,32 г (19 ммол) 2-(3-бензилоксифенил)этиламина, 7,9 мл (57 ммол) триэтиламина, 1,95 мл (19 ммол) 2-хлор-N,N-диметилацетамида и 332 мг (2 ммол) иодида калия в 110 мл сухого диметилформамида нагревают при 80°С в течение 3 часов. Растворитель удаляют в вакууме и неочищенную реакционную смесь очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:3:0,5, об.:об.:об.). Выделяют 3 г (выход 51%) указанного в заголовке соединения в виде желтого твердого вещества.
ЖХ-МС: MH+=313.
Стадия B. Гидрохлорид 2-[[2-(3-бензилоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида
Прибавляют порциями 1,06 г (22 ммол) NaBH4 к смеси 2,9 г (9,3 ммол) 2-[2-(3-бензилоксифенил)этиламино]-N,N-диметилацетамида, 1,28 мл (14,1 ммол) тетрагидрофуран-3-карбальдегида и 4 г молекулярных сит 4Å в 130 мл 1,2-дихлорэтана. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Прибавляют водный раствор хлорида аммония. Растворитель удаляют в вакууме, остаток экстрагируют этилацетатом и органическую фазу промывают насыщенным водным раствором K2CO3. Растворитель удаляют в вакууме, продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате. Растворитель удаляют и продукт растирают в диэтиловом эфире. Выделяют 3,2 г (выход 80%) указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (CDCl3): 7,45-7,32 (м, 5H); 7,23-7,19 (м, 1H); 6,89-6,85 (м, 3H); 5,05 (с, 2H); 4,14-2,98 (м, 14H); 2,92 и 2,86 (2с,
6H); 2,82-2,71 (м, 1H).
ЖХ-МС: MH+=397.
Пример 93. Гидрохлорид 2-[[2-[3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида
Вышеуказанное соединение синтезируют по схеме 18.
Схема 18
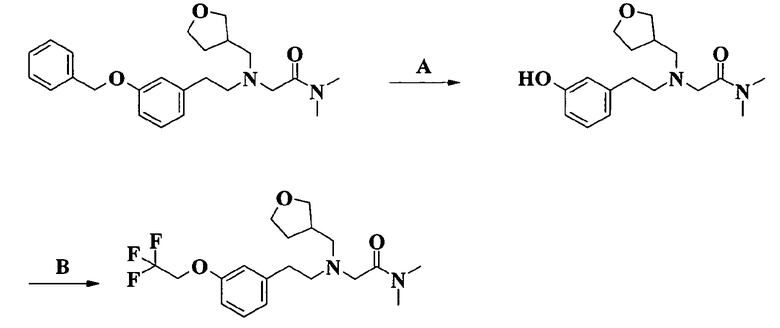
Стадия A. 2-[[2-(3-Гидроксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамид
Смесь 2,4 г (6,05 ммол) 2-[[2-(3-бензилоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида и 200 мг Pd/C 10% в смеси метанол/уксусная кислота (10:1) (70 мл) гидрируют в течение 18 часов при 60 фунт/кв.дюйм. Катализатор отфильтровывают и растворитель удаляют в вакууме. Необработанную реакционную смесь очищают флэш-хроматографией (дихлорметан/метанол/NН3 97:3:0,3, об.:об.:об.). Выделяют 1,63 г (выход 88%) указанного в заголовке соединения в виде желтого масла.
ЖХ-МС: MH+=307.
Стадия B. Гидрохлорид 2-[[2-[3-(2,2,2-
трифторэтокси)фенил]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамид
Смесь 100 мг (0,33 ммол) 2-[[2-(3-гидроксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида, 139 мг (0,66 ммол) 1,1,1-трифтор-2-йодэтана, 90 мг (0,66 ммол) K2CO3 и 5 мг иодида калия в 4 мл диметилформамида кипятят с обратным холодильником в течение ночи. Смесь фильтруют через целит и растворитель удаляют в вакууме. Неочищенную реакционную смесь очищают препаративной ВЭЖХ (дихлорметан/метанол/NН3 97:3:0,3, об.:об.:об.). Продукт растворяют в смеси этилацетат/хлористоводородная кислота, растворитель удаляют и продукт растирают в диэтиловом эфире. Выделяют 18 мг (выход 13%) указанного в заголовке соединения в виде желтого твердого вещества.
ЖХ-МС: MH+=389.
Пример 94. Гидрохлорид 2-[[2-(3-бутоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида.
Вышеуказанное соединение синтезируют по схеме 19
Схема 19
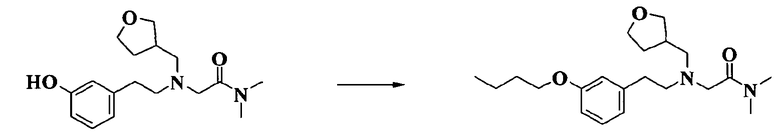
Раствор 2-[[2-(3-гидроксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида (1,0 г 3,3 ммол), 1-бромбутана (0,43 мл, 4 ммол), карбоната калия (680 мг, 5 ммол), иодида калия (50 мг, 0,3 ммол) в ДМФА (30 мл) кипятят с обратным холодильником в течение 16 часов. После фильтрования через слой целита раствор упаривают, удаляя растворитель, и полученное неочищенное масло очищают препаративной ВЭЖХ. Гидрохлоридную соль получают прибавлением HCl 1н. в AcOEt к свободному амину, растворенному в этиловом эфире. После фильтрования получают 690 мг (выход 50%) указанного в заголовке соединения в виде белого твердого вещества 99% чистоты.
1H-ЯМР (CDCl3): 12,54 (ушир. сигнал, 1H); 7,25-6,73 (м, 4H); 4,28-3,16 (м, 12H); 3,93 (т, 2H); 2,92 (с, 3H); 2,87 (с, 3H); 2,85-2,67 (м, 1H); 2,36-1,86 (м, 2H); 1,83-1,66 (м, 2H); 1,58-1,38 (м, 2H); 0,97 (т, 3H, J=8,3 Гц).
ЖХ-МС: MH+=363,43.
Пример 94-бис: (R)- и (S)-энантиомеры 2-[[2-(3-бутоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида
Рацемическую смесь гидрохлорида 2-[[2-(3-бутоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида, полученную по методике примера 94, разделяют на хиральной колонке CHIRALPAC® AD 20 мкм - 250×21 мм, подвижная фаза метанол/диэтиламин 100/0,1 (об./об.), скорость потока 20 мл/мин, УФ-детектирование (275 нм), температура 25°С.
Времена удерживания первого и второго элюируемых энантиомеров, полученных в виде похожих на мед желтоватых оснований, составляют 5,2 мин и 6,7 мин соответственно. [α]D первого элюируемого энантиомера составляет -10°, c=0,1, MeOH (20°C) и [α]D второго элюируемого энантиомера составляет +10°, c=0,1, MeOH (20°С).
Энантиомерный избыток для обоих >99,5%.
Пример 95. Гидрохлорид 2-[[2-(3-бутоксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида
Указанное соединение получают по методике, показанной на схеме 20, исходя из 2-[[2-(3-гидроксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида, полученного, как описано выше в примере 52.
ЖХ-МС: MH+=345.
Пример 96. Гидрохлорид 2-[[2-[4-фтор-3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида
Вышеуказанное соединение синтезируют по схеме 20
Схема 20
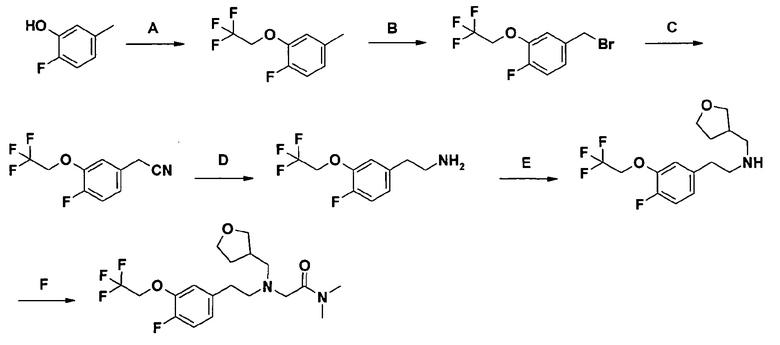
Стадия A. 1-Фтор-4-метил-2-(2,2,2-трифторэтокси)бензол
К раствору 2,9 г (23 ммол) 2-фтор-5-метилфенола в 15 мл сухого диметилформамида прибавляют порциями 1,05 г (26 ммол) NaH 60%. Смесь перемешивают при комнатной температуре в течение 30 минут. Прибавляют 6,35 г (25 ммол) 2,2,2-трифторэтилового эфира толуол-4-сульфоновой кислоты и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь выливают в воду и экстрагируют диэтиловым эфиром. Неочищенный продукт очищают флэш-хроматографией (гексан/этилацетат 9:1, об.:об.) и получают 2,7 г (выход 56%) указанного в заголовке соединения.
1H-ЯМР (CDCl3): 7,05-6,77 (м, 3H); 4,40 (кв, 2H, JH-F=8,48 Гц); 2,31 (с, 3H).
Стадия B. 4-Бромметил-1-фтор-2-(2,2,2-трифторэтокси)бензол
Смесь 2,68 г (12,8 ммол) 1-фтор-4-метил-2-(2,2,2-трифторэтокси)бензола, 2,3 г (12,9 ммол) NBS и 140 мг дибензоилпероксида в 60 мл CCl4 кипятят с обратным холодильником в течение 6 часов. Растворитель удаляют и неочищенный остаток используют без дополнительной очистки на следующей стадии.
Стадия C. [4-Фтор-3-(2,2,2-трифторэтокси)фенил]ацетонитрил
К раствору 3,3 г (11,5 ммол) 4-бромметил-1-фтор-2-(2,2,2-трифторэтокси)бензола в 30 мл сухого диметилсульфоксида прибавляют 900 мг (13,8 ммол) KCN. Реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Прибавляют воду и продукт экстрагируют диэтиловым эфиром. Растворитель удаляют в вакууме и неочищенный остаток очищают флэш-хроматографией (гексан/этилацетат 8:2 об.:об.). Выделяют 1,4 г (выход 52%) указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (CDCl3): 7,21-6,92 (м, 3H); 4,41 (кв, 2H, JH-F=8,48 Гц); 3,68 (с, 2H).
Стадия D. 2-[4-Фтор-3-(2,2,2-трифторэтокси)фенил]этиламин
К раствору 1,23 г (5,27 ммол) [4-фтор-3-(2,2,2-трифторэтокси)фенил]ацетонитрила в 50 мл сухого тетрагидрофурана прибавляют 800 мг (10 ммол) комплекса боран-метилсульфид. Реакционную смесь кипятят с обратным холодильником в течение 4 часов. Растворитель удаляют и затем прибавляют воду. Первая экстракция диэтиловым эфиром позволяет частично очистить неочищенную реакционную смесь. Полученный водный слой затем подщелачивают NH4OH и продукт экстрагируют дихлорметаном. После удаления растворителя выделяют 1 г (выход 80%) указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (CDCl3): 7,10-6,98 (м, 1H); 6,92-6,80 (м, 2H); 4,42 (кв, 2H, JH-F=8,5 Гц); 4,42 (кв, 2H); 2,95 (т, 2H, J=6,55 Гц); 2,70 (т, 2H, J=6,55 Гц).
Стадия E. [2-[4-Фтор-3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-илметил)амин
Смесь 500 мг (2,1 ммол) 2-[4-фтор-3-(2,2,2-трифторэтокси)фенил]этиламина, 210 мг (2,1 ммол) тетрагидрофуран-3-карбальдегида и 2 г молекулярных сит 4Å в 30 мл сухого дихлорметана перемешивают при комнатной температуре в течение 30 минут. Прибавляют порциями 630 мг (2,9 ммол) NaBH(OAc)3. Реакционную смесь перемешивают в течение 4 часов при комнатной температуре. Прибавляют водный 5% раствор NaHCO3 и продукт экстрагируют дихлорметаном. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:9:0,5 об.:об.:об.). Выделяют 300 мг (выход 44%) указанного в заголовке соединения в виде масла.
1H-ЯМР (CDCl3): 7,09-6,97 (м, 1H); 6,92-6,81 (м, 2H); 4,41 (кв, 2H, JH-F=8,7 Гц); 3,90-3,66 (м, 3H); 3,51-4,40 (м, 1H); 2,91-2,59 (м, 6H); 2,54-2,24 (м, 1H); 2,11-1,92 (м, 1H); 1,64-1,46 (м, 1H).
Стадия F. Гидрохлорид 2-[[2-[4-фтор-3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида
Смесь 85 мг (0,26 ммол) [2-[4-фтор-3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-илметил)амина, 0,06 мл триэтиламина и 48 мг (0,39 ммол) 2-хлор-N,N-диметилацетамида в 3 мл диметилформамида нагревают в течение 1 часа при 120°С микроволновым облучением. Растворитель удаляют в вакууме. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:3:0,3 об.:об.:об.). Продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате, растворитель удаляют при пониженном давлении, продукт растирают в диэтиловом эфире. Выделяют 74 мг (65% выход) указанного в заголовке соединения в виде коричневого твердого вещества.
1H-ЯМР (CDCl3): 7,07-6,80 (м, 3H); 4,41 (кв, 2H); 3,77 (м, 3H); 3,50 (м, 1H); 3,33 (ушир.м, 2H); 2,98-2,32 (м, 13H); 1,94 (м, 1H); 1,54 (м, 1H).
ЖХ-МС: MH+=407.
Пример 97. Гидрохлорид 2-[[2-[4-фтор-3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-илметил)амино]-1-пирролидин-1-илэтанона
Указанное соединение получают в виде коричневого твердого вещества с выходом 74% по методике, такой же, как описана схемой 20 для синтеза 2-[[2-[4-фтор-3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида, используя в качестве реагента 2-хлор-1-пирролидин-1-илэтанон, вместо 2-хлор-N,N-диметилацетамида.
1H-ЯМР (CDCl3): 7,05-6,81 (м, 3H), 4,43 (кв, 2H), 3,77 (м, 3H), 3,54-3,31 (м, 7H), 3,01-2,33 (м, 7H), 2,06-1,77 (м, 5H), 1,58 (м, 1H).
ЖХ-МС: MH+=433.
Пример 98. Гидрохлорид 2-[[2-[3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида
Вышеупомянутое соединение синтезируют по схеме 21
Схема 21
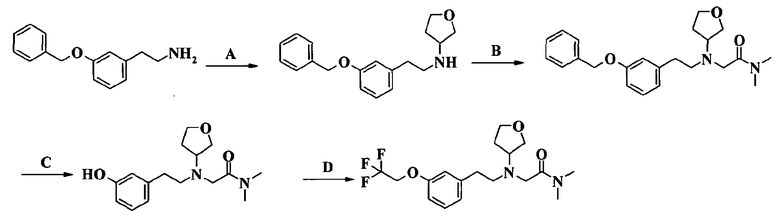
Стадия A. [2-(3-Бензилоксифенил)этил](тетрагидрофуран-3-ил)амин
Раствор дигидрофуран-3(2H)-она в 50 мл дихлорметана прибавляют к суспензии 1,99 г (7,5 ммол) 2-(3-бензилоксифенил)этиламина и 1,05 мл (7,5 ммол) триэтиламина в 70 мл 1,2-дихлорэтана. Смесь перемешивают при комнатной температуре в течение 15 минут, затем прибавляют порциями 3,2 г (15,1 ммол) NaBH(CH3COO)3. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Прибавляют воду и реакционную смесь экстрагируют дихлорметаном. Растворитель удаляют в вакууме и неочищенный остаток очищают флэш-хроматографией (дихлорметан/метанол/NH3 100:3:0,9 об.:об.:об.). Выделяют 1,35 г (60% выход) указанного в заголовке соединения в виде коричневого твердого вещества.
1H-ЯМР (CDCl3): 7,47-7,15 (м, 6H), 6,87-6,79 (м, 3H), 5,03 (с, 2H), 4,07-3,93 (м, 1H), 3,86-3,69 (м, 3H), 3,59-3,47 (м, 1H), 3,03-2,93 (м, 4H), 2,26-1,85 (м, 2H).
ЖХ-МС: MH+=298.
Стадия B. 2-[[2-[3-Бензилоксифенил)этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамид
Смесь 0,8 г (2,69 ммол) [2-(3-бензилоксифенил)этил](тетрагидрофуран-3-ил)амина, 0,56 мл (4,03 ммол) триэтиламина и 0,415 мл (4,03 ммол) 2-хлор-N,N- диметилацетамида в 5 мл сухого диметилформамида нагревают до 120°С в течение 2 часов микроволновым облучением. Растворитель удаляют в вакууме и неочищенный остаток очищают флэш-хроматографией (дихлорметан/метанол: 100:3, об.:об.). Выделяют 0,611 г (59% выход) указанного в заголовке соединения в виде желтого масла.
ЖХ-МС: MH+=383.
Стадия C. 2-[[2-(3-Гидроксифенил)этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамид
Смесь 0,61 г (1,59 ммол) 2-[[2-(3-бензилоксифенил)этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида и 60 мг Pd/C 10% в 10 мл этанола гидрируют в течение 2 часов при 40 фунт/кв.дюйм. Катализатор отфильтровывают
и растворитель удаляют в вакууме. Неочищенный остаток растирают в диэтиловом эфире и отфильтровывают. Выделяют 0,45 г (выход 96%) желтого твердого вещества.
1H-ЯМР (CDCl3): 7,18-7,06 (м, 1H); 6,78-6,65 (м, 3H); 6,50 (ушир.с, 1H); 4,04-3,90 (м, 1H); 3,84-3,64 (м, 4H); 3,59-3,35 (м, 2H); 3,03-2,87 (с+м, 7H); 2,80-2,67 (м, 2H); 2,13-1,83 (м, 2H).
ЖХ-МС: MH+=293.
Стадия D. Гидрохлорид 2-[[2-[3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида
Смесь 124 мг (0,424 ммол) [[2-(3-гидроксифенил)этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида, 178 мг (0,848 ммол) 1,1,1-трифтор-2-иодэтана, 117 мг (0,854 ммол) K2CO3 и 5 мг иодида калия в 4 мл диметилформамида нагревают микроволновым облучением в течение 2 часов при 120°С. Смесь фильтруют через целит и растворитель удаляют в вакууме. Неочищенный остаток очищают флэш-хроматографией (дихлорметан/метанол 100:0→100:2 градиент, об.:об.). Продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате, растворитель удаляют при пониженном давлении. Продукт растирают с диэтиловым эфиром. Выделяют 20 мг (выход 13%) указанного в заголовке соединения в виде белого твердого вещества.
ЖХ-МС: MH+=375.
Пример 99. 2-[[2-[3-(3,5-Диметилизоксазол-4-ил)фенил]этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамид
Вышеуказанное соединение синтезируют по схеме 22
Схема 22
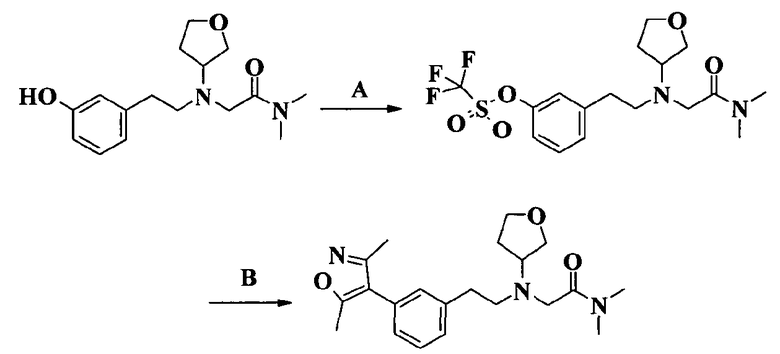
Стадия A. 2-[[2-(3-трифторметилсульфонилоксифенил)этил](тетрагидрофуран-2-ил)амино]-N,N-диметилацетамид
К смеси 120 мг (0,41 ммол) 2-[[2-(3-гидроксифенил)этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида и 113 мг (0,82 ммол) K2CO3 в 3 мл ацетонитрила в атмосфере азота прибавляют 164 мг (0,451 ммол) N-фенил-бис(трифторметансульфонимид) в 2 мл ацетонитрила. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют в вакууме, прибавляют воду и продукт экстрагируют этилацетатом. Выделяют 150 мг (выход 86%) указанного в заголовке соединения в виде желтого масла.
ЖХ-МС: MH+=425.
Стадия B. Гидрохлорид 2-[[2-[3-(3,5-диметилизоксазол-4-ил)фенил]этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида
Смесь 280 мг (0,66 ммол) 2-[[(3-трифторметилсульфонилоксифенил)этил](тетрагидрофуран-2-ил)амино]-N,N-диметилацетамида, 81 мг (0,57 ммол) 3,5- диметоксиизоксазол-4-бороновой кислоты, 80 мг (0,58 ммол) K2CO3 и 10 мг Pd(PPh3)4 в 4 мл этанола нагревают микроволновым облучением при 110°С в течение 15 минут. Смесь фильтруют через целит и растворитель удаляют в вакууме. Неочищенный продукт очищают флэш-хроматографией (дихлорметан/метанол 100:0→100:3 градиент). Продукт растворяют в растворе безводной хлористоводородной кислоты в этилацетате. Растворитель затем удаляют при пониженном давлении и продукт растирают в диэтиловом эфире. Выделяют 85 мг (0,21 ммол, выход 31%) указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (CDCl3): 7,44-7,12 (м, 4H); 4,57-4,39 (ушир.с, 1H); 4,39-3,72 (м, 7H); 3,61-3,05 (м, 3H); 2,99 (м, 6H); 2,74-2,51 (м, 1H); 2,41 (с, 3H); 2,37-2,24 (м, 1H); 2,27 (с, 3H).
ЖХ-МС: MH+=372.
Пример 100. Гидрохлорид 2-[[2-(3-пиперидин-1-илфенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида
Вышеуказанное соединение синтезируют по схеме 23
Схема 23
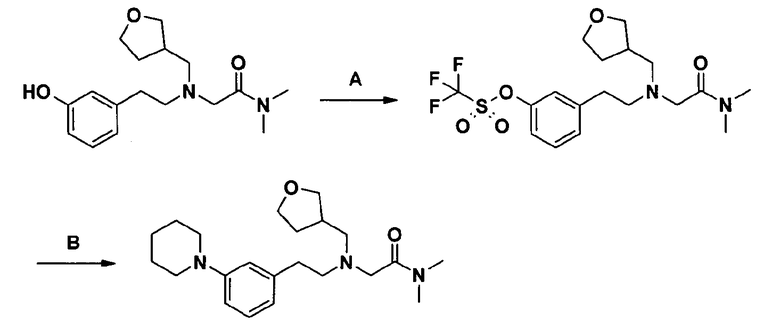
Стадия А. 2-[[2-(3-Трифторметилсульфонилоксифенил)этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамид
В атмосфере азота прибавляют 943 мг (2,64 ммол) N-фенил-бис(трифторметансульфонимид) в 4 мл ацетонитрила к смеси 735 мг (2,4 ммол) 2-[[2-(3-гидроксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида и 664 мг (4,8 ммол) K2CO3 в 15 мл ацетонитрила. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют в вакууме, прибавляют воду и экстрагируют продукт этилацетатом. Неочищенный продукт очищают флэш-хроматографией (гексан/этилацетат/триэтиламин 1:2:0,2, об.:об.:об.). Выделяют 399 мг (выход 38%) соединения, указанного в заголовке, в виде желтого масла.
ЖХ-МС: MH+=439.
Стадия В. 2-[[2-(3-Пиперидин-1-илфенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамид
Смесь 129 мг (0,29 ммол) 2-[[(3-трифторметилсульфонилоксифенил)этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида, 50 мг (0,58 ммол) пиперидина, 34 мг (0,35 ммол) трет-бутоксида натрия, 10 мг Pd(OAc)2 и 10 мг N-фенил-2-(ди-трет-бутилфосфинил)индола в 2 мл толуола нагревают до 110°С микроволновым облучением в течение 15 минут. Смесь фильтруют через целит и растворитель удаляют в вакууме. Неочищенный продукт очищают препаративной ВЭЖХ, растворяют в растворе безводного HCl в этилацетате и удаляют растворитель при пониженном давлении. Продукт растирают в диэтиловом эфире. Выделяют 23 мг (выход 19%) указанного в заголовке соединения в виде желтого твердого вещества.
1H-ЯМР (CDCl3): 7,94-7,91 (м, 2H); 7,49-7,29 (м, 2H); 4,00-3,10 (м, 17H); 3,01 (с, 3H); 2,99 (с, 3H); 2,80-2,60 (м, 3H); 2,29-2,19 (м, 1H); 1,97-1,90 (м, 4H).
ЖХ-МС: MH+=374.
Пример 101. Гидрохлорид 2-[[2-[3-(3,5-диметилизоксазол-4-ил)фенил]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамидa
Cмесь 135 мг (0,31 ммол) 2-[[(3-трифторметилсульфонилоксифенил)этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида, 61 мг (0,43 ммол) 3,5-диметилизоксазол-4-бороновой кислоты, 59 мг (0,43 ммол) K2CO3 и 10 мг Pd(PPh3)4 в 3 мл этанола нагревают микроволновым облучением при 110°С в течение 15 минут. Смесь фильтруют через целит и растворитель удаляют. Неочищенный остаток очищают препаративной ВЭЖХ. Продукт растворяют в смеси этилацетат/хлористоводородная кислота, растворитель удаляют и продукт растирают в диэтиловом эфире. Выделяют 44 мг (выход 34%) указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (CDCl3): 7,43-7,13 (м, 4H); 4,17-3,28 (м, 12H); 2,98 (с, 3H); 2,96 (с, 3H); 2,88-2,72 (м, 1H); 2,40 (с, 3H); 2,26 (с, 3H); 2,42-2,16 (м, 2H).
ЖХ-МС: MH+=386.
Пример 102. Анализ притока кальциевых каналов N-типа
Клетки нейробластомы человека IMR32 существенно экспрессируют каналы как L-, так и N-типа. В условиях дифференцировки клетки IMR32 преимущественно экспрессируют кальциевые каналы N-типа на поверхности мембран. Остающиеся
кальциевые каналы L-типа блокируют с помощью селективного блокатора каналов L-типа нифедипина. В указанных условиях эксперимента могут быть детектированы только каналы N-типа. Клетки IMR32 подвергают дифференцировке, используя 1 мM дибутирил-cAMP и 2,5 мкМ бромдеоксиуридин в течение 8 дней (4 раза) в колбе объемом 225 мл, затем отделяют, высевают 200000 клеток/лунка 96-луночного планшета, покрытого поли-L-лизином, и дополнительно инкубируют перед использованием в течение 18-24 часов в присутствии дифференцировочного буфера.
Для анализа используют набор для анализа Ca2+ (Molecular Devices), основанный на флуоресцентном индикаторе кальция, и он позволяет детектировать приток кальция, определяемый деполяризующими условиями. Дифференцированные клетки инкубируют красителем, загружаемым в течение 30 минут при 37°С, затем прибавляют нифедипин (1 мкМ), один или в присутствии ω-конотоксина (в качестве эталона), или испытываемое соединение в течение следующих 15 минут.
Флуоресценцию (возбуждение: длина волны 485 нм, испускание: длина волны 535 нм) измеряют до и после (30-40 с) автоматической инъекции 100 мкМ деполяризующего раствора KCl с помощью планшет-ридера Victor (Perkin Elmer).
Кривые ингибирования рассчитывают из 5 концентраций, для каждой три раза, и IC50 определяют, используя линейный регрессионный анализ.
Соединения по настоящему изобретению ингибируют кальциевые каналы N-типа с фармакологически значимыми значениями IC50.
Результаты, полученные с некоторыми соединениями, которые являются представителями целого класса соединений по изобретению, сравниваемые с внутренним стандартом ралфинамидом, приведены в таблице 1.
метоксифенил]этиламино]-1-(морфолин-4-ил)-2-фенилэтанона
Данные, выражаемые как значения IC50 при мкМ концентрации, показывают, что соединения по изобретению являются сильнодействующими в качестве ингибиторов кальциевых каналов N-типа.
Пример 103. Анализ притока кальциевых каналов L-типа клетки опухоли гипофиза мышей линии AtT20/D16v-F2 предпочтительно экспрессируют кальциевые каналы L-типа. Остающиеся кальциевые каналы N-типа блокируют с помощью селективного блокатора каналов N-типа ω-конотоксина. В указанных условиях эксперимента могут быть определены только каналы L-типа. Клетки AtT20 выращивают в DMEM с 10% FBS, 4 мM глутамина. Клетки высевают 200000 клеток/лунка 96-луночного планшета, покрытого поли-L-лизином, и дополнительно инкубируют перед использованием в течение 18-24 часов.
Для анализа используют набор для анализа Ca2+ (Мolecular Devices), основанный на флуоресцентном индикаторе кальция, и он позволяет детектировать приток кальция, определяемый деполяризующими условиями. Дифференцированные клетки инкубируют красителем, загружаемым в течение 30 минут при 37°С. Затем прибавляют ω-конотоксин, один (1 мкМ) или в присутствии нифедипина (в качестве эталона), или испытываемое соединение в течение следующих 15 минут.
Флуоресценцию (возбуждение: длина волны 485 нм, испускание: длина волны 535 нм) измеряют до и после (30-40 с) автоматической инъекции 100 мкМ деполяризующего раствора KCl с помощью планшет-ридера Victor (Perkin Elmer).
Кривые ингибирования рассчитывают из 5 концентраций, для каждой три раза, и IC50 определяют, используя линейный регрессионный анализ.
Результаты, полученные с некоторыми соединениями, которые являются представителями целого класса соединений по изобретению, сравниваемые с внутренним стандартом ралфинамидом, приведены в таблице 2.
[мкМ]
метоксифенил]этиламино]-1-(морфолин-4-ил)-2-фенилэтанона
Данные, выражаемые в значениях IC50 при мкМ концентрации, показывают, что соединения по изобретению в значительной степени препятствуют функционированию кальциевых каналов L-типа.
Пример 104. Анализ притока TTXs-натриевых каналов
Линия клеток ганглия заднего корешка спинного мозга крыс ND7/23 эндогенно экспрессирует смешанную популяцию TTXs натриевых каналов (таких как Navl.3, Navl.2, Navl.l, Navl.6). Эти клетки испытывают недостаток TTXr натриевых каналов, как показано отсутствием их соответствующих транскриптов.
Клетки ND 7/23 выращивают в DMEM с добавлением 10% FBS и 1 мM пирувата натрия. Клетки высевают 50000 клеток/лунка 96-лучного планшета, покрытого поли-L-лизином, и дополнительно инкубируют перед использованием в течение 18-24 часов.
Для анализа используют набор Membrane Potential Kit Assay (Molecular Devices), основанный на отрицательно заряженном флуоресцентном красителе, способном контролировать изменения в мембранном потенциале, вызванные притоком натрия, обусловленного открытием каналов.
Клетки инкубируют красителем, загружаемым в течение 30 минут при 25°С. Затем прибавляют 100 нМ токсина Anemonia sulcata (применяемого в качестве энхансера реакции открывателя каналов), одного или в присутствии TTX (в качестве эталона), или испытываемое соединение в течение дополнительных 15 минут.
Флуоресценцию (возбуждение: длина волны 530 нм, испускание: длина волны 565 нм) измеряют до и после (40-45 с) автоматической инъекции открывателя натриевых каналов вератридина (100 мкМ), используя планшет-ридер Victor (Perkin Elmer).
Кривые ингибирования рассчитывают из 5 концентраций, для каждой три раза, и IC50 определяют, используя линейный регрессионный анализ.
Соединения по настоящему изобретению ингибируют TTXs-натриевые каналы с фармакологически значимыми значениями IC50.
Результаты, полученные с некоторыми соединениями, которые являются представителями целого класса соединений по изобретению, сравниваемые с внутренним стандартом ралфинамидом, приведены в таблице 3.
Пример 105. Изучение ингибирования кальциевых токов методом «пэтч-кламп» («patch clamp»)
Клетки и методы:
Функциональное ингибирование Са токов N-типа исследуют методами пэтч-клэмп «целая клетка» (Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Pflugers Arch. (1981) 391: 85-100) на клетках HEK293, экспрессирующих рекомбинантные каналы N-типа человека, получаемых после транзиентной трансфекции субъединиц h α1B (hCav2.2)+β1b+α2δ-1.
Мембранные токи регистрируют и фильтруют при 5 кГц усилителем Axopatch 200B и преобразовывают в цифровую форму прибором Axon Digidata 1322A (Axon Instruments, CA, США). Фиксация напряжения мембранных потенциалов и сбор информации регулируют on-line с помощью Axon pCLAMP8 компьютерных программ. Электроды сравнения и измерения представляют собой AgCl-Ag электроды. Клетки имели первоначальные сопротивления контакта >1 GΩ и сопротивления доступа 4,2±0,2 MΩ. Клетки непрерывно поливают внеклеточными растворами, используя Biologic RSC-200.
Контрольный омывающий раствор для регистрации кальциевых токов содержит (мM): холинхлорид (70), MgCl2 (1), BaCl2 (20), TEA-Cl (50), Hepes (10), глюкозу (10). Раствор в пипетке состоит из (мM): CsCl (140), EGTA (10), MgCl2 (2), Hepes (10), MgATP (1), GTP Tris (0,3).
Соединения растворяют в ДМСО для приготовления 20 мM основных растворов и затем разбавляют до конечной концентрации в дополнительных растворах.
Протоколы напряжения и данные анализов:
Используют двухстадийный протокол для определения зависимости напряжения блока:
Ток N-типа активируют 600 мс ступенчатым импульсом до +10 мВ (тестовый импульс) от 5000 мс предварительного потенциала -110 мВ (состояние покоя) или -50/-55 мВ (половина максимального стационарного состояния) соответственно.
Амплитуду пиков кальциевых токов, вызванных соответствующими тестовыми импульсами с частотой 0,06 Гц, измеряют до и после воздействия испытываемого соединения. Toнический блок токов рассчитывают как разницу между пиком тока кальция, измеренным в конце периода стабилизации в контрольном внешнем омывающем растворе, и токами пиков, измеренными в конце периода перфузии испытываемого соединения (когда достигается устойчивое состояние), разделенную на пики контроля. Кривые концентрация лекарственного средства - ингибирование получают путем нанесения на график тонических блоков в зависимости от концентрации лекарственного средства. Кривые доза - ответ приводят в соответствие с данными тонического блока согласно логистическому уравнению: y=A2+(A1-A2)/[1+(x/IC50)p]. A1 и A2 представляют собой фиксированные значения 0 и 1, соответствующие 0 и 100% ингибированию токов, x представляет собой концентрацию лекарственного средства, IC50 представляет собой концентрацию лекарственного средства, которая вызывает 50% ингибирование токов, и p представляет собой соответствующий угловой коэффициент.
Соединения по настоящему изобретению ингибируют кальциевые каналы N-типа с фармакологически значимыми значениями IC50.
Результаты, полученные с некоторыми соединениями, которые являются представителями целого класса соединений по изобретению, сравниваемые с внутренним стандартом ралфинамидом, приведены в таблице 4.
(Ihalf)
Данные, выражаемые в значениях IC50 при мкМ концентрации, показывают, что соединения по изобретению обладают высокой активностью в качестве ингибиторов кальциевых каналов N-типа.
Пример 106. Изучение ингибирования натриевых каналов методом пэтч-кламп
Клетки и методы: Функциональное ингибирование натриевых токов изучают с помощью методов пэтч-клэмп «целая клетка» (Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Pflugers Arch. (1981) 391(2): 85-100) на клетках HEK293, экспрессирующих рекомбинантные каналы Nav 1.3.
Мембранные токи регистрируют, как описано в представленном выше примере.
Контрольный омывающий раствор для регистрации натриевых токов содержит (мM): NaCl (80), холинхлорид (38), CaCl2 (1,3), MgCl2 (2), KCl (2), CdCl2 (0,4), NiCl2 (0,3), TEA-Cl (20), Hepes (10), глюкозу (10). Раствор в пипетке состоит из (мM): EGTA (10), NaCl (10), CaCl2 (1,3), MgCl2 (2), Hepes (10), CsF (130), MgATP (1).
Соединения растворяют в ДМСО для приготовления 20 мM основных растворов и затем разбавляют до конечной концентрации в дополнительных растворах.
Протоколы потенциалов и данные анализов: Используют двухстадийный протокол для определения зависимости потенциала блока:
Натриевый ток активируют 30 мс ступенчатым импульсом до 10 мВ (тестовый импульс) от 2000 мс предварительного потенциала -100 мВ (состояние покоя) или -50 мВ (половина максимального стационарного состояния) соответственно.
Амплитуду пиков натриевых токов, вызванных соответствующими тестовыми импульсами с частотой 0,06 Гц, измеряют до и после воздействия испытываемого соединения. Toнический блок токов рассчитывают как разницу между пиком тока Na, измеренного в конце периода стабилизации в контрольном внешнем омывающем растворе, и пиком тока, измеренного в конце периода перфузии испытываемого соединения (когда достигается стационарное состояние), разделенную на пики контроля. Кривые концентрация лекарственного средства - ингибирование получают путем нанесения на график тонических блоков в зависимости от концентрации лекарственного средства. Кривые доза - ответ приводят в соответствие с данными тонического блока согласно логистическому уравнению: y=A2+(A1-A2)/[1+(x/IC50)p]. A1 и A2 представляют собой фиксированные значения 0 и 1, соответствующие 0 и 100% ингибированию токов, x представляет собой концентрацию лекарственного средства, IC50 представляет собой концентрацию лекарственного средства, которая вызывает 50% ингибирование токов, и p представляет собой соответствующий угловой коэффициент.
Соединения по настоящему изобретению ингибируют натриевые каналы с фармакологически значимыми значениями IC50.
Результаты, полученные с некоторыми соединениями, которые являются представителями целого класса соединений по изобретению, сравниваемые с внутренним стандартом ралфинамидом, приведены в таблице 5.
(Ihalf)
Данные, выражаемые в значениях IC50 при мкМ концентрации, показывают, что соединения по изобретению являются сильными ингибиторами натриевых каналов.
Пример 107. Ингибирование натриевых токов в кортикальных нейронах
Получение и культивирование клеток: кортикальные нейроны получают из эмбрионов крыс линии Вистар (E17-E19). Мозг крыс E17/E19 удаляют и помещают в ледяной раствор Хэнка (раствор Хэнка (Life tech. 14170-088) + глюкоза 30% + Pen-Strep 100x (Life Tech. 15140-122) 100U-100 мкг/мл и Hepes-NaOH 5 мM).
Выделяют кортикальный слой, разрезают на небольшие части и дважды промывают раствором Хэнка. Весь раствор, кроме 1-2 мл, удаляют и ткань механически разъединяют. После механического разъединения тканей прибавляют 5 мл полной DMEM (среда Игла, модифицированная Дульбекко) (Gibco 41966-029) + FBS (Hyclone) 10% + глутамин (Life tech. 25030-024) 2мM + Pen-Strep 100U-100 мкг/мл и суспензию клеток центрифугируют в течение 5 мин при скорости 1000 об./мин. Удаляют супернатант и прибавляют 5 мл полной среды Neurobasal (Neurobasal medium (Life tech. 21103-049) + B27 (Life tech. 17504-044) 2% + глутамин 2 мM + Pen-Strep 100U-100 мкг/мл).
Клетки подсчитывают и разводят в Neurobasal среде до концентрации 400000 клеток на чашку Петри, обработанную поли-D-лизином 5 мкг/мл.
Кортикальные нейроны используют с 6-го по 11-й день после посева и один раз в неделю меняют Neurobasal среду.
Снятие показаний методом «пэтч-кламп» (целая клетка): Эксперименты на кортикальных нейронах проводят стандартными методами «пэтч-кламп» (целая клетка) (Hamill et al., 1981). Мембранные токи регистрируют и фильтруют при 5 кГц усилителем Axopatch 200B и преобразовывают в цифровую форму прибором Axon Digidata 1322A (Axon Instruments, CA, США). Прогон протокола и сбор информации регулируют on-line с помощью Axon pClamp8 компьютерных программ. Электроды сравнения и измерения представляют собой AgCl-Ag электроды. Для вытягивания «пэтч» пипеток с сопротивлением 2-3 MΩ из борсиликатных стеклянных трубок Harward используют устройство Sutter Instrument P-87 Puller (CA, США). Клетки непрерывно поливают внеклеточными растворами, используя устройство для смены раствора Biologic RSC-200.
Растворы: Контрольный омывающий раствор для регистрации натриевых токов содержит (мM): NaCl 60, холинхлорид 60, CaCl2 1,3, MgCl2 2, KCl 2, CdCl2 0,4, NiCl2 0,3, TEACl 20, Hepes 10, глюкозу 10. Раствор в пипетке состоит из (мM): CsF 65, CsCl 65, NaCl 10, CaCl2 1,3, MgCl2 2, Hepes 10, EGTA 10, MgATP 1.
Протоколы напряжения и данные анализов: клетки присасываются при -90 мВ, затем используют двухстадийный протокол для определения зависимости напряжения блока. Натриевые токи активируют 30 мс ступенчатым импульсом до -10 мВ (тестовый импульс) от 2000 мс предварительного потенциала -110 мВ (стадия покоя) и потенциал приблизительно -50 мВ (половина максимального стационарного состояния).
Кривые концентрация лекарственного средства - ингибирование лекарственного средства получают путем нанесения на график тонических блоков в покоящемся и деполяризованном состоянии в зависимости от концентрации лекарственного средства. Кривые доза - ответ приводят в соответствие с данными тонического блока согласно логистическому уравнению: y=A2+(A1-A2)/[1+(x/IC50)p]. A1 и A2 представляют собой фиксированные значения 0 и 1, соответствующие 0 и 100% ингибированию токов, x представляет собой концентрацию лекарственного средства, IC50 представляет собой концентрацию лекарственного средства, которая вызывает 50% ингибирование токов, и p представляет собой соответствующий угловой коэффициент.
Соединения по настоящему изобретению ингибируют натриевые токи кортикальных нейронов с фармакологически значимыми значениями IC50.
Результаты, полученные с некоторыми соединениями, которые являются репрезентативными для целого класса соединений по изобретению, сравниваемые с внутренним стандартом ралфинамидом, приведены в таблице 6.
(Ihalf)
Пример 108. Анализ активности ферментов MAO-A и MAO-B in vitro
Препараты мембран (неочищенная митохондриальная фракция)
Крыс-самцов линии Вистар (Harlan, Италия - 175-200 г) умерщвляют при легкой анестезии, быстро удаляют мозг и гомогенизируют в 8 объемах ледяного 0,32 M сахарозного буфера, содержащего 0,1 M EDTA, pH 7,4. Неочищенный гомогенат центрифугируют при скорости 2220 об/мин в течение 10 минут и извлекают супернатант. «Tаблетку» гомогенизируют и повторно центрифугируют. Два супернатанта объединяют и центрифугируют при 9250 об/мин в течение 10 минут при +4°C. «Таблетку» повторно суспендируют в свежем буфере и центрифугируют при 11250 об/мин в течение 10 минут +4°C. Полученную таблетку хранят при температуре -80°C.
Анализ активности ферментов in vitro
Активность ферментов оценивают радиоферментным анализом, используя субстраты 14C-серотонин (5-HT) и 14C-фенилэтиламин (PEA) для MAO-A и MAO-B соответственно. Митохондриальную таблетку (500 мкг белка) повторно суспендируют в 0,1 M фосфатном буфере (pH 7,4). Прибавляют 500 мкл суспензии к 50 мкл раствора испытываемого соединения или буфера и инкубируют в течение 30 минут при 37°С (предварительное инкубирование), затем прибавляют субстрат (50 мкл). Инкубирование проводят в течение 30 минут при 37°С (14C-5-HT, 5 мкM) или в течение 10 минут при 37°C (14C-PEA, 0,5 мкM).
Реакцию останавливают, прибавляя 0,2 мл 37% HCl или перхлорной кислоты. После центрифугирования деаминированные метаболиты экстрагируют 3 мл диэтилового эфира (5-HT) или толуола (PEA) и радиоактивную органическую фазу измеряют жидкостно-сцинтилляционной спектрометрией при эффективности 90%. Количество нейтральных и/или кислых метаболитов, образованных в результате активности, получают измерением радиоактивности элюата.
Активность MAO в образце, соответствующую процентной доле радиоактивности, по сравнению с активностью контроля в отсутствие ингибитора, выражают в нмоль трансформированного субстрата/мг белка/мин.
Результаты, поскольку они касаются ингибирования MAO-B, получаемого с некоторыми соединениями, которые являются представителями целого класса соединений по изобретению, приведены в таблице 7.
МАО-В при 100 мкМ
этил](фуран-2-илметил)амино]-N-(2-амино-2-метилпропил)ацетамида
Данные, выражаемые в процентах ингибирования MAO-B, наблюдаемые в присутствии 100 мкМ соединения, демонстрируют, что соединения по изобретению являются слабыми ингибиторами MAO-B, если сравнивать с внутренним стандартом сафинамидом.
Пример 109. Модель хронической воспалительной боли с полным адъювантом Фрейнда
Moнoартрит индуцируют у крыс (массой 200 г) инъекцией в подушечку левой задней лапы 100 мкл полного адъюванта Фрейнда (CFA), содержащего убитый нагреванием и высушенный Mycobacterium tuberculosis в смеси парафинового масла и эмульгатора, mannide моноолеат. Инъекция CFA приводит к образованию локализованного отека и воспаления, начинающегося через несколько часов после инъекции, причем прогрессирующее уменьшение порога механического отдергивания.
В течение 8-9 дней до начала тестирования дают возможность артриту развиваться у каждого животного.
Mеханическая аллодиния
Пороги механической аллодинии определяют по методу Чаплана и сотр. (Chaplan S.R., Bach F.W., Pogrel J.W., Chung J.M., Yaksh T.L. J. Neurosci. Methods (1994) 53: 55-63). Крыс помещают в отдельные пластиковые коробки размером 24×10×15 см на сетчатый металлический пол и перед тестированием дают акклиматизироваться в течение 30 минут. Ряд калиброванных волосков Фрея (Stoelting, Wood Dale, IL) с логарифмически инкрементальной жесткостью в интервале от 2,83 до 5,88, выражаемой Log10 [10 × сила в (мг)], прикладывают к лапе модифицированным методом "вверх и вниз" (Dixon W.J. Am. Stat. Assoc. (1965) 60: 967-978). При отсутствии реакции отдергивания лапы на первоначально выбранный волосок более толстый волосок, соответствующий более сильному стимулу, прикладывают до тех пор, пока не зарегистрируют резкое отдергивание. Процедуру повторяют два раза. Каждый волосок помещают перпендикулярно лапе с силой, достаточной для того, чтобы вызвать слабое сгибание, и выдерживают 2-3 с. Так же и с такой же интенсивностью стимулируют заднюю лапу пять/шесть раз с интервалами несколько секунд. Механический порог выражают в Log10 [10 × сила в (мг)], указывая силу волоска Фрея, на которую животное реагирует (отдергивание лапы, облизывание или дрожание).
Пороги механической аллодинии измеряют до лечения (pre-drug) и через 30, 60, 90, 120, 240 и 360 минут после лечения. Измеряют также порог через 24 часа.
Соединения по изобретению вводят в дозах от 0,1 до 100 мг/кг.
Пример 110. Модель невропатической боли Беннета на крысах
Эффекты невропатической боли тестируют на модели хронического сжатия на крысах (Bennett, G.J. и Xie, Y.K., A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man, Pain, 33 (1988) 87-107). Под анестезией пентобарбиталом (Nembutal, 50 мг/кг, i.p.) односторонние множественные лигатуры накладывают на правый седалищный нерв крыс-самцов линии Sprague-Dawley (140-160 г). Делают надрез на уровне середины бедра и на седалищный нерв накладывают четыре не тугих лигатуры (хромированный кетгут 5-0), размещая их вокруг нерва таким образом, чтобы не нарушить эпиневральное кровообращение. После операции животным дают возможность выздоравливать в течение недели. У животных развивается холодная аллодиния, которая стабильна в течение, по меньшей мере, пяти недель. Холодную аллодинию тестируют на металлической пластине, охлажденной на водяной бане до постоянной температуры 4°С. Животных, произвольно включенных в состав групп из 10 крыс для каждой испытываемой дозы и носителя, наблюдают в течение 2 минут до и после применения испытываемого соединения и подсчитывают число сильных реакций отдергивания. Оценивают эффект в нескольких временных точках после применения. Максимально возможный эффект в процентах (% MPE) и стандартную погрешность среднего (SEM) для каждого значения времени определяют, используя значения до тестирования как 100% MPE. Область данных (AUD) рассчитывают для периода наблюдения и выражают как процент ингибирования по сравнению с контролем (носителем), как показано в таблице 8. Данные рассчитывают по значениям AUD в процентах.
[мг/кг] p.o.
амино]пропанамид (ралфинамид)
Пример 111. Тест максимального электрошока (MES) у мышей
Тест максимального электрошока (MES) обычно используют для скрининга противоэпилептических лекарственных средств на моделях грызунов.
Животные и аппаратура: Используют мышей-самцов линии CD1 массой 25 г. Применяют методику, описанную Уайтом с соавт. (White H.S., Woodhead J.H., Franklin M.R., Swinyard E.A., and Wolf H.H. Antiepileptic Drugs (1995) 4th ed: 99-110, Raven Press, Ltd., New York). Используют генератор стимуляции электрошоком An Ugo Basile (Model ECT UNIT 7801) для подачи электрического импульса, достаточного для того, чтобы вызвать тоническую экстензорную реакцию задней конечности по меньшей мере у 97% контрольных животных. Импульс подают интрааурально через электроды, прикрепленные клипсой к мышам (0,7 с электрошок 40 мA, последовательность импульсов с частотой 80 Гц и продолжительность импульса 0,4 мс). Сильное действие соединения, вводимого внутрибрюшинно или перорально за 15-60 минут до индукции MES, оценивают и сравнивают с контрольной группой (вводят носитель). Изучают группы из 10 мышей. Полное подавление судорожного компонента тонической экстензорной реакции задней конечности считают доказательством противосудорожной активности.
Соединения по изобретению вводят внутривенно, перорально или внутрибрюшинно в дозах от 0,1 до 100 мг/кг.
Результаты, полученные с соединениями, представляющими собой целый химический класс по изобретению, введенными внутривенно за 5 минут до тестирования, сравниваемые с внутренним стандартом сафинамидом и представленные в таблице 9, показывают, что указанные соединения активны в качестве противосудорожных лекарственных средств.
Пример 112. Локомоторная гиперактивность у мышей, индуцированная амфетамином и хлордиазепоксидом
В указанной модели мышей обрабатывают смесью d-амфетамина и анксиолитической дозы бензодиазепина, хлордиазепоксида (Rushton R., Steinberg H. Combined effects of chlordiazepoxide and d-amphetamine on activity of rats in an unfamiliar environment. Nature 1966; 211: 1312-3; R. Arban, G. Maraia, K. Brackenborough, L. Winyard, A. Wilson, P. Gerrard, C. Large. Evaluation of the effects of lamotrigine, valproate и carbamazepine in a rodent model of mania Behavioural Brain Research, 158: 123-132). Модель предназначена для имитации некоторых аспектов мании в биполярных расстройствах. Важно, что гиперактивность, индуцированная смесью d-амфетамина и хлордиазепоксида, можно предотвратить предварительным введением известного нормотимика - лития, а также других нормотимических лекарственных средств (например, валпроата магния и карбамазепина). Cледовательно, рассматриваемая модель имеет очевидную и предсказательную валидность в качестве модели биполярных расстройств и представляет собой ценный инструмент, чтобы определить, может ли испытываемое соединение быть потенциальным нормотимическим лекарственным средством.
Aмфетамин (AMP) (2,5 мг/кг) плюс гидрохлорид хлордиазепоксида (CDZ) (3 мг/кг/ip) вводят мышам-самцам Albino Swiss (25-32 г) в объеме 10 мл/кг. Локомоторную активность регистрируют с помощью системы Opto-M3 (Columbus Instruments), которая представляет собой многоканальный монитор. Система Opto-M3 содержит 10 инфракрасных излучателей и соответствующее количество ресиверов (0,5" beam интервал), присоединенных к персональному компьютеру, и рассчитывает как временную активность, так и суммарную активность. Таким образом, система дифференцирует поступательную локомоторику (передвижение) и стереотипное движение (суммарную активность). Мышей предварительно обрабатывают испытываемым соединением (5 мг/кг) и через 10 минут AMP (2,5 мг/кг) или AMP вместе с CDZ (3 мг/кг). После следующих 30 минут мышей опять обрабатывают такой же дозой испытываемого соединения и помещают по отдельности в клетки для двигательной активности. Локомоторную активность (передвижение и суммарную активность) оценивают в течение 30 минут. Каждая группа состоит из 8-10 мышей.
Статистический анализ: Данные оценивают, используя дисперсионный анализ (ANOVA), затем, если целесообразно, проводят отдельное сравнение с контролем, используя тест Даннетта.
Введение амфетамина-хлордиазепоксида вызывает значительное увеличение локомоторной активности.
Действие соединения гидрохлорида 2-[[2-(3-бутоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида, представителя целого класса химических соединений по настоящему изобретению, оценивают по его способности предотвращать индуцированное амфетамином-хлордиазепоксидом увеличение локомоторной активности, как показано на фиг.1.
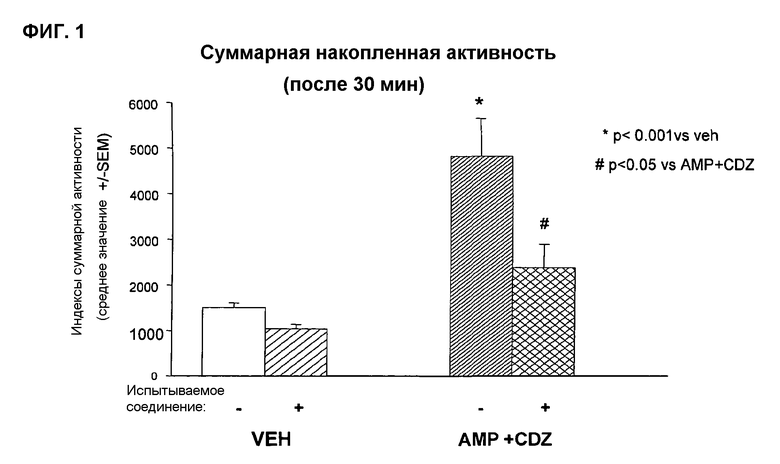
Пример 113. Когнитивные нарушения при шизофрении
Когнитивные нарушения часто связаны с шизофренией, и ее стали признавать как основной элемент нарушений, непосредственно влияющий на выздоровление пациента и его реинтеграцию в общество.
В последнее время особый интерес вызывает фармакологическая модель когнитивных дисфункций при шизофрении, которая базируется на действии антагонистов глутаматных NMDA рецепторов, таких как фенциклидин (PCP) и кетамин (Javitt et al., 1991), которые ослабляют внимание и увеличивают «импульсивность» и «навязчивую» персеверацию у мышей, выполняющих сложное задание (Greco et al., 2005).
Материалы и методы
Животные: Используют мышей-самцов линии DBA/2N (Charles River, Италия). Мышей массой 25-30 г в начале экспериментов помещают в термостатированные условия (21°С) с чередованием 12 часов света и 12 часов темноты (свет с 7.00 до 19.00). Питание (Rieper, Италия) доступно по желанию. Животные имеют два часа доступа к воде в конце каждого ежедневного тестирования.
Аппаратура для five-choice serial reaction time задачи:
Аппаратура для тестирования состоит из четырех камер 21,6×17,8×12,7 см (Med. Associates Inc. США), как было описано ранее [Greco, 2005 #26]. Стимулами и регистрацией реакций управляют с помощью SmartCtrlTM Package 8 In/16 Out (Med. Associates Inc. США) с дополнительным интерфейсом MED-PC для Windows (Med. Associates Inc. США). Управляющая программа для 5-CSRT задачи выполнена по заказу.
Поведенческие методики: привыкание к жидкой награде и засовыванию носа в отверстия. За мышами ухаживают в течение одной недели и регистрируют их массу тела. Затем их лишают воды и делают возможным 2-часовой доступ к воде в начале вечера до тех пор, пока масса тела не стабилизируется (8 дней). В течение следующих двух дней мышей приучают к «домашним клеткам», к награде (10% раствор сахарозы), используемой затем в оперантных методиках. В следующие два дня мыши привыкают к оперантным боксам. Во время этой стадии доступен 10% раствор сахарозы в небольшой чаше, расположенной ниже приемного отверстия бокса. Сначала мышам приходится приучаться к тому, что каждые 5 с жидкая награда доступна в небольшой чаше в приемном отверстии. Во время этого периода регистрируют входы головы. Во время следующего периода мышей обучают совать носы в освещенные отверстия. Сразу же после засовывания в емкость с водой включают LED позади одного из отверстий. Засовывание носа в освещенное отверстие гасит световой стимул, а приспособление для погружения обеспечивает подачу 0,01 мл награды в приемное отверстие. Любая реакция в одно из других четырех отверстий не имеет последствий и не регистрируется. Световой стимул имеет место во всех пяти отверстиях в произвольном порядке. Мышь переключают на задачу 5-CSRT после того, как она завершит по меньшей мере 50 вознагражденных попыток «совать нос» в течение одной 30-минутной сессии.
The five-choice serial reaction time задача. О начале сессии сигнализируют освещением и доставкой 0,01 мл жидкой награды. Засовыванием носа в приемное отверстие начинают первое испытание. После фиксированной задержки (интервал между испытаниями, ITI) LED позади одного из отверстий продолжают в течение короткого периода времени. Стимул LED имеет место одинаковое количество раз в каждом отверстии во время полной сессии, причем порядок освещения рандомизирован компьютером. Пока свет включен и в течение короткого периода времени после (ограниченная задержка), реакции в освещенное отверстие (правильные реакции) приводят в результате к жидкой награде. Реакции в отверстия, которые не освещены (неправильные реакции), или отсутствие реакций в ограниченный период времени приводят к тому, что освещение выключают на короткий период времени (пауза). Реакциями в отверстия при выключенном освещении начинается пауза. После доставки жидкой награды или в конце паузы мышь начинает следующее испытание, засовывая нос в приемное отверстие. Реакции в отверстия, сделанные после правильной реакции (персеверативные реакции) или после окончания паузы перед засовыванием носа в приемное отверстие, приводят в результате к периоду паузы. Реакции в отверстия во время ITI (антиципирующие реакции) также приводят в результате к периоду паузы. После антиципирующих реакций сованием носа в приемное отверстие повторно начинается текущее испытание. Ежедневная сессия состояла из 100 испытаний или 30 минут тестирования, независимо от того, что раньше завершится, затем весь свет отключают и дальнейшие реакции не дают эффекта. В первой сессии схемы тестирования стимул и ограниченная задержка длятся по 1 мин и, в зависимости от индивидуального выполнения, их постепенно уменьшают до 1 с. Продолжительность стимула уменьшают в следующей последовательности: 60, 30, 10, 5, 2,5, 2, 1,5 и 1 с (исходный уровень). Как ITI, так и пауза длятся 2 с в первой сессии, и ITI увеличивают до 5 с в последующих сессиях, время паузы не изменяют. На протяжении всего периода обучения и экспериментов для каждой мыши проводят одну сессию в день для выполнения задачи 5-CSRT.
Лекарственные средства и схемы лечения. Испытываемое соединение растворяют в воде и вводят внутрибрюшинно (IP) в дозе 10 мг/кг. Через 5 минут после лечения мышам делают инъекцию носителя (физиологического раствора) или PCP (1,5 мг/кг) и через 10 минут начинают тестовую сессию. В каждом эксперименте вводят различные комбинации испытываемого соединения и носителя или PCP в соответствии со схемой латинского квадрата. По меньшей мере 48 часов оставляют между днями тестирования лекарственного средства. Во время этих промежуточных дней мышей тестируют на 5-CSRT задание, чтобы повторно установить исходные характеристики и проверить любые остаточные эффекты лекарственных средств.
Статистический анализ: Основными зависимыми переменными, выбранными для анализа, были: (a) процент правильных реакций (сумма правильных реакций/сумма правильных + сумма неправильных реакций × 100); (b) процент пропусков (сумма пропусков/сумма правильных реакций + сумма неправильных реакций + сумма пропусков × 100); (c) число преждевременных реакций в отверстиях во время ITI; (d) число персеверативных реакций в отверстиях после правильного ответа. Правильные ответы и пропуски, выраженные в процентах, трансформируют по формуле 2arcsin(SQRT (%X/100)), чтобы нормализовать распределение в соответствии с моделью ANOVA (Winer, 1971).
Действие испытываемых соединений (n=12) на индуцированные PCP дефициты в задаче 5-CSRT анализируют независимо ANOVA (субъекты 2×2) с факторами лекарственного средства (испытываемое соединение) и PCP. Потом значения для группы, получавшей лечение, сравнивают с помощью post-hoc теста Tukey-Kramer. Статистической программой (SAS Institute Inc., США) управляют компьютером Micro VAX 3500 (Digital, США).
Как показано в таблице 10, PCP оказывает сильное действие на характеристики внимания мышей DBA/2N, что показано увеличенными антиципирующими и персеверативными реакциями. Репрезентативное соединение по настоящему изобретению, введенное в дозе 10 мг/кг i.p., может изменить в противоположном направлении индуцированное PCP увеличение антиципирующих и персеверативных реакций. Представленные результаты обосновывают применение соединений указанного типа для лечения психических заболеваний.
10 мг/кг
10 мг/кг + РСР
Пример 114. Тест поведенческой сенситизации, индуцированной кокаином
Аддикция к лекарственным средствам представляет собой патологическое поведение, характеризуемое навязчивой идеей поиска и приема лекарственных средств. Одной моделью таких поведенческих изменений на животных является длительное увеличение локомоторной активности, индуцируемое повторяющимся введением психостимуляторных средств грызунам (Robinson T.E. and Berridge K.C. Brain Res. Brain Res. Rev. (1993) 18, 247-91), известной как поведенческая сенситизация, индуцированная лекарственным средством. Действие испытываемых соединений оценивают на модели поведенческой сенситизации, индуцированной кокаином на крысах.
Аппаратура для определения локомоторной активности: используют крыс-самцов линии Вистар массой 200-250 г по прибытии.
Локомоторную активность определяют в шестнадцати одинаковых идентичных подвешенных клетках из металлической проволоки, каждая из которых имеет следующие размеры 36 см (L) × 25 см (W) × 20 см (H). Каждая клетка содержит два набора инфракрасных фотоэлементов излучатель-детектор, расположенных вдоль длинной оси на 1 см выше решетки пола и на расстоянии 8 см от передней и задней стенок клетки. Фоновый шум обеспечивается генератором белого шума. Движение внутри клеток приводит к прерываниям сигналов фотоэлементов, которые автоматически регистрируются IBM-совместимым компьютером.
Методика сенситизации и лечение: Животных приучают к камерам для изучения локомоторной активности в течение 2-3 дней подряд перед экспериментом. Крысы получают 5 ежедневных внутрибрюшинных (i.p.) инъекций кокаина (5 мг/кг), или физиологического раствора, или испытываемого соединения (0,1-100 мг/кг), или его носителя и регистрируют локомоторную активность в течение 3 часов. Через 10 дней после последней инъекции кокаина или физиологического раствора (день 15) животным дают 15 мг/кг кокаина в отсутствие испытываемого соединения и опять проводят мониторинг локомоторной активности в течение 3 часов.
К пятому дню обработки кокаином животные, предварительно обработанные носителем i.p., демонстрируют реакцию усиленной локомоторной активности (на 20% выше, чем в первый день, p<0,05). Через 10 дней после последней инъекции кокаина или физиологического раствора животным дают 15 мг/кг кокаина в отсутствие испытываемого соединения и опять проводят мониторинг локомоторной активности в течение 3 часов. Предполагается, что крысы, предварительно обработанные кокаином и не получавшие испытываемого соединения, показывают реакцию на кокаин - увеличение локомоторной активности (на 30% выше, чем в первый день, p<0,05). Если крысы, которых предварительно лечили испытываемым соединением во время 5-дневной обработки кокаином, не показывают усиления локомоторной активности, испытываемое соединение рассматривается как соединение, обладающее эффектом, предотвращающим привыкание к психостимуляторным средствам (Koob G.F., Sanna P.P., Bloom F.E. Neuron (1998) 21: 467-476; Robinson T.E., Berridge K.C. Brain Res Brain Res Rev (1993) 18: 247-291).
Статистический анализ: Данные (общее число прерываний за 3 часа) анализируют с помощью two way ANOVA с повторяющимися показателями на одном факторе, включающем четыре группы экспериментов (т.е. физиологический раствор/носитель, физиологический раствор/испытываемое соединение, кокаин/носитель и кокаин/испытываемое соединение) и две точки во времени (день 1 и день 5) с последующим простым анализом эффектов. Второй two way ANOVA с повторяющимися показателями на одном факторе используют для сравнения первого дня и дня проверки с последующим post hoc тестом Ньюмена-Кейсла.
Пример 115. Острое раздражение мочевого пузыря у крыс уксусной кислотой
Эксперименты проводят на взрослых анестезированных крысах-самках линии Sprague Dawley (170-200 г). Катетер (PE-50) вводят через абдоминальный по средней линии надрез в мочевой пузырь через свод мочевого пузыря и затем измеряют внутрипузырное давление, чтобы контролировать активность мочевого пузыря во время непрерывной инфузии 0,15% уксусной кислоты. Непрерывная внутрипузырная инфузия уксусной кислоты раздражает мочевой пузырь и уменьшает интервалы между сокращениями (ICI) в анестезированных крысах. У крыс, обработанных соединениями по изобретению, до и после внутрипузырной инфузии уксусной кислоты измеряют величины ICI, максимальное давление сокращений и пороги давления, индуцирующие рефлекторное сокращение мочевого пузыря.
Пример 116. Умеренное раздражение мочевого пузыря крыс циклофосфамидом (CYP)
Эксперименты проводят на взрослых бодрствующих и анестезированных крысах-самках линии Sprague Dawley (170-200 г). Химический цистит индуцируют CYP, который метаболизирует в акролеин, раздражитель выводится в мочу. CYP (150 мг/кг/i.p.) вводят за один день до начала эксперимента. Предварительная обработка CYP вызывает раздражение мочевого пузыря и очень частые выделения мочи, причем ICI между выделениями составляет около 150-200 секунд.
Активные соединения увеличивают ICI как у бодрствующих, так и у анестезированных крыс, используемых в экспериментальной модели.
Пример 117. Тест на мигрень на крысах
Животные и хирургия: Крыс-самцов линии Вистар массой 250-350 г подвергают анестезии пентобарбиталом натрия (50 мг/кг i.p.), растворенного в физиологическом растворе.
В трахею и левую бедренную артерию вводят трубку для искусственной вентиляции (55 тактов/мин) и для измерения среднего кровяного давления (MBP) соответственно. В бедренную вену помещают трубку для внутривенного введения испытываемых средств.
Температуру тела поддерживают при 37-38°С путем автоматического регулирования нагревательной пластины. Животных помещают в стереотаксическую раму и делают продольный надрез скальпа. Просверливают трепанационное отверстие и биполярный электрод из нержавеющей стали (Plastic One MS 306) опускают в левую глазную ветвь тригеминального ганглия (3,8 мм сзади брегмы, 2,5 мм в сторону от средней линии и на 9,5 мм ниже дуральной поверхности) и закрепляют зубным цементом. Правильное размещение электрода подтверждается краткой электрической стимуляцией, которая вызывает движение челюсти, обусловленное активированием тройничного нерва. Последующее удаление мозга, правильную установку электрода в нерв проверяют визуально в конце каждого эксперимента.
Второе отверстие просверливают ипсилатерально по отношению к электроду (1,5 мм рострально к брегме и 1,5 мм в сторону от сагиттального шва) и иглу-зонд (диаметр кончика 0,8 мм) лазерного флоуметра Доплера закрепляют, нацеливая кончик иглы на ветвь средней мозговой артерии (MCA), и регистрируют изменения церебрального кровотока (CBF) on-line системой PeriFlux 4001 Laser Doppler.
Артефактам показаний доплеровского лазера во время электрической стимуляции тригеминального ганглия, вследствие движений мышц препятствует болюсное внутривенное вливание нервно-мышечного блокатора панкуронийбромида (0,6 мг/кг i.v.).
Анестезию и нервно-мышечную блокаду сохраняют на протяжении всего эксперимента с инфузией пентобарбитала натрия и панкурония (12,5 мг/кг/ч + 2,4 мг/кг/ч, соответственно).
Протокол эксперимента: В конце хирургической операции имеет место 30-минутная пауза с целью стабилизировать измеряемые параметры.
Остаточный CBF увеличивают электрической стимуляцией прямоугольными импульсами длительностью 0,5 мс, 1-10 Гц, 0,5-1 мА в течение периодов 30 с. После двух средних pre-drug стимуляций вводят носитель или лекарственное средство.
Активные соединения уменьшают усиление кровотока, вызванное стимуляцией тройничного нерва.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ С ПОМОЩЬЮ СОЕДИНЕНИЙ 2,4-ПИРИМИДИНДИАМИНА | 2013 |
|
RU2659777C2 |
| ПРОИЗВОДНЫЕ 1,2-ДИ(ЦИКЛО)ЗАМЕЩЕННОГО БЕНЗОЛА | 2004 |
|
RU2340602C2 |
| ЗАМЕЩЕННЫЕ АМИНОАЛКИЛ- И АМИДОАЛКИЛБЕНЗОПИРАНПРОИЗВОДНЫЕ | 2006 |
|
RU2392276C2 |
| ПОЛИЗАМЕЩЕННЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ СЕРИНОВЫХ ПРОТЕАЗ | 2014 |
|
RU2678830C2 |
| МУЛЬТИЗАМЕЩЕННЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ТРОМБИНА | 2011 |
|
RU2639876C2 |
| ЗАМЕЩЕННЫЕ МЕТИЛФЕНИЛКЕТОНЫ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2007 |
|
RU2493149C2 |
| ИЗОИНДОЛИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ РАКА | 2009 |
|
RU2527952C2 |
| АНАЛОГИ НОЦИЦЕПТИНА | 2002 |
|
RU2426730C2 |
| АЛЬФА-АМИНОАМИДНЫЕ ПРОИЗВОДНЫЕ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ НИЖНИХ МОЧЕВЫВОДЯЩИХ ПУТЕЙ | 2005 |
|
RU2395504C2 |
Изобретение относится к новым 2-фенилэтиламинозамещенным производным карбоксамидов формулы (I)
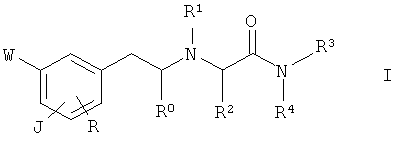
где J, W, R, R0, R1, R2, R3 и R4 имеют значения, такие, как определено в п.1 формулы изобретения, и их фармацевтически приемлемым солям, фармацевтическим композициям, содержащим их в качестве модуляторов натриевых и/или кальциевых каналов для предупреждения, ослабления и лечения широкого спектра патологий, включающих неврологические, психические, сердечно-сосудистые, воспалительные, офтальмологические, урологические и желудочно-кишечные заболевания. 2 н. и 20 з.п. ф-лы, 1 ил., 10 табл.
1. Соединение общей формулы I
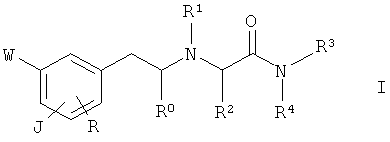
где (а)
J представляет собой группу А-[(СН2)n-О]r- в параположении по отношению к этиламиновому фрагменту, в которой n равно нулю или 1 и r равно 1;
А представляет собой трифторметил или фенил, необязательно замещенный галогеном;
W представляет собой (С1-С4)алкокси;
R представляет собой водород;
R0 представляет собой водород или (С1-С2) алкил;
R1 представляет собой водород; (С1-С4)алкил, необязательно замещенный гидроксигруппой; циклопропилметил; 2-пропин-1-ил; бензил, необязательно замещенный одной или двумя (С1-С2)алкоксигруппами в бензольном кольце; тиазолил; 5-6-членный насыщенный гетероциклил, содержащий атом азота, необязательно замещенный (С1-С2)алкильной группой; или гетероциклилметил, в котором гетероциклическая группа представляет собой 5-6-членный гетероциклил, содержащий 1-3 гетероатома, выбранных из азота, кислорода и серы, необязательно замещенный одной или двумя группами, выбранными из (С1-С2)алкила, гидроксиметила и (С1-С2)алкокси;
R2 представляет собой водород; (С1-С4)алкил или фенил;
R3 представляет собой водород или (С1-С4)алкил и
R4 представляет собой водород; (С1-С4)алкил, необязательно замещенный группой, выбранной из амино, (С1-С4) алкиламино, ди (С1-С4)алкиламино, имидазолила и пирролидинила, причем имидазолильная и пирролидинильная группы необязательно замещены (С1-С2)алкильной группой, или бензил или
R3 и R4 вместе с соседним атомом азота образуют пирролидинильное, морфолинильное или пиперазинильное кольцо, необязательно замещенное (С1-С2)алкильной группой;
или
(b)
J представляет собой водород;
W представляет собой группу А-[(СН2)n-О]r-, в которой n равно нулю, 1 или 2 и r равно нулю или 1;
А представляет собой (С1-С4)алкил; трифторметил;
циклопропил; циклопентил; фенил, необязательно замещенный группой, выбранной из галогена, метила, метокси, трифторметила, ацетиламино и диметиламинометила; тиенил, необязательно замещенный хлором; фуранил; изоксазолил, необязательно замещенный одной или двумя метальными группами; пиперидинил; морфолинил; пиридинил или пиримидинил, причем пиридинильное или пиримидинильное кольца необязательно замещены одной или двумя метоксигруппами;
R представляет собой водород или фтор;
R0 представляет собой водород или (С1-С2)алкил;
R1 представляет собой циклопропилметил; фуранилметил; тетрагидрофуранил или тетрагидрофуранилметил;
R2 представляет собой водород или (С1-С4)алкил;
R3 представляет собой водород или (С1-С4)алкил и
R4 представляет собой водород; (С1-С4)алкил, необязательно замещенный группой, выбранной из (С1-С4) алкокси, амино, (С1-С4) алкиламино и ди (C1-C4) алкиламино; или гетероциклил, где гетероциклил выбран из изоксазолила, пиразолила, имидазолила, тиазолила и 1,3,4-тиадиазолила и может быть необязательно замещен (С1-С2)алкильной группой; или
R3 и R4 вместе с соседним атомом азота образуют пирролидиновое кольцо; при условии, что когда А представляет собой (С1-С4)алкил, трифторметил, циклопропил или циклопентил, тогда r равно 1;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера либо в виде их смеси, и его фармацевтически приемлемые соли.
2. Соединение по п.1, группа (а), где
J представляет собой группу А-[(СН2)n-О]r- в параположении по отношению к этиламиновому фрагменту, в которой n равно 1 и r равно 1;
А представляет собой трифторметил, фенил или фенил, замещенный фтором или хлором;
W представляет собой метокси;
R представляет собой водород;
R0 представляет собой водород;
R1 представляет собой водород; (С1-С4)алкил; гидроксиэтил; циклопропилметил; 2-пропин-1-ил; бензил, необязательно замещенный одной или двумя метоксигруппами в бензольном кольце;
пиперидинил, необязательно замещенный метильной группой;
тиазолил или гетероциклилметил, где гетероциклическая группа выбрана из изоксазолила, необязательно замещенного метильной или метоксигруппой, имидазолила, необязательно замещенного метильной группой, фуранила, необязательно замещенного гидроксиметильной группой, тетрагидрофуранила, 1,2,3-тиадиазолила, пиразолила, необязательно замещенного одной или двумя метильными группами, пиридинила, необязательно замещенного метоксигруппой, тиенила и тиазолила;
R2 представляет собой водород; (С1-С4)алкил или фенил;
R3 представляет собой водород или (С1-С4)алкил; и
R4 представляет собой водород; (С1-С4)алкил, необязательно замещенный группой, выбранной из амино, диметиламино, имидазолила и пирролидинила, где пирролидинил необязательно замещен метильной группой, или бензил или
R3 и R4 вместе с соседним атомом азота образуют пирролидинильное, пиперазинильное или морфолинильное кольцо, необязательно замещенное метильной группой;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера либо в виде их смеси, и его фармацевтически приемлемые соли.
3. Соединение по п.1, группа (а), где
J представляет собой группу А-[(СН2)n-О]r- в параположении по отношению к этиламиновому фрагменту, в которой n равно 1 и r равно 1;
А представляет собой фенил или фенил, замещенный фтором;
W представляет собой метокси;
R представляет собой водород;
R0 представляет собой водород;
R1 представляет собой водород; (С1-С4)алкил;
циклопропилметил; бензил; или гетероциклилметил, где гетероциклическая группа выбрана из фуранила, тетрагидрофуранила и пиридинила, необязательно замещенного метоксигруппой;
R2 представляет собой водород; (С1-С4)алкил или фенил;
R3 представляет собой водород или (С1-С4)алкил и
R4 представляет собой водород; (С1-С4)алкил, необязательно замещенный группой, выбранной из амино, диметиламино и пирролидинил, где пирролидинил необязательно замещен метильной группой; или
R3 и R4 вместе с соседним атомом азота образуют пирролидинильное или морфолинильное кольцо;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера либо в виде их смеси, и его фармацевтически приемлемые соли.
4. Соединение по п.1, группа (b), где
J представляет собой водород;
W представляет собой группу А-[(СН2)nO]r-, в которой n равно нулю, 1 или 2; r равно нулю или 1;
А представляет собой (С1-С4)алкил; трифторметил;
циклопропил; циклопентил; фенил, необязательно замещенный группой, выбранной из фтора, хлора, метила, метокси, трифторметила, ацетиламино и диметиламинометила; тиенил, необязательно замещенный хлором; фуранил; изоксазолил, необязательно замещенный одной или двумя метильными группами;
пиперидинил; морфолинил; пиридинил или пиримидинил, причем пиридинильная и пиримидинильная группы необязательно замещены одной или двумя метоксигруппами;
R представляет собой водород или фтор;
R0 представляет собой водород;
R1 представляет собой циклопропилметил; фуранилметил; тетрагидрофуранил или тетрагидрофуранилметил;
R2 представляет собой водород или метил;
R3 представляет собой водород или (С1-С4)алкил и
R4 представляет собой водород; (С1-С4)алкил, необязательно замещенный группой, выбранной из метокси, амино, метиламино и диметиламино; изоксазолил, необязательно замещенный метильной группой, пиразолил; имидазолил; тиазолил; или 1,3,4-тиадиазолил;
или
R3 и R4 вместе с соседним атомом азота образуют пирролидинильное кольцо;
при условии, что когда А представляет собой (С1-С4)алкил, трифторметил, циклопропил или циклопентил, тогда r равно 1; в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера либо в виде их смеси, и его фармацевтически приемлемые соли.
5. Соединение по п.4, где
J представляет собой водород;
W представляет собой группу А-[(СН2)n-О]r-, в которой n равно 1 или 2; r равно 1;
А представляет собой (С1-С4)алкил; трифторметил;
циклопропил; циклопентил; фенил, необязательно замещенный группой, выбранной из фтора, хлора, метила, метокси и трифторметила; тиенил, необязательно замещенный хлором; изоксазолил, необязательно замещенный одной или двумя метильными группами; пиридинил; пиперидинил или морфолинил;
R представляет собой водород или фтор;
R0 представляет собой водород;
R1 представляет собой циклопропилметил; фуранилметил; тетрагидрофуранил или тетрагидрофуранилметил;
R2 представляет собой водород;
R3 представляет собой водород или (С1-С4)алкил и
R4 представляет собой водород; (С1-С4)алкил, необязательно замещенный группой, выбранной из метокси, амино, метиламино и диметиламино; изоксазолил, необязательно замещенный метильной группой; пиразолил; имидазолил; тиазолил или 1,3,4-тиадиазолил; или
R3 и R4 вместе с соседним атомом азота образуют пирролидинильное кольцо;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера либо в виде их смеси, и его фармацевтически приемлемые соли.
6. Соединение по п.4, где
J представляет собой водород;
W представляет собой группу А-[(СН2)n-О]r-, в которой n равно нулю; r равно 1;
А представляет собой циклопентил или фенил, необязательно замещенный фтором;
R представляет собой водород;
R1 представляет собой фуранилметил;
R2 представляет собой водород;
R3 представляет собой водород или (С1-С4)алкил и
R4 представляет собой водород или (С1-С4)алкил;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера либо в виде их смеси, и его фармацевтически приемлемые соли.
7. Соединение по п.4, где
J представляет собой водород;
W представляет собой группу А-[(СН2)n-О]r-, в которой n равно нулю; r равно нулю;
А представляет собой фенил, необязательно замещенный группой, выбранной из фтора, метокси, ацетиламино и диметиламинометила; тиенил; фуранил; изоксазолил, необязательно замещенный одной или двумя метильными группами; пиперидинил; пиридинил или пиримидинил, причем пиридинильная и пиримидинильная группы необязательно замещены одной или двумя метоксигруппами;
R представляет собой водород;
R0 представляет собой водород;
R1 представляет собой фуранилметил или тетрагидрофуранилметил;
R2 представляет собой водород;
R3 представляет собой водород или (С1-С4)алкил и
R4 представляет собой водород или (C1-C4)алкил;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера либо в виде их смеси, и его фармацевтически приемлемые соли.
8. Соединение по п.5, где
J представляет собой водород;
W представляет собой группу А-[(СН2)n-О]r-, в которой n равно 1; r равно 1;
А представляет собой (С1-С4)алкил;
R представляет собой водород;
R0 представляет собой водород;
R1 представляет собой фуранилметил или тетрагидрофуранилметил;
R2 представляет собой водород;
R3 представляет собой водород или (С1-С4)алкил и
R4 представляет собой (С1-С4)алкил;
в определенных случаях либо в виде индивидуального энантиомера или диастереоизомера либо в виде их смеси, и его фармацевтически приемлемые соли.
9. Соединение по п.1, выбранное из
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N-метилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]изобутиламино]-N-метилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](тетрагидрофуран-3-илметил)амино]-N-метилацетамида;
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N,N-диметилацетамида;
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметилпропионамида;
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметил-2-фенилацетамида;
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-1-(морфолин-4-ил)-2-фенилэтанона;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-1-(пирролидин-1-ил)этанона;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-(2-амино-2-метилпропил)ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-(2-диметиламиноэтил)ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-[2-(1-метилпирролидин-2-ил)этил]ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-этилацетамида;
2-[[2-[4-(бензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-метилацетамида;
2-[[2-(3-бензилоксифенил)этил](циклопропилметил)амино]-N-метилацетамида;
2-[[2-(3-бензилоксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида;
2-[[2-[3-(2-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-метилацетамида;
2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]ацетамида; (b)
2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(2-диметиламиноэтил)ацетамида;
2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(2-амино-2-метилпропил)ацетамида;
2-[[2-(3-бензилоксифенил)этил](циклопропилметил)амино]-N-метилпропионамида;
2-[[2-[3-метокси-4-(2,2,2-трифторэтокси)фенил]этил](циклопропилметил)амино]-N-метилацетамида;
2-[[2-(3′-фторбифенил-3-ил)этил](фуран-2-илметил)амино]-N-метилацетамида; 2-[[2-(3-бензилоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида;
2-[[2-(3-бутоксифенил)этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида;
2-[[2-(3-бутоксифенил)этил](фуран-2-илметил)амино]-N-метилацетамида;
2-[[2-[4-фтор-3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида;
2-[[2-[3-(2,2,2-трифторэтокси)фенил]этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида;
2-[[2-[3-(3,5-диметилизоксазол-4-ил)фенил]этил](тетрагидрофуран-3-ил)амино]-N,N-диметилацетамида;
2-[[2-[3-(пиперидин-1-ил)фенил]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-метилацетамида;
(S)-2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N-метил-4-метилвалерамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](фуран-3-илметил)амино]-N-метилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-этилацетамида и
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](6-метоксипиридин-3-илметил)амино]-N-метилацетамида;
в определенных случаях или в виде индивидуального энантиомера или в виде смеси энантиомеров, и его фармацевтически приемлемые соли.
10. Соединение по п.9, которое выбрано из
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]изобутиламино]-N-метилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](тетрагидрофуран-3-илметил)амино]-N-метилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N,N-диметилацетамида;
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N,N-диметил-2-фенилацетамида;
2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-1-(морфолин-4-ил)-2-фенилэтанона;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-1-(пирролидин-1-ил)этанона;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-(2-амино-2-метилпропил)ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-(2-диметиламиноэтил)ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-[2-(1-метилпирролидин-2-ил)этил]ацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-этилацетамида;
2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(2-диметиламиноэтил)ацетамида; 2-[[2-[3-(3-фторбензилокси)фенил]этил](фуран-2-илметил)амино]-N-(2-амино-2-метилпропил)ацетамида;
2-[[2-[3-бутоксифенил)]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](циклопропилметил)амино]-N-метилацетамида;
(S)-2-[2-[4-(3-фторбензилокси)-3-метоксифенил]этиламино]-N-метил-4-метилвалерамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](фуран-3-илметил)амино]-N-метилацетамида;
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил]бензиламино]-N-этилацетамида
и
2-[[2-[4-(3-фторбензилокси)-3-метоксифенил]этил](6-метоксипиридин-3-илметил)амино]-N-метилацетамида;
в определенных случаях или в виде индивидуального энантиомера или в виде смеси энантиомеров, и его фармацевтически приемлемые соли.
11. Соединение по п.10, которое представляет собой 2-[[2-(3-бутоксифенил)]этил](тетрагидрофуран-3-илметил)амино]-N,N-диметилацетамид и его фармацевтически приемлемые соли.
12. Соединение по п.11, которое представляет собой индивидуальный энантиомер, имеющий [α]D=-10°, с=0,1, МеОН (20°С), или индивидуальный энантиомер, имеющий [α]D=+10°, с=0,1, МеОН (20°С), или их смесь в любом соотношении, и его фармацевтически приемлемые соли.
13. Соединение по любому из пп.1-12, где фармацевтически приемлемые соли представляют собой гидрохлориды.
14. Соединение по любому из пп.1-12 для применения в качестве лекарственного средства, действующего как модулятор кальциевых и/или натриевых каналов против заболеваний, вызванных дисфункциями потенциалзависимых кальциевых и/или натриевых каналов.
15. Соединение по любому из пп.1-12 для применения в качестве лекарственного средства для лечения невропатической боли, хронической боли и/или острой боли.
16. Соединение по любому из пп.1-12 для применения в качестве лекарственного средства для лечения неврологических нарушений, таких, как головная боль.
17. Соединение по любому из пп.1-12 для применения в качестве лекарственного средства для лечения неврологических заболеваний, таких, как эпилепсия.
18. Соединение по любому из пп.1-12 для применения в качестве лекарственного средства для лечения когнитивных и/или психических расстройств.
19. Соединение, как в любом из пп.1-12, для применения в качестве лекарственного средства для лечения заболеваний, вызванных дисфункциями потенциалзависимых кальциевых и/или натриевых каналов, таких, как воспалительные процессы, влияющие на любые системы организма, заболевания мочеполового тракта и/или нейродегенеративные заболевания.
20. Соединение по любому из пп.1-12 для применения в качестве лекарственного средства, действующего как модулятор кальциевых и/или натриевых каналов, против заболеваний, вызванных дисфункциями потенциалзависимых кальциевых и/или натриевых каналов, отличающееся тем, что при дозировке указанного соединения, которая эффективна для лечения пациентов, страдающих заболеваниями, вызванными дисфункциями потенциалзависимых кальциевых и/или натриевых каналов, оно не обладает активностью в качестве ингибитора МАО или обладает существенно сниженной активностью в качестве ингибитора МАО.
21. Фармацевтическая композиция, действующая как модулятор кальциевых и/или натриевых каналов, содержащая соединение по любому из пп.1-13 в качестве активного ингредиента вместе с фармацевтически приемлемым эксципиентом.
22. Фармацевтическая композиция по п.21, содержащая дополнительное терапевтическое средство, выбранное из габапентина, L-допа, агониста допамина или ингибитора катехол-О-метилтрансферазы.
| Подъемник | 1987 |
|
SU1588704A1 |
| МЕТАЛЛОГАЛОГЕННАЯ ЛАМПА | 1991 |
|
RU2006978C1 |
| GB 586645 А, 26.03.1947 | |||
| WO 2004089353 А2, 21.10.2004 | |||
| WO 2005018627 A1, 03.03.2005 | |||
| WO 9935125 A1, 15.07.1999 | |||
| WO 9014334 A1, 29.11.1990 | |||
| WO 03091219 A1, 29.11.2003 | |||
| RU 9406144 A1, 27.09.1996. | |||

