Изобретение относится к области органической химии, конкретно к способу получения (4S,5S)-4,5-O-изопропилиденциклопент-2-ен-1-она формулы (I),
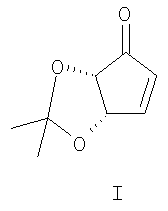
который находит применение в качестве хирального ключевого соединения в синтезе простагландинов [1], карбануклеозидов [2] и азасахаров [3].
Известен ряд способов получения соединения (I), ранние из которых можно разделить на 2 группы. Первая группа базировалась на использовании ахиральных исходных: подход из циклопентадиена [4] через (±)-(I) с последующим расщеплением с применением методики сульфоксииминного разрешения [5] и микробиологический подход [6] из толуола через хиральный цис-толуолдиол. Во второй группе использовались хиральные исходные - синтезы (I) и энт-(1) на основе D-изоаскорбиновой кислоты [7] и γ-лактона рибоновой кислоты [8].
К недостаткам 1-й группы способов получения соединения (I) следует отнести большую трудоемкость процесса сульфоксииминного разрешения на оптические антиподы [4] и падение воспроизводимости результатов на заключительной стадии циклизации в целевой (I) при «масштабировании» процесса [6]. Недостатки 2-й группы способов заключаются в использовании дорогостоящих и взрывоопасных реагентов [7-8].
Более эффективны подходы, исходящие из дешевых и доступных сахаров. Так, в трехстадийном синтезе (+)-(I) [9] метил-2,3-O-изопропилиден-β-D-рибофуранозид (II), легко получаемый реакцией доступной D-рибозы с диметоксипропраном в МеОН в присутствии НСlO4, окисляли 4-кратным избытком пиридинийхлорхромата (РСС) (кипячение в бензоле) в лактон (III). Последний после хроматографической очистки на колонке с силикагелем (SiO2) и взаимодействия с литиевым производным диметоксиметилфосфоната (ТГФ, -78°С) давал (+)-(I) с общим выходом 40%.
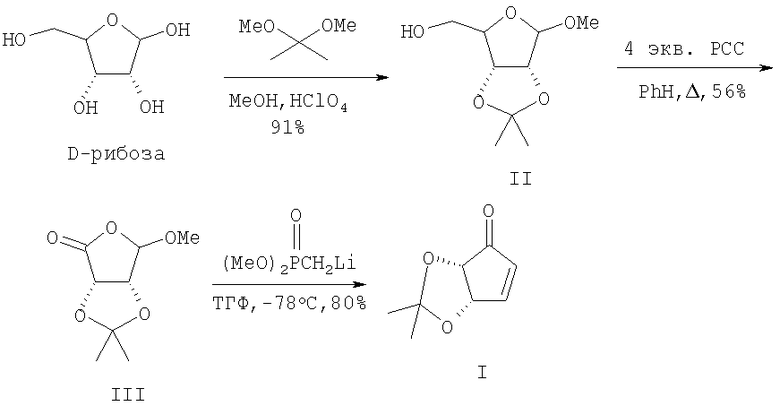
Несмотря на достаточно высокий общий выход циклопентенона (I) (40%) данная схема имеет ряд недостатков. Во-первых, это "капризные" трудновоспроизводимые стадии окисления спирта (II) до лактона (III) [используется большой избыток канцерогенного реагента (РСС)] и конденсации с литийпроизводным диметилметилфосфоната. Во-вторых, к недостаткам данного способа следует отнести необходимость использования на заключительной стадии [переход (III)→(I)] инертной атмосферы, низких температур, огне- и взрывоопасного н-BuLi и токсичного фосфорорганического реагента.
Следующий подход к (I) основан на использовании методологии метатезисного циклозамыкания подходящих диолефиновых субстратов, получаемых из сахаров [10].
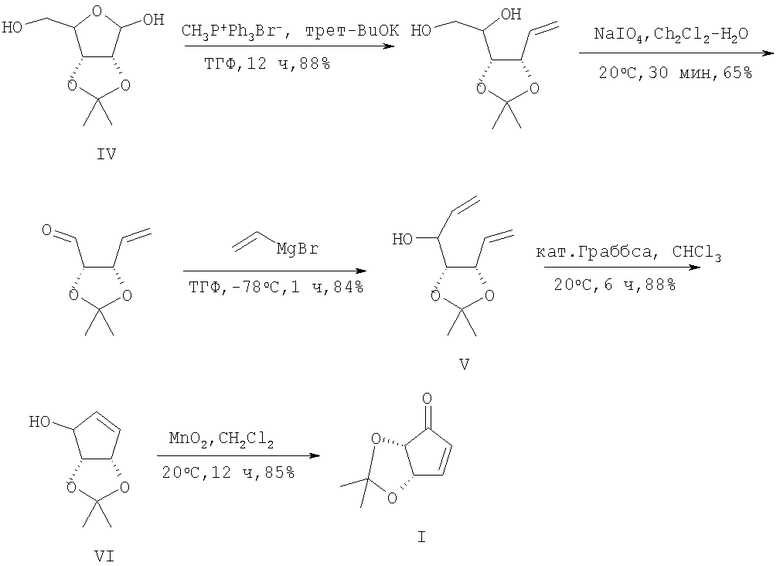
Так, авторы [10] в 3 стадии путем олефинирования по Виттигу метилентрифенилфосфораном легкодоступного ацетонида D-рибозы (IV), последующего окислительного расщепления NaIO4 в водном хлористом метилене и взаимодействия образующегося альдегида с винилмагнийбромидом (ТГФ, -78°С) получили α,ω-диеноацетонид (V), который в присутствии катализатора Граббса гладко трансформировался в циклопентенол (VI), окисленный далее MnO2 в (I). Общий выход целевого (I) в расчете на ацетонид (IV) составил 36%.
Основными недостатками данной схемы являются использование неудобных в технологии низких температур, необходимость применения инертной атмосферы и специальной аппаратуры, а также металлоорганических соединений (винилмагнийбромид) и нестабилизированных фосфорных илидов и исключительно дорогого рутениевого катализатора Граббса.
Ближайшим прототипом предлагаемого изобретения является способ получения циклопентенона (I) из метоксиацетонида D-рибозы (II) через метил-5-дезокси-4-метилен-2,3-O-изопропилиден-β-D-рибофуранозид (VII) [11]. Последний был получен из метоксиацетонида (II) в две стадии: иодирование системой иод-имидазол-трифенилфосфин с последующим дегидроиодированием под действием диазабициклоундецена (ДБУ) в кипящем бензоле. Катализируемое OsO4 периодатное расщепление (VII) приводило к лактону (III) с выходами 60-65%. Авторы отмечают, что заключительная стадия - циклизация лактона (III) под действием литийпроизводного диметилметилфосфоната, вопреки данным [9], протекает «капризно» и наилучший достигнутый выход не превышает 38%. Общий выход целевого циклопентенона (I) исходя из метоксиацетонида (II) составляет 17.7%.
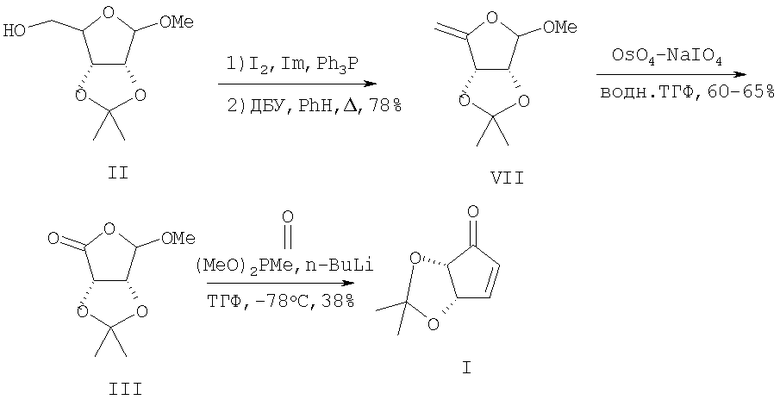
Недостатки данного метода включают использование на стадии окислительного расщепления двойной связи (VII) токсичного и дорогостоящего OsO4, а также отмеченные выше недостатки на переходе [(III)→(I)] в [9].
Задача, на решение которой направлено заявляемое техническое решение, заключается в упрощении процесса и повышении выхода целевого соединения.
По предлагаемому способу легко доступный из D-рибозы через метил-2,3-O-изопропилиден-β-D-рибофуранозид (II) метил-5-дезокси-4-метилен-2,3-О-изопропилиден-β-D-рибофуранозид (VII), согласно прототипу [11], иод гидроксилируют при комнатной температуре действием 1.5 эквивалентов I2 в растворе тетрагидрофуран-вода (3:1, v/v) в течение 5 минут. Взаимодействие полученных иодгидринов (VIII) и (IX) с 5-8 эквивалентами активированной цинковой пыли [12] при кипячении в бензоле приводит к циклопентенону (I) - продукту внутримолекулярной циклизации по Реформатскому, который выделяют путем перекристаллизации из петролейного эфира (т.кип. 40-70°С). Общий выход целевого циклопентенона (I) составляет 58-60% в расчете на енолэфир (VII), чистота - 97% (по данным ВЭЖХ). Общий выход целевого циклопентенона (I) исходя из метоксиацетонида (II) составляет 46-48.5%.
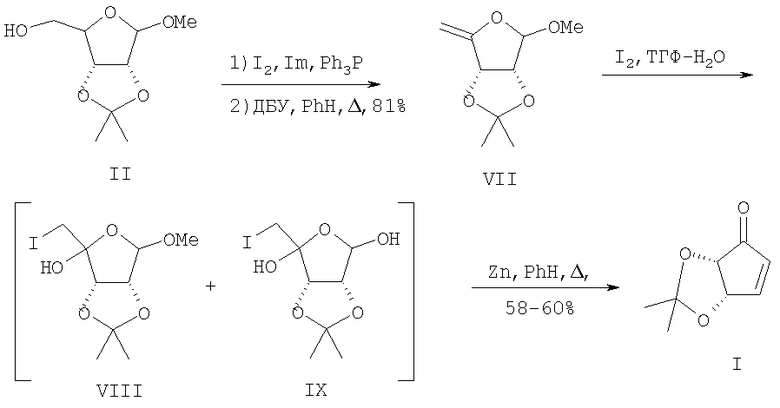
Преимущества предлагаемого метода, по сравнению с прототипом, состоят в исключении при осуществлении процесса малодоступных, дорогостоящих и токсичных реагентов, материало- и трудоемкой стадии хроматографической очистки промежуточных иодгидринов и конечного продукта и в более высоком общем выходе целевого продукта (46-48.5% против 17.7%) исходя из исходного метоксиацетонида (II).
Сущность изобретения подтверждается следующим примером.
Пример, а) Метил-5-дезокси-4-метилен-2,3-O-изопропилиден-β-D-рибофуранозид (VII). К смеси 2.00 г (9.78 ммоль) метил-2,3-O-изопропилиден-β-D-рибофуранозида (II), 3.20 г (12.22 ммоль) Ph3Р и 0.98 г (14.66 ммоль) имидазола в смеси 30 мл толуола и 5.0 мл ацетонитрила при 70°С добавляют порциями 3.12 г (19.5 ммоль) кристаллического иода. Реакционную массу перемешивают в течение 20 минут (ТСХ), добавляют этилацетат, промывают насыщенным раствором Nа2S2O3 и сушат Na2SO4. Остаток, полученный после упаривания раствора в вакууме, растворяют в 25 мл безводного бензола, добавляют 1.5 г ДБУ (10.75 ммоль) и перемешивают при кипении в течение 45 минут. Раствор концентрируют в вакууме водоструйного насоса, остаток перегоняют в вакууме. Получают 1.38 г (81.0%) енолэфира (VII), т.кип. 64-66°С (1 мм рт.ст.), Rƒ 0.22 (петролейный эфир - этилацетат, 9:1), [α]20 D +59.2° (с 1.15, СНСl3) (лит.89 [α]27d +68.2° (с 1.22, СНСl3). ИК-спектр, ν, см-1: 890 (С=СH2, 1060, 1085 (С-О-С), 1670, (С=СН2). Спектр ЯМР 1Н (СDСl3), δ, м.д. (J, Гц): 1.35 с (3Н, Me), 1.48 с (3Н, Me), 3.41 с (3Н, ОМе), 4.38 уш.с (1Н, Н5A), 4.49 д (1Н, Н3, 3J3,2 5.90), 4.59 уш.с (1Н, Н5B), 5.95 д (1Н, Н2, 3J2,3 5.90), 5.10 с (1Н, Н1). Спектр ЯМР 13С (СDСl3), δ, м.д.: 25.73 (Me), 26.73 (Me), 55.65 (ОМе), 78.69 (С3), 82.66 (С2), 88.70 (С5), 108.35 (С1), 113.20 (Сi-Pr), 161.23 (С4). Найдено, %: С 57.98; Н 7.39. С9Н14O4. Вычислено, %: С 58.05; Н 7.58.
б) (4S,5S)-4,5-O-изопропилиденциклопент-2-ен-1-он (I). К раствору 1.00 г (5.4 ммоль) енолэфира (VII) в 40 мл смеси ТГФ-Н2O (3:1) добавляют 2.04 г (8.0 ммоль) кристаллического иода и перемешивают 5 мин (ТСХ). Реакционную смесь концентрируют в вакууме водоструйного насоса и продукт экстрагируют СН2Сl2 (30 мл ×3). Объединенные органические экстракты промывают насыщенными растворами Nа2S2O3 (10 мл ×2), NaCl (10 мл ×2), сушат Na2SO4 и концентрируют в вакууме водоструйного насоса. Полученный остаток растворяют в 20 мл безводного бензола, добавляют 1.74 г (26.9 ммоль) цинковой пыли, предварительно активированной 2%-ной соляной кислотой, и перемешивают при кипячении до полной конверсии иодгидринов (VIII), (IX) (ТСХ, 1.5 ч). Реакционную смесь фильтруют через тонкий слой SiO2, концентрируют в вакууме водоструйного насоса и полученный остаток перекристаллизовывают из петролейного эфира (т.кип. 40-70°С). Получают 0.50 г (60%) циклопентенона (I). Белые кристаллы, т.пл. 66-67°С (лит. [7] т.пл. 68.5-69.5°С), Rƒ 0.22 (гексан - этилацетат, 7:3), [α]20 D +66.3° (с 1, СНСl3) (лит. [7] [α]20 D +69.1° (с 1.98, СНСl3)). ИК-спектр, ν, см-1: 1049, 1099 (С-О-С), 1583 (С=С), 1718 (С=О). Спектр ЯМР 1Н (СDСl3), δ, м.д.: 1.42 с (6Н, Me), 4.48 д (1Н, Н5, 3J5,4 5.5), 5.27 д.д (1Н, Н4, 3J4,3 2.2, 3J4,5 5.5), 6.22 д (1Н, Н2, 3J2,3 5.9), 7.10 д.д (1Н, Н3, 3J3,4 2.21, 3J3,2 5.9). Спектр ЯМР 13С (СDСl3), δ, м.д.: 26.33 (Me), 27.59 (Me), 76.69 (С2), 78.78 (С3), 115.69 (Сi-Pr), 134.49 (С5), 159.83 (С4), 203.17 (С=О). Найдено, %: С 62.28; Н 6.59. С8Н10О3. Вычислено, %: С 62.33; Н 6.54.
Литература
1. Johnson C.R., Chen Y.F. Development of a triply convergent aldol approach to prostanoids. // J. Org. Chem. - 1991. - Vol.56. - N.10. - P.3344-3351.
2. Chu C.K., Jin Y.H., Baker R.O., Huggins J. Antiviral activity of cyclopententenyl nucleosides against orthopox viruses (smallpox, monkeypox and cowpox) // Bioorg. and Med. Chem. Lett. - 2003. - Vol.13. - N.1. - P.9-12.
3. Tanaka K., Taniguchi Т., Ogasawara K. 7,7-Dimethyl-6,8-dioxabicyclo[3.3.0]oct-3-en-2-one as a synthetic equivalent of ketodicyclopentadiene: a new route to (-)-physostigmine, (-)-physovenine, and (-)-aphanorphine // Tetrahedron Lett. - 2001. - Vol.42. - N.6. - P.1049.
4. Johnson C.R., Penning T.D. Triply convergent synthesis of (-)-prostaglandin E2 methyl ester // J. Am. Chem. Soc. - 1988. - Vol.110. - N.14. - P.4726-4735.
5. Johnson C.R., Penning T.D. Triply convergent synthesis of (-)-prostaglandin Е2 // J. Am. Chem. Soc. - 1986. - Vol.108. - N.18. - P.5655-5656.
6. Hudlicky Т., Luna H., Barbieri G., Kwart L D. Enantioselective synthesis through Microbial Oxidation of arenes. Efficient preparation of terpen and prostaglandin synthons // J. Am. Chem. Soc. - 1988. - Vol.110. - N.14. - P.4735-4741.
7. Choi W.J, Park J.G, Yoo S.J, Kim H.O., Moon H.R, Chun M.W, Jung Y.H., Jeong L.S. Syntheses of D- and L-cyclopentenone derivatives using Ring-Closing Metathesis: versatile intermediates for the synthesis of D- and L-carbocyclic nucleosides // J. Org. Chem. - 2001. - Vol.66. - N.19. - P.6490-6494.
8. Belanger P., Prasit P. Carbocycles from carbohydrates: a simple route to an enantimerically pure prostaglandin intermediate // Tetrahedron Lett. - 1988. - Vol.29. - N.43. - P.5521-5524.
9. Ali S.M., Ramesh K., Borhart R.T. Efficient enantioselective synthesis of carbocyclic nucleoside and prostaglandin synthons // Tetrahedron Lett. - 1990. - V.31. - N.11. - P.1509-1512.
10. Moon H.R., Choi W.J., Kim H.O., Jeong L.S. Improved and alternative synthesis of D- and L-cyclopentenone derivatives, the versatile intermediates for the synthesis of carbocyclic nucleosides // Tetrahedron Asymmetry - 2002. - Vol.13. - N.11. - P.1189-1193.
11. Hill J. M., Hutchinson E. J., Le Gradnd D. M., Roberts S. M. Preparation of neplanocin-A from D-ribose by chemoenzymic methods. // J. Chem. Soc., Perkin Trans. I - 1994. - N.11. - P.1483-1487.
12. Физер Л., Физер M. Реагенты для органического синтеза. - M.: Мир, 1971. - Т.4. - С.285.
Изобретение относится к органической химии, конкретно к способу получения (4S,5S)-4,5-O-изопропилиденциклопент-2-ен-1-она, который заключается во взаимодействии метил-5-дезокси-4-метилиден-2,3-изопропилиден-β-D-рибофуранозида, полученного иодированием метил-2,3-О-изопропилиден-β-D-рибофуранозида комплексом иод-трифенилфосфин в присутствии имидазола с последующим дегидроиодированием с помощью диазабициклоундецена, с 1.5 эквивалентами I2 в растворе тетрагидрофуран-вода (3:1, v/v) при комнатной температуре в течение 5 минут с последующей обработкой смеси образующихся иодгидринов 5-8 эквивалентами цинковой пыли при кипячении в бензоле и выделением целевого продукта перекристаллизацией из петролейного эфира (т.кип. 40-70°С). Этот способ позволяет исключить использование малодоступных, дорогостоящих и токсичных реагентов, низких температур, материало- и трудоемкой стадии хроматографической очистки промежуточных иодгидринов и конечного продукта и повысить общий выход целевого продукта (46-48.5%) (против 17.7% в прототипе) исходя из исходного метил-2,3-O-изопропилиден-β-D-рибофуранозида. Полученное соединение может быть использовано в качестве хирального синтона в направленном синтезе биологически активных соединений (простагландинов, карбануклеозидов, циклопентаноидных антибиотиков, азасахаров и др.).
Способ получения (4S,5S)-4,5-O-изопропилиденциклопент-2-ен-1-она формулы (I)
из метил-2,3-O-изопропилиден-β-D-рибофуранозида через метил-5-дезокси-4-метилиден-2,3-изопропилиден-β-D-рибофуранозид путем иодирования метил-2,3-O-изопропилиден-β-D-рибофуранозида комплексом иод-трифенилфосфин в присутствии имидазола с последующим дегидроиодированием с помощью диазабициклоундецена, отличающийся тем, что метил-5-дезокси-4-метилиден-2,3-изопропилиден-β-D-рибофуранозид иодгидроксилируют действием 1,5 эквивалентов I2 в растворе тетрагидрофуран-вода (3:1, v/v) при комнатной температуре в течение 5 мин с последующей обработкой смеси образующихся иодгидринов 5-8 эквивалентами цинковой пыли при кипячении в бензоле и выделением целевого продукта перекристаллизацией из петролейного эфира (т.кип. 40-70°С).
| Hill J.M | |||
| et al, J.Chem.Soc., Perkin Trans | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WO 2006122128 A2, 16.11.2006 | |||
| DE 10063410 A1, 28.06.2001 | |||
| RU 2000129142 A, 27.01.2003. | |||

