Изобретение относится к органической химии, конкретно к способу получения энантиомерных производных лактона Кори формул (-)- и ((+)-Ia-c).
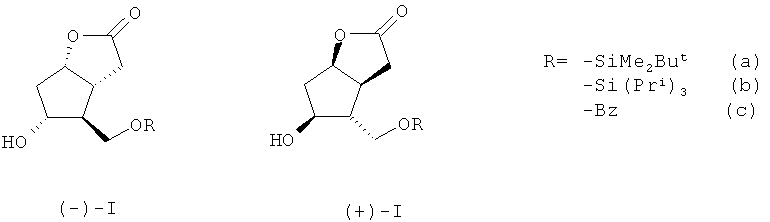
Получаемые из (±)-лактондиола (II) через ((-)-I) (R=SiR'3, Bz, Tr и др.) производные (III) зарекомендовали себя как базовые блок-синтоны в синтезе энантиомерно чистых простагландинов и аналогов [1-3].
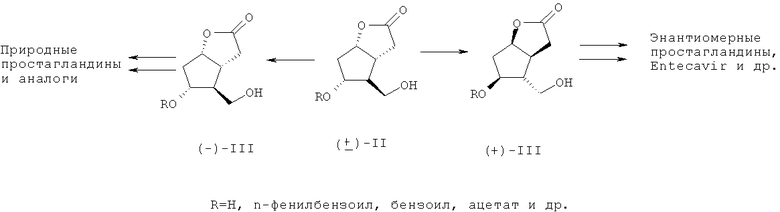
В частности, производные ((-)-III) использованы в синтезе простагландинов PGF2α и PGE2 [4, 5], клопростенола (синхронизация родовой деятельности животных) [6], латанопроста (лечение глаукомы) [7], кикапроста (аналога простациклина, для лечения сердечно-сосудистых заболеваний) [1] и мизо-простола (лечение язвенных заболеваний, а также родовспоможение и аборты у женщин) [8].
Лактондиол ((+)-III), получаемый из (+)-лактондиола (II) через ((+)-I) (R=SiR'3, Bz, Tr и др.), может быть использован в синтезе энантиомеров природных простагландинов для испытания их биологической активности [9], а также лекарственного препарата «Энтекавир», который проявляет высокую активность против вируса гепатита B [10].
Для получения энантиомерно чистых блоков ((-)-I) и ((+)-I) и далее соответственно ((-и ((+)-III) обычно исходят из индивидуальных энантиомеров лактондиола ((-)-II) и ((+)-II) соответственно [11], что затруднительно из-за их высокой стоимости. Синтез рацемического 5-гидрокси-4-(гидрокси-метил) гекса-гидро-2H-циклопента[b]фуран-2-она ((±)-II) детально описан в литературе [12]. Энантиомерно чистые блоки ((-)-I) и ((+)-I), на наш взгляд, практичнее синтезировать исходя именно из рацемического лактондиола ((±)-II). Так, авторы работы [13] осуществили синтез ((-)-Ia) энзиматическим гидролизом ацетильного производного ((±)-II) в присутствии липазы PS (Amano, Pseudomonas sp.). Для этого было проведено силилирование рацемического лактондиола ((±)-II) по первичной спиртовой группе с использованием 1.1 эквивалента диметил-трет-бутилхлоросилана в хлористом метилене в присутствии 2.2 эквивалентов имидазола при комнатной температуре в течение 3 ч. Далее полученное силильное производное ((±)-IIa) было обработано 1.1 эквивалента хлористого ацетила в безводном пиридине в течение суток. Полученное ацетильное производное рацемического ((±)-II) было помещено в децимолярный фосфатный буферный раствор со значением pH 7.6. После добавления каталитических количеств энзима - липазы PS (Amano, Pseudomonas sp.) смесь была нагрета до 30°C и выдержана при этой температуре в течение двух суток (48 ч). После стандартной обработки был выделен желаемый ((-)-Ia) с выходом 50% и энантиомерной чистотой >99%. Параллельно с такими же значениями выхода и энантиомерной чистоты было выделено ацетильное производное ((+)-II). Несмотря на высочайшую энантиомерную тиомерную чистоту продуктов ((-)-Ia) и ацетильного производного ((+)-Ia), а также почти количественные выходы для обоих соединений (50% для каждого из продуктов), данный метод является очень трудоемким, так как предполагает использование энзиматического гидролиза. Кроме того, следует отметить длительность всей процедуры получения ((-)-Ia) по данному методу, которая составляет в среднем около 4 суток.
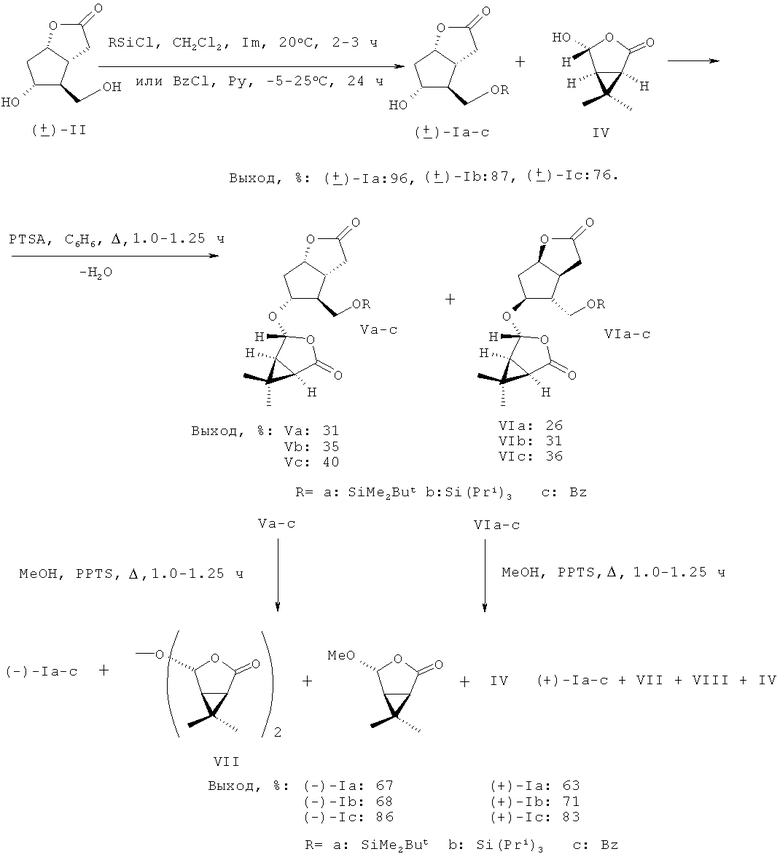
Задача, на решение которой направлено заявляемое изобретение, заключается в «энантиоконвергентном» получении энантиомерно чистых лактонов (-)- и ((+)-Ia-с) с хорошим выходом в пересчете на исходный рацемический лактондиол ((±)-II). По предлагаемому способу для получения (-)- и ((+)-Ia,b) рацемический лактондиол ((±)-II), полученный по методике, описанной в работе [12], взаимодействует с 1.1 эквивалента триалкилхлорсилана, выбранного из диметил-трет-бутилхлорсилана или триизопропилхлорсилана в безводном хлористом метилене в присутствии 2.20-2.60 эквивалента имидазола (Im) при комнатной температуре в течение 2-3 часов. В случае получения ((±)-Ib) рацемический 5-гидрокси-4-(гидроксиметил)гексагидро-2H-циклопента[b]фуран-2-он взаимодействует с 1.1 эквивалента триизопропил-хлорсилана в присутствии каталитических количеств N,N-диметиламинопиридина (DMAP).
Для получения ((±)-Ic) рацемический лактондиол ((±)-II) реагирует с 1.1 эквивалента хлористого бензоила (BzCl) в безводном пиридине (Ру) при -5-(+)25°С в течение 24 ч с образованием соответствующего производного ((±)-Ic). Производные ((±)-Ia), ((±)-Ib), ((±)-Ic) образуются с выходами - 96%, 87% и 76%, соответственно. Далее эти производные вводят в реакцию ацетализации с 1.1 эквивалента хирального полуацилаля (IV), синтезированного согласно методике [14]. Ацетализацию осуществляют кипячением в безводном бензоле в течение 1.0-1.25 ч в присутствии катализатора пара-толуолсульфокислоты (PTSA) в количестве 0.015-0.023 эквивалента и получают легко разделимые простой колоночной хроматографией на силикагеле (SiO2) соответствующие диастереомерные эфиры (Va-c) и (VIa-c).
Метанолиз эфиров (Va-c) и (VIa-c) кипячением в метаноле при катализе пиридиниевой солью пара-толуолсульфокислоты (PPTS), добавляемой в реакционную среду в количестве 0.008-0.020 эквивалента, в течение 1.0-1.25 ч дает желаемые монозамещенные производные ((-)-Ia-с) и ((+)-Ia-с) высокой энантиомерной чистоты (≥97%) с выходом 20-25% в пересчете на исходный рацемический лактондиол ((±)-II). При этом образующиеся при метанолизе эфиров (Va-c) и (VIa-c) целевые лактоны ((-)-Ia-с) и ((+)-Ia-с) очень легко отделимы хроматографией на SiO2 от побочных продуктов - димера (VII), основного побочного продукта метанолиза - метилового эфира (VIII) и источника хиральности - ацилаля (IV).
Преимущества предлагаемого способа:
1) хорошие суммарные выходы (20-25%) целевых лактонов ((-)-Ia-c) и ((+)-Ia-c) с высокой энантиомерной чистотой (≥97%);
2) одностадийное получение дифференцированно блокированных производных ((±)-Ia-c) с хорошими выходами от 76% для ((±)-Ic) до 96% для ((±)-Ia);
3) легкость разделения диастереомерных эфиров (Va-c) и (VIa-c) простой колоночной хроматографией на силикагеле;
4) возможность почти количественной рекуперации источника хиральности ацилаля (IV) после проведения метанолиза.
Сущность изобретения подтверждается следующими примерами.
Пример 1. Синтез (±)-Ia
К непрерывно перемешиваемой суспензии 225 мг (1.48 ммоль) рацемического лактондиола ((±)-II) в 5 мл безводного CH2Cl2 добавляют 222 мг (3.26 ммоль) имидазола и затем 246 мг (1.63 ммоль) трет-бутилдиметил-хлорсилана (ClSiMe2But). Смесь перемешивают в течение 2 ч до исчезновения исходного лактондиола ((±)-II) (контроль методом ТСХ). После окончания реакции белый творожистый осадок гидрохлорида имидазола отделяют на фильтре Шота и промывают CH2Cl2 (2×2 мл). Органическую фазу промывают водой и насыщенным водным раствором хлорида натрия. Хлористый метилен упаривают на роторном испарителе, остаток хроматографируют на SiO2 системой петролейный эфир - этилацетат, 4:1. Получают 359 мг (96%) соединения (±)-Ia.
(±)-(3aRS,4SR,5RS,6aSR)-4-({[трет-бутил(диметил)силил]окси}метил)-5-гидрокси-гексагидро-2H-циклопента[b]фуран-2-он ((±)-Ia). Бесцветные кристаллы, т.пл. 96-98°C; Rf 0.20 (гексан-этилацетат, 7:3). ИК-спектр, ν, см-1 (суспензия в нуйоле): 3385, 2944, 2899, 1759, 1465, 1255, 1226, 1109, 1089, 1008. Спектр ЯМР 1H, δ, м.д.: 0.02 с (6H, SiMe2), 0.85 с (9Н, Si-CMe3), 1.97 м (2Н, 4-Н, 6-Hendo), 2.38 м (1H, 6-Hexo), 2.49 д.д (1H, J=17.8, 2.0 Гц, 3-Hendo), 2.61 м (2Н, 3a-H, OH), 2.75 д.д (1-H, J=17.8, 10.0 Гц, 3-Hexo), 3.57 д.д (1Н, J=10.0, 6.5 Гц, CH2O), 3.65 д.д (1H, J=10.0, 5.0 Гц, CH2O), 4.09 к (1Н, J=6.4 Гц, 5-Н), 4.89 т.д (1H, J=6.9, 2.8 Гц, 6a-H). Спектр ЯМР 13С, δ, м.д.: -1.05 (SiMe2), 18.07 (Si-CMe3), 25.73 (Si-CMe3), 35.35 (C3), 39.54 (C3a), 40.72 (C6), 55.26 (C4), 63.83 (CH2O), 75.38 (C5), 83.99 (C6a), 177.41 (C2). Масс-спектр (APCI), m/z (%): 287 [MH+] (100), 269 [M-H2O]+ (18.9), 233 (14.4), 231 (9.0).
Пример 2. Синтез (±)-Ib
К непрерывно перемешиваемой суспензии 207 мг (1.20 ммоль) рацемического лактондиола ((±)-II) в 5 мл безводного CH2Cl2 добавляют 180 мг (2.64 ммоль) имидазола, затем 0.23 мл (1.32 ммоль) триизопропилхлорсилана (ClSi(Pri)3) и через 30 мин 0.020 ммоль N,N-диметил-2-аминопиридина (DMAP). Смесь перемешивают в течение 3 ч до исчезновения исходного лактондиола ((±)-II) (контроль методом ТСХ). После окончания реакции белый творожистый осадок гидрохлорида имидазола отделяют на фильтре Шотта и промывают СН2С2 (2×3 мл). Органическую фазу промывают водой и насыщенным водным раствором NaCl. Хлористый метилен упаривают на роторном испарителе, остаток хроматографируют на SiO2 системой петролейный эфир-этилацетат, 4:1. Получают 344 мг (87%) соединения (±)-Ib.
(±)-(3aRS,4SR,5RS,6aSR)-5-гидрокси-4-{[(триизопропилсилил)окси]метил}гексагидро-2H-циклопента[b]фуран-2-он ((±)-Ib). Бесцветные кристаллы, т.пл. 42-43°C; Rf 0.33 (гексан-этилацетат, 7:3). ИК-спектр, ν, см-1 (суспензия в нуйоле): 3448, 3433, 2941, 2866, 1772, 1475, 1176, 1107, 1037 см-1. Спектр ЯМР 1H, δ, м.д.: 1.03 с (3H, Si-CH), 1.05 с (18Н, 6Ме), 1.98 м (2Н, 4-Н, 6-Hendo), 2.34 уш.с (1H, OH), 2.47 м (2Н, 3-Hendo, 6-Hexo), 2.62 м (1H, 3a-H), 2.77 д.д (1Н, J=17.8, 10.0 Гц, 3-Hexo), 3.68 д.д (1Н, J=10.0, 6.6 Гц, CH2O), 3.81 д.д (1H, J=10.0, 5.0 Гц, CH2O), 4.15 к (1H, J=6.4 Гц, 5-H), 4.91 т.д (1Н, J=6.9, 2.8 Гц, 6a-H). Спектр ЯМР 13С, δ, м.д.: 11.71 (Si-CHMe2), 17.90 (Si-СНМе2), 35.26 (C3), 39.45 (C3a), 40.75 (С6), 55.31 (С4), 64.49 (CH2O), 75.62 (С5), 83.75 (C6a), 177.15 (С2). Масс-спектр (APCI), m/z (%): 329 [MH+] (47.7), 311 [М-H2O]+ (100).
Пример 3. Синтез (±)-Ic
К перемешиваемому при 0°C раствору 1000 мг (5.8 ммоль) рацемического лактондиола (II) в 10 мл безводного пиридина в атмосфере аргона прикапывают 0.74 мл (6.38 ммоль) хлористого бензоила в 5 мл пиридина. Температуру реакционной массы постепенно доводят до комнатной. Затем выдерживают смесь 24 ч. После удаления пиридина азеотропной отгонкой с гептаном остаток откачивают на вакуумном насосе, затем растворяют в 15 мл хлороформа (CHO3), органическую фазу промывают водой, сушат сульфатом магния (MgSO4), фильтруют, хлороформ упаривают. Сырой продукт хроматографируют на SiO2 в системе петролейный эфир-этилацетат, 2:1. Получают 1220 мг (76%) соединения (±)-Ic.
(±)-[(3aRS,4SR,5RS,6aSR)-5-гидрокси-2-оксогексагидро-2H-циклопента[b]фуран-4-ил]метил бензоат ((±)-Ic). Бесцветные кристаллы, т.пл. 84-86°C; Rf 0.40 (гексан-этилацетат, 1:1). ИК-спектр, ν, см-1 (пленка): 3385, 3034, 1749, 1703, 1454, 1375, 1355, 1278, 1116, 1101, 1037, 709. Спектр ЯМР 1H, δ, м.д.: 2.09 м (2Н, OH, 6-Hendo), 2.27 м (1H, 4-H), 2.46 м (1Н, 6-Hexo), 2.61 д.д (1Н, J=17.6, 1.9 Гц, 3-Hendo), 2.75 м (1Н, 3a-H), 2.87 д.д (1H, J=17.6, 9.9 Гц, 3-Hexo), 4.19 м (1H, 5-H), 4.30 д.д (1H, J=11.6, 5.8 Гц, CH2O), 4.43 д.д (1H, J=11.6, 6.1 Гц, CH2O), 4.98 т.д (1Н, J=6.9, 2.5 Гц, 6a-H), 7.46 м (2Н, Ph), 7.59 м (1Н, Ph), 8.00 д (2H, J=7.2 Гц, Ph). Спектр ЯМР 13С, δ, м.д.: 35.48 (C3), 40.13 (C3a), 40.40 (С6), 53.00 (С4), 64.66 (CH2O), 74.14 (С5), 83.64 (C6a), 128.59 (2CPh m), 129.55 (lCph i), 129.55 (2CPh o), 133.42 (1CPh p), 166.69 (PhCO), 177.40 (C2). Масс-спектр (APCI), m/z (%): 277 [MH+] (66.7), 259 [M-H2O]+ (100).
Пример 4. Синтез Va и VIa
К раствору 300 мг (1.049 ммоль) моносилильного производного (Ia) и 172 мг (1.21 ммоль) полуацилаля (IV) в 20 мл безводного бензола добавляют 2.5 мг (0.015 ммоль) пара-толуолсульфокислоты (PTSA). Смесь кипятят с насадкой Дина-Старка в течение 1.0 ч. По окончании реакции (контроль методом ТСХ) бензол отгоняют на роторном испарителе, остаток растворяют в 15 мл CHCl3, органическую фазу промывают водой, насыщенным водным раствором NaCl, хлороформ упаривают. Остаток хроматографируют на SiO2 в системе петролейный эфир - этилацетат, 5:1. Получают 134 мг (31%) диастереомера (Va) и 112 мг (26%) диастереомера (VIa).
(-)-(3aR,4S,5R,6aS)-4-({[трет-бутил(диметил)силил]окси}метил)-5-{[(1S,2R,5R)-6,6-диметил-4-оксо-3-оксабицикло[3.1.0]гекс-2-ил]окси}гекса-гидро-2H-циклопента[b]фуран-2-он (Va). Бесцветные кристаллы, mp=116-117°C; Rf 0.35 (гексан-этилацетат, 7:3); [α]22 D-93.65 (с 3.50, CHCl3). ИК-спектр, ν, см-1 (суспензия в нуйоле): 1768, 1460, 1377, 1352, 1188, 1149, 1118, 1103, 1080, 1056. Спектр ЯМР 1H, δ, м.д.: 0.02 с (6Н, SiMe2), 0.86 с (9Н, Si-СМе3), 1.13 с (3H, Me), 1.15 с (3H, Me), 1.97 д (1H, J=5.5 Гц, 1'-H), 2.01 д (1H, J=5.8 Гц, 5'-Н), 2.11-2.30 м (3H, 4-Н, 6-Hendo, 6-Нехо), 2.54 м (1Н, 3-Hendo), 2.84 м (2Н, 3a-Н, 3-Hexo), 3.56 м (2Н, CH2O), 4.26 м (1H, 5-Н), 5.02 т (1Н, J=5.8 Гц, 6a-H), 5.15 с (1H, 2'-Н). Спектр ЯМР 13С, δ, м.д.: -0.35 (SiMe2), 14.98 (Me), 18.04 (Si-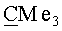
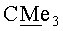
(-)-(3aS,4R,5S,6aR)-4-({[трет-бутил(диметил)силил]окси} метил)-5-{[(1S,2R,5R)-6,6-диметил-4-оксо-3-оксабицикло[3.1.0]гекс-2-ил]окси}гекса-гидро-2H-циклопента[b]фуран-2-он (VIa). Бесцветные кристаллы, mp=112-113°C; Rf 0.25 (гексан-этилацетат, 7:3); [α]22D-37.95 (с 3.50, CHCl3). ИК-спектр, ν, см-1 (суспензия в нуйоле): 1772, 1463, 1379, 1188, 1170, 1151, 1119, 1105, 1081, 1057. Спектр ЯМР 1H, δ, м.д.: 0.04 с (6Н, SiMe2), 0.88 с (9Н, Si-СМе3), 1.14 с (3H, Me), 1.16 с (3H, Me), 1.99 д (1H, J=5.8 Гц, 1'-H), 2.02 д (1Н, J=5.2 Гц, 5'-Н), 2.09 м (1H, 4-H), 2.20-2.40 м (2Н, 6-Hendo, 6-Hexo), 2.45 д.д (1H, J=17.8, 2.0 Гц, 3-Hendo), 2.67 м (1H, 3a-H), 2.82 д.д (1H, J=17.8, 10.1 Гц, 3-Hexo), 3.52 д.д (1H, J=10.2, 5.5 Гц, CH2O), 3.61 д.д (1Н, J=10.5, 5.0 Гц, CH2O), 4.18 м (1H, 5-Н), 4.94 т.д (1H, J=6.3, 1.8 Гц, 6a-H), 5.14 с (1H, 2'-Н). Спектр ЯМР 13С, δ, м.д.: -1.02 (SiMe2) 15.03 (Me), 18.13 (Si-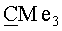
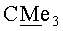
Пример 5. Синтез Vb и VIb
К раствору 300 мг (0.91 ммоль) моносилильного производного (Ib) и 149 мг (1.05 ммоль) полуацилаля (IV) в 20 мл безводного бензола добавляют 2.5 мг (0.015 ммоль) PTSA. Смесь кипятят с насадкой Дина-Старка в течение 1.0 ч. По окончании реакции (контроль методом ТСХ) бензол отгоняют на роторном испарителе, остаток растворяют в 10 мл CHCl3, органическую фазу промывают водой, насыщенным водным раствором NaCl, хлороформ упаривают. Остаток хроматографируют на SiO2 в системе петролейный эфир - этилацетат, 5:1. Получают 145 мг (35%) диастереомера (Vb) и 128 мг (31%) диастереомера (VIb).
(-)-(3aR,45S,5R,6aS)-5-{[(1S,2R,5R)-6,6-диметил-4-оксо-3-оксабицикло[3.1.0]гекс-2-ил]окси}-4-{[(триизопропилсилил)окси]метил}гекса-гидро-2H-циклопента[b]фуран-2-он (Vb). Бесцветные кристаллы, mp=87-88°C; Rf 0.43 (гексан-этилацетат, 7:3); [α]20 D-106.80 (с 1.50, CHCl3). ИК-спектр, ν, см-1 (суспензия в нуйоле): 2852, 1768, 1462, 1377, 1352, 1168, 1116, 1099, 1058, 1039. Спектр ЯМР 1H, δ, м.д.: 1.02 с (3H, Si-CH), 1.03 с (18Н, 6Ме), 1.13 с (3H, Me), 1.15 с (3H, Me), 1.97 д (1H, J=5.5 Гц, 1'-H), 2.01 д (1H, J=5.8 Гц, 5'-Н), 2.14-2.32 м (3H, 4-Н, 6-Hendo, 6-Hexo), 2.55 д (1Н, J=15.3 Гц, 3-Hendo), 2.79-2.97 м (2Н, 3a-H, 3-Нехо), 3.67 м (2Н, CH2O), 4.30 м (1H, 5-H), 5.04 т (1H, J=5.8 Гц, 6a-H), 5.16 с (1Н, 2'-Н). Спектр ЯМР 13С, δ, м.д.: 11.52 (Si-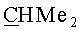
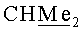
(-)-(3aS,4R,5S,6aR)-5-{[(1S,2R,5R)-6,6-диметил-4-оксо-3-оксабицикло[3.1.0]гекс-2-ил]окси}-4-{[(триизопропилсилил)окси]метил}гексагидро-2H-циклопента[b]фуран-2-он (VIb). Бесцветное масло, Rf 0.38 (гексан-этилацетат, 7:3); [α]26 D-19.60 (с 0.475, CHCl3). ИК-спектр, ν, см-1 (пленка): 2866, 1770, 1462, 1380, 1344, 1166, 1118, 1107, 1070, 1041. Спектр ЯМР 1H, 5, м.д.: 1.02 с (3H, Si-CH), 1.03 с (18Н, 6Ме), 1.12 с (3H, Me), 1.13 с (3H, Me), 1.97 д (1H, J=5.7 Гц, 1'-H), 2.00 д (1Н, J=5.7 Гц, 5'-Н), 2.11 м (1H, 4-Н), 2.25 м (1Н, 6-Hendo), 2.36 м (1H, 6-Hexo), 2.45 д.д (1Н, J=17.3, 1.2 Гц, 3-Hendo), 2.74 м (1Н, 3a-H), 2.82 д.д (1H, J=17.3, 10.2 Гц, 3-Hexo), 3.61 д.д (1H, J=10.2, 5.8 Гц, CH2O), 3.70 д.д (1H, J=10.2, 5.0 Гц, CH2O), 4.22 м (1H, 5-H), 4.93 т.д (1H, J=6.5, 2.1 Гц, 6a-H), 5.14 с (1H, 2'-Н). Спектр ЯМР 13С, δ, м.д.: 11.81 (Si-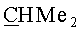
Пример 6. Синтез Vc и VIc
К раствору 400 мг (1.45 ммоль) монобензоильного производного (Ic) и 237 мг (1.61 ммоль) полуацилаля (IV) в 20 мл безводного бензола добавляют 4.0 мг (0.023 ммоль) PTSA. Смесь кипятят с насадкой Дина-Старка в течение 1.0 ч. По окончании реакции (контроль методом ТСХ) бензол отгоняют на роторном испарителе, остаток растворяют в 15 мл CHCl3, органическую фазу промывают водой, насыщенным водным раствором NaCl, хлороформ упаривают. Остаток хроматографируют на SiO2 в системе хлороформ-метанол, 50:1. Получают 232 мг (40%) диастереомера (Vc) и 209 мг (36%) диастереомера (VIc).
(-)-((3aR,4S,5R,6aS)-5-{[(1S,2R,5R)-6,6-диметил-4-оксо-3-оксабицикло[3.1.0]гекс-2-ил]окси}-2-оксогексагидро-2H-циклопента[b]фуран-4-ил)метил бензоат (Vc). Бесцветные кристаллы, mp=135-136°C; Rf 0.48 (гексан-этилацетат, 1:1); [α]20 D-122.50 (с 5.80, CHCl3). ИК-спектр, ν, см-1 (суспензия в нуйоле): 3072, 2852, 1768, 1716, 1463, 1455, 1379, 1336, 1279, 1186, 1171, 1099, 1071, 1042. Спектр ЯМР 1H, δ, м.д.: 1.12 с (3H, Me), 1.14 с (3H, Me), 1.96 д (1H, J=5.5 Гц, 1'-H), 2.01 д (1H, J=5.7 Гц, 5'-Н), 2.21 м (1Н, 6-Hexo), 2.35 м (1Н, 6-Hendo), 2.60 м (2Н, 4-Н, 3-Hendo), 2.86 м (2Н, 3a-Н, 3-Hexo), 4.23 м (2Н, CH2O), 4.36 м (1H, 5-H), 5.07 т (1H, J=5.7 Гц, 6a-H), 5.16 с (1H, 2'-Н), 7.43 м (2Н, Ph), 7.56 м (1H, Ph), 7.97 д (2Н, J=7.2 Гц, Ph). Спектр ЯМР 13C, δ, м.д.: 15.03 (Me), 24.55 (C6'), 25.26 (Me), 29.71 (C1'), 35.20 (С5'), 35.76 (C3), 36.96 (С6) 40.28 (C3a), 52.52 (С4), 64.57 (CH2O), 80.70 (С5), 83.97 (C6a), 99.08 (С2'), 128.59 (2CPh m), 129.55 (1CPh i) 129.55 (2CPh o), 133.37 (lCPh p), 166.24 (PhCO), 173.00 (С4'), 176.76 (С2). Масс-спектр (APCI), m/z (%): 401 [MH+] (100).
(-)-((3aS,4R,6aR)-5-{[(1S,2R,5R)-6,6-диметил-4-оксо-3-оксабицикло[3.1.0]гекс-2-ил]окси}-2-оксогексагидро-2H-циклопента[b]фуран-4-ил)метил бензоат (VIc). Бесцветное вязкое масло, Rf 0.44 (гексан-этилацетат, 1:1); [α]20 D - 34.77 (с 6.75, CHCl3). ИК-спектр, ν, см-1 (пленка): 3072, 2852, 1772, 1720. Спектр ЯМР 1H, 8, м.д.: 1.09 с (3H, Me), 1.11 с (3H, Me), 1.93 д (1Н, J=5.7 Гц, 1'-H), 1.99 д (1Н, J=5.7 Гц, 5'-Н), 2.26 м (1H, 6-Hendo), 2.39 м (1Н, 4-H), 2.52 м (1H, 6-Hexo), 2.54 д.д (1H, J=17.8, 1.6 Гц, 3-Hendo), 2.72 м (1H, 3a-H), 2.86 д.д (1Н, J=17.8, 9.8 Гц, 3-Hexo), 4.17 к (1H, J=5.9 Гц, 5-Н), 4.34 д (2Н, J=6.1 Гц, CH2O), 4.96 т.д (1H, J=6.7, 2.4 Гц, 6a-H), 5.13 с (1H, 2'-Н), 7.45 м (2Н, Ph), 7.59 м (1Н, Ph), 7.99 д (2Н, J=7.6 Гц, Ph). Спектр ЯМР 13C, δ, м.д.: 14.97 (Me), 24.45 (C6'), 25.26 (Me), 29.88 (C1'), 35.18 (С5'), 35.32 (C3), 39.37 (Сб), 39.72 (C3a), 50.85 (С4), 64.10 (CH2O), 81.56 (С5), 83.18 (C6a), 100.86 (С2'), 128.63 (2CPh m), 129.54 (1CPh i), 129.54 (2CPh o), 133.50 (1CPh p), 166.29 (PhCO), 172.82 (C4'), 176.22 (C2). Масс-спектр (APCI), m/z (%): 401 [MH+] (100).
Пример 7. Синтез энантиомера (-)-Ia
Раствор 130 мг (0.32 ммоль) диастереомера (Va) и 2 мг (0.008 ммоль) пиридиниевой соли пара-толуолсульфокислоты (PPTS) в 5 мл метанола кипятят до полной конверсии эфира (Va) (контроль методом ТСХ). Метанол упаривают, остаток растворяют в 3 мл CHCl3, органическую фазу промывают водой и насыщенным водным раствором NaCl. Хлороформ отгоняют, остаток хроматографируют на SiO2 в системе петролейный эфир - этилацетат, 3:1. Получают 61 мг (67%) (-)-энантиомера (Ia).
(-)-(3aR,4S,5S,6aS)-4-)-4-({[трет-бутил(диметил)силил]окси}метил)-5-гидроксигексагидро-2H-циклопента[b]фуран-2-он(-)-Ia. [α]20 D-14.7 (с 1.05, CHCl3).
Пример 8. Синтез энантиомера (+)-Ia
Раствор 100 мг (0.24 ммоль) диастереомера (VIa) и 2 мг (0.008 ммоль) пиридиниевой соли PPTS в 5 мл метанола кипятят до полной конверсии эфира (Via). Метанол упаривают, остаток растворяют в 3 мл CHCl3, органическую фазу промывают водой и насыщенным водным раствором NaCl. Хлороформ отгоняют, остаток хроматографируют на SiO2 в системе петролейный эфир-этилацетат, 3:1. Получают 44 мг (63%) (+)-энантиомера (Ia).
(+)-(3aS,4R,5S,6aR)-4-)-4-({[трет-бутил(диметил)силил]окси}метил)-5-гидрокси-гексагидро-2H-циклопента[b]фуран-2-он (+)-Ia. [α]20 D+14.9 (с 1.05, CHCl3).
Пример 9. Синтез энантиомера (-)-Ib
Раствор 130 мг (0.29 ммоль) диастереомера (Vb) и 3 мг (0.012 ммоль) PPTS в 6 мл метанола кипятят до полной конверсии эфира (Vb) (контроль методом ТСХ). Метанол упаривают, остаток растворяют в 3 мл CHCl3, органическую фазу промывают водой и насыщенным водным раствором NaCl. Хлороформ отгоняют, остаток хроматографируют на SiO2 в системе петролейный эфир-этилацетат, 3:1. Получают 64 мг (68%) (-)-энантиомера (Ib).
(-)-(3aR,4S,5R,6aS)-5-гидрокси-4-{[(триизопропилсилил)окси]метил}гексагидро-2H-циклопента[b]фуран-2-он(-)-Ib. [α]24 D-10.1 (с 1.725, CHCCl3).
Пример 10. Синтез энантиомера (+)-1b
Раствор 100 мг (0.22 ммоль) диастереомера (VIb) и 3 мг (0.012 ммоль) PPTS в 6 мл метанола кипятят до полной конверсии эфира (VIb) (контроль методом ТСХ). Метанол упаривают, остаток растворяют в 3 мл CHCl3, органическую фазу промывают водой и насыщенным водным раствором NaCl. Хлороформ отгоняют, остаток хроматографируют на SiO2 в системе петро-лейный эфир - этилацетат, 3:1. Получают 51 мг (71%) (+)-энантиомера (Ib).
(+)-(3aS,4R,5S,6aR)-5-гидрокси-4-{[(триизопропилсилил)окси]метил}гексагидро-2H-циклопента[b]фуран-2-он (+)-Ib. 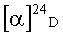
Пример 11. Синтез энантиомера (-)-Ic
Раствор 220 мг (0.55 ммоль) диастереомера (Vc) и 3 мг (0.012 ммоль) PPTS в 10 мл метанола кипятят до полной конверсии эфира (Vc) (контроль методом ТСХ). Метанол упаривают, остаток растворяют в 7 мл CHCl3, органическую фазу промывают водой и насыщенным водным раствором NaCl. Хлороформ отгоняют, остаток хроматографируют на SiO2 в системе петролейный эфир-этилацетат, 2:1. Получают 126 мг (83%) (-)-энантиомера (Ic).
(-)-[(3aR,4S,5R,6aS)-5-гидрокси-2-оксогексагидро-2H-циклопента[b]фуран-4-ил]метил бензоат (-)-Ic. 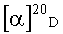
Пример 12. Синтез энантиомера (+)-Ic
Раствор 200 мг (0.50 ммоль) диастереомера (VIc) и 3 мг (0.012 ммоль) PPTS в 10 мл метанола кипятят до полной конверсии эфира (VIc) (контроль методом ТСХ). Метанол упаривают, остаток растворяют в 7 мл CHCl3, органическую фазу промывают водой и насыщенным водным раствором NaCl. Хлороформ отгоняют, остаток хроматографируют на SiO2 в системе петролейный эфир - этилацетат, 2:1. Получают 119 мг (86%) (+)-энантиомера (Ic).
(+)-[(3aS,4R,5S,6aR)-5-гидрокси-2-оксогексагидро-2H-циклопента[b]фуран-4-ил]-метил бензоат (+)-Ic. 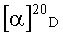
Литература
1. Collins P.W., Djuric S.W. Synthesis of Therapetically useful Prostaglandin and Prostacyclin Analogs. Chem. Rew. 1993, 93(4), p 1533.
2. Das S., Chandrasekhar S., Yadav G.S., Gree R. Recent Developments in the Synthesis of Prostaglandins and Analogues. Chem.Rev. 2007,107, 3286.
3. Durand Т., Guj A., Henry O., Roland A., Bernard S., Fangour S., Vidal J.-P., Rossi J.-C. Total Synthesis of iso-, neuro- and phytoprostanes new insight in lipid chemisrtry. Chem. Phys. Lipids. 2004, 128, 15.
4. Johuson F., Paul K.G., Favara D., Ciabatti R., Guzzi U. Prostaglandins 2. Synthesis of Prostaglandin F2α in optically active form from Chiral Precursors. J.Am.Chem.Soc. 1982, 704,2190.
5. Cooper EX., Vankee E.W. Enantiomeric Prostaglandins. J. Am. Chem. Soc. 1974, 96, 293.
6. Beeley N.R.A., Peel R., Sutherland J.K. Synthesis of PGF2α and Cloprostenol. Tetrahedron. 1981, 37, Supplement №1, p.441.
7. Martynow J.G., Jóźwik J., Szelejewski W., Achmatowicz O., Kutner A., Wisniewski K., Winiarski J., Zegrocka-Stendel O., Golibiewski P. A New Synthetic Approach to High-Purity (15R)-Latanoprost. Eur. J. Org. Chem. 2007, 689.
8. Park H., Lee V.S., Nam K.H., Lee K.-J., Jung S.H. Synthesis of Prostaglandins II. Convenient Synthesis of Misoprostol. Bull. Korean Chem. Soc. 1993, 14, №1, 2.
9. Cooper E.L., Vankee E.W. J. Am. Chem. Soc. 1974, 96, 5876.
10. Lai C.-L., Yuen M.-F. The Saga of entecavir. Hepatology International. 2009, 3, 1936.
11. Tömösközi L., Gruber L., Kovacs C., Szekely J., Simonidesz V. Regiospecific Prince reaction, a new way prostaglandin. Tetrahedron Lett. 1976, 50, 4639.
12. Толстиков Г.А., Мифтахов M.C., Валеев Ф.А., Востриков Н.С., Ахметвалеев P.P. 7α-Формилокси-6β-формилоксиметилен-цис-2-оксабицикло[3.3.0]октан-3-он как полезный синтон для простагландинов. ЖОрХ. 1984, 20, 1672.
13. Sugahara Т., Satoh I., Yamada О., Takano S. Efficient enzymatic preparation of (+)- and (-)-Corey Lactone derivatives. Chem. Pharm. Bull 1991, 39, 2758.
14. Галин Ф.З., Куковинец O.C., Шерешовец B.B., Сафиуллин Р.Л., Куковинец А.Г., Кабальнова Н.Н., Касрадзе В.Г., Зарипов Р.Н., Каргапольцева Т.А., Кашина Ю.А., Толстиков Г.А. Синтез 4α-гидрокси-6,6-диметил-3-оксабицикло[3.1.0]гекс-2-она из (+)-3-карена. ЖОрХ, 1996, 32, 1482.
Изобретение относится к области органической химии, конкретно к способу получения энантиомерных производных лактона Кори формул (-)- и ((+)-Ia,b), заключающийся во взаимодействии рацемического 5-гидрокси-4-(гидроксиметил)гексагидро-2H-циклопента[b]фуран-2-она с триалкил-хлорсиланом, с последующей ацетализацией полученных производных с (-)-(1R,4R,5S)-4-гидрокси-6,6-диметил-3-оксабицикло[3.1.0]гексан-2-оном при кипячении в безводном бензоле в присутствии катализатора пара-толуолсульфокислоты, с образованием разделимых колоночной хроматографией на силикагеле диастереомерных эфиров, метанолиз которых проводят кипячением в метаноле в присутствии катализатора пиридиниевой соли пара-толуолсульфокислоты в течение 1-1.25 ч. Заявленный способ позволяет получить энантиомерно чистые целевые соединения ((-)-Ia-с) и ((+)-Ia-с) с суммарным выходом 20-25% в пересчете на исходный (±)--лактондиол (II) и энантиомерной чистотой ≥97%. 2 н. и 1 з.п. ф-лы, 12 пр.
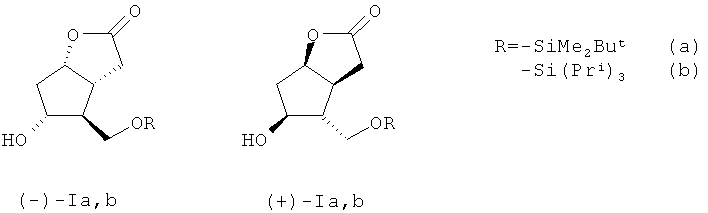
1. Способ получения энантиомерных производных лактона Кори формул (-)- и ((+)-Ia,b)
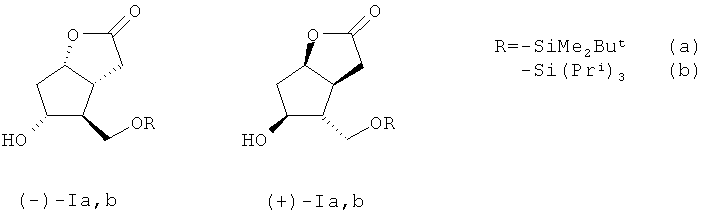
заключающийся во взаимодействии рацемического 5-гидрокси-4-(гидрокси-метил)гексагидро-2H-циклопента[b]фуран-2-она с 1,1 эквивалента триалкил-хлорсилана, выбранного из диметил-трет-бутилхлорсилана или триизопропилхлорсилана в безводном хлористом метилене в присутствии 2,20-2,60 эквивалента имидазола при комнатной температуре в течение 2-3 ч с последующей ацетализацией полученных производных ((±)-Ia,b) с 1.1 эквивалента (-)-(1R,4R,5S)-4-гидрокси-6,6-диметил-3-оксабицикло[3.1.0]гексан-2-она при кипячении в безводном бензоле в присутствии 0,015-0,023 эквивалента катализатора пара-толуолсульфокислоты в течение 1,0-1,25 ч с образованием разделимых колоночной хроматографией на силикагеле диастереомерных эфиров (Va,b) и (VIa,b)
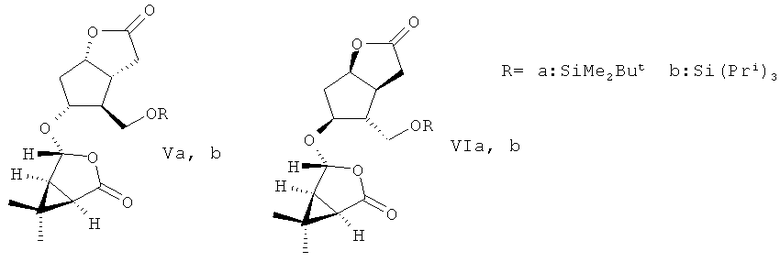
метанолиз которых кипячением в метаноле в присутствии 0,008-0,020 эквивалента катализатора пиридиниевой соли пара-толуолсульфокислоты в течение 1-1,25 ч приводит к образованию целевых продуктов, выделяемых колоночной хроматографией на силикагеле.
2. Способ по п.1, отличающийся тем, что взаимодействие рацемического 5-гидрокси-4-(гидроксиметил)гексагидро-2H-циклопента[b]фуран-2-она с 1,1 эквивалента триизопропилхлорсилана протекает в присутствии каталитических количеств N,N-диметиламинопиридина.
3. Способ получения энантиомерных производных лактона Кори формул (-)- и ((+)-Ic)
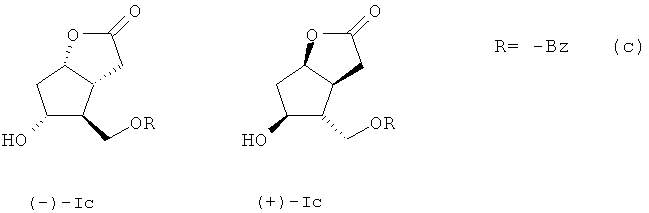
заключающийся в бензоилировании рацемического 5-гидрокси-4-(гидрокси-метил)гекса-гидро-2Н-циклопента[b]фуран-2-она
взаимодействием его с 1,1 эквивалента хлористого бензоила в безводном пиридине при температуре -5-(+)25°С в течение 24 ч с последующей ацетализацией полученного производного ((±)-Ic) кипячением его с 1,1 эквивалента (-)-(1R,4R,5S)-4-гидрокси-6,6-диметил-3-окса-бицикло[3.1.0]гексан-2-она в безводном бензоле в присутствии 0,015-0,023 эквивалента катализатора пара-толуолсульфокислоты в течение 1,0-1,25 ч с образованием разделимых колоночной хроматографией на силикагеле диастереомерных эфиров (Vc) и (VIc)
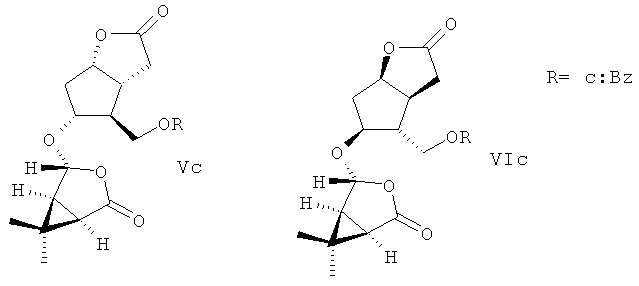
метанолиз которых кипячением в метаноле в присутствии 0,008-0,020 эквивалента катализатора пиридиниевой соли пара-толуолсульфокислоты в течение 1-1,25 ч приводит к образованию целевых продуктов, выделяемых колоночной хроматографией на силикагеле.
| Устройство для измерения моментов сил упругости в звеньях прокатного стана | 1972 |
|
SU501310A1 |
| JP 4267891 А, 24.09.1992 | |||
| Востриков Н.С | |||
| и др | |||
| Практичный вариантоптического разрешения производных лактондиола Кори, XIX Менделеевский съезд по общей и прикладной химии, Волгоград, 25-20 сентября 2011 г., Тезисы докладов | |||
| Химия и технология материалов, включая наноматериалы, т.2, с.227 (подписано в печать 10.08.2011.) | |||

