4В настоящем изобретении предлагаются органические соединения, способы их получения и применение в качестве лекарственных средств.
Одним объектом изобретения является соединение формулы I
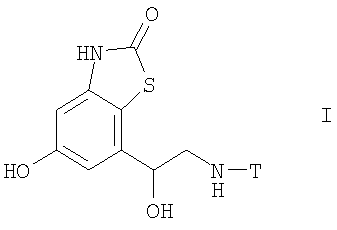
в свободной форме или в форме соли или сольвата, где
Т означает водород или C1-С10алкил, необязательно замещенный в одном, двух или трех положениях группой С1-С10алкокси, -NR1R2, 5- или 6-членным гетероциклическим циклом, содержащим в цикле по меньшей мере один гетероатом, выбранный из группы, включающей атомы азота, кислорода и серы, или С3-С15карбоциклической группой, причем указанная С3-С15карбоциклическая группа необязательно замещена в одном, двух или трех положениях группой галоген, С1-С10алкил, С3-С10циклоалкил, галоген(С1-С10)алкил, -NR3R4, 5- или 6-членным гетероциклическим циклом, содержащим в цикле по меньшей мере один гетероатом, выбранный из группы, включающей атомы азота, кислорода и серы, или группой С1-С10алкокси, необязательно замещенной в одном, двух или трех положениях С6-С10арилом,
или Т означает С3-С15карбоциклическую группу, необязательно замещенную в одном, двух или трех положениях группой галоген, C1-С10алкил, С3-С10циклоалкил, галоген(С1-С10)алкил, -NR5R6, 5- или 6-членным гетероциклическим циклом, содержащим в цикле по меньшей мере один гетероатом, выбранный из группы, включающей атомы азота, кислорода и серы, или группой С1-С10алкокси, необязательно замещенной в одном, двух или трех положениях С1-С4алкилом или С6-С10арилом, а
R1, R2, R3, R4, R5 и R6 независимо означают водород, С1-С10алкил, C1-С10алкокси, С3-С10циклоалкил или С6-С10арил.
Термины, используемые в описании, имеют следующие значения.
Термин "необязательно замещенный в одном, двух или трех положениях", используемый в описании, означает группу, замещенную в одном, двух или трех положениях любым одним из указанных ниже радикалов или их комбинацией.
Термин "галоген", используемый в описании, означает химический элемент, принадлжащиий к 17 группе (ранее группа VII) Периодической таблицы элементов, например фтор, хлор, бром или иод. Предпочтительным галогеном является хлор.
Термин "C1-С10алкил", используемый в описании, означает алкил с прямой или разветвленной цепью, содержащий от 1 до 10 атомов углерода. Если Т означает С1-С10алкил, то предпочтительно этот радикал означает С1-С8алкил, прежде всего н-пропил, изопропил, н-бутил, втор-бутил, -С(СН3)2С2Н5, -СН(СН3)С3Н7 или -СН(СН3)СН2С(СН3)3. Если Т означает С5-С10карбоциклическую группу, замещенную в одном, двух или трех положениях С1-С8алкилом, то C1-С10алкил предпочтительно означает С1-С4алкил, прежде всего этил или втор-бутил. Если любой из R1, R2, R3, R4, R5 и R6 означает С1-С10алкил, предпочтительно этот радикал означает С1-С4алкил, прежде всего метил.
Термин "С1-С10алкокси", используемый в описании, означает алкокси с прямой или разветвленной цепью, содержащий от 1 до 10 атомов углерода. Если Т означает C1-С10алкил, замещенный в одном, двух или трех положениях С3-С15карбоциклической группой, которая замещена в одном, двух или трех положениях группой C1-С10алкокси, то C1-С10алкокси предпочтительно означает С1-С4алкокси, прежде всего метокси или н-бутокси. Если Т означает С5-С15карбоциклическую группу, замещенную в одном, двух или трех положениях группой C1-С10алкокси, то C1-С10алкокси предпочтительно означает С1-С4алкокси, прежде всего этокси. Если любой из R1, R2, R3, R4, R5 и R6 означает C1-С10алкокси, то предпочтительно это С1-С4алкокси.
Термин "С3-С10циклоалкил", используемый в описании, означает циклоалкил, содержащий 3-10 атомов углерода в цикле, например моноцикличекую группу, такую, как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил или циклодецил, или бициклическую группу, такую, как бициклогептил или бициклооктил. Предпочтительно С3-С10циклоалкил означает С3-С6циклоалкил, прежде всего циклопентил или циклогексил.
Термин "галоген(С1-С10)алкил", используемый в описании, означает C1-С10алкил, указанный выше, замещенный одним или более атомами галогена, предпочтительно одним, двумя или тремя атомами галогена. Предпочтительно галоген(С1-С10)алкил означает фтор(С1-С4)алкил.
Термин "С6-С10арил", используемый в описании, означает одновалентную карбоциклическую ароматическую группу, содержащую 6-10 атомов углерода, например моноциклическую группу такую, как фенил, или бициклическую группу такую, как нафтил. Предпочтительно С6-С10арил означает С6-С8арил, прежде всего фенил.
Термин "С3-С15карбоциклическая группа", используемый в описании, означает карбоциклическую группу, содержащую 3-15 атомов углерода в цикле, например моноциклическую группу, ароматическую или неароматическую, такую, как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил или фенил, или бициклическую группу такую, как бициклооктил, бициклононил, бицилодецил, инданил или инденил.
Если Т означает C1-С10алкил, замещенный в одном, двух или трех положениях С5-С15карбоциклической группой, то С5-С15карбоциклическая группа предпочтительно означает С5-С10карбоциклическую группу, прежде всего моноциклическую группу такую, как фенил или циклогексил. Если Т означает С5-С15карбоциклическую группу, то предпочтительно этот радикал означает С5-С10карбоциклическую группу, прежде всего моноциклическую неароматическую группу такую, как циклопентил, или бициклическую группу такую, как инданил.
Термин "5- или 6-членный гетероциклический цикл, содержащий в цикле по меньшей мере один гетероатом, выбранный из группы, включающей атомы азота, кислорода и серы", используемый в описании, означает, например, пиррол, пирролидин, пиразол, имидазол, триазол, тетразол, фуран, тиадиазол, изотиазол, тиофен, оксадиазол, пиридин, оксазол, изоксазол, пиразин, пиридазин, пиримидин, пиперазин, морфолино, триазин, оксазин или тиазол. Предпочтительные 5- или 6-членные гетероциклические циклы включают ненасыщенные циклы такие, как пиридин, фуран и тиофен.
Термин "сольват", используемый в описании, означает комплекс, включающий соединение по настоящему изобретению и одну или более молекул фармацевтически приемлемого растворителя, например этанола. Термин "гидрат" используется в том случае, если растворителем является вода.
Если не указано иное, подразумевается, что термин "включать" или его варианты, такие как "включает" или "включающий", используемые в описании и в пунктах формулы изобретения, означает включение указанного целого числа или стадии, или группы целых чисел, или стадий, а не исключение любого такого целого числа или стадии, или группы целых чисел, или стадий.
Предпочтительные соединения по настоящему изобретению включают соединения формулы I в свободной форме, в форме соли или сольвата, где Т означает C1-С10алкил, необязательно замещенный в одном, двух или трех положениях группой -NR1R2 или С5-С15карбоциклической группой, причем указанная С5-С15карбоциклическая группа необязательно замещена в одном, двух или трех положениях группой галоген, -NR3R4 или C1-С10алкокси, или Т означает С5-С15карбоциклическую группу, необязательно замещенную в одном, двух или трех положениях группой C1-С10алкил, С3-С10циклоалкил, -NR5R6 или группой C1-С10алкокси, необязательно замещенной в одном, двух или трех положениях группой С6-С10арил, а
R1, R2, R3, R4, R5 и R6 независимо означают C1-С10алкил или С6-С10арил.
Более предпочтительные соединения по настоящему изобретению включают соединения формулы I, где
Т означает C1-С8алкил, необязательно замещенный в одном положении группой -NR1R2 или С5-С10карбоциклической группой, причем указанная С5-С10карбоциклическая группа необязательно замещена в одном или двух положениях группой галоген, -NR3R4 или С1-С4алкокси,
или Т означает С5-С10карбоциклическую группу, необязательно замещенную в одном или двух положениях группой С1-С8алкил, С3-С10циклоалкил, -NR5R6 или С1-С4алкокси, необязательно замещенной в одном положении группой С6-С8арил, прежде всего фенил, а
R1, R2, R3, R4, R5 и R6 независимо означают С1-С4алкил или С6-С8арил, прежде всего фенил.
Соединения формулы I могут образовывать кислотно-аддитивные соли, прежде всего фармацевтически приемлемые кислотно-аддитивные соли. Фармацевтически приемлемые кислотно-аддитивные соли соединения формулы I включают соли неорганических кислот, например галогенводородных кислот, таких как фтористоводородная кислота, хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота и соли органических кислот, например алифатических монокарбоновых кислот, таких как муравьиная кислота, уксусная кислота, трифторуксуная кислота, пропионовая кислота и масляная кислота, алифатических гидроксикислот, таких как молочная кислота, лимонная кислота, винная кислота или яблочная кислота, дикарбоновых кислот, таких как малеиновая кислота или янтарная кислота, ароматических карбоновых кислот, таких как бензойная кислота, пара-хлорбензойная кислота, дифенилуксусная кислота, пара-бифенилбензойная кислота или трифенилуксусная кислота, ароматических гидроксикислот, таких как орто-гидроксибензойная кислота, пара-гидроксибензойная кислота, 1-гидроксинафталин-2-карбоновая кислота или 3-гидроксинафталин-2-карбоновая кислота, коричных кислот, таких как 3-(2-нафталинил)пропеновая кислота, пара-метоксикоричная кислота или пара-метилкоричная кислота, и сульфоновых кислот, таких, как метансульфоновая кислота или бензолсульфоновая кислота. Указанные соли получают из соединений формулы I по известным методикам.
Соединения формулы I могут существовать в несольватированных и сольватированных формах. Фармацевтически приемлемые сольваты включают гидраты и сольваты, в которых растворитель, который использовался для кристаллизации, является замещенным изотопами, например D2O, d6-ацетон или d6-ДМСО.
Соединения формулы I включают по меньшей мере один асимметический атом углерода и, следовательно, могут существовать в виде индивидуальных оптически активных изомеров или в виде их смесей, например в виде рацемических или диастереомерных смесей. Настоящее изобретение включает индивидуальные оптически активные R и S изомеры, а также их смеси, например рацемические или диастереомерные смеси. Указанные изомеры разделяют известными методами, например фракционной кристаллизацией или хроматографией на колонке.
Наиболее предпочтительными соединеними по изобретению являются соединения, описанные ниже в разделе Примеры.
В настоящем изобретении предлагается также способ получения соединений формулы I в свободной форме, в форме соли или сольвата. Указанный способ включает:
(1) (А) взаимодействие соединения формулы II
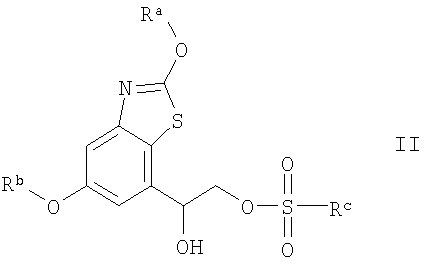
где Ra и Rb означают защитные группы, a Rc означает С1-С4алкил или С6-С10арил, с соединением формулы III

где Т имеет значения, указанные выше, или
(В) взаимодействие соединения IIA
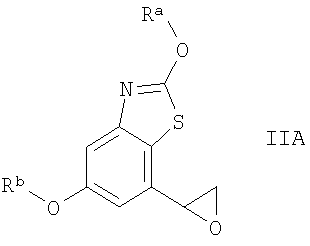
где Ra и Rb означают защитные группы, с соединением формулы III, где Т имеет значения, указанные выше,
(2) удаление защитных групп, и
(3) выделение полученного соединения формулы I в свободной форме или в форме соли или сольвата.
Способ по варианту А проводят по известным методикам взаимодействия эфиров сульфоновых кислот с аминами или аналогично тому, как описано в примерах. Rc предпочтительно означает С1-С4алкил, но прежде всего метил. Обычно реакцию проводят в органическом растворителе таком, как толуол. Обычно температура реакции составляет от 0°С до 200°С, предпочтительно от 70°С до 100°С, прежде всего от 80°С до 90°С. Реакцию обычно проводят при нагревании или при микроволновом излучении.
Способ по варианту В проводят по известным методикам взаимодействия эпоксидов с аминами или аналогично тому, как описано в примерах. Обычно реакцию проводят в органическом растворителе таком, как толуол. Обычно температура реакции составляет от 0°С до 200°С, предпочтительно от 70°С до 100°С, прежде всего от 80°С до 90°С. Реакцию обычно проводят при нагревании или при микроволновом излучении.
Защитные группы Ra и Rb выбирают в соответствии со строением функциональной группы, например, как описано в монографии Protective Groups in Organic Synthesis, T.W.Greene и P.G.M.Wuts, John Wiley & Sons Inc, Third Edition (1999), где также описаны методики, пригодные для замены защитных групп на атом водорода. Ra предпочтительно означает С1-С4алкил, прежде всего изопропил. Rb предпочтительно означает С1-С4алкил, прежде всего трет-бутил.
Защитные группы можно вводить или удалять по обычным методикам. Например, гидроксигруппы защищают бензильной группой, которую затем удаляют каталитическим гидрированием в присутствии палладия на угле по обычным методикам, например по методикам, описанным в примерах.
Соединения формулы I в свободной форме можно превратить в солевую форму и наоборот обычным способом. Соединения в свободной форме или в форме соли можно получить в форме гидратов или сольватов, содержащих растворитель, использованный для кристаллизации. Соединения формулы I можно выделить из реакционной смеси и очистить обычным способом. Изомеры, такие как энантиомеры, можно получить обычным способом, например фракционной кристаллизацией или асимметрическим синтезом из соответственно асимметрически замещенных, например, оптически активных исходных материалов.
Соединения формулы II являются новыми и их можно получить при взаимодействии соединения формулы IV
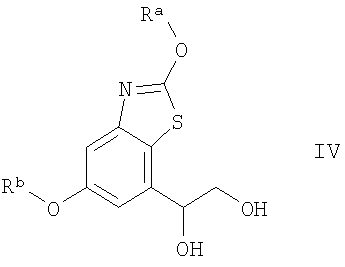
где Ra и Rb являются защитными группами, с сульфонилхлоридом, например метансульфонилхлоридом по известным методикам селективного моно-сульфонилирования, описанным в статье Zhou и др., J. Organic Letters, 4 (1), 43-46 (2002), или аналогично тому, как описано ниже в разделе Примеры. При взаимодействии сульфонилхлорида с (R)-1-(5-трет-бутокси-2-изопропоксибензотиазол-7-ил)этан-1,2-диолом получают R-энантиомер, при взаимодействии сульфонилхлорида с (S)-1-(5-трет-бутокси-2-изопропоксибензотиазол-7-ил)этан-1,2-диолом получают S-энатиомер. Обычно реакцию проводят в органическом растворителе, таком как пиридин. Температура реакционной смеси обычно составляет от -20°С до 30°С, предпочтительно приблизительно 0°С.
Соединения формулы IIА являются новыми и их можно получить с использованием известных методов получения оксиранил-замещенных гетероциклических соединений, например, как описано в международной заявке WO 04/016601. Например, соединения формулы IIA можно получить при нагревании соединений формулы II, например, от комнатной температуры до 150°С, предпочтительно при температуре от 50 до 100°С, в присутствии основания в растворителе, таком как толуол, тетрагидрофуран или дихлорэтан. Соединения формулы IIА можно также получить в качестве промежуточных соединений при проведении вышеуказанной реакции соединений формулы II с соединениями формулы III с образованием соединений формулы I.
Соединения формулы III являются известными соединениями или их можно получить по известным методикам или аналогично тому, как описано ниже в разделе Примеры.
Соединения формулы IV можно получить при взаимодействии соединения формулы V

где Ra и Rb являются защитными группами, с гидроксилирующим агентом, таким как тетроксид осмия, в присутствии или в отсутствии катализатора, например (DHQD)2PHAL (1,4-бис(дигидрохинидинил)фталазина) и реоксиданта, например K3Fe(CN)6, или в присутствии предварительно добавленных гидроксилирующих реагентов, таких как AD-mix-α или AD-mix-β, с использованием известных методик асимметрического гидроксилирования алкенов или аналогично тому, как описано ниже в разделе Примеры. Обычно реакцию проводят в органическом растворителе, например трет-бутаноле/воде, в присутствии тетроксида осмия, предпочтительно в присутствии катализатора, такого как (DHQD)2PHAL, и K3Fe(CN)6 в качестве реоксиданта. Температура реакционной смеси обычно составляет от -10°С до 10°С, предпочтительно приблизительно 0°С.
Соединения формулы V можно получить олефинированием соединения формулы VI
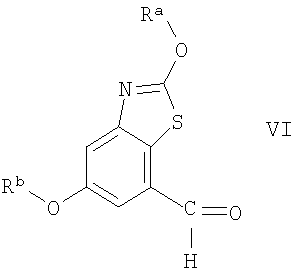
где Ra и Rb являются защитными группами, по известным методикам превращения альдегидов в алкены, например по реакции Виттига, или аналогично тому, как описано ниже в разделе Примеры. Обычно реакцию проводят в органическом растворителе, например ТГФ или ДХМ. Температура реакционной смеси обычно составляет от 10°С до 40°С, предпочтительно реакцию проводят при комнатной температуре.
Соединения формулы VI можно получить по реакции соединения формулы VII
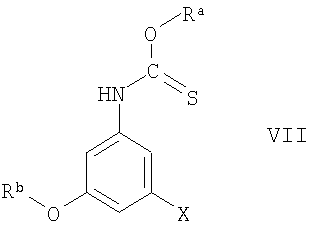
где Ra и Rb являются защитными группами, а X означает галоген, предпочтительно фтор, с сильным основанием, например трет-бутиллитием. Промежуточный анион нейтрализуют добавлением электрофильного соединения, например диметилформамида, а реакцию проводят по методике, описанной в статье Stanetty и др., J. Org. Chem., 61, 5130-5133 (1996), или аналогично тому, как описано ниже в разделе Примеры. Обычно реакцию проводят в органическом растворителе, например ТГФ. Температура реакционной смеси обычно составляет от -90°С до 20°С, предпочтительно от приблизительно -78°С до приблизительно -10°С.
Соединения формулы VII можно получить по реакции соединения формулы VIII
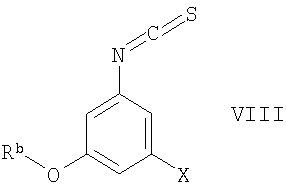
где Rb означает защитную группу, а X означает галоген, с соединением формулы IX

где Ra означает защитную группу, по известным методикам, например, по реакции изотиоцианатов со спиртами с образованием тиокарбаматов, или аналогично тому, как описано ниже в разделе Примеры. R1 предпочтительно означает С1-С4алкил, прежде всего изопропил. Обычно реакцию предпочтительно проводят в присутствии основания, например триэтиламина. Температура реакционной смеси обычно составляет от 0°С до 120°С, предпочтительно приблизительно 60°С.
Соединения формулы VIII можно получить известными методами превращения анилинов в изотиоцианаты, например, по реакции соединения формулы X
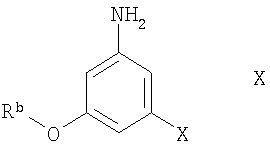
где Rb означает защитную группу, а X означает галоген, с тиофосгеном (тиокарбонилдихлоридом) с использованием известных методик превращения аминов в изотиоцианаты или аналогично тому, как описано ниже в разделе Примеры. Обычно реакцию проводят в органическом растворителе, таком как хлороформ, предпочтительно в присутствии основания, например карбоната натрия. Температура реакционной смеси обычно составляет от -20°С до 20°С, предпочтительно приблизительно 0°С.
Соединения формулы IX известны или их можно получить известными методами или аналогично тому, как описано ниже в разделе Примеры.
Соединения формулы I в свободной форме, в форме соли или сольвата, используются в качестве лекарственных средств. Соответственно в изобретении предлагается соединение формулы I в свободной форме, в форме соли или сольвата, в качестве лекарственного средства. Соединения формулы I в свободной форме, в форме соли или сольвата, обозначаемые в данном контексте как "агенты по изобретению", обладают высоким агонистическим действием на β2-адренорецептор. β2-Агонистическую активность, начало действия и продолжительность действия агентов по изобретению оценивают с использованием полосок трахеальной ткани морских свинок in vitro по методу, описанному в статье R.A.Coleman и А.Т.Nials, J. Pharmacol. Methods, 21, 71 (1989). Эффективность связывания и селективность в отношении β2-адренорецептора по сравнению с β1-адренорецептором определяют классическим анализом связывания на фильтрах по методу, описанному в руководстве Current Protocols in Pharmacology, S.J.Enna (гл. ред.) и др., John Wiley & Son, Inc (1998), или определяя уровень цАМФ в клетках, экспрессирующих β2- или β1-адренорецептор, по методу, описанному в статье В.January и др., Brit. J. Pharmacol., 123, 701 (1998).
Агенты по изобретению обычно оказывают быстрое действие и обладают продолжительным стимулирующим действием на β2-адренорецептор. Соединения, описанные ниже в примерах, характеризуются величинами Ki (β2) от 0,1 до 1000 нМ и продолжительностью действия от 1 ч до более 12 ч. Многие из соединений проявляют селективность в отношении β2-адренорецептора, в 1,5-500 раз превышающую селективность в отношении β1-адренорецептора. Соединения, описанные в примерах 2, 4, 9, 14 и 17, характеризуются эффективностью связывания с β2, определенную классическим анализом связывания на фильтрах, например, у указанных соединений значения Ki составляют 0,061, 0,027, 0,016, 0,056 и 0,002 мкМ соответственно.
Соединения, описанные в примерах 1 и 18, характеризуются временем Т(50%) (в мин) >672 при концентрации 100 нМ и 595 при концентрации 10 нМ соответственно, по данным анализа с использованием полосок трахеальной ткани морских свинок, где Т(50%) означает продолжительность ингибирования сокращения при снижении на 50% от максимального значения.
Благодаря β2-агонистической активности агенты по изобретению можно использовать при лечении любого состояния, которое предотвращается или облегчается при активации β2-адренорецептора. Благодаря продолжительному селективному β2-агонистическому действию агенты по изобретению можно использовать для релаксации бронхиальной гладкой мускулатуры и для снижения интенсивности бронхостеноза, который оценивают на моделях, таких как плетизмографические модели in vivo, как описано в статьях Chong и др., J. Pharmacol. Toxicol. Methods, 39, 163 (1998), Hammelmann и др., Am. J. Respir. Crit. Care Med., 156, 766 (1997), и на аналогичных моделях.
Таким образом, агенты по изобретению можно использовать при лечении обструктивных или воспалительных заболеваний дыхательных путей. Благодаря продолжительному действию агенты по изобретению можно вводить при лечении таких заболеваний однократно. Кроме того, агенты по изобретению обычно обладают слабыми побочными действиями, обычно характерными для β2-агонистов, такими как тахиакардия, тремор и беспокойное состояние, и, следовательно, такие агенты пригодны для применения при экстренной терапии, а также профилактической терапии обструктивных или воспалительных заболеваний дыхательных путей.
Лечение по настоящему изобретению является симптоматическим или профилактическим. Воспалительные или обструктивные заболевания дыхательных путей, в отношении которых может применяться настоящее изобретение, включают астму любого типа и любой этиологии, включая врожденную (неаллергическую) астму и приобретенную (аллергическую) астму. Лечение астмы включает лечение субъектов, например детей в возрасте младше 4 или 5 лет, у которых наблюдается симптоматическое свистящее дыхание или с диагнозом "дети с одышкой", и которые относятся к категории пациентов первостепенного медицинского значения, и которых в настоящее время относят к астматикам в ранней фазе. Такое астматическое состояние называют "синдромом детей со стерторозным дыханием".
Профилактическое действие при лечении астмы наглядно проявляется в снижении частоты или тяжести симптоматических приступов, например острых астматических приступов или бронхостеноза, в улучшении функционирования легких или повышении гиперреактивности дыхательных путей. Кроме того, профилактическое действие проявляется как снижение необходимости в другом симптоматическом лечении, предназначенном для подавления или остановки приступов астмы при их наличии, например необходимости в противовоспалительном (например, с применением кортикостероидов) или бронхолитическом лечении. Профилактика при лечении астмы прежде всего необходима для субъектов, подверженных "утренним приступам". "Утренние приступы" являются известным астматическим синдромом, свойственным большинству астматиков, который характеризуется астматическими приступами, например, между приблизительно 4 и 6 утра, т.е. через значительный промежуток времени после любого предшествующего введения симтоматического астматического терапевтического средства.
Другие воспалительные или обструктивные заболевания и состояния дыхательных путей, в отношении которых может применяться настоящее изобретение, включают острый респираторный дистресс-синдром у взрослых пациентов (ARDS), хроническое обструктивое заболевание легких и дыхательных путей (COPD или COAD), включая хронический бронхит, или ассоциированую с ним одышку, эмфизему, а также обострение гиперреактивности дыхательных путей вследствие применения других лекарственных средств, прежде всего другого ингаляционного медикаментозного лечения. Настоящее изобретение можно также использовать при лечении бронхитов любого типа или этиологии, включая, например, острый, арахноидальный, катаральный, крупозный, хронический или гнойный туберкулезный бронхит. Другие воспалительные или обструктивные заболевания дыхательных путей, в отношении которых может применяться настоящее изобретение, включают пневмокониоз (воспалительное заболевание легких, которое обычно связано с профессиональной деятельностью, сопровождающееся в большинстве случаев хронической или острой обструкцией дыхательных путей, а также возникающее при повторном вдыхании пыли) любого типа или этиологии, включая, например, алюминоз, антракоз, азбестоз, халикоз, птилоз, сидероз, силикоз, табакоз и биссиноз.
Благодаря β2-агонистической активности агенты по изобретению можно использовать при лечении состояния, требующего расслабления гладкой мускулатуры матки или сосудистой системы. Таким образом, их можно использовать для предотвращения или облегчения болей при преждевременных родах. Указанные агенты можно также использовать при лечении хронической и острой крапивницы, псориаза, аллергического конъюнктивита, актинита, сенной лихорадки и мастоцитоза.
Агенты по изобретению можно также использовать в качестве комбинированных терапевтических агентов для применения в сочетании с другими лекарственными препаратами такими, как противовоспалительные, бронхолитические, антигистаминные или противокашлевые лекарственные средства, прежде всего при лечении обструктивных или воспалительных заболеваний дыхательных путей, таких как заболевания, упомянутые выше, например, в качестве средств, потенциирующих терапевтическую активность указанных лекарственных препаратов, или в качестве средств, предназначенных для снижения требуемой дозы или возможных побочных действий указанных лекарственных препаратов. Агент по изобретению смешивают с другими лекарственными средствами в фиксированной фармацевтической композиции или его вводят отдельно перед введением другого лекарственного соединения, одновременно с ним или после него. Таким образом, изобретение включает комбинацию агента по изобретению, описанного выше, с противовоспалительным, бронхолитическим, антигистаминным или противокашлевым лекарственным средством, причем указанный агент по изобретению и указанное лекарственное средство могут находиться в одной и той же или в различных фармацевтических композициях.
Пригодные противовоспалительные средства включают стероиды, прежде всего глюкокортикостероиды, такие как будезонид, дипропионат бекламетазона, пропионат флутиказона, циклезонид или фуроат мометазона или стероиды, описанные в WO 02/88167, WO 02/12266, WO 02/100879, WO 02/00679 (прежде всего соединения, описанные в примерах 3, 11, 14, 17, 19, 26, 34, 37, 39, 51, 60, 67, 72, 73, 90, 99 и 101), WO 03/35668, WO 03/48181, WO 03/62259, WO 03/64445, WO 03/72592, WO 04/39827 и WO 04/66920, нестероидные агонисты рецептора глюкокортикоидов такие, как описанные в DE 10261874, WO 00/00531, WO 02/10143, WO 03/82280, WO 03/82787, WO 03/86294, WO 03/104195, WO 03/101932, WO 04/05229, WO 04/18429, WO 04/19935 и WO 04/26248, антагонисты LTB4, такие как BIIL 284, СР-195543, DPC11870, LTB4 этаноламид, LY 293111, LY 255283, CGS025019C, СР-195543, ONO-4057, SB 209247, SC-53228, и агенты, описанные в US 5451700, антагонисты LTD4, включающие монтелукаст, пранлукаст, зафирлукаст, акколат, SR2640, Wy-48252, ICI 198615, МК-571, LY-171883, Ro 24-5913 и L-648051, ингибиторы PDE4, такие как циломиласт (Ariflo® GlaxoSmithKline), рофлумиласт (фирма Byk Gulden),V-11294A (фирма Napp), BAY19-8004 (фирма Bayer), SCH-351591 (фирма Schering-Plough), арофиллин (фирма Almirall Prodesfarma), PD189659/PD168787 (фирма Parke-Davis), AWD-12-281 (фирма Asta Medica), CDC-801 (фирма Celgene), SelCID(TM) CC-10004 (фирма Celgene), VM554/UM565 (фирма Vernalis), T-440 (фирма Tanabe), KW-4490 (фирма Kyowa Hakko Kogyo), и агенты, описанные в WO 92/19594, WO 93/19749, WO 93/19750, WO 93/19751, WO 98/18796, WO 99/16766, WO 01/13953, WO 03/104204, WO 03/104205, WO 03/39544, WO 04/000814, WO 04/000839, WO 04/005258, WO 04/018450, WO 04/018451, WO 04/018457, WO 04/018465, WO 04/018431, WO 04/018449, WO 04/018450, WO 04/018451, WO 04/018457, WO 04/018465, WO 04/019944, WO 04/019945, WO 04/045607 и WO 04/037805, агонисты A2A, такие, как описанные в ЕР 1052264, ЕР 1241176, ЕР 409595А2, WO 94/17090, WO 96/02543, WO 96/02553, WO 98/28319, WO 99/24449, WO 99/24450, WO 99/24451, WO 99/38877, WO 99/41267, WO 99/67263, WO 99/67264, WO 99/67265, WO 99/67266, WO 00/23457, WO 00/77018, WO 00/78774, WO 01/23399, WO 01/27130, WO 01/27131, WO 01/60835, WO 01/94368, WO 02/00676, WO 02/22630, WO 02/96462 и WO 03/086408, и антагонисты А2В такие, как описанные в WO 02/42298.
Пригодные бронхолитические средства включают антихолинергические или антимускариновые агенты, прежде всего бромид ипратропия, бромид окситропия, соли тиотропия и CHF 4226 (фирма Chiesi), и гликопирролат, а также агенты, описанные в ЕР 424021, US 3714357, US 5171744, WO 01/04118, WO 02/00652, WO 02/51841, WO 02/53564, WO 03/00840, WO 03/33495, WO 03/53966, WO 03/87094, WO 04/018422 и WO 04/05285.
Пригодные бронхолитические средства с двойным действием включают агонист β2-адренорецептора/антагонисты мускаринового рецептора, такие, как описанные в US 2004/0167167, WO 04/74246 и WO 04/74812.
Пригодные антигистаминные средства включают гидрохлорид цетиризина, ацетаминофен, фумарат клемастина, прометазин, лоратидин, деслоратидин, дифенгидрамин и гидрохлорид фексофенадина, активастин, астемизол, азеластин, эбастин, эпинастин, мизоластин и тефенадин, а также агенты, описанные в JP 2004107299, WO 03/099807 и WO 04/026841.
Агенты по изобретению можно также использовать в качестве комбинированных терапевтических агентов для применения в сочетании с с другими агонистами β-адренорецептора, в качестве лекарственного средства при экстренной терапии. Пригодные агонисты β2-адренорецетора включают албутерол (салбутамол), метапротеренол, тербуталин, салметерол, фенотерол, кармотерол, прокатерол и прежде всего формотерол и их фармацевтически приемлемые соли, и соединения (в свободной форме, в форме соли или сольвата) формулы I, описанные в WO 0075114, включенной в описание заявки в качестве ссылки, предпочтительно соединения, описанные в примерах, прежде всего соединение формулы
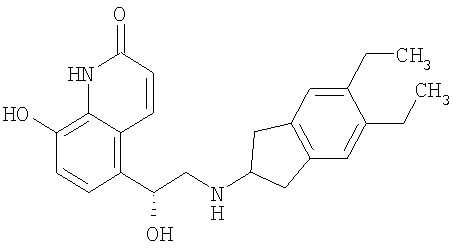
его фармацевтически приемлемые соли, а также соединения (в свободной форме, в форме соли или сольвата) формулы I, описанные в WO 04/16601, а также соединения, описанные в ЕР 1440966, JP 05025045, WO 93/18007, WO 99/64035, US 2002/0055651, WO 01/42193, WO 01/83462, WO 02/66422, WO 02/70490, WO 02/76933, WO 03/24439, WO 03/42160, WO 03/42164, WO 03/72539, WO 03/91204, WO 03/99764, WO 04/16578, WO 04/22547, WO 04/32921, WO 04/33412, WO 04/37768, WO 04/37773, WO 04/37807, WO 04/39762, WO 04/39766, WO 04/45618, WO 04/46083, WO 04/80964, EP1460064, WO 04/087142, WO 04/089892, EP 01477167, US 2004/0242622, US 2004/0229904, WO 04/108675, WO 04/108676, WO 05/033121, WO 05/040103 и WO 05/044787.
Другие комбинации агентов по изобретению с противоспалительными лекарственными средствами представляют собой комбинации с антагонистами рецепторов хемокина, например, CCR-1, CCR-2, CCR-3, CCR-4, CCR-5, CCR-6, CCR-7, CCR-8, CCR-9 и CCR10, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, прежде всего антагонистами CCR-5, такими как антагонисты SC-351125, SCH-55700 и SCH-D (фирма Schering-Plough), антагонистами, предлагаемыми фирмой Takeda, такими как хлорид N-[[4-[[[6,7-дигидро-2-(4-метилфенил)-5Н-бензоциклогептен-8-ил]карбонил]амино]фенил]метил]тетрагидро-N,N-диметил-2Н-пиран-4-амина (ТАК-770), и антагонистами CCR-5, описанными в US 6166037 (прежде всего в п.п.18 и 19), WO 00/66558 (прежде всего в п.8), WO 00/66559 (прежде всего в п.9), WO 04/018425 и WO 04/026873.
Комбинации агентов по изобретению и стероиды, ингибиторы PDE4, агонисты A2A, агонисты A2B или антагонисты LTD4 можно использовать, например, при лечении COPD или прежде всего астмы. Комбинации агентов по изобретению и антихолинергические или антимускариновые агенты, ингибиторы PDE4, агонисты A 2A, агонисты A2B, агонисты рецептора допамина или антагонисты LTB4 можно использовать, например, при лечении астмы или прежде всего COPD.
В соответствии с вышеизложенным в настоящем изобретении предлагается также способ лечения обструктивного или воспалительного заболевания дыхательных путей, который включает введение субъекту, прежде всего человеку, который нуждается в лечении, соединения формулы I или его фармацевтически приемлемой соли или сольвата, указанного выше. Другим объектом изобретения является соединение формулы I или его фармацевтически приемлемая соль или сольват, описанное выше, для применения при получении лекарственного средства, предназначенного для лечения обструктивного или воспалительного заболевания дыхательных путей.
Агенты по изобретению можно вводить любым обычным способом, например перорально, например, в форме таблетки или капсулы, парентерально, например внутривенно, местно на кожу, например, при лечении псориаза, интраназально, например, при лечении сенной лихорадки, или предпочтительно ингаляцией, прежде всего при лечении обструктивных или воспалительных заболеваний дыхательных путей.
Еще одним объектом изобретения является также фармацевтическая композиция, включающая соединение формулы I в свободной форме или в форме его фармацевтически приемлемой соли или сольвата, необязательно в смеси с фармацевтически приемлемым разбавителем или носителем. Такие композиции получают с использованием обычных разбавителей или эксципиентов известными в фармакологии методами. Таким образом, формы для перорального введения включают таблетки и капсулы. Формы для местного введения включают кремы, мази, гели или системы трансдермальной доставки, например пластыри. Композиции для ингаляции включают аэрозоль или другие распыляемые составы или порошкообразные составы.
Если композиция включает аэрозольный состав, то такая композиция предпочтительно содержит фторуглеводородный пропеллент (HFA), такой как HFA134a или HFA227, или их смесь, и может содержать один или более сорастворителей, известных в данной области техники, таких как этанол (до 20 мас.%) и/или один или более ПАВ, таких как олеиновая кислота или триолеат сорбита, и/или один или более наполнителей, таких как лактоза. Если композиция включает порошкообразный состав, то такая композиция предпочтительно содержит, например, соединение формулы I в виде частиц диаметром до 10 мкм, необязательно в смеси с разбавителем или носителем, таким как лактоза, с требуемым размером частиц и соединением, которое защищает продукт от повреждения за счет увлажнения, таким как стеарат магния, например, от 0,01 до 1,5%. Если композиция включает распыляемый состав, то такая композиция предпочтительно содержит, например, соединение формулы I, растворенное или суспендированное в носителе, содержащем воду, сорастворитель, такой как этанол или пропиленгликоль, и стабилизатор, который может означать ПАВ.
Изобретение также включает (А) соединение формулы I, указанное выше, в свободной форме или в форме его фармацевтически приемлемой соли или сольвата в форме для ингаляции, (В) лекарственное средство для ингаляции, включающее такое соединение в форме для ингаляции в смеси с фармацевтически приемлемым носителем в ингалируемой форме, (С) фармацевтический продукт, включающий такое соединение в ингалируемой форме вместе с ингалятором и (D) ингалятор, содержащий такое соединение в ингалируемой форме.
Дозы, используемые при осуществлении изобретения, изменяются в зависимости, например, от конкретного состояния, подлежащего лечению, требуемого результата, и способа введения. В общем случае пригодная суточная доза для введения ингаляцией составляет от 1 до 5000 мкг.
Изобретение иллюстрируется следующими примерами.
Примеры
Более предпочтительными соединениями формулы I являются также соединения формулы XI

где Т имеет значения, указанные в таблице, а способ получения описан ниже. Все соединения получают в форме соли или в свободной форме. Если не указано иное, спектры 1Н-ЯМР снимали при 400 МГц в CDCI3. Масс-спектры получали в условиях ионизации электроспреем в сочетании с ЖХ (элюент: градиент 5 ацетонитрил/вода + 1% муравьиная кислота, от 5% до 95%).

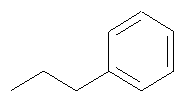
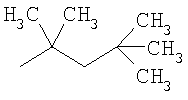
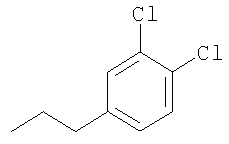

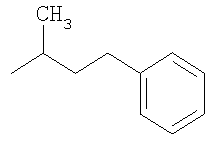
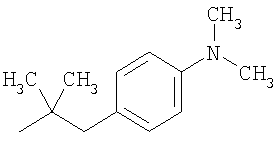

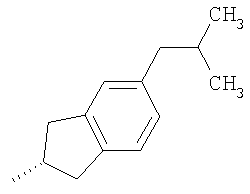
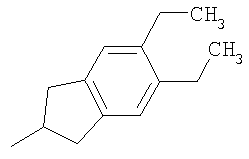
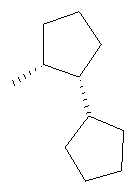


Получение промежуточных соединений
Список сокращений: ДХМ означает дихлорметан, ДМФА означает диметилформамид, ДМСО означает диметилсульфоксид, ТГФ означает тетрагидрофуран.
1-Бром-3-трет-бутокси-5-фторбензол
трет-Бутанол (28,2 г) растворяли в диметилацетамиде (200 мл) и в течение 15 мин добавляли NaH (15,6 г, 60% дисперсия в масле). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, а затем в течение 30 мин добавляли по каплям 3,5-дифторбромбензол (50 г). Реакционную смесь перемешивали при комнатной температуре до завершения реакции по данным ЖХВР. Реакцию останавливали при добавлении воды (10 мл), смесь промывали водой (1×), органическую фазу сушили над MgSO4, фильтровали и растворитель удаляли в вакууме, при этом получали указанное в заголовке соединение.
1Н-ЯМР (CDCl3, 400 МГц): δ 7,00 (ddd, 1Н), 6,95 (dd, 1Н), 6,68 (ddd, 1Н), 1,4 (s, 9Н).
3-трет-Бутокси-5-фторфениламин
1-Бром-3-трет-бутокси-5-фторбензол (56,1 г), бензофенон (50,9 г), NaOMe (50,5 г) и 2,2'-бис-дифенилфосфанил[1,1']бинафталинил (17,5 г) растворяли в толуоле (500 мл). Реакционную смесь продували аргоном, добавляли Pd2(dba)3 (5,4 г) и нагревали при 80°С в течение 40 ч. Реакцию останавливали при добавлении воды, органическую фазу отделяли, сушили над MgSO4, фильтровали и растворитель удаляли в вакууме. Промежуточное соединение получали после очистки экспресс-хроматографией на колонке с силикагелем (элюент: дихлорметан).
Полученный продукт растворяли в МеОН (1 л), добавляли NaOAc (46,1 г), гидрохлорид гидроксиламина (29,1 г) и реакционную смесь перемешивали при комнатной температуре в течение 2,5 ч. Реакцию останавливали при добавлении 0,1 М NaOH, смесь экстрагировали ДХМ (2×), экстракт сушили над MgSO 4, фильтровали и растворитель удаляли в вакууме, при этом получали указанное в заголовке соединение.
1Н-ЯМР (CDCl3, 400 МГц): δ 6,20 (m, 3Н), 3,75 (ушир. s, 2Н), 1,4 (s, 9Н).
1-трет-Бутокси-3-фтор-5-изотиоцианатобензол
В раствор 3-трет-бутокси-5-фторфениламина (42,9 г) в CHCl3 (350 мл) при 0°С раздельно или одновременно добавляли по каплям тиофосген (33,6 г) в CHCl3 (250 мл) и K2CO3 (64,7 г) в Н2О (450 мл) и реакционную смесь нагревали до комнатной температуры в течение ночи. Органическую фазу отделяли, промывали водой (3×), солевым раствором (1×), сушили над MgSO4, фильтровали и растворитель удаляли в вакууме. Указанное в заголовке соединение получали после очистки экспресс-хроматографией на колонке с силикагелем (элюент: дихлорметан/изогексан, 1:3).
1Н-ЯМР (CDCl3, 400 МГц): δ 6,70 (m, 3Н), 1,40 (s, 9Н).
О-Изопропиловый эфир (3-трет-бутокси-5-фторфенил)тиокарбаминовой кислоты
1-трет-Бутокси-3-фтор-5-изотиоцианатобензол (24,0 г) и триэтиламин (10,9 г) растворяли в изопропаноле (150 мл). Реакционную смесь кипятили с обратным холодильником в течение 18 ч и растворитель удаляли в вакууме. Неочищенный продукт растворяли в гексане/диэтиловом эфире (19:1), диэтиловый эфир удаляли в вакууме и раствор охлаждали до 0°С в течение 3 ч. Раствор фильтровали, при этом получали указанное в заголовке соединение.
1Н-ЯМР (CDCl3, 400 МГц): δ 8,10 (ушир. s, 1Н), 6,65 (ушир. s, 2Н), 6,45 (ddd, 1Н), 5,60 (септ, 1Н), 1,35 (d, 6Н), 1,30 (s, 9Н).
5-трет-Бутокси-2-изопропоксибензотиазол-7-карбальдегид
О-Изопропиловый эфир (3-трет-бутокси-5-фторфенил)тиокарбаминовой кислоты (2,2 г) растворяли в сухом тетрагидрофуране (20 мл), реакционную смесь охлаждали до -78°С и в течение 20 мин добавляли трет-бутиллитий (15,2 мл, 1,5 М раствор). Затем реакционную смесь нагревали до -10°С в течение 75 мин, снова охлаждали до -78°С, добавляли N,N-диметилформамид (1,5 г), медленно нагревали до комнатной температуры, а затем перемешивали при -10°С в течение 1 ч. Реакцию останавливали при добавлении 2 М соляной кислоты (5 мл), органическую фазу распределяли между этилацетатом/водой и растворитель удаляли в вакууме. Указанное в заголовке соединение получали после очистки экспресс-хроматографией на колонке с силикагелем (элюент: этилацетат/изогексан, 1:9). МС (ES+): m/e 294 (МН+), LCT50865.
5-трет-Бутокси-2-изопропокси-7-винилбензотиазол
Ph3PMe.Br (5,0 г) растворяли в сухом тетрагидрофуране (100 мл) в атмосфере аргона. Затем при комнатной температуре в течение 10 мин добавляли N-бутиллитий (8,8 мл, 1,6 М раствор) и реакционную смесь перемешивали в течение еще 30 мин. В реакционную смесь добавляли по каплям раствор 5-трет-бутокси-2-изопропоксибензотиазол-7-карбальдегида (1,25 г) в дихлорметане (40 мл) и смесь перемешивали при комнатной температуре в течение 4,5 ч. Растворитель удаляли в вакууме, остаток растворяли в этилацетате, промывали водой (3×), солевым раствором (1×), сушили над MgSO4, фильтровали и растворитель удаляли в вакууме. Указанное в заголовке соединение получали после очистки экспресс-хроматографией на колонке с силикагелем (элюент: этилацетат/изогексан, 1:9). MC (ES+): m/e 292 (MH+), LCT55980.
(R)-1-(5-трет-Бутокси-2-изопропоксибензотиазол-7-ил)этан-1,2-диол
K3Fe(CN)6 (1,2 г), K2CO3 (0,5 г), (DHQD)2PHAL (19 мг) растворяли в трет-бутаноле/воде (15 мл, 1:1) в атмосфере аргона и перемешивали в течение 15 мин. Реакционную смесь охлаждали до 0°с, добавляли OsO4 (3,1 мг), а затем 5-трет-бутокси-2-изопропокси-7-винилбензотиазол (0,35 г). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию останавливали при добавлении мета-бисульфата натрия (1 г) и перемешивали в течение 1,5 ч. В смесь добавляли этилацетат, органический слой отделяли, промывали водой (2×), солевым раствором (1×), сушили над MgSO4, фильтровали и растворитель удаляли в вакууме. Указанное в заголовке соединение получали после очистки экспресс-хроматографией на колонке с силикагелем (элюент: этилацетат/изогексан, 2:5). МС (ES+): m/е 326 (МH+), LCT56091.
(R)-2-(5-трет-Бутокси-2-изопропоксибензотиазол-7-ил)-2-гидроксиэтиловый эфир метансульфоновой кислоты
В раствор (R)-1-(5-трет-бутокси-2-изопропоксибензотиазол-7-ил)этан-1,2-диола (100 мг) в пиридине (2 мл) при 0°С добавляли метансульфонилхлорид (35 мг) и реакционную смесь перемешивали при 0°С в течение 3,5 ч. Растворитель удаляли в вакууме, остаток распределяли между 2 М соляной кислотой и эфиром. Органическую фазу промывали водой (1×) и солевым раствором (1×), сушили над MgSO4, фильтровали и растворитель удаляли в вакууме, при этом получали указанное в заголовке соединение.
1Н-ЯМР (CDCl3, 400 МГц): δ 7,20 (d, 1Н), 6,80 (d, 1Н), 5,30 (септ., 1H), 5,10 (t, 1Н), 4,30 (d, 2Н), 3,00 (s, 3Н), 1,40 (d, 6Н), 1,30 (s, 9Н).
(S)-1-(5-трет-Бутокси-2-изопропоксибензотиазол-7-ил)этан-1,2-диол
K3Fe(CN)6 (3,4 г), K2CO3 (1,4 г), (DHQ)2PHAL (53 мг) в трет-бутаноле/воде (40 мл, 1:1) в атмосфере аргона перемешивали в течение 20 мин. Реакционную смесь охлаждали до 0°С, добавляли OsO4 (8,6 мг), а затем 5-трет-бутокси-2-изопропокси-7-винилбензотиазол (1,0 г). Реакционную смесь перемешивали при комнатной температуре в течение ночи, реакцию останавливали при добавлении мета-бисульфата натрия (1,2 г) и смесь перемешивали в течение 1,5 ч. В смесь добавляли этилацетат, органическую фазу отделяли, промывали водой (2×) и солевыи раствором (1×), сушили над MgSO4, фильтровали и растворитель удаляли в вакууме. Указанное в заголовке соединение получали после очистки экспресс-хроматографией на колонке с силикагелем (элюент: этилацетат/изогексан, 1:3). МС (ES+): m/e 326,12, LCT60289.
(S)-2-(5-трет-Бутокси-2-изопропоксибензотиазол-7-ил)-2-гидроксиэтиловый эфир метансульфоновой кислоты
В раствор (S)-1-(5-трет-бутокси-2-изопропоксибензотиазол-7-ил)этан-1,2-диола (289 мг) в пиридине (2 мл) при 0°С добавляли метансульфонилхлорид (112 мг) и реакционную смесь перемешивали при 0°С в течение 3 ч. Растворитель удаляли в вакууме, остаток распределяли между 2 М соляной кислотой и эфиром. Органическую фазу промывали водой (2×) и солевым раствором (1×), сушили над MgSO4, фильтровали и растворитель удаляли в вакууме, при этом получали указанное в заголовке соединение.
1Н-ЯМР (CDCl3, 400 МГц): δ 7,20 (d, 1Н), 6,80 (d, 1Н), 5,30 (септ., 1Н), 5,10 (t, 1Н), 4,30 (d, 2Н), 3,00 (s, 3Н), 1,40 (d, 6Н), 1,30 (s, 9Н).
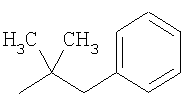
2-(4-Бутоксифенил)-1,1-диметилэтиламин
Указаное соединение получали по методике, описанной в международной заявке WO 01/83462. МС (ES+): m/е 222,20 (МН+), 20% LCT59933.
[4-(2-Амино-2-метилпропил)фенил]диметиламин
Указанное соединение получали по методике, описанной в международной заявке WO 01/83462, МС (ES+): m/e 193 (МН+), 2% LCT59932.
(S)-5-Изобутилиндан-2-иламин
(a) (8)-5-Броминдан-2-иламин
Указанное соединение получали по методикам, описанным в международной заявке WO 96/23760.
(b) Бензиловый эфир (8)-(5-изобутилиндан-2-ил)карбаминовой кислоты
Суспензию (S)-5-броминдан-2-иламина (1,0 г) в дихлорметане (10 мл) охлаждали до 0°С, добавляли по каплям бензилхлорформиат (0,74 мл) и реакционную смесь перемешивали в течение 0,5 ч. Раствор фильтровали, при этом получали бензиловый эфир (S)-(5-броминдан-2-ил)карбаминовой кислоты. В сухую колбу атмосфере аргона помещали PdCl2(dppf)2 (59 мг) и добавляли изобутилцинкбромид (50 мл, 0,5 М раствор в ТГФ). Бензиловый эфир (5-броминдан-2-ил)карбаминовой кислоты (2,50 г) растворяли в сухом ТГФ (2 мл), полученный раствор добавляли в указанную реакционную смесь и перемешивали при 50°С в течение 18 ч. Затем реакцию останавливали при добавлении 2 М соляной кислоты и смесь распределяли между этилцетатом и водой. Органический слой сушили над MgSO4, фильтровали и растворитель удаляли в вакууме. Указанное в заголовке соединение получали после очистки экспресс-хроматографией на колонке с силикагелем (элюент: этилацетат/изогексан, 1:4).
1H-ЯМР (CDCl3, 400 МГц): δ 7,35 (m, 5Н), 7,10 (d, 1Н), 7,00 (s, 1Н), 6,90 (d, 1H), 5,1 (s, 2Н), 4,55 (m, 1Н), 3,30 (т, 2Н), 2,75 (dt, 2Н), 2,45 (d, 2Н), 1,80 (m, 1Н), 0,90 (d, 6Н).
(c) (S)-5-Изобутилиндан-2-иламин
Бензиловый эфир (S)-(5-изобутилиндан-2-ил)карбаминовой кислоты растворяли в метаноле (100 мл), добавляли 10% Pd/C (200 мг) и колбу заполняли водородом (0,35 бар). Реакционную смесь перемешивали в течение 18 ч, катализатор отделяли фильтрованием и растворитель удаляли в вакууме, при этом получали указанное в заголовке соединение.
1H-ЯМР (CDCl3, 400 МГц): δ 7,10 (d, 1Н), 7,00 (s, 1Н), 6,90 (d, 1Н), 3,85 (m, 1H), 3,15 (dd, 2Н), 2,65 (dt, 2Н), 2,45 (d, 2Н), 1,80 (ушир. m, 3Н), 0,90 (d, 6Н).
5,6-Диэтилиндан-2-иламин
Указанное соединение получали по методике, описанной в международной заявке WO 03/76387.
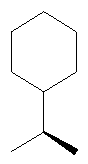
(R, R)-Бициклопентил-2-иламин
Указанное соединение получали из бициклопентил-2-она по методике, описанной в статье S.Hartmann и др., Eur. J. Med. Chem., 35, 377-392 (2000). МС (ES+): m/e 154,23 (MH+), LCT59419.
Пример 1
(R)-7-[2-(1,1-Диметил-2-фенилэтиламино)-1-гидроксиэтил]-5-гидрокси-3Н-бензотиазол-2-он
a) (R)-1-(5-трет-Бутокси-2-изопропоксибензотиазол-7-ил)-2-(1,1-диметил-2- фенилэтиламино)этанол
(R)-2-(5-трет-Бутокси-2-изопропоксибензотиазол-7-ил)-2-гидроксиэтиловый эфир метансульфоновой кислоты (122 мг) и фентермин (165 мг) растворяли в толуоле (2 мл) и реакционную смесь нагревали при 90°С в течение 20 ч. Растворитель удаляли в вакууме, (R)-1-(5-трет-Бутокси-2-изопропоксибензотиазол-7-ил)-2-(1,1-диметил-2-фенилэтиламино)этанол получали после очистки хроматографией на колонке с обращенной фазой с использованием системы для экспресс-хроматографии Jones Flashmaster Personal™ (колонка: ISOLUTE FLASH С18, элюент: градиент AcCN/вода, от 0% до 60%). МС (ES+): m/e 457,32 (МН+), LCT56716.
b) (R)-7-[2-(1,1-Диметил-2-фенилэтиламино)-1-гидроксиэтил]-5-гидрокси-3Н-бензотиазол-2-он
(R)-1-(5-трет-Бутокси-2-изопропоксибензотиазол-7-ил)-2-(1,1-диметил-2-фенилэтиламино)этанол (40 мг) перемешивали в муравьиной кислоте (2 мл) в течение 72 ч. Муравьиную кислоту удаляли в вакууме, указанное в заголовке соединение получали после очистки хроматографией на колонке с обращенной фазой с использованием системы для экспресс-хроматографии Jones Flashmaster Personal™ (колонка: ISOLUTE FLASH C18, элюент: градиент AcCN/вода, от 0% до 50%). МС (ES+): m/e 359,26 (МН+), LCT57144.
Примеры 2-17
Соединения, описанные в примерах 2-17, получали по методикам, аналогичным описанным в примере 1.
Пример 18
(S)-7-{2-[2-(4-Диметиламинофенил)-1,1-диметилэтиламино]-1-гидроксиэтил}-5-гидрокси-3Н-бензотиазол-2-он
a) (S)-1-(5-трет-Бутокси-2-изопропоксибензотиазол-7-ил)-2-[2-(4-диметиламинофенил)-1,1-диметилэтиламино]этанол
[4-(2-Амино-2-метилпропил)фенил]диметиламин (210 мг) и
(S)-2-(5-трет-бутокси-2-изопропоксибензотиазол-7-ил)-2-гидроксиэтиловый эфир метансульфоновой ислоты (120 мг) растворяли в толуоле (2 мл) и реакционную смесь нагревали при 80°С в течение 20 ч. Растворитель удаляли в вакууме, при этом получали (S)-1-(5-трет-бутокси-2-изопропоксибензотиазол-7-ил)-2-[2-(4-диметиламинофенил)-1,1-диметилэтиламино]этанол. МС (ES+): m/e 500.
b) (S)-7-{2-[2-(4-Диметиламинофенил)-1,1-диметилэтиламино]-1-гидроксиэтил}-5-гдрокси-3Н-бензотиазол-2-он
(S)-1-(5-трет-Бутокси-2-изопропоксибензотиазол-7-ил)-2-[2-(4-диметиламинофенил)-1,1-диметилэтиламино]этанол (40 мг) перемешивали в муравьиной кислоте (2 мл) в течение 72 ч. Муравьиную кислоту удаляли в вакууме, указанное в заголовке соединение получали после очистки хроматографией на колонке с обращенной фазой с использованием системы для экспресс-хроматографии Jones Flashmaster Personal™ (колонка: ISOLUTE FLASH С18, элюент: градиент AcCN/вода, от 0% до 50%). МС (ES+): m/e 402,15.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНИЯ В КАЧЕСТВЕ МУСКАРИНОВЫХ РЕЦЕПТОРОВ М3 | 2005 |
|
RU2412183C2 |
| КОМБИНАЦИИ ГЛИКОПИРРОЛАТА И АГОНИСТОВ В-2 АДРЕНОЦЕПТОРА | 2005 |
|
RU2388465C2 |
| КОНКРЕТНОЕ ГЛЮКОКОРТИКОСТЕРОИДНОЕ СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2004 |
|
RU2359973C2 |
| ЭНАНТИОСЕЛЕКТИВНЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ХИНОЛИНА | 2005 |
|
RU2383534C2 |
| СПОСОБ ПОДАВЛЕНИЯ РОСТА ОПУХОЛЕВЫХ КЛЕТОК, ОПОСРЕДОВАННОГО КИНАЗОЙ RAF, ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ МОЧЕВИНЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ) | 1998 |
|
RU2232015C2 |
| СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2244709C2 |
| ПРОИЗВОДНЫЕ БЕНЗОТИАЗОЛА, ХАРАКТЕРИЗУЮЩИЕСЯ АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ К БЕТА-2-АДРЕНОРЕЦЕПТОРАМ | 2003 |
|
RU2324687C2 |
| ВАРИАНТЫ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА ГАММА-АМИНОМАСЛЯНОЙ КИСЛОТЫ (GABA) ДЛЯ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ (ЖК) РАССТРОЙСТВ | 2005 |
|
RU2389722C2 |
| СПЕЦИФИЧЕСКОЕ ГЛЮКОКОРТИКОСТЕРОИДНОЕ СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2004 |
|
RU2348645C2 |
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ МОЧЕВИНЫ, СПОСОБ ИНГИБИРОВАНИЯ КИНАЗЫ RAF И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2265597C2 |
Изобретение относится к соединению формулы I, в свободной форме или в форме фармацевтически приемлемой соли, где Т означает C1-С10алкил, замещенный в одном положении группой -NR1R2 или С3-С15карбоциклической группой, выбранной из индана, фенила, С3-С8циклоалкила, причем указанная С3-С15карбоциклическая группа необязательно замещена в одном или двух положениях группой галоген, -NR3R4 или группой C1-С10алкокси, или Т означает С3-С15карбоциклическую группу, выбранную из индана, фенила, С5циклоалкила, необязательно замещенную в одном или двух положениях группой C1-С10алкил, С5циклоалкил или группой C1-С10алкокси, необязательно замещенной в одном положении фенилом, и R1, R2 независимо означают C1-С10алкил или фенил, а R3, R4 независимо означают C1-С10алкил. Соединение формулы I обладает агонистическим действием на β2-адренорецептор и предназначено для использования в качестве активного ингредиента фармацевтической композиции. Изобретение также относится к способу получения соединения формулы I. 4 н. и 3 з.п. ф-лы, 1 табл.

1. Соединение формулы I
в свободной форме или в форме фармацевтически приемлемой соли, где Т означает C1-С10алкил, замещенный в одном положении группой -NR1R2 или С3-С15карбоциклической группой, выбранной из индана, фенила, С3-С8циклоалкила, причем указанная С3-С15карбоциклическая группа необязательно замещена в одном или двух положениях группой галоген, -NR3R4 или группой C1-С10алкокси,
или Т означает С3-С15карбоциклическую группу, выбранную из индана, фенила, С5циклоалкила, необязательно замещенную в одном или двух положениях группой C1-С10алкил, С5циклоалкил или группой C1-С10алкокси, необязательно замещенной в одном положении фенилом, и
R1, R2 независимо означают C1-С10алкил или фенил, а
R3, R4 независимо означают C1-С10алкил.
2. Соединение по п.1, где
Т означает С1-С8алкил, замещенный в одном положении группой -NR1R2 или С5-С10карбоциклической группой, выбранной из индана, фенила, причем указанная С5-С10карбоциклическая группа необязательно замещена в одном или двух положениях группой галоген, -NR3R4 или С1-С4алкокси,
или Т означает С5-С10карбоциклическую группу, выбранную из индана, фенила, необязательно замещенную в одном или двух положениях группой С1-С8алкил, С5циклоалкил или С1-С4алкокси, необязательно замещенной в одном положении фенилом, и
R1, R2 независимо означают C1-С10алкил или фенил, а
R3, R4 независимо означают C1-С10алкил.
3. Соединение по п.1 или 2, которое означает соединение формулы XI
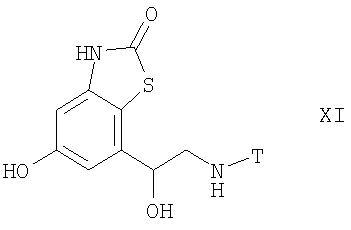
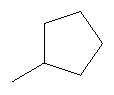
где Т имеет значения, указанные в таблице
4. Соединение по одному из пп.1-3, предназначенное для использования в качестве лекарственного средства, обладающего агонистическим действием на β2-адренорецептор.
5. Фармацевтическая композиция, обладающая агонистическим действием на β2-адренорецептор, включающая в качестве активного компонента соединение по любому из предшествующих пунктов или его фармацевтически приемлемую соль, в смеси с фармацевтически приемлемым носителем.
6. Применение соединения по любому из пп.1-3 для получения лекарственного средства, обладающего агонистическим действием на β2-адренорецептор.
7. Способ получения соединения формулы I по п.1 в свободной форме или в форме его соли, включающий:
(1) взаимодействие соединения формулы II
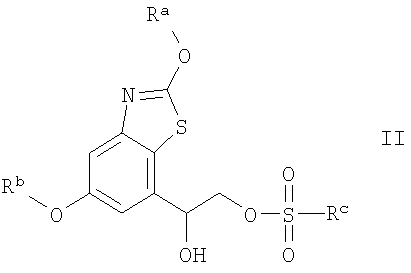
где Ra и Rb означают защитные группы С3-С4алкокси, a Rc означает С1-С4алкил, с соединением формулы III
где Т имеет значения, указанные в п.1,
(2) удаление защитных групп, и
(3) выделение полученного соединения формулы I в свободной форме или в форме его соли.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| SUZUKI H | |||
| Молокоотсос-шприц для парентерального введения стерильного молока | 1924 |
|
SU1319A1 |
| SUZUKI H | |||
| Молокоотсос-шприц для парентерального введения стерильного молока | 1924 |
|
SU1319A1 |

