Оральный путь введения часто является наиболее удобным путем введения лекарственных средств, но, к сожалению, многие терапевтические агенты не обладают активностью при оральном введении вследствие их слабой биологической доступности.
Биологическая доступность многих терапевтических агентов может уменьшаться в результате действия так называемых белков «откачивающего насоса», которые активно удаляют чужеродные субстанции из клетки, что приводит, например, к возникновению явления устойчивости ко многим лекарственным средствам. Эти белки, откачивающие лекарственное средство, преимущественно представляют собой белки-транспортеры типа MDR (белок обусловливающий устойчивость ко многим лекарственным средствам) и MRP (белок, ассоциированный с устойчивостью ко многим лекарственным средствам). Одними из наиболее хорошо изученных откачивающих белков являются Р-гликопротеин (Pgp или MDR1) и MRP2.
Хотя локализованные в мембране откачивающие белки хорошо известны в качестве факторов, участвующих в возникновении синдрома приобретенной устойчивости ко многим лекарственным средствам у многих страдающих раком пациентов после повторной химиотерапии, только в настоящее время было установлено, что, например, MDR1 присутствует также в здоровой ткани, например, в тонком кишечнике, ободочной кишке, печени и эндотелиальных клетках гематоэнцефалического барьера. Присутствие таких откачивающих белков в желудочно-кишечном (ЖК) тракте, прежде всего в тонком кишечнике и ободочной кишке, обусловливает слабую биологическую доступность многих лекарственных средств на основе природных продуктов (включая противораковые агенты винбластин и доксорубицин). Например, многие химиотерапевтические агенты при их введении оральным путем могут не проявлять противоопухолевой активности вследствие их слабой биологической доступности и неспособности проникать в ткани ЖК. Кроме того, откачивающие белки, присутствующие в гепатоцитах, могут дополнительно снижать биологическую доступность терапевтических агентов в результате элиминации с желчью (см. Faber и др., Adv. Drug Del. Rev., 55, 2003, cc.107-124).
Вводимые оральным путем терапевтические агенты должны преодолевать несколько барьеров перед тем, как они достигнут своей области-мишени. Первым основным препятствием, которое необходимо преодолеть, является эпителий тонкого кишечника. Хотя липофильные соединения легко могут диффундировать через апикальную плазматическую мембрану, это никоим образом не гарантирует их последующее прохождение через базолатеральную мембрану и попадание в кровь воротной вены. Белки откачивающего насоса, локализованные на апикальной мембране, которые включают различные транспортеры лекарственных средств, относящиеся к семейству АТФ-связывающих кассет (АВС), например, АВС-транспортеры, такие как MDR1, MRP1 и MRP2, могут переносить соединения изнутри клетки назад в полость кишечника, ограничивая их оральную биологическая доступность путем предотвращения их абсорбции кровью. Вторым основным барьером, с которым приходится сталкиваться, является печень, в которой лекарственные средства транспортируются пассивно или с помощью насыщаемых транспортных процессов из крови портальной вены через плазматические (синусоидальные) мембраны гепатоцитов и мембраны (канальцевые) клеток желчи в желчь. Локализованные на канальцевых мембранах белки откачивающего насоса, к которым также относятся различные транспортеры лекарственных средств, относящиеся к семейству АВС, например, АВС-транспортеры, такие как MDR1, белок, обусловливающие устойчивость к раку молочной железы (BCRP) и MRP2, могут переносить лекарственные соединения изнутри гепатоцитов в желчь, ограничивая их оральную биологическую доступность путем стимуляции элиминации с желчью. Например, было установлено, что MDR1 обладает способностью транспортировать большинство ингибиторов протеазы ВИЧ и снижает их оральную биологическую доступность и проникновение в лимфоциты, головной мозг, яичко и эмбрион, что, по-видимому, приводит к основным ограничивающим воздействиям на терапевтическую эффективность этих лекарственных средств.
Следовательно, один из подходов к повышению биологической доступности может заключаться в совместном введении ингибитора откачивающего белка, т.е. соединения, которое ингибирует функцию откачивающих белков, и лекарственной субстанции. Другими словами, когда ингибитор откачивающего белка вводят совместно с терапевтическим агентом, который является также субстратом для такой специфической откачивающей системы, можно увеличивать оральную биологическую доступность и/или обладающие фармакологической активностью концентрации в области-мишени терапевтического агента в результате ингибирования механизма откачивания изнутри клетки назад в полость кишечника и/или ингибирования секреции в желчь.
Однако откачивающие белки обладают низкой субстратной специфичностью и могут транспортировать много типов молекул. Специфичность не до конца изучена и невозможно предсказать на основе молекулярной структуры лекарственной субстанции может ли конкретное лекарственное средство являться субстратом для определенного белка-транспортера. Как правило, также невозможно предсказать, будет ли конкретное лекарственное средство или соединение подвергаться описанному выше действию откачивающего насоса. Также, если конкретное лекарственное средство обладает низкой оральной биологической доступностью, как правило, невозможно предсказать ни то, обусловлена ли низкая биологическая доступность полностью или частично описанными выше откачивающими белками, ни то, можно ли повысить низкую биологическую доступность путем совместного введения ингибитора откачивающего белка (см. Chan и др., Eur. J. Pharmaceut. Sci., 21, 2004, cc.25-51).
При создании изобретения неожиданно было установлено, что многие ингибиторы ренина, например, описанные в US 5559111, US 6197959 и US 6376672, полное содержание которых включено в настоящее описание в качестве ссылки, являются субстратами для известных откачивающих систем и активно транспортируются представителями АВС-семейства, прежде всего MDR1 и MRP2. Таким образом, биологическую доступность этих ингибиторов ренина можно повышать путем ингибирования откачивающего механизма, участвующего в этих системах, прежде всего путем ингибирования транспорта лекарственного средства, осуществляемого MDR1 и/или MRP2.
На чертежах представлено:
на фиг.1 - воздействие ингибитора ренина SPP100 на АТФазную активность в мембранных пузырьках, экспрессирующих высокие уровни MDR1,
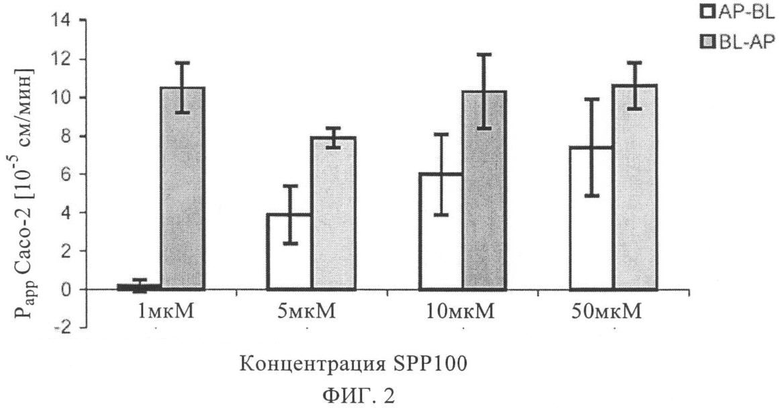
на фиг.2 - двунаправленный транспорт ингибитора ренина SPP100 через монослои клеток линии Сасо-2 в направлении от апикального слоя (АР) к базолатеральному слою (BL) и от BL к АР,
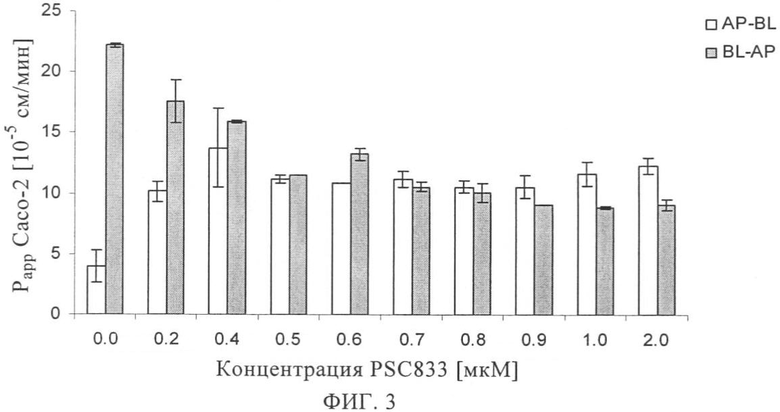
на фиг.3 - воздействие ингибитора MDR1 PSC833 на способность ингибитора ренина SPP100 проникать через монослои клеток линии Сасо-2,
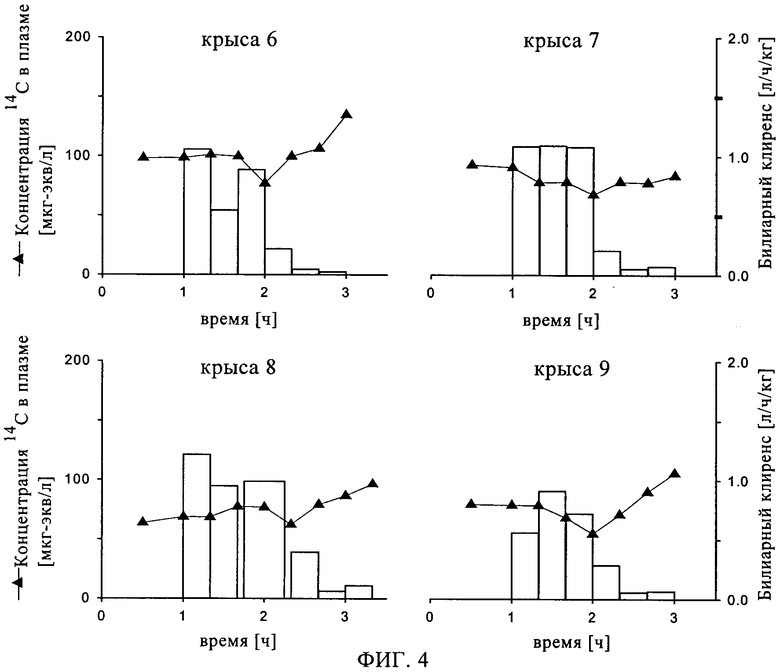
на фиг.4 - воздействие ингибитора MDR1 PSC833 на концентрацию ингибитора ренина SPP100 в плазме и билиарный клиренс у крыс в процессе непрерывной внутривенной инфузии,
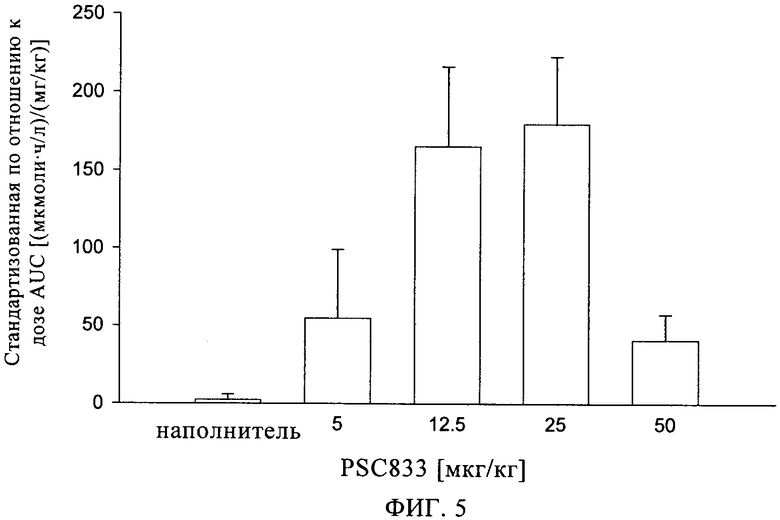
на фиг.5 - стандартизованные по отношению к дозе величины площади под кривой (AUC) концентрации ингибитора ренина SPP100 в плазме крыс после однократного введения оральным путем в присутствии PSC833 и без него.
В качестве ингибиторов ренина, применяемых согласно настоящему изобретению, можно использовать любые соединения, которые обладают активностью ингибитора ренина in vivo и, следовательно, являются полезными с фармацевтической точки зрения, например, в качестве терапевтических агентов для лечения гипертензии, застойной сердечной недостаточности, гипертрофии сердца, сердечного фиброза, постинфарктной кардиомиопатии, осложнений, обусловленных диабетом, таких как нефропатия, васкулопатия и нейропатия, заболеваний коронарных сосудов, рестеноза после пластической операции на сосудах, повышенного внутриглазного давления, глаукомы, аномального сосудистого роста, гиперальдостеронизма, состояний страха и когнитивных нарушений. В частности, настоящее изобретение относится к амидным производным δ-амино-γ-гидрокси-ω-арилалкановой кислоты, описанным в US 5559111.
Таким образом, в настоящем изобретении предложен способ повышения биологической доступности, предпочтительно оральной биологической доступности, ингибитора ренина, заключающийся в том, что млекопитающему, предпочтительно человеку, нуждающемуся в этом, совместно вводят комбинацию, содержащую ингибитор ренина и ингибитор откачивающего белка. Ингибитор откачивающего белка вводят в таком количестве, чтобы биологическая доступность ингибитора ренина повышалась по сравнению с биологической доступностью в отсутствии ингибитора откачивающего белка (например, на 10% при оральном введении человеку). Ингибитор откачивающего белка и ингибитор ренина предпочтительно вводят совместно, причем каждый в таком количестве, чтобы комбинация обладала требуемым терапевтическим действием, например, гипотензивным действием.
В частности, в настоящем изобретении предложен способ повышения биологической доступности производного δ-амино-γ-гидрокси-ω-арилалкановой кислоты, заключающийся в том, что млекопитающему, предпочтительно человеку, нуждающемуся в таком лечении, совместно вводят комбинацию, содержащую производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты или его фармацевтически приемлемую соль и ингибитор откачивающего белка.
Понятие «совместное введение» комбинации, содержащей ингибитор ренина, прежде всего производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты, и ингибитор откачивающего белка, означает, что два компонента можно вводить совместно в виде фармацевтической композиции или в виде части одной объединенной дозы лекарственного средства. Совместное введение включает также введение ингибитора ренина, прежде всего производного δ-амино-γ-гидрокси-ω-арилалкановой кислоты, и ингибитора откачивающего белка по отдельности, но в рамках единой лечебной схемы. Два компонента, если их вводят по отдельности, необязательно требуется вводить практически одновременно, хотя это можно делать при необходимости. Так, совместное введение включает, например, введение ингибитора ренина, прежде всего производного δ-амино-γ-гидрокси-ω-арилалкановой кислоты, и ингибитора откачивающего белка в виде раздельных доз или лекарственных форм, но в одно и то же время. Совместное введение включает также раздельное введение в различные моменты времени и в любом порядке.
Ингибитор ренина, прежде всего производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты, предлагаемое в настоящем изобретении, можно применять в форме его фармацевтически приемлемых солей, в безводной форме или в форме его гидрата или сольвата. Все такие формы подпадают под объем настоящего изобретения.
Понятие «ингибитор откачивающего белка» в контексте настоящего описания означает любое соединение, представляющее собой фармацевтическое соединение или эксципиент, которое обладает способностью ингибировать действие любого АВС-транспортера, например, из числа тех, которые описаны у Bakos и др., Mol Pharmacol., 57, 2002, cc.760-768 и у Maarten и др., AIDS, 16, 2002, cc.2295-2301.
Кроме того, следует отметить, что ингибитор откачивающего белка, повышающий биологическую доступность ингибитора ренина, может проявлять свою активность посредством одного или нескольких механизмов действия. Это означает, что, как хорошо известно, в данной области, он может представлять собой конкурентный или неконкурентный ингибитор или может иметь смешанный механизм действия. Может ли такой ингибитор воздействовать на откачивание конкретного ингибитора ренина, зависит среди прочего от относительных аффинностей ингибитора ренина и ингибитора откачивающего белка; относительных растворимостей в воде ингибитора ренина и ингибитора откачивающего белка, поскольку это может влиять на концентрацию обоих соединений в откачивающем насосе in vivo, когда они конкурируют друг с другом; от абсолютной растворимости в воде ингибитора откачивающего белка, поскольку необходимо обеспечивать достаточную концентрацию в откачивающем насосе in vivo для эффективного ингибирования откачивания; и от дозы ингибитора откачивающего белка. Для цели настоящего изобретения ингибитор откачивающего белка может представлять собой любое соединение, которое позволяет улучшать доступность ингибитора ренина при системном введении, когда ингибитор ренина вводят орально или любым другим путем, и которое является субстратом и/или ингибитором одного или нескольких откачивающих лекарственное средство белков/активностей эпителиальных клеток кишечника или присутствующих в гепатоцитах.
Как указано выше в настоящем описании, в настоящем изобретении предложен способ повышения биологической доступности ингибитора ренина, прежде всего производного δ-амино-γ-гидрокси-ω-арилалкановой кислоты, заключающийся в том, что осуществляют совместное введение ингибитора ренина и ингибитора откачивающего белка.
Предпочтительно ингибитор откачивающего белка, предлагаемый в настоящем изобретении, представляет собой ингибитор MDR1, например, PSC833.
Предпочтительно производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты, предлагаемое в настоящем изобретении, формулы
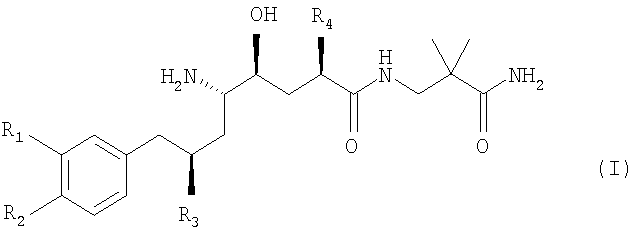
в которой R1 обозначает С1-С4алкокси-С1Салкоксигруппу или С1-С4алкокси-С1-С4алкил; R2 обозначает С1-С4алкил или С1-С4алкоксигруппу; и R3 и R4 независимо друг от друга обозначают разветвленный С1-С4алкил; или его фармацевтически приемлемую соль вводят совместно с ингибитором MDR1, например, PSC833.
Более предпочтительно производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты, предлагаемое в настоящем изобретении, формулы (I), в которой R1 обозначает 3-метоксипропоксигруппу; R2 обозначает метоксигруппу; и R3 и R4 обозначают изопропил; или его фармацевтически приемлемую соль вводят совместно с ингибитором MDR1, например, PSC833.
Наиболее предпочтительно производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты, предлагаемое в настоящем изобретении, представляющее собой гемифумарат (2-карбамоил-2-метилпропил)амида (2S,4S,5S,7S)-5-амино-4-гидрокси-2-изопропил-7-[4-метокси-3-(3-метоксипропокси)бензил]-8-метилнонановой кислоты, имеющий также название SPP100, вводят совместно с ингибитором MDR1, например, PSC833.
Как указано выше, ингибитор ренина, прежде всего производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты, и ингибитор откачивающего белка можно вводить совместно в виде фармацевтической композиции. Компоненты можно вводить совместно в виде любой общепринятой лекарственной формы, как правило, в сочетании также с фармацевтически приемлемым носителем или разбавителем.
Предназначенная для орального введения фармацевтическая композиция, содержащая ингибитор ренина, прежде всего производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты, и ингибитор откачивающего белка, может находиться в форме растворов, суспензий, таблеток, пилюль, капсул, порошков, микроэмульсий, пакетов, содержащих стандартную дозу, и т.п. Предпочтительными являются таблетки и желатиновые капсулы, содержащие действующее вещество в сочетании с: а) разбавителями, например, лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином; б) замасливателями, например, диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; для таблеток также с в) связующими веществами, например, алюмосиликатом магния, крахмальной пастой, желатином, трагакантом, метилцеллюлозой, натрийкарбоксиметилцеллюлозой и/или поливинилпирролидоном; при необходимости с г) разрыхлителями, например, крахмалами, агаром, альгиновой кислотой или ее натриевой солью, или шипучими смесями; и/или с д) абсорбентами, красителями, корригентами и подслащивающими веществами. Предназначенные для инъекции композиции предпочтительно представляют собой водные изотонические растворы или суспензии, а суппозитории преимущественно приготавливают из жирных эмульсий или суспензий.
Указанные композиции могут быть стерилизованными и/или содержать адъюванты, такие как консерванты, стабилизаторы, смачивающие или эмульгирующие агенты, вещества, повышающие растворимость, соли для регулирования осмотического давления и/или буферы. Кроме того, они могут содержать также другие обладающие терапевтической ценностью субстанции. Указанные композиции приготавливают с использованием общепринятых методов смешения, грануляции или нанесения покрытия соответственно, и они содержат примерно 0,1-75%, предпочтительно примерно 1-50% действующего вещества.
Более конкретно в настоящем изобретении предложена фармацевтическая композиция, содержащая в терапевтически эффективном количестве ингибитор ренина, предпочтительно производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты, в сочетании с ингибитором откачивающего белка, в которой ингибитор откачивающего белка присутствует в количестве, обеспечивающем после ее введения повышение биологической доступности ингибитора ренина по меньшей мере на 5%.
Предпочтительно фармацевтическая композиция, предлагаемая в настоящем изобретении, содержит ингибитор MDR1, например, PSC833.
Предпочтительно фармацевтическая композиция, предлагаемая в настоящем изобретении, содержит производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты формулы
в которой R1 обозначает С1-С4алкокси-С1-Салкоксигруппу или C1-С4алкокси-С1-С4алкил; R2 обозначает С1-С4алкил или С1-С4алкоксигруппу; и R3 и R4 независимо друг от друга обозначают разветвленный С1-С4алкил; или его фармацевтически приемлемую соль; в сочетании с ингибитором MDR1, например, PSC833.
Более предпочтительно фармацевтическая композиция, предлагаемая в настоящем изобретении, содержит производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты формулы (I), в которой R1 обозначает 3-метоксипропоксигруппу; R2 обозначает метоксигруппу; и R3 и R4 обозначают изопропил; или его фармацевтически приемлемую соль; в сочетании с ингибитором MDR1, например, PSC833.
Наиболее предпочтительно фармацевтическая композиция, предлагаемая в настоящем изобретении, содержит гемифумарат (2-карбамоил-2-метилпропил)амида (2S,4S,5S,7S)-5-амино-4-гидрокси-2-изопропил-7-[4-метокси-3-(3-метоксипропокси)бензил]-8-метилнонановой кислоты в сочетании с ингибитором MDR1, например, PSC833.
Предпочтительно биологическую доступность ингибитора ренина, прежде всего производного δ-амино-γ-гидрокси-ω-арилалкановой кислоты, например, SPP100, или его фармацевтически приемлемой соли повышают по меньшей мере на 5%.
Биологическую доступность лекарственного средства можно оценивать известным в данной области методом путем измерения AUC, где AUC представляет собой площадь под кривой графика, где по оси ординат (ось Y) отложена концентрация лекарственного средства в сыворотке или плазме, а по оси абсцисс (ось X) - время. Как правило, величины AUC вычисляют на основе большого количества результатов, полученных для всех индивидуумов, входящих в тестируемую популяцию, и следовательно, они представляют собой средние значения для всей тестируемой популяции.
Совместное введение ингибитора ренина и ингибитора откачивающего белка может также приводить к увеличению Сmax по сравнению с введением ингибитора ренина в отсутствии ингибитора откачивающего белка, и это является еще одним объектом изобретения. В данной области величина Сmax широко известна в качестве сокращения обозначения максимальной концентрации лекарственного средства в сыворотке или плазме тестируемого индивидуума.
Поскольку один из объектов настоящего изобретения относится к лечению с помощью комбинации соединений, предназначенных для совместного введения, которые можно вводить по отдельности, то изобретение относится также к отдельным фармацевтическим композициям, объединенным в форме набора. Набор содержит две индивидуальные фармацевтические композиции: (1) композицию, содержащую ингибитор ренина, прежде всего производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты, плюс фармацевтически приемлемый носитель или разбавитель; и (2) композицию, содержащую ингибитор откачивающего белка, плюс фармацевтически приемлемый носитель или разбавитель. Количества компонентов (1) и (2) должны быть такими, чтобы при совместном их применении путем раздельного введения биологическая доступность ингибитора ренина, прежде всего производного δ-амино-γ-гидрокси-ω-арилалкановой кислоты, повышалась по меньшей мере на 5%. Набор представляет собой содержащий индивидуальные композиции контейнер, такой как градуированная бутылка или разделенный на несколько компартментов пакет из фольги, где в каждом компартменте находится несколько лекарственных форм (например, таблеток), которые содержат компонент (1) или (2). В альтернативном варианте вместо индивидуальных содержащих действующее вещество лекарственных форм набор может иметь отдельные компартменты, каждый из которых содержит полную дозу, которая в свою очередь содержит индивидуальные лекарственные формы. Примером набора такого типа может служить блистерная упаковка, в которой каждый индивидуальный блистер содержит две (или более) таблеток, из которых одна (или несколько) таблетка(ок) содержит(ат) фармацевтическую композицию, указанную в подпункте (1), а вторая (или несколько) таблетка(ок) содержит(ат) фармацевтическую композицию, указанную в подпункте (2). Как правило, набор содержит инструкцию для введения индивидуальных компонентов. Форма в виде набора особенно предпочтительна в том случае, когда индивидуальные компоненты предпочтительно вводят в виде различных лекарственных форм (например, орально и парентерально), вводят с различными интервалами времени или когда лечащий врач считает необходимым осуществлять титрование индивидуальных компонентов комбинации. Таким образом, в настоящем изобретении предложен набор, включающий:
(1) композицию, содержащую в терапевтически эффективном количестве ингибитор ренина, прежде всего производное δ-амино-γ-гидрокси-ω-арилалкановой кислоты, например, SPP100, или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель или разбавитель, в качестве первой лекарственной формы;
(2) композицию, содержащую ингибитор откачивающего белка в количестве, обеспечивающем после введения повышение биологической доступности ингибитора ренина, прежде всего производного δ-амино-γ-гидрокси-ω-арилалкановой кислоты, например, SPP100, или его фармацевтически приемлемой соли, по меньшей мере на 5%, и фармацевтически приемлемый носитель или разбавитель, в качестве второй лекарственной формы; и
(3) контейнер, содержащий первую и вторую лекарственные формы.
Наконец, настоящее изобретение относится к применению ингибитора откачивающего белка, прежде всего, ингибитора MDR1, например, PSC833, для приготовления лекарственного средства, предназначенного для повышения биологической доступности, предпочтительно оральной биологической доступности, ингибитора ренина, предпочтительно производного δ-амино-γ-гидрокси-ω-арилалкановой кислоты, например, SPP100, или его фармацевтически приемлемой соли.
Можно идентифицировать откачивающий(ие) белок(ки), участвующий(ие) в откачивании лекарственной субстанции, и определять соответствующие кинетические параметры, т.е. константу Михаэлиса-Ментена и максимальный транспорт лекарственного средства (Кm и Jmax), с помощью методов, известных в данной области, например, с помощью анализа АТФазы с использованием мембранных пузырьков Sf9 (Spodopterafruigiperdd), обладающих высокими уровнями экспрессии выбранного АВС-транспортера. В этом анализе АВС-транспортеры удаляют субстраты из клеток, используя гидролиз АТФ в качестве источника энергии. Гидролиз АТФ приводит к образованию неорганического фосфата (Pi), который можно обнаруживать с помощью простой колориметрической реакции. Количество Pi, высвободившегося в результате воздействия транспортера, пропорционально активности транспортера. Препараты мембран, содержащие АВС-транспортеры, обладают исходной АТФазной активностью, которая варьируется для различных транспортеров. Транспортируемые субстраты повышают эту исходную АТФазную активность, в то время как ингибиторы ингибируют исходную АТФазную активность и/или АТФазную активность, измеряемую в присутствии стимулирующего агента. Можно проводить изучение, как активации, так и ингибирования. Как проиллюстрировано в примере 1 (фиг.1) настоящего описания, SPP100 увеличивает АТФазную активность в мембранных пузырьках, обладающих высокими уровнями экспрессии MDR1, при этом значение Кm составляет примерно 3 мкМ, это позволяет предположить, что откачивающая система, участвующая в транспорте SPP100, по-видимому, представляет собой MDR1.
В альтернативном варианте аффинность in vitro транспортера лекарственной субстанции можно определять и аппроксимировать с помощью анализа с использованием клеток линии Сасо-2 согласно методу, описанному, например, у Camenisch и др., Pharm. Act. Helv. 71, 1996, cc.309-327 или согласно методу, проиллюстрированому в настоящем описании в примерах. Идентификацию белка-транспортера и эффективность соединения в отношении ингибирования откачивающей системы, участвующей в транспорте, также можно осуществлять с помощью анализа с использованием клеток линии Сасо-2. Например, SPP100 идентифицируют как соединение, обладающее от низкой до средней проницаемостью (присущая ему проницаемость <80%), которое, кроме того, является субстратом для известной откачивающей системы (фиг.2). Однако в присутствии ингибитора MDR1 PSC833 проницаемость SPP100 существенно возрастает, т.е. PSC833 ингибирует откачивание SPP100, причем значение IC50 составляет примерно 0,1 мкМ (фиг.3).
Воздействие in situ совместного введения ингибитора откачивающего белка, например, ингибитора MDR1, на билиарную экскрецию ингибитора ренина можно исследовать путем сравнения количества соединения, экскретируемого в желчь в присутствии ингибитора откачивающего белка и без него. Например, в присутствии PSC833 билиарный клиренс SPP100 уменьшается на 97% по сравнению с контрольной группой (фиг.4).
Аналогично этому воздействие in vivo совместного введения ингибитора откачивающего белка, например, ингибитора MDR1, на биологическую доступность ингибитора ренина можно исследовать путем сравнения фармакокинетических параметров Сmax и AUC в присутствии ингибитора откачивающего белка и без него. Как проиллюстрировано в примере 4 (фиг.5) настоящего описания, у обработанных оральным путем крыс стандартизованная по отношению к дозе величина AUC(0-tlast) SPP100 возрастала в присутствии PSC833 примерно в 70 раз по сравнению с контрольной группой, которую обрабатывали только SPP100.
Приведенное выше описание полностью раскрывает изобретение, включая предпочтительные варианты его осуществления. Модификации и усовершенствования вариантов осуществления изобретения, конкретно представленных в настоящем описании, подпадают под объем приведенной ниже формулы изобретения. На основе предыдущего описания специалист в данной области может без дополнительных экспериментов осуществлять настоящее изобретение на практике в его полном объеме. Таким образом, приведенные в настоящем описании примеры служат только для иллюстрации и никоим образом не направлены на ограничение объема настоящего изобретения.
Пример 1
Анализ АТФазы
Опосредуемое человеческим MDR1, человеческим MRP1 или человеческим MRP2 откачивание оценивают путем инкубации очищенных мембранных пузырьков в присутствии стимулирующего агента и без него (верапамил [40 мкМ] для MDR1, NEM-GS [10мМ] для MRP1 и пробенецид [1 мМ] для MRP2) с различными концентрациями лекарственной субстанции [0,046, 0,137, 0,41, 1,23, 3,7, 11,1, 33,3 и 100 мкМ] в буфере для транспорта при значении рН 7,4 при 37°С.
5 мМ маточный раствор рассматриваемого терапевтического агента приготавливают в обычном органическом растворителе, например, диметилсульфоксиде, этаноле, метаноле и ацетонитриле, таким образом, чтобы после добавления маточного раствора или его разведении в смесь для анализа получать указанные выше конечные концентрации, и используемый органический растворитель должен составлять 2% общего объема (об./об.). Во всех растворах, используемых для анализа, значение рН поддерживают на уровне 7,4.
Для экспериментов с использованием АТФазы применяют мембранные пузырьки, хранившиеся при -80°С. Опосредуемое транспортером откачивание можно оценивать согласно описанному в литературе методу (Sarkadi В., Price E., Boucher R., Germann U., Scarborough G., J. Biol. Chem., 267, 1992, cc.4854-4858). В целом, метод состоит в следующем: суспензию мембран в присутствии тестируемого лекарственного средства и без него, стимулирующего агента, Na3VO4, 60 мМ, и глутатиона, 2 мМ, (только для транспортеров MRP1 и MRP2) вносят с помощью пипетки в 96-луночный планшет и подвергают предварительной инкубации при 37°С в течение 5 мин. АТФазную реакцию инициируют путем добавления 25 мМ раствора Mg-АТФ и последующей инкубации при 37°С (20 мин для MDR1, 60 мин для MRP1 и 30 мин для MRP2). Затем АТФазную реакцию прекращают путем добавления ДСН (5%) к каждому подвергнутому инкубации образцу. После добавления реагента для колориметрического обнаружения, молибдата/ацетата цинка, планшет инкубируют еще в течение 25 мин при 37°С.
После инкубации измеряют ОП при 730 нм. С использованием ранее построенной стандартной кривой для фосфата можно рассчитать количество высвободившегося Pi [нмолей/лунку]. Величины ОП представляют в виде средних значений±стандартных отклонений для проведенных экспериментов (n=2). Все статистические анализы проводят с помощью программы Microsoft EXCEL 5.0 с.
Для расчета так называемой удельной (чувствительной к Na3VO4) транспортерной АТФазной активности для каждого анализируемого лекарственного средства и концентрации лекарственного средства следует вычесть величины Pi, определенные в присутствии Na3VO4, из величин Pi, измеренных без Nа3VO4. Чувствительную к Nа3VO4 транспортерную активность, выраженную в виде количества высвободившегося Pi/мг мембранного белка мембраны/мин, можно определять путем деления полученных величин на количество мембранного белка, добавленного в каждую лунку и на время инкубации в минутах (фиг.1).
Пример 2
Анализ с использованием клеток линии Сасо-2
Для экспериментов по оценке транспорта используют монослои клеток линии Сасо-2, выращенные на полиэтилентерефталатных фильтрах (РЕТ-фильтры) в течение 21-25 дней. Поток соединений через монослои клеток линии Сасо-2, выращенные на РЕТ-фильтрах, а также только через РЕТ-фильтры без клеток линии Сасо-2, определяют следующим образом: Перед проведением эксперимента по оценке транспорта культуральную среду в акцепторном компартменте (0,2 мл для апикальной и 1,0 мл для базолатеральной сторон) заменяют акцепторным раствором (сбалансированный солевой раствор Хенкса (HBSS), содержащий, когда это требуется, рассматриваемый ингибитор), предварительно инкубированным при 37°С. Эксперимент начинают с того, что среду в донорном компартменте (0,35 мл для апикальной и 1,15 мл для базолатеральной сторон) заменяют донорным раствором (раствор соединения в HBSS, содержащий, когда это требуется, рассматриваемый ингибитор), предварительно инкубированным при 37°С. Через примерно 1 и 120 мин отбирают аликвоты по 150 мкл с донорной и акцепторной сторон. Эксперименты по оценке транспорта как в апикально-базолатеральном, так и в базолатерально-апикальном направлениях проводят в трех повторностях при 37°С в инкубаторе без встряхивания.
Пригодность клеток линии Сасо-2 для экспериментов по оценке транспорта исследуют путем измерения способности [3H]-маннита в концентрации <0,1 мкМ и [3Н]-пропрандолола в концентрации ≤0,1 мкМ проникать в течение 120 мин с апикальной на базолатеральную сторону в общей сложности для 6 репрезентативных клеточных монослоев (по 3 для каждого соединения) для одной и той же партии клеток.
Радиоактивные образцы анализируют с помощью жидкостного сцинтилляционного счетчика. Все другие не имеющие радиоактивной метки образцы хранят в замороженном состоянии при -20°С до проведения анализа с помощью жидкостной хроматографии/тандемной масс-спектрометрии (ЖХ-МС/МС).
Коэффициенты транспорта тестируемых соединений определяют с помощью следующего уравнения (Artwsson и др., Biochem. Biophys. Res. Comm. 175, 1991, cc.880-885):
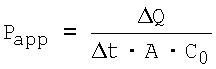
в котором Рaрр (см/мин) обозначает эффективный (кажущийся) коэффициент проницаемости, ΔQ обозначает количество соединения, обнаруженного в акцепторном компартменте в момент времени t, Δt (мин) обозначает период времени инкубации. С0 (мкг/мл) обозначает начальную концентрацию соединения в донорном компартменте и А (см2) обозначает площадь поверхности мембраны.
Для меченых образцов в качестве предела количественной оценки (LOQ) принимают величину наименьшей концентрации, получаемую с использованием радиоактивной шкалы, которая существенно превышает измеренную величину для контроля и для которой стандартная ошибка измерения составляет менее 20%. В условиях настоящего исследования LOQ абсолютной величины радиоактивности составляет 2 распада в минуту для меченного с помощью [14С] SPP100 с концентрацией, что соответствует 12 нмолей/л.
Величины Рарр выражают в виде средних значений ± стандартных отклонений для проведенных экспериментов по оценке транспорта (n=3). Статистическую значимость разницы между двумя рассматриваемыми рядами данных анализируют с помощью t-критерия. Уровень вероятности, при котором разница является значимой, составляет р<0,025. Все статистические анализы проводят с помощью программы Microsoft EXCEL 5.0 с.
Для концентрации SPP100 1 мкМ в течение периода времени 120 мин можно обнаружить уровень апикально-базолатерального транспорта, составляющий примерно 0,2·10-5 см/мин. С другой стороны, базолатерально-апикальный транспорт характеризуется величиной проницаемости, составляющей примерно 10·10-5 см/мин, что существенно превышает апикально-базолатеральный транспорт.
Для концентраций SPP100, составляющих 1,5, 10 и 50 мкМ, обнаружено постепенное увеличение апикально-базолатерального транспорта, величина проницаемости при 10 мкМ выходит на постоянный уровень, составляющий примерно 7-10-5 см/мин. С другой стороны, базолатерально-апикальный транспорт не претерпевает существенных изменений при увеличении концентраций.
Величины выхода при транспорте SPP100 (1,5, 10 и 50 мкМ) через клетки линии Сасо-2, как правило, являются очень большими (<100%), это свидетельствует о том, что SPP100 не связывается с основой фильтра или с находящимися в инкубационной камере пластиковыми материалами.
Апикально-базолатеральный поток парацеллюларного маркера маннита и трансцеллюларного маркера пропранолола всегда находятся ниже пороговых значений Рарр, составляющих 3·10-5 см/мин и 90·10-5 см/мин соответственно. Измеренные проницаемости при прохождении через фильтр в апикально-базолатеральном направлении, как правило, превышают соответствующие величины проницаемости, полученные при использовании клеток линии Сасо-2, это свидетельствует о том, что диффузия через фильтр не является ограничивающей скорость стадией транспорта через клетки линии Сасо-2 (фиг.2 и 3).
Пример 3
Эксперимент на крысах с использованием канюли, введенной in situ в желчный проток
Участие MDR1 и/или MRP2 в билиарной экскреции SPP100 можно выявлять с помощью следующего опыта на крысах. Так, билиарный клиренс SPP100 оценивают на самцах крыс с введенной в желчный проток канюлей и подвергнутых анестезии при непрерывной внутривенной инфузии меченного с помощью [14С] гемифумарата SPP100 до и после введения PSC833 (известный ингибитор MDR1 и MRP2) или пробенецида (известный ингибитор MRP2 селективного действия).
Животные, которых можно использовать для этой цели, представляют собой, например, самцов крыс-альбиносов линии HAN:WIST, по 4-5 животных в каждой обрабатываемой группе.
Меченный с помощью [14С] гемифумарат SPP100 вводят в 0,9%-ном растворе хлорида натрия, а PSC833 и пробенецид вводят в смеси этанол/полиэтиленгликоль 200/5%-ный раствор глюкозы (соотношение 1/3/1).
Во всех обрабатываемых группах постоянной концентрации исходного соединения в плазме достигают путем болюсной внутривенной инъекции (2 мл/кг) меченного с помощью [14С] раствора для болюсной инъекции гемифумарата SPP100 (0,015 мг основания/мл) и последующей внутривенной инфузии (6,67 мл/ч/кг) меченного с помощью [14С] раствора для инфузии гемифумарата SPP100 (0,022 мг основания/мл).
Через два часа осуществляют болюсное внутривенное введение следующих соединений:
обрабатываемая группа 1: смесь (1/3/1) этанол/полиэтиленгликоль 200/5%-ный раствор глюкозы (5 мл/кг; наполнитель);
обрабатываемая группа 2: PSC833 в дозе 10 мг/кг; и
обрабатываемая группа 3: пробенецид в дозе 50 мг/кг.
У животных во всех обрабатываемых группах через 0,5, 1, 1,33, 1,67, 2, 2,33, 2,67, 3 ч после обработки берут образцы крови и из крови сразу же получают плазму. Берут образцы желчи в течение промежутка времени 1-3 ч с 20-минутными интервалами.
Метод обнаружения:
для подсчета общей радиоактивности во всех образцах: жидкостно-твердая хроматография (LSC); порог количественного обнаружения LOQ 3,4 мкг/л для плазмы и желчи; и
для выявления состава метаболитов в выбранных пулах образцов: ЖХВР с радиоактивным обнаружением.
После внутривенного введения носителя (этанол/полиэтиленгликоль 200/5%-ный раствор глюкозы) не выявлено влияния на концентрацию 14С в плазме и на билиарный клиренс меченных с помощью [14С] соединений в процессе непрерывной инфузии меченного с помощью [14С] гемифумарата SPP100 крысам с введенной в желчный проток канюлей.
После введения PSC833 концентрация 14С имеет тенденцию к увеличению в зависимости от времени в плазме и к значительному уменьшению в желчи. Результирующий билиарный клиренс меченных с помощью [14С] соединений составляет примерно 7% от величины, полученной перед введением PSC833.
Введение пробенецида не оказывает влияния на концентрации в плазме меченных с помощью [14С] соединений у крыс с введенной в желчный проток канюлей. Однако пробенецид приводит к увеличению потока желчи (его величина после введения примерно на 65% больше, чем величина до введения) и к уменьшению концентрации [14С] в желчи (ее величина примерно на 40% меньше, чем величина до введения). Тем не менее, величины билиарного клиренса близки друг к другу в оба периода времени (до и после введения пробенецида).
На основании этих результатов (фиг.4) можно сделать вывод о том, что MDR1, по-видимому, играет важную роль в билиарном клиренсе SPP100 и родственных SPP100 соединений. Кроме того, по-видимому, MRP2 не участвует в билиарной экскреции SPP100 и родственных SPP100 соединений.
Пример 4
Эксперимент по изучению биологической доступности in vivo
Изучение относительной биологической доступности SPP100 при его введении оральным путем на самцах крыс после однократного введения оральным путем гемифумарата SPP100 в сочетании с совместным введением оральным путем PSC833 или без него можно проводить с помощью следующего исследования. Для выявления зависимости от дозы совместно вводимого PSC833 в сочетании с определенной дозой гемифумарата SPP100 (6 мг свободного основания/кг) вводят различные дозы PSC833 (0, 5, 12,5, 25 и 50 мг/кг).
В качестве животных можно использовать, например, самцов крыс-альбиносов линии HAN:WIST, по 5 животных в каждой обрабатываемой группе.
Гемифумарат SPP100 вводят в 0,9%-ном растворе хлорида натрия, а PSC833 вводят в смеси (1/3/1) этанол/полиэтиленгликоль 200/5%-ный раствор глюкозы (наполнитель).
Крысам вводят с помощью желудочного зонда сначала наполнитель (5 мл/кг), а затем гемифумарат SPP100 (2 мл/кг) (разница во времени: 2-3 мин). Всем крысам вводят гемифумарат SPP100 в дозе, составляющей 6 мг свободного основания/кг.Обработку PSC833 осуществляют с использованием следующих доз:
обрабатываемые группы 1 и 6: смесь (1/3/1) этанол/полиэтиленгликоль 200/5%-ный раствор глюкозы (5 мл/кг; наполнитель);
обрабатываемая группа 2: 50 мг/кг PSC833;
обрабатываемая группа 3: 25 мг/кг PSC833;
обрабатываемая группа 4: 12,5 мг/кг PSC833; и
обрабатываемая группа 5: 5 мг/кг PSC833.
Из подъязычной области берут образцы крови через 0,25, 0,5, 1, 2, 4, 8, 24 и 48 ч после введения. Плазму используют для биохимического анализа.
Аналитические методы:
ЖХВР-МС/МС с использованием APCI в SRM-позитивном режиме для SPP100, нижний LOQ в плазме составляет от 0,6 до 0,7 нг/мл.
У крыс, которым вводят оральным путем гемифумарат SPP100 (6 мг основания/кг) и наполнитель, SPP100 можно выявлять в плазме только в течение вплоть до 2 ч после введения дозы. Величины Сmax, составляющие примерно до 24,8±27 нг/мл, достигаются при tmax, составляющем 0,44±0,1 ч после введения дозы. Величина стандартизованной по отношению к дозе AUC(0-tlast) составляет 2,38±3,4 [(нг·ч/мл)/(мг/кг)].
У крыс, которым вводят оральным путем гемифумарат SPP100 (6 мг основания/кг) и PSC833 (5, 12,5, 25 и 50 мг/дозу), SPP100 можно обнаруживать в плазме в течение периода времени вплоть до 24 или 48 ч после введения дозы. По сравнению с крысами, которым вводят наполнитель, величины Сmax возрастают до 35,7±18, 115±93, 81,4±19 и 26,7±18 нг/мл, а величина tmax до 4,8±1,8, 4,95±4,2, 15,2±18 и 3,06±1,9 ч для доз PSC833 5, 12,5, 25 и 50 мг/кг соответственно. Величины стандартизованных по отношению к дозе AUC(0-tlast) примерно в 23, 69, 76 и 17 больше, чем в группе, обработанной наполнителем, при совместном введении PSC833 в дозах 5, 12,5, 25 и 50 мг/кг соответственно. Причина, обусловливающая относительно низкую величину frel для PSC833, вводимого в дозе 50 мг/кг по сравнению с дозой 25 мг/кг, в настоящее время не выяснена (фиг.5).
На основе этих результатов можно сделать вывод о том, что по меньшей мере один или оба из относящихся к семейству АВС откачивающих белков-транспортеров, MDR1 и MRP2, по-видимому, оказывают важное влияние на степень доступности SPP100 после орального введения.
Пример 5
Меченный с помощью [14С] гемифумарат SPP100, [14С]-(2-карбамоил-2-метилпропил)амид (2S,4S,5S,7S)-5-амино-4-гидрокси-2-изопропил-7-[4-метокси-3-(3-метоксипропокси)бензил]-8-метилнонановой кислоты
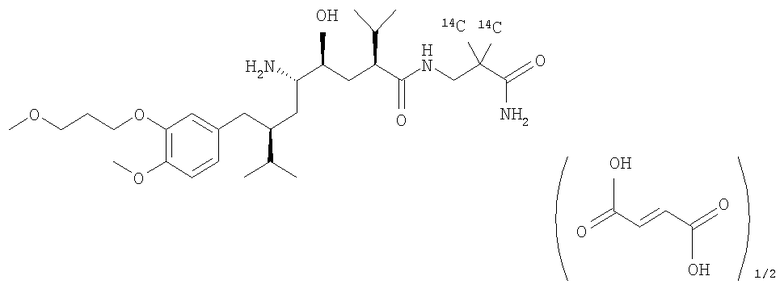
А. Метиловый эфир циан-[14С]-диметилуксусной кислоты
В ампулу вносят раствор метоксида натрия (119 мг, 2,2 ммоля) в метаноле (2 мл) при КТ. Добавляют при 0°С метилцианацетат (99 мг, 1,0 ммоля). Смесь встряхивают в течение нескольких минут при КТ и затем замораживают до -192°С в азоте и в реакционную смесь вносят в вакууме меченный с помощью [14С] метилйодид (3,7 ГБк при 2,04 ГБк/ммоль, 1,82 ммоля, полученный от фирмы Amersham Biosciences). Ампулу запечатывают в вакууме и дают медленно нагреться до КТ в течение периода времени, составляющего 1 ч. Ампулу встряхивают при 50°С в течение 16 ч. Затем ампулу повторно замораживают до -192°С и удаляют путем лиофилизации летучий растворитель и непрореагировавший меченный с помощью [14С] метилйодид. Затем неочищенный метиловый эфир циан-[14С]-диметилуксусной кислоты анализируют с помощью ГХ для гарантии того, что образовалось <0,2% монометилированного побочного продукта. После этого продукт используют на следующей стадии без дополнительной очистки.
Б. 2-циан-2,2-[14С]-диметилацетамид
Газообразным аммиаком барботируют находящийся в колбе безводный метанол в течение 30 мин при КТ или до достижения молярности 5М или более. К неочищенному указанному в заголовке раздела А соединению, метиловому эфиру циан-[14С]-диметилуксусной кислоты, добавляют свежеприготовленный 5,5М NН3 в МеОН (3 мл, 16,5 ммолей) при КТ. Реакционную смесь перемешивают при КТ в течение 2 ч, по истечении этого периода времени весь исходный продукт превращается в нитрил по данным ГХ-анализа, который проводят согласно следующему методу: 10 мин при 70°С, затем повышают температуру со скоростью 10°С/мин до 200°С, после чего выдерживают при 200°С в течение 10 мин. После завершения реакции растворитель удаляют путем лиофилизации и продукт очищают с помощью экспресс-хроматографии (этилацетат → этилацетат/10% метанола), получая 2-циан-2,2-[14С]-диметиламид, 2,813 ГБк.
В. 3-амино-2,2-[14С]-диметилпропионамид
К свежеочищенному указанному в заголовке раздела Б соединению, 2-циан-2,2-[14С]-диметиламиду (2,813 ГБк, 77 мг, 0,69 ммоля), добавляют свежеприготовленный 5,5М раствор NH2 в МеОН при КТ, а затем 5% Rh/Al2O3 (33 мг). Реакционную смесь перемешивают при 55°С в течение промежутка времени вплоть до 5 ч и каждый час осуществляют мониторинг с помощью ГХ до тех пор, пока весь исходный материал не превратится в продукт. ГХ-метод: 1 мин при 80°С, затем повышают температуру со скоростью 20°С/мин до 240°С, после чего выдерживают в течение 1 мин при 240°С. После завершения реакции смесь фильтруют через целит (фирма Hyflo), растворитель удаляют на роторном испарителе и продукт очищают с помощью экспресс-хроматографии (CH2Cl2/2% MeOH/NH3 → СН2Сl2/5%МеОН/NH3 → CH2Cl2/10% МеОН/NH3), получая 3-амино-[14С]-2,2-диметилпропионамид, 2,7 ГБк, в виде твердого вещества белого цвета. Этот продукт можно хранить в твердой форме в течение периодов времени, превышающих 1 неделю. Для более длительного хранения 3-амино-2,2-[С]-диметилпропионамид можно растворять в EtOH/20% толуола при -80°С при концентрации, не превышающей 50 МБк/мл.
Г. меченный с помощью [14С] (2-карбамоил-2-метилпропил)амид (2S,4S,5S,7S)-5-амино-4-гидрокси-2-изопропил-7-[4-метокси-3-(3-метоксипропокси)бензил]-8-метилнонановой кислоты, [14C]-SPP100, гемифумарат
Указанное в заголовке раздела В соединение, 3-амино-2,2-[14С]-диметилпропионамид, можно использовать для получения указанного в заголовке раздела Г соединения, меченного с помощью [14С] гемифумарата SPP100, согласно методам, известным в данной области, например, описанным в US 6730798.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИДЫ СИГМА-АМИНО-ГАММА-ГИДРОКСИ-ОМЕГА-АРИЛАЛКАНОВЫХ КИСЛОТ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕНИНА | 2004 |
|
RU2425027C2 |
| АМИДЫ δ-АМИНО-γ-ГИДРОКСИ-ω-АРИЛАЛКАНОВОЙ КИСЛОТЫ | 2004 |
|
RU2413716C2 |
| РАСКРЫТИЕ ЦИКЛОВ ЛАКТОНОВ И ЛАКТАМОВ | 2010 |
|
RU2550691C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРОВ РЕНИНА ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ДИАСТОЛИЧЕСКОЙ ДИСФУНКЦИИ ИЛИ ДИАСТОЛИЧЕСКОЙ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ | 2005 |
|
RU2407523C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ВАНИЛОИДНЫХ АНТАГОНИСТОВ | 2005 |
|
RU2449995C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ВАНИЛОИДНЫХ АНТАГОНИСТОВ | 2005 |
|
RU2396261C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА И ИНГИБИТОР БЕЛКА СЛИЯНИЯ RSV | 2005 |
|
RU2388476C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ИНГИБИТОРЫ NEP, ИНГИБИТОРЫ СИСТЕМЫ, ПРОДУЦИРУЮЩЕЙ ЭНДОГЕННЫЙ ЭНДОТЕЛИН, И ДИУРЕТИКИ | 2006 |
|
RU2409366C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ КОМПЛЕМЕНТОМ НАРУШЕНИЙ | 2015 |
|
RU2703995C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРОВ ПКС ПРИ ТРАНСПЛАНТАЦИИ | 2007 |
|
RU2494738C2 |
Изобретение относится к лекарственным средствам и касается способа повышения биологической доступности ингибитора ренина, заключающегося в том, что млекопитающему, нуждающемуся в таком лечении, совместно вводят комбинацию, содержащую ингибитор ренина и ингибитор откачивающего белка, в которой ингибитор откачивающего белка представляет собой ингибитор MDR1, такой как PSC833, а ингибитор ренина представляет собой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты, формулы (I). Также раскрыта фармацевтическая композиция, обладающая гипотензивным действием, содержащая в терапевтически эффективном количестве ингибитор ренина в сочетании с ингибитором откачивающего белка, и применение ингибитора откачивающего белка для приготовления лекарственного средства, предназначенного для повышения биологической доступности ингибитора ренина. 6 н. и 15 з.п. ф-лы, 5 ил.
где R1 R2, R3 и R4 имеют указанные в формуле значения
1. Способ повышения биологической доступности ингибитора ренина, заключающийся в том, что млекопитающему, нуждающемуся в таком лечении, совместно вводят комбинацию, содержащую ингибитор ренина и ингибитор откачивающего белка, в которой ингибитор откачивающего белка представляет собой ингибитор MDR1, а ингибитор ренина представляет собой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты, имеющее формулу
в которой R1 обозначает С1-С4алкокси-С1-С4алкоксигруппу или С1-С4алкокси-С1-С4алкил; R2 обозначает С1-С4алкил или С1-С4алкоксигруппу; и R3 и R4 независимо друг от друга обозначают разветвленный С1-С4алкил; или его фармацевтически приемлемую соль.
2. Способ по п.1, в котором производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой соединение формулы (I), в которой R1 обозначает 3-метоксипропоксигруппу; R2 обозначает метоксигруппу; и R3 и R4 обозначают изопропил; или его фармацевтически приемлемую соль.
3. Способ по п.2, в котором производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой гемифумарат (2-карбамоил-2-метилпропил)амида (2S,4S,5S,7S)-5-амино-4-гидрокси-2-изопропил-7-[4-метокси-3-(3-метоксипропокси)бензил]-8-метилнонановой кислоты.
4. Способ по п.1, в котором ингибитор MDR1 представляет собой PSC833.
5. Способ повышения биологической доступности ингибитора ренина, заключающийся в том, что млекопитающему, нуждающемуся в таком лечении, совместно вводят комбинацию, содержащую ингибитор ренина и ингибитор откачивающего белка, в которой ингибитор откачивающего белка представляет собой PSC833, а ингибитор ренина представляет собой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты, имеющее формулу
в которой R1 обозначает С1-С4алкокси-С1-С4алкоксигруппу или C1-С4алкокси-С1-С4алкил; R2 обозначает С1-С4алкил или С1-С4алкоксигруппу; и R3 и R4 независимо друг от друга обозначают разветвленный С1-С4алкил; или его фармацевтически приемлемую соль.
6. Способ по п.5, в котором производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой соединение формулы (I), в которой R1 обозначает 3-метоксипропоксигруппу; R2 обозначает метоксигруппу; и R3 и R4 обозначают изопропил; или его фармацевтически приемлемую соль.
7. Способ по п.6, в котором производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой гемифумарат (2-карбамоил-2-метилпропил)амида (2S,4S,5S,7S)-5-амино-4-гидрокси-2-изопропил-7-[4-метокси-3-(3-метоксипропокси)бензил]-8-метилнонановой кислоты.
8. Фармацевтическая композиция, обладающая гипотензивным действием, содержащая в терапевтически эффективном количестве ингибитор ренина в сочетании с ингибитором откачивающего белка, где ингибитор откачивающего белка представляет собой ингибитор MDR1, а ингибитор ренина представляет собой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты, имеющее формулу
в которой R1 обозначает С1-С4алкокси-С1-С4алкоксигруппу или C1-С4алкокси-С1-С4алкил; R2 обозначает С1-С4алкил или С1-С4алкоксигруппу; и R3 и R4 независимо друг от друга обозначают разветвленный С1-С4алкил; или его фармацевтически приемлемую соль.
9. Фармацевтическая композиция по п.8, в которой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой соединение формулы (I), в которой R1 обозначает 3-метоксипропоксигруппу; R2 обозначает метоксигруппу; и R3 и R4 обозначают изопропил; или его фармацевтически приемлемую соль.
10. Фармацевтическая композиция по п.9, в которой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой гемифумарат (2-карбамоил-2-метилпропил)амида (2S,4S,5S,7S)-5-аминно-4-гидрокси-2-изопропил-7-[4-метокси-3-(3-метоксипропокси)бензил]-8-метилнонановой кислоты.
11. Фармацевтическая композиция по п.8, в которой ингибитор MDR1 представляет собой PSC833.
12. Фармацевтическая композиция, обладающая гипотензивным действием, содержащая в терапевтически эффективном количестве ингибитор ренина в сочетании с ингибитором откачивающего белка, где ингибитор откачивающего белка представляет собой PSC833, а ингибитор ренина представляет собой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты, имеющее формулу
в которой R1 обозначает С1-С4алкокси-С1-С4алкоксигруппу или C1-С4алкокси-С1-С4алкил; R2 обозначает С1-С4алкил или С1-С4алкоксигруппу; и R3 и R4 независимо друг от друга обозначают разветвленный С1-С4алкил; или его фармацевтически приемлемую соль.
13. Фармацевтическая композиция по п.12, в которой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой соединение формулы (I), где R1 обозначает 3-метоксипропоксигруппу; R2 обозначает метоксигруппу; и R3 и R4 обозначают изопропил; или его фармацевтически приемлемую соль.
14. Фармацевтическая композиция по п.13, в которой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой гемифумарат (2-карбамоил-2-метилпропил)амида (2S,4S,5S,7S)-5-амино-4-гидрокси-2-изопропил-7-[4-метокси-3-(3-метоксипропокси)бензил]-8-метилнонановой кислоты.
15. Применение ингибитора откачивающего белка для приготовления лекарственного средства, предназначенного для повышения биологической доступности ингибитора ренина, где ингибитор откачивающего белка представляет собой ингибитор MDR1, а ингибитор ренина представляет собой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты, имеющее формулу
в которой R1 обозначает С1-С4алкокси-С1-С4алкоксигруппу или С1-С4алкокси-С1-С4алкил; R2 обозначает С1-С4алкил или С1-С4алкоксигруппу; и R3 и R4 независимо друг от друга обозначают разветвленный С1-С4алкил; или его фармацевтически приемлемую соль.где ингибитор ренина представляет собой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты, или его фармацевтически приемлемую соль.
16. Применение по п.15, в котором производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой соединение формулы (I), где R1 обозначает 3-метоксипропоксигруппу; R2 обозначает метоксигруппу; и R3 и R4 обозначают изопропил; или его фармацевтически приемлемую соль.
17. Применение по п.16, в котором производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой гемифумарат (2-карбамоил-2-метилпропил)амида (2S,4S,5S,7S)-5-амино-4-гидрокси-2-изопропил-7-[4-метокси-3-(3-метоксипропокси)бензил]-8-метилнонановой кислоты.
18. Применение по п.15, в котором ингибитор MDR1 представляет собой PSC833.
19. Применение ингибитора откачивающего белка для приготовления лекарственного средства, предназначенного для повышения биологической доступности ингибитора ренина или его фармацевтически приемлемой соли, где ингибитор откачивающего белка представляет собой PSC833, а ингибитор ренина представляет собой производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты, имеющее формулу
в которой R1 обозначает С1-С4алкокси-С1-С4алкоксигруппу или C1-С4алкокси-С1-С4алкил; R2 обозначает С1-С4алкил или С1-С4алкоксигруппу; и R3 и R4 независимо друг от друга обозначают разветвленный С1-С4алкил; или его фармацевтически приемлемую соль..
20. Применение по п.19, в котором производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой соединение формулы (I), в которой R1 обозначает 3-метоксипропоксигруппу; R2 обозначает метоксигруппу; и R3, R4 обозначают изопропил; или его фармацевтически приемлемую соль.
21. Применение по п.20, в котором производное амида δ-амино-γ-гидрокси-ω-арилалкановой кислоты представляет собой гемифумарат (2-карбамоил-2-метилпропил)амида (2S,4S,5S,7S)-5-амино-4-гидрокси-2-изопропил-7-[4-метокси-3-(3-метоксипропокси)бензил]-8-метилнонановой кислоты.
| 0 |
|
SU158470A1 | |
| Счетчик для измерения расстояния, пройденного трамвайным вагоном под током или без тока | 1924 |
|
SU833A1 |
| СПОСОБ ПОВЫШЕНИЯ БИОЛОГИЧЕСКОЙ ДОСТУПНОСТИ ФЕКСОФЕНАДИНА И ЕГО ПРОИЗВОДНЫХ | 1998 |
|
RU2197967C2 |
| US | |||

