ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Термин "множественная лекарственная устойчивость" (MDR) описывает явление, в соответствии с которым некоторые раковые опухолевые клетки вырабатывают устойчивость к широкому классу цитотоксических средств при воздействии на них отдельного цитотоксического средства. Другими словами, после некоторого периода лечения цитотоксическим средством, которое сначала показывает эффективность при борьбе с ростом опухоли, опухоль вырабатывает устойчивость не только к конкретному средству, которым воздействовали на опухоль, но также к обширным классам структурно и функционально не родственных средств. Недавно обнаружено, что опухолевые клетки с MDR сверхэкспрессируют определенный мембранный гликопротеин, известный как р-гликопротеин ("р" относится к проницаемости). Этот р-гликопротеин является членом суперсемейства переносчиков кассеты связывания АТР (ABC). Предполагается, что воздействие на опухолевые клетки с MDR цитотоксического средства вызывает индукцию этого р-гликопротеина, который опосредует систему обратного переноса, расположенную на мембране опухолевой клетки, которая выкачивает данное цитотоксическое средство, а также цитотоксические средства других классов, из опухолевых клеток, обеспечивая, таким образом, множественную лекарственную устойчивость клетки.
Р-гликопротеин обнаружен не только в опухолевых клетках. Он также экспрессируется во многих нормальных, нераковых, эпителиальных и эндотелиальных клетках, в том числе, в таких тканях как корковое вещество надпочечника, в щеточной каемке проксимального эпителия почечных канальцев, на люменальной поверхности билиарных гепатоцитов, в панкреатических проточках и в слизистой оболочке тонкой и толстой кишки. Для целей описания настоящего изобретения присутствие р-гликопротеина в тонкой и толстой кишке представляет особый интерес.
При приеме веществ внутрь они смешиваются с дигестивными веществами, секретируемыми организмом, и в конечном итоге образуют смесь в пространстве внутри кишки. Пространство внутри кишки соприкасается с некоторыми специализированными эпителиальными клетками, образующими слизистую оболочку кишки или интестинальной стенки. Питательные и другие вещества, находящиеся в интестинальном просвете, пассивно диффундируют к этим интестинальным эпителиальным клеткам, а затем диффундируют в портальный кровоток, который переносит питательные вещества током крови к печени. Таким образом, питательные вещества и другие вещества поглощаются организмом и становятся биологически доступными для использования другими тканями в организме.
Однако интестинальные эпителиальные клетки работают не только как носители в случае пассивной диффузии питательных и других веществ, принятых внутрь. Они являются также различными активными транспортными механизмами, расположенными на наружной мембране эпителиальных клеток, которые активно переносят различные питательные и другие вещества в клетку. В настоящее время считается, что один из активных транспортных механизмов, существующих в интестинальных эпителиальных клетках, представляет собой механизм переноса р-гликопротеина, который облегчает обратный перенос веществ, диффундировавших или привнесенных в клетку, назад в пространство внутри кишки. Предполагается, что р-гликопротеин, присутствующий в интестинальных эпителиальных клетках, может функционировать как защитный обратный насос, препятствующий попавшим внутрь токсическим веществам, продиффундировавшим или перенесенным в эпителиальную клетку, поглощаться системой кровообращения и становиться биологически доступный. Однако одним из неблагоприятных аспектов функции р-гликопротеина в интестинальной клетке является то обстоятельство, что он может также препятствовать биологической доступности веществ, которые полезны, таких как некоторые лекарственные средства, которые оказываются субсостояниями для системы обратного переноса р-гликопротеина.
В настоящее время обнаружено, что, к удивлению, антигистаминные средства настоящего изобретения, соответственно, также направляются системой обратного переноса р-гликопротеина в кишечные эпителиальные клетки и, таким образом, не являются полностью биологически доступными. Настоящее изобретение относится к успешному способу повышения биологической доступности таких антигистаминных средств.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу повышения биологической доступности антигистаминного средства пиперидиноалканола формулы I
где R представляет собой водород или (C1-C6)-алкил,
или его фармацевтически приемлемой соли, или его отдельного оптического изомера, в организме пациента, включающему совместное введение указанному пациенту эффективного антигистаминного количества указанного пиперидиноалканола и эффективного для ингибирования р-гликопротеина количества ингибитора р-гликопротеина. Настоящее изобретение также относится к способу лечения аллергических реакций у пациента, включающему совместное введение указанному пациенту эффективного антигистаминного количества антигистаминного средства пиперидиноалканола формулы I и эффективного для ингибирования р-гликопротеина количества ингибитора р-гликопротеина. Настоящее изобретение также относится к фармацевтической композиции, содержащей эффективное антигистаминное количество антигистаминного средства пиперидиноалканола формулы I и эффективное для ингибирования р-гликопротеина количество ингибитора р-гликопротеина.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу повышения биологической доступности антигистаминного средства пиперидиноалканола формулы I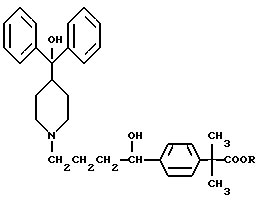
где R представляет собой водород или (C1-C6)-алкил,
или его фармацевтически приемлемой соли, или его отдельного оптического изомера.
Используемый термин "(C1-C6)-алкил" относится к насыщенному углеводородному радикалу с линейной или разветвленной конфигурацией цепи с 1-6 атомами углерода. Конкретно, подпадающими под объем термина "(C1-C6)-алкил" являются углеводородные радикалы метил, этил, пропил, изопропил, бутил, вторбутил, изобутил, третбутил, пентил, гексил и подобные радикалы. Специалист в этой области техники сразу сможет выяснить и представить, что соединения формулы I обладают хиральным центром и, в связи с этом, существуют в формах стереоизомеров. Настоящее изобретение относится к рацемической смеси таких стереоизомеров, так же, как и к отдельным выделенным стереоизомерам. Отдельные стереоизомеры можно выделить из рацемической смеси с помощью методов разделения, хорошо известных и признанных в технике, включая хроматографические методы и методы избирательной кристаллизации.
Соединения формулы I могут существовать в своей свободной форме или в виде фармацевтически приемлемых солей. Фармацевтически приемлемыми солями соединений формулы I являются соли любой подходящей неорганической или органической кислоты. Примерами подходящих неорганических кислот являются хлористоводородная, бромистоводородная, серная и фосфорная кислоты. Примерами подходящих органических кислот являются карбоновые кислоты, такие как, уксусная, пропионовая, гликолевая, молочная, пировиноградная, малоновая, янтарная, фумаровая, яблочная, винная, лимонная, цикламовая, аскорбиновая, малеиновая, гидроксималеиновая, дигидроксималеиновая, бензойная, фенилуксусная, 4-аминобензойная, 4-гидроксибензойная, антраниловая, коричная, салициловая, 4-аминосалициловая, 2-феноксибензойная, 2-ацетоксибензойная, миндальная кислоты, и сульфоновые кислоты, такие как метансульфоновая, этансульфоновая и β-гидроксиэтансульфоновая кислоты. Нетоксичные соли соединений формулы I, образованные с неорганическими или органическими основаниями, также входят в объем данного изобретения и включают, например, соли щелочноземельных металлов, например, кальция и магния, легких металлов группы IIIA, например, алюминия, органических аминов, таких как первичные, вторичные или третичные амины, например, циклогексиламина, этиламина, пиридина, метиламиноэтанола и пиперазина. Соли соединений формулы I можно получить обычными способами, например, посредством обработки соединения формулы I соответствующей кислотой или основанием. Предпочтительной фармацевтически приемлемой солью соединения формулы I является соль хлористоводородной кислоты.
Соединения формулы I можно получить так, как описано в патенте США 4254129, включенном в настоящее описание в качестве ссылки.
Предпочтительным соединением формулы I является соединение (±)-4-[1-гидрокси-4-[4-(гидроксидифенилметил)-1-пиперидинил]-α, α-диметилбензолуксусная кислота, которая известна также как фексофенадин, и ее отдельные стереоизомеры. Фексофенадин, в виде своей соли хлороводородной кислоты, недавно одобрен Управлением по контролю за продуктами и лекарствами США (FDA) для применения в качестве активного ингредиента в антигистаминном средстве, известном как аллеграТМ (AllegraТМ). Аллегра показана при лечении аллергического ринита при рекомендованной дозе 60 мг два раза в день.
Настоящее изобретение относится к способу повышения биологической доступности соединений формулы I. Совместное введение эффективного антигистаминного количества соединения формулы I с эффективным для ингибирования р-гликопротеина количеством ингибитора р-гликопротеина обеспечивает повышенную биологическую доступность соединений формулы I. Биологическая доступность лекарственного средства определяется как степень, до которой лекарственное средство после введения становится доступным для ткани-мишени, и обычно измеряется в виде общего количества лекарственного средства, доступного системно. Как правило, биологическую доступность оценивают путем измерения концентрации лекарственного средства в крови в разные моменты времени после введения лекарственного средства и последующего сложения полученных со временем величин, для того, чтобы получить общее количество лекарственного средства, циркулирующего в крови. Такое измерение, названное "площадью под кривой" (AUC), является прямым измерением биологической доступности лекарственного средства. С другой стороны, биологическую доступность в случае фексофенадина можно оценить путем измерения общего диуреза фексофенадина, так как известно, что фексофенадин не претерпевает существенного метаболизма после перорального введения.
Настоящее изобретение относится к повышению биологической доступность лекарственного средства формулы I за счет совместного введения с ингибитором р-гликопротеина. При совместном введении соединения формулы I и ингибитора р-гликопротеина общее количество соединения формулы I увеличивается по сравнению с количеством, которое могло бы циркулировать в крови в отсутствие ингибитора р-гликопротеина. Таким образом, совместное введение в соответствии с настоящим изобретением будет вызывать возрастание AUC соединения формулы I по сравнению с AUC, наблюдаемым при введении одного соединения формулы I.
Используемый здесь термин "пациент" относится к млекопитающему, такому как, например, человек, мышь, кролик, собака, кошка, и т.п., которое нуждается в лечении в случае аллергической реакции. Используемый здесь термин "аллергическая реакция" относится к опосредуемому гистамином аллергическому заболеванию, такому как, например, сезонный аллергический ринит, идиопатическая крапивница, и подобному заболеванию. Такие заболевания, как правило, различают по вызванному аллергеном выделению гистамина из консервированных клеток в тканях. Выделившийся гистамин связывает определенные H1-гистаминовые рецепторы, что приводит к проявлению хорошо известных аллергических симптомов, таких как чихание, зуд кожи, зуд глаз, ринорея и т.п. Антигистаминное средство, такое как соединения формулы I, будет блокировать проявление аллергических симптомов, вызванных выделением гистамина, посредством блокирования Н1-гистаминовых рецепторов в различных тканях организма, таких как кожа, легкие или назальная слизистая оболочка. Антигистаминные средства, такие как соединения формулы I, являются, таким образом, хорошо известным и эффективным лечением в случае аллергических реакций у пациентов.
Повышение биологической доступности соединения формулы I будет обеспечивать более действенное и эффективное лечение пациента, так как, для данной дозы, большее количество соединения будет доступно в участках тканей, в которых антигистаминное средство блокирует H1-гистаминовые рецепторы, чем в отсутствие такой повышенной биологической доступности.
Введение соединения формулы I относится к пероральному введению. Соединение формулы I можно вводить перорально в любой удобной лекарственной форме, включая, например, капсулу, таблетку, жидкую форму, суспензию и подобные формы.
Эффективное антигистаминное количество соединения формулы I является таким количеством, которое эффективно для обеспечения антигистаминного действия у пациента. Эффективное антигистаминное количество будет изменяться примерно от 1 мг до примерно 600 мг соединения формулы I в виде суточной дозы, в зависимости от типа заболевания, которое лечат, тяжести заболевания, вида пациента, которого лечат, схемы приема лекарственного средства и других факторов, которые специалист в области медицины может установить и оценить. Однако предпочтительное количество будет составлять, как правило, примерно от 10 мг до примерно 240 мг, более предпочтительное количество будет составлять, как правило, примерно от 20 мг до примерно 180 мг, и еще более предпочтительное количество будет составлять, как правило, примерно от 40 мг до примерно 120 мг. Наиболее предпочтительное количество соединения формулы I будет составлять от 60 мг до 120 мг. Вышеуказанные количества соединения формулы I можно вводить один или несколько раз в сутки. Как правило, дозы будут вводиться по схеме, требующей одной, двух или трех доз в сутки, причем предпочтительны одна и две. Более предпочтительной дозировкой и схемой приема будет прием 40 мг два раза в сутки, 60 мг два раза в сутки, 80 мг два раза в сутки, 80 мг один раз в сутки, 120 мг один раз в сутки и 180 мг один раз в сутки, причем наиболее предпочтительны прием 60 мг два раза в сутки и 120 мг один раз в сутки.
Используемый здесь термин "ингибитор р-гликопротеина" относится к органическим соединениям, ингибирующим активность системы активного переноса, опосредованной р-гликопротеином, имеющейся в кишке. Транспортная система активно переносит поглощенные лекарственные средства из пространства внутри кишки в эпителий и обратно в просвет. Ингибирование этой транспортной системы, опосредованной р-гликопротеином, будет приводить к меньшему переносу лекарственного средства обратно в просвет, и, таким образом, будет повышать общий перенос лекарственного средства через эпителий кишки и повышать количество лекарственного средства, имеющегося, в конечном итоге, в крови.
В технике хорошо известны и признаны различные ингибиторы р-гликопротеина. К ним относятся водорастворимый витамин Е; полиэтиленгликоль; полоксамеры, включая плюроник F-68; полиэтиленоксид; полиоксиэтилированные производные касторового масла, в том числе кремофор EL и кремофор RH 40; (+)-таксифолин; нарингенин; диосмин; кверцетин и подобные вещества.
Полиэтиленгликоли (ПЭГ) являются жидкими и твердыми полимерами общей формулы Н(ОСН2СН2)nОН, где n больше 4, с различной средней молекулярной массой в интервале примерно от 200 до примерно 20000. ПЭГи также известны как α-гидро-ω-гидроксиполи(окси-1,2-этандиил)полиэтиленгликоли. Например, ПЭГ 200 представляет собой полиэтиленгликоль, в котором средняя величина n равна 4, а средняя молекулярная масса составляет примерно от 190 до примерно 120. ПЭГ 400 представляет собой полиэтиленгликоль, в котором средняя величина n равна от 8,2 до 9,1, а средняя молекулярная масса составляет примерно от 380 до примерно 420. Подобным образом, ПЭГ 600, ПЭГ 1500 и ПЭГ 4000 имеют, соответственно, средние величины n 12,5-13,9, 29-36 и 68-84, и средние молекулярные массы 570-630, 1300-1600 и 3000-3700, соответственно, а ПЭГ 1000, ПЭГ 6000 и ПЭГ 8000 имеют, соответственно, средние молекулярные массы 950-1050, 5400-6600 и 7000-9000. Полиэтиленгликоли с различной средней молекулярной массой от 200 до 20000 хорошо известны и признаны в фармации и легко доступны.
Предпочтительные полиэтиленгликоли для использования в настоящем изобретении представляют собой полиэтиленгликоли со средней молекулярной массой примерно от 200 до примерно 20000. Более предпочтительные полиэтиленгликоли имеют среднюю молекулярную массу примерно от 200 до примерно 8000. Конкретнее, более предпочтительными полиэтиленгликолями для применения в настоящем изобретении являются ПЭГ 200, ПЭГ 400, ПЭГ 600, ПЭГ 1000, ПЭГ 1450, ПЭГ 1500, ПЭГ 4000, ПЭГ 4600 и ПЭГ 8000. Наиболее предпочтительными полиэтиленгликолями для применения в настоящем изобретении являются ПЭГ 400, ПЭГ 1000, ПЭГ 1450, ПЭГ 4600 и ПЭГ 8000.
Полисорбат 80 представляет собой эфир олеиновой кислоты и сорбита и его ангидриды, сополимеризованные приблизительно с 20 молями этиленоксида на каждый моль сорбита и сорбитангидридов. Полисорбат 80 состоит из производных поли(окси-1,2-этандиил)-сорбитанмоно-9-октадеканоата. Полисорбат 80, известный также как твин 80, хорошо известен и признан в фармации и легко доступен.
Водорастворимый витамин Е, известный также как d-α-токоферил(полиэтиленгликоль 1000)сукцинат [TPGS], является водорастворимым производным витамина Е из природных источников. TPGS можно получить посредством этерификации кислотной группы кристаллического сукцината d-α-токофериловой кислоты полиэтиленгликолем 1000. Этот продукт хорошо известен и признан в фармации, и легко доступен. Например, водорастворимый витамин Е доступен коммерчески от Eastman Corporation как TPGS витамин Е.
Нарингенин представляет собой бифлавоноид 2,3-дигидро-5,7-дигидрокси-2-(4-гидроксифенил)-4Н-1-бензопиран-4-он и известен также как 4',5,7-тригидроксифлавон. Нарингенин является аглюконом нарингена, который представляет собой природный продукт, обнаруженный в плодах и кожуре грейпфрута. Нарингенин широко доступен из коммерческих источников.
Кверцетин представляет собой биофлавоноид 2-(3,4-дигидроксифенил)-3,5,7-тригидрокси-4Н-1-бензопиран-4-он и известен также как 3,3',4',5,7-пентагидроксифлавон. Кверцетин является аглюконом кверцитрина, рутина и других гликозидов. Кверцетин широко доступен из коммерческих источников.
Диосмин представляет собой встречающийся в природе флавоновый глюкозид 7-[[6-0-6-дезокси-α-L-маннопиранозил)-β-D-глюкопиранозил] окси]-5-гидрокси-2-(3-гидрокси-4-метоксифенил)-4Н-1-бензопиран-4-он. Диосмин можно выделить из различных растительных источников, в том числе, из плодов цитрусовых. Диосмин широко доступен из коммерческих источников.
Хризин представляет собой встречающееся в природе соединение 5,7-дигидрокси-2-фенил-4Н-1-бензопиран-4-он, которое можно выделить из различных растительных источников. Хризин широко доступен из коммерческих источников.
Полоксамеры представляют собой блоксополимеры типа α-гидро-ω-гидроксиполи(оксиэтилен)-поли(оксипропилен)-поли(оксиэтилен). Полоксамеры представляют собой ряд близкородственных блоксополимеров этиленоксида и пропиленоксида, соответствующих общей формуле НО(С2H4O)a(С3Н6O)b(С2Н4O)aН.
Например, полоксамер 124 представляет собой жидкий полимер с "а", равным 12, "b", равным 20, и средний молекулярной массой примерно от 2090 до примерно 2360; полоксамер 188 представляет собой твердый полимер с "а", равным 80, "b", равным 27, и средний молекулярной массой примерно от 7680 до примерно 9510; полоксамер 237 представляет собой твердый полимер с "а", равным 64, "b", равным 37, и средней молекулярной массой примерно от 6840 до примерно 8830; полоксамер 338 представляет собой твердый полимер с "а", равным 141, "b", равным 44, и средней молекулярной массой примерно от 12700 до примерно 17400; и полоксамер 407 представляет собой твердый полимер с "а", равным 101, "b", равным 56, и средней молекулярной массой примерно от 9840 до примерно 14600. Полоксамеры хорошо известны и признаны в фармации, и легко доступы коммерчески. Например, плюроник F-68 является коммерчески доступным полоксамером, производимым BASF Corp. Предпочтительными полоксамерами для применения в настоящем изобретении являются такие полоксамеры, как полоксамер 188, плюроник F-68 и подобные.
Модифицированные полиэтиленоксидом производные касторового масла представляют собой ряд веществ, полученных посредством взаимодействия различного количества этиленоксида или с касторовым маслом или с гидрогенизированным касторовым маслом. Такие модифицированные полиэтиленоксидом производные касторового масла хорошо известны и признаны в фармации, и несколько их видов доступны коммерчески, в том числе, кремофоры, производимые BASF Corporation. Модифицированные полиэтиленоксидом производные касторового масла представляют собой сложные смеси различных гидрофобных и гидрофильных компонентов. Например, полиоксил-35-(касторовое масло) (известное также как кремофор EL) содержит гидрофобных составляющих примерно 83% от всей смеси, причем основным компонентом является полиэтиленгликольрицинолеат глицерина. Другими гидрофобными составляющими являются эфиры жирных кислот и полиэтиленгликоля вместе с некоторым количеством исходного касторового масла. Гидрофильная часть полиоксил-35-(касторового масла) (17%) состоит из полиэтиленгликолей и глицерилэтоксилатов.
В полиоксил-40-(гидрогенизированном касторовом масле) (кремофор RH 40) примерно 75% компонентов смеси являются гидрофобными. Эта часть состоит, главным образом, из эфиров жирных кислот и полиэтиленгликольглицерина и эфиров жирных кислот и полиэтиленгликоля. Гидрофильная часть состоит из полиэтиленгликолей и глицерилэтоксилатов. Предпочтительными модифицированными полиэтиленоксидом производными касторового масла для применения в настоящем изобретении являются полиоксил-35-(касторовое масло), такое как кремофор EL, и полиоксил-40-(гидрогенизированное касторовое масло), такое как кремофор RH 40. Кремофор EL и кремофор RH 40 коммерчески доступны от BASF Corporation.
Полиэтиленоксид представляет собой неионный гомополимер этиленоксида, соответствующий общей формуле (OCH2CH2)n, где n представляет среднее число оксиэтиленовых групп. Доступны различные сорта полиэтиленоксидов, которые хорошо известны и признаны специалистами в области фармации, и некоторые различные типы этого материала доступна коммерчески. Предпочтительным сортом полиэтиленоксида является NF и подобные сорта, доступные коммерчески.
(+)-Таксифолин представляет собой (2R-транс)-2-(3,4-дигидроксифенил)-2,3-дигидро-3,5,7-тригидрокси-4Н-1-бензопиран-4-он. Другими обычными названиями (+)-таксифолина являются (+)-дигидрокверцетин; 3,3',4',5,7-пентагидроксифлавонон; диквертин; таксифолиол и дистилин. (+)-Таксифолин хорошо известен и признан в фармации и легко доступен коммерчески.
Предпочтительными ингибиторами р-гликопротеина для применения в настоящем изобретении являются водорастворимый витамин Е, такой как TPGS витамин Е, и полиэтиленгликоли. Из полиэтиленгликолей наиболее предпочтительными ингибиторами р-гликопротеина являются ПЭГ 400, ПЭГ 1000, ПЭГ 1450, ПЭГ 4600 и ПЭГ 8000.
Введение ингибитора р-гликопротеина можно осуществить любым способом, с помощью которого ингибитор р-гликопротеина станет биологически доступным в эффективном количестве, в том числе, пероральным и парентеральным способами. Хотя пероральное введение предпочтительно, ингибиторы р-гликопротеина можно вводить также внутривенно, местно, подкожно, интраназально, рекстально, внутримышечно, или любым другим парентеральным способом. При пероральном введении ингибитор р-гликопротеина можно вводить в любой удобной лекарственной форме, включая, например, капсулу, таблетку, жидкую форму, суспензию и подобные формы.
Эффективным для ингибирования р-гликопротеина количеством ингибитора р-гликопротеина является количество, которое эффективно для обеспечения ингибирования активности активной транспортной системы, опосредованной р-гликопротеином, имеющейся в кишке. Эффективное для ингибирования р-гликопротеина количество ингибитора р-гликопротеина будет изменяться примерно от 5 мг до примерно 1000 мг ингибитора р-гликопротеина в виде суточной дозы, в зависимости от конкретно выбранного ингибитора р-гликопротеина, вида пациента, которого лечат, схемы приема лекарственного средства и других факторов, которые специалист в области медицины может учесть и оценить. Однако предпочтительное количество будет составлять, как правило, примерно от 50 мг до примерно 500 мг, а более предпочтительное количество будет составлять, как правило, примерно от 100 мг до примерно 500 мг. Вышеуказанные количества ингибитора р-гликопротеина можно вводить от одного до нескольких раз в сутки. Как правило, в случае перорального приема, дозы будут даваться по схеме, по которой требуется одна, две или три дозы в сутки, причем предпочтительны одна и две дозы.
Когда в качестве ингибитора р-гликопротеина выбирают водорастворимый витамин Е или полиэтиленгликоль, предпочтительное количество будет составлять, как правило, примерно от 5 мг до примерно 1000 мг, более предпочтительное количество будет составлять, как правило, примерно от 50 мг до примерно 500 мг, и еще более предпочтительное количество будет составлять, как правило, примерно от 100 мг до примерно 500 мг. Наиболее предпочтительное количество водорастворимого витамина Е или полиэтиленгликоля будет составлять примерно от 200 мг до примерно 500 мг. Вышеуказанные количества водорастворимого витамина Е или полиэтиленгликоля можно вводить от одного до нескольких раз в сутки. Как правило, дозы будут даваться по схеме, по которой требуется одна, две или три дозы в сутки, причем предпочтительны одна и две дозы.
Используемый термин "совместное введение" относится к введению пациенту как соединения формулы I, так и ингибитора р-гликопротеина, так, что фармакологическое действие ингибитора р-гликопротеина при ингибировании опосредованного р-гликопротеином переноса в кишке проявляется в то время, когда соединение формулы I поглощается из кишки. Конечно, соединение формулы I и ингибитор р-гликопротеина можно вводить в разное время как одновременно. Например, ингибитор р-гликопротеина можно ввести пациенту перед введением соединения формулы I, чтобы подготовить пациента к получению дозы соединения формулы I. Более того, для пациента может быть удобным предварительная обработка ингибитором р-гликопротеина, чтобы достичь стабильного содержания ингибитора р-гликопротеина до введения первой дозы соединения формулы I. Также предполагается, что соединение формулы I и ингибитор р-гликопротеина можно вводить, по существу, одновременно или в отдельных лекарственных формах или в одной и той же пероральной лекарственной форме.
Настоящее изобретение также предполагает, что соединение формулы I и ингибитор р-гликопротеина можно вводить в отдельных лекарственных формах или в одной и той же комбинированной пероральной лекарственной форме. Совместное введение соединения формулы I и ингибитора р-гликопротеина можно удобно осуществить путем перорального введения комбинированной лекарственной формы, содержащей как соединение формулы I, так и ингибитор р-гликопротеина.
Таким образом, еще одним вариантом воплощения настоящего изобретения является комбинированная фармацевтическая композиция для перорального введения, содержащая эффективное антигистаминное количество соединения формулы I (антигистаминное средство) и эффективное для ингибирования р-гликопротеина количество ингибитора р-гликопротеина (ингибитор). Эта комбинированная пероральная лекарственная форма может обеспечить немедленное высвобождение как соединения формулы I, так и ингибитора р-гликопротеина, или может обеспечить пролонгированное высвобождение одного или обоих компонентов - соединения формулы I и ингибитора р-гликопротеина. Специалист в этой области техники сможет легко определить соответствующие свойства комбинированной лекарственной формы, для того чтобы достичь нужного эффекта от совместного введения соединения формулы I и ингибитора р-гликопротеина.
Антигистаминное средство и ингибитор р-гликопротеина можно вводить одни или в форме фармацевтической композиции в смеси, или другими словами - в сочетании, с одним или несколькими фармацевтически приемлемыми носителями или эксципиентами, количество и природа которых определяются растворимостью и химическими свойствами выбранных антигистаминного средства и ингибитора р-гликопротеина, необходимой схемой приема лекарственных средств и обычной фармацевтической практикой. Антигистаминные средства, хотя они эффективны сами по себе, можно включать в композиции и вводить в форме их фармацевтически приемлемых солей присоединения кислот, таких как гидрохлориды, в целях устойчивости, удобства заключения в оболочку, повышенной растворимости и т.п. Одной из форм фармацевтической композиции по настоящему изобретению является комбинированная фармацевтическая композиция, в которой как антигистаминное средство, так и ингибитор находятся в одной и той же лекарственной форме. Фармацевтическую композицию можно получить способом, хорошо известным и принятым в фармации. Носитель или эксципиент является фармакологически инертным и может представлять собой твердое, полутвердое или жидкое вещество, которое может служить в качестве носителя или растворителя для антигистаминного средства и ингибитора. Подходящие носители и эксципиенты хорошо известны в технике. Фармацевтическую композицию можно приспособить для перорального введения в форме таблетки, капсулы, жидкого препарата, сиропа, вафли, жевательной резинки, суспензии или подобной формы. Такие препараты могут содержать по меньшей мере 4% активного ингредиента, т.е. 4 мас. процента антигистаминного средства и ингибитора, но обычно его количество может изменяться в зависимости от конкретной формы, так что активный ингредиент составляет примерно от 4 мас.% до 70 мас.% стандартной лекарственной формы.
Таблетки, капсулы и подобные формы могут содержать один или несколько из следующих носителей или эксципиентов: связующие вещества, такие как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; эксципиенты, такие как крахмал или лактоза; поверхностно-активные вещества, такие как полисорбат 80 и подобные; дисинтегрирующие вещества, такие как альгиновая кислота, примогельТМ, кукурузный крахмал, бикарбонат натрия, бикарбонат кальция и т.п.; смазывающие вещества, такие как стеарат магния или стеротексТМ; вещества, придающие скольжение, такие как коллоидный диоксид кремния; подслащивающие вещества, такие как сахароза или сахарин; ароматизаторы, такие как перечная мята, метилсалицилат или апельсиновая отдушка. Капсулы могут содержать, кроме ингредиентов, перечисленных выше для таблеток, жидкий носитель, такой как полиэтиленгликоль или жирное масло. Таблетки и капсулы могут содержать другие различные носители и эксципиенты, которые изменяют физическую форму стандартной лекарственной формы, например, такие как покрытия. Так, на таблетки в качестве покрытия могут быть нанесены сахар, шеллак или другие энтеросолюбильные покрытия. Сироп может содержать, кроме активных ингредиентов, стерильную воду, сахарозу как подсластитель, консерванты, красители и корригенты. Вещества, используемые при получении этих различных композиций, должны быть фармацевтически чистыми и нетоксичными в используемых количествах.
Для целей парентерального введения ингибитор можно включить в раствор или суспензию. Такие препараты должны содержать по меньшей мере 0,1% активного ингредиента, но его количество может изменяться примерно от 0,1 мас.% до 50 мас.%. Количество ингибитора в таких композициях необходимо регулировать таким образом, чтобы после введения получалась подходящая доза.
Растворы или суспензии также могут содержать один или несколько из следующих адъювантов: стерильные разбавители, такие как вода, физиологический раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоли или другие синтетические растворители; антибактериальные средства, такие как бензиловый спирт или метилпарабен; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; комплексообразующие вещества, такие как этилендиаминтетрауксусная кислота; буферные вещества, такие как ацетаты, цитраты или фосфаты; вещества для достижения изотоничности, такие как хлорид натрия или декстроза. Парентеральные препараты могут быть заключены в ампулы, одноразовые шприцы или многодозовые флаконы, изготовленные из стекла или пластика.
Точнее, комбинированная фармацевтическая композиция может находиться в форме таблетки, капсулы, жидкости, суспензии, сиропа и подобной форме. Комбинированная фармацевтическая композиция, в том числе, в форме таблетки, может представлять собой простую смесь антигистаминного средства, ингибитора и любых необходимых и соответствующих носителей и эксципиентов. С другой стороны, композиция может существовать в форме смеси различных неоднородных гранул, шариков или других неоднородных частиц, входящих в соответствующий состав. Кроме того, фармацевтическая композиция может находиться в форме сложной таблетки, полученной путем прессования, такой как многослойная таблетка или таблетка с покрытием, полученным путем прессования.
Комбинированные фармацевтические композиции, состоящие из разностных гранул, шариков или частиц (далее называемых "разнородные гранулы"), или состоящие из сложных таблеток, полученных путем прессования, полезны в случае назначения фармацевтических композиций, обеспечивающих различные характеристики высвобождения антигистаминного средства и ингибитора. Например, такие композиции могут обеспечить немедленное высвобождение ингибитора и пролонгированное высвобождение антигистаминного средства, или наоборот. Такие композиции получают в соответствии с обычными методами, хорошо известными и признанными в технике, такими как методы, описанные в патенте США 4996061, включенном в настоящее описание в качестве ссылки.
Приведенные далее примеры иллюстрируют особенно предпочтительный вариант воплощения настоящего изобретения. Эти примеры являются только иллюстративными и никоим образом не предназначены для ограничения объема изобретения.
ПРИМЕР 1
Влияние ПЭГ 400 на биологическую доступность фексофенадина у собак.
Влияние полиэтиленгликоля 400 (ПЭГ 400) на биологическую доступность фексофенадина определяют на двух голодных кобелях бигля. Обработка А заключается в пероральном введении одной таблетки с 120 мг гидрохлорида фексофенадина с пролонгированным высвобождением (SR), а обработка Б заключается в пероральном введении одной таблетки SR вместе с капсулой с 0,5 мл ПЭГ 400, которую дают в -1, 0, 2, 4, 6 и 8 часов до и после таблетки SR. Обработку А проводят за 10-17 дней до обработки Б. Анализируют концентрацию фексофенадина в плазме, чтобы определить биологическую доступность фексофенадина с сопутствующей обработкой ПЭГ 400 и без нее.
В среднем происходит 2-кратное повышение содержания фексофенадина в плазме (табл. I), когда вместе с фексофенадином вводят ПЭГ 400. Это повышение биологической доступности фексофенадина в два раза показано также на фигуре 1, которая иллюстрирует увеличение в среднем концентрации в плазме, полученное при совместном введении.
ПРИМЕР 2
Влияние водорастворимого витамина Е на биологическую доступность фексофенадина у собак
Влияние водорастворимого Е (d-α-токоферилполиэтиленгликольсукцината) на биологическую доступность фексофенадина определяют на двух голодных кобелях бигля в опыте двойным слепым методом. Обработка А заключается в пероральном введении водного раствора дозы 1 мг/кг меченного 14С одного фексофенадина, а обработка Б заключается в пероральном введении той же дозы меченного 14С фексофенадина и дозы 10 ME/кг водорастворимого витамина Е. Обработки даются вслепую в противоположном порядке двум собакам с перерывом между обработками в одну неделю. Определяют радиоактивность в плазме и моче, и представляют содержание неизменившегося фексофенадина у собак.
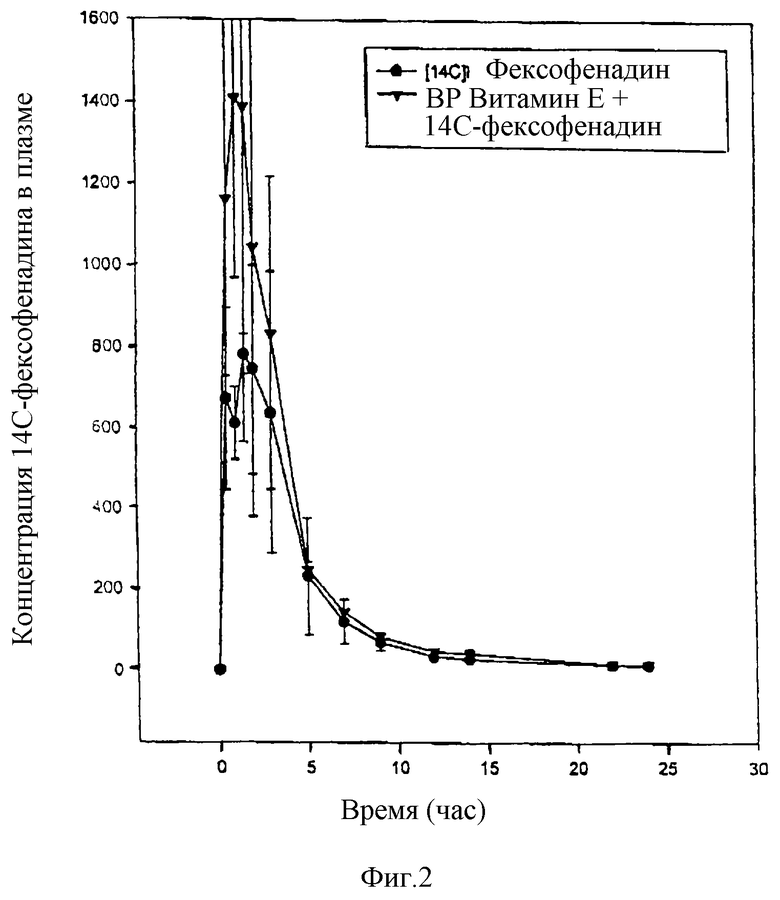
Результаты показывают 50% возрастание в плазме 14С AUC, которое имеет место, когда вместе с 14С-фексофенадином вводят водорастворимые витамин Е (табл.II). Иными словами, биологическая доступность фексофенадина повышается на 50% благодаря водорастворимому витамину Е. Фигура 2 показывает возрастание средней концентрации в плазме, вызванное совместным введением водорастворимого витамина Е.
Повышение поглощения и биологической доступности фексофенодина, наблюдающиеся при одновременном введении водорастворимого витамина Е, также очевидно из данных по выделению [14C]фексофенадина с мочой, которое в среднем возрастает в три раза (табл.III).
ПРИМЕР 3
Влияние ПЭГ 1000 на биологическую доступность фексофенадина у собак
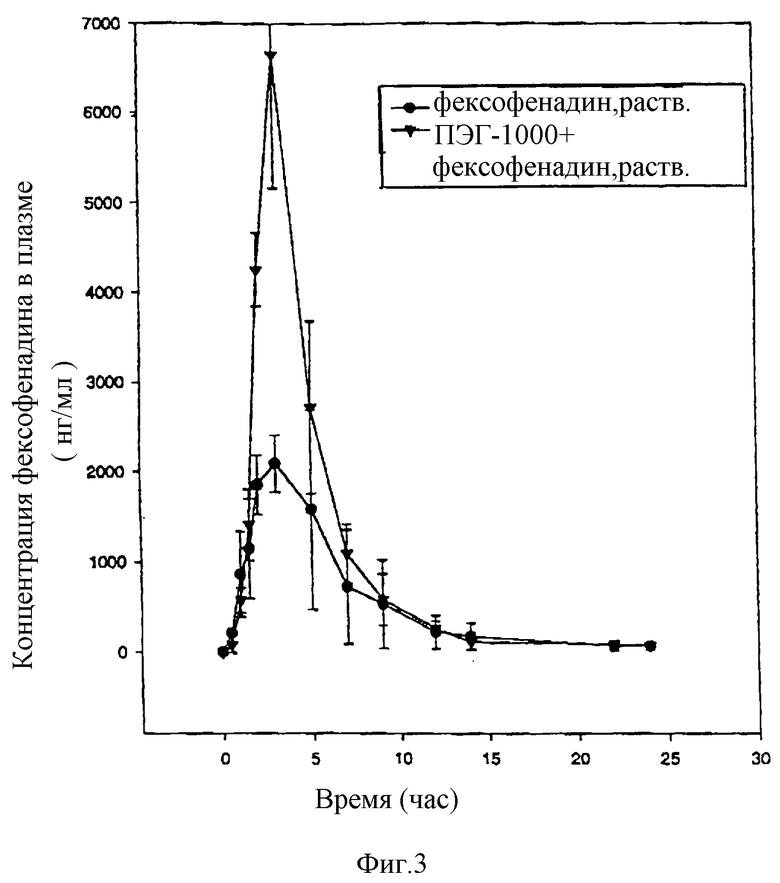
Влияние полиэтиленгликоля 1000 (ПЭГ 1000) на биологическую доступность фексофенадина определяют на двух голодных кобелях бигля. Обработка А заключается в пероральном введении одной таблетки со 120 мг гидрохлорида фексофенадина с пролонгированным высвобождением (SR), а обработка Б заключается в пероральном введении одной таблетки SR вместе с капсулой, содержащей с 0,5 г ПЭН 1000, растворенного в 2,5 мл воды, которую дают в -1, -0,1 и 4 часа до и после таблетки SR. Обработку А проводят за два месяца до обработки Б. Анализируют концентрацию фексофенадина в плазме, чтобы определить биологическую доступность фексофенадина с сопутствующей обработкой ПЭГ 1000 и без нее.
В среднем происходит 2-кратное повышение содержания фексофенадина в плазме [величины AUC (0-24 час) вычисляются из концентраций, приведенных в табл. IV] , когда вместе с фексофенадином вводят ПЭГ 1000. Пик концентрации повышается в среднем в 3 раза. Повышенная биологическая доступность в присутствии ПЭГ 1000 очевидна из графика средних концентраций фексофенадина (фигура 3).






Изобретение относится к медицине, и может быть использовано для лечения аллергических реакций. Изобретение представляет собой способ повышения биологической доступности антигистаминного средства пиперидиноалканола, включающий совместное введение указанному пациенту эффективного антигистаминного количества указанного антигистаминного средства пиперидиноалканола и эффективного для ингибирования р-гликопротеина количества ингибитора р-гликопротеина. Предложенное изобретение позволяет повысить эффективность лечения. 5 c. и 19 з.п. ф-лы, 4 табл., 3 ил.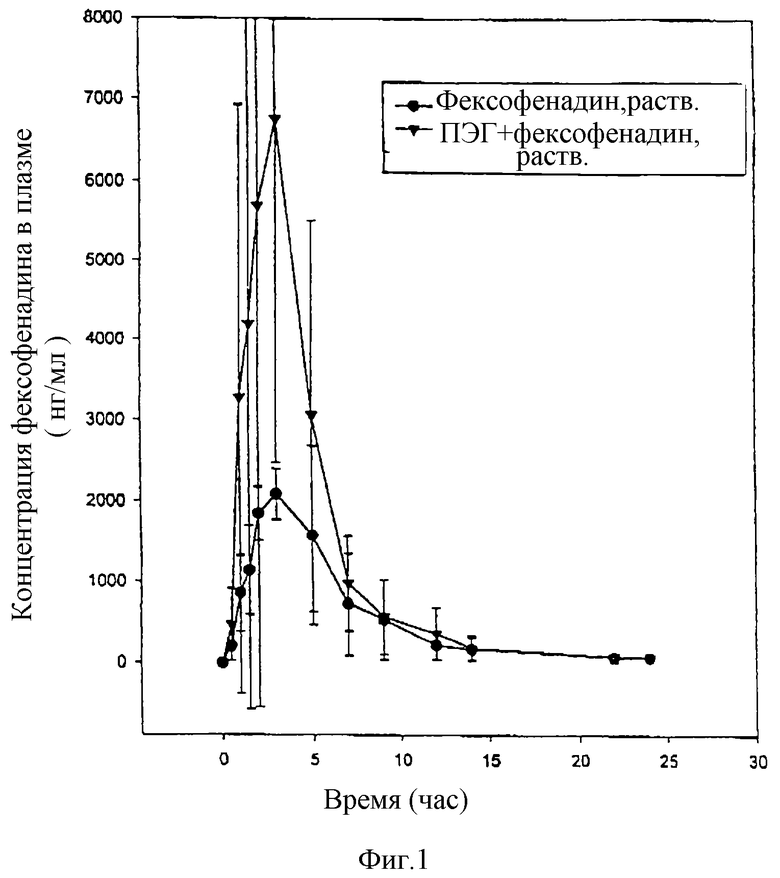

где R представляет собой водород или (С1-С6)алкил,
или его фармацевтически приемлемой соли, или его индивидуального оптического изомера у пациента, включающий совместное введение указанному пациенту эффективного антигистаминного количества указанного антигистаминного средства пиперидиноалканола и эффективного для ингибирования р-гликопротеина количества ингибитора р-гликопротеина.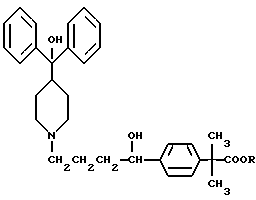
где R представляет собой водород или (С1-С6)алкил,
или его фармацевтически приемлемой соли, или его индивидуального оптического изомера и эффективного для ингибирования р-гликопротеина количества ингибитора р-гликопротеина.
где R представляет собой водород или (С1-С6)алкил,
или его фармацевтически приемлемой соли, или его индивидуального оптического изомера, и эффективное для ингибирования р-гликопротеина количество ингибитора р-гликопротеина.
где R представляет собой водород или (С1-С6)алкил,
или его фармацевтически приемлемой соли, или его индивидуального оптического изомера, где указанная композиция содержит эффективное антигистаминное количество указанного антигистаминного средства пиперидиноалканола и эффективное для ингибирования р-гликопротеина количество ингибитора р-гликопротеина.
| СПОСОБ РЕГУЛИРУЕМОГО ВЫДЕЛЕНИЯ АКТИВНОГО ВЕЩЕСТВА ИЗ СОСТАВА | 1990 |
|
RU2079301C1 |
| US 4254129, 03.03.1981 | |||
| WO 9640192, 19.12.1996 | |||
| US 5567592, 22.10.1996 | |||
| FLOREN LC et al, Tacrolimus oral bioavailability doubles with coadministration of ketoconazole | |||
| Clin | |||
| Pharmacol | |||
| Ther | |||
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |
| Способ крашения тканей | 1922 |
|
SU62A1 |

