Область техники настоящего изобретения
Эпителиальноклеточные раки, например рак простаты, рак молочной железы, рак толстой кишки, рак легких, рак поджелудочной железы, рак яичников, рак селезенки, рак яичек, рак тимуса и т.п., являются заболеваниями, характеризующимися аномальным, ускоренным ростом эпителиальных клеток. Такой ускоренный рост является первичной причиной образования опухоли. Впоследствии также может происходить метастазирование в участки других органов. Несмотря на то, что в диагностике и лечении разных раковых заболеваний произошел очевидный прогресс, указанные заболевания по-прежнему характеризуются высокой смертностью.
Рак легких лидирует среди причин смерти от рака в промышленно развитых странах. Раковые заболевания, начинающиеся в легких, подразделяют на два основных типа, немелкоклеточный рак легких и мелкоклеточный рак легких, в зависимости от того, как клетки выглядят под микроскопом. Немелкоклеточный рак легких (плоскоклеточный рак, аденокарцинома и крупноклеточный рак), как правило, распространяется на другие органы медленнее, чем мелкоклеточный рак легких. Приблизительно 75% случаев рака легких относятся к немелкоклеточному раку легких (например, к аденокарциномам), а остальные 25% относятся к мелкоклеточному раку легких. Немелкоклеточный рак легких (NSCLC) является основной причиной смертности от рака в Соединенных Штатах, Японии и Западной Европе. В случае запущенного заболевания химиотерапия позволяет несколько повысить выживание пациентов, однако она обладает высокой токсичностью, и поэтому существует настоятельная потребность в разработке терапевтических средств, специфично направленных на ключевые генетические нарушения, обуславливающие опухолевый рост (Schiller JH et al., N Engl J Med, 346: 92-98, 2002).
Рецептор эпидермального фактора роста (EGFR) представляет собой мембраносвязанный белок массой 170 килодальтон (кДа), экспрессирующийся на поверхности эпителиальных клеток. EGFR является членом рецепторного семейства тирозинкиназ, молекул, регулирующих клеточный цикл. (W.J.Gullick et al., 1986, Cancer Res., 46:285-292). EGFR активируется при связывании лиганда (либо EGF, либо TGF-α) с внеклеточным доменом, которое приводит к аутофосфорилированию внутриклеточного тирозинкиназного домена рецептора (S. Cohen et al., 1980, J. Biol. Chem., 255:4834-4842; A.B. Schreiber et al., 1983, J. Biol. Chem., 258:846-853).
EGFR является белковым продуктом онкогена, стимулирующего рост, erbB или ErbB1, который, кроме одного члена семейства, а именно, семейства протоонкогенов ERBB, предположительно играет важную роль в возникновении и развитии многих раковых заболеваний человека. В частности, повышенная экспрессия EGFR наблюдается при раке молочной железы, мочевого пузыря, легких, головы, шеи и желудка, а также при глиобластомах. Семейство онкогенов ERBB кодирует четыре структурно-родственных трансмембранных рецептора, а именно EGFR, HER-2/neu (erbB2), HER-3 (erbB3) и HER-4 (erbB4). С клинической точки зрения, амплификация онкогена и/или сверхэкспрессия рецептора ERBB в опухолях коррелирует с рецидивом заболевания и плохим прогнозом для пациента, а также с чувствительностью к терапии. (L. Harris et al., 1999, Int. J. Biol. Markers, 14:8-15; and J. Mendelsohn and J. Baselga, 2000, Oncogene, 19:6550-6565).
EGFR состоит из трех основных доменов, а именно внеклеточного домена (ECD), который находится в гликозилированном состоянии и содержит лиганд-связывающий карман с двумя обогащенными цистеином участками; короткий трансмембранный домен, и внутриклеточный домен, обладающий собственной тирозинкиназной активностью. Трансмембранный участок соединяет лиганд-связывающий домен с внутриклеточным доменом. Анализ аминокислотной и нуклеотидной последовательностей, а также исследования негликозилированных форм EGFR позволили установить, что EGFR имеет массу 132 кДа и содержит 1186 аминокислотных остатков (A.L.Ullrich et al., 1984, Nature, 307:418-425; J. Downward et al., 1984, Nature, 307:521-527; C. R. Carlin et al., 1986, Mol. Cell. Biol., 6:257-264; and F. L. V. Mayes and M. D. Waterfield, 1984, The EMBO J., 3:531-537).
Связывание EGF или TGF-α с EGFR активирует путь передачи сигнала и вызывает пролиферацию клеток. Димеризация, конформационные изменения и интернализация молекул EGFR инициирует внутриклеточные сигналы, регулирующие клеточный рост (G. Carpenter and S. Cohen, 1979, Ann. Rev. Biochem., 48:193-216). Генетические изменения, которые оказывают влияние на регуляцию функции рецептора факторов роста или приводят к сверхэкспрессии рецептора и/или лиганда, вызывают пролиферацию клеток. Кроме того, установлено, что EGFR участвует в дифференциации клеток, увеличении подвижности клеток, секреции белков, реваскуляризации, инвазии, метастазировании и формировании устойчивости раковых клеток к химиотерапевтическим средствам и облучению. (M.-J. Oh et al., 2000, Clin. Cancer Res., 6:4760- 4763).
Идентифицирован ряд ингибиторов EGFR, включающих в себя ингибиторы, уже прошедшие клинические испытания на применение для лечения разных раковых заболеваний. Один из последних обзоров опубликован de Bono, J. S. and Rowinsky, E.K. (2002), "The ErbB Receptor Family: A Therapeutic Target For Cancer", Trends in Molecular Medicine, 8, S 19-26.
Перспективный ряд мишеней для терапевтического вмешательства при лечении рака включает в себя членов линии HER-киназы. Повышающая регуляция данных киназ часто наблюдается в солидных эпителиальных опухолях таких органов, как, например, простата, легкие и молочная железа, а также в глиобластомных опухолях. Рецептор эпидермального фактора роста (EGFR) является членом линии HER-киназы и представляет собой предпочтительную мишень для разработки нескольких способов лечения разных раковых заболеваний. В число данных способов лечения входит применение ингибиторов тирозинкиназы EGFR (EGFR-TKI), поскольку обратимое фосфорилирование остатков тирозина необходимо для активации пути EGFR. Другими словами, EGFR-TKI блокирует клеточный поверхностный рецептор, отвечающий за инициацию и/или поддержание клеточного сигнального пути, который индуцирует рост и деление опухолевых клеток. А именно, полагают, что данные ингибиторы, называемые HER-I, препятствуют функционированию киназного домена EGFR. К наиболее перспективным EGFR-TKI относятся три ряда соединений: хиназолины, пиридопиримидины и пирролопиримидины.
Два из наиболее разработанных в клиническом отношении соединений включают в себя гефитиниб (соединение ZD 1839, разработанное AstraZeneca UK Ltd.; доступное под торговым наименованием IRESSA; далее называемое "IRESSA") и эрлотиниб (соединение OSI-774, разработанное Genentech, Inc. и OSI Pharmaceuticals, Inc.; доступное под торговым наименованием TARCEVA; далее называемое "TARCEVA"); указанные соединения дают обнадеживающие клинические результаты. Традиционный способ лечения рака с использованием как IRESSA, так и TARCEVA, включает ежедневное пероральное введение не более чем 500 мг соответствующих соединений. В мае 2003 первым из указанных продуктов на рынок Соединенных Штатов поступил IRESSA после утверждения его для лечения пациентов с запущенным немелкоклеточным раком легких.
IRESSA представляет собой хиназолин, активный после перорального введения, который функционирует путем непосредственного ингибирования фосфорилирования тирозинкиназы в молекуле EGFR. Он конкурирует за участок связывания аденозинтрифосфата (АТФ), подавляя линию HER-киназы. Точный механизм действия IRESSA полностью не известен, однако проведенные исследования позволяют предположить, что для его реализации необходимо присутствие EGFR.
Существенным ограничением при применении данных соединений является то, что у получающих их пациентов после первичного ответа на терапию может развиваться устойчивость к указанным соединениям, или такие пациенты могут совсем не отвечать на EGFR-TKI в какой-либо заметной степени. Процент отвечающих на EGFR-TKI варьирует среди разных этнических групп. На нижней границе отвечающих на EGFR-TKI находятся популяции, в которых только 10-15% пациентов с запущенным немелкоклеточным раком легких отвечают на ингибиторы киназы EGFR. Таким образом, более глубокое понимание молекулярных механизмов, лежащих в основе чувствительности к IRESSA и TARCEVA, является крайне важным при применении терапии к субъектам, у которых данная терапия с наибольшей вероятностью вызывает благоприятный эффект.
В данной области существует настоятельная потребность в удовлетворительном способе лечения рака, в особенности эпителиальноклеточного рака, такого как рак легких, яичников, молочной железы, мозга, толстой кишки и простаты, который включает в себя благоприятный эффект терапии TKI и преодоление устойчивости к данной терапии. Такое лечение может оказывать существенное воздействие на здоровье субъектов, особенно субъектов старшего возраста, среди которых рак встречается особенно часто.
Сущность изобретения
Авторы настоящего изобретения неожиданно обнаружили, что с помощью необратимых ингибиторов EGFR можно эффективно лечить рак у субъектов, которые больше не отвечают на лечение с использованием гефитиниба и/или эрлотиниба. Так, в одном воплощении настоящее изобретение предлагает способ лечения ракового заболевания, устойчивого к гефитинибу и/или эрлотинибу. В данном воплощении у субъекта после лечения гефитинибом и/или эрлотинибом регистрируют прогрессирование рака. Прогрессирование рака свидетельствует об устойчивости данного заболевания к лечению гефитинибом и/или эрлотинибом, и тогда субъекту вводят фармацевтическую композицию, содержащую необратимый ингибитор рецептора эпидермального фактора роста (EGFR).
В предпочтительных воплощениях в качестве необратимого ингибитора EGFR используют EKB-569, HKI-272 или HKI-357. Альтернативно необратимый ингибитор EGFR может представлять собой любое соединение, которое связывается с остатком цистеина в положении 797 молекулы EGFR (SEQ ID NO: 1).
Прогрессирование рака можно отслеживать с помощью способов, хорошо известных специалистам в данной области. Например, прогрессирование рака можно отслеживать путем визуального наблюдения с использованием рентгеновского излучения, срезов КТ или ЯМР-томографии. Альтернативно, прогрессирование можно отслеживать путем детекции опухолевых биомаркеров.
В одном воплощении состояние пациента регистрируют в разные моменты времени на протяжении всего лечения рака. Например, прогрессирование рака можно отслеживать путем анализа развития рака во второй момент времени и сравнения полученных результатов с результатами анализа, проведенного в первый момент времени. Первый момент времени может находиться до или после начала лечения гефитинибом и/или эрлотинибом, а второй момент времени наступает после первого. Повышенный рост раковой опухоли указывает на прогрессирование рака.
В одном воплощении прогрессирование рака отслеживают путем анализа размера раковой опухоли. В одном воплощении размер раковой опухоли анализируют путем визуального наблюдения с использованием рентгеновского излучения, срезов КТ или ЯМР-томографии. В одном воплощении размер раковой опухоли определяют путем детекции опухолевого биомаркера.
В одном воплощении раковое заболевание представляет собой эпителиальноклеточный рак. В одном воплощении раковое заболевание представляет собой рак желудочно-кишечного тракта, рак простаты, рак яичников, рак молочной железы, рак головы и шеи, рак пищевода, рак легких, немелкоклеточный рак легких, рак нервной системы, рак почек, рак сетчатки, рак кожи, рак печени, рак поджелудочной железы, рак мочеполовой системы и рак мочевого пузыря.
В одном воплощении размер раковой опухоли определяют в другие моменты времени, которые находятся после второго момента времени.
В одном воплощении более поздний момент времени находится, по меньшей мере, на 2 месяца позже предшествующего момента времени. В одном воплощении более поздний момент времени находится, по меньшей мере, на 6 месяцев позже предшествующего момента времени. В одном воплощении более поздний момент времени находится, по меньшей мере, на 10 месяцев позже предшествующего момента времени. В одном воплощении более поздний момент времени находится, по меньшей мере, на год позже предшествующего момента времени.
В другом воплощении настоящее изобретение предлагает способ лечения рака, включающий в себя введение субъекту, несущему мутацию последовательности EGFR, а именно замену метионина на треонин в положении 790 (Т790М) SEQ ID NO: 1, фармацевтической композиции, содержащей необратимый ингибитор EGFR. Мутация Т790М придает устойчивость к лечению гефитинибом и/или эрлотинибом.
Краткое описание чертежей
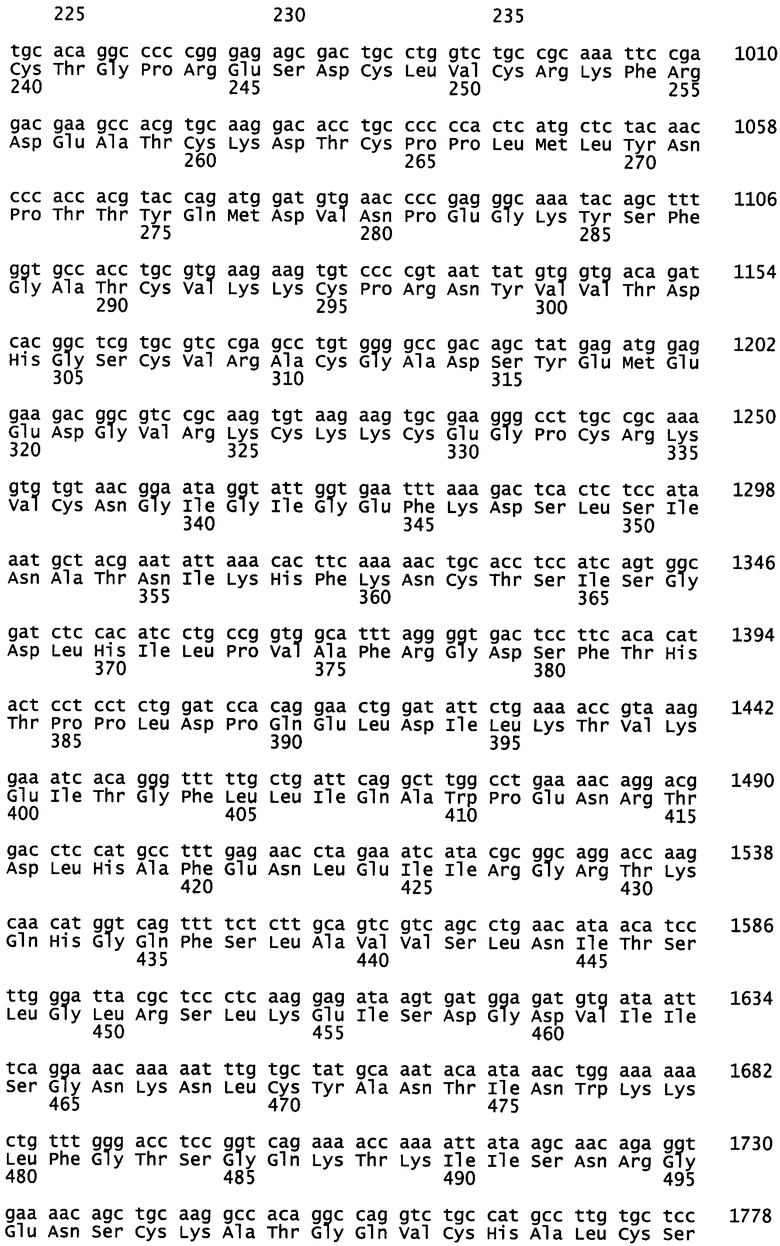
На фиг.1А-1В показаны результаты анализа последовательности EGFR из участков рецидивирующих метастатических опухолей двух пациентов NSCLC с приобретенной устойчивостью к гефитинибу. На фиг.1А показаны результаты анализа последовательности для случая 1. Мутация Т790М присутствует в EGFR при рецидивирующей опухоли печени после развития клинической устойчивости к гефитинибу. (Слева) Мутация не детектируется во время диагноза первичной опухоли печени.
(Справа) Как при первичной опухоли легкого, так и при рецидивирующей опухоли печени детектируют мутацию L858R, придающую чувствительность к гефитинибу. Следует отметить, что мутация L858R присутствует на уровне, характерном для гетерозиготной мутации как при первичном, так и при рецидивирующем заболевании, тогда как Т7 90М детектируется на низком уровне, сравнимом с аллелью дикого типа. Полиморфизм (G/A) показан в той же локализации, чтобы продемонстрировать эквивалентное представление двух аллелей в неклонированном продукте ПЦР (SEQ ID NO: 3 и 4, раскрытые соответственно в порядке приведения). На фиг.1В показаны результаты анализа последовательности для случая 2. Мутация Т790М присутствует в небольшом количестве клеток, устойчивых к гефитинибу. (Слева) В данном случае мутация Т790М не детектируется путем секвенирования неклонированных продуктов ПЦР ни в первичной опухоли легкого, ни в восьми рецидивирующих опухолях печени. Гетерозиготность по смежному полиморфизму (G/A) подтверждает амплификацию обоих аллелей EGFR из данных видов. Гетерозиготная мутация L861Q, обеспечивающая чувствительность к гефитинибу, детектируется на ожидаемом уровне в первичной опухоли легкого, а также в каждой из восьми рецидивирующих опухолей печени (SEQ ID NO: 3 и 5, раскрытые соответственно в порядке приведения).
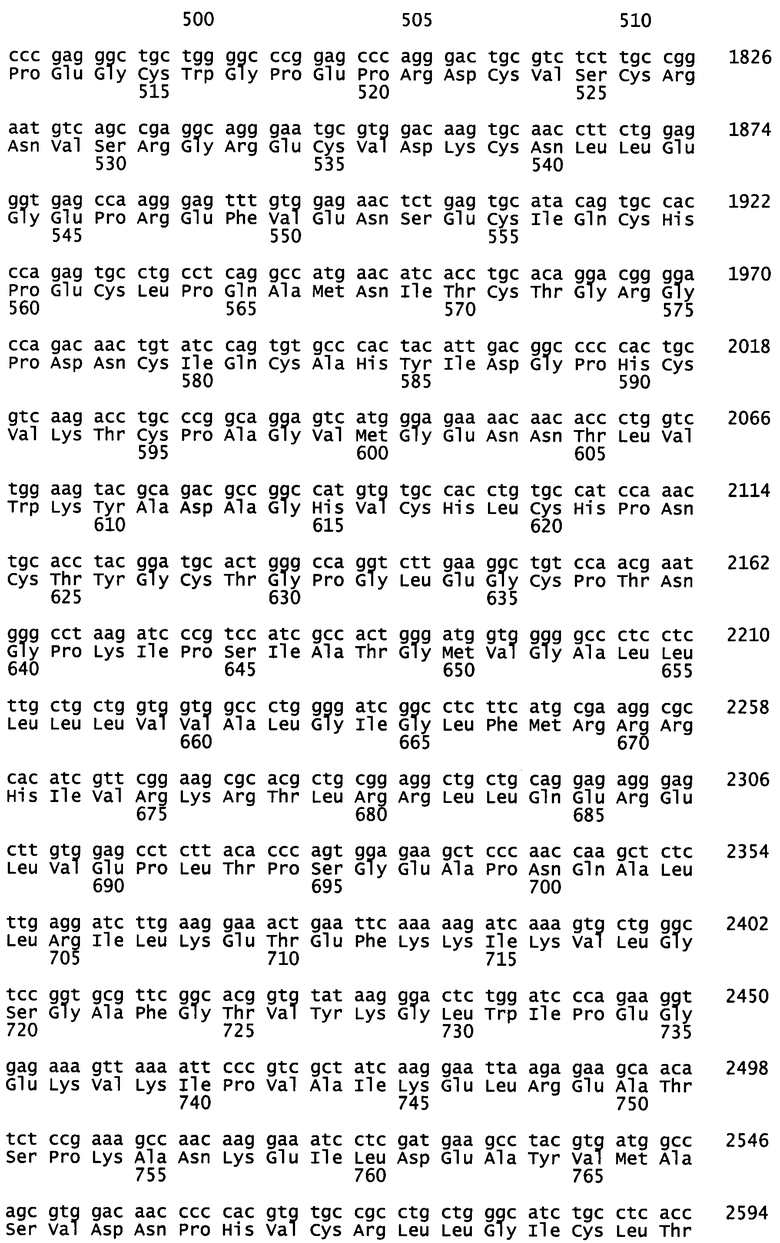
На фиг.2А-2С демонстрируется приобретенная устойчивость к гефитинибу в линиях клеток бронхоальвеолярного рака и стойкая чувствительность к необратимым ингибиторам семейства ERBB. На фиг.2А показано ингибирующее действие ингибиторов тирозинкиназы на пролиферацию клеточных линий бронхоальвеолярного рака, содержащих EGFR дикого типа (NCI-H1666), EGFR, несущий мутацию delE746-A750 (NCI-H1650), или два типичных гефитиниб-устойчивых субклона NCI-H1650 (G7 и C11). Действие обратимого ингибитора гефитиниба сравнивают с действием необратимого ингибитора HKI-357. Сравнимые результаты наблюдаются в случае других необратимых ингибиторов. Число клеток определяют путем окрашивания кристаллическим фиолетовым после культивирования в 5% FCS, содержащей 100 нг/мл EGFR, в течение 72 ч после воздействия определенных концентраций лекарственного средства. Каждая точка соответствует среднему значению, полученному от четырех образцов. На фиг.2В приведены химические структуры гефитиниба, обратимого ингибитора EGFR; ЕKВ-569, необратимого ингибитора EGFR; и HKI-272 и HKI-357, необратимых ингибиторов EGFR и ERBB2 двойного действия. На фиг.2С показано образование устойчивых к лекарственному средству клеток NCI-H1650 после обработки разными концентрациями гефитиниба или необратимого ингибитора ERBB EKB-569. Колонии окрашивают после 12 дней культивирования в присутствии ингибиторов.
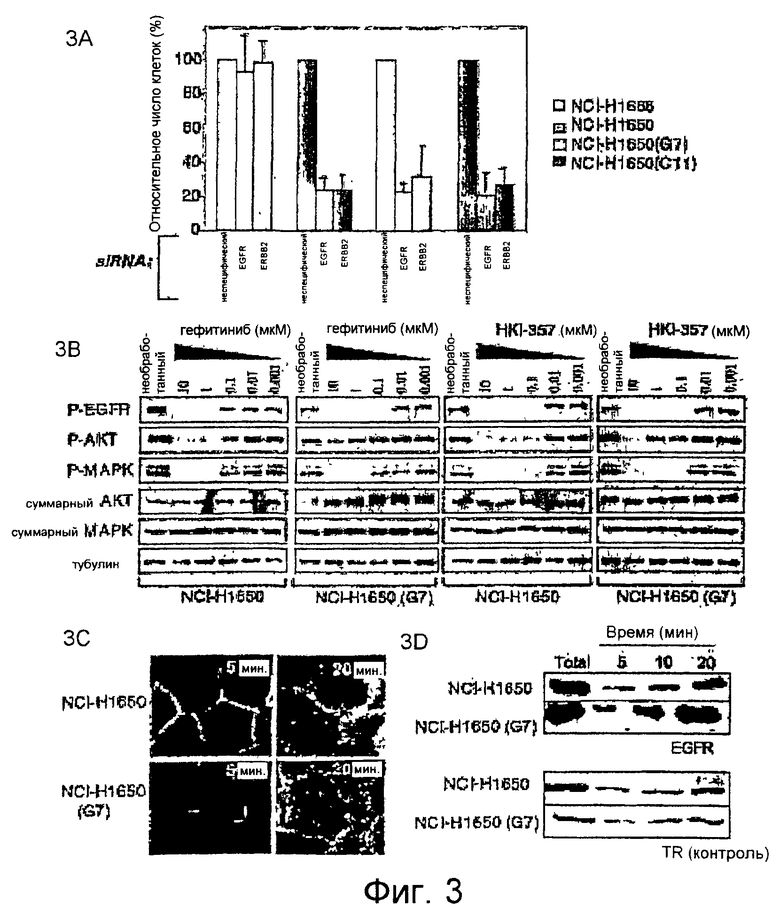
На фиг.3A-3D показана постоянная зависимость от сигнального пути EGFR и ERBB2 в гефитиниб-устойчивых клетках, а также изменение направленной миграции рецептора. На фиг.3A показана жизнеспособность клеток после siРНК-опосредованного нокдауна генов EGFR и ERBB2 в бронхоальвеолярных клеточных линиях, содержащих EGFR дикого типа (NCI-H1666), по сравнению с клетками, несущими активирующую мутацию delE746-A750 в последовательности EGFR (NCI-H1650) и двумя гефитиниб-устойчивыми производными линиями (G7 и C11). Жизнеспособные клетки считают через 72 часа после обработки двухцепочечной РНК и показывают в виде фракции, относящейся к клеткам, обработанным неспецифической siРНК, стандартное отклонение рассчитывают, используя тройные повторы образцов. На фиг.3B показано ингибирование аутофосфорилирования EGFR (Y1068) и фосфорилирования нижестоящих эффекторов AKT и MAPK (ERK) в клетках, обработанных увеличивающимися концентрациями гефитиниба или необратимого ингибитора HKI-357, с последующим 2-ч воздействием EGF. Исходную клеточную линию NCI-H1650 сравнивают с типичной гефитиниб-устойчивой линией, G7. Суммарные AKT и MAPK используют в качестве контроля; тубулин используют в качестве нагружающего контроля при определении общего уровня EGFR, который в данных клетках находится на нижнем пределе детекции. На фиг.3C показано изменение интернализации EGFR в гефитиниб-устойчивых клетках NCI-H1650 (G7), по сравнению с исходной чувствительной клеточной линией NCI-H1650. EGF с присоединенным родамином используют для мечения EGFR через 5 и 20 мин после добавления лиганда. Увеличение интернализации EGFR в клетках NCI-H1650 (G7) наиболее отчетливо наблюдается через 20 мин (микроскоп Цейса (Zeiss), увеличение ×63). На фиг.3D показаны результаты иммуноблоттинга интернализованного EGFR из исходных клеток NCI-H1650 и устойчивой производной линии G7 после импульсного мечения белков клеточной поверхности путем биотинилирования с мониторингом через 20 мин. Повышенный уровень внутриклеточного EGFR в клетках NCI-H1650 (G7) сравнивают с неизмененной интернализацией рецептора трансферрина (TR).
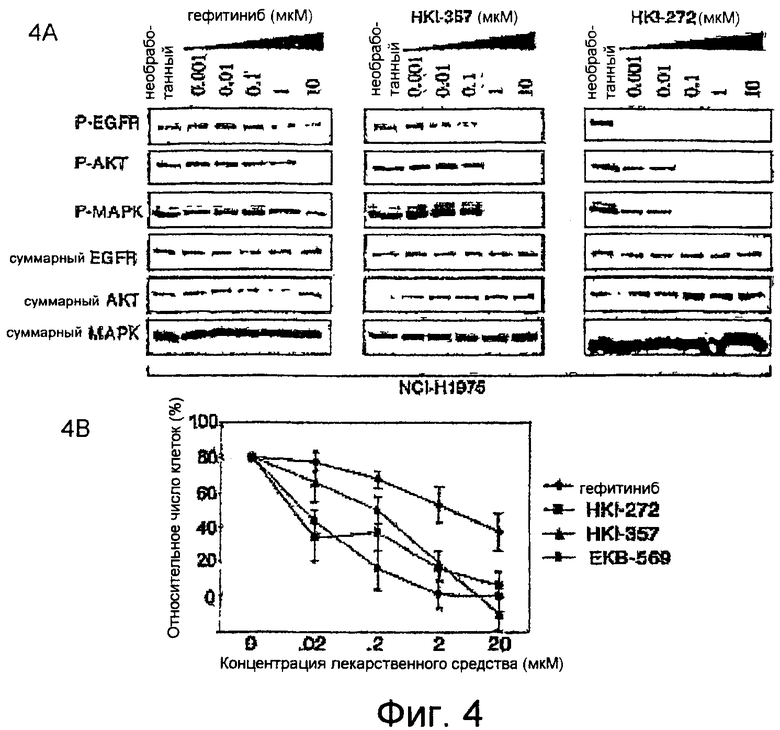
Фиг.4A-4B демонстрируют, что необратимые ингибиторы ERBB подавляют образование мутации T790M в EGFR. На фиг.4A приведены результаты сравнения способности гефитиниба и двух необратимых ингибиторов, HKI-357 и HKI-272, подавлять аутофосфорилирование EGFR (Y1068) и фосфорилирование нижестоящих эффекторов AKT и MAPK (ERK) в бронхоальвеолярной клеточной линии NCI-H1975, несущей как мутацию, придающую чувствительность (L858R), так и мутацию, связанную с устойчивостью (T790M). Суммарные EGFR, AKT и MAPK показаны как нагружающие контроли. На фиг.4B показано подавление пролиферации в клетках NCI-H1975, несущих мутации L858R и T790M, под действием трех необратимых ингибиторов семейства ERBB, по сравнению с гефитинибом.
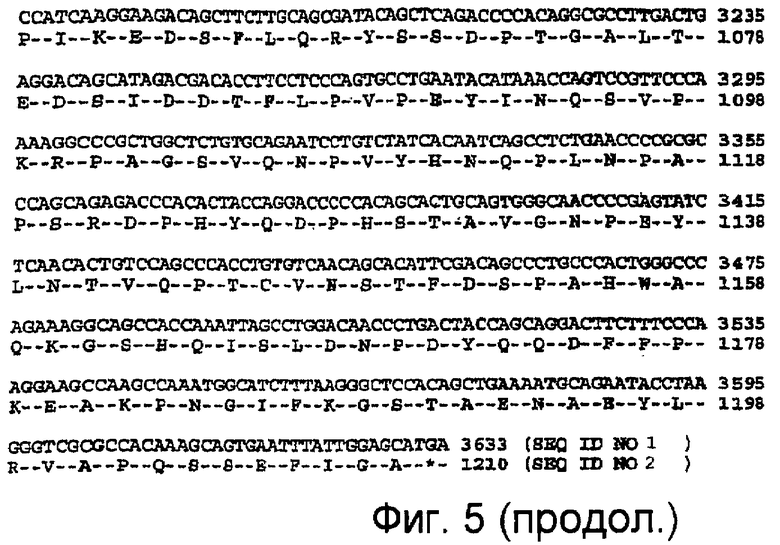
На фиг.5 показаны нуклеотидная (SEQ ID NO: 2) и аминокислотная последовательности (SEQ ID NO: 1) EGFR.
На фиг.6 показано, что, подобно гефитинибу, HKI-357 и EKB-569 (меченные "Wyeth") обладают повышенной способностью убивать клетки NSCLC, несущие мутацию EGFR, однако клоны, устойчивые к данным лекарственным средствам, образуются in vitro не так легко, как клоны, устойчивые к гефитинибу, и указанные соединения сохраняют способность эффективно подавлять гефитиниб-устойчивые клоны.
Подробное описание изобретения
Раковые заболевания, устойчивые к гефитинибу и эрлотинибу
Гефитиниб (соединение ZDl 839, разработанное AstraZeneca UK Ltd.; доступное под торговым наименованием IRESSA) и эрлотиниб (соединение OSI-774, разработанное Genentech, Inc. and OSI Pharmaceuticals, Inc.; доступное под торговым наименованием TARCEVA) вызывают интенсивные клинические ответы в случаях немелкоклеточного рака легких (NSCLC), несущего мутации, приводящие к активации рецептора EGF (EGFR) (1-3), на который и направлены данные конкурентные ингибиторы связывания АТФ (4, 5). Эффективность указанных ингибиторов тирозинкиназ может быть обусловлена как изменениями в расщеплении АТФ, связанными с данными мутациями, которые приводят к усилению ингибирования мутантных киназ под действием указанных соединений, так и биологической зависимостью раковых клеток от повышенных уровней сигналов выживания, передаваемых мутантными рецепторами, явлением, описанным как "зависимость от онкогена" (6, 7).
Хотя терапевтические ответы на гефитиниб и эрлотиниб могут продолжаться до 2-3 лет, средняя продолжительность ответа в большинстве случаев NSCLC составляет только 6-8 месяцев (8-10). Механизмы, лежащие в основе приобретенной устойчивости к лекарственным средствам, полностью не ясны. По аналогии с иматинибом (GLEEVEC), который ингибирует киназу BCR-ABL, участвующую в развитии хронических миелоидных лейкозов (CML), киназу C-KIT, участвующую в развитии желудочно-кишечных стромальных опухолей (GIST), и киназу FIP1L1-PDGFR-α, участвующую в развитии идиопатического синдрома гиперэозинофилии (HES), мутации во втором киназном домене потенциально могут подавлять связывание лекарственного средства (11-16). Однако при рецидивирующем NSCLC взятие образцов для биопсии затруднено; следовательно, для анализа доступно только ограниченное количество образцов. Недавно было обнаружено, что в трех из шести случаев рецидивирующего заболевания после лечения гефитинибом или эрлотинибом в киназном домене EGFR присутствует вторая одиночная мутация, T790M (17, 18). Кодон 315 BCR-ABL, аналогичный кодону 790 EGFR, часто мутирует при иматиниб-устойчивом CML (11, 12), а мутация соответствующего остатка в C-XJr (кодон 670) и FIP1L1-PDGFR-α(кодон 674) связана с иматиниб-устойчивыми GIST и HES соответственно (15, 16). Ранняя модель устойчивости к ингибиторам EGFR in vitro показывает, что мутация кодона 790 в рецепторе дикого типа подобным образом подавляет ингибирование под действием ингибитора тирозинкиназы EGFR (19). В последнее время было показано, что трансфицированные белки EGFR, содержащие активирующие мутации наряду с заменой T790M, меньше подвержены ингибированию под действием гефитиниба и эрлотиниба (17, 18). Хотя мутация T790M, судя по всему, участвует в приобретении устойчивости в некоторых случаях NSCLC, механизмы, лежащие в основе неудачного лечения при отсутствии вторичных мутаций EGFR, остаются невыясненными.
В противоположность цитоплазматической киназе BCR-ABL мембраносвязанный EGFR опосредует сигнальный путь, представляющий собой сложный каскад, который включает в себя связывание лиганда, гомодимеризацию рецептора и гетеродимеризацию с ERBB2 и другими членами семейства с последующими интернализацией и утилизацией связанного с лигандом рецептора или опосредованной убихитином деградацией рецептора (20). Полагают, что EGF-зависимый сигнальный путь в значительной мере осуществляется в процессе интернализации, которая связана с диссоциацией комплексов EGFR при низком значении рН, присутствующем во внутриклеточных везикулах. По существу, несколько факторов модулируют силу и качество сигнала, передаваемого рецептором, а изменения в направленной миграции EGFR тесно связаны с регуляцией EGF-зависимых клеточных ответов (20).
В основе настоящего изобретения лежит открытие, заключающееся в том, что устойчивые к гефитинибу раковые заболевания включают в себя заболевания, при которых EGFR, содержащий мутацию T790M, присутствует только в некоторых устойчивых опухолевых клетках, а также заболевания, при которых мутация T790M отсутствует, но наблюдается повышенная интернализация EGFR. Данное изобретение также основано на открытии, заключающемся в том, что необратимые ингибиторы EGFR, которые образуют ковалентные поперечные связи с рецептором, эффективно подавляют развитие раковых заболеваний, при которых присутствует мутация T790M, а также раковых заболеваний, при которых происходит изменение направленной миграции EGFR, что придает таким заболеваниям устойчивость к гефитинибу и/или эрлотинибу. Соответственно, настоящее изобретение предлагает способ лечения раковых заболеваний, устойчивых к гефитинибу и/или эрлотинибу, включающий в себя введение необратимых ингибиторов EGFR.
Способ лечения пациента
В одном воплощении настоящее изобретение предлагает способ лечения рака, устойчивого к гефитинибу/эрлотинибу. Данный способ включает в себя введение пациенту, нуждающемуся в таком лечении, эффективного количества определенных необратимых ингибиторов EGFR, включающих в себя EKB-569 (4-анилинохинолин-3-карбонитрил; Greenberger et al., 11th NCI-EORTC-AACR Symposium on New Drugs in Cancer Therapy, Amsterdam, November 7-10, 2000, abstract 388; Wyeth), HKI-357 (производное 4-анилинохинолин-3-карбонитрила; Tsou et al. J. Med. Chem. 2005, 48: 1107-1131; Wyeth) и/или HKI-272 (производное 4-анилинохинолин-3-карбонитрила; Rabindran et al., Cancer Res. 2004, 64, 3958-3965; Wyeth). В одном предпочтительном воплощении данное изобретение предлагает способ, включающий в себя введение пациенту, нуждающемуся в таком лечении, эффективного количества EKB-569. В другом предпочтительном воплощении данное изобретение предлагает способ, включающий в себя введение пациенту, нуждающемуся в таком лечении, эффективного количества HKI-357.
В данном способе лечения можно использовать сочетание разных средств, включающее в себя, без ограничения, сочетание ингибитора тирозинкиназы с другими ингибиторами тирозинкиназ, химиотерапевтическими средствами, лучевой терапией и др.
Раковые заболевания изначально могут быть диагностированы как чувствительные к гефитинибу/эрлотинибу, или предположительно имеющие чувствительность к гефитинибу/эрлотинибу, с помощью способов, описанных в Lynch et al., 2004; 350:2129-2139. Чувствительность к гефитинибу/эрлотинибу можно предсказать на основе присутствия в опухоли мутаций EGFR, включающих в себя, например, делецию остатков 747 (лизин) - 749 (глутаминовая кислота) в сочетании с мутацией в положении 750 (аланин), делецию остатков 747 (лизин) - 750 (аланин), замену аргинина на лейцин в положении 858, замену глутамина на лейцин в положении 861.
Раковые заболевания можно диагностировать как чувствительные к гефитинибу/эрлотинибу после начала лечения гефитинибом и/или эрлотинибом. Альтернативно, раковые заболевания можно диагностировать как чувствительные к гефитинибу/эрлотинибу перед началом лечения данными соединениями. Устойчивость опухоли к гефитинибу и/или эрлотинибу может начаться, например, через 6 месяцев после лечения гефитинибом и/или эрлотинибом, или позже. Альтернативно, устойчивость опухоли к гефитинибу и/или эрлотинибу можно диагностировать менее чем через 6 месяцев после начала лечения гефитинибом и/или эрлотинибом. Диагностирование устойчивости к гефитинибу и/или эрлотинибу можно проводить путем отслеживания развития опухоли в процессе лечения гефитинибом и/или эрлотинибом. Развитие опухоли можно детектировать путем сравнения состояния опухоли в момент времени после начала лечения и в момент времени до начала лечения гефитинибом и/или эрлотинибом. Развитие опухоли можно отслеживать в процессе лечения гефитинибом и/или эрлотинибом визуально, например, с помощью радиографических методов, таких как обследование с помощью рентгеновских лучей и компьютерная томография, или с помощью других методов наблюдения, известных специалистам в данной области, которые включают в себя пальпацию раковой опухоли или методы определения уровней опухолевых биомаркеров. Прогрессирование ракового заболевания в процессе лечения гефитинибом и/или эрлотинибом указывает на устойчивость к гефитинибу и/или эрлотинибу. Увеличение уровня опухолевых биомаркеров указывает на прогрессирование опухоли. Таким образом, увеличение уровней опухолевых биомаркеров в процессе лечения гефитинибом и/или эрлотинибом свидетельствует об устойчивости к гефитинибу и/или эрлотинибу. Наличие новых опухолей или метастазов также указывает на прогрессирование опухоли. Прекращение уменьшения опухоли свидетельствует о прогрессировании опухоли. Рост раковой опухоли определяют, например, по увеличению размера опухоли, наличию метастазов или новой раковой опухоли, и/или по повышению уровней опухолевых биомаркеров.
Развитие устойчивости к гефитинибу и/или эрлотинибу можно отслеживать путем тестирования опухолевых клеток, циркулирующих в кровотоке, на присутствие мутации, ассоциированной с устойчивостью к гефитинибу и/или эрлотинибу, указанные клетки можно получить из крови или других биологических жидкостей организма субъекта. Присутствие мутаций, ассоциированных с гефитинибом и/или эрлотинибом, в опухолевых клетках субъекта свидетельствует о том, что опухоль является устойчивой к гефитинибу и/или эрлотинибу.
В одном воплощении опухоль субъекта несет мутации, указывающие на чувствительность к гефитинибу и/или эрлотинибу, хотя при этом она является устойчивой к лечению гефитинибом и/или эрлотинибом. В одном воплощении опухоль субъекта несет мутации, указывающие на чувствительность к гефитинибу и/или эрлотинибу, и мутации, указывающие на устойчивость к гефитинибу и/или эрлотинибу, например мутацию T790M, то есть замену природного остатка треонина на метионин, в EGFR, например, приводящую к увеличению интернализации EGFR. В одном воплощении опухоль субъекта не несет мутации, указывающие на чувствительность к гефитинибу и/или эрлотинибу, и не несет мутации, указывающие на устойчивость к гефитинибу и/или эрлотинибу, такие как мутация T790M в EGFR, например, приводящая к увеличению интернализации EGFR.
В связи с введением лекарственного средства термин "эффективное количество" обозначает количество, которое, по меньшей мере, у статистически значимой группы пациентов вызывает благоприятный эффект, такой как улучшение симптомов, излечение, уменьшение тяжести заболевания, уменьшение массы опухоли или числа опухолевых клеток, увеличение продолжительности жизни, улучшение качества жизни, или другой эффект, который врачи, хорошо знакомые со способами лечения конкретного типа заболевания или состояния, обычно считают положительным.
Используемая эффективная доза активного ингредиента может варьировать в зависимости от конкретного используемого соединения, способа введения и тяжести состояния, подлежащего лечению. Опытному специалисту известно, что эффективная доза для каждого пациента может варьировать в зависимости от тяжести заболевания, индивидуальных генетических вариаций или скорости метаболизма. Однако, как правило, удовлетворительные результаты получают при использовании суточной дозы соединений данного изобретения, составляющей приблизительно от 0,5 до 1000 мг/кг массы тела, необязательно разделенной на два-четыре приема в день, или находящейся в виде формы, обеспечивающей замедленное высвобождение. Общая дневная доза может составлять приблизительно от 1 до 1000 мг, предпочтительно приблизительно от 2 до 500 мг. Лекарственные формы, подходящие для внутреннего применения, включают в себя приблизительно от 0,5 до 1000 мг активного соединения в однородной смеси с твердым или жидким фармацевтически приемлемым носителем. Данный дозировочный режим можно регулировать с получением оптимального терапевтического ответа. Например, можно вводить дозу, разделенную на несколько приемов в день, или дозу можно пропорционально уменьшить в зависимости от конкретной терапевтической ситуации.
Можно использовать внутривенный (I.V.), внутримышечный (I.M.), подкожный (S.C.), внутрикожный (I.D.), внутрибрюшинный (I.P.), интратекальный (I.T.), внутриплевральный, внутриматочный, ректальный, вагинальный, местный, внутриопухолевый и т.п. способ введения. Соединения данного изобретения можно вводить парентерально путем инъекции или путем непрерывной инфузии в течение некоторого времени, кроме того, они могут доставляться с помощью перистальтических средств.
Введение можно осуществлять через слизистую оболочку или через кожу. В состав композиции для введения через слизистую оболочку или через кожу вводят вещества, облегчающие проникновение через соответствующий барьер (пенетранты). Такие пенетранты широко известны в данной области и включают в себя, например, соли желчных кислот для введения через слизистую оболочку и производные фусидовой кислоты. Для облегчения проникновения также можно использовать детергенты. Введение через слизистую оболочку можно осуществлять, например, с использованием назальных спреев или суппозиториев. Композиции соединений данного изобретения для перорального введения получают в виде традиционных форм, таких как капсулы, таблетки и тоники.
Для местного введения фармацевтическую композицию (ингибитора киназной активности) получают в виде мазей, бальзамов, гелей или кремов, широко известных в данной области.
Терапевтические композиции данного изобретения, например необратимых ингибиторов EGFR, обычно вводят внутривенно, например, путем инъекции однократной дозы. Термин "однократная доза" в применении к терапевтической композиции настоящего изобретения относится к физически дискретным формам, подходящим для однократного введения субъекту, каждая из которых содержит предварительно определенное количество активного вещества, рассчитанное так, чтобы получить желательный терапевтический эффект, в сочетании с необходимыми разбавителями, например носителем или средой для лекарства.
Композиции вводят с помощью способа, соответствующего составу композиции, в терапевтически эффективном количестве. Вводимое количество и время введения зависят от субъекта, подлежащего лечению, способности организма субъекта утилизировать активный ингредиент и желательного уровня терапевтического эффекта. Точное количество вводимого активного ингредиента устанавливает лечащий врач индивидуально для каждого пациента.
В данном документе описывается терапевтическая композиция, используемая для осуществления способов настоящего изобретения, например, содержащая необратимые ингибиторы EGFR. Специалистам в данной области известно, что для доставки композиций или лекарственных средств можно использовать любые системы, содержащие активные ингредиенты, которые подходят для предполагаемого применения. Фармацевтически приемлемые носители, подходящие для перорального, ректального, местного или парентерального (в том числе ингаляционного, подкожного, внутрибрюшинного, внутримышечного и внутривенного) введения известны специалистам в данной области. Фармацевтически приемлемым является носитель, совместимый с другими ингредиентами композиции и не оказывающий вредного воздействия на реципиента.
В данном описании термины "фармацевтически приемлемый", "физиологически допустимый", а также их грамматические вариации в применении к композициям, носителям, разбавителям и реагентам используются как взаимозаменяемые и означают, что вещества при введении млекопитающему не вызывают нежелательных физиологических эффектов.
Композиции, подходящие для парентерального введения, как правило, включают в себя стерильные водные препараты активного соединения, которые предпочтительно являются изотоническими по отношению к крови реципиента. Такие композиции обычно содержат дистиллированную воду, 5% раствор декстрозы в дистиллированной воде или физиологическом растворе. Также можно использовать композиции, которые включают в себя концентрированные растворы или твердые вещества, содержащие активное соединение, которые после разбавления подходящим растворителем дают раствор, пригодный для указанного выше парентерального введения.
Для кишечного введения можно использовать дискретные формы, такие как капсулы, облатки, таблетки или пастилки, каждая из которых содержит инертный носитель и предварительно определенное количество активного соединения; в виде порошка или гранул; или в виде суспензии или раствора в водной или не водной жидкости, например в виде сиропа, эликсира, эмульсии или экстракта. Подходящими носителями, включающими в себя смазывающие средства, ароматизаторы, связующие средства и т.п., могут являться крахмалы или сахара.
Таблетки можно получить путем прессования или формования, необязательно с использованием одного или нескольких вспомогательных ингредиентов. Прессованные таблетки можно получить путем прессования в подходящей машине активного соединения в свободнотекучем виде, например в виде порошка или гранул, необязательно смешанного со вспомогательными ингредиентами, например, связующими средствами, смазывающими средствами, инертными разбавителями, поверхностно-активными или диспергирующими средствами. Формованные таблетки можно получить путем формования в подходящей машине смеси порошкообразного активного соединения и любого подходящего носителя.
Сироп или суспензию можно получить путем добавления активного соединения к концентрированному водному раствору сахара, например, сахарозы, к которому также могут быть добавлены любые вспомогательные ингредиенты. Такие вспомогательные ингредиенты могут включать в себя ароматизатор, средство, замедляющее кристаллизацию сахара, или средство, увеличивающее растворимость какого-либо другого ингредиента, такое как многоатомный спирт, например глицерин или сорбит.
Препаративные формы для ректального введения могут быть получены в виде суппозиториев с использованием традиционного носителя, например кокосового масла или Witepsol S55 (trademark of Dynamite Nobel Chemical, Germany), в качестве основы для суппозитория.
Препаративные формы для перорального введения могут быть получены с использованием усиливающего средства. Приемлемые для перорального применения средства, усиливающие абсорбцию, включают в себя поверхностно-активные вещества, такие как лаурилсульфат натрия, пальмитоилкарнитин, Laureth-9, фосфатидилхолин, циклодекстрин и их производные; соли желчных кислот, такие как дезоксихолат натрия, таурохолат натрия, гликохолат натрия и фусдат натрия; хелатообразующие средства, такие как ЭДТА, лимонная кислота и салицилаты; и жирные кислоты (например, олеиновая кислота, лауриновая кислота, ацилкарнитины, моно- и диглицероиды). Другие средства, усиливающие абсорбцию при пероральном применении, включают в себя хлорид бензалкония, хлорид бензетония, CHAPS (3-(3-холамидопропил)-диметиламмоний-1-пропансульфонат), Big-CHAPS (N,N-бис(3-D-глюконамидопропил)холамид), хлорбутанол, октоксинол-9, бензиловый спирт, фенолы, крезолы и алкильные спирты. Особенно предпочтительным средством согласно настоящему изобретению, усиливающим абсорбцию при пероральном применении, является лаурилсульфат натрия.
Альтернативно, соединение можно вводить в виде липосом или микросфер (или микрочастиц). Способы получения липосом и микросфер, использующихся для введения пациенту, хорошо известны специалистам в данной области. В патенте США №4789734, содержание которого включено в данное описание в качестве ссылки, описаны способы заключения биологических веществ в липосомы. Как правило, вещество растворяют в водном растворе, добавляют подходящие фосфолипиды и липиды, если нужно, добавляют также поверхностно-активные вещества, после чего смесь диализуют или обрабатывают ультразвуком, по необходимости. Обзор известных методов опубликован G. Gregoriadis, Chapter 14, "Liposomes," Drug Carriers in Biology and Medicine, pp.287-341 (Academic Press, 1979).
Микросферы из полимеров или белков хорошо известны специалистам в данной области и могут быть приспособлены для прохождения через желудочно-кишечный тракт непосредственно в кровоток. Альтернативно, микросферы, содержащие соединение, или смесь таких микросфер, имплантируют с целью замедленного высвобождения в течение периода времени, варьирующего от нескольких дней до нескольких месяцев. См., например, патенты США №№4906474, 4925673 и 3625214, а также Jein, TIPS 19:155-157 (1998), содержание которых включено в данное описание в качестве ссылки.
В одном воплощении ингибитор тирозинкиназы настоящего изобретения может быть заключен в липосому или микрочастицу, размер которой позволяет ей помещаться в капиллярном русле после внутривенного введения. Если липосома или микрочастица находится в капиллярном русле, окружающем ишемическую ткань, средства могут быть локально введены в участок, в котором они являются максимально эффективными. Липосомы, направленные на ишемическую ткань, как правило, имеют размер менее чем приблизительно 200 нанометров, и представляют собой однослойные везикулы, как описано, например, в патенте США №5593688, Baldescliweiler, озаглавленном "Liposomal targeting of ischemic tissue," содержание которого включено в данное описание в качестве ссылки.
Предпочтительные микрочастицы получают из биодеградируемых полимеров, таких как полигликолид, полилактид и их сополимеры. Специалисты в данной области могут легко определить подходящую систему-носитель, удовлетворяющую ряду факторов, таких как желательная скорость высвобождения лекарственного средства и доставка нужной дозы.
В одном воплощении композиции вводят через катетер непосредственно внутрь кровеносных сосудов. Введение может происходить, например, через отверстия в катетере. В тех воплощениях, где активные соединения имеют относительно большой период полужизни (например, от 1 дня до недели или больше), композиции могут быть включены в биодеградируемые полимерные гидрогели, такие как описанные в патенте США №5410016, Hubbell et al. Данные полимерные гидрогели могут доставляться внутрь тканевой полости с последующим постепенным высвобождением активных соединений по мере разрушения полимера. Иногда полимерные гидрогели могут включать в себя микрочастицы или липосомы, которые содержат распределенное в них активное соединение, при этом контролируемое высвобождение активных соединений протекает по другому механизму.
Как правило, композиции могут быть получены в виде стандартных лекарственных форм с помощью способов, хорошо известных в области фармацевтики. Все данные способы включают в себя стадию смешивания активного соединения с носителем, который состоит из одного или нескольких вспомогательных ингредиентов. Обычно композиции получают путем равномерного и однородного смешивания активного соединения с жидким носителем или мелкоизмельченным твердым носителем, при необходимости, с последующим формованием продукта с получением целевой стандартной лекарственной формы.
Препаративные формы могут дополнительно содержать один или несколько необязательных вспомогательных ингредиентов, используемых в области фармацевтических композиций, например разбавителей, буферов, ароматизаторов, связующих средств, поверхностно-активных веществ, загустителей, смазывающих средств, суспендирующих средств, консервантов (в том числе антиоксидантов) и т.п.
Соединения настоящего изобретения (т.е. необратимые ингибиторы EGFR) можно вводить через дыхательные пути в виде лекарственного порошка для вдыхания через нос, или аэрозоля, или раствора для распылителя, или мелкоизмельченного порошка для вдувания, отдельно или в сочетании с инертным носителем, таким как лактоза. В данном случае частицы активного соединения обычно имеют диаметр менее чем 50 микрон, предпочтительно менее чем 10 микрон, более предпочтительно от 2 до 5 микрон.
При назальном введении предпочтительно используют слабокислые значения рН. Предпочтительно композиции данного изобретения имеют рН в интервале приблизительно от 3 до 5, более предпочтительно приблизительно от 3,5 до 3,9 и наиболее предпочтительно рН составляет 3,7. pH доводят путем добавления подходящей кислоты, такой как хлористоводородная кислота.
Способы получения фармакологической композиции, содержащей растворенные или диспергированные в ней активные ингредиенты, хорошо известны в данной области и не ограничиваются составом композиции. Обычно такие композиции получают в виде форм, пригодных для инъекций, например, в виде жидких растворов или суспензий, однако они также могут быть получены в виде твердых форм, подходящих для растворения или суспендирования в жидкости перед применением. Такой препарат также может быть получен путем эмульгирования.
Активный ингредиент можно смешать с фармацевтически приемлемыми и совместимыми с активным ингредиентом наполнителями в количествах, подходящих для применения в терапевтических способах, описанных в данном документе. К подходящим наполнителям относятся, например, вода, физиологический раствор, декстроза, глицерин, этанол и т.п., а также их сочетания. Кроме того, если желательно, композиция может содержать минорные количества вспомогательных веществ, таких как увлажняющие или эмульгирующие средства, забуферивающие средства и т.п., которые повышают эффективность активного ингредиента.
Необратимые ингибиторы киназ настоящего изобретения могут включать в себя фармацевтически приемлемые соли входящих в их состав компонентов. Фармацевтически приемлемые соли включают в себя кислотно-аддитивные соли (образуемые свободными аминогруппами полипептида) с неорганическими кислотами, такими как, например, хлористоводородная или фосфорная кислоты, или с органическими кислотами, такими как уксусная, винная, миндальная и т.п. Также можно использовать соли, образуемые свободными карбоксильными группами с неорганическими основаниями, такими как, например, гидроксиды натрия, калия, аммония, кальция или железа, и органическими основаниями, такими как изопропиламин, триэтиламин, 2-этиламиноэтанол, гистидин, прокаин и т.п.
Физиологически приемлемые носители хорошо известны в данной области. Примерами жидких носителей являются стерильные водные растворы, которые не содержат других веществ кроме активных ингредиентов и воды, или содержат буфер, такой как натрий-фосфатный с физиологическим значением рН, физиологический раствор, или и то и другое, например забуференный фосфатом физиологический раствор. Кроме того, водные носители могут содержать более одной буферной соли, а также такие соли, как хлориды натрия и калия, декстрозу, полиэтиленгликоль и другие растворимые вещества.
Жидкие композиции также могут содержать жидкие фазы в добавление к воде и вместо воды. Примерами таких дополнительных жидких фаз являются глицерин, растительные масла, такие как хлопковое масло, и водно-масляные эмульсии.
Определения
Термины "ErbB1", "рецептор эпидермального фактора роста" и "EGFR" в данном описании используются как взаимозаменяемые и относятся к нативной последовательности EGFR, описанной, например, в Carpenter et al. Ann. Rev. Biochem. 56:881-914 (1987), в том числе, к ее вариантам (например, к делеционному мутанту EGFR, описанному в Humphrey et al. PNAS (USA) 87:4207-4211 (1990)). erbB1 обозначает ген, кодирующий белковый продукт EGFR. В данном описании под белком EGFR подразумевается белок, имеющий номер доступа в GenBank NP_005219 (SEQ ID NO: 1), который кодируется геном erbB1, имеющий номер доступа в GenBank NM_005228 (SEQ ID NO: 2). Нуклеотидная и аминокислотная последовательности erbB1/EGFR приведены на фиг.5.
Термин "изменение нуклеиновой кислоты, увеличивающее киназную активность" в данном описании относится к изменению (т.е. мутации) нуклеотидной последовательности гена, которое приводит к увеличению киназной активности. Увеличение киназной активности является непосредственным результатом изменения нуклеиновой кислоты и связано с изменением белка, который кодирует данный ген.
Термин "лекарственное средство" или "соединение" в данном описании относится к химической структурной единице или биологическому продукту, или к сочетанию химических структурных единиц, или биологических продуктов, вводимых субъекту для лечения или предотвращения, или подавления заболевания или состояния. Химическая структурная единица или биологический продукт предпочтительно, но необязательно, представляет собой низкомолекулярное соединение, однако они также могут представлять собой более крупное соединение, например олигомер из нуклеотидов, аминокислот или углеводов, включающий в себя, без ограничения, белки, олигонуклеотиды, рибозимы, ДНКзимы, гликопротеины, siРНК, липопротеины, аптамеры, а также их модификации и сочетания.
В данном описании термины "эффективный" и "эффективность" включают в себя как фармакологическую эффективность, так и физиологическую безопасность. Фармакологическая эффективность относится к способности лекарственного средства оказывать желаемое биологическое действие на пациента. Физиологическая безопасность относится к токсичности или к другим неблагоприятным физиологическим эффектам на уровне клеток, органов или всего организма (которые часто называют побочные эффекты), которые возникают в результате введения лекарственного средства. Термин "менее эффективный" означает, что с терапевтической точки зрения лечение имеет гораздо более низкую фармакологическую эффективность и/или более высокий уровень побочных физиологических эффектов.
Молекулы нуклеиновых кислот можно выделить из конкретного биологического образца с помощью любого из ряда способов, хорошо известных в данной области, причем конкретный способ выделения выбирают в зависимости от конкретного биологического образца. Например, для получения молекул нуклеиновых кислот из твердых веществ используют процедуры замораживания-оттаивания и щелочного лизиса; для получения молекул нуклеиновых кислот из мочи можно использовать процедуры нагревания и щелочного лизиса; и для получения молекул нуклеиновых кислот из крови можно использовать экстракцию с применением протеиназы К (Rolff, A. et al. PCR: Clinical Diagnostics and Research, Springer (1994)).
В данном описании термин "рак" относится к присутствию у субъекта или пациента клеток, обладающих свойствами, характерными для клеток, вызывающих образование раковой опухоли, такими как неконтролируемая пролиферация, бессмертие, метастатический потенциал, высокая скорость роста и пролиферации, а также некоторые характеристические морфологические признаки. В некоторых случаях раковые клетки находятся в виде опухоли, или такие клетки могут локально существовать в организме животного или циркулировать в кровотоке в виде самостоятельных клеток.
Примеры
Соединения. Используемые в настоящем изобретении соединения, в том числе EKB-569, HK1-357 и HK1-272, описаны в патенте США №6002008; Greenberger et al., Proc. 11th NCI EORTC-AACR Symposium on New Drugs in Cancer Therapy, Clinical Cancer Res. Vol.6 Supplement, Nov. 2000, ISSN 1078-0432; Rabindran et al., Cancer Res. 64: 3958-3965 (2004); Holbro and Hynes, Ann. Rev. Pharm. Tox. 44:195-217 (2004); и Tejpar et al., J. Clin. Oncol. ASCO Annual Meeting Proc. Vol.22, No. 14S: 3579 (2004).
Анализ рецидивирующего NSCLC и образования устойчивых к гефитинибу клеток NCI-H1650
Клинические образцы рецидивирующего NSCLC получают при вскрытии трупа, производимого после получения надлежащего разрешения. Весь киназный домен EGFR секвенируют после анализа неклонированных продуктов ПЦР. Несколько клонов экзона 20 секвенируют, чтобы определить кодон 790. Анализ мутаций EGFR (экзоны 1-28), ERBB2 (экзоны 1-24), PTEN (Qxons 1-9), Kras (кодоны 12, 13 и 61) и p53 (экзоны 5-8) в гефитиниб-устойчивых клонах и в исходной клеточной линии NCI-H1650 проводят путем автоматического секвенирования отдельных экзонов и фланкирующих последовательностей интронов (условия ПЦР предоставляются по запросу) в двух направлениях с использованием терминаторного красителя (BIGDYE, версия 1.1, Applied Biosystems). Секвенирование проводят на секвенаторе ABI3100 (Applied Biosystems), электроферограммы анализируют с помощью программного обеспечения SEQUENCE NAVIGATOR и FACTURA (Applied Biosystems).
Чтобы получить устойчивые субклоны, клетки NCI-H1 650 обрабатывают этилметансульфонатом (EMS; 600 мкг/мл), оставляют восстанавливаться в течение 72 ч и затем высевают с плотностью 6×104 клеток на чашки диаметром 10 см2 в присутствии 20 мкM гефитиниба. Чтобы определить относительную устойчивость данных клеток к гефитинибу по сравнению с необратимыми ингибиторами, клетки в количестве 5×104 высевают в шестилуночные планшеты в 5% FCS, содержащей 100 нг/мл EGF (Sigma), в присутствии разных концентраций лекарственных средств, затем через 72 ч клетки фиксируют 4% формальдегидом, окрашивают 0,1% кристаллическим фиолетовым и определяют количество клеток, используя инфракрасную систему визуализации Odyssey (LI-COR Biosciences, Lincoln, NE). Для проведения нокдауна в маленьких интерферирующих РНК (siРНК) клетки трансфицируют двухцепочечными РНК-олигонуклеотидами, направленными на EGFR, ERBB2 (оба SMARTpool от Dharmacon, Lafayette, CO) или неспецифический контроль (LRT1B), с использованием реагента для трансфекции X-treme GENE (Roche Applied Science). Через 72 ч клетки окрашивают кристаллическим фиолетовым и анализируют с помощью инфракрасного сканера Odyssey.
Иммуноблоттинг и исследование передачи сигнала. Чтобы определить ингибирование сигнального пути EGFR под действием повышающихся концентраций гефитиниба или необратимых ингибиторов, клетки в количестве 9×104 высевают в 24-луночные планшеты к среде, содержащей 5% FCS, в течение 15 мин добавляют лекарственные средства, затем в течение 2 ч подвергают воздействию 100 нг/мл EGF, после чего собирают лизаты. Лизаты получают в буфере, несущем 2-кратную гелевую нагрузку путем обработки ультразвуком, кипячения, разделения методом 10% SDS/PAGE с последующим электрофоретическим переносом на мембраны из поливинилиденфторида (PVDF) и иммуноблоттингом. Используют антитела против фосфо-EGFR Y1068 и фосфо-митоген-активированной протеинкиназы (MAPK) (Cell Signaling Technology, Beverly, MA), фосфо-AKT (BioSource International, Camarillo, CA) и полноразмерных EGFR, MAPK, AKT и тубулина (Santa Cruz Biotechnology).
Анализ интернализации EGFR. Чтобы продемонстрировать интернализацию EGFR с помощью флуоресцентной микроскопии, клетки выращивают на покровных стеклах и инкубируют в присутствии 1 нг/мл рекомбинантного человеческого (rh) EGF (Molecular Probes, Eugene, OR) для разных интервалов, после чего фиксируют в 4% параформальдегиде в течение 10 мин. Покровные стекла промывают PBS и закрепляют с помощью реагента, препятствующего затуханию изображения ProLong Gold (Molecular Probes). Чтобы количественно определить интернализацию путем клеточного поверхностного биотинилирования, клетки выращивают до слияния, предварительно обрабатывают циклогексамидом, инкубируют на льду в течение 1 часа в присутствии 1,5 мг/мл сульфосукцинимидил-2-(биотинамидо)этил-1,3-дитиопропионата (сульфо-NHS-SS-биотин; Pierce), промывают блокирующим буфером (50 нM NH4Cl/1 мM MgCl2/0,1 мM CaCl2 в PBS), чтобы погасить свободный сульфо-NHS-SS-биотин, и затем несколько раз промывают PBS. Затем, чтобы обеспечить интернализацию биотинилированных молекул, клетки в разных концентрациях инкубируют в культуральной среде при 37°C, дважды промывают в течение 20 минут раствором глутатиона (50 мM глутатион/75 мM NaCl/75 мM NaOH/1% БСА) на льду, чтобы удалить все биотинилированные группы с клеточной поверхности, соскабливают и лизируют в 500 мкМ буфере для радиоиммунопреципитационного анализа (RIPA) (25 мM Tris-HCl, pH 7,4, содержащий 150 мM NaCl/0,1% SDS/1% тритон X-100), дополненном NaF, ортованадатом Na и ингибиторами протеаз. Клеточные экстракты центрифугируют, супернатанты инкубируют в присутствии шариков стрептавидина (Sigma), чтобы собрать биотинилированные белки, которые затем анализируют методом SDS/PAGE и иммуноблоттинга с использованием антитела против EGFR (SC-03, Santa Cruz Biotechnology), или антитела против рецептора трансферрина (Santa Cruz Biotechnology).
Результаты и обсуждение
Анализ рецидивирующего рака легких с приобретенной устойчивостью к гефитинибу. Рецидивирующий гефитиниб-устойчивый NSCLC развивается у двух пациентов, несущих опухоли, которые на момент диагноза содержат мутацию, активирующую киназу EGFR, и демонстрирующих сильный начальный клинический ответ на лекарственное средство (1). В обоих случаях прогрессирующее метастатическое заболевание печени приводит к смерти пациентов через 1-2 года после начала лечения. В случае 1 анализ образцов основных метастазов печени, полученных во время вскрытия трупа, показывает наличие мутации EGFR (L858R), придающей чувствительность, а также наличие приобретенной мутации T790M (фиг.1A). Анализ неклонированных продуктов ПЦР показывает, что относительный уровень исходной мутации L858R свидетельствует о том, что это гетерозиготная мутация, присутствующая во всех опухолевых клетках, тогда как вторичная мутация T790M обнаруживается приблизительно в одной пятой части всего количества соответствующей аллели дикого типа. Таким образом, данная мутация, придающая устойчивость, обнаруживается только в части клеток, принадлежащих рецидивирующей опухоли.
Случай 2 включает в себя восемь разных рецидивирующих метастазов в печени после неудачного лечения гефитинибом. Во всех этих независимых пораженных участках придающая чувствительность мутация EGFR L861Q присутствует в соотношении, ожидаемом для гетерозиготной мутации. Вторичная мутация EGFR не детектируется с помощью анализа неклонированных продуктов ПЦР ни в одном из данных метастазов. Однако после субклонирования продуктов ПЦР мутация T790M обнаруживается на очень низком уровне в двух из четырех анализируемых метастатических опухолей (T790M, 2 из 50 секвенированных клонов в пораженном участке 1 и 1 из 56 в пораженном участке 2), но не в двух других рецидивирующих метастазах (0 из 55 клонов в пораженном участке 3 и 0 из 59 в пораженном участке 4) и не в первичной опухоли (0 из 75 клонов) (фиг.1В и таблица 1). В совокупности полученные результаты согласуются с опубликованными ранее данными, свидетельствующими о том, что мутация T790M присутствует в некоторых, но не во всех случаях приобретенной устойчивости к гефитинибу (три из семи опухолей; см. ссылки 17, 18 и 21). Кроме того, ранее описано (18), что даже в случаях, несущих ассоциированную с устойчивостью мутацию, данная мутация присутствует только в небольшой части опухолевых клеток пораженного участка. Данные наблюдения позволяют предположить, что в тех случаях, когда вторичная мутация EGFR отсутствует, устойчивость обеспечивается другими механизмами, которые действуют совместно с мутацией T790M в случае ее присутствия.
Получение гефитиниб-устойчивых клеточных линий, обладающих чувствительностью к необратимым ингибиторам.
Поскольку существует превосходная корреляция между клинической отвечаемостью NSCLC с мутацией в EGFR и повышенной чувствительностью клеточных линий NSCLC, содержащих указанные мутации, к гефитинибу (2, 6, 22, 23), и, с другой стороны, ограниченная доступность клинических образцов от пациентов с рецидивами, авторы настоящего изобретения смоделировали устойчивость к гефитинибу in vitro. Линию клеток бронхоальвеолярного рака NCI-H1650, содержащих делецию в рамке считывания киназы EGFR (delE746-A750), культивируют в присутствии 20 мкM гефитиниба либо с предварительной обработкой мутагеном этилметансульфонатом, либо без такой обработки. Чувствительность к гефитинибу в данной клеточной линии в 100 раз выше, чем в некоторых линиях NSCLC, экспрессирующих EGFR дикого типа (6). Хотя подавляющее большинство данных клеток погибает в присутствии 20 мкM гефитиниба, можно легко обнаружить устойчивые к лекарственному средству колонии с частотой ~10~5, независимо от обработки мутагеном. Выделяют сорок девять независимых устойчивых к лекарственному средству клонов, демонстрирующих в среднем 50-кратное уменьшение чувствительности к гефитинибу (фиг.2A). У всех этих клонов продолжает существовать мутация, обеспечивающая чувствительность, при неизменном уровне экспрессии EGFR, и ни один из клонов не содержит приобретенной вторичной мутации EGFR, или новых мутаций в ERBB2, p53, Kras или PTEN. Гефитиниб-устойчивые клоны имеют сравнимую устойчивость к родственным ингибиторам класса анилинохиназолинов. Однако они в значительной степени сохраняют чувствительность к трем ингибиторам семейства ERBB (фиг.2A): HKI-272 (24) и HKI-357 (соединение 7f, ссылка 25), которые являются двойными ингибиторами EGFR и ERBB2 (значения IC50 составляют 92 и 34 нM соответственно для EGFR и 59 и 33 нM соответственно для ERBB2), и EKB-569 (26), который является селективным ингибитором EGFR (значения IC50 составляют 39 нM для EGFR и 1,3 мкM для ERBB2) (Wyeth) (фиг.2B). Все три средства действуют как необратимые ингибиторы, по всей вероятности, посредством образования ковалентной связи с остатком cys773 в каталитическом домене EGFR или cys805 ERBB2. Подобно гефитинибу данные соединения демонстрируют повышенную способность уничтожать клетки NSCLC, несущие мутацию в EGFR, по сравнению с клетками, экспрессирующими рецептор дикого типа (фиг.2A). Однако в отличие от гефитиниба, устойчивые к которому клоны можно легко получить даже при высоких концентрациях лекарственного средства, авторы настоящего изобретения не смогли получить клоны, устойчивые к необратимым ингибиторам в концентрациях выше 10 мкM, даже после мутагенеза под действием этилметансульфоната (фиг.2C).
Зависимость гефитиниб-устойчивых клеток от экспрессии EGFR и ERBB2
Чтобы более детально изучить механизмы, лежащие в основе приобретения устойчивости к гефитинибу и сохранения чувствительности к необратимым ингибиторам, авторы настоящего изобретения вначале определяют, действительно ли сохраняется зависимость выживания устойчивых клеточных линий от EGFR. Ранее авторы показали, что siРНК-опосредованный нокдаун EGFR запускает апоптоз в клетках, несущих мутации в EGFR, но не в клетках, содержащих аллели дикого типа (6). Важно, что исходные клетки NCI-H1 650, а также их гефитиниб-устойчивые производные демонстрируют сравнимое уменьшение жизнеспособности после трансфекции siРНК, направленных против EGFR (фиг.3A). Таким образом, в приобретении устойчивости к гефитинибу не участвует EGFR-независимая активация нижестоящих эффекторов. Поскольку HKI-272 и HKI-357 направлены как на EGFR, так и на ERBB2, авторы также исследовали ингибирование данного родственного рецептора. Нокдаун ERBB2 в клетках NCI-H1 650 и их гефитиниб-устойчивых производных также приводит к снижению жизнеспособности (фиг.3A), что позволяет сделать вывод об участии гетеродимеров EGFR-ERBB2 в передаче необходимых сигналов выживания в опухолевых клетках, несущих мутации EGFR. Ингибирование только EGFR под действием необратимого ингибитора является достаточным условием для индуцирования апоптоза в гефитиниб-устойчивых клетках, о чем свидетельствует эффективность EKB-569, который в основном направлен на EGFR (26). Однако наличие потенциально взаимодополняющих эффектов подавления обоих EGFR и ERBB2 путем применения siRNA и доступность необратимых ингибиторов, направленных на обоих членов данного семейства, позволяют получать благоприятный эффект от двойного ингибирования.
Авторы настоящего изобретения сравнивают способность гефитиниба и необратимых ингибиторов семейства ERBB подавлять сигнальный путь через нижестоящие эффекторы EGFR, которые опосредуют процессы пролиферации и выживания. HKI-357 в 10 раз эффективнее, чем гефитиниб, подавляют аутофосфорилирование EGFR (измеряемое по остатку Y1068) и фосфорилирование AKT и MAPK в исходных клетках NCI-H1650, несущих мутацию delE746-A750 в EGFR (фиг.3B). В гефитиниб-устойчивой производной линии, NCI-H1650(G7), гефитиниб значительно менее эффективно подавляет фосфорилирование AKT, ключевого эффектора сигнального пути EGFR, связанного с чувствительностью к гефитинибу (6), тогда как HKI-357 сохраняет прежнюю активность (фиг.3B).
Изменение интернализации EGFR в гефитиниб-устойчивых клонах
С учетом отсутствия вторичных мутаций в EGFR и сохранения чувствительности гефитиниб-устойчивых клеток к siРНК-опосредованному ингибированию EGFR авторы определяют, действительно ли механизм, лежащий в основе разного ингибирования сигнального пути EGFR в гефитиниб-устойчивых клетках под действием обратимых и необратимых ингибиторов может коррелировать с изменениями в направленной миграции рецептора, хорошо описанного модулятора EGFR-зависимого сигнального пути (20). Действительно, анализ миграции EGFR в полученных из NCI-H1650 устойчивых клетках демонстрирует надежное увеличение интернализации EGFR по сравнению с исходными чувствительными к лекарственному средству клетками, которое определяют путем измерения интернализации меченого флуоресцеином EGF (фиг.3C) и количественного определения цитоплазматического биотинилированного EGFR (фиг.3D). Такой эффект не наблюдается в случае рецептора трансферрина, это позволяет предположить, что указанный эффект не является следствием изменения общего процессинга рецепторов. Хотя для определения точного механизма данного изменения миграции EGFR, сложного процесса, в котором участвует много регуляторных белков, требуется проведение дополнительных исследований, полученные результаты позволяют предположить, что способность гефитиниба ингибировать активацию EGFR в данных клетках находится под сомнением, тогда как детектируемые изменения в функционировании необратимых ингибиторов не наблюдаются.
Ингибирование сигнального пути T790M EGFR и увеличение гибели клеток под действием необратимых ингибиторов
Усиление подавления сигнального пути EGFR под действием необратимых ингибиторов ERBB повышает вероятность того, что данные лекарственные средства также могут сохранять активность в клетках, несущих вторичную мутацию T790M в EGFR. Поэтому авторы настоящего изобретения анализируют действие данных ингибиторов на клеточную линию бронхоальвеолярного рака NCI-H1975, которая несет мутации L858R и T790M в EGFR (18). Важно, что данную клеточную линию получают от пациента, не подвергавшегося лечению ингибитором EGFR, это означает, что данная мутация не связана однозначно с приобретенной устойчивостью к лекарственному средству. И HKI-357, и HKI-272 значительно более эффективно, чем гефитиниб, подавляют лиганд-индуцированное аутофосфорилирование EGFR и нижестоящий сигнальный путь, что определяют по фосфорилированию AKT и MAPK (фиг.4A). Подобным образом все три необратимых ингибитора подавляют пролиферацию в данной клеточной линии в условиях устойчивости к гефитинибу (фиг.4B). Таким образом, необратимые ERBB ингибиторы обладают эффективностью в клетках, несущих T790M EGFR, а также в клетках с измененной миграцией рецептора дикого типа.
Полученные результаты подтверждают, что мутации T790M в EGFR являются вторичными мутациями, возникающими в чувствительных NSCLC, несущих активирующую мутацию, связанную с возникновением приобретенной устойчивости (17, 18). Однако данная мутация присутствует только в некоторых случаях, и даже опухоли, несущие мутацию T790M, могут содержать только небольшую долю клеток с указанной мутацией. Данные наблюдения свидетельствуют о том, что в рецидивирующих опухолях после начального ответа на гефитиниб или подобные ему обратимые ингибиторы EGFR могут сосуществовать несколько механизмов устойчивости. Более того, полученные результаты позволяют предположить, что механизмы T790M-независимой устойчивости могут иметь такую же, если не более высокую, эффективность, как и замена T790M, в обеспечении устойчивости к лекарственному средству, и могут объяснять, почему в рецидивирующих опухолях редко возникают клоны, содержащие T790M (17, 18). In vitro механизмы приобретенной устойчивости к гефитинибу объясняются не значительной частотой вторичных мутаций EGFR, а скорее изменением миграции рецептора. Однако авторы данного изобретения не обнаружили миграции EGFR во всех устойчивых клонах, полученных in vitro, поэтому существует вероятность того, что в некоторых клонах вклад в формирование устойчивости к гефитинибу могут вносить другие механизмы. Тем не менее фактически все гефитиниб-устойчивые клоны обладают сравнимой чувствительностью к необратимым ингибиторам ERBB.
Полученные результаты показывают, что существуют убедительные различия между конкурентными ингибиторами EGFR, такими как гефитиниб, эффективность которых ограничена быстрым развитием устойчивости к лекарственному средству in vitro, и необратимыми ингибиторами, к которым редко возникает приобретенная устойчивость (фиг.2C). Авторы полагают, что увеличение интернализации лиганд-связанного EGFR в устойчивых клетках может быть обусловлено диссоциацией комплекса гефитиниб-EGFR при низких значениях pH внутриклеточных везикул. И наоборот, такие изменения в миграции рецептора не влияют на поперечные связи, образованные под действием необратимого ингибитора. Приобретенная устойчивость к гефитинибу стабильно сохраняется после пассажей клеток до 20 поколений в отсутствие лекарственного средства, позволяя предположить, что в основе данного явления лежат генетические или эпигенетические изменения в генах, которые модулируют метаболизм EGFR. Поскольку миграцию рецептора достаточно сложно исследовать с применением доступных клинических образцов, для установления клинических корреляций может потребоваться идентификация таких геномных изменений. Тем не менее такой механизм может вносить вклад в приобретенную in vivo устойчивость к гефитинибу у пациентов с рецидивирующим заболеванием, которые не несут вторичных мутаций в EGFR.
Необратимые ингибиторы ERBB также являются эффективными в преодолении устойчивости к гефитинибу, опосредованной мутацией T790M, данный эффект преимущественно обусловлен сохранением связывания ингибитора, несмотря на изменение данного критического остатка. Хотя данная работа еще не закончена, показано, что другой необратимый ингибитор EGFR [CL-387,785, Calbiochem (27)] ингибирует киназную активность мутанта T790M EGFR (17). Полагают, что эффективность CL-387785 в контексте T790M обусловлена отсутствием хлорида в положении 3 анилиновой группы, которая присутствует в гефитинибе, и стерическим препятствием связыванию с мутантным метионином в кодоне 790. Однако EKB-569, HKI-272 и HKI-357 имеют хлоридные фрагменты в анилиновом цикле, следовательно, их общая способность необратимо связывать EGFR скорее объясняет их эффективность, чем отсутствие специфического стерического взаимодействия с T790M (24-26). Таким образом, доказано, что указанные необратимые ингибиторы обладают широким спектром эффективности, позволяющим преодолевать разные механизмы устойчивости, не только опосредованные мутацией T790M.
Секвенирование большого числа клонированных продуктов ПЦР показывает, что меньшая часть аллелей в двух из четырех поврежденных участков печени содержит мутацию T790M.
Ссылки, приведенные в данной заявке, включены в данное описание в качестве ссылки во всей полноте.
Источники информации


















Группа изобретений относится к медицине, а именно к онкологии, и может быть использована при лечения рака, устойчивого к гефитинибу и/или эрлотинибу. Способы по изобретению включают введения субъекту, страдающему от рака, устойчивого к лечению гефитинибом и/или эрлотинибом, фармацевтической композиции, содержащей необратимый ингибитор рецептора эпидермального фактора роста (EGFR). Также вариант способа по изобретению касается введения субъекту, страдающему от рака и несущему мутацию в EGFR (SEQ ID NO: 1), где мутация представляет собой замену метионина в положении 790 на треонин, фармацевтической композиции, содержащей необратимый ингибитор EGFR. Использование изобретений позволяет лечить устойчивый к гефитинибу и/или эрлотинибу рак путем необратимого ингибирования EGFR за счет образования ковалентной связи ингибитора с рецептором. 3 н. и 24 з.п. ф-лы, 1 табл., 6 ил.
1. Способ лечения рака, устойчивого к гефитинибу и/или эрлотинибу, включающий в себя стадии: а) контролирования прогрессирования рака у субъекта в момент времени после начала лечения гефитинибом и/или эрлотинибом, где прогрессирование рака свидетельствует об устойчивости данного заболевания к лечению гефитинибом и/или эрлотинибом; и b) введения субъекту, страдающему от рака, устойчивого к лечению гефитинибом и/или эрлотинибом, фармацевтической композиции, содержащей необратимый ингибитор рецептора эпидермального фактора роста (EGFR).
2. Способ по п.1, где необратимый ингибитор EGFR выбран из группы, состоящей из ЕКВ-569, HKI-272 и HKI-357.
3. Способ по п.1, где необратимый ингибитор EGFR связывается с остатком цистеина в положении 797 молекулы EGFR (SEQ ID NO: 1).
4. Способ по п.1, где прогрессирование рака регистрируют путем визуального наблюдения.
5. Способ по п.4, где визуальное наблюдение рака осуществляют с помощью рентгеновского излучения, срезов КТ или ЯМР-томографии.
6. Способ по п.1, где прогрессирование рака отслеживают путем детекции опухолевых биомаркеров.
7. Способ по п.1, где контролирование прогрессирования рака включает сравнение состояния рака во второй момент времени и в первый момент времени, причем второй момент времени наступает после первого, а первый момент времени может находиться до или после начала лечения гефитинибом и/или эрлотинибом, где повышенный рост раковой опухоли указывает на прогрессирование рака.
8. Способ по п.7, где с указанными ранее моментами времени сравнивают другие моменты времени.
9. Способ по п.1, где рак представляет собой эпителиальноклеточный рак.
10. Способ по п.1, где рак представляет собой рак желудочно-кишечного тракта, рак простаты, рак яичников, рак молочной железы, рак головы и шеи, рак пищевода, рак легких, немелкоклеточный рак легких, рак нервной системы, рак почек, рак сетчатки, рак кожи, рак печени, рак поджелудочной железы, рак мочеполовой системы и рак мочевого пузыря.
11. Способ лечения рака, включающий в себя стадии: а) введения гефитиниба и/или эрлотиниба субъекту, страдающему от рака; b) контролирования прогрессирования рака у субъекта; и с) введения субъекту необратимого ингибитора EGFR в случае прогрессирования рака.
12. Способ по п.11, где необратимый ингибитор EGFR выбран из группы, состоящей из ЕКВ-569, HKI-272 и HKI-357.
13. Способ по п.11, где необратимый ингибитор EGFR связывается с остатком цистеина в положении 797 молекулы EGFR (SEQ ID NO: 1).
14. Способ по п.11, где прогрессирование рака регистрируют путем визуального наблюдения.
15. Способ по п.14, где визуальное наблюдение рака осуществляют с помощью рентгеновского излучения, срезов КТ или ЯМР-томографии.
16. Способ по п.11, где прогрессирование рака контролируют путем детекции опухолевых биомаркеров.
17. Способ по п.11, где контролирование прогрессирования рака включает сравнение состояния рака во второй момент времени и в первый момент времени, причем второй момент времени наступает после первого, а первый момент времени может находиться до или после начала лечения гефитинибом и/или эрлотинибом, где повышенный рост раковой опухоли указывает на прогрессирование рака.
18. Способ по п.17, где с указанными ранее моментами времени сравнивают другие моменты времени.
19. Способ по п.11, где рак представляет собой эпителиальноклеточный рак.
20. Способ по п.11, где рак представляет собой рак желудочно-кишечного тракта, рак простаты, рак яичников, рак молочной железы, рак головы и шеи, рак пищевода, рак легких, немелкоклеточный рак легких, рак нервной системы, рак почек, рак сетчатки, рак кожи, рак печени, рак поджелудочной железы, рак мочеполовой системы и рак мочевого пузыря.
21. Способ по п.11, где необратимый ингибитор EGFR вводят одновременно с обратимым ингибитором EGFR.
22. Способ по п.21, где обратимый ингибитор EGFR представляет собой гефитиниб или эрлотиниб.
23. Способ лечения рака, включающий введение субъекту, страдающему от рака и несущему мутацию в EGFR (SEQ ID NO: 1), где мутация представляет собой замену метионина в положении 790 на треонин, фармацевтической композиции, содержащей необратимый ингибитор EGFR.
24. Способ по п.23, где ингибитор EGFR выбран из группы, состоящей из ЕКВ-569, HKI-272 и HKI-357.
25. Способ по п.23, где необратимый ингибитор EGFR связывается с остатком цистеина в положении 797 молекулы EGFR (SEQ ID NO: 1).
26. Способ по п.23, где рак представляет собой эпителиальноклеточный рак.
27. Способ по п.23, где рак представляет собой рак желудочно-кишечного тракта, рак простаты, рак яичников, рак молочной железы, рак головы и шеи, рак пищевода, рак легких, немелкоклеточный рак легких, рак нервной системы, рак почек, рак сетчатки, рак кожи, рак печени, рак поджелудочной железы, рак мочеполовой системы и рак мочевого пузыря.
| ПОДДУБНАЯ И.В | |||
| Достижения современной химиотерапии | |||
| Consilium Medicum | |||
| Современная онкология, 2003, т.5, №2 |

