Настоящее изобретение относится к применению специфических производных имидазохинолина для лечения заболеваний, зависимых от членов семейства рецептора эпидермального фактора роста (EGFR) (включая EGFR1, известный так же как HER1, или Erb-B1; EGFR2, известный так же как HER2 или Erb-B2, и EGFR3, известный так же как HER3 или Erb-B3), или заболеваний, которые характеризуются приобретенной резистентностью к агентам, которые связываются с членами семейства EGFR, к применению указанных соединений для получения фармацевтических композиций, предназначенных для лечения указанных заболеваний, комбинаций указанных соединений с модуляторами EGFR для указанного применения, способам лечения указанных заболеваний указанными соединениями и к фармацевтическим препаратам, предназначенным для лечения указанных заболеваний и включающим указанные соединения в отдельности или в комбинации, прежде всего, с модулятором EGFR.
Соматические мутации в тирозинкиназном домене EGFR связаны с клинической ответной реакцией на ингибитор тирозинкиназ EGFR, такой как гефитиниб (иресса®) или эрлотиниб (тарцева®) (Paez и др., EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science, т.304, 1497-1500). Приобретенная резистентность к модуляторам EGFR развивается у пациентов, у которых первоначально наблюдалась клиническая ответная реакция на лечение, но затем развивались прогрессирующие опухоли. Примером резистентной ответной реакции на ингибиторы киназы EGFR является вторичная резистентная мутация Т790М (Kobayashi и др., EGFR mutation and resistance of non-small cell lung cancer to gefitinib, N.Engl. J.Med., т.352, 786-792), которая сравнима с резистентной мутацией, наблюдаемой при лечении препаратами гливек/дазатиниб пациентов, страдающих от хронического миелоидного лейкоза (ХМЛ) (Gorre и др., Bcr-Abl point mutants isolated from patients with imatinib mesylate resistant chronic leukemia reamin sensitive to inhibitors of the Bcr-Abl chaperone heat shock protein 90, Blood, т.100, 3041-3044) или пациентов, страдающих от стромальных опухолей желудочно-кишечного тракта (Antonescu и др.. Acquired resistance to Imatinib in gastrointestinal stromal tumors occurs through secondary gene mutation, Clin. Cancer Res., т.11, 4182-4190).
В литературе описаны данные, подтверждающие тот факт, что активация пути PI3K происходит после активации EGFR. Таким образом, генетическая абляция каталитической субъединицы PI3K (p110) в фибробластах мышиного эмбриона придает клеткам резистентность к трансформации активированными формами EGFR (Zhao и др.. The p110 alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation, PNAS, т.103, 16296-16300). HER3 (ErbB-3), один из четырех членов семейства EGFR и HER1 (EGFR1), часто характеризуется сверхэкспрессией в опухолях, чувствительных к ингибиторам EGFR, что коррелирует с накоплением и активацией конститутивного PI3K (Engelman и др., ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non small cell lung cancer cell lines, PNAS, т.102, 3788-3793; Sergina и др., Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER 3). Данные по генетической и биохимической характеристике биопсии и опухолей и опухолевых клеточных линий с амплификацией EGFR и резистентностью к ингибитору EGFR свидетельствуют о конститутивном статусе активации пути PI3K (Engelman и др., Allelic disruption obscures detection of a biologically significant resistance mutation in EGFR amplified lung cancer. The Journal of Clinical Investigation, т.116, 2695-2706).
Неожиданно было установлено, что специфические производные имидазохинолина, описанные в патенте WO 2006/122806, вызывают сильную антипролиферативную активность и противоопухолевую ответную реакцию in vivo в клеточных линиях рака молочной железы и легких с амплификацией EGFR и/или мутированным EGFR1 при введении указанного агента в отдельности и в комбинации с модуляторами киназы EGFR. Следовательно, указанные соединения можно использовать для лечения заболевания, зависимого от EGFR.
Специфические производные имидазохинолина, пригодные для применения по настоящему изобретению, их получение и пригодные фармацевтические композиции, содержащие указанные соединения, описаны в патенте WO 2006/122806 и включают соединения формулы I
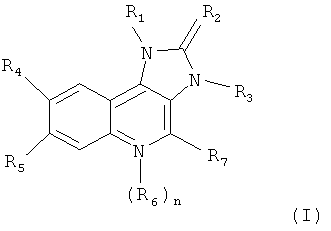
где R1 обозначает нафтил или фенил, где указанный фенил замещен одним или двумя заместителями, независимо выбранными из группы, включающей галоген, (низш.)алкил, незамещенный или замещенный галогеном, цианогруппой, имидазолилом или триазолилом, циклоалкил, аминогруппу, замещенную одним или двумя заместителями, независимо выбранными из группы, включающей (низш.)алкил, (низш.)алкилсульфонил, (низш.)алкоксигруппу и (низш.)алкокси(низш.)алкиламиногруппу, пиперазинил, незамещенный или замещенный одним или двумя заместителями, независимо выбранными из группы, включающей (низш.)алкил и (низш.)алкилсульфонил, 2-оксопирролидинил, (низш.)алкокси(низш.)алкил, имидазолил, пиразолил и триазолил,
R2 обозначает O или S,
R3 обозначает (низш.)алкил,
R4 обозначает пиридинил, незамещенный или замещенный галогеном, цианогруппой, (низш.)алкилом, (низш.)алкоксигруппой, или пиперазинил, незамещенный или замещенный (низш.)алкилом, пиримидинил, незамещенный или замещенный (низш.)алкоксигруппой, хинолинил, незамещенный или замещенный галогеном, хиноксалинил или фенил, замещенный алкоксигруппой,
R5 обозначает водород или галоген,
n равно 0 или 1,
R6 обозначает оксидо,
при условии, что если n=1, то атом N, содержащий радикал R6, положительно заряжен,
R7 обозначает водород или аминогруппу,
или их таутомер или фармацевтически приемлемую соль, или их гидрат или сольват.
Радикалы и символы, использованные при определении соединения формулы I, имеют значения, описанные в заявке WO 2006/122806, которая включена в настоящее описание в качестве ссылки.
Предпочтительное соединение по настоящему изобретению обозначает соединение, которое подробно описано в WO 2006/122806.
Наиболее предпочтительным соединением по настоящему изобретению является 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрил (соединение A) и его монотозилат. Синтез 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрила описан, например, в WO 2006/122806, в примере 1.
Другим наиболее предпочтительным соединением по настоящему изобретению является 8-(6-метоксипиридин-3-ил)-3-метил-1-(4-пиперазин-1-ил-3-трифторметилфенил)-1,3-дигидроимидазо[4,5-с]хинолин-2-он (соединение В) и его мономалат. Синтез 8-(6-метоксипиридин-3-ил)-3-метил-1-(4-пиперазин-1-ил-3-трифторметилфенил)-1,3-дигидроимидазо[4,5-с]хинолин-2-она описан, например, в WO2006/122806, в примере 86.
Соединения по настоящему изобретению, которые связываются с членами семейства EGFR, включают модуляторы семейства киназ EGFR, соединения, которые изменяют уровень экспрессии членов семейства EGFR или вызывают клеточный иммунный ответ, связанный с экспрессией членов семейства EGFR в опухолевых клетках. Предпочтительные модуляторы EGFR проявляют активность в качестве ингибиторов функциональной активности EGFR. Соединения, которые связываются с членами семейства EGFR по настоящему изобретению, включают, не ограничиваясь только ими, гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY334543, BIBW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональные антитела 806, анти-EGFR моноклональные антитела-Y90/Rе-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, MDX-214, CDX110, IMC11F8, пертузумаб, трастузумаб, земаб®, вакцину Her2 PX 1041 и ингибиторы HSP90, CNF1010, CNF2024, алвеспимицин, танеспимицина, IPI504, SNX5422 и NVP-AUY922.
Краткое описание фигур
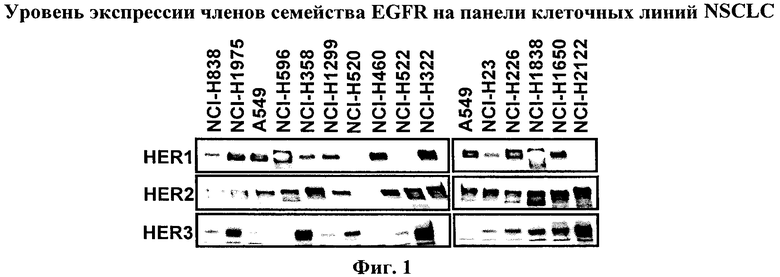
На фиг.1 показан уровень экспрессии белков семейства EGFR: HER-1, HER-2 и HER-3 на панели 15 клеточных линий опухоли человека NSCLC.
Указанные опухолевые линии культивировали в оптимальных условиях роста, из клеток в субконфлуентной фазе роста получали клеточные экстракты. Затем экстракты, содержащие эквивалентное количество общего белка, подвергали электрофорезу в ДСН-ПААГ, гели переносили на мембраны и мембраны инкубировали с антителами против белков, указанных на левой стороне панелей.
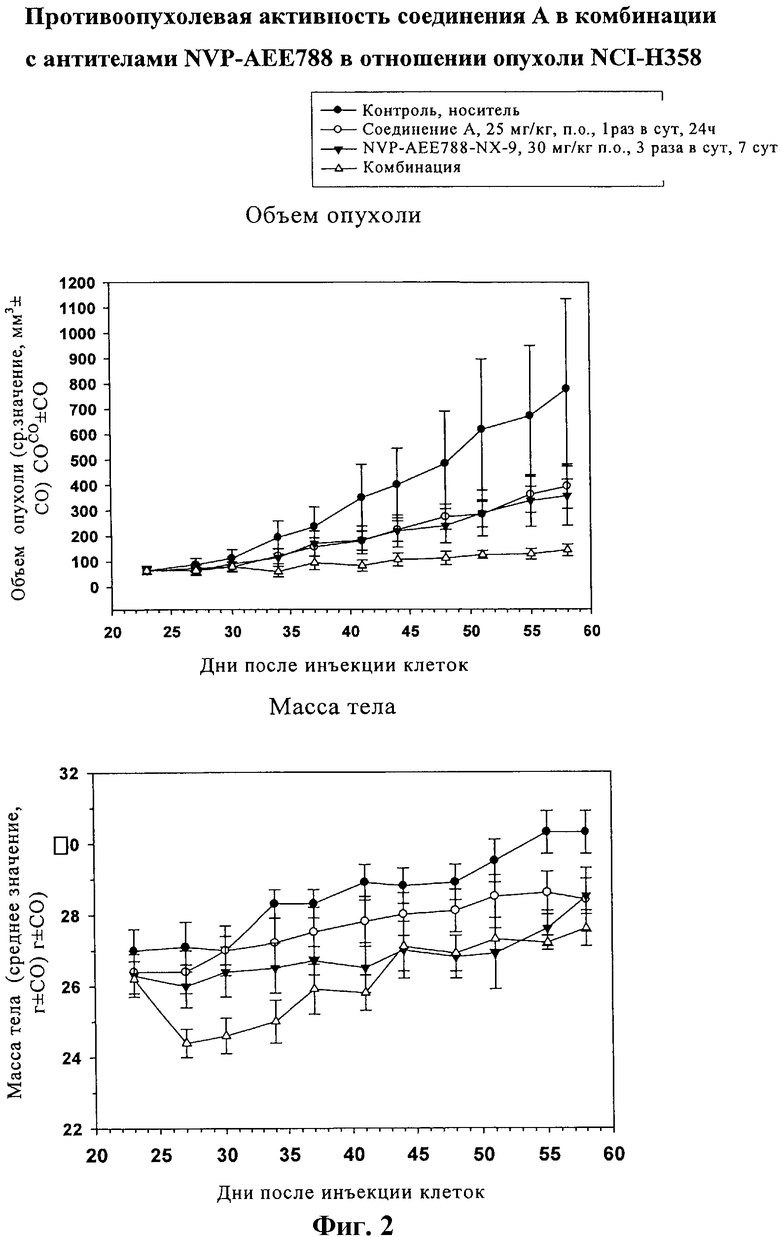
На фиг.2 показана противоопухолевая активность соединения А в комбинации с агентом NVP-AEE788 в отношении опухолей NCI-H358.
Самкам бестимусных мышей Harlan (n=8) с привитыми подкожно опухолями NCI-H358 вводили перорально в качестве инигибитора PI3K соединение A или ингибитор EGFR NVP-AEE788 или их комбинации согласно указанной схеме. *p<0.05 (критерий Даннета по сравнению с контролем).
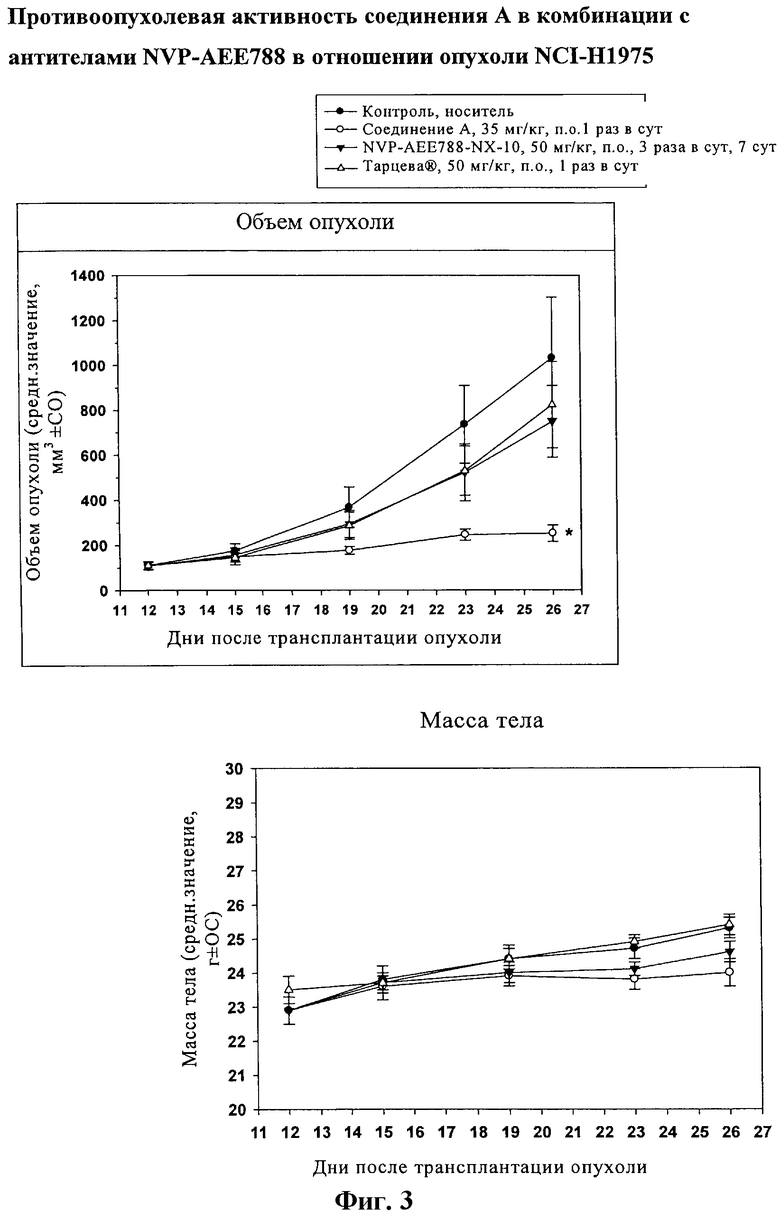
На фиг.3 показана противоопухолевая активность соединения A в отношении клеточной линии NSCLC, резистентной к ингибитору EGFR, NCI-H1975.
Самкам бестимусных мышей Harlan (n=8) с привитыми подкожно опухолями NCI-H1975 вводили перорально в качестве инигибитора PI3K соединение А или ингибитор EGFR NVP-AEE788 и эрлотиниб согласно указанной схеме. *p<0.05 (критерий Даннета по сравнению с контролем).
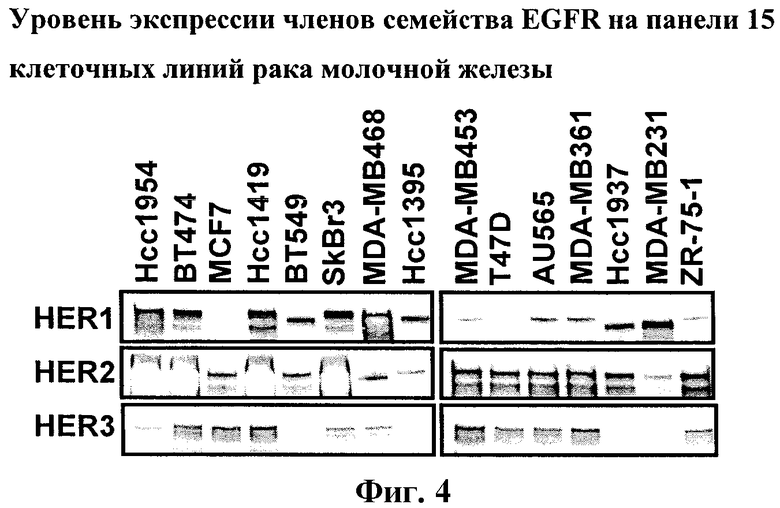
На фиг.4 показан уровень экспрессии белков семейства EGFR HER-1, -2 и -3 на панели 15 клеточных линий опухоли рака молочной железы человека.
Указанные клетки опухолевых линий культивировали в оптимальных условиях роста, из клеток в субконфлуентной фазе роста получали клеточные экстракты. Затем экстракты, содержащие эквивалентное количество общего белка, подвергали электрофорезу в ДСН-ПААГ, гели переносили на мембраны и мембраны инкубировали с антителами против белков, указанных на левой стороне панели.
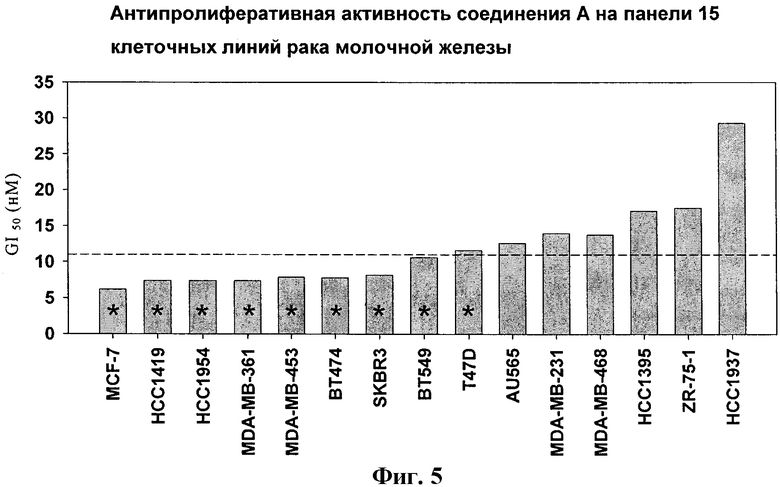
На фиг.5 показана противоопухолевая активность соединения А на панели клеточных линий рака молочной железы.
Указанные клеточные линии инкубировали с возрастающим количеством соединения A и влияние на пролиферацию оценивали по окрашиванию жизнеспособных клеток метиленовым синим. В обозначенных красной звездочкой клеточных линиях наблюдалась гибель клеток (то есть в этих клетках после обработки наблюдалось более низкое поглощение по сравнению с исходными клетками).
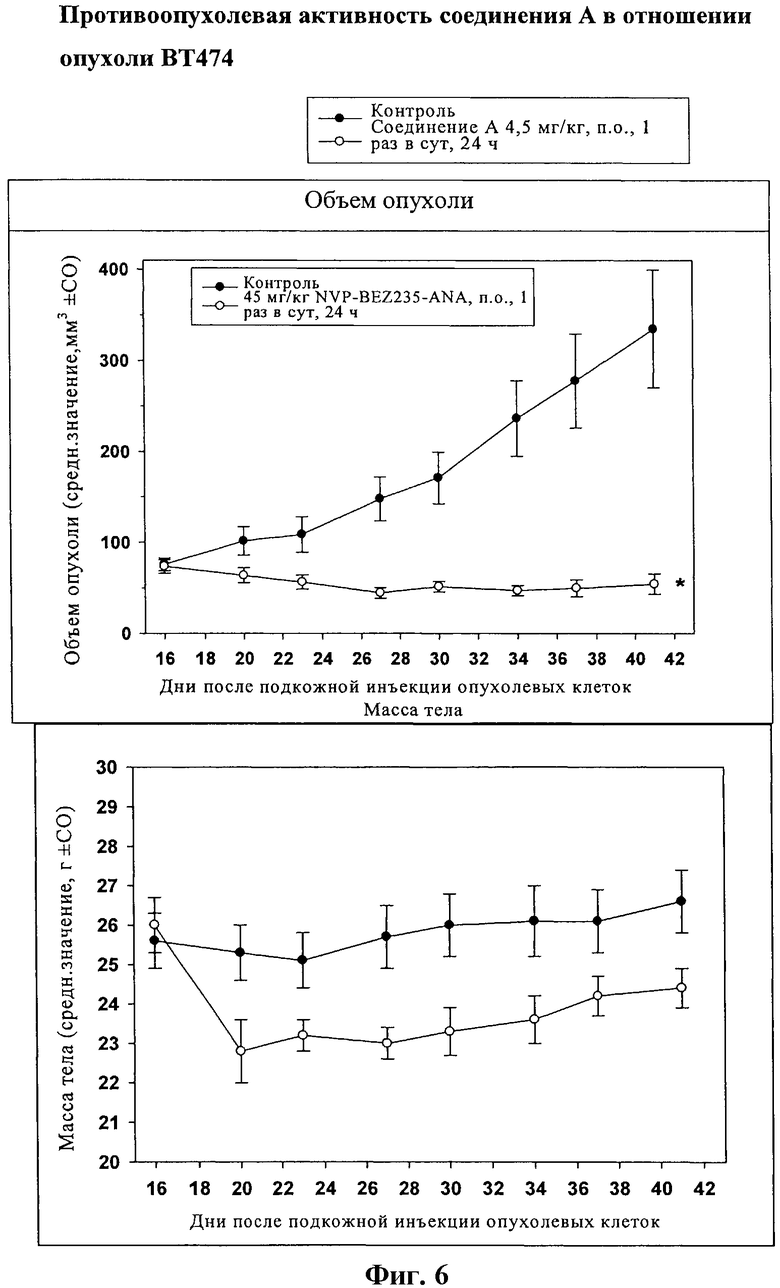
На фиг.6 представлена противоопухолевая активность соединения А в отношении опухолей BT474.
Самкам бестимусных мышей Harlan (n=8) с привитыми подкожно опухолями NCI-H1975 вводили перорально в качестве инигибитора PI3K соединение А согласно указанной схеме. *p<0.05 (критерий Даннета по сравнению с контролем).
Панель из 15 опухолевых клеточных линий NSCLC характеризовали по экспрессии членов семейства EGFR (фиг.1). Согласно данным, описанным в литературе, большинство этих линий характеризуются высоким уровнем HER1 и 3. Указанное относится и к клеточной линии NCI-H358, чувствительной к ингибиторам киназ EGFR. Указанную клеточную линию обрабатывали низкомолекулярными ингибиторами киназ EGFR и соединениями формулы I. Концентрация соединения, при которой наблюдается подавление роста на 50% (GI50), определенная для гефитиниба методом пролиферативного анализа с использованием метиленового синего для указанной клеточной линии, составляет 542 нМ и величина GI50 для соединения A составляет 31 нМ. Индекс комбинации соединения A и гефитиниба по данным пролиферативного анализа в присутствии опухолевых клеток NCI-H358 составляет 1,0, что свидетельствует об аддитивном действии обоих молекул на указанную клеточную линию опухоли легких. Аналогичные результаты были получены с использованием других ингибиторов EGFR, включая NVP-AEE788, эрлотиниб и лапатиниб. Более того, после введения in vivo комбинации таких производных имидазохинолина с ингибитором киназы EGFR, NVP-AEE788, наблюдается состояние опухоли, аналогичное состоянию, которое наблюдается при введении таких соединений животным с подкожно привитым ксенотрансплантатом NCI-H358 (фиг.2).
Клеточная линия немелкоклеточной карциномы легкого с амплифицированным EGFR, но резистентная к лечению ингибитором EGFR, является ценной моделью для оценки чувствительности к ингибиторам PI3K, подобным соединениям формулы I, в указанной генетической среде. Клеточная линия NCI-H1975 является моделью, которая характеризуется повышенной экспрессией HER1 и HER3 и высокой канцерогенностью in vivo. Более того, указанная линия клеток содержит HER1, характеризующийся мутацией T790M в гене-посреднике, что придает резистентность киназы к каталитическому ингибированию. Величина GI50 указанной клеточной линии составляет 11,4 нМ и 3645 нМ для соединения A и гефитиниба соответственно. Противоопухолевую активность in vivo ингибиторов PI3K, подобных соединениям формулы I, определяли с использованием указанной модели опухоли, индуцированной EGFR и резистентной к ингибиторам (фиг.3). Как ожидалось, EGFR ингибиторы киназы NVP-AEE788 и эрлотиниб не проявляют значительного подавления роста опухоли, но неожиданно после введения соединения A наблюдается подавление роста опухоли in vivo. Соединение A характеризуется достаточно высокой переносимостью - не наблюдается статистически значимого различия между массой тела в контрольной и леченой группах.
В клеточных линиях молочной железы и яичников в большинстве случаев наблюдается сверхэкспрессия ErB-B2 (HER2). Уровень экспрессии указанного белка на панели 15 клеточных линий рака молочной железы показан на фиг.4. Хотя в настоящее время существуют эффективные методы лечения с использованием ингибиторов ErbB2, у 50% пациентов, у которых наблюдается сверхэкспрессия HER2, не развивается ответная реакция на модуляторы ErbB2, такие как трастузумаб. С использованием панели клеточных линий рака молочной железы, характеризующихся или не характеризующихся амплификацией ErbB2, было установлено, что соединение A снижает клеточную пролиферацию (среднее значение GI50 составляет 11,1 нМ) и индуцирует гибель клеток в клеточных линиях, которые характеризуются сверхэкспрессией ErbB2 (фиг.5). Более того, подкожные трансплантаты ВТ474 проявляют чрезвычайно высокую чувствительность к лечению соединением A, поскольку регрессия опухоли наблюдается при лечении соединением в суточной дозе 45 мг/кг, перорально, один раз в сутки (фиг.6).
Следовательно, соединение формулы I, прежде всего, соединение A, можно использовать для лечения заболеваний, зависимых от EGFR, прежде всего, злокачественных заболеваний, или заболеваний с приобретенной резистентностью к членам семейства EGFR. Заболевания или злокачественные образования с установленной или потенциальной молекулярной взаимосвязью с разрегуляцией активности EGFR описаны, например, в статьях Mendelsohn и Baselga, Status of Epidermal Growth Factor Receptor Antagonists in the Biology and Treatment of Cancer, Journal of Clinical Oncology, 2787-2799; Mendelsohn и Baselga, Epidermal Growth Factor Receptor Targeting in Cancer, Seminars in Oncology, т.33, 369-385; Irmer и др., EGFR kinase domain mutations - functional impact and relevance for lung cancer therapy, Oncogene, 1-9, Roche-Lima и др., EGFR targeting of solid tumors, Cancer Control, т.14(3), 295-304 (2007), которые все, включая цитированные в данном контексте ссылки, включены в настоящее описание в качестве ссылок.
Согласно настоящему изобретению заболевания, зависимые от EGFR, прежде всего, злокачественные заболевания, которые можно лечить соединениями формулы I, прежде всего, соединением A или соединением В, предпочтительно включают:
- немелкоклеточный рак легких
- рак головы и шеи
- колоректальный рак
- рак молочной железы
- злокачественные заболевания мозга, включая глиобластому
- рак предстательной железы
- рак мочевого пузыря
- почечно-клеточную карциному
- рак поджелудочной железы
- рак шейки матки
- рак пищевода
- рак желудка
- рак яичников
или любую их комбинацию.
Настоящее изобретение относится к применению соединения формулы I, как описано выше, или их таутомеров, или их фармацевтически приемлемых соли или гидрата или сольвата для получения фармацевтического препарата, предназначенного для лечения заболевания, зависимого от EGFR.
Более того, настоящее изобретение относится к применению соединения формулы I, прежде всего, 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-с]хинолин-1-ил)фенил]пропионитрила (соединение A) или 8-(6-метоксипиридин-3-ил)-3-метил-1-(4-пиперазин-1-ил-3-трифторметилфенил)-1,3-дигидроимидазо[4,5-с]хинолин-2-она (соединение B), для получения фармацевтического препарата, предназначенного для лечения заболевания, зависимого от EGFR или злокачественного образования или заболевания с приобретенной резистентностью к агентам, которые связываются с членами семейства EGFR.
Резистентность к лечению модулятором EGFR развивается при лечении указанным модулятором EGFR или в результате одной или нескольких мутаций в белке.
Прежде всего, настоящее изобретение относится к лечению заболевания или злокачественного образования, которое зависит от членов семейства EGFR или характеризуется резистентностью, приобретенной в ходе лечения модулятором EGFR, содинениями формулы I, прежде всего соединением А или соединением В или их фармацевтически приемлемой солью. Возможными модуляторами EGFR, к которым при лечении может развиваться резистентность, являются, например, гефитиниб, эрлотиниб, лапатиниб, цетуксимаб, нимотузумаб, панитумумаб и трастузумаб.
Соединение формулы (I) можно также использовать для лечения зависимых от EGFR
заболеваний или заболеваний с приобретенной резистентностью к EGFR в комбинации с другими активными соединениями, например, в комбинации компонентов, описанных в WO2006/122806, более предпочтительно с агентами, связывающимися с семейством EGFR, такими как, но не ограничиваясь только ими, гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY334543, BIBW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональные антитела 806, анти-EGFR моноклональные антитела-Y90/Re-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, MDX-214, CDX110, IMC11F8, пертузумаб, трастузумаб, земаб®, вакцина Нег2 РХ 1041 и ингибиторы HSP90: CNF1010, CNF2024, танеспимицин, алвеспимицин, IPI504, SNX5422 и NVP-AUY922.
Настоящее изобретение также относится к комбинированному лечению заболеваний, зависимых от EGFR, соединениями формулы I, прежде всего соединениями, выбранными из группы, включающей 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-с]хинолин-1-ил)фенил]пропионитрил (соединение А) и 8-(6-метоксипиридин-3-ил)-3-метил-1-(4-пиперазин-1-ил-3-трифторметилфенил)-1,3-дигидроимидазо[4,5-с]хинолин-2-он (соединение В), и модулятором EGFR, выбранным из группы, включающей гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY334543, BIBW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональные антитела 806, анти-EGFR моноклональные антитела Y90/Re-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, пертузумаб, MDX-214, CDX110, IMC11F8, пертузумаб, трастузумаб, земаба®, вакцину Her2 PX1041 и ингибиторы HSP90: CNF1010, CNF2024, танеспимицин, алвеспимицин, IPI504, SNX5422 и NVP-AUY922, при этом активные ингредиенты присутствуют в каждом случае в свободной форме или в форме фармацевтически приемлемой соли, и указанные комбинации необязательно включают по крайней мере один фармацевтически приемлемый носитель и предназначены для совместного, раздельного или последовательного применения при лечении немелкоклеточного рака легких, рака головы и шеи, колоректального рака, рака молочной железы, злокачественных заболеваний мозга, включая глиобластому, рака предстательной железы, рака мочевого пузыря, почечно-клеточной карциномы, рака поджелудочной железы, рака шейки матки, рака пищевода, рака желудка и/или рака яичников.
Прежде всего, настоящее изобретение относится к комбинации соединения формулы I, выбранного из группы, включающей 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрил и 8-(6-метоксипиридин-3-ил)-3-метил-1-(4-пиперазин-1-ил-3-трифторметилфенил)-1,3-дигидроимидазо[4,5-c]хинолин-2-он, и модулятора EGFR, выбранного из группы, включающей гефитиниб, эрлотиниб, лапатиниб, цетуксимаб, нимотузумаб, панитумумаб и трастузумаб, в которой активные ингредиенты присутствуют в каждом случае в свободной форме или в форме фармацевтически приемлемой соли, и необязательно включающей по крайней мере один фармацевтически приемлемый носитель, и указанная комбинация предназначена для совместного, раздельного или последовательного применения при лечении немелкоклеточного рака легких, рака головы и шеи, колоректального рака, рака молочной железы, злокачественных заболеваний мозга, включая глиобластому, рака предстательной железы, рака мочевого пузыря, почечно-клеточной карциномы, рака поджелудочной железы, рака шейки матки, рака пищевода, рака желудка и рака яичников.
В другом варианте осуществления настоящего изобретения предлагается способ лечения заболевания или злокачественного образования, зависимого от EGFR, предпочтительно, злокачественного образования, которое характеризуется приобретенной резистентностью к модуляторам киназ EGFR в ходе лечения указанным модулятором EGFR, и указанный способ включает введение терапевтически эффективного количества специфического производного имидазохинолина формулы I, прежде всего предпочтительно 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрила (соединение A) или 8-(6-метоксипиридин-3-ил)-3-метил-1-(4-пиперазин-1-ил-3-трифторметилфенил)-1,3-дигидроимидазо[4,5-c]хинолин-2-она (соединения B) или их фармацевтически приемлемой соли, в отдельности или в комбинации с модулятором EGFR, теплокровному животному, нуждающемуся в таком лечении.
Заболеваниями, которые можно лечить указанным способом, предпочтительно включают немелкоклеточный рак легких, рак головы и шеи, колоректальный рак, рак молочной железы, злокачественные заболевания мозга, включая глиобластому, рак предстательной железы, рак мочевого пузыря, почечно-клеточную карциному, рак поджелудочной железы, рак шейки матки, рак пищевода, рак желудка и рак яичников.
В другом варианте настоящего изобретения предлагается фармацевтический препарат, предназначенный для лечения заболевания, зависимого от EGFR, или заболевания, которое характеризуется приобретенной резистентностью в ходе лечения модулятором EGFR, и указанный препарат включает соединение формулы I, прежде всего, предпочтительно 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрил (соединение A) или 8-(6-метоксипиридин-3-ил)-3-метил-1-(4-пиперазин-1-ил-3-трифторметилфенил)-1,3-дигидроимидазо[4,5-c]хинолин-2-он (соединение B), или их фармацевтически приемлемую соль и по крайней мере один фармацевтически приемлемый носитель, в отдельности или в комбинации с модулятором EGFR.
Заболевания, которые можно лечить указанным фармацевтическим препаратом, предпочтительно включают немелкоклеточный рак легких, рак головы и шеи, колоректальный рак, рак молочной железы, злокачественные заболевания мозга, включая глиобластому, рак предстательной железы, рак мочевого пузыря, почечно-клеточную карциному, рак поджелудочной железы, рак шейки матки, рак пищевода, рак желудка и рак яичников.
В еще одном варианте настоящего изобретения предлагается применение соединения формулы I, прежде всего, предпочтительно 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрила (соединения A) или 8-(6-метоксипиридин-3-ил)-3-метил-1-(4-пиперазин-1-ил-3-трифторметилфенил)-1,3-дигидроимидазо[4,5-c]хинолин-2-она (соединения В), или их фармацевтически приемлемой соли для лечения заболевания, зависимого от EGFR, или заболевания, которое характеризуется резистентностью, приобретенной в ходе лечения модулятором EGFR.
Заболеваниями, которые можно лечить указанными соединениями в отдельности или в комбинации с модулятором EGFR, предпочтительно включают немелкоклеточный рак легких, рак головы и шеи, колоректальный рак, рак молочной железы, злокачественные заболевания мозга, включая глиобластому, рак предстательной железы, рак мочевого пузыря, почечно-клеточную карциному, рак поджелудочной железы, рак шейки матки, рак пищевода, рак желудка и рак яичников.
Соединения формулы (I) можно также использовать для усиления действия в комбинации с известными способами лечения, например, при введении гормонов или, прежде всего, при лучевой терапии. Соединения формулы (I), прежде всего, можно использовать в качестве радиосенсибилизаторов, прежде всего, для лечения опухолей, которые характеризуются слабой чувствительностью к лучевой терапии.
Лечение по изобретению включает симптоматическое или профилактическое лечение.
Термин «комбинация», использованный в данном контексте, обозначает фиксированную комбинацию в виде единой лекарственной формы, либо в виде набора компонентов для совместного введения, в котором соединение формулы (I) и комбинацию компонентов можно вводить независимо, в одно и то же время или каждый в отдельности через определенные промежутки времени, при этом прежде всего обеспечивается кооперативный, например, синергетический эффект комбинации компонентов.
Соединение формулы I можно вводить в отдельности или в комбинации с одним или более других терапевтических соединений, причем комбинированное лечение проводят фиксированными комбинациями или при введении соединения по изобретению и одного или более других терапевтических соединений поочередно или независимо друг от друга, или при совместном введении фиксированных комбинаций и одного или более других терапевтических соединений.
Дозировка активного ингредиента зависит от различных факторов, включая тип, вид, возраст, массу тела, пол и состояние здоровья пациента, тяжесть состояния, которое предназначено для лечения, способ введения, функции почек и печени пациента и, прежде всего, конкретное использованное соединение. Терапевт, клиницист или ветеринар может определить и прописать эффективное количество лекарственного средства, требуемое для предотвращения, контроля или остановки прогрессирования состояния. Оптимальная точность достижения концентрации лекарственного средства в крови, которая находится в интервале эффективных концентраций, определяют на основе кинетики доступности лекарственного средства в участках-мишенях. При этом необходимо учитывать распределение, равновесие и экскрецию лекарственного средства.
Соединения по изобретению можно вводить любым стандартным способом, прежде всего, парентерально, например в форме растворов или суспензий для инъекции, энтерально, например перорально, например в форме таблеток или капсул, местным способом, например в форме лосьонов, гелей, мазей или кремов, или в назальной форме или в форме суппозитория. Местное введение включает, например, введение на кожу. Другой формой местного введения является введение в глаза. Фармацевтические композиции, включающие соединение по изобретению в смеси по крайней мере с одним фармацевтически приемлемым носителем или разбавителем, можно получать стандартным способом при смешивании с фармацевтически приемлемым носителем или разбавителем.
Фармацевтические композиции для лечения указанных выше нарушений включают эффективное количество соединения формулы I или его N-оксида или таутомера в смеси с фармацевтически приемлемыми носителями, которые пригодны для местного, энтерального, например перорального или ректального, или парентерального введения и которые могут представлять собой неорганические или органические, твердые или жидкие вещества.
Фармацевтические композиции, предназначенные для перорального введения, прежде всего, таблетки или желатиновые капсулы, включают активный ингредиент в смеси с разбавителем, например лактозой, декстрозой, маннитом и/или глицерином и/или замасливателями и/или полиэтиленгликолем. Таблетки также могут включать связующие агенты, например алюмосиликат магния, крахмалы, такие как кукурузный, пшеничный или рисовый крахмал, желатин, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы и/или поливинилпирролидон, и, при необходимости, дезинтегрирующие агенты, например крахмалы, агар, альгиновую кислоту или ее соль, такую как альгинат натрия, и/или шипучие смеси, или адсорбенты, красители, ароматизаторы и подсластители. Можно также использовать фармакологически активные соединения по настоящему изобретению в форме парентеральных композиций или в форме растворов для вливания. Фармацевтические композиции можно стерилизовать и/или они могут включать эксципиенты, например консерванты, стабилизаторы, увлажняющие соединения и/или эмульгаторы, солюбилизаторы, соли для регуляции осмотического давления и/или буферные вещества. Фармацевтические композиции по настоящему изобретению, которые при необходимости могут включать другие фармакологически активные вещества, получают стандартным способом, например при стандартном смешивании, грануляции, переработке, растворении или лиофилизации, и указанные фармацевтические композиции включают приблизительно от 1% до 99%, предпочтительно от приблизительно 1% до приблизительно 20% активного ингредиента(ов).
Настоящее изобретение относится к применению соединения 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-с]хинолин-1-ил)фенил]пропионитрила (соединение А) или его фармацевтически приемлемой соли для получения фармацевтического препарата, предназначенного для лечения заболевания, зависимого от рецептора эпидермального фактора роста (EGFR), связанного с амплификацией EGFR, мутацией EGFR1, мутацией Т790М EGFR или их комбинацией. Изобретения также относится к комбинациям указанного соединения с другими активными соединениями и к способам лечения указанных заболеваний указанными соединениями и фармацевтическими препаратами. Фармацевтические препараты включают указанное соединение в отдельности или в комбинации, прежде всего, с модулятором EGFR. При использовании комбинаций обеспечивается синергетический эффект. 4 н. и 12 з.п. ф-лы, 6 ил.
1. Применение соединения 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-с]хинолин-1-ил)фенил]пропионитрил (соединение А)
или его фармацевтически приемлемая соль,
для получения фармацевтического препарата, предназначенного для лечения заболевания, зависимого от рецептора эпидермального фактора роста (EGFR), связанного с амплификацией EGFR, мутацией EGFR1, мутацией Т790М EGFR, или их комбинацией.
2. Применение по п.1, где заболеванием является злокачественное образование.
3. Применение по п.1, где указанное заболевание характеризуется резистентностью к лечению модулятором EGFR.
4. Применение по п.3, где резистентность к лечению модулятором EGFR приобретена в ходе лечения указанным модулятором EGFR.
5. Применение по п.3, где резистентность вызвана мутацией или мутациями в белке.
6. Применение по п.3, где модулятор EGFR выбран из группы, включающей гефитиниб, эрлотиниб, лапатиниб, цетуксимаб, нимотузумаб, панитумумаб и трастузумаб.
7. Применение по п.1 совместно с модулятором EGFR.
8. Применение по п.7, где модулятор EGFR выбран из группы, включающей гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY334543, BIBW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональные антитела 806, анти-EGFR моноклональные антитела Y90/Re-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, MDX-214, CDX110, IMC11F8, пертузумаб, трастузумаб, земаб®, вакцину Неr2 РХ 1041 и ингибиторы HSP90, CNF1010, CNF2024, танеспимицин, алвеспимицин, IPI504, SNX5422 и NVP-AUY922.
9. Применение по любому из пп.1 или 8, где заболевание, предназначенное для лечения, включает:
- немелкоклеточный рак легких
- рак головы и шеи
- колоректальный рак
- рак молочной железы
- злокачественные заболевания мозга, включая глиобластому
- рак предстательной железы
- рак мочевого пузыря
- почечно-клеточную карциному
- рак поджелудочной железы
- рак шейки матки
- рак пищевода
- рак желудка
- рак яичников
или любую их комбинацию.
10. Комбинация 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-с]хинолин-1-ил)фенил]пропионитрила (соединение А) и модулятора EGFR, выбранного из группы, включающей гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY334543, BIBW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональные антитела 806, анти-EGFR моноклональные антитела Y90/Re-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, MDX-214, CDX110, IMC11F8, пертузумаб, трастузумаб, земаб®, вакцину Her2 РХ 1041 и ингибиторов HSP90, CNF1010, CNF2024, танеспимицин, алвеспимицин, IPI504, SNX5422 и NVP-AUY922, при этом активные ингредиенты присутствуют в каждом случае в свободной форме или в форме фармацевтически приемлемой соли, и указанная комбинация необязательно включает по крайней мере один фармацевтически приемлемый носитель и предназначена для одновременного, раздельного или последовательного применения при лечении немелкоклеточного рака легких, рака головы и шеи, колоректального рака, рака молочной железы, злокачественных заболеваний мозга, включая глиобластому, рака предстательной железы, рака мочевого пузыря, почечно-клеточной карциномы, рака поджелудочной железы, рака шейки матки, рака пищевода, рака желудка и/или рака яичников, связанного с амплификацией EGFR, мутацией EGFR1, мутацией Т790М EGFR, или их комбинацией.
11. Способ лечения заболевания, зависимого от EGFR, или заболевания, характеризующегося резистентностью, приобретенной в ходе лечения модулятором EGFR, который заключается во введении терапевтически эффективного количества соединения по п.1 теплокровному животному, нуждающемуся в таком лечении, где указанное заболевание связано с амплификацией EGFR, мутацией EGFR1, мутацией Т790М EGFR, или их комбинацией.
12. Способ по п.11, где заболевание, которое предназначено для лечения, это
- немелкоклеточный рак легких
- рак головы и шеи
- колоректальный рак
- рак молочной железы
- злокачественные заболевания мозга, включая глиобластому
- рак предстательной железы
- рак мочевого пузыря
- почечно-клеточную карциному
- рак поджелудочной железы
- рак шейки матки
- рак пищевода
- рак желудка
- рак яичников.
13. Способ по п.11, где 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-с]хинолин-1-ил)фенил]пропионитрил (соединение А) вводят в смеси с модулятором EGFR, выбранным из группы, включающей гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY3 34543, BIBW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональные антитела 806, анти-EGFR моноклональные антитела Y90/Re-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, MDX-214, CDX110, IMC11F8, пертузумаб, трастузумаб, земаб®, вакцину Her2 РХ 1041 и ингибиторов HSP90, CNF1010, CNF2024, танеспимицин, алвеспимицин, IPI504, SNX5422 и NVP-AUY922.
14. Фармацевтический препарат для лечения заболевания, зависимого от EGFR, или заболевания, характеризующегося резистентностью, приобретенной в ходе лечения модулятором EGFR, включающий соединение по п.1 или его фармацевтически приемлемую соль и по крайней мере один фармацевтически приемлемый носитель, где указанное заболевание связано с амплификацией EGFR, мутацией EGFR1, мутацией Т790М EGFR, или их комбинацией.
15. Фармацевтический препарат по п.14, где заболевание, которое предназначено для лечения, это
- немелкоклеточный рак легких
- рак головы и шеи
- колоректальный рак
- рак молочной железы
- злокачественные заболевания мозга, включая глиобластому
- рак предстательной железы
- рак мочевого пузыря
- почечно-клеточную карциному
- рак поджелудочной железы
- рак шейки матки
- рак пищевода
- рак желудка
- рак яичников.
16. Фармацевтический препарат по п.14 или 15, включающий модулятор EGFR, выбранный из группы, включающей гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY334543, BIBW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональные антитела 806, анти-EGFR моноклональные антитела Y90/Re-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, MDX-214, CDX110, IMC11F8, пертузумаб, трастузумаб, земаб®, вакцину Her2 РХ 1041 и ингибиторов HSP90, CNF1010, CNF2024, танеспимицин, алвеспимицин, IPI504, SNX5422 и NVP-AUY922.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| НОВИК А.А | |||
| и др | |||
| Введение в молекулярную биологию канцерогенеза | |||
| - М., 2004, с.188 | |||
| SUSUMI KABAYASHI ET AL.: «EGFR Mutation and Resistance of Non-Small-Cell Lung Cancer to Gefitinib» // N | |||
| Engl | |||
| J | |||
| Med., 24 February 2005, vol.352, P.P.786-792 | |||
| BIANCO ET AL: «Rational bases for the development of EGFR | |||

