Изобретение относится к новым соединениям формулы (I). Изобретение также имеет отношение к родственным аспектам, включающим способы получения соединений, фармацевтические композиции, содержащие соединение формулы (I), и особенно их применение как ингибиторов ренина в случаях сердечно-сосудистых нарушений и почечной недостаточности.
В системе ренин-ангиотензин (RAS) биологически активный ангиотензин II (Ang II) образуется по двухступенчатому механизму. Высокоспецифический фермент ренин расщепляет ангиотензиноген до ангиотензина I (Ang I), который затем дополнительно подвергается обработке до Ang II менее специфическим ферментом (АСЕ), превращающим ангиотензин. Известно, что Ang II воздействует, по меньшей мере, на два подтипа рецепторов, называемых как АТ1 и АТ2. В то время как АТ1, по-видимому, передает большинство известных функций Ang II, роль АТ2 является еще неизвестной.
Модуляция RAS-системы представляет собой главное достижение в лечении сердечно-сосудистых заболеваний. АСЕ-ингибиторы и АТ1-блокаторы были приняты для лечения гипертонии (Waeber B. et al., “The renin-angiotensin system: role in experimental and human hypertension”, in Birkenhager W. H., Reid J. L., (eds): Hypertension, Amsterdam, Elsevier Science Publishing Co., 1986, 489-519; Weber M. A., Am. J. Hypertens., 1992, 5, 247S). Кроме того, АСЕ-ингибиторы применяются для защиты почек (Rosenberg M. E. et al., Kidney International, 1994, 45, 403; Breyer J. A. et al., Kidney International, 1994, 45, S156), для профилактики застойной сердечной недостаточности (Vaunghan D. E. et al., Cardiovasc. Res., 1994, 28, 159; Fouad-Tarazi F. et al., Am. J. Med., 1988, 84 (Suppl. 3A), 83) и инфаркта миокарда (Pfeffer M. A. et al., N. Engl. J. Med., 1992, 327, 669).
Рациональный подход для разработки ингибиторов рeнина состоит в специфичности ренина (Kleinert H. D., Cardiovasc. Drugs, 1995, 9, 645). Единственный субстрат, известный для ренина, представляет собой ангиотензиноген, который может быть обработан только ренином (при физиологических условиях). Напротив, АСЕ может также расщеплять брадикинин помимо Ang I и обходным путем может быть дублирована химазой, серинпротеазой (Husain A., J. Hypertens., 1993, 11, 1155). У пациентов ингибирование АСЕ тем самым ведет к аккумуляции брадикинина, вызывающей кашель (5-20%) и потенциально угрожающий жизни ангионевротический отек (0,1-0,2%) (Israili Z. H. et al., Annals of Internal Medicine, 1992, 117, 234). Химаза не ингибируется АСЕ-ингибиторами. Поэтому образование Ang II все же возможно в пациентах, пролеченных АСЕ-ингибиторами. Блокада АТ1-рецептора (например, лозартаном), с другой стороны, чрезмерно воздействует на другие подтипы АТ-рецепторов (например, АТ2) до Ang II, концентрация которых значительно повышается блокадой АТ1-рецепторов. В итоге ингибиторы ренина, как ожидают, будут демонстрировать фармацевтический профиль, отличный от профиля ингибиторов АСЕ и блокаторов AT1 с точки зрения эффективности в блокировании RAS и аспектов безопасности.
Только ограниченный клинический опыт (Azizi M. et al., J. Hypertens., 1994, 12, 419; Neutel J. M. et al., Am. Heart, 1991, 122, 1094) был cоздан с ингибиторами ренина по причине их недостаточной пероральной активности из-за их пептидомиметической природы (Kleinert H. D., Cardiovasc. Drugs, 1995, 9, 645). Клиническая разработка нескольких соединений была прекращена из-за данной проблемы и высокой стоимости продуктов. Только одно соединение, содержащее четыре хиральных центра, вошло в клинические испытания (Rahuel J. et al., Chem. Biol., 2000, 7, 493; Mealy N. E., Drugs of the Future, 2001, 26, 1139). Таким образом, требуются ингибиторы ренина с хорошей пероральной биодоступностью и длительным периодом действия. Недавно были описаны первые непептидные ингибиторы ренина, которые показывают высокую активность in vitro (Oefner C. et al., Chem. Biol., 1999, 6, 127; Patent Application WO 97/09311; Marki H. P. et al., Il Farmaco, 2001, 56, 21). Однако состояние разработки данных соединений неизвестно.
Данное изобретение относится к ингибиторам ренина непептидной природы и низкой молекулярной массы. Описаны перорально активные ингибиторы ренина формулы (I), которые имеют длительный период действия и которые являются активными при симптомах за пределами регулирования кровяного давления, где тканевая система ренин/химаза может быть активирована, что приводит к патофизиологически измененным локальным функциям, таким как почечная, кардиальная и сосудистая перестройка, атеросклероз и возможно рестеноз. Таким образом, настоящее изобретение описывает данные непептидные ингибиторы ренина формулы (I).
В частности, данное изобретение относится к новым соединениям формулы (I)
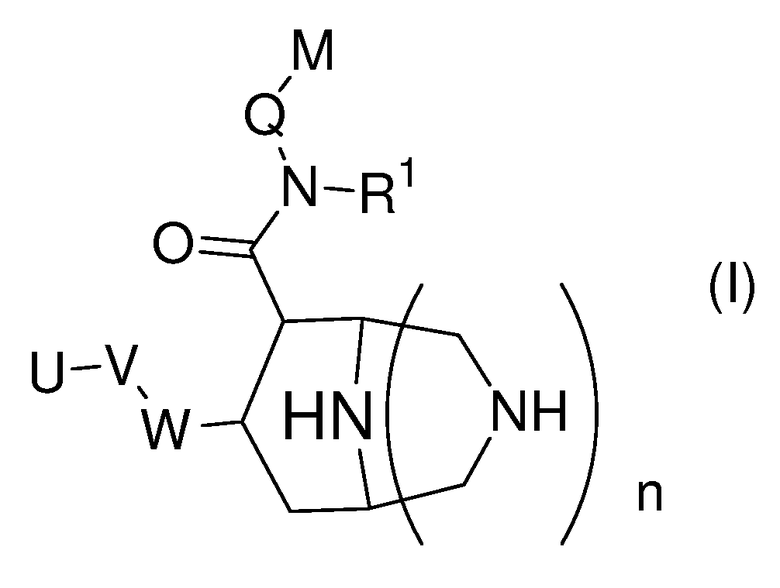
в которой
W представляет собой фенильный цикл или шестичленный, не бензоконденсированный ароматический цикл, содержащий от одного до четырех атомов азота, где указанные циклы замещены в пара-положении посредством V;
V представляет собой связь; -(СН2)r-; -A-(CH2)s-; -CH2-A-(CH2)t-; -(CH2)s-A-; -(CH2)2-A-(CH2)u-; -A-(CH2)v-B-; -CH2-CH2-CH2-A-CH2-; -A-CH2-CH2-B-CH2-; -CH2-A-CH2-CH2-B-; -CH2-CH2-CH2-A-CH2-CH2-; -CH2-CH2-CH2-CH2-A-CH2-; -A-CH2-CH2-B-CH2-CH2-; -CH2-A-CH2-CH2-B-CH2-; -CH2-A-CH2-CH2-CH2-B-; -CH2-CH2-A-CH2-CH2-B-; -O-CH2-CH(OCH3)-CH2-O-; -O-CH2-CH(CH3)-CH2-O-; -O-CH2-CH(CF3)-CH2-O-; -O-CH2-C(CH3)2-CH2-O-; -O-CH2-C(CH3)2-O-; -O-C(CH3)2-CH2-O-; -O-CH2-CH(CH3)-O-; -O-CH(CH3)-CH2-O-; -O-CH2-C(CH2CH2)-O-; или -O-C(CH2CH2)-CH2-O-;
А и В независимо представляют собой -О- или -S-, предпочтительно -О-;
U представляет собой незамещенный арил; моно-, ди-, три- или тетразамещенный арил, в котором заместители независимо выбраны из группы, состоящей из галогена, алкила, алкокси и -СF3; или моно-, ди-, тризамещенный гетероарил, в котором заместители независимо выбраны из группы, состоящей из галогена, алкила, алкокси и -СF3;
Q представляет собой метилен или этилен, предпочтительно метилен;
М представляет собой арильную, хинолинильную, изохинолинильную, дигидрохинолинильную или тетрагидрохинолинильную группу, где указанные группы могут быть необязательно моно- или дизамещенными заместителями, независимо выбранными из группы, cостоящей из алкила; алкокси; -OCF3; -CF3; гидроксиалкила; галогена; алкил-О-(СН2)0-4-СН2-; алкил-О-(СН2)2-4-О-; R'2N-(СН2)0-4-СН2-, где R' независимо выбран из группы, cостоящей из водорода, алкила (необязательно замещенного одним, двумя или тремя атомами фтора), циклопропила, циклопропилметила, -С(=О)О-R'', и -С(=О)-R'', где R'' означает С1-С4-алкил, -CF3, -CH2-CF3 или циклопропил; и R'''NH-C(=O)-(O)0-1-(CH2)0-4-CH2-, где R''' означает алкил или циклопропил;
R1 представляет собой алкил или циклоалкил, предпочтительно циклоалкил, а именно, особенно циклопропил;
n представляет собой целое число 0 или 1;
r представляет собой целое число 3, 4, 5 или 6;
s представляет собой целое число 2, 3, 4 или 5;
t представляет собой целое число 1, 2, 3 или 4;
u представляет собой целое число 1, 2 или 3; и
v представляет собой целое число 2, 3 или 4;
и к оптически чистым энантиомерам, смесям энантиомеров, таким как рацематы, диастереомерам, смесям диастереомеров, диастереомерным рацематам, смесям диастереомерных рацематов, и мезоформам, а также к солям и сольватам таких соединений и к морфологическим формам.
Общие термины, использованные выше и ниже в данном документе, имеют в рамках данного раскрытия следующие значения, если не оговорено особо.
Там, где применяется форма множественного числа для соединений, солей, фармацевтических композиций, заболеваний и тому подобного, она предполагается также для обозначения отдельного соединения, соли или тому подобного.
Любая ссылка на соединение формулы (I) должна быть воспринята как относящаяся также к оптически чистым энантиомерам, смесям энантиомеров, таким как рацематы, диастереомерам, смесям диастереомеров, диастереомерным рацематам, смесям диастереомерных рацематов и мезоформам, а также к солям (особенно фармацевтически приемлемым солям) и сольватам (включающим гидраты) таких соединений и морфологическим формам, как уместным и практически целесообразным.
В определениях формулы (I) - если не оговорено особо - термин алкил, сам по себе или в комбинации с другими группами, означает насыщенные, прямые и разветвленные цепочечные группы с одним-семью атомами углерода, предпочтительно с одним-четырьмя атомами углерода, т.е. С1-С4-алкил. Примеры алкильных групп представляют собой метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, пентил, гексил и гептил. Предпочитают метильные, этильные и изопропильные группы.
Термин алкокси, сам по себе или в комбинации с другими группами, относится к R-O-группе, в которой R означает алкильную группу. Примеры алкоксигрупп представляют собой метокси, этокси, пропокси, изо-пропокси, изо-бутокси, втор-бутокси и трет-бутокси.
Термин гидроксиалкил, сам по себе или в комбинации с другими группами, относится к НО-R-группе, в которой R означает алкильную группу. Примеры гидроксиалкильных групп представляют собой НО-СН2-, НО-СН2СН2-, НО-СН2СН2СН2- и СН3СН(ОН)-.
Термин галоген означает фтор, хлор, бром или иод, предпочтительно фтор, хлор или бром, особенно фтор или хлор.
Термин циклоалкил, сам по себе или в комбинации, означает насыщенную циклическую углеводородную кольцевую систему с 3-7 атомами углерода, например, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Циклопропильная группа является предпочтительной группой.
Термин арил, сам по себе или в комбинации, относится к фенильной, нафтильной или инданильной группе, предпочтительно к фенильной группе.
Термин гетероарил, сам по себе или в комбинации, означает шестичленные ароматические циклы, содержащие от одного до четырех атомов азота; бензоконденсированные шестичленные ароматические циклы, содержащие от одного до трех атомов азота; пятичленные ароматические циклы, содержащие один атом кислорода, один атом азота или один атом серы; бензоконденсированные пятичленные ароматические циклы, содержащие один атом кислорода, один атом азота или один атом серы; пятичленные ароматические циклы, содержащие два гетероатома, независимо выбранные из кислорода, азота и серы, и бензоконденсированные производные таких циклов; пятичленные ароматические циклы, содержащие три атома азота и их бензоконденсированные производные; тетразолильный цикл; триазинильный цикл; или кумаринил. Примеры таких циклических систем представляют собой фуранил, тиенил, пирролил, пиридинил, пиримидинил, индолил, хинолинил, изохинолинил, имидазолил, триазинил, тиазолил, изотиазолил, пиридазинил, пиразолил, оксазолил, изоксазолил, бензотиенил, хиназолинил и хиноксалинил.
Соли предпочтительно представляют собой фармацевтически приемлемые соли соединений формулы (I).
Выражение фармацевтически приемлемые соли охватывает соли как с неорганическими кислотами или с органическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота, азотная кислота, фосфористая кислота, азотистая кислота, лимонная кислота, муравьиная кислота, уксусная кислота, щавелевая кислота, малеиновая кислота, молочная кислота, винная кислота, фумаровая кислота, бензойная кислота, миндальная кислота, коричная кислота, пальмитиновая кислота, стеариновая кислота, глутаминовая кислота, аспарагиновая кислота, метансульфоновая кислота, этансульфоновая кислота, этандисульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, янтарная кислота, трифторуксусная кислота и тому подобное, которые нетоксичны для живых организмов или, в случае соединения формулы (I), являются кислотными по природе, так и с неорганическим основанием, таким как основание щелочного или щелочноземельного элемента, например, гидроксид натрия, гидроксид калия, гидроксид кальция и тому подобное. На другие примеры фармацевтически приемлемых солей можно сделать отсылку к “Salt selection for basic drugs”, Int. J. Pharm. (1986), 33, 201-217.
Соединения формулы (I) могут содержать асимметрические атомы углерода и могут быть приготовлены в форме оптически чистых энантиомеров, смесей энантиомеров, таких как рацематы, диастереомеров, смесей диастереомеров, диастереомерных рацематов, смесей диастереомерных рацематов, или мезоформ. Настоящее изобретение охватывает все данные формы. Смеси разделяют способом, известным для разделения веществ в чистом виде, например, колоночной хроматографией, тонкослойной хроматографией (ТСХ/TLC), высокоэффективной жидкостной хроматографией (ВЭЖХ/HPLC), или кристаллизацией.
Соединения изобретения также включают нитрозированные соединения формулы (I), которые были нитрозированы по одному или нескольким сайтам, таким как кислород (гидроксильная конденсация), сера (сульфгидрильная конденсация) и/или азот. Нитрозированные соединения данного изобретения могут быть приготовлены традиционными способами, известными специалисту в данной области. Например, известные способы по нитрозирующим соединениям описаны в U.A. Pat. Nos. 5380758, 5703073, 5994294, 6242432 и 6218417; WO 98/19672; и Oae et al., Org. Prep. Proc. Int., 15(3): 165-198 (1983).
Группа предпочтительных соединений формулы (I) является группой, в которой n представляет собой целое число 1 и заместители в цикле, -СОN(R1)-Q-M и -W-V-U, находятся в транс-положении друг к другу.
Группа особенно предпочтительных соединений формулы (I) является группой, в которой n представляет собой целое число 0 и конфигурации в положениях 3 и 4 пиперидинового цикла формулы (I) представляют собой 3R и 4S, соответственно.
Группа предпочтительных соединений формулы (I) является группой, в которой М представляет собой арильную, хинолинильную, изохинолинильную, дигидрохинолинильную или тетрагидрохинолинильную группу, где указанные группы необязательно могут быть моно- или дизамещены заместителями, независимо выбранными из группы, состоящей из алкила; алкокси; -OCF3; -CF3; гидроксиалкила; галогена; алкил-О-(СН2)0-4-СН2-; алкил-О-(СН2)2-4-О-; и R'2N-(СН2)0-4-СН2-, где R' независимо выбран из группы, cостоящей из водорода, алкила, циклопропила, и -С(=О)-R'', где R'' означает С1-С4-алкил, -CF3, -CH2-CF3 или циклопропил.
Другая группа предпочтительных соединений формулы (I) является группой, в которой М представляет собой арил, предпочтительно фенил, который, в частности, необязательно моно- или дизамещен заместителями, независимо выбранными из группы, состоящей из алкила, алкокси, -OCF3, -CF3, гидроксиалкила и галогена, предпочтительно из группы, состоящей из алкила, алкокси и галогена.
Другая группа особенно предпочтительных соединений формулы (I) является группой, в которой М представляет собой следующий радикал:
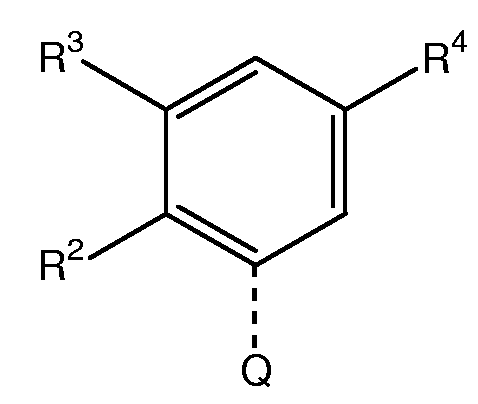 , где R2 означает метил или хлор, R3 означает водород, и R4 означает водород, -СН2СН2-О-СН3, -СН2СН2СН2-О-СН3, или R'NH-(CH2)0-1-CH2-, где R' означает -СН2-СНF2, -CH2-CF3, циклопропил, -СО-СН3, -СО-СН2-СF3, -CO-CH2-CH3, или циклопропилкарбонил, при условии, когда R4 означает водород, то R3 представляет собой метил, метокси, хлор, или -ОСН2СН2-О-СН3.
, где R2 означает метил или хлор, R3 означает водород, и R4 означает водород, -СН2СН2-О-СН3, -СН2СН2СН2-О-СН3, или R'NH-(CH2)0-1-CH2-, где R' означает -СН2-СНF2, -CH2-CF3, циклопропил, -СО-СН3, -СО-СН2-СF3, -CO-CH2-CH3, или циклопропилкарбонил, при условии, когда R4 означает водород, то R3 представляет собой метил, метокси, хлор, или -ОСН2СН2-О-СН3.
Очень предпочтительная группа соединений формулы (I) является группой, в которой М представляет собой следующий радикал:
, где R2 означает хлор, R3 означает водород и R4 означает -СН2СН2-О-СН3.
Очень предпочтительная группа соединений формулы (I) является группой, в которой М представляет собой следующий радикал:
, где R2 означает водород, R3 означает метоксиэтокси и R4 означает -СН2СН2СН2-О-СН3.
Следующая группа предпочтительных соединений формулы (I) является группой, в которой Q представляет собой метилен.
Следующая группа предпочтительных соединений формулы (I) является группой, в которой R1 представляет собой циклопропил.
Другая группа предпочтительных соединений формулы (I) является группой, в которой W представляет собой фенил, замещенный на V в пара-положении, или следующий радикал:
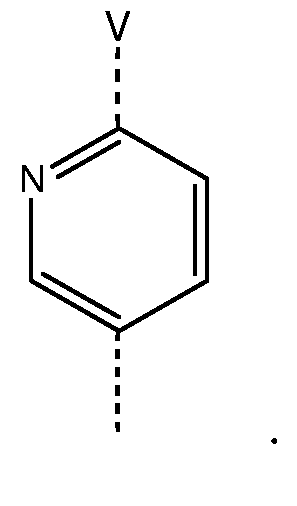
Другая группа предпочтительных соединений формулы (I) является группой, в которой W представляет собой фенил, замещенный на V в пара-положении.
Другая группа особенно предпочтительных соединений формулы (I) является группой, в которой W представляет собой следующий радикал:
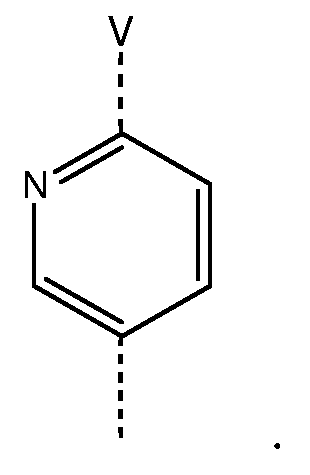
Следующая группа предпочтительных соединений формулы (I) является группой, в которой V представляет собой -CH2CH2O- или -CH2CH2СН2O-, где в обоих случаях бивалентный радикал присоединен к группе U формулы (I) через атом кислорода, или -ОСН2СН2О-.
Очень предпочтительная группа соединений формулы (I) является группой, в которой V представляет собой -ОСН2СН2О-.
Другая группа предпочтительных соединений формулы (I) является группой, в которой V представляет собой -OCH2CH2O- или особенно -CH2CH2СН2O- [предпочтительно, где -СН2-часть -CH2CH2СН2O-фрагмента присоединена к группе W формулы (I)].
Следующая группа предпочтительных соединений формулы (I) является группой, в которой U представляет собой моно-, ди-, три- или тетразамещенный арил, предпочтительно моно-, ди-, или тризамещенный фенил, где заместители независимо выбраны из группы, состоящей из галогена, алкила, алкокси и -СF3, особенно из группы, состоящей из галогена или алкила.
Другая группа особенно предпочтительных соединений формулы (I) является группой, в которой U представляет собой 2,6-дихлор-4-метилфенил.
Другая группа особенно предпочтительных соединений формулы (I) является группой, в которой U представляет собой 2-хлор-3,6-дифторфенил.
Предпочтительный вариант осуществления данного изобретения относится к соединению формулы (I), где W представляет собой фенил, замещенный на V в пара-положении, или следующий радикал:
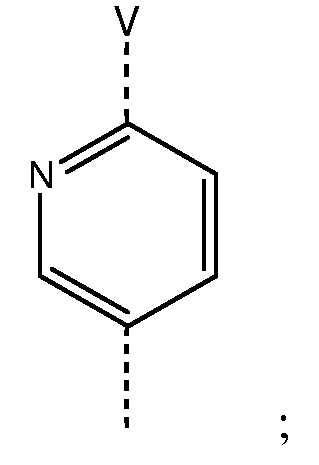
V представляет собой -OCH2CH2O- или -CH2CH2СН2O-, где -СН2-часть -CH2CH2СН2O-фрагмента присоединена к группе W формулы (I);
U представляет собой тризамещенный фенил, в котором заместители независимо выбраны из атомов галогена (в частности, фтора и хлора) и алкила (в частности, метила);
Q представляет собой метилен;
М представляет собой следующий радикал:
, где R2 означает метил или хлор, R3 означает водород, и R4 означает водород, -СН2СН2-О-СН3, -СН2СН2СН2-О-СН3, или R'NH-(CH2)0-1-CH2-, где R' означает алкил (необязательно замещенный одним или двумя атомами фтора), циклопропил, циклопропилметил, -СО-СН3, или -СО-СН2-СF3, при условии, когда R4 означает водород, то R3 представляет собой метил, метокси, или хлор;
R1 представляет собой циклопропил; и
n представляет собой целое число 0 или 1.
Данное изобретение также относится к соединениям формулы (I), где значения одного или нескольких заместителей или символов, определенные для формулы (I), или предпочтительного варианта формулы (I), заменены на их предпочтительные значения, определенные здесь, а именно, значения, определенные для вышеданных предпочтительных групп соединений.
Особенно предпочтительные соединения формулы (I) представляют собой соединения, выбранные из группы, включающей:
циклопропил(2,3-дихлорбензил)амид (1R∗, 5S∗, 6R∗, 7S∗)-7-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}-3,9-диаза-бицикло[3.3.1]нонан-6-карбоновой кислоты,
циклопропил(3-метокси-2-метилбензил)амид 4-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
циклопропил(3-метокси-2-метилбензил)амид 4-{4-[3-(2,6-дихлор-4-метилфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
циклопропил(3-метокси-2-метилбензил)амид 4-{4-[3-(2,3,6-трифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2,6-дихлор-4-метилфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2-хлор-6-фтор-3-метилфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты, и
циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2,3,6-трифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты.
Следующие особенно предпочтительные соединения формулы (I) представляют собой соединения, выбранные из группы, включающей:
циклопропил(2,3-диметилбензил)амид (3R∗, 4S∗)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[2-хлор-5-(3-метоксипропил)бензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[2-хлор-5-(2-метоксиэтил)бензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[2-хлор-5-(3-метоксипропил)бензил]циклопропиламид (3R, 4S)-6-[2-(2,6-дихлор-4-метилфенокси)этокси]-1',2',3',4',5',6'-гексагидро[3,4']бипиридинил-3'-карбоновой кислоты, и
[2-хлор-5-(2-метоксиэтил)бензил]циклопропиламид (3R, 4S)-6-[2-(2,6-дихлор-4-метилфенокси)этокси]-1',2',3',4',5',6'-гексагидро[3,4']бипиридинил-3'-карбоновой кислоты.
Другая группа особенно предпочтительных соединений формулы (I) представляет собой соединения, выбранные из группы, включающей:
{2-хлор-5-[(2,2-дифторэтиламино)метил]бензил}циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
{2-хлор-5-[(3,3,3-трифторпропиониламино)метил]бензил}циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
{2-хлор-5-[(циклопропилметиламино)метил]бензил}циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[5-(ацетиламинометил)-2-хлорбензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[2-хлор-5-(2-метиламиноэтил)бензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
(2-хлор-5-метиламинометилбензил)циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
{2-хлор-5-[2-(3,3,3-трифторпропиониламино)этил]бензил}циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[2-хлор-5-(2-этиламиноэтил)бензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[5-(2-ацетиламиноэтил)-2-хлорбензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
{2-хлор-5-[(2-фторэтиламино)метил]бензил}циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
(2-хлор-5-этиламинометилбензил)циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
(2-хлор-5-циклопропиламинометилбензил)циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты, и
[2-хлор-5-(2-циклопропиламиноэтил)бензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты.
Cоединения формулы (I) применимы для лечения и/или профилактики заболеваний, таких как (или связанных с ними) гипертония, застойная сердечная недостаточность, легочная гипертензия, почечная недостаточность, почечная ишемия, почечная декомпенсация, фиброз почек, сердечная недостаточность, гипертрофия сердца, фиброз сердца, миокардиальная ишемия, кардиомиопатия, гломерулонефрит, почечная колика, осложнения, полученные от диабета, а именно, нефропатия, васкулопатия и невропатия, глаукома, повышенное внутриглазное давление, атеросклероз, рестеноз после пластической операции на сосудах, осложнения после хирургической операции на сосудах и сердце, эректильная дисфункция, гиперальдостеронизм, пневмофиброз, склеродерма, страх, расстройство познавательной способности, осложнения от лечения иммунодепрессантами, и других заболеваний, которые известны, как связанные с системой ренин-ангиотензин.
Соединения формулы (I) особенно применимы для лечения и/или профилактики заболеваний, таких как (или связанных с ними) гипертония, застойная сердечная недостаточность, легочная гипертензия, почечная недостаточность, почечная ишемия, почечная декомпенсация, фиброз почек, сердечная недостаточность, гипертрофия сердца, фиброз сердца, миокардиальная ишемия, кардиомиопатия, осложнения, полученные от диабета, а именно, нефропатия, васкулопатия и невропатия.
В одном варианте осуществления изобретение относится к способу лечения или профилактики заболеваний, которые связаны с нарушением регулирования системы ренин-ангиотензин, в частности, к способу лечения или профилактики вышеупомянутых заболеваний, при этом указанный способ включает введение пациенту фармакологически активного количества соединения формулы (I).
Следующий аспект данного изобретения относится к фармацевтическим композициям, содержащим соединение формулы (I) и фармацевтически приемлемый материал в качестве носителя. Данные фармацевтические композиции можно применять для лечения и/или профилактики вышеупомянутых заболеваний. Фармацевтические композиции можно применять для энтерального, парентерального или местного введения. Их можно вводить, например, перорально, например, в виде таблеток, таблеток с покрытием, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий, ректально, например, в виде суппозиториев, парентерально, например, в виде растворов для инъекций или растворов для инфузий, или локально, например, в виде мазей, кремов или масел.
Изобретение также относится к применению соединения формулы (I) для приготовления фармацевтических композиций для лечения или профилактики вышеупомянутых заболеваний.
Изготовление фармацевтических композиций может быть осуществлено способом, который будет знаком любому специалисту в данной области (см., например, Mark Gibson, Editor, Pharmaceutical Preformulation and Formulation, IНS Health Group, Englewood, CO, USA, 2001; Remington, The Science and Practice of Pharmacy, 20th Edition, Philadelphia College of Pharmacy and Science), внесением описанных соединений формулы (I) или их фармацевтически приемлемых солей, необязательно в комбинации с другими терапевтически полезными веществами, в галеновую форму для введения вместе с подходящими, нетоксичными, инертными, терапевтически совместимыми твердыми или жидкими носителями и, если желательно, полезными фармацевтическими адъювантами.
Соединения формулы (I) или вышеупомянутые фармацевтические композиции также применимы в комбинации с другими фармакологически активными соединениями, такими как АСЕ-ингибиторы, ингибиторы нейтральной эндопептидазы, антагонисты альдостерона, антагонисты рецептора ангиотензина II, антагонисты рецепторов эндотелина, вазодилататоры, антагонисты кальция, активаторы калия, диуретики, симпатолитики, бета-адренергические антагонисты, альфа-адренергические антагонисты и/или другие лекарственные средства, полезные для предотвращения или лечения вышеупомянутых заболеваний, такие как ингибиторы 11-бета-гидроксистероидной дегидрогеназы типа 1 и активаторы растворимой гуанилатциклазы.
Данное изобретение также относится к пролекарствам соединения формулы (I), которые превращаются in vivo в соединение формулы (I) как таковое. Любая ссылка на соединение формулы (I) поэтому должна быть воспринята как относящаяся также к соответствующим пролекарствам соединения формулы (I), как уместным и практически целесообразным.
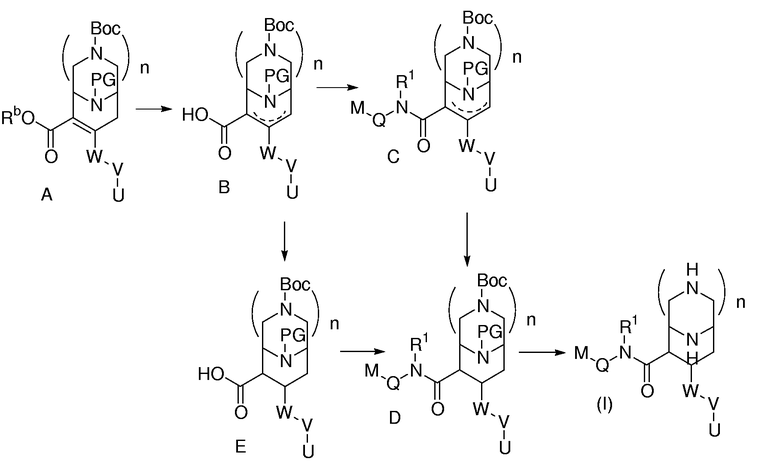
Соединения формулы (I) могут быть приготовлены из соединения вида А (WO 03/093267; WO 04/002957), как описано на схеме 1, где W, V, U и n принимают значения, определенные для формулы (I). PG означает подходящую защитную группу. Rb означает подходящий сложный эфир, который затем можно разложить, например, омылением или гидрированием. В течение омыления соединения вида А образуется соединение вида В с 20-100% выходом в зависимости от условий, в котором двойная связь частично смещена до деконьюгированного положения. Данное соединение можно отделять или можно не отделять от соответствующего аналога с двойной еще коньюгированной связью, и затем, например, амидная конденсация, приводит к соединению вида С, где R1, Q и М принимают значения, определенные для формулы (I). В течение амидной конденсации с помощью оптимизации условий двойная связь может смещаться почти полностью. Соединение вида С затем восстанавливается в соединение вида D. Наконец, удаление защиты приводит к соединению формулы (I). Конечно, восстановление может также происходить с соединением вида В, которое в данном случае отделяют от его региоизомера с еще коньюгированной двойной связью. В данном случае получается соединение вида Е, которое превращается в соединение вида D после амидной конденсации. Если n=0, то коньюгированное соединение вида А или В может восстанавливаться до соответствующего насыщенного соединения магнием в метаноле. После равновесия выделяется транс-пространственно расположенный стереоизомер. Омыление (если необходимо) приводит к соединению вида Е.
Cпециалисту в данной области будет очевидно, что последовательность данных стадий может быть изменена или модифицирована в большинстве случаев. Смещение двойной связи в соединении вида В и его восстановление в соединения D и Е приводят к смесям стереоизомеров. Данные стереоизомеры могут быть выделены стандартными методами, такими как флэш-колоночная хроматография, ВЭЖХ или хиральная ВЭЖХ.
Схема 1
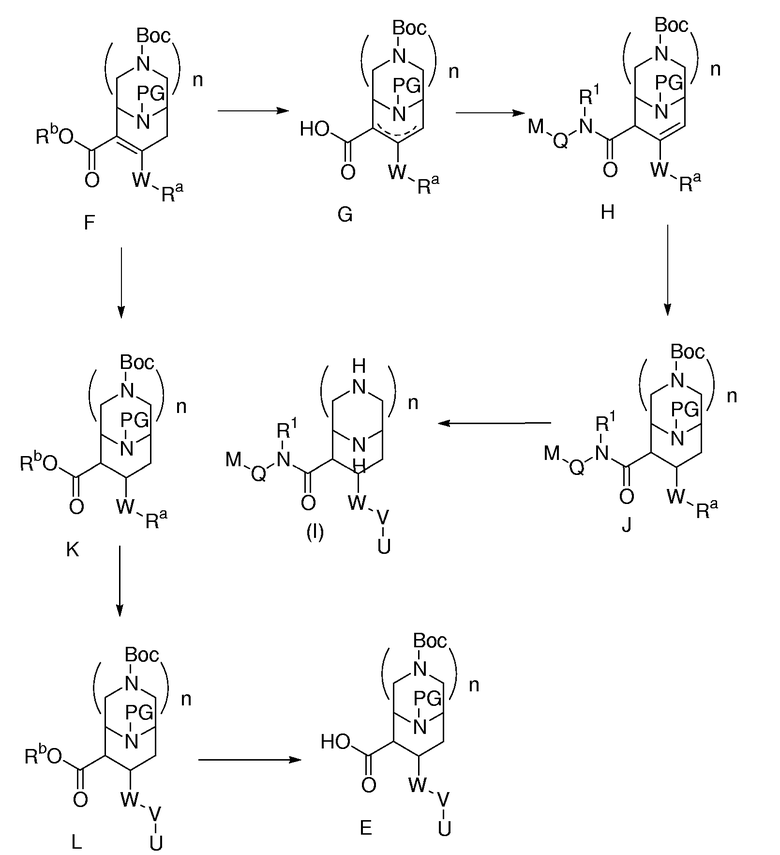
Иное соединение вида F можно использовать, как описано на схеме 2, где Rb означает подходящий сложноэфирный заместитель, и Ra означает предшественник заместителя V, определенного для формулы (I). Данное соединение вида F превращается в соединение вида G, которое, в свою очередь, превращается в соединение вида Н, при применении условий амидной конденсации, которые способствуют смещению двойной связи. Восстановление двойной связи ведет к соединению вида J, которое затем превращается в соединение формулы (I). Также соединение вида F может восстанавливаться в соединение вида К. U-V-W-цепь завершается с образованием соединения вида L, затем сложный эфир гидролизуется до соединения вида Е.
Схема 2
Примеры
Сокращения (использованные в данном документе):
ВЭЖХ- или ЖХ-МС-условия (если не оговорены особо)
Аналитические: Zorbax 59 SB Aqua колонка, 4,6 × 50 мм от Agilent Technologies. Элюент: А: ацетонитрил; В: Н2О + 0,5% TFA. Градиент: 90% В → 5% В в течение 2 мин. Поток: 1 мл/мин. Детектирование: УФ/вид. + МС.
Препаративные: Zorbax SB Aqua колонка, 20 × 500 мм от Agilent Technologies. Элюент: А: ацетонитрил; В: Н2О + 0,05% аммоний гидроксид (25% водным). Градиент: 80% В → 10% В в течение 6 мин. Поток: 40 мл/мин. Детектирование: УФ + МС, или УФ + ELSD.
Хиральные, аналитические: Regis Whelk колонка, 4,6 × 250 мм, 10 мкм. Элюент А: EtOH + 0,05% Et3N. Элюент В: гексан. Изократические условия, 1 мл/мин. Изократическая смесь может меняться в зависимости от соединений.
Хиральные, препаративные: такие как аналитические условия, но на Regis Whelk 01 колонке, 50 × 250 мм, на потоке при 100 мл/мин.
Общая методика для Mitsunobu конденсации и удаления Вос-защиты
Требуемый фенол (2 экв.) помещают в реакционную колбу. Добавляют соединение J3 или J4 (примерно 80 мг, 1 экв.), растворенное в толуоле (1 мл), с последующим добавлением дипиперидида азодикарбоновой кислоты (2 экв.), растворенного в толуоле (1 мл). Реакционную смесь дегазируют с помощью N2 и в конце добавляют PВu3 (3 экв.). Реакционную смесь нагревают при 90°С (в предварительно нагретой системе) в течение 1 час, затем охлаждают снова до к.т. Добавляют Et2O (5 мл), смесь фильтруют, и растворители выпаривают при пониженном давлении. Сырой промежуточный продукт очищают ВЭЖХ с обращенными фазами. Очищенный продукт растворяют в диоксане (1 мл) и добавляют HCl в диоксане (4 М; 1 мл). Смесь перемешивают при к.т. в течение 2 час. Растворители удаляют при пониженном давлении, получая гидрохлоридную соль конечного соединения формулы (I).
Общие условия А для амидной конденсации
HOBt (16,9 мг, 0,125 ммоль), DMAP (3,05 мг, 0,025 ммоль), DIPEA (0,068 мл, 0,40 ммоль) и EDC.HCl (28,8 мг, 0,150 ммоль) добавляли к смеси карбоновой кислоты (0,100 ммоль) и амина (0,100 ммоль) в CH2Cl2 (1,00 мл). Смесь перемешивали в течение 4 дней при к.т. Смесь фильтровали через Isolute® (0,6 г сорбента, приготовленного с 1 мл водной 1 М НCl). Органический слой упаривали при пониженном давлении. Сырой продукт затем использовали без очистки.
Общие условия В для удаления Вос-защиты
Исходный продукт растворяли в CH2Cl2 (1 мл), и раствор охлаждали до 0°С. HCl (4 M в диоксане, 0,5 мл), и смесь перемешивали в течение 1 час, в то время как смесь нагревалась до к.т. Добавляли водный 1 М NaOH, и смесь перемешивали еще в течение 5 мин. Слои разделяли, и органический слой упаривали при пониженном давлении. Очистка с помощью ВЭЖХ (ацетонитрил с 0,05% водным концентрированным NH3/вода 10:90→90:10 по всей Х-мостиковой колонке) давала указанное в заголовке соединение.
Общие условия С для амидной конденсации через хлорангидрид кислоты
Исходную карбоновую кислоту (1,00 экв.) растворяли в толуоле (14 мл/ммоль карбоновой кислоты) и добавляли ДМФА (каталитическое количество). Добавляли оксалилхлорид (1,30 экв.), и смесь перемешивали в течение 1 час при к.т. Растворители удаляли при пониженном давлении, и сырой хлорангидрид кислоты разделяли на 0,100 ммоль-порции для последующих амидных конденсаций. Такую порцию хлорангидрида кислоты (0,100 ммоль) растворяли в СН2Cl2 (1,00 мл), и к данному раствору добавляли Et3N (0,014 мл, 0,10 ммоль) и раствор целевого амина (0,100 ммоль) в СН2Сl2 (0,50 мл). Смесь перемешивали в течение 1 час при к.т. и фильтровали через Isolute®, предварительно промытый водной 1 М НСl. После элюирования с помощью СН2Сl2 органический слой упаривали при пониженном давлении и остаток использовали на следующей стадии без очистки.
3-трет-Бутиловый эфир 6-этиловый эфир (рац.)-(1R∗, 5S∗)-9-метил-7-трифторметансульфонилокси-3,9-диазабицикло[3.3.1]нон-6-ен-3,6-дикарбоновой кислоты
3-трет-Бутиловый эфир 6-этиловый эфир (рац.)-(1R∗, 5S∗)-9-метил-7-оксо-3,9-диазабицикло[3.3.1]нонан-3,6-дикарбоновой кислоты (WO 2003/093267, 99,58 г, 305 ммоль) растворяют в сухом ТГФ (1450 мл) в атмосфере азота и смесь охлаждают до 0°С. NaH (16,64 г; 55% дисперсия в минеральном масле, 381 ммоль) добавляют порциями по 2 г на протяжении 35 мин, поддерживая температуру в интервале от 0 до 4°С. Выделяется Н2 газ. После добавления смесь становится желто-зеленой и представляет собой слабую суспензию. Реакционную смесь перемешивают в течение 75 мин при температуре от 0 до 4°С. Затем добавляют Tf2NPh (128,6 г, 360 ммоль) в виде твердого вещества в течение 5 мин. Реакционная смесь становится коричневой. Охлаждающую баню удаляют и реакционную смесь перемешивают на протяжении выходного дня при к.т. Реакционную смесь выливают в 1 л смеси лед/вода и растворители удаляют при пониженном давлении. Оставшуюся водную фазу экстрагируют с помощью EtOAc (3 × 500 мл). Объединенные органические слои промывают водой (500 мл) и насыщенным солевым раствором (500 мл). Органическую фазу затем сушат над MgSO4, фильтруют и упаривают при пониженном давлении. К сырому коричневому остатку (174 г) добавляют 50 мл пентана и смесь перемешивают при 4°С в течение ночи. Кристаллы отфильтровывают и промывают холодным гексаном (70 мл) и холодной смесью гексан/Et2O (4:1, 100 мл). Это приводит к 84 г продукта, содержащего некоторое количество TfNНPh. Данный продукт фильтруют через силикагель (75 г). TfNНPh вымывают с СН2Сl2. Данный продукт затем вымывают с помощью EtOAc (3 раза 1 л), получая указанное в заголовке соединение после упаривания при пониженном давлении. Указанное в заголовке соединение получают в трех фракциях: а) 44,45 г не совсем белых кристаллов, b) 27,98 г слегка коричневых кристаллов и с) 15 г желтого масла, содержащего продукт и TfNНPh. Через 2 дня TfNНPh, содержащийся во фракции с), кристаллизуется. Ее фильтруют, получая 9,43 г продукта в виде коричневого масла.
Обработка маточных растворов
Объединенные маточные растворы, полученные выше, концентрируют в вакууме. Коричневый маслянистый остаток (75 г) очищают с помощью FC (1500 г силикагеля), применяя градиентное элюирование (EtOAc/гептан 1-9 → EtOAc). Колонку затем промывают смесью EtOAc/MeOH 9:1. Указанное в заголовке соединение выделяют в виде 25,44 г не совсем белого твердого вещества в качестве чистого продукта. ЖХ-МС: tR = 0,87 мин; ES+: 459,24.
1-Аллил-4-бромбензол
Mg (8,76 г, 360 ммоль) суспендируют в ТГФ (90 мл) в атмосфере азота в трехгорлой колбе, снабженной холодильником и капельной воронкой. Капельную воронку наполняют 1,4-дибромбензолом (77,3 г, 327 ммоль) в ТГФ (40 мл). Примерно 5% раствора 1,4-дибромбензола осторожно добавляют к суспензии Mg и реакцию запускают с помощью теплового металлизатора. Когда реакция начинается, добавляют раствор 1,4-дибромбензола с такой скоростью, чтобы реакционная смесь слабо кипела с обратным холодильником (примерно 20 мин). Смесь перемешивают в течение еще 30 мин и охлаждают до 0°С. Добавляют ТГФ (100 мл) и капельную воронку наполняют раствором аллилбромида (30,5 мл, 360 ммоль) в ТГФ (50 мл). Аллилбромид добавляют медленно, поддерживая температуру реакционной смеси ниже 20°С. Когда добавление заканчивается, смесь перемешивают в течение еще 30 мин, при одновременном охлаждении до 0°С. Добавляют водную 1 M НСl. Смесь разбавляют Et2O и промывают водной 1 М НСl и насыщенным солевым раствором. Объединенные водные экстракты снова экстрагируют Et2O. Объединенные органические экстракты сушат над MgSO4, фильтруют и растворители удаляют при пониженном давлении. Перегонка остатка (11 мбар, 88-92°С) дает указанное в заголовке соединение (39,3 г, примерно 61%) вместе с другой неидентифицированной примесью.
3-(4-Бромфенил)пропан-1-ол
ВН3 (1 М в ТГФ, 412 мл, 412 ммоль) добавляют к раствору 1-аллил-4-бромбензола (204 г, 1,03 ммоль) в ТГФ (1,00 л) в атмосфере азота при 0°С. Смесь перемешивают в течение ночи, в то время как она нагревается до к.т. Добавляют водный NaOH (2,5 М, 1,65 л, 4,12 моль) и смесь охлаждают до 0°С. Осторожно добавляют по каплям Н2О2 (35%, 480 мл, 5,15 мол), и смесь перемешивают в течение 3 час. Добавляют Et2O и фазы отделяют. Органический слой промывают водой (1×) и насыщенным солевым раствором (1×). Органический слой сушат над MgSO4, фильтруют и растворители удаляют при пониженном давлении. Очистка с помощью FC (Et2O/петролейный эфир 1:1 → Et2O) дает указанное в заголовке соединение (97,4 г, 44%).
[3-(4-Бромфенил)пропокси]трет-бутилдиметилсилан
Раствор 3-(4-бромфенил)пропан-1-ола (49,4 г, 230 ммоль) в ДМФА (500 мл) охлаждают до 0°С и добавляют имидазол (23,77 г, 349 ммоль) и TBDMS-Cl (52,6 г, 349 ммоль). Смесь перемешивают в течение ночи, в то время как она нагревается до к.т. Смесь разбавляют гептаном (1,0 л) и водным насыщенным NH4Cl (800 мл), и смесь встряхивают. Слои разделяют. Водный слой экстрагируют гептаном, и объединенные органические экстракты промывают насыщенным солевым раствором. Органические экстракты сушат над MgSO4, фильтруют и растворители удаляют при пониженном давлении. Очистка с помощью FC (Et2O/гептан 1:99 → 1:19) дает указанное в заголовке соединение (68,1 г, 90%). ЖХ-МС: tR = 1,24 мин.
2-(2,6-Дихлор-4-метилфенокси)этанол
В трехгорлой колбе, снабженной газовым капельным счетчиком и эффективной системой охлаждения, нагревали смесь 2,6-дихлор-п-крезола (20,0 г, 113 ммоль), [1,3]диоксолан-2-она (9,95 г, 113 ммоль) и имидазола (115 мг, 1,70 ммоль) при 160°С в течение 25 час. Смеси позволяли охлаждаться до к.т. Очистка с помощью FC (Et2O/гептан 1:1) давала указанное в заголовке соединение (18,7 г, 75%). ЖХ-МС: tR = 0,88 мин.
5-Бром-2-[2-(2,6-дихлор-4-метилфенокси)этокси]пиридин
Раствор 2-(2,6-дихлор-4-метилфенокси)этанола (18,6 г, 84 ммоль) в ТГФ (360 мл) охлаждали до 0°С. NaH (примерно 55% в масле, 6,60 г, примерно 153 ммоль) добавляли порциями, и смесь перемешивали при к.т. в течение 30 мин. Раствор 2,5-дибромпиридина (18,0 г, 76,3 ммоль) в ТГФ (60 мл) добавляли по каплям, и смесь нагревали при кипении с обратным холодильником в течение 90 мин. Смеси позволяли охлаждаться до к.т., и к ней осторожно добавляли лед. Растворители частично удаляли при пониженном давлении, и остаток разбавляли с помощью EtOAc. Данную смесь промывали водным насыщенным NH4Cl. Водный слой экстрагировали снова с помощью EtOAc (2×). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtОАс/гептан 3:97) давала указанное в заголовке соединение (22,7 г, 79%). ЖХ-МС: tR = 1,13 мин; ES+: 378,08.
5-Бром-2-хлор-N-циклопропилбензамид
К суспензии 5-бром-2-хлорбензойной кислоты (17,6 г, 75 ммоль) в СН2Сl2 (180 мл) добавляли оксалилхлорид (7,0 мл, 83 ммоль). Добавляли ДМФА (8 капель), и смесь перемешивали в течение 2 час при к.т. (выделение газа). Смесь концентрировали при пониженном давлении, и сырой остаток разбавляли с помощью СН2Сl2 (530 мл). Смесь охлаждали до 0°С, и раствор циклопропиламина (5,8 мл, 83 ммоль) в СН2Сl2 (45 мл) добавляли по каплям. Смесь перемешивали в течение 10 мин при 0°С и позволяли нагреваться до к.т. Добавляли DIPEA (14,3 мл, 84 ммоль), и смесь перемешивали в течение 2 час при к.т. Смесь разбавляли большим количеством СН2Сl2 и промывали водной 10% HCl, водой и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Остаток растирали в гексане, фильтровали и сушили в высоком вакууме, получая указанное в заголовке соединение (19,3 г, 94%). ЖХ-МС: tR = 0,84 мин; ES+: 316,89.
2-Хлор-N-циклопропил-5-(3-метоксипропенил)бензамид
5-Бром-2-хлор-N-циклопропилбензамид (19,3 г, 70,5 ммоль) растворяли в смеси ДМФА (350 мл) и 1-пропанола (210 мл). Добавляли Pd(OAc)2(PPh3)3 (2,64 г, 3,53 ммоль). Добавляли с помощью шприца (E)-2-(3-метоксипропенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (15 мл, 70,5 ммоль) и добавляли водный 2 М Na2CO3 (123 мл). Смесь перемешивали в течение 2 час при 80°С, и ей позволяли нагреваться до к.т. Водную 10% НСl осторожно добавляли до тех пор, пока рН не становился равным 2. Растворители частично удаляли при пониженном давлении, и остаток экстрагировали с помощью Et2O (3Ч). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (EtОАс/гептан 1:2 → 1:1 → EtOAc) давала указанное в заголовке соединение (12,7 г, 68%). ЖХ-МС: tR = 0,82 мин; ES+: 266,17.
2-Хлор-N-циклопропил-5-(3-метоксипропил)бензамид
2-Хлор-N-циклопропил-5-(3-метоксипропенил)бензамид (12,6 г, 47,6 ммоль) растворяли в сухом толуоле (650 мл). Добавляли сухой ДМФА (70 мл) и смесь нагревали до 110°С. Бензолсульфонилгидразин (24,5 г, 143 ммоль) добавляли тремя порциями в течение 3 час. Смесь нагревали в течение общего периода времени 3 час, и ей позволяли охлаждаться до к.т. Растворители удаляли при пониженном давлении и остаток разбавляли Et2О. Смесь промывали водой и водную фазу экстрагировали cнова Et2O (3×). Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (EtОАс/гептан 1:9 → 1:4 → 1:3 → 1:1) давала указанное в заголовке соединение (10,0 г, 78%). ЖХ-МС: tR = 0,83 мин; ES+: 268,19.
[2-Хлор-5-(3-метоксипропил)бензил]циклопропиламин
LiAlH4 (5,70 г, 151 ммоль) добавляли порциями к раствору 2-хлор-N-циклопропил-5-(3-метоксипропил)бензамида (10 г, 37,7 ммоль) в ТГФ (230 мл) при 0°С. Ледяную баню удаляли, и смесь нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и охлаждали ее до 0°С. Осторожно добавляли воду (7,5 мл), водный 15% NaOH (17 мл) и воду (5,7 мл). Смесь фильтровали, и осадок промывали с помощью EtOAc. Фильтрат упаривали при пониженном давлении. Остаток разбавляли EtOAc, и полученную смесь промывали водой и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (EtОАс/гептан 1:4 → 1:3 → 1:2) давала указанное в заголовке соединение (5,60 г, 59%). ЖХ-МС: tR= 0,86 мин; ES+: 254,19.
Общая методика для восстановительного аминирования замещенных бензальдегидов с циклопропиламином:
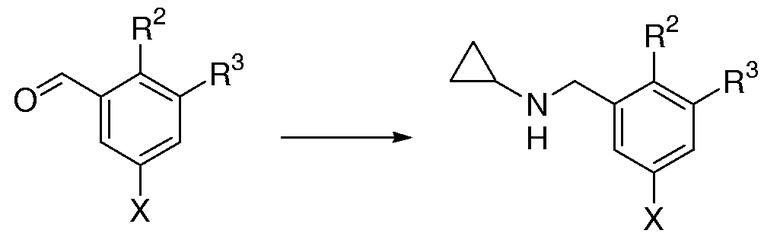 Х = галоген, в частности, Br
Х = галоген, в частности, Br
Раствор замещенного бензальдегида (17,8 ммоль, 1,0 экв.), циклопропиламина (3,13 мл, 44,5 ммоль, 2,5 экв.) и цианоборогидрида натрия (1,34 г, 21,4 ммоль, 1,2 экв.) в МеОН (100 мл) обрабатывали капельным добавлением ледяной уксусной кислоты (3,06 мл, 53,4 ммоль, 3,0 экв.). Полученный раствор перемешивали при к.т. в течение 16 час в течение ночи. Реакционную смесь гасили капельным добавлением насыщенного водного NaHCO3 и концентрировали при пониженном давлении для удаления МеОН. Сырой остаток выливали в 250 мл разделительную воронку, содержащую насыщенный водный раствор NaHCO3 (150 мл), и экстрагировали с помощью EtOAc (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка с помощью FC давала бензаминовый продукт.
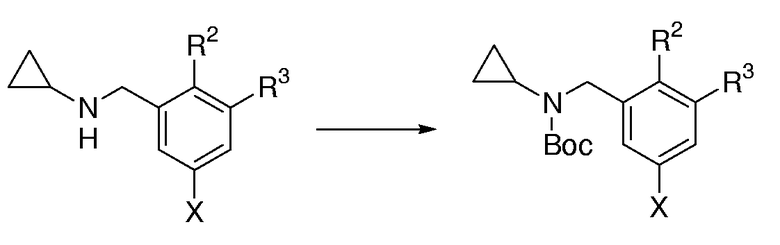
Общая методика для Вос-защиты циклопропилбензаминов:
 Х = галоген, в частности, Br
Х = галоген, в частности, Br
Раствор циклопропилбензамина (43,7 ммоль, 1,0 экв.) в бифазной смеси СН2Сl2 (50 мл) и 1 М водного NaOH (50 мл) обрабатывали с помощью Вос2О (15,1 мл, 65,6 ммоль, 1,5 экв.). Смесь интенсивно перемешивали при к.т. в течение 16 час. Смесь выливали в 500 мл разделительную воронку, содержащую Н2О (300 мл), и экстрагировали с помощью CH2Cl2 (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка с помощью FC давала амин, защищенный Вос-группой.
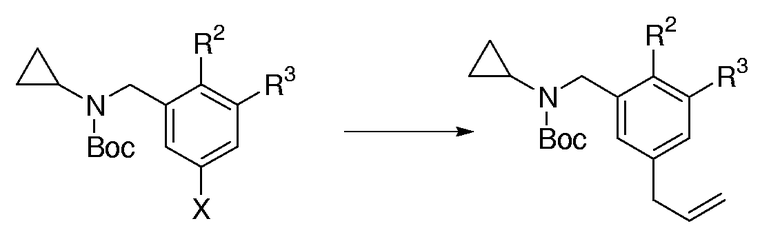
Общая методика для аллилирования Вос-защищенных циклопропилбензаминов:
 Х = галоген, в частности, Br
Х = галоген, в частности, Br
В круглодонную колбу, высушенную пламенем, или сосуд Шленка, в атмосфере N2 добавляли Pd[PCy3]2 (0,05 экв.), CsF (2,0 экв.) и соответствующий арилбромид (1,0 экв.). Если использовали арилхлорид в качестве исходного продукта, то применяли (Pd[PtBu3]Br)2 димер (0,025 экв.) вместо Pd[PCy3]2 катализатора. Колбу откачивали при пониженном давлении (0,1 мм Hg) и снова наполняли N2 (повторяли 3 раза). Полученные твердые вещества растворяли в безводном ТГФ или диоксане (0,15 М раствор), и к данному раствору добавляли три-н-бутилаллилолово (1,5 экв.), затем полученную смесь кипятили с обратным холодильником в течение 8-16 час, пока ТСХ не показывала полное потребление исходного продукта. Реакционную смесь охлаждали до к.т. и фильтровали через пад силикагеля на воронке со спекшимся стеклом, промывая с помощью Et2O. Фильтрат концентрировали и очищали с помощью FC, получая соответствующее аллилбензамидное производное.
Общая методика для гидроборирования/окисления аллилбензаминов:
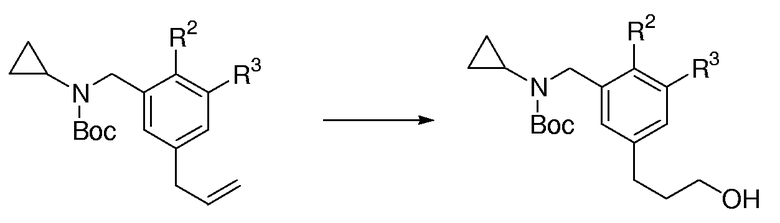
В высушенную пламенем круглодонную колбу, снабженную магнитной мешалкой, добавляли аллилбензамин (1,0 экв.) и безводный ТГФ (0,3 М раствор). Раствор охлаждали до 0°С и борандиметилсульфидный комплекс (1,1 экв.) добавляли по каплям в течение 20 мин. Раствор перемешивали при 0°С в течение 1 час, затем ему позволяли нагреваться до к.т., и перемешивали в течение дополнительных 2 час. Раствор охлаждали до 0°С, и к нему добавляли по каплям 1 М водный NaOH (ПРЕДОСТЕРЕЖЕНИЕ - ЭКЗОТЕРМИЧЕСКАЯ РЕАКЦИЯ) с последующим добавлением по каплям 30% водной Н2О2. Смеси позволяли нагреваться до к.т. и перемешивали ее в течение 2 час. Смесь выливали в разделительную воронку, содержащую Н2О и экстрагировали с помощью Et2O (3 раза). Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка с помощью FC давала целевой спиртовый продукт.
Общая методика для окислительного расщепления/восстановления аллилбензаминов:
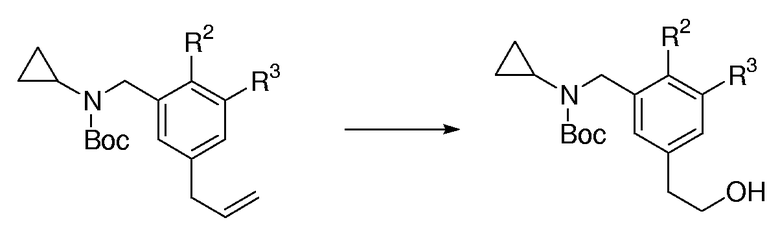
Раствор аллилбензамина (1,0 экв.) в CH2Cl2 (0,4 M раствор) охлаждали до -78°С и вводили в раствор О3 газ, применяя трубку для распределения газа. Озоновый газ вводили до тех пор, пока не был израсходован исходный продукт, определяемый с помощью ТСХ, и реакционная смесь сохраняла слабо голубое окрашивание. Реакционную смесь перемешивали при -78°С в течение 20 мин, затем к ней добавляли EtOH (0,5 М раствор) и NaBH4 (2,5 экв.). Смеси позволяли нагреваться при к.т. в течение ночи (16 час). Реакционную смесь гасили капельным добавлением насыщенного водного NH4Cl (5 мл) и выливали в разделительную воронку, содержащую насыщенный водный NH4Cl. Смесь экстрагировали с помощью Et2O (3 раза). Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка с помощью FC давала целевой спирт.
Общая методика для этерификации ароматических первичных спиртов с метилиодидом:
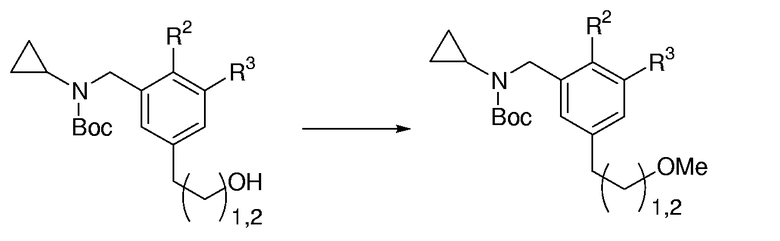
Суспензию первичного спирта (1,0 экв.) в ТГФ (0,25 М раствор) охлаждали до 0°С и обрабатывали с помощью NaH (60% в масле, 2,0 экв.). Полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. еще в течение 30 мин. Суспензию снова охлаждали до 0°С и затем MeI (8,0 экв.) добавляли одной порцией. Реакционную смесь перемешивали при 0°С в течение 30 мин, при к.т. в течение 30 мин, и затем нагревали при кипении с обратным холодильником в течение 4 час до тех пор, пока не был израсходован исходный продукт, определяемый с помощью ТСХ. Охлажденную реакционную смесь гасили капельным добавлением насыщенного водного NH4Cl и выливали в разделительную воронку, содержащую насыщенный водный NH4Cl, и экстрагировали с помощью EtOАс (3 раза). Объединенные органические слои промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Очистка с помощью FC давала метиловый эфир.
Общая методика для удаления защиты с циклопропилбензаминов, защищенных Вос-группой:
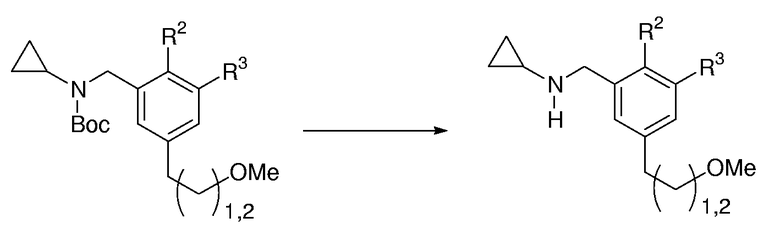
К раствору циклопропилбензамина (1,0 экв.), защищенного Вос-группой, в CH2Cl2 (0,1-0,5 М раствор) добавляли 4 М HCl в диоксане (5,0 экв.). Реакционную смесь перемешивали при к.т. в течение 8-16 час до тех пор, пока ТСХ не показывала полное превращение исходного продукта. Реакционную смесь выливали в разделительную воронку, содержащую 1 М водный NaOH, и экстрагировали с помощью СH2Cl2 (3 раза). Очистка с помощью FC давала соответствующий свободный амин.
(5-Бром-2-хлорбензилокси)трет-бутилдиметилсилан
TBDMS-Cl (10,6 г, 66,7 ммоль) добавляли к раствору (5-бром-2-хлорфенил)метанола (12,8 г, 55,6 ммоль) и имидазола (9,42 г, 138 ммоль) в ДМФА (190 мл) при 0°С. Смесь перемешивали в течение 2 час при 0°С, и к ней добавляли водный насыщенный NH4Cl. Смесь экстрагировали гептаном (2×). Объединенные органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (гептан → EtOAc/гептан 1:49) давала указанное в заголовке соединение (18,0 г, 96%). ЖХ-МС: tR = 1,22 мин.
3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорбензальдегид
BuLi (1,6 M в гексане, 46,6 мл, 74,6 ммоль) добавляют к раствору (5-бром-2-хлорбензилокси)трет-бутилдиметилсилана (16,7 г, 49,7 ммоль) в ТГФ (500 мл). Смесь перемешивали в течение 30 мин при -78°С, и к ней добавляли ДМФА (19,2 мл, 249 ммоль) с такой скоростью, чтобы температура не поднималась выше -70°С. Смесь перемешивали в течение 30 мин при -78°С, и ей позволяли нагреваться до к.т. Смесь выливали в водный насыщенный NH4Cl. Полученную смесь экстрагировали несколько раз с помощью EtOАс. Объединенные органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (EtOAc/гептан 1:4) давала указанное в заголовке соединение (11,2 г, 79%). ЖХ-МС: tR = 1,15 мин.
трет -Бутиловый эфир [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]-(2,2-дифторэтил)карбаминовой кислоты
2,2-Дифторэтиламин (660 мг, 7,90 ммоль) добавляли к раствору 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензальдегида (1,50 г, 5,27 ммоль) в МеОН (53 мл). Смесь нагревали при кипении с обратным холодильником в течение 4 час, и позволяли ей охлаждаться до температуры ниже к.т. К смеси порциями осторожно добавляли NaBH4 (300 мг, 7,90 ммоль), и смесь перемешивали в течение 1 час. Растворители удаляли при пониженном давлении, и оставшееся масло разбавляли с помощью EtOAc. Смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении, получая [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]-(2,2-дифторэтил)амин (1,86 г) в виде светлого сырого масла, которое затем использовали непосредственно. Сырой продукт растворяли в СН2Сl2 (53 мл), и к раствору добавляли DIPEA (2,7 мл, 15,6 ммоль), затем Вос2О (1,70 г, 7,90 ммоль). Смесь перемешивали при к.т. в течение 1 час. Смесь разбавляли с помощью СН2Сl2 (50 мл) и промывали водной 1 М HCl, водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 5:95) давала указанное в заголовке соединение (2,27 г, 96%). ЖХ-МС: tR = 1,22 мин.
трет -Бутиловый эфир (4-хлор-3-гидроксиметилбензил)-(2,2-дифторэтил)карбаминовой кислоты
TBAF (1 M в ТГФ, 9,57 мл, 9,57 ммоль) добавляли по каплям к раствору трет-бутилового эфира [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]-(2,2-дифторэтил)карбаминовой кислоты (2,15 г, 4,78 ммоль) в ТГФ (48 мл) при 0°С. Смесь перемешивали в течение 1 час, в то время как она нагревалась до к.т. Смесь разбавляли с помощью EtOAc и полученную смесь промывали водным насыщенным NH4Cl (×2) и насыщенным солевым раствором (1×). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 30:70) давала указанное в заголовке соединение (591 мг, 37%). ЖХ-МС: tR = 0,97 мин; ES+: 336,09.
трет -Бутиловый эфир (4-хлор-3-формилбензил)-(2,2-дифторэтил)карбаминовой кислоты
MnO2 (830 мг, 8,59 ммоль) добавляли к раствору трет-бутилового эфира (4-хлор-3-гидроксиметилбензил)-(2,2-дифторэтил)карбаминовой кислоты (577 мг, 1,72 ммоль) в СН3СN (35 мл). Смесь перемешивали при к.т. в течение 4,5 час, и к ней снова добавляли MnO2 (830 мг, 8,59 ммоль). Смесь перемешивали в течение 1 час. Смесь фильтровали через целит, и осадок промывали с помощью CH3CN и CH2Cl2. Фильтрат упаривали при пониженном давлении, получая сырое указанное в заголовке соединение (563 мг, 98%), которое затем использовали без очистки. ЖХ-МС: tR = 1,03 мин.
трет -Бутиловый эфир (4-хлор-3-циклопропиламинометилбензил)-(2,2-дифторэтил)карбаминовой кислоты
Смесь трет-бутилового эфира (4-хлор-3-формилбензил)-(2,2-дифторэтил)карбаминовой кислоты (563 мг, 1,69 ммоль) и циклопропиламина (0,180 мл, 2,52 ммоль) в МеОН (18 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т. Добавляли порциями NaBH4 (96 мг, 2,53 ммоль), и реакционную смесь перемешивали в течение 1 час. Растворители удаляли при пониженном давлении, и полученное масло разбавляли с помощью EtOAc.
Смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (CH2Cl2/MeOH 95:5) давала указанное в заголовке соединение (558 мг, 88%). ЖХ-МС: tR= 0,76 мин; ES+: 375,17.
Оксим 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензальдегида
3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорбензальдегид (12,7 г, 44,6 ммоль) растворяли в CH3CN (53 мл). К данному раствору добавляли NaHCO3 (11,2 г, 134 ммоль), и смесь перемешивали интенсивно в течение 5 мин. Добавляли воду (96 мл) и смесь перемешивали в течение 10 мин. NH2OH.HCl (6,20 г, 89,2 ммоль) добавляли по каплям, затем добавляли TBAC (622 мг, 2,24 ммоль). Смесь перемешивали при к.т. в течение 1 час, и к ней добавляли по каплям АсОН (4,00 мл) до рН 6-7. Смесь разбавляли водой (100 мл), и данную смесь экстрагировали с помощью Et2O (×3). Объединенные органические экстракты сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Сушка остатка в высоком вакууме давала неочищенное указанное в заголовке соединение (15,1 г, 98%), которое затем использовали без очистки. ЖХ-МС: tR = 1,09 мин; ES+: 341,13.
3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорбензиламин
LiAlH4 (4,11 г, 108 ммоль) добавляли порциями к раствору оксима 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензальдегида (13,0 г, 43,4 ммоль) в Et2O (433 мл). Смесь перемешивали при к.т. в течение 1 час. Водный насыщенный раствор тартрата калия натрия (400 мл) осторожно добавляли к смеси. Смесь перемешивали в течение 3 час и экстрагировали с помощью Et2O (3×). Объединенные органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Сушка остатка в высоком вакууме давала неочищенное указанное в заголовке соединение (12,4 г, количественный выход), которое затем использовали без очистки. ЖХ-МС: tR = 0,84 мин; ES+: 327,37.
N-[3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорбензил]-3,3,3-трифторпропионамид
TBTU (3,37 г, 10,5 ммоль) добавляли к раствору 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензиламина (2,00 г, 7,00 ммоль), DIPEA (4,80 мл, 28,0 ммоль) и 3,3,3-трифторпропионовой кислоты (0,927 мл, 10,5 ммоль) в CH2Cl2 (70 мл). Смесь перемешивали при к.т. в течение 1 час. CH2Cl2 (30 мл) добавляли, и смесь промывали водным насыщенным NH4Cl (2×), водным 1 М NaOH (1×) и насыщенным солевым раствором (1×). Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 40:60) давала указанное в заголовке соединение (1,80 г, 65%). ЖХ-МС: tR = 1,10 мин; ES+: 396,15.
N-(4-хлор-3-гидроксиметилбензил)-3,3,3-трифторпропионамид
TBAF (1 M в ТГФ, 9,10 мл, 9,10 ммоль) добавляли к раствору N-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]-3,3,3-трифторпропионамида (1,80 г, 4,55 ммоль) в ТГФ (45 мл) при 0°С. Смесь перемешивали в течение 1 час, в то время как она нагревалась до к.т. EtOAc (100 мл) добавляли, и смесь промывали водным насыщенным NH4Cl (3×) и водой (4×). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 70:30) давала указанное в заголовке соединение (835 мг, 65%). ЖХ-МС: tR = 0,76 мин; ES+: 323,02.
N-(4-хлор-3-формилбензил)-3,3,3-трифторпропионамид
MnO2 (1,44 г, 14,9 ммоль) добавляли к раствору N-(4-хлор-3-гидроксиметилбензил)-3,3,3-трифторпропионамида (841 мг, 2,99 ммоль) в CH3CN (60 мл). Смесь перемешивали при к.т. в течение 3 час. Снова добавляли MnO2 (1,44 г, 14,9 ммоль), и смесь перемешивали в течение 90 мин. Смесь фильтровали через целит, и осадок промывали с помощью CH3CN и CH2Cl2. Растворители удаляли при пониженном давлении, и остаток сушили в высоком вакууме, получая сырое указанное в заголовке соединение (840 мг, количественный выход), которое использовали без очистки. ЖХ-МС: tR = 0,84 мин; ES+: 341,21.
N-(4-Xлор-3-циклопропиламинометилбензил)-3,3,3-трифторпропионамид
Смесь N-(4-хлор-3-формилбензил)-3,3,3-трифторпропионамида (840 мг, 3,00 ммоль) и циклопропиламина (0,320 мл, 4,51 ммоль) в МеОН (30 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т. Добавляли порциями NaBH4 (170 мг, 4,51 ммоль). Смесь перемешивали в течение 1 час, и растворители удаляли при пониженном давлении. Остаток разбавляли с помощью EtOAc, и полученную смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (CH2Cl2/MeOH 90:10) давала указанное в заголовке соединение (929 мг, 96%). ЖХ-МС: tR = 0,64 мин; ES+: 321,05.
трет -Бутиловый эфир [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]циклопропилметилкарбаминовой кислоты
Смесь 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензиламина (2,00 г, 7,00 ммоль) и циклопропанкарбоксальдегида (0,784 мл, 10,5 ммоль) в МеОН (70 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т. К смеси порцией добавляли NaBH4 (397 мг, 10,5 ммоль), и смесь перемешивали в течение 1 час при к.т. Растворители удаляли при пониженном давлении, и полученное масло разбавляли с помощью EtOAc. Полученную смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении, получая сырой продукт [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]циклопропилметиламин. Данный сырой продукт растворяли в СН2Сl2 (70 мл). К раствору добавляли DIPEA (3,60 мл, 21,0 ммоль) и Вос2О (2,30 г, 10,5 ммоль). Смесь перемешивали при к.т. в течение 2 час, и смесь разбавляли с помощью СН2Сl2 (30 мл). Полученную смесь промывали водной 1 М HCl, водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (гептан → EtOAc/гептан 10:90) давала указанное в заголовке соединение с примесью Вос2О (3,60 г, количественный выход). ЖХ-МС: tR = 1,25 мин; ES+: 440,80.
трет -Бутиловый эфир (4-хлор-3-гидроксиметилбензил)циклопропилметилкарбаминовой кислоты
TBAF (1 M в ТГФ, 14,0 мл, 14,0 ммоль) добавляли к раствору трет-бутилового эфира [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]циклопропилметилкарбаминовой кислоты (3,08 г, 7,00 ммоль) в ТГФ (68 мл) при 0°С. Смесь перемешивали в течение 1 час, в то время как она нагревалась до к.т. добавляли EtOAc, и смесь промывали водным насыщенным NH4Cl и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2 → CH2Cl2/MeOH 9:1) давала указанное в заголовке соединение (1,00 г, 44%). ЖХ-МС: tR = 0,99 мин; ES+: 326,10.
трет -Бутиловый эфир (4-хлор-3-формилбензил)циклопропилметилкарбаминовой кислоты
MnO2 (1,48 г, 15,3 ммоль) добавляли к раствору трет-бутилового эфира (4-хлор-3-гидроксиметилбензил)циклопропилметилкарбаминовой кислоты (1,00 г, 3,07 ммоль) в СН3СN (61 мл). Смесь перемешивали при к.т. в течение 4 час, и к ней снова добавляли MnO2 (1,48 г, 15,3 ммоль). Смесь перемешивали в течение 1 час и фильтровали через целит. Осадок промывали с помощью CH3CN и CH2Cl2. Фильтрат упаривали при пониженном давлении. Сушка остатка в высоком вакууме давала сырое указанное в заголовке соединение (1,00 г, количественный выход), которое затем использовали без очистки. ЖХ-МС: tR = 1,06 мин.
трет -Бутиловый эфир (4-хлор-3-циклопропиламинометилбензил)циклопропилметилкарбаминовой кислоты
Смесь трет-бутилового эфира (4-хлор-3-формилбензил)циклопропилметилкарбаминовой кислоты (1,00 г, 3,09 ммоль), Et3N (0,646 мл, 4,64 ммоль) и циклопропиламина (0,325 мл, 4,64 ммоль) в МеОН (10,5 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли NaBH4 (292 мг, 7,71 ммоль). Смесь перемешивали в течение 1 час, и к ней добавляли водный насыщенный NaHCO3. Cмесь экстрагировали с помощью EtOAc (2×). Объединенные органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (МеОН/CH2Cl2 1:49) давала указанное в заголовке соединение (535 мг, 48%). ЖХ-МС: tR = 0,81 мин; ES+: 406,20.
N-[3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорбензил]ацетамид
AcCl (0,547 мл, 7,70 ммоль) добавляли к раствору 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензиламина (2,00 г, 7,00 ммоль) и DIPEA (4,80 мл, 28,0 ммоль) в CH2Cl2 (70 мл) при 0°С. Смесь перемешивали в течение 1 час, в то время как она нагревалась до к.т. CH2Cl2 (30 мл) добавляли, и смесь промывали водным насыщенным NH4Cl (2×), водным 1 М NaOH (1×) и насыщенным солевым раствором (1×). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 60:40) давала указанное в заголовке соединение (2,10 г, 91%). ЖХ-МС: tR = 1,07 мин; ES+: 369,19.
N-(4-Хлор-3-гидроксиметилбензил)ацетамид
TBAF (1 M в ТГФ, 12,0 мл, 12,0 ммоль) добавляли к раствору N-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]ацетамида (2,05 г, 6,00 ммоль) в ТГФ (60 мл) при 0°С. Смесь перемешивали в течение 2 час, в то время как она нагревалась до к.т. EtOAc (100 мл) добавляли, и смесь промывали водным насыщенным NH4Cl (1×) и водой (4×1). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/MeOH 90:10) давала указанное в заголовке соединение (750 мг, 59%). ЖХ-МС: tR = 0,63 мин; ES+: 237,09.
N-(4-Хлор-3-формилбензил)ацетамид
NMO (1,15 г, 8,26 ммоль) добавляли к раствору N-(4-хлор-3-гидроксиметилбензил)ацетамида (588 мг, 2,75 ммоль) в CH3CN (27 мл). Смесь перемешивали при к.т. в течение 30 мин, и к ней добавляли перрутенат тетрапропиламмония (97 мг, 0,28 ммоль). Смесь перемешивали в течение 1 час при к.т. и фильтровали через целит. Осадок промывали с помощью CH3CN. Фильтрат упаривали при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/MeOH 95:5) давала указанное в заголовке соединение (382 мг, 66%). ЖХ-МС: tR = 0,71 мин; ES+: 253,07.
N-(4-Xлор-3-циклопропиламинометилбензил)ацетамид
Смесь N-(4-хлор-3-формилбензил)ацетамида (382 мг, 1,81 ммоль) и циклопропиламина (0,194 мл, 2,71 ммоль) в МеОН (18 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли порциями NaBH4 (102 мг, 2,71 ммоль). Смесь перемешивали в течение 1 час, и растворители удаляли при пониженном давлении. Добавляли EtOAc (50 мл), и полученную смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (CH2Cl2/MeOH 95:5) давала указанное в заголовке соединение (371 мг, 81%). ЖХ-МС: tR = 0,53 мин; ES+: 253,11.
трет -Бутил-[2-хлор-5-(2-нитровинил)бензилокси]диметилсилан
Смесь 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензальдегида (14,0 г, 49,1 ммоль) и ацетата аммония (3,79 г, 49,1 ммоль) в нитрометане (8,19 мл, 152 ммоль) и АсОН (39 мл) нагревали при кипении с обратным холодильником в течении 3 час. Смеси позволяли охлаждаться до к.т., и смесь выливали в воду. Полученную смесь экстрагировали несколько раз с помощью EtOAc. Объединенные органические экстракты промывали водой и водным насыщенным NaHCO3 несколько раз. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Остаток сушили в высоком вакууме в течение ночи и растворяли в ДМФА (217 мл). Раствор охлаждали до 0°С, и к нему добавляли имидазол (8,36 г, 123 ммоль) и TBDMS-Cl (8,84 г, 58,6 ммоль). Смесь перемешивали в течение 2 час при 0°С и выливали в водный насыщенный NH4Cl. Полученную смесь экстрагировали несколько раз с помощью EtOAc. Объединенные органические экстракты промывали водой и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 2:8) давала указанное в заголовке соединение (9,50 г, 59%). ЖХ-МС: tR = 1,18 мин.
2-[3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорфенил]этиламин
LiAlH4 (1,09 г, 28,7 ммоль) добавляли к раствору трет-бутил-[2-хлор-5-(2-нитровинил)бензилокси]диметилсилана (3,95 г, 11,5 ммоль) в Et2O (115 мл). Смесь перемешивали при к.т. в течение 1 час, и к ней добавляли водный насыщенный раствор тартрата калия натрия. Смесь перемешивали в течение 1 час, и слои разделяли. Водный слой экстрагировали несколько раз с помощью Et2O. Объединенные органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Сушка остатка в высоком вакууме давала неочищенное указанное в заголовке соединение (3,20 г, 93%), которое затем использовали без очистки. ЖХ-МС: tR = 0,90 мин; ES+: 341,18.
трет -Бутиловый эфир {2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}метилкарбаминовой кислоты
К2СО3 (728 мг, 5,27 ммоль) и метилхлорформиат (0,405 мл, 5,27 ммоль) добавляли к раствору 2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этиламина (1,05 г, 3,51 ммоль) в СН2Сl2 (36 мл). Смесь перемешивали при к.т. в течение 1 час и снова добавляли метилхлорформиат (0,304 мл, 3,95 ммоль). Смесь перемешивали при к.т. в течение 1 час и снова добавляли метилхлорформиат (0,304 мл, 3,95 ммоль). Смесь перемешивали при к.т. в течение 1 час и добавляли CH2Cl2 (30 мл). Полученную смесь промывали водным насыщенным NH4Cl (2×), водным 10% К2СО3 (1×) и насыщенным солевым раствором (1×). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении, получая сырой метиловый эфир {2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}карбаминовой кислоты. Сырой продукт растворяли в ТГФ (36 мл). Раствор охлаждали до 0°С, и к нему осторожно порциями добавляли LiAlH4 (400 мг, 10,5 ммоль). Смеси позволяли нагреваться до к.т., и перемешивали ее при к.т. в течение ночи. Смесь выливали в водный насыщенный тартрат калия натрия (50 мл), и смесь перемешивали при к.т. в течение 2 час. Смесь экстрагировали с помощью Et2O (3×). Объединенные органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении, получая сырой {2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}метиламин. Данный сырой продукт растворяли в СН2Сl2 (36 мл). К раствору добавляли DIPEA (1,80 мл, 10,5 ммоль), затем Вос2О (2,30 г, 10,5 ммоль). Смесь перемешивали при к.т. в течение 1 час, и к ней добавляли СН2Сl2 (50 мл). Смесь промывали водной 1 М HCl, насыщенным солевым раствором, водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 4:1) давала указанное в заголовке соединение (1,00 г, 91%). ЖХ-МС: tR = 0,84 мин; ES+: 355,23.
трет -Бутиловый эфир [2-(4-хлор-3-гидроксиметилфенил)этил]метилкарбаминовой кислоты
TBAF (1 M в ТГФ, 2,90 мл, 2,90 ммоль) добавляли к раствору трет-бутилового эфира {2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}метилкарбаминовой кислоты (600 мг, 1,45 ммоль) в ТГФ (14,2 мл) при 0°С. Смесь перемешивали в течение 1 час, в то время как она нагревалась до к.т. EtOAc добавляли, и смесь промывали водным насыщенным NH4Cl и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/MeOH 19:1) давала указанное в заголовке соединение (435 мг, количественный выход). ЖХ-МС: tR = 0,93 мин.
трет -Бутиловый эфир [2-(4-хлор-3-формилфенил)этил]метилкарбаминовой кислоты
NMO (607 мг, 4,35 ммоль) добавляли к раствору трет-бутилового эфира [2-(4-хлор-3-гидроксиметилфенил)этил]метилкарбаминовой кислоты (435 мг, 1,45 ммоль) в CH2Cl2 (30 мл). Смесь перемешивали в течение 30 мин, и к ней добавляли перрутенат тетрапропиламмония (51,0 мг, 0,145 ммоль). Смесь перемешивали в течение 1 час и фильтровали через целит. Фильтрат упаривали при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/MeOH 19:1) давала указанное в заголовке соединение (330 мг, 76%). ЖХ-МС: tR = 1,00 мин.
трет -Бутиловый эфир [2-(4-хлор-3-циклопропиламинометилфенил)этил]метилкарбаминовой кислоты
Смесь трет-бутилового эфира [2-(4-хлор-3-формилфенил)этил]метилкарбаминовой кислоты (325 мг, 1,09 ммоль), Et3N (0,228 мл, 1,64 ммоль) и циклопропиламина (0,115 мл, 1,64 ммоль) в МеОН (3,66 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли NaBH4 (103 мг, 2,73 ммоль). Смесь перемешивали в течение 1 час при к.т., и к ней добавляли водный насыщенный NaHCO3. Смесь экстрагировали с помощью EtOAc несколько раз. Объединенные органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC давала указанное в заголовке соединение (186 мг, 50%). ЖХ-МС: tR = 0,79 мин; ES+: 339,39.
трет -Бутиловый эфир [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]метилкарбаминовой кислоты
К2СО3 (363 мг, 2,63 ммоль) и метилхлорформиат (0,202 мл, 2,63 ммоль) добавляли к раствору 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензиламина (500 мг, 1,76 ммоль) в СН2Сl2 (18 мл). Смесь перемешивали при к.т. в течение 1 час, и к ней снова добавляли метилхлорформиат (0,151 мл, 1,97 ммоль). Смесь перемешивали при к.т. в течение 1 час, и к ней добавляли СН2Сl2 (30 мл). Полученную смесь промывали водным насыщенным NH4Cl (2×), водным 10% К2СО3 (1×) и насыщенным солевым раствором (1×). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении, получая сырой метиловый эфир [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]карбаминовой кислоты. Данный сырой продукт растворяли в ТГФ (18 мл). Раствор охлаждали до 0°С, и к нему осторожно порциями добавляли LiAlH4 (200 мг, 5,27 ммоль). Смеси позволяли нагреваться до к.т., и перемешивали ее при к.т. в течение ночи. Смесь выливали в водный насыщенный тартрат калия натрия (50 мл), и смесь перемешивали при к.т. в течение 2 час. Смесь экстрагировали с помощью Et2O (3×). Объединенные органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении, получая сырой [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]метиламин. Данный сырой продукт растворяли в СН2Сl2 (18 мл). К раствору добавляли DIPEA (0,900 мл, 5,26 ммоль), затем Вос2О (1,15 г, 5,27 ммоль). Смесь перемешивали при к.т. в течение ночи, и к ней добавляли СН2Сl2 (50 мл). Смесь промывали водной 1 М HCl, водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 50:50) давала смесь (0,900 г) указанного в заголовке соединение, Boc2O и трет-бутиловый эфир [3-(трет-бутилдиметилсиланилоксиметил)бензил]метилкарбаминовой кислоты. Данную сырую смесь, в которой на указанное в заголовке соединение приходилось 439 мг (83%), затем использовали без очистки. ЖХ-МС: tR = 1,10 мин; ES+: 344,13.
трет -Бутиловый эфир (4-хлор-3-гидроксиметилбензил)метилкарбаминовой кислоты
TBAF (1 M в ТГФ, 2,25 мл, 2,25 ммоль) добавляли к раствору трет-бутилового эфира [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]метилкарбаминовой кислоты (450 мг, 1,13 ммоль) в ТГФ (11 мл) при 0°С. Смесь перемешивали в течение 1 час, в то время как она нагревалась до к.т. EtOAc (50 мл) добавляли, и полученную смесь промывали водным насыщенным NH4Cl (2×) и насыщенным солевым раствором (1×). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 40:60) давала указанное в заголовке соединение (110 мг, 34%). ЖХ-МС: tR = 0,91 мин; ES+: 271,10.
трет -Бутиловый эфир (4-хлор-3-формилбензил)метилкарбаминовой кислоты
MnO2 (372 мг, 3,85 ммоль) добавляли к раствору трет-бутилового эфира (4-хлор-3-гидроксиметилбензил)метилкарбаминовой кислоты (110 мг, 0,385 ммоль) в СН3СN (7,70 мл). Смесь перемешивали при к.т. в течение 4,5 час, и к ней снова добавляли MnO2 (186 мг, 1,93 ммоль). Смесь перемешивали в течение 1 час, и смесь фильтровали через целит. Осадок промывали с помощью CH3CN и CH2Cl2. Фильтрат упаривали при пониженном давлении. Сушка остатка в высоком вакууме давала сырое указанное в заголовке соединение (95 мг, 87%), которое затем использовали без очистки. ЖХ-МС: tR = 0,99 мин.
трет -Бутиловый эфир (4-хлор-3-циклопропиламинометилбензил)метилкарбаминовой кислоты
Смесь трет-бутилового эфира (4-хлор-3-формилбензил)метилкарбаминовой кислоты (95 мг, 0,335 ммоль) и циклопропиламина (0,036 мл, 0,50 ммоль) в МеОН (3,30 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли порциями NaBH4 (19 мг, 0,50 ммоль). Смесь перемешивали в течение 1 час, и растворители удаляли при пониженном давлении. Полученное масло разбавляли EtOAc (20 мл), и данную смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (СН2Сl2/МеОН 90:10) давала указанное в заголовке соединение (81 мг, 75%). ЖХ-МС: tR = 0,73 мин; ES+: 366,20.
N-{2-[3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}-3,3,3-трифторпропионамид
TBTU (2,09 г, 6,50 ммоль) добавляли к раствору 2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этиламина (1,30 г, 4,33 ммоль), 3,3,3-трифторпропионовой кислоты (0,574 мл, 6,5 ммоль) и DIPEA (2,97 мл, 17,3 ммоль) в CH2Cl2 (43 мл). Смесь перемешивали в течение 1 час, и к ней добавляли CH2Cl2. Смесь промывали водным насыщенным NH4Cl, водным 10% Na2CO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 60:40) давала указанное в заголовке соединение (500 мг, 28%). ЖХ-МС: tR = 1,12 мин; ES+: 410,14.
N-[2-(4-Хлор-3-гидроксиметилфенил)этил]-3,3,3-трифторпропионамид
TBAF (1 M в ТГФ, 2,46 мл, 2,46 ммоль) добавляли к раствору N-{2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}-3,3,3-трифторпропионамида (500 мг, 1,22 ммоль) в ТГФ (15,8 мл) при 0°С. Смесь перемешивали в течение 1 час при 0°С, и добавляли к ней водный насыщенный NH4Cl. Смесь экстрагировали с помощью EtOAc (3×). Объединенные органические экстракты промывали водой (4Ч), сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (CН2Cl2/MeOH 19:1) давала указанное в заголовке соединение (230 мг, 64%). ЖХ-МС: tR = 0,79 мин.
N-[2-(4-Хлор-3-формилфенил)этил]-3,3,3-трифторпропионамид
NMO (318 мг, 2,28 ммоль) добавляли к раствору N-[2-(4-Хлор-3-гидроксиметилфенил)этил]-3,3,3-трифторпропионамида (225 мг, 0,761 ммоль) в CH2Cl2 (16 мл). Смесь перемешивали в течение 30 мин, и к ней добавляли перрутенат тетрапропиламмония (26,7 мг, 0,076 ммоль). Смесь перемешивали в течение 1 час при к.т. и фильтровали через целит. Фильтрат упаривали при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/MeOH 19:1) давала указанное в заголовке соединение (180 мг, 81%). ЖХ-МС: tR= 0,88 мин; ES+: 335,28.
N-[2-(4-Xлор-3-циклопропиламинометилфенил)этил]-3,3,3-трифторпропионамид
Смесь N-[2-(4-хлор-3-формилфенил)этил]-3,3,3-трифторпропионамида (175 мг, 0,596 ммоль), Et3N (0,125 мл, 0,896 ммоль) и циклопропиламина (0,063 мл, 0,90 ммоль) в МеОН (2,00 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли порциями NaBH4 (56,4 мг, 1,49 ммоль). Смесь перемешивали в течение 1 час и добавляли водный насыщенный NaНCO3. Смесь экстрагировали с помощью EtOAc несколько раз, и объединенные органические экстракты промывали насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (CH2Cl2/MeOH 95:5) давала указанное в заголовке соединение (130 мг, 65%). ЖХ-МС: tR = 0,65 мин; ES+: 335,12.
{2-[3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}этиламин
Смесь 2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этиламина (1,05 г, 3,50 ммоль) и ацетальдегида (1,19 мл, 21,0 ммоль) в МеОН (35 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней порциями добавляли NaBH4 (198 мг, 5,25 ммоль). Смесь перемешивали при к.т. в течение 1 час, и растворители удаляли при пониженном давлении. Полученное масло разбавляли с помощью EtOAc, и данную смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/МеОН 19:1 → 9:1) давала указанное в заголовке соединение (320 мг, 28%). ЖХ-МС: tR = 0,90 мин; ES+: 369,22.
трет -Бутиловый эфир {2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}этилкарбаминовой кислоты
Смесь {2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}этиламина (320 мг, 0,976 ммоль), DIPEA (0,501 мл, 2,93 ммоль) и Вос2О (320 мг, 1,47 ммоль) в СН2Сl2 (10 мл) перемешивали при к.т. в течение 2 час. Добавляли СН2Сl2 (30 мл), и смесь промывали водной 1 М НСl, водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 20:80) давала указанное в заголовке соединение (298 мг, 71%). ЖХ-МС: tR = 1,25 мин; ES+: 428,19.
трет -Бутиловый эфир [2-(4-хлор-3-гидроксиметилфенил)этил]этилкарбаминовой кислоты
TBAF (1 M в ТГФ, 1,39 мл, 1,39 ммоль) добавляли к раствору трет-бутилового эфира {2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}этилкарбаминовой кислоты (298 мг, 0,696 ммоль) в ТГФ (6,82 мл) при 0°С. Смесь перемешивали в течение 1 час, в то время как она нагревалась до к.т. EtOAc добавляли, и смесь промывали водным насыщенным NH4Cl и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/MeOH 19:1) давала указанное в заголовке соединение (110 мг, 50%). ЖХ-МС: tR = 0,97 мин; ES+: 314,11.
трет -Бутиловый эфир [2-(4-хлор-3-формилфенил)этил]этилкарбаминовой кислоты
NMO (133 мг, 0,956 ммоль) добавляли к раствору трет-бутилового эфира [2-(4-хлор-3-гидроксиметилфенил)этил]этилкарбаминовой кислоты (110 мг, 0,319 ммоль) в CH2Cl2 (7,0 мл). Смесь перемешивали в течение 30 мин, и к ней добавляли перрутенат тетрапропиламмония (11,2 мг, 0,032 ммоль). Смесь перемешивали в течение 1 час и фильтровали через целит. Фильтрат упаривали при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/MeOH 49:1) давала указанное в заголовке соединение (80 мг, 80%). ЖХ-МС: tR = 1,07 мин.
трет -Бутиловый эфир [2-(4-хлор-3-циклопропиламинометилфенил)этил]этилкарбаминовой кислоты
Смесь трет-бутилового эфира [2-(4-хлор-3-формилфенил)этил]этилкарбаминовой кислоты (80,1 мг, 0,257 ммоль), Et3N (0,054 мл, 0,386 ммоль) и циклопропиламина (0,027 мл, 0,386 ммоль) в МеОН (0,86 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли порциями NaBH4 (24,2 мг, 0,642 ммоль). Смесь перемешивали в течение 1 час, и к ней добавляли водный насыщенный NaHCO3. Смесь экстрагировали с помощью EtOAc несколько раз, и объединенные органические экстракты промывали насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (СН2Сl2/MeOH 95:5) давала указанное в заголовке соединение (40 мг, 44%). ЖХ-МС: tR = 0,81 мин; ES+: 353,22.
N-{2-[3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}ацетамид
AcCl (0,063 мл, 0,88 ммоль) добавляли к раствору 2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этиламина (252 мг, 0,840 ммоль) и DIPEA (0,575 мл, 3,36 ммоль) в CH2Cl2 (8,4 мл). Смесь перемешивали при к.т. в течение 30 мин, и к ней добавляли водный насыщенный NH4Cl. Слои разделяли, и органический слой промывали водным 1 М NaOH, сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (CH2Cl2/MeOH 19:1) давала указанное в заголовке соединение (190 г, 66%). ЖХ-МС: tR = 1,09 мин; ES+: 342,19.
N-[2-(4-Хлор-3-гидроксиметилфенил)этил]ацетамид
TBAF (1 M в ТГФ, 1,12 мл, 1,12 ммоль) добавляли к раствору N-{2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}ацетамида (190 мг, 0,555 ммоль) в ТГФ (7,10 мл) при 0°С. Смесь перемешивали в течение 1 час, в то время как она нагревалась до к.т. Водный насыщенный NH4Cl добавляли, и смесь экстрагировали с помощью EtOAc (3×). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/MeOH 19:1) давала указанное в заголовке соединение (100 мг, 79%). ЖХ-МС: tR= 0,67 мин; ES+: 284,11.
N-[2-(4-Xлор-3-формилфенил)этил]ацетамид
NMO (184 мг, 1,32 ммоль) добавляли к раствору N-[2-(4-хлор-3-гидроксиметилфенил)этил]ацетамида (100 мг, 0,439 ммоль) в CH2Cl2 (9,22 мл). Смесь перемешивали в течение 30 мин, и к ней добавляли перрутенат тетрапропиламмония (15,5 мг, 0,044 ммоль). Смесь перемешивали в течение 1 час при к.т. и фильтровали через целит. Фильтрат упаривали при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/MeOH 49:1) давала указанное в заголовке соединение (50 мг, 50%). ЖХ-МС: tR = 0,75 мин; ES+: 267,10.
N-[2-(4-Xлор-3-циклопропиламинометилфенил)этил]ацетамид
Смесь N-[2-(4-хлор-3-формилфенил)этил]ацетамида (50,1 мг, 0,222 ммоль), Et3N (0,046 мл, 0,332 ммоль) и циклопропиламина (0,023 мл, 0,332 ммоль) в МеОН (0,50 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли порциями NaBH4 (21,0 мг, 0,554 ммоль). Смесь перемешивали в течение 1 час и добавляли водный насыщенный NaHCO3. Cмесь экстрагировали с помощью EtOAc несколько раз, и объединенные органические экстракты промывали насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (CH2Cl2/MeOH 95:5) давала указанное в заголовке соединение (35 мг, 59%). ЖХ-МС: tR = 0,59 мин; ES+: 267,17.
трет -Бутиловый эфир [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]-(2-фторэтил)карбаминовой кислоты
Cмесь 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензальдегида (1,50 г, 5,27 ммоль), DIPEA (1,80 мл, 10,5 ммоль) и гидрохлорида 2-фторэтиламина (873 мг, 7,90 ммоль) в МеОН (53 мл) нагревали при кипении с обратным холодильником в течение 4 час. Реакционной смеси позволяли охлаждаться до к.т., и к ней осторожно порциями добавляли NaBH4 (300 мг, 7,91 ммоль). Смесь перемешивали в течение 1 час, и растворители удаляли при пониженном давлении. К остатку добавляли EtOAc (100 мл), и смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении, получая сырой [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]-(2-фторэтил)амин. Данный сырой продукт растворяли в СН2Сl2 (70 мл). К раствору добавляли DIPEA (2,70 мл, 15,8 ммоль), затем Вос2О (1,70 г, 7,91 ммоль). Смесь перемешивали при к.т. в течение 1 час. Смесь разбавляли с помощью СН2Сl2 (50 мл) и промывали водной 1 М HCl, водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 10:90) давала указанное в заголовке соединение (1,92 г, 85%). ЖХ-МС: tR = 1,22 мин; ES+: 417,17.
трет -Бутиловый эфир (4-хлор-3-гидроксиметилбензил)-(2-фторэтил)карбаминовой кислоты
TBAF (1 M в ТГФ, 8,84 мл, 8,84 ммоль) добавляли к раствору трет-бутилового эфира [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]-(2-фторэтил)карбаминовой кислоты (1,91 г, 4,42 ммоль) в ТГФ (44,2 мл) при 0°С. Смесь перемешивали в течение 1 час, в то время как она нагревалась до к.т. Добавляли EtOAc (100 мл), и полученную смесь промывали водным насыщенным NH4Cl (2×) и насыщенным солевым раствором (1×). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 30:70) давала указанное в заголовке соединение (901 мг, 64%). ЖХ-МС: tR = 0,64 мин; ES+: 318,07.
трет -Бутиловый эфир (4-хлор-3-формилбензил)-(2-фторэтил)карбаминовой кислоты
MnO2 (1,22 г, 12,6 ммоль) добавляли к раствору трет-бутилового эфира (4-хлор-3-гидроксиметилбензил)-(2-фторэтил)карбаминовой кислоты (801 мг, 2,52 ммоль) в СН3СN (50 мл). Смесь перемешивали при к.т. в течение 4,5 час, и к ней снова добавляли MnO2 (1,22 г, 12,6 ммоль). Смесь перемешивали в течение 1 час и фильтровали через целит. Осадок промывали с помощью CH3CN и CH2Cl2. Фильтрат упаривали при пониженном давлении. Сушка остатка в высоком вакууме давала сырое указанное в заголовке соединение (800 мг, количественный выход), которое затем использовали без очистки. ЖХ-МС: tR = 1,01 мин.
трет -Бутиловый эфир (4-хлор-3-циклопропиламинометилбензил)-(2-фторэтил)карбаминовой кислоты
Смесь трет-бутилового эфира (4-хлор-3-формилбензил)-(2-фторэтил)карбаминовой кислоты (850 мг, 2,69 ммоль) и циклопропиламина (0,290 мл, 4,05 ммоль) в МеОН (27 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли порциями NaBH4 (153 мг, 4,40 ммоль). Смесь перемешивали в течение 1 час. Растворители удаляли при пониженном давлении, и к остатку добавляли EtOAc. Полученную смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (CH2Cl2/MeOH 95:5) давала указанное в заголовке соединение (845 мг, 88%). ЖХ-МС: tR= 0,76 мин; ES+: 357,19.
трет -Бутиловый эфир [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]этилкарбаминовой кислоты
Ацетальдегид (2,4 мл, 42 ммоль) добавляли к раствору 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензиламина (2,00 г, 7,00 ммоль) в МеОН (70 мл). Смесь нагревали при кипении с обратным холодильником в течение 4 час, и ей позволяли охлаждаться до к.т. К смеси осторожно порциями добавляли NaBH4 (397 мг, 10,5 ммоль) в течение 1 час. Растворители удаляли при пониженном давлении. Остаток разбавляли с помощью EtOAc (100 мл), и полученную смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении, получая сырой [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]этиламин (3,07 г). Данный сырой продукт растворяли в СН2Сl2 (70 мл). К данному раствору добавляли DIPEA (3,60 мл, 21,0 ммоль), затем Вос2О (2,29 г, 10,5 ммоль). Смесь перемешивали при к.т. в течение 2 час. Смесь разбавляли с помощью СН2Сl2 (30 мл) и промывали водной 1 М HCl, водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 5:95) давала указанное в заголовке соединение (850 мг, 29%). ЖХ-МС: tR = 1,23 мин; ES+: 414,23.
трет -Бутиловый эфир (4-хлор-3-гидроксиметилбензил)этилкарбаминовой кислоты
TBAF (1 M в ТГФ, 3,03 мл, 3,03 ммоль) добавляли по каплям к раствору трет-бутилового эфира [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]этилкарбаминовой кислоты (628 мг, 1,52 ммоль) в ТГФ (14,9 мл) при 0°С. Смесь перемешивали в течение 1 час, в то время как она нагревалась до к.т. Добавляли EtOAc, и смесь промывали водным насыщенным NH4Cl и насыщенным солевым раствором). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (МеОН/CH2Cl2 19:1) давала указанное в заголовке соединение (300 мг, 66%). ЖХ-МС: tR= 0,98 мин; ES+: 300,28.
трет -Бутиловый эфир (4-хлор-3-формилбензил)этилкарбаминовой кислоты
MnO2 (475 мг, 4,92 ммоль) добавляли к раствору трет-бутилового эфира (4-хлор-3-гидроксиметилбензил)этилкарбаминовой кислоты (295 мг, 0,984 ммоль) в СН3СN (20 мл). Смесь перемешивали при к.т. в течение 5 час, и к ней снова добавляли MnO2 (475 мг, 0,984 ммоль). Смесь перемешивали в течение 1 час, и смесь фильтровали через целит и промывали с помощью CH3CN и CH2Cl2. Выпаривание растворителей при пониженном давлении давало сырое указанное в заголовке соединение (240 мг, 82%), которое затем использовали без очистки. ЖХ-МС: tR = 1,02 мин.
трет -Бутиловый эфир (4-хлор-3-циклопропиламинометилбензил)этилкарбаминовой кислоты
Смесь трет-бутилового эфира (4-хлор-3-формилбензил)этилкарбаминовой кислоты (240 мг, 0,806 ммоль), циклопропиламина (0,085 мл, 1,2 ммоль) и Et3N (0,169 мл, 1,21 ммоль) в МеОН (2,7 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли NaBH4 (76,2 мг, 2,01 ммоль). Смесь перемешивали в течение 1 час при к.т., и к ней добавляли водный насыщенный NaHCO3. Cмесь несколько раз экстрагировали с помощью EtOAc. Объединенные органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (МеОН/CH2Cl2 1:49) давала указанное в заголовке соединение (125 мг, 46%). ЖХ-МС: tR = 0,78 мин; ES+: 380,20.
трет -Бутиловый эфир [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]циклопропилкарбаминовой кислоты
Смесь 3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензальдегида (1,73 г, 6,06 ммоль) и циклопропиламина (0,64 мл, 9,1 ммоль) в МеОН (60 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней порциями добавляли NaBH4 (344 мг, 9,09 ммоль). Смесь перемешивали, и к ней добавляли воду (20 мл). Растворители частично удаляли при пониженном давлении. Полученную водную суспензию разбавляли водой (50 мл) и экстрагировали с помощью EtOAc. Органические экстракты промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении, получая сырой [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]циклопропиламин (1,54 г). Данный сырой продукт растворяли в СН2Сl2 (60 мл). К раствору добавляли DIPEA (3,1 мл, 18 ммоль), затем Вос2О (1,98 г, 9,09 ммоль). Смесь перемешивали при к.т. в течение 2 час. Добавляли СН2Сl2 (40 мл), и смесь промывали водной 1 М HCl, водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (гептан →EtOAc/гептан 5:95) давала указанное в заголовке соединение (1,54 г, 78%). ЖХ-МС: tR= 1,26 мин; ES+: 426,14.
трет -Бутиловый эфир (4-хлор-3-гидроксиметилбензил)циклопропилкарбаминовой кислоты
Водный 1 М NaOH (16 мл) добавляли к раствору трет-бутилового эфира [3-(трет-бутилдиметилсиланилоксиметил)-4-хлорбензил]циклопропилкарбаминовой кислоты (700 мг, 1,64 ммоль) в МеОН (32 мл). Смесь нагревали при кипении с обратным холодильником в течение 2 час, и позволяли ей охлаждаться до к.т. Растворители частично удаляли при пониженном давлении, и полученный водный слой разбавляли водой (100 мл). Смесь экстрагировали с помощью Et2O (3×). Объединенные органические слои сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtОАс/гептан 40:60) давала указанное в заголовке соединение (550 мг, количественный выход). ЖХ-МС: tR = 0,98 мин; ES+: 312,04.
трет -Бутиловый эфир (4-хлор-3-формилбензил)циклопропилкарбаминовой кислоты
MnO2 (815 мг, 8,44 ммоль) добавляли к раствору трет-бутилового эфира (4-хлор-3-гидроксиметилбензил)циклопропилкарбаминовой кислоты (526 мг, 1,69 ммоль) в СН3СN (34 мл). Смесь перемешивали при к.т. в течение 3 час, фильтровали через целит и промывали с помощью CH3CN и CH2Cl2. Выпаривание растворителей при пониженном давлении давало сырое указанное в заголовке соединение (563 мг, количественый выход), которое затем использовали без очистки. ЖХ-МС: tR = 1,05 мин; ES+: 310,04.
трет -Бутиловый эфир (4-хлор-3-циклопропиламинометилбензил)циклопропилкарбаминовой кислоты
Смесь трет-бутилового эфира (4-хлор-3-формилбензил)циклопропилкарбаминовой кислоты (563 мг, 1,82 ммоль) и циклопропиламина (0,195 мл, 2,73 ммоль) в МеОН (18 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли порциями NaBH4 (103 мг, 2,73 ммоль). Смесь перемешивали в течение 1 час, и растворители удаляли при пониженном давлении. Полученное масло разбавляли с помощью EtOAc (100 мл), и полученную смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (CH2Cl2/МеОН 95:5) давала указанное в заголовке соединение (343 мг, 54%). ЖХ-МС: tR = 0,78 мин; ES+: 351,39.
трет -Бутил-(2-хлор-5-винилбензилокси)диметилсилан
Pd(PPh3)4 (173 мг, 0,149 ммоль) добавляли к раствору (5-бром-2-хлорбензилокси)трет-бутилдиметилсилана (1,00 г, 2,98 ммоль) в диметоксиэтане (30 мл). Смесь перемешивали при к.т. в течение 20 мин, и к ней добавляли К2СО3 (411 мг, 2,98 ммоль), воду (10 мл) и 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолан (0,53 мл, 2,98 ммоль). Смесь быстро нагревали до кипения с обратным холодильником и перемешивали при кипении с обратным холодильником в течение 2 час. Смеси позволяли охлаждаться до к.т., и разбавляли ее с помощью Et2O (100 мл). Смесь промывали водой, и водный слой экстрагировали снова с помощью Et2O (3×). Объединенные органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtОАс/гептан 5:95) давала указанное в заголовке соединение (822 мг, 98%). ЖХ-МС: tR = 1,22 мин.
2-[3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорфенил]этанол
9-BBN (0,5 М в ТГФ, 34,0 мл, 17,0 ммоль) добавляли по каплям в течение 30 мин к раствору трет-бутил-(2-хлор-5-винилбензилокси)диметилсилана (800 мг, 2,83 ммоль) в ТГФ (28 мл) при 0°С. Смесь перемешивали в течение 30 мин при 0°С и в течение 4 час при к.т. Смесь снова охлаждали до 0°С, и к ней добавляли по каплям водный 1 М NaOH (39,0 мл) и Н2О2 (33%, 9,8 мл, 113 ммоль). Смесь перемешивали в течение 2 час, в то время как она нагревалась до к.т., и охлаждали до 0°С. Осторожно добавляли водный насыщенный Na2S2O3 (100 мл), и данной смеси позволяли осторожно нагреваться при к.т. в течение ночи. Растворители частично удаляли при пониженном давлении, и водный остаток экстрагировали с помощью EtOAc (3×). Объединенные органические экстракты промывали насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtОАс/гептан 40:60) давала указанное в заголовке соединение (677 мг, 80%). ЖХ-МС: tR = 1,10 мин; ES+: 301,08.
2-[3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорфенил]этиловый эфир метансульфоновой кислоты
К раствору 2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этанола (2,00 г, 6,65 ммоль) в СН2Сl2 (66 мл) при 0°С добавляли по каплям Et3N (1,02 мл, 7,31 ммоль) и метансульфонилхлорид (0,57 мл, 7,31 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 час и разбавляли с помощью СН2Сl2 (40 мл). Полученную смесь промывали водным насыщенным NH4Cl (2×). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении, получая сырое указанное в заголовке соединение (2,55 г, количественный выход), которое затем использовали без очистки. ЖХ-МС: tR = 1,13 мин; ES+: 379,29.
{2-[3-(трет-Бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}циклопропиламин
Циклопропиламин (1,14 мл, 16,3 ммоль) добавляли к раствору 2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этилового эфира метансульфоновой кислоты (1,76 г, 4,65 ммоль) в EtOH (46 мл). Смесь нагревали при кипении с обратным холодильником в течение 2 ч и снова добавляли циклопропиламин (0,57 мл, 8,2 ммоль). Смесь нагревали при кипении с обратным холодильником в продолжении ночи, и ей позволяли охлаждаться до к.т. Растворители удаляли при пониженном давлении, и остаток очищали с помощью FC (EtОАс/гептан 50:50 → 7 М NH3/MeOH), получая указанное в заголовке соединение (865 мг, 68%). ЖХ-МС: tR = 0,92 мин; ES+: 340,39.
трет -Бутиловый эфир {2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}циклопропилкарбаминовой кислоты
DIPEA (2,61 мл, 5,60 ммоль) и Вос2О (1,22 г, 5,60 ммоль) добавляли к раствору {2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}циклопропиламина (1,73 г, 5,09 ммоль) в CH2Cl2 (50 мл). Смесь перемешивали при к.т. в течение 4 час. Смесь разбавляли CH2Cl2 (50 мл), промывали водным насыщенны NaHCO3, водным насыщенным NH4Cl и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtОАс/гептан 10:90) давала указанное в заголовке соединение (2,14 г, 96%). ЖХ-МС: tR = 1,26 мин.
трет -Бутиловый эфир [2-(4-хлор-3-гидроксиметилфенил)этил]циклопропилкарбаминовой кислоты
Водный 1 М NaOH (48 мл) добавляли к суспензии трет-бутилового эфира {2-[3-(трет-бутилдиметилсиланилоксиметил)-4-хлорфенил]этил}циклопропилкарбаминовой кислоты (2,10 г, 4,77 ммоль) в МеОН (96 мл). Смесь нагревали при кипении с обратным холодильником в течение 90 мин. Смеси позволяли охлаждаться до к.т., и растворители частично удаляли при пониженном давлении. Полученную водную смесь разбавляли водой (100 мл) и экстрагировали с помощью Et2O (3×). Объединенные органические экстракты сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtОАс/гептан 40:60) давала указанное в заголовке соединение (1,34 г, 86%). ЖХ-МС: tR = 0,98 мин; ES+: 326,30.
трет -Бутиловый эфир [2-(4-хлор-3-формилфенил)этил]циклопропилкарбаминовой кислоты
MnO2 (1,97 г, 20,4 ммоль) добавляли к раствору трет-бутилового эфира [2-(4-хлор-3-гидроксиметилфенил)этил]циклопропилкарбаминовой кислоты (1,33 г, 4,08 ммоль) в СН3СN (41 мл). Смесь перемешивали при к.т. в течение ночи. Смесь фильтровали через целит и промывали с помощью CH3CN и CH2Cl2. Упаривание фильтрата при пониженном давлении давало сырое указанное в заголовке соединение (1,32 г, количественый выход), которое затем использовали без очистки. ЖХ-МС: tR = 1,05 мин.
трет -Бутиловый эфир [2-(4-хлор-3-циклопропиламинометилфенил)этил]циклопропилкарбаминовой кислоты
Смесь трет-бутилового эфира [2-(4-хлор-3-формилфенил)этил]циклопропилкарбаминовой кислоты (1,32 г, 4,08 ммоль) и циклопропиламина (0,438 мл, 6,1 ммоль) в МеОН (41 мл) нагревали при кипении с обратным холодильником в течение 4 час. Смеси позволяли охлаждаться до к.т., и к ней добавляли порциями NaBH4 (232 мг). Смесь перемешивали в течение 1 час, и растворители удаляли при пониженном давлении. Добавляли EtOAc (100 мл), и смесь промывали водным насыщенным NaHCO3 и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (CH2Cl2/МеОН 95:5) давала указанное в заголовке соединение (883 мг, 59%). ЖХ-МС: tR = 0,82 мин; ES+: 365,38.
3,9-Ди-трет-бутиловый эфир 6-этиловый эфир (рац.)-(1R∗, 5S∗)-7-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}-3,9-диазабицикло[3.3.1]нон-6-ен-3,6,9-трикарбоновой кислоты (А1)
Смесь соединения F3 (56,0 г, 106 ммоль), 2-хлор-3,6-дифторфенола (34,8 г, 211 ммоль), дипиперидида азадикарбоновой кислоты (53,4 г, 211 ммоль) и PВu3 (85%, 83 мл, 317 ммоль) в толуоле (1,20 л) нагревают при кипении с обратным холодильником в атмосфере азота в течение 1 час. Смеси позволяют охлаждаться до к.т. Смесь разбавляют с помощью EtOAc (2,00 л), и смесь промывают водным 1 М NaOH (2×900 мл). Органические экстракты сушат над MgSO4, фильтруют, и растворители удаляют при пониженном давлении. Очистка остатка с помощью FC (EtOAc/гептан 1:19 → 1:1) дает указанное в заголовке соединение (67,5 г, 94%). ЖХ-МС: tR = 1,24 мин; ES+: 617,25.
1'-трет-Бутиловый эфир 3'-метиловый эфир 6-[2-(2,6-дихлор-4-метилфенокси)этокси]-5',6'-дигидро-2'H-[3,4']бипиридинил-1',3'-дикарбоновой кислоты (А2)
BuLi (1,6 М в гексане, 39,3 мл, 45,2 ммоль) добавляли к раствору 5-бром-2-[2-(2,6-дихлор-4-метилфенокси)этокси]пиридина (13,0 г, 34,5 ммоль) в ТГФ (700 мл) при -78°С. Смесь перемешивали в течение 1 час при -78°С, и к ней добавляли ZnCl2 (0,72 М в ТГФ, 78,6 мл, 56,5 ммоль). Смеси позволяли нагреваться до к.т. К смеси добавляли 1-трет-бутиловый эфир 3-метиловый эфир 4-трифторметансульфонилокси-5,6-дигидро-2Н-пиридин-1,3-дикарбоновой кислоты (WO 2004/002957, 11,0 г, 28,3 ммоль) и Pd(PPh3)4 (813 мг, 0,704 ммоль). Смесь перемешивали в течение 30 мин при 70°С, затем в течение ночи при к.т. К смеси добавляли водный насыщенный NH4Cl, и смесь экстрагировали с помощью EtOAc (2×). Органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (гептан/EtOAc 9:1 → 5:5) давало указанное в заголовке соединение (10,7 г, 70%). ЖХ-МС: tR = 1,16 мин; ES+: 537,33.
3,9-Ди-трет-бутиловый эфир (рац.)-(1R∗, 5S∗)-7-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}-3,9-диазабицикло[3.3.1]нон-7-ен-3,6,9-трикарбоновой кислоты (В1)
В 250 мл круглодонной колбе, снабженной магнитной мешалкой, растворяют соединение А1 (4,72 г, 6,97 ммоль) в EtOH (100 мл). К данной смеси добавляют водный 1 М NaOH (50 мл), и полученный бесцветный раствор нагревают при 80°С в течение 14 час. Полученному светло-розовому раствору позволяют охлаждаться до к.т., и раствор осторожно подкисляют до рН 1,5. Летучие компоненты удаляют в вакууме, и полученный остаток распределяют между EtOAc (300 мл) и водой (300 мл). Водную фазу отделяют и снова экстрагируют с помощью EtOAc (3×300 мл). Объединенные органические экстракты затем промывают насыщенным солевым раствором (500 мл), обесцвечивают активированным углем и сушат над MgSO4. Фильтрование через слой целита и концентрирование бесцветного фильтрата в вакууме дает белое пенистое вещество. Разделение 3,9-ди-трет-бутилового эфира (рац.)-(1R∗, 5S∗)-7-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}-3,9-диазабицикло[3.3.1]нон-6-ен-3,6,9-трикарбоновой кислоты [1,01 г, 22%; Rf = 0,55 (гексан:EtOAc + 1% АсОН)] и указанного в заголовке соединения (2,77 г, 61%) может быть достигнуто с помощью FC (SiO2, 2:1 гексан: EtOAc + 1% АсОН). Остаточная АсОН может быть удалена количественно сначала азеотропной отгонкой с гептаном и затем с толуолом. Rf = 0,50 (1:1 гексан: EtOAc + 1% АсОН); ES- = 646,8.
Cмесь четырех возможных диастереоизомеров 3,9-ди-трет-бутилового эфира 7-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}-3,9-диазабицикло[3.3.1]нонaн-3,6,9-трикарбоновой кислоты (E1)
В 250 мл круглодонной колбе, снабженной магнитной мешалкой, суспендируют соединение В1 (1,17 г, 1,80 ммоль) и Pd/C (10% мас./мас., 1,20 г) в МеОН (100 мл). Полученную суспензию дезоксигенируют и затем через нее пропускают Н2 газ в течение 15 мин. Наконец, реакционную смесь эффективно перемешивают при к.т. в статической атмосфере H2, регулируемой баллоном. Через 2 час может быть отмечено количественное потребление соединения В1. Затем реакционный сосуд эффективно продувают с помощью N2, и суспензию фильтруют через слой целита и SiO2. После промывания нерастворимых веществ обильными количествами СН2Сl2 и EtOAc фильтрат концентрируют в вакууме до получения указанного в заголовке соединения в виде смеси четырех диастереомеров (1,116 г, 95% выход сырого продукта, ES- = 648,9). Данный продукт используют затем без очистки.
Ди-трет-бутиловый эфир (рац.)-(1R∗, 5S∗, 6R∗, 7S∗)-7-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}-6-[циклопропил-(2,3-дихлорбензил)карбамоил]-3,9-диазабицикло[3.3.1]нонан-3,9-дикарбоновой кислоты (D2) и ди-трет-бутиловый эфир (рац.)-(1R∗, 5S∗, 6R∗, 7R∗)-7-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}-6-[циклопропил-(2,3-дихлорбензил)карбамоил]-3,9-диазабицикло[3.3.1]нонан-3,9-дикарбоновой кислоты (D1).
В 100 мл круглодонной колбе, снабженной магнитной мешалкой, смешивают соединение Е1 (905 мг, 1,39 ммоль), циклопропил-(2,3-дихлорбензил)амин (полученный из 2,3-дихлорбензальдегида и циклопропиламина восстановительным аминированием, 600 мг, 2,78 ммоль), HOBt (282 мг, 2,08 ммоль) и DMAP (509 мг, 4,17 ммоль) в ДМФА (10 мл). Затем добавляют EDC.HCl (400 мг, 2,08 ммоль), и полученный желтый раствор перемешивают при к.т., и ход реакции отслеживают с помощью ЖХ-МС. По прошествии реакции, полностью завершившейся через 56 час с 86% конверсией, реакционную смесь разбавляют 150 мл Et2O и гасят холодной 10% водной HCl (150 мл). Органическую фракцию отделяют, и водную фракцию снова экстрагируют с помощью Et2O (3×100 мл). Объединенные органические экстракты последовательно промывают водным 1 М NaOH (100 мл), водой (150 мл) и насыщенным солевым раствором (150 мл). Сушка над MgSO4, фильтрование и концентрирование фильтрата в вакууме дает белый пенистый продукт. Дальнейшая очистка методом колоночной хроматографии (SiO2, 1,5:1 гексан: Et2O) дает указанное в заголовке соединение (0,8591 г, 73%) в виде 16:80:4 смеси диастереомеров. 200 мг смеси, полученной таким образом, затем растворяют в cмеси 10 мл 95% СН3СN + 4,9% Н2О + 0,1% муравьиная кислота. Данный раствор затем вводят 500 мкл пробами на ВЭЖХ, снабженную Phenomenex Synes 4 мк max RP 80 ангстрем 100 × 21,20 мм колонкой. Элюирование при скорости 20 мл/мин изократической смесью 95% СН3СN + 4,9% Н2О + 0,1% муравьиная кислота дает два основных диастереомера:
соединение D1: tR = 5,04 мин, ES- = 848,1, белое пенистое вещество, 40,0 мг (выделено с выходом 20%);
соединение D2: tR = 6,64 мин, ES- = 848,1, белое пенистое вещество, 140,4 мг (выделено с выходом 70%).
трет -Бутиловый эфир (рац.)-(3R∗, 4S∗)-3-[циклопропил-(2,3-диметилбензил)карбамоил]-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D3)
Раствор соединения Е2 (90 мг, 0,17 ммоль), (2,3-диметилбензил)циклопропиламина (приготовленного восстановительным аминированием 2,3-диметилбензальдегида и циклопропиламина, 90 мг, 0,51 ммоль), DMAP (5 мг, 0,04 ммоль), DIPEA (0,118 мл, 0,69 ммоль), HOBt (29 мг, 0,21 ммоль) и EDC.HCl (49 мг, 0,26 ммоль) в СН2Сl2 (3 мл) перемешивали в течение 18 час. Смесь разбавляли с помощью EtOAc, промывали водной 1 М HCl и насыщенным солевым раствором. Органическую фазу сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Указанное в заголовке соединение далее не очищали. ЖХ-МС: tR = 1,25 мин, ES+: = 681,37.
трет -Бутиловый эфир (3R, 4S)-3-{[5-хлор-2-(3-метоксипропил)бензил]циклопропилкарбамоил}-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D4)
Раствор соединения Е3 (0,052 г, 0,10 ммоль), [5-хлор-2-(3-метоксипропил)бензил]циклопропиламина (0,073 г, 0,30 ммоль), DMAP (0,003 г, 0,03 ммоль), DIPEA (0,068 мл, 0,40 ммоль), HOBt (0,017 г, 0,13 ммоль) и EDC.HCl (0,029 г, 0,15 ммоль) в СН2Сl2 (2 мл) перемешивали в течение 48 час. Смесь разбавляли с помощью EtOAc, промывали водной 1 М HCl и насыщенным солевым раствором. Органическую фазу сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Указанное в заголовке соединение далее не очищали. ЖХ-МС: tR = 1,27 мин, ES+: = 761,44.
трет -Бутиловый эфир (3R, 4S)-3-{[2-хлор-5-(2-метоксиэтил)бензил]циклопропилкарбамоил}-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D5)
Смесь соединения Е3 (200 мг, 0,383 ммоль), [2-хлор-5-(2-метоксиэтил)бензил]циклопропиламина (120 мг, 0,473 ммоль), HOBt (64,7 мг, 0,479 ммоль), DMAP (11,7 мг, 0,096 ммоль), DIPEA (0,262 мл, 1,53 ммоль) и EDC.HCl (110 мг, 0,574 ммоль) в СН2Сl2 (4 мл) перемешивали в течение 48 час при к.т. Cмесь фильтровали через Isolute®, предварительно промытый водной 1 М HCl. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка с помощью FC (EtOAc) давала указанное в заголовке соединение (280 мг, 98%). ЖХ-МС: tR= 1,26 мин, ES+: = 747,54.
трет -Бутиловый эфир (рац.)-(3R∗, 4S∗)-3'-{[2-хлор-5-(3-метоксипропил)бензил]циклопропилкарбамоил}-6-[2-(2,6-дихлор-4-метилфенокси)этокси]-3',4',5',6'-тетрагидро-2'Н-[3,4']бипиридинил-1'-карбоновой кислоты (D6)
Смесь соединения Е4 (1,00 г, 1,90 ммоль), [5-хлор-2-(3-метоксипропил)бензил]циклопропиламина (531 мг, 2,09 ммоль), DIPEA (1,30 мл, 7,61 ммоль), DMAP (58,1 мг, 0,476 ммоль), HOBt (321 мг, 2,38 ммоль) и EDC.HCl (547 мг, 2,86 ммоль) в СН2Сl2 (4 мл) перемешивали в течение 4 дней при к.т. EDC.HCl (100 мг, 0,506 ммоль) добавляли снова, и смесь перемешивали в течение 24 час. Добавляли СН2Сl2, и смесь промывали водной 1 М HCl (1×), водой (1×) и насыщенным солевым раствором (1×). Органический слой сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2 → СН2Сl2/МеОН 97:3) давала указанное в заголовке соединение (1,14 г, 79%). ЖХ-МС: tR = 1,26 мин, ES+: = 762,50.
трет -Бутиловый эфир (рац.)-(3R∗, 4S∗)-3'-{[2-хлор-5-(2-метоксиэтил)бензил]циклопропилкарбамоил}-6-[2-(2,6-дихлор-4-метилфенокси)этокси]-3',4',5',6'-тетрагидро-2'Н-[3,4']бипиридинил-1'-карбоновой кислоты (D7)
Смесь соединения Е4 (1,00 г, 1,90 ммоль), [5-хлор-2-(3-метоксиэтил)бензил]циклопропиламина (533 мг, 2,09 ммоль), DIPEA (1,30 мл, 7,61 ммоль), DMAP (58,1 мг, 0,476 ммоль), HOBt (321 мг, 2,38 ммоль) и EDC.HCl (547 мг, 2,86 ммоль) в СН2Сl2 (4 мл) перемешивали в течение 5 дней при к.т. EDC.HCl (100 мг, 0,506 ммоль) добавляли снова, и смесь перемешивали в течение 24 час. Добавляли СН2Сl2, и смесь промывали водной 1 М HCl (1×), водой (1×) и насыщенным солевым раствором (1×). Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2 → СН2Сl2/МеОН 97:3) давала указанное в заголовке соединение (1,12 г, 79%). ЖХ-МС: tR = 1,24 мин, ES+: = 746,16.
трет -Бутиловый эфир (3R, 4S)-3-[(5-{[трет-бутоксикарбонил-(2,2-дифторэтил)амино]метил}-2-хлорбензил)циклопропилкарбамоил]-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D8)
Получали, следуя общим условиям А, из соединения Е3 и трет-бутилового эфира (4-хлор-3-циклопропиламинометилбензил)-(2,2-дифторэтил)карбаминовой кислоты. ЖХ-МС: tR = 1,28 мин, ES+: = 880,26.
трет -Бутиловый эфир (3R, 4S)-3-({2-хлор-5-[(3,3,3-трифторпропиониламино)метил]бензил}циклопропилкарбамоил)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D9)
Получали, следуя общим условиям А, из соединения Е3 и N-(4-хлор-3-циклопропиламинометилбензил)-3,3,3-трифторпропионамида. ЖХ-МС: tR = 1,21 мин, ES+: = 828,20.
трет -Бутиловый эфир (3R, 4S)-3-({5-[(трет-бутоксикарбонилциклопропилметиламино)метил]-2-хлорбензил}циклопропилкарбамоил)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D10)
Получали, следуя общим условиям А, из соединения Е3 и трет-бутилового эфира (4-хлор-3-циклопропиламинометилбензил)циклопропилметилкарбаминовой кислоты. ЖХ-МС: tR = 1,31 мин, ES+: = 872,31.
трет -Бутиловый эфир (3R, 4S)-3-{[5-(ацетиламинометил)-2-хлорбензил]циклопропилкарбамоил}-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D11)
Получали, следуя общим условиям А, из соединения Е3 и N-(4-хлор-3-циклопропиламинометилбензил)ацетамида. ЖХ-МС: tR = 1,19 мин, ES+: = 758,22.
трет -Бутиловый эфир (3R, 4S)-3-({5-[2-(трет-бутоксикарбонилметиламино)этил]-2-хлорбензил}циклопропилкарбамоил)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D12)
Получали, следуя общим условиям А, из соединения Е3 и трет-бутилового эфира [2-(4-хлор-3-циклопропиламинометилфенил)этил]метилкарбаминовой кислоты. ЖХ-МС: tR = 1,29 мин, ES+: = 844,33.
трет -Бутиловый эфир (3R, 4S)-3-({5-[(трет-бутоксикарбонилметиламино)метил]-2-хлорбензил}циклопропилкарбамоил)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D13)
Получали, следуя общим условиям А, из соединения Е3 и трет-бутилового эфира (4-хлор-3-циклопропиламинометилбензил)метилкарбаминовой кислоты. ЖХ-МС: tR= 1,28 мин, ES+: = 832,31.
трет -Бутиловый эфир (3R, 4S)-3-({2-хлор-5-[2-(3,3,3-трифторпропиониламино)этил]бензил}циклопропилкарбамоил)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D14)
Получали, следуя общим условиям А, из соединения Е3 и N-[2-(4-хлор-3-циклопропиламинометилфенил)этил]-3,3,3-трифторпропионамида. ЖХ-МС: tR = 1,22 мин, ES+: = 840,25.
трет -Бутиловый эфир (3R, 4S)-3-({5-[2-(трет-бутоксикарбонилэтиламино)этил]-2-хлорбензил}циклопропилкарбамоил)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D15)
Получали, следуя общим условиям А, из соединения Е3 и трет-бутилового эфира [2-(4-хлор-3-циклопропиламинометилфенил)этил]этилкарбаминовой кислоты. ЖХ-МС: tR = 1,30 мин.
трет -Бутиловый эфир (3R, 4S)-3-{[5-(2-ацетиламиноэтил)-2-хлорбензил]циклопропилкарбамоил}-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D16)
Получали, следуя общим условиям А, из соединения Е3 и N-[2-(4-хлор-3-циклопропиламинометилфенил)этил]ацетамида. ЖХ-МС: tR = 1,22 мин, ES+: = 774,29.
трет -Бутиловый эфир (3R, 4S)-3-[(5-{[трет-бутоксикарбонил-(2-фторэтил)амино]метил}-2-хлорбензил)циклопропилкарбамоил]-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D17)
Получали, следуя общим условиям А, из соединения Е3 и трет-бутилового эфира (4-хлор-3-циклопропиламинометилбензил)-(2-фторэтил)карбаминовой кислоты. ЖХ-МС: tR = 1,28 мин, ES+: = 864,27.
трет -Бутиловый эфир (3R, 4S)-3-({5-[(трет-бутоксикарбонилэтиламино)метил]-2-хлорбензил}циклопропилкарбамоил)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D18)
Получали, следуя общим условиям С, из соединения Е3 и трет-бутилового эфира (4-хлор-3-циклопропиламинометилбензил)этилкарбаминовой кислоты. ЖХ-МС: tR = 1,30 мин, ES+: = 846,32.
трет -Бутиловый эфир (3R, 4S)-3-({5-[(трет-бутоксикарбонилциклопропиламино)метил]-2-хлорбензил}циклопропилкарбамоил)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D19)
Получали, следуя общим условиям С, из соединения Е3 и трет-бутилового эфира (4-хлор-3-циклопропиламинометилбензил)циклопропилкарбаминовой кислоты. ЖХ-МС: tR = 1,31 мин, ES+: = 856,37.
трет -Бутиловый эфир (3R, 4S)-3-({5-[2-(трет-бутоксикарбонилциклопропиламино)этил]-2-хлорбензил}циклопропилкарбамоил)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1-карбоновой кислоты (D20)
Получали, следуя общим условиям С, из соединения Е3 и трет-бутилового эфира [2-(4-хлор-3-циклопропиламинометилфенил)этил]циклопропилкарбаминовой кислоты. ЖХ-МС: tR = 1,31 мин, ES+: = 872,35.
1-трет-Бутиловый эфир (рац.)-(3R∗, 4S∗)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1,3-дикарбоновой кислоты (Е2)
К раствору соединения L2 (0,15 г, 0,27 ммоль) в МеОН (1 мл) добавляли водный 1 М NaOH (1 мл). Смесь перемешивали при 70°С в течение 2 час. Добавляли воду, и смесь экстрагировали с помощью EtOAc. Органическую фазу промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Сырой остаток очищали на паде силикагеля, получая указанное в заголовке соединение (93 мг, 65%). ЖХ-МС: tR= 1,12 мин, ES+: = 524,24.
1-трет-Бутиловый эфир (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1,3-дикарбоновой кислоты (Е3)
Соединение Е2 (4,46 г, 8,5 ммоль) разделяли, используя препаративную ВЭЖХ, снабженную хиральной колонкой, описанной в данном документе выше. Применяли изократический элюент, состоящий из 97% гексана, 3% этанола и 0,1% ТГФ. Получали указанное в заголовке соединение (1,35 г, 30%). Аналитическая хиральная ВЭЖХ (элюент, тождественный препаративному): tR = 29,00 мин.
1'-трет-Бутиловый эфир (рац.)-(3R∗, 4S∗)-6-[2-(2,6-дихлор-4-метилфенокси)этокси]-3',4',5',6'-тетрагидро-2'H-[3,4']бипиридинил-1',3'-дикарбоновой кислоты (Е4)
Cмесь соединения L4 (4,92 г, 9,12 ммоль) в водном 1 М NaOH (75 мл) и МеОН (75 мл) перемешивали при 70°С в течение 4,5 час. Добавляли водную 1 М HCl до тех пор, пока рН не достигал 4. Смесь экстрагировали с помощью EtOAc (3×). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Сушка остатка в высоком вакууме давала сырое указанное в заголовке соединение (4,63 г, 97%), которое затем использовали без очистки. ЖХ-МС: tR = 1,08 мин, ES+: = 525,40.
1-трет-Бутиловый эфир 3-метиловый эфир 4-{4-[2-(трет-бутилдиметилсиланилокси)этокси]фенил}-5,6-дигидро-2Н-пиридин-1,3-дикарбоновой кислоты (F1)
К раствору 4-[2-(трет-бутилдиметилсиланилокси)этокси]бромбензола (WO 03/093267, 7,95 г, 24 ммоль) в ТГФ (200 мл) при -78°С добавляли BuLi (1,6 М в гексане, 17,12 мл, 27,4 ммоль). Раствор перемешивали при -78°С в течение 30 мин, затем добавляли ZnCl2 (1 М в ТГФ, 30 мл, 30 ммоль). Полученному раствору позволяли охлаждаться до к.т., и к нему добавляли 1-трет-бутиловый эфир 3-метиловый эфир 4-трифторметансульфонилокси-5,6-дигидро-2Н-пиридин-1,3-дикарбоновой кислоты (WO 2004/002957, 7,79 г, 20 ммоль) в ТГФ (20 мл) и Pd(PPh3)4 (0,69 г, 0,60 ммоль). Реакционную смесь нагревали при 50°С в течение 1 час и перемешивали 16 час при к.т. Смесь охлаждали до 0°С, и к ней добавляли водный насыщенный NH4Cl. Добавляли EtOAc, и органическую фазу промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (EtOAc/гептан 2:8 → 1:0) давала указанное в заголовке соединение (8,1 г, 82%). ЖХ-МС: tR = 1,23 мин, ES+: = 506,47.
3-трет-Бутиловый эфир 6-этиловый эфир (рац.)-(1R∗, 5S∗)-7-{4-[3-(трет-бутилдиметилсиланилокси)пропил]фенил}-9-метил-3,9-диазабицикло[3.3.1]нон-6-ен-3,6-дикарбоновой кислоты (F2)
Раствор [3-(4-бромфенил)пропокси]трет-бутилдиметилсилана (102,1 г, 310 ммоль) в ТГФ (1,50 л) в атмосфере азота охлаждают до -78°С. Добавляют BuLi (1,5 М в гексане, 212 мл, 318 ммоль). Через 30 мин добавляют ZnCl2 (1 М в ТГФ, 465 мл, 465 ммоль). Смеси позволяют нагреться до к.т. К ней добавляют 3-трет-бутиловый эфир 6-этиловый эфир (рац.)-(1R∗, 5S∗)-9-метил-7-трифторметансульфонилокси-3,9-диазабицикло[3.3.1]нон-6-ен-3,6-дикарбоновой кислоты (83,6 г, 155 ммоль) в ТГФ (100 мл) и затем Pd(PPh3)4 (4,48 г, 3,87 ммоль). Смесь нагревают при кипении с обратным холодильником в течение 30 мин, и позволяют ей охлаждаться до к.т. Смесь разбавляют с помощью EtOAc и промывают водным 1 М NaOH (1×). Органические экстракты сушат над MgSO4, фильтруют, и растворители удаляют при пониженном давлении. Очистка остатка с помощью FC (МеОН/СН2Cl2 1:49 → 1:24 → 3:47 → 2:23) дает указанное в заголовке соединение (88,6 г, фактически количественный выход). ЖХ-МС: tR = 0,98 мин, ES+: = 559,24.
3,9-Ди-трет-бутиловый эфир 6-этиловый эфир (рац.)-(1R∗, 5S∗)-7-[4-(3-гидроксипропил)фенил]-3,9-диазабицикло[3.3.1]нон-6-ен-3,6,9-трикарбоновой кислоты (F3)
Соединение F2 (32,0 г, 57,3 ммоль) растворяют в сухом 1,2-дихлорэтане (590 мл). К данному раствору добавляют NaHCO3 (48,2 г, 573 ммоль) и 1-хлорэтилхлорформиат (62,5 мл, 573 ммоль), и суспензию нагревают при 80°С. Через 3 час реакционной смеси позволяют охлаждаться до к.т. Смесь фильтруют, и фильтрат упаривают при пониженном давлении. Остаток сушат 15 мин в высоком вакууме. Продукт разбавляют с помощью МеОН (400 мл), и смесь нагревают при 50°С в течение 20 мин. Реакционной смеси позволяют охлаждаться до к.т., и растворители удаляют при пониженном давлении. Желтое твердое вещество сушат в высоком вакууме в течение 1 час. Твердое вещество растворяют в СН2Сl2 (190 мл), и раствор охлаждают до 0°С. Добавляют DIPEA (49,1 мл, 287 ммоль) и Вос2О (37,5 г, 172 ммоль). Смесь перемешивают в течение ночи, в то время как она нагревается до к.т. Реакционную смесь разбавляют с помощью СН2Сl2 (110 мл). Органический слой промывают водной 1 М НСl (2×300 мл) и водным насыщенным NaHCO3 (300 мл). Органический слой сушат над MgSO4, фильтруют, и растворители удаляют при пониженном давлении. Очистка остатка с помощью FC (СН2Cl2/МеОН 100:0 → 2:98 → 5:95) дает указанное в заголовке соединение (26,2 г, 86%). ЖХ-МС: tR = 1,05 мин, ES+: = 531,33.
1-трет-Бутиловый эфир 4-{4-[3-(трет-бутилдиметилсиланилокси)пропил]фенил}-5,6-дигидро-2Н-пиридин-1,3-дикарбоновой кислоты (G1)
1-трет-Бутиловый эфир 3-метиловый эфир 4-{4-[3-(трет-бутилдиметилсиланилокси)пропил]фенил}-5,6-дигидро-2Н-пиридин-1,3-дикарбоновой кислоты (WO 2004/002957, 17,1 г 35,0 ммоль) растворяют в смеси ДМСО (300 мл) и ТГФ (100 мл). Добавляют водный NaOH (1 М, 70 мл и 2 М, 50 мл). Смесь нагревают при 50°С в течение 7 час, затем охлаждают до 0°С и добавляют воду (100 мл). рН доводят до 2 добавлением водной HCl (1 М, 100 мл), и реакционную смесь экстрагируют с помощью EtOAc (2×250 мл). Объединенные органические слои сушат над MgSO4, фильтруют, и растворители удаляют при пониженном давлении. Светло-коричневое масло сушат в высоком вакууме (18 г). ЖХ-МС: tR = 0,86 мин, [М+Н]+: = 362,05.
Данное сырое соединение (17,98 г) растворяют в ДМФА (300 мл) с последующим добавлением имидазола (6,84 г, 97,7 ммоль) и TBDMS-Cl (11,1 г, 73,8 ммоль). Смесь перемешивают при к.т. в течение 14 час. Добавляют насыщенный водный NH4Cl (250 мл) и воду (250 мл), и смесь экстрагируют с помощью Et2O (2×250 мл). Объединенные органические слои сушат над MgSO4, фильтруют, и растворители выпаривают при пониженном давлении. Остаток растворяют в ТГФ (300 мл) и добавляют МеОН (125 мл), воду (125 мл) и К2СО3 (3,00 г). Смесь перемешивают при к.т. в течение 10 мин. Добавляют насыщенный водный NH4Cl (250 мл) и воду (25 мл), и смесь экстрагируют с помощью EtОАс (2×250 мл). Объединенные органические слои сушат над MgSO4, фильтруют, и растворители выпаривают при пониженном давлении. Очистка остатка с помощью FC (гексан/EtOAc = 4/1-1/1) дает указанное в заголовке соединение (5,1 г, 32%). ЖХ-МС: tR = 1,16 мин; ES+: = 476,35.
трет -Бутиловый эфир 4-{4-[3-(трет-бутилдиметилсиланилокси)пропил]фенил}-3-[циклопропил-(3-метокси-2-метилбензил)карбамоил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (Н1)
Соединение G1 (2 г, 4,21 ммоль) растворяют в СН2Сl2 (30 мл), и к раствору добавляют (1-хлор-2-метилпропенил)диметиламин (618 мг, 4,63 ммоль). Смесь перемешивают при к.т. в течение 5 мин, и к ней добавляют циклопропил(3-метокси-2-метилбензил)амин (полученный восстановительным аминированием из 3-метокси-2-метилбензальдегида, Сomins, D. L.; Brown, J. D., J. Org. Chem., 1989, 54, 3730, и циклопропиламина, 844 мг, 4,42 ммоль) и DIPEA (815 мг, 6,32 ммоль). Смесь перемешивают при к.т. в течение 15 мин, разбавляют с помощью СН2Сl2 (100 мл) и экстрагируют водной 10% лимонной кислотой (100 мл). Водный слой промывают с помощью СН2Сl2 (50 мл). Объединенные органические слои сушат над MgSO4, фильтруют, и растворители выпаривают при пониженном давлении. Очистка сырого продукта с помощью FC (СН2Сl2/МеОН 19/1) дает указанное в заголовке соединение (3,36 г, количественный выход). ЖХ-МС: tR = 1,28 мин; ES+: = 649,42.
трет -Бутиловый эфир 4-{4-[3-(трет-бутилдиметилсиланилокси)пропил]фенил}-3-[циклопропил-(2,3-диметилбензил)карбамоил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (Н2)
Следуя методике для соединения Н1, но используя циклопропил-(2,3-диметилбензил)амин (полученный восстановительным аминированием 2,3-диметилбензальдегида и циклопропиламина) вместо циклопропил-(3-метокси-2-метилбензил)амина, соединение Н2 (2,84 г, количественный выход) получали из соединения G1 (2,00 г). ЖХ-МС: tR = 1,30 мин; ES+: = 633,45.
трет -Бутиловый эфир 4-{4-[3-(трет-бутилдиметилсиланилокси)пропил]фенил}-3-[циклопропил-(3-метокси-2-метилбензил)карбамоил]пиперидин-1-карбоновой кислоты (J1)
Pd/C (10%; 340 мг) суспендируют в атмосфере азота в EtOH (25 мл). Добавляют раствор соединения Н1 (3,36 г, 5,19 ммоль) в EtOH (25 мл). Смесь выдерживают в атмосфере Н2 (3 бар) в течение 3 час, фильтруют через целит и концентрируют в вакууме. Остаток сушат в высоком вакууме, что дает указанное в заголовке соединение (2,20 г, 65%), которое затем используют без очистки. ЖХ-МС: tR = 1,31 мин; ES+: = 651,46.
трет -Бутиловый эфир 4-{4-[3-(трет-бутилдиметилсиланилокси)пропил]фенил}-3-[циклопропил-(2,3-диметилбензил)карбамоил]пиперидин-1-карбоновой кислоты (J2)
Следуя методике для соединения J1, получают указанное в заголовке соединение (2,42 г, 85%) из соединения Н2 (2,84 г, 4,47 ммоль). ЖХ-МС: tR = 1,32 мин; ES+: = 635,47.
трет -Бутиловый эфир 3-[циклопропил-(3-метокси-2-метилбензил)карбамоил]-4-[4-(3-гидроксипропил)фенил]пиперидин-1-карбоновой кислоты (J3)
Cоединение J1 (2,20 г, 3,38 ммоль) растворяют в ТГФ (50 мл) с последующим добавлением TBAF (1,21 г, 3,84 ммоль). Реакционную смесь перемешивают при к.т. в течение 2,5 час, разбавляют с помощью EtOAc (250 мл) и промывают водой (200 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют в вакууме. Очистка остатка с помощью FC (гексан/EtOAc 1/1) дает указанное в заголовке соединение (0,91 г, 50%). ЖХ-МС: tR = 1,07 мин; ES+: = 537,37.
трет -Бутиловый эфир 3-[циклопропил-(2,3-диметилбензил)карбамоил]-4-[4-(3-гидроксипропил)фенил]пиперидин-1-карбоновой кислоты (J4)
Следуя методике для соединения J3, получают указанное в заголовке соединение (0,91 г, 46%) из соединения J2 (2,42 г). ЖХ-МС: tR = 1,08 мин; ES+: = 521,36.
1-трет-Бутиловый эфир 3-метиловый эфир 4-{4-[2-(трет-бутилдиметилсиланилокси)этокси]фенил}пиперидин-1,3-дикарбоновой кислоты (К1)
Mg (1,40 г, 58 ммоль) добавляли к раствору соединения F1 (8,10 г, 17 ммоль) в МеОн (40 мл) в атмосфере Ar. Смесь перемешивали в течение 1 час, в то время как температура поддерживалась ниже 30°С. Добавляли по каплям водную 1 М НСl (115 мл, 115 ммоль), и смесь перемешивали в течение 1 час. Данную смесь экстрагировали с помощью EtOAc (2×). Объединенные органические слои промывали водой, насыщенным солевым раствором, сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (EtOAc/гептан 2:1) давала 2:3 транс/цис смесь указанного в заголовке соединения (7,6 г, 93%). ЖХ-МС: tR = 1,23 мин; ES+: = 508,47.
1-трет-Бутиловый эфир 3-метиловый эфир 4-[4-(2-(гидроксиэтокси)фенил]пиперидин-1,3-дикарбоновой кислоты (К2)
К раствору соединения К1 (7,60 г, 15,4 ммоль) в ТГФ (150 мл) при 0°С и в атмосфере Ar добавляли TBAF (4,86 г, 15,4 ммоль). После перемешивания смеси в течение 1 час добавляли водный насыщенный NH4Cl (100 мл), и реакционную смесь экстрагировали с помощью EtOAc (2×). Органический слой промывали водой, насыщенным солевым раствором, сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (EtOAc/гептан 2:1 → 1:0) давала указанное в заголовке соединение (5,06 г, 87%). ЖХ-МС: tR = 0,91 мин; ES+: = 380,30.
1-трет-Бутиловый эфир 3-метиловый эфир 4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1,3-дикарбоновой кислоты (L1)
Смесь соединения К2 (5,50 г, 15 ммоль), 2,6-дихлор-п-крезола (3,08 г, 18 ммоль), дипиперидида азодикарбоновой кислоты (7,31 г, 29 ммоль) и PВu3 (14 мл, 58 ммоль) в толуоле (150 мл) нагревали при 50°С в течение 16 час. Смеси позволяли охлаждаться до к.т., фильтровали, и осадок промывали толуолом. Фильтрат разбавляли с помощью EtOAc, промывали водой (2×) и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка сырого продукта с помощью FC (EtOAc/гептан 0:1 → 1:9 → 2:8) давала указанное в заголовке соединение в виде бесцветного масла (7,3 г, 90%). ЖХ-МС: tR = 1,18 мин; ES+: = 538,34.
1-трет-Бутиловый эфир 3-метиловый эфир (рац.)-(3R∗, 4S∗)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-1,3-дикарбоновой кислоты (L2)
К раствору соединения L1 (0,21 г, 0,38 ммоль) в МеОН (2 мл) в атмосфере Ar добавляли NaOMe (6 мг, 0,11 ммоль). Смесь перемешивали в течение 3 суток при 70°С. Добавляли воду, и смесь экстрагировали с помощью EtOAc. Органическую фазу промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и растворители удаляли при пониженном давлении. Указанное в заголовке соединение (150 мг, 72%) дополнительно не очищали. ЖХ-МС: tR = 1,18 мин; ES+: = 538,32.
Смесь всех возможных стереоизомеров 1'-трет-бутилового эфира 3'-метилового эфира 6-[2-(2,6-дихлор-4-метилфенокси)этокси]-3',4',5',6'-тетрагидро-2'H-[3,4']бипиридинил-1',3'-дикарбоновой кислоты (L3)
Соединение А2 (7,00 г, 13,0 ммоль) растворяли в МеОН (130 мл). К данному раствору медленно добавляли Mg (500 мг, 20,6 ммоль). Смесь перемешивали в течение 1 час, и к ней снова добавляли Mg (608 мг, 25,0 ммоль). Смесь перемешивали 3 час. Медленно добавляли водную 1 М НСl, и смесь перемешивали в течение 1 час при к.т. Растворители выпаривали при пониженном давлении, и смесь экстрагировали с помощью EtOAc. Органические экстракты сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Сушка в высоком вакууме давала сырое указанное в заголовке соединение (4,92 мг, 70%). ЖХ-МС: tR = 1,16 мин; ES+: = 539,43.
1'-трет-Бутиловый эфир 3'-метиловый эфир (рац.)-(3R∗, 4S∗)-6-[2-(2,6-дихлор-4-метилфенокси)этокси]-3',4',5',6'-тетрагидро-2'H-[3,4']бипиридинил-1',3'-дикарбоновой кислоты (L4)
Смесь соединения L3 (5,40 г, 10,0 ммоль) и MeONa (540 мг, 10,0 ммоль) в МеОН (250 мл) перемешивали в течение ночи при 70°С. MeONa (54 мг, 1,00 ммоль) добавляли снова, и смесь перемешивали в течение 5 час при 70°С. MeONa (54 мг, 1,00 ммоль) добавляли снова, и смесь перемешивали в течение ночи при 70°С. Водную 1 М НСl добавляли до установления рН 6-7. Растворители выпаривали при пониженном давлении, и смесь экстрагировали с помощью EtOAc. Объединенные органические экстракты промывали водой и насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FC (гептан/EtOAc 8:2 с 1% Et3N) давала указанное в заголовке соединение (4,92 г, 91%). ЖХ-МС: tR = 1,16 мин; ES+: = 539,43.
Пример 1
Циклопропил-(2,3-дихлорбензил)амид (рац.)-(1R∗, 5S∗, 6R∗, 7R∗)-7-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}-3,9-диазабицикло[3.3.1]нонан-6-карбоновой кислоты
В 25 мл круглодонной колбе, снабженной магнитной мешалкой, растворяют соединение D1 (31 мг, 0,037 ммоль) в СН2Сl2 (1 мл). При 0°С добавляют по каплям 4 М НСl (диоксан, 0,36 мл), и полученный раствор нагревают при к.т. в течение 2 час, и затем перемешивают при к.т. в течение следующих 8 час. Летучие компоненты удаляют в вакууме, и полученный остаток распределяют между СН2Сl2 (10 мл) и водным 1 М NaOH (10 мл). Органический слой отделяют и водную промывку снова экстрагируют с помощью СН2Сl2 (3×10 мл). Объединенные органические экстракты промывают насыщенным солевым раствором (10 мл), сушат над MgSO4, фильтруют, и фильтрат концентрируют в вакууме. Полученный таким образом продукт затем очищают колоночной хроматографией (SiO2, 95:5 CH2Cl2:2 M NH3 в МеОН), выделяя указанное в заголовке соединение (15,2 мг, 63%). tR = 2,0 мин на Agilent Zorbax RX-C18 4,6 × 150 мм ВЭЖХ колонке (изократическая смесь 49,8% СН3СN + 49,9% Н2О + 0,1% муравьиная кислота); ES+: = 648,1.
Пример 2
Циклопропил-(2,3-дихлорбензил)амид (рац.)-(1R∗, 5S∗, 6R∗, 7S∗)-7-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}-3,9-диазабицикло[3.3.1]нонан-6-карбоновой кислоты
В 25 мл круглодонной колбе, снабженной магнитной мешалкой, растворяют соединение D2 (54 мг, 0,064 ммоль) в СН2Сl2 (1 мл). При 0°С добавляют по каплям 4 М НСl (диоксан, 0,47 мл), и полученный раствор нагревают при к.т. в течение 2 час, и затем перемешивают при к.т. в течение следующих 8 час. Летучие компоненты удаляют в вакууме, и полученный остаток распределяют между СН2Сl2 (10 мл) и водным 1 М NaOH (10 мл). Органический слой отделяют, и водную промывку снова экстрагируют с помощью СН2Сl2 (3×10 мл). Объединенные органические экстракты промывают насыщенным солевым раствором (10 мл), сушат над MgSO4, фильтруют, и фильтрат концентрируют в вакууме. Сырой продукт затем очищают колоночной хроматографией (SiO2, 95:5 CH2Cl2:2 M NH3 в МеОН), получая указанное в заголовке соединение (36,1 мг, 87%). tR = 3,1 мин на Agilent Zorbax RX-C18 4,6 × 150 мм ВЭЖХ колонке (изократическая смесь 49,8% СН3СN + 49,9% Н2О + 0,1% муравьиная кислота); ES+: 648,1.
Пример 3
Циклопропил(3-метокси-2-метилбензил)амид 4-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты
Указанное в заголовке соединение получают по общей методике, описанной выше, из соединения J3 и 2-хлор-3,6-дифторфенола. ЖХ-МС: tR = 0,99 мин; ES+: 583,23.
Пример 4
Циклопропил(3-метокси-2-метилбензил)амид 4-{4-[3-(2,6-дихлор-4-метилфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты
Указанное в заголовке соединение получают по общей методике, описанной выше, из соединения J3 и 2,6-дихлор-п-крезола. ЖХ-МС: tR = 1,02 мин; ES+: 595,43.
Пример 5
Циклопропил(3-метокси-2-метилбензил)амид 4-{4-[3-(2,3,6-трифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты
Указанное в заголовке соединение получают по общей методике, описанной выше, из соединения J3 и 2,3,6-трифторфенола. ЖХ-МС: tR= 0,98 мин; ES+: 567,48.
Пример 6
Циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты
Указанное в заголовке соединение получают по общей методике, описанной выше, из соединения J4 и 2-хлор-3,6-дифторфенола. ЖХ-МС: tR = 1,00 мин; ES+: 567,26.
Пример 7
Циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2,6-дихлор-4-метилфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты
Указанное в заголовке соединение получают по общей методике, описанной выше, из соединения J4 и 2,6-дихлор-п-крезола. ЖХ-МС: tR = 1,04 мин; ES+: 579,44.
Пример 8
Циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2-хлор-6-фтор-3-метилфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты
Указанное в заголовке соединение получают по общей методике, описанной выше, из соединения J4 и 2-хлор-6-фтор-3-метилфенола. ЖХ-МС: tR = 1,04 мин; ES+: 563,47.
Пример 9
Циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2,3,6-трифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты
Указанное в заголовке соединение получают по общей методике, описанной выше, из соединения J4 и 2,3,6-трифторфенола. ЖХ-МС: tR= 1,00 мин; ES+: 551,48.
Пример 10
Циклопропил(2,3-диметилбензил)амид (рац.)-(3R∗, 4S∗)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Соединение D3 (0,068 г, 0,10 ммоль) растворяли в СН2Сl2 (1 мл), и данный раствор охлаждали до 0°С. Добавляли 4 М НСl в диоксане (1 мл), и реакционную смесь перемешивали в течение 1 час при к.т. Растворители выпаривали при пониженном давлении. Очистка остатка с помощью ВЭЖХ давала указанное в заголовке соединение (37 мг, 63%). ЖХ-МС: tR = 0,95 мин; ES+: 581,44.
Пример 11
[2-Хлор-5-(3-метоксипропил)бензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Соединение D4 (0,076 г, 0,10 ммоль) растворяли в СН2Сl2 (1 мл), и данный раствор охлаждали до 0°С. Добавляли 4 М НСl в диоксане (1 мл), и реакционную смесь перемешивали в течение 1 час при к.т. Растворители выпаривали при пониженном давлении. Очистка остатка с помощью ВЭЖХ давала указанное в заголовке соединение (44 мг, 67%). ЖХ-МС: tR = 0,99 мин; ES+: 661,41.
Пример 12
[2-Хлор-5-(2-метоксиэтил)бензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Соединение D5 (279 мг, 0,375 ммоль) растворяли в СН2Сl2 (3 мл). Данный раствор охлаждали до 0°С, и к нему добавляли НСl (4 М в диоксане, 1,00 мл). Смесь перемешивали при 0°С в течение 1 час. Снова добавляли НСl (4 М в диоксане, 1,00 мл), и смесь перемешивали при к.т. в течение 2 час. Снова добавляли НСl (4 М в диоксане, 1,00 мл), и смесь перемешивали при к.т. в течение 3 час. Водный 1 М NaOH добавляли до тех пор, пока смесь не стала слабо щелочной. Смесь фильтровали через Isolute®. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью FС (MeOH/CH2Cl2 1:19) давала указанное в заголовке соединение (120 мг, 50%). ЖХ-МС: tR = 0,96 мин; ES+: 645,49.
Пример 13
[2-Хлор-5-(3-метоксипропил)бензил]циклопропиламид (3R, 4S)-6-[2-(2,6-дихлор-4-метилфенокси)этокси]-1',2',3',4',5',6'-гексагидро[3,4']бипиридинил-3'-карбоновой кислоты
Раствор соединения D6 (1,14 г, 1,50 ммоль) в СН2Сl2 (4,13 мл) охлаждали до 0°С, и к нему добавляли НСl (4 М в диоксане, 3,75 мл). Смесь перемешивали при к.т. в течение 3 час, и растворители удаляли при пониженном давлении. Добавляли СН2Сl2 и смесь промывали водным 1 М NaOH. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью ВЭЖХ давала рацемическое указанное в заголовке соединение (621 мг, 63%). Данный рацемат разделяли с помощью ВЭЖХ (Regis Whelk, элюент В 75% → 30% в течение 30 мин, затем изократическая смесь). Получали указанное в заголовке соединение (167 мг, 27%). ЖХ-МС: tR = 0,95 мин; ES+: 684,49. Хиральная ВЭЖХ колонка: tR = 17,0 мин.
Пример 14
[2-Хлор-5-(2-метоксиэтил)бензил]циклопропиламид (3R, 4S)-6-[2-(2,6-дихлор-4-метилфенокси)этокси]-1',2',3',4',5',6'-гексагидро[3,4']бипиридинил-3'-карбоновой кислоты
Раствор соединения D7 (1,12 г, 1,50 ммоль) в СН2Сl2 (4,13 мл) охлаждали до 0°С, и к нему добавляли НСl (4 М в диоксане, 3,75 мл). Смесь перемешивали при к.т. в течение 3 час, и растворители удаляли при пониженном давлении. Добавляли СН2Сl2, и смесь промывали водным 1 М NaOH. Органический слой сушили над MgSO4, фильтровали, и растворители удаляли при пониженном давлении. Очистка остатка с помощью ВЭЖХ давала рацемическое указанное в заголовке соединение (392 мг, 40%). Данный рацемат разделяли с помощью ВЭЖХ (Regis Whelk, элюент В 75% → 30% в течение 30 мин, затем изократическая смесь). Получали указанное в заголовке соединение (167 мг, 27%). ЖХ-МС: tR = 0,94 мин; ES+: 646,50. Хиральная ВЭЖХ колонка: tR = 17,4 мин.
Пример 15
{2-Хлор-5-[(2,2-дифторэтиламино)метил]бензил}циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D8 получали указанное в заголовке соединение (17,8 мг, 26% от двух стадий). ЖХ-МС: tR = 0,83 мин; ES+: 682,21.
Пример 16
{2-Хлор-5-[(3,3,3-трифторпропиониламино)метил]бензил}циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D9 получали указанное в заголовке соединение (30,0 мг, 42% от двух стадий). ЖХ-МС: tR = 0,95 мин; ES+: 727,81.
Пример 17
{2-Хлор-5-[(циклопропилметиламино)метил]бензил}циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D10 получали указанное в заголовке соединение (20,3 мг, 30% от двух стадий). ЖХ-МС: tR = 0,84 мин; ES+: 672,30.
Пример 18
[5-(ацетиламинометил)-2-хлорбензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D11 получали указанное в заголовке соединение (30 мг, 46% от двух стадий). ЖХ-МС: tR = 0,91 мин; ES+: 658,25.
Пример 19
[2-Хлор-5-(2-метиламиноэтил)бензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D12 получали указанное в заголовке соединение (34,3 мг, 53% от двух стадий). ЖХ-МС: tR = 0,82 мин; ES+: 646,25.
Пример 20
(2-Хлор-5-метиламинометилбензил)циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D13 получали указанное в заголовке соединение (24,1 мг, 38% от двух стадий). ЖХ-МС: tR = 0,81 мин; ES+: 630,26.
Пример 21
{2-Хлор-5-[2-(3,3,3-трифторпропиониламино)этил]бензил}циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D14 получали указанное в заголовке соединение (30,0 мг, 40% от двух стадий). ЖХ-МС: tR = 0,96 мин; ES+: 742,24.
Пример 22
[2-Хлор-5-(2-этиламиноэтил)бензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D15 получали указанное в заголовке соединение (11,8 мг, 18% от двух стадий). ЖХ-МС: tR = 0,83 мин; ES+: 660,28.
Пример 23
[5-(2-ацетиламиноэтил)-2-хлорбензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D16 получали указанное в заголовке соединение (11,8 мг, 18% от двух стадий). ЖХ-МС: tR = 0,93 мин; ES+: 672,27.
Пример 24
{2-Хлор-5-[(2-фторэтиламино)метил]бензил}циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D17 получали указанное в заголовке соединение (21,2 мг, 32% от двух стадий). ЖХ-МС: tR = 0,82 мин; ES+: 682,21.
Пример 25
(2-Хлор-5-этиламинометилбензил)циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D18 получали указанное в заголовке соединение (38,7 мг, 60% от двух стадий). ЖХ-МС: tR = 0,81 мин; ES+: 644,20.
Пример 26
(2-Хлор-5-циклопропиламинометилбензил)циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D19 получали указанное в заголовке соединение (41,1 мг, 63% от двух стадий). ЖХ-МС: tR = 0,81 мин; ES+: 656,24.
Пример 27
[2-Хлор-5-(2-циклопропиламиноэтил)бензил]циклопропиламид (3R, 4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты
Следуя общим условиям В, из соединения D20 получали указанное в заголовке соединение (45,7 мг, 68% от двух стадий). ЖХ-МС: tR = 0,82 мин; ES+: 670,24.
Биологические испытания
1. Иммуноферментный анализ (ЕIA) для оценки AngI аккумуляции и ингибирования ренина
1.1. Приготовление AngI-BSA коньюгата
1,3 мг (1 мкмоль) AngI [1-10 (Bachem, H-1680)] и 17 мг (0,26 мкмоль) BSA (Fluka, 05475) растворяли в 4 мл 0,1 М фосфатного буфера, рН 7,4, после чего добавляли по каплям 2 мл глутаральдегида в Н2О при разбавлении 1:100 (Sigma G-5882). Cмесь инкубировали в течение ночи при 4°С, затем диализировали на фоне 2 литров 0,9% NaCl, дважды в течение 4 час при к.т., с последующим диализом на фоне 2 литров PBS 1X в течение ночи при к.т. Раствор затем фильтровали через Syringe фильтр, 0,45 мкм (Nalgene, Cat. No. 194-2545). Коньюгат можно хранить в полиэтиленовых тубах в 0,05% азиде натрия при 4°С в течение, по меньшей мере, 12 месяцев.
1.2. Приготовление МТР, покрытых с помощью BSA-AngI
Микротитрационные планшеты (МРТ384, MaxiSorp™, Nunc) инкубировали в течение ночи при 4°С с 80 мкл AngI (1-10)/BSA коньюгата, разбавленного 1:100000 в PBS IX, в тефлоновом стакане (точное разбавление в зависимости от загрузки коньюгата), осушали, наполняли 90 мкл блокирующего раствора [0,5% BSA (Sigma A-2153) в PBS 1X, 0,02% NaN3], инкубировали в течение, по меньшей мере, 2 час при к.т., или в течение ночи при 4°С. 96-луночные МТР (MaxiSorp™, Nunc) покрывали 200 мкл коньюгатом и блокировали 250 мкл блокирующим раствором, указанным выше, за исключением того, что блокирующий раствор содержал 3% BSA. Планшеты можно хранить в блокирующем растворе при 4°С в течение 1 месяца.
1.3. AngI-EIA в 384-луночных МТР
МТР, покрытые с помощью AngI (1-10)/BSA, промывали 3 раза промывным буфером (PBS 1X, 0,01% твин 20) и наполняли 75 мкл раствором первичного антитела (анти-AngI антисыворотка, предварительно разбавленная 1:10 в сыворотке лошади), разбавленным до конечной концентрации 1:100000 в оценочном буфере (PBS 1X, 1 mM EDTA, 0,1% BSA, pH 7,4). 5 мкл смеси от взаимодействия ренина (или стандартов в оценочном буфере) (см. ниже) добавляли к раствору первичного антитела, и планшеты инкубировали в течение ночи при 4°С. После инкубирования планшеты промывали 3 раза промывным буфером и инкубировали с вторичным антителом [антикроличий IgG, связанный с пероксидазой хрена обыкновенного (Amersham Bioscience, NA 934V), разбавленный 1:2000 в промывном буфере] в течение 2 час при к.т. Планшеты промывали 3 раза промывным буфером и затем инкубировали в течение 1 час при к.т. с раствором субстрата [1,89 mM ABTS (2,2'-азино-ди(3-этилбензтиазолинсульфонат))] (Roche Diagnostics, 102 946) и 2,36 мМ Н2О2 [30%, (Fluka, 95300)] в субстратном буфере (0,1 М ацетат натрия, 0,05 М дигидрофосфат натрия, рН 4,2). OD планшета читали при 405 нм c помощью считывающего устройства для микропланшетов (FLUOStar Optima от BMG). Образование AngI в течение взаимодействия ренина количественно определяли сравнением OD образца с OD cтандартной кривой AngI(1-10), измеренной параллельно.
2. Анализ первичного ингибирования ренина: IC 50 в буфере, 384 луночный МТР
Анализ ренина был адаптирован с анализа, описанного ранее (Fischli W. et al., Hypertension, 1991, 18:22-31), и cостоит из двух стадий: на первой стадии, рекомбинантный человеческий ренин инкубируют с его субстратом (коммерческий человеческий тетрадекапептидный рениновый субстрат) для образования продукта ангиотензина I (AngI). На второй стадии аккумулированный AngI измеряют с помощью иммунологического анализа (иммуноферментный анализ, EIA). Подробное описание данного анализа находится ниже. EIA является очень чувствительным и хорошо подходит для измерений активности ренина в буфере или в плазме. Вследствие малой концентрации ренина, применяемой в данном анализе (2 фмоль на анализируемую пробирку или 10 пМ), можно измерять сходства ингибиторов в данном первичном испытании ниже пМ концентрации.
2.1 Методология
Рекомбинантный человеческий ренин (3 пг/мкл) в оценочном буфере (PBS 1X, 1 mM EDTA, 0,1% BSA, pH 7,4), человеческий тетрадекапептидный (1-14) субстрат (Bachem, M-1120) [5 мкМ в 10 мМ HCl], гидроксихинолинсульфат (Fluka, 55100) [30 мM в Н2О] и оценочный буфер предварительно смешивали при 4°С в соотношении 100:30:10:145. 47,5 мкл на лунку предварительно приготовленной смеси переносили в полипропиленовые планшеты (МТР384, Nunc). Тестируемые соединения растворяли и разбавляли в 100% ДМСО и 2,5 мкл добавляли к предварительно приготовленной смеси, затем инкубировали при 37°С в течение 3 час. В конце инкубационного периода 5 мкл содержимого ренинового взаимодействия (или стандартов в оценочном буфере) переносили в EIA анализы (как описано выше) и количественно определяли AngI, произведенный ренином. Процент ингибирования ренина (снижение AngI) вычисляли для каждой концентрации соединения и определяли концентрацию ингибирования ренина, которая ингибировала активность фермента на 50% (IC50). Cоединения формулы (I) имеют IC50 величины от 0,1 нМ до 300 нМ, особенно от 1 нМ до 30 нМ.
Примеры ингибирования:
| название | год | авторы | номер документа |
|---|---|---|---|
| ВТОРИЧНЫЕ АМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕНИНА | 2007 |
|
RU2425032C2 |
| МОСТИКОВЫЕ ПРОИЗВОДНЫЕ СПИРО[2.4]ГЕПТАНА В КАЧЕСТВЕ АГОНИСТОВ РЕЦЕПТОРА ALX И/ИЛИ FPRL2 | 2010 |
|
RU2540274C2 |
| ФТОРИРОВАННЫЕ АМИНОТРИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ | 2010 |
|
RU2544862C2 |
| 2-МЕТОКСИ-ПИРИДИН-4-ИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2012 |
|
RU2588141C2 |
| ПИРИДИН-2-ИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИММУНОМОДУЛИРУЮЩИХ АГЕНТОВ | 2009 |
|
RU2494099C2 |
| ПРОИЗВОДНЫЕ ПИРИДИН-3-ИЛА В КАЧЕСТВЕ ИММУНОМОДУЛИРУЮЩИХ АГЕНТОВ | 2007 |
|
RU2454413C2 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛИЛМЕТИЛОВОГО ЭФИРА В КАЧЕСТВЕ АГОНИСТОВ РЕЦЕПТОРА ALX | 2011 |
|
RU2588567C2 |
| ПРОИЗВОДНЫЕ 2-АЗА-БИЦИКЛО[3.3.0]ОКТАНА | 2008 |
|
RU2478099C2 |
| ПРОИЗВОДНЫЕ 1-[М-КАРБОКСАМИДО(ГЕТЕРО)АРИЛ-МЕТИЛ]-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДА | 2013 |
|
RU2644761C2 |
| ПРОИЗВОДНЫЕ 4-ГИДРОКСИ-1,2,3,4-ТЕТРАГИДРОНАФТАЛИН-1-ИЛ-МОЧЕВИНЫ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ, СРЕДИ ПРОЧЕГО, ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНОГО ТРАКТА | 2011 |
|
RU2586333C1 |
Изобретение относится к новым соединениям, выбранным из группы, состоящей из амидов пиперидинкарбоновой кислоты формулы (I)
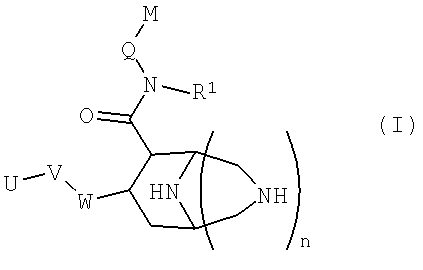
в которой W представляет собой фенильный цикл или шестичленный, не бензоконденсированный ароматический цикл, содержащий один атом азота, где указанные циклы замещены в пара-положении посредством V; V представляет собой связь; -A-(CH2)s- или -A-(CH2)v-B-; А и В независимо представляют собой -O-; U представляет собой моно-, ди-, три- или тетразамещенный арил, в котором заместители независимо выбраны из группы, состоящей из галогена, алкила и -CF3; Q представляет собой метилен; М представляет собой арильную группу, где указанная группа может быть необязательно моно- или дизамещенной заместителями, независимо выбранными из группы, состоящей из алкила; алкокси; -CF3; галогена; алкил-O-(СН2)0-4-СН2- и R'2N-(СН2)0-4-СН2-, где R' независимо выбран из группы, состоящей из водорода, алкила (необязательно замещенного одним, двумя или тремя атомами фтора), циклопропила, циклопропилметила, -C(=O)-R'', где R'' означает С1-С4-алкил или -СН2-CF3; R1 представляет собой циклоалкил; n представляет собой целое число 0 или 1; s представляет собой целое число 3; v представляет собой целое число 2; и заместители в цикле, -CON(R1)-Q-M и -W-V-U, находятся в транс-положении друг к другу, если n представляет собой целое число 1, и где конфигурации в положениях 3 и 4 пиперидинового цикла формулы (I) представляют собой 3R и 4R соответственно, если n представляет собой целое число 0; и к оптически чистым энантиомерам, смеси энантиомеров, такие как рацематы, диастереомеры, смеси диастереомеров, диастереомерные рацематы, смеси диастереомерных рацематов, и мезоформы, а также к солям таких соединений. Изобретение также относится к фармацевтической композиции. Технический результат - получение новых биологически активных соединений, обладающих активностью непептидных ингибиторов ренина. 2 н. и 10 з.п. ф-лы, 1 табл.
1. Соединение, выбранное из группы, состоящей из амидов пиперидинкарбоновой кислоты формулы (I)
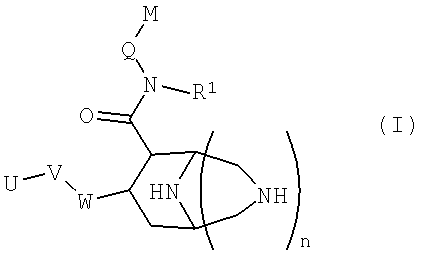
в которой W представляет собой фенильный цикл или шестичленный, небензоконденсированный ароматический цикл, содержащий один атом азота, где указанные циклы замещены в параположении посредством V;
V представляет собой связь, -A-(CH2)s- или -A-(CH2)v-B-;
А и В независимо представляют собой -O-;
U представляет собой моно-, ди-, три- или тетразамещенный арил, в котором заместители независимо выбраны из группы, состоящей из галогена, алкила и -CF3;
Q представляет собой метилен;
М представляет собой арильную группу, где указанная группа может быть необязательно моно- или дизамещенной заместителями, независимо выбранными из группы, состоящей из алкила; алкокси; -CF3; галогена; алкил-O-(СН2)0-4-СН2- и R'2N-(CH2)0-4-CH2-, где R' независимо выбран из группы, состоящей из водорода, алкила (необязательно замещенного одним, двумя или тремя атомами фтора), циклопропила, циклопропилметила, -C(=O)-R'', где R'' означает С1-С4-алкил или -CH2-CF3;
R1 представляет собой циклоалкил;
n представляет собой целое число 0 или 1;
s представляет собой целое число 3;
v представляет собой целое число 2;
и заместители в цикле, -CON(R1)-Q-M и -W-V-U, находятся в трансположении друг к другу, если n представляет собой целое число 1, и где конфигурации в положениях 3 и 4 пиперидинового цикла формулы (I) представляют собой 3R и 4R соответственно, если n представляет собой целое число 0;
и оптически чистые энантиомеры, смеси энантиомеров, такие, как рацематы, диастереомеры, смеси диастереомеров, диастереомерные рацематы, смеси диастереомерных рацематов и мезоформы, а также соли таких соединений.
2. Соединение по п.1, в котором М представляет собой арильную группу, где указанная группа может быть необязательно моно- или дизамещенной заместителями, независимо выбранными из группы, состоящей из алкила; алкокси; -CF3; галогена; алкил-O-(СН2)0-4-СН2- и R'2N-(СН2)0-4-СН2-, где R' независимо выбран из группы, состоящей из водорода, алкила, циклопропила, и -C(=O)O-R'', где R'' означает С1-С4-алкил или -СН2-CF3; или соль такого соединения.
3. Соединение по п.1, в котором М представляет собой следующий радикал:
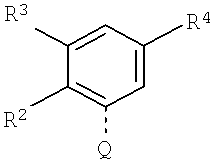
где R2 означает метил или хлор, R3 означает водород и R4 означает водород, -CH2CH2-О-СН3, -СН2СН2СН2-О-СН3 или R'NH-(CH2)0-1-CH2-, где R' означает -CH2-CHF2, -CH2-CF3, циклопропил, -СО-СН3, -СО-СН2-CF3 или -СО-СН2-СН3, при условии, когда R4 означает водород, то R3 представляет собой метил, метокси, хлор;
или соль такого соединения.
4. Соединение по любому одному из пп.1-3, в котором W представляет собой фенил, замещенный на V в параположении, или следующий радикал:
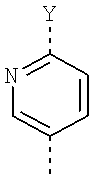
или соль такого соединения.
5. Соединение по любому одному из пп.1-3, в котором V представляет собой -CH2CH2CH2O-, где в обоих случаях бивалентный радикал присоединен к группе U формулы (I) через их атом кислорода, или -OCH2CH2O-; или его соль.
6. Соединение по любому одному из пп.1-3, в котором U представляет собой моно-, ди- или тризамещенный фенил, где заместители независимо выбраны из группы, состоящей из галогена, алкила, алкокси и -CF3; или его соль.
7. Соединение по любому одному из пп.1-3, в котором R' представляет собой циклопропил или его соль.
8. Соединение по п.1, выбранное из группы, включающей:
циклопропил(2,3-дихлорбензил)амид (1R*,5S*,6R*,7S*)-7-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}-3,9-диаза-бицикло[3.3.1]нонан-6-карбоновой кислоты,
циклопропил(3-метокси-2-метилбензил)амид 4-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
циклопропил(3-метокси-2-метилбензил)амид 4-{4-[3-(2,6-дихлор-4-метилфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
циклопропил(3-метокси-2-метилбензил)амид 4-{4-[3-(2,3,6-трифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2-хлор-3,6-дифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2,6-дихлор-4-метилфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2-хлор-6-фтор-3-метилфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты, и
циклопропил(2,3-диметилбензил)амид 4-{4-[3-(2,3,6-трифторфенокси)пропил]фенил}пиперидин-3-карбоновой кислоты,
или его соль.
9. Соединение по п.1, выбранное из группы, включающей:
циклопропил(2,3-диметилбензил)амид(3R*,4S*)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[2-хлор-5-(3-метоксипропил)бензил]циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[2-хлор-5-(2-метоксиэтил)бензил]циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[2-хлор-5-(3-метоксипропил)бензил]циклопропиламид(3R,4S)-6-[2-(2,6-дихлор-4-метилфенокси)этокси]-1',2',3',4',5',6'-гексагидро[3,4']бипиридинил-3'-карбоновой кислоты, и
[2-хлор-5-(2-метоксиэтил)бензил]циклопропиламид(3R,4S)-6-[2-(2,6-дихлор-4-метилфенокси)этокси]-1',2',3',4',5',6'-гексагидро[3,4']бипиридинил-3'-карбоновой кислоты, или его соль.
10. Соединение по п.1, выбранное из группы, включающей:
{2-хлор-5-[(2,2-дифторэтиламино)метил]бензил}циклопропиламид (3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил)пиперидин-3-карбоновой кислоты,
{2-хлор-5-[(3,3,3-трифторпропиониламино)метил]бензил}циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
{2-хлор-5-[(циклопропилметиламино)метил]бензил}циклопропиламид (3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[5-(ацетиламинометил)-2-хлорбензил]циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[2-хлор-5-(2-метиламиноэтил)бензил]циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
(2-хлор-5-метиламинометилбензил)циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
{2-хлор-5-[2-(3,3,3-трифторпропиониламино)этил]бензил}циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[2-хлор-5-(2-этиламиноэтил)бензил]циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
[5-(2-ацетиламиноэтил)-2-хлорбензил]циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
{2-хлор-5-[(2-фторэтиламино)метил]бензил}циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
(2-хлор-5-этиламинометилбензил)циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты,
(2-хлор-5-циклопропиламинометилбензил)циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты, и
[2-хлор-5-(2-циклопропиламиноэтил)бензил]циклопропиламид(3R,4S)-4-{4-[2-(2,6-дихлор-4-метилфенокси)этокси]фенил}пиперидин-3-карбоновой кислоты, или его соль.
11. Фармацевтическая композиция, которая обладает активностью непептидных ингибиторов ренина, содержащая соединение по любому одному из пп.1-10 или его фармацевтически приемлемую соль, и фармацевтически приемлемый продукт в виде носителя.
12. Соединение по пп.1-10, или его фармацевтически приемлемая соль, или композиция по п.11 для применения в качестве лекарственного средства, которое обладает активностью непептидных ингибиторов ренина.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| GULLER R | |||
| et al | |||
| BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Резервуар для керосиновых ламп типа "Примус" | 1924 |
|
SU1403A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 2167865 C2, 27.05.2001. | |||

