Эта заявка утверждает преимущество предварительной заявки на патент США №60/569510, поданной 6 мая 2004 г. и включенной в данное изобретение путем ссылки.
Это изобретение относится к химическим составам, которые являются ингибиторами одного или более митотических кинезинов и могут быть использованы для лечения клеточно-пролиферативных заболеваний, например рака, гиперплазии, рестеноза, кардиальной гипертрофии, нарушений иммунитета, грибковых заболеваний и воспаления.
Среди терапевтических агентов, используемых для лечения рака можно назвать таксаны и винкоалкалоиды, которые действуют на микротрубочки. Микротрубочки представляют собой первичный структурный элемент митотического веретена. Митотическое веретено отвечает за распределение реплицированных копий генома каждой из двух дочерних клеток, возникающих при делении клетки. Считается, что разрыв митотического веретена этими лекарственными препаратами вызывает ингибирование деления раковой клетки и индуцирует гибель раковых клеток. Однако микротрубочки формируют другие типы клеточных структур, включая пути для внутриклеточного транспорта в процессах нервной деятельности. Неспецифичность воздействия указанных терапевтических агентов на митотические веретена приводит к возникновению побочных эффектов, ограничивающих возможности применения этих агентов.
Значительный интерес к проблеме повышения специфичности агентов, используемых для лечения рака, объясняется предполагаемым благотворным влиянием, которое может быть получено в результате уменьшения побочных эффектов, связанных с назначением этих агентов. Традиционно значительные успехи в лечении рака связывают с идентификацией терапевтических агентов, действующих через новые механизмы. В качестве примеров можно назвать не только таксаны, но также и класс камптотецинов, являющихся ингибиторами топоизомеразы I. В свете обеих этих перспектив митотические кинезины представляются привлекательными мишенями для новых противораковых агентов.
Митотические кинезины - это ферменты, необходимые для образования и функционирования митотического веретена, но, как правило, не только в составе других структур из микротрубочек, типа участвующих в процессах нервной деятельности. Митотические кинезины играют важнейшие роли на всех фазах митоза. Эти ферменты являются "молекулярными моторами", преобразующими энергию, выделяемую в результате гидролиза АТР, в механическую силу, которая обеспечивает направленное движение клеточных грузов по микротрубочкам. Каталитический домен, достаточный для выполнения этой задачи, представляет собой компактную структуру приблизительно из 340 аминокислот. В процессе митоза кинезины организовывают микротрубочки в биполярную структуру, являющуюся митотическим веретеном. Кинезины участвуют в движении хромосом по микротрубочкам веретена, а также в структурных изменениях в митотическом веретене, связанных с определенными фазами митоза. Экспериментальное нарушение митотической функции кинезина вызывает мальформацию или дисфункцию митотического веретена, часто заканчивающуюся арестом клеточного цикла и гибелью клеток.
В одном аспекте изобретение относится к способам лечения клеточно-пролиферативных заболеваний и лечения нарушений путем ингибирования активности одного или более митотческих кинезинов.
Обеспечивается по крайней мере один химический состав, выбранный из соединений по формуле I
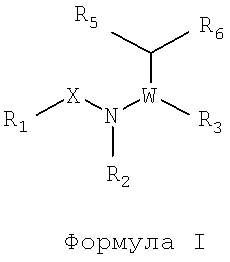
и фармацевтические приемлемых солей, сольватов, хелатов, нековалентных комплексов, пролекарств и их смесей, в которой
R1 - возможно замещенный арил, возможно замещенный гетероциклоалкил или возможно замещенный гетероарил;
Х - СО или -SO2-;
R2 - водород или возможно замещенный низший алкил;
W - CR4-, -CH2CR4- или N;
R3 - CO-R7, водород, возможно замещенный алкил, возможно замещенный гетероциклил, циано, возможно замещенный сульфонил или возможно замещенный арил;
R4 - водород или возможно замещенный алкил;
R5 - водород, гидроксильная группа, возможно замещенная амино группа, возможно замещенный гетероциклил или возможно замещенный низший алкил;
R6 - водород, возможно замещенный алкил, возможно замещенная алкокси группа, возможно замещенная арилоксигруппа, возможно замещенная гетерарилокси группа, возможно замещенный алкоксикарбонил-, возможно замещенный аминокарбонил-, возможно замещенный арил, возможно замещенный гетероарил, возможно замещенный гетероциклил или возможно замещенный аралкил и
R7 - возможно замещенный низший алкил, возможно замещенный арил, гидроксильная группа, возможно замещенная амино группа, возможно замещенная аралкокси группа или возможно замещенная алкокси группа.
В некоторых примерах осуществления изобретения, если W является N, тогда R5 не является гидроксильной группой или возможно замещенным амино, и R6 не является возможно замещенной алкокси группой, возможно замещенной аралкокси группой, возможно замещенной гетероаралкокси группой или возможно замещенным амино.
Также представлен по крайней мере один химический состав, выбранный из соединений по формуле II
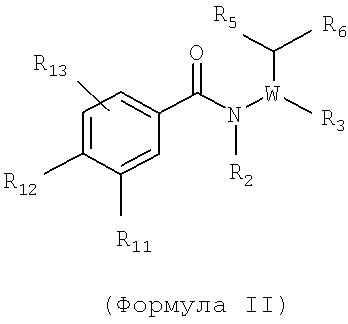
и фармацевтически приемлемых солей, сольватов, хелатов, нековалентных комплексов, пролекарств и их смесей, в которой R2, R3, R5, R6 и W такие же, как описано для соединений по формуле I и при этом
R11 - возможно замещенный гетероциклил, возможно замещенный низший алкил, нитро, циано, водород, сульфонил или гало;
R12 - водород, гало, возможно замещенный алкил, возможно замещенный амино, возможно замещенный сульфанил, возможно замещенная алкокси группа, возможно замещенный арилокси, возможно замещенный гетероциклил или возможно замещенный гетероарилокси и
R13 - водород, ацил, возможно замещенный алкил-, возможно замещенный алкокси, гало, гидроксильная группа, нитро, циано, возможно замещенный амино, алкилсульфонил-, алкилсульфонамидо-, алкилсульфонил-, карбоксиалкил-, аминокарбонил-, возможно замещенный арил или возможно замещенный гетероарил.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле III
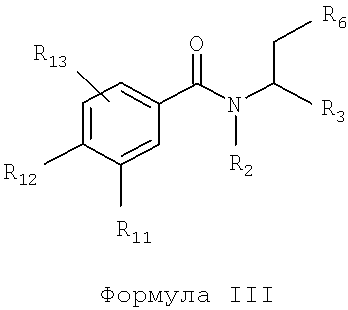
и фармацевтически приемлемых солей, сольватов, хелатов, нековалентных комлексов, пролекарств и их смесей, в которой R2, R3, R6, R11, R12 и R13 такие же, как описано для соединений по формуле II.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле IV
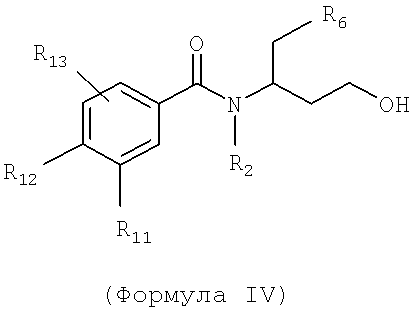
и фармацевтически приемлемых солей, сольватов, хелатов, нековалентных комплексов, пролекарств и их смесей, где R2, R6, R11, R12 и R13 такие же, как описанные для соединений по формуле III.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле V
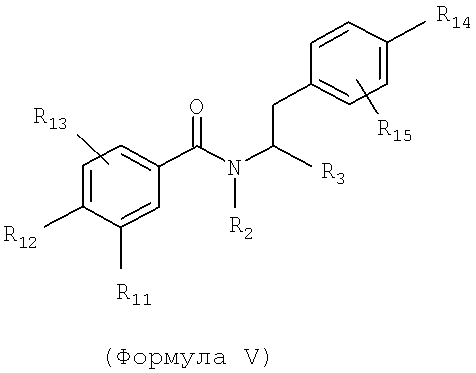
и фармацевтически приемлемых солей, сольватов, хелатов, нековалентных комлексов, пролекарств и их смесей, в которых R2, R3, R11, R12 и R13 такие же, как описано для соединений по формуле III, при этом
R14 - возможно замещенный гетероарил и
R15 - выбран из водорода, гало, гидроксила и низшего алкила.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле VI
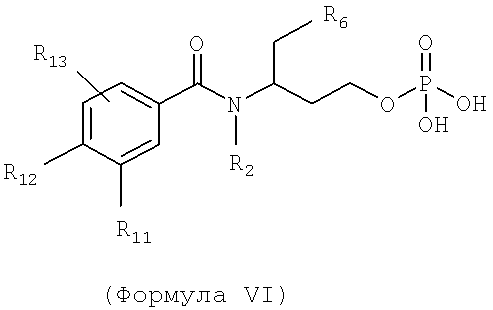
и фармацевтически приемлемых солей, сольватов, хелатов, нековалентных комплексов, пролекарств и их смесей, в которой R2, R6, R11, R12 и R13 такие же, как описано для соединений по формуле III.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле VII
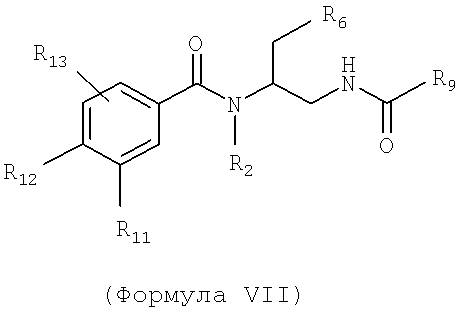
и фармацевтически приемлемых солей, сольватов, хелатов, нековалентных комплексов, пролекарств и их смесей, в которой R2, R6, R11, R12 и R13 такие же, как и описанные для соединений по формуле III, при этом
R9 выбран из возможно замещенной алкокси группы, возможно замещенной циклоалкокси группы, возможно замещенной арилалкокси группы, возможно замещенной амино группы и возможно замещенного низшего алкила.
Также предусмотрена композиция, включающая фармацевтический наполнитель и по крайней мере один описанный здесь химический состав.
Также предусматривется способ модулирования активности кинезина CENP-E, включающий взаимодействие вышеупомянутого кинезина с эффективным количеством по крайней мере одного описанного здесь химического состава.
Также предусматривается способ ингибирования CENP-E, включающий вступление вышеупомянутого кинезина в контакт с эффективным количеством по крайней мере одного описанного здесь химического состава.
Также предусматривается способ лечения клеточно-пролиферативного заболевания, включающий назначение нуждающемуся в этом субъекту по крайней мере одного описанного здесь химического состава.
Также предусматривается способ лечения клеточно-пролиферативного заболевания, включающий назначение нуждающемуся в этом субъекту композиции, включающей фармацевтический наполнитель и по крайней мере один описанный здесь химический состав.
Также предусматривается использование по крайней мере одного описанного здесь химического состава при изготовлении медикамента для лечения клеточно-пролиферативного заболевания.
Также предусматривается использование по крайней мере одного описанного здесь химического состава для изготовления медикамента для лечения нарушения, связанного с активностью кинезина CENP-E.
При использовании в данном описании следующие слова и фразы, как правило, имеют указываемые ниже значения, за исключением специально оговариваемых случаев. Следующие сокращения и термины имеют указанные значения по всему тексту.
При использовании в данном тексте в случае, если любая переменная встречается более одного раза в химической формуле, ее определение в любом случае употребления не зависит от ее определения в каждом другом случае.
Следующие сокращения и термины имеют указанные значения по всему тексту:
Черточка (« - »), расположенная не между двумя буквами или символами, используется для указания точки присоединения для заместителя. Например, -CONH2 присоединяется через атом углерода.
Возможный или возможно означает, что описываемое впоследствии событие или обстоятельства могут произойти или не произойти и что описание включает в себя случаи, в которых указанное событие или обстоятельства происходят, и случаи, в которых этого не происходит.Например, "возможно замещенный алкил" включает в себя, как определено в данном изобретении, "алкил" и "замещенный алкил".
Специалистам в данной области техники должно быть очевидно относительно любой группы, содержащей один или более заместителей, что такие группы не предназначены для введения любого замещения или замещающих структур, стерические свойства которых невозможно использовать и/или которые нельзя синтезировать и/или которые являются существенно нестабильными.
Алкил включает в себя линейные и разветвленные цепи, имеющие указанное число атомов углерода, обычно от 1 до 20 аотомов углерода, например от 1 до 8 атомов углерода, подобно от 1 до 6 атомов углерода. Например, C1-С6 алкил включает как линейные, так и разветвленные цепи алкила с от 1 до 6 атомами углерода. В качестве примеров алкильных групп можно назвать метил, этил, пропил, изопропил, n-бутил-, sec-бутил, tert-бутил, пентил, 2-пентил, изопентил, неопентил, гексил, 2-гексил, 3-гексил, 3-метилпентил и т.п. Алкилен является другой подгруппой алкила, относящийся к тем же остаткам, что и алкил, но имеющий две точки присоединения. Алкиленовые группы обычно имеют от 2 до 20 атомов углерода, например от 2 до 8 атомов углерода, подобно от 2 до 6 атомов. Например алкилен С0 указывает ковалентную связь и алкилен C1 алкилен - метиленовая группа. Когда указан остаток алкила, имеющий конкретное число атомов углерода, предполагается, что включаются все геометрические изомеры, имеющие это число атомов углерода; таким образом, например, предполагается, что «бутил» включает n-бутил, sec-бутил, изобутил и t-бутил; «пропил» включает n-пропил и изопропил. «Низший алкил» относится к алкильным группам, имеющим от одного до четырех атомов углерода.
Циклоалкил указывает на насыщенные циклические углеводородные группы с определенным количеством атомов углерода, обычно от 3 до 7. В качестве примеров циклоалкильных групп можно назвать циклопропил, циклобутил, циклопентил и циклогексил, а также мостовые и клеточные насыщенные циклические группы, такие как норборнан.
Алкоксигруппа относится к алкильной группе, включающей в свой состав указанное число атомов углерода, присоединенных к основной структуре через кислород, как, например, метоксигруппа, этоксигруппа, пропоксигруппа, изопропоксигруппа, n-бутоксигруппа, sec-бутоксигруппа, терт-бутоксигруппа, пентоксигруппа, 2-пентилоксигруппа, изопентоксигруппа, неопентоксигруппа, гексоксигруппа, 2 - гексоксигруппа, 3 - гексоксигруппа, 3-метилпентоксигруппа и т.п. Алкоксигруппы обычно имеют от 1 до 6 атомов углерода, присоединенных через кислород. «Низшая алкоксигруппа» относится к алкоксигруппам, содержащим от одного до четырех углеродов.
Ацил относится к группам (алкил)-С(О)-; (циклоалкил)-С(О); (арил)-С(О)-; (гетероарил)-С(О)- и (гетероциклоалкил)-С(О), при этом группа присоединена к основной структуре через карбонильную функциональную группу и при этом алкил, циклоалкил, арил, гетероарил и гетероциклоалкил соответствуют приводимому здесь описанию. Ацильные группы имеют указанное число атомов углерода, при этом углерод кетогруппы включен в пронумерованные атомы углерода. Например, ацильная группа С2 представляет собой ацетильную группу, имеющую формулу СН3(С=O)-.
Алкоксикарбонил означает эфирную группу формулы (алкокси группа) (С=O)-, присоединяемую через карбонил углерод, при этом алкокси группа имеет указанное число атомов углерода. Таким образом алкоксикарбонильная группа C1-С6 представляет собой алкокси группу, имеющую от 1 до 6 актомов углерода, присоединяемых через ее кислород к карбонильному связывающему звену.
Аминогруппа относится к группе -NH2.
Термин «аминокарбонил» относится к группе -CONRbRc, где
Rb выбран из Н, возможно замещенного C1-С6 алкила возможно замещенного арила и возможно замещенного гетероарила;
Rc выбран из водорода и возможно замещенного С1-C4 алкила или
Rb и Rc, взятые вместе с азотом, к которому они присоединяются, образуют возможно замещенный от 5 до 7 членный азотосодержащий гетероциклил, который возможно включает 1 или 2 дополнительных гетероатома, выбранных из О, N и S в гетероциклоалкильном кольце,
при этом каждая замещенная группа независимо замещается одним или более заместителей, независмо выбранных из С1-C4 алкила, арила, гетероарила, арил-С1-С4 алкила-, гетероарил-С1-С4 алкила-, С1-C4 галоалкила, -ОС1-C4алкила, -ОС1-C4алкилфенила, -С1-C4 алкил-ОН, -ОС1-C4 галоалкила, гало, -ОН, -NH2, -С1-C4 алкил - NH2, -N(С1-C4 алкил) (С1-C4алкил), - NH(С1-C4 алкил), -N(С1-C4 алкил), (С1-C4 алкилфенил), -NH(С1-C4 алкилфенил), цианогруппы, нитрогруппы, оксогруппы (в качестве заместителя для гетероарила), -СО2Н, -C(O)OC1-C4 алкила, -CON(С1-C4 алкил)(C1-C4 алкил), CONH(С1-C4 алкил), -CONH2, -NHC(O)(С1-C4 алкил), -NHC(O)(фенил). -N(С1-C4 алкил)С(O)(С1-C4 алкил), -N(С1-C4 алкил)С(O)(фенил), -C(O)С1-C4 алкила, -С(O)С1-C4 фенила, -C(O)С1-C4 галогеналкила, -OC(O)С1-C4 алкила, - SO2(С1-C4 алкил), -SO2(фенил), -SO2(С1-C4 галогеналкил), -SO2NH2, -SO2NH(С1-C4 алкил), -SO2NH(фенил), -NHSO2(С1-C4 алкил), -NHSO2(фенил) и -NHSO2(С1-C4 галогеналкил).
Арил включает
5- или 6-членные карбоциклические ароматические кольца, например бензол; бициклические кольцевые системы, в которых только одно из колец является карбоциклическим ароматическим кольцом, например нафталином, инданом и тетралином; трициклические кольцевые системы, в которых по крайней мере одно кольцо является карбоциклическим ароматическим кольцом, например, фтором.
Например, арил включает 5- и 6-членные карбоциклические ароматические кольца, присоединенные к 5-7-членному кольцу гетероциклоалкила, содержащему 1 или более гетероатомов, выбранных из N, O и S. Для подобных присоединенных бициклических кольцевых систем, в которых только одно из колец является карбоциклическим ароматическим кольцом, точка присоединения может находиться у карбоциклического ароматического кольца или гетероциклоалкильного кольца. Бивалентные радикалы, образованные из замещенных производных бензола и имеющие свободную валентность у атомов кольца, называются замещенными радикалами фенилина. Бивалентные радикалы, полученные из унивалентных полициклических радикалов углеводорода, названия которых оканчиваются на «-ил», путем удаления одного атома водорода их атома углерода со свободной валентностью, называются путем добавления «-идин» к имени, соответствующему унивалентному радикалу, например группа нафтила с двумя точками присоединения называется нафтилидин. Однако арил не включает или не перекрывается никаким образом с гетероарилом, который определен далее отдельно. Следовательно, если одно или более карбоциклических ароматических колец объединяются с гетероциклоалкильным ароматическим кольцом, получаемая в результате кольцевая система является гетероарилом, а не арилом, по приведенному здесь определению
Термин «арилокси группа» относится к группе -O-арил.
Аралкил- относится к остатку, в котором арильный компонент присоединен к основной структуре через алкильный остаток. В качестве примеров можно назвать бензил-, фенетил-, фенилвинил-, фенилаллил- и т.п.
Гетероаралкил относится к остатку, в котором гетероарильный компонент присоединен к основной структуре через алкильный остаток. В качестве примеров можно назвать фуранилметил-, пиридинилметил-, пиримидинилэтил- и т.п.
Галогруппа (гало) относится к фторогруппе, хлорогруппе, бромогруппе и иодогруппе, и термин «галоген» включает фтор, хлор, бром или иод.
Галоалкил указывает на определенный выше алкил, имеющий указанное число атомов углерода, замещенных одним или более атомов галогена, до максимально допустимого числа атомов галогена. Примеры галоалкила включают трифторометил, дифторометил, 2-фтороэтил и пентафтороэтил, но не ограничиаются ими.
Гетероарил включает:
от 5 до 7-членные ароматические моноциклические кольца, содержащие один или более, например от 1 до 4, или в некоторых примерах осуществления изобретения от 1 до 3 гетероатомов, выбранных из N, О и S, при этом остальные атомы кольца являются углеродом, и
бициклические гетероциклоалкильные кольца, содержащие один или более, например от 1 до 4, или в некоторых примерах осуществления изобретения от 1 до 3 гетероатомов, выбранных из N, О и S, при этом остальные атомы кольца являются углеродом; и в которых по крайней мере один гетероатом присутствует в ароматическом кольце.
Например, гетероарил включает от 5 до 7-членное ароматическое кольцо гетероциклоалкила, объединенное с 5 до 7-членным кольцом циклоалкила. Для подобных объединенных бициклических гетероарильных кольцевых систем, в которых только одно из колец содержит один или более гетероатомов, точка присоединения может быть у гетероароматического кольца или циклоалкильного кольца. Когда общее число атомов S и О в гетероарильной группе превышает 1, эти гетероатомы не примыкают друг к другу. В некоторых примерах осуществления изобретения общее число атомов S и О в гетероарильной группе равно не более 2. В некоторых примерах осуществления изобретения общее число атомов S и О в гетероарильной группе равно не более 1. Примеры гетероарильных групп включают (пронумерованные по назначенному приоритету от позиции связи) 2-пиридил, 3-пиридил, 4-пиридил, 2,3-пиразинил, 3,4-пиразинил, 2,4-пиримидинил, 3,5-пиримидинил, 2,4-имидазолинил, изоксазолинил, оксазолинил, тиазолинил, тиадиазолинил, тетразолил, тиенил, бензотиофенил, фуранил, бензофуранил, бензоимидазолинил, индолинил, пиридизинил, хинолинил, пиразолил, имидазопиридинил и 5,6,7,8-тетрагидроизохинолин, но не ограничиваются ими. Бивалентные радикалы, полученные от унивалентных радикалов гетероарила, чье название оканчивается на «ил», при удалении одного атома водорода из атома со свободной валентностью называются прибавлением окончания «-идин» - к названию соответствующего унивалентного радикала, например группа пиридила с двумя точками присоединения является пиридилидином. Гетероарил не включает и не перекрывается с арилом, определенным выше.
В термине «гетероаралкил» гетероарил и алкил соответствуют приведенному выше определению и точкой присоединения является алкильная группа. Термин включает пиридилметил, тиофнилметил и (пирролил)1-этил, но не ограничиваются ими.
Уходящая (отщепляемая) группа или уходящий атом - любая группа или атом, которые отщепляются в условиях реакции от исходного материала, обеспечивая таким образом продвижение реакции на определенном участке. Подходящими примерами таких групп, если не определено другого, являются атомы галогена, мезилоксигруппа, р-нитробензолсульфонилоксигруппа и тозилоксигруппа.
Возможный или возможно означает, что впоследствии описываемое событие или обстоятельства могут произойти или не произойти и что описание включает в себя случаи, в которых указанное событие или обстоятельства происходят, и случаи, в которых этого не происходит. Например, "возможно замещенный алкил" включает в себя, как определено в данном изобретении, "алкил" и "замещенный алкил". Специалистам в данной области техники должно быть очевидно относительно любой группы, содержащей один или более заместителей, что такие группы не предназначены для введения любого замещения или замещающих структур, стерические свойства которых невозможно использовать, и/или которые нельзя синтезировать, и/или которые являются существенно нестабильными.
Защитная группа имеет значение, традиционно ассоциируемое с такой группой в органическом синтезе, т.е. группой, которая избирательно блокирует один или более активных участков в многофункциональном соединении так, чтобы химическая реакция могла бы протекать избирательно на другом незащищенном активном участке и чтобы группу можно было легко удалить после завершения избирательной реакции. Множество защитных групп рассмотрено, например, в книге Т.Х.Грина и П.Г.Ватса, Защитные группы в органическом синтезе, третье издание, Джон Уили и Сыновья, Нью-Йорк, 1999 г. (Т.Н.Greeene and P.G. Wuts, Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons, New York, 1999), которая включена в данный материал полностью путем ссылки. Например, гидроксильная защитная форма это форма, в которой по крайней мере одна гидроксильная группа, присутствующая в растворе, защищена гидроксильной защитной группой. Подобным же образом амины и другие группы реакций могут быть подобным же образом защищены.
Гетероциклоалкил означает отдельное алифатическое кольцо, обычно с 3 по 7 атомов кольца, содержащее по крайней мере 2 атома углерода в дополнении к 1-3 гетероатомам, независимо выбранным из кислорода, серы и азота, а также комбинации, включающие по крайней мере один из ранее упомянутых гетероатомов. Приемлемые группы гетероциклоалкила включают, например (пронумерованные по приоритету 1 назначенному по позиции связи), 2-пирролинил, 2,4-имидазолидинил, 2,3-пиразолидинил, 2-пиперидил, 3-пиперидил, 4-пиперидил и 2,5-пиперзинил. Также предусматриваются морфолиниловые группы, включающие 2-морфолинил и 3-морфолинил (пронумерованные по приоритету 1 назначенному кислороду).
При использовании в данной работе термин модуляция относится к изменению активности CENP-E в качестве прямого или косвенного отклика на присутствие по крайней мере одного описанного здесь химического состава относительно активности CENP-E в отсутствие химического состава. Изменением может быть увеличение активности или снижение активности и это может быть вызвано косвенным вхаимодействием химического состава с CENP-E или взаимодействием соединения с одним или более факторов, которые влияют на активность CENP-E.
Сульфанил относится к группам -S-(возможно замещенный (C1-С6) алкил), -S-(возможно замещенный арил), -S-(возможно замещенный гетероарил) и -S-(возможно замещенный гетероциклоалкил). Следовательно, сульфанил включает группу C1-С6алкилсульфанил.
Сульфинил относится к группам -S(O)-H, -S(O)-(возможно замещенный (C1-С6) алкил), -S(O)-(возможно замещенный арил), -S(O)-(возможно замещенный гетероарил), -S(O)-(возможно замещенный гетероциклоалкил) и -S(O)-(возможно замещенная аминогруппа).
Сульфонил относится к группам -S(O2)-H, -S(O2)-(возможно замещенный (C1-С6) алкил), -S(O2)-(возможно замещенный арил), -S(O2)-(возможно замещенный гетероарил), -S(O2)-(возможно замещенный гетероциклоалкил), -S(O2)-(возможно замещенная алкоксигруппа), -S(O2)-(возможно замещенная арилоксигруппа), -S(O2)-(возможно замещенная гетероарилоксигруппа), -S(O2)-(возможно замещенная гетероциклилоксигруппа) и -S(O2)-(возможно замещенная аминогруппа).
Термин «замещенный» при использовании в данной работе означает, что любой один или более атомов водорода в обозначенном атоме или группе замещается выбором из указанной группы при условии, что не превышается нормальная валентность обозначенного атома. Когда заместителем является оксо (т.е. =О), тогда замещается 2 водорода на атоме. Комбинации заместителей и/или переменных допустимы только тогда, когда подобные комбинации приводят к стабильным соединениям или полезным синтетическим посредникам. Стабильное соединение или стабильная структура подразумевает соединение, которое является достаточно прочным, чтобы пережить выделение из реактивной смеси и последующее формирование в качестве агента, имеющего по крайней мере практическую пользу. Если не определено иначе, заместители именуются в корневой структуре. Например, следует понимать, что когда (циклоалкил)алкил перечислен в качестве возможного заместителя, точкой присоединения этого заместителя к корневой структуре является алкильная часть.
Термины «замещенный» алкил, циклоалкил, арил, гетероциклоалкил и гетероарил, если не определено иначе, относятся к алкилу, циклоалкилу, арилу, гетероциклоалкилу или гетероарилу, где один или более (до 5, например до 3) атомов водорода замещаются заместителем независимо выбранным из
-Ra, -ORb, -O(C1-C2алкил)O- (например, метилендиокси-), -SRb, гуанидин, гуанидин, в котором один или более водородов гуанидина замещен группой низшего алкила,
-NRbRc, гало, циано, нитрогруппой, -CORb, -CO2Rb, -CONRbRc, -OCORb, -OCO2Ra, -OCONRbRc, -NRCCORb, -NRcCO2Ra, -NRcCONRbRc, -CO2Rb, -CONRbRc, -NRcCORb, -SORa, -SO2Ra, SO2NRbRc, -NRcSO2Ra,
при этом Ra выбрана из возможно замещенного C1-С6алкила, возможно замещенного арила и возможно замещенного гетероарила;
Rb выбрана из Н, возможно замещенного C1-С6алкила, возможно замещенного арила и возможно замещенного гетероарила и
Rc выбран из водорода и возможно замещенного C1-C4 алкила,
где каждая возможно замещенная группа незамещена или независимо замещена одним или более, подобно одному, двум или трем, заместителями, независимо выбранными из C1-C4 алкила, арила, гетероарила, арил-C1-C4 алкила, гетероарил-C1-C4 алкила; C1-C4 галоалкила-, -OC1-C4 алкила, OC1-C4 алкилфенила, -C1-C4 алкил-ОН, OC1-C4 галоалкила, гало, ОН, -NH2, -С1-С4 алкил-NH2-N(С1-С4 алкил) (C1-C4 алкил), -NH(C1-C4 алкил), -N(C1-C4 алкил)(С1-С4 алкилфенил), -NH(C1-C4 алкилфенил), циано, нитрогруппа, оксо (в качестве заместителя для гетероарила), СО2Н, -C(O)OC1-C4 алкил, -CON(C1-C4 алкил)(C1-C4 алкил), -CONH(C1-C4 алкил), -CONH2, -NHC(O)(C1-C4 алкил), -NHC(O)(фенил), -N(C1-C4 алкил)C(O)(C1-C4 алкил), -Н(С1-С4 алкил)С(O)(фенил), -С(O)C1-C4 алкил, -C(O)(C1-C4 фенил, -С(O)C1-C4 галоалкил, -ОС(O)C1-C4 алкил, -SO2(С1-С4 алкил), -SO2(фенил), -SO2(С1-С4 галоалкил), -SO2NH2, -SO2NH(C1-C4 алкил), SO2NH(фенил), -NHSO2(C1-C4 алкил), -NHSO2(фенил) и -NHSO2(C1-C4 галоалкил).
Термин «замещенный ацил» относится к группам (замещенный алкил)-С(О)-; (замещенный циклоалкил)-С(О)-; (замещенный арил)-С(О)-; (замещенный гетероарил)-С(O) и (замещенный гетероциклоалкил)-С(О)-, в которых группа присоединяется к основной структуре через карбонильную функциональную группу и в которых алкил, циклоалкил, арил, гетероарил и гетероциклоалкил соответственно относятся к алкилу, циклоалкилу, арилу, гетероарилу и гетероциклоалкилу и при этом один или более (как до 5, например до 3) атомов водорода замещаются заместителем независимо выбранным из:
-Ra, -ORb, -О(C1-C2алкил)O- (например, метилендиокси-), -SRb, гуанидин, гуанидин, в котором один или более атомов водорода гуанидина замещены группой низшего алкила, -NRbRc, гало, циано, нитро группой, -CORb, -CO2Rb, -CONRbRc, -OCORb, -OCO2Ra, -OCONRbRc, -NRcCORb, -NRcCO2Ra, -NRcCONRbRc, -CO2Rb, -CONRbRc, -NRcCORb, -SORa, -SO2Ra, SO2NRbRc, -NRcSO2Ra,
где Ra выбрана из возможно замещенного C1-С6 алкила, возможно замещенного арила и возможно замещенного гетероарила;
Rb выбрана из Н, возможно замещенного C1-С6 алкила, возможно замещенного арила и возможно замещенного гетероарила и
Rc выбран из водорода и возможно замещенного С1-С6 алкила,
при этом каждая возможно замещенная группа незамещена или независимо замещена одним или более, таким как один, два или три заместителя, независимо выбранным из C1-C4 алкила, арила, гетероарила, арил-C1-C4 алкила, гетероарил-C1-C4 алкила; C1-C4 галоалкила-, -OC1-C4 алкила, OC1-C4 алкилфенила, -C1-C4 алкил-ОН, OC1-C4 галоалкила, гало, ОН, -NH2, -С1-С4 алкил-NH2-N(С1-С4 алкил) (C1-C4 алкил), -NH(C1-C4 алкил), -N(C1-C4 алкил)(С1-С4 алкилфенил), -NH(C1-C4 алкилфенил), циано, нитро группа, оксо (в качестве заместителя для гетероарила), СО2Н, -C(O)OC1-C4 алкил, -CON(C1-C4 алкил)(C1-C4 алкил), -CONH(C1-C4 алкил), -CONH2, -NHC(O)(C1-C4 алкил), -NHC(O)(фенил), -N(C1-C4 алкил)C(O)(C1-C4 алкил), -N(С1-С4 алкил)С(O)(фенил), -С(O)C1-C4 алкил, -C(O)(C1-C4 фенил, -С(O)C1-C4 галоалкил, -ОС(O)C1-C4 алкил, -SO2(С1-С4 алкил), -SO2(фенил), -SO2(С1-С4 галоалкил), -SO2NH2, -SO2NH(C1-C4 алкил), SO2NH(фенил), -NHSO2(C1-C4 алкил), -NHSO2(фенил) и -NHSO2(C1-C4 галоалкил).
Термин «замещенная алкокси группа» относится к алкокси группе, при этом алкильная составляющая является замещенной (например, -O-(замещенный алкил)), где «замещенный алкил» относится к алкилу в котором один или более (подобно до 5, например до 3) атомов водорода замещены заместителем, незаивисимо выбранным из:
-Ra, -ORb, -О(C1-C2алкил)O- (например, метилендиокси-), -SRb, гуанидин, гуанидин, в котором один или более атомов водорода гуанидина замещены группой низшего алкила, -NRbRc, гало, циано, нитро группой, -CORb, -CO2Rb, -CONRbRc, -OCORb, -OCO2Ra, -OCONRbRc, -NRcCORb, -NRcCO2Ra, -NRcCONRbRc, -CO2Rb, -CONRbRc, -NRcCORb, -SORa, -SO2Ra, SO2NRbRc, -NRcSO2Ra,
где Ra выбрана из возможно замещенного C1-С6 алкила, возможно замещенного арила и возможно замещенного гетероарила;
Rb выбрана из Н, возможно замещенного C1-С6 алкила, возможно замещенного арила и возможно замещенного гетероарила и
Rc выбран из водорода и возможно замещенного С1-C4 алкила,
при этом каждая возможно замещенная группа незамещена или независимо замещена одним или более, таким как один, два или три заместителя, независимо выбранным из C1-C4 алкила, арила, гетероарила, арил-C1-C4 алкила, гетероарил-C1-C4 алкила; C1-C4 галоалкила-, -OC1-C4 алкила, OC1-C4 алкилфенила, -C1-C4 алкил-ОН, OC1-C4 галоалкила, гало, ОН, -NH2, -С1-С4 алкил-NH2-N(С1-С4 алкил) (C1-C4 алкил), -NH(C1-C4 алкил), -N(C1-C4 алкил)(С1-С4 алкилфенил), -NH(C1-C4 алкилфенил), циано, нитро группа, оксо (в качестве заместителя для гетероарила), СО2Н, -C(O)OC1-C4 алкил, -CON(C1-C4 алкил)(C1-C4 алкил), -CONH(C1-C4 алкил), -CONH2, -NHC(O)(C1-C4 алкил), -NHC(O)(фенил), -N(C1-C4 алкил)C(O)(C1-C4 алкил), -N(С1-С4алкил)С(O)(фенил), -С(O)C1-C4 алкил, -C(O)(C1-C4 фенил, -С(O)C1-C4 галоалкил, -ОС(O)C1-C4 алкил, -SO2(С1-С4алкил), -SO2(фенил), -SO2(С1-С4 галоалкил), -SO2NH2, -SO2NH(C1-C4 алкил), SO2NH(фенил), -NHSO2(C1-C4 алкил), -NHSO2(фенил) и -NHSO2(C1-C4 галоалкил). В некоторых примерах изобретения замещенная алкокси группа является «полиалкокси» или -O-(возможно замещенный алкилен)-(возможно замещенная алкокси группа), и включает такие группы как -ОСН2СН2ОСН3 и остатки эфиров гликоля, таких как полиэтиленгликоль и -O(CH2CH2O)х СН3, где х является целым числом 2-20, подобным 2-10 и, например, 2-5. Другая замещенная алкокси группа является гидроксиалкокси группой или -ОСН2(СН2)у ОН, где у является целым числом 1-10, подобно 1-4.
Термин «замещенный алкоксикарбонил» относится к группе (замещенный алкил)-О-С(О)-, в которой группа присоединяется к основной структуре через карбонильную функциональную группу и в которой замещенный относится к алкилу и при этом один или более (подобно до 5, например до 3) атомов водорода замещаются заместителем, независимо выбранным из:
-Ra, -ORb, -О(C1-C2алкил)O- (например, метилендиокси-), -SRb, гуанидин, гуанидин, в котором один или более атомов водорода гуанидина замещены группой низшего алкила, -NRbRc, гало, циано, нитро группой, -CORb, -CO2Rb, -CONRbRc, -OCORb, -OCO2Ra, -OCONRbRc, -NRcCORb, -NRcCO2Ra, -NRcCONRbRc, -CO2Rb, -CONRbRc, -NRcCORb, -SORa, -SO2Ra, SO2NRbRc и -NRcSO2Ra,
где Ra выбрана из возможно замещенного C1-С6 алкила, возможно замещенного арила и возможно замещенного гетероарила;
Rb выбрана из Н, возможно замещенного C1-С6 алкила, возможно замещенного арила и возможно замещенного гетероарила и
Rc выбран из водорода и возможно замещенного C1-C4 алкила,
где каждая возможно замещенная группа незамещена или независимо замещена одним или более, таким как один, два или три заместителя, независимо выбранных из C1-C4 алкила, арила, гетероарила, арил-C1-C4 алкила, гетероарил-C1-C4 алкила; C1-C4 галоалкила-, -OC1-C4 алкила, OC1-C4 алкилфенила, -C1-C4 алкил-ОН, OC1-C4 галоалкила, гало, ОН, -NH2, -С1-С4 алкил-NH2-N(С1-С4 алкил) (C1-C4 алкил), -NH(C1-C4 алкил), -N(C1-C4 алкил)(С1-С4 алкилфенил), -NH(C1-C4 алкилфенил), циано, нитро группа, оксо (в качестве заместителя для гетероарила), СО2Н, -C(O)OC1-C4 алкил, -CON(C1-C4 алкил)(C1-C4 алкил), -CONH(C1-C4 алкил), -CONH2, -NHC(O)(C1-C4 алкил), -NHC(O)(фенил), -N(C1-C4 алкил)C(O)(C1-C4 алкил), -Н(С1-С4 алкил)С(O)(фенил), -С(O)C1-C4 алкил, -C(O)(C1-C4 фенил, -С(O)C1-C4 галоалкил, -ОС(O)C1-C4 алкил, -SO2(С1-С4 алкил), -SO2(фенил), -SO2(С1-С4 галоалкил), -SO2NH2, - SO2NH(C1-C4 алкил), SO2NH(фенил), -NHSO2(C1-C4 алкил), -NHSO2(фенил) и -NHSO2(C1-C4 галоалкил).
Термин «замещенная амино группа» относится к группе -NHRd или -NRdRd, в которой каждое Rd незаивисимо выбрано из возможно замещенного алкила, возможно замещенного циклоалкила, возможно замещенного ацила, возможно замещенного арила, возможно замещенного гетероарила, возможно замещенного гетероциклоалкила, алкосикарбонила, сульфинила и сульфонила, при этом замещенный алкил, циклоалкил, арил, гетероциклоалкил и гетероарил соответственно относятся к алкилу, циклоалкилу, арилу, гетероциклоалкилу и гетероарилу, в которых один или более (подобно до 5, например до 3) атомов водорода независимо выбраны из:
-Ra, -ORb, -О(C1-C2 алкил)O- (например, метилендиокси-), -SRb, гуанидина, гуанидина, в котором один или более атомов водорода гуанидина замещены группой низшего алкила, -NRbRc, гало, циано, нитро группой, -CORb, -CO2Rb, -CONRbRc, -OCORb, -OCO2Ra, -OCONRbRc, -NRcCORb, -NRcCO2Ra, -NRcCONRbRc, -CO2Rb, -CONRbRc, -NRcCORb, -SORa, -SO2Ra, SO2NRbRc, -NRcSO2Ra,
где Ra выбран из возможно замещенного C1-С6 алкила, возможно замещенного арила и возможно замещенного гетероарила;
Rb выбран из Н, возможно замещенного C1-С6 алкила, возможно замещенного арила и возможно замещенного гетероарила и
Rc выбран из водорода и возможно замещенного С1-С4 алкила;
при этом каждая возможно замещенная группа незамещена или независимо замещена одним или более, таким как один, два или три заместителя, независимо выбранных из C1-C4 алкила, арила, гетероарила, арил-C1-C4 алкила, гетероарил-C1-C4 алкила; C1-C4 галоалкила-, -OC1-C4 алкила, OC1-C4 алкилфенила, -C1-C4 алкил-ОН, OC1-C4 галоалкила, гало, ОН, -NH2, -С1-С4 алкил-NH2-N(С1-С4 алкил) (C1-C4 алкил), -NH(C1-C4 алкил), -N(C1-C4 алкил)(С1-С4 алкилфенил), -NH(C1-C4 алкилфенил), циано, нитро группа, оксо (в качестве заместителя для гетероарила), СО2Н, -C(O)OC1-C4 алкил, -CON(C1-C4 алкил)(C1-C4 алкил), -CONH(C1-C4 алкил), -CONH2, -NHC(O)(C1-C4 алкил), -NHC(O)(фенил), -N(C1-C4 алкил)C(O)(C1-C4 алкил), -N(С1-С4 алкил)С(O)(фенил), -С(O)C1-C4 алкил, -C(O)(C1-C4 фенил, -С(O)C1-C4 галоалкил, -ОС(O)C1-C4 алкил, -SO2(С1-С4 алкил), -SO2(фенил), -SO2(С1-С4 галоалкил), -SO2NH2, -SO2NH(C1-C4 алкил), SO2NH(фенил), -NHSO2(C1-C4 алкил), -NHSO2(фенил) и -NHSO2(C1-C4 галоалкил), и
в которой возможно замещенный ацил, алкоксикарбонил, сульфинил и сульфонил соответствуют данному здесь определению.
Термин «замещенная амино группа» также относится к группе -NReRf, в которой Re и Rf, вместе с азотом, с которым они связаны, образуют возможно замещенный 5-7-членный азотсодержащий неароматический гетероциклил, который возможно содержит 1 или 2 дополнительных гетероатома, выбранных из азота, кислорода и серы.
Соединения по формуле I-XIII включают оптические изомеры соединений по формуле I-XIII, рацематы и их смеси, но не ограничиваются ими. В этих ситуациях отдельные энантиомеры или диастериомеры, т.е. оптически активные формы, могут быть получены путем асимметрического синтеза или расщеплением рацематов. Расщепление рацематов может быть осуществлено, например, такими обычными способами, как кристаллизация в присутствие расщепляющего агента, или хроматография с использованием, например, хиральной колонки жидкостной хромотографии высокого разрешения (ЖХВР). В дополнении к этому соединения по формуле I-XIII включают Z- и Е- формы (или формы cis и trans) соединений с двойной связью углерод-углерод. В случаях, когда соединения по формуле I-XIII существуют в различных таутомерных формах, химические составы по настоящему изобретению включают все таутрмерные формы соединения. Соединения по формуле I-XIII также включают кристаллические формы, такие как полиморфные формы и клатраты.
Химические составы по настоящему изобретению включают соединения по формуле I-XIII и все их фармацевтически приемлемые формы, но не ограничиваются ими. Фармацевтически приемлемые формы соединений, приведенные здесь, включают фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси. В некоторых примерах осуществления изобретения описанные здесь соединения имеют форму фармацевтически приемлемых солей. Следовательно термины химический состав и химические составы также включает фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси.
Фармацевтически приемлемые соли включают соли с неорганическими кислотами, такие как гидрохлорат, фосфат, дифосфат, гидробромат, сульфат, сульфинат, нитрат и подобные соли, а также соли органических кислот, такие как малат, малеат, фумарат, тартрат, сукцинат, цитрат, ацетат, лактат, метансульфонат, р-толуолсульфонат, 2-гидроксиэтилсульфонат, бензоат, салицилат, стеарат и алканоат, подобный ацетату, НООС-(СН2)n-СООН, в которых n представляет собой 0-4, и подобные соли, но не ограничиваются ими. Подобным же образом фармацевтически приемлемые катионы включают натрий, калий, кальций, алюминий, литий и аммоний, но не ограничиваются ими.
В дополнение, если соединение по формуле I-XIII получено в качестве соли с присоединением кислоты, свободная база может быть получена путем превращения в основание раствора кислой соли. И наоборот, если продукт является свободным основанием, соль присадки, в частности фармацевтически приемлемая соль присадки, может быть получена растворением свободного основания в приемлемом органическом растворителе и путем обработки раствора кислотой в соответствии с обычными процедурами приготовления солей с присоединением кислоты из соединений основы. Специалисты в данной области распознают различные методики синтеза, которые могут быть использованы для приготовления нетоксических фармацевтически приемлемых солей с присоединением кислоты.
Как отмечалось ранее, пролекарства также попадают в объем химических составов, например эфирные или амидные производные соединений по формуле I-XIII. Термин пролекарство включает любое соединение, которое становится соединением по формуле I-XIII при назначении пациенту, например, при метаболической обработке пролекарства. Примеры пролекарств включают ацетат, формат и бензоат и подобные производные функциональных групп (таких как спиртовые или аминовые группы) в соединениях по формуле I-XIII, но не ограничиваются ими. В некоторых примерах осуществления изобретения пролекарство является эфиром фосфата. Подробное рассмотрение пролекарств представлено в книге Т.Хигучи и В.Стела, Пролекарства в качестве новой системы доставки, том 14 серии симпозиумов A.C.S., 1987, в издании Эдварда Б.Роше (Т.Higuchi and V.Stella, Pro-drugs as Novel Delivery Systems, Vol.14 of the A.C.S. Symposium Series, in Edward B. Roche, ed.; Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, and Design of Prodrugs, ed.H.Bundgaad, Elsevier, 1985), Биореверсивные носители в конструировании лекарств, Американская фармацевтическая ассоциация и Пергамон Пресс, 1987, и в Конструировании пролекарств, издание X. Бундгаард, Элсевьер, 1985, каждая из которых включена в данный материал путем ссылки.
Термин «сольват» относится к химическому составу, получаемому в результате взаимодействия растворителя и соединения. Подходящие сольваты - фармацевтически приемлемые сольваты, такие как гидраты, включая моногидраты и хеми-гидраты.
Термин «хелат» относится к химическому составу, образованному координацией соединения с ионом металла в двух (или более) точках.
Термин «нековалентный комплекс» относится к химическому составу, образованному путем взаимодействия соединения и другой молекулы, при этом ковалентная связь между соединением и молекулой не формируется. Например, создание комплекса может произойти путем взаимодействий ван дер Ваалса (van der Waals), водородного связывания и электростатических взаимодействий (также называемых ионным связыванием).
Термин «активный агент» используется для указания химического состава, имеющего биологическую активность. В некоторых примерах осуществления изобретения активный агент может быть противораковым терапевтическим агентом.
Термин «антимитотический» относится к лекарственному средству для ингибирования или предотвращения митоза, например путем метафазного ареста. Некоторые противоопухолевые лекарственные препараты блокируют пролиферацию и считаются антимитотическими.
Термин «терапевтически эффективное количество химического состава» по данному изобретению означает количество, эффективное при назначениии человеку или пациенту, не принадлежащему к человеческому роду, для обеспечения терапевтических преимуществ, таких как ослабевание симптомов, замедление хода заболевания или профилактики заболевания, например, терапевтически эффективное количество может быть количеством, достаточным для снижения симптомов заболевания в ответ на ингибирование CENP-E. В некоторых примерах осуществления изобретения терапевтически эффективное количество - это количество, достаточное для снижения симптомов рака. В некоторых примерах осуществления изобретения терапевтически эффективное количество это количество, достаточное для снижения числа детектируемых раковых клеток в организме, заметно замедляющее или останавливающее рост раковой опухоли. В некоторых примерах осуществления изобретения терапевтически эффективное количество, это количество, достаточное для сужения раковой опухоли.
Термин «ингибирование» указывает на значительное уменьшение исходной активности биологическое деятельности или процесса. Ингибирование активности CENP-E относится к уменьшению активности CENP-E в качестве прямого или косвенного отклика на присутствие по крайней мере одного описанного здесь химического состава относительно активности CENP-E в случае отсутствия по крайней мере одного химического состава. Снижение активности может быть в результате прямого взаимодействия химического состава с CENP-E или в результате взаимодействия описанных здесь химических составов с одним или более факторов, влияющих на активность CENP-E. Например, присутствие химического состава(ов) может снизить активность CENP-E путем прямого связывания с CENP-E, вызывая (напрямую или косвенно) снижение активности CENP-E с помощью другого фактора или (напрямую или косвенно) путем уменьшения количества CENP-E, присутствующего в клетке или организме.
Заболевание, чувствительное к ингибированию CENP-E, представляет собой заболевание, при котором ингибирование активности CENP-E обеспечивает терапевтические преимущества, такие как ослабление симптомов, снижение прогрессирования заболевания, профилактики или замедления начала заболевания или ингибирования аберрантной активности некоторых типов клеток.
Лечение или проведение лечения означает любое лечение заболевания пациента, включающее:
а) профилактику заболевания, то есть прекращение развития клинических симптомов заболевания;
b) ингибирование болезни;
c) замедление или арест развития клинических симптомов и/или
d) облегчение заболевания, то есть способствование регрессии клинических симптомов.
Пациент относится к таким животным, как млекопитающее, которое было или будет объектом лечения, наблюдения или опытов. Способы изобретения могут быть полезны как при терапии человека, так и в ветеринарных применениях. В некоторых примерах осуществления изобретения пациент является млекопитающим; в некоторых примерах осуществления изобретения пациент является человеком и в некоторых примерах осуществления изобретения пациент выбран из кошек и собак.
Настоящее изобретение касается класса новых химических составов, являющихся ингибиторами одного или более митотических кинезинов. В соответствии с некотрыми примерами осуществления изобретения описываемые в данном изобретении химические составы ингибируют митотический кинезин, CENP-E, в частности человеческий CENP-E. CENP-E является направленным на конечный результат двигателем микротрубочек, имеющим существенное значение для достижения выравнивания метафазы хромосом. CENP-E накапливается во время промежуточной фазы и деградирует вслед за завершением митоза. Микроинъецирование антитела, направленного против CENP-E или чрезмерной экспрессии доминантного отрицательного мутанта CENP-E, вызывает митотческий арест при рассеянии прометафазы хромосом на биполярном веретене. Конечный домен CENP-E ослабляет локализацию кинетохор и также взаимодействует с киназой митотической контрольной точки hBubR1. CENP-E также связан с активными формами киназы MAP. Клонирование человеческого CENP-E было отмечено в отчете Йен и др., Природа, 359(6395): 536-9, 1992 (Yen et al., Nature, 359(6395): 536-9 (1992)). В работе Троуера и др., журнал ЕМВО, 14:918-26 (1995) (Thrower et al., EMBO J., 14:918-26 (1995) был отмечен частично очищенный природный человеческий CENP-E. Более того, исследования показывают, что CENP-E был направленным на конечный результат двигателем микротрубочек со знаком минус. Вуд и др., Клетка, 91: 357-66 (1997) (Wood et al., Cell, 91:357-66 (1997) раскрывают экспрессированный ксентопус CENP-E в E.coli и то, что XCENP-E имеет мотильность в качестве плюсового направленного на конечный результат мотора in vitro. CENP-E, см. публикацию РСТ № WO 99/13061, включенную в данную работу путем ссылки.
В некоторых примерах осуществления изобретения химические составы ингибируют митотический кинезин, CENP-E, а также модулируют один или более митотических человеческих кинезинов из числа HSET (см. патент США №6361993, включенный в данное изобретение путем ссылки); МСАК (см. патент США №6331424, включенный в данное изобретение путем ссылки); RabK-6 (см. патент США №6544766, включенный в данное изобретение пуьем ссылки); Kif4 (см. патент США №6440684, включенный в данное изобретение путем ссылки); MKLP1 (см. патент США №6448025, включенный в данное изобретение путем ссылки); Kif15 (см. патент США №6355466, включенный в данное изобретение путем ссылки); Kid (см. патент США №6387644, включенный в данное изобретение путем ссылки); Мрр1, CMKrp, KinI-3 (см. патент США №6461855, включенный в данное изобретение путем ссылки); Kip3a (см. публикацию РСТ № WO 01/96593, включенную в данное изобретение путем ссылки); Kip3d (см. патент США №6492151, включенный в данное изобретение путем ссылки) и KSP (см. патент США №6617115, который включен в данное изобретение путем ссылки).
Способы ингибирования митотического кинезина включают в себя обеспечение контакта ингибитора, соответствующего настоящему изобретению, с одним или более митотических кинезинов, в частности с человеческим кинезином или его фрагментами и разновидностями. Ингибированию может быть подвергнута активность гидролиза АТР митотического кинезина и/или активность образования митотического веретена, что в результате может приводить к разрыву митотических веретен.
В настоящем изобретении предлагаются ингибиторы одного или более митотических кинезинов, в частности одного или более человеческого митотического кинезина для лечения нарушений, связанных с пролиферацией клеток. Композиции химических составов и способы их использования, описываемые в данном изобретении, могут отличаться по своей избирательности и используются для лечения клеточной пролиферации, включая, рак, гиперплазии, рестеноз, кардиальную гипертрофию, нарушения иммунитета, грибковые заболевания, воспаление и др.
Следовательно, настоящее изобретение относится к способам, использующим по крайней мере один химический состав, представленный формулой I
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси, в которой:
R1 - возможно замещенный арил, возможно замещенный гетероциклоалкил или возможно замещенный гетероарил;
Х - СО или -SO2-;
R2 - водород или возможно замещенный низший алкил;
W - CR4, -CH2CR4 или N;
R3 - водород, возможно замещенный алкил, возможно замещенный гетероциклил, циано, возможно замещенный сульфонил или возможно замещенный арил;
R4 - водород или возможно замещенный алкил;
R5 - водород, гидроксильная группа, возможно замещенная амино группа, возможно замещенный гетероциклил или возможно замещенный низший алкил;
R6 - водород, возможно замещенный алкил, возможно замещенная алкокси группа, возможно замещенная арилокси группа, возможно замещенная гетерарилокси группа, возможно замещенный алкоксикарбонил-, возможно замещенный аминокарбонил-, возможно замещенный арил, возможно замещенный гетероарил, возможно замещенный гетероциклил или возможно замещенный аралкил и
R7 - возможно замещенный низший алкил, возможно замещенный арил, гидроксильная группа, возможно замещенная амино группа, возможно замещенная аралкокси группа или возможно замещенная алкокси группа;
при условии, что если W является N, тогда R5 не является гидроксильной группой или возможно замещенной аминогруппой, a R6 не является возможно замещенной алкокси группой, возможно замещенной аралкокси группой, возможно замещенной гетераралкокси группой или возможно замещенной аминогруппой.
В некоторых примерах осуществления изобретения R1 является возможно замещенным арилом или возможно замещенным гетероарилом. В некоторых примерах осуществления изобретения R1 является возможно замещенным арилом. В некоторых примерах осуществления изобретения R1 является возможно замещенным фенилом. В некоторых примерах осуществления изобретения R1 является фенилом, замещенным одной, двумя или тремя группами, независимо выбранными из возможно замещенного гетероциклила, возможно замещенного алкила, сульфонила, гало, возможно замещенной амино группы, возможно замещенного сульфанила, возможно замещенной алкокси группы, возможно замещенной арилокси группы, возможно замещенной гетероарилокси группы; ацила, гидроксильной группы, нитро, циано, возможно замещенного арила и возможно замещенного гетероарила-. В некоторых примерах осуществления изобретения R1 выбран из 3-гало-4-изопропокси-фенила, 3-циано-4-изопропокси-фенила, 3-циано-4-изопропиламино-фенила, 3-хлоро-4-изопропиламино-фенила, 3-циано-4-трифторопропилоксифенила, 3-хлоро-4-трифтороизопропилоксифенила, 3-циано-4-циклобутил-оксифенила, 3-хлоро-4-циклобутилоксифенила 3-циано-4-циклопропилоксифенила, и 3-хлоро-4-цилопропилоксифенила. В некоторых примерах осуществления изобретения R1 является 3-гало-4-изопропокси-фенилом или 3-циано-4-изопропокси-фенилом.
В некоторых примерах осуществления изобретения R2 является водородом.
В некоторых примерах осуществления изобретения Х является -СО-.
В некоторых примерах осуществления изобретения W является -CR4- и R4 является водородом.
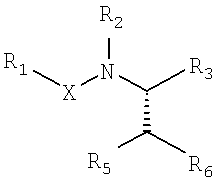
В некоторых примерах осуществления изобретения описанные здесь соединения обладают потенциально хиральным центром, например, когда W является -CR4. Изобретение предусматривает использование чистых энантиомеров и смесей энантиомеров, включая рацемические смеси, хотя использование практически оптически чистого энантиомера обычно является предпочтительным. Термин «практически оптически чистый» или «энантиомерно чистый» означает наличие по крайней мере около 95% описанного энантиомера, в котором никакая отдельная примесь не превышает 1% и, в частности, по крайней мере содержит приблизительно 97,5% энантиомерного излишка. В некоторых примерах осуществления изобретения стереогенный центр в W соответствует показанному ниже:
В некоторых примерах осуществления изобретения R3 является -CO-R7; водородом; возможно замещенным низшим алкилом; циано; возможно замещенным сульфонилом; возможно замещенным арилом или возможно замещенным гетероциклилом. В некоторых примерах осуществления изобретения R3 является возможно замещенным низшим алкилом. В некоторых примерах осуществления изобретения R3 является низшим алкилом, который возможно замещается гидроксильной группой или ее эфиром фосфата, низшим алкилом, который возможно замещен низшей алкокси группой, низшим алкилом, возможно замещенным возможно замещенной амино группой или низшим алкилом, который возможно замещен COR8, где R8 является гидроксильной группой или возможно замещенной амино группой.
В некоторых примерах осуществления изобретения R5 является водородом, гидроксильной группой или возможно замещенным низшим алкилом. В некоторых примерах осуществления изобретения R5 является водородом.
В некоторых примерах осуществления изобретения описанные здесь соединения обладают потенциально хиральным центром, например, когда R5 не является водородом. Изобретение предусматривает использование чистых энантиомеров и смесей энантиомеров, включая рацемические смеси, хотя обычно предпочтительно использование практически оптически чистого энантиомера. Термин «практически оптически чистый» или «энантиомерно чистый» означает наличие по крайней мере около 95% описанного энантиомера, в котором никакая отдельная примесь не превышает 1% и, в частности, по крайней мере содержит приблизительно 97,5% энантиомерного излишка.
В некоторых примерах осуществления изобретения R6 является возможно замещенным арилом, возможно замещенным гетероарилом, возможно замещенным гетероциклилом или возможно замещенным алкилом (таким, в котором алкильная группа замещается возможно замещенной амино группой или в котором алкильная группа является возможно замещенным циклоалкилом-). В некоторых примерах осуществления изобретения R6 является фенилом, замещенным одним или двумя из следующих заместителей: возможно замещенным гетероарилом, возможно замещенным амино, аралкокси, гало, гидрометилом-, гидрокси, циано, алкокси, фенилом, фенокси, метилендиокси, этилендиокси, сульфонилом, аминокарбонилом, карбокси, алкоксикарбонилом, нитро, гетероаралкокси, аралкокси и возможно замещенным гетероциклилом.
Также предусматрвается по крайней мере один химический состав из соединений по формуле II
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси, в которой R2, R3, R5, R6 и W такие же, как и описанные для соединений по формуле I, и в которой
R11 является возможно замещенным гетероциклилом, возможно замещенным низшим алкилом, нитро, циано, водородом, сульфонилом или гало;
R12 является водородом, гало, возможно замещенным алкилом, возможно замещенной амино группой, возможно замещенным сульфанилом, возможно замещенной алкокси группой, возможно замещенной арилокси группой, возможно замещенным гетероциклилом или возможно замещенной гетероарилокси группой, и
R13 является водородом, ацилом, возможно замещенным алкилом-, возможно замещенной алкокси группой, гало, гидроксильной группой, нитро, циано, возможно замещенной амино группой, алкилсульфонилом-, алкилсульфонамидо-, алкилсульфонилом-, карбоксиалкилом-, аминокарбонилом-, возможно замещенным арилом или возможно замещенным гетероарилом-.
В некоторых примерах осуществления изобретения R11 является водородом, циано, нитро или гало. В некоторых примерах осуществления изобретения R11 является хлоро или циано.
В некоторых примерах осуществления изобретения R11 является возможно замещенной низшей алкокси группой, возможно замещенным низшим алкилом или возможно замещенным амино-. В некоторых примерах осуществления изобретения R12 выбран из изопропокси группы, изопропиламино группы, трифтороизопропилокси группы, цилобутилокси группы и циклопропилокси группы. В некоторых примерах осуществления изобретения R12 является низшей алкокси группой (такой как пропокси группа) или 2,2,2-трифторо-1-метилэтокси. В некоторых примерах осуществления изобретения R12 является пропокси группой или 2,2,2-трифторо-1-метилэтокси. В некоторых примерах осуществления изобретения R12 не является -O-(CH2)nNH2 или -O-(CH2)4NH(CH3), в котором n является 4 или 5.
В некоторых примерах осуществления изобретения R11 и R12, взятые вместе, образуют возможно замещенное карбоциклическое или гетероциклическое кольцо. В некоторых примерах осуществления изобретения R11 и R12, взятые вместе, образуют метилендиокси или этилендиокси кольцо. В некоторых примерах осуществления изобретения R12 и R13, взятые вместе, образуют возможно замещенное карбоциклическое или гетероциклическое кольцо. В некоторых примерах осуществления изобретения R11 и R13, взятые вместе, образуют возможно замещенное карбоциклическое или гетероциклическое кольцо.
В некоторых примерах осуществления изобретения R13 является водородом.
В некоторых примерах осуществления изобретения R2 и R13, взятые вместе, образуют возможно замещенное карбоциклическое или гетероциклическое кольцо, т.е. R1, X, N и R2, взятые вместе, образуют возможно замещенное карбоциклическое или гетероциклическое кольцо. В некоторых примерах осуществления изобретения образуется замещенное 2,4-диоксо-1,4-дигидро-2Н-хиназолин-3-ил кольцо, например
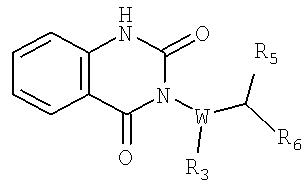
где фенильное кольцо является возможно замещенным. В других примерах осуществления изобретения образуется 4-оксо-4Н-хиназолин-3-ил кольцо, например
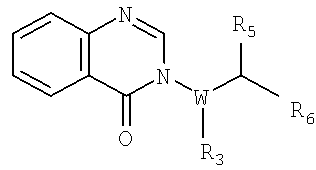
в котором фенильное кольцо является возможно замещенным. В других примерах осуществления изобретения образуется 4-оксо-4Н-пиридопиримидин-3-ил кольцо, например
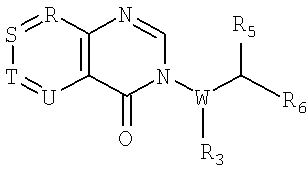
в котором R, S, Т и U являются азотом, при этом остальные являются -СН и где пиридиновое кольцо является возможно замещенным.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле III
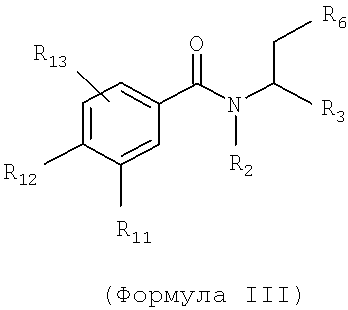
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси, в которой R2, R3, R6, R11, R12 и R13 такие же, как и описанные для соединений по формуле II.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле IV
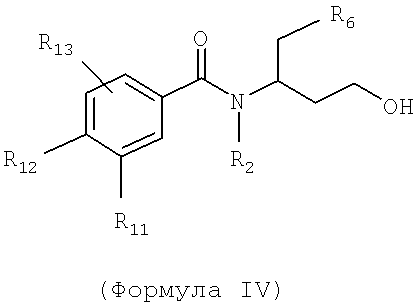
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси, в которой R2, R6, R11, R12 и R13 такие же, как и описанные для соединений по формуле III.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле V
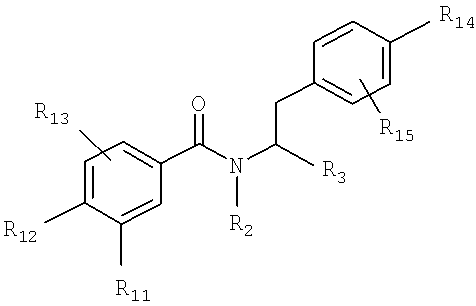
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси, в которой R2, R3, R6, R11, R12 и R13 такие же, как и описанные для соединений по формуле III, и где
R14 - возможно замещенный гетероарил и
R15 выбран из водорода, гало, гидроксильной группы и низшего алкила.
В некоторых примерах осуществления изобретения R14 выбран из
7,8-дигидро-имидазо[1,2-с][1,3]оксазин-2-ила,
3а,7а-дигидро-1Н-бензоимидазол-2-ила,
имидазо[2,1-b]оксазол-6-ила,
оксазол-4-ила,
5,6,7,8-тетрагидро-имидазо[1,2-а]пиридин-2-ила,
1Н-[1,2,4]триазол-3-ила,
2,3-дигидро-имидазол-4-ила,
1Н-имидазол-2-ила,
имидазо[1,2-а]пиридин-2-ила,
тиазол-2-ила,
тиазол-4-ила,
пиразол-3-ила и
1Н-имидазол-4-ила,
каждый из которых возможно замещен одной, двумя или тремя группами, выбранными из возможно замещенного низшего алкила, гало, ацила, сульфонила, циано, нитро, возможно замещенной амино группы и возможно замещенного гетероарила.
В некоторых примерах осуществления изобретения R14 выбран из
1Н-имидазол-2-ила,
имидазо[1,2-а]пиридин-2-ила и
1Н-имидазол-4-ила,
каждый из которых возможно замещен одной или двумя группами, выбранными из возможно замещенного низшего алкила, гало и ацила.
В некоторых примерах осуществления изобретения R15 - водород.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле VI
и фармацевтически приемлемые соли, сольваты, хелаты, нековалеитные комплексы, пролекарства и их смеси, в которой R2, R6, R11, R12 и R13 такие же, как и описанные для соединений по формуле III.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле VII
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси, в которой R2, R6, R11, R12 и R13 такие же, как и описанные для соединений по формуле III, и где
R9 выбран из возможно замещенной алкокси группы, возможно замещенной циклоалкокси группы, возможно замещенной арилалкокси группы, возможно замещенной амино группы и возможно замещенного низшего алкила.
В некоторых примерах осуществления изобретения R9 - низший алкил, замещенный гидроксильной группой или возможно замещенной амино группой. В некоторых примерах осуществления изобретения R9 - низший алкил, замещенный гидроксильной группой, амино группой, N-метиламино группой или N,N-диметиламино группой.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле VIII
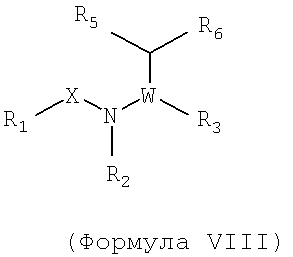
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси,
в которых R1, X, W, R3, R4, R6, и R7 такие же, как определено для формулы I, и в которых R2 и R5, вместе с атомами, к которым они присоединены, образуют возможно замещенный 5-7 членный гетероцикл, который возможно может включать один или два дополнительных гетероатома.
В некоторых примерах осуществления изобретения R2 и R5, взятые вместе, образуют возможно замещенное пирролидиниловое кольцо или возможно замещенное пиперидиниловое кольцо.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле IX
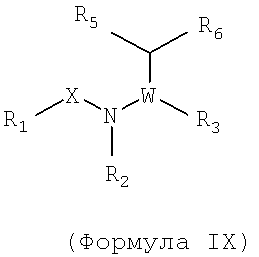
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси,
где R1, X, W, R2, R3, R4 и R7 такие же, как определено для формулы I, и где R5 и R6, вместе с атомами, к которым они присоединены, образуют возможно замещенный 5-7 членный гетероцикл, который возможно может включать один или два гетероатома.
В некоторых примерах осуществления изобретения R5 и R6 вместе с атомами, к которым они присоединены, образуют возможно замещенный 2H-[1,2,3]триазол-4-ил; возможно замещенный 1H-бензоимидазол-2-ил; возможно замещенное кольцо пиперазинила; возможно замещенное кольцо морфолинила; возможно замещенное кольцо 1H-имидазол-4-ила; возможно замещенное кольцо изоксазол-4-ила.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле Х
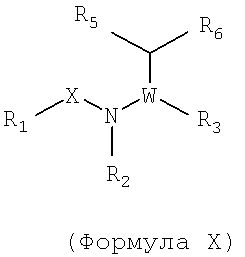
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси, в которой R1, X, W, R4, R5, R6 и R7 такие же, как и описанные для соединений по формуле I, и где
R2 и R3 вместе с атомами, к которым они присоединены, образуют возможно замещенное 5-7-членное кольцо гетероцикла.
В некоторых примерах осуществления изобретения R2 и R3 вместе с атомами, к которым они присоединены, образуют возможно замещенное 3-7-членное кольцо гетероцикла. В некоторых примерах осуществления изобретения они образуют кольцо азиридинила.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле XI
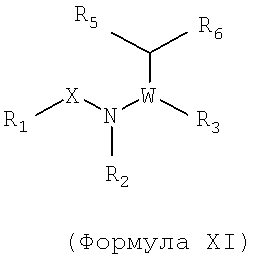
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси, в которой W, R3, R4, R5, R6 и R7 такие же, как и описанные для соединений по формуле I, и где
R1, X, N и R2, взятые вместе, образуют замещенный 2,4-диоксо-1,4-дигидро-2Н-хиназолин-3-ил, 4-оксо-4Н-хиназолин-3-ил или кольцо 4-оксо-4Н-пиридопиримидин-3-ила.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле XII
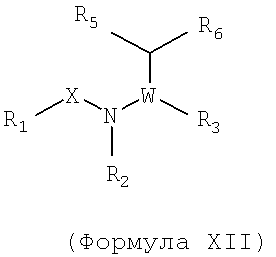
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси, в которой R1, W, R4, R5 и R6 такие же, как и описанные для соединений по формуле I, и в которой
-X-N(R2)- является -C=N- и
X, взятое вместе с R3, образует возможно замещенное кольцо гетероцикла.
В каждом случае при условии, если W является N, тогда R5 не является гидроксильной или возможно замещенной амино группой, и
R6 не является возможно замещенной алкокси группой, возможно замещенной аралкокси группой, возможно замещенной гетероаралокси группой или возможно замещенной амино группой.
В некоторых примерах осуществления изобретения -X-N(R2) является - C=N- и Х, взятый вместе с R3, образует возможно замещенное кольцо гетероцикла, включая 3Н-[1,3,4]оксадиазол-2-он; 4,5-дигидро-оксазол; тиазол; имидазол; 3,5-дигидро-имидазол-4-он или 3Н-пиримидин-4-он, каждый из которых возможно замещен, но не ограничиваясь ими.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по формуле XIII
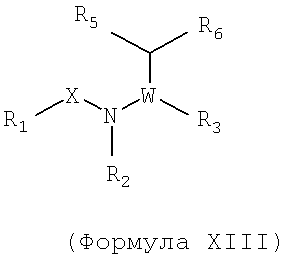
и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси,
в которых R1, X, W, R2, R4, R5 и R6 такие же, как определено для формулы I, и в которых R3 и R6, вместе с атомами, к которым они присоединены, образуют возможно замещенный 5-7-членный гетероцикл, который возможно может включать один или два дополнительных гетероатома.
В некоторых примерах осуществления изобретения R3 и R6, взятые вместе с атомами, к которым они присоединены, образуют возможно замещенное пирролидиниловое кольцо, возможно замещенное пиперидиниловое кольцо или возможно замещенное 1,2,3,4-тетрагидро-хинолин-3-иловое кольцо.
Также предусматривается по крайней мере один химический состав, выбранный из соединений по таблицам 1, 2, 3, 4, 5 или 6, и фармацевтически приемлемые соли, сольваты, хелаты, нековалентные комплексы, пролекарства и их смеси.
Названия соединений и нумерация атомов этих соединений могут быть даны с использованием AutoNom версии 2.1 в ChemDraw Ultra 6.0, Cambridgesoft, Кембридж, Массачусетс; Struc <=> название алгоритма по ChemDraw Ultra 9.0, Cambridgesoft, Кембридж, Массачусетс; или ISIS-DRAW.
Далее перечислены названия соединений, следующих после таблицы 1, которая приведена в конце описания изобретения. (S)-3-Хлоро-N-(1-(4-(1-этил-2-(2-гидроксипропан-2-ил)-1H-имидазол-4-ил)фенил-4-гидроксибутан-2-ил)-4-изопропоксибензамид
(S)-N-(1-(4-(2-Ацетил-1-этил-1H-имидазол-4-ил)фенил)-4-гидроксибутан-2-ил)-3-хлоро-4-изопропоксибензамид
N-((S)-1-(4-(2-(1-Ацетамидоэтил)-1-этил-1H-имидазол-4-ил)фенил)-4-гидроксибутан-2ил)-3-хлоро-4-изопропоксибензамид
3-Хлоро-N-((S)-1-(4-(1-этил-2-(2-гидроксипропан-2-ил)-1H-+имидазол-4-ил)фенил)-4-гидроксибутан-2-ил)-4(1,1,1-трифторопропан-2-илокси)бензамид
N-((S)-1-(4-(2-(1-Ацетамидоэтил)-1-этил-1H-имидазол-4-ил)фенил)-4-гидроксибутан-2-ил)-3-хлоро-4-(1,1,1-трифторопропан-2-илокси)бензамид
(S)-N-(1-(4-(2-Ацетил-1-(2,2,2-трифтороэтил)-1H-имидазол-4-ил)фенил)-4-гидроксибутан-2-ил)-3-хлоро-4-изопропоксибензамид
3-Хлоро-N-((S)-4-гидрокси-1-(4-(8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил)фенил)бутан-2-ил)-4-изопропоксибензамид
(S)-3-Хлоро-N-(1-(2-(диметиламино)ацетамидо)-3-(4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил)пропан-2-ил)-4-изопропоксибензамид
3-Хлоро-N-((S)-1-(2-(диметиламино)ацетамидо)-3-(4-(8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил)фенил)пропан-2-ил)-4-изопропоксибензамид
3-Циано-N-((1S)-3-гидрокси-1-{[4-(8-метилимидазо[1,2-а]пиридн-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]-фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-2[(N,N-диметилглицил)амино]-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид
N-((1S)-2-(D-Аланиламино)-1-{[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((S)-2-[(2-метилаланил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-2-[(N,N-диметилглицил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(метилэтил)окси]бензамид
N-((1R)-4-Амино-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-4-оксобутил-3-хлоро-4-[(1-метилэтил)окси]бензамид
N-((1R)-1-{[4-(2-Ацетил-1-метил-1H-имидазол-4-ил)фенил]метил}-4-амино-4-оксобутил-3-хлоро-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидроксиэтил)имидазо [1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-3-гидрокси-1-{[4-(8-метилимидазо[1,2-а-пирид-1Н-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-2-[(N,N-диметилглицил)амино]-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-2-[(N,N-диметилглицил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
N-((1R)-1-{[4-(2-Ацетил-1-метил-1H-имидазол-4-ил)фенил]метил}-4-амино-4-оксобутил-3-циано-4-[(1-метилэтил)окси]бензамид
N-[(1R)-4-Амино-1-({4-[2-(1-гидрокси-1-метилэтил)-1-метил-1H-имидазол-4-ил]фенил}метил)-4-оксобутил]-3-циано-4-[(1-метилэтил)окси]бензамид
N-[(1S)-2-(D-Аланиламино)-1-({4-[1-(2-аминоэтил)-2-(1,1-диметилэтил)-1H-имидазол-4-ил]фенил}метил)этил]-3-хлоро-4-[(1-метилэтил)окси]бензамид
N-((1S)-2-{4-[1-(2-Аминоэтил)-2-(1,1-диметилэтил)-1H-имидазол-4-ил]фенил}-1-{[2-метилаланил)амино]метил}этил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
N-[(1S)-2-(D-Аланиламино)-1-({4-[1-(2-аминоэтил)-2-(1,1-диметил)-1H-имидазол-4-ил]фенил}метил)этил]-3-циано-4-[(1-метилэтил)окси]бензамид
N-((1S)-2-{4-[1-(2-Аминоэтил)-2-(1,1-диметилэтил)-1H-имидазол-4-ил]фенил}-1-{[гидроксиацетил)амино]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
N-((1S)-2-{4-[1-(2-Аминоэтил)-2-(1,1-диметилэтил)-1H-имидазол-4-ил]фенил}-1-{[2-метилаланил)амино]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
N-((1S)-2-{4-[1-(2-Аминоэтил)-2-(1,1-диметилэтил)-1H-имидазол-4-ил]фенил}-1-{[(N,N-диметилглицил)амино]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-2-{4-[8-(1-гидроксиэтил)имидазо[1,2-a]пиридин-2-ил]фенил}-1-({[2R)-2-гидроксипропаноил]амино}метил)этил]-4-[(1-метилэтил)окси]бензамид
N-((1S)-2-[(Аминокарбонил)амино]-1-{[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[1-метилэтил)окси]бензамид
N-{(1S)-2-[4-(8-Бромоимидазо[1,2-а]пиридин-2-ил)фенил]-1-[(2-оксотетрагидро-1-(2Н)-пиридинил)метил]этил}-3-хлоро-4-[(1-метилэтил)окси]бензамид
N-{(1S)-2-[4-(8-Бромоимидазо[1,2-а]пиридин-2-ил)фенил]-1-[(2-оксогексагидро-1H-1,3-диазепин-1-ил)метил]этил}-3-хлоро-4-[(1-метилэтил)окси]бензамид
N-((1S)-2-[(Аминокарбонотиоил)амино]-1-{[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[1-метилэтил)окси]бензамид
2-(4-{(2S)-2-[({3-Циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-3-[(1,2,3-тиадиазол-4-илкарбонил)амино]пропил}фенил)имидазо[1,2-а]пиридин-8-карбоксамид
N-{(1S)-2-[(Аминосульфонил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
(3S)-3-[({3-Хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-{4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]фенил}бутановая кислота
N-[(1S)-2-[(Аминосульфонил)амино]-1-({4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]фенил}метил)этил]-3-циано-4-[(1-метилэтил)окси]бензамид
N-((1S)-1-{[4-(1Н-Бензимидазол-2-ил)фенил]метил}-3-гидроксипропил-3-хлоро-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[5-(трифторометил)-1H-бензимидазол-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-1-{[4-(5,6-диметил-1H-бензимидазол-2-ил)фенил]метил}-3-гидроксипропил)-4-[(метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[5-(метилокси)-1H-бензимидазол-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-1-{[4-(5-хлоро-1H-бензимидазол-2-ил)фенил]метил}-3-гидрокси-пропил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-3-гидрокси-1{[4-(4-метил-1H-бензимидазол-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-1-{[4-(6-хлоро-1H-имидазо[4,5-b]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
Этил 2-(4-{(2S)-2-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)-1H-бензимидазол-5-карбоксилат
2-(4-{(2S)-2-[({3-Хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)-1H-бензимидазол-5-карбоновая кислота
N-((1S)-3-Амино-1-{[4-(1H-бензимидазол-2-ил)фенил]метил}пропил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-1-{[4-(8-цианоимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
N-((1S)-1-{[4-(8-Хлороимидазо[1,2-а]пиридин-2-ил)фенйл]метил}-3-гидроксипропил-3-циано-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(трифторометил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-3-гидрокси-1-({4-[8-гидроксиимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
2-(4-{(2S)-2-[({3-Циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)имидазо[1,2-а]пиридин-7-карбоксамид
Этил 2-(4-{(2S)-2-[({3-циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)имидазо[1,2-а]пиридин-7-карбоксилат
3-Циано-N-((1S)-3-гидрокси-1-{[4-(8-нитроимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метил)окси]бензамид
N-((1S)-1-{[4-(8-Аминоимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси]бензамид
2-(4-{(2S)-2-[({3-Циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)имидазо[1,2-а]пиридин-8-карбоксамид
3-Циано-N-[(1S)-3-гидрокси-1-({4-[9-(гидроксиметил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
N-[(1S)-1-({4-[8-(Аминометил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)-3-гидроксипропил]-3-циано-4-[(1-метилэтил)окси]бензамид
N-((1S)-1-{[4-(8-Ацетилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидрокси-1-метилэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-3-гидрокси-1-{[4-(8-метил-5,6,7,8-тетрагидроимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-1-({4-[2-(1,1-диметилэтил)-1-(2-гидроксиэтил)-1H-имидазол-4-ил]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
N-[(1S)-1-({4-[1-[2-Ацетиламино)этил]-2-(1,1-диметилэтил)-1H-имидазол-4-ил]фенил}метил)-3-гидроксипропил]-3-циано-4-[(1-метилэтил)окси]бензамид
3-Циано-N-{(1S)-3-гидрокси-1-[(4-{8-[(1R)-1-гидроксиэтил]имидазо[1,2-а]пиридин-2-ил}фенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
3-Циано-N-{(1S)-3-гидрокси-1-[(4-{8-[(1S)-1-гидроксиэтил]имидазо[1,2-а]пиридин-2-ил}фенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1R)-3-гидрокси-1-({4-[8-(1-гидроксипропил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
N-((1S)-1-{[4-{8-Бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-1-{[4-(8-хлороимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидрокси-2-метилпропил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
N-[(1R)-4-Амино-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)-4-оксобутил]-3-хлоро-4-[(1-метилэтил)окси]бензамид
N-[(1R)-4-Амино-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)-4-оксобутил]-3-циано-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-1-{[4-(3-фторо-8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-1-{[4-(3-фторо-8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-2-гидрокси-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-4-[(1-метилэтил)окси]-N-[(1S)-2-[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]-1-(4-морфолинилметил)этил]бензамид
3-Хлоро-N-[(1S)-2-(4-гидрокси-1-пиперидинил)-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-2-(3-гидрокси-1-пирролидинил)-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-2-[(2S)-2-(гидроксиметил)-1-пирролидинил]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-2-[(2R)-2-(гидроксиметил)-1-пирролидинил]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-4-[(1-метилэтил)окси]-N-((1S)-2-[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]-1-{[2,2,2-трифтороэтил)амино]метил}этил)бензамид
3-Хлоро-N-((1S)-2-[(2-гидроксиэтил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-1-{[4-(8-этил-5-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
Метил (3S)-3-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-{4-[(фенилкарбонил)амино]фенил}бутаноат
3-Хлоро-N-[(1S)-2-(3-гидрокси-1-({4-[(фенилкарбонил)амино]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-{(1S)-1-[(4-{[(4-хлорофенил)карбонил]амино}фенил)метил]-3-гидроксипропил}-4-[(1-метилэтил)окси]бензамид
Фенилметил (4-{(2S)-2-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)карбамат
3-Хлоро-N-{(1S)-3-гидрокси-1-{[4-({[2-(метиламино)фенил]карбонил}амино)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
N-(4-{(2S)-2-[({3-Хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)-4-пиридинкарбоксамид
3-Хлоро-N-[(1S)-1-({4-[(циклогексилкарбонил)амино]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-1-({4-[(3,3-диметилбутаноил)амино]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[[(фенилацетил)амино]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-{(1S)-3-гидрокси-1-[(4-{[(фениламино)карбонил]амино}фенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-3-гидрокси-1-{[4-(8-метил-5-оксо-5,6-дигидроимидазо[1,2-с]пиримидин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-3-гидрокси-1-{[1-метил-3-оксо-2,3-дигидро-1H-имидазо[1,2-а]имидазол-6-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-3-гидрокси-1-{[4-(8-оксо-7,8-дигидроимидазо[1,2-а]пиразин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
2,3-Дихлоро-N-((1S)-3-гидрокси-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
N-((1S)-3-Гидрокси-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]-3-нитробензамид
3-Хлоро-N-[(1S)-2-[(гидроксиацетил)амино]-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-2-{4-[8-(1-гидроксиэтил)имидазо[1,2-a]пиридин-2-ил]фенил}-1-({[(2R)-2-гидроксипропаноил]амино}метил)этил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-2-{4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}-1-({[(2S)-2-гидроксипропаноил]амино}метил)этил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-2-[(N,N-диметилглицил)амино]-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид
N-[(1S)-2-(D-Аланиламино)-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-3-хлоро-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-2-{4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}-1-{[(2-метилаланил)амино]метил}этил)-4-[(1-метилэтил)окси]бензамид
(3S)-3-[({3-Хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-{4-[(фенилкарбонил)амино]фенил}бутановая кислота
3-Хлоро-N-{(1S)-3-гидрокси-1-[(4-имидазо[1,2-а]пиридин-6-илфенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-1-({4-[2-(1,1-диметилэтил)имидазо[1,2-а]пиридин-6-ил]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-{(1S)-3-гидрокси-1-[(4-имидазо[1,2-a]пиридин-2илфенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-{(1S)-3-гидрокси-1-[(4-имидазо[1,2-а]пиридин-2илфенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-3-гидрокси-1-{[4-(5-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-3-гидрокси-1-{[4-(7-метилимидазо[1,2-а]пиримидин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-{(1S)-3-гидрокси-1-[(4-имидазо[2,1-b][1,3]тиазол-6-илфенил)метил]бутил}-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-3-гидрокси-1-{[4-(3-метилимидазо[2,1-b][1,3]тиазол-6-ил)фенил]метил}бутил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-1-{[4-(2,3-дигидроимидазо[2,1-b][1,3]тиазол-6-ил)фенил]метил}-3-гидроксибутил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-1-{[4-(1,1-диоксидо-2,3-дигидроимидазо[2,1-b][1,3]тиазол-6-ил)фенил]метил}-3-гидроксибутил)-4-[(1-метилэтил)окси]бензамид
N-[(1S)-1-({4-[1-(3-Аминопропил)-2-(1,1-диметилэтил)-1H-имидазол-4-ил]фенил}метил)-3-гидроксипропил]-3-циано-4-[(1-метилэтил)окси]бензамид
3-Циано-4-[(1-метилэтил)окси]-N-[(1S)-2-[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]-1-(5-метил-1,2,4-оксадиазол-3-ил)этил]бензамид
3-Циано-N-[(1S)-1-({4-[8-(3,5-диметил-4-изоксазолил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-3-гидрокси-1-{[4-(8-фенилимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(1H-пиразол-4-ил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(4-изоксазолил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
N-((1S)-1-{[4-(8-Ацетилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
Этил (2E)-3-[2-(4-{(2S)-2-[({3-циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)имидазо[1,2-а]пиридин-8-ил]-2-пропеноат
(2E)-3-[2-(4-{(2S)-2-[({3-Циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)имидазо[1,2-а]пиридин-8-ил]-2-акриловая кислота
N-{(1S)-1-[(4-{8-[(1Е)-3-Амино-3-оксо-1-пропен-1-ил]имидазо[1,2-а]пиридин-2-ил}фенил)метил]-3-гидроксипропил}-3-циано-4-[(1-метилэтил)окси]бензамид
N-[(1S)-1-({4-[8-(3-Амино-3-оксопропил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)-3-гидроксипропил]-3-циано-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-1-{[4-(3-хлоро-8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
N-((1S)-1-{[4-(3-Хлоро-8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-1-({3-фторо-4-[2-(1-гидрокси-1-метилэтил)-1-метил-1H-имидазол-4-ил]фенил)метил]-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-2-гидрокси-1-{[5-(8-метилимидазо[1,2-а]пиридин-2-ил)-2-пиридинил]метил}этил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-2-гидрокси-1-{[5-(8-метилимидазо[1,2-а]пиридин-2-ил)-2-тиенил]метил}этил-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-1-({4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]-2-фторофенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-1-({4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]-2,6-дифторофенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-1-({2-хлоро-4-[2-(1,1-диметил)-1-метил-1H-имидазол-4-ил]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-1-({5-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]-2-пиридинил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-1-{[2-хлоро-4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-1-{[2-хлоро-4-(8-хлороимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-1-{[2,5-дифторо-4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-1-{[3-хлоро-4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-1-({4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]фенил}метил)-3-(метиламино)-3-оксопропил]-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-1-({4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]фенил}-1-({[(фениламино)карбонил]амино}метил)этил]-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-1-({4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]фенил}-1-({[(этиламино)карбонил]амино}метил)этил]-4-[(1-метилэтил)окси]бензамид
N-[(1S)-2-(Аминосульфонил)-1-({4-[2-(1,1-диметилэтил)-1-метил-1Н-имидазол-4-ил]фенил}метил)этил]-3-хлоро-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-2-{4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]фенил}-1-{[(метилсульфонил)амино]метил}этил)-4-[(1-метилэтил)окси]бензамид
3-Циано-N-((1S)-2-{4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]фенил}-1-[({[(2-гидроксиэтил)амино]карбонил}амино)метил]этил}-4-[(1-метилэтил)окси]бензамид
N-[(S)-1-[4-(2-терт-Бутил-1-метил-1Н-имидазол-4-ил)бензил]-2-(2 Метокси-этаноиламино)этил]-3-циано-4-изопропокси-бензамид
(4R)-4-[({3-Циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-5-{4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]фенил}пентановая кислота
3-Циано-N-{(1S)-2-{4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]фенил}-1-[(2-оксо-1-имидазолидинил)метил]этил}-4-[(1-метилэтил)окси]бензамид
N-((1S)-2-Амино-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
N-((1S)-2-(Ацетиламино)-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил-3-циано-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((1S)-2-{[(2R)-2-гидроксипропаноил]амино}-1-{[4-(8-диметилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-[(1S)-2-[(N,N-диметилглицил)амино]-1-({4-[2-(1-гидрокси-1-метилэтил)-1-метил-1H-имидазол-4-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид
3-Циано-N-[(1S)-2-[(N,N-диметилглицил)амино]-1-({4-[2-(1-гидрокси-1-метилэтил)-1-метил-1H-имидазол-4-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид
3-Хлоро-N-((S)-4-гидрокси-1-(4-(1-метил-2-(R)-1-(2-оксопирролидин-1-ил)этил)-1H-имидазол-4-ил)фенил)бутан-2-ил)-4-изопропоксибензамид
3-Хлоро-N-((S)-4-гидрокси-1-(4-(1-метил-2-(R)-1-(2-оксопирролидин-1-ил)этил)-1H-имидазол-4-ил)фенил)бутан-2-ил)-4-(1,1,1-трифторопропан-2-илокси)бензамид
3-Хлоро-N-((S)-4-гидрокси-1-(4-(1-метил-2-(R)-1-(2-оксооксазолидин-3-ил)этил)-1H-имидазол-4-ил)фенил)бутан-2-ил)-4-изопропоксибензамид
3-Хлоро-N-((S)-4-гидрокси-1-(4-(1-метил-2-(R)-1-(2-оксооксазолидин-3-ил)этил)-1Н-имидазол-4-ил)фенил)бутан-2-ил)-4-(1,1,1-трифторопропан-2-илокси)бензамид
Отдельные соединения показаны в таблицах 2-6, см. в конце описания.
В некоторых примерах осуществления изобретения химический состав является пролекарством, подобным эфиру фосфата, одного из соединений, приведенных в таблицах 1, 2, 3, 4, 5 или 6. В некоторых примерах осуществления изобретения химический состав выбран из первичного кислого фосфата (3S)-4-[4-(2-ацетил-1-метил-1H-имидазол-4-ил)фенил]-3-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]бутила и первичного кислого фосфата (3S)-3-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]бутила.
Химические составы, описанные в данной работе, могут быть приготовлены с помощью процедур, изложенных, например, в РСТ WO 99/13061, патенте США №6420561 и РСТ WO 98/56756, каждый из которых включен в данный материал путем ссылки. Стартовые материалы и другие реагенты имеются в продаже, например, у компании Олдридж Кемикал Компани (Aldrich Chemical Company), Милуоки, Висконсин, или могут быть приготовлены специалистами в данной области, используя обычные методики синтеза.
Если не определено другого, то термины "растворитель", "инертный органический растворитель" или "инертный растворитель" означают растворитель, инертный в условиях реакции, описываемой со ссылками на эти условия, включая, например, бензол, толуол, ацетонитрил, тетрагидрофуран (THF), диметилформамид (DMF), хлороформ, метиленхлорид (или дихлорметан), диэтиловый эфир, метанол, пиридин и т.п. Если не определено противоположного, растворители, используемые в реакциях, рассматриваемых в настоящем изобретении, - инертные органические растворители.
В общем, эфиры карбоновых кислот могут быть приготовлены путем проведения обычных операций эстерификации, например алкиловые эфиры могут быть подготовлены путем обработки требуемой карбоновой кислоты подходящим алканолом, как правило, в кислотных условиях. Таким же образом, амиды могут быть приготовлены с использованием обычных операций амидирования, например амиды могут быть приготовлены путем обработки соответствующей карбоновой кислоты подходящим амином. В качестве альтернативы низший алкиловый эфир типа метилового эфира кислоты можно обработать амином для получения требуемого амида, возможно в присутствии триметилалюминия, следуя процедуре, описанной в Tetrahedron Lett. 48. 4171-4173 (1977). Карбоксильные группы могут быть защищены как алкиловые эфиры, например метиловые эфиры, которые могут быть приготовлены и удалены с использованием обычных операций; одним удобным способом для преобразования карбометоксигруппы в карбоксил является использование водного гидроксида лития.
Соли и сольваты соединений, упоминаемых в данном изобретении, могут быть произведены в случае необходимости обычными технологическими способами. Например, если патентуемое соединение - кислота, требуемая соль с присоединением основания может быть приготовлена путем обработки свободной кислоты неорганическим или органическим основанием типа амина (первичного, вторичного или третичного); гидроксида щелочного или щелочноземельного металла или т.п. Иллюстрируемые примеры подходящих солей включают в свой состав органические соли - производные аминокислот типа глицина и аргинина; аммиака; первичных, вторичных и третичных аминов типа этилендиамина и циклических аминов типа циклогексиламина, пиперидина, морфолина и пиперазина, а также неорганические соли - производные натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
Если соединение - основание, требуемая соль с присоединением кислоты может быть приготовлена любым подходящим способом, известным в данной области, включая обработку свободного основания неорганической кислотой типа хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты и т.п. или органической кислотой типа уксусной кислоты, малеиновой кислоты, янтарной кислоты, миндальной кислоты, фумаровой кислоты, малоновой кислоты, пировиноградной кислоты, щавелевой кислоты, гликолевой кислоты, салициловой кислоты, пиранозидиловой кислоты типа глюкуроновой кислоты или галактуроновой кислоты, алфа-гидроксикислоты типа лимонной кислоты или винной кислоты, аминокислоты типа аспарагиновой кислоты или глутаминовой кислоты, ароматической кислоты типа бензойной кислоты или коричной кислоты, сульфоновой кислоты типа р-толуолсульфоновой кислоты, метансульфоновой кислоты, этансульфоновой кислоты или т.п.
Выделение и очистка соединений и промежуточных продуктов, описываемых в данном изобретении, могут быть проведены в случае необходимости с использованием любой подходящей операции разделения или очистки типа, например, фильтрации, экстракции, кристаллизации, колоночной хроматографии, тонкослойной хроматографии или толстослойной хроматографии или комбинации этих операций. Конкретные иллюстрации подходящих операций разделения и выделения можно получить путем ссылки на приводимые ниже примеры. Однако могут также использоваться и другие эквивалентные операции разделения или выделения.
Схема реакции 1
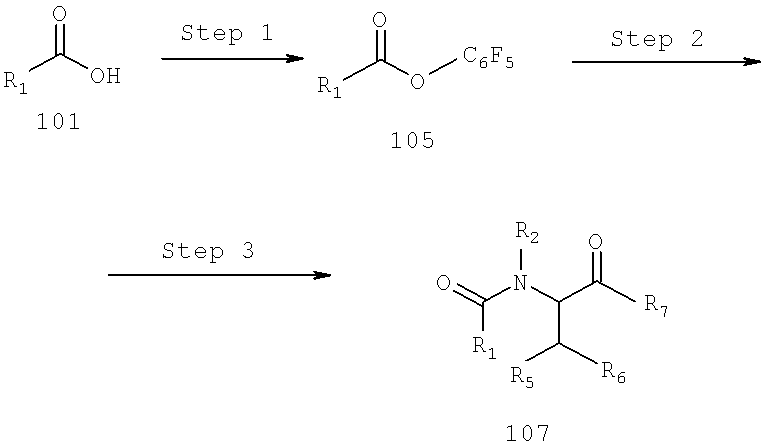
Как показано в схеме реакции 1 на этапе 1, к раствору соединения формулы 103 в инертном растворителе типа дихлорметана (DCM) добавляется избыток (подобный около 1,2 эквивалента) пентафторотрифтороацетата и основание типа триэтиламина при температуре около 0°С. Реакционную смесь перемешивают в течение около 1 часа. Продукт, соединение формулы 105, выделяют и очищают.
Как показано в схеме реакции 1 на этапе 2, к раствору соединения формулы 105, растворенному в полярном апротонном растворителе, добавляется избыток (подобный около 1,2 эквивалента) соединения по формуле R7(CO)-CH(NHR2)-CH(R5)(R6) и основание типа N,N-диизопропилэтиламина. Реакция контролируется, например, жидкостной хроматографией/масс-спектрометрией (LC/MS) и дает соединение по формуле 107, в которой R7 является NH2, которое выделяют и возможно очищают.
Схема реакции 2
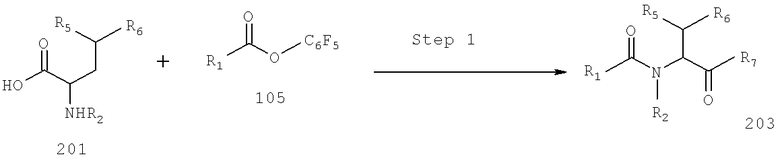
Как показано в схеме реакции 2, к раствору соединения формулы 201, растворенному в полярном апротонном растворителе типа DMF, добавляется избыток (подобный около 1,2 эквивалента) соединения формулы 105 и основание типа диизопропилэтиламина при комнатной температуре. Реакция контролируется, например, жидкостной хроматографией/масс-спектрометрией (LC/MS). После завершения к реакционному раствору добавляется первичный или вторичный амин в инертном растворителе типа THF (тетрагидрофуран) и шаровые пептидные реагенты (HBTU). Реакционную смесь перемешивают в течение около 2 дней. Продукт, соединение формулы 203, в которой R7 является возможно замещенной аминогруппой, выделяют и очищают.
В некоторых примерах осуществления изобретения R6 в соединении формулы 203 является галидом, алкил галидом или арил галидом. Этот галид может быть преобразован в различные другие заместители, используя разнообразные реакции при использовании методик, известных в данной области, которые более подробно описаны далее в примерах.
В других примерах осуществления изобретения R6 в соединении формулы 203 является алкилом или арил амином. Вновь аминная составляющая может быть алкилирована, ацилирована, преобразована в сульфонамид и тому подобное, при использовании методик, известных в данной области, которые более подробно описаны ниже.
В еще одном примере осуществления изобретения R6 в соединении формулы 203 является алкиловым спиртом или ариловым спиртом. Гидроксильная составляющая может быть преобразована в соответствующий простой эфир или сложный эфир, используя известные в данной области методики.
Схема реакции 3
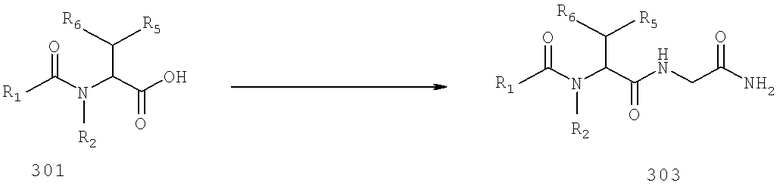
Как показано в схеме реакции 3, к раствору соединения формулы 301, растворенному в полярном апротонном растворителе типа DMF, добавляется глицинамида гидрохлорид, основание типа диизопропилэтиламина и HBTU. Реакционную смесь перемешивают в течение приблизительно 15 часов. Продукт, соединение формулы 303, выделяют и очищают.
Схема реакции 4
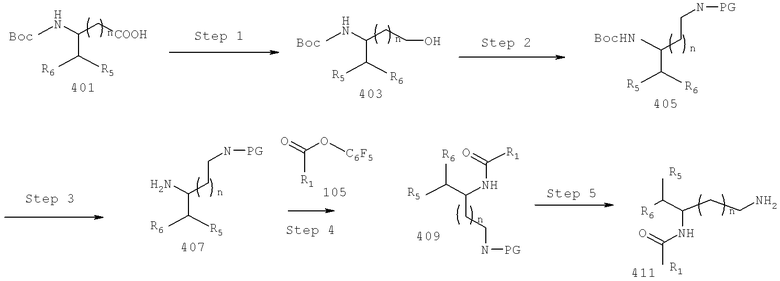
Как показано в схеме реакции 4, этап 1, к промешанному раствору соединения формулы 401, в которой n является 0, 1 или 2, в инертном растворителе типа THF (тетрагидрофуран) при температуре около 0°С добавляется избыток (типа приблизительно 2 эквивалентов) LAH (типа раствора 1,0 М в тетрагидрофуране). После перемешивания в течение 2 часов продукт, соединение формулы 403, выделяют и используют без дальнейшей очистки.
Как показано в схеме реакции 4, этап 2, гидроксильная группа преобразуется в защитную аминогруппу. Если защитная группа является фталамидом, то это может быть сделано следующим образом. К промешанному раствору соединения формулы 403 в инертном растворителе типа THF добавляется избыток (типа приблизительно 1,1 эквивалента) изоиндол-1,3-диона и трифенилфосфина. Избыток (типа приблизительно 1,1 эквивалента) DEAD затем добавляется по каплям и реакция перемешивается приблизительно 30 минут. Продукт, соединение формулы 405, выделяют и очищают.
Как показано в схеме реакции 4, этап 3, защитная группа Вос затем удаляется и образует соответствующий свободный амин. Специалисту в данной области будет понятно, что это должно выполняться таким образом, чтобы оставить другую защитную аминогруппу нетронутой. Например, к раствору соединения формулы 405 в неполярном апротонном растворителе типа дихлорметана (DCM) при комнатной температуре добавляется кислота типа TFA. Реакционную смесь перемешивают в течение приблизительно 20 минут. Продукт, соединение формулы 407, выделяют и используют без дальнейшей очистки.
Как показано в схеме реакции 4, этап 4, к раствору соединения формулы 407, растворенному в полярном апротонном растворителе типа DMF (N,N-диметилфорамид), добавляется соединение формулы 105 и основание типа диизопропилэтиламина при комнатной температуре. Реакционную смесь перемешивают в течение ночи. Продукт, соединение формулы 409, выделяют и очищают.
Как показано в схеме реакции 4, этап 5, аминная защитная группа PG затем удаляется. Если аминная защитная группа PG является фталамидом, она может быть удалена следующим образом. К раствору соединения формулы 409 в полярном протонном растворе типа метанола добавляется избыток (приблизительно 10 эквивалентов) гидрата гидразина. Реакционная смесь перемешивается при приблизительно 50°С около 5 часов и затем охлаждается до комнатной температуры. Продукт, соединение формулы 411, изолируется и возможно очищается. Условия удаления других защитных групп известны специалистам в данной области.
Свободный амин соединения формулы 411 может быть ацелирован, алкилирован, редуктивно алкилирован или сульфонилирован с использованием способов, известных специалистам в данной области.
Схема реакции 5
В некоторых соединениях изобретения определенная стереоконфигурация может быть предпочтительна для соединений по формуле I-XIII. Для краткости в остальном описании синтеза соединений формул I-XIII следует понимать, что для получения соответствующего продукта может быть использован либо отдельный изомер, либо смесь изомеров.
Определенные стереоизомеры могут быть получены из смесей, используя способы, известные в данной области. Например, в некоторых примерах осуществления изобретения свободный амин формулы 605 растворяется в инертном органическом растворителе (типа IPA) и подогревается до 60°С. В отдельном сосуде расстворяется расщепляющий агент (типа дибензоил-D-винной кислоты) в таком же теплом растворителе и затем быстро добавляется (без взбалтывания) к теплому аминному раствору. Реакционная смесь оставляется для кристаллизации путем охлаждения до комнатной температуры в течение 16 часов при постоянном помешивании. Желаемый изомер выделяется и очищается обычным образом.
В некоторых примерах осуществления изобретения оптически активный амин формулы 607 может быть приготовлен из соответствующего альдегида арила, как показано в схеме реакции 5.
Как показано в схеме реакции 5, этап 1, раствор соединения формулы 601 и избыток ацетата амония в нитроэтане нагревается в сосуде с обратным холодильником около 8 часов. Продукт, соединение формулы 603, выделяется и возможно очищается.
Как показано в схеме реакции 5, этап 2, при приблизительно °С к раствору редуцирующего агента типа борогидрида натрия в инертном растворителе типа тетрагидрофурана добавляется избыток (типа около 1,2 эквивалента) боран-тетрагидрофуранового комплекса. Результирующий раствор перемешивается при комнатной температуре около 15 минут. Соединение формулы 603 в инертном растворителе типа тетрагидрофурана добавляется по каплям и результирующий раствор выдерживается в колбе с обратным холодильником около 4 часов. Продукт, соединение формулы 605 выделяется и возможно очищается.
Амин формулы 605 затем может быть расщеплен, используя способы, известные в данной области. Например, при 0°С раствор амина формулы 605 в инертном растворителе типа этилацетата насыщается хлористоводородной кислотой (газом). Результирующая соль собирается фильтрацией и сушится в условиях ваккума. Натриевая соль L-N-ацетиллейкина медленно добавляется к перемешанному раствору вышеупомянутой соли в воде. За ночь образуются кристаллы и они удаляются фильтрацией, смываются небольшим количеством холодной воды и рекристаллизуются из абсолютного метанола. Кристаллическая соль формулы 607 выделется и возможно очищается.
Материнские растворы, которые были богаты соединением формулы 607b, комбинируются, делаются сильно щелочными и три раза промываются диэтиловым эфиром. Комбинированные органические слои промываются водой и высушиваются сульфатом натрия. Хлористоводородная кислота пропускается через раствор до завершения выпадения в осадок соли гидрохлорида. Такая же процедура, как и описанная выше, применяется к соли D-N-ацетиллейцина. Кристаллическое соединение формулы 607b выделяется и возможно очищается.
Схема реакции 6
Как показано в схеме реакции 6, этап 1, к раствору соединения формулы 701 в полярном протонном растворителе типа метанола добавляется избыток (подобный приблизительно 2 эквивалентам) SOCl2. После перемешивания, в течение ночи при температуре окружающей среды, продукт, соединение формулы 703, выделяется и используется без дальнейшей очистки.
Как показано в схеме реакции 6, этап 2, к раствору соединения формулы 703 в полярном протонном растворителе добавляется избыток (подобный около 5 эквивалентам) N2H4·Н2О. Реакционная смесь нагревается до флегмы и перемешивается приблизительно 3 часа. После охлаждения продукт, соединение формулы 705, выделяется и очищается.
Как показано в схеме реакции 6, этап 3, к раствору соединения формулы 705 в инертном растворителе типа THF добавляется избыток (подобный около 1,1 эквивалентам) карбонилдиимидазола. Реакционная смесь нагревается в сосуде с обратным холодильником и перемешивается в течение 1,5 часов. После охлаждения продукт, соединение формулы 707, выделяется и очищается.
Как показано в схеме реакции 6, этап 4, к раствору соединения формулы 707 в инертном растворителе типа ацетонитрила добавляется избыток (подобный около 1,1 эквивалентам) R5R6CH-Z, где Z - уходящая группа, и основание типа K2CO3. Реакционная смесь нагревается до приблизительно 80°С под микроволновым облучением приблизительно 30 минут, за чем следует фильтрация и концентрация в условиях вакуума. Продукт, соединение формулы 709, выделяется и возможно очищается.
Как показано в схеме реакции 6, этап 5, к раствору соединения формулы 709 добавляется избыток первичного амина в инертном растворителе, подобном THF. Реакционная смесь нагревается до около 100°С под микроволновым облучением приблизительно 4 часа. Продукт, соединение формулы 711, выделяется и очищается.
Схема реакции 7
Как показано в схеме реакции 7, этап 1, суспензия цинкового порошка в сухом дегазированном апротонном растворителе типа DMF была активирована, используя способы, известные в данной области, далее описанные в примере следующим образом. 1,2-Дибромоэтан был добавлен к цинковому раствору в атмосфере азота. Смесь нагревалась, используя тепловую пушку приблизительно 30 секунд до начала выхода газа из раствора, указывая на активацию цинка. Смесь затем охлаждалась до комнатной температуры, за чем следовало добавление TMSCl и производилось перемешивание при комнатной температуре в течение 30 минут. Раствор соединения формулы 701 в сухом дегазированном апротонном растворителе, подобном DMF, добавлялся к раствору цинка и реакционная смесь перемешивалась в течение 1 часа при комнатной температуре. Раствор формулы 702 использовался для следующего этапа.
Как показано в схеме реакции 7, этап 2, к раствору 702 добавлялось соединение формулы 703 (где X1 является Br или I) в сухом дегазированном полярном апротонном растворителе типа DMF и палладиевый катализатор, и лиганд типа Pd2(dba3), и три-о-толилфосфин. Реакционная смесь перемешивалась 3 часа. Продукт, соединение формулы 704, выделялся и очищался.
Приготовленные химические составы, соответствующие изобретению, предназначены для самых различных областей применения, затрагивающих изменение митоза. Специалистам ясно, что существуют различные пути изменения митоза, то есть можно воздействовать на митоз как повышением, так и снижением активности компонента, участвующего в процессе митоза. Другими словами, на митоз можно воздействовать (например, прерывать) путем нарушения равновесия или в результате ингибирования или активизации определенных компонентов. Подобные же подходы могут быть использованы и для изменения мейоза.
В некоторых примерах осуществления химические составы согласно изобретению используются для ингибирования образования митотического веретена, вызывающего в результате длительный арест клеточного цикла в митозе. Термин "ингибирование" в контексте изобретения используется в смысле замедления процесса образования митотического веретена или создания условий, препятствующих протеканию этого процесса, или в смысле инициирования дисфункции митотического веретена. Под "образованием митотического веретена" в изобретении понимается предполагаемая организация микротрубочек в биполярные структуры митотическими кинезинами. "Дисфункция митотического веретена" означает предполагаемый митотический арест.
Химические составы, соответствующие изобретению, могут быть использованы для связывания и/или ингибирования активности митотического кинезина. В некоторых примерах осуществления изобретения митотический кинезин человеческий, однако химические составы могут быть использованы для связывания или ингибирования активности кинезинов от других организмов. В этом контексте "ингибирование" означает или ускорение или замедление расхождения полюсов веретена, вызывающего мальформацию, т.е. расширение полюсов митотического веретена, или, другими словами, нарушение морфологического строения митотического веретена. В определение митотический кинезин для этих целей включены также варианты и/или фрагменты подобного белка и более конкретно моторная область подобного белка.
Химические составы, соответствующие настоящему изобретению, используются для лечения клеточно-пролиферативных заболеваний. Среди таких заболеваний, при лечении которых можно использовать предлагаемые в данном изобретении соединения, композиции на их основе и способы их использования, можно назвать раковые заболевания (дополнительно рассматриваемые ниже), аутоиммунные заболевания, грибковые заболевания, артрит, отторжение трансплантата, воспаления кишечника, клеточную пролиферацию вследствие медицинских операций, включая хирургические операции, ангиопластику и т.п. Лечение состоит в ингибировании клеточной пролиферации. Понятно, что в некоторых случаях лечение требуется даже, если клетки еще не перешли в патологическое состояние. Поэтому в одном примере осуществления изобретение может быть использовано применительно к клеткам или организмам, пораженным любым из этих заболеваний или находящимся в состояниях, предшествующих этим заболеваниям.
Предлагаемые в данном изобретении химические составы, фармацевтические рецептуры и способы их использования считаются чрезвычайно полезными для лечения рака, включая твердые опухоли типа кожи и груди, мозговые цервикальные карциномы, тестикулярные карциномы и т.д. В частности, среди раковых образований, которые можно лечить, можно назвать опухоли:
- Сердца: саркому (ангиосаркому, фибросаркому, рабдомиосаркому, липосаркому), миксому, рабдомиому, фиброму, липому и тератому;
- Легких: бронхогенную карциному (клетки сквамозного эпителия, мелкие недифференцированные клетки, крупные недифференцированные клетки, аденокарциному), альвеолярную (бронхиолярную) карциному, бронхиальную аденому, саркому, лимфому, хондроматозную гамартому, мезотелиому;
- Желудочно-кишечного тракта: пищевода (сквамозно-клеточную карциному, аденокарциному, лейомиосаркому, лимфому), желудка (карциному, лимфому, лейомиосаркому), поджелудочной железы (дуктальную аденокарциному, инсулиному, глюкагоному, гастриному, карциноидные опухоли, випому), тонкой кишки (аденокарциному, лимфому, карциноидные опухоли, саркому Капоши, лейомиому, гемангиому, липому, нейрофиброму, фиброму), толстой кишки (аденокарциному, трубчатую аденому, ворсинчатую аденому, гамартому, лейомиому);
- Мочеполового тракта: почек (аденокарциному, опухоль Вилмса (нефробластому), лимфому, лейкемию), мочевого пузыря и уретры (сквамозно-клеточную карциному, переходно-клеточный рак, аденокарциному), простаты (аденокарциному, саркому), яичек (семиному, тератому, эмбриональную карциному, тератокарциному, хориокарциному, саркому, внутритканевую клеточную карциному, фиброму, фиброаденому, аденоматоидные опухоли, липому);
- Печени: гепатому (злокачественную гепатому), холангиокарциному, гептобластому, ангиосаркому, печеночно-клеточную аденому, гемангиому;
- Костей: остеогенную саркому (остеосаркому), фибросаркому, злокачественную волокнистую гитиоцитому, хондросаркому, саркому Эвинга, злокачественную лимфому (саркому клеток ретикулярной ткани), многократную миеломную болезнь, злокачественную гигантоклеточную хордому, остеохондрому (костно-хрящевые экзостозы), доброкачественную хондрому, хондробластому, хондромиксофиброму, остеому остеоида и гигантоклеточные опухоли;
- Нервной системы: черепа (остеому, гемангиому, гранулему, ксантому, деформирующий остит), мозговых оболочек (менингиому, менингиосаркому, глиоматоз), мозга (астроцитому, медуллобластому, глиому, эпендимому, гоноцитому (пинеалому), многообразную глиобластому, олигодендроглиому, шванному, ретинобластому, врожденные опухоли), нейрофиброму спинного мозга, менингиому, глиому, саркому);
- Женской половой сферы: матки (внутриматочную карциному), шейки (цервикальную карциному, предопухолевую цервикальную дисплазию), яичников (овариальную карциному (серозную цистаденокарциному, муцинозную цистаденокарциному, неклассифицированную карциному), гранулезо-текаклеточные опухоли, опухоли клеток Сертоли-Лейдига, дисгерминому, злокачественную тератому), вульвы (сквамозно-клеточную карциному, интраэпителиальную карциному, аденокарциному, фибросаркому, меланому), влагалища (гипернефрому, сквамозно-клеточную карциному, ботриоидную саркому (эмбриональную рабдомиосаркому), маточных труб (карциному);
- Кроветворной системы: крови (миелоидную лейкемию (острую и хроническую), острую лимфобластную лейкемию, хроническую лимфоцитарную лейкемию, миелопролиферативные заболевания, многократную миеломную болезнь, миелодиспластический синдром), болезнь Ходжкина, неходжкинскую лимфому (злокачественную лимфому);
- Кожи: злокачественную меланому, базально-клеточный рак, сквамозно-клеточную карциному, саркому Капоши, диспластические невусы, липому, ангиому, дерматофиброму, келоиды, псориаз и
- Надпочечников: нейробластому.
Таким образом, лечение рака включает лечение "раковых клеток", включая в себя клетки, пораженные любым из идентифицированных выше заболеваний. Таким образом, термин "раковая клетка" в данном изобретении включает в себя клетку, пораженную любым из идентифицированных выше заболеваний.
Другим ценным аспектом изобретения является комплект, включающий по крайней мере один описанный здесь химический состав и листок-вкладыш в упаковке или другую маркировку, включающие инструкции по лечению клеточно-пролиферативного заболевания путем назначения эффективного количества по крайней мере одного химического состава. Химический состав, входящий в комплект изобретения, предпочтительно поставляется в одной или более дозах для курса лечения клеточно-пролиферативного заболевания, каждая из доз при этом представляет собой фармацевтический состав, содержащий фармацевтический наполнитель и по крайней мере один описанный здесь химический состав.
Для анализа активности модуляции митотического кинезина, как правило, или митотический кинезин, или по крайней мере один химический состав, соответствующий изобретению, связывают с нерастворимой основой, не вызывающей диффузии, имеющей площадки для размещения выделенных проб (например, с пластиной микротитратора, область для выделенной пробы (например, пластины микротитратора, культуральные планшеты и т.д.). Нерастворимая основа может быть выполнена из любой композиции, с которой проба может быть связана, которая легко отделяется от растворимого материала и, другими словами, совместима с методом скрининга от начала до конца. Поверхность таких основ может быть твердой или пористой и иметь любую удобную форму. В качестве примеров подходящих нерастворимых основ можно назвать пластины микротитратора, культуральные планшеты, мембраны и бусины. Они, как правило, выполнены из стекла, пластмассы (например, полистирола), полисахаридов, нейлона или нитроцеллюлозы, Teflonтм и т.д. Пластины микротитратора и культуральные планшеты особенно удобны для большого количества анализов, которые можно выполнять одновременно с использованием малых количеств реагентов и проб. Специфический способ связывания пробы не является критическим до тех пор, пока остается совместимым с реагентами и всеми рассматриваемыми в данном изобретении способами, поддерживает активность проб и приводит к возникновению диффузии. Среди предпочтительных способов связывания можно назвать использование антител (которые не дают стерических блоков ни с участками связи лигандов, ни с последовательностью активации, когда белок связан с основой), прямого связывания с "липкими" или ионными основами, химического сшивания, синтеза белка или агента на поверхности и т.д. После связывания пробы избыток несвязанного материала удаляют путем промывания. Площадки для размещения проб могут быть затем блокированы путем инкубации с бычьим сывороточным альбумином (BSA), казеином или другим безвредным белком или другим компонентом.
Химические составы, соответствующие настоящему изобретению, могут быть использованы сами по себе для ингибирования активности митотического кинезина. В некоторых примерах осуществления по крайней мере один химический состав, соответствующий настоящему изобретению, объединен с митотическим кинезином и проведен анализ активности митотического кинезина. Активность кинезина, как известно специалистам, проявляется в виде одного или более эффектов: способность влиять на гидролиз АТР; связывание микротрубочек; скольжение и полимеризацию/деполимеризацию (влияние на динамику микротрубочек); связывание с другими белками веретена; связывание с белками, участвующими в управлении клеточным циклом; использование в качестве субстрата для других ферментов типа киназ или протеаз и специфическое воздействие кинезина на клетки, которое приводит к расхождению полюсов веретена.
Способы выполнения анализов подвижности хорошо известны специалистам (см., например, Hall с соавт. (1996), Biophys. J., 71: 3467-3476, Turner с соавт., 1996, AnaL Biochem. 242 (I): 20-5; Gittes с соавт., 1996, Biophys. J. 70 (I): 418-29; Shirakawa с соавт., 1995, J. Exp.BioL 198: 1809-15; Winkelmann с соавт., 1995, Biophys. J. 68: 2444-53; Winkelmann с соавт., 1995, Biophys. J. 68:72S.).
Могут быть также использованы известные специалистам способы определения активности гидролиза АТРазы. В предпочтительном варианте использованы анализы на основе растворов. Патент США №6410254, в котором описываются такие анализы, включен в данное изобретение путем ссылки. В качестве альтернативы использованы обычные способы. Например, может быть количественно определено выделение Pi из кинезина. В одном случае осуществления для анализа активности гидролиза АТРазы использованы 0,3 М РСА (перхлорной кислоты) и реагент малахитовой зелени (8,27 мМ молибдата натрия II, 0,33 мМ оксалата малахитовой зелени и 0,8 мМ Triton X-100). Для выполнения анализа 10 мкл реакционной смеси охлаждают в 90 мкл холодной 0,3 М РСА. Использование фосфатных стандартов позволяет осуществить приведение данных к выделяемым неорганическим фосфатам в мМ. После охлаждения всех реакционных смесей и стандартов в РСА 100 мкл реагента малахитовой зелени добавляют в соответствующие лунки, например, пластины микротитратора. Смесь выдерживают в течение 10-15 минут и затем считывают оптическую плотность пластины на длине волны 650 нм. При использовании фосфатных стандартов результаты считывания оптической плотности могут быть приведены к Pi в мМ и вычерчены в виде временных графиков. Среди известных специалистам анализов АТРазы можно назвать также анализ люциферазы.
Активность АТРазы кинезиновых моторных доменов может быть также использована для контроля воздействий агентов и известна специалистам. В одном примере осуществления анализы АТРазы кинезина выполнены в отсутствие микротрубочек. В другом случае осуществления анализы АТРазы выполнены в присутствии микротрубочек. В рассмотренных выше анализах могут быть обнаружены различные типы агентов. В некоторых примерах осуществления воздействие агента не зависит от концентрации микротрубочек и АТР. В некоторых примерах осуществления воздействие агентов на АТРазу кинезина может быть ослаблено путем увеличения концентраций АТР, микротрубочек или и того и другого. В некоторых случаях осуществления воздействие агента усилено путем увеличения концентрации АТР, микротрубочек или и того и другого.
Химические составы, ингибирующие биохимическую активность митотического кинезина in vitro, могут быть затем подвергнуты скринингу in vivo. Среди способов скрининга in vivo можно назвать анализы распределения клеточного цикла, жизнеспособности клеток или присутствия, морфологии, активности, распределения или числа митотических веретен. Способы контроля распределения клеточной популяции по клеточному циклу, например поточная цитометрия, известны специалистам, также как и способы определения жизнеспособности клетки. См., например, патент США №6437115, включенный в данное изобретение путем ссылки. Хорошо известны специалистам и микроскопические способы контроля образования и мальформации веретена (см., например, Whitehead и Rattner (1998), J. Cell Sci. 111: 2551-61; Galgio с соавт. (1996), J. Cell Biol, 135: 399-414), каждая из статей включена в данное изобретение путем ссылки.
Соответствующие изобретению химические составы ингибируют один или более митотических кинезинов. Один из показателей ингибирования - IC50, определяемый как концентрация соединения, при которой активность митотического кинезина уменьшается на пятьдесят процентов относительно контрольного значения. В некоторых примерах осуществления по крайней мере один химический состав имеет IC50 менее 1 мМ. В некоторых случаях осуществления по крайней мере один химический состав имеет IC50 менее 100 мкМ. В некоторых случаях осуществления по крайней мере один химический состав имеет IC50 менее 10 мкМ. В некоторых примереах осуществления по крайней мере один химический состав имеет IC50 даже менее около 1 мкМ. В некоторых примерах осуществления IC50 составляет менее 100 нМ. В некоторых примерах осуществления IC50 составляет менее 10 нМ. Измерение IC50 было проведено с использованием описываемого в данном изобретении анализа АТРазы.
Другой показатель ингибирования - Ki. Для химических составов с IC50 менее 1 мкМ, Ki или Kd определяют как константу скорости диссоциации для взаимодействия описываемых в данном изобретении соединений с митотическим кинезином. В некоторых примерах осуществления по крайней мере один химический состав имеет Ki менее 100 мкМ. В некоторых примерах по крайней мере один химический состав имеет Ki 10 мкМ. В некоторых примерах осуществления по крайней мере один химический состав имеет Ki менее 1 мкМ. В некоторых примерах осуществления по крайней мере один химический состав имеет Ki менее 100 нМ. В некоторых примерах осуществления по крайние мере один химический состав имеет Ki менее 10 нМ.
Кi для химического состава определяют по IC50 на основе трех допущений и уравнения Михаэлиса-Ментен. Во-первых, только одна молекула соединения связывается с ферментом и при этом нет никакой кооперативности. Во вторых, концентрации активного фермента и испытуемого соединения известны (т.е. препараты не содержат сколько-нибудь существенных количеств примесей или неактивных форм). В-третьих, скорость ферментативной реакции при образовании комплекса между ферментом и ингибитором равна нулю. Данные скорости (т.е. концентрация соединения) удовлетворяют уравнению:
где V - наблюдаемая скорость, Vmax - скорость, определяемая свободным ферментом, I0 - концентрация ингибитора, Е0 - концентрация фермента, а Kd - константа диссоциации комплекса между ингибитором и ферментом.
Другим показателем ингибирования является GI50, определяемый как концентрация химического состава, при которой происходит снижение скорости роста клеток на пятьдесят процентов. В некоторых примерах осуществления изобретения по крайней мере один химический состав имеет GI50 менее 1 мМ. В некоторых примерах осуществления изобретения по крайней мере один химический состав имеет GI50 менее 20 мкМ. В некоторых примерах осуществления изобретения по крайней мере один химический состав имеет GI50 менее 10 мкМ. В некоторых примерах осуществления изобретения по крайней мере один химический состав имеет GI50 менее 1 мкМ. В некоторых примерах осуществления изобретения по крайней мере один химический состав имеет GI50 менее 100 нМ. В некоторых примерах осуществления изобретения по крайней мере один химический состав имеет GI50 менее 10 нМ. Измерения GI50 были проведены с использованием описываемого в данном изобретении анализа пролиферации клеток. Было установлено, что химические составы этого класса ингибируют пролиферацию клеток.
Эффективность in vitro ингибиторов с молекулами малого размера была определена, например, путем анализа жизнеспособности овариальных раковых клеток (SKOV3) человека после 72-часовой выдержки в серии 9-кратных разведений соединения. Жизнеспособность клеток определяли путем измерения оптической плотности формазона - продукт, получаемого путем биоредукции MTS/PMS, являющегося доступным для приобретения реагентом. Каждая точка кривой зависимости от дозы была рассчитана в виде процента от необработанных контрольных клеток после 72-часовой выдержки минус оптическая плотность фона (полное умерщвление клеток).
Значения GI50 для антипролиферативных соединений, которые были с успехом применены в клинике для лечения рака (при химиотерапии рака), варьируются в широком диапазоне. Например, в клетках А549 значение GI50 для паклитаксела составляет 4 нМ, для доксорубицина 63 нМ, для 5-флюороурацила 1 мкМ, а для гидроксимочевины 500 мкМ (данные предоставлены National Cancer Institute, Developmental Therapeutic Programm, http://dtp.nci.nih.gov/). Поэтому, соединения, способные ингибировать клеточную пролиферацию, могут быть полезными в клинических условиях независимо от концентрации проявления эффекта ингибирования.
Для использования соответствующих данному изобретению химических составов в способе скрининга на соединения, которые связывают митотический кинезин, митотический кинезин связывали с основой и для анализа добавляли соответствующее данному изобретению соединение. В качестве альтернативы рассматриваемый в данном изобретении химический состав связывали с основой и добавляли митотический кинезин. В составе классов соединений, среди которых можно найти новые связывающие агенты, можно назвать специфические антитела, искусственные связывающие агенты, идентифицированные в результатах скрининга химических библиотек, пептидные аналоги и т.д. Особый интерес представляют анализы скрининга кандидатов в агенты, которые имеют низкую токсичность для человеческих клеток. Для этой цели может быть использован целый ряд анализов, включая анализы белок-белкового связывания in vitro, анализы изменения электрофоретической подвижности, иммунологические анализы для белкового связывания, функциональные анализы (анализы фосфорилирования и т.д.) и т.п.
Для определения связывания рассматриваемых в данном изобретении химических составов с митотическим кинезином можно использовать ряд способов. В некоторых случаях осуществления химический состав метят, например, с помощью флуоресцентного или радиоактивного компонента и непосредственно определяют связывание. Например, это может быть сделано путем присоединения всего или части митотического кинезина к твердой основе, добавления меченого испытуемого соединения (например, рассматриваемого в настоящем изобретении химического состава, в котором по меньшей мере один атом был заменен обнаружимым изотопом), смывания избытка реагента и определения количества меченого соединения на твердой основе.
Термин "меченый" в данном изобретении означает, что соединение или непосредственно или опосредованно помечено маркером, который обеспечивает обнаружимый сигнал, например радиоизотопным, флуоресцентным маркером, ферментом, антителом, частицами типа магнитных, хемилюминесцентным маркером или специфическими связывающими молекулами и т.д. Среди специфических связывающих молекул можно назвать пары типа биотина и стрептавидина, дигоксин и антидигоксин и т.д. Для специфических связывающих партнеров дополнительный партнер обычно метят молекулой, обеспечивающей возможность обнаружения в соответствии с известными операциями, как изложено выше. Метка может обеспечивать обнаружимый сигнал непосредственно или опосредованно.
В некоторых случаях осуществления метят только один из компонентов. Например, белки кинезина могут быть помечены в тирозиновых позициях с помощью 125I или флуорофорами. В качестве альтернативы не один, а несколько компонентов могут быть помечены различными метками: с помощью 125I, например, для белков и флуорофора для антимитотических агентов.
Соответствующие данному изобретению химические составы могут быть также использованы в качестве конкурентов в скрининге на дополнительные кандидаты в лекарственные средства. "Кандидат в агенты" или "кандидат в лекарственные средства" или грамматические эквиваленты этих терминов используются в данном изобретении для описания любой молекулы, например белка, олигопептида, органической молекулы малого размера, молекулы полисахарида, полинуклеотида и т.д., для проверки на биоактивность. Они могут обладать способностью непосредственного или опосредованного изменения клеточно-пролиферативного фенотипа или экспрессии клеточно-пролиферативной последовательности, включая как последовательности нуклеиновых кислот, так и белковые последовательности. В других случаях проводят скрининг изменения клеточно-пролиферативного белкового связывания и/или активности. Скрининг этого вида может быть выполнен или в присутствии или в отсутствие микротрубочек. В случае проведения скрининга белкового связывания или активности в предпочтительных примерах осуществления исключают молекулы, уже известные способностью связывания со специфическим белком, например полимерных структур типа микротрубочек и источников энергии типа АТР. В предпочтительных примерах осуществления данного изобретения анализ проводят для кандидатов в агенты, которые не связывают клеточно-пролиферативный белок в его природном эндогенном состоянии, называемых в данном изобретении "экзогенными" агентами. В некоторых примерах осуществления из состава экзогенных агентов дополнительно исключают антитела к митотическому кинезину.
Кандидаты в агенты могут охватывать многочисленные химические классы, однако обычно это органические молекулы в предпочтительном варианте органические соединения с молекулами малых размеров, имеющие молекулярную массу не менее 100, но не более 2500 дальтон. Кандидаты в агенты содержат функциональные группы, необходимые для структурного взаимодействия с белками, в частности для водородного и липофилического связывания, и обычно включают в свой состав по меньшей мере амин, карбонил-, гидроксил-, эфирную группу или карбоксильную группу, в предпочтительном варианте по меньшей мере две функциональные химические группы. Кандидаты в агенты часто содержат циклические углеродные или гетероциклические структуры и/или ароматические или полиароматические структуры, замещенные одним или более упомянутыми выше функциональными группами. Кандидаты в агенты были также найдены среди биомолекул, включая пептиды, сахариды, жирные кислоты, стероиды, пурины, пиримидины, их производные, структурные аналоги или комбинации.
Кандидаты в агенты были получены из самых различных источников, включая библиотеки синтетических или природных соединений. Например, многочисленные средства доступны для случайного и направленного синтеза самых различных органических соединений и биомолекул, включая экспрессию усредненных олигонуклеотидов. В качестве альтернативы доступны или могут быть легко изготовлены библиотеки природных соединений в форме бактериальных, грибковых, растительных и животных экстрактов. Кроме того, природные или синтетически изготовленные библиотеки и соединения могут быть легко модифицированы с помощью обычных химических, физических и биохимических средств. Известные фармакологические агенты могут быть подвергнуты направленным или случайным химическим модифицированиям типа ацилирования, алкилирования, эстерификации и/или амидификации для изготовления структурных аналогов.
Анализы конкурентного скрининга могут быть проведены путем объединения митотического кинезина и кандидата в лекарственные средства в первой пробе. Вторая проба содержит по крайней мере один химический состав, соответствующий данному изобретению, митотический кинезин и кандидат в лекарственные средства. Это может быть выполнено или в присутствии, или в отсутствие микротрубочек. Связывание кандидата в лекарственные средства определяют для обеих проб и изменение или различие в связывании между этими двумя пробами указывает на присутствие кандидата в лекарственные средства, способного к связыванию с митотическим кинезином и возможного ингибирования его активности. То есть различие связывания кандидата в лекарственные средства во второй пробе относительно первой пробы указывает на способность кандидата в лекарственное средство к связыванию с митотическим кинезином.
В некоторых примерах осуществления связывание кандидата в агенты с митотическим кинезином определяют путем использования анализов конкурентного связывания. В этом случае осуществления конкурент - связывающий компонент, известный способностью связывания с митотическим кинезином, типа антитела, пептида, связывающего партнера, лиганда и т.д. При определенных обстоятельствах кандидат в агенты и связывающий компонент могут конкурировать за связывание, причем связывающий компонент замещает кандидата в агенты.
В некоторых примерах осуществления кандидата в агенты помечают. Или кандидата в агенты, или конкурента, или обоих сначала добавляли в митотический кинезин на время, достаточное для связывания. Инкубации можно осуществлять при любой температуре, способствующей оптимальной активности, обычно между 4 и 40°С.
Выбор инкубационных периодов осуществляют исходя из оптимальной активности, но можно также оптимизировать этот выбор с точки зрения обеспечения высокой скорости сплошного скрининга. Обычно между 0,1 и 1 часом бывает достаточно. Избыток реагента, как правило, удаляют или вымывают. Затем добавляют второй компонент и последующее присутствие или отсутствие меченого компонента укажет на связывание.
В некоторых примерах осуществления сначала добавляют конкурента, а затем - кандидата в агенты. Замещение конкурента указывает на связывание кандидата в агенты с митотическим кинезином и, следовательно, на его способность к связыванию и потенциальному ингибированию активности митотического кинезина. В этом случае осуществления любой компонент может быть помечен. Таким образом, например, если конкурент помечен, присутствие метки в промывочном растворе указывает на замещение агентом. В случае же, если помечен кандидат в агенты, то на замещение указывает присутствие метки на основе.
В некоторых примерах осуществления сначала добавляют кандидата в агенты с инкубацией и промыванием, а затем - конкурента. Отсутствие связывания конкурентом может указывать на связывание кандидата в агенты с митотическим кинезином с более высоким сродством. Таким образом, если кандидат в агенты помечен, то присутствие метки на основе, связанное с недостаточным связыванием конкурентом, может указывать на способность кандидата в агенты к связыванию с митотическим кинезином.
Ингибирование проверяют путем скрининга кандидатов в агенты, способных к ингибированию активности митотического кинезина, содержащего этапы объединения кандидата в агенты с митотическим кинезином, как было указано выше, и определения изменения биологической активности митотического кинезина. Таким образом, в этом случае осуществления кандидат в агенты должен связываться с митотическим кинезином (хотя это может и не понадобиться) и изменять свою биологическую или биохимическую активность, как определено в данном изобретении. Способы включают в себя скрининг клеток как in vitro, так и in vivo для определения изменений в распределении клеточного цикла, жизнеспособности клеток или присутствия, морфологии, активности, распределения или числа митотических веретен, как в общих чертах было описано выше.
В качестве альтернативы дифференциальный скрининг может быть использован для идентификации кандидатов в лекарственные средства, которые связываются с природным митотическим кинезином, но не могут связываться с модифицированным митотическим кинезином.
При проведении анализов можно использовать положительные и отрицательные контрольные образцы. Для получения статистически значимых результатов в предпочтительном варианте все контрольные и пробы для анализа были выполнены по меньшей мере в трех экземплярах. Инкубацию всех проб проводили в течение промежутка времени, достаточного для связывания агента с белком. По окончании инкубации все пробы подвергали промывке до свободного от неспецифически связанного материала состояния и определяли количество связанного, как правило, меченого агента. Например, при использовании радиоактивной метки для определения количества связанного соединения пробы могут быть просчитаны в сцинтилляционном счетчике.
Анализы скрининга можно проводить с использованием ряда других реагентов. В их состав входят реагенты, подобные солям, нейтральным белкам, например альбумин, детергенты и т.д., которые можно использовать для обеспечения оптимального белок-белкового связывания и/или уменьшения неспецифических или фоновых взаимодействий. Кроме того, могут быть использованы реагенты, улучшающие эффективность анализа в других отношениях, типа ингибиторов протеазы, ингибиторов нуклеазы, антимикробных агентов и т.д. Смесь компонентов можно добавлять в любом порядке, который обеспечивает необходимое связывание.
Таким образом, рассматриваемые в данном изобретении составы могут быть назначены применительно к клеткам. Слово "назначенный" здесь означает назначение терапевтически эффективной дозы по крайней мере одного рассматриваемого в данном изобретении состава применительно к клетке как в клеточной культуре, так и у пациента. "Терапевтически эффективная доза" - это доза, применение которой приводит к результатам, достижение которых предполагалось при назначении. Точная доза зависит от цели лечения и может быть установлена специалистом с использованием известных методик. Как известно специалистам может потребоваться подбор системного, а не локального способа доставки лекарственного средства, пути поступления лекарственного вещества в организм, возраста, массы тела, общего состояния здоровья, пола, диеты, времени назначения, природы состава, взаимодействия лекарственных средств и серьезности условия и все это может быть установлено специалистом путем проведения обычных экспериментов. Под термином "клетка" понимается любая клетка, в которой митоз или мейоз может быть изменен.
Термин "пациент" для целей настоящего изобретения включает в себя как людей, так и животных, в частности млекопитающих, и другие организмы. Поэтому рассматриваемые способы пригодны для применения как в терапии человека, так и в ветеринарии. В предпочтительном примере осуществления пациент - это млекопитающее, а в более предпочтительном примере осуществления пациентом является человек.
Рассматриваемые в настоящем изобретении химические составы, имеющие требуемую фармакологическую активность, могут быть назначены пациенту в некоторых примерах осуществления, в качестве фармацевтически приемлемой композиции, содержащей фармацевтический наполнитель. В зависимости от способа введения соединения могут быть приготовлены, как описано ниже, с использованием целого ряда способов. Концентрация терапевтически активного соединения в составе может варьироваться приблизительно от 0,1÷100 мас.%.
Агенты могут быть назначены сами по себе или в комбинации с другими видами терапии, т.е. в комбинации с радиотерапией или другими хемотерапевтическими агентами типа агентов класса таксанов, которые, по-видимому, воздействуют на образование микротрубочек, или класса камптотецинов - ингибиторов топоизомеразы I. При использовании другие хемотерапевтические агенты могут быть назначены перед, одновременно или после назначения по крайней мере одного химического состава, рассматриваемого в настоящем изобретении. В одном объекте изобретения по крайней мере один химический состав, рассматриваемый в настоящем изобретении, назначают совместно с одним или более другими хемотерапевтическими агентами. Термин "совместное назначение" означает, что рассматриваемые в настоящем изобретении соединения назначают пациенту так, что по крайней мере один состав, а также совместно назначаемое соединение могут быть обнаружены в крови пациента в одно и то же время, независимо от фактического назначения соединений, включая одновременное.
Назначение химических составов по настоящему изобретению может быть реализовано целым рядом способов, включая пероральный, подкожный, внутривенный, интраназальный, чрескожный, интраперитонеальный, внутримышечный, внутрилегочный, вагинальный, ректальный или внутриглазной и др. В некоторых случаях, например в для лечения ран и воспаления, соединение или композиция на его основе могут быть использованы непосредственно в виде раствора или аэрозоля.
Фармацевтические лекарственные формы включают в себя по крайней мере один описанный здесь химический состав и один или более фармацевтических наполнителей. Специалистам известно, что фармацевтические наполнители - вторичные компоненты, функцией которых является обеспечение или улучшение доставки лекарственного средства или препарата в виде целого ряда лекарственных форм (например, форм, для перорального приема типа таблеток, капсул и жидкостей; форм для местного применения в виде кожных, глазных и ушных форм; свечей; инъекций; форм для лечения заболеваний респираторного тракта и т.п.). Фармацевтические наполнители включают в свой состав инертные или неактивные компоненты, синергически действующие лекарственные средства или химикалии, которые существенно дополняют лекарственное действие активного компонента. Например, фармацевтические наполнители могут быть использованы для улучшения реологических характеристик, однородности, стабильности, вкуса или внешнего вида готовых лекарственных форм, облегчения обработки и назначения дозы, для удобства использования или контроля биологической усвояемости. Обычно фармацевтические наполнители описывают как инертные или неактивные, однако специалистам понятно, что выбор фармацевтических наполнителей оказывает влияние на свойства содержащих их лекарственных форм.
Фармацевтические наполнители, подходящие для использования в качестве переносчиков или разбавителей известны специалистам и могут быть использованы в целом ряде составов. См., например, Remington's Pharmaceutical Sciences, 18-е издание, А.R.Gennaro, редактор, Mack Publishing Company (1990); Remington: The Science and Practice of Pharmacy, 20-е издание, A.R.Gennaro, Editor, Lippincott Williams & Wilkins (200,0); Handbook of Pharmaceutical Excipients, 3-е издание, А.Н.Kibbe, редактор, American Pharmaceutical Association, and Pharmaceutical Press (2000); и Handbook of Pharmaceutical Additives, компилированный Michael и Irene Ash, Gower (1995), включенные в данное изобретение путем ссылки для всех целей.
Твердые лекарственные формы для перорального приема типа таблеток будут как правило, содержать один или более фармацевтических наполнителей, которые смогут, например, придать таким формам свойства, способствующие их удовлетворительной обработке, и возможность сжатия, или обеспечить таблетке дополнительные требуемые физические характеристики. Такие фармацевтические наполнители могут быть выбраны из числа разбавителей, связующих компонентов, глидантов, смазывающих веществ, дисинтегрантов, красителей, ароматизаторов, подсластителей, полимеров, воска или других материалов с пролонгированной растворимостью.
Композиции для внутривенного введения будут, как правило, содержать специальные жидкости, т.е. стерильные растворы простых химикалий типа сахара, аминокислот или электролитов, которые могут легко переноситься кровеносной системой и ассимилироваться. Такие жидкости приготавливают с помощью воды для инъекций USP.
Лекарственные формы для парентерального введения обычно включают жидкости, особенно внутривенные жидкости, т.е. стерильные растворы простых химикатов, таких как сахара, аминокислоты или электролиты, которые могут легко быть доставлены циркуляторной системой и усвоены. Такие жидкости приготавливают с помощью воды для инъекций USP. Жидкости, используемые обычно для внутривенного введения (IV), приведены в книге Remington, the Science and Practice of Pharmacy [см. выше], и среди них:
- спирт, например 5% спирт (например, в декстрозе и воде (D/W) или D/W в физиологическом растворе (NSS), включая в 5% декстрозе и воде (D5/W) или D5/W в NSS);
- синтетическая аминокислота типа Aminosyn, FreAmine, Travasol, например 3,5 или 7; 8,5; 3,5; 5,5 или 8,5% соответственно;
- хлорид аммония, например 2,14%;
- декстран 40 в NSS, например 10%, или в D5/W, например 10%;
- декстран 70 в NSS, например 6%, или в D5/W, например 6%;
- декстроза (глюкоза, D5/W), например, 2,5÷50%;
- декстроза и хлорид натрия, например 5÷20% декстроза и 0,22÷0,9% NaCl;
- раствор Лактат-Рингера (по Хартману), например NaCl 0,6%, KCl 0,03%, CaCl2 0,02%;
- лактат 0,3%;
- маннитол, например 5%, возможно в комбинации с декстрозой, например 10% или NaCl, например 15 или 20%;
- многочисленные растворы электролитов с различными комбинациями электролитов, декстрозы, фруктозы, инвертированного сахара Рингера, например NaCl 0,86%, KCl 0,03%, CaCl2 0,033%;
- бикарбонат натрия, например 5%;
- хлорид натрия, например 0.45, 0.9, 3 или 5%;
- лактат натрия, например 1/6 М, и
- стерилизованная вода для инъекции.
Показатель рН таких жидкостей IV может меняться и обычно составляет, как известно специалистам, от 3,5 до 8.
Химические составы по данному изобретению могут назначаться самостоятельно или в комбинации с другим лечением, таким как облучение, или другими терапевтические средствами, подобными агентам класса таксанов, которые действуют на образование микротрубочек, или ингибиторами топоизмеразы I класса камптотецинов. При подобном использовании терапевтические средства могут назначаться до, одновременно (либо отдельной дозой или в комбинации) или после приема активного агента по настоящему изобретению.
Рассматриваемые ниже примеры служат для более полного описания способа использования изложенного выше изобретения, а также для описания лучших способов реализации различных особенностей изобретения. Следует понимать, что эти примеры никоим образом не ограничивают истинный объем этого изобретения и носят скорее иллюстративный характер. Все публикации, включая патенты и заявки на патенты, цитируемые в этом описании изобретения, включены в данное изобретение путем ссылки, как если бы каждая отдельная публикация была бы специально и отдельно отмечена как включенная в данное изобретение путем ссылки полностью.
Примеры
Рассматриваемые ниже примеры служат для более полного описания способа использования изложенного выше изобретения, а также для описания лучших способов реализации различных особенностей изобретения. Следует понимать, что эти примеры никоим образом не ограничивают истинный объем этого изобретения и носят скорее иллюстративный характер. Все публикации, цитируемые здесь, полностью включены в данное изобретение путем ссылки.
Пример 1
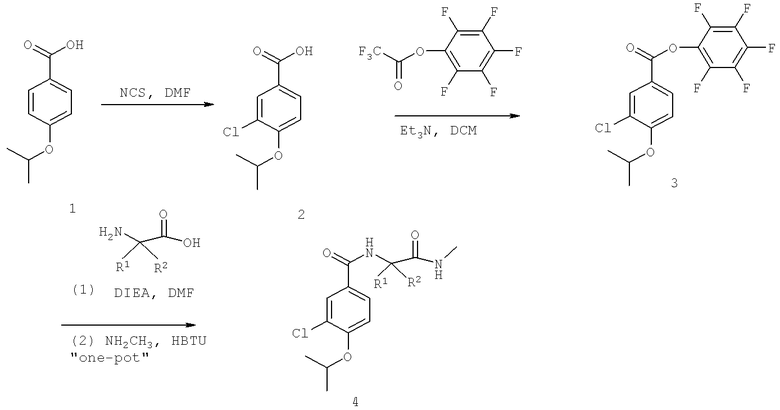
К раствору 4-изопропоксибензоловой кислоты 1 (25 г, 140 ммоль) в DMF был добавлен NCS (24 г, 182 ммоль). Реакционная смесь перемешивалась в течение ночи. В реакционную смесь добавлялся H2O (500 мл). Осадок собирался, промывался водой и высушивался в условиях ваккума для получения 2 (26,4 г, 88%) в виде белого твердого вещества, которое использовалось на следующем этапе без дальнейшей очистки. LRMS (M+H+) m/z 213,0.
К раствору 2 (20 г, 93 ммоль) в дихлорметане (DCM) добавлался пентафторотрифтороацетат (20 мл, 112 ммоль) и триэтиламин (17 мл, 112 ммоль) при 0°С. Реакционная смесь перемешивалась в течение 1 часа. Раствор концентрировался и смесь очищалась методом колоночной флеш-хроматографии (100% дихлорметана) для получения 3 (35 г, quant.) в виде белого твердого вещества.
К раствору 3 в DMF (0,2 М) была добавлена аминокислота (1,2 эквивалента) и N,N-диизопропилэтиламин (3 эквивалента). Реакционная смесь контролировалась LC/MS (жидкостной хроматографией/масс-спектрометрией). После завершения метиламин (2М в THF, 1,5 эквивалента) и HBTU (1,5 эквивалента) были добавлены к реакционному раствору. Реакционная смесь перемешивалась в течение 4 часов. Продукт очищался либо жидкостной хромотаграфией высокого разрешения (ЖХВР) или методом колоночной флеш-хроматографии для получения требуемого продукта 4.
Пример 2
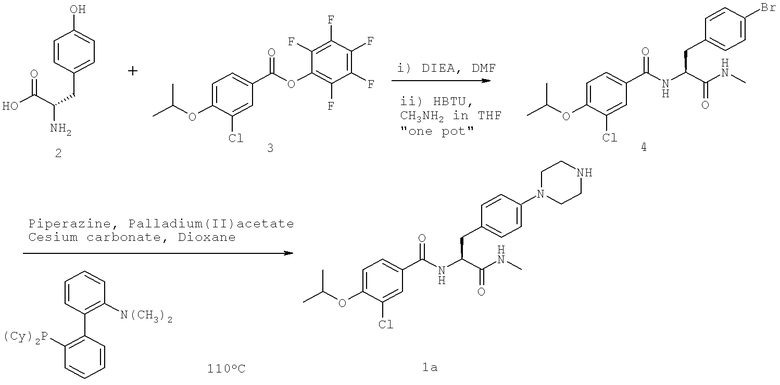
К раствору H-Phe(4-Br)-OH (2, 2,5 г, 10 ммоль) в DMF (20 мл) при комнатной температуре был добавлен 3 (4,7 г, 12 ммоль) и диизопропилэтиламин (5,4 мл, 30 ммоль). Реакционная смесь контролировалась LC/MS (жидкостной хроматографией/масс-спектрометрией). После завершения к реакционному раствору был добавлен метиламин (2М в THF, 7,7 мл, 15 ммоль) и HBTU (5,8 г, 15 ммоль). Реакционная смесь перемешивалась в течение 2 дней. Смесь отфильтровывалась и фильтрат очищался на RP-HPLC (программой обеспечения надежности при жидкостной хроматографии высокого разрешения) с использованием смеси ацетонитрила и H2O для получения 4 (2,3 г, 50%). LRMS (М+Н+) m/z 455,0.
К суспензии 4 (71 мг, 0,16 ммоль) в диоксане (1 мл) был добавлен пиперазин (16 мг, 0,19 ммоль), ацетат палладия (II) (4 мг, 0,016 ммоль), дициклогексилфосфино-2'-(N,N'-диметиламино)-бифенил (6 мг, 0,016 ммоль) и карбонат цезия (104 мг, 0,32 ммоль). Полученная в результате смесь перемешивалась в течение 36 часов при 110°С. Реакционная смесь была разбавлена EtOAc. Органический слой был промыт насыщенным NaHCO3 (20 мл) и соляным раствором, высушен над Na2SO4 и концентрирован. Осадок был очишен методом RP-HPLC (программой обеспечения надежности при жидкостной хроматографии высокого разрешения) с использованием смеси ацетонитрила и H2O для получения 1а (6 мг, 8%). LRMS (М+Н+) m/z 459,2.
Пример 3
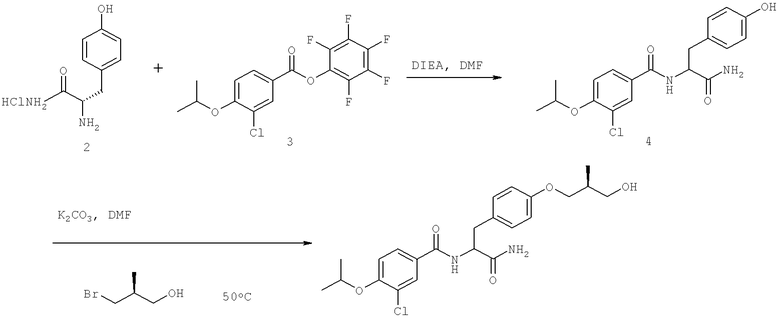
К раствору H-Tyr-NH2HCl (2, 830 мг, 3,8 ммоль) в DMF (5 мл) был добавлен 3 (1,8 г, 4,5 ммоль) и диизопропилэтиламин (3,4 мл, 19 ммоль) при комнатной температуре. Реакционная смесь перемешивалась в течение 20 часов и была отфильтрована после добавления воды. Белый осадок был рекристаллизован в дихлорметане и метаноле и был получен 4 в виде белых кристаллов (1,120 г, 78%). LRMS (М+Н+) m/z 377,1.
К раствору 4 (50 мг, 0,13 ммоль) в DMF (1 мл) был добавлен (s)-(+)-3-бромо-2-метил-1-пропанол (0,083 мл, 0,8 ммоль) и карбонат калия (110 мг, 0,8 ммоль). Полученная в результате смесь перемешивалась в течение 15 часов при 50°С. Смесь была отфильтрована, и фильтрат очищался методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 1b (30 мг, 51%).
LRMS (М+H+) m/z 449,1.
Пример 4
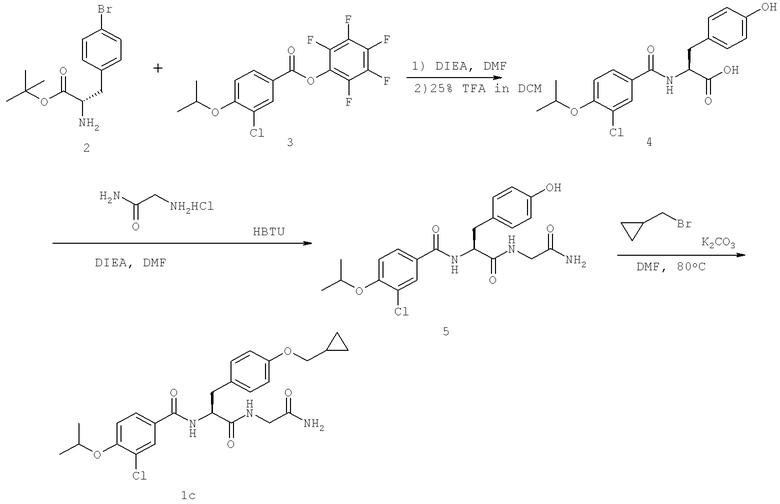
К раствору H-Tyr-OBut (2, 1,9 г, 8 ммоль) в DMF (50 мл) при комнатной температуре был добавлен 3 (2,4 г, 6,2 ммоль) и диизопропилэтиламин (3,3 мл, 19 ммоль). Реакционная смесь перемешивалась в течение 2 часов. Полученный в результате раствор был разбавлен EtOAc (200 мл) и промыт насыщенным NaHCO3 (50 мл). Органический слой был отделен, промыт соляным раствором, высушен над Na2SO4 и сконцентрирован для получения желтого твердого вещества. К раствору желтого твердого вещества в дихлорометане (10 мл) была добавлена трифтороуксуная кислота (30 мл). Смесь перемешивалась при комнатной температуре в течение 12 часов и затем была сконцентрирована при пониженном давлении. Осадок был высушен в условиях вакуума для получения 4 (3,1 г), который был использован в следующем этапе без дальнейшей очистки. LRMS (М-Н+) m/z 376,1.
К раствору 4 (3,1 г, 8 ммоль) в DMF был добавлен глицинамид гидрохлорид (1,1 г, 9,6 ммоль), диизопропилэтиламин (7 мл, 40 ммоль) и HBTU (3,6 г, 9,6 ммоль). Реакционная смесь перемешивалась в течение 15 часов, после чего раствор был разбавлен этилацетатом и промыт насыщенным NaHCO3. Органический слой был отделен, промыт соляным раствором, высушен над Na2SO4 и сконцентрирован. Полученный в результате неочищенный продукт был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 5 (38 мг, 35%). LRMS (M+H+) m/z 434,1.
К раствору 5 (100 мг, 0,23 ммоль) в DMF (1 мл) был добавлен циклопропилметил бромид (0,18 мл, 1,84 ммоль) и карбонат калия (317 мг, 2,3 ммоль). Полученная в результате смесь перемешивалась в течение 10 часов при 80°С. Смесь была отфильтрована и фильтрат был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 1 с (36 мг, 34%). LRMS (M+H+) m/z 488,1.
Пример 5
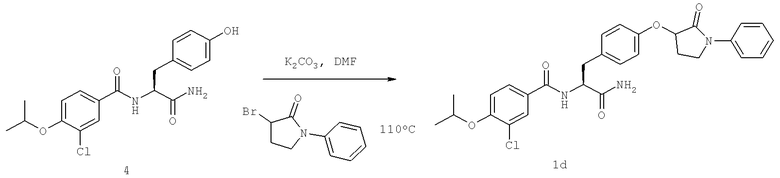
К раствору 4 (80 мг, 0,2 ммоль) в DMF (1 мл) был добавлен (±)-3-бромо-1-фенил-2-пирролидинон (250 мг, 1 ммоль) и карбонат калия (235 мг, 1,7 ммоль). Полученная в результате смесь была подвергнута перемешиванию в течение 10 часов при 110°С. Смесь была отфильтрована и фильтрат был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хроматографии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 5 (38 мг, 35%).
LRMS (M+H+) m/z 536,1.
Пример 6
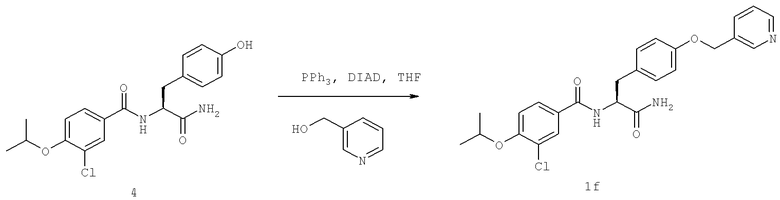
К раствору 4 (70 мг, 0,19 ммоль) в DMF (1 мл) был добавлен 3-(гидроксиметил)пиридин (0,023 мл, 0,23 ммоль), трифенилфосфин (100 мг, 0,38 ммоль) и диизопропилазодикарбоксилат (0,055 мл, 0,38 ммоль). Полученная в результате смесь была подвергнута перемешиванию в течение 20 часов при комнатной температуре. Реакционный раствор был сконцентрирован и очищен через колоночную флеш-хроматографию с использованием смеси этилацетата и гексана в качестве элюента для получения 1f (22 мг, 25%). LRMS (M+H+) m/z 468,2.
Пример 7
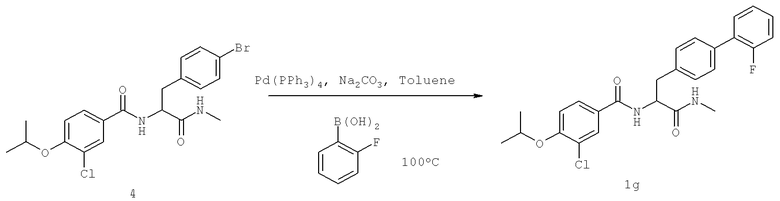
К раствору 4 (50 мг, 0,12 ммоль) в толуоле (2 мл) была добавлена 2-(фторофенил) бороновая кислота (20 мг, 0,14 ммоль), тетракис(трифенилфосфин)паладдий (0) (42 мг, 0,04 ммоль) и 2М карбоната натрия (0,18 мл, 0,36 ммоль). Реакционная смесь перемешивалась в течение 90 минут при 100°С. Полученный в результате раствор был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и H2O для получения 5 (22 мг, 40%). LRMS (M+H+) m/z 469,2.
Пример 8

К раствору 4 (45 мг, 0,1 ммоль) в DMF (1 мл) был добавлен бис(пиноколат)дибор (30 мг, 0,12 ммоль), 1,1'-бис(дифенилфосфино)ферроцен-палладий (II) дихлорид дихлорметановый комлекс (17 мг, 0,02 ммоль) и ацетат калия (39 мг, 0,4 ммоль). Реакционная смесь премешивалась около 1 часа при 80°С. К получаемой в результате смеси был добавлен 4-бромо-3,5-диметилизоксазол (35 мг, 0,2 ммоль) и 2М карбонат натрия (0,4 мл, 0,8 ммоль). Смесь перемешивалась при 80°С в течение 90 минут. Получаемый в результате остаток был отфильтрован и фильтрат был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и H2O для получения 1h (23 мг, 49%). LRMS (М+Н+) m/z 470,1.
Пример 9

К раствору 4 (62 мг, 0,14 ммоль) в DMF (1 мл) был добавлен бис(пинаколат)дибор (42 мг, 0,16 ммоль), 1,1'-бис(дифенилфосфино)ферроцен-палладий(II) дихлорид дихлорметановый комлекс (34 мг, 0,04 ммоль) и ацетат калия (54 мг, 0,55 ммоль). Реакционная смесь премешивалась около 1 часа при 80°С. К получаемой в результате смеси был добавлен N-метил-2-бромбензимидазол (58 мг, 0,27 ммоль) и 2М карбонат натрия (0,54 ммоль, 1,08 ммоль). Смесь перемешивалась при 80°С в течение 60 минут.
Полученный в результате раствор был разбавлен этилацетатом (20 мл) и промыт насыщенным NaHCO3. Органический слой был отделен, промыт соляным раствором, высушен над Na2SO4 и сконцентрирован. Полученный в результате осадок был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 1i (44 мг, 64%). LRMS (M+H+) m/z 505,1.
Пример 10

К раствору 4 (50 мг, 0,13 ммоль) в DMF (1 мл) был добавлен бис(пинаколат)дибор (34 мг, 0,13 ммоль), 1,1'-бис(дифенилфосфино)ферроцен - палладий (II) дихлорид дихлорметановый комплекс (27 мг, 0,03 ммоль) и ацетат калия (43 мг, 0,44 ммоль). Реакционная смесь промешивалась около 1 часа при 80°С. К получаемой в результате смеси был добавлен 3-бромтиофен-2-карбонитрил (41 мг, 0,22 ммоль) и 2М карбонат натрия (0,44 мл, 0,88 ммоль). Смесь перемешивалась при 80°С в течение 90 минут. Получаемый в результате остаток был отфильтрован и фильтрат был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 5 (20 мг, 37%). LRMS (M+H+) m/z 482,1.
Пример 11
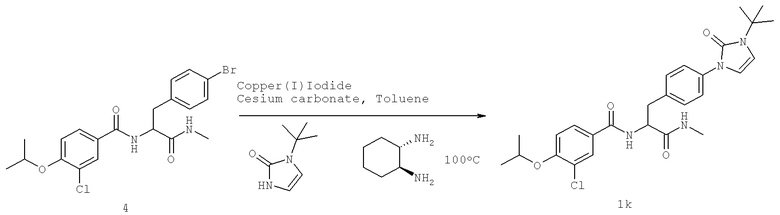
К раствору 4 (85 мг, 0,2 ммоль) в толуоле (2 мл) был добавлен 1-t-бутил-1,3-дигидро-имидазол-2-он (53 мг, 0,4 ммоль), иодид меди (I) (18 мг, 0,1 ммоль), транс-1,2-диамино-циклогексан (11 мг, 0,1 ммоль) и крабонат цезия (124 мг, 0,4 ммоль). Реакционная смесь премешивалась 4 часа при 100°С. Смесь была отфильтрована и фильтрат сконцентрирован. Получаемый в результате остаток был отфильтрован и фильтрат был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и H2O для получения 1k (27 мг, 28%). LRMS (M+H+) m/z 513,1.
Пример 12
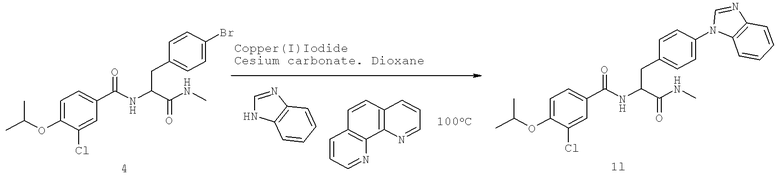
К раствору 4 (100 мг, 0,2 ммоль) в диоксане (2 мл) был добавлен бензимидазол (39 мг, 0,33 ммоль), иодид меди (I) (8,4 мг, 0,04 ммоль), 1,10-фенантролин (16 мг, 0,1 ммоль) и карбонат цезия (144 мг, 0,44 ммоль). Реакционная смесь премешивалась 15 часов при 100°С. Смесь была отфильтрована и фильтрат сконцентрирован. Получаемый в результате остаток был отфильтрован и фильтрат был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хроматографии высокого разрешения) с использованием смеси ацетонитрила и H2O для получения 11 (5,7 мг, 6%). LRMS (M+H+) m/z 491,1.
Пример 13
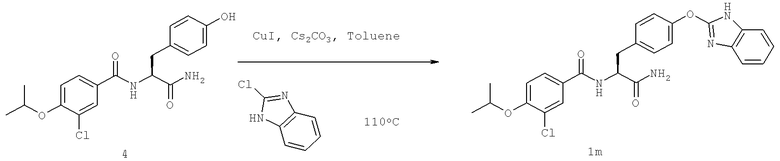
К раствору 4 (60 мг, 0,16 ммоль) в толуоле (1 мл) был добавлен 2-хлоробензимидазол (50 мг, 0,32 ммоль), иодид меди (I) (9 мг, 0,05) и карбонат цезия (105 мг, 0,32 ммоль). Реакционная смесь премешивалась 28 часов при 110°С. Смесь была отфильтрована и фильтрат был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 1m (8 мг, 10%).
LRMS (M+H+) m/z 493,0.
Пример 14
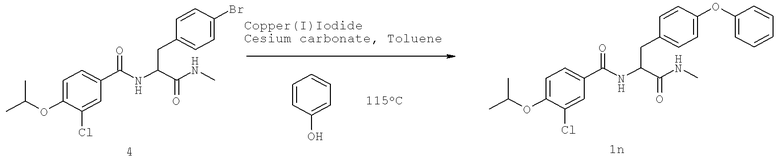
К раствору 4 (50 мг, 0,1 ммоль) в толуоле (1 мл) был добавлен фенол (21 мг, 0,2 ммоль), иодид меди (I) (6 мг, 0,03 ммоль) и карбонат цезия (72 мг, 0,2 ммоль). Реакционная смесь перемешивалась 5 часов при 115°С. Смесь была отфильтрована и фильтрат сконцентрирован. Получаемый в результате осадок был очищен методом колоночной флеш-хроматографии, используя смесь этилацетата и гексана в качестве элюэента для получения 1n (23 мг, 45%). LRMS (М+Н+) m/z 467,1.
Пример 15
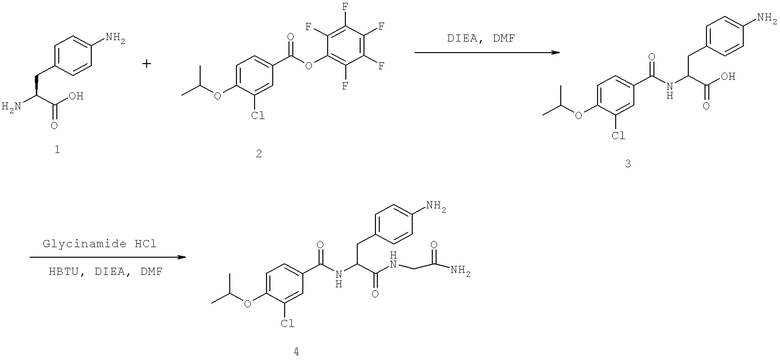
К раствору 1 (2,3 г, 12,6 ммоль) в DMF (30 мл) был добавлен 2 (4,0 г, 10,5 ммоль) и N,N-диизопропилэтиламин (5,2 мл, 30 ммоль). Реакция контролировалась LC/MS (жидкостной хромотаграфией/масс-спектрометрией). Получаемый в результате раствор использовался в следующем этапе без дальнейшей очистки. LRMS (M+H+) m/z 377,1.
К раствору неочищенного 3 в DMF (6 мл, ~2 ммоль) был добавлен глицинамид HCl (330 мг, 3 ммоль), HBTU (1,14 г, 3 ммоль) и N,N-диизопропилэтиламин (522 мкл, 3 ммоль). Реакционная смесь перемешивалась в течение ночи. Полученный в результате неочищенный продукт был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 4 (600 мг, 69% от 2). LRMS (M+H+) m/z 433,1.
Пример 16
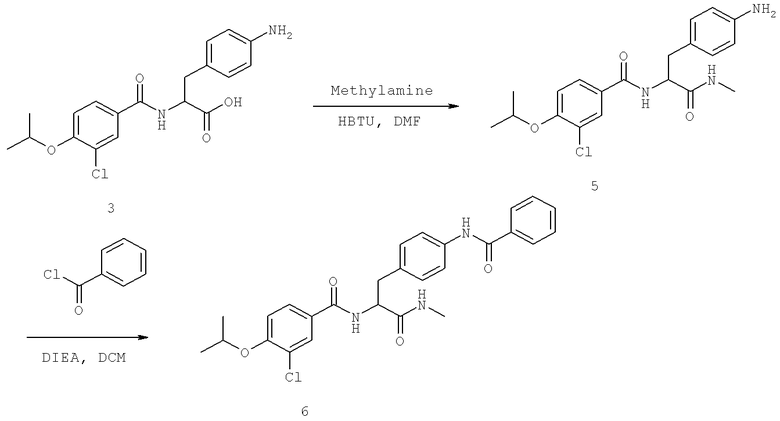
К раствору неочищенного 3 в DMF (15 мл, ~5,25 ммоль) был добавлен метиаламин (2 М в THF, 4 мл, 8 ммоль) и HBTU (3 г, 7,9 ммоль). Реакция была подвергнута перемешиванию в течение ночи. Смесь была разбавлена этилацетатом (200 мл). Органический слой был промыт Н2О, соляным раствором, высушен над сульфатом натрия и сконцентирован. Полученный в результате неочищенный 5 был использован в следующем этапе без дальнейшей очистки. LRMS (М+Н+) m/z 390,1.
К раствору неочищенного 5 (75 мг, ~0,2 ммоль) в DCM (2 мл) был добавлен хлорид бензоила (23 мкл, 0,2 ммоль) и N,N-диизопропилэтиламин (35 мкл, 0,2 ммоль). Реакционная смесь была подвергнута перемешиванию в течение ночи. Раствор был сконцентрирован и очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 6 (36 мг, 40% от 2). LRMS (M+H+) m/z 494,1.
Пример 17
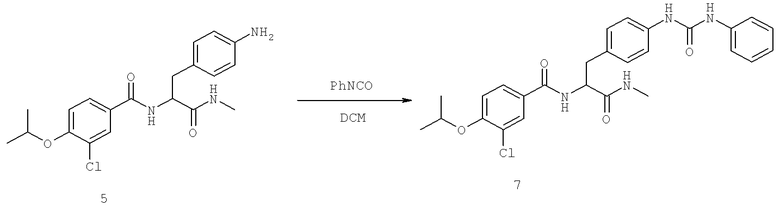
К раствору 5 (75 мг, ~0,2 ммоль) в DCM (2 мл) был добавлен фенилизоцианат (26 мкл, 0,24 ммоль). Реакционная смесь была подвергнута перемешиванию в течение ночи. Раствор был сконцентрирован и очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 7 (40 мг, 39% от 2). LRMS (M+H+) m/z 509,1.
Пример 18
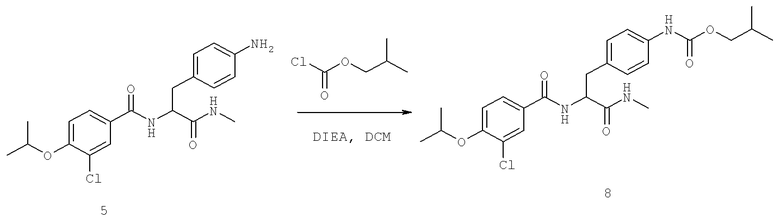
К раствору 5 (75 мг, 0,19 ммоль) в DCM (3 мл) был добавлен изобутил хлороформат (38 мкл, 0,29 ммоль) и N,N-диизопропилэтиламин (50 мл, 0,29 ммоль). Реакционная смесь была подвергнута перемешиванию в течение ночи. Раствор был сконцентрирован и очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 8 (45 мг, 48% от 2). LRMS (M+H+) m/z 490,1.
Пример 19
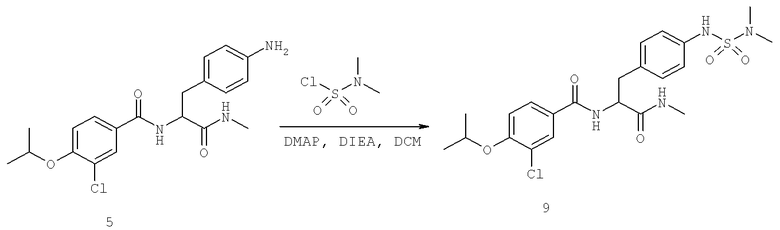
К раствору 5 (75 мг, 0,19 ммоль) в DCM (5 мл) был добавлен диметилсульфамоил хлорид (30 мкл, 0,29 ммоль), N,N-диизопропилэтиламин (50 мкл, 0,29 ммоль) и DMAP (50 мг, 0,4 ммоль). Реакционная смесь была подвергнута перемешиванию в течение ночи. Реакционная смесь затем была нагрета до 30°С и перемешивание продолжилось в течение 8 часов. Полученный в результате раствор был сконцентрирован и очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 9 (30 мг, 32%). LRMS (M+H+) m/z 497,1.
Пример 20
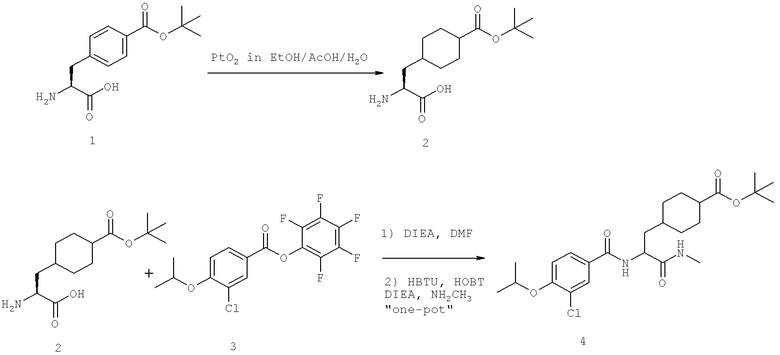
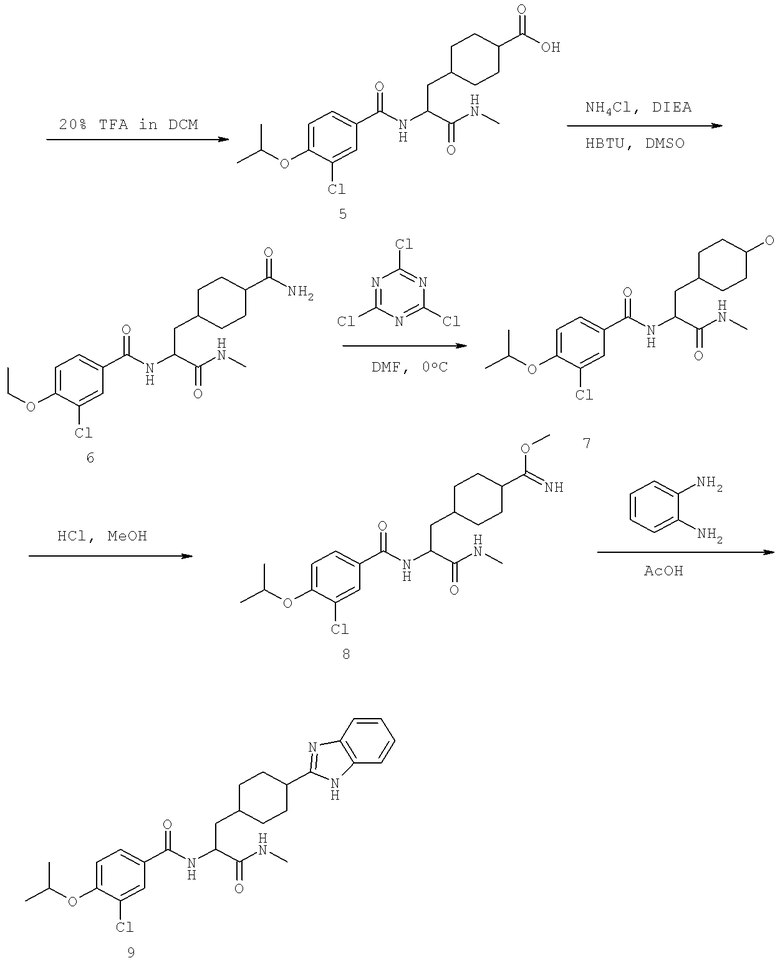
К раствору H-Phe(4-COOtBU)-OH (5,8 г, 22 ммоль) в этилацетате (60 мл) и воде (20 мл) была добавлен оксид платины (IV) (400 мг, 1,8 ммоль) и уксусная кислота (50 мл). Реакционная смесь перемешивалась под потоком H2 (60 psi) в течение 20 часов. Катализатор был удален фильтрацией через политетрафторэтиленовый (PTFE) фильтр (0,45 мкм) и растворитель был выпарен для получения 2 (5,9 г), который использовался в следующем этапе без дальнейшей очистки. LRMS (M+H+) m/z 272,1.
К раствору 2 (6,9 г, 18 ммоль) в DMF (30 мл) был добавлен 3 (5,9 г, 21,8 ммоль) и N,N-диизопропилэтиламин (9,5 мл, 54,3 ммоль). Реакция контролировалась LC/MS(жидкостной хромотаграфией/масс-спектрометрией). После завершения к реакционному раствору был добавлен 2 М метиламин в THF (13,6 мл, 27 ммоль), HOBt (4 г, 27 ммоль) и HBTU (10 г, 27 ммоль). Реакция перемешивалась в течение 4 часов. Смесь была разбавлена этилацетатом (60 мл) и промывалась насыщенным NaHCO3 (20 мл). Органический слой был отделен, а водная фаза экстрагировалась этилацетатом (2×50 мл). Комбинированные органические слои были промыты соляным раствором, высушены над сульфатом натрия и сконцентированы. Полученный в результате сырой продукт был очищен через колоночную флеш-хроматографию с использованием смеси этилацетата и гексана в качестве элюэнта для получения 4 (изомер cis 808 мг 1,68 ммоль, транс-изомер 300 мг, 0,63 ммоль). LRMS (M+H+) m/z 481,1.
К раствору 4 (290 мг, 0,6 ммоль) в дихлорметане (20 мл) была добавлена трифтороуксусная кислота (5 мл). Полученный в результате раствор перемешивался при комнатной температуре в течение 1 часа и затем концентрировался при пониженном давлении. Остаток был очищен через колоночную флеш-хроматографию с использованием смеси 99% этилацетата и 1% уксусной кислоты в качестве элюэнта для получения 5 в виде белого твердого вещества (140 мг, 55%). LRMS (M+H+) m/z 425,1.
К раствору 5 (330 мг, 0,7 ммоль) в DMSO (5 мл) был добавлен хлорид аммония (83 мг, 1,5 ммоль), диизопропилэтиламин (0,27 мл, 1,5 ммоль) и HBTU (580 мг, 1 ммоль). Полученный в результате раствор перемешивался при комнатной температуре в течение 15 часов. Был добавлен дополнительный хлорид аммония (37 мг, 0,7 ммоль), диизопропилэтиламин (0,12 мл, 0,7 ммоль) и HBTU (266 мг, 0,7 ммоль). Перемешивание продолжалось в течение дополнительных 5 часов, смесь была разбавлена этилацетатом (50 мл) и промыта насыщенным NaHCO3 (20 мл). Органический слой был отделен и водная фаза экстрагирована этилацетатом (2×50 мл). Комбинированные органические слои были промыты соляным раствором, высушены над сульфатом натрия и сконцентрированы для получения слегка желтоватого неочищенного продукта. Остаток был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 6 (128 мг, 30%). LRMS (М+H+) m/z 424,1.
К раствору 6 (94 мг, 0,2 ммоль) в DMF (2 мл) был добавлен цианурхлорид (45 мг, 0,2 ммоль) при 0°С и реакционная смесь перемешивалась в атмосфере азота. После 1 часа реакционный раствор был сконцентрирован для получения 7 (79 мг), который использовался в следующем этапе без дальнейшей очистки. LRMS (М+H+) m/z 406,1.
Раствор 7 (17 мг, 0,04 ммоль) в метаноле (2 мл) перемешивался в потоке HCl в течение 15 минут.Поток азота был затем барботирован через реакционную смесь. После 1 часа реакционный раствор был сконцентирован для получения 8, который был использован в следующем этапе без дальнейшей очистки. LRMS (М+Н+) m/z 438.1.
К раствору неочищенного 8 (17 мг, ~0,04 ммоль) в уксусной кислоте (2 мл) был добавлен о-фенилиндиамин (50 мг, 0,46 ммоль) и полученный в результате раствор перемешивался при 80°С в течение 1 часа. Реакционная смеь была сконцентрирована и очищена через препаративную тонкослойную хроматографию с использованием 5% метанола в дихлорметане в качестве элюента для получения белого твердого вещества.
Твердое вещество было очищено методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 9 (9 мг, 45%). LRMS (М+Н+) m/z 497,1.
Пример 21
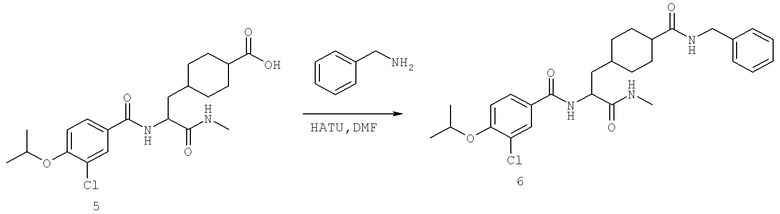
К раствору 5 (50 мг, 0,12 ммоль) в DMF (1 мл) был добавлен бензиламин (16 мг, 0,14 ммоль) и HATU (57 мг, 0,14 ммоль). Реакционная смесь перемешивалась при комнатной температуре в течение 3 часов. Полученный раствор был отфильтрован и фильтрат был очищен методом RP-HPLC (программой обеспечения надежности при жидкостной хромотаграфии высокого разрешения) с использованием смеси ацетонитрила и Н2О для получения 6 (16 мг, 26%). LRMS (М+Н+) m/z 514,1.
Примеры 22-24
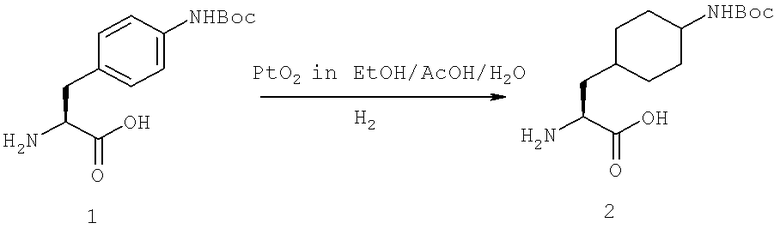
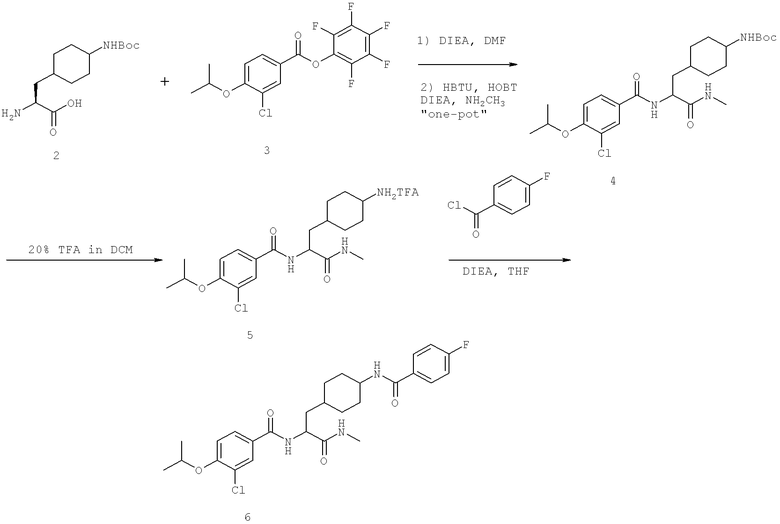
К раствору H-Phe(4-NHBoc)-OH (4,8 г, 17,1 ммоль) в этаноле (60 мл), метаноле (20 мл), уксусной кислоте (60 мл) и воде (30 мл) была добалена окись платины (IV) (360 мг, 1,6 ммоль). Реакционная смесь перемешивалась под потоком H2 (45 psi (фунт/кв.дюйм)) в течение 20 часов. Катализатор был удален фильтрацией через фильтр ПТФЭ (политетрафтороэтилен) (0,45 мкм) и растворитель выпаривался для получения 2 (5 г), который использовался в следующем этапе без дальнейшей очистки. LRMS (М+H+) m/z 287,1.
К раствору неочищенного 2 (3,2 г, 11,2 ммоль) в DMF (20 мл) был добавлен 3 (3,8 г, 10 ммоль) и N,N-диизопропилэтиламин (5,2 мл, 30 ммоль). Реакция контролировалась LC/MS. После завершения к реакционному раствору был добавлен метиламин (2 М в THF, 7,5 мл, 15 ммоль) и HBTU (5,7 г, 15 ммоль). Реакция перемешивалась в течение ночи. Смесь была разбавлена этилацетатом (60 мл) и промыта насыщенным NaHCO3. Органический слой был отделен и водная фаза была экстрагирована этилацетатом (2×50 мл). Комбинированные органические слои были промыты соляным раствором, высушены над сульфатом натрия и сконцентрированы. Полученное неочищенное вещество было очищено через RP-HPLC с использованием смеси ацетонитрила и Н2О для получения 4 (1,0 г, 18% от 1). LRMS (М+Н+) m/z 496,1.
К раствору 4 (1,0 г, 2,0 ммоль) в дихлорометане (25 мл) была добавлена трифтороуксусная кислота (8 мл). Полученный в результате раствор перемешивался при комнатной температуре в течение 4 часов и затем сконцентрирован при пониженном давлении. Остаток был очищен через RP-HPLC с использованием смеси ацетонитрила и Н2О для получения 5 (820 мг, 79%). LRMS (M+H+) m/z 396,1.
К раствору 5 (75 мг, 0.16 ммоль) в THF (3 мл) был добавлен 4-фторобензоил хлорид (28 мкл, 0,23 ммоль) и N,N-диизопропилэтиламин (100 мкл, 0,57 ммоль). Реакционная смесь перемешивалась в течение ночи. Полученный в результате раствор был сконцентрирован и очищен на RP-HPLC с использованием смеси ацетонитрила и Н2О для получения 6 (65 мг, 78%). LRMS (M+H+) m/z 518,1.
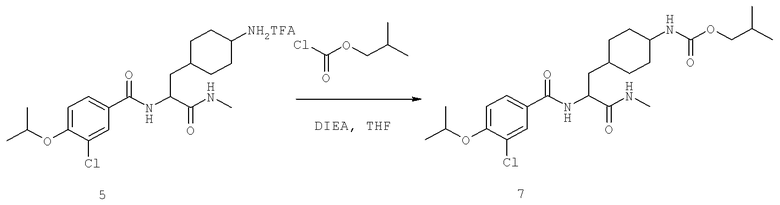
К раствору 5 (75 мг, 0,16 ммоль) в THF (3 мл) был добавлен изобутил хлороформат (30 мкл, 0,23 ммоль) и N,N-диизопропилэтиламин (100 мкл, 0,57 ммоль).
Реакционная смесь перемешивалась в течение ночи. Полученный в результате раствор был сконцентрирован и очищен на RP-HPLC с использованием смеси ацетонитрила и H2O для получения 7 (62 мг, 78%). LRMS (М+H+) m/z 496,1.
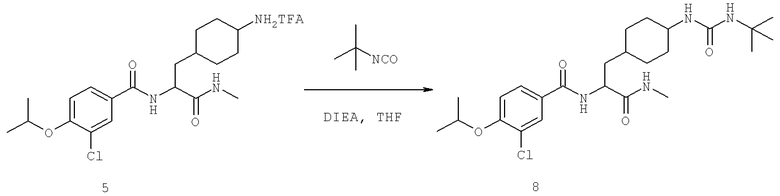
К раствору 5 (75 мг, 0,16 ммоль) в THF (3 мл) был добавлен терт-бутил изоцианат (26 мкл, 0,23 ммоль) и N,N-диизопропилэтиламин (100 мкл, 0,57 ммоль). Реакционная смесь перемешивалась в течение ночи. Полученный в результате раствор был сконцентрирован и очищен на RP-HPLC с использованием смеси ацетонитрила и H2O для получения 7 (55 мг, 69%). LRMS (M+H+) m/z 495,1.
Пример 25
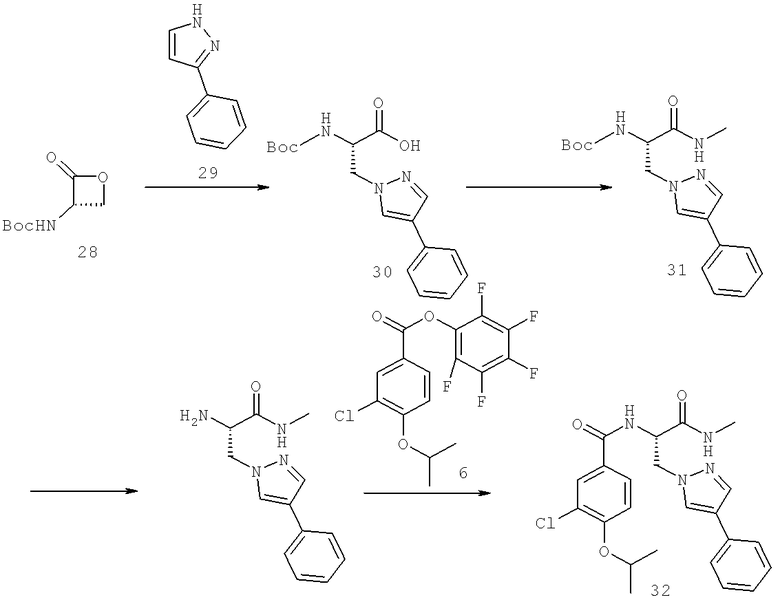
К раствору Boc-L-серин-бета-лактона 28 (200 мг, 1,07 ммоль) в ацетонитриле (5 мл) был добавлен 29 (154 мг, 1,07 ммоль). Смесь перемешивалась при 56°С в течение ночи. Она была сконцентрирована при пониженном давлении для получения 30.
Неочищенный 30 был повторно растворен в DMF (1 мл), обработанном метиламином (2М в THF) (0, 54 мл, 1,08 ммоль) и HBTU (404 мг, 1,07 ммоль). Смесь перемешивалась в течение 1 часа, после чего она была отфильтрована и фильтрат был очищен с помощью обратнофазовой HPLC (С 18), используя смесь ацетонитрила и Н2О для получения 31 (50,0 г, 14%).
К раствору 31 (50,0 г, 0,145 ммоль) в DCM (5 мл) был добавлен TFA (5 мл) при комнатной температуре. Реакционная смесь перемешивалась в течение 20 минут. Растворители выпаривались при пониженном давлении и остаток повторно осаждался в DMF (100 мл), за чем следовало добавление 6 (66,3 мг, 0,174 ммоль) и диизопропилэтиламина (51 мкл, 0,290 ммоль) при комнатной температуре. Реакционная смесь перемешивалась в течение 1 часа, затем концентриовалась при пониженном давлении и осадок очищался на колонке для отгонки легких фракций с использованием силикагеля (гексан : EtOAc, 1:1) для получения 32 (50,0 мг, 78,2%). LCMS (M+H+) m/z 441,1.
Пример 26
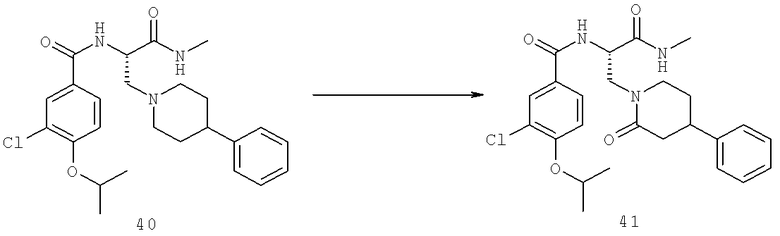
К раствору 40 (50,0 г, 0,0874 ммоль) в воде (1 мл) и метаноле (1 мл) был добавлен EDTA натрия (88,1 мг, 0,262 ммоль) и Hg(OAc)2 (83,7 мг, 0,262 ммоль). Реакционная смесь перемешивалась при 100°С в течение 1 часа и затем концентировалась при пониженном давлении. Осадок очищался на колонке для отгонки легких фракций с использованием силикагеля (DCM:MeOH, 10:1) для получения 41 (29 мг, 71%). LCMS (М+H+) m/z 472,4.
Пример 27
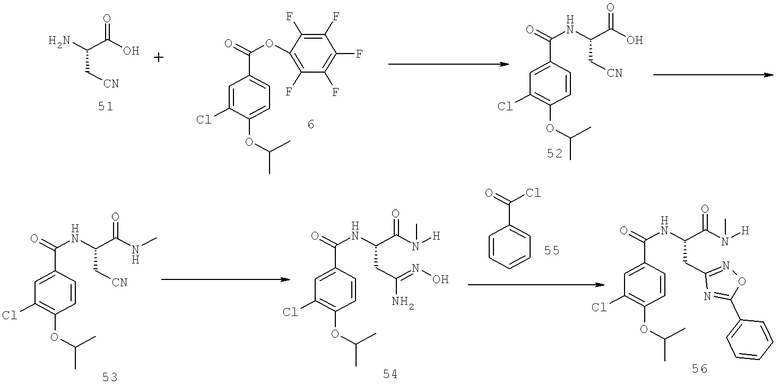
К перемешиваемому раствору 51 (1,0 г, 8,76 ммоль) в DMF (20 мл) был добавлен 6 (3,34 г, 8, 76 ммоль) и диизопропилэтиламин (2,30 мл, 13,1 ммоль) при комнатной температуре. Реакционная смесь контролировалась с помощью обратнофазовой HPLC/MS. После завершения 2 М метиламина в THF (8,80 мл, 17,5 ммоль) и HBTU (4,97 г, 13,1 ммоль) было добавлено к реакционному раствору. После перемешивания в течение 1 часа реакционная смесь концентрировалась и очищалась на колонке для отгонки легких фракций с использованием силикагеля (гексан : EtOAc, 1:1) для получения 53 (1,0 мг, 35,3%).
К раствору 53 (1,0 г, 3,09 ммоль) в этаноле (20 мл) был добавлен триэтиламин (0,517 мл, 3,71 ммоль) и гидроксиамин гидрохлорид (258 мг, 3,71 ммоль). После перемешивания в противотоке в течение 24 часов растворители были выпарены при пониженном давлении. Осадок был очищен на обратнофазовой HPLC (С 18) с использованием смеси ацетонитрила и Н2О для получения 54 (280 мг, 25%).
К перемешиваемому раствору 54 (280 мг, 0,785 ммоль) в THF (50 мл) был добавлен диизопропилэтиламин (164 мкл, 0,942 ммоль) и бензоилхлорид (100 мкл, 0,864 ммоль) при комнатной температуре. После перемешивания в течение 30 минут при комнатной температуре реакционная смесь была сконцентрирована, осадок был растворен в НОАс (100 мл) и смесь перемешивалась в сосуде с обратным холодильником в течение 5 часов. Растворители удалялись при пониженном давлении и остаток очищался на колонке для отгонки легких фракций с использованием силикагеля (гексан : EtOAc, 1:1) для получения 56 (49 мг, 14,1%). LCMS (M+H+) m/z 443,1.
Пример 28
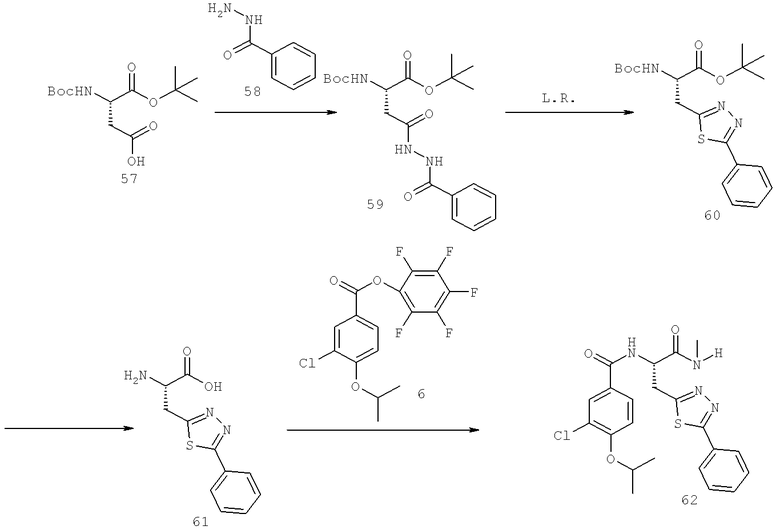
К раствору 57 (3,0 г, 10,4 ммоль) в DMF (50 мл) был добавлен 58 (1,69 г, 12,4 ммоль) и HBTU (5,92 г, 15,6 ммоль). Реакционная смесь контролировалась обратнофазовой HPLC/MS. После перемешивания в течение 5 часов растворители были выпарены при пониженном давлении и осадок очищался на колонке для отгонки легких фракций с использованием силикагеля (гексан : EtOAc, 1:1) для получения 59 (3,50 г, 82%).
К раствору 59 (200 мг, 0,491 ммоль) в толуоле (5 мл) был добавлен реагент Лоуэссона (Lawesson's Reagent) (109 мг, 0,270 ммоль). После перемешивания при 100°С в течение 30 минут растворители были выпарены при пониженном давлении. Осадок очищался на колонке для отгонки легких фракций с использованием силикагеля (гексан : EtOAc, 1:1) для получения 60 (160 мг, 80%).
К раствору 60 (150 мг, 0,431 ммоль) в DCM (5 мл) был добавлен TFA (5 мл) при комнатной температуре. Реакционная смесь перемешивалась в течение 2 часов. Растворители были выпарены при пониженном давлении и осадок 61 (121 мг, 100%) высушивался в вакууме в течение ночи.
К перемешиваемому раствору 61 (0,395 ммоль) в DMF (5 мл) был добавлен 6 (180 мг, 0,473 ммоль) и диизопропилэтиленамин (138 мкл, 0,790 ммоль) при комнатной температуре. Реакционная смесь контролировалась HPLC/MS. После завершения 2 М метиламина в THF (395 мкл, 0,790 ммоль) и HBTU (225 мг, 0,593 ммоль) были добавлены к реакционному раствору. Реакционная смесь перемешивалась в течение 30 мин, после чего смесь была отфильтрована и фильтрат был очищен обратнофазовой HPLC с использованием смеси ацетонитрила и Н2О для получения 62 (70,0 мг, 38,6%). LCMS (M+H+) m/z 459,0.
Пример 29
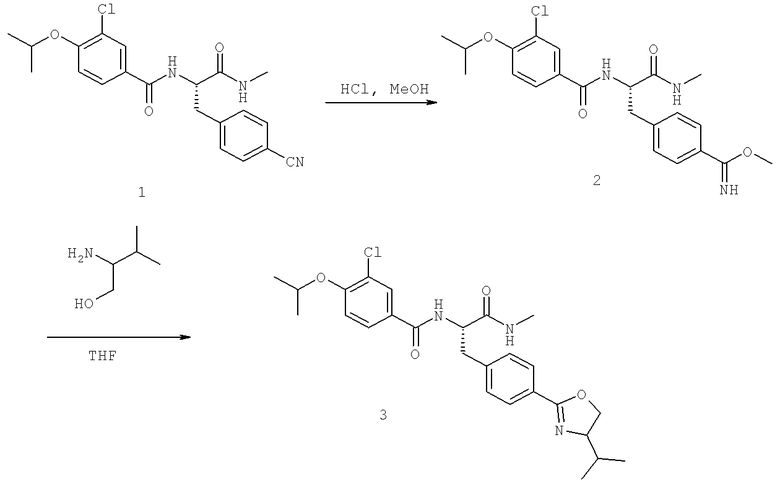
Раствор нитрила 1 (640 мг, 1,6 ммоль) и МеОН (25 мл) при 0°С был насыщен газом HCl. Сосуд для проведения реакции был нагрет до 23°С. После 2 часов при 23°С реакционный раствор был сконцентрирован в условиях ваккума и полученный в результате остаток 2 использовался без дальнейшей очистки.
Раствор неочищенного имидата 2 (50 мг, 0,12 ммоль), 2-амино-3-метил-пропанола (36 мг, 0,35 ммоль) и THF (1 мл) перемешивался при 80°С в течение 30 минут. Реакционный раствор был затем сконцентрирован в условиях ваккума и полученный в результате остаток растворен в EtOAc (10 мл) и промыт 1 N NaOH (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 100% EtOAc) и получено количество 25 мг (43%) оксазола 3. LRMS (M+H+) m/z 486,3.
Пример 30
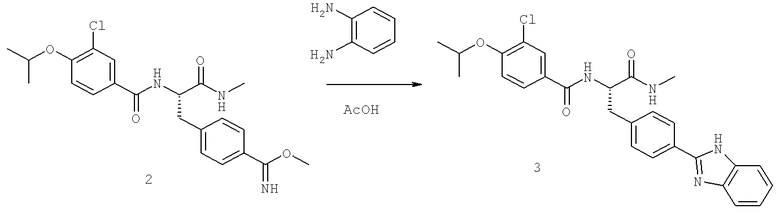
Раствор неочищенного имидата 2 (50 мг, 0,12 ммоль), фенилен диамина (36 мг, 0,32 ммоль) и уксусной кислоты (1 мл) перемешивался при 80°С в течение 30 минут. Реакционная смесь была сконцентрирована в условиях ваккума и полученный в результате остаток растворен в EtOAc (10 мл) и промыт 1 N NaOH (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:3 гексаны : EtOAc) и получено 20 мг (34%) бензимидазола 3. LRMS (M+H+) m/z 491,2.
Пример 31
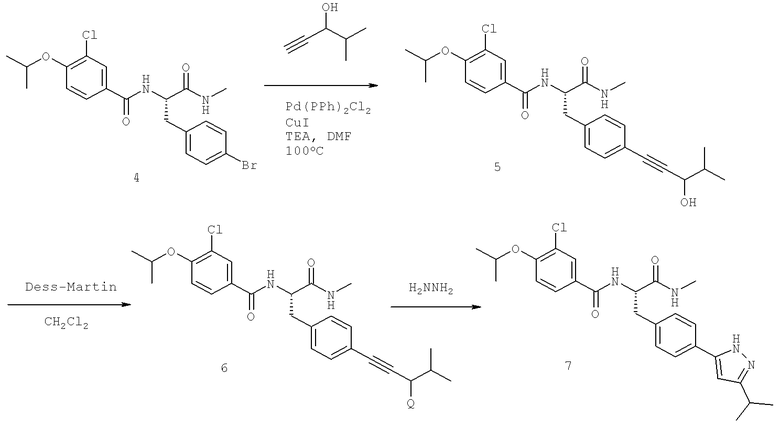
Раствор бромида 4 (500 мг, 1,1 ммоль), 4-метил-1-пентил-3-ола (0,15 мл, 1,32 ммоль), бис(трифенилфосфин)палладий (II) хлорида (390 мг, 0,55 ммоль), иодида меди (52 мг, 0,28 ммоль), триэтиламина (5 мл) и DMF (10 мл) перемешивался при 100°С в течение 3 часов. Реакционная смесь была сконцентрирована в условиях ваккума и полученный в результате остаток растворен в EtOAc (50 мл) и промыт 0,1 N HCl (3×20 мл) и соляным раствором (20 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:1 гексаны : EtOAc) и было получено 200 мг (42%) ацетилена 5. LRMS (M+H+) m/z 471,2.
Раствор спирта 5 (50 мг, 0,12 ммоль), периодинан Десс-Мартина (90 мг, 0,21 ммоль) и CH2Cl2 (3 мл) перемешивался при 23°С в течение 2 часов. Реакционная смесь была растворена в EtOAc (20 мл) и промыта насыщенным водным NaHCO3 (10 мл) и соляным раствором (20 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный остаток использовался напрямую.
Раствор неочищенного кетона 6 (30 мг, 0,06 ммоль), гидразина (0,13 мл, 1,0 М в THF)
и DMF (1 мл) перемешивался при 23°С в течение 12 часов. Реакционная смесь была затем растворена в EtOAc (10 мл) и промыта 0, 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный остаток был очищен обратнофазовой HPLC (С 18, ацетонитрил/вода) для получения выхода 5 мг (18%) пиразина 7. LRMS (M+H+) m/z 483,2.
Пример 32
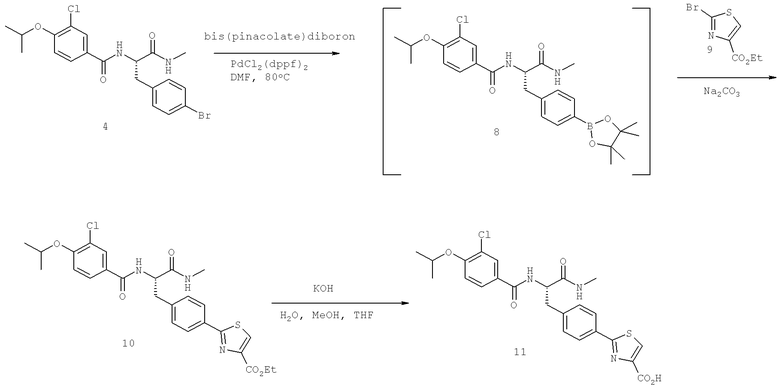
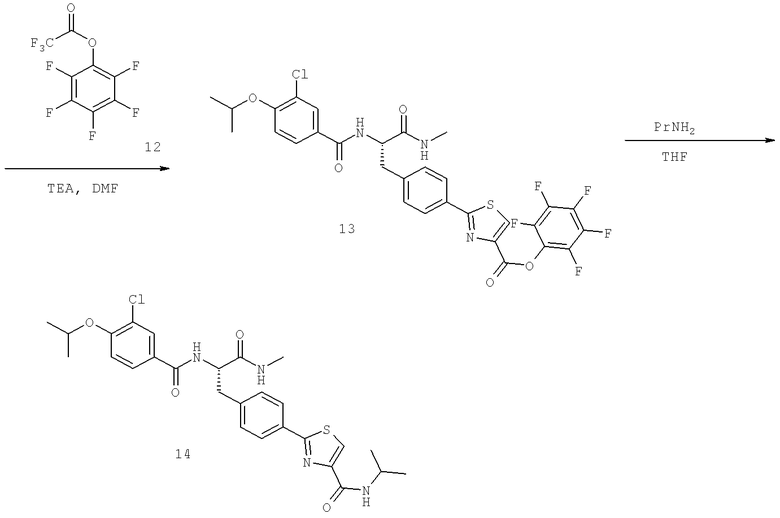
Раствор бромида 4 (500 мг, 1,1 ммоль), бис(пинаколат)дибора (420 мг, 1,65 ммоль), ацетата калия (433 мг, 4,4 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлор палладия (II) (180 мг, 0,22 ммоль) и DMF перемешивался при 80°С в течение 3 минут. Затем был добавлен бромид 9 (366 мг, 4,65 ммоль) и Na2CO3 (4,4 мл, 2,0 М в Н2О) и смесь перемешивалась при 80°С в течение 2 часов. Реакционная смесь затем была разбавлена в EtOAc (50 мл), слои были отделены и органический слой был промыт 0,1 N HCl (10 мл) и соляным раствором (10 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученый в результате остаток был очищен обратнофазовой HPLC (С 18, ацетонитрил/вода) для получения 165 мг (28%) тиазола 10. LRMS (M+H+) m/z 531,2.
Раствор эфира 10 (165 мг, 0,31 ммоль), гидроокиси калия (35 мг, 0,62 ммоль), Н2О (1 мл), МеОН (1 мл) и THF (2 мл) перемешивался при 50°С в течение 2 часов. Реакционная смесь затем была разбавлена EtOAc (20 мл) и промыта 1 N HCl (5 мл) и соляным раствором (10 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума, а полученный в результате остаток использовался напрямую.
Раствор кислоты 11 (140 мг, 0,28 ммоль), пентафторофенол трифтороацетата 12 (96 мкл, 0,56 ммоль), триэтиламина (77 мкл, 0,56 ммоль) и DMF (4 мл) перемешивался при 23°С в течение 2 часов. Реакционная смесь затем была разбавлена EtOAc (20 мл) и промыта 1 N HCl (5 мл), насыщенным водным NaHCO3 (5 мл) и соляным раствором (10 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:1 гексаны : EtOAc) для получения выхода 80 мг эфира 13.
Раствор эфира 13 (20 мг, 0,03 ммоль), изопропиламина (5 мкл, 0,06 ммоль) и THF (1 мл) перемешивался при 23°С в течение 12 часов. Реакционная смесь затем была разбавлена EtOAc (20 мл) и промыта 1 N HCl (5 мл), насыщенным водным NaHCO3 (5 мл) и соляным раствором (10 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:3 гексаны : EtOAc) для получения 9 мг (55%) амида 14. LRMS (M+H+) m/z 543,2.
Пример 33
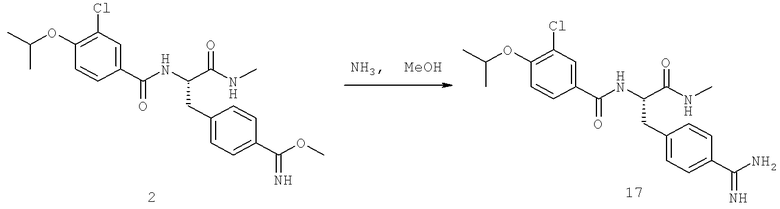
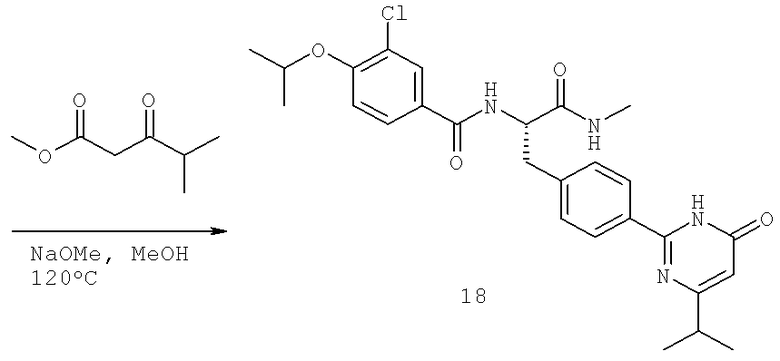
Раствор имидата 2 (1,6 г, 4,0 ммоль) и 2,0 М NH3 в МеОН (10 мл) перемешивался при 23°С в течение 12 часов. Реакционная смесь была затем сконцентрирована в условиях вакуума и полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:10 CH2Cl2:МеОН) для получения количества 1,3 г (78%) амидина 17. LRMS (М+Н+) m/z 417,2.
Раствор амидина 17 (50 мг, 0,12 ммоль), метил изобутирилацетата (17 мкл, 0,12 ммоль) и NaOMe (0,5 М в МеОН, 0,72 мл) перемешивался при 120°С в течение 1 часа. Реакционная смесь была затем сконцентрирована в условиях вакуума и полученный в результате остаток был очищен обратнофазовой HPLC (С 18, ацетонитрил/вода) для получения 10 мг (16%) пиримидина 18. LRMS (M+H+) m/z 511,2.
Пример 34
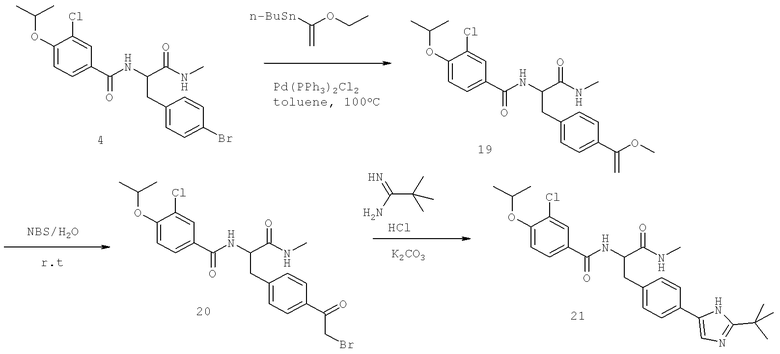
Раствор бромида 4 (1,0 г, 2,20 моль), дихлоробис(трифенилфосфин) палладия (II) (154 мг, 0,220 моль), трибутил(1-этоксивинил) олова (1,49 мл, 4,41 ммоль) и толуола (15 мл) перемшивался в атмосфере N2 при 100°С в течение 6 часов. После завершения контролируемая LCMS реакционная смесь была охлаждена, отфильтрована через Целит (Celite) и сконцентрирована в условиях ваккума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 2:1:0,1 EtOAc : гексаны : триэтиламин) для получения 540 мг (55%) стирола 19. LRMS (M+H+) m/z 445,2.
Раствор соединения 19 (540 мг, 1,21 ммоль), THF:H2O (3:1, 12 мл) и N-бромо-сукцинимида (216 мг, 1,21 ммоль) перемешивался при 23°С в течение 15 минут. Реакционная смесь затем была сконцентрирована в условиях ваккума и неочищенный остаток разбавлен EtOAc (30 мл), промыт соляным раствором (10 мл) и сконцентрирован в условиях ваккума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:1 EtOAc : гексаны) для получения 210 мг (35%) бромокетона 20. LRMS (M+H+) m/z 495,1.
Раствор бромокетона 20 (210 мг, 0,42 ммоль), K2CO3 (174 мг, 1,26 ммоль), терт-бутилкарбамидин гидрохлорида (115 мг, 0,84 ммоль) и DMF (4 мл) перемешивался при 23°С в атмосфере N2 в течение 18 часов. Реакционная смесь была затем сконцентрирована под высоким ваккумом (0,1 мм Hg) и полученный в результате остаток был очищен колоночной хроматографией (силикагель, 4:1 EtOAc : гексаны) для получения 50 мг (24%) имидазола 21. LRMS (M+H+) m/z 497,2.
Пример 35
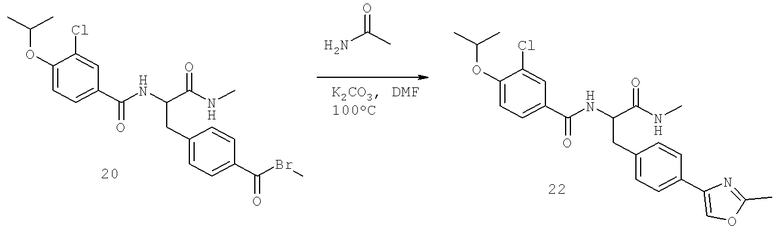
Раствор бромокетона 20 (25 мг, 0,05 ммоль), K2CO3 (14 мг, 0,1 ммоль), ацетамида (6 мг, 0,1 ммоль) и DMF (1 мл) перемешивался при 100°С в течение 4 часов. Реакционная смесь была разбавлена EtOAc (15 мл), промыта соляным раствором (3×10 мл) и сконцентрирована в условиях вакуума. Полученный остаток был очищен колоночной хромотографией (силикагель, 2:1 EtOAc : гексаны) для получения 5 мг (22%) оксазола 22. LRMS (M+H+) m/z 446,2.
Пример 36
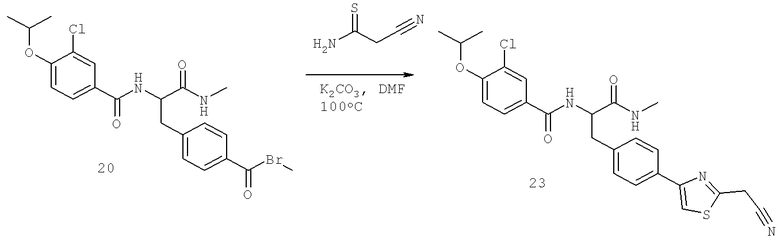
Раствор бромокетона 20 (25 мг, 0,05 ммоль), K2CO3 (14 мг, 0,1 ммоль), цианотиоацетамида (10 мг, 0,1 ммоль) и DMF (1 мл) перемешивался при 100°С в течение 4 часов. Реакционная смесь была разбавлена EtOAc (15 мл), промыта соляным раствором (3×10 мл) и сконцентрирована в условиях вакуума. Полученный остаток был очищен обратнофазовой HPLC (С 18, ацетонитрил/вода) для получения 5 мг (20%) тиазола 23. LRMS (M+H+) m/z 497,2.
Пример 37
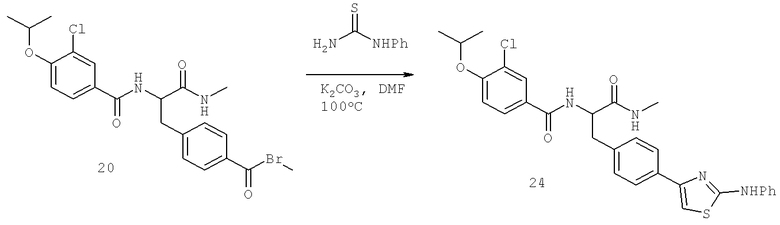
Раствор бромокетона 20 (30 мг, 0,06 ммоль), K2CO3 (25 мг, 0,18 ммоль), фенилтиомочевины (18 мг, 0,12 ммоль) и DMF (1 мл) перемешивался при 100°С в течение 4 часов. Реакционная смесь была разбавлена EtOAc (15 мл), промыта соляным раствором (3×10 мл) и сконцентрирована в условиях вакуума. Полученный остаток был очищен обратнофазовой HPLC (С 18, ацетонитрил/вода) для получения 10 мг (30%) аминотиазола 24. LRMS (М+Н+) m/z 549,2.
Пример 38
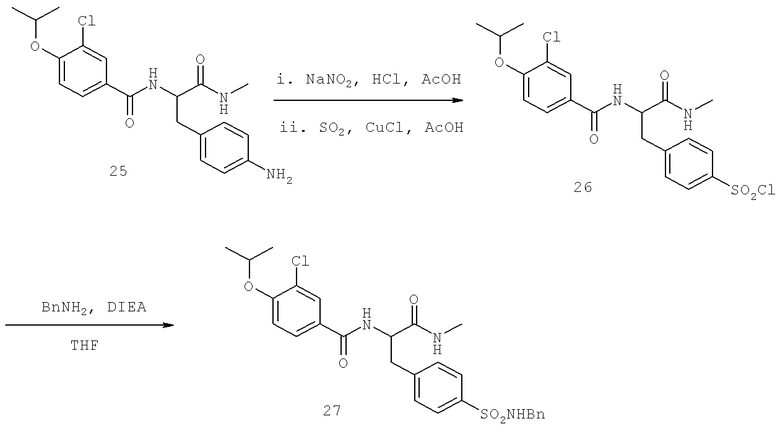
Раствор анилина 25 (100 мг, 0,25 ммоль) концентрированной HCl (1 мл) и АсОН (1 мл) был охлажден до -5°С. Затем к раствору в течение 1 минуты был медленно добавлен NaNO2 (20 мг, 0,29 ммоль). Реакционный раствор перемешивался при -5°С в течение 45 минут для получения раствора соли диазония.
В другом реакционном сосуде SO2 был барботирован через раствор АсОН (1 мл) и хлорида меди (I) (6 мг, 0,06 ммоль) до преобладания сине-зеленого цвета. Раствор диазония затем был медленно добавлен в течение 1 минуты к раствору SO2/CuCl. Внутренняя температура реакционного раствора была перемешена до ниже 30°С. Полученная в результате реакционная смесь была затем вылита в холодную Н2О (10 мл), экстрагирована диэтил эфиром (3×10 мл) и органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Неочищенный сульфонилхлорид 26 использовался без дальнейшей очистки.
Раствор сульфонилхлорида 26 (74 мг, 0,16 ммоль), диизопропил этиламина (81 мл, 0,16 ммоль), бензиламина (17 мл, 0,16 ммоль) и THF (1 мл) перемешивался при 23°С в течение 18 часов. Реакционная смесь была разбавлена EtOAc (15 мл), промыта соляным раствором (3×10 мл) и сконцентрирована в условиях вакуума. Полученный остаток был очищен колоночной хроматографией (силикагель, 1:1 EtOAc : гексаны) для получения 25 мг (29%) сульфонамида 27. LRMS (M+H+) m/z 544,2.
Пример 39
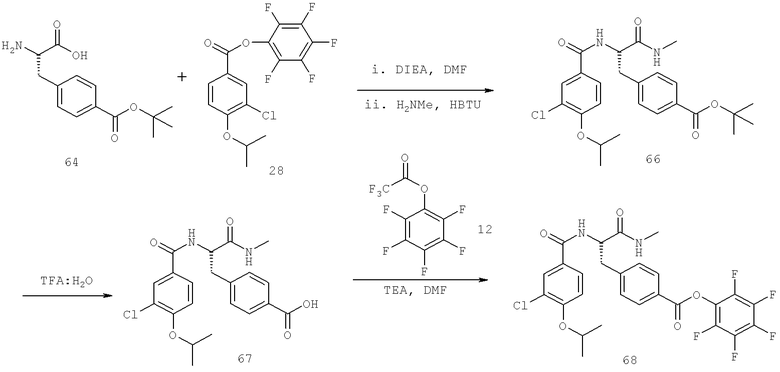
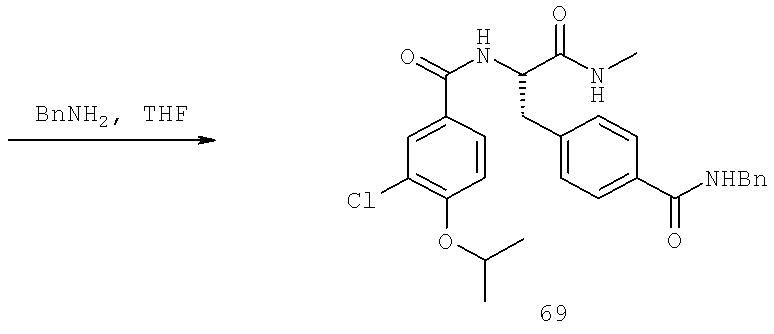
Раствор аминокислоты 64 (2,20 г, 8,27 ммоль), эфира пентафторофенила 28 (3,0 г, 7,88 ммоль), диизопропилэтиламина (5,5 мл, 31,5 ммоль) и DMF (30 мл) перемешивался при 23°С. После 18 часов были добавлены H2NMe (2,0 М в THF, 3,94 мл), O-бензотиазол-N,N,N',N'-тетраметил-уроний-гексафторо-фосфат (HBTU, 6,0 г, 15,76 ммоль). После 4 часов реакционный раствор был растворен в EtOAc (200 мл), промыт соляным раствором (3×200 мл) и сконцентрирован в условиях ваккума. Полученный остаток был очищен колоночной хроматографией (силикагель, 2:1 EtOAc : гексаны) для получения 3,5 мг (93%) амида 30. LRMS (M+H+) m/z 475,2.
Раствор эфира 66 (3,5 г, 7,36 ммоль), TFA: H2O (97,5:2,5, 10 мл) и CH2Cl2 (10 мл) перемешивался при 23°С в течение 3 часов. Реакционный раствор был сконцентрирован в условиях ваккума и полученный в результате остаток был помещен под высокий вакуум на 2 часа и затем использовался без дальнейшей очистки.
Раствор кислоты 67 (4,7 г, 11,2 ммоль), пентафторофенол трифторацетата 12 (3,86 мл, 22,4 ммоль), триэтиламина (4,7 мл, 33,6 ммоль) и DMF (25 мл) перемешивался при 23°С в течение 18 часов. Реакционный раствор был разбавлен EtOAc (200 мл) и промыт 1 N HCL (50 мл), насыщенным водным NaHCO3 (50 мл) и соляным раствором (100 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 2:1 гексаны : EtOAc) для получения 1,1 г (17%) эфира 68.
Раствор эфира 68 (20 мг, 0,03 ммоль), бензиламина (6 мкл, 0,05 ммоль) и THF (0,5 мл) перемешивался при 23°С в течение 18 часов. Реакционная смесь была разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл), насыщенным водным NaHCO3 (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен обратнофазовой HPLC (С 18, ацетонитрил/вода) для получения 13 мг (85%)амида 14. LRMS (M+H+) m/z 508,2.
Пример 40
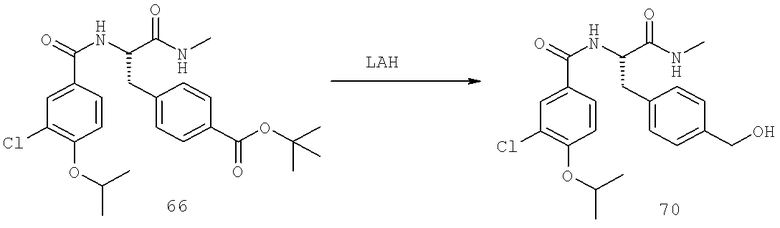
Раствор эфира 66 (50 мг, 0,1 ммоль), LiAlH4 (1,0 М в THF, 0,21 мл) и THF (1 мл) перемешивался при 23°С в течение 2 часов. Реакционная смесь была погашена МеОН (1 мл), затем разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен обратнофазовой HPLC (С 18, ацетонитрил/вода) для получения 2 мг (5%) спирта 70. LRMS (M+H+) m/z 405,2.
Пример 41
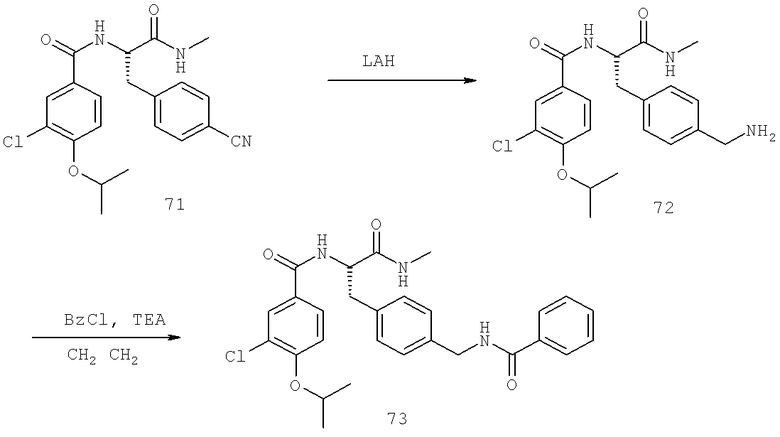
Раствор нитрила 71 (108 мг, 0,27 ммоль), LiAlH4 (1,0 М в THF, 0,1 мл) и THF (2 мл) перемешивался при 23°С в течение 2 часов. Реакционная смесь была погашена МеОН (1 мл), затем разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен обратнофазовой HPLC (С 18, ацетонитрил/вода) для получения 30 мг (28%) амина 72. LRMS (M+H+) m/z 404,2.
Раствор амина 72 (10 мг, 0,02 ммоль), бензоилхлорида (3,2 мкл, 0,03 ммоль), триэтиламина (50 мкл, 0,36 ммоль) и CH2Cl2 (0,5 мл) перемешивался при 23°С в течение 2 часов. Реакционная смесь была затем разбавлена EtOAc (15 мл) и промыта 1 N HCl (2 мл), насыщенным водным NaHCO3 (2 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 2:1 гексаны : EtOAc) для получения 5 г (49%) амида 73. LRMS (M+H+) m/z 508,2.
Пример 42
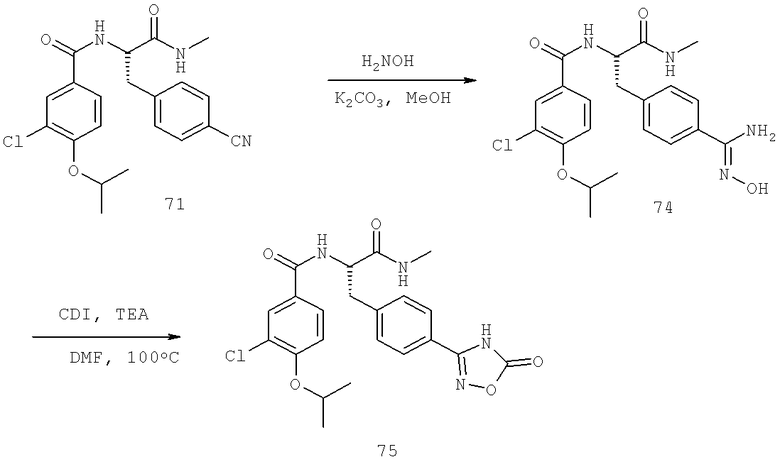
Раствор нитрила 71 (300 мг, 0,75 ммоль), гидроксиламин гидрохлорида (156 мг, 2,25 ммоль), K2CO3 (726 мг, 5,25 ммоль) и EtOH (10 мл) перемешивался при 80°С в течение 18 часов. Реакционная смесь была сконцентрирована в условиях вакуума. Полученный в результате отстаток был разбавлен EtOAc (15 мл) и промыт соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:20 МеОН : EtOAc) для получения 80 мг (25%) гидроксиамидина 74. LRMS (M+H+) m/z 433,2.
Раствор гидроксиамидина 74 (20 мг, 0,05 ммоль), карбонилдиимидазола (CDI, 15 мг, 0,09 ммоль), триэтиламина (13 мкл, 0,09 ммоль) и DMF (1 мл) перемешивался при 100°С в течение 2 часов. Полученный отстаток был разбавлен EtOAc (15 мл) и промыт соляным раствором (3×5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:20 МеОН: EtOAc) для получения 10 мг (44%) оксадиазолона 75. LRMS (M+H+) m/z 459,2.
Пример 43
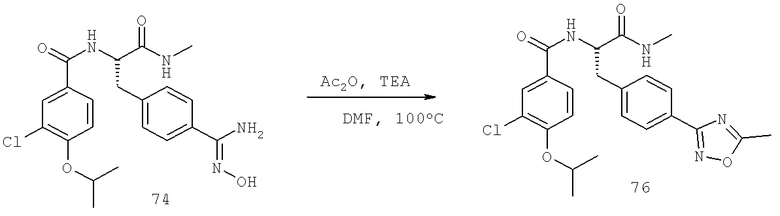
Раствор гидроксиамидина 74 (20 мг, 0,05 ммоль), триэтиламина (13 мкл, 0,09 ммоль), уксусного ангидрида (0,5 мл) и DMF (0,5 мл) перемешивался при 100°С в течение 2 часов. Полученный отстаток был разбавлен EtOAc (15 мл) и промыт соляным раствором (3×5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:20 MeOH : EtOAc) для получения 13 мг (57%) оксадиазола 75. LRMS (M+H+) m/z 457,2.
Пример 44
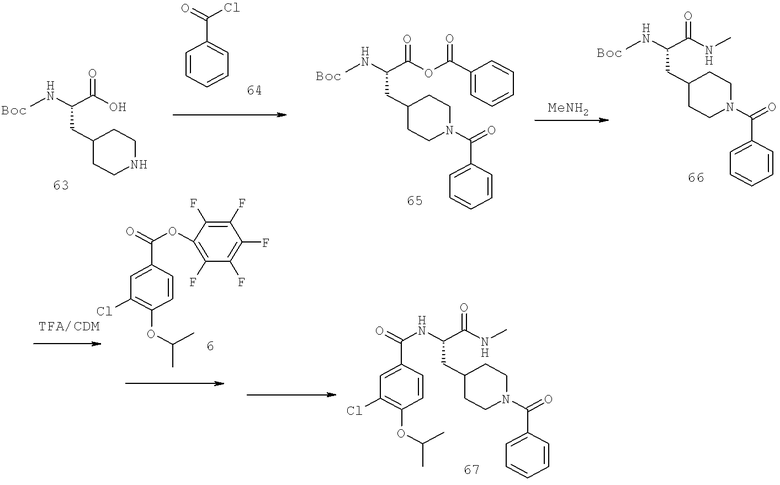
К раствору 63 (100 мг, 0,367 ммоль) в DCM (10 мл) был добавлен хлорид бензойной кислоты 64 (93,8 мкл, 0,808 ммоль) и диизопропилэтиламин (192 мкл, 1,10 ммоль). После перемешивания в течение 10 минут к реакционному раствору было добавлено 2 М метиламина в THF (550 мкл, 1,10 ммоль). Реакционная смесь перемешивалась в течение 30 минут и была сконцентрирована. Остаток был очищен на колонке для отгонки легких фракций с силикагелем (гексан : EtOAc, 1:1) для получения 66 (100 мг, 70%).
К раствору 66 (100 мг, 0,257 ммоль) в DCM (5 мл) был добавлен TFA (5 мл) при комнатной температуре. Реакционная смесь перемешивалась в течение 100 минут. Растворители были выпарены при пониженном давлении и осадок высушен в условиях высокого вакуума в течение ночи. Остаток был растворен в DMF (2 мл) и затем перемешен с 6 (117 мг, 0,308 ммоль) и диизопропилэтиламином (90,0 мкл, 0,515 ммоль) при комнатной температуре. Реакционная смесь контролировалась обратнофазовой HPLC/MS. Реакционная смесь перемешивалась в течение 30 минут, после чего она была отфильтрована и фильтрат очищен обратнофазовой HPLC (С 18) с использованием смеси ацетонитрила и Н2О для получения 67 (100 мг, 52,9%). LCMS (M+H+) m/z 486,2.
Пример 45
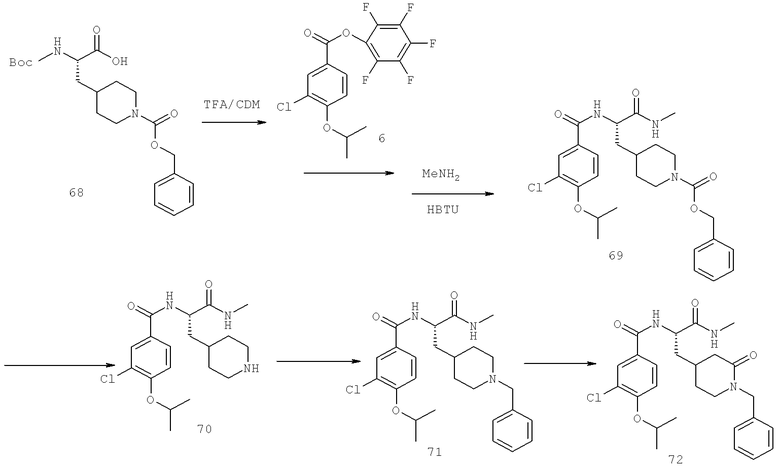
К раствору 68 (500 мг, 1,23 ммоль) в DCM (10 мл) был добавлен TFA (10 мл) при комнатной температуре. Реакционная смесь перемешивалась в течение 10 минут и затем растворители были выпарены при пониженном давлении. Полученный остаток (~1,23 ммоль) был повторно осажден в DMF (50 мл), к которому при комнатной температуре был добавлен 6 (562 мг, 1,48 ммоль) и диизопропилэтиламин (429 мкл, 2,46 ммоль). Реакционная смесь контролировалась обратнофазовой HPLC/MS. После того как более не наблюдалось начального материала, к раствору было добавлено 2 М метиламина в THF (1,23 мл, 2, 46 ммоль) и HBTU (702 мг, 1,85 ммоль). После перемешивания в течение 30 минут растворители были выпарены при пониженном давлении и остаток очищен на колонке для отгона легких фракций с силикагелем (гексан : EtOAc, 1:1) для получения 69 (450 г.71%). LCMS (М+H+) m/z 516,2.
69 (400 мг, 0,775 ммоль) был растворен в растворе HBr/НОАс (10 мл). После перемешивания в течение 10 минут растворители были удалены. Остаток был растворен в растворе бикарбоната натрия (50 мл) и три раза экстрагирован DCM (50 мл). Комбинированные слои DCM были высушены над сульфатом натрия и отфильтрованы, и фильтрат сконцентрирован при пониженном давлении для получения 70 (285 мг, 96%).
К раствору 70 (82,5 г, 216 ммоль) в DMF (2 мл) был добавлен бромид бензила (30,8 мкл, 0,259 ммоль) и диизопропилэтиламин (75,3 мкл, 0,432 ммоль). Реакционная смесь контролировалась обратнофазовой HPLC/MS. После перемешивания в течение 2 часов смесь была отфильтрована и фильтрат очищен обратнофазовой HPLC (С 18) с использованием смеси ацетонитрила и Н2О для получения 71 (183 мг, 90%). LCMS (M+H+) m/z 472,1.
К раствору 71 (128 мг, 0,271 ммоль) в воде (1 мл) и метаноле (1 мл) была добавлена этилендиаминтетрауксусная кислота (EDTA) натрия (273 мг, 0,814 ммоль), НОАс (20 мкл) и Hg(OAc)2 (259 мг, 0,814 ммоль). Реакционная смесь перемешивалась при 100°С в течение 8 часов, после чего растворители выпаривались при пониженном давлении. Остаток был повторно растворен в DMF (2 мл) и отфильтрован, а фильтрат очищен обратнофазовой HPLC (С18) с использованием смеси ацетонитрила и Н2О для получения 72 (37,6 мг, 28,5%). LCMS (M+H+) m/z 486,1.
Пример 46
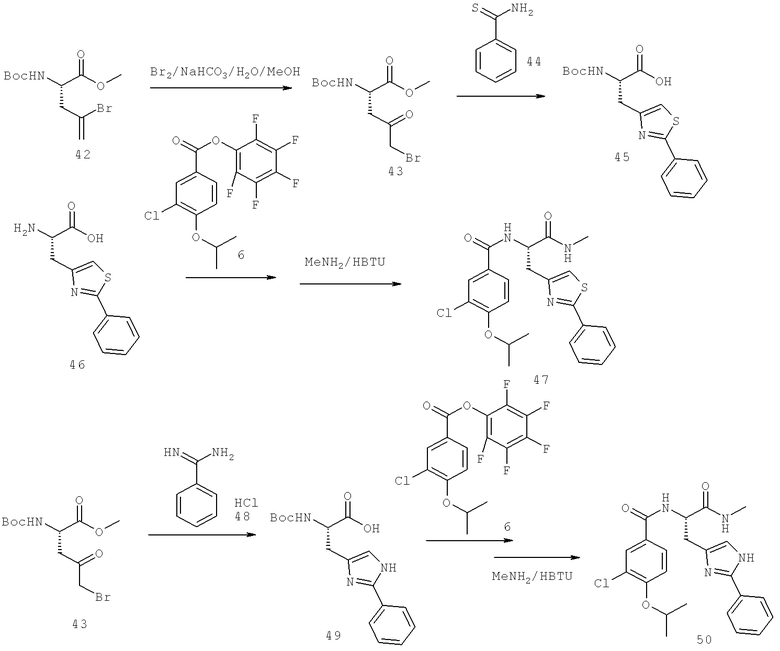
К раствору 42 (1,0 г, 3,24 ммоль) в метаноле/воде (2/1, 20 мл) был добавлен бикарбонат натрия (327 мг, 3,24 ммоль). В то время как производилось перемешивание над ледяной баней, в реакционную смесь был по каплям добавлен бром (200 мкл, 3,89 ммоль). Реакционная смесь контролировалась обратнофазовой HPLC/MS. Когда более не было заметно начального материала, растворители были выпарены при пониженном давлении. Остаток был растворен в воде (100 мл) и три раза экстрагирован DCM (50 мл). Комбинированные слои DCM были высушены над сульфатом натрия, смесь отфильтрована и фильтрат скноцентрирован для получения 43.
К раствору неочищенного 43 (3,24 ммоль) в метаноле (50 мл) был добавлен диизопропилэтиламин (1,14 мл, 6,48 моль) и бензил тиоамид 44 (445 мг, 3,24 ммоль). После перемешивания в обратном потоке в течение 24 часов растворители были выпарены при пониженном давлении. Остаток был очищен обратнофазовой HPLC (С 18) с использованием смеси ацетонитрила и Н2О для получения 45 (200 мг, 18%).
К раствору 45 (150 мг, 0,431 ммоль) в DCM (5 мл) был добавлен TFA (5 мл) при комнатной температуре. Реакционная смесь перемешивалась в течение 10 минут. Затем растворители были выпарены при пониженном давлении и остаток 46 (107 мг, 100%) был высушен в условиях высокого вакуума в течение ночи.
К перемешанному раствору 46 (0,431 ммоль) в DMF (20 мл) был добавлен 6 (197 мг, 0,517 ммоль) и диизопропилэтиламин (150 мкл, 0,862 ммоль) при комнатной температуре. Реакционная смесь контролировалась обратнофазовой HPLC/MS. Когда более не стало заметно начального материала, к раствору было добавлено 2М метиламина в THF (0,431 мл, 0,862 ммоль) и HBTU (245 мг, 0,647 ммоль). Реакционная смесь перемешивалась в течение 30 минут, после чего смесь было отфильтрована и фильтрат был очищен обратнофазовой HPLC (С 18) с использованием смеси ацетонитрила и H2O для получения 47 (80,0 мг, 40,5%). LCMS (M+H+) m/z 458,1.
Такие же процедуры применялись к 47 для получения 50 (58,3 мг). LCMS (M+H+) m/z 441,4.
Пример 47
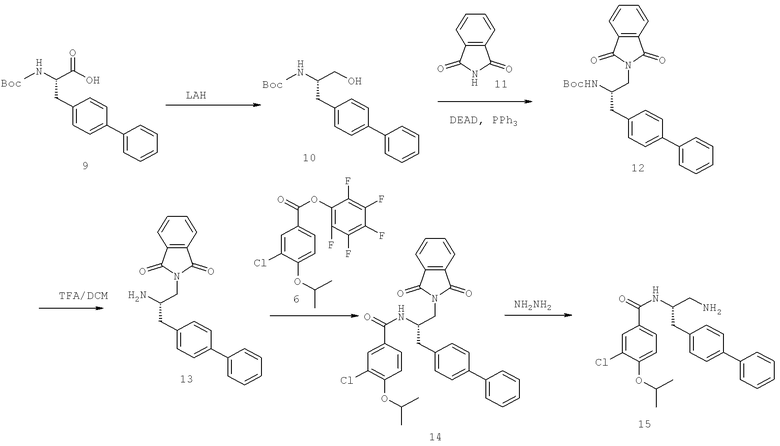
К перемешанному раствору Вос-р-бифиниламина 9 (3,0 г, 8,79 ммоль) в THF (200 мл) над водяной баней был добавлен LAH (1,0 М в THF, 17,6 ммоль). После перемешивания в течение 2 часов реакция была погашена с помощью МеОН (10 мл) с последующим применением раствора NaOH (17,6 мл, 35,1 ммоль). Смесь была отфильтрована через Целит (Celite) и фильтрат был сконцентрирован при пониженном давлении. Остаток был растворен в воде (200 мл) и три раза экстрагирован DCM (200 мл). Комбинированные слои DCM были высушены (Na2SO4), отфильтрованы и сконцентированы при пониженном давлении для получения 10 (2,85 г, 98%).
К перемешиваемому раствору 10 (2,85 г, 8,80 ммоль) в THF (250 мл) был добавлен 11 (1,54 г, 10,4 ммоль) и трифенилфосфин (2,51 г, 9,57 ммоль). Затем по каплям был добавлен DEAD (1,49 мл, 9,57 ммоль), реакция перемешивалась в течение 30 минут и была сконцентриована в условиях вакуума, а остаток был очищен на колонке для отгона легких фракций с силикагелем (гексан : EtOAc, 6:1) для получения продукта 12 (2,0 г, 50%).
К раствору 12 (2,0 г, 4,38 ммоль) в DCM (50 мл) был добавлен TFA (50 мл) при комнатной температуре. Реакционная смсеь перемешивалась в течение 20 минут и затем была сконцентрирована в условиях вакуума для получения 13 (1,56 г, 100%).
К раствору 13 в DMF (100 мл) был добавлен 6 (2,0 г, 5,26 ммоль) и диизопропилэтиламин (1,53 мл, 8,76 ммоль) при комнатной температуре. Реакционная смесь была подвергнута перемешиванию в течение ночи. Растворители выпаривались при пониженном давлении и остаток очищался над силикагелем (гексан : EtOAc = 2:1) для получения 14 (1,5 г, 61,9%). LRMS (М+Н+) m/z 553,1.
К раствору 14 (1,5 г, 2,71 ммоль) в метаноле (20 мл) был добавлен гидрат гидразина (0,845 мл, 27,1 ммоль). Реакционная смесь перемешивалась при 50°С в течение 5 часов и затем охлаждалась до комнатной температуры. Твердое вещество было отфильтровано и фильтрат был сконцентрирован при пониженном давлении для получения 15 (1,0 г, 87,2%). LCMS (M+H+) m/z 423,1.
Пример 48
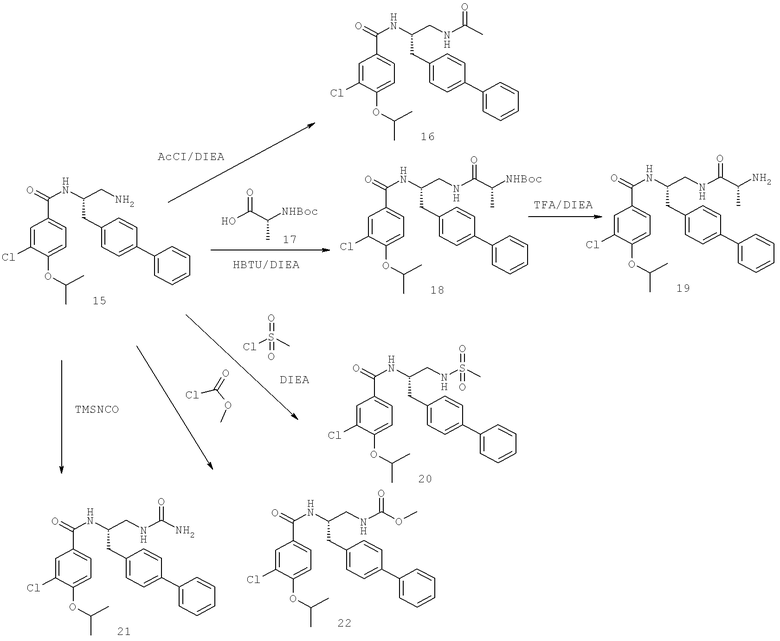
К раствору 15 (20,0 мг, 0,0473 ммоль) в DCM (10 мл) был добавлен диизопропилэтиламин (24,7 мкл, 0,142 ммоль) и ацетилхлорид (5,0 мкл, 0,0709 ммоль). Реакционная смесь перемешивалась в течение 10 минут, затем была сконцентрирована при пониженном давлении и очищена на обратнофазовой HPLC (С 18) с использованием смеси ацетонитрила и Н2О для получения 16 (8,0 мг, 36,4%). LCMS (M+H+) m/z 465,2.
К раствору 15 (60,0 мг, 0,142 ммоль) в DCM (2 мл) был добавлен диизопропилэтиламин (49,5 мкл, 0,282 ммоль), 17 (32,2 мг, 0,170 ммоль) и HBTU (80,8 мг, 0,213 ммоль). Реакционная смесь перемешивалась в течение 10 минут и затем была сконцентрирована при пониженном давлении. Полученный в результате остаток был растворен в DCM (1 мл) и TFA (1 мл) и перемешивался в течение 10 минут, сконцентрирован при пониженном давлении, а продукт был очищен на обратнофазовой HPLC (С 18) с использованием смеси ацетонитрила и H2O для получения 19 (25,0 мг, 35,6%). LCMS (M+H+) m/z 494,2.
К раствору 15 (35,0 мг, 0,0828 ммоль) в DCM (2 мл) был добавлен диизопропилэтиламин (28,8 мкл, 0,166 ммоль) и метаносульфонил хлорид (9,64 мкл, 0,124 ммоль). Реакционная смесь перемешивалась в течение 10 минут, затем концентрировалась при пониженном давлении, а продукт был очищен на обратнофазовой HPLC (C18) с использованием смеси ацетонитрила и Н2О для получения 20 (25,0 мг, 60,3%). LCMS (М+Н+) m/z 501,2.
К раствору 15 (60,0 мг, 0,142 ммоль) в DCM (2 мл) был добавлен диизопропилэтиламин (49,5 мкл, 0,282 ммоль) и триметилсилилизоцианид (19,6 мг, 0,170 ммоль). Реакционная смесь перемешивалась в течение 10 минут, затем концентрировалась при пониженном давлении, а продукт был очищен на обратнофазовой HPLC (C18) с использованием смеси ацетонитрила и Н2О для получения 21 (20,6 мг, 31,1%). LCMS (M+H+) m/z 466,1.
К раствору 15 (60,0 мг, 0,142 ммоль) в DCM (2 мл) был добавлен диизопропилэтиламин (49,5 мкл, 0,282 ммоль) и метил хлороформат (13,1 мкл, 0,170 ммоль). Реакционная смесь перемешивалась в течение 10 минут, затем концентрировалась при пониженном давлении, а продукт очищался на обратнофазовой HPLC (C18) с использованием смеси ацетонитрила и H2O для получения 22 (19,9 мг, 29,1%). LCMS (M+H+) m/z 481,1.
Пример 49
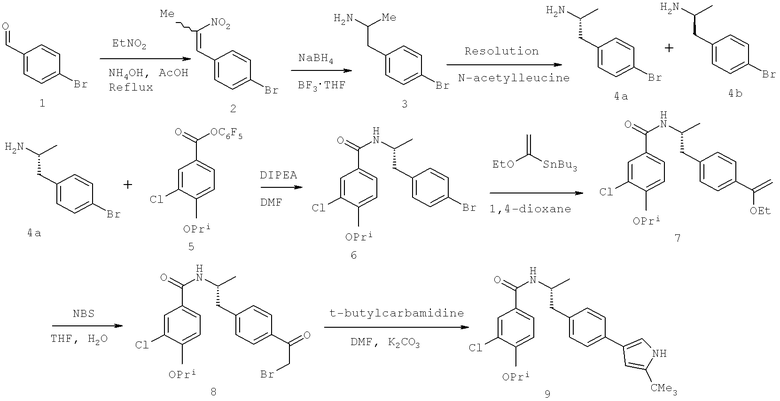
Раствор 4-бромобензальдегида (14,8 г, 80 ммоль) и ацетат амония (14,0 г, 180 ммоль) в нитроэтане (50,0 г) нагревался в противотоке в течение 8 часов. Затем он был охлажден до комнатной температуры, разделен между дихлорометаном (150 мл) и водой (30 мл). Фазы были разделены, после чего органический слой был высушен над сульфатом натрия и сконцентрирован в условиях вакуума. Остаток был пропущен через закупоренную колонку силикагеля (этилацетат/гексан в качестве элюента), за чем следовала рекристаллизация из метанола для получения промежуточного 2 (9,8 г, 51%), который, как было определено, был достаточно чист для использования в последующих трансформациях. LC/MS (LRMS (M+H+) m/z 240,97.
К раствору борогидрида натрия (4,6 г, 124 ммоль) в тетрагидрофуране (100 мл) при 0°С был добален боран-тетрагидрофурановый комплекс (150 мл, 150 ммоль, 1,0 М). Полученный в результате раствор перемешивался при комнатной температуре в течение дополнительных 15 минут. По каплям был добавлен промежуточный 2 (6,5 г, 27 ммоль) в тетрагидрофуране (30 мл) и полученный в результате раствор нагревался в сосуде с обратным холодильником в течение 4 часов. Далее был охлажден до комнатной температуры, реакция гасилась водой и экстрагировалась дихлорометаном (3×80 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях ваккума, а остаток был очищен флеш-хроматографией (силикагель, гексан/этилацетат) для получения промежуточного 3 (5,2 г, 90%), который характеризовался LC/MS. LRMS (М+Н+) m/z 213, 02.
Раствор амина 3 (4,0 г, 16 ммоль) в этилацетате (30 мл) при 0°С был насыщен хлористоводородной кислотой (газ). Полученная в результате соль собиралась фильтрацией и высушивалась в условиях вакуума.
Соль L-N-ацетиллейцина натрия (8,0 ммоль) (приготовленная добавлением раствора 1N гидроокиси натрия к суспензии L-N-ацетиллейцина (1,39 г, 8,0 ммоль) в 5 мл воды до достижения рН=7) была медленно добавлена к перемешиваемому раствору вышеупомянутой 3 соли гидрохлорида в воде (10 мл). Образованные за ночь кристаллы были удалены фильтрацией, промыты небольшим количеством холодной воды и рекристаллизованы из абсолютного метанола. Кристаллическая соль 4а была собрана и высушена в условиях вакуума.
Материнские растворы, которые были богаты (S)-3, были объединены, сделаны сильнощелочными с помощью раствора 5N гидроксида натрия и три раза промыты диэтил эфиром. Комбинированные органические слои были промыты водой и высушены над сульфатом натрия. После удаления сульфата натрия хлористоводородная кислота пропускалась через раствор до окончания выпадения соли гидрохлорида в осадок. Такая же процедура применялась к соли D-N-ацетиллейцина. Кристаллическая соль 4b была собрана и высушена в условиях вакуума.
Диастереометрическая соль каждого энантиомера была разделена между 20 мл воды, которая была сделана сильнощелочной с помощью растворов 5N гидроокиси натрия, и экстрагирована диэтил эфиром. Комбинированные органические слои были промыты водой и высушены над сульфатом натрия. Растворители были удалены и оба продукта были определены как достаточно чистые для использования в последующих трансформациях (4а: 1,3 г, 32%; 4b: 0,9 г, 22%). 1H-NMR и LC/MS (LRMS (M+H+) m/z 213,02). Капиллярный электрофорез показал >98% ее.
К раствору амина 4а (111 мг, 0, 52 ммоль) в диметилформамиде (5 мл) при комнатной температуре был добавлен диизопропилэтиламин (99 мкл, 0,57 ммоль). Полученный в результате раствор перемешивался в течение 5 минут и был добавлен промежуточный 5 (217 мг, 0,57 ммоль). Реакционная смесь перемешивалась в атмосфере азота в течение 30 минут и растворители были удалены в условиях вакуума. Остаток был разделен между этилацетатом (20 мл) и водным раствором лимонной кислоты (20 мл, 10%). Слои были разделены, а орагническая фаза была промыта водным раствором лимонной кислоты (20 мл, 10%) и водным растором гидроксида калия (2×20 мл, 0,1 М). Затем они были высушены над сульфатом натрия и сконцентрированы в условиях вакуума для получения 6 (212 мг, 100%), который был определен достаточно чистым для использования в последующих трансформациях. (LRMS (M+H+) m/z 410,1).
К раствору бромида 6 (212 мг, 0,53 ммоль) в диоксане (10 мл) при комнатной температуре были последовательно добавлены транс-дихлоробис(трифенил фосфин)палладий (II) (37 мг, 10 мол.%) и 1-этоксивинилтри-n-бутилин (481 мг, 1,33 ммоль). Полученный в резульате раствор нагревался до 100°С в течение 4 часов. Он был охлажден до комнатной температуры и растворители были удалены в условиях вакуума.
Остаток был затем очищен флеш-хроматографией (силикагель, этилацетат плюс 5% триэтиламин) для получения промежуточного 7 (250 мг), который был нестабилен и определен как достаточно чистый для использования в последующих трансформациях LC/MS (LRMS (М+Н+) m/z 402,8).
Промежуточный 7 в тетрагидрофуране (10 мл) и вода (3 мл) перемешивались с N-бромосуцинимидом (190 мг, 1,1 ммоль) при 50°С в течение 2 часов. Растворители были удалены в условиях вакуума, а полученный в результате остаток был разделен между водой (10 мл) и экстрагирован этилацетатом (3×50 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентированы в условиях вакуума. Остаток был затем очищен флеш-хромотаграфией (силикагель, гексан/этилацетат) для получения промежуточного 8 (55 мг, 23%), который был охарактеризован LC/MS (LRMS (М+Н+) m/z 452,1).
К раствору промежуточного 9 (55 мг, 0,12 ммоль) в диметилформамиде (3 мл) при комнатной температуре был добавлен карбонат калия (34 мг, 0,24 ммоль) и терт-бутилкарбамид (31 мг, 0,30 ммоль). Реакционная смесь перемешивалась в атмосфере азота при 50°С в течение 1,5 часов. Она была охлаждена до комнатной температуры и растворители были удалены в условиях вакуума. Остаток был разделен между этилацетатом (15 мл) и водой (15 мл). Слои были разделены и водная фаза экстрагировалась этилацетатом (2×20 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях вакуума. Остаток был затем очищен флеш-хроматографией (силикагель, гексан/этилацетат) для получения 9 (35 мг, 67%), который был охарактеризован 1Н-NMR и LC/MS (LRMS (М+Н+) m/z 452,1).
Пример 50
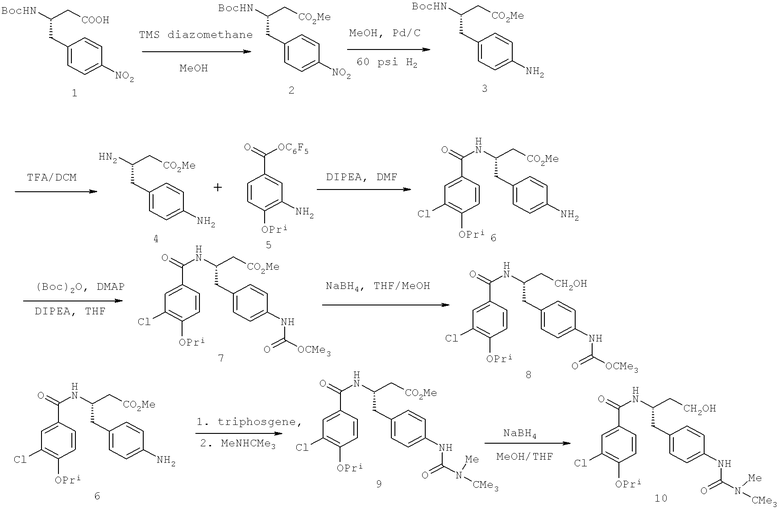
К раствору кислоты 1 (2,98 г, 9,2 ммоль) в метаноле (15 мл) при комнатной температуре был добавлен по каплям раствор TMS диазометана в гексане (9,2 мл, 18,4 ммоль, 2,0 М). Полученный в результате желтый раствор перемешивался при температуре окружающей среды в течение 30 минут и растворители удалялись в условиях вакуума. Полученное в результете вязкое масло 2 (3,10 г, 9,2 ммоль) было высушено и определено как достаточно чистое для использования в дальнейших трансформациях. LC/MS (LRMS (M+H+) m/z 339,10).
Смесь промежуточного 2 (3,10 г, 9,2 ммоль) и палладия на меди (310 мг) в метаноле (30 мл) перемешивалась в атмосфере водорода при комнатной температуре в течение 2 часов. Она была затем отфильтрована через Целит (Celite) и сконцентрирована для получения анилина 3 (2,47 г, 8,0 ммоль) в качестве вязкого масла, которое было высушено и определено как достаточно чистое для использования в последующих трансформациях. LC/MS (LRMS (М+H+) m/z 309,20).
К раствору анилина 3 (2,47 г, 8,0 ммоль) в дихлорометане (20 мл) при комнатной температуре была добавлена трифтороуксусная кислота (20 мл). Полученный в результате раствор перемешивался в течение 45 минут и растворители были удалены в условиях вакуума. Остаток был разделен между дихлорометаном (75 мл) и насыщенным водным раствором бикабоната натрия (25 мл) и слои были отделены. Водная фаза была насыщена хлоридом натрия и экстрагирована с помощью дихлорометана (3×75 мл) и тетрагидрофурана (2×50 мл). Комбинированные органические слой были высушены в атмосфере сульфата натрия и сконцентрированы в условиях вакуума для получения 4 (1,30 г, 6,3 ммоль) в качестве вязкого масла, которое было охарактеризовано LC/MS (LRMS (MH) m/z 209,30).
Раствор амина 4 (1,30 г, 6,25 ммоль) в диметилформамиде (20 мл) перемешивался с диизопропилэтиламином (3,27 мл, 18,80 ммоль) при комнатной температуре в течение 5 минут, за чем следовало добавление промежуточного 5 (2,38 г, 6.25 ммоль). Реакционная смесь была дополнительно перемешена в течение 30 минут и растворы были удалены в условиях вакуума. Остаток был разделен между этилацетатом (50 мл) и водой (50 мл). Слои были отделены и водная фаза была экстрагирована с помощью этилацетата (3×50 мл). Комбинированные органические слои были высушены в атмосфере сульфата натрия и сконцентриованы в условиях вакуума. Остаток был очищен флеш-хроматографией (силикагель, гексан/этилацетат) для получения 6 (1,47 г, 58%) в качестве пенистого белого твердого вещества, которое было охарактеризовано с помощью LC/MS (LRMS (M+H+) m/z 405,15).
К раствору анилина 6 (131 мг, 0,32 ммоль) в тетрагидрофуране (5 мл) при комнатной температуре был добавлен диизопропилэтиламин (85 мкл, 0,48 ммоль), 4-(диметиламино)пиридин (15 мг, 0,12 ммоль) и ди-терт-бутилдикарбонат (85 мг, 0,39 ммоль). Полученный в результате раствор перемешивался в течение ночи и затем разбавлялся этилацетатом (20 мл), промыт водным раствором хлористоводородной кислоты (2×15 мл, 0,1 М) и просушен над сульфатом натрия. Удаление растворителей дало карбамат 7 (139 мг, 88%) в виде стекловидного твердого вещества, которое было определено как достаточно чистое для использования в последующих трансформациях. LC/MS (LRMS (М+Н+) m/z 505,10).
К раствору карбамата 7 (139 мг, 0,28 ммоль) в метаноле (2 мл) и тетрагидрофуране (2 мл) при комнатной температуре был добавлен борогидрид натрия (261 мг, 6,9 ммоль). Полученная в результате смесь перемешивалась в течение 2 часов, после чего растворители были удалены в условиях вакуума. Остаток был разделен между этилацетатом (15 мл) и водой (15 мл), слои были разделены и водная фаза была экстрагирована с помощью этилацетата (3×15 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентриованы в условиях вакуума. Остаток был очищен обратнофазовой HPLC с использованием мобильного фазового градиента, состоящего из ацетонитрила и воды. Чистый продукт 8 (47 мг, 36%) был выделен и охарактеризован с помощью 1H-NMR и LC/MS (LRMS (M+Н+) m/z 477,20).
К раствору трифосгена (37 мг, 0,13 ммоль) в тетрагидрофуране (15 мл) при 0°С был добавлен по каплям 6 (145 мг, 0,36 ммоль) и диизопропилэтиламин (130 мкл, 0,75 ммоль) в тетрагидрофуране (5 мл). Полученная в результате смесь выдерживалась в атомосфере азота при той же температуре в течение 30 минут и гасилась с помощью метил-терт-бутиламина (215 мкл, 1,80 ммоль). Реакционная смесь перемешивалась в течение дополнительных 30 минут, за чем следовало удаление растворителей в условиях вакуума. Остаток был разделен между этилацетатом (15 мл) и водным раствором хлористоводородной кислоты (15 мл, 0,1 М), слои были разделены и водная фаза экстрагирована этилацетатом (2×15 мл). Комбинированные органические слои были высушены в атмосфере сульфата натрия и сконцентрированы в условиях вакуума для получения мочевины 9 (152 мг, 0,29 ммоль) в качестве стекловидного твердого вещества, которое было определено как достаточно чистое для использования в дальнейших трансформациях. (LC/MS (LRMS (М+Н+) m/z 518,2).
К раствору мочевины (150 мг, 0,29 ммоль) в метаноле (2 мл) и тетрагидрофуране (2 мл) при комнатной температуре был добавлен борогидрид натрия (260 мг, 6,90 ммоль). Полученная в результате смесь перемешивалась в атмосфере азота при комнатной температуре в течение 2 часов. Растворители были удалены и остаток был разделен между этилацетатом (15 мл) и водой (15 мл). Слои были разделены и водная фаза экстрагирована этилацетатом (3×15 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях вакуума. Остаток был очищен обратнофазовой HPLC с использованием градиента мобильной фазы, состоящего из ацетонитрила и воды. Чистый продукт 10 (4 мг, 28%) был выделен и охарактеризован с помощью 1H-NMR и LC/MS (LRMS (М+Н+) m/z 490,1).
Пример 51
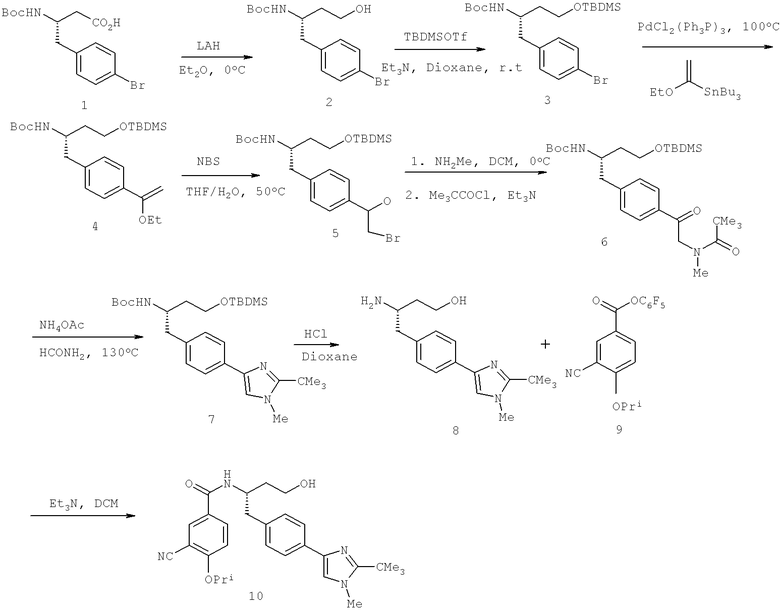
К раствору карбоновой кислоты 1 (10,0 г,28 ммоль) в безводном диэтил эфире (200 мл) при 0°С был по каплям добавлен раствор литиевого гидрида алюминия в тетрагидрофуране (40 мл, 40 ммоль, 1 М). Полученный в результате раствор затем перемешивался в течение дополнительных 2 часов при той же температуре. Он был осторожно погашен водой (2,5 мл), водной гидроокисью натрия (2,5 мл, 1 М) и водой (3,0 мл). Затем раствор был высушен над сульфатом натрия и удаление растворителей дало промежуточный продукт 2 (9,2 г, 96%), который был определен как достаточно чистый для использования в дальнейших трансформациях с помощью 1H-NMR и LC/MS (LRMS (М+H+) m/z 344,08).
При комнатной температуре к раствору промежуточного продукта 2 в безводном диоксане (200 мл) был добавлен триэтиламин (6 мл, 40 ммоль) и терт-бутилдиметилсилилтрифторометансульфонат (8,6 г, 32 ммоль). Полученный в результате раствор затем перемешивался в течение ночи и гасился насыщенным водным раствором бикарбоната натрия. Он был экстрагирован дихлорометаном (3×100 мл), а комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях вакуума. Остаток был очищен флеш-хроматографией (силикагель, гексан/этилацетат) для получения промежуточного 3 (9,2 г, 72% всего), который был охарактеризован LC/MS (LRMS (M+H+) m/z 458,16).
При комнатной температуре к раствору бромида 3 (6,0 г, 13 ммоль) в диоксане (100 мл) был последовательно добавлен транс-дихлоробис(трифенилфосфин)палладий (II) (500 мг) и 1- этоксивинилтри-n-бутилин (12,3 г, 34 ммоль). Полученный в результате раствор нагревался до 100°С в течение 4 часов. За удалением растворителей в условиях вакуума последовало очищение флеш-хроматографией (силикагель, гексан/этилацетат плюс 5% триэтиламин) для получения промежуточного 4 (5,4 г), который был охарактеризован LC/MS (LRMS (М+Н+) m/z 450,30). Было обнаружено, что продукт нестабилен и он немедленно использовался в последующих трансформациях.
Промежуточный продукт 4 в метаноле (100 мл) и воде (50 мл) перемешивался с N-бромосуцинимидом (5,9 г, 33 ммоль) при 50 С в течение 4 часов. Растворители удалялись в условиях вакуума и полученный в результате остаток был экстрагирован этилацетатом (3×50 мл). Комбинированные органические слои были высушены в атмосфере сульфата натрия и сконцентрированы в условиях вакуума. Остаток был очищен флеш-хроматографией (силикагель, гексан/этилацетат) для получения промежуточного 5 (4,5 г, 69% всего), который был охарактеризован LC/MS (LRMS (М+Н+) m/z 500,54).
В атмосфере азота выравнивающая давление капельная воронка, заряженная бромометилкетоном 5 (2,5 г, 5,0 ммоль) в дихлорометане (40 мл) была присоединена к 150 мл колбе, содержащей раствор метиламина (15 мл, 30 ммоль, 1 М в THF). Колба была охлаждена до 0°С и по каплям в течение 2 часов добавлялся раствор бромида. Полученный в результате раствор перемешивался в течение еще одного часа, после чего были добавлены триэтиламин (1 мл) и раствор триметилацетил хлорида (4,8 мл, 40 ммоль) в дихлорометане (10 мл). Полученная в результате смесь перемешивалась в течение еще 2 часов и затем гасилась насыщенным раствором бикарбоната натрия. Смесь была экстрагирована этилацетатом (3×50 мл) и комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях вакуума. Очищение остатка флеш-хроматографией (силикагель, этилацетат/гексан) дало эфир 6 (1,3 г, 49% всего), который был охарактеризован с помощью анализа 1H-NMR и LC/MS анализом (LRMS (M+H+) m/z 535,35).
Раствор 6 (1,3 г, 2,6 ммоль) в избытке ацетата аммония в формамиде (10 мл) нагревался до 130°С в атмосфере азота в течение 3 часов. Полученная в результате смесь охлаждалась до комнатной температуре, разделена водой и экстрагирована дихлорометаном (3×50 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях вакуума. Остаток был очищен флеш-хроматографией (силикагель, этилацетат/гексан) для получения имидазола 7 (0,8 г, 60%), который был охарактеризован с помощью анализа 1H-NMR и LC/MS (LRMS (М+Н+) m/z 516,35).
Раствор 7 (800 мг, 1,55 ммоль) в тетрагидрофуране (10 мл) перемешивался с хлоридом водорода в 1,4-диоксане (10 мл, 4,0 М) при комнатной температуре в течение одного часа. Растворители были удалены в условиях вакуума и остаток был высушен при высоком вакууме в течение ночи для получения промежуточного продукта 8 (600 мг), который был определен как достаточно чистый для следующей тренсформации. 1Н-NMR и LC/MS (LRMS (М+Н+) m/z 302,22).
При комнатной температуре к раствору амина 8 (60 мг, 0,02 ммоль) в диметилформамиде (3 мл) был добавлен диизопропилэтиламин (53 мкл, 0,30 ммоль) и полученный в результате раствор перемешивался при комнатной температуре в течение 5 минут. Затем был добавлен промежуточный продукт 9 (23 мг, 0,06 ммоль) и реакционная смесь перемешивалась в атмосфере азота в течение 30 минут. Растворители были удалены в условиях вакуума и остаток был очищен флеш-хроматографией (силикагель, метанол/дихлорометан) для получения 10 (25 г, 26%) в качестве стекловидного твердого вещества, которое было охарактеризовано с помощью анализа 1H-NMR и LC/MS (LRMS (M+H+) m/z 489,28).
Пример 52
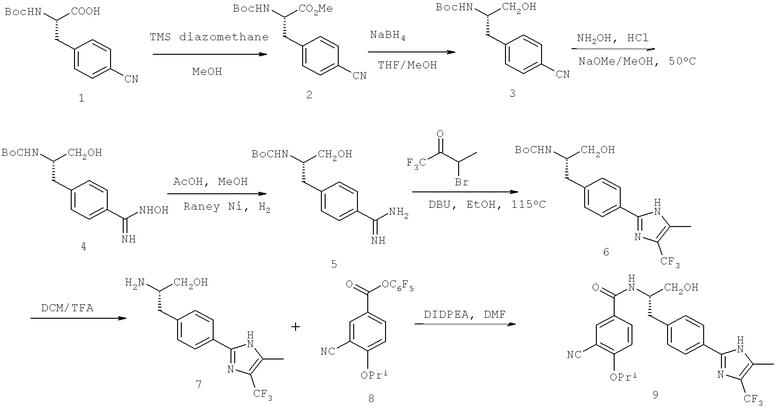
При комнатной температуре к раствору 1 (4,96 г, 17 ммоль) в метаноле (15 мл) по каплям был добавлен раствор TMS диазометана в гексанах (17,0 мл, 34 ммоль, 2 М). Полученный в результате желтый раствор перемешивался при температуре окружающей среды в течение 30 минут. Растворители были удалены в условиях вакуума и вязкое масло 2 (5,19 г, 17 ммоль) было высушено при высоком вакууме и оно было определено как достаточно чистое для использования в дальнейших трансформациях. LC/MS (LRMS (М+Н+) m/z 305,3).
Промежуточный продукт 2 (5,19 г, 17 ммоль) перемешивался с борогидридом натрия (3,23 г, 85 ммоль) в метаноле (50 мл) и тетрагидрофуране (50 мл) при комнатной температуре в течение 2 часов. Растворители были удалены в условиях вакуума и остаток был разделен между этилацетатом (50 мл) и водой (50 мл). Слои были разделены и водная фаза была экстрагирована этилацетатом (3×50 мл), а комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях вакуума для получения 3 (4,71 г, 17 ммоль) в качестве белого твердого вещества, которое было определено как достаточно чистое для использования в дальнейших трансформациях LC/MS (LRMS (М+Н+) m/z 277,3). Нитрил 3 (1,92 г, 6,9 ммоль) перемешивался с метоксидом натрия в метаноле (27,7 мл, 13,9 ммоль, 0,5 М) и гидроксиламин гидрохлориде (964 мг, 13,9 ммоль) в атмосфере азота при 50°С в течение 2 часов. Затем он охлаждался до комнатной температуры и растворители были удалены в условиях вакуума. Остаток был разделен между насыщенным водным раствором хлорида аммония (30 мл) и этилацетатом (30 мл). Слои были разделены и водная фаза экстрагировалась этилацетатом (2×30 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях вакуума. Остаток был очищен флеш-хроматографией (силикагель, гексан/этилацетат) для получения промежуточного продукта 4 (1,08 г, 51%), который был охарактеризован с помощью анализа 1H-NMR и LC/MS (LRMS (М+Н+) m/z 310,2).
При комнатной температуре к раствору 4 (1,08 г, 3,5 ммоль) в метаноле (30 мл) был добавлен скелетный никелевый катализатор гидрирования Raney nickel (200 мг) и уксусная кислота (1 мл). Полученная в результате смесь перемешивалась в атмосфере водорода при комнатной температуре в течение 2 часов. Она была отфильтрована через Целит (Celite) и сконцентрировна в условиях вакуума для получения 5 (1,02 г, 100%), в качестве белого твердого вещества, которое как было определено как достаточно чистое для использования в дальнейших трансформациях. LC/MS (LRMS (M+H+) m/z 294,3).
При комнатной температуре к раствору амидина 5 (304 мг, 1,0 ммоль) в безводном этаноле (15 мл) был добавлен 1,8-диазабицикло[5.4.0]ундек-7-ин (622 мкл, 4,2 ммоль) и 3-бромо-1,1,1-трифторо-2-бутанон (424 мг, 2,1 ммоль). Полученная в результате смесь перемешивалась в атмосфере азота при 115°С в течение 30 минут. Затем она была охлаждена до комнатной температуры и растворители были удалены в условиях вакуума. Остаток был очищен обратнофазовой HPLC с использованием градиента мобильной фазы, состоящего из ацетонитрила и воды. Соединение 6 (76 мг, 20%) было выделено и охарактеризовано с помощью анализа 1H-NMR и LC/MS (LRMS (M+H+) m/z 400,1).
Раствор 6 (76 мг, 0,2 ммоль) в дихлорметане (2 мл) перемешивался с трифтороуксусной кислотой (2 мл) при комнатной температуре в течение 45 минут. Растворители были удалены в условиях вакуума для получения 7, который, как было определено, был достаточно чистым для использования в дальнейших трансформациях. LC/MS. LRMS (М+Н+) m/z 300,3.
При комнатной температуре к раствору амина 7 (25 мг, 0,08 ммоль) в диметилформамиде (3 мл) был добавлен диизопропилэтиламин (87 мкл, 0,50 ммоль). Полученный в результате раствор перемешивался при комнатной температуре в течение 5 минут и был добавлен промежуточный продукт 8 (32 мг, 0, 08 ммоль). Реакционная смесь перемешивалась в атмосфере азота при комнатной температуре в течение 30 минут, и растворители были удалены в условиях вакуума. Остаток был разделен между этилацетатом (5 мл) и водой (5 мл), после чего слои были отделены и водная фаза экстрагирована этилацетатом (3×10 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях ваккума. Остаток был очищен флеш-хроматографией (силикагель, этилацетат) для получения 9 (7 мг, 18%), в качестве стеклообразного твердого вещества, которое было охарактеризовано с помощью анализа 1H-NMR и LC/MS (LRMS (М+Н+) m/z 496,4).
Пример 53
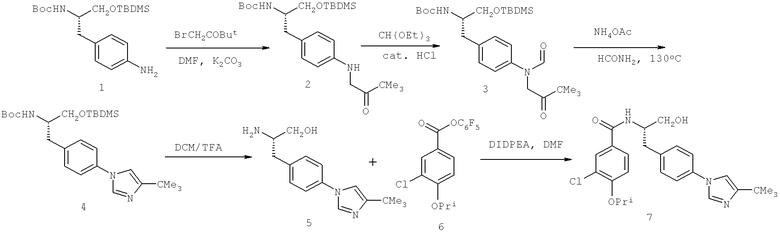
При комнатной температуре к раствору анилина 1 (500 мг, 1,3 ммоль) в диметилформамиде (3 мл) был добавлен карбонат калия (1,5 г) и 1-бромопинаколон (500 мг, 2,8 ммоль). Реакционная смесь перемешивалась при 50°С в течение 4 часов и растворители удалялись в условиях вакуума. Остаток был разделен между этилацетатом (50 мл) и водой (15 мл), слои были отделены и водная фаза экстрагирована этилацетатом (3×50 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях ваккума. Остаток был очищен флеш-хроматографией (силикагель, этилацетат) для получения 2 (320 мг, 51%), который был охарактеризован с помощью анализа 1H-NMR и LC/MS (LRMS (М+Н+) m/z 479,74).
При комнатной температуре к раствору 2 (307 мг, 0,64 ммоль) в триэтил ортоформате (20 мл) была добавлена концентрированная водная HCl (25 мкл). Полученная в результате смесь перемешивалась при 90°С в течение 3 часов и затем охлаждалась до комнатной температуры. Растворители были удалены в условиях вакуума и остаток разделен между водой (15 мл) и этилацетатом (50 мл). Слои были отделены и органический слой был промыт водой (3×20 мл) и соляным раствором (3×20 мл) и высушены над сульфатом натрия. Удаление растворителей дало промежуточный продукт 3 (326 мг, 100%) в качестве вязкого масла, которое было охарактеризовано LC/MS (LRMS (М+Н+) m/z 507,1).
Промежуточный продукт 3 (207 мг, 0,41 ммоль) перемешивался с ацетатом аммония (1,57 г, 20,40 ммоль) в формамиде под атмосферой азота при 130°С в течение 4,5 часов. Полученный в результате раствор был охлажден до комнатной температуры и разделен между этилацетатом (50 мл) и водой (10 мл). Слои были отделены, и органический слой был промыт водой (4×10 мл) и соляным раствором (20 мл) и высушены над сульфатом натрия. Растворители были удалены в условиях вакуума и остаток был очищен обратнофазовой HPLC с использованием градиента мобильной фазы, состоящего из ацетонитрила и воды, для получения имидазола 4 (159 мг, 80%), который был выделен и охарактеризован 1H-NMR и LC/MS (LRMS (М+Н+) m/z 488,2).
При комнатной температуре к раствору 4 (159 мг, 0,33 ммоль) в дихлорометане (4 мл) была добавлена трифтороуксусная кислота (4 мл) и полученный в результате раствор перемешивался при комнатной температуре в течение ночи. Растворители были удалены в условиях вакуума для получения амина 5 (89 мг) в качестве стеклообразного твердого вещества, которое было определено достаточно чистым для использования в последующих трансформациях. LC/MS (LRMS (М+Н+) m/z 274,1).
Неочищенный амин 5 (72 мг, 0,26 ммоль) перемешивался с диизопропилэтиламином (197 мкл, 1,1 ммоль) в диметилформамиде (3 мкл) при комнатной температуре в течение 5 минут, после чего был добавлен промежуточный продукт 6 (100 мг, 0,26 ммоль). Полученный в результате раствор перемешивался еще в течение 30 минут и растворители были удалены в условиях вакуума. Неочищенный остаток очищался обратнофазовой HPLC с использованием градиента мобильной фазы, состоящего из ацетонитрила и воды, для получения 7 (10 мг, 8%) в виде стеклообразного твердого вещества, которое было охарактеризовано 1H-NMR и LC/MS (LRMS (М+Н+) m/z 470,2).
Пример 54
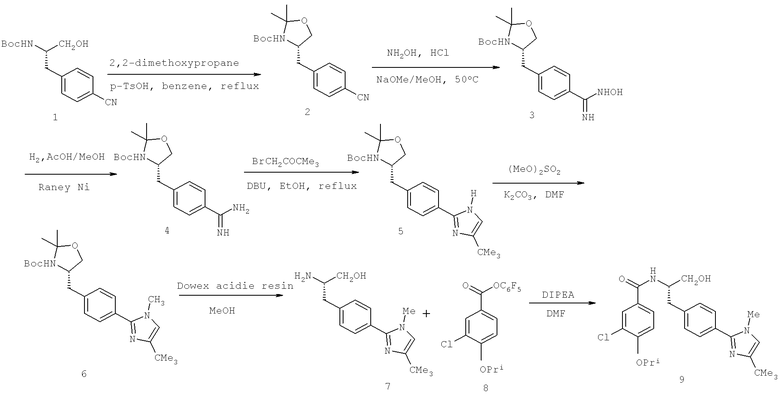
При комнатной температуре к раствору спирта 1 (2,59 г, 9,4 ммоль) в бензоле (50 мл) был добавлен 2,2-диметоксипропан (1,75 мл, 14,1 ммоль) и p-толуолсульфоновая кислота (179 мг, 0,94 ммоль). Полученный в результате раствор перемешивался под атмосферой азота при 110°С в течение 1,5 часов. Растворители были удалены в условиях вакуума и остаток был очищен с использованием флеш-хроматографии (силикагель, этилацетат/гексаны) для получения 2 (765 мг, 27%), который был охарактеризован LC/MS (LRMS (М+Н+) m/z 317,4).
Нитрил 2 (765 мг, 2,4 ммоль) перемешивался с метоксидом натрия в метаноле (10,0 мл, 5,0 ммоль, 0,5 М) и гидрохлориде гидроксиламина (336 мг, 4,8 ммоль) под атмосферой азота при 50°С в течение 3 часов. Затем он был охлажден до комнатной температуры и растворители были удалены в условиях вакуума. Остаток был разделен между насыщенным водным раствором хлорида аммония (30 мл) и этилацетатом (30 мл); слои были отделены и водная фаза экстрагирована этилацетатом (2×30 мл).
Комбинированные органические экстракты были высушены над сульфатом натрия и сконцентрированы в условиях вакуума. Остаток был очищен с использованием флеш-хроматографии (силикагель, гексан/этилацетат) для получения 3 (314 мг, 38%), который был охарактеризован 1H-NMR и LC/MS (LRMS (М+Н+) m/z 350,1).
При комнатной температуре к раствору 3 (314 мг, 0,9 ммоль) в метаноле (15 мл) был добавлен скелетный никелевый катализатор (Raney nickel) гидрирования (50 мг) и уксусная кислота (300 мкл). Полученная в результате смесь перемешивалась под атмосферой азота при комнатной температуре в течение 2 часов. Она была отфильтрована через Целит (Celite) и сконцентрирована в условиях вакуума для получения 4 (275 мг, 0,83 ммоль) в качестве белого твердого вещества, которое было определено достаточно чистым для использования в последующих трансформациях. LC/MS (LRMS (M+H+) m/z 414,1).
При комнатной температуре к раствору амидина 4 (138 мг, 0,4 ммоль) в безводном этаноле (15 мл) был добавлен 1,8-диазабицикло[5.4.0]ундек-7-ен (622 мкл, 4,2 ммоль) и 1-бромопинаколон (84 мкл, 0,6 ммоль). Полученная в результате смесь перемешивалась под атмосферой азота при 115°С в течение 30 минут. Затем она была охлаждена до комнатной температуры и растворители удалены в условиях вакуума. Остаток очищался обратнофазовой HPLC с использованием градиента мобильной фазы, состоящего из ацетонитрила и воды, для получения 5 (29 мг, 17%), который был охарактеризован 1Н-NMR и LC/MS (LRMS (М+H+) m/z 414,1).
При комнатной температуре к раствору имидазола 5 (29 мг, 0,07 ммоль) в безводном диметилформамиде (5 мл) был добавлен карбонат калия (39 мг, 0,28 ммоль) и диметилсульфат (133 мкл, 1,40 ммоль). Полученная в результате смесь перемешивалась под атмосферой азота при 50°С в течение 24 часов, после чего растворители были удалены в условиях вакуума. Остаток очищался с использованием флеш-хроматографии (силикагель, этилацетат/гексаны) для получения 6 (15 мг, 43%) в виде стеклообразного твердого вещества, которое было охарактеризовано 1H-NMR и LC/MS (LRMS (M+H+) m/z 428,3).
Раствор 6 (15 мг, 0,04 ммоль) в безводном метаноле (3 мл) и вода (300 мкл) перемешивались с ионообменной смолой DOWEX 50WX8-400 (100 мг) при комнатной температуре в течение 16 часов. Смола была удалена фильтрацией и промыта триэтиламином (3 мл). Растворители были удалены под высоким вакуумом для получения 7 (12 мг, 0,04 ммоль), которое было определено достаточно чистым для использования в последующих трансформациях. (LRMS (M+H+) m/z 288,2).
При комнатной температуре к раствору амина 7 (12 мг, 0,04 ммоль) в диметилформамиде (3 мл) был добавлен диизопропилэтиламин (20 мкл, 0,10 ммоль).
Полученный в результате раствор перемешивался при комнатной температуре в течение 5 минут, после чего был добавлен промежуточный продукт 8 (16 мг, 0,04 ммоль). Реакционная смесь перемешивалась еще в течение 30 минут. Растворители были удалены в условиях вакуума и остаток очищен флеш-хроматографией (силикагель, метанол/дихлорометан) для получения 9 (10 мг, 50%) в качестве стеклообразного твердого вещества, которое было охарактеризовано 1H-NMR и LC/MS (LRMS (M+H+) m/z 484,2).
Пример 55
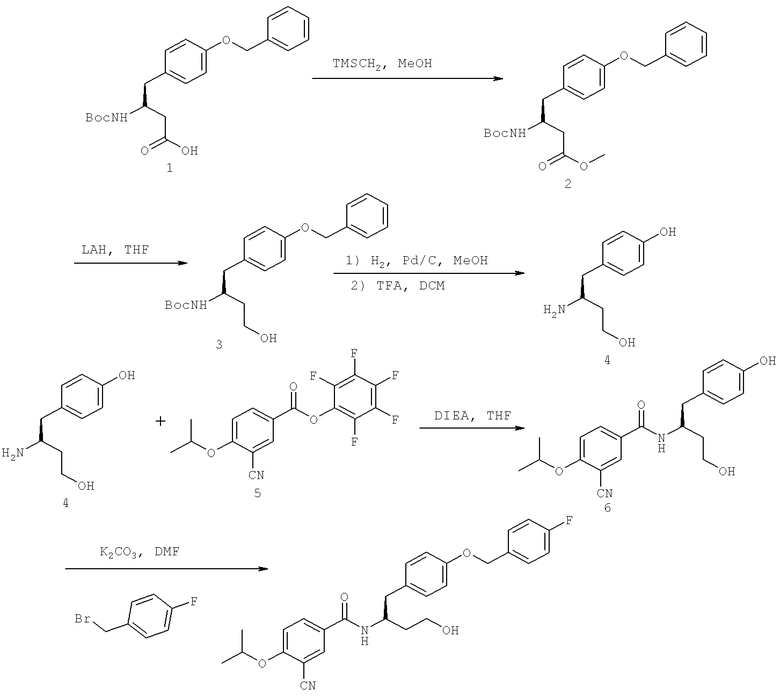
К раствору Boc-L-β-гомотирозина (OBzl) (5 г, 13 ммоль) в метаноле (200 мл) был по каплям добавлен триметилсилилдиазометан (2 М в гексанах, 40 мл, 78 ммоль).
Реагент добавлялся непрерывно, если необходимо, до тех пор пока не прекратилось выделение пузырьков. Смесь была сконцентрирована для получения 2, который использовался в последующем этапе без дальнейщей очистки. LRMS (М+Н+) m/z 300,3.
К раствору 2 (5,5 г, 13,76 ммоль) в THF (100 мл) был добавлен LAH (1 М в THF, 13,7 мл, 13,7 ммоль) при 0°С. Полученный в результате раствор перемешивался в течение 2 часов и был добавлен метанол для того, чтобы погасить реакцию. Растворители были выпарены и было получено желтоватое твердое тело, которое было растворено в этилацетате и промыто в насыщенном NaHCO3. Органический слой был промыт соляным рствором, высушен над Na2SO4 и сконцентрирован при пониженном давлении. Остаток был очищен через колоночную флеш-хроматографию с использованием смеси этилацетата и гексана в качестве элюентов для получения 3 в качестве белого твердого тела (3,5 г, 70%). LRMS (М+Н+) m/z 394,4).
Раствор 3 (1,9 г, 5 ммоль) в МеОН (40 мл) перемешивался под потоком H2 (50 psi фунт/кв. дюйм) в присутствии 10% Pd/C (200 мг) в течение 30 часов. Катализатор был удален фильтрацией через фильтр ПТФЭ (политетрафторэтила) (0,45 мкм), а растворители были выпарены для получения белого твердого вещества (1,5 г), который перемешивался в смеси TFA (1 мл) и DCM (9 мл) в течение 2 часов. Полученный в результате раствор был сконцентрирован и использован в следующем этапе без дальнейшей очистки. LRMS (М+Н+) m/z 182,3.
К раствору 4 (926 мг, 5 ммоль) в THF (10 мл) был добавлен 5 (950 мг, 2,6 ммоль) и N,N-диизопропилэтиламин (4,5 мл, 25,5 ммоль). Реакция перемешивалась при комнатной температуре в течение 10 часов. Смесь была сконцентрирована и высушена на высоком вакууме. Полученный в результате неочищенный продукт очищался через колоночную флеш-хроматографию с использованием этилацетата в качестве элюента для получения 6 (710 мг, 74%). LRMS (М+Н+) m/z 370,4.
К раствору 6 (70 мг, 0,2 ммоль) в DMF (1 мл) был добавлен 4-фторобензил бромид (0,15 мл, 1,2 ммоль) и карбонат калия (166 мг, 1,2 ммоль). Полученная в результате смесь перемешивалась в течение 12 часов при комнатной температуре. Смесь была отфильтрована и фильтрат очищен на RP-HPLC с использованием ацетонитрила и H2O для получения 1е (35 мг, 37%). LRMS (M+H+) m/z 477,5.
Пример 56
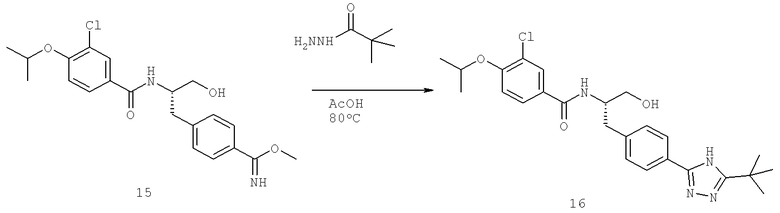
Раствор имидата 15 (20 мг, 0,05 ммоль), гидразида пивалиновой кислоты (9 мг, 0,08 ммоль) и уксусная кислота (1 мл) перемешивались при 80°С в течение 1 часа. Реакционная смесь была затем сконцентрирована в условиях вакуума и полученный в результате остаток был очищен обратнофазовой HPLC (С 18, ацетонитрил/вода) для получения 10 мг (43%) тетразола 16. LRMS (M+H+) m/z 471,2.
Пример 57
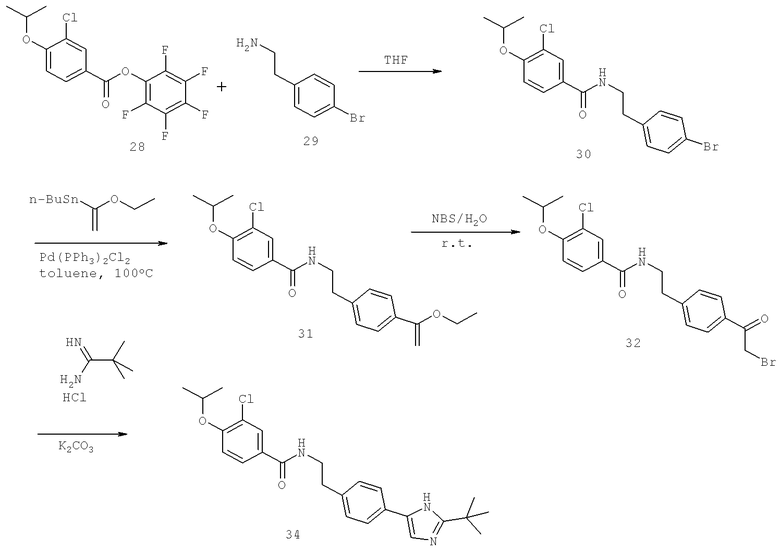
Раствор эфира пентафторофенила 28 (1,0 г, 2,62 ммоль), амина 29 (0,49 мл, 3,15 ммоль) и THF (10 мл) перемешивался при 23°С в течение 4 часов. Реакционный раствор был сконцентирован в условиях вакуума и полученный в результате остаток был очищен колоночной хроматографией (силикагель, 1:1 EtOAc : гексаны) для получения 1,1 г (88%) амида 30. LRMS (М+Н+) m/z 396,1.
Раствор бромида 30 (200 мг, 0,51 моль), дихлоробис(трифенилфосфин) палладия (II) (35 мг, 0,05 моль), трибутил(1-этоксивинил)тина (0,34 мл, 1,0 ммоль) и толуола (2 мл) перемешивались при 100°С в атмосфере N2 в течение 4 часов. После завершения, при контроле LCMS, реакционная смесь была охлаждена, профильтрована через хлопок и сконцентирована в условиях вакуума. Полученный в результате остаток был очищен колоночной хроматографией (силикагель, 1:4:0,1 EtOAc : гексаны : триэтиламин) для получения 100 мг (52%) стирола 31. LRMS (М+Н+) m/z 388,2.
Раствор соединения 31 (100 мг, 0,25 ммоль), ТНР : Н2О (3:1,4 мл) и N-бромо-сукцинамида (46 мг, 0,25 ммоль) перемешивались при 23°С в течение 15 минут.
Реакционная смесь была затем сконцентрирована в условиях вакуума, неочищенный остаток был разбавлен EtOAc (30 мл), промыт соляным раствором (10 мл) и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 4:1 EtOAc : гексаны) для получения 50 мг (46%) бромокетона 32. LRMS (М+Н+) m/z 438,1.
Раствор бромокетона 32 (50 мг, 0,11 ммоль), K2CO3 (47 мг, 0,34 ммоль), терт-бутилфарбамидин гидрохлорида (21 мг, 0,23 ммоль) и DMF (2 мл) перемешивался при 23°С в атмосфере N2 в течение 18 часов. Реакционная смесь была сконцентирована под высоким вакуумом (0,1 мм Hg), и полученный в результате остаток был очищен колоночной хроматографией (силикагель, 2:1 EtOAc : гексаны) для получения 35 мг (72%) имидазола 34. LRMS (M+H+) m/z 440,2.
Пример 58
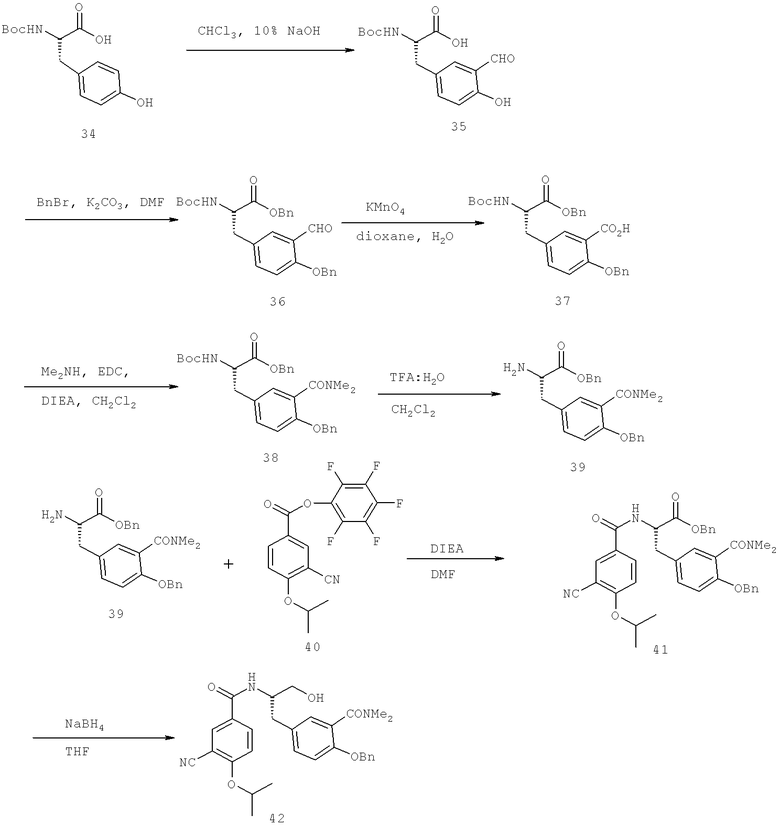
Хлороформ (20 мл) медленно добавлялся в течение 2 часов к раствору Вос-тирозина (20 г, 71 ммоль) и 10% NaOH в Н2О (400 мл) при 85°С. После истечения общего количества 4 часов реакционный раствор был подкислен 3 N HCl (200 мл) и экстрагирован EtOAc (3×150 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:1:0,1 гексаны : EtOAc : AcOH) для получения 6,3 г смеси альдегида 35 и немного восстановленного 34.
Раствор альдегида 35 (загрязненный 34, 6:3 г, 20 ммоль), K2CO3 (5,8 г, 42 ммоль), бромида бензила (5,0 мл, 42 ммоль) и DMF (100 мл) перемешивался при 23°С в течение 18 часов. Реакционная смесь была разбавлена EtOAc (200 мл) и промыта 1 N HCl (3×200 мл) и соляным раствором (3×200 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:4 EtOAc : гексаны) для получения 2,2 г (22%) эфира 36. LRMS (M+H+) m/z 490,2.
Раствор альдегида 36 (570 мг, 1,16 ммоль), KMnO4 (368 мг, 2,32 ммоль), диоксана (3 мл) и Н2О (1 мл) перемешивался при 23°С в течение 3 часов. Реакционная смесь была сконцентрирована в условиях вакуума и полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:1 EtOAc : гексаны) для получения 350 мг (60%) кислоты 37. LRMS (М+Н+) m/z 506,2.
Раствор кислоты 37 (115 мг, 0,23 ммоль), диметиламина (0,23 мл, 2,0 М в THF), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорида (65 мг, 0,34 ммоль), диизопропил этил амина (0,12 мл, 0,68 ммоль) и CH2Cl2 (1 мл) перемешивались при 23°С в течение 4 часов. Реакционная смесь была затем разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:1 гексаны : EtOAc) для получения 60 мг (49%) амида 38. LRMS (M+H+) m/z 533,3.
Раствор амида 38 (60 мг, 0,11 ммоль), TFA:H2O (97,5:2,5, 1 мл) и CH2Cl2 (1 мл) перемешивался при 23°С в течение 30 минут. Реакционный раствор был сконцентрирован в условиях вакуума и полученный в результате остаток был помещен под высокий вакуум на 2 часа и затем использовался без дальнейшей очистки.
Раствор амина 39 (69 мг, 0,16 ммоль), эфира пентафторофенола 40 (71 мг, 0,19 ммоль), диизопропилэтиламина (83 мкл, 0,68 ммоль) и DMF (1 мл) перемешивались при 23°С в течение 4 часов. Реакционная смесь была затем разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:1 гексаны : EtOAc) для получения 60 мг (60%) эфира амида 41. LRMS (М+Н+) m/z 620,3.
Раствор эфира 41 (50 мг, 0,08 ммоль), NaBH4 (30 мг, 0,81 ммоль), THF (0,5 мл) и МеОН (0,5 мл) перемешивались при 23°С в течение 2 часов. Реакционная смесь была затем разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:50 МеОН : EtOAc) для получения 31 мг (75%) спирта 42. LRMS (М+Н+) m/z 516,3.
Пример 59
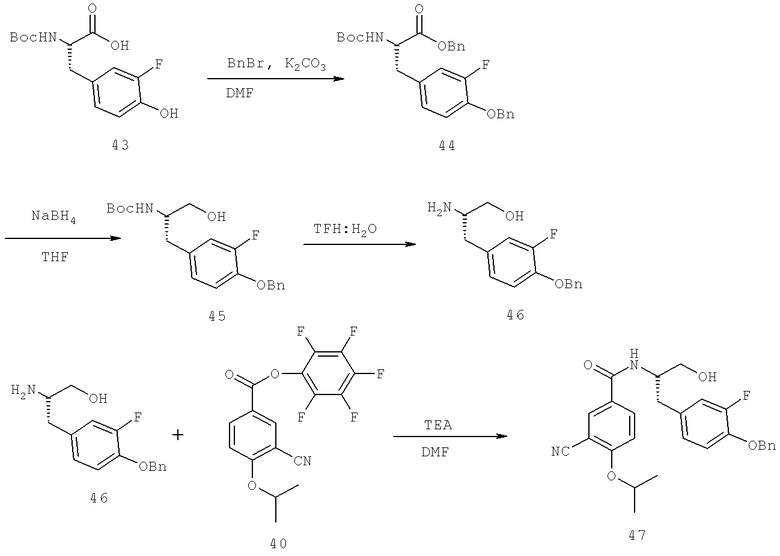
Раствор кислоты 43 (300 мг, 1,0 ммоль), K2CO3 (276 мг, 2,0 ммоль), бромид бензила (0,24 мл, 2,0 ммоль) и DMF (4 мл) перемешивался при 23°С в течение 18 часов.
Реакционная смесь была разбавлена EtOAc (30 мл) и промыта 1 N HCl (10 мл) и соляным раствором (3×15 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:3 EtOAc : гексаны) для получения 400 мг (83%) эфира 44. LRMS (M+H+) m/z 480,2.
Раствор эфира 44 (100 мг, 0,21 ммоль), NaBH4 (24 мг, 0,63 ммоль), THF (1 мл) и МеОН (1 мл) перемешивался при 23°С в течение 18 часов. Реакционная смесь была затем разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток использовался без дальнейшей очистки.
Раствор спирта 45 (100 мг, 0,27 ммоль) и TFA:H2O (97,5:2,5, 1 мл) перемешивался при 23°С в течение 30 минут. Реакционный раствор был сконцентрирован в условиях вакуума, и полученный в результате остаток был помещен под высокий вакуум на 2 часа и затем использовался без дальнейшей очистки.
Раствор амина 46 (40 мг, 0,15 ммоль), эфир пентафторофенила 40 (43 мг, 0,12 ммоль), триэтиламина (51 мкл, 0,29 ммоль) и DMF (0,6 мл) перемешивался при 23°С в течение 4 часов. Реакционная смесь была разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:2 гексаны : EtOAc) для получения 30 мг (43%) эфира амида 47. LRMS (M+H+) m/z 463,2.
Пример 60
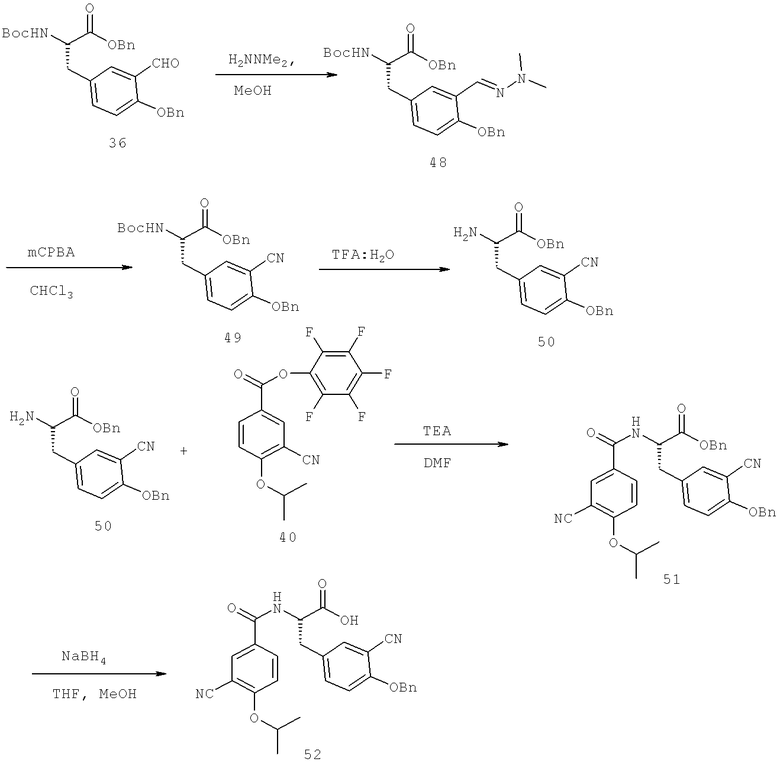
Раствор альдегида 36 (300 мг, 0,6 ммоль), диметилгидразина (47 мкл, 0,6 ммоль) и МеОН (2,5 мл) перемешивался при 0°С в течение 2 часов, затем ему было дано нагреться до 23°С и он дополнительно перемешивался в течение 15 часов. Реакционный раствор был сконцентрирован в условиях вакуума и полученный в результате остаток использовался без дальнейшей очистки.
К раствору неочищенного гидразона 48 (325 мг, 0,6 ммоль) и CHCl3 (2 мл) при -5°С по каплям был добавлен раствор m-хлоропероксибензоловой кислоты (212 мг, 1,23 ммоль) и CHCl3 (2 мл). Реакционный раствор нагревали до 23°С и перемешивали в течение 2 дней. Реакционная смесь была затем разбавлена EtOAc (10 мл) и промыта насыщенным водным NaHCO3 (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:2 гексаны : EtOAc) для получения 100 мг (34%) нитрила 49.
LRMS (М+Н+) m/z 487,2.
Раствор нитрила 49 (60 мг, 0,27 ммоль) и TFA:H2O (97,5:2,5, 2 мл) перемешивался при 23°С в течение 30 минут. Реакционный раствор был сконцентрирован в условиях вакуума, и полученный в результате остаток был помещен под высокий вакуум на 2 часа и затем использовался без дальнейшей очистки.
Раствор амина 50 (100 мг, 0,25 ммоль), эфира пентафторофенила 40 (85 мг, 0,22 ммоль), триэтиламина (96 мкл, 0,74 ммоль) и DMF (1 мл) перемешивался при 23°С в течение 4 часов. Реакционная смесь была затем разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:1 гексаны : EtOAc) для получения 60 мг (42%) нитрила 51. LRMS (M+H+) m/z 574,2.
Раствор эфира 51 (60 мг, 0,1 ммоль), NaBH4 (12 мг, 0,3 ммоль), THF (1 мл) и МеОН (1 мл) поддерживался при 23°С в течение 18 часов. Реакционная смесь была затем разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл).
Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:2 гексаны : EtOAc) для получения 30 мг (64%) спирта 52. LRMS (M+H+) m/z 470,2.
Пример 61
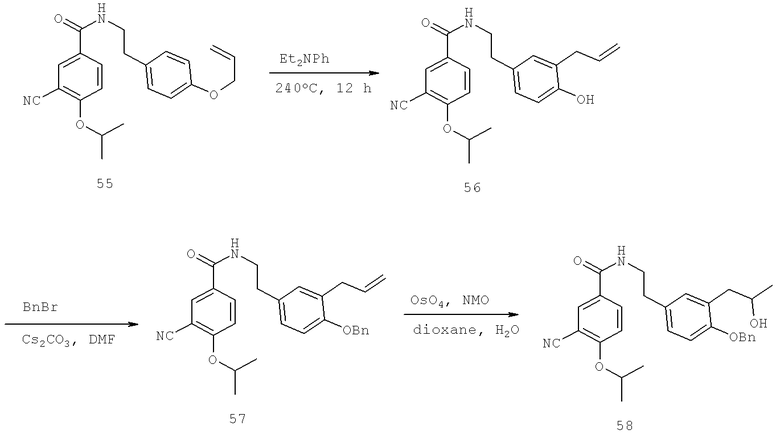
Раствор амида 55 (1,6 г, 4,38 ммоль) и диэтиланилина (5 мл) поддерживался при 240°С в течение 18 часов. Реакционный раствор был охлажден до 23°С, разбавлен EtOAc (30 мл) и промыт 1 N HCl (3×50 мл) и соляным раствором (2×50 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 2:1 гексаны : EtOAc) для получения 1 г (63%) фенола 56. LRMS (M+H+) m/z 365,2.
Раствор фенола 56 (700 мг, 1,92 ммоль), Cs2CO3 (1,25 мг, 3,84 ммоль), бромида бензила (0,46 Мл, 3,84 ммоль) и DMF (10 мл) поддерживались при 50°С в течение 2 часов. Реакционная смесь была разбавлена EtOAc (30 мл) и промыта 1 N HCl (20 мл) и соляным раствором (3×50 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 1:3 EtOAc : гексаны) для получения 500 мг (57%) амида 57. LRMS (M+H+) m/z 455,2.
Раствор амида 57 (150 мг, 0,33 ммоль), тетраоксида осмия (8 мг, 0,03 ммоль), N-метилморфолин-N-оксида (182 мг, 1,55 ммоль), пиридина (2,4 мкл, 0,03 ммоль), THF (2 мл) и Н2О (2 мл) поддерживался при 23°С. После 3 часов были добавлены Целит (Celit) (1 г), NaHSO3 (1 г) и EtOAc (20 мл) и полученная в результате смесь перемешивалась. После 30 минут реакционная смесь была отфильтрована и полученный в результате фильтрат сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 3:1 EtOAc : гексаны) для получения 100 мг (62%) диола 58. LRMS (M+H+) m/z 489,2.
Раствор диола 58 (52 мг, 0,11 ммоль), Pb(OAc)4 и CH2Cl2 (2 мл) поддерживался при 23°С в течение 30 минут. Реакционная смесь была затем отфильтрована через пробку Целита и фильтрат сконцентрирован для получения альдегида в качестве бесцветного масла.
Раствор неочищенного альдегида (~50 мг, ~0,11 ммоль), NaBH4 (24 мг, 0,63 ммоль), THF (1 мл) и МеОН (1 мл) поддерживался при 23°С в течение 30 минут.
Реакционная смесь была затем разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 2:1 EtOAc : гексаны) для получения 20 мг (40%) спирта 59. LRMS (M+H+) m/z 459,2.
Пример 62
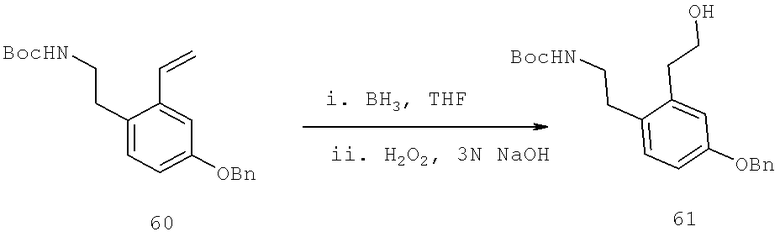
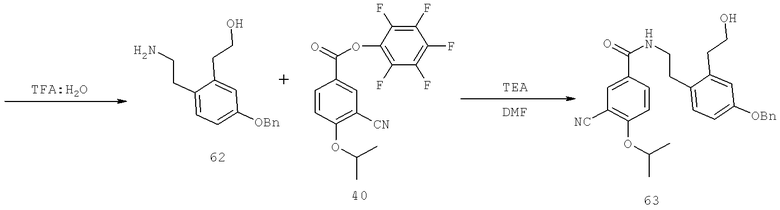
Раствор стирола 60 (190 мг, 0,54 ммоль), боран-THF (1,0 М, 0,54 мл) поддерживался при 23°С в течение 2 часов. Затем было добавлено дополнительное количество боран-THF (0,54 мл). Еще после 2 часов была добавлена третья порция. Реакционный раствор выдерживался в течение 18 часов, был охлажден до 0°С, а затем был добавлен 3 N NaOH (0,5 мл) и H2O (0,5 мл). После 2 часов при 23°С реакционная смесь была разбавлена EtOAc (20 мл) и промыта соляным раствором (20 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 2:1 EtOAc : гексаны) для получения 150 мг (75%) спирта 61. LRMS (M+H+) m/z 372,2.
Раствор спирта 61 (120 мг, 0,32 ммоль), TFA : H2O (97,5:2,5, 4 мл) поддерживался при 23°С в течение 30 минут. Реакционный раствор был сконцентрирован в условиях вакуума, и полученный в результате остаток был помещен под высокий вакуум на 2 часа и затем использовался без дальнейшей очистки.
Раствор вышеупомянутого амина 62 (50 мг, 0,18 ммоль), эфира пентафторофенола 40 (82 мг, 0,22 ммоль), триэтиламина (96 мкл, 0,55 ммоль) и DMF (1 мл) поддерживался при 23°С в течение 2 часов. Реакционная смесь была затем разбавлена EtOAc (10 мл) и промыта 1 N HCl (5 мл) и соляным раствором (5 мл). Органический слой был высушен (MgSO4), отфильтрован и сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен обратнофазовой HPLC (С 18, ацетонитрил/вода) для получения 6 мг (7%) амида 63. LRMS (М+Н+) m/z 459,2.
Пример 63
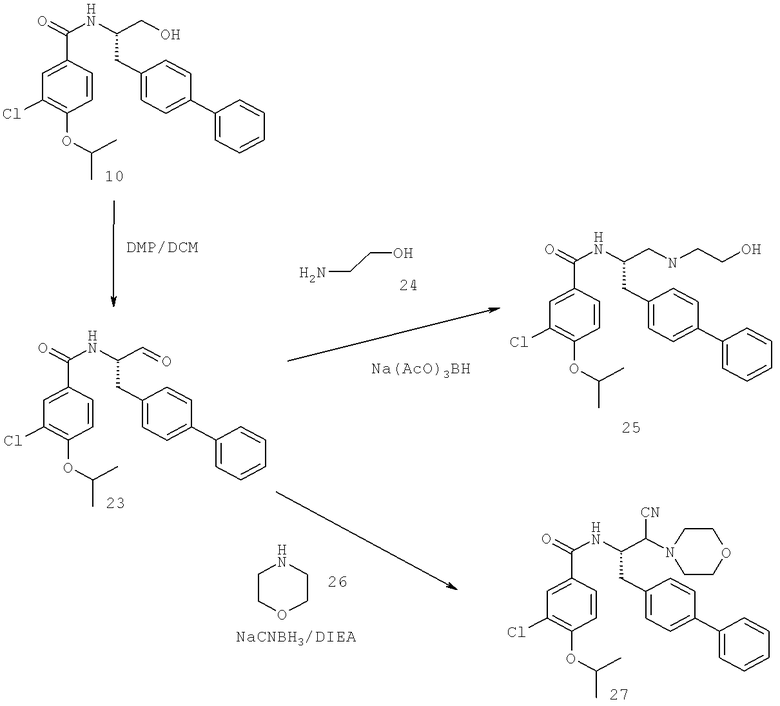
К раствору 10 (1,15 г, 2,71 ммоль) в DCM (100 мл) был добавлен периодинан Десс-Мартина (Dess-Martin) (2,30 г, 5,42 ммоль). Реакционная смесь перемешивалась в течение 1 часа, после чего раствор DCM был промыт раствором тиосульфата натрия и бикарбонатом натрия и высушен над сульфатом натрия. Смесь была отфильтрована и фильтрат сконцентрирован при пониженном давлении для получения 23 (1,0 г, 87%).
К раствору 23 (30,0 мг, 0,0711 ммоль) в DCM (2 мл) были добавлены диизопропилэтиламин (37,0 мкл, 0,213 ммоль), 24 (12,9 мкл, 0,213 ммоль) и триацетоксиборогидрид натрия (20,0 мг, 0,142 ммоль). Реакционная смесь перемешивалась в течение ночи, и затем была сконцентрирована при пониженном давлении. Остаток был очищен обратнофазовой HPLC (С 18) с использованием смеси ацетонитрила и H2O для получения 25 (5,6 мг, 16,9%). LRMS (М+H+) m/z 467,4.
К раствору 23 (50,0 мг, 0,119 ммоль) в метаноле (2 мл) были добавлены диизопропилэтиламин (62,0 мкл, 0,356 ммоль), 26 (31,1 мкл, 0,356 ммоль) и цианоборогидрид натрия (22,4 мг, 0,356 ммоль). Реакционная смесь перемешивалась в течение ночи, затем была сконцентрирована при пониженном давлении и остаток был очищен обратнофазовой HPLC (С 18) с использованием смеси ацетонитрила и H2O
для получения 27 (31,0 мг, 22,9%). LRMS (M+H+) m/z 518,5.
Пример 64
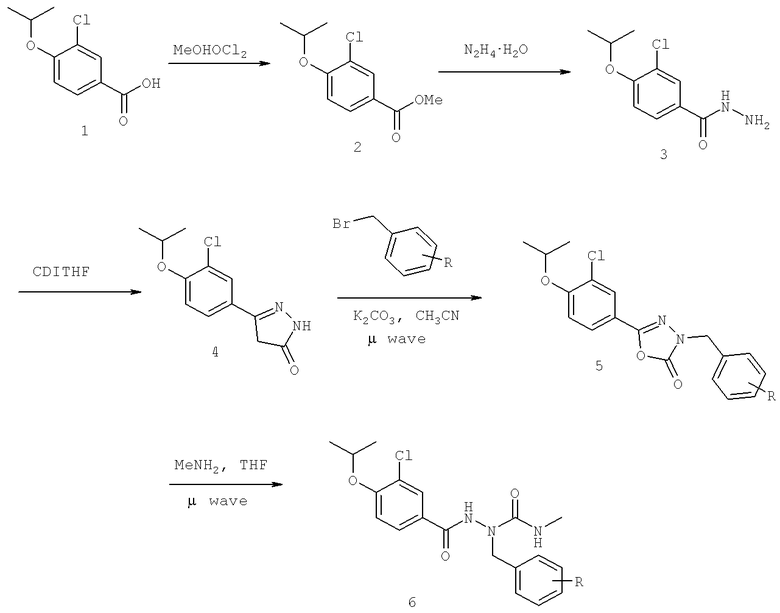
К раствору 1 (1,0 г, 4,66 ммоль) в МеОН (10,0 мл) был добавлен SOCl2 (0,68 мл, 9,32 ммоль). После перемешивания в течение ночи при температуре окружающей среды раствор был сконцентрирован в условиях вакуума и использовался далее без дальнейшей очистки.
К раствору 2 (~1,065 г неочищенного продукта, 4,66 ммоль) в EtOH (1,5 мл) был добавлен N2H4×H2O (1,13 мл, 23,3 ммоль). Реакционная смесь была нагрета до флегмы и перемешивалась в течение 3 часов. После охлаждения раствор был обработан H2O, трижды экстрагирован с помощью EtOAc, высушен над MgSO4, отфильтрован и сконцентрирован. Рекристаллизация из CH2Cl2 дала 1,01 г 3 в качестве белых кристаллов; 95% выход, 2 этапа.
К раствору 3 (0,477 г, 2,09 ммоль) в THF (8,0 мл) был добавлен карбонилдиимидазол (0,379 г, 2,29 ммоль). Реакционная смесь была нагрета в сосуде с обратным холодильником и перемешивалась в течение 1,5 часа. После охлаждения раствор был сконцентрирован в условиях вакуума и очищен через колоночную флеш-хроматаграфию (10-40% EtOAc/Hex) для получения 0,515 г 4 в качестве белого твердого вещества; выход 97%.
К раствору 4 (1,0 эквивалента; обычно 0.3-1,0 ммоль) в CH2Cl2 (2,0 мл) был добавлен электрофил (1.1 эквивалента) и K2CO3 (1,1 эквивалента). Реакционная смесь нагревалась до 80°С под микроволновым излучением в течение 30 минут, за чем следовала фильтрация и концентрация в условиях вакуума. Продукт может быть использован без дальнейшей очистки или очищен через колоночную флеш-хроматографию (обычно 10-40% EtOAc/Hex) для получения 5 при обычном выходе более 90%.
К 5 (1,0 эквивалента; обычно 0,3-1,0 ммоль) был добавлен метиламин (2,0 М раствор в THF, 10,0 эквивалента). Реакционная смесь была нагрета до 100°С под микроволновым излучением в течение 4 часов, за чем следовала концентрация в условиях вакуума. Продукт был очищен через колоночную флеш-хроматографию (обычно 40-80% EtOAc/Hex) для получения 6 при обычном выходе 70-85%.
Пример 65
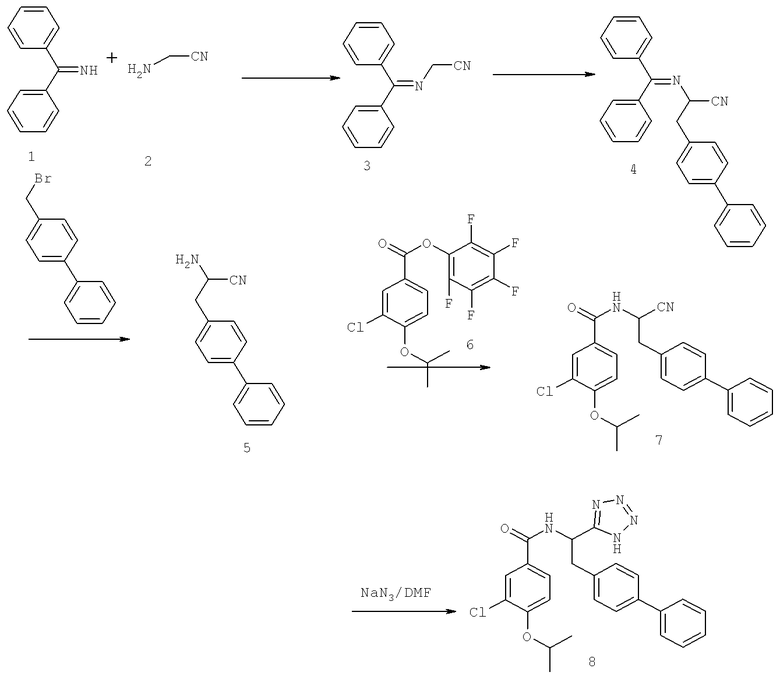
К перемешиваемому раствору бисульфата 2-аминоацетонитрила (2,9 г, 0,013 ммоль) в DCM был добавлен бензофенон (3,48 мл, 0,0208 ммоль) за чем следовал DIEA (4,53 мл, 0,026 ммоль). После перемешивания в течение 18 часов раствор DCM был промыт водой (50 мл), высушен над сульфатом натрия, отфильтрован и фильтрат сконцентрирован при пониженном давлении. Остаток был очищен на флеш-колонке с силикагелем (гексаны EtOAc, 1:1) для получения 3 (2,40 г, 82%).
Бис(триметилсилил)амид лития (раствор 1М в THF) медленно добавлялся в перемешиваемый раствор 3 (1,2 г, 0,00545 моль) и р-фенилбензил бромида (1,08 г, 0.00435 моль) в THF (50 мл) над ледяной баней из сухого ацетона в атмосфере азота.
После 1 часа реакция гасилась добавлением метанола и растворитель выпаривался при пониженном давлении. Остаток был очищен на флеш-колонке с силикагелем (гекасаны : EtOAc, 1:1) для получения 4. 4 был повторно осажден в EtOAc (100 мл) и обработан концентрированной HCl (5 мл). После перемешивания в течение 1 часа растворители были выпарены при пониженном давлении и полученное в результате твердое вещество 5 было три раза промыто эфиром этила (50 мл) и высушено в условиях вакуума (0,39 г, 32,1%).
К раствору 5 (0,39 г, 1,75 ммоль) в DMF (10 мл) были добавлены 6 (0,801 г, 2,11 ммоль) и диизопропилэтиламин (0,61 мл, 3,50 ммоль) при комнатной температуре. Реакционная смесь перемешивалась в течение ночи. Затем растворители были выпарены при пониженном давлении и остаток очищен на флеш-колонке с силикагелем (гексан : EtOAc, 3:1) для получения 7 (0,40 г, 54,5%). LCMS (M+H+) m/z 419,1.
К раствору 7 (50 мг, 0,119 ммоль) в DMF (2 мл) был добавлен азид натрия (15,5 мг, 0,239 ммоль) и хлорид аммония (12,8 мг, 0,238 ммоль). Смесь перемешивалась при 80°С в течение ночи и затем была отфильтрована. Фильтрат был очищен на обратнофазовой HPLC (С 18) с использованием смеси ацетонитрила и H2O для получения 8 (6,40 мг, 11,6%). LCMS (M+H+) m/z 462,4.
Пример 66
Схема А
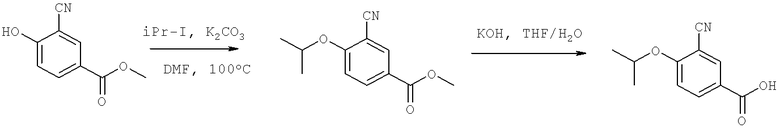
Метил 3-циано-4-[(метилэтил)окси]бензоат:
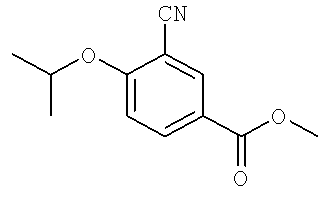
К раствору метил 3-циано-4-гидроксибензоата (82 г, 463 ммоль; Журнал медицинской химии, 2002, 45, 5769) в диметилформамиде (800 мл) был добавлен 2-иодопропан (93 мл, 926 ммоль) и карбонат калия (190 г, 1,4 моль). Полученная в результате смесь нагревалась при 50°С в течение 16 часов, после чего ей было дано охладиться до комнатной температуры. Реакционная смесь была отфильтрована и материнский раствор был разбавлен 0,5 N гироокиси натрия (1 л). Полученная в результате смесь была экстрагирована эфиром (2×1 л) и орагнические вещества были промыты 1 N HCl (1 л) и соляным раствором (700 мл), высушены (MgSO4) и сконцентрированы для получения 100 г (~100%) метил 3-циано-4-[(1-метилэтил)окси]бензоата в качестве желтого твердого вещества.
3-Циано-4-[(1-метилэтил)окси]бензойная кислота:
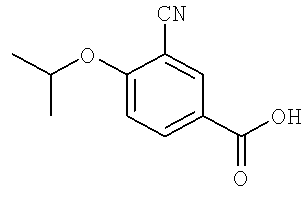
К охлажденному раствору (0°С) метил 3-циано-4-[(1-метилэтил)окси]бензоата (100 г, 463 ммоль) в тетрагидрофуране (500 мл) была добавлена 10% гидроокись калия (500 мл). Полученному в результате раствору было дано нагреться до комнатной температуры и она поддерживалась в течение 16 часов, когда он был сконцентрирован для удаления тетрагидрофурана. Остаток был разбавлен водой (500 мл) и промыт эфиром (2×500 мл). Водяной слой был затем окислен 3 N HCl и был оставлен на 2 часа. Твердые вещества были собраны фильтрацией и промыты несколько раз водой и затем растворены в метиленхлориде (1 л). В основном однородная смесь была отфильтрована через Целит и сконцентрирована до минимального объема метиленхлорида. Сбор твердых веществ путем фильтрации дал 82 г (87%) 3-циано-4-[(1-метилэтил)окси]бензойной кислоты в качестве белого твердого вещества.
Схема В
Реагенты и условия: а) 4 N HCl/диоксан, комнатная температура; б) HBTU, i-Pr2NEt, DMF, комнатная температура; в) 1-этоксивинилтри-n-бутилтин, PdCl2(PPh3)2, диоксан, 100°С; г) NBS, THF/H2O (3:1), комнатная температура; д) 2-амино-3-пиколин, NaHCO3, i-PrOH, 80°C.
(3S)-3-Амино-4-(4-бромофенил)-1-бутанол гидрохлорид.
1,1-Диметилэтил{(1S)-1-[(4-бромофенил)метил]-3-гидроксипропил}карбамат (4,4 г, 12,8 ммоль) был растворен в 4N HCl/диоксане (20 мл). После 2 часов реакционная смесь была сконцентрирована в условиях вакуума для получения 3,69 г (94%) титульного соединения в качестве белого твердого вещества. LC/MS (ES) m/e 244,0 (М+Н)+.
N-{(1S)-1-[(4-Бромофенил)метил]-3-гидроксипропил}-3-циано-4-[(1-метилэтил)окси]бензамид.
К суспензии (3S)-3-амино-4-(4-бромофенил)-1-бутанол гидрохлорида (1,80 г, 6,42 ммоль) в сухом DMF (32 мл) был добавлен N,N-диизопропилэтиламин (2,49 г, 19,3 ммоль) и полученный в результате чистый раствор перемешивался в течение 3 мин. Были добавлены 3-циано-4-[(1-метилэтил)окси]бензойная кислота (1,45 г, 7,06 ммоль) и HBTU (2,68 г, 7,06 ммоль) и реакционная смесь перемешивалась при комнатной температуре в атмосфере азота. После 1,5 часов реакционная смесь была погашена водой (50 мл) и экстрагирована с помощью EtOAc (3×30 мл). Экстракты были высушены (Na2SO4), отфильтрованы и сконцентрированы при пониженном давлении. Остаток был очищен хроматографией с силикагелем (75% EtOAс/гексаны) для получения 2,18 г (78%) титульного соединения в качестве белого твердого вещества. LC/MS (ES) m/e 431,2 (М+Н)+.
N-((1S)-1-{[4-(Бромоацетил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси]бензамид
Колба, высушеная тепловой пушкой при аргоновой чистке, была заряжена N-{(1S)-1-[(4-бромофенил)метил]-3-гидроксипропил}-3-циано-4-[(1-метилэтил)окси]бензамидом (1,0 г, 2,32 моль), дихлоробис(трифенилфосфин)-палладием (II) (81 мг, 0.116 моль), трибутил(1-этоксивинил)тином (1,68 г, 4,64 ммоль) и 1,4-диоксаном (15 мл). Смесь перемешивалась при 100°С в атмосфере аргона. После завершения, при контроле LCMS, реакция была сконцентрирована при пониженном давлении и остаток был немедленно очищен на деактивированном силикагеле (65% EtOAc/гексаны с 5% триэтиламином) для получения 720 мг (1,70 ммоль) промежуточного эфира энола в качестве бесцветной пены, которая была немедленно растворена в THF:H2O (3:1, 18 мл) и обработана N-бромосукцинимидом (318 мг, 1,79 ммоль). После 15 минут при комнатной температуре реакционная смесь была сконцентрирована при пониженном давлении и неочищенный остаток был разбавлен EtOAc (30 мл), промыт соляным раствором (10 мл) и сконцентрирован при пониженном давлении. Остаток был очищен хроматографией с силикагелем (80% EtOAc/гексаны) для получения 651 мг (59%) N-((1S)-1-{[4-(бромоацетил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси] бензамида в качестве белого клейкого твердого вещества. LC/MS (ES) m/e 473,2 (M+H)+.
3-Циано-N-((1S)-3-гидрокси-1-{[4-(6-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
К раствору N-((1S)-1-{[4-(бромоацетил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси]бензамида (300 мг, 0,634 ммоль) в i-PrOH (6 мл) был добавлен 2-амино-3-пиколин (Олдрич, 69 мг, 0,634 ммоль), за чем последовал твердый NaHCO3 (64 мг, 0,761 ммоль). Полученная в результате суспензия была нагрета до 80°С. После 7 часов большинство i-PrOH было удалено при пониженном давлении и остаток был растворен в 3% MeOH/EtOAc (30 мл) и промыт водой (10 мл) и соляным раствором (10 мл). Комбинированные водные слои экстрагировались 3% MeOH/EtOAc (30 мл) и комбинированные экстракты были высушены (Na2SO4), отфильтрованы и сконцентрированы при пониженном давлении. Остаток был очищен обратнофазовой HPLC (MeCN/H2O с 0,1% TFA) и чистые фракции были доведены до рН~8 насыщенным водным NaHCO3 и экстрагированы 3% MeOH/EtOAc (3×30 мл). Экстракты были высушены (Na2SO4), отфильтрованы и сконцентрированы при пониженном давлении для получения 215 мг (70%) титульного соединения в качестве бледно-желтого твердого вещества. LC/MS (ES) m/e 483,2 (М+Н)+.
Схема С
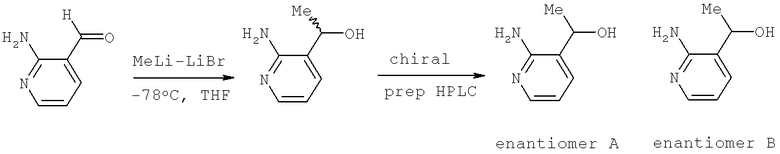
1-(2-амино-3-пиридинил)этанол:
В сухую колбу (высушенную тепловой пушкой при аргоновой чистке) был добавлен сухой THF (400 мл) и MeLi-LiBr (137 мл раствора 1,5 М в Et2O, 204,9 ммоль) через канюлю. Этот раствор был охлажден до -78°С, когда по каплям был добавлен раствор 2-аминопиридин-3-карбоксальдегида (10,0 г, 82,0 ммоль) в THF через выравнивающую давление воронку над ~45 при сильном помешивании (экзотерма наблюдалась, преобладал оранжевый цвет). После завершения добавления раствор продолжали помешивать в течение 1 часа при -78°С, когда TLC (пятно KMnO4 при нагревании) указывало, что большинство начального вещества было преобразовано в продукт. Реакция была очень тщательно погашена водой (200 мл; первоначально по каплям), разбавлена EtOAc (200 мл) и доведена до комнатной температуры. Слои были отделены и водный слой экстрагирован с помощью 3% МеОН в EtOAc. Комбинированные экстракты были высушены над сульфатом натрия, отфильтрованы и сконцентрированы при пониженном давлении. Остаток был очищен хроматографией с силикагелем (Аналоджикс; от 0 до 5% МеОН в EtOAc) для получения 7,78 г (68%) искомого рацемического продукта в качестве желтого масла, которое уплотнялось при высоком вакууме в течение нескольких дней. Этот материал был разделен на соответствующие энантиомеры (более 98% ее) при помощи SFC (сверхкритической флюидной хроматорграфии) с колонкой фирмы Chiralcel с наружным диаметром - высотой (20×250 мм) (10% EtOH/0,1% изопропиламина в гептане/0,1% изопропиламин).
Схема D
1,1-Диметилэтил[(S)-1-({4-[1-(этилокси)этенил]фенил}метил-3-гидроксипропил]карбамат:
К раствору 1,1-диметилэтил {(1S)-1-[(4-бромофенил)метил]-3-гидроксипропил}карбамата (20 г, 58 ммоль) в диоксане (500 мл) был добавлен трибутил[1-(этилокси)этенил]станнан (39 мл, 116 ммоль) и PdCl2(PPh3)2. Полученный в результате раствор нагревался при 100°С в течение 5 часов. Реакция затем была сконцентрирована и остаток очищен флеш-хроматографией (47,5% EtOAc, 47,5% гексаны, 5% триэтиламин) для получения 15 г (77%) 1,1-диметилэтил[(1S)-1-({4-[1-(этилокси)этенил] фенил} метил)-3-гидроксипропил]карбамата в качестве коричневого твердого вещества.
1,1-Диметилэтил((1S)-1-{[4-(бромоацетил)фенил]метил}-3-гидроксипропил)карбамат:
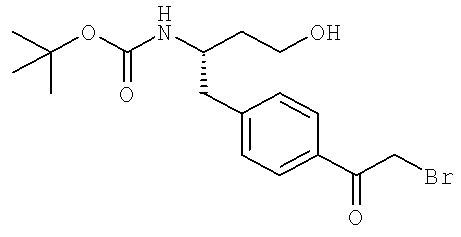
К охлажденному (0°С) раствору 1,1-диметилэтил[(1S)-1-({4-[1-(этилокси) этенил]фенил}метил)-3-гидроксипропил]карбамата (15 г, 44 ммоль) в тетрагидрофуране (450 мл) и воде (150 мл) был добавлен N-бромосуцинамид. Полученный в результате раствор нагревался до комнатной температуры и выдерживался в течение 90 минут. Затем реакция была сконцентрирована и разбавлена этилацетатом (1 л). Полученный в результате раствор был промыт водой (1 л) и соляным раствором (500 мл), высушен (MgSO4) и сконцентрирован для получения 19,5 г (~100%) 1,1-диметилэтил ((1S)-1-{[4-(бромоацетил)фенил] метил }-3-гидроксипропил)карбамата в качестве слегка желтоватого твердого вещества. ESMS [М+Н]+:386,2.
1,1-Диметилэтил[(1S)-3-гидрокси-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]карбамат
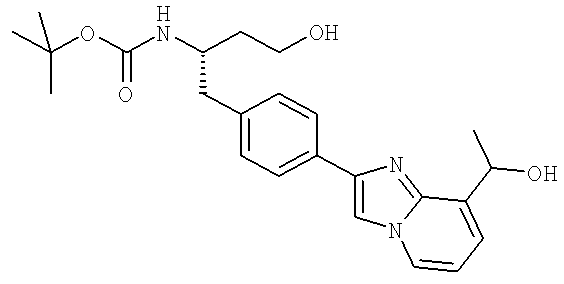
Смесь 1,1-диметилэтил((1S)-1-{[4-(бромоацетил)фенил]метил}-3-гидрокси пропил)карбамата (1,0 г, 2,59 ммоль), 1-(2-амино-3-пиридинил)этанола (0,358 г, 2,59 ммоль) и твердого бикарбоната натрия (0,272 г, 3,24 ммоль) в изопропаноле (25 мл) нагревалась в противотоке в течение 3,5 часов и концентрировалась в условиях вакуума. Остаток был растворен этилацетатом, промыт водой и соляным раствором, высушен (Na2SO4), отфильтрован и сконцентрирован. Полученное в результате бледно-желтое твердое вещество использовалось в следующей реакции без дальнейшей очистки. MS (ES+) m/e 426 [М+H]+.
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидроксиэтил)имидазо [1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
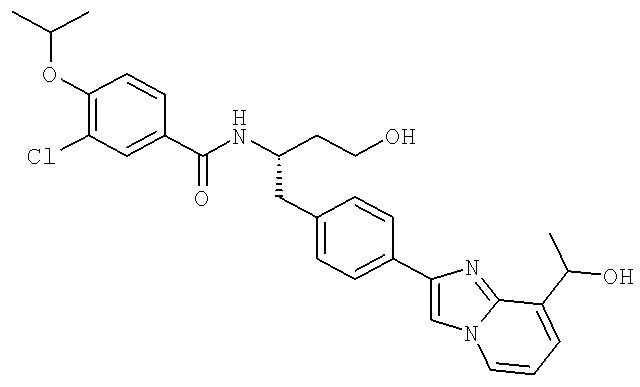
Смесь 1,1-диметилэтил[(1S)-3-гидрокси-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]карбамата (1,08 г, 2,54 ммоль) и 4М HCl в 1,4-диоксане (8,0 мл, 32 ммоль) перемешивалась при комнатной температуре в течение 30 минут. Реакция была сконцентрирована до сухости, повторно растворена в DMF (25 мл) и к данному раствору был добавлен N,N-диизопропилэтиламин (1,64 г, 12,7 ммоль) и пентафторофенил-3-хлоро-4-[(1-метилэтил)окси]бензоат (0,963 г, 2,54 ммоль). Смесь перемешивалась в течение 3,0 часов при комнатной температуре, разбавлена водой и экстрагирована в этилацетат. Экстракты были промыты водой и насыщенным хлоридом натрия, высушены (Na2SO4) и сконцентрированы в условиях вакуума. Остаток был очищен флеш-хроматографией на силикагеле (2% MeOH : EtOAc) для получения титульного соединения (0,7 г, 53%) в качестве бледно-желтого порошка. MS (ES+) m/e 522 [М+Н]+.
Схема Е
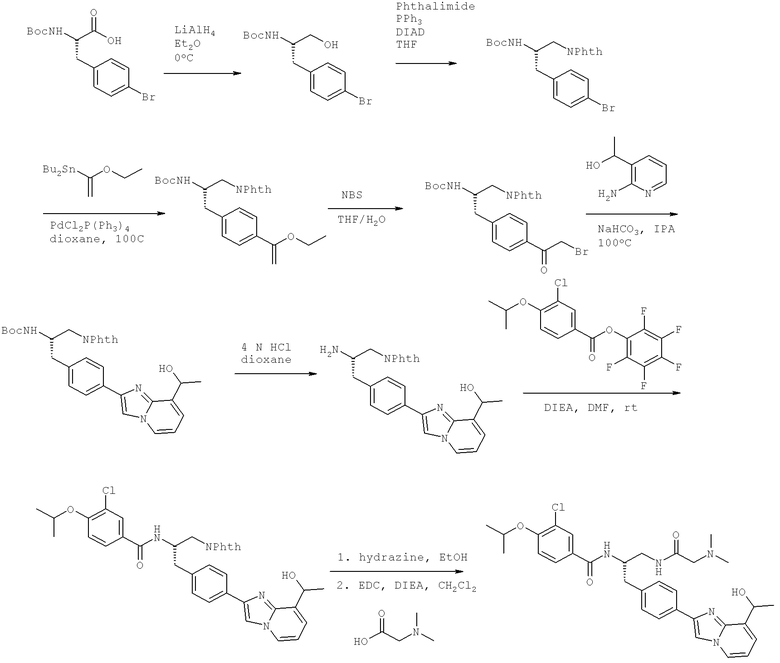
1,1-Диметилэтил[(1S)-2-(4-бромофенил)-1-(гидроксиметил)этил]карбамат:
К раствору 4-бромо-N-{[(1,1-диметилэтил)окси]карбонил}-L-фенилаланина (72,6 ммоль) в безводном диэтил эфире (550 мл) при 0°С медленно добавлялся алюминиевый гидрид лития, 95% (108,9 ммоль). Полученный в результате раствор перемешивался дополнительно в течение 2 часов при 0°С. Затем реакция была осторожно погашена насыщенным водным раствором бикарбоната натрия (73 мл), который перемешивался при комнатной температуре в течение получаса. Алюминиевые соли лития выпадали из раствора и удалялись фильтрацией. Фильтрат был сконцентрирован и в течение 24 часов накачивался вакуум для получения титульного продукта в качестве белого твердого вещества (97%). ESMS [М+Н]+: 331,2.
1,1-Диметилэтил[(1S)-2-(4-бромофенил)-1-[(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)метил]этил}карбамат:
К раствору 1,1-диметилэтил[(1S)-2-(4-бромофенил)-1-(гидроксиметил)этил] карбамата (70,6 ммоль), трифейфосфина (84,7 ммоль) и фталимида (84,7 ммоль) в безводном тетрагидрофуране (550 мл) при 0°С по каплям добавлялся диизопропил азодикарбоксилат (84,7 ммоль) в течение 10 минут. Реакции продолжалась при перемешивании, давая нагреться до комнатной температуры в течение 5 часов. Реакция затем была сконцентрирована в условиях вакуума и продукт был титрирован из раствора, используя ацетат (500 мл). Осадок был отфильтрован, промыт этилацетатом (3×100 мл) и высушен для получения титульного продукта в качестве белого твердого вещества (57%). ESMS [М+H]+: 460,4.
1,1-Диметилэтил {(1S)-2-[4-(бромоацетил)фенил]-1-[(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)метил]этил}карбамат:
Раствор 1,1-диметилэтил {(1S)-2-[4-(бромофенил]-1-[(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)метил]этил}карбамата (21,7 ммоль), 1-этоксивинилтри-п-бутилина (43,5 ммоль) и транс-дихлоробис(трифенилфосфин)палладия (II) (5 мол.%) перемешивались в безводном диоксане (300 мл) при 100°С в течение 3 часов. Затем реакция была сконцентирована в условиях вакуума и повторно растворена в растворе тетрагидрофурана и воды (3:1, 400 мл) и обработана N-бромосукцинимидом (108,8 ммоль) и перемешивалась при комнатной температуре в течение получаса. Реакционный раствор затем был сконцентрирован до сухости и повторно растворен в этилацетате (150 мл), образовался осадок при добавлении гексанов (350 мл). Осадок был отфильтрован и высушен для получения титульного продукта в качестве желтого твердого вещества (71%). ESMS [M+H]+: 502,4.
1,1-Диметилэтил[(1S)-2-(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)-1-({4-[8-1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]карбамат:
Смесь 1,1-диметилэтил{(1S)-2-[4-(бромоацетил)фенил]-1-[(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)метил]этил}карбамата (1,90 г, 3,79 ммоль), 1-(2-амино-3-пиридинил) этанола (0,523 г, 3,79 ммоль) и твердого бикарбоната натрия (0,398 г, 4,72 ммоль) в изопропаноле (24 мл) пропускалась в противотоке в течение 3,0 часов и была сконцентрирована в условиях вакуума. Остаток был растворен в этилацетате, промыт водой и насыщенным хлоридом натрия, высушен (Na2SO4) и сконцентрирован для получения титульного соединения (1,79 г, 87%) в качестве слегка розоватого твердого вещества. MS (ES+) m/e 541 [М+Н]+.
3-Хлоро-N-[(1S)-2-(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид:
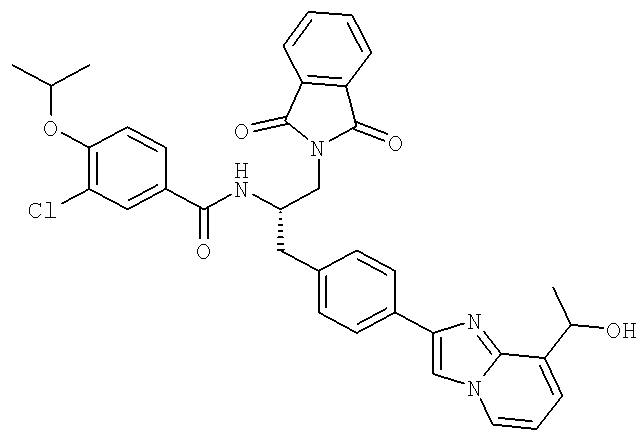
Смесь 1,1-диметилэтил[(1S)-2-(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]карбамата (1,79 г, 3,31 ммоль) и 4М HCl в 1,4-диоксане (20 мл, 80 ммоль) перемешивалась при комнатной температуре в течение 45 минут. Реакция была сконцентрирована до сухости, повторно растворена в DMF (30 мл) и к данному раствору был добавлен N,N-диизопропилэтиламин (2,14 г, 16,55 ммоль) и пентафторофенил 3-хлоро-4-[(1-метилэтил)окси]бензоат (1,36 г, 3,31 ммоль). Смесь перемешивалась в течение ночи при комнатной температуре, разбавлена водой и экстрагирована в этилацетат. Экстракты были промыты водой, высушены (Na2SO4) и сконцентрированы в условиях вакуума для получения титульного соединения (2,10 г, 100%) в качестве золотистого твердого вещества. MS (ES+) m/e 637 [М+Н]+.
N-[(1S)-2-Амино-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-3-хлоро-4-[(1-метилэтил)окси]бензамид:
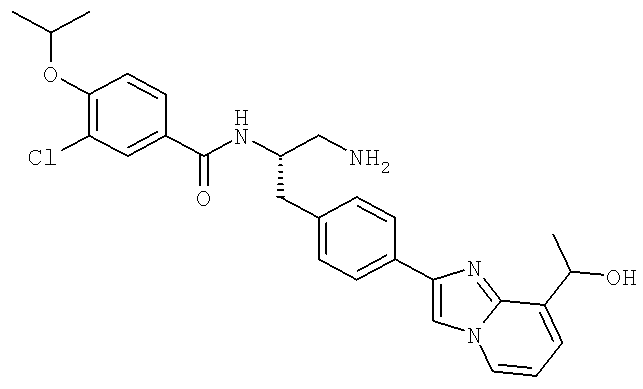
Смесь 3-хлоро-N-(1S)-2-(1,3-диоксо-1,3-дигидро-2H-изоиндол-2-ил)-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамида (2,10 г, 3,30 ммоль) и гидразин моногидрата (0,83 г, 16,5 ммоль) в этаноле (30 мл) нагревалась при 57°С в течение ночи. Реакция была охлаждена, разбавлена этанолом, отфильтрована и сконцентрирована для получения титульного соединения (1,67 г, 100%) в качестве бледно желтого порошка.
MS (ES+) m/e 507 [М+Н]+.
3-Хлоро-N-[(1S)-2-[(N,N-диметилглицил)амино]-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид:
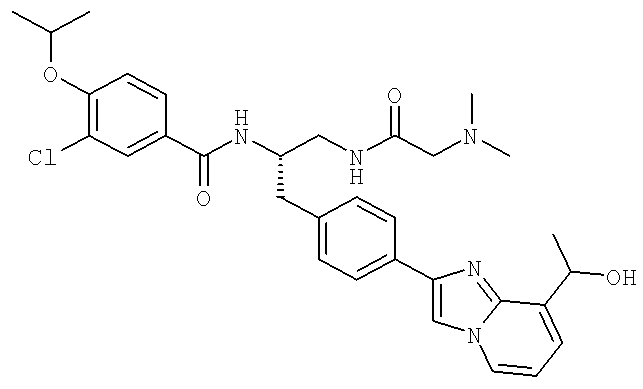
Смесь N-[(1S)-2-амино-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-3-хлоро-4-[(1-метилэтил)окси]бензамида (0,912 г, 1,80 ммоль), EDCN (0,69 г, 3,6 ммоль), N,N-диизопропилэтиламина (0,466 г, 3,6 ммоль) и N,N-диметилглицина (0,372 г, 3,6 ммоль) в метиленхлориде (17 мл) перемешивалась в течение ночи при комнатной температуре. Реакция была разбавлена водой, промыта соляным раствором, высушена (Na2SO4) и сконцентрирована. Остаток был очищен флеш-хроматографией на силикагеле (8%-10% MeOH : CH2Cl2) для получения титульного соединения (0,515 г, 48%) в качестве бледно-желтого твердого вещества. MS (ES+) m/e 592 [М+Н]+.
1,1-Диметил{(1S)-2-[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]-1-[(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)метил]этил}карбамат:
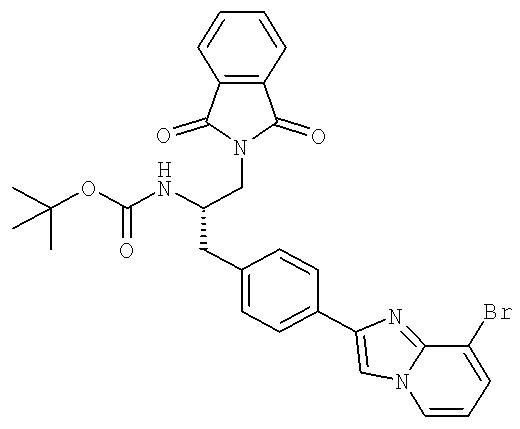
Раствор 1,1-диметил{(1S)-2-[4-(бромоацетил)фенил]-1-[(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)метил]этил}карбамата (6,9 ммоль), 3-бромо-2-пиридинамина (8,4 ммоль) и бикарбоната натрия (10,4 ммоль) в изопропаноле (70 мл) перемешивался при температуре 80°С в течение 18 часов. Реакция затем была охлаждена до комнатной температуры и образовавшийся осадок был отфильтрован, промыт холодными гексанами (3×100 мл) и высушен для получения титульного соединения в качестве бледно-серого твердого вещества (72%). ESMS [М+Н+]: 576,2.
1,1-Диметилэтил((1S)-2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)карбамат:
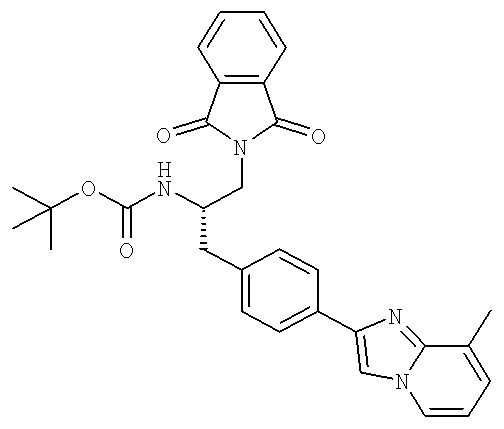
Следуя описанной выше процедуре с 3-метил-2-пиридинамином вместо 3-бромо-2-пиридинамина, был получен названный продукт в качестве слегка розоватого твердого тела. ESMS [М+Н]+: 511,0.
N-{(1S)-2-[4-(8-Бромоимидазо[1,2-а]пиридин-2-ил)фенил]-1-[(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)метил]этил}-3-хлоро-4-[(1-метилэтил)окси]бензамид:
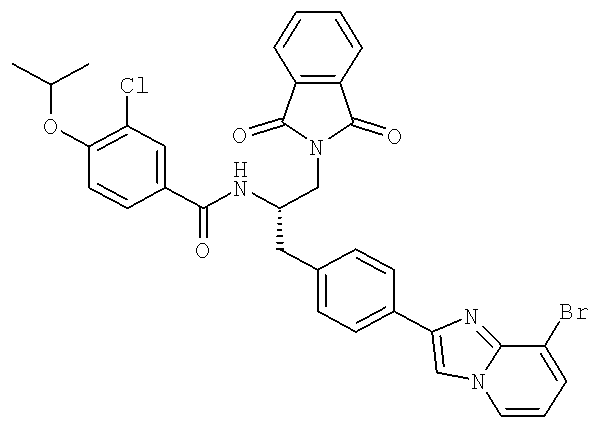
Раствор 1,1-диметилэтил{(1S)-2-[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]-1-[(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)метил]этил}карбамата (3,5 ммоль) и хлорида водорода в 1,4-диоксане (20 мл, 4,0М) подвергался перемешиванию в течение 1 часа при комнатной температуре. Реакция была сконцентрирована до сухости и повторно растворена в N,N-диметилформамиде (35 мл). К раствору был добавлен диизопропилэтиламин (10,5 ммоль) и пентафторофенил 3-хлоро-4-[(1-метилэтил) окси]бензоат (3,8 ммоль), которые перемешивались при комнатной температуре в течение получаса. Реакция была растворена в этилацетате (80 мл) и промыта водой (3×50 мл) и соляным раствором (1×50 мл). К отделенному органическому слою были добавлены гексаны (150 мл), после чего был образован осадок. Он был отфильтрован и высушен для получения титульного соединения в качестве беловатого твердого вещества (65%). ESMS [М+Н]+: 672,2.
3-Хлоро-N-((1S)-2-(1,3-диоксо-1,3 -дигидро-2Н-изоиндол-2-ил)-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид:
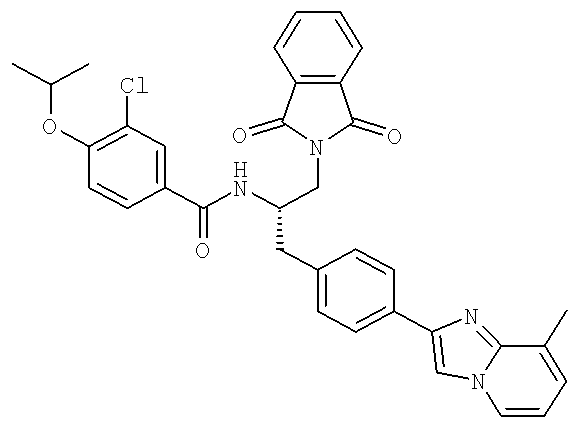
Следуя описанной выше процедуре с 1,1-диметилэтил((1S)-2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил} этил)карбаматом, был получен титульный продукт в качестве беловатого твердого вещества. ESMS [М+Н]+: 608,2.
N-((1S)-2-Амино-1-{[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[(1-метилэтил)окси]бензамид:
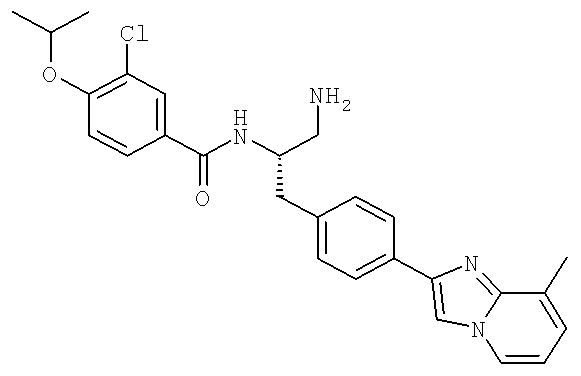
К раствору N-{(1S)-2-[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]-1-[(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)метил]этил}-3-хлоро-4-[(1-метилэтил)окси] бензамида (1,5 ммоль) в этаноле (10 мл) был добавлен гидразин моногидрат (7,6 ммоль). Реакция перемешивалась в течение 18 часов при 50°С, после чего был образован белый осадок и он был отфильтрован. Фильтрат был сконцентрирован в условиях вакуума. Полученное в результате слегка желтоватое твердое вещество непосредственно использовалось в следующей реакции без дальнейшей очистки. ESMS [М+Н]+: 533,2.
N-((1S)-2-Амино-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[(1-метилэтил)окси]бензамид:
Следуя процедуре, описанной выше с 3-хлоро-N-((1S)-2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамидом, был получен титульный продукт в качестве беловатого твердого вещества. ESMS [М+Н]+: 478,2.
N-((1S)-2-(D-Аланиламино)-1-{[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[(1-метилэтил)окси]бензамид:
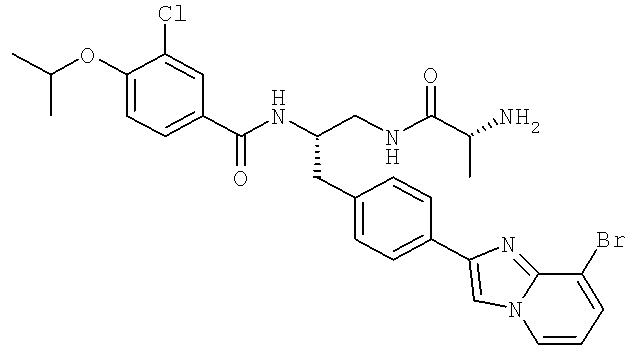
Раствор N-((1S)-2-амино-1-{[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[(1-метилэтил)окси]бензамида (0,28 ммоль), N-{[(1,1-диметилэтил) окси]карбонил}-D-аланина (0,56 ммоль), EDCI (0,56 ммоль) и TEA (1,12 ммоль) перемешивался в метиленхлориде (2 мл) при комнатной температуре в течение 18 часов. Реакция была затем обработана 4М HCl в 1,4-диоксане (2 мл) и перемешивалась при комнатной температуре в течение 1 часа, а затем была сконцентрирована в условиях ваккума, повторно растворена в этилацетате (25 мл) и промыта насыщенным водным раствором бикарбоната натрия (1×10 мл). Органический слой был сконцентрирован в условиях ваккума. Очистка остатка обратнофазовой HPLC Гильсона дала титульный продукт в качестве белого твердого вещества (25%). ESMS [М+Н]+: 613,2.
3-Хлоро-N-((1S)-2-[(2-метилаланил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид:
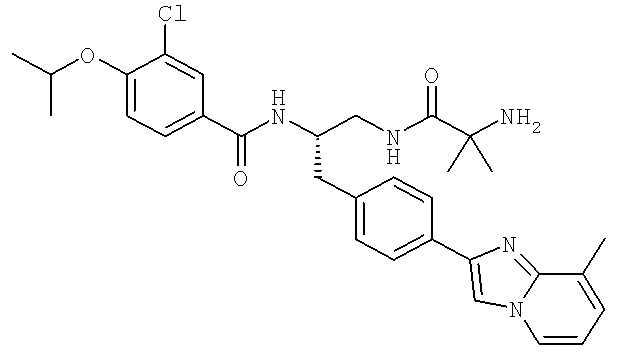
Следуя описанной выше процедуре с N-((1S)-2-амино-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[(1-метилэтил)окси]бензамидом и N-{[(1,1-диметилэтил)окси]карбонил}-2-метилаланином был получени титульный продукт в качестве белого твердого вещества. ESMS [М+Н]+: 563,2.
3-Хлоро-N-((1S)-2-[N,N-диметилглицил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид:
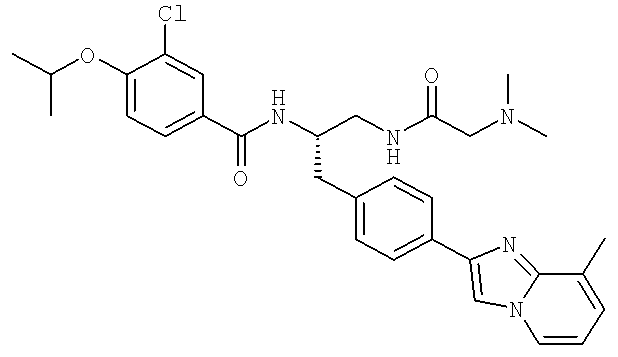
Следуя описанной выше процедуре с N-((S)-2-амино-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[(1-метилэтил)окси] бензамидом и N,N-диметилглицином был получен названный продукт в качестве белого твердого вещества. ESMS [М+Н]+: 563,2.
Схема F
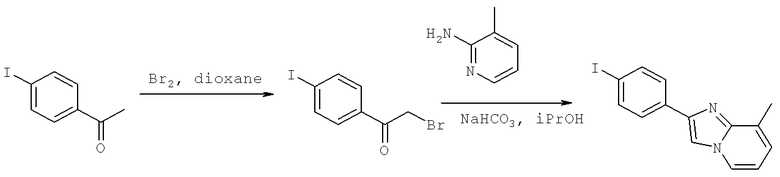
2-Бромо-1-(4-иодофенил)этанон:
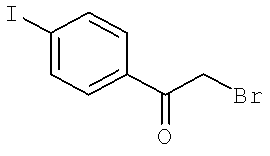
Раствор 1-(4-иодофенил)этанона (55,9 ммоль) в диоксане (160 мл) был охлажден до 10°С. Бром (1,1 эквивалента, 61,6 ммоль) добавлялся по каплям к реакционной смеси. После 10 мин охлаждающая баня была удалена и реакционная смесь перемешивалась при комнатной температуре. После 1,5 часов реакционная смесь была сконцентрирована в условиях вакуума, налита в воду (100 мл) и экстрагирована с помощью (3×100 мл) этилацетата. Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы в условиях ваккума в качестве желтовато-коричневого твердого вещества (18,2 г), которое напрямую использовалось в следующем этапе.
2-(4-Иодофенил)-8-метилимидазо[1,2-а]пиридин:
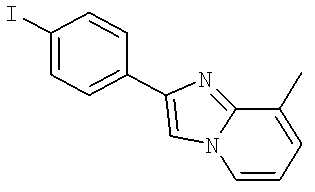
Смесь неочищенного 2-бромо-1-(4-иодофенил)этанона (18,2 г), 2-амино-3-пиколина (1,1 эквивалента, 61,6 ммоль) и бикарбоната натрия (1,3 эквивалента, 72,8 ммоль) в изопропаноле (160 мл) нагревалась при 80°С в течение 16 часов. После концентрации реакционной смеси в условиях вакуума была добавлена вода (100 мл) и полученная в результате желтовато-коричневая кашица была отфильтрована, промыта водой (2×50 мл). Коричневое твердое вещество было кристаллизовано из горячего изопропанола и далее высушено в условия вакуума для получения титульного продукта в качестве коричневого твердого вещества. ESMS [М+Н]+: 335,0.
Схема G
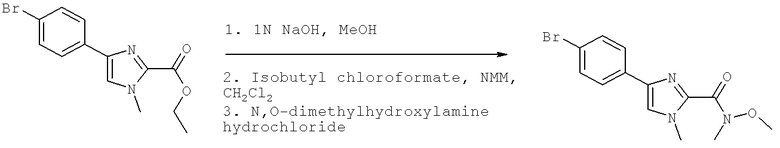
4-(4-Бромофенил)-N,1-диметил-N-(метилокси)-1Н-имидазол-2-карбоксамид:
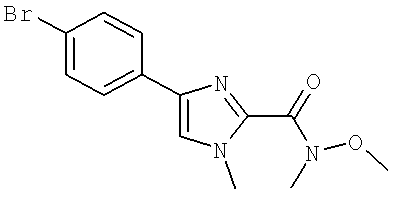
К раствору этил 4-(4-бромофенил)-1-метил-1H-имидазол-2-кабоксилата (1,66 г, 5,37 ммоль) в МеОН (38 мл) был добавлен раствор 1N NaOH (19 мл). Реакция стала дымчато-белой и подверглась перемешиванию при комнатной температуре в течение 30 минут. Реакционная смесь была сконцентрирована в условиях вакуума и прокачана под высоким вакуумом в течени ночи для получения соли натрия 4-(4-бромофенил)-1-метил-1H-имидазол-2-карбоновой кислоты в качестве белого твердого вещества. Натриевая соль 4-(4-бромофенил)-1-метил-1H-имидазол-2-карбоновой кислоты была растворена в безводном CH2Cl2 (40 мл) в атмосфере азота при -15°С (лед/метаноловая баня) и был добавлен N-метилморфолин (1,1 эквивалента, 5,91 ммоль), за которым последовал изобутил хлороформат (1,1 эквивалент, 5,91 ммоль). Реакционная смесь перемешивалась при -15°С в течение 15 минут и затем был добавлен N,O-диметилгидроксиламин гидрохлорид (1,0 эквивалент, 5,37 ммоль). Реакция была оставлена нагреваться до комнатной температуры и перемешивалась в течение 17 часов. Реакция была погашена H2O (10 мл). Продукт был экстрагирован с использованием EtOAc (3×30 мл) и комбинированные органические слои были промыты соляным раствором (20 мл), высушены над MgSO4 и сконцентрированы в условиях вакуума. Очистка хроматографией с силикагелем (Аналоджикс IF280, 20-100% EtOAc/гексаны) дала титульное соединение в качестве желтовато-коричневого твердого вещества (32%). ESMS [M+H]+: 324,2.
Схема H
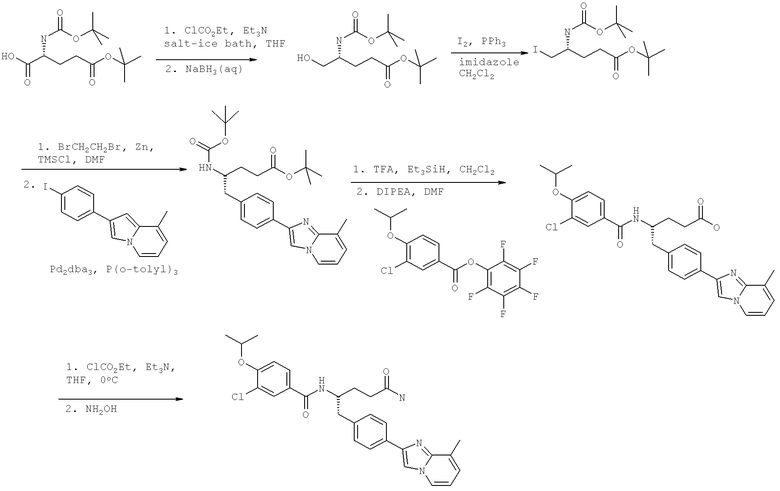
1,1-Диметилэтил(4R)-4-({[(1,1-диметилэтил)окси]карбонил}амино)-5-гидрокси пентаноат:
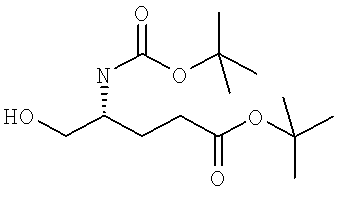
Триэтиламин (11,49 мл, 82,4 ммоль) и этил хлороформат (8,27 мл, 86,5 ммоль) добавлялись последовательно с помощью шприца к 5-терт-бутиловому эфиру N-t-BOC-D-глютаминовой кислоты (25 г, 82,4 ммоль) в THF (588 мл) при <0°С (ледово-соляная баня). После перемешивания в холодной бане в течение 40 минут твердые вещества были отфильтрованы и промыты THF (150 мл). Фильтрат был перемещен в 250 мл дополнительную воронку и добавлен к раствору борогидрида натрия (8,42 г, 222,5 ммоль) в Н2О (114 мл) при <0°С в течение 1 часа. Реакционная смесь поддерживалась при <0°С в течение 1,5 часов и затем перемешивалась в течение 16 часов (0°С до комнатной температуры). После того, как основные растворители были удалены ротационным выпариванием, концентрат был погашен ледяной водой (50 мл) и 1N HCl (50 мл). После экстрагирования с помощью EtOAc (4×100 мл) экстракты были промыты 100 мл 0,5 М лимонной кислоты, насыщены NaHCO3, H2O и соляным раствором и сконцентрированы в условиях вакуума для получения титульного соединения, которое напрямую использовалось в следующем этапе. ESMS [M+H]+=290,4, [2M+H]+=579,4 (литература по подготовке: Журнал медицинской химии, 1999, 42(1), 95-108 по вопросу другого изомера).
1,1-Диметилэтил(4R)-4-({[(1,1-диметил)окси]карбонил}амино)-5-иодопентаноат:
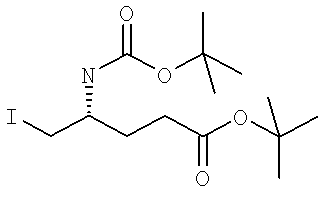
К раствору неочищенного 1,1-диметилэтил(4R)-4-({[(1,1-диметилэтил)окси] карбонил}амино)-5-гидроксипентаноата (23,8 г, 82,4 ммоль), трифенилфосфина (32,42 г, 123,6 ммоль) и имидазола (8,41 г, 123,6 ммоль) в 515 мл безводного CH2Cl2 в атмосфере N2 при 0°С по порциям добавлялся иод в течение 15 минут. Ледяная баня была удалена и реакция была оставлена нагреваться до комнатной температуры и перемешивалась в течение 30 минут. Реакция была погашена 200 мл H2O. Водный слой был экстрагирован диэтил эфиром (2×150 мл). Комбинированные органические слои были промыты насыщенным водным раствором Na2SO3 (2×15 мл) и соляным раствором (25 мл), высушены над MgSO4 и сконцентрированы в вакууме. Очистка хроматографией с силикагелем (Аналоджикс IF280, 5%-50% EtOAc/гексаны) дала титульное соединение в качестве белого твердого вещества (25,34 г, 77%).
ESMS [М+Н]+=400,4.
1,1-Диметилэтил(4R)-4-({[(1,1-диметилэтил)окси]карбонил}амино-5-[4-(8-метил имидазо[1,2-а]пиридин-2-ил)фенил]пентаноат:
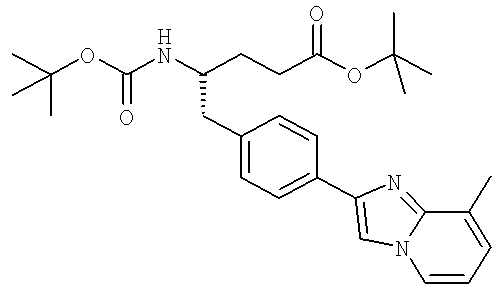
Колба, содержащая цинковую пыль (6,0 эквивалента, 325 меш, Strem) была нагрета тепловой пушкой при эвакуации и наполнении азотом (3 раза). В атмосфере азота дегазированный DMF (14 мл) был добавлен через шприц, за чем последовал 1,2-бромоэтан (0,35 эквивалента). Серая реакционная смесь перемешивалась в масляной ванне при 100°С в течение 15 мнут и затем была охлаждена до комнатной температуры. Хлоротриметилсилан (0,25 эквивалента) был добавлен к смеси через шприц и реакция перемешивалась при комнатной температуре в течение 30 минут. Раствор 1,1-диметилэтил(4R)-4-({[1(-диметилэтил)окси]карбонил}амино-5-иодопентаноата (2,0 г, 1,2 эквивалента) в дегазированном DMF (14 мл) был добавлен к реакционной смесь через канюлю. Колба, содержащая раствор, была промыта дегазированном DMF (4 мл) и он был пропущен через канюлю в реакционную смесь. Реакция перемешивалась при комнатной температуре в течение 1 часа. Затем через верх все вместе были добавлены трис(дибензилидинацетон)дипалладий (0) (2,5 мол.%), три-о-толилфосфин (10 мол.%) и 2-(4-иодофенил)-8-метилимидазо[1,2-а]пиридин (1,4 г, 1,0 эквивалент). Реакционная смесь перемешивалась при комнатной температуре в течение 17 часов. Реакция была разбавлена EtOAc (40 мл) и отфильтрована через Целит (Celite)®. Фильтрат был промыт H2O (20 мл) и соляным раствором (20 мл), органический слой был высушен над MgSO4 и сконцентрирован в условиях вакуума. Очистка хроматографией с силикагелем (Аналоджикс IF 280, 5-90% EtOAc/гексаны) дало титульное соединение в качестве белого твердого вещества (90%). ESMS [М+Н]+=480,4.
1,1-Диметилэтил(4R)-4-({[(1,1-диметилэтил)окси]карбонил}амино)-5-[4-(1-метил-2-{[метил(метилокси)амино]карбонил}-1Н-имидазол-4-ил)фенил]пентаноат:
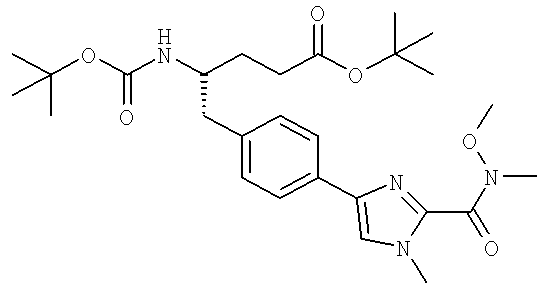
Следуя описанной выше процедуре с использованием 4-(4-бромофенил)-N,1-диметил-N-(метилокси)-1Н-имидазол-2-карбоксамида, был получен титульный продукт в качестве твердого вещества (82%). ESMS [M+H]+=517,2.
(4R)-4-[({3-Хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-5-[4-(8-метил имидазо[1,2-а]пиридин-2-ил)фенил]пентановая кислота:
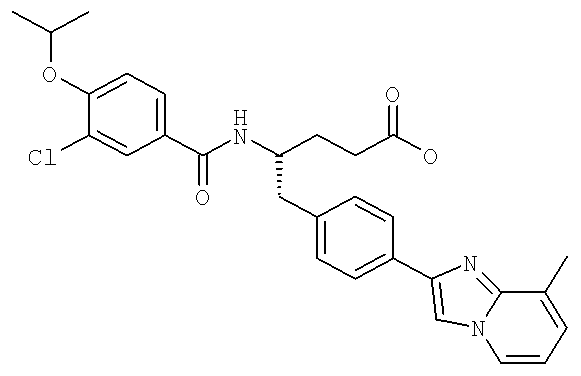
К раствору 1,1-диметилэтил(4R)-4-({[(1,1-диметилэтил)окси]карбонил}амино)-5-[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]пентаноата (1,35 г, 2,82 ммоль) в CH2Cl2 (14 мл) была добавлена трифтороуксусная кислота (10 мл), после чего последовал триэтилсилан (2,5 эквивалента, 7,04 ммоль). Реакция перемешивалась в течение 45 минут при комнатной температуре и затем была сконцентрирована в условиях вакуума. DMF (35 мл) был добавлен к остатку, за чем последовал диизопропиламин (14,7 мл, 84,51 ммоль) в атмосфере азота. Реакция перемешивалась в течение 5 минут, и был добавлен пентафторофенил 3-хлоро-4-[(1-метилэтил)окси]бензоат (1,1 эквивалента, 3,10 ммоль). Реакция перемешивалась в течение 45 минут и затем была сконцентрирована в условиях вакуума. К остатку был добавлен этилацетат (50 мл) и он был промыт H2O (30 мл). Водный слой был экстрагирован EtOAc (20 мл), комбинированные органические слои были высушены над MgSO4 и сконцентрированы в условиях вакуума. Очистка хроматографией с силикагелем (Аналоджикс IF280, 25-100% EtOAc/гексаны) дала титульное соединение в качестве белого пенистого твердого вещества. ESMS [M+H]+=520,2.
(4R)-4-[({3-Хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-5-[4-(1-метил-2-{[метил(метилокси)амино]карбонил}-1Н-имидазол-4-ил)фенил]пентановая кислота:
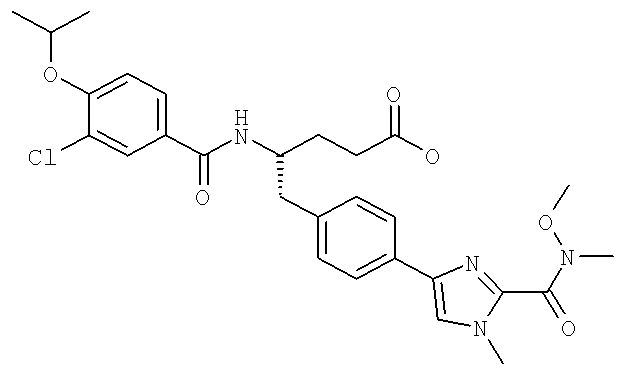
Следуя описанной выше процедуре с 1,1-диметилэтил(4R)-4-({[(1,1-диметилэтил)окси]карбонил}амино)-5-[4-(1-метил-2-{[метил(метилокси)амино]карбонил}-1Н-имидазол-4-мл)фенил]пентаноатом и вышеупомянутой очистке, было получено титульное соединение. ESMS [М+H]+=557,5.
(4R)-5-[4-(2-Ацетил-1-метил-1Н-имидазол-4-ил)фенил]-4-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]пентановая кислота:
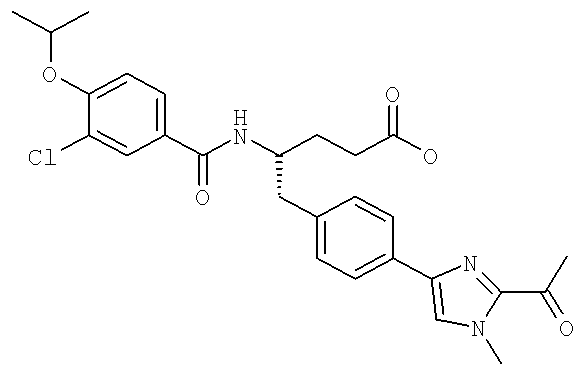
К раствору неочищенной (4R)-4-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-5-[4-(1-метил-2-{[метил(метилокси)амино]карбонил}-1Н-имидазол-4-ил)фенил]пентановой кислоты (3,18 ммоль) в безводном THF (16 мл) в атмосфере азота при 0°С был добавлен бромид метилмагния (10,6 мл, 10 эквивалентов, 3,0М в эфире) по каплям с помощью шприца. Реакция перемешивалась в течение 30 минут при 0°С и затем осторожно была погашена насыщенным водным раствором NH4Cl, за чем последовал раствор 1N HCl (60 мл) таким образом, чтобы рН водного слоя был ~5,5. Продукт был экстрагирован EtOAc (4×40 мл), комбинированные органические слои были высушены над MgSO4 и сконцентрированы в условиях вакуума для получения титульного соединения, которое напрямую использовалось в следующей реакции. ESMS [М+Н]+=512,4.
N-((1R)-4-Амино-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-4-оксобутил)-3-хлоро-4-[(1-метилэтил)окси]бензамид:
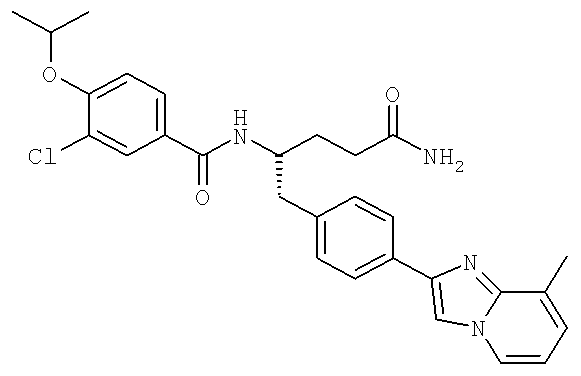
К раствору (4R)-4-[({3-хлоро-4-[(1-метилэтил)окси] фенил} карбонил)амино]-5-[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]пентановой кислоты (900 мг, 1,73 ммоль) в безводном THF (12,4 мл) при 0°С в атмосфере азота был добавлен триэтиламин (242 мкл, 1,73 ммоль), за которым последовал хлороформат (174 мкл, 1,82 ммоль). Реакция перемешивалась в течение 40 минут при 0°С и затем твердые вещества были отфильтрованы и промыты 5 мл THF. Фильтрат был добавлен к колбе, содержащей NH4OH (5 мл) при комнатной температуре, и реакционная смесь перемешивалась в течение 1 часа. Продукт был экстрагирован из реакционной смеси с помощью EtOAc (50 мл). Водный слой был экстрагирован с помощью EtOAc (20 мл) и затем окислен раствором 1N HCl (30 мл) и повторно экстрагирован с помощью EtOAc (10 мл). Комбинированные органические слои были высушены над MgSO4 и сконцентрированы в условиях вакуума для получения белого твердого вещества. Очистка рекристаллизацией из горячего изопропанола дала титульное соединение в качестве белого твердого вещества (90%). ESMS [М+Н]+=519,4.
N-((1R)-1-{[4-(2-Ацетил-1-метил-1Н-имидазол-4-ил)фенил]метил}-4-амино-4-оксобутил)-3-хлоро-4-[(1-метилэтил)окси]бензамид:
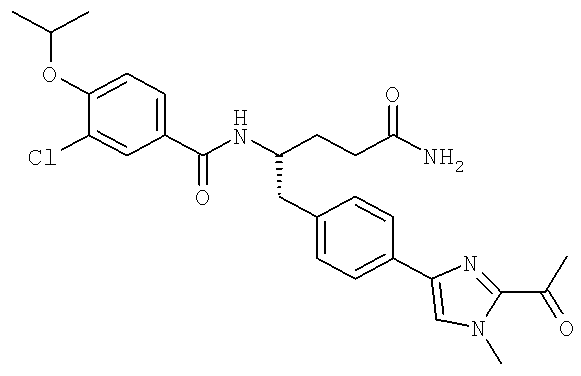
Следуя процедуре, описанной выше, с (4R)-5-[4-(2-ацетил-1-метил-1Н-имидазол-4-ил)фенил]-4-[({3-хлоро-4-[(1-метилэтил)окси]фенил} карбонил)амино]пентановой кислотой и очистке с помощью обратнофазовой HPLC Гильсена было получено титульное соединение в качестве белого твердого вещества. ESMS [M+H]+=511,2.
Схема I
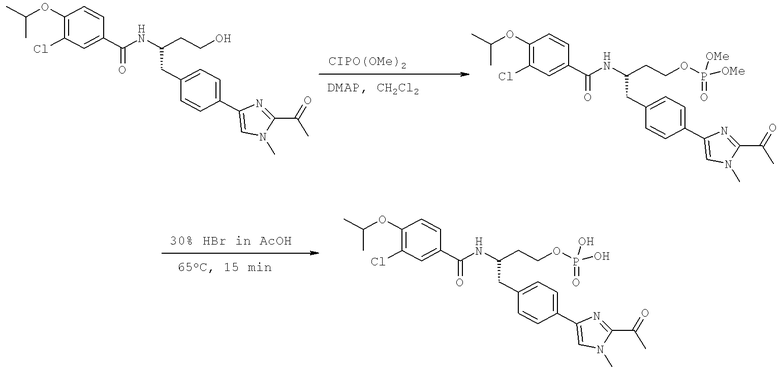
(3S)-4-[4-Ацетил-1-метил-1Н-имидазол-4-ил)фенил]-3-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]бутил диметил фосфат:
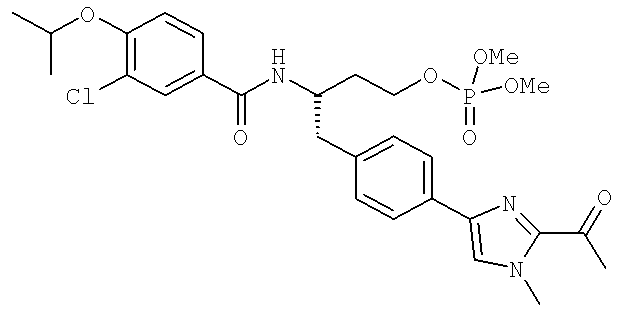
К раствору N-((1S)-1-{[4-(2-ацетил-1-метил-1Н-имидазол-4-ил)фенил]метил}-3-гидроксипропил)-3-хлоро-4-[(1-метилэтил)окси]бензамида (500 мг, 1,04 ммоль) в сухом CH2Cl2 (10 мл) в атмосфере N2 был добавлен диметил хлорофосфат (748 мг, 5,18 ммоль), за чем последовал DMAP (660 мг, 5,41 ммоль) при комнатной температуре. После 30 минут TLC (95:5 EtOAc/MeOH) продемонстрировала ~50% конверсию, так что была добавлена дополнительная порция диметилфосфата (748 мг, 5,18 ммоль) и DMAP (660 мг, 5,41 ммоль). После дополнительных 30 минут реакция была погашена насыщенным водным NH4Cl и разбавлена CH2Cl2, а комбинированные органические слои были высушены (Na2SO4), отфильтрованы и сконцентрированы при пониженном давлении. Остаток был очищен хроматографией с силикагелем (100% EtOAc; изократический на Аналоджикс) для получения 475 мг (77%) титульного соединения в качестве бледно-желтого масла. LC/MS(ES) m/e 592,4 (М+Н)+. Обратите внимание, что продукт был загрязнен ~1 эквивалентом стартового реагента диметилхлорофосфат/диметилгидрогенфосфата и использовался как таковой.
Первичный кислый фосфат (3S)-4-[4-(2-ацетил-1-метил-1Н-имидазол-4-ил)фенил]-3-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]бутила:
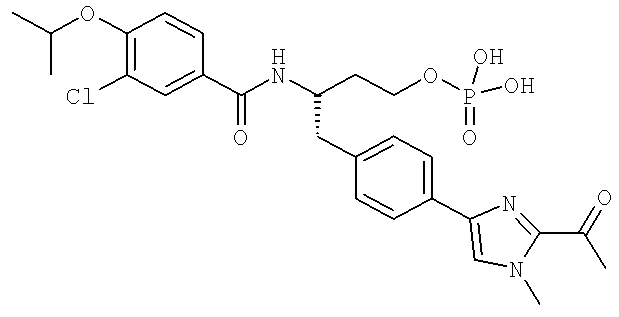
Желтый раствор (3S)-4-[4-(2-ацетил-1-метил-1Н-имидазол-4-ил)фенил]-3-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]бутил диметил фосфата (475 мг, 0,804 ммоль) в 30% HBr в АсОН был помещен в предварительно нагретую (60°С) ванну на 10 минут и затем был немедленно доведен до комнатной температуры. Реакционная смесь была сконцентрирована при пониженном давлении и остаток был растворен в DMSO (6 мл), отфильтрован и очищен обратнофазовой HPLC Гильсена (MeCN/Н2О с 0,1% TFA). MeCN чистых фракций был удален при пониженном давлении и оставшийся водный раствор был заморожен и лиофилизирован в течение ночи для получения 84 мг (19%) титульного соединения в качестве желтого порошка. LC/MS(ES) m/e 564,2 (М+Н)+.
Первичный кислый фосфат (3S)-3-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]бутила:
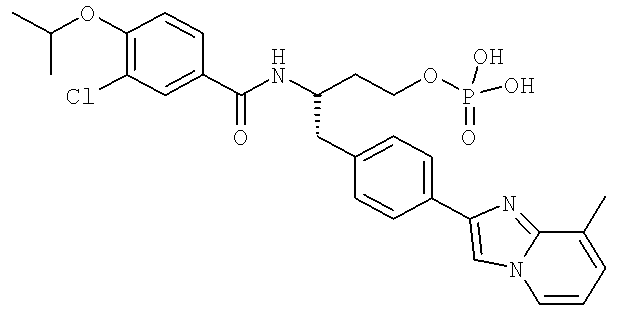
Следуя описанным выше процедурам, за исключением замены 3-xnopo-N-((1S)-3-гидрокси-1-{{4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метил этил)окси]бензамида на N-((1S)-1-{[4-(2-ацетил-1-метил-1Н-имидазол-4-ил)фенил]метил}-3-гидроксипропил)-3-хлоро-4-[(1-метилэтил)окси]бензамид и терт-бутоксида калия на DMAP, было приготовлено титульное соединение в качестве белого порошка (35% выдачи). LC/MS(ES) m/e 563 (М+Н)+.
Схема J
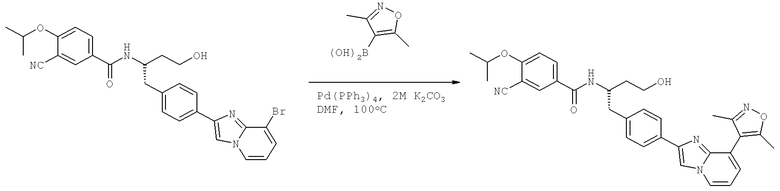
3-Циано-N-[(1S)-1-({4-[8-(3,5-диметил-4-изоксазолил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид:
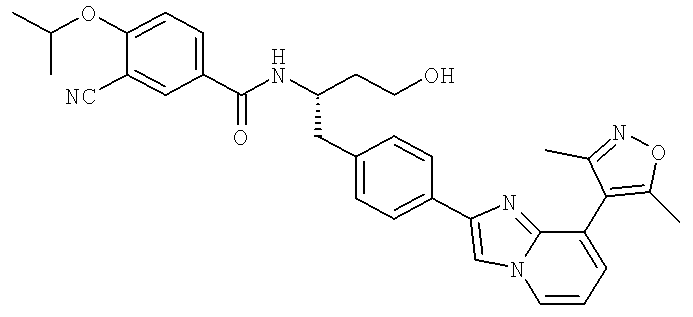
К раствору N-((1S)-1-{[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси]бензамида (200 мг, 0,366 ммоль) в сухом DMF (2 мл) были добавлены 3,5-диметил-изоксазол-4-борная кислота (63 мг, 0,439 ммоль), тетракистрифенилфосфин палладия (0) (21 мг, 0,018 ммоль) и 2,0 М водного K2CO3 (0,46 мл) последовательно при комнатной температуре. Реакционная смесь была очищена аргоном и нагрета до 100°С, была подвергнута перемешиванию в течение 22 часов, охлаждена до комнатной температуры и напрямую очищена обратнофазовой HPLC (MeCN/H2O с 0,1% TFA). Чистые фракции были откорректированы до рН ~8 с помощью насыщенного водного NaHCO3 и экстрагированы 3% MeOH/EtOAc (3×30 мл). Экстракты были высушены (Na2SO4), отфильтрованы и сконцентрированы при пониженном давлении для получения 45 мг (22%) титульного соединения в качестве грязно-беловатого твердого вещества. LC/MS(ES) m/e 564,2 (М+Н)+.
Схема K
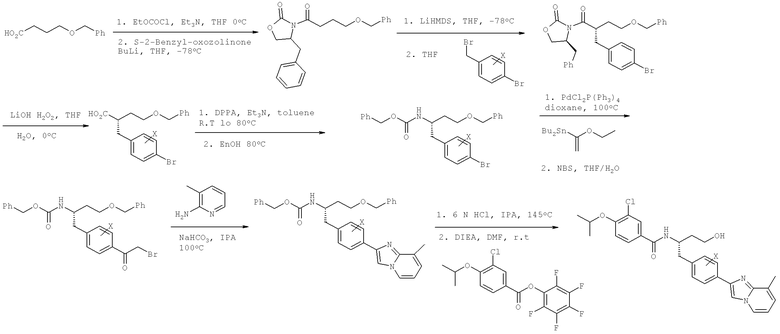
3-Хлоро-N-((1S)-1-{[3-хлоро-4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид:
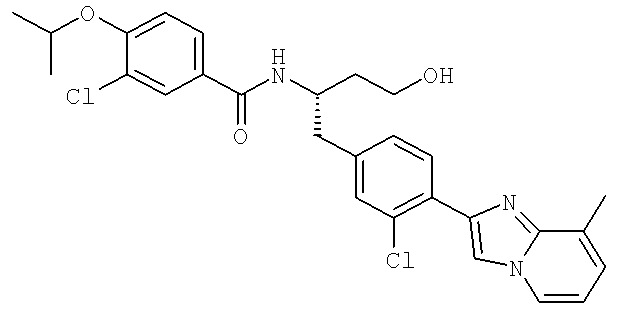
Следуя процедурам, описанным в литературе (Журнал органической химии, 2003, 68, 4215; Журнал органической химии, 2002, 67, 1738; Журнал американского химического общества, 1972, 94, 6203), а также описанным выше процедурам, было приготовлено титульное соединение в качестве белого твердого вещества. LC/MS(ES) m/e 526 (М+Н)+.
Следующие соединения были приготовлены, используя описанные выше процедуры.
Структура
Название
(М+H)+.
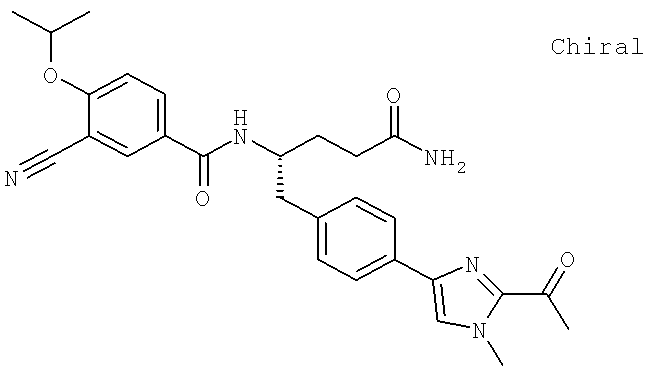
N-((1R)-1-{[4-(2-Ацетил-1-метил-1Н-имидазол-4-ил)фенил]метил}-4-амино-4-оксобутил)-3-циано-4-[(1-метилэтил)окси]бензамид:
502,4
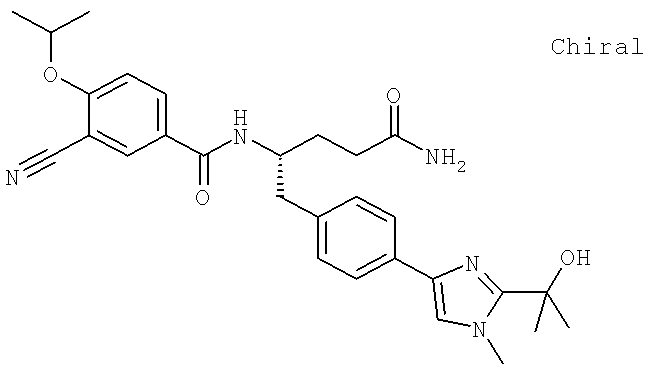
N-[(1R)-4-Амино-1-({4-[2-(1-гидрокси-1-метилэтил)-1-метил-1Н-имидазол-4-ил]фенил}метил)-4-оксобутил]-3-циано-4-[(1-метилэтил)окси]бензамид:
518,4
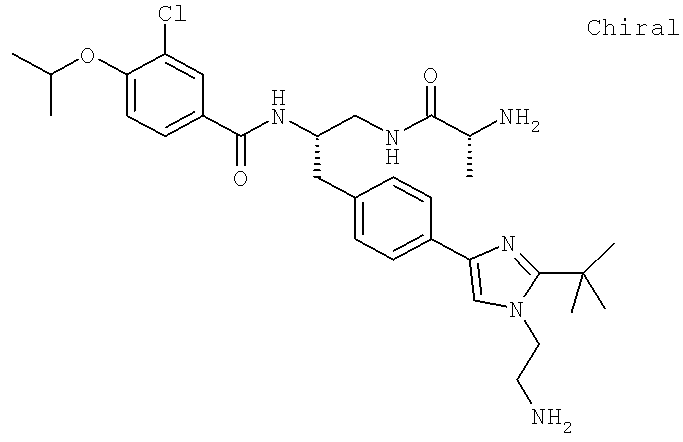
N-[(1S)-2-(D-Аланиламино)-1-({4-[1-(2-аминоэтил)-2-(1,1-диметилэтил)-1Н-имидазол-4-ил]фенил}метил)этил]-3-хлоро-4-[(1-метилэтил)окси]бензамид:
583,6
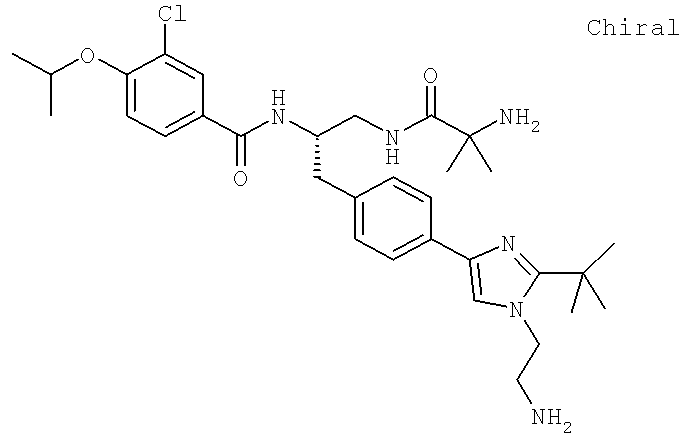
N-((1S)-2-{4-[1-(2-Аминоэтил)-2-(1,1-диметилэтил)-1Н-имидазол-4-ил]фенил}-1-{[(2-метилаланил)амино]метил}этил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
597,6
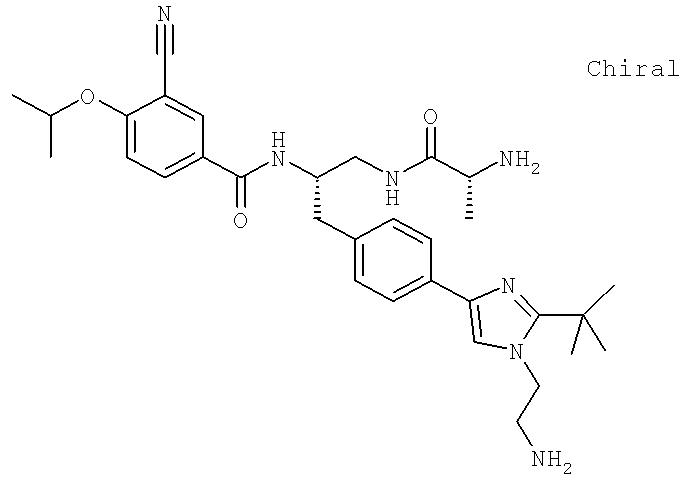
N-[(1S)-2-(D-Аланиламино)-1-({4-[1-(2-аминоэтил)-2-(1,1-диметилэтил)-1Н-имидазол-4-ил]фенил}метил)этил]-3-циано-4-[(1-метилэтил)окси]бензамид:
574,4
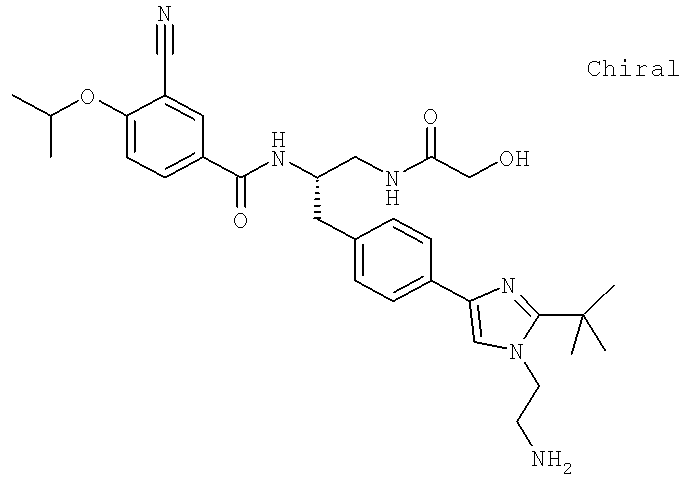
N-((1S)-2-{4-[1-Аминоэтил)-2-(1,1-диметилэтил)-1Н-имидазол-4-ил]фенил}-1-{[гидроксиацетил)амино]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
561,4
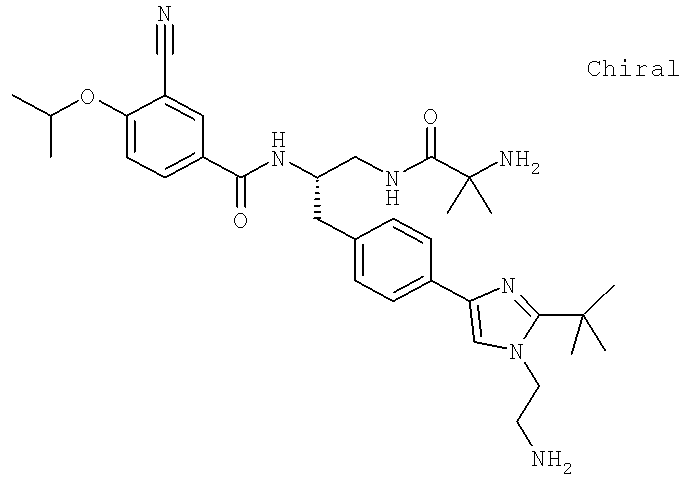
N-((1S)-2-{4-[1-(2-Аминоэтил)-2-(1,1-диметилэтил)-1Н-имидазол-4-ил]фенил}-1-{[(2-метилаланил)амино]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
588,2
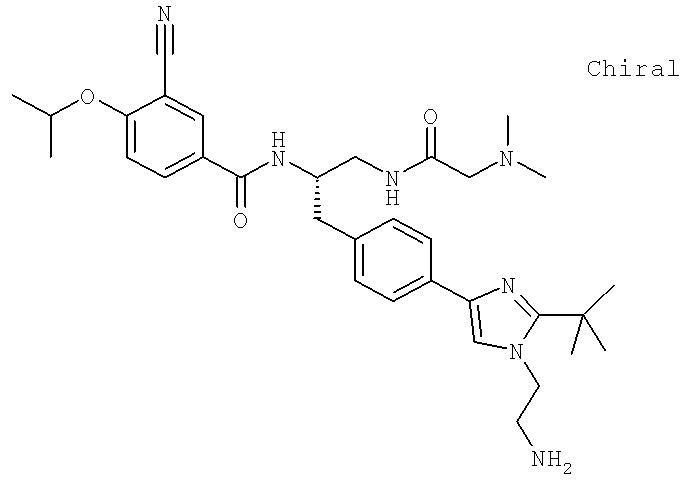
N-((1S)-2-{4-[1-(2-Аминоэтил)-2-(1,1-диметилэтил)-1Н-имидазол-4-ил]фенил}-1-{[N,N-диметилглицил)амино]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
588,4
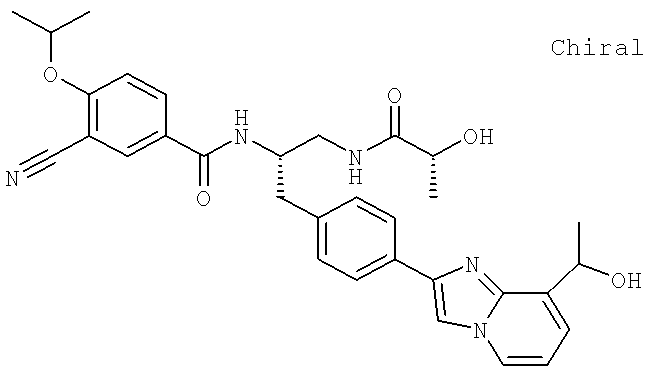
3-Циано-N-[(1S)-2-{4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}-1-({[(2R)-2-гидроксипропаноил]амино}метил)этил]-4-[(1-метилэтил)окси]бензамид
569,4
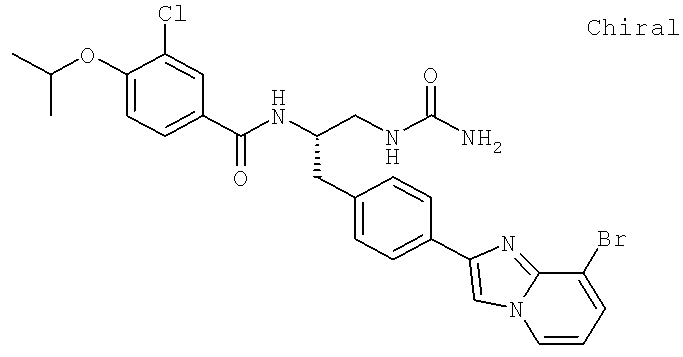
N-((1S)-2-[Аминокарбонил)амино]-1-{[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
584,2
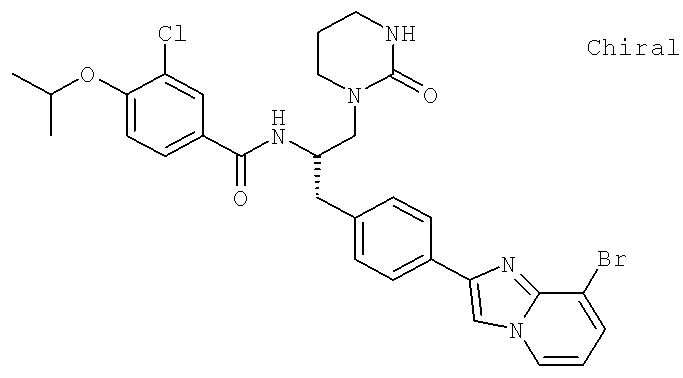
N-{(1S)-2-[4-(8-Бромоимидазо[1,2-а]пиридин-2-ил)фенил]-1-[2-оксотетрагидро-1-(2Н)-пиримидинил)метил]этил}-3-хлоро-4-[(1-метилэтил)окси]бензамид
625,1
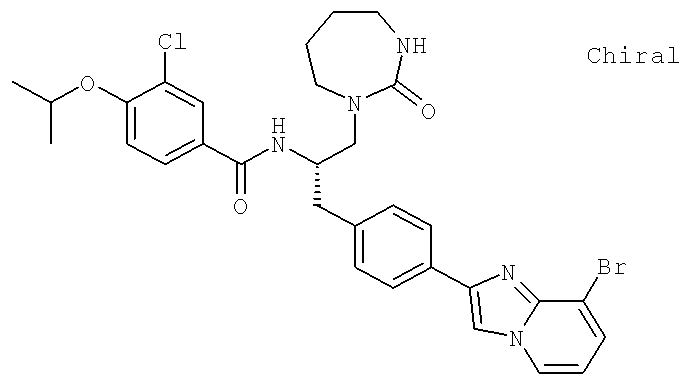
N-{(1S)-2-[4-(8-Бромоимидазо[1,2-а]пиридин-2-ил)фенил]-1-[(2-оксогексагидро-1Н-1,3-диазепин-1-ил)метил]этил}-3-хлоро-4-[(1-метилэтил)окси]бензамид
639,2
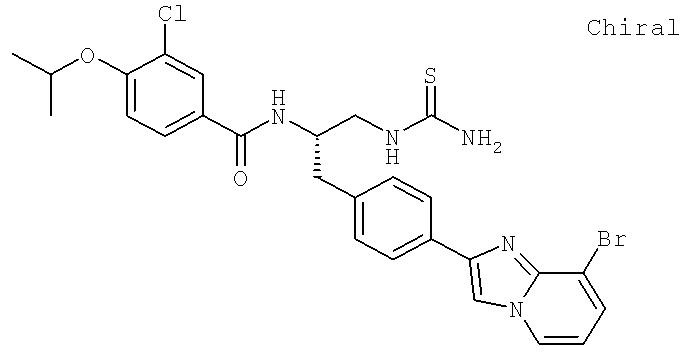
N-((1S)-2-[(Аминокарбонотиоил)амино]-1-{[4-(8-бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
601,2
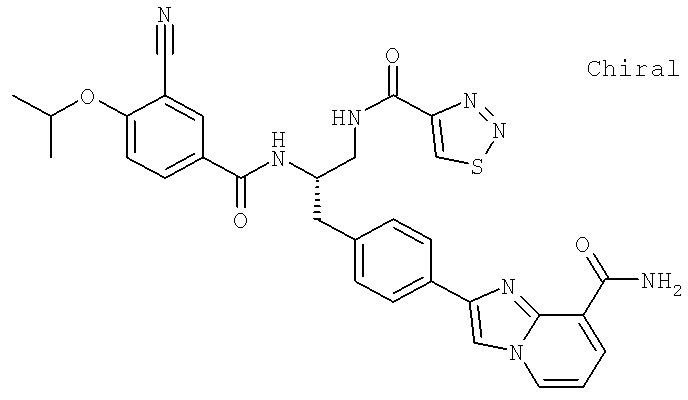
2-(4-{(2S)-2-[({3-Циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-3-[(1,2,3-тиадиазол-4-илкарбонил)амино]пропил}фенил)имидазо[1,2-а]пиридин-8-карбоксамид
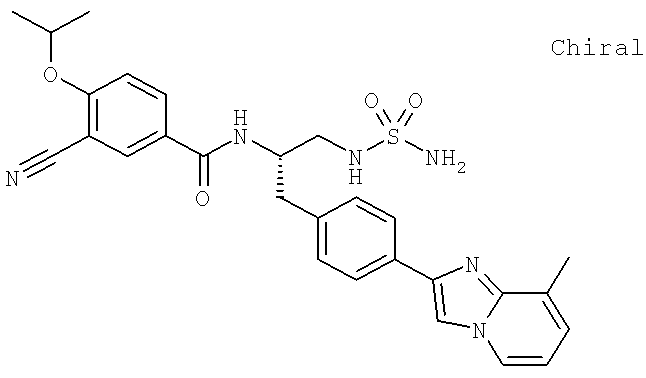
N-((1S)-2-[(Аминосульфонил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
547,2
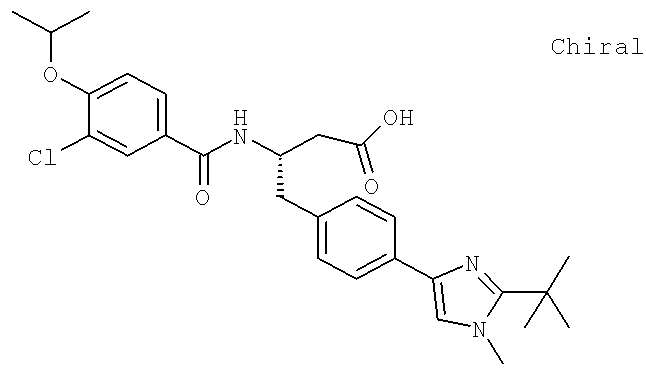
(3S)-3-[({3-Хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-{4-[2-(1,1-диметилэтил)-1-метил-1-метил-1Н-имидазол-4-ил]фенил}бутановая кислота
512,4
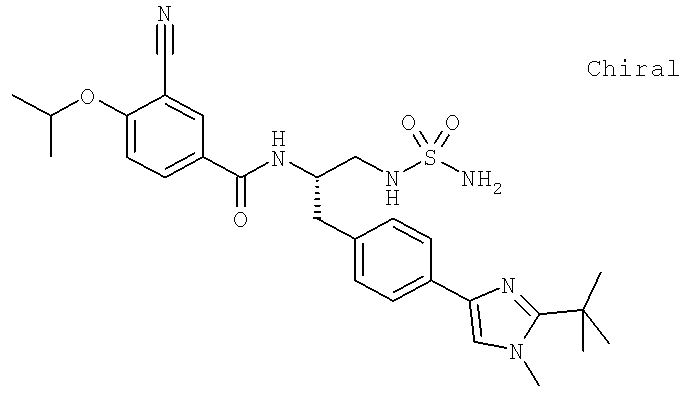
N-[(1S)-2-[(Аминосульфонил)амино]-1-({4-[2-(1,1-диметилэтил)-1-метил-1Н-имидазол-4-ил]фенил}метил)этил]-3-циано-4-[(1-метилэтил)окси]бензамид
553,2
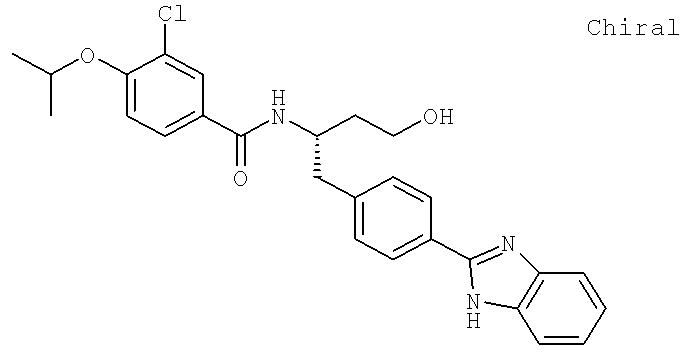
N-((1S)-1-{[4-(1H-Бензимидазол-2-ил)фенил]метил}-3-гидроксипропил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
477,8
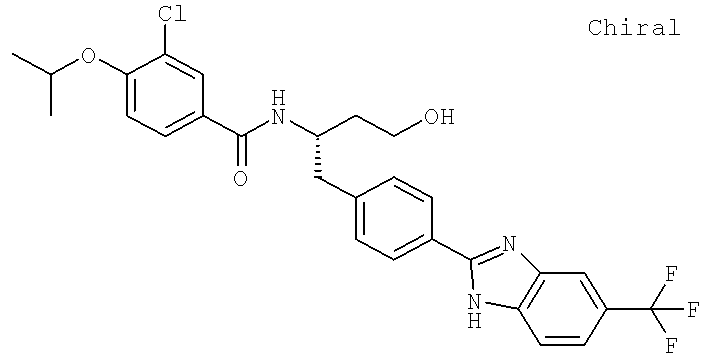
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[5-(трифторометил)-1H-бензимидазол-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
546,2
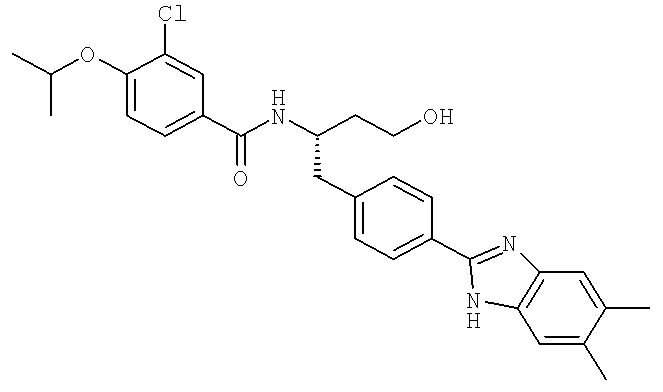
3-Хлоро-N-((1S)-1-{[4-(5,6-диметил-1H-бензимидазол-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
506,2
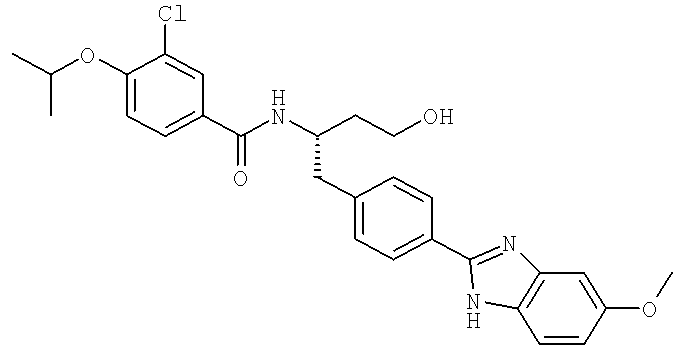
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[5-(метилокси)-1H-бензимидазол-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси] бензамид
508,2
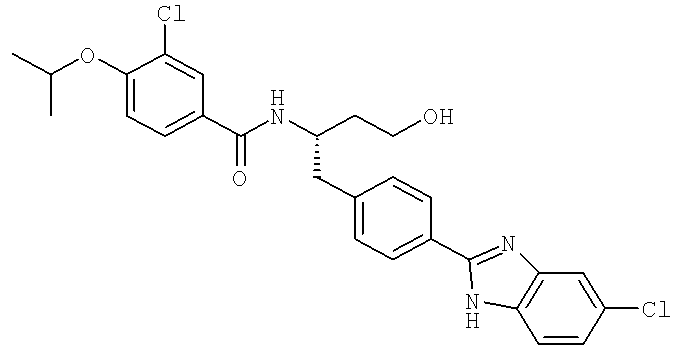
3-Хлоро-N-((1S)-1-{[4-(5-хлоро-1H-бензимидазол-2-ил)фенил]метил}-3-гидрокси пропил)-4-[(1-метилэтил)окси]бензамид
512,0
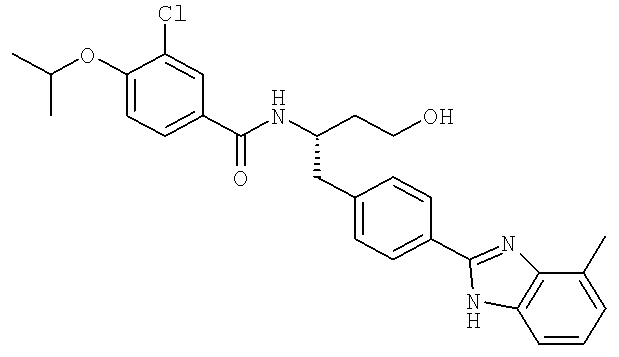
3-Хлоро-N-((1S)-3-гидрокси-1-{[4-(4-метил-1H-бензимидазол-2-ил)фенил]метил}пропил-4-[(1-метилэтил)окси]бензамид
492,2
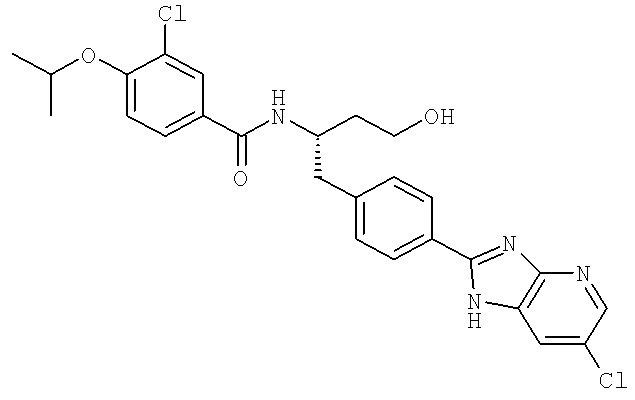
3-Хлоро-N-((1R)-1-{[4-(6-хлоро-1H-имидазо[4,5-b]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси] бензамид
513,2
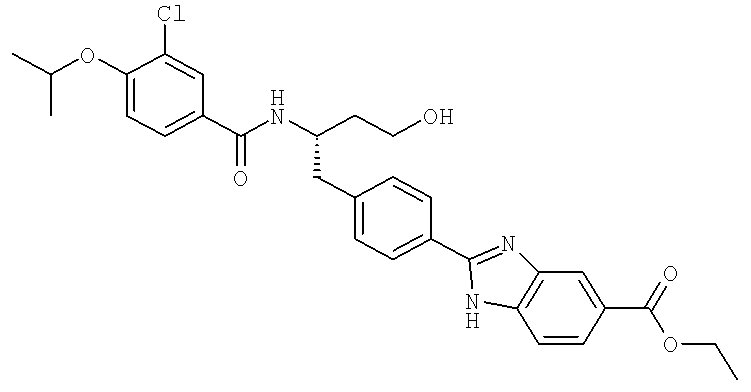
Этил-2-(4-{(2S)-2-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)-1H-бензимидазол-5-карбоксилат
550,2
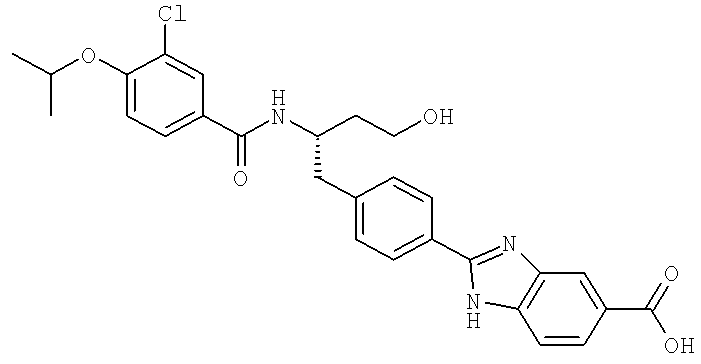
2-(4-{(2S)-2-[({3-Хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)-1Н-бензимидазол-5-карбоновая кислота
522,2
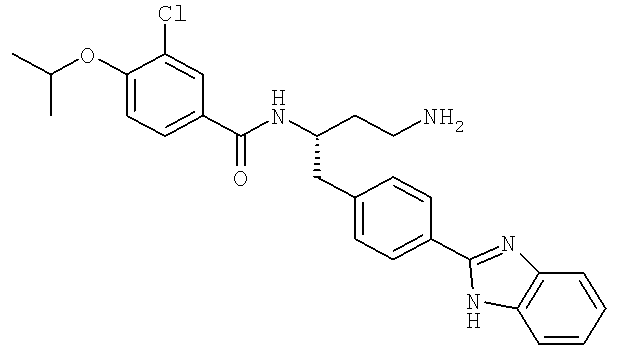
N-((1S)-3-Амино-1-{[4-(1H-бензимидазол-2-ил)фенил]метил}пропил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
477,0
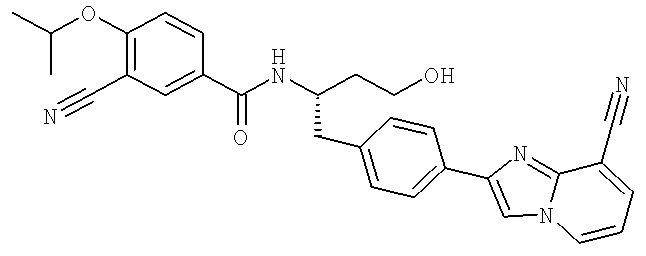
3-Циано-N-((1S)-1-{[4-(8-цианоимидазо[1,2-a]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
494,4
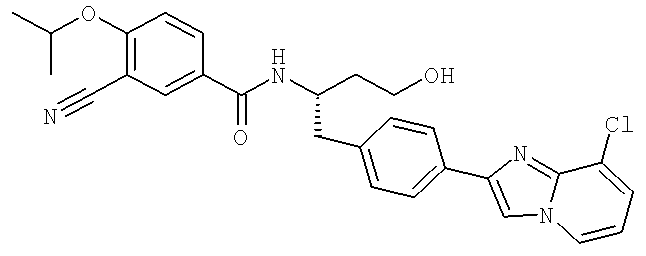
N-((1S)-1-{[4-(8-хлороимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси]бензамид
504,2
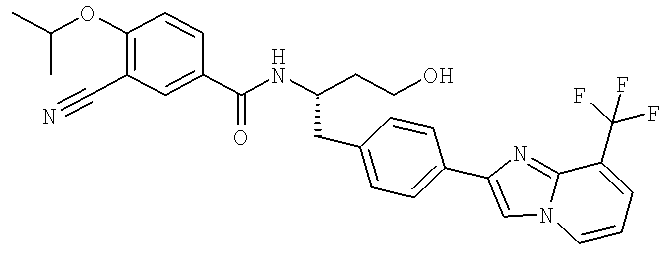
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(трифторометил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил-4-[(1-метилэтил)окси]бензамид
537,2
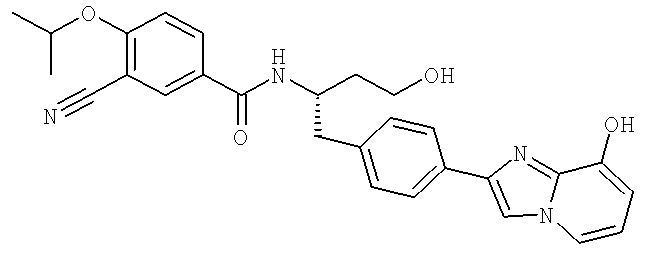
3-Циано-N-((1S)-3-гидрокси-1-{[4-(8-гидроксимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
485,2
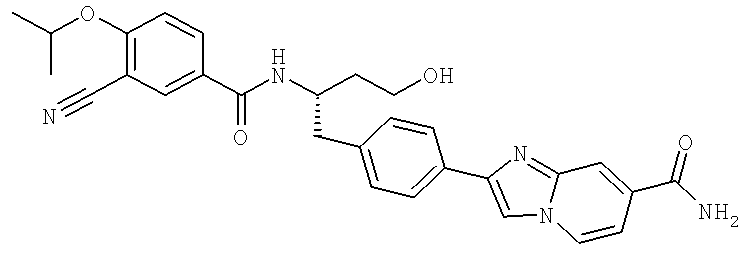
2-(4-{(2S)-2-[({3-Циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)имидазо[1,2-а]пиридин-7-карбоксамид
512,2
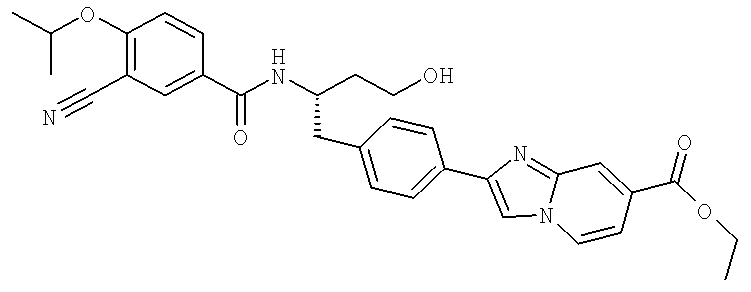
Этил-2-(4-{(2S)-2-[({3-циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)имидазо[1,2-а]пиридин-7-карбоксилат
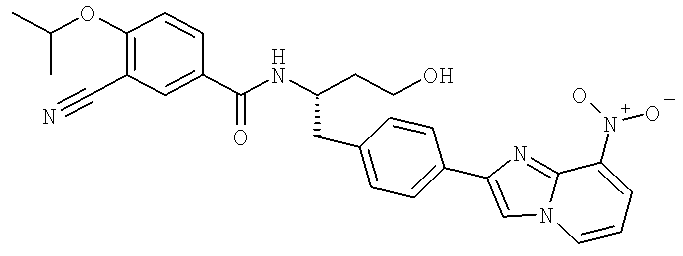
3-Циано-N-((1S)-3-гидрокси-1-{[4-(8-нитроимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
514,4
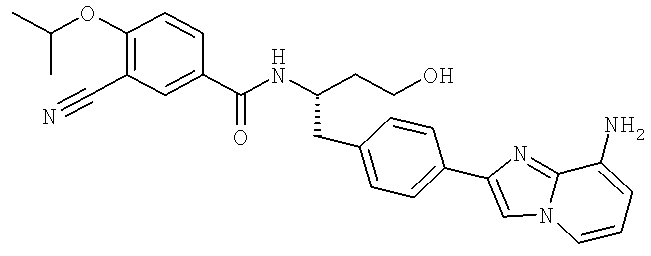
N-((1S)-1-{[4-(8-Аминоимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси]бензамид
484,2
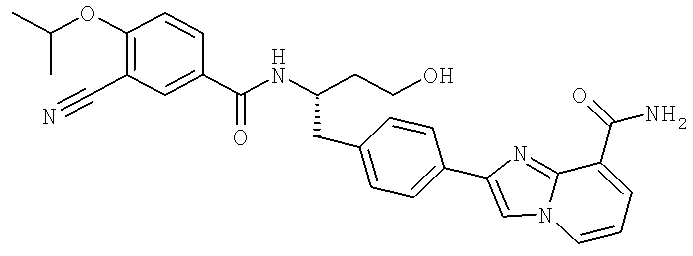
2-(4-{(2S)-2-[({3-Циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)имидазо[1,2-а]пиридин-8-карбоксамид
512,2
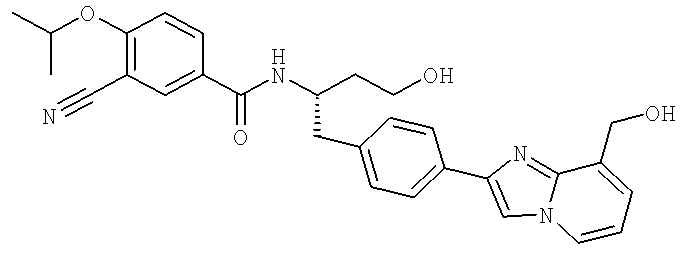
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(гидроксиметил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
499,4
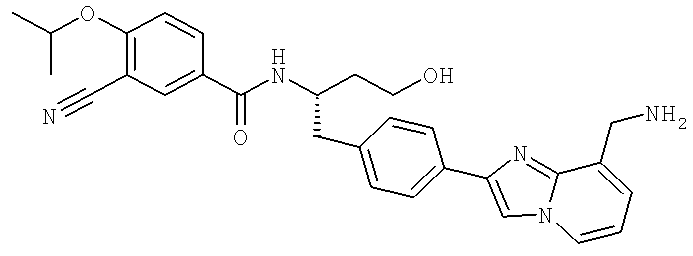
N-[(1S)-1-({4-[8-(Аминометил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)-3-гидроксипропил]-3-циано-4-[(1-метилэтил)окси]бензамид
498,4
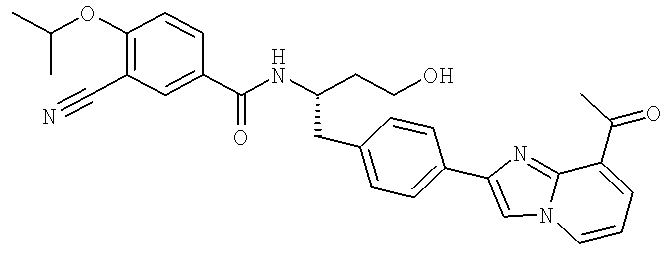
N-((1S)-1-{[4-(8-Ацетилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси]бензамид
511,2
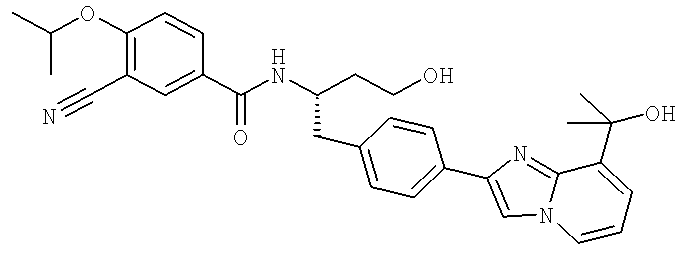
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидрокси-1-метилэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
527,4
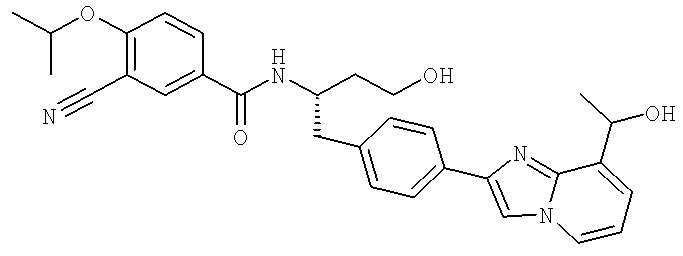
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидроксиэтил)имидазо [1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
513,4
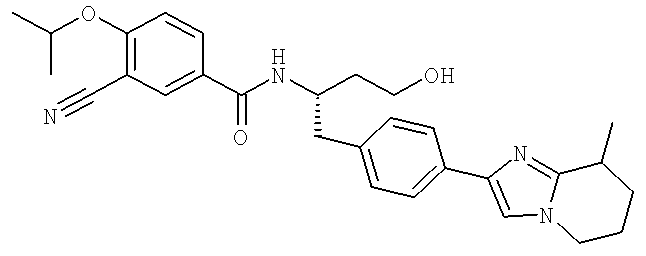
3-Циано-N-[(1S)-3-гидрокси-1-{[4-(8-метил-5,6,7,8-тетрагидроимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
487,2
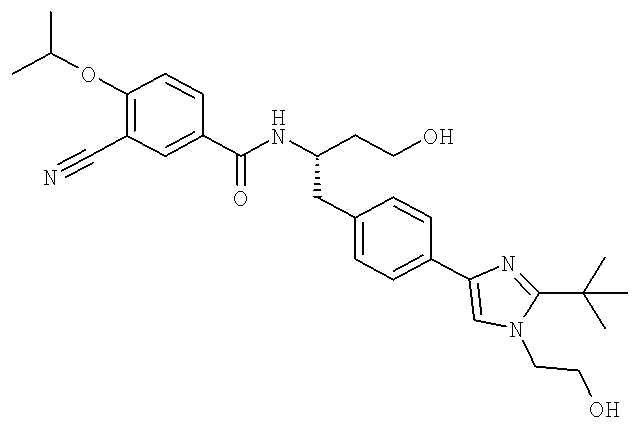
3-Циано-N-[(1S)-1-({4-[2-(1,1-диметилэтил)-1-(2-гидроксиэтил)-1H-имидазол-4-ил]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
519,4
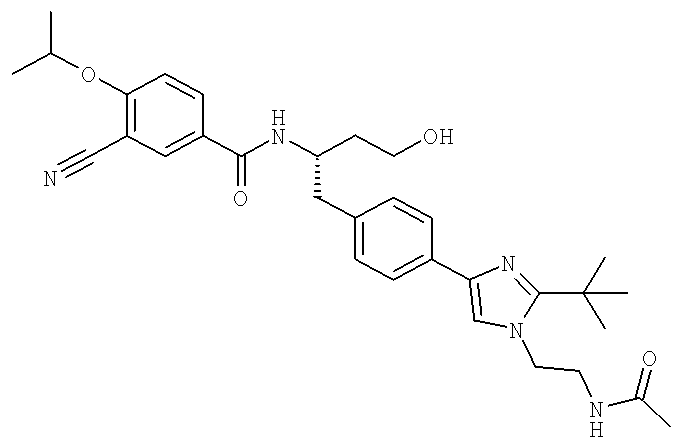
N-[(1S)-1-({4-[1-[2-(Ацетиламино)этил]-2-(1,1-диметилэтил)-1H-имидазол-4-ил]фенил}метил)-3-гидроксипропил]-3-циано-4-[(1-метилэтил)окси]бензамид
560,4
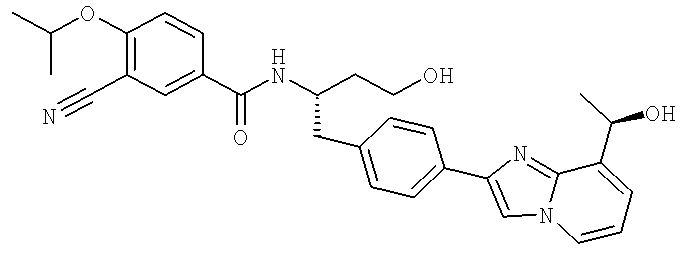
3-Циано-N-{(1S)-3-гидрокси-1-[(4-{8-[(1R)-1-гидроксиэтил]имидазо[1,2-а]пиридин-2-ил}фенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
513,4
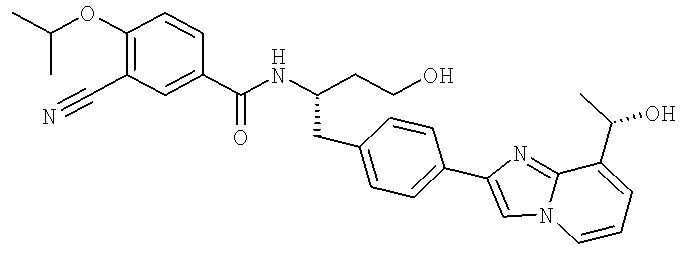
3-Циано-N-{(1S)-3-гидрокси-1-[(4-{8-[(1S)-1-гидроксиэтил]имидазо[1,2-а]пиридин-2-ил}фенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
513,4
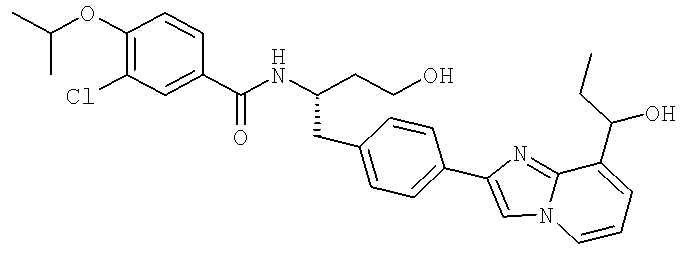
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидроксипропил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
536,2
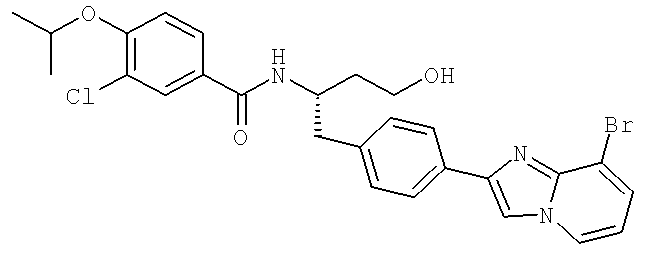
N-((1S)-1-{[4-(8-Бромоимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
556,2
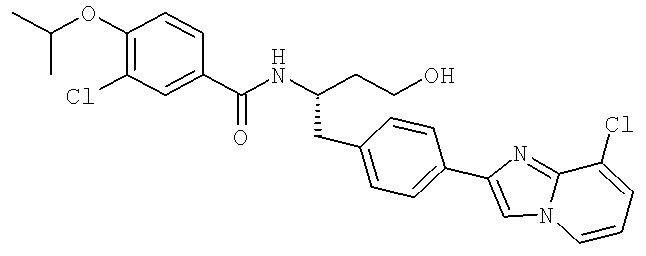
3-Хлоро-Н-((1S)-1-{[4-(8-хлороимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
512,4
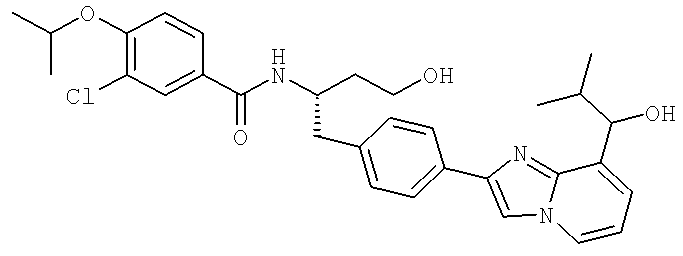
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидрокси-2-метилпропил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
550,4
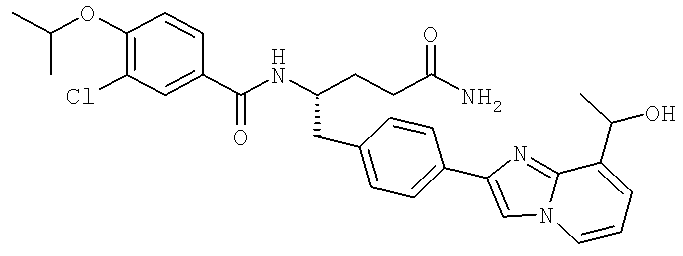
N-[(1R)-4-Амино-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)-4-оксобутил]-3-хлоро-4-[(1-метилэтил)окси]бензамид
549,2
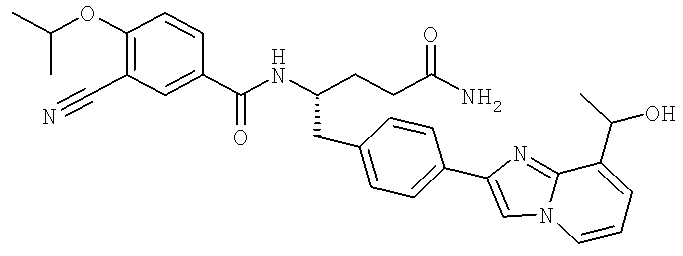
N-[(1R)-4-Амино-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-илфенил}-метил)-4-оксобутил]-3-циано-4-[(1-метилэтил)окси]бензамид
540,6
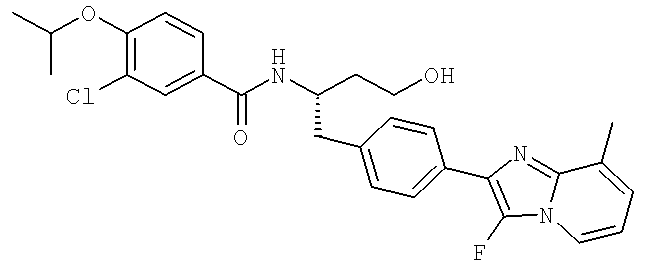
3-Хлоро-N-((1S)-1-{[4-(3-фторо-8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
510,2
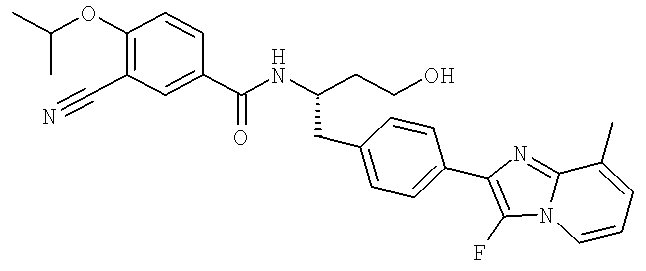
3-Циано-N-((1S)-1-{[4-(3-фторо-8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
501,2
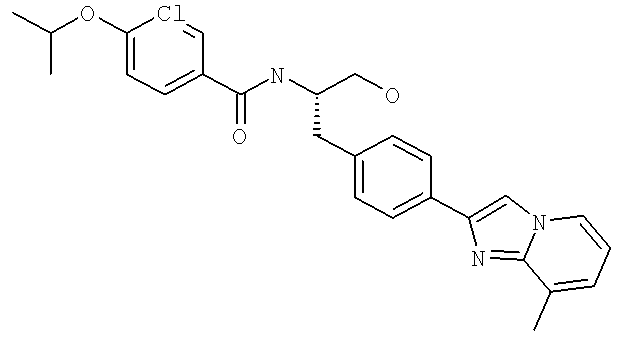
3-Хлоро-N-((1S)-2-гидрокси-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
478,2
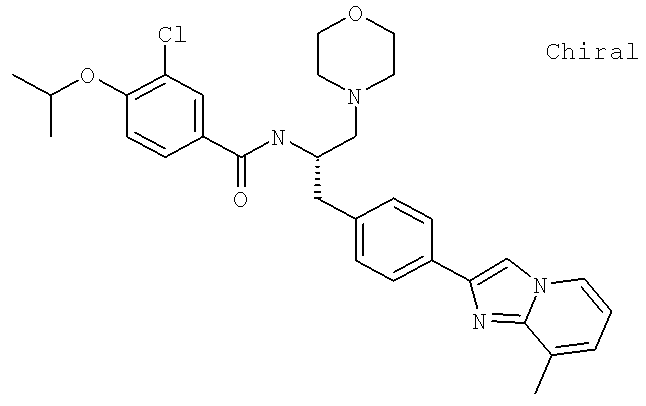
3-Хлоро-4-[(1-метилэтил)окси]-N-[(1S)-2-[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]-1-(4-морфолинилметил)этил]бензамид
547,2
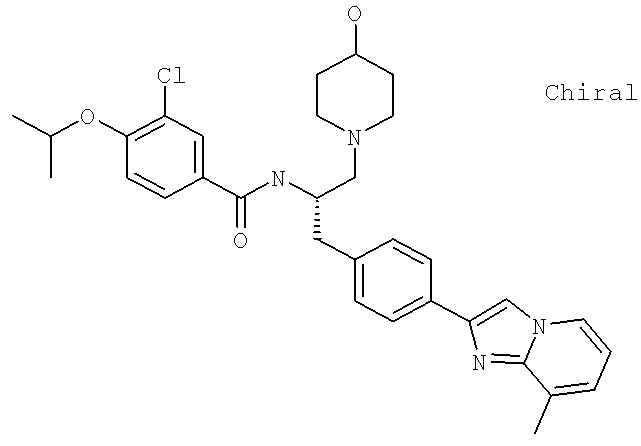
3-Хлоро-N-((1S)-2-(4-гидрокси-1-пиперидинил)-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
561,2
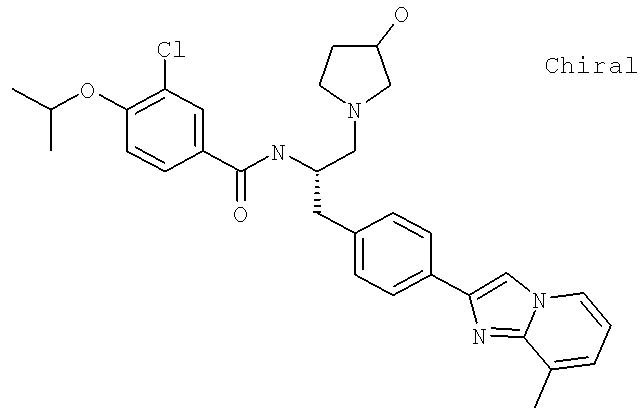
3-Хлоро-N-((1S)-2-(3-гидрокси-1-пирролидинил)-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
547,2
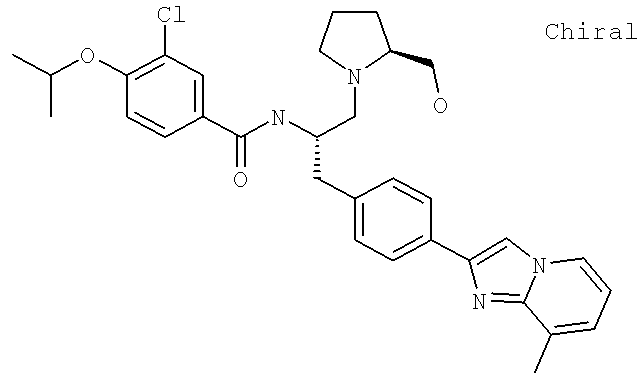
3-Хлоро-N-((1S)-2-[(2S)-2-(гидроксиметил)-1-пирролидинил]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
561,2
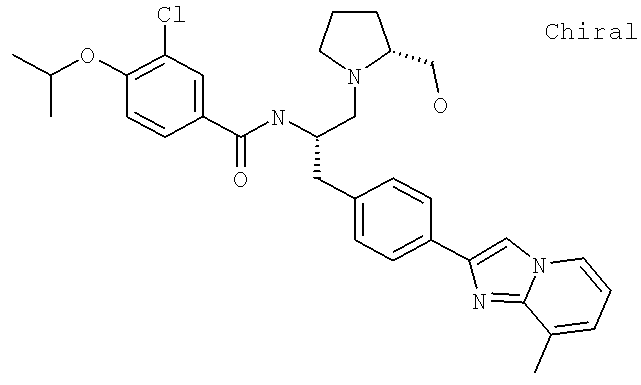
3-Хлоро-N-((1S)-2-[(2R)-2-(гидроксиметил)-1-пирролидинил]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
561,2
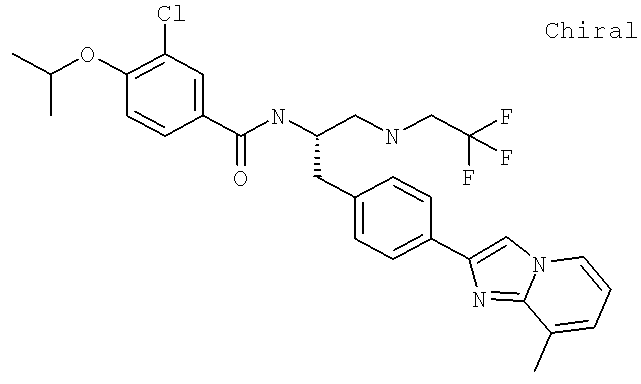
3-Хлоро-4-[(1-метилэтил)окси]-N-((1S)-2-[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]-1-{[(2,2,2-трифтороэтил)амино]метил}этил)бензамид
559,2
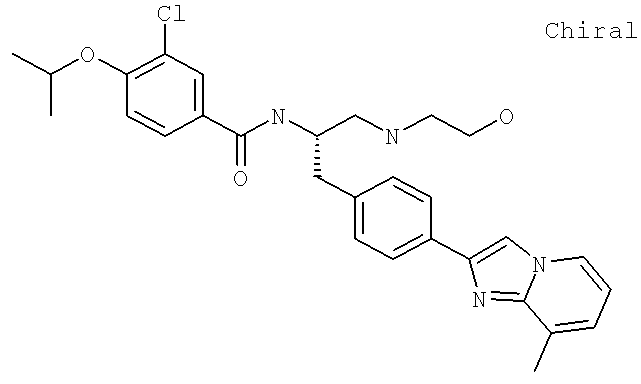
3-Хлоро-N-((1S)-2-[(2-гидроксиэтил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
521,2
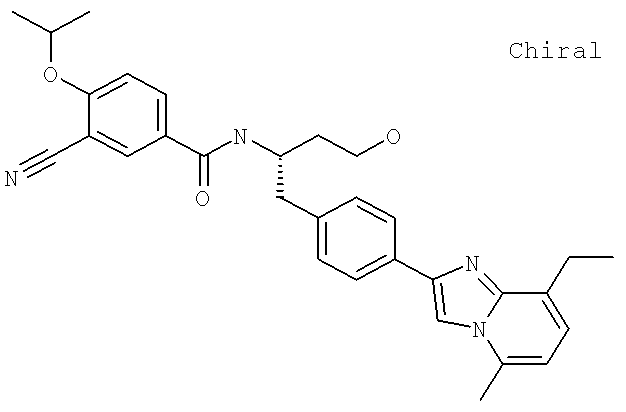
3-Циано-N-((1S)-1-{[4-(8-этил-5-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
511,2
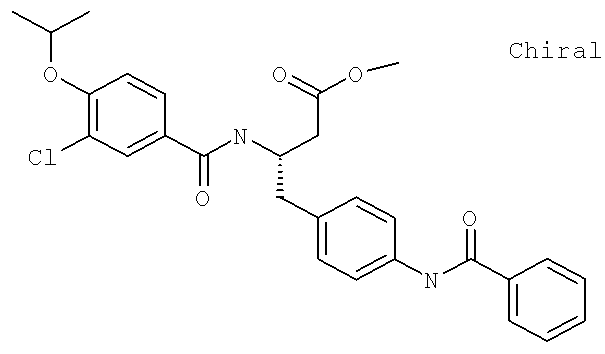
Метил(3S)-3-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-{4-[(фенилкарбонил)амино]фенил}бутаноат
509
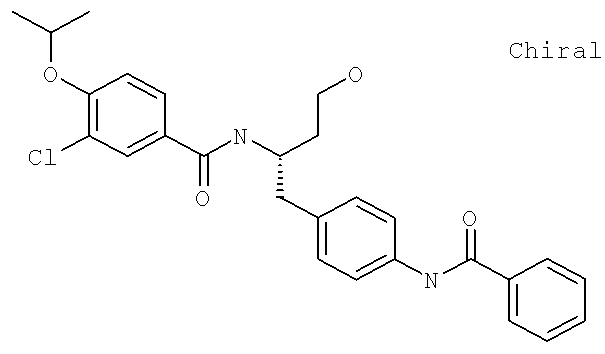
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[(фенилкарбонил)амино]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
481
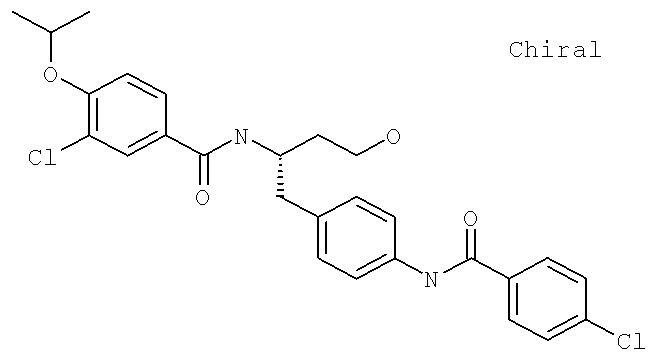
3-Хлоро-N-{(1S)-1-[(4-{[(4-хлорофенил)карбонил]амино}фенил)метил]-3-гидроксипропил}-4-[(1-метилэтил)окси]бензамид
515
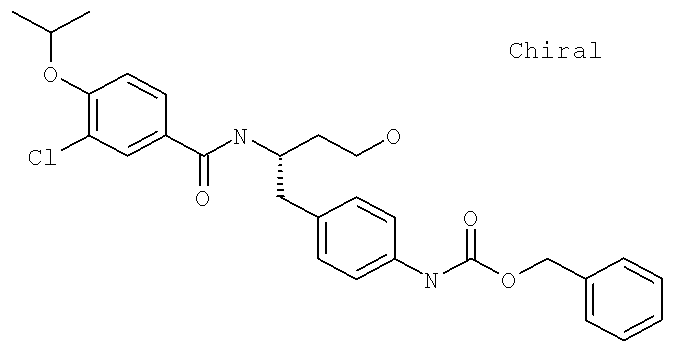
Фенилметил(4-{(2S)-2-[({3-хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино-4-гидроксибутил}фенил)карбамат
511
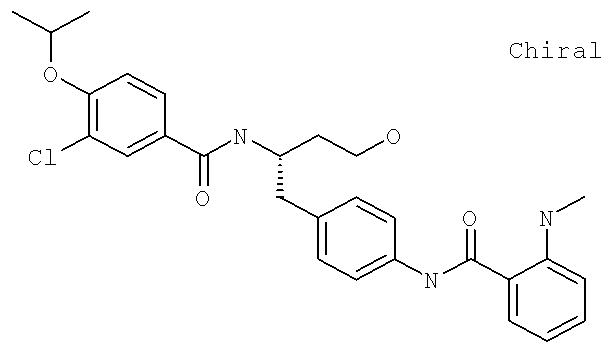
3-Хлоро-N-((1S)-3-гидрокси-1-{[4-({[2-метиламино)фенил]карбонил}амино)фенил]метил}пропил-4-[(1-метилэтил)окси]бензамид
510
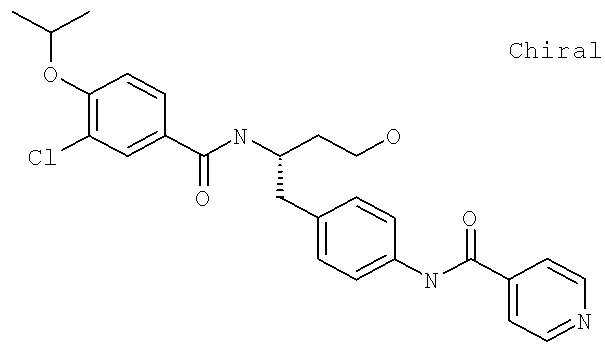
N-(4-{(2S)-2-[({3-Хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)-4-пиридинкарбоксамид
482
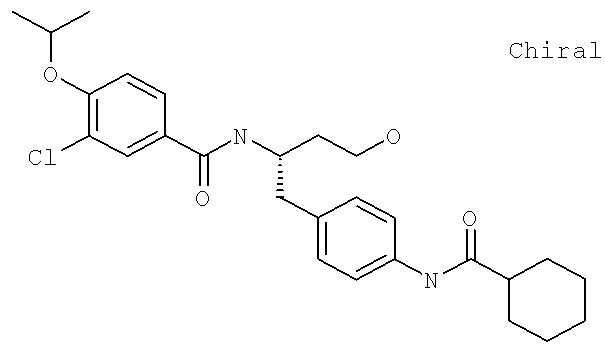
3-Хлоро-N-[(1S)-1-({4-[(циклогексилкарбонил)амино]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
487
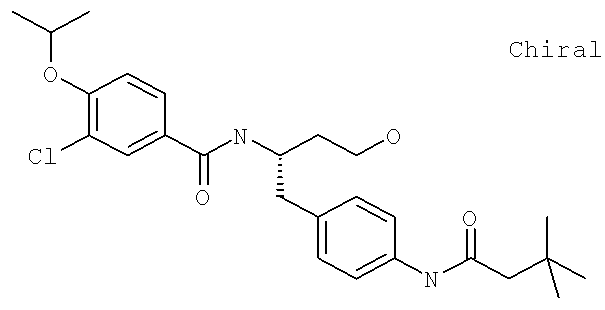
3-Хлоро-N-[(1S)-1-({4-[(3,3-диметилбутаноил)амино]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
475
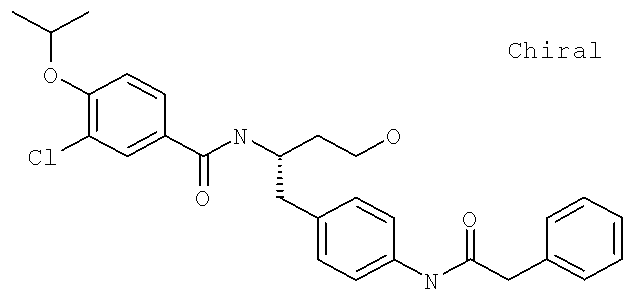
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[(фенилацетил)амино] фенил} метил)пропил]-4-[(1-метилэтил)окси]бензамид
495
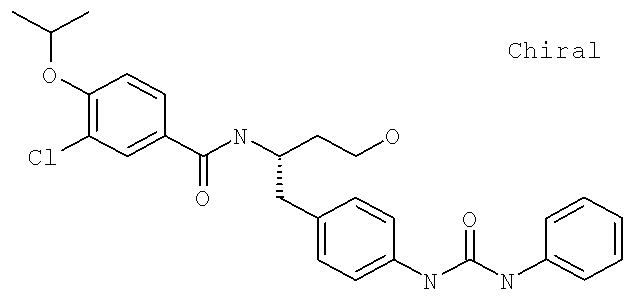
3-Хлоро-N-{(1S)-3-гидрокси-1-[(4-{[(фениламино)карбонил]амино}фенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
496
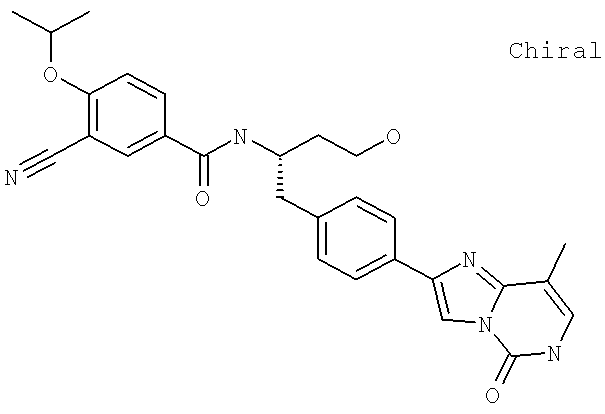
3-Циано-N-((1S)-3-гидрокси-1-{[4-(8-метил-5-оксо-5,6-дигидроимидазо[1,2-с]пиримидин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
500
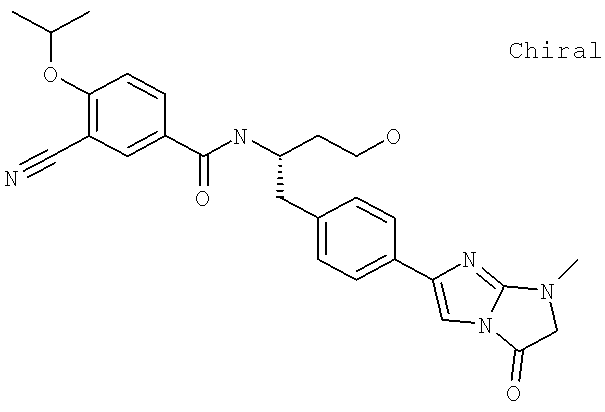
3-Циано-N-((1S)-3-гидрокси-1-{[4-(1-метил-3-оксо-2,3-дигидро-1H-имидазо[1,2-а]имидазол-6-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
488
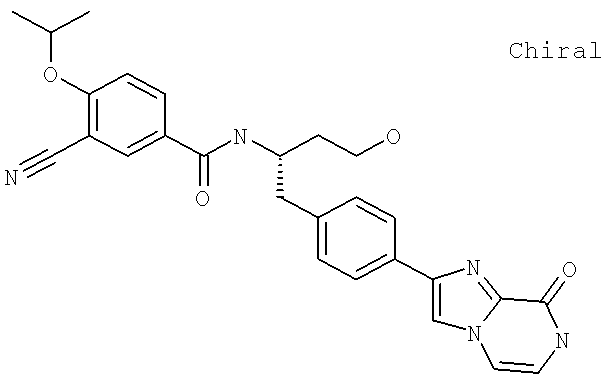
3-Циано-N-((1S)-3-гидрокси-1-{[4-(8-оксо-7,8-дигидроимидазо[1,2-а]пиразин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
486
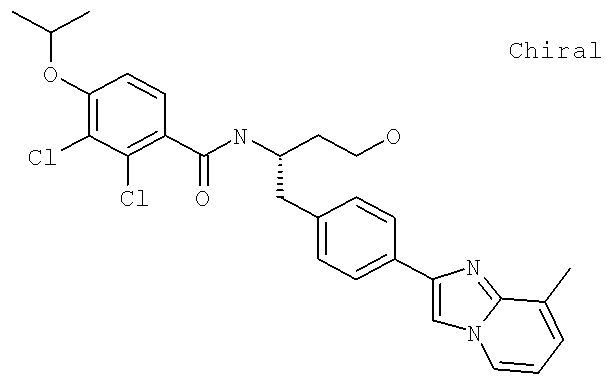
2,3-Дихлоро-N-((1S)-3-гидрокси-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
526
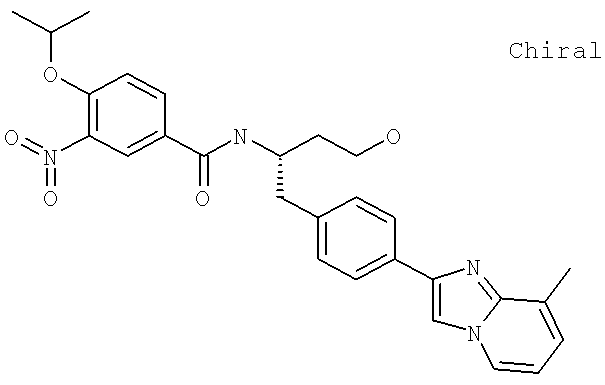
N-((1S)-3-Гидрокси-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]-3-нитробензамид
503
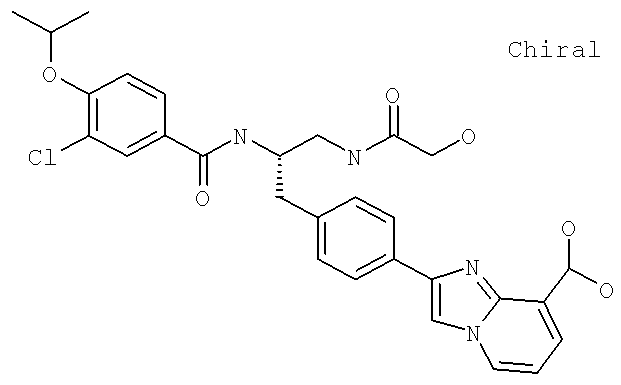
3-Хлоро-N-[(1S)-2-[(гидроксиацетил)амино]-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид
565
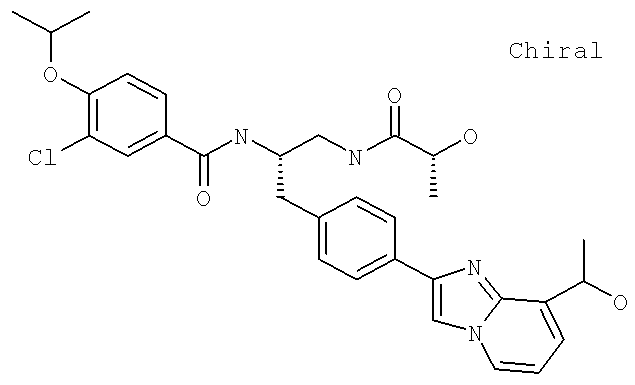
3-Хлоро-N-[(1S)-2-{4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}-1-({[(2R)-2-гидроксипропаноил]амино}метил)этил]-4-[(1-метилэтил)окси]бензамид
579
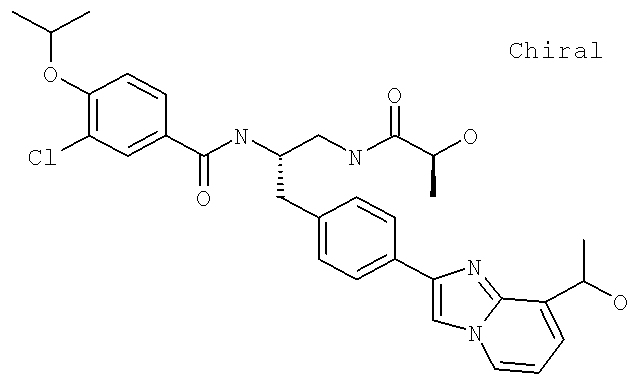
3-Хлоро-N-[(1S)-2-{4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}-1-({[(2S)-2-гидроксипропаноил]амино}метил)этил]-4-[(1-метилэтил)окси]бензамид
579
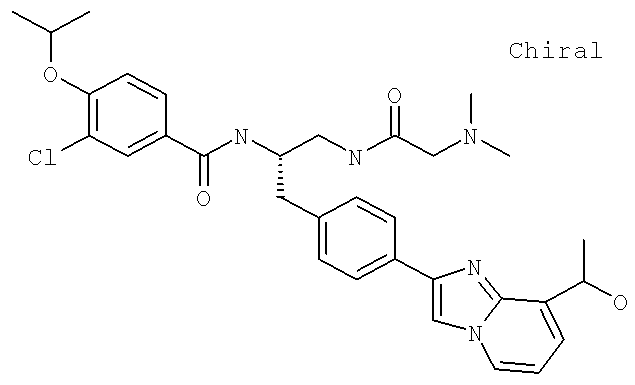
3-Хлоро-N-[(1S)-2-[(N-диметилглицил)амино]-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид
592
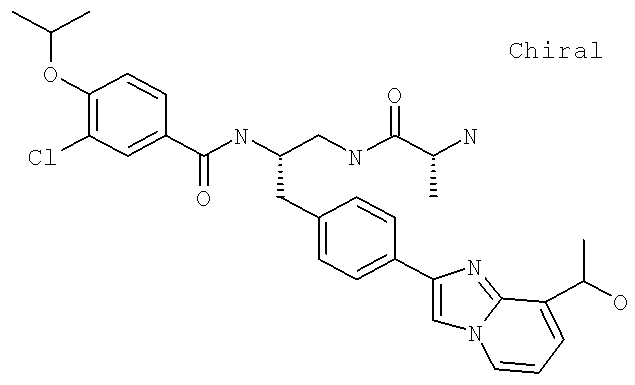
N-[(1S)-2-(D-Аланиламино)-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-3-хлоро-4-[(1-метилэтил)окси]бензамид
578
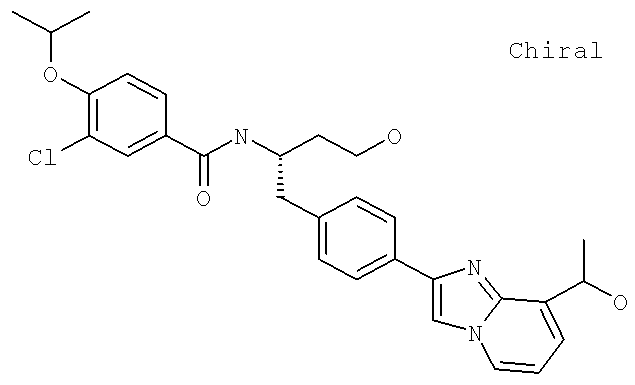
3-Хлоро-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
522
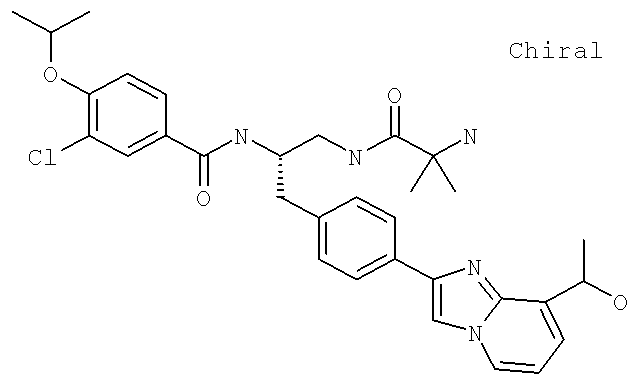
3-Хлоро-N-((1S)-2-{4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}-1-{[(2-метилаланил)амино]метил}этил)-4-[(1-метилэтил)окси]бензамид
592
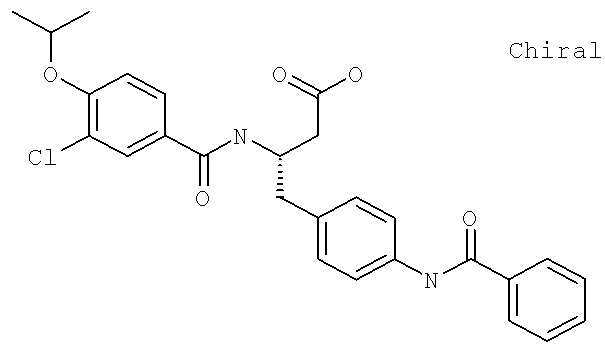
(3S)-3-[({3-Хлоро-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-{4-[(фенилкарбонил)амино]фенил}бутановая кислота
495
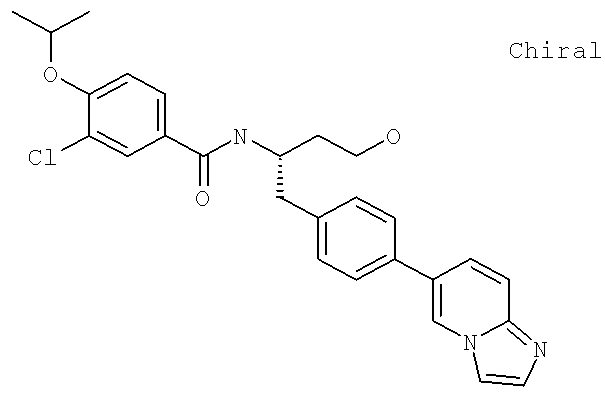
3-Хлоро-N-{(1S)-3-гидрокси-1-[(4-имидазо[1,2-а]пиридин-6-илфенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
477,8
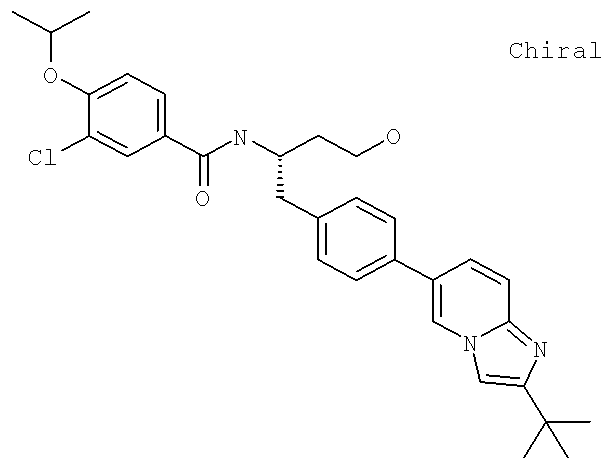
3-Хлоро-N-[(1S)-1-({4-[2-(1,1-диметилэтил)имидазо[1,2-а]пиридин-6-ил]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
534,2
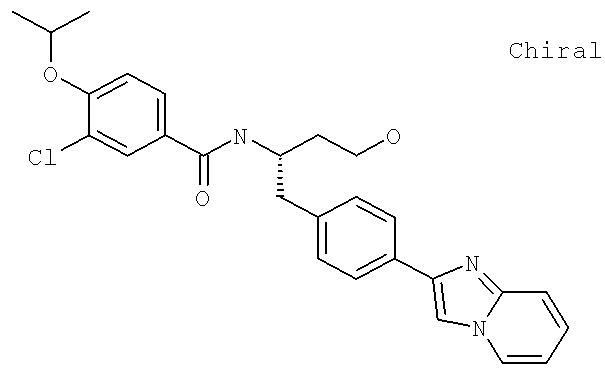
3-Хлоро-N-{(1S)-3-гидрокси-1-[(4-имидазо[1,2-а]пиридин-2-илфенил)метил]пропил-4-[(1-метилэтил)окси]бензамид
478,2
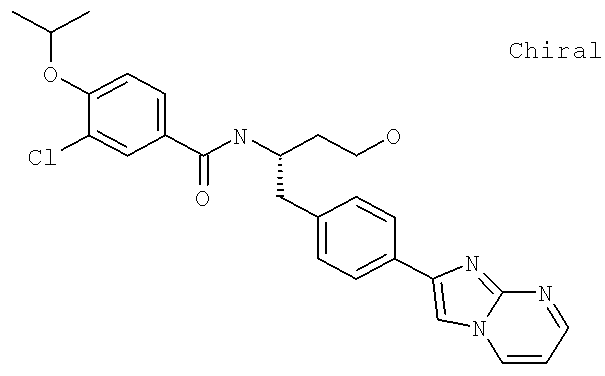
3-Хлоро-N-{(1S)-3-гидрокси-1-[(4-имидазо[1,2-а]пиримидин-2-илфенил)метил]пропил}-4-[(1-метилэтил)окси]бензамид
479,2
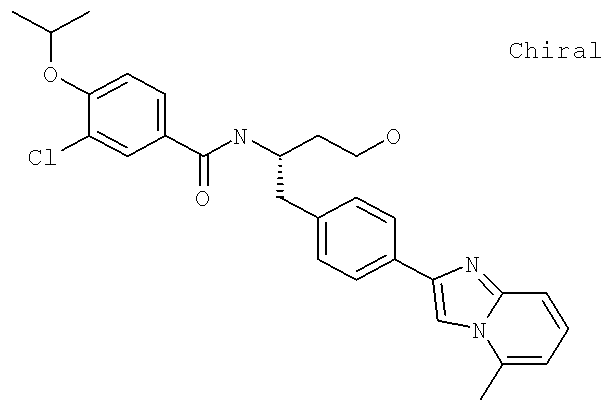
3-Хлоро-N-((1S)-3-гидрокси-1-{[4-(5-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
492,2
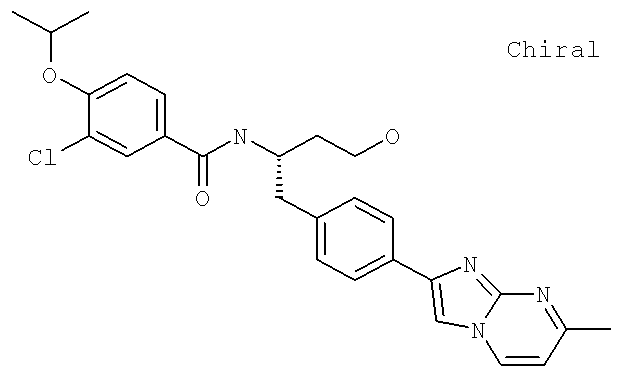
3-Хлоро-N-((1S)-3-гидрокси-1-{[4-(7-метилимидазо[1,2-а]пиримидин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
494,2
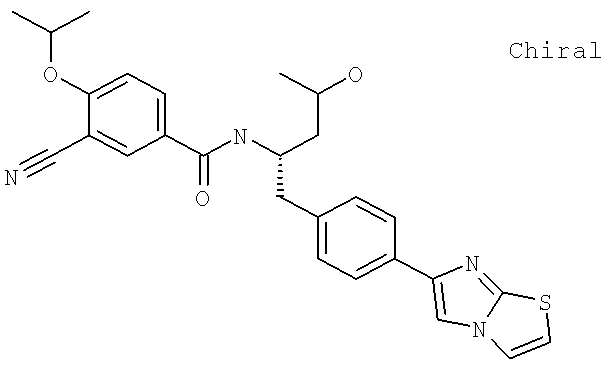
3-Циано-N-{(1S)-3-гидрокси-1-[(4-имидазо[2,1-b][1,3]тиазол-6-илфенил)метил]бутил}-4-[(1-метилэтил)окси]бензамид
489,0
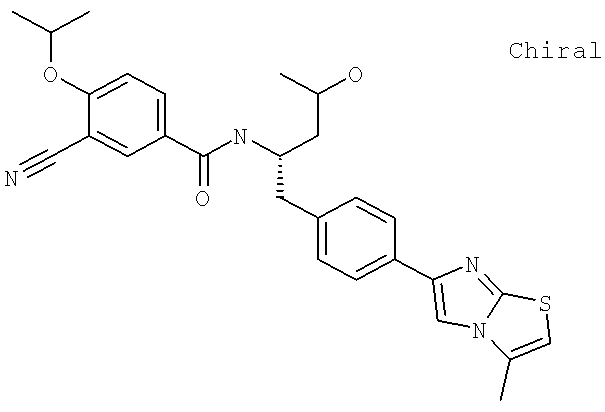
3-Циано-N-((1S)-3-гидрокси-1-{[4-(3-метилимидазо[2,1-b][1,3]тиазол-6-ил)фенил]метил}бутил)-4-[(1-метилэтил)окси]бензамид
503,2
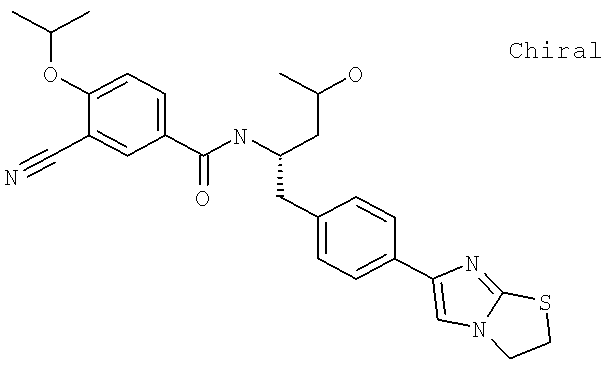
3-Циано-N-((1S)-1-{[4-(2,3-дигидроимидазо[2,1-b][1,3]тиазол-6-ил)фенил]метил}-3-гидроксибутил)-4-[(1-метилэтил)окси]бензамид
491,2
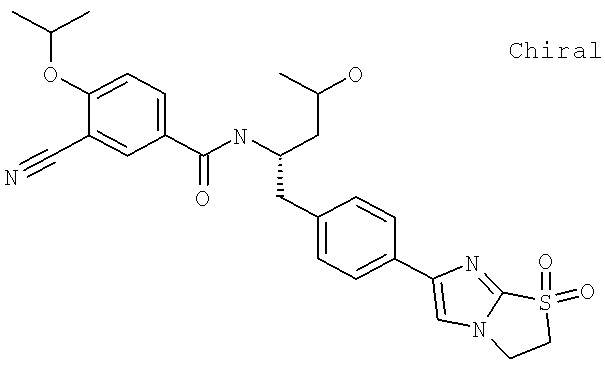
3-Циано-N-((1S)-1-{[4-(1,1-диоксидо-2,3-дигидроимидазо[2,1-b][1,3]тиазол-6-ил)фенил]метил}-3-гидроксибутил-4-[(1-метилэтил)окси]бензамид
523,2
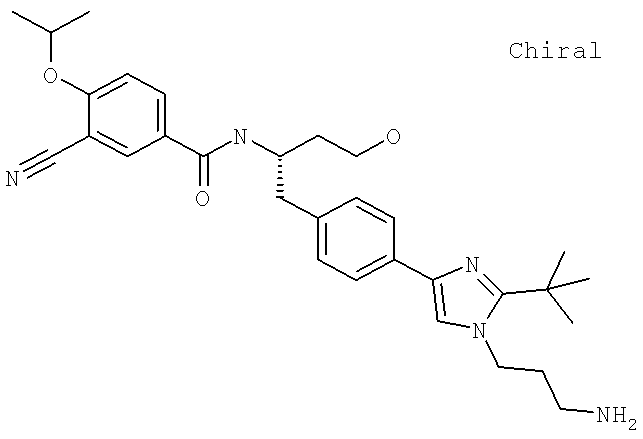
N-[(1S-1-({4-[1-(3-Аминопропил)-2-(1,1-диметилэтил)-1Н-имидазол-4-ил]фенил}метил)-3-гидроксипропил]-3-циано-4-[(1-метилэтил)окси]бензамид
532,4
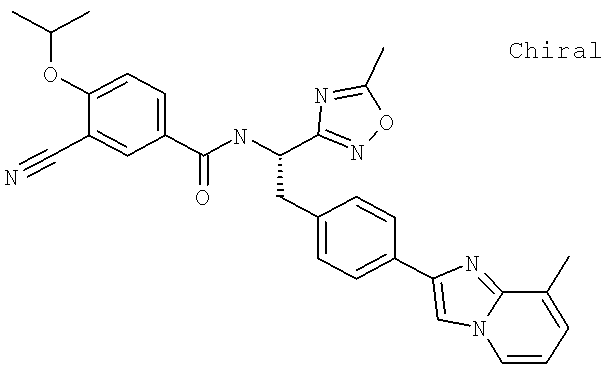
3-Циано-4-[(1-метилэтил)окси]-N-[(1S)-2-[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]-1-(5-метил-1,2,4-оксадиазол-3-ил)этил]бензамид
521,4
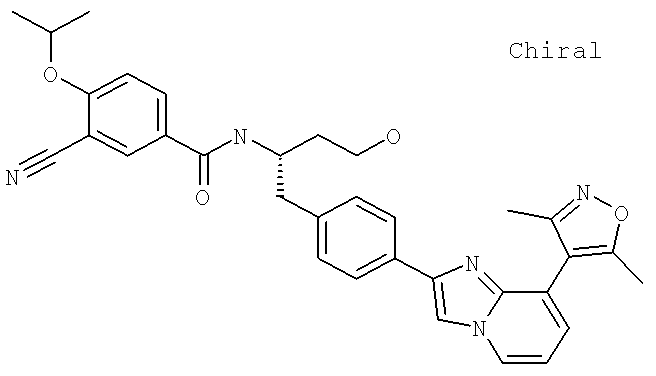
3-Циано-N-[(1S)-1-({4-[8-(3,5-диметил-4-изоксазолил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
564,2
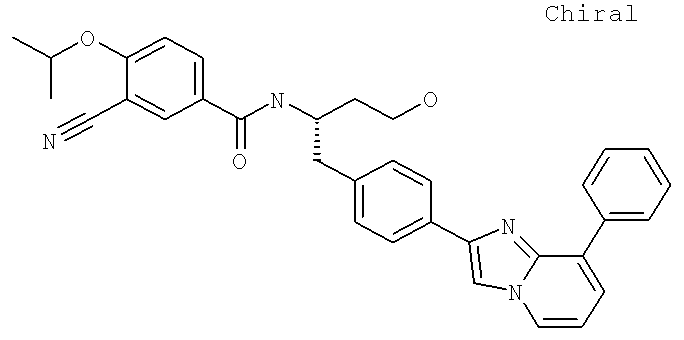
3-Циано-N-((1S)-гидрокси-1-{[4-(8-фенилимидазо[1,2-а]пиридин-2-ил)фенил]метил}пропил)-4-[(1-метилэтил)окси]бензамид
545,4
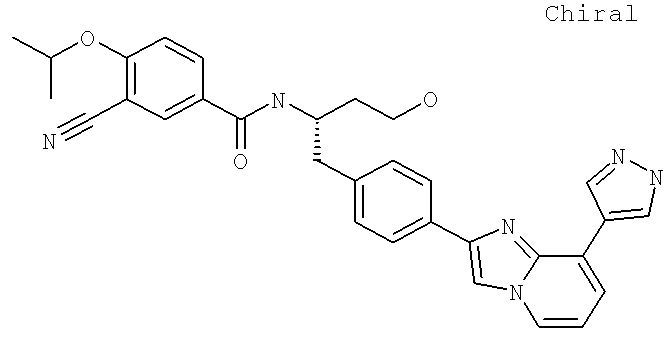
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(1H-пиразол-4-ил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
535,4
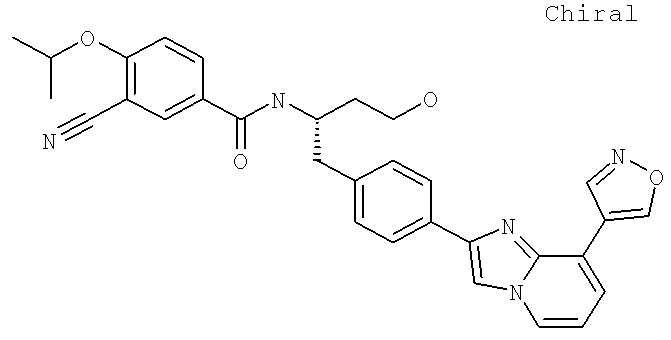
3-Циано-N-[(1S)-3-гидрокси-1-({4-[8-(4-изоксазолил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид
536,2
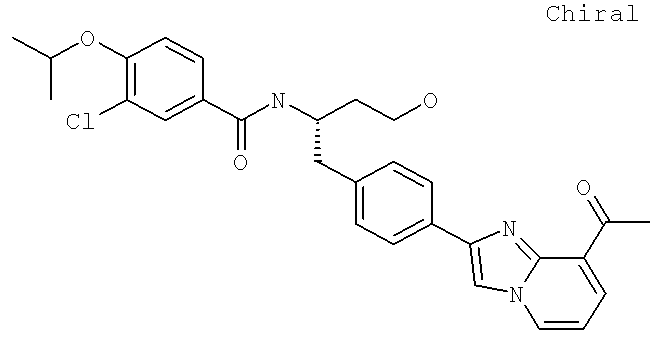
N-((1S)-1-{[4-(8-Ацетилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-хлоро-4-[(1-метилэтил)окси]бензамид
520,2
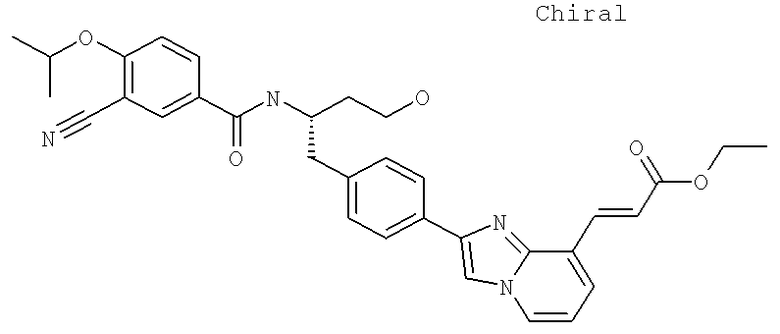
Этил(2E)-3-[2-(4-{(2S)-2-[({3-циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)имидазо[1,2-а]пиридин-8-ил]-2-пропеноат
567,6
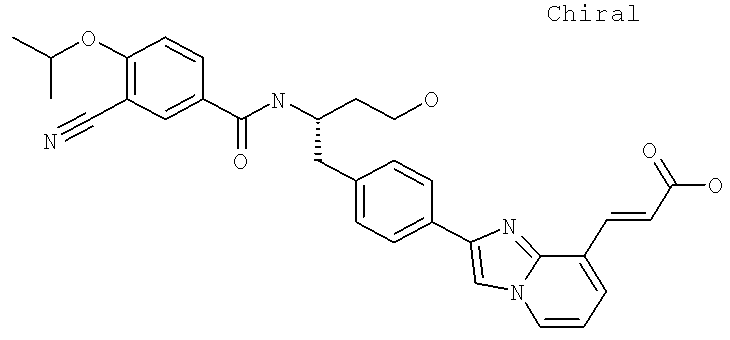
(2Е)-3-[2-(4-{(2S)-2-[({3-Циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-4-гидроксибутил}фенил)имидазо[1,2-а]пиридин-8-ил]-2-пропеновая кислота
539,4
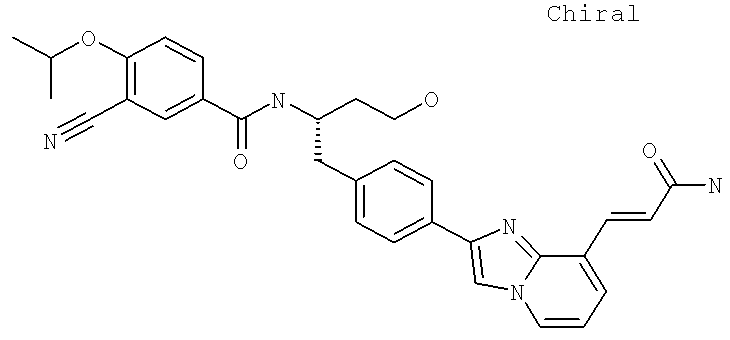
N-{(1S)-1-[(4-{8-[(1E)-3-амино-3-оксо-1-пропен-1-ил]имидазо[1,2-a]пиридин-2-ил}фенил)метил]-3-гидроксипропил}-3-циано-4-[(1-метилэтил)окси]бензамид
538,4
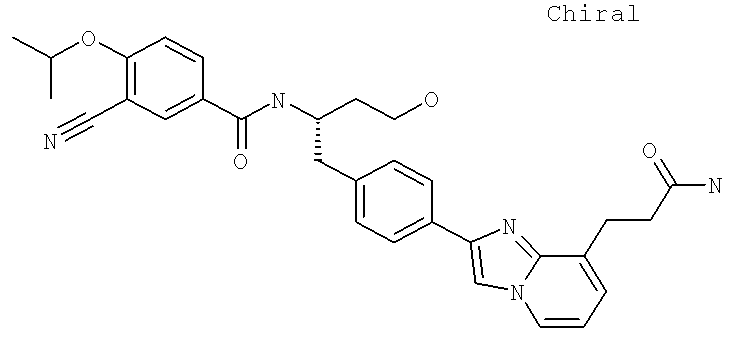
N-[(1S)-1-({4-[8-(3-Амино-3-оксопропил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)-3-гидроксипропил]-3-циано-4-[(1-метилэтил)окси]бензамид
540,4
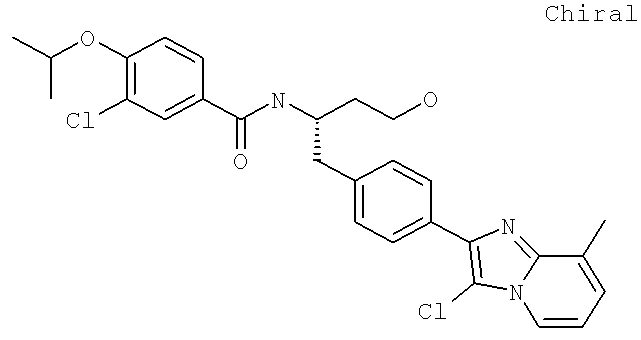
3-Хлоро-N-((1S)-1-{[4-(3-хлоро-8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
526,2
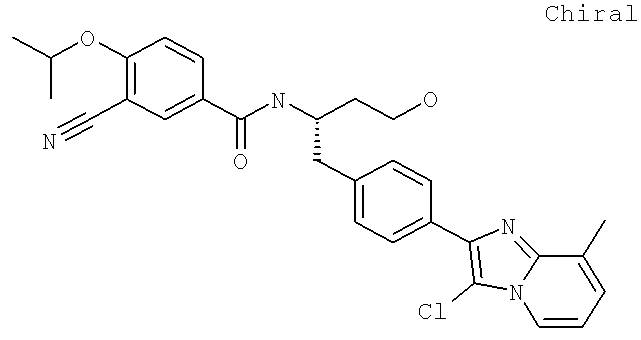
N-((1S)-1-{[4-(3-Хлоро-8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-3-циано-4-[(1-метилэтил)окси]бензамид
517,2
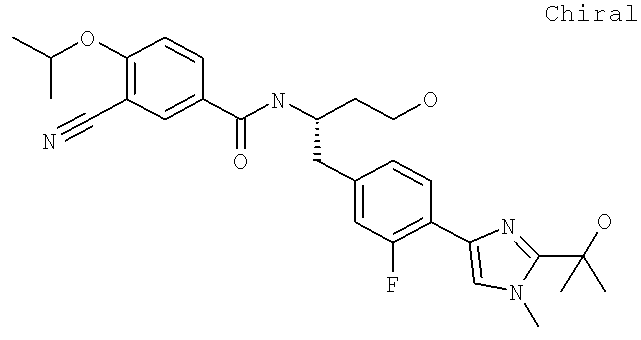
3-Циано-N-[(1S)-1-({3-фторо-4-[2-(1-гидрокси-1-метилэтил)-1-метил-1H-имидазол-4-ил]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
509,2
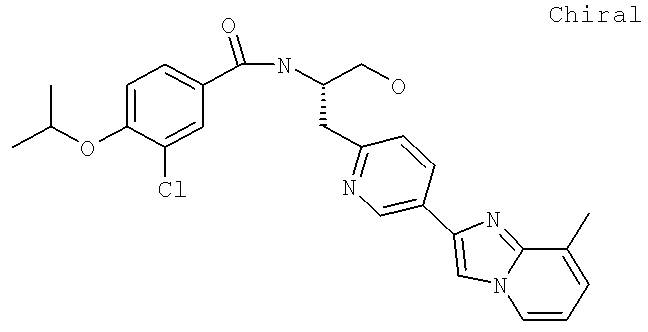
3-Хлоро-N-((1S)-2-гидрокси-1-{[5-(8-метилимидазо[1,2-а]пиридин-2-ил)-2-пиридинил]метил}этил)-4-[(1-метилэтил)окси]бензамид
479
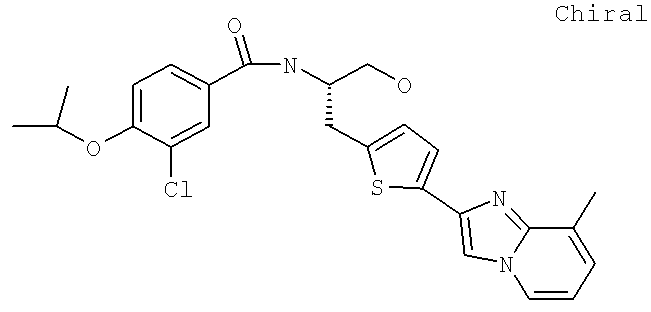
3-Хлоро-N-((1S)-2-гидрокси-1-{[5-(8-метилимидазо[1,2-а]пиридин-2-ил)-2-тиэнил]метил}этил)-4-[(1-метилэтил)окси]бензамид
484
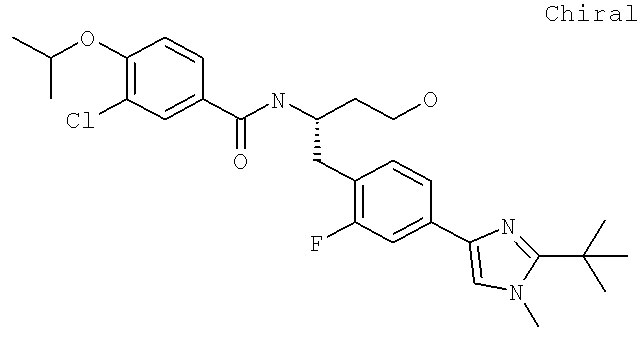
3-Хлоро-N-[(1S)-1-({4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]-2-фторофенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
516
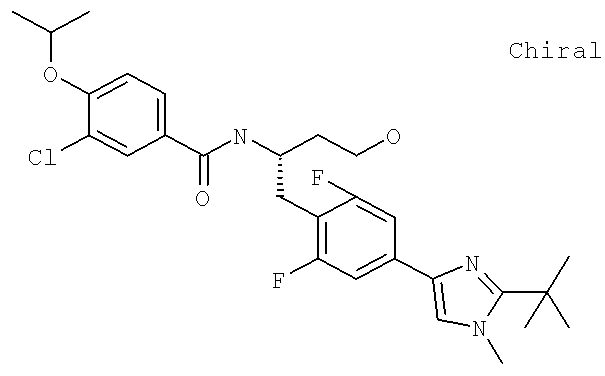
3-Хлоро-N-[(1S)-1-({4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]-2,6-дифторофенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
534
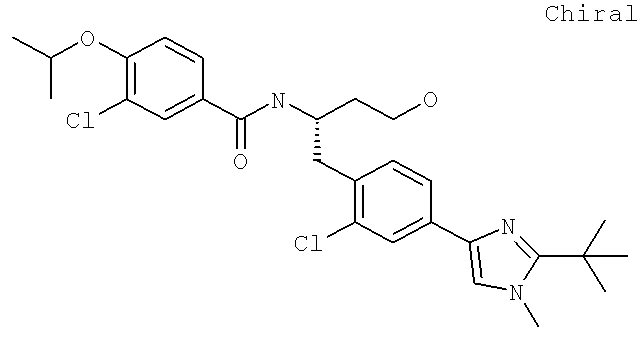
3-Хлоро-N-[(1S)-1-({2-хлоро-4-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]фенил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
532
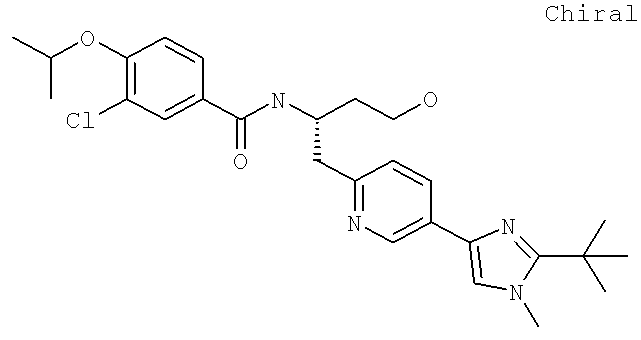
3-Хлоро-N-[(1S)-1-({5-[2-(1,1-диметилэтил)-1-метил-1H-имидазол-4-ил]-2-пиридинил}метил)-3-гидроксипропил]-4-[(1-метилэтил)окси]бензамид
499
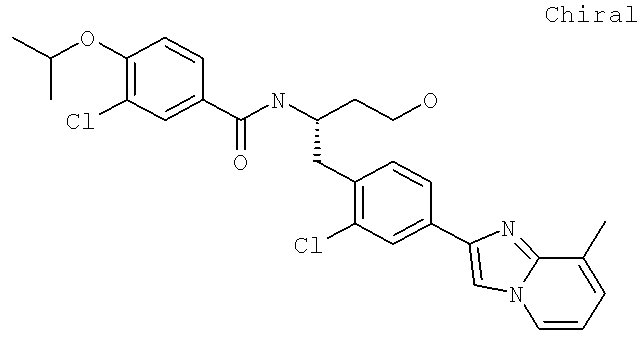
3-Хлоро-N-((1S)-1-{[2-хлоро-4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
526
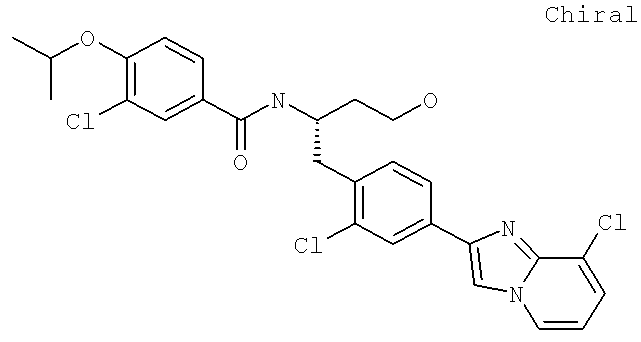
3-Хлоро-N-((1S)-1-{[2-хлоро-4-(8-хлороимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
546
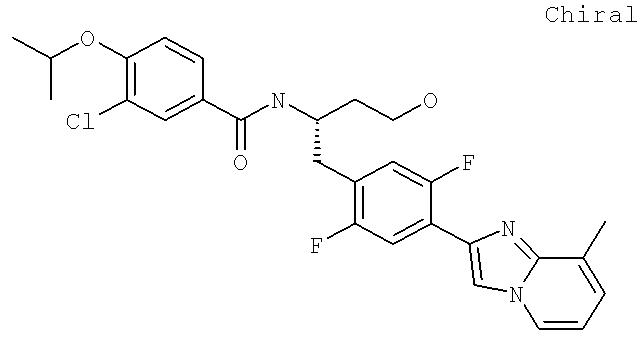
3-Хлоро-N-((1S)-1-{[2,5-дифторо-4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}-3-гидроксипропил)-4-[(1-метилэтил)окси]бензамид
528
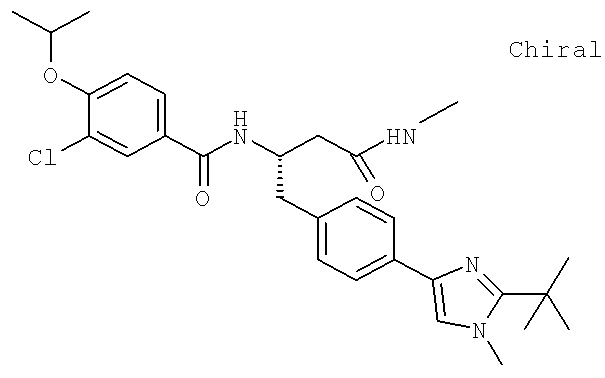
3-Хлоро-N-[(1S)-1-({4-[2-(1,1-диметилэтил)-1-метил-1Н-имидазол-4-ил]фенил}метил)-3-(метиламино)-3-оксопропил]-4-[(1-метилэтил)окси]бензамид
525,4
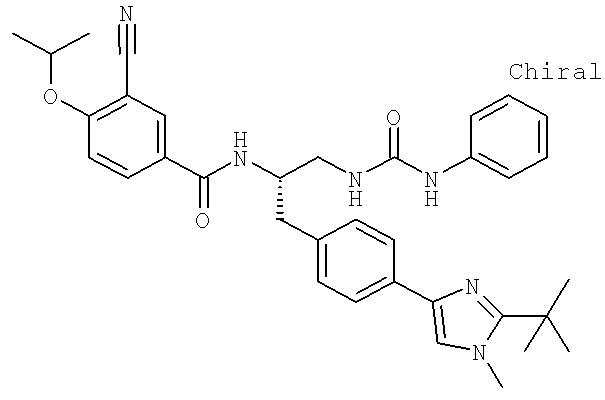
3-Циано-N-[(1S)-2-{4-[2-(1,1-диметилэтил)-1-метил-1Н-имидазол-4-ил]фенил}-1-({[(фениламино)карбонил]амино}метил)этил]-4-[(1-метилэтил)окси]бензамид
593,2
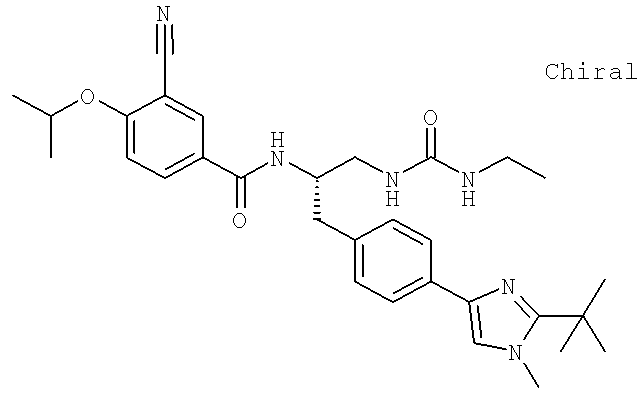
3-Циано-N-[(1S)-2-{4-[2-(1,1-диметилэтил)-1-метил-1Н-имидазол-4-ил]фенил}-1-({[(этиламино)карбонил]амино}метил)этил]-4-[(1-метилэтил)окси]бензамид
545,2
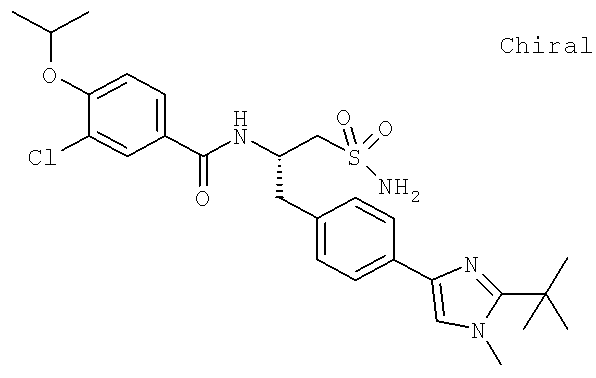
N-[(1S)-2-(Аминосульфонил)-1-({4-[2-(1,1-диметилэтил)-1-метил-1Н-имидазол-4-ил]фенил}метил)этил]-3-хлоро-4-[(1-метилэтил)окси]бензамид
548,2
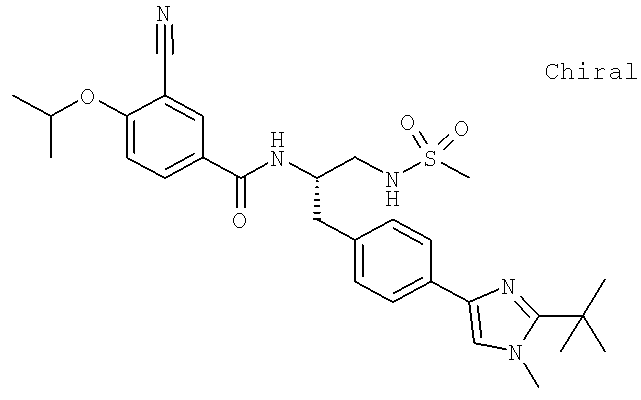
3-Циано-N-((1S)-2-{4-[2-(1,1-диметилэтил)-1-метил-1Н-имидазол-4-ил]фенил}-1-{[(метилсульфонил)амино]метил}этил)-4-[(1-метилэтил)окси]бензамид
552,4
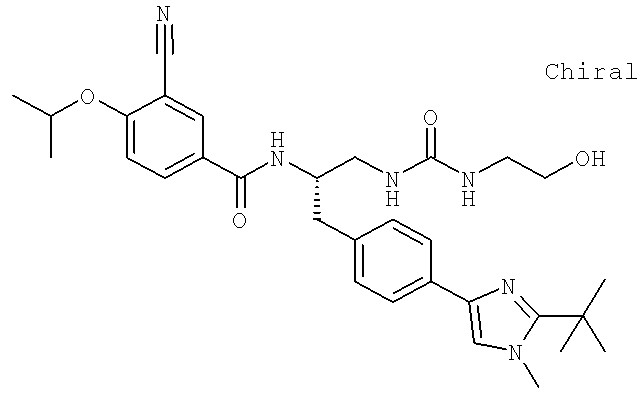
3-Циано-N-{(1S)-2-{4-[2-(1,1-диметилэтил)-1-метил-1Н-имидазол-4-ил]фенил}-1-{({[(2-гидроксиэтил)амино]карбонил}амино)метил]этил}-4-[(1-метилэтил)окси]бензамид
561,2
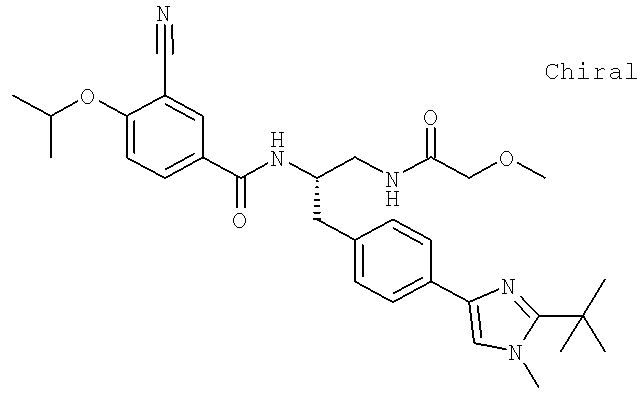
N-[(S)-1-[4-(2-терт-Бутил-1-метил-1Н-имидазол-4-ил)-бензил]-2-(2-метокси-этаноиламино)-этил]-3-циано-4-изопропокси-бензамид
546,2
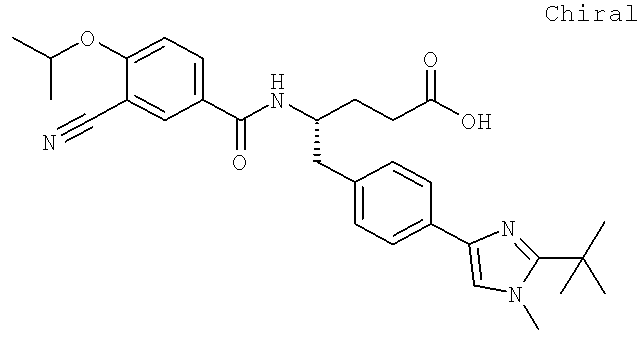
(4R)-4-[({3-Циано-4-[(1-метилэтил)окси]фенил}карбонил)амино]-5-{4-[2-(1,1-диметилэтил)-1-метил-1Н-имидазол-4-ил]фенил}пентановая кислота
517,4
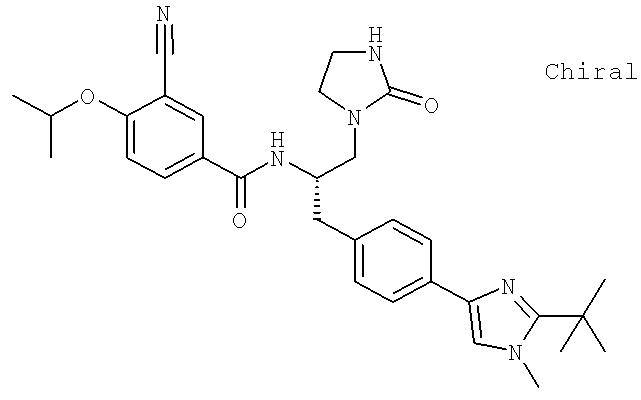
3-Циано-N-{(1S)-2-{4-[2-(1,1-диметилэтил)-1-метил-1Н-имидазол-4-ил]фенил}-1-[(2-оксо-1-имидазолидинил)метил]этил}-4-[(1-метилэтил)окси]бензамид
543,4
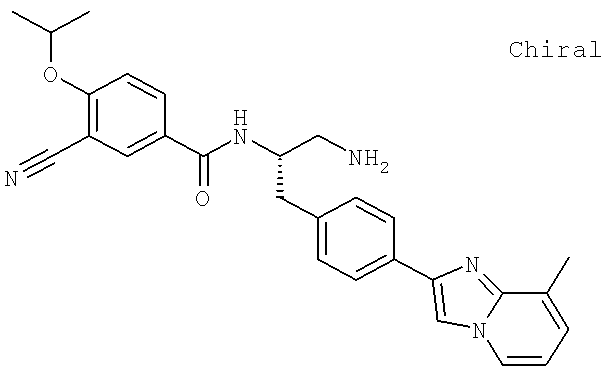
N-((1S)-2-Амино-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
468,2
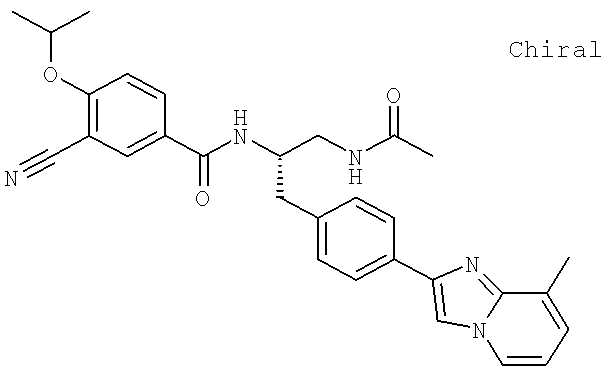
N-((1S)-2-(Ацетиламино)-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-3-циано-4-[(1-метилэтил)окси]бензамид
510,4
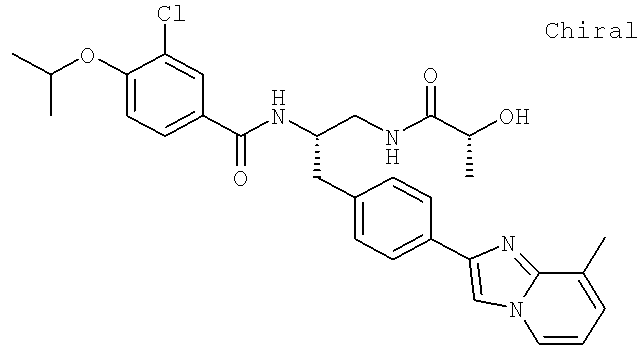
3-Хлоро-N-((1S)-2-{[(2R)-2-гидроксипропаноил]амино}-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид
549,2
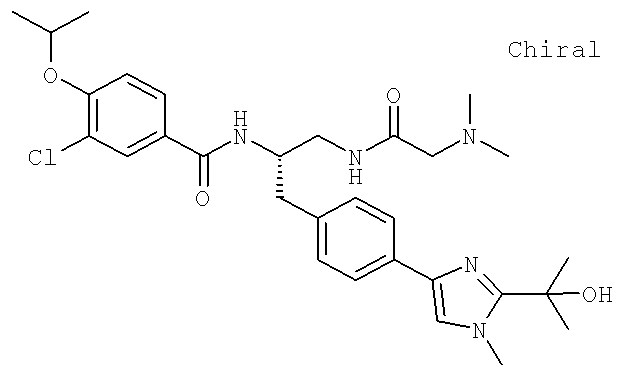
3-Хлоро-N-[(1S)-2-[N,N-диметилглицил)амино]-1-({4-[2-(1-гидрокси-1-метилэтил)-1-метил-1Н-имидазол-4-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид
570,4
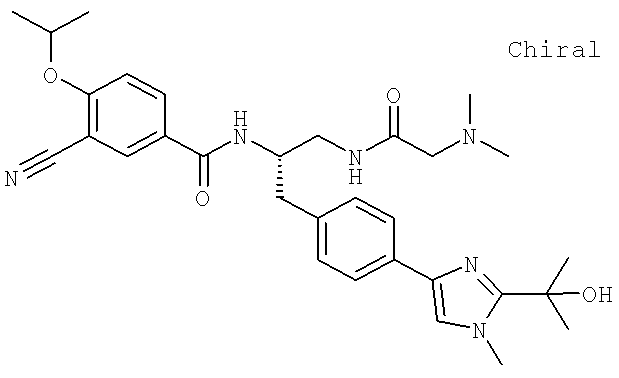
3-Циано-N-[(1S)-2-[N,N-диметилглицил)амино]-1-({4-[2-(1-гидрокси-1-метилэтил)-1-метил-1Н-имидазол-4-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид
561,4
Пример 67
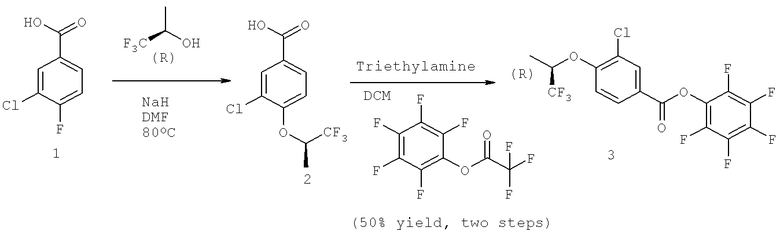
К раствору соединения 1 (10,7 г, 61,37 ммоль), (R)-1,1,1-трифторопропанола (3,5 г, 30,68 ммоль) в диметилформамиде (200 мл) при 0°C был добавлен гидрид натрия (3,7 г, 92,05 ммоль) по порциям в течение 5 минут. После 10 минут ледяная баня была удалена и реакционная смесь перемешивалась во время нагревания до комнатной температуры. Реакционная смесь была нагрета до 80°С и перемешивалась в течение ночи. Реакция контролировалась LC/MS. После выполнения реакции она была охлаждена до комнатной температуры. Реакционная смесь была погашена HCl (0,5 N, 200 мл) и экстрагирована этилацетатом (3×250 мл). Органический слой был высушен над сульфатом натрия, отфильтрован и фильтрат был сконцентрирован в условиях вакуума, дав неочищенное соединение 2 (8,2 г), которое напрямую использовалось в следующем этапе без дальнейшей очистки.
К неочищенному раствору соединения 2 (4,1 г, 15,34 ммоль), триэтиламина (6,4 мл, 56,02 ммоль) в дихлорометане (200 мл) при 0°С был добавлен пентафторофенил трифтороацетат (6, 35 мл, 36,82 ммоль) через шприц в течение 3 минут. После 5 минут ледяная баня была удалена и реакционная смесь перемешивалась во время нагревания до комнатной температуры в течение еще 2 часов. Реакционная смесь была сконцентрирована в условиях вакуума. Полученный в результате остаток был очищен флеш-хроматографией (силикагель, гексаны/этилацетат = 1:0, 50:1) для получения 3 (3,5 г, 50% выдача).
Пример 68
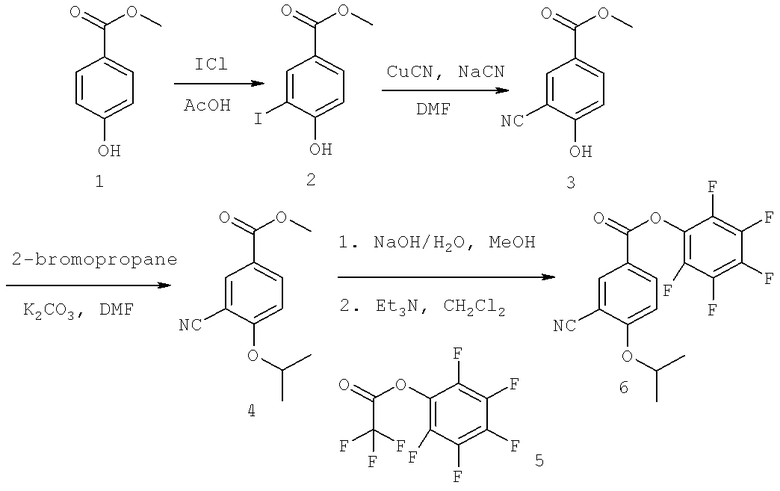
Метил-4-гидрокси-3-иодобензоат 2. Метил 4-гидроксибензоат (35,5 г, 0,233 моль) был растворен в 200 мл уксусной кислоты и перемешиваемая смесь была нагрета до 65°С. Раствор ICl (37,8 г, 0,233 моль) в 50 мл АсОН добавлялся по каплям в течение 40 минут. Смесь перемешивалась при 65°С в течение 5 часов и затем дополнительно перемешивалась в течение 16 часов при комнатной температуре. Выпавший в осадок продукт был выделен фильтрацией, промыт водой и высушен в условиях вакуума для получения 27,5 г (99% чистого при проверке LCMS и HNMR) желаемого продукта. Материнские растворы были выпарены и полученный в результате остаток был промыт водой и высушен в условиях вакуума для получения 31 г (95% чистоты при проверке LCMS и HNMR) желаемого продукта. Комбинированный выход метил 4-гидрокси-3-иодобензоата составил 58,5 г (90,3% выход).
Метил-3-циано-4-гидроксибензоат 3. 28 г (0,1 моль) метил 4-гидрокси-3-иодобензоата 2, растворенного в 100 мл DMF, было обработано 9,92 г (0,11 моль) CuCN и 0,49 г (0,11 моль) NaCN. Система была промыта азотом, после чего смесь была нагрета до 105°С и перемешивалась в течение 18 часов. Смеси было дано охладиться до комнатной температуры, и осадки были удалены фильтрацией и промыты EtOAc. Комбинированные органические слои были растворены 200 мл воды и затем экстрагированы EtOAc (2×200 мл). Комбинированные слои были высушены над сульфатом натрия, отфильтрованы и выпарены до сухости. После высушивания в условиях вакуума полученные 18 г (100% производство) 3 были охарактеризованы LCMS и HNMR.
Метил-3-циано-4-изопропоксибензоат 4. Метил-3-циано-4-гидроксибензоат 3 (18 г, 0,1 моль) были растворены в 100 мл DMF и обработаны 14,2 мл (0,15 моль) 2-бромопропана и 41,9 г (0,3 моль) безводного карбоната калия. Система была промыта азотом и смесь была нагрета до 90°С и перемешивалась в течение ночи. После охлаждения до комнатной температуры смесь была разбавлена 200 мл воды и экстрагирована с помощью CH2Cl2 (2×200 мл). Комбинированные органические слои были высушены над сульфатом натрия, отфильтрованы и выпарены до сухости для получения 20,5 г (99% производство) 4 в качестве масла, которое было охарактеризовано LCMS и HNMR.
Перфторофенил 3- циано-4-изопропоксибензоат 6. 20,5 г (0,093 моль) метил 3-циано-4-изопропоксибензоата 4 было растворено в 200 мл 6:4 смеси метанола и воды. К этому было добавлено 5,61 г (0,14 моль) NaOH, и смесь перемешивалась в течение 2 часов при комнатной температуре. Раствор был затем отфильтрован через силикагелиевую пробку и растворители были удалены в условиях вакуума. Полученное в результате твердое вещество было повторно растворено в 200 мл CH2Cl2 и обработано 19,3 мл (0,11 моль) перфторофенил 2,2,2-трифороацетата 5 и 19,5 мл (0,14 моль) триэтиламина. После перемешивания в течение ночи раствор был отфильтрован и любые твердые вещества промыты CH2Cl2. Комбинированные органические смеси были прогнаны через короткую колонку с силикагелем и затем выпарены до сухости для получения 29 г (83,5% выход) 6, который был охарактеризован LCMS и HNMR.
Пример 69
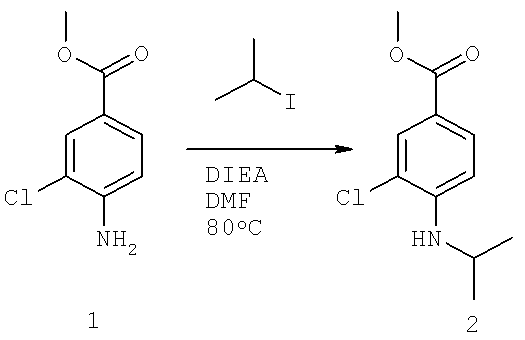
К раствору соединения 1 (200 мг, 1,077 ммоль) и 2-иодопропана (322 мкл, 3,23 ммоль) в DMF (10 мл) был добавлен DIEA (750 мкл, 4,31 ммоль). Реакиционная смесь была нагрета до 80°С и перемешивалась в течение ночи. Реакиця контролировалась LC/MS. После выполнения реакции она была охлаждена до комнатной температуры. Реакицонная смесь была погашена HCl (0,5 N, 30 мл) и экстрагирована этилацетатом (50 мл × 3). Органический слой был высушен над сульфатом натрия, сконцентрирован и высушен при высоком вакууме. Полученный в результате остаток был очищен обратнофазовой хроматографией (используя смесь ацетонитрила и воды) для получения соединения 2 (50 мг, 20%).
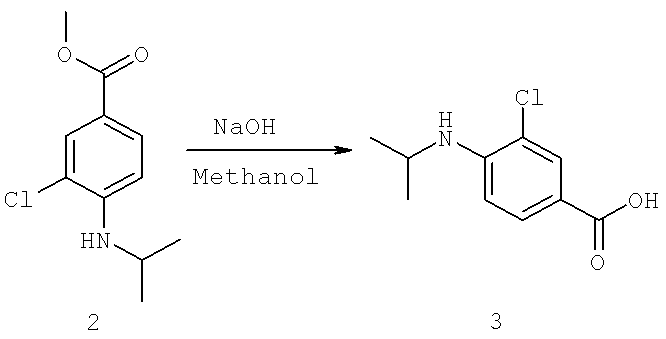
К раствору соединения 2 (50 мг, 0,22 ммоль) в МЕОН был добавлен NaOH в воде (1,0 М, 330 мкл, 0,330 ммоль). Реакционная смесь перемешивалась при температуре окружающей среды в течение 2 часов. Реакция контролировалась LC/MS. Реакционная смесь была погашена HCl (0,5 N, 5 мл) и экстрагирована этилацетатом (10 мл × 3). Органический слой был высушен над сульфатом натрия и сконцентрирован для получения 2 (45 мг). LRMS (M-H+) m/z 212,0.
Пример 70
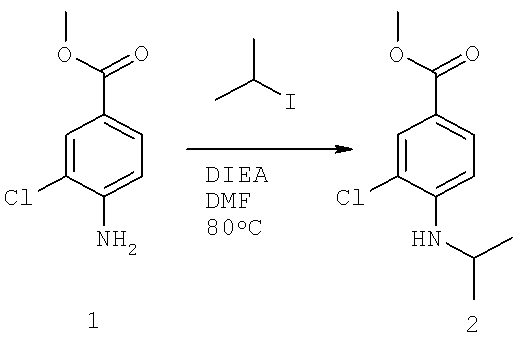
К раствору соединения 1 (200 мг, 1,077 ммоль) и 2-иодопропана (322 мкл, 3,23 ммоль) в DMF (10 мл) был добавлен DIEA (750 мкл, 4,31 ммоль). Реакционная смесь была нагрета до 80°С и перемешивалась в течение ночи. Реакция контролировалась LC/MS. После выполнения реакции она была охлаждена до комнатной температуры. Реакционная смесь была погашена HCl (0,5 N, 30 мл) и экстрагирована этилацетатом (50 мл × 3). Органический слой был высушен над сульфатом натрия, сконцентрирован и высушен при высоком вакууме. Полученный в результате остаток был очищен обратнофазовой хроматографией с использованием смеси ацетонитрила и воды для получения 2 (50 мг, 20%).
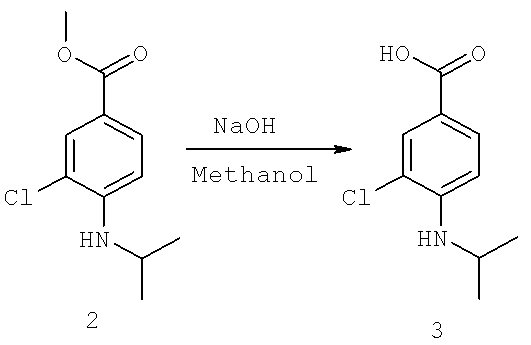
К раствору соединения 2 (50 мг, 0,22 ммоль) в МЕОН (1,0 мл) был добавлен NaOH в воде (1,0 М, 330 мкл, 0,330 ммоль). Реакционная смесь перемешивалась при температуре окружающей среды в течение 2 часов. Реакция контролировалсь LC/MS. Реакционная смесь была погашена HCl (0,5 N, 5 мл) и экстрагирована этилацетатом (10 мл × 3). Органический слой был высушен над сульфатом натрия и сконцентрирован для получения 2 (45 мг). LRMS (M-H+) m/z 212,0.
Пример 71
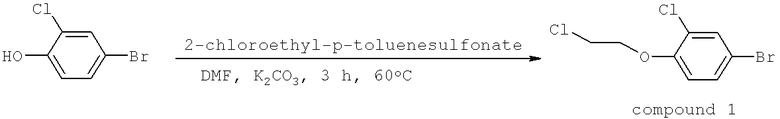
4-Бромо-2-хлорофенол (5,04 г, 24,3 ммоль) был растворен в DMF (30 мл) и был добавлен K2CO3 (10,10 г, 72,9 ммоль), за которым последовал 2-хлороэтил-р-толуолсульфонат (4,86 мл, 26,7 ммоль). Полученная в результате смесь нагревалась до 60°С и затем была охлаждена до комнатной температуры. Реакция была разбавлена EtOAc (350 мл) и промыта водой (5×150 мл). Органическая фаза была высушена (Na2SO4) и сконцентрирована до вязкого масла, которое затвердело до белого твердого вещества в условиях высокого вакуума. Соединение 1 (6,46 г, 24,1 ммоль, количественный выход) было охарактеризовано с использованием 1H NMR и использовалось в последующем этапе без дальнейшей очистки.
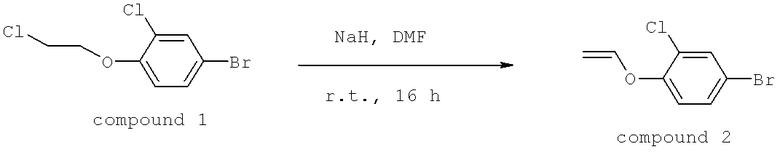
Соединение 1 (6,46 г, 24,1 ммоль) было растворено в DMF (30 мл) и был добавлен гидрид натрия (1,94 г от 60% дисперсии в минеральном масле, 48,6 ммоль). Полученная в результате смесь перемешивалась при комнатной температуре в течение 16 часов. Реакция была разбавлена водой (100 мл) и EtOAc (350 мл). Слои были разделены и органический слой был промыт водой (4×150 мл). Органическая фаза была высушена (Na2SO4) и сконцентрирована до белого твердого вещества. Соединение 2 (5,56 г, 24,0 ммоль, количественный выход) было высушено в условиях высокого вакуума и охарактеризовано с использованием 1Н NMR. Оно использовалось в последующем этапе без дальнейшей очистки.
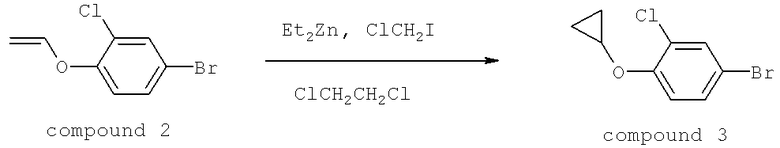
Соединение 2 (5,56 г, 24,0 ммоль) было объединено с хлороиодометаном (5,59 мл, 76,8 ммоль) и растворено в 1,2-дихлороэтане (35 мл) в атмосфере азота. Раствор был охлажден до 0°С ледяной баней и через 10 минут был добавлен диэтиловый цинк (38,4 мл, 1,0 М в гексанах, 38,4 ммоль). Полученная в результате смесь перемешивалась в течение 30 минут и была доведена до комнатной температуры. Она была охлаждена до 0°С ледяной баней и был добавлен насыщенный водный NH4Cl (150 мл), за которым последовал концентрированный водный NH4OH (25 мл) и EtOAc (200 мл). Слои были разделены и водная фаза экстрагирована дополнительным EtOAc (2×100 мл). Органические фазы были объединены, высушены (Na2SO4) и сконцентрированы до неочищенного масла, которое было очищено с использованием силикагеля (100% гексаны). Соединение 3 (1,76 г, 7,2 ммоль, 30% выход) было бесцветным маслом, которое было охарактеризовано с использованием 1Н NMR.
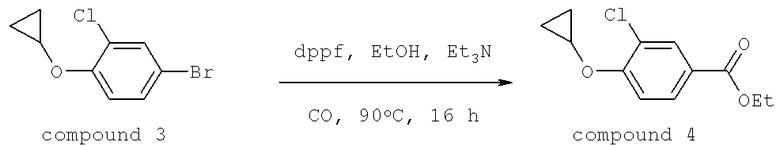
В реакторе высокого давления соединение 3 (1,76 г, 7,2 ммоль) было растворено в EtOH (40 мл). Был добавлен триэтиламин (5,0 мл, 35,8 ммоль), за чем последовал [1,1-бис (дифенилфосфино)ферроцене]дихлоропалладий (II) (188 мг, 0,36 ммоль). Реакционный сосуд был герметизирован моноокисью углерода (100 фунт/кв.дюйм), эвакууирован и повторно герметизирован моноокисью углерода (100 фунт/кв.дюйм). Сосуд был эвакууирован и затем вновь герметизирован моноокисью углерода (350 фунт/кв.дюйм). Реакция была нагрета до 90°С и перемешивалась в течение 16 часов. Она была охлаждена до комнатной температуры, разгерметизирована и отфильтрована через целит. Растворители были выпарены и оставшийся остаток был разделен между дихлорометаном (150 мл) и 1М водного KHSO4 (75 мл). Слои были разделены и органическая фаза была промыта дополнительным 1М водного KHSO4 (1×75 мл). Органическая фаза была высушена (Na2SO4) и сконцентрирована до масла, которое было очищено с использованием силикагеля (EtOAC/гексаны), дав соединение 4 (648 мг, 2,70 ммоль, 38% выход) в качестве белого вещества, охарактеризованного с использованием 1Н NMR.
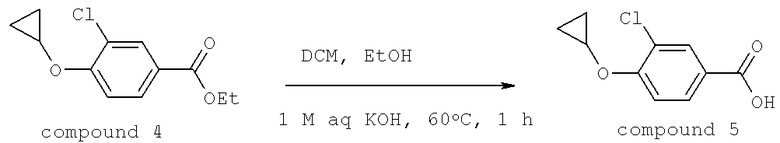
К раствору соединения 4 (648 мг, 2,70 ммоль) в дихлорометане (3 мл) и EtOH (15 мл) было добавлено 1М водного КОН (7 мл, 7 ммоль). Полученная в результате дымчатая смесь была нагрета до 60°С в течение 1 часа. Дихлорометан и EtOH были выпарены при пониженном давлении и оставшийся водный раствор был окислен с использованием концентрированной HCl. Полученный в результате осадок был отфильтрован. После фильтрации белое твердое вещество было чистым соединением 5 (506 мг, 2,39 ммоль, 88% выход), и оно было охарктеризовано с использованием LC/MS (LRMS (M-H) 211,1 m/z).
Пример 72
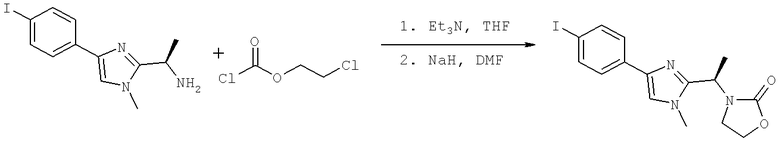
К раствору амина (580 мг, 1,7 ммоль) и триэтиламина (449 мкл, 3,4 ммоль, 2 эквивалента) в THF (8,5 мл, 0,2 М) был добавлен хлороэтил хлороформат (278 мкл, 2,6 ммоль, 1,5 эквивалента). Смесь перемешивалась в течение 30 минут при комнатной температуре. Затем она была разбавлена этилацетатом, промыта 1 N HCl и соляным раствором. Органический слой был высушен, отфильтрован и сконцентрирован в условиях вакуума для получения неочищенного вещества в качестве желтого масла (900 мг). К раствору неочищенного вещества в DMF (10 мл) был добавлен NaH (272 мг, 6,8 ммоль, 4 эквивалента) и смесь перемшивалась при комнатной температуре в течение 16 часов. Смесь была разбавлена в этилацетате (100 мл) и промыта соляным раствором (5×50 мл). Органический слой был высушен, отфильтрован и сконцентрирован в условиях вауума для получения неочищенного вещества в качестве масла. Очистка колоночной хроматографией (1:1 этилацетат : гексаны) дала 800 мг (24%) желаемого продукта, m/z (+1)=398,0.
Пример 73
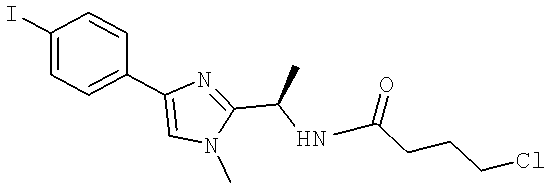
(R)-4-Хлоро-N-(1-(4-(4-иодофенил)-1-метил-1Н-имидазол-2-ил)этил)бутанамид. В 100 мл колбу с круглым основанием был добавлен (R)-бензил-1-(4-(4-иодофенил)-1-метил-1Н-имидазол-2-ил)этилкарбамат (1,50 г, 3,27 ммоль, 1,0 эквивалент), CH3CN (20 мл) и TMSI (900 мкл, 6,3 ммоль, 1,9 эквивалента). Реакционная смесь была закрыта крышкой и перемешивалась в течение 2 часов. Затем в колбу был добавлен метанол (40 мл) и смесь была сконцентрирована, растворена в EtOAc (100 мл) и промыта водой. Органический слой был высушен над Na2SO4, отфильтрован и сконцентрирован. Остаток был растворен в DCM и очищен хроматографией с силикагелем (35-60% CH3CN/CH2Cl2, затем 20% МеОН/CH2Cl) для получения 950 мг (90%) желаемого первичного амина в качестве масла. (М+Н (m/z)=328).
К данному амину были добавлены CH2Cl2 (20 мл) и пиридин (260 мкл, 1,1 эквивалента), за чем последовал 4-хлоробутирил хлорид (344 мкл, 1,05 эквивалента), который добавлялся по каплям. Реакция перемешивалась в течение 15 минут, за чем последовало добавление EtOAc (50 мл) и воды (10 мл). Органический слой был отделен, высушен над Na2SO4, отфильтрован и сконцентрирован. Остаток был растворен в DCM и очищен хроматографией с силикагелем (5-35% CH3CN/CH2Cl2) для получения 747 мг (60%) (R)-4-хлоро-N-(1-(4-(4-иодофенил)-1-метил-1Н-имидазол-2-ил)этил)бутанамида в качестве беловатого твердого вещества. (М+Н (m/z)=432).
Пример 74
(R)-1-(1-(4-(4-Иодофенил)-1-метил-1Н-имидазол-2-ил)этил)пирролидин-2-он. В 20-граммовую пробирку был добавлен (R)-4-хлоро-N-(1-(4-(4-иодофенил)-1-метил-1Н-имидазол-2-ил)этил)бутанамид и THF (10 мл). Пробирка была затем охлаждена до 0°С в атмосфере азота и был добавлен t-бутоксид калия (214 мг, 1,91 ммоль). Реакция перемешивалась в течение 1,5 часов. К реакционной смеси был добавлен EtOAC (50 мл) и вода (10 мл). Органический слой был отделен, высушен над Na2SO4, отфильтрован и сконцентрирован. Остаток был растворен в DCM и очищен хроматографией с силикагелем (5-50% CH3CN/CH2Cl2 для получения 593 мг (86%) (R)-1-(1-(4-(4-иодофенил)-1-метил-1Н-имидазол-2-ил)этил)пирролидин-2-она в качестве белого твердого вещества. (М+Н (m/z)=396).
Пример 75
К раствору 1 (10 г, 45,7 ммоль) в DMF (150 мл) был добавлен HBTU (26 г, 68,5 ммоль) и диметилгидроксиламиновая соль HCl (5,35 г, 54,8 ммоль) и DIEA (9,6 мл, 55,0 ммоль) при 0°С. После перемешивания в течение 2 часов смесь была оставлена нагреваться до комнатной температуры. Перемешивание продолжалось в течение 2 дней. Реакционная смесь была разделена между EtOAc (500 мл) и H2O (200 мл). Органический слой был промыт NaOH (2N, 200 мл), HCl (2N, 200 мл), H2O, соляным раствором, высушен над Na2SO4 и сконцентрирован для получения 2 (9,6 г), который использовался без дальнейшей очистки. LRMS (M+H+) m/z 262,0.
К раствору 2 (9,6 г, ~36,8 ммоль) в Et2O (100 мл) был добавлен MeMgBr (3 М в Et2O, 27 мл) при 0°С. Полученная в результате смесь перемешивалась в течение 4 часов и она была доведена до комнатной температуры. Реакицонная смесь была погашена насышенным NH4Cl (100 мл). Органический слой был промыт H2O, соляным раствором, высушен над Na2SO4 и сконцентрирован для получения 3 (7 г, 71% от 1), который был охарактеризован NMR.
К раствору 3 (6,5 г, 30 ммоль) в DCM (200 мл) и МеОН (100 мл) был добавлен трибромид тетрабутиламмония (14,5 г, 30 ммоль). Реакционная смесь перемешивалась в течение 14 часов. Смесь была сконцентрирована и высушена в условиях высокого ваккума для получения 5 (охарактеризованного NMR), который использовался в последующем этапе без дальнейшей очистки.
К растовору 4 (5 г, ~16,9 ммоль) в DCM (50 мл) был добавлен гексаметилентетрамин (2,6 г, 18,5 ммоль). Реакционная смесь перемешивалась в течение 2 часов. Смесь была разбавлена DCM (500 мл). Осадок был собран, промыт DCM (500 мл × 2) и высушен в условиях высокого вакуума. К полученому в результате остатку был добавлен EtOH (60 мл) и сконцентрирован HCl (30 мл). Реакционная смесь перемешивалась в течение 2 часов. Смесь была сконцентрирована, высушена для получения 5, который использовался без дальнейшей очистки. LRMS (M+H+) m/z 231,9.
К раствору неочищенного 5 (~16,9 ммоль) в диоксане (50 мл) были добавлены NaOAc (6,93 г, 84,5 ммоль), НОАс (4,8 мл, 84,5 ммоль) и 5.1 (5,93 г, 84,5 ммоль). После одного часа реакционная смесь была нагрета до 80°С и перемешивалась в течение 3 часов. Реакционная смесь была разделена между EtOAc (500 мл) и насыщенным NaHCO3 (200 мл). Водный слой был экстрагирован с помощью EtOAc (300 мл × 2). Комбинированные органические слои были промыты соляным раствором, высушены над Na2SO4 и сконцентрированы. Полученный в результате остаток был очищен на силикагеле (гексан/EtOAc, 1:0, 1:2, 1:1, 0:1) для получения 6 (1,2 г, 23% от 4). LRMS (M+H+) m/z 312,9.
Пример 76
К раствору этил тиооксамата (10,0 г, 75 ммоль) в дихлорметане (400 мл) был медленно добавлен тетрафтороборат триметилоксония (13,1 г, 89 ммоль) при 0°С. После 10 минут ледяная баня была удалена и реакционная смесь перемешивалась в течение ночи. Растворитель был удален для получения 18,0 г продукта 2 в качестве белого твердого вещества, которое использовалось без дальнейшей очистки.
Смесь 2-амино-4'-бромоацетофен гидрохлорида (10,0 г, 40 ммоль), ацетата натрия (16, 4 г, 200 ммоль), уксусной кислоты (11,5 мл, 200 ммоль) и соединения 2 (19,2 г, 80 ммоль) в диоксане (70 мл) перемешивалась при 65°С пока TLC не показало, что больше не осталось соединения 2 (приблизительно 2 часа). Реакционная смесь была осторожно нейтрализована насыщенным раствором NaHCO3 и была экстрагирована этилацетатом. Органический раствор был высушен над Na2SO4 и сконцентрирован. Очистка колоночной флеш-хроматографией (EtOAc : гексаны 1:1) дала продукт 3 (9,11 г, 79%) в качестве белого твердого вещества.
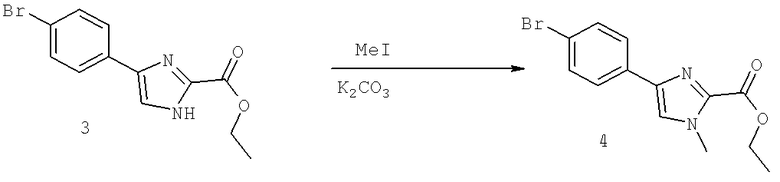
В колбе с круглым дном продукт 3 (2,00 г, 6,8 ммоль) был растворен в DMF (20 мл), за чем последовало добавление иодометана (5,1 мл, 10,1 ммоль) и K2CO3 (1,4 г, 10,1 ммоль). Смесь была оставлена перемешиваться при 60°С в течение 3 часов, поко это не было завершено TLC. Раствор был погашен соляным раствором, три раза экстрагирован с помощью EtOAc, высушен над сульфатом натрия и сконцентрирован. Очистка колоночной флеш-хроматографией с использованием EtOAc : гексаны 1:1 дала 1,381 г (66% выход) продукта 4.

К раствору соединения 3 (3, 174 г, 10,8 ммоль) в DMF (15 мл) был добавлен K2CO3 (4,478 г, 32,4 ммоль) и (2-бромоэтокси)-терт-бутилдиметилсилан (2,780 мл, 13,0 ммоль). Полученная в результате смесь перемешивалась при 55°С в течение ночи. Раствор был сконцентрирован, разбавлен водой и экстрагирован с помощью EtOAc (3×50 мл). Органические слои были объединены и высушены над Na2SO4. Растворитель был удален для получения вязкого масла (4,805 г, 10,6 ммоль, 98,4%), которое использовалось в последующем этапе без дальнейшей очистки.
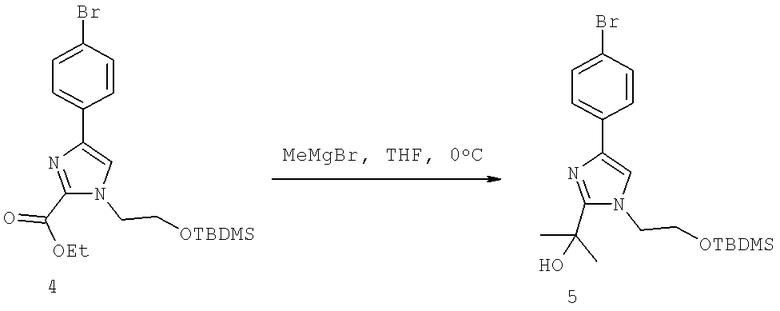
К раствору соединения 4 (2,174 г, 4,8 ммоль) в безводном THF (25 мл) по каплям был добавлен бромид метилмарганца (4,8 мл, 3М в диэтил эфире, 14,4 ммоль) в атмосфере азота при 0°С. Реакция перемешивалась в течение 15 минут. Реакция была осторожно погашена насыщенным раствором хлорида аммония (5 мл) и водой (30 мл) и экстрагирована EtOAc (3×50 мл). Органические слои были объединены, высушены над Na2SO4 и сконцентрированы для получения неочищенного масла. Очистка на колоночной флеш-хроматографии (15% EtOAc/гексаны) дала искомый продукт 5 (1,371 г, 65%) в качестве белого аморфного твердого вещества.
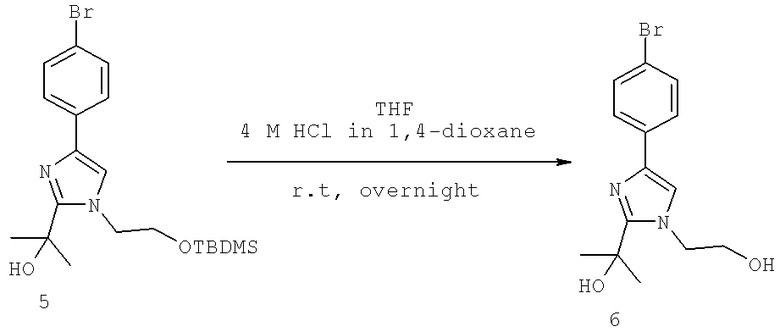
К раствору соединения 5 (1,371 г, 3,1 ммоль) в THF (5 мл) было добавлено 35 мл HCl (4М в 1,4-диоксане). Полученный в результате раствор перемешивался при комнатной температуре в течение ночи. Растворители были удалены для получения продукта 6 (1,0 г, 99%) в качестве белого твердого вещества.
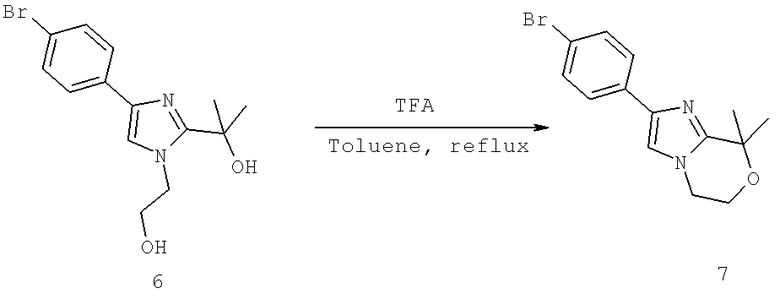
Смесь соединения 6 (0,5 г, 1,54 ммоль) и 1 мл TFA в толуоле (60 мл) дефлегмировалась в течение ночи. Твердое вещество 6 не растворялось до достижения практически точки кипения толуола. Растворитель был удален. Остаток был разбавлен EtOAc, промыт водным раствором NaHCO3, высушен над Na2SO4 и сконцентрирован. Очистка колоночной флеш-хроматографией (EtOAc : гексаны) дала продукт 7 (0,348 г, 74%) в качестве белого твердого вещества.
Пример 77
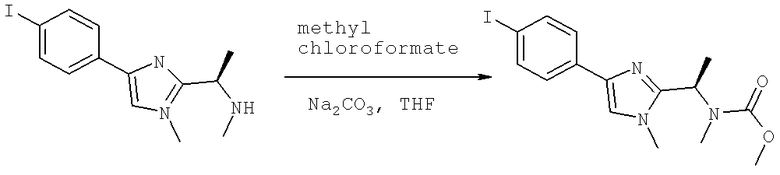
(R)-метил 1-(4-(4-иодофенил)-1-метил-1Н-имидазол-2-ил)этил(метил)карбамат. В 250 мл колбу с круглым дном был добавлен (R)-1-(4-(4-иодофенил)-1-метил-1Н-имидазол-2-ил)-N-метилэтанамин (3,1 г, 9,1 ммоль), метил хлороформат (0,84 мл, 10,9 ммоль), Na2CO3 (1,15 г, 10,9 ммоль) и THF (100 мл). Реакция перемшивалась в течение 2 часов, за чем последовало добавление EtOAc (50 мл) и воды (10 мл). Органический слой был высушен над Na2SO4, отфильтрован и сконцентрирован для получения 1,50 г (41%) (R)-метил 1-(4-(4-иодофенил)-1-метил-1Н-имидазол-2-ил)этил(метил)карбамата в качестве беловатого твердого вещества. (М+Н) (m/z)=400).
Пример 78
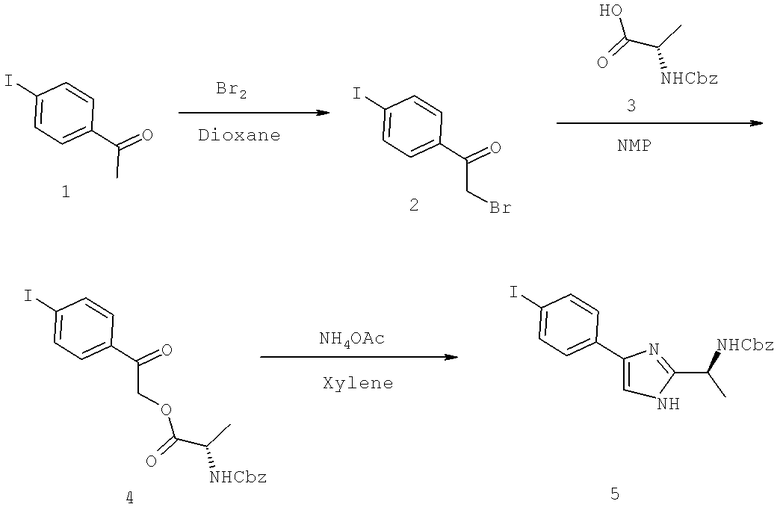
Ссылка: Журнал медицинской химии, 2001, 44, 2990-3000.
К перемешиваемому раствору p-иодоацетофенона 1 (30,0 г, 122 ммоль) в диоксане (200 мл) над ледяной баней по каплям был добавлен бром (6,56 мл, 128 ммоль). Реакционная смесь перемешивалась при комнатной температуре и контролировалась LC/MS. После завершения (около 1 часа) растворитель был выпарен ротационным вапоризатором и остаток был высушен в условиях вакуума до получения твердого вещества 2 (40 г, 100%).
(Основано на Журнале медицинской химии, 2001, 44, 2990-3000). К раствору Cbz-D-Ala-OH 3 (5,0 г, 22,4 ммоль) в NMP (100 мл) был добавлен карбонат цезия (3,72 г, 11,4 ммоль). После перемешивания при комнатной температуре в течение 1 часа был добавлен 2 (7,60 г, 22,4 ммоль). Реакционная смесь перемешивалась при комнатной температуре и контролировалась LC/MS до формирования 4. Реакционный раствор был разбавлен ксилолом (100 мл) и ацетатом амония (9,25 г, 120 ммоль) и затем перемешивался при 120°С в течение 4 часов. Может потребоваться до 50 эквивалентов ацетата аммония в зависимости от хода реакции. Ключом является постоянное наличие твердого вещества в колбе. После охлаждения до комнатной температуры реакционная смесь была разбавлена этилацетатом (200 мл). Раствор EtOAc был дважды промыт насыщенным раствором бикарбоната натрия (200 мл) и высушен сульфатом натрия, затем отфильтрован, а фильтрат был сконцентрирован при пониженном давлении. Остаток был растворен в DCM (100 мл) и перемешивался в течение 1 часа для получения осадка, а твердое вещество 5 (4,0 г) было отфильтровано и высушено в условиях вакуума. Материнский раствор был сконцентрирован ротационным вапоризатором, остаток был очищен на Bio-tage для получения 5 (гексан : EtOAC = 1:1, к EtOAc 100%). Два продукта были объединены и высушены в условиях вакуума для получения 5 (5,8 г, 58%).
Пример 79
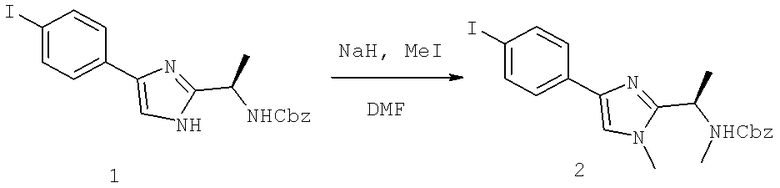
(R)-Бензил 1-(4-иодофенил)-1-метил-1Н-имидазол-2-ил)этил(метил)карбамат 2. Перемешанная смесь (R)-бензил 1-(4-(4-иодофенил)-1Н-имидазол-2-ил)этилкарбомата 1 (5 г, 11 ммоль) в 55 мл DMF была охлаждена до 0°С и обработана NaH (1,33 г, 60% дисперсия в масле, 33 ммоль) небольшими порциями для избежания пенообразования. Когда образование пузырьков от последней порции прекратилось, сразу же был добавлен MeI (2,1 мл, 34 ммоль) и смесь перемешивалась дополнительно в течение 30 минут. Растворители были удалены в условиях вакуума и остаток растворен в 200 мл EtOAc. Раствор был промыт насыщенным NH4Cl (4×100 мл) и насыщенным NaCl (4×100 мл), затем отфильтрован и выпарен до сухости. Неочищенный остаток был очищен через колоночную флеш-хроматографию над силикагелем (60:40, EtOAc/гексаны) для получения 5,13 г (97% получения) 2, который был охарактеризован LCMS.
Пример 80
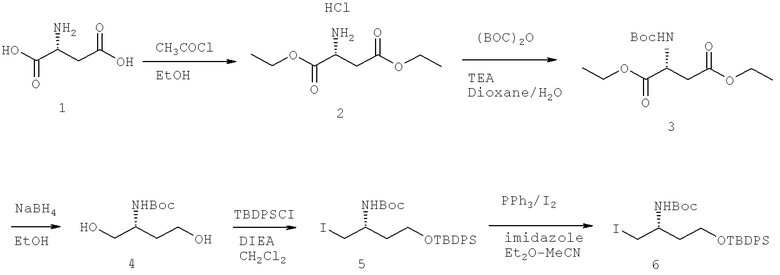
Ацетилхлорид (54,6 мл, 0,75 моль) добавлялся по каплям в этанол (316 мл) при 0-5°С. Когда добавление было завершено, ледяная баня была удалена и раствор был оставлен перемешиваться при нагревании до комнатной темепературы еще на 30 минут. Затем была добавлена D-аспаргиновая кислота 1. Реакционная смесь подвергалась нагреву в сосуде с обратным холодильником в течение 2 часов. Реакционный раствор затем был сконцентрирован в условиях вакуума и помещен под высокий вакуум (0,4 мм Hg) на ночь. Было получено соединение 2 в качестве белого твердого вещества (42 г, 99%) и оно непосредственно использовалось в следующем этапе.
(BOC)2O (44,7 г, 0,21 моль) добавлялся по порциям в течение 10 минут при 0°С к раствору соединения 1 (42 г, 0,19 моль), триметиламина (51,9 мл, 0,37 моль), диоксана (140 мл) и воде (56 мл). Еще после 10 минут ледяная баня была удалена и реакционная смесь перемешивалась при нагревании до комнатной температуры еще в течение 2 часов. Реакционная смесь была разбавлена этилацетатом (150 мл) и промыта 0,5 N HCl (200 мл × 3). Органический слой был высушен над сульфатом магния, отфильтрован и фильтрат был сконцентрирован в условиях вакуума, дав соединение 3 (52 г, выход 97%), которое напрямую использовалось в следующем этапе.
NaBH4 (54,4 г, 1,44 моль) добавлялся по порциям в течение 30 минут к раствору соединения 3 (952 г, 86,4 ммоль) и этанола при 0°С. Реакционная смесь была чрезвычайно экзотермичной и следует быть очень осторожным во время добавления редуцирующего агента. После того, как добавление было завершено, реакицонная смесь нагревалась в противотоке в течение 1 часа. Раствор был охлажден до температуры окружающей среды и реакционная смесь затвердевала. Твердое вещество было разбито до шлама, который затем был добавлен в соляной раствор (250 мл). Полученная в результате смесь была отфильтрована и фильтрат сконцентрирован в условиях вакуума. Полученный в результате остаток был активно перемешен с эфиром (200 мл × 5). Органические слои были последовательно отделены от остатка. Комбинированные экстракты эфира были высушены над сульфатом магния, отфильтрованы и фильтрат был сконцентрирован в условиях вакуума для получения соединения 4 в качестве твердого белого вещества (25,2 г, выход 68%). [Примечание: выход был 89% при выполнении на 25 г (соединение 3)].
t-Бутилфенилхлоросилан (31,9 мл, 0,123 моль) был добавлен к раствору соединения 4 (25,2 г, 0, 123 моль), диизопропилэтиламина (42,8 мл, 0, 245 моль) и CH2Cl2 (500 мл). Реакционный раствор перемешивался при температуре окружающей среды в течение 24 часов. Реакционный раствор был затем промыт 0,5 N HCl (150 мл × 3) и соляным раствором (150 мл). Органический слой был высушен над сульфатом магния, отфильтрован и фильтрат был сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен флеш-хроматографией (силикагель, 4:1 гексаны : EtOAc) до получения соединения 5 (42 г, выход 77%). [Примечание: выход был 85% при выполнении на 15 г (соединения 4)].
Иод (24 г, 94,7 ммоль) добавлялся по порциям в течение 15 минут к 0°С раствору соединения 5 (28 г, 63,1 ммоль), Ph3P (24,8 г, 94,7 ммоль), имидазола (6,4 г, 94,7 ммоль), диэтил эфира (450 мл) и ацетонитрила (150 мл). Ледяная баня была удалена и реакционный раствор был оставлен нагреваться до температуры окружающей среды в течение 30 минут. Реакция была завершена судя по данным анализа TLC (4:1 гексаны : EtOAc). Реакция была погашена водой (400 мл). Слои были отделены и водный слой был экстрагирован диэтил эфиром (100 мл). Комбинированные органические слои были промыты насыщенным водным Na2SO3 (100×2) и соляным раствором (100 мл). Органический слой был высушен над сульфатом магния, отфильтрован и фильтрат был сконцентрирован в условиях вакуума. Полученный в результате остаток был очищен колоночной флеш-хроматографией (силикагель, 4:1 гексаны : EtOAc) до получения соединения 6 (33 г, 92%).
Пример 81
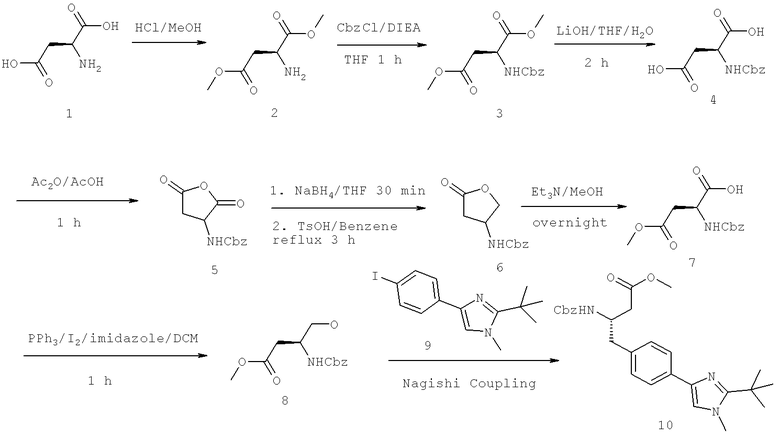
Под ледяной баней к раствору D-аспаргиновой кислоты 1 (59 г, 0,376 моль) в метаноле (200 мл) был барботирован газ HCl в течение 10 минут. После того как реакция перемешивалась при комнатной температуре в течение ночи, растворитель был выпарен. Полученный в результате остаток был высушен под вакуумом для получения продукта 2 в качестве соли HCl (0, 376 моль).
К перемешиваемому раствору 2 (0,376 моль), DIEA (196 мл, 1,13 моль) и THF (200 мл) был по каплям добавлен бензил хлороформат (59,0 мл, 0,414 моль). После того, как реакционный раствор был подвергнут перемешиванию при комнатной температуре в течение 1 часа, раствор был сконцентрирован на ротационном вапоризаторе. Затем остаток был растворен в растворе NaHCO3 (300 мл) и экстрагирован с помощью DCM (100 мл × 3). Комбинированный раствор DCM был высушен над сульфатом натрия, отфильтрован и фильтрат был сконцентрирован в условиях вакуума для получения продукта 3 (0,376 моль).
К раствору 3 (0,376 моль), THF (200 мл) и воде (100 мл) была добавлена гидроокись лития (31,6 г, 0,752 моль) и они были подвергнуты перемешиванию в течение 2 часов. Реакционная смесь была отфильтрована через пробку из силикагеля (рН фильтрата был около 7) и сконцентрирована. Остаток был высушен в условиях вакуума для получения 4 (0, 376 моль).
После того, как раствор 4 (0,376 моль) и уксусный ангидрид (200 мл) перемешивались в течение 1 часа, реакционная смесь была сконцентрирована. Остаток был высушен в условиях вакуума для получения 5 (0,376 моль).
Под ледяной баней к раствору 5 (0,376 моль) и THF (1000 мл) в течение 30 минут добавлялся борогидрид натрия (14,2 г, 0,376 моль) в течение 30 минут и они перемешивались в течение 3 часов. Затем раствор HCl (4N) по каплям вводился в реакционный раствор до тех пор, пока рН не составил около 2. Раствор был сконцентрирован до тех пока, пока не осталось около четверти, разбавлен водой (300 мл) и экстрагирован этил эфиром (200 мл × 3). Комбинированный раствор эфира был высушен над сульфатом натрия, отфильтрован и фильтрат был сконцентрирован в условиях вакуума. Полученный в результате остаток был растворен в бензоле (300 мл) и был добавлен TsOH (500 мг). Затем реакционная смесь перемешивалась в сосуде с обратным холодильником в течение 3 часов. Раствор был сконцентрирован до около 100 мл и был добавлен эфир (200 мл) для образования осадка. Белое твердое вещество 6 (57,5 г, 65% от 1) было отфильтровано, промыто эфиром и высушено в условиях вакуума.
К раствору 6 (30,0 г, 0,128 моль) и метанола (200 мл) был добавлен триэтиламин (142 мл, 1,02 моль) и они перемешивались в течение ночи. Реакционная смесь была сконцентрирована. Остаток был высушен в условиях вакуума для получения 7 (анализ LC/MS показал, что осталось около 20 мол.% 6), который напрямую использовался в следующем этапе.
Под ледяной баней раствор соединения 7 (0,128 моль), Ph3P (50,4 г, 0,192 моль), имидазола (13,1 г, 0,192 моль) и DCM (300 мл) перемешивался в течение 10 минут. По порциям был добавлен иод (48,7 г, 0,192 моль) в течение 15 минут. Ледяная баня была удалена и реакционный раствор перемешивался при комнатной температуре в течение 1 часа. Твердое вещество было отфильтровано. Фильтрат был промыт насыщенным водным Na2SO3 (200 мл × 2) и соляным раствором (200 мл). Органический слой был высушен над сульфатом натрия, отфильтрован и фильтрат был сконцентрирован. Полученный в результате остаток был очищен колоночной флеш-хроматографией (гексаны : EtOAc 4:1 по 1:1) до получения соединения 8 (27,5 г, 59,2% от 6) в качестве белого твердого вещества.
Смесь порошка цинка (Strem, 10,1 г, 0,154 моль) и DMF (15 мл) очищалась в атмосфере азота в течение 10 минут и к ней был добавлен 1,2-дибромоэтан (0,758 мл, 8,80 ммоль). Смесь нагревалась тепловой пушкой в течение около 2 минут, охлаждалась в течение 5 минут и снова нагревалась тепловой пушкой и была охлаждена до комнатной температуры. К смеси был добавлен TMSCl (281 мкл, 2,20 ммоль). После того как смесь перемешивалась в течение 30 минут, был добавлен 8 (9,98 г, 26,5 ммоль). После 1 часа анализ LCMS показал полное потребление 8. К вышеупомянутому реакционному раствору был добавлен иодид арила 9 (7,50 г, 22,0 ммоль), Pd2(dba)3 (50,4 мг, 0,55 ммоль) и три-о-толилфосфат (670 мг, 2,20 ммоль). Реакционная смесь поддерживалась при 50°С в течение 1 часа (контролируемая анализом LC-MS). Реакционная смесь была напрямую загружена через пробку из силикагеля и промыта гексанами : EtOAc (3:1 по 1:1) для получения соединения 10 (5,0 г, 49%).
Пример 82
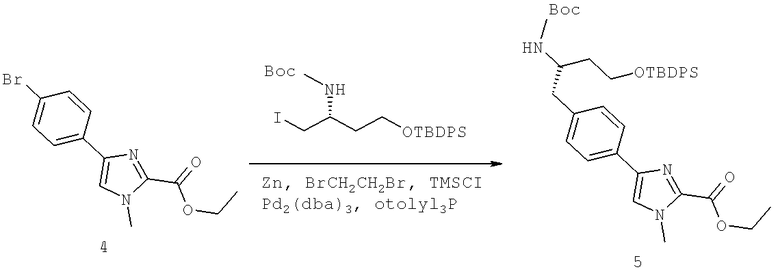
К высушеной в печи колбе с круглым дном был добавлен порошок цинка (1753 мг, 27 ммоль), за которым последовала DMF (15 мл). Колба была закрыта крышкой и прочищена азотом в течение 10 минут. К раствору был добавлен 1,2-дибромоэтан (0,139 мл, 1,6 ммоль). Смесь затем нагревалась с использованием тепловой пушки в течение 30 секунд до тех пор, пока не стал появляться газ, указывая на активацию цинка. Смесь была оставлена охлаждаться при помешивании в течение 1 минуты перед тем, как она была снова нагрета с использованием тепловой пушки до появления газа. Затем смесь была оставлена охлаждаться до комантной температуры, за чем последовало добавление TMSCl (0,042 мл, 0,33 ммоль) и перемешивание в течение 30 минут. Реактив А был затем растворен в DMF, барботирован азотом, добавлен к раствору цинка и был подвергнут перемешиванию в течение 1 часа при комнатной температуре. Бромид 4 (1,381 г, 4,5 ммоль) был растворен в DMF, барботирован с азотом и затем был впрыснут в раствор, за чем последовало добавление Pd2(aba)3 (102 мг, 0,11 ммоль) и три-о-толилфосфина (136 мг, 0,44 ммоль). Смесь была пробарботирована азотом и выдержана в атмосфере азота при перемешивании в течение одного часа при комнатной температуре. После 1 часа перемешиваемый раствор нагревался до 40°С в течение 2 часов, пока это не было завершено TLC и LC/MS. Раствор был погашен с использованием соляного раствора и пять раз экстрагирован с использованием EtOAc. Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы. Полученный в результате неочищенный продукт 5 был очищен колоночной флеш-хроматографией с использованием EtOAc : гексанов 1:1 для получения 2,0 г (70% выход) чистого продукта 5.
Пример 83
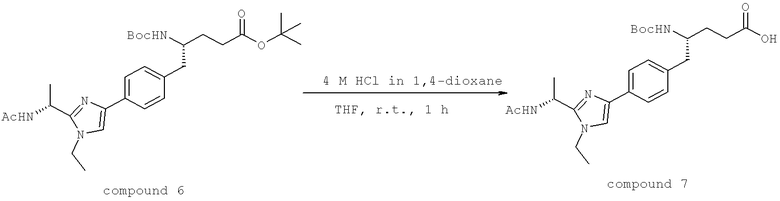
К раствору соединения 6 (940 мг, 1,78 ммоль) в THF (5 мл) была добавлена 4 М HCl в 1,4-диоксане (20 мл, 80 ммоль). Полученная в результате смесь перемешивалась при комнатной температуре в течение 1 часа. Растворители были выпарены и оставшееся соединение 7 (726 мг, 1,78 ммоль, количественный выход) было тщательно высушено под высоким вакуумом. Соединение 7 было охарактеризовано с использованием LCMS (LRMS (МН) 373 m/z) и использовалось в последующем этапе без дальнейшей очистки.
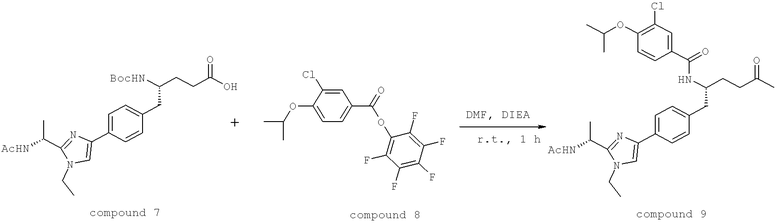
К раствору соединения 7 (662 мг, 1,62 ммоль) в DMF (5 мл) был добавлен DIEA (930 мкл, 5,34 ммоль) и соединение 8 (678 мг, 1,78 ммоль). Полученный в результате раствор перемешивался при комнатной температуре в течение 1 часа. DMF была выпарена и неочищенный остаток напрямую очищен с использованием препаративного HPLC для получения соединения 9 (435 мг, 0,77 ммоль, выход 48%), которое было охарактеризовано 1Н NMR и LC/MS (LRMS (МН) 569 m/z).
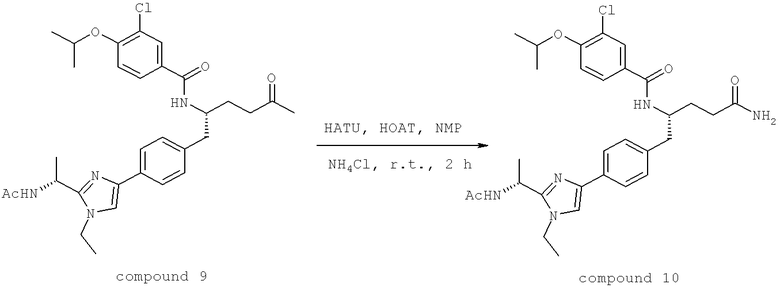
Соединение 9 (100 мг, 0,176 ммоль) было объединено с HATU (134 мг, 0,352 ммоль), НОАТ (48 мг, 0,352 ммоль) и NH4Cl (50 мг, 0,944 ммоль). Твердые вещества были растворены в N-метилпирролидиноне (5 мл) и был добавлен DIEA (93 мкл, 0,528 ммоль). Полученная в результате смесь перемешивалась при комнатной температуре в течение 2 часов и затем напрямую очищена препаративным HPLC для получения соединения 10 (16 мг, 0,028 ммоль, выход 16%) в качестве стекловидного твердого вещества, которое было охарактеризовано 1Н NMR и LC/MS (LRMS (МН) 568 m/z).
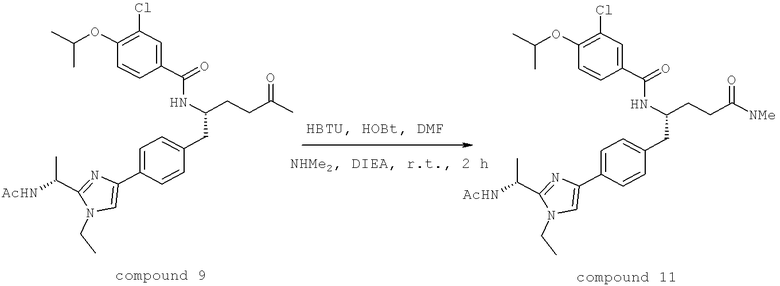
Соединение 9 (91 мг, 0,160 ммоль) было объединено с HBTU (121 мг, 0,320 ммоль) и HOBt (43 мг, 0,320 ммоль). Твердые вещества были растворены в DMF (3 мл) и диметиламине (400 мкл, 0,800 ммоль, 2М в THF) и был добавлен DIEA (93 мкл, 0,528 ммоль). Полученный в результате раствор перемешивался при комнатной температуре в течение 2 часов. Неочищенный продукт был напрямую очищен препаративным HPLC
для получения соединения 11 (25 мг, 0,042 ммоль, выход 25%) в виде стекловидного тведого вещества, которое было охарактеризовано 1Н NMR и LC/MS (LRMS (MH) 596 m/z).
Пример 84
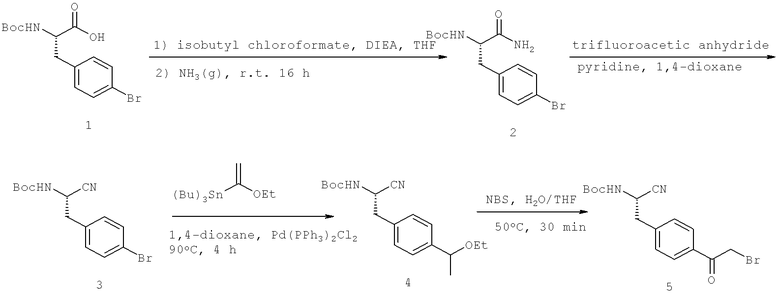
При 0°С к раствору 1 (40,2 г, 117 ммоль) в THF (250 мл) и DIEA (11,4 мл, 175 ммоль) был добавлен изобутил хлороформат (21,2 мл, 163 ммоль). Полученная в результате смесь перемешивалась при комнатной температуре в течение 3 часов. Реакция очищалась газообразным аммиаком в течение 1 часа и затем перемешивалась при комнатной температуре в течение 16 часов. Она была разбавлена водой (200 мл), этилацетатом (200 мл) и отфильтрована. Белое, отфильтрованное твердое вещество было искомым продуктом. Дополнительный продукт был получен переносом двухфазного фильтрата в делительную воронку с разделением слоев. Водная фаза была экстрагирована дополнительным этилацетатом (3×150 мл). Органические фазы были объединены, высушены (Na2SO4) и сконцентрированы до белого твердого вещества, которое было рекристаллизовано из этилацетата для получения искомого продукта. Чистый продукт 2 (20,6 г, 60 ммоль) был охарактеризован 1H-NMR и LC/MS (LRMS (МН) 343,1 m/z).
Амид 2 (18,1 г, 53 ммоль) был суспендирован в 1,4-диоксане (200 мл) и пиридине (10,7 мл, 132 ммоль). Был добавлен трифтороуксуный ангидрид (22,0 мл, 158 ммоль) и белое суспендированное твердое вещество немедленно растворилось. Однородный раствор перемешивался при комнатной температуре в течение 30 минут. Растворители были удалены при пониженном давлении, оставшийся остаток был растворен в этилацетате (200 мл) и промыт 1 М водного KHSO4 (2×100 мл) и насыщенным водным NaHCO3 (1×100 мл). Органическая фаза была высушена над Na2SO4 и сконцентрирована в условиях вакуума. Оставшийся искомый продукт 3 (14,9 г, 46 ммоль) был определен как достаточно чистый для следующей трансформации. (LC/MS (LRMS (МН) 198,0 m/z).
Нитрил 3 (14,9 г, 46 ммоль) был растворен в 1,4-диоксане (100 мл) и был добавлен трибутил (1-этоксивинил)тин, за чем последовал Pd(PPh3)2Cl2 (1,6 г, 5 мол.%). Полученная в результате смесь была нагрета до 90°С и перемешивалась в течение 4 часов. Она была охлаждена до комнатной температуры и растворители были удалены при пониженном давлении. Оставшийся остаток был напрямую очищен с использованием силикагеля, приготовленного в шламе с использованием 95% гексана/триэтиламина.
Элюирование происходило по этапам, начиная с 95% гексана/триэтиламина и изменяясь до 50% этилацетата/гексана-5% триэтиламина. Искомый продукт 4 был элюирован последней мобильной фазой и являлся вязким маслом желтого цвета, охарактеризованным LC/MS (LRMS (МН) 317,1 m/z). Продукт немедленно использовался в последующей трансформации.
К раствору винил эфира 4 (14,5 г, 46 ммоль) в THF (60 мл) и воде (20 мл) был добавлен N-бромосукцинимид (12,3 г, 69 ммоль). Полученная в результате смесь перемешивалась при 50°С в течение 30 минут. Она была охлаждена до комнатной температуры и разбавлена 2М водного Na2CO3. Смесь была экстрагирована этилацетатом (2×150 мл), органические экстракты были комбинированы, высушены над Na2SO4 и сконцентрированы до аморфного твердого вещества, которое было очищено с использованием силикагеля (дихлорметан/этилацетат). Искомый продукт 5 (10,6 г, 29 ммоль) был желтым твердым веществом, охарактеризованным 1H-NMR и LC/MS (LRMS (МН) 239,9 m/z).
Пример 85
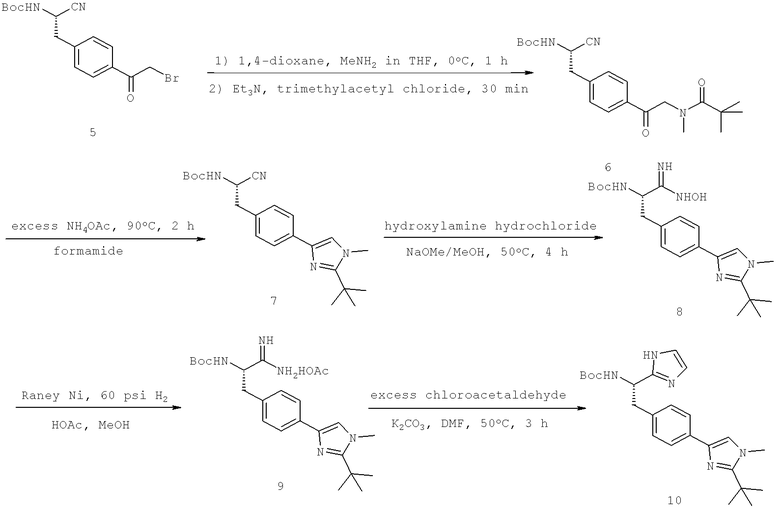
Раствор кетона 5 (10,6 г, 29 ммоль) в 1,4-диоксане (50 мл) капался в раствор матиламина (72 мл, 144 ммоль, 2 М в THF) в течение 45 минут при 0°С. Полученный в результате дымчатый раствор дополнительно перемешивался в течение 15 минут при комнатной температуре. THF и метиламин были выпарены при пониженном давлении, и была приложена осторожность, чтобы не выпарить 1,4-диоксан. К полученной в результате смеси при комнатной температуре был добавлен триэтиламин (12 мл, 87 ммоль), за чем последовал триметилацетил хлорид (15 мл, 144 ммоль). Полученная в результате суспензия перемешивалась при комнатной температуре в течение 30 минут. Она была разбавлена водой (125 мл) и экстрагирована этилацетатом (3×100 мл). Органические фазы были объединены, высушены над Na2SO4 и сконцентрированы в условиях вакуума. Неочищенный продукт 6 был охарактеризован LC/MS (LRMS (МН) 402,1 m/z) и использовался далее без дальнейшей очистки.
Амид 6 (11,6 г, 29 ммоль) был объединен с ацетатом аммония (55 г, 723 ммоль) и формамидом (150 мл). Полученная в результате смесь была нагрета до 100°С и перемешивалась в течение 3 часов. Она была охлаждена до комнатной температуры, разбавлена этилацетатом (500 мл) и промыта водой (3×200 мл). Органическая фаза была высушена над Na2SO4 и сконцентрирована при пониженном давлении. Оставшийся неочищенный остаток был очищен с использованием силикагеля (диэтил эфир/гексаны) для получения 7 в качестве пенистого желтого твердого вещества, охарактеризованного 1H-NMR и LC/MS (LRMS (МН) 383,2 m/z).
Имидазол 7 (1,316 г, 3,4 ммоль) был объединен с гидроксиламин гидрохлоридом (478 мг, 6,9 ммоль) и растворен в растворе метоксида натрия в метаноле (14 мл, 6,9 ммоль, 0,5 М). Полученный в результате раствор премешивался при 50°С в течение 4 часов. Он был сконцентрирован при пониженном давлении и напрямую был очищен с использованием силикагеля (5% метанол/дихлорметан) для получения искомого амидоксима 8 (913 мг, 2,2 ммоль) в качестве белого твердого вещества, охарактеризованного LC/MS (LRMS (МН) m/z: 416,1).
К раствору амидоксима 8 (652 мг, 1,6 ммоль) в метаноле (10 мл) был добавлен ренейский никель (скелетный никелевый катализатор гидрирования) (50 мг) и уксусная килота (250 мкл). Смесь перемешивалась при комнатной температуре при 60 фунт/кв. дюйм Н2 в течение 3 часов и затем была отфильтрована через слой целита. Концентрация при пониженном давлении дала чистый амид 9 в качестве белого твердого вещества (638 мг, 1,6 ммоль), охарактеризованного LC/MS (LRMS (MH) m/z: 400,2).
Хлороацетальдегид (360 мл, 5,7 ммоль) был добавлен к раствору амида 9 (283 мг, 0,71 ммоль) в DMF (4 мл) и K2CO3 (195 мг, 1,4 ммоль). Смесь была нагрета до 50°С и перемешивалась в течение 4 часов. Реакция была отфильтрована и напрямую очищена обратнофазовой HPLC с использованием градиента мобильной фазы, состоящего из ацетонитрила и воды. Чистый продукт 10 (25 мг, 0,06 ммоль) был стекловидным твердым веществом, охарактеризованным 1H NMR и LC/MS (LRMS (MH) m/z: 424,1).
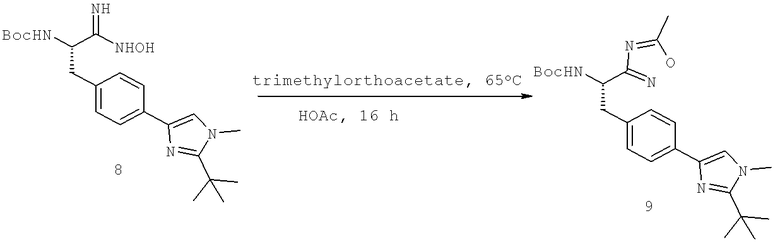
К раствору амидоксима 8 (148 мг, 0,35 ммоль) в триметилортоацетате (5 мл) была добавлена ледяная уксусная кислота (100 мкл). Полученный в результате раствор перемешивался при 65°С в течение 16 часов. Растворители были выпарены и остаток был напрямую очищен силикагелем (5% метанол/дихлорометан) для получения искомого продукта 9 в качестве стекловидного твердого вещества, охарактеризованного LC/MS (LRMS (MH) m/z: 440,1.
Пример 86
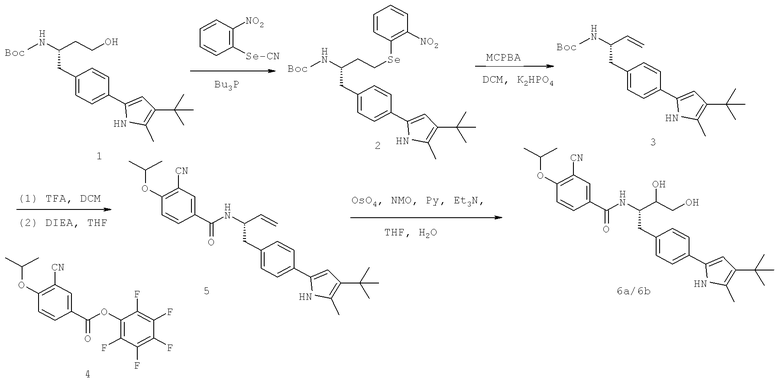
К раствору 1 (200 мг, 0,5 ммоль) в THF (3 мл) был добавлен Bu3P (150 мкл, 0,6 ммоль) и 2-нитрофенилселеноцианат (136 мг, 0,6 ммоль) при комнатной температуре. Реакционная смесь перемешивалась в течение 14 часов. Смесь была разделена между EtOAc (200 мл) и H2O (50 мл). Органический слой был промыт соляным раствором, высушен над Na2SO4 и сконцентрирован. Полученный в результате остаток использовался без дальнейшей очистки. LRMS (MH+) m/z, 587,1.
К раствору 2 (~0,5 ммоль) в DCM (5 мл) был добавлен водный KH2PO4 (2 М, 1 мл) и МСРВА (77%, 135 мг, 0,6 ммоль). Полученная в результате смесь перемешивалась в течение 6 часов. Реакционная смесь была погашена насыщенным Na2S2O3 (10 мл). Органический слой был промыт насыщенным NaHCO3, H2O, соляным раствором, высушен над Na2SO4 и сконцентрирован. Остаток был очищен на RP-HPLC с использованием смеси ацетонитрила и Н2О для получения 3 (150 мг, 65% от 1). LRMS (MH+) m/z 384,2.
К раствору 3 (150 мг, 0,39 ммоль) в DCM (8 мл) был добавлен TFA (1 мл). Реакционная смесь перемешивалась в течение 4 часов. Смесь была сконцентрирована и высушена под высоким вакуумом. К полученному в результате остатку (90 мг, 0,32 ммоль) в THF (4 мл) был добавлен DIEA (165 мкл, 0,95 ммоль) и 4 (140 мг, 0,38 ммоль). Полученная в результате смесь перемешиваласьв течение 14 часов. Реакционная смесь была сконцентрирована. Остаток был очищен на RP-HPLC с использованием смеси ацетонитрила и H2O для получения 5 (120 мг, 65%). LRMS (MH+) m/z 471,2.
К раствору 5 (90 мг, 0,19 ммоль) в THF/H2O (2 мл/2 мл) был добавлен OsO4 (4,8 мг, 0,019 ммоль), NMO (117 мг, 0,95 ммоль) и пиридин (1,5 мкл, 0,019 ммоль). Полученная в результате смесь перемешивалась в течение 6 часов. Был добавлен NaHSO3 (300 мг). Реакционная смесь была сконцентрирована. Полученное в результате твердое вещество было промыто EtOAc (100 мл × 3). Фильтрат был сконцентрирован. Полученный в результате остаток был очищен на препаративной пластине TLC (силикагель, 5:1 EtOAc. MeOH) для получения диастероизомеров 6а (23 мг, 24%) и 6b (2 мг, 2%). LRMS (MH+) m/z 505,2.
Пример 87
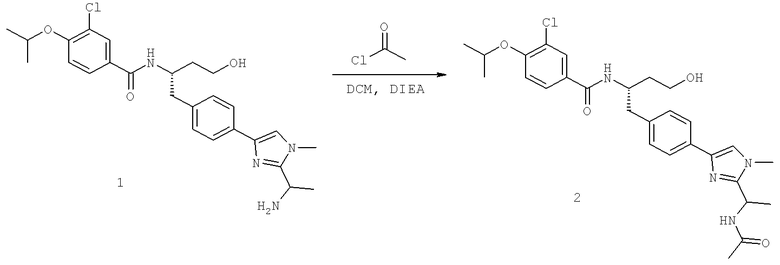
К раствору амина 1 (150 мг, 0,309 ммоль), DCM (2 мл) и DIEA (53,8 мкл, 0,309 ммоль) был добавлен ацетилхлорид (53,8 мкл, 0,309 ммоль). Полученный в результате раствор перемешивался при комнатной температуре в течение 10 минут. Растворитель был выпарен и оставшийся остаток был очищен на обратнофазовой Prep-HPLC (ацетонитрил/вода) для получения 2 (43,7 мг, 26,8%). MS (MW+1): 527,2.
Пример 88
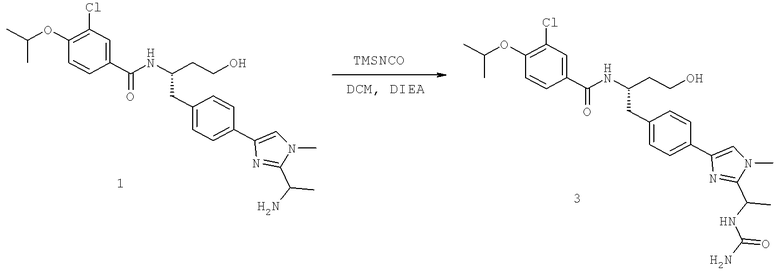
К раствору амина 1 (150 мг, 0,309 ммоль), DCM (2 мл) и DIEA (53,8 мкл, 0,309 ммоль) был добавлен триметилсилил изоцианит (35,6 мкл, 0,309 ммоль). Полученный в результате раствор перемешивался при комнатной температуре в течение ночи. Растворитель был выпарен и оставшийся остаток был очищен на обратнофазовой Prep-HPLC (ацетонитрил/вода) для получения 3 (30,3 мг, 18,6%). MS (MW+1): 528,2.
Пример 89
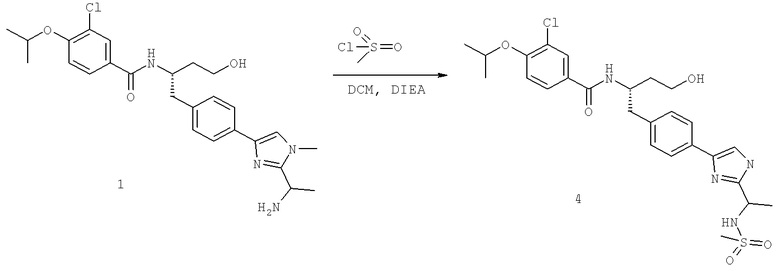
К раствору амина 1 (150 мг, 0,309 ммоль), DCM (2 мл) и DIEA (53,8 мкл, 0,309 ммоль) был добавлен метансульфонил хлорид (24 мкл, 0,309 ммоль). Полученный в результате раствор перемешивался при комнатной температуре в течение 30 минут.
Растворитель был выпарен и оставшийся остаток был очищен на обратнофазовой Prep-HPLC (ацетонитрил/вода) для получения 4 (18,4 мг, 10,6%). MS (MW+1): 563,1.
Пример 90
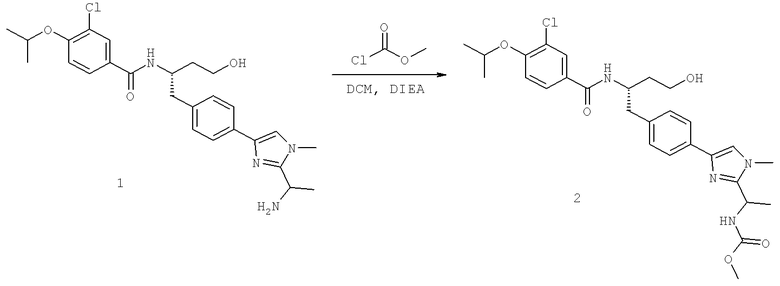
К раствору амина 1 (150 мг, 0,309 ммоль), DCM (2 мл) и DIEA (53,8 мкл, 0,309 ммоль) был добавлен метил хлороформат (24 мкл, 0,309 ммоль). Полученный в результате раствор перемешивался при комнатной температуре в течение 30 минут. Растворитель были выпарен и оставшийся остаток был очищен на обратнофазовой Prep-HPLC (ацетонитрил/вода) для получения 5 (СК1828648) (25,7 мг, 15,3%). MS (MW+1): 543,1.
Пример 91
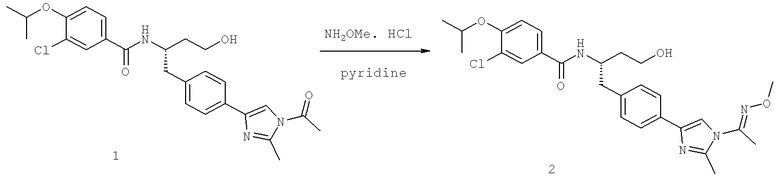
(S)-3-Хлоро-N-(4-гидрокси-1-(4-(2-(1-(метоксиимино)этил)-1-метил-1Н-имидазол-4-ил)фенил)бутан-2-ил)-4-изопропоксибензамид 2. 80 мг (0,031 ммоль) (S)-N-(1-(4-(2-ацетил-1-метил-1Н-имидазол-4-ил)фенил)-4-гидроксибутан-2-ил)-3-хлоро-4-изопропоксибензамида в 2 мл пиридина были обработаны 27,6 мг (0,033 ммоль) гидпрохлоридом гидроксиламин метил эфира. Реакция перемешивалась в течение ночи, после чего растворители были выпарены и оставшийся остаток был очищен на обратнофазовой HPLC (ацетонитрил/вода). Было получено 11,2 мг (70% выход) 2 и охарактеризовано LCMS и HNMR.
Пример 92
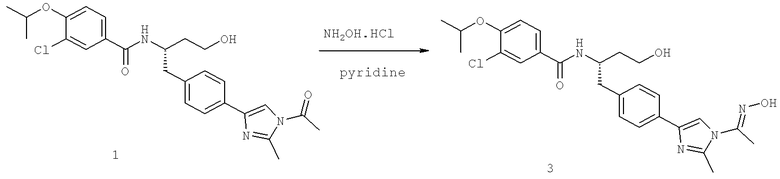
(S)-3-Хлоро-N-(4-гидрокси-1-(4-(2-(1-(гидроксиимино)этил)-1-метил-1Н-имидазол-4-ил)фенил)бутан-2-ил)-4-изопропоксибензамид 3. 100 мг (0,21 ммоль) (S)-Н-(1-(4-(2-ацетил-1-метил-1Н-имидазол-4-ил)фенил)-4-гидроксибутан-2-ил)-3-хлоро-4-изопропоксибензамида в 2 мл пиридина было обработано 71,8 мг (1,0 ммоль) гидроксиламин гидрохлорида. Реакция перемешивалась в течение ночи, после чего растворители были выпарены и оставшийся остаток был очищен на обратнофазовой HPLC (ацетонитрил/вода). Было получено 69,7 мг (67% выход) 3 и охарактеризовано LCMS и HNMR.
Пример 93
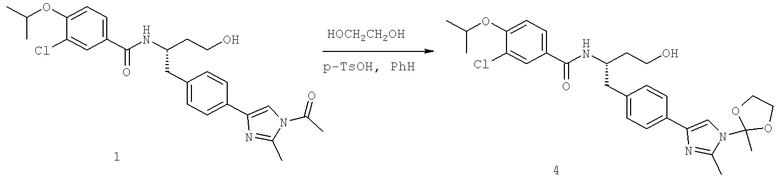
(S)-3-Хлоро-N-(4-гидрокси-1-(4-(1-метил-2-(2-метил-1,3-диоксолан-2-ил)-1Н-4-ил) фенил)бутан-2-ил)-4-изопропоксибензамид 4. 150 мг (0,31 ммоль) (S)-N-(1-(4-(2-ацетил-1-метил-1Н-имидазол-4-ил)фенил)-4-гидроксибутан-2-ил)-3-хлоро-4-изопропоксибензамида в 2 мл бензола были обработаны 34,6 мкл (0,62 ммоль) этан-1,2-диола и 59 мг (0,31 ммоль) моногидрата р-толуолсульфоновой кислоты. Реакция перемешивалась при 70°С в течение 2 часов, после чего растворители были выпарены и оставшийся остаток был очищен на обратнофазовой HPLC (ацетонитрил/вода). Было получено 25,5 мг (16% выход) 4 и охарактеризовано LCMS и HNMR.
Пример 94
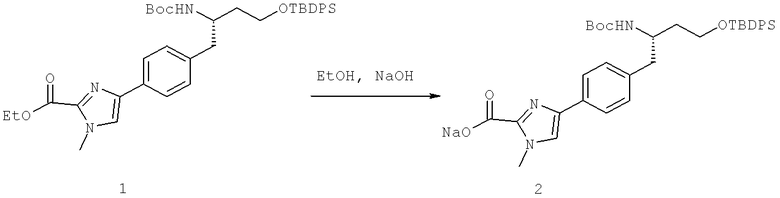
К раствору 1 (1,5 г, 2,29 ммоль) в этаноле (5,0 мл) был добавлен NaOH в воде (1,0 М, 3,7 мл, 2,80 ммоль). Реакционная смесь перемешивалась при температуре окружающей среды в течение 2 часов. После выполнения реакции она была сконцентрирована для получения 2 (1,49 г), который напрямую использовался в следующем этапе без дальнейшей очистки.
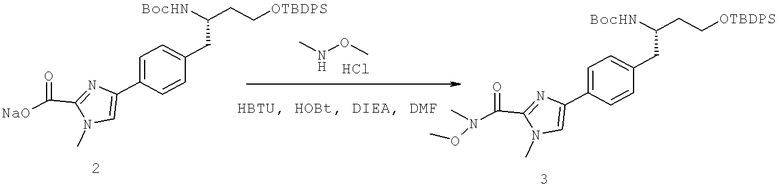
К раствору 2 (1,49 г, 2,29 ммоль), HBTU (1,3 г, 3,44 ммоль), HOBt (530 мг, 3,44 ммоль) и N,О-диметилгидроксиламиновой соли HCl (340 мг, 3,44 ммоль) в DMF (20 мл) был добавлен DIEA (785 мкл, 4,58 ммоль). Полученная в результате смесь перемешивалась при комнатной температуре в течение 2 часов. Реакционная смесь была сконцентрирована. Полученный в результате остаток был очищен с использованием силикагеля (гексаны/EtOAc = 1:3) для получения соединения 3 (1,20 г, 78%) в качестве беловатого пенистого твердого вещества.
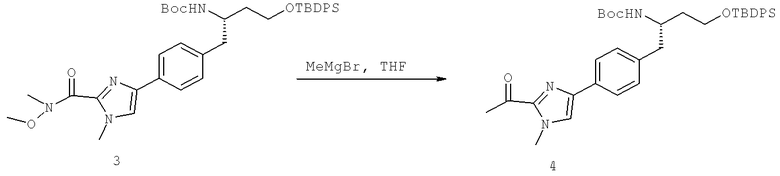
При 0°С к раствору 3 (1,20 г, 1,79 ммоль) в безводном THF (20 мл) был добавлен бромид метилмагния (3М в Et2O, 2,38 мл). Реакционная смесь перемешивалась при 0°С в течение 1 часа. Реакционная смесь была погашена насыщенной NH4Cl (5 мл) и водой (20 мл). Был добавлен EtOAc (50 мл) и слои были разделены. Водная фаза была экстрагирована дополнительным EtOAc (50 мл × 2). Органические фазы были объединены, высушены (Na2SO4) и сконцентрированы до неочищенного масла, которое было очищено с использованием силикагеля (50% EtOAc/гексаны). Искомое соединение 4 (0,83 г, 73%) было вязким маслом, которое стало белым пенистым твердым веществом во время сушки в условиях высокого вакуума.
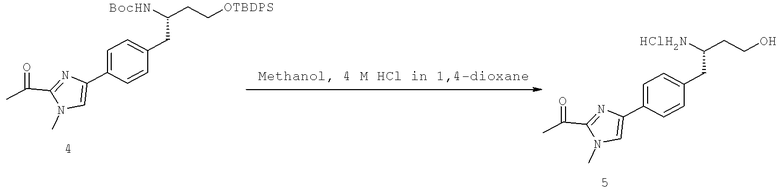
К раствору 4 (0,82 г, 1,31 ммоль) в метаноле (10 мл) была добавлена HCl (4 М в 1,4-диоксане, 20 мл). Реакционная смесь перемешивалась при комнатной температуре в течение ночи. Смесь была сконцентрирована и высушена в условиях высокого вакуума для получения 5, которое использовалось в последующем этапе без дальнейшей очистки.
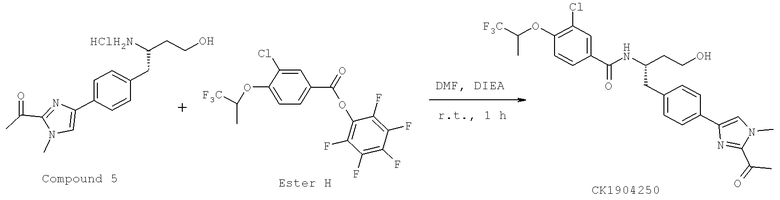
К раствору 5 (350 мг, 1,09 ммоль) в DMF (5 мл) был добавлен DIEA (280 мкл, 1,63 ммоль) и эфир Н (472 мг, 1,09 ммоль). Полученный в результате раствор перемешивался при комнатной температуре в течение 1 часа. Неочищенный раствор был отфильтрован и затем очищен обратнофазовой хроматографией (используя смесь ацетонитрила и воды) для получения СК 1904250 в качестве пенистого белого вещества (400 мг, 68%). LRMS (MH+) m/z 538,1.
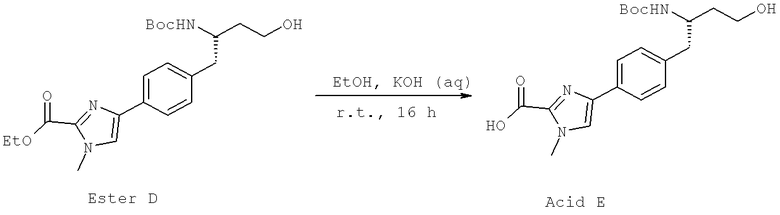
Эфир D (10,2 г, 24,5 ммоль) был растворен в EtOH (150 мл) и воде (50 мл). Была добавлена гидроокись калия (4,1 г, 73,5 ммоль) и реакция перемешивалась при комнатной температуре в течение ночи. Реакционная смесь была охлаждена до 0°С и нейтрализована концентрированной HCl. Была предпринята большая осторожность, чтобы не позволить рН стать <7 во время нейтрализации. Растворители были выпарены в условиях вакуума и остаток был очищен под высоким вакуумом. Кислота Е (9,5 г, 24,5 ммоль) использовалась в следующем этапе без дальнейшей очистки.
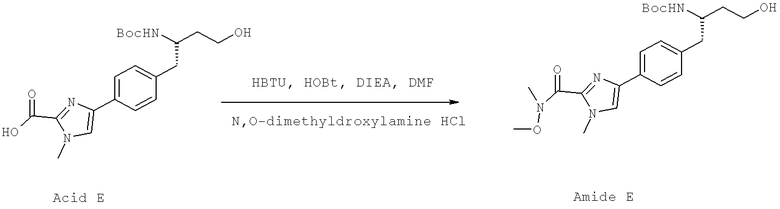
Кислота Е (9,5 г, 24,5 ммоль) была объединена с HBTU (18,5 г, 48,7 ммоль), HOBt (6,6 г, 48,7 ммоль) и N,O-диметилгидроксиламиновой HCl (4,8 г, 48,7 ммоль). К твердым веществам был добавлен DMF (150 мл) и DIEA (12,7 мл, 73,1 ммоль). Полученная в результате смесь перемешивалась при комнатной температуре в течение 4 часов. Большая часть DMF была выпарена и оставшийся остаток был разбавлен этилацетатом (300 мл) и водой (300 мл). Слои были отделены и водная фаза экстрагирована EtOAc (1×200 мл). Органические фазы были объединены, промыты насыщенным водным бикарбонатом натрия (2×250 мл) и высушены над Na2SO4. Концентрация при пониженном давлении дала неочищенный амид Е, который был очищен с использованием силикагеля (3% MeOH/DCM) для получения чистого амида Е (6,73 г, 17,4 ммоль) в качестве беловатого пенистого твердого вещества.
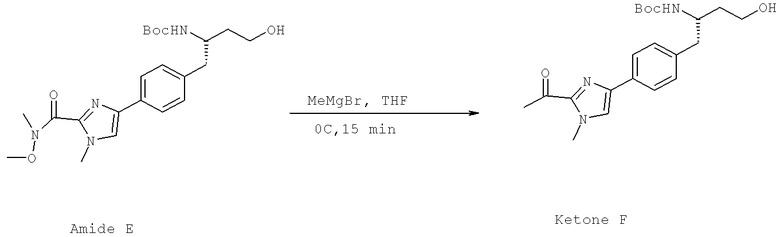
Растовор амида Е (6,73 г, 17,4 ммоль) в безводном THF (250 мл) был охлажден до 0°С ледяной баней. Был добавлен бромид метилмагния (3 М в диэтиловом эфире, 52,2 мл, 156,6 ммоль) и реакция перемешивалась при 0°С в течение 15 минут. Реакция была осторожно погашена насыщенным раствором хлорида аммония (20 мл) и водой (100 мл). Был добавлен EtOAc (200 мл) и слои были разделены. Водная фаза была экстрагирована дополнительным EtOAc (2×200 мл). Органическая фаза была объединена, высушена (Na2SO4) и сконцентрирована до неочищенного масла, которое было очищено силикагелем (50% EtOAc/гексаны). Искомый кетон F (4,45 г, 11,5 ммоль) был вязким маслом, которое стало белым пенистым твердым веществом при сушке в условиях высокого вакуума.
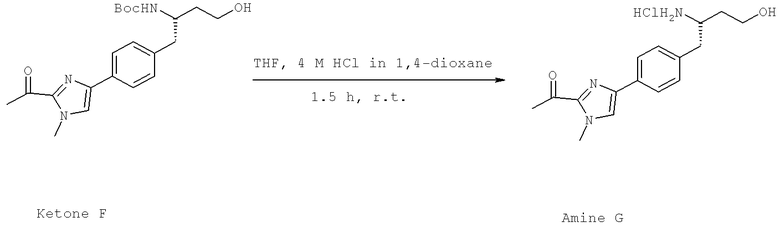
Кетон F (4,45 г, 11,5 ммоль) был растворен в THF (25 мл) и была добавлена 4 М HCl в 1,4-диоксане (75 мл). Реакция перемешивалась при комнатной температуре в течение 1,5 часов. Растворители были выпарены в условиях вакуума и остаток был тщательно высушен в условиях высокого вакуума для получения амина G. Амин G использовался в последующем этапе без дальнейшей очистки.
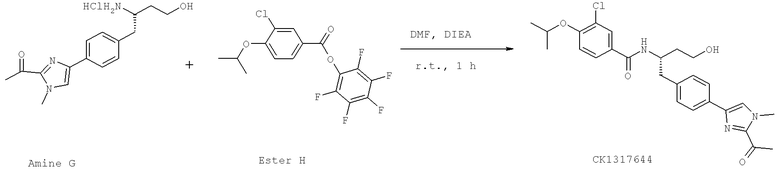
К раствору амина G (3,30 г, 11,5 ммоль) в DMF (50 мл) был добавлен DIEA (8,0 мл, 46,0 ммоль) и эфир Н (5,25 г, 13,8 ммоль). Полученный в результате раствор перемешивался при комнатной температуре в течение 1 часа. Большая часть DMF была выпарена и оставшийся остаток был разбавлен EtOAc (250 мл) и водой (200 мл). Слои были разделены, органическая фаза промыта дополнительной водой (2×150 мл) и соляным раствором (2×150 мл). Органическая фаза была высушена (Na2SO4) и сконцентрирована. Оставшееся неочищенное вязкое масло было очищено с использованием силикагеля (100% EtOAc) для получения СК 1317644 в качестве пенистого белого твердого вещества (2,98 г, 6,2 ммоль).
Пример 95
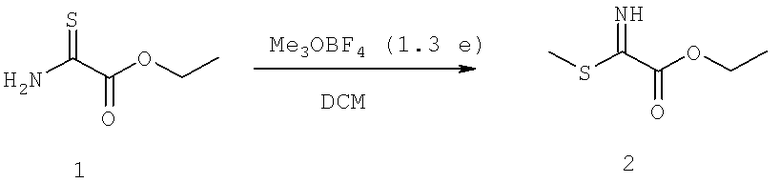
К раствору этил тиооксамата (10,0 г, 75 ммоль) в дихлорометане (400 мл) был медленно добавлен тетрафтороборат триметилоксония (13,1 г, 89 ммоль) при 0°С. После 10 минут ледяная баня была удалена и реакционная смесь перемешивалась в течение ночи. Растворитель был удален для получения 18,0 г продукта 2 в качестве белого твердого вещества, которое использовалось без дальнейшей очистки.
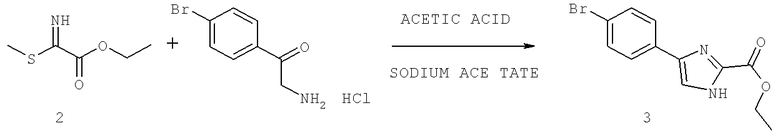
Смесь 2-амино-4'-бромоацетофен гидрохлорида (10,0 г, 40 ммоль), ацетата натрия (16,4 г, 200 ммоль), уксусной кислоты (11,5 мл, 200 ммоль) и соединения 2 (19,2 г, 80 ммоль) в диоксане (70 мл) перемешивалась при 65°С до тех пор, пока TLC не показывал на отсутствие соединения 2 (приблизительно 2 часа). Реакционная смесь была осторожно нейтрализована насыщенным раствором NaHCO3 и экстрагирована этилацетатом. Органический раствор был высушен над Na2SO4 и сконцентрирован. Очистка колоночной флеш-хроматографией (EtOAc : гексаны 1:1) дала продукт 3 (9,11 г, 79%) в качестве белого твердого вещества.

К раствору соединения 3 (3,174 г, 10,8 ммоль) в DMF (15 мл) был добавлен K2CO3 (4,478 г, 32,4 ммоль) и (2-бромоэтокси)-терт-бутилметилсилан (2,780 мл, 13,0 ммоль). Полученная в результате смесь перемешивалась при 55°С в течение ночи. Раствор был сконцентрирован, разбавлен водой и экстрагирован EtOAc (3×50 мл). Органические слои были комбинированы и высушены над Na2SO4. Растворитель был удален для получения вязкого масла (4,805 г, 10,6 ммоль, 98,4%), которое использовалось в последующем этапе без дальнейшей очистки.
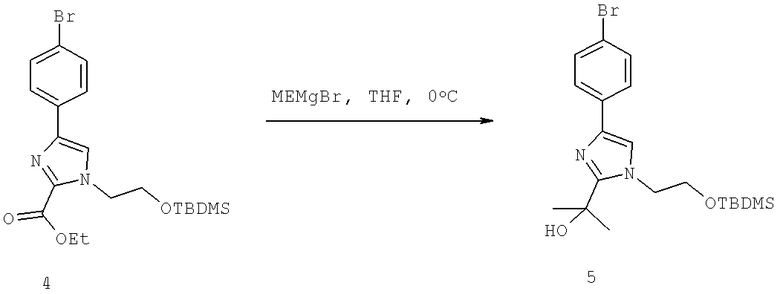
К раствору соединения 4 (2,174 г, 4,8 ммоль) в безводном THF (25 мл) по каплям был добавлен бромид метилмагния (4,8 мл, 3 М в диэтил эфире, 14,4 ммоль) в атмосфере азота при 0°С. Реакция перемешивалась при 0°С в течение 15 минут. Реакция была тщательно погашена насыщенным раствором хлорида аммония (5 мл) и воды (30 мл) м экстрагирована EtOAc (3×50 мл). Органические слои были объединены и высушены над Na2SO4 и сконцентрированы до неочищенного масла. Очистка колоночной флеш-хроматографией (15% EtOAc/гексаны) дала искомый продукт 5 (1,371 г, 65%) в виде аморфного твердого вещества.
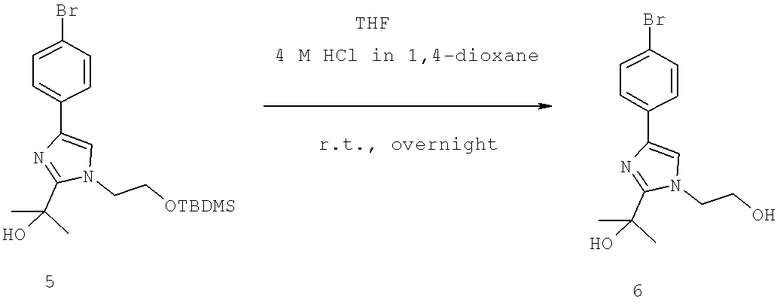
К раствору соединения 5 (1,371 г, 3,1 ммоль) в THF (5 мл) было добавлено 35 мл HCl (4М в 1,4-диоксане). Полученный в результате раствор перемешивался при комнатной температуре в течение ночи. Растворители были удалены для получения продукта 6 (1,0 г, 99%) в качестве белого твердого вещества.
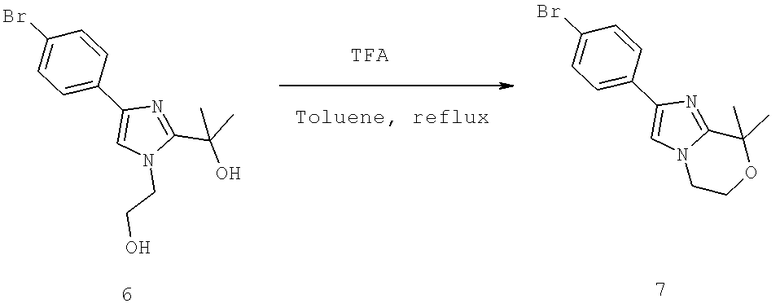
Смесь соединения 6 (0,5 г, 1,54 ммоль) и 1 мл TFA в толуоле (60 мл) была обработана в противотоке в течение ночи. Твердое вещество 6 не растворялось до приблизительно точки кипения толуола. Растворитель был удален. Остаток был разбавлен EtOAc, промыт водным раствором NaHCO3, высушен над Na2SO4 и сконцентрирован. Очистка колоночной флеш-хроматографией (EtOAc/гексаны 1:1) дала продуки 7 (0,348 г, 74%) в виде белого твердого вещества.
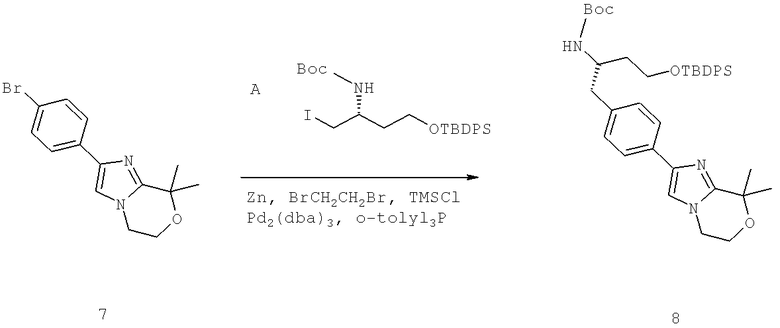
К суспензии порошка цинка (255 мг, 3,9 ммоль) в сухом обезгаженном DMF (15 мл) был добавлен 1,2-дибромоэтан (0,020 мл, 0,23 ммоль) в атмосфере азота. Смесь нагревалась с использованием тепловой пушки в течение 30 секунд до тех пор, пока из раствора не стал выходить газ, указывая на активацию цинка. Смесь затем была оставлена охлаждаться до комнатной температуры, за чем последовало добавление TMSCl (6 мкл, 0,05 ммоль) и смесь была оставлена перемешиваться при комнатной температуре в течение 30 минут. К раствору цинка был добавлен раствор иодо соединения А в обезгаженном DMF и реакционная смесь перемешивалась при комнатной температуре в течение 1 часа. Затем через шприц был добавлен раствор соединения 7 (200 мг, 0,65 ммоль) в обезгаженном DMF, за чем последовало добавление Rd2(dba3) (14,9 мг, 0,016 ммоль) и три-о-толилфосфина (19,8 мг, 0,065 ммоль). Реакционная смесь перемешивалась при комнатной температуре в течение 1 часа, затем при 40°С в течение 2 часов. Реакция была завершена, как было показано на TLC. Раствор был погашен соляным раствором и экстрагирован EtOAc (5×50 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы. Очистка колоночной флеш-хроматографией (EtOAc/гексаны 1:1) дала продукт 8 (373 мг, 88%) в виде бесцветного масла.
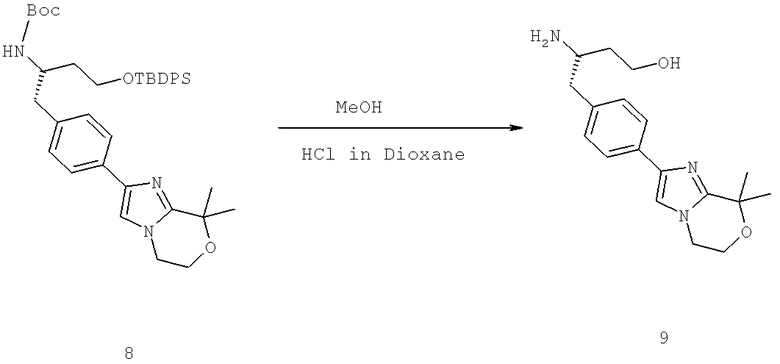
К раствору соединения 8 (373 мг, 0,57 ммоль) в МеОН (10 мл) было добавлено 2 мл HCl (4,0 М в диоксане). Раствор был оставлен перемешиваться при комнатной температуре в течение 2 часов. Растворитель был удален для получения неочищенного продукта 9 (180 мг, 99%), который использовался без дальнейшей очистки.
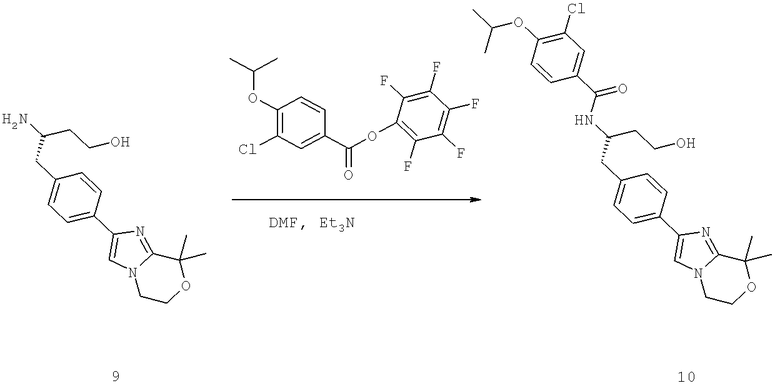
Смесь соединения 9 (180 мг, 0,57 ммоль) и реактива эфира В (260 мг, 0,68 ммоль) в DMF (10 мл), содержащая триэтиламин (0,24 мл, 1,71 ммоль) перемешивалась при комнатной температуре в течение ночи. Реакционный раствор был разбавлен соляным раствором и экстрагирован EtOAc (3×50 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы. Очистка HPLC (С 18 колонка) дала продукт 10 (141 г, 50%) в виде белого твердого вещества.
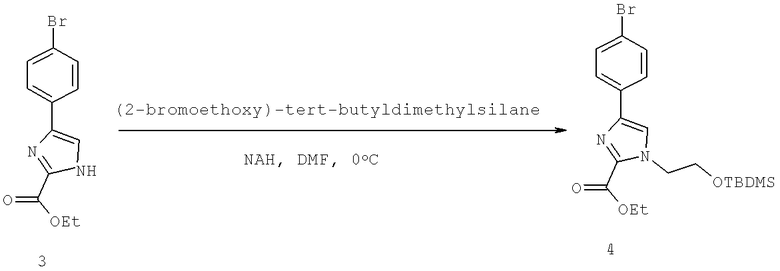
К суспензии NaH (0,39 г, 9,3 ммоль) в DMF (15 мл) был добавлен раствор 3 (1,9 г, 6,5 ммоль) в DMF (10 мл) при 0°С в атмосфере азота. Реакция перемешивалась в течение 1, 5 часов, затем был добавлен (2-бромоэтокси)-терт-бутилдиметилсилан (2,09 мл, 9,7 ммоль). Реакционная смесь перемешивалась в течение ночи, была разбавлена EtOAc, погашена водным раствором хлорида аммония и экстрагирована EtOAc (3×50 мл). Органические слои были объединены и высушены над Na2SO4. Очистка биотажем (EtOAc) дала продукт 4 (1,2 г, 41%) в виде светло-желтого твердого вещества.
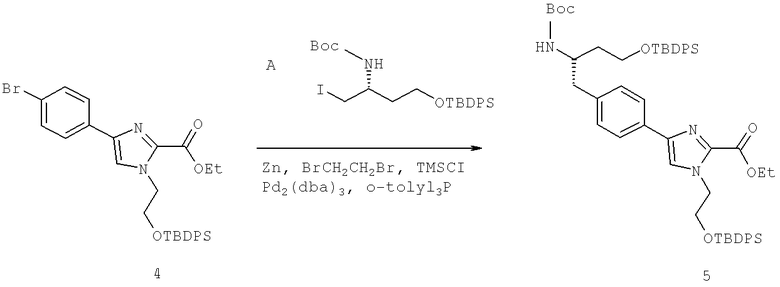
К суспензии порошка цинка (1,2 г, 18,4 ммоль) в сухом обезгаженном DMF (15 мл) был добавлен 1,2-дибромоэтан (0,13 мл, 1,5 ммоль) в атмосфере азота. Смесь нагревалась с использованием тепловой пушки в течение около 30 секунд, пока газ не стал выделяться из раствора, указывая на активацию цинка. Смесь была затем оставлена охлаждаться до комнатной температуры, за чем последовало добавление TMSCl (100 мкл), и она перемешивалась при комнатной температуре в течение 30 минут. Раствор иодо соединения А (1,71 г, 3,1 ммоль) в обезгаженном DMF был добавлен к раствору цинка, и реакционная смесь перемешивалась в течение 1 часа при комнатной температуре. Затем раствор соединения 4 (1,0 г, 2,2 ммоль) в обезгаженном DMF был добавлен через шприц, за чем последовало добавление Pd2(dba3) (0,14 г, 0,015 ммоль) и три-о-толилфосфина (0,18 г, 0,06 ммоль). Реакционная смесь перемешивалась в течение одного часа при комнатной температуре, а затем при 60°С в течение ночи. Раствор был погашен соляным раствором и экстрагирован EtOAc (5×50 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы. Очистка колоночной флеш-хроматографией (EtOAc : гексаны 1:1) дала продукт 5 (346 мг, 20%) в виде бесцветного масла.
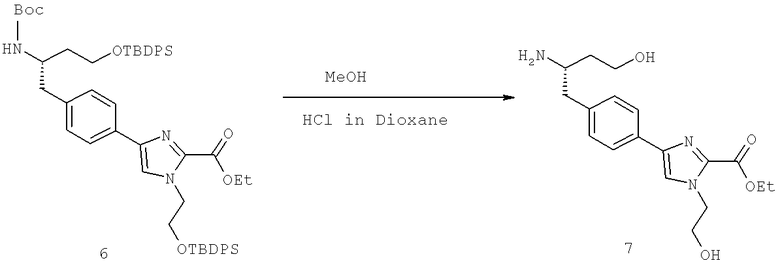
К раствору соединения 7 (346 мг) в МеОН (10 мл) было добавлено 2 мл HCl (4,0 М в диоксане). Раствор был оставлен перемешиваться при комнатной температуре в течение 2 часов. Растворитель был удален для получения неочищенного продукта 7, который использовался в следующем этапе без дальнейшей очистки.
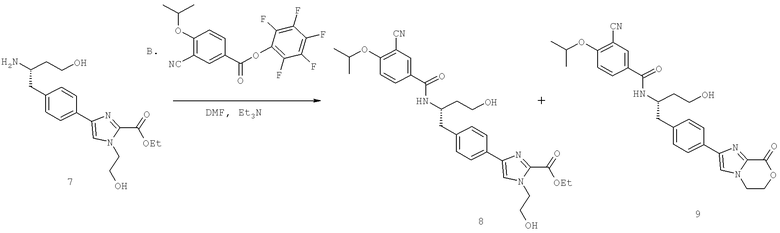
Смесь соединения 7, реактив эфира В (200 мг, 0,52 ммоль) в DMF (10 мл), содержащая триэтиламин (0,15 мл, 1,08 ммоль), перемешивалась при комнатной температуре в течение ночи. Реакционный раствор был растворен соляным раствором, экстрагирован EtOAc (3×50 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы. Очистка HPLC (С 18 колонка) дала продукт 8 (0,2 г, 87%) в виде белого твердого вещества и продукт лактон 9 (15,4 мг, 7,3%) в виде белого твердого вещества. LC-MS (CI) m/z 489,1 (MH+).
Пример 96
К раствору этил тиооксамата (10,0 г,75 ммоль) в дихлорометане (400 мл) был медленно добавлен тетрафтороборат триметилоксония (13,1 г, 89 ммоль) при 0°С. После 10 минут ледяная баня была удалена и реакционная смесь перемешивалась в течение ночи. Растворитель был удален для получения 18,0 г продукта 2 в виде белого твердого вещства, которое использовалось без дальнейшей очистки.
Смесь 2-амино-4'-бромоацетофен гидрохлорида (10,0 г, 40 ммоль), ацетата натрия (16,4 г, 200 ммоль), уксусной кислоты (11,5 мл, 200 ммоль) и соединения 2 (19,2 г, 80 ммоль) в диоксане (70 мл) перемешивалась при 65°С, пока TLC не показал на отсутствие соединения 2 (около 2 часов). Реакционная смесь была осторожно нейтрализована раствором NaHCO3 и экстрагирована с помощью этилацетата. Органический раствор был высушен над Na2SO4 и сконцентрирован. Очистка колоночной флеш-хроматографией (EtOAc : гексаны 1:1) дала продукт 3 (9,11 г, 79%) в виде белого твердого вещества.
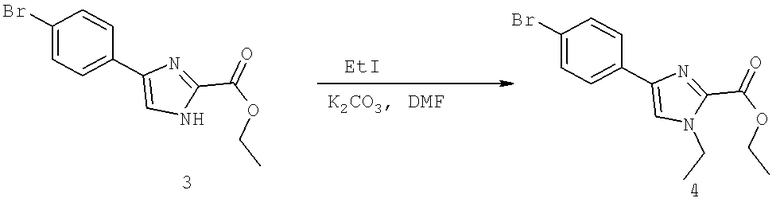
К раствору соединения 3 (5,307 г, 18 ммоль) в DMF (15 мл) был добавлен K2CO3 (3,73 г, 27 ммоль) и иодоэтан (3,5 мл, 43,2 ммоль). Полученная в результате смесь была перемешана при 60°С в течение трех часов. Смесь была разбавлена водой и экстрагирована EtOAc (3×50 мл). Органические слои были объединены, высушены над Na2SO4 и сконцентрированы. Очистка колоночной флеш-хроматографией (гексаны/EtOAc 50:50) дала продукт 4 (3,2 г, 55%) в виде белого твердого вещества.
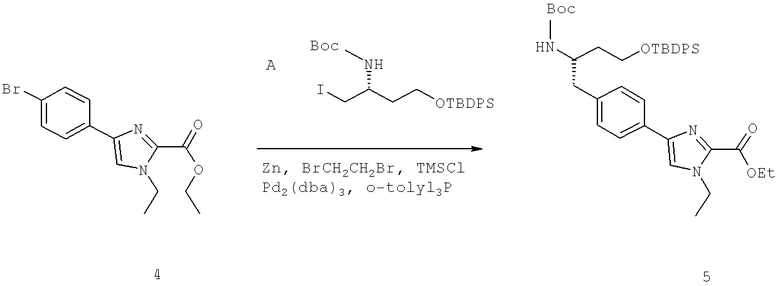
К суспензии порошка цинка (3,90 г, 59,6 ммоль) в сухом обезгаженном DMF (10 мл) был добавлен 1,2-дибромоэтан (308 мкл, 3,58 ммоль) в атмосфере азота. Смесь нагревалась тепловой пушкой в течение около 30 секунд, пока из раствора не стал выделяться газ, указывая на активацию цинка. Смесь была оставлена охлаждаться до комнатной температуры, за чем последовало добавление TMSCl (92 мкл, 0,735 ммоль) и она перемешивалась при комнатной температуре в течение 30 минут. Раствор иодо соединения А (6,6 г, 11,9 ммоль) в обезгаженном DMF был добавлен к раствору цинка и реакционная смесь перемешивалась в течение 1 часа при комнатной температуре. Затем через шприц был добавлен раствор соединения 4 (3,2 г, 9,93 ммоль) в обезгаженном DMF, за чем последовало добавление Pd2(dba3) (223 мг, 0,244 ммоль) и три-о-толилфосфина (302 мг, 0,992 ммоль). Реакционная смесь перемешивалась в течение одного часа при комнатной температуре, затем при 60°С в течение 2 часов. Реакция была завершена, как показал TLC. Раствор был погашен соляным раствором и экстрагирован EtOAc (3×80 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы. Очистка колоночной флеш-хроматографией (EtOAc : гексаны 1:1) дала продукт 5 (5,43 мг, 82%) в виде бесцветного масла.
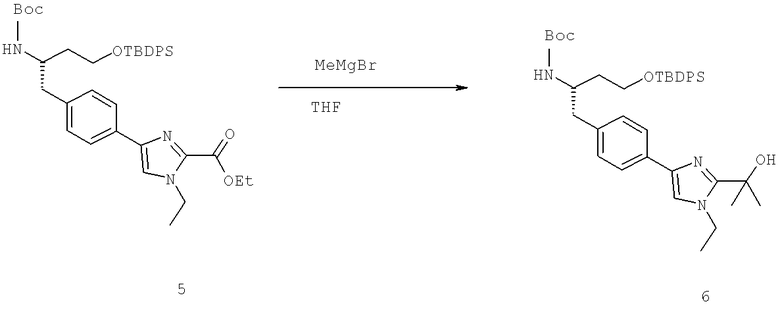
К раствору соединения 5 (5,43 мг, 82%) в THF (50 мл) был по каплям добавлен раствор бромида MeMgBr в эфире (9,0 мл, 27 ммоль) при 0°С в атмосфере азота. Реакция была завершена за 10 минут через TLC. Раствор был погашен водным раствором хлорида аммония, находящимся во льду, экстрагирован EtOAc (3×60 мл). Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы. Очистка колоночной флеш-хроматографией (гексаны/EtOAc 1:1) дала продукт 6 (4,86 мг, 91%) в виде бесцветного масла.
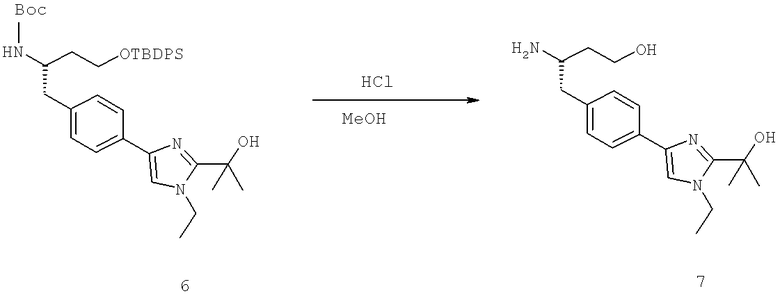
Смесь соединения 6 (4,86 г, 7,4 ммоль) и 18 мл HCl (4М в диоксане) в МеОН (10,0 мл) перемешивалась при комнатной температуре в течение 1 часа, за чем последовало нагревание при 60°С в течение 30 минут. Реакция была завершена через TLC и LC/MS. Растворитель был удален для получения продукта 7, который напрямую использовался в следующем этапе.
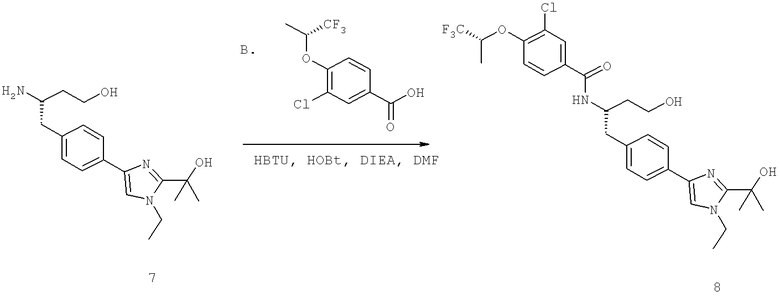
Смесь кислоты В (677 мг, 2,51 ммоль), HBTU (3,6 г, 9,49 ммоль), НОВТ (1,45 г, 9,46 ммоль) и DIEA (2,20 мл, 12,6 ммоль) в DMF (40 мл) перемешивалась при комнатной температуре в течение 1 часа, за чем последовало добавление 7 (1,0 г, 3,14 ммоль). Реакция была завершена через TLC и LC/MS. Раствор был разделен в EtOAc и соляном растворе и экстрагирован EtOAc. Комбинированные органические слои были высушены над сульфатом натрия и сконцентрированы. Очистка HPLC дала продукт 8 (390 мг) в виде белого твердого вещества.
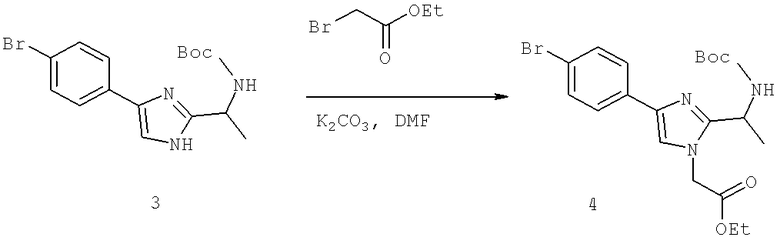
К раствору соединения 3 (2,66 г, 7,27 ммоль) в DMF (15 мл) был добавлен K2CO3 (2,00 г, 15 ммоль) и этил бромоацетат (1,61 мл, 14,5 ммоль). Полученная в результате смесь перемешивалась при 60°С в течение 3 часов. Смесь была разбавлена водой и экстрагирована EtOAc (3×50 мл). Органические слои были объединены, высушены над Na2SO4 и сконцентрированы. Очистка колоночной флеш-хроматографией (гексаны/EtOAc 50:50) дала продукт 4 (3,02 г, 91%).
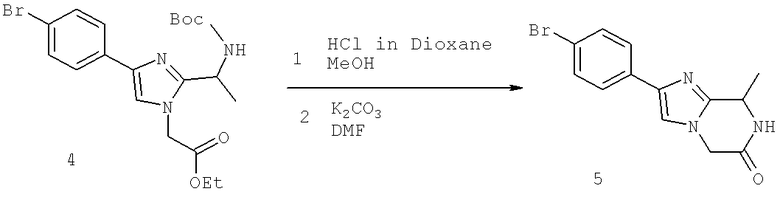
К раствору соединения 4 (3,02 г, 6,7 ммоль) в МеОН (20 мл) была добавлена HCl (4,0 М) в диоксане (7,0 мл) и они перемешивались при 60°С в течение одного часа. Смесь была сконцентрирована и не выполнялась очистка. Полученное в результате масло было растворено в DMF (15 мл), был добавлен K2CO3 (2,0 г, 14,7 ммоль) и они перемешивались при 60°С в течение ночи. Смесь была разбавлена водой и экстрагирована EtOAc (3×50 мл). Органические слои были обединены, высушены над Na2SO4 и сконцентрированы. Очистка колоночной флеш-хроматографией (гексаны/EtOAc 50:50) дала продукт 5 (1,80 г, 88%).
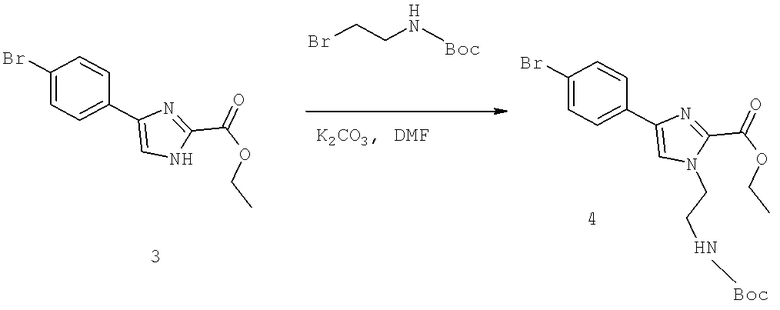
К растору соединения 3 (5,000 г, 17 ммоль) в DMF (15 мл) был добавлен K2CO3 (3,51 г, 26 ммоль) и бромид Вос-2-амино этила (4,56 г, 20,35 ммоль). Полученная в результате смесь перемешивалась при 60°С в течение трех часов. Смесь была разбавлена водой и экстрагирована EtOAc (3×50 мл). Органические слои были обединены, высушены над Na2SO4 и сконцентрированы. Очистка колоночной флеш-хроматографией (гексаны/EtOAc 50:50) дала продукт 4 (4,08 г, 55%) в виде белого твердого вещества.
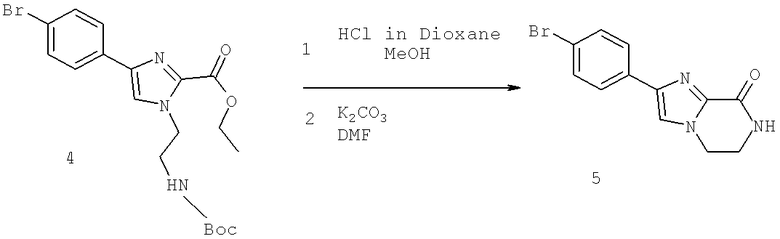
Пример 97
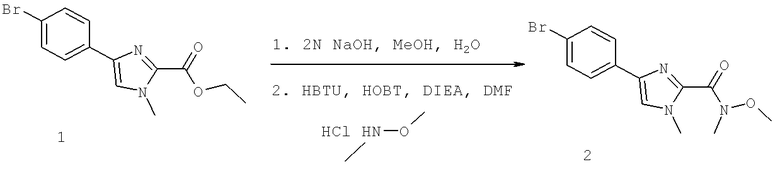
К раствору 1 (10,7 г, 34,6 ммоль) в МеОН/Н2О (60 мл/20 мл) был добавлен NaOH (2N, 20,8 мл, 41,6 ммоль). После того как смесь перемешивалась при 50°С в течение 2 часов, раствор был сконцентрирован в условиях высокого вакуума и было получено 10,3 г светло-желтого твердого вещества (LRMS (M-H+) m/z 278,9), которое использовалось в следующем этапе без дальнейшей очистки. К раствору неочищенной смеси в DMF (50 мл) последовательно добавлялись N,O-диметилгидроксиламин гидрохлорид (4,0 г, 40,7 ммоль), HBTU (4,0 г, 40,7 ммоль), НОВТ (6,2 г, 40,7 ммоль) и DIEA (6,0 мл, 40,7 ммоль). Смесь перемешивалась при комнатной температуре в течение ночи. Затем раствор был разделен между EtOAc и Н2О. Органические слои были промыты NaOH (1N), соляным раствором, высушены над Na2SO4, отфильтрованы и сконцентрированы. Остаток был очищен колоночной флеш-хроматографией с использованием смеси гексанов и EtOAc для получения 2 (8 г, 72%). LRMS (М+Н+) m/z 324,0.
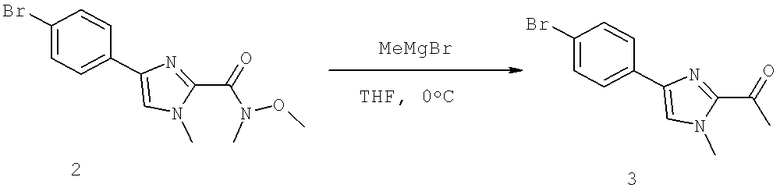
К раствору 2 (3,7 г, 11,4 ммоль) в THF (40 мл) по каплям был добавлен MeMgBr в Et2O (3M, 11,4 мл, 34, 2 ммоль) при 0°С. Смесь перемешивалась при 0°С в течение 30 минут. Раствор был погашен насыщенным NH4Cl при 0°С и разделен между EtOAc и Н2О. Органический слой был промыт соляным раствором, высушен над Na2SO4, отфильтрован и сконцентрирован для получения 3 (3,0 г, 94%) без дальнейшей очистки. LRMS (М+Н+) m/z 279,0.
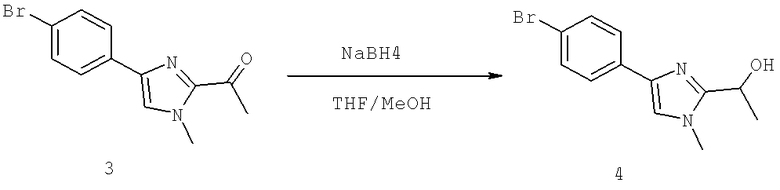
К раствору 3 (3,0 г, 10,8 ммоль) в THF/MeOH (10 мл/10 мл) был медленно добавлен NaBH4 (407 мг, 10,8 ммоль). Смесь перемешивалась в течение 10 минут, была погашена насыщенным NH4Cl и разделена между EtOAc и Н2О. Органический слой был промыт насыщенным NaHCO3, соляным раствором, высушен над Na2SO4, отфильтрован и сконцентрирован для получения 4 (3,0 г, 99%), который использовался без дальнейшей очистки. LRMS (M+H+) m/z 281,0.
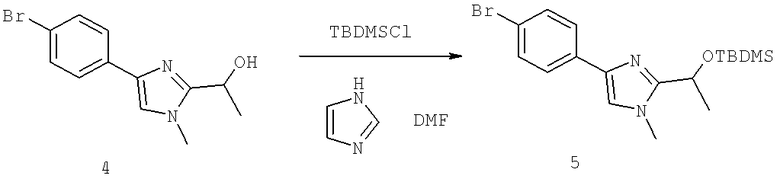
К раствору 4 (3,0 г, 10,7 ммоль) в DMF (20 мл) был добавлен TBDMSCl (1,6 г, 10,7 ммоль), имидазол (726 мг, 10,7 ммоль) и DMAP (271 мг, 21,3 ммоль). Смесь перемешивалась при комнатной температуре в течение ночи. Раствор был разделен между EtOAc и Н2О. Органический слой был промыт насыщенным NaHCO3, H2O, соляным раствором, высушен над Na2SO4, отфильтрован и сконцентрирован. Остаток был очищен колоночной флеш-хроматографией с использованием смеси гексанов и EtOAc для получения 5 (3,5 г, 83%). LRMS (M+H+) m/z 395,1.
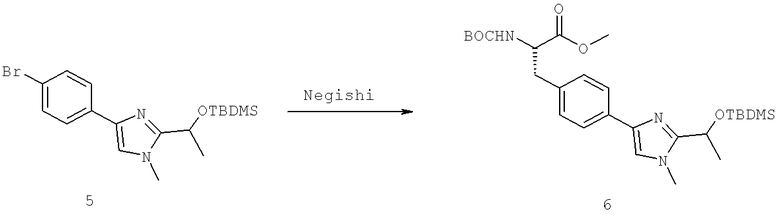
К суспензии Zn (4,8 г, 74,4 ммоль) в DMF (20 мл) был добавлен BrCH2CH2Br (320 мкл, 3,7 ммоль). Смесь нагревалась тепловой пушкой в течение 4 минут. После того, как раствор был охлажден, был добавлен триметилхлоросилан (95 мкл, 0,74 ммоль). После 30 минут был добавлен ВОС-β-иодо-Ala-ОМе (5,2 г, 16,0 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение 1 часа. Затем к этой смеси был добавлен Pd2(dba3) (243 мг, 0,27 ммоль), (O-Tol)3Р (269 мг, 0,88 ммоль) и 5 (3,5 г, 8, 9 ммоль). Смесь нагревалась при 50°С в течение 2 часов, была охлаждена и отфильтрована через целит. Раствор был разделен между EtOAc и Н2О. Органический слой был промыт соляным раствором, высушен над Na2SO4, отфильтрован и сконцентрирован. Остаток был очищен колоночной флеш-хроматографией с использованием смеси гексанов и EtOAc для получения 6 (3,3 г, 72%). LRMS (M+H+) m/z 518,2.
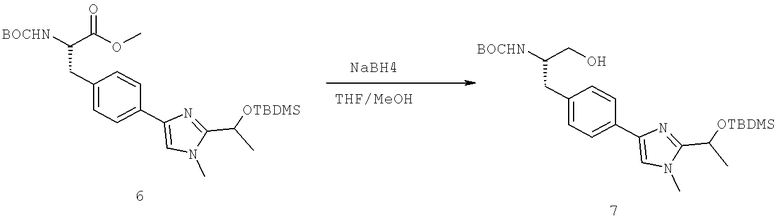
К раствору 6 (3,3 г, 6,4 ммоль) в THF (20 мл) был медленно добавлен LAH (1 М, 6,4 мл, 6,4 ммоль) при 0°С. Смесь перемешивалась при 0°С в течение 20 минут, была погашена Н2О (240 мкл), NaOH (3 N, 240 мкл), H2O (720 мкл) и отфильтрована. Органический слой был высушен над Na2SO4, отфильтрован и сконцентрирован. Остаток был очищен колоночной флеш-хроматографией с использованием смеси гексанов и EtOAc для получения 7 (1,57 г, 50%). LRMS (M+H+) m/z 490,2.
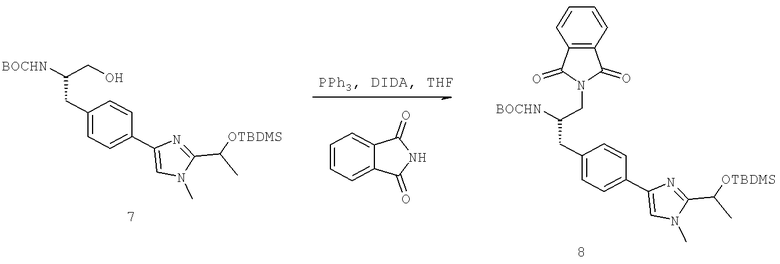
К раствору 7 (1,57 г, 3,2 ммоль) в THF (20 мл) был добавлен PPh3 (1,0 г, 3,9 ммоль), DIAD (746 мкл, 3,9 ммоль) и фталимид (567 мг, 3,9 ммоль). После того как раствор перемешивался при комнатной температуре в течение 4 часов, что контролировалось LCMS, реакция была разделена между EtOAc и H2O. Органический слой был промыт соляным раствором, высушен над Na2SO4, отфильтрован и сконцентрирован. Остаток был очищен колоночной флеш-хроматографией с использованием смеси гексанов и EtOAc для получения 8 (2,0 г, 99%). LRMS (M+H+) m/z 619,2.
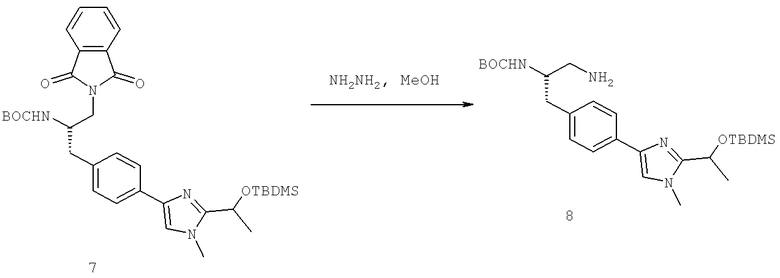
К раствору 7 (2 г, 3,2 ммоль) в МеОН (15 мл) был добавлен NH2 NH2 (1,01 мл, 32,3 ммоль). После того как реакция перемешивался при комнатной температуре в течение 4 часов, раствор был осажден, отфильтрован и промыт CH2Cl2, МеОН. Органический слой был сконцентрирован для получения 8 (2,5 г), который использовался без дальнейшей очистки. LRMS (М+Н+) m/z 489,2.
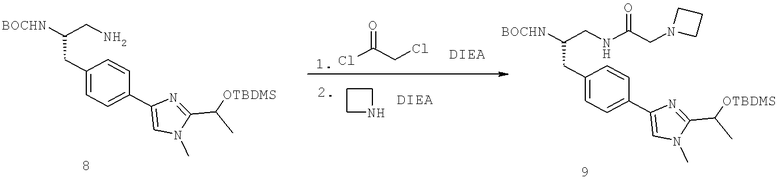
К раствору 8 (1,5, 3,1 ммоль) в CH2Cl2/CH3CN (15 мл/15 мл) были добавлены DIEA (588 мкл, 3,4 ммоль) и хлороацетил хлорид (269 мкл, 3,4 ммоль). После того как реакцию перемешивали при комнатной температуре в течение 10 минут, были добавлены азетидин (2 мл, 30,7 ммоль) и DIEA (2,7 мл, 15,3 ммоль). Реакционная смесь перемешивалась в течение ночи. Раствор был сконцентрирован и разделен между EtOAc и H2O. Органический слой был промыт соляным раствором, высушен над Na2SO4, отфильтрован и сконцентрирован. Остаток был очищен колоночной флеш-хроматографией с использованием смеси гексанов и EtOAc для получения 9 (900 мг, 51%). LRMS (M+H+) m/z 586,3.
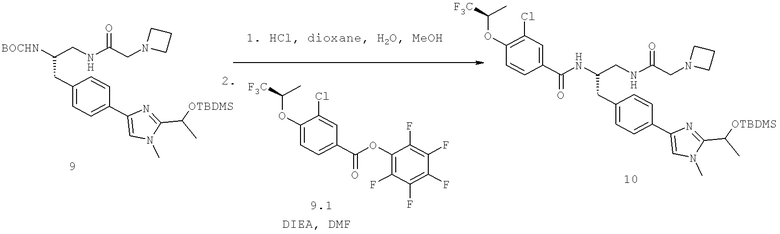
К раствору 9 (900 мг, 1,53 ммоль) в МеОН (1 мл) была добавлена HCl в диоксане (4 N, 2 мл) и HCl в H2O (2 N, 1 мл). Раствор перемешивался при комнатной температуре в течение ночи, был сконцентрирован для получения белого тведого вещества и направлен для последующего соединяющего этапа. К раствору неочищенного соединения в DMF (10 мл) были добавлены 9,1 (665 мг, 1,53 ммоль) и DIEA (800 мкл, 4,59 ммоль). Смесь перемешивалась при комнатной температуре в течение 1 часа и была разделена между EtOAc и Н2О. Органический слой был промыт соляным раствором, высушен над Na2SO4, отфильтрован и сконцентрирован. Остаток был очищен обратнофазовой HPLC для получения 10 (600 мг, 63%). LRMS (M+H+) m/z 622,2.
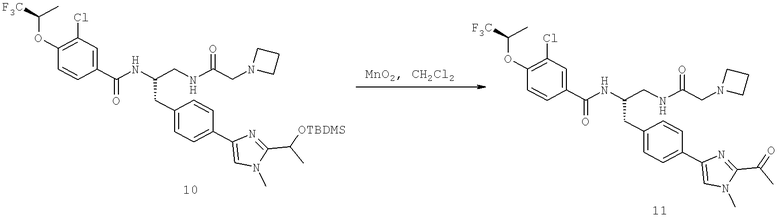
К раствору 10 (160 мг, 0,24 ммоль) в DCM (10 мл) был добавлен MnO2 (416 мг, 4,8 ммоль). Суспензия перемешивалась в течение 14 часов. Реакционная смесь была отфильтрована, фильтрат был сконцентрирован и очищен обратнофазовой HPLC с использованием смеси ацетонитрила и Н2О для получения 11 (90 мг, 60%). LRMS (M+H+) m/z 620,1.
Пример 98
Следующие соединения были приготовлены с использованием описанных выше процедур.
Пример 99
Ингибирование жизнеспособности клеток в опухолевых клеточных линиях путем обработки ингибиторами митотического кинезина
Материалы и растворы
- Клетки: SKOV3, овариальный рак (человека).
- Среда: RPMI (без фенолового красного) + 5% фетальная бычья сыворотка + 2 мМ L-глутамина.
- Колориметрический агент для определения жизнеспособности клеток: тетразолиевое соединение Promega MTS.
- Контрольное соединение для максимального умерщвления клеток: Topotecan, 1 мкМ
Процедура: день 1 - посев клеток:
Прикрепленные клетки SKOV3 были подвергнуты промывке с помощью 10 мл PBS, затем в клетки было добавлено 2 мл 0,25% трипсина и была проведена инкубация в течение 5 минут при 37°С. Клетки были смыты из колбы с использованием 8 мл среды (RPMI без фенолового красного+5% FBS) и помещены в новую колбу. Концентрацию клеток определяли счетчиком Coulter и при этом был рассчитан подходящий объем клеток для достижения 1000 клеток/100 мкл. 100 мкл суспензии клеток в среде (доведенной до 1000 клеток/100 мкл) было добавлено во все лунки 96-луночных планшетов и затем была проведена инкубация в течение от 18 до 24 часов при 37°С, влажности 100% и 5% CO2, что позволило клеткам прочно прикрепиться к планшетам.
Процедура: день 2 - добавление соединений
В одну колонку лунок обработанного в автоклаве блока анализа было добавлено по 2,5 мкл испытуемого соединения(й) с 400-кратной допустимой концентрацией. В другие лунки было добавлено 1,25 мкл топотекана с 400-кратной концентрацией (400 мкМ) (измерение оптической плотности этих лунок используется для вычитания фоновой оптической плотности умерщвленных клеток и разбавителя). 500 мкл среды без DMSO было добавлено в лунки с испытуемым соединением и 250 мкл в лунки с топотеканом. 250 мкл среды + 0,5% DMSO было добавлено во все остальные лунки, в которых испытуемое соединение(я) было последовательно разбавлено. В ряду среда, содержащая соединение, представляла собой копию, высеянную (в двух экземплярах) из блока анализа в соответствующие планшеты с клетками. Инкубация планшетов с клетками проводилась в течение 72 часов при 37°С, влажности 100% и 5% СО2.
Процедура: день 4 - добавление MTS и считывание оптической плотности
Планшеты были извлечены из термостата и в каждую лунку было добавлено 40 мкл MTS/PMS. Затем планшеты были выдержаны в инкубаторе в течение 120 минут при 37°С, влажности 100% и 5% CO2, после чего в 96-луночном спектрофотометре было проведено считывание оптических плотностей на длине волны 490 нм, которому предшествовал 5 с - цикл встряхивания.
Анализ данных
Был рассчитан нормализованный % контроля (оптическая плотность - фон) и с использованием XLfit была построена кривая зависимости концентрации соединения от дозы, по которой была определена концентрация соединения, требуемого для ингибирования жизнеспособности клеток на 50%. При тестировании этим методом соединения, соответствующие настоящему изобретению, показали активность.
Пример 100
Пименения ингибитора митотического кинезина
Опухолевые клетки человека Skov-3 (овариальные) были высеяны в 96-луночные планшеты при плотностях 4000 клеток на лунку, выдержаны для прикрепления в течение 24 часов и обработаны различными концентрациями соединений хроменона в течение 24 часов. Клетки были закреплены в 4% формальдегиде и окрашены с помощью антитубулиновых антител (впоследствии распознаваемых с помощью флуоресцентно меченых вторичных антител) и красителя Hoechst (который окрашивает ДНК).
Визуальный осмотр показал, что соединения вызывали арест клеточного цикла в стадии прометафазы митоза.
Пример 101
Ингибирование клеточной пролиферации в опухолевых клеточных линиях, обработанных ингибиторами митотического кинезина
Клетки были высеяны в 96-луночные планшеты при плотностях 1000-2500 клеток/лунка и выдержаны для прикрепления/роста в течение 24 часов. Затем эти клетки были обработаны различными концентрациями лекарственного средства в течение 48 часов. Время, в течение которого добавлялись соединения, обозначено Т0. Для определения числа жизнеспособных клеток при То и числа клеток, оставшихся после выдержки в соединении в течение 48 часов, был использован анализ на основе тетразолия с использованием реагента 3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий (MTS) (патент №5185450) (см. Promega product catalog #G3580, CellTiter 96® AQueous One Solution Cell Proliferation Assay). Было проведено сравнение числа клеток, оставшихся по истечении 48 часов, с числом жизнеспособных клеток при добавлении лекарственного средства, позволившее осуществить расчет ингибирования роста.
Рост клеток в контрольных лунках, обработанных только разбавителем (0,25% DMSO), за период 48 часов был принят за 100% и использован для сравнения роста клеток в лунках с соединениями. Ингибиторы митотического кинезина ингибируют пролиферацию клеток в овариальных опухолевых клеточных линиях человека (SKOV-3).
Путем построения графика зависимости концентрации соединения в мкМ от процента роста клеток в обработанных лунках был рассчитан показатель Gi50. Gi50, рассчитанный для соединений, представляет собой оценочную концентрацию, при которой наблюдается ингибирование роста на 50% по сравненнию с контролем, т.е. концентрацию, при которой:
100 × [(Treated48-T0)/(Control48-T0)]=50.
Все концентрации соединений были проверены дважды и контрольные значения были усреднены по 12 лункам. Очень похожая разметка 96-луночных плашетов и схема расчета Gi50 были использованы National Cancer Institute (см. Monks с соавт., J. Natl. Cancer Inst. 83: 757-766 (1991). Однако способ, по которому в National Cancer Institute расчитывают число клеток, основан не на MTS, вместо этого используются альтернативные способы.
Пример 102
Расчет IC50
При измерении IC50 соединений был использован анализ АТРазы. Были использованы следующие растворы. Раствор 1 состоит из 3 мМ фосфоенолпирувата калия (Sigma Р-7127), 2 мМ АТР (Sigma А-3377), 1 мМ IDTT (Sigma D-9779), 5 мкМ паклитаксела (Sigma Т-7402), 10 млн-1 противовспенивателя 289 (SigmaA-8436), 25 мМ Pipes/KOH, pH 6,8 (Sigma P6757), 2 мМ MgCl2 (VWR JT400301) и 1 мМ EGTA (Sigma E3889). Раствор 2 состоит из 1 мМ NADH (Sigma N8129), 0,2 мг/мл BSA (Sigma A7906), пируваткиназы 7 Eд/мл, L-лактата дегидрогеназы 10 Eд/мл (Sigma P0294), 100 нМ моторного домена митотического кинезина, 50 мкг/мл микротрубочек, 1 мМ DTT (Sigma D9779), 5 мкМ паклитаксела (Sigma T-7402), 10 млн-1 противовспенивателя 289 (Sigma А-8436), 25 мМ Pipes/KOH, pH 6,8 (Sigma P6757), 2 мМ MgCl2 (VWRJT4003-01) и 1 мМ EGTA (Sigma E3889). Последовательные разбавления (8÷12-кратные) соединений были сделаны в пластине микротитратора с 96 лунками (Corning Costar 3695) с использованием раствора 1. После последовательного разбавления каждая лунка имела 50 мкл раствора 1. Реакция была инициирована путем добавления 50 мкл раствора 2 в каждую лунку. Это может быть сделано многоканальным пипеттором или вручную, или с помощью автоматизированых манипуляторов для работы с жидкостями. Затем пластина микротитратора была помещена в устройство для считывания оптической плотности микротитратора и были проведены многократные считывания оптической плотности на длине волны 340 нм для каждой лунки в кинетическом режиме. Затем наблюдаемая скорость изменения, пропорциональная скорости АТРазы, была вычерчена в виде графика функции концентрации соединения. Для определения стандартного показателя IC50 собранные данные могут быть использованы в следующем уравнении с четырмя параметрами, использующем программу нелинейного сглаживания (например, Grafit 4):
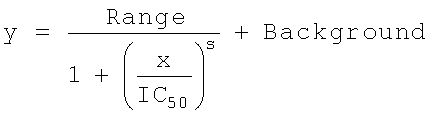
где Range - диапазон значений, Background - фон,
у - наблюдаемая скорость, х - концентрация соединения.
Было обнаружено, что другие химические составы данного класса ингибируют пролиферацию клеток, хотя значения GI50 варьировались. Значения GI50 для тестированных химических составов колебалось в диапазоне от 200 нм до превышающих наивысшую протестированную концентрацию. Под этим мы имеем в виду, что хотя большинство химических составов, ингибирующих активность митотического кинезина биохимически, не ингибировали пролиферацию клетки, для некоторых, при наивысшей протестированной концентрации (обычно около 20 мкМ), рост клетки ингибировался на менее чем 50%. Многие химические составы имеют значения GI50, меньшие чем 10 мкМ, и несколько имеют значения GI50 ниже 1 мкМ. Противопролиферативные соединения, которые успешно применялись в клинике для лечения рака (раковая химиотерапия) имеют очень разные значения GI50. Например, в клектах А 549, паклитаксель GI50 составляет 4 нМ, доксорубицин равен 63 нМ, 5-фтороурацил 1 мкМ и гироксимочевина 500 мк М (данные предоставлены Национальным институтом рака, отделом развития терапевтических программ, http://dtp.nci.nih.gov/). Следовательно, соединения, которые ингибируют пролиферацию клетки, могут быть полезны при практически любой концентрации.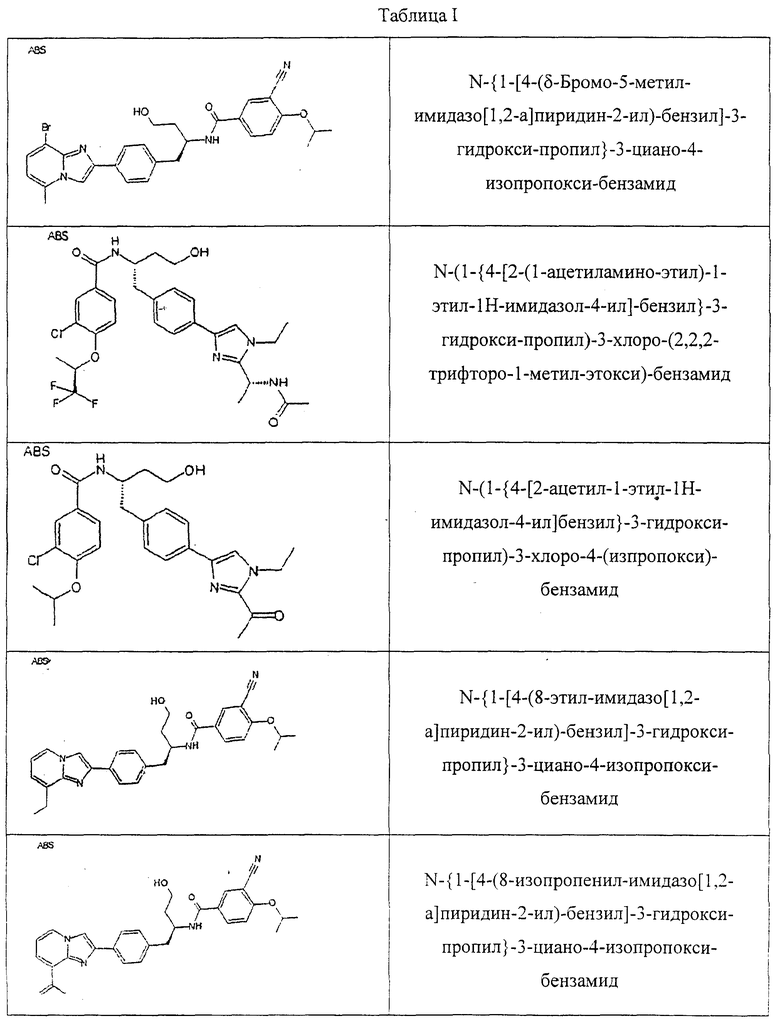
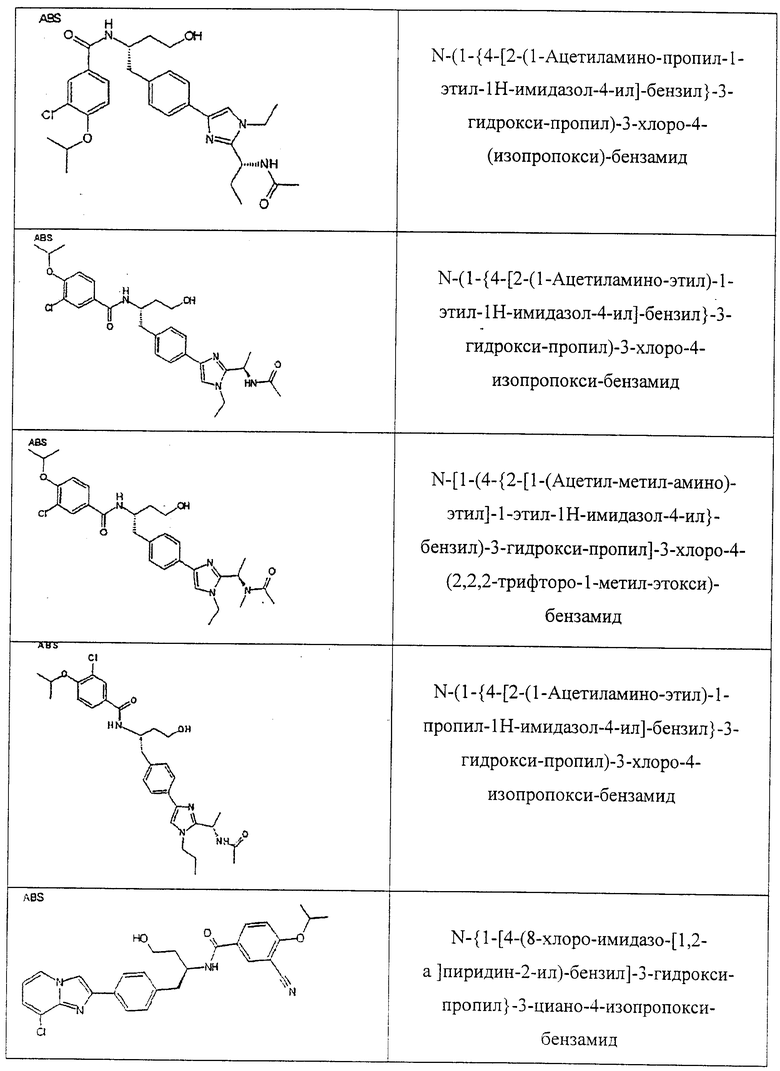
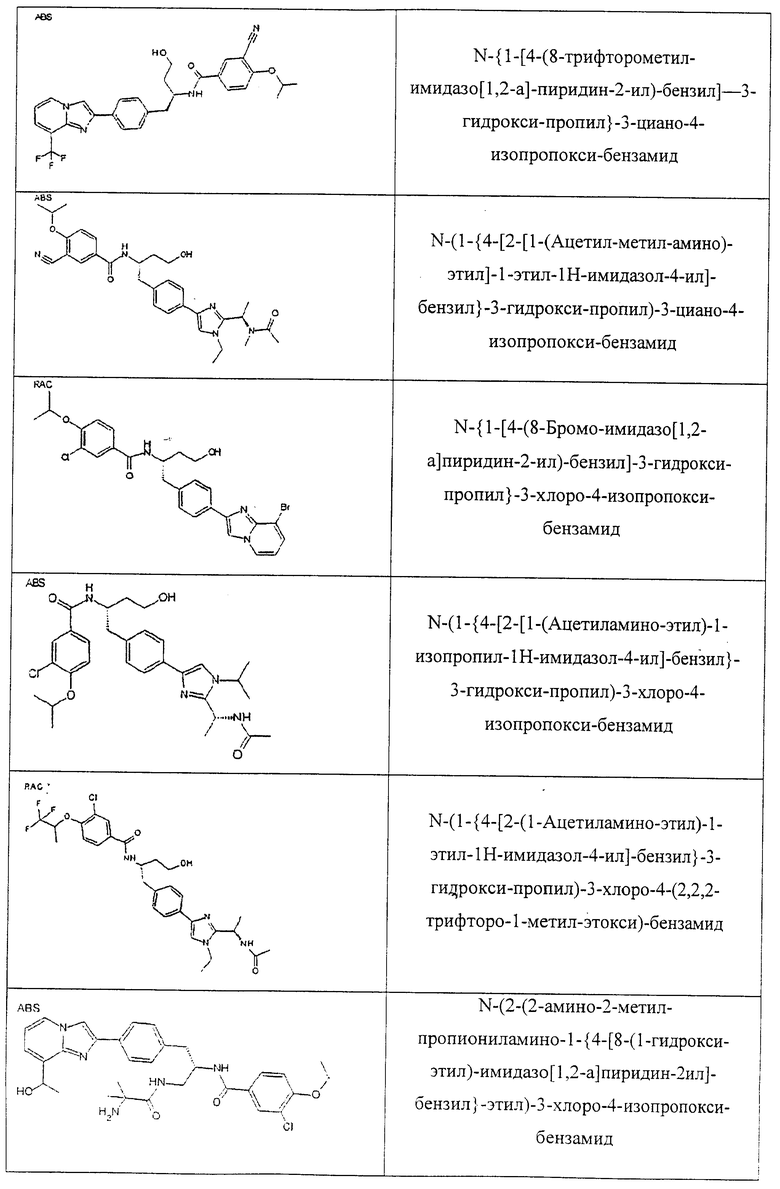
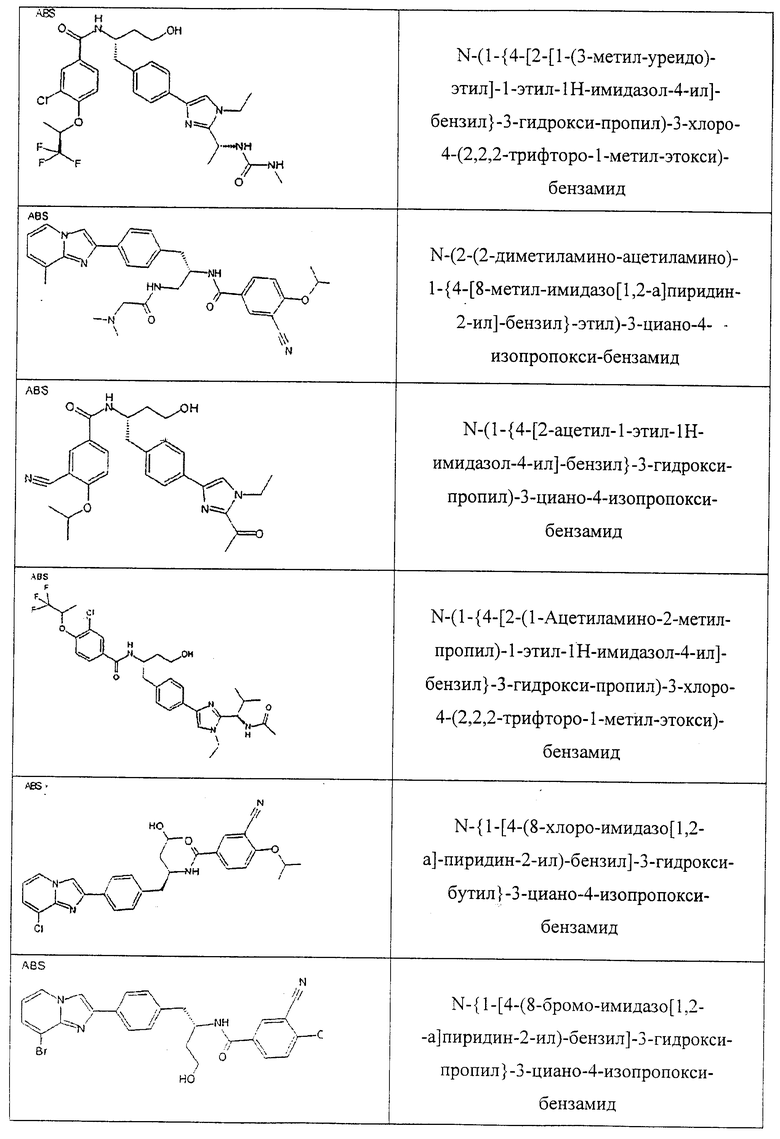
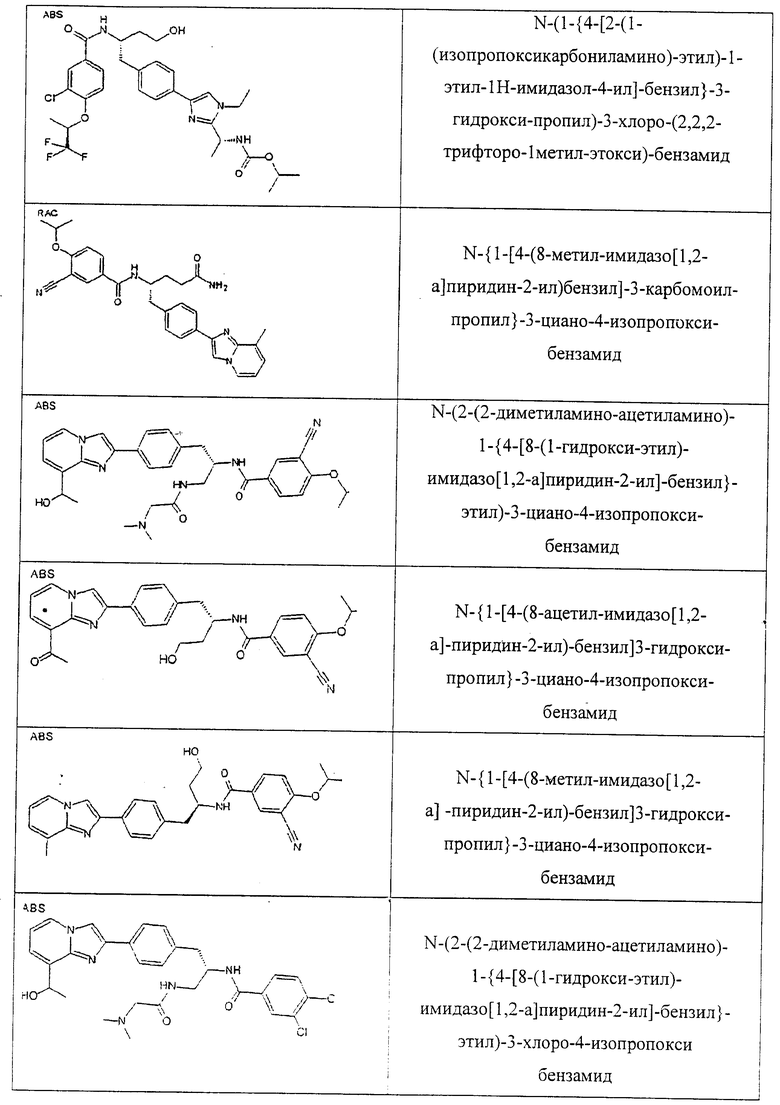
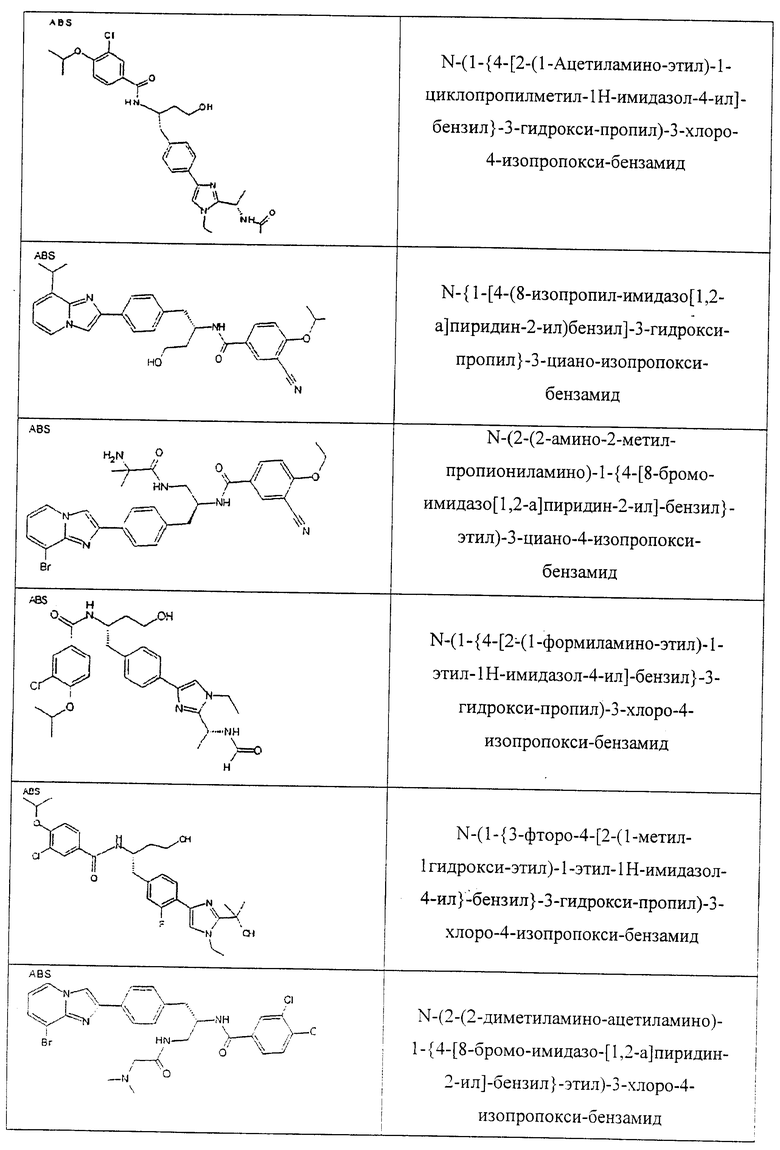
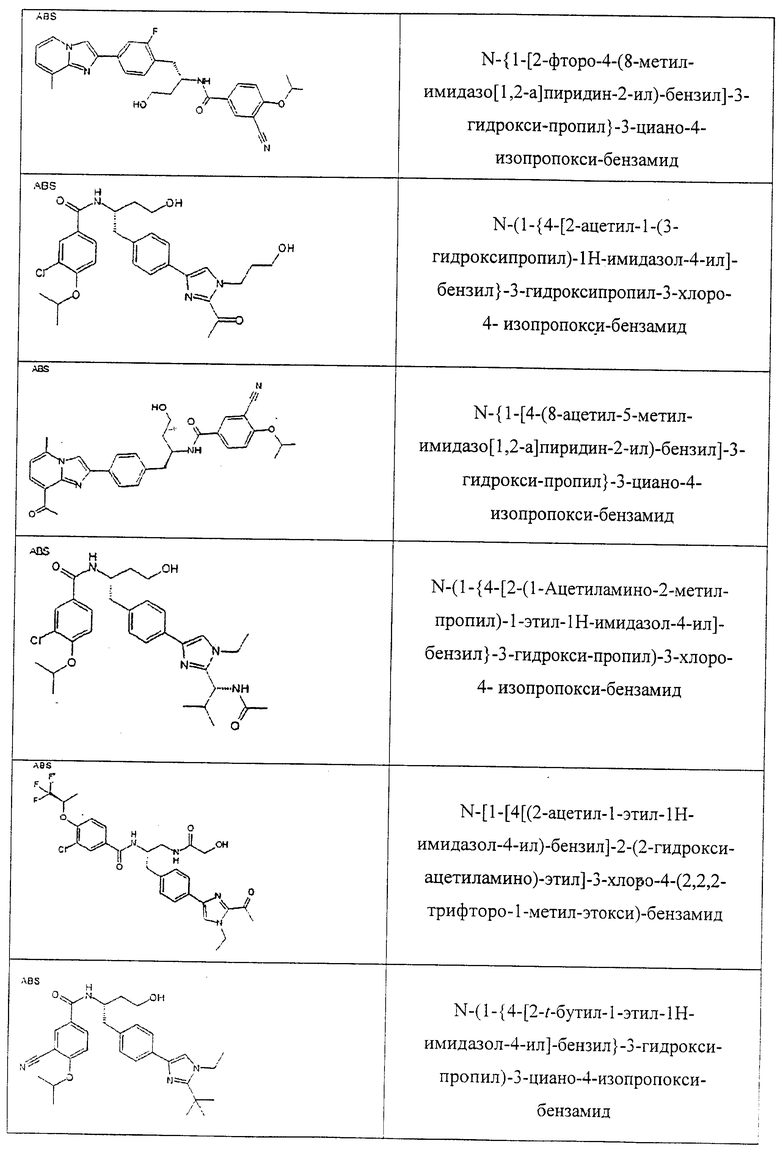
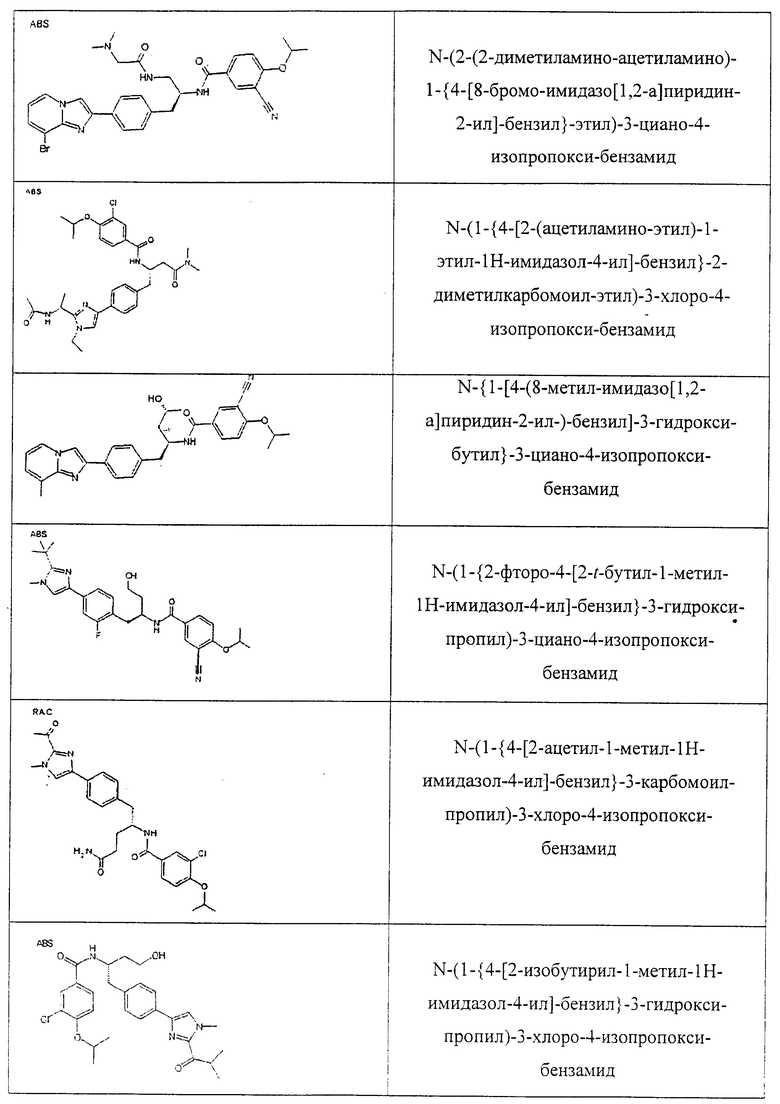
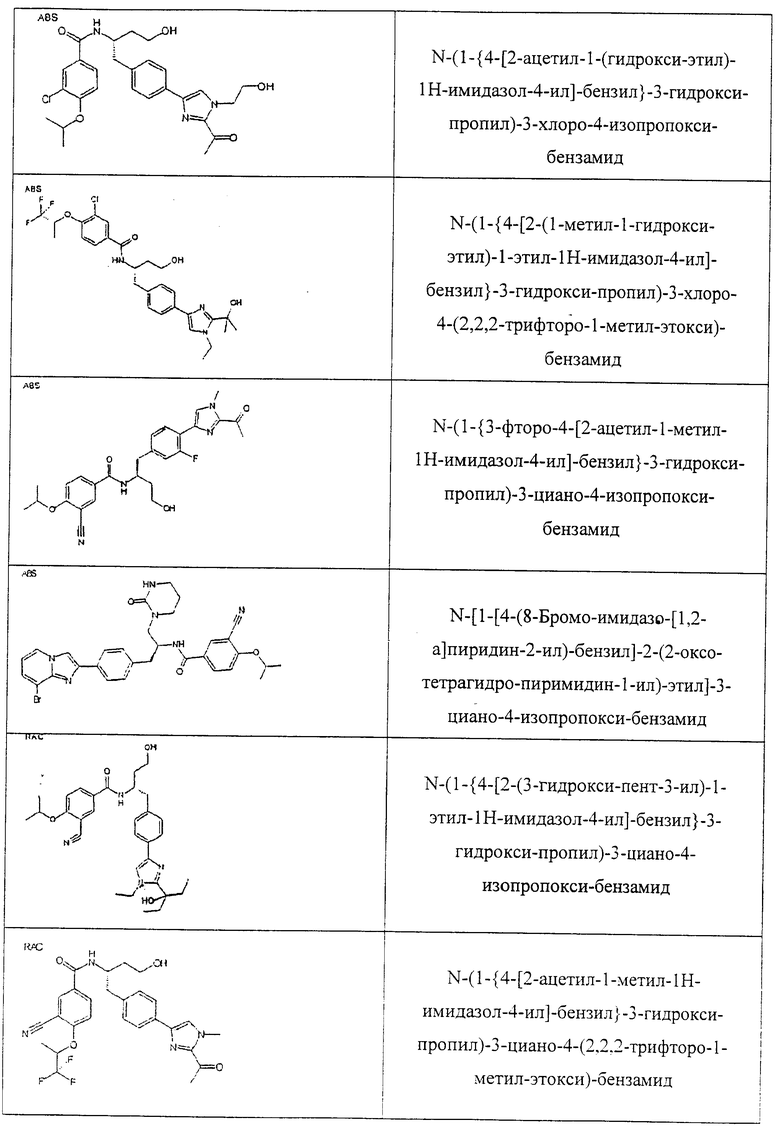
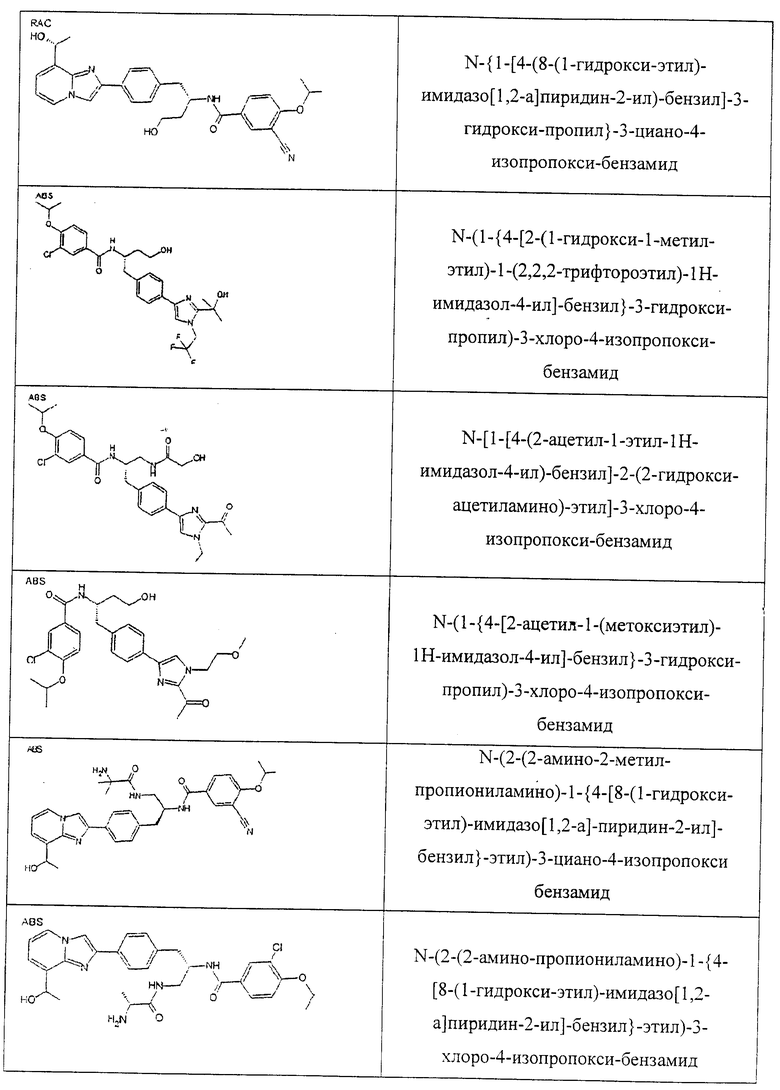
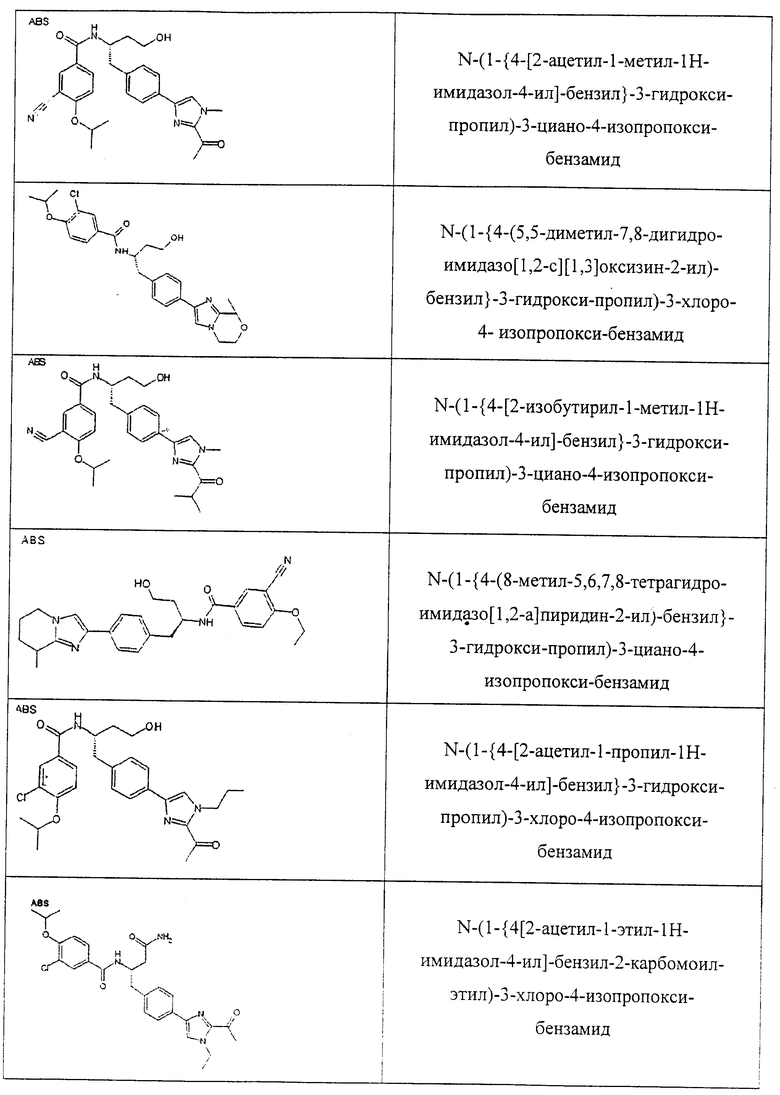
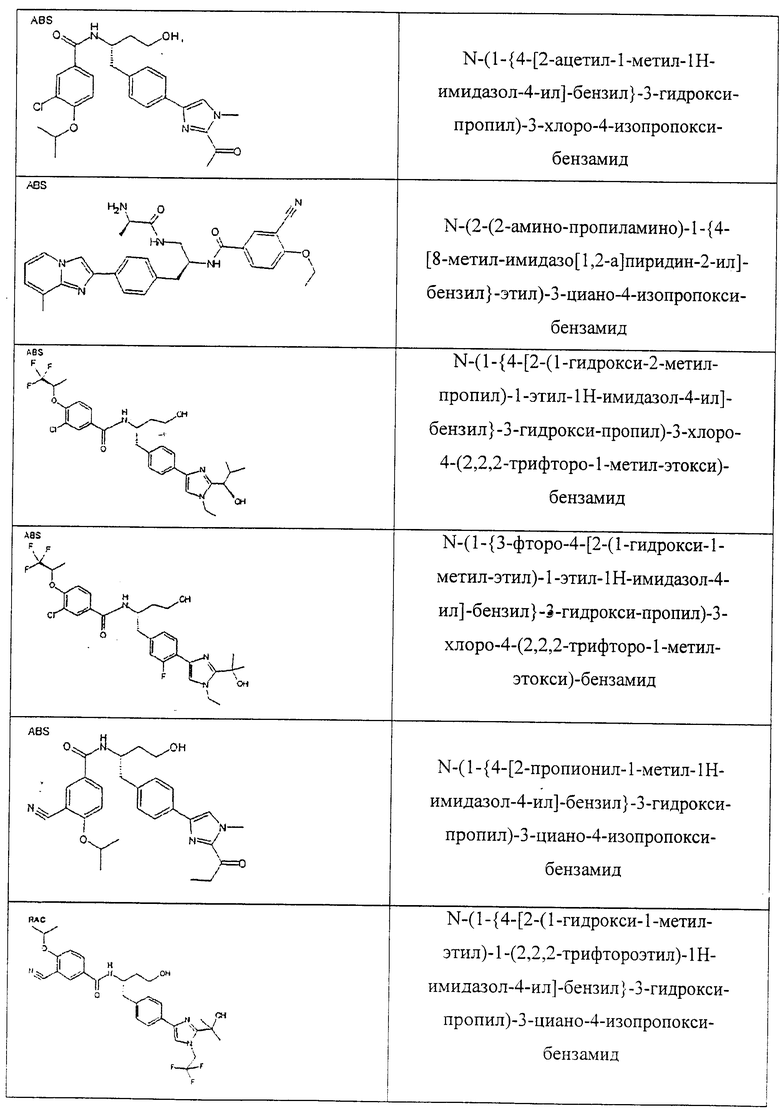
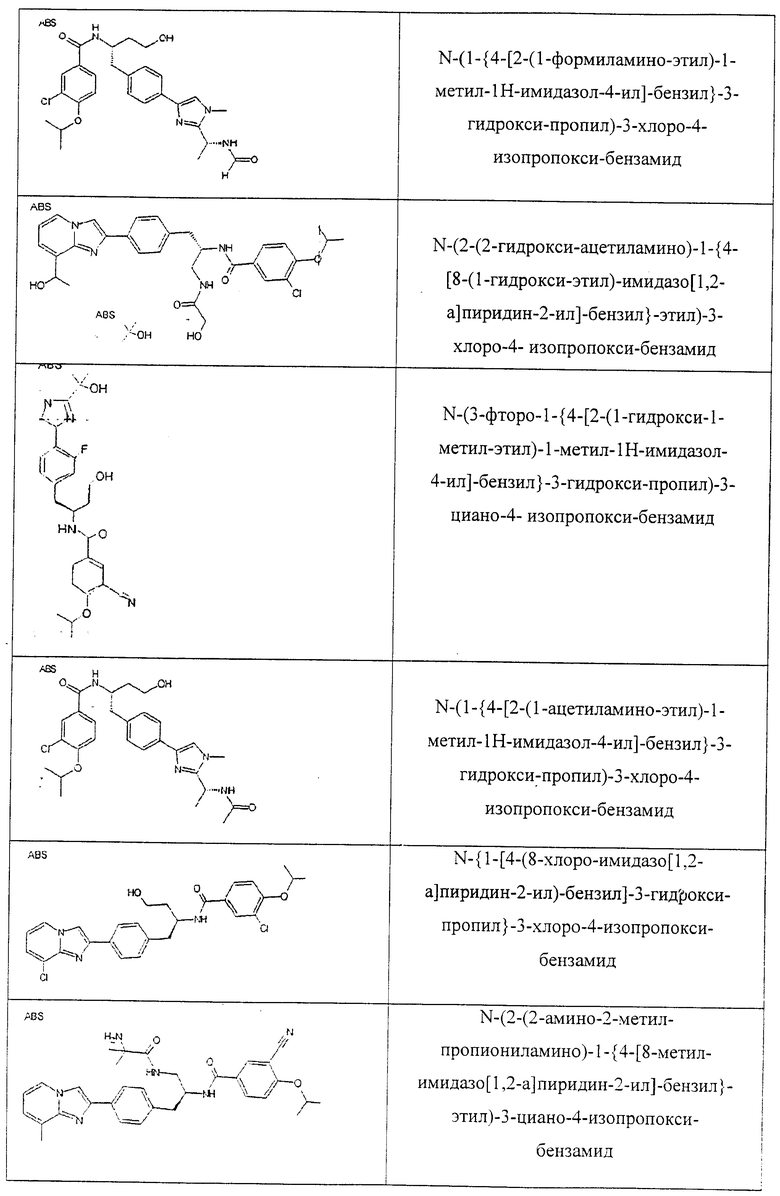
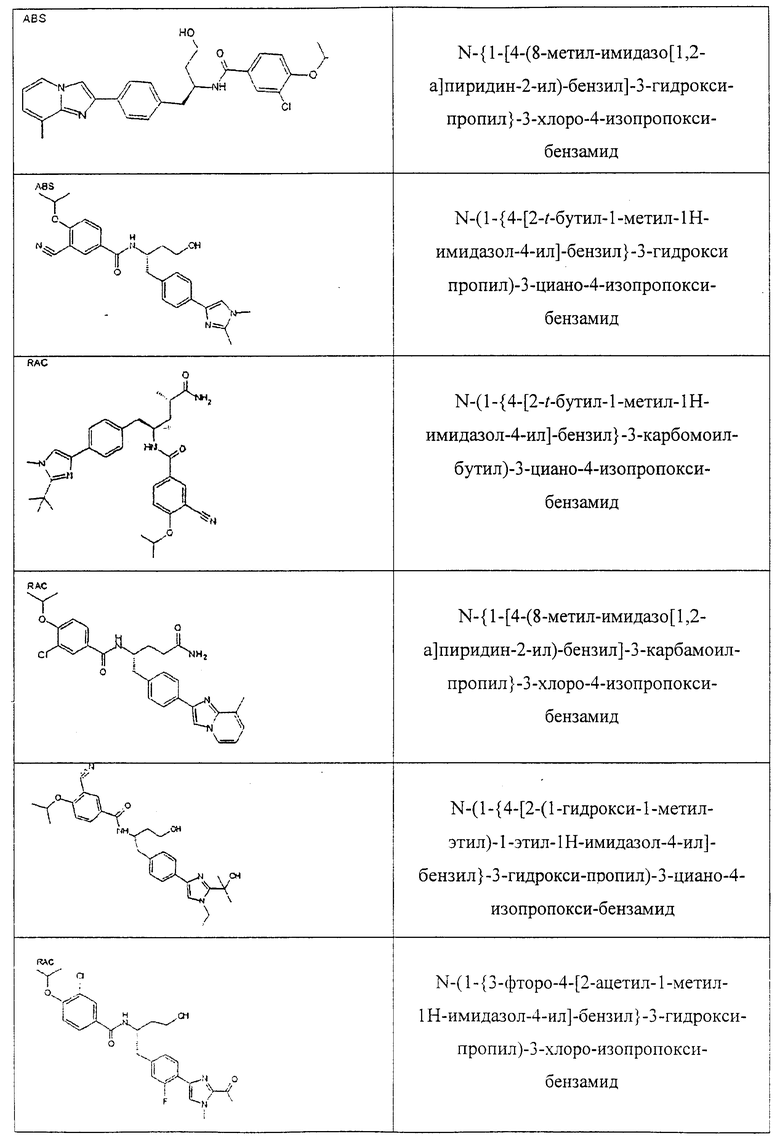
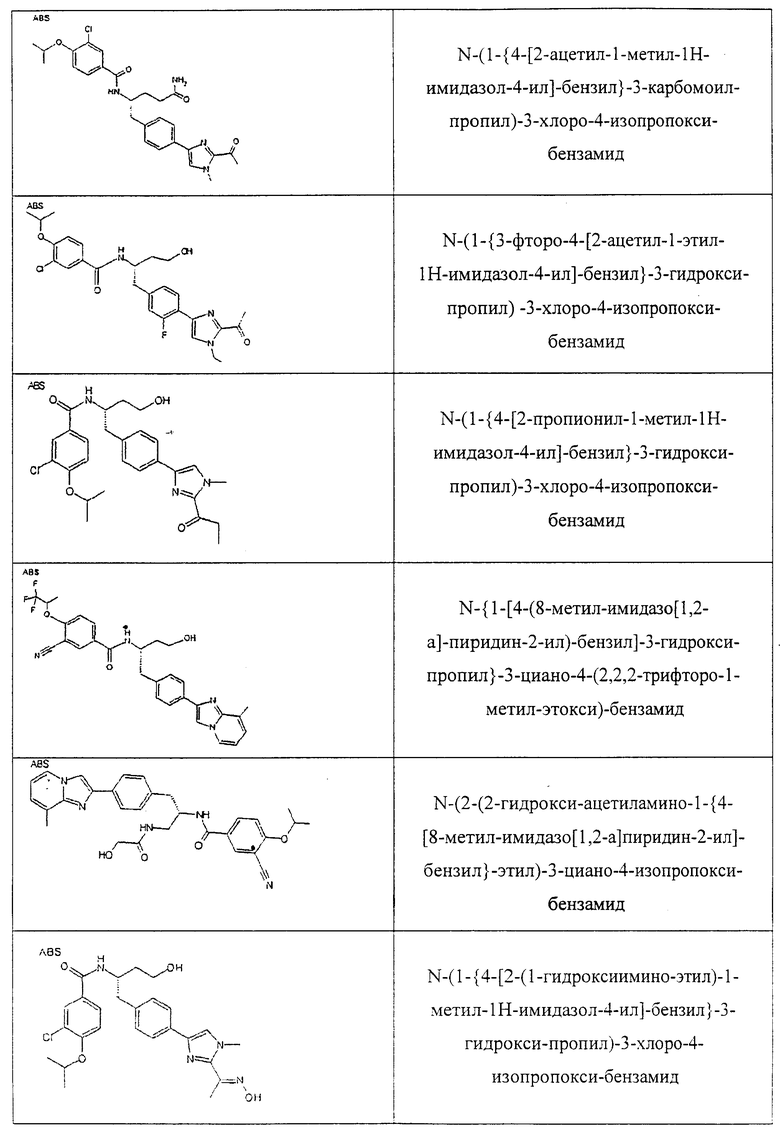
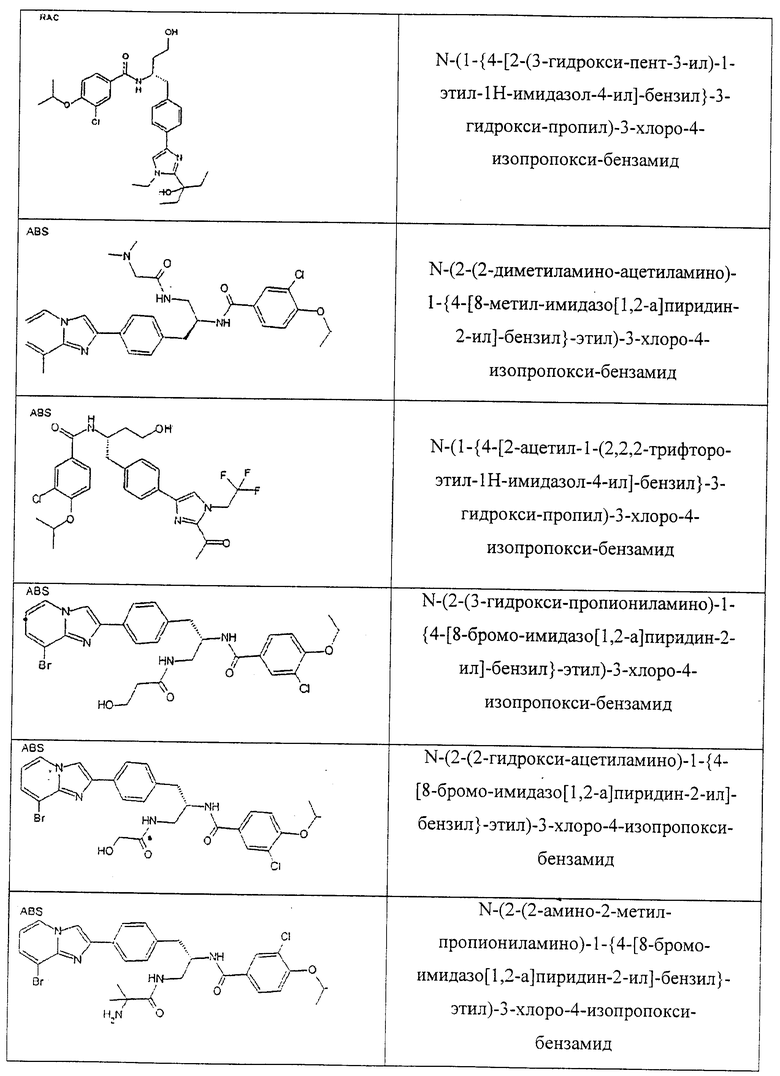
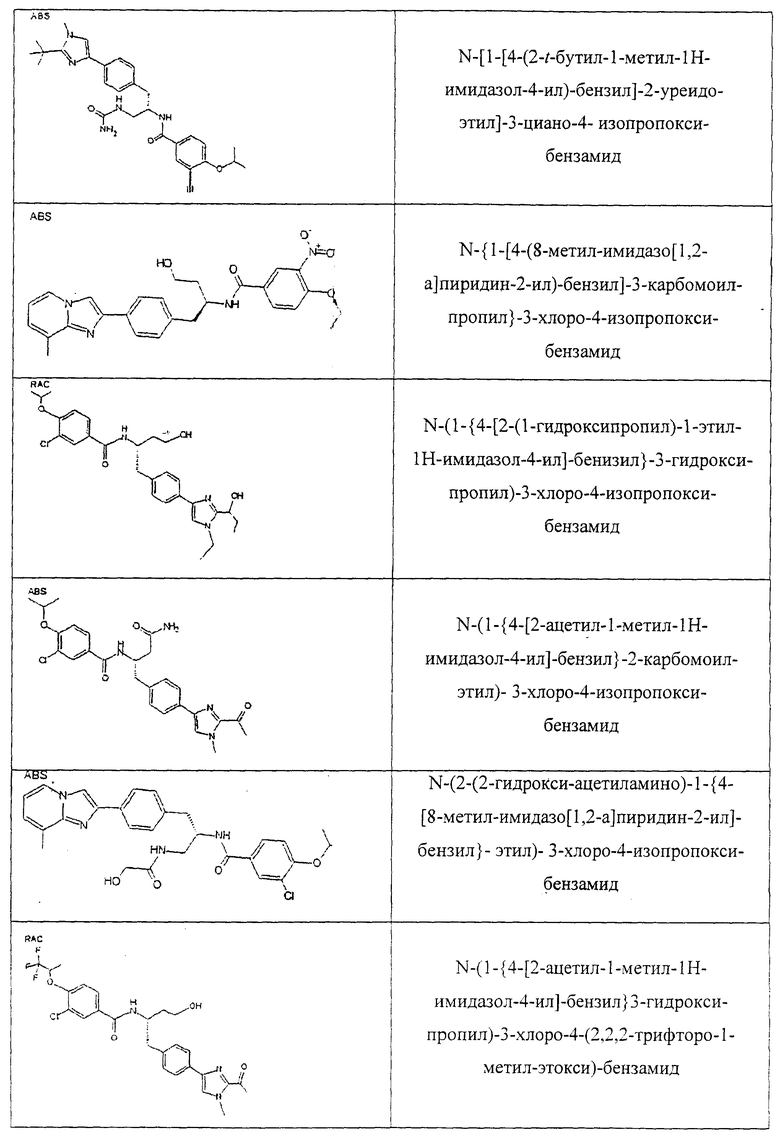
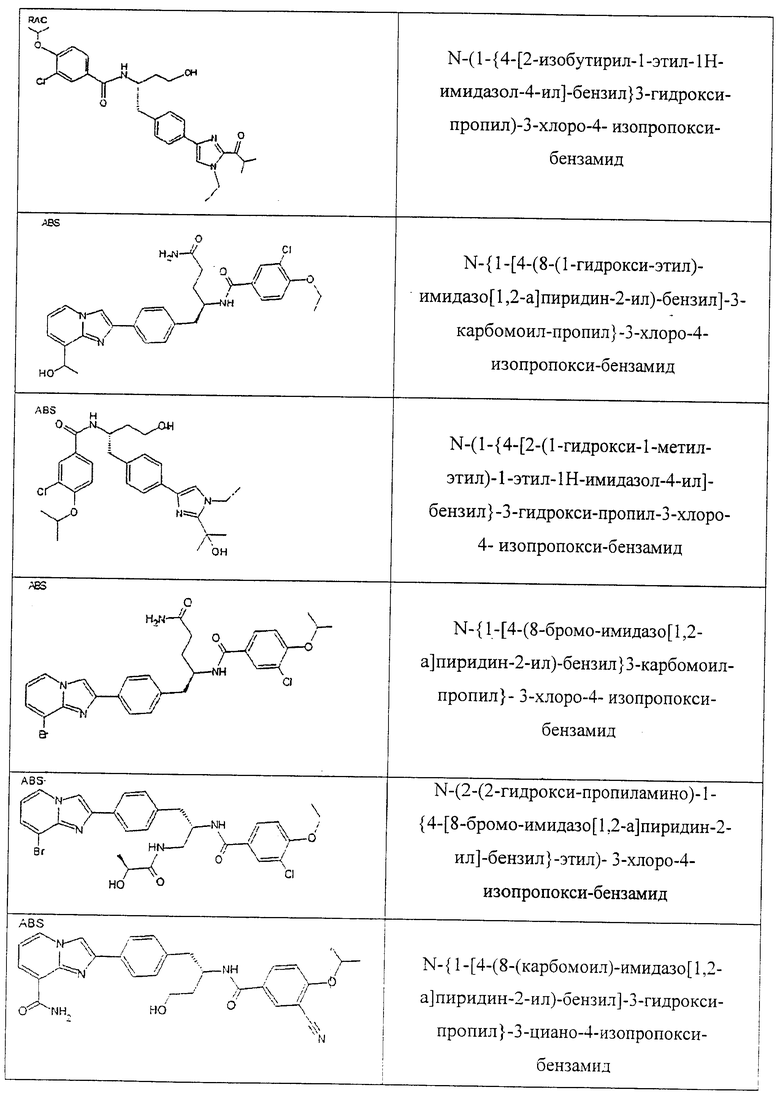
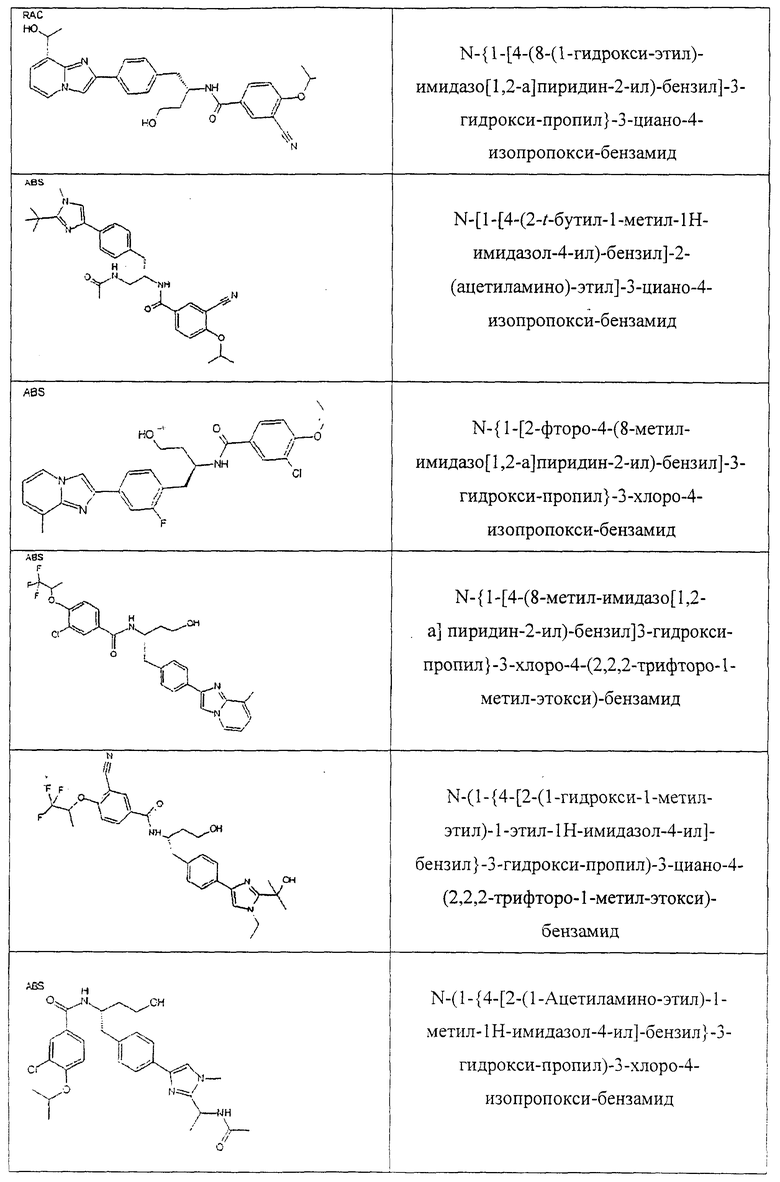
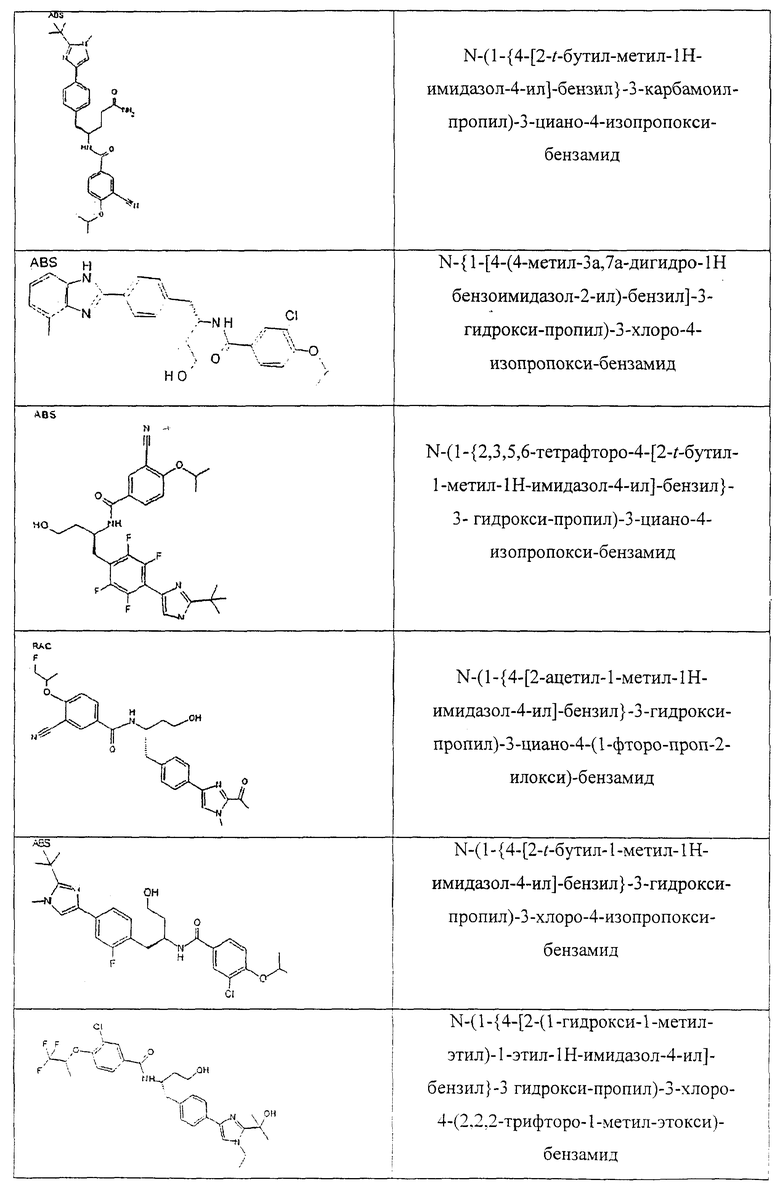
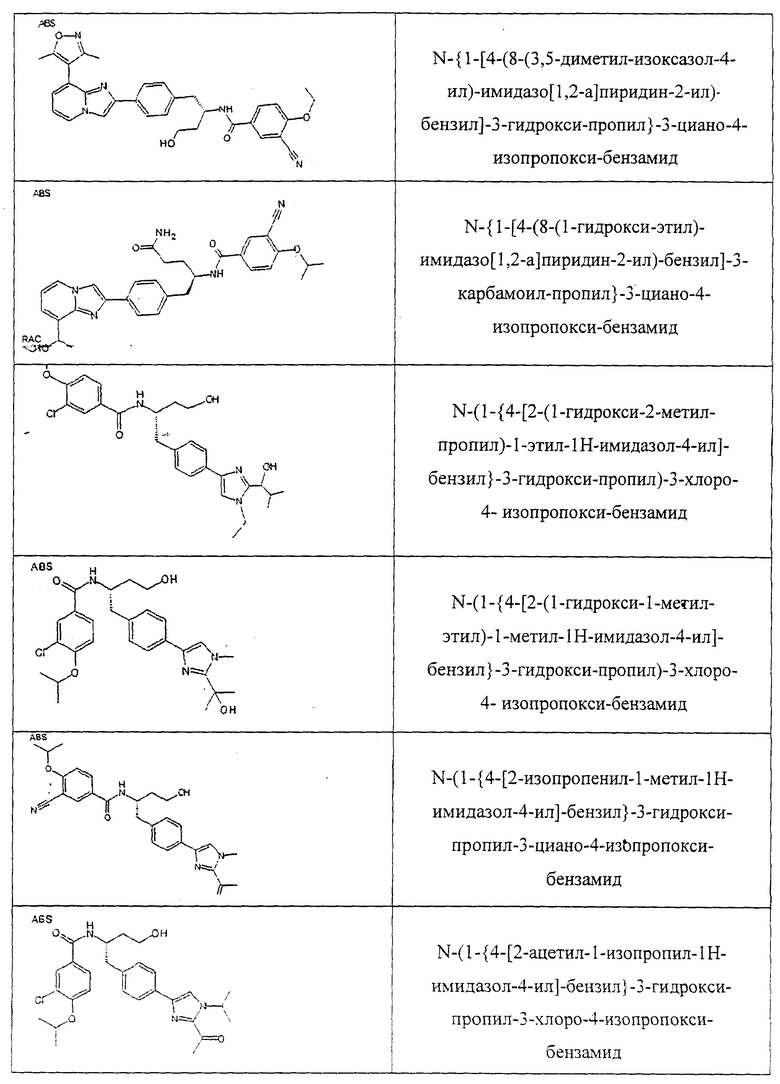
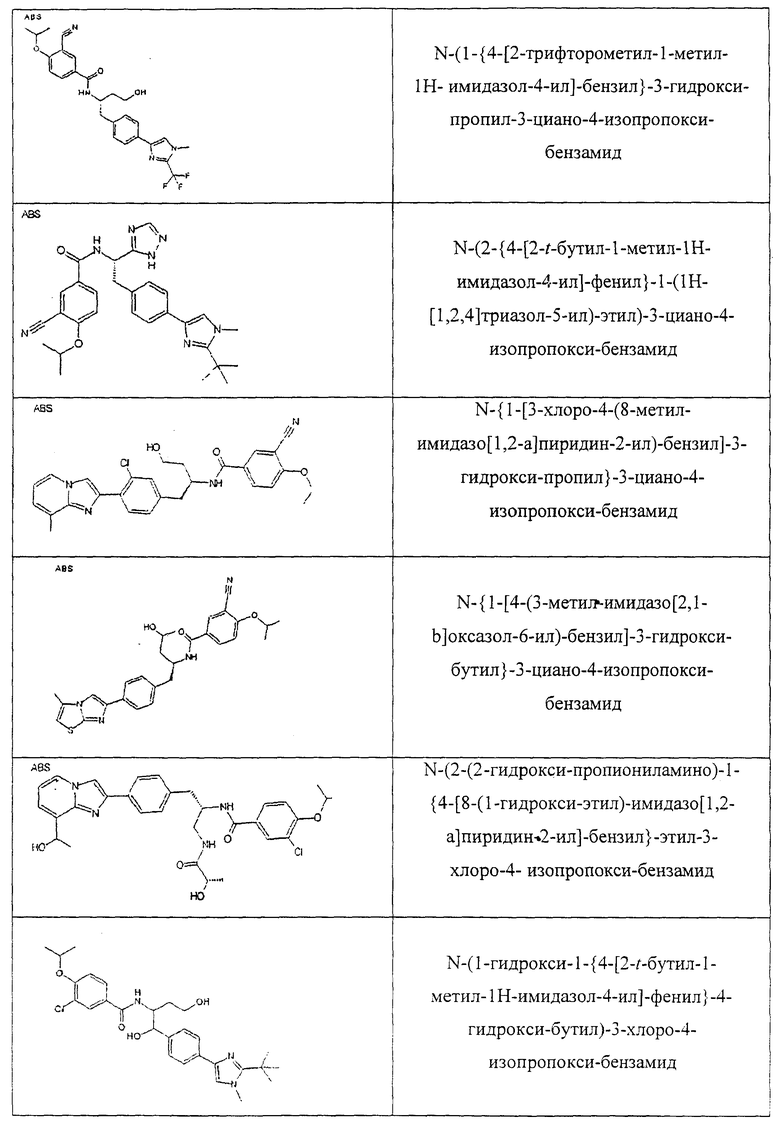
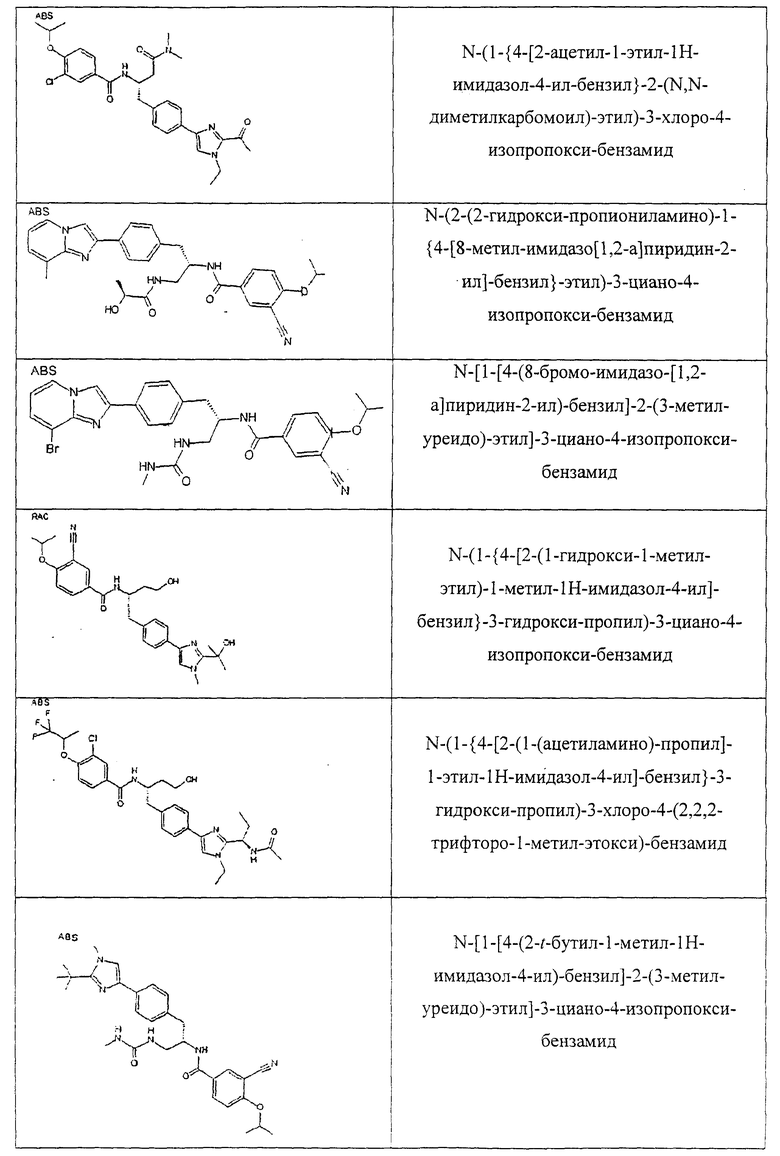
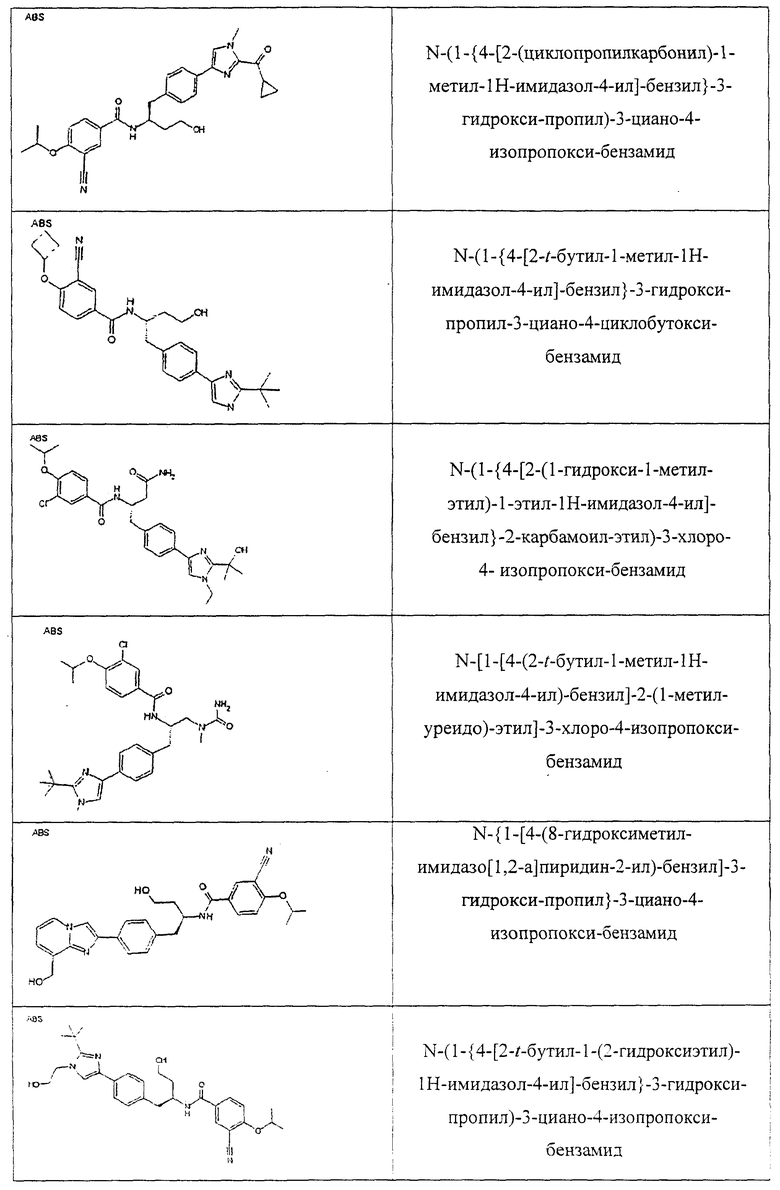
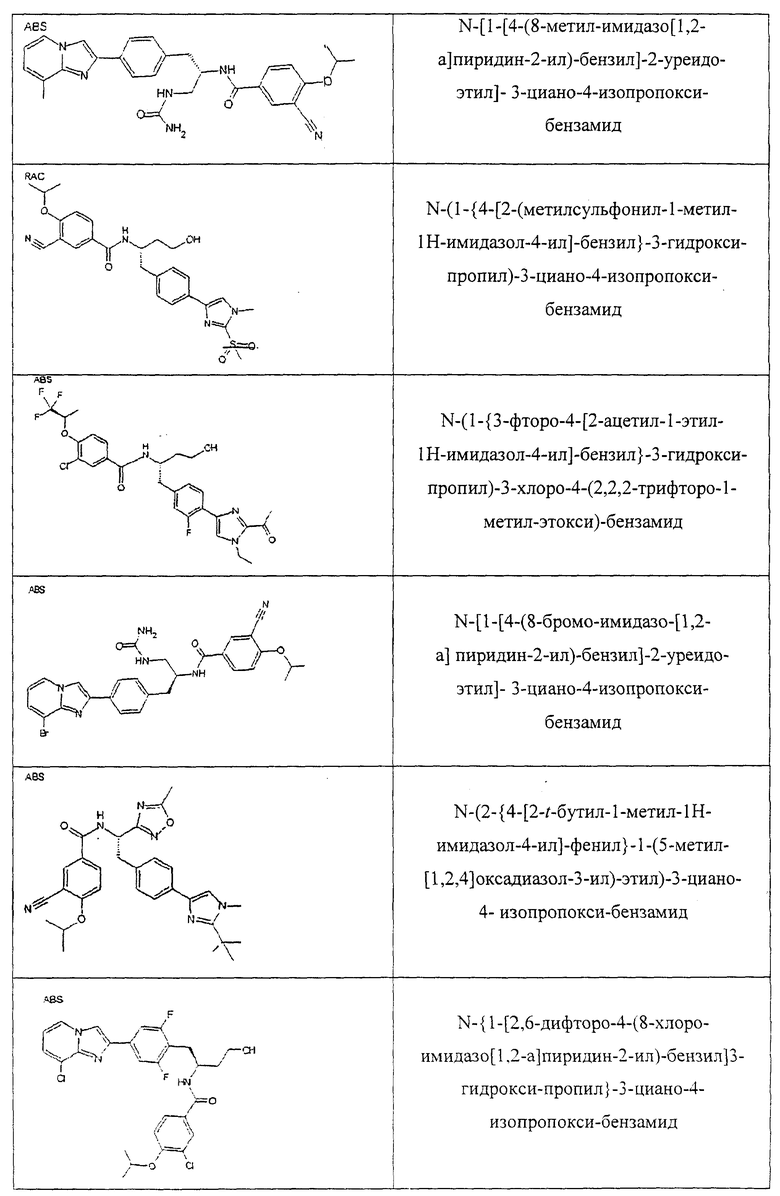
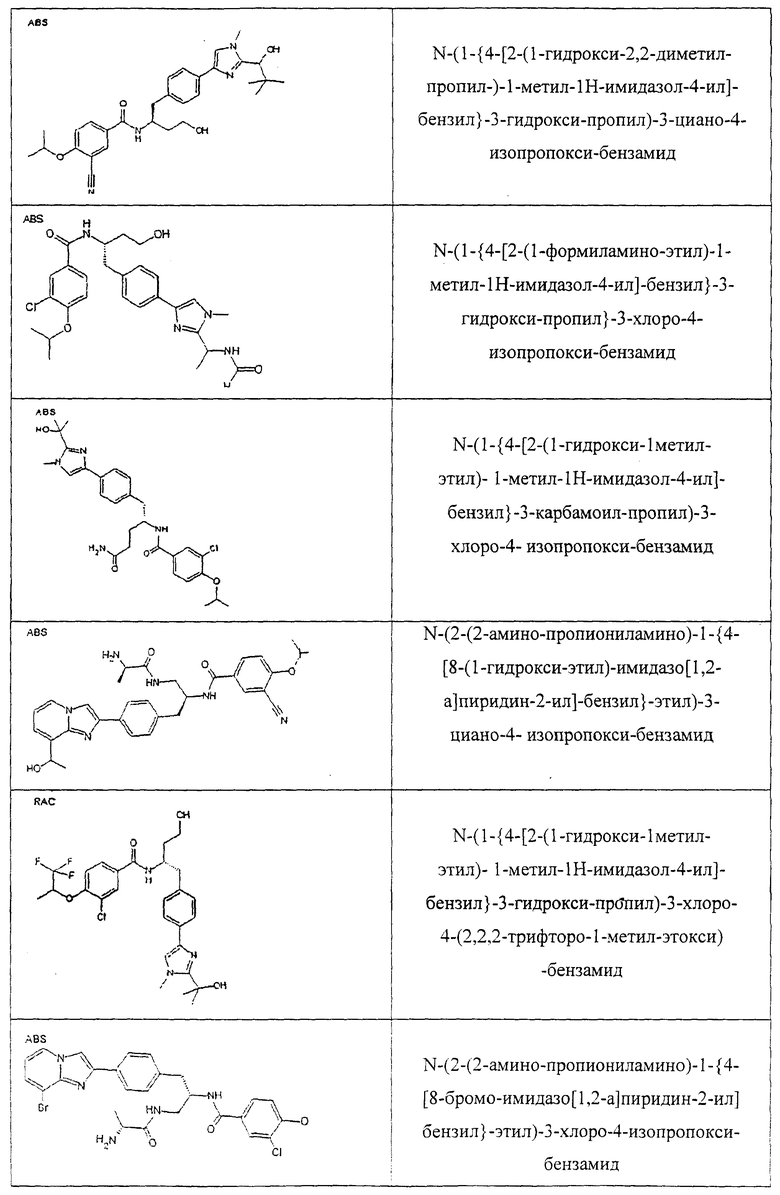
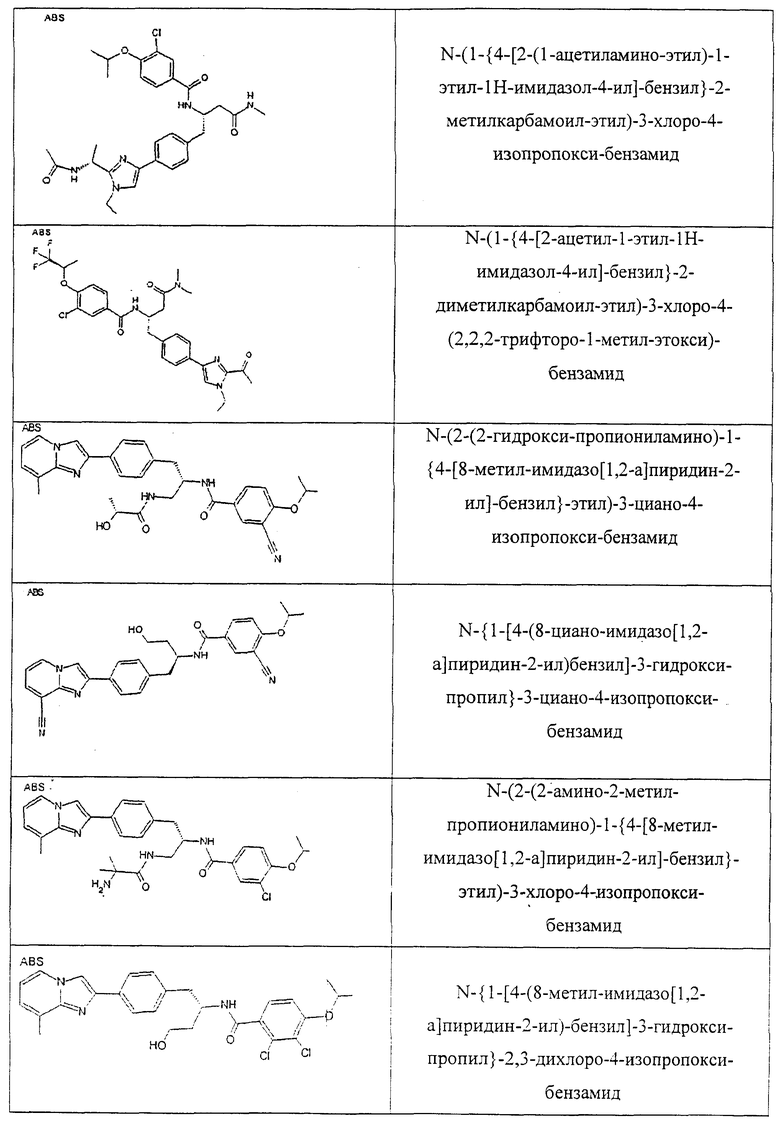
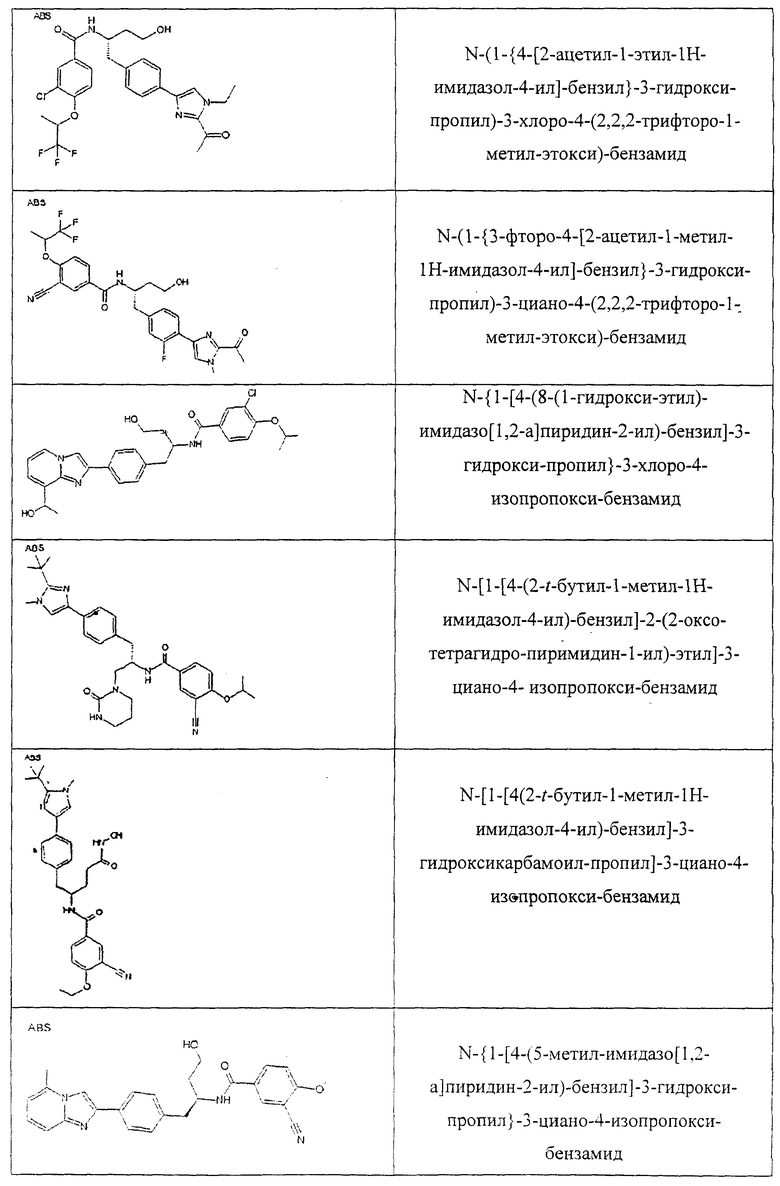
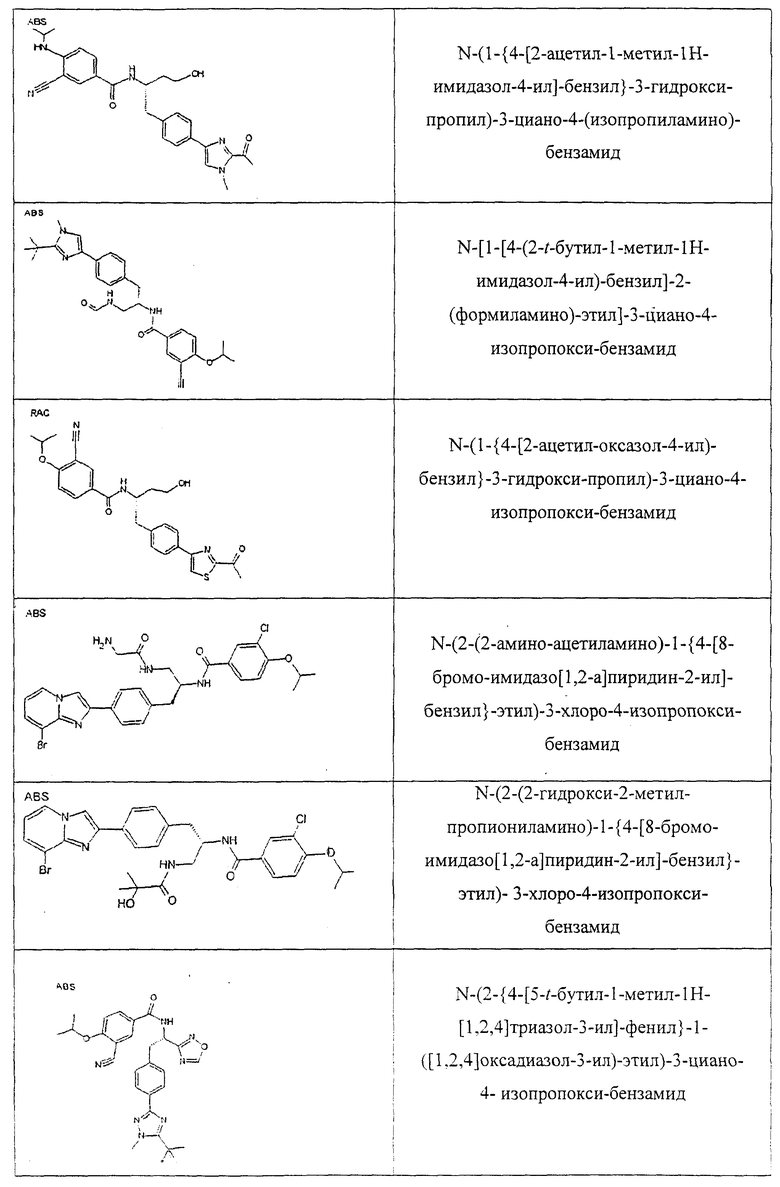
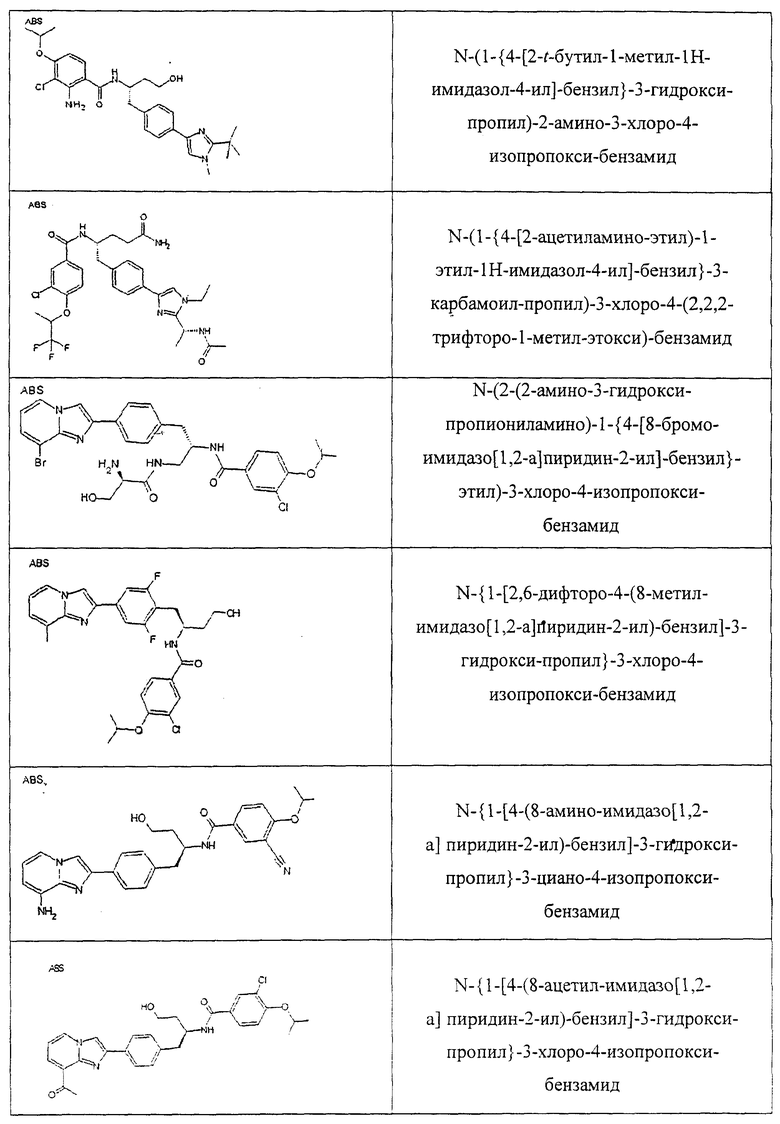
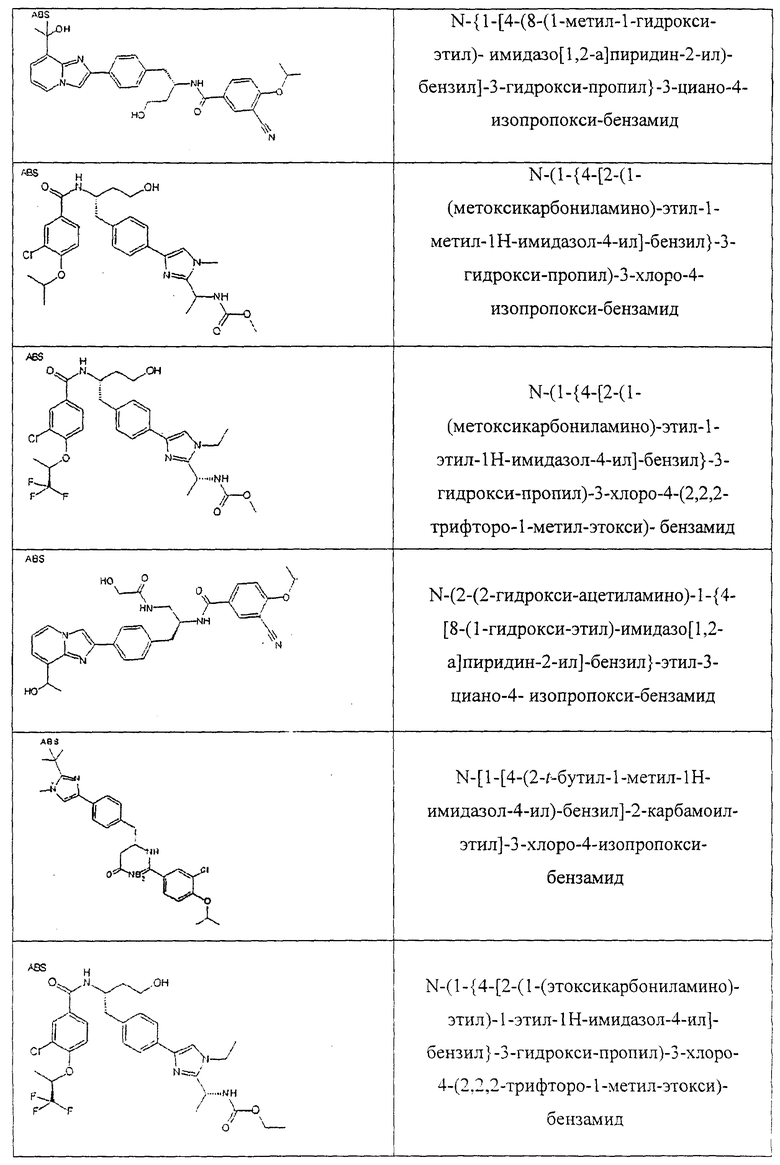
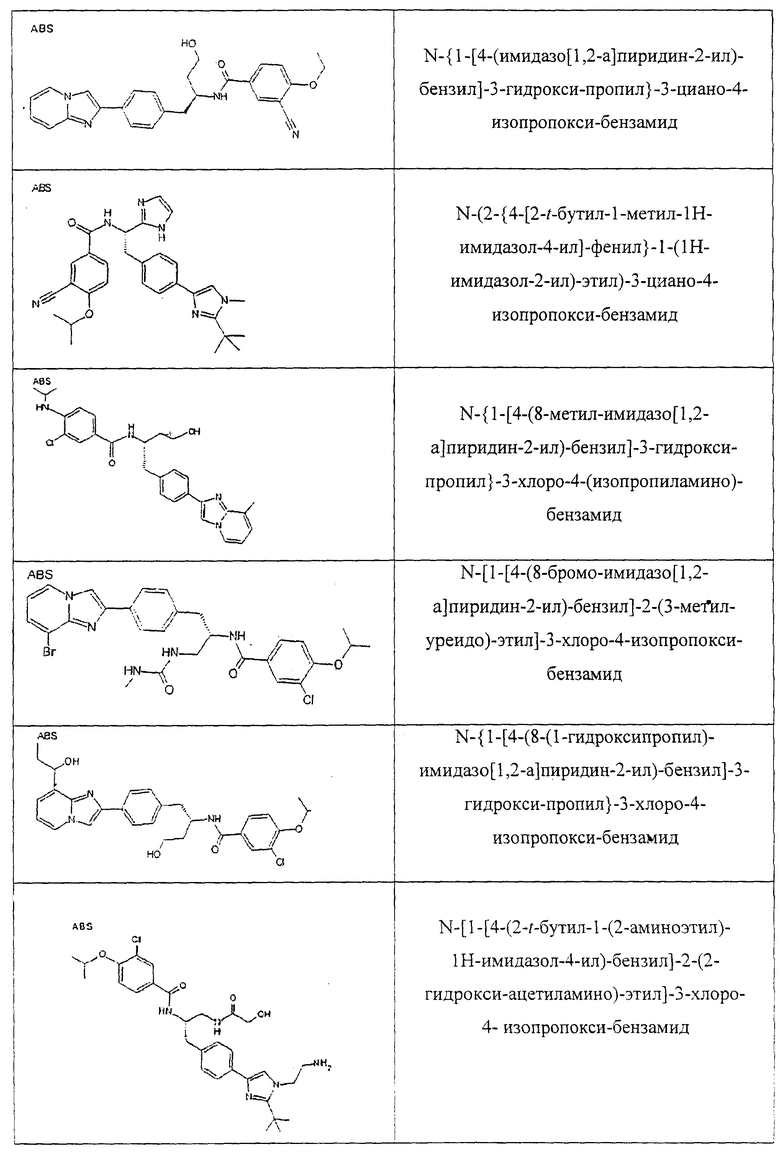
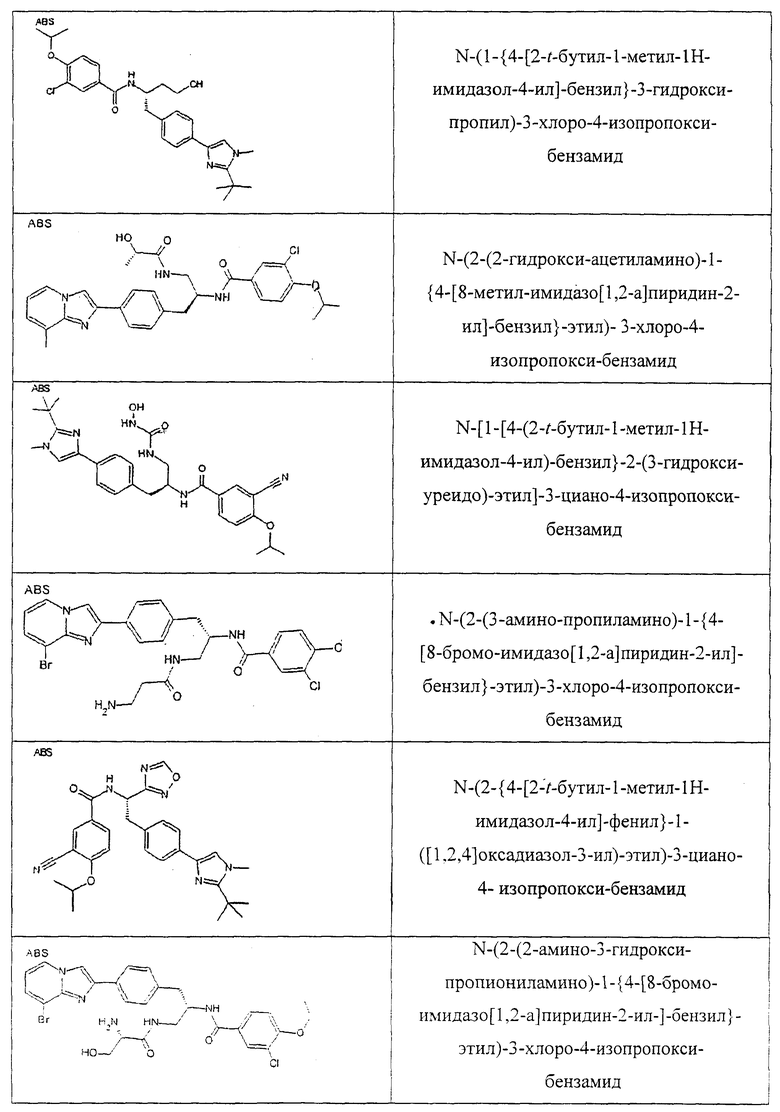
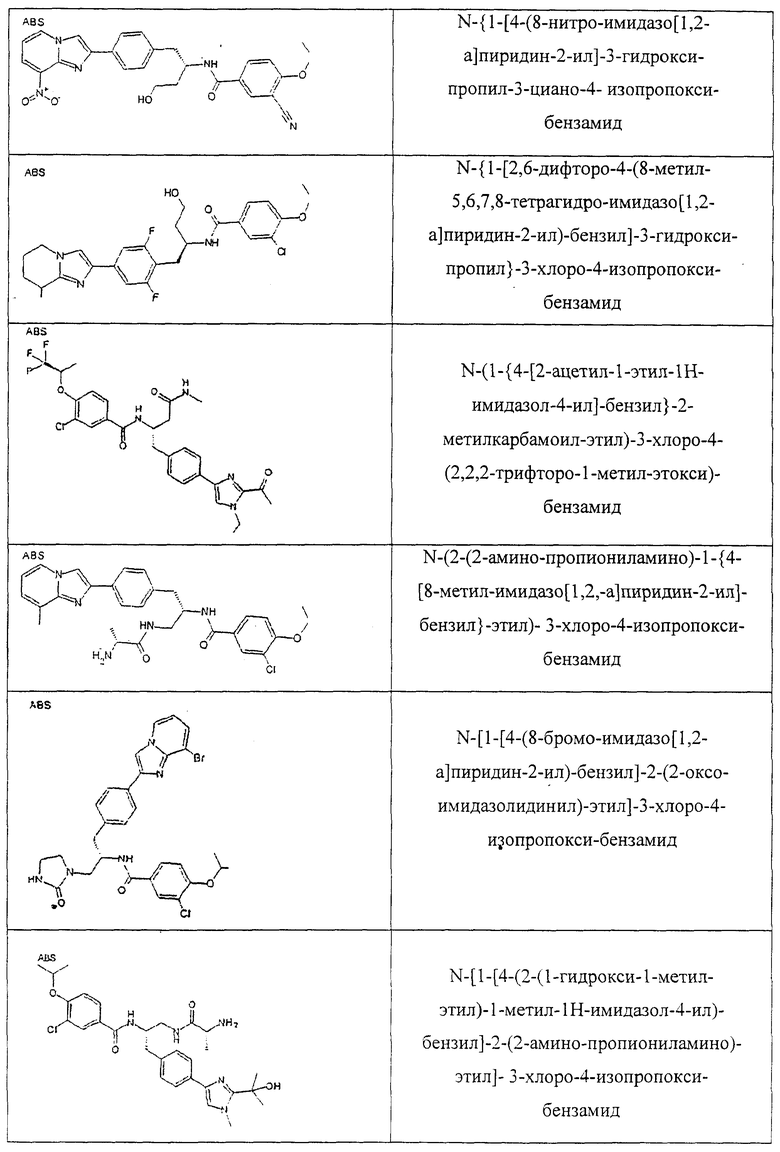
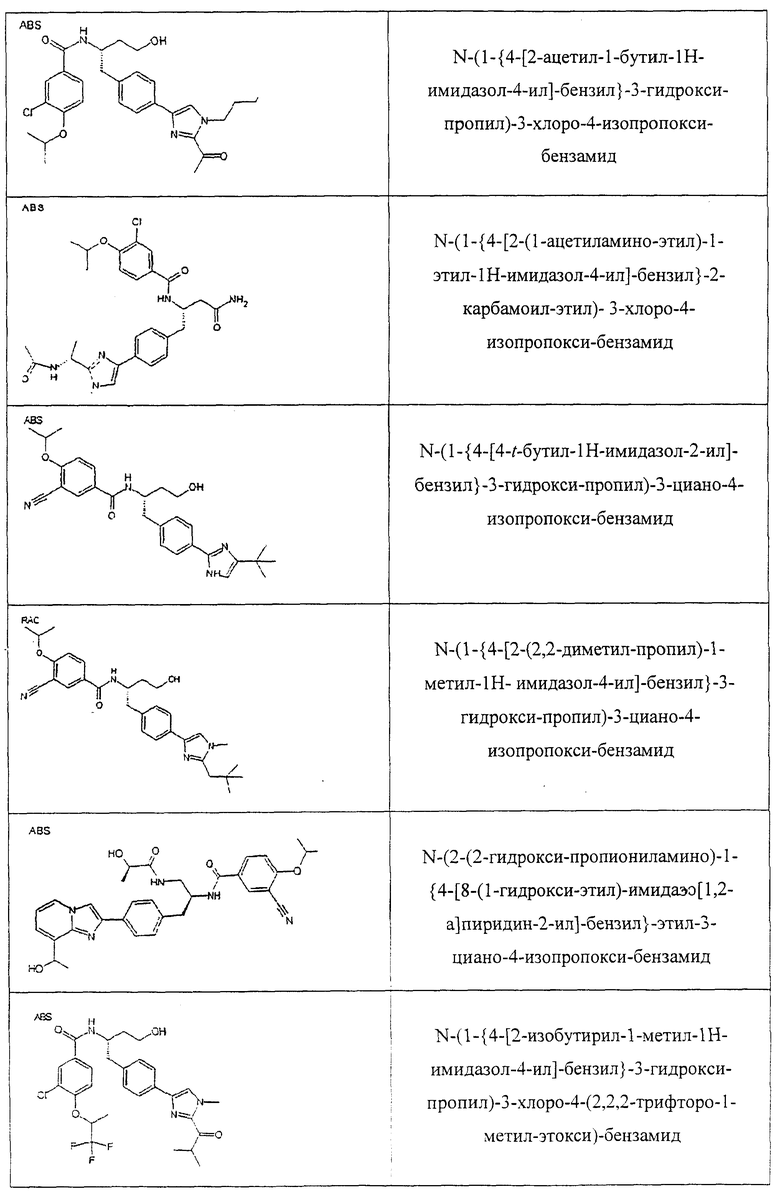
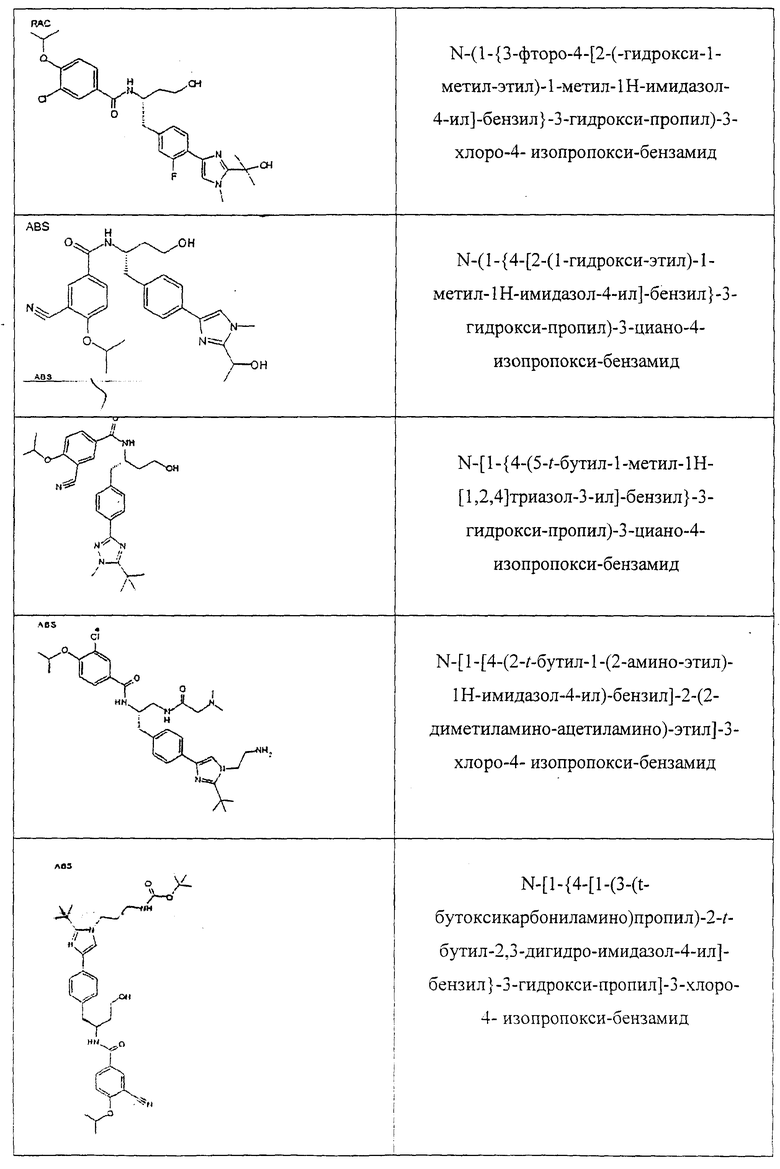
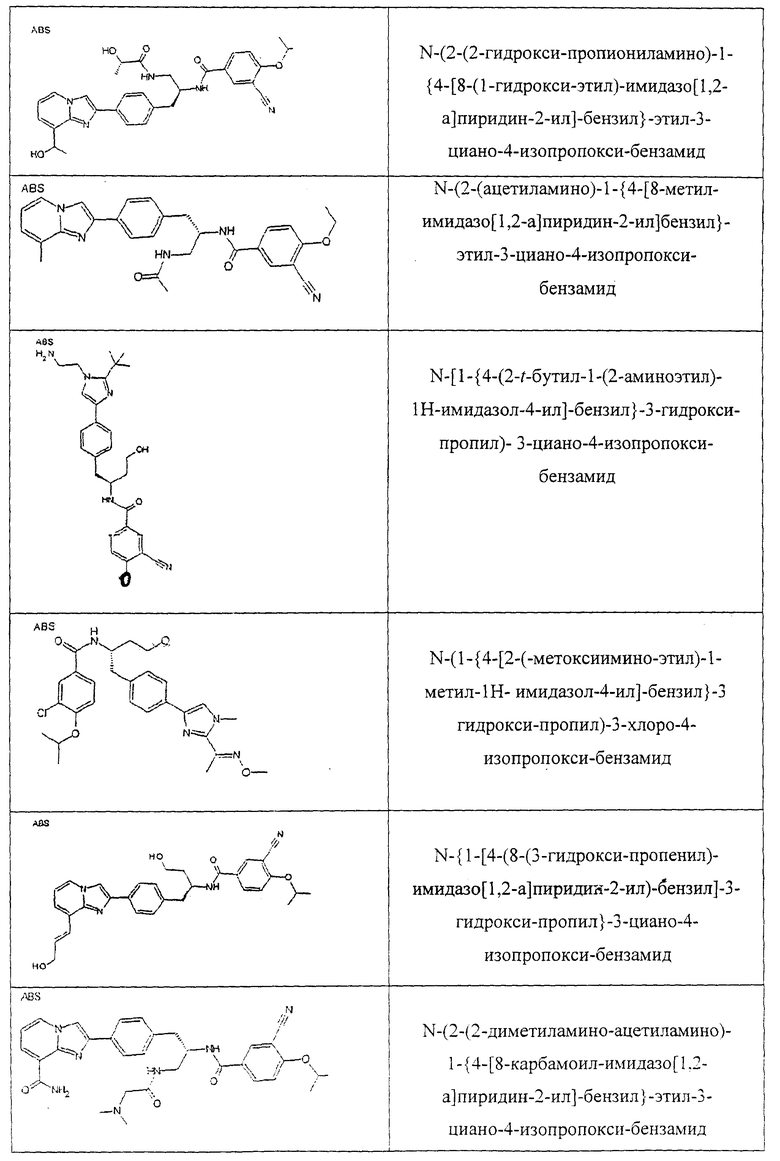
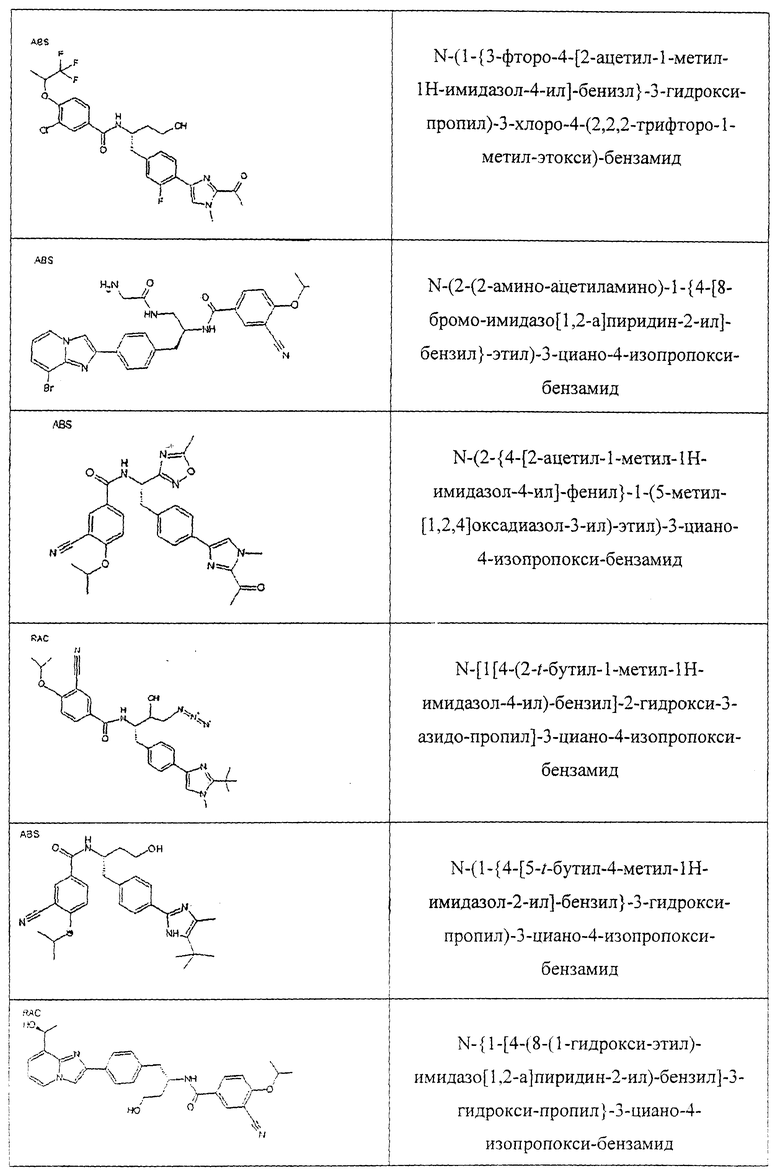
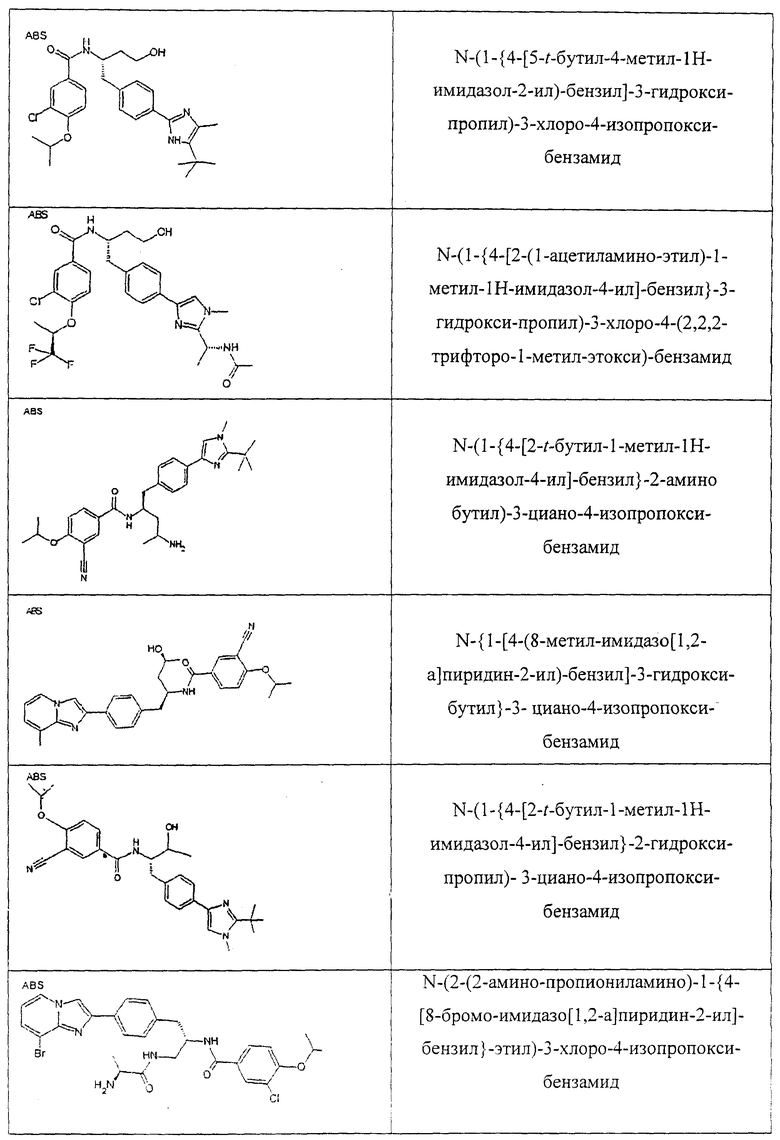
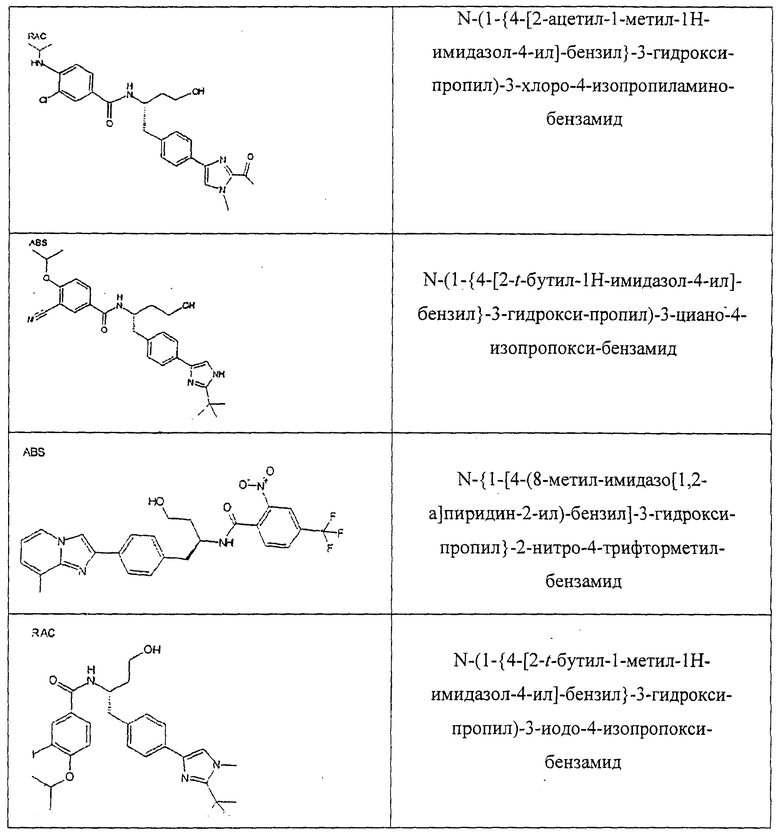
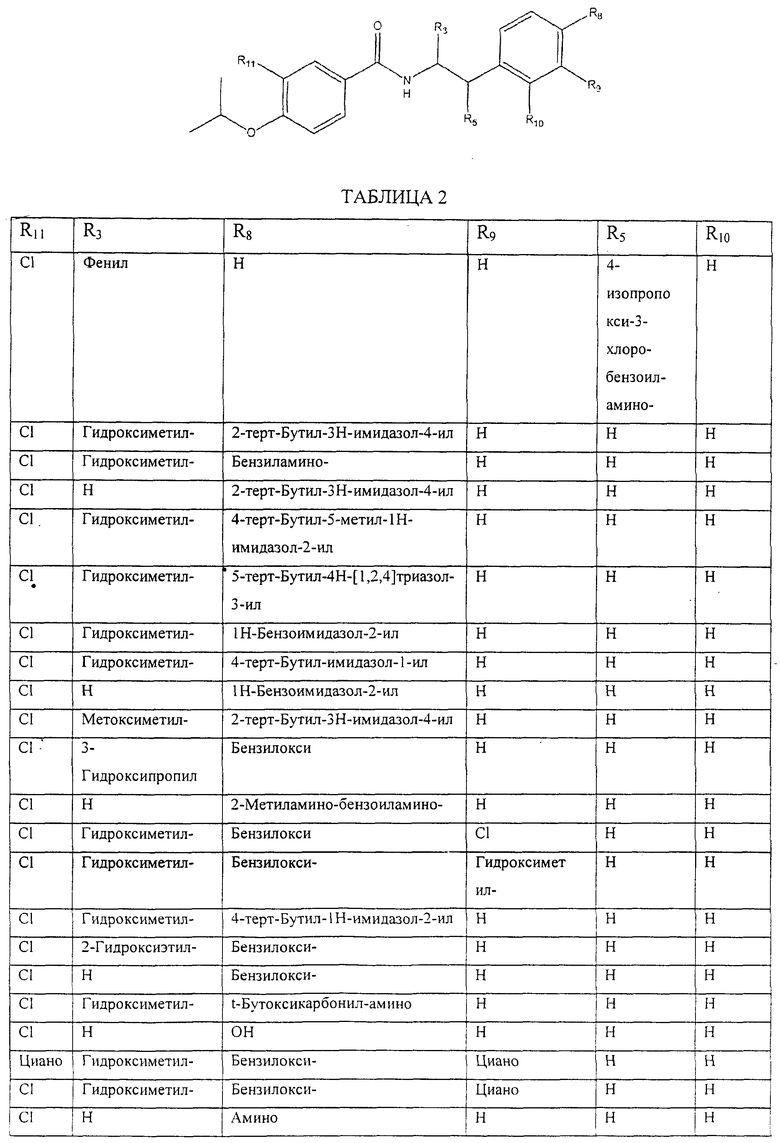
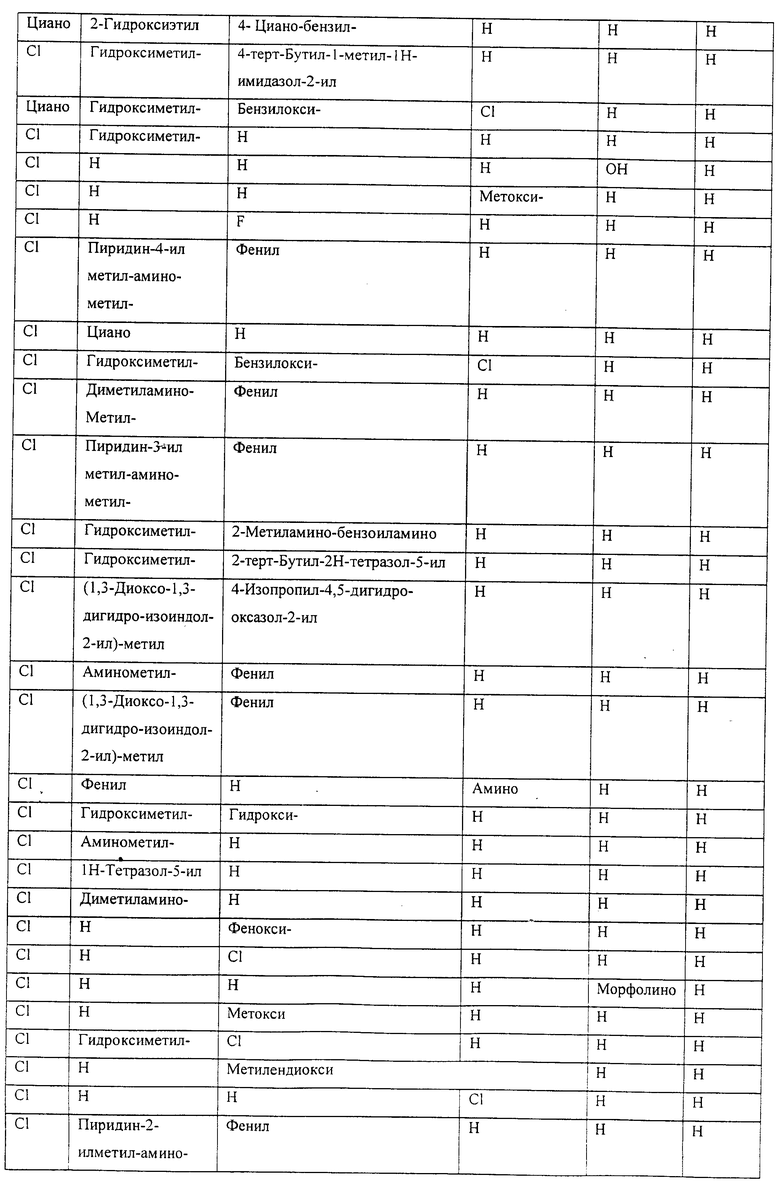
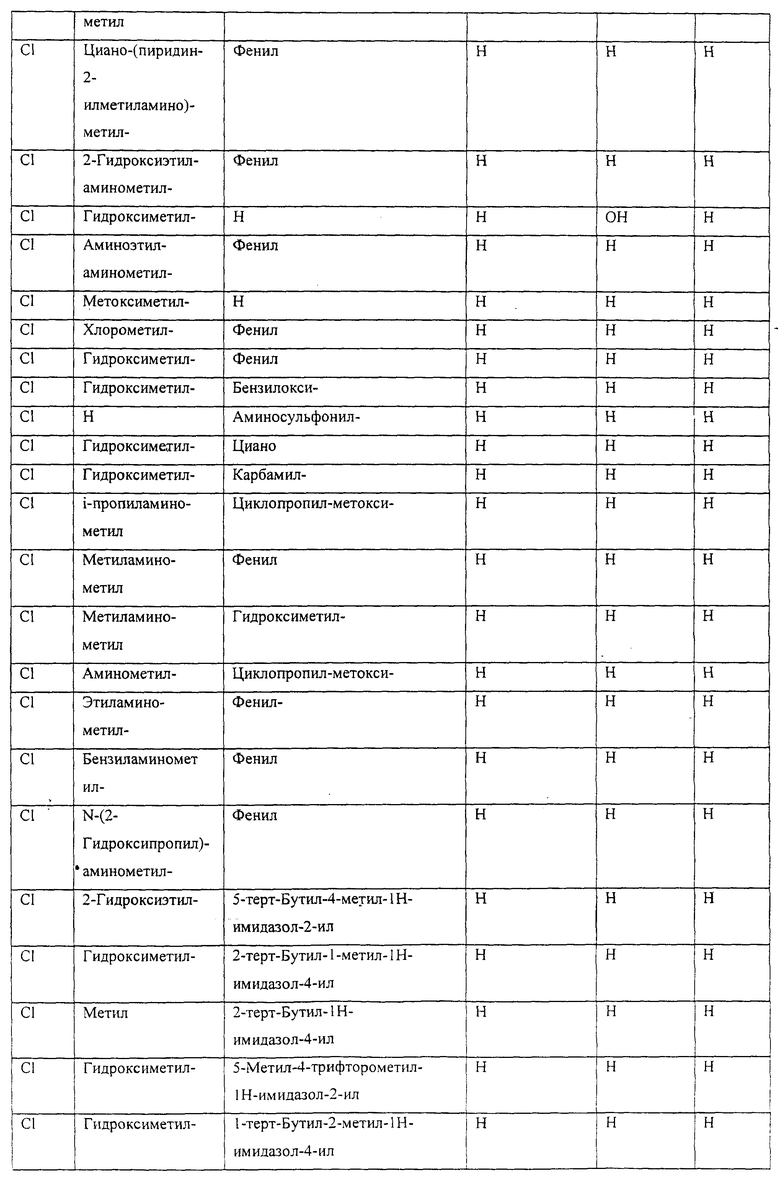
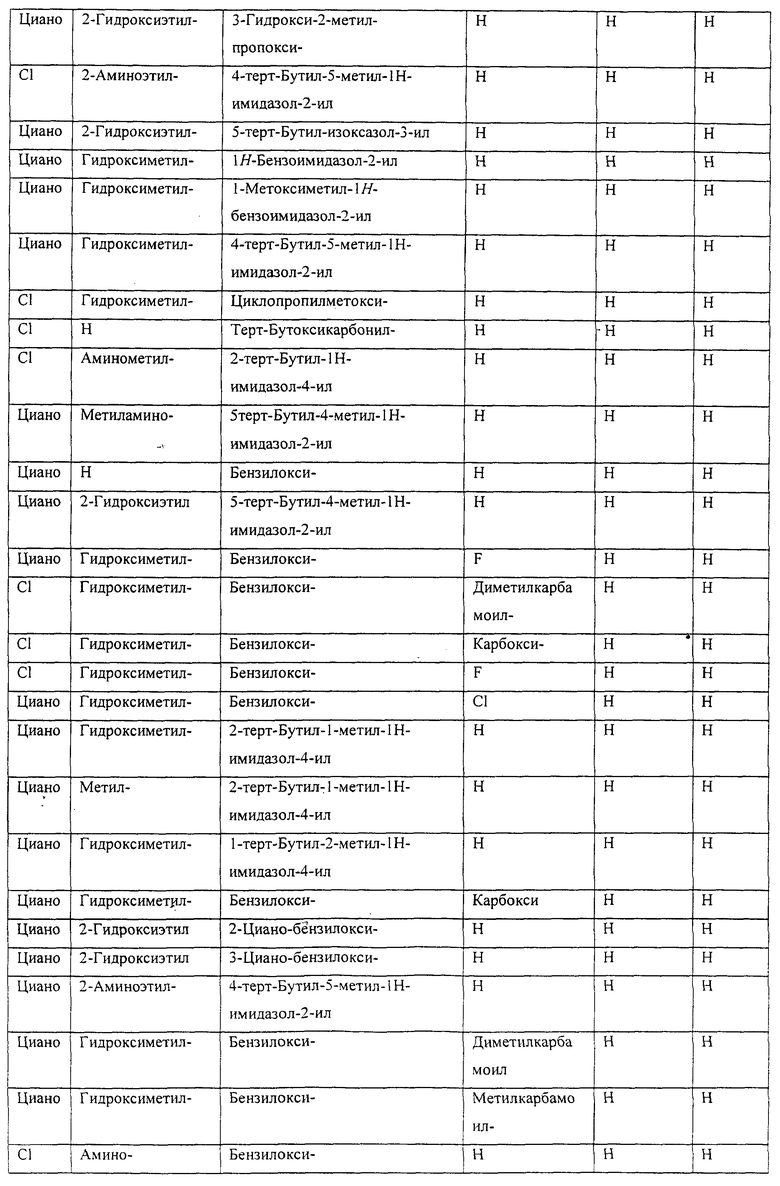
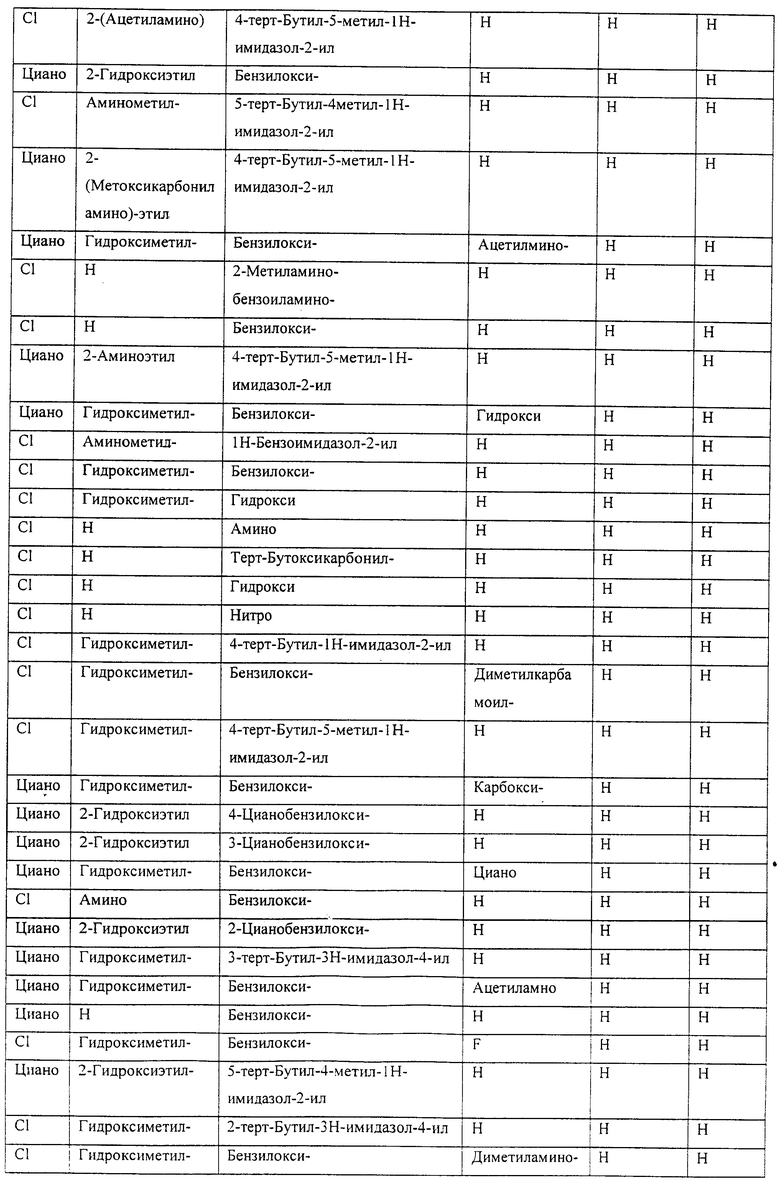
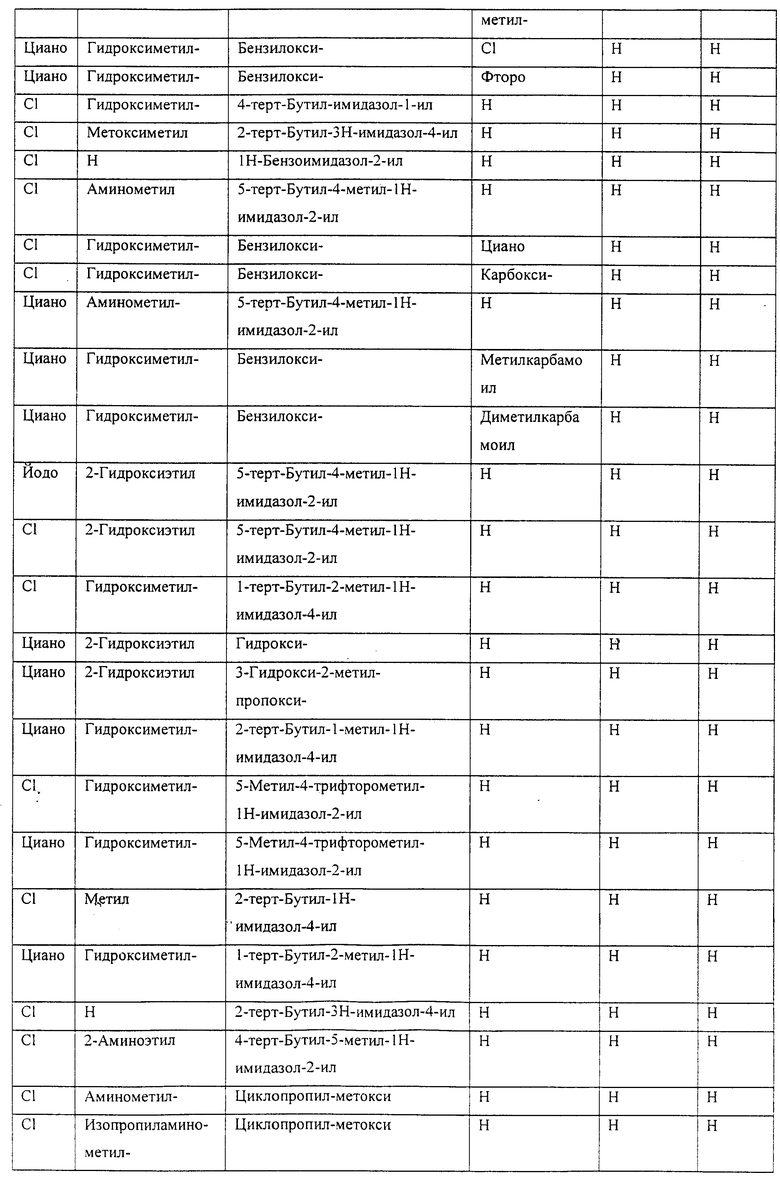
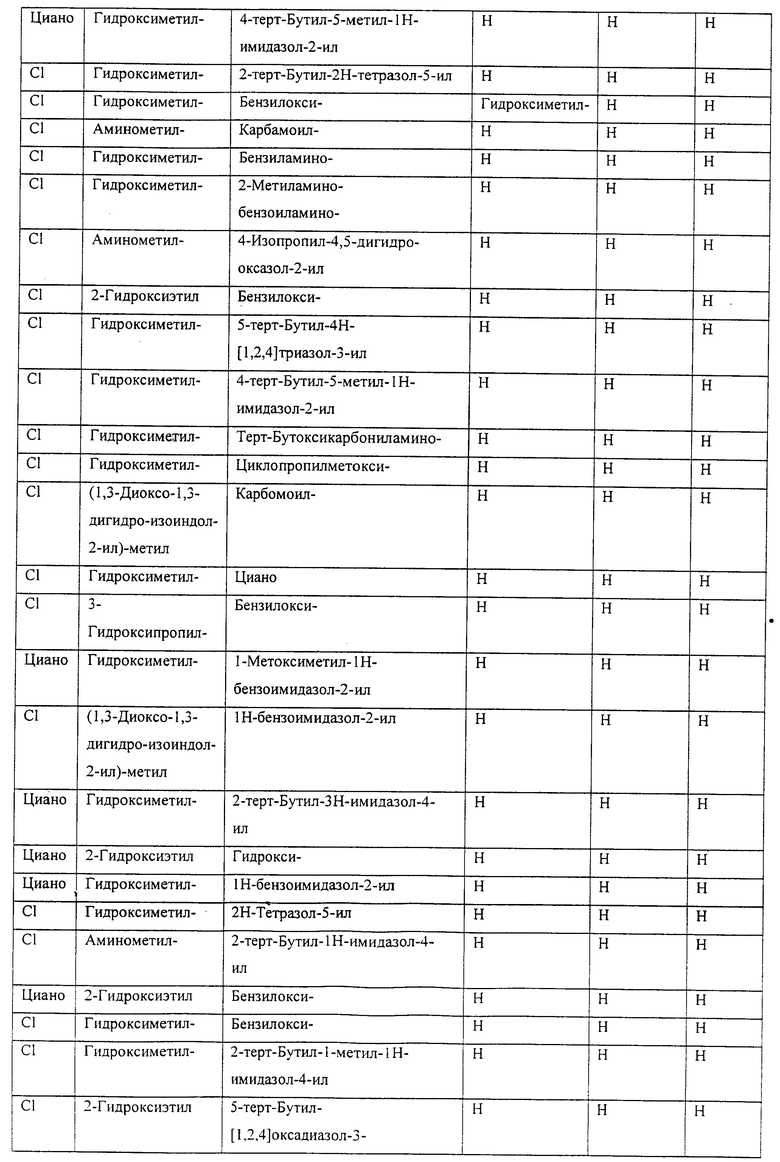
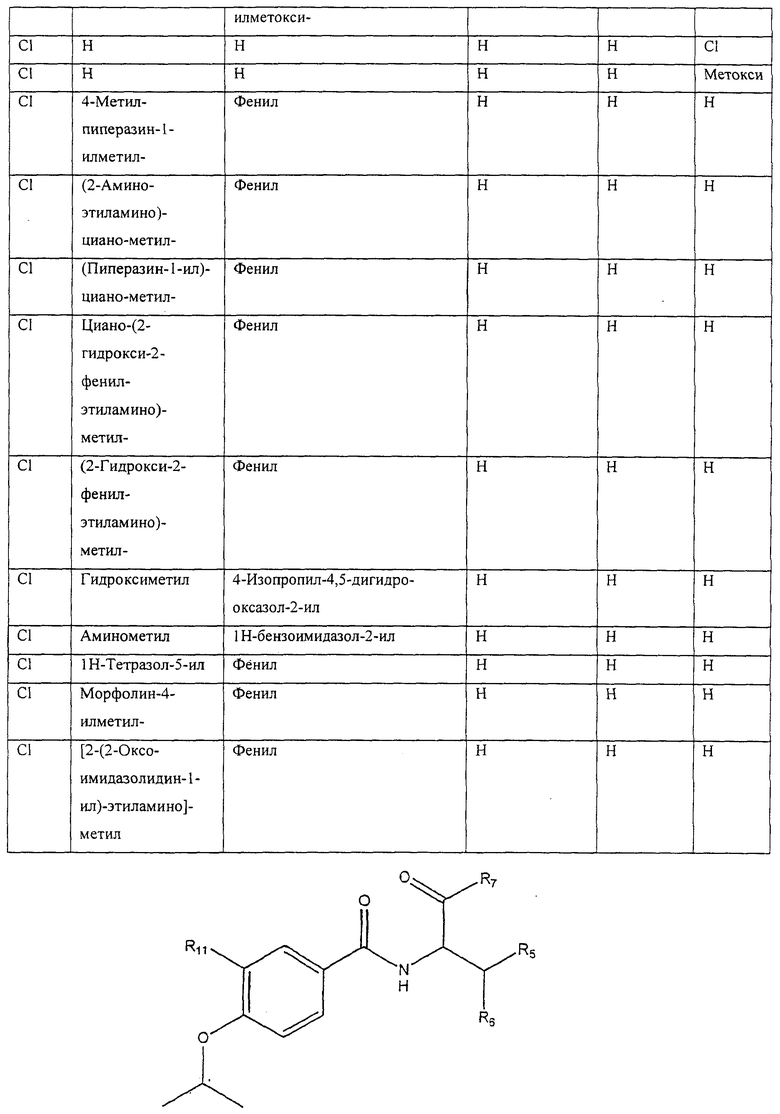


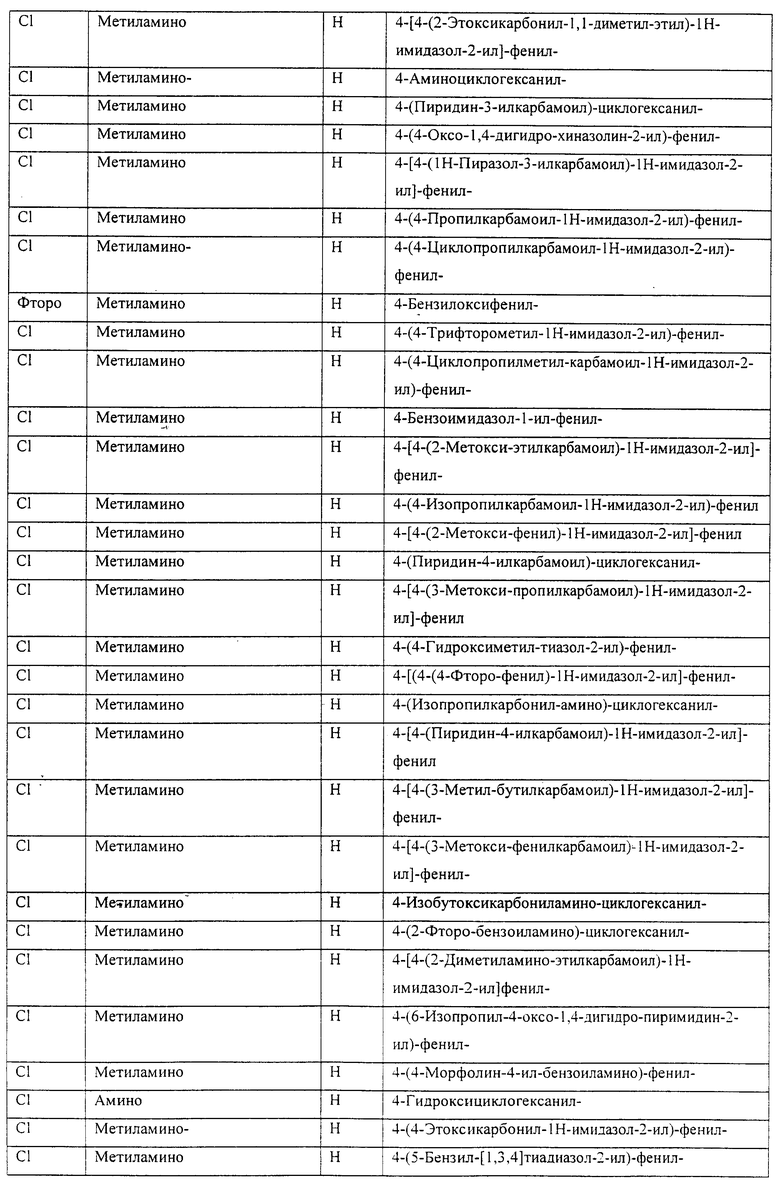



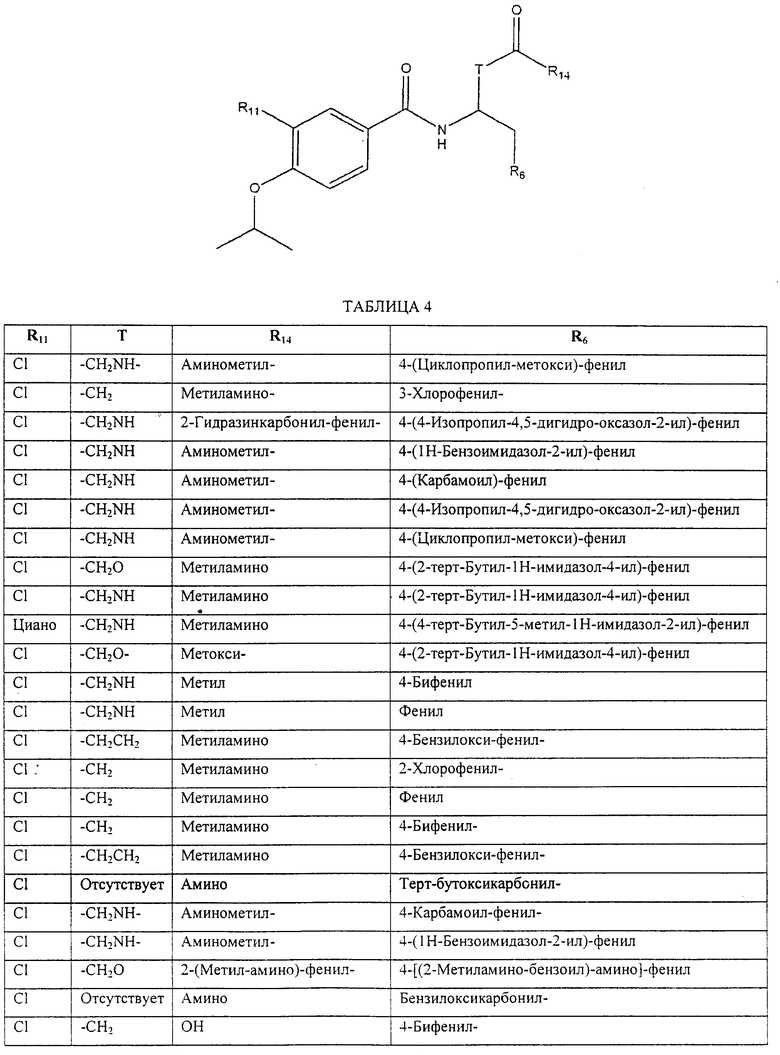

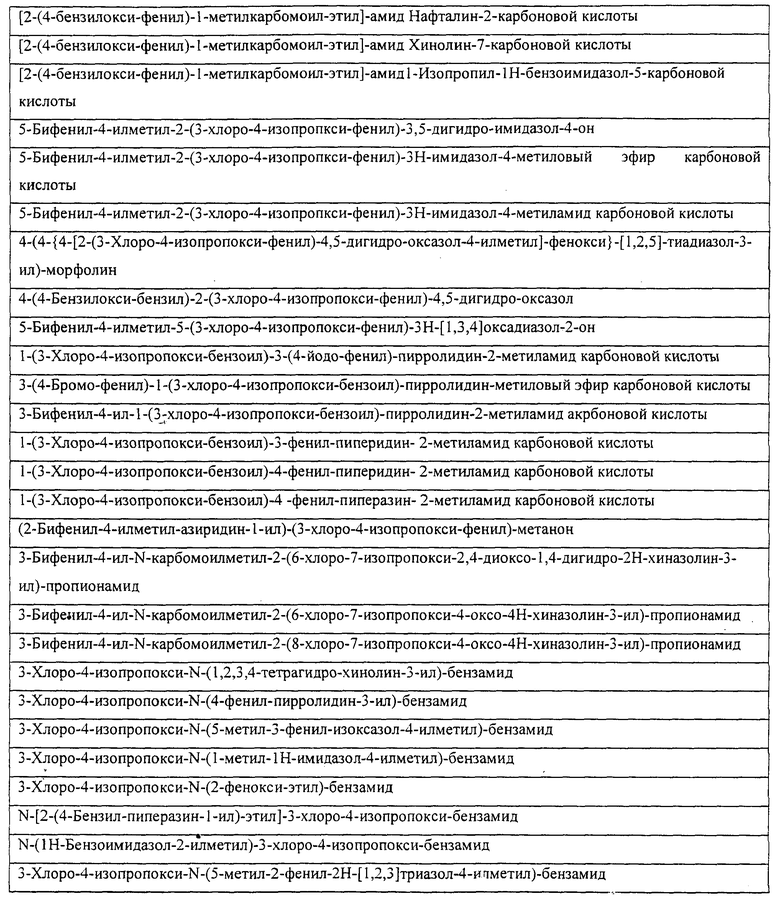
| название | год | авторы | номер документа |
|---|---|---|---|
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ | 2006 |
|
RU2408584C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| ФЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АГОНИСТОВ КАННАБИНОИДНОГО РЕЦЕПТОРА 2 | 2016 |
|
RU2728823C1 |
| 1-ГИДРОКСИИМИНО-3-ФЕНИЛ-ПРОПАНЫ | 2011 |
|
RU2579114C9 |
| ГЕМИНАЛЬНО-ЗАМЕЩЕННЫЕ ЦИАНОЭТИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ JANUS КИНАЗ | 2014 |
|
RU2664533C2 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИЧЕСКИХ ПРЕПАРАТАХ | 2013 |
|
RU2701156C2 |
| ПРОИЗВОДНЫЕ 3-АМИНОПИРРОЛИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ХЕМОКИНОВ | 2003 |
|
RU2355679C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ 11БЕТА-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ 1 | 2009 |
|
RU2531272C2 |
Настоящее изобретение относится к следующим соединениям: N-(1-{4-[2-(1-ацетиламиноэтил)-1-этил-1Н-имидазол-4-ил]-бензил}-3-гидроксипропил)-3-хлоро-4-(2,2,2-трифтор-1-метилэтокси)-бензамид, N-(1-{4-[2-(1-метил-1-гидроксиэтил)-1-этил-1Н-имидазол-4-ил]-бензил)-3-гидроксипропил)-3-хлоро-4-(2,2,2-трифтор-1-метилэтокси)-бензамид, N-(1-{4-[2-(1-гидрокси-1-метилэтил)-1-метил-1Н-имидазол-4-ил]-бензил}-3-гидроксипропил)-3-хлоро-4-(2,2,2-трифтор-1-метилэтокси)-бензамид, 3-хлоро-N-[2-[(N,N-диметилглицил)амино]-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид, 3-хлоро-N-(1-(2-(диметиламино)ацетамидо)-3-(4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил)пропан-2-ил)-4-изопропоксибензамид, 3-хлоро-N-(2-[(2-метилаланил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид, 3-хлоро-N-[(3-гидрокси)-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид, а также к их фармацевтически приемлемым солям. Такие соединения и соли могут быть полезны для лечения клеточно-пролиферативных заболеваний и нарушений путем модуляции активности митотического кинезина CENP-E. 4 н. и 22 з.п. ф-лы, 6 табл.
1. Соединение
N-(1-{4-[2-(1-Ацетиламино-этил)-1-этил-1Н-имидазол-4-ил]-бензил}-3-гидрокси-пропил)-3-хлоро-4-(2,2,2-трифтор-1-метил-этокси)-бензамид:
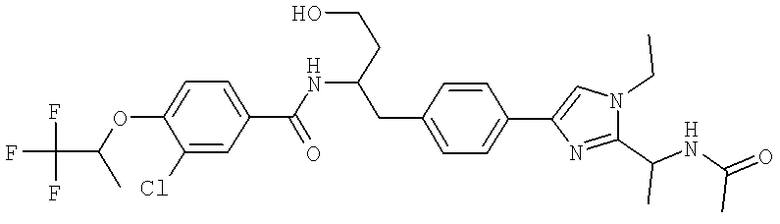 ,
,
N-(1-{4-[2-(1-метил-1-гидрокси-этил)-1-этил-1Н-имидазол-4-ил]-бензил}-3-гидрокси-пропил)-3-хлоро-4-(2,2,2-трифтор-1-метил-этокси)-бензамид:
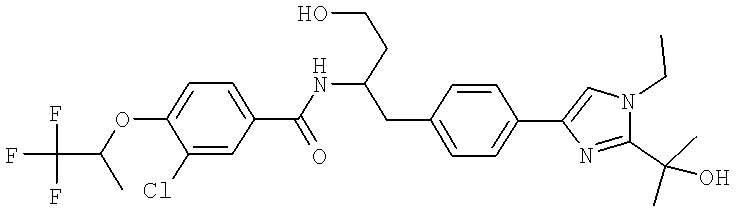 ,
,
N-(1-{4-[2-(1-гидрокси-1-метил-этил)-1-метил-1Н-имидазол-4-ил]-бензил}-3-гидрокси-пропил)-3-хлоро-4-(2,2,2-трифтор-1-метил-этокси)-бензамид:
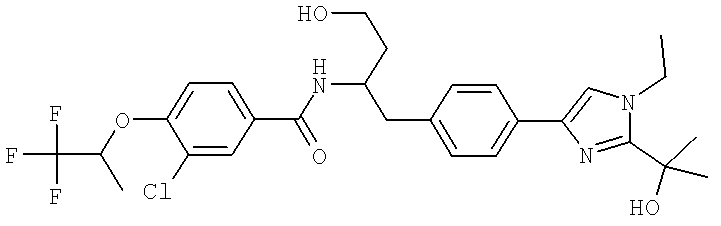 ,
,
3-хлоро-N-[2-[(N,N-диметилглицил)амино]-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид:
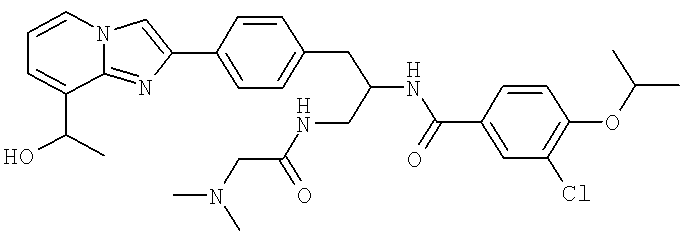 ,
,
3-хлоро-N-(1-(2-(диметиламино)ацетамидо)-3-(4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил)пропан-2-ил)-4-изопропоксибензамид:
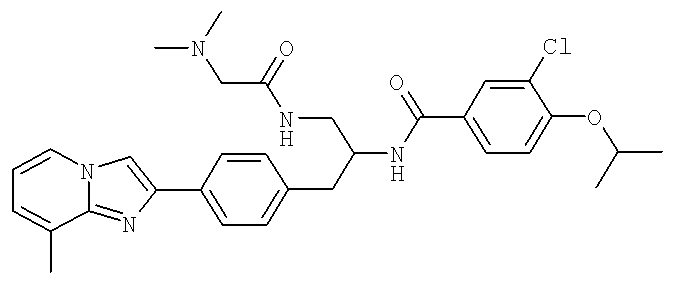 ,
,
3-хлоро-N-(2-[(2-метилаланил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид:
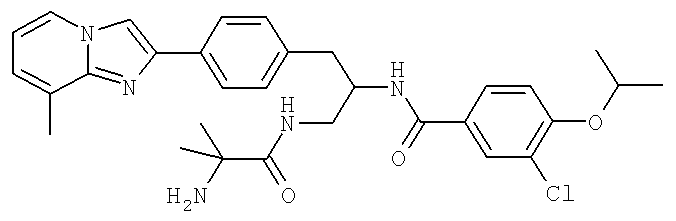 , и
, и
3-хлоро-N-[(3-гидрокси)-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид:
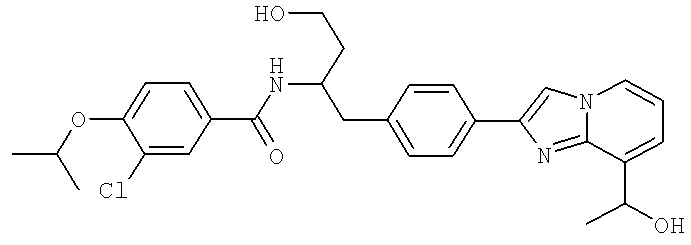 ;
;
или его фармацевтически приемлемая соль.
2. Фармацевтическая композиция для ингибирования активности кинезина CENP-E, включающая, по крайней мере, один фармацевтический наполнитель и соединение по п.1, или его фармацевтически приемлемую соль.
3. Композиция по п.2, отличающаяся тем, что она выполнена в форме, пригодной для пути введения, выбранного из группы, включающей пероральный, подкожный, внутривенный, внутриназальный, трансдермальный, внутрибрюшинный, внутримышечный, внутрипульмонарный, вагинальный, ректальный и интраокулярный пути введения.
4. Композиция по п.3, отличающаяся тем, что она выполнена в форме, пригодной для перорального введения.
5. Композиция по п.4, отличающаяся тем, что она выполнена в виде таблетки, капсулы или жидкости.
6. Композиция по п.4, отличающаяся тем, что, по крайней мере, один фармацевтический наполнитель выбран из разбавителей, связующих веществ, глидантов, лубрикантов, разрыхлителей, пигментных красителей, отдушек, подсластителей, полимеров, восков и других замедляющих растворимость веществ.
7. Композиция по п.3, отличающаяся тем, что она выполнена в форме, пригодной для внутривенного введения.
8. Композиция по п.7, отличающаяся тем, что, по крайней мере, один фармацевтический наполнитель включает стерильный раствор сахаров, аминокислот или электролитов.
9. Композиция по п.7, отличающаяся тем, что, по крайней мере, один фармацевтический наполнитель является водой для инъекции USP.
10. Композиция по п.2, отличающаяся тем, что она выполнена в форме, пригодной для парентерального введения.
11. Композиция по п.10, отличающаяся тем, что, по крайней мере, один фармацевтический наполнитель включает стерильный раствор сахаров, аминокислот или электролитов.
12. Композиция по п.10, отличающаяся тем, что, по крайней мере, один фармацевтический наполнитель является водой для инъекции USP.
13. Соединение или фармацевтически приемлемая соль по п.1 для использования в производстве фармацевтической композиции для лечения рака, связанного с активностью кинезина CENP-E.
14. Соединение
3-хлоро-N-[(1S)-2-[(N,N-диметилглицил)амино]-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)этил]-4-[(1-метилэтил)окси]бензамид:
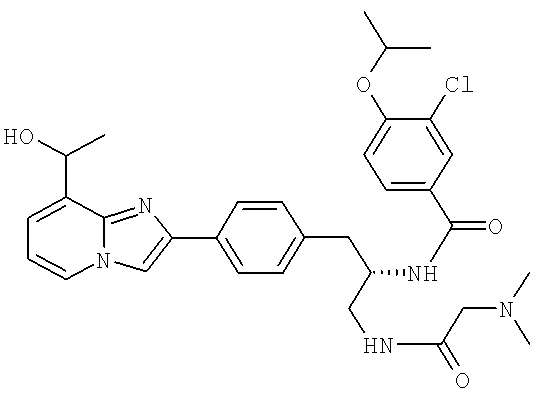 ,
,
(S)-3-хлоро-N-(1-(2-(диметиламино)ацетамидо)-3-(4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил)пропан-2-ил)-4-изопропоксибензамид:
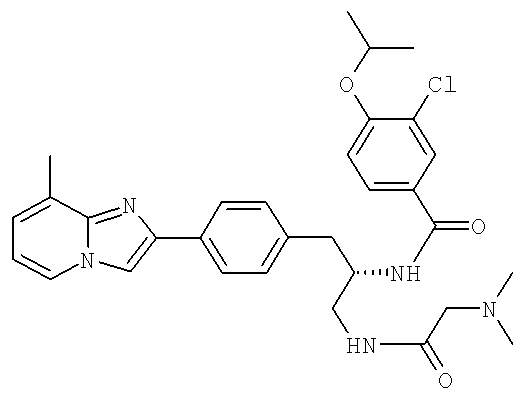 ,
,
3-хлоро-N-((1S)-2-[(2-метилаланил)амино]-1-{[4-(8-метилимидазо[1,2-а]пиридин-2-ил)фенил]метил}этил)-4-[(1-метилэтил)окси]бензамид:
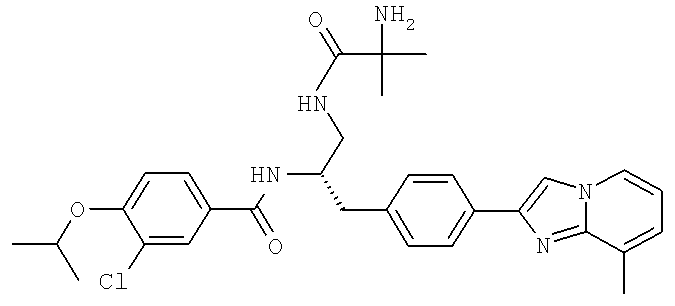
3-хлоро-N-[(1S)-3-гидрокси-1-({4-[8-(1-гидроксиэтил)имидазо[1,2-а]пиридин-2-ил]фенил}метил)пропил]-4-[(1-метилэтил)окси]бензамид:
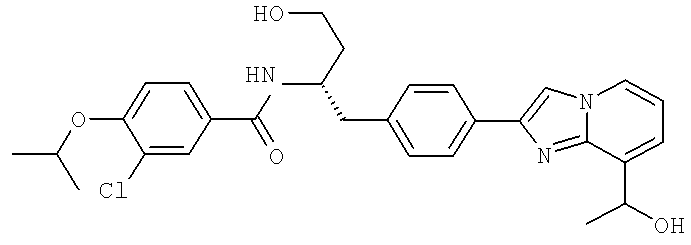 ,
,
N((1S)-1-{4-[2-(1-ацетиламино-этил)-1-этил-1Н-имидазол-4-ил]-бензил}-3-гидрокси-пропил)-3-хлоро-4-(2,2,2-трифторо-1-метил-этокси)-бензамид:
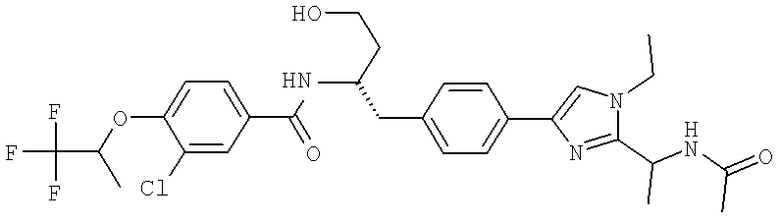 ,
,
N-((1S)-1-{4-[2-(1-метил-1-гидрокси-этил)-1-этил-1Н-имидазол-4-ил]-бензил}-3-гидрокси-пропил)-3-хлоро-4-(2,2,2-трифторо-1-метил-этокси)-бензамид;
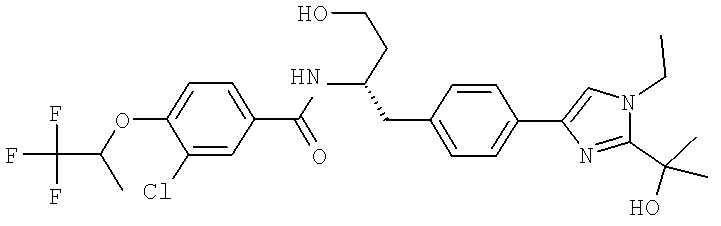 ,
,
(S,R)-N-(1-{4-[2-(1-метил-1-гидрокси-этил)-1-этил-1Н-имидазол-4-ил]-бензил}-3-гидрокси-пропил)-3-хлоро-4-(2,2,2-трифторо-1-метил-этокси)-бензамид:
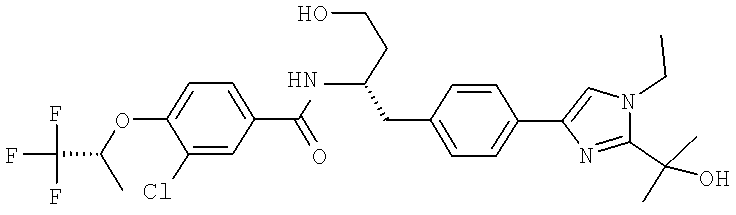 , и
, и
N-((1S)-1-{4-[2-(1-гидрокси-1-метил-этил)-1-метил-1Н-имидазол-4-ил]-бензил}-3-гидрокси-пропил)-3-хлоро-4-(2,2,2-трифторо-1-метил-этокси)-бензамид:
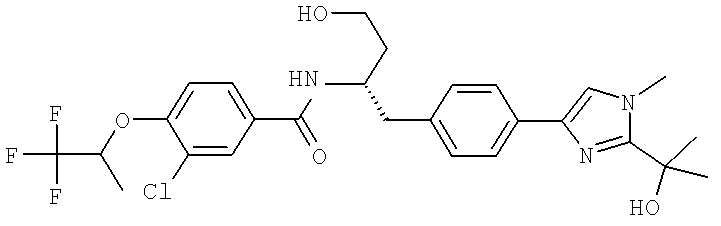
или его фармацевтически приемлемая соль.
15. Фармацевтическая композиция для ингибирования активности кинезина CENP-E, включающая, по крайней мере, один фармацевтический наполнитель и соединение по п.14 или его фармацевтически приемлемая соль.
16. Композиция по п.15, отличающаяся тем, что она выполнена в форме, пригодной для пути введения, выбранного из группы, включающей пероральный, подкожный, внутривенный, внутриназальный, трансдермальный, внутрибрюшинный, внутримышечный, внутрипульмонарный, вагинальный, ректальный и интраокулярный пути введения.
17. Композиция по п.16, отличающаяся тем, что она выполнена в форме, пригодной для перорального введения.
18. Композиция по п.17, отличающаяся тем, что она выполнена в виде таблетки, капсулы или жидкости.
19. Композиция по п.17, отличающаяся тем, что, по крайней мере, один фармацевтический наполнитель выбран из разбавителей, связующих веществ, глидантов, лубрикантов, разрыхлителей, пигментных красителей, отдушек, подсластителей, полимеров, восков и других замедляющих растворимость веществ.
20. Композиция по п.16, отличающаяся тем, что она выполнена в форме, пригодной для внутривенного введения.
21. Композиция по п.20, отличающаяся тем, что, по крайней мере, один фармацевтический наполнитель включает стерильный раствор сахаров, аминокислот или электролитов.
22. Композиция по п.20, отличающаяся тем, что, по крайней мере, один фармацевтический наполнитель является водой для инъекции USP.
23. Композиция по п.15, отличающаяся тем, что она выполнена в форме, пригодной для парентерального введения.
24. Композиция по п.23, отличающаяся тем, что, по крайней мере, один фармацевтический наполнитель включает стерильный раствор сахаров, аминокислот или электролитов.
25. Композиция по п.23, отличающаяся тем, что, по крайней мере, один фармацевтический наполнитель является водой для инъекции USP.
26. Соединение или фармацевтически приемлемая соль по п.14 для использования в производстве фармацевтической композиции для лечения рака, связанного с активностью кинезина CENP-E.
| US 4431851 14.02.1984 | |||
| Hyun M.H | |||
| et al "Improved chiral stationary phase derived from (s)-naproxen for the liquid chromatographic resolution of enantiomers" | |||
| Journal of Chromatography, 1996, v.732, pp.209-214 | |||
| ИНГИБИТОРЫ ПРЕНИЛТРАНСФЕРАЗ | 1997 |
|
RU2186776C2 |

