Настоящее изобретение относится к новым производным изохинолона и изохинолина, как описано в формуле изобретения, их получению и применению для лечения и/или предотвращения заболеваний, связанных с ингибированием Rho-киназы и/или активируемого Rho-киназой фосфорилирования фосфатазы легких цепей миозина.
Активирование малой ГТФазы RhoA под действием агонистического воздействия приводит к превращению RhoA из неактивной ГДФ-связанной формы в активную ГТФ-связанную форму с последующим связыванием c и активированием Rho-киназы. Известны две изоформы, Rho-киназа 1 и Rho-киназа 2. Rho-киназа 2 экспрессируется в гладких мышечных клетках кровеносных сосудов и эндотелиальных клетках. Активация Rho-киназы 2 активной ГТФ-связанной RhoA приводит к кальциевой сенсибилизации клеток гладкой мускулатуры за счет вызванного фосфорилированием ингибирования активности фосфатазы легких цепей миозина и, таким образом, повышающей регуляции активности регуляторных легких цепей миозина (Uehata et al., Nature 1997, 389, 990-994).
Известно, что Rho-киназа задействована в сужении кровеносных сосудов, включая развитие миогенного тонуса и гиперсокращаемость гладкой мускулатуры (Gokina et al. J. Appl. Physiol. 2005, 98, 1940-8), сокращаемость гладкой мускулатуры бронхов (Yoshii et al. Am. J. Resp. Cell Mol. Biol. 20, 1190-1200), астму (Setoguchi et al. Br J Pharmacol. 2001, 132, 111-8; Nakahara, et al. Eur J 2000,389, 103) и хроническое обструктивное заболевание легких (COPD, Maruoka, Nippon Rinsho, 1999, 57, 1982-7), гипертензию, легочную гипертензию (Fukumoto et al. Heart, 91, 391-2, 2005, Mukai et al. Nature 1997,389, 990-4) и офтальмологическую гипертензию и регулирование внутриглазного давления (Honjo et al. Invest. Ophthalmol. Visual Sci. 2001, 42, 137-144), дисфункцию эндотелия (Steioff et al. Eur. J. Pharmacol. 2005, 512, 247-249), стенокардию (Masumoto et al. Circ 2002, 105, 1545-47, Shimokawa et al. JCP, 2002, 40, 751-761), нефропатию, включая индуцированную гипертензией, не индуцированную гипертензией и диабетическую нефропатии, почечную недостаточность и периферическое окклюзионное поражение артерии (PAOD) (Wakino et al. Drug News Perspect. 2005, 18, 639-43), инфаркт миокарда (Demiryurek et al. Eur J Pharmacol. 2005, 527, 129-40, Hattori et al. Circulation, 2004, 109, 2234-9), гипертрофию сердца и сердечную недостаточность (Yamakawa, et al. Hypertension 2000, 35, 313-318, Liao et al. Am J Physiol Cell Physiol. 2006, 290, C661-8, Kishi et al. Circ 2005, 111, 2741-2747), коронарную болезнь сердца, атеросклероз, реностеноз (Pacaud et al. Arch. Mal. Coeur 2005, 98, 249-254, Retzer, et al. FEBS Lett 2000, 466, 70, Negoro, et al. Biochem Biophys Res Commun 1999, 262, 211), диабеты, осложнения диабетов, утилизацию глюкозы и метаболический синдром (Sandu, et al.Diabetes 2000, 49, 2178, Maeda et al. Cell Metab. 2005, 2, 119-29), сексуальные дисфункции, например нарушение эрективной способности пениса (Chitaley et al. Nature Medicine 2001, 7, 119-122), ретинопатии, воспаление, иммунологические заболевания, СПИД, остеопороз, эндокринные нарушения, например гиперальдостеронизм, нарушения центральной нервной системы, такие как слабоумие и повреждение спинного мозга (Hara, et al. J Neurosurg 2000, 93, 94), ишемия головного мозга (Uehata, et al. Nature 1997, 389, 990; Satoh et al. Life Sci. 2001, 69, 1441-53; Hitomi, et al. Life Sci 2000, 67, 1929; Yamamoto, et al. J Cardiovasc Pharmacol. 2000, 35, 203-11), спазм сосудов головного мозга (Sato, et al. Circ Res 2000, 87, 195; Kim, et al. Neurosurgery 2000, 46, 440), боль, например невропатическая боль (Tatsumi, et al. Neuroscience 2005, 131, 491, Inoue, et al. Nature medicine 2004, 10, 712), бактериальную инфекцию пищеварительного тракта (WO 98/06433), развитие и прогрессию рака, неоплазию, где было показано, что ингибирование Rho-киназы приводит к ингибированию роста опухолевых клеток и метастазов (Itoh, et al. Nature Medicine 1999, 5, 221; Somlyo, et al. Res Commun 2000, 269, 652), ангиогенез (Uchida, et al. Biochem Biophys Res 2000, 269, 633-40; Gingras, et al. Biochem J 2000, 348, 273), пролиферацию и сократительную способность клеток гладкой мускулатуры кровеносных сосудов (Tammy et al. Circ. Res. 1999, 84, 1186-1193; Tangkijvanich et al. Atherosclerosis 2001, 155, 321-327), пролиферацию эндотелиальных клеток, сокращение и сократительную способность эндотелиальных клеток (Oikawa et al.Biochem. Biophys. Res. Commun. 2000, 269, 633-640), образование тонофибрилл (Kimura et al. Science 1997, 275, 1308-1311; Yamashiro et al. J. Cell Biol. 2000, 150, 797-806), тромбозные нарушения (Kikkawa, et al. FEBS Lett. 2000, 466, 70-74; Bauer et al. Blood 1999, 94, 1665-1672, Klages, et al. J Cell Biol. 1999, 144, 745; Retzer, et al. Cell Signal 2000, 12, 645) и агрегацию лейкоцитов (Kawaguchi, et al. Eur J Pharmacol. 2000, 403:203-8; Sanchez-Madrid, et al. J Immunol. 2003, 171:1023-34, Sanchez-Madrid, et al. J Immunol. 2002, 168:400-10), и резорбцию кости (Chellaiah, et al. J Biol. Chem. 2003, 278:29086-97), активацию Na/H обменной транспортной системы (Kawaguchi, et al. Eur J Pharmacol. 2000, 403:203-8), болезнь Альцгеймера (Zhou et al. Science 2003, 302, 1215-1217), активацию аддуцина (Fukata et al. J. Biol. Chem., 1998, 273, 5542-5548), и в SREB (чувствительный к стеролу связывающий элемент) сигнальной системе и ее действии на метаболизм липидов (Lin et al. Circ. Res., 92, 1296-304, 2003).
Таким образом, соединение, обладающее ингибирующим воздействием на Rho-киназу и/или активируемое Rho-киназой фосфорилирование фосфатазы легких цепей миозина пригодно для лечения и/или предотвращения сердечно-сосудистых или не сердечно-сосудистых заболеваний, в которых Rho-киназа является первичной или вторичной причиной заболеваний, таких как гипертензия, легочная гипертензия, офтальмологическая гипертензия, ретинопатия и глаукома, нарушение периферического кровообращения, периферическое окклюзионное поражение артерии (PAOD), коронарная болезнь сердца, стенокардия, гипертрофия сердца, сердечная недостаточность, ишемические заболевания, ишемическая недостаточность органа (повреждение органа-мишени), пневмофиброз, фиброз печени, печеночная недостаточность, нефропатия, включая вызванную гипертензией, не вызванную гипертензией, и диабетическую нефропатии, почечная недостаточность, фиброз почек, почечный гломерулосклероз, гипертрофия органа, астма, хроническое обструктивное заболевание легких (COPD), синдром расстройства дыхания у взрослых, тромботические нарушения, удар, спазм сосудов головного мозга, ишемия сосудов головного мозга, боль, например невропатическая боль, нервная деградация, повреждение спинного мозга, болезнь Альцгеймера, преждевременные роды, нарушение эрекции, эндокринные нарушения, атеросклероз, гипертрофия простаты, диабеты и осложнения диабетов, метаболический синдром, рестеноз кровеносных сосудов, атеросклероз, воспаление, аутоиммунные заболевания, СПИД, остеопатия, такая как остеопороз, бактериальная инфекция пищеварительного тракта, сепсис, развитие и прогрессия рака, например рака груди, толстой кишки, простаты, яичников, мозга и легкого и их метастазы.
В WO 01/64238 описаны производные изохинолин-5-сульфонамида, необязательно замещенные -(CH2)1-6-O-(CH2)0-6-, -(CH2)1-6-S-(CH2)0-6- или -(CH2)0-6-связанной гетероциклической группой, пригодные в качестве нейропротекторных агентов.
В WO 2004/106325 (Schering AG) описаны пролекарства ингибитора Rho-киназы фазудила, имеющие простую или сложную эфирную группу в положении-1 изохинолинового кольца.
В WO 2001/039726 в общем описаны -O-(C0-C10)алкил-гетероарил замещенные циклогексильные производные, пригодные для лечения микробных инфекций.
В JP 10087629 A описаны производные изохинолина, пригодные для лечения заболеваний, вызванных Heliobacter pylori, таких как, например, гастрит, рак или язва. Производные изохинолина могут быть замещены ОН в положении-1 и предпочтительно представляют собой 5-замещенные X-[(C1-C6)алкилен)]0-1-Y, где X может представлять собой кислород и Y может представлять собой арил или гетероциклическую группу.
Yoshida et al. (Bioorg. Med. Chem. 1999, 7, 2647-2666) описывают 6-бензилокси-изохинолин для лечения инфекций, вызванных Heliobacter pylori.
В патенте США 5480883 в общем описаны в качестве ингибиторов EGF и/или PDGF рецептора, пригодных для ингибирования клеточной пролифирации, соединения формулы “Ar I - X - Ar II”, где X может представлять собой (CHR1)m-Z-(CHR1)n, например, Z-CH2, где Z может представлять собой O, R1 представляет собой водород или алкил, Ar I может среди прочего представлять собой необязательно замещенный изохинолон, и Ar II может среди прочего представлять собой необязательно замещенную C3-7 моноциклическую насыщенную гетероциклическую систему.
В WO 2005/030791 (Merck & Co.) в общем описаны в качестве ингибиторов калиевых каналов для лечения аритмий сердца, удара, острой сердечной недостаточности и т.д. производные изохинолона, которые необязательно могут быть замещены в положении-6 группой (CReRf)pOR43, где p может быть равно нулю и R43 представляет собой, например, группу R81, определенную как 4-6 членное ненасыщенное или насыщенное моноциклическое гетероциклическое кольцо с 1, 2, 3 или 4 гетероатомами, выбранными из N, O или S; и замещенные непосредственно связанным в положении-4 с необязательно замещенным арильным или гетероарильным кольцом.
В WO 2005/030130 (Merck & Co.) в общем описаны в качестве ингибиторов калиевых каналов для лечения аритмий сердца, удара, острой сердечной недостаточности и т.д. производные изохинолина, которые могут быть замещены гидроксилом в положении-1 и необязательно замещены в положении-6 группой (CReRf)pOR43, где p может быть равно нулю и R43 представляет собой, например, группу R81, определенную как 4-6-членное ненасыщенное или насыщенное моноциклическое гетероциклическое кольцо с 1, 2, 3 или 4 гетероатомами, выбранными из N, O или S; и замещенные непосредственно связанным в положении-4 с необязательно замещенным арильным или гетероарильным кольцом.
В WO 03/053330 (Ube) описаны производные изохинолона формулы
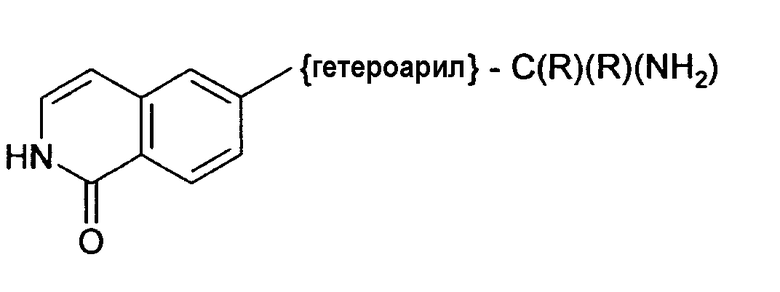
в качестве ингибиторов Rho-киназы.
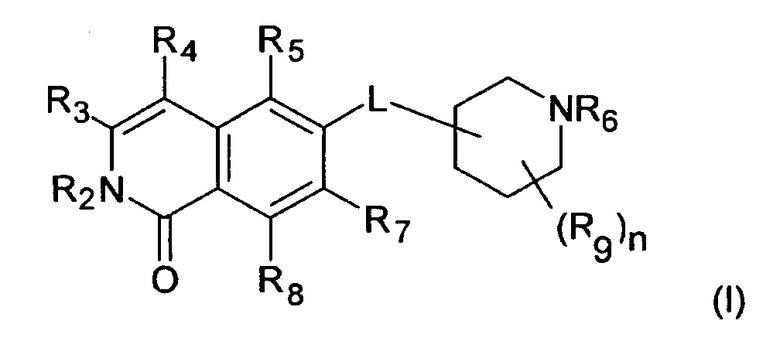
Один из вариантов настоящего изобретения представляет собой соединение формулы (I)
 ,
,
где
R2 представляет собой H, (C1-C6)алкил, [(C1-C6)алкилен]0-1-R', [(C1-C6)алкилен]0-1-O-(C1-C6)алкил, [(C1-C6)алкилен]0-1-O-R', [(C1-C6)алкилен]0-1-NH2, [(C1-C6)алкилен]0-1-NH(C1-C6)алкил, [(C1-C6)алкилен]0-1-N[(C1-C6)алкил]2, [(C1-C6)алкилен]0-1-CH[R']2, [(C1-C6)алкилен]0-1-C(O)-R', [(C1-C6)алкилен]0-1-C(O)NH2, [(C1-C6)алкилен]0-1-C(O)NH-R' или [(C1-C6)алкилен]0-1-C(O)N[R']2;
R3 представляет собой H, галоген, CN, (C1-C6)алкил, (C1-C6)алкилен-R', OH, O-R”, NH2, NHR”, NR”R” или NH-C(O)-R”,
R4 представляет собой H, галоген, гидрокси, CN, (C1-C6)алкил, (C3-C8)циклоалкил, (C1-C6)алкилен-R';
R5 представляет собой H, галоген, CN, NO2, (C1-C6)алкил, (C2-C6)алкенил, R', (C1-C6)алкилен-(C6-C10)арил, (C2-C6)алкенилен-(C6-C10)арил, (C1-C6)алкилен-(C5-C10)гетероциклил, NH2, NH-R', NH-SO2H, NH-SO2-(C1-C6)алкил, NH-SO2-R', NH-C(O)-(C1-C6)алкил, NH-C(O)-R', C(O)N[(C1-C6)алкил]2, C(O)OH или C(O)O-(C1-C6)алкил;
R6 представляет собой H, R', (C1-C8)алкил,(C1-C6)алкилен-R', (C1-C6)алкилен-O-(C1-C6)алкил, (C1-C6)алкилен-O-R', (C1-C6)алкилен-CH[R']2, (C1-C6)алкилен-C(O)-R', (C1-C6)алкилен-C(O)NH2, (C1-C6)алкилен-C(O)NH-R' или (C1-C6)алкилен-C(O)N[R']2;
R7 и R8 независимо друг от друга представляют собой H, галоген, CN, NO2, (C1-C6)алкил, O-(C1-C6)алкил, O-[(C1-C6)алкилен]0-1-R', (C2-C6)алкенил, R', (C2-C6)алкенилен-(C6-C10)арил, (C1-C6)алкилен-R', NH2, NH-R', NH-SO2H, NH-SO2-(C1-C6)алкил, NH-SO2-R', SO2-NH2, SO2-NHR', NH-C(O)-(C1-C6)алкил, NH-C(O)-R', C(O)N[(C1-C6)алкил]2, C(O)OH или C(O)O-(C1-C6)алкил;
R9 представляет собой галоген или (C1-C6)алкил;
n равно 0, 1, 2, 3 или 4; и
L представляет собой O или O-(C1-C6)алкилен;
где R' представляет собой (C3-C8)циклоалкил, (C5-C10)гетероциклил или (C6-C10)арил; и
R” представляет собой (C3-C8)циклоалкил, (C5-C10)гетероциклил, (C6-C10)арил, (C1-C6)алкил, (C1-C6)алкилен-R', (C1-C6)алкилен-O-(C1-C6)алкил, (C1-C6)алкилен-O-R' или (C1-C6)алкилен-NRxRy; и
где Rx и Ry независимо друг от друга представляют собой (C1-C6)алкил, (C5-C10)гетероциклил, (C6-C10)арил, (C1-C4)алкилен-(C5-C10)гетероциклил,(C1-C4)алкилен-(C6-C10)арил, (C1-C4)алкилен-NH(C1-C6)алкил, (C1-C4)алкилен-N[(C1-C6)алкил]2, (C1-C4)алкилен-N[(C6-C10)арил]2 или (C1-C4)алкилен-N[(C5-C10)гетероциклил]2; и
где в группах R4, R5, R7 и R8 один алкильный или алкиленовый атом водорода может быть необязательно заменен на OH, OCH3, COOH, COOCH3, NH2, NHCH3, N(CH3)2, CONH2, CONHCH3 или CON(CH3)2 или алкил или алкилен может быть галогенирован в одном или более положениях;
или их фармацевтически приемлемые соли и/или стереоизомерные формы и/или физиологические функциональные производные.
В другом варианте осуществления соединения формулы (I) в группах R4, R5, R7 и R8 один алкильный или алкиленовый атом водорода может быть необязательно заменен на OH, F, OCH3, COOH, COOCH3, NH2, NHCH3, N(CH3)2, CONH2, CONHCH3 или CON(CH3)2.
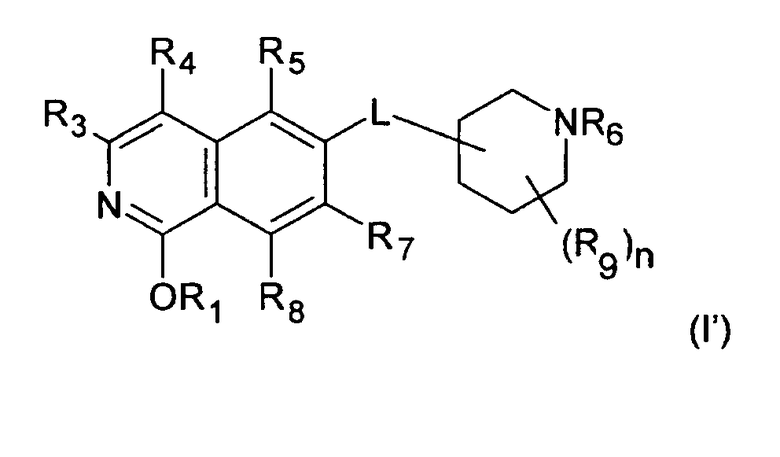
Стереоизомерные формы производных изохинолина формулы (I) включают соответствующие таутомерные 1-гидрокси-замещенные изохинолиновые производные формулы (I')
 ,
,
где R1 представляет собой H, (C1-C6)алкил, (C2-C6)алкенил, (C2-C6)алкинил, [(C1-C6)алкилен]0-1-(C3-C8)циклоалкил, [(C1-C6)алкилен]0-1-(C5-C10)гетероциклил, [(C1-C6)алкилен]0-1-(C6-C10)арил, C(O)-(C1-C6)алкил, C(O)(C2-C6)алкенил, C(O)(C2-C6)алкинил, C(O)-[(C1-C6)алкилен]0-1-(C3-C8)циклоалкил, C(O)-[(C1-C6)алкилен]0-1-(C5-C10)гетероциклил или C(O)-[(C1-C6)алкилен]0-1-(C6-C10)арил, и
где R3, R4, R5, R6, R7, R8, R9, n и L представляют собой такие же, как описано выше.
В предпочтительном варианте осуществления изобретения R2 в соединении формулы (I) представляет собой Н, соединение, таким образом, характеризуется формулой (II)
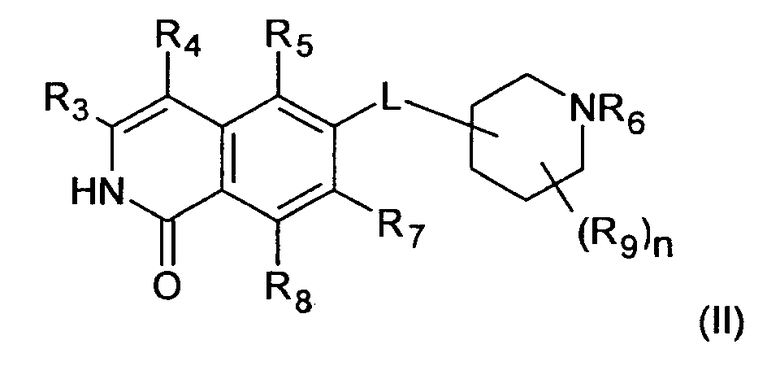
В дополнительном предпочтительном варианте осуществления изобретения R1 в соединении формулы (I')представляет собой Н, соединение, таким образом, характеризуется формулой (II')
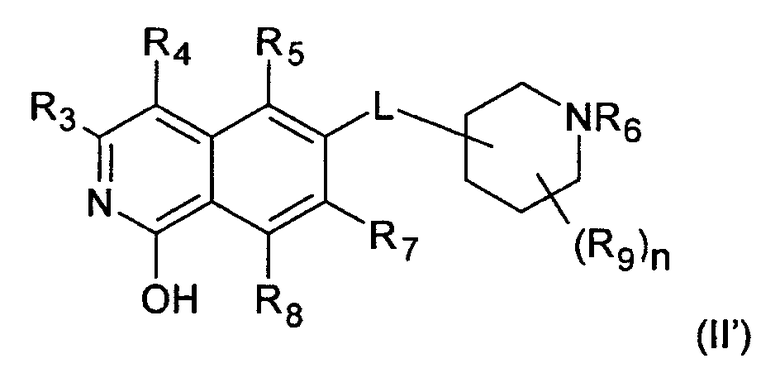 .
.
Соединения формулы (II) и (II') представляют собой взаимно таутомерные формы. Например, соединение формулы
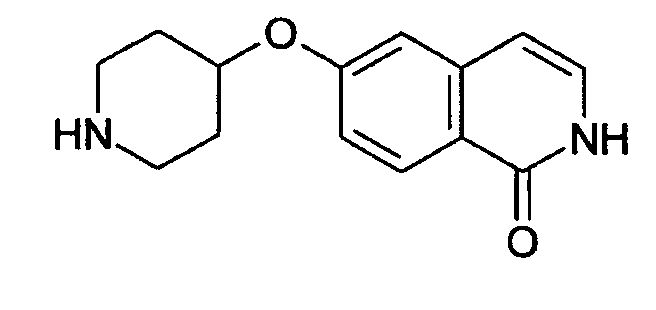
представляет собой таутомерную форму соединения формулы
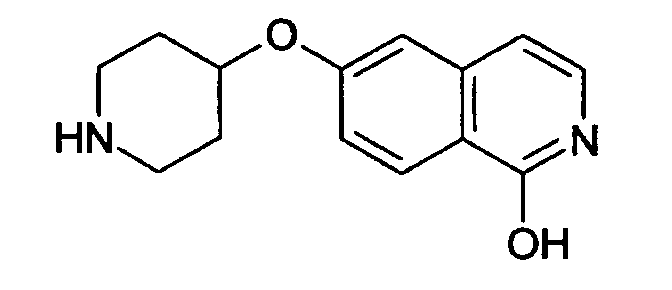 .
.
Следующие предпочтительные варианты осуществления относятся к формулам (I), (I'), (II) и (II'):
R3 предпочтительно представляет собой Н, галоген, (С1-С6)алкил, (С1-С4)алкилен-R', O-R” или NRH”. Более предпочтительно R3 представляет собой H, (С1-С6)алкил или NRH”. Более предпочтительно, R3 представляет собой Н, (С1-С4)алкил, NH-(C5-C6)гетероциклил или NH-фенил, в особенности предпочтительно R3 представляет собой Н, (С1-С4)алкил, NH-(C5-C6)гетероарил, содержащий один или несколько атомов N, или NH-фенил. Более предпочтительно R3 представляет собой Н.
Предпочтительно R4 представляет собой H, галоген, CN, (C1-C6)алкил, NH-(C6-C10)арил или (C1-C6)алкилен-R'. Более предпочтительно, R4 представляет собой H, галоген, (C1-C6)алкил, NH-(C6-C10)арил или (C1-C6)алкилен-R'. В дополнительном предпочтительном варианте осуществления R4 представляет собой H, галоген, (C1-C6)алкил, NH-(C6-C10)арил или (C1-C2)алкилен-(C6-C10)арил. Наиболее предпочтительно, R4 представляет собой H, галоген или (C1-C6)алкил. В особенности предпочтительно, R4 представляет собой H, галоген или (C1-C6)алкил. Наиболее предпочтительно, R4 представляет собой H.
Предпочтительно, R5 представляет собой H, галоген, CN, (C1-C6)алкил, R', NH-(C6-C10)арил или (C1-C6)алкилен-R'. Более предпочтительно, R5 представляет собой H, галоген, (C1-C6)алкил, R', NH-(C6-C10)арил или (C1-C6)алкилен-R'. В дополнительном предпочтительном варианте осуществления R5 представляет собой H, галоген, (C6-C10)арил, NH-(C6-C10)арил, (C1-C2)алкилен-(C6-C10)арил, (C1-C6)алкил или (C5-C10)гетероарил. Наиболее предпочтительно, R5 представляет собой H, галоген, фенил, (C1-C6)алкил или (C5-C6)гетероарил. В особенности предпочтительно, R5 представляет собой H, галоген или (C1-C6)алкил. В большей степени предпочтительно R5 представляет собой H или галоген. Наиболее предпочтительно, R5 представляет собой H.
Предпочтительно, R6 представляет собой H, (C1-C6)алкил, R', (C1-C4)алкилен-(C3-C8)циклоалкил, (C1-C4)алкилен-(C5-C10)гетероциклил, (C1-C4)алкилен-C(O)-(C5-C10)гетероциклил, (C1-C4)алкилен-C(O)-(C6-C10)арил или (C1-C6)алкилен-(C6-C10)арил. В дополнительном предпочтительном варианте осуществления R6 представляет собой H, (C1-C6)алкил, (C5-C10)гетероциклил, (C3-C8)циклоалкил, (C1-C4)алкилен-(C3-C8)циклоалкил, (C1-C4)алкилен-(C5-C10)гетероциклил или (C1-C6)алкилен-(C6-C10)арил. Более предпочтительно, R6 представляет собой H, (C1-C6)алкил, (C3-C8)циклоалкил, (C1-C4)алкилен-(C3-C8)циклоалкил, (C1-C4)алкилен-(C5-C10)гетероциклил или (C1-C6)алкилен-(C6-C10)арил. В более предпочтительном варианте осуществления изобретения R6 представляет собой H, (C1-C6)алкил, (C3-C8)циклоалкил, (C1-C4)алкилен-(C3-C8)циклоалкил, (C1-C4)алкилен-(C5-C10)гетероциклил, в котором гетероциклил является незамещенным или замещен (C1-C4)алкилом или представляет собой (C1-C4)алкилен-(C6-C10)арил, в котором арил является незамещенным или замещен, предпочтительно в одном-трех положениях, галогеном, (C1-C4)алкилом, в особенности метилом, этилом, изопропилом, или 3,3,3-трифторметилом, О-(C1-C4)алкилом, в особенности метокси, SO2-(C1-C4)алкилом, в особенности SO2-CH3 или SO2-CF3, или N[(C1-C4)алкилом]2, в особенности N(CH3)2. В большей степени предпочтительном варианте осуществления R6 представляет собой H, (C1-C6)алкил, (C3-C8)циклоалкил, (C1-C4)алкилен-(C3-C8)циклоалкил, (C1-C2)алкилен-тиенил, (C1-C2)алкилен-пиридил, (C1-C2)алкилен-пиперидинил, (C1-C2)алкилен-пирролидинил, (C1-C2)алкилен-1-метил-пирролил, (C1-C2)алкилен-1-метил-пиразолил, (C1-C2)алкилен-фуранил, (C1-C2)алкилен-тетрагидрофуранил или (C1-C2)алкилен-1Н-индазолил, (C1-C2)алкилен-нафтил или (C1-C2)алкилен-фенил, где фенил является незамещенным или замещен галогеном, метилом, этилом, изопропилом, 3,3,3-трифторметилом, метокси, SO2-CH3, SO2-CF3 или N(CH3)2; предпочтительно (C1-C2)алкилен представляет собой метилен. В более предпочтительном варианте осуществления R6 представляет собой H, (C1-C6)алкил, (C3-C8)циклоалкил или (C1-C4)алкилен-(C3-C6)циклоалкил. В дополнительном более предпочтительном варианте осуществления R6 представляет собой H, (C1-C6)алкил. В наиболее предпочтительном варианте осуществления R6 представляет собой H. Примерами R6 групп являются водород, метил, этил, пропил, изопропил, циклопропил, 3-метилбутил, бутил, изобутил, 3,3,3-трифторпропил или заместители, выбранные из группы, состоящей из
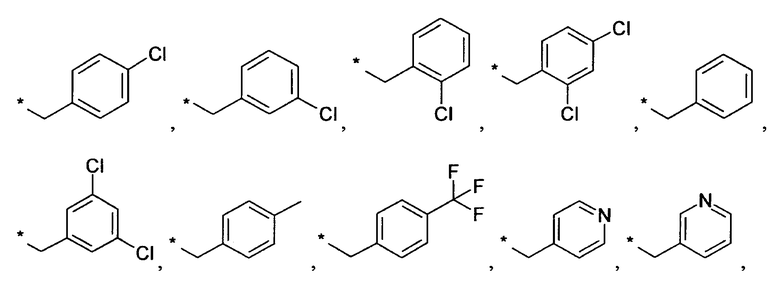
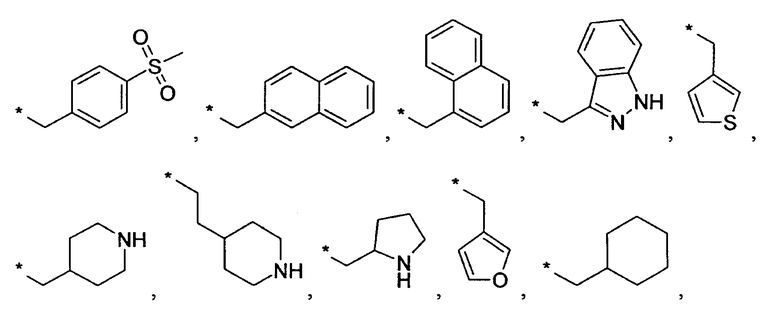
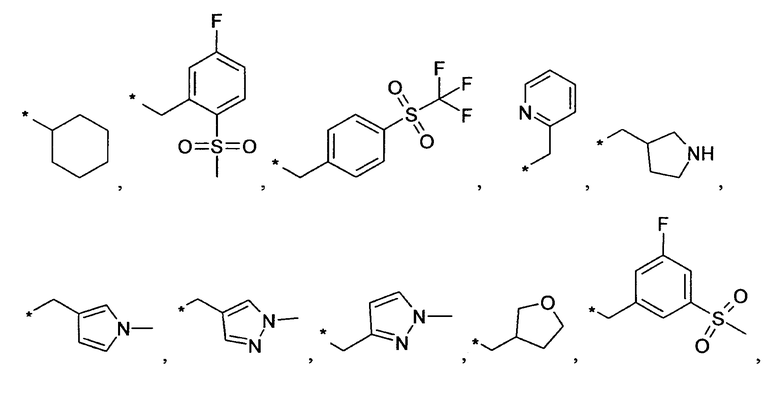
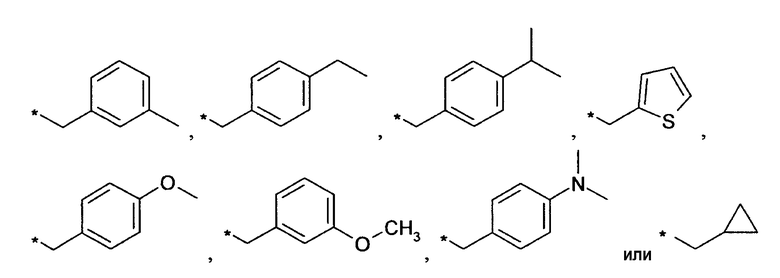 .
.
Звездочка (*) обозначает место присоединения к N-атому пиперидина.
Предпочтительно, R7 и R8, каждый независимо друг от друга, представляет собой H, галоген, CN, (C1-C6)алкил, O-(C1-C6)алкил, (C2-C6)алкенил, R' или (C1-C6)алкилен-(C3-C8)циклоалкил. Более предпочтительно, R7 и R8 независимо друг от друга представляют собой H, галоген, CN, (C1-C4)алкил, O-(C1-C4)алкил, (C2-C4)алкенил, фенил, (C3-C6)циклоалкил, (C1-C4)алкилен-(C3-C6)циклоалкил или (C5-C6)гетероарил. Еще более предпочтительно, R7 и R8 независимо друг от друга представляют собой H, галоген, (C1-C4)алкил, O-(C1-C4)алкил или (C3-C6)циклоалкил. Наиболее предпочтительно, R7 представляет собой H, галоген, (C1-C4)алкил или O-(C1-C4)алкил, и R8 представляет собой H. В еще одном даже более предпочтительном варианте осуществления, R7 и R8 независимо друг от друга представляют собой H, галоген, (C1-C4)алкил, O-(C1-C4)алкил или фенил. Особенно предпочтительно, R7 и R8 представляют собой H.
R9 предпочтительно представляет собой галоген или (C1-C4)алкил. Более предпочтительно, R9 представляет собой Cl, F, метил или этил. Более предпочтительно, R9 представляет собой метил.
Предпочтительно, n равно 0, 1, 2 или 3. Более предпочтительно, n равно 0 или 1. Более предпочтительно, n равен 0.
Линкерная группа L может быть связана с пиперидинильным кольцом в любом положении через атом углерода пиперидинильного кольца и может, таким образом, образовывать (R)- или (S)- стереоизомер соединения согласно изобретению.
В предпочтительном варианте осуществления L присоединена в положении-4 пиперидинильного кольца
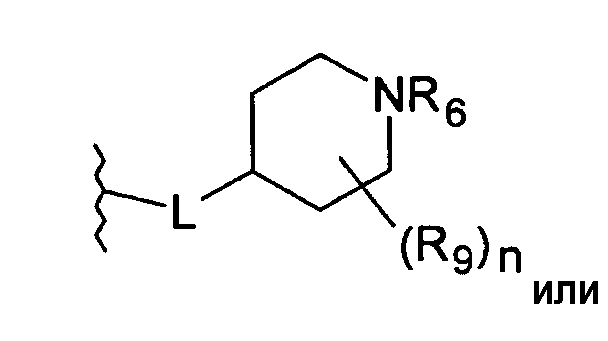
L присоединена в положении-3 пиперидинильного кольца
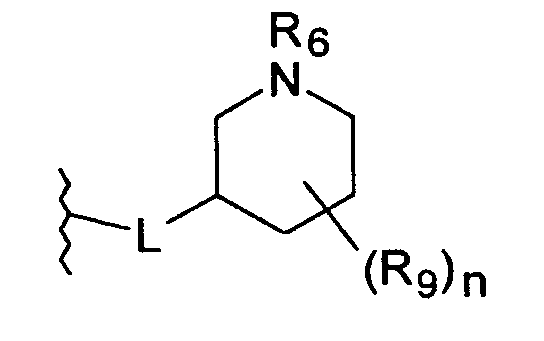
во всех их стереохимических формах.
В особенности предпочтительном варианте осуществления L присоединена в положении-4 пиперидинильного кольца.
Предпочтительно, L представляет собой О-метилен, О-этилен или О. Более предпочтительно, L представляет собой О-метилен, О-этилен или, наиболее предпочтительно, О, присоединенные в положении-4 пиперидинильного цикла.
Наиболее предпочтительно L представляет собой О.
В предпочтительных вариантах осуществления настоящего изобретения одна или несколько, или все группы, содержащиеся в соединениях формул (I) или (I'), могут независимо друг от друга иметь предпочтительные, более предпочтительные или наиболее предпочтительные определения групп, указанные выше, или любое или некоторое конкретное значение, которое включено в определение групп и указано выше, все комбинации предпочтительных определений, более предпочтительных или наиболее предпочтительных и/или конкретных значений представляют собой предмет настоящего изобретения. Также по отношению ко всем предпочтительным вариантам осуществления изобретение включает соединения формул (I) или (I') во всех стереоизомерных формах и смеси стереоизомерных форм в любых соотношениях и/или их физиологически приемлемые соли.
Предпочтительный вариант осуществления настоящего изобретения представляет собой соединение формулы (I), (I'), (II) или (II'), где
R3 представляет собой H, галоген, CN, (C1-C6)алкил, (C1-C6)алкилен-R', OH, O-R”, NH2 или NHR”;
R4 представляет собой H, галоген, гидрокси, CN, (C1-C6)алкил, (C3-C8)циклоалкил, (C1-C6)алкилен-R';
R5 представляет собой H, галоген, CN, NO2, (C1-C6)алкил, (C2-C6)алкенил, R', (C1-C6)алкилен-(C6-C10)арил, (C2-C6)алкенилен-(C6-C10)арил, (C1-C6)алкилен-(C5-C10)гетероциклил, NH2, NH-R', NH-SO2H, NH-SO2-(C1-C6)алкил, NH-SO2-R', NH-C(O)-(C1-C6)алкил, NH-C(O)-R', C(O)N[(C1-C6)алкил]2, C(O)OH или C(O)O-(C1-C6)алкил;
R6 представляет собой H, (C3-C8)циклоалкил, (C1-C8)алкил, (C1-C6)алкилен-R', (C1-C6)алкилен-O-(C1-C6)алкил, (C1-C6)алкилен-O-R', (C1-C6)алкилен-CH[R']2, (C1-C6)алкилен-C(O)NH2, (C1-C6)алкилен-C(O)NH-R' или (C1-C6)алкилен-C(O)N[R']2;
R7 и R8 независимо друг от друга представляют собой H, галоген, CN, NO2, (C1-C6)алкил, (C2-C6)алкенил, R', (C2-C6)алкенилен-(C6-C10)арил, (C1-C6)алкилен-R', NH2, NH-R', NH-SO2-(C1-C6)алкил, NH-SO2-R', SO2-NH2, SO2-NHR', NH-C(O)-(C1-C6)алкил, NH-C(O)-R', C(O)N[(C1-C6)алкил]2, C(O)OH или C(O)O-(C1-C6)алкил;
R9 представляет собой галоген или (C1-C6)алкил;
n равно 0, 1, 2; и
L представляет собой O или O-(C1-C3)алкилен;
где R1, R2, R', R'', Rx и Ry такие же, как определено выше;
или их фармацевтически приемлемые соли и/или стереоизомерные формы и/или физиологические функциональные производные.
Дополнительный предпочтительный вариант осуществления настоящего изобретения представляет собой соединение формулы (I), (I'), (II) или (II'), где
R3 представляет собой H, галоген, CN, (C1-C6)алкил, (C1-C2)алкилен-R' или NHR”;
R4 представляет собой H, галоген, CN, (C1-C6)алкил, (C3-C8)циклоалкил, (C1-C2)алкилен-R';
R5 представляет собой H, галоген, CN, NO2, (C1-C6)алкил, (C2-C6)алкенил, R', (C1-C6)алкилен-(C6-C10)арил, (C2-C6)алкенилен-(C6-C10)арил, (C1-C6)алкилен-(C5-C10)гетероциклил, NH2, NH-R', NH-C(O)-(C1-C6)алкил или C(O)N[(C1-C6)алкил]2;
R6 представляет собой H, (C3-C8)циклоалкил, (C1-C8)алкил или (C1-C3)алкилен-R';
R7 и R8 независимо друг от друга представляют собой H, галоген, CN, NO2, (C1-C6)алкил, (C2-C6)алкенил, R', (C2-C3)алкенилен-(C6-C10)арил, (C1-C3)алкилен-R', NH-R', NH-SO2-(C1-C6)алкил или SO2-NH2;
R9 представляет собой галоген или (C1-C6)алкил;
n равно 0 или 1; и
L представляет собой O или O-метилен;
где R1, R2, R', R'', Rx и Ry такие же, как определено выше;
или их фармацевтически приемлемые соли и/или стереоизомерные формы и/или физиологические функциональные производные.
Наиболее предпочтительный вариант осуществления настоящего изобретения представляет собой соединение формулы (I), (I'), (II) или (II'), где
R3 представляет собой H, галоген, CN, (C1-C6)алкил, (C1-C2)алкилен-R' или NHR”;
R4 представляет собой H, галоген, CN, (C1-C4)алкил, (C3-C6)циклоалкил, (C1-C2)алкилен-R';
R5 представляет собой H, галоген, CN, NO2, (C1-C6)алкил, (C2-C6)алкенил, R', (C1-C6)алкилен-(C6-C10)арил, (C2-C6)алкенилен-(C6-C10)арил, (C1-C6)алкилен-(C5-C10)гетероциклил, NH-R';
R6 представляет собой H, (C3-C8)циклоалкил или (C1-C4)алкил;
R7 и R8 независимо друг от друга представляют собой H, галоген, CN, NO2, (C1-C6)алкил, (C2-C6)алкенил, R', (C2-C3)алкенилен-(C6-C10)арил, (C1-C3)алкилен-R', NH-SO2-(C1-C6)алкил или SO2-NH2;
n равно 0, и R9 отсутствует, или
n равно 1, и R9 представляет собой галоген или (C1-C4)алкил; и
L представляет собой O;
где R1, R2, R', R'', Rx и Ry такие же, как определено выше;
или их фармацевтически приемлемые соли и/или стереоизомерные формы и/или физиологические функциональные производные.
Другой предпочтительный вариант осуществления настоящего изобретения представляет собой соединение формулы (I), (I'), (II) или (II'), где
R3 представляет собой H, галоген, CN, (C1-C6)алкил, (C1-C6)алкилен-R', ОН, O-R”, NH2 или NHR”;
R4 представляет собой H, галоген, гидрокси, CN, (C1-C6)алкил, (C3-C8)циклоалкил, (C1-C6)алкилен-R';
R5 представляет собой H, галоген, CN, NO2, (C1-C6)алкил, (C2-C6)алкенил, R', (C1-C6)алкилен-(C6-C10)арил, (C2-C6)алкенилен-(C6-C10)арил, (C1-C6)алкилен-(C5-C10)гетероциклил, NH2, NH-R', NH-SO2H, NH-SO2-(C1-C6)алкил, NH-SO2-R', NH-C(O)-(C1-C6)алкил, NH-C(O)-R', C(O)N[(C1-C6)алкил]2, C(O)OH или C(O)O-(C1-C6)алкил;
R6 представляет собой H, (C3-C8)циклоалкил, (C1-C8)алкил, (C1-C6)алкилен-R', (C1-C6)алкилен-O-(C1-C6)алкил, (C1-C6)алкилен-O-R', (C1-C6)алкилен-CH[R']2, (C1-C6)алкилен-C(O)NH2, (C1-C6)алкилен-C(O)NH-R' или (C1-C6)алкилен-C(O)N[R']2;
R7 и R8 независимо друг от друга представляют собой H, галоген, CN, NO2, (C1-C6)алкил, O-(C1-C6)алкил, (C2-C6)алкенил, R', (C2-C6)алкенилен-(C6-C10)арил, (C1-C6)алкилен-R', NH2, NH-R', NH-SO2-(C1-C6)алкил, NH-SO2-R', SO2-NH2, SO2-NHR', NH-C(O)-(C1-C6)алкил, NH-C(O)-R', C(O)N[(C1-C6)алкил]2, C(O)OH или C(O)O-(C1-C6)алкил;
R9 представляет собой галоген или (C1-C6)алкил;
n равно 0, 1, 2; и
L представляет собой O или O-(C1-C4)алкилен;
где R1, R2, R',Rʺ, Rx и Ry такие же, как определено выше;
или их фармацевтически приемлемые соли и/или стереоизомерные формы и/или физиологические функциональные производные.
Дополнительный предпочтительный вариант осуществления настоящего изобретения представляет собой соединение формулы (I), (I'), (II) или (II'), где
R3 представляет собой H, галоген, CN, (C1-C6)алкил или (C1-C2)алкилен-R';
R4 представляет собой H, галоген, CN, (C1-C6)алкил или (C1-C2)алкилен-R';
R5 представляет собой H, галоген, CN, NO2, (C1-C6)алкил;
R6 представляет собой H, (C3-C8)циклоалкил, (C1-C8)алкил, (C1-C3)алкилен-(C3-C6)циклоалкил, (C1-C3)алкилен-(C6-C10)арил или (C1-C3)алкилен-(C5-C10)гетероциклил;
R7 и R8 независимо друг от друга представляют собой H, галоген, CN, NO2, (C1-C6)алкил, O-(C1-C6)алкил, (C3-C6)циклоалкил или фенил;
R9 представляет собой галоген или (C1-C6)алкил;
n равно 0 или 1; и
L представляет собой O или O-метилен;
где R1, R2, R' такие же, как определено выше;
или их фармацевтически приемлемые соли и/или стереоизомерные формы и/или физиологические функциональные производные.
Наиболее предпочтительный вариант осуществления настоящего изобретения представляет собой соединение формулы (I), (I'), (II) или (II'), где
R3 представляет собой H;
R4 представляет собой H, галоген или (C1-C4)алкил;
R5 представляет собой H, галоген или (C1-C6)алкил;
R6 представляет собой H, (C3-C6)циклоалкил, (C1-C4)алкил, (C1-C2)алкилен-(C3-C6)циклоалкил, (C1-C4)алкилен-(C5-C10)гетероциклил, в котором гетероциклил представляет собой незамещенный или замещенный (С1-С4)алкилом или представляет собой (C1-C4)алкилен-(C6-C10)арил, в котором арил незамещенный или замещенный галогеном, (С1-С4)алкилом, О-(С1-С4)алкилом, SO2-(C1-C4)алкилом или N[(C1-C4)алкилом]2;
R7 и R8 независимо друг от друга представляют собой H, галоген, (C1-C4)алкил, O-(C1-C4)алкил или фенил;
R9 представляет собой (C1-C4)алкил;
n равно 0 или 1; и
L представляет собой O;
где R1, R2 такие же, как определено выше, предпочтительно R1 представляет собой Н, и R2 представляет собой Н;
или их фармацевтически приемлемые соли и/или стереоизомерные формы и/или физиологические функциональные производные.
Как и в любом варианте осуществления изобретения, в предшествующих вариантах осуществления, которые содержат предпочтительные, более предпочтительные, наиболее предпочтительные или приведенные в качестве примера определения соединений согласно изобретению, один или несколько или все группы могут иметь любые из их предпочтительных, более предпочтительных, наиболее предпочтительных определений, указанных выше, или любое одно или некоторые конкретные значения, которые включены в их определения и указаны выше.
Физиологические приемлемые соли соединений формул (I) и (I') означают как их органические, так и неорганические соли, как описано в Remington's Pharmaceutical Sciences (17th edition, p.1418 (1985)). Из-за физической и химической стабильности и растворимости для кислотных групп предпочтение отдают, среди прочих, натриевым, калиевым, кальциевым и аммониевым солям; для основных групп предпочтение отдают солям малеиновой кислоты, янтарной кислоты, яблочной кислоты, винной кислоты, метилсульфоновой кислоты, соляной кислоты, серной кислоты, фосфорной кислоты или карбоновых кислот, или сульфоновых кислот, например, в виде гидрохлоридов, гидробромидов, фосфатов, сульфатов, метансульфонатов, ацетатов, лактатов, малеатов, фумаратов, малатов, глюконатов и солей аминокислот, природных оснований или карбоновых кислот. Получение физиологически приемлемых солей соединений формул (I) или (I'), которые способны к образованию солей, включая их стереоизомерные формы, проводят известными способами. Соединения формулы (I) образуют стабильные соли щелочных металлов, щелочноземельных металлов или необязательно замещенные аммониевые соли с основными реагентами, такими как гидроксиды, карбонаты, бикарбонаты, алкоголяты и аммиак или органические основания, например триметил- или триэтиламин, этаноламин, диэтаноламин или триэтаноламин, трометамол или другие основные аминокислоты, например, лизин, орнитин или аргинин. Если соединения формул (I) и (I') содержат основные группы, могут также быть получены стабильные кислотно-аддитивные соли с сильными кислотами. Пригодные фармацевтически приемлемые кислотно-аддитивные соли соединений по изобретению представляют собой соли неорганических кислот, таких как соляная кислота, бромистоводородная, фосфорная, метафосфорная, азотная и серная кислота, и органических кислот, таких как, например, уксусная кислота, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изетионовая, молочная, лактобионовая, малеиновая, яблочная, метансульфоновая, янтарная, п-толуолсульфоновая и винная кислота.
Соли физиологически неприемлемых анионов, такие как, например, трифторацетат, также входят в объем применения изобретения как важные промежуточные соединения для получения или очистки фармацевтически приемлемых солей и/или для использования в нетерапевтических целях, например, in vitro.
Термин «физиологические функциональные производные», используемый здесь, относится к любым физиологически переносимым производным соединения формул (I) и (I') по изобретению, например N-оксиду, который при введении млекопитающему, такому как, например, человек, способен образовывать (непосредственно или через промежуточную стадию) соединение формулы (I) и (I') или их активный метаболит.
Физиологические функциональные производные включают пролекарства соединений по изобретению, как описано, например, в H. Okada et al., Chem. Pharm. Bull. 1994, 42, 57-61. Такие пролекарства могут подвергаться метаболизму in vivo с образованием соединения по изобретению. Такие пролекарства могут быть активными или неактивными.
Изобретение относится к соединениям формулы (I) или (I') в форме их рацематов, рацемических смесей и чистых энантиомеров, и их диастереомерам, и их смесям.
Если радикалы или заместители присутствуют более чем в одном положении в соединении формул (I) и (I'), они могут все, независимо один от другого, иметь указанные значения и быть одинаковыми или различными.
Соединения по изобретению могут также существовать в виде различных полиморфных формах и/или сольватов, например в виде аморфных и кристаллических полиморфных форм. Все полиморфные формы соединений по изобретению входят в объем применения изобретения и представляют собой дополнительный объект изобретения.
Все ссылки на «соединение(я) формулы (I)» или «соединение(я) формулы (I')» здесь и далее относятся к соединениям формулы (I) и (I'), как описано выше, и их физиологически приемлемым солям, сольватам и физиологическим функциональным производным, как описано здесь.
Термин «алкил» и соответствующие алкиленовые заместители обозначают углеводородный остаток, который может быть линейным, т.е. с прямой цепью, или разветвленным и имеет 1, 2, 3, 4, 5 или 6 атомов углеводорода, соответственно, если применимо. Этот термин также применяют, если алкильная группа присутствует в качестве заместителя другой группы, например, в алкокси-группе (О-алкил), S-алкил или -О(С1-С6)алкилен-О-, алкоксикарбонильной группе или арилалкильной группе. Примерами алкильных групп являются метил, этил, пропил, бутил, пентил или гексил, н-изомеры всех указанных групп, изопропил, изобутил, 1-метилбутил, изопентил, неопентил, 2,2-диметилбутил, 2-метилпентил, 3-метилпентил, изогексил, втор-бутил, трет-бутил или трет-пентил. Алкильные группы могут быть, если не указано иначе, галогенированными в одном или нескольких положениях, например, алкильные группы могут быть фторированными, например перфторированными. Примерами галогенированных алкильных групп являются CF3 и CH2CF3, OCF3, SCF3 или -O-(CF2)2-O.
Алкенил представляет собой, например, винил, 1-пропенил, 2-пропенил (=аллил), 2-бутенил, 3-бутенил, 2-метил-2-бутенил, 3-метил-2-бутенил, 5-гексенил или 1,3-пентадиенил. Алкинил представляет собой, например, этинил, 1-пропинил, 2-пропинил (=пропаргил) или 2-бутинил.
Галоген обозначает фтор, хлор, бром или йод.
(С3-С8)циклоалкильные группы представляют собой циклические алкильные группы, содержащие 3, 4, 5, 6, 7 или 8 атомов углерода в цикле, как циклопропил, циклобутил, циклопентил, циклогексил или циклооктил, которые также могут быть замещенными и/или содержат 1 или 2 двойные связи (незамещенный циклоалкильные группы), как, например, циклопентенил или циклогексенил могут быть связаны через любой атом углерода.
(С6-С10)арильная группа обозначает ароматический цикл или циклическую систему, которая включает два ароматических кольца, которые конденсированы или связаны другим способом, например, фенил, нафтил, бифенил, тетрагидронафтил, альфа- или бета-тетралон-, инданил- или индан-1-он-ил группа. Предпочтительная (С6-С10)арильная группа представляет собой фенил.
(С5-С10)гетероциклическая группа обозначает моно- или бициклическую кольцевую систему, которая включает кроме углерода один или несколько гетероатомов, таких как, например, 1, 2 или 3 атома азота, 1 или 2 атома кислорода, 1 или 2 атома серы или комбинацию различных гетероатомов. Гетероциклические остатки могут быть связаны в любом положении, например в положении-1, положении-2, положении-3, положении-4, положении-5, положении-6, положении-7 или положении-8. (С5-С10)гетероциклические группы могут представлять собой (1) ароматическую [=гетероарильные группы] или (2) насыщенную или (3) смешанную ароматическую/насыщенную.
Пригодные (С5-С10)гетероциклические группы включают акридинил, азоцинил, бензимидазолил, бензофурил, бензоморфолинил, бензотиенил, бензотиофенил, бензоксазолил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, карбазолил, 4аН-карбазолил, карболинил, фуранил, хиназолинил, хинолинил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, хроманил, хроменил, хромен-2-онил, циннолинил, декагидрохинолинил, 2Н,6Н-1,5,2-дитиазинил, дигидрофуро[2,3,b]-тетрагидрофуран, фурил, фуразанил, гомоморфолинил, гомопиперазинил, имидазолидинил, имидазолинил, имидазолил, 1Н-индазолил, индолинил, индолизинил, индолил, 3Н-индолил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил (бензимидазолил), изотиазолил, изоксазолил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, оксазолидинил, пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, феноксатиинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пролинил, птеридинил, пуринил, пиранил, пиразинил, пироазолидинил, пиразолинил, пиразолил, пиридазинил, пиридонил, пиридооксазолы, пиридоимидазолы, пиридотиазолы, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, 2Н-пирролил, пирролил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, 6Н-1,2,5-тиадазинил, тиазолил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиенил, триазолил, тетразолил и ксантенил. В качестве пиридила понимают как 2-, так 3- и 4-пиридил. Тиенил означает как 2-, так и 3-тиенил. Фурил означает как 2-, так и 3-фурил. Также включены соответствующие N-оксиды указанных соединений, например, 1-окси-2-, 3- или 4-пиридил.
Заместители в (С5-С10)гетероциклических остатках могут располагаться на свободных атомах углерода или на атомах азота.
Предпочтительными примерами (С5-С10)гетероциклических остатков являются пиразинил, пиридил, пиримидинил, пиразолил, морфолинил, пирролидинил, пиперазинил, пиперидинил, тиенил, бензофурил, хинолинил, тетразолил и триазолил.
(С6-С10)арильные и (С5-С10)гетероциклические группы являются незамещенными или, если не указано иначе, замещены пригодными группами в одном или нескольких положениях пригодными группами, независимо выбранными из галогена, CF3, NO2, N3, CN, C(O)-(C1-C6)алкила, C(O)-(C1-C6)арила, COOH, COO(C1-C6)алкила, CONH2, CONH(C1-C6)алкила, CON[(C1-C6)алкила]2, (C3-C8)циклоалкила, (C1-C6)алкила, (C1-C6)алкилен-OH, (C1-C6)алкилен-NH2, (C1-C6)алкилен-NH(C1-C6)алкила, (C1-C6)алкилен-N[(C1-C6)алкила]2, (C2-C6)алкенила, (C2-C6)алкинила, O-(C1-C6)алкила, O-C(O)-(C1-C6)алкила, O-C(O)(C6-C10)арила, O-C(O)-(C5-C10)гетероциклила, PO3H2, SO3H, SO2-NH2, SO2NH(C1-C6)алкила, SO2N[(C1-C6)алкила]2, S-(C1-C6)алкила, S-(C1-C6)алкилен-(C6-C10)арила, S-(C1-C6)алкилен-(C5-C10)гетероциклила, SO-(C1-C6)алкила, SO-(C1-C6)алкилен-(C6-C10)арила, SO-(C1-C6)алкилен-(C5-C10)гетероциклила, SO2-(C1-C6)алкила, SO2-(C1-C6)алкилен-(C6-C10)арила, SO2-(C1-C6)алкилен-(C5-C10)гетероциклила, SO2-NH(C1-C6)алкилен-(C6-C10)арила, SO2-NH(C1-C6)алкилен-(C5-C10)гетероциклила, SO2-N[(C1-C6)алкил][(C1-C6)алкилен-(C6-C10)арила], SO2-N[(C1-C6)алкил][(C1-C6)алкилен-(C5-C10)гетероциклила], SO2-N[(C1-C6)алкилен-(C6-C10)арила]2, SO2-N[(C1-C6)алкилен-(C5-C10)гетероциклила]2, C(NH)(NH2), NH2, NH-(C1-C6)алкила, N[(C1-C6)алкила]2, NH-C(O)-(C1-C6)алкила, NH-C(O)O-(C1-C6)алкила, NH-C(O)-(C6-C10)арила, NH-C(O)-(C5-C10)гетероциклила, NH-C(O)O-(C6-C10)арила, NH-C(O)O-(C5-C10)гетероциклила, NH-C(O)-NH-(C1-C6)алкила, NH-C(O)-NH-(C6-C10)арила, NH-C(O)-NH-(C5-C10)гетероциклила, NH-SO2-(C1-C6)алкила, NH-SO2-(C6-C10)арила, NH-SO2-(C5-C10)гетероциклила, N(C1-C6)алкил-C(O)-(C1-C6)алкила, N(C1-C6)алкил-C(O)O-(C1-C6)алкила, N(C1-C6)алкил-C(O)-(C6-C10)арила, N(C1-C6)алкил-C(O)-гетероциклила, N(C1-C6)алкил-C(O)O-(C6-C10)арила, N(C1-C6)алкил-C(O)O-(C5-C10)гетероциклила, N(C1-C6)алкил-C(O)-NH-(C1-C6)алкила], N(C1-C6)алкил-C(O)-NH-(C6-C10)арила, N(C1-C6)алкил-C(O)-NH-(C5-C10)гетероциклила, N[(C1-C6)алкил]-C(O)-N[(C1-C6)алкила]2, N[(C1-C6)алкил]-C(O)-N[(C1-C6)алкил]-(C6-C10)арила, N[(C1-C6)алкил]-C(O)-N[(C1-C6)алкил]-(C5-C10)гетероциклила, N[(C1-C6)алкил]-C(O)-N[(C6-C10)арила]2, N[(C1-C6)алкил]-C(O)-N[(C5-C10)гетероциклила]2, N[(C6-C10)арил]-C(O)-(C1-C6)алкила, N[(C5-C10)гетероциклил]-C(O)-(C1-C6)алкила, N[(C6-C10)арил]-C(O)O-(C1-C6)алкила, N[(C5-C10)гетероциклил]-C(O)O-(C1-C6)алкила, N(арил)-C(O)-(C6-C10)арила, N[(C5-C10)гетероциклил]-C(O)-(C6-C10)арила, N[(C6-C10)арил]-C(O)O-(C6-C10)арила, N[(C5-C10)гетероциклил]-C(O)O-(C6-C10)арила, N[(C6-C10)арил]-C(O)-NH-(C1-C6)алкила, N[(C5-C10)гетероциклил]-C(O)-NH-(C1-C6)алкила, N(арил)-C(O)-NH-(C6-C10)арила, N[(C5-C10)гетероциклил]-C(O)-NH-(C6-C10)арила, N[(C6-C10)арил]-C(O)-N[(C1-C6)алкила]2, N[(C5-C10)гетероциклил]-C(O)-N[(C1-C6)алкила]2, N[(C6-C10)арил]-C(O)-N[(C1-C6)алкил]-(C6-C10)арила, N[(C5-C10)гетероциклил]-C(O)-N[(C1-C6)алкил]-(C6-C10)арила, N[(C6-C10)арил]-C(O)-N[(C6-C10)арила]2, N[(C5-C10)гетероциклил]-C(O)-N[(C6-C10)арила]2, (C6-C10)арила, (C1-C6)алкилен-(C6-C10)арила, O-(C1-C6)алкилен-(C6-C10)арила, (C5-C10)гетероциклила, (C1-C6)алкилен-(C5-C10)гетероциклила, O-(C1-C6)алкилен-(C5-C10)гетероциклила, где (C6-C10)арил или (C5-C10)гетероциклил может быть замещенным в от одного до трех положениях галогеном, OH, NO2, CN, O-(C1-C6)алкилом, (C1-C6)алкилом, NH2, NH(C1-C6)алкилом, N[(C1-C6)алкилом]2, SO2CH3, COOH, C(O)O-(C1-C6)алкилом, CONH2, (C1-C6)алкилен-O-(C1-C6)алкилом, (C1-C6)алкилен-O-(C6-C10)арилом, O-(C1-C6)алкилен-(C6-C10)арилом; или где (C6-C10)арил замещен в вицинальном положении O-(C1-C4)алкилен-O группой, за счет чего образуется 5-8-членный цикл вместе с атомами углерода, к которым присоединены атомы кислорода. Арильные или гетероциклильные заместители (C6-C10)арильной и (C5-C10)гетероциклильной групп не могут иметь дополнительных заместителей, содержащих арильную или гетероциклильную группу.
В случае замещения предпочтительными заместителями (C6-C10)арильных групп являются (C1-C4)алкил, O-(C1-C4)алкил, O-фенил, C(O)O-(C1-C6)алкил, C(O)OH, C(O)-(C1-C4)алкил, галоген, NO2, SO2NH2, CN, SO2-(C1-C4)алкил, NH-SO2-(C1-C4)алкил, NH2, NH-C(O)-(C1-C4)алкил, (C3-C8)циклоалкил, (C1-C4)алкил-OH, C(O)N[(C1-C4)алкил]2, C(O)NH2, N[(C1-C4)алкил]2, (C1-C4)алкенилен-(C6-C10)арил, где (C6-C10)арил может быть дополнительно замещенным (C1-C4)алкилом, (C1-C4)алкилен-O-(C1-C6)алкилом, O-(C1-C6)алкил-(C6-C10)арилом или может быть замещен в вицинальном положении O-(C1-C4)алкилен-O группой, за счет чего образуется 5-8-членный цикл вместе с атомами углерода, к которым присоединены атомы кислорода. Более предпочтительно, заместители (C6-C10)арила представляют собой галоген, (C1-C4)алкил, в особенности метил, этил, изопропил или 3,3,3-трифторметил, O-(C1-C4)алкил, в особенности метокси, SO2-(C1-C4)алкил, в особенности SO2-CH3 или SO2-CF3, или N[(C1-C4)алкил]2, в особенности N[(CH3)2.
В монозамещенных фенильных группах заместитель может находиться в положении-2, положении-3 или в положении-4, где предпочтительны положение-3 и положение-4. В фенильных группах, несущих два заместителя, они могут располагаться в положениях-2,3, положениях-2,4, положениях-2,5, положениях-2,6, положениях- 3,4 или положениях-3,6. В фенильных группах, несущих три заместителя, заместители могут располагаться в положениях-2,3,4, положениях-2,3,5, положениях-2,3,6, положениях-2,4,5, положениях-2,4,6 или положениях-3,4,6. Изложенные выше положения, касающиеся фенильных групп, соответствующим образом применимы к двухвалентным группам, полученным из фенильных групп, т.е. фенилену, который может быть незамещенным или замещенным 1,2-фениленом, 1,3-фениленом или 1,4-фениленом. Изложенные выше положения также соответствующим образом применимы к арильным подгруппам в арилалкиленовых группах. Примеры арилалкиленовых групп, которые также могут быть замещенными или незамещенными в арильной подгруппе, как и в алкиленовой подгруппе, представляют собой бензил, 1-фенилэтилен, 2-фенилэтилен, 3-фенилпропилен, 4-фенилбутилен, 1-метил-3-фенилпропилен.
В случае наличия заместителей предпочтительные заместители для (C5-C10)гетероциклических групп представляют собой (C1-C4)алкил, O-(C1-C4)алкил, (C1-C4)алкилен-фенил, галоген, (C1-C4)алкилен-О-(C1-C4)алкил, (C5-C10)гетероциклил, (C1-C4)алкилен-N[(C1-C4)алкил]2 или (C6-C10)арил, в которых (C6-C10)арил может быть дополнительно замещен (C1-C4)алкилом, (C1-C4)алкилен-O-(C1-C6)алкилом, O-(C1-C6)алкил-(C6-C10)арилом или может быть замещен в вицинальном положении O-(C1-C4)алкилен-O группой, за счет чего образуется 5-8-членный цикл вместе с атомами углерода, к которым присоединены атомы кислорода. Более предпочтительно, заместители (C5-C10)гетероциклила представляют собой (C1-C4)алкил.
Общие и предпочтительные заместители (C6-C10)арильной и (C5-C10)гетероциклильной групп можно комбинировать с общими и предпочтительными определениями R1, R2, R3, R4, R5, R6, R7, R8, R9, n и L, как описано выше.
Настоящее изобретение таким образом также относится к соединениям формул (I) или (I') или к их физиологически приемлемым солям и/или стереоизомерным формам для применения в качестве фармацевтических средств (или лекарственных средств), к применению соединений формул (I) или (I') или их физиологически приемлемых солей и/или стереоизомерных форм для получения фармацевтических средств для лечения и/или предотвращения заболеваний, связанных с Rho-киназой и/или активируемым Rho-киназой фосфорилированием фосфатазы легких цепей миозина, т.е. для лечения и/или предотвращения гипертензии, легочной гипертензии, офтальмологической гипертензии, ретинопатии, глаукомы, нарушения периферического кровообращения, периферического окклюзионного поражения артерии (PAOD), коронарной болезни сердца, стенокардии, гипертрофии сердца, сердечной недостаточности, ишемических болезней, ишемической недостаточности органа (повреждение органа-мишени), пневмофиброза, фиброза печени, печеночной недостаточности, нефропатии, включая вызванную гипертензией, не вызванную гипертензией и диабетическую нефропатии, почечной недостаточности, фиброза почек, почечного гломерулосклероза, гипертрофии органа, астмы, хронического обструктивного заболевания легких (COPD), синдрома расстройства дыхания у взрослых, тромботических нарушений, удара, спазма сосудов головного мозга, ишемии сосудов головного мозга, боли, например невропатической боли, нервной деградации, повреждения спинного мозга, болезни Альцгеймера, преждевременных родов, нарушения эрекции, эндокринных нарушений, атеросклероза, гипертрофии простаты, диабетов и осложнений диабетов, метаболического синдрома, рестеноза кровеносных сосудов, атеросклероза, воспаления, аутоиммунных заболеваний, СПИДа, остеопатии, такой как остеопороз, бактериальной инфекции пищеварительного тракта, сепсиса, развития и прогрессии рака, например рака груди, толстой кишки, простаты, яичников, мозга и легкого и их метастазов.
Лечение и/или предотвращение заболеваний у человека представляет собой предпочтительный вариант осуществления, но при использовании соединений по настоящему изобретению можно также лечить теплокровных животных, таких как кошки, собаки, крысы, лошади и т.д.
Настоящее изобретение дополнительно относится к фармацевтическим составам (или фармацевтическим композициям), которые содержат эффективное количество по крайней мере одного соединения формулы (I) или (I') или их физиологически приемлемых солей и/или стереоизомерных форм и фармацевтически приемлемый носитель, т.е. одно или несколько фармацевтически приемлемых веществ носителя (или наполнителя) и/или добавок (или эксципиентов).
Необязательно, физиологические функциональные производные, включая пролекарства, соединения формулы (I) или (I') можно использовать для указанных выше применений и фармацевтических составов.
Фармацевтические средства можно вводить перорально, например в форме пилюль, таблеток, таблеток с гладким покрытием, таблеток с покрытием, гранул, твердых и мягких желатиновых капсул, растворов, сиропов, эмульсий, суспензий или аэрозольных смесей. В то же время введение можно проводить ректально, например, в форме суппозиториев, или парентерально, например внутривенно, внутримышечно или подкожно, в форме растворов для инъекций или растворов для инфузии, микрокапсул, имплантантов или стержневидных капсул, или чрескожно или местно, например, в форме мазей, растворов или настоек, или другими способами, например, в форме аэрозолей или назальных спреев.
Фармацевтические составы согласно изобретению получают при использовании способов, известных в данной области и специалисту в данной области техники, дополнительно к соединению(ям) формул (I) или (I') или его(их) физиологически приемлемым солям и/или его(их) стереоизомерным формам, а также их пролекарствам используют фармацевтически приемлемые инертные неорганические и/или органические соединения носителя и/или добавки. Для получения пилюль, таблеток, покрытых таблеток и твердых желатиновых капсул можно использовать, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и т.д. Соединения носителей для мягких желатиновых капсул и суппозиториев представляют собой, например, жиры, воска, полутвердые и жидкие полиолы, природные или гидрированные масла и т.д. Пригодные соединения носителей для приготовления растворов, например растворы для инъекций или эмульсий или сиропы, представляют собой, например, воду, физиологический раствор, спирты, глицерин, полиолы, сахарозу, инвертированный сахар, глюкозу, растительные масла и т.д. Пригодные соединения носителей для микрокапсул, имплантантов или стержневидных капсул представляют собой, например, сополимеры гликолевой кислоты и молочной кислоты. Фармацевтические составы обычно содержат от примерно 0,5 до примерно 90% по массе соединений формул (I) или (I') или их физиологически приемлемых солей и/или стереоизомерных форм. Количество активного ингредиента формул (I) или (I') и/или их физиологически приемлемых солей и/или стереоизомерных форм в фармацевтических составах обычно составляет от примерно 0,5 до примерно 1000 мг, предпочтительно от примерно 1 до примерно 500 мг.
Дополнительно к активным ингредиентам формул (I) или (I') и/или их физиологически приемлемых солей и/или стереоизомерных форм и соединениям носителя фармацевтические составы могут содержать одну или несколько добавок, таких как, например, наполнители, агенты, вызывающие дезинтеграцию, связующие, смазывающие агенты, смачивающие агенты, стабилизаторы, эмульгаторы, консерванты, подсластители, красители, вкусовые добавки, ароматизаторы, загустители, разбавители, буферные соединения, растворители, солюбилизаторы, агенты для достижения депо-эффекта, соли, изменяющие осмотическое давление, глазировочные средства и антиоксиданты. Фармацевтические составы также могут содержать два или более соединения формул (I) и/или (I') и/или их физиологически приемлемые соли и/или их стереоизомерные формы. В случае фармацевтических составов, содержащих два или более соединений формул (I) и/или (I'), выбор конкретного соединения может основываться на конкретном общем фармацевтическом профиле фармацевтического препарата. Например, высокоактивное соединение с коротким периодом действия можно объединять с соединением с низкой активностью, действующим в течение продолжительного времени. Легкость с точки зрения выбора заместителей в соединениях формул (I) или (I') позволяет в значительной степени контролировать биологические и физико-химические свойства соединений, таким образом позволяя выбирать такие целевые соединения. Более того, дополнительно к по крайней мере одному соединению формул (I) или (I') и/или их физиологически приемлемым солям и/или стереоизомерным формам, фармацевтические составы также могут содержать один или несколько других терапевтически или профилактически активных ингредиентов.
При использовании соединений формулы (I) или (I') доза может изменяться в широком диапазоне и, как принято и хорошо известно терапевту, ее следует подбирать, исходя из индивидуальных условий в каждом конкретном случае. Доза зависит, например, от конкретного используемого соединения, природы и тяжести заболевания, подвергаемого лечению, от типа и графика введения и зависит от того, острое или хроническое состояние подвергается лечению или осуществляется профилактика. Подходящую дозу можно определить при использовании клинических способов, хорошо известных в медицине. В общем дневная доза для достижения заданных результатов для взрослого человека весом примерно 75 кг составляет от примерно 0,01 до примерно 100 мг/кг, предпочтительно от примерно 0,1 до примерно 50 мг/кг, в особенности от примерно 0,1 до примерно 10 мг/кг (в каждом случае мг на кг массы тела). Дневная доза может быть разделена, в особенности в случае введения относительно больших количеств, на несколько, например 2, 3 или 4, приемов. Как обычно, в зависимости от индивидуального состояния может оказаться необходимым отклонение в большую или меньшую сторону от указанной дневной дозы.
Более того, соединения формул (I) или (I') могут использоваться в качестве синтетических промежуточных соединений для получения других соединений, в особенности других фармацевтических активных ингредиентов, которые можно получить из соединения формулы I, например, путем введения заместителя или модификацией функциональных групп.
Необходимо понимать, что модификации, которые не оказывают значительного влияния на активность в различных вариантах осуществления изобретения, включены в описываемое здесь изобретение.
Не ограничивая объем изобретения, соединения формул (I) или (I') могут быть получены сходным образом со следующими соединениями, приведенными в качестве примера.
В общем случае, защитные группы, которые могут оставаться в продуктах, полученных в реакциях сочетания, затем удаляют при использовании стандартных способов. Например, трет-бутильные защитные группы в особенности трет-бутоксикарбонильная группа, которая представляет собой защитную группу для аминогруппы, может быть снята, т.е. превращена в аминогруппы при помощи обработки трифторуксусной кислотой. Как уже объяснялось выше, после реакции сочетания также могут быть получены функциональные группы из подходящих групп-предшественников. Дополнительно, затем можно проводить превращение соединения формулы (I) или (I') в физиологически приемлемую соль или пролекарство, при использовании известных способов.
В общем случае обрабатывают реакционную смесь, содержащую конечное соединение формул (I) или (I') или промежуточное соединение, и, если желательно, продукт затем очищают при использовании обычных способов, известных специалистам в данной области техники. Например, синтезированное соединение может быть очищено при использовании хорошо известных способов, таких как кристаллизация, хроматография или высокоэффективная жидкостная хроматография с обращенной фазой (RP-HPLC), или других способов разделения, основанных, например, на размере, заряде или гидрофобности соединения. Сходным образом, для характеризации соединения по изобретению можно использовать хорошо известные способы, такие как анализ аминокислотной последовательности, ЯМР, ИК и масс-спектрометрия (MС).
Изохинолиноны могут быть синтезированы при использовании различных способов. Следующая общая схема иллюстрирует некоторые возможные пути получения изохинолонов, но не ограничивает настоящее изобретение.
Схема 1
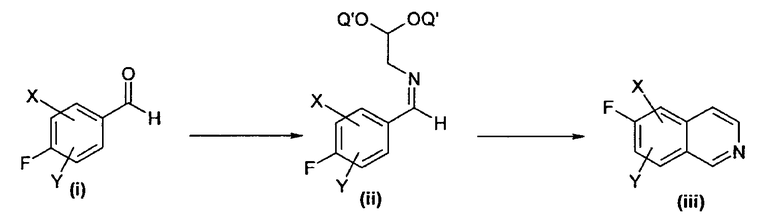
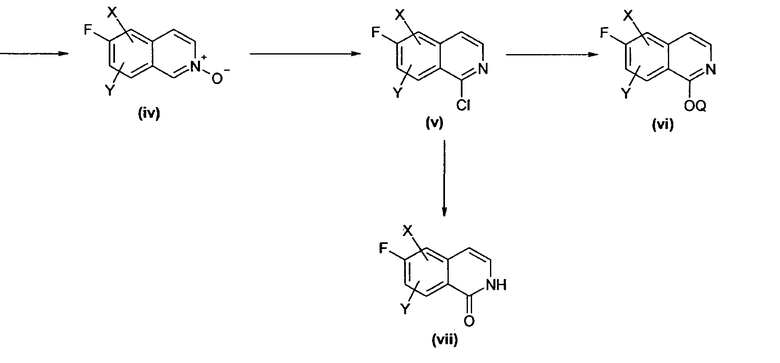
Подходящим образом замещенный альдегид, например, замещенный X или Y, представляющие собой, независимо друг от друга, водород, алкил, алкокси или галоген, присоединенные в подходящем положении, можно вводить в реакцию с подходящим соединением, таким как, например, ацеталь аминоацетальдегида в растворителе, таком как ТГФ, хлороформ или толуол при кислотном катализе толуолсульфоновой кислотой или другой подходящей кислотой с получением имина (ii), где Q' может представлять собой, например, метил или этил, который, в свою очередь, может быть циклизован различными способами в изохинолин (iii). Например, циклизацию можно осуществить катализом кислотой Льюиса под действием пригодных кислот Льюиса, таких как тетрахлорид титана, хлориды железа, хлориды алюминия и т.д., при температурах, изменяющихся в диапазоне от комнатной до 100°С, или путем восстановления имина в соответствующий амин под действием пригодного восстановителя, такого как боргидрид натрия, превращением амина в амид или сульфонамид реакцией с подходящим хлоридом кислоты и последующей циклизацией в изохинолин под действием подходящей кислоты Льюиса. Изохинолин (iii) сам по себе затем может быть превращен в соответствующий N-оксид (iv) под действием подходящего окислителя, такого как пероксид водорода, м-хлор пербензойной кислоты или других, при комнатной температуре или при повышенной температуре. N-оксид (iv) затем может быть превращен в 1-хлоризохинолиновое производное (v) путем его реакции с реагентом, таким как оксихлорид фосфора в присутствии или в отсутствие пентахлорида фосфора. Производное (v) затем может быть превращено в подходящее 1-алкокси производное путем его реакции с различными спиртами Q-OH, такими как метанол, этанол или бензиловый спирт, в присутствии подходящего основания, такого как гидрид натрия, и в подходящем растворителе, таком как диметилформамид, диметилацетамид или другие. В качестве альтернативы, (v) может быть непосредственным образом превращено в изохинолиноновое производное (vii) его реакцией с реагентом, таким как ацетат аммония.
Схема 2
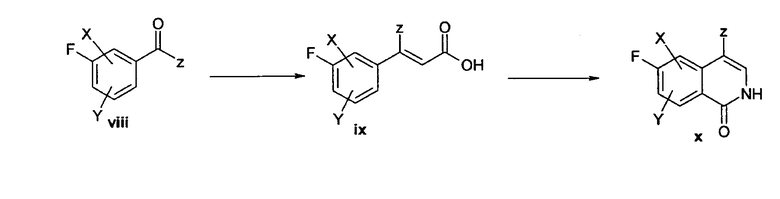
В качестве альтернативы изохинолины могут быть получены реакцией подходящих 3-формилированных или ацилированных фторбензолов (viii), где z представляет собой, например, Н или алкил, такой как метил или этил, с реагентом, таким как триэтилфосфоноацетат в присутствии подходящего основания, такого как гидрид натрия, с получением соответствующего эфира коричной кислоты, который затем расщепляют под действием подходящего основания, такого как гидроксид калия, гидроксид натрия или гидроксид лития, в подходящем растворителе с получением кислоты (ix). Кислота (ix) затем может быть превращена в соответствующий хлорангидрид при использовании хорошо известных способов, который может быть превращен в азид кислоты реакцией с азидом натрия в подходящем растворителе, таком как эфир, хлороформ или ацетон, в присутствии или в отсутствие воды. Соответствующий азид затем может быть превращен в изохинолинон (x) его реакцией в подходящем растворителе, таком как дифенилметан или дифениловый эфир, при подходящей температуре.
Схема 3
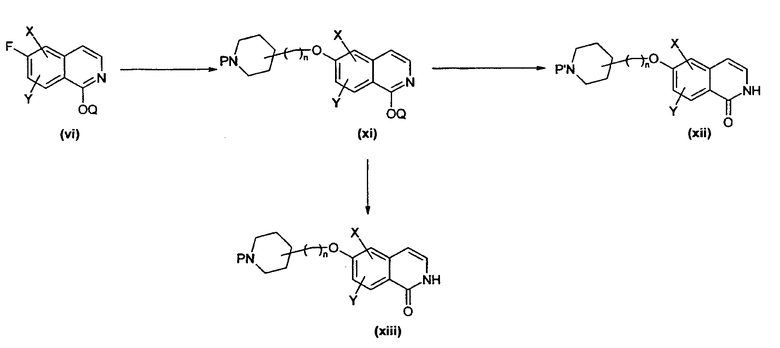
Полученные выше 6-фторизохинолоны, например, (vi), можно вводить в реакцию с подходящим образом Р-замещенными аминоспиртами, где Р представляет собой, например, водород, алкил или защитную группу, такую как, например, Boc, в присутствии основания, такого как DBU, карбонат цезия или гидрид натрия, с получением соответствующих алкокси-замещенных производных (xi). При соответствующих условиях это превращение можно проводить уже на ранних стадиях синтеза (например, путем реакции с подходящим промежуточным соединением). Следует понимать, что, в случае незащищенных изохинолонов, это может требовать защиты азота или кислорода изохинолонового фрагмента при помощи известных способов, таких как реакция с подходящим образом замещенными алкил или бензилгалогенидами в присутствии основания.
Продукты, такие как (xi), полученные при использовании указанного способа, можно затем, если присутствует подходящая аминофункциональная группа, вводить в реакцию с подходящими альдегидами или кетонами в присутствии восстановителя, такого как триацетокси боргидрид натрия, боргидрид натрия или цианоборгидрид натрия в подходящем растворителе и в присутствии водопоглощающего агента, такого как молекулярные сита или подходящий ортоэфир. С указанной аминогруппы возможно следует снять защитную группу в начальной стадии, как, например, удалением Boc-групп под действием кислоты. В случае использования защищенных изохинолонов, для высвобождения заданного изохинолона (xii) необходимо расщепление использованных защитных групп. Указанное высвобождение, однако, можно проводить до или после стадии восстановительного аминирования, в зависимости от природы используемого альдегида/кетона и используемой защитной группы.
Изохинолоновые производные, такие как (xii), могут быть получены в виде свободных оснований или в виде различных солей, таких как гидрохлориды, гидробромиды, фосфаты, сульфаты или фумараты. Полученные соли могут быть затем превращены в соответствующие свободные основания или путем проведения ионообменной хроматографии, или, например, обработкой водной щелочью и последующей экстракцией пригодными органическими растворителями, такими как, например, метиловый трет-бутиловый простой эфир, хлороформ, этилацетат или смесями изопропанол/дихлорметан и последующим выпариванием досуха.
Общие способы получения замещенных производных изохинолона, как описано выше, могут быть легко адаптированы для получения соединений формулы (I) или формулы (I'). В следующих примерах более подробно описано получение соединений по настоящему изобретению. Таким образом, следующие примеры представляют собой часть настоящего изобретения и предназначены для иллюстрации, но не ограничивают настоящее изобретение.
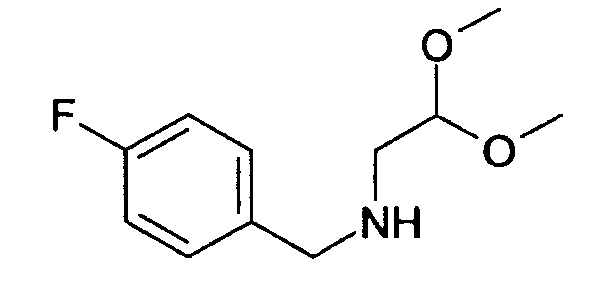
(2,2-диметоксиэтил)-(4-фторбензил)амин (1)
12,4 г 4-фторбензальдегида растворяли в 100 мл толуола и вводили в реакцию с
10,5 г 2-аминоацетальдегид диметилацеталя и 1,90 г (10 ммоль) моногидрата п-толуолсульфоновой кислоты в течение двух часов в аппарате Дина Старка. Раствор оставляли охлаждаться, экстрагировали насыщенным бикарбонатом натрия, водой и рассолом, сушили над сульфатом магния и упаривали досуха. Неочищенный продукт растворяли в 100 мл этанола. Добавляли порциями 1,89 г боргидрида натрия. Перемешивание продолжали в течение ночи. Для обработки добавляли уксусную кислоту до прекращения выделения газа. Затем раствор упаривали досуха, поглощали дихлорметаном и дважды промывали водой. Органический слой экстрагировали рассолом, сушили над сульфатом магния и упаривали досуха. Полученный неочищенный продукт (20 г) использовали для последующих реакций без очистки. Rt=0,86 мин (способ В). Детектированная масса: 182,1 (М-ОМе-), 214,2 (М+Н+).
N-(2,2-диметоксиэтил)-N-(4-фторбензил)-4-метилбензолсульфонамид (2)
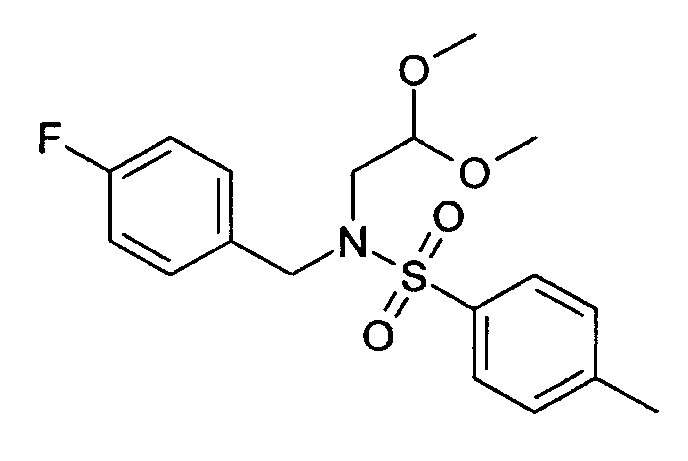
20 г (2,2-диметоксиэтил)-(4-фторбензил)амин (1) растворяли в 120 мл дихлорметана. Добавляли 20 мл пиридина. При 0°С по каплям добавляли раствор 23,8 г хлорида п-толуолсульфоновой кислоты в дихлорметане. Реакцию затем оставляли охлаждаться до комнатной температуры и продолжали перемешивание до полной конверсии. Для обработки реакционную смесь дважды экстрагировали 2М соляной кислоты, дважды бикарбонатом натрия и один раз рассолом. Органический слой сушили над сульфатом магния, упаривали досуха и полученный неочищенный продукт очищали хроматографией на силикагеле с получением 22,95 г соединения 2 в виде оранжевого масла. Rt=1,71 мин (способ С). Детектированная масса: 336,1 (М-ОМе-).
6-фторизохинолин (3)
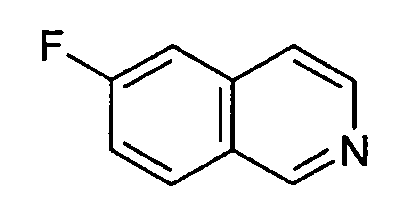
41,6 г AlCl3 суспендировали в 400 мл дихлорметана. При комнатной температуре добавляли 22,95 г раствора N-(2,2-диметоксиэтил)-N-(4-фторбензил)-4-метилбензолсульфонамид (2) в 150 мл дихлорметана. Перемешивание продолжали при комнатной температуре в течение ночи, раствор выливали на лед, органический слой отделяли, водную фазу дважды экстрагировали дихлорметаном и объединенные органические слои дважды экстрагировали бикарбонатом натрия. Органический слой сушили над сульфатом магния, упаривали досуха и полученный неочищенный продукт (8,75 г) очищали хроматографией на силикагеле с получением 2,74 г соединения (3). Rt=0,30 мин (способ С). Детектированная масса: 148,1 (М+Н+).
7-хлор-6-фторизохинолин (4)
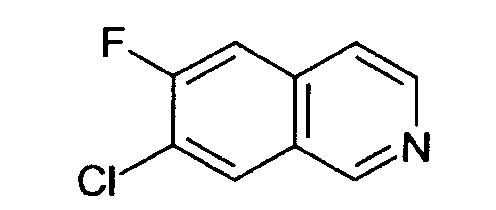
Исходя из 3-хлор-4-фторбензальдегида, требуемое соединение было получено той последовательностью реакций, что и использованная для синтеза 6-фторизохинолина (3). Rt=0,77 мин (способ А). Детектированная масса: 181,1/184,1 (М+Н+).
7-хлор-6-фторизохинолин 2-оксид (5)
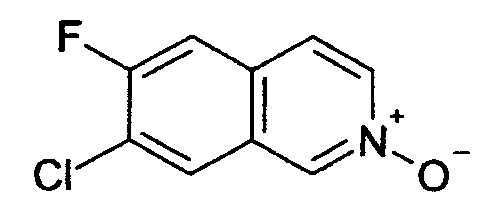
25 г (137,7 ммоль) 7-хлор-6-фторизохинолина (4) растворяли в 500 мл дихлорметана. Добавляли 50,9 г (206,5 ммоль) м-хлор пербензойной кислоты (70%) при комнатной температуре, и смесь перемешивали при комнатной температуре до достижения полной конверсии. Для обработки осадок фильтровали и промывали дихлорметаном. Фильтрат промывали дважды раствором бикарбоната натрия. Слои разделяли и водную фазу дважды экстрагировали дихлорметаном. Органические фазы сушили над MgSO4 и упаривали. Полученное таким образом твердое вещество (18,4 г) использовали без дальнейшей очистки. Rt=0,87 мин (способ С). Детектированная масса: 198,1/200,1 (М+Н+).
1,7-дихлор-6-фторизохинолин (6)
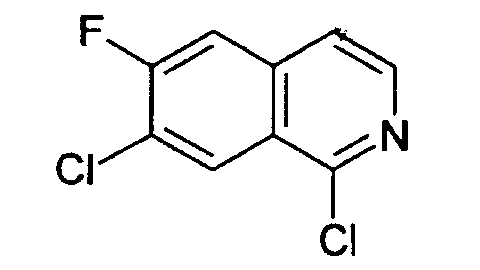
2,6 г (12,0 ммоль) 7-хлор-6-фторизохинолин 2-оксида (5) нагревали в 40 мл РОСl3 и кипятили с обратным холодильником в течение 4 ч. После того как смесь остывала до комнатной температуры, ее выливали на лед. Водный раствор экстрагировали три раза дихлорметаном. Объединенные органические слои сушили над MgSO4 и упаривали с получением 2,91 г целевого соединения, которое использовали без дополнительной очистки. Rt=2,34 мин (способ А). Детектированная масса: 216,0/218,0 (М+Н+)
Трет-бутиловый эфир 4-(изохинолин-6-илокси)пиперидин-1-карбоновой кислоты (7)
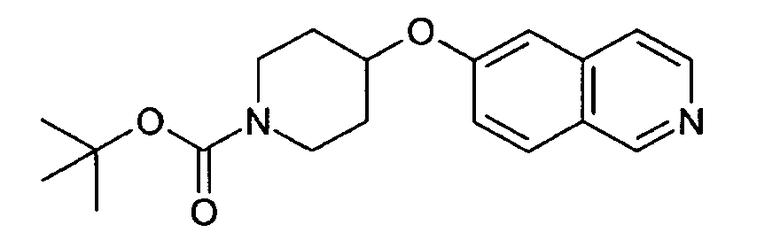
7,49 г трет-бутилового эфира 4-гидроксипиперидин-1-карбоновой кислоты растворяли в 20 мл безводного диметилацетамида. Добавляли 1,49 г гидрида натрия (60%). Затем добавляли по каплям 3,65 г 6-фторизохинолина (3) в диметилацетамиде. Раствор нагревали при 80°С в течение 2 часов, затем растворитель удаляли и осадок поглощали дихлорметаном. Органический слой дважды экстрагировали водой и затем рассолом, сушили над сульфатом магния и упаривали досуха. Неочищенный продукт очищали хроматографией на силикагеле с получением 6,22 г трет-бутилового эфира 4-(изохинолин-6-илокси)пиперидин-1-карбоновой кислоты (7). Rt=1,32 мин (способ В). Детектированная масса: 329,1 (М+Н+).
Трет-бутиловый эфир 4-(2-окси-изохинолин-6-илокси)пиперидин-1-карбоновой кислоты (8)
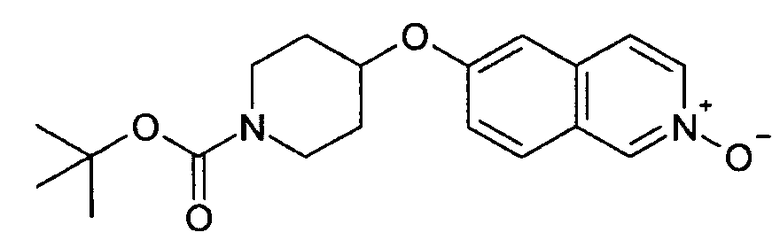
Растворяли 3,97 г (12,1 ммоль) трет-бутилового эфира 4-(изохинолин-6-илокси)пиперидин-1-карбоновой кислоты (7) в 100 мл дихлорметана и при комнатной температуре добавляли 4,47 г (18,1 ммоль) м-хлор пербензойной кислоты (70%). Реакционную смесь перемешивали в течение 1 ч и затем промывали насыщенным раствором бикарбоната натрия. Водную фазу отделяли и экстрагировали дихлорметаном. Объединенные органические фазы сушили над MgSO4 и упаривали с получением 4,19 г неочищенного вещества, которое может быть использовано для последующего превращения без очистки. Rt=1,46 мин (способ В). Детектированная масса: 345,2 (М+Н+).
1-хлор-6-(пиперидин-4-илокси)изохинолингидрохлорид (9)
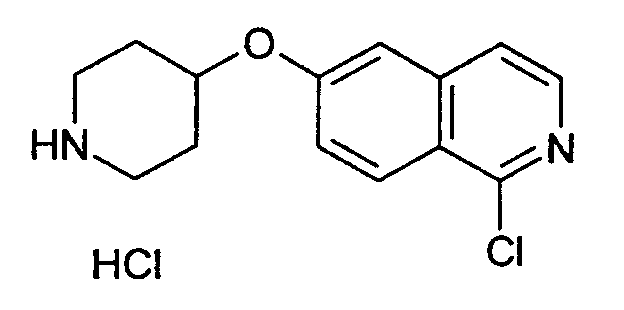
Растворяли при 50°С 3,5 г (10,16 ммоль) трет-бутилового эфира 4-(2-окси-изохинолин-6-илокси)пиперидин-1-карбоновой кислоты (8) в 250 мл этанола, насыщенного НСl. Прозрачный раствор концентрировали под вакуумом и остаток кипятили с обратным холодильником в 50 мл РОCl3. После 3 ч РОCl3 удаляли под вакуумом и остаток поглощали водой. Устанавливали рН 11 путем добавления гидроксида натрия и водный раствор дважды экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом магния и упаривали досуха. Остаток очищали препаративной ВЭЖХ, при помощи которой целевое соединение получали в виде трифторацетата. Полученное вещество превращали в соответствующую соль НСl путем растворения продукта в 2Н HCl, с последующей лиофилизацией. Выход: 950 мг. Rt=1,03 мин (способ В). Детектированная масса: 263,1/265,1 (М+Н+).
Трет-бутиловый эфир 4-(1-хлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (10)
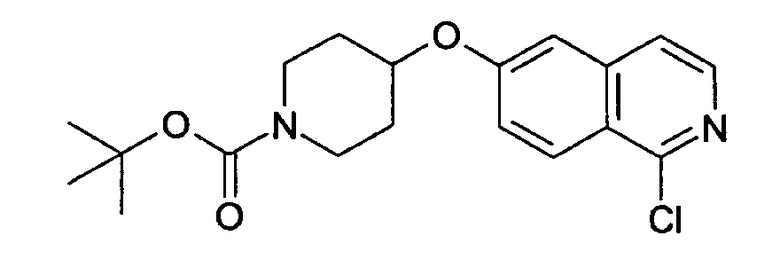
Растворяли 1,23 г (4,11 ммоль) 1-хлор-6-(пиперидин-4-илокси)изохинолингидрохлорида (9) в 50 мл дихлорметана и добавляли 0,85 мл (6,15 ммоль) диэтиламина. При 0°С добавляли по каплям 1,09 г (5,0 ммоль) ди-трет-бутилдикарбоната в 10 мл дихлорметана и смесь оставляли при комнатной температуре в течение ночи. Для обработки смесь дважды промывали водой, сушили над сульфатом магния и выпаривали с получением 1,1 г целевого продукта, который можно было использовать без дальнейшей очистки. Rt=1,86 мин (способ С). Детектированная масса: 363,1/365,2 (М+Н+).
Трет-бутиловый эфир 4-(1-бензилоксиизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11)
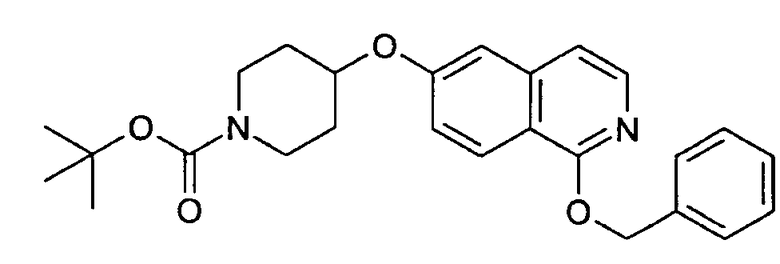
Растворяли 0,70 г (6,49 ммоль) бензилового спирта в 10 мл диметилацетамида. Добавляли 260 мг (6,49 ммоль) гидрида натрия (60%) и раствор перемешивали при комнатной температуре. Через 30 минут добавляли раствор 1,57 г (4,33 ммоль) трет-бутилового эфира 4-(1-хлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (10) в 10 мл диметилацетамида и полученную смесь нагревали при 90°С (температура бани). Через 8 ч и выдерживания при комнатной температуре в течение ночи добавляли 1,0 дополнительные эквиваленты бензилового спирта и гидрида натрия и продолжали нагревание при 90°С в течение 8 ч. Для обработки растворитель удаляли под вакуумом и остаток растворяли в дихлорметане. Органический раствор дважды промывали водой, сушили над MgSO4 и упаривали. Полученный неочищенный продукт очищали препаративной ВЭЖХ. Rt=2,13 мин (способ С). Детектированная масса: 435,2 (М+Н+).
6-(пиперидин-4-илокси)-2Н-изохинолин-1-он (12)
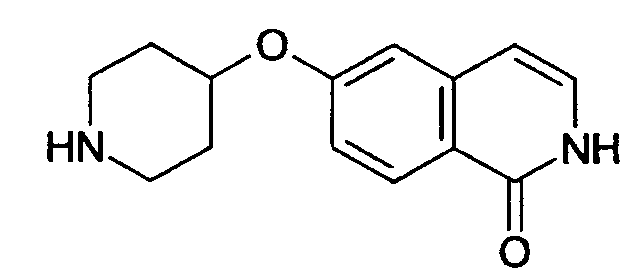
Соединение (11) растворяли в смеси этанол/2Н HCl (1:1) и перемешивали при комнатной температуры до достижения полной конверсии. Растворитель удаляли под вакуумом и осадок очищали препаративной ВЭЖХ. Полученный трифторацетат растворяли в 2Н HCl и лиофилизировали. После последующей лиофилизации из воды может быть получено 850 мг целевого соединения в виде соли HCl. Rt=0,75 мин (способ В). Детектированная масса: 245,1 (М+Н+).
Альтернативный способ синтеза:
6-фтор-изохинолинон (13)
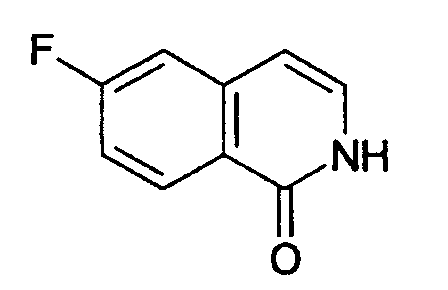
4,8 мл (90,3 ммоль, 1,5 экв.) тионил хлорида добавляли по частям к раствору 10 г (60,2 ммоль) 3-фторкоричной кислоты в 44 мл хлороформа и 1 мл ДМФА. Реакцию нагревали до кипения с обратным холодильником в течение 2,5 ч. Затем растворители перегоняли с получением 11,4 г неочищенного хлорангидрида, который использовали без дополнительной очистки.
Хлорид кислоты растворяли в 45 мл ацетона. Добавляли по частям при 0°С 8,03 г NaN3 (123,5 ммоль, 2 экв.). Затем добавляли 41 мл воды и при этом температуру поддерживали ниже 5°С. Реакцию перемешивали еще 1,5 ч. Затем добавляли 55 мл хлороформа. Смесь экстрагировали 80 мл воды и затем 40 мл рассола. После сушки над NaSO4 и фильтрации добавляли 14 мл дифенилового простого эфира и большую часть хлороформа удаляли под вакуумом (без нагревания). Необходимо избегать полного удаления хлороформа.
Раствор, содержащий азид, дифениловый простой эфир и оставшийся хлороформ, добавляли по каплям при 260°С в течение 15 минут к раствору 10 мл трибутил амина в 97 мл дифенилового простого эфира. При добавлении наблюдалась интенсивная реакция. Реакцию перемешивали в течение дополнительных 20 минут при 260°С. После охлаждения до комнатной температуры добавляли 270 мл н-гептана. Осажденный продукт фильтровали и промывали простым эфиром с получением 5,65 г целевого соединения. МС (DCI) Детектированная масса: 164,0 (М+Н+).
6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-он (14)
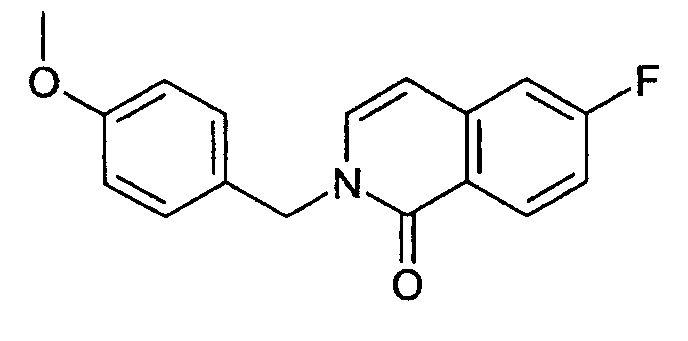
Добавляли 169 мкл п-метоксибензилхлорида (1,24 ммоль, 1,1 экв.) к суспензии 200 мг 6-фторизохинолинона (13) (1,13 ммоль) и 368 мг Cs2CO3 (1,36 ммоль, 1,2 экв.) в 3 мл ДМФА. Смесь перемешивали в течение 2 ч и затем выливали на лед. Осадок фильтровали, промывали водой и сушили с получением 300 мг целевого соединения. ЖХ/МС способ В, время удерживания 1,76 мин, детектированная масса:
284,14 (М+Н+).
6-(пиперидин-4-илокси)-2Н-изохинолин-1-он (12)
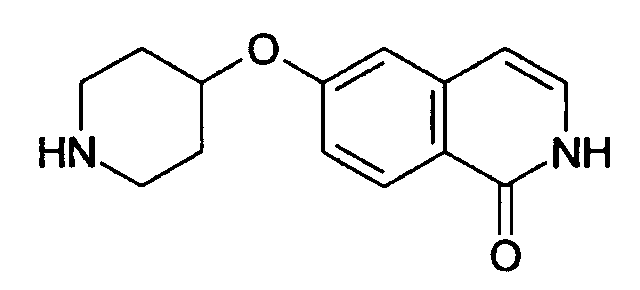
Растворяли 117 мг (0,58 ммоль) трет-бутилового эфира 4-гидроксипиперидин-1-карбоновой кислоты в 2 мл N,N-диметилацетамида. Добавляли в атмосфере аргона 63,6 мг (2,7 ммоль) гидрида натрия и перемешивали смесь при комнатной температуре. Через 30 минут добавляли 150 мг (0,53 ммоль) 6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-она (14) и раствор нагревали до 80°С в течение 1 ч. Смесь выливали в воду и экстрагировали хлороформом. Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали. Неочищенное промежуточное соединение очищали препаративной ВЭЖХ. Защитные группы удаляли путем растворения защищенного промежуточного соединения в 2 мл ТФУ (трифторуксусная кислота) и нагревали реакцию до 150°С в течение 2 ч в микроволновом реакторе. Реакционную смесь гасили метанолом и упаривали досуха. Оставшийся осадок поглощали дихлорметаном, экстрагировали три раза 1Н HCl и комбинированный водный слой один раз экстрагировали дихлорметаном. Комбинированный водный слой лиофилизировали, остаток дважды поглощали водой и вновь лиофилизировали с получением продукта в виде соли HCl. Чистота полученного продукта представляет собой достаточную, но в некоторых случаях присутствующие примеси могут быть удалены при использовании хроматографии на силикагеле или ВЭЖХ.
7-бром-6-фторизохинолин (15)
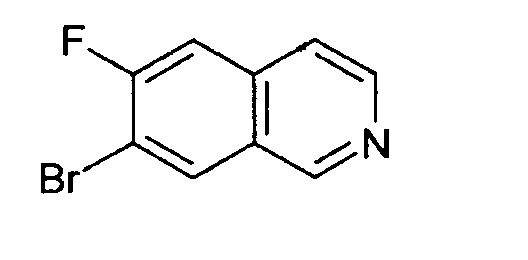
Исходя из 3-бром-4-фторбензальдегида, целевое соединение получали согласно той же последовательности реакций, как и использованной для синтеза 6-фторизохинолина (3). Rt=0,91 мин (способ В). Детектированная масса: 226,0/228,0
(М+Н+).
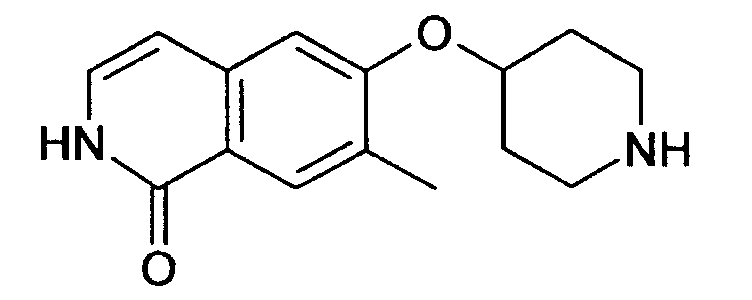
7-метил-6-(пиперидин-4-илокси)-2Н-изохинолин-1-он (16)
а) 6-фтор-7-метил-2Н-изохинолин-1-он
К раствору 10,0 г (55 ммоль) 3-фтор-4-метил-коричной кислоты в 80 мл ацетона последовательно добавляли при 0°С 6,74 г (66,6 ммоль) триэтиламина в 10 мл ацетона, затем 7,83 г (72,2 ммоль) этилхлорформиата. После перемешивания в течение 2 ч при от 0 до 5°С добавляли раствор 4,0 г (61,1 ммоль) азида натрия в 9,5 мл воды. После перемешивания в течение одного дополнительного часа реакционную смесь вливали в 200 мл ледяной воды и дважды экстрагировали хлороформом. Органическую фазу сушили над сульфатом магния, добавляли 40 мл дифенилового эфира и хлороформ аккуратно удаляли под вакуумом. Остаток затем добавляли по каплям в 50 мл дифенилового эфира, который предварительно нагревали до 245°С. После полного добавления смесь перемешивали в течение одного дополнительного часа при 230-250°С. После охлаждения до 150°С реакционную смесь вливали в 270 мл гептана и, после дополнительного охлаждения на ледяной бане осажденный продукт фильтровали с отсасыванием и получали 4,1 г 6-фтор-7-метил-2Н-изохинолин-1-она.
b) 6-фтор-2-(4-метоксибензил)-7-метил-2Н-изохинолин-1-он
К раствору 9,17 г (51,8 ммоль) 6-фтор-7-метил-2Н-изохинолин-1-она в 80 мл ДМФА добавляли 20,2 г (62,1 ммоль) карбоната цезия и затем 8,92 г (56,9 ммоль) 4-метоксибензилхлорида. После перемешивания при комнатной температуре в течение 90 минут реакционную смесь вливали в 600 мл воды, перемешивали в течение 1 ч и затем осажденный продукт фильтровали с отсасыванием. Из маточного раствора выделяли дополнительный продукт при помощи хроматографии смесью гептан/этилацетат (80:20). Комбинированные продукты перекристаллизовывали из этилацетата и получали 8,39 г 6-фтор-2-(4-метоксибензил)-7-метил-2Н-изохинолин-1-она.
с) трет-бутиловый эфир 4-[2-(4-метоксибензил)-7-метил-1-оксо-1,2-дигидроизохинолин-6-илокси]пиперидин-1-карбоновой кислоты
Раствор 3,2 г (15,9 ммоль) 1-трет-бутоксикарбонил-4-гидроксипиперидина в 110 мл диметилацетамида перемешивали с 1,36 г (45,4 ммоль) 80% гидрида натрия в течение 1 ч при комнатной температуре. Затем добавляли суспензию 4,5 г (15,1 ммоль) 6-фтор-2-(4-метоксибензил)-7-метил-2Н-изохинолин-1-она в диметилацетамиде. Реакционную смесь нагревали до 80°С в течение 2 ч, в течение которых был получен прозрачный раствор. Реакционную смесь медленно добавляли к 160 мл воды и после 1 ч перемешивания продукт выделяли фильтрацией и сушили в течение ночи под вакуумом. Получали 6,4 г трет-бутилового эфира 4-[2-(4-метоксибензил)-7-метил-1-оксо-1,2-дигидроизохинолин-6-илокси]пиперидин-1-карбоновой кислоты.
d) 7-метил-6-(пиперидин-4-илокси)-2Н-изохинолин-1-он гидрохлорид
6,4 г (13,4 ммоль) трет-бутилового эфира 4-[2-(4-метоксибензил)-7-метил-1-оксо-1,2-дигидроизохинолин-6-илокси]пиперидин-1-карбоновой кислоты растворяли в 30,5 г (267,4 ммоль) трифторуксусной кислоты. После 1 ч при комнатной температуре смесь нагревали в течение 2 ч в микроволновой печи при 150ºС. Затем избыток трифторуксусной кислоты отгоняли под вакуумом и остаток разбавляли 130 мл 1М соляной кислоты. Водную фазу промывали 3 раза хлористым метиленом и затем ее подвергали лиофильной сушке для получения гидрохлорида, который кристаллизовали из изопропанола. Это привело к получению 3,2 г 7-метил-6-(пиперидин-4-илокси)-2Н-изохинолин-1-она (16) в виде гидрохлорида. Rt=1,24 мин (способ В). Детектированная масса: 259,1 (М+Н+).
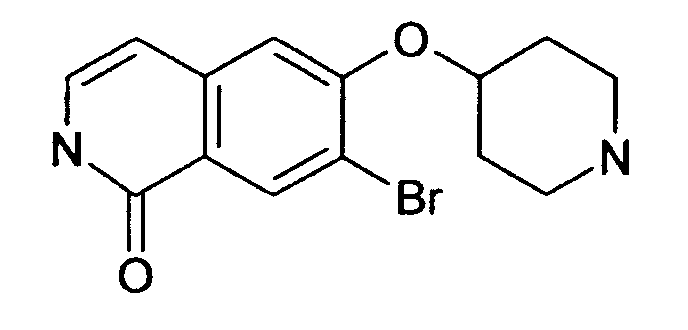
7-бром-6-(пиперидин-4-илокси)-2Н-изохинолин-1-он (17)
а) этиловый эфир 3-(4-бром-3-фторфенил)акриловой кислоты
К раствору 13,4 г (60 ммоль) триэтилфосфонацетата в 80 мл толуола добавляли 1,8 г (60 ммоль) 80% гидрид натрия при 0°С. После 30 минут добавляли 11,0 г (54 ммоль) 4-бром-3-фторбензальдегида в 40 мл толуола и полученную густую смесь перемешивали в течение ночи при использовании механической мешалки. После разбавления 500 мл этилацетата и 200 мл воды органическую фазу отделяли и промывали раствором бикарбоната натрия и рассолом. После высушивания над сульфатом магния с последующим упариванием и очисткой флэш-хроматографией получали 10,6 г этилового эфира 3-(4-бром-3-фторфенил)акриловой кислоты.
b) 3-(4-бром-3-фторфенил)акриловая кислота
10,5 г (38,6 ммоль) этилового эфира 3-(4-бром-3-фторфенил)акриловой кислоты растворяли в 100 мл метанола и перемешивали в течение ночи с 97 мл водного 1М раствора гидроксида натрия. После удаления метанола под вакуумом остаток подкисляли концентрированной соляной кислотой. Осадок выделяли отсасыванием и сушкой под вакуумом при 5°С, что приводило к получению 8,0 г 3-(4-бром-3-фторфенил)акриловой кислоты.
с) 7-бром-6-фтор-2Н-изохинолин-1-он
К раствору 4,0 г (16,3 ммоль) 3-(4-бром-3-фторфенил)акриловой кислоты в 60 мл ацетона последовательно добавляли при от 0 до 5°С 2,0 г (19,6 ммоль) триэтиламина в 10 мл ацетона и затем 2,3 г (21,2 ммоль) этилхлорформиата в 10 мл ацетона. После перемешивания в течение 1 ч при от 0 до 5°С добавляли 1,6 г (24,5 ммоль) азида натрия в 9 мл воды. После перемешивания в течение дополнительного 1 ч реакционную смесь вливали в 200 мл ледяной воды и дважды экстрагировали хлороформом. Органическую фазу сушили над сульфатом магния, добавляли 24 мл дифенилового простого эфира и аккуратно удаляли хлороформ под вакуумом. Остаток затем добавляли по каплям в 60 мл дифенилового эфира, который предварительно нагревали до 250°С. После полного добавления реакционную смесь перемешивали в течение дополнительных 30 минут при 230-250°С. После охлаждения до 100°С реакционную смесь вливали в 100 мл гептана и, после дополнительного охлаждения на ледяной бане, осажденный продукт фильтровали с отсасыванием и получали 2,4 г неочищенного 7-бром-6-фтор-2Н-изохинолин-1-она.
d) 7-бром-6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-он
Из 2,4 г неочищенного 7-бром-6-фтор-2Н-изохинолин-1-она, 3,9 г (11,9 ммоль) карбоната цезия и 1,7 г (10,9 ммоль) 4-метоксибензилхлорида получали 0,93 г 7-бром-6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-она аналогично способу, описанному в стадии b) примера 16.
е) 7-бром-6-(пиперидин-4-илокси)-2Н-изохинолин-1-он гидрохлорид
Из 0,93 г (2,6 ммоль) 7-бром-6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-она и 0,54 г (2,7 ммоль) 1-трет-бутоксикарбонил-4-гидроксипиперидина получали 0,35 г 7-бром-6-(пиперидин-4-илокси)-2Н-изохинолин-1-она в виде гидрохлорида аналогично способам, описанным в стадиях с и d примера 16. Rt=0,84 мин (способ А). Детектированная масса: 323,1/325,1 (М+Н+).
Цис и транс N-Вос-2-метилпиперидин-4-ол (18 и 19)
Добавляли по частям 213 мг (5,6 ммоль) NaBH4 при 0°С к раствору 1,0 г (4,7 ммоль) 1-Boc-2-метилпиперидин-4-она в 10 мл этанола. Смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли перегонкой и остаток растворяли в воде и этилацетате. Водный раствор дважды экстрагировали этилацетатом, и объединенные органические слои сушили над Na2SO4. После фильтрации растворитель удаляли перегонкой, и неочищенный продукт очищали колоночной хроматографией (смесью н-гептан/этилацетат 1/1) с получением 367 мг (36%) цис-изомера 18 и 205 мг (20%) транс-изомера 19 дополнительно к 97 мг (10%) смеси обоих изомеров.
цис-изомер (18):
1H-ЯМР (CDCl3): δ = 4,28 (1H, м), 4,17 (1H, м), 3,82 (1H, м), 3,26 (1H, м), 1,85 (1H, ддд, J=14,7, 6,6, ниже 3,4 Гц), 1,77 (1H, м), 1,66 (2H, м), 1,33 (3H, д, J=7,1 Гц).
транс-изомер (19):
1H-ЯМР (CDCl3): δ = 4,50 (1H, м), 4,04 (1H, м), 3,95 (1H, м), 2,87 (1H, дт, J=2,9 ниже 13,6 Гц), 1,93 (1H, м), 1,83 (1H, м), 1,53 (1H, м), 1,32 (1H, м), 1,14 (3H, д, J=7,1 Гц).
1-циклопропилпиперидин-4-ол (20)

5 г 4-гидроксипиперидина растворяли в метаноле. Добавляли 23,8 мл 1[(1-этоксициклопропил)окси]триметилсилана и 5,8 г цианборгидрида натрия и смесь вводили в реакцию при 60°С в течение 12 ч. Затем добавляли такие же количества двух реагентов и продолжали перемешивание при 60°С в течение дополнительных 12 ч. Смесь разбавляли метанолом, фильтровали через целит и упаривали досуха. Остаток поглощали этилацетатом, дважды экстрагировали 2Н гидроксидом натрия и один раз рассолом, сушили над сульфатом натрия и упаривали досуха. Остаток очищали хроматографией на силикагеле с получением 2 г продукта 20, МС: 141 (М+).
Следующие соединения получали в виде их НCl солей сходным образом, как описано в синтезах 12, 16 или 17, исходя из кислот и аминов, приведенных в последующей таблице 1.
Использованные акриловые кислоты или представляли собой коммерчески доступные или были синтезированы из соответствующих альдегидов способами, сходными с описанными в литературе (см., например, J. Med. Chem. 2005, 48, 71-90). Один из примеров описан в синтезе 17 стадия а.
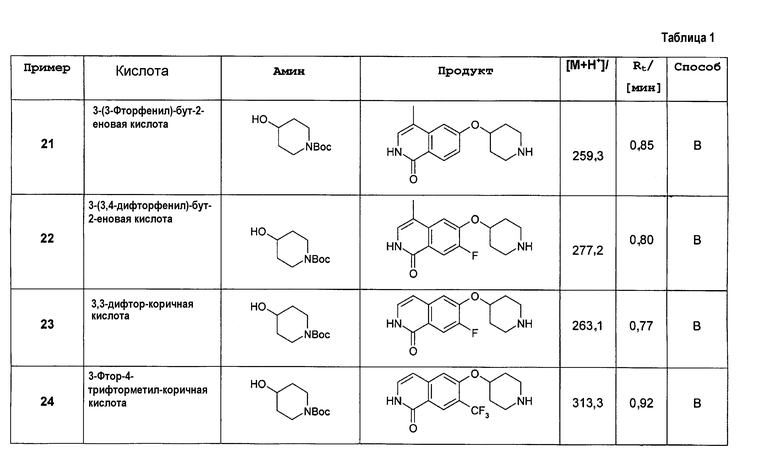
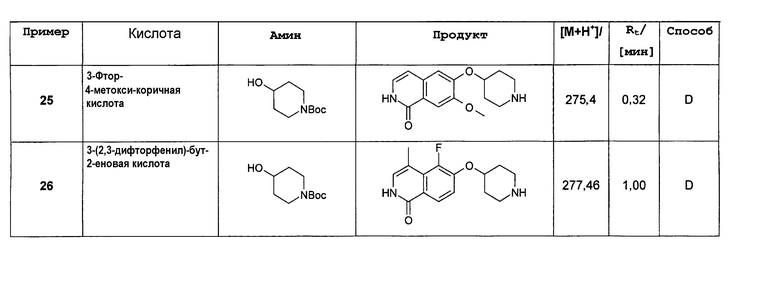
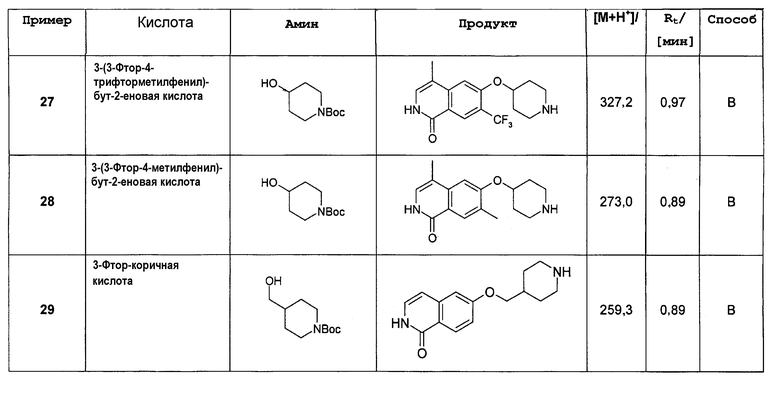
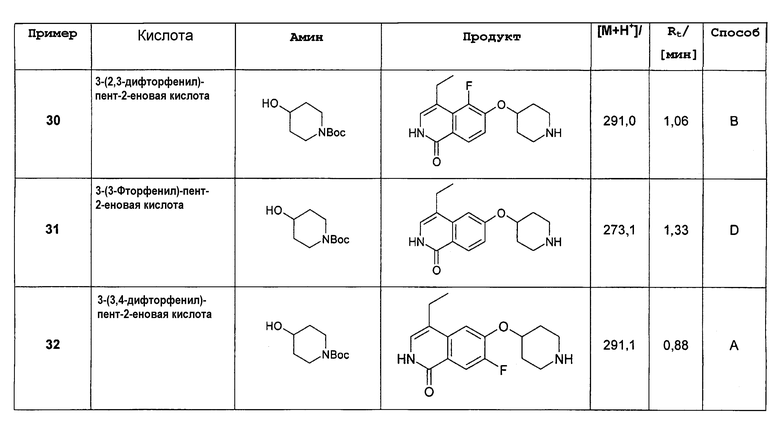
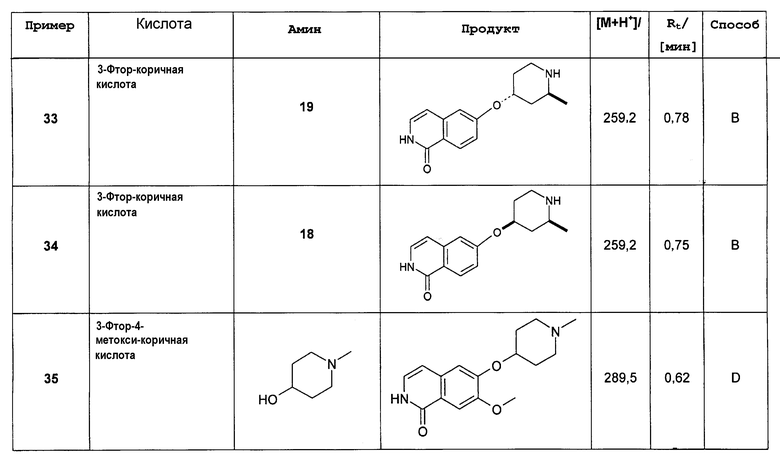
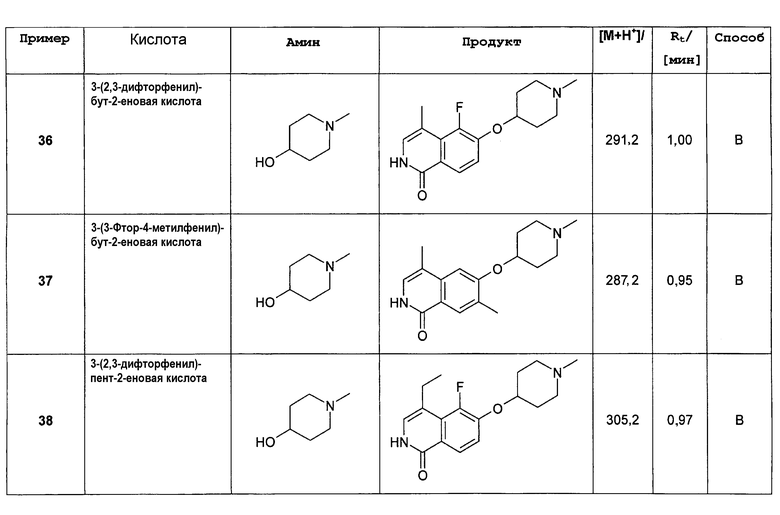
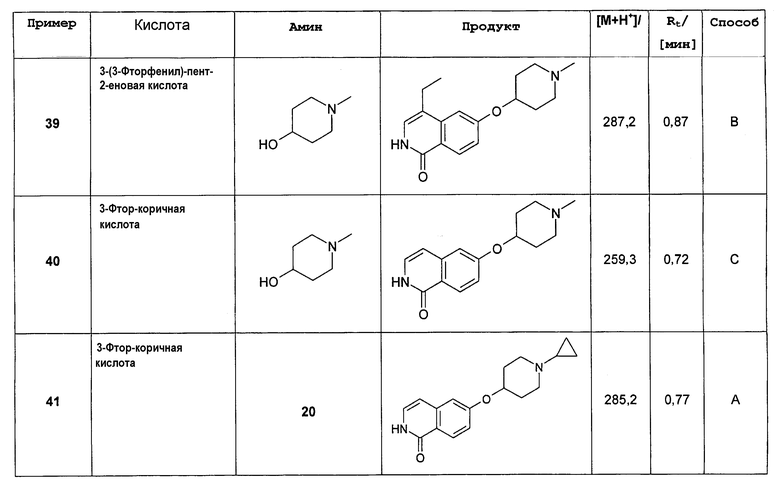
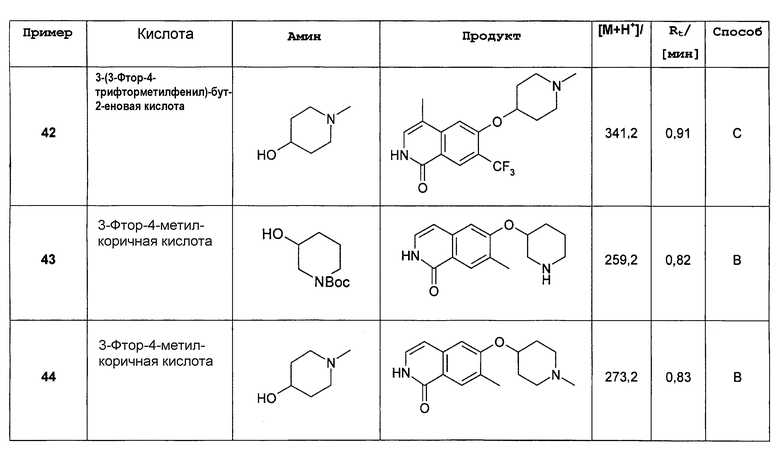
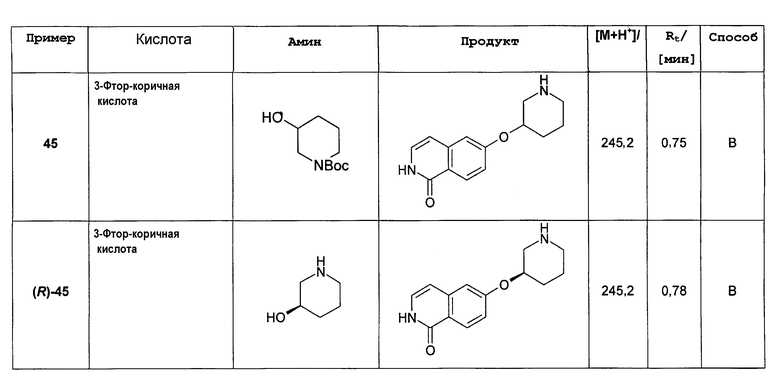

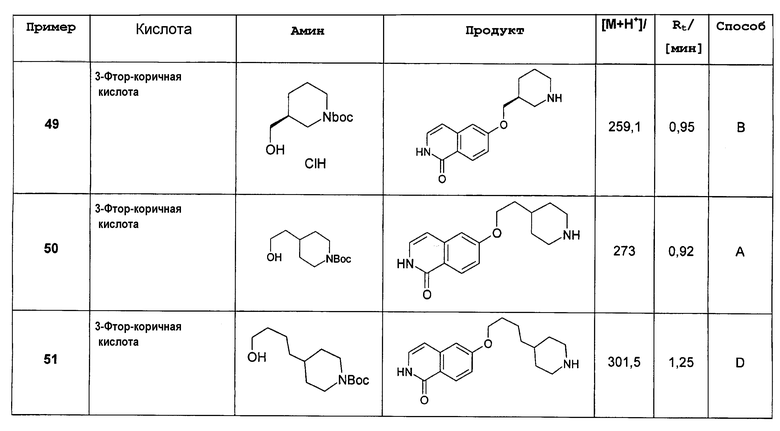
4-бром-6-(пиперидин-4-илокси)-2Н-изохинолин-1-он (52)
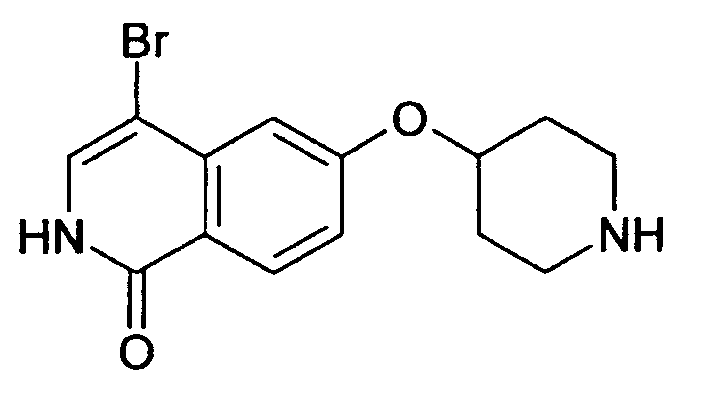
Суспендировали 200 мг (12) в 25 мл хлороформа. Добавляли 100 мкл триэтиламина и продолжали перемешивание в течение 2 ч. Раствор упаривали и остаток очищали хроматографией на силикагеле (дихлорметан:метанол:триэтиламин 10:1:0,1). Другая очистка ВЭЖХ и замена аниона на HBr привели к выходу 73 мг продукта в виде гидробромида. Rt=1,35 мин (способ А). Детектированная масса: 407,1/409,1 (М+Н+).
Трет-бутиловый эфир 4-(1-бензилокси-7-хлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (53)
Растворяли 289,8 мг (1,44 ммоль) трет-бутилового эфира 4-гидроксипиперидин-1-карбоновой кислоты в 10 мл диметилацетамида и добавляли 57,6 мг (1,44 ммоль) гидрида натрия (60%). Реакционную смесь перемешивали при комнатной температуре. Через 30 минут добавляли раствор 310 мг (1,44 ммоль) 1,7-дихлор-6-фторизохинолина (6) в 3 мл диметилацетамида и смесь перемешивали при комнатной температуре в течение 1 ч до полного превращения. Затем добавляли 155,7 мг (1,44 ммоль) бензилового спирта с последующим добавлением 57,6 мг (1,44 ммоль) гидрида натрия (60%) и продолжали перемешивание при комнатной температуре. Для достижения полной конверсии дважды добавляли по 0,5 эквивалента бензилового спирта и гидрида натрия через 2 ч и после выдерживания в течение ночи. Для обработки растворитель упаривали, а остаток поглощали дихлорметаном, дважды промывали водой, сушили над MgSO4 и упаривали. Окончательную очистку осуществляли при помощи препаративной ВЭЖХ.
7-хлор-6-(пиперидин-4-илокси)-2Н-изохинолин-1-он (54)
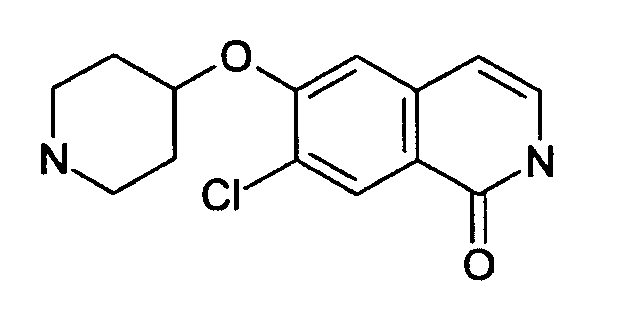
Перемешивали 254 мг (0,52 ммоль) трет-бутилового эфира 4-(1-бензилокси-7-хлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (53) в смеси метанол/2Н HCl (1:1) при комнатной температуре в течение ночи. Растворитель упаривали под вакуумом, и остаток очищали препаративной ВЭЖХ. Фракции продукта упаривали и растворяли в 2Н HCl. Лиофилизация привела к получению 57 мг целевого соединения в виде гидрохлорида. Rt=0,95 мин (способ В). Детектированная масса: 279,1 (М+Н+).
7-хлор-6-(1-изопропилпиперидин-4-илокси)-2Н-изохинолин-1-он (55)
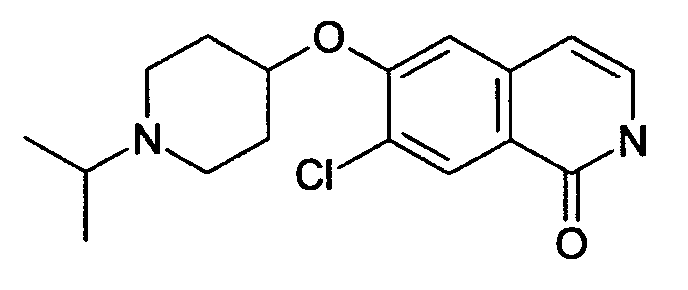
Растворяли 64 мг (0,23 ммоль) 7-хлор-6-(пиперидин-4-илокси)-2Н-изохинолин-1-она (54) в 5 мл метанола. Добавляли 41,4 мг (0,41 ммоль) триэтиламина и смесь перемешивали при комнатной температуре в течение 10 минут. После добавления свежевысушенных молекулярных сит, 122,4 мг (2,04 ммоль) уксусной кислоты, 26,7 мг (0,46 ммоль) ацетона и 43,3 мг (0,69 ммоль) цианборгидрида натрия реакционную смесь кипятили с обратным холодильником в течение 8 ч. После добавления 2 экв. ацетона и 2 экв. цианборгидрида натрия при комнатной температуре реакцию кипятили с обратным холодильником в течение дополнительных 2 ч до полной конверсии. Для обработки смесь фильтровали и фильтрат упаривали. Остаток растворяли в дихлорметане, дважды промывали 2Н NaOH и водой и сушили над MgSO4. После упаривания растворителя и очистки препаративной ВЭЖХ получали 13 мг целевого соединения в виде трифторацетата. Rt=0,96 мин (способ В). Детектированная масса: 321,1/323,2 (М+Н+).
Общий способ А реакции восстановительного аминирования:
В 3 мл HC(OMe)3 перемешивают в течение 1 ч при комнатной температуре: 0,243 ммоль гидрохлорида 6-(пиперидин-4-илокси)-2Н-изохинолин-1-она (12) или другого пригодного амина, 0,243 ммоль альдегида и 0,365 ммоль триэтиламина. Смесь охлаждают до -10°С, добавляют 1,75 мл свежеполученного раствора ДМФА, содержащего 1,215 ммоль NaHB(OAc)3 и 1,215 ммоль НОАс. Перемешивание продолжают при -10°С в течение 30 мин, затем смесь оставляют нагреваться до комнатной температуры и оставляют при комнатной температуре в течение ночи. Добавляют 0,5 мл воды и смесь упаривают, растворяют в ДМФА и очищают препаративной ВЭЖХ. Очищенные продукты растворяют в 1 мл HCl в изопропаноле (5-6 М) и оставляют в течение ночи при комнатной температуре (расщепление Boc/tBu сложноэфирных групп некоторых продуктов). Добавляют 2 мл воды и раствор подвергают лиофильной сушке с получением гидрохлоридов продуктов.
Следующие соединения, показанные в последующей таблице 2, синтезировали способом, сходным с описанным общим способом, и получали в виде солей гидрохлорида.
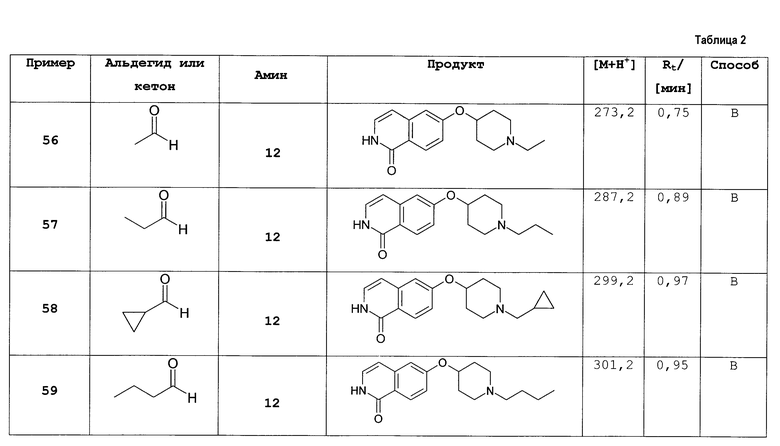
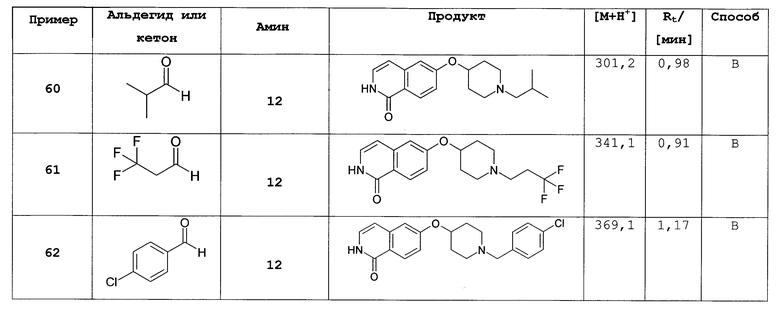
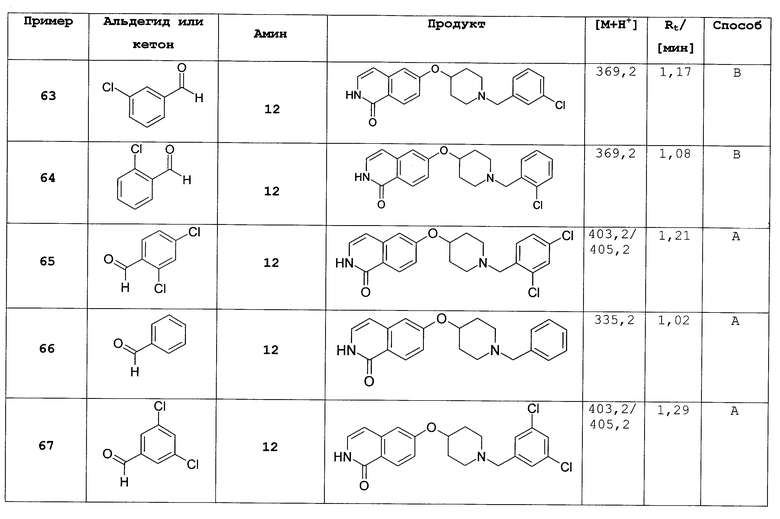

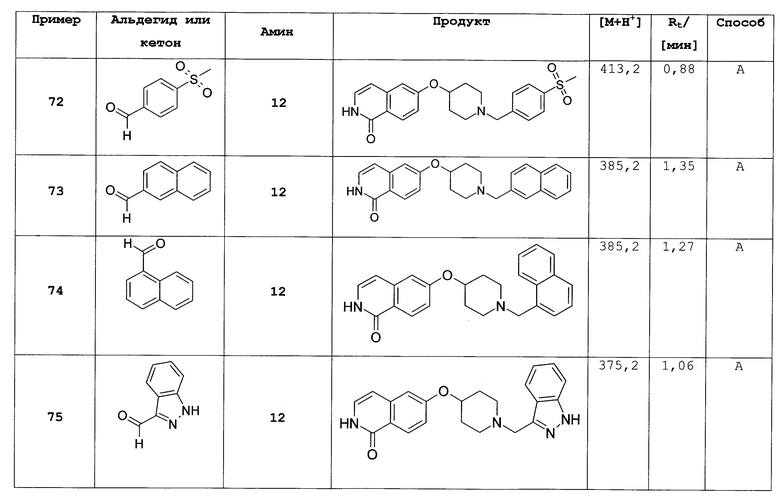
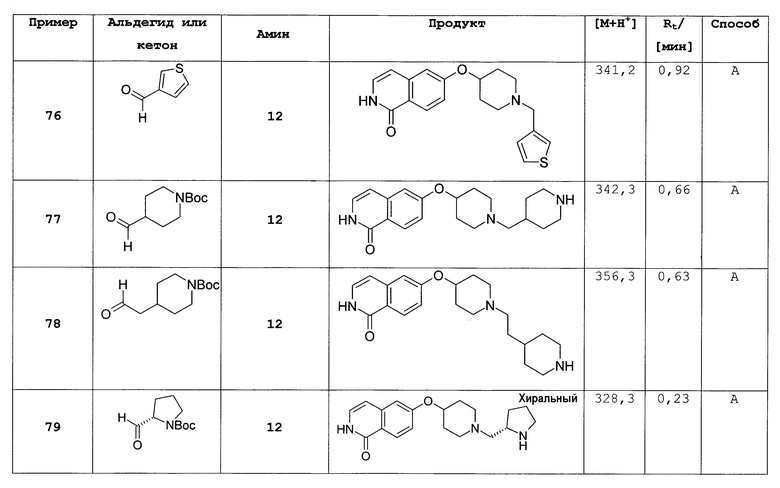
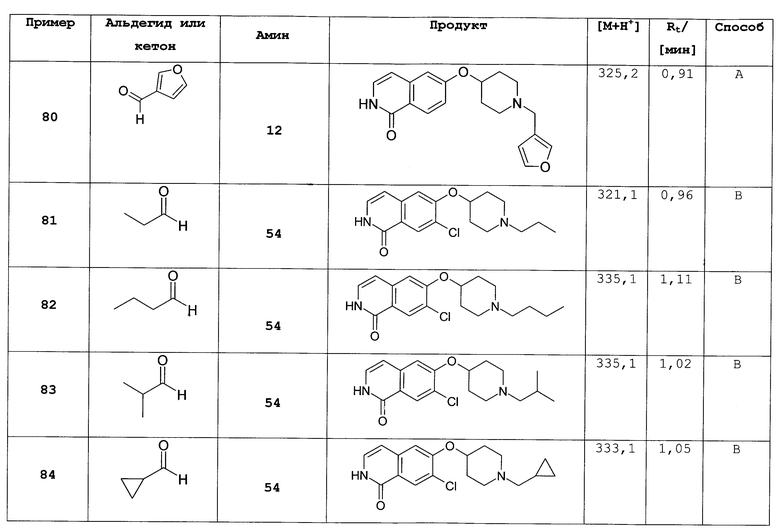
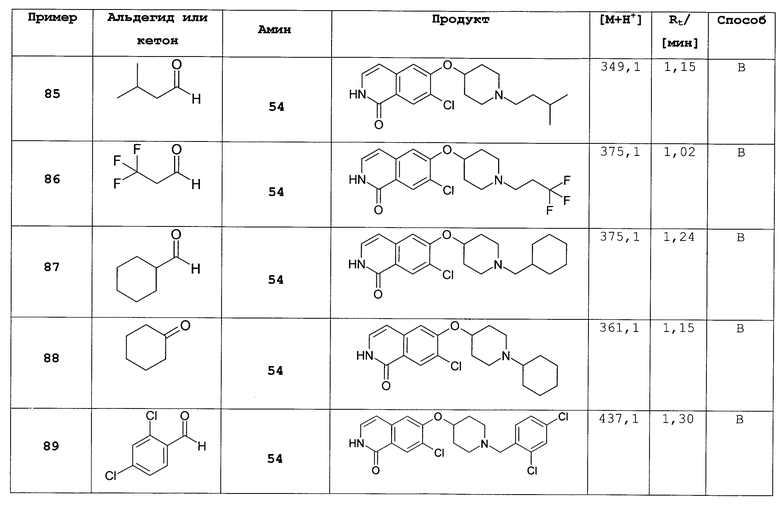
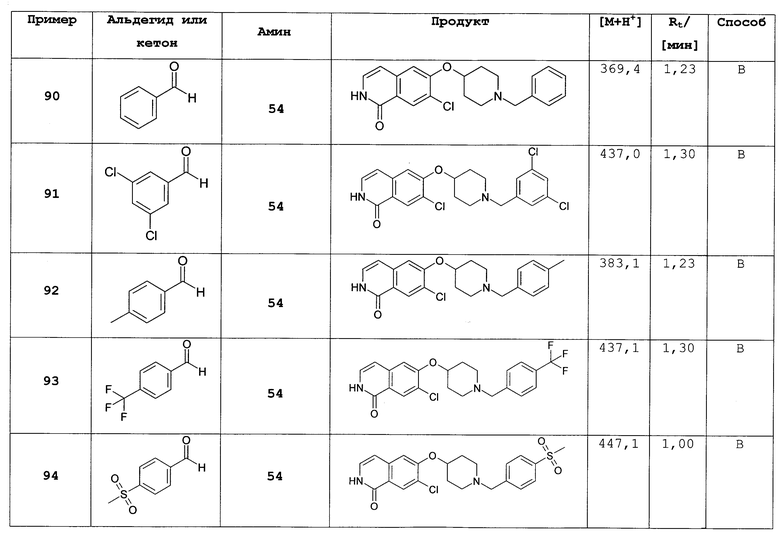
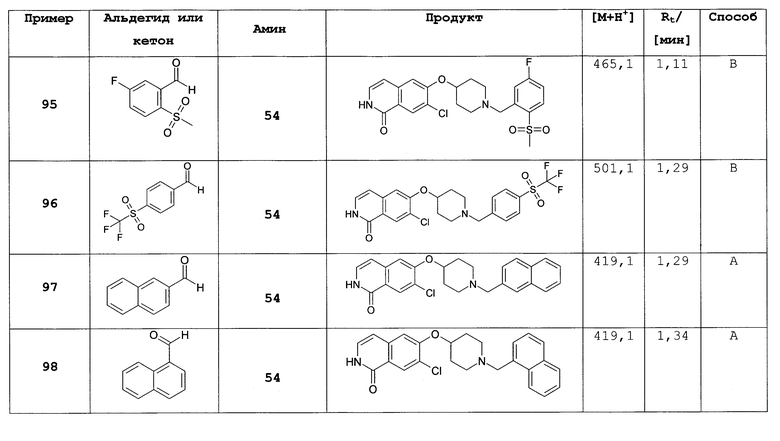
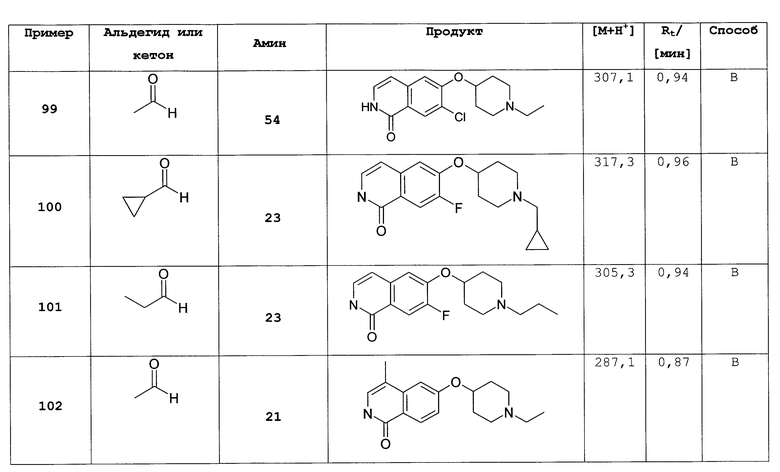
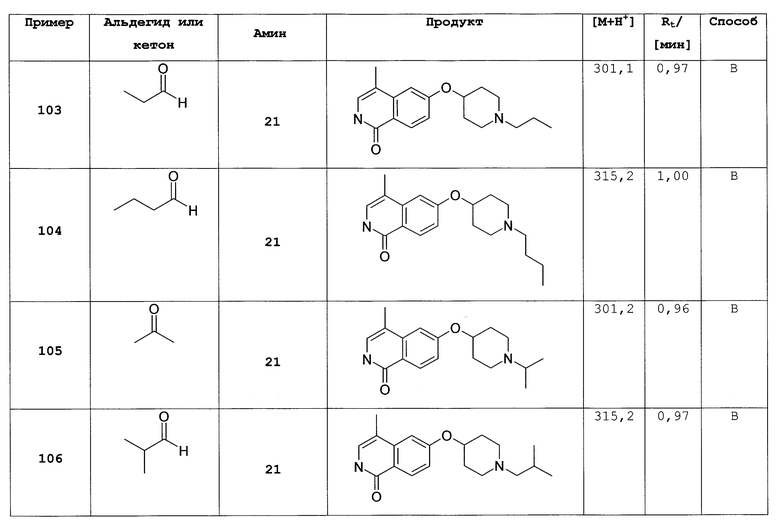
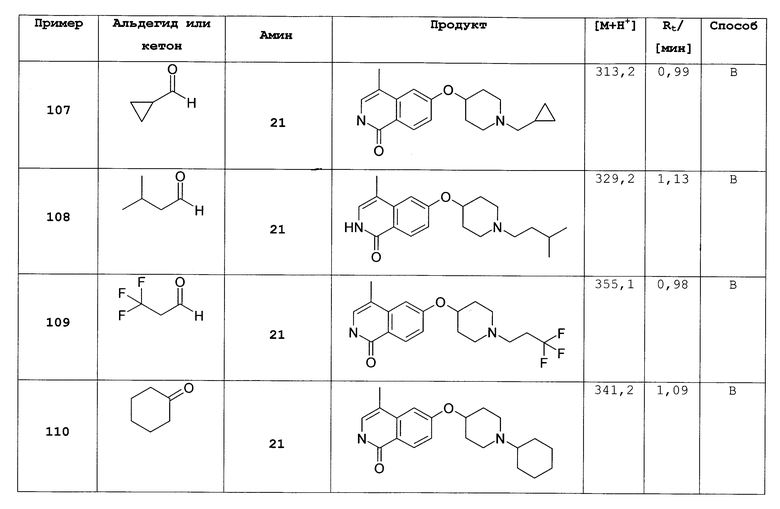
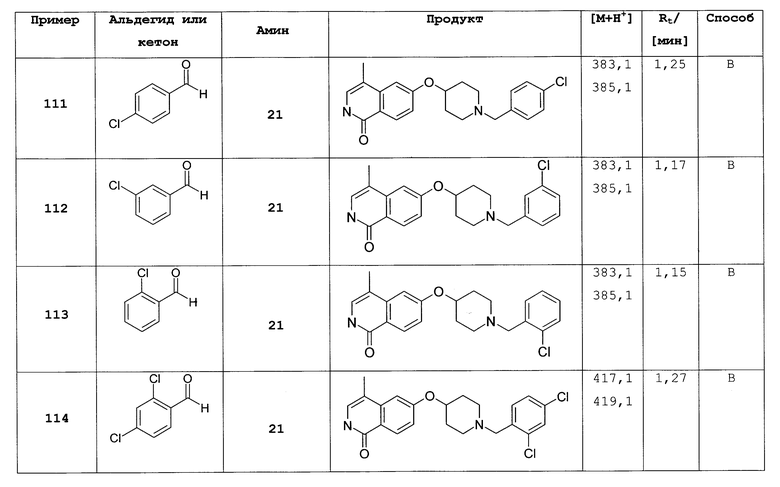
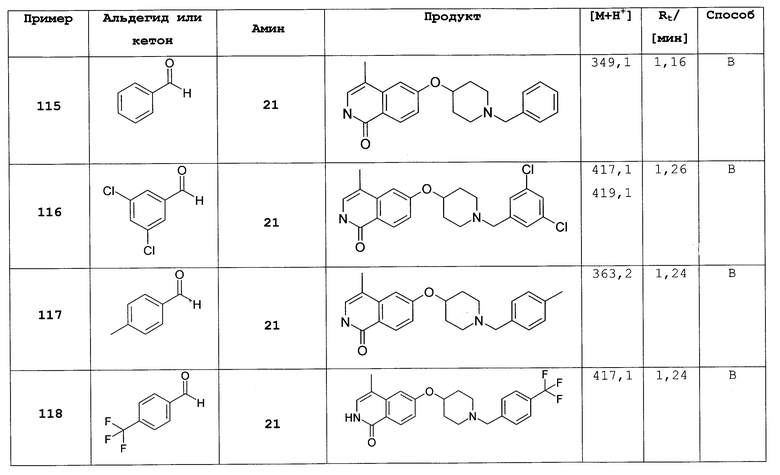
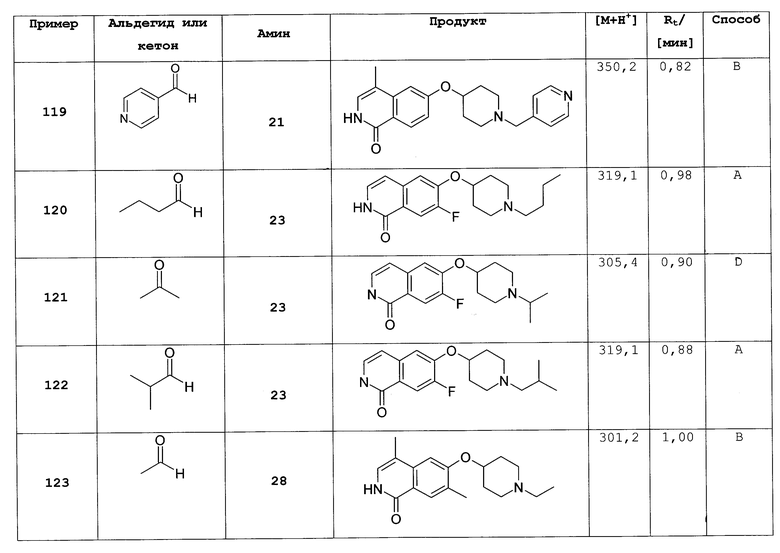
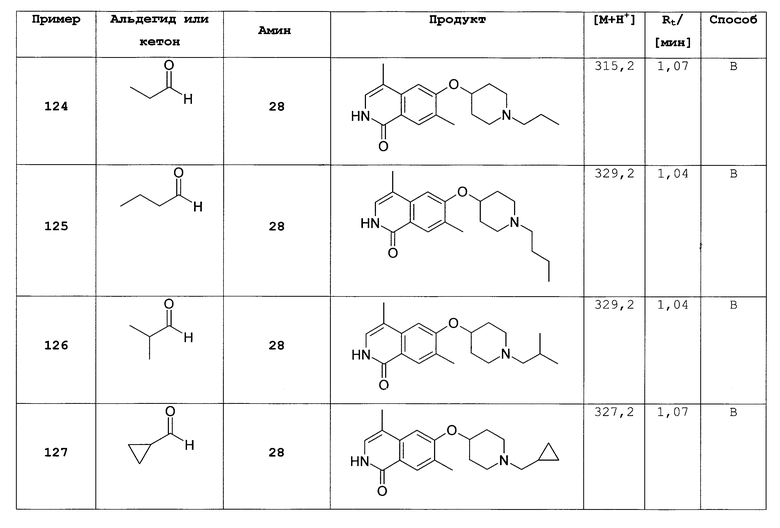
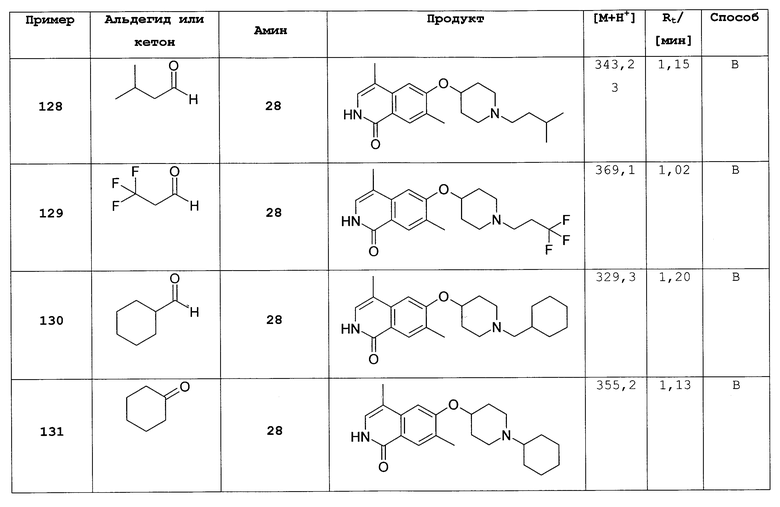
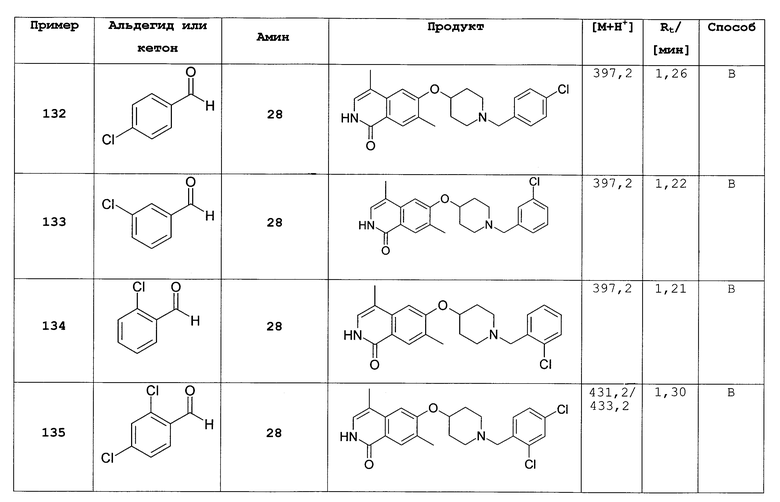
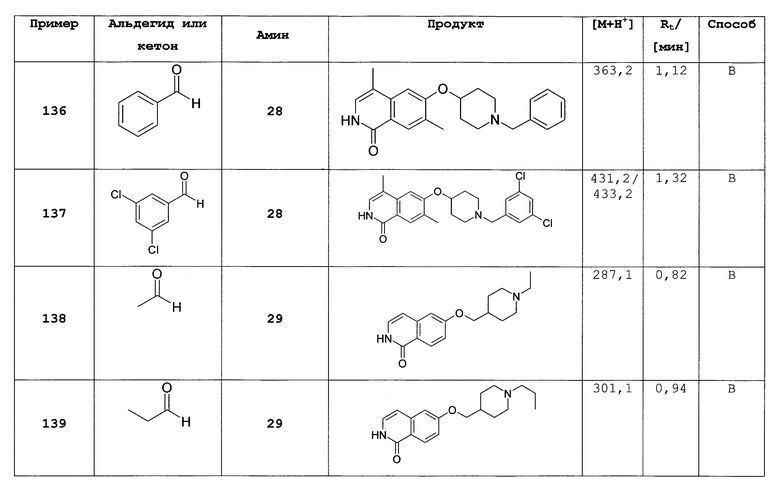
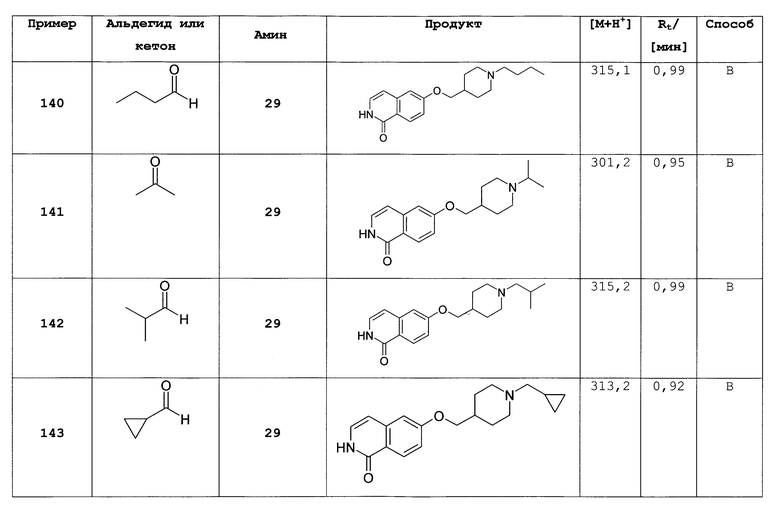
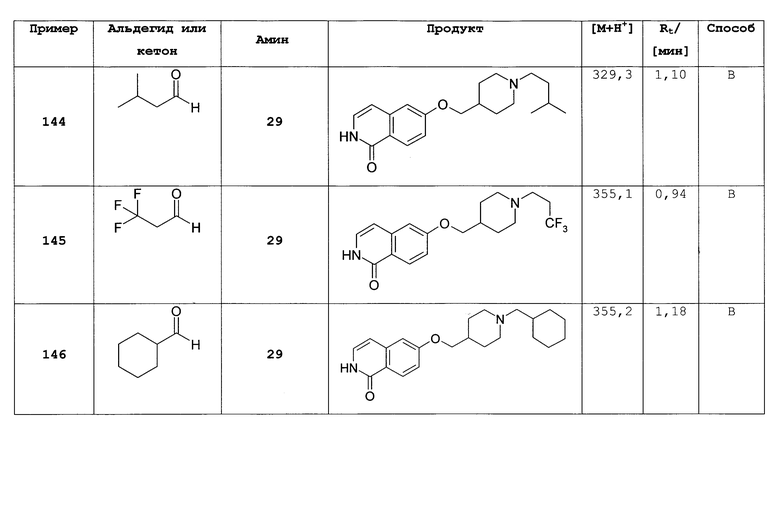
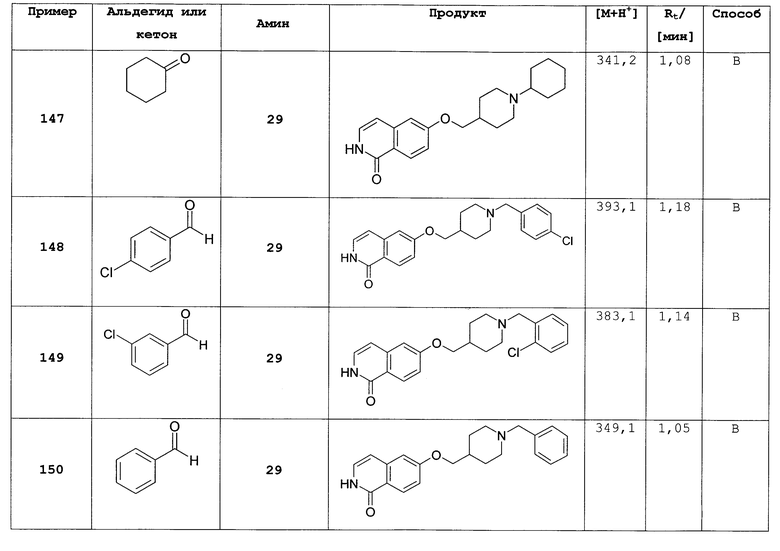
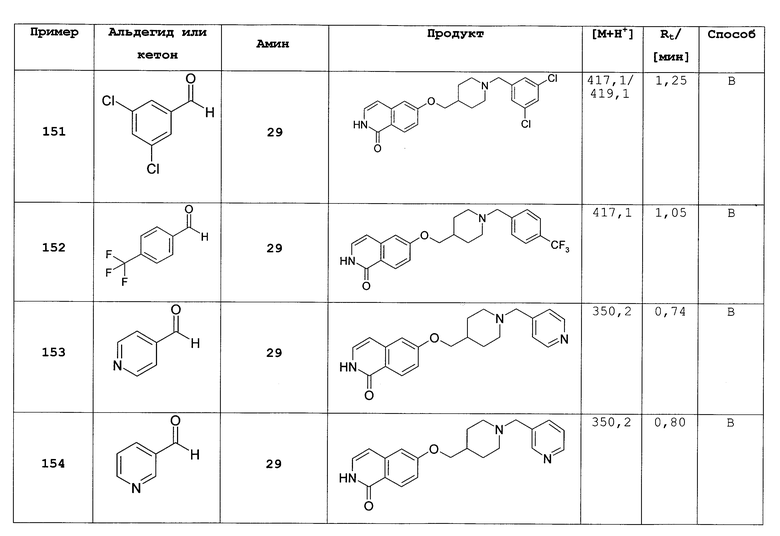
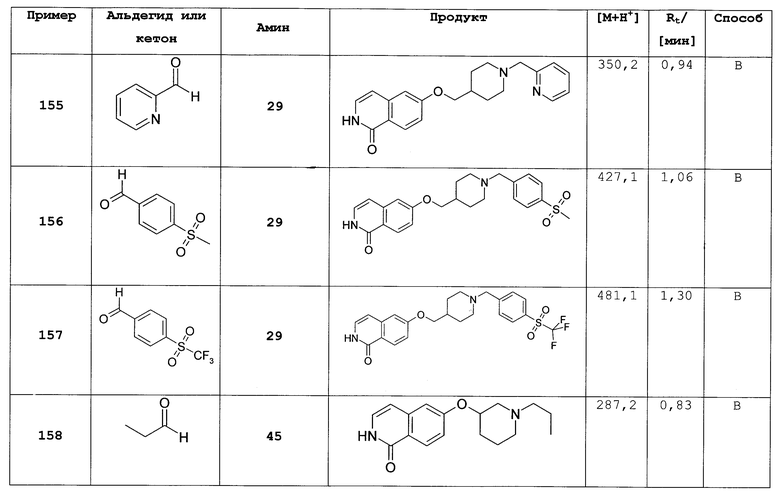

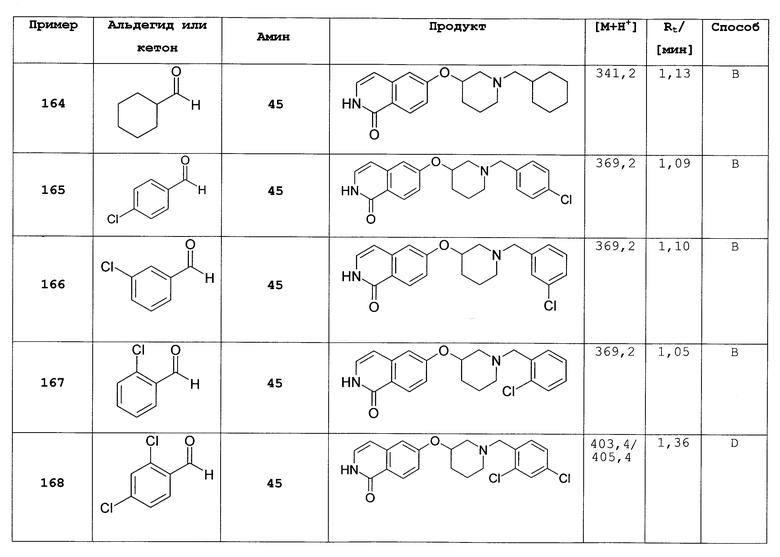
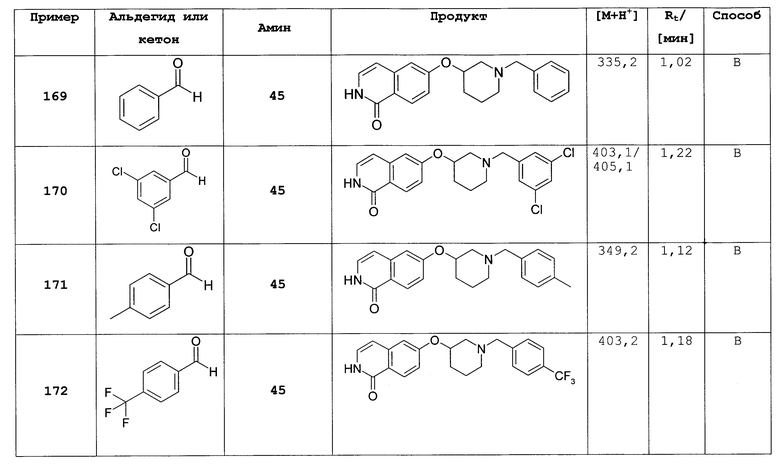

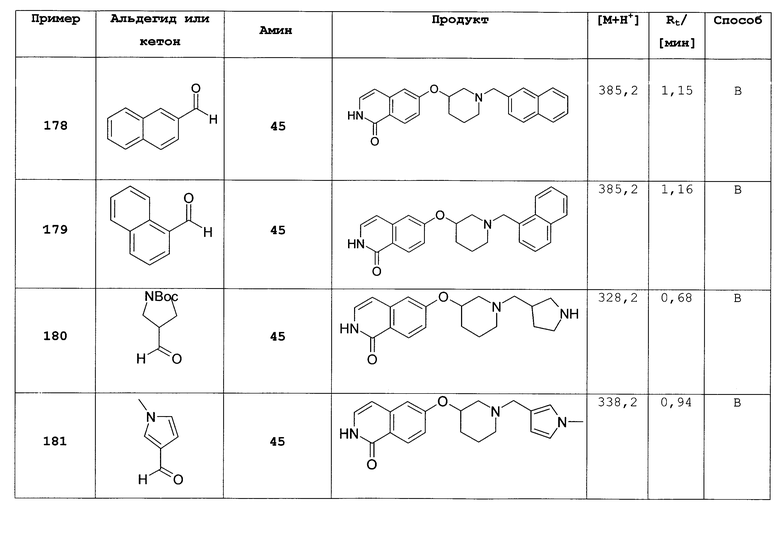
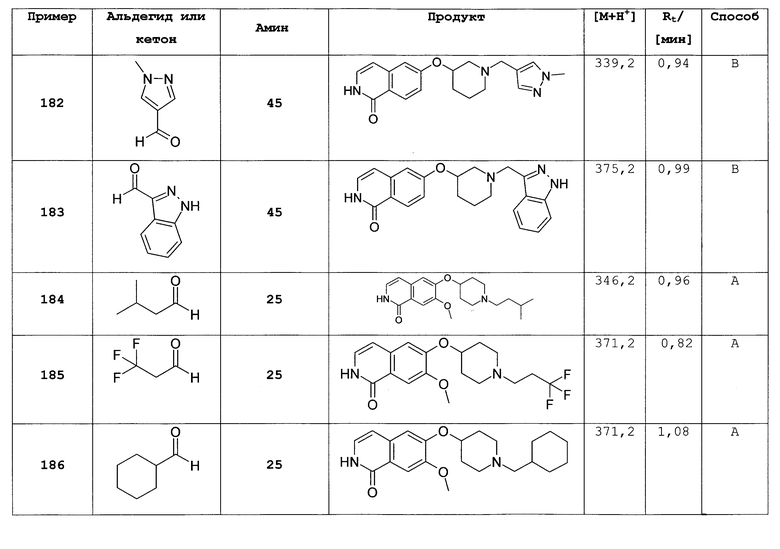
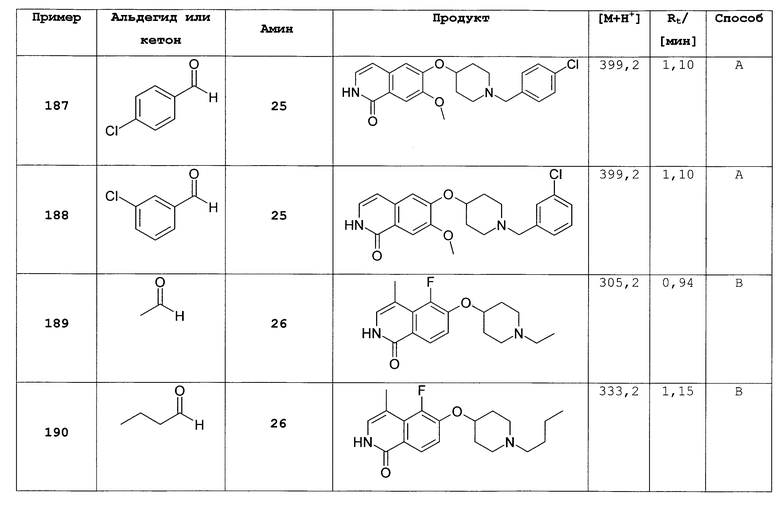
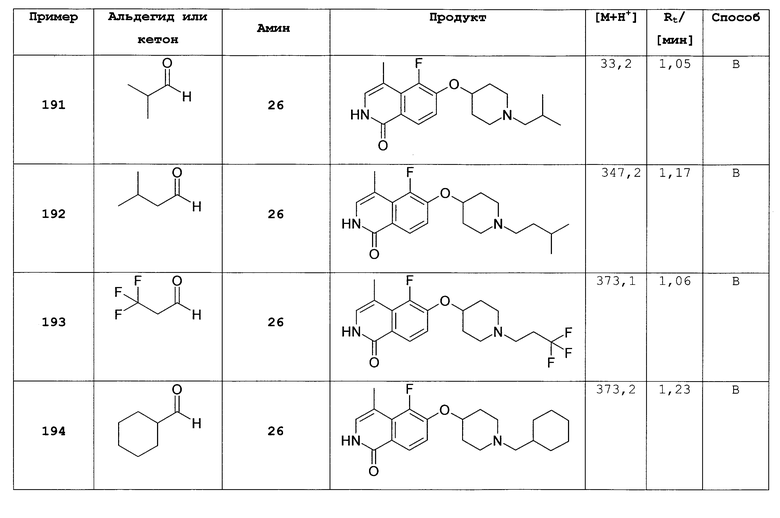
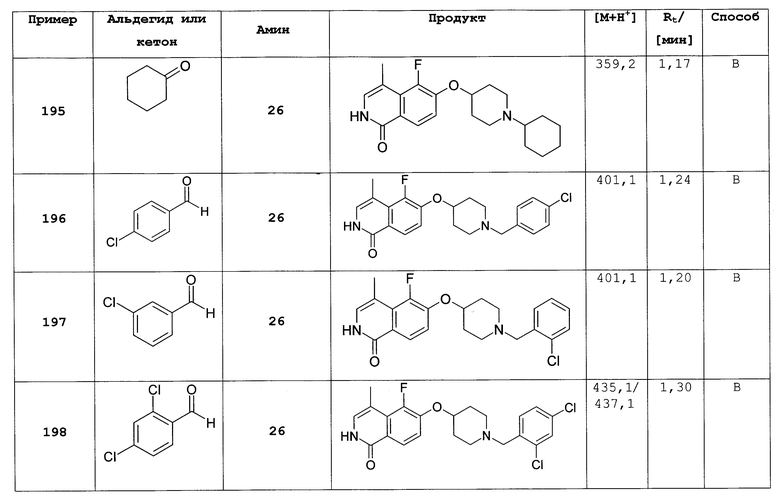
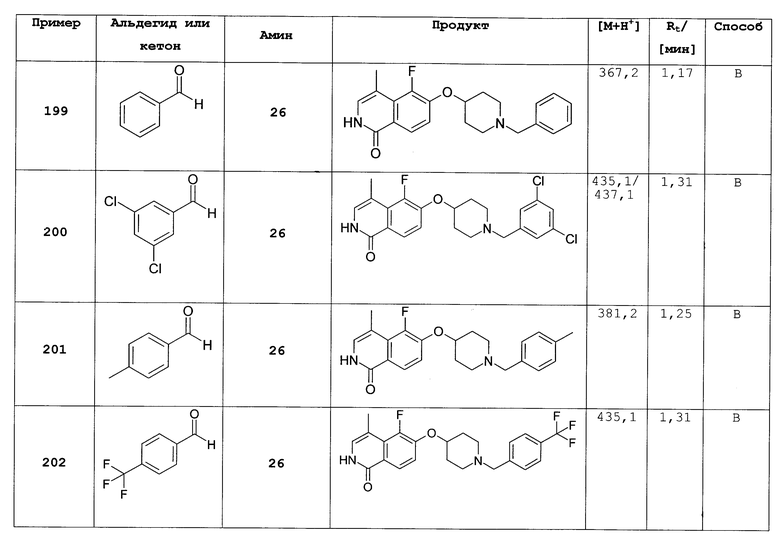
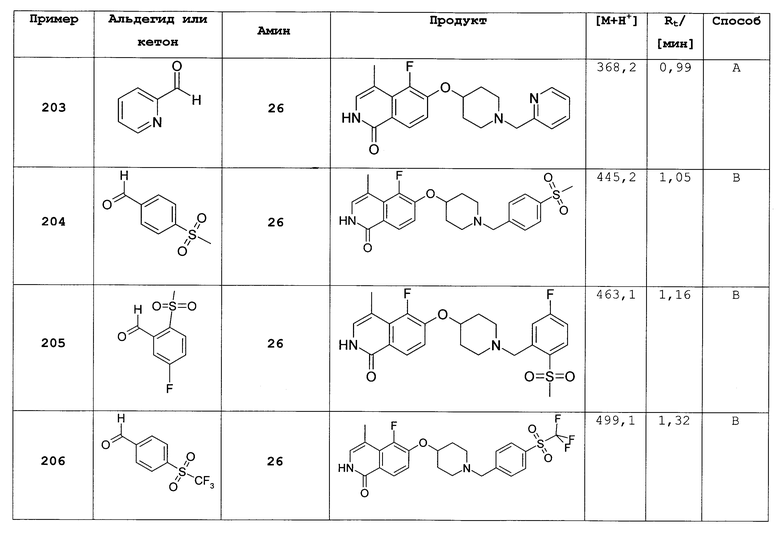
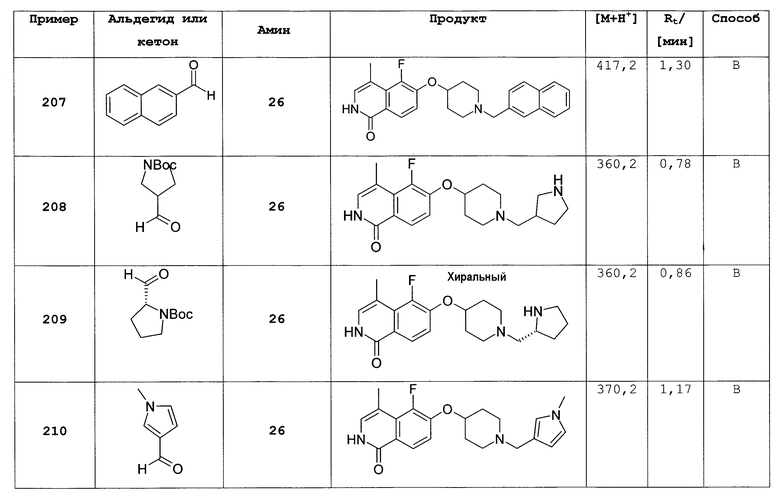
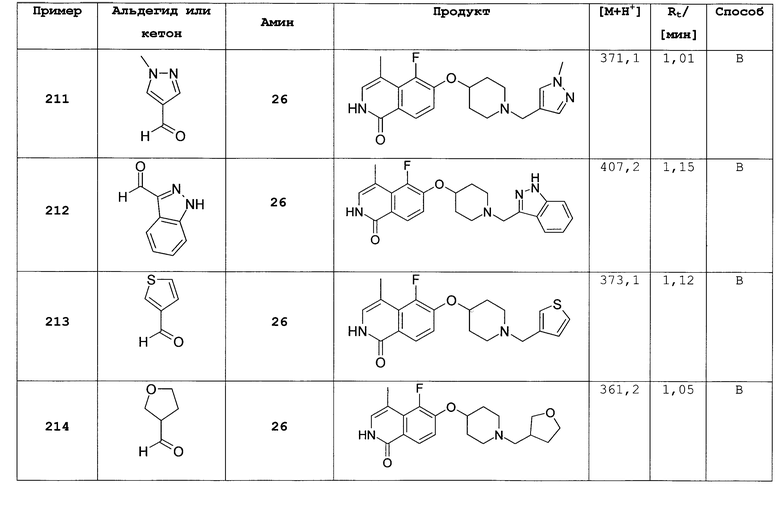
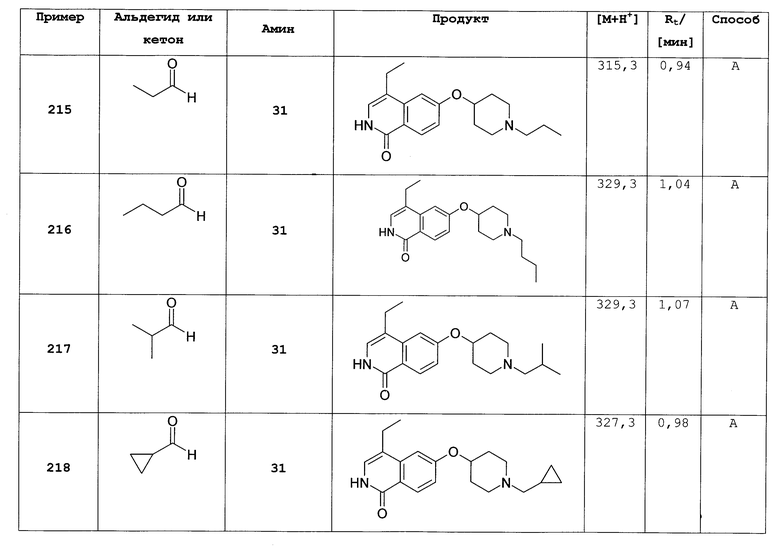
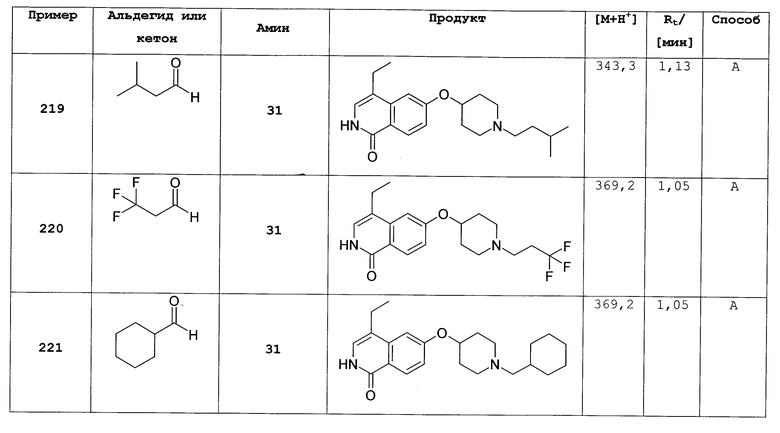
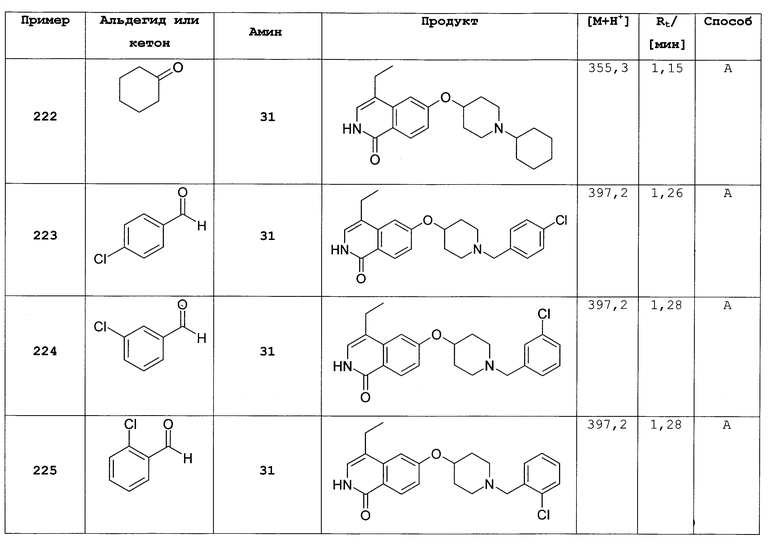
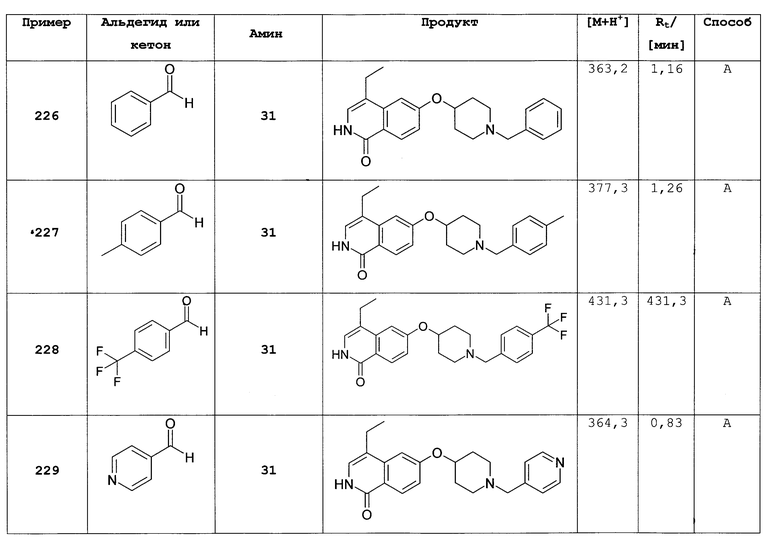
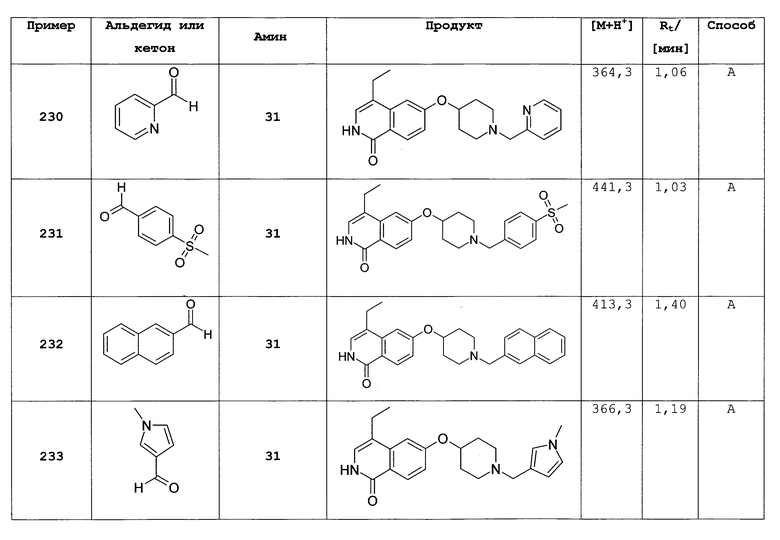
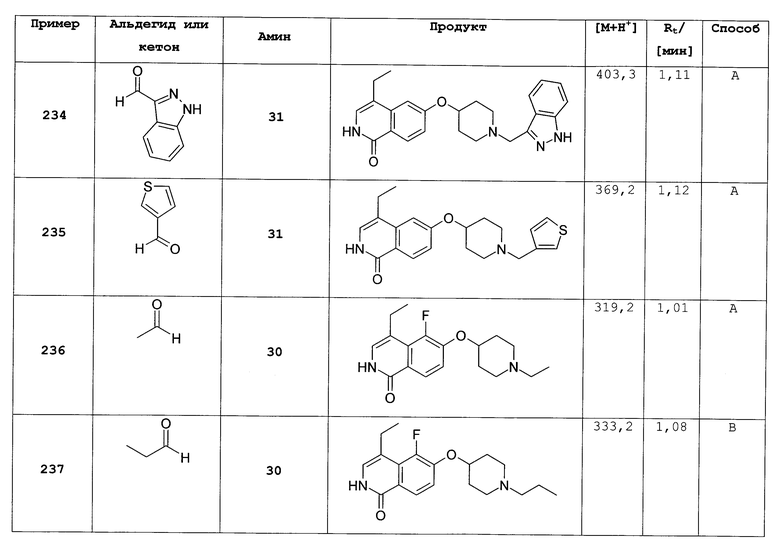
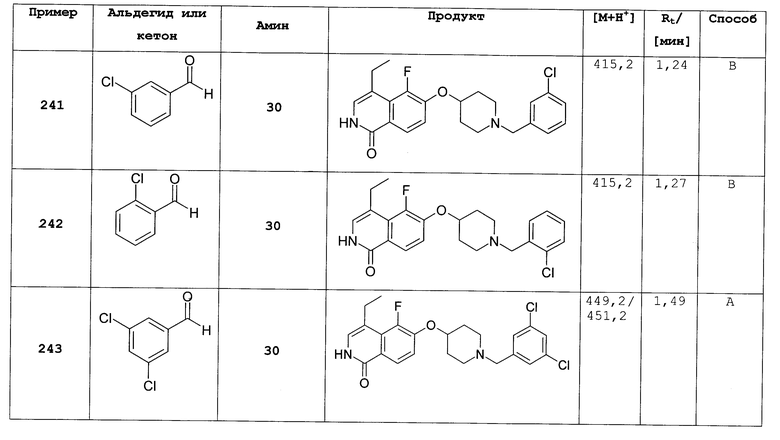
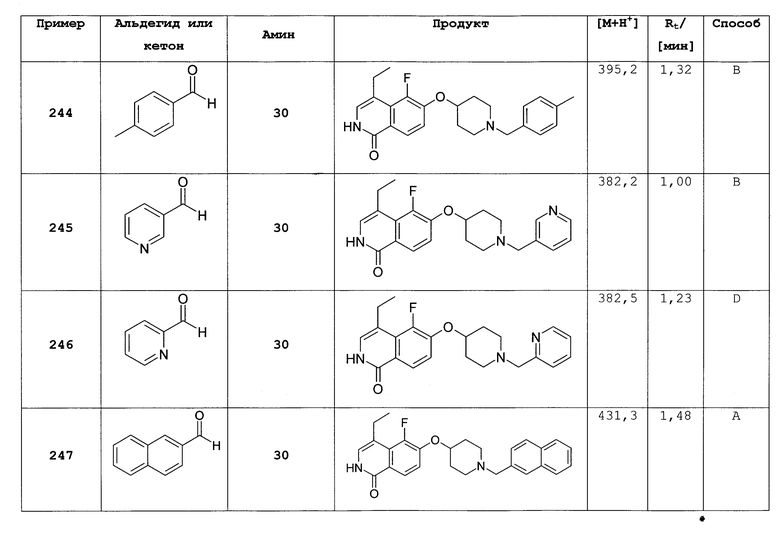
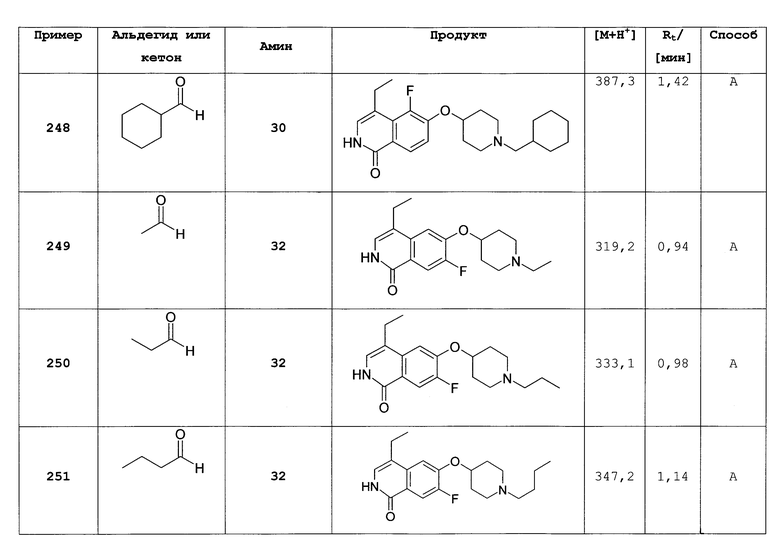
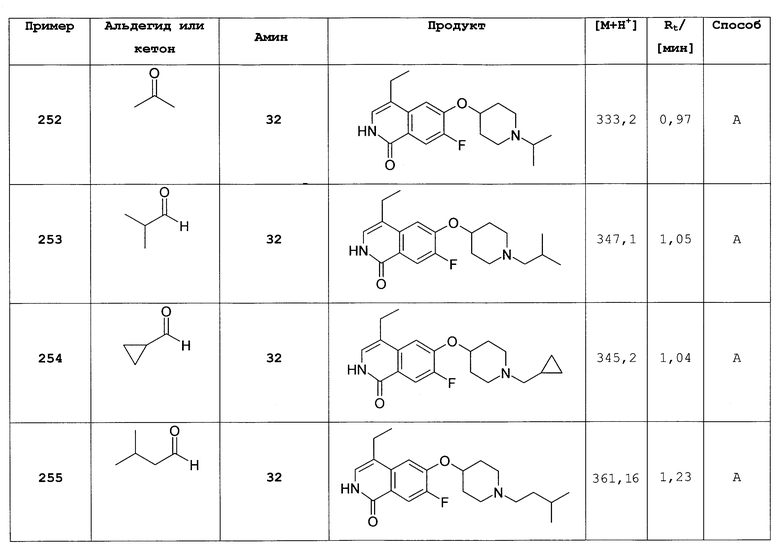
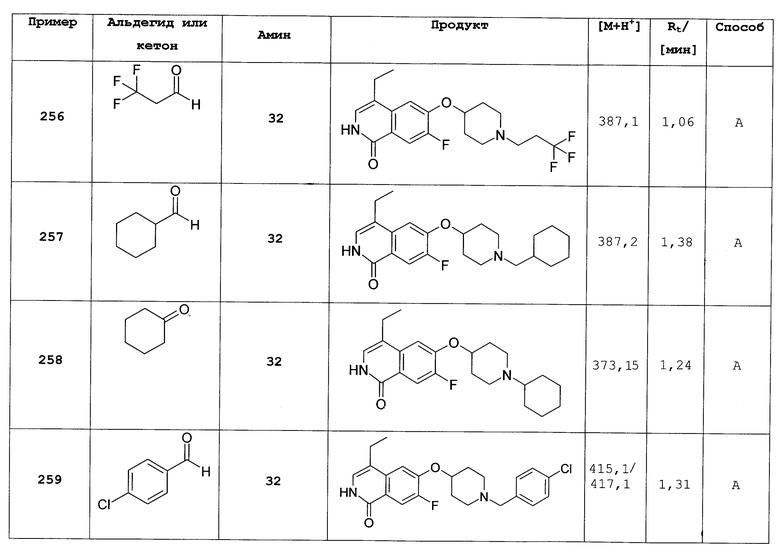
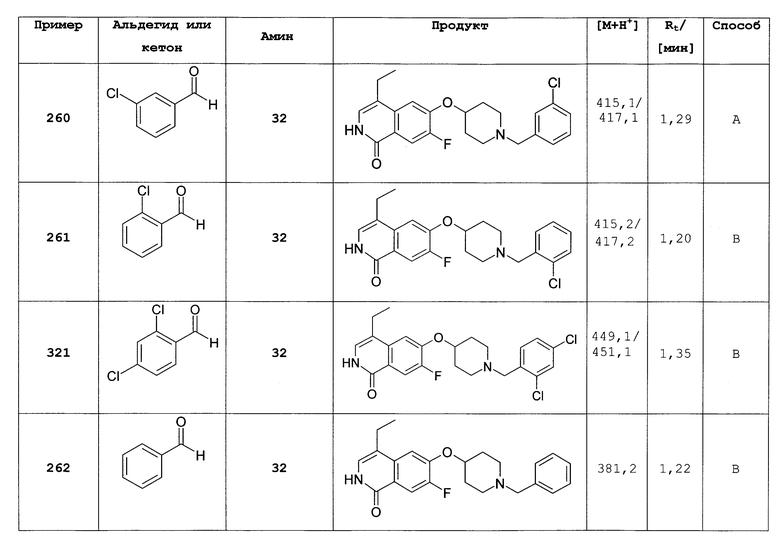
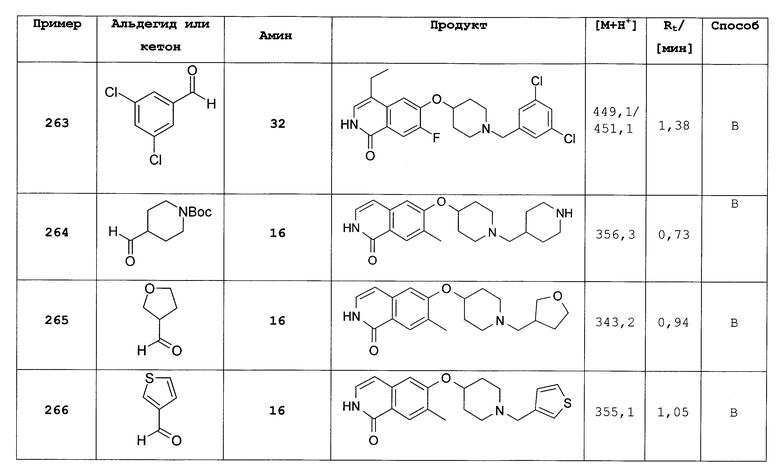
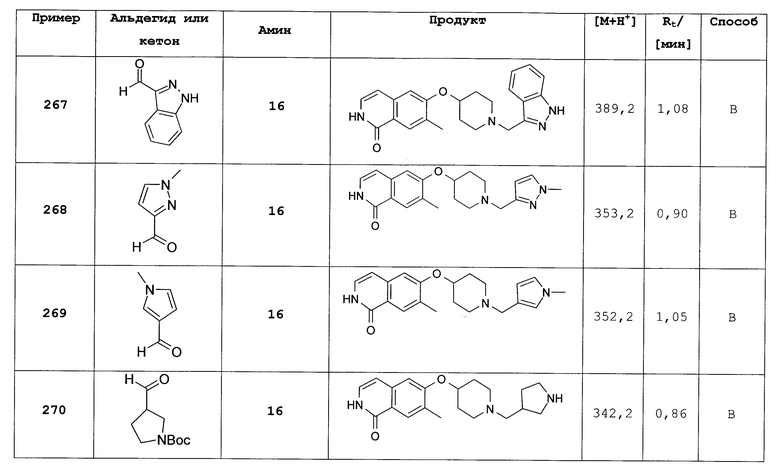
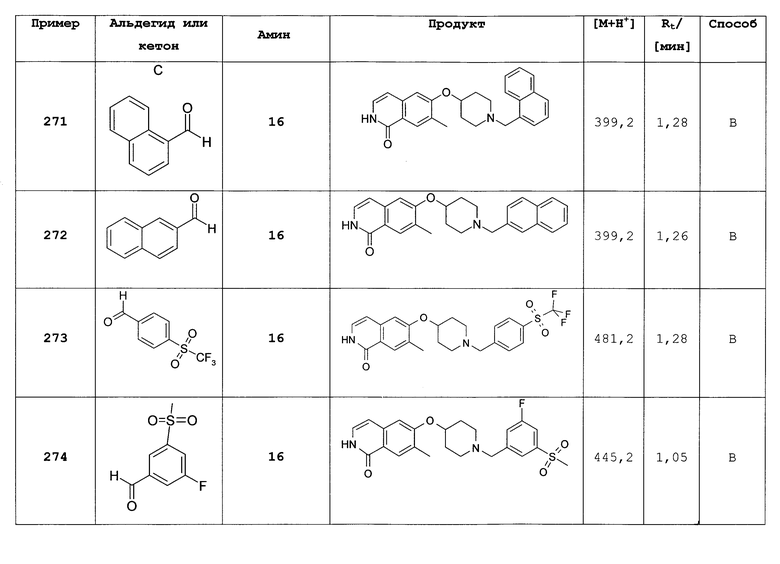
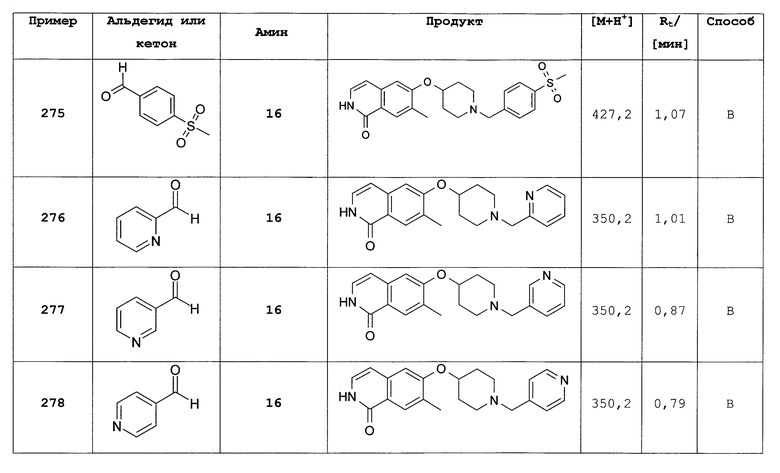
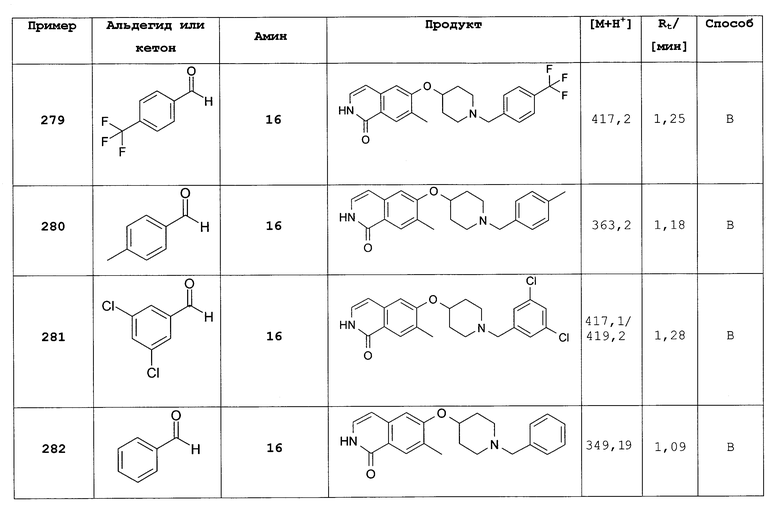
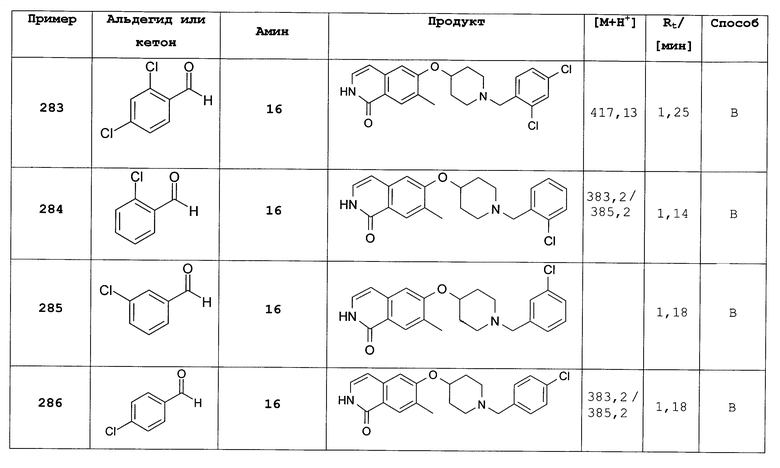
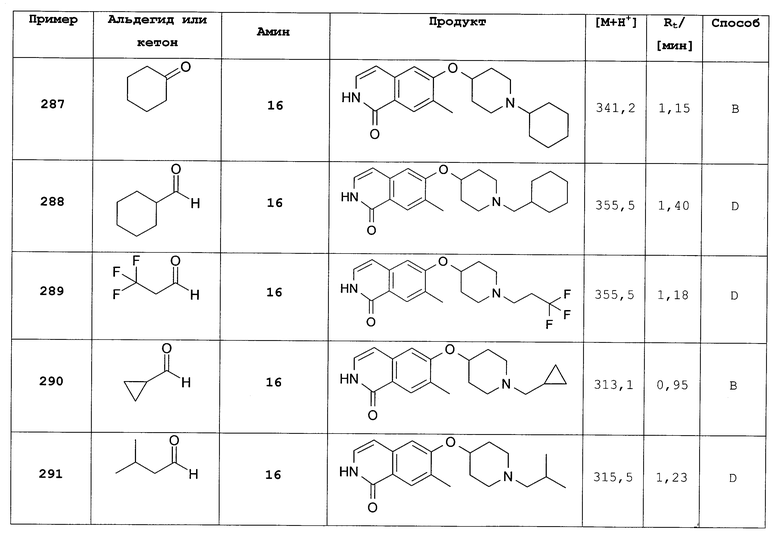
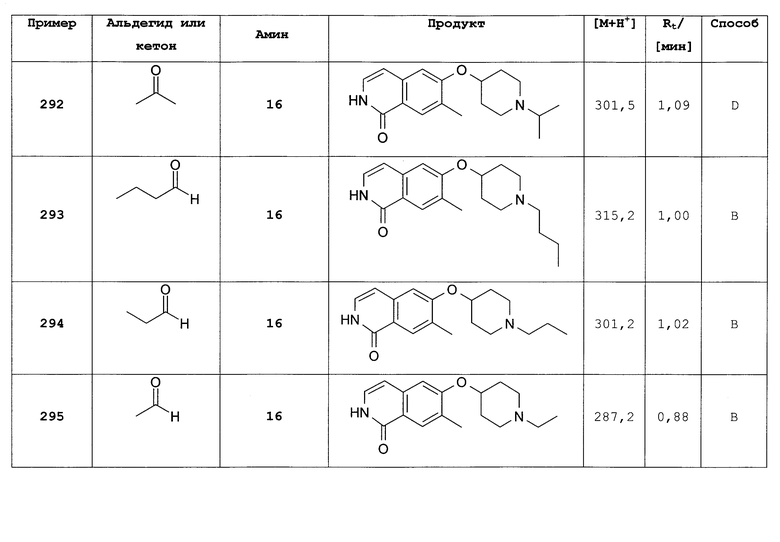
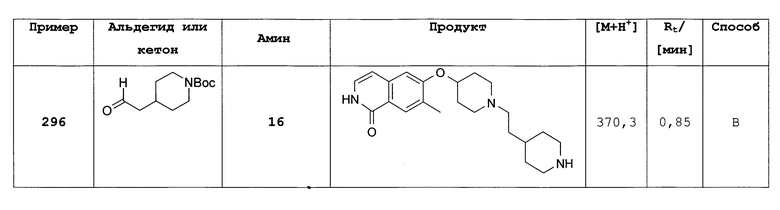
Общий способ В реакции восстановительного аминирования.
100 мг (0,25 ммоль) трифторацетата 7-хлор-6-(пиперидин-4-илокси)-2Н-изохинолин 1-она (54, трифторацетат) растворяли в 5 мл метанола. После добавления молекулярных сит 4 Å, 51,5 мг (0,51 ммоль) триэтиламина, 152,9 мг (2,55 ммоль) уксусной кислоты и 0,32 ммоль соответствующего альдегида добавляли по каплям раствор 48,0 мг (0,76 ммоль) цианоборгидрида натрия и полученную смесь перемешивали при комнатной температуре до достижения полного превращения. В некоторых случаях необходимо нагревать смесь до 60ºС до достижения полного превращения. Для выделения продуктов раствор отфильтровывали и растворитель удаляли в вакууме. Остаток растворяли в дихлорметане, промывали 1Н NaOH и насыщенным раствором NaCl, сушили над MgSO4 и выпаривали. Неочищенный продукт очищали с помощью препаративной ВЭЖХ. Полученные трифторацетаты перемешивали в смеси 2Н HCl/метанол, упаривали, растворяли в воде и подвергали лиофилизации с получением целевых продуктов в виде гидрохлоридов.
Следующие соединения в таблице 3 были синтезированы и получены в виде гидрохлоридов с помощью этой процедуры с использованием соединения 54.
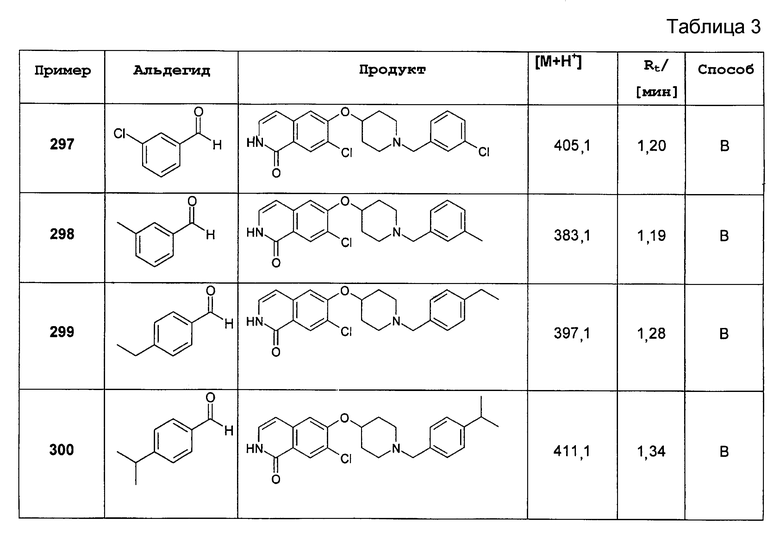
7-фтор-6-(1-изопропилпиперидин-4-илокси)-4-метил-2Н-изохинолин-1-он (308)
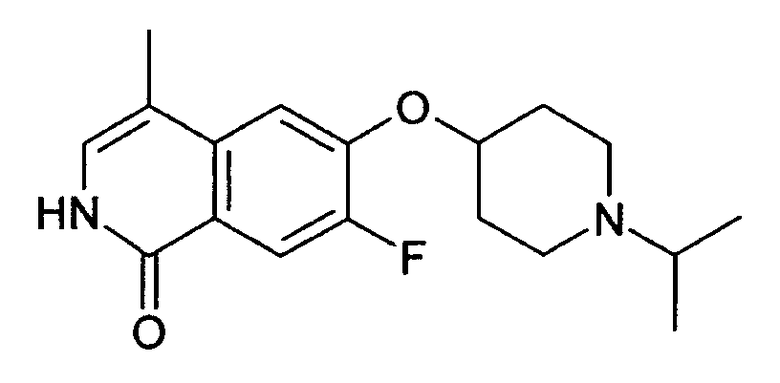
Путем алкилирования 50 мг 7-фтор-4-метил-6-(пиперидин-4-илокси)-2Н-изохинолин-1-она (22) изопропилбромидом в присутствии триэтиламина в ДМФА при 60°С получали 31 мг 7-фтор-6-(1-изопропилпиперидин-4-илокси)-4-метил-2Н-изохинолин-1-она. Rt=0,93 мин (способ В). Детектированная масса: 319,2 (М+Н+).
5-хлор-6-(пиперидин-4-илокси)-2Н-изохинолин-1-он (309)
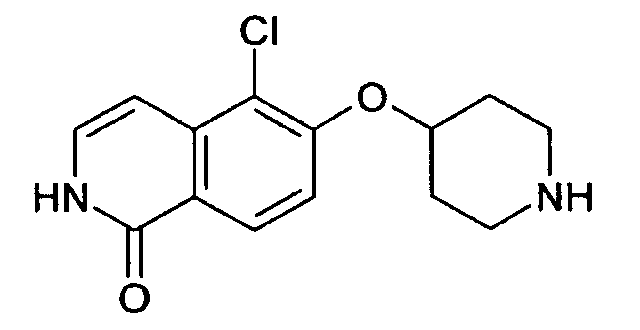
Растворяли 60 мг (0,21 ммоль) гидрохлорид 6-(пиперидин-4-илокси)-2Н-изохинолин-1-она (12) в 5 мл концентрированной серной кислоты. Добавляли при 0°С 28,6 мг (0,21 ммоль) N-хлорсукцинимида и смесь перемешивали при 50°С. Через 2 ч раствор выливали на лед и рН доводили до примерно 12 путем добавления твердого NaOH. Водный раствор дважды экстрагировали дихлорметаном. Органические слои сушили над MgSO4 и упаривали. Неочищенный продукт очищали препаративной ВЭЖХ. Полученный трифторацетат растворяли в 2н HCl и растворитель удаляли под вакуумом. Растворение остатка в воде с последующей лиофилизацией привело к получению целевого продукта в виде соли HCl. Rt=0,86 мин (способ А). Детектированная масса: 279,1/281,1 (М+Н+).
7-бром-6-фторизохинолин 2-оксид (310)
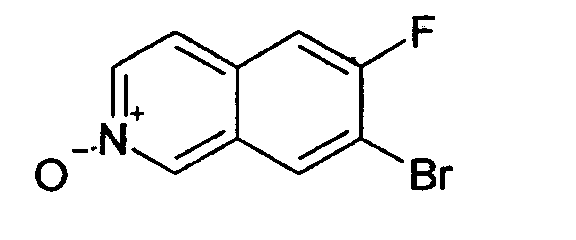
Исходя из 7-бром-6-фторизохинолина (15) было получено целевое соединение согласно способу, описанному для 7-хлор-6-фторизохинолин 2-оксида (5). Rt=0,93 мин (способ С). Детектированная масса: 242,2/244,2 (М+Н+).
7-бром-1-хлор-6-фторизохинолин (311)
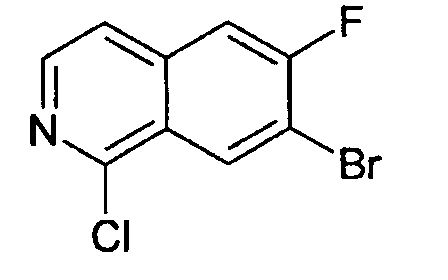
Исходя из 7-бром-6-фторизохинолин 2-оксида (310) было получено целевое соединение, согласно протоколу, описанному для 1,7-дихлор-6-фторизохинолина (6). Rt=1,70 мин (способ С). Детектированная масса: 260,0/262,0 (М+Н+).
7-бром-6-фтор-2Н-изохинолин-1-он (312)
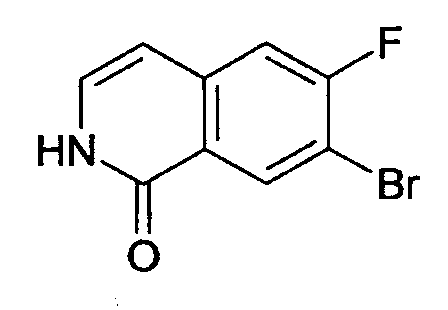
Растворяли 12,9 г (49,5 ммоль) 7-бром-1-хлор-6-фторизохинолина (311) в 250 мл уксусной кислоты. После добавления 38,7 г (0,5 моль) аммоний ацетата раствор перемешивали при 100°С. Через 3 часа растворитель удаляли под вакуумом и остаток выливали в воду. Осадок фильтровали и сушили с получением 9,91 г (83%) целевого соединения. Rt=1,15 мин (способ С). Детектированная масса: 242,2/244,1 (М+Н+).
7-бром-6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-он (313)
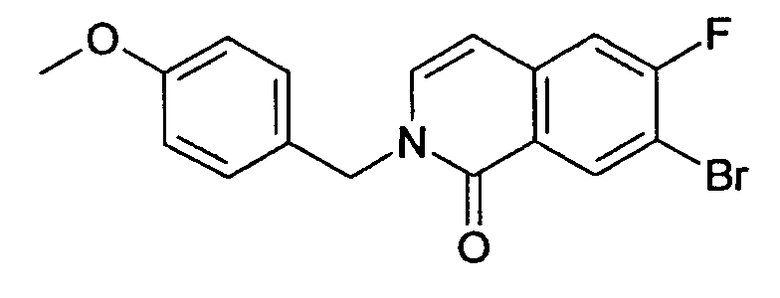
Растворяли 9,66 г (39,9 ммоль) 7-бром-6-фтор-2Н-изохинолин-1-она (312) в 180 мл диметилацетамида и добавляли 1,92 г (48,0 ммоль) гидрида натрия (60%). Через 1 ч при комнатной температуре добавляли раствор 7,50 г (48,0 ммоль) 4-метоксибензилхлорида в 25 мл диметилацетамида. Смесь перемешивали при комнатной температуре до достижения полной конверсии. Для проведения выделения растворитель удаляли под вакуумом, остаток поглощали раствором насыщенного бикарбоната натрия и три раза экстрагировали дихлорметаном. Органические слои сушили над MgSO4 и упаривали с получением 16,8 г неочищенного продукта в виде темного масла, который затем перемешивали в метаноле. Фильтрация осадка привела к 6,56 г целевого соединения в виде желтого твердого вещества. Маточный раствор упаривали и остаток очищали препаративной ВЭЖХ, что привело к получению дополнительных 2,62 г целевого продукта. Rt=1,71 мин (способ С). Детектированная масса: 362,3/364,3 (М+Н+).
Трет-бутиловый эфир 4-[7-бром-2-(4-метоксибензил)-1-оксо-1,2-дигидроизохинолин-6-илокси]пиперидин-1-карбоновой кислоты (314)
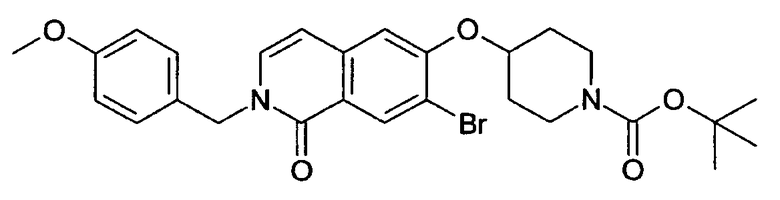
126 мг (0,625 ммоль) трет-бутилового сложного эфира 4-гидроксипиперидин-1-карбоновой кислоты растворяли в 2,5 мл диметилацетамида и добавляли 30 мг (0,75 ммоль) NaH (60% чистоты) при комнатной температуре. Через 15 минут добавляли 181 мг (0,5 ммоль) 7-бром-6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-она (313) и продолжали перемешивание при комнатной температуре. Через 5,5 ч растворитель удаляли под вакуумом. После добавления насыщенного раствора бикарбоната натрия смесь дважды экстрагировали дихлорметаном. Органические слои сушили над MgSO4 и упаривали. После конечной очистки препаративной ВЭЖХ выделяли 182 мг продукта. Rt=1,93 мин (способ С). Детектированная масса: 543,5/545,5 (М+Н+).
7-бром-6-(пиперидин-4-илокси)-2Н-изохинолин-1-он (17)
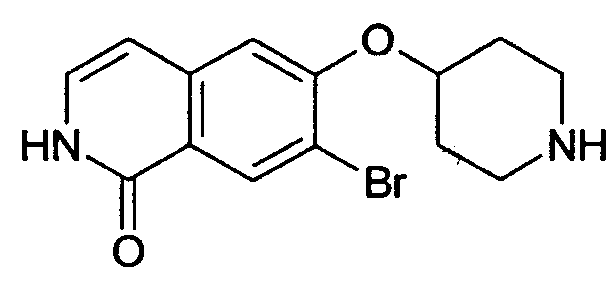
Растворяли 182 мг трет-бутилового эфира 4-[7-бром-2-(4-метоксибензил)-1-оксо-1,2-дигидроизохинолин-6-илокси]пиперидин-1-карбоновой кислоты (314) в 5 мл трифторуксусной кислоты. Через 2 ч при комнатной температуре смесь нагревали до 140°С в микроволновой печи в течение 2 ч. Растворитель удаляли под вакуумом и остаток растворяли в 2Н HCl. Водный раствор дважды промывали дихлорметаном и органические слои экстрагировали 2Н HCl. Объединенные водные растворы упаривали под вакуумом и остаток растворяли в воде. После лиофилизации выделяли целевое соединение в виде соли HCl. Rt=0,80 мин (способ В). Детектированная масса: 323,1/325,1 (М+Н+).
6-фтор-2-(4-метоксибензил)-7-фенил-2Н-изохинолин-1-он (315)
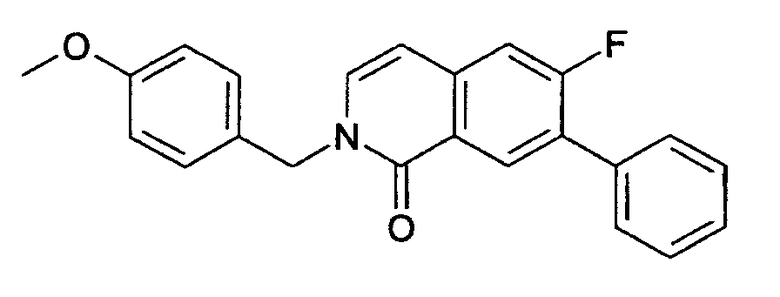
Растворяли 453 мг (1,25 ммоль) 7-бром-6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-она (313), 432 мг (3,125 ммоль) K2CO3 и 190,5 мг (1,56 ммоль) фенилбороновой кислоты в 12,5 мл толуола. В атмосфере аргона добавляли 72 мг (0,062 ммоль) Pd(Ph3)4 и раствор перемешивали при 100°С. После полной конверсии растворитель удаляли под вакуумом и добавляли насыщенный раствор бикарбоната натрия. Органические раствор экстрагировали три раза дихлорметаном и органические слои сушили над MgSO4. После упаривания неочищенный продукт очищали препаративной ВЭЖХ. Rt=1,80 мин (способ С). Детектированная масса: 360,4 (М+Н+).
Трет-бутиловый эфир 4-[2-(4-метоксибензил)-1-оксо-7-фенил-1,2-дигидроизохинолин-6-илокси]пиперидин-1-карбоновой кислоты (316)
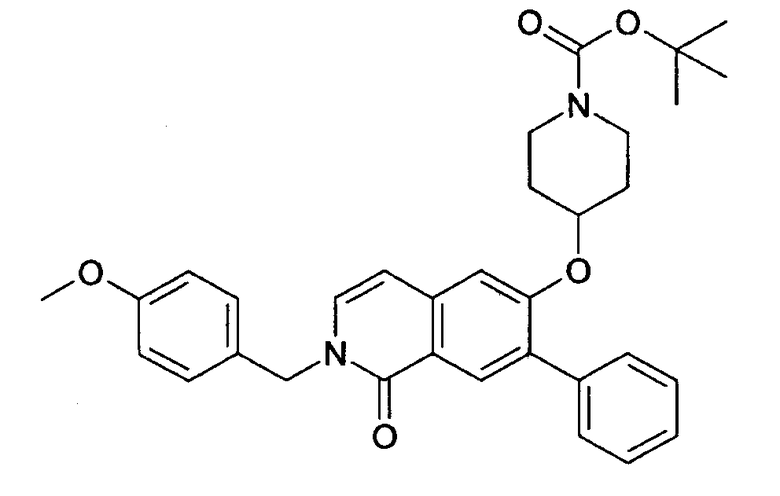
Растворяли 168 мг (0,83 ммоль) трет-бутилового эфира 4-гидроксипиперидин-1-карбоновой кислоты в 5 мл диметилацетамида и добавляли 20 мг (0,83 ммоль) гидрида натрия (60%). Смесь перемешивали при комнатной температуре. Через 30 минут добавляли раствор 240 мг (0,67 ммоль) 6-фтор-2-(4-метоксибензил)-7-фенил-2Н-изохинолин-1-она (315) в 5 мл диметилацетамида и продолжали перемешивание при комнатной температуре. После выдерживания в течение ночи добавляли 20 мг (0,83 ммоль) натрия гидрида (60%) и раствор перемешивали при 100°С. Через 1 ч растворитель удаляли под вакуумом и добавляли раствор насыщенного бикарбоната натрия. Водную фазу трижды экстрагировали дихлорметаном. Органические слои сушили над MgSO4 и упаривали. Неочищенный продукт очищали препаративной ВЭЖХ. Rt=1,98 мин (способ С). Детектированная масса: 541,7 (М+Н+).
7-фенил-6-(пиперидин-4-илокси)-2Н-изохинолин-1-он (317)
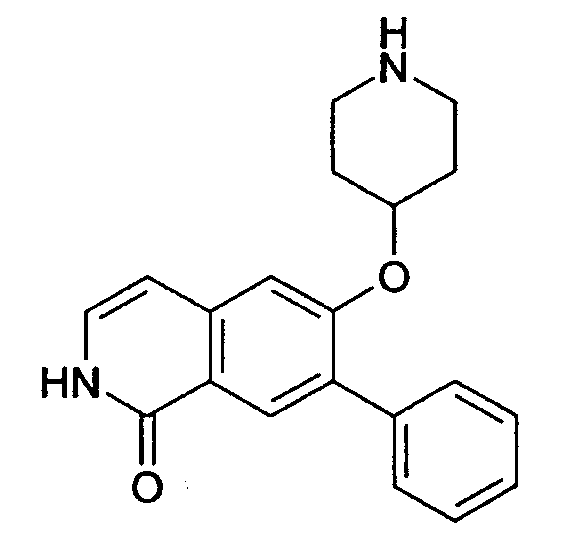
С трет-бутилового эфира 4-[2-(4-метоксибензил)-1-оксо-7-фенил-1,2-дигидроизохинолин-6-илокси]-пиперидин-1-карбоновой кислоты (316) снимали защиту, следуя способу, описанному для 7-бром-6-(пиперидин-4-илокси)-2Н-изохинолин-1-она (17). Обычная обработка позволяет получить целевое соединение в виде гидрохлорида. Rt=1,05 мин (способ В). Детектированная масса: 321,1 (М+Н+).
7-этил-6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-он (318)
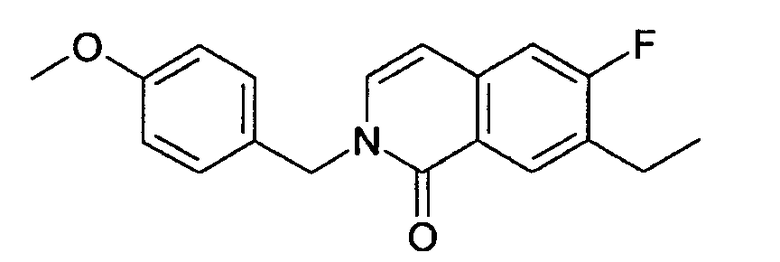
Целевое соединение синтезировали при использовании способа, описанного для 6-фтор-2-(4-метоксибензил)-7-фенил-2Н-изохинолин-1-она (315), исходя из 7-бром-6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-она (313) и этилбороновой кислоты. Rt=1,69 мин (способ С). Детектированная масса: 312,4 (М+Н+).
Трет-бутиловый эфир 4-[7-этил-2-(4-метоксибензил)-1-оксо-1,2-дигидроизохинолин-6-илокси]пиперидин-1-карбоновой кислоты (319)
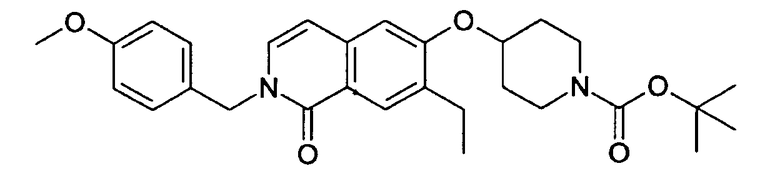
Целевое соединение синтезировали согласно способу, описанному для трет-бутилового эфира 4-[2-(4-метоксибензил)-1-оксо-7-фенил-1,2-дигидроизохинолин-6-илокси]пиперидин-1-карбоновой кислоты (316), исходя из 7-этил-6-фтор-2-(4-метоксибензил)-2Н-изохинолин-1-она (318) и трет-бутилового эфира 4-гидроксипиперидин-1-карбоновой кислоты. Rt=1,91 мин (способ С). Детектированная масса: 493,6 (М+Н+).
7-этил-6-(пиперидин-4-илокси)-2Н-изохинолин-1-он (320)
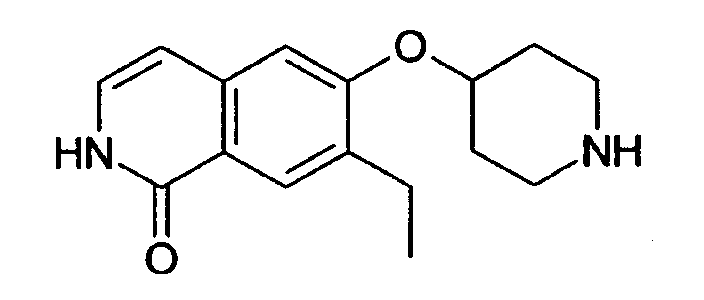
С трет-бутилового эфира 4-[7-этил-2-(4-метоксибензил)-1-оксо-1,2-дигидроизохинолин-6-илокси]пиперидин-1-карбоновой кислоты (319) снимают защиту, следуя способу, описанному для 7-бром-6-(пиперидин-4-илокси)-2Н-изохинолин-1-она (17). Конечная очистка при помощи препаративной ВЭЖХ дает целевое соединение в виде трифторацетата. Rt=0,92 мин (способ А). Детектированная масса: 273,2 (М+Н+).
Определение ингибирования Rho-киназы
Для измерения ингибирования Rho-киназы, определяли значения IC50 согласно следующему протоколу:
Буфер: 25 мМ Tris рН 7,5; 0,02% BSA; 5% глицерин; 0,008% Triton × 100; 2% ДМСО, 1 мM DTT; 1 мM MgCl2; 0,5 мкКи/ячейка γ33P АТФ
Фермент: ROCKII или ROKα) (Upstate, каталожный № 14-451 партия № 24880U) 0,1 нг/мкл
Конечная концентрация АТФ в реакционной смеси 40мкM
Биотинилированный субстрат, разбавленный описанным выше буфером (без АТФ) до 0,25 мкM
1. 10 мкл Tris буфера (± Ингибитор)
2. Добавляют 30 мкл раствора фермента
3. Начинают реакцию с 30 мкл смеси субстрат/АТФ/АТФ33
4. Инкубируют в течение 20 мин при комнатной температуре
5. Останавливают реакцию 30 мкл 50 мМ этилендиаминтриуксусной кислоты
6. Переносят 50 мкл остановленного раствора на Streptavidin Flash Plate plus, Perkin Elmer, SMP 103A
7. Инкубируют в течение 30 мин при комнатной температуре
8. Промывают 4 раза 300 мкл PBS/0,1% Tween 20
9. Определяют радиоактивность в ячейке
Следующие продукты испытывали в указанном анализе при использовании соответствующей формы (соли или основания), полученной согласно примерам, описанным выше, и были измерены следующие активности.
Полученные активности представляли в виде отрицательного десятичного логарифма IC50 (pIC50) следующим образом:
+: pIC50≤3,0
++: 3,0≤pIC50<4,0
+++: 4,0≤pIC50<5,0
++++: 5,0≤pIC50<6,0
+++++: 6,0≤pIC50
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ИЗОХИНОЛИНОВЫЕ И ИЗОХИНОЛИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2007 |
|
RU2455302C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОАЛКИЛАМИНОМ ПРОИЗВОДНЫЕ ИЗОХИНОЛОНА | 2007 |
|
RU2457203C2 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2006 |
|
RU2443688C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСИЛАМИНИЗОХИНОЛОНА В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2006 |
|
RU2440988C2 |
| БИ- И ПОЛИЦИКЛИЧЕСКИЕ ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА И ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2009 |
|
RU2532481C2 |
| 6-ЗАМЕЩЕННЫЕ ИЗОХИНОЛИНЫ И ИЗОХИНОЛИНОНЫ ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2009 |
|
RU2528229C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОАЛКИЛАМИНОМ ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА И ИЗОХИНОЛИНОНА | 2007 |
|
RU2468011C2 |
| ЗАМЕЩЕННЫЕ ИЗИХИНОЛИНЫ И ИЗОХИНОЛИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2009 |
|
RU2538588C2 |
| МОНОМЕРНЫЕ ПРОИЗВОДНЫЕ ГЛИКОПЕПТИДНОГО АНТИБИОТИКА | 2005 |
|
RU2424248C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ 1β-ДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2006 |
|
RU2415855C2 |
Изобретение относится к новым соединениям формулы (I)
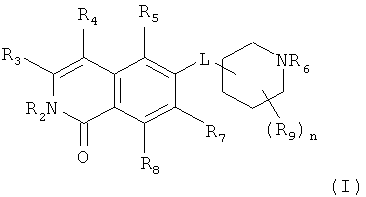
где R3 представляет собой Н, [(C1-С6)алкилен]0-1-R'; R3 представляет собой Н; R4 представляет собой Н, галоген или (C1-С6)алкил; R5 представляет собой Н или галоген; R6 представляет собой Н, (C1-C8)алкил, R', (C1-С6) алкилен-R'; R7 и R8 независимо друг от друга представляют собой Н, галоген, (C1-С6) алкил, О-(С1-С6)алкил, R'; R9 представляет собой (С1-С6)алкил; n равно 0 или 1; L представляет собой О или O-(С1-С6)алкилен; где R' представляет собой (С3-C8)циклоалкил; (С5-С10)гетероциклил, который обозначает ароматическую или насыщенную моно- или бициклическую кольцевую систему, которая включает кроме атома углерода один или несколько гетероатомов, таких как атомы азота, кислорода и серы; или (С6-С10) арил; причем гетероциклил является незамещенным или замещен (С1-С6)алкилом, а арил является незамещенным или замещен галогеном, (C1- С4)алкилом, -O-(С1-С4)алкилом, SO2-(C1-C4) алкилом или N[(С1-С4)алкил]2; и где в группах R4, R6 и R7 алкил может быть галогенирован в одном или более положениях; или их фармацевтически приемлемым солям и/или стереоизомерным формам. Изобретение также относится к применению соединений формулы (I), а также к лекарственному средству. Технический результат - получение новых биологически активных соединений, обладающих активностью ингибитора Rho-киназы. 3 н. и 18 з.п. ф-лы, 3 табл.
1. Соединение формулы (I)
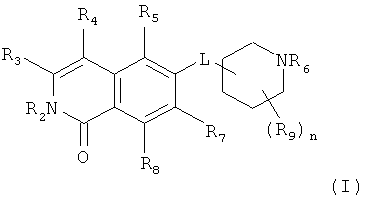
где R2 представляет собой Н, [(C1-С6)алкилен]0-1-R';
R3 представляет собой Н;
R4 представляет собой Н, галоген или (C1-С6)алкил;
R5 представляет собой Н или галоген;
R6 представляет собой Н, (C1-C8) алкил, R', (C1-C6) алкилен-R';
R7 и R8 независимо друг от друга представляют собой Н, галоген, (C1-C6) алкил, О-(C1-C6) алкил, R';
R9 представляет собой (C1-C6)алкил;
n равно 0 или 1;
L представляет собой О или О-(C1-C6)алкилен;
где R' представляет собой
(С3-C8)циклоалкил;
(С5-С10)гетероциклил, который обозначает ароматическую или насыщенную моно- или бициклическую кольцевую систему, которая включает кроме атома углерода один или несколько гетероатомов, таких как атомы азота, кислорода и серы; или
(С6-С10)арил;
причем гетероциклил является незамещенным или замещен (C1-С6)алкилом, а арил является незамещенным или замещен галогеном, (C1-С4)алкилом, -O-(С1-С4)алкилом, SO2-(C1-C4) алкилом или N[(C1-C4)алкил]2;
и
где в группах R4, R6 и R7 алкил может быть галогенирован в одном или более положениях;
или его фармацевтически приемлемые соли и/или стереоизомерные формы.
2. Соединение формулы (I) по п.1, в котором R2 представляет собой Н.
3. Соединение по п.1, в котором R6 представляет собой Н, (C1-С6)алкил, (С3-С6)циклоалкил или (C1-C4) алкилен-(С3-С6)циклоалкил.
4. Соединение по п.1, в котором R6, представляет собой Н или (C1-С6)алкил.
5. Соединение по п.1, в котором R6 представляет собой Н.
6. Соединение по п.1, в котором R5 представляет собой Н.
7. Соединение по п.1, в котором R4 представляет собой Н.
8. Соединение по п.1, в котором R7 представляет собой Н, галоген, (C1-C4)алкил, O-(C1-C4) алкил или фенил.
9. Соединение по п.1, в котором R7 и R8 представляют собой Н.
10. Соединение по п.1, в котором R9 представляет собой метил.
11. Соединение по п.1, в котором n равно 0.
12. Соединение по п.1, в котором L присоединен в положении-4 пиперидинильного кольца
или L присоединен в положении-3 пиперидинильного кольца
13. Соединение по п.12, в котором L присоединен в положении-4 пиперидинильного кольца.
14. Соединение по п.1, в котором L представляет собой O-метилен, O-этилен или О.
15. Соединение по п.1, в котором L представляет собой O-метилен, O-этилен или О, присоединенный в положении-4 пиперидинильного кольца.
16. Соединение по п.1, в котором L представляет собой О.
17. Соединение по п.1, выбранное из группы, состоящей из
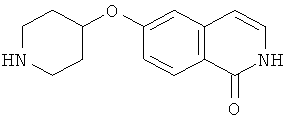 ,
,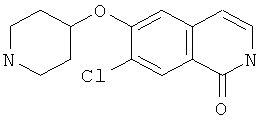 и
и 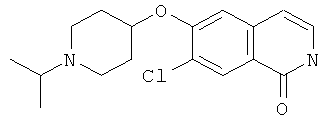 ,
,
или его фармацевтически приемлемые соли и/или стереоизомерные формы.
18. Соединение по п.1, выбранное из группы, состоящей из
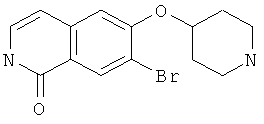 ,
, 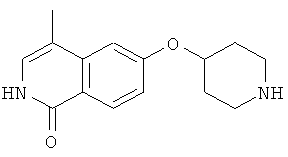 ,
, 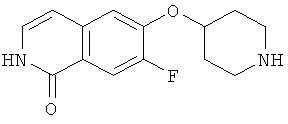 ,
,
 ,
,  ,
, 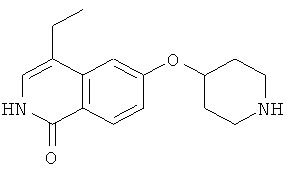 ,
,
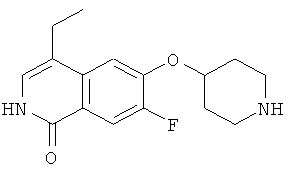 ,
, 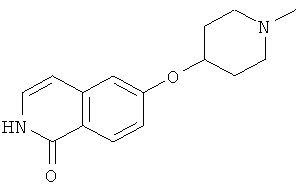 ,
, 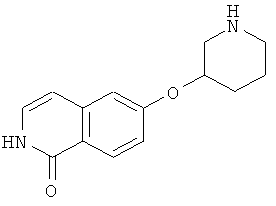 ,
, 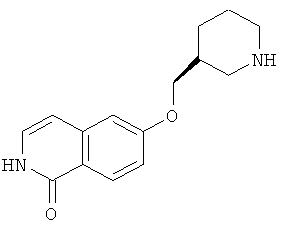 ,
, 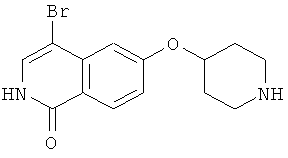 ,
,
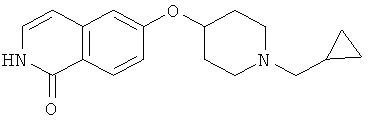 ,
, 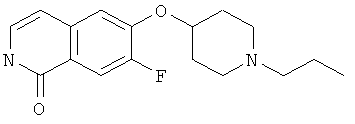 ,
, 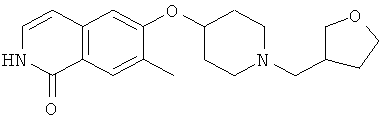 ,
, 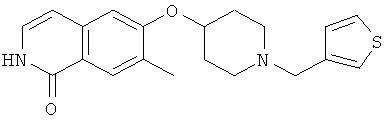 ,
,
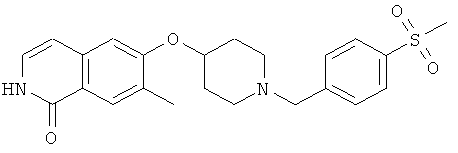 ,
, 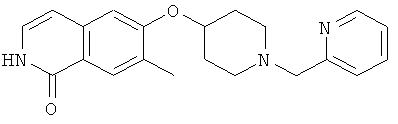 и
и 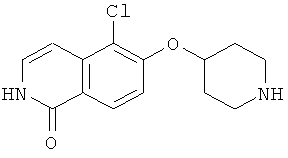 ,
,
или его фармацевтически приемлемые соли и/или стереоизомерные формы.
19. Соединение формулы (I) или его физиологически приемлемые соли и/или стереоизомерные формы по любому из пп.1-18 для применения в качестве лекарственного средства, обладающего активностью ингибитора Rho-киназы.
20. Применение соединения формулы (I) или его физиологически приемлемых солей и/или стереоизомерных форм по любому из пп.1-18 для получения лекарственного средства, обладающего активностью ингибитора Rho-киназы.
21. Лекарственное средство, обладающее активностью ингибитора Rho-киназы, включающее эффективное количество соединения формулы (I) или его фармацевтически приемлемых солей и/или стереоизомерных форм по любому из пп.1-18, физиологически приемлемые эксципиенты и носители.
| Устройство для управления механизмом резания листорезальной машины | 1988 |
|
SU1541559A1 |
| Устройство для комплектования пластин в пакет магнитопровода | 1986 |
|
SU1403255A1 |
| RU 2167865 С2, 27.05.2001. | |||

