Настоящее изобретение относится к новым производным изохинолина, их получению и применению для лечения и/или профилактики заболеваний, которые связаны с ингибированием Rho-киназы и/или опосредованного Rho-киназой фосфорилирования фосфатазы легкой цепи миозина.
Некоторая активация RhoA GTPазы при стимуляции агонистом приводит к конверсии RhoA из неактивной GDP-связанной формы в активную GTP-связанную форму с последующим связыванием с и активацией Rho-киназы. Известны две изоформы, Rho-киназа 1 и Rho-киназа 2. Rho-киназа 2 экспрессируется в клетках гладких мышц сосудов и эндотелиальных клетках. Активация Rho-киназы 2 активной GTP-связанной RhoA приводит к сенсибилизации кальция в клетках гладких мышц через опосредованное фосфорилированием ингибирование активности фосфатазы легкой цепи миозина и посредством этого к повышенной регуляции активности регуляторной легкой цепи миозина (Uehata et al. Nature 1997, 389, 990-994).
Известно, что Rho-киназа вовлечена в сужение кровеносных сосудов, включая развитие миогенного тонуса (J. Appl. Physiol. 2005, 1940-8, 98), сокращение гладкой мускулатуры бронхов (Am. J. Resp. Cell Mol. Biol. 20, 1190-1200), гипертензию, например легочную гипертензию (Heart, 91, 391-2, 2005) и глазную гипертензию (Invest. Ophthalmol. Visual Sci. 2001, 42, 137-144), эндотелиальную дисфункцию (Eur. J. Pharmacol. 2005, 512, 247-249), атеросклероз, рестеноз (Arch. Mal. Coeur 2005, 98, 249-254), утилизацию глюкозы, сердечную гипертрофию (Hypertension 2000, 35, 313-318), эректильную дисфункцию (Nature Medicine 2001, 7, 119-122), ретинопатию, воспаление, иммунные заболевания, СПИД, остеопороз, нарушения функций мозга, бактериальную инфекцию пищеварительного тракта (WO 98/06433), развитие раковых заболеваний, пролиферацию и сократительную способность гладких мышц сосудов (Circ. Res. 1999, 84, 1186-1193; Atherosclerosis 2001, 155, 321-327), эндотелиальную пролиферацию и сократительную способность (Biochem. Biophys. Res. Commun. 2000, 269, 633-640), образование стрессовых волокон (Science 1997, 275, 1308-1311; J. Cell Biol. 2000, 150, 797-806), агрегацию тромбоцитов (FEBS Lett. 2000, 466, 70-74; Blood 1999, 94, 1665-1672), активацию транспортной системы обмена Na/H (EMBO J. 1998, 17, 4712-4722), болезнь Альцгеймера (Science 2003, 302, 1215-1217), активацию аддуцина (J. Biol. Chem., 273, 5542-5548, 1998), и в передачу сигнала SREB (связывающий элемент стерольного ответа) и его действие на липидный метаболизм (Circ. Res., 92,1296-304, 2003).
Поэтому соединение, обладающее эффектом ингибирования Rho-киназы и/или опосредованного Rho-киназой фосфорилирования фосфатазы легкой цепи миозина, является полезным для лечения и/или профилактики заболеваний с вовлечением Rho-киназы как основной причины заболевания, например гипертензии, т.е. легочной гипертензии и глазной гипертензии, нарушения периферического кровообращения, стенокардии, церебрального вазоспазма, астмы, преждевременных родов, гиперагрегации тромбоцитов, окклюзивного заболевания периферических артерий (PAOD), хронического обструктивного легочного заболевания (COPD), развития рака и эректильной дисфункции, или как вторичной причины заболевания, например артериосклероза, ишемического заболевания органов (с конечным повреждением органа), фиброидного легкого, фиброидной печени, печеночной недостаточности, фиброидной почки, почечного гломерулосклероза, почечной недостаточности, гипертрофии органов, гипертрофии простаты, осложнений диабета, рестеноза кровеносных сосудов, атеросклероза, рака, сердечной гипертрофии, сердечной недостаточности; ишемических заболеваний; воспаления; аутоиммунных заболеваний; СПИДа, остеопатии, такой как остеопороз, нарушения функций мозга, бактериальных инфекций пищеварительного тракта, сепсиса, респираторного дистресс-синдрома взрослых, ретинопатии, глаукомы и болезни Альцгеймера.
WO 01/64238 описывает производные изохинолин-5-сульфонамида, необязательно замещенные -(CH2)1-6-O-(CH2)0-6-, -(CH2)0-6-S-(CH2)0-6- или -(CH2)0-6-связанной гетероциклической группой, полезные в качестве нейрозащитных средств.
JP 10087629 A описывает производные изохинолина, полезные для лечения инфекций, вызванных Heliobacter pylori, таких как, например, гастрит или язва. Производные изохинолина предпочтительно являются 5-замещенными группой X-[(C1-C6)алкилен)]0-1-Y, где X может представлять собой кислород, а Y может представлять собой арил или гетероциклическую группу.
Hagihara et al. (Bioorg. Med. Chem. 1999, 7, 2647-2666) раскрывает 6-бензилоксиизохинолин для лечения инфекций, вызванных Heliobacter pylori.
US 5480883 в общем виде раскрывает в качестве ингибиторов рецептора EGF и/или PDGF соединения, полезные для ингибирования клеточной пролиферации, имеющие формулу “Ar I - X - Ar II”, где X может представлять собой (CHR1)m-Z-(CHR1)n, например Z-CH2, где Z может представлять собой O, R1 представляет собой водород или алкил, Ar I может, среди прочего, представлять собой необязательно замещенную C5-12 бициклическую гетероарильную кольцевую систему и Ar II может, среди прочего, представлять собой необязательно замещенную C3-7 моноциклическую насыщенную гетероциклическую систему.
WO 03/053330 описывает производные изохинолина формулы
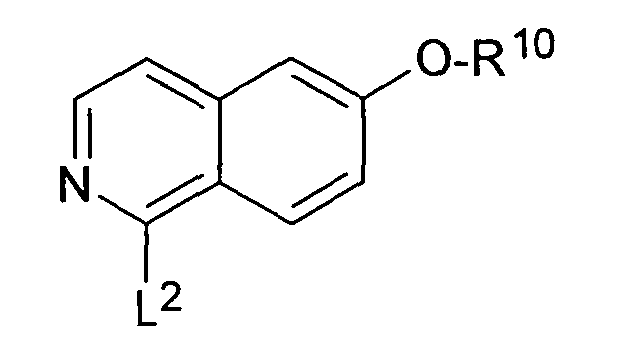
где L2 представляет собой галоген и R10 может представлять собой C1-5алкилен-C5-6гетероциклическую группу, в качестве промежуточных соединений в синтезе GSK-3 ингибиторов.
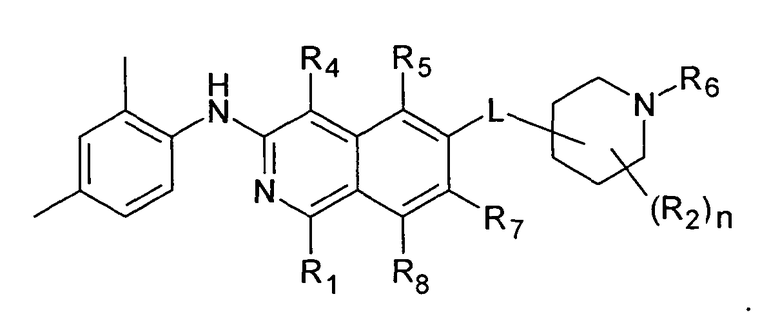
Вариант воплощения настоящего изобретения представляет соединение формулы (I)
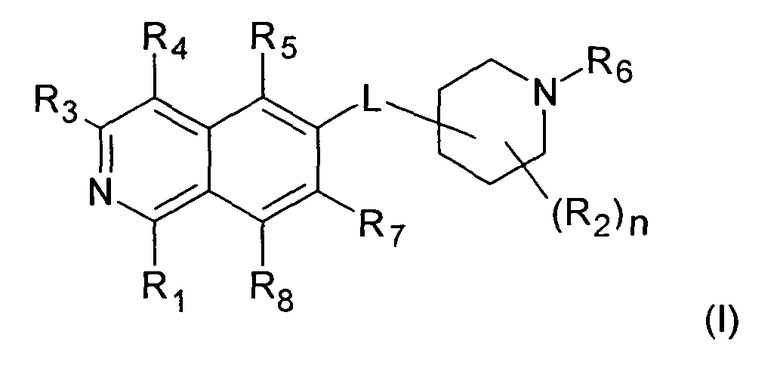
где
R1 представляет собой
H,
(C1-C6)алкил,
R',
NH-(C1-C6)алкил,
NH-R' или
N[(C1-C6)алкил]2;
R2 представляет собой водород, галоген или (C1-C6)алкил;
R3 представляет собой
H,
галоген,
(C1-C6)алкил,
(C1-C6)алкилен-R',
OH,
O-R'',
NH2,
NHR'',
NR''R'' или
NH-C(O)-R'',
R4 представляет собой
H,
галоген,
гидрокси,
CN,
(C1-C6)алкил,
R',
(C1-C6)алкилен-R';
R5 представляет собой
H,
галоген,
CN,
NO2,
(C1-C6)алкил,
(C2-C6)алкенил,
R',
(C1-C6)алкилен-(C6-C10)арил,
(C1-C6)алкенилен-(C6-C10)арил,
(C1-C6)алкилен-(C5-C10)гетероциклил,
CH(OH)-(C1-C6)алкил,
NH2,
NH-R',
NH-SO2H,
NH-SO2-(C1-C6)алкил,
NH-SO2-R',
NH-C(O)-(C1-C6)алкил,
NH-C(O)-R',
C(O)N[(C1-C6)алкил]2,
C(O)OH или
C(O)O-(C1-C6)алкил;
R6 представляет собой
H,
R',
(C1-C8)алкил,
(C1-C6)алкилен-R',
(C1-C6)алкилен-O-(C1-C6)алкил,
(C1-C6)алкилен-O-R',
(C1-C6)алкилен-CH[R']2,
(C1-C6)алкилен-C(O)-R',
(C1-C6)алкилен-C(O)NH2,
(C1-C6)алкилен-C(O)NH-R' или
(C1-C6)алкилен-C(O)N[R']2;
R7 представляет собой
H,
галоген,
CN,
NO2,
(C1-C6)алкил,
(C2-C6)алкенил,
R',
(C1-C6)алкенилен-(C6-C10)арил,
(C1-C6)алкилен-R',
CH(OH)-(C1-C6)алкил,
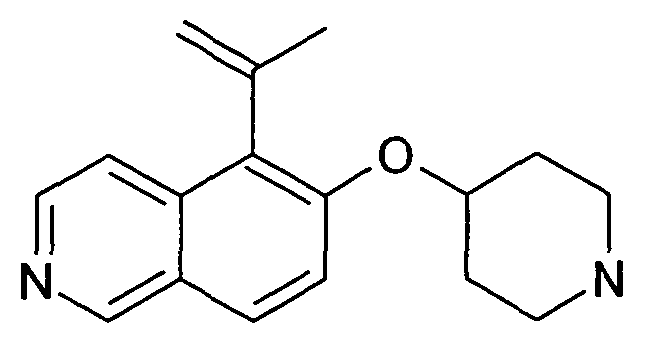
CH(OH)-(C6-C10)арил,
CH(OH)-(C5-C10)гетероциклил,
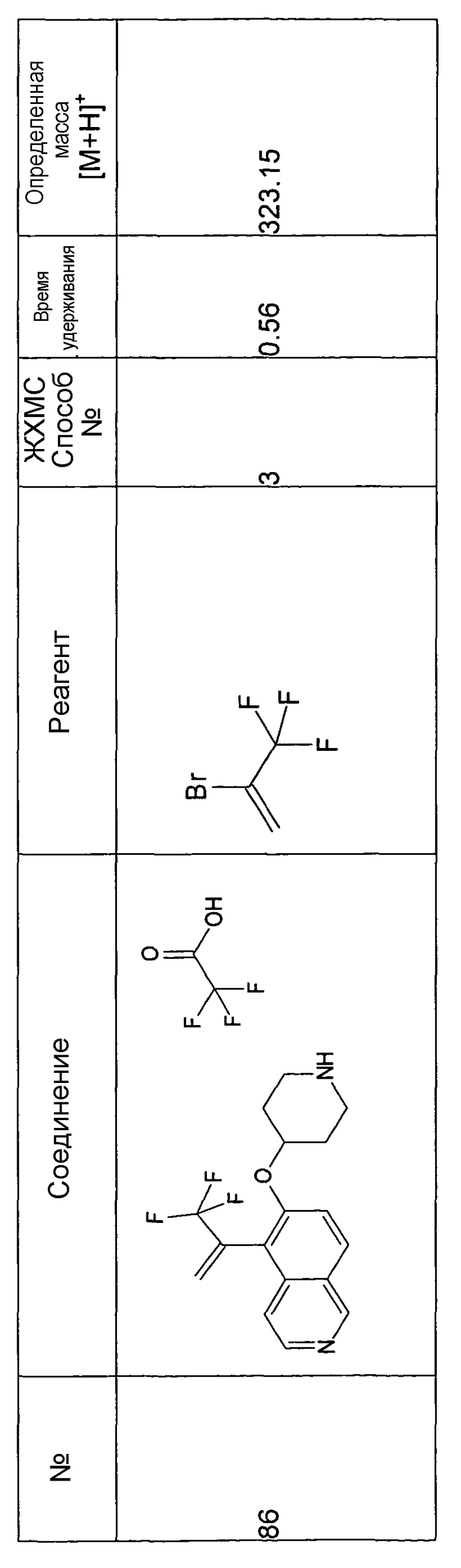
NH2,
NH-R',
NH-SO2H,
NH-SO2-(C1-C6)алкил,
NH-SO2-R',
SO2-NH2,
SO2-NHR',
NH-C(O)-(C1-C6)алкил,
NH-C(O)-R',
C(O)N[(C1-C6)алкил]2,
C(O)OH или
C(O)O-(C1-C6)алкил;
R8 представляет собой H, галоген или (C1-C6)алкил;
n имеет значение 1, 2, 3 или 4; и
L представляет собой O или O-(C1-C6)алкилен;
где
R' представляет собой
(C3-C8)циклоалкил,
(C5-C10)гетероциклил,
(C6-C10)арил; и
R'' представляет собой
(C3-C8)циклоалкил,
(C5-C10)гетероциклил,
(C6-C10)арил,
(C1-C6)алкил,
(C1-C6)алкилен-R',
(C1-C6)алкилен-O-(C1-C6)алкил,
(C1-C6)алкилен-O-R' или
(C1-C6)алкилен-NRxRy; и
где Rx и Ry независимо друг от друга представляют собой
(C1-C6)алкил,
(C5-C10)гетероциклил,
(C6-C10)арил,
(C1-C4)алкилен-(C5-C10)гетероциклил,
(C1-C4)алкилен-(C6-C10)арил,
(C1-C4)алкилен-NH(C1-C6)алкил,
(C1-C4)алкилен-N[(C1-C6)алкил]2,
(C1-C4)алкилен-N[(C6-C10)арил]2 или
(C1-C4)алкилен-N[(C5-C10)гетероциклил]2;
где в остатках R4, R5, R7 и R8 один атом водорода алкила или алкилена необязательно может быть замещен OH, F, OCH3, COOH, COOCH3, NH2, NHCH3, N(CH3)2, CONH2, CONHCH3 или CON(CH3)2;
и их фармацевтически приемлемые соли и/или физиологически функциональные производные.
Предпочтительно, R1 представляет собой H, (C1-C6)алкил, (C6-C10)арил, NH-(C1-C6)алкил, NH-(C6-C10)арил или N[(C1-C6)алкил]2. Более предпочтительно, R1 представляет собой H, галоген, (C1-C4)алкил, NH-(C1-C4)алкил, N[(C1-C4)алкил]2 или NH-фенил. Наиболее предпочтительно, R1 представляет собой H, (C1-C2)алкил или NH-(C1-C2)алкил, особенно предпочтителен H.
Предпочтительно, R2 представляет собой H, галоген или (C1-C4)алкил. Предпочтительно, R2 представляет собой H или (C1-C4)алкил. Более предпочтительно, R2 представляет собой H, (C1-C2)алкил. R2 может быть связан с любым атом углерода пиперидинового кольца, включая положение, где связана линкерная группа L.
R3 предпочтительно представляет собой H, галоген, (C1-C4)алкилен-R', O-R'' или NHR''. Более предпочтительно, R3 представляет собой Н или NHR''. Наиболее предпочтительно, R3 представляет собой H, NH-(C5-C6)гетероциклил или NH-фенил, особенно предпочтительными являются H, NH-(C5-C6)гетероарил, содержащий один или несколько N атомов или NH-фенил. Наиболее предпочтительно, R3 представляет собой H. Примеры заместителей R3 представлены в конце описания.
Предпочтительно, R4 представляет собой H, галоген или (C1-C6)алкил. Более предпочтительно, R4 представляет собой H, галоген или (C1-C4)алкил. Наиболее предпочтительно, R4 представляет собой H.
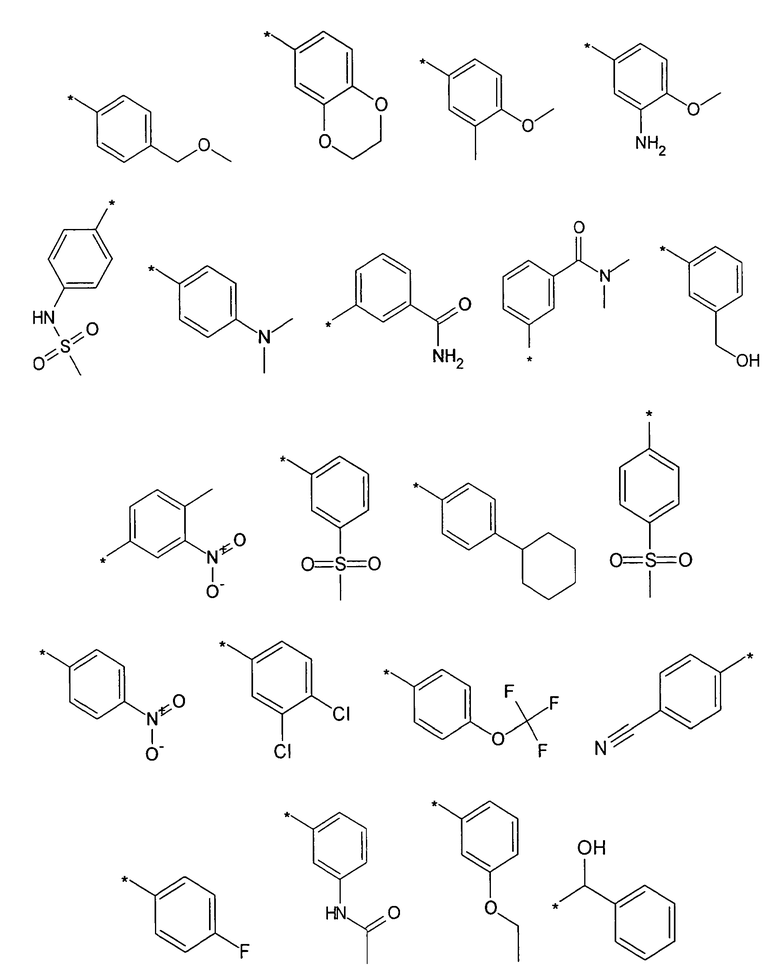
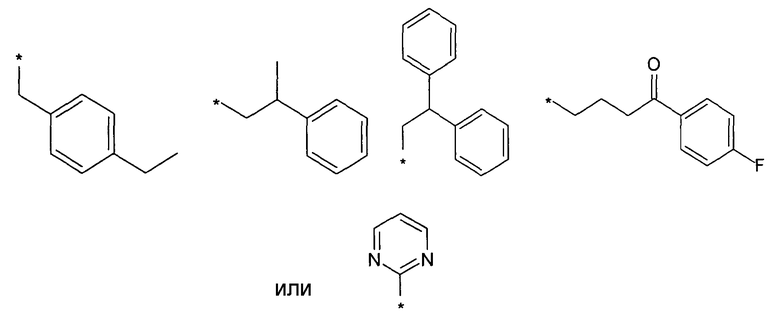
Предпочтительно, R5 представляет собой H, галоген, CN, (C1-C6)алкил, R', NH-(C6-C10)арил или (C1-C6)алкилен-R'. Более предпочтительно, R5 представляет собой H, галоген, (C1-C6)алкил, R', NH-(C6-C10)арил или (C1-C6)алкилен-R'. Наиболее предпочтительно, R5 представляет собой H, галоген, (C6-C10)арил, NH-(C6-C10)арил, (C1-C2)алкил-(C6-C10)арил, (C1-C6)алкил или (C5-C10)гетероарил. Особенно предпочтительно, R5 представляет собой H, галоген, фенил, (C1-C6)алкил или (C5-C6)гетероарил. Примерами R5 являются водород, фтор, хлор, бром, йод, нитрил, нитро, (пара-метокси)фенил, N-анилин, фенил, бензил, метил, этил, винил, 2-пропенил, втор-бутенил, циклопропил, тиенил, тетразол, амино, 4-метоксианилин, N-ацетил или заместитель группы, состоящей из
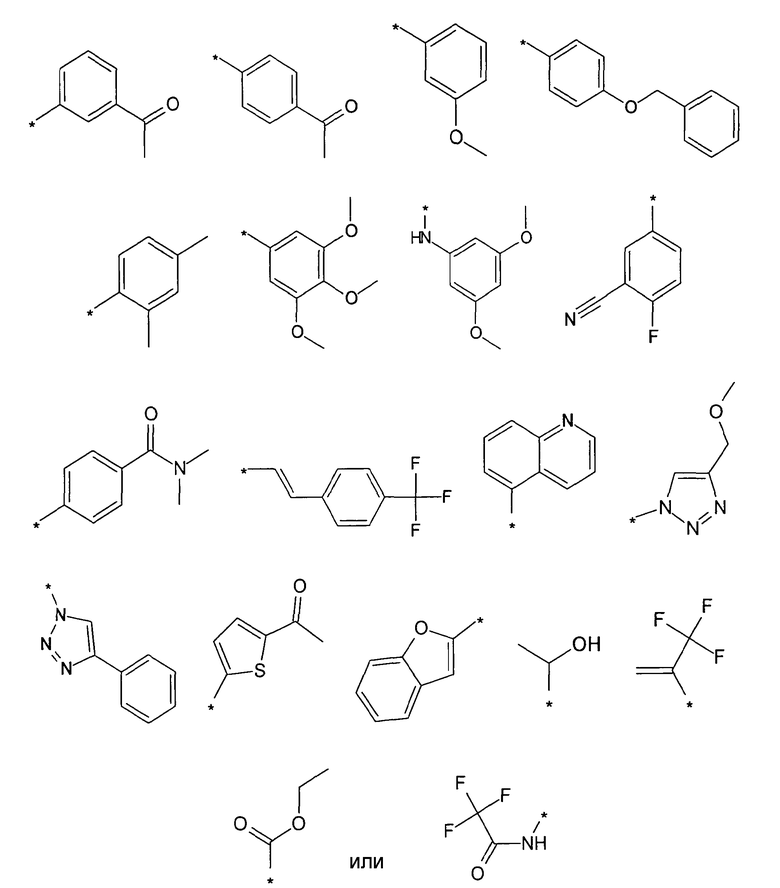
Предпочтительно, R6 представляет собой H, (C1-C6)алкил, R', (C1-C4)алкилен-(C3-C8)циклоалкил, (C1-C4)алкилен-(C5-C10)гетероциклил, (C1-C4)алкилен-C(O)-(C5-C10)гетероциклил, (C1-C4)алкилен-C(O)-(C6-C10)арил или (C1-C6)алкилен-(C6-C10)арил. Более предпочтительно, R6 представляет собой H, (C1-C6)алкил, (C5-C10)гетероциклил, (C1-C4)алкилен-(C5-C10)гетероциклил или (C1-C6)алкилен-(C6-C10)арил. Примерами R6 являются H, метил, этил, пропил, бутил, s-бутил, пентил, 3-метилбутил, изопропил, трифторметил, 3,3,3-трифторбутил, циклопропил, метилен циклопропил, 2-пиримидинил, бензил или заместитель группы, состоящей из
Предпочтительно, R7 представляет собой H, галоген, CN, (C1-C6)алкил, (C2-C6)алкенил, R' или (C1-C6)алкилен-(C3-C8)циклоалкил. Более предпочтительно, R7 представляет собой H, галоген, CN, (C1-C4)алкил, (C1-C4)алкенил, фенил, циклопропил или (C5-C6)гетероарил. Наиболее предпочтительно, R7 представляет собой H, фтор, хлор, бром, метил, этил, фенил, нитрил, циклопропил, тиенил или винил.
R8 предпочтительно представляет собой H, галоген или (C1-C4)алкил. Более предпочтительно, R8 представляет собой H, Cl, F, метил или этил.
Предпочтительно, n имеет значение 1, 2 или 3. Более предпочтительно, n равно 1.
Линкерная группа L может быть связана с пиперидиновым кольцом в любом положении через атом углерода пиперидинового кольца. В предпочтительном воплощении L присоединен к положению 4 пиперидинового кольца
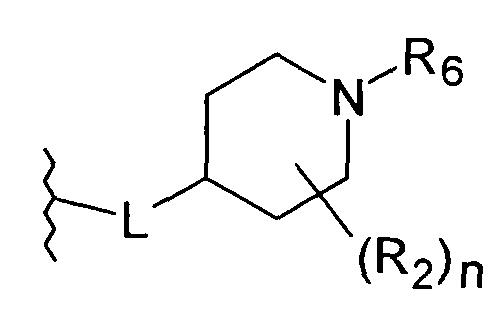 или
или
L присоединен к положению 3 пиперидинового кольца
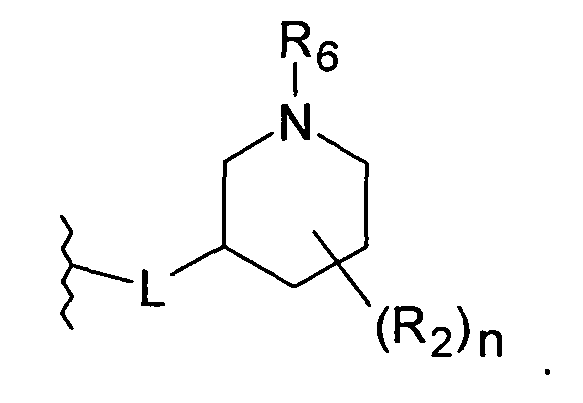
В наиболее предпочтительном воплощении L присоединен к положению 4 пиперидинового кольца.
В более предпочтительном воплощении L представляет собой O-метилен, O-этилен или, предпочтительно, O. Более предпочтительно, L представляет собой O-метилен, O-этилен или O, присоединенный к 4-положению пиперидинового кольца.
В предпочтительном воплощении настоящего изобретения одна, или несколько, или все группы, содержащиеся в соединениях формулы (I), могут независимо друг от друга иметь любое из предпочтительных, более предпочтительных или наиболее предпочтительных определений групп, указанных выше, или любое одно или несколько из конкретных значений, которые включены в определения групп и указаны выше, все комбинации предпочтительных определений, более предпочтительных или наиболее предпочтительных и/или конкретных определений, являются объектом настоящего изобретения. Также относительно всех предпочтительных вариантов воплощения настоящее изобретение включает соединения формулы (I) во всех стереоизомерных формах и смесях стереоизомерных форм в любых соотношениях и их физиологически приемлемые соли.
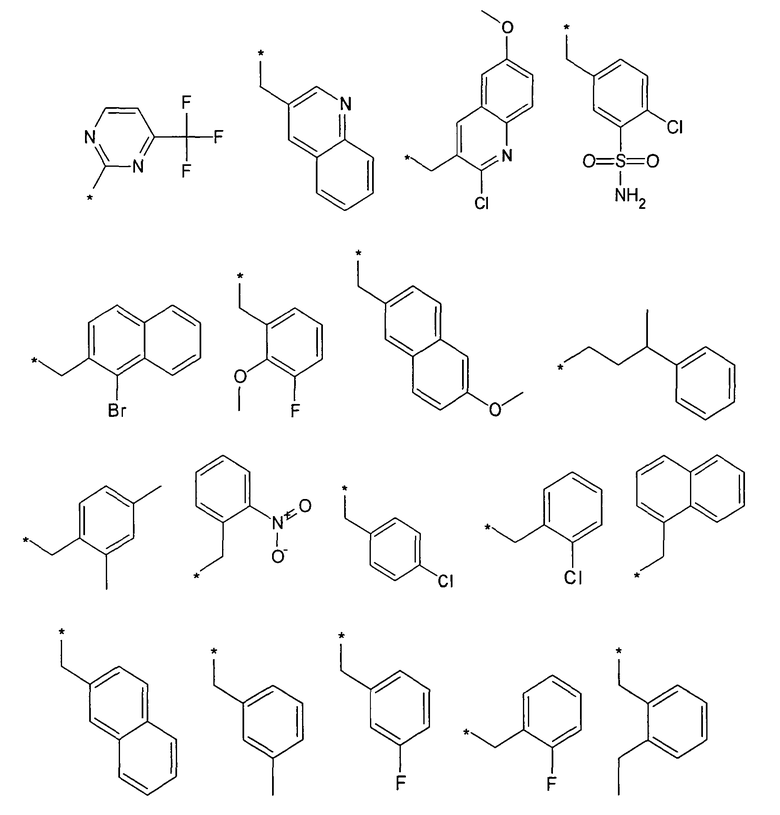
Термин “*-“ в приведенных в примерах заместителей (см. выше) указывает точку присоединения заместителя, которая обозначает, например, для заместителя R3
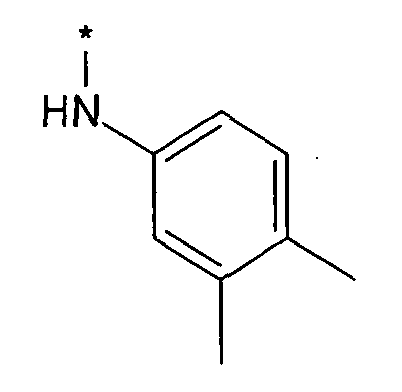
соединение формулы
Предпочтительный вариант воплощения представляет собой соединение формулы (I), где
R1 представляет собой H, (C1-C6)алкил, (C6-C10)арил, NH-(C1-C6)алкил, NH-(C6-C10)арил или N[(C1-C6)алкил]2;
R2 представляет собой водород, галоген или (C1-C6)алкил;
R3 представляет собой H, галоген, (C1-C4)алкилен-R', O-R'' или NHR'', где R' и R'' определены выше;
R4 представляет собой H, галоген или (C1-C6)алкил;
R5 представляет собой H, галоген, (C1-C6)алкил, CN, (C6-C10)арил, NH-(C6-C10)арил, (C1-C6)алкилен-(C6-C10)арил, (C5-C10)гетероциклил или (C1-C6)алкилен-(C5-C10)гетероциклил;
R6 представляет собой H, R', (C1-C4)алкилен-(C5-C10)гетероциклил, (C1-C6)алкилен-C(O)-(C6-C10)арил, (C1-C4)алкилен-C(O)-(C5-C10)гетероциклил, (C1-C6)алкилен-(C6-C10)арил или (C1-C6)алкил.
R7 представляет собой H, галоген, CN, (C1-C6)алкил, (C2-C6)алкенил или R';
R8 представляет собой H, галоген или (C1-C6)алкил;
n имеет значение 1, 2 или 3, и
L представляет собой O, O-метилен или O-этилен;
и их фармацевтически приемлемые соли и/или физиологически функциональные производные.
Следующий предпочтительный вариант воплощения представляет собой соединение формулы (I), где
R1 представляет собой H, (C1-C6)алкил, (C6-C10)арил, NH-(C1-C6)алкил, NH-(C6-C10)арил или N[(C1-C6)алкил]2;
R2 представляет собой H или (C1-C4)алкил;
R3 представляет собой H, галоген или NHR'', где R'' имеет значения, определенные выше;
R4 представляет собой H, галоген или (C1-C4)алкил;
R5 представляет собой H, галоген, (C1-C6)алкил, (C6-C10)арил, NH-(C6-C10)арил, (C1-C6)алкилен-(C6-C10)арил или (C5-C10)гетероциклил;
R6 представляет собой H, (C1-C6)алкил, R', (C1-C4)алкилен-(C5-C10)гетероциклил или (C1-C6)алкилен-(C6-C10)арил;
R7 представляет собой H, галоген, CN, (C1-C6)алкил, (C2-C6)алкенил или R';
R8 представляет собой H, галоген или (C1-C6)алкил;
n имеет значение 1, 2 или 3; и
L представляет собой O;
и их фармацевтически приемлемые соли и/или физиологически функциональные производные.
Особенно предпочтительный вариант воплощения представляет собой соединение формулы (I), где
R1 представляет собой H, (C1-C4)алкил, NH-(C1-C4)алкил, N[(C1-C4)алкил]2 или NH-фенил;
R2 представляет собой H, (C1-C4)алкил;
R3 представляет собой H, NH-(C5-C6)гетероарил или NH-фенил;
R4 представляет собой H, галоген или (C1-C4)алкил;
R5 представляет собой H, галоген, (C1-C4)алкил, (C6-C10)арил, NH-(C6-C10)арил, (C1-C2)алкил-(C6-C10)арил или (C5-C10)гетероарил;
R6 представляет собой H, (C1-C6)алкил, (C5-C10)гетероциклил, (C1-C4)алкилен-(C5-C10)гетероциклил, (C6-C10)арил или (C1-C6)алкилен-(C6-C10)арил;
R7 представляет собой H, галоген, CN, (C1-C4)алкил, (C1-C4)алкенил, фенил, циклопропил, (C5-C6)гетероарил;
R8 представляет собой H, галоген или (C1-C4)алкил;
n имеет значение 1; и
L представляет собой O;
и их фармацевтически приемлемые соли и/или физиологически функциональные производные.
Как в любом варианте воплощения изобретения, в описанных выше вариантах воплощения, которые содержат предпочтительные, более предпочтительные, наиболее предпочтительные или приведенные в качестве примера определения соединений по настоящему изобретению, одна, или несколько, или все группы могут иметь любое из их предпочтительных, более предпочтительных или наиболее предпочтительных определений, указанных выше, или любое одно или несколько конкретных значений, которые включены в определения групп и указаны выше.
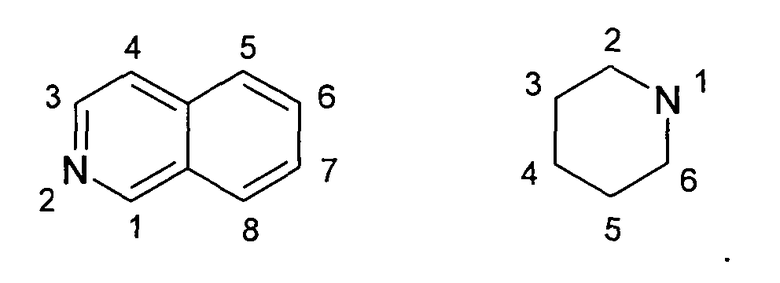
Представленная картина замещения изохинолина и пиперидила пронумерована согласно правилам IUPAC:
Физиологически приемлемые соли соединений формулы (I) представляет собой как их органические, так и неорганические соли, как описано в Remington's Pharmaceutical Sciences (17th edition, page 1418 (1985)). Из-за физической и химической стабильности и растворимости предпочтительными являются кислотные группы, среди прочего, для солей натрия, калия, кальция и аммония; предпочтительными являются оснόвные группы, среди прочего, для солей малеиновой кислоты, фумаровой кислоты, янтарной кислоты, яблочной кислоты, винной кислоты, метилсульфоновой кислоты, хлористоводородной кислоты, серной кислоты, фосфорной кислоты или карбоновых кислот или сульфоновых кислот, например таких как гидрохлориды, гидробромиды, фосфаты, сульфаты, метансульфонаты, ацетаты, лактаты, малеаты, фумараты, малаты, глюконаты, и соли аминокислот, природных оснований или карбоновых кислот. Получение физиологически приемлемых солей из соединений формулы (I) и (II), которые являются способными к образованию соли, включая их стереоизомерные формы, осуществляют способом, известным per se. Соединения формулы (I) образуют стабильные соли со щелочными металлами, щелочноземельными металлами или необязательно замещенные аммониевые соли с оснόвными реагентами, такими как гидроксиды, карбонаты, бикарбонаты, алкоголяты и аммониевые или органические основания, например триметил- или триэтиламин, этаноламин, диэтаноламин или триэтаноламин, трометамол, а также оснόвные аминокислоты, например лизин, орнитин или аргинин. Когда соединения формулы (I) имеют оснόвные группы, также могут быть получены устойчивые кислотно-аддитивные соли с сильными основаниями. Подходящие фармацевтически приемлемые кислотно-аддитивные соли соединений по настоящему изобретению представляют собой соли неорганических кислот, таких как хлористоводородная кислота, бромистоводородная, фосфорная, метафосфорная, азотная и серная кислота, и органических кислот, таких как, например, уксусная кислота, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изетионовая, молочная, лактобионовая, малеиновая, яблочная, метансульфоновая, янтарная, п-толуолсульфоновая и винная кислота.
Соли с физиологически неприемлемым анионом, таким как, например, трифторацетат, также являются подходящими в рамках данного изобретения в качестве полезных промежуточных соединений для получения или очистки фармацевтически приемлемых солей и/или для использования в нетерапевтических применениях, например in vitro.
Термин “физиологически функциональное производное”, используемый в данном описании, относится к любому физиологически толерантному производному соединения формулы (I) по настоящему изобретению, например N-оксиду, которое при введении млекопитающему, такому как, например, человек, может образовывать (прямо или опосредованно) соединение формулы (I) или его активный метаболит.
Физиологически функциональные производные включают пролекарства соединений по настоящему изобретению, такие как описанные, например, в H. Okada et al., Chem. Pharm. Bull. 1994, 42, 57-61. Такие пролекарства могут быть метаболизированы in vivo в соединения настоящего изобретения. Эти пролекарства могут быть активными как таковые или неактивными.
Изобретение относится к соединениям формулы (I) в форме их рацематов, рацемических смесей и чистых энантиомеров и к их диастереомерам и их смесям.
Если радикалы или заместители встречаются более одного раза в соединениях формулы (I), то все они могут, независимо один от другого, иметь указанное значение и быть одинаковыми или отличными друг от друга.
Соединения настоящего изобретения также могут существовать в различных полиморфных формах, например таких как аморфные и кристаллические полиморфные формы. Все полиморфные формы соединений настоящего изобретения являются подходящими в рамках настоящего изобретения и представляют собой еще один аспект настоящего изобретения.
Все ссылки на “соединение(соединения) формулы (I)”, приведенные далее, относятся к соединению(ям) формулы (I), как описано выше, и их физиологически приемлемым солям, сольватам и физиологически функциональным производным, как описано в настоящей заявке.
Должно быть понятно, что термины (C1-C2)алкил, (C1-C4)алкил, (C1-C6)алкил, (C1-C8)алкил и соответствующие алкиленовые заместители означают углеводородный остаток, который может быть линейным, т.е. с прямой цепью, или разветвленным и содержит 1, 2, 3, 4, 5, 6, 7 или 8 атомов углерода соответственно. Это также применимо, когда алкильная группа присутствует в качестве заместителя в другой группе, например в алкоксигруппе (O-алкил), S-алкил или -O(C1-C6)алкилен-O-, алкоксикарбонильной группе или арилалкильной группе. Примерами алкильных групп являются метил, этил, пропил, бутил, пентил или гексил, н-изомеры всех этих групп, изопропил, изобутил, 1-метилбутил, изопентил, неопентил, 2,2-диметилбутил, 2-метилпентил, 3-метилпентил, изогексил, втор-бутил, трет-бутил или трет-пентил. Алкильные группы могут, если не указано иное, быть галогенированными один или более раз, т.е. алкильные группы могут быть фторированы, например, перфторированы. Примерами галогенированных алкильных групп являются CF3 и CH2CF3, OCF3, SCF3 или -O-(CF2)2-O-.
Алкенил представляет собой, например, винил, 1-пропенил, 2-пропенил (= аллил), 2-бутенил, 3-бутенил, 2-метил-2-бутенил, 3-метил-2-бутенил, 5-гексенил или 1,3-пентадиенил.
Алкинил представляет собой, например, этинил, 1-пропинил, 2-пропинил (= пропаргил) или 2-бутинил.
Галоген означает фтор, хлор, бром или йод.
(C3-C8)циклоалкильные группы представляют собой циклические алкильные группы, содержащие 3, 4, 5, 6, 7 или 8 атомов углерода в кольце, такие как циклопропил, циклобутил, циклопентил, циклогексил или циклооктил, которые также могут быть замещенными и/или содержать 1 или 2 двойные связи (ненасыщенные циклоалкильные группы), например циклопентенил или циклогексенил могут быть связаны через любой углеродный атом.
(C6-C10)арильные группы означают ароматическое кольцо или кольцевую систему, которая содержит два ароматических кольца, которые являются конденсированными или связанными иным образом, например фенильная, нафтильная, бифенильная, тетрагидронафтильная, альфа- или бета-тетралон-, индалин- или индан-1-он-ильная группа. Предпочтительной (C6-C10)арильной группой является фенил.
(C5-C10)гетероциклильные группы означают моно- или бициклическую кольцевую систему, которая включает в себя помимо углерода один или несколько гетероатомов, таких как, например, 1, 2 или 3 атома азота, 1 или 2 атома кислорода, 1 или 2 атома серы или комбинации различных гетероатомов. Гетероциклильные остатки могут быть связаны в любых положениях, например в 1-положении, 2-положении, 3-положении, 4-положении, 5-положении, 6-положении, 7-положении или 8-положении. (C5-C10)гетероциклильные группы могут быть (1) ароматическими [= гетероарильные группы], или (2) насыщенными, или (3) смешанными ароматическими/насыщенными.
Подходящие (C5-C10)гетероциклильные группы включают акридинил, азоцинил, бензимидазолил, бензофурил, бензоморфолинил, бензотиенил, бензoтиофенил, бензоксазолил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоxазолил, бензизотиазолил, карбазолил, 4aH-карбазолил, карболинил, фуранил, хиназолинил, хинолинил, 4H-хинолизинил, хиноксалинил, хинуклидинил, хроманил, хроменил, хромен-2-онил, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]-тетрагидрофуран, фурил, фуразанил, гомоморфолинил, гомопиперазинил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индолинил, индолизинил, индолил, 3H-индолил, изобензoфуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил (бензимидазолил), изотиазолил, изоксазолил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, оксазолидинил, пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, феноксатиинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пролинил, птеридинил, пуринил, пиранил, пиразинил, пироазолидинил, пиразолинил, пиразолил, пиридазинил, пиридонил, пиридооксазолы, пиридоимидазолы, пиридотиазолы, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, 2H-пирролил, пирролил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, 6H-1,2,5-тиадазинил, тиазолил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиенил, триазолил, тетразолил и ксантенил. Пиридил означает как 2-, 3-, так и 4-пиридил. Тиенил означает как 2-, так и 3-тиенил. Фурил означает как 2-, так и 3-фурил. Также включены соответствующие N-оксиды этих соединений, например 1-окси-2-, 3- или 4-пиридил.
Замещения в (C5-C10)гетероциклильных остатках могут иметь место по свободным атомам углерода или по атомам азота.
Предпочтительными примерами (C5-C10)гетероциклильных остатков являются пиразинил, пиридил, пиримидинил, пиразолил, морфолинил, пирролидинил, пиперазинил, пиперидинил, тиенил, бензофурил, хинолинил, тетразолил и триазолил.
(C6-C10)арильные и (C5-C10)гетероциклильные группы являются незамещенными или замещенными один или более раз подходящими группами, независимо выбранными из галогена, CF3, NO2, N3, CN, C(O)-(C1-C6)алкила, C(O)-(C1-C6)арила, COOH, COO(C1-C6)алкила, CONH2, CONH(C1-C6)алкила, CON[(C1-C6)алкил]2, (C3-C8)циклоалкила, (C1-C6)алкила, (C1-C6)алкилен-OH, (C1-C6)алкилен-NH2, (C1-C6)алкилен-NH(C1-C6)алкила, (C1-C6)алкилен-N[(C1-C6)алкил]2, (C2-C6)алкенила, (C2-C6)алкинила, O-(C1-C6)алкила, O-C(O)-(C1-C6)алкила, O-C(O)-(C6-C10)арила, O-C(O)-(C5-C10)гетероциклила, PO3H2, SO3H, SO2-NH2, SO2NH(C1-C6)алкила, SO2N[(C1-C6)алкил]2, S-(C1-C6)алкила; S-(C1-C6)алкилен-(C6-C10)арила, S-(C1-C6)алкилен-(C5-C10)гетероциклила, SO-(C1-C6)алкила, SO-(C1-C6)алкилен-(C6-C10)арила, SO-(C1-C6)алкилен-(C5-C10)гетероциклила, SO2-(C1-C6)алкила, SO2-(C1-C6)алкилен-(C6-C10)арила, SO2-(C1-C6)алкилен-(C5-C10)гетероциклила, SO2-NH(C1-C6)алкилен-(C6-C10)арила, SO2-NH(C1-C6)алкилен-(C5-C10)гетероциклила, SO2-N[(C1-C6)алкил][(C1-C6)алкилен-(C6-C10)арил], SO2-N[(C1-C6)алкил][(C1-C6)алкилен-(C5-C10)гетероциклил], SO2-N[(C1-C6)алкилен-(C6-C10)арил]2, SO2-N[(C1-C6)алкилен-(C5-C10)гетероциклил]2, C(NH)(NH2), NH2, NH-(C1-C6)алкила, N[(C1-C6)алкил]2, NH-C(O)-(C1-C6)алкила, NH-C(O)O-(C1-C6)алкила, NH-C(O)-(C6-C10)арила, NH-C(O)-(C5-C10)гетероциклила, NH-C(O)O-(C6-C10)арила, NH-C(O)O-(C5-C10)гетероциклила, NH-C(O)-NH-(C1-C6)алкила, NH-C(O)-NH-(C6-C10)арила, NH-C(O)-NH-(C5-C10)гетероциклила, NH-SO2-(C1-C6)алкила, NH-SO2-(C6-C10)арила, NH-SO2-(C5-C10)гетероциклила, N(C1-C6)алкил-C(O)-(C1-C6)алкила, N(C1-C6)алкил-C(O)O-(C1-C6)алкила, N(C1-C6)алкил-C(O)-(C6-C10)арила, N(C1-C6)алкил-C(O)-гетероциклила, N(C1-C6)алкил-C(O)O-(C6-C10)арила, N(C1-C6)алкил-C(O)O-(C5-C10)гетероциклила, N(C1-C6)алкил-C(O)-NH-(C1-C6)алкил], N(C1-C6)алкил-C(O)-NH-(C6-C10)арила, N(C1-C6)алкил-C(O)-NH-(C5-C10)гетероциклила, N[(C1-C6)алкил]-C(O)-N[(C1-C6)алкил]2, N[(C1-C6)алкил]-C(O)-N[(C1-C6)алкил]-(C6-C10)арила, N[(C1-C6)алкил]-C(O)-N[(C1-C6)алкил]-(C5-C10)гетероциклила, N[(C1-C6)алкил]-C(O)-N[(C6-C10)арил]2, N[(C1-C6)алкил]-C(O)-N[(C5-C10)гетероциклил]2, N[(C6-C10)арил]-C(O)-(C1-C6)алкила, N[(C5-C10)гетероциклил]-C(O)-(C1-C6)алкила, N[(C6-C10)арил]-C(O)O-(C1-C6)алкила, N[(C5-C10)гетероциклил]-C(O)O-(C1-C6)алкила, N(арил)-C(O)-(C6-C10)арила, N[(C5-C10)гетероциклил]-C(O)-(C6-C10)арила, N[(C6-C10)арил]-C(O)O-(C6-C10)арила, N[(C5-C10)гетероциклил]-C(O)O-(C6-C10)арила, N[(C6-C10)арил]-C(O)-NH-(C1-C6)алкила, N[(C5-C10)гетероциклил]-C(O)-NH-(C1-C6)алкила, N(арил)-C(O)-NH-(C6-C10)арила, N[(C5-C10)гетероциклил]-C(O)-NH-(C6-C10)арила, N[(C6-C10)арил]-C(O)-N[(C1-C6)алкил]2, N[(C5-C10)гетероциклил]-C(O)-N[(C1-C6)алкил]2, N[(C6-C10)арил]-C(O)-N[(C1-C6)алкил]-(C6-C10)арила, N[(C5-C10)гетероциклил]-C(O)-N[(C1-C6)алкил]-(C6-C10)арила, N[(C6-C10)арил]-C(O)-N[(C6-C10)арил]2, N[(C5-C10)гетероциклил]-C(O)-N[(C6-C10)арил]2, (C6-C10)арила, (C1-C6)алкилен-(C6-C10)арила, O-(C1-C6)алкилен-(C6-C10)арила, (C5-C10)гетероциклила, (C1-C6)алкилен-(C5-C10)гетероциклила, O-(C1-C6)алкилен-(C5-C10)гетероциклила, где (C6-C10)арил или (C5-C10)гетероциклил может быть замещен от одного до 3 раз галогеном, OH, NO2, CN, O-(C1-C6)алкилом, (C1-C6)алкилом, NH2, NH(C1-C6)алкилом, N[(C1-C6)алкил]2, SO2CH3, COOH, C(O)O-(C1-C6)алкилом, CONH2, (C1-C6)алкилен-O-(C1-C6)алкилом, (C1-C6)алкилен-O-(C6-C10)арилом, O-(C1-C6)алкилен-(C6-C10)арилом; или где (C6-C10)арил является вицинально замещенным O-(C1-C4)алкилен-O группой с образованием, таким образом, 5-8-членного кольца вместе с атомами углерода, с которыми связываются атомы кислорода. Арильные или гетероциклильные заместители (C6-C10)арильных и (C5-C10)гетероциклильных групп могут не быть дополнительно замещенными арил- или гетероциклилсодержащей группой.
Предпочтительными заместителями для (C6-C10)арильных групп являются (C1-C4)алкил, O-(C1-C4)алкил, O-фенил, C(O)O-(C1-C6)алкил, C(O)OH, C(O)-(C1-C4)алкил, галоген, NO2, SO2NH2, CN, SO2-(C1-C4)алкил, NH-SO2-(C1-C4)алкил, NH2, NH-C(O)-(C1-C4)алкил, (C3-C8)циклоалкил, (C1-C4)алкил-OH, C(O)N[(C1-C4)алкил]2, C(O)NH2, N[(C1-C4)алкил]2, (C1-C4)алкенилен-(C6-C10)арил, где (C6-C10)арил может быть дополнительно замещен (C1-C4)алкилом, (C1-C4)алкилен-O-(C1-C6)алкилом, O-(C1-C6)алкил-(C6-C10)арилом или может быть вицинально замещен O-(C1-C4)алкилен-O группой с образованием, таким образом, 5-8-членного кольца вместе с атомами углерода, с которыми связываются атомы кислорода.
В монозамещенных фенильных группах заместитель может находиться в 2-положении, 3-положении или 4-положении, предпочтительно, в 3-положении и 4-положении. Если фенильная группа содержит два заместителя, они могут находиться в 2,3-положении, 2,4-положении, 2,5-положении, 2,6-положении, 3,4-положении или 3,5-положении. В фенильных группах с тремя заместителями заместители могут находиться в 2,3,4-положении, 2,3,5-положении, 2,3,6-положении, 2,4,5-положении, 2,4,6-положении или 3,4,5-положении.
Приведенные выше указания, относящиеся к фенильным группам, соответственно применимы к двухвалентным группам, являющимся производными фенильных групп, т.е. к фенилену, который может представлять собой незамещенный или замещенный 1,2-фенилен, 1,3-фенилен или 1,4-фенилен. Приведенные выше указания также соответственно применимы к арильной подгруппе в арилалкиленовых группах. Примерами арилалкиленовых групп, которые также могут быть незамещенными или замещенными как в арильной подгруппе, так и в алкиленовой подгруппе, являются бензил, 1-фенилэтилен, 2-фенилэтилен, 3-фенилпропилен, 4-фенилбутилен, 1-метил-3-фенилпропилен.
Предпочтительными заместителями для (C5-C10)гетероциклильных групп являются (C1-C4)алкил, O-(C1-C4)алкил, (C1-C4)алкилен-фенил, галоген, (C1-C4)алкилен-O-(C1-C4)алкил, (C5-C10)гетероциклил, (C1-C4)алкилен-N[(C1-C4)алкил]2 или (C6-C10)арил, где (C6-C10)арил может быть дополнительно замещен (C1-C4)алкилом, (C1-C4)алкилен-O-(C1-C6)алкилом, O-(C1-C6)алкил-(C6-C10)арилом или может быть вицинально замещен O-(C1-C4)алкилен-O группой с образованием, таким образом, 5-8-членного кольца вместе с атомами углерода, с которыми связываются атомы кислорода.
Основные и предпочтительные заместители (C6-C10)арильных и (C5-C10)гетероциклильных групп могут быть объединены с основными и предпочтительными определениями R1, R2, R3, R4, R5, R6, R7, R8, n и L, как описано выше.
Настоящее изобретение, следовательно, также относится к соединениям формулы (I) и/или их физиологически приемлемым солям и/или их пролекарствам для применения в качестве фармацевтических средств (или лекарственных средств), к применению соединений формулы (I) и/или их физиологически приемлемых солей и/или их пролекарств для получения фармацевтических средств для лечения и/или профилактики заболеваний, связанных с Rho-киназой и/или опосредованным Rho-киназой фосфорилированием фосфатазы легкой цепи миозина, т.е. для лечения и/или профилактики гипертензии, например легочной гипертензии и глазной гипертензии, нарушения периферического кровообращения, стенокардии, спазмов сосудов мозга, астмы, преждевременных родов, гиперагрегации тромбоцитов, окклюзивного заболевания периферических артерий (PAOD), хронического обструктивного заболевания легких (COPD), развития рака, эректильной дисфункции, атеросклероза, ишемического поражения органов, фиброза легкого, фиброза печени, печеночной недостаточности, фиброзной опухоли почек, почечного гломерулосклероза, почечной недостаточности, гиперторофии органов, гипертрофии простаты, осложнений диабета, рестеноза кровеносных сосудов, атеросклероза, рака, сердечной гипертрофии, сердечной недостаточности; ишемических заболеваний; воспалений; аутоиммунных заболеваний; СПИДа, остеопатии, такой как остеопороз, нарушений функций мозга, бактериальных инфекций пищеварительного тракта, сепсиса, респираторного дистресс-синдрома взрослых, ретинопатии, глаукомы и болезни Альцгеймера.
Настоящее изобретение, кроме того, относится к фармацевтическим препаратам (или фармацевтическим композициям), которые содержат эффективное количество, по меньшей мере, одного соединения формулы (I) и/или его физиологически приемлемых солей и/или его пролекарств и фармацевтически приемлемый носитель, т.е. один или несколько фармацевтически приемлемых материалов носителя (или растворителей) и/или добавки (или эксципиенты).
Фармацевтические средства можно вводить орально, например в форме пилюль, таблеток, таблеток с глянцевым покрытием, покрытых оболочкой таблеток, гранул, твердых и мягких желатиновых капсул, растворов, сиропов, эмульсий, суспензий или аэрозольных смесей. Однако введение также может быть ректальным, например в форме суппозиториев, или парентеральным, например внутривенным, внутримышечным или подкожным, в форме инъекционных растворов или инфузионных растворов, микрокапсул, имплантатов или штифтов, или подкожным или местным, например в форме мазей, растворов или настоек, или другими способами, например в форме аэрозолей или назальных спреев.
Фармацевтические препараты согласно настоящему изобретению получают способом, известным per se и известным специалистам в данной области техники, с использованием фармацевтически приемлемых инертных неорганических и/или органических веществ-носителей и/или или добавок в дополнение к соединению(ям) формулы (I) и/или его (их) физиологически приемлемым солям и/или его (их) пролекарствам. Для получения пилюль, таблеток, таблеток с глянцевым покрытием, покрытых оболочкой таблеток, гранул, твердых и мягких желатиновых капсул можно использовать, например, лактозу, кукурузный крахмал или их производные, тальк, стеариновую кислоту или ее соли и т.п. Вещества-носители для мягких желатиновых капсул и суппозиториев представляют собой, например, жиры, воски, полутвердые и жидкие полиолы, натуральные или отвержденные масла и т.п. Подходящие вещества-носители для получения растворов представляют собой, например, инъекционные растворы, или эмульсии, или сиропы, например вода, насыщенный солевой раствор, спирты, глицерин, полиолы, сахароза, инвертированные сахара, глюкоза, растительные масла и т.п. Подходящие вещества-носители для микрокапсул, имплантов или штифтов представляют собой, например, сополимеры гликолевой кислоты и молочной кислоты. Фармацевтические препараты обычно содержат от около 0,5 до около 90 мас.% соединений формулы (I) и/или их физиологически приемлемых солей и/или их пролекарств. Количество активного ингредиента формулы (I) и/или его физиологически приемлемых солей и/или его пролекарств в фармацевтических препаратах обычно составляет от около 0,5 до около 1000 мг, предпочтительно от около 1 до около 500 мг.
В дополнение к активным ингредиентам формулы (I) и/или их физиологически приемлемым солям и/или пролекарствам и к веществам-носителям фармацевтические препараты могут содержать одну или более добавок, таких как, например, наполнители, дезинтегранты, связующие вещества, лубриканты, увлажняющие вещества, стабилизаторы, эмульгаторы, консерванты, подсластители, красители, отдушки, ароматизаторы, загустители, разбавители, буферные вещества, растворители, солюбилизаторы, агенты для достижения эффекта депо, соли для изменения осмотического давления, агенты для покрытия или антиоксиданты. Они также могут содержать два или более соединений формулы (I) и/или их физиологически приемлемых солей и/или их пролекарств. В случае если фармацевтический препарат содержит два или более соединений формулы (I), выбор конкретных соединений может осуществляться с целью получения специфического общего фармакологического профиля фармацевтического препарата. Например, высокоактивное соединение с более короткой продолжительностью действия можно сочетать с соединением продолжительного действия с более низкой активностью. Допускаемая гибкость в отношении выбора заместителей соединений формулы (I) обеспечивает возможность более лучшего контроля биологических и физико-химических свойств соединений и, таким образом, обеспечивает возможность отбора таких желаемых соединений. Более того, в дополнение к, по меньшей мере, одному соединению формулы (I) и/или его физиологически приемлемым солям и/или его пролекарствам фармацевтические препараты также могут содержать один или несколько других терапевтически или профилактически активных ингредиентов.
При использовании соединений формулы (I) доза может варьировать в широком диапазоне и обычно, как это известно лечащим врачам, должна быть подходящей для конкретных условий в каждом конкретном случае. Это зависит, например, от конкретного применяемого соединения, от природы и тяжести заболевания, подвергаемого лечению, от способа и схемы введения, или от того, является ли заболевание острым или хроническим, или от того, проводилась ли профилактика. Подходящую дозу можно определить с использованием клинических подходов, хорошо известных в области медицины. Как правило, суточная доза для достижения желаемых результатов для взрослых с массой тела около 75 кг составляет от около 0,01 до около 100 мг/кг, предпочтительно от около 0,1 до около 50 мг/кг, в частности от около 0,1 до около 10 мг/кг (в каждом случае в мг на кг массы тела). Суточную дозу можно разделить, в частности, в случае введения относительно больших количеств, на несколько, например 2, 3 или 4, дробных введений. Как правило, в зависимости от индивидуального поведения может быть необходимым отклонение от указанной суточной дозы в большую или меньшую сторону.
Кроме того, соединения формулы (I) можно использовать в качестве промежуточных соединений синтеза для получения других соединений, в частности других фармацевтически активных ингредиентов, которые можно получить из соединений формулы I, например, путем введения заместителей или модификации функциональных групп.
Соединения формулы (I) можно получить в соответствии со следующими примерами соединений, не ограничивая объема притязаний.
Как правило, защитные группы, которые все еще могут присутствовать в продуктах, полученных в реакции сочетания, затем можно удалить, используя стандартные процедуры. Например, можно снять защиту трет-бутильных защитных групп, в частности трет-бутоксикарбонильной группы, которая представляет собой защищенную форму амидиногруппы, т.е. преобразовать в амидиногруппу, путем обработки трифторуксусной кислотой. Как объяснялось выше, после реакции сочетания могут также образовываться функциональные группы из подходящих групп-предшественников. Кроме того, затем можно осуществить преобразование в физиологически приемлемую соль или пролекарство соединения формулы (I) известными способами.
Как правило, реакционную смесь, содержащую конечное соединение формулы (I) или промежуточное соединение, подвергают обработке и, если это желательно, продукт затем очищают традиционными способами, известными специалистам в данной области. Например, синтезированное соединение можно очистить, используя хорошо известные способы, такие как кристаллизация, хроматография или обращенно-фазовая высокоэффективная жидкостная хроматография (ОФ-ВЭЖХ), или другими способами разделения, основанными, например, на размере, заряде или гидрофобности соединения. Аналогичным образом, можно использовать хорошо известные способы, такие как анализ аминокислотной последовательности, ЯМР, ИК и масс-спектрометрия (МС), для идентификации соединения по настоящему изобретению.
Должно быть понятно, что модификации, существенно не влияющие на активность различных вариантов воплощения настоящего изобретения, включены в объем изобретения, раскрываемого в настоящей заявке. Соответственно, представленные ниже примеры предназначены для иллюстрации, а не для ограничения настоящего изобретения.
Методы ЖХМС
Способ №1
Колонка: YMC J'shere 33×2 4 мкм
градиент (AcN+0,05% ТФУК): H2O+0,05% ТФУК; 5:95 (0 мин) до 95:5 (2,5 мин) до 95:5 (3 мин)
Способ №2
Колонка: YMC J'shere 33x2 4 мкм
градиент (AcN+0,05% ТФУК): H2O+0,05% ТФУК, 5:95 (0 мин) до 95:5 (3,4 мин) до 95:5 (4,4 мин)
Способ №3
Колонка: YMC J'shere 33x2 4 мкм
градиент AcN+0,08% ФУК: H2O+0,1% ФУК; 5:95 (0 мин) до 95:5(2,5 мин) до 95:5(3 мин)
Способ № Top
Колонка: YMC YMC J`sphere ODS H80 20×2 14 мкм
градиент 0 мин 96% H2O(0,05% ТФУК) 2,0 мин - 95% ACN; 95% ACN повторно 2,4 мин; 4% ACN 2,45 мин
Синтез основной структуры
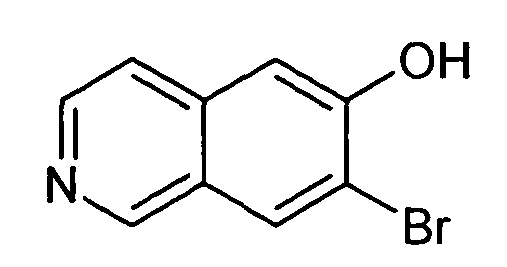
7-Бромизохинолин-6-ол (1)
25 г (116,3 ммоль) 3-бром-4-метоксибензальдегида, 19,0 мл (18,3 г, 174,5 ммоль) аминоацетальдегиддиметилацеталя и 250 мл толуола нагревали до температуры кипения с обратным холодильником в течение 6 ч, используя аппарат Дина-Старка. Растворитель и избыток реагента отгоняли и неочищенный продукт (приблизит. 37 г) использовали на следующей стадии без дополнительной очистки.
Имин растворяли в 240 мл ТГФ. Добавляли по каплям 11,1 мл (12,6 г, 116,3 ммоль) этилхлорформиата при 0°C. После перемешивания в течение 5 минут добавляли по каплям 24,3 мл (23,2 г, 139,2 ммоль) триэтилфосфита. Смесь перемешивали в течение 18 ч при комнатной температуре. Затем растворители отгоняли. Избыточное количество реагента удаляли повторным добавлением 100 мл толуола и выпариванием растворителей. P,N-ацеталь (приблизит. 62 г ) использовали на следующей стадии без дополнительной очистки.
P,N-ацеталь, 51,3 мл (88,2 г, 465,2 ммоль) тетрахлорида титана и 300 мл хлороформа нагревали до температуры кипения с обратным холодильником в течение 48 ч. Смесь выливали на лед и pH доводили до 9, используя водный раствор аммиака. Повторная экстракция этилацетатом с последующим удалением растворителей давала в результате 14,8 г (53%) 7-бром-6-метоксиизохинолина.
1H-ЯМР (d6-ДМСО): δ = 9,16 (1H, с), 8,46 (1H, д, J=5,9 Гц), 8,46 (1H, с), 7,76 (1H, д, J=5,9 Гц), 7,51 (1H, с), 4,01 (3H, с).
MS: m/z = 238 (MH+).
3,6 мл (9,5 г, 37,8 ммоль) BBr3 добавляли при 0°C к раствору 4,5 г (18,9 ммоль) 7-бром-6-метоксиизохинолина в 30 мл дихлорметана и перемешивали в течение 18 ч при комнатной температуре. Водный раствор NaHCO3 добавляли до достижения pH 8. Экстракция хлороформом/изопропанолом (3/1) с последующей сушкой над сульфатом натрия и удаление растворителей давали в результате 2,7 г (64%) соединения 1.
1H-ЯМР (d6-ДМСО): δ = 9,19 (1H, с), 8,49 (1H, с), 8,38 (1H, д, J=6,1 Гц), 7,78 (1H, д, J=6,1 Гц), 7,34 (1H, с).
MS: m/z = 224 (MH+).
Синтезировали следующие промежуточные соединения, используя данную процедуру:
8-Фторизохинолин-6-ол (2)
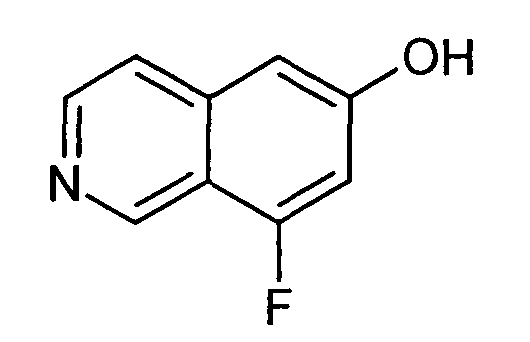
1H-ЯМР (d6-ДМСО): δ = 10,84 (1H, с), 9,21 (1H, с), 8,40 (1H, д, J=5,8 Гц), 7,67 (1H, д, J=5,8 Гц), 7,01 (2H, м).
MS: m/z = 164 (MH+).
7-Фторизохинолин-6-ол (3)
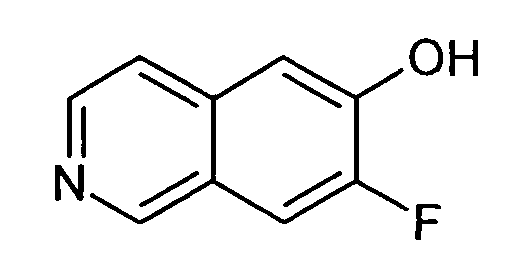
1H-ЯМР (d6-ДМСО): δ = 11,06 (1H, с), 9,07 (1H, с), 8,33 (1H, д, J=5,6 Гц), 7,88 (1H, д, J=11,4 Гц), 7,64 (1H, д, J=5,6 Гц), 7,31 (1H, д, J=8,6 Гц).
MS: m/z = 164 (MH+).
8-Метилизохинолин-6-ол (4)
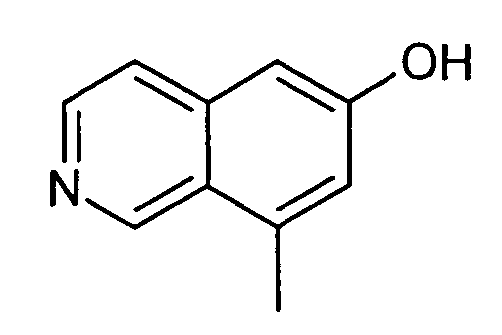
1H-ЯМР (d6-ДМСО): δ = 11,55 (1H, с), 9,47 (1H, с), 8,42 (1H, д, J=6,5 Гц), 8,11 (1H, д, J=6,5 Гц), 7,31 (1H, с), 7,25 (1H, с), 2,76 (3H, с).
MS: m/z = 160 (MH+).
7,8-Диметилизохинолин-6-ол (5)
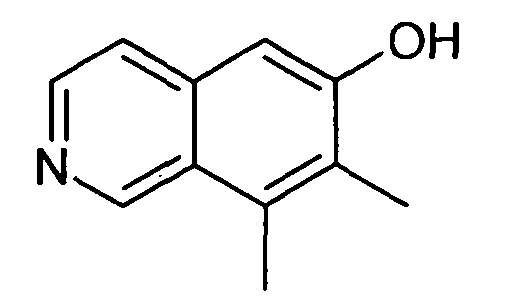
1H-ЯМР (d6-ДМСО): δ = 11,87 (1H, с), 9,58 (1H, с), 8,41 (1H, д, J=6,5 Гц), 8,18 (1H, д, J=6,5 Гц), 7,35 (1H, с), 7,25 (1H, с), 2,71 (3H, с), 2,35 (3H, с).
MS: m/z = 174 (MH+).
5,8-Диметилизохинолин-6-ол (6)
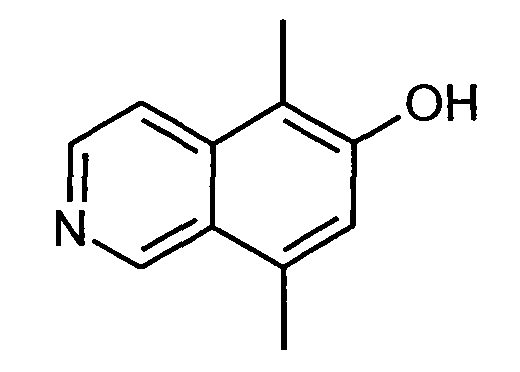
1H-ЯМР (d6-ДМСО): δ = 11,55 (1H, с), 9,52 (1H, с), 8,47 (1H, д, J=6,8 Гц), 8,26 (1H, д, J=6,8 Гц), 7,42 (1H, с), 2,76 (3H, с), 2,42 (3H, с).
MS: m/z = 174 (MH+).
6-Гидроксиизохинолин (7)
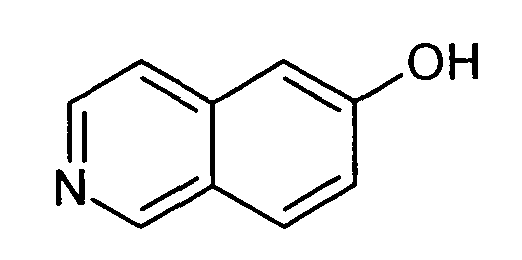
ЖХМС Способ № 1, время удерживания 0,14 мин, определенная масса 146,08 [M+H]+
5-Хлоризохинолин-6-ол (8)
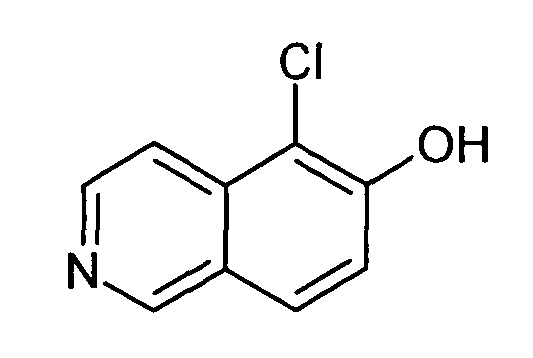
0,61 мл (1,02 г, 7,6 ммоль) сульфурилхлорида добавляли к раствору 1,0 г (6,9 ммоль) соединения 7 в 30 мл дихлорметана. Добавляли три капли диэтилового эфира и реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Растворители удаляли с помощью дистилляции и остаток обрабатывали водным раствором NaHCO3. Осадок отфильтровывали, промывали водой и сушили с получением 1,1 г (89%) соединения 8 в виде твердого вещества желто-зеленого цвета.
1H-ЯМР (d6-ДМСО): δ = 11,37 (1H, с), 9,18 (1H, с), 8,50 (1H, д, J=6 Гц), 8,00 (1H, д, J= 8,8 Гц), 7,83 (1H, J=6 Гц), 7,44 (1H, д, J=8,7 Гц).
MS: m/z = 180 (MH+).
5-Бромизохинолин-6-ол (9)
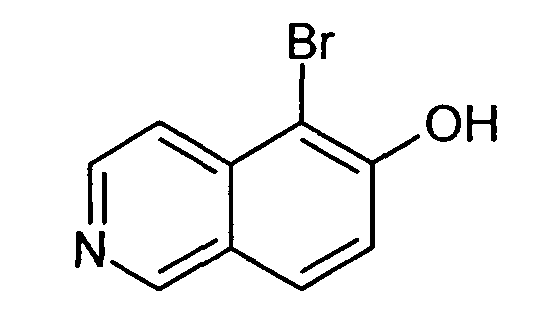
7,9 мл (19,18 г, 120 ммоль) брома добавляли по каплям к суспензии 17,42 г (120 ммоль) соединения 7 в 250 мл хлороформа при комнатной температуре. После перемешивания в течение 2 ч добавляли этилацетат. Осадок отфильтровывали, промывали этилацетатом и сушили. Осторожно добавляли водный раствор NaHCO3. Осадок отфильтровывали и промывали водным раствором NaHCO3 до достижения pH фильтрата, равного 8. Сушка давала в результате 23,78 г (88%) соединения 9 в виде твердого вещества беловатого цвета.
1H-ЯМР (d6-ДМСО): δ = 11,30 (1H, с), 9,13 (1H, с), 8,48 (1H, д, J=5,9 Гц), 8,02 (1H, д, J= 8,8 Гц), 7,78 (1H, J=5,9 Гц), 7,40 (1H, д, J=8,8 Гц).
MS: m/z = 224 (MH+).
5-Йодизохинолин-6-ол (10)
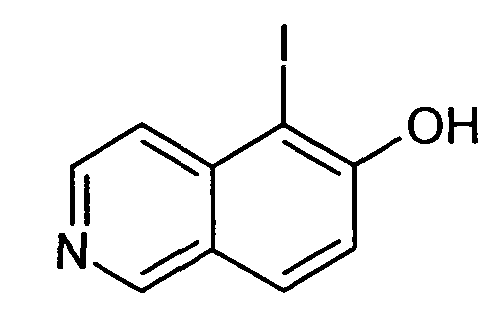
В атмосфере аргона 1,77 г (12,2 ммоль) соединения 7 добавляли к раствору 5,0 г (13,5 ммоль) бис(пиридин)йодонийтетрафторбората в 100 мл сухого дихлорметана. Добавляли по каплям раствор 2,4 мл (4 г, 26,8 ммоль) трифторметансульфоновой кислоты в 20 мл сухого дихлорметана при 0°C и смесь перемешивали в течение 3 часов при комнатной температуре. Растворители удаляли с помощью дистилляции и остаток обрабатывали водным раствором NaHCO3. Осадок отфильтровывали, промывали водой и сушили с получением 3,2 г (97%) соединения 10 в виде твердого вещества бежевого цвета.
1H-ЯМР (d6-ДМСО): δ = 9,09 (1H, с), 8,47 (1H, д, J=6,1 Гц), 8,04 (1H, д, J= 8,8 Гц), 7,76 (1H, J=6,1 Гц), 7,37 (1H, д, J=8,8 Гц).
MS: m/z = 272 (MH+).
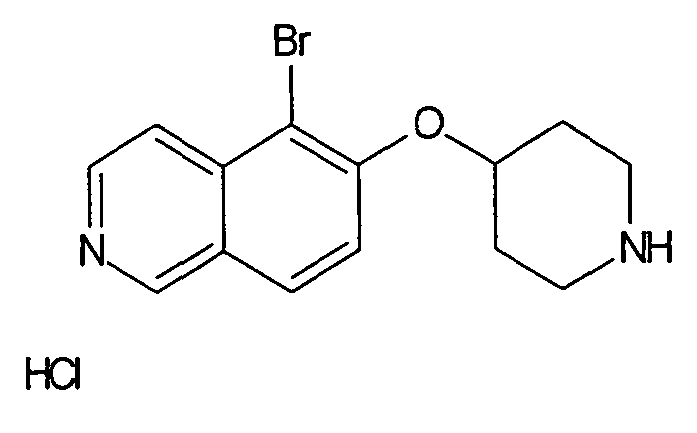
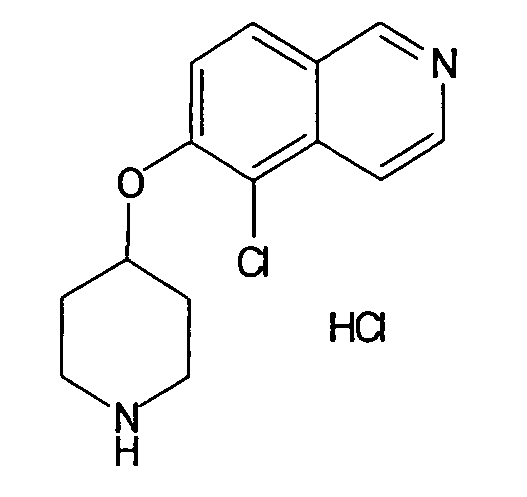
Трет-бутиловый эфир 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11)
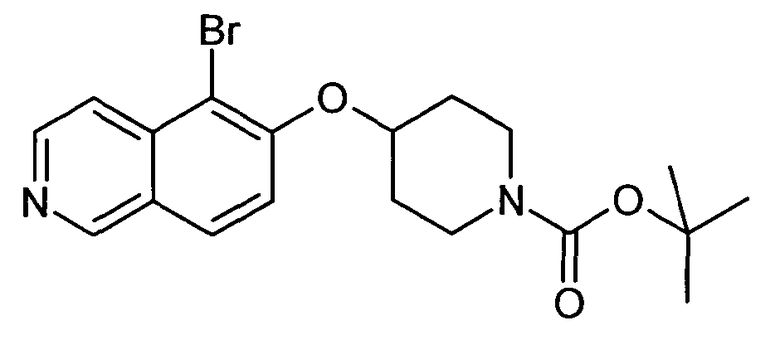
3,75 мл (4,15 г, 23,8 ммоль) диэтилазодикарбоксилата добавляли к 12,7 г (19,9 ммоль) связанного на полимере трифенилфосфина (PS-PPh3, приблизит. 1,6 ммоль/г, Argonaut) в 250 мл дихлорметана при 0°C и перемешивали в течение 15 мин. Добавляли 4,45 г (19,9 ммоль) 5-бромизохинолин-6-ола (9), 4,0 г (19,9 ммоль) Boc-(4-гидрокси)пиперидина и 4,1 мл (3,0 г, 29,8 ммоль) триэтиламина. Смесь встряхивали в течение 16 ч. Полимер удаляли фильтрацией через целит и растворители отгоняли. Добавляли 20 мл дихлорметана и осадок выделяли фильтрацией. Неочищенный продукт (8 г) очищали с помощью флэш-хромотографии с использованием этилацетата/н-гептана в качестве элюента с получением 4,78 г (60%) соединения 11.
1H-ЯМР (d6-ДМСО): δ = 9,24 (1H, с), 8,97 (1H, с), 8,56 (1H, д, J=6 Гц), 8,20 (1H, д, J=9 Гц), 7,85 (1H, д, J= 6 Гц), 7,75 (1H, д, J=9 Гц), 5,02 (1H, м), 3,58 (2H, м), 3,40 (2H, м), 1,91 (2H, м), 1,70 (2H, м), 1,41 (9H, с).
MS: m/z = 407 (MH+).
Следующие основные структуры синтезировали согласно данному способу:
Трет-бутиловый эфир 4-(5-иодизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (12)
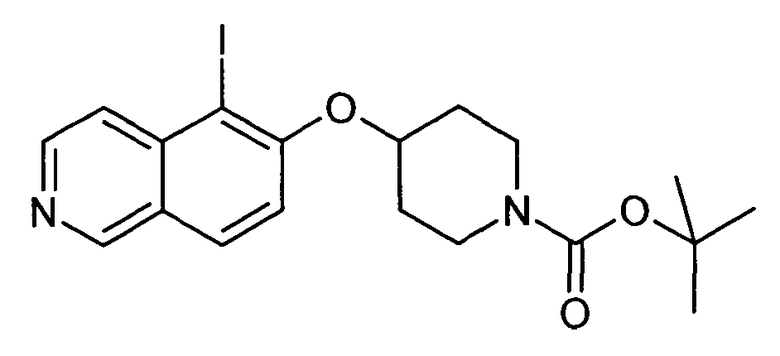
используя соединение 10 в качестве исходного вещества
1H-ЯМР (CDCl3): δ = 9,04 (1H, с), 8,55 (1H, д, J=6 Гц), 7,93 (1H, д, J=9 Гц), 7,86 (1H, д, J=6 Гц), 7,27 (1H, д, J= 9 Гц), 4,87 (1H, м), 3,66 (4H, м), 1,93 (4H, м), 1,48 (9H, с).
MS: m/z = 455 (MH+).
Трет-бутиловый эфир 4-(7-Бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (13)
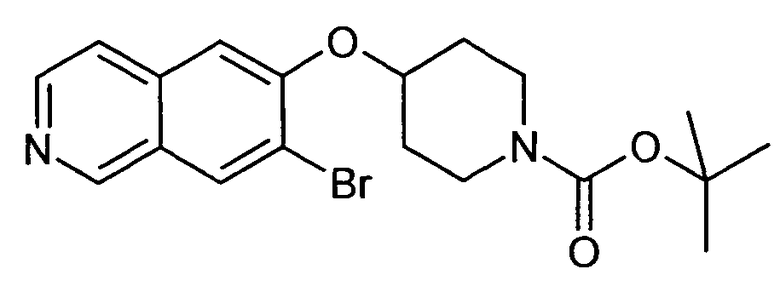
используя соединение 1 в качестве исходного вещества
ЖХМС Способ № 4, время удерживания 1,13 мин, определенная масса 407,4 [M+H]+
Трет-бутиловый эфир 4-[5-(4,4,5,5-Тетраметил-[1,3,2]диоксаборолан-2-ил)изохинолин-6-илокси]пиперидин-1-карбоновой кислоты (14)
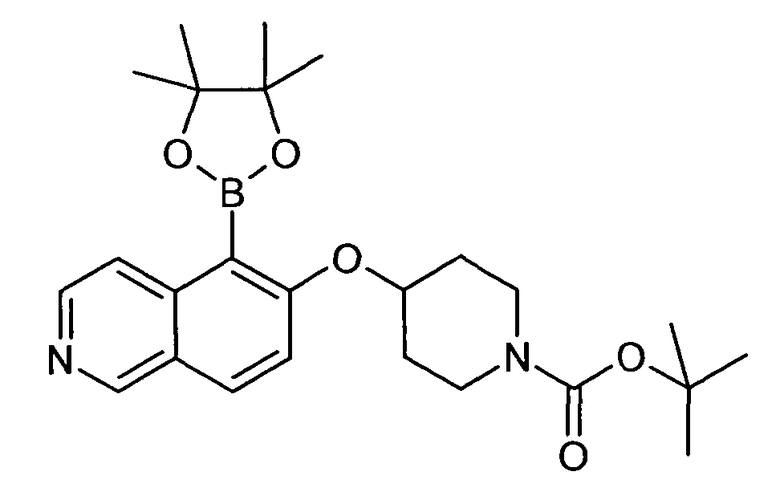
Раствор 0,55 г (1,34 ммоль) трет-бутилового эфира 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11) в 14 мл ДМСО добавляли к смеси 1,0 г (4,0 ммоль) бис(пинаколато)диборона, 0,78 г (8,0 ммоль) K2CO3 и 29 мг (0,03 экв.) Pd(dppf)Cl2. Через смесь барботировали аргон в течение 30 мин и затем реакционную смесь нагревали в микроволновом реакторе (CEM Discovery) до 100°C в течение 60 мин. После охлаждения до комнатной температуры добавляли воду. Смесь экстрагировали этилацетатом. После удаления растворителя продукт выделяли с помощью флэш-хроматографии (этилацетат/н-гептан) с получением 269 мг (44%) соединения 14 в виде твердого вещества белого цвета.
ЖХМС Способ № 4, время удерживания 1,30 мин, определенная масса 433,3 [M+H]+
Трет-бутиловый эфир 4-(5-цианоизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (15)
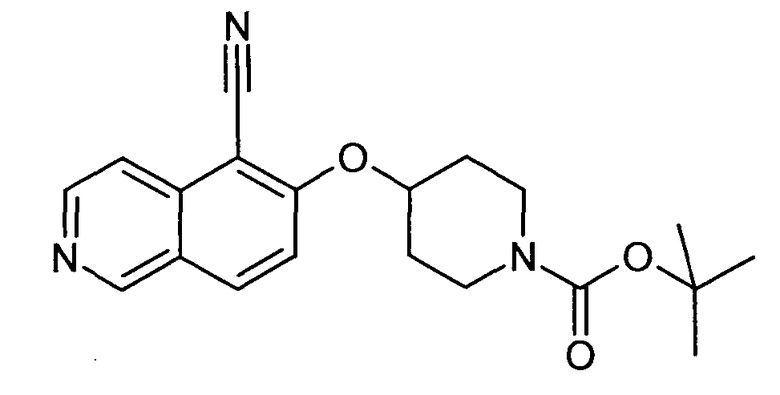
В атмосфере аргона 47 мг (0,4 ммоль) Zn(CN)2 и 23 мг (0,02 экв.). Pd(PPh3)4 добавляли к раствору 62 мг (0,4 ммоль) трет-бутилового эфира 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11) в ДМФ. Реакционную смесь нагревали в течение 5 минут до 150°C в микроволновом реакторе (CEM Discovery). После охлаждения до комнатной температуры добавляли воду и этилацетат. Смесь фильтровали через целит, промывали этилацетатом и концентрировали с получением 176 мг соединения 15.
MS: m/z = 354 (MH+).
Трет-бутиловый эфир 4-(7-Цианоизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (16)
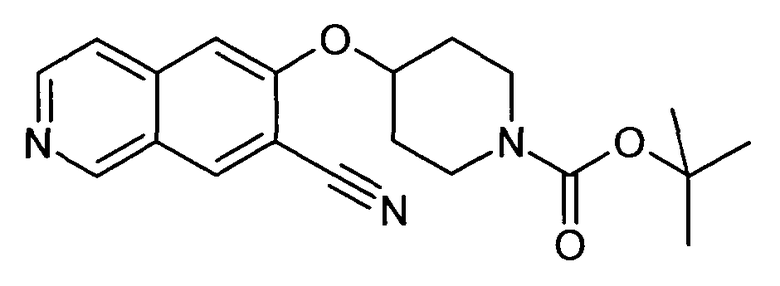
В атмосфере аргона 35 мг (0,3 ммоль) Zn(CN)2 и 17 мг (0,05 экв.). Pd(PPh3)4 добавляли к раствору 122 мг (0,3 ммоль) трет-бутилового эфира 4-(7-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (13) в ДМФ. Реакционную смесь нагревали в течение 5 минут до 150°C в микроволновом реакторе (CEM Discovery). После охлаждения до комнатной температуры добавляли воду и этилацетат. Смесь фильтровали через целит, промывали этилацетатом и концентрировали. Неочищенный продукт очищали препаративной ВЭЖХ с получением 77 мг соединения 16.
ЖХМС Способ № 4, время удерживания 1,06 мин, определенная масса 354,5 [M+H]+
Трет-бутиловый эфир 4-(5-азидоизохинолин-6-илокси)пиперидин1-карбоновой кислоты (17)

В атмосфере аргона 40 мкл (0,04 ммоль) 1 н. NaOH, 4,6 мг (0,04 ммоль) L-пролина, 3,8 мг (0,02 ммоль) CuI и 15,6 мг (0,24 ммоль) NaN3 добавляли к раствору 91 мг (0,2 ммоль) трет-бутилового эфира 4-(5-йодизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (12) в 2 мл ДМСО. Смесь нагревали до 60°C в течение 18 ч. Снова добавляли NaN3, NaOH и L-пролин в тех же количествах и реакционную смесь нагревали до 60°C в течение 5 ч. После охлаждения до комнатной температуры добавляли воду. Осадок отфильтровывали, промывали водой и сушили в вакууме с получением 74 мг соединения 17, которое использовали без дополнительной очистки.
MS: m/z = 370 (MH+).
Трет-бутиловый эфир 4-(5-Аминоизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (18)
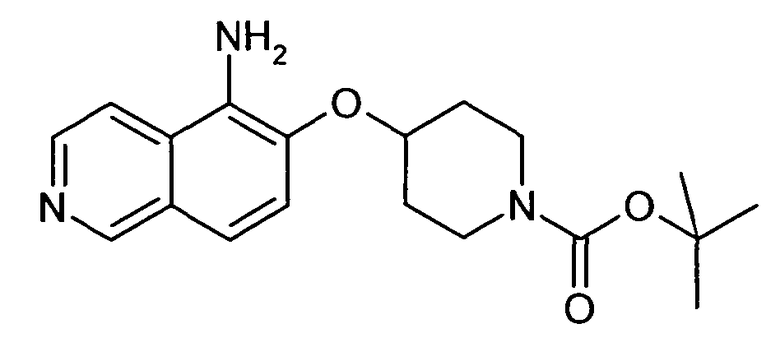
В атмосфере аргона 600 мкл (0,6 ммоль) 1 н. NaOH, 13,8 мг (0,12 ммоль) L-пролина, 7,6 мг (0,04 ммоль) CuI и 52 мг (0,8 ммоль) NaN3 добавляли к раствору 163 мг (0,4 ммоль) трет-бутилового эфира 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11) в 0,6 мл воды. Смесь нагревали до 95°C в течение 3 ч в микроволновом реакторе (CEM Discovery). После охлаждения до комнатной температуры добавляли воду и этилацетат. Смесь фильтровали через целит, промывали этилацетатом и концентрировали. Неочищенный продукт очищали препаративной ВЭЖХ с получением 42 мг соединения 18 (содержащего некоторое количество 11 в виде примеси)
ЖХМС Способ № 4, время удерживания 0,97 мин, определенная масса 344,5 [M+H]+
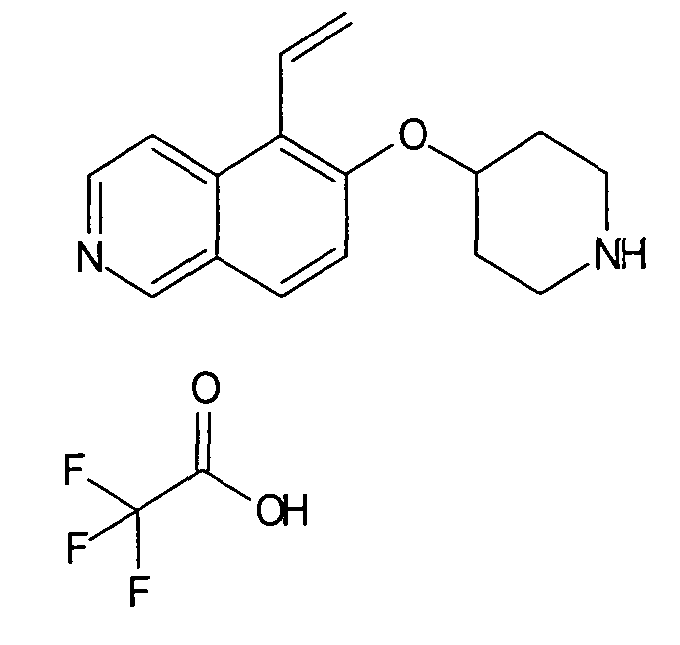
Трет-бутиловый эфир 4-(7-винилизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (19)
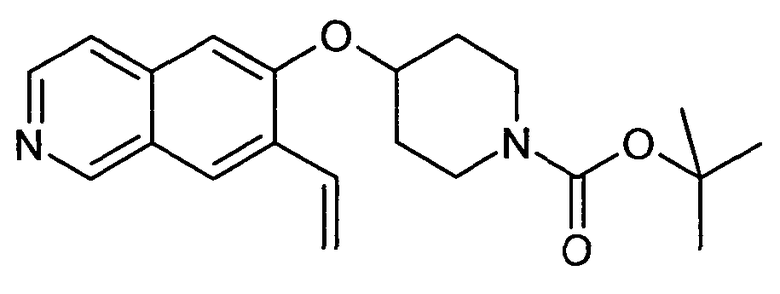
В атмосфере аргона 340 мг трибутилвинилстаннана (1,07 ммоль, 1,2 экв.) и 103 мг Pd(PPh3)4 (0,1 экв.) добавляли к раствору 364 мг трет-бутилового эфира 4-(7-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (13) (0,98 ммоль) в 4 мл толуола. Реакционную смесь нагревали до 100°C в микроволновом реакторе (CEM Discovery) в течение 1 ч.
После охлаждения до комнатной температуры добавляли воду и этилацетат. Смесь фильтровали через картридж с целитом, промывали этилацетатом и концентрировали. Неочищенный продукт очищали препаративной ВЭЖХ с получением 256 мг (81%) соединения 19.
ЖХМС Способ № 4, время удерживания 1,19 мин, определенная масса 355,5 [M+H]+
Следующие структуры синтезировали согласно данному способу:
Трет-бутиловый эфир 4-(5-Винилизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (19A)
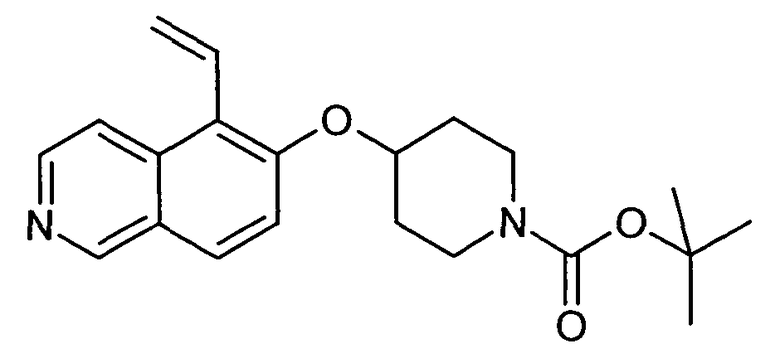
используя соединение 11 в качестве исходного вещества
ЖХМС Способ № 4, время удерживания 1,11 мин, определенная масса 355,4 [M+H]+
Трет-бутиловый эфир 4-(7-тиофен-2-илизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (20)
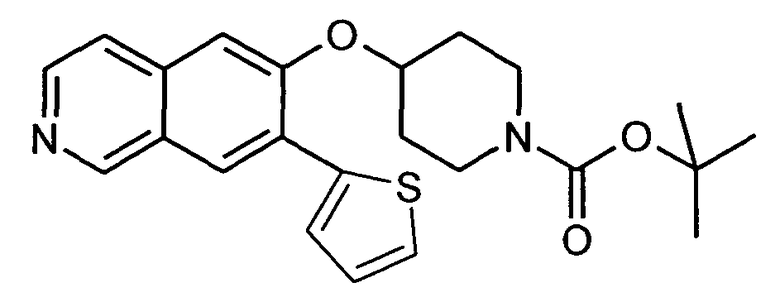
используя соединение 13 и трибутилтиофен-2-илстаннан в качестве исходных материалов.
ЖХМС Способ № 4, время удерживания 1,26 мин, определенная масса 411,5 [M+H]+
(2,2-Диметоксиэтил)-(4-фторбензил)амин (21)
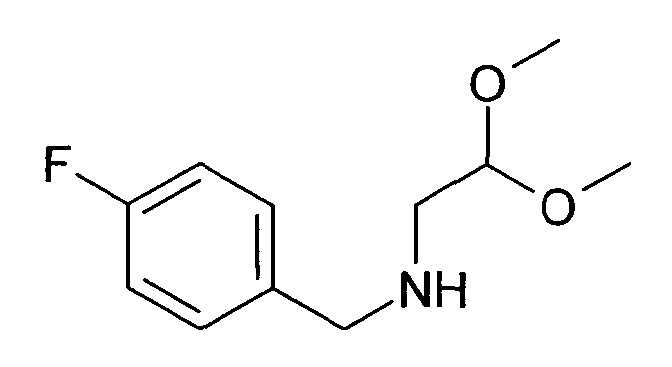
12,4 г 4-Фторбензальдегида растворяли в 100 мл толуола и подвергали взаимодействию с 10,5 г 2-аминоацетальдегиддиметилацеталя и 1,90 г (10 ммоль) моногидрата п-толуолсульфоновой кислоты в течение двух часов в аппарате Дина-Старка. Раствору давали охладиться, экстрагировали при помощи насыщенного раствора бикарбоната натрия, воды и насыщенного солевого раствора, сушили над сульфатом магния и упаривали досуха. Неочищенный продукт растворяли в 100 мл этанола. Добавляли по порциям 1,89 г борогидрида натрия. Перемешивание продолжали в течение ночи. Для обработки добавляли уксусную кислоту до тех пор, пока не наблюдали прекращение выделения газа. Затем раствор упаривали досуха, забирали в дихлорметан и дважды промывали водой. Органический слой экстрагировали насыщенным солевым раствором, сушили над сульфатом магния и упаривали досуха. Полученный неочищенный продукт (20 г) использовали для следующих реакций без очистки. Rt=0,86 мин (Способ №1). Определенная масса: 182,1 (M-OMe-), 214,2 (M+H+).
N-(2,2-Диметоксиэтил)-N-(4-фторбензил)-4-метил-бензолсульфонамид (22)
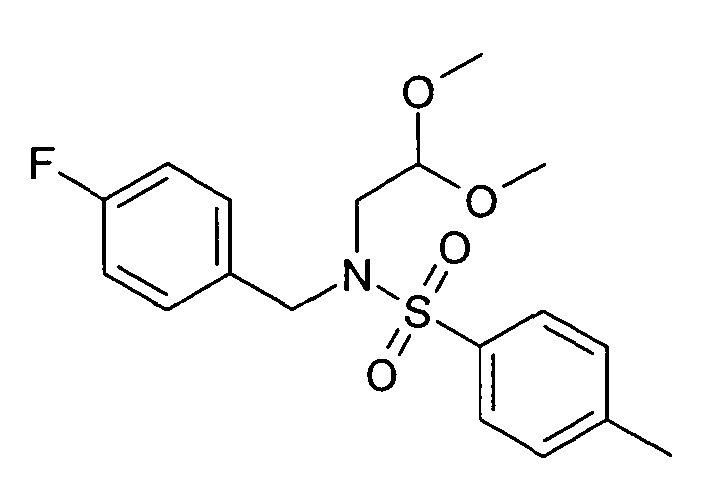
20 г (2,2-Диметоксиэтил)-(4-фторбензил)амина (21) растворяли в 120 мл дихлорметана. Добавляли 20 мл пиридина. При 0°C добавляли по каплям раствор 23,8 г хлорида п-толуолсульфоновой кислоты в дихлорметане. Реакционную смесь нагревали до комнатной температуры и продолжали перемешивание до завершения превращения. Для обработки реакционную смесь дважды экстрагировали 2 M хлористоводородной кислотой, дважды бикарбонатом натрия и один раз насыщенным солевым раствором. Органический слой сушили над сульфатом магния, упаривали досуха и полученный неочищенный продукт очищали хроматографией на силикагеле с получением 22,95 г соединения 22 в виде оранжевого масла. Rt=1,71 мин (Способ № 4). Определенная масса: 336,1 (M-OMe-).
6-Фторизохинолин (23)
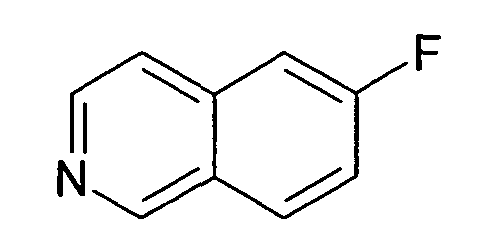
41,6 г AlCl3 суспендировали в 400 мл дихлорметана. При комнатной температуре добавляли раствор 22,95 г N-(2,2-диметоксиэтил)-N-(4-фторбензил)-4-метилбензолсульфонамида (22) в 150 мл дихлорметана. Перемешивание продолжали при комнатной температуре в течение ночи, раствор выливали на лед, органический слой отделяли, водную фазу дважды экстрагировали дихлорметаном и объединенные органические слои затем дважды экстрагировали бикарбонатом натрия. Органический слой сушили над сульфатом магния, упаривали досуха и полученный неочищенный продукт очищали хроматографией на силикагеле с получением 2,74 г соединения 23. Rt=0,30 мин (Способ № 4). Определенная масса: 148,1 (M+H+).
4-Хлор-6-фторизохинолин (24)
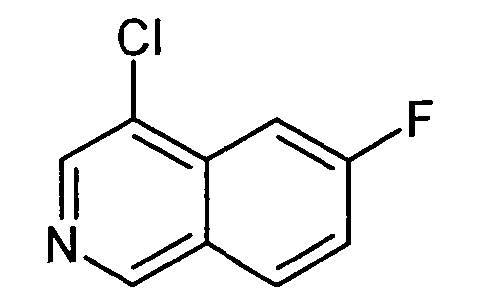
Раствор 1,5 г 6-фторизохинолина (23) в 4,5 мл сульфурилхлорида нагревали до 60°C в микроволновом реакторе (CEM Discovery) в течение 8 ч. После охлаждения до комнатной температуры смесь выливали на лед и экстрагировали три раза при помощи CHCl3. После сушки над Na2SO4 растворитель отгоняли путем дистилляции и неочищенный продукт очищали при помощи флэш-хроматографии с получением 930 мг соединения 24.
ЖХМС Способ № 1, время удерживания 1,37 мин, определенная масса 182,01 [M+H]+
Цис - и транс-N-Boc-2-метилпиперидин-4-ол (25 и 26)
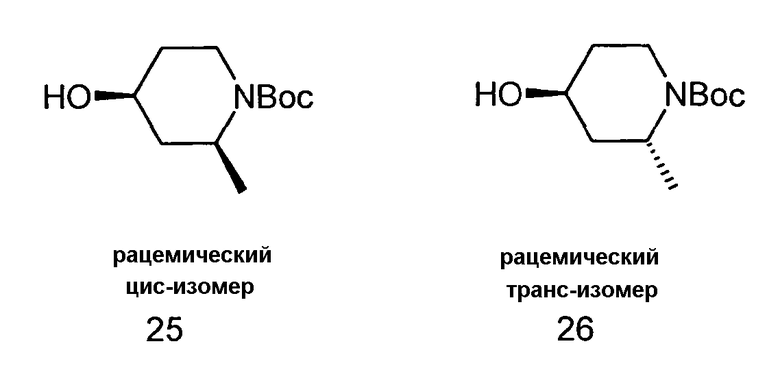
213 мг (5,6 ммоль) NaBH4 добавляли по порциям при 0°C к раствору 1,0 г (4,7 ммоль) 1-Boc-2-метилпиперидин-4-она в 10 мл EtOH. Смесь перемешивали при комнатной температуре в течение еще 2 ч. Растворитель удаляли дистилляцией и остаток растворяли в воде и этилацетате. Водный слой экстрагировали дважды этилацетатом и объединенные органические слои сушили над Na2SO4. После фильтрации растворитель удаляли с помощью дистилляции и неочищенный продукт очищали колоночной хроматографией с использованием н-гептана/этилацетата (1/1) с получением 367 мг (36%) цис-изомера 25 и 205 мг (20%) транс-изомера в дополнение к 97 мг (10%) смеси обоих изомеров.
Цис-изомер (25):
1H-ЯМР (CDCl3): δ = 4,28 (1H, м), 4,17 (1H, м), 3,82 (1H, м), 3,26 (1H, м), 1,85 (1H, ддд, J= 14,7, 6,6, и 3,4 Гц), 1,77 (1H, м), 1,66 (2H, м), 1,33 (3H, д, J=7,1 Гц).
Транс-изомер (26):
1H-ЯМР (CDCl3): δ = 4,50 (1H, м), 4,04 (1H, м), 3,95 (1H, м), 2,87 (1H, дт, J=2,9 и 13,6 Гц), 1,93 (1H, м), 1,83 (1H, м), 1,53 (1H, м), 1,32 (1H, м), 1,14 (3H, д, J=7,1 Гц).
Фенил-[6-(пиперидин-4-илокси)изохинолин-5-ил]амин (27)
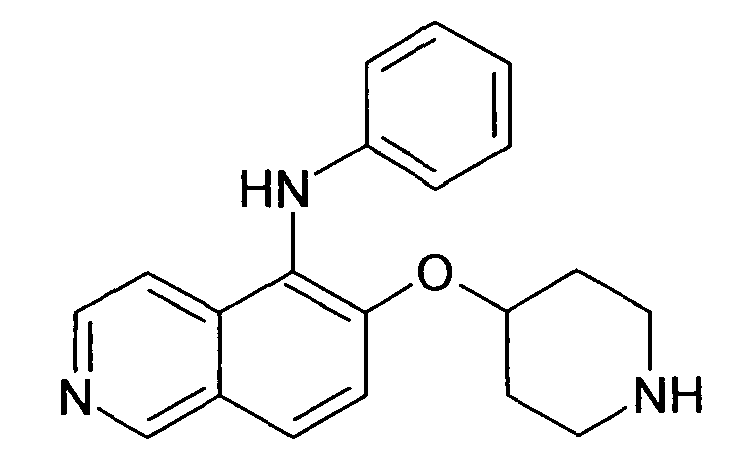
В атмосфере аргона к раствору 27 мг (0,28 ммоль) NaOtBu в 3 мл толуола добавляли 81 мг (0,2 ммоль) трет-бутилового эфира 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11) и 24 мг (0,26 ммоль) анилина. После перемешивания при комнатной температуре в течение 10 мин добавляли 9 мг (0,05 экв.) Pd2dba3 и смесь нагревали до 100°C в микроволновом реакторе (CEM Discovery) в течение 1 ч. После охлаждения до комнатной температуры добавляли воду и этилацетат. Органический слой отделяли, сушили над Na2SO4 и концентрировали. Очистка с помощью ВЭЖХ давала Boc-защищенное промежуточное соединение, которое обрабатывали 2 мл 5-6 н. раствора HCl в изопропаноле в течение 2 ч. Гидрохлорид фильтровали и подвергали еще одной очистке ВЭЖХ хроматографией с получением соединения 27 в виде трифторацетата (31,3 мг).
ЖХМС Способ № 2, время удерживания 0,78 мин, определенная масса 320,26 [M+H]+
5-Метил-6-(пиперидин-4-илокси)изохинолин гидрохлорид (28)
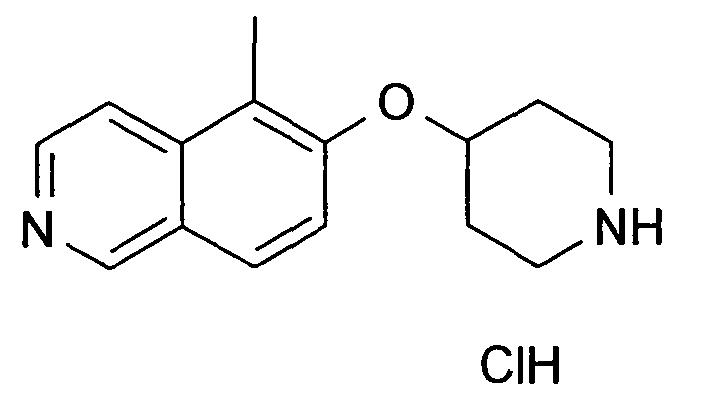
В атмосфере аргона к раствору 100 мг (0,24 ммоль) трет-бутилового эфира 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11) и 10 мг (1,1'-бис(дифенилфосфино)ферроцен)палладий(II)хлорида (0,056 экв. Pd(dppf)Cl2) в 3 мл диоксана добавляли 2 M раствор диметилцинка (0,5 мл, 93,7 мг, 4 экв.) в толуоле. Реакционную смесь нагревали до 100°C в течение 5 ч. После охлаждения растворители удаляли дистилляцией и остаток подвергали обработке препаративной ВЭЖХ с получением Boc-защищенного промежуточного соединения, которое обрабатывали 5-6 н. раствором HCl в изопропаноле в течение 2 ч при комнатной температуре. Удаление растворителей давало 13,7 мг (18%) соединения 28.
ЖХМС Способ № 1, время удерживания 0,67 мин, определенная масса 243,24 [M+H]+
5-Бензил-6-(пиперидин-4-илокси)изохинолин (29)
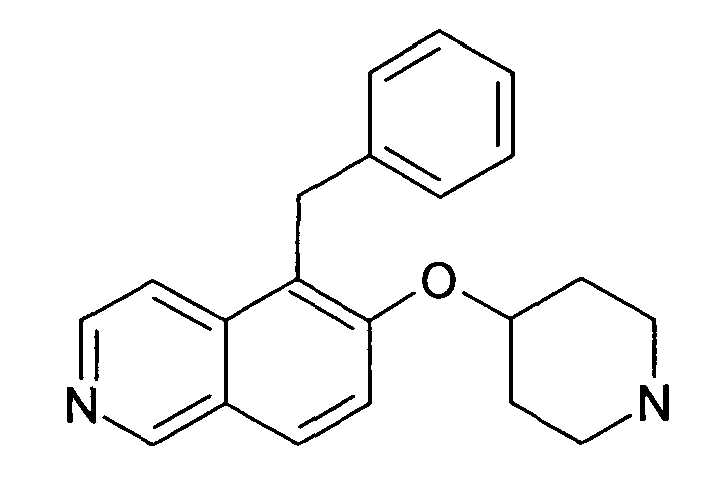
К раствору 81 мг (0,2 ммоль) трет-бутилового эфира 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11), 195 мг (0,6 ммоль) Cs2CO3, 14,6 мг (0,02 ммоль) Pd(dppf)Cl2 и 51 мг (0,26 ммоль) бензилтрифторбората калия в 3 мл ТГФ добавляли 0,3 мл воды. Через смесь барботировали аргон в течение 10 минут и затем реакционную смесь нагревали до температуры кипения с обратным холодильником в течение 16 ч (неполная конверсия). После охлаждения до комнатной температуры добавляли воду и этилацетат. Органический слой отделяли, сушили над Na2SO4. После удаления растворителей добавляли 2 мл 5-6 н. раствора HCl в изопропаноле. Через 2 ч растворители отгоняли с помощью дистилляции и остаток дважды подвергали обработке препаративной ВЭЖХ с получением 3,5 мг соединения 29 в виде трифторацетата.
ЖХМС Способ № 3, время удерживания 0,56 мин, определенная масса 319,23 [M+H]+
Этиловый эфир 6-(пиперидин-4-илокси)изохинолин-5-карбоновой кислоты (30)
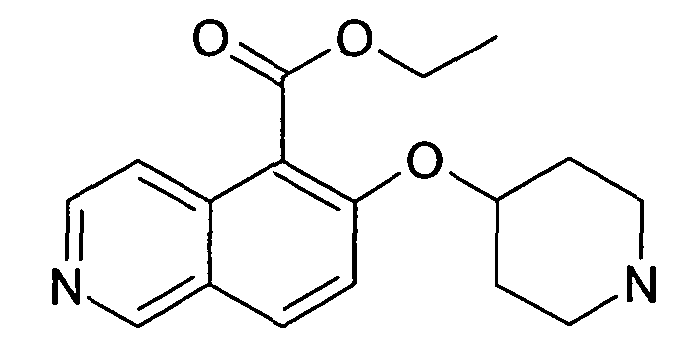
Раствор 200 мг (0,44 ммоль) трет-бутилового эфира 4-(5-йодизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (12), 107 мг (0,88 ммоль) DMAP, 4,7 мг (0,1 экв.) Pd на углероде (10%), 150 мкл (0,88 ммоль) триэтиламина и 58 мг (0,22 ммоль) Mo(CO)6 в 3 мл этанола нагревали до 135°C в течение 1 ч в микроволновом реакторе (CEM Discovery). Затем добавляли воду и этилацетат и смесь фильтровали через картридж с целитом. После удаления растворителей остаток подвергали обработке препаративной ВЭЖХ с получением 7,4 мг Boc-защищенного промежуточного соединения. Для того чтобы удалить Boc группу, промежуточное соединение обрабатывали 2 мл 5-6 н. раствора HCl в изопропаноле при комнатной температуре в течение 2 ч. Очистка при помощи препаративной ВЭЖХ давала 2,5 мг соединения 30 в виде соли ТФУК.
ЖХМС Способ № 3, время удерживания 0,14 мин, определенная масса 301,29 [M+H]+
6-(Пиперидин-4-илокси)изохинолин-5-иламин (31)
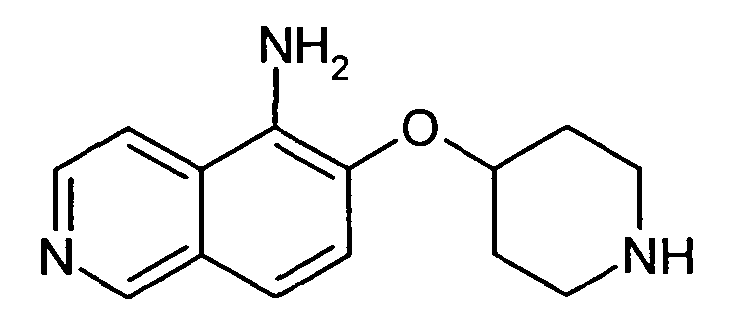
60 мкл 1 н. раствора NaOH, 6,9 мг (0,3 экв.) L-пролина, 3,8 мг (0,1 экв.) CuI и 26 мг (0,4 ммоль) NaN3 добавляли к раствору 82 мг (0,2 ммоль) трет-бутилового эфира 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11) в 2 мл смеси этанол/вода (7/3). Смесь нагревали до 95°C в течение 3 ч в микроволновом реакторе (CEM Discover). После охлаждения добавляли воду и этилацетат и смесь фильтровали через картридж с целитом. После удаления растворителей при помощи дистилляции остаток подвергали обработке препаративной ВЭЖХ. Удаляли защиту у N-Boc-защищенного промежуточного соединения, обрабатывая 2 мл 5-6 н. раствора HCl в изопропаноле в течение 2 ч при комнатной температуре. Затем добавляли воду и все растворители удаляли при помощи сушки вымораживанием с получением 18 мг соединения 31 в виде гидрохлорида.
1H-ЯМР (d6-ДМСО): δ = 9,60 (1H, с), 8,95 (2H, шир. с), 8,56 (1H, д, J=7,1 Гц), 8,41 (1H, д, J=7,1 Гц), 7,85 (1H, д, J=9,0 Гц), 7,81 (1H, д, J=9,0 Гц), 5,03 (1H, м), 3,13 (1H, м), 2,92 (1H, м), 2,15 (2H, м), 1,99 (2H, м), 1,84 (1H, м), 1,55 (1H, м).
ЖХМС Способ № 1, время удерживания 0,35 мин, определенная масса 244,25 [M+H]+
6-(Пиперидин-4-илокси)-5-(1H-тетразол-5-ил)изохинолин (32)
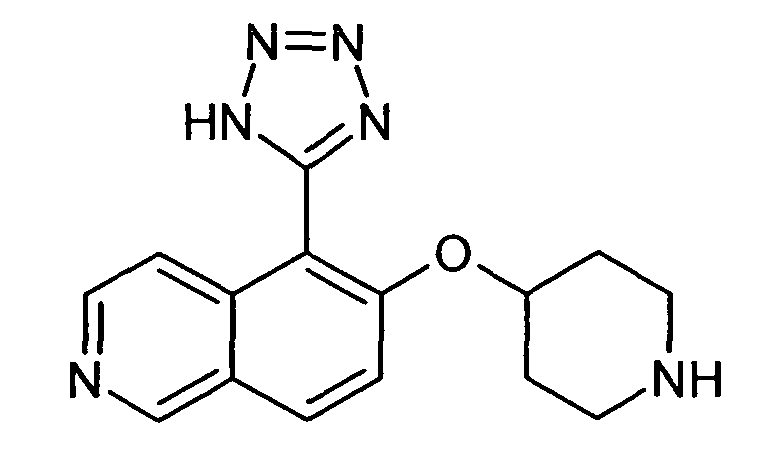
В атмосфере аргона к раствору 35 мг (0,1 ммоль) трет-бутилового эфира 4-(5-цианоизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (15) в 1 мл ДМФ добавляли 78 мг (1,2 ммоль) NaN3 и 64 мг (1,2 ммоль) NH4Cl. Реакционную смесь нагревали до приблизительно 160°C и при давлении 7 бар в течение 3 ч в микроволновом реакторе (CEM Discovery). После охлаждения до комнатной температуры добавляли водный раствор NH4Cl и дихлорметан. Смесь фильтровали через картридж фазового разделения и водный слой дважды промывали дихлорметаном. Органические слои объединяли и растворители отгоняли с помощью дистилляции. Остаток подвергали обработке препаративной ВЭЖХ с получением 4 мг (8%) соединения 32 в виде трифторацетата.
ЖХМС Способ № 3, время удерживания 0,90 мин, определенная масса 297,04 [M+H]+
5-(4-Метоксиметил-[1,2,3]триазол-1-ил)-6-(пиперидин-4-илокси)изохинолин (33)
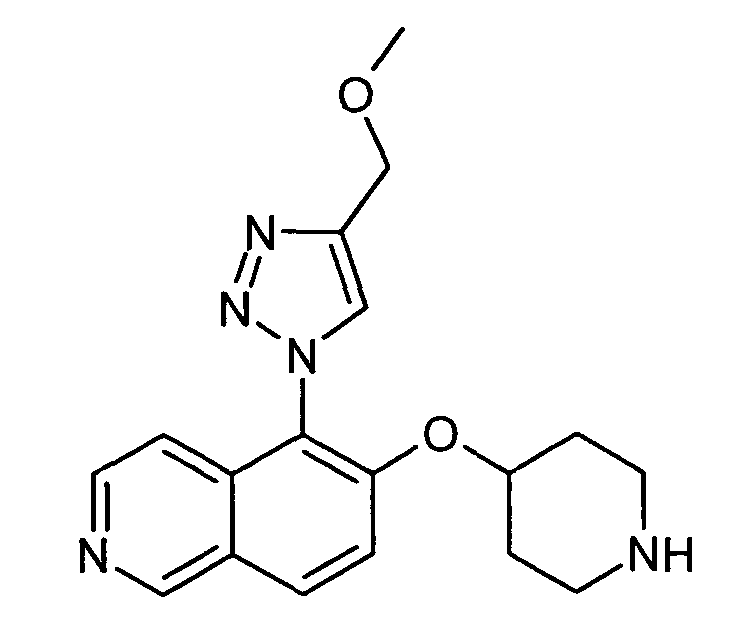
К раствору 73 мг (0,2 ммоль) трет-бутилового эфира 4-(5-азидоизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (17) и 14 мг (0,2 ммоль) метилпропаргилового эфира в 4 мл раствора вода/трет-бутанол (1/1) добавляли 4 мг (0,1 экв.) аскорбата натрия и 0,5 мг (0,01 экв.) сульфогидрата меди (II). Смесь перемешивали в течение 18 ч при комнатной температуре. Затем добавляли этилацетат и смесь фильтровали через картридж с целитом. После удаления растворителей остаток подвергали обработке препаративной ВЭЖХ. У N-Boc-защищенного промежуточного соединения удаляли защиту, обрабатывая 2 мл 5-6 н. раствора HCl в изопропаноле в течение 2 ч при комнатной температуре. Затем растворитель выпаривали и продукт отделяли при помощи препаративной ВЭЖХ с получением 2,8 мг соединения 33 в виде трифторацетата.
ЖХМС Способ № 3, время удерживания 0,08 мин, определенная масса 340,17 [M+H]+
5-(4-Фенил-[1,2,3]триазол-1-ил)-6-(пиперидин-4-илокси)изохинолин (34)
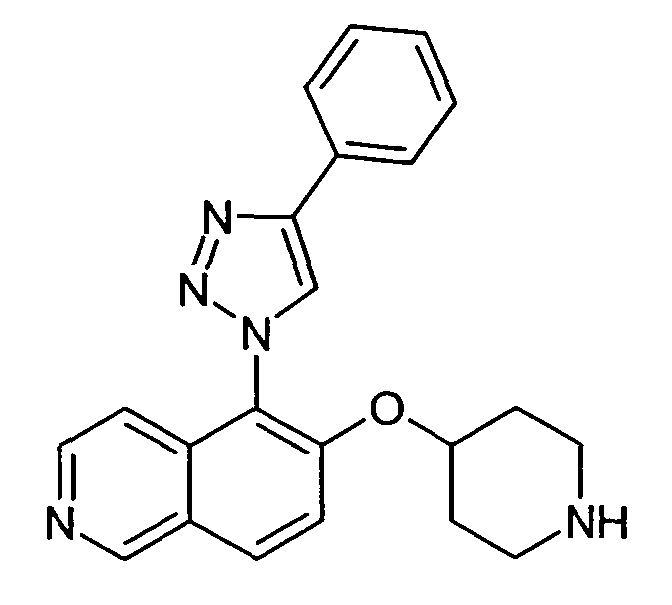
Указанное в заголовке соединение получали в соответствии с процедурой, описанной для соединения 33, используя 20 мг (0,2 ммоль) фенилацетилена. Получали 2,5 мг соединения 34 в виде трифторацетата.
ЖХМС Способ № 3, время удерживания 0,14 мин, определенная масса 372,2 [M+H]+
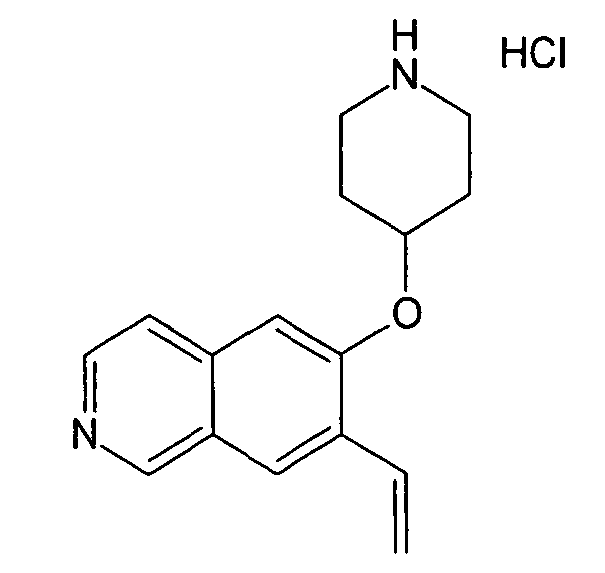
7-Этил-6-(пиперидин-4-илокси)изохинолин гидрохлорид (35)
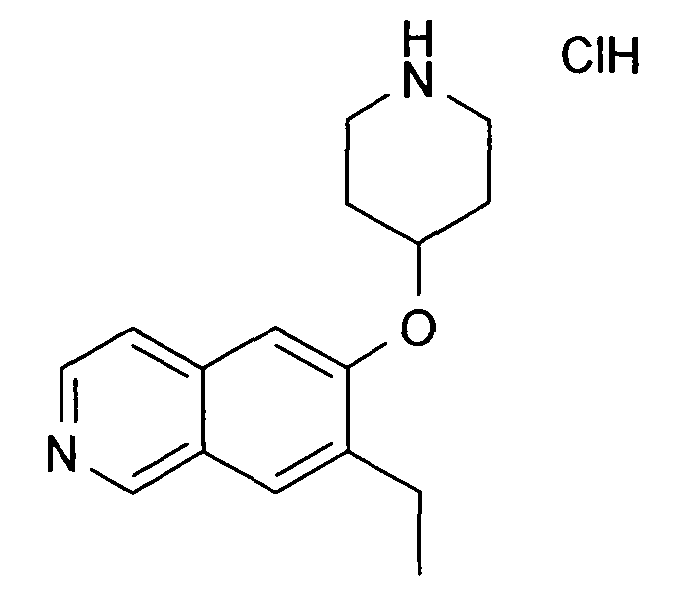
К раствору 174 мг трет-бутилового эфира 4-(7-винилизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (19) (0,49 ммоль, 1 экв.) в 15 мл метанола добавляли 1 мг 5% палладия на углероде (0,02 экв.). Олефин гидрировали при давлении 5 бар H2 при температуре окружающей среды в течение ночи. Наблюдали только частичную конверсию, поэтому катализатор удаляли при помощи фильтрации и добавляли свежий катализатор. Реакцию завершали при помощи еще одной обработки при тех же условиях гидрирования. Затем катализатор удаляли при помощи фильтрации и неочищенный продукт очищали препаративной ВЭЖХ с получением 97 мг Boc-защищенного промежуточного соединения.
Защитную группу удаляли обработкой при помощи 5-6 н. раствора HCl в изопропаноле в течение 2 ч при комнатной температуре. Растворитель отгоняли с помощью дистилляции и добавляли воду и ацетонитрил. Сушка вымораживанием смеси давала 53 мг соединения 35.
ЖХМС Способ № 1, время удерживания 0,71 мин, определенная масса 257,18 [M+H]+
Следующее иллюстративное соединение синтезировали согласно данному способу:
5-Этил-6-(пиперидин-4-илокси)изохинолин трифторацетат (36)
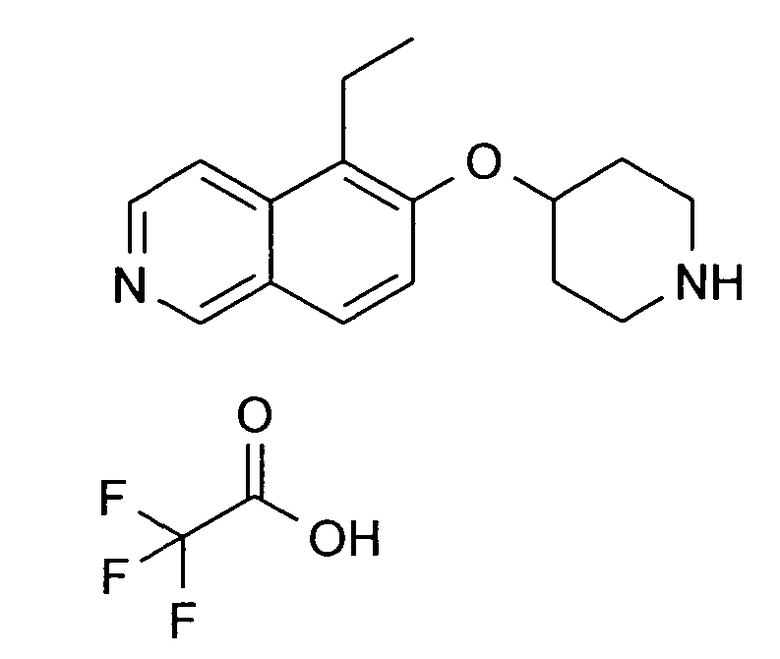
используя в качестве исходного вещества соединение 19A
ЖХМС Способ № 2, время удерживания 0,17 мин, определенная масса 257,21 [M+H]+
Фенил-[6-(пиперидин-4-илокси)изохинолин-5-ил]метанол гидрохлорид (37)
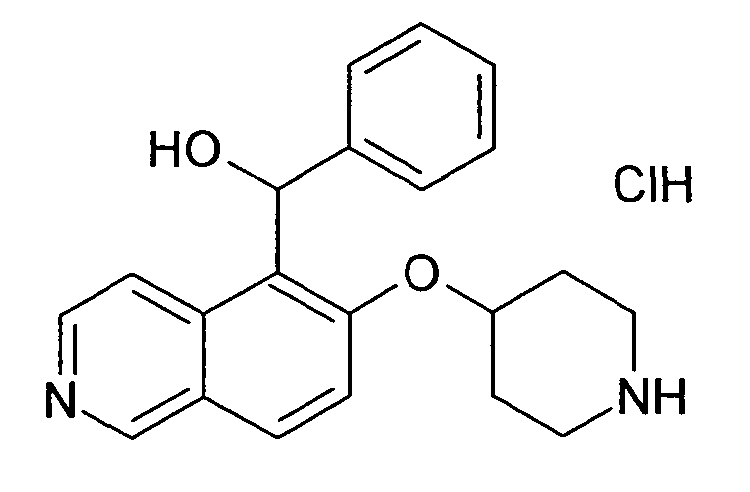
К раствору 200 мг (0,49 ммоль, 1 экв.) трет-бутилового эфира 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11) в 3 мл ТГФ при -78°C добавляли 0,6 мл (0,98 ммоль, 1,6 M в гексане) н-бутиллития. Через 30 мин добавляли 110 мкл (115 мг, 1,08 ммоль) бензальдегида и смесь оставляли для нагревания до температуры окружающей среды. Через 2 ч перемешивания при комнатной температуре добавляли воду и этилацетат. Слои разделяли и органический слой промывали водой и насыщенным солевым раствором. После сушки над Na2SO4 и выпаривания растворителя остаток подвергали обработке препаративной ВЭЖХ с получением Boc-защищенного промежуточного соединения.
Boc-группу удаляли, растворяя промежуточное соединение в изопропаноле и добавляя 5-6 н. раствор HCl в изопропаноле. Осажденный гидрохлорид выделяли при помощи фильтрации с получением 5,2 мг соединения 37 (3%).
1H-ЯМР (d6-ДМСО): δ = 9,43 (1H, с), 8,50 (1H, шир. с), 8,40 (1H, шир. с), 8,30 (3H, м), 7,87 (1H, д, J=9,2 Гц), 7,33 (2H, д, J=7,4 Гц), 7,28 (2H, т, J= 7,4 Гц), 7,19 (1H, т, J=7,4 Гц), 6,74 (1H, с), 6,34 (1H, с), 5,10 (1H, м), 3,25 (2H, м), 3,15 (2H, м), 2,19 (2H, м), 1,93 (2H, м).
ЖХМС Способ № 1, время удерживания 0,80 мин, определенная масса 335,22 [M+H]+
Следующее соединение примера также синтезировали согласно данному способу:
1-[6-(Пиперидин-4-илокси)изохинолин-5-ил]этанол гидрохлорид (38)
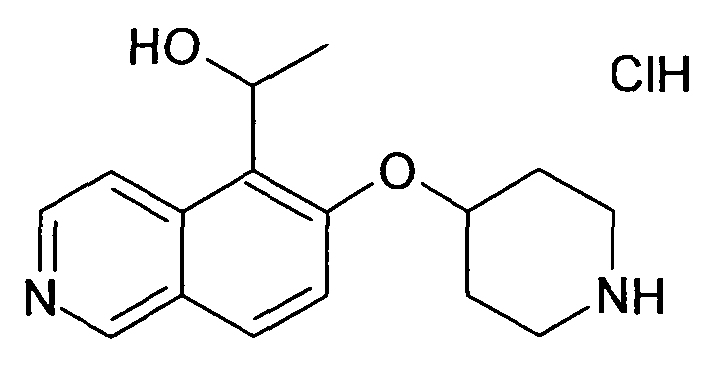
ЖХМС Способ № 1, время удерживания 0,55 мин, определенная масса 273,2 [M+H]+
2,2,2-Трифтор-N-[6-(пиперидин-4-илокси)изохинолин-5-ил]ацетамидтрифторацетат (39)
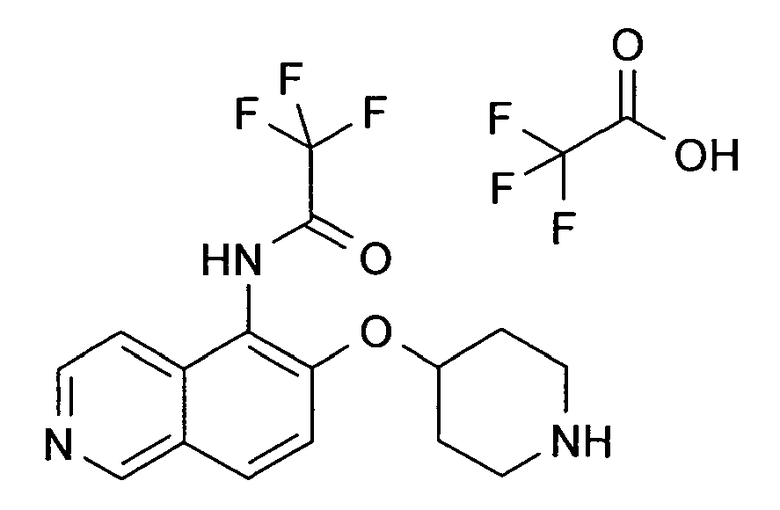
К раствору 39 мг (0,11 ммоль) трет-бутилового эфира 4-(5-аминоизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (18) (содержащего некоторое количество соединения 11) в 3 мл ДМФ добавляли 62,8 мг карбоната калия (0,46 ммоль, 4 экв.) и 10,7 мкл метансульфонилхлорида (0,13 ммоль, 1,2 экв.). Реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Затем добавляли воду и этилацетат. Смесь фильтровали через целит, промывали этилацетатом и концентрировали с получением только одного продукта. У N-Boc-защищенного промежуточного соединения удаляли защиту, обрабатывая 2 мл 5-6 н. раствора HCl в изопропаноле в течение 2 ч при комнатной температуре. Затем растворитель выпаривали и продукт выделяли с помощью препаративной ВЭЖХ с получением 18,5 мг соединения 39.
ЖХМС Способ № 1, время удерживания 0,39 мин, определенная масса 340,15 [M+H]+
N-[6-(Пиперидин-4-илокси)изохинолин-5-ил]ацетамидтрифторацетат (40)
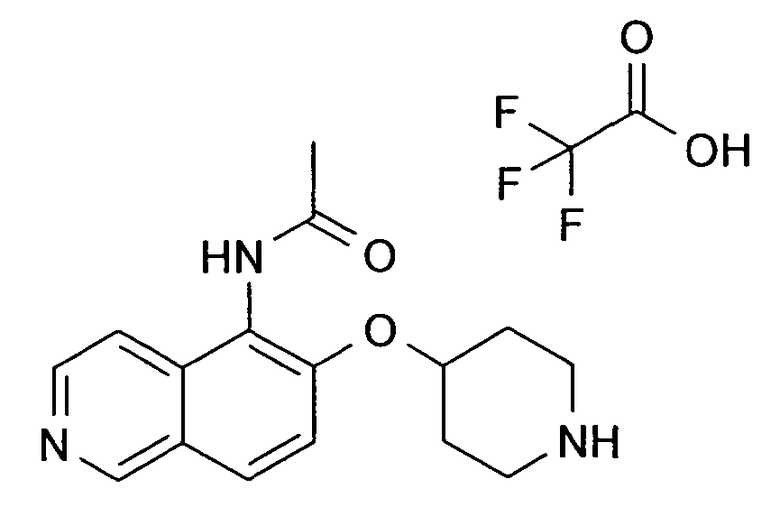
В атмосфере аргона к раствору 27 мг (0,28 ммоль, 1,4 экв.) NaOtBu в 3 мл толуола добавляли 81,5 мг (0,2 ммоль) трет-бутилового эфира 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11) и 14,2 мг ацетамида (0,24 ммоль, 1,2 экв.). После перемешивания в течение 10 минут при комнатной температуре добавляли 9,1 мг (0,01 ммоль, 0,05 экв.) Pd2(dba)3 и 11,9 мг (0,04 ммоль, 0,2 экв.) 2-(дитрет-бутилфосфино)бифенила. Реакционную смесь нагревали до 120°C в течение 2 ч в микроволновом реакторе (CEM Discovery). Затем добавляли воду и этилацетат. Смесь фильтровали через целит, промывали этилацетатом и концентрировали. Остаток дважды подвергали обработке препаративной ВЭЖХ с получением N-Boc-защищенного промежуточного соединения. У N-Boc-защищенного промежуточного соединения удаляли защиту, обрабатывая 2 мл 5-6 н. раствора HCl в изопропаноле в течение 2 ч при комнатной температуре. Затем растворитель выпаривали и продукт выделяли при помощи препаративной ВЭЖХ с получением 2,5 мг соединения 40.
ЖХМС Способ № 3, время удерживания 0,15 мин, определенная масса 286,15 [M+H]+
Основная процедура удаления защитной Boc-группы в основной структуре:
Соответствующие N-Boc-защищенные соединения обрабатывали 5-6 н. раствором HCl в изопропаноле в течение 2 ч при комнатной температуре. Осажденные гидрохлориды выделяли с помощью фильтрации и сушили. Если было необходимо, проводили дополнительную очистку при помощи препаративной ВЭЖХ.
№
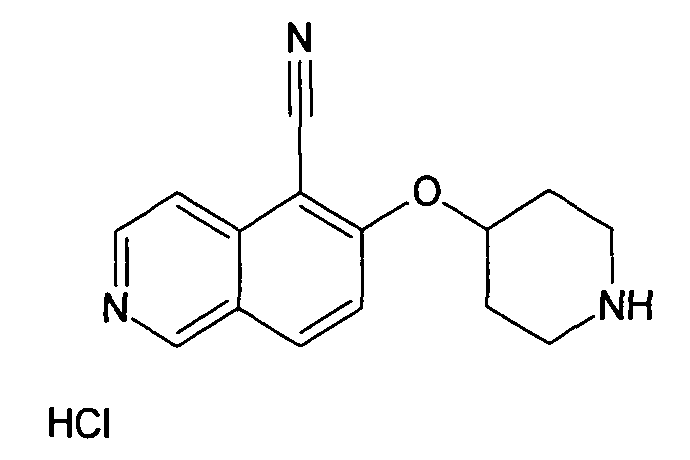
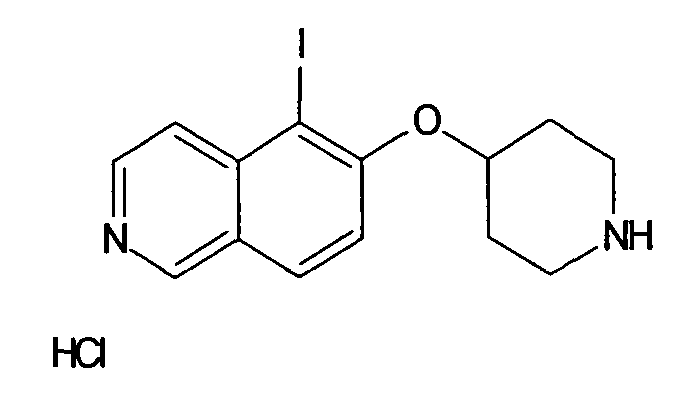
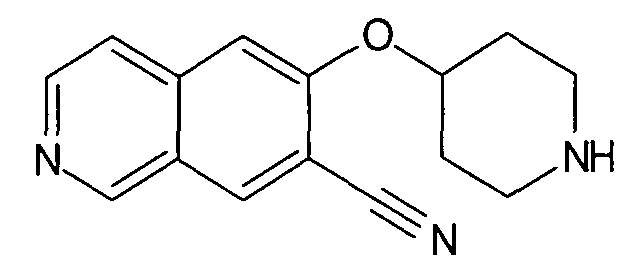
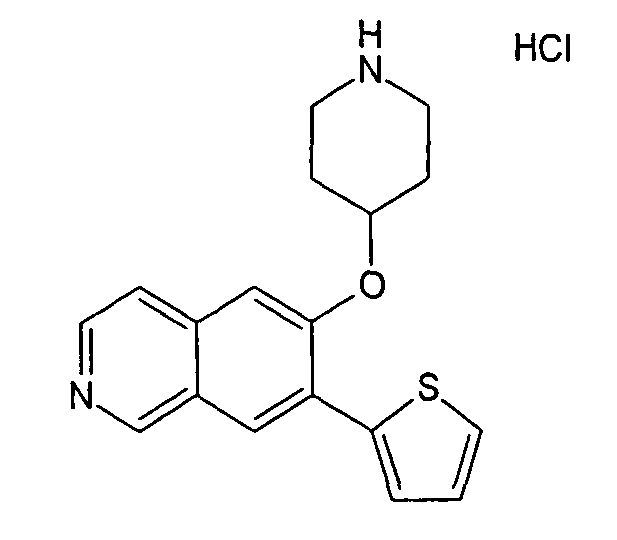
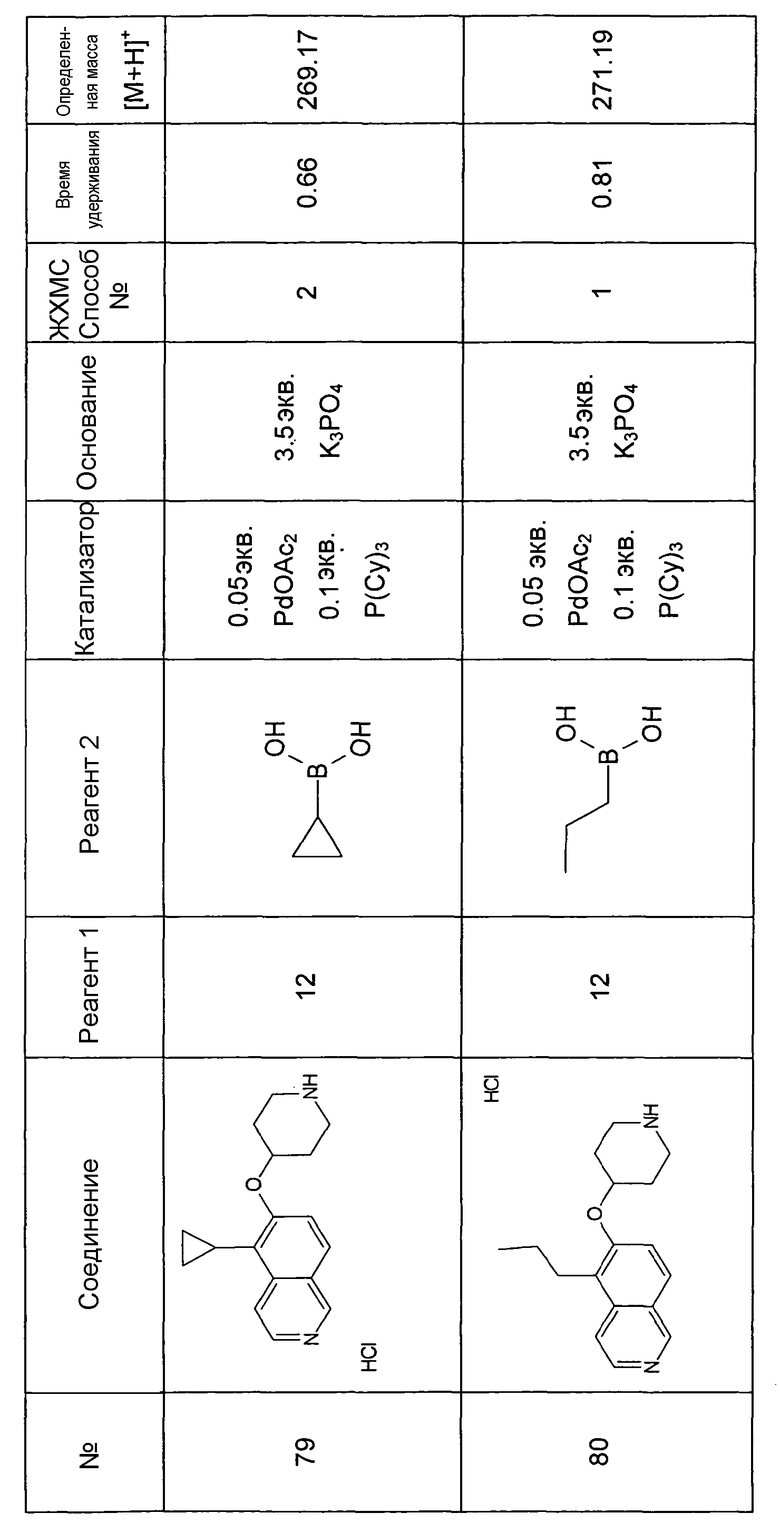
Основная процедура для сочетания способом Suzuki с трет-бутиловым эфиром 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11)
Водный раствор Na2CO3 (0,2 мл, 0,4 ммоль, 2 экв. 2 M) добавляли к раствору 81 мг (0,2 ммоль, 1 экв.) трет-бутилового эфира 4-(5-бромизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (11) и 1,5 экв. (0,3 ммоль) соответствующей бороновой кислоты (реагент 2) в 3 мл DME. Через реакционную смесь в течение 10 мин барботировали аргон. Затем добавляли 23 мг (0,1 экв.) Pd(PPh3)4 и реакционную смесь перемешивали при 95°C в течение ночи в атмосфере аргона. После охлаждения добавляли 2 мл воды и 10 мл этилацетата. Органический слой отделяли, сушили и растворитель отгоняли с помощью дистилляции. Остаток подвергали обработке препаративной ВЭЖХ.
Boc-группу удаляли, растворяя промежуточное соединение в изопропаноле и добавляя 5-6 н. раствор HCl в изопропаноле. Осадок выделяли при помощи фильтрации.
В некоторых реакциях гидрохлорид не осаждался или чистота осадка была неудовлетворительной. В таких случаях растворитель отгоняли с помощью дистилляции и остаток очищали при помощи препаративной ВЭЖХ.
Примеры 49-78 синтезировали, используя данный способ (см. в конце описания).
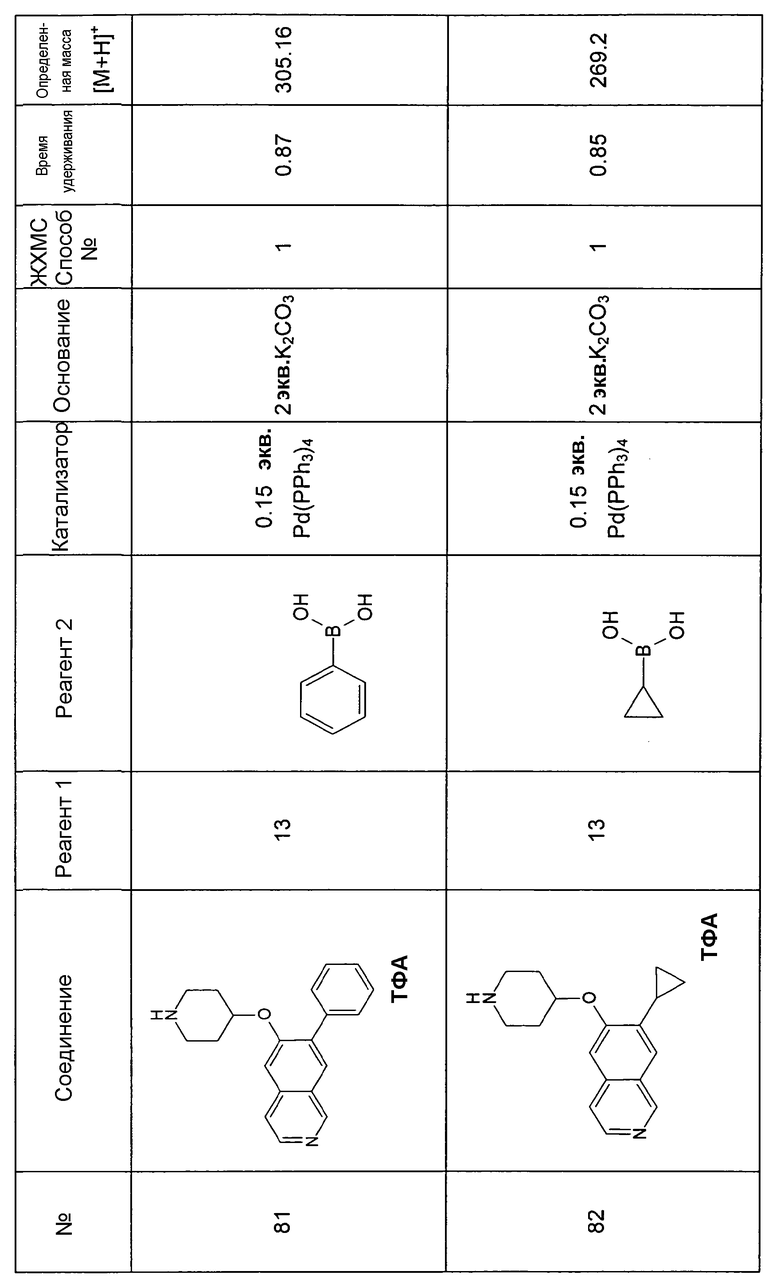
Процедура реакции сочетания Suzuki для вариантов в положении 5 и 7
К раствору реагента 1 (типично 0,2 ммоль) и реагента 2 в DME добавляли основание. Через реакционную смесь в течение 10 мин барботировали аргон. Затем добавляли катализатор и реакционную смесь перемешивали при температуре кипения с обратным холодильником в течение ночи в атмосфере аргона. После охлаждения добавляли 2 мл воды и 10 мл этилацетата. Смесь фильтровали через картридж с целитом. Растворители отгоняли с помощью дистилляции и остаток подвергали обработке препаративной ВЭЖХ. У N-Boc-защищенного промежуточного соединения удаляли защиту, обрабатывая 2 мл 5-6 н. раствора HCl в изопропаноле в течение 2 ч при комнатной температуре. Растворители удаляли с помощью дистилляции и осадок выделяли фильтрацией. В некоторых реакциях гидрохлорид не осаждался или чистота осадка была неудовлетворительной. В таких случаях растворитель отгоняли с помощью дистилляции и остаток очищали при помощи препаративной ВЭЖХ.
Следующие примеры синтезировали, используя данный способ:
5-Изопропенил-6-(пиперидин-4-илокси)изохинолин (83)
К раствору 22 мг (0,18 ммоль) 2-бромпропена в 2 мл ДМФ добавляли 85 мг (0,62 ммоль) K2CO3 и 70 мг (0,15 ммоль) трет-бутилового эфира 4-[5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)изохинолин-6-илокси]пиперидин-1-карбоновой кислоты (14). В атмосфере аргона добавляли 5,6 мг (0,05 экв.) Pd(dppf)Cl2 и реакционную смесь нагревали до 100°C в течение 16 ч. После охлаждения до комнатной температуры добавляли воду и дихлорметан. Смесь фильтровали через картридж с целитом. Растворители отгоняли с помощью дистилляции и остаток подвергали обработке препаративной ВЭЖХ. У N-Boc-защищенного промежуточного соединения удаляли защиту, обрабатывая 2 мл 5-6 н. раствора HCl в изопропаноле в течение 2 ч при комнатной температуре. Растворители удаляли с помощью дистилляции и остаток подвергали препаративной ВЭЖХ. У выделенного промежуточного соединения удаляли защиту, обрабатывая 2 мл 5-6 н. раствора HCl в изопропаноле в течение 2 ч при комнатной температуре. Растворители отгоняли с помощью дистилляции и выделяли соединение 83 при помощи препаративной ВЭЖХ с получением 3,2 мг в виде трифторацетата.
ЖХМС Способ № 3, время удерживания 0,15 мин, определенная масса 269,15 [M+H]+
Следующие примеры получали, используя данный способ:
6-Метокси-4-(4,4,4-трифторбутил)изохинолин (87)
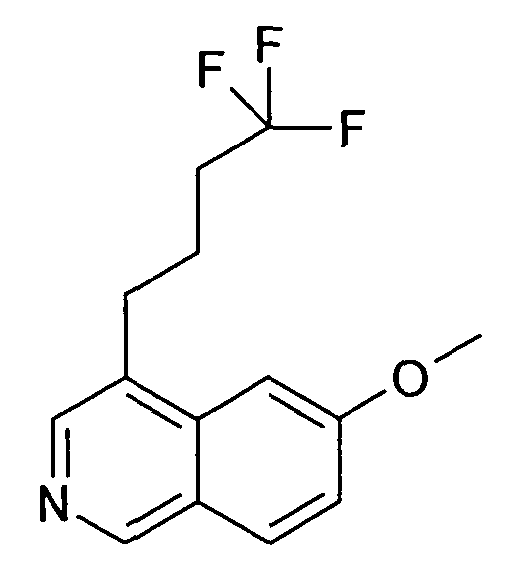
2 г 6-Метоксиизохинолин растворяли в 25 мл сухого тетрагидрофурана. По каплям добавляли 12,56 мл 1 M раствора триэтилборогидрата калия. Раствор оставляли перемешиваться при комнатной температуре в течение 2 ч, затем добавляли по каплям 3,29 г 4,4,4-трифтор-1-йодбутана. Раствор оставляли перемешиваться в течение ночи, затем добавляли раствор 32 мл 1 M гидроксида натрия и 12 мл пероксида натрия (35%). Перемешивание продолжали в течение еще 3 часов, затем раствор разбавляли дихлорметаном, экстрагировали водой и насыщенным солевым раствором и органический слой сушили над сульфатом натрия и упаривали досуха. Хроматография силикагелем давала 1,03 г желаемого продукта.
ЖХМС Способ № 1, время удерживания 1,20 мин, определенная масса 270,06 [M+H]+
6-Гидрокси-4-(4,4,4-трифторбутил)изохинолин (88)
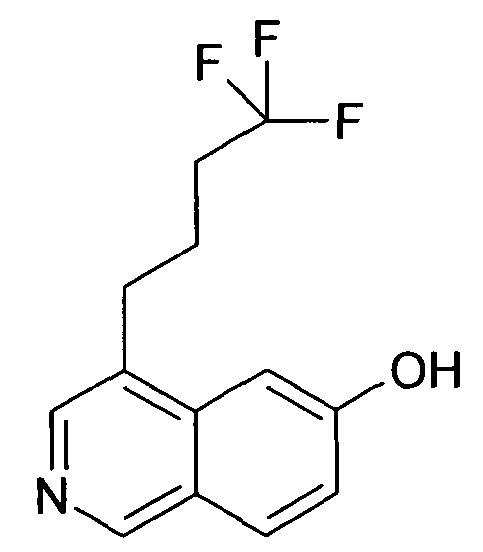
1,02 г 6-Метокси-4-(4,4,4-трифторбутил)изохинолин (87) обрабатывали трибромидом бора, как описано для синтеза соединения 1, с получением 410 мг желаемого продукта 88.
ЖХМС Способ № 1, время удерживания 1,04 мин, определенная масса 256,00 [M+H]+
Трет-бутиловый эфир 4-[4-(4,4,4-трифторбутил)изохинолин-6-илокси]пиперидин-1-карбоновой кислоты (89)
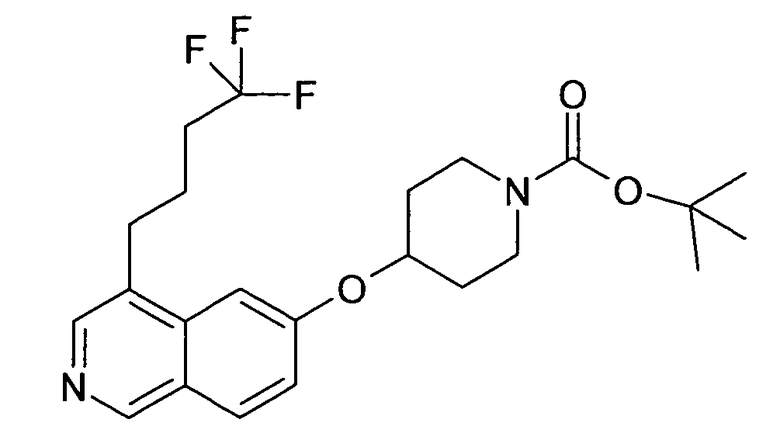
100 мг соединения 88, 118 мг Boc-(4-гидрокси)пиперидина и 416 мг дифенил-[4-[1H,1H,2H,2H-перфтордецил]фенил]фосфина растворяли в 5 мл сухого тетрагидрофурана. Добавляли 208 мг бис (1H,2H,2H,3H,3H-перфторононил)азодикарбоксилата и реакционную смесь оставляли перемешиваться в течение ночи. Смесь выпаривали досуха и фильтровали над 5 г картриджа Fluor-Flash. Полученный неочищенный продукт очищали препаративной ВЭЖХ с получением 46 мг желаемого продукта.
ЖХМС Способ № 1, время удерживания 1,51 мин, определенная масса 439,13 [M+H]+
6-(Пиперидин-4-илокси)-4-(4,4,4-трифторбутил)изохинолин (90)
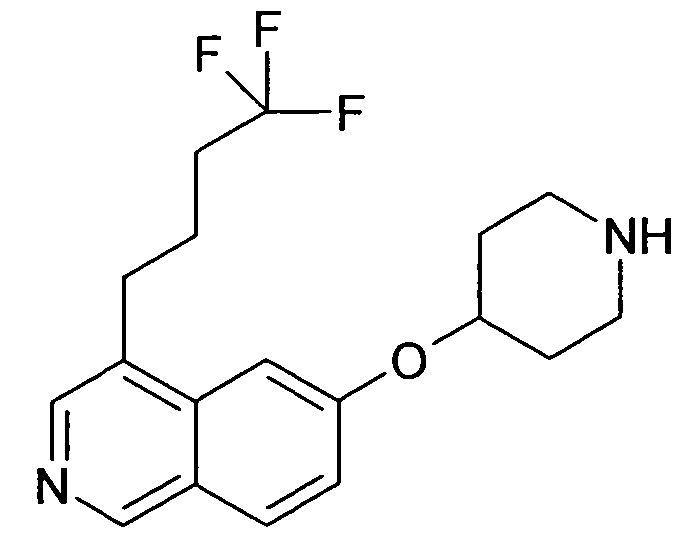
42 мг соединения 89 растворяли в 5 M хлористоводородной кислоты в изопропаноле. Раствор перемешивали при комнатной температуре в течение 2 часов и еще 2 часа при 40°C, упаривали досуха и помещали в воду и лиофилизировали три раза с получением 32 мг желаемого продукта в виде гидрохлоридной соли.
ЖХМС Способ № 1, время удерживания 0,98 мин, определенная масса 338,16 [M+H]+
Следующие изохинолины синтезировали таким же способом, как описано для соединения 90, используя подходящие алкилгалогениды:
[M+H]+
Основная процедура реакции восстановительного аминирования:
1,5 экв. альдегида растворяли в 1 мл метанола и 50 мг соединения 124 и добавляли 27 мг безводного ацетата натрия, растворенного в метаноле. Добавляли 0,250 мл раствора 1M цианоборогидрида натрия в ТГФ. Смесь оставляли для взаимодействия в течение ночи, затем раствор фильтровали, выпаривали досуха и остаток забирали в этилацетат. Органический слой экстрагировали раствором 5% карбоната натрия в воде, затем 5% хлорида натрия в воде. Органический слой сушили, упаривали досуха и очищали при помощи ОФ хроматографии.
Данную процедуру использовали для получения соединений 93-123 (см. в конце описания).
Все данные ЖХМС в данной таблице получали, используя ЖХМС Способ №2.
Основная процедура для реакции boc-защищенных аминоспиртов с 6-гидроксиизохинолинами (реакция Mitsunobu):
AAV1:
К 500 мг (1,5 ммоль) трифенилфосфина (связанного с полистиролом, 3 ммоль/г) и 10 мл дихлорметана добавляли 0,195 мл (1,2 ммоль) диэтилазодикарбоксилата (или, альтернативно, диизопропилазодикарбоксилат). Реакционную смесь встряхивали в течение 10 мин и затем добавляли 0,14 мл триэтиламина, 145 мг 6-гидроксиизохинолина (7) (или равное количество другого подходящего изохинола) (реагент 1) и 1 ммоль желаемого boc-защищенного аминоспирта (реагент 2). Реакционную смесь встряхивали при комнатной температуре до прекращения какого-либо дальнейшего превращения согласно данным ЖХМС. Для дальнейшей обработки раствор фильтровали, остаток промывали дихлорметаном и органический слой дважды промывали 1 н. раствором гидроксида натрия, дважды водой и один раз насыщенным солевым раствором, сушили над сульфатом натрия и упаривали. Неочищенный продукт очищали при помощи препаративной ВЭЖХ с получением boc-защищенного связанного продукта.
Основная процедура для удаления boc-группы (AAV2):
Исходное вещество растворяли в 2 M хлористоводородной кислоты и оставляли реагировать в течение ночи. К соединениям с плохой растворимостью в воде добавляли метанол или диоксан до получения гомогенного раствора. Альтернативно, использовали 4 M хлористоводородной кислоты в изопропаноле для взаимодействия с соединением. Реакционную смесь лиофилизировали и получали незащищенный продукт в виде соответствующего гидрохлорида свободного амина.
Примеры соединений 124-149 представлены в конце описания.
Хроматографическое разделение соединений 140 и 143:
N-Boc-защищенное промежуточное соединение, полученное в виде энантиомерной смеси соединения 140 и соединения 143, разделяли на энантиомеры на хиральной колонке (Chiralcel OD-H/56 250×4,6 мм). Удаление защитной группы как завершающую стадию осуществляли, как описано в основной процедуре.
Абсолютная конфигурация стереоцентров не была определена.
Соединение 140: более раннее элюирование Boc-защитного промежуточного соединения;
Соединение 143: более позднее элюирование Boc-защитного промежуточного соединения.
Хроматографическое разделение соединений 141 и 142:
N-Boc-защищенное промежуточное соединение, полученное в виде энантиомерной смеси соединения 141 и соединения 142, разделяли на энантиомеры на хиральной колонке (Chiralpak AD-H/40 250×4,6 мм). Удаление защитной группы как завершающую стадию осуществляли, как описано в основной процедуре.
Абсолютная конфигурация стереоцентров не была определена.
Соединение 141: более раннее элюирование Boc-защитного промежуточного соединения;
Соединение 142: более позднее элюирование Boc-защитного промежуточного соединения.
7-Хлор-6-фторизохинолин (150)
7-Хлор-6-фторизохинолин получали при той же последовательности реакций, как использовали для синтеза 6-фторизохинолина (23), исходя из 3-хлор-4-фторбензальдегида. Rt=0,77 мин (Способ №2). Определенная масса: 182,1/184,1 (M+H+).
5-Хлор-6-фторизохинолин-трифторацетат (151)
7,0 г (38,1 ммоль) 6-Фторизохинолина (23) растворяли в 60 мл концентрированной серной кислоты. При 0°C добавляли 10,18 г N-хлорсукцинимид. Через 1 ч добавляли еще 5,2 г N-хлорсукцинимида и раствор нагревали до 50°C. Последовательно добавляли две дополнительные порции 5,2 г N-хлорсукцинимида и перемешивание продолжали при 50°C до тех пор, пока реакция не прекращалась. Реакционную смесь охлаждали до комнатной температуры, выливали на лед и доводили значение pH до 10, добавляя гидроксид натрия. Осадок отфильтровывали, забирали в дихлорметан и промывали водным раствором гидроксида натрия. Органический слой сушили над сульфатом натрия, упаривали и неочищенный продукт очищали препаративной ВЭЖХ с получением 4,04 г 5-хлор-6-фторизохинолин (151) в виде трифторацетата. Rt=0,97 мин (Способ №2). Определенная масса: 182,0/184,0 (M+H+).
6-Фтор-5-нитроизохинолин (152)
К 2,8 мл серной кислоты при охлаждении добавляли 2,0 мл концентрированной азотной кислоты. Последовательно добавляли 350 мг 6-фторизохинолина (23), реакционную смесь нагревали до комнатной температуры и оставляли перемешиваться в течение ночи. Реакционную смесь выливали на лед, экстрагировали дихлорметаном и доводили значение pH до щелочного, добавляя гидроксид натрия. Водный слой снова экстрагировали дихлорметаном. Слой дихлорметана сушили над сульфатом натрия и упаривали с получением 90 мг 6-фтор-5-нитроизохинолина, который можно было использовать без дополнительной очистки. Rt=1,03 мин (Способ №1). Определенная масса: 193,0 (M+H+).
Трет-бутиловый эфир 4-(изохинолин-6-илокси)пиперидин-1-карбоновой кислоты (154)
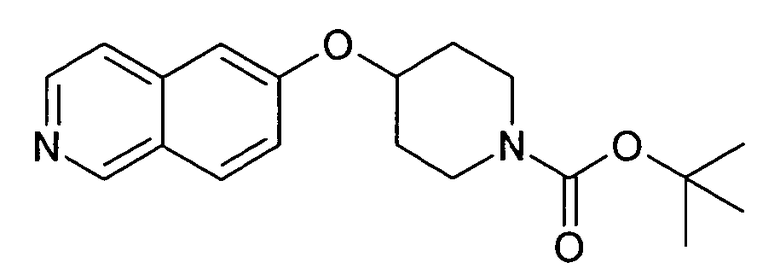
7,49 г трет-бутилового эфира 4-гидроксипиперидин-1-карбоновой кислоты растворяли в 20 мл сухого диметилацетамида. Добавляли 1,49 г гидрида натрия (60%). Затем добавляли по каплям раствор 3,65 г 6-фторизохинолина (23). Раствор нагревали при 80°C в течение 2 часов, затем растворитель удаляли и остаток забирали в дихлорметан. Органический слой дважды экстрагировали водой и затем насыщенным солевым раствором, сушили над сульфатом натрия и упаривали досуха. Неочищенный продукт очищали хроматографией на силикагеле с получением 6,22 г трет-бутилового эфира 4-(изохинолин-6-илокси)пиперидин-1-карбоновой кислоты. Rt=1,32 мин (Способ №1). Определенная масса: 329,1 (M+H+).
6-(Пиперидин-4-илокси)изохинолин гидрохлорид (124)
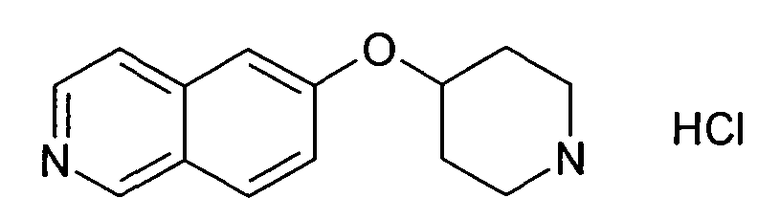
Удаляли защиту у трет-бутилового эфира 4-(изохинолин-6-илокси)пиперидин-1-карбоновой кислоты (154), используя основную процедуру, описанную в AAV2, с получением указанного в заголовке соединения в виде HCl-соли. Rt=0,20 мин. (Способ №2). Определенная масса: 229,1 (M+H+).
Следующий пример синтезировали в соответствии с данным способом:
4-Хлор-6-(пиперидин-4-илокси)изохинолин гидрохлорид (156)
используя 4-хлор-6-фторизохинолин (24) в качестве исходного вещества
ЖХМС Способ № 2, время удерживания 0,56 мин, определенная масса 263,12 [M+H]+
6-(1-Пиримидин-2-ил-пиперидин-4-илокси)изохинолингидрохлорид (157)
150 мг 6-(Пиперидин-4-илокси)изохинолин гидрохлорида (124) растворяли в 10 мл безводного пиридина. Добавляли 177 мг триэтиламина и 69 мг 4-хлорпиримидина и раствор перемешивали при 65°C в течение 6 часов. Реакционную смесь выливали в насыщенный солевой раствор и экстрагировали три раза этилацетатом. Объединенные органические слои сушили над сульфатом натрия, упаривали досуха и неочищенный продукт очищали препаративной ВЭЖХ. Продукт преобразовывали в соответствующую соль HCl, помещая продукт в 20 мл 1 н. раствора хлористоводородной кислоты, с последующей лиофилизацией. Выход: 47 мг. Rt=1,05 мин (Способ №2). Определенная масса: 307,1 (M+H+).
6-[1-(4-Трифторметилпиримидин-2-ил)пиперидин-4-илокси]изохинолин гидрохлорид (158)
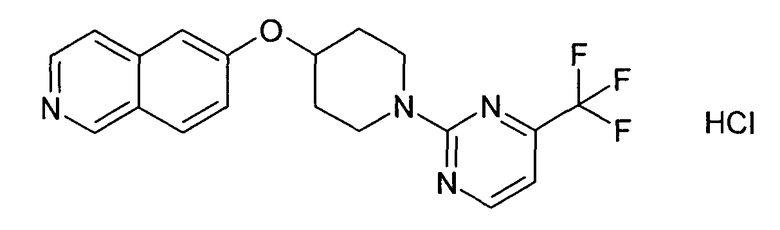
75 мг 6-(Пиперидин-4-илокси)изохинолингидрохлорида (124) растворяли в 5 мл безводного пиридина и 5 мл ДМФ. Добавляли 55 мг 2-хлор-4-трифторметилпиримидина и раствор перемешивали при 60°C в течение 3 часов. Растворители удаляли в вакууме и остаток забирали в насыщенный солевой раствор и экстрагировали три раза этилацетатом. Объединенные органические слои сушили над сульфатом натрия, упаривали досуха и неочищенный продукт очищали препаративной ВЭЖХ. Продукт превращали в соответствующую соль HCl, помещая продукт в 20 мл 1 н. хлористоводородной кислоты с последующей лиофилизацией. Выход: 29 мг. Rt=1,69 мин (Способ №2). Определенная масса: 375,1 (M+H+).
6-(1-Циклопропилпиперидин-4-илокси)изохинолингидрохлорид (159)
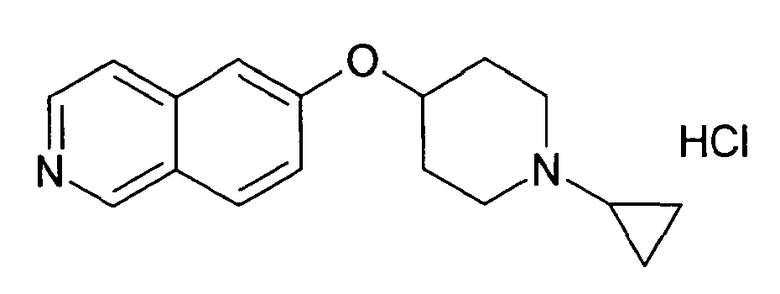
300 мг (1,13 ммоль) 6-(Пиперидин-4-илокси)изохинолингидрохлорида (124) растворяли в 10 мл метанола. Последовательно добавляли 202 мг триэтиламина, молекулярные сита 4A, 600 мг ледяной уксусной кислоты, 871 мг (1-этоксициклопропилокси)триметилсилана и 101 мг цианоборогидрида натрия и реакционную смесь нагревали при кипячении с обратным холодильником в течение 6 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли 6 мл 2 н. раствора гидроксида натрия и реакционную смесь фильтровали. Фильтрат упаривали, остаток забирали в дихлорметан, экстрагировали 2 н. раствором гидроксидом натрия и насыщенным солевым раствором, сушили над сульфатом натрия, упаривали досуха и неочищенное вещество очищали препаративной ВЭЖХ. Фракции продукта упаривали, продукт забирали в 2 н. соляную кислоту и лиофилизировали.
Выход: 60 мг. Rt=0,50 мин (Способ №1). Определенная масса: 269,2 (M+H+).
Трет-бутиловый эфир 4-(2-оксиизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (160)
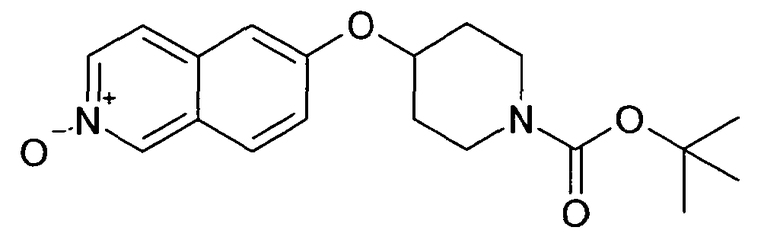
3,97 г (12,1 ммоль) трет-бутилового эфира 4-(изохинолин-6-илокси)пиперидин-1-карбоновой кислоты (154) растворяли в 100 мл дихлорметана и добавляли 4,47 г (18,1 ммоль) 3-хлорпербензойной кислоты (70%) при комнатной температуре. Реакционную смесь перемешивали в течение 1 ч и затем промывали насыщенным раствором бикарбоната натрия. Водную фазу отделяли и экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом натрия и упаривали с получением 4,19 г неочищенного продукта, который можно было использовать без дополнительной очистки. Rt=1,46 мин (Способ №1). Определенная масса: 345,2 (M+H+).
1-Хлор-6-(пиперидин-4-илокси)изохинолингидрохлорид (161)
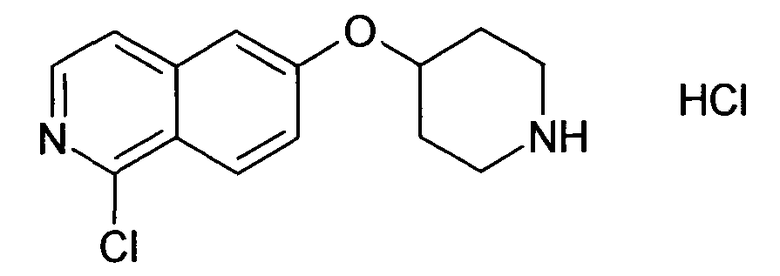
3,5 г (10,16 ммоль) Трет-бутилового эфира 4-(2-оксиизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (160) растворяли в 250 мл насыщенного HCl этанола при 50°C. Прозрачный раствор концентрировали в вакууме и остаток нагревали при кипячении с обратным холодильником в 50 мл POCl3. Через 3 ч POCl3 удаляли в вакууме и остаток помещали в H2O. Доводили значение pH до 11, добавляя гидроксид натрия, и водный раствор экстрагировали дважды дихлорметаном. Объединенные органические слои сушили над сульфатом натрия и упаривали досуха. Остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения в виде трифторацетата. Данный продукт превращали в соответствующую соль HCl, растворяя продукт в 2 н. растворе хлористоводородной кислоты с последующей лиофилизацией. Выход: 950 мг. Rt=1,03 мин (Способ №1). Определенная масса: 263,1/265,1 (M+H+).
Трет-бутиловый эфир 4-(1-Хлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (162)
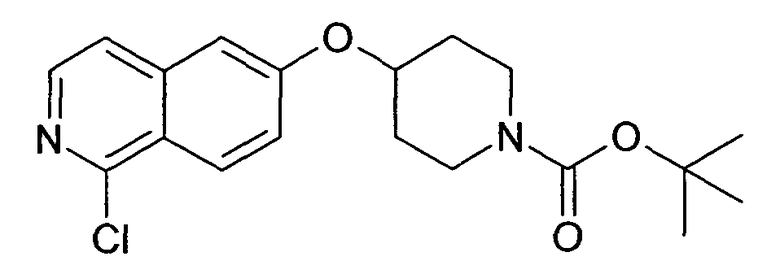
1,23 г (4,11 ммоль) 1-Хлор-6-(пиперидин-4-илокси)изохинолингидрохлорида (161) растворяли в 50 мл дихлорметана и добавляли 0,85 мл (6,15 ммоль) триэтиламина. При 0°C добавляли по каплям раствор 1,09 г (5,0 ммоль) трет-бутилкарбоната в 10 мл дихлорметана и смесь оставляли выстаиваться при комнатной температуре в течение ночи. Для дальнейшей обработки смесь дважды промывали H2O, сушили над сульфатом натрия и упаривали с получением 1,1 г желаемого продукта, который можно было использовать без дополнительной очистки. Rt=1,86 мин (Способ № 4). Определенная масса: 363,1/365,2 (M+H+).
Трет-бутиловый эфир 4-(1-метиламиноизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (163)
154 мг (0,42 ммоль) трет-бутилового эфира 4-(1-хлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (162) нагревали в 15 мл водного раствора метиламина (41%) при 110°C в запаянной трубке. Через 7 ч реакционную смесь выпаривали и остаток помещали в насыщенный раствор бикарбоната натрия и экстрагировали этилацетатом. Органический слой отделяли, сушили над сульфатом натрия и растворитель удаляли в вакууме. Остаток очищали хроматографией на силикагеле (этилацетат/метанол 5:1). Выход: 45 мг. Rt=1,14 мин (Способ № 4). Определенная масса: 358,3 (M+H+).
Метил-[6-(пиперидин-4-илокси)изохинолин-1-ил]амингидрохлорид (164)
Трет-бутиловый эфир 4-(1-метиламиноизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (163) превращали в указанное в заголовке соединение без защитной группы, применяя основную процедуру, описанную в AAV2, с получением 34 мг соответствующей соли HCl. Rt=0,69 мин (Способ №1). Определенная масса: 258,3 (M+H+).
Следующие соединения получали, следуя процедуре синтеза, описанной для соединения 164, исходя из трет-бутилового эфира 4-(1-хлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (162) и соответствующих аминов:
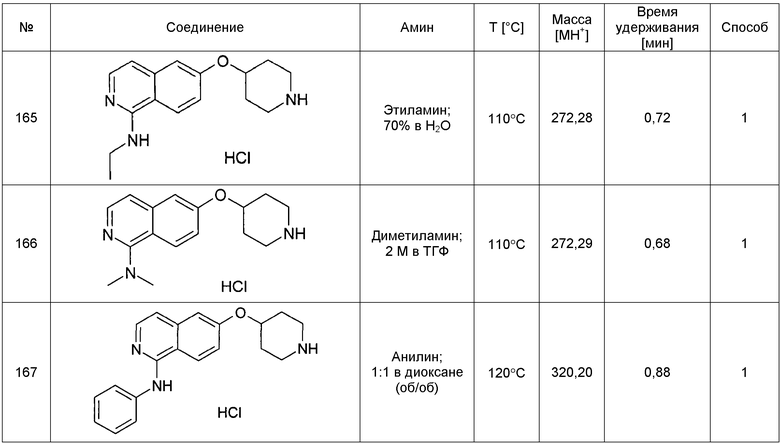
2-Хлор-N-Диметиламинометилен-5-формилбензолсульфонамид (168)
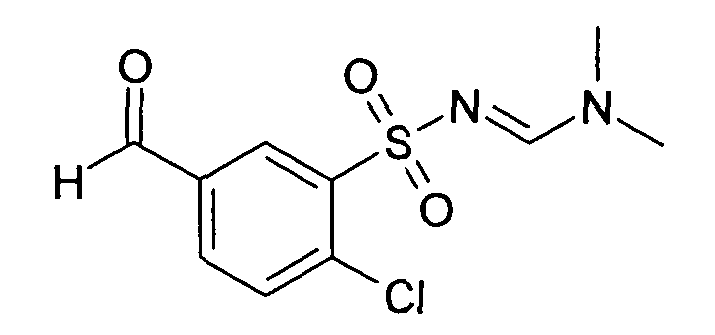
5,0 г (22,8 ммоль) 2-хлор-5-формилбензолсульфонамида растворяли в 50 мл дихлорметана. Добавляли 4,08 г (34,3 ммоль) диметилформамиддиметилацеталя и смесь нагревали при кипячении с обратным холодильником в течение 2 ч. После охлаждения до комнатной температуры раствор промывали дважды H2O, сушили над сульфатом натрия и упаривали. Получали 5,16 г неочищенного продукта и использовали на следующей стадии без дополнительной очистки. Rt=1,14 мин (Способ №1). Определенная масса: 275,1/277,1 (M+H+).
2-Хлор-N-диметиламинометилен-5-[4-(изохинолин-6-илокси)пиперидин-1-илметил]бензолсульфонамидтрифторацетат (169)
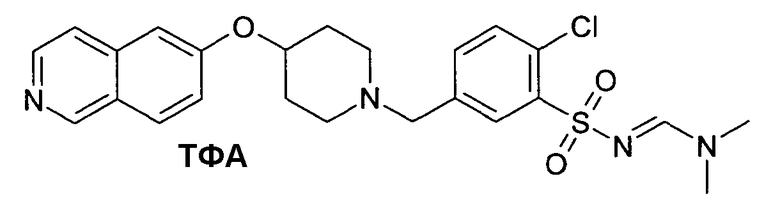
200 мг (0,88 ммоль) 6-(пиперидин-4-илокси)изохинолингидрохлорида (124) растворяли в 20 мл метанола и добавляли 158 мг (1,56 ммоль) триэтиламина. После перемешивания в течение 15 минут при комнатной температуре добавляли 467 мг (7,78 ммоль) ледяной уксусной кислоты, 482 мг (1,76 ммоль) 2-хлор-N-диметиламинометилен-5-формилбензолсульфонамида (168), 166 мг (2,64 ммоль) цианоборогидрида натрия и свежевысушенные молекулярные сита и смесь нагревали при кипячении с обратным холодильником в течение 3 ч. После перемешивания в течение ночи при комнатной температуре смесь фильтровали и фильтрат упаривали. Остаток растворяли в дихлорметане и промывали дважды насыщенным раствором бикарбоната натрия и насыщенным солевым раствором. После сушки над сульфатом магния и выпаривания растворителя неочищенный продукт очищали препаративной ВЭЖХ с выделением 133 мг желаемого продукта в виде трифторацетата. Rt=0,87 мин (Способ №2). Определенная масса: 487,2/489,2 (M+H+).
2-Хлор-5-[4-(изохинолин-6-илокси)пиперидин-1-илметил]бензолсульфонамид (170)
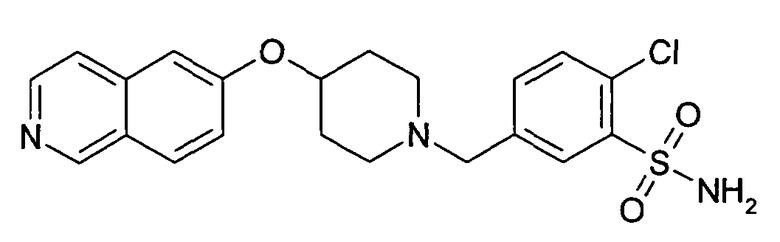
133 мг (0,27 ммоль) 2-Хлор-N-диметиламинометилен-5-[4-(изохинолин-6-илокси)пиперидин-1-илметил]бензолсульфонамида (169) растворяли в этаноле. После добавления 50 мл 2 н. раствора NaOH, раствор нагревали до 60°C в течение 6 ч. После охлаждения до комнатной температуры смесь нейтрализовали добавлением водного раствора HCl и растворитель удаляли в вакууме. Остаток перемешивали с этанолом, неорганические соли отфильтровывали и фильтрат выпаривали. Rt=0,78 мин (Способ №2). Определенная масса: 432,1 (M+H+).
Трет-бутиловый эфир 4-(5-нитроизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (171)
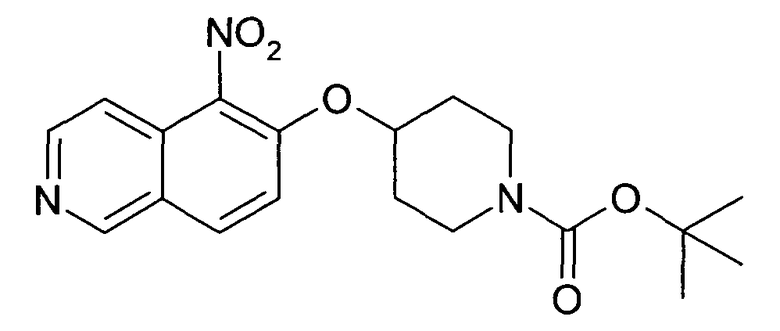
90 мг (0,47 ммоль) 6-фтор-5-нитроизохинолина (152) обрабатывали трет-бутиловым эфиром 4-гидроксипиперидин-1-карбоновой кислоты, следуя способу, описанному для получения соединения 154.
5-Нитро-6-(пиперидин-4-илокси)изохинолингидрохлорид (172)
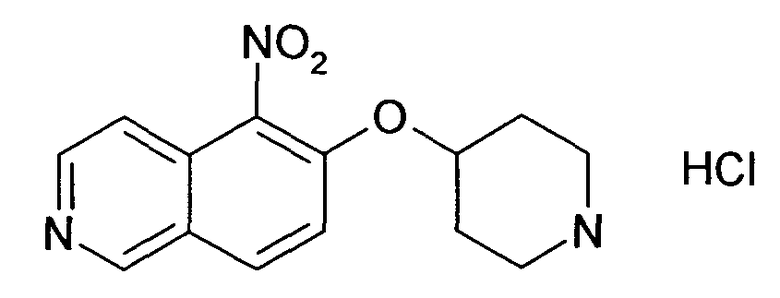
18,5 мг (0,05 ммоль) трет-бутилового эфира 4-(5-нитроизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (171) подвергали процедуре удаления защиты, описанной в AAV2, с выделением 12,5 мг указанного в заголовке соединения в виде соли HCl. Rt=0,57 мин (Способ №1). Определенная масса: 274,2 (M+H+).
7-Хлор-6-(пиперидин-4-илокси)изохинолингидрохлорид (173)
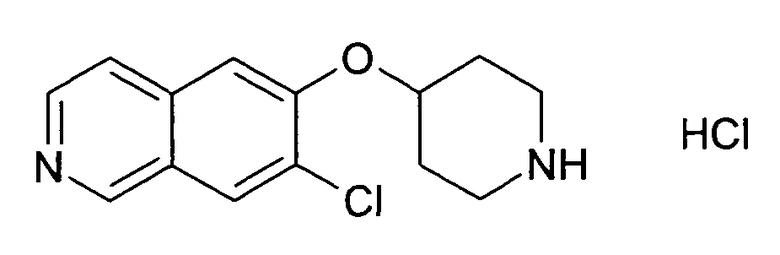
Исходя из 7-хлор-6-фторизохинолина (150), получали указанное в заголовке соединение, используя ту же процедуру синтеза, как для соединения 124. Rt=0,66 мин (Способ №1), определенная масса: 263,1/265,1 (M+H+).
Процедура синтеза для получения 3,6-дизамещенных изохинолинов
Общая схема реакции:
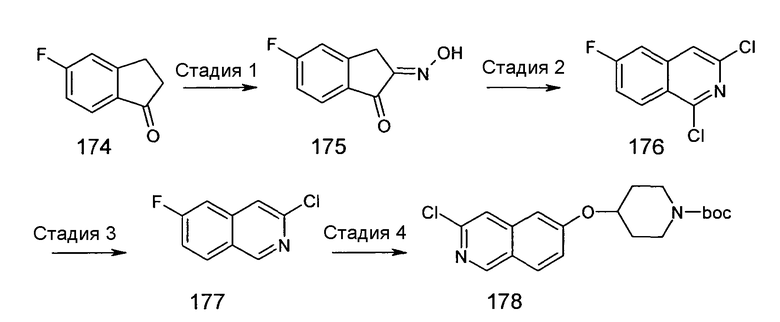
Стадия 1:
188 г 5-Фторинданона-1 (174) растворяли в 1,8 л диэтилового эфира, добавляли при 0°C 50 мл EtOH, насыщенного HCl, и 1,1 л 15% раствора этилнитрита в простом эфире в течение 1 часа.
Раствор оставляли перемешиваться еще в течение 3 часов до достижения комнатной температуры, затем частично удаляли растворитель и осажденный продукт собирали фильтрацией.
Стадия 2:
129 г продукта Стадии 1 добавляли к смеси 170 г PCl5 в 2 л POCl3. Затем добавляли ледяную HCl при 0°C до тех пор, пока раствор не становился насыщенным. Оставшуюся смесь нагревали до 60°C в течение 6 ч, частично удаляли растворитель в вакууме и остаток гидролизовали на смеси толченого льда и воды. Осажденный продукт выделяли фильтрацией.
Стадия 3:
155 г продукта Стадии 2 добавляли к смеси 740 мл HOAc и 330 мл HI (57%), содержащей 53 г красного фосфора. После кипячения с обратным холодильником в течение 4 часов раствор обрабатывали концентрированным NaOH (до значения pH 8) и осажденный продукт выделяли фильтрацией.
Стадия 4:
16,5 г N-Boc-4-гидроксипиперидина растворяли в 210 мл диглима и обрабатывали 4,1 г 50% NaH в атмосфере азота. Полученную в результате смесь перемешивали в течение 1 ч при комнатной температуре, затем добавляли 14,8 г продукта Стадии 4. Смесь оставляли перемешиваться в течение 1 дня при комнатной температуре, затем добавляли 100 мл толуола и полученную смесь 3 раза промывали водой. Органические фазы собирали и растворитель удаляли в вакууме.
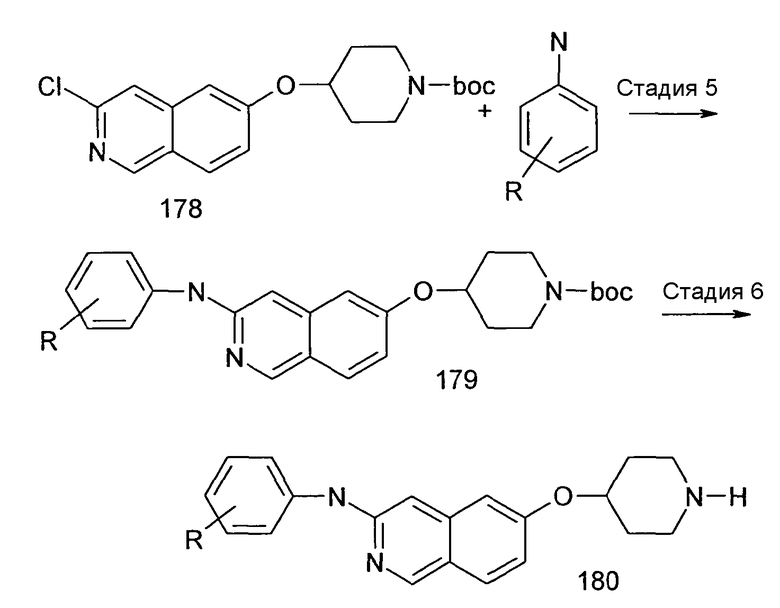
Стадия 5:
100 мг соединения 178 и 1,1 эквивалентов соответствующего анилина растворяли в 5 мл диоксана, добавляли 350 мг Cs2CO3, 20 мг Pd(OAc)2 и 60 мг XANTHPHOS и полученную смесь кипятили с обратным холодильником в атмосфере азота до полного расходования исходного вещества (реакцию контролировали при помощи ЖХМС). Растворитель удаляли в вакууме и остаток подвергали хроматографии с использованием системы ВЭЖХ.
Стадия 6:
Продукты Стадии 5 растворяли в 5 мл этанола, насыщенного газообразным HCl. После перемешивания в течение 5 ч желаемый продукт выделяли удалением растворителей в вакууме.
Все 3,5,6-тризамещенные производные синтезировали в соответствии с процедурой, проиллюстрированной для синтеза соединения 184. Для синтеза соединения 185 использовали ацетамид в качестве аминового компонента на стадии Pd связывания.
Синтез трет-бутилового эфира 4-(5-Бром-3-хлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (181)
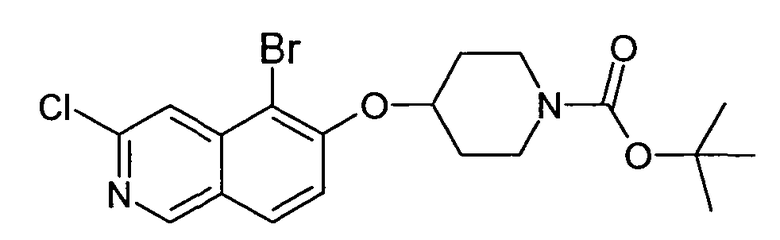
200 мг трет-бутилового эфира 4-(3-хлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (178) растворяли в 5 мл CH3CN и нагревали до 85°C. Затем добавляли смесь 148 мг N-бромсукцинимида и 9 мг AIBN в виде твердого вещества и полученную смесь нагревали при кипячении с обратным холодильником в течение 1 ч. Растворитель удаляли в вакууме и остаток подвергали колоночной флэш-хроматографии. Выход выделенного продукта составил 41%.
ЖХМС: определенная масса: 441,03, Rt=2,41 мин (Способ №1)
Синтез трет-бутилового эфира 4-[3-Хлор-5-(4-фторфенил)изохинолин-6-илокси]пиперидин-1-карбоновой кислоты (182)
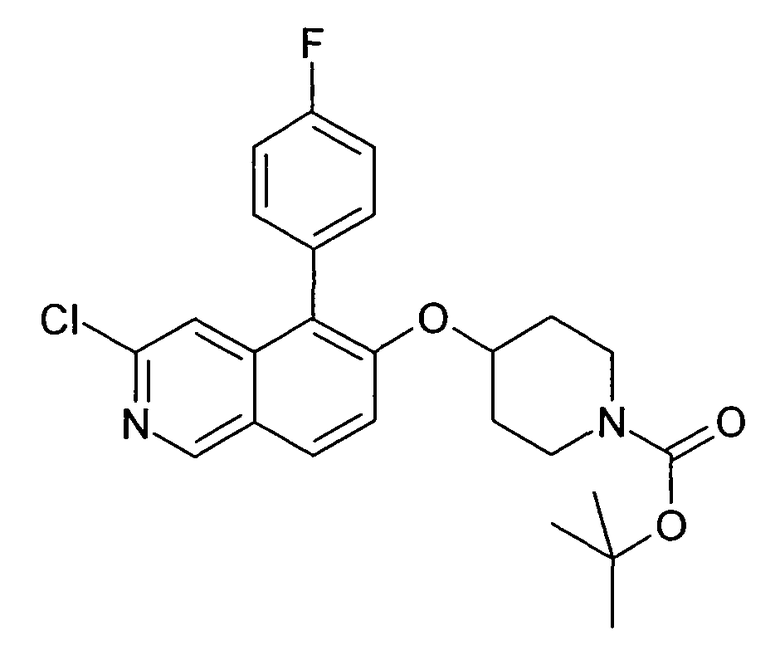
150 мг трет-бутилового эфира 4-(5-бром-3-хлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (181) растворяли в смеси 9 мл диоксана и 3 мл воды, добавляли 47 мг 4-фторбензолбороновой кислоты, 47 мг Na2CO3 и 40 мг Pd(PPh3)4 и полученную смесь нагревали до 100°C в течение 6 ч. Растворитель удаляли в вакууме и остаток подвергали хроматографии с использованием системы ВЭЖХ. Выход: 44%.
ЖХМС: определенная масса: 457,22, Rt=2,45 мин (Способ №1)
Синтез трет-бутилового эфира 4-[5-(4-фторфенил)-3-(3,4,5-триметоксифениламино)изохинолин-6-илокси]пиперидин-1-карбоновой кислоты (183)
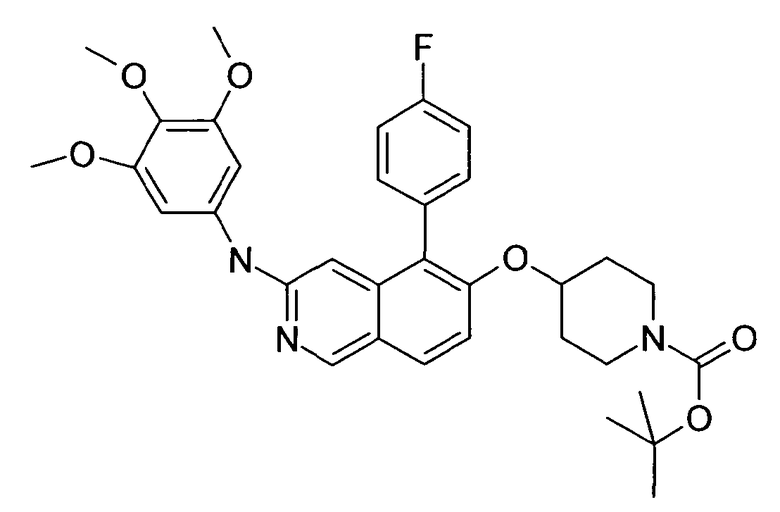
70 мг трет-бутилового эфира 4-[3-Хлор-5-(4-фторфенил)изохинолин-6-илокси]пиперидин-1-карбоновой кислоты (182) растворяли в 7 мл толуола, добавляли 20 мг Pd(OAc)2, 60 мг XANTHPHOS, 400 мг Cs2CO3 и 30 мг 3,4,5 триметоксианилина и полученную смесь нагревали до 100°C в течение 6 ч. Затем растворитель удаляли в вакууме и остаток подвергали хроматографии с использованием системы ВЭЖХ. Выход выделенного продукта был 24%.
ЖХМС: определенная масса: 604,17, Rt=1,81 мин (Способ №1)
Синтез [5-(4-фторфенил)-6-(пиперидин-4-илокси)изохинолин-3-ил]-(3,4,5-триметоксифенил)амина (184)
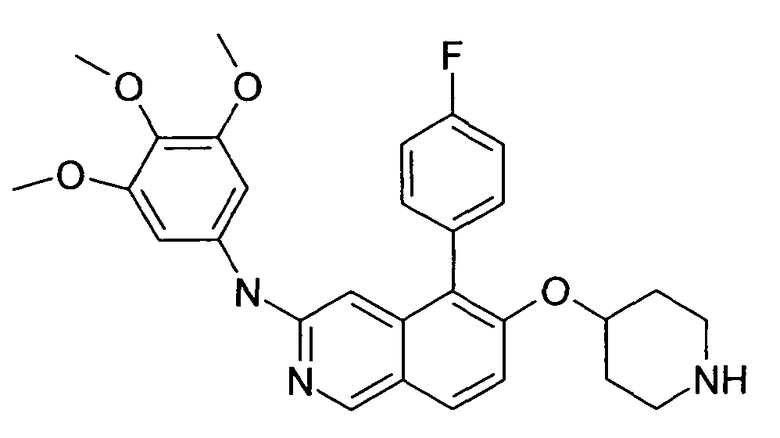
20 мг трет-бутилового эфира 4-[5-(4-фторфенил)-3-(3,4,5-триметоксифениламино)изохинолин-6-илокси]пиперидин-1-карбоновой кислоты (183) растворяли в 5 мл этанола, насыщенного HCl (газ). Полученную смесь перемешивали в течение 1 ч, затем растворитель выпаривали и собирали продукт. Выход: 85%
ЖХМС: определенная масса: 503,22, Rt=1,22 мин (Способ №1)
Примеры соединений 185-265 представлены в конце описания.
4-Этил-6-(пиперидин-4-илокси)-изохинолин (266)
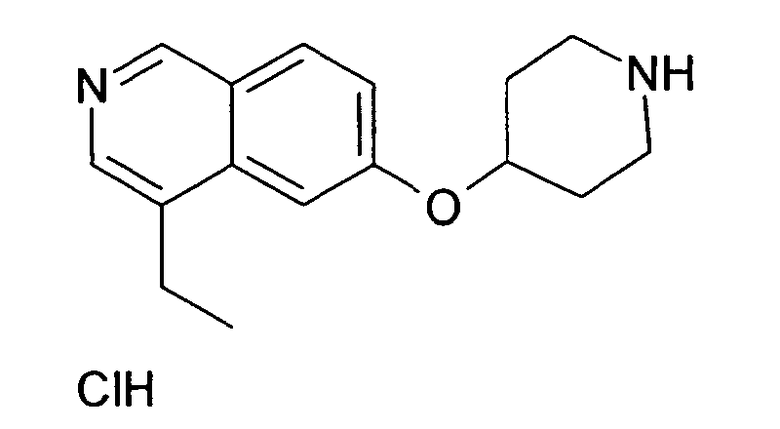
Соединение 266 синтезировали тем же способом, как описано для соединения 90, используя этилйодид. ЖХМС Способ № 1, время удерживания 0,98 мин, определенная масса 257,31 [M+H]+
Используя ту же самую последовательность реакций, как для синтеза 6-фторизохинолина (23), получали следующие два соединения:
8-Хлор-6-фторизохинолин (267)
Rt=0,83 мин (Способ №1). Определенная масса: 182,12 (M+H+).
6-Фтор-7-метилизохинолин (268)
Rt=0,70 мин (Способ №TOP). Определенная масса: 162,3 (M+H+).
8-Хлор-6-(пиперидин-4-илокси)изохинолингидрохлорид (269)
8-Хлор-6-(пиперидин-4-илокси)изохинолингидрохлорид (269) получали тем же способом, как описано для синтеза (124), исходя из 267.
Rt=0,63 мин (Способ №1). Определенная масса: 263,14 (M+H+).
6-(Пиперидин-4-илокси)-7-метилизохинолингидрохлорид (270)
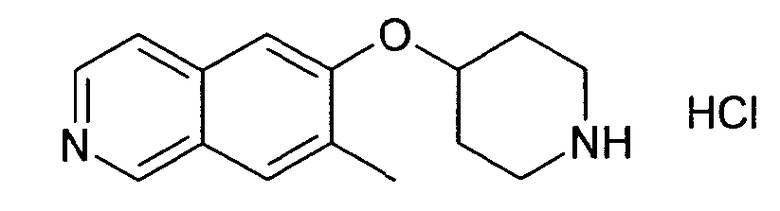
6-(Пиперидин-4-илокси)-7-метилизохинолингидрохлорид (270) получали тем же способом, как описано для синтеза (124), исходя из 268.
Rt=0,64 мин (Способ №1). Определенная масса: 243,18 (M+H+).
Следующий ряд соединений 271-372, представленный в таблице в конце описания, получали тем же способом, следуя процедуре восстановительного аминирования, используемой для получения соединений примеров 93-123, используя 269, 129 или 270 соответственно и подходящие альдегиды в качестве исходных веществ. Все данные ЖХМС получали, используя ЖХМС Способ №2.
5,6,7-Трифторизохинолин (373)
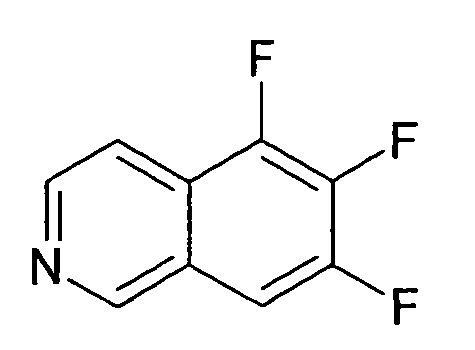
5,6,7-Трифторизохинолин (373) получали, используя ту же самую последовательность реакций, как для синтеза 6-фторизохинолина (23), исходя из 3,4,5-трифторбензальдегида. Конечная очистка при помощи препаративной ВЭЖХ давала требуемый изохинолин в виде трифторацетата. Rt=1,15 мин (Способ №2). Определенная масса: 183,0.
Трет-бутиловый эфир 4-(5,7-дифторизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (374)
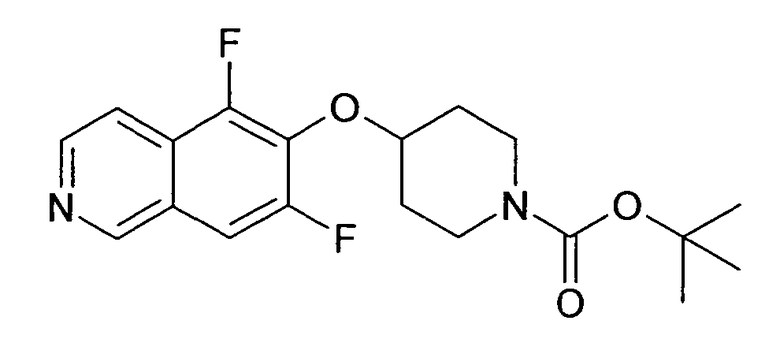
Указанное в заголовке соединение синтезировали, следуя протоколу, описанному для трет-бутилового эфира 4-(изохинолин-6-илокси)пиперидин-1-карбоновой кислоты (154). Rt=1,27 мин (Способ №TOP). Определенная масса: 365,2 (M+H+).
5,7-Дифтор-6-(пиперидин-4-илокси)изохинолин (375)
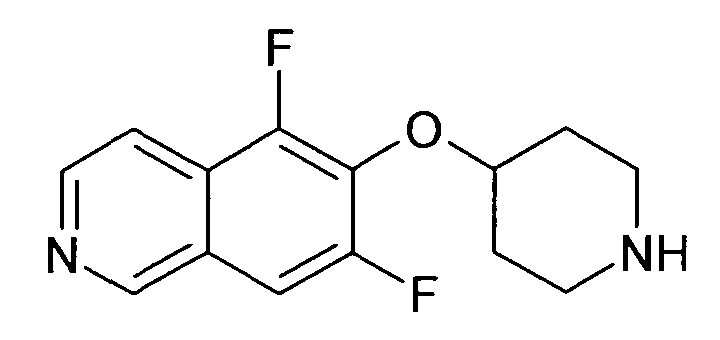
Трет-бутиловый эфир 4-(5,7-дифторизохинолин-6-илокси)пиперидин-1-карбоновой кислоты (374) подвергали процедуре удаления защиты в смеси метанол/2 н. раствор HCl, используя процедуру, описанную в AAV2, с получением указанного в заголовке соединение в виде соли HCl. Rt=0,43 мин (Способ №TOP). Определенная масса: 265,1 (M+H+).
5,7-Дихлор-6-фторизохинолин (376)
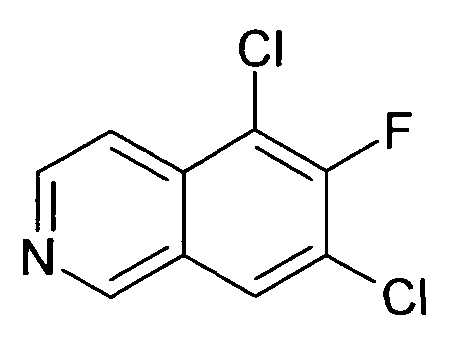
5,0 г (27,5 ммоль) 7-Хлор-6-фторизохинолина (150) растворяли в 90 мл концентрированной серной кислоты. При комнатной температуре добавляли 7,35 г (55,0 ммоль) N-хлорсукцинимида и смесь перемешивали при 50°C. После выстаивания в течение ночи при комнатной температуре добавляли дополнительные 3 экв. N-хлорсукцинимида и снова добавляли 5 экв. N-хлорсукцинимида и температуру повышали до 80°C. После того как дальнейшую конверсию больше не наблюдали, смесь охлаждали до комнатной температуры и выливали на лед. Водный раствор доводили до щелочного значения pH, добавляя твердый NaOH. Осадок отфильтровывали и промывали три раза дихлорметаном. Желаемый изохинолин, 1,09 г, выделяли после сушки органическими фильтратами с MgSO4 и выпаривания растворителя. Rt=1,26 мин (Способ №TOP). Определенная масса: 216,0/218,0 (M+H+).
Трет-бутиловый эфир 4-(5,7-дихлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (377)
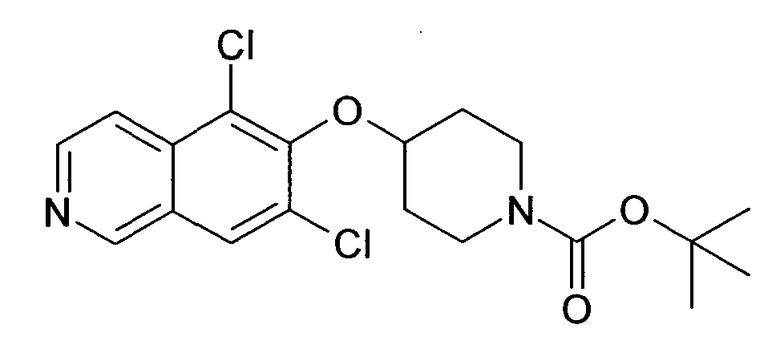
Указанное в заголовке соединение синтезировали, следуя протоколу, описанному для трет-бутилового эфира 4-(изохинолин-6-илокси)пиперидин-1-карбоновой кислоты (154). После конечной очистки при помощи препаративной ВЭЖХ и упаривания фракциий продукта Boc-группа частично отщеплялась. Rt=1,71 мин (Способ №2). Определенная масса: 397,2/399,2 (M+H+).
5,7-Дихлор-6-(пиперидин-4-илокси)изохинолин (378)
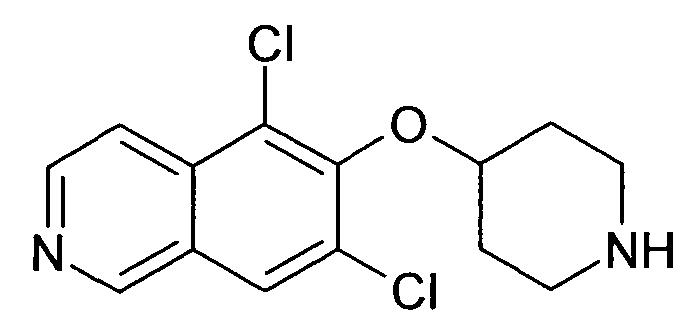
150 мг Трет-бутилового эфира 4-(5,7-дихлоризохинолин-6-илокси)пиперидин-1-карбоновой кислоты (377, с уже частично удаленной защитной группой) растворяли в 10 мл дихлорметана и при 0°C добавляли 1 мл трифторуксусной кислоты. Раствор перемешивали в течение 2 ч при комнатной температуре. Для дальнейшей обработки добавляли 50 мл дихлорметана и раствор промывали насыщенным раствором NaHCO3. Слои разделяли и водную фазу экстрагировали один раз дихлорметаном. Объединенные органические слои снова промывали насыщенным раствором NaHCO3, сушили MgSO4 и упаривали. Остаток очищали при помощи препаративной ВЭЖХ. Фракции продукта упаривали и остаток растворяли в 2 н. растворе HCl. После упаривания указанное в заголовке соединение выделяли в виде соли HCl. Rt=0,90 мин (Способ №2). Определенная масса: 297,1/299,1 (M+H+).
Определение ингибирования Rho киназы
Для измерения ингибирования Rho-киназы определяли значения IC50 в соответствии со следующим протоколом:
Буфер: 25 мМ Tris pH7,5; 0,02% BSA; 5% глицерина; 0,008% Triton X100; 2% ДМСО, 1 мМ DTT; 1 мМ MgCl2; 0,5 мкКи/лунка γ33P АТФ
Фермент: ROCKII или ROKα) (Upstate, Catalog № 14-451 Lot № 24880U) 0,1 нг/мкл
Конечная концентрация АТФ в реакционной смеси 40 мкм
Биотинилированный субстрат, разбавленный до 0,25 мкм буферным раствором, описан выше (без АТФ)
1. 10 мкл Tris буфера (± ингибитор)
2. Добавить 30 мкл раствора фермента
3. Начать реакцию с 30 мкл смеси субстрат/АТФ/АТФ33
4. Инкубировать в течение 20 мин при комнатной температуре
5. Остановить реакцию при помощи 30 мкл 50 мМ EDTA
6. Перенести 50 мкл полученного раствора в Streptavidin Flash Plate plus, Perkin Elmer, SMP 103A
7. Инкубировать в течение 30 мин при комнатной температуре
8. Промыть 4 раза 300 мкл PBS/0,1% Tween 20
9. Измеряли радиоактивность в лунках
Полученную активность обозначали как отрицательный логарифм IC50 (pIC50) следующим образом:
+: 3,0 ≤ pIC50 < 4,0
++ 4,0 ≤ pIC50 < 5,0
+++: 5,0 ≤ pIC50 < 6,0
++++: 6,0 ≤ pIC50
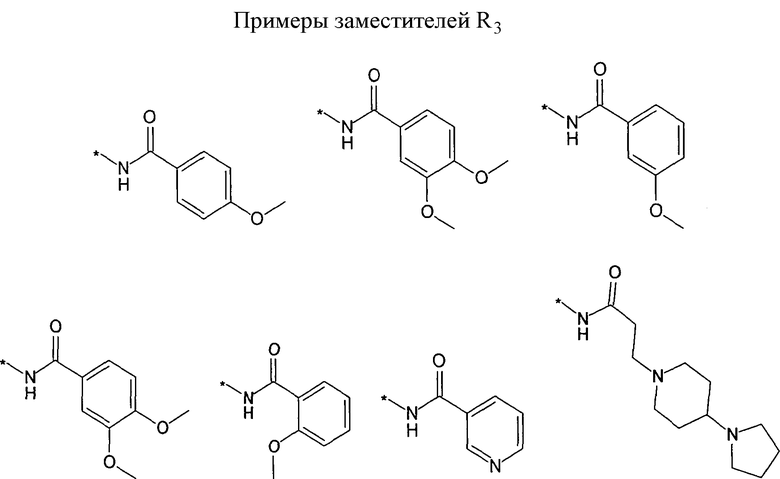
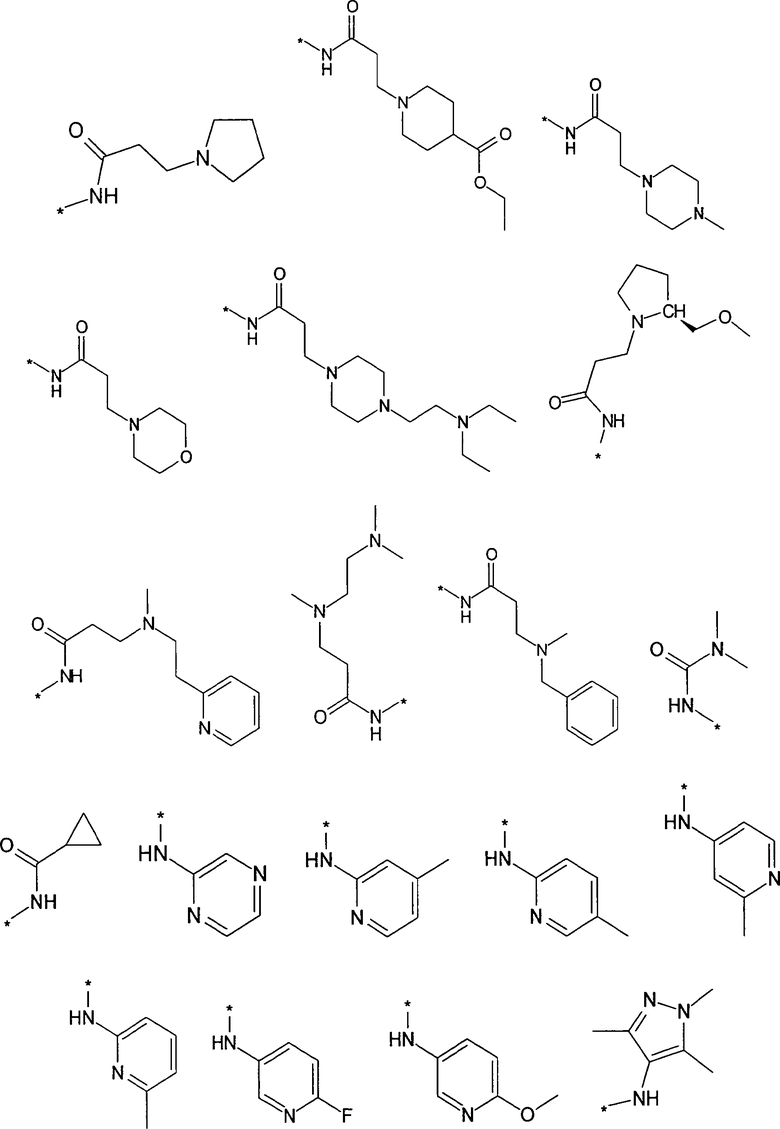
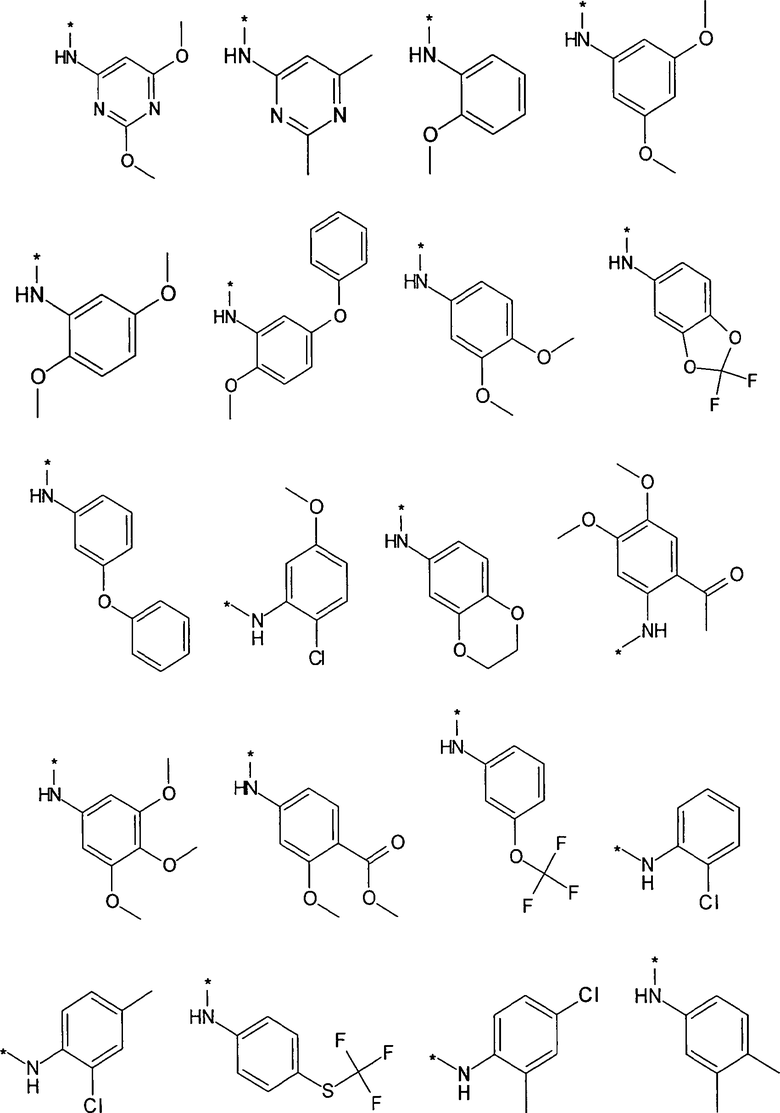
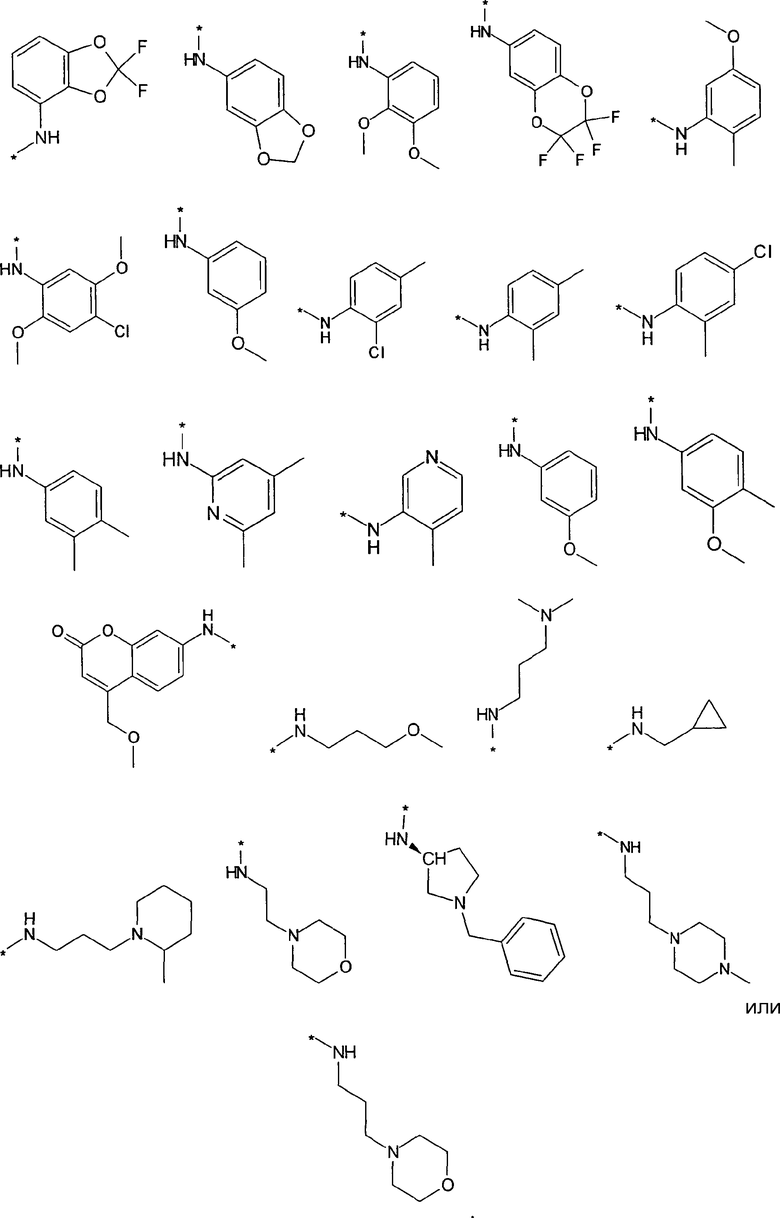
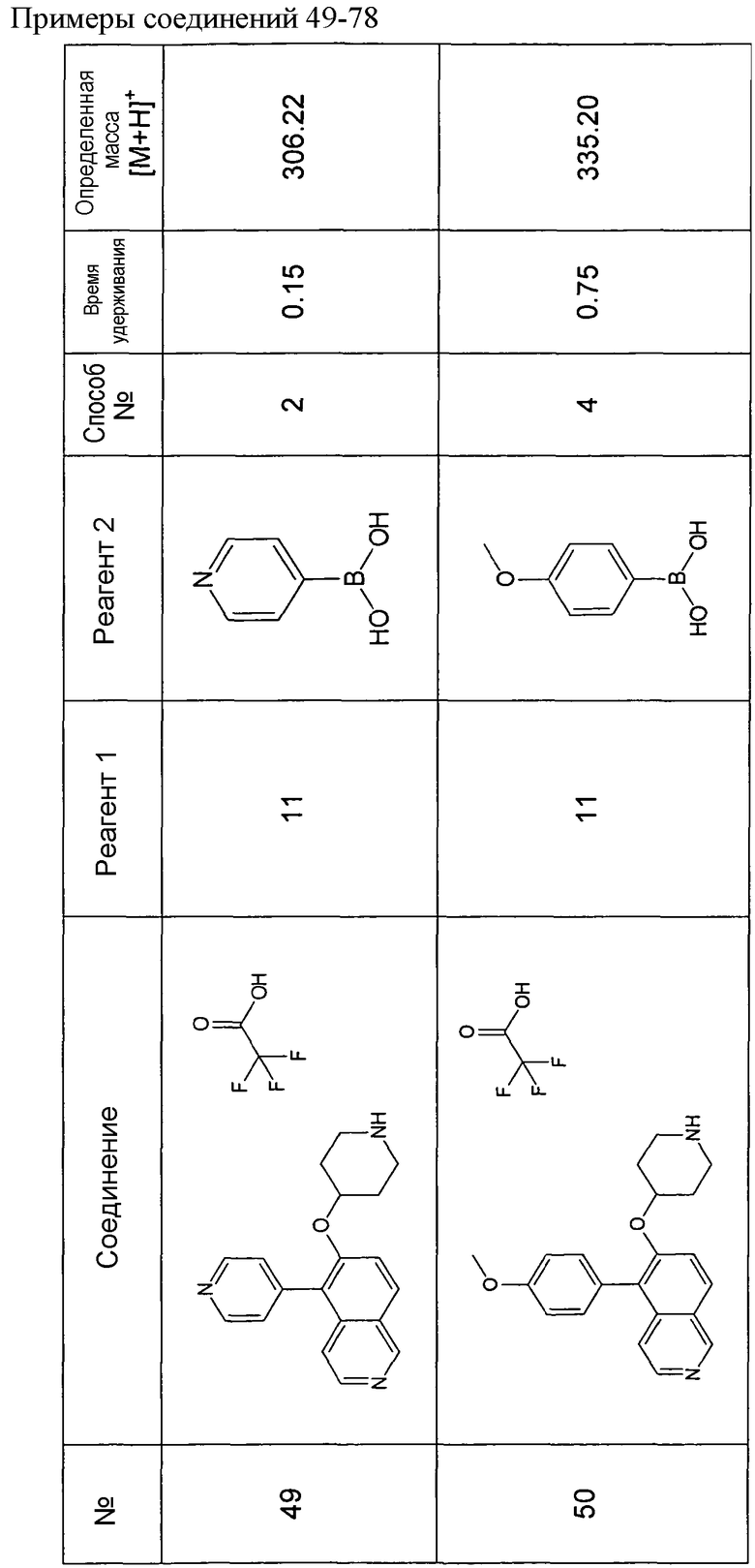
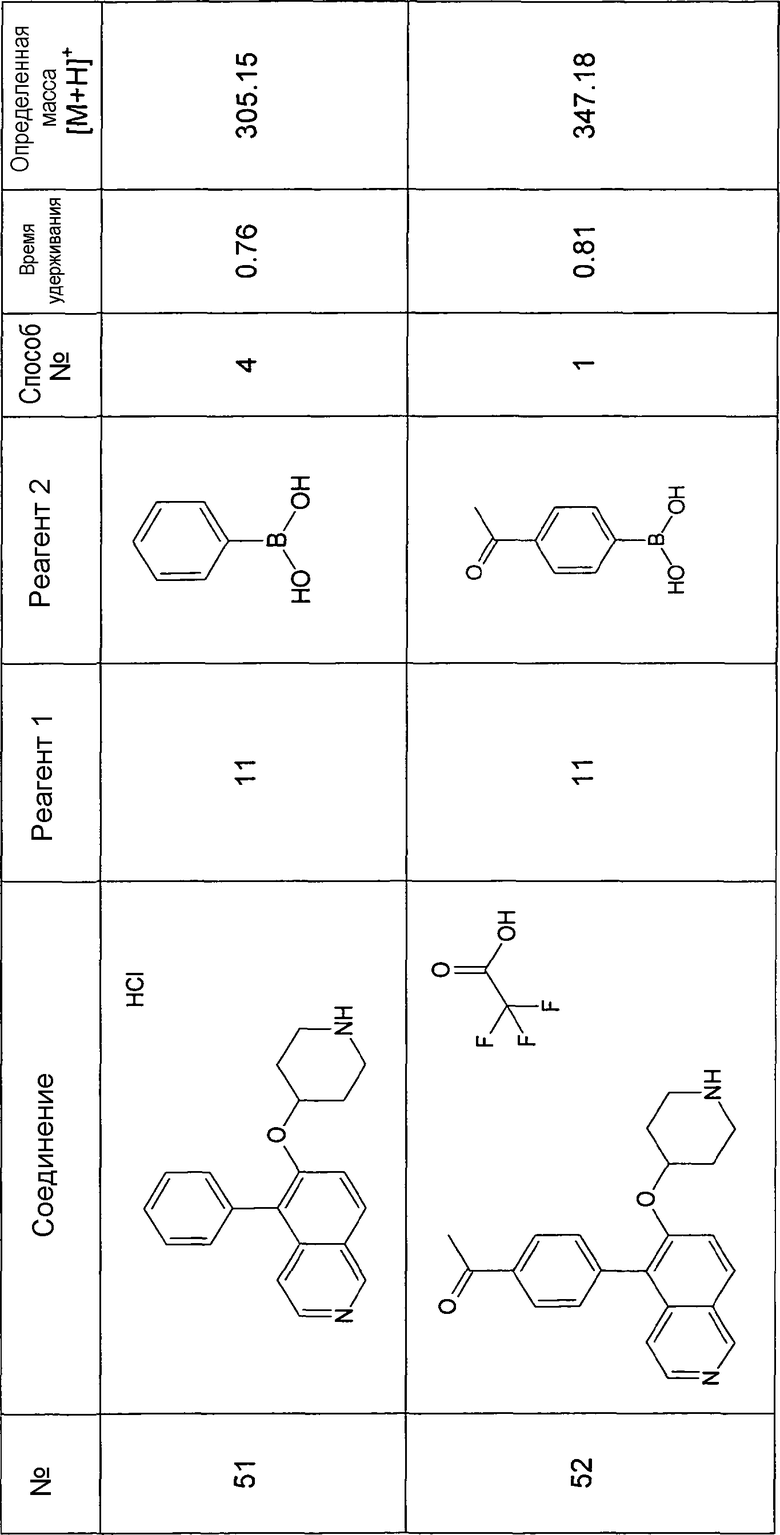
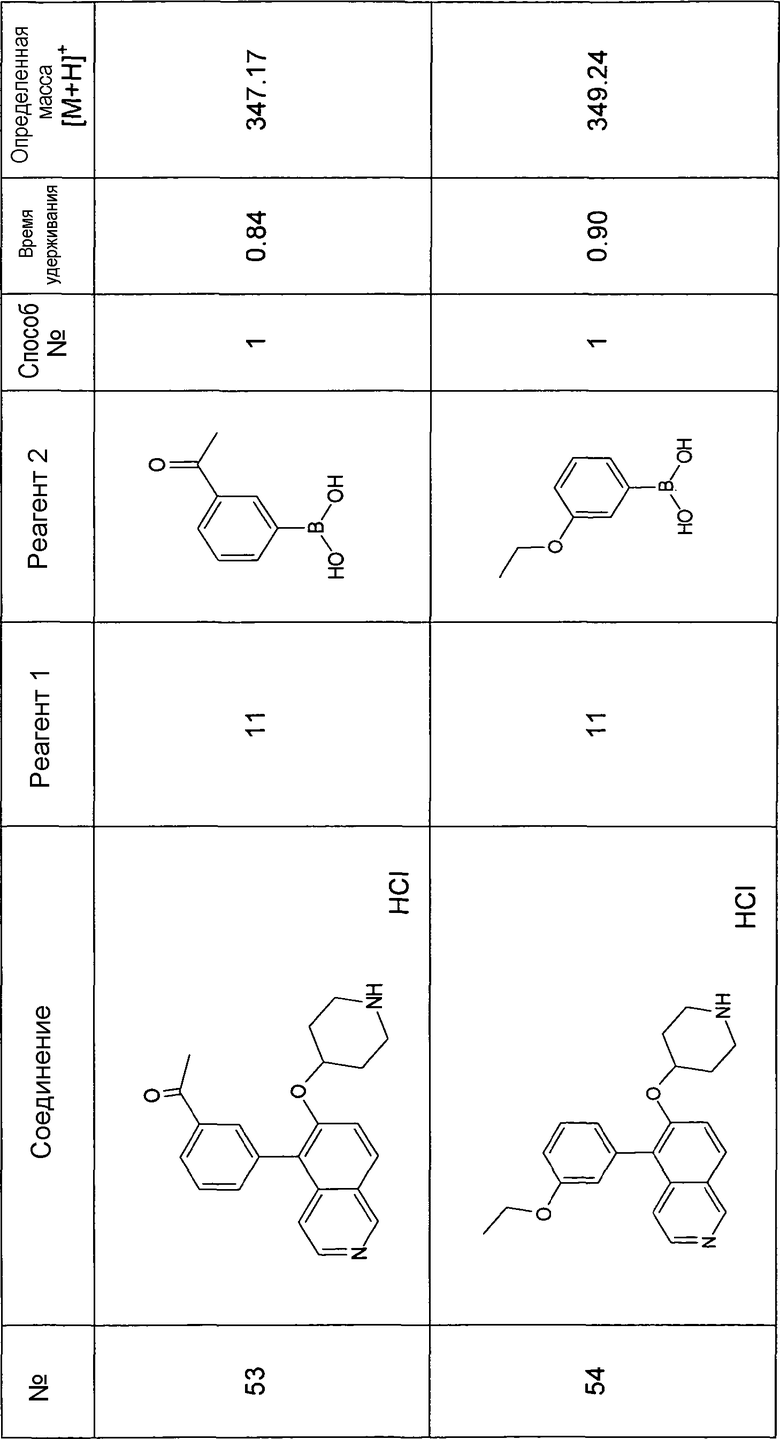
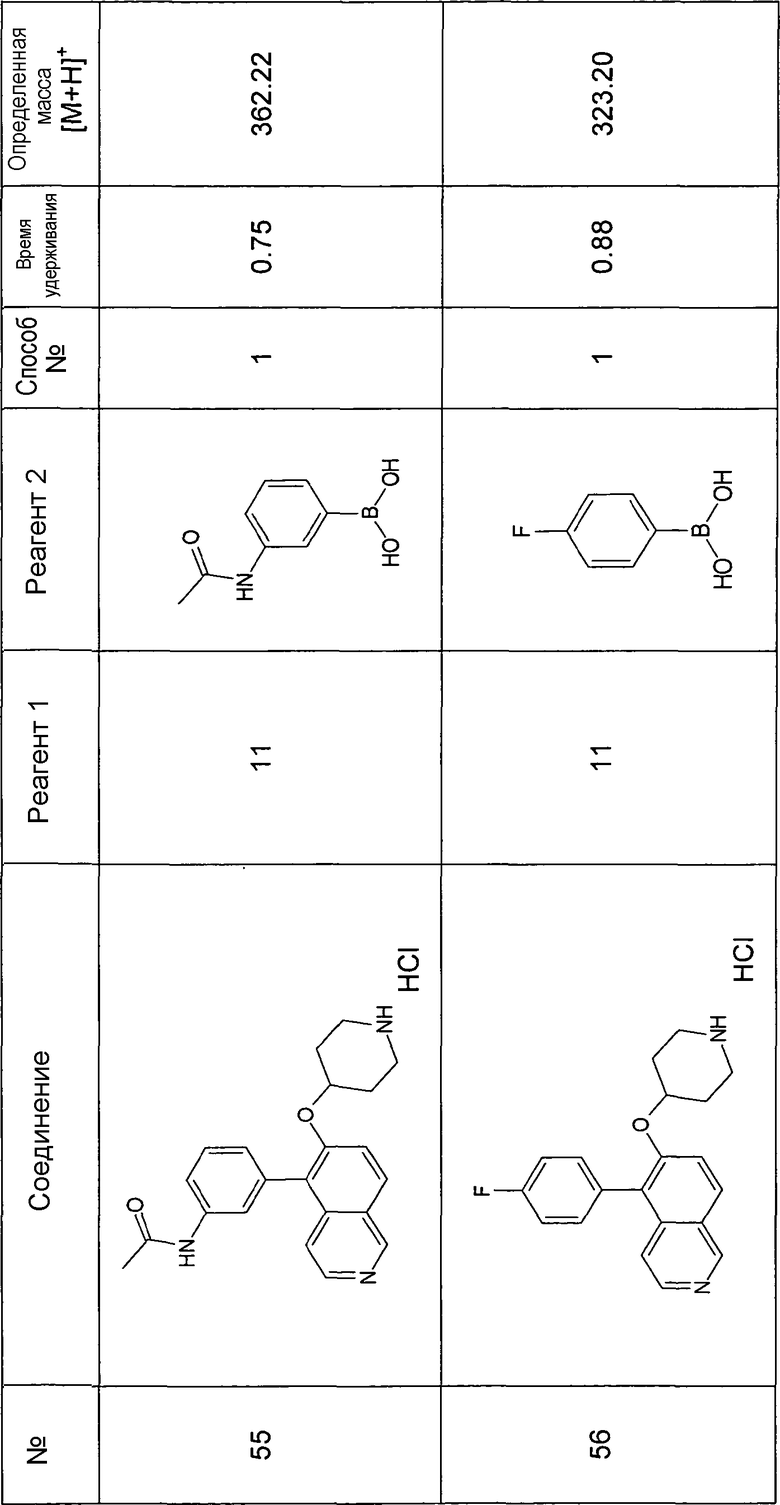
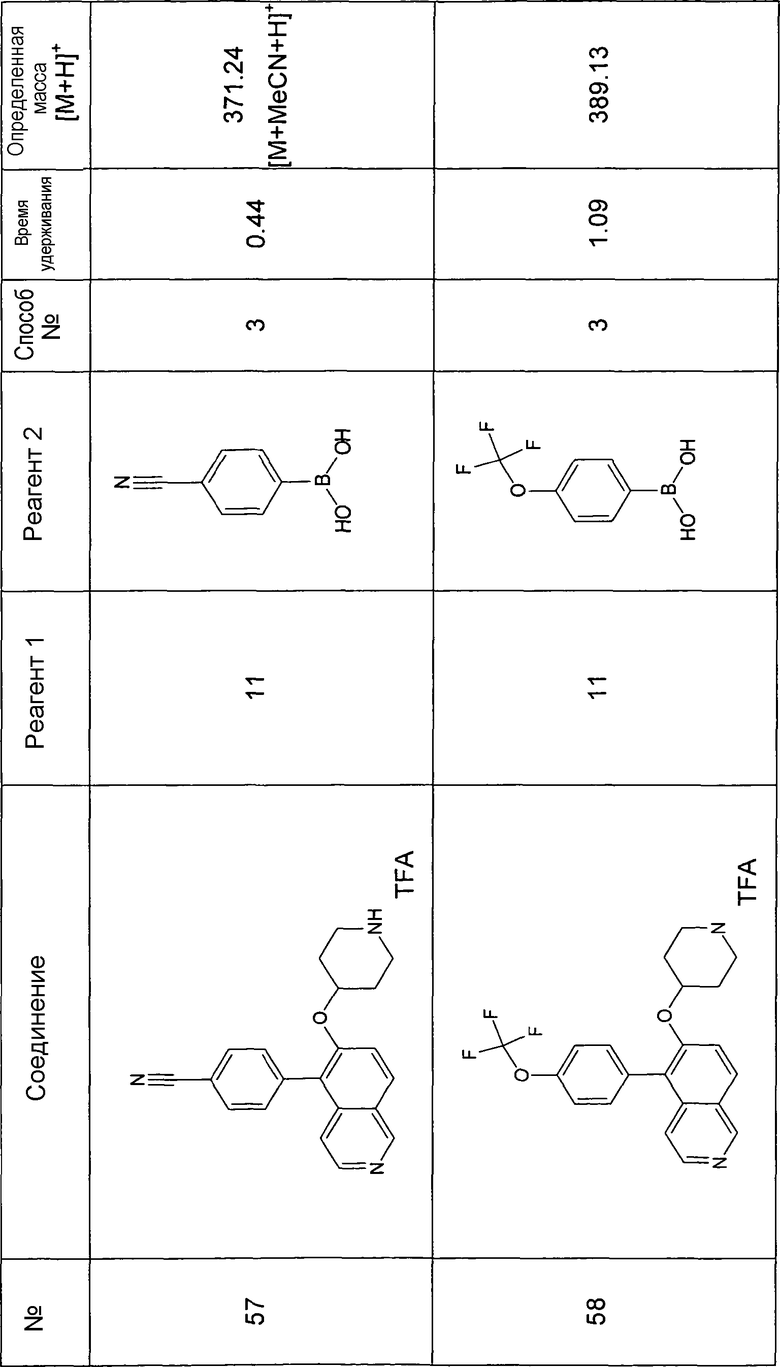
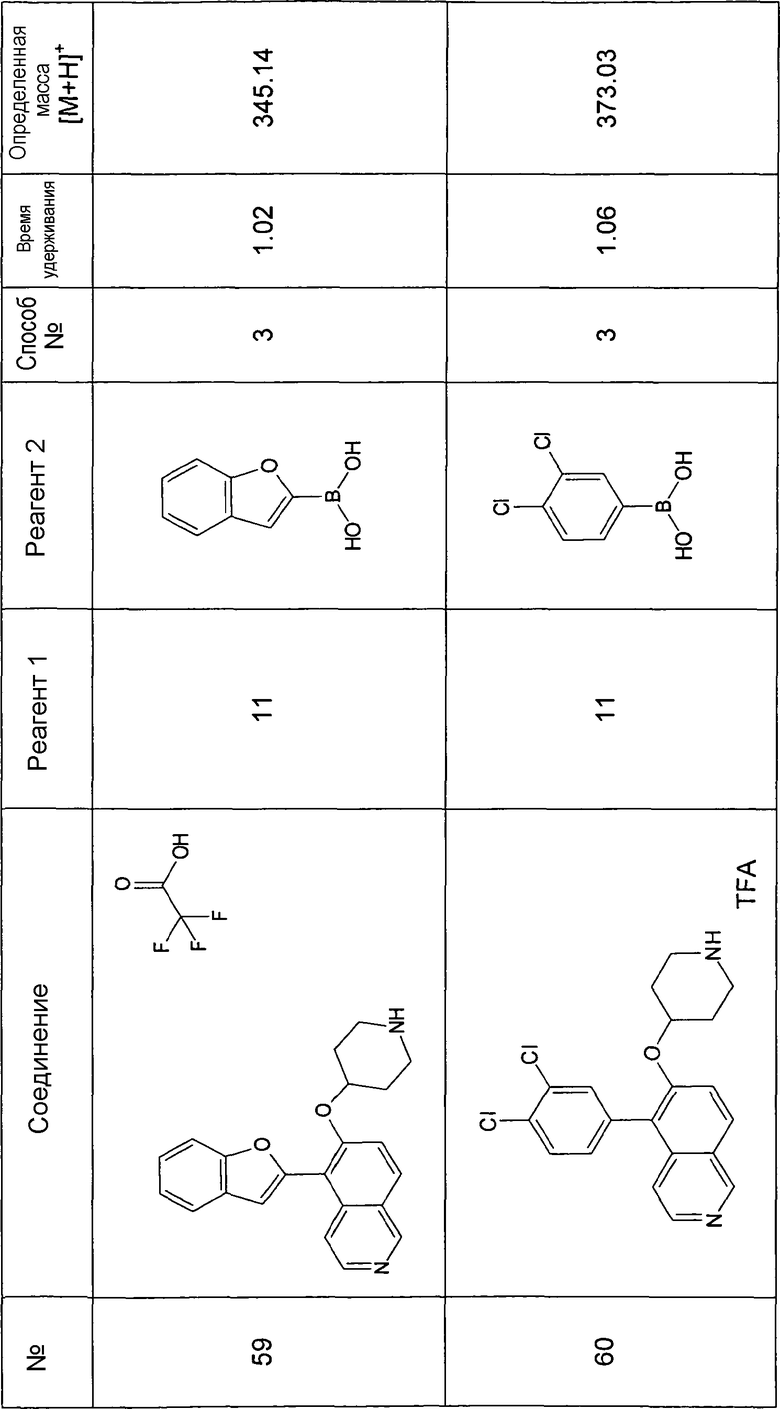
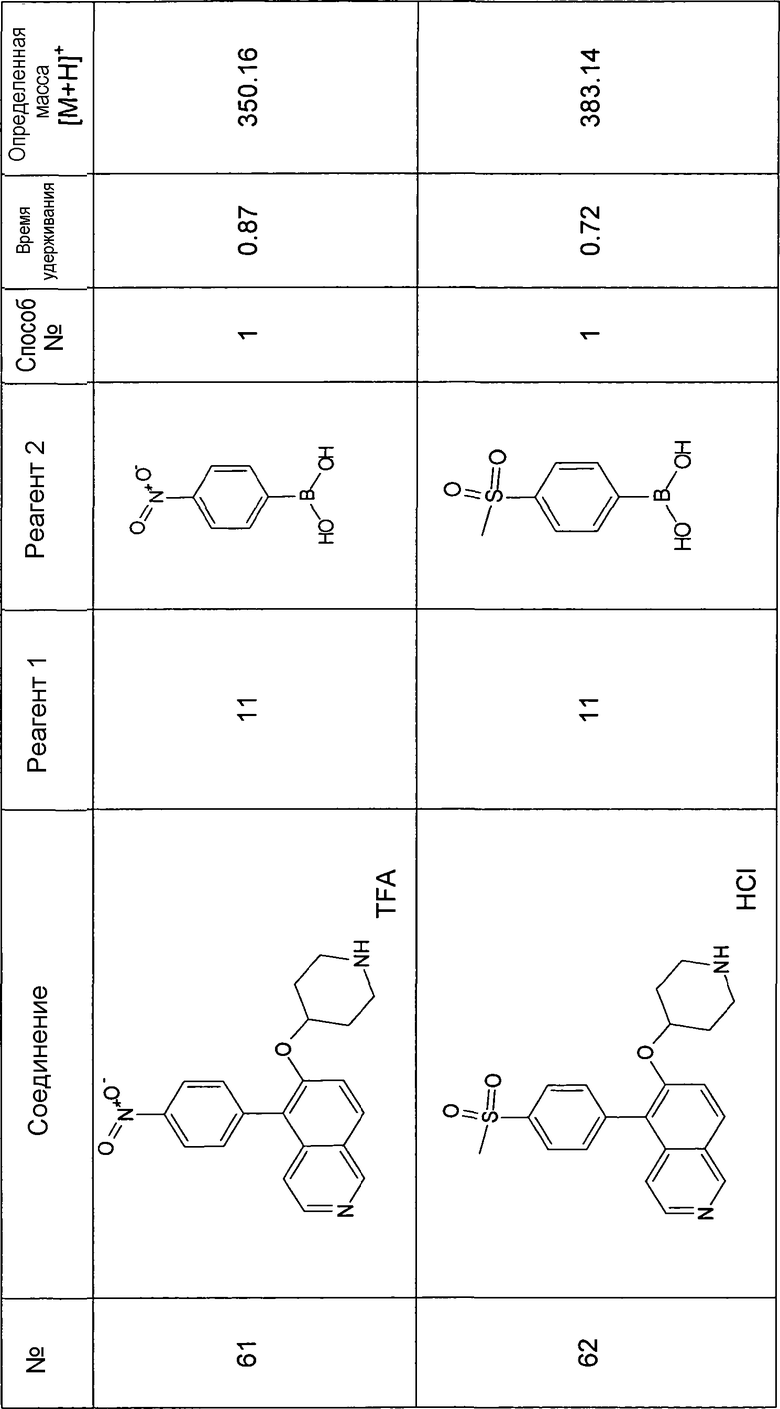

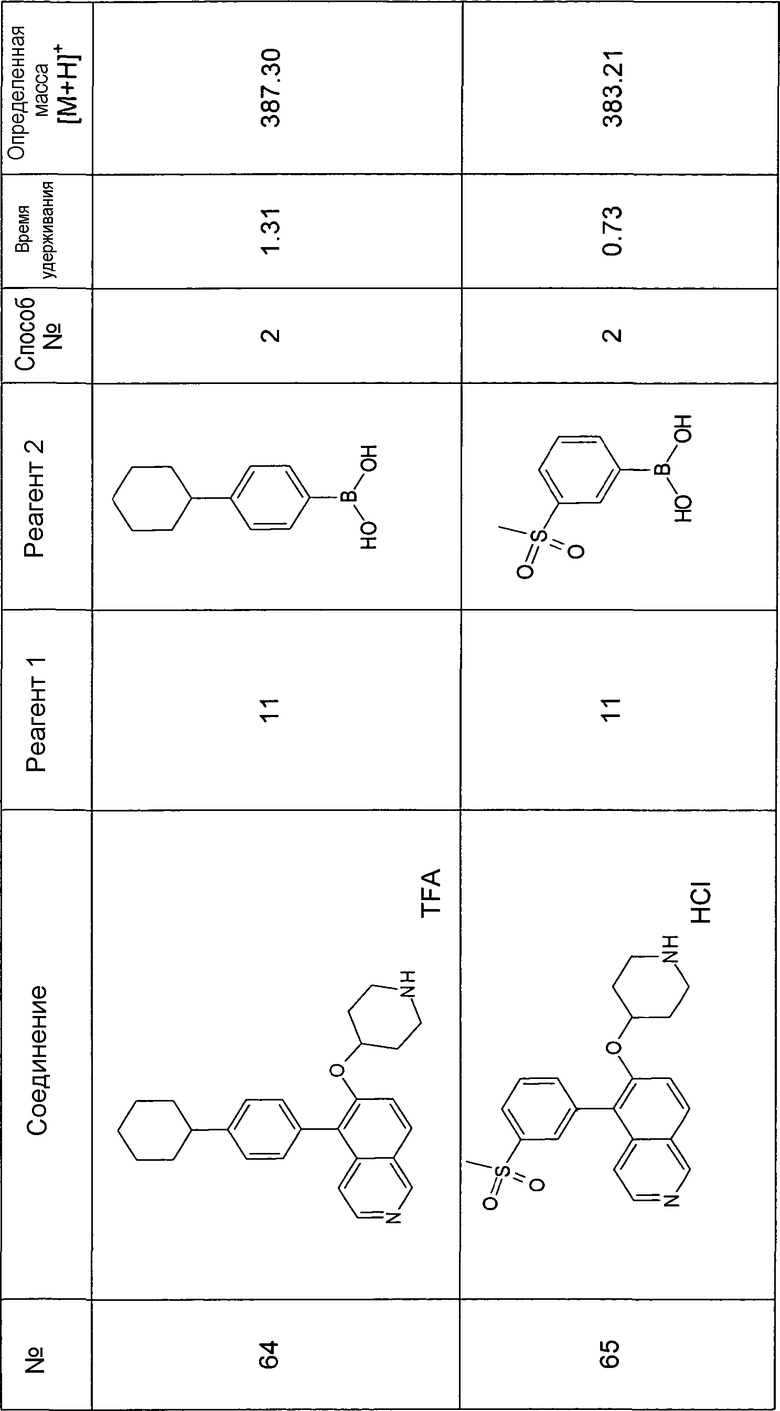
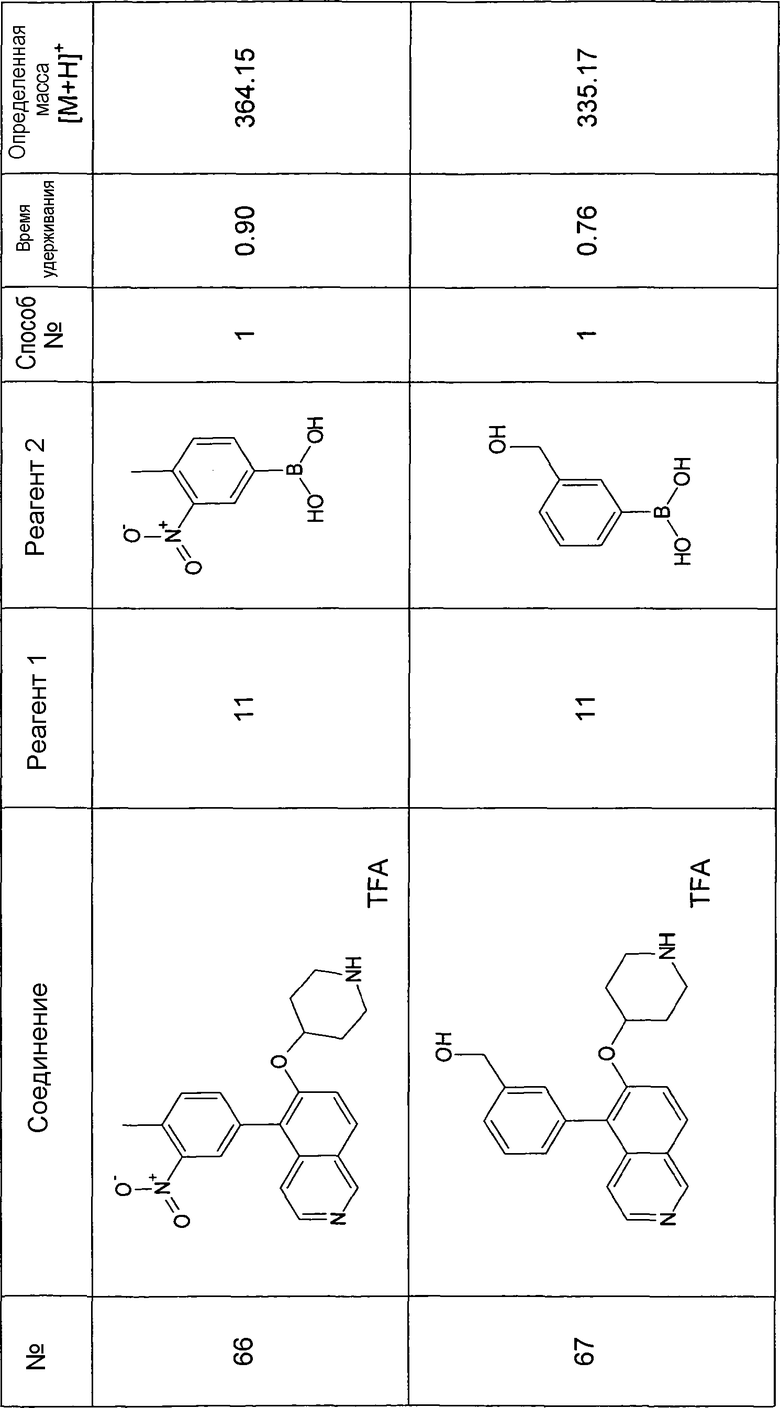
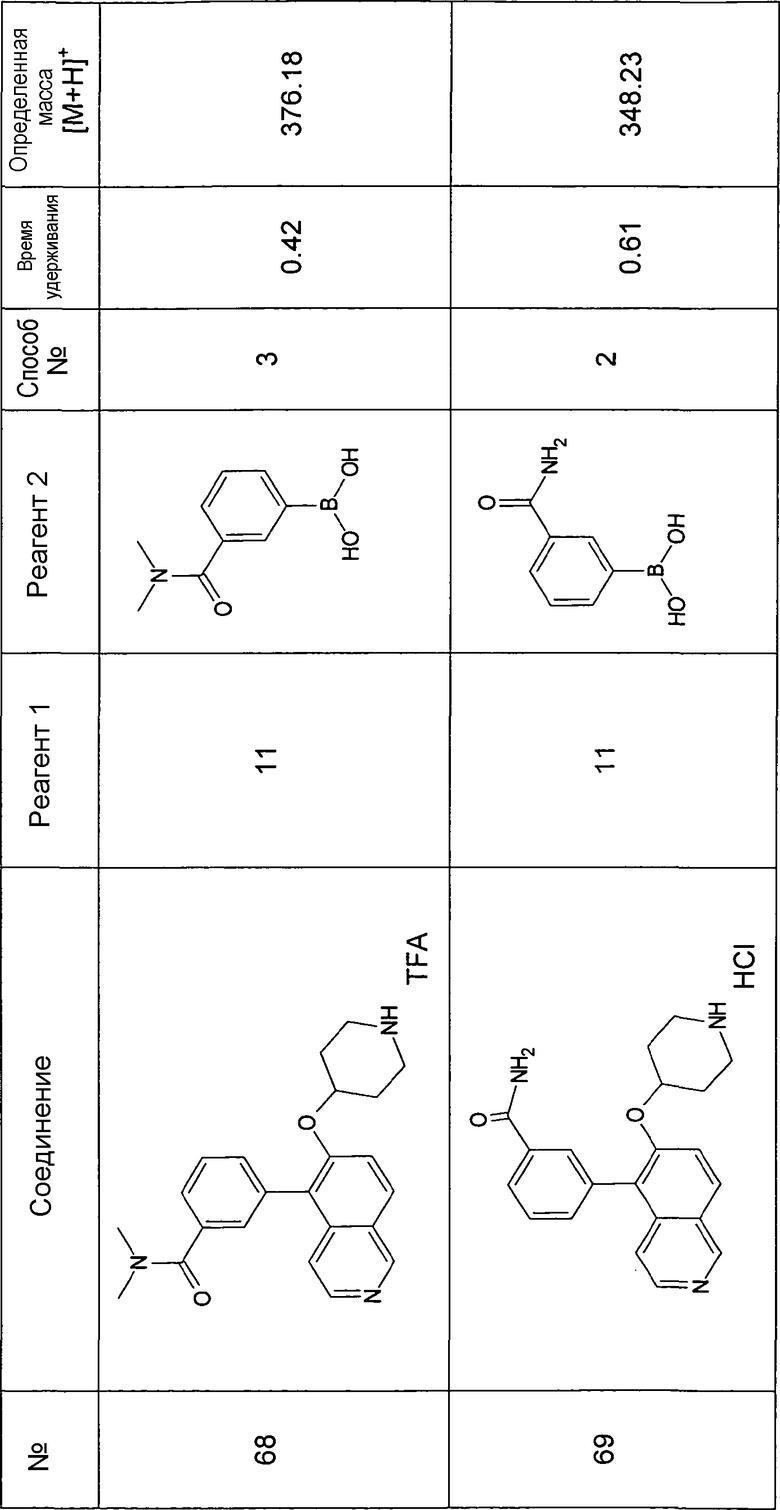
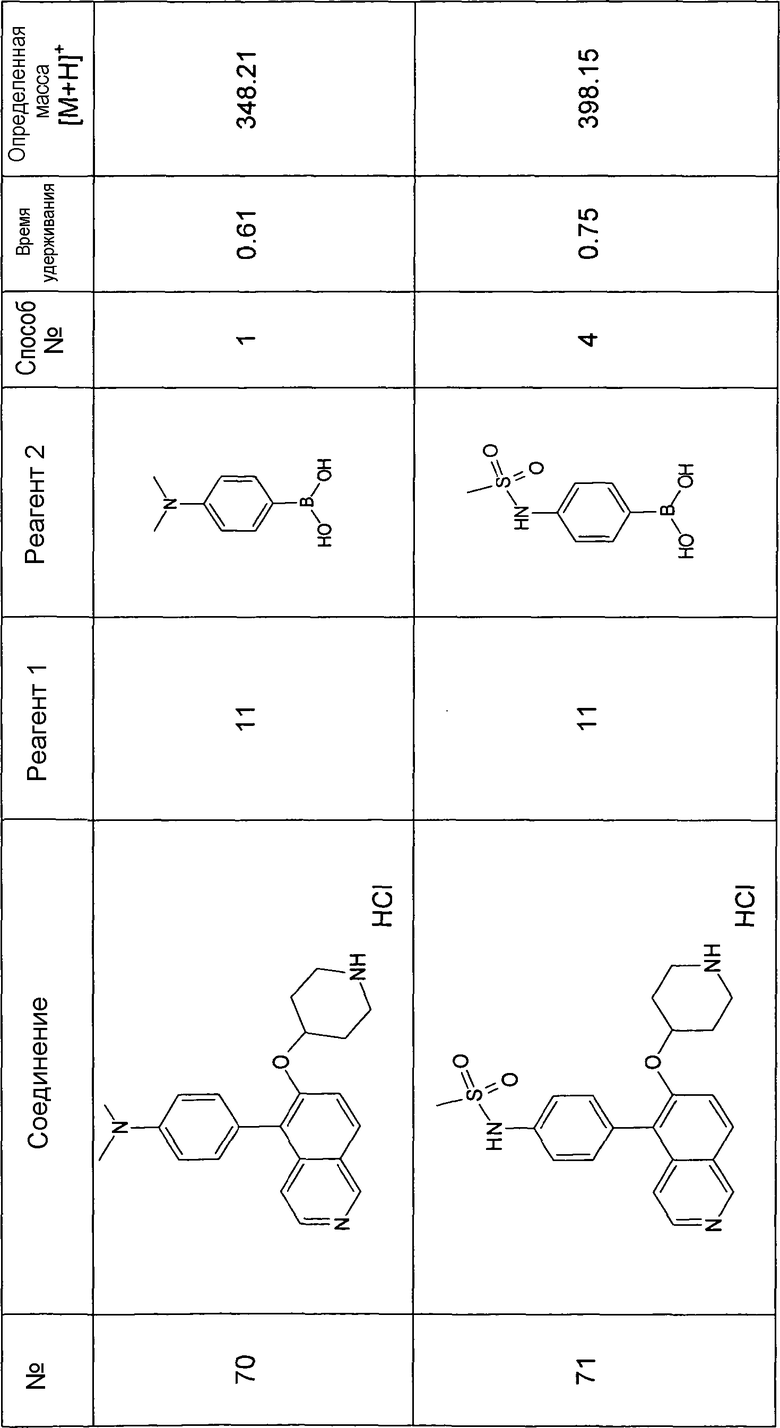
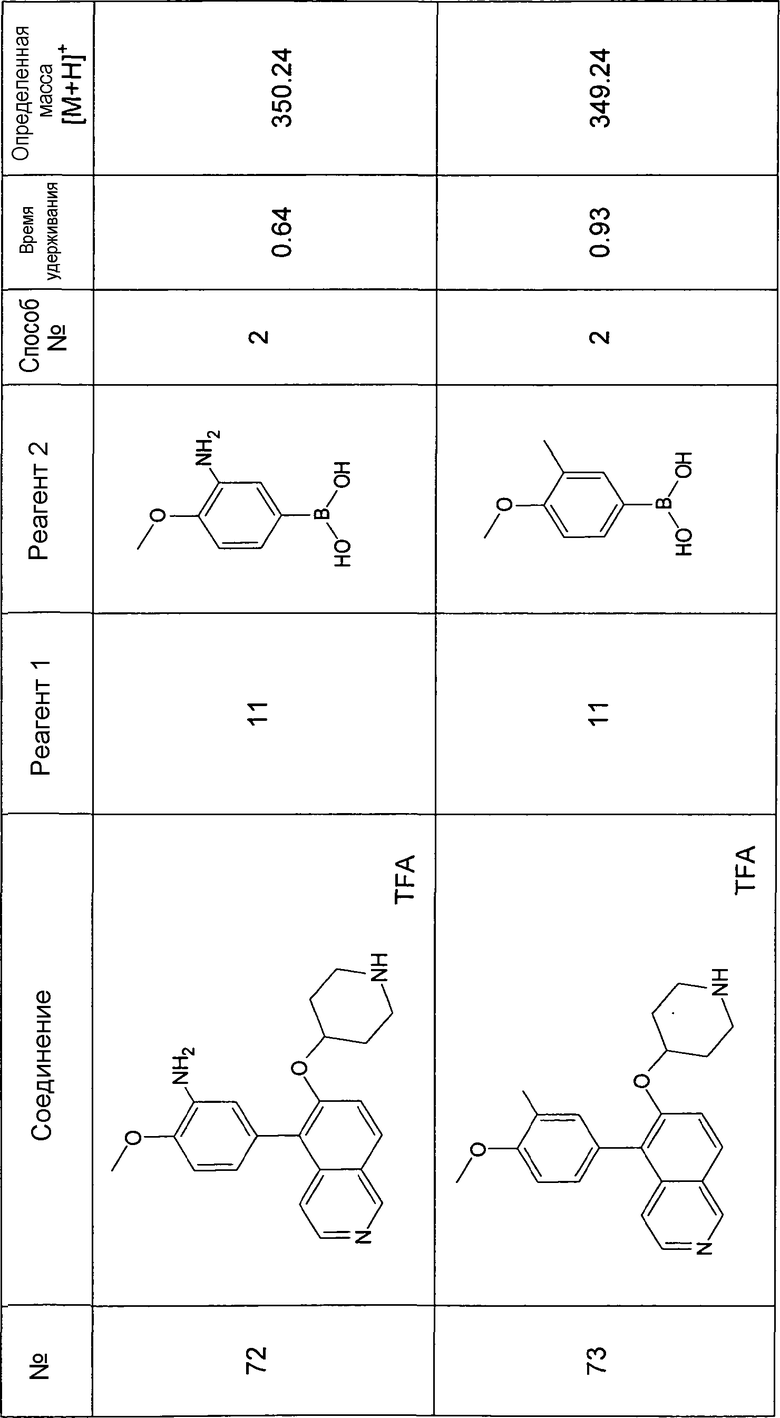
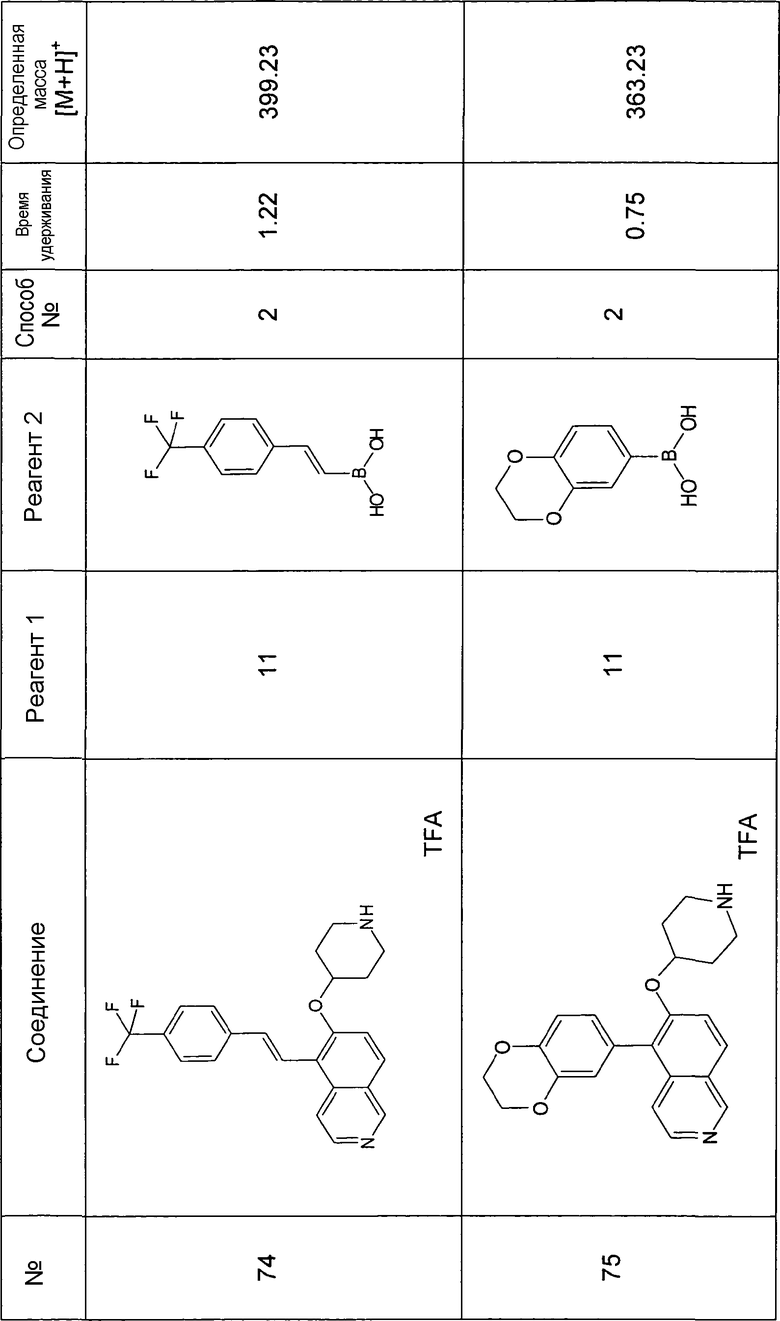
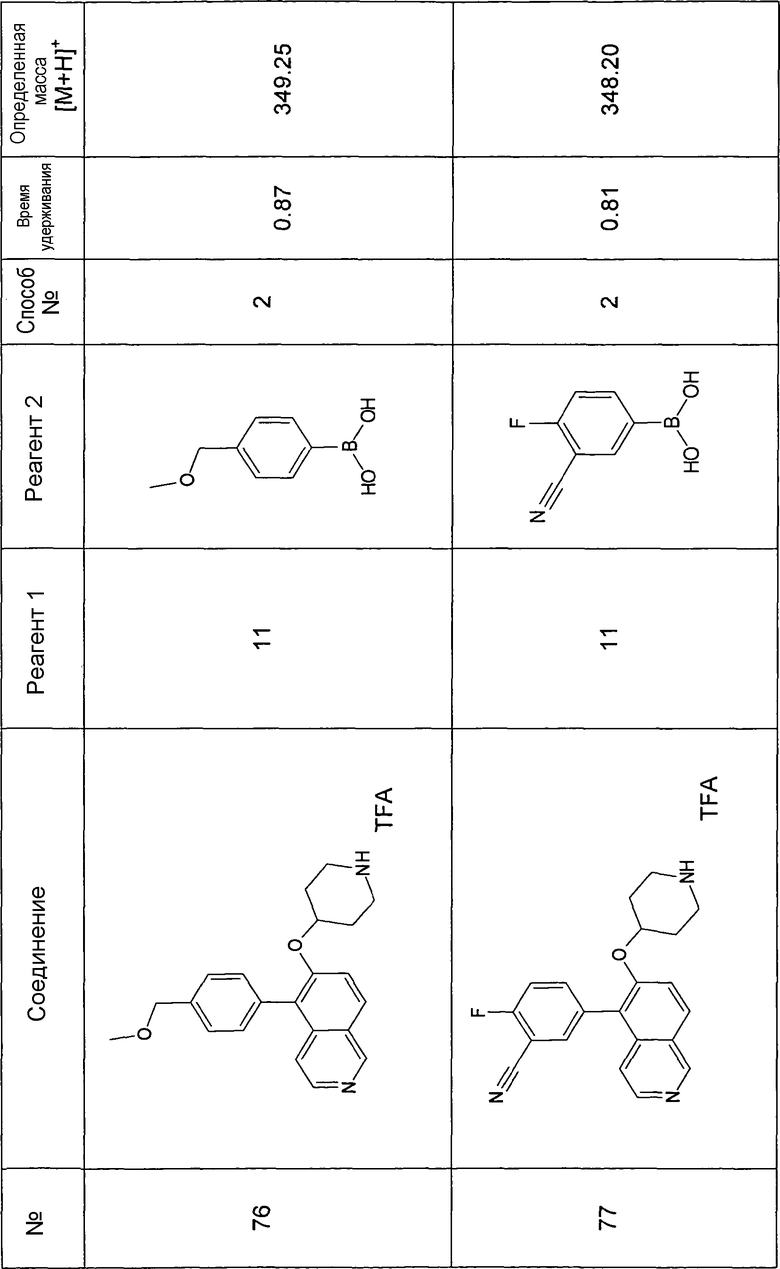
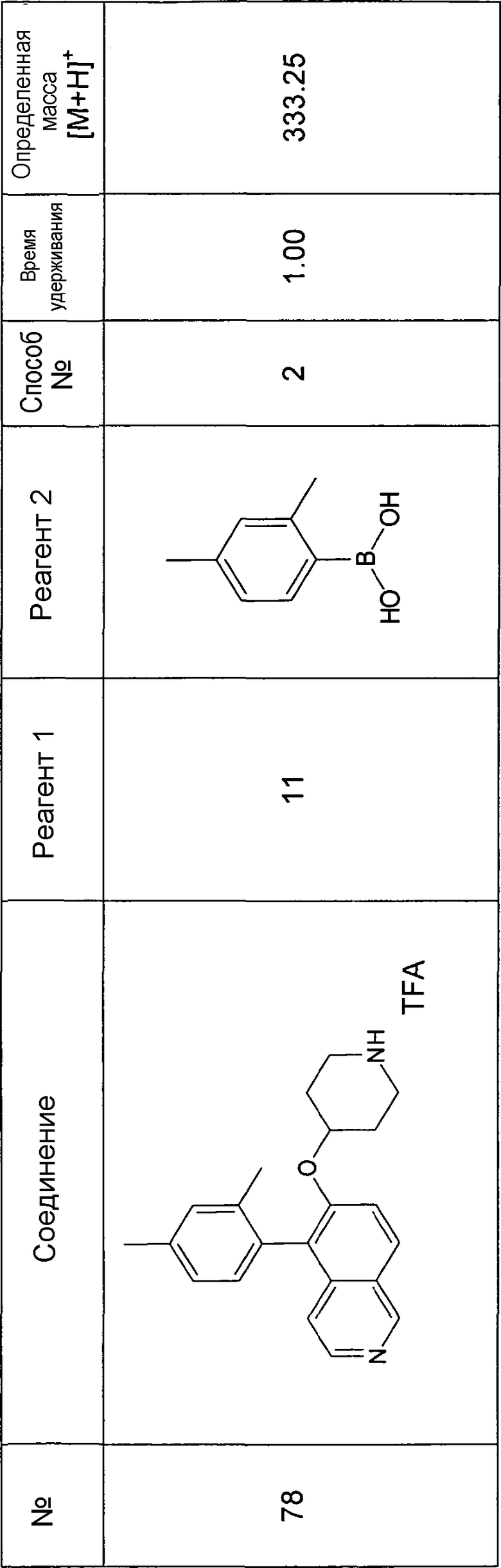
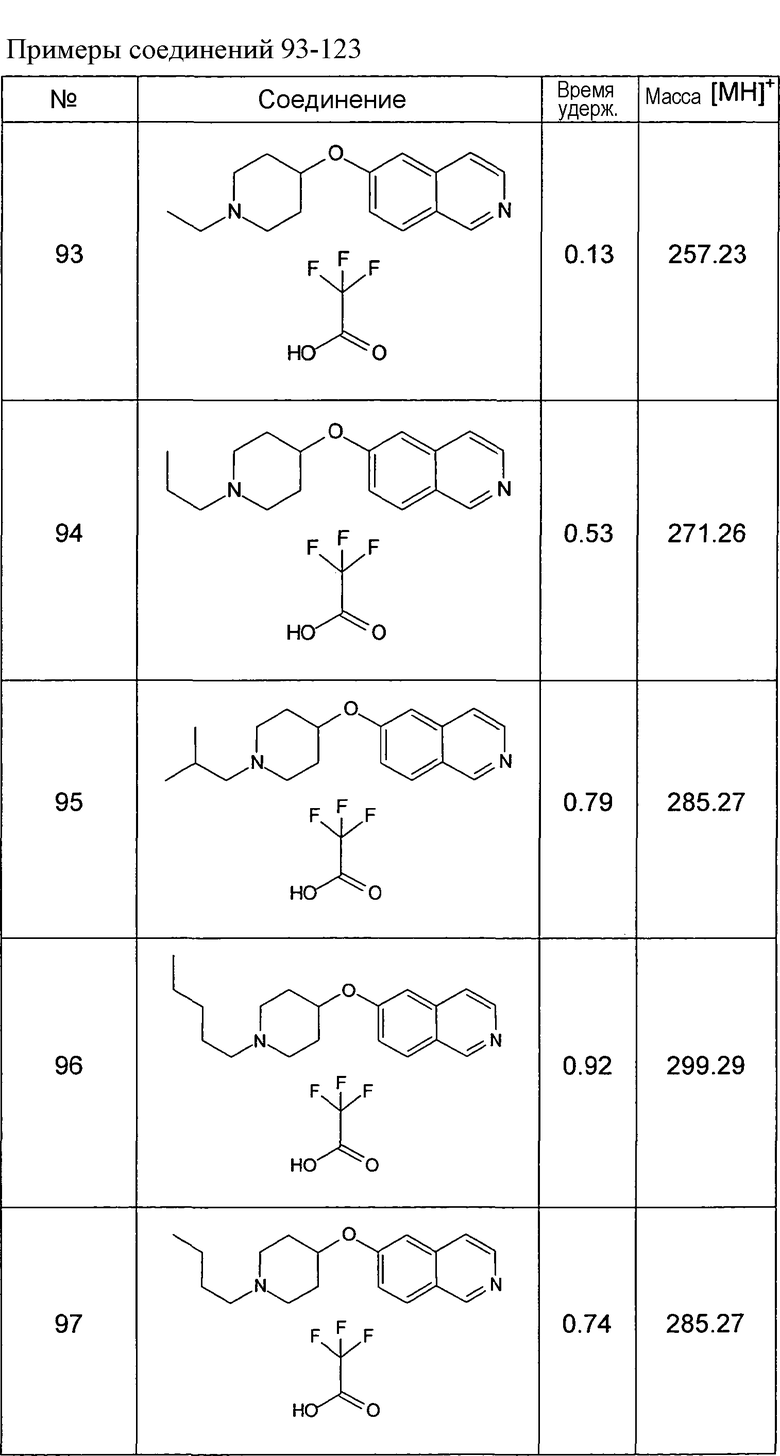
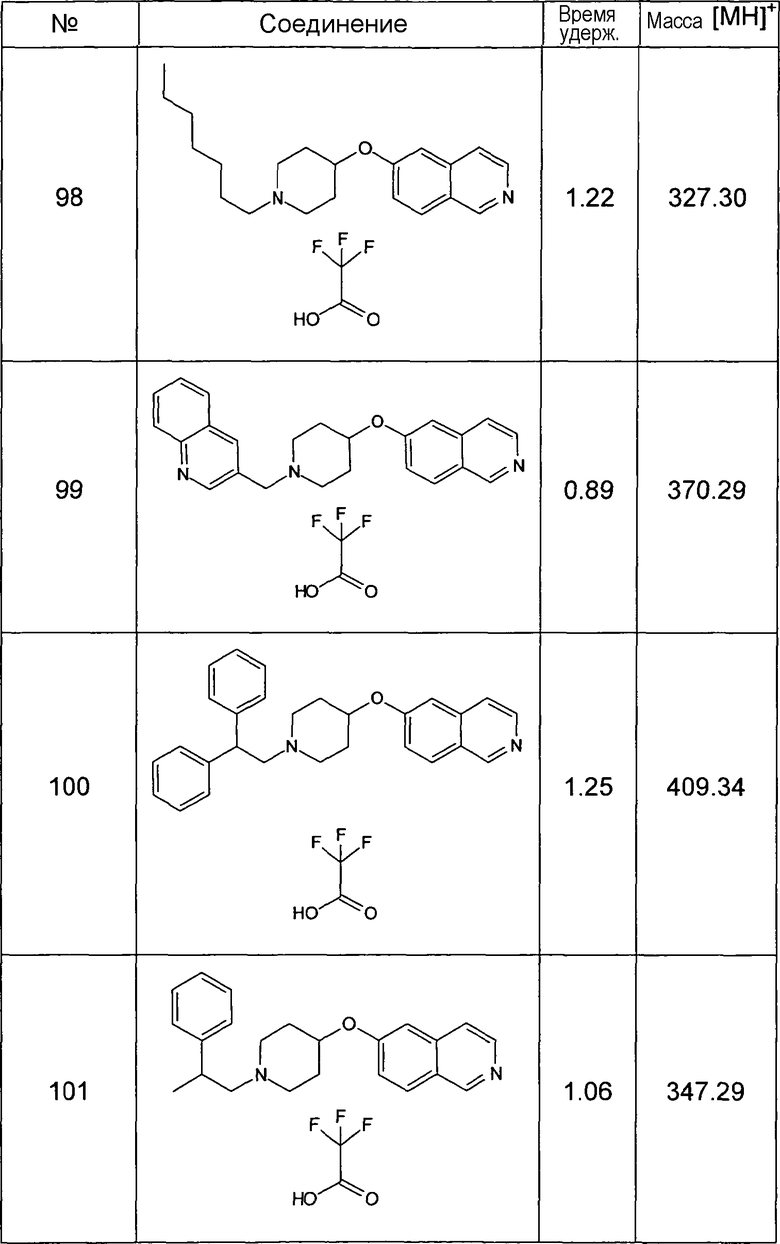
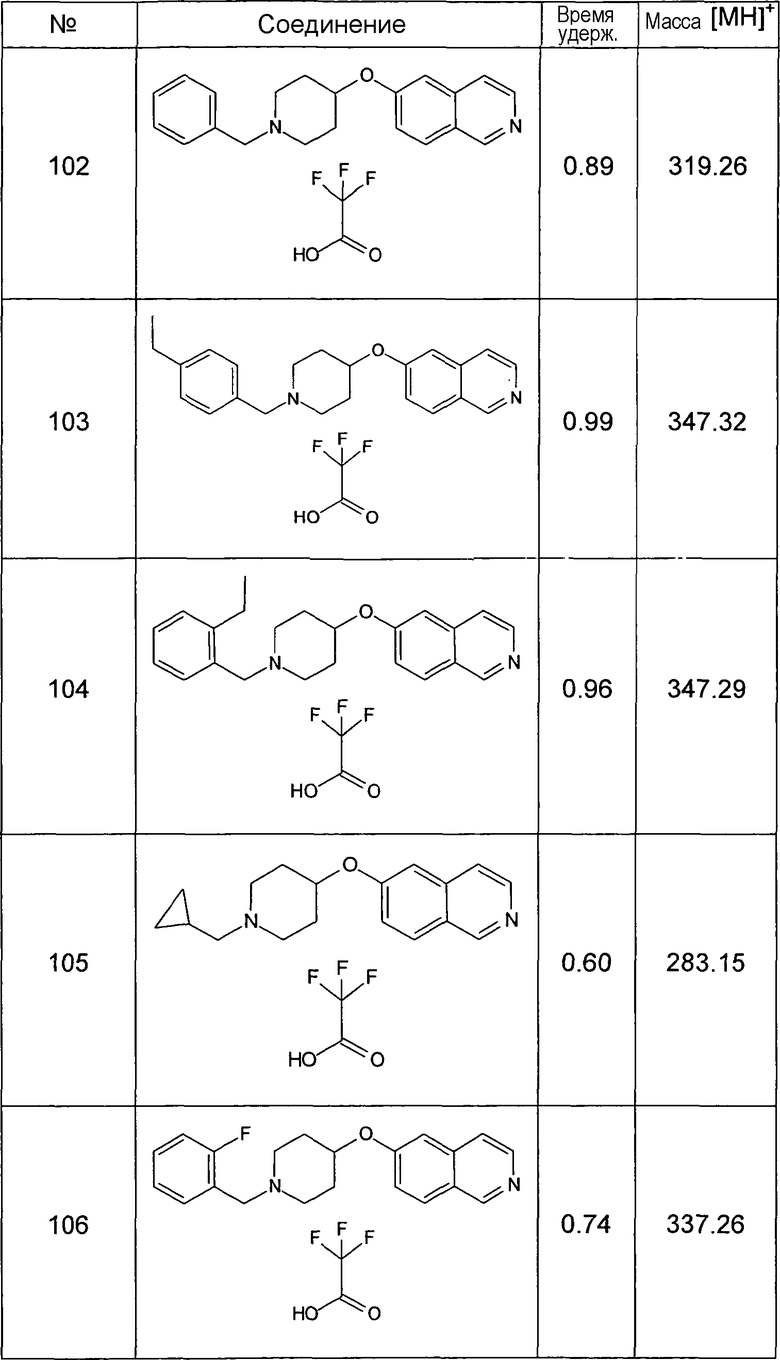
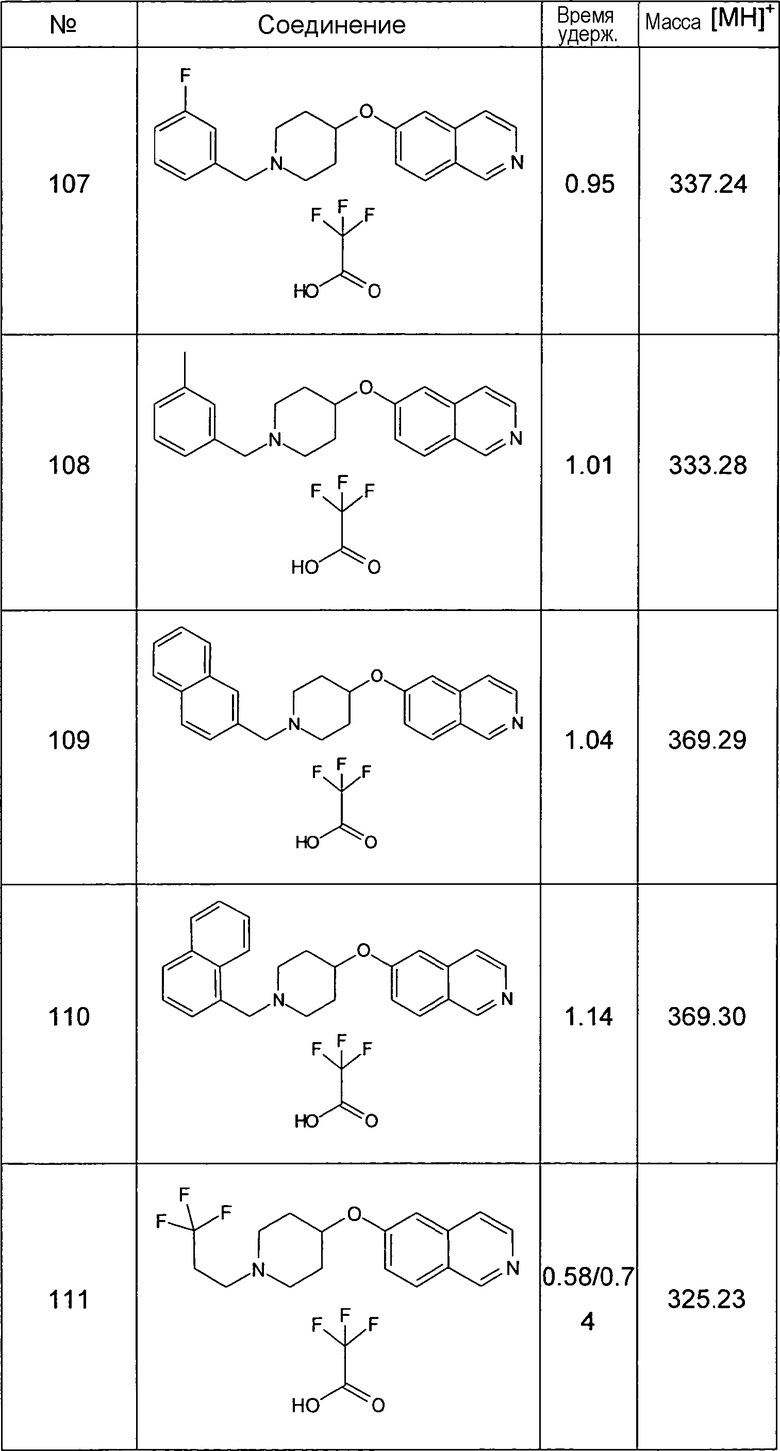
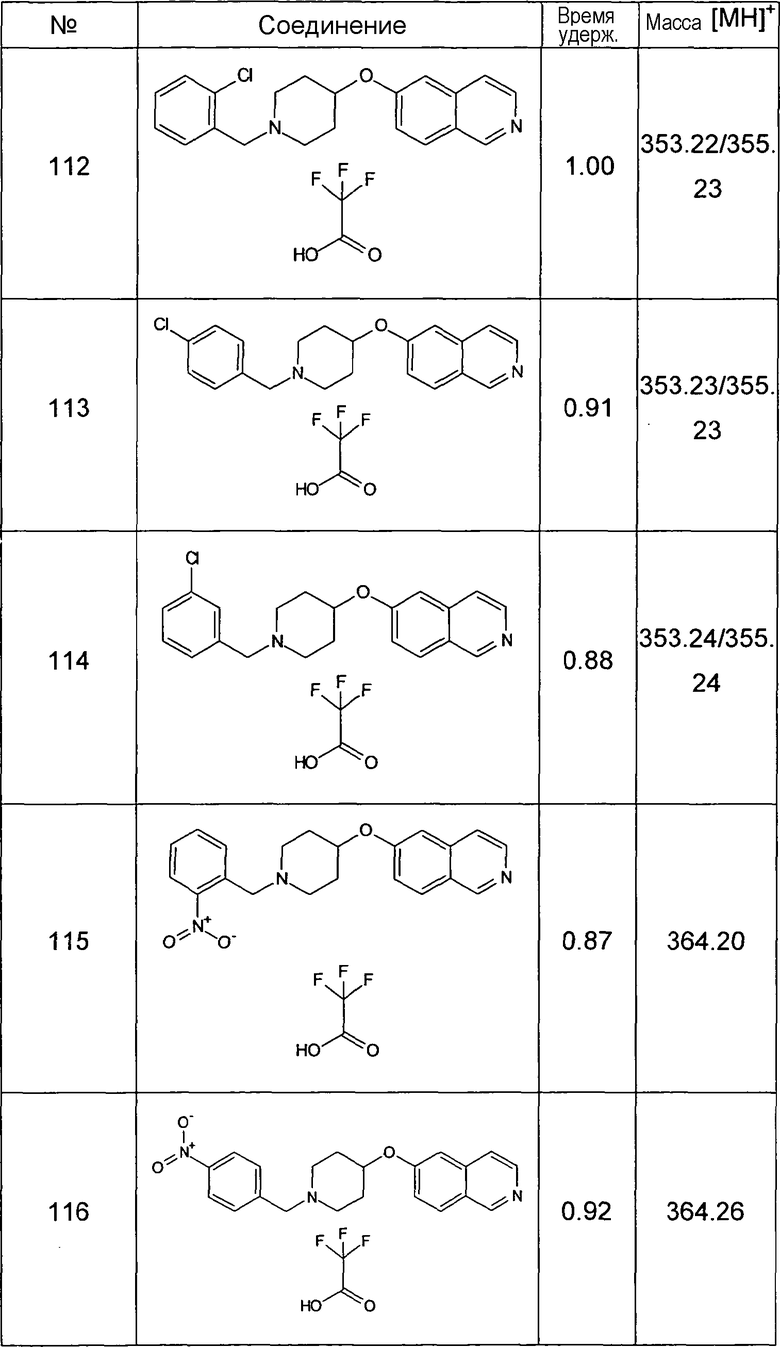
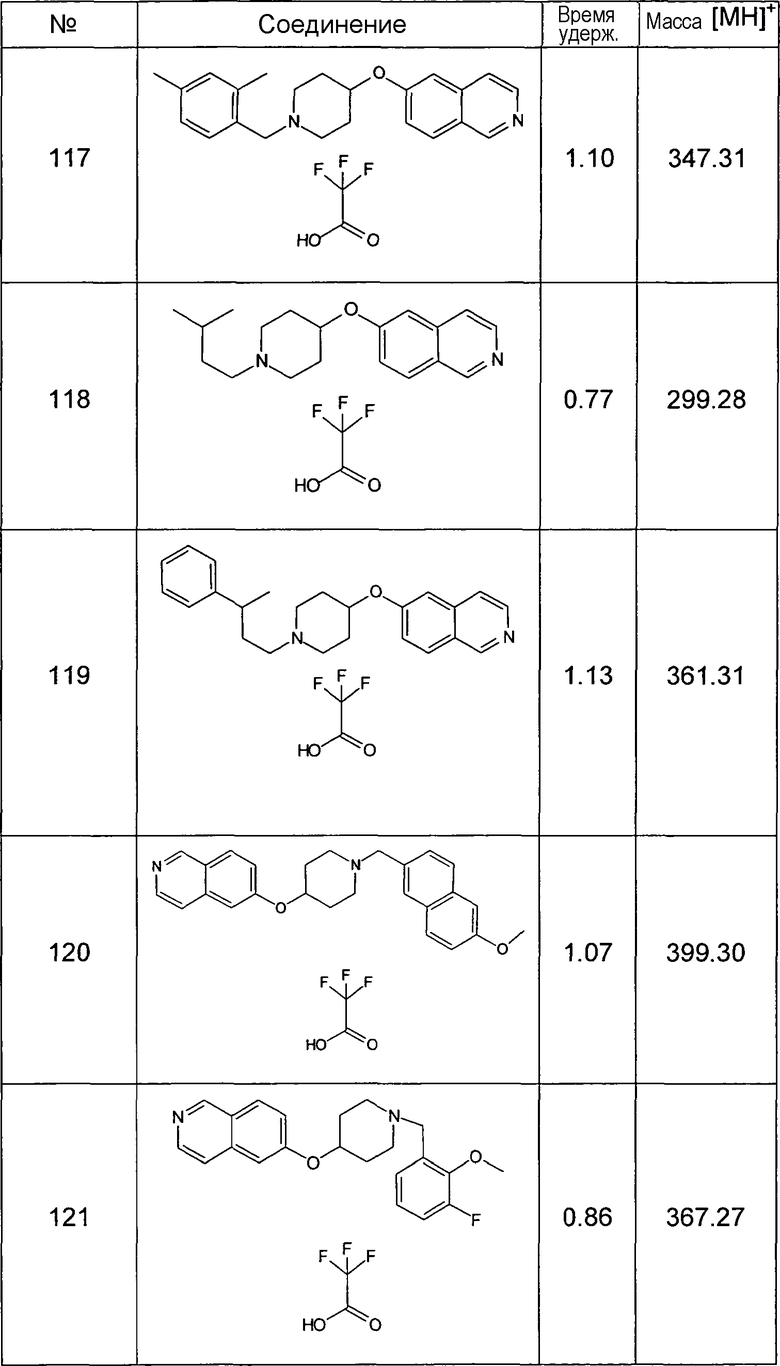
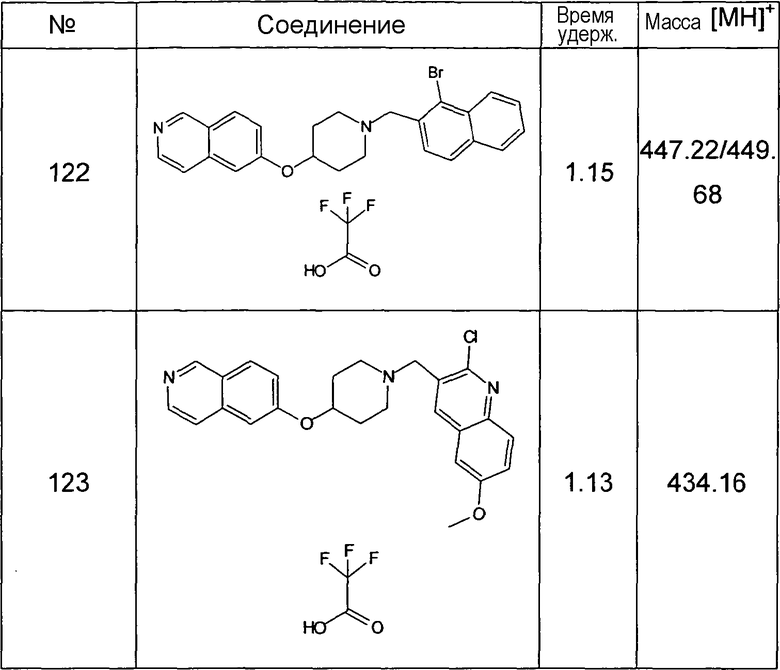
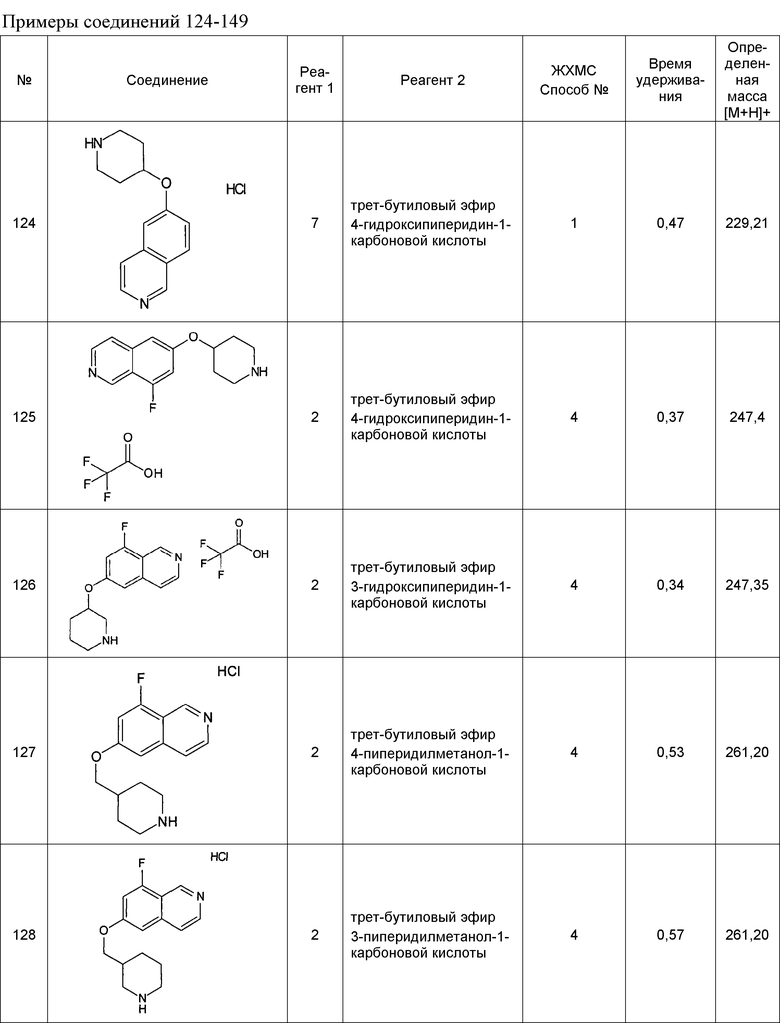
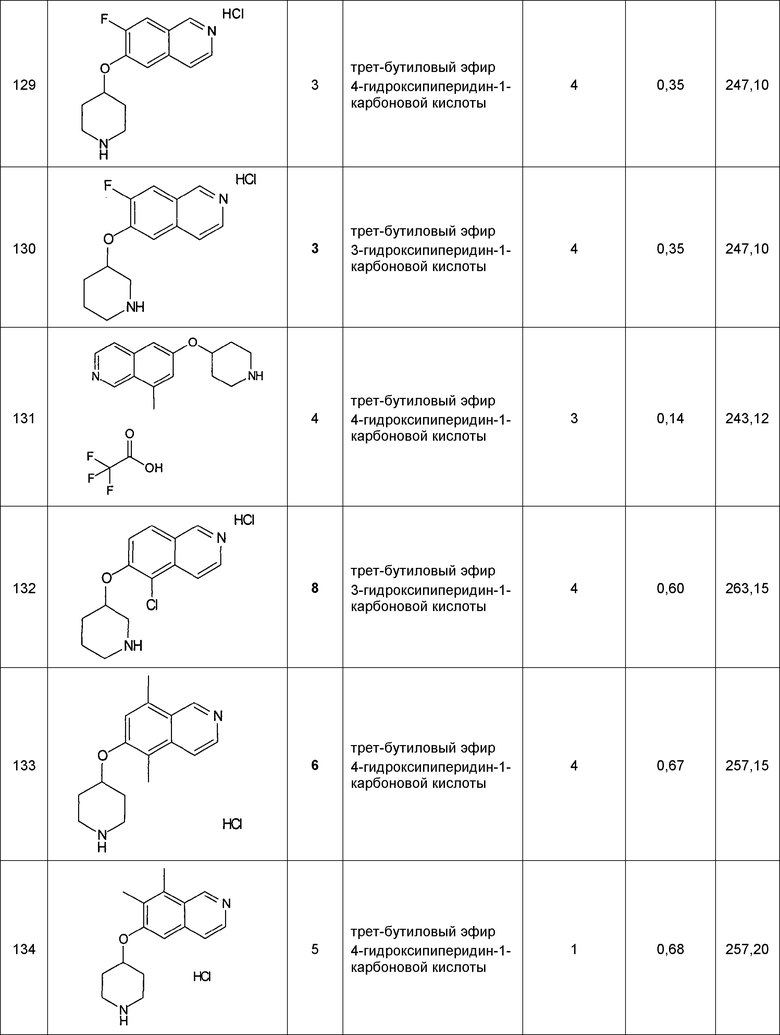
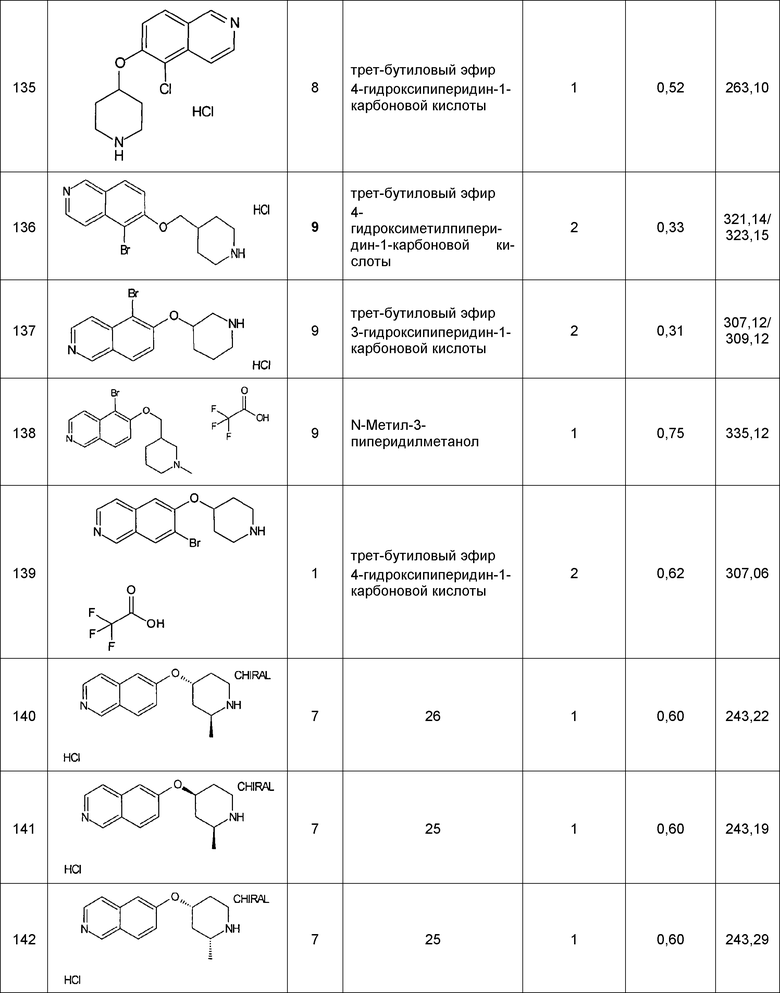
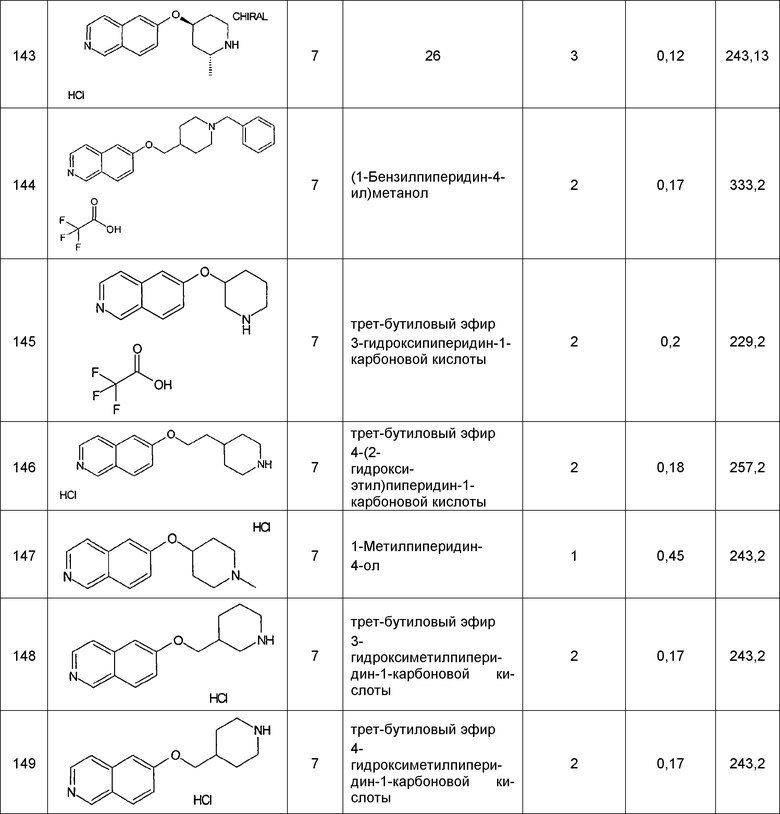
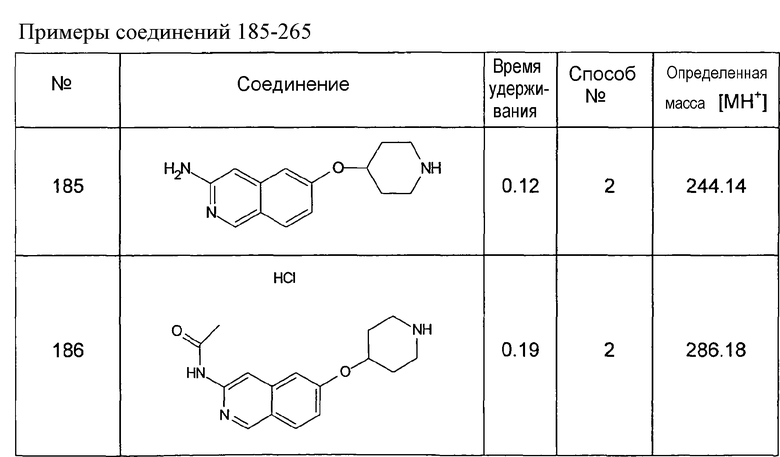
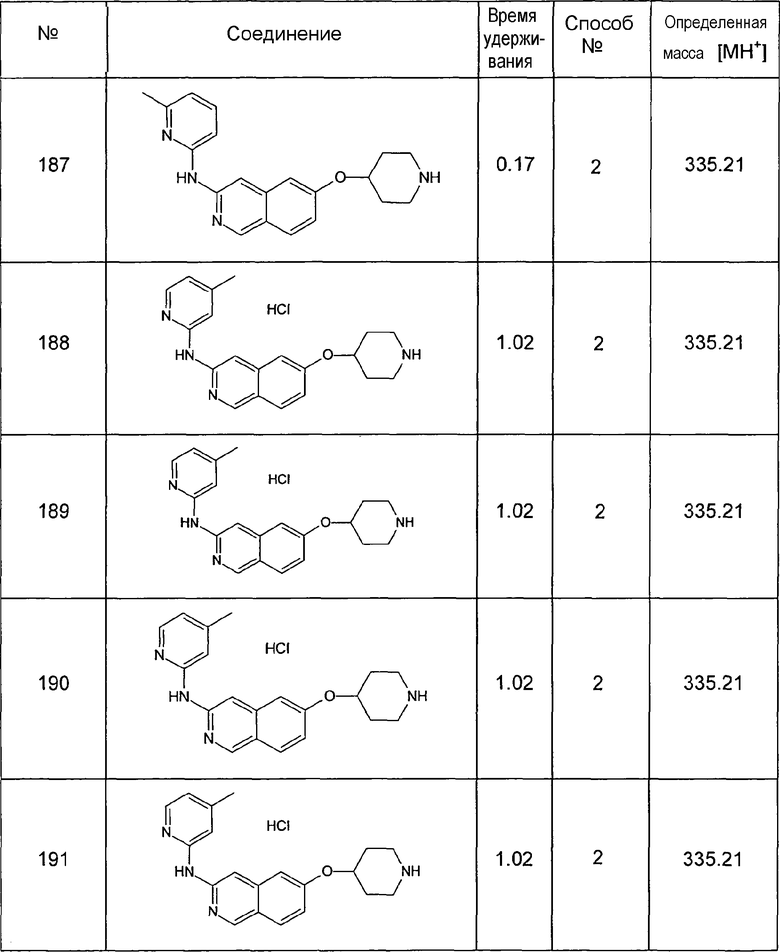
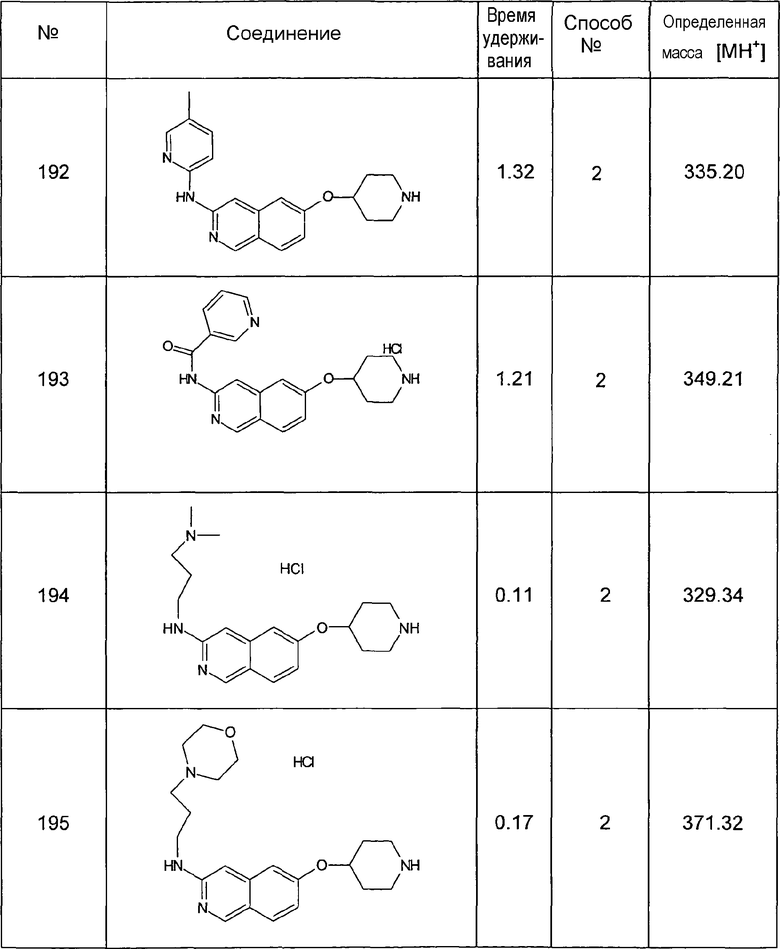
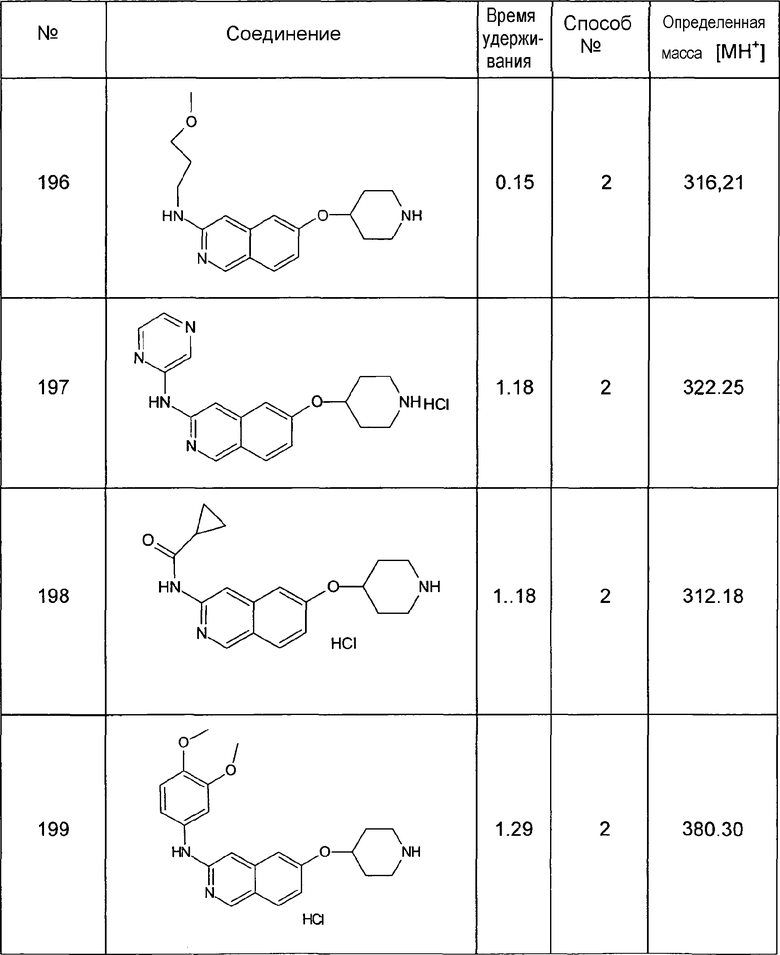
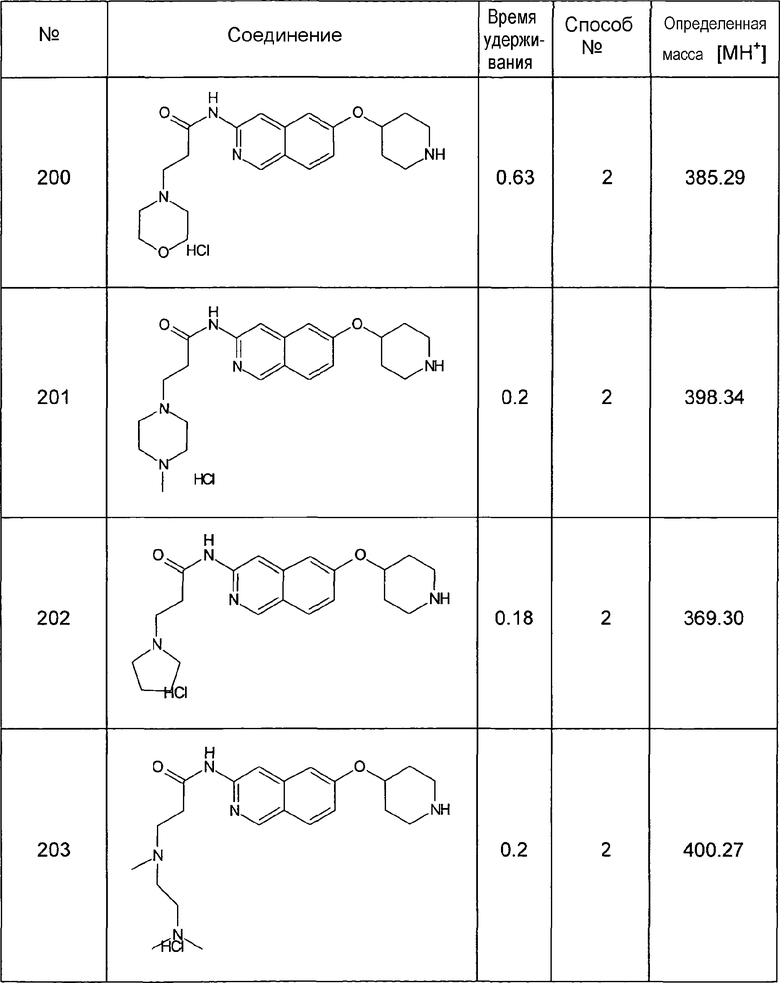
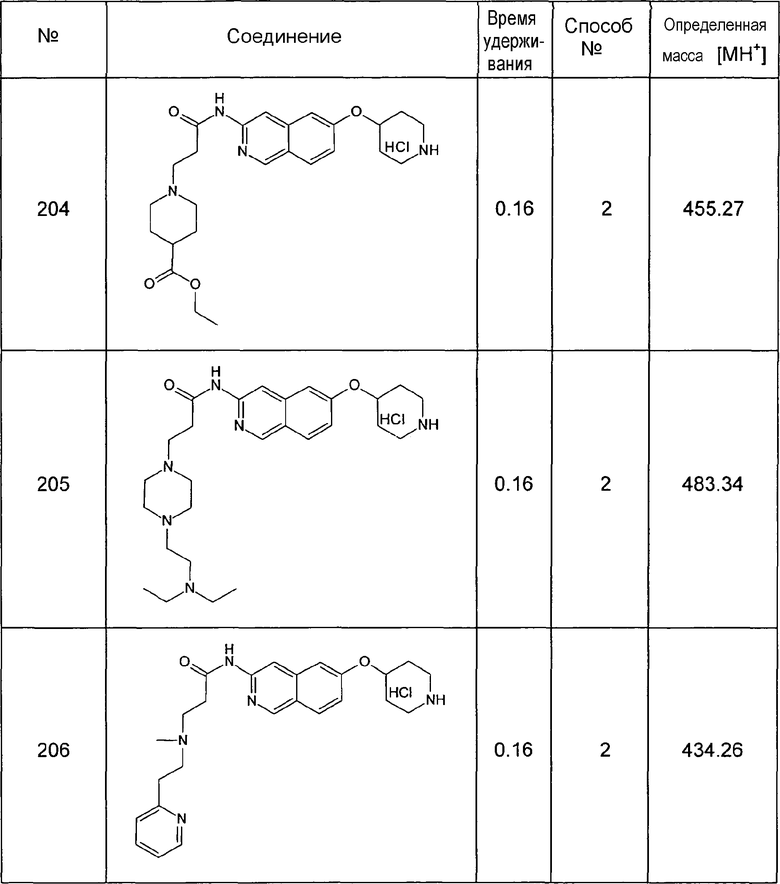
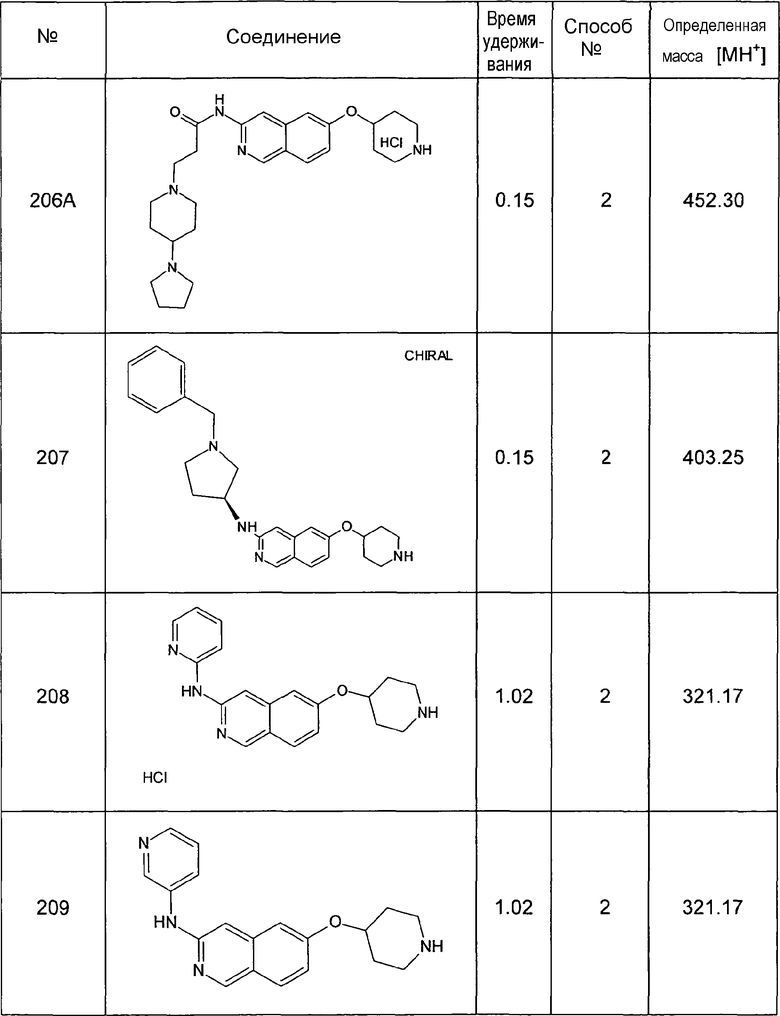
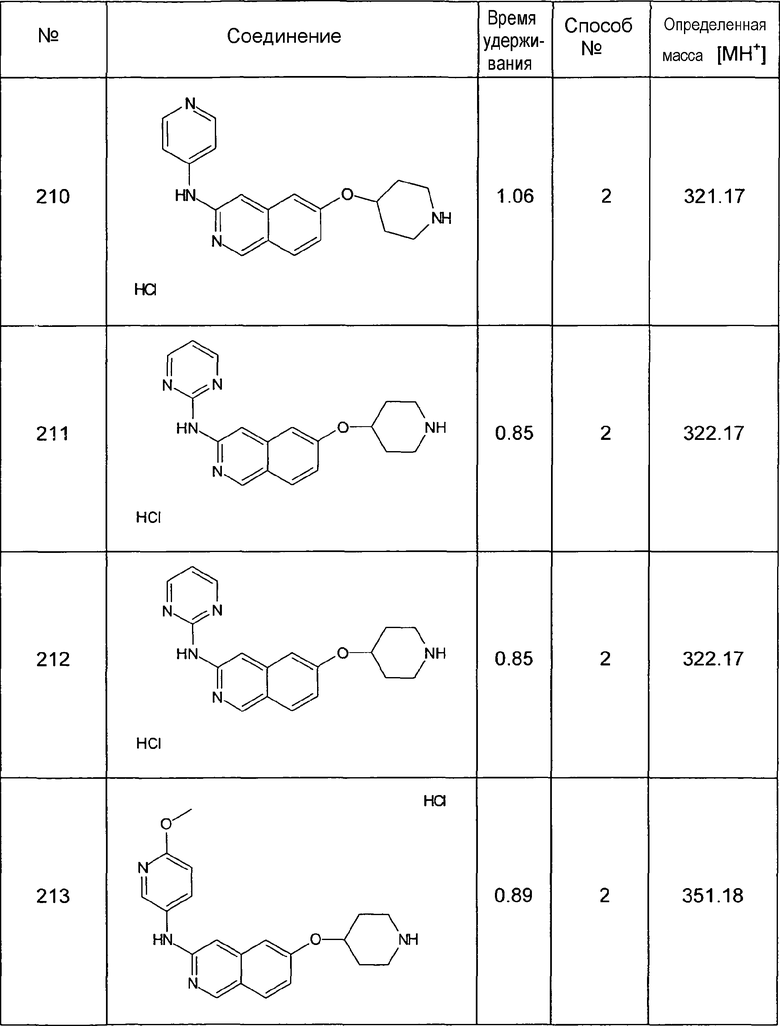
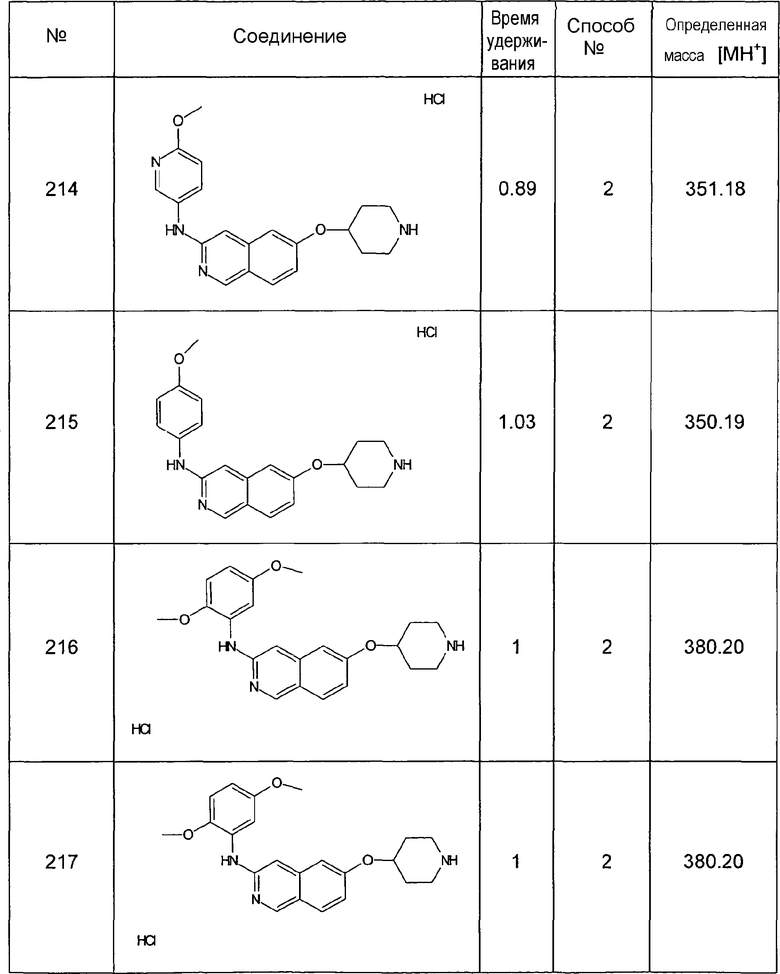
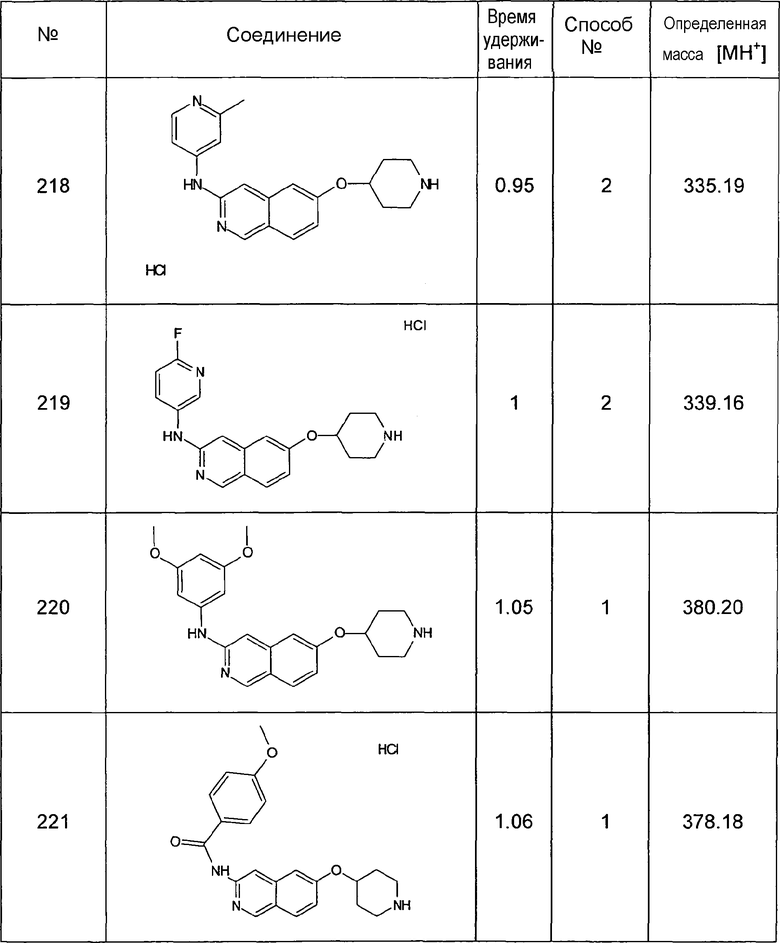
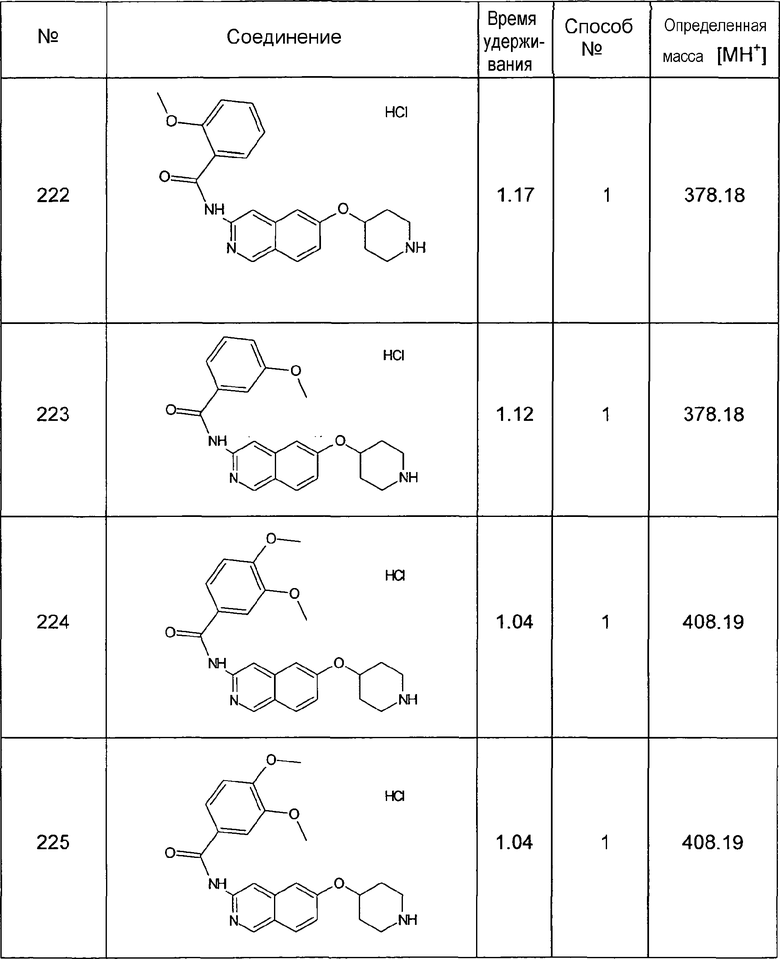
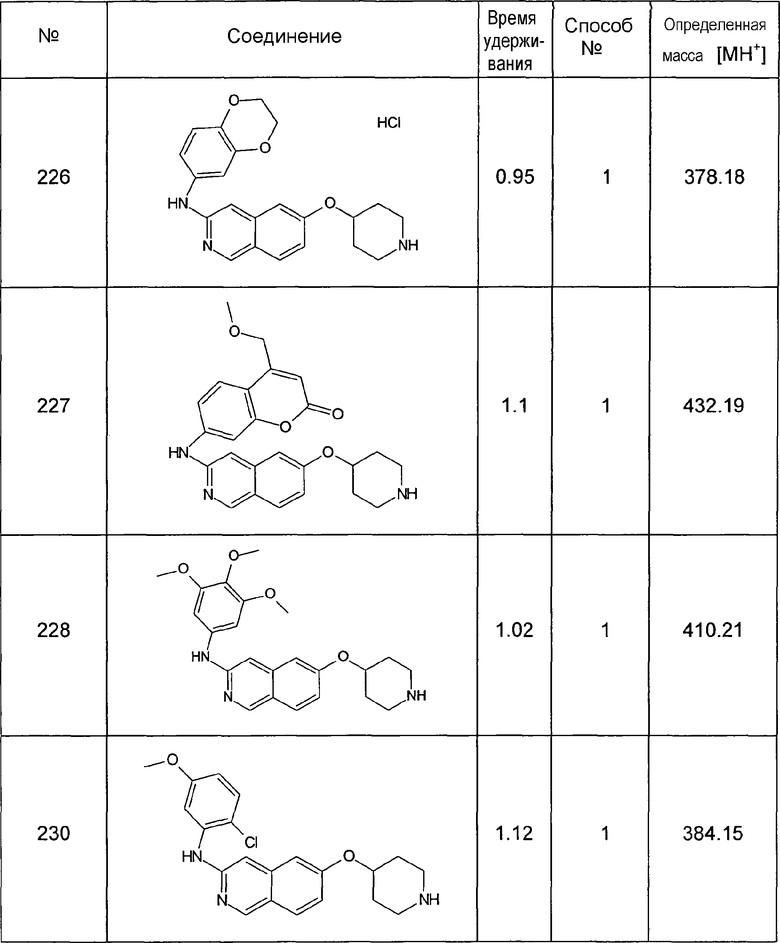
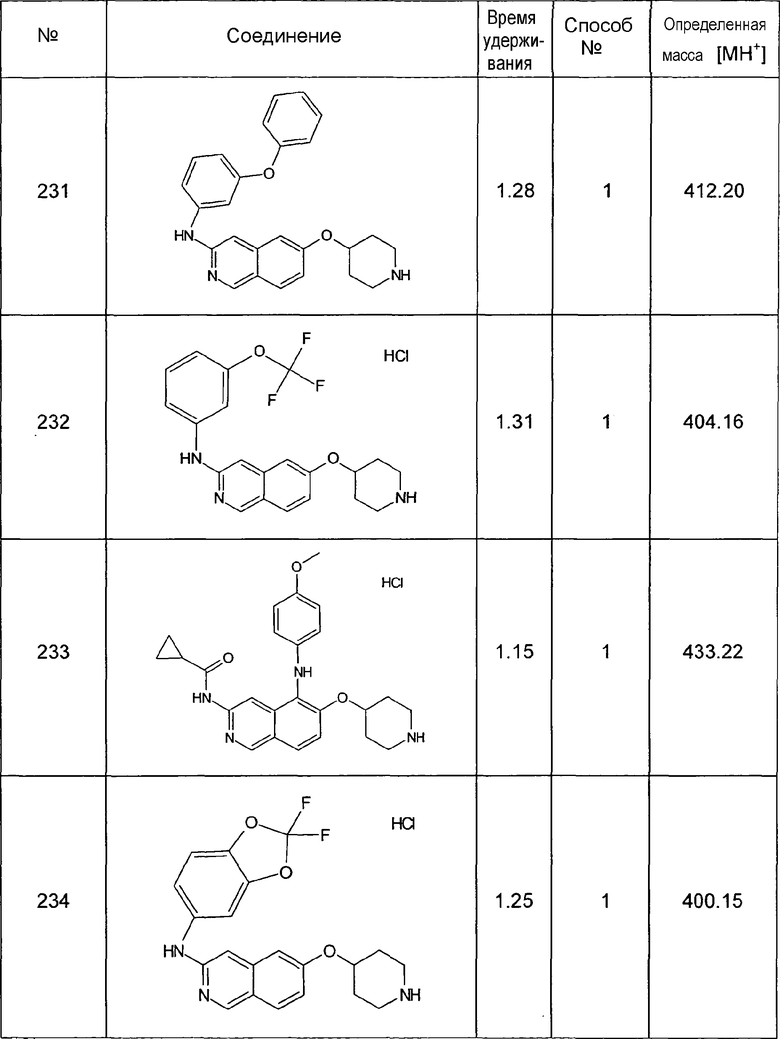
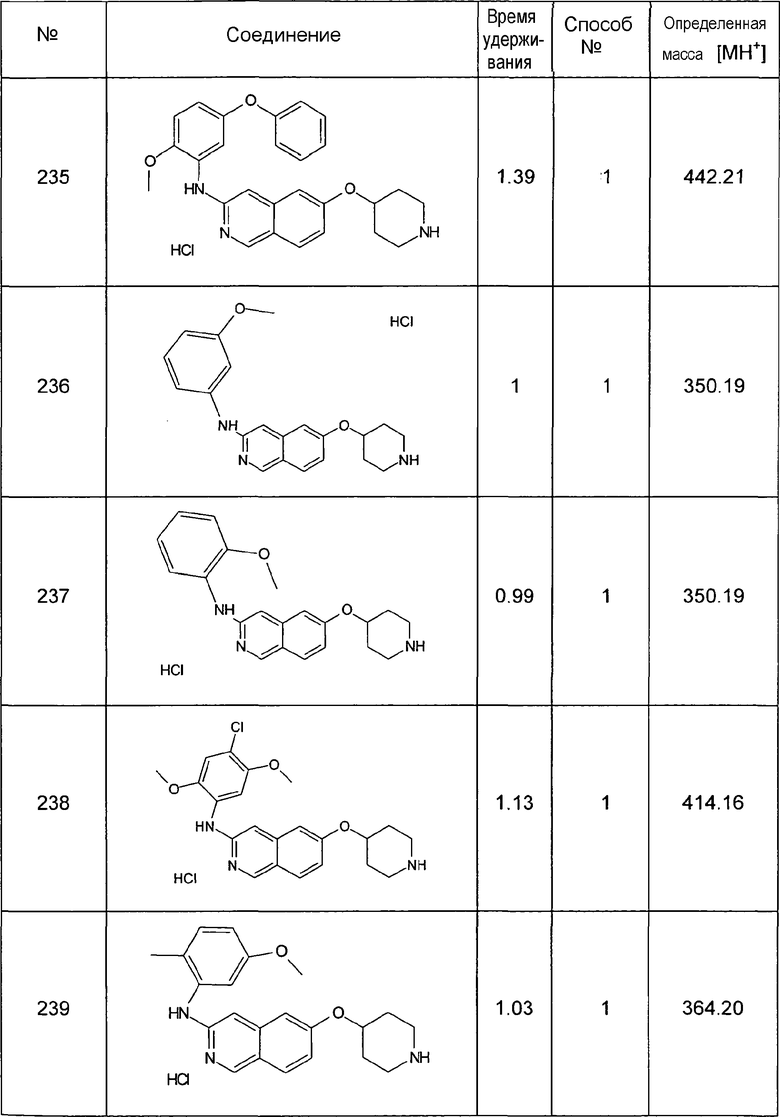
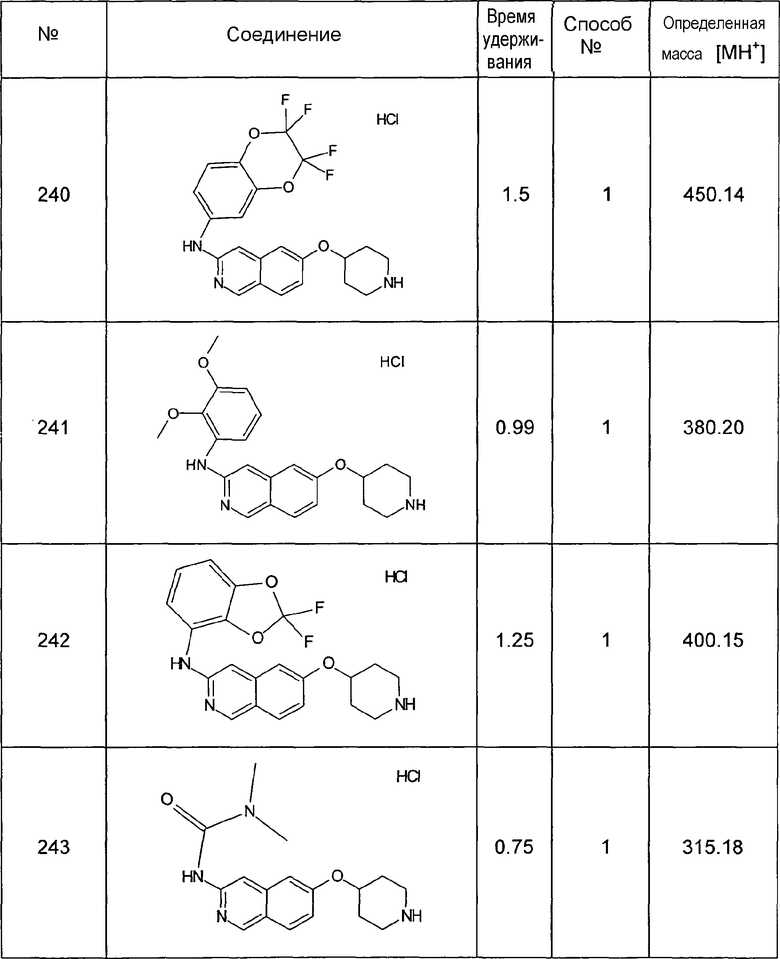
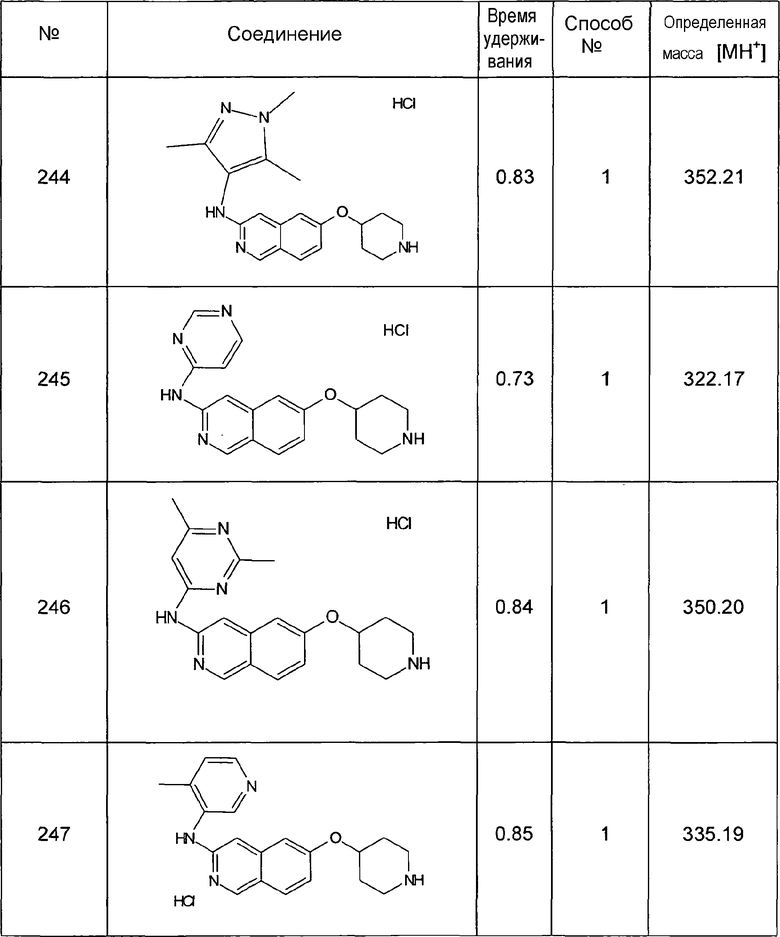
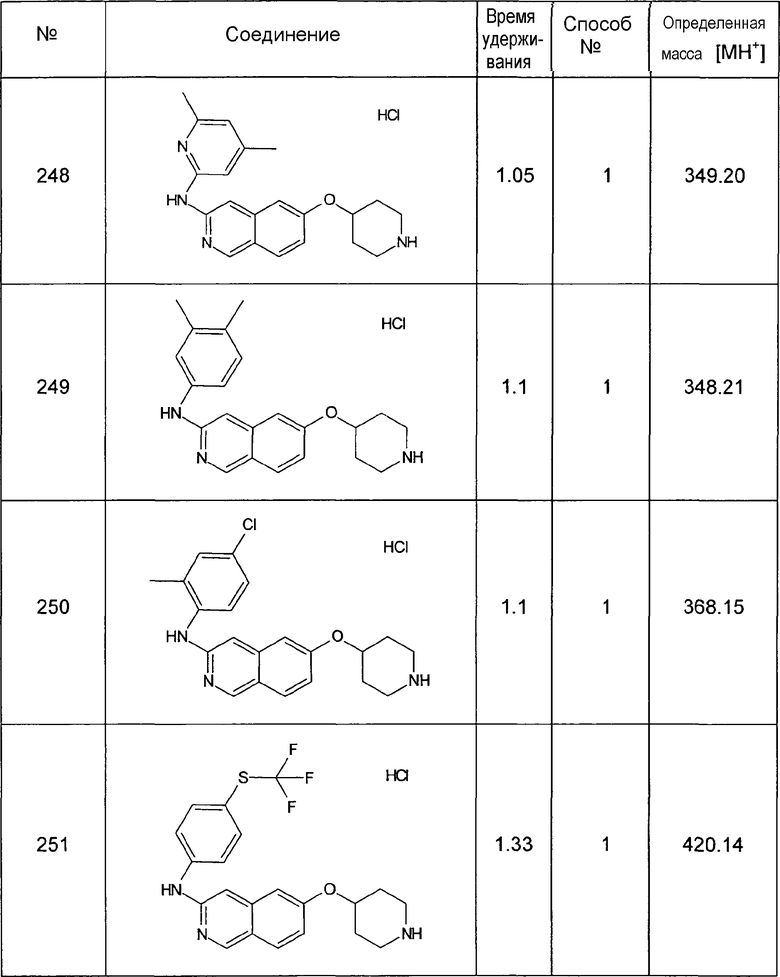
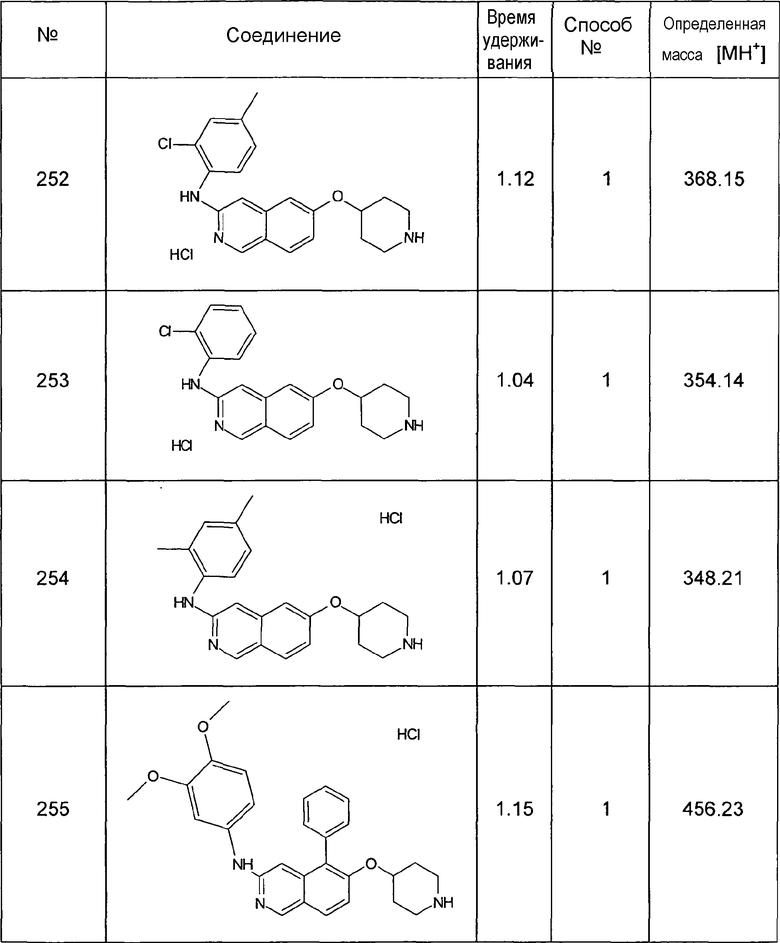
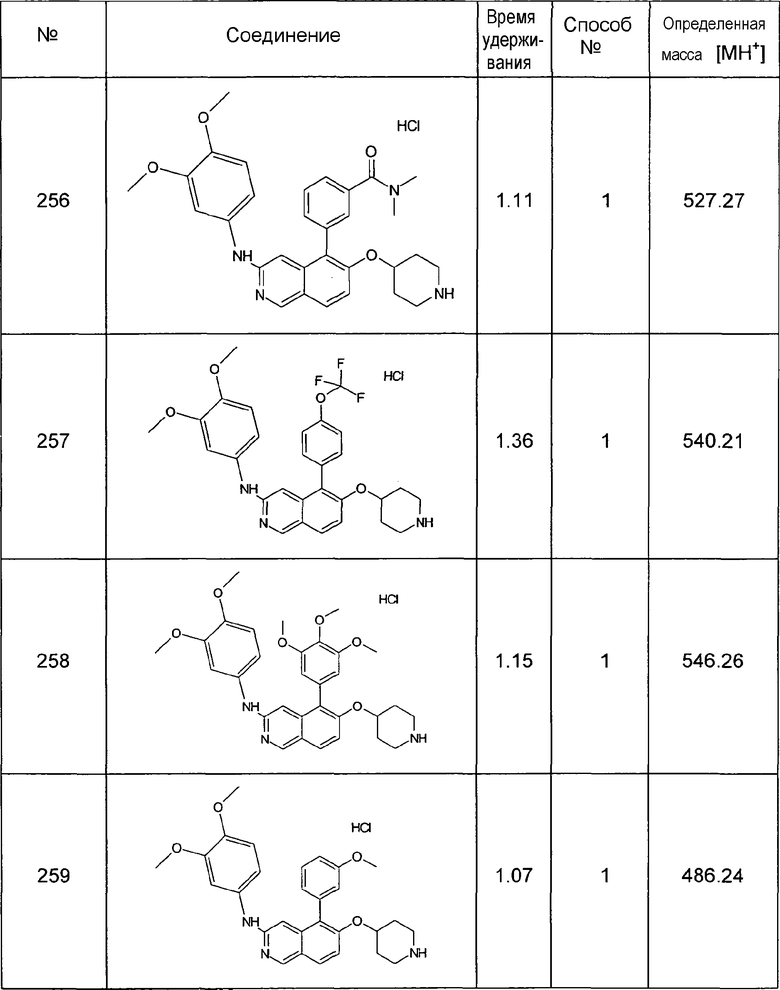
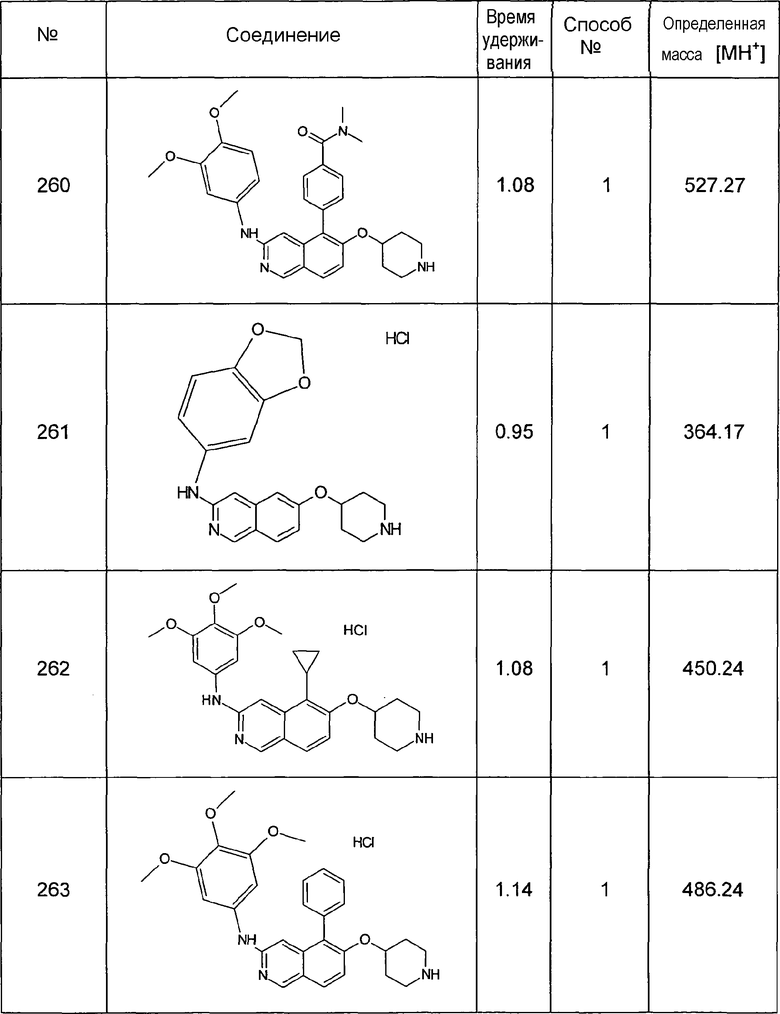
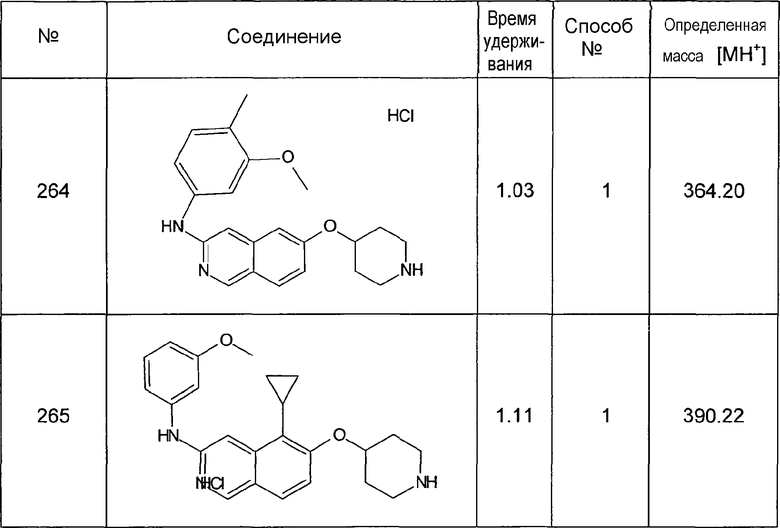
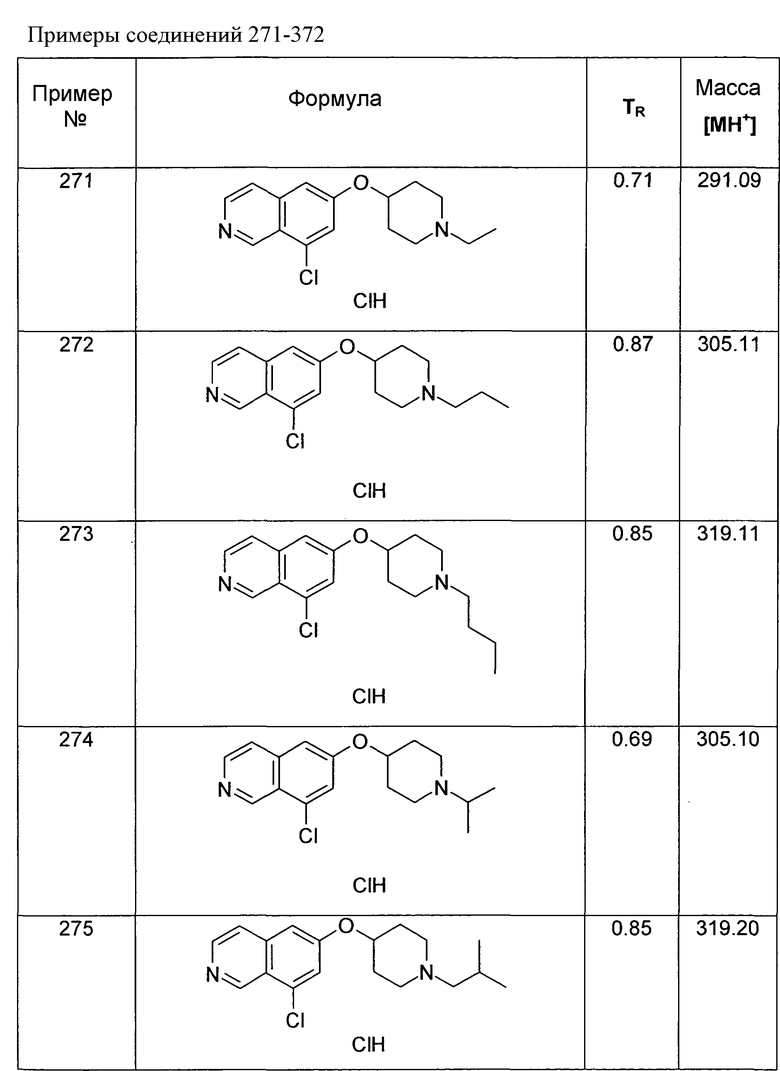
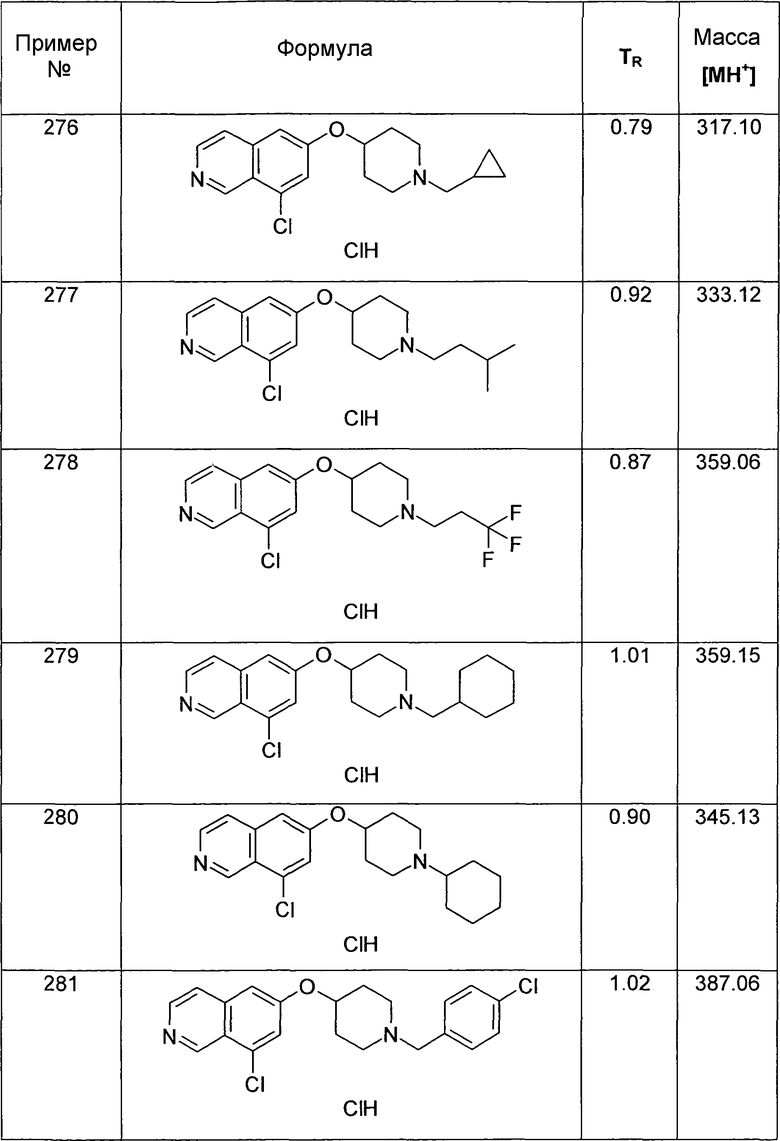
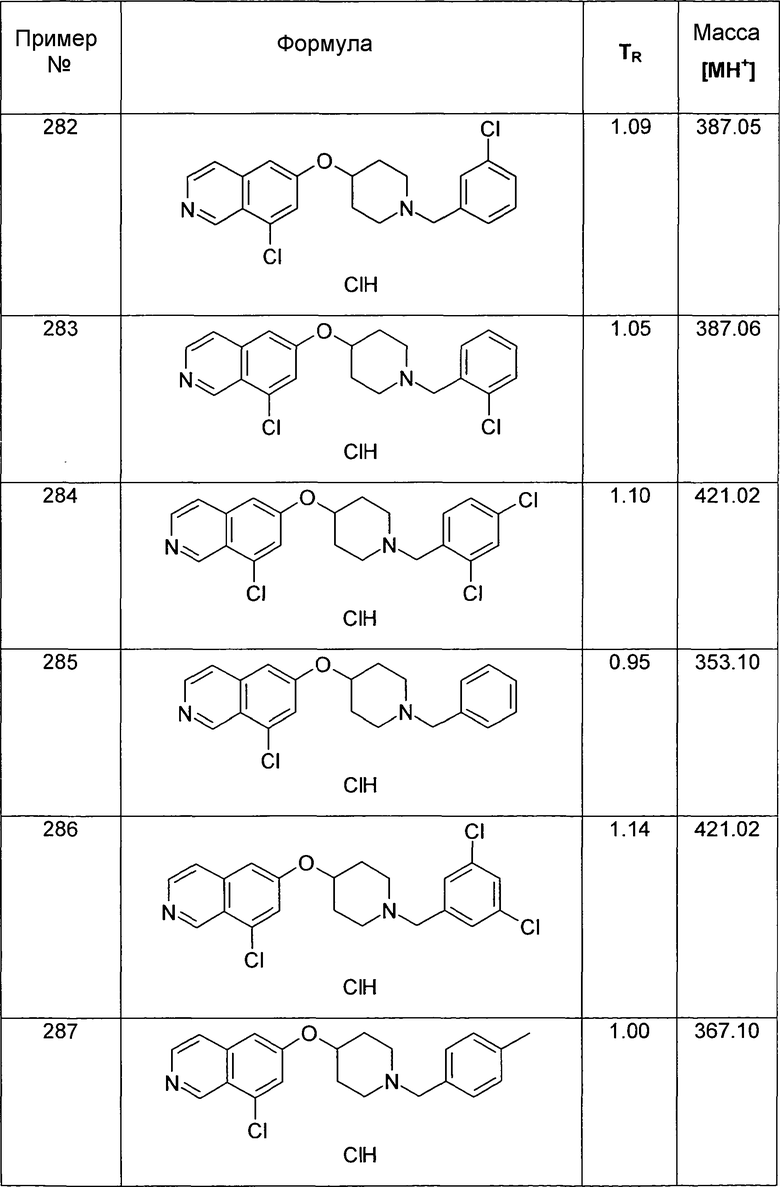
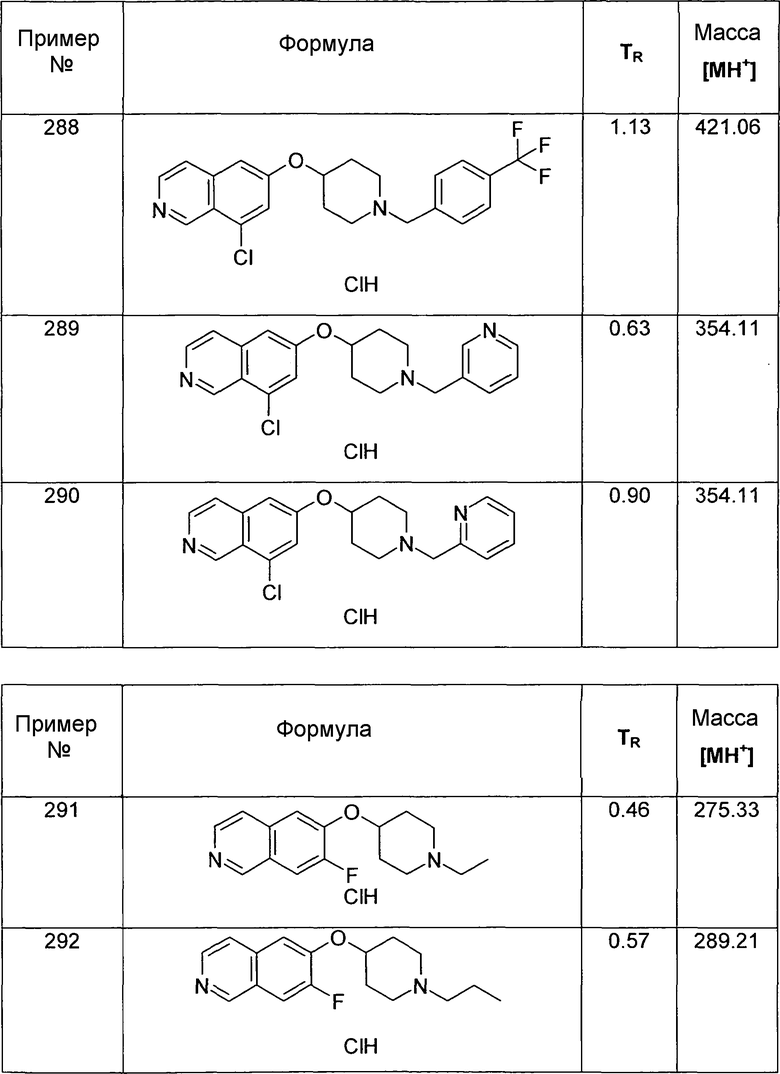

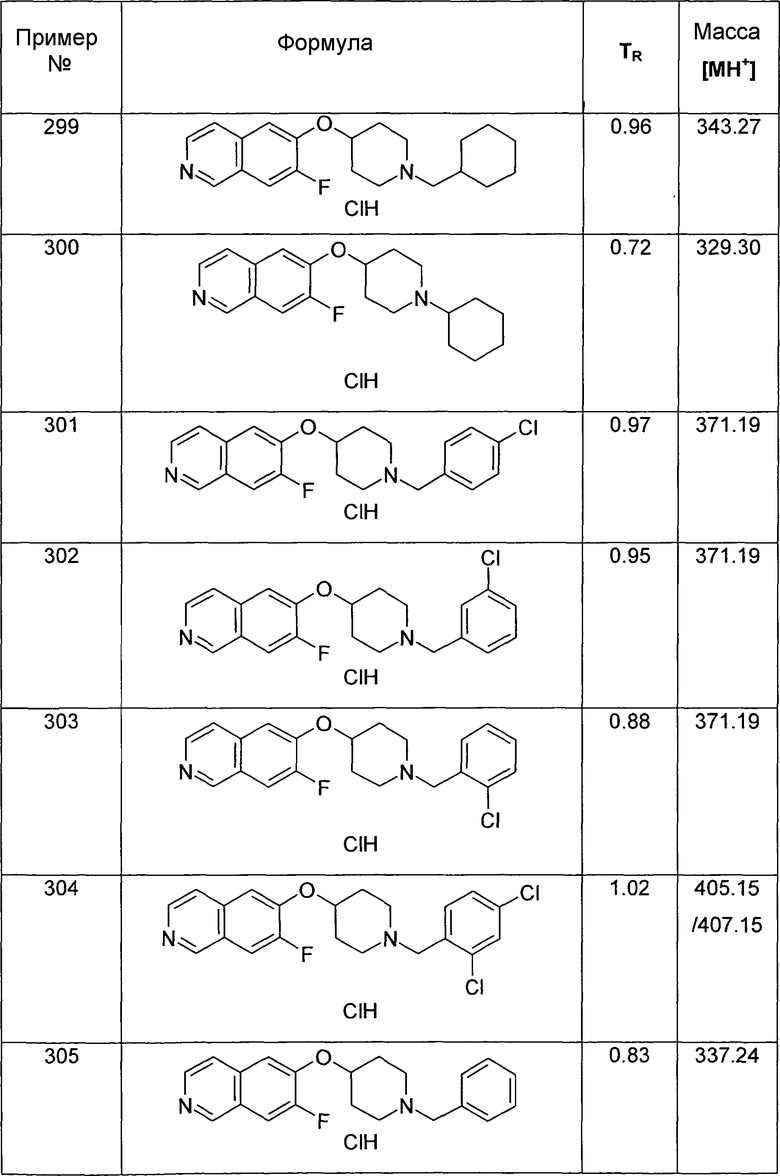
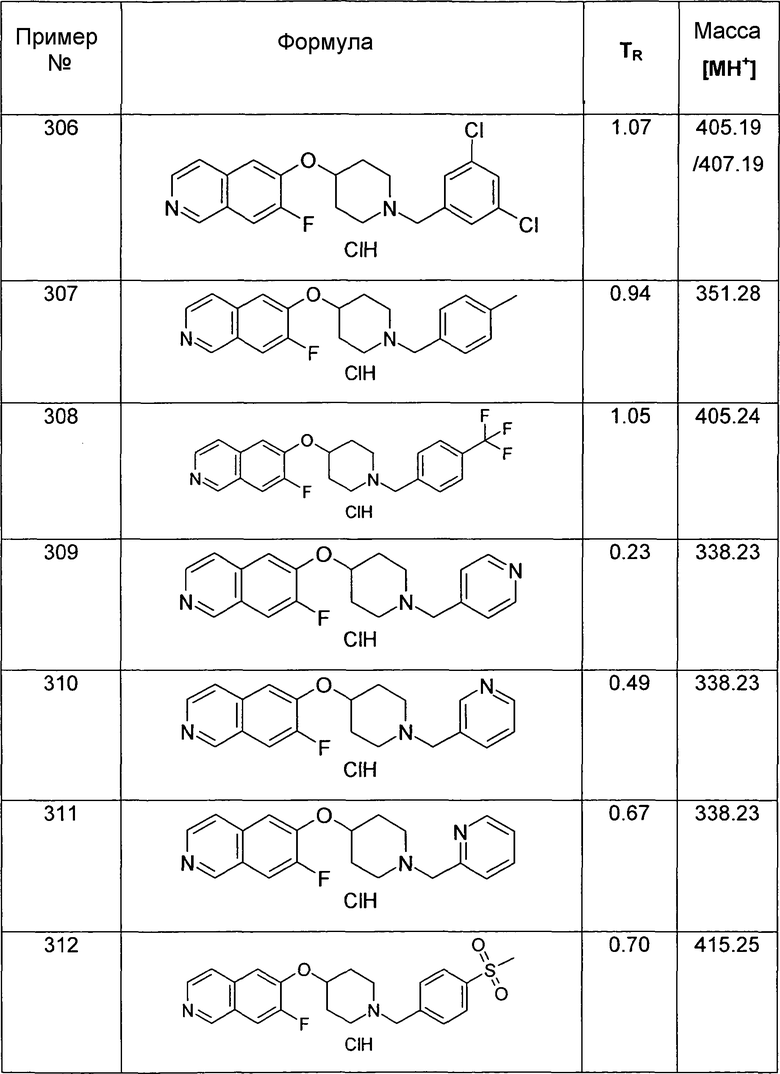
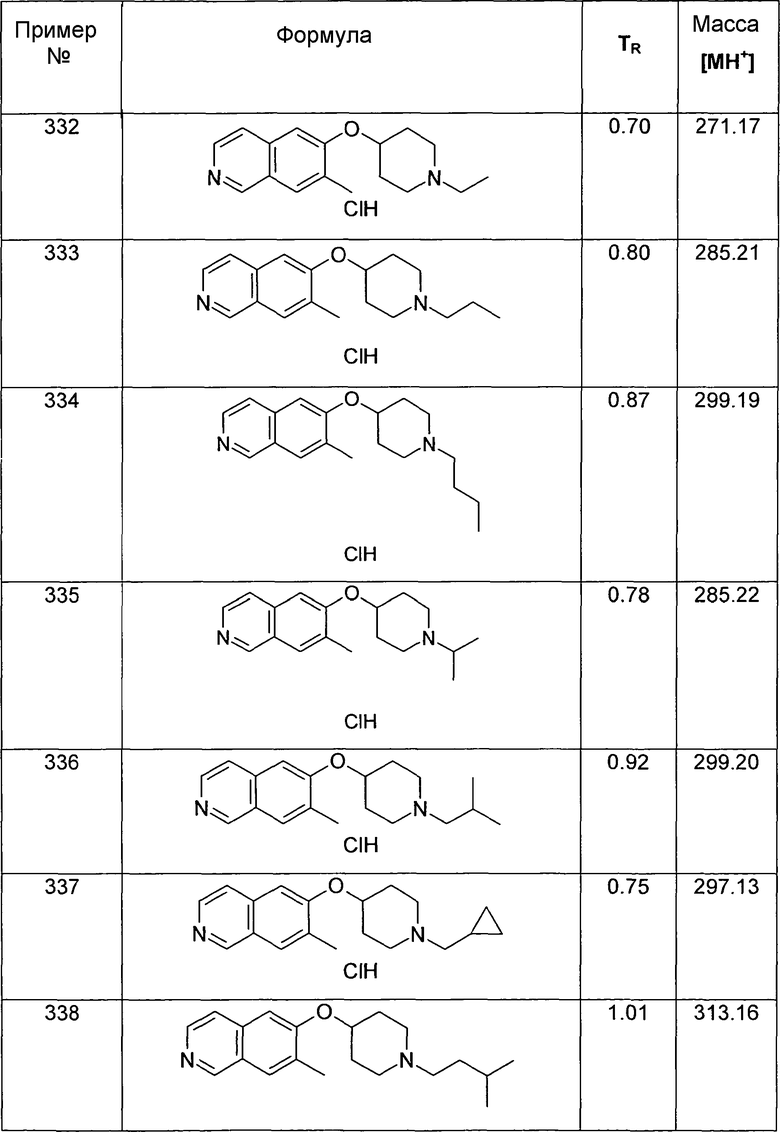
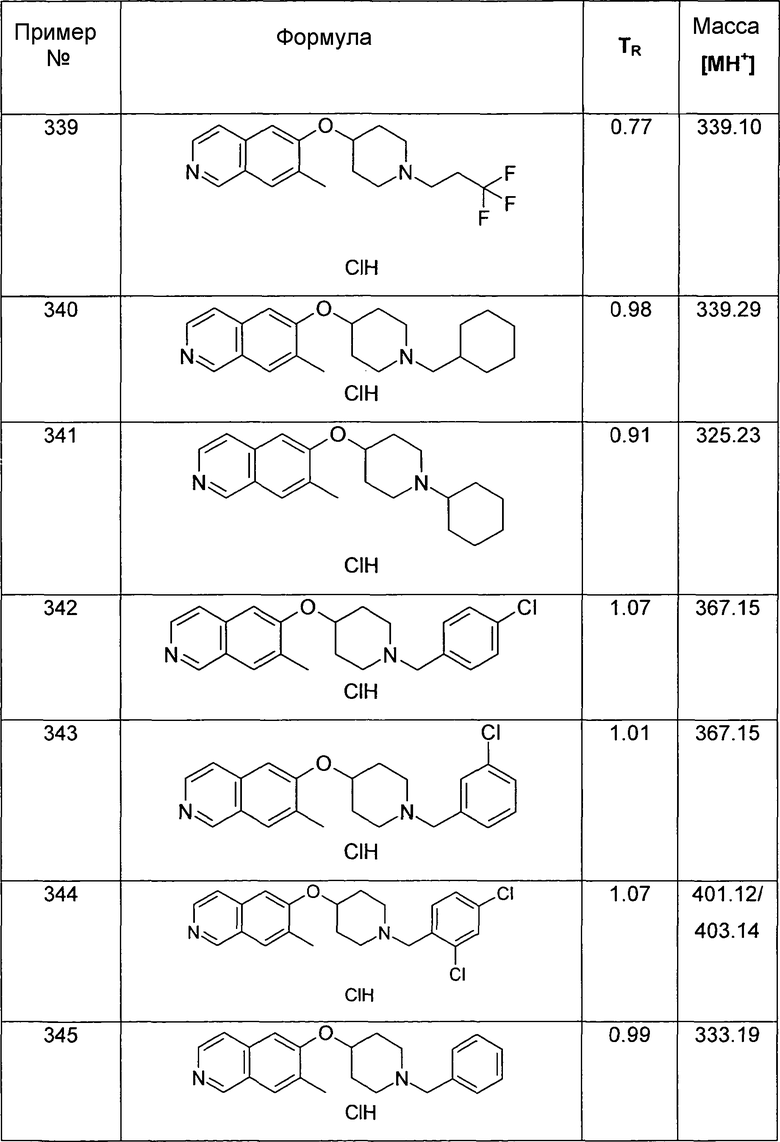
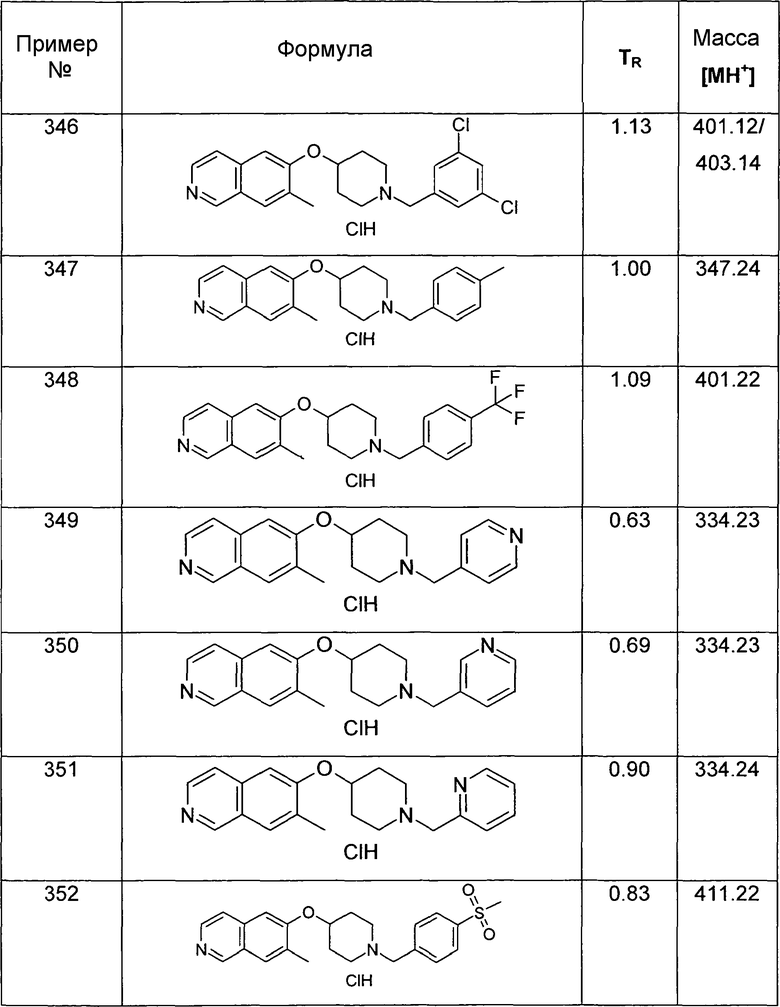
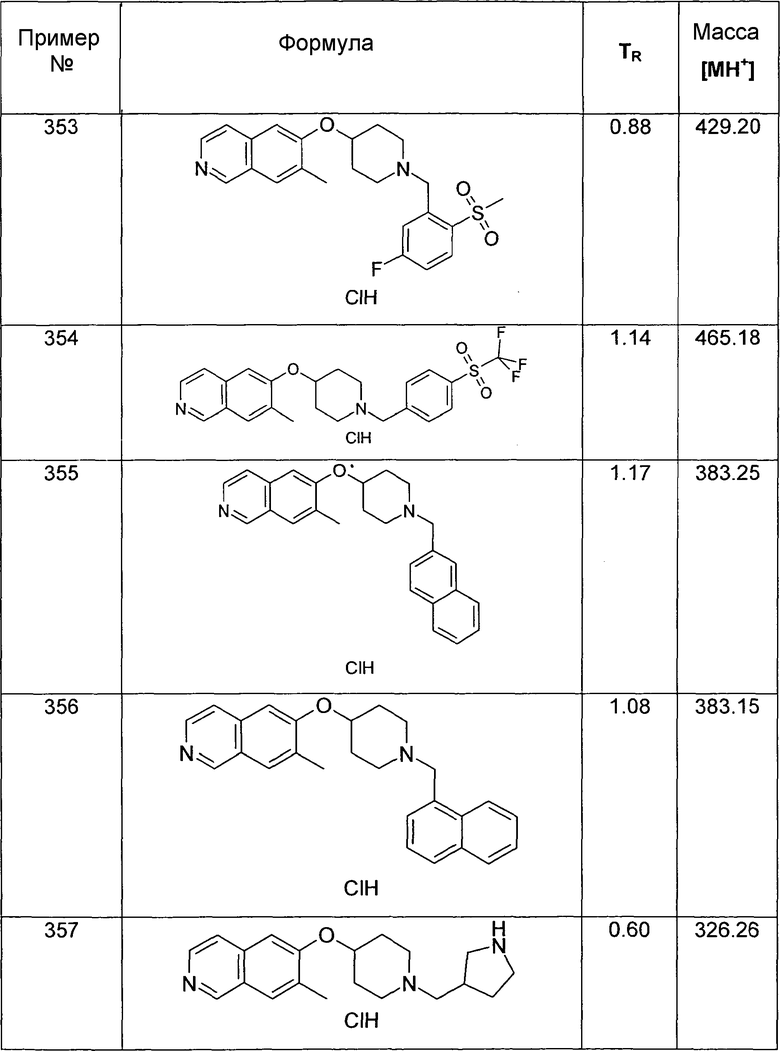
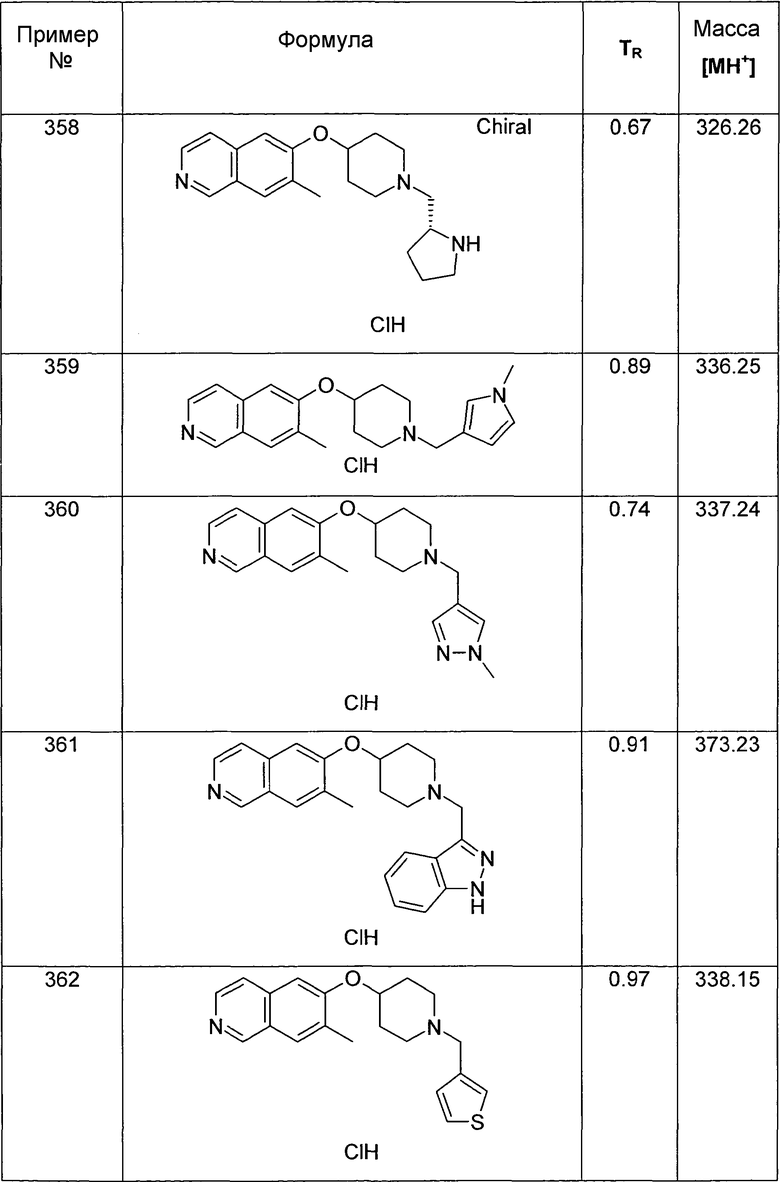
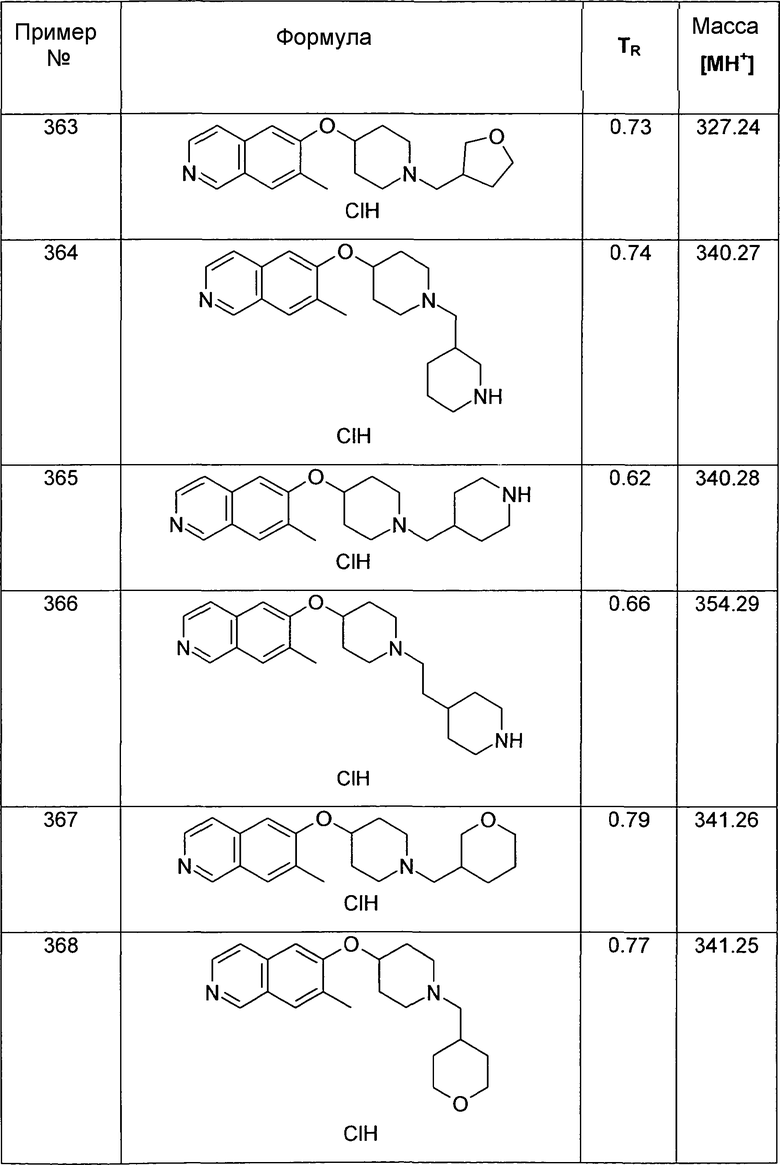
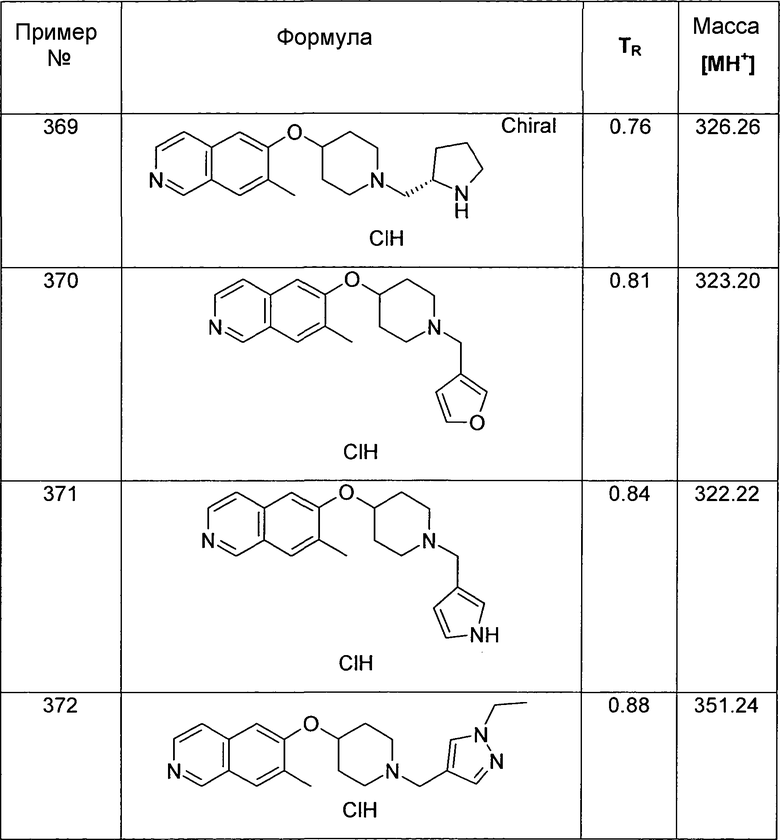
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИПЕРИДИНИЛ-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛОНА КАК ИНГИБИТОРЫ Rho-КИНАЗЫ | 2006 |
|
RU2414467C2 |
| ЗАМЕЩЕННЫЕ ИЗОХИНОЛИНОВЫЕ И ИЗОХИНОЛИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2007 |
|
RU2455302C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОАЛКИЛАМИНОМ ПРОИЗВОДНЫЕ ИЗОХИНОЛОНА | 2007 |
|
RU2457203C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСИЛАМИНИЗОХИНОЛОНА В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2006 |
|
RU2440988C2 |
| 6-ЗАМЕЩЕННЫЕ ИЗОХИНОЛИНЫ И ИЗОХИНОЛИНОНЫ ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2009 |
|
RU2528229C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОАЛКИЛАМИНОМ ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА И ИЗОХИНОЛИНОНА | 2007 |
|
RU2468011C2 |
| БИ- И ПОЛИЦИКЛИЧЕСКИЕ ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА И ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2009 |
|
RU2532481C2 |
| ЗАМЕЩЕННЫЕ ИЗИХИНОЛИНЫ И ИЗОХИНОЛИНОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2009 |
|
RU2538588C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ 1β-ДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2006 |
|
RU2415855C2 |
| АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Xa | 2004 |
|
RU2330853C2 |
Настоящее изобретение относится к 6-пиперидинил-замещенным изохинолиновым производным формулы (I)
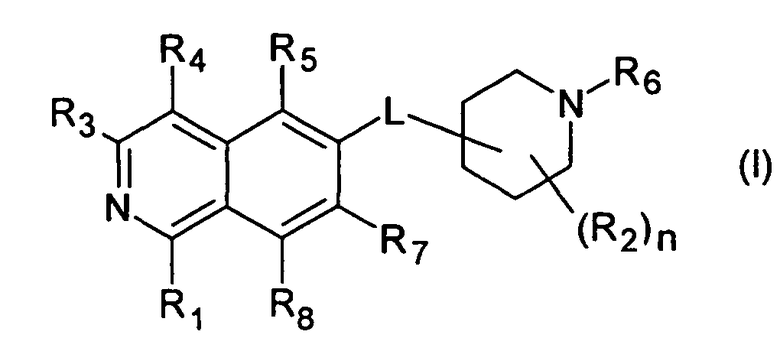
где значения радикалов раскрыты в формуле изобретения, и к композициям, содержащим такие соединения. Обнаружено, что такие соединения и композиции могут быть полезны для лечения и/или профилактики заболеваний, связанных с Rho-киназой и/или опосредованным Rho-киназой фосфорилированием фосфатазы легкой цепи миозина. 4 н. и 27 з.п. ф-лы, 378 пр., 12 табл.
1. Соединение формулы (I)
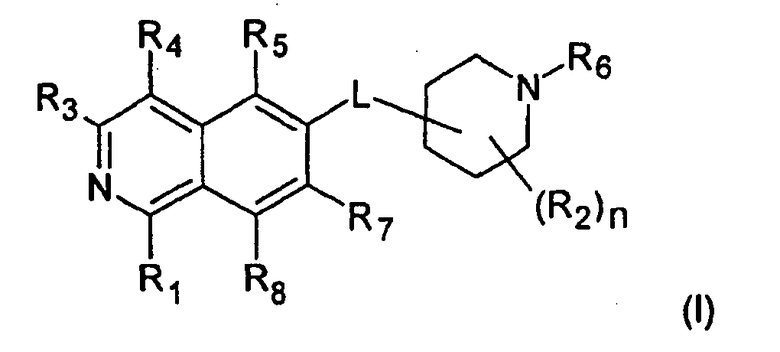
где R1 представляет собой
Н,
(С1-С6)алкил,
NH-(С1-С6)алкил,
NHR' или
N[(С1-С6)алкил]2;
R2 представляет собой водород или (С1-С6)алкил;
R3 представляет собой
Н,
NH2,
NHR'' или
NH-C(O)-R'',
R4 представляет собой
Н,
галоген, (С1-С6)алкил,
или
(С1-С6)алкилен-R';
R5 представляет собой
Н,
галоген,
CN,
NO2,
(С1-С6)алкил,
(С2-С6)алкенил,
R',
(С1-С6)алкилен-(С6-С10)арил,
(С2-С6)алкенилен-(С6-С10)арил,
СН(ОН)-(С1-С6)алкил,
NH2,
NH-R',
NH-C(O)-(C1-C6)алкил,
или
С(O)O-(C1-С6)алкил;
R7 представляет собой Н,
R',
(С1-С8)алкил,
(C1-C8)алкилен-R' или
(C1-C6)алкилен-С(O)N[R']2;
R7 представляет собой
Н,
галоген,
CN,
(С1-С6)алкил,
(С2-С6)алкенил,
R',
(С1-С6)алкилен-R',
СН(ОН)-(С1-С6)алкил,
NH2,
NH-R',
NН-С(O)-(С1-С6)алкил или
С(O)O-(С1-С6)алкил;
R8 представляет собой Н, галоген или (С1-С6)алкил;
n имеет значение 1, 2, 3 или 4; и
L представляет собой О или O-(С1-С6)алкилен;
где R' представляет собой
(С3-С8)циклоалкил,
(С5-С10)гетероциклил, в котором 1-4 атома углерода могут быть заменены на атом азота, кислорода или серы,
(С6-С10)арил; и
R'' представляет собой
(С3-С8)циклоалкил,
(С5-С10)гетероциклил, в котором 1-4 атома углерода могут быть заменены на атом азота, кислорода или серы,
(С6-С10)арил
(С1-С6)алкил,
(С1-С6)алкилен-R',
(С1-С6)алкилен-O-(С1-С6)алкил или
(С1-С6)алкилен-NRxRy; и
где Rx и Ry независимо друг от друга представляют собой
(С1-С6)алкил или
(С1-С4)алкилен-(С5-С10)гетероциклил;
где в остатках R3, R5 и R6 (С6-С10)арил и (С5-С10)гетероциклил являются незамещенными или замещенными от одного до трех раз группой, независимо выбранной из
галогена, CF3, OCF3, NO2, CN, С(O)-(С1-С6)алкила, С(O)O(С1-С6)алкила, CONH2, C(O)N[(C1-C6)алкил]2, (С3-С8)циклоалкила, (С1-С6)алкила, (С1-С6)алкил-ОН, (C1-С6)алкилен-N[(С1-С6)алкил]2, O-(С1-С6)алкила, SO2NH2, S-(С1-С6)алкила, SO2-(C1-С6)алкила, SO2-N=СН-N((С1-С6)алкил)2, NH2, N[(С1-C6)алкил]2, NH-C(O)-(C1-С6)алкила, NH-SO2-(С1-С6)алкила, (С6-С10)арила, (С1-С6)алкилен-(С6-С10)арила, (С5-С10)гетероциклила или O-фенила,
или (С6-С10)арил может быть вицинально замещен группой O-(С1-С4)алкилен-O, образуя, таким образом, вместе с атомами углерода, с которыми связаны атомы кислорода, 5-8-членное кольцо;
где в остатках R4, R5, R7 и R8 один атом водорода алкила или алкилена необязательно может быть замещен ОН или ОСН3; и
где в остатках R3, R4, R5, R7 и R8 алкил или алкилен необязательно может быть замещен одним или несколькими атомами F;
и их фармацевтически приемлемые соли.
2. Соединение по п.1, где R1 представляет собой Н, (С1-С6)алкил, NH-(C1-С6)алкил, NH-(С6-С10)арил или N[(С1-С6)алкил]2.
3. Соединение по п.1, где R1 представляет собой Н, (С1-С4)алкил, NH-(C1-С4)алкил, N[(С1-С4)алкил]2 или NH-фенил.
4. Соединение по п.1, где R1 представляет собой Н, (C1-C2)алкил или NH-(C1-C2)алкил.
5. Соединение по п.1, где R1 представляет собой Н.
6. Соединение по п.1, где R3 представляет собой Н или NHR''.
7. Соединение по п.1, где R3 представляет собой Н; NH-(С5-С6)гетероциклил, предпочтительно NH-(С5-С6)гетероарил, содержащий один или несколько N атомов; или NH-фенил.
8. Соединение по п.1, где R8 представляет собой Н, галоген или (С1-С4)алкил.
9. Соединение по п.1, где R8 представляет собой Н, Cl, F, метил или этил.
10. Соединение по п.1, где R4 представляет собой Н, галоген или (С1-С6)алкил.
11. Соединение по п.1, где R4 представляет собой Н, галоген или (С1-С4)алкил.
12. Соединение по п.1, где R4 представляет собой Н.
13. Соединение по п.1, где R5 представляет собой Н, галоген, (С1-С6)алкил, R', NH-(С6-С10)арил или (С1-С6)алкилен-С6-С10)арил.
14. Соединение по п.1, где R5 представляет собой Н, галоген, (С1-С6)алкил, (С6-С10)арил, NH-(С6-С10)арил, (С1-С2)алкил-(С6-С10)арил или (С5-С10)гетероарил.
15. Соединение по п.1, где R7 представляет собой H, галоген, CN, (С1-С6)алкил, (С2-С6)алкенил, R' или (С1-С6)алкилен-(С3-С8)циклоалкил.
16. Соединение по п.1, где R7 представляет собой H, галоген, CN, (С1-С4)алкил, (С2-С4)алкенил, фенил, циклопропил или (С5-С6)гетероарил.
17. Соединение по п.1, где R7 представляет собой H, фтор, хлор, бром, метил, этил, фенил, нитрил, циклопропил, тиенил или винил.
18. Соединение по п.1, где n имеет значение 1, 2 или 3.
19. Соединение по п.1, где n имеет значение 1.
20. Соединение по п.1, где R2 представляет собой Н или (С1-С4)алкил.
21. Соединение по п.1, где R2 представляет собой Н, (С1-С2)алкил.
22. Соединение по п.1, где R6 представляет собой H, (С1-С6)алкил, R', (C1-С4)алкилен-(С3-С8)циклоалкил, (С1-С4)алкилен-(С5-С10)гетероциклил или (С1-С6)алкилен-(С6-С10)арил.
23. Соединение по п.1, где R6 представляет собой Н, (С1-С6)алкил, (C5-С10)гетероциклил, (С1-С4)алкилен-(С5-С10)гетероциклил или (С1-С6)алкилен-(С6-С10)арил.
24. Соединение по п.1, где L присоединен к 3-положению или 4-положению пиперидинового кольца.
25. Соединение по п.1, где L присоединен к 4-положению пиперидинового кольца.
26. Соединение по п.1, где L представляет собой O-метилен, O-этилен или предпочтительно О.
27. Соединение по п.1, где
R1 представляет собой H, (С1-С4)алкил, NH-(С1-С4)алкил, N[(С1-С4)алкил]2 или NH-фенил;
R2 представляет собой Н, (С1-C4)алкил;
R3 представляет собой H, NH-(С5-С6)гетероарил или NH-фенил;
R4 представляет собой H, галоген или (С1-С4)алкил;
R5 представляет собой H, (С1-С4)алкил, галоген, (С6-С10)арил, NH-(С6-С10)арил, (С1-С2)алкил-(С6-С10)арил или (С5-С10)гетероарил;
R6 представляет собой H, (С1-С6)алкил, (С5-С10)гетероциклил, (С1-С4)алкилен-(С5-С10)гетероциклил, (С6-С10)арил или (С1-С6)алкилен-(С6-С10)арил;
R7 представляет собой H, галоген, CN, (С1-С4)алкил, (С2-С4)алкенил, фенил, циклопропил, (С5-С6)гетероарил;
R8 представляет собой H, галоген или (С1-С4)алкил;
n имеет значение 1; и
L представляет собой О.
28. Соединение формулы (I) по п.1, выбранное из группы:
 ,
,  ,
,
 ,
, 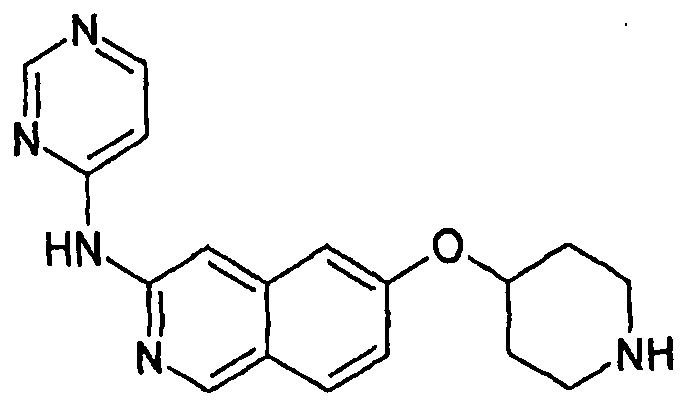 ,
,
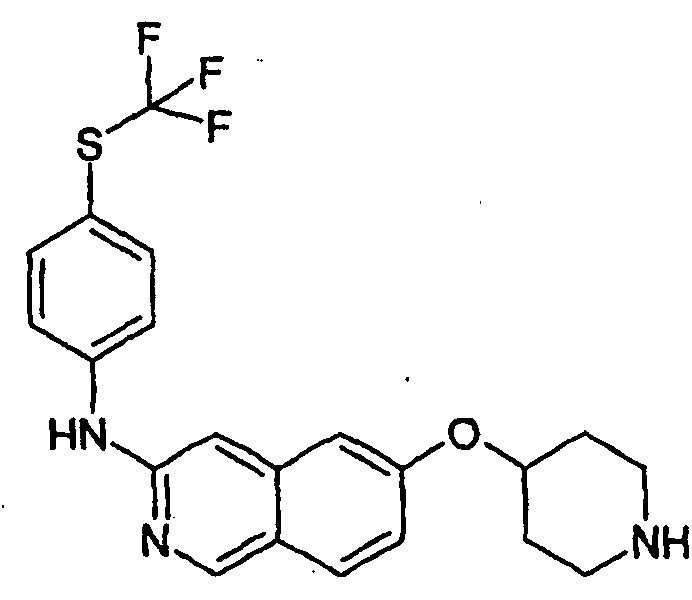 ,
, 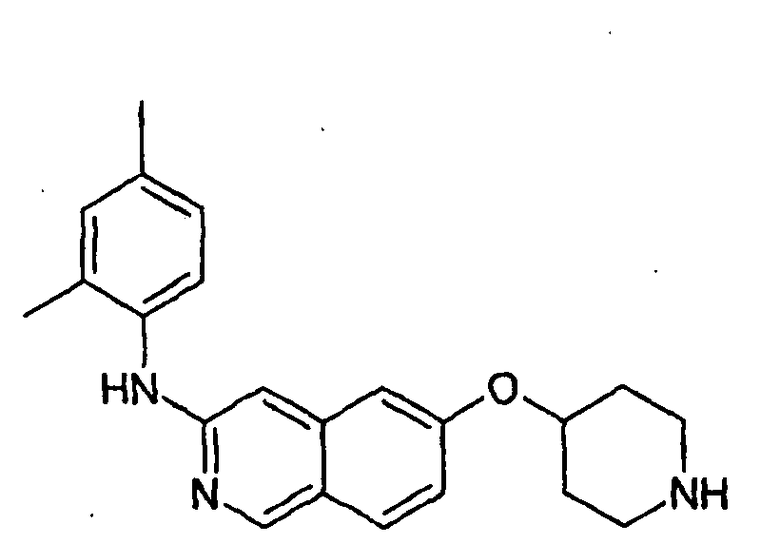 ,
,
и их фармацевтически приемлемых солей.
29. Применение, по меньшей мере, одного из соединений формулы (I) и/или их физиологически приемлемых солей по пп.1-28 для получения лекарственного средства.
30. Применение, по меньшей мере, одного из соединений формулы (I) и/или их физиологически приемлемых солей по пп.1-28 для получения лекарственного средства для лечения и/или профилактики гипертензии, легочной гипертензии, глазной гипертензии, нарушения периферического кровообращения, стенокардии, церебрального вазоспазма, астмы, преждевременных родов, гиперагрегации тромбоцитов, окклюзивного заболевания периферических артерий (PAOD), хронического обструктивного легочного заболевания (COPD), развития рака, эректильной дисфункции, артериосклероза, ишемического заболевания органов (с конечным повреждением органа), фиброидного легкого, фиброидной печени, печеночной недостаточности, фиброидной почки, почечного гломерулосклероза, почечной недостаточности, гипертрофии органов, гипертрофии простаты, осложнений диабета, рестеноза кровеносных сосудов, атеросклероза, рака, сердечной гипертрофии, сердечной недостаточности при ишемических заболеваниях; воспаления; аутоиммунных заболеваний; СПИДа, остеопатии, такой как остеопороз, нарушения функций мозга, бактериальных инфекций пищеварительного тракта, сепсиса, респираторного дистресс-синдрома взрослых, ретинопатии, глаукомы и болезни Альцгеймера.
31. Лекарственное средство, содержащее эффективное количество, по меньшей мере, одного соединения по любому из пп.1-28 и/или его фармакологически приемлемой соли, физиологически толерантные эксципиенты и носители и, если это является подходящим, дополнительные добавки и/или другие активные ингредиенты, предназначенное для лечения или профилактики гипертензии, легочной гипертензии, глазной гипертензии, нарушения периферического кровообращения, стенокардии, церебрального вазоспазма, астмы, преждевременных родов, гиперагрегации тромбоцитов, окклюзивного заболевания периферических артерий (PAOD), хронического обструктивного легочного заболевания (COPD), развития рака, эректильной дисфункции, артериосклероза, ишемического заболевания органов (с конечным повреждением органа), фиброидного легкого, фиброидной печени, печеночной недостаточности, фиброидной почки, почечного гломерулосклероза, почечной недостаточности, гипертрофии органов, гипертрофии простаты, осложнений диабета, рестеноза кровеносных сосудов, атеросклероза, рака, сердечной гипертрофии, сердечной недостаточности при ишемических заболеваниях; воспаления; аутоиммунных заболеваний; СПИДа, остеопатии, такой как остеопороз, нарушения функций мозга, бактериальных инфекций пищеварительного тракта, сепсиса, респираторного дистресс-синдрома взрослых, ретинопатии, глаукомы и болезни Альцгеймера.
| Устройство для управления механизмом резания листорезальной машины | 1988 |
|
SU1541559A1 |
| Takami, Atsuya et al, Bioorganic & Medicinal Chemistry, 2004, 12(9), 2115-2137 | |||
| RU 2003103606 A, 27.08.2004. | |||

