Изобретение относится к производным пиразола, которые ингибируют или модулируют активность циклин-зависимых киназ (CDK), киназ гликоген-синтазы (GSK) и Aurora киназ, к применению этих соединений в лечении или профилактике опосредованных киназами заболеваний или состояний, а также к новым соединениям, обладающим ингибирующей или модулирующей активностью в отношении киназ. Кроме того, разработаны фармацевтические композиции, содержащие указанные соединения, а также новые химические промежуточные соединения.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Протеинкиназы образуют большое семейство структурно родственных ферментов, которые отвечают за управление самыми разнообразными процессами передачи сигналов в клетке (Hardie, G. and Hanks, S. (1995) The Protein Kinase Facts Book. I and II, Academic Press, San Diego, CA). Киназы могут быть классифицированы на семейства по субстратам, которые они фосфорилируют (например, протеин-тирозин, протеин-серин/треонин, липиды и т.д.). Были идентифицированы характерные фрагменты последовательностей, которые, как правило, соответствуют каждому из этих семейств киназ (например, Hanks, S.K., Hunter, T., FASEB J., 9:576-596 (1995); Knighton, et al., Science, 253: 407-414 (1991); Hiles, et al., Cell, 70: 419-429 (1992); Kunz, et al., Cell, 73: 585-596 (1993); Garcia-Bustos, et al., EMBO J., 13: 2352-2361 (1994)).
Протеинкиназы могут быть охарактеризованы механизмами их регулирующего действия. Эти механизмы включают, например, аутофосфорилирование, трансфосфорилирование другими киназами, взаимодействия белок-белок, взаимодействия белок-липид и взаимодействия белок-полинуклеотид. Отдельные протеинкиназы могут регулироваться более чем одним механизмом.
Киназы регулируют большое количество различных процессов в клетках, включая, но не ограничиваясь перечисленным, пролиферацию, дифференцировку, апоптоз, подвижность, транскрипцию, трансляцию и другие процессы передачи сигнала, путем присоединения фосфатной группы к целевым белкам. Эти акты фосфорилирования действуют как молекулярные переключатели включено/выключено, которые могут модулировать или регулировать биологическую функцию целевого белка. Фосфорилирование целевых белков происходит в ответ на различные внеклеточные сигналы (гормоны, нейромедиаторы, факторы роста и дифференцировки и т.д.), события клеточного цикла, стрессы, связанные с окружающей средой или питанием и т.д. Присущие протеинкиназам функции в путях передачи сигнала состоят в том, чтобы активировать или инактивировать (либо прямо, либо косвенно), например, метаболический фермент, регуляторный белок, рецептор, белок цитоскелета, ионный канал или насос, или фактор транскрипции. Неуправляемая передача сигнала из-за нарушенного управления фосфорилированием белков вовлечена в ряд заболеваний, включая, например, воспаление, рак, аллергию/астму, заболевания и состояния иммунной системы, заболевания и состояния центральной нервной системы и ангиогенез.
Циклин-зависимые киназы
Процесс деления эукариотических клеток может быть в общих чертах разделен на ряд последовательных фаз, именуемых G1, S, G2 и M. Было показано, что правильное прохождение через различные фазы клеточного цикла решающим образом зависит от пространственной и временной регуляции семейства белков, известных как циклин-зависимые киназы (CDK) и многообразной группы родственных им белковых партнеров, именуемых циклинами. CDK являются CDC2 (также известными как CDK1)-гомологичными белками серин-треонин киназы, которые способны использовать АТФ как субстрат при фосфорилировании различных полипептидов в зависимости от окружающей последовательности. Циклины представляют собой семейство белков, характеризующихся гомологичной областью, содержащей приблизительно 100 аминокислот и называемой «циклиновый бокс», которая используется для связывания с конкретными CDK белковыми партнерами и определяет селективность по отношению к ним.
Модуляция уровней экспрессии, скоростей распада и уровней активации различных CDK и циклинов во время клеточного цикла ведет к образованию циклинами ряда комплексов CDK/циклин, в которых CDK являются ферментно-активными. Образование этих комплексов управляет прохождением через отдельные контрольные точки клеточного цикла и, тем самым, делает возможным продолжение процесса деления клетки. Неспособность удовлетворить предварительно установленным биохимическим критериям в данной контрольной точке клеточного цикла, т.е. невозможность образования требуемого комплекса CDK/циклин, может привести к остановке клеточного цикла и/или клеточному апоптозу. Аномальная клеточная пролиферация, которая проявляется при раке, часто может быть отнесена к потере правильного управления клеточным циклом. Следовательно, ингибирование ферментной активности CDK предоставляет средство, с помощью которого можно остановить деление аномально делящихся клеток или убить их. Многообразие CDK и комплексов CDK и их решающая роль в опосредовании клеточного цикла обеспечивают широкий спектр потенциальных терапевтических мишеней, выбранных на основе определенного биохимического обоснования.
Движение от фазы G1 к фазе S клеточного цикла в основном регулируется CDK2, CDK3, CDK4 и CDK6 за счет связывания с циклинами типов D и E. Циклины типа D, по-видимому, делают возможным прохождение через ограничивающую точку G1, тогда как комплекс CDK2/циклин E является ключевым для перехода от фазы G1 к фазе S. Дальнейшее прохождение через фазу S и вхождение в G2, как считается, требует наличия комплекса CDK2/циклин A. Как митоз, так и переход от фазы G2 к фазе M, который запускает его, регулируются комплексами CDK1 и циклинов типов A и B.
Во время фазы G1 белок ретинобластомы (Rb) и родственные pocket-белки («карманные» белки), как, например, p130, являются субстратами для комплексов CDK(2, 4 и 6)/циклин. Прохождение через фазу G1 частично облегчается гиперфосфорилированием и, таким образом, инактивацией Rb и p130 комплексами CDK(4/6)/циклин-D. Гиперфосфорилирование Rb и p130 вызывает высвобождение факторов транскрипции, таких как E2F, и, таким образом, экспрессию генов, как, например, гена циклина E, необходимую для прохождения через G1 и для вхождения в S-фазу. Экспрессия циклина E облегчает образование комплекса CDK2/циклин E, который увеличивает или поддерживает уровни E2F путем дополнительного фосфорилирования Rb. Комплекс CDK2/циклин E также фосфорилирует другие белки, необходимые для репликации ДНК, такие как NPAT, который вовлечен в биосинтез гистона. Прохождение G1 и переход G1/S также регулируются митоген-стимулированным сигнальным путем Myc, который входит в сигнальный путь комплекса CDK2/циклин E. Кроме того, CDK2 связан с опосредованным p53 путем отклика на повреждение ДНК за счет регулирования уровней p21 при действии p53. p21 представляет собой белковый ингибитор комплекса CDK2/циклин E и за счет этого он способен блокировать или задерживать переход G1/S. Комплекс CDK2/циклин E может, следовательно, представлять собой точку, в которой, до некоторой степени, объединены биохимические стимулы для сигнальных путей Rb, Myc и p53. Поэтому CDK2 и/или комплекс CDK2/циклин E являются хорошими мишенями для терапевтических способов, предназначенных для остановки клеточного цикла или возобновления его управлением в аномально делящихся клетках.
Точная роль CDK3 в клеточном цикле неясна. До сих пор не был идентифицирован родственный циклиновый партнер, но доминирующей негативной формой CDK3-задержанных клеток является G1, позволяя, тем самым, предположить, что CDK3 играет определенную роль в управлении переходом G1/S.
Хотя большая часть CDK вовлечена в регулирование клеточного цикла, существует доказательство того, что некоторые члены семейства CDK вовлечены в другие биохимические процессы. Примером таких CDK является CDK5, которая необходима для правильного развития нейронов и которая, кроме того, вовлечена в фосфорилирование нескольких нейронных белков, таких как Tau, NUDE-1, синапсин1, DARPP32 и комплекса Munc18/синтаксин1А. Нейронная CDK5 обычно активируется связыванием с белками p35/p39. Однако активность CDK5 может быть разрегулирована связыванием с p25, т.е. усеченным вариантом p35. Превращение p35 в p25 и последующее нарушение регулирования активности CDK5 может быть вызвано ишемией, эксцитотоксичностью и β-амилоидным пептидом. Следовательно, p25 вовлечен в патогенез нейродегенеративных заболеваний, таких как болезнь Альцгеймера и, следовательно, представляет интерес в качестве мишени для терапевтических способов, направленных против этих заболеваний.
CDK7 представляет собой ядерный белок, который обладает CDC2 CAK активностью, и связывается с циклином H. CDK7 была идентифицирована в качестве компонента TFIIH транскрипционного комплекса, который обладал активностью в отношении C-концевого домена (CTD) РНК полимеразы II. Это было связано с регулированием транскрипции ВИЧ-1 с помощью Tat-опосредованного биохимического сигнального пути. CDK8 связывает циклин C и вовлечена в фосфорилирование CTD РНК полимеразы II. Подобным же образом комплекс CDK9/циклин-T1 (комплекс P-TEFb) вовлечен в управление элонгацией РНК полимеразы II. Кроме того, P-TEFb необходим для активации транскрипции генома ВИЧ-1 вирусным трансактиватором Tat за счет его взаимодействия с циклином T1. Следовательно, CDK7, CDK8, CDK9 и комплекс P-TEFb являются потенциальными мишенями для способов антивирусной терапии.
Опосредование активности комплекса CDK/циклин на молекулярном уровне требует ряда актов стимулирующего и ингибирующего фосфорилирования или дефосфорилирования. Фосфорилирование CDK осуществляется группой CDK-активирующих киназ (CAK) и/или такими киназами, как wee1, Myt1 и Mik1. Дефосфорилирование осуществляется такими фосфатазами, как cdc25(a и c), pp2a или KAP.
Далее активность комплекса CDK/циклин может регулироваться двумя семействами эндогенных клеточных белковых ингибиторов: семейством Kip/Cip или семейством INK. Белки INK специфично связывают CDK4 и CDK6. p16ink4 (известный также как MTS1) является геном, подавляющим потенциальные опухоли, который подвергнут мутации или удален в большом количестве первичных раковых опухолей. Семейство Kip/Cip включает такие белки как, например, p21Cip,Waf1, p27Kip1 и p57Kip2. Как обсуждалось ранее, p21 индуцируется p53, причем он способен инактивировать комплексы CDK2/циклин(E/A) и CDK4/циклин(D1/D2/D3). Атипически низкие уровни экспрессии p27 наблюдались при раке груди, ободочной кишки и простаты. Напротив, избыточная экспрессия циклина E в солидных опухолях, как было показано, коррелирует с неблагоприятным для пациента прогнозом. Избыточную экспрессию циклина D1 связывали с карциномой пищевода, груди, сквамозной карциномой и немелкоклеточной карциномой легкого.
Ключевая роль CDK и связанных с ними белков в координировании и продвижении клеточного цикла в пролиферирующих клетках была подчеркнута выше. Кроме того, были описаны некоторые из биохимических сигнальных путей, в которых CDK играют ключевую роль. Следовательно, разработка способов монотерапии для лечения пролиферативных расстройств, как, например, различных видов рака, использующих разновидности терапии, нацеленные на CDK в общем, или на конкретные CDK, потенциально является в высшей степени желательной. Ингибиторы CDK, предположительно, могли бы также применяться для лечения других состояний, как, например, в числе прочего, вирусных инфекций, аутоиммунных заболеваний и нейродегенеративных заболеваний. Кроме того, терапевтические способы, нацеленные на CDK, могут обеспечить клинические преимущества при лечении указанных выше заболеваний при применении в рамках комбинированной терапии совместно с либо существующими, либо новыми терапевтическими средствами. Нацеленные на CDK способы противораковой терапии потенциально могли бы иметь преимущества над многими существующими противоопухолевыми средствами, т.к. они не взаимодействовали бы непосредственно с ДНК и, следовательно, должны были бы снизить риск развития вторичной опухоли.
Диффузные B-крупноклеточные лимфомы (DLBCL)
Развитие клеточного цикла регулируется комбинированным действием циклинов, циклин-зависимых киназ (CDK) и ингибиторов CDK (CDKi), которые являются отрицательными регуляторами клеточного цикла. p27KIP1 является ключевым CDKi в регулировании клеточного цикла, и его распад необходим для перехода G1/S. Несмотря на отсутствие экспрессии p27KIP1 в пролиферирующих лимфоцитах, сообщалось, что некоторые агрессивные B-клеточные лимфомы демонстрируют аномальное окрашивание p27KIP1. Аномально высокая экспрессия p27KIP1 была обнаружена в лимфомах этого типа. Анализ клинической значимости этих данных показал, что высокий уровень экспрессии p27KIP1 в опухолях данного типа является неблагоприятным прогностическим признаком как при одномерном, так и при многомерном анализе. Эти результаты показывают, что в диффузных B-крупноклеточных лимфомах (DLBCL) имеется аномальная экспрессия p27KIP1, с неблагоприятной клинической значимостью, в предположении, что этот аномальный белок p27KIP1 может приводиться в нефункциональное состояние при взаимодействии с другими белками, регулирующими клеточный цикл (Br. J. Cancer. 1999 Jul; 80(9): 1427-34. p27KIP1 аномально экспрессируется в диффузных B-крупноклеточных лимфомах, и он связан с неблагоприятным клиническим исходом. Saez A, Sanchez E, Sanchez-Beato M, Cruz MA, Chacon I, Munoz E, Camacho FI, Martinez-Montero JC, Mollejo M, Garcia JF, Piris MA. Department of Pathology, Virgen de la Salud Hospital, Toledo, Spain).
Хронический лимфоцитарный лейкоз
B-клеточный хронический лимфоцитарный лейкоз (CLL) является наиболее обычной разновидностью лейкемии в западном полушарии, причем ежегодно диагностируется приблизительно 10000 новых случаев (Parker SL, Tong T, Bolden S, Wingo PA: Cancer statistics, 1997. Ca. Cancer. J. Clin. 47:5, (1997)). По отношению к другим формам лейкемии прогноз CLL в целом является благоприятным, причем даже пациенты с наиболее развитой стадией имеют среднюю продолжительность жизни, равную 3 годам.
Включение флударабина в первичную терапию пациентов с симптомами CLL привело к более высоким показателям полной реакции (27% против 3%) и продолжительности жизни без развития заболевания (33 месяца против 17) по сравнению с применявшимися ранее способами лечения на основе алкилирующих средств. Хотя достижение полной клинической реакции после терапии является первым шагом по направлению к улучшению выживаемости при CLL, большинство пациентов либо не достигают полной ремиссии, либо не реагируют на флударабин. Кроме того, у всех пациентов с CLL, подвергавшихся лечению флударабином, в конце концов возникал рецидив, что делает роль флударабина в качестве единственного средства лечения чисто паллиативной (Rai KR, Peterson B, Elias L, Shepherd L, Hines J, Nelson D, Cheson B, Kolitz J, Schiffer CA: A randomized comparison of fludarabine and chlorambucil for patients with previously untreated chronic lymphocytic leukemia. A CALGB SWOG, CTG/NCI-C and ECOG Inter-Group Study. Blood 88:141a, 1996 (abstr 552, suppl 1). Следовательно, если должен реализоваться дальнейший прогресс в лечении данного заболевания, будет необходимо выявлять новые средства с новыми механизмами действия, которые дополняют цитотоксичность флударабина и устраняют устойчивость CLL к действию лекарственных средств, вызванную внутренними факторами.
Наиболее широко изученным и имеющим постоянную предсказательную силу фактором плохой реакции на терапию и низкой выживаемости CLL-пациентов является аномальная функция p53, характеризующаяся точечными мутациями или делециями хромосомы 17p13. Действительно фактическое отсутствие реакции на терапию как алкилирующими средствами, так и аналогами пурина, для таких CLL-пациентов с аномальной функцией p53 было документально подтверждено в целом ряде отдельных установленных историй болезни. Разработка терапевтического средства, которое способно преодолеть устойчивость к лекарственным средствам, связанную с мутацией p53 при CLL, могло бы потенциально стать значительным прогрессом в лечении данного заболевания.
Флавопиридол и CYC 202, т.е. ингибиторы циклин-зависимых киназ, вызывают in vitro апоптоз злокачественных клеток B-клеточного хронического лимфоцитарного лейкоза (B-CLL).
Действие флавопиридола приводит к стимулированию активности каспазы 3 и к зависимому от каспазы расщеплению p27(kip1), т.е. отрицательного регулятора клеточного цикла, который избыточно экспрессируется при B-CLL (Blood. 1998 Nov 15;92(10):3804-16 Flavopiridol induces apoptosis in chronic lymphocytic leukemia cells via activation of caspase-3 without evidence of bcl-2 modulation or dependence on functional p53. Byrd JC, Shinn C, Waselenko JK, Fuchs EJ, Lehman TA, Nguyen PL, Flinn IW, Diehl LF, Sausville E, Grever MR).
Aurora киназы
Относительно недавно было обнаружено новое семейство серин/треонин киназ, известных как Aurora киназы, которые вовлечены в фазы G2 и M клеточного цикла, и которые являются важными регуляторами митоза. Точная роль Aurora киназ еще должна быть объяснена, но они принимают участие в регулировании контрольной точки митоза, динамике хромосом и цитокинезе (Adams et al., Trends Cell Biol., 11: 49-54 (2001)). Aurora киназы находятся на центросомах интерфазных клеток, на полюсах биполярного веретена и в клеточной пластинке в плоскости экватора веретена митотического аппарата.
До настоящего времени у млекопитающих было обнаружено три члена семейства Aurora киназ (E.A. Nigg, Nat. Rev. Mol. Cell Biol. 2: 21-32, (2001)). А именно:
Aurora A (также именуемая в литературе Aurora 2);
Aurora B (также именуемая в литературе Aurora 1); и
Aurora C (также именуемая в литературе Aurora 3).
Aurora киназы включают высокогомологичные каталитические домены, но значительно отличаются в своих N-концевых частях (Katayama H, Brinkley WR, Sen S.; The Aurora kinases: role in cell transformation and tumorigenesis; Cancer Metastasis Rev. 2003 Dec; 22(4):451-64).
Было установлено, что субстраты Aurora киназ A и B включают подобные кинезину моторные белки, белки аппарата веретена, белок гистон H3, белок кинетохора и подавляющий опухоли белок p53.
Полагают, что Aurora A киназы вовлечены в формирование веретена и во время ранней фазы G2 локализуются на центросоме, где они фосфорилируют белки, связанные с веретеном (Prigent et al., Cell, 114:531-535 (2003). Hirota и соавторы (Hirota et al, Cell, 114: 585-598, (2003)) обнаружили, что клетки, обедненные Aurora A протеинкиназой, были неспособны начать митоз. Кроме того, было найдено (Adams, 2001), что мутация или разрушение гена Aurora A у различных видов ведет к митотическим аномалиям, включая разделение центросом и дефекты созревания, аномалии веретена и дефекты сегрегации хромосом.
Aurora киназы, как правило, экспрессируются в незначительных количествах в большинстве нормальных тканей, причем исключениями являются ткани с высокой долей делящихся клеток, такие как вилочковая железа и яички. Однако повышенные уровни Aurora киназ были обнаружены в большинстве раковых опухолей человека (Giet et al., J. Cell. Sci. 112: 3591-361, (1999) и Katayama (2003)). Кроме того, Aurora A киназа отображает область хромосомы 20ql3, которая, как часто обнаруживается, усилена во многих раковых опухолях человека.
Так, например, значительная избыточная экспрессия Aurora A была обнаружена в раковых опухолях груди, яичников и поджелудочной железы человека (см. Zhou et al, Nat. Genet. 20: 189-193, (1998), Tanaka et al., Cancer Res., 59: 2041-2044, (1999) и Han et al, cancer Res., 62: 2890-2896, (2002).
Кроме того, Isola, American Journal of Pathology 147, 905-911 (1995) сообщил, что амплификация локуса Aurora A (20ql3) коррелирует с неблагоприятным прогнозом для пациентов с раком груди без поражения лимфатических узлов.
Амплификация и/или избыточная экспрессия Aurora A наблюдается при раковых опухолях мочевого пузыря человека, и амплификация Aurora A связана с анеуплодией и агрессивным клиническим поведением, см. Sen et al., J. Natl. Cancer Inst, 94: 1320-1329 (2002).
Увеличенная экспрессия Aurora A была обнаружена более чем в 50% случаев колоректального рака (см. Bischoff et al., EMBO J., 17: 3052-3065, (1998) и Takahashi et al., Jpn. J. Cancer Res., 91: 1007-1014 (2000)), раковых опухолей яичников (см. Gritsko et al., Clin. Cancer Res., 9: 1420-1426 (2003)) и раковых опухолей желудка (Sakakura et al., British Journal of Cancer, 84: 824-831 (2001)).
Tanaka et al., Cancer Research, 59: 2041-2044 (1999) обнаружил доказательства избыточной экспрессии Aurora A в 94% случаев инвазивной аденокарциномы протоков молочной железы.
Кроме того, высокие уровни Aurora A киназы были обнаружены в клеточных линиях опухолей почек, шейки матки, поджелудочной железы и простаты, а также клеточных линиях нейробластомы, меланомы, лимфомы Bischoff et al. (1998), EMBO J., 17: 3052-3065 (1998); Kimura et al. J. Biol. Chem., 274: 7334-7340 (1999); Zhou et al., Nature Genetics, 20: 189-193 (1998); Li et al., Clin Cancer Res. 9 (3): 991-7 (2003)].
Aurora B в значительных количествах экспрессируется во многих линиях клеток опухолей человека, включая лейкемические клетки [Katayama et al., Gene 244:1-7]. При первичных колоректальных раковых опухолях уровни данного фермента возрастают как функция стадии по Дюку [Katayama et al., J. Natl Cancer Inst., 91: 1160-1162 (1999)].
Высокие уровни Aurora-3 (Aurora-C) были обнаружены в нескольких линиях опухолевых клеток, даже несмотря на то, что в нормальных тканях присутствие этой киназы имеет тенденцию ограничиваться зародышевыми клетками (см. Kimura et al., Journal of Biological Chemistry, 274: 7334-7340 (1999)). Кроме того, Takahashi et al., Jpn. J. Cancer Res., 91: 1007-1014 (2001) сообщалось об избыточной экспрессии Aurora-3 приблизительно в 50% случаев колоректального рака.
Другие сообщения о роли Aurora киназ в пролиферативных расстройствах могут быть найдены в Bischoff et al., Trends in Cell Biology 9: 454-459 (1999); Giet et al. Journal of Cell Science, 112: 3591-3601 (1999) и Dutertre, et al. Oncogene, 21: 6175-6183 (2002).
Royce и сотрудники сообщают, что экспрессия гена Aurora 2 (известного как STK15 или BTAK) была отмечена приблизительно в одной четвертой части случаев первичных опухолей груди (Royce ME, Xia W, Sahin AA, Katayama H, Johnston DA, Hortobagyi G, Sen S, Hung MC; STK15/Aurora-A expression in primary breast tumours is correlated with nuclear grade but not with prognosis; Cancer. 2004 Jan 1; 100(1): 12-9).
Внутриматочная карцинома (EC) включает как минимум два типа рака: эндометриоидные карциномы (EEC) представляют собой опухоли, связанные с эстрогеном, которые часто являются эуплоидными и имеют благоприятный прогноз. Неэндометриоидные карциномы (NEEC; серозная и светлоклеточная формы) не связаны с эстрогеном, часто бывают анэуплоидными и являются клинически агрессивными. Также было обнаружено, что уровень Aurora увеличивался на 55,5% при NEEC, но не при всех EEC (P≤0,001) (Moreno-Bueno G, Sanchez-Estevez C, Cassia R, Rodriguez-Perales S, Diaz-Uriarte R, Dominguez O, Hardisson D, Andujar M, Prat J, Matias-Guiu X, Cigudosa JC, Palacios J. Cancer Res. 2003 Sep 15; 63(18): 5697-702).
Reichardt и соавторы (Oncol Rep. 2003 Sep-Oct; 10 (5): 1275-9) сообщали, что количественный анализ ДНК с помощью PCR для поиска амплификации Aurora в глиомах показал, что пять из 16 опухолей (31%) в различных стадиях по классификации ВОЗ (1 в стадии II, 1 в стадии III, 3 в стадии IV) показали амплификацию ДНК гена Aurora 2. Была сформулирована гипотеза, что амплификация гена Aurora 2 может быть неслучайным генетическим изменением в глиомах человека, играющим определенную роль в генетических путях образования опухолей.
Результаты Hamada и сотрудников (Br. J. Haematol. 2003 May; 121 (3): 439-47) также наводят на мысль, что Aurora 2 является эффективным кандидатом для выявления не только активности заболевания, но также и путей образования неходжкинских лимфом. Замедление роста опухолевых клеток, являющееся следствием ограничения функций этого гена, могло бы стать терапевтическим подходом к лечению неходжкинских лимфом.
В исследовании, выполненном Gritsko и соавторами (Clin Cancer Res. 2003 Apr; 9 (4): 1420-6) у 92 пациентов с первичными опухолями яичников были исследованы активность киназ и уровни белка Aurora A. Анализ киназ in vitro выявил повышенную активность Aurora A киназы в 44 случаях (48%). Повышенные уровни белка Aurora A были обнаружены в 52 образцах (57%). Высокие уровни белка Aurora A хорошо коррелировали с повышенной активностью киназы.
Результаты, полученные Li и соавторами (Clin. Cancer Res. 2003 Mar; 9(3): 991-7), показали, что ген Aurora A подвергается избыточной экспрессии в опухолях поджелудочной железы и линиях раковых клеток, и предположили, что повышенная экспрессия Aurora A может играть определенную роль в образовании раковых опухолей поджелудочной железы.
Аналогично было показано, что амплификация гена Aurora A и связанное с этим увеличение экспрессии митотической киназы, которую кодирует этот ген, ассоциируется с анеуплоидией и агрессивным клиническим поведением рака мочевого пузыря человека (J. Natl. Cancer Inst. 2002 Sep 4; 94 (17): 1320-9).
Исследования нескольких групп (Dutertre S, Prigent C., Aurora-A overexpression leads to override of the microtubule-kinetochore attachment checkpoint; Mol. Interv. 2003 May; 3(3): 127-30 и Anand S, Penrhyn-Lowe S, Venkitaraman AR., Aurora-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol, Cancer Cell. 2003 Jan; 3 (1): 51-62) наводят на мысль, что избыточная экспрессия активности Aurora киназы связана с устойчивостью к некоторым современным способам лечения рака. Например, избыточная экспрессия Aurora A в фибробластах мышиных зародышей может понизить чувствительность этих клеток к цитотоксическому действию производных таксана. Следовательно, ингибиторы Aurora киназы могут найти конкретное применение у пациентов с развившейся устойчивостью к существующим видам терапии.
На базе исследований, выполненных к настоящему времени, представляется, что ингибирование Aurora киназ, в частности Aurora киназы A и Aurora киназы B, обеспечит эффективные средства замедления развития опухолей.
Harrington и сотрудники (Nat Med. 2004 Mar; 10 (3) 262-7) продемонстрировали, что ингибитор Aurora киназ подавляет рост опухоли и вызывает регресс опухоли in vivo. В этом исследовании ингибитор Aurora киназы блокировал пролиферацию раковых клеток, и, кроме того, запускал процесс клеточной смерти у целого ряда линий раковых клеток, включая лейкемические и колоректальные клеточные линии и линию клеток рака груди. В дополнение к сказанному показана возможность лечения лейкемии путем индуцирования апоптоза лейкемических клеток. VX-680 эффективно убивает устойчивые к лечению клетки первичной острой миелогенной лейкемии (AML) у пациентов (Andrews, Oncogene, 2005, 24, 5005-5015).
Разновидности рака, которые могут быть особенно подвержены действию ингибиторов Aurora, включают рак груди, мочевого пузыря, колоректальный рак, рак поджелудочной железы, яичников, неходжкинскую лимфому, глиомы и неэндометриоидные внутриматочные карциномы. Разновидности лейкемии, особенно подверженные действию ингибиторов Aurora, включают острую миелогенную лейкемию (AML), хроническую миелогенную лейкемию (CML), B-клеточную лимфому (клетки мантийной ткани) и острый лимфобластный лейкоз (ALL).
Киназа гликоген-синтазы
Киназа-3 гликоген-синтазы (GSK3) представляет собой серин-треонин киназу, которая встречается у людей в виде двух изоформ, экспрессируемых во всех тканях (GSK3α и GSK3β). GSK3 привлекалась к рассмотрению в качестве фермента, обладающего определенной ролью в эмбриональном развитии, синтезе белка, клеточной пролиферации, дифференцировке клеток, динамике микроканальцев, клеточной подвижности и клеточном апоптозе. Как таковая, GSK3 вовлечена в развитие таких болезненных состояний, как диабет, рак, болезнь Альцгеймера, удар, эпилепсия, боковой амиотрофический склероз и/или травма головы. Филогенетически GSK3 наиболее тесно связана с циклин-зависимыми киназами (CDK).
Консенсусная пептидная последовательность субстрата, распознаваемая GSK3, является последовательностью (Ser/Thr)-X-X-X-(pSer/pThr), где X представляет собой любую аминокислоту (в положениях (n+1), (n+2), (n+3)) и pSer и pThr представляют собой фосфосерин и фосфотреонин, соответственно, в положении (n+4). GSK3 фосфорилирует первый серин или треонин в положении (n). Фосфосерин или фосфотреонин в положении (n+4), по-видимому, необходим для стимулирования GSK3 к достижению максимальной скорости превращения субстрата. Фосфорилирование GSK3α по Ser21 или GSK3β по Ser9 приводит к ингибированию GSK3. Исследования мутагенеза и конкурентных реакций с пептидами привели к модели, согласно которой фосфорилированный N-конец GSK3 способен конкурировать с фосфопептидным субстратом (S/TXXXpS/pT) за счет механизма аутоингибирования. Кроме того, имеются данные, дающие возможность предположить, что GSK3α и GSK3β могут в незначительной степени регулироваться фосфорилированием остатков тирозина 279 и 216, соответственно. Мутация этих остатков в Phe вызывала восстановление активности киназы in vivo. Установление кристаллографической структуры GSK3β с помощью рентгеновских лучей помогло пролить свет на все аспекты активации и регулирования GSK3.
GSK3 формирует часть сигнального пути реакции на инсулин у млекопитающих, причем фермент способен фосфорилировать и, тем самым, инактивировать гликоген-синтазу. Таким образом, увеличение активности гликоген-синтазы и за счет этого синтеза гликогена, с помощью ингибирования GSK3, рассматривалось в качестве потенциального средства борьбы с сахарным диабетом типа II или инсулинонезависимым сахарным диабетом (NIDDM): состоянием, при котором ткани тела становятся устойчивыми к стимулированию инсулином. Реакция клеток на инсулин в печени, жировой или мускульной тканях запускается за счет связывания инсулина с внеклеточным рецептором инсулина. Это вызывает фосфорилирование и последующее поступление на плазматическую мембрану белков-субстратов рецептора инсулина (IRS). Дальнейшее фосфорилирование белков IRS инициирует появление фосфоинозитид-3-киназы (PI3K) на плазматической мембране, где она способна высвободить вторичный мессенджер - фосфатидилинозитил 3,4,5-трифосфат (PIP3). Это содействует совместной локализации 3-фосфоинозитид-зависимой протеинкиназы 1 (PDK1) и протеинкиназы B (PKB или Akt) на мембране, где PDK1 активирует PKB. PKB способна фосфорилировать и, тем самым, ингибировать GSK3α и/или GSK3β путем фосфорилирования остатков Ser9 или Ser21, соответственно. Затем ингибирование GSK3 инициирует увеличение активности гликоген-синтазы. Таким образом, терапевтические средства, способные ингибировать GSK3, могут вызвать реакции клеток, сходные с реакциями, наблюдаемыми при стимуляции инсулином. Дополнительным in vivo субстратом GSK3 является эукариотический фактор инициации синтеза белка 2B (eIF2B). eIF2B инактивируется при фосфорилировании и, таким образом, приобретает способность подавлять биосинтез белка. Ингибирование GSK3, например, путем инактивации белка, представляющего собой «молекулу-мишень рапамицина у млекопитающих» (mTOR), может, таким образом, увеличивать биосинтез белка. Наконец, существуют доказательства в пользу регулирования активности GSK3 через сигнальный путь митоген-активированной протеинкиназы (MAPK) за счет фосфорилирования GSK3 киназами, как, например, митоген-активированной протеинкиназой, активированной протеинкиназой (MAPKAP-K1 или RSK). Эти данные позволяют предположить, что активность GSK3 может модулироваться митогенными, инсулиновыми и/или аминокислотными стимулирующими воздействиями.
Кроме того, было показано, что GSK3β является ключевым компонентом в Wnt сигнальном пути позвоночных. Было показано, что этот биохимический путь имеет решающее значение для нормального эмбрионального развития, а также регулирует пролиферацию клеток в нормальных тканях. GSK3 подвергается ингибированию в ответ на стимулирующее воздействие Wnt. Это может вести к дефосфорилированию субстратов GSK3, как, например, аксина, т.е. продукта гена аденоматозного полипоза толстой кишки (APC), и β-катенина. Аномальное регулирование пути Wnt связывалось со многими видами рака. Мутации в APC и/или β-катенине являются обычными при колоректальном раке и других опухолях. Кроме того, было показано, что β-катенин играет важную роль в клеточной адгезии. Таким образом, GSK3 также до некоторой степени может модулировать процессы клеточной адгезии. Помимо уже описанных биохимических путей, существуют данные, которые позволяют предположить участие GSK3 в регулировании деления клеток за счет фосфорилирования циклина-D1, в фосфорилировании факторов транскрипции, таких как c-Jun, CCAAT/энхансер-связывающий белок α (C/EBPα), c-Myc и/или других субстратов, таких как ядерный фактор активированных T-клеток (NFATc), фактор теплового шока-1 (HSF-1) и белок, связывающийся с c-AMP зависимым элементом (CREB). Кроме того, GSK3, по-видимому, играет определенную, хотя и тканеспецифичную, роль в регулировании клеточного апоптоза. Роль GSK3 в модулировании клеточного апоптоза через про-апоптический механизм может иметь особое значение для медицинских состояний, в которых может наблюдаться апоптоз нейронов. Примерами таких состояний являются травма головы, удар, эпилепсия, болезнь Альцгеймера и боковой амиотрофический склероз, прогрессирующий супрануклеарный паралич, кортико-базальная дегенерация и болезнь Пика. Было показано in vitro, что GSK3 способна гиперфосфорилировать микроканальцы, связанные с тау-белком. Гиперфосфорилирование тау-белка прекращает его нормальное связывание с микроканальцами и может также приводить к образованию внутриклеточных нитей тау-белка. Полагают, что последовательное накопление этих нитей ведет к возможной дисфункции и дегенерации нейронов. Ингибирование фосфорилирования тау-белка за счет ингибирования GSK3 может, таким образом, предоставить средства ограничения и/или предупреждения нейродегенеративных эффектов.
УРОВЕНЬ ТЕХНИКИ
В WO 02/34721, поданной Du Pont, раскрыт класс инден[1,2-c]пиразол-4-онов, как ингибиторов циклин-зависимых киназ.
В WO 01/81348, поданной Bristol Myers Squibb, раскрыто применение 5-тио-, сульфинил- и сульфонилпиразол[3,4-b]пиридинов, как ингибиторов циклин-зависимой киназы.
В WO 00/62778, также поданной Bristol Myers Squibb, раскрыт класс ингибиторов тирозиновой протеинкиназы.
В WO 01/72745A1, поданной Cyclacel, описаны 2-замещенные 4-гетероарилпиримидины и их получение, содержащие их фармацевтические композиции и их применение в качестве ингибиторов циклин-зависимых киназ (CDK) и, следовательно, их применение в лечении пролиферативных расстройств, таких как рак, лейкемия, псориаз и т.п.
В WO 99/21845, поданной Agouron, описаны производные 4-аминотиазола, предназначенные для ингибирования циклин-зависимых киназ (CDK), таких как CDK1, CDK2, CDK4 и CDK6. Данное изобретение также направлено на терапевтическое и профилактическое применение фармацевтических композиций, содержащих такие соединения, и на способы лечения злокачественных новообразований и других расстройств путем введения эффективных количеств указанных соединений.
В WO 01/53274, поданной Agouron, в качестве ингибиторов CDK киназы раскрыт класс соединений, которые могут включать амидзамещенное бензольное кольцо, связанное с N-содержащей гетероциклической группой.
В WO 01/98290 (Pharmacia & Upjohn) раскрыт класс производных 3-аминокарбонил-2-карбоксамидотиофена в качестве ингибиторов протеинкиназ. Установлено, что эти соединения обладают активностью в отношении большого числа протеинкиназ.
В WO 01/53268 и WO 01/02369, поданных Agouron, раскрыты соединения, которые опосредуют или ингибируют пролиферацию клеток путем ингибирования протеинкиназ, как, например, циклин-зависимых киназ или тирозиновых киназ.
В WO 00/39108 и WO 02/00651 (обе поданы Du Pont Pharmaceuticals) описаны обширные классы гетероциклических соединений, которые являются ингибиторами трипсиноподобных ферментов сериновых протеаз, в особенности фактора Xa и тромбина. Установлено, что эти соединения применимы в качестве антикоагулянтов или для профилактики тромбоэмболических расстройств.
В каждой из заявок US 2002/0091116 (Zhu et al.), WO 01/1978 и WO 01/64642 раскрыты различные группы гетероциклических соединений, которые обладают активностью против фактора Xa.
В WO 03/035065 (Aventis) раскрыт обширный класс производных бензимидазола в качестве ингибиторов протеинкиназы, но не раскрыта активность против CDK киназ или GSK киназ.
В WO 97/36585 и US 5874452 (заявка и патент принадлежат Merck) раскрыты бигетероарильные соединения, которые являются ингибиторами фарнезил трансферазы.
В WO 03/066629 (Vertex) раскрыты бензимидазолилпиразоламины как ингибиторы GSK-3.
В WO 97/12615 (Warner Lambert) раскрыты бензимидазолы как ингибиторы 15-липоксигеназы.
В WO 2004/54515 (SmithKline Beecham Corporation) раскрыт класс бензимидазолов в качестве миметиков тромбопоэтина.
В WO 2004/41277 (Merck) раскрыт класс аминобензимидазолов как модуляторов рецептора андрогенов.
В WO 2005/028624 (Plexxikon) раскрыты каркасы молекул соединений, обладающих активностью против протеинкиназ.
В поданной ранее авторами настоящего изобретения международной заявке на патент PCT/GB2004/002824 (WO 2005/002552) раскрыт класс замещенных [(1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевин в качестве ингибиторов CDK, Aurora киназ и GSK киназ. Одним из соединений, конкретно названных и приведенных в качестве примера в WO 2005/002552, является 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина. В экспериментальном разделе WO 2005/002552 описано получение 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем изобретении разработаны соединения, которые обладают активностью в отношении ингибирования или модулирования циклин-зависимых киназ, активностью в отношении ингибирования или модулирования киназы-3 гликоген-синтазы (GSK3), а также активностью в отношении ингибирования или модулирования Aurora киназы, и которые, как представляется, будут применимы в предупреждении или лечении заболеваний или состояний, опосредованных перечисленными киназами.
Так, например, предусматривается, что соединения по настоящему изобретению будут применимы для облегчения симптомов или сокращения числа случаев раковых заболеваний.
В первом аспекте изобретение относится к соединению формулы (I):
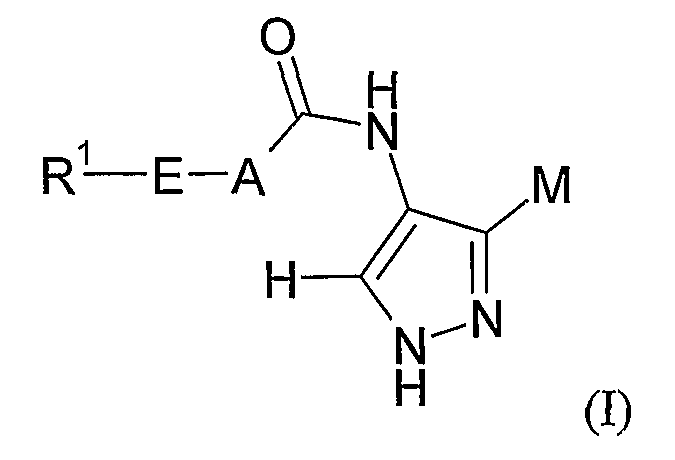
или его соли, сольвату, таутомеру или N-оксиду,
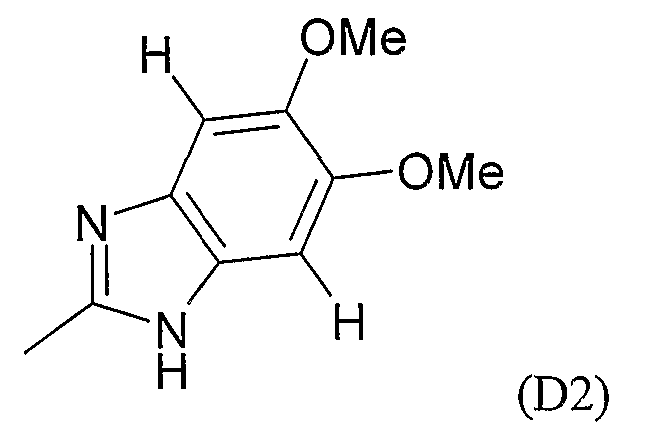
где M выбран из группы D1 и группы D2:
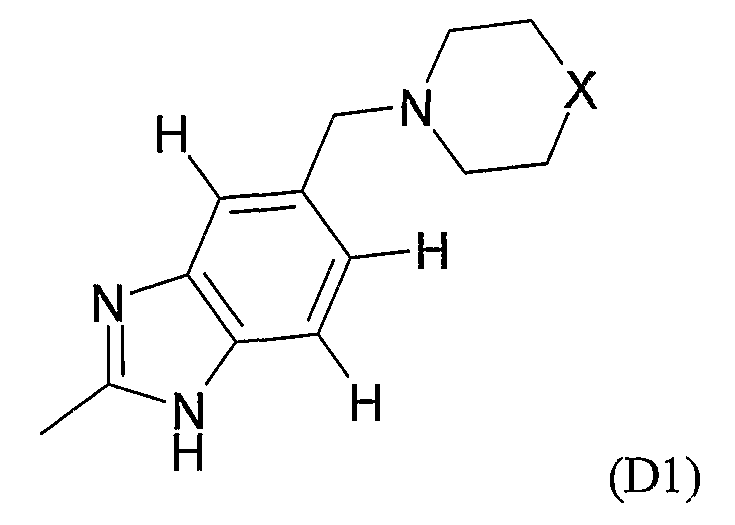
и где (A) если M представляет собой группу D1:
X выбран из O, NH и NCH3;
A выбран из связи и группы NR2, где R2 представляет собой водород или метил;
E выбран из связи CH2, CH(CN) и C(CH3)2;
R1 выбран из:
(i) 3-5-членной циклоалкильной группы, необязательно замещенной гидрокси, фтором, амино, метиламино, метилом или этилом;
(ii) 4-6-членной насыщенной гетероциклической группы, включающей 1 или 2 гетероатома, выбранных из O, N, S и SO2, причем гетероциклическая группа необязательно замещена C1-4алкилом, амино или гидрокси; но исключая незамещенный 4-морфолинил, незамещенный тетрагидропиран-4-ил, незамещенный 2-пирролидинил, а также незамещенный и 1-замещенный пиперидин-4-ил;
(iii) 2,5-замещенной фенильной группы формулы:
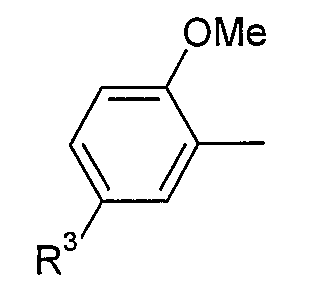
в которой: (a) если X представляет собой NH или N-CH3, R3 выбран из хлора и циано; и (b) если X представляет собой O, R3 представляет собой CN;
(iv) группы CR6R7R8, в которой каждый из R6 и R7 выбран из водорода и метила, и R8 выбран из водорода, метила, C1-4алкилсульфонилметила, гидроксиметила и циано;
(v) пиридазин-4-ильной группы, необязательно замещенной одним или двумя заместителями, выбранными из метила, этила, метокси и этокси;
(vi) замещенной имидазотиазольной группы, в которой заместители выбраны из метила, этила, амино, фтора, хлора, амино и метиламино; и
(vii) необязательно замещенных 1,3-дигидроизоиндол-2-ильной или 2,3-дигидроиндол-1-ильной групп, в которых необязательно присутствующие заместители в каждом случае выбраны из галогена, циано, амино, C1-4моно- и диалкиламино, CONH2 или CONH-C1-4алкила, C1-4алкила и C1-4алкокси, где C1-4алкильная и C1-4алкоксигруппы необязательно замещены гидрокси, метокси или амино;
(viii) 3-пиридила, необязательно замещенного одним или двумя заместителями, выбранными из гидрокси, галогена, циано, амино, C1-4моно- и диалкиламино, CONH2 или CONH-C1-4алкила, C1-4алкила и C1-4алкокси, где C1-4алкильная и C1-4алкоксигруппы необязательно замещены гидрокси, метокси или амино; но исключая такие соединения, как [3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амид 2-оксо-1,2-дигидропиридин-3-карбоновой кислоты и 2,6-диметокси-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]никотинамид;
(ix) тиоморфолина или его S-оксида, или S,S-диоксида, необязательно замещенного одним или двумя заместителями, выбранными из галогена, циано, амино, C1-4моно- и диалкиламино, CONH2 или CONH-C1-4алкила, C1-4алкила и C1-4алкокси, где C1-4алкильная и C1-4алкоксигруппы необязательно замещены гидрокси, метокси или амино; и
если E-A представляет собой NR2, R1 дополнительно выбран из:
(x) 2-фторфенила, 3-фторфенила, 4-фторфенила, 2,4-дифторфенила, 3,4-дифторфенила, 2,5-дифторфенила, 3,5-дифторфенила, 2,4,6-трифторфенила, 2-метоксифенила, 5-хлор-2-метоксифенила, циклогексила, незамещенного 4-тетрагидропиранила и трет-бутила;
(xi) группы NR10R11, где каждый из R10 и R11 представляет собой C1-4алкил, или же R10 и R11 соединены таким образом, что NR10R11 образует 4-6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом в цикле, выбранный из O, N, S и SO2, причем гетероциклическая группа необязательно замещена C1-4алкилом, амино или гидрокси;
(xii) пиридона, необязательно замещенного одним или двумя заместителями, выбранными из гидрокси, галогена, циано, амино, C1-4моно- и диалкиламино, CONH2 или CONH-C1-4алкила, C1-4алкила и C1-4алкокси, где C1-4алкильная и C1-4алкоксигруппы необязательно замещены гидрокси, метокси или амино;
если E-A представляет собой C(CH3)2NR2 или CH2-NR2, R1 дополнительно выбран из:
(xiii) незамещенного 2-фурила и 2,6-дифторфенила; и
если E-A представляет собой C(CH3)2NR2, R1 дополнительно выбран из:
(xiv) незамещенного фенила; и
если E представляет собой CH2, R1 дополнительно выбран из:
(xv) незамещенного тетрагидропиран-4-ила; и
(B) если M представляет собой группу D2:
A выбран из связи и группы NR2, где R2 представляет собой водород или метил;
E выбран из связи, CH2, CH(CN) и C(CH3)2;
R1 выбран из:
(xvi) 2-замещенной 3-фурильной группы формулы:
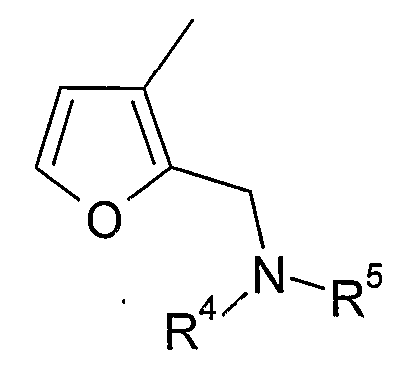
в которой R4 и R5 являются одинаковыми или различными и выбраны из водорода и C1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом, или группу, выбранную из O, NH, NMe, S или SO2, причем 5- или 6-членный насыщенный цикл необязательно замещен гидрокси, фтором, амино, метиламино, метилом или этилом;
(xvii) 5-замещенной 2-фурильной группы формулы:
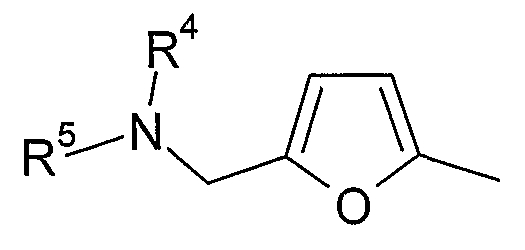
в которой R4 и R5 являются одинаковыми или различными и выбраны из водорода и C1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом, или группу, выбранную из O, NH, NMe, S или SO2, причем 5- или 6-членная насыщенная гетероциклическая группа необязательно замещена гидрокси, фтором, амино, метиламино, метилом или этилом; при условии, что соединение не является [3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амидом 5-пиперидин-1-илметилфуран-2-карбоновой кислоты;
(xviii) группы формулы:
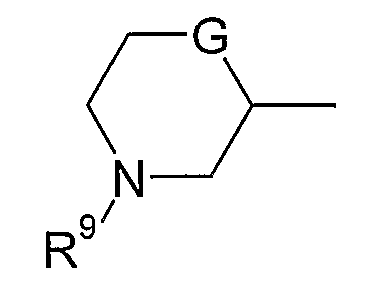
в которой R9 представляет собой водород, метил, этил или изопропил; G представляет собой CH, O, S, SO, SO2 или NH, и указанная группа необязательно замещена одним, двумя или тремя заместителями, выбранными из C1-4гидрокарбила, гидрокси, C1-4гидрокарбилокси, фтора, амино, моно- и ди-C1-4алкиламино, и где каждая из C1-4гидрокарбильных и C1-4гидрокарбилоксигрупп необязательно замещена гидрокси, фтором, амино, моно- или ди-C1-4алкиламино; и
(xix) 3,5-дизамещенной фенильной группы формулы:
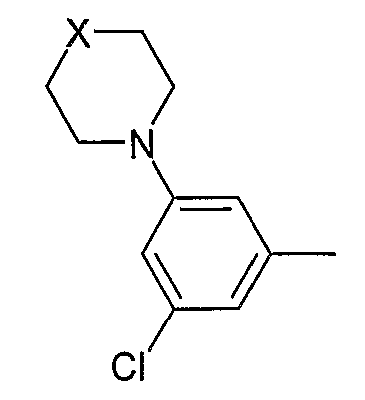
в которой X выбран из O, NH и NCH3;
(C) если M представляет собой группу D1:
и X представляет собой O; A представляет собой группу NR2, где R2 является водородом; E представляет собой связь; и R1 представляет собой 2,6-дифторфенил; тогда соединение формулы (I) является кислотно-аддитивной солью, выбранной из солей, образованных кислотой, которая выбрана из группы, состоящей из уксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), аспарагиновой (например, L-аспарагиновой), бензолсульфоновой, бензойной, камфорной (например, (+)камфорной), каприновой, каприловой, угольной, лимонной, цикламовой, додекановой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, хлористоводородной, изетионовой, изомасляной, молочной (например, (+)-L-молочной и (±)-DL-молочной), лактобионовой, лаурилсульфоновой, малеиновой, яблочной, (-)-L-яблочной, малоновой, метансульфоновой, слизевой, нафталинсульфоновой (например, нафталин-2-сульфоновой), нафталин-1,5-дисульфоновой, никотиновой, олеиновой, оротовой, щавелевой, пальмитиновой, памовой, фосфорной, пропионовой, себациновой, стеариновой, янтарной, серной, винной (например, (+)-L-винной), тиоциановой, толуолсульфоновой (например, п-толуолсульфоновой), валериановой и ксинафовой кислот.
В одном из вариантов осуществления группа M представляет собой группу D1 или D2, определенную в описании подгрупп (A) и (B) приведенной выше формулы (I).
В другом варианте осуществления группа M представляет собой группу D1 и соединение формулы (I) представляет собой кислотно-аддитивную соль 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, как определено в описании подгруппы (С) приведенной выше формулы (I).
В отдельном варианте осуществления изобретение относится к соли свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и, конкретно, к лактату указанного соединения.
Кроме того, изобретение относится к новым применениям 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и ее солей (например, кислотно-аддитивных солей), сольватов, таутомеров или N-оксидов.
В числе прочего изобретение также относится к:
- Применению соединений формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, для изготовления лекарственного средства для профилактики или лечения заболевания или состояния, опосредованного циклин-зависимой киназой или киназой-3 гликоген-синтазы.
- Способу профилактики или лечения заболевания или состояния, опосредованного циклин-зависимой киназой или киназой-3 гликоген-синтазы, причем указанный способ включает введение субъекту в случае необходимости соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке.
- Способу облегчения симптомов или уменьшения частоты появления заболевания или состояния, опосредованных циклин-зависимой киназой или киназой-3 гликоген-синтазы, причем указанный способ включает введение субъекту в случае необходимости соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке.
- Способу лечения заболевания или состояния, включающего или возникающего из-за аномального роста клеток у млекопитающего, причем указанный способ включает введение млекопитающему соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, в количестве, эффективном для ингибирования аномального роста клеток.
- Способу облегчения симптомов или уменьшения частоты появления заболеваний или состояний, включающих или возникающих из-за аномального роста клеток у млекопитающего, причем указанный способ включает введение млекопитающему соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, в количестве, эффективном для ингибирования аномального роста клеток.
- Способу лечения заболевания или состояния, включающего или возникающего из-за аномального роста клеток у млекопитающего, причем указанный способ включает введение млекопитающему соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, в количестве, эффективном для ингибирования активности CDK киназы (как, например, CDK1 или CDK2) или киназы-3 гликоген-синтазы.
- Способу облегчения симптомов или уменьшения частоты появления заболеваний или состояний, включающих или возникающих из-за аномального роста клеток у млекопитающего, причем указанный способ включает введение млекопитающему соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, в количестве, эффективном для ингибирования активности CDK киназы (как, например, CDK1 или CDK2) или киназы-3 гликоген-синтазы.
- Способу ингибирования циклин-зависимой киназы или киназы-3 гликоген-синтазы, причем способ включает приведение во взаимодействие указанной киназы с ингибирующим киназу соединением формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке.
- Способу модулирования клеточных процессов (например, деления клеток) за счет ингибирования активности циклин-зависимой киназы или киназы-3 гликоген-синтазы с применением соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке.
- Применению соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, для изготовления лекарственного средства для профилактики или лечения заболеваний или состояний, характеризующихся повышающей регуляцией Aurora киназы (например, Aurora A киназы и/или Aurora B киназы).
- Применению соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, для изготовления лекарственного средства для профилактики или лечения ракового заболевания, причем раковое заболевание является таким раковым заболеванием, которое характеризуется повышающей регуляцией Aurora киназы (например, Aurora A киназы и/или Aurora B киназы).
- Применению соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, для изготовления лекарственного средства для профилактики или лечения ракового заболевания у пациента, выбранного из подгруппы, обладающей вариантом Ile31 гена Aurora A.
- Применению соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, для изготовления лекарственного средства для профилактики или лечения ракового заболевания у пациента, который, по результатам диагностики, является членом подгруппы, обладающей вариантом Ile31 гена Aurora A.
- Способу профилактики или лечения заболевания или состояния, характеризующегося повышающей регуляцией Aurora киназы (например, Aurora A киназы и/или Aurora B киназы), причем способ включает введение соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке.
- Способу облегчения симптомов или уменьшения частоты появления заболеваний или состояний, характеризующихся повышающей регуляцией Aurora киназы (например, Aurora A киназы и/или Aurora B киназы), причем способ включает введение соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке.
- Способу профилактики или лечения (или облегчения симптомов или уменьшения частоты появления) раковых заболеваний у пациентов, страдающих от рака или у которых предполагают наличие рака; причем указанный способ включает (i) проведение диагностического тестирования пациента для определения, обладает ли пациент вариантом Ile31 гена Aurora A; и (ii) если пациент действительно обладает указанным вариантом, введение пациенту после этого соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, имеющих ингибирующую активность в отношении Aurora киназы.
- Способу профилактики или лечения (или облегчения симптомов или уменьшения частоты появления) заболеваний или состояний, характеризующихся повышающей регуляцией Aurora киназы (например, Aurora A киназы и/или Aurora B киназы); причем указанный способ включает (i) проведение диагностического тестирования пациента для обнаружения маркеров, характерных для повышающей регуляции Aurora киназы; и (ii) если диагностический тест указывает на повышающую регуляцию Aurora киназы, введение пациенту после этого соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, имеющих ингибирующую активность в отношении Aurora киназы.
- Способу профилактики или лечения (или облегчения симптомов или уменьшения частоты появления) заболеваний или состояний, характеризующихся (a) избыточной активацией CDK киназы; и/или (b) увеличением чувствительности сигнального пути к нормальной активности CDK; и/или (c) повышающей регуляцией циклина E; причем указанный способ включает (i) проведение диагностического тестирования пациента для обнаружения маркеров, характерных для (a) и/или (b) и/или (c); и (ii) если диагностический тест указывает на (a) и/или (b) и/или (c), введение пациенту после этого соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, имеющих ингибирующую активность в отношении CDK киназы.
- Способу лечения, применению в медицине или соединению для применения, где соединение формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельные примеры соединений указанных формул, определенных в настоящей заявке, вводят (например, в терапевтически эффективных количествах) подгруппе пациентов, которые с помощью одного или нескольких диагностических тестов, описанных в настоящей заявке, идентифицированы как имеющие заболевание или состояние, которое должно поддаваться лечению указанным соединением.
- Применению соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, для изготовления лекарственного препарата для профилактики или лечения болезненного состояния, описанного в настоящей заявке.
- Соединению формул (I), (II), (III) или (XXX) или любой подгруппе, или отдельным примерам соединений указанных формул, определенных в настоящей заявке, для применения в профилактике или лечении болезненных состояний, описанных в настоящей заявке.
- Способу профилактики или лечения (или облегчения симптомов или уменьшения частоты появления) заболеваний или состояний, описанных в настоящей заявке, причем способ включает введение млекопитающему терапевтически эффективного количества соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке.
- Фармацевтической композиции, включающей соединение формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельные примеры соединений указанных формул, определенных в настоящей заявке, а также фармацевтически приемлемый носитель.
- Фармацевтической композиции, предназначенной для введения в форме водного раствора, причем фармацевтическая композиция включает соединение формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельные примеры соединений указанных формул, определенных в настоящей заявке, в форме соли, имеющей растворимость в воде более 1 мг/мл, типично, более 5 мг/мл, более типично, более 15 мг/мл, более типично, более 20 мг/мл и предпочтительно более 25 мг/мл.
- Соединению формул (I), (II), (III) или (XXX) или любой подгруппе, или отдельным примерам соединений указанных формул, определенных в настоящей заявке, для применения в медицине.
- Соединению, определенному в настоящей заявке, предназначенному для любого применения и способа, из числа изложенных выше, и как описано еще где-либо в настоящей заявке.
- Соединению формул (I), (II), (III) или (XXX) или любой подгруппе, или отдельным примерам соединений указанных формул, определенных в настоящей заявке, или их солям (например, кислотно-аддитивным солям), сольватам, таутомерам или N-оксидам для применения в лечении B-клеточной лимфомы.
- Соединению формул (I), (II), (III) или (XXX) или любой подгруппе, или отдельным примерам соединений указанных формул, определенных в настоящей заявке, или их солям (например, кислотно-аддитивным солям), сольватам, таутомерам или N-оксидам для применения в лечении хронического лимфоцитарного лейкоза.
- Соединению формул (I), (II), (III) или (XXX) или любой подгруппе, или отдельным примерам соединений указанных формул, определенных в настоящей заявке, или их солям (например, кислотно-аддитивным солям), сольватам, таутомерам или N-оксидам для применения в лечении диффузной B-крупноклеточной лимфомы.
- Способу лечения B-клеточной лимфомы, диффузной B-крупноклеточной лимфомы или хронического лимфоцитарного лейкоза с помощью введения пациенту в случае необходимости такого лечения соединения формул (I), (II), (III) или (XXX) или любой подгруппы, или отдельных примеров соединений указанных формул, определенных в настоящей заявке, или их солей (например, кислотно-аддитивных солей), сольватов, таутомеров или N-оксидов.
- Соединению формул (I), (II), (III) или (XXX) или любой подгруппе, или отдельным примерам соединений указанных формул, определенных в настоящей заявке, или их солям (например, кислотно-аддитивным солям), сольватам, таутомерам или N-оксидам для применения в лечении лейкемии, в частности повторной или рефрактерной острой миелогенной лейкемии, миелодиспластического синдрома, острого лимфоцитарного лейкоза и хронической миелогенной лейкемии.
- Кислотно-аддитивной соли свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и, конкретно, лактату, предназначенному для любых применений и способов, изложенных выше, а также описанных еще где-либо в настоящей заявке.
Предпочтительные варианты и определения
К каждому из фрагментов D1, D2, A, E, X, Xa и R1-R9, а также к различным их подгруппам, к определениям внутри подгрупп, примерам и вариантам осуществления будут применяться следующие общие предпочтения и определения, если контекст не будет указывать на иное.
Все ссылки на формулу (I) в настоящей заявке будут также относиться к формулам (II)-(VIII) и любой другой подгруппе соединений, входящих в число соединений формулы (I), если контекст не требует другого понимания.
Термин «повышающая регуляция Aurora киназы» в настоящем описании определяется, как включающий повышенную экспрессию или избыточную экспрессию Aurora киназы, включая амплификацию гена (т.е. умножение копий гена) и увеличение экспрессии под действием транскрипционного эффекта, а также гиперактивность и активацию Aurora киназы, в т.ч. активацию под влиянием мутаций.
Термин «насыщенный» в настоящем описании относится к циклам, в которых отсутствуют кратные связи между циклическими атомами.
Термин «гидрокарбил» в настоящем описании, независимо от того, использован ли он отдельно или в виде части сложного термина, как, например, «гидрокарбилокси», является общим термином, охватывающим алифатические и алициклические группы, скелет которых полностью состоит из углерода. Примеры гидрокарбильных групп включают алкил, циклоалкил, циклоалкенил, алкенил, алкинил, циклоалкилалкил, циклоалкенилалкил. Отдельными гидрокарбильными группами являются насыщенные группы, такие как алкильные и циклоалкильные группы.
Примеры гидрокарбилокси групп включают алкокси, циклоалкокси, циклоалкенокси, алкенилокси, алкинилокси, циклоалкилалкилокси, циклоалкенилалкилокси. Отдельными гидрокарбилокси группами являются насыщенные группы, такие как алкокси.
Префикс “C1-n” (где n означает целое число) в настоящем описании относится к числу атомов углерода в данной группе. Так, C1-4гидрокарбильная группа содержит от 1 до 4 атомов углерода, тогда как C1-3гидрокарбильная группа содержит от 1 до 3 атомов углерода и т.д.
Примеры C1-4гидрокарбильных групп включают C1-3гидрокарбильные группы или C1-2гидрокарбильные группы, причем конкретными примерами являются любые индивидуальные группы или комбинации групп, выбранные из гидрокарбильных групп C1, C2, C3 и C4.
Термин «алкил» охватывает как алкильные группы с линейной цепью, так и разветвленные алкильные группы. Примерами алкильных групп являются метил, этил, пропил, изопропил, н-бутил, изобутил и трет-бутил.
Примерами циклоалкильных групп служат группы, являющиеся производными циклопропана, циклобутана и циклопентана.
Примерами алкенильных групп являются этенил (винил), 1-пропенил, 2-пропенил (аллил), изопропенил, бутенил и бута-1,4-диенил.
Примерами циклоалкенильных групп служат циклопропенил и циклобутенил.
Примерами алкинильных групп являются этинильная и 2-пропинильная (пропаргильная) группы.
Примеры циклоалкилалкильных и циклоалкенилалкильных групп включают циклопропилметил.
Примерами алкоксигрупп являются метокси, этокси, н-пропилокси, изопропилокси, н-бутокси, изобутокси и трет-бутокси.
Если алкильная группа является составной частью моноалкиламино или диалкиламино групп, алкильная группа может являться любой из алкильных групп, приведенных выше в качестве примеров. Конкретными алкиламино и диалкиламино группами являются метиламино, диметиламино, этиламино, диэтиламино, н-пропиламино, изопропиламино, бутиламино, изобутиламино и трет-бутиламино. Особыми алкил- и диалкиламино группами являются метиламино и диметиламино.
Термин «насыщенная гетероциклическая группа» в настоящем описании относится к гетероциклической группе, не содержащей кратных связей между соседними атомами в цикле. Насыщенные гетероциклические группы могут включать 1 или 2 циклических гетероатома, выбранных из O, S и N.
В зависимости от контекста гетероциклические группы могут содержать, например, циклические фрагменты простого эфира (например, как в тетрагидрофуране или диоксане), циклические фрагменты тиоэфира (например, как в тетрагидротиофене и дитиане), циклические фрагменты амина (например, как в пирролидине), циклические амидные фрагменты (например, как в пирролидоне), циклические тиоамиды, циклические сложные тиоэфиры, циклические мочевины (например, как в имидазолидин-2-оне), циклические фрагменты сложного эфира (например, как в бутиролактоне), циклические сульфоны (например, как в сульфолане и сульфолене), циклические сульфоксиды, циклические сульфонамиды и комбинации перечисленных фрагментов (например, тиоморфолин).
Насыщенные гетероциклические группы, как правило, являются моноциклическими и обычно содержат 4, 5 или 6 атомов в цикле, если не указано иное.
Конкретным примером насыщенной гетероциклической группы, содержащей 4 атома в цикле, является азетидиновая группа.
Примеры насыщенных гетероциклических групп, содержащих 5 атомов в цикле, включают пирролидин (например, 1-пирролидинил, 2-пирролидинил и 3-пирролидинил), пирролидон, тетрагидрофуран и тетрагидротиофен.
Примеры насыщенных гетероциклических групп, содержащих 6 атомов в цикле, включают морфолин, тиоморфолин, S-оксид тиоморфолина, S,S-диоксид тиоморфолина, пиперидин (например, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и 4-пиперидинил), пиперидон, диоксан, тетрагидропиран (например, 4-тетрагидропиранил), пиперазон, пиперазин, а также N-алкилпиперазины, как, например, N-метилпиперазин.
Конкретные варианты осуществления и предпочтения для D1, D2, A, E, R 1 -R 9 и X в подгруппах (A) и (B) формулы (I)
В одном из общих вариантов осуществления M представляет собой группу D1.
В другом общем варианте осуществления M представляет собой группу D2.
X выбран из O, NH и NCH3. В одном конкретном варианте осуществления X представляет собой O.
A выбран из связи и группы NR2, где R2 является водородом или метилом.
В одном из вариантов осуществления A представляет собой связь.
В другом варианте осуществления A представляет собой группу NR2, где R2 является водородом или метилом.
E выбран из связи, CH2, CH(CN) и C(CH3)2.
В одной подгруппе соединений E представляет собой связь.
В другой подгруппе соединений E представляет собой CH2.
В еще одной подгруппе соединений E представляет собой CH(CN).
В другой подгруппе соединений E представляет собой C(CH3)2.
Если M представляет собой группу D1, R1 может быть выбран из групп (i), (ii), (iii), (iv), (v), (vi), (vii), (viii), (ix), (x), (xi) и (xii).
Если M представляет собой группу D1, и E-A представляет собой C(CH3)2NR2 или CH2-NR2, то R1 может быть дополнительно выбран из:
(xiii) незамещенного 2-фурила и 2,6-дифторфенила.
Если M представляет собой группу D1, и E-A представляет собой C(CH3)2NR2, R1 может быть дополнительно выбран из:
(xiv) незамещенного фенила.
Если M представляет собой группу D1, и E представляет собой CH2, R1 может быть дополнительно выбран из:
(xv) незамещенного тетрагидропиран-4-ила.
Каждая отдельная группа в перечне групп (i)-(xv) представляет собой отдельный вариант осуществления настоящего изобретения.
В варианте осуществления (i) R1 является 3-5-членной циклоалкильной группой, необязательно замещенной гидрокси, фтором, амино, метиламино, метилом или этилом.
Отдельные циклоалкильные группы необязательно замещены циклопропильной и циклобутильной группами, более типично необязательно замещены циклопропильными группами. В предпочтительном варианте осуществления R1 является незамещенной циклопропильной группой.
В варианте осуществления (ii) R1 является 4-6-членной насыщенной гетероциклической группой, включающей 1 или 2 гетероатома в цикле, выбранных из O, N, S и SO2, причем гетероциклическая группа необязательно замещена C1-4алкилом, амино или гидрокси; но из числа этих групп исключаются незамещенный 4-морфолинил, незамещенный тетрагидропиран-4-ил, незамещенный 2-пирролидинил, а также незамещенный и 1-замещенный пиперидин-4-ил.
Примеры насыщенных гетероциклических групп приведены выше в разделе «Общие предпочтения и определения».
Отдельные примеры насыщенных гетероциклических групп включают:
- пятичленные циклы, содержащие единственный входящий в цикл гетероатом, выбранный из O, N и S (отличные от незамещенного 2-пирролидинила);
- шестичленные циклы, содержащие два входящих в цикл гетероатома, выбранных из O, N и S (отличные от незамещенного 4-морфолинила).
Насыщенные гетероциклические группы могут быть замещенными или незамещенными. В одном из вариантов осуществления они являются незамещенными. В другом варианте осуществления они замещены одной или двумя C1-4алкильными группами, например одной или двумя метильными группами.
Одной из конкретных насыщенных гетероциклических групп является необязательно замещенная группа тетрагидрофурана (например, тетрагидрофуран-2-ил или тетрагидрофуран-3-ил), более предпочтительно незамещенная группа тетрагидрофурана.
В варианте осуществления (iii) R1 представляет собой 2,5-замещенную фенильную группу формулы:
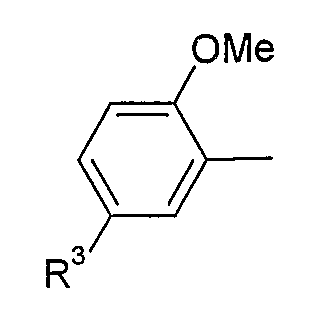
в которой (a) если X представляет собой NH или N-CH3, R3 выбран из хлора и циано; и (b) если X представляет собой O, R3 представляет собой CN.
В одной из подгрупп соединений, входящих в вариант осуществления (iii), X представляет собой N-CH3 и R3 выбран из хлора и циано.
В другой подгруппе соединений, входящих в вариант осуществления (iii), X представляет собой O и R3 представляет собой CN.
В варианте осуществления (iv) R1 является группой CR6R7R8, в которой каждый из R6 и R7 выбран из водорода и метила, а R8 выбран из водорода, метила, C1-4алкилсульфонилметила, гидроксиметила и циано.
В варианте осуществления (iv) конкретными примерами R1 являются метил, цианометил, HOCH2C(CH3)2- и 2-метилсульфонилэтил.
В варианте осуществления (iv) другими конкретными примерами R1 являются метил и изопропил.
В варианте осуществления (v) R1 представляет собой пиридазин-4-ильную группу, необязательно замещенную одним или двумя заместителями, выбранными из метила, этила, метокси и этокси. Пиридазинильная группа может быть пиридазин-3-ильной или пиридазин-4-ильной группой, но, как правило, является пиридазин-4-илом. Конкретными заместителями являются метоксигруппы, и, например, пиридазинильная группа может нести два метокси заместителя.
В варианте осуществления (vi) R1 представляет собой замещенную имидазотиазольную группу, в которой заместители выбраны из метила, этила, амино, фтора, хлора, амино и метиламино. Конкретным заместителем является метил.
В варианте осуществления (vii) R1 представляет собой необязательно замещенную 1,3-дигидроизоиндол-2-ильную или 2,3-дигидроиндол-1-ильную группу, в которых необязательно присутствующие заместители в каждом случае выбраны из галогена, циано, амино, C1-4моно- и диалкиламино, CONH2 или CONH-C1-4алкила, C1-4алкила и C1-4алкокси, где C1-4алкильная и C1-4алкоксигруппы необязательно замещены гидрокси, метокси или амино.
Конкретные заместители выбраны из метила, этила, фтора, хлора (предпочтительно только на арильном цикле дигидроиндола или дигидроизоиндола), CONH2, амино, метиламино, диметиламино и метокси.
В одной подгруппе соединений в варианте осуществления (vii) каждый из фрагментов дигидроизоиндола или дигидроиндола является незамещенным.
В варианте осуществления (viii) R1 представляет собой 3-пиридил, необязательно замещенный одним или двумя заместителями, выбранными из гидрокси, галогена, циано, амино, C1-4моно- и диалкиламино, CONH2 или CONH-C1-4алкила, C1-4алкила и C1-4алкокси, где C1-4алкильная и C1-4алкоксигруппы необязательно замещены гидрокси, метокси или амино; при условии, что не образуются такое соединение, как 2,6-диметокси-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]никотинамид или такое соединение, как [3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амид 2-оксо-1,2-дигидропиридин-3-карбоновой кислоты.
В одном из вариантов осуществления R1 представляет собой 3-пиридил, необязательно замещенный одним или двумя заместителями, выбранными из гидрокси, галогена, циано, амино, C1-4моно- и диалкиламино, CONH2 или CONH-C1-4алкила, C1-4алкила и C1-4алкокси, где C1-4алкильная и C1-4алкоксигруппы необязательно замещены гидрокси, метокси или амино, но если R1 представляет собой 3-пиридил, X означает O, A является связью и E является связью, пиридил имеет один или два заместителя, выбранных из галогена, циано, амино, C1-4моно- и диалкиламино, CONH2 или CONH-C1-4алкила, C1-4алкила и C1-4алкокси, где C1-4алкильная и C1-4алкоксигруппы необязательно замещены гидрокси, метокси или амино.
Конкретные заместители выбраны из метила, этила, фтора, хлора, CONH2, амино, метиламино, диметиламино и метокси. Более конкретно заместители выбраны из метила, этила, фтора, хлора, CONH2, амино, метиламино и диметиламино.
В одной подгруппе соединений 3-пиридильная группа является незамещенной.
В варианте осуществления (ix) R1 представляет собой тиоморфолин или его S-оксид, или S,S-диоксид, необязательно замещенный одним или двумя заместителями, выбранными из галогена, циано, амино, C1-4моно- и диалкиламино, CONH2 или CONH-C1-4алкила, C1-4алкила и C1-4алкокси, где C1-4алкильная и C1-4алкоксигруппы необязательно замещены гидрокси, метокси или амино.
В одной из подгрупп соединений тиоморфолин или его S-оксид, или S,S-диоксид являются незамещенными.
В варианте осуществления (x) E-A представляет собой NR2, и R1 выбран из: 2-фторфенила, 3-фторфенила, 4-фторфенила, 2,4-дифторфенила, 3,4-дифторфенила, 2,5-дифторфенила, 3,5-дифторфенила, 2,4,6-трифторфенила, 2-метоксифенила, 5-хлор-2-метоксифенила, циклогексила, незамещенного 4-тетрагидропиранила и трет-бутила;
В варианте осуществления (xi) E-A представляет собой NR2, и R1 представляет собой группу NR10R11, где каждый из R10 и R11 представляет собой C1-4алкил, или же R10 и R11 соединены таким образом, что NR10R11 образует 4-6-членную насыщенную гетероциклическую группу, необязательно содержащую в цикле второй гетероатом, выбранный из O, N, S и SO2, причем гетероциклическая группа необязательно замещена C1-4алкилом, амино или гидрокси.
В этом варианте осуществления одной из подгрупп соединений является группа соединений, в которых каждый из R10 и R11 представляет собой C1-4алкил, в частности метил.
Другой подгруппой соединений является группа соединений, в которой R10 и R11 соединены таким образом, что NR10R11 образует 4-6-членную насыщенную гетероциклическую группу, необязательно содержащую в цикле второй гетероатом, выбранный из O, N, S и SO2, причем гетероциклическая группа необязательно замещена C1-4алкилом, амино или гидрокси. Насыщенная гетероциклическая группа может быть любой из азотсодержащих насыщенных гетероциклических групп, перечисленных выше в разделе «предпочтительные варианты и определения», но конкретные насыщенные гетероциклические группы включают пирролидинил, морфолинил, пиперазинил и N-C1-4алкилпиперазинил. Эти группы, как правило, являются незамещенными или замещены одной или двумя метильными группами и, в одном конкретном варианте осуществления, являются незамещенными.
В варианте осуществления (xii) E-A представляет собой NR2, и R1 представляет собой пиридоновую группу, необязательно замещенную одним или двумя заместителями, выбранными из гидрокси, галогена, циано, амино, C1-4моно- и диалкиламино, CONH2, CONH-C1-4алкила, C1-4алкила и C1-4алкокси, где C1-4алкильная и C1-4алкоксигруппы необязательно замещены гидрокси, метокси или амино.
Пиридоновая группа может быть N-замещенной, например алкильной группой, такой как метил, и в другом случае может быть незамещенной.
В варианте осуществления (xiii) E-A представляет собой C(CH3)2NR2 или CH2-NR2 и R1 выбран из незамещенного 2-фурила и 2,6-дифторфенила.
В варианте осуществления (xiv) E-A представляет собой C(CH3)2NR2 и R1 является незамещенным фенилом.
В варианте осуществления (xv) E представляет собой CH2 и R1 является незамещенным тетрагидропиран-4-илом.
Если M представляет собой группу D2, R1 может быть выбран из групп (xvi), (xvii), (xviii) и (xix).
Каждая индивидуальная группа в перечне групп (xvi)-(xix) представляет собой отдельный вариант осуществления изобретения.
В варианте осуществления (xvi) R1 представляет собой 2-замещенную 3-фурильную группу формулы:
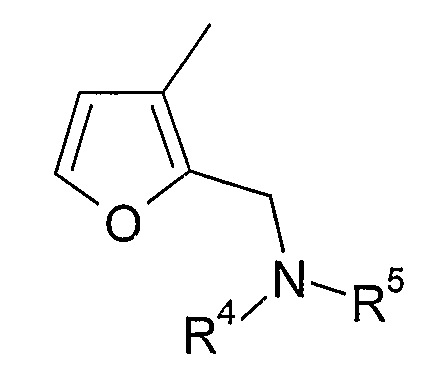
в которой R4 и R5 являются одинаковыми или различными и выбраны из водорода и C1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом или группу, выбранную из O, NH, NMe, S или SO2, причем 5- или 6-членный насыщенный цикл необязательно замещен гидрокси, фтором, амино, метиламино, метилом или этилом. В одном из вариантов осуществления R1 представляет собой 2-замещенную 3-фурильную группу формулы:
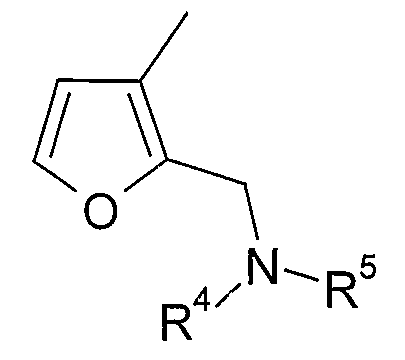
в которой R4 и R5 являются одинаковыми или различными и выбраны из водорода и C1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом или группу, выбранную из O, NH, NMe, S или SO2, причем 5- или 6-членный насыщенный цикл необязательно замещен гидрокси, фтором, амино, метиламино, метилом или этилом, но где A является связью и E является связью, R4 и R5 связаны таким образом, что NR4R5 образует незамещенный пиперидин.
Примеры насыщенных гетероциклических групп соответствуют изложенному выше в разделе «предпочтительные варианты и определения», но конкретные насыщенные гетероциклические группы включают пирролидинил, морфолинил, пиперазинил и N-C1-4алкилпиперазинил. Такие группы, как правило, являются незамещенными или замещены одной или двумя метильными группами и, в одном конкретном варианте осуществления, являются незамещенными.
Конкретными примерами соединений, в которых R4 и R5 выбраны из водорода и C1-4алкила, являются метиламино и диметиламино группы, более типично диметиламино группа.
В варианте осуществления (xvii) R1 представляет собой 5-замещенную 2-фурильную группу формулы:
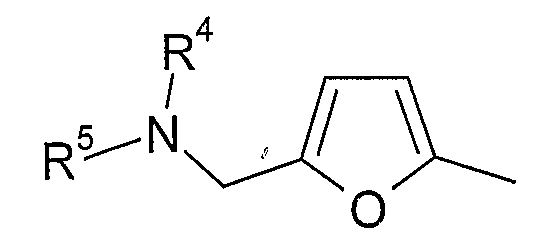
в которой R4 и R5 являются одинаковыми или различными и выбраны из водорода и C1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом, или группу, выбранную из O, NH, NMe, S или SO2, причем 5- или 6-членная насыщенная гетероциклическая группа необязательно замещена гидрокси, фтором, амино, метиламино, метилом или этилом; при условии, что соединение не является [3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амидом 5-пиперидин-1-илметилфуран-2-карбоновой кислоты.
Примеры насыщенных гетероциклических групп соответствуют изложенному выше в разделе «предпочтительные варианты и определения», но конкретные насыщенные гетероциклические группы включают пирролидинил, морфолинил, пиперазинил и N-C1-4алкилпиперазинил. Такие группы, как правило, являются незамещенными или замещены одной или двумя метильными группами и, в одном конкретном варианте осуществления, являются незамещенными.
В варианте осуществления (xviii) R1 представляет собой группу формулы:
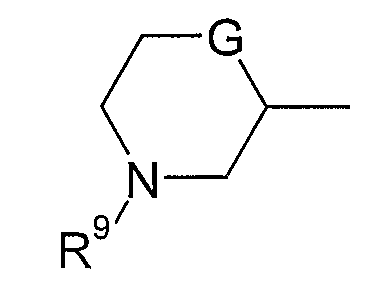
в которой R9 представляет собой водород, метил, этил или изопропил; G представляет собой CH, O, S, SO, SO2 или NH, и группа необязательно замещена одним, двумя или тремя заместителями, выбранными из C1-4гидрокарбила, гидрокси, C1-4гидрокарбилокси, фтора, амино, моно- и ди-C1-4алкиламино, и где каждая из групп C1-4гидрокарбил и C1-4гидрокарбилокси необязательно замещена гидрокси, фтором, амино, моно- или ди-C1-4алкиламино.
В одной подгруппе соединений, входящих в вариант осуществления (xviii), G выбран из O и CH.
В варианте осуществления (xviii) группа R1, как правило, является незамещенной или замещена одной или двумя метильными группами, и более типично является незамещенной.
В варианте осуществления (xix) R1 представляет собой 3,5-дизамещенную фенильную группу формулы:

в которой Xa, как и X, выбран из O, NH и NCH3.
Предпочтительно Xa представляет собой N-CH3.
Конкретные примеры фрагмента R1-A- показаны в таблице 1, причем звездочка означает точку присоединения к карбонильной группе C=O в группе R1-E-A-C(=O)-NH-.
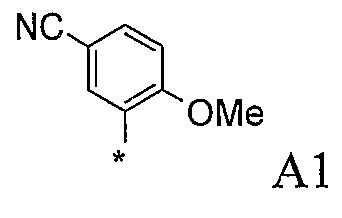
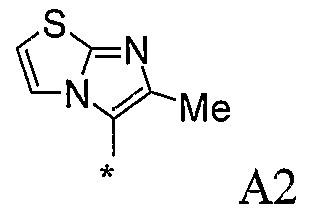
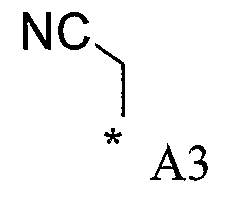
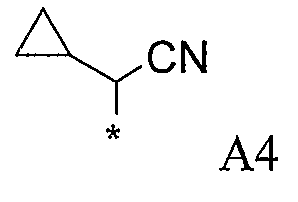
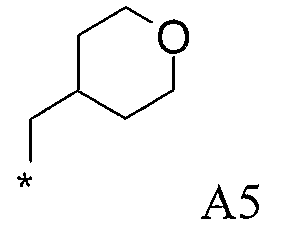
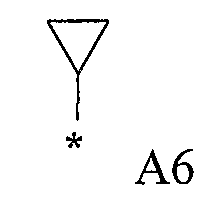
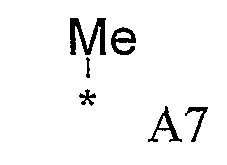
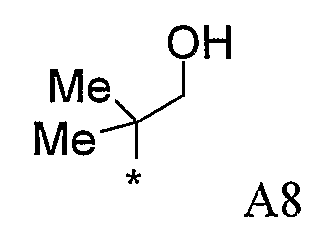
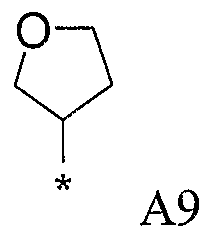
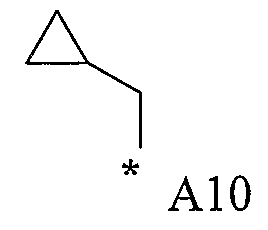
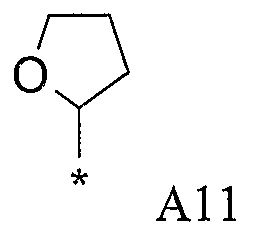
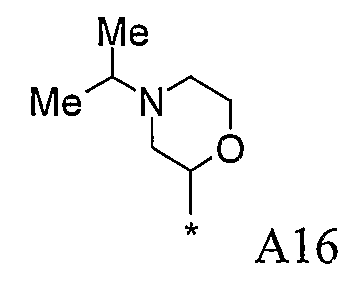
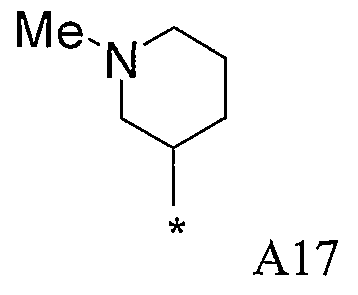
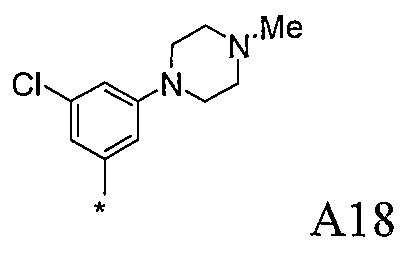
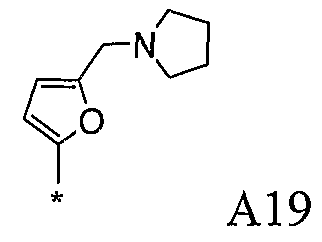
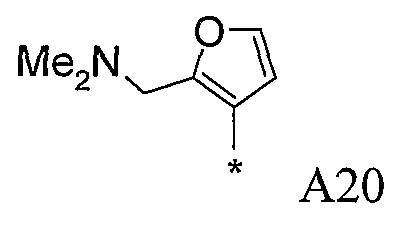
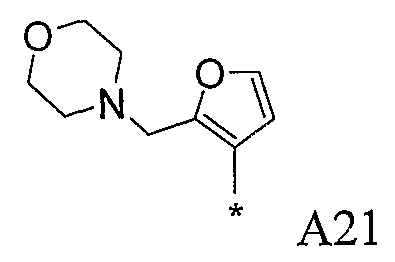

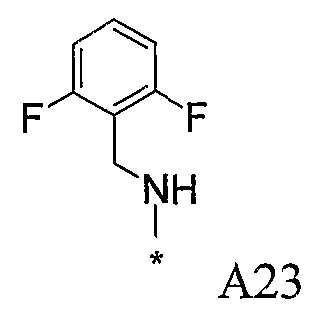
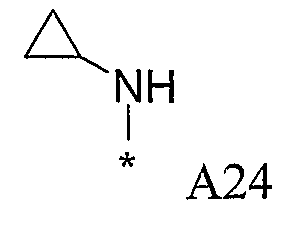
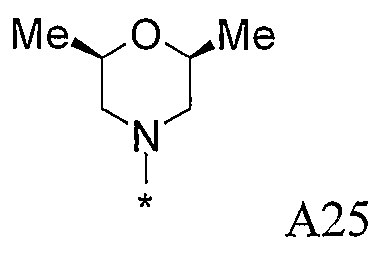
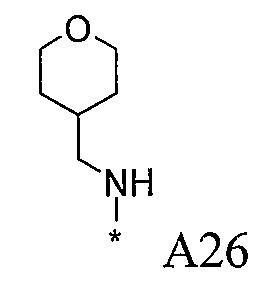
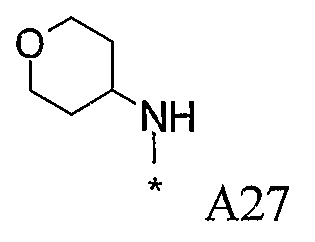
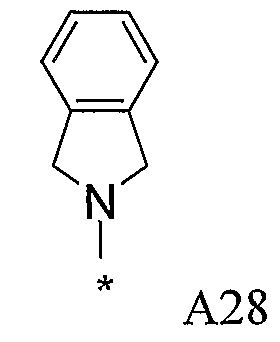
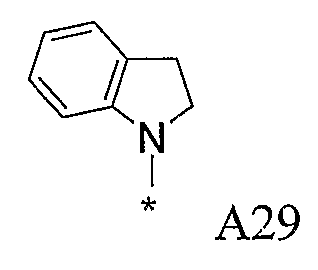
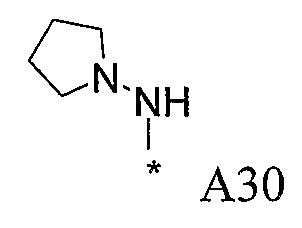
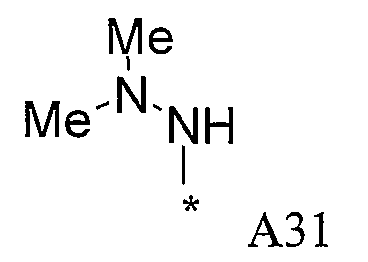
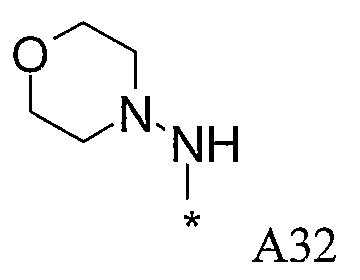
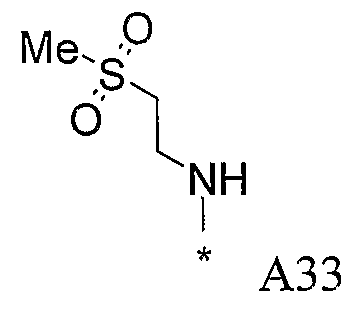
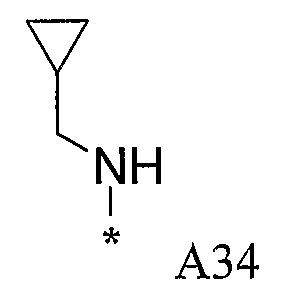
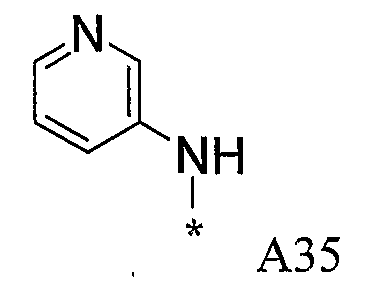
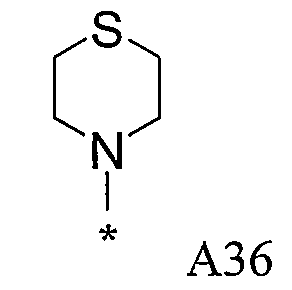
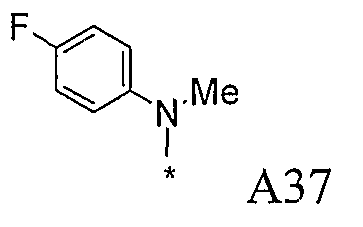
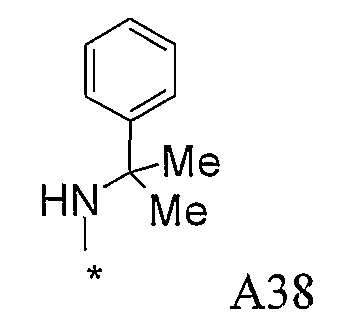
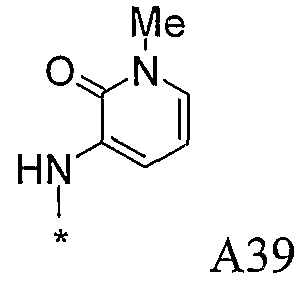

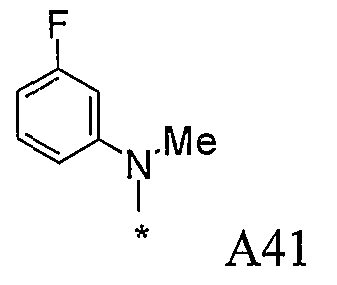
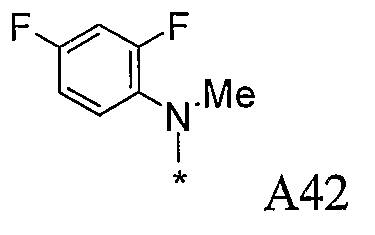
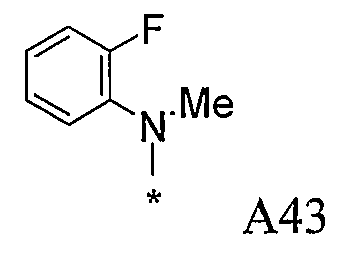
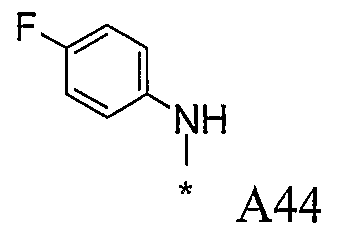
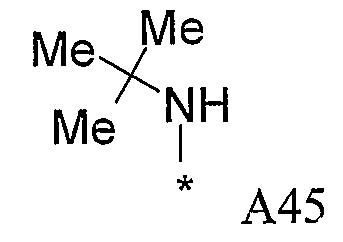
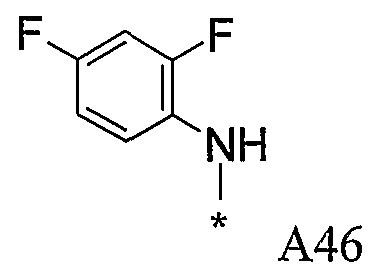
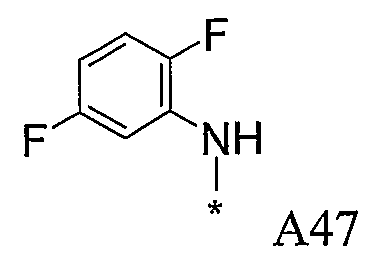
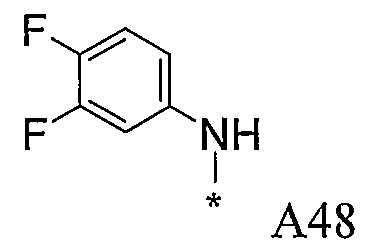
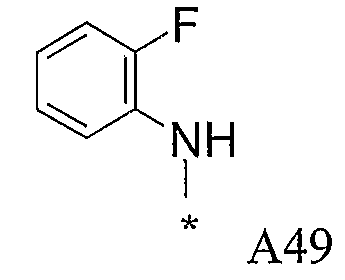
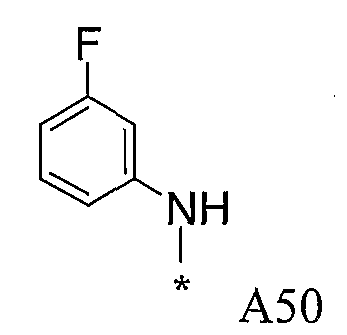
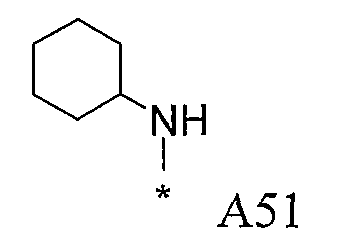
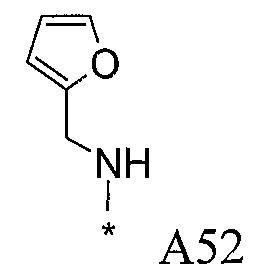
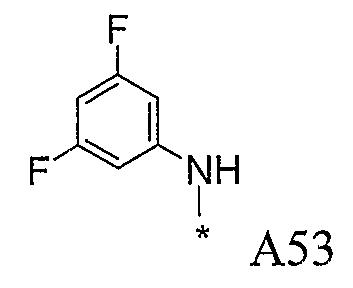
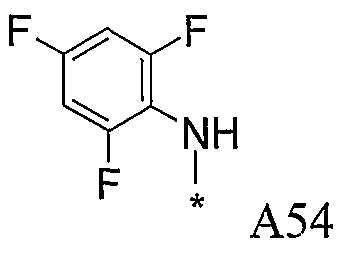
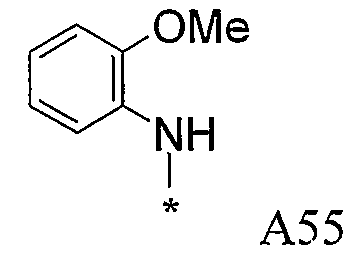
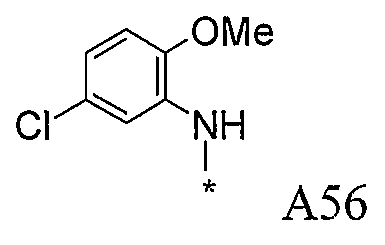
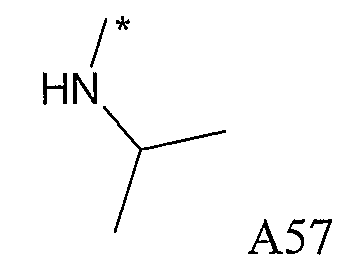
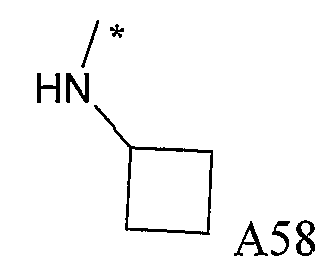
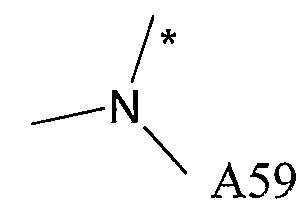
В таблице 1 предпочтительные группы R1-E-A- включают A1, A4, A10, A11, A13, A20, A22, A23, A24, A29, A30, A31, A32, A38, A42, A43, A44, A46, A47, A49, A54 и A56.
В другом варианте осуществления группа R1-E-A- представляет собой A57, A58 или A59.
Предпочтительное подмножество групп R1-E-A- включает A1, A4, A20, A24, A30, A44, A46 и A54. В этом подмножестве одной особой группой R1-E-A- является группа A24.
Одна подгруппа соединений по настоящему изобретению представлена формулой (II):
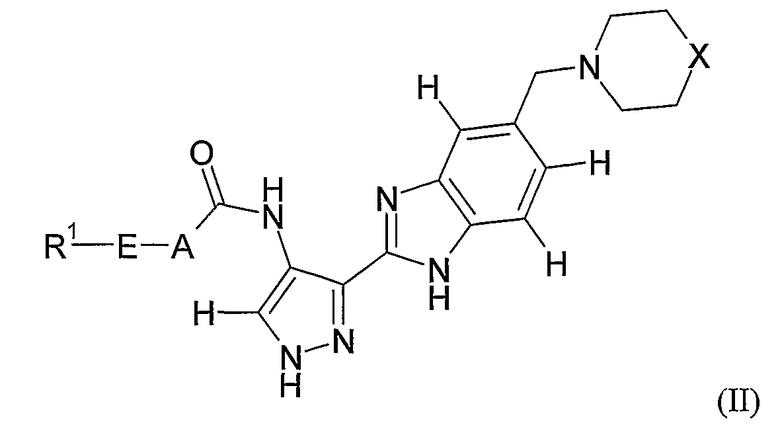
где R1, E, A и X соответствуют приведенным в настоящем описании определениям.
В формуле (II) одним из подмножеств соединений является подмножество, в котором X представляет собой O.
Одна подгруппа соединений формулы (II) может быть представлена формулой (III):
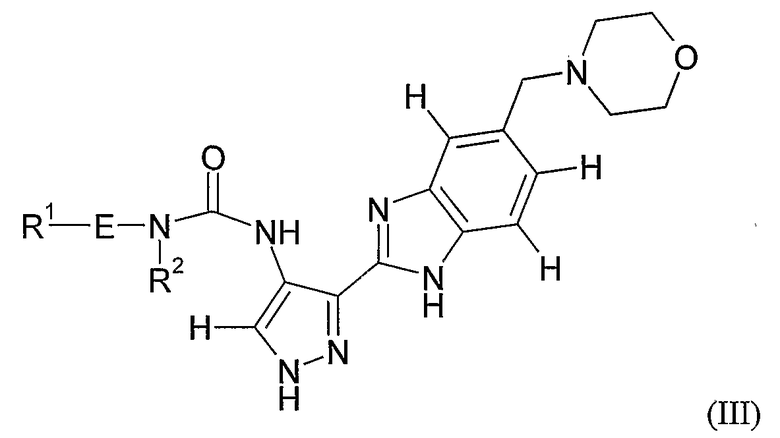
а также их солями, в частности лактатом.
В формуле (III) одним из подмножеств соединений является подмножество, в котором E является связью.
Другим подмножеством соединений из числа соединений формулы (III) является подмножество, в котором E представляет собой CH2 или C(CH3)2.
В одном особенно предпочтительном варианте осуществления соединения формулы (III) E представляет собой связь, R2 представляет собой H и R1 является циклоалкильной группой (i), определенной в настоящей заявке. В одном из вариантов осуществления циклоалкильная группа может быть циклопропилом или циклобутилом. Более предпочтительно, R1 представляет собой циклопропильную группу.
Чтобы избежать сомнений, следует понимать, что каждый общий и конкретный предпочтительный вариант, вариант осуществления и пример группы R1 может сочетаться с каждым общим и конкретным предпочтением, вариантом осуществления и примером группы R2 и/или R3, и/или R4, и/или R5, и/или R6, и/или R7, и/или R8, и/или R9, и/или R10, и/или R11, и/или D1, и/или D2, и/или A, и/или E, и/или X, и/или Xa и любыми их подгруппами, определенными в настоящей заявке, если контекст не указывает на другое, и что все такие комбинации охвачены настоящей заявкой.
Различные функциональные группы и заместители, из которых составлены соединения формулы (I), как правило, выбраны таким образом, что молекулярная масса соединения формулы (I) не превышает 1000. Чаще молекулярная масса соединений будет менее 750, например, менее 700 или менее 650, или менее 600, или менее 550. Более предпочтительно, молекулярная масса имеет значение менее 525 и, например, составляет 500 или меньше.
Конкретными соединениями по настоящему изобретению являются соединения, которые приведены в качестве иллюстрации в примерах ниже.
Одним предпочтительным соединением по настоящему изобретению является 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина, а также ее соли, сольваты и таутомеры.
Соли, сольваты, таутомеры, изомеры, N-оксиды, сложные эфиры, пролекарства и формы, содержащие изотопы, соединений подгрупп (A) и (B) соединений формулы (I), а также их подгруппы и варианты осуществления
Если не указано иначе, упоминания конкретного соединения также включают ионные формы, соли, сольваты и их защищенные формы, например, как обсуждается ниже.
Многие соединения формулы (I) могут существовать в форме солей, например, кислотно-аддитивных солей или, в некоторых случаях, солей органических и неорганических оснований, например в форме карбоксилатных, сульфонатных и фосфатных солей. Все такие соли включены в объем настоящего изобретения, и ссылки на соединения формулы (I) включают солевые формы этих соединений. Как и в предыдущем разделе настоящей заявки, все ссылки на формулу (I) следует воспринимать, как относящиеся к формулам (II), (III) и их подгруппам, определенным в настоящей заявке, если контекст не указывает на другое.
Соли по настоящему изобретению могут быть синтезированы из исходных соединений, которые содержат основный или кислотный фрагмент, с помощью стандартных химических способов, как, например, способов, описанных в Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, в твердом переплете, 388 страниц, август 2002. В основном такие соли могут быть получены взаимодействием свободных форм кислоты или основания рассматриваемых соединений с подходящим основанием или кислотой в воде или органическом растворителе, или их смеси; как правило, применяются неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Кислотно-аддитивные соли могут быть образованы с широким кругом кислот как неорганических, так и органических. Примеры кислотно-аддитивных солей включают соли, образованные кислотами, выбранными из группы, состоящей из уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), L-аспарагиновой, бензолсульфоновой, бензойной, 4-ацетамидобензойной, бутановой, (+)камфорной, камфорсульфоновой, (+)-(1S)-камфор-10-сульфоновой, каприновой, капроновой, каприловой, коричной, лимонной, цикламовой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, бромистоводородной, хлористоводородной, иодистоводородной, изетионовой, (+)-L-молочной, (±)-DL-молочной, лактобионовой, малеиновой, яблочной, (-)-L-яблочной, малоновой, (±)-DL-миндальной, метансульфоновой, нафталин-2-сульфоновой, нафталин-1,5-дисульфоновой, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памовой, фосфорной, пропионовой, L-пироглутамовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, дубильной, (+)-L-винной, тиоциановой, п-толуолсульфоновой, ундециленовой и валериановой кислот, а также ацилированных аминокислот и катионообменных смол.
Кроме того, кислотно-аддитивные соли могут быть выбраны из солей аспарагиновой (например, D-аспарагиновой), карбоновой, додекановой, изомасляной, лаурилсульфоновой, слизевой, нафталинсульфоновой (например, нафталин-2-сульфоновой), толуолсульфоновой (например, п-толуолсульфоновой) и ксинафовой кислот.
Одна отдельная группа солей состоит из солей, образованных хлористоводородной, иодистоводородной, фосфорной, азотной, серной, лимонной, молочной, янтарной, малеиновой, яблочной, изетионовой, фумаровой, бензолсульфоновой, толуолсульфоновой, метансульфоновой, этансульфоновой, нафталинсульфоновой, валериановой, уксусной, пропановой, бутановой, малоновой, глюкуроновой и лактобионовой кислотами.
Одна подгруппа солей состоит из солей, образованных хлористоводородной, уксусной, адипиновой, L-аспарагиновой и DL-молочной кислотами.
Другая подгруппа солей состоит из ацетата, мезилата, этансульфоната, DL-лактата, адипата, D-глюкуроната, D-глюконата и гидрохлорида.
Соли, как, например, кислотно-аддитивные соли, обладают рядом преимуществ перед соответствующими свободными основаниями. Например, соли будут отличаться одним или несколькими из следующих преимуществ перед свободными основаниями тем, что они будут:
- обладать большей растворимостью и, следовательно, будут лучше подходить для внутривенного введения (например, с помощью инфузии) и будут обладать лучшей фармакокинетикой;
- обладать большей стабильностью (например, увеличенным сроком хранения);
- обладать лучшей термической стабильностью;
- менее основными и, следовательно, будут лучше для внутривенного введения;
- иметь преимущества для получения;
- иметь лучшие метаболические свойства; и
- проявлять меньшее изменение клинических свойств от пациента к пациенту.
Предпочтительными солями для применения в получении жидких (например, водных) композиций соединений формул (I), (II), (III), (XXX), а также подгрупп и отдельных примеров указанных соединений, описанных в настоящей заявке, являются соли, имеющие растворимость в данном жидком носителе (например, воде), превышающую 25 мг/мл жидкого носителя (например, воды), более типично, превышающую 50 мг/мл и, предпочтительно, превышающую 100 мг/мл.
В других вариантах осуществления предпочтительными солями для применения в получении жидких (например, водных) композиций соединений формул (I), (II), (III), (XXX), а также подгрупп и отдельных примеров указанных соединений, описанных в настоящей заявке, являются соли, имеющие растворимость в данном жидком носителе (например, воде или буферных системах), превышающую 1 мг/мл, типично превышающую 5 мг/мл жидкого носителя (например, воды), более типично, превышающую 15 мг/мл, более типично, превышающую 20 мг/мл и, предпочтительно, превышающую 25 мг/мл.
В другом варианте осуществления предпочтительными кислотно-аддитивными солями являются мезилат, этансульфонат, D- или L-лактат и гидрохлорид. В одном отдельном варианте осуществления кислотно-аддитивная соль представляет собой лактат, в частности L-лактат или D-лактат, предпочтительно L-лактат.
В одном из вариантов осуществления настоящего изобретения разработана фармацевтическая композиция, включающая водный раствор, который содержит соединения формул (I), (II), (III), (XXX), а также подгруппы и отдельные примеры указанных соединений, описанных в настоящей заявке, в форме соли, в концентрации превышающей 25 мг/мл, типично превышающей 50 мг/мл и предпочтительно превышающей 100 мг/мл.
В другом варианте осуществления настоящего изобретения разработана фармацевтическая композиция, включающая водный раствор, который содержит соединения формул (I), (II), (III), (XXX), а также подгруппы и отдельные примеры указанных соединений, описанных в настоящей заявке, в форме соли, в концентрации превышающей 1 мг/мл, типично превышающей 5 мг/мл жидкого носителя (например, воды), более типично, превышающей 15 мг/мл, более типично, превышающей 20 мг/мл и предпочтительно превышающей 25 мг/мл.
Если соединение является анионным или включает функциональную группу, которая может быть анионной (например, -COOH может иметь форму -COO-), тогда может быть образована соль с подходящим катионом. Примеры подходящих неорганических катионов включают, не ограничиваясь перечисленными, ионы щелочных металлов как, например, Na+ и K+, катионы щелочноземельных металлов как, например, Ca2+ и Mg2+, и другие катионы, такие как Al3+. Примеры подходящих органических катионов включают, не ограничиваясь перечисленными, ион аммония (т.е. NH4 +) и замещенные аммониевые ионы (например, NH3R+, NH2R2 +, NHR3 +, NR4 +). Примерами некоторых подходящих замещенных аммониевых ионов служат аммониевые ионы, являющиеся производными: этиламина, диэтиламина, дициклогексиламина, триэтиламина, бутиламина, этилендиамина, этаноламина, диэтаноламина, пиперазина, бензиламина, фенилбензиламина, холина, меглумина и трометамина, а также аминокислот, таких как лизин и аргинин. Примером обычного четвертичного аммониевого иона является N(CH3)4 +.
Если соединения формулы (I) содержат аминогруппу, они могут образовывать четвертичные аммониевые соли, например, при взаимодействии с алкилирующим реагентом, в соответствии со способами, которые хорошо известны специалистам. Такие четвертичные аммониевые соединения охвачены формулой (I).
Солевые формы соединений по настоящему изобретению, как правило, являются фармацевтически приемлемыми солями, и примеры фармацевтически приемлемых солей обсуждаются в Berge et al., 1977, “Pharmaceutically Acceptable Salts”, J. Pharm. Sci., Vol. 66, pp.1-19. Однако соли, которые не являются фармацевтически приемлемыми, также могут быть получены в качестве промежуточных форм, которые затем могут быть превращены в фармацевтически приемлемые соли. Такие формы фармацевтически неприемлемых солей, которые могут применяться, например, при очистке или разделении соединений по настоящему изобретению, также входят в объем изобретения.
Соединения формулы (I), содержащие аминогруппу, также могут образовывать N-оксиды. В настоящей заявке ссылки на соединения формулы (I), которые содержат аминогруппы, также включают N-оксиды.
Если соединение содержит несколько аминогрупп, один или несколько атомов азота могут быть окислены с образованием N-оксидов. Конкретными примерами N-оксидов являются N-оксиды третичных аминов или атомов азота азотсодержащих гетероциклов.
N-оксиды могут быть получены обработкой соответствующих аминов окисляющим реагентом, таким как пероксид водорода или пероксикислота (например, пероксикарбоновая кислота), см., например, Advanced Organic Chemistry, by Jerry March, 4th Edition, Wiley Interscience, pp. Более конкретно, N-оксиды могут быть получены по методике L.W. Deady (Syn. Comm. 1977, 7, 509-514) в которой соединение, содержащее аминогруппу, вводят во взаимодействие с м-хлорпероксибензойной кислотой (MCPBA), например, в инертном растворителе, таком как дихлорметан.
Соединения формулы (I) могут существовать в виде ряда различных геометрически изомерных и таутомерных форм, и упоминание соединения формулы (I) включают все такие формы. Во избежание сомнений, если соединение может существовать в одной из нескольких геометрически изомерных или таутомерных форм, и конкретно описана или показана только одна из них, все остальные формы, тем не менее, охвачены формулой (I).
Например, в соединениях формулы (I) бензимидазольная группа может принимать любую из двух следующих таутомерных форм A, A', B и B'. Для простоты общая формула (I) показывает формы A и A', но следует воспринимать данную формулу, как охватывающую все четыре таутомерные формы.
Пиразольный цикл также может проявлять таутомерию, причем он может существовать в двух таутомерных формах C или D, приведенных ниже:
Другие примеры таутомерных форм включают, например, кетонные формы, енольные формы и формы енолятов, как, например, в следующих таутомерных парах: кетон/енол (показаны ниже), имин/енамин, амид/иминоспирт, амидин/амидин, нитрозо/оксим, тиокетон/ентиол и нитро/аци-нитро:
Если соединения формулы (I) содержат один или несколько хиральных центров и могут существовать в форме двух или нескольких оптических изомеров, упоминание соединения формулы (I) включает все его оптически изомерные формы (например, энантиомеры, эпимеры и диастереомеры) либо как индивидуальные оптические изомеры, либо как смеси (например, рацемические смеси) двух или нескольких оптических изомеров, если контекст не требует иного.
Например, группа A может включать один или несколько хиральных центров. Так, если E и R1 оба присоединены к одному и тому же атому углерода связывающей группы A, указанный атом углерода, как правило, является хиральным и, следовательно, соединение формулы (I) будет существовать в виде пары энантиомеров (или более чем одной пары энантиомеров, если в соединении присутствует более чем один хиральный центр).
Оптические изомеры могут быть охарактеризованы и идентифицированы по их оптической активности (т.е. как + и - изомеры, или d и l изомеры) или же они могут быть охарактеризованы в терминах их абсолютной стереохимической конфигурации, с использованием номенклатуры “R и S”, разработанной Канном, Ингольдом и Прелогом, см. Advanced Organic Chemistry, by Jerry March, 4th Edition, John Wiley & Sons, New York, 1992, pages 109-114, и, кроме того, см. Cahn, Ingold & Prelog, Angew. Chem. Int. Ed. Engl., 1966, 5, 385-415.
Оптические изомеры могут быть разделены с применением ряда методик, включая хиральную хроматографию (хроматографию на хиральной неподвижной фазе), и такие методики хорошо известны специалисту в данной области техники.
В качестве альтернативы хиральной хроматографии оптические изомеры могут быть разделены с помощью образования диастереомерных солей с хиральными кислотами, как, например, (+)-винной кислотой, (-)-пироглутамовой кислотой, (-)-дитолил-L-винной кислотой, (+)-миндальной кислотой, (-)-яблочной кислотой и (-)-камфорсульфоновой кислотой, разделения диастереомерных солей селективной кристаллизацией и затем разложения солей с получением индивидуальных энантиомеров свободного основания.
Если соединения формулы (I) существуют в виде двух или нескольких оптически изомерных форм, один из энантиомеров энантиомерной пары может проявлять преимущества перед другим энантиомером, например, с точки зрения биологической активности. Таким образом, в определенных обстоятельствах может быть желательно применение в качестве терапевтического средства только одного энантиомера из пары или только одного из многих диастереомеров. Соответственно изобретение относится к композициям, содержащим соединение формулы (I), которое имеет один или несколько хиральных центров и в которых не менее 55% (например, не менее 60, 65, 70, 75, 80, 85, 90 или 95%) соединения формулы (I) присутствует в виде одного оптического изомера (например, энантиомера или диастереомера). В одном из основных вариантов осуществления 99% или более от общего количества соединения формулы (I) (например, практически все соединение) может присутствовать в виде одного оптического изомера (например, энантиомера или диастереомера).
Соединения по настоящему изобретению включают соединения, в которых один или несколько атомов заменены их изотопами, и упоминание конкретного элемента включают в себя все изотопы этого элемента. Например, упоминание водорода включает в себя 1H, 2H(D) и 3H(T). Подобным же образом упоминания углерода и кислорода включают в себя соответственно 12C, 13C и 14C, и 16O и 18O.
Эти изотопы могут быть радиоактивными или нерадиоактивными. В одном из вариантов осуществления настоящего изобретения соединения не содержат радиоактивных изотопов. Такие соединения являются предпочтительными для терапевтического применения. Однако в другом варианте осуществления соединения могут содержать один или несколько радиоактивных изотопов. Соединения, содержащие такие радиоактивные изотопы, могут быть полезны в диагностических применениях.
Сложные эфиры, как, например, эфиры карбоновых кислот и ацилоксиэфиры соединений формулы (I), включающих группу карбоновой кислоты или гидроксильную группу, также охвачены формулой (I). Примерами сложных эфиров являются соединения, содержащие группу -C(=O)OR, где R представляет собой заместитель сложноэфирной группы, например, C1-7алкильную группу, C3-20гетероциклическую группу или C5-20арильную группу, предпочтительно C1-7алкильную группу. Конкретные примеры сложноэфирных групп включают, не ограничиваясь перечисленными, -C(=O)OCH3, -C(=O)OCH2CH3, -C(=O)OC(CH3)3 и -C(=O)OPh. Примеры ацилокси (обращенных эфирных) групп представлены формулой -OC(=O)R, где R представляет собой заместитель ацилоксигруппы, например C1-7алкильную группу, C3-20гетероциклическую группу или C5-20арильную группу, предпочтительно C1-7алкильную группу. Конкретные примеры ацилоксигрупп включают, не ограничиваясь перечисленными, -OC(=O)CH3 (ацетокси), -OC(=O)CH2CH3, -OC(=O)C(CH3)3, -OC(=O)Ph и -OC(=O)CH2Ph.
Кроме того, формулой (I) охвачены любые полиморфные формы соединений, сольваты (например, гидраты), комплексы (например, комплексы включения или клатраты с такими соединениями, как циклодекстрины, или комплексы с металлами) соединений или пролекарства соединений. Под «пролекарством» подразумевается, например, любое соединение, которое in vivo превращается в биологически активное соединение формулы (I).
Например, одним из видов пролекарств являются сложные эфиры активных соединений (например, физиологически приемлемые, способные к метаболизму сложные эфиры). В процессе метаболизма сложноэфирная группа (-C(=O)OR) расщепляется, что приводит к образованию активного лекарственного вещества. Такие сложные эфиры могут быть получены этерификацией, например, любой из групп карбоновой кислоты (-C(=O)OH) в исходном соединении, при условии предварительной защиты, если есть такая возможность, любых других реакционноспособных групп, имеющихся в исходном соединении, с последующим снятием защиты в случае необходимости.
Примеры таких способных к метаболизму сложных эфиров включают эфиры формулы -C(=O)OR, в которых R представляет собой:
C1-7алкил
(например, -Me, -Et, -н-Pr, -изо-Pr, -н-Bu, -втор-Bu, -изо-Bu, -трет-Bu);
C1-7аминоалкил
(например, аминоэтил; 2-(N,N-диэтиламино)этил; 2-(4-морфолино)этил); и
ацилокси-C1-7алкил
(например, ацилоксиметил;
ацилоксиэтил;
пивалоилоксиметил;
ацетоксиметил;
1-ацетоксиэтил;
1-(1-метокси-1-метил)этилкарбонилоксиэтил;
1-(бензоилокси)этил; изопропоксикарбонилоксиметил;
1-изопропоксикарбонилоксиэтил; циклогексилкарбонилоксиметил;
1-циклогексилкарбонилоксиэтил;
циклогексилоксикарбонилоксиметил;
1-циклогексилоксикарбонилоксиэтил;
(4-тетрагидропиранилокси)карбонилоксиметил;
1-(4-тетрагидропиранилокси)карбонилоксиэтил;
(4-тетрагидропиранил)карбонилоксиметил; и
1-(4-тетрагидропиранил)карбонилоксиэтил).
Кроме того, некоторые пролекарства активируются под действием ферментов, образуя активные соединения, или соединения, которые в результате дальнейших химических реакций образуют активные соединения (например, как в случае ADEPT, GDEPT, LIDEPT и т.п.). Например, пролекарство может быть производным сахара или другим конъюгатом гликозида, или может быть сложноэфирным производным аминокислоты.
1-Циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина и ее соли
Одним из конкретных соединений формулы (I), относящихся к подгруппе (A), является 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина.
Соответственно, в одном из предпочтительных вариантов осуществления изобретение относится к форме свободного основания или кислотно-аддитивной соли 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины.
Форма свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, из которой образуются соли, имеет формулу (XXX):
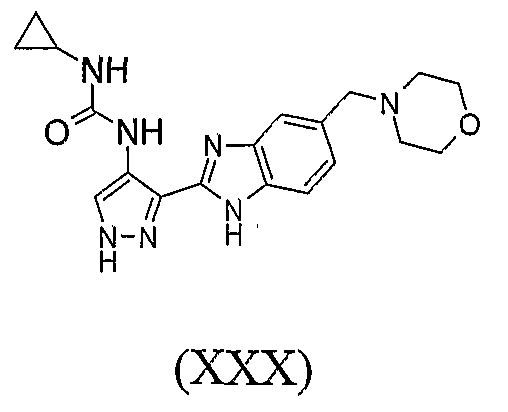
Соединение формулы (XXX) может называться в настоящей заявке его химическим наименованием, т.е. 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевиной, или, для удобства, «соединением XXX», «соединением формулы (XXX)» или соединением примера 24. Каждый из этих синонимов относится к соединению, показанному формулой (XXX) выше, называемому 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевиной.
Упоминания формы свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и его кислотно-аддитивных солей включают в себя все его сольваты, таутомеры и изотопы, и где это допускается контекстом, N-оксиды, другие ионные формы и пролекарства. Следовательно, упоминания другого таутомера соединения формулы (XXX), т.е. 1-циклопропил-3-[3-(6-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, следует понимать, как упоминания соединения (XXX).
Кислотно-аддитивные соли соединения формулы (XXX) могут быть выбраны из солей, образованных широким кругом кислот, как неорганических, так и органических. Примеры кислотно-аддитивных солей включают соли, образованные кислотами, которые выбраны из группы, состоящей из уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), аспарагиновой (например, L-аспарагиновой), бензолсульфоновой, бензойной, 4-ацетамидобензойной, бутановой, (+)камфорной (например, (+)-камфарной), камфорсульфоновой, (+)-(1S)-камфор-10-сульфоновой, каприновой, капроновой, каприловой, угольной, коричной, лимонной, цикламовой, додекановой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, бромистоводородной, хлористоводородной, иодистоводородной, изетионовой, молочной (например,(+)-L-молочной и (-)-D-молочной), лактобионовой, лаурилсульфоновой, малеиновой, яблочной, (-)-L-яблочной, малоновой, (±)-DL-миндальной, метансульфоновой, слизевой, нафталинсульфоновой (например, нафталин-2-сульфоновой), нафталин-1,5-дисульфоновой, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памовой, фосфорной, пропионовой, L-пироглутамовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, дубильной, винной (например, (+)-L-винной), тиоциановой, толуолсульфоновой (например, п-толуолсульфоновой), ундециленовой, валериановой и ксинафовой кислот, а также ацилированных аминокислот и катионообменных смол.
Одна отдельная группа солей состоит из солей, образованных хлористоводородной, иодистоводородной, фосфорной, азотной, серной, лимонной, молочной, янтарной, малеиновой, яблочной, изетионовой, фумаровой, бензолсульфоновой, толуолсульфоновой, метансульфоновой, этансульфоновой, нафталинсульфоновой, валериановой, уксусной, пропановой, бутановой, малоновой, глюкуроновой и лактобионовой кислотами.
Одна подгруппа солей состоит из солей, образованных хлористоводородной, уксусной, адипиновой, L-аспарагиновой и D- или L-молочными кислотами.
Другая подгруппа солей состоит из ацетата, мезилата, этансульфоната, D- или L-лактата, адипата, D-глюкуроната, D-глюконата и гидрохлорида. В другом варианте осуществления предпочтительными кислотно-аддитивными солями являются мезилат, этансульфонат, D- или L-лактат и гидрохлорид.
В одном отдельном варианте осуществления кислотно-аддитивная соль является DL-лактатом, в частности L-лактатом или D-лактатом, предпочтительно L-лактатом.
В другом варианте осуществления свободное основание или соль соединения формулы (XXX) выбраны из L-лактата, дигидрата свободного основания, эзилата, свободного основания и гидрохлорида.
В еще одном и предпочтительном варианте осуществления соль соединения формулы (XXX) выбрана из лактата и цитрата, а также их смеси и, более предпочтительно, выбрана из L-лактата и цитрата и их смеси, причем L-лактат является особенно предпочтительным. Особые и предпочтительные варианты осуществления настоящего изобретения, относящиеся к L-лактату и цитрату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, выделены и более подробно описаны ниже.
В другом варианте осуществления соединение формулы (XXX) является свободным основанием.
Соли по настоящему изобретению, такие как лактаты (например, L-лактаты) и цитраты, могут быть получены из исходного соединения, т.е. 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины по обычным химическим методикам, как, например, способам, описанным в Pharmaceutical Salts: Properties, Selection, and Use, P.Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, в твердом переплете, 388 страниц, август 2002. Как правило, эти соли могут быть получены взаимодействием исходного соединения, т.е. 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины с соответствующей кислотой в воде или органическом растворителе, или смеси воды и органического растворителя; в основном используются неводные среды, как, например, эфир, этилацетат, этанол, изопропанол или ацетонитрил.
В другом аспекте настоящее изобретение относится к способу получения кислотно-аддитивной соли 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, такой как лактат (например, L-лактат) и цитрат, причем указанный способ включает получение раствора 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания в растворителе (как правило, органическом растворителе) или смеси растворителей и обработку полученного раствора кислотой для образования осадка кислотно-аддитивной соли.
Кислота может быть добавлена в виде раствора в растворителе, обладающем способностью смешиваться с растворителем, в котором растворено свободное основание. Растворитель, в котором первоначально растворяют свободное основание, может являться одним из растворителей, в которых нерастворима кислотно-аддитивная соль свободного основания. С другой стороны, растворитель, в котором первоначально растворяют свободное основание, может являться одним из растворителей, в которых кислотно-аддитивная соль растворима хотя бы частично, причем впоследствии добавляют другой растворитель, в котором кислотно-аддитивная соль растворима меньше, так что соль выпадает из раствора.
В альтернативном способе получения кислотно-аддитивной соли, такой как лактат (например, L-лактат) или цитрат, 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину растворяют в растворителе, содержащем летучую кислоту и необязательно дополнительный растворитель, тем самым, получая раствор кислотно-аддитивной соли с летучей кислотой, и затем полученный раствор концентрируют или упаривают для выделения соли. Другим примером кислотно-аддитивной соли, которая может быть получена таким способом, является ацетат.
В другом аспекте изобретение относится к способу получения кислотно-аддитивной соли 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, как определено в настоящем описании, такой как лактат (например, L-лактат) и цитрат, причем указанный способ включает обработку соединений формулы (XXX):
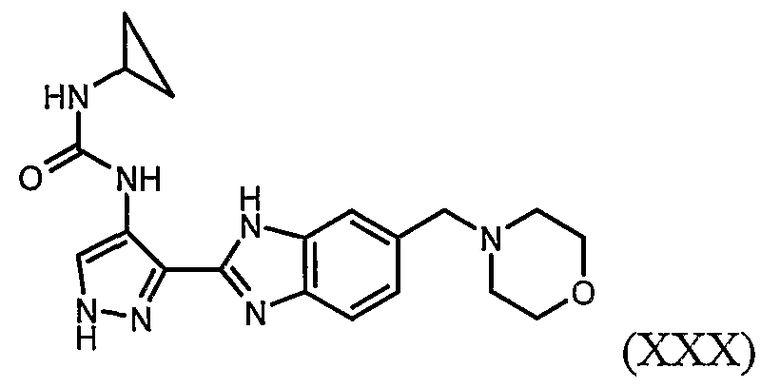
органической или неорганической кислотой, указанной в настоящем описании, в органическом растворителе для получения кислотно-аддитивной соли 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины с органической или неорганической кислотой, и, необязательно, выделение полученной таким образом кислотно-аддитивной соли.
Соль, как правило, выпадает в осадок из органического растворителя после своего образования и, следовательно, может быть выделена отделением твердого вещества от раствора, например фильтрованием.
Одна солевая форма соединения по настоящему изобретению может быть превращена в свободное основание и, необязательно, в другую солевую форму с помощью способов, хорошо известных специалисту. Например, свободное основание может быть получено при прохождении раствора соли через колонку с неподвижной фазой, содержащей аминогруппы (например, колонку Strata-NH2). С другой стороны, раствор соли в воде может быть обработан бикарбонатом натрия для разложения соли и выпадения в осадок свободного основания. Затем свободное основание можно ввести в реакцию с другой кислотой по одному из способов, описанных выше или еще где-либо в настоящей заявке.
Предпочтительные соли, такие как кислотно-аддитивные соли, например лактат (например, L-лактат) и цитрат, имеют ряд преимуществ. Например, соли будут отличаться одним или несколькими из следующих преимуществ, в том, что они:
- будут иметь большую растворимость, в частности будут иметь лучшую растворимость в водном растворе и, следовательно, будут лучше для внутривенного введения (например, инфузией);
- дадут возможность регулировать pH раствора и, следовательно, будут лучше для внутривенного введения;
- могут иметь лучшую противораковую активность; и
- могут иметь лучший терапевтический индекс.
Другие преимущества солей заключаются в том, что они:
- будут обладать лучшей устойчивостью, например термической устойчивостью (например, увеличенным сроком хранения);
- будут иметь преимущества при изготовлении; и
- будут иметь лучшие физико-химические свойства.
Лактат (например, L-лактат) по настоящему изобретению обладает особыми преимуществами, т.к. он имеет хорошую растворимость в воде и демонстрирует лучшую растворимость в буферных системах.
Предпочтительными солями для применения в приготовлении жидких (например, водных) фармацевтических композиций являются кислотно-аддитивные соли (как, например, лактаты) имеющие растворимость в данном жидком носителе (например, воде) более 1 мг/мл, типично более 5 мг/мл жидкого носителя (например, воды), более типично более 15 мг/мл, более типично более 20 мг/мл и предпочтительно более 25 мг/мл.
Водные растворы солей (например, в форме фармацевтических композиций) представляют собой еще один аспект настоящего изобретения. Такие растворы могут быть забуференными или незабуференными. В растворе эти соли, как правило, будут диссоциировать, образуя протонированную форму 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины совместно с одним или несколькими противоионами. Следовательно, в другом аспекте настоящее изобретение также относится к водным растворам 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в протонированной форме в сочетании с одним или несколькими противоионами и, необязательно, одним или несколькими дополнительными противоионами (например, противоионами, образовавшимися из других солей, таких как хлорид натрия или буферные реагенты).
Соли 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, как правило, являются фармацевтически приемлемыми солями, и примеры фармацевтически приемлемых солей обсуждаются Berge et al., 1977, “Pharmaceutically Acceptable Salts”, J. Pharm. Sci., Vol. 66, pp. 1-19. Однако соли, которые не являются фармацевтически приемлемыми, также могут быть получены в качестве промежуточных форм, которые затем могут быть превращены в фармацевтически приемлемые соли. Следовательно, такие формы фармацевтически неприемлемых солей также входят в объем настоящего изобретения.
Кроме того, 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина может образовывать N-оксиды. N-оксиды могут быть получены способами, обсуждавшимися выше.
Как и другие соединения по настоящему изобретению, 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина и ее кислотно-аддитивные соли могут существовать в виде ряда различных таутомерных форм и упоминание этих соединений в настоящей заявке включает все эти формы.
Более конкретно, в 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевине и ее солях бензимидазольная группа может принимать любую из двух таутомерных форм A” и B”, показанных ниже. Для простоты общая формула (I) показывает форму A”, но эту формулу следует воспринимать, как охватывающую все таутомерные формы

Следовательно упоминания альтернативного таутомера, т.е. 1-циклопропил-3-[3-(6-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины очевидно указывает на то же самое соединение 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину.
Пиразольный цикл также может демонстрировать таутомерию и может существовать в двух таутомерных формах C” и D”, показанных ниже
Кроме того, как показано ниже, возможны цис- и транс-конформации мочевины
Упоминания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и ее солей также включают варианты соединений с одним или несколькими изотопными замещениями, и упоминание того или иного конкретного элемента включает в себя все изотопы этого элемента. Например, упоминание водорода включает в себя 1H, 2H(D) и 3H(T). Подобным же образом упоминания углерода и кислорода включают в себя соответственно 12C, 13C и 14C, и 16O и 18O.
Эти изотопы могут быть радиоактивными или нерадиоактивными. В одном из вариантов осуществления настоящего изобретения соединения не содержат радиоактивных изотопов. Такие соединения являются предпочтительными для терапевтического применения. Однако в другом варианте осуществления соединения могут содержать один или несколько радиоактивных изотопов. Соединения, содержащие такие радиоактивные изотопы, могут быть полезны в диагностических применениях.
Кроме того, упоминанием 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и ее солей охвачены любые полиморфные формы, сольваты (например, гидраты) и комплексы (например, комплексы включения или клатраты с такими соединениями, как циклодекстрины, или комплексы с металлами) данного соединения.
Лактаты и цитраты, их смеси и кристаллы
Как должно быть ясно из предыдущих разделов настоящей заявки предпочтительными солями 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины являются кислотно-аддитивные соли, образованные молочной кислотой (более предпочтительно L-молочной кислотой), лимонной кислотой или их смесями.
Для удобства соли, образованные молочной кислотой, L-молочной кислотой и лимонной кислотой, могут называться в настоящей заявке лактатами, L-лактатами и цитратами, соответственно.
В одном конкретном варианте осуществления соль представляет собой L-лактат или D-лактат, предпочтительно L-лактат.
В другом варианте осуществления соль представляет собой соль, образованную лимонной кислотой.
Более конкретно, соли являются смесями L-лактатов и цитратов.
В твердом состоянии лактат (в частности, L-лактат) или цитрат по настоящему изобретению может быть кристаллическим или аморфным, или смесью этих двух форм.
В одном из вариантов осуществления лактат (в частности, L-лактат) или цитрат являются аморфными.
В аморфном твердом веществе отсутствуют трехмерные структуры, которые обычно существуют в кристаллической форме, и положения молекул относительно друг друга в аморфной форме являются, по существу, случайными (см., например, Hancock et al. J. Pharm. Sci. (1997), 86, 1).
В другом варианте осуществления лактат (в частности, L-лактат) или цитрат являются в основном кристаллическими, т.е. они могут быть на 50-100% кристаллическими и, более конкретно, они могут быть кристаллическими по меньшей мере на 50%, или кристаллическими по меньшей мере на 60%, или кристаллическими по меньшей мере на 70%, или кристаллическими по меньшей мере на 80%, или кристаллическими по меньшей мере на 90%, или кристаллическими по меньшей мере на 95%, или кристаллическими по меньшей мере на 98%, или кристаллическими по меньшей мере на 99%, или кристаллическими по меньшей мере на 99,5%, или кристаллическими по меньшей мере на 99,9%, например кристаллическими на 100%.
В другом варианте осуществления лактат (в частности, L-лактат) или цитрат выбраны из группы, состоящей из лактата (в частности, L-лактата) или цитрата, которые являются кристаллическими на 50-100%, например кристаллическими по меньшей мере на 50%, кристаллическими по меньшей мере на 60%, кристаллическими по меньшей мере на 70%, кристаллическими по меньшей мере на 80%, кристаллическими по меньшей мере на 90%, кристаллическими по меньшей мере на 95%, кристаллическими по меньшей мере на 98%, кристаллическими по меньшей мере на 99%, кристаллическими по меньшей мере на 99,5% и кристаллическими по меньшей мере на 99,9%, например кристаллическими на 100%.
Более предпочтительно, лактат (в частности, L-лактат) или цитрат могут быть такими солями (или быть выбраны из группы, состоящей из таких солей), которые являются кристаллическими на 95-100%, например кристаллическими по меньшей мере на 98%, или кристаллическими по меньшей мере на 99%, или кристаллическими по меньшей мере на 99,5%, или кристаллическими по меньшей мере на 99,6%, или кристаллическими по меньшей мере на 99,7%, или кристаллическими по меньшей мере на 99,8%, или кристаллическими по меньшей мере на 99,9%, например кристаллическими на 100%.
Одним примером в основном кристаллической соли является кристаллическая соль, образованная L-молочной кислотой.
Другим примером в основном кристаллической соли является кристаллическая соль, образованная лимонной кислотой.
Соли по настоящему изобретению в твердом состоянии могут быть сольватированными (например, гидратированными) или несольватированными (например, безводными).
В одном из вариантов осуществления соли являются несольватированными (например, безводными).
Другим примером несольватированной соли является кристаллическая соль, образованная молочной кислотой (в частности, L-молочной кислотой), как определено в настоящей заявке.
Термин «безводный» в настоящей заявке не исключает возможности наличия некоторого количества воды на или внутри соли (например, кристаллов соли). Например, вода может присутствовать в некотором количестве на поверхности соли (например, кристаллов соли), или незначительные количества воды могут иметься в объеме соли (например, кристаллов). Как правило, безводная форма содержит менее чем 0,4 молекулы воды на молекулу соединения, более предпочтительно содержит менее чем 0,1 молекулу воды на молекулу соединения, например 0 молекул воды.
В другом варианте осуществления лактат (в частности, L-лактат) или цитрат являются сольватированными. Если соли гидратированы, они могут содержать, например, до трех молекул кристаллизационной воды, более обычно до двух молекул воды, например одну молекулу воды или две молекулы воды. Также могут образовываться нестехиометрические гидраты, в которых имеющееся количество молекул воды менее одной или, в другом случае, не является целым. Например, в случае наличия менее одной молекулы воды может присутствовать, например, 0,4 или 0,5, или 0,6, или 0,7, или 0,8, или 0,9 молекул воды на молекулу соединения.
Другие сольваты включают алкоголяты, такие как этаноляты и изопропаноляты.
В одном из вариантов осуществления соль молочной кислоты (в частности, соль L-молочной кислоты) сольватирована, например, водой и/или этанолом.
L-лактаты или цитраты могут быть получены способами, описанными в предыдущих разделах настоящей заявки и в любом месте настоящей заявки.
Преимущества L-лактатов и цитратов включают общие преимущества, изложенные выше в предыдущих разделах настоящей заявки. Однако кристаллический лактат по настоящему изобретению обладает особыми преимуществами, в том, что он:
- негигроскопичен;
- является безводным и не образует гидратов;
- существует в одной кристаллической форме и, как считается, не проявляет полиморфизма;
- является кристаллическим;
- устойчив при хранении;
- обладает четко выраженной температурой плавления и не демонстрирует изменений форм, при анализе с помощью DSC (дифференциальная сканирующая калориметрия);
- обладает хорошей растворимостью в воде; и
- демонстрирует лучшую растворимость в буферных системах.
Таким образом, L-лактат существует в устойчивой кристаллической форме, которая не образует гидратов, и не подвергается изменениям формы при типовых операциях, переработке и условиях хранения.
Термин «стабильный» («устойчивый») или «стабильность» («устойчивость») при использовании в настоящем описании включает химическую стабильность и (физическую) стабильность твердого состояния. Термин «химическая стабильность» означает, что соединение может храниться в чистом виде или в виде состава, в котором оно включено в смесь, например, с фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами, описанными в настоящей заявке, в нормальных условиях хранения, с незначительным или отсутствующим химическим распадом или разложением. «Стабильность твердого состояния» означает, что соединение может храниться в виде выделенной твердой формы или в виде твердого состава, в котором оно включено в смесь, например, с фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами, описанными в настоящей заявке, в нормальных условиях хранения, с незначительным или отсутствующим изменением твердого состояния (например, гидратацией, дегидратацией, сольватацией, десольватацией, кристаллизацией, рекристаллизацией или фазовым переходом в твердом состоянии).
L-лактат и цитрат, а также их смеси имеют хорошую растворимость в воде и, следовательно, могут применяться для приготовления водных растворов, содержащих относительно высокие концентрации солей. Соответственно в другом варианте осуществления разработан водный раствор (например, в форме фармацевтической композиции), содержащий L-лактат или цитрат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, или их смесь в концентрации, превышающей 1 мг/мл, типично, превышающей 5 мг/мл жидкого носителя (например, воды или буферной системы), более типично превышающей 15 мг/мл, более типично превышающей 20 мг/мл и предпочтительно превышающей 25 мг/мл. В данном варианте осуществления водные растворы (например, в форме фармацевтической композиции), содержащие (i) L-лактат или (ii) смеси L-лактата и цитрата, являются особенно предпочтительными.
Водные растворы L-лактата или цитрата, или их смесей могут быть получены в виде водных растворов, имеющих значение pH в диапазоне от 2 до 6, например от 2 до 5, и более конкретно от 4 до 6, как, например, от 4 до 5.
Водные растворы L-лактата или цитрата, или их смесей могут быть забуференными или незабуференными, но, в одном из вариантов осуществления они являются забуференными, например, до значений pH в указанном выше диапазоне.
Предпочтительными буферами являются те, которые способны буферировать раствор до значения pH приблизительно 4,5 и которые не являются летучими в условиях, применяемых для лиофилизации раствора.
В ситуации, когда соль образована L-молочной кислотой, предпочтительным буфером является буфер, полученный из лимонной кислоты и доведенный до нужного значения pH с помощью NaOH или HCl, например, до значения pH раствора приблизительно 4,5. При этом значении pH и в цитратном буфере свободное основание имеет растворимость примерно 80 мг/мл.
Водные растворы L-лактата или цитрата, или их смесей будут содержать 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину в протонированной форме совместно с L-лактатными и/или цитратными противоионами. Кроме них могут присутствовать другие противоионы, и они могут быть образованы, например, реагентами, регулирующими тоничность, такими как солевой раствор (т.е. противоионы хлора) и/или буферными реагентами, как, например, цитратными буферами. Например, если L-лактат смешан в водном растворе с цитратным буфером, будут присутствовать противоионы как L-молочной, так и лимонной кислоты, причем природа цитратного противоиона зависит от pH раствора. Кроме того, водные растворы L-лактата или цитрата или их смесей могут содержать один или несколько других наполнителей, как правило, имеющихся в составах для внутривенного введения, как, например, реагентов для регулирования тоничности, примеры которых подробно описаны в фармакопее Соединенных Штатов и национальном фармакологическом справочнике и включают гексозы, такие как глюкозу, например, декстрозу (D-глюкозу).
Следовательно, в другом варианте осуществления изобретение относится к водному раствору 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в протонированной форме, содержащему один или несколько противоионов, выбранных из L-лактата, цитрата, и их смесей; и, необязательно, (i) одного или нескольких других противоионов, как, например, хлорид-ионов и/или (ii) одного или нескольких наполнителей для внутривенных составов, как, например, средств, регулирующих тоничность (например, гексоз, таких как глюкоза, предпочтительно, D-глюкоза).
Водные растворы, в числе прочих способов, могут быть получены растворением лактата в растворе цитрат-ионов (например, цитратном буфере) или растворением цитрата в растворе лактат-ионов. Лактат- и цитрат-ионы могут присутствовать в растворе в соотношении лактат:цитрат от 10:1 или менее, например от 10:1 до 1:10, более предпочтительно менее чем 8:1 или менее чем 7:1, или менее чем 6:1, или менее чем 5:1, или менее чем 4:1, или менее чем 3:1, или менее чем 2:1, или менее чем 1:1, более конкретно от 1:1 до 1:10. В одном из вариантов осуществления лактат- и цитрат-ионы присутствуют в растворе в соотношении лактат:цитрат от 1:1 до 1:10, например, от 1:1 до 1:8, или 1:1 до 1:7, или 1:1 до 1:6, или 1:1 до 1:5, например приблизительно 1:4,4.
Каждый из водных растворов, описанных в данном разделе заявки, а также в любом месте заявки, может быть подвергнут лиофилизации для получения твердого состава, который при необходимости может быть легко восстановлен с образованием водного раствора (предпочтительно, стерильного раствора) добавлением воды (предпочтительно, стерильной воды) или водной среды, содержащей наполнитель для внутривенного введения, такой как солевой раствор и/или декстрозу.
Соответственно настоящее изобретение также относится к лиофилизованному составу (например, в форме фармацевтической композиции), включающему L-лактат или цитрат, или их смеси, как определено в настоящей заявке, например, где состав при растворении в воде имеет pH от 2 до 6, например, от 2 до 5 и более конкретно от 4 до 6, как, например от 4 до 5.
В другом варианте осуществления изобретение относится к лиофилизованному составу (например, в форме фармацевтической композиции), включающему 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину в протонированной форме, совместно с одним или несколькими противоионами, выбранными из L-лактата, цитрата и их смесей; и необязательно (i) один или несколько других противоионов, таких как хлорид-ионы и/или (ii) один или несколько наполнителей для внутривенного введения, таких как средства регулирования тоничности (например, гексозы, такие как глюкоза, предпочтительно D-глюкоза).
Соотношение L-лактат-ионов к цитрат-ионам в каждом из лиофилизованных составов может находиться в пределах, установленных выше для ионных растворов.
Кристаллические структуры 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и ее солей
Как указано выше, лактат (в частности, L-лактат) или цитрат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины могут быть аморфными или в основном кристаллическими. В одном отдельном варианте осуществления лактат (в частности, L-лактат) или цитрат являются по существу кристаллическими, причем термин «по существу кристаллический» имеет значение, определенное выше. В частности, лактат (в т.ч. L-лактат) 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины является по существу кристаллическим.
1-Циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина в форме свободного основания также может быть аморфной или по существу кристаллической. В одном отдельном варианте осуществления свободное основание является по существу кристаллическим, причем термин «по существу кристаллический» имеет значение, определенное выше. В одном из вариантов осуществления свободное основание 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины существует в форме кристаллического дигидрата.
Кристаллы, описанные в настоящей заявке, и структуры кристаллов образуют еще один аспект настоящего изобретения.
Как отмечено выше, считается, что лактат по настоящему изобретению существует в единственной кристаллической форме, которая имеет характеристики, изложенные в данном описании. Эта кристаллическая форма представляет собой предпочтительный вариант осуществления изобретения. Однако в случае, если другие кристаллические формы действительно существуют, они не исключаются из объема настоящего изобретения.
Таким образом, если лактат (в частности, L-лактат) 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины является по существу кристаллическим, может преобладать одна единственная форма (например, кристаллическая форма, определенная и охарактеризованная в настоящем описании), хотя другие кристаллические формы могут присутствовать в незначительных и, предпочтительно, пренебрежимо малых количествах.
Кристаллические формы (например, кристаллические формы, определенные и охарактеризованные в настоящем описании) 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины (или ее солей) содержат другие кристаллические формы 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины (или ее солей) в количестве, меньшем или равном примерно 5% по массе, в частности содержание других кристаллических форм (или их солей) меньше или равно примерно 1% по массе.
В предпочтительном варианте осуществления изобретение относится к в основном кристаллической соли (например, лактату, такому как L-лактат, как описано в настоящей заявке) 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, содержащей единственную кристаллическую форму (например, кристаллическую форму, определенную и охарактеризованную в настоящем описании) соли и не более 5% по массе других кристаллических форм соли.
Предпочтительно наряду с единственной кристаллической формой (например, кристаллической формой, определенной и охарактеризованной в настоящем описании) присутствует менее 4% или менее 3%, или менее 2% других кристаллических форм и, в частности, количество других кристаллических форм меньше или равно примерно 1% по массе. Более предпочтительно единственной кристаллической форме (например, кристаллической форме, определенной и охарактеризованной в настоящем описании) сопутствует менее 0,9% или менее 0,8%, или менее 0,6%, или менее 0,5%, или менее 0,4%, или менее 0,3%, или менее 0,2%, или менее 0,1%, или менее 0,05%, или менее 0,01% по массе других кристаллических форм, например 0% по массе других кристаллических форм.
Кристаллы и их кристаллическая структура могут быть охарактеризованы с применением ряда методик, включая рентгеноструктурный анализ монокристаллов, дифракцию рентгеновских лучей на порошке (XRPD), дифференциальную сканирующую калориметрию (DSC) и инфракрасную спектроскопию, например инфракрасную спектроскопию с преобразованием Фурье (FTIR). Поведение кристаллов в условиях изменяющейся влажности может быть проанализировано гравиметрическими исследованиями по поглощению паров воды и, кроме того, XRPD.
Определение кристаллической структуры соединения может быть осуществлено рентгеноструктурным анализом, который может проводиться согласно стандартным методикам, как, например, описанным в настоящей заявке, а также описанным в Fundamentals of Crystallography, C. Giacovazzo, H.L. Monaco, D. Viterbo, F. Scordari, G. Gilli, G. Zanotti and M. Catti, (International Union of Crystallography/Oxford University Press, 1992 ISBN 0-19-855578-4(p/b), 0-19-85579-2(h/b)). Эти методики включают анализ и интерпретацию дифракции рентгеновских лучей на монокристалле.
Кристаллические структуры дигидрата свободного основания и L-лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины были определены рентгеноструктурным анализом (см. ниже по тексту примеры 69 и 71, соответственно).
В таблицах 2 и 4 примеров 69 и 71, соответственно, приведены данные о координатах атомов для кристаллов 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и ее L-лактата в формате Crystallographic Information File (CIF) (см. Hall, Allen and Brown, Acta Cryst. (1991). A47, 655-685; http://www.iucr.ac.uk/iucr-top/cif/home.html). Другими специалистами в данной области техники могут применяться или предпочитаться альтернативные форматы файлов, как, например, формат PDB (например, формат, совместимый с форматом EBI Macromolecular Structure Database (Hinxton, UK)). Однако должно быть ясно, что применение к настоящим данным другого формата файла или преобразования координат в таблицах попадают в объем настоящего изобретения. В таблицах числа в скобках представляют собой отклонение (s.u., стандартная неопределенность). Кристаллическая структура лактата показана на фиг. 4 и 5.
В одном из вариантов осуществления изобретение относится к дигидрату свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который является кристаллическим и (i) имеет кристаллическую структуру, определенную координатами в таблице 2 настоящей заявки; и/или (ii) кристаллы которого принадлежат к моноклинной пространственной группе P2l/n (#14) с параметрами кристаллической решетки a=7,66(10), b=15,18(10), c=17,71(10)Е, β=98,53(2)°, α=γ=90°.
В другом варианте осуществления изобретение относится к L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который является кристаллическим и имеет кристаллическую структуру, определенную координатами в таблице 4 настоящего описания.
В другом варианте осуществления изобретение относится к L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который является кристаллическим и имеет кристаллическую структуру, приведенную на фиг.4 и 5.
В другом варианте осуществления изобретение относится к L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который является кристаллическим и имеет кристаллическую структуру, принадлежащую к орторомбической пространственной группе P2l2l2l(#19) и имеет параметры кристаллической решетки при 97(2)K a=9,94(10), b=15,03(10), c=16,18(10)Е, α=β=γ=90°.
В другом варианте осуществления изобретение относится к L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который является кристаллическим и имеет параметры кристаллической решетки при комнатной температуре a=10,08(10), b=15,22(10), c=16,22(10)Е, α=β=γ=90°.
Согласно еще одному варианту осуществления настоящее изобретение относится к L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который является кристаллическим и:
(a) имеет кристаллическую структуру, показанную на фиг. 4 и 5; и/или
(b) имеет кристаллическую структуру, определяемую координатами в таблице 4 настоящего описания; и/или
(c) имеет параметры кристаллической решетки при 97(2)K a=9,94(10), b=15,03(10), c=16,18(10)Е, α=β=γ=90°; и/или
(d) имеет параметры кристаллической решетки при комнатной температуре a=10,08(10), b=15,22(10), c=16,22(10)Е, α=β=γ=90°; и/или
(e) имеет кристаллическую структуру, принадлежащую к орторомбической пространственной группе P2l2l2l(#19).
С другой стороны, кристаллическая структура соединения может быть проанализирована с помощью применимой для твердого вещества методики дифракции рентгеновских лучей на порошке (XRPD). XRPD может проводиться согласно общепринятым способам, как, например, описанным в настоящей заявке (см. примеры 70 и 72), а также описанным в Introduction to X-Ray Powder Diffraction, Ron Jenkins and Robert L. Snyder (John Wiley & Sons, New York, 1996). Наличие выраженных пиков (в противоположность случайному фоновому шуму) на дифрактограмме показывает, что соединение обладает определенной степенью кристалличности.
Порошковая рентгенограмма соединения характеризуется такими параметрами рентгеновского дифракционного спектра, как угол дифракции (2θ) и межплоскостные расстояния (d). Эти параметры связаны уравнением Брэгга nλ=2dsin θ (где n=1; λ=длина волны применяемого катода, d=межплоскостное расстояние; и θ=дифракционный угол). В данном способе межплоскостные расстояния, дифракционные углы и общий вид картины являются важными для идентификации кристалла по порошковой рентгенограмме, благодаря характерности этих данных. Относительная интенсивность не должна подвергаться точной интерпретации, т.к. она может изменяться в зависимости от направления роста кристаллов, размера частиц и условий регистрации спектра. Кроме того, дифракционные углы обычно означают углы, которые совпадают в диапазоне 2θ±0,2°. Пики означают основные пики и включают пики, не превышающие средних при дифракционных углах, отличных от углов, указанных выше.
Как L-лактат, так и форма свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины были охарактеризованы XRPD. В каждом случае порошковая рентгенограмма выражена в величинах дифракционных углов (2θ), межплоскостных расстояний (d) и/или относительных интенсивностей. В таблицах 3, 5 и 6 в примерах 70 и 72 показаны величины межплоскостных расстояний (d) спектра дифракции рентгеновских лучей, которые соответствуют величинам дифракционных углов в свободном основании, L-лактате и дигидрате свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины.
Следовательно, 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина и ее соли имеют картины дифракции рентгеновских лучей, в основных чертах показанные на фиг.3, 6, 7 или 8 и/или в таблицах 3, 5 или 6 в примерах 70 и 72.
В соответствии с одним из вариантов осуществления настоящее изобретение относится к кристаллам 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания и его L-лактата, демонстрирующих порошковую рентгенограмму, включающую пики при тех же самых дифракционных углах, что и пики на порошковой рентгенограмме, показанной на фиг. 3, 6, 7 или 8 и/или в таблице 3, и/или таблице 5, и/или таблице 6, причем эти пики необязательно имеют ту же относительную интенсивность. Более конкретно кристаллы указанных солей являются такими, что они имеют порошковые рентгенограммы, в основном сходные с показанными на фиг. 3, 6, 7 или 8.
В предпочтительном варианте осуществления изобретение относится к кристаллам соли 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и L-молочной кислоты, порошковая рентгенограмма которых в основном соответствует показанной на фиг.6.
В другом варианте осуществления изобретение относится к в основном кристаллическому L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который демонстрирует пики, соответствующие тем же самым дифракционным углам, что и в порошковой рентгенограмме, показанной на фиг.6. Предпочтительно пики имеют ту же относительную интенсивность, что и пики на фиг.6.
Изобретение также относится к по существу кристаллической соли L-молочной кислоты и 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, порошковая рентгенограмма которой в основном соответствует показанной на фиг.6.
Порошковая рентгенограмма соли L-молочной кислоты может быть охарактеризована наличием пиков, соответствующих дифракционным углам (2θ) и межплоскостным расстояниям (d) и, предпочтительно, интенсивностями, показанными в таблице 5 примера 72.
Следовательно, изобретение относится к кристаллам L-лактата циклопропил-3-[3-(6-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, которые демонстрируют порошковую рентгенограмму, имеющую характеристические пики, соответствующие дифракционным углам (2θ±1,0°, как, например, ±0,2°, в частности ±0,1°) из таблицы 5 в примере 72.
Кроме того, изобретение относится к кристаллам L-лактата циклопропил-3-[3-(6-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, дифракция рентгеновских лучей на порошке которых демонстрирует основные пики, соответствующие дифракционным углам 2θ, равным 17,50, 18,30, 19,30, 19,60 и 21,85±1,0°, как, например, ±0,2°, в частности ±0,1°. Эти кристаллы могут быть дополнительно охарактеризованы пиками в картине дифракции рентгеновских лучей при углах 2θ 12,40, 15,20, 15,60, 17,50, 18,30, 18,50, 19,30, 19,60, 21,85 и 27,30±1,0°.
Кристаллы L-лактата циклопропил-3-[3-(6-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины также отличаются тем, что характеристическая порошковая рентгенограмма представлена расстояниями между плоскостями кристаллической решетки d(Е), указанными в таблице 5 в примере 72.
В другом варианте осуществления изобретение относится к кристаллам лактата циклопропил-3-[3-(6-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, порошковая рентгенограмма которых включает характеристические пики, появляющиеся при расстояниях в кристаллической решетке (d) 5,06, 4,85, 4,60, 4,53 и 4,07, и более конкретно включает дополнительные характеристические пики, появляющиеся при расстояниях в кристаллической решетке (d) 7,13, 5,83, 5,68, 5,06, 4,85, 4,79, 4,60, 4,53, 4,07 и 3,26 ангстрем.
В другом варианте осуществления изобретение относится к по существу кристаллическому L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, порошковая рентгенограмма которого характеризуется наличием основных пиков, соответствующих дифракционным углам (2θ) 17,50, 18,30, 19,30, 19,60 и 21,85°, более конкретно 12,40, 15,20, 15,60, 17,50, 18,30, 18,50, 19,30, 19,60, 21,85 и 27,30° и межплоскостными расстояниями (d), равными 5,06, 4,85, 4,60, 4,53 и 4,07, более конкретно 7,13, 5,83, 5,68, 5,06, 4,85, 4,79, 4,60, 4,53, 4,07 и 3,26 ангстрем.
В другом варианте осуществления изобретение относится к по существу кристаллическому L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, порошковая рентгенограмма которого характеризуется наличием пиков, соответствующих дифракционным углам (2θ), межплоскостных расстояний (d) и, предпочтительно, интенсивностей, показанных в таблице 5 в примере 72.
Кроме того, изобретение относится к кристаллам 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания, порошковая рентгенограмма которых включает характеристические пики, соответствующие дифракционным углам (2θ±1,0°, как, например, ±0,2°, в частности ±0,1°) из таблицы 2.
В другом варианте осуществления изобретение относится к кристаллам 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания, в порошковой рентгенограмме которых присутствуют характеристические пики, появляющиеся при межплоскостных расстояниях (d) из таблицы 2.
Кристаллические соли по настоящему изобретению также могут быть охарактеризованы дифференциальной сканирующей калориметрией (DSC).
L-лактат был проанализирован способом DSC и проявил пик (температура плавления и разложения) при 190°C.
Соответственно, в другом аспекте изобретение относится к L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который является безводным и демонстрирует эндотермический пик при 190°C при исследовании способом DSC.
Другим аспектом настоящего изобретения является L-лактат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который демонстрирует пики, соответствующие тем же дифракционным углам, которые присутствуют на картине дифракции рентгеновских лучей, показанной на фиг. 6, 7 или 8, и, кроме того, проявляет эндотермический пик, сопровождающий разложение, вблизи 190°C согласно термическому анализу (DSC).
Форма свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины демонстрирует пики при тех же самых дифракционных углах, что и пики в порошковой рентгенограмме, показанной на фиг.3 и/или в таблице 2 и, кроме того, проявляет экзотермический пик, сопровождающий разложение, вблизи 193°C согласно термическому анализу (DSC).
Поведение солей по настоящему изобретению в условиях высокой влажности может быть исследовано стандартными способами гравиметрического исследования поглощения паров (GVS), например, как описано в разделе E примера 68.
L-лактат может существовать в устойчивой безводной кристаллической форме в условиях высокой относительной влажности, и в таких условиях кристаллическая структура не подвергается изменениям.
Соли 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины могут быть, кроме того, охарактеризованы инфракрасной спектроскопией, например FTIR. Инфракрасный спектр L-лактата (способ с таблеткой KBr) включает характеристические пики при 3229, 2972 и 1660 см-1.
Соответственно в другом варианте осуществления изобретение относится к (предпочтительно по существу кристаллическому) L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который демонстрирует инфракрасный спектр, при использовании способа с таблеткой KBr, включающий характеристические пики при 3229, 2972 и 1660 см-1.
Как должно быть ясно из предыдущих параграфов, L-лактаты по настоящему изобретению могут быть охарактеризованы рядом различных физико-химических параметров. Соответственно в предпочтительном варианте осуществления настоящее изобретение относится к L-лактату 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, который является кристаллическим и характеризуется одним или несколькими (в любом сочетании), или всеми параметрами из числа следующих, а именно что соль:
(a) имеет кристаллическую структуру, показанную на фиг. 4 и 5; и/или
(b) имеет кристаллическую структуру, определяемую координатами в таблице 4 примера 71 настоящего описания; и/или
(c) имеет параметры кристаллической решетки при 97(2)K a=9,94(10), b=15,03(10), c=16,18(10)Е, α=β=γ=90°; и/или
(d) имеет параметры кристаллической решетки при комнатной температуре a=10,08(10), b=15,22(10), c=16,22(10)Е, α=β=γ=90°; и/или
(e) имеет кристаллическую структуру, которая принадлежит к орторомбической пространственной группе P2l2l2l(#19); и/или
(f) имеет порошковую рентгенограмму, характеризующуюся наличием основных пиков, соответствующих дифракционным углам (2θ) 17,50, 18,30, 19,30, 19,60 и 21,85°, и более конкретно дополнительно при 12,40, 15,20, 15,60, 17,50, 18,30, 18,50, 19,30, 19,60, 21,85 и 27,30° и/или межплоскостными расстояниями (d), равными 5,06, 4,85, 4,60, 4,53 и 4,07, и более конкретно дополнительно 7,13, 5,83, 5,68, 5,06, 4,85, 4,79, 4,60, 4,53, 4,07 и 3,26 ангстрем; и/или
(g) демонстрирует пики, соответствующие там же дифракционным углам, которые имеются в картине дифракции рентгеновских лучей, показанной на фиг.6 или в таблице 5 примера 72 и, необязательно, где пики имеют такую же относительную интенсивность, как пики на фиг.6 или в таблице 5; и/или
(h) имеет порошковую рентгенограмму, по существу соответствующую показанной на фиг.6; и/или
(i) является безводной и демонстрирует эндотермический пик при 190°C при исследовании способом DSC; и/или
(j) при исследовании с применением методики с таблеткой KBr, имеет инфракрасный спектр, который содержит характеристические пики при 3229, 2972 и 1660 см-1.
Соединения подгруппы (C) формулы (I)
В одной из подгрупп соединений формулы (I) (т.е. в подгруппе (C) формулы (I)), M представляет собой группу D1; X представляет собой O; A является группой NR2, где R2 является водородом; E представляет собой связь; R1 является 2,6-дифторфенилом; и соединение представляет собой кислотно-аддитивную соль, образованную избранной группой кислот.
Соответственно в одном из вариантов осуществления изобретение относится к кислотно-аддитивной соли 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, которая является солью, образованной кислотой, выбранной из группы, состоящей из уксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), аспарагиновой (например, L-аспарагиновой), бензолсульфоновой, бензойной, камфорной (например,
(+)камфорной), каприновой, каприловой, угольной, лимонной, цикламовой, додекановой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, фумаровой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, хлористоводородной, изетионовой, изомасляной, молочной (например, (+)-L-молочной и (±)-DL-молочной), лактобионовой, лаурилсульфоновой, малеиновой, яблочной, (-)-L-яблочной, малоновой, метансульфоновой, слизевой, нафталинсульфоновой (например, нафталин-2-сульфоновой), нафталин-1,5-дисульфоновой, никотиновой, олеиновой, оротовой, щавелевой, пальмитиновой, памовой, фосфорной, пропионовой, себациновой, стеариновой, янтарной, серной, винной (например, (+)-L-винной), тиоциановой, толуолсульфоновой (например, п-толуолсульфоновой), валериановой и ксинафовой кислот.
В одном из вариантов осуществления кислотно-аддитивная соль образована кислотой, выбранной из группы, состоящей из адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой), аспарагиновой (например, L-аспарагиновой), бензойной, камфорной (например, (+)камфорной), каприновой, каприловой, угольной, цикламовой, додекановой, додецилсерной, этан-1,2-дисульфоновой, галактаровой, гентизиновой, глюкогептоновой, D-глюконовой, глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, изомасляной, лаурилсульфоновой, слизевой, нафталин-1,5-дисульфоновой, никотиновой, олеиновой, оротовой, щавелевой, пальмитиновой, памовой, себациновой, стеариновой, винной (например, (+)-L-винной), тиоциановой и ксинафовой кислот.
В другом варианте осуществления кислотно-аддитивная соль образована кислотой, выбранной из группы, состоящей из уксусной, адипиновой, аскорбиновой, аспарагиновой, лимонной, DL-молочной, фумаровой, глюконовой, глюкуроновой, гиппуровой, хлористоводородной, глутаминовой, DL-яблочной, п-толуолсульфоновой, метансульфоновой (соль-мезилат), этансульфоновой (соль-эзилат), себациновой, стеариновой, янтарной и винной кислот.
В другом варианте осуществления кислотно-аддитивная соль образована кислотой, выбранной из группы, состоящей из адипиновой, аскорбиновой, аспарагиновой, глюконовой, гиппуровой, глутаминовой, себациновой, стеариновой и винной кислот.
В другом отдельном варианте осуществления соединение является кислотно-аддитивной солью, образованной хлористоводородной кислотой.
Предпочтительными солями являются соли, имеющие растворимость в данном жидком носителе (например, воде), превышающую 25 мг/мл жидкого носителя (например, воды), более типично, превышающую 50 мг/мл и предпочтительно превышающую 100 мг/мл. Такие соли являются особенно предпочтительными для введения в жидкой форме, например, инъекцией или инфузией.
В другом аспекте настоящего изобретения разработана композиция (например, фармацевтическая композиция), включающая водный раствор, содержащий описанную выше соль, в концентрации, превышающей 25 мг/мл, типично превышающей 50 мг/мл и предпочтительно превышающей 100 мг/мл.
Соли по настоящему изобретению, которые обладают растворимостью, превышающей 25 мг/мл, включают D-глюкуронат, мезилат, эзилат и DL-лактат, причем три последние обладают растворимостью более 100 мг/мл.
Соответственно в одном конкретном варианте осуществления разработан мезилат 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины.
В другом конкретном варианте осуществления разработан эзилат (этансульфонат) 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины.
В еще одном конкретном варианте осуществления разработан DL-лактат 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины. В одном из вариантов осуществления лактат представляет собой L-лактат.
Свободное основание или исходное соединение, из которого получены указанные соединения (т.е. кислотно-аддитивные соли) подгруппы (C) формулы (I), имеет формулу (IA):
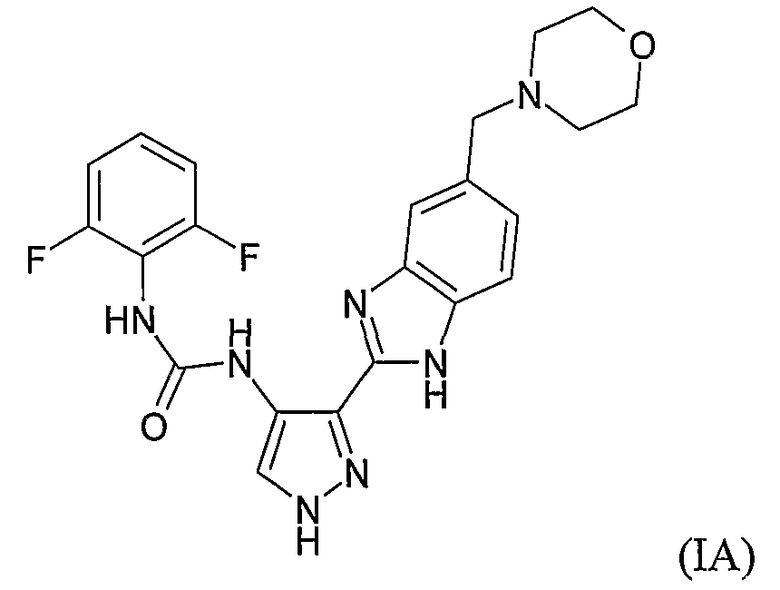
Соли соединения (IA) могут быть аморфными или кристаллическими.
В одном из вариантов осуществления соль имеет аморфную форму.
В другом варианте осуществления соединение имеет кристаллическую форму.
Соединение может быть несольватированным (например, безводным) или сольватированным.
В одном из вариантов осуществления соли несольватированы.
В другом варианте осуществления соли сольватированы, например гидратированы.
Если соединения являются гидратированными, они могут содержать, например, до трех молекул кристаллизационной воды, чаще до двух молекул воды, например одну молекулу воды или две молекулы воды.
Соли по настоящему изобретению имеют преимущества над формой свободного основания, т.е. 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевиной.
Например, соли обладают более высокой растворимостью в воде и, следовательно, лучше подходят для применения в приготовлении парентеральных составов для инъекции или инфузии (например, внутривенной инфузии). Соли по настоящему изобретению, кроме того, обладают одним или несколькими другими преимуществами, выбранными из:
- лучшей фармакокинетики;
- более высокой стабильности, например, увеличенного срока хранения;
- более низкой основности, что делает их более пригодными для внутривенного применения;
- преимуществ для изготовления;
- лучших метаболических свойств; и
- меньших различий в клинической эффективности от пациента к пациенту.
Соли могут быть получены любым из способов, изложенных в предыдущих разделах настоящей заявки, описывающих соли соединений подгрупп (A) и (B) формулы (I).
Соединения подгруппы (C) формулы (I), как правило, являются фармацевтически приемлемыми солями. Однако соли, которые не являются фармацевтически приемлемыми, также могут быть получены в качестве промежуточных форм, которые затем могут быть превращены в фармацевтически приемлемые соли. Такие формы фармацевтически неприемлемых солей, которые могут быть полезны, например, при очистке или разделении соединений по настоящему изобретению, также входят в объем изобретения.
Соединения подгруппы (C) формулы (I) могут существовать в виде ряда различных таутомерных форм, и упоминания соединений по настоящему изобретению включают все такие формы. Во избежание сомнений, если соединение может существовать в одной из нескольких геометрически изомерных или таутомерных форм, и конкретно описана или показана только одна из них, все остальные, тем не менее, также охвачены настоящей заявкой.
Например, в соединениях по настоящему изобретению бензимидазольная группа может принимать одну из двух таутомерных форм A” и B”, показанных выше.
Пиразольный цикл также может проявлять таутомерию и может существовать в двух таутомерных формах C”' и D”', показанных ниже.
Соединения по настоящему изобретению включают соединения с одним или несколькими изотопными замещениями, и упоминания того или иного конкретного элемента включают в себя все изотопы этого элемента. Например, упоминание водорода включает в себя 1H, 2H(D) и 3H(T). Подобным же образом упоминания углерода и кислорода включают в себя соответственно 12C, 13C и 14C, и 16O и 18O.
Эти изотопы могут быть радиоактивными или нерадиоактивными. В одном из вариантов осуществления настоящего изобретения соединения не содержат радиоактивных изотопов. Такие соединения являются предпочтительными для терапевтического применения. Однако в другом варианте осуществления соединения могут содержать один или несколько радиоактивных изотопов. Соединения, содержащие такие радиоактивные изотопы, могут быть полезны в диагностических применениях.
Кроме того, настоящим изобретением охвачены любые полиморфные формы соединений, а также комплексы (например, комплексы включения или клатраты с такими веществами, как циклодекстрины, или комплексы с металлами) соединений и пролекарства соединений. Под «пролекарствами» подразумеваются, например, любые соединения, которые превращаются in vivo в биологически активные соединения по настоящему изобретению.
Биологическая активность
Соединения по настоящему изобретению обладают ингибирующей или модулирующей активностью в отношении циклин-зависимой киназы и ингибирующей или модулирующей активностью в отношении киназы-3 гликоген-синтазы (GSK3), и/или ингибирующей или модулирующей активностью в отношении Aurora киназы, причем эта активность, как представляется, будет полезной в предотвращении или лечении состояний или заболеваний, опосредованных указанными киназами.
Так, например, представляется, что соединения по настоящему изобретению будут применимы для облегчения симптомов или сокращения частоты случаев рака.
Более конкретно соединения формулы (I) и ее подгрупп, являются ингибиторами циклин-зависимых киназ. Например, соединения по настоящему изобретению обладают активностью в отношении киназ CDK1, CDK2, CDK3, CDK4, CDK5, CDK6 и CDK7, и, в частности, циклин-зависимых киназ, выбранных из CDK1, CDK2, CDK3, CDK4, CDK5 и CDK6.
Предпочтительными соединениями являются соединения, которые ингибируют одну или несколько CDK киназ, выбранных из CDK1, CDK2, CDK3, CDK4 и CDK5, например, CDK1 и/или CDK2.
Кроме того, интерес могут представлять CDK4, CDK8 и/или CDK9.
Лактаты или цитраты 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины обладают активностью в отношении CDK2, CDK4, CDK5, CDK6 и CDK9 киназ, и, в частности, CDK2.
Соединения по настоящему изобретению также обладают активностью в отношении киназы-3 гликоген-синтазы (GSK-3).
Кроме того, соединения по настоящему изобретению обладают активностью против Aurora киназ. Предпочтительными соединениями по настоящему изобретению являются соединения, имеющие величины IC50, меньшие, чем 0,1 мкМ.
В частности, лактаты или цитраты 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины являются ингибиторами Aurora киназ и, например, ингибируют Aurora A и/или Aurora B.
Многие из соединений по настоящему изобретению проявляют селективность в отношении Aurora A киназы по сравнению с CDK1 и CDK2, и такие соединения представляют собой один предпочтительный вариант осуществления настоящего изобретения. Например, многие соединения по настоящему изобретению имеют величины IC50 в отношении Aurora A, которые лежат в диапазоне от одной десятой до одной сотой от значения IC50, относящегося к CDK1 и CDK2.
Вследствие активности соединений по настоящему изобретению в модулировании или ингибировании CDK, Aurora киназ и киназ гликоген-синтазы они, как ожидается, применимы при создании средств для замедления клеточного цикла или восстановления контроля над ним в случае аномально делящихся клеток. Следовательно, ожидается, что эти соединения действительно будут применимы в лечении или предупреждении пролиферативных расстройств, таких как раковые заболевания. Кроме того, представляется, что соединения по настоящему изобретению будут применимы при лечении таких состояний, как вирусные инфекции, сахарный диабет типа II или инсулинонезависимый сахарный диабет, аутоиммунные заболевания, травмы головы, удар, эпилепсия, нейродегенеративные расстройства, как, например, болезнь Альцгеймера, заболевания двигательных нейронов, прогрессирующий супрануклеарный паралич, кортикобазальная дегенерация и болезнь Пика, например, при лечении аутоиммунных заболеваний и нейродегенеративных заболеваний.
Одна подгруппа заболеваний и болезненных состояний, в отношении которой, как представляется, будут применимы соединения по настоящему изобретению, состоит из вирусных инфекций, аутоиммунных заболеваний и нейродегенеративных заболеваний.
CDK играют определенную роль в регулировании клеточного цикла, апоптозе, транскрипции, дифференцировке и деятельности ЦНС. Следовательно, ингибиторы CDK могли бы быть полезны в лечении заболеваний, при которых имеют место расстройства пролиферации, апоптоза или дифференцировки как, например, раковых заболеваний. В частности RB+ve опухоли могут быть особенно чувствительными к ингибиторам CDK. RB-ve опухоли также могут быть чувствительными к ингибиторам CDK.
Примеры раковых заболеваний, в отношении которых проявляется ингибирующее действие, включают, не ограничиваясь перечисленными, рак, например рак мочевого пузыря, груди, ободочной кишки (например, колоректальные виды рака, как, например, аденокарцинома ободочной кишки и аденома ободочной кишки), почек, эпидермиса, печени, легких, например аденокарциному, мелкоклеточный рак легких и немелкоклеточные раки легких, пищевода, желчного пузыря, яичника, поджелудочной железы, например экзокринный рак поджелудочной железы, желудка, шейки матки, щитовидной железы, простаты или кожи, например рак чешуйчатых клеток; гематопоэтические опухоли лимфоидного происхождения, например лейкемию, острый лимфоцитарный лейкоз, B-клеточную лимфому, T-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, лимфому ворсистых клеток или лимфому Беркетта; гематопоэтические опухоли миелоидного происхождения, например острую и хроническую миелогенную лейкемию, миелодиспластический синдром или промиелоцитарную лейкемию; фоликуллярный рак щитовидной железы; опухоли мезенхимального происхождения, например фибросаркому или габдомиосаркому; опухоли центральной или периферической нервной системы, например, астроцитому, нейробластому, глиому или шванному; меланому; семиному; тератокарциному; остеосаркому, пигментную ксеродерму; кератоксантому; фоликуллярный рак щитовидной железы; или саркому Капоши.
Раковые заболевания могут быть разновидностями рака, которые чувствительны к ингибированию любой или любых нескольких циклин-зависимых киназ, выбранных из CDK1, CDK2, CDK3, CDK4, CDK5 и CDK6, например одной или нескольких CDK киназ, выбранных из CDK1, CDK2, CDK4 и CDK5, например CDK1 и/или CDK2.
Является или нет конкретный случай рака заболеванием, чувствительным к ингибированию циклин-зависимой киназы или Aurora киназы, можно определить с помощью анализа роста клеток, как показано ниже в примерах 79 и 80, или способом, который описан в разделе под названием «способы диагностики».
Кроме того, известно, что CDK играют определенную роль в апоптозе, пролиферации, дифференцировке и транскрипции и, следовательно, ингибиторы CDK могли бы также быть применимы в лечении следующих заболеваний, помимо рака: вирусных инфекций, например вируса герпеса, вируса оспы, вируса Эпштейна-Барр, вируса Синдбис, аденовируса, ВИЧ, HPV (вируса папилломы), HCV (вируса гепатита C) и HCMV (цитомегаловируса); предотвращении развития СПИД у ВИЧ-инфицированных пациентов; хронических воспалительных заболеваний, например системной красной волчанки, аутоиммунно-опосредованного гломерулонефрита, ревматоидного артрита, псориаза, воспалительных заболеваний кишечника и аутоиммунного сахарного диабета; сердечно-сосудистых заболеваний, например гипертрофии сердца, рестеноза, атеросклероза; нейродегенеративных расстройств, например болезни Альцгеймера, связанной со СПИД деменции, болезни Паркинсона, бокового амиотрофического склероза, пигментного ретинита, спинной мышечной атрофии и церебральной дегенерации; гломерулонефрита; миелодиспластических синдромов, инфарктов миокарда, связанных с ишемическим поражением, ударов и поражений, связанных с реперфузией, аритмии, атеросклероза, заболеваний печени, вызванных токсинами или связанных с алкоголем, гематологических заболеваний, например хронической анемии и апластической анемии; дегенеративных заболеваний скелетно-мышечной системы, например остеопороза и артрита, аспирин-чувствительного риносинусита, кистозного фиброза, рассеянного склероза, заболеваний почек и раковой боли.
Кроме того, было обнаружено, что некоторые ингибиторы циклин-зависимой киназы могут применяться в сочетании с другими противораковыми реагентами. Например, такой ингибитор циклин-зависимой киназы, как флавопиридол применяли в комбинированной терапии с другими противораковыми средствами.
Таким образом, для фармацевтических композиций, применений или способов по настоящему изобретению с целью лечения заболевания или состояния, включающего аномальный рост клеток, этим заболеванием или состоянием, включающим аномальный рост клеток, в одном из вариантов осуществления является рак.
Одна группа раковых заболеваний включает рак груди человека (например, первичные опухоли груди, рак груди без поражения лимфатических узлов, инвазивные аденокарциномы протоков молочной железы, неэндометриоидные формы рака груди); и лимфомы клеток мантийной ткани. Кроме того, другими видами раковых заболеваний являются колоректальные и внутриматочные виды рака.
Другое подмножество раковых заболеваний включает рак груди, рак яичников, рак ободочной кишки, рак простаты, рак пищевода, рак чешуйчатых клеток и немелкоклеточные карциномы легких.
Для соединений, обладающих активностью против Aurora киназы, конкретные примеры раковых заболеваний, в случае которых, как предполагается, будут применимы соединения по настоящему изобретению, ингибирующие Aurora киназу, включают:
различные виды рака груди человека (например, первичные опухоли груди, рак груди без поражения лимфатических узлов, инвазивные аденокарциномы протоков молочной железы, неэндометриоидные формы рака груди);
различные виды рака яичников (например, первичные опухоли яичников);
различные виды рака поджелудочной железы;
различные виды рака мочевого пузыря человека;
различные виды колоректального рака (например, первичные колоректальные раковые заболевания);
опухоли желудка;
различные виды рака почек;
различные виды рака шейки матки;
нейробластомы;
меланомы;
лимфомы;
различные виды рака простаты;
лейкемия;
неэндометриоидные раковые заболевания матки;
глиомы; и
неходжкинская лимфома.
Раковые заболевания, которые могут особенно успешно поддаваться действию ингибиторов Aurora киназы, включают рак груди, мочевого пузыря, колоректальный рак, рак поджелудочной железы, яичников, неходжкинскую лимфому, глиомы и неэндометриоидные раковые заболевания матки.
Отдельное подмножество разновидностей рака, которые могут особенно хорошо поддаваться действию ингибиторов Aurora киназы, состоит из рака груди, яичников, ободочной кишки, печени, желудка и простаты.
Другое подмножество раковых заболеваний, для лечения которых могут особенно хорошо подходить ингибиторы Aurora киназы, состоит из гематологических раковых заболеваний, в частности лейкемии. Следовательно, в другом варианте осуществления соединения формулы (I) применяются для лечения гематологических раковых заболеваний, конкретно лейкемии. Конкретные разновидности лейкемии выбраны из острой миелогенной лейкемии (AML), хронической миелогенной лейкемии (CML), B-клеточной лимфомы (клеток мантийной ткани) и острой лимфобластной лейкемии (ALL) (с другой стороны известной, как острый лимфоцитарный лейкоз). В одном из вариантов осуществления разновидности лейкемии выбраны из повторной или рефрактерной острой миелогенной лейкемии, миелодиспластического синдрома и хронической миелогенной лейкемии.
Одна группа раковых заболеваний включает различные виды рака груди человека (например, первичные опухоли груди, рак груди без поражения лимфатических узлов, инвазивные аденокарциномы протоков молочной железы, неэндометриоидные формы рака груди); и лимфомы клеток мантийной ткани. Кроме того, другими видами рака является колоректальный рак и различные виды рака матки.
Другое подмножество раковых заболеваний включает гематопоэтические опухоли лимфоидного происхождения, например лейкемию, хроническую лимфоцитарную лейкемию, лимфому клеток мантийной ткани и B-клеточную лимфому (как, например, диффузную B-крупноклеточную лимфому).
Одним конкретным видом рака является хроническая лимфоцитарная лейкемия.
Другим конкретным видом рака является лимфома клеток мантийной ткани.
Еще одним конкретным видом рака является диффузная B-крупноклеточная лимфома.
Далее предусматривается, что соединения по настоящему изобретению и, в частности, те соединения, которые обладают ингибирующей активностью в отношении Aurora киназы, будут, в частности, применимы в лечении или предупреждении тех типов раковых заболеваний, которые связаны с, или характеризуются наличием повышенных уровней Aurora киназ, например раковых заболеваний, упомянутых в этом контексте во вводной части настоящей заявки.
Активность соединений по настоящему изобретению в качестве ингибиторов циклин-зависимых киназ, Aurora киназ и киназы-3 гликоген-синтазы может быть измерена с помощью аналитических методик, изложенных ниже в примерах, и уровень активности, демонстрируемый данным соединением, может быть выражен в величинах IC50. Предпочтительными соединениями по настоящему изобретению являются соединения, имеющие значения IC50 менее 1 мкМ, более предпочтительно менее 0,1 мкМ.
Преимущества соединений по настоящему изобретению
Соединения по настоящему изобретению (например, соединения примеров 24, 62, 63 и 64) обладают рядом преимуществ перед соединениями известного уровня техники. Например, соединения по настоящему изобретению (см. таблицу A) демонстрируют улучшенную селективность и эффективность, в частности, в отношении Aurora A и B киназ.
Соединения по настоящему изобретению также обладают преимуществами перед соединениями известного уровня техники в том, что они имеют другую чувствительность к ферментам P450 (см.таблицу B ниже и пример 81).
Кроме того, соединения по настоящему изобретению обладают преимуществами над соединениями известного уровня техники в том, что они демонстрируют улучшенные свойства, относящиеся к метаболизму лекарственных средств, и фармакокинетические свойства. В частности, соединения по настоящему изобретению обладают уменьшенным связыванием с белком плазмы. Связывание соединений примеров 24, 62, 63 и 64 с белками плазмы являлось относительно умеренным для всех исследованных видов, изменяясь от 61% в плазме крыс до 82% в плазме мышей. Это могло бы предоставить преимущество, заключающееся в наличии в общем кровообращении большего количества доступного свободного лекарственного средства для достижения надлежащего места действия с целью проявления его терапевтического эффекта. Увеличенная доля свободного лекарственного средства для проявления фармакологического действия в опухолях потенциально приводит к увеличению эффективности, в силу чего появляется возможность снижения вводимых дозировок.
Соединения по настоящему изобретению (например, соединения примеров 24, 62, 63 и 64) также демонстрируют повышенную активность по отношению к клеткам в пролиферационных и клоногенных анализах (например, в анализах, описанных в примерах 79 и 80) против более широкого диапазона клеточных линий солидных опухолей, показывая, тем самым, улучшенную противораковую активность (таблица C). Данные показывают, что обработка соединениями по настоящему изобретению иначе воздействует на опухолевые клетки по сравнению с нормальными клетками. Обработка соединениями по настоящему изобретению опухолевых клеток с аномальной контрольной точкой ведет к формированию многих ядер, из-за прерывания митоза, ингибирования цитокинеза и блокирования контрольной точки образования веретена, вследствие ингибирования Aurora киназы. Именно возникновение мультиядерности ведет, по-видимому, к смерти клеток. В противоположность этому в нормальных клетках с правильной контрольной точкой, обработанных соединениями по настоящему изобретению, меньшая часть клеток становится мультиядерной или погибает через 24 часа после обработки, тогда как большая часть претерпевает обратимую задержку G2/M и затем повторно входит в клеточный цикл, как только соединение перестает действовать. Эти различия в результатах, по-видимому, отражают тот факт, что нормальные клетки имеют контрольные точки в таких местах, чтобы остановить клеточный цикл, если не происходит точной сегрегации хромосом, как, например, пост-митотическую p-53 зависимую контрольную точку. В опухолевых клетках эти контрольные точки отсутствуют, что делает возможным продолжение митоза и возникновение мультиядерности.
Кроме того, солевые формы соединений по настоящему изобретению демонстрируют повышенную растворимость в водной среде и лучшие физико-химические свойства, например, более низкий logD.
Способы получения соединений формулы (I)
В этом разделе, как и во всех других разделах настоящей заявки, если контекст не указывает на другое, упоминания формулы (I) также включают формулы (II), (III), (XXX) и все другие подгруппы и отдельные примеры соединений, определенных в настоящей заявке.
Соединения формулы (I) могут быть получены в соответствии с синтетическими методиками, хорошо известными специалисту.
Например, соединения формулы (I), в которых A представляет собой связь (т.е. где A и карбонильная группа образуют амидную связь), могут быть получены взаимодействием соединения формулы (X):
с карбоновой кислотой R1-E-CO2H или ее реакционноспособными производными в стандартных условиях образования амидов.
Реакция конденсации между карбоновой кислотой и амином (X) может проводиться в присутствии некоторых типов реагентов, обычно применяемых при формировании пептидных связей. Примеры таких реагентов включают 1,3-дициклогексилкарбодиимид (DCC) (Sheehan et al., J. Amer. Chem. Soc. 1955, 77, 1067), 1-этил-3-(3'-диметиламинопропил)карбодиимид (EDC) (Sheehan et al, J. Org. Chem., 1961, 26, 2525), конденсирующие реагенты на основе урониевых соединений, как, например, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) (L.A. Caprino, J, Amer. Chem. Soc., 1993, 115, 4397) и конденсирующие реагенты на основе фосфониевых соединений, как, например, гексафторфосфат 1-бензотриазолилокситрис(пирролидино)фосфония (PyBOP) (Castro et al, Tetrahedron Letters, 1990, 31, 205). Конденсирующие реагенты на основе карбодиимида преимущественно применяются в комбинации с 1-гидроксиазабензотриазолом (HOAt) или 1-гидроксибензотриазолом (HOBt) (Konig et al., Chem. Ber., 103, 708, 2024-2034). Предпочтительные конденсирующие реагенты включают EDC и DCC в сочетании с HOAt или HOBt.
Реакцию конденсации, как правило, проводят в неводном, апротонном растворителе, таком как ацетонитрил, диоксан, диметилсульфоксид, дихлорметан, диметилформамид или N-метилпирролидон, или в водном растворителе, необязательно в сочетании с одним или несколькими смешивающимися дополнительными растворителями. Реакцию можно проводить при комнатной температуре, или, если реагенты обладают меньшей реакционной способностью (например, в случае обедненных электронной плотностью анилинов, несущих оттягивающие электроны группы, как, например, сульфонамидные группы), при надлежащей повышенной температуре. Реакцию можно осуществлять в присутствии основания, которое не мешает ее протеканию, например, третичного амина, такого как триэтиламин или N,N-диизопропилэтиламин.
В качестве альтернативы могут применяться реакционноспособные производные карбоновых кислот, например ангидриды или хлорангидриды. Взаимодействие с реакционноспособными производными, как, например, ангидридами, в основном осуществляют при перемешивании амина и ангидрида при комнатной температуре в присутствии основания, такого как пиридин.
Амины формулы (X) могут быть получены восстановлением соответствующих нитросоединений формулы (XI) в стандартных условиях. Восстановление можно провести, например, каталитическим гидрированием в присутствии катализатора, такого как палладий на угле, в полярном растворителе, как, например, этаноле или диметилформамиде при комнатной температуре.
Нитросоединения формулы (XI) могут быть получены взаимодействием нитропиразолкарбоновой кислоты формулы (XII):
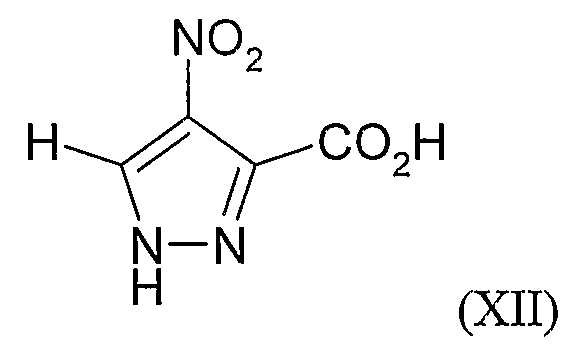
либо с 4-морфолин-4-илметилбензол-1,2-диамином (для получения соединений, в которых M представляет собой D1), либо с 4,5-диметоксибензол-1,2-диамином (для получения соединений, в которых M представляет собой D2).
Взаимодействие между диамином и карбоновой кислотой (XII) может проводиться в присутствии таких реагентов, как DCC или EDC в присутствии HOBt, как описано выше, в условиях реакции конденсации для получения амидов, описанных ранее, с образованием в качестве промежуточного соединения орто-аминофениламида (не показан), который затем подвергают циклизации с образованием бензимидазольного цикла. Заключительную стадию циклизации, как правило, выполняют при кипячении с обратным холодильником в присутствии уксусной кислоты.
Иллюстративная схема реакции, показывающая получение соединений формулы (X), где M означает группу D1, представлена на схеме 1.

Типовые условия для каждой стадии на схеме 1 можно найти в разделе «примеры» ниже по тексту.
Соединения, в которых M означает группу D2, могут быть получены аналогичным способом, но с применением 4,5-диметоксибензол-1,2-диамина вместо диамина (XVI) на схеме 1.
В альтернативном способе синтеза соединений формулы (I), в которых A представляет собой связь, диамины, т.е. 4-морфолин-4-илметилбензол-1,2-диамин и 4,5-диметоксибензол-1,2-диамин, также могут быть введены во взаимодействие с карбоновыми кислотами формулы (XVII), где A означает связь, с получением соединений формулы (I).
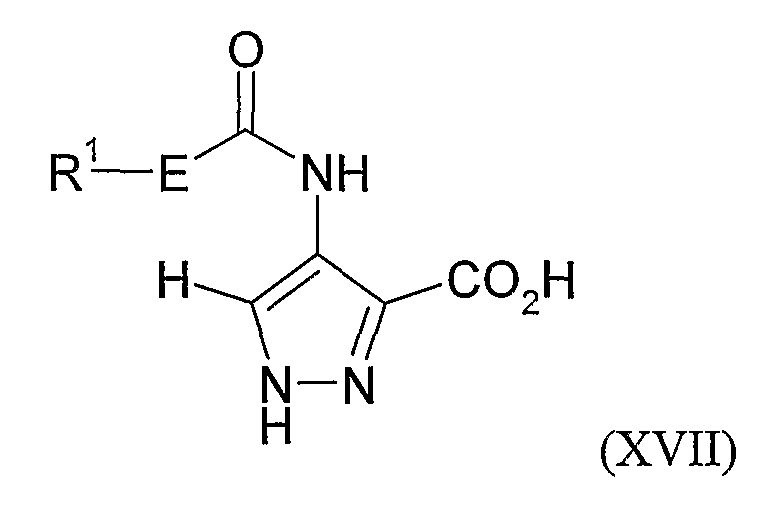
Взаимодействие диамина с карбоновой кислотой (XVII) можно проводить в условиях, аналогичных описанным выше для получения нитросоединений (XI). Карбоновые кислоты формулы (XVII) могут быть получены в результате последовательности реакций, показанной на схеме 2.
Как видно из схемы 2, замещенная или незамещенная 4-нитро-3-пиразолкарбоновая кислота (XVIII) может быть этерифицирована взаимодействием с тионилхлоридом с образованием хлорангидрида кислоты в качестве промежуточного соединения и последующим взаимодействием с этанолом, ведущим к получению этилового эфира (XIX). С другой стороны, этерификация может быть проведена взаимодействием спирта и карбоновой кислоты в присутствии кислотного катализатора, одним из примеров которого является тионилхлорид. Эту реакцию, как правило, проводят при комнатной температуре с использованием этерифицирующего спирта (например, этанола) в качестве растворителя. Затем нитрогруппа может быть восстановлена с применением палладия на угле согласно стандартным методикам с получением амина (XX). Амин (XX) вводят в реакцию конденсации с соответствующей карбоновой кислотой R1-E-CO2H в условиях получения амидов, которые совпадают с описанными выше условиями или аналогичны им, с получением амида (XXI). Затем сложноэфирная группа амида (XXI) может быть гидролизована с применением гидроксида щелочного металла, такого как гидроксид натрия, в полярном смешивающемся с водой растворителе, таком как метанол, как правило, при комнатной температуре.
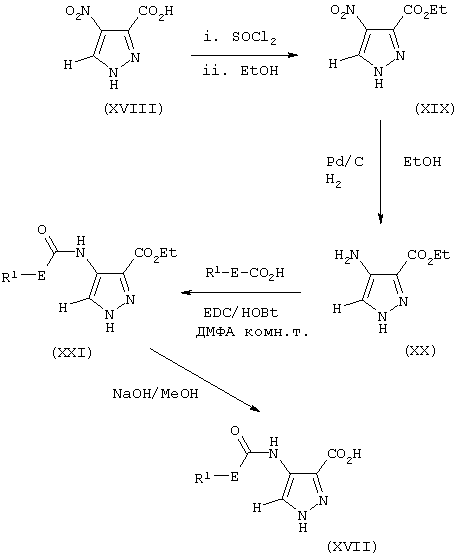
Соединения формулы (I), в которых A означает NR2, могут быть получены с применением стандартных способов синтеза мочевин.
Например, такие соединения можно получить взаимодействием производного аминопиразола формулы (X) с подходяще замещенным изоцианатом формулы R1-E-N=C=O в полярном растворителе, как, например, ДМФА. Реакцию обычно проводят при комнатной температуре.
По другому способу мочевины формулы (I) могут быть получены взаимодействием амина формулы (X) с амином формулы R1-E-NH2 в присутствии карбонилдиимидазола (CDI). Реакцию, как правило, проводят в полярном растворителе, как, например, ТГФ, нагревая (например, применяя микроволновое нагревающее устройство) при температуре примерно до 150°C.
Вместо использования CDI реакцию конденсации двух аминов с образованием мочевины можно проводить с применением трифосгена (бис(трихлорметил)карбоната) в присутствии не участвующего в реакции основания, как, например, триэтиламина, в таком растворителе как дихлорметан при комнатной или более низкой температуре.
В качестве другой альтернативы CDI вместо трифосгена можно применять фосген.
Во многих из описанных выше реакций может быть необходима защита одной или нескольких групп для предотвращения протекания реакций по нежелательным фрагментам молекул. Примеры защитных групп, а также способы защиты функциональных групп и снятия защиты могут быть найдены в Protective Groups in Organic Synthesis (T.Green and P.Wuts; 3rd Edition; John Wiley and Sons, 1999). Гидроксигруппа может быть защищена, например, образованием эфирной (-OR) или сложноэфирной (OC(=O)R) группы, например: образованием трет-бутилового простого эфира; бензильного, бензгидрильного (дифенилметилового) или тритилового (трифенилметилового) простого эфира; триметилсилилового или трет-бутилдиметилсилилового простого эфира; или ацетилового сложного эфира (-OC(=O)CH3, -OAc). Альдегидная или кетонная группы могут быть защищены, например, образованием, соответственно, ацеталя (R-CH(OR)2)) или кеталя (R2C(OR)2), в которых карбонильную группу (>C=O) превращают в диэфир (>C(OR)2), взаимодействием, например, с первичным спиртом. Альдегидная или кетонная группа легко регенерируется гидролизом с применением большого избытка воды в присутствии кислоты. Аминогруппу можно защитить, например, образованием амида (-NRCO-R) или уретана (-NRCO-OR), например, метиламида (-NHCO-CH3); бензилоксиамида (-NHCO-OCH2C6H5, -NH-Cbz); трет-бутоксиамида (-NHCO-OC(CH3)3, -NH-Boc); 2-бифенил-2-пропоксиамида (-NHCO-OC(CH3)2C6H4C6H5, -NH-Bpoc), 9-флуоренилметоксиамида (-NH-Fmoc), 6-нитровератрилоксиамида (-NH-Nvoc), 2-триметилсилилэтоксиамида (-NH-Teoc), 2,2,2-трихлорэтилоксиамида (-NH-Troc), аллилоксиамида (-NH-Alloc) или 2-(фенилсульфонил)этилоксиамида (-NH-Psec). Другие защитные группы для аминов, как, например, циклических аминов и гетероциклических N-H групп, включают толуолсульфонильную (тозильную) и метансульфонильную (мезильную) группы, а также бензильные группы, такие как пара-метоксибензильную (PMB)группу. Карбоксильная группа может быть защищена образованием сложного эфира, например C1-7алкилового эфира (например, метилового эфира, трет-бутилового эфира); C1-7галогеналкилового эфира (например, C1-7тригалогеналкилового эфира); триC1-7алкилсилил-C1-7алкилового эфира; или C5-20арил-C1-7алкилового эфира (например, бензилового эфира; нитробензилового эфира); или образованием амида, например метиламида. Тиольная группа может быть защищена, например, образованием простого тиоэфира (-SR), например бензилового тиоэфира; ацетамидометилового эфира (-S-CH2NHC(=O)CH3).
Кислотно-аддитивные соли, образующие подгруппу (C) соединений формулы (I), могут быть получены в ходе синтеза самого соединения формулы (I), т.е. 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины или превращением исходного соединения в форме свободного основания в желаемую соль, или же превращением одной соли исходного соединения в другую желаемую соль исходного соединения. Исходное соединение, т.е. 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина (соединение формулы (IA)) может быть получено способом, показанным ниже на схеме 3.
Как показано на схеме 3, 3,4-динитрокарбоновую кислоту (XIII), являющуюся коммерчески доступным соединением, превращают в морфолид (XXI). Получение амида может быть осуществлено превращением кислоты (XIII) в активное производное, как, например, хлорангидрид, с применением стандартных способов. Например, хлорангидрид может быть получен нагреванием с избытком тионилхлорида при температуре его кипения с последующим удалением избытка тионилхлорида азеотропной отгонкой с толуолом.
Морфолид (XXI) может быть восстановлен в динитробензилморфолин (XXIII) обработкой подходящим восстанавливающим реагентом, как, например, боргидридом натрия в комбинации с трифторидом бора. Реакцию восстановления обычно проводят в безводном растворителе, таком как тетрагидрофуран при пониженной температуре, например при температуре 0-5°C. После этого динитробензилморфолин (XXIII) может быть восстановлен в диаминобензилморфолин (XXIV) в стандартных условиях, например каталитическим гидрированием в присутствии катализатора, такого как палладий на угле в полярном растворителе, как, например, этаноле при комнатной температуре.
Затем диаминобензилморфолин (XXIV) вводят в реакцию с коммерчески доступной 4-нитропиразол-3-карбоновой кислотой для получения нитропиразолилбензимидазола (XXV). Получение нитропиразолилбензимидазола (XXV) может быть достигнуто, во-первых, формированием амидной связи между карбоновой кислотой и диаминобензилморфолином (XXIV) с использованием реагента для пептидной конденсации, как, например, тетрафторбората O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), способного содействовать образованию амидной связи с ароматической аминогруппой. Промежуточный амид (не показан) затем циклизуют в нитропиразолилбензимидазол (XXV) нагреванием в избытке ледяной уксусной кислоты, например, при температуре приблизительно 65°C.
Нитропиразолилбензимидазол (XXV) может быть восстановлен в соответствующий амин (XXVI) в стандартных условиях. Восстановление может быть проведено, например, каталитическим гидрированием в присутствии катализатора, как, например, палладия на угле, в полярном растворителе, таком как этанол или диметилформамид при комнатной температуре.
Амин (XXVI) может быть, в свою очередь, превращен в мочевину (IA) с применением стандартных методик синтеза мочевин, например, взаимодействием амина (XXVI) с 2,6-дифторфенилизоцианатом в полярном растворителе, таком как ТГФ, при комнатной или более низкой температуре, например при температуре 0-5°C.
Мочевина (IA) в форме свободного основания может применяться для получения кислотно-аддитивных солей по настоящему изобретению.
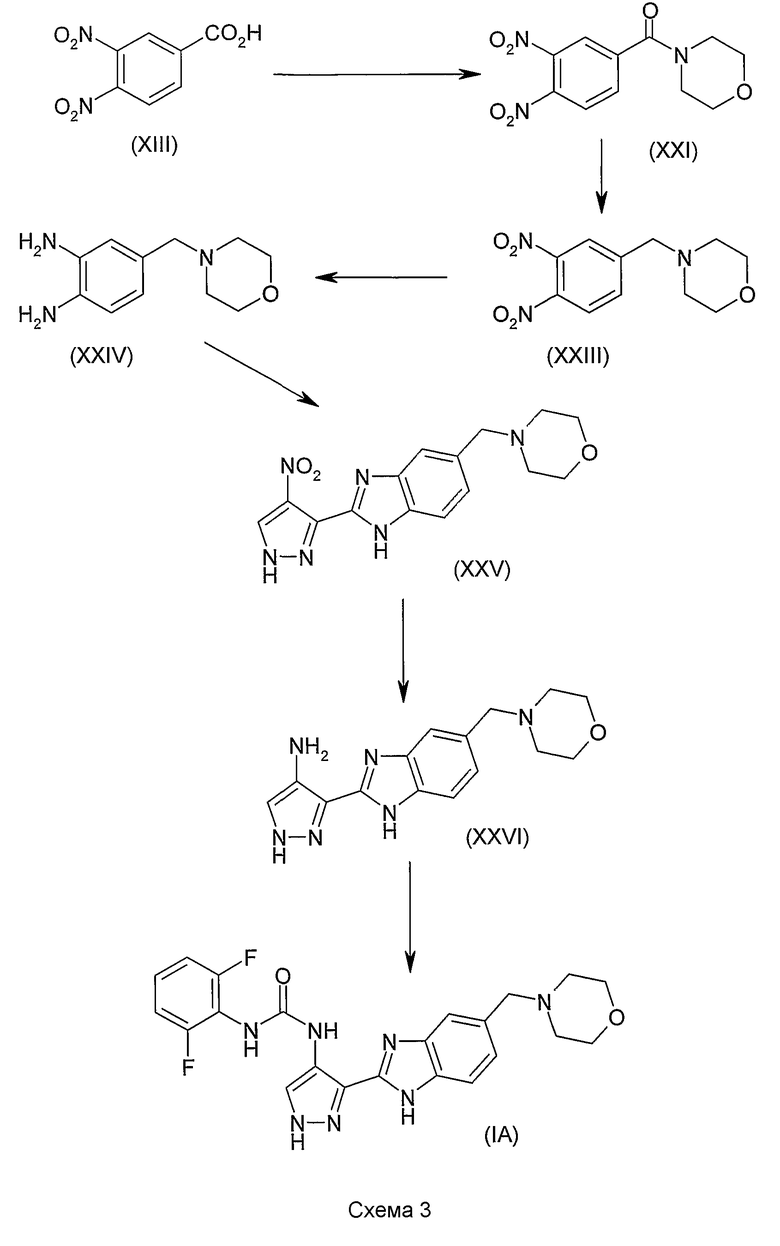
Соли по настоящему изобретению могут быть получены из свободных оснований стандартными способами, как, например, способами, описанными в: Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, в твердом переплете, 388 страниц, август 2002. Например, соли могут быть получены взаимодействием свободного основания с подходящей кислотой в воде или органическом растворителе, или в их смеси; в основном применяют неводную среду, такую как эфир, этилацетат, метанол, этанол, изопропанол или ацетонитрил.
В другом аспекте изобретение относится к способу получения кислотно-аддитивных солей 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, причем данный способ включает получение раствора 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания в растворителе (как правило, органическом растворителе) или смеси растворителей и обработку полученного раствора кислотой для образования осадка кислотно-аддитивной соли.
Кислоту обычно добавляют в виде раствора в растворителе, который смешивается с растворителем, в котором растворено свободное основание.
Растворитель, в котором вначале растворяют свободное основание, может являться растворителем, в котором нерастворима кислотно-аддитивная соль. С другой стороны, растворитель, в котором вначале растворяют свободное основание, может быть растворителем, в котором кислотно-аддитивная соль растворима хотя бы частично, причем добавляют другой растворитель, в котором кислотно-аддитивная соль растворима в меньшей степени, так что соль выпадает из раствора в виде осадка.
Например, в одном из способов получения солей по настоящему изобретению свободное основание растворяют в первом растворителе (который может быть этилацетатом или смесью этилацетата и спирта, такого как метанол) и затем добавляют раствор (например, концентрированный или насыщенный раствор) кислоты, как, например, соляной кислоты, во втором растворителе (который может быть эфиром, таким как диэтиловый эфир или диоксин), так что образуется осадок кислотно-аддитивной соли, после чего осадок собирают, например, фильтрованием.
Способы получения 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
В примерах более ранней заявки авторов WO 2005/002552 и на схемах 1 и 3, выше, показано, что [3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амид может быть получен в результате осуществления последовательности стадий, включающих:
(i) взаимодействие 4-морфолин-4-илметилбензол-1,2-диамина с 4-нитро-1H-пиразол-3-карбоновой кислотой в присутствии 1-этил-3-(3'-диметиламинопропил)карбодиимида (EDC) и 1-гидроксибензотриазола (HOBt) в N,N-диметилформамиде (ДМФА) с получением 5-морфолин-4-илметил-2-(4-нитро-1H-пиразол-3-ил)-1H-бензимидазола; и
(ii) восстановление нитрогруппы обработкой палладием на угле в атмосфере водорода;
или
(i) взаимодействием 4-амино-1H-пиразол-3-карбонового эфира с соответствующей карбоновой кислотой в присутствии 1-этил-3-(3'-диметиламинопропил)карбодиимида (EDC) и 1-гидроксибензотриазола (HOBt) в N,N-диметилформамиде (ДМФА) или с хлорангидридом соответствующей кислоты в присутствии триэтиламина, с образованием 4-амида-1H-пиразолкарбоновой кислоты; и
(ii) взаимодействие 4-морфолин-4-илметилбензол-1,2-диамина с соответствующей 4-амид-1H-пиразолкарбоновой кислотой в присутствии 1-этил-3-(3'-диметиламинопропил)карбодиимида (EDC) и 1-гидроксибензотриазола (HOBt) в N,N-диметилформамиде (ДМФА) с получением [3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амида.
В настоящей заявке обнаружено, что вместо взаимодействия нитропиразольного соединения с диамином и последующего восстановления нитрогруппы до амина, или реакции амидопиразола с диамином, аминопиразол может быть введен во взаимодействие с диамином, при условии, что аминогруппа аминопиразола защищена надлежащим образом. Продукт этой реакции может быть затем циклизован с образованием бензимидазола. Кроме того, было найдено, что удаление аминозащитной группы и циклизация с образованием бензимидазола могут быть проведены в одну стадию.
Соответственно в другом аспекте в настоящем изобретении разработан способ получения соединения формулы (XXVII), или (XXVIII), или их солей:
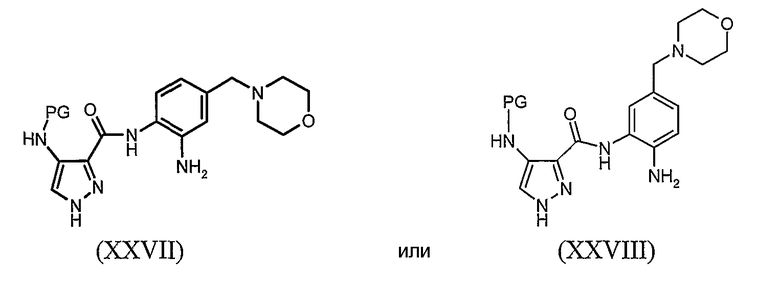
причем этот способ включает:
(i) взаимодействие соединения формулы (XXIX):
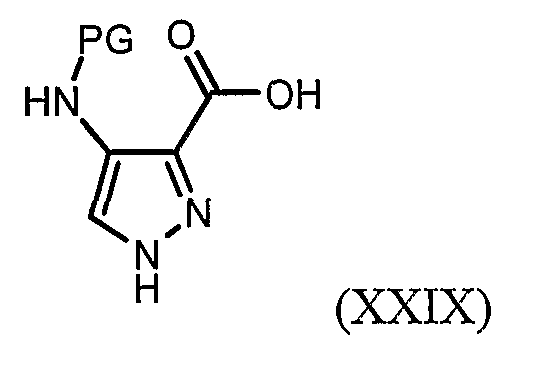
где PG означает аминозащитную группу:
(ii) с соединением формулы (XXXI):
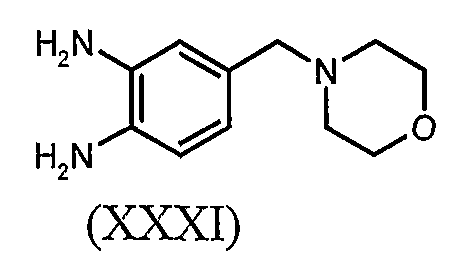
в органическом растворителе в присутствии конденсирующего реагента, как, например, EDC и HOBt:
Соединение формулы (XXVIII) является региоизомером соединения (XXVII).
Аминозащитная группа PG может являться любой известной защитной группой, применяемой для защиты аминогрупп в условиях, применяемых в описанном выше способе, см., например, работу Green et al., упомянутую выше. Так, например, азот может быть защищен образованием амида (NCO-R) или уретана (NCO-OR), например метиламида (NCO-CH3); бензилоксиамида (NCO-OCH2C6H5, -NH-Cbz); трет-бутоксиамида (-NCO-OC(CH3)3, N-Boc); 2-бифенил-2-пропоксиамида (NCO-OC(CH3)2C6H4C6H5, N-Bpoc), 9-флуоренилметоксиамида (N-Fmoc), 6-нитровератрилоксиамида (N-Nvoc), 2-триметилсилилэтоксиамида (N-Teoc), 2,2,2-трихлорэтилоксиамида (N-Troc), аллилоксиамида (N-Alloc) или 2-(фенилсульфонил)этилоксиамида (-N-Psec). Другие защитные группы для аминов включают бензильные группы как, например, пара-метоксибензильную (PMB) группу. Предпочтительными аминозащитными группами являются уретаны (NCO-OR), например бензилоксиамид (NCO-OCH2C6H5, -NH-Cbz) или трет-бутоксиамид (-NCO-OC(CH3)3, N-Boc); или аллилоксиамид (N-Alloc). В одном из вариантов осуществления защитная группа PG является защитной группой APG, которая представляет собой аминозащитную группу, которую можно удалить в условиях кислой среды. Такие группы включают уретаны. Особенно предпочтительной уретановой защитной группой является трет-бутилоксикарбонил, который может быть удален в кислой среде.
В одном из вариантов осуществления защитную группу PG затем удаляют из соединений формул (XXVII) или (XXVIII) и заменяют защитной группой APG, получая соединения формул (XXVIIa) или (XXVIIIa).
Одним особенно предпочтительным соединением формулы (XXIX) является соединение следующей формулы (XXXII):

Кроме того, в изобретении разработано новое химическое промежуточное соединение формулы (XXXII) как таковое.
Изобретение также относится к новым химическим промежуточным соединениям формул (XXVII) или (XXVIII) как таковым, например, новым химическим промежуточным соединениям формул (XXVIIa) или (XXVIIIa), ниже. Следовательно, в настоящем изобретении разработаны (2-амино-4-морфолин-4-илметилфенил)амид 4-амино-1H-пиразол-3-карбоновой кислоты или (2-амино-5-морфолин-4-илметилфенил)амид 4-амино-1H-пиразол-3-карбоновой кислоты, а также их защищенные формы в качестве новых химических промежуточных соединений. Одним особенно предпочтительным новым химическим промежуточным соединением формулы (XXVII) является трет-бутиловый эфир [3-(2-амино-4-морфолин-4-илметилфенилкарбамоил)-1H-пиразол-4-ил]карбаминовой кислоты. Одним особенно предпочтительным новым химическим промежуточным соединением формулы (XXVIII) является трет-бутиловый эфир [3-(2-амино-5-морфолин-4-илметилфенилкарбамоил)-1H-пиразол-4-ил]карбаминовой кислоты.
Если защитная группа PG является трет-бутоксикарбонильной группой, общий выход описанного способа превышает 85%. Кроме того, способ обладает тем преимуществом, что в нем применяются относительно простые и недорогие реагенты и растворители, и, кроме того, способ является выгодным в отношении легкости очистки продуктов.
В другом аспекте изобретение относится к способу получения [3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амина или его солей, причем способ включает:
(i) обработку соединений формул (XXVIIa) или (XXVIIIa):
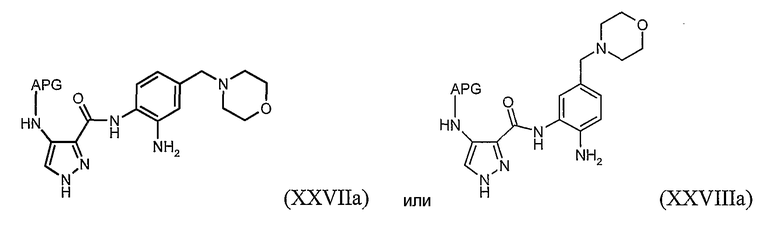
кислотой в растворителе, необязательно при нагревании; и
(ii) нейтрализацию реакционной смеси.
Аминозащитной группой APG может быть любая известная защитная группа, применяемая для защиты аминогрупп, как определено выше применительно к соединениям формул (XXVII) или (XXVIII), и которая может быть удалена в условиях, применяемых в предыдущем способе.
На стадии (i) реакция с кислотой может проводиться при нагревании, например, до температуры в диапазоне от 80 до 100°C. Растворитель, в котором проводится реакция на стадии (i), является спиртовым растворителем, и может быть, например, этанолом.
Защитная группа на стадии (i) предпочтительно является такой защитной группой, как группа Boc, которую можно удалить обработкой кислотой, причем кислоту выбирают так, чтобы она подходила для протонирования промежуточного соединения с целью активации карбонильной группы для реакции циклизации. Подходящие кислоты включают сильные кислоты, такие как серная кислота, метансульфоновая кислота или хлористоводородная кислота, и одной конкретной кислотой является хлористоводородная кислота.
После завершения реакции на стадии (i), о котором судят, например, по исчезновению исходного вещества (XIIIa), можно нейтрализовать реакционную смесь.
На стадии (ii) применяют не мешающее основание. Термин «не мешающее основание» в данном контексте означает основание, как, например, карбонат натрия, которое не будет вступать в реакцию с полученным соединением. Стадию (ii) обычно проводят при комнатной температуре.
На стадии (ii) реакционную смесь нейтрализуют, например, до насыщения реакционной смеси нейтрализующим реагентом и при pH 8,5.
Вслед за стадией (ii) соединение может быть введено во взаимодействие с карбонилирующим реагентом, как, например, 1,1'-карбонилдиимидазолом (CDI) или эквивалентом фосгена, и затем обработано циклопропиламином. Эквиваленты фосгена включают трифосген или фосген. Предпочтительным карбонилирующим реагентом является 1,1'-карбонилдиимидазол (CDI).
В качестве альтернативы мочевина может быть получена взаимодействием аминопиразола, т.е. 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина с фенилхлорформиатом в присутствии основания, например пиридина, в таком растворителе как, например, ТГФ, с получением циклической мочевины и последующей обработкой циклопропиламином, или взаимодействием аминопиразола с циклопропилизоцианатом, который может быть получен перегруппировкой азида циклопропанкарбоновой кислоты по Курциусу (как описано в US 4313755 и US 4299778).
Таким образом, еще одним аспектом настоящего изобретения является способ получения 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины или ее солей, причем способ включает
(i) обработку соединения формулы (XXVIIa) кислотой в растворителе, необязательно при нагревании;
(ii) нейтрализацию реакционной смеси;
(iii) взаимодействие продукта стадии (ii) с карбонилирующим реагентом;
(iv) взаимодействие продукта стадии (iii) с циклопропиламином.
Стадию (iii) обычно проводят при кипячении с обратным холодильником, например, при температуре вплоть до примерно 100°C, более типично до 70-75°C. На стадии (iii) реакцию можно проводить в полярном апротонном растворителе, таком как тетрагидрофуран. Карбонилирующий реагент может быть таким соединением, как 1,1'-карбонилдиимидазол (CDI) или эквивалент фосгена, такой как трифосген или фосген. Предпочтительным карбонилирующим реагентом является 1,1'-карбонилдиимидазол (CDI).
Стадию (iv), как правило, проводят при нагревании, например, при температуре до примерно 100°C.
После стадии (iv) продукт может быть подвергнут превращению в соль или перекристаллизации (например, с применением 2-пропанола или этанола в качестве растворителя) для увеличения чистоты и с целью получения кристаллической формы.
Стадия (iii) приводит к получению промежуточного соединения формулы (XXXIII) и/или его региоизомера (XXXIIIa):
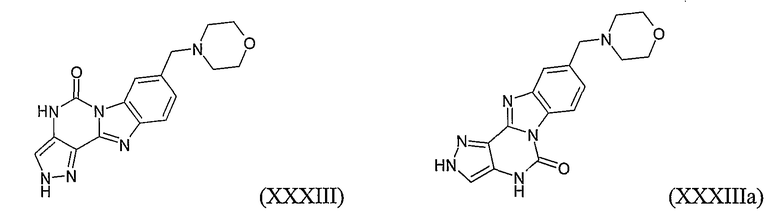
Промежуточные соединения формул (XXXIII) и (XXXIIIa), которые при необходимости могут быть выделены, затем вводят в реакцию с циклопропиламином, получая соединение формулы (XXX).
Соответственно в другом аспекте изобретение относится к способу получения соединения формулы (XXX), определенного в настоящей заявке, причем способ включает взаимодействие соединения формулы (XXXIII) или (XXXIIIa) с циклопропиламином и затем необязательное получение кислотно-аддитивной соли соединения формулы (XXX). Реакцию обычно проводят в полярном апротонном растворителе, таком как N-метилпирролидон, предпочтительно при повышенной температуре, как, например, при температуре, превышающей 80°C, более типично, превышающей 90°C, например от 95 до 105°C.
Предыдущий способ также может применяться для получения других соединений формулы (I) и их подгрупп, описанных в настоящей заявке, в которых A в формуле (I) представляет собой группу NH.
Соответственно в еще одном аспекте изобретение относится к способу получения соединения формулы (I), определенной в настоящей заявке, в котором A в формуле (I) представляет собой группу NH; причем этот способ включает взаимодействие (i) соединения формулы (XXXIII) и/или его региоизомера (XXXIIIa), или (ii) соединения формулы (XXXIV) и/или его региоизомера (XXXIVa):
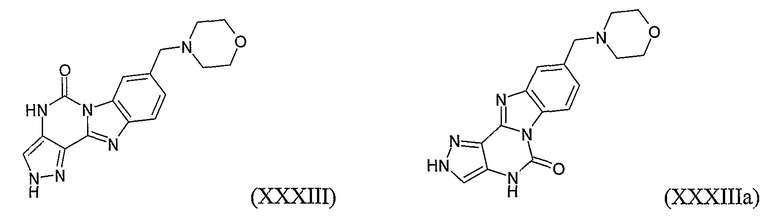
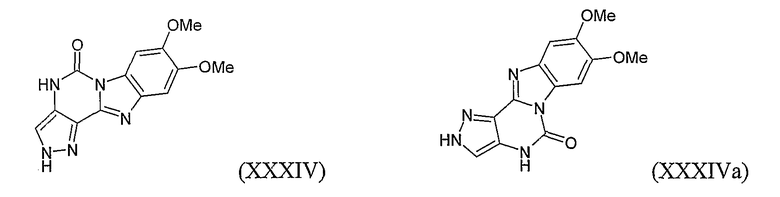
с соединением формулы R1-E-NH2, предпочтительно в полярном апротонном растворителе, таком как N-метилпирролидон, предпочтительно при повышенной температуре, как, например, при температуре, превышающей 80°C, более типично, превышающей 90°C, например, от 95 до 105°C, и затем необязательно образование кислотно-аддитивной соли соединения формулы (I).
Кроме того, в изобретении разработаны новые химические промежуточные соединения формул (XXXIII), (XXXIIIa), (XXXIV) и (XXXIVa).
В другом варианте осуществления могут быть получены соединение формулы (XXVIIa) в описанном выше способе получения 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина или его соли, или способе получения 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины или ее солей, способом, который включает:
(i) взаимодействие соединения формулы (XXIX), в котором PG представляет собой аминозащитную группу, которая может быть удалена кислотой, т.е. APG;
(ii) с соединением формулы (XXXI) в органическом растворителе в присутствии конденсирующего реагента, такого как EDC и HOBt.
Способ, описанный выше, необязательно включает дополнительную стадию перекристаллизации соли для получения кристаллической формы, например кристаллической формы, описанной в настоящей заявке.
Способы очистки
Соединения по настоящему изобретению могут быть выделены и очищены радом способов, хорошо известных специалистам в данной области техники, и примеры таких способов включают хроматографические методики, как, например, колоночную хроматографию (например, флэш-хроматографию) и ВЭЖХ. Препаративная ЖХ-МС является стандартной и эффективной методикой, применяемой для очистки небольших органических молекул, таких как молекулы соединений, описанных в настоящей заявке. Можно изменять методики жидкостной хроматографии (ЖХ) и масс-спектрометрии (МС) для обеспечения лучшего разделения неочищенных веществ и лучшего выявления состава образцов с помощью МС. Оптимизация препаративных градиентных методик ЖХ будет включать подбор колонок, летучих растворителей и модификаторов, а также градиентов. Способы оптимизации препаративных ЖХ-МС методик и их применение для очистки соединений хорошо известны в технике. Такие способы описаны в Rosentreter U, Huber U.; Optimal fraction collecting in preparative LC/MS; J Comb Chem.; 2004; 6(2), 159-64 и Leister W, Strauss K, Wisnoski D, Zhao Z, Lindsley C., Development of a custom high-throughput preparative liquid chromatography/mass spectrometer platform for the preparative purification and analytical analysis of compound libraries; J Comb Chem.; 2003; 5(3); 322-9.
Одна из таких систем для очистки соединений препаративной ЖХ-МС описана ниже в экспериментальном разделе, хотя специалист в данной области техники примет во внимание, что могли бы применяться альтернативные системы и способы, описанные для этого. В частности могли бы применяться способы, основанные на препаративной ЖХ на нормальной фазе, вместо описанных здесь методик на обращенной фазе. Большинство препаративных ЖХ-МС систем используют ЖХ на обращенной фазе и летучие кислотные модификаторы, т.к. данный подход является весьма эффективным для очистки небольших молекул, и применяемые растворители совместимы с методикой масс-спектрометрии с электрораспылением положительных ионов. С другой стороны, для очистки соединений могли бы применяться другие решения, разработанные в хроматографии, например ЖХ на нормальной фазе, альтернативное буферирование подвижной фазы, основные модификаторы и т.д., что кратко изложено в аналитических методиках, описанных выше.
Перекристаллизация
Перекристаллизация соединений формулы (I) и их солей, в частности 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и ее солей, может выполняться способами, хорошо известными специалисту - см., например (P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Chapter 8, Publisher Wiley-VCH). Продукты, получаемые в результате органических реакций, редко являются чистыми при выделении непосредственно из реакционной смеси. Если соединение (или его соль) является твердым, оно может быть очищено и/или получено в кристаллическом виде перекристаллизацией из подходящего растворителя. Хороший растворитель для перекристаллизации должен в умеренном количестве растворять подлежащее перекристаллизации вещество при повышенной температуре и лишь небольшое количество этого вещества при более низкой температуре. Он должен легко растворять примеси при низких температурах или вообще не растворять их. Наконец, растворитель должен легко удаляться из очищенного продукта. Это обычно означает, что он имеет относительно низкую температуру кипения, и специалист в данной области техники должен знать растворители для перекристаллизации конкретного вещества, или, если эта информация недоступна, исследует несколько растворителей. Для достижения хорошего выхода очищенного вещества используют минимальное количество горячего растворителя, способное растворить все загрязненное вещество. На практике применяют на 3-5% больше растворителя, чем необходимо, так что раствор является ненасыщенным. Если загрязненное вещество содержит примесь, которая нерастворима в растворителе, после этого ее можно удалить фильтрованием и затем оставить раствор кристаллизоваться. Кроме того, если загрязненное вещество содержит следы окрашенных веществ, которые не являются собственной окраской соединения, они могут быть удалены добавлением к горячему раствору небольшого количества обесцвечивающего активированного угля с последующим фильтрованием и кристаллизацией раствора. Обычно кристаллизация происходит самопроизвольно при охлаждении раствора. Если этого не случается, кристаллизацию можно вызвать охлаждением раствора до температуры ниже комнатной или добавлением одного кристалла чистого вещества (кристаллической затравки). Также можно проводить перекристаллизацию и/или оптимизировать выход с применением антирастворителя. В этом случае соединение растворяют в подходящем растворителе при повышенной температуре, фильтруют и затем для содействия кристаллизации добавляют дополнительный растворитель, в котором подвергаемое очистке соединение имеет плохую растворимость. После этого, обычно с применением вакуумного фильтрования, выделяют кристаллы, промывают их и затем сушат, например в печи, или путем высушивания.
Другие примеры способов кристаллизации включают кристаллизацию из парообразного состояния, которая включает стадию испарения, например, в запаянной пробирке или в потоке воздуха, а также кристаллизацию из расплава (Crystallization Technology Handbook 2nd Edition, edited by A. Mersmann, 2001).
В частности, соединения формулы (I) могут подвергаться перекристаллизации (например, с применением 2-пропанола или этанола в качестве растворителя) для увеличения чистоты и получения их в кристаллической форме.
Как правило, полученные кристаллы исследуют способами дифракции рентгеновских лучей, как, например, дифракции рентгеновских лучей на порошке (XRPD) или рентгеноструктурного анализа кристаллов.
Фармацевтические составы
Хотя имеется возможность вводить действующие соединения и их соли в чистом виде, предпочтительно применять их в виде фармацевтических композиций (например, составов), включающих хотя бы одно действующее соединение по настоящему изобретению в сочетании с одним или несколькими фармацевтически приемлемыми носителями, вспомогательными веществами, наполнителями, разбавителями, буферными соединениями, стабилизаторами, консервантами, смазывающими средствами или другими веществами, хорошо известными специалисту в данной области техники, и необязательно другими терапевтическими или профилактическими средствами, например средствами, которые снижают или облегчают некоторые из побочных эффектов, связанных с химиотерапией. Конкретные примеры таких средств включают противорвотные средства и средства, которые предотвращают или уменьшают продолжительность связанной с химиотерапией нейтропении, а также предотвращают осложнения, которые появляются из-за пониженных уровней эритроцитов или лейкоцитов, например эритропоэтин (EPO), гранулоцит макрофаг-колониестимулирующий фактор (GM-CSF) и гранулоцит-колониестимулирующий фактор (G-CSF).
Таким образом, настоящее изобретение помимо прочего относится к фармацевтическим композициям, определенным выше, а также способам получения фармацевтических композиций, включающим смешивание хотя бы одного действующего соединения, определенного выше, с одним или несколькими фармацевтически приемлемыми носителями, наполнителями, буферами, вспомогательными веществами, стабилизаторами или другими веществами, описанными в настоящей заявке.
Термин «фармацевтически приемлемый» в настоящем описании относится к соединениям, веществам, композициям и/или дозированным лекарственным формам, которые, в рамках обоснованного мнения медицины, подходят для применения в контакте с тканями субъекта (например, человека), не вызывая избыточной токсичности, раздражения, аллергических реакций, или других проблем или осложнений, и соответствуют разумному соотношению польза/риск. Каждый из носителей, наполнителей и т.д. также должен быть «приемлемым» в смысле наличия совместимости с другими ингредиентами состава.
Соответственно в еще одном аспекте настоящее изобретение относится к соединениям формулы (I) и их подгруппам, как, например, соединениям формул (II) и (III), а также их подгруппам, определенным в настоящей заявке, в форме фармацевтических композиций.
Фармацевтические композиции могут иметь любую форму, подходящую для перорального, парентерального, местного, интраназального, глазного, ушного, ректального, интравагинального или трансдермального способов введения. Если композиции предназначены для парентерального введения, они могут быть предназначены для внутривенного, внутримышечного, интраперитонеального, подкожного введения или для непосредственной доставки в целевой орган или ткань с помощью инъекции, инфузии или других средств доставки. Доставка может происходить путем болюсной инъекции, кратковременной инфузии или более долговременной инфузии и может осуществляться в виде пассивного вливания или с применением подходящего насоса для инфузии.
Фармацевтические составы, предназначенные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферные соединения, бактериостатики и растворенные вещества, придающие составу изотоничность с кровью предполагаемого реципиента; а также водные и неводные стерильные суспензии, которые могут включать суспендирующие средства и загустители. Их примеры описаны в R.G. Strickly, Solubilizing Excipients in oral and injectable formulations, Pharmaceutical Research, Vol 21(2) 2004, p.201-230. Кроме того, они могут содержать дополнительные растворители, смеси органических растворителей, циклодекстриновые комплексообразующие реагенты, эмульгирующие средства (для образования и стабилизации эмульсионных составов), липосомальные компоненты для образования липосом, желирующие полимеры для образования полимерных гелей, защитные средства для лиофилизации и комбинации средств, для, в числе прочего, стабилизации активного ингредиента в растворимой форме и придания составу изотоничности с кровью предполагаемого реципиента. Эти составы могут помещаться в контейнеры, содержащие одну или несколько доз, например, запаянные ампулы или сосуды, и могут храниться в высушенном при замораживании (лиофилизованном) состоянии, требуя только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением.
Растворимость лекарственного средства, молекулы которого подвержены ионизации, может быть повышена до желаемой величины регулированием pH, если pKa лекарственного средства значительно отличается от значения pH состава. Приемлемый диапазон pH для внутривенного и внутримышечного введения составляет 2-12, но для подкожного введения находится в диапазоне 2,7-9,0. pH раствора регулируют либо солями лекарственного соединения с сильными кислотами/основаниями, как, например, соляной кислотой или гидроксидом натрия, либо растворами буферных соединений, которые включают, не ограничиваясь перечисленными, буферные растворы, полученные из глицина, цитратов, ацетатов, малеатов, сукцинатов, гистидина, фосфатов, трис(гидроксиметил)аминометана (TRIS) или карбонатов.
В составах, пригодных для инъекций, часто применяется комбинация водного раствора и водорастворимого органического растворителя/ПАВ(поверхностно-активного вещества) (т.е. дополнительного растворителя). Водорастворимые органические растворители и ПАВ, применяемые в пригодных для инъекций составах, включают, не ограничиваясь перечисленным, пропиленгликоль, этанол, полиэтиленгликоль 300, полиэтиленгликоль 400, глицерин, диметилацетамид (DMA), N-метил-2-пирролидон (NMP; Pharmasolve), диметилсульфоксид (ДМСО), Solutol HS 15, Cremophor EL, Cremophor RH 60 и polysorbat 80. Такие составы обычно, но не всегда, можно разбавлять перед инъекцией.
Пропиленгликоль, ПЭГ 300, этанол, Cremophor EL, Cremophor RH 60 и polysorbat 80 являются полностью органическими смешивающимися с водой растворителями и ПАВ, применяемыми в коммерчески доступных составах для инъекций, и они могут применяться в комбинациях друг с другом. Полученные органические составы обычно разбавляют, как минимум, два раза перед внутривенной болюсной инъекцией или инфузией.
С другой стороны, повышенная растворимость в воде может быть достигнута путем образования молекулярных комплексов с циклодекстринами.
Липосомы являются почти сферическими пузырьками, состоящими из внешних липидных двухслойных мембран и внутреннего водного ядра, с общим диаметром <100 мкм. В зависимости от уровня гидрофобности умеренно гидрофобные лекарственные средства могут быть солюбилизированы липосомами, если лекарственное средство инкапсулируется или включается в липосому. Гидрофобные лекарственные средства также могут быть солюбилизированы липосомами, если молекула лекарственного средства становится составной частью двухслойной липидной мембраны, и в этом случае гидрофобное лекарственное средство растворяется в липидной части липидного двойного слоя. Типовой липосомальный состав содержит воду с фосфолипидом при концентрации 5-20 мг/мл, изотоническое средство, буфер с pH 5-8 и, необязательно, холестерин.
Составы могут помещаться в контейнеры, рассчитанные на одну или несколько доз, например запаянные ампулы или сосуды, и могут храниться в высушенном при замораживании (лиофилизованном) состоянии, требуя лишь добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением.
Фармацевтический состав может быть получен лиофилизацией соединения формулы (I) или его кислотно-аддитивной соли. Лиофилизацией называют способ высушивания композиции при замораживании. Следовательно, термины «высушивание при замораживании» и «лиофилизация» используются в настоящей заявке как синонимы. Типовой способ заключается в том, чтобы солюбилизировать соединение, после чего полученный состав осветляют, фильтруют в стерильных условиях и асептически переносят в емкости, подходящие для лиофилизации (например, бутылочки). Что касается бутылочек, они частично закрыты лиофилизационными крышками. Состав может быть охлажден до замерзания, подвергнут лиофилизации в стандартных условиях и затем герметично закупорен, в результате чего получается стабильный сухой лиофильный состав. Композиция, как правило, будет иметь низкое остаточное содержание воды, например менее 5%, например менее 1% по массе, от массы лиофилизованного вещества.
Лиофилизованные составы могут содержать другие наполнители, например загустители, диспергирующие средства, буферные соединения, антиоксиданты, консерванты и регуляторы тоничности. Типовые буферные соединения включают фосфаты, ацетаты, цитраты и глицин. Примеры антиоксидантов включают аскорбиновую кислоту, бисульфит натрия, метабилсульфит натрия, монотиоглицерин, тиомочевину, бутилированный гидрокситолуол, бутилированный гидроксианизол и соли этилендиаминтетрауксусной кислоты. Консерванты могут включать бензойную кислоту и ее соли, сорбиновую кислоту и ее соли, алкиловые эфиры пара-гидроксибензойной кислоты, фенол, хлорбутанол, бензиловый спирт, тимеросал, хлорид бензалкония и хлорид цетилпиридиния. Упомянутые ранее буферные соединения, а также декстроза и хлорид натрия, могут применяться для регулирования тоничности в случае необходимости.
В технологии лиофилизации, как правило, применяются средства для придания объема с целью облегчения процесса и/или обеспечения объема и/или механической целостности лиофилизованной массы. Под средством для придания объема понимается свободно растворимый в воде наполнитель, состоящий из твердых частиц, который при совместной лиофилизации с соединением или его солью приводит к получению физически стабильной лиофилизованной массы, обеспечивает наиболее оптимальное протекание процесса лиофилизационной сушки, а также быстрое и полное восстановление. Средство для придания объема может также применяться с целью придания раствору изотоничности.
Водорастворимое средство для придания объема может быть любым из фармацевтически приемлемых инертных твердых веществ, обычно применяемых для лиофилизации. Такие средства для придания объема включают, например, сахара, такие как глюкоза, мальтоза, сахароза и лактоза; полиспирты, такие как сорбит или маннит; аминокислоты, такие как глицин; полимеры, такие как винилпирролидон; и полисахариды, такие как декстран.
Соотношение массы средства для придания объема к массе действующего соединения, как правило, находится в диапазоне от примерно 1 до примерно 5, например, от примерно 1 до примерно 3, например, в диапазоне от примерно 1 до примерно 2.
С другой стороны, можно получать составы в форме раствора, который можно концентрировать и герметически упаковывать в подходящие сосуды. Стерилизация дозированных форм может осуществляться путем фильтрования или нагревания в автоклаве сосудов вместе с их содержимым на подходящей стадии процесса получения составов. При применении составов могут потребоваться дополнительное разбавление или подготовка составов перед введением, например разбавление в подходящих стерильных упаковках для инфузии.
Растворы и суспензии для инъекций немедленного приготовления могут быть получены из стерильных порошков, гранул или таблеток.
В одном из предпочтительных вариантов осуществления настоящего изобретения фармацевтическая композиция имеет форму, подходящую для внутривенного введения, например инъекцией или инфузией.
Фармацевтические композиции по настоящему изобретению для парентеральных инъекций могут, кроме того, включать фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для восстановления в стерильные растворы или дисперсии для инъекций непосредственно перед применением. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или основ включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.п.), карбоксиметилцеллюлозу и ее подходящие смеси, растительные масла (как, например, оливковое масло) и пригодные для инъекций органические сложные эфиры, такие как этилолеат. Необходимая подвижность может поддерживаться, например, применением таких материалов для покрытия, как лецитин, подержанием требуемого размера частиц в случае дисперсий и применением ПАВ.
Композиции по настоящему изобретению могут также содержать вспомогательные вещества, такие как консерванты, средства для смачивания, эмульгаторы и диспергирующие средства. Предотвращение деятельности микроорганизмов может обеспечиваться включением различных антибактериальных и противогрибковых средств, например парабена, хлорбутанола, фенола, сорбиновой кислоты и т.п. Кроме того, может быть желательным включение изотонических средств, таких как сахара, хлорид натрия и т.п. Более продолжительная абсорбция фармацевтических форм, предназначенных для инъекции, может быть достигнута включением средств, которые замедляют абсорбцию, как, например, моностеарата алюминия и желатина.
Если соединение нестабильно или имеет низкую растворимость в водной среде, на его основе может быть получен концентрат в органических растворителях. Этот концентрат затем может быть разбавлен до более низкой концентрации в водной системе, и он может быть в основном стабильным в течение короткого периода времени введения препарата. Следовательно, в другом аспекте разработаны фармацевтические композиции, включающие неводный раствор, целиком составленный на основе одного или нескольких органических растворителей, который может вводиться сам по себе или, чаще, разбавленным перед введением подходящим внутривенным наполнителем (солевым раствором, декстрозой; забуференным или незабуференным) (Solubilizing excipients in oral and injectable formulations, Pharmaceutical Research, 21(2), 2004, p.201-230). Примерами растворителей и ПАВ являются пропиленгликоль, ПЭГ 300, ПЭГ 400, этанол, диметилацетамид (DMA), N-метил-2-пирролидон (NMP; Pharmasolve), глицерин, Cremophor EL, Cremophor RH 60 и polysorbate. Конкретные неводные растворы состоят из 70-80% пропиленгликоля и 20-30% этанола. Один из конкретных неводных растворов состоит из 70% пропиленгликоля и 30% этанола. Другой включает 80% пропиленгликоля и 20% этанола. Как правило, эти растворители используют в комбинации и обычно разбавляют, как минимум, в два раза перед внутривенной болюсной инъекцией или внутривенной инфузией. Типичными количествами для болюсных внутривенных составов являются ~50% для глицерина, пропиленгликоля, ПЭГ300, ПЭГ400 и ~20% для этанола. Типичными количествами для внутривенных составов для инфузии являются ~15% для глицерина, 3% для DMA и ~10% для пропиленгликоля, ПЭГ300, ПЭГ400 и этанола.
В одном из предпочтительных вариантов осуществления настоящего изобретения фармацевтическая композиция имеет форму, подходящую для внутривенного введения, например, инъекцией или инфузией. В случае внутривенного введения раствор можно вводить в исходном виде или первоначально можно ввести раствор в мешок для жидкостей (содержащий фармацевтически приемлемый наполнитель, как, например, 0,9% раствор соли или 5% декстрозу), перед введением пациенту.
В другом предпочтительном варианте осуществления фармацевтическая композиция имеет форму, подходящую для подкожного (s.c.) введения.
Фармацевтические дозированные лекарственные формы, удобные для перорального введения, включают таблетки, капсулы, таблетки в форме капсул, пилюли, ромбовидные таблетки, сиропы, растворы, порошки, гранулы, эликсиры и суспензии, сублингвальные таблетки, облатки или пластыри, а также буккальные пластыри.
Фармацевтические композиции, содержащие соединения формулы (I), могут создаваться в соответствии с известными методиками, см., например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, USA.
Так, например, композиции таблеток могут содержать единичную дозу действующего соединения совместно с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например лактоза, сахароза, сорбит или маннит; и/или с разбавителем, не являющимся сахаром, как, например, карбонатом натрия, фосфатом кальция, карбонатом кальция или целлюлозой или ее производными, такими как метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза, а также крахмалами, например, кукурузным крахмалом. Кроме того, таблетки могут содержать такие стандартные ингредиенты, как связующие или гранулирующие средства, как, например, поливинилпирролидон, дезинтегрирующие средства (например, способные к разбуханию поперечно сшитые полимеры, такие как поперечно сшитая карбоксиметилцеллюлоза), средства для скольжения (например, стеараты), консерванты (например, парабены), антиоксиданты (например, BHT), буферные средства (например, фосфатные или цитратные буферы), а также средства для придания шипучести, такие как смеси цитрат/бикарбонат. Подобные наполнители хорошо известны, и нет необходимости подробно обсуждать их здесь.
Капсулы могут иметь разновидности из твердого желатина и мягкого желатина и могут содержать действующий компонент в твердой, полутвердой или жидкой форме. Желатиновые капсулы могут быть получены из животного желатина или его эквивалентов синтетического или растительного происхождения.
Твердые дозированные формы (например, таблетки, капсулы и т.д.) могут быть или не быть покрытыми, но, как правило, имеют покрытие, например, покрыты защитной пленкой (например, из воска или глазури), или покрытием, регулирующим высвобождение лекарственного средства. Это покрытие (например, полимер типа EudragitTM) может быть предназначено для высвобождения действующего компонента в желаемом месте желудочно-кишечного тракта. Так, например, это покрытие можно выбрать таким образом, чтобы оно разрушалось при определенных значениях pH в желудочно-кишечном тракте, тем самым, селективно высвобождая соединение в желудке или в подвздошной кишке, или в двенадцатиперстной кишке.
Вместо покрытия или дополнительно к нему лекарственное средство может быть включено в твердую матрицу, содержащую средство, регулирующее высвобождение, например, средство, замедляющее высвобождение, которое может быть приспособлено для селективного высвобождения соединения в условиях изменяющейся кислотности или щелочности в желудочно-кишечном тракте. С другой стороны, материал матрицы или замедляющее высвобождение покрытие может принимать форму разрушаемого полимера (например, полимера малеинового ангидрида), который подвергается непрерывному разрушению при прохождении лекарственной формы через желудочно-кишечный тракт. В качестве еще одной альтернативы действующее соединение может быть включено в систему доставки, которая обеспечивает осмотическое регулирование высвобождения соединения. Составы с осмотическим высвобождением, а также другие составы с замедленным или продолжительным высвобождением могут быть получены способами, хорошо известными специалистам в данной области техники.
Фармацевтические композиции включают от примерно 1% до примерно 95%, предпочтительно от примерно 20% до примерно 90% активного ингредиента. Фармацевтические композиции по настоящему изобретению могут быть получены, например, в виде дозированных лекарственных форм, таких как ампулы, флаконы, суппозитории, драже, таблетки или капсулы.
Фармацевтические композиции для перорального введения могут быть получены смешиванием активного ингредиента с твердыми носителями, при необходимости, гранулированием полученной смеси, и дальнейшей переработкой полученной смеси, если это желательно или необходимо после добавления подходящих наполнителей, в таблетки, ядра драже или капсулы. Кроме того, имеется возможность включения активных ингредиентов в полимерные носители, что позволяет активным ингредиентам диффундировать или высвобождаться в рассчитанных количествах.
Композиции для местного нанесения включают мази, кремы, спреи, пластыри, гели, жидкие капли и вставки (например, внутриглазные вставки). Такие композиции можно создавать согласно известным способам.
Композиции для парентерального введения, как правило, представляют собой стерильные водные или масляные растворы, или тонкодисперсные суспензии, или же они могут иметь форму тонкодисперсного стерильного порошка, предназначенного для приготовления раствора для инъекций в стерильной воде непосредственно перед применением.
Примеры составов для ректального или интравагинального введения включают пессарии и суппозитории, которые могут быть изготовлены, например, из формованного пластичного или воскообразного вещества, содержащего действующее соединение.
Композиции для введения ингаляцией могут принимать форму пригодных для ингаляции порошкообразных композиций, или жидких, или порошкообразных спреев, и могут вводиться в стандартной форме с применением порошковых ингаляторов или устройств для дозирования аэрозолей. Такие устройства являются хорошо известными. Порошкообразные составы для введения ингаляцией, как правило, включают действующее соединение совместно с инертным твердым порошкообразным разбавителем, таким как лактоза.
Фармацевтические составы могут передаваться пациенту в «упаковках пациента», содержащих полный курс лечения в одной упаковке, обычно блистерной упаковке. Упаковка пациента имеет преимущество по сравнению с традиционными рецептами, где фармацевт отделяет потребление пациентом лекарственного средства от его общего количества, в том, что пациент всегда имеет доступ к вкладышу, содержащемуся в упаковке пациента, который обычно отсутствует в рецептах пациентов. Было показано, что включение вкладыша в упаковку улучшает готовность пациента выполнять указания врача.
Соединения по настоящему изобретению в основном будут представлены в дозированных лекарственных формах, которые, как правило, будут содержать достаточное количество соединения для обеспечения желаемого уровня биологической активности. Например, состав, предназначенный для перорального введения, может включать от 0,1 миллиграмма до 2 граммов активного ингредиента, например, от 1 нанограмма до 2 миллиграммов активного ингредиента. В этом диапазоне отдельными поддиапазонами содержания соединения являются от 0,1 миллиграмма до 2 граммов активного ингредиента (более обычно от 10 миллиграммов до 1 грамма, например, от 50 миллиграммов до 500 миллиграммов, или от 1 микрограмма до 20 миллиграммов (например, от 1 микрограмма до 10 миллиграммов, например, от 0,1 миллиграмма до 2 миллиграммов активного ингредиента)).
Для пероральных композиций одна единица дозированной лекарственной формы может содержать от 1 миллиграмма до 2 граммов, более типично от 10 миллиграммов до 1 грамма, например, от 50 миллиграммов до 1 грамма, например от 100 миллиграммов до 1 грамма действующего соединения.
Действующее соединение будет вводиться пациенту в случае необходимости (например, пациенту, являющемуся животным или человеком) в количестве, достаточном для достижения желаемого терапевтического эффекта.
Способы лечения
Предполагается, что соединения формул (I), (II), (III), (XXX) и их подгруппы, определенные в настоящей заявке, будут применимы в профилактике или лечении ряда заболеваний или болезненных состояний, опосредованных циклин-зависимыми киназами, киназой-3 гликоген-синтазы и Aurora киназами. Примеры таких заболеваний и состояний приведены выше.
Эти соединения, как правило, вводят субъекту, например человеку или животному, предпочтительно человеку, в случае необходимости такого введения.
Соединения, как правило, должны вводиться в количествах, которые являются терапевтически или профилактически полезными и которые, в основном, являются нетоксичными. Однако в некоторых ситуациях (например, в случае заболеваний, угрожающих жизни), польза от введения соединений формулы (I) может превзойти вред любых токсических или побочных эффектов, и в этом случае может оказаться желательным вводить соединения в количествах, которые связаны с определенной степенью токсичности.
Соединения могут вводиться в течение продолжительного срока для поддержания благоприятных терапевтических результатов или же могут вводиться лишь в течение короткого периода. С другой стороны, их можно вводить в пульсирующем или непрерывном режиме.
Типовая дневная доза соединения может находиться в пределах от 100 пикограммов до 100 миллиграммов на килограмм массы тела, более типично от 5 нанограммов до 25 миллиграммов на килограмм массы тела и более обычно от 10 нанограммов до 15 миллиграммов на килограмм (например, от 10 нанограммов до 10 миллиграммов и, более типично, от 1 микрограмма на килограмм до 20 миллиграммов на килограмм, как, например, от 1 микрограмма до 10 миллиграммов) массы тела, хотя при необходимости могут вводиться более высокие или более низкие дозы.
Описываемые соединения (например, такое соединение как 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина или ее соли, как, например, лактат или цитрат) могут вводиться ежедневно или введение может повторяться, например, каждые 2 или 3, или 4, или 5, или 6, или 7, или 10, или 14, или 21, или 28 дней. Однако, в конечном счете, количество вводимого соединения и тип применяемой композиции должны соответствовать природе заболевания или физиологического состояния, подвергаемого лечению, и должны выбираться по усмотрению врача.
Пример режима ежедневной дозировки для такого соединения, как 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина или ее солей, таких как лактат (в частности, L-лактат) или цитрат, включает введение указанного соединения (например, в форме L-лактата) при начальной дозировке, равной 1 мг/м2/день - 100 мг/м2/день, в частности 1 мг/м2/день - 10 мг/м2/день, более конкретно 3-6 мг/м2/день (что эквивалентно 2,5-5 мг свободного основания/м2/день) или при эффективной дозировке лактата, равной 2,5 мг/м2/день - 1,5 г/м2/день, в частности 25 мг/м2/день - 600 мг/м2/день, более конкретно 200-500 мг/м2/день, как, например, 250 мг/м2/день или 45-200 мг/м2/день, например, 45-150 мг/м2/день или 56-185 мг/м2/день (что эквивалентно 45-150 мг свободного основания /м2/день), хотя при необходимости могут вводиться более высокие или более низкие дозы.
В одной конкретной схеме введения пациент будет получать непрерывную внутривенную инфузию 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины или ее соли, как, например, лактата (в частности, L-лактата) или цитрата на протяжении периодов от 2 до 120 часов, например от 2 до 96 часов, в частности от 24 до 72 часов, и введение повторяют через желаемый промежуток времени, как, например, один раз в одну-три недели.
Более конкретно, пациент может получать непрерывную внутривенную инфузию 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины или ее соли, как, например, лактата (в частности, L-лактата) или цитрата на протяжении периодов 24 часа ежедневно в течение 5 дней, причем введение повторяют каждую неделю, или в течение периодов 48 часов, причем введение повторяют каждые две недели или в течение периодов 72 часа, и введение повторяют каждые три недели.
В другой конкретной схеме введения пациент получает инфузию 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины или ее соли, как, например, лактата (в частности, L-лактата) или цитрата, в виде внутривенного болюса на протяжении 2 часов один день в неделю каждые 1, 2 или 3 недели или один раз на протяжении 2 часов каждую 1, 2 или 3 недели.
Более высокие дозы, как, например, 1,5 г/м2/день, могли бы вводиться с применением режимов дозировки с частыми периодами перерывов, как, например, 24-48 часовая непрерывная внутривенная инфузия раз в одну-две недели. Более низкие дозы также могли бы вводиться с применением режимов дозировки с более продолжительным дозированием (но по-прежнему циклических есть/нет), как, например, 48-72 часовая непрерывная внутривенная инфузия каждые две-три недели.
В частности, соединения формулы (I') или 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина или все ее соли, как, например, лактат или цитрат, в частности, лактат, могли бы вводиться пациенту в дозировке 250 мг/м2/день в течение 72 часов путем непрерывной внутривенной инфузии каждые три недели.
В другом варианте осуществления соединения формулы (I') или 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина или все ее соли, как, например, лактат или цитрат, в частности лактат, могли бы вводиться пациенту в течение пятидневного цикла введения.
В конечном счете количество вводимого соединения и тип применяемой композиции должны соответствовать природе заболевания или физиологического состояния, подвергаемого лечению, и выбираться по усмотрению врача.
Соединения формул (I), (II), (III), (XXX) и подгруппы этих соединений, определенные в настоящей заявке, могут вводиться в качестве единственного терапевтического средства или же они могут вводиться в комбинированной терапии с одним или несколькими другими соединениями для лечения конкретного болезненного состояния, например новообразования, такого как рак, как определено выше по тексту настоящей заявки. Примеры других терапевтических средств или видов лечения, которые могут вводиться или применяться совместно (или одновременно, или в другие промежутки времени) с соединениями по настоящему изобретению, включают, не ограничиваясь перечисленным, ингибиторы топоизомеразы, алкилирующие средства, антиметаболиты, ДНК-связывающие средства, ингибиторы микротубулина (нацеливающие на тубулин средства), причем конкретными примерами являются цисплатин, циклофосфамид, доксорубицин, иринотекан, флударабин, 5FU, таксаны, митомицин C и радиационная терапия.
Другие примеры терапевтических средств, которые могут вводиться совместно (или одновременно, или в другие промежутки времени) с соединениями формул (I), (II), (III), (XXX) и их подгруппами, определенными в настоящей заявке, включают моноклональные антитела и ингибиторы сигнальной трансдукции.
Для случая ингибиторов CDK или Aurora киназы, сочетаемых с другими видами терапии, пациенту можно давать два или несколько видов лечения при индивидуальном изменении схем дозировки и при различных путях введения.
Если соединения формулы (I) вводят в комбинированной терапии вместе с одним, двумя, тремя, четырьмя или большим количеством других терапевтических средств (предпочтительно, одним или двумя, более предпочтительно одним), эти соединения могут вводиться одновременно (либо в одном и том же, либо в различных фармацевтических составах) или последовательно. В случае последовательного введения они могут вводиться через небольшие интервалы (например, в течение периода 5-10 минут) или через более продолжительные интервалы (например, через 1, 2, 3, 4 или более часов, или, при необходимости, даже через более длительные периоды времени), причем точные режимы дозировки согласуются со свойствами терапевтического средства (средств).
Кроме того, соединения по настоящему изобретению могут применяться совместно с нехимиотерапевтическими способами лечения, такими как радиационная терапия, фотодинамическая терапия, генная терапия; хирургия и регулируемые диеты.
Для применения в комбинированной терапии с другими химиотерапевтическими средствами соединение формулы (I) и одно, два, три, четыре или большее число других терапевтических средств может быть, например, совместно включено в лекарственную форму, содержащую два, три, четыре или большее количество терапевтических средств. В качестве альтернативы индивидуальные терапевтические средства могут быть включены в отдельные составы и предоставляться совместно в форме комплекта, необязательно с инструкцией по применению.
Специалист в данной области техники из своей обычной общеобразовательной подготовки знает, какие режимы дозировки и комбинированные виды терапии следует применять.
Способы диагностики
Перед введением соединения формулы (I) пациент может быть подвергнут исследованию с целью определения, является ли заболевание или состояние, от которого страдает или может страдать пациент, заболеванием или состоянием, которое могло бы быть чувствительно к лечению соединением, обладающим активностью против Aurora и/или циклин-зависимых киназ.
Например, можно подвергнуть анализу взятый у пациента биологический образец с целью определения, является ли состояние или заболевание, такое как рак, от которого страдает или может страдать пациент, состоянием или заболеванием, которое характеризуется генетической аномалией или аномальной экспрессией белка, которая ведет к избыточной активации CDK или к увеличению чувствительности сигнального пути к нормальной активности CDK. Примеры таких аномалий, которые приводят к активации или увеличению чувствительности к сигналу CDK2, включают повышающую регуляцию циклина E (Harwell RM, Mull BB, Porter DC, Keyomarsi K.; J. Biol. Chem. 2004 Mar 26; 279(13): 12695-705) или утрату p21 или p27 или наличие вариантов CDC4 (Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C.; Nature. 2004 Mar 4; 428(6978): 77-81). Опухоли с мутантами CDC4 или повышающей регуляцией, в частности избыточной экспрессией, циклина E, или утратой p21 либо p27 могут быть особенно чувствительными к ингибиторам CDK. В качестве альтернативы или дополнения биологический образец, взятый у пациента, может быть проанализирован с целью определения, является ли состояние или заболевание, такое как рак, от которого страдает или может страдать пациент, состоянием или заболеванием, которое характеризуется повышающей регуляцией Aurora киназы и, таким образом, может быть особенно чувствительно к ингибиторам Aurora киназы. Термин «повышающая регуляция» включает повышенную или избыточную экспрессию, включая амплификацию гена (т.е. многочисленные копии гена) и увеличенную экспрессию под влиянием транскрипционного эффекта, а также гиперактивность и активацию, в т.ч. активацию в результате мутаций.
Так, например, пациент может быть подвергнут диагностическому тесту для обнаружения маркера, характерного для избыточной экспрессии, повышающей регуляции или активации Aurora киназы, или же пациент может быть подвергнут диагностическому тесту для обнаружения маркера, характерного для повышающей регуляции циклина E, или утраты p21 или p27, или присутствия вариантов CDC4. Термин «диагностика» включает тестирование. В число маркеров авторы включают генетические маркеры, в т.ч., например, исследование структуры ДНК для выявления мутаций Aurora или CDC4. Кроме того, термин «маркер» включает маркеры, которые являются характерными для повышающей регуляции Aurora киназы или циклина E, включая активность ферментов, уровни ферментов, состояние ферментов (например, фосфорилированы или нет), а также уровни мРНК упомянутых выше белков. Опухоли с повышающей регуляцией циклина E, или утратой p21 или p27 могут быть особенно чувствительны к ингибиторам CDK. Перед лечением опухоли могут быть преимущественно исследованы на повышающую регуляцию циклина E или утрату p21 или p27. Так, пациент может быть подвергнут диагностическому тестированию с целью обнаружения маркера, характерного для повышающей регуляции циклина E, или утраты p21 или p27.
Диагностические тесты, как правило, проводят на биологических образцах, выбранных из образцов биопсии опухоли, образцов крови (выделение и обогащение отделившимися опухолевыми клетками), биопсий стула, мокроты, хромосомных анализов, плевральной жидкости, перитонеальной жидкости или мочи.
Было обнаружено, см. Ewart-Toland et al., (Nat Genet. 2003 Aug; 34(4): 403-12), что индивидуумы, образующие часть группы населения, обладающей вариантом Ile31 гена STK (гена Aurora киназы A), могут иметь повышенную восприимчивость к некоторым формам рака. Следовательно, представляется, что таким индивидуумам, страдающим от рака, будет полезно введение соединений, обладающих ингибирующей активностью в отношении Aurora киназы. Следовательно, пациент, страдающий от рака или подозреваемый в наличии рака, может быть протестирован для определения, является ли он или она частью группы населения с вариантом Ile31. Кроме того, Rajagopalan и соавторы (Nature. 2004 Mar 4; 428 (6978): 77-81) обнаружили, что при колоректальных раковых заболеваниях и раковых заболеваниях матки человека, в CDC4 (известном также, как Fbw7 или архипелаг) присутствуют мутации (Spruck et al, Cancer Res. 2002 Aug 15; 62 (16): 4535-9). Выявление индивидуума, имеющего мутацию в CDC4, может означать, что пациент мог бы особенно хорошо подходить для лечения ингибиторами CDK. Перед лечением опухоли преимущественно могли бы быть исследованы на наличие варианта CDC4. Способ исследования обычно будет включать прямое секвенирование, олигонуклеотидный микроматричный анализ или мутант-специфичные антитела.
Опухоли с активирующими мутантами Aurora или повышающей регуляцией Aurora, включая любую из изоформ фермента, могут быть особенно чувствительными к ингибиторам Aurora киназы. Перед лечением опухоли могут быть преимущественно исследованы на повышающую регуляцию Aurora или на ген Aurora, обладающий вариантом Ile31 (Ewart-Toland et al., Nat Genet. 2003 Aug; 34(4): 403-12). Ewart-Toland и соавторы идентифицировали распространенный генетический вариант в STK15 (приводящий к аминокислотному замещению F31I), который преимущественно амплифицирован и связан со степенью анеуплоидии в опухолях ободочной кишки человека. Эти результаты согласуются с важной ролью варианта Ile31 STK15 в восприимчивости людей к раку. В частности, предполагалось, что этот полиморфизм в Aurora A является генетическим модификатором для развивающейся карциномы груди (Sun et al., Carcinogenesis, 2004, 25 (11), 2225-2230).
Ген Aurora A отображает область хромосомы 20q13, которая часто амплифицирована при многих раковых заболеваниях, например груди, мочевого пузыря, ободочной кишки, яичников, поджелудочной железы. Пациенты с опухолями, которые имеют амплификацию этого гена, могли бы быть особенно чувствительными к лечению, нацеленному на ингибирование Aurora киназы.
Способы идентификации и анализа мутаций и повышающей регуляции белка, например изоформ Aurora, а также амплификации хромосомы 20q13 известны специалисту в данной области техники. Способы тестирования могли бы включать, не ограничиваясь перечисленным, стандартные способы, такие как полимеразная цепная реакция с обратной транскриптазой (RT-PCR) или гибридизация in-situ.
При тестировании способом RT-PCR уровень мРНК в опухоли определяют путем создания кДНК копии мРНК с последующей амплификацией кДНК с помощью ПЦР. Способы PCR-амплификации, выбора праймеров и условий амплификации известны специалисту в данной области техники. Операции с нуклеиновыми кислотами и PCR выполняют по стандартным методикам, описанным, например, в Ausubel, F.M. et al., eds. Current Protocols in Molecular Biology, 2004, John Wiley & Sons Inc. или Innis, M.A. et al., eds. PCR Protocols: a guide to methods and applications, 1990, Academic Press, San Diego. Реакции и операции, включающие методики, которые относятся к нуклеиновым кислотам, описаны также в Sambrook et al., 2001, 3rd Ed, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press. С другой стороны, могут применяться коммерчески доступный набор для RT-PCR (например, Roche Molecular Biochemicals) или методики, изложенные в патентах США 4666828, 4683202, 4801531, 5192659, 5272057, 5882864 и 6218529 и включенные в настоящую заявку с помощью ссылки.
Примерами методик гибридизации in-situ для оценки экспрессии мРНК могла бы служить флуоресцентная гибридизация in-situ (FISH) (см. Angerer, 1987 Meth. Enzymol., 152: 649).
Как правило, гибридизация in situ включает следующие основные стадии: (1) фиксацию ткани, которую предполагается анализировать; (2) предгибридизационную обработку образца для увеличения доступности целевой нуклеиновой кислоты и снижения неспецифического связывания; (3) гибридизацию смеси нуклеиновых кислот с нуклеиновой кислотой в биологической структуре ткани; (4) послегибридизационную промывку для удаления фрагментов нуклеиновых кислот, не связанных во время гибридизации, и (5) определение гибридизованных фрагментов нуклеиновых кислот. Зонды, применяемые в таких приложениях, как правило, метят, например, радиоактивными изотопами или флуоресцентными репортерами. Предпочтительные зонды обладают значительной длиной, например, от примерно 50, 100 или 200 нуклеотидов до примерно 1000 или более нуклеотидов, чтобы сделать возможной специфичную гибридизацию с целевой нуклеиновой кислотой (кислотами) в жестких условиях. Стандартные способы проведения FISH описаны в Ausubel, F.M. et al., eds. Current Protocols in Molecular Biology, 2004, John Wiley & Sons Inc и Fluorescence In Situ Hybridization: Technical Overview by John M.S. Bartlett in Molecular Diagnosis of Cancer, Methods and Protocols, 2nd ed.; ISBN: 1-59259-760-2; March 2004, pps. 077-088; Series Methods in Molecular Medicine.
С другой стороны, белковые продукты, экспрессируемые из мРНК, могут быть исследованы иммуногистохимией образцов опухолей, твердофазным иммуноанализом на микротитровальных планшетах, Вестерн-блоттингом, 2-мерным электрофорезом на SDS-полиакриламидном геле, ELISA, цитометрией в потоке, а также другими способами, известными в технике, для обнаружения конкретных белков. Способы обнаружения могли бы включать применение сайт-специфичных антител. Специалист должен отдавать себе отчет, что все такие хорошо известные методики выявления повышающей регуляции циклина E, или утраты p21 или p27, или обнаружения вариантов CDC4, повышающей регуляции Aurora и мутантов Aurora могли бы быть применимы в данном случае.
Следовательно, все эти методики также могли бы применяться для отождествления опухолей, особенно подходящих для лечения соединениями по настоящему изобретению.
Опухоли с мутантами CDC4 или повышающей регуляцией, в частности избыточной экспрессией циклина E, или утратой p21 или p27, могут быть особенно чувствительны к ингибиторам CDK. Перед началом лечения опухоли могут быть преимущественно исследованы на повышающую регуляцию, в частности избыточную экспрессию циклина E (Harwell RM, Mull BB, Porter DC, Keyomarsi K.; J. Biol. Chem. 2004 Mar 26; 279 (13): 12695-705) или утрату p21 или p27, или на наличие вариантов CDC4 (Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C.; Nature. 2004 Mar 4; 428 (6978): 77-81).
Пациенты с лимфомой мантийной ткани (MCL) могут быть выбраны для лечения соединениями по настоящему изобретению с применением диагностических тестов, описанных в настоящей заявке. MCL является особой клинико-патологической разновидностью неходжкинской лимфомы, характеризующейся пролиферацией лимфоцитов от небольшого до среднего размера с коэкспрессией CD5 и CD20, агрессивным и неизлечимым клиническим течением и частой транслокацией t(11; 14)(q13; q32). Избыточная экспрессия мРНК циклина D1, найденная в лимфоме мантийных клеток (MCL), является важнейшим диагностическим маркером. Yatabe и соавторы (Blood. 2000 Apr 1; 95 (7): 2253-61) предположили, что наличие циклина D1 должно учитываться как один из стандартных критериев для диагностики MCL, и необходимо исследовать инновационные способы терапии этого неизлечимого заболевания, основанные на указанном новом критерии. Jones и соавторы (J. Mol. Diagn. 2004 May; 6 (2): 84-9) разработали количественный PCR анализ с обратной транскрипцией в режиме реального времени для экспрессии циклина D1 (CCND1) для помощи в диагностике лимфомы мантийных клеток (MCL). Howe и соавторы (Clin Chem. 2004 Jan; 50 (1): 80-7) применяли количественный RT-PCR анализ в реальном времени для оценки экспрессии мРНК циклина D1 и обнаружили, что количественный RT-PCR для мРНК циклина D1, нормированный по мРНК CD19, может применяться для диагностики MCL в крови, костном мозге и тканях. С другой стороны, пациенты с раком груди для лечения ингибиторами CDK могли бы выбираться с применением изложенных выше диагностических тестов. Опухолевые клетки обычно экспрессируют циклин E в избыточных количествах, и было показано, что циклин E избыточно экспрессируется в раковых опухолях груди (Harwell et al, Cancer Res, 2000, 60, 481-489). Следовательно, рак груди можно лечить, в частности, ингибиторами CDK, разработанными в настоящем изобретении.
Противогрибковое применение
В еще одном аспекте настоящее изобретение относится к применению соединений формул (I), (II), (III), (XXX) и их подгрупп, определенных в настоящей заявке, в качестве противогрибковых средств.
Соединения формул (I), (II), (III), (XXX) и их подгруппы, определенные в настоящей заявке, могут применяться в ветеринарии (например, при лечении млекопитающих, как, например, людей), или для обработки растений (например, в сельском хозяйстве и садоводстве), или как противогрибковые средства общего действия, например, в качестве консервантов и дезинфицирующих средств.
В одном из вариантов осуществления настоящее изобретение относится к соединениям формул (I), (II), (III), (XXX) и их подгруппам, определенным в настоящей заявке, для применения в профилактике или лечении грибковых инфекций у млекопитающих, как, например, людей.
Кроме того, разработано применение соединений формул (I), (II), (III) и их подгрупп, определенных в настоящей заявке, для изготовления лекарственного средства, предназначенного для применения в профилактике или лечении грибковых инфекций у млекопитающих, как, например, людей.
Например, соединения по настоящему изобретению могут вводиться пациентам из числа людей, страдающих или у которых есть риск возникновения местных грибковых инфекций, вызванных, в числе прочих микроорганизмов, видами Candida, Trichophyton, Microsporum или Epidermophyton, или инфекций слизистых оболочек, вызванных Candida albicans (т.е. молочницы и вагинального кандидоза). Кроме того, соединения по настоящему изобретению могут вводиться для лечения или профилактики системных грибковых инфекций, вызванных, например, Candida albicans, Cryptococcus neoformans, Aspergillus flavus, Aspergillus fumigatus, Coccidiodies, Paracoccidiodies, Histoplasma или Blastomyces.
В другом аспекте изобретение относится к противогрибковым композициям для сельскохозяйственного (включая садоводство) применения, содержащим соединения формул (I), (II), (III), (XXX) и их подгрупп, описанных в настоящей заявке, совместно с приемлемым для сельскохозяйственного применения разбавителем или носителем.
Кроме того, изобретение относится к способу лечения животных (включая млекопитающих, как, например, человека), обработки растений или семян, имеющих грибковую инфекцию, который включает обработку указанных млекопитающих, растений или семян или очага инфекции указанных растений или семян эффективным количеством соединений формул (I), (II), (III), (XXX) и их подгрупп, описанных в настоящей заявке.
Помимо этого изобретение относится к способу борьбы с грибковой инфекцией растений или семян, который включает обработку растений или семян эффективным против грибков количеством фунгицидной композиции, содержащей соединение формул (I), (II), (III), (XXX) и их подгрупп, описанных в настоящей заявке.
Для выбора соединений по настоящему изобретению, обладающих специфичностью в отношении нечеловеческих ферментов CDK, могут применяться различные скрининговые исследования. Соединения, которые специфично воздействуют на ферменты CDK эукариотических патогенов, могут применяться в качестве противогрибковых и антипаразитарных средств. Ингибиторы CDK киназы Candida, т.е. CKSI, могут применяться в лечении кандидозов. Противогрибковые агенты могут применяться против инфекций указанных выше типов, или условно-патогенных инфекций, которые обычно встречаются у ослабленных и иммунодепрессивных пациентов, как, например, пациентов с различными видами лейкемии и лимфомы, людей, получающих иммунодепрессивную терапию, и пациентов с предрасполагающими состояниями, такими как сахарный диабет или СПИД, а также у неиммунодепрессивных пациентов.
Описанные в технике способы анализа могут применяться для отбора средств, которые могут быть полезны для ингибирования хотя бы одного грибка, вовлеченного в грибковое заболевание, такое как кандидоз, аспергиллез, мукормикоз, бластомикоз, геотрихоз, криптококкоз, хромобластомикоз, кокцидиоидомикоз, конидиоспороз, гистоплазмоз, мадуромикоз, риноспоридиоз, нокардиоз, пара-актиномикоз, пенициллиоз, монолиаз или споротрихоз. Для идентификации противогрибковых средств, которые могут иметь терапевтическую ценность при лечении аспергиллеза, могут применяться различные скрининговые исследования с использованием генов CDK, клонированных из дрожжевых грибков, как, например, Aspergillus fumigatus, Aspergillus flavus, Aspergillus niger, Aspergillus nidulans или Aspergillus terreus, или, если грибковая инфекция представляет собой mucon-nycosis, анализ CDK может быть проведен на основе таких дрожжевых грибков как Rhizopus arrhizus, Rhizopus oryzae, Absidia corymbifera, Absidia ramose или Mucorpusillus. Источники других ферментов CDK включают патогенный микроорганизм Pneumocystis carinii.
В качестве примера in vitro оценка противогрибковой активности соединений может быть проведена с помощью определения минимальной ингибирующей концентрации (M.I.C.), которая представляет собой концентрацию тестируемого соединения в подходящей среде, при которой наблюдается ослабление роста конкретного микроорганизма. На практике засевают стандартной культурой, например Candida albicans, ряд чашек с агаром, каждая из которых содержит тестируемое соединение в определенной концентрации, и затем инкубируют каждую чашку в течение соответствующего периода времени при 37°C. Затем чашки исследуют на наличие или отсутствие роста грибков и отмечают соответствующее значение M.I.C. С другой стороны, может быть выполнен анализ мутности жидких культур, причем протокол, описывающий пример этого анализа, может быть найден в примере 64.
Оценка активности соединений in vivo может быть осуществлена в результате введения соединений при различных уровнях дозировки с помощью интраперитонеальных или внутривенных инъекций, или перорального приема, мышам, которые были заражены грибками, например штаммом Candida albicans или Aspergillus flavus. Активность соединений можно оценить, наблюдая за развитием грибковой инфекции в группах мышей, которым вводили и не вводили соединения (с помощью гистологии или путем отбора грибков у инфицированных животных). Активность может измеряться уровнем дозировки, при которой соединение обеспечивает 50% защиту против летального исхода инфекции (PD50).
Для противогрибкового применения у человека соединения формул (I), (II), (III), (XXX) и их подгруппы, определенные в настоящей заявке, могут вводиться сами по себе или в виде смеси с фармацевтическим носителем, выбранным в соответствии с предполагаемым путем введения и стандартной фармацевтической практикой. Так, например, они могут вводиться перорально, парентерально, внутривенно, внутримышечно или подкожно, в виде составов, описанных выше в разделе, озаглавленном «фармацевтические составы».
Для перорального или парентерального введения людям уровень дневной дозировки противогрибковых соединений по настоящему изобретению может составлять от 0,01 до 10 мг/кг (в виде отдельных доз) в зависимости, в том числе, от эффективности соединений при введении либо пероральным, либо парентеральным путем. Таблетки или капсулы соединений могут содержать, например, от 5 мг до 0,5 г действующего соединения для введения одной, двух или нескольких таблеток или капсул в соответствующее время. В любом случае врач должен определить фактическую дозировку (эффективное количество), которая будет в наибольшей степени подходить для данного пациента, и эта дозировка будет изменяться в зависимости от возраста, веса и реакции конкретного пациента.
С другой стороны, противогрибковые соединения могут вводиться в форме суппозитория или пессария, или их можно наносить топически в форме лосьона, раствора, крема, мази или пудры. Например, их можно включать в состав крема, состоящего из водной эмульсии полиэтиленгликолей или жидкого парафина; или они могут быть включены в концентрации от 1 до 10% в мазь, состоящую из основы, например отбеленного воска или белого мягкого парафина, а также тех стабилизаторов и консервантов, которые могут быть необходимы.
Кроме описанного выше терапевтического применения противогрибковые средства, отобранные с помощью различных скрининговых исследований, могут применяться, например, в качестве консервантов в продуктах питания, пищевых добавок для ускорения набора веса крупного рогатого скота, в дезинфицирующих составах для обработки неживой материи, например для удаления загрязнений больничного оборудования и помещений. Подобным же образом параллельное сравнение ингибирования CDK млекопитающих и CDK насекомых, как, например, гена CDK5 дрозофилы (Hellmich et al. (1994) FEBS Lett 356:317-21), позволит осуществить среди соединений по настоящей заявке выбор ингибиторов, которые по-разному действуют на ферменты человека/млекопитающих и насекомых. Соответственно в настоящем изобретении специально рассматриваются применение и включение соединений по настоящему изобретению в состав инсектицидов, как, например, для применения в регулировании числа насекомых, таких как плодовая мушка.
В еще одном варианте осуществления некоторые из рассматриваемых ингибиторов CDK могут быть выбраны на основе ингибирующей специфичности к CDK растений, относительно соответствующих ферментов животных. Например, растительная CDK может быть передана на различные тесты с одним или несколькими человеческими ферментами, для выбора тех соединений, которые обладают наибольшей селективностью в отношении ингибирования растительного фермента. Таким образом, в настоящем изобретении конкретно рассматриваются составы на основе описываемых ингибиторов CDK для применения в сельском хозяйстве, например, в форме дефолиантов и т.п.
Для целей сельского хозяйства и садоводства соединения по настоящему изобретению могут применяться в форме композиций, состав которых соответствует конкретному применению и намечаемой цели. Так, соединения могут применяться в форме дустов или гранул, протрав для семян, водных растворов, дисперсий или эмульсий, пропиток, спреев, аэрозолей или средств для окуривания. Кроме того, композиции могут поставляться в форме дисперсных порошков, гранул или зерен, или концентратов для разбавления перед применением. Подобные композиции могут содержать такие общепринятые носители, разбавители или вспомогательные вещества, которые известны и приемлемы в сельском хозяйстве и садоводстве, и они могут производиться согласно общепринятым способам. Кроме того, эти композиции могут включать другие активные ингредиенты, например, соединения, обладающие гербицидной или инсектицидной активностью, или другой фунгицид. Соединения и композиции могут применяться рядом способов, например их можно наносить непосредственно на листву, стебли, ветви, семена или корни растения, или на грунт, или другую ростовую среду, и они могут применяться не только для ликвидации заболевания, но также с целью профилактики, для защиты растений или семян от поражений. В качестве примера композиции могут содержать от 0,01 до 1% масс. активного ингредиента. Для применения в полевых условиях вероятные нормы расхода активного ингредиента могут быть от 50 до 5000 г/гектар.
Настоящее изобретение также предусматривает применение соединений формул (I), (II), (III), (XXX) и их подгрупп, определенных в настоящей заявке, для контроля над грибками, разрушающими древесину, и в обработке грунта, на котором растут растения, полей со всходами риса или воды для орошения. Кроме того, настоящим изобретением предусмотрено применение соединений формул (I), (II), (III), (XXX) и их подгрупп, определенных в настоящей заявке, для защиты хранящегося зерна и других веществ нерастительного происхождения от грибкового заражения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 представляет собой изображение молекулы дигидрата свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины с атомами в виде тепловых эллипсоидов, как описано в примере 69 ниже.
На фиг.2 показана схема кристаллической упаковки дигидрата свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, как описано в примере 69 ниже.
На фиг.3 показана картина XRPD 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания, как описано ниже в примере 70.
На фиг.4 показано изображение молекулы L-лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины с атомами в виде тепловых эллипсоидов, как описано в примере 71 ниже.
На фиг.5 показана схема кристаллической упаковки L-лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, как описано в примере 71 ниже.
На фиг.6 показаны картины XRPD исходного образца L-лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и образца, испытанного на устойчивость, как описано ниже в примере 72.
На фиг.7 показаны картины XRPD исходного образца 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания (FB1) и образца, испытанного на устойчивость, как описано ниже в примере 72.
На фиг.8 показаны картины XRPD исходного образца 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме дигидрата свободного основания (FB2) и образца, испытанного на устойчивость, как описано ниже в примере 72.
ПРИМЕРЫ
В этом разделе настоящее изобретение будет проиллюстрировано, но не ограничено, ссылками на конкретные варианты осуществления, описанные в следующих примерах.
В примерах используются следующие сокращения:
Система анализа ЖХ-МС и описание методик
В примерах полученные соединения характеризовали жидкостной хроматографией и масс-спектроскопией, используя описанные ниже системы и условия работы. Если в состав соединения входили атомы элементов, обладающих различными изотопами, и приведено единственное значение массы, приведенная для соединения масса соответствует массе соединения с одним изотопом (т.е. 35Cl; 79Br и т.д.). Применялось несколько описанных ниже систем, и они были оборудованы и настроены таким образом, чтобы функционировать в очень сходных рабочих режимах. Применявшиеся режимы работы также описаны ниже.
ЖХ-МС система очистки, управляемая молекулярной массой
Препаративная ЖХ-МС является стандартной и эффективной методикой, применяемой для очистки органических соединений с небольшими молекулами, таких, как соединения, описанные в настоящей заявке. Методики жидкостной хроматографии (ЖХ) и масс-спектрометрии (МС) могут изменяться для обеспечения лучшего разделения неочищенных веществ и лучшего детектирования образцов с помощью МС. Оптимизация препаративных градиентных методик ЖХ будет включать подбор колонок, летучих элюентов и модификаторов, а также градиентов. В технике хорошо известны способы оптимизации препаративных ЖХ-МС методик и их последующего применения для очистки соединений. Такие способы описаны в Rosentreter U, Huber U.; Optimal fraction collecting in preparative LC/MS; J Comb Chem.; 2004; 6(2), 159-64 и Leister W, Strauss K, Wisnoski D, Zhao Z, Lindsley C, Development of a custom high-throughput preparative liquid chromatography/mass spectrometer platform for the preparative purification and analytical analysis of compound libraries; J Comb Chem.; 2003; 5(3); 322-9.
Одна из таких систем для очистки соединений препаративной ЖХ-МС описана ниже, хотя специалист в данной области техники поймет, что могли бы применяться альтернативные описанным системы и методики. В частности, могли бы применяться методики, основанные на препаративной ЖХ на нормальной фазе вместо описанных здесь методик на обращенной фазе. Большинство препаративных ЖХ-МС систем используют ЖХ на обращенной фазе и летучие кислотные модификаторы, поскольку такой подход является весьма эффективным для очистки молекул небольшого размера и поскольку растворители совместимы с масс-спектроскопией с электрораспылением положительных ионов. В качестве альтернативы для очистки соединений могли бы применяться другие хроматографические методики, например, ЖХ на нормальной фазе, другое буферирование подвижной фазы, основные модификаторы и т.д., как показано в описанных выше способах анализа.
Препаративные системы ЖХ-МС:
Система Waters Fractionlynx:
- Оборудование:
2767 Dual Loop Autosampler/Fraction Collector
2525 preparative pump
CFO (column fluidic organiser) для выбора колонок
RMA (Waters reagent manager) как подкачивающий насос
Waters ZQ Mass Spectrometer
Waters 2996 Photo Diode Array detector
Waters ZQ Mass Spectrometer
- Программное обеспечение:
Masslynx 4,0
ЖХ-МС препаративная система Agilent 1100
- Оборудование:
Устройство авт.подачи образцов: 1100 series “prepALS”
Насос: 1100 series “PrepPump” для препаративной подачи градиентного растворителя и 1100 series “QuatPump” для подачи модификатора в поток растворителя
УФ-детектор: 1100 series “MWD” Multi Wavelength Detector
МС-детектор: 1100 series “LC-MSD VL”
Коллектор фракций: 2 × “Prep-FC”
Подкачивающий насос: “Waters RMA”
Agilent Active Splitter
- Программное обеспечение:
Chemstation: Chem 32
Условия проведения хроматографии:
- Колонки:
1. Хроматография при низких значениях pH:
Phenomenex Synergy MAX-RP, 10µ, 100×21,2 мм (в качестве альтернативы применялась Thermo Hypersil-Keystone HyPurity Aquastar, 5µ, 100 × 21,2 мм для более полярных соединений)
2. Хроматография при высоких значениях pH:
Phenomenex Luna С18(2), 10µ, 100×21,2 мм (в качестве альтернативы применялась Phenomenex Gemini, 5µ, 100×21,2 мм)
-Элюенты:
1. Хроматография при низких значениях pH:
Растворитель A: H2O + 0,1% муравьиная кислота, pH~1,5
Растворитель B: CH3CN + 0,1% муравьиная кислота
2. Хроматография при высоких значениях pH:
Растворитель A: H2O + 10 мМ NH4HCO3 + NH4OH, pH 9,2
Растворитель B: CH3CN
3. Дополнительный растворитель
MeOH + 0,2% Муравьиная кислота (для хроматографии обоих типов)
- Методики:
В соответствии с аналитическими признаками выбирали наиболее подходящий тип препаративной хроматографии. Типовая процедура заключалась в осуществлении аналитической ЖХ-МС с использованием наиболее подходящего для структуры соединения типа хроматографии (при низких или высоких значениях pH). Если аналитические признаки указывали на хороший результат хроматографии, выбирали подходящую препаративную методику того же типа. Типовые условия реализации хроматографических методик при низких и высоких значениях pH были следующими:
Скорость потока: 24 мл/мин
Градиент: В основном все градиенты включали исходную 0,4-мин стадию, когда подавалась смесь 95% A + 5% B. Затем в соответствии с аналитическим признаками выбирали 3,6-мин градиент для достижения хорошего разделения (например, от 5 до 50% B для плохо удерживаемых соединений, от 35 до 80% для среднеудерживаемых соединений и т.д.)
Промывка: В конце градиента проводили 1,2-мин стадию промывки
Возврат к равновесному состоянию: для подготовки системы к следующему запуску проводили 2,1-мин стадию возврата к равновесному состоянию
Скорость дополнительного потока: 1 мл/мин
- Растворитель:
Все соединения обычно растворяли в 100% MeOH или 100% ДМСО
На основе предоставленной информации кто-либо из специалистов в данной области техники мог бы очистить соединения, описанные в настоящей заявке, с помощью препаративной ЖХ-МС.
Исходные вещества в каждом из примеров являются коммерчески доступными, если не указано иначе.
Пример 1
Синтез 5-циано-2-метокси-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]бензамида
1A. Синтез (3,4-динитрофенил)морфолин-4-илметанона
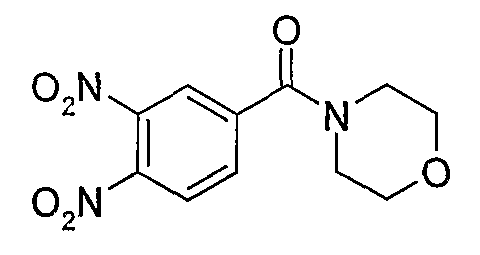
Смесь 3,4-динитробензойной кислоты (10,0 г) и тионилхлорида (30 мл) кипятили с обратным холодильником в течение 2 часов, охлаждали до комнатной температуры и удаляли избыток тионилхлорида азеотропной отгонкой с толуолом. Остаток растворяли в ТГФ (100 мл) и при 0°C одновременно добавляли к смеси морфолин (4,1 мл) и Et3N (7,2 мл). Смесь перемешивали в течение 3 часов, добавляли воду (100 мл) и затем экстрагировали EtOAc. Органическую фракцию промывали насыщенным раствором соли, высушивали (MgSO4) и упаривали в вакууме. Перекристаллизация остатка из MeOH давала (3,4-динитрофенил)морфолин-4-илметанон (8,23 г) в виде твердого вещества желтого цвета. (1H ЯМР (300 МГц, ДМСО-d6) δ 8,3 (д, 1H), 8,3 (с, 1H), 8,0 (д, 1H), 3,7-3,5 (м, 8H)).
1B. Синтез (3,4-диаминофенил)морфолин-4-илметанона
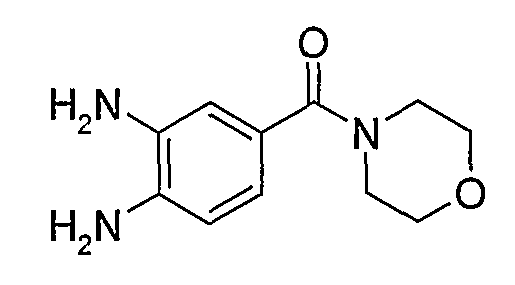
Смесь (3,4-динитрофенил)морфолин-4-илметанона (1,0 г) и 10% Pd/C (150 мг) в MeOH (30 мл) встряхивали в атмосфере водорода при комнатной температуре в течение 10 часов, затем фильтровали через слой целита и упаривали в вакууме, получая (3,4-диаминофенил)морфолин-4-илметанон (900 мг) (1H ЯМР (300 МГц, ДМСО-d6) δ 6,6 (с, 1H), 6,5 (с, 2H), 4,8 (с, 1,5H), 4,6 (с, 1,5H), 4,1 (с, 1H), 3,6 (м, 4H), 3,4 (м, 4H)).
1C. Синтез 4-морфолин-4-илметилбензол-1,2-диамина
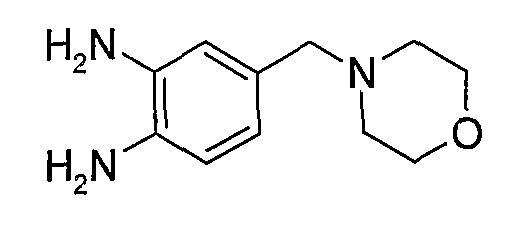
К смеси (3,4-динитрофенил)морфолин-4-илметанона (2,84 г) и сухого ТГФ (50 мл) добавляли NaBH4 (954 мг) и затем по каплям BF3 .Et2O (3,2 мл). Смесь перемешивали при комнатной температуре в течение 3 часов и затем гасили добавлением MeOH. Смесь упаривали в вакууме, распределяли между EtOAc и водой, органическую фракцию промывали насыщенным раствором соли, высушивали (MgSO4) и упаривали в вакууме. Остаток очищали колоночной флэш-хроматографией, элюируя EtOAc, и получали 4-(3,4-динитробензил)морфолин (1,08 г).
Смесь 4-(3,4-динитробензил)морфолина (550 мг) и 10% Pd/C (75 мг) в MeOH (10 мл) встряхивали в атмосфере водорода при комнатной температуре в течение 4 часов, затем фильтровали через слой целита и упаривали в вакууме, получая 4-морфолин-4-илметилбензол-1,2-диамин (483 мг) в качестве основного компонента смеси.
1D. Синтез 5-морфолин-4-илметил-2-(4-нитро-1H-пиразол-3-ил)-1H-бензимидазола
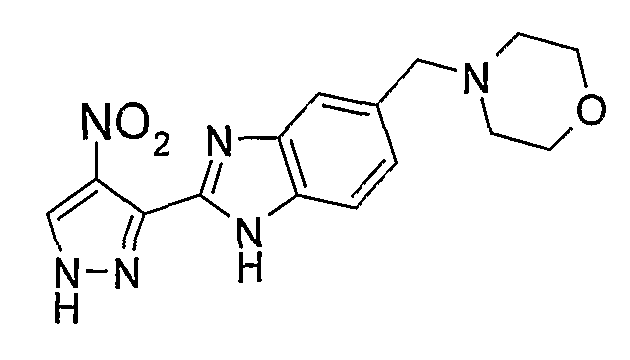
Смесь 4-морфолин-4-илметилбензол-1,2-диамина (2,3 г, 11,1 ммоль), 4-нитро-1H-пиразол-3-карбоновой кислоты (1,57 г, 10,0 ммоль), EDC (2,13 г, 11,1 ммоль) и HOBt (1,50 г, 11,1 ммоль) в сухом ДМФА (25 мл) перемешивали при комнатной температуре в течение 24 часов. Смесь упаривали в вакууме, неочищенный остаток растворяли в AcOH (40 мл) и кипятили с обратным холодильником в течение 3 ч. Растворитель удаляли в вакууме и остаток очищали колоночной флэш-хроматографией, элюируя 0-20% MeOH в EtOAc, получая 5-морфолин-4-илметил-2-(4-нитро-1H-пиразол-3-ил)-1H-бензимидазол в виде твердого желтого вещества (1,0 г, 61%). (ЖХ/МС: Rt 1,83, [M+H]+ 329).
1E. Синтез 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина

Палладий на угле (10%, 0,08 г) добавляли к раствору 5-морфолин-4-илметил-2-(4-нитро-1H-пиразол-3-ил)-1H-бензимидазола (0,82 г, 2,5 ммоль) в ДМФА (30 мл) в атмосфере азота. Смесь встряхивали в атмосфере водорода в течение 4 часов, затем фильтровали через целит, промывая метанолом. Фильтрат концентрировали в вакууме, получая 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламин в виде твердого коричневого вещества (530 мг, 71%). (ЖХ/МС: Rt 1,94, [M+H]+ 299).
1F. Синтез 5-циано-2-метоксибензойной кислоты
К смеси метил-2-гидрокси-5-цианобензоата (2 г, 5,6 ммоль) и K2CO3 (4,68 г, 16,8 ммоль) в ацетоне (50 мл) добавляли метилиодид (0,7 мл, 5,6 ммоль). После этого реакционную смесь нагревали при 65°C в течение ночи, получая твердое вещество, которое отфильтровывали, пока оно было горячим, и промывали ацетоном, получая метиловый эфир 5-циано-2-метоксибензойной кислоты (0,45 г). Неочищенный продукт растворяли в ТГФ (5 мл), затем обрабатывали раствором LiOH (0,108 г, 0,26 ммоль) в воде (5 мл) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь подкисляли 2М HCl и экстрагировали EtOAc (×2). Органическую фракцию высушивали (MgSO4) и упаривали в вакууме, получая 5-циано-2-метоксибензойную кислоту (0,277 г). (ЖХ/МС кислотн.: Rt 2,92, [M+H]+ 178).
1G. Синтез 5-циано-2-метокси-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]бензамида (способ с применением хлорангидрида кислоты)
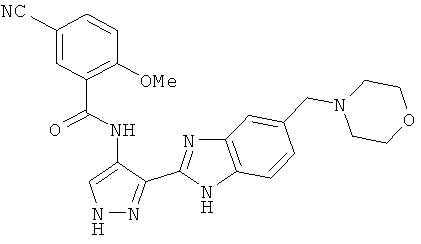
5-Циано-2-метоксибензойную кислоту (пример 1F) (40 мг, 0,22 ммоль) растворяли в дихлорметане (5 мл), после чего по каплям добавляли оксалилхлорид (34,4 мг, 0,264 ммоль) и затем ДМФА (1 каплю). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, упаривали в вакууме, затем повторно упаривали с применением толуола (×2). Смесь 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина (100 мг, 0,33 ммоль), 5-циано-2-метоксибензоилхлорида и диизопропилэтиламина (1,83 мкл, 0,9 ммоль) в ТГФ (5 мл) перемешивали при 0°C и затем давали нагреться до комнатной температуры в течение 2 часов. Затем реакционную смесь концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (SiO2, 5-7% MeOH-DCM), получая 5-циано-2-метокси-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]бензамид (12 мг). (ЖХ/МС кислотн.: Rt 2,02 мин, [M-H]+ 458).
Пример 2
Синтез [3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амида 6-метилимидазол[2.1-b]тиазол-5-карбоновой кислоты

Смесь 6-метилимидазол[2.1-b]тиазол-5-карбоновой кислоты (Bionet) (61 мг, 0,33 ммоль), 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина (100 мг, 0,33 ммоль), EDC (77 мг, 0,39 ммоль) и HOAt (54 мг, 0,39 ммоль) перемешивали в ДМФА (3 мл) при 80°C в течение 1 ч, затем при комнатной температуре в течение 20 ч. Смесь упаривали в вакууме и остаток распределяли между EtOAc и насыщенным NaHCO3. Органическую часть промывали насыщенным раствором соли, высушивали (MgSO4) и упаривали в вакууме. Остаток очищали препаративной ЖХ/МС, получая [3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амид 6-метилимидазо[2.1-b]тиазол-5-карбоновой кислоты (29 мг). (ЖХ/МС основн.: Rt 2,56, [M+H]+ 463).
Пример 3
Синтез 2-циано-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]ацетамида
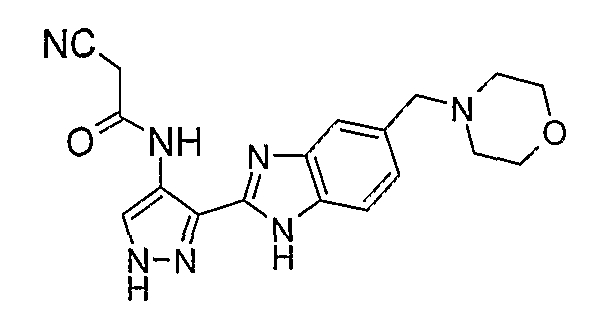
Смесь цианоуксусной кислоты (23 мг, 0,28 ммоль), 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина (70%, 100 мг, 0,23 ммоль), TBTU (89 мг, 0,28 ммоль) и ДМФА (2 мл) перемешивали при 25°C в течение ночи. Затем смесь упаривали в вакууме. После очистки флэш-хроматографией, при элюировании DCM-6% MeOH/DCM, получали 2-циано-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]ацетамид в виде твердого вещества желтого цвета (65 мг, 77%). (ЖХ/МС (кислотный способ/конечное соединение): Rt 4,61, [M+H]+ 366).
Пример 4
2-Циано-2-циклопропил-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]ацетамид
4A. Синтез цианоциклопропилуксусной кислоты
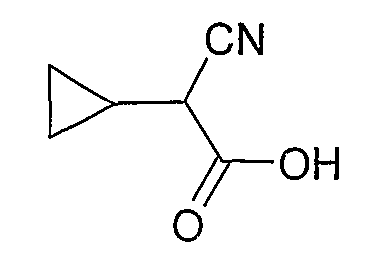
1н. NaOH (3,26 мл, 3,26 ммоль) добавляли к раствору этилового эфира цианоциклопропилуксусной кислоты (0,5 г, 3,26 ммоль) в ТГФ (15 мл). После 4-часового перемешивания при 25°C реакционную смесь упаривали в вакууме, повторно растворяли в воде (20 мл) и нейтрализовали добавлением 1н. раствора HCl (3,26 мл). Затем смесь экстрагировали EtOAc (3×20 мл) и объединенные, высушенные (Na2SO4) органические фракции упаривали в вакууме, получая загрязненную цианоциклопропилуксусную кислоту в виде прозрачного масла. Полученное вещество без всякой дальнейшей очистки использовали в синтезе примера 4B.
4B. 2-Циано-2-циклопропил-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]ацетамид
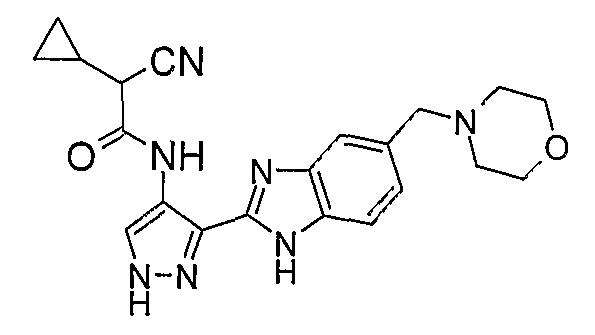
Продукт примера 4A вводили во взаимодействие с 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламином согласно способу, описанному в примере 3, за исключением того, что неочищенный продукт распределяли между дихлорметаном и насыщенным водным NaHCO3 и затем очищали растиранием с Et2O. ЖХ/МС (кислотн. способ): Rt 1,79, [M+H]+ 406.
Примеры 5-14
Получали соединения примеров 5-14, следуя способам примеров 1-3, модифицированных в тех случаях, которые показаны в таблице ниже.
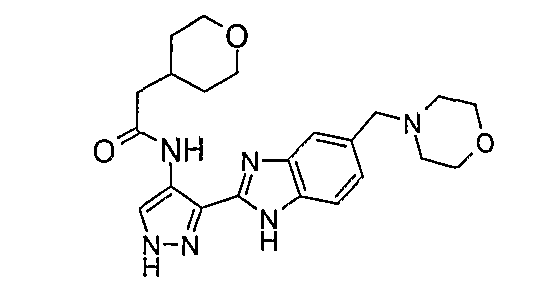
Rt 1,77 кислотн.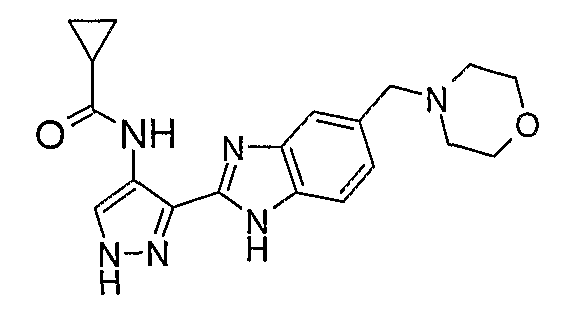
ЖХ/МС
Rt 2,45 основн.
ЖХ/МС
Rt 2,21 основн.
ЖХ/МС
Rt 1,74 кислотн.
ЖХ/МС
Rt 1,64 кислотн.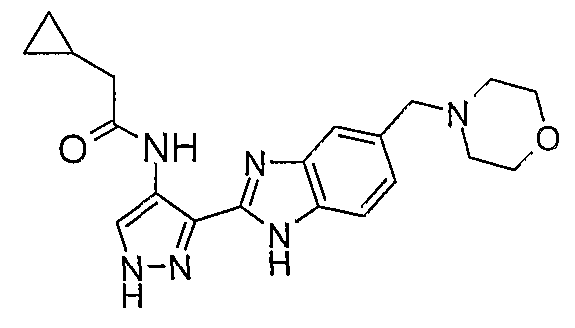
[SiO2, элюирование DMAW 240-120]
Rt 1,85
кислотн.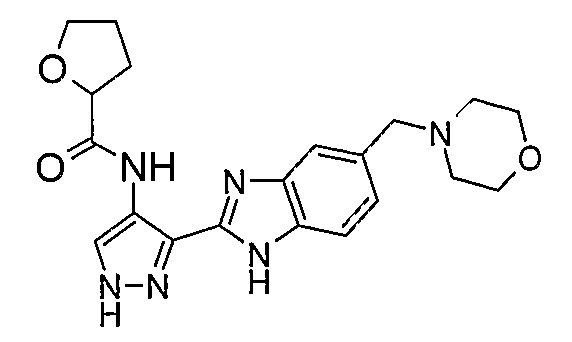
[SiO2, элюирование DMAW 240-120]
Rt 1,76
кислотн.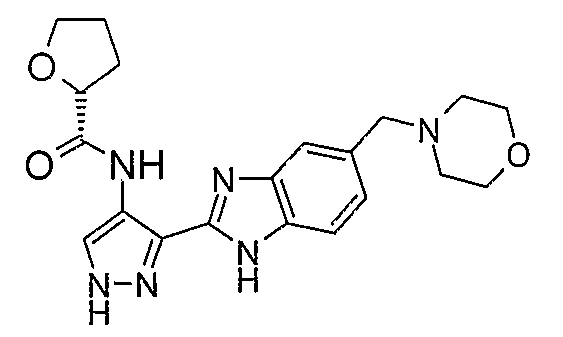
[SiO2, элюирование DMAW 240-120]
Rt 1,76
кислотн.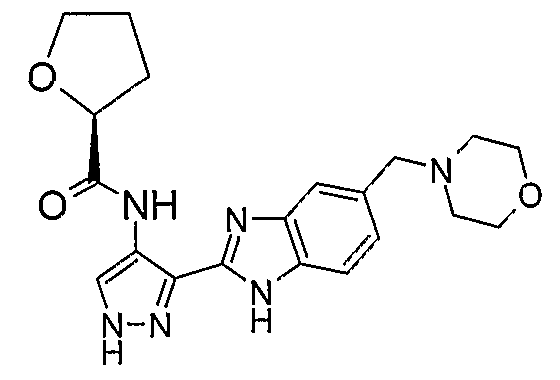
[SiO2, элюирование DMAW 240-120]
Rt 1,79
кислотн.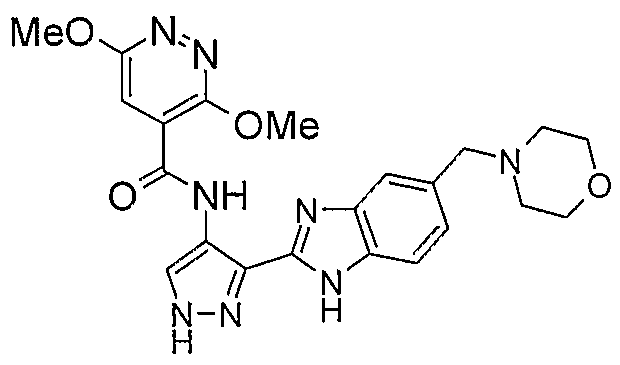
[SiO2, элюирование DMAW 240-120]
Rt 1,99
кислотн.
Пример 15
Синтез трифторацетата N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-2-морфолинкарбоксамида
15A. Синтез 5,6-диметокси-2-(4-нитро-1H-пиразол-3-ил)-1H-бензимидазола
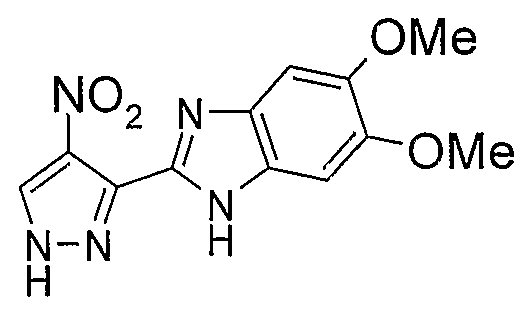
К раствору EDC (4,81 г, 25 ммоль), HOBt (3,40 г, 25 ммоль) и триэтиламина (4,67 г, 46 ммоль) в ДМФА (100 мл) добавляли 4-нитро-1H-пиразол-3-карбоновую кислоту (3,63 г, 23,09 ммоль) и дигидрохлорид 4,5-диметоксибензол-1,2-диамина (5,06 г, 20,99 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в вакууме и оставшееся твердое вещество распределяли между EtOAc (50 мл) и насыщенным водным NaHCO3 (50 мл). Образовавшийся осадок удаляли фильтрованием. Фильтрат промывали водой, диэтиловым эфиром и затем подвергали азеотропной отгонке с MeOH и толуолом, получая (2-амино-4,5-диметоксифенил)амид 4-нитро-1H-пиразол-3-карбоновой кислоты (2,35 г, 36%). (2-Амино-4,5-диметоксифенил)амид 4-нитро-1H-пиразол-3-карбоновой кислоты (2,35 г, 7,65 ммоль) растворяли в уксусной кислоте (150 мл) и кипятили с обратным холодильником при 140°C в течение 5 часов. Раствор оставляли охлаждаться и растворитель удаляли в вакууме. Полученное твердое вещество распределяли между EtOAc (25 мл) и насыщенным раствором соли (25 мл). Органический слой отделяли, высушивали (MgSO4), фильтровали и удаляли растворитель в вакууме, получая 5,6-диметокси-2-(4-нитро-1H-пиразол-3-ил)-1H-бензимидазол (2,08 г, 94%).
15B. Синтез 3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина
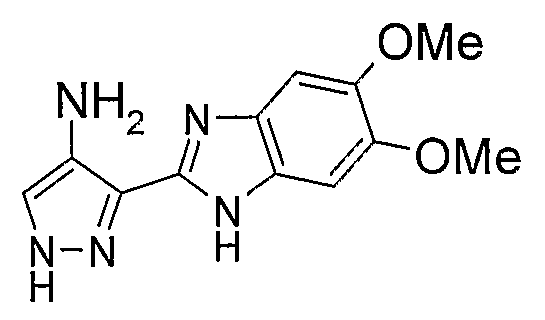
Смесь 5,6-диметокси-2-(4-нитро-1H-пиразол-3-ил)-1H-бензимидазола (2,08 г, 7,2 ммоль) и 10% палладия на угле (200 мг) в этаноле (150 мл) и ДМФА (50 мл) гидрировали при комнатной температуре и давлении в течение ночи. Реакционную смесь фильтровали через целит и удаляли растворитель в вакууме. Полученное твердое вещество подвергали азеотропной отгонке с метанолом и толуолом и удаляли растворитель в вакууме. Неочищенное вещество очищали флэш-хроматографией, элюируя смесью DCM:MeOH:уксусная кислота:вода (120:18:3:2) [DMAW120] и затем смесью DCM:MeOH:уксусная кислота:вода (90:18:3:2) [DMAW90]. Затем объединяли фракции, содержащие продукт, и удаляли растворитель в вакууме, получая 3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-иламин (~1 г, 53%).
15C. Синтез N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-4-BOC-2-морфолин карбоксамида
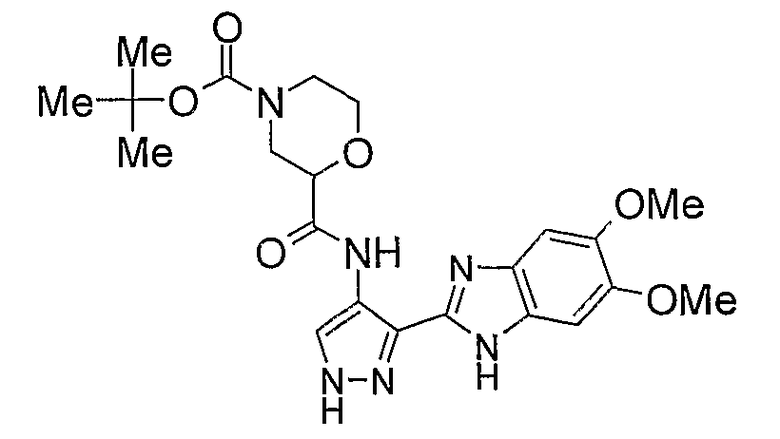
К раствору EDC (125 мг, 0,54 ммоль) и HOAt (74 мг, 0,54 ммоль) в ДМФА (2 мл) добавляли 3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-иламин (117 мг, 0,45 ммоль) (пример 15B) и (rac)-BOC-2-карбоксиморфолин (125 мг, 0,54 ммоль) и смесь перемешивали в течение ночи при комнатной температуре. Затем смесь распределяли между EtOAc и водой. После этого органический слой последовательно промывали насыщенным водным раствором бикарбоната натрия, насыщенным раствором соли и затем высушивали (MgSO4). Раствор выпаривали в вакууме досуха и очищали остаток колоночной флэш-хроматографией [SiO2, градиентное элюирование: EtOAc-гексан (1:1)- EtOAc-MeOH (80:20)], получая N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-4-BOC-2-морфолин карбоксамид (65 мг) в виде бесцветного твердого вещества. (ЖХ/МС (кислотн. способ): Rt 2,65 мин, [M+H]+ 473).
15D. Синтез трифторацетата N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-2-морфолинкарбоксамида
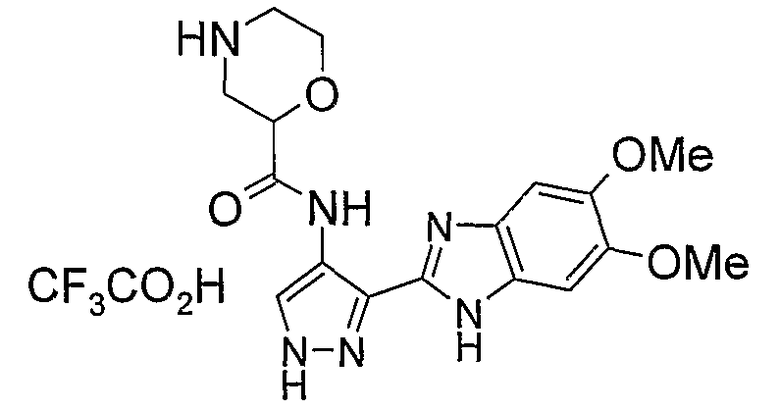
N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-4-BOC-2-морфолин карбоксамид (65 мг, 0,14 ммоль) и анизол (60 мкл, 0,56 ммоль) растворяли в смеси трифторуксусной кислоты и дихлорметана (1:1; 2 мл). Через три часа при комнатной температуре полученную смесь упаривали досуха, получая трифторацетат N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-2-морфолинкарбоксамида (73 мг) в виде бесцветного твердого вещества (ЖХ/МС (кислотн. способ): Rt 1,42 мин, [M-H]+ 371).
Пример 16
Синтез N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-4-изопропил-2-морфолин карбоксамида
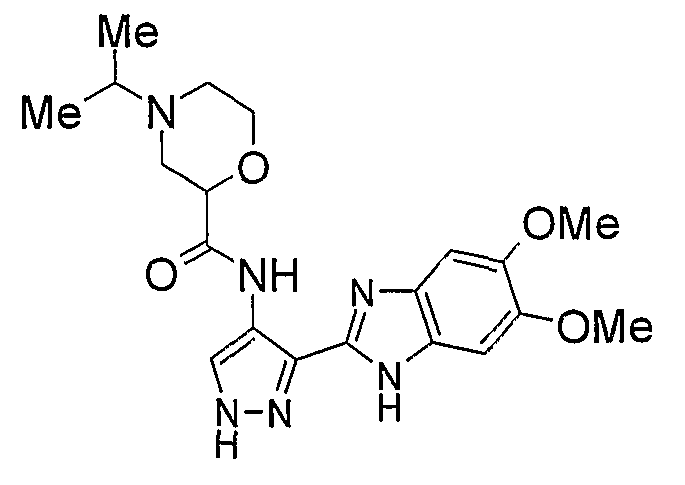
К трифторацетату N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-2-морфолинкарбоксамида (пример 15D) (34 мг, 0,07 ммоль) и K2CO3 (20 мг, 0,14 ммоль) в MeCN (1 мл) добавляли 2-иодпропан (17 мкл, 0,15 ммоль). Смесь перемешивали при 80°C в течение примерно 48 часов, после чего смесь концентрировали и очищали остаток флэш-хроматографией [SiO2, элюирование градиентом: DCM:MeOH (98:2) - DCM:MeOH:конц. водн. NH3 (90:10:1)], получая N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-4-изопропил-2-морфолинкарбоксамид (12 мг) в виде бесцветной смолы (ЖХ/МС (основн. способ): Rt 2,52 мин, [M+H]+ 415).
Пример 17
Синтез N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-1-метилпиперидин-3-карбоксамида
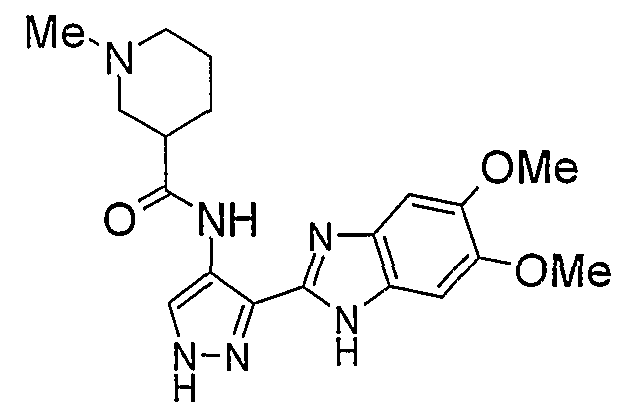
К раствору 3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина (65 мг, 0,25 ммоль) (пример 15B), гидрохлорида (rac)-1-метилпиперидин-3-карбоновой кислоты (50 мг, 0,27 ммоль) и диизопропилэтиламина (50 мкл, 0,27 ммоль) в ДМФА (1 мл) добавляли TBTU (97 мг, 0,30 ммоль). Смесь перемешивали при комнатной температуре в течение примерно 16 часов, после чего добавляли 1н. водный NaOH (1 мл) и смесь перемешивали еще 1 час. Затем смесь упаривали досуха в вакууме, остаток очищали колоночной флэш-хроматографией (SiO2, элюирование градиентом: DCM:MeOH (98:2) - DCM:MeOH:конц. водн. NH3 (70:30:3)), получая N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-rac-1-метилпиперидин-3-карбоксамид (20 мг) в виде бесцветной смолы (ЖХ/МС (основн. способ): Rt 2,35 мин, [M+H]+ 385).
Пример 18
Синтез 3-хлор-N-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил-5-(4-метилпиперазин-1-ил)бензамида
18A. Синтез 3-хлор-5-(4-метилпиперазин-1-ил)бензонитрила
5-Фтор-3-хлорбензонитрил (1 г, 6,4 ммоль) растворяли в ДМСО (20 мл) с последующим добавлением K2CO3 (1,3 г, 9,6 ммоль) и 1-метилпиперазина (1,4 мл, 12,8 ммоль). Реакционную смесь нагревали при 80°C в течение 20 часов. К неочищенному веществу добавляли диэтиловый эфир (10 мл), затем подкисляли 1н. HCl. От неочищенной реакционной смеси отфильтровывали осадок, получая 3-хлор-5-(4-метилпиперазин-1-ил)бензонитрил (1,4 г, выход 93%) в виде белого твердого вещества (ЖХ/МС: Rt 1,83 мин, [M+H]+ 236, кислотн. способ).
18B. Синтез 3-хлор-5-(4-метилпиперазин-1-ил)бензойной кислоты
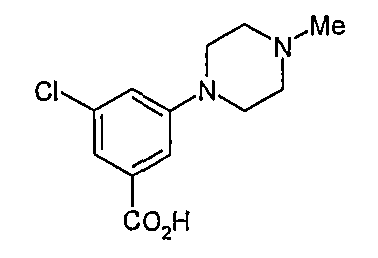
К 3-хлор-5-(4-метилпиперазин-1-ил)бензонитрилу (1,4 г, 5,9 ммоль), растворенному в этаноле (10 мл), добавляли 2М NaOH (20 мл) и реакционную смесь кипятили с обратным холодильником в течение 20 часов. Смесь упаривали в вакууме, подкисляли неочищенный продукт 1н. HCl до pH 6 и распределяли между EtOAc и H2O. Органический слой упаривали в вакууме досуха, получая 0,7 г указанного в заголовке соединения в виде белого твердого вещества (ЖХ/МС: Rt 1,67 мин, [M+H]+ 256, кислотн. способ).
18С. Синтез [3-хлор-N-[3-(5,6-диметокси-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-5-(4-метилпиперазин-1-ил)бензамида
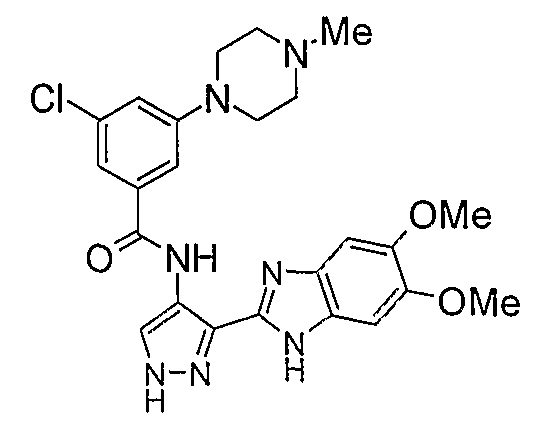
Соединение получают способом, аналогичным примеру 15C, но с применением 3-хлор-5-(4-метилпиперазин-1-ил)бензойной кислоты (200 мг, 0,78 ммоль) в качестве реагента в методике примера 15C вместо (rac)-BOC-2-карбоксиморфолина. Неочищенный продукт очищали колоночной флэш-хроматографией [SiO2, элюируя DMAW 240-90], получая 92 мг (выход 25%) указанного в заголовке соединения в виде светло-коричневого твердого вещества (ЖХ/МС: Rt 2,07, [M+H]+ 496).
Примеры 19-21
Следуя методике, изложенной в примере 15, измененной в тех случаях, где это указано, получали соединения примеров 19-21.
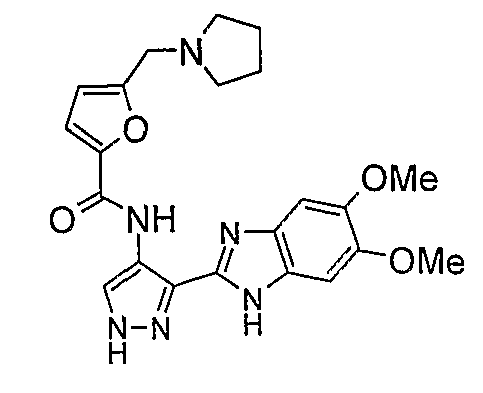
Rt 2,62
основн.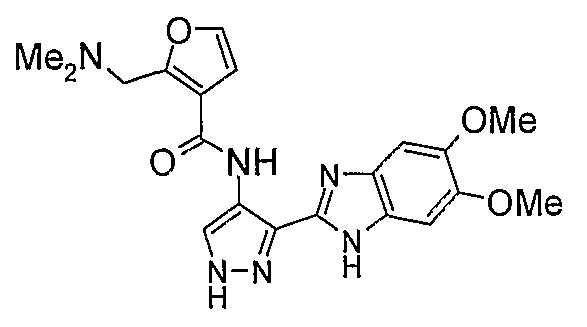
Очистка препаративной ЖХ/МС
Rt 1,6
кислотн.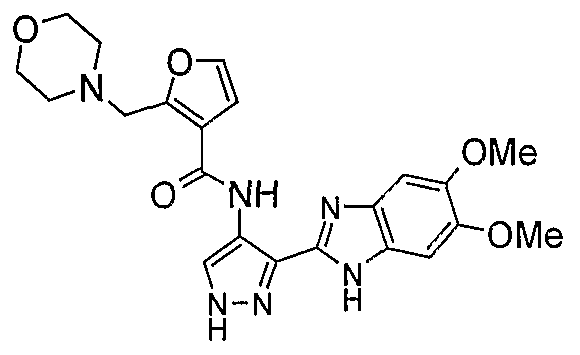
Очистка колоночной
хроматографией
[SiO2, элюирование DMAW 240] дальнейшая очистка препаративной ЖХ/МС
Rt 1,75
кислотн.
Пример 22
Синтез 5-хлор-2-метокси-N-{3-[5-(4-метилпиперазин-1-илметил)-1H-бензимидазол-2-ил]-1H-пиразол-4-ил}бензамида
22A. Синтез (3,4-динитрофенил)-(4-метилпиперазин-1-ил)метанона
3,4-Динитробензойную кислоту (50 г, 0,24 моль) кипятили с обратным холодильником в SOCl2 (160 мл) в течение 6 часов. Затем смесь упаривали досуха в вакууме. Продукт растворяли в ТГФ и охлаждали до 5°C. К этому раствору по каплям добавляли N-метилпиперазин (26,2 мл, 0,24 моль) и Et3N (42 мл) в виде раствора в ТГФ (50 мл). После перемешивания в течение ночи при комнатной температуре раствор выливали в воду (1,5 л) и перемешивали приблизительно при 5°C в течение 0,5 часа. Образовавшийся твердый осадок собирали и высушивали, получая (3,4-динитрофенил)-(4-метилпиперазин-1-ил)метанон (40 г) в виде твердого вещества желтого цвета.
22B. Синтез 1-(3,4-диаминобензил)-4-метилпиперазина
К охлажденному раствору (5°C) (3,4-динитрофенил)-(4-метилпиперазин-1-ил)метанона (12,2 г, 0,041 моль) в ТГФ добавляли порошкообразный NaBH4 с последующим добавлением по каплям BF3·Et2O, поддерживая все это время температуру ниже 5°C. Давали подняться температуре смеси до комнатной в течение 2 часов, затем перемешивали еще 2 часа при комнатной температуре. После этого к реакционной смеси осторожно добавляли MeOH (вызывая бурное выделение газа), продолжали перемешивание в течение 10 минут и затем концентрировали смесь. Остаток распределяли между EtOAc и насыщенным водным раствором NaHCO3. Органический слой промывали водой, насыщенным раствором соли и затем высушивали (MgSO4). Раствор упаривали в вакууме, затем остаток очищали флэш-хроматографией на SiO2, элюируя градиентом DCM:MeOH (98:2) - DCM:MeOH:конц. водн. NH3 (90:10:1), получая оранжевое кристаллическое твердое вещество (3,7 г). Перекристаллизация из MeOH позволила получить 1-(3,4-динитробензил)-4-метилпиперазин (1 г) в виде твердого кристаллического вещества оранжевого цвета.
22C. Синтез 1-(3,4-диаминобензил)-4-метилпиперазина
1-(3,4-динитробензил)-4-метилпиперазин (1 г) растворяли в смеси ДМФА:MeOH (1:1, 20 мл) и перемешивали с 10% Pd/C (50 мг) в атмосфере H2 в течение 6 часов. Затем смесь фильтровали и упаривали, получая темное твердое вещество, которое немедленно применяли в следующей стадии без какой-либо дальнейшей очистки.
22D. Синтез 5-хлор-2-метокси-N-{3-[5-(4-метилпиперазин-1-илметил)-1H-бензимидазол-2-ил]-1H-пиразол-4-ил}бензамида
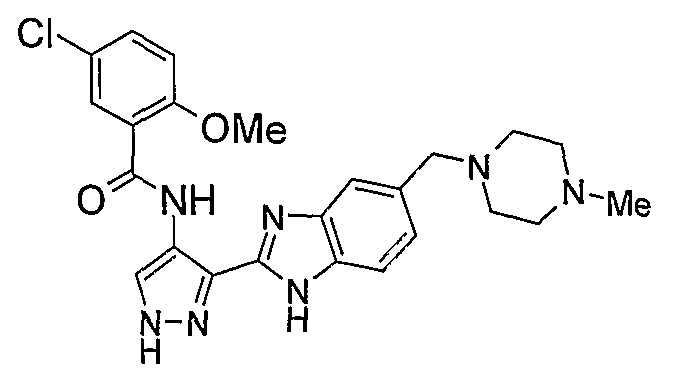
4-(5-Хлор-2-метоксибензоиламино)-1H-пиразол-3-карбоновую кислоту (1,17 г), неочищенный диамин, т.е. 1-(3,4-диаминобензил)-4-метилпиперазин (0,87 г) и TBTU (1,52 г) растворяли в ДМФА (15 мл) и перемешивали приблизительно 16 часов. Затем смесь упаривали досуха получая темное твердое вещество. Это темное твердое вещество (100 мг) растворяли в AcOH (4 мл) и смесь нагревали при 80°C в течение 3 часов. Реакционную смесь упаривали в вакууме и остаток очищали флэш-хроматографией (SiO2, элюируя DMAW 120), получая 5-хлор-2-метокси-N-{3-[5-(4-метилпиперазин-1-илметил)-1H-бензимидазол-2-ил]-1H-пиразол-4-ил}бензамид в виде диуксуснокислой соли (35 мг). (ЖХ/МС(кислотный способ/конечное соединение): Rt 6,63 [M+H]+ 480).
Пример 23
Синтез 1-(2,6-дифторбензил)-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
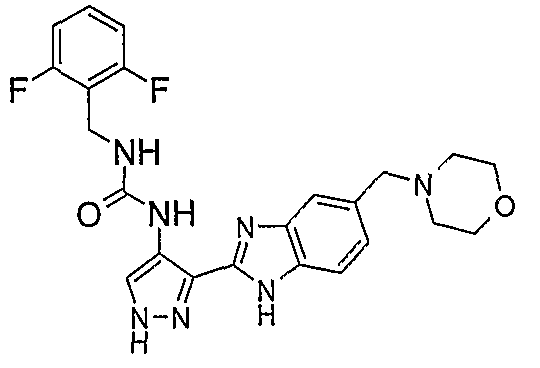
Смесь 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина (пример 1E), (100 мг, 0,33 ммоль) и CDI (217 мг, 1,34 ммоль) в ТГФ (2 мл) облучали микроволновым излучением (150°C, 150 Вт) в течение 15 минут. После этого добавляли 2,6-дифторбензиламин (384 мг, 2,68 ммоль), и реакционную смесь вновь облучали в тех же условиях в течение еще 15 минут. После охлаждения гетерогенную смесь фильтровали, фильтрат концентрировали, остаток очищали колоночной хроматографией (SiO2, элюируя градиентом DCM:MeOH:AcOH:H2O (240:20:3:2) (DMAW240) - (120:18:3:2) (DMAW120)) и получали 1-(2,6-дифторбензил)-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину (30 мг, 19%). (ЖХ/МС кислотн.: Rt 1,84 [M+H]+ 468).
Примеры 24-34
Следуя общей методике, изложенной в примере 23, но с модификациями, показанными в таблице ниже, получали соединения примеров 24-34.
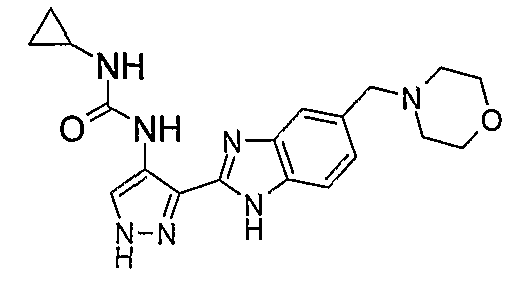
Rt 1,59
кислотн.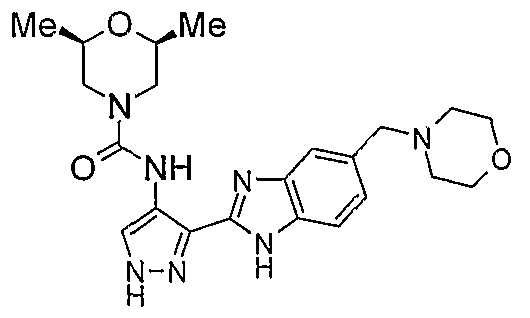
Rt 1,84
кислотн.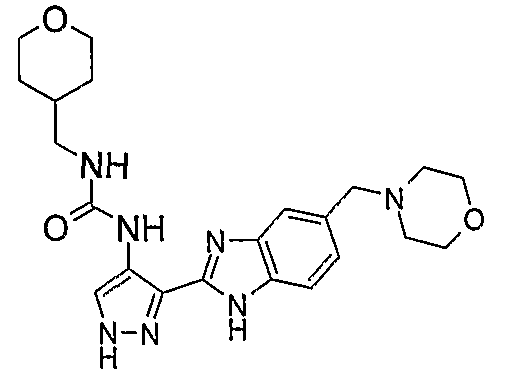
Rt 2,20
полярн.
Rt 1,57
кислотн.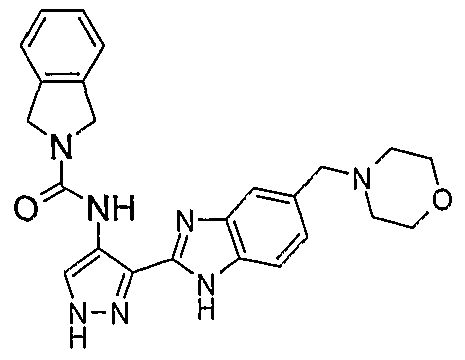
Очищали колоночной флэш-хроматографией на оксиде кремния, элюируя 100% EtOAc-10% MeOH
Rt 6,67
кислотн.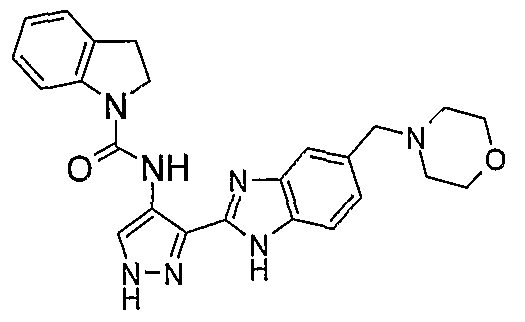
Очищали колоночной флэш-хроматографией на оксиде кремния, элюируя 100% EtOAc-10% MeOH
Rt 6,98
кислотн.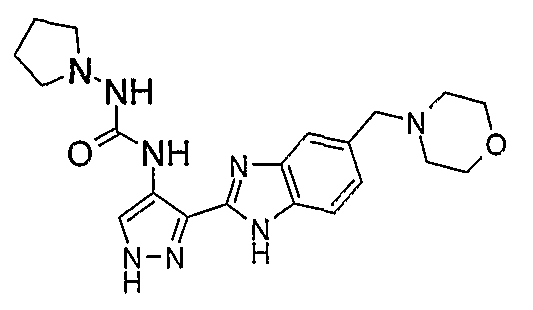
Очищали колоночной флэш-хроматографией 2-5% MeOH-дихлорметан, затем ЖХ/МС
Rt 2,45
основн.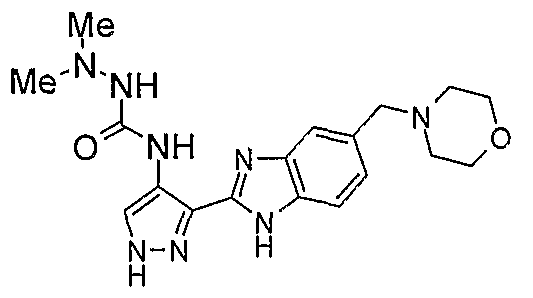
Очищали препаративной ЖХ/МС
Rt 1,60
кислотн.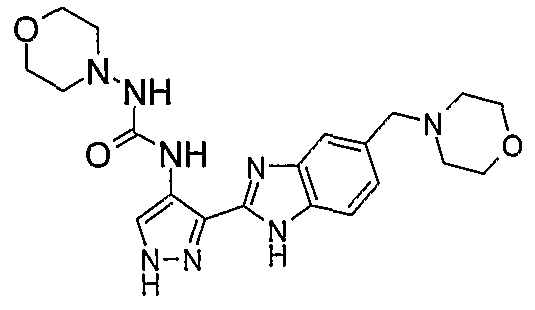
Очищали препаративной ЖХ/МС
Rt 1,69
кислотн.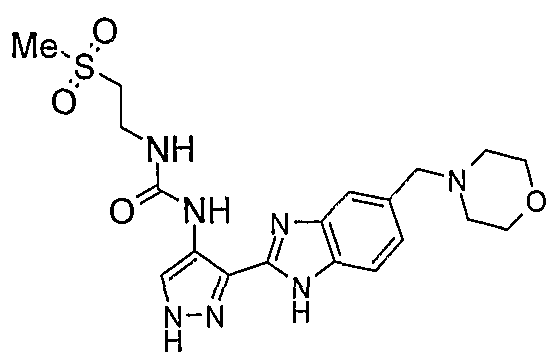
Обрабатывали распределением между EtOAc и насыщ. NaHCO3.
Очищали колоночной хроматографией на оксиде кремния (DMAW240-120), затем препаративной ЖХ/МС
Rt 2,07
основн.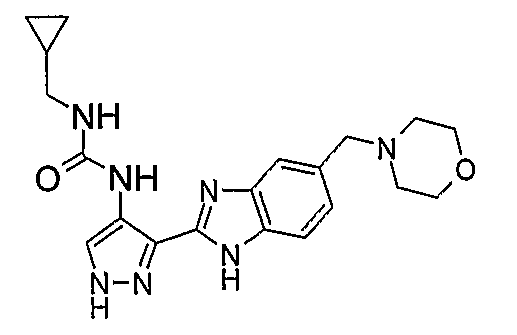
Обрабатывали распределением между EtOAc и насыщ. NaHCO3.
Очищали препаративной ЖХ/МС
Rt 1,74
основн.
Пример 35
Синтез 1-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-3-пиридин-3-илмочевины
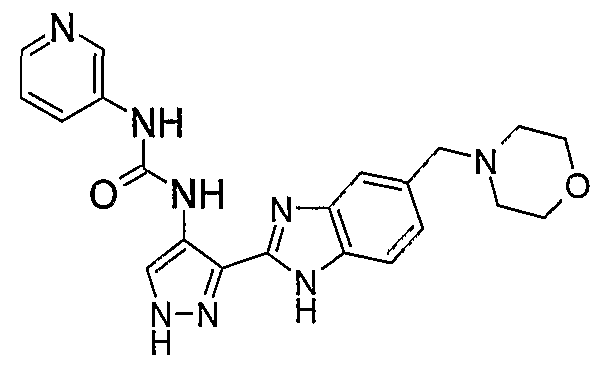
Смесь 3-аминопиридина (31,5 мг, 0,33 ммоль), Et3N (0,195 мл, 1,32 ммоль) в дихлорметане (3 мл) охлаждали до 0°C и затем обрабатывали трифосгеном (85 мг, 0,28 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем добавляли 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламин (100 мг, 0,33 ммоль) и перемешивали при комнатной температуре до завершения реакции. Смесь обрабатывали 2М NaOH в MeOH в течение 30 минут и затем упаривали в вакууме. Остаток очищали колоночной флэш-хроматографией [SiO2, 2-20% MeOH/DCM], после чего растиранием с дихлорметаном и затем диэтиловым эфиром получали 1-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]-3-пиридин-3-илмочевину (20 мг). (ЖХ/МС основн.: Rt 2,29, [M+H]+ 419).
Пример 36
Синтез [3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амида тиоморфолин-4-карбоновой кислоты
Фосген (20% в толуоле) (0,3 мл) при 0°C добавляли к раствору 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина (100 мг, 0,33 ммоль) в смеси толуол/дихлорметан (1:1). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем избыток фосгена выдували током азота. Добавляли тиоморфолин (35 мг, 0,33 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем при 60°C в течение 1 часа. Затем реакционную смесь концентрировали в вакууме и остаток очищали препаративной ЖХ/МС, получая [3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]амид тиоморфолин-4-карбоновой кислоты. (ЖХ/МС полярн.: Rt 2,58, [M+H]+ 428).
Пример 37
Синтез 1-(4-фторфенил)-1-метил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
Методика, примененная для синтеза указанного в заголовке соединения, аналогична методике, описанной в примере 35, но с использованием 4-фтор-N-метиламина вместо 3-аминопиридина, кроме того, реакцию проводили при 50°C в течение 2 часов. Неочищенный продукт выделяли в виде осадка из охлажденной реакционной смеси и затем очищали колоночной флэш-хроматографией [SiO2, элюирование смесью DCM:MeOH:AcOH:вода(240:20:3:2)]. Полученный продукт растирали с диэтиловым эфиром, получая 1-(4-фторфенил)-1-метил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину (3 мг) в виде твердого бесцветного вещества. (ЖХ/МС кислотн.: Rt 2,12, [M+H]+ 450).
Примеры 38-43
Следуя методикам, описанным в примерах 35 и 37, модифицированным в случаях, указанных в таблице ниже, получали соединения примеров 38-43.
Очищали препаративной ЖХ/МС
Rt 2,03
кислотн.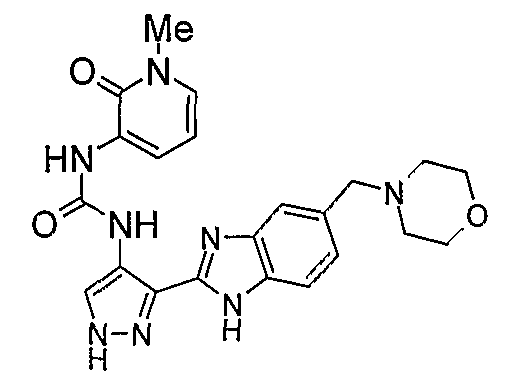
Очищали препаративной ЖХ/МС
Rt 2,32
полярн.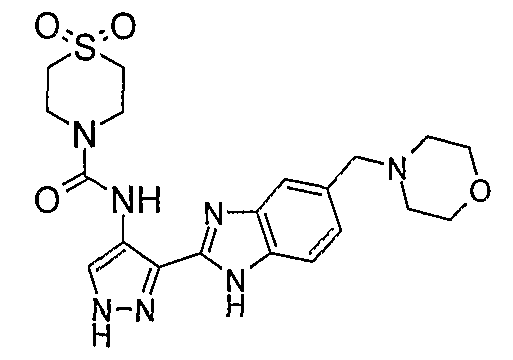
Очищали препаративной ЖХ/МС
Rt 2,22
основн.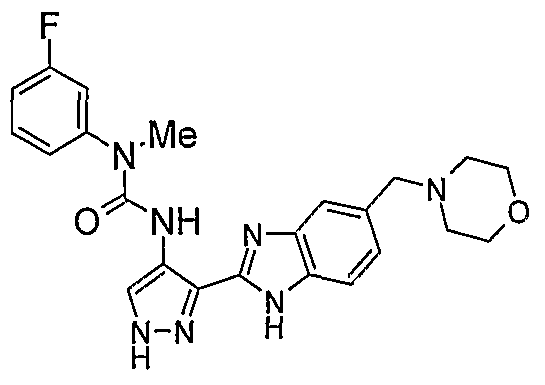
Rt 2,09
кислотн.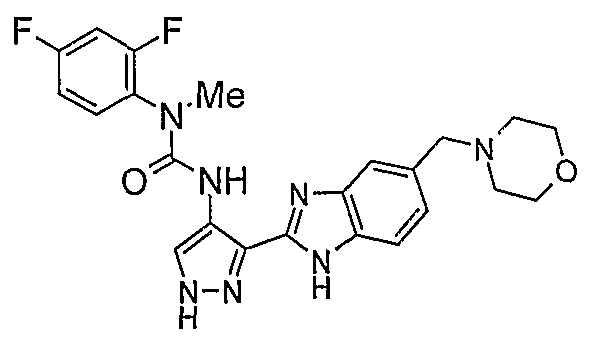
Rt 1,99
кислотн.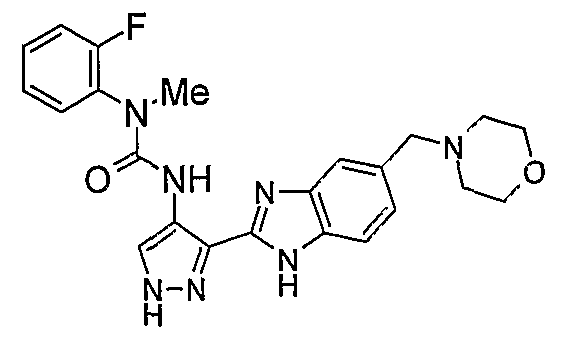
Rt 2,68
основн.
Пример 44
Синтез 1-(4-фторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины

К 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламину (пример 1E) (100 мг, 0,33 ммоль) в ТГФ (2 мл) добавляли 4-фторфенилизоцианат и смесь перемешивали в течение примерно 16 часов при комнатной температуре. Добавляли трис-амин на полимерной смоле (800 мг, 4 ммоль/г) и взбалтывали смесь в течение еще 4 часов. Смолу удаляли фильтрованием, фильтрат обрабатывали 1н. KOH (2 мл, MeOH:ТГФ 1:3) и раствор перемешивали в течение приблизительно 16 часов. Затем смесь распределяли между EtOAc и H2O. Затем экстрагировали водный слой EtOAc и после этого объединенные органические фракции промывали насыщенным раствором соли, высушивали (MgSO4) и упаривали досуха. Неочищенное твердое вещество растворяли в дихлорметане и растирали с гексаном, получая твердое вещество, которое собирали фильтрованием. Это твердое вещество очищали колоночной флэш-хроматографией [SiO2, EtOAc-MeOH (90:10)], получая 1-(4-фторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину (30 мг, 20%) в виде твердого вещества желтого цвета (ЖХ/МС (кислотн. способ): Rt 2,01 мин, [M-H]+ 434).
Примеры 45-56
Следуя методике, описанной в примере 44, но модифицируя условия в случаях, указанных в таблице ниже, получали соединения примеров 45-56.
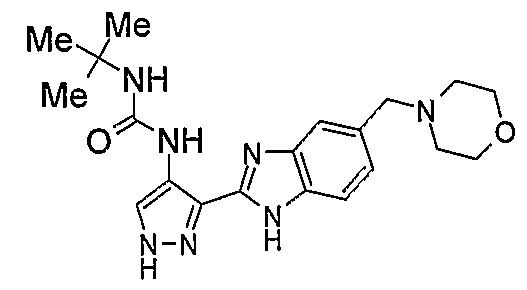
Хроматография не требовалась
Rt 1,79
кислотн.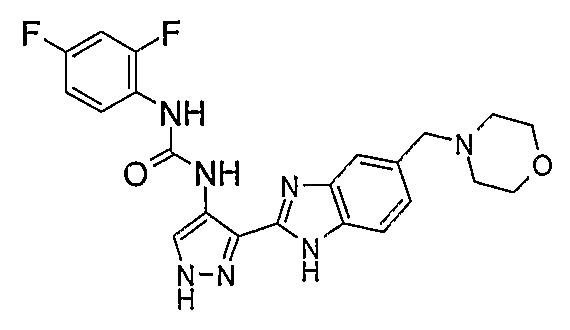
Rt 1,95
кислотн.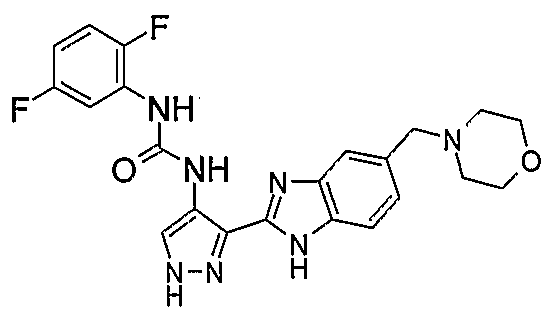
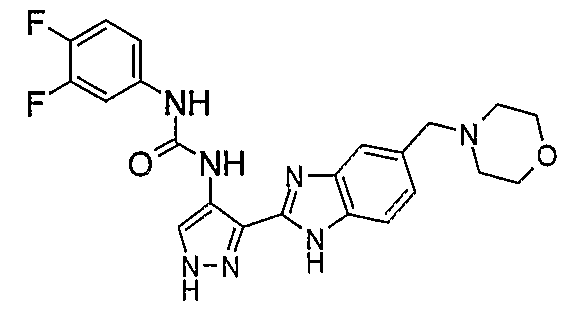
Rt 2,09
кислотн.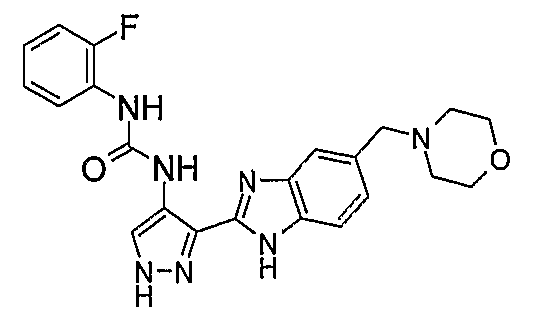
Rt 2,68
основн.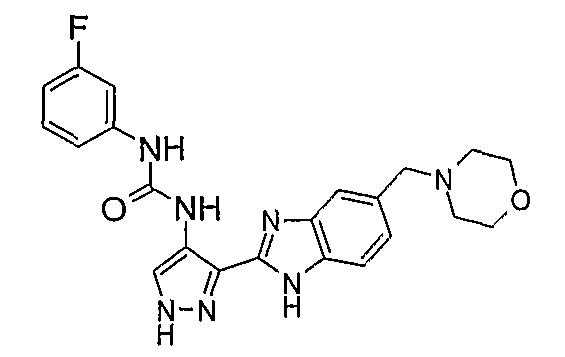
Rt 2,77
основн.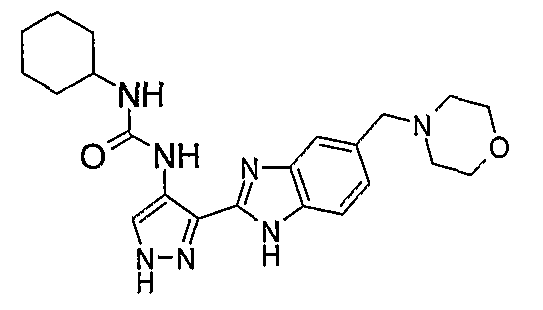
Rt 1,89
кислотн.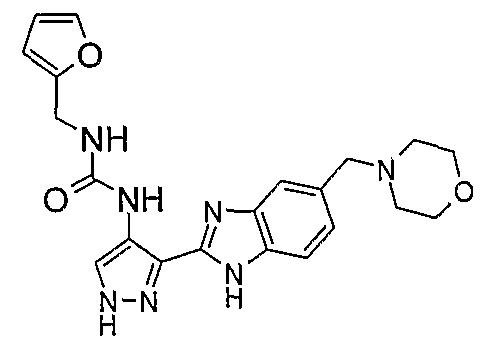
Rt 1,65
кислотн.
Rt 2,21
кислотн.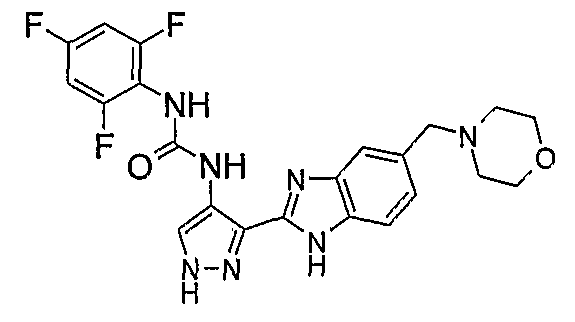
Rt 1,89
кислотн.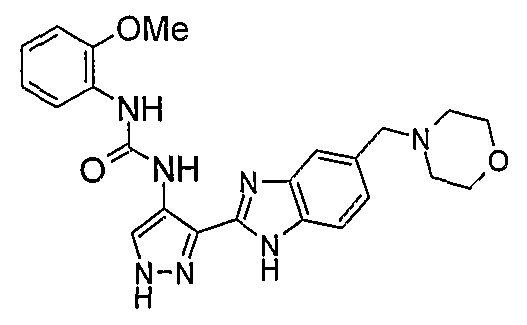
Rt 6,28
кислотн.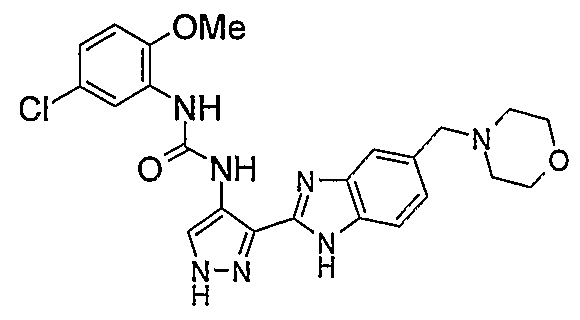
Rt 7,28
кислотн.
Примеры 57-59
Следуя общей методике, описанной в примере 23, но модифицированной в случаях, указанных в таблице ниже, получали соединения примеров 57-59.
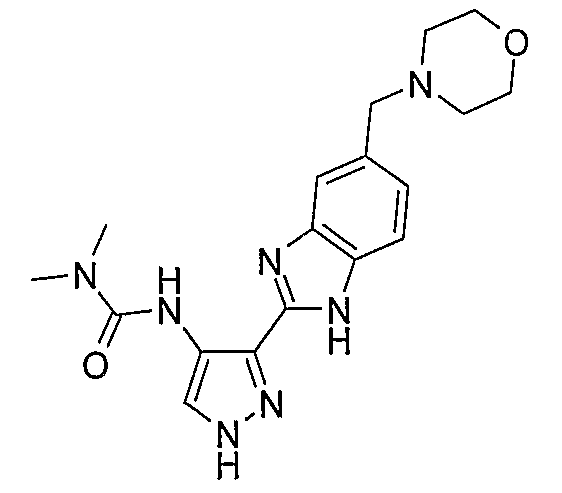
Rt 2,39
(основн. способ)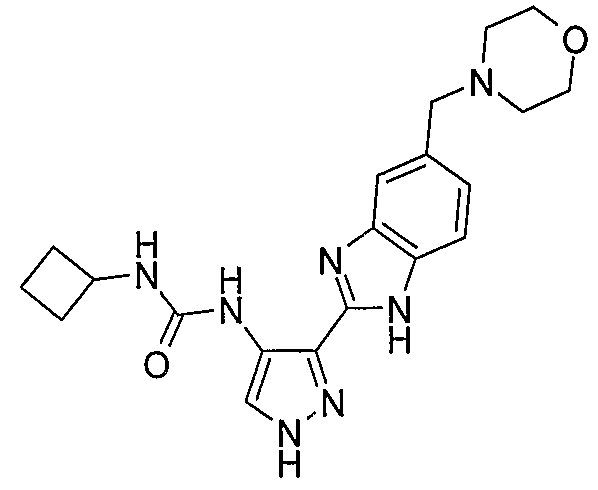
Rt 2,48
(основн. способ)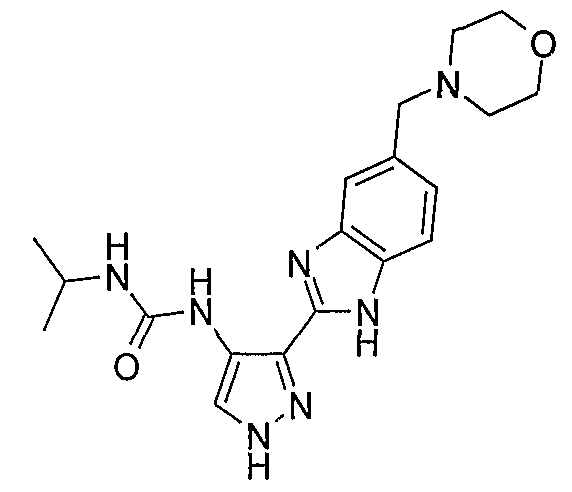
Rt 2,40
(основн. способ)
Пример 60
Синтез гидрохлорида 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
60A. Синтез (3,4-динитрофенил)морфолин-4-илметанона
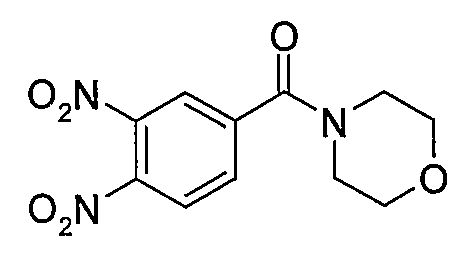
Смесь 3,4-динитробензойной кислоты (1 мол. экв.) и тионилхлорида (9,2 мол. экв.) кипятили с обратным холодильником в течение 6 часов, охлаждали до комнатной температуры и избыток тионилхлорида удаляли азеотропной отгонкой с толуолом. Остаток растворяли в ТГФ (8 объемов), затем одновременно к смеси добавляли морфолин (1,0 мол. экв.) и Et3N (1,1 молю экв.) при 0-5°C. Полученную смесь перемешивали в течение 1 часа при комнатной температуре, после чего выливали в воду (25 объемов). Смесь охлаждали до 3-7°C и оставляли стоять в течение 0,5 часа, причем за это время продукт выделялся в виде осадка. Осадок собирали фильтрованием, промывали водой и высушивали, получая (3,4-динитрофенил)морфолин-4-илметанон (75%) в виде твердого вещества желтого цвета (1H ЯМР(300 МГц, ДМСО-d6) δ 8,3 (д, 1H), 8,3 (с, 1H), 8,0 (д, 1H), 3,7-3,5 (м, 8H).
60B. Синтез 4-(3,4-динитробензил)морфолина
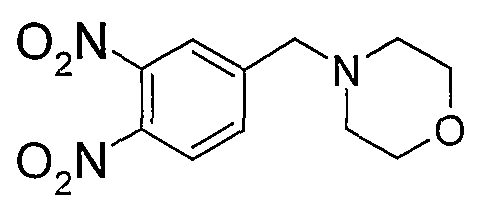
К смеси (3,4-динитрофенил)морфолин-4-илметанона (1 мол. экв.) и сухого тетрагидрофурана (ТГФ) (25 объемов) при 0-5°C добавляли NaBH4 (2 мол. экв.) и затем по каплям BF3·Et2O (1,01 мол. экв.) так, чтобы температура сохранялась в пределах 0-5°C. Затем смесь перемешивали при комнатной температуре в течение 3 часов и после этого гасили добавлением метанола. Затем смесь упаривали в вакууме, распределяли между этилацетатом и насыщенным водным раствором NaHCO3. Полученную смесь перед разделением слоев быстро перемешивали в течение 30 минут. Органический слой последовательно промывали водой и насыщенным раствором соли, после чего упаривали в вакууме. Полученный продукт кристаллизовали из метанола, получая 4-(3,4-динитробензил)морфолин (85%). (ЖХ/МС (основн. способ): Rt 2,80, [M+H]+ 268).
60C. Синтез 4-морфолин-4-илметилбензол-1,2-диамина
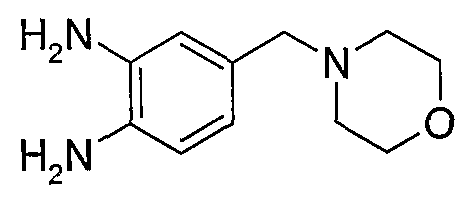
Смесь 4-(3,4-динитробензил)морфолина (1 мол. экв.) и 5% Pd/C (0,05 масс. экв.) в IMS (33 объема) перемешивали при 0-5°C и одновременно подавали в сосуд водород. Температуру смеси осторожно поднимали до 15-20°C при перемешивании, пока реакция не завершилась (<24 часов). Смесь фильтровали и фильтрат упаривали досуха, получая 4-морфолин-4-илметилбензол-1,2-диамин (90%). Полученное вещество немедленно использовали в следующей стадии. (ЖХ/МС (основн. способ): Rt 1,64, [M-N(CH2CH2)2O-]+ 121).
60D. Синтез 5-морфолин-4-илметил-2-(4-нитро-1H-пиразол-3-ил)-1H-бензимидазола
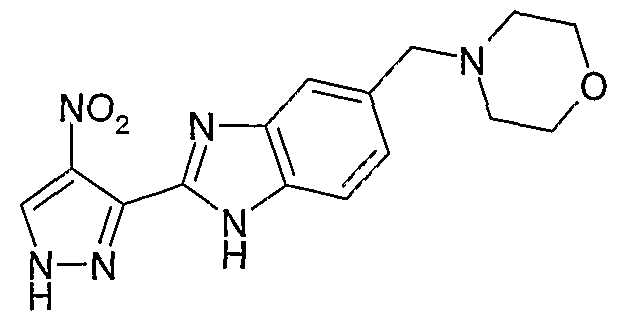
4-Морфолин-4-илметилбензол-1,2-диамин (1 мол. экв.) 4-нитро-1H-пиразол-3-карбоновую кислоту (1 мол. экв.) растворяли в диметилформамиде (ДМФА) (10 объемов). Добавляли тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU) (1,2 мол. экв.) и перемешивали смесь при комнатной температуре в течение 24 часов. Смесь концентрировали в вакууме до момента, когда дальнейшая отгонка растворителя переставала быть заметной. Затем осадок растворяли в ледяной уксусной кислоте (10 объемов) и нагревали при 65°C в течение ~12 часов. Смесь концентрировали в вакууме и затем растворяли в воде (6 объемов) при 75°C. Полученный раствор черного цвета охлаждали до 0-5°C в течение 2 часов, причем в это время образовывалось твердое вещество. Это твердое вещество удаляли фильтрованием, и водный фильтрат разбавляли этилацетатом (4 объема) и тетрагидрофураном (2 объема). К перемешиваемой смеси медленно добавляли твердый NaHCO3 до тех пор, пока не переставало наблюдаться выделение газа и не достигалось значение pH 6,8. Затем смесь перемешивали до образования осадка. Смесь оставляли стоять в течение 2 часов при 0-5°C, после этого собирали твердое вещество фильтрованием и промывали водой (2 объема), этилацетатом (2 объема) и высушивали, получая 5-морфолин-4-илметил-2-(4-нитро-1H-пиразол-3-ил)-1H-бензимидазол в виде твердого вещества коричневого цвета (40%). (ЖХ/МС (основн. способ): Rt 1,93, [M-H+]- 327).
60E. Синтез 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина
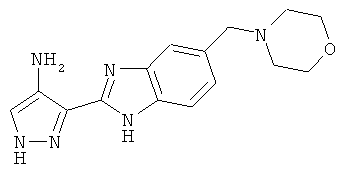
К 5-морфолин-4-илметил-2-(4-нитро-1H-пиразол-3-ил)-1H-бензимидазолу (1 мол. экв.) в ДМФА (36 объемов) в атмосфере азота добавляли 5% Pd/C (0,1 масс. экв.). Реакционный сосуд заполняли водородом и перемешивали при комнатной температуре в течение 24 часов. Затем смесь фильтровали через целит, промывая метанолом. Фильтрат концентрировали в вакууме, получая 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламин в виде коричневого твердого вещества (90%). (ЖХ/МС (основн. способ): Rt 1,94, [M-H+]- 297). Продукт использовали без дополнительной очистки.
60F. Синтез гидрохлорида 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
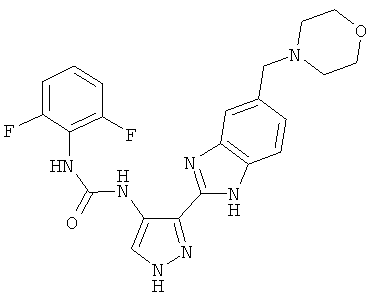
К смеси 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина (1 мол. экв.) и ТГФ (10 объемов) при 0-5°C и перемешивании добавляли 2,6-дифторфенилизоцианат (1,3 мол. экв.). Затем смесь перемешивали в течение 16 часов при комнатной температуре и по прошествии этого времени обрабатывали 1М водн. KOH (4 объема). После перемешивания на протяжении еще 2 часов смесь концентрировали в вакууме и распределяли между этилацетатом и насыщенным водным раствором NaHCO3. Органический слой промывали насыщенным раствором соли, высушивали (MgSO4), упаривали досуха и затем очищали остаток колоночной флэш-хроматографией [SiO2, элюируя градиентом CH2Cl2-MeOH (98:2)-(90:10)], получая 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину.
60G. Перекристаллизация и характеристики свободного основания
После хроматографии на оксиде кремния, описанной в примере 60E, продукт растворяли в минимальном количестве горячего этилацетата, фильтровали и оставляли охлаждаться. Таким способом получали свободное основание в виде чистого кристаллического твердого вещества.
Полученное соединение, т.е. 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина, обладало следующими физико-химическими параметрами.
60H. Получение хлористоводородной соли
Полученный продукт растворяли в этилацетате и обрабатывали избытком насыщенного раствора HCl в диэтиловом эфире. Полученный осадок собирали фильтрованием, промывали диэтиловым эфиром и высушивали, получая гидрохлорид 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины (59%) в виде бесцветного твердого вещества. (ЖХ/МС (кислотн. способ): Rt 1,80, [M+H]+ 454).
Заменяя хлористый водород другими кислотами (например, DL молочной кислотой, этансульфоновой кислотой и метансульфоновой кислотой) и изменяя при необходимости характер растворителя, можно получить другие соли 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины.
60J. Сравнение растворимостей свободного основания и гидрохлорида
Измеряли и сравнивали растворимости свободного основания и гидрохлорида. Было найдено, что растворимость свободного основания при pH 7,4 (буферный водный раствор) составляла <0,001 мг/мл, тогда как растворимость гидрохлорида при pH 7,1 (в буферном водном растворе), как было обнаружено, составляла 0,093 мг/мл. Таким образом, с точки зрения растворимости, хлористоводородная соль обладала значительным преимуществом относительно свободного основания.
Пример 61
Определение растворимостей кислотно-аддитивных солей 1-(2,6-дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
1-(2,6-Дифторфенил)-N-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину в форме свободного основания вводили во взаимодействие с различными кислотами по описанной ниже методике с целью оценки растворимостей образующихся кислотно-аддитивных солей.
Методика
В пробирку объемом 8 мл добавляли свободное основание (59 мг, 0,13 ммоль) и воду (0,59 мл). В пробирку добавляли соответствующую кислоту (1 экв, 0,13 ммоль) и встряхивали пробирки при комнатной температуре в течение 16 часов. По завершении этого времени подвергали пробирки визуальному осмотру. Если наблюдалось образование гомогенного раствора, эксперимент прекращали и делали вывод, что полученная описанным способом соль обладала растворимостью, превышающей 100 мг/мл.
Если оставалось твердое вещество, добавляли еще 0,59 мл воды и пробирки встряхивали в течение 4 часов. Если на этой стадии образовывался гомогенный раствор, приходили к выводу, что соль имеет растворимость, превышающую 50 мг/мл.
Если твердое вещество оставалось и в этой ситуации, добавляли еще 1,18 мл воды и встряхивали пробирку при комнатной температуре. Если это приводило к образованию гомогенного раствора, приходили к заключению, что растворимость превышает 25 мг/мл. Если твердое вещество все еще оставалось, делали вывод, что растворимость соли менее 25 мг/мл. Свободное основание регенерировали пропусканием раствора соли через колонку Strata-NH2.
Результаты описанных экспериментов приведены в таблице ниже.
Этансульфонат
DL-лактат
Адипат
L-(+)-аспартат
D-глюконат
L-глутамат
гидрохлорид
тозилат
свободное основание
На основе результатов, показанных в таблице, можно прийти к выводу, что мезилат, этансульфонат и DL-лактат должны оказаться особенно пригодными для получения жидких композиций на водной основе, например, для парентерального введения.
Пример 62
Свободное основание и соли 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
Соединение примера 24, т.е. 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина, может быть выделено в форме свободного основания или кислотно-аддитивной соли приведенными ниже или аналогичными способами.
Свободное основание
После проведения хроматографии на оксиде кремния (см. пример 24) продукт примера 24 растворяли в минимальном объеме горячего MeOH, фильтровали и оставляли охлаждаться. После примерно ~16 ч собирали продукт в виде бесцветного кристаллического твердого вещества.
Хлористоводородная соль (общая методика)
После проведения хроматографии на оксиде кремния продукт (2,05 г) растворяли в смеси MeOH:EtOAc (1:10, 100 мл) и обрабатывали 4н. HCl в диоксане (1,1 мол. экв.). Образовавшийся осадок собирали фильтрованием и высушивали, получая гидрохлорид 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины (1,5 г). Продукт растворяли в минимальном объеме MeOH и затем растирали с Et2O до помутнения, сохранявшегося в течение нескольких секунд. После выдерживания в течение ночи в охлажденном состоянии собирали продукт в виде бесцветного кристаллического твердого вещества.
Мезилат
Получали продукт в виде бесцветного кристаллического твердого вещества, применяя описанную выше общую методику, но используя метансульфоновую кислоту вместо хлористоводородной.
Другие соли
Ожидается, что другие представляющие интерес соли могли бы быть получены по описанной выше общей методике.
Пример 63
Определение растворимости свободного основания и солей 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
Соединение примера 24, т.е. 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину, вводили во взаимодействие с различными кислотами по описанной ниже методике с целью оценки растворимостей образующихся кислотно-аддитивных солей.
Методика
В пробирку объемом 8 мл добавляли соединение примера 24 в форме свободного основания (50 мг, 0,131 ммоль) и воду (0,50 мл). В пробирку добавляли соответствующую кислоту (1 экв., 0,131 ммоль) и встряхивали пробирку при комнатной температуре в течение 14-16 часов. По завершении этого времени подвергали пробирки визуальному осмотру. Если наблюдалось образование гомогенного раствора, эксперимент прекращали и делали вывод, что полученная описанным способом соль обладала растворимостью, превышающей 100 мг/мл.
Если оставалось твердое вещество, добавляли еще 0,5 мл воды и пробирки встряхивали в течение 6 часов. Если на этой стадии образовывался гомогенный раствор, приходили к выводу, что соль имеет растворимость, превышающую 50 мг/мл.
Если твердое вещество оставалось и в этой ситуации, добавляли еще 1 мл воды и встряхивали пробирку при комнатной температуре. Если это приводило к образованию гомогенного раствора, приходили к заключению, что растворимость превышает 25 мг/мл. Если твердое вещество все еще оставалось, делали вывод, что растворимость соли менее 25 мг/мл.
Свободное основание регенерировали пропусканием раствора соли через колонку Strata-NH2.
Результаты описанных экспериментов приведены в таблице ниже.
мезилат
этансульфонат
DL-лактат
адипат
D-глюкуронат
D-глюконат
гидрохлорид
L-глутамат
свободное основание
На основе результатов, показанных в таблице, можно прийти к выводу, что ацетат, мезилат, этансульфонат, DL-лактат, адипат, D-глюкуронат, D-глюконат и гидрохлорид должны оказаться особенно пригодными для получения жидких композиций на водной основе, например, для парентерального введения.
Из данных, собранных на сегодняшний день, представляется, что соединения по настоящему изобретению и, в особенности, 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина в форме свободного основания и солей (в частности, L-лактата), будут иметь ряд преимуществ перед соединениями известного уровня техники. В частности, такие преимущества включают одно или несколько из числа следующих:
- большая растворимость в водном растворе;
- лучшие физико-химические свойства, в частности более низкий logD;
- различная чувствительность к ферментам P450;
- улучшенный метаболизм лекарственных препаратов и фармакокинетические свойства;
- лучшая стабильность, например более длительный срок хранения и/или лучшая термическая стабильность;
- требуется более низкая дозировка;
- лучшая эффективность в отношении мишеней терапии и, в частности, Aurora A и B;
- лучшая активность в отношении клеток, например, в пролиферационном и клоногенном анализах;
- более высокая противораковая активность; и
- лучший терапевтический индекс.
Пример 64
Получение лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
К раствору 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины (0,7 г, 1,83 ммоль) в EtOAc-MeOH добавляли L-молочную кислоту (166 мг, 1,85 ммоль). Смесь перемешивали при комнатной температуре, затем упаривали в вакууме. Полученное твердое вещество очищали перекристаллизацией из кипящего EtOH (20 мл), получая после высушивания L-лактат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины (0,48 г).
Пример 65
Синтез L-лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
L-лактат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины может быть получен по способу, показанному на схеме ниже.

Стадия 1: Синтез (3,4-динитрофенил)морфолин-4-илметанона
Раствор 3,4-динитробензойной кислоты (10 г, 47 ммоль, 1 экв.) и ДМФА (0,1 мл) в ТГФ (100 мл) обрабатывали тионилхлоридом (4,5 мл, 62 ммоль, 1,3 экв.) и затем кипятили с обратным холодильником в течение 2,5 ч. Смесь охлаждали во льду, затем в течение 20 мин добавляли триэтиламин (10 мл, 71 ммоль, 1,1 экв.), поддерживая внутреннюю температуру <5°C. К полученной густой желтой суспензии в течение 15 минут добавляли морфолин (6,2 мл, 71 ммоль, 1,5 экв.), поддерживая внутреннюю температуру <10°C. Удаляли баню со льдом и давали смеси нагреться до комнатной температуры. Через 15 минут добавляли дополнительную порцию морфолина (1 мл, 11 ммоль, 0,24 экв.) и смесь перемешивали в течение ночи. Разбавляли смесь водой (250 мл) и охлаждали на льду. Бежевое твердое вещество отфильтровывали на вакууме, промывали дополнительной порцией холодной воды (25 мл) и высушивали в вакууме, получая соединение, указанное в заголовке примера (12,7 г, 96%).
Стадия 2: Синтез 4-(3,4-динитробензил)морфолина
Боргидрид натрия (3,36 г, 89 ммоль, 2,1 экв.) измельчали, помещали в заполненную азотом колбу и суспендировали в ТГФ (120 мл). После охлаждения до ~0°C через шприц добавляли комплекс трехфтористого бора с диэтиловым эфиром (11,3 мл, 89 ммоль, 2,1 экв.). Данная реакция умеренно экзотермическая, причем отмечалось некоторое выделение водорода. Добавляли 4-(3,4-динитробензол)морфолин (11,91 г, 42 ммоль, 1,0 экв.) одной порцией в виде твердого вещества, причем сосуд промывали дополнительной порцией ТГФ (20 мл). Удаляли баню со льдом и перемешивали суспензию при комнатной температуре в течение 3 часов, после чего вновь охлаждали на льду. Осторожно добавляли метанол (100 мл) (выделение водорода), затем смесь кипятили с обратным холодильником в течение 1 ч. Смесь концентрировали в вакууме, затем распределяли остаток между этилацетатом (100 мл) и смесью 1:1 насыщенный раствор бикарбоната натрия/вода (100 мл). Отделяли органическую фазу, промывали водой (50 мл), затем насыщенным раствором соли (100 мл) и высушивали (MgSO4). Раствор бикарбоната, которым осуществляли первую промывку, второй раз экстрагировали этилацетатом (50 мл), причем этот экстракт впоследствии промывали теми же водными составами, которые применяли для первого экстракта, и затем высушивали (MgSO4), объединение экстрактов и концентрирование привели к получению 10,97 г неочищенного вещества. Перекристаллизация из метанола (45 мл, 10 мл промывка) приводила к получению указанного в заголовке соединения (9,34 г, 83%).
Стадия 3: Синтез 4-морфолин-4-илметилбензол-1,2-диамина
4-(3,4-Динитробензил)морфолин (21 г, 101 ммоль) суспендировали в этаноле (0,9 л) и продували сосуд азотом. 10% палладий на активированном угле (1,05 г) суспендировали в этаноле (25 мл) и добавляли к субстрату. Смесь охлаждали на льду, затем газ в сосуде заменяли водородом. Смеси давали нагреться до 15-20°C и продолжали гидрирование при атмосферном давлении в течение 2 дней. Сосуд продували азотом, затем смесь фильтровали через целит, промывая этанолом (0,3 л), разделенным на порции. Концентрирование дало возможность получить указанное в заголовке соединение (15,8 г, 97%).
Стадия 4: Синтез метилового эфира 4-нитро-1H-пиразол-3-карбоновой кислоты
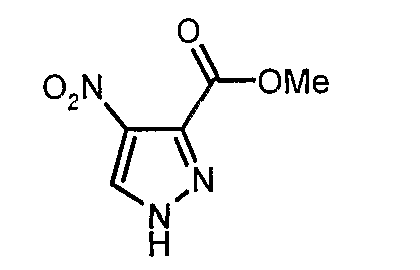
В реакционный сосуд объемом 20 л, снабженный цифровым термометром и мешалкой, загружали 4-нитро-1H-пиразол-3-карбоновую кислоту (1,117 кг, 7,11 моль, 1 мас.ч.) и метанол (8,950 л, 8 объемов). Реакционную смесь перемешивали в атмосфере азота, охлаждали до температуры 0-5°C, в течение 180 минут добавляли тионилхлорид (0,581 л, 8,0 моль, 0,52 объема), полученной смеси давали нагреться до 18-22°C и перемешивали в течение ночи, после чего анализ спектроскопией 1H ЯМР (d6-ДМСО) показал завершение реакции. Реакционную смесь концентрировали при пониженном давлении при температуре 40-45°C, остаток обрабатывали толуолом и повторно концентрировали (3×2,250 л, 3×2 объема) при пониженном давлении при температуре 40-45°C, получая метиловый эфир 4-нитро-1H-пиразол-3-карбоновой кислоты в виде не совсем белого твердого вещества (1,210 кг, 99,5%).
Стадия 5: Синтез метилового эфира 4-амино-1H-пиразол-3-карбоновой кислоты
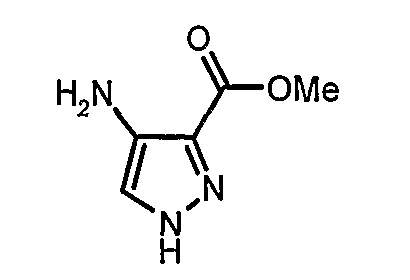
В реакционный сосуд объемом 20 л, снабженный цифровым термометром и мешалкой, в атмосфере азота загружали палладий на угле (10% влажная паста, 0,170 кг, 0,14 мас.ч.). В отдельном сосуде нагревали суспензию метилового эфира 4-нитро-1H-пиразол-3-карбоновой кислоты (1,210 кг, 7,07 моль, 1 мас.ч.) в этаноле (12,10 л, 10 объемов) до 30-35°C, добиваясь растворения, и раствор добавляли к катализатору в атмосфере азота. После продувки сосуда газами в последовательности азот-водород в реакционный сосуд вводили водород и реакционную смесь выдерживали при 28-30°C до завершения реакции (5-10 часов), которое определяли спектроскопией 1H ЯМР (d6-ДМСО). После цикла продувки реакционную смесь фильтровали в атмосфере азота и концентрировали жидкий остаток при пониженном давлении, получая метиловый эфир 4-амино-1H-пиразол-3-карбоновой кислоты (0,987 кг, 98,9%).
Стадия 6: Синтез 4-трет-бутоксикарбониламино-1H-пиразол-3-карбоновой кислоты

К смеси метилового эфира 4-амино-1H-пиразол-3-карбоновой кислоты (50,0 г, 355 ммоль) с диоксаном (500 мл) добавляли 2М водный раствор NaOH (213 мл, 426 ммоль), смесь нагревали до 50°C и перемешивали в течение 5 ч. Затем к этой смеси добавляли (BOC)2O (81,4 г, 373 ммоль), осуществляя промывание диоксаном (100 мл), и смесь нагревали при 50°C еще 5 ч, а затем перемешивали при комнатной температуре в течение 14 ч. Диоксан удаляли в вакууме и добавляли воду (1 л). pH смеси доводили до ~2, используя концентрированный водный раствор HCl, образовавшееся твердое вещество собирали фильтрованием и высушивали на фильтре. Полученное твердое вещество дополнительно высушивали азеотропной отгонкой с толуолом (×3) и в вакуумной печи, получая 4-трет-бутоксикарбониламино-1H-пиразол-3-карбоновую кислоту (70,0 г, 87%) в виде твердого вещества фиолетового цвета.
Стадия 7: Синтез трет-бутилового эфира [3-(2-амино-4-морфолин-4-илметилфенилкарбамоил)-1H-пиразол-4-ил]карбаминовой кислоты
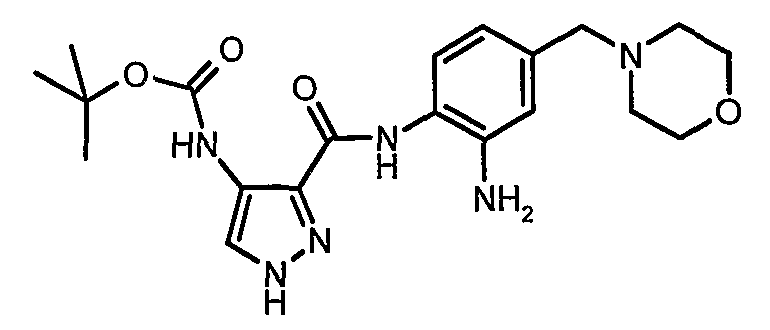
Смесь 4-трет-бутоксикарбониламино-1H-пиразол-3-карбоновой кислоты (10,0 г, 44,1 ммоль), 4-морфолин-4-илметилбензол-1,2-диамина (10,0 г, 48,5 ммоль), EDC (10,14 г, 52,9 ммоль) и HOBt (7,15 г, 52,9 ммоль) в ДМФА (150 мл) перемешивали при комнатной температуре в течение 20 ч и затем удаляли большую часть растворителя в вакууме. Остаток распределяли между EtOAc (150 мл) и насыщенным водным раствором NaHCO3 (150 мл), слои разделяли и органическую часть промывали насыщенным раствором соли, высушивали над MgSO4 и упаривали в вакууме, получая трет-бутиловый эфир [3-(2-амино-4-морфолин-4-илметилфенилкарбамоил)-1H-пиразол-4-ил]карбаминовой кислоты (17,6 г, 96%) в виде твердого вещества коричневого цвета. Как показал анализ ЖХ/МС, продукт содержал ~15% диамида. Этот продукт показал примерно 5% уровень в спектре 1H ЯМР. Диамид расщеплялся в последующих стадиях.
Стадия 8: Синтез 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина
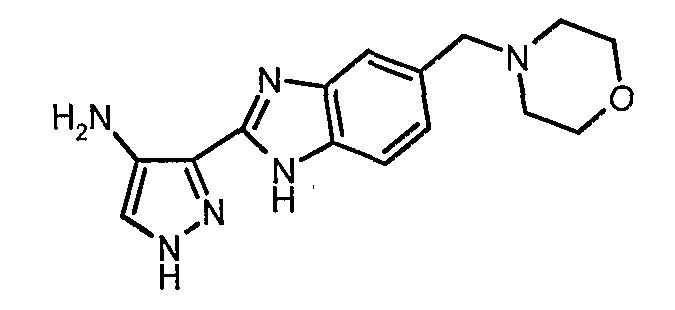
Смесь трет-бутилового эфира [3-(2-амино-4-морфолин-4-илметилфенилкарбамоил)-1H-пиразол-4-ил]карбаминовой кислоты (12,0 г, 28,8 ммоль) и 2М водного раствора HCl (50 мл) нагревали при 85°C в течение 14 часов, затем давали остыть до комнатной температуры. Осторожно добавляли твердый Na2CO3 до достижения pH смеси ~8,5 и раствор становился насыщенным. Образовывалась вязкая жидкость темной окраски. Смеси давали отстояться и растворитель декантировали. К остатку добавляли EtOH (60 мл), смесь кипятили с обратным холодильником в течение 1 ч и затем фильтровали в горячем состоянии, промывая EtOH (2Ч20 мл) для удаления остатков неорганических соединений. Фильтрат упаривали в вакууме, получая стеклообразное твердое вещество, которое затем перемешивали в Et2O (60 мл) в течение 1 часа, полученный порошок пурпурной окраски собирали фильтрованием и высушивали в вакууме, получая 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламин (6,8 г, 80%, ~90% чистота).
Стадия 9: Синтез 7-морфолин-4-илметил-2,4-дигидро-1,2,4,5a,10-пентаазациклопента[a]флуорен-5-она
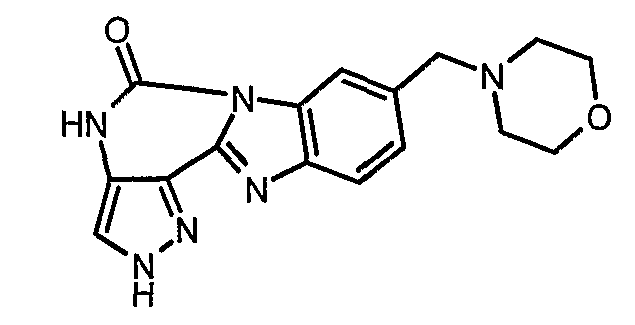
К смеси 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина (3,2 г, 10,7 ммоль) с безводным ТГФ (50 мл) при комнатной температуре и перемешивании добавляли 1,1'-карбонилдиимидазол (1,78 г, 11 ммоль). Смесь кипятили с обратным холодильником в течение 14 ч и затем охлаждали до комнатной температуры. Образовавшееся твердое вещество собирали фильтрованием, промывали ТГФ (20 мл) и высушивали в вакууме, получая 7-морфолин-4-илметил-2,4-дигидро-1,2,4,5a,10-пентаазациклопента[a]флуорен-5-он (2,34 г, 67%) в виде твердого розового вещества.
Стадия 10: Синтез 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
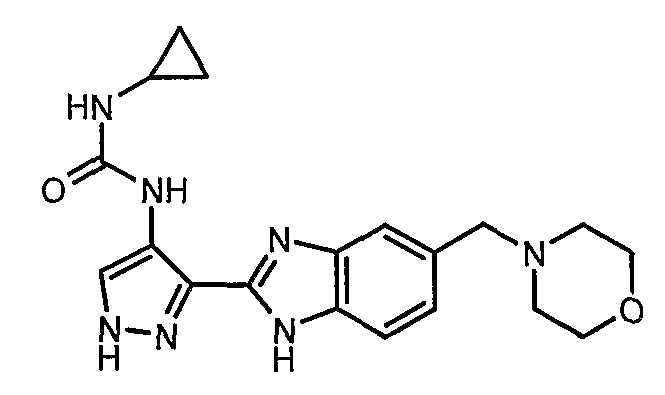
К смеси 7-морфолин-4-илметил-2,4-дигидро-1,2,4,5a,10-пентаазациклопента[a]флуорен-5-она (10,7 г, 32,9 ммоль) и N-метилпирролидона (65 мл) добавляли циклопропиламин (6,9 мл, 99 ммоль). Смесь нагревали при 100°C в течение 5 часов. ЖХ/МС анализ показал степень превращения исходного вещества в продукт ~75%, поэтому добавляли дополнительную порцию циклопропиламина (2,3 мл, 33 ммоль), смесь нагревали при 100°C в течение 4 ч и затем охлаждали до комнатной температуры. Смесь разбавляли водой (100 мл) и экстрагировали EtOAc (100 мл). Органическую фракцию промывали насыщенным водным раствором NH4Cl (2×50 мл), насыщенным раствором соли (50 мл) и затем водную фракцию повторно экстрагировали EtOAc (3×100 мл). Объединенные органические фракции высушивали над MgSO4 и упаривали в вакууме, получая 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину в виде оранжевого стеклообразного твердого вещества (9,10 г).
Стадия 11: Синтез L-лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
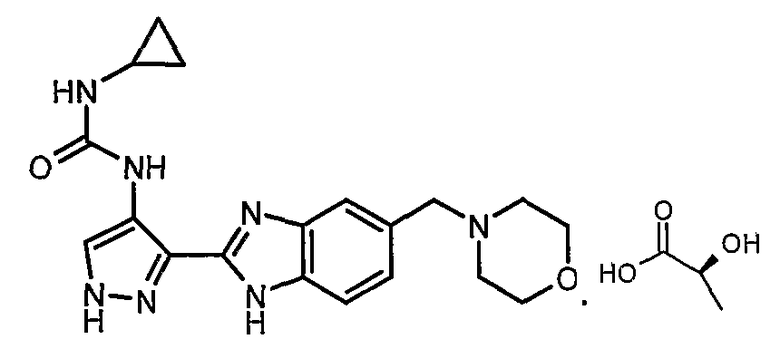
К раствору 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины (9,10 г, 24 ммоль) в смеси EtOAc-iPrOH (1:1, 90 мл) добавляли L-молочную кислоту (2,25 г, 25 ммоль). Смесь перемешивали при комнатной температуре в течение 24 ч и затем упаривали в вакууме. Остаток последовательно суспендировали в толуоле (100 мл) и Et2O (100 мл) и полученное твердое вещество собирали и высушивали (8,04 г).
Это твердое вещество очищали перекристаллизацией из кипящего iPrOH (200 мл), получая после высушивания L-лактат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины (5,7 г) в виде твердого вещества бежевого цвета.
Пример 66
Стадия 1: Получение (3,4-динитрофенил)морфолин-4-илметанона
В колбу в атмосфере азота загружали 3,4-динитробензойную кислоту (1,000 кг, 4,71 моль, 1 мас.ч.), тетрагидрофуран (10,00 л, 10,0 объемов) и диметилформамид (0,010 л, 0,01 объема). Добавляли тионилхлорид (0,450 л, 6,16 моль, 0,45 объема) при температуре от 20 до 30°C и реакционную смесь нагревали до 65-70°C. Завершение реакции, которое, как правило, происходило через 3 часа, определяли спектроскопией 1H ЯМР (d6-ДМСО). Реакционную смесь охлаждали до 0-5°C и при 0-10°C добавляли триэтиламин (1,25 л, 8,97 моль, 1,25 объема). При 0-10°C в реакционную смесь добавляли морфолин (0,62 л, 7,07 моль, 0,62 объема) и перемешивали образовавшуюся суспензию в течение 30 минут при 0-10°C. Завершение реакции определяли спектроскопией 1H ЯМР (d6-ДМСО). Реакционную смесь нагревали до 15-20°C и добавляли воду (4,00 л, 4,0 объема). Эту смесь затем загружали в 40 л колбу с фланцем, содержащую воду (21,00 л, 21,0 объем) при температуре 15-25°C для осаждения продукта. Содержимое колбы охлаждали до 0-5°C, выдерживали при этой температуре в течение 1 часа и собирали твердые вещества фильтрованием. Лепешку осадка на фильтре промывали водой (4×5,00 л, 4×5,0 объемов), причем, как было найдено, pH последней порции промывочной воды составлял 7. Влажную лепешку осадка на фильтре анализировали 1H ЯМР на наличие гидрохлорида триэтиламина. Затем отфильтрованное вещество высушивали при 40-45°C в вакууме до содержания воды по KF (по Карлу Фишеру) <0,2% масс./масс., получая (3,4-динитрофенил)морфолин-4-илметанон (1,286 кг, 97%, KF 0,069% масс./масс.) в виде твердого вещества желтого цвета.
Стадия 2: Получение 4-(3,4-динитробензил)морфолина

В колбу в атмосфере азота загружали (3,4-динитрофенил)морфолин-4-илметанон (0,750 кг, 2,67 моль, 1,0 мас.ч.) и тетрагидрофуран (7,50 л, 10,0 объема) и охлаждали до 0-5°C. При температуре 0-5ºC добавляли эфират трехфтористого бора (0,713 л, 5,63 моль, 0,95 объема), и суспензию перемешивали при этой же температуре в течение 15-30 минут. На протяжении 90-120 минут добавляли боргидрид натрия в виде шести равных порций (0,212 кг, 5,60 моль, 0,282 мас.ч.). (Отсроченное начало экзотермической реакции наблюдали через 10-15 минут после добавления первой порции. Если экзотермическая реакция начиналась и реакционную смесь вновь охлаждали, следующие порции реагента добавляли с 10-15-мин интервалами, позволяя реакционной смеси охладиться между добавлениями). Реакционную смесь перемешивали при 0-5°C в течение 30 минут. Завершение реакции определяли, анализируя реакционную смесь спектроскопией 1H ЯМР (d6-ДМСО). При 0-10°C по каплям добавляли метанол (6,30 л, 8,4 объема) для гашения реакционной смеси (бурное выделение газа, образование некоторого количества пены). После гашения реакционную смесь перемешивали при 0-10°C в течение 25-35 минут, затем нагревали до 20-30°C и перемешивали при этой температуре (экзотермическая реакция, выделение газа/паров эфира при растворении твердого вещества) до тех пор, пока выделение газа не замедлялось. Полученную смесь нагревали до 65-70°C и перемешивали при этой температуре 1 час. Смесь охлаждали до 30-40°C и концентрировали в вакууме при 40-45°C, получая неочищенный 4-(3,4-динитробензил)морфолин (0,702 кг, 98,4%) в виде желтого/оранжевого твердого вещества.
4-(3,4-Динитробензил)морфолин (2,815 кг, 10,53 моль, 1,0 мас.ч.) и метанол (12,00 л, 4,3 объема) загружали в колбу в атмосфере азота и нагревали до 65-70°C. Эту температуру поддерживали до полного растворения. Затем смесь охлаждали до 0-5°C и выдерживали при этой температуре в течение 1 часа. Твердые вещества выделяли фильтрованием. Лепешку осадка на фильтре промывали метанолом (2×1,50 л, 2×0,5 объема) и высушивали в вакууме при 35-45°C, получая 4-(3,4-динитробензил)морфолин (2,353 кг, 83,5% относительно исходного вещества стадии 2, 82,5% общий выход, относительно исходного вещества стадии 1) в виде твердого вещества желтого цвета.
Стадия 3: Получение 4-морфолин-4-илметилбензол-1,2-диамина
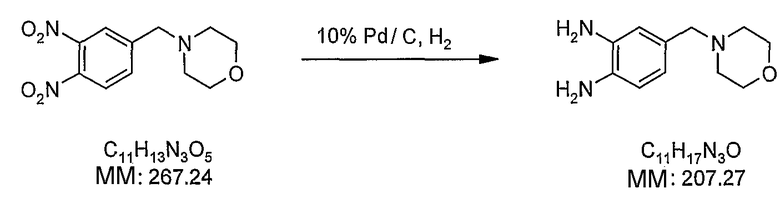
4-(3,4-Динитробензил)морфолин (0,800 кг, 2,99 моль, 1,0 мас.ч.) и этанол (11,20 л, 14,0 объемов) загружали в подходящую колбу, перемешивали при 15-25°C и три раза осуществляли цикл вакуумирование/заполнение азотом. 10% палладий на угле (10% Pd/C, паста 50% влажности, 0,040 кг, сухая масса 0,05 мас.ч.) суспендировали в этаноле (0,80 л, 1,0 объем) и добавляли к реакционной смеси. Смесь охлаждали до 10-20°C и три раза осуществляли цикл вакуумирование/заполнение азотом. Три раза осуществляли цикл вакуумирование/заполнение водородом и перемешивали реакционную смесь в атмосфере водорода при 10-20°C. Завершение реакции определяли, анализируя реакционную смесь спектроскопией 1H ЯМР (d6-ДМСО), как правило, для этого требовалось 14-20 часов. Три раза осуществляли цикл вакуумирование/заполнение азотом, и реакционную смесь в атмосфере азота фильтровали через бумагу из микростекловолокна. Осадок на фильтре промывали этанолом (3×0,80 л, 3×1,0 объем), объединенный фильтрат и промывочную жидкость концентрировали в вакууме при 35-45°C досуха, получая 4-морфолин-4-илметилбензол-1,2-диамин (0,611 кг, 98,6%) в виде твердого вещества коричневого цвета.
Стадия 4: Получение метилового эфира 4-нитро-1H-пиразол-3-карбоновой кислоты
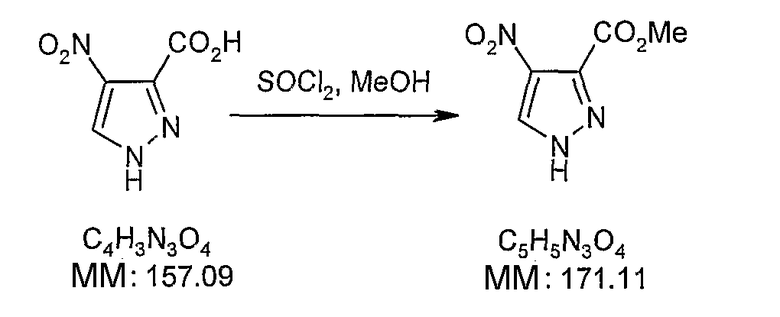
В колбу с фланцем, снабженную механической мешалкой, обратным холодильником и термометром, загружали 4-нитро-1H-пиразол-3-карбоновую кислоту (1,00 кг, 6,37 моль, 1,0 мас.ч.) и метанол (8,00 л, 8,0 объемов). Суспензию охлаждали в атмосфере азота до 0-5°C и при этой температуре добавляли тионилхлорид (0,52 л, 7,12 моль, 0,52 объема). Температуру смеси поднимали до 15-25°C в течение 16-24 часов. Завершение реакции определяли, анализируя реакционную смесь спектроскопией 1H ЯМР (d6-ДМСО). Смесь концентрировали в вакууме при 35-45°C. К остатку добавляли толуол (2,00 л, 2,0 объема) и удаляли в вакууме при 35-45°C. Азеотропную отгонку повторяли дважды, используя толуол (2,00 л, 2,0 объема), и получали метиловый эфир 4-нитро-1H-пиразол-3-карбоновой кислоты (1,071 кг, 98,3%) в виде не совсем белого твердого вещества.
Стадия 5: Получение метилового эфира 4-амино-1H-пиразол-3-карбоновой кислоты
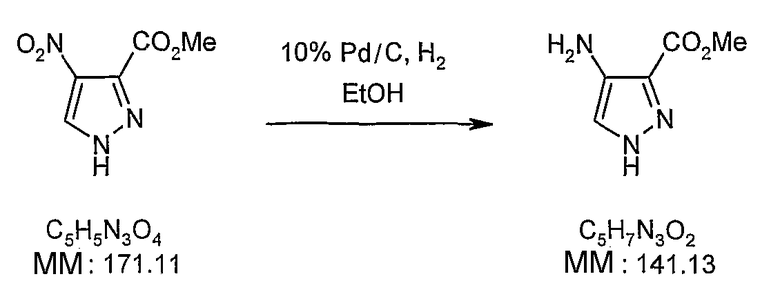
Суспензию метилового эфира 4-нитро-1H-пиразол-3-карбоновой кислоты (1,084 кг, 6,33 моль, 1,0 мас.ч.) и этанола (10,84 л, 10,0 объемов) нагревали до 30-35°C и выдерживали при этой температуре до полного растворения. 10% палладий на угле (10% Pd/C, влажная паста, 0,152 кг, 0,14 мас.ч.) вносили в отдельную колбу в атмосфере азота и три раза осуществляли цикл вакуумирование/заполнение азотом. К катализатору добавляли раствор метилового эфира 4-нитро-1H-пиразол-3-карбоновой кислоты в этаноле и три раза осуществляли цикл вакуумирование/заполнение азотом. Три раза осуществляли цикл вакуумирование/заполнение водородом и реакционную смесь помещали в атмосферу водорода. Реакционную смесь перемешивали при 28-30°C до предполагаемого по данным спектроскопии 1H ЯМР (d6-ДМСО) завершения реакции. Полученную смесь фильтровали в атмосфере азота и концентрировали в вакууме при 35-45°C, получая метиловый эфир 4-амино-1H-пиразол-3-карбоновой кислоты (0,883 кг, 98,9%) в виде твердого вещества пурпурного цвета.
Стадия 6: Получение 4-трет-бутоксикарбониламино-1H-пиразол-3-карбоновой кислоты
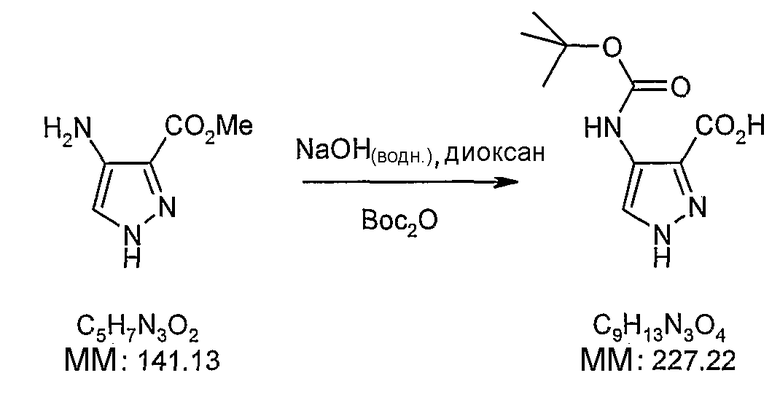
Метиловый эфир 4-амино-1H-пиразол-3-карбоновой кислоты (1,024 кг, 7,16 моль, 1,0 мас.ч.) и диоксан (10,24 л, 10,0 объемов) загружали в колбу с фланцем, снабженную механической мешалкой, обратным холодильником и термометром. При температуре 15-25°C добавляли раствор гидроксида натрия (4,36 л, 8,72 моль, 4,26 объема), и нагревали смесь до 45-55°C. Температуру поддерживали на уровне 45-55°C до завершения реакции, которое определяли спектроскопией 1H ЯМР (d6-ДМСО). При 45-55°C добавляли ди-трет-бутилдикарбонат (Boc ангидрид, 1,667 кг, 7,64 моль, 1,628 мас.ч.), и смесь перемешивали 55-65 минут. Анализ 1H ЯМР IPC (d6-ДМСО) указал на присутствие 9% не вступившего в реакцию промежуточного соединения. При 55°C добавляли дополнительную порцию ди-трет-бутилдикарбоната (Boc ангидрид, 0,141 кг, 0,64 моль, 0,14 мас.ч.), и смесь перемешивали в течение 55-65 минут. Завершение реакции определяли спектроскопией 1H ЯМР (d6-ДМСО). Удаляли диоксан в вакууме при 35-45°C и к остатку добавляли воду (17,60 л, 20,0 объемов). Используя 2М водный раствор хлористоводородной кислоты (4,30 л, 4,20 объемов), доводили значение pH до 2 и фильтровали смесь. Отфильтрованное вещество суспендировали в воде (10,00 л, 9,7 объемов) в течение 20-30 минут и эту смесь фильтровали. Осадок на фильтре промывали гептаном (4,10 л, 4,0 объема) и высушивали на прокладке в течение 16-20 часов. Полученное твердое вещество подвергали азеотропной отгонке с толуолом (5×4,00 л, 5×4,6 объема), затем высушивали в вакууме при 35-45°C, получая 4-трет-бутоксикарбониламино-1H-пиразол-3-карбоновую кислоту (1,389 кг, 85,4%) в виде твердого вещества пурпурного цвета.
Стадия 7: Получение трет-бутилового эфира [3-(2-амино-4-морфолин-4-илметилфенилкарбамоил)-1H-пиразол-4-ил]карбаминовой кислоты
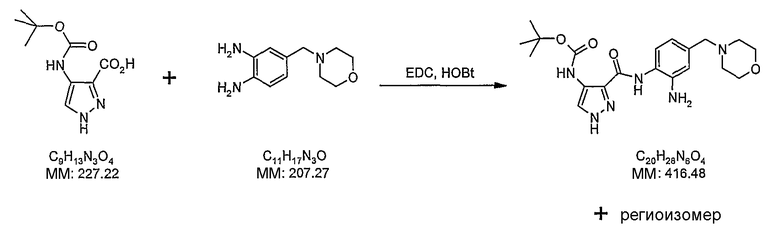
4-трет-Бутоксикарбониламино-1H-пиразол-3-карбоновую кислоту (0,750 кг, 3,30 моль, 1,0 мас.ч.), 4-морфолин-4-илметилбензол-1,2-диамин (0,752 кг, 3,63 моль, 1,0 мас.ч.) и N,N'-диметилформамид (11,25 л, 15,0 объемов) в атмосфере водорода загружали в колбу с фланцем, снабженную механической мешалкой и термометром. При 15-25°C добавляли 1-гидроксибензотриазол (HOBt, 0,540 кг, 3,96 моль, 0,72 мас.ч.). При 15-25°C добавляли N-(3-диметиламинопропил)-N'-этилкарбодиимид (EDC, 0,759 кг, 3,96 моль, 1,01 мас.ч.), и смесь перемешивали при этой температуре в течение 16-24 часов. Завершение реакции определяли спектроскопией 1H ЯМР. Реакционную смесь концентрировали в вакууме при 35-45°C. Остаток распределяли между этилацетатом (7,50 л, 10,0 объемов) и насыщенным водным раствором гидрокарбоната натрия (8,03 л, 10,7 объемов) и разделяли слои. Органическую фазу промывали насыщенным водным раствором соли (3,75 л, 5,0 объемов), высушивали над сульфатом магния (1,00 кг, 1,33 мас.ч.) и фильтровали. Осадок вещества на фильтре промывали этилацетатом (1,50 л, 2,0 объемов). Объединенные фильтрат и промывочную жидкость концентрировали в вакууме при 35-45°C, получая трет-бутиловый эфир [3-(2-амино-4-морфолин-4-илметилфенилкарбамоил)-1H-пиразол-4-ил]карбаминовой кислоты (1,217 кг, 88,6%) в виде темно-коричневого твердого вещества.
Стадия 8: Получение 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламина
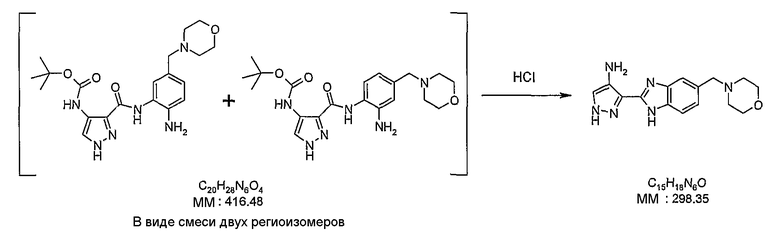
трет-Бутиловый эфир [3-(2-амино-4-морфолин-4-илметилфенилкарбамоил)-1H-пиразол-4-ил]карбаминовой кислоты (1,350 кг, 3,24 моль, 1,0 мас.ч.) и этанол (6,75 л, 5,0 объема) загружали в колбу с фланцем, снабженную механической мешалкой, обратным холодильником и термометром. При 15-30°C в атмосфере азота добавляли концентрированную водную хлористоводородную кислоту (1,10 л, 13,2 моль, 0,80 объема), затем содержимое колбы нагревали до 70-80°C и выдерживали при этой температуре в течение 16-24 часов. При 70-80°C добавляли вторую порцию хлористоводородной кислоты (0,11 л, 1,32 моль, 0,080 объема) и реакционную смесь нагревали еще 4 часа. Завершение реакции определяли анализом ВЭЖХ. Реакционную смесь охлаждали до 10-20°C и при этой температуре порциями добавляли карбонат калия (1,355 кг, 9,08 моль, 1,0 мас.ч.). Полученную суспензию перемешивали до прекращения выделения газа и затем фильтровали. Лепешку вещества на фильтре промывали этанолом (1,35 л, 1,0 объем) и фильтраты сохраняли. Отфильтрованное вещество суспендировали в этаноле (4,00 л, 3,0 объема) при 15-25°C в течение 20-40 минут и фильтровали смесь. Лепешку вещества на фильтре промывали этанолом (1,35 л, 1,0 объема), все фильтраты объединяли и концентрировали в вакууме при 35-45°C. К остатку добавляли этанол (4,00 л, 3,0 объема) и удаляли в вакууме при 35-45°C. К остатку добавляли тетрагидрофуран (5,90 л, 4,4 объема) и перемешивали в течение 10-20 минут при 15-25°C. Полученный раствор фильтровали, осадок на фильтре промывали тетрагидрофураном (1,35 л, 1,0 объема), фильтраты объединяли и концентрировали в вакууме при 35-45°C. К концентрату добавляли тетрагидрофуран (5,40 л, 4,0 объема) и удаляли в вакууме при 35-45°C. К концентрату добавляли тетрагидрофуран (5,40 л, 4,0 объема) и удаляли в вакууме при 35-45°C, получая желаемый продукт, т.е. 3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламин (0,924 кг, 95,5%, 82,84% по площади на хроматограмме ВЭЖХ) в виде пурпурной пены.
Стадия 9: Получение 7-морфолин-4-илметил-2,4-дигидро-1,2,4,5a,10-пентаазациклопента[a]флуорен-5-она
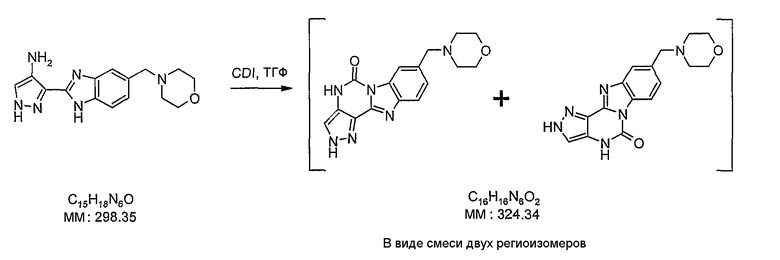
3-(5-Морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-иламин (0,993 кг, 3,33 моль, 1,0 мас.ч.) и тетрагидрофуран (14,0 л, 15,0 объемов) загружали в колбу с фланцем, снабженную механической мешалкой, обратным холодильником и термометром. Содержимое перемешивали в атмосфере азота при 15-25°C и добавляли 1,1'-карбонилдиимидазол (0,596 кг, 3,67 моль, 0,60 мас.ч.). После этого содержимое нагревали до 60-70°C и перемешивали при этой температуре в течение 16-24 часов. Завершение реакции определяли анализом ТСХ. Смесь охлаждали до 15-20°C и фильтровали. Лепешку вещества на фильтре промывали тетрагидрофураном (4,00 л, 4,0 объема) и высушивали 15-30 минут. Полученное твердое вещество высушивали в вакууме при 35-45ºC, получая 7-морфолин-4-илметил-2,4-дигидро-1,2,4,5a,10-пентаазациклопента[a]флуорен-5-он (0,810 кг, 75,05%, 92,19% по площади на хроматограмме ВЭЖХ) в виде твердого вещества пурпурного цвета.
Стадия 10: Получение 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
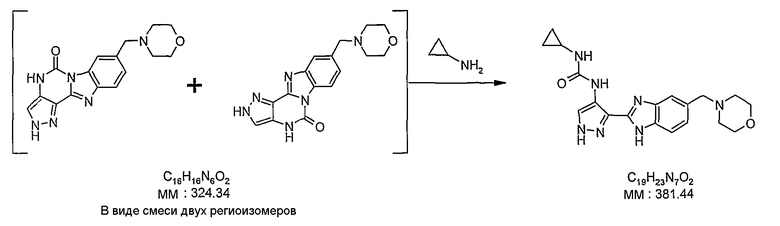
7-Морфолин-4-илметил-2,4-дигидро-1,2,4,5a,10-пентаазациклопента[a]флуорен-5-он (0,797 кг, 2,46 моль, 1,0 мас.ч.) и 1-метил-2-пирролидинон (2,40 л, 3,0 объема) загружали в колбу с фланцем, снабженную механической мешалкой, обратным холодильником и термометром. При 15-30°C в атмосфере азота добавляли циклопропиламин (0,279 кг, 4,88 моль, 0,351 мас.ч.). Реакционную смесь нагревали до 95-105°C и перемешивали при этой температуре в течение 16-24 часов. Завершение реакции определяли спектроскопией 1H ЯМР. Реакционную смесь охлаждали до 10-20°C, добавляли этилацетат (8,00 л, 10,0 объемов) и насыщенный водный раствор хлорида натрия (2,50 л, 3,0 объема), смесь перемешивали в течение 2-5 минут и разделяли слои. Органическую фазу перемешивали с насыщенным водным раствором хлорида натрия (5,00 л, 6,0 объемов) в течение 25-35 минут, смесь фильтровали и осадок на фильтре промывали этилацетатом (0,40 л, 0,5 объема). Отфильтрованный осадок сохраняли, фильтраты переносили в делительную воронку и разделяли слои. Эту процедуру повторяли еще три раза, сохраненные порции твердого вещества объединяли с органической фазой и смесь концентрировали досуха в вакууме при 35-45°C. Концентрат растворяли в пропан-2-оле (8,00 л, 10,0 объемов) при 45-55°C и добавляли активированный уголь (0,080 кг, 0,1 мас.ч.). Смесь перемешивали при 45-55°C в течение 30-40 минут и затем фильтровали в горячем состоянии при 45-55°C. Осадок на фильтре промывали пропан-2-олом (0,40 л, 0,5 объема). К объединенным фильтратам и промывной жидкости добавляли активированный уголь (0,080 кг, 0,1 мас.ч.), и смесь перемешивали при 45-55°C в течение 30-40 минут. Смесь фильтровали в горячем состоянии при 45-55°C и осадок на фильтре промывали пропан-2-олом (0,40 л, 0,5 объема). Фильтраты и промывную жидкость концентрировали в вакууме при 35-45°C. К концентрату при 25-35°C добавляли этилацетат (8,00 л, 10,0 объемов) и воду (2,20 л, 3,0 объема) и смесь перемешивали в течение 1-2 минут. Слои разделяли и органическую фазу концентрировали в вакууме при 35-45°C. К остатку добавляли этилацетат (4,00 л, 5,0 объемов) и концентрировали в вакууме при 35-45°C. К остатку добавляли этилацетат (4,00 л, 5,0 объемов) и смесь перемешивали в течение 2-20 часов при 15-25°C. Смесь охлаждали до 0-5°C, выдерживали при этой температуре 90-120 минут и затем фильтровали. Лепешку вещества на фильтре промывали этилацетатом (0,80 л, 1,0 объем) и высушивали 15-30 минут. Полученное твердое вещество высушивали в вакууме при 35-45°C, получая 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину (0,533 кг, 56,8%, 93,20% по площади пика на хроматограмме ВЭЖХ) в виде твердого коричневого вещества.
Несколько порций продукта стадии 9 обрабатывали описанным способом, и подробности относительно количеств исходного вещества и продукта для каждой загрузки приведены в таблице 1A.
загрузки
55,2%, 64,9%масс./масс.
47,0%, 56,6%масс./масс.
43,0%, 50,6%масс./масс.
56,8%, 66,8%масс./масс.
Стадия 11: Получение L-лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
1-Циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину (1,859 кг, 4,872 моль, 1,0 мас.ч.), пропан-2-ол (9,00 л, 5,0 объема) и этилацетат (8,00 л, 4,5 объема) загружали в колбу с фланцем, снабженную механической мешалкой и термометром. Содержимое колбы перемешивали в атмосфере азота и при 15-25°C добавляли L-молочную кислоту (0,504 кг, 5,59 моль, 0,269 мас.ч.), после чего установку промывали этилацетатом (0,90 л, 0,5 объема). Смесь перемешивали при 15-25°C в течение 120-140 минут. Твердое вещество отделяли фильтрованием, лепешку осадка на фильтре промывали этилацетатом (2×2,00 л, 2×1,0 объем) и высушивали 20-40 минут. Отфильтрованное вещество растворяли в этаноле (33,00 л, 17,7 объемов) при 75-85°C, охлаждали до 65-70°C и очищали раствор фильтрованием через бумагу из микростекловолокна. Фильтраты охлаждали до 15-25°C и выдерживали при этой температуре в течение 2-3 часов. Кристаллизовавшееся твердое вещество выделяли фильтрованием, вещество на фильтре промывали этанолом (2×1,00 л, 1×0,5 объема) и высушивали не менее 30 минут. Полученное твердое вещество высушивали в вакууме при 35-45°C, получая L-лактат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины (1,386 кг, 58,7%, 99,47% по площади на хроматограмме ВЭЖХ) в виде темно-розового однородного твердого вещества.
Инфракрасный спектр лактата (способ с таблеткой KBr) содержал характеристические пики при 3229, 2972 и 1660 см-1.
Не желая ограничиваться какой-либо теорией, полагают, что пики в инфракрасном спектре могут быть отнесены к структурным фрагментам полученной соли следующим образом:
Пример 67
Получение кристаллического свободного основания и кристаллических форм солей 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
A. Получение 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
Получали образец неочищенной 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания, как описано в примере 60, и на первом этапе очищали соединение колоночной хроматографией на силикагеле, элюируя градиентом EtOAc-MeOH (98:2-80:20). Полученный образец свободного основания затем перекристаллизовывали из горячего метанола, получая свободное основание 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в виде кристаллического вещества.
B. Получение дигидрата свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
Образец неочищенной 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания растворяли в ТГФ и затем концентрировали в вакууме до минимального объема (~4 объемов). К раствору по каплям добавляли воду (2-4 объема), пока он не становился мутным. Добавляли небольшое количество ТГФ для восстановления прозрачности и смесь оставляли стоять в течение ночи, получая кристаллическое вещество, которое высушивали на воздухе и получали дигидрат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания.
C. Получение гидрохлорида 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
Образец неочищенной 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания растворяли в минимальном количестве MeOH и затем разбавляли EtOAc. К раствору при 0°C медленно добавляли 1,1 эквивалента HCl (4М раствор в диоксане). После добавления из раствора осаждалось твердое вещество, которое собирали фильтрованием. К этому твердому веществу добавляли метанол и упаривали смесь в вакууме. Для удаления следов остаточного метанола остаток растворяли в воде, упаривали и затем высушивали при 60°C/0,1 мбар, получая гидрохлорид.
D. Получение этансульфоната 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
К раствору 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания в смеси MeOH-EtOAc добавляли 1 эквивалент этансульфоновой кислоты. Смесь перемешивали при комнатной температуре и затем упаривали в вакууме. Остаток растворяли в метаноле и к раствору добавляли Et2O. Смесь оставляли стоять в течение 72 часов, образовавшееся твердое вещество собирали фильтрованием и высушивали, получая этансульфонат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины.
E. Получение метансульфоната 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
К раствору 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания (394 мг) в смеси MeOH-EtOAc добавляли 1 эквивалент метансульфоновой кислоты (67 мкл). Образовывалось твердое вещество, которое собирали фильтрованием, промывая EtOAc. Это твердое вещество растворяли в минимальном количестве горячего MeOH, давая охладиться, и затем растирали с Et2O. Твердое вещество оставляли стоять в течение 72 часов и затем собирали фильтрованием, промывая MeOH, и получали метансульфонат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины.
Пример 68
Определение характеристик 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания и солей
Определяли характеристики различных форм 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины. Формы, выбранные для определения характеристик, были идентифицированы с помощью исследований, которые первоначально были посвящены исследованию степени полиморфизма и стабильности солей. Солями, выбранными для дальнейшего определения характеристик, являлись L-лактат, дигидрат свободного основания, эзилат, свободное основание и гидрохлорид.
A. Дифференциальная сканирующая калориметрия (DSC)
Термограммы регистрировали на приборе TA instrument Q1000, снабженном устройством автоматической подачи на 50 образцов. Калибровочным стандартом по энергии и температуре служил индий. Образцы нагревали со скоростью 10°C/минута от 10 до 250°C. Над образцом пропускали ток азота 30 мл/мин. Использовали от 2 до 10 мг образцов (если не указано иначе), и все образцы помещали в алюминиевые кюветы с точечным отверстием в крышке.
B. Термогравиметрический анализ (TGA)
Термограммы регистрировали на приборе TA instruments Q500. Образцы нагревали со скоростью 10°C/минута. Над образцом пропускали ток азота со скоростью 100 мл/мин. Обычно во взвешенную открытую алюминиевую кювету помещали 5-20 мг образца.
C. Микроскопия в поляризованном свете
Образцы исследовали на микроскопе Leica LM/DM с цифровой камерой для регистрации изображений. Небольшое количество образца в иммерсионном масле помещали на предметное стекло и закрывали покровным стеклом. Как можно лучше разделяли индивидуальные частицы и наблюдали при увеличениях 50-500× и частично повернутых относительно друг друга поляризационных фильтрах, совместно укрепленных на λ-волновой пластине.
D. XRPD (Дифракция рентгеновских лучей на порошке)
D5000
Исследования XRPD выполняли на дифрактометре Siemens D5000, используя излучение CuKα (40 кВ, 40 мА), θ-θ гониометр, автоматические отклоняющие и приемные щели, графитовый вторичный монохроматор и сцинтилляционный счетчик. Данные регистрировали в диапазоне углов 2θ от 2 до 30° в режиме непрерывного сканирования, используя размер шага либо 0,02° 2θ, либо 0,005° 2θ и время шага, равное 1 секунде.
Образцы, которые исследовали в комнатных условиях, приготавливали в виде плоских пластин, используя порошки, в том виде, в котором они были получены, без измельчения. Приблизительно 25-50 мг образца осторожно помещали в углубление диаметром 12 мм и глубиной 0,5 мм в полированной кремниевой пластине с нулевой подложкой (510) (The Gem Dugout, 1652 Princeton Drive, Pennsylvania State College, PA 16803, USA). Все исследования по методике XRPD осуществляли с применением программы Diffrac Plus XRD Commander v.2.3.1.
Дифрактометр Brucker AXS C2 GADDS (применялся для образцов, возвращенных из GVS)
Картины дифракции рентгеновских лучей на порошке для образцов получали на дифрактометре Brucker AXS C2 GADDS, используя CuKα-излучение (40 кВ, 40 мА), автоматический XYZ координатный стол, лазерный видеомикроскоп для автоматического позиционирования образцов и 2-мерный детектор поверхности HiStar. Оптика для рентгеновских лучей состояла из одного многослойного зеркала Гебеля, спаренного с точечным коллиматором диаметром 0,3 мм.
Расхождение пучка, т.е. эффективный размер пучка рентгеновских лучей на образце, составлял примерно 4 мм. Применяли режим непрерывного сканирования по θ-θ, при расстоянии от образца до детектора, равным 20 см, что давало эффективный диапазон 2θ, равный 3,2-29,8°. Типовое время регистрации дифракционной картины образца составляло 120 с.
Образцы приготавливали в виде плоских пластин, используя порошки в том виде, как они были получены, без измельчения. Приблизительно 1-2 мг образца подвергали легкому давлению на предметном стекле для получения плоской поверхности.
Картину XRPD регистрировали для L-лактата и свободного основания. На дифракционной картине наблюдалось хорошее соотношение сигнал/шум, и она указывала на кристаллическую природу веществ.
E. Гравиметрическое исследование адсорбции пара (GVS)
Все образцы исследовали на анализаторе адсорбции влаги Hiden IGASorp, применяя программу CFRSorp. Масса образца составляла приблизительно 10-25 мг. Изотерму адсорбции/десорбции паров воды регистрировали, как описано ниже. Образцы загружали и выгружали при комнатной влажности и температуре (приблизительно 40% влажность, 25°C) и позднее анализировали способом XRPD (используя систему Brucker AXS C2 GADDS).
Стандартным способом регистрации изотермы являлся единичный цикл, начинавшийся при относительной влажности 40%.
Влажность ступенчато изменяли следующим образом:
40, 50, 60, 70, 80, 90
85, 75, 65, 55, 45, 35, 25, 15, 5, 0
10, 20, 30, 40
(i) L-лактат
Изотерма GVS для L-лактата показала, что образец не проявлял гигроскопических свойств и не образовывал гидрата. Порошковая рентгенограмма дифракции XRPD для образцов, зарегистрированная после экспериментов по GVS, соответствовала рентгенограмме для исходного вещества, показывая отсутствие фазовых изменений во время эксперимента.
(ii) Свободное основание
Во время эксперимента масса образца при влажности 0% и влажности 95% различалась примерно на 9%. Эти данные показали, что образец по своей природе являлся гигроскопичным.
Пример 69
Определение кристаллической структуры дигидрата свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины рентгеноструктурным анализом
Кристалл, использованный для рентгеноструктурного анализа, был бесцветным и обладал неправильной формой с размерами 0,2×0,2×0,2 мм3. Его получали осаждением водного раствора эзилата с помощью ТГФ в эксперименте по диффузии жидкость-жидкость. Эквивалентность этого образца и образца той же кристаллической формы, полученной из свободного основания (при использовании воды в качестве антирастворителя в сочетании с рядом растворителей, таких как спирты, например, этанол, кетоны, например метилэтилкетон и простые эфиры, такие как ТГФ и диоксан) была установлена сравнением порошковой рентгенограммы обоих образцов. Кристаллографические данные регистрировали при 101(2)K, используя излучение CuKα (λ=1,5418 Å) вращающегося анода Rigaku RU3HR, синюю конфокальную оптику Osmic, гониометр AFC9 1/4χ и детектор Rigaku Jupiter CCD. Изображения регистрировали в результате четырех сканирований по ω, одном при 2θ=30° и трех при 2θ=90°, при расстоянии от кристалла до детектора 67 мм. Регистрация данных проходила под управлением программы CrystalClear, и изображения обрабатывали и масштабировали с помощью Dtrek. Хотя коэффициент поглощения был умеренным (µ=0,82 мм-1), данные корректировали с применением коррекции поглощения Фурье 4-го порядка для компенсации поглощения клея и кристаллодержателя (микродержателя). Было обнаружено, что кристаллы относятся к моноклинной пространственной группе P2l/n (#14) с параметрами кристаллической решетки a=7,66(10), b=15,18(10), c=17,71(10) Å, β=98,53(2)°, α=γ=90°. Числа в скобках означают отклонение (s.u., стандартная неопределенность).
Кристаллическая структура была расшифрована с применением прямых методов, включенных в SHELXS-97. Данные по интенсивности для, в общей сложности, 2822 уникальных отражений в диапазоне разрешения 11,5-0,89 Å (3,85<θ<60,01) использовали для выявления 274 кристаллографических параметров с помощью SHELXL-97. Итоговые статистические параметры были следующими: wR2=0,2416 (все данные), RF=0,0866 (данные с I>2σ(I)) и степень согласия S=1,145.
Было найдено, что одна молекула свободного основания и две молекулы воды расположены в асимметрической ячейке. Элементный состав асимметрической ячейки C19H26N7O4 и вычисленная плотность кристаллов составляли 1,36 мг/м3. Атомы водорода были генерированы на геометрических площадках, в то время как положение атомов водорода, связанных с гетероатомами, было подтверждено исследованием карт разности Fo-Fc. Позиционные и тепловые параметры атомов водорода ограничивали в зависимости от соответствующих неводородных атомов. Тепловое движение неводородных атомов моделировали анизотропными тепловыми факторами (см. фиг.1).
Кристаллическая структура включала одну внутримолекулярную (N22-H…N14 2,898 Å) и семь межмолекулярных водородных связей (см. фиг.2). Молекулы 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины соединены друг с другом в цепи вдоль кристаллографической оси b двумя водородными связями: N7-H…O24 2,761 Å и N25-H…N2 3,310 Å. Бензимидазольные фрагменты двух цепей уложены в кристаллической структуре на расстоянии 3,5-3,6 Å друг от друга. Кристаллическая решетка молекул 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины образует карманы, занятые четырьмя молекулами воды, которые попарно связаны с центром симметрии. Три водородные связи соединяют молекулы 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины с молекулами воды, одна из них с первой молекулой воды (O1W1-H…N16 2,845 Å), и оставшиеся две со второй молекулой воды (N1-H…O1W2 2,875 Å и O1W2-H…O19 2,746 Å). Молекулы воды взаимодействуют друг с другом за счет двух других водородных связей: O1W1-H…O1W2 2,884 Å и O1W2-H…O1W1 2,771 Å.
Изображение структуры, полученной по результатам рентгеноструктурного анализа, в виде тепловых эллипсоидов приведено на фиг.1, и схема кристаллической упаковки изображена на фиг.2.
Координаты атомов, из которых состоит структура дигидрата свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, приведены в таблице 2. Числа в скобках представляют собой отклонение (s.u., стандартная неопределенность).
Положение атома_обозначение атома
Положение атома_химический символ
Положение атома_дробная часть x
Положение атома_дробная часть y
Положение атома_дробная часть z
Положение атома_U изо или эквивалент
Положение атома_тип adp
Положение атома_занятость положения
Положение атома_кратность симметрии
Положение атома_отметка о расчете


Пример 70
Дифракционная картина XRPD 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в форме свободного основания
Образцы для регистрации данных по дифракции рентгеновских лучей на порошке (XRPD) осторожно измельчали в мраморной ступке и помещали в кристаллографический капилляр (Hampton Research, Quartz or Glass Type 10, диаметр 0,4 или 0,7 мм). Дифракционные картины регистрировали при комнатной температуре, используя излучение CuKα (λ=1,5418 Å) вращающегося анода Rigaku RU3HR, синюю конфокальную оптику Osmic, 1/4χ гониометр и детектор сигнала в анодной цепи Rigaku HTC. Двумерные изображения регистрировали при вращении вокруг оси φ, при расстоянии детектор-кристалл 250 мм. Регистрацией данных управляла программа CrystalClear, и двумерные изображения преобразовывали в одномерную кривую (в координатах 2θ - интенсивность) с помощью Datasqueeze (интенсивность усреднялась по азимутальному углу 0<χ<360° для диапазона 2θ 3-30° с шагом 0,01 или 0,02°). Для обработки и визуализации одномерных дифракционных картин XRPD использовали самостоятельно созданную программу AstexXRPD.
Картина XRPD и относительные интенсивности сигналов не менялись для различных партий кристаллических образцов, что согласуется с наличием лишь одной кристаллической формы.
Дифракционная картина XRPD для формы FB1 свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины приведена на фиг.3, и подробности, касающиеся основных пиков, суммированы в таблице 3.
Пример 71
Определение кристаллической структуры лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
Идентифицировали монокристаллическую форму L-лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины. Кристалл, использованный для дифракционного исследования, был бесцветной призмой с размерами 0,1×0,1×0,1 мм3, полученной осаждением из этанола при его выпаривании. Кристаллографические данные регистрировали при 97 K, используя излучение CuKα (λ=1,5418 Å) вращающегося анода Rigaku RU3HR, синюю конфокальную оптику Osmic, гониометр AFC9 1/4χ и детектор Rigaku Jupiter CCD. Изображения регистрировали в результате пяти сканирований по ω, одном при 2θ=15° и четырех при 2θ=90°, при расстоянии от кристалла до детектора 67 мм. Регистрация данных проходила под управлением программы CrystalClear и изображения обрабатывали и масштабировали с помощью Dtrek. Хотя коэффициент поглощения был умеренным (µ=0,78 мм-1), данные корректировали с применением коррекции поглощения Фурье 4-го порядка для компенсации поглощения в клее и кристаллодержателе (микродержателе). Было обнаружено, что кристаллы относятся к орторомбической пространственной группе P2l2l2l (#19) с параметрами кристаллической решетки a=9,94(10), b=15,03(10), c=16,18(10) Å, α=β=γ=90°. Числа в скобках означают отклонение (s.u., стандартная неопределенность). Проводили одно короткое сканирование при комнатной температуре для проверки параметров и симметрии кристаллической решетки. Обнаружили, что симметрия была той же, что и при 97(2)K, и параметры кристаллической решетки являлись похожими (a=10,08, b=15,22, c=16,22 Å).
Кристаллическая структура была расшифрована с применением прямых методов, включенных в SHELXS-97. Абсолютную конфигурацию выбирали таким образом, чтобы она соответствовала конфигурации L-молочной кислоты, применявшейся в эксперименте по получению кристаллов. Данные по интенсивности для, в общей сложности, 3417 уникальных отражений в диапазоне разрешения 11-0,9 Å (4,01<θ<58,92) использовали для выявления 308 кристаллографических параметров с помощью SHELXL-97. Итоговые статистические параметры были следующими: wR2=0,2275 (все данные), RF=0,0817 (данные с I>2σ(I)) и степень согласия S=1,076.
Было найдено, что в асимметрической ячейке расположены одна молекула свободного основания и один анион L-молочной кислоты. Элементный состав асимметрической ячейки C22H29N7O5 и вычисленная плотность кристаллов составляла 1,30 мг/м3. Атомы водорода были генерированы на геометрических площадках, в то время как положение атомов водорода, связанных с гетероатомами, было подтверждено исследованием карт разности Fo-Fc. Позиционные и тепловые параметры атомов водорода ограничивали, чтобы они зависели от соответствующих неводородных атомов. Тепловое движение неводородных атомов моделировали анизотропными тепловыми факторами (см. фиг.4).
Кристаллическая структура включала одну внутримолекулярную (N22-H…N14 2,852 Å) и семь межмолекулярных водородных связей, образующих сложную трехмерную решетку (см. фиг.5). Две межмолекулярные водородные связи, а именно N7-H…O24 2,800 Å и N25-H…N2 3,004 Å связывают молекулы 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в цепи вдоль кристаллографической оси c. Анионы L-молочной кислоты связаны в цепи вдоль кристаллографической оси a водородными связями O3L-H…O1L 2,626 Å. Две раздвоенные водородные связи соединяют катионы 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины и анионы L-молочной кислоты. Протонированный атом азота морфолинового фрагмента взаимодействует с обоими карбоксильными атомами кислорода (N16-H…O1L 3,125 Å и N16H…O2L 2,625 Å), тогда как пиразольный атом азота N1 является донором H для атомов O2L и O3L (N1-H…O2L 2,882 Å, N1-H…O3L 2,740 Å).
Изображение структуры, полученной по результатам рентгеноструктурного анализа, в виде тепловых эллипсоидов приведено на фиг.4, и схема кристаллической упаковки изображена на фиг.5.
Координаты атомов, из которых состоит структура лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, приведены в таблице 4. Числа в скобках представляют собой отклонение (s.u., стандартная неопределенность).
Положение атома_обозначение атома
Положение атома_химический символ
Положение атома_дробная часть x
Положение атома_дробная часть y
Положение атома_дробная часть z
Положение атома_U изо или эквивалент
Положение атома_тип adp
Положение атома_занятость положения
Положение атома_кратность симметрии
Положение атома_отметка о расчете


Пример 72
Устойчивость соли 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины при 40°C и относительной влажности 75%
Приблизительно 15 мг образцов для исследования устойчивости осторожно измельчали в мраморной ступке и переносили в чашку Петри в виде тонкого слоя. Затем образцы помещали в плотно закрытые емкости, содержащие насыщенный раствор NaCl с избытком нерастворенного NaCl. Эти емкости, в свою очередь, помещали в инкубатор, где поддерживали температуру 40°C для получения окружающей среды с температурой 40°C и относительной влажностью (RH)≈75%. Образцы регулярно анализировали способом дифракции рентгеновских лучей на порошке (XRPD).
Для получения данных XRPD образцы помещали в кристаллографический капилляр (фирмы Hampton Research, изготовленный из кварца, диаметр=0,4 мм). Дифракционные картины регистрировали при комнатной температуре, используя излучение CuKα (λ=1,5418 Å) вращающегося анода Rigaku RU3HR, синюю конфокальную оптику Osmic, 1/4χ гониометр и детектор сигнала в анодной цепи Rigaku HTC. Двумерные изображения регистрировали при вращении вокруг оси φ, при расстоянии детектор-кристалл 250 мм. Регистрацией данных управляла программа CrystalClear, и двумерные изображения преобразовывали в одномерную кривую (в координатах 2θ - интенсивность) с помощью Datasqueeze (интенсивность усредняли по азимутальному углу 0<χ<360° для диапазона 2θ 3-30° с шагом 0,01°). Для обработки и визуализации одномерных дифракционных картин XRPD использовали самостоятельно созданную программу AstexXRPD.
Дифракционные картины XRPD лактата, свободного основания (FB1) и дигидрата свободного основания (FB2) не изменялись на протяжении периода 1-2 месяцев, при действии среды с температурой 40°C и относительной влажностью 75%. Дифракционные картины исходных образцов, а также образцов лактата, свободного основания (FB1) и дигидрата свободного основания (FB2), полученных в результате исследования устойчивости, приведены на фиг. 6-8.
Дифракционная картина XRPD L-лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины приведена на фиг.6 и подробные сведения об основных пиках суммированы в таблице 5.
Дифракционная картина XRPD формы FB2, т.е. дигидрата свободного основания 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины, приведена на фиг.8, и подробные сведения об основных пиках суммированы в таблице 6.
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
Пример 73
Измерения активированного комплекса CDK2/циклин A в рамках исследования ингибирующей активности в отношении киназы (IC 50 )
Соединения по настоящему изобретению тестировали на ингибирующую активность в отношении киназы, используя следующий протокол.
Активированный комплекс CDK2/циклин A (Brown et al, Nat.Cell Biol., 1, pp.438-443,1999; Lowe, E.D., et al, Biochemistry, 41, pp. 15625-15634, 2002) разбавляли до 125 пМ в аналитическом буфере 2,5× концентрации (50 мМ MOPS pH 7,2, 62,5 мМ β-глицерофосфата, 12,5 мМ EDTA, 37,5 мМ MgCl2, 112,5 мМ АТФ, 2,5 мМ DTT, 2,5 мМ ортованадата натрия, 0,25 мг/мл альбумина бычьей сыворотки) и 10 мкл смешивали с 10 мкл субстратной смеси, содержащей гистон (60 мкл бычьего гистона H1 (Upstate Biotechnology, 5 мг/мл), 940 мкл H2O, 35 мкКюри γ33P-АТФ) и добавляли в 96-луночные планшеты вместе с 5 мкл раствора исследуемого вещества в ДМСО в различных разбавлениях (до 2,5%). Давали реакции пройти в течение 2-4 часов, после чего останавливали ее избытком ортофосфорной кислоты (5 мкл конц. 2%). γ33P-АТФ, который не соединился с гистоном H1, отделяли от фосфорилированного гистона H1 на фильтровальной пластине Millipore MAPH. Ячейки на пластине MAPH смачивали 0,5% ортофосфорной кислотой и затем через эти ячейки фильтровали продукты реакции с помощью прибора для вакуумного фильтрования Millipore. После фильтрования остаток дважды промывали 200 мкл 0,5% ортофосфорной кислоты. Когда фильтры высыхали, добавляли 20 мкл сцинтиллятора Microscint 20 и затем производили подсчет на Packard Topcount в течение 30 секунд.
Рассчитывали ингибирование активности CDK2 в % и наносили на график для определения концентрации тестируемого соединения, требуемой для ингибирования 50% активности CDK2 (IC50).
Соединения примеров 1, 10, 11, 18, 20, 22, 30, 31, 32, 46, 47 и 54 в анализе ингибирования CDK2 имели значения IC50 менее 1 мкМ, в то время как соединения примеров 44, 45, 48, 51 и 53 имели значения IC50 менее 10 мкМ.
Пример 74
Измерения активированного комплекса CDK1/циклин B в рамках исследования ингибирующей активности в отношении киназы (IC 50 )
Анализ комплекса CDK1/циклин B идентичен описанному выше анализу комплекса CDK2/циклин A за исключением того, что использовали CDK1/циклин B (Upstate Discovery) и фермент разбавляли до 6,25 нМ.
Соединения примеров 1, 4, 6, 10, 11, 13, 22, 42, 47 и 54 в анализе ингибирования CDK1 имели значения IC50 менее 1 мкМ, в то время как соединения примеров 3, 8, 9, 16, 17, 20, 24, 28, 29, 31, 32, 34, 39, 41, 45, 46, 48, 49, 50, 51, 52, 53 и 56 имели значения IC50 менее 10 мкМ и соединения примеров 2, 23, 26, 27, 33, 37 и 43 имели значения IC50 менее 50 мкМ.
Пример 75
Исследования Aurora A киназы
Активность Aurora A киназы может быть определена с применением диссоциативно-усиленного лантанидного иммуноанализа (DELFIA) с полученным из GSK-3 биотинилированным пептидом. Количество образовавшегося фосфорилированного пептида измеряют с помощью фосфо-специфичного первичного антитела и меченного европием анти-кроличьего IgG антитела, используя флуоресценцию с временным разрешением при λex=337 нм, λcm=620 нм.
Реакция киназы
Аналитические реакции проводили в 96-луночных планшетах при общем объеме реакционной смеси 25 мкл с 0,5 нМ Aurora A (Upstate Discovery), 3 мкМ биотин-CGPKGPGRRGRRRTSSFAEG, 15мкМ АТФ и при различных разбавлениях тестируемого соединения в смеси 10 мМ MOPS, pH 7,0, 0,1 мг/мл BSA, 0,001% Brij-35, 0,5% глицерина, 0,2 мМ EDTA, 10 мМ MgCl2, 0,01% β-меркаптоэтанола и 2,5% ДМСО. Реакции давали пройти в течение 60 минут при комнатной температуре, затем останавливали ее добавлением 100 мкл буфера STOP, содержащего 100 мМ EDTA, 0,05% Surfact-Amps20 (Pierce) и 1× BlockerTM BSA в TBS (Pierce).
Стадия детектирования
После этого реакционную смесь переносили в 96-луночный планшет, покрытый Neutravidin (Pierce) и инкубировали в течение 30 минут для захвата биотинилированного пептида. После 5-кратного промывания TBST буфером в количестве 200 мкл на лунку в каждую лунку добавляли смесь антитела анти-фосфо(Ser/Thr)-AKT субстрата (Cell Signalling Technology) и Eu-N1 анти-кроличий IgG (Perkin Elmer) и оставляли на 1 час. После стадии дополнительной промывки во все лунки добавляли DELFIA усиливающий раствор (Perkin Elmer). После инкубирования в течение 5 минут проводили подсчет лунок на считывателе планшетов Fusion.
В описанном выше анализе соединения примеров 1-56 имели значение IC50 менее 1 мкМ. Хлористоводородная соль примера 60H имела значение IC50, равное 0,0025 мкМ.
Пример 76
Исследования Aurora B киназы
Реакция киназы
Аналитические реакции проводили в 96-луночных планшетах, при общем объеме реакционной смеси 25 мкл с 0,5 нМ Aurora B (ProQinase), 3 мкМ биотин-CGPKGPGRRGRRRTSSFAEG, 15 мкМ АТФ и при различных разбавлениях тестируемого соединения в смеси 25 мМ TRIS pH 8,5, 0,1 мг/мл BSA, 0,025% Surfact-Amps20, 5 мМ MgCl2, 1 мМ DTT и 2,5% ДМСО. Реакции давали пройти в течение 90 минут при комнатной температуре, затем останавливали ее добавлением 100 мкл буфера STOP, содержащего 100 мМ EDTA, 0,05% Surfact-Amps20 (Pierce) и 1× BlockerTM BSA в TBS (Pierce).
Стадию детектирования приводили, как описано для Aurora A.
В анализе ингибирования Aurora B хлористоводородная соль примера 60H продемонстрировала 57% ингибирование при концентрации 0,003 мкМ.
Пример 77
Анализ ингибирующей активности в отношении киназы GSK3-B
GSK3-β (Upstate Discovery) разбавляли до концентрации 7,5 нМ в смеси 25 мМ MOPS, pH 7,0, 25 мг/мл BSA, 0,0025% Brij-35, 1,25% глицерина, 0,5 мМ EDTA, 25 мМ MgCl2, 0,025% β-меркаптоэтанола, 37,5 мМ АТФ, и 10 мкл смешивали с 10 мкл субстратной смеси. Субстратная смесь для GSK3-β представляла собой 12,5 мМ пептид-2 фосфо-гликоген синтазы (Upstate Discovery) в 1 мл воды с 35 мкКюри γ33P-АТФ. Фермент и субстрат помещали в 96-луночные планшеты вместе с 5 мкл раствора тестируемого соединения в ДМСО в различных разбавлениях (до 2,5%). Реакции давали пройти в течение 3 часов (GSK3-β), после чего останавливали ее избытком ортофосфорной кислоты (5 мкл, конц. 2%). Методика фильтрования соответствовала приведенной выше методике из анализа активированного комплекса CDK2/циклин A.
Пример 78
Исследования селективности в отношении CDK
78A. Протокол A
Соединения по настоящему изобретению могут быть испытаны на ингибирующую активность в отношении киназ на примере ряда различных киназ с использованием общего протокола, описанного выше в примере 3, но модифицированного, как указано ниже.
Киназы разбавляли до 10× рабочего запасного раствора в смеси 20 мМ MOPS, pH 7,0, 1 мМ EDTA, 0,1% γ-меркаптоэтанола, 0,01% Brij-35, 5% глицерина, 1 мг/мл BSA. Одна единица соответствовала включению 1 нмоль фосфата в минуту в 0,1 мг/мл гистон H1 или в пептид, являющийся субстратом CDK7, при 30°C, с итоговой концентрацией АТФ 100 мкМ.
Субстратом для всех исследований CDK (кроме CDK7) являлся гистон H1, перед использованием разбавленный до 10× запасного рабочего раствора в 20 мМ буфере MOPS pH 7,4. Субстратом для CDK7 являлся определенный пептид, разбавленный до 10× запасного рабочего раствора в деионизированной воде.
Методика анализа для CDK1/циклин B, CDK2/циклин A, CDK2/циклин E, CDK3/циклин E, CDK5/p35, CDK6/циклин D3:
При конечном объеме реакционной смеси 25 мкл, фермент (5-10 мЕд.) инкубировали с 8 мМ MOPS, pH 7,0, 0,2 мМ EDTA, 0,1 мг/мл гистона H1, 10 мМ ацетата Mg и [γ33P-АТФ] (удельная активность приблизительно 500 имп/мин/пмоль, концентрация - как требовалось по условиям эксперимента). Реакцию инициировали добавлением Mg2+ [γ33P-АТФ]. После инкубирования в течение 40 минут при комнатной температуре реакцию останавливали добавлением 5 мкл 3% раствора фосфорной кислоты. 10 мкл реакционной смеси наносили на плоский фильтр P30 и три раза в течение 5 минут промывали 75 мМ фосфорной кислотой и один раз метанолом, после чего высушивали и производили подсчет.
Методика анализа для CDK7/циклин H/MAT1
При конечном объеме реакционной смеси 25 мкл фермент (5-10 мЕд.) инкубировали с 8 мМ MOPS, pH 7,0, 0,2 мМ EDTA, 500 мкМ пептида, 10 мМ ацетата Mg и [γ33P-АТФ] (удельная активность приблизительно 500 имп/мин/пмоль, концентрация - как требовалось по условиям эксперимента). Реакцию инициировали добавлением Mg2+ [γ33P-АТФ]. После инкубирования в течение 40 минут при комнатной температуре реакцию останавливали добавлением 5 мкл 3% раствора фосфорной кислоты. 10 мкл реакционной смеси наносили на плоский фильтр P30 и три раза в течение 5 минут промывали 75 мМ фосфорной кислотой и один раз метанолом, после чего высушивали и производили подсчет.
78В. Протокол B
Ингибирующую активность в отношении указанных ферментов исследовали в Upstate Discovery Ltd. Получали растворы ферментов в конечной концентрации 10× в ферментном буфере (состав указан в таблице ниже). Затем ферменты инкубировали в аналитическом буфере с различными субстратами и 33P-АТФ (~500 имп/мин/пмоль), как показано в таблице.
Реакцию инициировали добавлением Mg/АТФ. Затем реакции давали пройти в течение 40 минут при комнатной температуре, после чего останавливали добавлением 5 мкл 3% раствора фосфорной кислоты. Десять мкл реакционной смеси наносили на плоский фильтр A или P30, три раза промывали 75 мМ фосфорной кислотой и один раз метанолом, после чего высушивали и производили подсчет сцинтилляции.
Тестируемые соединения испытывали при концентрациях, подробно описанных ниже в дубликате, со всеми киназами, и рассчитывали имеющуюся активность по сравнению с контролем. Если соединения демонстрировали значительное ингибирование, определяли IC50.
LSEEEQEMFRDFDYIADWC
Использованные ферментные буферы имели следующий состав:
A: 20 мМ MOPS, pH 7,0, 1 мМ EDTA, 0,1% β-меркаптоэтанол, 0,01% Brij-35, 5% глицерин, 1 мг/мл BSA.
Использованные аналитические буферы имели следующий состав:
A: 8 мМ MOPS, pH 7,0, 0,2 мМ EDTA, 10 мМ ацетат Mg.
Пример 79
Антипролиферативная активность
Антипролиферативную активность соединений по настоящему изобретению можно определить путем измерения способности соединений ингибировать рост клеток ряда клеточных линий. Измеряли ингибирование роста клеток, используя анализ с тест-системой Alamar Blue (Nociari, M.M., Shalev, A., Benias, P., Russo, C. Journal of Immunological Methods 1998, 213, 157-167). Способ основан на способности жизнеспособных клеток восстанавливать резазурин до его флуоресцирующего продукта резоруфина. Для каждого исследования пролиферации клетки наносили на 96-луночные планшеты и давали им восстановиться в течение 16 часов, после чего на следующие 72 часа добавляли ингибирующие соединения. В конце периода инкубации добавляли 10% (объем/объем) Alamar Blue и инкубировали еще 6 часов, после чего определяли содержание флуоресцентного продукта при длинах волн 535 нм возбуждение/590 нм эмиссия. В случае исследования непролиферирующих клеток скопление клеток выдерживали 96 часов, после этого добавляли ингибирующие соединения на последующие 72 часа. Количество жизнеспособных клеток определяли анализом с Alamar Blue, как указано выше. Кроме того, фиксировали все морфологические изменения. Линии клеток могут быть получены из ECACC (Европейской коллекции клеточных культур).
В исследовании, в котором использовалась клеточная линия HCT-116, хлористоводородная соль примера 60H имела IC50, равную 0,070 мкМ.
В частности, была испытана активность соединений по настоящему изобретению против клеток линии HCT-116 (ECACC, рег. номер: 91091005), полученных из раковой опухоли ободочной кишки человека.
Было обнаружено, что многие соединения по настоящему изобретению в этом анализе обладают величинами констант IC50 менее 25 мкМ, и предпочтительные соединения имеют значения IC50 менее 1 мкМ. С другой стороны, было найдено, что для многих соединений минимальная концентрация, при которой наблюдается полиплодия или многоядерность клеток, составляет менее 10 мкМ, и предпочтительные соединения имеют значения IC50 менее 100 нМ.
Было найдено, что такое соединение, как 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевина, в описанном анализе имеет значение IC50 менее 1 мкМ. Кроме того, для этого соединения было обнаружено, что минимальная концентрация, при которой наблюдается полиплодия или появление нескольких ядер, составляет менее 100 нМ.
Пример 80
A. Общий протокол исследования образования колоний
Воздействие различных соединений на линии адгезивных опухолевых клеток оценивали в клоногенном анализе.
Клетки высевали при концентрации 75-100 клеток/мл соответствующей культуральной среды на 6- или 24-луночные планшеты для культур тканей и давали восстановиться в течение 16 часов.
Соединение или носитель в случае контрольного опыта (ДМСО) добавляли в парные лунки до достижения конечной концентрации ДМСО 0,1%. После добавления соединения колониям давали возможность прорастать в течение 10-14 дней для оптимального дискретного подсчета колоний. Колонии фиксировали 2 мл фиксирующего состава Carnoys (25% уксусной кислоты, 75% метанола) и окрашивали в 2 мл 0,4% масса/объем кристаллического фиолетового. Подсчитывали количество колоний в каждой лунке. Значения IC50 рассчитывали с помощью сигмоидальных IC50 кривых зависимости реакции от дозы (переменный наклон), применяя программу Prism Graphpad.
B. Протокол исследования образования колоний для 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины
Воздействие 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины на клеточные линии А2780, A549, HCT 116, HCT 116 N7, HT-29, MCF7, MIA-Pa-Ca-2, SW620 оценивали в клоногенном анализе.
Клетки высевали при концентрации 75-100 клеток/мл соответствующей культуральной среды на 6- или 24-луночные планшеты для культур тканей и давали восстановиться в течение 16 часов.
1-Циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевину или, в случае контрольного опыта, носитель (ДМСО) добавляли в парные лунки до достижения конечной концентрации ДМСО 0,1%. После добавления соединения колониям давали возможность прорастать в течение 10-14 дней для оптимального дискретного подсчета колоний. Колонии фиксировали 2 мл фиксирующего состава Carnoys (25% уксусной кислоты, 75% метанола) и окрашивали в 2 мл 0,4% масса/объем кристаллического фиолетового. Подсчитывали количество колоний в каждой лунке. Учитывали только многоклеточные колонии, включавшие приблизительно 50 или более клеток, которые продемонстрировали пролиферацию от одной клетки до колонии из многих клеток (т.е. полный клеточный цикл, включавший успешный цитокинез). Отдельные мультиядерные (полиплоидные) клетки не учитывали. Значения IC50 рассчитывали с помощью сигмоидальных IC50 кривых зависимости реакции от дозы (переменный наклон), применяя программу Prism Graphpad.
Результаты описанных исследований приведены в таблице C в разделе, озаглавленном «Преимущества соединений по настоящему изобретению».
Пример 81
Определение эффективности в отношении цитохрома P450
Эффективность соединения примера 24 в отношении CYP450 1A2, 2C9, 2C19, 3A4 и 2D6 определяли с применением исследовательского набора Pan Vera Vivid Cyp450, который можно приобрести у Invitrogen (Paisley, UK). CYP вводили в реакционную смесь в форме бакулосом, содержащих CYP450 и НАДФН редуктазу. Субстратами являлись флуоресцентные субстраты Vivid.
Итоговые реакционные смеси содержали следующие компоненты:
1A2
100 мМ фосфат калия, pH 8, 1% метанол, субстрат 2 мкМ 1A2 blue vivid, 100 мкМ НАДФ+, 4 нМ CYP450 1A2, 2,66 мМ глюкоза-6-фосфат, 0,32 Ед/мл глюкоза-6-фосфат дегидрогеназы.
2C9
50 мМ фосфат калия, pH 8, 1% метанол, субстрат 2 мкМ green vivid, 100 мкМ НАДФ+, 8 нМ CYP450 2C9, 2,66 мМ глюкоза-6-фосфат, 0,32 Ед/мл глюкоза-6-фосфат дегидрогеназы.
2C19
50 мМ фосфат калия, pH 8, 1% метанол, субстрат 8 мкМ blue vivid, 100 мкМ
НАДФ+, 4 нМ CYP450 2C19, 2,66 мМ глюкоза-6-фосфат, 0,32 Ед/мл глюкоза-6-фосфат дегидрогеназы.
3A4
100 мМ фосфат калия, pH 8, 1% метанол, субстрат 10 мкМ 3A4 blue vivid, 100 мкМ НАДФ+, 2,5 нМ CYP450 3A4, 2,66 мМ глюкоза-6-фосфат, 0,32 Ед/мл глюкоза-6-фосфат дегидрогеназы.
2D6
100 мМ фосфат калия, pH 8, 1% метанол, субстрат 5 мкМ 2D6 blue vivid, 100 мкМ НАДФ+, 5 нМ CYP450 2D6, 2,66 мМ глюкоза-6-фосфат, 0,32 Ед/мл глюкоза-6-фосфат дегидрогеназы.
Флуоресценцию регистрировали в течение 20 мин через 30 с интервалы на Molecular Devices Spectramax Gemini reader. Длины волн возбуждения и эмиссии равнялись 390 нм и 460 нм для 1A2, 2C19 и 3A4, 390 нм и 485 нм для 2D6 и 485 нм и 530 нм для 2C9. Исходные скорости определяли из кривых хода реакции.
Готовили растворы тестируемых соединений в метаноле и определяли их эффективность в отношении CYP450 при концентрации 10 мкМ. Результаты представлены в Таблице B.
ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ
Пример 82
(i) Рецептура таблеток
Композицию таблеток, содержащую соединение формулы (I), получают смешиванием 50 мг соединения с 197 мг лактозы (BP) в качестве разбавителя и 3 мг стеарата магния, в качестве средства для скольжения, и прессованием смеси известным способом для формирования таблеток.
(ii) Рецептура капсул
Состав в форме капсул получают смешиванием 100 мг соединения формулы (I) со 100 мг лактозы и заполнением полученной смесью стандартных непрозрачных твердых желатиновых капсул.
(iii) Пригодный для инъекции состав I
Парентеральная композиция для введения с помощью инъекции может быть получена растворением соединения формулы (I) (например, в форме соли) в воде, содержащей 10% пропиленгликоля, для получения концентрации действующего соединения 1,5% по массе. Затем полученный раствор стерилизуют фильтрованием, наполняют им ампулы и запаивают.
(iv) Пригодный для инъекции состав II
Парентеральную композицию для инъекций получают растворением в воде соединения формулы (I) (например, в форме соли) (2 мг/мл) и маннита (50 мг/мл), стерильным фильтрованием раствора и заполнением этим раствором плотно закрываемых 1 мл пузырьков или ампул.
(v) Пригодный для инъекции состав III
Состав для внутривенного введения инъекцией или инфузией может быть получен растворением соединения формулы (I) (например, в форме соли) в воде в количестве 20 мг/мл. Затем флакон плотно закрывают и стерилизуют обработкой в автоклаве.
(vi) Пригодный для инъекции состав IV
Состав для внутривенного введения инъекцией или инфузией может быть получен растворением соединения формулы (I) (например, в форме соли) в воде, содержащей буфер (например, 0,2М ацетатный буфер pH 4,6), в количестве 20 мг/мл. Затем флакон плотно закрывают и стерилизуют обработкой в автоклаве.
(vii) Лиофилизованный состав I
Аликвоты соединения формулы (I) или его соли, определенной в настоящей заявке, смешанные с другими необходимыми компонентами, помещают в 50 мл флаконы и лиофилизуют. Во время лиофилизации композиции замораживают, используя одностадийную методику заморозки при (-45°C). Температуру поднимают до -10°C для отжига, затем снижают для замораживания при -45°C, с последующей первичной сушкой при +25ºC в течение примерно 3400 минут, и следующей за ней вторичной сушкой со ступенчатым повышением температуры до 50°C. Во время первичной и вторичной сушки устанавливают давление 80 миллитор.
(viii) Лиофилизованный состав II
Аликвоты соединения формулы (I) или его соли, определенной в настоящей заявке, смешанные с другими необходимыми компонентами, помещают в 50 мл флаконы и лиофилизуют. Во время лиофилизации композиции замораживают, используя одностадийную методику заморозки при (-45°C). Температуру поднимают до -10°C для отжига, затем снижают для замораживания при -45°C, с последующей первичной сушкой при +25°C в течение примерно 3400 минут и следующей за ней вторичной сушкой со ступенчатым повышением температуры до 50°C. Во время первичной и вторичной сушки устанавливают давление 80 миллитор.
(ix) Лиофилизованный состав, применяемый для внутривенного введения III
Забуференный водный раствор получают растворением L-лактата 1-циклопропил-3-[3-(5-морфолин-4-илметил-1H-бензимидазол-2-ил)-1H-пиразол-4-ил]мочевины в концентрации 12,86 мг/мл в 0,02М цитратном буфере, pH которого скорректировано до 4,5 добавлением гидроксида натрия или хлористоводородной кислоты.
Полученным забуференным раствором заполняют, с промежуточным фильтрованием для удаления частиц вещества, емкость (как, например, стеклянный флакон класса 1), которую затем частично закрывают (например, с помощью пробки Florotec). Если соединение и состав в основном стабильны, состав стерилизуют обработкой в автоклаве при 121°C в течение необходимого периода времени. Если состав недостаточно стабилен для обработки в автоклаве, его можно стерилизовать применением подходящего фильтра и в стерильных условиях наполнить им стерильные флаконы. Раствор подвергают лиофилизации с применением подходящего цикла, например:
Замораживание - охлаждают до -40°C в течение 2 часов и выдерживают при -40°C в течение 3 часов.
Первичная сушка - поднимают температуру с -40°C до -30°C в течение 8 часов и выдерживают при -30°C в течение 7 часов.
Вторичная сушка - поднимают температуру до +30°C в течение 4 часов и выдерживают при +30°C в течение 8-10 часов.
По завершении цикла лиофилизации емкости вновь заполняют азотом до атмосферного давления, закрывают и защищают крышку (например, алюминиевым обжимным колпачком). Для внутривенного введения лиофилизованный твердый состав может быть восстановлен с использованием фармацевтически приемлемого наполнителя, как, например, 0,9% физиологического раствора или 5% декстрозы. Раствор можно вводить в таком виде или перед введением больному впрыснуть его в мешок для хранения жидкостей (содержащий фармацевтически приемлемый наполнитель, как, например, 0,9% физиологический раствор или 5% декстрозу).
(x) Состав для подкожной инъекции
Композицию для подкожного введения получают смешиванием соединения формулы (I) с кукурузным маслом фармацевтической категории, достигая концентрации 5 мг/мл. Композицию подвергают стерилизации и заполняют ей подходящую емкость.
Пример 83
Определение противогрибковой активности
Противогрибковую активность соединений формулы (I) можно определить, применяя следующий протокол.
Испытывали активность соединений против набора грибков, включавшего Candida parpsilosis, Candida tropicalis, Candida albicans-ATCC 36082 и Cryptococcus neoformans. Исследуемые микроорганизмы содержали на скошенных питательных средах Sabourahd Dextrose Agar при 4°C. Синглетные суспензии каждого микроорганизма получали выращиванием грибков в течение ночи при 27°C на вращающемся барабане в питательном бульоне на азотно-дрожжевой основе (YNB) с аминокислотами (Difco, Detroit, Mich.), pH 7,0 с 0,05М морфолинпропансульфоновой кислотой (MOPS). После этого суспензии центрифугировали и дважды промывали 0,85% NaCl, после чего промытые суспензии клеток обрабатывали ультразвуком в течение 4 секунд (Branson Sonifier, model 350, Danbury, Conn.). Синглетные бластоспоры подсчитывали в гемоцитометре, и доводили до желаемой концентрации в 0,85% NaCl.
Активность тестируемого соединения определяли, используя модификацию методики микроразведения в бульоне. Тестируемые соединения разбавляли ДМСО до соотношения 1 мг/мл, затем разводили до 64 мкг/мл в бульоне YNB pH 7,0 с MOPS (в качестве контроля применяли флюконазол), получая рабочий раствор каждого соединения. При использовании 96-луночного планшета лунки 1 и 3-12 заполняли бульоном YNB, в лунках 2-11 делали десять разведений раствора тестируемого соединения (диапазон концентраций от 64 до 0,125 мкг/мл). Лунка 1 служила контролем стерильности и холостым опытом для спектрофотометрического анализа. Лунка 12 служила контролем роста. Микротитровальные планшеты засевали по 10 мкл в каждую из лунок 2-11 (итоговое засеянное количество составляло 104 микроорганизмов/мл). Засеянные планшеты инкубировали в течение 48 часов при 35°C. Значения IC50 определяли спектрофотометрически измерением поглощения при 420 нм (Automatic Microplate Reader, DuPont Instruments, Wilmington, Del.) после перемешивания содержимого планшетов в течение 2 минут с помощью вихревой мешалки (Vorte-Genie 2 Mixer, Scientific Industries, Inc., Bolemia, N.Y.). Конечную точку IC50 определяли как наименьшую концентрацию препарата, вызывающую примерно 50% (или более сильное) замедление роста по сравнению с контрольной лункой. С помощью турбидиметрического анализа ее определяли как наименьшую концентрацию препарата, при которой мутность в лунке составляла <50% контроля (IC50). Минимальные цитолитические концентрации (MCC) определяли путем пересева всех лунок из 96-луночного планшета на планшет с Sabourahd Dextrose Agar (SDA), инкубирования в течение 1-2 дней при 35°C и затем проверяя жизнеспособность.
Пример 84
Протокол биологической оценки регулирования in vivo грибковой инфекции целого растения
Соединения формулы (I) растворяли в ацетоне и затем проводили последовательные разбавления в ацетоне для получения желаемого диапазона концентраций. Конечные объемы составов для обработки получали добавлением девяти объемов 0,05% водного Tween-20TM или 0,01% Triton X-100TM в зависимости от патогена.
Затем полученные композиции использовали для исследования активности соединений по настоящему изобретению против болезни томатов (Phytophthora infectans), применяя следующий протокол. Томаты (сорта Rutgers) выращивали из семян на беспочвенной горшечной смеси на основе торфа, пока сеянцы не достигали высоты 10-20 см. Затем растения опрыскивали до стекания раствора с листьев тестируемыми соединениями в концентрации 100 частей на млн. Через 24 часа исследуемые растения заражали опрыскиванием водной суспензией спорангий Phytophthora infectans и держали в камере с орошением в течение ночи. Затем растения переносили в теплицу до развития заболевания на необработанных контрольных растениях.
Аналогичные протоколы применялись для тестирования активности соединений по настоящему изобретению против бурой ржавчины пшеницы (возбудитель Puccinia), настоящей мучнистой росы пшеницы (Ervisiphe vraminis), пшеницы (сорта Monon), пятнистости листьев пшеницы (Septoria tritici) и септориоза колосковой чешуи пшеницы (Leptosphaeria nodorum).
Эквиваленты
Описанные выше примеры приведены с целью иллюстрации изобретения, и не должны рассматриваться, как налагающие какое бы то ни было ограничение на объем изобретения. Должно быть легко понятно, что в конкретные варианты осуществления изобретения, описанные выше и проиллюстрированные в примерах, могут быть внесены многочисленные модификации и изменения без отступления от принципов, лежащих в основе изобретения. Предполагается, что все такие модификации и изменения охвачены настоящей заявкой.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПОЛИЦИКЛИЧЕСКИЕ ПИРАЗОЛЬНЫЕ ИНГИБИТОРЫ КИНАЗНОЙ АКТИВНОСТИ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2655921C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2332415C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2355688C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2006 |
|
RU2368602C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2006 |
|
RU2387653C2 |
| ПРОИЗВОДНОЕ АМИНОПИРАЗОЛА | 2010 |
|
RU2580543C9 |
| 5-ЗАМЕЩЕННЫЕ ИНДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2008 |
|
RU2487873C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ НАФТИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2010 |
|
RU2573390C2 |
| ПРОИЗВОДНЫЕ 4-ГИДРОКСИ-1,2,3,4-ТЕТРАГИДРОНАФТАЛИН-1-ИЛ-МОЧЕВИНЫ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ, СРЕДИ ПРОЧЕГО, ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНОГО ТРАКТА | 2011 |
|
RU2586333C1 |
| СОЕДИНЕНИЯ | 2008 |
|
RU2461559C2 |
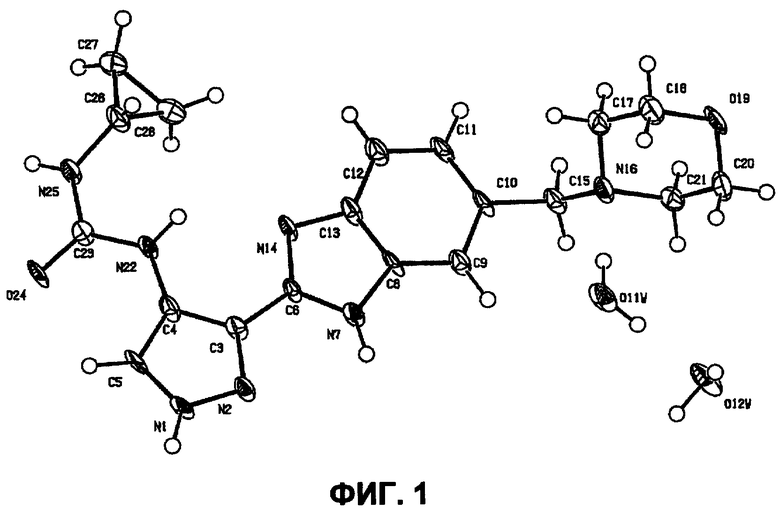
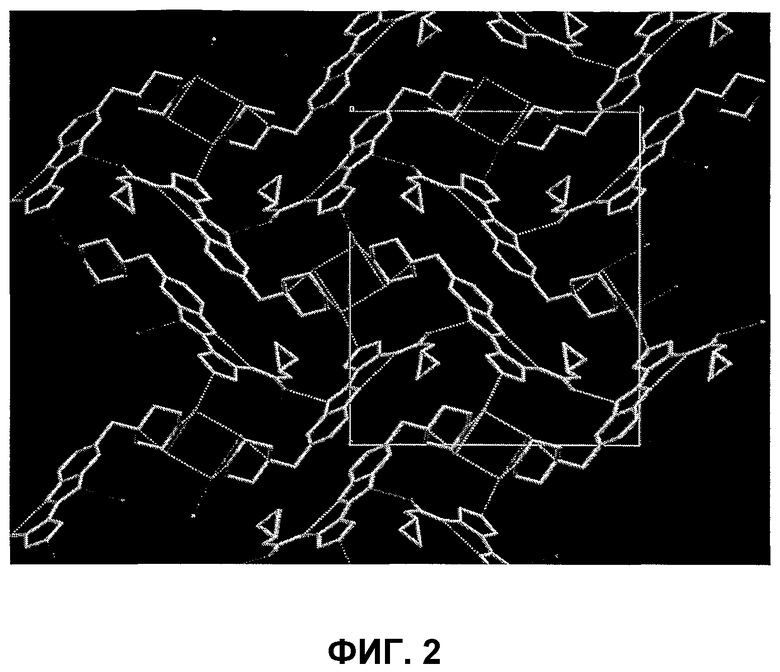
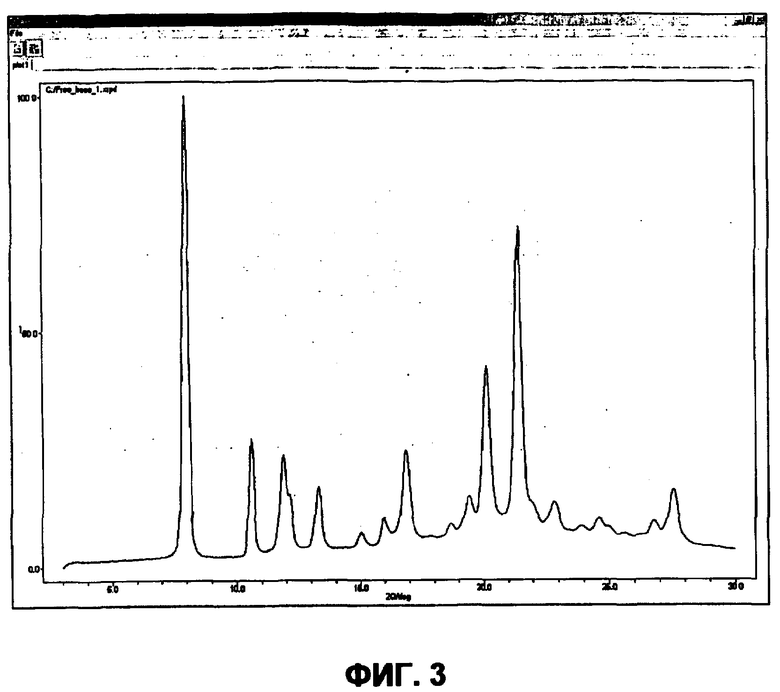
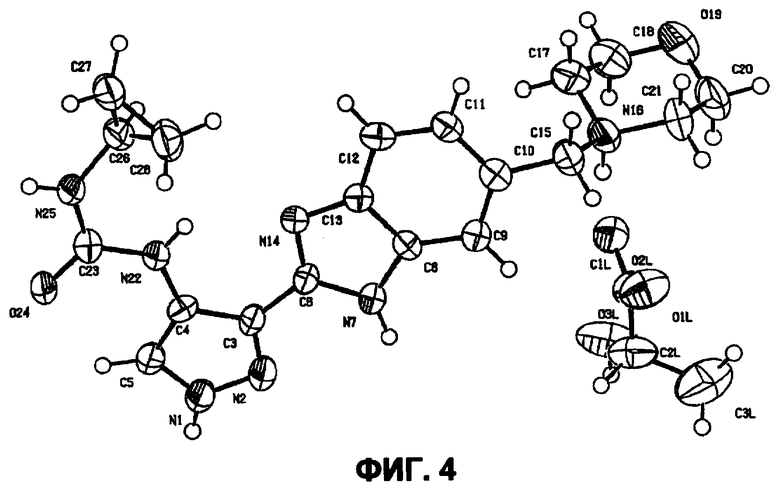
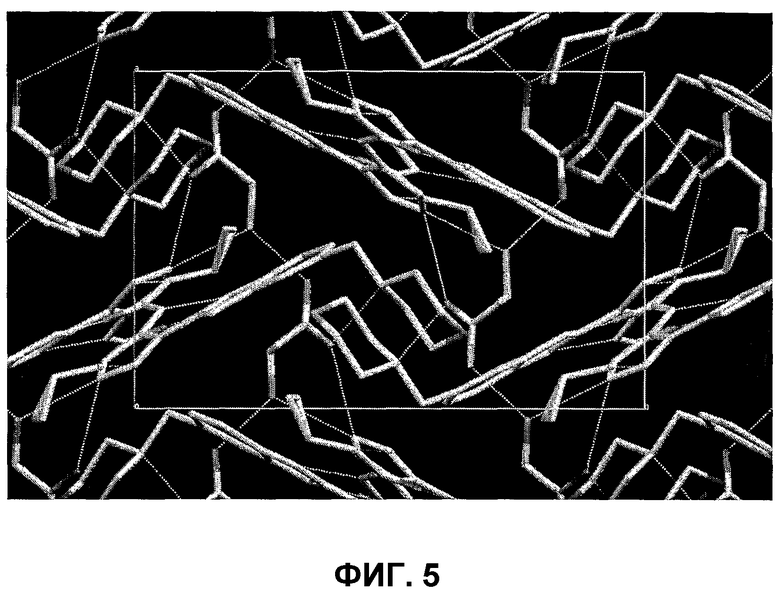
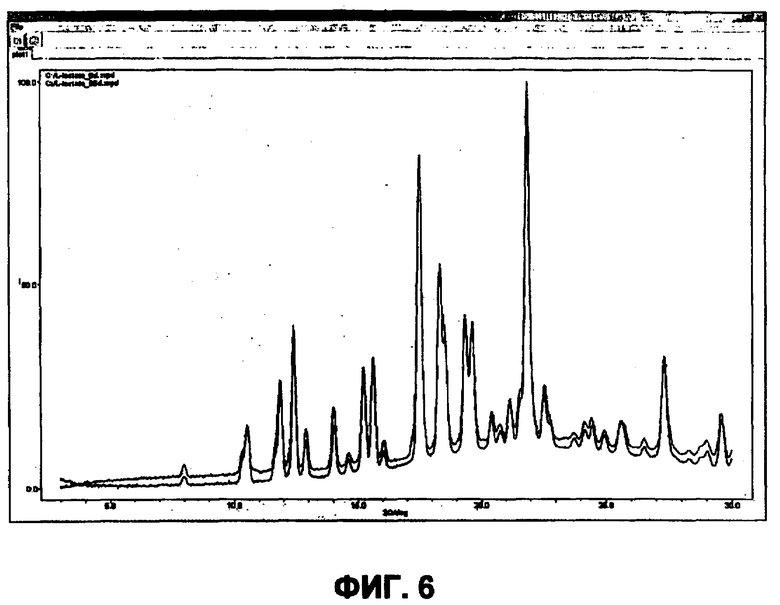
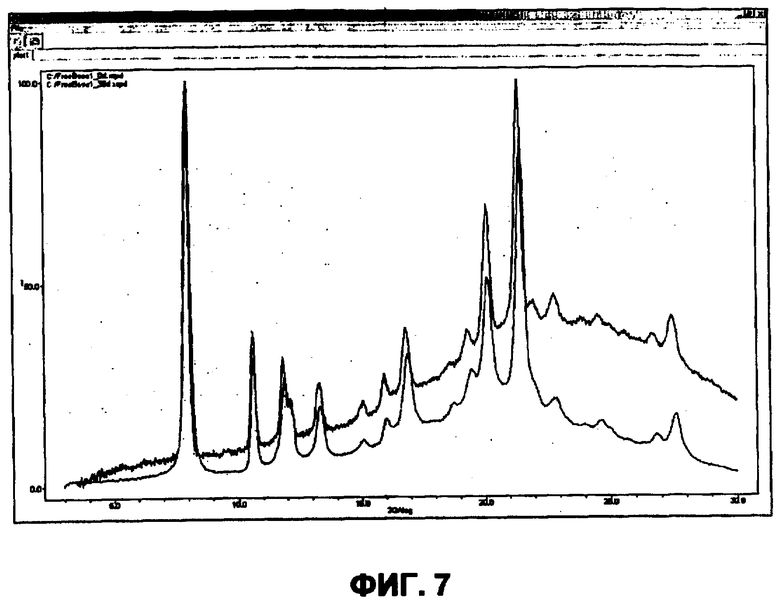
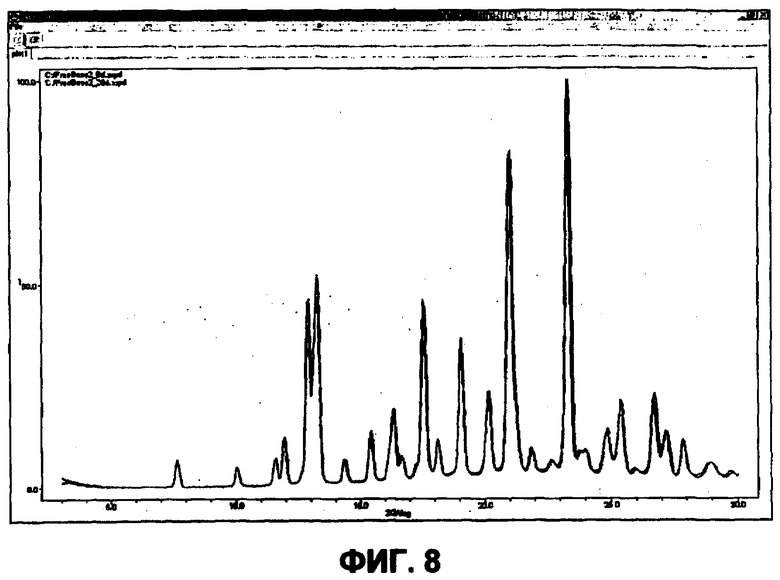
Изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям, сольватам или таутомерам, где заместитель М выбран из групп D1 и D2, имеющих структурные формулы, приведенные ниже, и R1, Е, А и X соответствуют определениям, приведенным в формуле изобретения. Кроме того, раскрыты фармацевтические композиции, содержащие эти соединения, способы получения этих соединений, промежуточные соединения и способы их получения, а также применение соединений формулы (I) в профилактике или лечении заболеваний, опосредованных CDK киназами, GSK-3 киназами или Aurora киназами. 15 н. и 25 з.п. ф-лы, 8 ил., 18 табл.
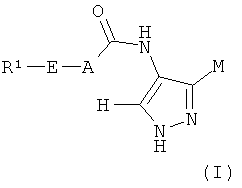
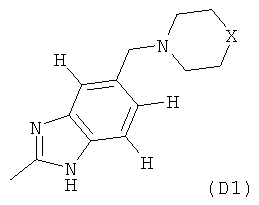
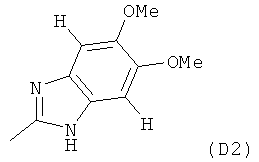
1. Соединение формулы (I):

или его фармацевтически приемлемая соль, сольват или таутомер,
где М выбран из группы D1 и группы D2:
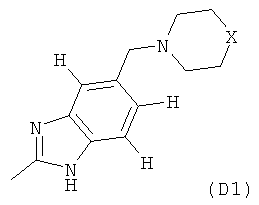
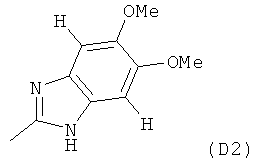
и где (А) если М представляет собой группу D1:
X выбран из О, NH и NCH3;
А выбран из связи и группы NR2, где R2 представляет собой водород или метил;
Е выбран из связи, СН2, CH(CN) и С(СН3)2;
R1 выбран из
(i) 3-5-членной циклоалкильной группы;
(ii) 4-6-членной насыщенной гетероциклической группы, включающей 1 или 2 гетероатома, выбранных из О, N, S и SO2, причем гетероциклическая группа необязательно замещена С1-4алкилом; но исключая незамещенный 4-морфолинил, незамещенный тетрагидропиран-4-ил, незамещенный 2-пирролидинил, а также незамещенный и 1-замещенный пиперидин-4-ил;
(iii) 2,5-замещенной фенильной группы формулы:
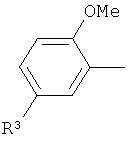
в которой (а) если X представляет собой NH или N-CH3, R3 представляет собой хлор; и (b) если X представляет собой О, R3 представляет собой CN;
(iv) группы CR6R7R8, в которой каждый из R6 и R7 выбран из водорода и метила, и R8 выбран из водорода, метила, С1-4алкилсульфонилметила, гидроксиметила и циано;
(v) пиридазин-4-ильной группы, необязательно замещенной одним или двумя заместителями, выбранными из метокси и этокси;
(vi) замещенной имидазотиазольной группы, в которой заместители выбраны из метила и этила; и
(vii) 1,3-дигидроизоиндол-2-ильной или 2,3-дигидроиндол-1-ильной групп;
(viii) 3-пиридила;
(ix) тиоморфолина или его S-оксида, или S,S-диоксида; и
если Е-А представляет собой NR2, R1 дополнительно выбран из
(x) 2-фторфенила, 3-фторфенила, 4-фторфенила, 2,4-дифторфенила, 3,4-дифторфенила, 2,5-дифторфенила, 3,5-дифторфенила, 2,4,6-трифторфенила, 2-метоксифенила, 5-хлор-2-метоксифенила, циклогексила, незамещенного 4-тетрагидропиранила и трет-бутила;
(xi) группы NR10R11, где каждый из R10 и R11 представляет собой С1-4алкил, или же R10 и R11 соединены таким образом, что NR10R11 образует 4-6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом в цикле, выбранный из О, N, S и SO2, причем гетероциклическая группа необязательно замещена С1-4алкилом;
(xii) пиридона, необязательно замещенного С1-4алкилом;
если Е-А представляет собой C(CH3)2NR2 или CH2-NR2, R1 дополнительно выбран из
(xiii) незамещенного 2-фурила и 2,6-дифторфенила; и
если Е-А представляет собой С(СН3)2NR2, R1 дополнительно выбран из
(xiv) незамещенного фенила; и
если Е представляет собой СН2, R1 дополнительно выбран из
(xv) незамещенного тетрагидропиран-4-ила; и
(В) если М представляет собой группу D2:
А представляет собой связь;
Е представляет собой связь;
R1 выбран из
(xvi) 2-замещенной 3-фурильной группы формулы:
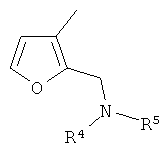
в которой R4 и R5 являются одинаковыми или различными и выбраны из водорода и С1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом или группу, выбранную из О, NH, NMe, S или SO2;
(xvii) 5-замещенной 2-фурильной группы формулы:
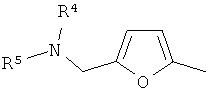
в которой R4 и R5 являются одинаковыми или различными и выбраны из водорода и С1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом или группу, выбранную из О, NH, NMe, S или SO2; при условии, что соединение не является [3-(5,6-диметокси-1Н-бензимидазол-2-ил)-1Н-пиразол-4-ил]амидом 5-пиперидин-1-илметилфуран-2-карбоновой кислоты;
(xviii) группы формулы:
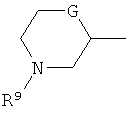
в которой R9 представляет собой водород, метил, этил или изопропил; и G представляет собой СН, О, S, SO, SO2 или NH; и
(xix) 3,5-дизамещенной фенильной группы формулы:
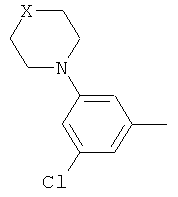
в которой X выбран из NH и NCH3; и
(С) если М представляет собой группу D1:
и X представляет собой О; А представляет собой группу NR2, где R2 является водородом; Е является связью; и R1 является 2,6-дифторфенилом; тогда соединение формулы (I) представляет собой фармацевтически приемлемую кислотно-аддитивную соль, выбранную из солей, образованных кислотами, которые выбраны из группы, состоящей из уксусной, адипиновой, аспарагиновой (например L-аспарагиновой), бензолсульфоновой, этансульфоновой, D-глюконовой, глюкуроновой (например D-глюкуроновой), глутаминовой (например L-глутаминовой), хлористоводородной, молочной (например (+)-L-молочной и (±)-DL-молочной), метансульфоновой и толуолсульфоновой (например п-толуолсульфоновой) кислот.
2. Соединение по п.1 или его фармацевтически приемлемая соль, сольват или таутомер, в котором М выбран из группы D1 и группы D2:
и в котором
(I) если М представляет собой группу D1:
X выбран из О, NH и NCH3;
А выбран из связи и группы NR2, где R2 представляет собой водород или метил;
Е выбран из связи, СН2, CH(CN) и С(СН3)2;
R1 выбран из
(i) 3-5-членной циклоалкильной группы;
(ii) 4-6-членной насыщенной гетероциклической группы, включающей 1 или 2 гетероатома, выбранных из О, N, S и SO2, причем гетероциклическая группа необязательно замещена С1-4алкилом; но исключая незамещенный 4-морфолинил, незамещенный тетрагидропиран-4-ил, незамещенный 2-пирролидинил, а также незамещенный и 1-замещенный пиперидин-4-ил;
(iii) 2,5-замещенной фенильной группы формулы:
в которой (а) если X представляет собой NH или N-CH3, R3 представляет собой хлор; и (b) если X представляет собой О, R3 представляет собой CN;
(iv) группы CR6R7R8, в которой каждый из R6 и R7 выбран из водорода и метила, и R8 выбран из водорода, метила, С1-4алкилсульфонилметила, гидроксиметила и циано;
(v) пиридазин-4-ильной группы, необязательно замещенной одним или двумя заместителями, выбранными из метокси и этокси;
(vi) замещенной имидазотиазольной группы, в которой заместители выбраны из метила и этила; и
(vii) 1,3-дигидроизоиндол-2-ильной или 2,3-дигидроиндол-1-ильной групп;
(ix) тиоморфолина или его S-оксида, или S,S-диоксида; и
если Е-А представляет собой NR2, R1 дополнительно выбран из
(x) 2-фторфенила, 3-фторфенила, 4-фторфенила, 2,4-дифторфенила, 3,4-дифторфенила, 2,5-дифторфенила, 3,5-дифторфенила, 2,4,6-трифторфенила, 2-метоксифенила, 5-хлор-2-метоксифенила, циклогексила, незамещенного 4-тетрагидропиранила и трет-бутила;
(xi) группы NR10R11, где каждый из R10 и R11 представляет собой С1-4алкил, или же R10 и R11 соединены таким образом, что NR10R11 образует насыщенную 4-6-членную гетероциклическую группу, необязательно содержащую второй циклический гетероатом, выбранный из О, N, S и SO2, причем гетероциклическая группа необязательно замещена С1-4алкилом;
(xii) пиридона, необязательно замещенного С1-4алкилом;
если Е-А представляет собой C(CH3)2NR2 или CH2-NR2, R1 дополнительно выбран из
(xiii) незамещенного 2-фурила и 2,6-дифторфенила; и
если Е-А представляет собой C(CH3)2NR2, R1 дополнительно выбран из
(xiv) незамещенного фенила; и
если Е представляет собой СН2, R1 дополнительно выбран из
(xv) незамещенного тетрагидропиран-4-ила; и
(II) если М представляет собой группу D2:
А представляет собой связь;
Е представляет собой связь;
R1 выбран из
(xvi) 2-замещенной 3-фурильной группы формулы:
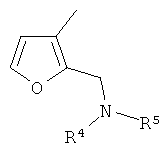
в которой R4 и R5 являются одинаковыми или различными, и выбраны из водорода и С1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом или группу, выбранные из О, NH, NMe, S или SO2;
(xvii) 5-замещенной 2-фурильной группы формулы:
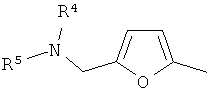
в которой R4 и R5 являются одинаковыми или различными, и выбраны из водорода и С1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом или группу, выбранные из О, NH, NMe, S или SO2;
(xviii) группы формулы:
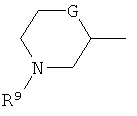
в которой R9 представляет собой водород, метил, этил или изопропил; и G представляет собой СН, О, S, SO, SO2 или NH; и
(xix) 3,5-дизамещенной фенильной группы формулы:
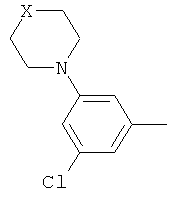
в которой X выбран из NH и NCH3.
3. Соединение по п.1 или его фармацевтически приемлемая соль, сольват или таутомер, где М выбран из группы D1 и группы D2:
и где (А) если М представляет собой группу D1:
X выбран из О, NH и NCH3;
А выбран из связи и группы NR2, где R2 представляет собой водород или метил;
Е выбран из связи, СН2, CH(CN) и С(СН3)2;
R1 выбран из
(i) 3-5-членной циклоалкильной группы;
(ii) 4-6-членной насыщенной гетероциклической группы, включающей 1 или 2 гетероатома, выбранных из О, N, S и SO2, причем гетероциклическая группа необязательно замещена С1-4алкилом; но исключая незамещенный 4-морфолинил, незамещенный тетрагидропиран-4-ил, незамещенный 2-пирролидинил, а также незамещенный и 1-замещенный пиперидин-4-ил;
(iii) 2,5-замещенной фенильной группы формулы:
в которой (а) если X представляет собой NH или N-CH3, R3 представляет собой хлор; и (b) если X представляет собой О, R3 представляет собой CN;
(iv) группы CR6R7R8, в которой каждый из R6 и R7 выбран из водорода и метила, и R8 выбран из водорода, метила, С1-4алкилсульфонилметила, гидроксиметила и циано;
(v) пиридазин-4-ильной группы, необязательно замещенной одним или двумя метокси заместителями;
(vi) замещенной имидазотиазольной группы, в которой заместитель представляет собой метил; и
(vii) 1,3-дигидроизоиндол-2-ильной или 2,3-дигидроиндол-1-ильной групп;
(viii) 3-пиридила;
(ix) тиоморфолина или его S-оксида, или S,S-диоксида; и
если Е-А представляет собой NR2, R1 дополнительно выбран из
(x) 2-фторфенила, 3-фторфенила, 4-фторфенила, 2,4-дифторфенила, 3,4-дифторфенила, 2,5-дифторфенила, 3,5-дифторфенила, 2,4,6-трифторфенила, 2-метоксифенила, 5-хлор-2-метоксифенила, циклогексила, незамещенного 4-тетрагидропиранила и трет-бутила;
(xi) группы NR10R11, где каждый из R10 и R11 представляет собой С1-4алкил, или же R10 и R11 соединены таким образом, что NR10R11 образует 4-6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом в цикле, выбранный из О, N, S и SO2, причем гетероциклическая группа необязательно замещена С1-4алкилом;
(xii) пиридона, необязательно замещенного С1-4алкилом;
если Е-А представляет собой C(CH3)2NR2 или CH2-NR2, R1 дополнительно выбран из
(xiii) незамещенного 2-фурила и 2,6-дифторфенила; и
если Е-А представляет собой С(СН3)2NR2, R1 дополнительно выбран из
(xiv) незамещенного фенила; и
если Е представляет собой СН2, R1 дополнительно выбран из
(xv) незамещенного тетрагидропиран-4-ила; и
(В) если М представляет собой группу D2:
А представляет собой связь;
Е представляет собой связь;
R1 выбран из
(xvi) 2-замещенной 3-фурильной группы формулы:
в которой R4 и R5 являются одинаковыми или различными и выбраны из водорода и С1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом или группу, выбранную из О, NH, NMe, S или SO2;
(xvii) 5-замещенной 2-фурильной группы формулы:
в которой R4 и R5 являются одинаковыми или различными и выбраны из водорода и С1-4алкила, или R4 и R5 связаны таким образом, что NR4R5 образует 5- или 6-членную насыщенную гетероциклическую группу, необязательно содержащую второй гетероатом или группу, выбранную из О, NH, NMe, S или SO2; при условии, что соединение не является [3-(5,6-диметокси-1Н-бензимидазол-2-ил)-1Н-пиразол-4-ил]амидом 5-пиперидин-1-илметилфуран-2-карбоновой кислоты;
(xviii) группы формулы:
в которой R9 представляет собой водород, метил, этил или изопропил; и G представляет собой СН, О, S, SO, SO2 или NH; и
(xix) 3,5-дизамещенной фенильной группы формулы:
в которой X представляет собой NCH3.
4. Соединение по п.1 формулы (II):
где R1, Е, А и X такие, как определено в п.1.
5. Соединение по п.3 формулы (III):
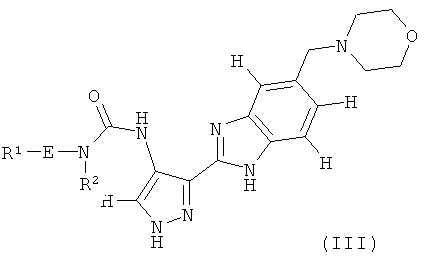
или его фармацевтически приемлемая соль, сольват или таутомер, где R1, R2 и Е такие, как определено в п.3.
6. Соединение по п.5 формулы (III):
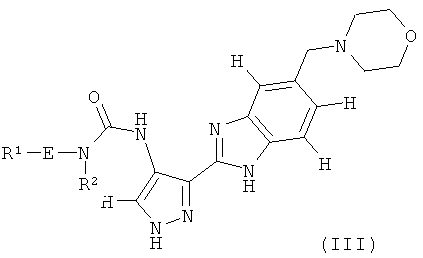
или его фармацевтически приемлемая соль, сольват или таутомер, в которой Е является связью, R2 представляет собой водород, и R1 представляет собой циклоалкильную группу.
7. Соединение по п.6, в котором R1 является циклопропильной группой, причем указанное соединение представляет собой 1-циклопропил-3-[3-(5-морфолин-4-илметил-1Н-бензимидазол-2-ил)-1Н-пиразол-4-ил]мочевину или ее фармацевтически приемлемую соль, сольват или таутомер.
8. Соединение по п.7, где соединение является фармацевтически приемлемой солью, выбранной из ацетата, мезилата, этансульфоната, DL-лактата, адипата, D-глюкуроната, D-глюконата и гидрохлорида.
9. Соединение по п.7 в форме свободного основания.
10. Соединение по п.7 или его фармацевтически приемлемая соль, которое является, по существу, кристаллическим.
11. Соединение по п.9, которое является кристаллическим дигидратом, кристаллы которого относятся к моноклинной пространственной группе P21/n (#14) со следующими параметрами кристаллической решетки при 101(2)K: размер ячейки а=7,66(10), размер ячейки b=15,18(10), размер ячейки с=17,71(10)Å, угол ячейки α=90°, угол ячейки β=98,53(2)°, угол ячейки γ=90°.
12. Соединение по п.7 в форме фармацевтически приемлемой соли, выбранной из лактата и цитрата, а также их смесей.
13. Соединение по п.12, которое является L-лактатом.
14. Соединение по п.12, которое является цитратом.
15. Соединение по п.12, которое является смесью L-лактатов и цитратов.
16. Соединение по п.13, которое представляет собой L-лактат 1-циклопропил-3-[3-(5-морфолин-4-илметил-1Н-бензимидазол-2-ил)-1Н-пиразол-4-ил]мочевины, который является кристаллическим и характеризуется одним или несколькими (в любой комбинации) или всеми параметрами из числа следующих, а именно, что данная соль
(a) имеет кристаллическую структуру, показанную на фиг.4 и 5; и/или
(b) имеет кристаллическую структуру, которая относится к орторомбической пространственной группе Р212121 (#19) со следующими параметрами кристаллической решетки при 97(2)K: размер ячейки а=9,94(10), размер ячейки b=15,03(10), размер ячейки с=16,18(10)Å, углы ячейки α=β=γ=90°; и/или
(c) имеет порошковую рентгенограмму, характеризующуюся наличием основных пиков, соответствующих дифракционным углам (2θ) 17,50, 18,30, 19,30, 19,60 и 21,85 градусов, и более конкретно дополнительно при 12,40, 15,20, 15,60, 17,50, 18,30, 18,50, 19,30, 19,60, 21,85 и 27,30 градусах, и/или межплоскостными расстояниями (d), равными 5,06, 4,85, 4,60, 4,53 и 4,07, и более конкретно дополнительно 7,13, 5,83, 5,68, 5,06, 4,85, 4,79, 4,60, 4,53, 4,07 и 3,26 ангстрем; и демонстрирует пики при тех же дифракционных углах, что и пики в порошковой рентгенограмме, показанной на фиг.6, и необязательно где пики имеют такую же относительную интенсивность, как пики на фиг.6;
причем соль является безводной и демонстрирует эндотермический пик при 190°С при исследовании способом DSC; и/или при исследовании с применением методики с таблеткой KBr, имеет инфракрасный спектр, который содержит характеристические пики при 3229, 2972 и 1660 см-1.
17. Необязательно забуференный водный раствор для внутривенного введения, содержащий L-лактат или цитрат, или смесь этих солей 1-циклопропил-3-[3-(5-морфолин-4-илметил-1Н-бензимидазол-2-ил)-1Н-пиразол-4-ил]мочевины в концентрации, превышающей 1 мг/мл, имеющий рН в интервале от 2 до 6.
18. Водный раствор для внутривенного введения, содержащий 1-циклопропил-3-[3-(5-морфолин-4-илметил-1Н-бензимидазол-2-ил)-1Н-пиразол-4-ил]мочевину в протонированной форме, совместно с одним или несколькими противоионами, выбранными из L-лактата и цитрата, а также их смесей; и необязательно (i) один или несколько других противоионов, таких как хлорид-ионы и/или (ii) один или несколько наполнителей для внутривенных препаратов.
19. Антипролиферативная фармацевтическая композиция, включающая 1-циклопропил-3-[3-(5-морфолин-4-илметил-1Н-бензимидазол-2-ил)-1Н-пиразол-4-ил]мочевину или ее фармацевтически приемлемую соль, сольват или таутомер и фармацевтически приемлемый носитель.
20. Фармацевтическая композиция по п.19 в высушенной форме, предназначенной для растворения в воде.
21. Фармацевтическая композиция по п.19 в лиофилизованной форме, включающая 1-циклопропил-3-[3-(5-морфолин-4-илметил-1Н-бензимидазол-2-ил)-1Н-пиразол-4-ил]мочевину в протонированной форме, совместно с одним или несколькими противоионами, выбранными из L-лактата и цитрата, а также их смесей; и необязательно (i) один или несколько других противоионов, таких как хлорид-ионы и/или (ii) один или несколько наполнителей для внутривенных препаратов.
22. Соединение по любому из пп.1-16 или его фармацевтически приемлемая соль, сольват или таутомер для использования при лечении у млекопитающего заболевания или состояния, причем (i) указанное заболевание или состояние включает аномальный рост клеток или (ii) указанное заболевание или состояние возникает из-за аномального роста клеток.
23. Применение соединения по любому из пп.1-16 или его фармацевтически приемлемой соли, сольвата или таутомера для изготовления лекарственного средства для лечения заболевания или состояния, включающего аномальный рост клеток или возникающего из-за аномального роста клеток, у млекопитающего.
24. Применение по п.23, где заболевание или состояние представляет собой рак.
25. Применение соединения по п.24, где раковое заболевание характеризуется повышающей регуляцией Aurora киназы.
26. Применение по п.23, где заболевание или состояние является раковым заболеванием, выбранным из рака мочевого пузыря, груди, ободочной кишки, почек, эпидермиса, печени, легких, пищевода, желчного пузыря, яичника, поджелудочной железы, желудка, шейки матки, щитовидной железы, простаты или кожи; гематопоэтической опухолью лимфоидного происхождения, выбранной из лейкемии, острого лимфоцитарного лейкоза, В-клеточной лимфомы, Т-клеточной лимфомы, лимфомы Ходжкина, неходжкинской лимфомы, лимфомы ворсистых клеток или лимфомы Беркетта; гематопоэтической опухолью миелоидного происхождения, выбранной из острой и хронической миелогенной лейкемии, миелодиспластического синдрома или промиелоцитарной лейкемии; фоликуллярным раком щитовидной железы; опухолью мезенхимального происхождения, выбранной из фибросаркомы или габдомиосаркомы; опухолью центральной или периферической нервной системы, выбранной из астроцитомы, нейробластомы, глиомы или шванномы; меланомой; семиномой; тератокарциномой; остеосаркомой; пигментной ксеродермой; кератоксантомой; фоликуллярным раком щитовидной железы; или саркомой Капоши.
27. Применение по п.26, где заболевание или состояние представляет собой лейкемию.
28. Применение по п.27, где лейкемия выбрана из повторной или рефракторной острой миелогенной лейкемии, миелодиспластического синдрома, острой лимфоцитарной и хронической миелогенной лейкемии.
29. Применение соединения по любому из пп.1-16 для изготовления лекарственного средства для профилактики или лечения ракового заболевания у пациента, который, по результатам диагностики, входит в подгруппу людей, обладающих вариантом I1e31 гена Aurora А.
30. Применение соединения по любому из пп.1-16 для изготовления лекарственного средства для профилактики или лечения рака, характеризующегося повышающей регуляцией Aurora киназы.
31. Способ получения соединения формулы (I), определенного в п.2, где А является связью, включающий взаимодействие соединения формулы (X)
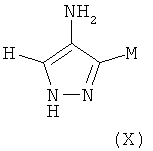
с карбоновой кислотой R1-E-CO2H или ее реакционноспособным производным в условиях образования амидов.
32. Способ получения соединения формулы (I), определенного в любом из пп.2-16, где А представляет собой NH, включающий взаимодействие соединения формулы (X)
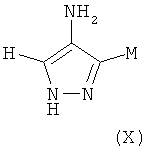
с изоцианатом формулы R1-E-N=C=O в условиях образования производных мочевины.
33. Способ получения соединения формулы (I), определенного в любом из пп.2-16, где А представляет собой NR2, включающий взаимодействие соединения формулы (X)
с амином формулы R1-E-NR2H в присутствии реагента, образующего карбонилсодержащую мочевину, такого как 1,1′-карбонилдиимидазол (CDI), фосген или трифосген.
34. Способ получения соединения формулы (XXVII) или (XXVIII) или его соли:
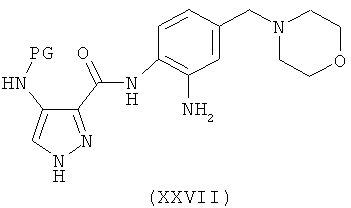 или
или 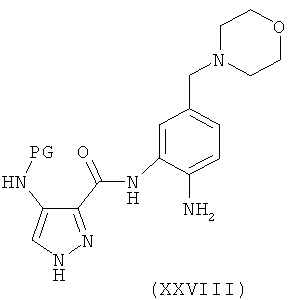
включающий взаимодействие соединения формулы (XXIX)
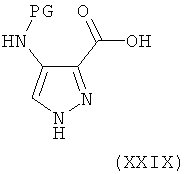
где PG является аминозащитной группой, с соединением формулы (XXXI):
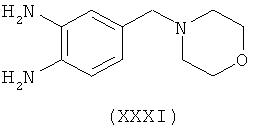
в органическом растворителе в присутствии конденсирующего реагента, такого как 1-этил-3-(3′-диметиламинопропил)карбодиимид (EDC) и 1-гидрокси-бензотриазол (HOBt).
35. Способ по п.34, в котором соединение формулы (XXIX) представляет собой соединение формулы (XXXII) ниже:
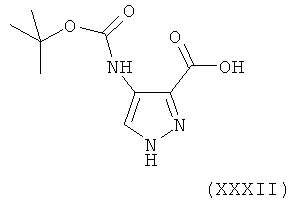
36. Способ получения 3-(5-морфолин-4-илметил-1Н-бензимидазол-2-ил)-1Н-пиразол-4-иламина или его соли, включающий
(i) обработку соединения формулы (XXVIIa) или (XXVIIIa):
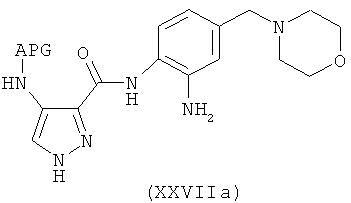 или
или 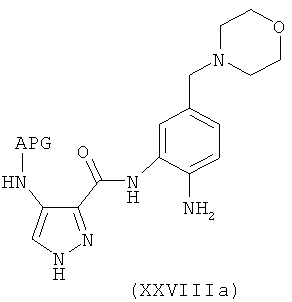
где APG представляет собой аминозащитную группу, которая может быть удалена в условиях кислой среды,
кислотой в растворителе, необязательно при нагревании; и
(ii) нейтрализацию реакционной смеси.
37. Способ получения 1-циклопропил-3-[3-(5-морфолин-4-илметил-1Н-бензимидазол-2-ил)-1Н-пиразол-4-ил]мочевины или ее соли, сольвата или таутомера, включающий:
(i) обработку соединения формулы (XXVIIa) по п.36 кислотой в растворителе, необязательно при нагревании;
(ii) нейтрализацию реакционной смеси;
(iii) взаимодействие продукта стадии (ii) с карбонилирующим реагентом; и
(iv) взаимодействие продукта стадии (iii) с циклопропиламином.
38. Способ по п.37, где карбонилирующий реагент представляет собой 1,1′-карбонилдиимидазол (CDI), трифосген или фосген.
39. Способ получения 1-циклопропил-3-[3-(5-морфолин-4-илметил-1Н-бензимидазол-2-ил)-1Н-пиразол-4-ил]мочевины или ее соли, сольвата или таутомера, включающий взаимодействие соединения формулы (XXXIII) или (XXXIIIa):
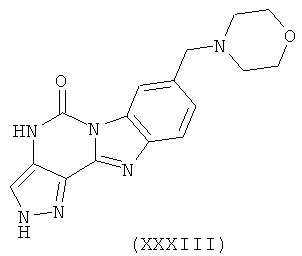
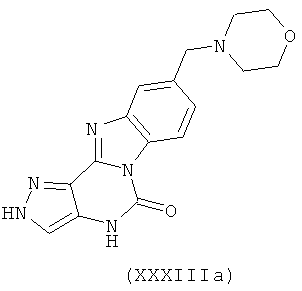
с циклопропиламином и затем необязательно образование кислотно-аддитивной соли.
40. Промежуточное соединение формулы (XXXII), (XXVIIa), (XXVIIIa), (XXXIII) или (XXXIIIa):
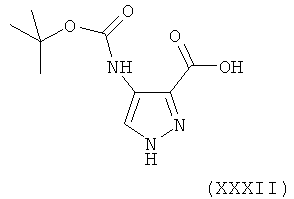
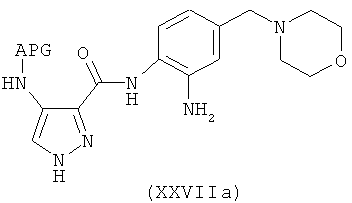 или
или 
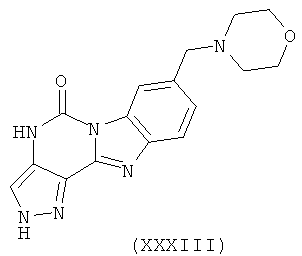
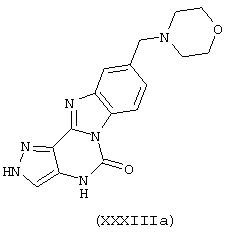
где APG представляет собой трет-бутоксикарбонильную группу.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| RU 2003122210 A, 27.12.2004 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

