Настоящее изобретение относится к новым способам фторирования, в частности [18F]фторирования некоторых ароматических соединений, и к новым предшественникам, используемым в этом способе. Данное изобретение особенно пригодно для класса бензотиазольных производных, которые известны как in vivo визуализирующие агенты.
Фторирование ароматических соединений может быть осуществлено путем электрофильного взаимодействия с молекулярным фтором, однако электрофильное фторирование ароматических соединений фтором является в общем случае плохим и неселективным способом. Были разработаны разные электрофильные фторирующие реагенты, получаемые из молекулярного фтора, такие как CH3COOF "AcOF", однако они имеет некоторые недостатки. В отношении [18F]радиофторирования, жесткие условия и малая пригодность электрофильных способов [18F]фторирования, а также низкая удельная активность полученных продуктов означают, что такой подход не является полезным для промышленного получения [18F]-меченых продуктов. Чаще используются способы нуклеофильного фторирования с использованием фторида. [18F]Фторид является более широкодоступным реагентом, чем электрофильные реагенты, и полученные продукты имеют более высокую удельную активность, что является преимуществом в области in vivo визуализации. Нуклеофильное фторирование ароматических колец, особенно тех, которые богаты электронами, может быть проблематичным. Например, трудно осуществимо нуклеофильное фторирование анилинов, где аминогруппа добавляется к электронной плотности ароматического кольца. В настоящем изобретении предложен предшественник, пригодный для нуклеофильного фторирования с получением фторированного анилина, который объединяет ряд полезных эффектов, включая улучшенную активацию ароматического кольца для фторирования, некоторые стерические эффекты на реакцию фторирования, и легкое превращение во фторированный анилиновый продукт.
В соответствии с изобретением предложен способ получения соединения формулы (I):
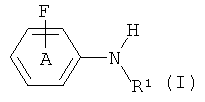
где фенильное кольцо А возможно замещено 1-4 заместителями;
R1 выбран из C1-6алкила, С2-6алкенила и С2-6алкинила,
который включает:
(1) взаимодействие соответствующего соединения формулы (II):
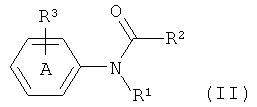
где кольцо А возможно замещено, как определено для соединения формулы (I);
R2 выбран из водорода, C1-10алкила, C1-10 галогеналкила, С6-14арила, С6-14арилалкила, -(CH2CH2O)q-CH3, где q представляет собой целое число от 1 до 10;
R1 является таким, как он определен для соединения формулы (I); и
R3 представляет собой уходящую группу;
с фторидом, с получением соединения формулы (III)
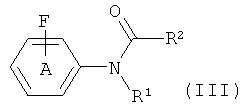
где R1 и R2 такие, как они определены для соединения формулы (I), и фенильное кольцо А замещено, как определено для соединения формулы (I);
с последующей стадией (2) и возможно стадией (3) в любом порядке:
(2) превращение группы -C(O)R2 в водород, подходяще посредством гидролиза;
(3) удаление любой дополнительной защитной группы.
Фенильное кольцо А возможно замещено 1-4 органическими заместителями, например выбранными из фторо, хлоро, бромо, йодо, циано, нитро, -R, -OR, -OC(O)R, -C(O)R, -SR, -NR2, -C(O)NR2, где R в каждом случае выбран из С1-6алкила, С2-6алкенила, С2-6алкинила, С1-6алкокси-С1-6алкила, C1-6галогеналкила, С2-6галогеналкенила, С2-6галогеналкинила, С1-6галогеналкокси-С1-6алкила, С5-12арила, С5-12гетероарила, где указанные арильные и гетероарильные заместители могут быть дополнительно замещены неарильными и негетероарильными заместителями, перечисленными для фенильного кольца А, и защищенного производного любого из них.
R2 в соединении формулы (II) выбран из водорода, C1-10алкила (более подходяще С1-6алкила, еще более подходяще метила), С1-10 алогеналкила (более подходяще C1-6галогеналкила, такого как С1-6фторалкил, например трифторметил), С6-14арила (подходяще фенила), С6-14арилалкила (подходяще фенил-С1-4алкила, например бензила) и -(CH2CH2O)q-СН3 где q представляет собой целое число от 1 до 10. Соединения формулы (II), где R2 представляет собой С4-10алкил или -(CH2CH2O)q-CH3, где q представляет собой целое число от 1 до 10, можно использовать в способе, где желательно увеличить растворимость соединения формулы (II), и, как таковые, эти соединения и способ по изобретению с их использованием образуют отдельные аспекты изобретения. Предпочтительно, R2 в соединении формулы (II) выбран из водорода и C1-6алкила, более предпочтительно R2 представляет собой водород.
R3 в соединении формулы (II) представляет собой уходящую группу, которая может быть заменена фторидом, и подходящим образом выбрана из
нитро,
-N2 +,
хлоро,
бромо,
йодо,
-NR4(C1-6алкил)2 +, где R4 представляет собой С1-6алкил или группу формулы (X):

-OSO2R5, где R5 выбран из С1-6алкила, C1-6 галогеналкила, такого как C1-6перфторалкил, арила, такого как фенил или толил (например, пара-толил), и группы формулы (X), как определено выше; и
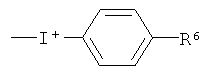
где R6 выбран из водорода, C1-6алкила, галогено, нитро и группы формулы (X), как определено выше.
R3 в соединении формулы (II) подходяще выбран из:
нитро,
-N2 +,
хлоро,
бромо,
йодо,
-NR4(C1-6алкил)2 +, где R4 представляет собой C1-6алкил;
-OSO2R5, где R5 выбран из C1-6алкила, C1-6галогеналкила, такого как C1-6перфторалкил, арила, такого как фенил или толил (например, пара-толил); и
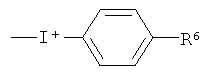
где R6 выбран из водорода, С1-6алкила, галогено и нитро.
В одном конкретном аспекте изобретения R3 представляет собой нитро.
В способе по изобретению применение соединений формулы (II), в которых R3 содержит группу формулы (X), позволяет осуществить фторирование в твердой фазе, что может упростить очистку фторированного продукта, так как любой непрореагировавший предшественник остается связанным с твердой подложкой и может быть удален из жидкой фазы продукта фильтрацией.
В группе формулы (X) твердой подложкой может являться любая подходящая твердофазная подложка, которая нерастворима в любых растворителях, используемых в данном способе, но к которой может быть ковалентно присоединен линкер и/или соединение формулы (II). Примеры подходящей твердой подложки включают полимеры, такие как полистирол (который может быть блок-привитым, например полиэтиленгликолем), полиакриламид или полипропилен, или стекло или силикон, покрытые таким полимером. Твердая подложка может находиться в форме небольших дискретных частиц, таких как шарики или иглы, или в виде покрытия на внутренней поверхности картриджа, или на изготовленном посредством микротехнологий сосуде.
В группе формулы (X) линкер может представлять собой любую подходящую органическую группу, которая служит для того, чтобы в достаточное мере отделить в пространстве реакционноспособный сайт от структуры твердой подложки так, чтобы максимизировать реакционную способность. Подходяще линкер содержит от нуля до четырех арильных групп (подходяще фенил) и/или С1-6алкил или C1-6галогеналкил (подходяще С1-6фторалкил), и возможно от одной до четырех дополнительных функциональных групп, таких как амидные или сульфонамидные группы. Примеры таких линкеров хорошо известны специалистам в области твердофазной химия, но включают:
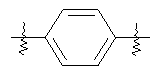
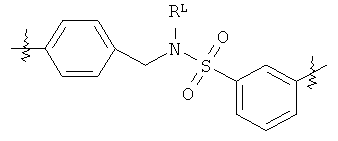
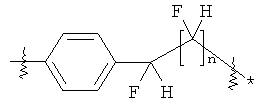
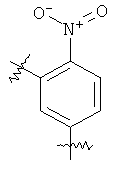
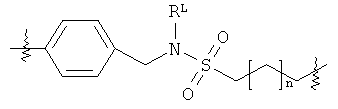
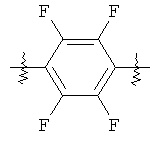
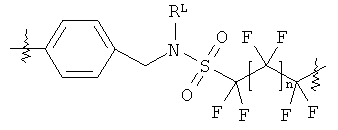
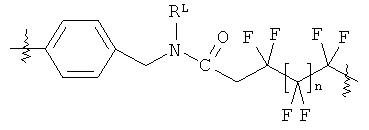
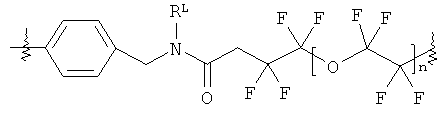
где в каждом случае n представляет собой целое число от 0 до 3, и RL представляет собой водород или C1-6алкил.
Стадия (1) способа по изобретению, то есть взаимодействие соединения формулы (II) с фторидом, подходяще [18F]фторидом, может быть осуществлена с использованием источника фторида, такого как NaF, KF, CsF, тетраалкиламмония фторид, или тетраалкилфосфония фторид, подходяще источника [18F]фторида, такого как Na18F, K18F, Cs18F, тетраалкиламмония [18F]фторид (например, тетрабутиламмония [18F]фторид) или тетраалкилфосфония 18F-фторид. Для увеличения реакционной способности фторида может быть добавлен катализатор фазового перехода, такой как аминополиэфир или краун-эфир, например 4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8,8,8]гексакозан (Kryptofix 2.2.2), и взаимодействие может осуществляться в подходящем растворителе. В таких условиях получаются реакционноспособные фторидные ионы. Для увеличения выхода при фторировании возможно может быть использована ловушка свободных радикалов, как описано в WO 2005/061415. Термин "ловушка свободных радикалов" определяется как любой агент, который взаимодействует со свободными радикалами и инактивирует их. Подходящая ловушка свободных радикалов для данной цели может быть выбрана из 2,2,6,6-тетраметилпиперидин-N-оксида (TEMPO), 1,2-дифенилэтилена (DPE), аскорбата, пара-аминобензойной кислоты (РАВА), α-токоферола, гидрохинона, ди-трет-бутилфенола, β-каротина и гентизиновой кислоты. Предпочтительными ловушками свободных радикалов для применения в способе по изобретению являются TEMPO и DPE, причем TEMPO является наиболее предпочтительным.
Обработка фторидом, подходяще [18F]фторидом на стадии (1), может быть осуществлена в присутствии подходящего органического растворителя, такого как ацетонитрил, диметилформамид, диметилсульфоксид, диметилацетамид, тетрагидрофуран, диоксан, 1,2-диметоксиэтан, сульфолан, N-метилпирролидинон, или в ионной жидкости, такой как производное имидазолия (например, 1-этил-3-метилимидазолия гексафторфосфат), производное пиридиния (например, 1-бутил-4-метилпиридиния тетрафторборат), фосфониевое соединение или тетраалкиламмониевое соединение, при неэкстремальной температуре, например от 15°С до 180°С, предпочтительно при повышенной температуре, например от 80°С до 150°С, например примерно 120°С. Органический растворитель подходящим образом является безводным, но в некоторых случаях может содержать низкие уровни воды.
В одном аспекте изобретения группа фторо в соединении формулы (I) представляет собой [18F]фторо, и фторид, используемый на стадии (1) данного способа, представляет собой [18F]фторид. Существует особая необходимость в новых способах радиофторирования, особенно радиофторирования богатых электронами ароматических систем.
Стадию (2) способа, то есть превращение группы -C(O)R2 в водород подходяще осуществляют посредством кислотного или щелочного гидролиза, используя органическую или неорганическую кислоту, при неэкстремальной температуре, например от температуры окружающей среды до температуры дефлегмации. Это взаимодействие может быть осуществлено в присутствие водного растворителя или органического растворителя, например С1-4спирта, такого как метанол или этанол, или ацетонитрила, или смеси водных и органических растворителей.
Подходящие кислоты, используемые на стадии (2), включают бромистоводородную, трифторуксусную, фосфорную и соляную.
Подходящие основания, используемые на стадии (2), включают гидроксид натрия или гидроксид калия. Применение гидроксида натрия в органическом растворителе, таком как ацетонитрил, при повышенной температуре, например примерно 100°С, может привести к хорошим радиохимическим выходам и облегчить очистку фторированного продукта.
Альтернативные основания, используемые на стадии (2), включают ненуклеофильные основания, такие как гидрид натрия. Данный способ приводит к хорошим радиохимическим выходам, а также облегчает очистку фторированного продукта. Обработку натрия гидридом можно осуществить в подходящем апротонном растворителе, таком как ацетонитрил или пропионитрил, и при повышенной температуре, например от 40°С до 120°С, обычно примерно 100°С. Натрия боргидрид, лития боргидрид и лития алюминийгидрид также являются подходящими основаниями для применения на стадии (2).
Как очевидно специалисту в данной области техники, иногда необходимо применять стратегии использования защитных групп для предупреждения нежелательных побочных взаимодействий в процессе органического синтеза. Примеры таких стратегий можно найти в Protecting Groups in Organic Synthesis, Theodora W. Greene and Peter G.M. Wuts, опубликованного John Wiley & Sons Inc., описывающем способы введения и удаления защитных групп. Чтобы избежать лишних стадий синтеза, особенно полезно удалять любые защитные группы, оставшиеся в соединении формулы (III), в условиях стадии (2) для того, чтобы избежать отдельной стадии удаление защиты.
При использовании здесь термин "алкил", используемый сам по себе или как часть другой группы, означает насыщенный углеводородный радикал с прямой или разветвленной цепью, такой как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, н-пентил или н-гексил.
При использовании здесь термин "алкенил", используемый сам по себе или как часть другой группы, означает ненасыщенный углеводородный радикал с прямой или разветвленный цепью, содержащий по меньшей мере одну углерод-углеродную двойную связь, такой как этенил, пропенил, изопропенил, бутенил, изобутенил, трет-бутенил, н-пентенил и н-гексенил.
При использовании здесь термин "алкинил", используемый сам по себе или как часть другой группы, означает ненасыщенный углеводородный радикал с прямой или разветвленной цепью, содержащий по меньшей мере одну углерод-углеродную тройную связь, такой как этинил, пропинил, изопропинил, бутинил, изобутинил, трет-бутинил, н-пентинил и н-гексинил.
При использовании здесь термин "галогено", используемый сам по себе или как часть другой группы, означает фторо, хлоро, бромо или йодо.
При использовании здесь термин "арил", используемый сам по себе или как часть другой группы, означает ароматическую углеводородную систему с одним кольцом или конденсированными кольцами, такую как фенил или нафтил.
При использовании здесь термин "гетероарил", используемый сам по себе или как часть другой группы, означает ароматическую углеводородную систему с одним кольцом или конденсированными кольцами, которая дополнительно включает 1 или более чем один гетероатом, выбранный из серы, азота и кислорода, такую как пиридил, тиофенил, бензотиазолил, бензоксазолил или фурил.
Способ по изобретению особенно пригоден для синтеза соединений формулы (I), в которых R1 представляет собой C1-6алкил, и в частности метил, таким образом способы, где R1 в соединениях формулы (I), (II), (III) представляет собой C1-6алкил, и в частности метил, образуют отдельный аспект данного изобретения.
Способ по изобретению особенно пригоден, когда группа фторо в соединении формулы (I) находится в орто- или пара-положении к группе -N(R1)C(O)R2, поскольку -N(R1)C(O)R2, расположенная в орто- или пара-положении к R3 в соответствующем соединении формулы (II), может активировать R3 для нуклеофильного замещения фторидом. Предпочтительно группа фторо в соединении формулы (I) находится в орто-положении к группе -N(R1)C(O)R2, и группа R3 в соответствующем соединении формулы (II) находится в орто-положении к группе -N(R1)C(O)R2.
Известно, что некоторые соединения формулы (I) применяются в диагностических и терапевтических способах, например бензотиазольные производные, описанные для in vivo визуализации амилоида в соответствии со способами, описанными в WO 02/16333 и WO 2004/083195. Способы, ранее описанные для получения этих бензотиазольных производных, хотя и подходят для получения небольших количеств таких соединений, имеют такие недостатки, как небольшие радиохимические выходы и плохая воспроизводимость, так что существует необходимость в улучшенных способах их получения, в частности для промышленного получения. Как упомянуто выше, нуклеофильное фторирование ароматического кольца может быть проблематичным, когда кольцо богато электронами. В соединениях формулы (Ia) и (Ib) ниже схема замещения затрудняет фторирование ароматического кольца. Попытки создания подходящего предшественника для фторирования, который был бы стабильным, мог быть фторирован с хорошим выходом и затем легко мог быть превращен в конечный продукт, были проблематичными, как продемонстрировано ниже в Примере 3. Таким образом, в дополнительном аспекте изобретения предложен способ получения соединения формулы (Ia):
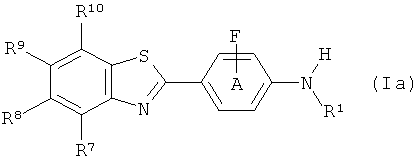
где R1 выбран из C1-6алкила, С2-6алкенила и С2-6алкинила;
R7, R8, R9 и R10 каждый независимо выбран из водорода, фторо, хлоро, бромо, йодо, C1-6алкила, С2-6алкенила, С2-6алкинила, (СН2)mOR11 (где m=1, 2 или 3), CF3, CH2-CH2Y, O-CH2-CH2Y, CH2-CH2-CH2Y, O-CH2-CH2-CH2Y (где Y выбран из фторо, хлоро, бромо и йодо), CN, (C=O)-R11, N(R11) 2, NO2, (C=O)N(R11)2, O(CO)R11, OR11, SR11, COOR11, Rph, CR11=CR11-Rph, CR11 2-CR11 2-Rph (где Rph представляет собой незамещенную или замещенную фенильную группу с заместителями фенила, выбранными из любого из нефенильных заместителей, определенных для R7-R10, и где R11 представляет собой Н или C1-6алкил) и защищенного производного любого из них; и
фенильное кольцо А замещено 1-3 заместителями, выбранными из любого из нефенильных заместителей, определенных для R7-R10,
который включает:
(1) взаимодействие соответствующего соединения формулы (IIa):
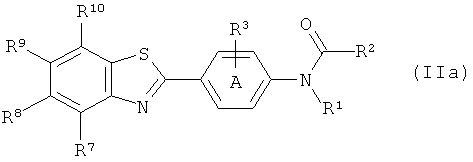
где R1 определен для соединения формулы (Ia),
фенильное кольцо А замещено, как определено для соединения формулы (Ia); и
R2 выбран из водорода, С1-10алкила, C1-10галогеналкила, С6-14арила, С6-14арилалкила, -(CH2CH2O)q-CH3, где q представляет собой целое число от 1 до 10;
R3 представляет собой уходящую группу, как определено для соединения формулы (II);
R7, R8, R9 и R10 такие, как они определены для соединения формулы (Ia);
с фторидом с получением соединения формулы (IIIa)
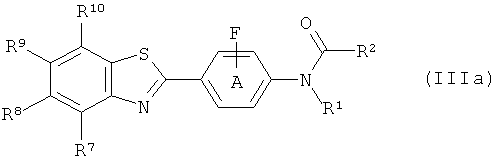
где R1 и R2 такие, как они определены для соединения формулы (IIa), фенильное кольцо А замещено, как определено для соединения формулы (Ia);
R7, R8, R9 и R10 такие, как они определены для соединения формулы (Ia); с последующей стадией (2) и возможно стадией (3) в любом порядке;
(2) превращение группы -C(O)R2 в водород, подходяще посредством гидролиза;
(3) удаление любых дополнительных защитных групп.
В соединениях формулы (Ia), (IIa), и (IIIa) и соответствующем способе по изобретению R7, R8, R9 и R10 подходяще выбраны из водорода, гидрокси, -NO2, -CN, -COOR11, -OCH2OR11 (где R11 выбран из водорода и С1-6алкила), С1-6алкила, С2-6алкенила, С2-6алкинила, C1-6алкокси, галогено и защищенного производного любого из них. Подходящие защищенные производные заместителей R7, R8, R9 и R10 очевидны специалисту в данной области техники, и описаны в Theodora W. Greene and Peter G.M. Wuts, упомянутых здесь выше. Например, когда R7, R8, R9 или R10 представляет собой гидрокси, функциональная группа гидрокси подходяще защищена как C1-6алкоксиметоксигруппа, например этоксиметокси или метоксиметокси.
Одним классом предпочтительных соединений формулы (Ia) для применения в in vivo визуализации амидоида являются соединения формулы (Ib)
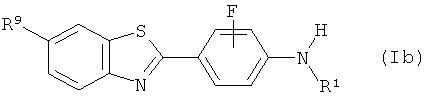
где R1 выбран из C1-6алкила, С2-6алкенила и С2-6алкинила; и
R9 выбран из гидрокси, -NO2, -CN, -COOR11, -OCH2OR11 (где R11 выбран из водорода и C1-6алкила), С1-6алкила, С2-6алкенила, С2-6алкинила, С1-6алкокси, галогено и защищенного производного любого из них и предпочтительно выбран из гидрокси, С1-6алкокси и защищенного производного любого из них и более предпочтительно выбран из гидрокси, метокси и защищенного производного любого из них. Следовательно, в соответствии с одним предпочтительным аспектом изобретения предложен способ получения соединения формулы (Ib):
где R1 выбран из C1-6алкила, C2-6алкенила и С2-6алкинила;
R9 выбран из гидрокси, -NO2, -CN, -COOR, -OCH2OR, C1-6алкила, С2-6алкенила, С2-6алкинила, C1-6алкокси, галогено и защищенного производного любого из них и предпочтительно выбран из гидрокси, C1-6алкокси и защищенного производного любого из них и более предпочтительно выбран из гидрокси, метокси и защищенного производного любого из них,
который включает:
(1) взаимодействие соответствующего соединения формулы (IIb):
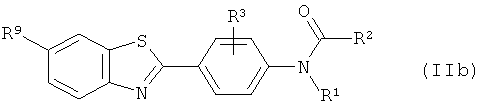
где R1 выбран из C1-6алкила, С2-6алкенила и С2-6алкинила, и R2 выбран из водорода, С1-10алкила, C1-10галогеналкила, С6-14арила, С6-14арилалкила, -(CH2CH2O)q-CH3, где q представляет собой целое число от 1 до 10;
R3 представляет собой уходящую группу, как определено для соединения формулы (II);
R9 такой, как он определен для соединения формулы (Ib);
с фторидом с получением соединения формулы (IIIb)

где R1 и R2 такие, как они определены для соединения формулы (IIb);
R9 такой, как он определен для соединения формулы (Ib); с последующей стадией (2) и возможно стадией (3) в любом порядке;
(2) превращение группы -C(O)R2 в водород, подходяще посредством гидролиза
(3) удаление любых дополнительных защитных групп в заместителе R9.
Соединения формул (IIa) и (IIb), как они определены выше, являются важными предшественниками, полезными для получения in vivo визуализирующих агентов, и, следовательно, образуют дополнительные аспекты изобретения.
Предпочтительные предшественники формулы (IIa) и (IIb) включают те, в которых R2 представляет собой водород или C1-6алкил, подходяще метил, более подходяще R2 представляет собой водород; R1 представляет собой C1-6алкил, подходяще метил; из них особенно полезными могут быть предшественники, в которых R3 представляет собой нитро. Соединения формулы (IIa) и (IIb), в которых R9 представляет собой гидрокси или C1-6алкокси или их защищенное производное, также могут быть особенно полезны. Одним таким предпочтительным предшественником является 2-[3-нитро-4-(метилформиламино)фенил]-6-этоксиметокси-бензотиазол.
Предшественник формулы (II), (IIa) или (IIb) может быть удобно представлен как часть набора, например, для применения в радиофармации. Набор может содержать картридж, который может быть помещен в подходящим образом адаптированный автоматический синтезатор. Картридж может содержать, помимо предшественника, колонку для удаления нежелательного фторид-иона и подходящий сосуд, присоединенный таким образом, чтобы позволить выпариваться реакционной смеси и позволить продукту иметь тот состав, который требуется. Также могут быть включены реагенты и растворители и другие расходные материалы, требующиеся для синтеза, вместе с компакт-диском с программным обеспечением, которое позволяет работать синтезатору таким образом, чтобы удовлетворять требованиям покупателя в отношении радиоактивной концентрация, объемов, времени доставки и так далее. Удобно, чтобы все компоненты набора являлись одноразовыми, чтобы минимизировать возможности загрязнения между прогонами, и они могут быть стерильными и с гарантированным качеством.
В изобретении дополнительно предложен радиофармацевтический набор для получения 18F-меченой радиометки для применения в PET (позитрон-эмиссионной томографии), включающий:
(1) сосуд, содержащий соединение формулы (II), (IIa) или (IIb); и
(2) средства для элюирования из сосуда с источником 18F-;
(3) ионообменный картридж для удаления избытка 18F-; и возможно
(4) картридж для удаления защиты с полученного продукта формулы (I), (Ia) или (Ib).
В изобретении дополнительно предложен картридж для радиофармацевтического набора для получения 18F-меченой радиометки для применения в PET, который включает:
(1) сосуд, содержащий соединение формулы (II), (IIa) или (IIb); и
(2) средство для элюирования из сосуда с источником 18F-.
Соединения формулы (II), (IIa) и (IIb) могут быть получены из имеющихся в продаже исходных веществ или с использованием исходных веществ, описанных в WO 02/16333 и WO 2004/083195, стандартными способами органической химии, например способами, описанными ниже и в примерах.
Соединения формулы (II), (IIa) и (IIb), где R3 представляет собой нитро, могут быть получены способами, аналогичными описанным в Примере 1.
Соединения формулы (II), (IIa) и (IIb), где R3 представляет собой хлоро, бромо, йодо, тозилат или йодониевую соль, могут быть получены способами, аналогичными показанным на Схемах 1-4 соответственно.
На Схемах 1-6 R2 такой, как он определен для соединения формулы (I) выше, R на Схеме 1 представляет собой алкильный или арильный заместитель, Ас представляет собой ацил, Ts представляет собой тозил, NaHDMS представляет собой натрия гексаметилдисилазид, TFA представляет собой трифторуксусную кислоту, Pd2dba3 на Схеме 6 представляет собой трис-(дибензилиденацетон)дипалладий(О), а другие сокращения являются такими, как они определены в Примерах.
Схема 1
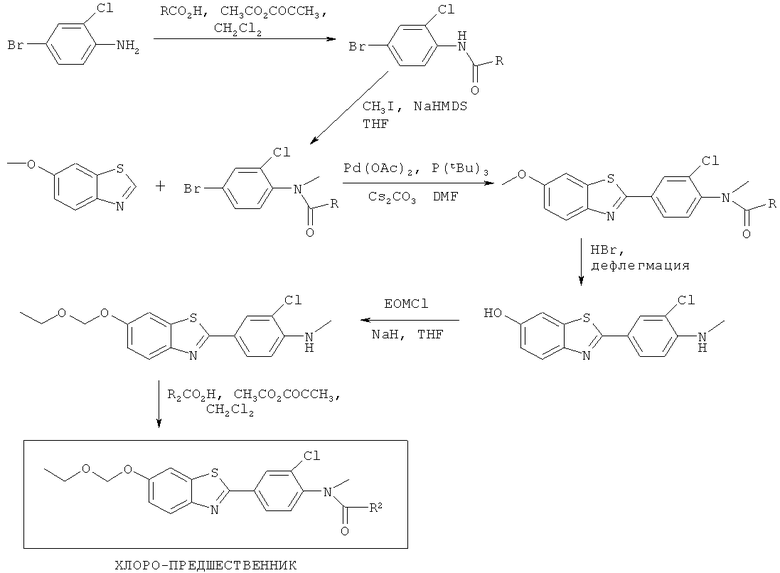
Схема 2
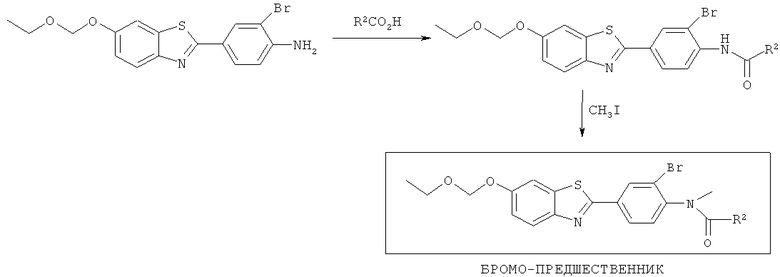
Схема 3
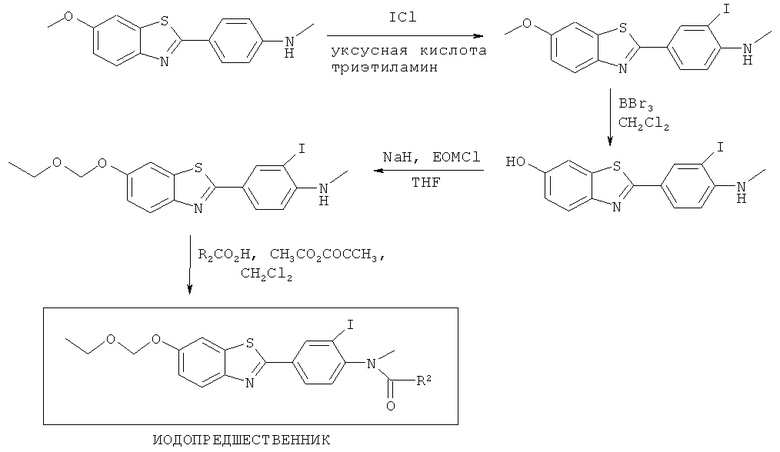
Схема 4
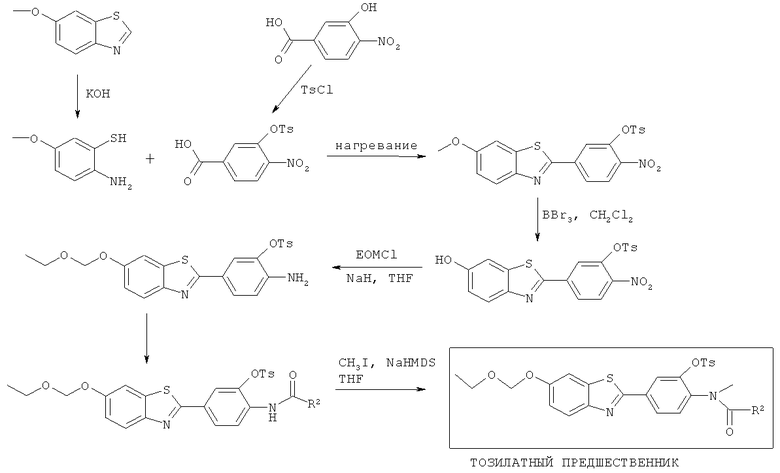
Схема 5
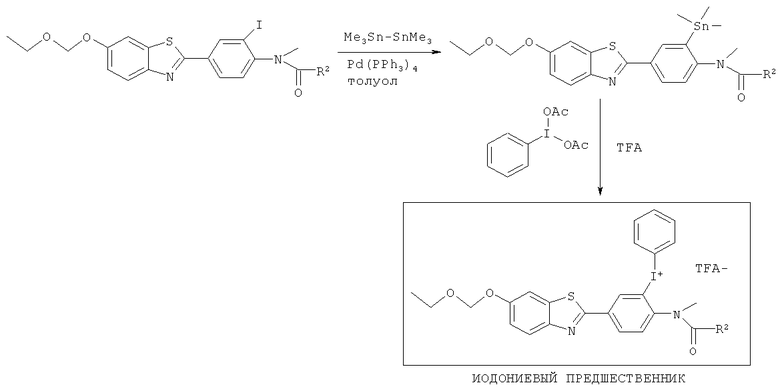
Соединения формулы (II), (IIa) и (IIb), где R3 представляет собой -N2 +, могут быть получены из соответствующего соединения, где R3 представляет собой нитро, путем восстановления нитрогруппы до амино, например используя водород и Pd/C в качестве катализатора, и затем диазотизацию с использованием NaNO2.
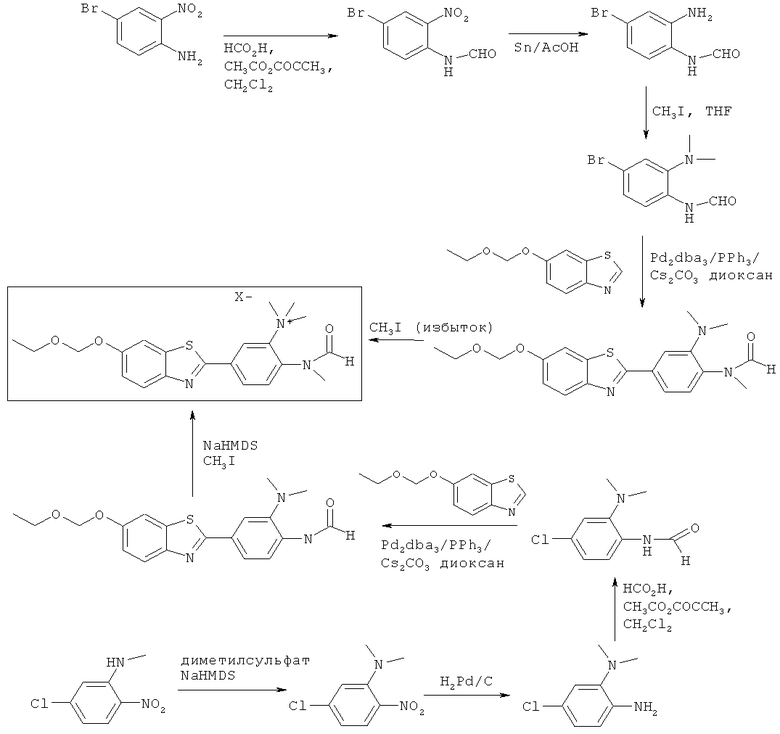
Соединения формулы (II), (IIa) и (IIb), где R3 представляет собой -NR4(C1-6алкил)2 +, могут быть получены в соответствии со Схемой 6.
Схема 6
Изобретение далее проиллюстрировано посредством следующих Примеров, в которых использованы следующие сокращения:
DMF: N,N-диметилформамид
DCM: дихлорметан
EOMCl: этоксиметоксихлорид
DMAP: диметиламинопиридин
RT: комнатная температура
THF: тетрагидрофуран
IMS: денатураты, используемые в промышленности
Т.пл.: точка плавления
eq.: эквиваленты
EtOAc: этилацетат
QMA: четвертичный аммоний
HPLC: высокоэффективная жидкостная хроматография
мл или мл: миллилитр(ы)
ТСХ: тонкослойная хроматография
об./об.: объем/объем
NMR: ядерный магнитный резонанс
MS: масс-спектрометрия
Пример 1: Синтез 2-[3-нитро-4-(метилформиламино)фенил]-6-этоксиметоксибензотиазола
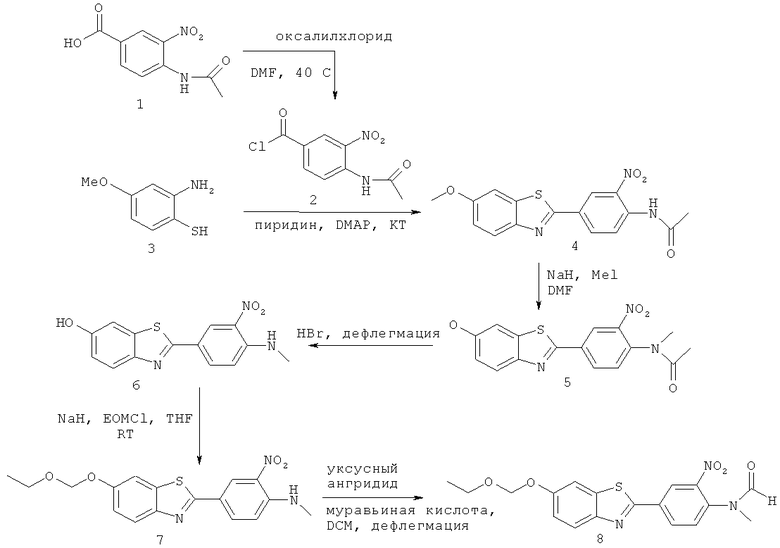
Пример 1(1): 4-Ацетамидо-3-нитробензоилхлорид (2)
4-Ацетамидо-3-нитробензойную кислоту 1 (Alfa Aesar, 5,6 г, 25 ммоль), оксалилхлорид (4,76 г, 38 ммоль), хлороформ (50 мл), DMF (несколько капель) перемешивали при 40°С в течение 3 часов. Растворитель удаляли в вакууме с получением желтого твердого вещества, которое использовали на следующей стадия без дополнительной очистки.
Пример 1(1а): 5-метокси-2-аминобензолтиол (3)
2-Амино-6-метоксибензотиазол 10 г (55,6 ммоль) суспендировали в 25%-ном водном гидроксиде калия и смесь нагревали с обратным холодильником в течение 24 ч. Бледно-желтый раствор охлаждали и подкисляли до рН 6 сначала водной 6 н. HCl, затем уксусной кислотой. Осажденное твердое вещество отфильтровывали, промывали водой (3×100 мл), сушили (глубокий вакуум) с получением требуемого вещества в виде бледно-желтого порошка, 8,18 г, 95%.
Пример 1(2): 2-(4-Ацетамидо-3-нитрофенил)-6-метоксибензотиазол (4)
5-Метокси-2-аминобензолтиол 3 (3,88 г, 25 ммоль), пиридин (100 мл) и DMAP (несколько кристаллов) перемешивали при комнатной температуре.
4-Ацетамидо-3-нитробензоилхлорид (25 ммоль, как получено выше) добавляли одной порцией при температуре ниже 30°С. Смесь перемешивали в течение еще 1 часа. Смесь нагревали до 80°С и перемешивали в течение выходных. Смесь охлаждали. Кристаллы отфильтровывали и промывали IMS с получением 2,2 г (26%-ный выход) 2-(4-ацетамидо-3-нитрофенил)-6-метоксибензотиазола.
Пример 1(3): 2-(4-N-Метилацетамидо-3-нитрофенил)-6-метоксибензотиазол (5)
Натрия гидрид (6,33 г, 157 ммоль) и DMF (400 мл) перемешивали при комнатной температуре. Одной порцией добавляли 2-(4-ацетамидо-3-нитрофенил)-6-метоксибензотиазол 4 (45 г, 131 ммоль). Смесь перемешивали в течение 1 часа. Смесь охлаждали на ледяной бане и одной порцией добавляли метилиодид (23,1 г, 164 ммоль), температура оставалась ниже 20°С.
Смесь перемешивали в течение 3 часов, добавляли воду (900 мл), смесь фильтровали и промывали водой. Твердое вещество перекристаллизовывали из IMS с получением 43,7 г (93% выход) 2-(4-N-метилацетамидо-3-нитрофенил)-6-метоксибензотиазола. Т.пл. 168-172°С.
Пример 1(4): 2-(4-Метиламино-3-нитрофенил)-6-гидроксибензотиазол (6)
Смесь 2-(4-N-метилацетамидо-3-нитрофенил)-6-метоксибензотиазола (58 г, 162 ммоль), бромистоводородной кислоты (500 мл, 48%-ный водный раствор) и бромистоводородной кислоты (500 мл, 45% в уксусной кислоте) перемешивали при 135°С в течение 5 часов. Смесь охлаждали до комнатной температуры и твердое вещество отфильтровали и промывали небольшим количеством воды. Твердое вещество суспендировали с водой и рН доводили примерно до 10 концентрированным раствором аммиака. Твердое вещество отфильтровывали и промывали водой.
Твердые вещества растирали с IMS (200 мл), фильтровали, смесь кипятили с IMS (500 мл), затем охлаждали до комнатной температуры, потом фильтровали. Твердое вещество опять кипятили с IMS (500 мл), затем охлаждали до комнатной температуры, затем фильтровали. Твердое вещество растворяли в горячем DMF (200 мл), фильтровали и добавляли воду (100 мл). Твердое вещество отфильтровывали и промывали IMS. Твердое вещество кипятили с водой (300 мл) в течение 5 минут, охлаждали, фильтровали, промывали водой, затем IMS, с получением 45,9 г (94%-ный выход) 2-(4-метиламино-3-нитрофенил)-6-гидроксибензотиазола. Т.пл. 269-272°С.
Пример 1(5): 2-[3-нитро-4-(метиламино)фенил-6-этоксиметокси-бензотиазол (7)
3-горлую 250 мл круглодонную колбу сушили в термостате при 80°С в течение ночи. Суспензию 6 (16,6 ммоль, 5 г) в сухом THF (180 мл) вливали по каплям в суспензию 60%-ной дисперсии NaH в минеральном масле (33,2 ммоль, 1,26 г, 2 эквивалента) в сухом THF (20 мл). Сразу после окончания добавления добавляли чистый этоксиметилхлорид (16,6 ммоль, 1,54 мл, 1 экв.) и реакционную смесь перемешивали в течение ночи. Темно-коричневую смесь фильтровали под вакуумом и фильтрат концентрировали в глубоком вакууме.
Неочищенный продукт наносили на диоксид кремния и очищали посредством флэш-хроматографии в DCM/EtOAc:3% EtOAc.
Нужную фракцию выделяли, концентрировали в глубоком вакууме с получением 60% не совсем красного твердого вещества с 95%-ной чистотой.
Пример 1(4): 2-[3-нитро-4-(метилформиламино)фенил]-6-этоксиметокси-бензотиазол (8)
Всю стеклянную посуду сушили в термостате при 80°С в течение ночи.
В 1 л трехгорлую круглодонную колбу, снабженную холодильником и термометром, по каплям добавляли уксусный ангидрид (15 мл, 160 ммоль, 22 экв.) к раствору муравьиной кислоты (160 ммоль, 6 мл, 22 экв.) при 0°С. Смесь перемешивали в течение 15 минут при 60°С.
Раствор 7 (7,2 ммоль, 2,6 г) в сухом DCM (310 мл) по каплям добавляют при 0°С к перемешиваемому ангидриду. Перемешивание продолжали при данной температуре в течение одного часа и чистый оранжевый раствор перемешивали 5 суток при 40°С. За реакцией следили при помощи HPLC: через 5 суток наблюдали 60%-ное превращение в нужный продукт.
Условия HPLC:
Колонка Phenomenex Luna 150Х 4,6 мн
Поток 1 мл/мин
Растворитель: ацетонитрил (В) и вода (А)
Детекция: 254-214
Градиент 5-95% В в течение 8 минут
Время удерживания: 9,5 минут
Чистый оранжевый раствор промывали 1 н. водным NaOH (3×100 мл), водой (3×100 мл), сушили над сульфатом магния и концентрировали в глубоком вакууме.
Ярко-оранжевый неочищенный продукт наносили на диоксид кремния и очищали посредством флэш-хроматографии в DCM/EtOAc: 3-10% EtOAc.
Нужную фракцию выделяли, концентрировали в глубоком вакууме с получением 54,2% не совсем желтого твердого вещества с 98%-ной чистотой.
Пример 1(7): Получение 2-[3-[18F]фтор-4-(метиламино)фенил]-6-гидрокси-бензотиазол (11) - см. Схему ниже (Подход 1)
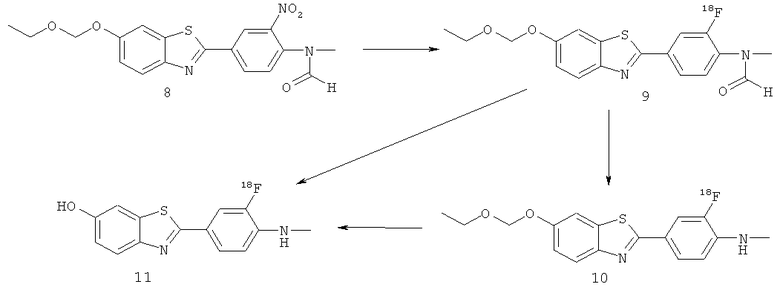
[18F]фторид (в 200 мкл обогащенной 95% 18О воды), 2,5 мг Kryptofix 2.2.2 (в 0,5 мл ацетонитриле) и 50 мкл 0,1 М K2CO3 добавляли в стеклоуглеродный реакционный сосуд. Затем раствор упаривали досуха, используя поток азота и нагревая реакционный сосуд до 100°С в течение 15 минут. 2×1 мл ацетонитрила добавляли в реакционный сосуд через 5 минут и 10 минут соответственно, чтобы способствовать азеотропной сушке. Реакционный сосуд охлаждали до комнатной температуры и добавляли 2-[3-нитро-4-(метилформиламино)фенил]-6-этоксиметоксибензотиазол (8) (5,0 мг) в 1 мл безводного диметилсульфоксида. Реакционную смесь герметично закрывали и нагревали в течение 10 минут при 130°С. Неочищенную смесь анализировали посредством HPLC и TLC.
0,25 мл 6 М HCl и 0,5 мл DMSO добавляли к неочищенному реакционному раствору 2-[3-[18F]фтор-4-(метилформиламино)фенил]-6-этоксиметоксибензотиазола (9) и нагревали при 125°С в течение 10 минут. Реакционную смесь затем охлаждали до комнатной температуры и нейтрализовали, используя 2 М ацетат натрия, что приводило к синтезу 2-[3-[18F]фтор-4-(метиламино)фенил]-6-гидроксибензотиазола (11). Неочищенную смесь анализировали посредством HPLC и TLC.
HPLC-очистка и изготовление препарата
2-[3-[18F]фтор-4-(метиламино)фенил]-6-гидроксибензотиазол (11) очищали при помощи HPLC, используя препаративную Phenomenex Prodigy ODS preparative 10 мкм 250 мм×10 мм (номер 00G-4088-N0) препаративную колонку (колонку элюируют смесью 40/60 ацетонитрил/триэтиламин-фосфатный буферный раствор рН 7 (об./об.)). Регулируемый способ представляет собой 0-15 минут 5 мл/минута, 15,5-39,9 минут 8 мл/минута, 40 минут 5 мл/минута. Продукт элюируется с временем удерживания 22-23 минут (в объеме 8 мл).
Очищенное при помощи HPLC "отсечение" разбавляли до 50 мл, добавляя дистиллированную воду. Продукт затем "удерживали" на C8-sep-pak картридже и затем элюировали картридж 1 мл этанола. Этанол затем удаляли под вакуумом и конечный продукт готовили в виде препарата в смеси 10% этанола/90% фосфатного буферного физиологического раствора.
Пример 2: Получение 2-[3-[18F]фтор-4-(метиламино)фенил]-6-гидрокси-бензотиазол (11) - см. Схему выше (Подход 2)
Пример 2(1): Получение [K/K2.2.2]+ 18F- (используя обогащенную 95% 18О воду).
После облучения целевое содержимое пропускали через колонку, заполненную смолой QMA (четвертичная аминометиловая смола). Колонку продували гелием в течение 5 минут, [18F]фторид, адсорбированный на смоле, элюировали в реакционный сосуд при помощи 4 мл смеси 96:4 (по объему) ацетонитрил-вода, содержащей 19,1 мг Kryptofix 2.2.2 и 2,9 мг K2CO3; раствор затем упаривали и выпаривали досуха совместно с безводным ацетонитрилом (2×1 мл) в потоке азота при 110°С.
Пример 2(2): Получение 2-[3-[18F]фтор-4-(метилформиламино)фенил]-6-этоксиметоксибензотиазол (9) и 2-[3-[18F]фтор-4-(метиламино)фенил]-6-этоксиметоксибензотиазол (10).
Раствор 2-[3-нитро-4-(метилформиламино)фенил]-6-этоксиметокси-бензотиазола (8) (3,0 мг) в безводном ацетонитриле (0,1 мл) добавляли к раствору [K/K2.2.2]+18F- в безводном ацетонитриле (0,25 мл). Реакционную смесь нагревали при 150°С в течение 15 минут. Неочищенную смесь анализировали при помощи аналитической HPLC.
Пример 2(3): Превращение 2-[3-[18F]фтор-4-(метилформиламино)фенил]-6-этоксиметоксибензотиазола (9) в 2-[3-[18F]фтор-4-(метиламино)фенил]-6-этоксиметоксибензотиазол (10).
Примерно 0,2 мл предыдущей реакционной смеси добавляли к раствору NaH (3,2 мг) в безводном ацетонитриле (0,2 мл) при комнатной температуре. Полученную смесь нагревали при 100°С в течение 5 минут. Неочищенную смесь анализировали при помощи аналитической HPLC.
Получение 2-[3-[18F]фтор-4-(метиламино)фенил]-6-гидроксибензотиазола (11).
Раствор концентрированной HCl в МеОН (1:2) (0,25 мл) добавляли к предыдущей реакционной смеси и нагревали при 100°С в течение 5 минут. Неочищенную смесь анализировали при помощи аналитической HPLC.
Пример 3: Сравнительное [18F]фторирование разных предшественников.
Радиофторирование различных бензотиазольных соединений-предшественников с использованием способов, аналогичных описанным в Примере 1(7), приводило к результатам, показанным в Таблице 1. Примерный выход рассчитывали исходя из радиохимической чистоты, измеренной при помощи HPLC, корректировали с учетом потери продукта при удерживании на HPLC и в реакционном сосуде.

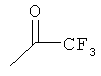
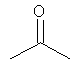

25-30% включение
Пример 4: Автоматизированный синтез 2-[3-[18F]фтор-4-(метиламино)фенил]-6-гидроксибензотиазола (11)
В положения для реагентов на TRACERlab FXFN (GE Healthcare Ltd) автоматическом синтезаторе загружали следующие растворы:
1. 0,1 М калия карбонат в воде (0,5 мл)
2. 0,13 М Kryptofix 2.2.2 в ацетонитриле (0,5 мл)
3. Раствор предшественника: 0,1 М 2-[3-нитро-4-(метилформиламино)фенил]-6-этоксиметоксибензотиазол (8) в DMSO (1,0 мл)
4. 4 М соляная кислота (0,25 мл)
5. Этанол (1,0 мл)
6. 0,01 М фосфатный буфер, рН 7,4 (13,1 мл)
Когда раствор [18F]фторида в [18O]-обогащенной воде (121 МБк) загружали в исходное положение синтезатора, оператор запускал программу, которая привела к следующей последовательности событий.
Раствор фторида пропускали через QMA картридж (предварительно уравновешенный 10 мл 0,5 М водного карбоната калия и 20 мл воды), улавливая фторид и отправляя обогащенную воду в отходы. QMA картридж затем элюировали 0,1 М раствором калия карбоната с получением фторида и элюат направляли в реакционный сосуд. Раствор Kryptofix 2.2.2 добавляли в реактор и смесь нагревали при 60°С в течение 5 минут под слабым потоком азота при пониженном давлении. Температуру затем повышали до 120°С и выдерживали под вакуумом в течение 7 минут, чтобы высушить содержимое реактора. После охлаждения до 50°С раствор предшественника добавляли в реактор и температуру повышали до 135°С в течение 10 минут. Данная стадия позволяет включить [18F]фторид в органическую молекулу. Раствор охлаждали до 50°С и добавляли 4 М соляную кислоту. Смесь нагревали до 125°С в течение 5 минут, чтобы вызвать удаление защиты с промежуточного соединения, и после охлаждения до 40°С раствор неочищенного продукта инъецировали на колонку Phenomenex Gemini С18 HPLC (250×21,2 мм, 5 мкм). Колонку элюировали смесью 6 мМ соляная кислота - ацетонитрил (53:47, об.:об.) при 10 мл/мин. Нужный продукт идентифицировали радиодетекцией и собирали отсечением. Полученный раствор разбавляли водой (150 мл) и пропускали через картридж Sep-Pak® Plus C8 (предварительно уравновешенный 10 мл этанола и 10 мл воды) так, что продукт удерживался на картридже. Картридж элюировали этанолом в сосуд для продукта, который содержал пропиленгликоль (0,9 мл). Фосфатный буфер также пропускали через картридж в сосуд с продуктом с получением продукта в виде препарата. Выход продукта составлял 10,8% (нескорректированный, на основании [18F]-исходной активности) и радиохимическая чистота составляла >99%.
Пример 5: Альтернативный синтез 2-[3-нитро-4-(метилформиламино)фенил]-6-этоксиметоксибензотиазола (8)
Пример 5(1): Синтез 4-хлор-N-(4-гидроксифенил)-3-нитробензамида
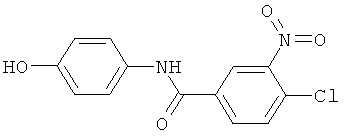
4-Аминофенол (12 г, 0,11 моль, Acros и Aldrich) растворяли в инертной атмосфере при перемешивании в сухом DMF (50 мл) и охлаждали в ледяной бане. Добавляли триэтиламин (TEA, 11 г, 0,11 моль) и перемешивание продолжали в течение 1 часов. 4-Хлор-3-нитробензоилхлорид (22,2 г, 0,1 моль, Acros и Aldrich) медленно добавляли и перемешивали в течение ночи. Осажденную соль, триэтиламина гидрохлорид, отфильтровывали и DMF удаляли при пониженном давлении. Остаток экстрагировали EtOAc (3×100 мл) и лимонной кислотой (1 М, 3×100 мл). Органическую фазу сушили сульфатом магния, фильтровали и упаривали досуха при пониженном давлении. Указанный в заголовке продукт перекристаллизовывали из смеси метанол/вода (1:1, 250 мл), выход 85% и анализировали посредством NMR и MS.
Пример 5(2): Синтез 4-хлор-N-(4-этоксиметоксифенил)-3-нитробензамида
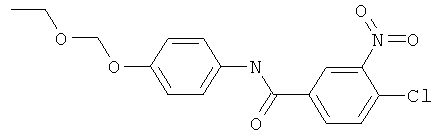
4-Хлор-N-(4-гидроксифенил)-3-нитробензамид (14,6 г, 0,05 моль) помещали в высушенную в термостате 2-горлую 500 мл круглодонную колбу и продували N2. Добавляли достаточное количество диметоксиэтана (DME, 100 мл) для растворения амида. Смесь охлаждали на ледяной бане и небольшими порциями добавляли натрия гидрид (NaH, 50% в масле, всего 3,6 г, 0,075 моль) при энергичном перемешивании. Через час после окончания добавления по каплям добавляли хлорметоксиэтан (7,13 г, 0,075 моль, имеется в продаже) через уравнивающую давление капельную воронку. За реакцией следили при помощи TLC (дихлорметан, DCM,: метанол, МеОН, 95:5). Реакционную смесь вливали в ледяную воду и экстрагировали EtOAc (3×50 мл). Органическую фазу сушили (MgSO4) и упаривали при пониженном давлении. Неочищенный продукт перекристаллизовывали из смеси гексан/этилацетат 1:4 с получением 81% указанного в заголовке соединения.
Пример 5(3): Синтез 4-хлор-N-(4-этоксиметоксифенил)-3-нитро-тиобензамида
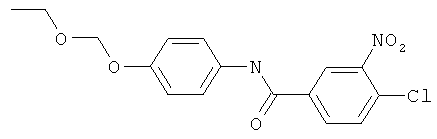
4-Хлор-М-(4-этоксиметоксифенил)-3-нитробензамид (3,5 г, 10 ммоль), фосфора пентасульфид P4S10 (0,81 г, 1,83 ммоль, имеется в продаже), гексаметилдисилоксан (2,7 г, 16,7 ммоль, имеется в продаже) и толуол (10 мл) добавляли в 100 мл круглодонную колбу и продували азотом. Смесь нагревали с обратным холодильником и контролировали при помощи TLC. Нагревание продолжали, пока больше не оставалось исходного бензамида. Также может быть использовано микроволновое нагревание. Реакционную смесь охлаждали до комнатной температуры. Добавляли раствор калия карбоната (4 мл 5,3 М раствора). Добавляли ацетон (10 мл) и смесь перемешивали в течение 1 часа в ледяной бане, экстрагировали толуолом и водой. Органическую фазу сушили (MgSO4) и толуол удаляли при пониженном давлении и очищали флэш-хроматографией со смесью этилацетат/гексан в качестве элюента.
Пример 5(3а): Альтернативный метод тиоамидирования: синтез N-(4-бензилоксифенил)-4-хлор-3-нитротиобензамида
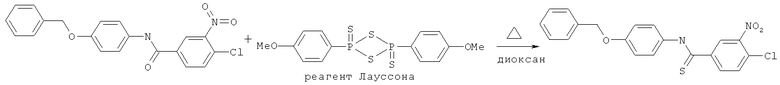
N-(4-Бензилоксифенил)-4-хлор-3-нитробензамид (19,15 г, 50 ммоль), реагент Лауссона (11 г, 27 ммоль, имеется в продаже) и диоксан (150 мл) перемешивали вместе и нагревали с обратным холодильником в течение 4 ч. Когда, как показала TLC, исходный амид более не присутствовал, реакционную смесь охлаждали и растворитель удаляли при пониженном давлении. Неочищенный продукт растворяли в минимальном количестве кипящего толуола для перекристаллизации. Очищенный продукт отфильтровывали и промывали холодным толуолом и холодным гексаном с получением тиоамида, с 77%-ным выходом.
Пример 5(4а): Синтез 6-бензилокси-2-(4-хлор-3-нитрофенил)-бензотиазола
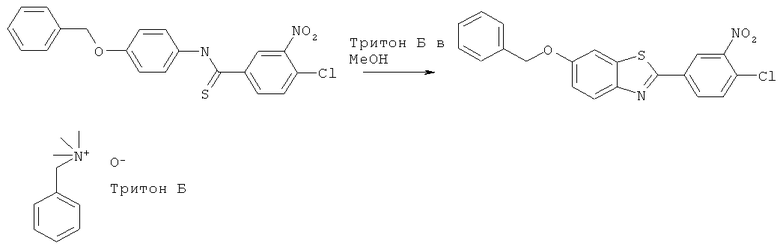
N-(4-Бензилоксифенил)-4-хлор-3-нитротиобензамид (2 г, 5 ммоль) растворяли в метаноле (100 мл). Добавляли натрия гидроксид (1,6 г в 5 мл воды), затем Тритон Б (2,1 мл, 5 ммоль, имеется в продаже). Смесь охлаждали на ледяной бане. По каплям при энергичном перемешивании добавляли калия ферри(III)цианид (13,2 г в 50 мл воды). Реакционную смесь оставляли нагреваться в течение ночи и дополнительно нагревали до 130°С в течение 1 часа. Реакционную смесь охлаждали и экстрагировали смесью этилацетат/вода. Органическую фазу сушили и растворитель удаляли при пониженном давлении. Продукт, соединение, очищали флэш-хроматографией со смесью гексан/этилацетат в качестве элюента.
Пример 5(4): Синтез 6-этоксиметокси-2-(4-хлор-3-нитрофенил)-бензотиазола
При использовании способов, аналогичных описанным в Примере 5 (4а), тиобензамид, полученный в Примере 5 (3) может быть циклизован с образованием указанного в заголовке соединения.
Пример 5(5): Синтез 6-бензилокси-2-(4-метиламино-3-нитрофенил)-бензотиазола и 6-этоксиметокси-2-(4-метиламино-3-нитрофенил)-бензотиазола (7)
Соединения из Примеров 5 (4а) и 5 (4) соответственно взаимодействуют с метиламином в водном растворе при нагревании до 130°С, например в микроволновой печи. Реакционную смесь экстрагируют смесью этилацетат/вода и органическую фазу сушат, затем при пониженном давлении удаляют растворитель. Указанные в заголовке продукты очищают флэш-хроматографией, используя гексан/этилацетат.
Пример 5(6): Синтез 2-[3-нитро-4-(метилформиламино)фенил]-6-бензилоксибензотиазола и 2-[3-нитро-4-(метилформиламино)фенил]-6-этоксиметоксибензотиазола (8)
Указанные в заголовке соединения получают из соединений Примера 5 (5) соответственно, используя способы формилирования, аналогичные описанным в Примере 1 (6).
Пример 6: Альтернативный синтез 2-[3-нитро-4-(метилформиламино)фенил]-6-этоксиметоксибензотиазола (8)
Синтез осуществляли аналогично Примеру 5, но начиная с 4-амино-3-хлорфенола с получением 6-этоксиметокси-2-(4-хлор-3-нитрофенил)-бензотиазола через 4-хлор-N-(4-гидрокси-2-хлорфенил)-3-нитробензамид, 4-хлор-N-(4-этоксиметокси-2-хлорфенил)-3-нитробензамид и 4-хлор-N-(4-этоксиметокси-2-хлорфенил)-3-нитротиобензамид. Циклизацию 4-хлор-N-(4-этоксиметокси-2-хлорфенил)-3-нитротиобензамида с образованием 6-этоксиметокси-2-(4-хлор-3-нитрофенил)бензотиазола осуществляют, используя способы, известные из литературы, например Bowman et al Tetrahedron, 47 (48), 10119-10128 (1991); Couture and Glandclaudon, Heterocycles, 22 (6) 1984; Hutchinson et al. Tetrahedron Lett. 2000, 41 (3), 425-8. Затем осуществляют метилирование и формилирование, как описано в Примере 5.
| название | год | авторы | номер документа |
|---|---|---|---|
| РАДИОФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2008 |
|
RU2475267C2 |
| АГЕНТЫ ДЛЯ ВИЗУАЛИЗАЦИИ | 2004 |
|
RU2355702C2 |
| НУКЛЕОФИЛЬНОЕ ФТОРИРОВАНИЕ В ТВЕРДОЙ ФАЗЕ | 2002 |
|
RU2315769C9 |
| НОВЫЕ ПРЕДШЕСТВЕННИКИ ПРОИЗВОДНЫХ ГЛУТАМАТА | 2012 |
|
RU2600981C2 |
| ДИАГНОСТИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2396272C9 |
| НОВЫЕ ПРЕКУРСОРНЫЕ МОЛЕКУЛЫ ДЛЯ МЕЧЕННЫХ F-18 ПЭТ РАДИОАКТИВНЫХ ИНДИКАТОРОВ | 2010 |
|
RU2538276C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛАМИНОПИРИМИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ОБОГАЩЕННОЙ ЛЕЙЦИНОВЫМИ ПОВТОРАМИ КИНАЗЫ 2 (LRRK2) ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ БОЛЕЗНИ ПАРКИНСОНА | 2013 |
|
RU2637948C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛПИРРОЛИДИНИЛ- И ПИПЕРИДИНИЛКЕТОНА | 2007 |
|
RU2479575C2 |
| СПОСОБ СИНТЕЗА | 2011 |
|
RU2572554C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-4H-3,1-БЕНЗОКСАЗИН-4-ОНА ДЛЯ ПРЕДОТВРАЩЕНИЯ ИЛИ ЛЕЧЕНИЯ ОЖИРЕНИЯ ИЛИ СОПУТСТВУЮЩЕГО НАРУШЕНИЯ | 2000 |
|
RU2244711C2 |
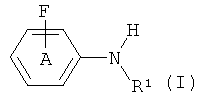
Изобретение относится к способу получения соединений формулы
где группа фторо возможно представляет собой [18F]фторо; кольцо А замещено бензотиазол-2-илом, который замещен гидрокси, С1-6алкилом, C1-6алкокси, галогено, OCH2OR11, где R11 представляет собой C1-6алкил; R1 представляет собой C1-6алкил. Способ заключается во взаимодействии соответствующего соединения формулы

где кольцо А замещено, как определено для соединения формулы (I); R2 выбран из водорода, С1-10алкила, С1-10галогеналкила, -(СН2СН2О)q-СН3, где q означает целое число от 1 до 10; R1 такой, как определен для соединения формулы (I); R3 представляет собой уходящую группу, такую как нитро, с фторидом щелочного металла или тетра-алкиламмония, возможно с [18F]фторидом, с получением соединения формулы
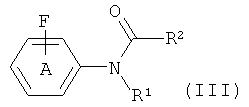
где R1 и R2 такие, как они определены для соединений формул (I) и (II), группа фторо возможно представляет собой [18F]фторо, и кольцо А замещено, как определено для соединения формулы (I); с последующим превращением группы -C(O)R2 в водород посредством гидролиза и возможным удалением любых дополнительных защитных групп. Способ позволяет получать соединения формулы (I) более простым и эффективным методом. Изобретение относится также к новым соединениям формулы
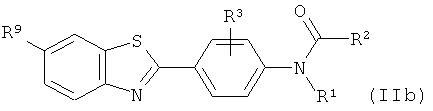
где R1-R3, R9 определены выше, являющимся предшественниками соединений формулы (I), а также к радиофармацевтическому набору и картриджу для использования в позитронно-эмиссионной томографии, которые содержат указанные предшественники. 6 н. и 5 з.п. ф-лы, 1 табл.
1. Способ получения соединения формулы (I):
,
где группа фторо возможно представляет собой [18F]фторо;
фенильное кольцо А замещено бензотиазол-2-илом, который замещен гидрокси, C1-6алкилом, C1-6алкокси, галогено, OCH2OR11, где R11 представляет собой С1-6алкил;
R1 представляет собой С1-6алкил;
который включает:
(1) взаимодействие соответствующего соединения формулы (II):
,
где кольцо А замещено, как определено для соединения формулы (I);
R2 выбран из водорода, С1-10алкила, С1-10галогеналкила, -(CH2CH2O)q-СН3, где q представляет собой целое число от 1 до 10;
R1 такой, как он определен для соединения формулы (I); и
R3 представляет собой уходящую группу, такую как нитро;
с фторидом щелочного металла или тетраалкиламмония, возможно с [18F]фторидом, с получением соединения формулы (III)
,
где R1 и R2 такие, как они определены для соединений формул (I) и (II), группа фторо возможно представляет собой [18F]фторо, и фенильное кольцо А замещено, как определено для соединения формулы (I);
и затем стадию (2) и возможно стадию (3) в любом порядке:
(2) превращение группы -C(O)R2 в водород посредством гидролиза
(3) удаление любых дополнительных защитных групп.
2. Способ по п.1, где группа фторо в соединении формулы (I) представляет собой [18F]фторо, и фторид, используемый на стадии (1) способа, представляет собой [18F]фторид.
3. Способ по п.1, где R1 представляет собой метил.
4. Способ по п.1, где R2 в соединении формулы (II) выбран из водорода и C1-6алкила, и более предпочтительно представляет собой водород.
5. Способ по п.1, где стадию (2) осуществляют путем взаимодействия с ненуклеофильным основанием, таким как гидрид натрия, в апротонном растворителе, таком как ацетонитрил или пропионитрил, и при повышенной температуре, например 40°С-120°С, обычно примерно 100°С.
6. Соединение формулы (IIb):
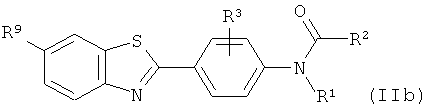 ,
,
где R1 представляет собой С1-6алкил; R2 выбран из водорода, С1-10алкила, С1-10галогеналкила, -(СН2СН2О)q-СН3, где q представляет собой целое число от 1 до 10;
R3 представляет собой нитро; и
R9 выбран из гидрокси, C1-6алкила, С1-6алкокси, галогено, OCH2OR11, где R11 представляет собой C1-6алкил.
7. Соединение формулы (IIb):
,
где R2 представляет собой водород или C1-6алкил;
R1 представляет собой С1-6алкил;
R3 представляет собой нитро; и
R9 представляет собой гидрокси или C1-6алкокси.
8. Соединение формулы (IIb) по п.6 или 7, где R2 представляет собой водород.
9. 2-[3-Нитро-4-(метилформиламино)фенил]-6-этоксиметоксибензотиазол.
10. Радиофармацевтический набор для получения 18F-меченой радиометки для применения в PET (позитронно-эмиссионной томографии), который включает:
(1) сосуд, содержащий соединение формулы (IIb), как оно определено в любом из пп.6-9; и
(2) средство для элюирования из сосуда с источником 18F-;
(3) ионообменный картридж для удаления избытка 18F-; и возможно
(4) картридж для удаления защиты с полученного продукта формулы (I), как оно определено в любом из пп.1 и 2.
11. Картридж для радиофармацевтического набора для получения 18F-меченой радиометки для применения в PET, который включает:
(1) сосуд, содержащий соединение формулы (IIb), как оно определено в любом из пп.6-9; и
(2) средство для элюирования из сосуда с источником 18F-.
| D.A.FORSYTH et al, Intrinsic and Equilibrium NMR Isotope Shift Evidence for Negative Hyperconjugation, J | |||
| AM | |||
| CHEM | |||
| SOC., 1986, 108, 2157-2161 | |||
| A.ZAKRZEWSKA et al, 4-Fluoroanilines: synthesis and decomposition, J | |||
| FLUORINE CHEM., 2001, 111, 1-10 | |||
| E.D.BERGMANN et al, An improved method for the preparation of fluorine-substituted aromatic amines, J. |

