РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка претендует на приоритет предварительной заявки на патент США номер 60/877836, поданной 29 декабря 2006 Theuer et al. под названием "ANTIMETABOLITE AGENT COMBINATIONS IN THE TREATMENT OF CANCER" ("КОМБИНАЦИИ АНТИМЕТАБОЛИЧЕСКОГО АГЕНТА В ЛЕЧЕНИИ РАКА"), предварительной заявки на патент США номер 60/883266, поданной 3 января 2007 Theuer et al. под названием "ANTIMETABOLITE AGENT COMBINATIONS IN THE TREATMENT OF CANCER" и предварительной заявки на патент США номер 60/883959, поданной 8 января 2007 Theuer et al. под названием "ANTIMETABOLITE AGENT COMBINATIONS IN THE TREATMENT OF CANCER", все из которых включены посредством ссылки в данное описание изобретения во всей их полноте, включая любые графические материалы, в полной мере дозволенной законом.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение в основном относится к соединениям, имеющим различные применения, включая применения в исследовании, диагностике и терапии. Более конкретно, в данном описании изобретения описаны и предложены композиции, содержащие метоксиамин и антиметаболический противораковый агент, и способы лечения некоторых видов рака посредством введения таких композиций.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Рак представляет собой проблему мирового масштаба. По этой причине представляет жизненный интерес нахождение новых композиций и способов лечения рака. Лечение рака разделяется на три главные категории: химиотерапию, радиационную терапию и хирургию. Обычно терапии являются комбинированными, так как комбинация терапий часто увеличивает вероятность того, что рак будет уничтожен по сравнению с лечебными стратегиями, использующими единственную терапию. Обычно за хирургическим удалением больших опухолевых масс следует химиотерапия и/или радиационная терапия.
Химиотерапевтические агенты могут действовать разным образом. Например, химиотерапевтические средства могут действовать вмешиваясь в последовательность клеточного цикла или генерируя разрывы нитей ДНК. Если раковая клетка не способна преодолеть блокирование клеточного цикла или повреждение клетки, вызванное терапевтическим соединением, то часто клетка погибает из-за апоптозных механизмов. Применение одного химиотерапевтического агента в лечении рака в присутствии или в отсутствие хирургии или радиации имеет несколько недостатков. Обычно раковые клетки развивают устойчивость к химиотерапевтическому агенту. Такая устойчивость приводит либо к потребности в более высоких дозировках лекарственного средства и/или к возобновлению распространения рака. Химиотерапевтические агенты могут быть токсичными для пациента. Следовательно, существует основанная на практике верхняя граница количества, которое пациент может получать. Однако если может быть разработан второй агент для подавления направления, вызывающего устойчивость, раковые клетки могут стать чувствительными к действиям химиотерапевтического агента.
К созданию лекарственного средства для преодоления устойчивости к химиотерапевтическому лечению рака следует подходить с целью 1) нахождения комбинации, которая реверсирует устойчивость, а не только усиливает активность химиотерапевтического средства в отношении действия на опухоль, и 2) нахождения второго лекарственного средства, которое не усиливает токсических эффектов первого химиотерапевтического агента. Эти условия требуют большого количества эмпирического тестирования агентов, которые, как известно, обладают противораковыми свойствами, с агентами, которые либо могут иметь противораковые свойства либо могут дополнять первый агент другим образом. К сожалению, такие подходы оказываются в значительной степени безуспешными для комбинаций многих противораковых агентов.
Следовательно, существуют недостаток терапий, которые реверсируют устойчивость к химиотерапии для лечения рака.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Изобретения, описанные и заявленные в данном описании изобретения, имеют много признаков и воплощений, включая, но без ограничения ими, те, что изложены или описаны, или упоминаются в данном кратком изложении сущности изобретения. Изобретения, описанные и заявленные в данном описании изобретения, не ограничены признаками или воплощениями, указанными в данном кратком изложении сущности изобретения, которые включены только для иллюстрации, а не для ограничения.
Эти и другие аспекты и воплощения изобретений, описанных и заявленных в данном описании изобретения, будут очевидны из и по всему тексту заявки и формулы изобретения, все из которых следует считать частью их письменного описания.
В данном описании изобретения раскрыты композиции и способы, полезные в лечении некоторых видов рака. Частично данная заявка основана на ранее неизвестном признании того, что некоторые молекулы, которые нацелены на абазические повреждения или АР (апуриновые/апиримидиновые) сайты в ДНК улучшают, увеличивают или потенцируют эффективность антиметаболических противораковых агентов. В другом воплощении ингибитор пути удаления оснований, такой как метоксиамин, объединяют с антиметаболическим противораковым агентом. Антиметаболический противораковый агент представляет собой химиотерапевтическое средство со структурой, аналогичной структуре вещества (метаболита), требующегося для нормальных биохимических реакций, однако достаточно отличающимся для того, чтобы вмешиваться в нормальные функции клеток, включая деление клеток. Антифолаты являются предпочтительным классом антиметаболических агентов. Антифолатный противораковый агент представляет собой химиотерапевтическое средство со структурой, аналогичной структуре фолиевой кислоты, однако достаточно отличающейся для того, чтобы блокировать активность фолиевой кислоты и нарушать фолат-зависимые механизмы, необходимые для репликации клеток. Такие антифолатные противораковые агенты включают пеметрексед, капецитабин, эдатрексат, метотрексат, лометрексол, нолатрексед, ралтитрексид, РТ523 и триметрексат. Рассмотрено применение любого антифолатного противоракового агента в комбинации с ингибитором BER (эксцизионной репарации путем удаления оснований). В одном аспекте в способе берут 1) субъекта, у которого диагностирован рак, 2) первую композицию, содержащую антифолатный противораковый агент, и 3) вторую композицию, содержащую метоксиамин; вводят указанную первую композицию указанному субъекту; и вводят указанную вторую композицию указанному субъекту, причем метоксиамин вводят в количестве, достаточном для улучшения или усиления действия антифолатного противоракового агента. Вторую композицию можно вводить перорально. В еще одном аспекте в способе берут 1) пациента, у которого диагностирован рак, причем указанный рак является по меньшей мере частично устойчивым к лечению одним пеметрекседом, 2) первую композицию, содержащую пеметрексед; и 3) вторую композицию, содержащую метоксиамин; вводят указанную первую композицию указанному пациенту; и вводят указанную вторую композицию указанному пациенту, причем метоксиамин вводят в количестве, достаточном для потенцирования активности указанного пеметрекседа и преодоления указанной устойчивости. В данных способах метоксиамин и антифолатный противораковый агент можно вводить в виде композиции. Кроме того, метоксиамин и антифолатный противораковый агент можно вводить последовательно, в любом порядке. Также метоксиамин можно вводить перорально, и антифолатный противораковый агент можно вводить или перорально или внутривенно. Также количество указанного метоксиамина может количеством, достаточным для сенсибилизации раковых клеток без чрезмерной сенсибилизации нормальных клеток. Также метоксиамин и антифолатный противораковый агент можно вводить, чтобы достигнуть синергического эффекта. Также антифолатный противораковый агент может быть введен перорально или внутривенно, и указанный метоксиамин может быть введен перорально, не более чем два раза в сутки, в количестве, достаточном для потенцирования активности указанного антифолатного противоракового агента. Также может быть выбран пациент, имеющий рак, по меньшей мере частично устойчивый к лечению одним антифолатным противораковым агентом, и где указанную вторую композицию, содержащую метоксиамин, вводят в количестве, эффективном для потенцирования активности указанного антифолатного противоракового агента и преодоления указанной устойчивости. Кроме того, отношение указанного метоксиамина к указанному антифолатному противораковому агенту может составлять от 1:5 до 1:500, более предпочтительно от 1:15 до 1:40, и еще более предпочтительно от примерно 1:20 до примерно 1:30. Кроме того, раковое заболевание может быть выбрано из группы, состоящей из карцином, меланом, сарком, лимфом, лейкемий, астроцитом, глиом, злокачественных меланом, хронического лимфолейкоза, раковых заболеваний легких, колоректальных раковых заболеваний, раковых заболеваний яичников, раковых заболеваний поджелудочной железы, раковых заболеваний почек, раковых заболеваний эндометрия, раковых заболеваний желудка, раковых заболеваний печени, раковых заболеваний головы и шеи и раковых заболеваний молочной железы. В предпочтительных воплощениях антифолатный противораковый агент представляет собой пеметрексед.
В еще одном воплощении раскрыт улучшенный способ в способе лечения рака у пациента, у которого диагностировано раковое заболевание, включающий введение антифолатного противоракового агента пациенту, улучшение, включающее введение метоксиамина пациенту в количестве, достаточном для потенцирования токсичности указанного антифолатного противоракового агента. Также раскрыты противораковые композиции, содержащие лекарственную форму, содержащую пеметрексед, и лекарственную форму, содержащую синергическое количество метоксиамина, и способы применения такой композиции согласно раскрытым способам лечения. В еще одном воплощении раскрыто улучшенное применение метоксиамина в применении антифолатного противоракового агента для лечения рака у пациента, улучшение, включающее применение метоксиамина в количестве, достаточном для потенцирования токсичности указанного антифолатного противоракового агента у указанного пациента.
В одном аспекте настоящее изобретение основано на ранее неизвестном признании того, что некоторые молекулы, такие как метоксиамин, которые нацелены на АР-сайты, являются полностью перорально биодоступными и поддерживают минимальные эффективные концентрации при приеме один раз или два раза в сутки посредством перорального введения. Противораковые агенты обычно вводят в виде внутривенного болюса, так как они редко хорошо абсорбируются из желудочно-кишечного тракта. Внутривенное дозирование имеет недостатки. Во-первых, внутривенная инъекция при химиотерапии требует лечения в кабинете врача или в больнице. Во-вторых, внутривенная терапия обычно назначается в виде болюса, что приводит к очень высокой, но кратковременной экспозиции лекарственного средства. Некоторые противораковые агенты могут быть наиболее активными после продолжительной экспозиции, которая может быть достигнута посредством повторных пероральных доз. Это особенно справедливо для агентов, которые ингибируют механизмы устойчивости к химиотерапевтическим лекарственным средствам, где продолжительное ингибирование путей устойчивости может быть необходимым для нужного лечебного эффекта. Продолжительная экспозиция лекарственного средства может быть осуществлена с использованием непрерывного внутривенного введения. Однако для введения противораковых агентов в виде непрерывных инфузий требуется сложный аппарат для инфузии лекарственного средства и внутривенной катетеризации. Пероральное введение избавляет от необходимости непрерывной внутривенной инфузии и представляет собой способ введения, предпочитаемый пациентами. Однако, насколько известно авторам изобретения, ингибиторы ВЕR, которые реверсируют устойчивость к химиотерапии и обладают почти полной пероральной биодоступностью, как предложено в данном описании изобретения, не были созданы до настоящего времени.
Пеметрексед представляет собой многоцелевой антифолат, который действует способом, механизм которого отличен от 5-FU (5-фторурацила) и другого более раннего поколения антиметаболитов. Пеметрексед уникален в том, что он является антифолатным аналогом пирролопиримидина, который внутриклеточно метаболизируется в более высокие полиглутаматные формы с помощью фолил-полиглутамат-синтетазы (FPGS). Пентаглутаматная форма является превалирующей внутриклеточной разновидностью, и пеметрексед-полиглутаматы приблизительно более чем в 60 раз более активны чем родительское моноглутаматное соединение; пеметрексед-полиглутаматы также продолжительно удерживаются клеткой. Следовательно, фармакологические эффекты пеметрекседа сохраняются в течение многих дней после внутривенного введения болюса.
Пеметрексед подавляет тимидилат-синтазу (TS), дигидрофолат-редуктазу (DHFR) и глицинамид-рибонуклеотид-формилтрансферазу (GARFT), все фолат-зависимые ферменты, вовлеченные в биосинтез de novo тимидиновых и пуриновых нуклеотидов. В отличие от этого, 5-FU и другие более ранние поколения антиметаболитов главным образом ингибируют только TS. Точный механизм, посредством которого пеметрексед вызывает гибель клетки, все еще не установлен, но включает больше, чем просто ингибирование TS. Следовательно, в то время как в гетерогенной неселективной панели клеточной линии рака толстого кишечника человека лучшим прогностическим показателем в отношении чувствительности к 5FU являлась активность TS, для пеметрекседа были важны детерминанты множественной чувствительности, включая активность FPGS и ферментативную кинетику TS (van Triest В, Pinedo HM, van Hensbergen Y. Thymidylate synthase level as the main predictor parameter for sensitivity to 5-FU, but not for Folate-based Thymidylate Synthase Inhibitors, in 13 Nonselected Colon Cancer Cell Lines. Clin. Cancer. Res. 1999; 5:643-54). Дополнительное исследование подтвердило, что чувствительность желудочно-кишечных клеточных линий к пеметрекседу не может быть спрогнозирована с помощью экспрессии TS (Kim JH, Lee KW, Jung Y et al. Cytotoxic effects of pemetrexed in gastric cancer cells. Cancer Sci. 2005; 96:365-71).
Уникальная фармакологическая активность пеметрекседа проявилась в in vitro исследовании активности на многих линиях раковых клеток в сравнении с 5-FU. В сериях из 13 клеточных линий колоректального рака, например, пеметрексед был от 18 до 627 раз более действенным, чем 5-FU (van Triest et al, 1999). Данная уникальная фармакология и тот факт, что пеметрексед, как предполагается, имеет ряд механизмов действия, которые полностью не поняты, создают трудность для понимания того, насколько эффективным он может быть при комбинировании с другими конкретными противораковыми агентами для лечения конкретных видов рака.
Одним аспектом настоящего изобретения является обнаружение неожиданного улучшения в лечении раковых заболеваний посредством комбинированного введения метоксиамина с антифолатным соединением. Таким образом, одно воплощение, описанное в данном описании изобретения, направлено на способы, при которых берут 1) пациента, у которого диагностировано раковое заболевание, 2) первую композицию, содержащую антифолатный противораковый агент, и 3) вторую композицию, содержащую метоксиамин; вводят первую композицию пациенту; и вводят вторую композицию указанному пациенту, причем метоксиамин может быть введен в количестве, достаточном для улучшения или усиления действия (например потенцирование активности) антифолатного противоракового агента. Может быть использован любой антифолатный противораковый агент при условии, что в конкретных воплощениях способа 5-FU специально исключен. В типичном воплощении противораковый агент может быть выбран из группы, состоящей из пеметрекседа, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, 5,10-дидиазатетрагидрофолиевой кислоты, (DDATHF), пиритрексима, ралтитрексида, GW1843 [(S)-2-[5-[(1,2-дигидро-3-метил-1-оксобензо[f]хиназолин-9-ил)метил]амино-1-оксо-2-изоиндолин]-глутаровой кислоты], их фармацевтических солей и любых комбинаций. В более типичном воплощении противораковый агент может быть выбран из группы, состоящей из пеметрекседа, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, их фармацевтических солей и любых комбинаций. В наиболее типичном воплощении противораковый агент может представлять собой пеметрексед и его фармацевтически приемлемые соли. Например, пеметрексед может представлять собой динатриевую соль. В типичном воплощении пеметрексед может представлять собой гептагидрат динатриевой соли.
В типичном, но не ограничивающем воплощении, антифолатный противораковый агент представлять собой пеметрексед. Метоксиамин и антифолатный противораковый агент могут быть введены последовательно (в любом порядке) или введены вместе в виде композиции. Пеметрексед может, например, быть введен внутривенно в дозе 200-1000 мг/м2 площади поверхности тела в сутки, или в дозе 500-600 мг/м2 площади поверхности тела в сутки. В еще одном воплощении отношение метоксиамина к антифолатному противораковому агенту может составлять от 1:5 до 1:500.
В еще одном аспекте метоксиамин может быть введен перорально в количестве, достаточном для сенсибилизирования рака без вызывания чрезмерной сенсибилизации нормальной ткани. В неограничивающем предпочтительном воплощении метоксиамин вводят перорально, так что он имеет существенно усиленную биодоступность по сравнению с другими противораковыми агентами, введенными перорально. В других неограничивающих предпочтительных воплощениях метоксиамин вводят перорально так, что он поддерживает минимальную эффективную концентрацию при дозировании один или два раза в сутки. Одним способом измерения пероральной биодоступности является сравнение достигнутых уровней по сравнению с внутривенно введенным метоксиамином. Таким образом, в еще одном аспекте настоящего изобретения метоксиамин вводят перорально с получением биодоступности по меньшей мере 50% по сравнению с внутривенным введением, по меньшей мере 60% по сравнению с внутривенным введением, по меньшей мере 70% по сравнению с внутривенным введением, по меньшей мере 75% по сравнению с внутривенным введением, по меньшей 80% по сравнению с внутривенным введением, по меньшей мере 85% по сравнению с внутривенным введением, по меньшей мере 90% по сравнению с внутривенным введением, по меньшей мере 95% по сравнению с внутривенным введением, или приблизительно эквивалентной биодоступности при внутривенном введении. Важно признать, что в дополнение к неожиданной высокой степени биодоступности, достигнутой посредством перорального введения, по сравнению с внутривенным введением метоксиамина получают более желательный профиль рК по сравнению с внутривенным введением метоксиамина. В еще одном аспекте настоящего изобретения перорально введенный метоксиамин поддерживает минимальные эффективные концентрации после введения один или два раза в сутки благодаря периоду полувыведения > 4 часов в плазме. Данное преимущество обеспечивает возможность желательного режима перорального дозирования для метоксиамина, включая введение один раз или два раза в сутки. В то время как внутривенное введение пеметрекседа, комбинированное с пероральным введением метоксиамина, представляют собой предпочтительные неограничивающие воплощения, для каждого противоракового агента предполагаются другие пути введения.
В еще одном аспекте изобретения предложены способы лечения некоторых видов рака, которые устойчивы к лечению одним противораковым агентом. В соответствии с этим, также предложены способы, при которых:
берут 1) пациента, у которого диагностировано раковое заболевание, где указанный рак может быть устойчивым к лечению одним пеметрекседом, 2) первую композицию, содержащую антифолатный противораковый агент; и 3) вторую композицию, содержащую метоксиамин;
вводят первую композицию пациенту; и
вводят вторую композицию пациенту, причем метоксиамин можно вводить в количестве, достаточном для улучшения или усиления воздействия (то есть потенцирование токсичности) антифолатного противоракового агента. В одном воплощении антифолатный противораковый агент может представлять собой пеметрексед. Метоксиамин и антифолатный противораковый агент можно вводить последовательно (в любом порядке) или вводить вместе в виде композиции. Пеметрексед можно вводить внутривенно в дозе 200-1000 мг/м2 площади поверхности тела в сутки, или в дозе 500-600 мг/м2 площади поверхности тела в сутки. Отношение пеметрекседа к метоксиамину может составлять от 1:5 до 1:500. В еще одном воплощении количество метоксиамина можно вводить перорально в количестве, достаточном для сенсибилизации рака без вызывания чрезмерной сенсибилизации нормальной ткани. В еще одном воплощении данное количество метоксиамина можно вводить перорально либо один раз в сутки либо два раза в сутки в количестве, достаточном для сенсибилизации рака без вызывания чрезмерной сенсибилизации нормальной ткани. В то время как пероральное введение метоксиамина неожиданно представляет собой предпочтительный способ введения, возможны другие типы введения.
Еще одно воплощение направлено на способ лечения рака, при котором берут первую и вторую композицию, где указанная первая композиция содержит антифолат и вторая композиция содержит перорально вводимый метоксиамин. Первая композиция, содержащая антифолат, может быть введена обычными способами введения, включая внутривенное. Неограничивающий предпочтительный антифолат представляет собой пеметрексед. В соответствии с этим, в одном воплощении антифолат представляет собой пеметрексед, и вторую композицию, содержащую метоксиамин, вводят перорально в количестве, которое является синергетически эффективным по сравнению с лечением одним пеметрекседом.
В еще одном аспекте способ можно применять для лечения раковых заболеваний, которые устойчивы к одному пеметрекседу. Согласно этим воплощениям пеметрексед вводят в количестве, которое реверсирует устойчивость (и следовательно, является синергическим) к одному антифолату. Таким образом, в одном воплощении метоксиамин вводят перорально в количестве, эффективном для улучшения или увеличения токсичности пеметрекседа, и преодолевают устойчивость рака к лечению пеметрекседом. Например, эффективность пеметрекседа для лечения рака может быть понижена из-за развития устойчивости во время лечебного цикла. Введение метоксиамина может обойти развившуюся устойчивость, обеспечивая более чем аддитивный эффект на лечение рака или одним метоксиамином или одним пеметрекседом.
Также предложены способы, при которых:
берут 1) пациента, у которого диагностировано раковое заболевание, где рак может быть выбран из группы, состоящей из карцином, меланом, сарком, лимфом, лейкемий, астроцитом, глиом, злокачественных меланом, хронического лимфоцитарного лейкоза, раковых заболеваний легкого, колоректальных раковых заболеваний, раковых заболеваний яичников, раковых заболеваний поджелудочной железы, раковых заболеваний почек, раковых заболеваний эндометрия, раковых заболеваний желудочно-кишечного тракта, раковых заболеваний печени, раковых заболеваний головы и шеи и раковых заболеваний молочной железы, и где рак может быть устойчивым к лечению одним пеметрекседом, 2) первую композицию, содержащую антифолатный противораковый агент и 3) вторую композицию, содержащую метоксиамин;
вводят первую композицию указанному пациенту; и
вводят вторую композицию указанному пациенту, причем метоксиамин можно вводить в количестве, достаточном для улучшения или увеличения воздействия антифолатного противоракового агента. Метоксиамин и антифолатный противораковый агент можно вводить последовательно или вводить вместе в виде композиции. Например, метоксиамин можно вводить первым и затем последним можно вводить антифолатный противораковый агент, или антифолатный противораковый агент можно вводить первым и последним можно вводить метоксиамин.
В типичном воплощении антифолатный противораковый агент может представлять собой пеметрексед. Пеметрексед можно вводить внутривенно в дозе 200-1000 мг/м2 площади поверхности тела в сутки, или в дозе 500-600 мг/м2 площади поверхности тела в сутки. Отношение пеметрекседа к метоксиамину может составлять от 1:5 до 1:500. В еще одном воплощении это количество метоксиамина можно вводить перорально в количестве, достаточном, чтобы обеспечить восприимчивость раковых клеток к лечению противораковым агентом (то есть сенсибилизировать), не вызывая неоправданного повреждения нормальных клеток. В еще одном воплощении это количество метоксиамина можно вводить перорально либо один, либо два раза в сутки в количестве, достаточном, чтобы сенсибилизировать рак, не вызывая чрезмерной сенсибилизации нормальной ткани.
Еще одно воплощение может представлять собой композицию, содержащую метоксиамин и антифолатный противораковый агент, где метоксиамин можно вводить в количестве, достаточном, чтобы потенцировать токсичность антифолатного противоракового агента. Предпочтительно антифолатный противораковый агент представляет собой пеметрексед.
В еще одном воплощении отношение метоксиамина к антифолатному противораковому агенту может составлять от 1:5 до 1:500 в любом из способов, описанных выше.
В еще одном воплощении второй противораковый агент можно вводить до или после лечения метоксиамином и антифолатным противораковым агентом в любом из способов, описанных выше.
В еще одном воплощении описан способ лечения рака у пациента, у которого диагностировано раковое заболевание, включающий введение антиметаболитного противоракового агента пациенту, имеющий следующее улучшение: введение метоксиамина пациенту в количестве, достаточном, чтобы потенцировать токсичность указанного антиметаболитного противоракового агента. Антиметаболический противораковый агент может представлять собой антифолатный противораковый агент. Антифолатный противораковый агент может представлять собой пеметрексед, и отношение указанного метоксиамина к антифолатному противораковому агенту может составлять от 1:5 до 1:500. Рак может быть устойчивым к лечению одним пеметрекседом.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг.1А-Б показано влияние пеметрекседа и MX на разрыв нитей ДНК, с использованием щелочного (Фиг.1А) и нейтрального (Фиг.1Б) кометного анализа.
На Фиг.1В-Г показано сравнение длины кометных хвостов клеток, обработанных одним пеметрекседом или одним MX и пеметрекседом плюс MX, в клетках, подвергнутых щелочному (ФИГ.1В) и нейтральному (ФИГ.1Г) кометному анализу.
На Фиг.2 представлен график, показывающий среднее значение концентрации MX в плазме самцов крыс Sprague-Dawley в разные моменты времени после дозирования одного болюса MX посредством внутривенного и перорального введения в концентрации 20 мг/кг массы тела.
На Фиг.3 представлен график, показывающий значение концентрации MX в плазме, полученной от самок крыс Sprague-Dawley в разные моменты времени после дозирования одного болюса MX посредством внутривенного и перорального введения в концентрации 20 мг/кг массы тела.
На Фиг.4А представлена диаграмма, показывающая относительное количество АР-сайтов, обнаруженных в клетках Н460 через 24 часа после лечения пеметрекседом и MX.
На Фиг.4Б представлена диаграмма, показывающая относительное количество АР-сайтов, обнаруженных в клетках Н460 через 24 ч, 48 ч и 72 ч.
На Фиг.5А и 5Б показано, что МХ-связанные АР-сайты не поддаются репарации АР-эндонуклеазой
На Фиг.5А показана схема получения субстратов ДНК с регулярными АР-сайтами или МХ-АР-сайтами. Структурная схема показывает получение позитивных специфических субстратов олигонуклеотидов, содержащих АР-сайт или MX связанный АР-сайт
На Фиг.5Б показано, что МХ-связанные АР-сайты резистентны к расщеплению АР-эндонуклеазой (АРЕ). АРЕ обладает реакционной способностью для расщепления АР-сайта, но не АР-МХ-сайта, что указывает на то, что АР-МХ-сайт не поддается репарации посредством АРЕ
На Фиг.6 показано влияние комбинации пеметрекседа и MX на разрывы двойных нитей ДНК и апоптоз. Комбинация пеметрекседа и TRC2 усиливает двунитевые разрывы ДНК и апоптоз, который не зависит от Вс12-пути. Клетки обрабатывали MX с концентрацией 6 мМ и собрали в 24 ч.
На Фиг.7 показано влияние комбинации пеметрекседа и MX на белковые уровни при ВЕR в клетках Н460. Представлены белковые уровни ВЕR в клетках Н460 перед и после обработки пеметрекседом или комбинацией. Клетки обрабатывали MX в концентрации 6 мМ и собрали в 24 ч.
На Фиг.8 показано влияние пеметрекседа и MX на средний объем опухолей NCI-H460, опухолей А549, опухолей НСТ116 и опухолей MDA-MB-468, выросших у "голых мышей".
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Некоторые воплощения изобретения в основном относятся к новым композициям, содержащим метоксиамин и антифолатный противораковый агент, и к лечению некоторых видов рака с применением данных композиций.
Определения
Если не указано иное, следующие термины имеют следующие значения при использовании в данном описании изобретения и в прилагаемой формуле изобретения. Те термины, которые не определены ниже или где-либо еще в описании, имеют свое признанное в уровне техники значение.
Термины "агент" и "лекарственное средство" при использовании в данном описании изобретения для целей описания и формулы изобретения означает химические соединения, смеси химических соединений, биологические макромолекулы, или экстракты, полученные из биологических материалов (таких как бактерии, растения, грибы или животное, в частности млекопитающее), клеток или тканей, которые, как предполагается, обладают терапевтическими свойствами. Агент или лекарственное средство могут быть очищенными, по существу, очищенными или частично очищенными.
Термин "антиметаболит" используется в данном описании изобретения для целей описания и формулы изобретения для обозначения химиотерапевтического средства со структурой, аналогичной структуре вещества (метаболита, например нуклеозида), требующегося для нормальных биохимических реакций, однако достаточно отличающегося для того, чтобы вмешиваться в нормальные функции клеток, включая клеточное деление.
Термин "антифолат" используется в данном описании изобретения для целей подробного описания и формулы изобретения, для обозначения химиотерапевтического средства со структурой, аналогичной структуре фолиевой кислоты, однако достаточно отличающегося для того, чтобы блокировать активность фолиевой кислоты и нарушить фолат-зависимые механизмы, необходимые для клеточной репликации. При использовании в данном описании изобретения антифолаты представляют собой один класс антиметаболитов.
Термин "антинеопластический" используется в данном описании изобретения для целей подробного описания и формулы изобретения, для обозначения химиотерапевтического средства, предназначенного для ингибирования или предотвращения созревания и пролиферации неоплазм (опухолей), которые могут становиться злокачественными посредством ДНК-нацеливания.
Термин "окрашивание" используется в данном описании изобретения для целей подробного описания и формулы изобретения, для обозначения любого количества способов, известных в данной области, которые используются для лучшего наглядного представления, различения или идентификации конкретного(ых) компонента(ов) и/или характеристик(и) клетки или клеток.
Термин "в функциональной комбинации", "в функциональном порядке" и "функционально связанный" используется в данном описании изобретения для целей подробного описания и формулы изобретения для обозначения соединения нуклеиновокислотных последовательностей таким образом, что получается молекула нуклеиновой кислоты, способная управлять транскрипцией данного гена и/или синтезом нужной белковой молекулы. Этот термин также относится к соединению аминокислотных последовательностей таким образом, что получается функциональный белок.
Термин "антиген" используется в данном описании изобретения для целей описания и формулы изобретения для обозначения белка, гликопротеина, липопротеина, липида или другого вещества, которое обладает реакционной способностью в отношении антитела, специфичного к участку молекулы.
Термин "морфология" используется в данном описании изобретения для целей описания и формулы изобретения для обозначения визуальных характеристик клетки или организма при рассматривании невооруженным глазом, посредством оптической микроскопии, конфокальной микроскопии или электронной микроскопии в зависимости от целесообразности.
Термин "субъект", "индивидуум" и "пациент" используется в данном описании изобретения для целей описания и формулы изобретения для обозначения человека или другого животного, такого как сельскохозяйственные животные или лабораторные животные (например, морская свинка или мышь), способного иметь заболевания, определяемые клеточным циклом (на которые влияет клеточный цикл), либо естественные, либо индуцированные, включая рак, но без ограничения им.
Термин "реверсирует устойчивость" означает, что использование второго агента в комбинации с основным химиотерапевтическим средством способно вызывать значительное уменьшение объема опухоли при уровне статистической значимости (например, р<0,05), если сравнивать с объемом необработанной опухоли в случае, когда главное химиотерапевтическое средство не обладает способностью вызывать статистически значимое уменьшение объема опухоли по сравнению с объемом необработанной опухоли. Термин в основном применяется к измерениям объемов опухолей, выполненных в период времени, когда необработанная опухоль растет логарифмически ритмично.
Термин "потенцировать" при использовании в данном описании изобретения означает улучшение или увеличение полезной активности или эффективности противоракового агента по сравнению с тем, который следовало бы ожидать от одного противоракового агента или одного потенцирующего агента.
Термин "сенсибилизация" при использовании в данном описании изобретения означает изменение раковых клеток или опухолевых клеток таким образом, который обеспечивает возможность более эффективного лечения ассоциированного неопластического заболевания противораковым агентом или радиационной терапией. В некоторых воплощениях нормальные клетки не подвергают воздействию в той степени, которая приводит к излишнему травмированию нормальных клеток посредством химиотерапии или радиационной терапии.
Термин "синергический эффект" при использовании в данном описании изобретения означает, что комбинированный эффект двух или более противораковых агентов или химиотерапевтических лекарственных средств может быть более значительным, чем сумма отдельных эффектов противораковых агентов или химиотерапевтических лекарственных средств самих по себе. Например, комбинированный эффект ингибитора ВЕR, такого как метоксиамин, и противоракового агента, такого как пеметрексед, может быть больше, чем сумма отдельных эффектов метоксиамина и пеметрекседа самих по себе.
Термин "терапевтически эффективное количество" означает количество рассматриваемого соединения, которое будет вызывать нужный ответ, например биологический или медицинский ответ ткани, системы, животного или человека, к которому стремится, например, исследователь, ветеринар, доктор или другой врач-клиницист.
Термин "дикий тип" (wt) клетки или клеточной линии используется в данном описании изобретения для целей описания и формулы изобретения для обозначения клетки или клеточной линии, которые сохраняют свойства, обычно ассоциированные с этим типом клетки или клеточной линии, в отношении физиологического процесса или морфологической характеристики, которые проверяют. Допускается, чтобы клетка или клеточная линия имела свойства недикого типа в отношении физиологического процесса или морфологических свойств, которые не проверялись, до тех пор, пока они существенно не влияют на проверяемый процесс или свойство.
Термин "фармацевтически приемлемая соль" относится к соли соединения, которая не вызывает значительного раздражения в организме, в который ее вводят, и не аннулирует биологическую активность и свойства соединения. В некоторых воплощениях соль представляет собой соль присоединения кислоты к соединению. Фармацевтические соли можно получить путем взаимодействия соединения с неорганическими кислотами, такими как галогенводородная кислота (например, соляная кислота или бромистоводородная кислота), серная кислота, азотная кислота, фосфорная кислота и тому подобное. Фармацевтические соли также можно получить путем взаимодействия соединения с органической кислотой, такой как алифатические или ароматические карбоновые или сульфоновые кислоты, например уксусная, янтарная, молочная, малеиновая, винная, лимонная, аскорбиновая, никотиновая, метансульфоновая, этансульфоновая, пара-толуолсульфоновая, салициловая или нафталинсульфоновая кислота. Фармацевтические соли можно также получить путем взаимодействия соединения с основанием с образованием соли, такой как соль аммония, соль щелочного металла, такая как натриевая или калиевая соль, соль щелочноземельного металла, такая как кальциевая или магниевая соль, соль огранических оснований, таких как дициклогексиламин, N-метил-D-глюкамин, трис(гидроксиметил)метиламин, С1-С7алкиламин, циклогексиламин, триэтаноламин, этилендиамин и соли с аминокислотами, такими как аргинин, лизин и тому подобное.
Повреждение ДНК минимизируют с помощью ферментов, которые узнают ошибки, удаляют их и заменяют поврежденную ДНК правильными нуклеотидами. Повреждение ДНК происходит, когда вводят одноцепочечный разрыв, основание удаляют, оставляя его прежнего партнера неспаренным, основание ковалентно модифицируют, основание превращают в другое, которое не является надлежащим образом спаренным с парным основанием, или вводят ковалентную связь между основаниями на противоположных нитях. Системы эксцизионной репарации удаляют неправильно спаренное или поврежденное основание из нити ДНК и затем синтезируют новую ДНК для ее замены. Эксцизионная репарация путем удаления оснований (ВЕR) инициируется при репликации ДНК и позволяет исправить поврежденные основания/неправильно спаренные основания до окончания репликации.
Эксцизионную репарацию путем удаления оснований (ВЕR) инициируют посредством ДНК-гликозилазы, которая устраняет N-гликозидные (основание-сахар) связи, освобождая поврежденное основание и образуя абазический сайт (например, апуриновый или апиримидиновый (АР) сайт). Апуриновый или апиримидиновый (АР) сайт возникает в результате потери пуринового или пиримидинового остатка соответственно из ДНК (дезоксирибонуклеиновой кислоты). Урациловые остатки могут образоваться из спонтанного дезаминирования цитозина и могут привести к перемещению С→Т, если не репарированы. Также существует гликозилаза, которая узнает и удаляет гипоксантин, продукт дезаминирования аденина. Другие гликозилазы удаляют алкилированные основания (такие как 3-метиладенин, 3-метилгуанин и 7-метилгуанин), пурины с раскрытым кольцом, окислительно поврежденные основания и в некоторых организмах UV (ультрафиолетовые) фотодимеры. Урацил-ДНК-гликозилаза (UDG) является примером ДНК-гликозилазы. На белковые уровни BER UDG влияет обработка комбинацией пеметрекседа и MX (Фиг.7).
Затем процессируется с помощью 5'-3' эндонуклеазы (АР-эндонуклеаза (АРЕ)), которая разрезает фосфодиэфирную связь с обеих сторон поврежденного пуринового или пиримидинового основания. АР-эндонуклеазы вводят разрывы цепи посредством расщепления фосфодиэфирных связей на АР-сайтах.
PARP помогает процессировать разрывов нитей ДНК, индуцированные во время BER. PARP представляет собой белок, контролирующий одноцепочечные разрывы ДНК, который слабо связывается с промежуточными продуктами BER, когда однонуклеотидная BER протекает нормально до завершения. В отличие от этого, когда однонуклеотидную BER останавливают путем блокирования на стадии удаления, PARP сильно связывается с промежуточными продуктами BER, а также с АР-эндонуклеазой (АРЕ), ДНК pol β и FEN-1.
В клетках млекопитающих 5'-дезоксирибозный сахар-фосфат удаляют с помощью эндогенной АР-лиазной (dRP)-активности (транскрипционной) ДНК-полимеразы β (pol β). Фермент ДНК-полимераза также заполняет пробелы новыми нуклеотидами.
В результате ДНК-лигаза ковалентно связывает 3'-конец нового вещества со старым веществом. Таким образом восстанавливается последовательность дикого типа.
Топоизомеразы I и II также вовлечены в репарацию ДНК, так как они узнают спонтанные АР-сайты и образовывают стабильные расщепляемые комплексы. Ингибиторы топоизомеразы II стимулируют расщепление ДНК и другие хромосомные аберрации, включая сестринские хроматидные обмены.
Как описано в данном описании изобретения, некоторые воплощения могут быть направлены на способы, при которых:
берут 1) пациента, у которого диагностировано раковое заболевание, 2) первую композицию, содержащую антифолатный противораковый агент и 3) вторую композицию, содержащую метоксиамин;
вводят первую композицию пациенту; и вводят вторую композицию указанному пациенту, причем метоксиамин можно вводить в количестве, достаточном для улучшения или усиления воздействия антифолатного противоракового агента. Можно использовать любой антифолатный противораковый агент, при условии что в некоторых воплощениях способа конкретно исключен 5-FU.
В типичном воплощении противораковый агент может быть выбран из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, 5,10-дидезазатетрагидрофолиевой кислоты (DDATHF), пиритрексима, ралитрекседа, GW1843 [(S)-2-[5-[(1,2-дигидро-3-метил-1-оксобензо[f]хиназолин-9-ил)метил]амино-1-оксо-2-изоиндолин]-глутаровой кислоты], их фармацевтических солей и любых комбинаций. В более типичном воплощении противораковый агент может быть выбран из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, их фармацевтических солей и любых комбинаций. В наиболее типичном воплощении противораковый агент может представлять собой пеметрексед и его фармацевтически приемлемые соли. Например, пеметрексед может представлять собой динатриевую соль. В типичном воплощении пеметрексед может представлять собой гептагидрат динатриевой соли.
В некоторых воплощениях настоящего изобретения предполагается применение противоракового агента, который индуцирует образование АР-сайтов, и ингибитор ВЕR
В типичном воплощении противораковый агент может быть выбран из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, 5,10-дидезазатетрагидрофолиевой кислоты (DDATHF), пиритрексима, ралитрекседа, GW1843 [(S)-2-[5-[(1,2-дигидро-3-метил-1-оксобензо[f]хиназолин-9-ил)метил]амино-1-оксо-2-изоиндолин]-глутаровой кислоты], их фармацевтических солей и любых комбинаций. В более типичном воплощении противораковый агент может быть выбран из группы, состоящей из пеметрексед, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, их фармацевтических солей и любых комбинаций. В наиболее типичном воплощении противораковый агент может представлять собой пеметрексед и его фармацевтически приемлемые соли. Например, пеметрексед может представлять собой динатриевую соль. В типичном воплощении пеметрексед может представлять собой гептагидрат динатриевой соли.
В типичном воплощении ингибитор ВЕR может быть выбран из группы, состоящей из метоксиамина, этопозида (VP-16, VP-16-123), мезо-4,4'-(2,3-бутандиил)-бис-(2,6-пиперазиндион) (ICRF-193, бис-диоксопиперазин), доксорубицина (DOX), амсаркина (4′,9-акридиниламинометансульфон-m-анисидид; mAMSA), пазеллиптина, налидиксовой кислоты, оксолиновой кислоты, новобиоцина, кумермицина А1, фостриецина, тенипозида, митоксантрона, даунорубицина, N-[2-диметиламино)этил]акридин-4-карбоксамида (DACA), мербарона, хинакрина, эллиптицинов, эпиподофилотоксинов, этидия бромида, эпирубицина, пирарубицина, 3′-дезамино-3′-морфолино-13-деоксо-10-гидрокси-карминомицина; 2′′,3′′-бис-пентафторфеноксиацетил-4′,6′-этилиден-бета-D-гликозида 4′-фосфат-4′-диметилэпиподофиллотоксина 2N-метил-глюкаминовой соли (F11782; фторированный липофильный эпиподофиллоид), адриамицина, актиномицина D, антрациклинов (таких как 9-амино-антрациклин), пиразолоакридина (PZA), камптотецина, топотекана, их фармацевтических солей и сольватов и любых комбинаций. В более типичном воплощении ингибитор ВЕR может быть выбран из группы, состоящей из метоксиамина (MX), N-этилмалеимида, O6-бензилгуанина, их фармацевтически приемлемых солей и любых комбинаций. В наиболее типичном воплощении ингибитор ВЕR может представлять собой метоксиамин (MX) или его соли.
В одном воплощении ингибитор ВЕR может представлять собой соединения, имеющие структуры формулы I:
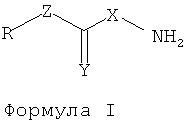
где Х представляет собой О или NH,
Y представляет собой О, S или NH,
Z отсутствует или представляет собой О, S или NH,
R представляет собой водород или углеводородную группировку.
и их фармацевтически приемлемые соли.
В некоторых воплощениях ингибитор BER можно применять для лечения пациента или субъекта, имеющего неопластическое заболевание. Например, неопластическое заболевание может представлять собой рак, выбранный из группы, состоящей из карцином, меланом, сарком, лимфом, лейкемий, астроцитом, глиом, злокачественных меланом, хронического лимфолейкоза, раковых заболеваний легкого, рака простаты, колоректальных раков, раковых заболеваний яичников, раковых заболеваний поджелудочной железы, раковых заболеваний почек, раковых заболеваний эндометрия, раковых заболеваний желудочно-кишечного тракта, раковых заболеваний печени, раковых заболеваний головы и шеи.
В некоторых воплощениях ингибитор BER можно применять для лечения пациента или индивидуума, имеющего неопластическое заболевание, которое лечится противораковым агентом.
В типичном воплощении ингибитор BER может быть выбран из группы, состоящей из метоксиамина, этопозида (VP-16, VP-16-123), мезо-4,4′-(2,3-бутандиил)-бис-(2,6-пиперазиндион) (ICRF-193, бис-диоксопиперазина), доксорубицина (DOX), амсаркина (4′,9-акридиниламинометансульфон-мета-анисидида; mAMSA), пазеллиптина, налидиксовой кислоты, оксолиновой кислоты, новобиоцина, кумермицина А1, фостриецина, тенипозида, митоксантрона, даунорубицина, N-[2-диметиламино)этил]акридин-4-карбоксамида (DACA), мербарона, хинакрина, эллиптицинов, эпиподофилотоксинов, этидия бромида, эпирубицина, пирарубицина, 3′-деамино-3′-морфолино-13-деоксо-10-гидрокси-карминомицина; 2′′,3′′-бис-пентафторфеноксиацетил-4′,6′-этилиден-бета-D-гликозида 4′-фосфат-4′-диметилэпиподофиллотоксина 2N-метил-глюкаминовой соли (F11782; фторированный липофильный эпиподофиллоид), адриамицина, актиномицина D, антрациклинов (таких как 9-амино-антрациклин), пиразолоакридин (PZA), камптотецин, топотекан, их фармацевтических солей и любых комбинаций. В более типичном воплощении ингибитор BER может быть выбран из группы, состоящей из метоксиамин (MX), N-этилмалеимида, O6-бензилгуанина, их фармацевтически приемлемых солей и любых комбинаций. В наиболее типичном воплощении ингибитор BER может представлять собой метоксиамин (MX) или его соли.
В типичном воплощении противораковый агент может быть выбран из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, 5,10-дидезаза-тетрагидрофолиевой кислоты (DDATHF), пиритрексима, ралитрекседа, GW1843 [(S)-2-[5-[(1,2-дигидро-3-метил-1-оксобензо[f]хиназолин-9-ил)метил]амино-1-оксо-2-изоиндолин]-глутаровой кислоты], их фармацевтических солей и любых комбинаций. В более типичном воплощении противораковый агент может быть выбран из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, их фармацевтических солей и любых комбинаций. В наиболее типичном воплощении противораковый агент может представлять собой пеметрексед и их фармацевтически приемлемые соли и сольваты. Например, пеметрексед может представлять собой динатриевую соль. В типичном воплощении пеметрексед может представлять собой гептагидрат динатриевой соли.
В некоторых воплощениях ингибитор BER и противораковый агент можно вводить индивидууму в комбинации. Например, ингибитор BER и противораковый агент можно вводить индивидууму вместе в парентеральной композиции. Альтернативно ингибитор BER и противораковый агент можно вводить индивидууму вместе в пероральной композиции, такой как твердая дозированная композиция.
В некоторых воплощениях ингибитор BER и противораковый агент можно вводить индивидууму последовательно, причем индивидуум сначала получает противораковый агент и затем получает ингибитор BER. Например, индивидууму может быть дан противораковый агент в парентеральной композиции, такой как внутривенная композиция, или в пероральной композиции, такой как твердая дозированная композиция, и затем дают ингибитор BER в парентеральной композиции, такой как внутривенная композиция, или в пероральной композиции, такой как твердая дозированная композиция.
Альтернативно в некоторых воплощениях ингибитор BER и противораковый агент можно вводить индивидууму последовательно, причем сначала индивидууму дают ингибитор BER и затем дают противораковый агент. Например, индивидууму может быть дан ингибитор BER в парентеральной композиции, такой как внутривенная композиция, или в пероральной композиции, такой как твердая дозированная композиция, и затем дается противораковый агент в парентеральной композиции, такой как внутривенная композиция, или в пероральной композиции, такой как твердая дозированная композиция.
В некоторых воплощения противораковый агент и ингибитор BER могут вызывать противораковый эффект более значительный, чем отдельные противораковые эффекты отдельных агентов. Например, комбинированный противораковый эффект противоракового агента и ингибитора BER может быть более значительным, чем суммарный противораковый эффект противоракового агента и ингибитора BER при индивидуальном применении.
Таким образом, соединения, полезные в качестве ингибиторов BER, такие как метоксиамин (MX), N-этилмалеимид, O6-бензилгуанин и соединения, имеющие структуры формулы I:
где Y представляет собой О, S, или NH,
Z представляет собой отсутствие или присутствие О, S, или NH, и
R представляет собой водород или углеводородную группировку,
и их фармацевтически приемлемые соли.
В однонуклеотидной BER дезоксирибозу-фосфат (dRP) в абазическом сайте удаляют посредством лиазной активности ДНК pol β. Соединения, такие как метоксиамин, взаимодействуют с альдегидом абазического сайта, делая их невосприимчивыми к стадии β-элиминации dRP-лиазного механизма, блокируя таким образом однонуклеотидную BER.
В некоторых воплощениях подходящие соединения могут предохранять субстрат АР-эндонуклеазы от подверженности расщеплению. Противораковые агенты могут действовать посредством связывания с АР-сайтами и предотвращения АРЕ-опосредованного расщепления фосфодиэфирных связей. Другие соединения, которые могут связываться с АР-сайтами и предотвращать АРЕ-опосредованное расщепление фосфодиэфирных связей, включают O-бензилгидроксиламин; этиламинооксиацетат; аминооксиуксусную кислоту; этил-амино-оксиацетат; H2N-OCHMeCO2H; карбоксиметоксиамин; аминооксиуксусную кислоту; HN=C(NH2)SCH2CH2ONH2; H2N-O(CH2)3SC(NH2)=NH; MeOC(O)CH(NH2)CH2O-NH2; H2NOCH2CH(NH2)CO2H; каналин; H2N-O(CH2)4O-NH2; O-(пара-нитробензил)гидроксиламин; 2-амино-4-(аминооксиметил)тиазол; 4-(аминооксиметил)тиазол; O,O'-(орто-фенилендиметилен)дигидроксиламин; 2,4-динитрофеноксиамин; O,O'-(мета-фенилендиметилен)дигидроксиламин; O,O'-(пара-фенилендиметилен)дигидроксиламин; Н2С=CHCH2O-NH2; H2N-O(CH2)4O-NH2; H3C(CH2)15O-NH2, 2,2'-(1,2-этандиил)бис(3-аминоокси)фумаровой кислоты диметилдиэтиловый эфир; соединения, имеющие любую из следующих структур:
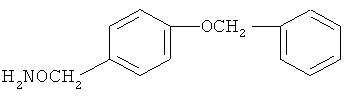
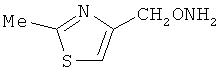
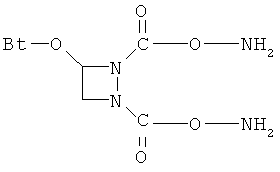
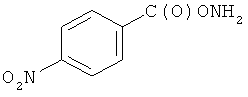
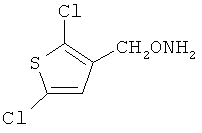
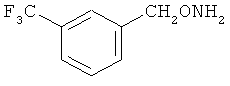
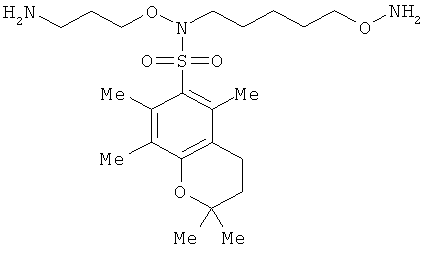
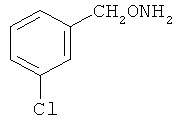
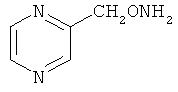
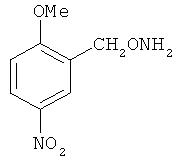
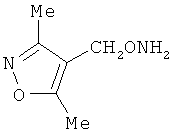
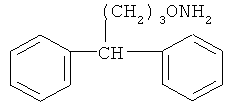
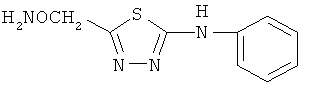
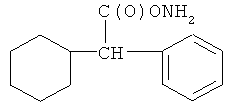
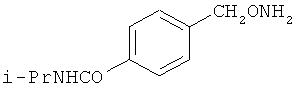
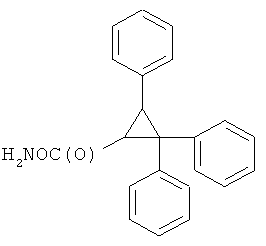
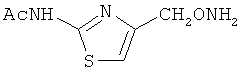
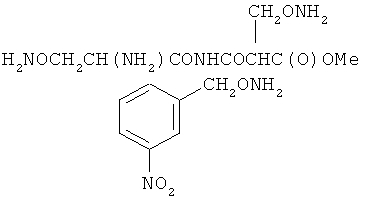
и фармацевтически приемлемые соли любого из этих соединений.
Соединения, полезные в качестве ингибиторов BER, включают ингибиторы PARP, такие как 4-амино-1,8-нафталимид (ANI), PD128763, 3-АВ, 6-AN и 8-гидрокси-2-метил-хиназолин-4-[3Н]он (NU-1025).
Соединения, полезные в качестве ингибиторов BER, включают ингибиторы ДНК-полимеразы (например, ДНК-полимераз S β, γ или ε), такие как пруназин, афидиколин, 2′,3′-дидезоксицитидина трифосфат (ddCTP), 2′,3′-дидезокситимидина трифосфат (ddTTP), 2′,3′-дидезоксиаденозина трифосфат (ddATP), 2′,3′-дидезоксигуанозина трифосфат (ddGTP), 1-бета-D-арабинофуранозилцитозин (Ara-С), кофеин, арабиноцитидин и блеомицин.
Соединения, полезные в качестве ингибиторов BER, включают ингибиторы ДНК-лигазы (например, ДНК-лигазы I, II, или III), такие как урсоловая и олеаноловая кислоты, алейритоловая кислота, протолихестериновая кислота, свертифранчесид (swertifrancheside), фулвопумерин, фагаронина хлорид и блеомицин. XRCC1 представляет собой белковый партнер ДНК-лигазы III, и ингибиторы XRCC1, такие как 3-АВ, также полезны в качестве ингибиторов BER.
Ингибиторы топоизомеразы II индуцируют расщепление ДНК и другие хромосомные аберрации, включая сестринские хроматидные обмены. Соединения, полезные в качестве ингибиторов BER, также включают ингибиторы топоизомеразы II, такие как этопозид (VP-16, VP-16-123), мезо-4,4′-(2,3-бутандиил)-бис-(2,6-пиперазиндион) (ICRF-193, бис-диоксопиперазин), доксорубицин (DOX), амсаркин (4′,9-акридиниламинометансульфон-мета-анисидид; mAMSA), пазеллиптин, налидиксовую кислоту, оксолиновую кислоту, новобиоцин, кумермицин А1, фостриецин, тенипозид, митоксантрон, даунорубицин, N-[2-диметиламино)этил]акридин-4-карбоксамид (DACA), мербарон, хинакрин, эллиптицины, эпиподофилотоксины, этидия бромид, эпирубицин, пирарубицин, 3′-дезамино-3′-морфолино-13-дезоксо-10-гидрокси-карминомицин; 2′′,3′′-бис-пентафторфеноксиацетил-4′,6′-этилиден-бета-D-гликозид 4′-фосфат-4′-диметилэпиподофиллотоксина 2N-метил-глюкаминовой соли (F11782; фторированный липофильный эпиподофиллоид), адриамицин, актиномицин D, антрациклины (такие как 9-аминоантрациклин) и пиразолоакридин (PZA). Ингибиторы топоизомеразы I, такие как камптотецин и топотекан, также можно использовать в качестве ингибиторов BER.
В некоторых воплощениях другие ингибиторы ферментов, либо известные в данной области техники, либо идентифицированные ниже, а также ингибиторы других элементов BER-пути, таких как ДНК-алкилтрансфераза, можно применять в композициях и способах, не выходя за пределы объема и сущности настоящих воплощений.
В некоторых воплощениях настоящее изобретение предполагает применение противоракового агента, такого как пеметрексед, который индуцирует образование АР-сайтов, и ингибитора BER (отличного от ингибитора топоизомеразы), такого как метоксиамин.
В типичном воплощении противораковый агент может быть выбран из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523 и триметрексата. В более типичном воплощении противораковый агент может представлять собой пеметрексед и его фармацевтически приемлемые соли. Например, пеметрексед может представлять собой динатриевую соль. В типичном воплощении пеметрексед может представлять собой гептагидрат динатриевой соли.
В некоторых воплощениях противораковый агент можно вводить в дозе от примерно 25 мг/м2 до примерно 5000 мг/м2 площади поверхности тела. Например, доза может составлять от примерно 25 мг/м2 до примерно 200 мг/м2 площади поверхности тела; доза может составлять от примерно 150 мг/м2 до примерно 500 мг/м2 площади поверхности тела; доза может составлять от примерно 400 мг/м2 до примерно 1000 мг/м2 площади поверхности тела; доза может составлять от примерно 900 мг/м2 до примерно 5000 мг/м2 площади поверхности тела; доза может составлять от примерно 200 мг/м2 до примерно 1000 мг/м2 площади поверхности тела; или доза может составлять от примерно 500 мг/м2 до примерно 600 мг/м2 площади поверхности тела. Антифолаты представляют собой неограничивающий предпочтительный класс противораковых агентов. В некоторых воплощениях противораковый агент можно выбирать из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523 и триметрексата. В более типичном воплощении противораковый агент может представлять собой пеметрексед и его фармацевтически приемлемые соли. Например, пеметрексед может представлять собой динатриевую соль. В типичном воплощении пеметрексед может представлять собой гептагидрат динатриевой соли.
В некоторых воплощения отношение ингибитора BER к противораковому агенту может составлять от примерно 1:2 до примерно 1:10000. Например, отношение ингибитора BER к противораковому агенту может составлять от примерно 1:2 до примерно 1:100; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:50 до примерно 1:500; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:450 до примерно 1:10000; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:5 до примерно 1:500; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:10 до примерно 1:50; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:15 до примерно 1:40; или отношение ингибитора BER к противораковому агенту может составлять от примерно 1:20 до примерно 1:30. В типичном воплощении ингибитор BER может быть выбран из группы, состоящей из метоксиамина (MX), N-этилмалеимида, O6-бензилгуанина, их фармацевтически приемлемых солей и любых комбинаций. В более типичном воплощении ингибитор BER может представлять собой метоксиамин (MX).
В некоторых воплощениях ингибитор BER вводят в количестве, достаточном для улучшения или усиления эффекта противоракового агента.
В типичном воплощении противораковый агент может быть выбран из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, 5,10-дидезаза-тетрагидрофолиевой кислоты (DDATHF), пиритрексима, ралитрекседа, GW1843 [(S)-2-[5-[(1,2-дигидро-3-метил-1-оксобензо[f]хиназолин-9-ил)метил]амино-1-оксо-2-изоиндолин]-глутаровой кислоты], их фармацевтических солей и любых комбинаций. В более типичном воплощении противораковый агент можно выбирать из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, их фармацевтических солей и любых комбинаций. В наиболее типичном воплощении противораковый агент может представлять собой пеметрексед и его фармацевтически приемлемые соли. Например, пеметрексед может представлять собой динатриевую соль. В типичном воплощении пеметрексед может представлять собой гептагидрат динатриевой соли.
В типичном воплощении ингибитор BER может быть выбран из группы, состоящей из метоксиамина, этопозида (VP-16, VP-16-123), мезо-4,4′-(2,3-бутандиил)-бис-(2,6-пиперазиндион) (ICRF-193, бис-диоксопиперазина), доксорубицина (DOX), амсакрина (4′,9-акридиниламинометансульфон-м-анисидида; mAMSA), пазеллиптина, налидиксовой кислоты, оксолиновой кислоты, новобиоцина, кумермицина А1, фостриецина, тенипозида, митоксантрона, даунорубицина, N-[2-диметиламино)этил]акридин-4-карбоксамида (DACA), мербарона, хинакрина, эллиптицинов, эпиподофилотоксинов, этидия бромида, эпирубицина, пирарубицина, 3′-дезамино-3′-морфолино-13-деоксо-10-гидрокси-карминомицина; 2′′,3′′-бис-пентафторфеноксиацетил-4′,6′-этилиден-бета-D-гликозида 4′-фосфат-4′-диметилэпиподофиллотоксина 2N-метил-глюкаминовой соли (F11782; фторированный липофильный эпиподофиллоид), адриамицина, актиномицина D, антрациклинов (таких как 9-аминоантрациклин), пиразолоакридина (PZA), камптотецина, топотекана, их фармацевтических солей и любых комбинаций. В более типичном воплощении ингибитор BER может быть выбран из группы, состоящей из метоксиамина (MX), N-этилмалеимида, O6-бензилгуанина, их фармацевтически приемлемых солей и любых комбинаций. В наиболее типичном воплощении ингибитор BER может представлять собой метоксиамин (MX) или его соли. Например, ингибитор BER может представлять собой метоксиамина гидрохлорид (MX).
В некоторых воплощениях отношение ингибитора BER к противораковому агенту может составлять от примерно 1:2 до примерно 1:10000. Например, отношение ингибитора BER к противораковому агенту может составлять от примерно 1:2 до примерно 1:100: отношение ингибитора BER к противораковому агенту может составлять от примерно 1:50 до примерно 1:500; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:450 до примерно 1:10000; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:5 до примерно 1:500; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:10 до примерно 1:50; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:15 до примерно 1:40; или отношение ингибитора BER к противораковому агенту может составлять от примерно 1:20 до примерно 1:30. В типичном воплощении ингибитор BER можно выбирать из группы, состоящей из метоксиамина (MX), N-этилмалеимида, O6-бензилгуанина, их фармацевтически приемлемых солей и любых комбинаций. В более типичном воплощении ингибитор BER может представлять собой метоксиамин (MX). В наиболее типичном воплощении ингибитор BER может представлять собой метоксиамин (MX), и противораковый агент может представлять собой пеметрексед. Например, пеметрексед может представлять собой динатриевую соль пеметрекседа. В типичном воплощении пеметрексед может представлять собой гептагидрат динатриевой соли.
В некоторых воплощениях предложен способ лечения рака, при котором
берут первую композицию, содержащую противораковый агент, и вторую композицию, содержащую ингибитор BER, которые можно вводить по отдельности или в виде комбинированной композиции;
выбирают субъекта, у которого диагностировано раковое заболевание, где указанный рак является устойчивым к лечению противораковым агентом одним или в комбинации с другими противораковыми агентами;
вводят указанную первую композицию и указанную вторую композицию;
где количество указанной первой композиции и количество указанной второй композиции может быть таким, что при введении указанному субъекту противораковый эффект может быть более значительным, чем противораковый эффект только одной первой композиции.
В некоторых воплощениях первая композиция может содержать противораковый агент, выбранный из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, их фармацевтических солей и любых комбинаций. В типичном воплощении противораковый агент может представлять собой пеметрексед. В некоторых воплощениях вторая композиция может содержать ингибитор BER, выбранный из группы, состоящей из метоксиамина (MX), N-этилмалеимида, O6-бензилгуанина, их фармацевтически приемлемых солей и любых комбинаций. В типичном воплощении ингибитор BER может представлять собой метоксиамин.
В некоторых воплощениях противораковый агент можно вводить в дозе от примерно 25 мг/м2 до примерно 5000 мг/м2 площади поверхности тела. Например, доза может составлять от примерно 25 мг/м2 до примерно 200 мг/м2 площади поверхности тела; доза может составлять от примерно 150 мг/м2 до примерно 500 мг/м2 площади поверхности тела; доза может составлять от примерно 400 мг/м2 до примерно 1000 мг/м2 площади поверхности тела; доза может составлять от примерно 900 мг/м2 до примерно 5000 мг/м2 площади поверхности тела; доза может составлять от примерно 200 мг/м2 до примерно 1000 мг/м2 площади поверхности тела; или доза может составлять от примерно 500 мг/м2 до примерно 600 мг/м2 площади поверхности тела. В некоторых воплощениях противораковый агент может быть выбран из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523 и триметрексата. В более типичном воплощении противораковый агент может представлять собой пеметрексед и его фармацевтически приемлемые соли. Например, пеметрексед может представлять собой динатриевую соль. В типичном воплощении пеметрексед может представлять собой гептагидрат динатриевой соли.
В некоторых воплощениях отношение ингибитора BER к противораковому агенту может составлять от примерно 1:2 до примерно 1:10000. Например, отношение ингибитора BER к противораковому агенту может составлять от примерно 1:2 до примерно 1:100; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:50 до примерно 1:500; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:450 до примерно 1:10000; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:5 до примерно 1:500; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:10 до примерно 1:50; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:15 до примерно 1:40; или отношение ингибитора BER к противораковому агенту может составлять от примерно 1:20 до примерно 1:30. В типичном воплощении ингибитор BER можно выбирать из группы, состоящей из метоксиамина (MX), N-этилмалеимида, O6-бензилгуанина, их фармацевтически приемлемых солей и любых комбинаций. В более типичном воплощении ингибитор BER может представлять собой метоксиамин (MX).
В некоторых воплощениях предложен способ лечения рака, при котором берут первую композицию, содержащую противораковый агент, и вторую композицию, содержащую ингибитор BER, которые можно вводить по отдельности или в виде комбинированной композиции:
выбирают субъекта, у которого диагностировано раковое заболевание;
вводят указанную первую композицию и указанную вторую композицию;
где количество указанной первой композиции и количество указанной второй композиции может быть таким, что при введении указанному субъекту противораковый эффект может быть значительно больше, чем суммарный противораковый эффект первой композиции, содержащей противораковый агент, и второй композиции, содержащей ингибитор BER.
В некоторых воплощениях первая композиция может содержать противораковый агент, выбранный из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523, триметрексата, аминоптерина, их фармацевтических солей и любых комбинаций. В типичном воплощении противораковый агент может представлять собой пеметрексед. В некоторых воплощениях вторая композиция может содержать ингибитор BER, выбранный из группы, состоящей из метоксиамина (MX), N-этилмалеимида, O6-бензилгуанина, их фармацевтически приемлемых солей и любых комбинаций. В типичном воплощении ингибитор BER может представлять собой метоксиамин.
В некоторых воплощениях противораковый агент можно вводить в дозе от примерно 25 мг/м2 до примерно 5000 мг/м2 площади поверхности тела. Например, доза может составлять от примерно 25 мг/м2 до примерно 200 мг/м2 площади поверхности тела; доза может составлять от примерно 150 мг/м2 до примерно 500 мг/м2 площади поверхности тела; доза может составлять от примерно 400 мг/м2 до примерно 1000 мг/м2 площади поверхности тела; доза может составлять от примерно 900 мг/м2 до примерно 5000 мг/м2 площади поверхности тела; доза может составлять от примерно 200 мг/м2 до примерно 1000 мг/м2 площади поверхности тела; или доза может составлять от примерно 500 мг/м2 до примерно 600 мг/м2 площади поверхности тела. В некоторых воплощениях противораковый агент может быть выбран из группы, состоящей из пеметрекседа, капецитабина, эдатрексата, метотрексата, лометрексола, нолатрекседа, ралитрекседа, РТ523 и триметрексата. В более типичном воплощении противораковый агент может представлять собой пеметрексед и его фармацевтически приемлемые соли. Например, пеметрексед может представлять собой динатриевую соль. В типичном воплощении пеметрексед может представлять собой гептагидрат динатриевой соли.
В некоторых воплощениях отношение ингибитора BER к противораковому агенту может составлять от примерно 1:2 до примерно 1:10000. Например, отношение ингибитора BER к противораковому агенту может составлять от примерно 1:2 до примерно 1:100; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:50 до примерно 1:500; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:450 до примерно 1:10000; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:5 до примерно 1:500: отношение ингибитора BER к противораковому агенту может составлять от примерно 1:10 до примерно 1:50; отношение ингибитора BER к противораковому агенту может составлять от примерно 1:15 до примерно 1:40; или отношение ингибитора BER к противораковому агенту может составлять от примерно 1:20 до примерно 1:30. В типичном воплощении ингибитор BER можно выбирать из группы, состоящей из метоксиамина (MX), N-этилмалеимида, O6-бензилгуанина, их фармацевтически приемлемых солей и любых комбинаций. В более типичном воплощении ингибитор BER может представлять собой метоксиамин (MX).
Еще одним аспектом настоящего воплощения является неожиданная демонстрация того, что данные, что некоторые ингибиторы BER действуют синергически в комбинации с некоторыми антифолатами с неожиданным реверсированием устойчивости к некоторым антифолатам. Таким образом, в неограничивающих предпочтительных воплощениях антиметаболический противораковый агент представляет собой антифолатный противораковый агент. Эти антифолаты прерывают фолат-зависимые метаболические процессы, существенные для клеточной репликации. Антифолаты отличаются от других химиотерапевтических средств тем, что они действуют, прерывая клеточные процессы, вовлеченные в фолатный метаболизм. Это действие включает ингибирование фолат-зависимых ферментов, включая тимидилатсинтазу (TS), но не ограничиваясь ей. Прерывание фолат-зависимых процессов приводит к неправильной репликации ДНК и апоптозу быстро делящихся клеток, включая раковые клетки. В одном воплощении отношение MX к антиметаболитному противораковому агенту составляет от примерно 1:5 до 1:500. В некоторых воплощениях отношение MX к антиметаболитному противораковому агенту составляет от примерно 1:10 до примерно 1:100, от примерно 1:25 до примерно 1:75, от примерно 1:15 до примерно 1:40, или от примерно 1:20 до примерно 1:30. Кроме того, второй противораковый агент может быть введен до или после комбинирования MX и антиметаболитного противоракового агента.
Фармацевтические композиции
Следует понимать, что композиции, предложенные в данном описании изобретения, могут находиться в любой форме, которая позволяет вводить композицию пациенту. Например, композиция может быть в форме твердого тела, жидкости или газа (например, аэрозоля). Другие подходящие способы введения включают, без ограничения ими, пероральный, местный, парентеральный (например, сублингвально или буккально), сублингвальный, ректальный, вагинальный и интраназальный. Термин парентеральный при использовании в данном описании изобретения включает подкожные инъекции, внутривенную, внутримышечную, интрастернальную, внутрикавернозную, интратекальную, внутриканальную, внутриуретральную инъекцию или инфузионные способы. Фармацевтическую композицию готовят в виде препарата таким образом, чтобы обеспечить биодоступность активных ингредиентов, содержащихся в ней, при введении композиции пациенту. Композиции, которые подлежат введению пациенту, принимают форму одной или более дозированных единиц, где, например, таблетка может представлять собой однократную дозированную единицу, и контейнер для одного или больше соединений по изобретению в аэрозольной форме может заключать в себе множество дозированных единиц.
В еще одном аспекте настоящее описание относится к фармацевтической композиции, содержащей физиологически приемлемые поверхностно-активные агенты, носители, разбавители, эксципиенты, сглаживающие агенты, суспензионные агенты, пленкообразующие вещества и покрывающие вспомогательные вещества или их комбинацию; и соединение, раскрытое в данном описании изобретения. Приемлемые носители или разбавители для терапевтического использования хорошо известны в фармацевтической области и описаны, например, в Remington′s Pharmaceutical Sciences, 18th ed., Mack Publishing Co., Easton, PA (1990), который включен в данное описание изобретения посредством ссылки во всей его полноте. Консерванты, стабилизаторы, красители, подсластители, ароматизаторы, вкусоароматизирующие агенты и тому подобное могут быть представлены в фармацевтической композиции. Например, бензоат натрия, аскорбиновая кислота и эфиры пара-гидроксибензойной кислоты могут быть добавлены в качестве консервантов. Кроме того, можно использовать антиоксиданты и суспендирующие агенты. В различных воплощениях можно использовать в качестве поверхностно-активных агентов спирты, эфиры, сульфатированные алифатические спирты и тому подобное; в качестве эксципиентов можно использовать сахар, глюкозу, лактозу, крахмал, кристаллическую целлюлозу, маннит, легкий безводный силикат, алюминат магния, алюмометасиликат магния, синтетический силикат алюминия, карбонат кальция, гидрокарбонат натрия, гидрофосфат кальция, карбоксиметилцеллюлозу кальция и тому подобное; стеарат магния, тальк, гидрогенизированный жир и тому подобное можно использовать в качестве сглаживающих агентов; кокосовое масло, оливковое масло, кунжутное масло, арахисовое масло и сою можно использовать в качестве суспензионных агентов или смазывающих веществ; в качестве суспензионных агентов можно использовать ацетатфталат целлюлозы в виде производного углевода, такого как целлюлоза или сахар, или метилацетат-метакрилатный сополимер в виде производного поливинила; и пластификаторы, такие как эфиры фталаты и тому подобное, можно использовать в качестве суспензионных агентов.
Термин "фармацевтическая композиция" относится к смеси соединения, раскрытого в данном описании изобретения, с другими химическими компонентами, такими как разбавители или носители. Фармацевтическая композиция облегчает введение соединения в организм. В данной области техники существуют многочисленные способы введения соединения, включая, но не ограничиваясь ими, пероральное, инъекционное, аэрозольное, парентеральное и местное введение. Фармацевтические композиции также могут быть получены с помощью взаимодействия соединений с неорганическими или органическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфоновая кислота, салициловая кислота и тому подобное.
Термин "носитель" определяет химическое соединение, которое облегчает включение соединения в клетки или ткани. Например, диметилсульфоксид (DMSO) является обычно используемым носителем, так как он облегчает поглощение многих органических соединений клетками или тканями организма.
Термин "разбавитель" определяет химические соединения, разбавленные водой, которая растворяет представляющие интерес соединения, а также стабилизирует биологически активную форму соединения. Соли, растворенные в буферизованных растворах, используют в качестве разбавителей в данной области техники. Одним обычно используемым буферизованным раствором является фосфатно-солевой буферный раствор, так как он имитирует солевые условия крови человека. Так как буферные соли могут контролировать рН раствора при низких концентрациях, буферизованный разбавитель редко изменяет биологическую активность соединения.
Термин "физиологически приемлемые" определяет носитель или разбавитель, которые не аннулируют биологическую активность и свойства соединения.
Фармацевтические композиции, описанные в данном описании изобретения, можно вводить пациенту-человеку сами по себе, или в фармацевтических композициях, где их смешивают с другими активными ингредиентами, как в комбинированной терапии, или с подходящими носителями или эксципиентом (эксципиентами). Способы приготовления в виде препарата и введения соединений по настоящей заявке можно найти в "Remington's Pharmaceutical Sciences," Mack Publishing Co., Easton, PA, 18-е издание, 1990.
Подходящие способы введения могут, например, включать пероральное, ректальное, трансмукозальное, местное или интестинальное введение; парентеральную доставку, включая внутримышечные, подкожные, внутривенные, интрамедуллярные инъекции, а также интратекальные, прямые внутрижелудочковые, внутрибрюшинные, интраназальные или внутриглазные инъекции. Соединения также можно вводить в виде лекарственных форм с замедленным или контролируемым высвобождением, включая депо-инъекции, осмотические насосы, таблетки, трансдермальные (включая электроперенос) пластыри и тому подобное, для пролонгированного и/или регулируемого по времени, пульсирующего введения с заранее установленной скоростью.
Фармацевтические композиции по настоящему изобретению могут быть изготовлены способом, который известен сам по себе, например, с помощью обычного смешивания, растворения, гранулироввания, изготовления драже, растирания в порошок, эмульгирования, инкапсулирования, процессов захвата или таблетирования.
Таким образом, фармацевтические композиции для применения в соответствии с настоящим изобретением могут быть изготовлены в виде препарата обычным способом с использованием одного или более физиологически приемлемых носителей, содержащих эксципиенты и вспомогательные вещества, которые облегчают переработку активных соединений в препараты, которые можно использовать фармацевтически. Подходящая композиция зависит от выбранного способа введения. Любые хорошо известные способы, носители и эксципиенты можно использовать в качестве подходящих и как понимается в данной области техники, например в Remington's Pharmaceutical Sciences выше.
Годные для инъекции композиции можно получить в обычных формах или в виде жидких растворов или суспензий, твердых формах, подходящих для раствора или суспензии в жидкости перед инъецированием, или в виде эмульсий. Подходящими эксципиентами являются, например, вода, физиологический раствор, декстроза, маннит, лактоза, децитин, альбумин, глутамат натрия, цистеина гидрохлорид и тому подобное. Кроме того, если требуется, инъецируемые фармацевтические композиции могут содержать минорные количества нетоксичных вспомогательных веществ, таких как увлажняющие агенты, буферизующие рН агенты и тому подобное. Физиологически совместимые буферы включают, но без ограничения ими, раствор Ханка, раствор Рингера или физиологический раствор. Если требуется, можно использовать препараты, усиливающие абсорбцию (например, липосомы).
Для трансмукозального введения в композиции можно использовать проникающие жидкости, подходящие к барьеру, через который надо проникать.
Фармацевтические композиции для парентерального введения, например посредством болюсной инъекции или непрерывной инфузии, содержат водные растворы активных соединений в водорастворимой форме. Кроме того, суспензии активных соединений можно приготовить в виде подходящих масляных инъецируемых суспензий. Подходящие липофильные растворители или среды включают жирные кислоты, такие как кунжутное масло, или другие органические масла, такие как соевое, грейпфрутовое или миндальное масла, или синтетические эфиры жирных кислот, такие как этилолеат, или триглицериды, или липосомы. Водные инъецируемые суспензии могут содержать вещества, которые увеличивают вязкость суспензии, такие как натрия карбоксиметилцеллюлоза, сорбит или декстран. Возможно суспензия также может содержать подходящие стабилизаторы или агенты, которые увеличивают растворимость соединений, чтобы обеспечить возможность получения растворов с высокой концентрацией. Композиции для инъекции могут быть представлены в стандартной дозированной форме, например в ампулах или в многодозовых контейнерах с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных средах, и могут содержать препаратообразующие агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент перед использованием может находиться в форме порошка для разбавления подходящей средой, например стерильной апирогенной водой.
Для перорального введения соединения могут быть легко приготовлены в виде препарата путем комбинирования активных соединений с фармацевтически приемлемыми носителями, хорошо известными в данной области техники. Такие носители обеспечивают возможность приготовить соединения по изобретению в виде препарата, такого как таблетки, пилюли, драже, капсулы, жидкости, гели, сиропы, пасты, суспензии и тому подобное, для перорального приема внутрь пациентом, подлежащим лечению. Фармацевтические препараты для перорального применения можно получать путем комбинирования активных соединений с твердым эксципиентом, возможно растирая полученную смесь и обрабатывая смесь гранул после добавления подходящих вспомогательных веществ, если требуется, с получением таблеток или ядер драже. Подходящими эксципиентами являются, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; препараты целлюлозы, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрия карбоксиметилцеллюлоза и/или поливинилпирролидон (PVP). Если требуется, могут быть добавлены разрыхляющие агенты, такие как сшитый поливинипирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия. Ядра драже предложены с подходящими покрытиями. Для этой цели можно использовать концентрированные растворы сахара, которые возможно могут содержать аравийскую камедь, тальк, поливинипирролидон, карбопол, полиэтиленгликоль, и/или диоксид титана, лакирующие растворы и подходящие органические растворители или смеси растворителей. Красители или пигменты могут быть добавлены к покрытиям таблеток или драже для идентификации или для различения разных комбинаций доз активного соединения. С этой целью можно использовать концентрированные растворы сахара, которые возможно могут содержать аравийскую камедь, тальк, поливинилпирролидон, карбопол, полиэтиленгликоль, и/или диоксид титана, лакирующие растворы, и подходящие органические растворители или смеси растворителей. Красители или пигменты могут быть добавлены к покрытиям таблеток или драже для идентификации или для различения разных комбинаций доз активного соединения.
Фармацевтические препараты, которые можно применять перорально, включают плотно заполненные капсулы, изготовленные из желатина, а также мягкие, укупоренные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Плотно заполненные капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующие вещества, такие как крахмаль), и/или смазывающие вещества, такие как тальк или стеарат магния, и возможно стабилизаторы. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того можно добавлять стабилизаторы. Все композиции для перорального введения должны быть в количествах, подходящих для такого введения.
Композиции для буккального введения могут принимать форму таблеток или пастилок, изготовленных обычным способом.
Для введения посредством ингаляции соединения для применения по настоящему изобретению удобно доставлять в форме подаваемого аэрозольного спрея из находящейся под давлением упаковки или небулайзера, с использованием подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае находящегося под давлением аэрозоля единица дозировки может определяться с помощью клапана для выпускания отмеренного количества. Капсулы и ампулы из, например, желатина для использования в ингаляторе или инсуффляторе могут быть приготовлены содержащими порошковую смесь соединения и подходящей порошковой основы, такой как лактоза или крахмал.
Кроме того, в данном описании изобретения раскрыты различные фармацевтические композиции, хорошо известные в фармацевтической области для применений, которые включают внутриглазную, интраназальную и внутриушную доставку. Подходящие проникающие вещества для этих применений в основном известны в данной области техники. Фармацевтические композиции для внутриглазной доставки включают водные глазные растворы активных соединений в водорастворимой форме, такой как глазные капли, или в геллановой камеди (Shedden et al., Clin. Ther., 23(3):440-50 (2001)) или гидрогели (Mayer et al., Ophtalmologica, 210(2):101-3 (1996)); глазные мази; глазные суспензии, такие как микрочастицы, содержащее лекарственное средство небольшие полимерные частицы, которые суспендированы в жидкой среде-носителе (Joshi, A., J. Ocul. Pharmacol., 10(1):29-45 (1994)), липидорастворимые композиции (Alm et al., Prog. Clin. Biol. Res., 312:447-58 (1989)), и микросферы (Mordenti, Toxicol. Sci., 52(1):101-6 (1999)); и окулярные вкладки. Все из указанных выше ссылок включены в данное описание изобретения посредством ссылки во всей их полноте. Такие подходящие фармацевтические композиции чаще всего и предпочтительно приготавливают стерильными, изотоническими и буферизованными для стабильности и комфорта. Фармацевтические композиции для интраназальной доставки также могут включать капли и спреи, часто получаемые для имитирования во многих отношениях назальных секретов, чтобы обеспечить сохранение нормального цилиарного эффекта. Как раскрыто в Remington's Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), которое включено в данное описание изобретения посредством ссылки во всей его полноте и хорошо известно специалистам в данной области техники, подходящие композиции наиболее часто и предпочтительно являются изотоническими, слегка буферизованными для поддержания рН от 5,5 до 6,5, и наиболее часто и предпочтительно содержат противомикробные консерванты и подходящие стабилизаторы лекарственного средства. Фармацевтические композиции для внутриаурикулярной доставки включают суспензии и мази для местного нанесения в ухо. Обычные растворители для таких ушных композиций содержат глицерин и воду.
Соединения могут также быть приготовлены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, например содержащие обычные суппозиторные основы, такие как какао-масло или другие глицериды.
В дополнение к композициям, описанным ранее, соединения также могут быть приготовлены в виде депо-препарата. Такие композиции с длительным действием могут быть введены путем имплантации (например, подкожно или внутримышечно) или посредством внутримышечной инъекции. Таким образом, например, соединения могут быть приготовлены в виде препарата с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или с ионообменными смолами, или в виде труднорастворимых производных, например, таких как труднорастворимая соль.
Для гидрофобных соединений подходящий фармацевтический носитель может представлять собой систему сорастворителей, содержащую бензиловый спирт, неполярное поверхностно-активное вещество, смешивающийся с водой органический полимер и водную фазу. Обычно используемой системой сорастворителей является система сорастворителей VPD, которая представляет собой раствор 3% масс./об. бензилового спирта, 8% масс./об. неполярного поверхностно-активного вещества Polysorbate 80™ и 65% масс./об. полиэтиленгликоля 300, доведенный до нужного объема абсолютным этанолом. Конечно, пропорции в системе сорастворителей могут существенно варьироваться без аннулирования их характеристик растворимости и токсичности. Кроме того, характеристики компонентов сорастворителей могут варьироваться: например, могут использоваться другие низкотоксичные неполярные поверхностно-активные вещества вместо POLYSORBATE 80™; может варьироваться фракционный размер полиэтиленгликоля; другие биосовместимые полимеры, например поливинилпирролидон, могут заменять полиэтиленгликоль; и другие сахара или полисахариды могут замещать декстрозу.
Альтернативно для гидрофобных фармацевтических соединений можно использовать другие системы доставки. Липосомы и эмульсии являются хорошо известными примерами средств доставки или носителей для гидрофобных лекарственных средств. Также можно использовать некоторые органические растворители, такие как диметилсульфоксид, хотя обычно за счет большей токсичности. Кроме того соединения могут быть доставлены с использованием системы с замедленным высвобождением, такой как полупроницаемые матрицы твердых гидрофобных полимеров, содержащие терапевтический агент. Были созданы и хорошо известны специалистам в данной области техники различные вещества для замедленного высвобождения. Капсулы с замедленным высвобождением могут в зависимости от их химической природы высвобождать соединения в течение от нескольких недель вплоть до свыше 100 суток. В зависимости от химической природы и биологической стабильности терапевтического реагента можно использовать дополнительные стратегии стабилизации белка.
Агенты, предназначенные для внутриклеточного введения, могут быть введены с использованием методик, хорошо известных специалистам в данной области техники. Например, такие агенты могут быть инкапсулированы в липосомы. Все молекулы, присутствующие в водном растворе в момент образования липосомы, включены в водное внутреннее пространство. Липосомальное содержимое и защищено от наружной микросреды, и так как липосомы сливаются с клеточными мембранами, эффективно доставляется в клеточную цитоплазму. Липосома может быть покрыта ткане-специфичным антителом. Липосомы будут нацелены на нужный орган и избирательно захватываться им. Альтернативно, небольшие гидрофобные органические молекулы могут быть непосредственно введены внутриклеточно.
В фармацевтические композиции могут быть включены дополнительные терапевтические или диагностические агенты. Альтернативно или дополнительно фармацевтические композиции можно комбинировать с другими композициями, которые содержат другие терапевтические или диагностические агенты.
Способы введения
Соединения или фармацевтические композиции могут быть введены пациенту любыми подходящими способами. Неограничивающие примеры способов введения включают среди прочих (а) введение пероральными путями, причем введение включает введение в капсуле, таблетке, грануле, спрее, сиропе или других подобных формах; (б) введение непероральными путями, такими как ректальный, вагинальный, внутриуретральный, внутриглазной, интраназальный или внутриаурикулярный, причем введение включает введение в виде водной суспензии, масляного препарата или подобного, или в виде капель, спрея, суппозитория, крема, мази или подобного; (в) введение посредством инъекции подкожно, внутрибрюшинно, внутривенно, внутримышечно, внутрикожно, внутриглазнично, внутрисуставно, интраспинально, интрастернально или подобно, включая доставку инфузионным насосом; (г) введение локально, такое как посредством инъекции непосредственно в область почек или сердца, например, с помощью депо-имплантации; а также (д) введение местно; как кажется целесообразным специалистами в данной области техники для приведения соединения по изобретению в контакт с живой тканью.
Фармацевтические композиции, подходящие для введения, включают композиции, где активные ингредиенты содержатся в количестве, эффективном для достижения их предназначения. Терапевтически эффективное количество соединений, раскрытых в данном описании изобретения, требующееся в качестве дозы, будет зависеть от способа введения, вида животного, включая человека, подлежащего лечению, и физических характеристик конкретного рассматриваемого животного. Доза может быть рассчитана для достижения требуемого эффекта, но будет зависеть от таких факторов, как масса, рацион, сопутствующего лечения и других факторов, которые узнают специалисты в медицинской области. Более конкретно, терапевтически эффективное количество означает количество соединения, эффективное для предупреждения, смягчения или облегчения симптомов заболевания или продлевания жизни субъекта, подлежащего лечению. Определение терапевтически эффективного количества действительно находится в рамках квалификации специалистов в данной области техники, особенно с учетом подробного описания, предложенного в данном описании изобретения.
Специалисту в данной области техники совершенно очевидно, что полезная in vivo вводимая дозировка и конкретный способ введения будет варьироваться в зависимости от возраста, массы и вида млекопитающего, подлежащего лечению, конкретных используемых соединений и конкретной пользы, для которой используют данные соединениям. Определение эффективных уровней дозировки, которые являются уровнями дозировки, необходимыми для достижения требуемого результата, может быть выполнено специалистом в данной области техники с использованием обычных фармакологических способов. Обычно клинические применения продуктов к человеку начинают при более низких уровнях дозировки, с увеличением уровня дозировки вплоть до достижения нужного эффекта. Альтернативно, приемлемые in vitro исследования можно использовать для установления полезных доз и путей введения композиций, идентифицированных с помощью настоящих способов с использованием установленных фармакологических способов.
В исследованиях не на человеке применение потенциальных продуктов начинают при более высоких уровнях дозировки, с уменьшением дозировки вплоть до того, когда желательный эффект более не достигается или до исчезновения вредных побочных эффектов. Дозировка может широко меняться в зависимости от требуемых эффектов и терапевтического показания. Обычно дозировки могут составлять от примерно 10 микрограмм/кг до 100 мг/кг массы тела, предпочтительно от примерно 100 микрограмм/кг до 10 мг/кг массы тела. Альтернативно дозировки могут быть основаны и рассчитаны на основании площади поверхности пациента, как понимается специалистами в данной области техники.
Точное приготовление, способ введения и дозировка фармацевтических композиций по настоящему изобретению могут быть выбраны лечащим врачом с учетом состояния пациента (см. например, Fingl et al. 1975, в "The Pharmacological Basis of Therapeutics", который включен в данное описание изобретения посредством ссылки во всей его полноте, с конкретной ссылкой к гл.1, стр.1). Обычно диапазон доз композиции, вводимых пациенту, может составлять от примерно 0,5 до 1000 мг/кг массы тела пациента. Дозировка может быть однократной или серией двух или более, даваемым курсом из одного или более дней, как необходимо пациенту. В случаях, когда дозировки соединений для человека были установлены для по меньшей мере некоторых состояний, в настоящем изобретении применяются те же самые дозировки, или дозировки, которые составляют примерно 0,1%-500%, более предпочтительно примерно 25%-250% установленной дозировки для человека. Если дозировка для человека не установлена, как будет в случае вновь открытых фармацевтических соединений, подходящая дозировка для человека может быть выведена из значений ED50 или ID50, или других подходящих значений, полученных из исследований in vitro или in vivo, как пригодная в исследованиях токсичности и исследованиях эффективности на животных.
Следует отметить, что лечащий врач должен знать как и когда прекратить, прервать или скорректировать введение из-за токсичности или дисфункций органов. С другой стороны, лечащий врач также должен знать как скорректировать лечение до более высоких уровней, если клинический ответ не был адекватным (исключая токсичность). Величина введенной дозы в управлении интересующим нарушением будет варьироваться в зависимости от тяжести состояния, подлежащего лечению, и от способа введения. Тяжесть состояния может, например, частично может быть оценена с помощью стандартных прогностических методов анализа. Кроме того, доза и, возможно, частота доз также будут варьироваться в зависимости от возраста, массы тела и иммунного ответа отдельного пациента. Программа, сравнимая с обсуждаемой выше, может использоваться в ветеринарной медицине.
Хотя точную дозировку будут определять в зависимости от конкретного лекарственного средства, в большинстве случаев могут быть сделаны некоторые обобщения, касающиеся дозировки. Суточная схема приема для взрослого пациента-человека может представлять собой, например, пероральную дозу от 0,1 мг/м2 до 2000 мг/м2 площади поверхности тела в сутки каждого активного ингредиента, обычно от 1 мг/м2 до 500 мг/м2 площади поверхности тела в сутки, например от 5 мг/м2 до 200 мг/м2 площади поверхности тела в сутки. В других воплощениях можно применять внутривенную, подкожную или внутримышечную дозу каждого активного ингредиента от 0,01 мг/м2 до 100 мг/м2 площади поверхности тела в сутки, обычно от 0,1 мг/м2 до 60 мг/м2 площади поверхности тела в сутки, например от 1 мг/м2 до 40 мг/м2 площади поверхности тела в сутки. В случаях введения фармацевтически приемлемой соли дозировки можно рассчитывать на свободное основание. В некоторых воплощениях композицию вводят 1-4 раза в сутки. Альтернативно композиции по изобретению могут быть введены посредством непрерывной внутривенной инфузии, предпочтительно с дозой каждого активного ингредиента вплоть до 1000 мг/м2 площади поверхности тела в сутки. Специалистам в данной области техники будет понятно, что в конкретных ситуациях может быть необходимо вводить соединения, раскрытые в данном описании изобретения, в количествах, которые превышают или даже значительно превышают вышеуказанный предпочтительный диапазон дозировок, чтобы эффективно и активно лечить особенно агрессивные заболевания или инфекции. В некоторых воплощениях соединения будут вводить во время периода непрерывного лечения, например в течение недели или более, или в течение месяцев или лет.
Величину дозировки и интервал можно регулировать индивидуально, чтобы получить концентрации активной группировки в плазме, достаточные для поддержания модулирующих эффектов, или минимальную эффективную концентрацию (МЕС). МЕС будет варьироваться для каждого соединения, но может быть определена из данных in vitro. Дозировки, необходимые для достижения МЕС, будут зависеть от индивидуальных характеристик и способа введения. Однако для определения концентраций в плазме можно использовать анализ HPLC или биоанализ.
Интервалы дозирования также могут быть определены с использованием значения МЕС. Композиции следует вводить, используя режим, который поддерживает уровни в плазме выше МЕС в течение 10-90% времени, обычно 30-90% и наиболее типично 50-90%.
В случаях местного введения или селективного захвата эффективная местная концентрация лекарственного средства может не быть связанной с концентрацией в плазме.
Количество введенной композиции может зависеть от субъекта, подлежащего лечению, массы субъекта, тяжести заболевания, способа введения и суждения назначающего лечение врача.
Соединения, раскрытые в данном описании изобретения, можно оценивать в отношении эффективности и токсичности, используя известные способы. Например, токсикология конкретного соединения или подгруппы соединений, имеющих некоторые общие химические фрагменты, может быть установлена путем определения токсичности in vitro в отношении клеточной линии, такой как клеточная линия млекопитающего и предпочтительно человека. Результаты таких исследований часто являются прогностическими в отношении токсичности для животных, таких как млекопитающие, или более конкретно для людей. Альтернативно, токсичность конкретных соединений в животной модели, такой как мыши, крысы, кролики или обезьяны, можно определить, используя известные способы. Эффективность конкретного соединения может быть установлена с использованием различных общепризнанных методов, таких как методы in vitro, животные модели или клинические исследования на людях. Общепризнанные in vitro модели существуют для почти каждого класса состояния, включая, но не ограничиваясь ими, рак, сердечно-сосудистое заболевание и различные иммунные дисфункции. Аналогично, приемлемые животные модели можно использовать для установления эффективности химических веществ при лечении таких состояний. При выборе модели для определения эффективности специалист в данной области техники может руководствоваться предшествующим уровнем техники для выбора подходящей модели, дозы и способа введения и режима. Конечно, для определения эффективности соединения у человека также можно использовать клинические исследования на людях.
Композиции могут быть представлены, если требуется, в упаковке или распыляющем устройстве, которое может содержать одну или более чем одну дозированную лекарственную форму, содержащую активный ингредиент. Упаковка может, например, содержать металлическую или пластиковую фольгу, примером является блистерная упаковка. Упаковка или распыляющее устройство может сопровождаться инструкциями по введению. Упаковка или распылитель также могут сопровождаться уведомлением, связанным с контейнером, в форме, установленной государственным учреждением, регулирующим изготовление, применение или продажу фармацевтических средств, причем уведомление отражает одобрение этим учреждением этой формы лекарственного средства для введения человеку или применения в ветеринарии. Такое уведомление, например, может представлять собой этикетку, утвержденную Управлением по контролю качества продуктов и лекарственных средств США для отпускаемых по рецепту лекарственных средств, или утвержденный вкладыш. Также можно получить композиции, содержащие соединение по изобретению, приготовленные в виде препарата в совместимом фармацевтическом носителе, помещенные в подходящий контейнер, и маркированные для лечения указанного состояния.
На протяжении всего описания изобретения любое упоминание конкретного соединения следует понимать как охватывающее это соединение и его любые (другие) фармацевтически приемлемые соли.
На первой стадии BER ряд глюкозидаз распознает неправильные основания, такие как N3 mA и N7 mG (O′Connor et al. "Isolation and structure of a cDNA expressing a mammalian 3-methyladenine-DNA glycosylase" EMBO J. 9:3337-3342, 1990; Samson et al. "Cloning and characterization of a 3-methyladenine DNA glycosylase с DNA from human cells whose gene maps to chromosome 16" Proc. Natl. Acad. Sci. USA 88:9127-9131, 1991), ошибочное спаривание оснований T:G (Neddermann et al. "Functional expression of soluble human interleukin-11 (IL-11) receptor alpha and stoichiometry of in vitro IL-11 receptor complexes with gp130" J. Biol. Chem. 271:12767-12774, 1996), и дезаминированные основания, такие как гипоксантин/окисленный 8-оксо-7,8-дигидрогуанин или урацил: A (Vollberg et al. "Isolation and characterization of the human uracil DNA glycosylase gene" Proc. Natl. Acad. Sci. USA 86: 8693-8697, 1989; Olsen et al. "Molecular cloning of human uracil-DNAglycosylase, a highly conserved DNA repair enzyme" EMBO J. 8:3121-3125. 1989; Radicella et al. "Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae" Proc. Natl. Acad. Sci. USA 94.8010-8015, 1997; Rosenquist et al. "Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase" Proc. Natl. Acad. Sci. USA 94:7429-7434, 1997). После ферментативного или самопроизвольного гидролиза N-гликозидной связи и высвобождения неправильного основания, АР (апуриновая/апиримидиновая) эндонуклеаза гидролизует фосфодиэфирный остов 5′ до повреждения, и dRpase (ДНК-дезоксирибофосфодиэстераза и ее активность связана с полимеразой β) удаляет остаточную dRp, генерируя пропуск одного нуклеотида. ДНК-полимераза β заполняет пропуск, и ДНК-лигаза сшивает одноцепочечный разрыв. Такой путь называется BER короткими последовательностями. Альтернативный путь для BER включает синтез ДНК для заполнения разрыва из 2-13 нуклеотидов. Такая репарация длинными последовательностями нуждается в ядерном антигене пролиферирующих клеток (PCNA) и PCNA-зависимой ДНК-полимеразе (Wilson "Mammalian base excision repair and DNA polymerase beta" Mutation Res. 407:203-215, 1998).
Поли-(АДФ-рибоза)-полимераза (РАRP) действует в качестве сенсора одноцепочечного разрыва нитей ДНК самостоятельно или посредством взаимодействия с XRCC1 и вовлечена в BER. PARP связывает поврежденную ДНК, что приводит к авторибозилированию. Затем модифицированный белок высвобождается и позволяет другим белкам достичь и репарировать разрывы нитей ДНК (Wilson "Mammalian base excision repair и DNA polymerase beta" Mutation Res. 407:203-215, 1998; Molinete et al. "Over production of the poly (ADP-ribose) polymerase DNA-binding domain blocks alkylation-induced DNA repair synthesis in mammalian cells" EMBO J. 12:2109-2117, 1993; Caldecott et al. "XRCCI polypeptide interacts with DNA polymerase β and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular 'nick-sensor' in vitro" Nucleic Acids Res. 24:4387-4394, 1996). Таким образом, PARP участвует в BER после образования одноцепочечного разрыва как в репарации короткими, так и длинными последовательностями. Она оказывается наиболее активной в альтернативном (репарация длинными последовательностями) пути BER. На Фиг.6 показано, что комбинация пеметрекседа и MX усиливает образование разрезанной PARP. Увеличение разрывов двойных нитей ДНК и апоптоз не зависят от пути Bcl-2.
В общем случае, терминология, употребляемая ниже, и лабораторные методики для клеточной культуры, тканевой культуры, биологии опухолей и молекулярной генетики, описанные ниже, хорошо известны и обычно используются в данной области техники. Общепринятые методики используют для методов культивирования клеток, дизайна эксперимента и изготовления соединения в виде препарата и терминологии. В общем случае химические взаимодействия и стадии очистки осуществляют согласно инструкциям производителя. Методики и методы в общем случае выполняют согласно общепринятым методам в данной области техники и различным общим ссылкам (см., в основном Sambrook et al. Molecular Cloning: A Laboratory Manual, 2d ed. (1989) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., и Current Protocols in Molecular Biology (1996) John Wiley and Sons, Inc., N.Y., которые включены в данное описание изобретения посредством ссылки), которые представлены на протяжении данного документа. Вся информация, содержащаяся в нем, включена в данное описание изобретения посредством ссылки.
Пеметрексед (2-[4-[2-(4-амино-2-оксо-3,5,7-триазабицикло[4.3.0]нона-3,8,10-триен-9-ил)этил]бензоил]аминопентадикарбоновая кислота) (ALIMTA™; Eli Lilly & Со.) представляет собой антиметаболический химиотерапевтический агент, который имеет следующую структуру:
Пеметрексед представляет собой антифолатный антинеопластический агент, который обнаруживает свое действие посредством нарушения фолат-зависимых механизмов, необходимых для репликации клеток. Пеметрексед ингибирует тимидилат-синтазу (TS), дигидрофолат-редуктазу (DHFR) и глицинамид-рибонуклеотид-формилтрансферазу (GARFT), ферменты, вовлеченные в de novo биосинтез тимидиновых и пуриновых нуклеотидов. Пеметрексед утвержден FDA для лечения немелкоклеточного рака легких и утвержден в комбинации с цисплатином (лекарственным средством на основе платины для химиотерапии) для лечения злокачественной плевральной мезотелиомы. Рекомендованная доза для лечения мезотелиомы (в комбинации с цисплатином) или немелкоклеточного рака легкого составляет примерно 500 мг/м2 площади поверхности тела в сутки, вводимых в виде внутривенной инфузии в течение 10 минут в 1 сутки каждого 21-суточного цикла. Для использования в комбинированном лечении с метоксиамином типичные диапазоны дозировок пеметрекседа составляют в общем случае от 200 мг/м2 до 1000 мг/м2 площади поверхности тела в сутки, или от 500 мг/м2 до 600 мг/м2 площади поверхности тела в сутки; и типичные диапазоны дозировок метоксиамина составляют от 1 до 200 мг/м2 площади поверхности тела в сутки, или 6-120 мг/м2 площади поверхности тела в сутки. В одном воплощении пеметрексед вводят таким же образом, как описано выше; однако внутривенная инфузия может быть выполнена в течение 15 мин, 20 мин, 30 мин, 45 мин, 60 мин или более, и может быть выполнена в 1 сутки, плюс один или больше дополнительных дней данного цикла, который может составлять 1 неделю, 2 недели, три недели, один месяц или дольше.
ПРИМЕРЫ
Вещества и способы
Химические вещества и реагенты. Метоксиамин (MX) приобретали у Sigma (ST. Louis, Mo.). MX растворяли в стерилизованной воде (рН 7,0).
Вестерн-блоттинг для определения расщепления PARP. Клеточные экстракты разделяли с помощью SDS-PAGE (электрофорез на полиакриламидном геле с додецилсульфатом натрия) (12%-ный полиакриламд) в минигелевом приборе Bio-Rad при 150 В в течение 1 ч. Белки переносили на мембраны PVDV, используя мини-трансблоттер Bio-Rad в течение 1 ч при 100 В. Блоттированные мембраны блокировали 5%-ным сухим молоком в буфере 15 TBS и затем испытывали в течение 2 ч с анти-PARP антителом С2-10 (Trevigen, Gaithersburg, Md.). После трех 5-минутных промываний с помощью TBS-Твин 20 (0,05%) блоты инкубировали с вторичными антителами, анти-lgC против мышиных HRP, в течение 1 ч (Amersham Life Science, Arlington Height III.). Связывание антител наблюдали с помощью ECL (электрогенерированной хемилюминесценции) в соответствии с инструкциями производителя (Amersham Life Science, Arlington Heights, III.).
Опухоли "голых" мышей. Опухолевые клетки (5×106) инъецировали в бочок самки бестимусной HSD голой мыши возрастом 6-8 недель. Животные получали пять ежесуточных внутрибрюшинных инъекций либо физиологического раствора, одного пеметрекседа (150 мг/кг), одного MX (4 мг/кг), или комбинации пеметрекседа (150 мг/кг) и MX (4 мг/кг) с отношением пеметрекседа к MX 26,7:1,0. Опухоли измеряли с помощью кронциркулей, используя формулу Национального института рака: V=L (мм) × I2 (мм)/2, где L представляет собой наибольший диаметр и I представляет собой наименьший диаметр опухоли. Когда объем узелков достигал примерно 100-150 мм3, мышей-носителей опухоли произвольно распределяли по контрольным или лечебным группам (6-9 мышей/группа).
ПРИМЕР 1
На Фиг.8 показано, что во всех группы, получающих метоксиамин (MX) + пеметрексед, было продемонстрировано более значительное подавление роста опухоли по сравнению с группами, получающими только пеметрексед, в модели на клеточной линии человека NCl-H460 NSCLC, модели на клеточной линии А549 NSCLC, модели на клеточной линии колоректального рака НСТ116 и модели на клеточной линии рака молочной железы MDA-MB-468. MX реверсировал устойчивость клеточной линии NCl-H460 NSCLC и клеточной линии колоректального рака НСТ116 к воздействиям химиотерапии пеметрекседом.
ПРИМЕР 2
Чтобы определить влияние пеметрекседа + метоксиамина (MX) по сравнению с одним пеметрекседом на число генерированных АР-сайтов, для измерения АР-сайтов, образованных с помощью пеметрекседа и блокированных MX, использовали реагент-альдегидная реактивная проба (ARP). ARP и MX имеют аналогичную способность вступать в реакцию с АР-сайтами и взаимодействуют конкретно с альдегидной группой, формой с открытым кольцом АР-сайтов. Анализ описан в Liu et al. (Molecular Cancer Therapeutics 2:1061-1066, 2003) и был выполнен, по существу, как описано у Nakamura et al. (Cancer Res. 58:222-225, 1998). Клетки Н460 обрабатывали пеметрекседом (0, 100, 200 или 400 мкм) в течение 24 часов. Затем ДНК (15 мкг) экстрагировали из клеток и инкубировали с 1 мМ ARP при 37°С в течение 10 мин. ДНК осаждали и промывали этанолом, затем ресуспендировали в буфере ТЕ (10 мМ Трис-HCl, рН 7,2, 1 мМ EDTA) и денатурировали при 100°С в течение 5 мин. Затем ДНК быстро охлаждали на льду и смешивали с эквивалентным количеством ацетата аммония (2 М). Затем однонитевую ДНК иммобилизовали на нитроцелюлозной мембране, используя вакуум-фильтрующее устройство. Мембрану инкубировали с стрептавидин-конъюгированной пероксидазой хрена при комнатной температуре в течение 30 мин и промывали промывающим буфером (20 мМ Трис-HCl, 1 мМ EDTA, 0,26 М NaCl, 1% Tween-20). ARP-AP-сайты визуализировали с помощью реагентов ECL (Amersham, Piscataway, NJ). Результаты показаны на Фиг.4А. Пеметрексед в дозах 100, 200 и 400 мкМ индуцировал образование АР-сайтов со степенью индукции АР-сайтов, пропорциональной дозе пеметрекседа (Фиг.4А). Добавление MX к пеметрекседу значительно снижало количество определяемых АР-сайтов по сравнению с одним пеметрекседом. Воздействие комбинации 200 мкМ пеметрекседа и 6 мМ MX оценивали в 24 ч, 48 ч и 72 ч. Результаты показаны на Фиг.4Б. С течением времени количество определяемых АР-сайтов снижалось как с помощью одного пеметрекседа, так и с помощью пеметрекседа в комбинации с MX.
Таким образом, пеметрексед индуцирует образование АР-сайтов, хотя комбинация пеметрекседа и 100 мкМ MX снижала определяемые АР-сайты до контрольных уровней, что происходит не из-за отсутствия АР-сайтов, но благодаря оккупированию АР-сайтов MX, что делает их недоступными для ARP. Удерживание АР-сайтов зависит от времени, указывая на то, что для максимального эффекта необходимы постоянные уровни MX.
ПРИМЕР 3
Анализ разрыва нитей ДНК выполняли для определения способности пеметрекседа и метоксиамина (MX) усиливать гибель опухолевых клеток, опосредованную апоптозом и разрывом нитей ДНК. Кометный анализ описан в Liu et al. (supra.) и основан на способности фрагментов денатурированной, разрезанной ДНК перемещаться из клетки под действием электрического поля. Неповрежденная ДНК перемещается медленнее и остается в пределах ядра при пропускании электрического тока. Повреждение ДНК оценивали в клетке на основе определения формы «кометного» хвоста ДНК и расстояния перемещения (Helma et al., Mutat. Res. 466:9-15, 2000). Клетки собирали и промывали PBS (фосфатно-солевой буферный раствор) после воздействия 200 мкМ пеметрекседа, 6 мМ MX, или 200 мкМ пеметрексед+6 мМ MX, каждого в течение 4 часов. Суспензию клеток Н460 (1×105/мл холодный PBS) смешивали с 1%-ной легкоплавкой агарозой при 42°С в отношении 1:10 (об/об), и 75 мкл сразу же отмеряли пипеткой на CometSlide (Trevigen, Inc., Gaithersburg, MD). Когда легкоплавкая агароза застывала, предметные стекла погружали в предварительно охлажденный лизирующий буфер (10 мМ Трис-HCl, рН 10,5-11,5, 2,5 М NaCl, 100 мМ EDTA, содержащий 1% Тритон Х-100, добавленный непосредственно перед использованием) при 4°С в течение 1 ч. После лизиса предметные стекла промывали дистиллированной водой, располагали продольно в резервуаре для электрофореза и погружали в щелочной буфер (50 мМ NaOH, рН 12-12,5, 1 мМ EDTA) на 30 мин. Затем предметные стекла подвергали электрофорезу как в щелочном (рН>13, 300 мМ NaOH, 1 мМ EDTA), так и в нейтральном растворе (1 Х ТВЕ) в течение 25 мин при 18 В (0,6 В/см), 250 мА. Щелочной электрофорез определяет и одно- и двунитевые разрывы ДНК, возникающие из-за АР-сайтов, а также другие неустойчивые в щелочной среде ДНК-аддукты, в то время как нейтральный электрофорез определяет в основном двунитевые разрывы ДНК. Предметные стекла убирали и промывали нейтрализующим буфером (0,5 М Трис-HCl, рН 7,5) в течение 10 мин, затем PBS, затем оставляли сушиться на воздухе в течение ночи при комнатной температуре. ДНК окрашивали, используя набор для окрашивания серебром (Trevigen), следуя инструкциям производителя. «Кометы» визуализировали, используя микроскоп Olympus. Изображения фиксировали с помощью цифровой камеры и анализировали, используя программу обработки изображений NIH.
Кометные изображения показаны на Фиг.1А (щелочной анализ) и 1Б (нейтральный анализ). После обработки пеметрекседом и MX наблюдались четко различимые «кометы», и длина хвоста была примерно в 4 раза больше, чем в случае одного MX, и примерно в два раза больше, чем в случае одного пеметрекседа (Фиг.1В-1Г).
Результаты исследования эффективности ксенотрасплантата, анализа АР-сайтов и кометного анализа показывают, что MX действует в качестве структурного модулятора АР-сайтов, усиливая терапевтический эффект антиметаболического агента пеметрекседа путем реверсирования устойчивости к химиотерапии, создавая тем самым синергический эффект.
ПРИМЕР 4
Анализ воздействия АР-сайта или МХ-АР-сайта на расщепление ДНК, опосредованное топоизомеразой II. Позиционно-специфичный апуриновый сайт включали путем замены отдельного нуклеозида дезоксиуридином в сайте расщепления топоизомеразой II и затем удаляя урациловое основание с помощью урацил-ДНК-гликозилазы, образуя АР-сайты, которые затем инкубируют с MX для получения МХ-АР-сайтов (Фиг.5А).
Сначала определяли, оказывает ли АРЕ разный эффект для регулярных АР-сайтов и МХ-АР-сайтов, расположенных в позиционно-специфическом сайте в отношении расщепления топоизомеразой II. Результаты показывают, что АРЕ способна расщеплять регулярные АР-сайты быстрее, чем МХ-связанные АР-сайты (Фиг.5Б), однако, и АР, и МХ-АР-сайты расщепляются топоизомеразой II, свидетельствуя от том, что МХ-АР-сайты способны стимулировать опосредованное топоизомеразой II расщепление ДНК.
ПРИМЕР 5
Изучение пероральной и внутривенной биодоступности MX посредством введения единичного болюса крысам Sprague Dawley (не-GLP). Исследования, описанные ниже, выполняли, чтобы оценить биодоступность метоксиамина (MX) при безопасном уровне дозы путем сравнения фармакокинетических параметров после одноболюсного перорального и внутривенного введений MX.
Исследуемые животные. Во время исследования использовали тридцать самцов и тридцать самок крыс Sprague Dawley, 250-350 г с возрастом 7-10 недель.
Получение и концентрация дозы. Однодозовый раствор получали в день введения дозы, корректировали в соответствии с 98%-ной чистотой исследуемого препарата, для достижения концентрации 4,00 мг/мл "активного" MX, 816,77 мг MX растворяли в 5%-ной декстрозе в 200-мл мерной колбе. Полученный раствор поровну разделяли в два желтых сосуда, предназначенных либо для перорального либо для внутривенного введения. Аликвоты отбирали для анализа при получении и после дозирования, переносили на сухой лед и хранили при температуре менее -70°С.
Введение дозы. Всех животных взвешивали в день введения дозы. Индивидуальные дозы для животных были основаны на массе его тела. Использовали постоянный объем дозы 5 мл/кг. Внутривенные дозы вводили с помощью единичной болюсной инъекции в хвостовую вену, используя 3 мл шприц, прикрепленный к игле 26G×1". Внутривенную дозу вводили со скоростью приблизительно 2 мл/мин. Пероральные дозы вводили в виде единичного болюса с помощью питающей иглы 18G×2", прикрепленной к 3 мл шприцу.
Отбор проб крови. Пробы крови отбирали в 5, 15, 30 мин и 1, 2, 4, 6, 8, 12 и 24 ч после введения, с отбором в два момента времени для каждого животного. Пробы крови отбирали из яремной вены для более раннего момента времени и из абдоминальной вены при умерщвлении для более позднего момента времени. Кровь переносили из шприца для отбора крови в 2 мл пробирку для сбора крови, содержащую К3-EDTA в качестве антикоагулянта, и переворачивали для смешивания. Отбор крови из абдоминальной вены выполняли сразу же после эвтаназии с помощью СО2.
Получение образца плазмы и условия хранения. Пробирку с кровью помещали на влажный лед, затем центрифугировали с получением плазмы. Образцы цельной крови центрифугировали со скоростью 3000 об/мин при 4°С в течение 10 мин. Плазму отбирали в пробирки в пробирки и сначала помещали на сухой лед и затем хранили при температуре ниже -70°С.
Масс-спектрометрия (MS). Масс-спектрометрическое определение выполняли с помощью ионизации электрораспылением посредством положительного турбораспыления, согласно техническим условиям, перечисленным ниже.
Прибор для MS: Applied Biosystems 3000
Прибор для HPLC (высокоэффективной жидкостной хроматографии): бинарный насос Agilent 1100 Series
Автоматический пробоотборник: LEAP Technologies CTC-PAL
Условия ионизации электрораспылением (ESI):
Температура: 500°С
Газ-распылитель: азот
газ для CAD (столкновительной диссоциации): азот
DP: 40
Защитный газ (CUR): 10
Столкновительный газ: 6
Напряжение ионного распыления (IS): 5000
Выходное напряжение (ЕР): 10
NEB: 12
Монитор:
Анализируемое вещество: 207,0/149,3 и 207,0/178,4 а.е.м.
IS (аценокумарол): 354,2/296,0 и 354,2/163,1 а.е.м.
Условия HPLC (на Agilent 1100):
Подвижная фаза А: 0,1%-ная муравьиная кислота в воде
Подвижная фаза В: 0,1%-ная муравьиная кислота в ацетонитриле
Колонка: Thermo Aquasil С18, 50×3 мм
Защитная колонка: Thermo Aquasil C18, 10×4 мм
Расход газа: 1,0 мл/мин
Объем введенной пробы: 50 мкл
Градиент:
Проба плазмы для анализа LC-MS/MS. Индивидуальные пробы плазмы размораживали при температуре окружающей среды, и аликвоту 250 мкл отбирали для анализа, если не требовалось разбавление. Пробы, отобранные в период времени от 5 мин до 1 ч от животных с внутривенным введением, или от 15 мин до 8 ч от животных с пероральной дозой, требовали 5-40-кратного разбавления, чтобы попасть в линейный диапазон метода (1-1000 нг/мл). Для проб, требующих разбавления, отбирали подходящий объем и смешивали с плазмой необработанной крысы, содержащей такой же антикоагулянт, с получением итогового объема 250 мкл. Количественные результаты корректировали с помощью коэффициента разбавления. Плазму для анализа LC-MS/MS готовили следующим образом:
- Аликвоты плазмы перемешивали на вортексе в течение 30 с и центрифугировали при 14000 об/мин в течение 10 мин для осаждения интерферирующих частиц.
- Аливоту 100 мкл отбирали из надосадочной жидкости и помещали в 1,5 мл микроцентрифужную пробирку.
- К 100 мкл аликвоте плазмы добавляли 310 мкл смеси H2O: муравьиная кислота (2:1), 30 мкл аценокумарола (IS) в H2O (10 мкг/мл) и 100 мкл диэтиламинобензальдегида в растворе 2:1 H2O: муравьиная кислота (10 мг/мл) и хорошо смешивали.
- Затем смесь инкубировали на водяной бане при 80°С в течение 2 ч. После инкубирования надосадочную жидкость переносили во флакон для HPLC для количественного определения посредством LC-MS/MS.
Анализ фармакокинетики и биодоступности метоксиамина (MX). Фармакокинетический анализ
Фармакокинетические (РК) профили и пероральную биодоступность метоксиамина (MX) определяли на самце и самке крыс Sprague Dawley после единичного болюсного дозирования MX посредством внутривенного и перорального введения в концентрации 20 мг/кг массы тела. В фармакокинетическом анализе использовали тридцать крыс (15 самцов и 15 самок) для каждого способа дозирования. С целью определения фармакокинетических характеристик MX пробы плазмы получали в заданные моменты отбора проб перед дозированием 5, 15, 30 мин и через 1, 2, 4, 6, 8, 12 и 24 ч после введения дозы. Для поддержания нормального состояния здоровья у каждой крысы отбирали кровь максимум два раза в заданный момент времени для получения плазмы. Типичную концентрацию MX получали путем усреднения значений в каждый момент времени для трех крыс одного и того же пола и способа дозирования. Для средней величины дозы среднее фактическое время отбора проб получали соответственно от трех крыс одного и того же пола и способа дозирования для каждого номинального момента времени. Средняя концентрация MX в плазме, средняя величина дозы и среднее действительное время отбора проб использовали для фармакокинетического анализа каждого способа дозирования.
Кривые средней концентрации MX в плазме от времени усредненной пробы строили для каждого пола и способа дозирования, используя Microsoft Excel 2000-SR1™ (Фиг.2-3). Концентрации MX в плазме, зафиксированные как ниже количественно определяемых пределов (BQL) или неопределяемые, если это имеет место, в целях фармакокинетического моделирования считают равными 0,00 нг/мл.
Анализ фармакокинетических параметров выполняли, используя некомпартментное моделирование с помощью WinNonlin 5.1 (Pharsight Corporation, Mountain View, CA). Фармакокинетические параметры включали максимальную плазменную концентрацию (Cmax), время максимальной плазменной концентрации (Tmax), период полувыведения (t1/2), площадь под кривой концентрации в плазме в зависимости от времени от момента времени ноль до последней измеряемой концентрации в плазме (AUCпосл.), и площадь под кривой концентрации в плазме в зависимости от времени от момента времени ноль, экстраполированной до бесконечности (AUC0-∞). Для сравнения AUC0-∞ приводили к номинальной общей величине дозы MX 5 мг (AUC0-∞5). Для фармакокинетических параметров использовали сокращения, указанные ранее.
Анализ биодоступности. Абсолютную пероральную биодоступность определяли с помощью отношений перорального введения метоксиамина (MX) к внутривенному AUC0-∞ (приведенных к общей величине дозы 5 мг MX) посредством Microsoft Excel 2000-SR1™, используя следующую формулу (MX=TRC102):

Фактические уровни доз MX составляли 20,1 мг/кг, 20,1 мг/кг для самцов и крыс в группе внутривенного дозирования, и 19,9 мг/кг, 20,0 мг/кг соответственно для самцов и самок крыс в группах перорального дозирования.
Фармакокинетика и биодоступность. В Таблице 1 представлены фармакокинетические параметры метоксиамина (MX) для самца и самки крыс Sprague Dawley, после дозирования единичного болюса MX посредством внутривенного и перорального введения в концентрации 20 мг/кг массы тела.
Для самцов крыс измеряли концентрации MX в плазме на протяжении полного 24-часового контролируемого периода как для внутривенного, так и для перорального введений. Визуальный осмотр кривой зависимости внутривенной средней концентрации MX в плазме самцов от среднего времени свидетельствует о фазе быстрого распределения, которая была завершена в течение приблизительно 2 ч после дозирования. Cmax при внутривенном пути составляла 15,510 нг/мл и была достигнута сразу же по завершении болюсного введения. Системное воздействие MX, на что указывают AUCпосл. и AUC0-∞, составляло 12,518 нг/мл·ч и 12,706 нг/мл·ч соответственно. Внутривенная AUC0-∞, приведенная к общей номинальной величине дозы 5 мг (AUC0-∞5), составляла 11,284 нг/мл·ч.
Для самцов крыс в пероральном способе дозирования абсорбция MX была быстрой, с Tmax 1,0 ч. Значение Cmax 2,205 нг/мл было значительно меньше, чем при внутривенном пути дозирования. Системное воздействие MX, на что указывают пероральная AUCпосл. и AUC0-∞, составляло соответственно 13,596 нг/мл·ч и 13,811 нг/мл·ч. Пероральная AUC0-∞, приведенная к номинальной общей величине дозы 5 мг (AUC0-∞5), составляла 12,420 нг/мл·ч. Периоды полувыведения были короткими и аналогичными для двух путей дозирования (ВВ: 5,2 ч, пероральный: 4,2 ч). Рассчитанная абсолютная биодоступность при пероральном введении у самцов крыс Sprague Dawley составляла 110 процентов.
Для самок крыс имелись количественно определяемые концентрации MX в плазме на протяжении полного 24-часового периода отбора проб как для внутривенного, так и для перорального способа дозирования. Визуальный осмотр кривой зависимости внутривенной средней концентрации MX в плазме самок от среднего времени говорит о фазе быстрого распределения, которая завершается так же как у самцов крыс, в течение приблизительно 2 ч после дозирования. Cmax для внутривенного способа Cmax составляла 10,965 нг/мл и наступала сразу же по завершении болюсного введения. Системное воздействие MX, на что указывают AUCпосл. и AUC0-∞, составляло соответственно 12,971 нг/мл·ч и 13,142 нг/мл·ч. Внутривенная AUC0-∞, приведенная к номинальной общей величине дозы 5 мг (AUC0-∞5), составляла 13,892 нг/мл·ч.
Для самок крыс при пероральном пути дозирования абсорбция MX была быстрой, с Tmax 0,5 ч. Значение Cmax 2959 нг/мл было значительно меньше, чем при внутривенном способе, однако аналогично значению для самцов крыс при пероральном способе дозирования. Системное воздействие MX, на что указывают AUCпосл. и AUC0-∞, составляло соответственно 11,643 нг/мл·ч и 12,029 нг/мл·ч соответственно. Пероральная AUC0-∞, приведенная к номинальной общей дозе 5 мг (AUC0-∞5), составляла 13,047 нг/мл·ч. Период полувыведения был коротким и аналогичным для двух способов дозирования (ВВ: 4,6 ч, пероральный: 5,7 ч) и сравнимым с таковыми у самцов крыс. Рассчитанная абсолютная биодоступность у самок крыс Sprague Dawley составляла 94 процента.
У крыс, которым вводили дозы 20 мг/кг массы тела (BW) как внутривенно так и перорально, отсутствовали клинических признаки токсичности.
И у самцов и у самок крыс Sprague Dawley MX, введенный в концентрации 20 мг/кг массы тела посредством единичного болюсного перорального введения, быстро (Tmax 0,5-1,0 ч) и полностью абсорбировался с системной абсолютной биодоступностью приблизительно 100 процентов. Хотя пероральная Cmax MX значительно меньше, чем ВВ Cmax. системное воздействие MX, на что указывают AUCпосл. и AUC0-∞, аналогично при двух способах дозирования. Кроме того, уровни в сыворотке, превышающие целевые Cmax, связаны с активностью в мышиных моделях рака человека (50 нг/мл) после перорального дозирования в моменты времени, что позволяет использовать схему перорального дозирования один или два раза в сутки.
Эти данные имеют важное значение, поскольку они указывают на то, что метоксиамин является полностью биодоступным при пероральном введении и имеет период полувыведения 4-6 ч, что допускает достижение минимально эффективных концентраций при дозировании однократно или два раза в сутки. Оба эти наблюдения являются неожиданными. Большинство противораковых агентов не являются перорально биодоступным в количествах, достаточных для того чтобы позволить пероральное дозирование. Следует заметить, что попытки вводить другие конкретные противораковые лекарственные средства перорально приводили к значительно более низкой биодоступности, чем достигнутая в данном случае. Смотри, например, блеомицин, карбоплатин, цисплатин, оксалиплатин, пакситаксел, ралитрексед (антифолат), топотекан, винбластин, винкристин, винорелбин, все из которых имели биодоступность менее 50% (Chu E and DeVita VT. Physicians′ Cancer Chemotherapy Drug manual 2002. Boston: Jones and Bartlett Publishers, 2002). Во-вторых, продемонстрированный период полувыведения длиннее, чем ожидаемый для небольшой молекулой с молекулярной массой менее 100 Д, которая легко взаимодействует с альдегидами, которые могут присутствовать в плазме, и более длинный, чем ожидаемый период полувыведения из плазмы, допускающий поддерживание уровней лекарственного средства (выше минимальной эффективной концентрации) со схемой перорального дозирования один раз или два раза в сутки. Полная биодоступность и период полувыведения 4-6 ч может обеспечивать возможность перорального дозирования с использованием схемы введения один или два раза в сутки, которая может быть удобной раковым пациентам.
Специалистам в данной области техники понятно из вышеизложенного, что различные модификации вышеописанных способов и композиций могут быть произведены без отклонения от сущности и объема изобретения. Соответственно, изобретение может быть воплощено в других конкретных формах без отклонения от сущности или его существенных особенностей. Настоящие воплощения и примеры, таким образом, следует рассматривать в качестве иллюстративных и неограничивающих во всех отношениях, и все изменения, которые попадают в значение и диапазон эквивалентности формулы изобретения, следовательно, как предполагается, охвачены ею.
Таким образом, следует понимать, что отсутствует намерение использовать такие термины и выражения для исключения любых эквивалентов признаков, показанных и описанных, или их элементов, но следует признать, что возможны различные модификации в рамках объема изобретения, как оно заявлено. Также следует понимать, что каждый из более узких вариантов и субродовых группировок, входящих в общее описание изобретения, также образует часть изобретения. Это включает основное описание изобретения с условием или негативным ограничением, исключающим любой объект из класса объектов, несмотря на то, является ли исключаемый материал конкретно упоминаемым здесь.
Все патенты, публикации, научные статьи, веб-сайты и другие документы и материалы, упоминаемые или отмеченные в данном описании изобретения, являются признаком компетентности специалистов в данной области техники, к которой изобретение принадлежит, и каждый такой ссылочный документ и материал включен в данное описание изобретения посредством ссылки в той же степени, как если были они были включены посредством ссылки во всей их полноте индивидуально или изложены в данном описании изобретения во всей их полноте.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНАЦИЯ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2009 |
|
RU2516027C2 |
| СПОСОБЫ ПРИМЕНЕНИЯ (+)-1,4-ДИГИДРО-7-[(3S, 4S)-3-МЕТОКСИ-4-(МЕТИЛАМИНО)-1-ПИРРОЛИДИНИЛ]-4-ОКСО-1-(2-ТИАЗОЛИЛ)-1,8-НАФТИРИДИН-3-КАРБОНОВОЙ КИСЛОТЫ ДЛЯ ЛЕЧЕНИЯ РАКА | 2006 |
|
RU2592231C2 |
| СОКРИСТАЛЛЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2014 |
|
RU2675270C2 |
| КОМБИНИРОВАННАЯ ХИМИОТЕРАПИЯ | 2006 |
|
RU2429838C2 |
| КОМБИНИРОВАННАЯ ХИМИОТЕРАПИЯ | 2010 |
|
RU2587013C2 |
| СПОСОБЫ ПРИМЕНЕНИЯ (+)-1,4-ДИГИДРО-7-[(3S,4S)-3-МЕТОКСИ-4-(МЕТИЛАМИНО)-1-ПИРРОЛИДИНИЛ]-4-ОКСО-1-(2-ТИАЗОЛИЛ)-1,8-НАФТИРИДИН-3-КАРБОНОВОЙ КИСЛОТЫ ДЛЯ ЛЕЧЕНИЯ РАКА | 2006 |
|
RU2428983C2 |
| СО-КРИСТАЛЛЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2014 |
|
RU2823603C2 |
| СИНЕРГИЧЕСКИЕ СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ РАКА | 2001 |
|
RU2264217C2 |
| СПОСОБ ЛЕЧЕНИЯ ПУТЕМ ПРИМЕНЕНИЯ КОМБИНИРОВАННОЙ ТЕРАПИИ | 2010 |
|
RU2543348C2 |
| АНТИНЕОПЛАСТИЧЕСКИЕ КОМБИНАЦИИ, СОДЕРЖАЩИЕ HKI-272 И ВИНОРЕЛБИН | 2013 |
|
RU2670974C2 |


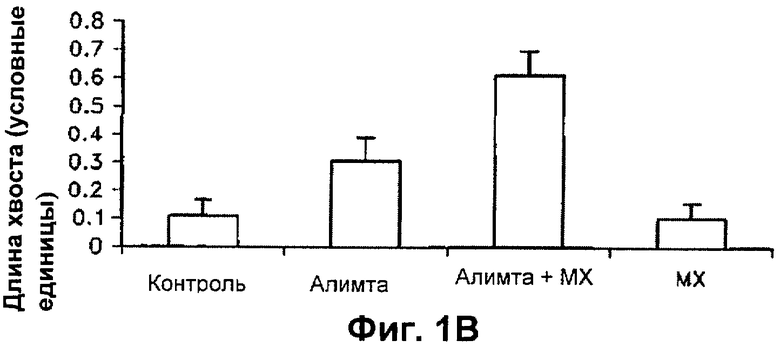
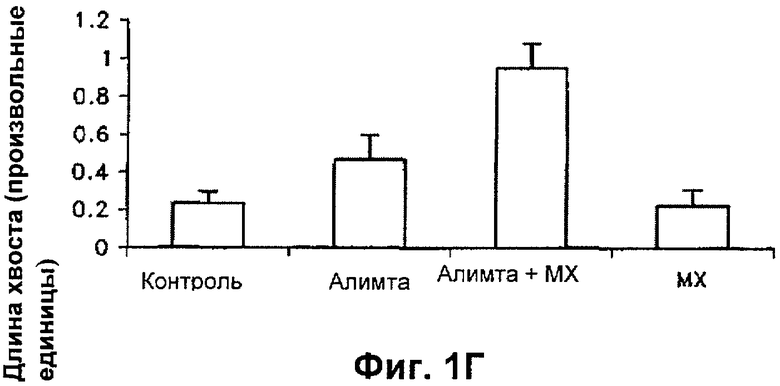
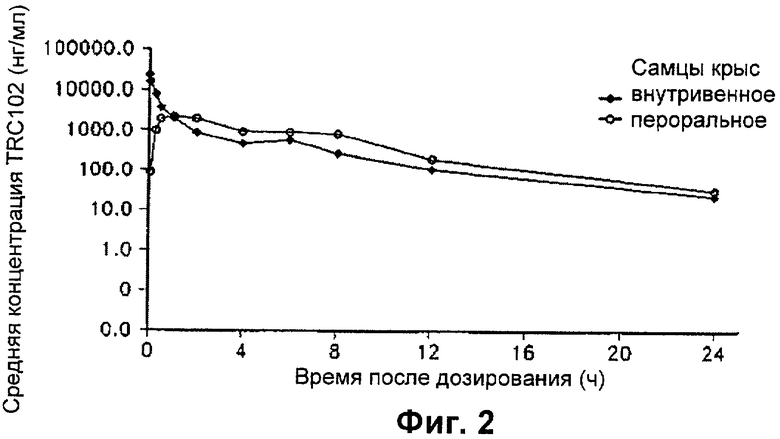
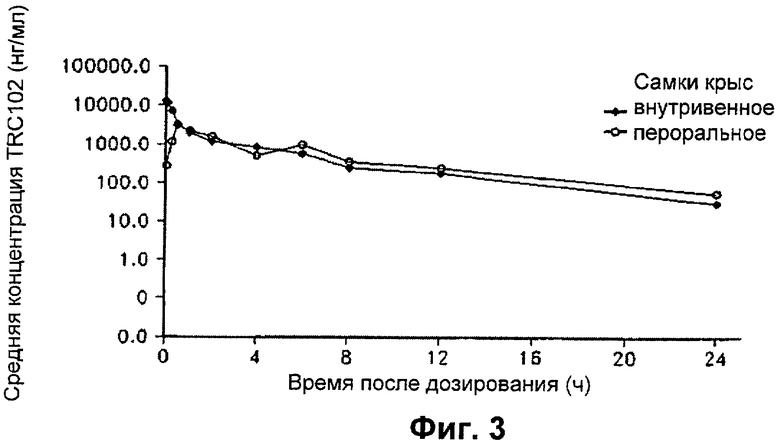
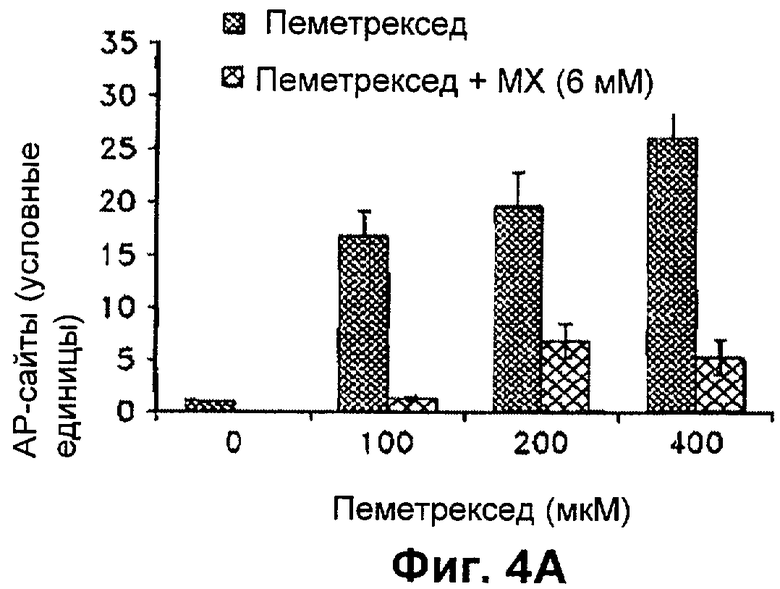
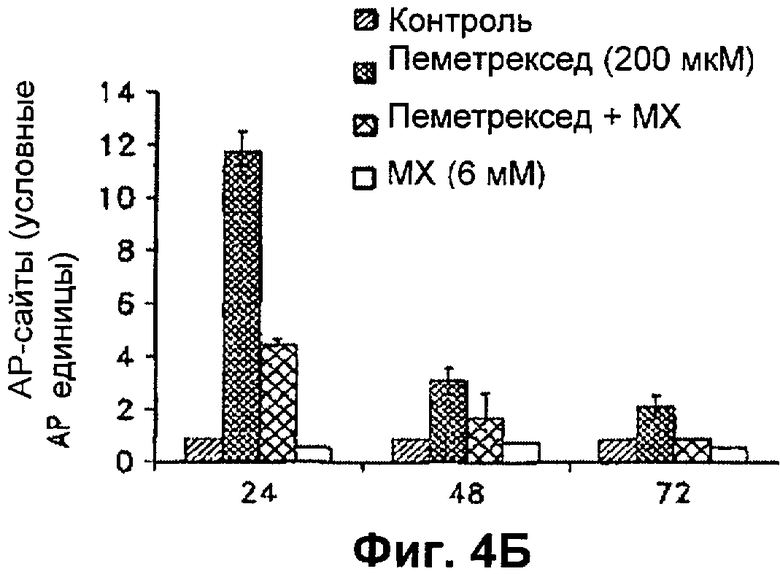
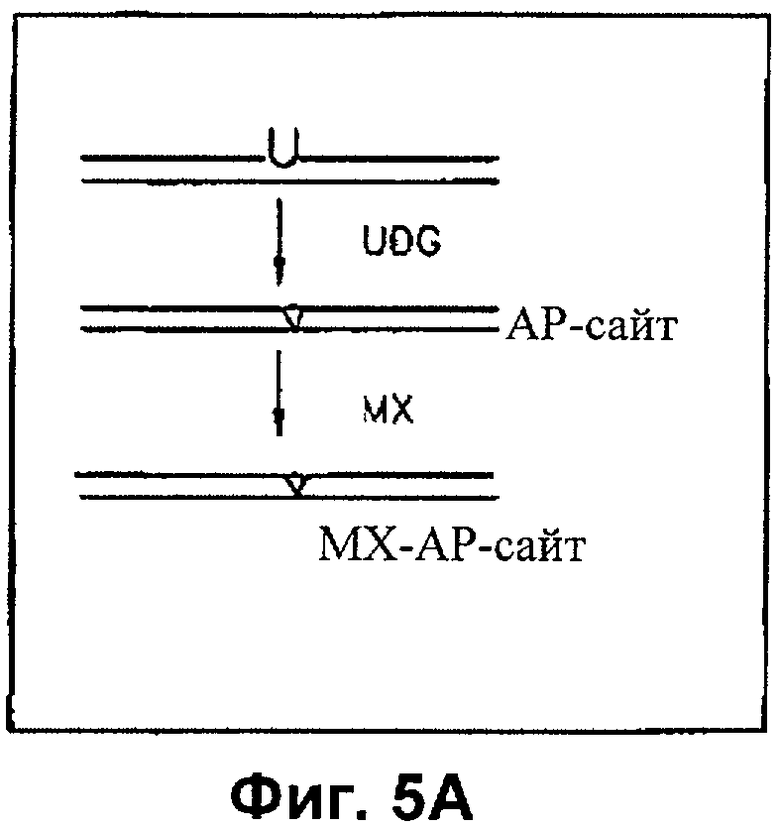
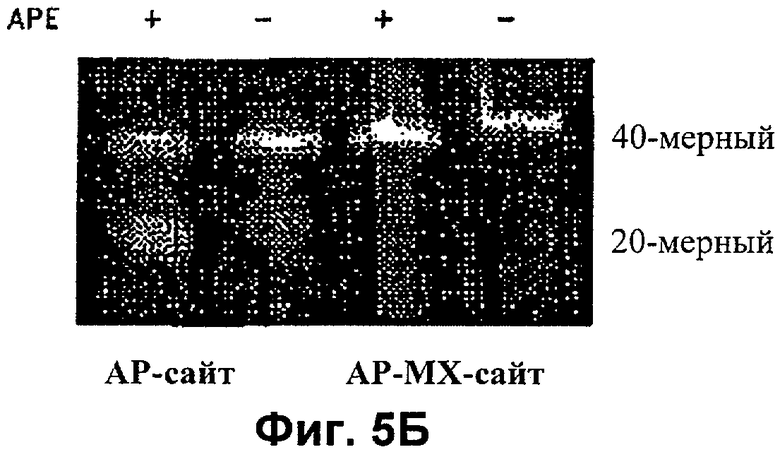


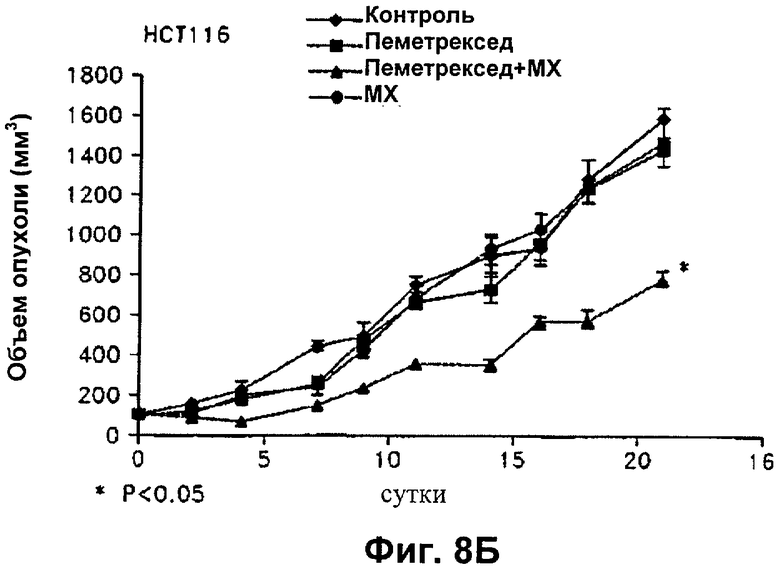
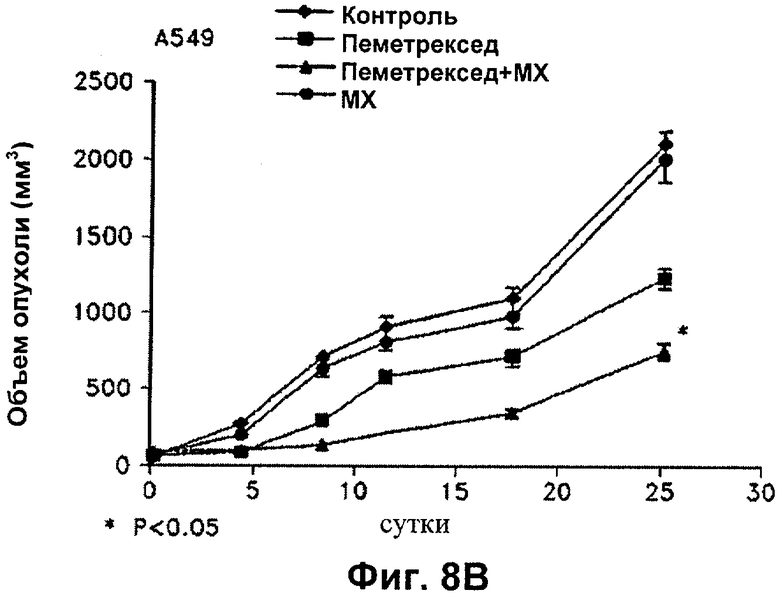
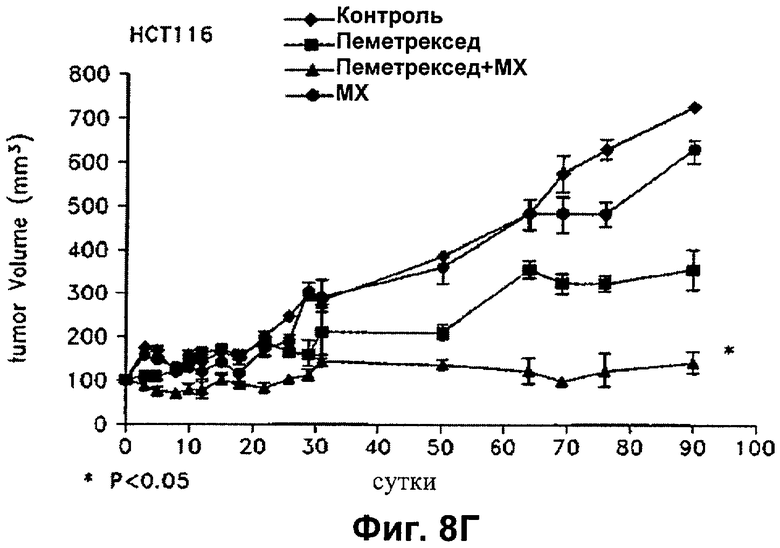
Предложено применение первой композиции, содержащей пеметрексед, и второй композиции, содержащей метоксиамин, для лечения пациента, у которого диагностирован рак, выбранный из рака легкого, колоректального рака, рака яичника, рака поджелудочной железы, рака почки, рака эндометрия, рака желудка, рака печени и рака молочной железы. Показано, что метоксиамин усиливал, увеличивал или потенцировал химиотерапевтический эффект антифолатного противоракового агента. Показано неожиданное реверсирование устойчивости к пеметрекседу. 15 з.п. ф-лы, 8 ил.,1 табл.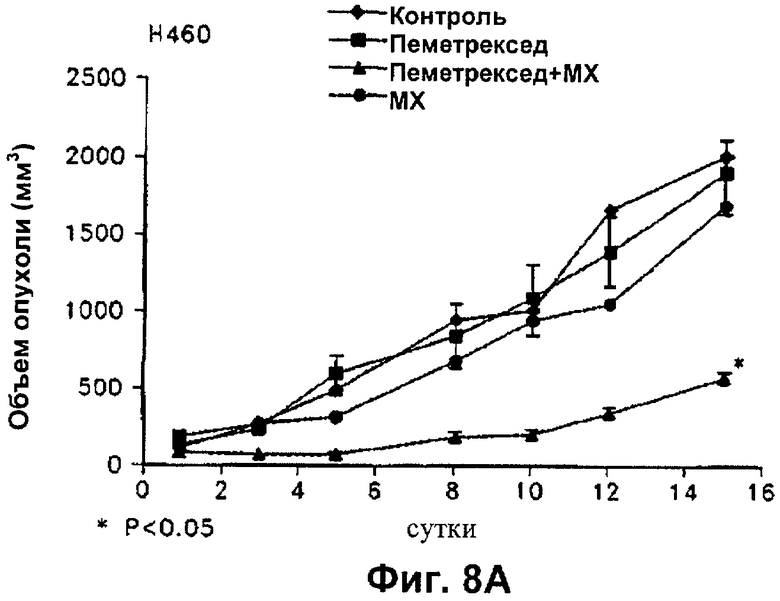
1. Применение 1) первой композиции, содержащей пеметрексед, и 2) второй композиции, содержащей метоксиамин, для лечения пациента, у которого диагностирован рак, выбранный из рака легкого, колоректального рака, рака яичника, рака поджелудочной железы, рака почки, рака эндометрия, рака желудка, рака печени и рака молочной железы.
2. Применение по п.1, где указанный рак представляет собой немелкоклеточный рак легких, колоректальный рак или рак молочной железы.
3. Применение по п.1, где указанный пеметрексед готовят в виде препарата для внутривенного или перорального введения.
4. Применение по п.1, где указанный метоксиамин готовят в виде препарата для внутривенного или перорального введения.
5. Применение по п.1, где указанный метоксиамин готовят в виде препарата для перорального введения.
6. Применение по п.1, где указанный метоксиамин и указанный пеметрексед вводят последовательно.
7. Применение по п.1, где указанный метоксиамин вводят перед пеметрекседом.
8. Применение по п.1, где указанный пеметрексед вводят перед метоксиамином.
9. Применение по п.1, где указанный пеметрексед в указанной первой композиции представляет собой гептагидрат динатриевой соли пеметрекседа.
10. Применение по п.1, где указанный метоксиамин готовят в виде препарата в количестве, достаточном для сенсибилизации указанного рака к указанному пеметрекседу.
11. Применение по п.1, где указанный метоксиамин и указанный пеметрексед достигают синергического эффекта при введении пациенту.
12. Применение по п.1, где указанный метоксиамин вводят один или два раза в сутки.
13. Применение по п.1, где указанный пациент выбран как имеющий рак, частично или полностью устойчивый к лечению одним пеметрекседом, и где указанное второе лекарственное средство, содержащее метоксиамин, готовят в виде препарата в количестве, эффективном для того, чтобы потенцировать активность пеметрекседа и преодолеть указанную устойчивость при введении пациенту.
14. Применение по п.1, где отношение метоксиамина к пеметрекседу представляет собой отношение, выбранное из: от примерно 1:2 до примерно 1:10000; от примерно 1:2 до примерно 1:100; от примерно 1:50 до примерно 1:500; от примерно 1:450 до примерно 1:10000; от примерно 1:10 до примерно 1:50; от примерно 1:5 до примерно 1:500; от примерно 1:15 до примерно 1:40 и от примерно 1:20 до примерно 1:30.
15. Применение по п.1, где отношение метоксиамина к пеметрекседу составляет от примерно 1:2 до примерно 1:100.
16. Применение по любому из предшествующих пп.1-15, где указанный пеметрексед приготавливают в виде препарата в диапазоне дозировок, выбранном из: от примерно 25 мг/м2 до примерно 200 мг/м2 площади поверхности тела; от примерно 150 мг/м2 до примерно 500 мг/м2 площади поверхности тела; от примерно 400 мг/м2 до примерно 1000 мг/м2 площади поверхности тела; от примерно 900 мг/м2 до примерно 5000 мг/м2 площади поверхности тела; от примерно 200 мг/м2 до примерно 1000 мг/м2 площади поверхности тела и от примерно 500 мг/м2 до примерно 600 мг/м2 площади поверхности тела.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| van der Bogaert DP et al | |||
| Pemetrexed maintenance therapy in patients with malignant pleural mesothelioma | |||
| J Thorac Oncol | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Прибор с двумя призмами | 1917 |
|
SU27A1 |
| Rollins KD et al | |||
| Pemetrexed: a multitargeted antifolate | |||
| Clin Ther | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Реферат | |||

