Гепатит С, вызываемый вирусом гепатита С (ВГС), является одним из наиболее распространенных заболеваний печени и широко распространен по всему миру. На основе ежегодных докладов Всемирной организации здравоохранения (ВОЗ), более 130-150 миллионов человек инфицированы ВГС и более 700 тысяч лиц умирают от ВГС [WHO. Hepatitis C. WHO fact sheet №164. Updated July 2016, http://www.who.int/mediacentre/factsheets/fs164/en/]. ВГС демонстрирует высокое генетическое разнообразие и характеризуется региональными вариациями генотипов (gT) ВГС. Генотип 1 (gT1) является наиболее распространенным во всем мире (83,4 миллиона человек, что составляет 46,2% всех случаев инфицированных ВГС), приблизительно одна треть из которых находятся в Восточной Азии. Следующим наиболее распространенным является генотип 3 (gT3), Этим генотипом инфицированы во всем мире 54,3 миллиона человек (30,1%). Генотипы 2, 4, 6 составляют 22,8% всех случаев инфицированных ВГС человек, а генотип 5 (gT5) составляет оставшиеся <1%. В то время как генотипы 1 и 3 доминируют в большинстве стран, независимо от экономического статуса, наибольшие доли генотипы 4 и 5 находятся в странах с низким уровнем дохода [Messina, J.P. at al. Global Distribution and Prevalence of Hepatitis С Virus Genotypes. Hepatology 2015, 61(1), 77-87.].
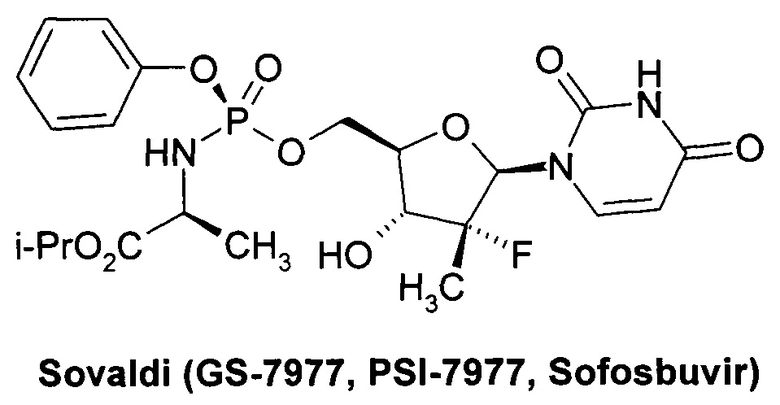
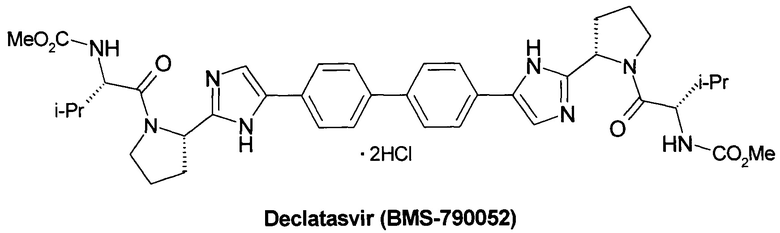
Значительный прогресс в лечении гепатита С был достигнут в последние годы, который связан в первую очередь с открытием Софосбувира (Sofosbuvir, Sovaldi, PSI-7977, GS-7977) - ингибитора нуклеозида NS5B HCV [Sofia, M.J. et al. Discovery of a β-D-20-Deoxy-20-rfluoro-20-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis С Virus. J. Med. Chem. 2010, 53, 7202-7218. Sofia, M.J. et al. Nucleoside phosphoramidate prodrugs. Patent US 7964580, 2011. Sofia, M.J. Nucleoside phosphoramidate prodrugs. Patent US 8334270, 2012.] и Даклатасвира (Daclatasvir, BMS-790052) - ингибитора inhibitor of NS5A HCV [Belema, M. et al. Discovery and Development of Hepatitis С Virus NS5A Replication Complex Inhibitors. J. Med. Chem. 2014, 57, 1643-1672. Bachand, C. et al. Hepatitis С virus inhibitors. Patent WO 2008/021927, 2008. Bachand, C. et al. Hepatitis с virus inhibitors. Patent WO 2008/021928, 2008. Bachand, C. et al. Hepatitis С virus inhibitors. Patent WO 2008/021936, 2008.
http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_Public_assessment report/human/003768/WC500172849.pdf.].
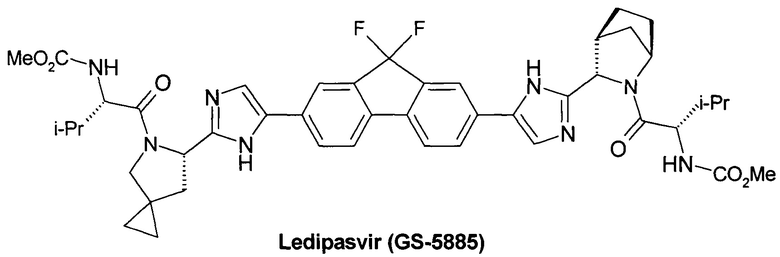
Несколько позже были получены и другие ингибиторы NS5A включая Ледипасвир (Ledipasvir, GS-5885) [Link, J.О. et al. Discovery of ledipasvir (GS-5885): a potent, once-daily oral NS5A inhibitor for the treatment of hepatitis С virus infection J. Med. Chem. 2014, 57, 2033-2046. Guo, H. et al. Antiviral compounds. Patent WO 2010/132601, 2010. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_Public_assessment_report/human/003850/WC500177996.pdf. http://www.hcvdruginfo.ca/downloads/HCV_ledipasvir.pdf],
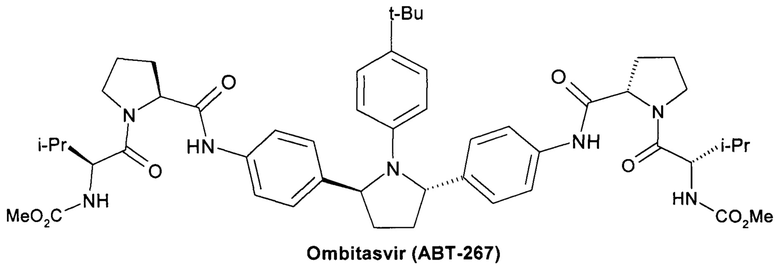
Омбитасвир (Ombitasvir, АВТ-267) [Gardelli, С. et al. Phosphoramidate Prodrugs of 20-C-Methylcytidine for Therapy of Hepatitis С Virus Infection. J. Med. Chem. 2014, 57, 2047-2057. Patent WO 2010/144646, 2010.],
Элбасвир (Elbasvir, MK-8742) [Coburn, C.A. et al. ChemMedChem. 2013, 8, 1930-1940. Patent WO 2012/040923, 2012. Patent WO 2012/041014, 2012],
Велапатасвир (Velpatasvir, VEL, GS-5816) [Patent WO 2015/110048, 2015. http://www.accessdata.fda.gov/drugsatfdadocs/nda/2016/208341O rig1s000PharmR.pdf]. http://www.gilead.com/~/media/files/pdfs/medicines/liver-disease/epclusa/epclusa_pi.pdf],
Гепавивир (Hepavivir, AV-4025) Ivachtchenko, A.V. et al. Discovery of Novel Highly Potent Hepatitis С Virus NS5A Inhibitor (AV4025). J. Med. Chem. 2014, 57, 7716-7730. Patent WO 2012/074437, 2012. Patent US 9428491, 2016.], AV-4056 and AV-4058 [US 9428491.], AV-4067 and AV-4084 [Pat. Appl. US 14/845,333.],


Саматасвир (Samatasvir, IDX 18719, IDX 719) [Patent WO 2011075615, 2011. Bilello J.P. et al. In vitro activity and resistance profile of samatasvir, a novel NS5A replication inhibitor of hepatitis С virus. Antimicrobobial Agents Chemother. 2014, 58(8), 4431-4442],
Odalasvir (ACH-3102) [Nakamoto S. et al. Hepatitis С virus NS5A inhibitors and drug resistance mutations. World J. Gastroenterol. 2014, 20(11), 2902-2912].
Комбинации ингибиторов NS5A и NS5B позволяют не только исключить использование интерферона и рибавирина для лечения гепатита С, но и радикально повысить эффективность лечения. Например, Клинические испытания комбинированного препарата Epclusa (таблетка Epclusa содержит 400 мг Софосбувира и 100 мг Велапатасвира) показали высокую эффективность (SVR12≥95%) у субъектов без цирроза и субъектов с компенсированным циррозом генотип 1, 2, 3, 4, 5 и 6 ВГС-инфекции у взрослых [http://www.gilead.com/~/media/files/pdfs/medicines/liver-disease/epclusa/epclusa_pi.pdf].
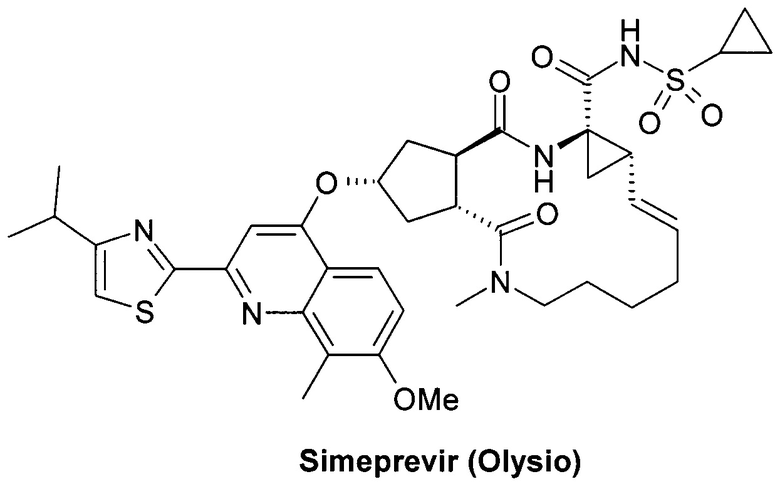
В комбинированной терапии гепатита С до настоящего времени успешно используются также протеазные ингибиторы, в том числе, Нарлапревир (Narlaprevir, SCH 900518) - ингибитор неструктурированного белка 3 ВГС (NS3 HCV) [Arasappan A. et al. Discovery of Narlaprevir (SCH 900518): A Potent, Second Generation HCV NS3 Serine Protease Inhibitor. ACS Med. Chem. Lett., 2010, 1(2), 64-69] и Симепревир (Simeprevir, Olysio) ингибитор неструктурированного белка NS3/NS4 ВГС (NS3/4A HCV) [https://www.tga.gov.au/sites/default/files/auspar-simeprevir-141027-pi.docx].
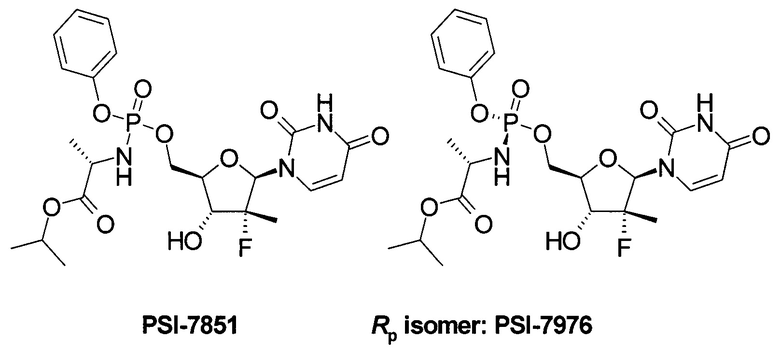
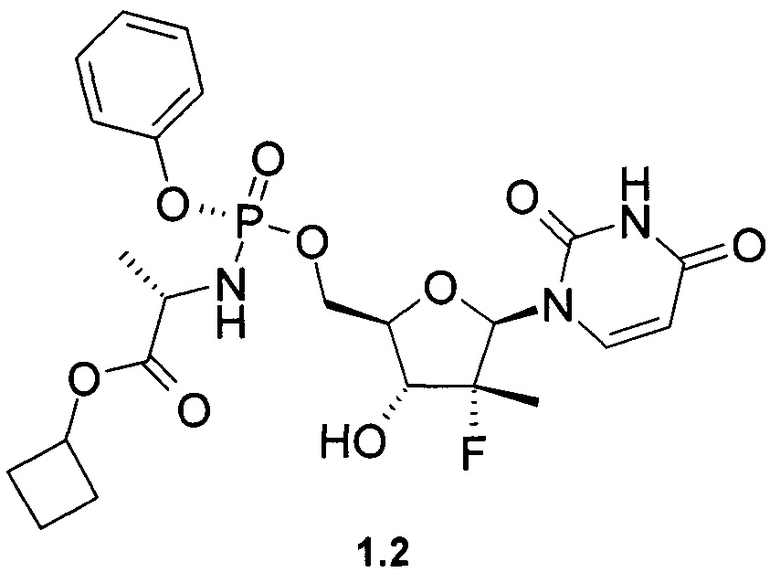
Недавно была предложена противовирусная композиция, содержащая в качестве пролекарства ингибитора NS5B HCV полимеразы, фосфорамидатный нуклеотид PSI-7851, представляющий собой изопропил (S)-2-{[(2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-4-фтор-3-гидрокси-4-метил-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропионат или его фосфорные стереоизомеры (Rp isomer: PSI-7976 и Sp isomer: PSI-7977) [Патент US 7964580 (2011), RU 2478104 (2013)].
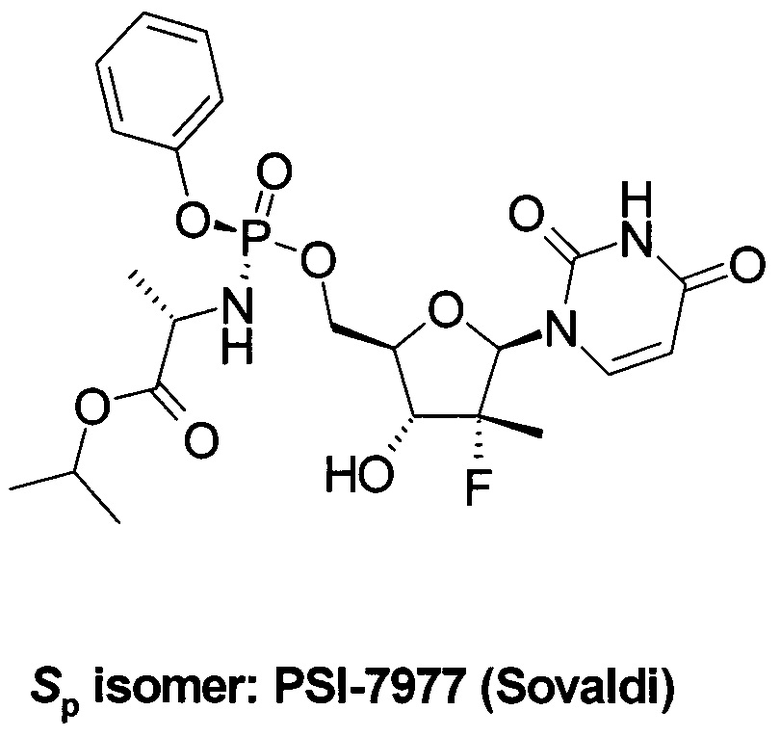
Противовирусная композиция, включающая PSI-7977, известный как Sovaldi® и Sofosbuvir http://www.gilead.com/~/media/Files/pdfs/medicines/liver-disease/sovaldi/sovaldi_pi.pdf] в настоящее время широко используется для комбинированного лечения гепатита С, в том числе, совместно с ингибиторами NS5A HCV, NS3, NS3/4 и другими противовирусными компонентами. PSI-7851 и его стереоизомеры PSI-7976 и PSI-7977 метаболизируют до трифосфата PSI-7409, который и являются ингибитором NS5B HCV полимеразы [Е. Murakami et al. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 2010, 285(45), 34337-34347].

Препарат Sovaldi® [М.J. Sofia et al. Discovery of a β-D-20-Deoxy-20-r-fluoro-20-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis С Virus. J. Med. Chem. 2010, 53, 7202-7218. M.J. Sofia et al. Nucleoside phosphoramidate prodrugs. US 7964580 (2011).] стал первым нуклеотидом, одобренным обоими регулирующими органами FDA и ЕС для комбинированного лечения больных гепатитом С, инфицированных различными генотипами (gT) вируса гепатита С. В клинических испытаниях он показал высокую эффективность против шести генотип HCV (gT1-gT6) [I.M. Jacobson et al. Sofosbuvir for hepatitis С genotype 2 or 3 in patients without treatment options. Engl. J. Med. 2013, 368, 1867-1877. E. Lewirz et al. Sofosbuvir for previously untreated chronic hepatitis С infection. Engl. J. Med. 2013, 368, 1878-1887].
Несмотря на прогресс, достигнутый в последние годы в терапии гепатита С, остается актуальной задача поиска новых противовирусных композиций, включающих в свой состав пролекарство ингибиторов NS5B ВГС, с более эффективным метаболизмом пролекарства в трифосфат PSI-7409 и улучшенными свойствами.
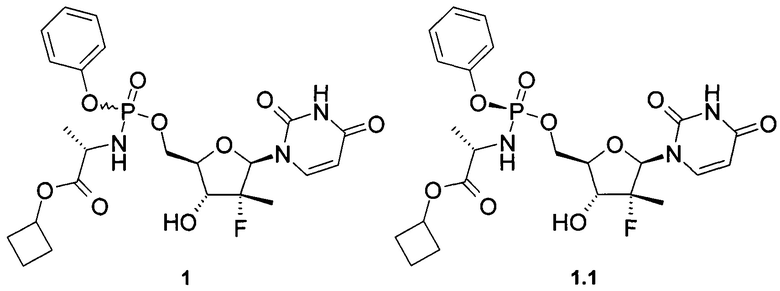
Авторы неожиданно обнаружили что противовирусные композиции, содержащие в качестве пролекарства ингибитора NS5B HCV полимеразы неизвестное ранее соединение общей формулы формулы 1 или его фосфорный стереоизомер формулы 1.1 или 1.2 (Sp стереоизомер: 1.1 и Rp стереоизомер: 1.2), являются эффективным противовирусным средством, в том числе для лечения гепатита С
Изобретатели установили, что суммарная концентрация трифосфата PSI-7409 образующегося в результате метаболизма в печени крыс в течение всего времени наблюдения неизвестного ранее пролекарства циклобутил (S)-2-{(S)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидро-фуран-2-илметокси]-фенокси-фосфориламино}-пропаноата (1.1) имеет значение AUC0-t=27079 нг⋅ч/г, которое в 1.6 раза выше суммарной концентрации PSI-7409 (AUC0-t=16796 нг⋅ч/г), образующегося в результате метаболизма Sovaldi® в печени крыс [М.J. Sofia et al. Discovery of a β-D-20-Deoxy-20-r-fluoro-20-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis С Virus. J. Med. Chem. 2010, 53, 7202-7218].
Отметим, что в настоящее время в комбинированном лечении гепатита С используется противовирусная композиция в форме таблеток, включающая 400 мг Sovaldi®, которая является наиболее эффективной дневной дозой. Новая противовирусная композиция более эффективна при лечении гепатита С, поскольку включает активный компонент, в частности, пролекарство формулы 1.1, которое более эффективно метаболизируется в печени млекопитающего в трифосфат PSI-7409, который и является ингибитором NS5B HCV полимеразы. Это позволяет снизить дневную дозу пролекарства формулы 1.1 по сравнению с дозой Sovaldi®. Последнее в свою очередь приведет к снижению побочных эффектов.
Приведенные выше данные убедительно подтверждают новизну изобретения и его изобретательский уровень (эффективность).
Ниже приведены определения различных терминов, используемых для описания данного изобретения. Эти определения применимы к терминам, как они использованы в данном описании и формуле изобретения, если иным не ограничены в конкретных случаях, либо по отдельности, либо как часть большей группы.
Термин «активный компонент» (лекарственное вещество) относится к физиологически активному веществу синтетического или иного (биотехнологического, растительного, животного, бактерицидного и так далее) происхождения, обладающему фармакологической активностью, которое является активным ингредиентом фармацевтической композиции.
Термин «кристаллическая форма» - структура вещества, характеризующаяся упаковкой образующих их молекул в один из видов кристаллической решетки.
Термин «поликристаллическая форма» - структура вещества, имеющая поликристаллическое строение, т.е. состоящая из множества мелких монокристаллов, т.е. кристаллитов определенной кристаллической формы.
Термин «лекарственный препарат» означает вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
Термин «фармацевтическая композиция» обозначает композицию, включающую в себя соединение общей формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
Термин «инертный наполнитель», используемый в данном описании, относится к соединению, которое используют для получения фармацевтической композиции, и, как правило, безопасному, нетоксичному, ни биологически, ни иным образом нежелательному, и включает в себя вспомогательные вещества, которые являются приемлемыми для применения в ветеринарии, а также фармакологически приемлемыми для использования человеком. Соединения по данному изобретению могут быть введены отдельно, но обычно их будут вводить в смеси с одним или более фармацевтически приемлемыми эксципиентами, разбавителями или носителями, выбранными с учетом предполагаемого пути введения и стандартной фармацевтической практики.
Термин «терапевтически эффективное количество», используемый здесь, означает количество субстанции, пролекарства или лекарства, необходимое для уменьшения симптомов заболевания у субъекта. Доза субстанции, пролекарства или лекарства будет соответствовать индивидуальным требованиям в каждом конкретном случае. Эта доза может варьироваться в широких пределах в зависимости от многочисленных факторов, таких как тяжесть заболевания, подлежащего лечению, возраста и общего состояния здоровья пациента, других лекарственных средств, с помощью которых пациент проходит лечение, способа и формы введения и опыта лечащего врача. Для перорального введения суточная доза составляет приблизительно от 0,01 до 10 г, включая все значения между ними, в день, в монотерапии и/или в комбинированной терапии. Предпочтительная суточная доза составляет примерно от 0,1 до 7 г в день. Как правило, лечение начинают с большой начальной «нагрузочной дозы», чтобы быстро уменьшить или устранить вирус, сопровождающей убывающую дозу до уровня, достаточного для предотвращения всплеска инфекции.
Термин «субъект» означает млекопитающее, которое включает, но не ограничивается ими, крупный рогатый скот, свиней, овец, куриц, индеек, буйволов, лам, страусов, собак, кошек и человека, предпочтительно субъектом является человек. Предполагается, что в способе лечения субъекта может быть любое из пролекарств общей формулы 1, его стереоизомеры, изотопно-обогащенный аналоги, его фармацевтически приемлемые соли, гидраты, сольваты, кристаллические и полиморфные формы, либо в сочетании их с другим соединением, в том числе с ингибитором NS5A HCV.
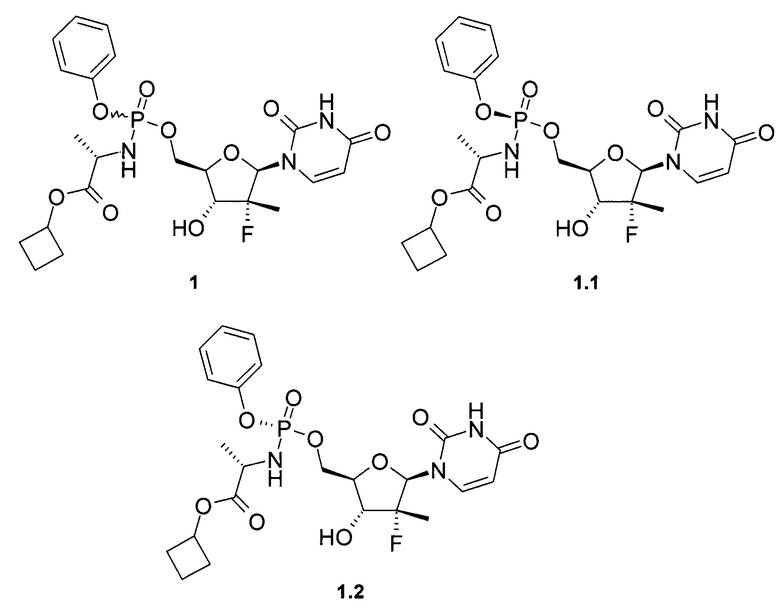
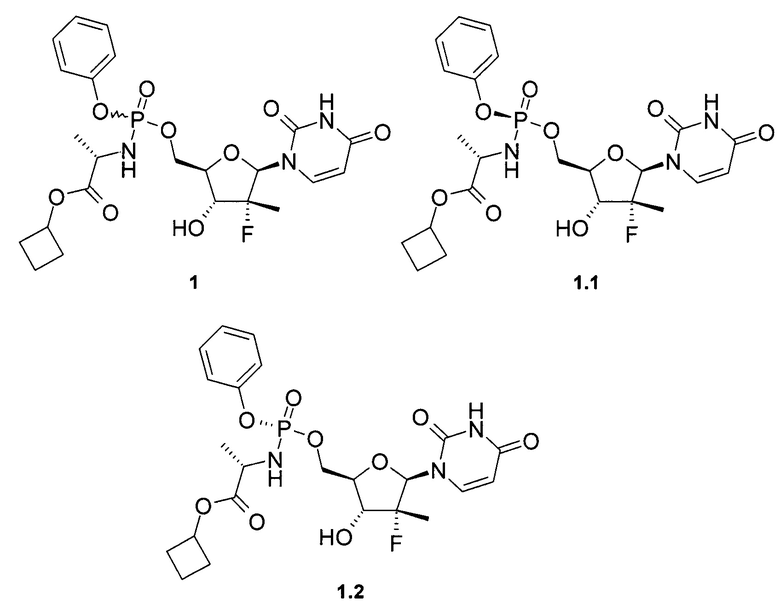
Предметом данного изобретения является противовирусная фармацевтическая композиция, содержащая в качестве пролекарства ингибитора NS5B HCV полимеразы циклобутил (S)-2-{[(2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-4-фтор-3-гидрокси-4-метил-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат общей формулы 1, и/или его фосфорный стереоизомер (Sp стереоизомер: формулы 1.1 или Rp стереоизомер: формулы 1.2), их изотопно-обогащенный аналог, кристаллическую или поликристаллическую форму.
Предпочтительная противовирусная фармацевтическая композиция содержит в качестве пролекарства ингибитор NS5B HCV полимеразы циклобутил (S)-2-{(S)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидро-фуран-2-илметокси]-фенокси-фосфориламино}-пропаноат формулы 1.1, его изотопно-обогащенный аналог, кристаллическую или поликристаллическую форму.
Новая противовирусная фармацевтическая композиция, как показано выше, является более эффективной чем известные композиции, в том числе содержащая в качестве ингибитора NS5B HCV полимеразы Sovaldi®.
Предметом настоящего изобретения является противовирусная фармацевтическая композиция для лечения вируса гепатита С у млекопитающих, содержащая пролекарство общей формулы 1 или его стереоизомер, изотопно-обогащенный аналог, кристаллическую или поликристаллическую форму, необязательно в комбинации с фармацевтически приемлемым наполнителем, носителем, добавкой или разбавителем.
Противовирусная фармацевтическая композиция может быть в виде самых разнообразных перорально вводимых лекарственных форм и носителей, в том числе в форме таблеток, таблеток с покрытием, твердых и мягких желатиновых капсул, растворов, эмульсий, сиропа или суспензии. Противовирусная фармацевтическая композиция по настоящему изобретению является эффективной при введении в виде суппозитория. Наиболее удобным способом введения обычно является пероральный с использованием обычной суточной схемы приема лекарственных доз, который можно регулировать в зависимости от тяжести заболевания и реакции пациента на противовирусное и противоопухолевое лекарство.
Противовирусная фармацевтическая композиция с одним или несколькими обычными эксципиентами, носителями или разбавителями может быть представлены в форме фармацевтических композиций и их единичных доз. Фармацевтические композиции и стандартные лекарственные формы могут состоять из обычных ингредиентов в обычных пропорциях с или без дополнительных активных соединений и лекарственных форм. Противовирусная композиция может содержать любое соответствующее эффективное количество активного ингредиента, соразмерное с назначенной суточной дозой. Противовирусная композиция может быть использована в виде твердых веществ, таких как таблетки или заполненные капсулы, в виде полутвердых порошков, препаратов с замедленным высвобождением или жидкостей, таких как суспензии, эмульсии или заполненные капсулы для перорального применения; или в форме суппозиториев для ректального или вагинального введения. Типичный препарат будет содержать примерно от 5% вес. до 95% вес. активного соединения или соединения. Термин «препарат» или «лекарственная форма» предназначен для включения как твердых, так и жидких композиций активного соединения, и специалисту в данной области техники будет понятно, что активный ингредиент может существовать в виде различных препаратов в зависимости от требуемой дозы и фармакокинетических параметров.
Препараты в твердой форме включают порошки, таблетки, пилюли, капсулы, суппозитории и диспергируемые гранулы. Твердый носитель может представлять собой одно или несколько веществ, которые могут также действовать как разбавители, корригенты, солюбилизаторы, смазывающие вещества, суспендирующие агенты, связующие вещества, консерванты, дезинтегрирующих таблетки агентов или инкапсулирующий материал. В порошках носитель обычно представляет собой тонко измельченное твердое вещество, которое представляет собой смесь с тонкоизмельченным активным компонентом. В таблетках активный компонент обычно смешивают с носителем, имеющим необходимую связывающую способность, в подходящих пропорциях и спрессовывают в желаемую форму желаемого размера. Подходящие носители включают, но не ограничиваются ими, карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, натрийкарбоксиметилцеллюлозу, легкоплавкий воск, масло какао и тому подобное. Препараты в твердой форме могут содержать в дополнение к активному компоненту красители, ароматизаторы, стабилизаторы, буферы, искусственные и природные подсластители, диспергаторы, загустители, солюбилизирующие агенты и тому подобное.
Жидкие составы также пригодны для перорального введения. Жидкие лекарственные формы представляют собой эмульсии, сиропы, эликсиры и водные суспензии. Они включают твердые формы препаратов, которые предназначены для превращения в жидкие препараты непосредственно перед использованием. Эмульсии могут быть приготовлены в растворах, например, в водных растворах пропиленгликоля или могут содержать эмульгаторы, такие как лецитин, моноолеат сорбита или гуммиарабик. Водные суспензии могут быть приготовлены диспергированием тонко измельченного активного компонента в воде с вязкими материалами, такими как природные или синтетические камеди, смолы, метилцеллюлоза, натрийкарбоксиметилцеллюлоза и другими хорошо известными суспендирующими агентами.
Противовирусная фармацевтическая композиция может быть приготовлена для введения в виде суппозиториев. Низкоплавкий воск, такой как смесь глицеридов жирных кислот или масло какао, сначала расплавляют и активный компонент гомогенно диспергируют, например, перемешиванием. Расплавленную гомогенную смесь затем выливают в формы удобного размера, дают остыть и затвердеть.
Противовирусная фармацевтическая композиция может быть приготовлена для вагинального введения. Применение суппозиториев, тампонов, кремов, гелей, паст, пен или спреев, содержащих в дополнение к активному ингредиенту такие носители, которые известны в данной области техники, будет уместно.
Более предпочтительной является противовирусная фармацевтическая композиция, которая вместе с новым пролекарством общей формулы 1 или его стереоизомером формулы 1.1, изотопно-обогащенным аналогом, кристаллической или поликристаллической формой дополнительно включает в себя антивирусное или противораковое средство в терапевтически эффективном количестве.
Более предпочтительной является противовирусная фармацевтическая композиция, которая вместе с новым пролекарством общей формулы 1 или его стереоизомером формулы 1.1, изотопно-обогащенным аналогом, кристаллической или поликристаллической формой дополнительно содержит терапевтически эффективное количество ингибитора NS5A HCV, выбраного из группы, включающей Даклатасвир (Daclatasvir) (Daklinza, BMS790052) Гепавивир (AV-4025), AV-4056 and AV-4058, Омбитасвир (Ombitasvir) (АВТ-267), Элбасвир (Elbasvir) (MK-8742) или Велпатасвир (Velpatasvir) (VEL, GS-5816) или Нарлапревир (Narlaprevir, SCH 900518) - ингибитор неструктурированного белка 3 ВГС (NS3 HCV или Симепревир (Simeprevir, Olysio) ингибитор неструктурированного белка NS3/NS4 ВГС.
Более предпочтительной является также противовирусная фармацевтическая композиция, которая вместе с новым пролекарством общей формулы 1 или его стереоизомером формулы 1.1, изотопно-обогащенным аналогом, кристаллической или поликристаллической формой дополнительно содержит терапевтически эффективное количество ингибитора полимеразы ДНК HBV и ингибитор ВИЧ-1 обратной транскриптазы (RT).
Предметом настоящего изобретения является способ комбинированного лечения вирусных и раковых заболеваний у субъекта, нуждающегося в этом, причем указанный способ включает в себя последовательное или одновременное введение терапевтически эффективного количества противовирусной фармацевтической композиции, включающей пролекарство общей формулы 1 или его стереоизомер формулы 1.1, изотопно-обогащенный аналог, кристаллическая или поликристаллическая форма и другого противовирусного или противоракового средства. Понятно, что время между раздельным введением препаратов может находиться в диапазоне, который включает в себя любой диапазон времени.
Предметом настоящего изобретения является способ комбинированного лечения и/или профилактики субъекта, нуждающегося в этом, причем указанный способ включает введение субъекту терапевтически эффективного количества противовирусной фармацевтическая композици содержащей в качестве пролекарства ингибитора NS5B HCV полимеразы циклобутил (S)-2-{[(2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-4-фтор-3-гидрокси-4-метил-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат общей формулы 1, и/или его фосфорный стереоизомер (Sp стереоизомер: формулы 1.1 или Rp стереоизомер: формулы 1.2), их изотопно-обогащенный аналог, кристаллическую или поликристаллическую форму, по данному изобретению, которая может включать дополнительно терапевтически эффективное количество одного или нескольких других противовирусных или противораковых агентов, в котором используют параллельное или альтернативное введение агентов. Понятно, что время между последовательным введением агентов может находиться в любом временном диапазоне.
Примеры «других противовирусных агентов» включают в себя, но не ограничиваются ими: ингибиторы HCV NS3 протеазы [US US 20140296136, US 8,987,195, US 7973040, US 2012214783], ингибиторы NS4 HCV [EP 1497282], ингибиторы NS3/NS4 HCV [ЕР 2364984], ингибиторы NS5A HCV [С. Wang et al. Hepatitis С virus RNA elimination and development of resistance in replicon cells treated with BMS-790052. Antimicrob. Agents Chemother. 2012, 56, 1350-1358. https://en.wikipedia.org/wiki/Daclatasvir; A.V. Ivachtchenko et al. Discovery of Novel Highly Potent Hepatitis С Virus NS5A Inhibitor (AV4025). J. Med. Chem. 2014, 57, 7716-7730; Pat. Appl. US 14/845,333); Toll-like receptor agonists (see WO 2015023958, WO 2012097012); and other inhibitors (see WO 2014106019, WO 2014033176, WO 2014033170, WO 2014033167, WO 2013006394, US 20090163545].
Предполагается, что другой антивирусный агент представляет собой, но не ограничивается ими: интерферон-альфа, интерферон-бета, пегилированный интерферон-альфа, рибавирин, левовирин, вирамидин, другой нуклеозидный HCV полимеразный ингибитор, ненуклеозидный HCV полимеразный ингибитор, HCV протеазный ингибитор, HCV хеликазный ингибитор или HCV ингибитор слияния, ингибитор полимеразы ДНК HBV и ингибитор ВИЧ-1 обратной транскриптазы (RT). Когда пролекарство общей формулы 1 или его стереоизомер, изотопно-обогащенные аналоги, кристаллические или поликристаллические формы вводят в комбинации с другим антивирусным или противораковым агентом, активность может быть увеличена по сравнению с исходным пролекарством. Когда лечение комбинированное, то введение препаратов может быть одновременным или последовательным по отношению к пролекарству общей формулы 1 или его стереоизомерам, изотопно-обогащенному аналогам, кристаллическим или поликристаллическим формам. Понятие «одновременное введение», используемое здесь, таким образом, включает в себя введение агентов в то же время или в разное время. Прием двух или более агентов в одно и то же время может быть достигнут с помощью одной препаративной формы, содержащей два или более активных ингредиентов, или, по существу, одновременного введения двух или более лекарственных форм с одним активным агентом. Следует понимать, что ссылки здесь на лечение распространяются и на профилактику. Кроме того, термин «лечение» вирусной инфекции, как он использован здесь, включает также лечение или профилактику заболевания или состояния, связанного с опосредованной вирусной инфекцией, или их клинических симптомов.
Авторы неожиданно обнаружили что новая противовирусная фармацевтическая композиция, включающая пролекарство общей формулы 1 и/или его стереоизомер формулы 1.1, является более эффективной, чем известная противовирусная композиция, в том числе более эффективна чем содержащая Sovaldi®.
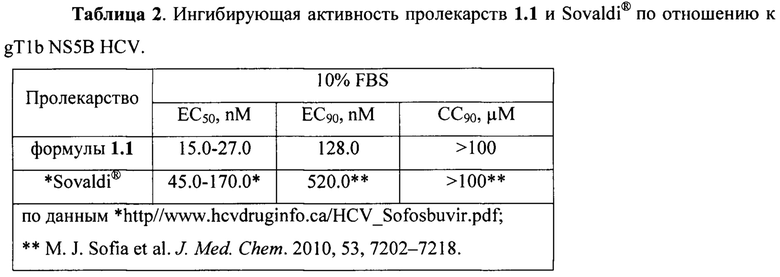
В частности Sovaldi® по отношению к генотипу 1b (gT1b) HCV имеет ЕС50 0.045-0.170 μM [http://www.hcvdruginfo.ca/downloads/HCV_Sofosbuvir.pdf] и EC90 0.59 μM, а новое пролекарство формулы 1.1 имеет ЕС50 15.0-27.0 nM и ЕС90 128.0 nM (Таблица 2), т.е. новое пролекарство формулы 1.1 более чем в три раза активнее, чем Sovaldi®. Время полураспада пролекарства формулы 1.1 в S9 фракции микросом печени человека составляет T1/2hS9=0,05 ч, а у Sovaldi® соответственно T1/2hS9=0,57 ч (Таблица 3), т.е. новое пролекарство формулы 1.1 в 11 раз быстрее метаболизирует в S9 фракции микросом печени человека, чем Sovaldi®. Кроме того, концентрация и AUC24 ч трифосфат PSI-7409, образующегося при метаболизме пролекарства формулы 1.1 в печени крыс, составляет Сmax=3,224.0 нг/г и AUC24 ч=30,487.0 нг⋅ч/г, в то время как при аналогичном метаболизме Sovaldi® имеет Сmax=1,934.0 нг/г и AUC24 ч=16,796.0 нг⋅ч/г (Таблице 4). Это свидетельствует о том, что новое пролекарство почти в 2 раза более эффективно метаболизирует в печени в нужный трифосфат PSI-7409 (лекарство).
Настоящее изобретение далее будет описано в связи с определенными вариантами осуществления, которые не предназначены для ограничения его объема. Напротив, настоящее изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем формулы изобретения. Таким образом, следующие примеры, которые включают в себя конкретные варианты, иллюстрируют, но не ограничивают настоящее изобретение.
Пример 1. Протокол синтеза пролекарства циклобутил (S)-2-{[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидро-фуран-2-илметокси]-фенокси-фосфориламино}-пропаноата общей формулы (1) и его стереоизомеров формулы 1.1 и формулы 1.2 (Схема 1).
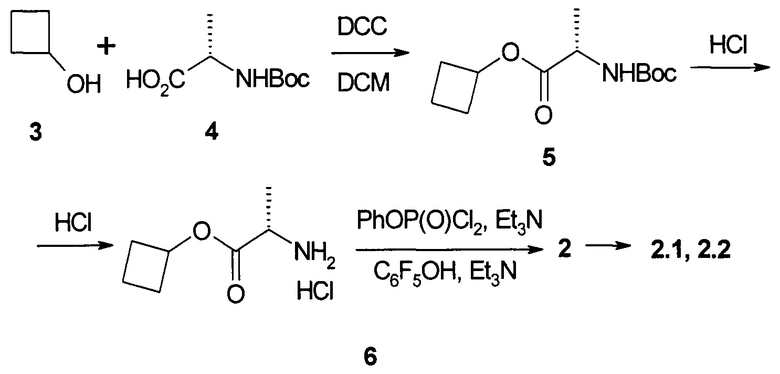
К раствору N-Boc-L-аланина (4: 15,5 г, 81,9 ммоль) в 300 мл дихлорметана при 0°С добавляют DCC (16,9 г, 81,9 ммоль) и через 5 мин циклобутанол (3: 5,6 г, 78,0 ммоль) и DMAP (2,0 г, 16,4 ммоль). Смесь оставляют перемешиваться на ночь при комнатной температуре, упаривают в вакууме и остаток обрабатывают 300 мл этилацетата. Осадок отфильтровывают и промывают этилацетатом. Фильтрат промывают 5% раствором лимонной кислоты (2×100 мл), насыщенным раствором NaHCO3 (2×100 мл), рассолом, сушат над Na2SO4 и упаривают в вакууме. Получают 19,6 г (98%) (S)-циклобутил 2-(трет-бутоксикарбониламино)-пропаноат (5) в виде белого порошка. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7.22 (d, J=7.2 Hz, 0.85Н), 6.87 (m, 0.15Н), 4.89 (р, J=7.2 Hz, 1H), 3.94 (m, 1H), 2.26 (m, 2H), 1.98 (m, 2H), 1.74 (m, 1H), 1.59 (m, 1H), 1.38 (s, 7.5H), 1.34 (brs, 1.5H), 1.22 (d, J=7.2 Hz, 3H).
К раствору соединения 5 (19,6 г, 80,6 ммоль) в 50 мл диоксана добавляют 230 мл 3М HCl в диоксане, смесь оставляют перемешиваться на ночь и затем упаривают в вакууме. Остаток обрабатывают 400 мл эфира и оставляют перемешиваться на ночь. Осадок отфильтровывают, промывают эфиром и сушат в вакууме. Получают 14,1 г (97%) (S)-циклобутил 2-амино-пропаноат гидрохлорид (6) в виде белого порошка. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8.56 (brs, 3Н), 5.00 (р, J=7.6 Hz, 1Н), 4.02 (q, J=7.2 Hz, 1H), 2.31 (m, 2H), 2.07 (m, 2H), 1.78 (m, 1H), 1.62 (m, 1H), 1.41 (d, J=7.2 Hz, 3H).
К раствору соединения 6 (14,4 г, 80,2 ммоль) в 214 мл дихлорметана добавляют фенилдихлорфосфат (16,9 г, 80,2 ммоль). Смесь охлаждают до -75-70°С и по каплям добавляют раствор триэтиламина (16,2 г, 160,4 ммоль) в 16 мл дихлорметана, поддерживая температуру -75-70°С. Смесь перемешивают при -70°С в течение 30 мин и затем нагревают до -20°С. Добавляют раствор пентафторфенола (14,6 г, 79,4 ммоль) в 105 мл дихлорметана при -20-10°С, затем по каплям раствор триэтиламина (8,1 г, 80,2 ммоль) в 8 мл дихлорметана и смесь оставляют перемешиваться на ночь при комнатной температуре. Смесь упаривают в вакууме, добавляют 500 мл этилацетата и 500 мл воды, органический слой отделяют, промывают 5% раствором NaHCO3, рассолом, сушат над Na2SO4 и упаривают в вакууме. К остатку добавляют 200 мл смеси гексан/этилацетат 6:1 и оставляют перемешиваться на ночь. Образовавшийся осадок отфильтровывают, промывают 50 мл смеси гексан/этилацетат 6:1 и сушат на воздухе. Получают 16,7 г циклобутил (S)-2-((перфторфенокси)(фенокси)фосфориламино)-пропаноат (2).
Полученный продукт 2 перекристаллизовывают из 500 мл смеси гексан/этилацетат 4:1 получают 13,8 г (37%) циклобутил (S)-2-((S)-(перфторфенокси)-(фенокси)-фосфориламино)пропаноат формулы (2.1) в виде белого рыхлого порошка. 1Н ЯМР (300 МГц, ДМСО-d6) δ 7.42 (m, 2Н), 7.24 (m, 3Н), 6.87 (dd, J1=14.1 Hz, J2=10.2 Hz, 1H), 4.87 (p, J=7.5 Hz, 1H), 3.94 (m, 1H), 2.23 (m, 2H), 1.94 (m, 2H), 1.71 (m, 1H), 1.58 (m, 1H), 1.27 (d, J=7.2 Hz, 3Н). Маточный раствор от промывки смесью гексан/этилацетат 6:1 при перекристаллизации соединения формулы 2.1 упаривают в вакууме и трижды перекристаллизовывают из гексана. Получают циклобутил (S)-2-((R)-(перфторфенокси)-(фенокси)-фосфориламино)-пропаноат (2.2) в виде белого рыхлого порошка. 1Н ЯМР (300 МГц, ДМСО-d6) δ 7.42 (m, 2Н), 7.26 (m, 3Н), 6.85 (dd, J1=13.8 Hz, J2=10.2 Hz, 1H), 4.88 (p, J=7.5 Hz, 1H), 3.95 (m, 1H), 2.24 (m, 2H), 1.93 (m, 2H), 1.72 (m, 1H), 1.59 (m, 1H), 1.28 (d, J=6.9 Hz, 3H).
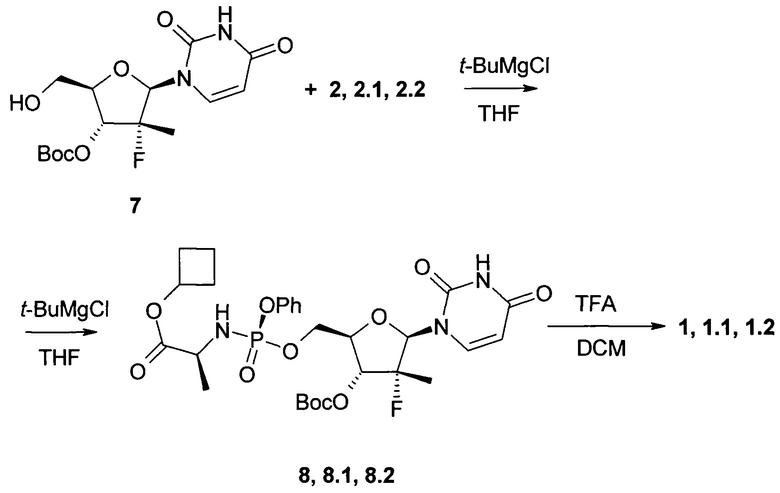
К раствору трет-бутил (2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-(гидроксиметил)-4-метил-4-фтор-тетрагидрофуран-3-ил карбоната (7: 5 г, 13,9 ммоль) в 165 мл ТГФ добавляют 1М раствор трет-бутилмагния хлорида в ТГФ (31,3 мл, 31,3 ммоль) под аргоном при 0°С и смесь перемешивают в течение 30 мин при комнатной температуре. Затем при помощи шприца добавляют раствор соединения формулы 2.1 (7,8 г, 16,7 ммоль) в 30 мл ТГФ при 0-5°С и смесь перемешивают под аргоном в течение суток. К смеси добавляют 10 мл метанола и упаривают в вакууме. Остаток растворяют в 500 мл этилацетата, промывают 5% раствором лимонной кислоты, 5% раствором NaHCO3, сушат над Na2SO4 и упаривают в вакууме. Остаток хроматографируют на силикагеле (элюент гексан/этилацетат 1:2). Получают 5,89 г (66%) циклобутил (S)-2-((S)-(((2R,3R,4R,5R)-3-(трет-бутоксикарбонилокси)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-4-метил-4-фтор-тетрагидрофуран-2-ил)метокси)(фенокси)фосфориламино)пропаноат (8.1) в виде бесцветной застывшей пены. LC-MS (ESI) 642 (М+Н)+.
Аналогично получают циклобутил (S)-2-((((2R,3R,4R,5R)-3-(трет-бутоксикарбонилокси)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-4-метил-4-фтор-тетрагидрофуран-2-ил)метокси)(фенокси)фосфориламино)пропаноат (8). Выход 52%. LC-MS (ESI) 642 (M+H)+, исходя из полупродуктов 7 и 2, и циклобутил (S)-2-((R)-(((2R,3R,4R,5R)-3-(трет-бутоксикарбонилокси)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-4-метил-4-фтор-тетрагидрофуран-2-ил)метокси)(фенокси)фосфориламино)пропаноат (8.2). Выход 59%. LC-MS (ESI) 642 (М+Н)+, исходя из полупродуктов 7 и 2.2.
К раствору соединения формулы 8.1 (4,45 г, 6,9 ммоль) в 60 мл дихлорметана при 0°С добавляют 60 мл трифторуксусной кислоты, смесь перемешивают в течение 15 ч при комнатной температуре, упаривают в вакууме, растворяют в 250 mL of DCM, промывают 300 мл of 5% NaHCO3, сушат над Na2SO4 и упаривают в вакууме. Перекристаллизовывают из смеси этилацетата с метил-трет-бутиловым эфиром (1:1). Получают 2.7 g (71%) циклобутил (S)-2-((S)-(((2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-гидрокси-4-метил-4-фтор-тетрагидрофуран-2-ил)метокси)-(фенокси)-фосфориламино)-пропаноат формулы (1.1) в виде белого кристаллического вещества. LC-MS (ESI) 542 (М+Н)+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11.51 (brs, 1H), 7.56 (d, J=8.0 Hz, 1H), 7.38 (m, 2H), 7.23 (m, 2H), 7.19 (m, 1H), 6.03 (m, 2H), 5.84 (d, J=6.8 Hz, 1H), 5.55 (dd, J1=8.0 Hz, J2=1.2 Hz, 1H), 4.85 (p, J=7.2 Hz, 1H), 4.37 (m, 1H), 4.27 (m, 1H), 4.01 (m, 1H), 3.83 (m, 2H), 2.23 (m, 2H), 1.95 (m, 2H), 1.71 (m, 1H), 1.56 (m, 1H), 1.25 (d, J=22.8 Hz, 3H), 1.23 (d, J=6.8 Hz, 3H).
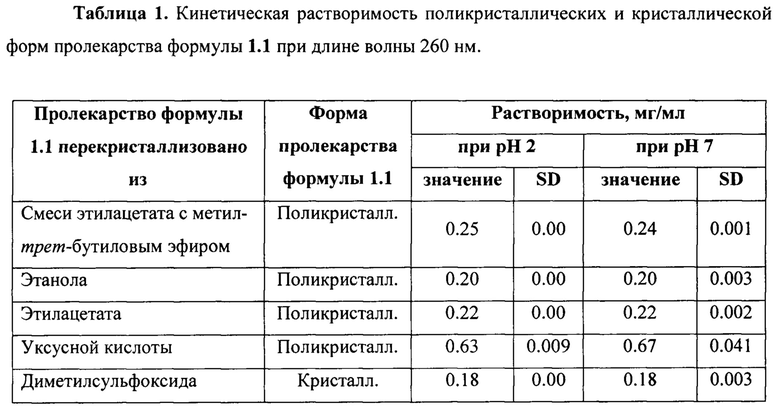
Перекристаллизация пролекарства формулы 1.1 из различных растворителей приводит к получению поликристаллических или кристаллических форм. При перекристаллизации из смеси этилацетата с метил-трет-бутиловым эфиром (1:1), этанола, этилацетата и смеси уксусной кислоты с водой получают пролекарство формулы 1.1 в поликристаллических формах, включающих в основном ромбическую фазу с параметрами элементарной ячейки а=28.1056(8)А, b=16.8998(4)А, с=5.25380(12)А и моноклинную фазу с параметрами элементарной ячейки а=16.2770(6)А, b=16.9117(8)А, с=5.20429(15)А, в=117.822(2)°.
При перекристаллизации пролекарства формулы 1.1 из смеси диметилсульфоксида с водой получают вещество в белой кристаллической форме, состоящей из ромбической фазы с параметрами элементарной ячейки а=28.1056(8)А, b=16.8998(4)А, с=5.25380(12)А.
Растворимость кристаллической и поликристаллических форм после перекристаллизации из различных растворителей при pH 2 и рН7 близка и составляет 0,18-0,25 мг/мл. Исключение составляет поликристаллический образец, полученный при перекристаллизации из диметилсульфоксида, растворимость которого несколько выше и имеет значение 0,63-0,67 мг/мл (Таблица 1).
Аналогично получают циклобутил (S)-2-((R)-(((2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-гидрокси-4-метил-4-фтор-тетрагидрофуран-2-ил)метокси)-(фенокси)-фосфориламино)-пропаноат (1) с выходом 58%. LC-MS (ESI) 542 и циклобутил (S)-2-((R)-(((2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-гидрокси-4-метил-4-фтор-тетрагидрофуран-2-ил)метокси)-(фенокси)-фосфориламино)-пропаноат (1.2) с выходом 64%. LC-MS (ESI) 542 (М+Н)+. 1Н ЯМР (300 МГц, ДМСО-d6) δ 11.53 (brs, 1Н), 7.56 (d, J=8.1 Hz, 1H), 7.38 (m, 2H), 7.19 (m, 3H), 6.08 (m, 2H), 5.91 (d, J=6.3 Hz, 1H), 5.57 (d, J=8.1 Hz, 1H), 4.85 (p, J=7.5 Hz, 1H), 4.41 (m, 1H), 4.27 (m, 1H), 4.05 (m, 1H), 3.79 (m, 2H), 2.23 (m, 2H), 1.94 (m, 2H), 1.70 (m, 1H), 1.56 (m, 1H), 1.24 (d, J=23.4 Hz, 3H), 1.21 (d, J=6.0 Hz, 3H).
Пример 2. Получение противовирусной композиции в виде таблетки. Крахмал (500 мг), молотую лактозу (800 мг), тальк (200 мг), и 1500 мг пролекарства формулы 1.1 смешивали друг с другом и прессовали в бар. Полученный брусок измельчали в гранулы и просеивали через сито, чтобы собрать гранулы размером 14-16 меш. Полученные таким образом гранулы были сформированы в таблетки подходящей формы весом 400 или 800 мг каждая.
Пример 3. Получение противовирусной композиции в виде капсул. Пролекарство формулы 1.1 тщательно смешивали с порошком лактозы в соотношении 1:1. Полученную порошкообразную смесь упаковывали в желатиновые капсулы подходящего размера по 200 мг 400 мг в каждой капсуле.
Пример 4. Получение противовирусной композиции в форме композиций для внутримышечных, внутрибрюшинных или подкожных инъекций. Смешивали 500 мг пролекарства формулы 1.1, 300 мл хлорбутанола, 2 мл пропиленгликоля и 100 мл воды для инъекций. Полученный раствор фильтровали, помещали в ампулы по 5 мл и запечатывали.
Пример 5. Определение анти-HCV активности и цитотоксичности композиций, включающих пролекарства формулы 1.1 и Sovaldi®. Для определения противовирусной активности тестируемых композиций, включающих пролекарства, использовалась клеточная линия гепатоцеллюлярной карциномы человека Huh7, стабильно трансфицированная репликоном вируса гепатита С (HCV). По 50 мкл клеточной суспензии в полной культуральной среде (DMEM 1X, Cellgro; cat. # 10-013-CV) переносили в 96-луночные плашки с итоговой плотностью 7500 клеток на лунку. Серийное разведение тестируемых пролекарств готовили из свежеприготовленного 200-кратного исходного раствора в ДМСО с 11 концентрационными точками с шагом разведения 3 от 20 нМ в полной среде и использовали в качестве 2-кратных растворов. Не менее чем через 4 часа после посадки клеток добавляли по 50 мкл растворов серийных разведений пролекарств. Конечная концентрация пролекарств составляла от 10 нМ до 0.1 пМ, а ДМСО - 0.5%. Плашку с клетками инкубировали в течение 3 дней при 37°С в увлажненной атмосфере 5% CO2. После инкубации среда была удалена переворачиванием плашки с аккуратным встряхиванием. Клетки фиксировали 100 мкл 1:1 раствора ацетон : метанол в течение 1 минуты, промывали 3 раза фосфатным буфером (PBS) и затем блокировали 150 мкл/лунку 10% бычьей фетальной сывороткой (FBS) в PBS или в течение часа при комнатной температуре. Далее клетки промывали 3 раза PBS и инкубировали в течение 2 часов при 37°C с 100 мкл/лунку антителами к неструктурному белку 5В (NS5B) HCV (Affinity BioReagents; cat. # МА1-080, исходный раствор (1 мг/мл) разводили 1:4000 в 10% FBS-PBS). Клетки промывались 3 раза PBS проявлялись 100 мкл/лунку раствора OPD (на 1 плашку: 1 таблетка OPD, растворенная в 12 мл цитрат/фосфатного буфера с добавлением 5 мкл 30% Н2О2) в течение 30 минут в темноте при комнатной температуре. Реакцию останавливали 100 мкл/лунку 2N H2SO4, после чего измеряли OD490 с помощью мультифункционального ридера Victor3 V 1420 (Perkin Elmer). Значения ЕС50 тестируемых пролекарств определяли построением кривой активности в программе GraphPad Prizm. В частности, по отношению к генотипу 1b (gT1b) HCV в среде 10% бычьей фетальной сыворотки (10% FBS) в сопоставимых условиях Sovaldi® имеет ЕС50 45.0-170.0 nM и ЕС90 520.0.0 nM, а новое пролекарство формулы 1.1 имеет ЕС50 15.0-27.0 nM и ЕС90 128.0 nM (Таблица 2). Таким образом, новое пролекарство формулы 1.1 более активно чем Sovaldi® более чем в три раза.
Цитотоксичность тестируемых композиций, включающих пролекарства, определялась параллельно на той же клеточной линии Huh7 с помощью набора ATPLite (Perkin-Elmer, Boston, USA) в соответствии с инструкцией производителя. По 50 мкл клеточной суспензии в полной культуральной среде (DMEM 1X, Cellgro; cat. # 10-013-CV) переносили в 96-луночные плашки с черными стенками и прозрачным дном с итоговой плотностью 7500 клеток на лунку. Через 18 часов после посадки клеток добавляли по 50 мкл растворов серийных разведений соединений. Плашку с клетками инкубировали в течение 4 дней при 37°С в увлажненной атмосфере 5% CO2. Далее клетки промывали 2 раза 200 мкл/лунку PBS и лизировали добавлением 50 мкл/лунку лизирующего буфера (все реактивы из набора ATPLite). После помешивания в течение 5 минут на шейкере добавляли 50 мкл/лунку субстрата. После дополнительной 5-минутной инкубации плашку хранили 10 минут в темноте и считывали люминесценцию в лунках с помощью мультифункционального ридера Victor3 V 1420 (Perkin Elmer). Значения СС50 тестируемых соединений определяли построением кривой цитотоксичности в программе GraphPad Prizm. В частности, для композиции, включающей пролекарство формулы 1.1 и Sovaldi® найдены значения цитотоксичности СС50>100 мкМ (Таблица 2) и терапевтического окна (терапевтический индекс TI=ЕС50/СС50) TI>6,000.0
Пример 6. Определение кинетической растворимости веществ.
Принцип метода. Изучаемое вещество растворяют в ДМСО до концентрации 10 мМ, затем вносят в водный растворитель (фосфатный буфер, вода, универсальные буферы с различными pH) до концентрации 200 мкМ. Полученный раствор инкубируют в течение часа при комнатной температуре на шейкере в 96-луночной фильтровальной плашке (Millipore's MultiScreen Solubility Filter Plate), после этого осадок отфильтровывают под вакуумом. Спектр поглощения вещества регистрируют на спектрофотометре в диапазоне 240-400 нм с шагом 10 нм. Для количественных расчетов растворимости используют калибровочную кривую стандартных растворов (0-200 мкМ) с содержанием 40% ацетонитрила. Диапазон определяемых концентраций 3-200 мкМ. Тестирование проводится в дубликатах.
Приготовление калибровочных стандартов. Калибровочные стандарты готовили из 50-кратных сток-растворов в ДМСО с последующим разбавлением их в буфере с 40% содержанием ацетонитрила, который добавлялся, чтобы обеспечить полную растворимость тестируемого соединения в калибровочном образце. 6 стандартных образцов с концентрациями 0, 3,125, 12,5, 50, 100 и 200 мкМ были приготовлены в лунках 96-луночного UV-планшета путем добавления 4 мкл соответствующих 50х сток-растворов в ДМСО к 196 мкл буфера с 40% содержанием ацетонитрила. При этом концентрация ДМСО во всех точках оставалась постоянной и была равна 2% (v/v).
Для построения калибровочных кривых сняли оптический спектр UV-планшета в диапазоне длин волн от 250 нм до 400 нм с шагом 10 нм. Из полученных спектров для каждого соединения были выбраны длины волн, отвечающие следующим критериям:
- При минимальной концентрации вещества OD>0.1 (AU)
- При максимальной концентрации вещества OD<2.0
Калибровочная кривая была построена для каждого соединения по зависимости OD при выбранной длине волны от концентрации.
Определение кинетической растворимости соединений. Растворимость определяли в фильтровальном планшете MultiScreen Solubility (Millipore Corp.) следующим образом:
- В лунку фильтровального планшета MultiScreen Solubility добавили 196 мкл буфера (без ацетонитрила) и 4 мкл 10 мМ вещества в ДМСО либо 4 мкл ДМСО (для холостого образца). Планшет инкубировали в течение часа на шейкере (400 rpm) при комнатной температуре.
- После инкубации растворы отфильтровали через фильтровальный планшет с помощью вакуума (10'' Hg) в полипропиленовый планшет с U-образным дном.
- Из планшета с U-образным дном перенесли 120 мкл/лунку фильтрата в новый UV-планшет, после чего добавили туда 80 мкл/лунку ацетонитрила.
- Измерили оптическую плотность полученных растворов при выбранной ранее длине волны для каждого соединения.
Расчеты. Финальная концентрация вещества в фильтрате рассчитывалась по формуле: Сфильтрат=(ODλFiltrate-AλBlank)/Slope×1,67,
где:
- ODλFiltrate - оптическая плотность фильтрата при выбранной длине волны,
- ODλBlank - OD холостого образца,
- Slope - тангенс угла наклона калибровочной прямой,
- 1.67 - коэффициент разбавления фильтрата ацетонитрилом.
Полученные результаты представлены в Таблице 1.
Пример 7. Порошковые рентгенофазовые исследования образцов пролекарств. Все дифрактограммы зарегистрированы на дифрактометре Bruker D8 Advance Vario, оснащенном рентгеновской трубкой с медным анодом и Ge(111)-монохроматором (CuKo∧) и позиционно-чувствительным детектором LynxEye, в установках на просвет. Интервал съемки составил 3-90° 26 для образца s5 и 5.7-90° 26 для остальных образцов, шаг 0.01° 26. Анализ проводили в программе Bruker Topas5 ['Bruker TOPAS 5 User Manual. - Karlsruhe, Germany: Bruker AXS GmbH, 2015.].
Образцы, полученные перекристаллизацией 1B.7 из смеси этилацетата с метил-трет-бутиловым эфиром (1:1), этанола, этилацетата и смеси уксусной кислоты с водой имеют поликристаллическую форму. Порошковые рентгенофазовые исследования этих образцов показывают, что образцы имеют одинаковый качественный фазовый состав и незначительно различаются соотношением фаз. Образцы содержат ромбическую фазу с параметрами элементарной ячейки а=28.1056(8)А, b=16.8998(4)А, с=5.25380(12)А. Анализ систематических погасаний позволяет предположить пространственную группу P212121. Объем элементарной ячейки 2495.45(11) А3 соответствует заявленному составу и Z'=1. Образцы содержат также моноклинную фазу с параметрами элементарной ячейки а=16.2770(6)А, b=16.9117(8)А, с=5.20429(15)А, в=117.822(2)°. Систематические погасания позволяют предположить пространственную группу Р21. Объем элементарной ячейки 1266.98(9) А3 соответствует заявленному составу и Z'=1. Оценка соотношения фаз, основанная на сравнении интегральных интенсивностей пиков, предсказывает содержание моноклинной фазы от 30 до 50%.
Образец, полученный перекристаллизацией 1В.7 из смеси диметилсульфоксида с водой представляет собой белое вещество кристаллической формы. Образец этой формы по данным порошковые рентгенофазовые исследования однофазен и состоит из ромбической фазы с параметрами элементарной ячейки а=28.1056(8)А, b=16.8998(4)А, с=5.25380(12)А. Анализ систематических погасаний позволяет предположить пространственную группу P212121. Объем элементарной ячейки 2495.45(11) А3 соответствует заявленному составу и Z'=1.
Пример 8. Определение стабильности в биологических средах композиций, включающих пролекарства формулы 1.1. Исходные композиции включающие пролекарства формулы 1.1 (тестируемое соединение) были приготовлены с концентрацией 10 мМ в ДМСО, из которых затем готовили 100-кратные рабочие растворы с концентрацией 100 мкМ в смеси ацетонитрил: вода с объемным отношением 1:1.
а) Стабильность в S9 фракции. Реакционная смесь была приготовлена в 0,1 М калия фосфатном буфере (pH 7,4 BD Gentest) в общем финальном объеме 250 мкл и содержала 1 мМ NADPH-тетранатриевую соль (AppliChem), 7 мМ глюкозо-6-фосфат натриевую соль (Sigma), 1.5 U/мл глюкозо-6-фосфат дегидрогеназу (Sigma), 3.3 мМ MgCl2 (Sigma), 5 мМ уридин-5-дифосфат-глюкуроновой кислоты тринатриевую соль (УДФГК, Sigma) и 1 мкМ тестируемого соединения (указаны финальные концентрации). Метаболическая реакция была инициирована путем добавления суспензии S9 фракции печени человека (BD Gentest), финальная концентрация белка составляла 1 мг/мл. Реакционная смесь инкубировалась при 37°С на шейкере (Vortemp56) с перемешиванием при 400 об/мин. Через определенные промежутки времени (0, 0.25, 0.5, 1, 2, 4, 6 8, 24 ч), проводился отбор проб в объеме 30 мкл, реакция останавливалась путем добавления к отобранной пробе 180 мкл холодного ацетонитрила, содержащего внутренний стандарт. Осаждение белков проводилось на льду в течение 15 мин. После чего образцы центрифугировались и в течение 10 мин при 3000 об/мин. 150 мкл супернатанта отбиралось для анализа. Инкубация проводилась в двух повторах, каждая проба измерялась дважды.
б) Стабильность в искусственном желудочном и кишечном соках. Тестируемая композиция, включающих пролекарство формулы 1.1 в конечной концентрации 1 мкМ инкубировали в искусственном желудочном соке (0,2% NaCl в 0,7% v/v HCl) и искусственном кишечном соке (0,05М KH2PO4, pH б) 6,75). Инкубацию проводили в шейкере-инкубаторе (Vortemp56) при 37°С и перемешивании при 300 об/мин. Через определенные промежутки времени (0, 0.25, 0.5, 1, 2, 4, 6 8, 24 ч), проводился отбор проб в объеме 30 мкл, реакция останавливалась путем добавления к отобранной пробе 180 мкл холодного ацетонитрила, содержащего внутренний стандарт. После чего образцы центрифугировались и в течение 10 мин при 3000 об/мин. 150 мкл супернатанта отбиралось для анализа. Инкубация проводилась в двух повторах, каждая проба измерялась дважды.
в) Стабильность в плазме крови. Тестируемое соединение в конечной концентрации 1 мкМ инкубировали в пулированной плазме крови человека (Innovative Research). Инкубацию проводили в шейкере-инкубаторе (Vortemp56) при 37°С и перемешивании при 300 об/мин. Через определенные промежутки времени (0, 0.25, 0.5, 1, 2, 4, 6 8, 24 ч), проводился отбор проб в объеме 30 мкл, реакция останавливалась путем добавления к отобранной пробе 180 мкл холодного ацетонитрила, содержащего внутренний стандарт. После чего образцы центрифугировались и в течение 10 мин при 3000 об/мин. 150 мкл супернатанта отбиралось для анализа. Инкубация проводилась в двух повторах, каждая проба измерялась дважды.
Анализ проб. Анализ проб проводился методом ВЭЖХ-МС/МС, разработанным для каждого тестируемого пролекарства, с использованием хроматографической системы 1290 Infinity II (Agilent Technologies), сопряженной с тандемным масс-спектрометром QTRAP5500 (АВ Sciex). При разработке условий масс-спектрометрического детектирования растворы тестируемых соединений в смеси ацетонитрил-вода 1:1 с концентрацией 100 нг/мл анализировали путем прямого ввода в масс-спектрометр при помощи шприцевого насоса при ионизации электрораспылением в режиме регистрации положительных ионов. При сканировании в режиме полного ионного тока (MS1) определяли молекулярный ион для каждого соединения, основные ионы-продукты регистрировали в режиме MS2. Далее была проведена оптимизация МС/МС метода в режиме MRM для достижения максимальной чувствительности. При количественной обработке хроматограмм использовали наиболее интенсивный MRM-переход для аналита и внутреннего стандарта. Хроматографическое разделение проводили на колонке YMC Triart С18, 50×2 мм, 1,9 мкм в градиентном режиме элюирования в подвижной фазе состава 0,1% муравьиная кислота в воде - 0.1% муравьиная кислота в ацетонитриле. В качестве внутреннего стандарта использовали толбутамид (Fluka).
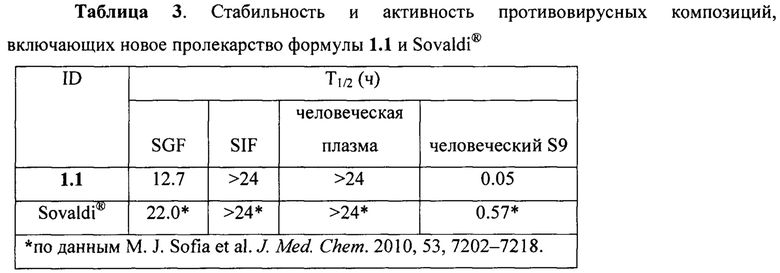
Вычисления. По кинетике убыли тестируемого пролекарства в противовирусной композиции в процессе инкубации в биологической среде рассчитывали время полураспада (Т1/2). Для расчетов использовали нормированные на сигнал внутреннего стандарта значения площадей хроматографических пиков веществ в опытных образцах. По линейной зависимости лог-нормированных площадей хроматографических пиков от времени рассчитывали константу скорости элиминации k - наклон линейного участка). Далее рассчитывали время полураспада: Т1/2=0.693/k. В частности, было найдено (Таблица 3), что пролекарство формул 1.1, 1.2 и Sovaldi® в противовирусной композиции имеют сопоставимую стабильность в человеческом желудочном соке (T1/2SGF=12.7 ч - 15 ч), в человеческом кишечном соке (T1/2SIF>24 ч) и в человеческой плазме (Т1/2HPL>24 ч). В то же время пролекарство формулы 1.1 более быстро метаболизируют ет в S9 фракции микросом печени человека и имеет время полураспада T1/2HS9=0.05 ч, в то время как его прототип Sovaldi® имеет T1/2HS9=0.54 ч (Таблица 3), т.е. пролекарство формулы 1.1 в 11 раз быстрее метаболизирует в S9 фракции микросом печени человека чем Sovaldi®.
Пример 9. Изучение фармакокинетики (РК) композиций, включающих пролекарство формулы 1.1 и Sovaldi® в печени крыс.
Приготовления композиций, включающих пролекарство формулы 1.1 и Sovaldi®, для введения крысам. Исследуемую композицию вводили в дозе 50 мг/кг. Для этого готовили композиции с концентрацией пролекарства формулы 1.1 или Sovaldi®_5,0 мг/мл в 0,5% р-ре гидроксипропил-метилцеллюлозе (НРМС) с добавкой 5% этанола следующим образом: к навеске пролекарства формулы 1.1 или Sovaldi®_добавляли необходимое количество НРМС и насухую перетирали в ступке, затем постепенно порционно добавляли необходимое количество 5%-го этанола в дистиллированной воде., тщательно перемешивали до получения суспензии, пригодной для внутрижелудочного введения.
Введение композиций, включающих пролекарство формулы 1.1 и Sovaldi®, животным. Получение образцов плазмы крови и печени. Исследование проводилось на лабораторных крысах линии Sprague Dawley. Крыс разделили на группы, соответственно выбранным временным точкам (1, 2, 4, 6, 8, 10, 12, 16 и 24 ч), по 6 животных в группе. Животных взвешивали, рассчитывали объем вводимой композиции, включающих пролекарство формулы 1.1 или Sovaldi®, для каждого животного из расчета 10 мл/кг. Введение композиций, включающих пролекарство формулы 1.1 или Sovaldi®, проводили внутрижелудочно через зонд. Между введениями животным одной временной точки делали промежутки, достаточные для взятия образца печени и крови. Через необходимый промежуток времени после введения крысу эвтаназировали ингаляцией CO2. Немедленно после эвтаназии животное как можно быстрее вскрывали, отрезали верхнюю долю печени и сразу бросали ее в жидкий азот. Замороженный фрагмент печени помещали в промаркированную охлажденную в жидком азоте пробирку. До завершения эксперимента образцы хранили в жидком азоте, затем перемещали в низкотемпературный морозильник на -80°С.
Пробоподготовка. Навеску печени массой около 1 г перетирали в ступке при охлаждении жидким азотом, полученный порошок заливали трехкратным объемом метанола с 70% метанола с ЭДТА и гомогенизировали с помощью гомогенизатора Omni Bead Ruptor 24 на скорости 6.3 м/сек по 45 сек 2 раза с интервалом 10 сек. К 360 мкл полученного гомогената добавляли 40 мкл десятикратного стандартного раствора, содержащего PSI-7409 и Н027-4261 (или метанола в случае экспериментальных проб) и 100 мкл раствора внутреннего стандарта (5-бромуридин трифосфата) с концентрацией 25т нг/мл. После перемешивания и центрифугирования 400 мкл супернатанта разбавляли 400 мкл раствора 1% муравьиной кислотой в смеси метанол - вода (1:1). Проводили твердофазную экстракцию на картриджах Waters Oasis WAX, элюировали 800 мкл 5% раствора аммиака в метаноле, элюат упаривали и перерастворяли в 200 мкл метанола.
Условия ВЭЖХ-МС/МС анализа. Анализ проводили методом ВЭЖХ-МС/МС с помощью ВЭЖХ системы Agilent 1290 Infinity II соединенной с масс-спектрометром АВ Sciex QTrap 5500. Разделение проводили на колонке Thermo Hypercarb (50×3 мм, 5 мкм), в качестве подвижной фазы А (ПФА) использовали раствор 25 мМ ацетата аммония с добавкой 0.5% аммиака, в качестве подвижной фазы Б (ПФБ) - раствор 25 мМ ацетата аммония в смеси вода-изопропанол-ацетонитрил (1:1:3) с добавкой 0.5% аммиака. Разделение проводили в градиентном режиме: 0-0.3 мин - 5% ПФБ; 3-3.4 мин - 50% ПФБ; 3.6-4.5 мин - 5% ПФБ, Регистрацию PSI-7409 и Н027-4261 проводили в MRM режиме по ионным переходам 499/159 и 410/150 соответственно.
Фармакокинетический анализ. Фармакокинетический анализ данных «концентрация в печени - время» выполнялся модельно-независимым методом с помощью программы Phoenix™ WinNonlin® 6.3 (Pharsight Corp.) и программы GraphPad Prizm. Рассчитывались следующие фармакокинетические параметры: максимальная концентрация в печени (Cmax) и время ее достижения (Tmax), период полувыведения (T1/2), площадь под ФК кривой (AUC0-t, AUC0-inf). Полученные данные представлены в Таблице 4. Как видно из Таблицы 4 концентрация и AUC24 ч трифосфата PSI-7409, образующегося при метаболизме пролекарства формулы 1.1 в печени крыс, составляет Сmax=3,224.0 нг/г и AUC24 ч=30,487.0 нг⋅ч/г, в то время как при аналогичном метаболизме Sovaldi® имеет Cmax=1,934.0 нг/г и AUC24 ч=16,796.0 нг⋅ч/г (Таблице 4). Это свидетельствует о том, что новое пролекарство почти в 2 раза более эффективно метаболизирует в печени в нужный трифосфат PSI-7409 (лекарство).

Группа изобретений относится к химико-фармацевтической промышленности и представляет собой противовирусную фармацевтическую композицию для лечения вируса гепатита С у субъекта, нуждающегося в этом, которая содержит эффективное количество пролекарства, выбранного из циклобутил (S)-2-{[(2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-4-фтор-3-гидрокси-4-метил-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноата общей формулы 1, и/или циклобутил (S)-2-{(S)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидро-фуран-2-илметокси]-фенокси-фосфориламино}-пропаноата формулы 1.1, и/или циклобутил (S)-2-{(R)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидро-фуран-2-илметокси]-фенокси-фосфориламино}-пропаноата формулы 1.2, их кристаллическую или поликристаллическую форму и фармацевтически приемлемый носитель.
Группа изобретений также включает способ лечения гепатита С у субъекта, нуждающегося в этом, включающий в себя последовательное или одновременное введение терапевтически эффективного количества противовирусной фармацевтической композиции, описанной выше. Данная новая противовирусная композиция является более эффективной, чем известные фармацевтические противовирусные композиции. 2 н. и 9 з.п. ф-лы, 4 табл., 9 пр.
1. Противовирусная фармацевтическая композиция для лечения вируса гепатита С у субъекта, нуждающегося в этом, которая содержит эффективное количество пролекарства, выбранного из циклобутил (S)-2-{[(2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-4-фтор-3-гидрокси-4-метил-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноата общей формулы 1, и/или циклобутил (S)-2-{(S)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидро-фуран-2-илметокси]-фенокси-фосфориламино}-пропаноата формулы 1.1, и/или циклобутил (S)-2-{(R)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидро-фуран-2-илметокси]-фенокси-фосфориламино}-пропаноата формулы 1.2, их кристаллическую или поликристаллическую форму и фармацевтически приемлемый носитель
2. Противовирусная фармацевтическая композиция по п. 1, которая дополнительно включает терапевтически эффективное количество ингибитора NS5A HCV.
3. Противовирусная фармацевтическая композиция по п. 1, которая дополнительно включает терапевтически эффективное количество ингибитора NS3 HCV.
4. Противовирусная фармацевтическая композиция по п. 1, которая дополнительно включает терапевтически эффективное количество ингибитора NS3/4A HCV.
5. Противовирусная фармацевтическая композиция по п. 1, которая дополнительно включает терапевтически эффективное количество ингибитора полимеразы ДНК HBV.
6. Противовирусная фармацевтическая композиция по п. 1, которая дополнительно включает терапевтически эффективное количество ингибитора ВИЧ-1 обратной транскриптазы.
7. Противовирусная фармацевтическая композиция по п. 1, которая дополнительно включает терапевтически эффективное количество ингибитора полимеразы ДНК HBV и ингибитора ВИЧ-1 обратной транскриптазы.
8. Противовирусная фармацевтическая композиция по п. 1 или 2, включающая в качестве ингибитора NS5A Даклатасвира (Daclatasvir, BMS-790052) или Гепавивир (Hepavivir, AV-4025), или AV-4067, или AV-4084, или AV-4056, или AV-4058, или Омбитасвир (Ombitasvir, АВТ-267), или Элбасвир (Elbasvir, МК-8742), или Велапатасвир (Velpatasvir, VEL, GS-5816).
9. Противовирусная фармацевтическая композиция по п. 1 или 2, включающая Нарлапревир (Narlaprevir, SCH 900518) в качестве ингибитора NS3.
10. Противовирусная фармацевтическая композиция по п. 1 или 2, включающая Симепревир (Simeprevir, Olysio) в качестве ингибитора NS3/4A.
11. Способ лечения гепатита С у субъекта, нуждающегося в этом, включающий в себя последовательное или одновременное введение терапевтически эффективного количества противовирусной фармацевтической композиции по п. 1.
| НУКЛЕОЗИДФОСФОРАМИДАТЫ В КАЧЕСТВЕ ПРОТИВОВИРУСНЫХ АГЕНТОВ | 2008 |
|
RU2478104C2 |
| WO 2016141092 A1, 09.09.2016 | |||
| EA 200400986 A1, 24.02.2005 | |||
| RU 2010123928 A, 20.12.2011 | |||
| И.С.Ажгихин | |||
| Технология лекарств | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| и доп.- М.: Медицина | |||
| Способ получения фтористых солей | 1914 |
|
SU1980A1 |
| Способ обогащения руд | 1915 |
|
SU440A1 |
| Д.А | |||
| Харкевич | |||
| Фармакология | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| и испр | |||
| - М.: ГЭОТАР- МЕДИА, 2006 | |||
| ЦУГАЛЬТНЫЙ ВИСЯЧИЙ ЗАМОК | 1923 |
|
SU736A1 |

