ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к области L-нуклеозидов.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
В последние несколько десятилетий значительные усилия были затрачены на исследование возможных применений аналогов D-нуклеозидов в качестве противовирусных агентов. Отчасти эта работа принесла плоды, и в настоящее время на рынок в качестве противовирусных лекарств поставляются многие аналоги нуклеозидов, включая ингибиторы обратной транскриптазы ВИЧ (AZT, ddl, ddC, d4T и 3ТС).
Аналоги нуклерозидов исследованы также на их применение в качестве модуляторов иммунной системы (Bennet, P.A. et al., J. Med. Chem., 36. 635, 1993), но опять же с не вполне удовлетворительными результатами. Например, аналоги гуанозина, такие как 8-бром-, 8-меркапто-, 7-метил-8-оксогуанозин (Goodman, M. G. Immunopharmacology, 21. 51-68, 1991) и 7-тиа-8-оксогуанозин (Nagahara, К., J. Med. Chem., 33, 407-415, 1990; патент США 5041426) годами исследовались на их способность активировать иммунную систему. Эти производные гуанозина показывают прекрасную противовирусную и/или противоопухолевую активность in vivo. Но эти С8-замещенные гуанозины были неспособны активировать Т-клетки (Sharma, R.B. et al., Clin. Exp. Metastasis, 9, 429-439, 1991). То же самое оказалось справедливым в отношении 6-арилпиримидинонов (Wierenga, A. Ann. N. Y. Acad. Sci., 685. 296-300, 1993). В других исследованиях синтезировали ряд 3-деазапуриновых нуклеозидов и оценили их в качестве иммуномодулирующих агентов. Патент США 4309419 описывает применение 3-деазааденозина в качестве ингибитора иммунной системы. β-D-нуклеозид, β-2'-дезокси-3-деазагуанозин (Патент США 4950647) проявляет наиболее сильное иммуноактивирующее действие на реакции активированных Т-клеток. Для некоторых 2'-дезоксинуклезидов (заявка ЕРО 0038569) также раскрыта противовоспалительная и иммуносупрессорная активность. Однако эти соединения in vivo легко подвергаются метаболическому расщеплению по их гликозильной связи, что эффективно инактивирует их биологическую действенность. Производные аденозина, раскрытые в Патенте США 4148888, также легко катаболизируются in vivo ферментами деаминазами. В еще одном исследовании оказалось, что иммуностимулирующий тимомиметик левамизол (Levamisole) (Hadden et al., Immunol. Today, 14, 275-280, 1993) действует на линии Т-клеток подобно гормонам тимуса. Тукаресол (Tucaresol), еще один стимулятор Т-клеток (Reitz et al, Nature, 377, 71-75, 1995), сейчас проходит клинические испытания. В более недавнее время в качестве перспективного иммуностимулятора, который может быть направлен на болезненные состояния, требующие усиления реакций цитотоксических лимфоцитов или реакций типа Тh1, был описан пурин, который в 6-м положении замещен через линкер аминокислотой (Zacharie et al., J. Med. Chem., 40, 2883-1894, 1997).
Одна из возможных мишеней иммуномодуляции включает в себя стимуляцию или подавление Тh1- и Тh2-лимфокинов. Клетки типа I (Тh1) продуцируют интерлейкин 2 (1L-2), фактор некроза опухолей (TNFα) и интерферон-гамма (INFγ), и они отвечают главным образом за клеточные иммунные реакции, такие как гиперчувствительность замедленного типа и противовирусный иммунитет. Клетки типа II (Th2) продуцируют интерлейкины IL-4, IL-5, IL-6, IL-9, IL-10 и IL-13 и вовлечены главным образом в ассистирование гуморальным иммунным реакциям, таким как наблюдаемые при реакциях на аллергены, например переключение изотипов антител IgE и lgG4 (Mosmann, 1989, Аnnu Rev Immunol, 7:145-173). Показано, что аналоги D-гуанозина производят разные эффекты на лимфокины IL-1, IL-6, INFα и TNFα (не прямо) in vitro (Goodman, 1988, Int J Immunopharmacol, 10, 579-88) и in vivo (Smee et al., 1991, Antiviral Res 15:229). Однако способность аналогов D-гуанозина, таких как 7-тио-8-оксогуанозин, модулировать цитокины типа I и типа II непосредственно в Т-клетках была слабая или не была описана.
Важно, что исследования малых молекул в большинстве были сфокусированы на синтезе и оценке D-нуклеозидов. К их числу относятся рибавирин (Witkowski, J. Т. et al., J. Med. Chem., 15, 1150, 1972), AZT (De Clerq, E, Adv. Drug. Res., 17, 1, 1988), DDI (Yarchoan, R. et al., Science (Washington, D. C. ), 245. 412, 1989), DDC (Mitsuya, H. et al., Proc. Natl. Acad. Sci. U.S.A., 83, 1911, 1986), d4T (Mansuri, M.M. et al., J. Med. Chem. . 32, 461, 1989) и 3ТС (Doong, S.L. et al., Proc. Natl. Acad. Sci. U.S.A., 88, 8495-8599, 1991). В этой небольшой группе терапевтических агентов только 3ТС содержит не природную модифицированную L-рибозную группировку, энантиомер природной D-рибозы.
После того как 3ТС был одобрен в FDA (Управление США по контролю за качеством пищевых продуктов, медикаментов и косметических средств), были сообщения о многих нуклеозидах с неприродной L-конфигурацией как о сильных химиотерапевтических агентах против вируса иммунодефицита (ВИЧ), вируса гепатита Б (ВГБ) и некоторых форм рака. К их числу относятся (-)-β-L-1-[2-(гидроксиметил)-1,3-оксатиолан-4-ил] -5-фторцитозин (FTC, Furman, P. А. et al. , Antimicrob. Agents Chemother., 36, 2686-2692, 1992), (-)-β-L-2',3'-дидезоксипентофуранозил-5-фторцитозин (L-FddC; Gosselin, G., et al., Antimicrob. Agents Chemother. 38, 1292-1297, 1994), (-)-β-L-1-[2-(гидроксиметил)-1,3-оксатиолан-4-ил]-цитозин [(-)-OddC; Grove, К. L., et al. , Cancer Res., 55, 2008-3011, 1995), 2',3'-дидезокси-β-L-цитидин (β-L-ddC, Lin, T. S., et al., J. Med. Chem., 37, 798-803, 1994), 2'-фтор-5-метил-β-L-арабинофуранозилурацил (L-FMAU; Патент США 5567688), 2'3'-дидезокси-2',3'-дидегидро-β-L-цитидин ((β-L-d4C, Lin, Т. S. , et al., J. Med. Chem., 39, 1757-1759, 1996), 2',3'-дидезокси-2',3'-дидегидро-β-L-5-фторцитидин ((β-L-Fd4C, Lin, Т. S., et al., J. Med. Chem., 39, 1757-1759, 1996), карбоциклические L-циклопентилнуклеозиды (Wang et al., Tetrahedron Letts., 38, 4207-4210, 1996) и множество разных 9-(2'-дезокси-2'-фтор-β-L-арабинофуранозил)-пуриновых нуклеозидов (Ма, Т.' et al., J. Med. Chem., 40, 2750-2754, 1997).
Также сообщалось о других исследованиях L-нуклеозидов. Патент США 5009698, например, описывает синтез и применение L-аденозина для стимуляции роста растений. WO 92/08727 описывает определенные L-2'-дезоксиуридины и их применение в отношении вирусов. Spadari, S., et al., J. Med. Chem., 35, 4214-4220, 1992, описывает синтез определенных L-β-нуклеозидов, полезных для лечения вирусных инфекций, включая вызываемые вирусом простого герпеса типа I. Патент США 5559101 описывает синтез α- и β-L-рибофуранозильных нуклеозидов, способы их получения, фармацевтические композиции, которые их содержат, и способы их применения для лечения различных болезней у млекопитающих. Германский патент (DE 19518216) описывает синтез 2'-фтор-2'-дезокси-L-β-арабинофуранозилпиримидиновых нуклеозидов. Патенты США 5565438 и 5567688 описывают синтез и использование L-FMAU. Патент WO 95/20595 описывает синтез 2'-дезокси-2'-фтор-L-β-арабинофуранозилпуриновых и -пиримидовых нуклеозидов и способ лечения вирусного гепатита Б и инфицирования вирусом Эпштейна-Барра. Патент США 5567689 описывает способы повышения уровней уридина с помощью L-нуклеозидов. Патент WO 96/28170 описывает способ снижения токсичности D-нуклеозидов путем совместного введения эффективного количества L-нуклеозидных соединений.
Важно, что хотя показано, что некоторые известные L-нуклеозиды имеют сильную противовирусную активность с более низкими профилями токсичности, чем у соответствующих D-соединений, ни у одного их этих L-нуклеозидных соединений не было продемонстрировано иммуномодулирующих свойств. Более того, в настоящее время нет эффективного способа лечения для модулирования иммунной системы, когда имеют значение профили лимфокинов (подтипов Th1 и Тh2). Таким образом, сохраняется потребность в новых L-нуклеозидных аналогах, особенно потребность в L-нуклеозидных аналогах, которые модулируют иммунную систему, и более всего потребность в L-нуклеозидных аналогах, которые специфичным образом модулируют Th1 и Th2.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на новые L-нуклеозидные соединения, их терапевтические применения и синтез.
По одному из аспектов данное изобретение предлагает новые L-нуклеозидные соединения в соответствии с нижеследующей формулой: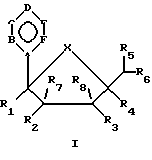
где А независимо выбирают из N или С;
В, С, Е, F выбраны независимо из СН, СО, N, S, Se, О, NR1, CCONH2, ССН3, С-R2 или Р; R1 представляет собой независимо Н, низший алкил, низшие алкиламины, СОСНз, низший алкилалкенил, низший алкилвинил или низшие алкиларилы; R2 представляет собой независимо Н, ОН, галогены, CN, N3, NH2, C(=O)NH2, C(= S)NH2, C(= NH)NH2•HCl, C(=NOH)NH2, C(=NH)OMe, низший алкил, низшие алкиламины, низший алкилалкенил, низший алкилвинил, низшие алкиларилы или замещенные гетероциклы;
D независимо выбирают из СН, СО, N, S, Se, О, NR1, ССОNН2, ССН3, C-R2, Р или отсутствия; где R1 представляет собой независимо Н, О, низший алкил, низшие алкиламины, СОСН3, низший алкилалкенил, низший алкилвинил или низшие алкиларилы, и R2 представляет собой независимо Н, ОН, галогены, CN, N3, NH2, низший алкил, низшие алкиламины, низший алкилалкенил, низший алкилвинил, низшие алкиларилы или замещенные гетероциклы;
Х представляет собой независимо О, S, СН2 или NR, где R представляет собой СОСН3;
R1 и R4 выбраны независимо из Н, CN, N3, CH2OH, низшего алкила и низших алкиламинов;
R2, R3, R5, R6, R7 и R8 выбраны независимо из Н, ОН, CN, N3, галогенов, СН2OН, NH2, ОСН3, NНСН3, ONHCH3, SСН3, SPh, алкенила, низшего алкила, низших алкиламинов и замещенных гетероциклов; и
R1, R2, R3, R4, R5, R6, R7 и R8 не все замещены одновременно, так что
когда R2= R3= Н, то тогда R7 и R8 представляют собой водород или отсутствуют;
когда R1, R4 или R5 замещены, то тогда R7= R8=H и R2= R3=ОН;
когда R2 или R3 замещены, то тогда R7 и R8 представляют собой Н или ОН;
когда R7 или R8 замещены, то тогда R2 и R3 представляют собой Н или ОН;
когда R7 и R8 представляют собой гидроксил, то тогда R2 и R3 не представляют собой ОН;
когда A=N, B=CO, C=N или NH; D=CO или C-NH2; E представляет собой СН или имеет заместитель по С, F=CH, Х=O, S или СН2, то тогда R2 не будет представлять собой Н, ОН, СН3, галогены, N3, CN, SH, SPh, CH2OH, СН2ОСН3, CH2SH, CH2F, CH2N3, арил, арилокси или гетероциклы;
когда A= N, B=CO, C=N или NH, D=CO или C-NH2, Е представляет собой СН, С-СН3 или галоген, F=CH, X=N-COCH3, то тогда R2 не будет представлять собой Н или ОН;
когда A=N, В=СН, С=СН или СН3, D=CH или С-СН3, Е представляет собой СН, С-СН3 или C-CONH2, F= CH, X=O или CH2, то тогда R2 не будет представлять собой Н или ОН;
когда A= N; B= N, СО или СН, С=СН, C-Cl или С-ОСН3, D=CH или С-Рh, Е представляет собой СН, C-Cl или C-Ph, F=N или СО; Х=O, то тогда R2 не будет представлять собой Н или ОН;
когда A=N, В=СО или CS, C=N или NH, D=CO или C-NH2, Е представляет собой СН или N, F=N или СН, Х=0, то тогда R2 не будет представлять собой Н или ОН, и
когда А= С, В=СН, C=NH, D=CO, CS или C-NH2, E представляет собой N или NH, F=CO или СН, Х=O, то тогда R2 не будет представлять собой Н или ОН.
По одному из классов предпочтительных воплощений данного изобретения соединение по данному изобретению включает в себя рибофуранозильную группировку, а в особо предпочтительном воплощении такое соединение представляет собой L-рибавирин.
По еще одному аспекту данного изобретения фармацевтическая композиция включает в себя терапевтически эффективное количество соединения формул 1 и 3-5 или его фармацевтически приемлемого эфира или соли в смеси по меньшей мере с одним фармацевтически приемлемым носителем.
По еще одному аспекту данного изобретения соединение, соответствующее формулам 1 и 3-5, применяют в лечении любого состояния, которое положительно реагирует на введение такого соединения в любом составе и по любому протоколу, которые приводят к положительным изменениям. Среди прочего предполагается, что соединения формулы I можно применять, чтобы лечить инфекцию, инвазию, рак или опухоль или аутоиммунное заболевание.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фигуры 1-12 являются схематическими представлениями стадий химического синтеза, которые можно применять для получения соединений из приведенного ниже раздела примеров.
Фигуры 13-14 являются графическими представлениями эффекта D-рибавирина и L-рибавирина на уровни IL-2, TNFα, INF-γ, IL-4 и IL-5 активированных Т-клеток.
Фигура 15 является графическим представлением, изображающим результаты другой серии экспериментов, в которой определяли эффекты L-рибавирина на воспаление уха в ответ на динитрофторбензол.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
При использовании в данном описании нижеследующих терминов они используются так, как определено ниже.
Термин "нуклеозид" относится к соединению, образованному любой пентозной или модифицированной пентозной группировкой, которая присоединена в определенном положении гетероцикла или в естественном положении пурина (положение 9) или пиримидина (положение 1) или в эквивалентном положении в аналоге.
Термин "нуклеотид" относится к фосфатному эфиру, замещающему нуклеозид в положении 5'.
Термин "гетероцикл" относится к моновалентному насыщенному или ненасыщенному карбоциклическому радикалу, имеющему по меньшей мере один гетероатом, такой как N, О или S, в кольце, каждое доступное положение которого могут возможно и независимо замещать, например, гидрокси, оксо, амино, имино, низший алкил, бромо, хлоро и/или циано. В этот класс заместителей включены пурины, пиримидины.
Термин "пурин" относится к азотсодержащим бициклическим гетероциклам.
Термин "пиримидин" относится к азотсодержащим моноциклическим гетероциклам.
Термин "D-нуклеозиды" при использовании в настоящем изобретении описывает нуклеозидные соединения, которые имеют D-рибозную сахарную группировку (например, аденозин).
Термин "L-нуклеозиды" при использовании в настоящем изобретении описывает нуклеозидные соединения, которые имеют L-рибозную сахарную группировку.
Термин "L-конфигурация" используется в настоящем изобретении, чтобы описывать химическую конфигурацию рибофуранозильной группировки соединений, которая присоединена к нуклеиновым основаниям. L-конфигурация сахарной группировки соединений по данному изобретению отличается от D-конфигурации рибозных сахарных группировок нуклеозидов, встречающихся в природе, таких как цитидин, аденозин, тимидин, гуанозин и уридин.
Термин "С-нуклеозиды" используется в данном описании, чтобы обозначать тип связи, которая образована между рибозной сахарной группировкой и гетероциклическим основанием. В С-нуклеозидах эта связь идет от положения С-1 рибозной сахарной группировки к углероду гетероциклического основания. Эта связь, которая образована в С-нуклеозидах, относится к типу углерод-углерод.
Термин "N-нуклеозиды" используется в данном описании, чтобы обозначать тип связи, которая образована между рибозной сахарной группировкой и гетероциклическим основанием. В N-нуклеозидах эта связь идет от положения С-1 рибозной сахарной группировки к азоту гетероциклического основания. Эта связь, которая образована в N-нуклеозидах, относится к типу углерод-азот.
Термин "защитная группа" относится к химической группе, которая присоединена к атому кислорода или азота, чтобы предотвращать его дальнейшее реагирование в ходе процесса дериватизации других группировок в молекуле, где расположен этот кислород или азот. Специалистам по органическому синтезу известно широкое разнообразие защитных групп для кислорода и азота.
Термин "низший алкил" относится к метилу, н-пропилу, изопропилу, н-бутилу, трет-бутилу, изобутилу или н-гексилу. Этот термин также применим к циклическим, разветвленным или простым цепям, содержащим от одного до шести атомов углерода.
Термин "арил" относится к моновалентному ненасыщенному ароматическому карбоциклическому радикалу, имеющему одно кольцо (например, фенил) или два конденсированных кольца (например, нафтил), которые могут возможно быть замещены гидроксилом, низшим алкилом, хлоро и/или циано.
Термин "гетероцикл" относится к моновалентному насыщенному или ненасыщенному карбоциклическому радикалу, имеющему по меньшей мере один гетероатом, такой как N, О, S, Se или Р, в кольце, каждое доступное положение которого может возможно и независимо быть замещено или не замещено, например, гидрокси, оксо, амино, имино, низшим алкилом, бромо, хлоро и/или циано.
Термин "моноциклический" относится к моновалентному насыщенному карбоциклическому радикалу, имеющему по меньшей мере один гетероатом, такой как N, О, S, Se или Р, в кольце, каждое доступное положение которого могут возможно и независимо замещать сахарная группировка или любые другие группы, как то: бромо, хлоро и/или циано, так что эта моноциклическая кольцевая система в конечном счете ароматизируется [например, тимидин, 1-(2'-дезокси-β-D-эритропентофуранозил)-тимин].
Термин "иммуномодуляторы" относится к природным или синтетическим продуктам, способным модифицировать нормальную или нарушенную иммунную систему посредством стимуляции или подавления.
Термин "эффективное количество" относится к количеству соединения формулы (I), которое восстанавливает иммунную функцию до нормальных уровней или усиливает иммунную функцию сверх нормальных уровней, чтобы ликвидировать инфекцию.
Соединения Формулы I могут иметь по несколько центров асимметрии. Соответственно, их можно получать в любой из оптически активных форм или в виде рацемической смеси. Объем данного изобретения, как оно описано и заявлено, охватывает индивидуальные оптические изомеры и их нерацемические смеси, а также рацемические формы соединений Формулы I.
Термин "α" или "β" указывает специфическую стереохимическую конфигурацию заместителя при асимметричном атоме углерода в химической структуре, как она выведена. Все описанные здесь соединения находятся в L-фуранозильной конфигурации.
Термин "энантиомеры" относится к паре стереоизомеров, которые представляют собой несовместимые зеркальные отражения друг друга. Смесь пары энатиомеров в соотношении 1:1 является "рацемической" смесью.
Термин "изомер" относится к разным соединениям, имеющим одну формулу. "Стереоизомеры" представляют собой изомеры, которые различаются только пространственным расположением атомов.
"Фармацевтически приемлемыми солями" могут быть любые соли, образованные неорганическими или органическими кислотами или основаниями.
Соединения по настоящему изобретению названы в соответствии с условиями Формулы II: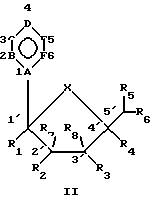
Соединения
Соединения по настоящему изобретению в общем описаны Формулой I.
Однако имеется несколько подгрупп таких соединений, которые представляют особый интерес, включая соединения, соответствующие нижеприведенным Формулам III, IV и V.
Соединения формулы III имеют следующую структуру: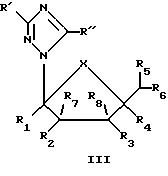
где X представляет собой независимо О, S, CH2 и NR, где R представляет собой СОСН3;
R' и R" выбраны независимо из Н, CN, С(=O)NН2, NH2, С(=S)NН2, C(= NH)NH2•HCl, C(= NOH)NH2, C(=NH)OMe, гетероциклов, галогенов, низшего алкила или низшего алкиларила;
R1 и R4 выбраны независимо из Н, CN, N3, CH2OH, низшего алкила или низших алкиламинов; и
R2, R3, R5, R6, R7 и R8 выбраны независимо из Н, ОН, CN, N3, галогенов, СН2ОН, NH2, ОСН3, NHCH3, ONHCH3, SСН3, SPh, алкенила, низшего алкила, низших алкиламинов или замещенных гетероциклов так, чтобы,
когда R2= R3= H, то тогда R7 и R8 представляют собой водород или отсутствуют.
В соединениях Формулы III R' предпочтительно представляет собой карбоксамид или CN, a R" представляет собой водород или галогены; R1=R4=R5=R7= R8=H и R2=R3=ОН и Х предпочтительно представляет собой кислород.
Соединения, соответствующие Формуле IV, имеют следующую структуру: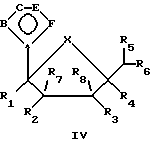
где А выбирают независимо из N или С;
В, С, Е и F выбраны независимо из СН, СО, N, S, Se, О, NR1, ССОNН2, ССН3, С-R2 или Р; R1 представляет собой независимо Н, низший алкил, низшие алкиламины, СОСН3, низший алкилалкенил, низший алкилвинил или низшие алкиларилы; R2 представляет собой независимо Н, ОН, галогены, CN, N3, NH2, C(= O)NH2, C(=S)NH2, C(=NH)NH2•HCl, С(=NОН)NН2, C(=NH)OMe, низший алкил, низшие алкиламины, низший алкилалкенил, низший алкилвинил, низшие алкиларилы или замещенные гетероциклы;
Х представляет собой независимо О, S, CH2 или NR, где R представляет собой СОСН3;
R1 и R4 выбраны независимо из Н, CN, N3, СН2OН, низшего алкила или низших алкиламинов; и
R2, R3, R5, R6, R7 и R8 выбраны независимо из Н, ОН, CN, N3, галогенов, NH2, СН2OН, ОСН3, NHCH3, ONHCH3, SСН3, SPh, алкенила, аллила, низшего алкила, низших алкиламинов или замещенных гетероциклов так, чтобы:
когда R2= R3= Н, то тогда R7 и R8 представляют собой водород или отсутствуют;
когда А представляет собой углерод, B=E=N, С представляет собой N-Ph, то тогда F не представляет собой СН;
когда A= N, С представляет собой СН, В=Е=С-СН3, то тогда F не является азотом, и
когда А представляет собой углерод, B=N, C=C-CONH2, E=CH; F=S, то тогда Х не представляет собой СН2.
В соединениях Формулы IV R1 предпочтительно представляет собой Н, низший алкил или аллил; R2 предпочтительно представляет собой Н, ОН, галогены, CN, N3, NH2, C(=O)NH2, C(=S)NH2, C(=NH)NH2•HCl, C(=NOH)NH2 или C(=NH)OMe; и, когда R1=R4=R5=R7=R8=H, то тогда предпочтительно R2=R3=ОН, а Х предпочтительно представляет собой кислород.
Соединения Формулы V имеют следующую структуру: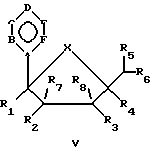
где А выбирают независимо из N или С;
В, С, Е, F выбраны независимо из СН, СО, N, S, Se, О, NR1, CCONH2, ССН3, С-R2 или Р; R1 представляет собой независимо Н, низший алкил, низшие алкиламины, СОСН3, низший алкилалкенил, низший алкилвинил или низшие алкиларилы; R2 представляет собой независимо Н, ОН, галогены, CN, N3, NH2, C(=O)NH2, C(= S)NH2, C(= NH)NH2•HCl, C(=NOH)NH2, C(=NH)OMe, низший алкил, низшие алкиламины, низший алкилалкенил, низший алкилвинил, низшие алкиларилы или замещенные гетероциклы;
D выбирают независимо из СН, СО, N, S, Se, О, NR1, CCONH2, ССН3, C-R2, Р или отсутствия; R1 представляет собой независимо Н, О, низший алкил, низшие алкиламины, СОСН3, низший алкилалкенил, низший алкилвинил или низшие алкиларилы; R2 представляет собой независимо Н, ОН, галогены, CN, N3, NH2, низший алкил, низшие алкиламины, низший алкилалкенил, низший алкилвинил, низшие алкиларилы или замещенные гетероциклы;
Х представляет собой независимо О, S, СН2 или NR, где R представляет собой СОСН3;
R1 и R4 выбраны независимо из Н, CN, N3, СН2ОН, низшего алкила или низших алкиламинов; и
R2, R3, R5, R6, R7 и R8 выбраны независимо из Н, ОН, CN, N3, галогенов, СН2OН, NH2, ОСН3, NHCH3, ONHCH3, SСН3, SPh, алкенила, низшего алкила, низших алкиламинов и замещенных гетероциклов так, чтобы:
когда R2=R3=H, то тогда R7 и R8 представляют собой водороды или отсутствуют;
когда A=N, B=CO, C=N или NH, D=CO или C-NH2, E представляет собой СН или замещен по С, F=CH, Х=O, S или СН2, то тогда R2 не будет представлять собой Н, ОН, СН3, галогены, N3, CN, SH, SPh, CH2OH, СН2OСН3, CH2SH, CH2F, CH2N3, арил, арилокси или гетероциклы;
когда A= N, B=CO, C=N или NH, D=CO или C-NH2, Е представляет собой СН, С-СН3 или галоген, F=CH, X=N-COCH3, то тогда R2 не будет представлять собой Н или ОН;
когда A=N, B=CH, С=СН или СН3, D=CH или С-СН3, Е представляет собой СН, С-СН3 или C-CONH2, F=CH, Х=O или СН2, то тогда R2 не будет представлять собой Н или ОН;
когда А= N, В= N, СО или СН, С=СН, С-Сl или С-ОСН3, D=CH или C-Ph, E представляет собой СН, С-С1 или C-Ph, F=N или СО, Х=O, то тогда R2 не будет представлять собой Н или ОН;
когда А=N, В=СО или CS, C=N или NH, D=CO или С-NН2, Е представляет собой СН или N, F=N или СН; Х=O, то тогда R2 не будет представлять собой Н или ОН, и
когда А= С, В=СН, C=NH, D=CO, CS или C-NH2, Е представляет собой N или NH, F=CO или СН, Х=O, то тогда R2 не будет представлять собой Н или ОН.
Особый класс обсуждаемых здесь соединений включает в себя нуклеозидные аналоги, имеющие рибофуранозильную группировку, в которой сахар имеет L-конфигурацию, а не природную D-конфигурацию. Этот класс включает в себя соединения, которые содержат модифицированные природные основания нуклеиновых кислот и/или синтетические нуклеозидные основания, как то: триазол, 3-циано-1,2,4-триазол, метиловый эфир 1,2,4-триазол-3-карбоновой кислоты, 3-бром-5-нитро-1,2,4-триазол, имидазол, 2-нитроимидазол, 2-бром-4(5)-аминоимидазол, пиразол, 3(5)-аминопиразол-4-карбоксамид, триазины, пиррол, пиридин, азапиридин, тиазол, 1,2,5-тиадиазол, селенадиазол, 4-амино-1,2,5-тиадиазол-3-карбоновую кислоту, метиловый эфир 4-оксо(5Н)-1,2,5-тиадиазол-3-карбоновой кислоты, 4-амино-1,2,5-селенадиазол-3-карбоновую кислоту, тетразол, азафосфол, 4-амино-1,3-азафосфол-5-карбонитрил, 4-бром-1,3-азафосфол-5-карбонитрил, 2-аминофосфин-3-карбонитрил, метиловый эфир 2-амино-3-цианофосфол-4-карбоновой кислоты, 4,5-дициано-1,3-диазафосфол, диазафосфол, изооксазол, 3-оксо(2Н)-изотиазол-3-карбоновую кислоту, 5-амино-3-хлоризотиазол-4-карбонитрил, 5-метилтио-3-оксо(2Н)-изотиазол-4-карбонитрил, изотиазол, пиримидин и другие замещенные производные этих оснований. Соединения этого класса также могут содержать независимо другие гетеромоноциклические основания и их производные, определенные модификации рибофуранозильной группировки и L-нуклеозиды, присоединенные как по N, так и по С.
Особо предпочтительные соединения этого класса включают в себя L-рибавирин, 1-β-L-рибофуранозил-1,2,4-триазол-3-карбоксамид. L-рибавирин изображен на Фигуре I, где А, В и Е представляют собой азот; С представляет собой C-C(O)NH2; D отсутствует; F представляет собой СН; Х представляет собой кислород; R1, R4, R5, R7 и R8 представляют собой водороды; a R2, R3 и R5 представляют собой гидроксил.
Рибавирин (1-β-D-рибофуранозил-1,2,4-триазол-3-карбоксамид) представляет собой моноциклический синтетический D-нуклеозид, который проявил активность против множества разных вирусных заболеваний (Huffman et al., Antimicrob. Agents Chemother., 3, 235, 1975; Sidwell et al., Science, 177, 705, 1972) и в настоящее время проходит клинические испытания в сочетании с γ-интерфероном для лечения вирусного гепатита С. В последние два десятилетия было исследовано множество D-нуклеозидных аналогов рибавирина, и многие из них проявляют исключительную противовирусную и противоопухолевую активность. Однако нет сообщений о синтезе L-изомера аналогов рибавирина и об их биологической активности. В единственном рентгеноструктурном исследовании рибавирина (Prusiner et al., Nature, 244, 116, 1973) он оказался структурно похожим на гуанонзин. Из-за сходства рибавирина с гуанозином мы предполагали, что нуклеозидные аналоги рибавирина в дополнение к противовирусной активности должны показывать сходную или более высокую иммуномодулирующую активность, чем гуанозиновые аналоги (Robbins et al., патент США 5041426).
Применения
Предполагается, что соединения по формулам I, III, IV и V, соединения по настоящему изобретению, будут применяться для лечения разнообразных состояний и, фактически, любого состояния, которое будет положительно реагировать на введение одного или более чем одного из этих соединений. Среди прочего в особенности предполагается, что соединения по данному изобретению можно применять для лечения инфекции, инвазии, рака или опухоли или аутоиммунного заболевания.
Предполагается, что инфекции, которые можно лечить соединениями по настоящему изобретению, включают в себя вызываемые респираторно-синтициальным вирусом (RSV), вирусом гепатита В (HBV), вирусом гепатита С (HCV), вирусами простого герпеса типа 1 и 2, генитального герпеса, герпетического кератита, герпетического энцефалита, опоясывающего лишая, вирусом иммунодефицита человека (HIV), вирусом гриппа А, хантавирусом (геморрагическая лихорадка), вирусом папилломы человека (HPV), вирусом кори, а также грибками.
Предполагается, что инвазии, которые можно лечить соединениями по данному изобретению, включают в себя протозойные инфекции, а также заражения гельминтами и другими паразитами.
Предполагается, что рак и опухоли, которые можно лечить, включают в себя вызываемые каким-либо вирусом, а эффект может включать в себя ингибирование трансформации инфицированных вирусом клеток в неопластическое состояние, ингибирование распространения вирусов от трансформированных клеток к другим, нормальным, клеткам и/или остановку роста трансформированных вирусом клеток.
Предполагается, что аутоиммунные и другие болезни, которые можно лечить, включают в себя артрит, псориаз, кишечное заболевание, юношеский диабет, волчанку, рассеянный склероз, подагру и подагрический артрит, ревматоидный артрит, отторжение трансплантата, аллергию и астму.
Другие предположительные применения соединений по настоящему изобретению включают в себя применение в качестве промежуточных соединений химического синтеза других нуклеозидных и нуклеотидных аналогов, которые в свою очередь полезны в качестве терапевтических агентов или для других целей.
По еще одному аспекту способ лечения млекопитающего включает в себя введение терапевтически и/или профилактически эффективного количества фармацевтического препарата, содержащего соединение по настоящему изобретению. В этом аспекте эффект может относиться к модуляции некоторой части иммунной системы млекопитающего, особенно к модуляции профилей Тh1- и Тh2-лимфокинов. Когда происходит модуляция Тh1- и Тh2-лимфокинов, предполагается, что такая модуляция может включать в себя стимуляцию как Тh1, так и Тh2, подавление как Тh1, так и Тh2, стимуляцию одного, либо Тh1, либо Тh2, и подавление другого или бимодальную модуляцию, при которой одно воздействие на уровни Th1/Th2 (такое как генерализованное подавление) происходит при низкой концентрации, тогда как другое воздействие (такое как стимуляция одного, либо Тh1, либо Тh2, и подавление другого) происходит при более высокой концентрации.
Вообще, наиболее предпочтительными применениями по настоящему изобретению являются такие, при которых активные соединения соответственно менее цитотоксичны для клеток хозяина, не являющихся клетками-мишенями, и соответственно более активны в отношении мишени. В этом отношении преимущество может состоять в том, L-нуклеозиды могут быть более стабильными, чем D-нуклеозиды, что может приводить к лучшей фармакокинетике. Этого результата можно достичь, потому что L-нуклеозиды могут не распознаваться ферментами и поэтому могут иметь более длительные периоды полужизни.
Предполагается, что соединения по настоящему изобретению можно будет вводить в любом подходящем фармацевтическом составе и по любому подходящему протоколу. Так, введение может происходить перорально, парентерально (включая методики подкожной, внутривенной, внутримышечной, интрастернальной инъекции или вливания), с помощью ингаляционного аэрозоля или ректальным или местным образом и так далее, а также в составах стандартной дозировки, содержащих общепринятые нетоксичные фармацевтически приемлемые носители, адъюванты и растворители для введения.
Например, можно предполагать, что соединения по настоящему изобретению можно приготавливать в виде состава в смеси с фармацевтически приемлемым носителем. Например, соединения по настоящему изобретению можно вводить перорально в виде фармакологически приемлемых солей. Поскольку соединения по настоящему изобретению по большей части растворимы в воде, их можно вводить внутривенно в физиологическом солевом растворе (например, забуференном до рН около 7,2-7,5). Для этой цели можно использовать общепринятые буферы, такие как фосфаты, бикарбонаты или цитраты. Разумеется, специалист может модифицировать эти составы в соответствии с требованиями, чтобы получать многочисленные составы для определенного пути введения без того, чтобы делать композиции по настоящему изобретению нестабильными или нарушать их терапевтическую активность. В частности, модификацию данных соединений с целью сделать их более растворимыми в воде или другом растворителе для введения можно, например, легко осуществить путем минимальных модификаций (образование соли, этерификация и т.д), что вполне находится в пределах навыков, обычных для данной области. Также, не выходя за пределы навыков, обычных для данной области, можно модифицировать пути введения и режимы дозировки определенного соединения, чтобы контролировать фармакокинетику данных соединений для получения максимально полезного эффекта у пациентов.
В определенных фармацевтических лекарственных формах предпочтительны про-лекарственные формы соединений, особенно такие, которые включают в себя ацилированные (ацетилированные и другие) производные, пиридиновые эфиры и различные солевые формы данных соединений. Специалист в данной области может определить, как с легкостью модифицировать данные соединения до пролекарственных форм, чтобы способствовать доставке активных соединений в целевые участки организма-хозяина или пациента. При наличии навыков, обычных в данной области, можно пользоваться преимуществами благоприятных фармакокинетических параметров пролекарственных форм, в случаях их применимости, для доставки данных соединений в целевые участки в организме-хозяине или в пациенте, чтобы максимизировать желаемые эффекты соединения.
В дополнение, соединения по данному изобретению можно вводить отдельно или в комбинации с другими агентами для лечения вышеуказанных инфекций или состояний. Способы комбинированной терапии по данному изобретению включают в себя введение по меньшей мере одного соединения по настоящему изобретению или функционального производного такого соединения и по меньшей мере одного другого фармацевтически активного ингредиента. Активный(е) ингиредиент(ы) и фармацевтически активные агенты можно вводить по отдельности или совместно и при введении по отдельности это может происходить одновременно или порознь в любом порядке. Количество активных(ого) ингредиентов(а) и фармацевтически активных(ого) агентов(а) и относительный временной порядок введения выбирают, чтобы достичь желаемого комбинированного терапевтического эффекта. Комбинированная терапия предпочтительно включает в себя введение одного соединения по настоящему изобретению или физиологически функционального производного этого соединения и одного из агентов, упомянутых здесь ниже.
Примеры таких дополнительных терапевтических агентов включают в себя агенты, которые эффективны для модулирования иммунной системы или связанных состояний, такие как AZT, 3ТС, 8-замещенные гуанозиновые аналоги, 2',3'-дидезоксинуклеозиды, интерлейкин II, интерфероны, такие как γ-интерферон, тукаресол, левамизол, изопринозин и циклолигнаны. Определенные соединения по настоящему изобретению могут быть эффективными для усиления биологической активности определенных агентов по настоящему изобретению путем снижения метаболизма или инактивации других соединений и как таковые они подлежат введению совместно с последними для достижения этого желаемого эффекта.
Что касается дозировки, то обычному специалисту в данной области понятно, что терапевтически эффективное количество варьирует в зависимости от инфекции или состояния, которые подлежат лечению, от тяжести заболевания, от применяемого режима лечения, от фармакокинетики используемого агента, также как от пациента (животного или человека), который подлежит лечению. Эффективные дозировки могут быть в пределах от 1 мг/кг веса тела или меньше до 25 мг/кг веса тела или больше. В общем, терапевтически эффективное количество данного соединения в лекарственной форме обычно находится в пределах от чуть меньше примерно 1 мг/кг до примерно 25 мг/кг веса пациента, в зависимости от используемого соединения, состояния или инфекции, которые лечатся, и пути введения. Такие пределы дозировки обычно создают эффективные концентрации активного соединения в крови, находящиеся в диапазоне от примерно 0,04 до примерно 100 микрограмм/сс крови пациента. Однако предполагается, что будет разработан эффективный режим, предусматривающий введение небольшого количества с последующим увеличением количества до тех пор, пока либо побочные эффекты не станут чрезмерно нежелательными, либо не будет достигнут желаемый эффект.
Введение активного соединения можно варьировать в диапазоне от непрерывного (внутривенная капельница) до нескольких пероральных введений в день (например 4 раза в день) и оно может включать в себя пероральное, местное, парентеральное, внутримышечное, внутривенное, подкожное, чреcкожное (что может включать в себя применение агента, способствующего проникновению), трансбуккальное и суппозиторное введение наряду с другими путями введения.
Для приготовления фармацевтических композиций по настоящему изобретению терапевтически эффективное количество одного или более чем одного соединения по настоящему изобретению предпочтительно смешивать до однородности с фармацевтически приемлемым носителем в соответствии с общепринятыми фармацевтическими методиками для смешивания, чтобы получить дозу. Носитель может принимать широкое разнообразие форм в зависимости от формы препарата, желательной для ведения, например перорального или парентерального. При приготовлении фармацевтических композиций в пероральной лекарственной форме можно использовать любую из обычных фармацевтических сред. Так, для жидких пероральных препаратов, таких как суспензии, эликсиры и растворы, можно использовать подходящие носители и добавки, включая воду, гликоли, масла, спирты, корригенты, консерванты, красители и тому подобное. Для твердых пероральных препаратов, таких как порошки, таблетки, капсулы, и для твердых препаратов, таких как суппозитории, можно использовать подходящие носители и добавки, включая крахмалы, сахарные носители, такие как декстроза, маннит, лактоза, и родственные носители, разбавители, гранулирующие агенты, смазывающие вещества, связывающие вещества, разрыхлители и тому подобное. При желании таблетки или капсулы можно с помощью стандартных методик покрывать энтеросолюбильной оболочкой или изготавливать как препараты для непрерывного высвобождения.
Носитель для парентеральных составов обычно включает в себя стерильную воду или водный раствор хлорида натрия, хотя можно включать в него другие ингредиенты, в том числе такие, которые помогают диспергированию. Разумеется, когда надо использовать стерильную воду и поддерживать ее стерильность, композиции и носители также надо стерилизовать. Также можно готовить инъецируемые суспензии, в каковом случае можно применять подходящие жидкие носители, суспендирующие агенты и тому подобное.
Результаты тестирования
С соединением Формулы I, L-рибавирином, были выполнены тесты in vitro и in vivo, и их результаты представлены ниже.
В первой серии экспериментов мононуклеарные клетки периферической крови (МКПК) выделяли из светлого слоя после центрифугирования 60 мл крови здоровых доноров в градиенте плотности Ficoll-Hypaque. Затем из МКПК выделяли Т-клетки, используя Lymphokwik, реагент для выделения лимфоцитов, специфичный для Т-клеток (LK-25T, One Lambda, Canoga Park CA). Затем полученные при среднем выходе 40-60•106 Т-клетки инкубировали в течение ночи при 37oС в 20-30 мл среды RPMI-AP5 (среда RPMI-1640, ICN, Costa Mesa, Са), содержащей 20 мМ буфер HEPES (рН 7,4), 5% аутологичной плазмы, 1% L-глутамина, 1% пенициллина/стрептомицина и 0,05% 2-меркаптоэтанола для удаления любых примесных прилипающих клеток. Во всех экспериментах Т-клетки промывали средой RPMI-AP5, а затем высевали их на микротитровальные планшеты с 96 лунками при концентрации клеток 1•106 клеток/мл.
Эти Т-клетки активировали добавлением 500 нг иономицина и 10 нг 12-миристата, 13-ацетата форбола (МАФ) (Calbiochem, La Jolla, Са) и инкубировали в течение 48-72 часов при 37oС. Т-клетки, активированные иономицином и МАФ, обрабатывали либо рибавирином (D-рибавирин), либо L-рибавирином в концентрации 0,5-50 мкМ или контрольным противовирусным интерфероном-альфа (Accurate, Westbury, NY) в концентрации 250-10000 ед./мл немедленно после активации и еще раз через 24 часа. Т-клетки с каждого планшета использовали для анализа методом иммунофлюоресценции, а супернатанты использовали для определения внеклеточных цитокинов. После активации 900 мкл супернатанта от клеток с каждого микропланшета переносили на другой микропланшет для анализа продукции произведенных клетками цитокинов. Клетки затем использовали для иммунофлюоресцентного анализа уровней внутриклеточных цитокинов и экспрессии рецепторов к цитокинам.
Концентрации произведенных клетками цитокинов определяли в супернатантах от клеток с каждого микропланшета. Вызванные активацией изменения уровней интерлейкина-2 (IL-2) определяли с использованием имеющихся в продаже наборов для твердофазного иммуноферментного анализа ELISA (R&D systems Quantikine kit, Minneapolis, MN) или биологическим методом с использованием зависимой от IL-2 клеточной линии CTLL-2 (АТСС, Rockville, MD). Вызванные активацией изменения уровней интерлейкина-4 (IL-4), фактора некроза опухолей (TNFα), интерлейкина-8 (IL-8) (R&D systems Quantikine kit, Minneapolis, MN) и интерферона-гамма (INF-γ) (Endogen, Cambridge, MA) определяли с использованием наборов для иммуноферментного анализа. Все результаты иммуноферментного анализа выражали в пг/мл, а результаты биологического определения с CTLL-2 выражали в импульсах в минуту, что отражало зависимое от IL-2 включение 3H-тимидина (ICN, Costa Mesa, CA) клетками CTLL-2.
Сравнение эффектов D-рибавирина и L-рибавирина (выражено в процентах от активированного контроля) на уровни IL-2, TNFα, IFN-γ, IL-4 и IL-5 представлено на Фигурах 13 и 14.
В другой серии экспериментов определяли эффекты L-рибавирина на воспалительную реакцию уха в ответ на динитрофторбензол. Результаты этих экспериментов показаны на Фигуре 15.
Синтез
Соединения по настоящему изобретению можно получать в соответствии со способами синтеза, о каждом из которых хорошо известно специалистам в данной области. В общем, соединения по настоящему изобретению синтезируют конденсацией подходящего нуклеозидного основания с необходимым сахарным синтоном, что приводит к получению защищенного L-нуклеозида, что при дальнейших манипуляциях и снятии защиты с защищенных гидроксильных групп сахара в конечном счете дает нуклеозидный аналог, имеющий желаемую рибофуранозильную группировку в L-конфигурации.
Во время химического синтеза различных композиций по настоящему изобретению обычный специалист может применять настоящее изобретение без излишнего экспериментирования. В частности, обычному специалисту понятны разные стадии, которые следует выполнить, чтобы ввести определенный заместитель в желаемое положение сахарной группировки. Вдобавок, в качестве подходящих для условий синтеза можно распознать химические стадии, которые следует выполнить, чтобы защитить функциональные группы, такие как, среди прочих, гидроксильные или аминогруппы, а также, чтобы снять защиту с этих же самых групп.
Данное изобретение определено далее отсылкой к нижеследующим примерам, которые предназначены для того, чтобы быть иллюстрацией, но не ограничением. Обычный специалист поймет, что эти примеры никоим образом не являются ограничивающими и что детали можно варьировать без отхода от сути и области настоящего изобретения.
Соединения по настоящему изобретению можно получать в соответствии с хорошо известными в данной области процедурами. Особенно полезны нижеследующие схемы синтеза.
Схема 1 (см. в конце описания): Синтез рибофуранозильных (R1, R4, R5, R7 и R8 являются водородами; R2, R3 и R6 являются гидроксилом) нуклеозидов формулы (II): Триазол-L-рибофуранозильные нуклеозиды получали в процедуре присоединения в условиях кислого катализа (Sato, Т., et al., Nippon Kagaku Zasshi, 81, 1440, 1960). Соответственно, триазолы (1) смешивали с 1,2,3,5-тетра-O-ацетил-L-рибозой (2) и каталитическим количеством бис(пара-нитрофенил)фосфата и нагревали при 160-165oС в течение 30 минут под пониженным давлением с получением требуемых нуклеозидов, которые при последующем снятии защиты давали триазоловые L-рибонуклеозиды (3) формулы (II).
Схема 2 (см. в конце описания): Синтез L-рибофуранозильных (R1, R4, R5, R7 и R8 являются водородами; R2, R3 и R6 являются гидроксилом) нуклеозидов формулы (III): Триазольные, пиразольные и другие 5-членные гетероциклические L-рибофуранозильные нуклеозиды по настоящему изобретению получали с помощью процедуры Ворбрюггена (Vorbruggen), включающей в себя обработку гетероциклов (4) хлортриметилсиланом с получением силильного промежуточного соединения, который при конденсации с защищенной рибозой (5) в присутствии хлорида олова в инертном растворителе дает требуемые нуклеозиды (6). После конденсации эти продукты снятием защиты общепринятыми способами, известными специалистам, переводят в соединения формулы (III).
Большинство соединений формулы (III) можно получать с помощью вышеописанной процедуры конденсации. Требуемые 1,2,3,5-тетра-O-ацетил-L-рибозу и 1-O-ацетил-2,3,5-три-O-бензоил-L-рибозу получали, как показано в Примере 2 и Примере 13 соответственно. Гетеромоноциклические основания есть в продаже у фирм Aldrich, Fluka, ICN, Acros, Alfa, Lancaster и TCI America или их готовили, следуя процессам, описания которых можно найти в опубликованных статьях (Robbins R. К., et al., Nucleosides & Nucleotides, 13, 17, 76, 1994). Получение пиррольных, пиразольных и других типов триазольных L-нуклеозидов формулы (IV) осуществляли, следуя процедурам, описанным для получения соответствующих D-нуклеозидов в Chemistry of Nucleosides and Nucleotides, Edited by Leroy B. Townsend, New York, Plenum Press, 3, 1-105, 1994. Различные имидазольные L-нуклеозиды готовили, следуя описанным (Shaw, G., in Chemistry of Nucleosides and Nucleotides, Edited by Leroy B. Townsend, New York, Plenum Press, 3, 263-420, 1994) способам для имидазольных D-нуклеозидов.
Схема 3 (см. в конце описания): Соединения формулы (I) можно получать в реакции гетероциклов (7) с L-рибозой (5), следуя процедуре Ворбрюггена (Niedballa, U. , et al., J. Org. Chem., 39, 3654, 1974), описанной выше для получения соединений формулы (III).
С-нуклеозиды (где А в формулах I и III является углеродом) в L-конфигурации получали, применяя к конкретному примеру процедуру, описанную (Watanabe, K.A., in Chemistry of Nucleosides and Nucleotides, Edited by Leroy B. Townsend, New York, Plenum Press, 3, 421-435, 1994) для приготовления соответствующих им С-нуклеозидов в D-конфигурации.
Схема 4 (см. в конце описания): Получение L-арабинофуранозильных нуклеозидов (R1, R2, R4, R5 и R8 являются водородами; R3, R6 и R7 являются гидроксилом): β-аномеры арабинозильных L-нуклеозидов формул (I-III) можно получать в реакции 2,3,5-три- -бензил-L-арабинофуранозилбромида (9), Baker, R., et al., J. Org. Chem., 26, 4605-4609, 1961) и триметилсилильных производных этого основания с получением промежуточного L-нуклеозида (10). Удаление блокирующих групп у 10 должно давать требуемые β-L-арабинофуранозильные нуклеозиды. В случае пиррольных β-L-арабинонуклеозидов следовали процедуре гликозилирования натриевых солей (Ravenkar G. R. , et al., Nucleosides & Nucleotides, 6, 261-264, 1987).
-бензил-L-арабинофуранозилбромида (9), Baker, R., et al., J. Org. Chem., 26, 4605-4609, 1961) и триметилсилильных производных этого основания с получением промежуточного L-нуклеозида (10). Удаление блокирующих групп у 10 должно давать требуемые β-L-арабинофуранозильные нуклеозиды. В случае пиррольных β-L-арабинонуклеозидов следовали процедуре гликозилирования натриевых солей (Ravenkar G. R. , et al., Nucleosides & Nucleotides, 6, 261-264, 1987).
Схема 5 (см. в конце описания): Получение L-ксилофуранозильных нуклеозидов (R1, R3, R4, R5 и R7 являются водородами; R2, R6 и R8 являются гидроксилом): β-аномеры ксилофуранозильных L-нуклеозидов формул (I-III) можно получать из 1,2-ди ацетил-3,5-ди
ацетил-3,5-ди бензил-L-ксилофуранозы (11); Gosselin, G., et al., J. Heterocyclic Chem., 30, 1229-1233, 1993) и соответствующего основания, следуя способу, который аналогичен тому, что описан на схеме 4.
бензил-L-ксилофуранозы (11); Gosselin, G., et al., J. Heterocyclic Chem., 30, 1229-1233, 1993) и соответствующего основания, следуя способу, который аналогичен тому, что описан на схеме 4.
Схема 6 (см. в конце описания): Получение L-2'-дезоксифуранозильных нуклеозидов (R1, R2, R4, R5, R7 и R8 являются водородами; R3 и R6 являются гидроксилом): β-аномеры 2'-дезоксирибофуранозильных L-нуклеозидов формул (I-III) можно получать в реакции 3',5'-ди пара-толуил-2'-дезоксиэритро-β-L-пентофуранозилхлорида (13) (Smejkal, J. Et al., Collect. Czec. Chem. Commun. 29, 2809-2813, 1964) с силильными производными этих гетероциклов в присутствии кислоты Бронстеда с получением исключительно β-изомеров (14) с хорошим выходом (Fujimori, S., et al., Nucleosides & Nucleotides, 11, 341-349, 1992; Aoyama, H., Bull. Chem. Soc., 60, 2073, 1987). Те же β-L-2'-дезоксифуранозильные нуклеозиды также готовили в реакции хлоруглевода (13) с натриевой солью основания (Kazimierczuk, Z., et al., J. Аmer. Chem. Soc. 106, 6379-6382, 1984) в сухом ацетонитриле. Промежуточный продукт (14) при обработке раствором аммиака в метаноле дал требуемые β-L-2'-дезоксиэритропентофуранозильные нуклеозиды.
пара-толуил-2'-дезоксиэритро-β-L-пентофуранозилхлорида (13) (Smejkal, J. Et al., Collect. Czec. Chem. Commun. 29, 2809-2813, 1964) с силильными производными этих гетероциклов в присутствии кислоты Бронстеда с получением исключительно β-изомеров (14) с хорошим выходом (Fujimori, S., et al., Nucleosides & Nucleotides, 11, 341-349, 1992; Aoyama, H., Bull. Chem. Soc., 60, 2073, 1987). Те же β-L-2'-дезоксифуранозильные нуклеозиды также готовили в реакции хлоруглевода (13) с натриевой солью основания (Kazimierczuk, Z., et al., J. Аmer. Chem. Soc. 106, 6379-6382, 1984) в сухом ацетонитриле. Промежуточный продукт (14) при обработке раствором аммиака в метаноле дал требуемые β-L-2'-дезоксиэритропентофуранозильные нуклеозиды.
Схема 7 (см. в конце описания): Получение L-3'-дезоксирибофуранозильных нуклеозидов (R1, R3, R4, R5, R6, R7 и R8 являются водородами; R2 и R6 являются гидроксилом): β-аномеры 3'-дезоксирибофуранозильных L-нуклеозидов формул (I-III) можно получать в реакции 1,2-ди ацетил-5
ацетил-5 бензоил-3-дезокси-L-эритропентозы (15) с силильными производными этих гетероциклов в присутствии кислоты Льюиса с получением β-изомеров (16), которые при деблокировании посредством аммония в метаноле должны давать β-L-3'-дезокси-эритро-пентофуранозильные нуклеозиды. Те же соединения можно также получать в реакции соответствующих 1-хлор-производных соединения (15) с натриевой солью гетероциклического основания, как в случае 2'-дезокси-L-нуклеозидов, описанных на схеме 6.
бензоил-3-дезокси-L-эритропентозы (15) с силильными производными этих гетероциклов в присутствии кислоты Льюиса с получением β-изомеров (16), которые при деблокировании посредством аммония в метаноле должны давать β-L-3'-дезокси-эритро-пентофуранозильные нуклеозиды. Те же соединения можно также получать в реакции соответствующих 1-хлор-производных соединения (15) с натриевой солью гетероциклического основания, как в случае 2'-дезокси-L-нуклеозидов, описанных на схеме 6.
Схема 8 (см. в конце описания): Получение L-2',3'-дидезоксирибофуранозильных нуклеозидов (R1, R2, R3, R4, R5, R7 и R8 являются водородами; R6 является гидроксилом): β-аномеры 2',3'-дидезоксирибофуранозильных L-нуклеозидов формул (I-III) можно получать обработкой их соответствующих  трифенилметил-2', 3'-бис(метансульфонат)-β-L-рибофуранозильных нуклеозидов (17) водородотеллуридом натрия (Clive, D. L. et al., J. Org. Chem., 61, 7426-7437, 1996) в СН3СN при комнатной температуре, как показано ниже. По конец из (18) удаляют тритильную группу в мягких условиях с получением 2',3'-дидезоксирибофуранозил-β-L-нуклеозидов.
трифенилметил-2', 3'-бис(метансульфонат)-β-L-рибофуранозильных нуклеозидов (17) водородотеллуридом натрия (Clive, D. L. et al., J. Org. Chem., 61, 7426-7437, 1996) в СН3СN при комнатной температуре, как показано ниже. По конец из (18) удаляют тритильную группу в мягких условиях с получением 2',3'-дидезоксирибофуранозил-β-L-нуклеозидов.
Более того, замещенные сахара, такие как 1-бром-2-дезокси-2-фтор-3,6 бензоил-L-арабинофураноза (Ма, Т., et al., J. Mod. Chem., 39, 2835-2843, 1996) и другие модифицированные сахара в L-конфигурации известны по Патенту США 5473063; WO 96/13512; WO 96/13498; WO 96/22778; WO 95/20595; Патенту США 5473063; Патенту США 5567688; Walczak, К., et. al., Monatsh. Fur Chemie, 123, 349-354 (1992); Wengel, L., et al., J. Org. Chem., 56, 3591-3594 (1991); Genu-Dellac, С., et al., Tetrahedron Letts., 32, 79-82 (1991) и Czernecki, S. , et al., Synthesis, 783 (1991). Вдобавок, получение модифицированных cахаров и нуклеозидов в D-конфигурации описано в Патенте США 5192749; WO 94/22890; Uteza, V., et al., Tetrahedron, 49, 8579-8588 (1993); Thrane, H. , et al., Tetrahedron, 51, 10389-10403 (1995); Yoshimura, Y., et al. , Nucleosides & Nucleotides, 14, 427-429 (1993); Lawrence, A.J., et al., J. Org. Chem. , 61, 9213-9222 (1996); Ichikawa, S., et al., J. Org. Chem., 62, 1368-1375 (1997); ЕР 0457326 А1; Патенте США 3910885; WO 96/13498 и Karpeisky, M.Y., et al., Nucleic Acids Res. Symposium Series, 9, 157 (1981). Применяя процедуры (схемы) синтеза, которые были описаны в этих статьях для получения D-нуклеозидов, можно получить соответствующие модифицированные L-нуклеозиды.
бензоил-L-арабинофураноза (Ма, Т., et al., J. Mod. Chem., 39, 2835-2843, 1996) и другие модифицированные сахара в L-конфигурации известны по Патенту США 5473063; WO 96/13512; WO 96/13498; WO 96/22778; WO 95/20595; Патенту США 5473063; Патенту США 5567688; Walczak, К., et. al., Monatsh. Fur Chemie, 123, 349-354 (1992); Wengel, L., et al., J. Org. Chem., 56, 3591-3594 (1991); Genu-Dellac, С., et al., Tetrahedron Letts., 32, 79-82 (1991) и Czernecki, S. , et al., Synthesis, 783 (1991). Вдобавок, получение модифицированных cахаров и нуклеозидов в D-конфигурации описано в Патенте США 5192749; WO 94/22890; Uteza, V., et al., Tetrahedron, 49, 8579-8588 (1993); Thrane, H. , et al., Tetrahedron, 51, 10389-10403 (1995); Yoshimura, Y., et al. , Nucleosides & Nucleotides, 14, 427-429 (1993); Lawrence, A.J., et al., J. Org. Chem. , 61, 9213-9222 (1996); Ichikawa, S., et al., J. Org. Chem., 62, 1368-1375 (1997); ЕР 0457326 А1; Патенте США 3910885; WO 96/13498 и Karpeisky, M.Y., et al., Nucleic Acids Res. Symposium Series, 9, 157 (1981). Применяя процедуры (схемы) синтеза, которые были описаны в этих статьях для получения D-нуклеозидов, можно получить соответствующие модифицированные L-нуклеозиды.
Применяя указания приведенных здесь схем, а также конкретных примеров и других схем, которые будут приведены ниже, можно синтезировать другие соединения в пределах, охватываемых данным изобретением. В дополнение к приведенным здесь указаниям умелый специалист сразу поймет, как получать соединения, охватываемые данным изобретением, применяя известные методики, такие как описанные в Nucleic Acid Chemistry, Improved and New Synthetic Procedures, Methods and Techniques, Edited by Leroy B. Townsend and R. Stuart Tipson, John Wiley & Sons, New York, Plenum Press (1978-1991), Chemistry of Nucleosides and Nucleotides, Edited by Leroy B. Townsend, New York, Plenum Press (1988-1994) и Nucleosides and Nucleotides as Antitumor and Antiviral Agents, Edited by Chung K. Chu and David C. Baker, New York, Plenum Press (1993). Подходящие способы для того, чтобы делать замещения в сахарной группировке заявленных здесь соединений, известны специалистам и описаны в различных публикациях, включая Патент США 5559101; Патент США 5192749; Патент США 5473063; Патент США 5565438. Подходящие способы для того, чтобы получать различные гетероциклические соединения и замещения в них, даны в Chemistry of Nucleosides and Nucleotides, Edited by Leroy B. Townsend, New York, Plenum Press, 2, 161-398 (1991) и Chemistry of Nucleosides and Nucleotides, Edited by Leroy B. Townsend, New York, Plenum Press, 3, 1-535 (1994).
ПРИМЕРЫ
Данное изобретение можно понять лучше, обратившись к нижеследующим примерам, где номера соединений, выделенные жирным шрифтом, соответствуют подобным же номерам на Фигурах 1-12.
ПРИМЕР 1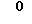 Метил-2,3,5-три
Метил-2,3,5-три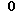 ацетил-β-L-рибофураноза (19)
ацетил-β-L-рибофураноза (19)
L-Рибозу (15,0 г; 100 ммоль) растворяли в сухом метаноле (200 мл) и охлаждали до 0oС. К этому холодному перемешиваемому раствору медленно добавляли H2SO4 (2 мл) и перемешивали реакционную смесь при температуре ниже 20oС в атмосфере аргона. Добавляли сухой пиридин (75 мл) и упаривали досуха. Добавляли сухой пиридин (100 мл) и упаривали под пониженным давлением до маслянистого остатка. Этот остаток растворяли в сухом пиридине (150 мл) и обрабатывали уксусным ангидридом (50 мл) при 0oС в атмосфере аргона. Добавляли TEA (триэтаноламин) (41 мл), реакционную смесь перемешивали при 0oС в течение 1 часа и при комнатной температуре в течение 36 часов и упаривали досуха. Остаток растворяли в воде (200 мл) и медленно добавляли твердый NaHCO3, чтобы довести рН раствора до 7. Водную смесь экстрагировали в CH2Cl2 (250 мл), промывали водой (150 мл) и соляным раствором (100 мл), сушили и концентрировали. Маслянистый остаток фильтровали на слое силикагеля (200 г) и промывали смесью CH2Cl2:EtOAc (8:2, 100 мл). Фильтрат упаривали и масло использовали как таковое в следующей реакции.
ПРИМЕР 2
1,2,3,5-Тетра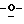 ацетил-β-L-рибофураноза (2)
ацетил-β-L-рибофураноза (2)
Сироп (19) (20,0 г; 100 ммоль) из вышеописанной реакции упаривали вместе с сухим толуолом (2х100 мл) и сушили в течение ночи под твердым NaOH при комнатной температуре в вакууме. Высушенный сироп растворяли в ледяной уксусной кислоте (150 мл) и охлаждали до 0oС в атмосфере аргона. К этому холодному раствору добавляли уксусный ангидрид (35 мл), а затем очень медленно в течение 15 минут добавляли Н2SО4 (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи и вливали при перемешивании в лед (200 г). Эту смесь экстрагировали двумя порциями СНСl3 по 200 мл и органический экстракт промывали водой (200 мл), насыщенным NаНСО3 (200 мл) и соляным раствором (150 мл), сушили над безводным Na2SO4 и упаривали досуха. Эти 30 г сиропа (94%) были определены как достаточно чистые для реакций гликозилирования.
ПРИМЕР 3А
Метиловый эфир 1-(2,3,5-три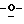 ацетил-β-L-рибофуранозил)-1,2,4-триазол-3-карбоновой кислоты (20)
ацетил-β-L-рибофуранозил)-1,2,4-триазол-3-карбоновой кислоты (20)
Смесь метилового эфира 1,2,4-триазол-3-карбоновой кислоты (0,64 г; 5 ммоль), 1,2,3,5-тетра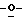 ацетил-β-L-рибофуранозы (2) (1,5 г; 4,72 ммоль) и бис(р-нитрофенил)-фосфата (20 мг) помещали в грушевидную колбу, которую помещали в предварительно прогретую масляную баню при 160-165oС. Колбу подсоединяли к водоструйному насосу и держали при 160-165oС (температура масляной бани) при пониженном давлении и перемешивании в течение 25 минут. Реакционную смесь извлекали, охлаждали и разводили в ЕtOАс (150 мл) и насыщенном NаНСО3 (100мл). Продукт экстрагировали, используя ЕtOАс. Органический экстракт промывали водой (100 мл) и соляным раствором (50 мл), сушили и упаривали досуха. Полученный остаток чистили на колонке для быстрой хроматографии с силикагелем, используя СН3Сl-->ЕtOАс в качестве элюента. Чистые фракции собирали и упаривали досуха с получением 1,2 г (66%) чистого продукта: 1Н-ЯМР (СDСl3) δ 2.10 (3s, 9H, 3 СОСН3), 3.98 (s, 3H, ОСН3), 4.22 (m. 1H), 4,46 (m, 2Н), 5,54 (t, 1H), 5.76 (m, 1Н), 6.04 (d, 1H, C1•H), 8,38 (s, 1H, C3h). Анализ. Рассчитано для С15Н19N3О9 (385,22): С 46,75; Н 4,97; N 10,91. Найдено: С 46,82; Н 4,57; N=10,71.
ацетил-β-L-рибофуранозы (2) (1,5 г; 4,72 ммоль) и бис(р-нитрофенил)-фосфата (20 мг) помещали в грушевидную колбу, которую помещали в предварительно прогретую масляную баню при 160-165oС. Колбу подсоединяли к водоструйному насосу и держали при 160-165oС (температура масляной бани) при пониженном давлении и перемешивании в течение 25 минут. Реакционную смесь извлекали, охлаждали и разводили в ЕtOАс (150 мл) и насыщенном NаНСО3 (100мл). Продукт экстрагировали, используя ЕtOАс. Органический экстракт промывали водой (100 мл) и соляным раствором (50 мл), сушили и упаривали досуха. Полученный остаток чистили на колонке для быстрой хроматографии с силикагелем, используя СН3Сl-->ЕtOАс в качестве элюента. Чистые фракции собирали и упаривали досуха с получением 1,2 г (66%) чистого продукта: 1Н-ЯМР (СDСl3) δ 2.10 (3s, 9H, 3 СОСН3), 3.98 (s, 3H, ОСН3), 4.22 (m. 1H), 4,46 (m, 2Н), 5,54 (t, 1H), 5.76 (m, 1Н), 6.04 (d, 1H, C1•H), 8,38 (s, 1H, C3h). Анализ. Рассчитано для С15Н19N3О9 (385,22): С 46,75; Н 4,97; N 10,91. Найдено: С 46,82; Н 4,57; N=10,71.
ПРИМЕР 3Б
1-β-L-Рибофуранозил-1,2,4-триазол-3-карбоксамид (21)
Субстрат (20) (1,1 г) растворяли в СН3ОН/NН3 при 0oС и помещали в стальной баллон. Баллон закрывали и перемешивали при комнатной температуре в течение 18 часов. Стальной баллон охлаждали, открывали и его содержимое выпаривали досуха. Остаток пробовали кристаллизовать с небольшим количеством этанола. Продукт кристаллизовался, но при фильтрации кристаллы набирали воду и превращались в пасту. Кристаллизацию повторяли несколько раз. В конце концов кристаллизация проходила из смеси метанола с этанолом. Бесцветные кристаллы отфильтровывали, промывали метанолом и сушили в вакууме. Фильтрат упаривали снова, и при стоянии он образовывал еще кристаллы. Общий выход 0,5 г (72%); т. пл.: 177-179oС; [а]D=+38,33 (при 3 мг/мл H2O); D-форма рибавирина [а]D=-36,0 (при 3 мг/мл H2O); 1H-ЯМР (Me2SO4-d6) δ 3.46 (m, 1H, C5•H), 3.60 (m, 1H, С5•Н), 3.92 (q, 1H, C4•H), 4.12 (q, 1H), 4.34 (q, 1H), 4.88 (t, 1H, C5•OH), 5.20 (d, 1H), 5.58 (d, 1H), 5.80 (d, 1H, C1•H), 7.60 (bs, 1H, NH), 7.82 (bs, 1H, NH), 8.82 (s, 1H, С3Н). Анализ. Рассчитано для C8H12N4O5 (244.20): С, 39.34; Н, 4.95; N, 22.94. Найдено: С 39,23; Н 4,97; N 22,91.
ПРИМЕР 4 Изопропилиден-L-рибоза (22)
Изопропилиден-L-рибоза (22)
К перемешиваемой суспензии L-рибозы (30,0 г; 260 ммоль) в сухом ацетоне (200 мл) при комнатной температуре в атмосфере аргона добавляли йод (1,27 г; 10 ммоль). Эту реакционную смесь перемешивали 1 час (в течение этого времени раствор становится гомогенным) и гасили раствором тиосульфата (1 М). Этот раствор упаривали досуха. Остаток растворяли в CH2Cl2 (250 мл), сушили над безводным MgSO4, фильтровали и твердое вещество промывали с помощью CH2Cl2 (150 мл). Объединенный фильтрат упаривали досуха. Остаток наносили сверху на колонку с кремнеземом (8•116 см), заполненную в СНСl3. Колонку элюировали, используя СНСl3 (500 мл), СНСl3:ЕtOАс (9:1, 1000 мл) и СНСl3:ЕtOАс (7:3, 1500 мл). Чистый продукт, элюированный в СНСl3:ЕtOАс (7:3), собирали и упаривали с получением 34,5 г (90%) маслянистого остатка. Этот маслянистый продукт использовали как таковой в следующей реакции. 1H-ЯМР (СDСl3) δ 1.30 and 1.38 (2s, 6H, isopropylidene СН3), 3.70 (m, 3Н). 4.08 (m, 1Н), 4.38 (m, 1H). 4.55 (d, 1H), 4.81 (d, 1H) and 5.38 (m, 1H).
ПРИМЕР 5
1-Дезокси-1-гидразинил-2,3 изопропилиден-L-рибоза (23)
изопропилиден-L-рибоза (23)
Раствор 2,3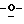 изопропилиден-L-рибозы (22) (34,5 г, 182 ммоль) в абсолютном метаноле (200 мл) в течение 30 минут при комнатной температуре в атмосфере аргона обрабатывали по каплям раствором безводного гидразина (42,0 г 1313 ммоль) в абсолютном метаноле (100 мл). Почти бесцветный раствор перемешивали при комнатной температуре и в безводных условиях в течение 18 часов. Этот раствор упаривали в вакууме с получением бесцветного сиропа. Сироп несколько раз упаривали с абсолютным метанолом (5•100 мл). Полученный сироп мгновенно нагревали (70oС) под давлением от вакуумного насоса (0,1 торр) и затем держали под этим давлением в течение 12 часов для высушивания. Выход был 35 г (95%). Этот материал использовали как таковой без дальнейшей очистки для следующей стадии.
изопропилиден-L-рибозы (22) (34,5 г, 182 ммоль) в абсолютном метаноле (200 мл) в течение 30 минут при комнатной температуре в атмосфере аргона обрабатывали по каплям раствором безводного гидразина (42,0 г 1313 ммоль) в абсолютном метаноле (100 мл). Почти бесцветный раствор перемешивали при комнатной температуре и в безводных условиях в течение 18 часов. Этот раствор упаривали в вакууме с получением бесцветного сиропа. Сироп несколько раз упаривали с абсолютным метанолом (5•100 мл). Полученный сироп мгновенно нагревали (70oС) под давлением от вакуумного насоса (0,1 торр) и затем держали под этим давлением в течение 12 часов для высушивания. Выход был 35 г (95%). Этот материал использовали как таковой без дальнейшей очистки для следующей стадии.
ПРИМЕР 6
5-Амино-4-циано-1-(2',3'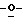 изопропилиден-β-L-рибофуранозил)-пиразол (24)
изопропилиден-β-L-рибофуранозил)-пиразол (24)
Раствор 1-дезокси-1-гидразанил-2,3 изопропилиден-L-рибозы (23) (16,3 г; 79.90 ммоль) в абсолютном этаноле (100 мл) продували постоянным током аргона в течение 30 минут. К этому быстро перемешиваемому раствору добавляли по каплям в течение часа при комнатной температуре продутый аналогичным образом раствор (этоксиметилен)-маланонитрила (10,37 г; 85 ммоль) в абсолютном этаноле. Раствор перемешивали под аргоном еще 30 минут и затем грели при температуре образования флегмы в течение 12 часов. Оранжевый раствор охлаждали до комнатной температуры и упаривали в вакууме досуха. Этот материал растворяли в этилацетате (100 мл) и затем обрабатывали силикагелем (50 г). Эту смесь упаривали досуха в вакууме и полученный порошок наносили сверху на колонку с 500 г кремнезема (6х30 см, заполненную в сухом виде). Эту колонку элюировали градиентом CH2Cl2-->EtOAc. Фракции, содержащие чистый продукт, объединяли и упаривали до пены: Выход 17 г (76%); 1Н-ЯМР (CDCl3) δ 1.30 and 1.52 (2s, 6H, isopropylidene СН3), 3.86 (m, 2Н, C5•H), 4.40 (m, 1Н, С4•Н), 4.80 (bs, 2H, NH2), 5.00 (d, 1H), 5.20 (m, 1H), 5.80 (d, 1H, C1•H) and 7.54 (bs, 1H, С3Н). Анализ. Рассчитано для C12H16N4O4 (280.28): С 51,43; Н 5,75; N 19,99. Найдено: С 51,20; Н 5,63; N 19,98.
изопропилиден-L-рибозы (23) (16,3 г; 79.90 ммоль) в абсолютном этаноле (100 мл) продували постоянным током аргона в течение 30 минут. К этому быстро перемешиваемому раствору добавляли по каплям в течение часа при комнатной температуре продутый аналогичным образом раствор (этоксиметилен)-маланонитрила (10,37 г; 85 ммоль) в абсолютном этаноле. Раствор перемешивали под аргоном еще 30 минут и затем грели при температуре образования флегмы в течение 12 часов. Оранжевый раствор охлаждали до комнатной температуры и упаривали в вакууме досуха. Этот материал растворяли в этилацетате (100 мл) и затем обрабатывали силикагелем (50 г). Эту смесь упаривали досуха в вакууме и полученный порошок наносили сверху на колонку с 500 г кремнезема (6х30 см, заполненную в сухом виде). Эту колонку элюировали градиентом CH2Cl2-->EtOAc. Фракции, содержащие чистый продукт, объединяли и упаривали до пены: Выход 17 г (76%); 1Н-ЯМР (CDCl3) δ 1.30 and 1.52 (2s, 6H, isopropylidene СН3), 3.86 (m, 2Н, C5•H), 4.40 (m, 1Н, С4•Н), 4.80 (bs, 2H, NH2), 5.00 (d, 1H), 5.20 (m, 1H), 5.80 (d, 1H, C1•H) and 7.54 (bs, 1H, С3Н). Анализ. Рассчитано для C12H16N4O4 (280.28): С 51,43; Н 5,75; N 19,99. Найдено: С 51,20; Н 5,63; N 19,98.
ПРИМЕР 7
5-Амино-1-(β-L-рибофуранозил)-пиразол-4-карбонитрил (25)
Раствор 5-амино-1-(2', 3'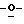 иэопропилиден-β-L-рибофуранозил)-пиразол-4-карбонитрила (24) (4,6 г; 16,43 ммоль) в 90% трифторуксусной кислоте (30 мл) перемешивали при комнатной температуре в течение 4 часов. Эту реакционную смесь упаривали досуха и остаток упаривали вместе с метанолом (3х35 мл). Остаток использовали как таковой для дальнейших реакций.
иэопропилиден-β-L-рибофуранозил)-пиразол-4-карбонитрила (24) (4,6 г; 16,43 ммоль) в 90% трифторуксусной кислоте (30 мл) перемешивали при комнатной температуре в течение 4 часов. Эту реакционную смесь упаривали досуха и остаток упаривали вместе с метанолом (3х35 мл). Остаток использовали как таковой для дальнейших реакций.
ПРИМЕР 8
5-Амино-1-(β-L-рибофуранозил)-пиразол-4-карбоксамид (26)
К раствору (25) (4,60 г) в гидроксиде аммония (35 мл) добавляли 30% перекиси водорода (2 мл). Эту смесь перемешивали в толстостенном флаконе при комнатной температуре в течение 18 часов, толстостенный флакон охлаждали, осторожно открывали и летучие продукты выпаривали досуха. Полученный таким образом остаток упаривали с этанолом (3•20 мл). Неочищенный продукт при кристаллизации с этанолом/водой давал чистое соединение (3,5 г, 71%): 1Н-ЯМР (DMSO-d6) δ 3.57 (m, 2H, C5•CH2), 3.86 (q, 1H, С4•Н), 4.11 (q, 1H, С3•Н), 4.43 (q, 1H, C2•OH), 5.63 (d, 1H, J=3.99 Hz, C1•H), 6.51 (br s, 2H, NH2), 6.71 and 7.26 (2br s, 2H, CONH2) and 7.69 (s, 1H, С3Н). Анализ. Рассчитано для C9H14N4O5 (258,23): С 41,86; Н 5.46; N 21,69. Найдено: С 41,57; H 5,40; N 21,61.
ПРИМЕР 9
5-Амино-1-(2', 3' изопропилиден-β-L-рибофуранозил)-пиразол-3,4-дикарбонитрил (27)
изопропилиден-β-L-рибофуранозил)-пиразол-3,4-дикарбонитрил (27)
Раствор тетрацианоэтилена (24,32 г; 190 ммоль) в абсолютном EtoH (100 мл) добавляли по каплям в течение 30 мин при температуре 0oС и при перемешивании к раствору 1-дезокси-1-гидразинил-2,3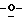 изопропилиден-L-рибозы (223,0 г; 113,0 ммоль) в ЕtOН (100 мл). Эту реакционную смесь перемешивали при температуре ледяной бани еще 2 часа и затем упаривали досуха. Остаток растворяли в МеОН (50 мл), сорбировали на силикагель (90 г) и наносили сверху на колонку с силикагелем (10•25 см), заполненную с CH2Cl2. Колонку элюировали смесью CH2Cl2/EtOAc (10:1, объем/объем), гомогенные фракции объединяли и упаривали досуха. Оставшуюся желтую пену кристаллизовали из этанола длительным выдерживанием при комнатной температуре с получением 15 г (44%) чистого (27): т. пл. oС; 1H-ЯМР (Me2SO-d6) δ 1.31 and 1.48 (2s, 6Н, isopropylidene-СН3), 3.29 (m, 2H, С5•СН2), 4.13 (m, 1H, С4•Н), 4.83 (m, 1H, С3•Н), 4.97 (t, 1H, С5•ОН). 5.24 (m, 1H, C2•H), 6.12 (s, 1H, C1•H), 7.65 (s, 2H, NH2). Анализ. Рассчитано для C13H15N5O4 (305,29): С 51,14; Н 4,95; N 22,94. Найдено: С 51,20; Н 5,04; N 22,61.
изопропилиден-L-рибозы (223,0 г; 113,0 ммоль) в ЕtOН (100 мл). Эту реакционную смесь перемешивали при температуре ледяной бани еще 2 часа и затем упаривали досуха. Остаток растворяли в МеОН (50 мл), сорбировали на силикагель (90 г) и наносили сверху на колонку с силикагелем (10•25 см), заполненную с CH2Cl2. Колонку элюировали смесью CH2Cl2/EtOAc (10:1, объем/объем), гомогенные фракции объединяли и упаривали досуха. Оставшуюся желтую пену кристаллизовали из этанола длительным выдерживанием при комнатной температуре с получением 15 г (44%) чистого (27): т. пл. oС; 1H-ЯМР (Me2SO-d6) δ 1.31 and 1.48 (2s, 6Н, isopropylidene-СН3), 3.29 (m, 2H, С5•СН2), 4.13 (m, 1H, С4•Н), 4.83 (m, 1H, С3•Н), 4.97 (t, 1H, С5•ОН). 5.24 (m, 1H, C2•H), 6.12 (s, 1H, C1•H), 7.65 (s, 2H, NH2). Анализ. Рассчитано для C13H15N5O4 (305,29): С 51,14; Н 4,95; N 22,94. Найдено: С 51,20; Н 5,04; N 22,61.
ПРИМЕР 10
5-Амино-1β-L-рибофуранозилпиразол-3,4-дикарбонитрил (28)
Суспензию 5-амино-1-(2', 3'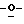 изопропилиден-β-L-рибофуранозил)-пиразол-3,4-дикарбонитрила (4,6 г; 15,0 ммоль) в 90% TFA (трифторуксусная кислота )/воде (50 мл) перемешивали при комнатной температуре в течение 12 часов. Растворитель выпаривали и остаток упаривали с ЕtOН (3•50 мл). Полученный таким образом светло-коричневый остаток использовался как таковой для дальнейшей реакции.
изопропилиден-β-L-рибофуранозил)-пиразол-3,4-дикарбонитрила (4,6 г; 15,0 ммоль) в 90% TFA (трифторуксусная кислота )/воде (50 мл) перемешивали при комнатной температуре в течение 12 часов. Растворитель выпаривали и остаток упаривали с ЕtOН (3•50 мл). Полученный таким образом светло-коричневый остаток использовался как таковой для дальнейшей реакции.
ПРИМЕР 11
5-Амино-1-β-L-рибофуранозилпиразол-3,4-дикарбоксамид (29)
Соль 5-амино-1β-L-рибофуранозилпиразол-3,4-дикарбонитрила (28) с TFA растворяли в конц. NН4OН (28%, 100 мл) и обрабатывали перекисью водорода (30%, 15 мл). Эту реакционную смесь 12 часов перемешивали при комнатной температуре в толстостенном флаконе и затем упаривали досуха. Остаток упаривали с МеОН (3х50 мл). Неочищенный продукт кристаллизовали из смеси ЕtOН с водой с получением 2,0 г (68%) (29): 1H-ЯМР (Me2SO-d6) δ 3.60 (m, 2H, С5•СH2), 3.87 (m, 1H, C4•H), 4.18 (m, 1H, С3•Н), 4.54 (m, 1H, C2•H), 4.91 (t, 1H, C5•OH), 5.03 and 5.38 (2d, 2H, С2',3ОН), 5.69 (d, 1H, С1•Н), 6.99 (br s, 3H, NH2 and CONH(H)), 7.72 and 7.78 (2s, 2H, CONH2) and 9.65 (d, 1H, CON(H)H). Анализ. Рассчитано для C10H15N5O6 (301, 26): С 39,87; Н 5,03; N 23,25. Найдено: С 39,72; Н5,40; N 23,61.
ПРИМЕР 12
Диметиловый эфир 1,2,3-триазол-4,5-дикарбоновой кислоты (30)
К перемешиваемой суспензии азида натрия (5,03 г; 77,39 ммоль) в DMF (диметилформамиде) (120 мл) добавляли по каплям при 0oС в течение 30 минут раствор ацетилендикарбоксилата (10,0 г; 70,36 ммоль) в DMF (100 мл). После 30 минут растворитель удаляли в вакууме при 30oС с получением светлого пурпурно-коричневого твердого вещества. Это твердое вещество дважды промывали эфиром и собирали в воду (100 мл). Водный раствор подкисляли концентрированной HCl до рН 2. Водный слой сначала экстрагировали эфиром (100 мл), а затем хлороформом (100 мл). Объединенные органические слои упаривали с получением светло-красного твердого вещества: 128-130oС. Это твердое вещество промывали горячим гексаном и кристаллизовали из бензола: выход 11,0 г (85%); 1H-ЯМР (CDCl3) δ 4.00 (s, 6H), 11.87 (br s, 1H, NH).
ПРИМЕР 13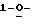 Ацетил-2,3,5-три
Ацетил-2,3,5-три бензоил-β-L-рибофураноза (5)
бензоил-β-L-рибофураноза (5)
К раствору L-рибозы (25,0 г, 166,66 ммоль) в МеОН (300 мл) добавляли 25 мл насыщенного раствора хлористого водорода в метаноле и перемешивали при комнатной температуре в течение 6 часов. Реакция завершалась через 6 часов, как свидетельствовали результаты ТСХ с использованием CH2Cl2/MeOH 9:1. По завершении реакции добавляли сухой пиридин (30 мл) и выпаривали растворители. К остатку добавляли еще 30 мл пиридина и выпаривали досуха. Остаток растворяли в сухом пиридине (200 мл) и Cl2Cl2 (150 мл), а затем охлаждали до 0oС. По каплям добавляли бензоилхлорид (96,26 мл; 830,12 ммоль) и перемешивали при комнатной температуре в течение ночи. ТСХ с использованием гексана/атилацетата (7: 3) свидетельствовала о завершении реакции. Растворители выпаривали и остаток растворяли в СНСl3 (300 мл), промывали водой (200 мл) и насыщенным NаНСО3 (200 мл) и сушили над безводным Na2SO4. После выпаривания СНСl3 остаток упаривали с толуолом с получением маслянистого остатка. Этот остаток растворяли в АсОН (200 мл), уксусном ангидриде (85,0 мл; 770,9 ммоль) и серной кислоте (4,46 мл; 83,29 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение ночи, после чего ТСХ (гексан/этилацетат 7:3) свидетельствовала о завершении реакции. Растворители выпаривали в вакууме и полученный остаток упаривали с толуолом. Коричневый остаток перетирали с ЕtOН с получением светло-коричневых кристаллов. Фильтрация этого твердого вещества и перекристаллизация из ЕtOН дали 1 ацетил-2,3,5-три
ацетил-2,3,5-три бензоил-L(+)-глюкофуранозу (40,5 г; 48%) в виде белых кристаллов: т. пл. 125-125oС; 1H-ЯМР (СDСl3) δ 4.49 (m, 1Н, С5•Н), 4.77 (m, 2Н, С4•Н and C5•H), 5.80 (d, 1H), 5.93 (m, 1H, C2•H), 6.43 (d, 1H, C1•H, J1,2=1.5 Hz) and 7.30-8.09 (m, 15H, PhH).
бензоил-L(+)-глюкофуранозу (40,5 г; 48%) в виде белых кристаллов: т. пл. 125-125oС; 1H-ЯМР (СDСl3) δ 4.49 (m, 1Н, С5•Н), 4.77 (m, 2Н, С4•Н and C5•H), 5.80 (d, 1H), 5.93 (m, 1H, C2•H), 6.43 (d, 1H, C1•H, J1,2=1.5 Hz) and 7.30-8.09 (m, 15H, PhH).
ПРИМЕР 14
Диметиловый эфир 2-(2', 3', 5'-три бензоил-β-L-рибофуранозил)-1,2,3-триазол-4,5-дикарбоновой кислоты (31)
бензоил-β-L-рибофуранозил)-1,2,3-триазол-4,5-дикарбоновой кислоты (31)
Смесь сухого диметилового эфира 1,2,3-триазол-β-L-дикарбоновой кислоты (3,70 г; 20,0 ммоль), гексаметилдисилазана (HMDS, 60 мл) и (NH4)2SO4 (0,1 г) грели с образованием флегмы (температура масляной бани 140oС) в течение 12 часов в условиях, исключающих увлажнение. Избыток HMDS удаляли дистилляцией в вакууме с получением триметилсилильного производного, которое растворяли в безводном CH3CN (100 мл).
К вышеуказанному прозрачному раствору добавляли 1 ацетил-2,3,5
ацетил-2,3,5 бензоил-L-рибофуранозу (10,12 г; 20 ммоль) и смесь перемешивали в течение 10 минут. К этому перемешиваемому раствору добавляли триметилсилиловый эфир трифторметансульфоновой кислоты (4,6 мл; 26,0 моль) и продолжали перемешивание 12 часов при температуре окружающей среды. Эту реакционную смесь упаривали и остаток растворяли в СH2Сl2 (500 мл). Органический слой последовательно промывали насыщенным водным раствором NaHCO3 (3•100 мл), насыщенным водным раствором NaCl (3•100 мл) и водой (3х50 мл) и сушили над безводным Na2SO4. Растворитель выпаривали с получением 12,0 г (95%) (31): 1H-ЯМР (Me2SO4-d6) δ 3.88 (s, 6H, 2 ОСН3), 4.65 (m, 2H, С5•Н), 5.01 (m, 1H, С4•Н), 6.10 (m, 1H, С3•Н), 6.32 (m, 1H, C2•H), 6.88 (d, 1H, C1•H, J1,2=2.75 Hz) and 7.45-7.95 (m, 15H, PhH).
бензоил-L-рибофуранозу (10,12 г; 20 ммоль) и смесь перемешивали в течение 10 минут. К этому перемешиваемому раствору добавляли триметилсилиловый эфир трифторметансульфоновой кислоты (4,6 мл; 26,0 моль) и продолжали перемешивание 12 часов при температуре окружающей среды. Эту реакционную смесь упаривали и остаток растворяли в СH2Сl2 (500 мл). Органический слой последовательно промывали насыщенным водным раствором NaHCO3 (3•100 мл), насыщенным водным раствором NaCl (3•100 мл) и водой (3х50 мл) и сушили над безводным Na2SO4. Растворитель выпаривали с получением 12,0 г (95%) (31): 1H-ЯМР (Me2SO4-d6) δ 3.88 (s, 6H, 2 ОСН3), 4.65 (m, 2H, С5•Н), 5.01 (m, 1H, С4•Н), 6.10 (m, 1H, С3•Н), 6.32 (m, 1H, C2•H), 6.88 (d, 1H, C1•H, J1,2=2.75 Hz) and 7.45-7.95 (m, 15H, PhH).
ПРИМЕР 15
2-β-L-Рибофуранозил-1,2,3-триазол-4,5-дикарбоксамид (32)
Соединение (31) (6,0 г 9,5 ммоль) растворяли в МеОН/NН3 (сухой метанол, насыщенный безводным NH3 при 0oС, 60 мл) и помещали в стальной реакционный сосуд. Этот сосуд 16 часов нагревали при 95oС. Реакционный сосуд охлаждали, осторожно открывали и давали аммиаку испариться при комнатной температуре. МеОН выпаривали досуха и остаток перетирали с горячим толуолом (3•50 мл) и отфильтровывали. Коричневый остаток кристаллизовали из водного ЕtOН (95%) с получением 2,40 г (89%) (32): т. пл. 210-212oС; 1Н-ЯМР (Me2SO4-d6) δ 3.45-3.59 (m, 2H, C5•H), 3.98 (m, 1H, C4•H), 4.25 (m, 1H, С3•Н), 4.54 (m, 1H, C2•H), 4.78 (t, 1H, C5•OH, D2O exchangeable), 5.27 and 5.67 (2d, 2H, С2,3•ОН, D2O exchangeable), 5.89 (d, 1H, J1',2'=3.85 Hz, C1•H), 8.05 and 9.05 (2br s, 4H, 2 CONH2). Анализ. Рассчитано для C9H13N5O6 (287,23): С 37,63; Н 4,56; N 24,38. Найдено: С 37,52; Н 4,19; N 24,49.
ПРИМЕР 16
1-(2', 3',5'-Три бензоил-β-L-рибофуранозил)-пиридин-4-он-3-карбоксамид (33)
бензоил-β-L-рибофуранозил)-пиридин-4-он-3-карбоксамид (33)
К смеси гексаметилдисилазана (50 мл; 239,77 ммоль) и хлортриметилсилана (1,0 мл; 21,43 ммоль) добавляли пиридин-4-он-3-карбоксамид (1,38 г; 10,00 ммоль) (получен по процедуре, описанной в: W.C.J. Ross, J. Chem. Soc., С. 1816 (1966); W. Herz and D.R.K. Murty, J. Org. Chem., 26, 122, 1961). Эту смесь в течение 2 часов выдерживали в условиях образования флегмы при перемешивании и затем упаривали досуха под вакуумом и далее сушили под глубоким вакуумом в течение 2 часов при 60oС. Сухой смолистый остаток суспензировали в свежеперегнанном 1,2-дихлорэтане (50 мл) и к этой суспензии добавляли 1 ацетил-2,3,5-три
ацетил-2,3,5-три  бензоил-L-рибофуранозу (5,06 г; 10,0 ммоль) и свежеперегнанный SnCl4 (1,0 мл; 8,52 ммоль). Эту реакционную смесь грели до образования флегмы в течение 1,5 часов, охлаждали и разводили в СН2Сl2 (100 мл) и насыщенном водном NаНСО3 (25 мл). Смесь фильтровали через целит и слой целита промывали с помощью CH2Cl2 (20 мл). Органическую фазу отделяли, промывали водой, пока промывная жидкость не становилась нейтральной, и сушили над безводным сульфатом натрия. Органический экстракт упаривали досуха с получением смолистого остатка. Этот остаток чистили быстрой хроматографией на силикагеле, используя CH2Cl2-->EtOAc в качестве элюента. Чистые фракции объединяли и концентрировали с получением 0,50 г (9%) (33) в виде белой пены: 1H-ЯМР (СDСl3) δ 4.94 (m, 3Н, С4•Н and С5•Н), 6,12 (m, 1H). 6.20 (m, 1H), 6.32 (d, 1H) and 7.20-8.30 (m, 20H).
бензоил-L-рибофуранозу (5,06 г; 10,0 ммоль) и свежеперегнанный SnCl4 (1,0 мл; 8,52 ммоль). Эту реакционную смесь грели до образования флегмы в течение 1,5 часов, охлаждали и разводили в СН2Сl2 (100 мл) и насыщенном водном NаНСО3 (25 мл). Смесь фильтровали через целит и слой целита промывали с помощью CH2Cl2 (20 мл). Органическую фазу отделяли, промывали водой, пока промывная жидкость не становилась нейтральной, и сушили над безводным сульфатом натрия. Органический экстракт упаривали досуха с получением смолистого остатка. Этот остаток чистили быстрой хроматографией на силикагеле, используя CH2Cl2-->EtOAc в качестве элюента. Чистые фракции объединяли и концентрировали с получением 0,50 г (9%) (33) в виде белой пены: 1H-ЯМР (СDСl3) δ 4.94 (m, 3Н, С4•Н and С5•Н), 6,12 (m, 1H). 6.20 (m, 1H), 6.32 (d, 1H) and 7.20-8.30 (m, 20H).
ПРИМЕР 17
1-β-L-рибофуранозилпиридин-4-он-3-карбоксамид (34)
Соединение (33) (0,5 г, 0,86 моль) растворяли в сухом растворе аммиака в метаноле (50 мл) и 15 часов перемешивали в баллоне при комнатной температуре. Затем этот раствор концентрировали до небольшого объема и охлаждали в течение ночи при температуре 4oС. Образовавшийся кристаллический продукт отфильтровывали и промывали холодным метанолом. Твердое вещество перекристаллизовывали из абсолютного этанола с получением 0,23 г (87%) чистого продукта: т. пл. 209-211oС; 1Н-ЯМР (Me2SO4-d6) δ 3.60 (m, 2H, С5•Н), 3.93 (m, 1H, С4•Н), 4.09 (m, 1H, С3•Н), 4.34 (m, 1H, C2•H), 5.11 (m, 1H, C5•OH, D2O exchangeable), 5.22 and 5.47 (2m, 2H, С2',3•ОН, D2O exchangeable), 5.84 (d, 1H, J1',2'=6.3 Hz, C1•H), 7.21 (m, 2H, PhH), 7.64 (m, 2H, PhH and CONH2) and 8.44 (s, 1H, CONH2). Анализ. Рассчитано для С11Н14N2O6 (270,24): С 48,89; Н 5,22; N 10,37. Найдено: С 48,89; Н 5,42; N 10,51.
ПРИМЕР 18
2,3,5-Три  бензоил-β-L-рибофуранозилазид (35)
бензоил-β-L-рибофуранозилазид (35)
Сухой хлористый водород пропускали через суспензию тонко измельченной и высушенной 1 ацетил-2,3,5-три-O-бензоил-L-рибозы (20,0 г; 39,52 ммоль) в эфире (300 мл) при 0oС до получения прозрачного раствора (2-3 часа). Затем эту смесь оставляли при 0oС на ночь. Растворитель удаляли и оставшуюся смолу последовательно упаривали с сухим бензолом (2•25 мл) и толуолом (25 мл). Остаток растворяли в метилцианиде (250 мл). К тому, что получилось, добавляли азид натрия (20,0 г, 307,6 ммоль) и нагревали реакционную смесь до образования флегмы в атмосфере аргона в течение 2 часов. После завершения реакции, как это было определено с помощью ТСХ с гексаном/этилацетатом (7: 3), раствор фильтровали и упаривали с получением маслянистого продукта (14,6 г) с количественным выходом. Продукт при сушке в вакууме образовывал белую пену. Сухой материал использовали как таковой в последующей реакции. 1H-ЯМР (СDСl3) δ 4.54 (m, 1H), 4.76 (m, 2H), 5.57-5.58 (dd, 1H), 5.68(d, 1H, J= 1.65Hz), 5.84-5.86 (m, 1H) and 7.34-8.12 (m, 15H, PhH).
ацетил-2,3,5-три-O-бензоил-L-рибозы (20,0 г; 39,52 ммоль) в эфире (300 мл) при 0oС до получения прозрачного раствора (2-3 часа). Затем эту смесь оставляли при 0oС на ночь. Растворитель удаляли и оставшуюся смолу последовательно упаривали с сухим бензолом (2•25 мл) и толуолом (25 мл). Остаток растворяли в метилцианиде (250 мл). К тому, что получилось, добавляли азид натрия (20,0 г, 307,6 ммоль) и нагревали реакционную смесь до образования флегмы в атмосфере аргона в течение 2 часов. После завершения реакции, как это было определено с помощью ТСХ с гексаном/этилацетатом (7: 3), раствор фильтровали и упаривали с получением маслянистого продукта (14,6 г) с количественным выходом. Продукт при сушке в вакууме образовывал белую пену. Сухой материал использовали как таковой в последующей реакции. 1H-ЯМР (СDСl3) δ 4.54 (m, 1H), 4.76 (m, 2H), 5.57-5.58 (dd, 1H), 5.68(d, 1H, J= 1.65Hz), 5.84-5.86 (m, 1H) and 7.34-8.12 (m, 15H, PhH).
ПРИМЕР 19
5-Амино-1-(2',3',5'три ацетил-β-L-рибофуранозил)-триазол-4-карбоксамид (36)
ацетил-β-L-рибофуранозил)-триазол-4-карбоксамид (36)
N, N-Диметилформамид (60 мл) добавляли к холодному (0oС) раствору гидроксида калия (1,72 г; 30,7 ммоль) в воде (10 мл) и раствор перемешивали при указанной температуре в течение 15 минут. К этому раствору добавляли цианоацетамид (2,58 г; 30,68 ммоль) и затем смесь перемешивали при 0oС, пока твердый материал не растворялся. К этому раствору одной порцией добавляли 2,3,5-три бензоил-β-L-рибофуранозилазид (10,0 г; 20,5 ммоль) и 14 часов перемешивали реакционную смесь при -5oС. Янтарный раствор упаривали в вакууме (водяная баня при 50oС) с получением оранжевого полутвердого вещества, которое последовательно упаривали с абсолютным этанолом (2•50 мл) и толуолом (3•50 мл) в вакууме с получением густой оранжевой смолы. Эту смолу растворяли в безводном метаноле (150 мл), добавляли метоксид натрия (1 н.; 25 мл) и перемешивали раствор при комнатной температуре в безводных условиях в течение 6 часов. Янтарный раствор обрабатывали ионообменной смолой Dowex 50X в протонированной форме (примерно 35 мл влажной смолы), чтобы довести рН до 6. Этот раствор фильтровали, слой смолы промывали дополнительной порцией метанола (50 мл) и объединенные фильтраты упаривали в вакууме досуха (водяная баня при 80oС) с получением оранжевой смолы. Эту смолу несколько раз перетирали с этилацетатом (6х50 мл) и каждую порцию по очереди декантировали, пока смола не затвердевала в светло-коричневое аморфное твердое вещество. Беловатый неочищенный продукт (2,5 г; 32%) был хроматографически чист. После нескольких кристаллизаций продукт содержал примеси и его переводили в ацетатную форму, как описано ниже.
бензоил-β-L-рибофуранозилазид (10,0 г; 20,5 ммоль) и 14 часов перемешивали реакционную смесь при -5oС. Янтарный раствор упаривали в вакууме (водяная баня при 50oС) с получением оранжевого полутвердого вещества, которое последовательно упаривали с абсолютным этанолом (2•50 мл) и толуолом (3•50 мл) в вакууме с получением густой оранжевой смолы. Эту смолу растворяли в безводном метаноле (150 мл), добавляли метоксид натрия (1 н.; 25 мл) и перемешивали раствор при комнатной температуре в безводных условиях в течение 6 часов. Янтарный раствор обрабатывали ионообменной смолой Dowex 50X в протонированной форме (примерно 35 мл влажной смолы), чтобы довести рН до 6. Этот раствор фильтровали, слой смолы промывали дополнительной порцией метанола (50 мл) и объединенные фильтраты упаривали в вакууме досуха (водяная баня при 80oС) с получением оранжевой смолы. Эту смолу несколько раз перетирали с этилацетатом (6х50 мл) и каждую порцию по очереди декантировали, пока смола не затвердевала в светло-коричневое аморфное твердое вещество. Беловатый неочищенный продукт (2,5 г; 32%) был хроматографически чист. После нескольких кристаллизаций продукт содержал примеси и его переводили в ацетатную форму, как описано ниже.
Вышеуказанный неочищенный материал (0,4 г; 1,54 ммоль) растворяли в пиридине (10 мл). Этот раствор охлаждали до 0oС в атмосфере аргона и обрабатывали уксусным ангидридом (0,95 г, 9,26 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и гасили метанолом (1,0 мл). Растворитель удаляли и остаток растворяли в CH2Cl2 (100 мл). Органический слой промывали насыщенным NаНСО3 (100 мл) и соляным раствором (50 мл), сушили и упаривали досуха. Неочищенный продукт чистили быстрой хроматографией на силикагеле, используя EtOAc в качестве элюента: выход 0,52 г (88%); 1H-ЯМР (СDСl3) δ 2.12 (3s, 9H, 3 СОСН3), 4.32-4.52 (m, 3H), 5.64 (m, 1H, С3•Н), 5.85 (m, 1Н, С2•Н), 6.00 (br s, 2H, NH2), 6.32 (d, 1H, С1•Н) and 8.73 (br s, 2H, CONH2).
ПРИМЕР 20
5-Амино-1-β-L(+)-рибофуранозилтриазол-4-карбоксамид (37)
Соединение 36 (0,5 г; 1,29 ммоль) растворяли в растворе аммиака в метаноле (50 мл; насыщенный при oС). Эту реакционную смесь 16 часов перемешивали при комнатной температуре и упаривали досуха. Остаток трижды перетирали с EtOAc и твердое вещество кристаллизовали из минимального количества сухого ЕtOН с получением бесцветного твердого вещества: т. пл. 159-161oС; 1H-ЯМР (Me2SO4-d6) δ 3.40-3.52 (m, 2H, C5•H), 3.93 (m, 1H, С4•Н), 4.19 (m, 1Н, С3•Н), 4.46 (m, 1H, С2•Н), 4.74, 5.22, 5.62 (m, 3Н, 3 ОН, D2O exchangeable), 5.84 (d, 1H, J=3.90 Hz, C1•H), 7.95 (br s, 2H) and 9.02 (br s, 2H). Анализ. Рассчитано для C6H13N5O5 (259,22): С 37,07; Н 5,05; N 27,02. Найдено: С 37,36; Н 5,14; N 27,01.
ПРИМЕР 21 Ацетил-1-(2', 3', 5'-три
Ацетил-1-(2', 3', 5'-три  ацетил-β-L-рибофуранозил)-триазол-4-карбоксамид (38)
ацетил-β-L-рибофуранозил)-триазол-4-карбоксамид (38)
N,N-Диметилформамид (40 мл) добавляли к холодному (0oС) раствору гидроксида калия (1,16 г; 20,82 ммоль) в воде (20 мл) и раствор перемешивали при указанной температуре в течение 10 минут. К этому раствору добавляли этилмалонамат (2,73 г; 20,82 ммоль) и затем перемешивали смесь при 0oС, пока твердый материал не растворялся. К этому раствору одной порцией добавляли 2,3,5-три бензоил-β-L-рибофуранозилазид (6,76 г; 13,88 ммоль) и перемешивали реакционную смесь при -5oС в течение 14 часов. Янтарного цвета раствор упаривали в вакууме (водяная баня при 80oС) с получением оранжевого полутвердого вещества, которое последовательно упаривали с абсолютным этанолом (2•50 мл) и толуолом (3•50 мл) в вакууме с получением густой оранжевой смолы. Эту смолу растворяли в безводном метаноле (150 мл), добавляли метоксид натрия (0,5 н.; 10 мл) и перемешивали раствор при комнатной температуре в безводных условиях в течение 6 часов. Янтарного цвета раствор обрабатывали ионообменной смолой Dowex 50 X в протонированной форме (примерно 35 мл влажной смолы), чтобы довести рН до 6. Раствор фильтровали, смoлу промывали дополнительной порцией 50 мл метанола и объединенные фильтраты упаривали досуха в вакууме (водяная баня при 80oС) с получением оранжевой смолы. Эту смолу несколько раз перетирали с этилацетатом (6•50 мл) и каждую порцию по очереди декантировали, пока смола не затвердевала в светло-коричневое аморфное твердое вещество. Это твердое вещество суспензировали в безводном
бензоил-β-L-рибофуранозилазид (6,76 г; 13,88 ммоль) и перемешивали реакционную смесь при -5oС в течение 14 часов. Янтарного цвета раствор упаривали в вакууме (водяная баня при 80oС) с получением оранжевого полутвердого вещества, которое последовательно упаривали с абсолютным этанолом (2•50 мл) и толуолом (3•50 мл) в вакууме с получением густой оранжевой смолы. Эту смолу растворяли в безводном метаноле (150 мл), добавляли метоксид натрия (0,5 н.; 10 мл) и перемешивали раствор при комнатной температуре в безводных условиях в течение 6 часов. Янтарного цвета раствор обрабатывали ионообменной смолой Dowex 50 X в протонированной форме (примерно 35 мл влажной смолы), чтобы довести рН до 6. Раствор фильтровали, смoлу промывали дополнительной порцией 50 мл метанола и объединенные фильтраты упаривали досуха в вакууме (водяная баня при 80oС) с получением оранжевой смолы. Эту смолу несколько раз перетирали с этилацетатом (6•50 мл) и каждую порцию по очереди декантировали, пока смола не затвердевала в светло-коричневое аморфное твердое вещество. Это твердое вещество суспензировали в безводном
пиридине (30 мл) и уксусном ангидриде (7,8 мл; 83,28 ммоль) и перемешивали в безводных условиях при комнатной температуре в течение 18 часов. Реакционную смесь фильтровали через тонкий слой уплотненного целита. Слой целита промывали свежим пиридином (50 мл) и объединенные экстракты упаривали досуха в вакууме с получением коричневой смолы. Эту смолу растворяли в CH2Cl2 (150 мл). Органический слой промывали насыщенным NаНСО3 (100 мл) и соляным раствором (50 мл), сушили и упаривали досуха. Неочищенный продукт чистили быстрой хроматографией на силикагеле, используя CH2Cl2-->EtOAc в качестве элюента. Чистые фракции собирали и упаривали с получением 1,5 (42%) чистого продукта (38). 1H-ЯМР (СDСl3) δ 2.14 (3s, 9Н, 3 СОСН3), 2.60 (s, 3H, СОСН3), 4.15-4.58 (m, 3H, C4•H and C5•H), 5.62 (m, 1H, С3•Н), 5.82 (m, 1H, C2•H), 6.28 (d, 1H, C1•H) and 10.63 (br s, 2H, CONH2).
ПРИМЕР 22
5-Гидрокси-1-β-L(+)-рибофуранозилтриазол-4-карбоксамид (39)
Соединение (38) (1,5 г; 3,50 ммоль) растворяли в растворе аммиака в метаноле (50 мл, насыщенный при oС). Эту реакционную смесь 16 часов перемешивали при комнатной температуре и упаривали досуха. Остаток трижды перетирали с ЕtOАс и твердое вещество кристаллизовали из минимального количества сухого EtOH с получением 0,70 г (77%) (39): т. пл. 162-164oС; 1H-ЯМР (Me2SO4-d6) δ 3.40-3.50 (m, 2H, С5•Н). 3.84 (m, 1H, С4•Н), 4.17 (m, 1H, С3•Н), 4.42 (m, 1H, C2•H), 4.90 (t, 1H, С5•ОН), 5.20, 5.58 (2d, 2H, 2 ОН, D2O exchangeable), 5.76 (d, 1H, J=3.90 Hz, C1•H), 7.58 and 7.80 (2br s, 2H, CONH2) and 8.82 (s, 1H, C5OH). Анализ. Рассчитано для C8H12N4O6 (260,21): С 39,92; Н 4,65; N 21,53. Найдено: С 36,90; Н 4,79; N 21,43.
ПРИМЕР 23
1-(2',3',5'-Три бензоил-β-L-рибофуранозил)-6-метилурацил (40)
бензоил-β-L-рибофуранозил)-6-метилурацил (40)
Смесь 6-метилурацила (2,52 г, 20,00 ммоль), гексаметилдисилазина (50 мл) и сульфата аммония (100 мг) 6 часов грели с образованием флегмы при 135oС. Растворитель удаляли в вакууме и полученный остаток дважды упаривали с сухим толуолом (2•50 мл), чтобы удалить последние следы гексаметилдисилазина. Полученное таким образом твердое вещество сушили под вакуумом в течение 6 часов. К раствору этого 2,4-бис(триметилсилилокси)-6-метилпиримидина (20,0 ммоль) в сухом ацетонитриле (100 мл) добавляли  ацетил-2,3,5-три
ацетил-2,3,5-три бензоил-L-рибофуранозу (10,12 г, 20 ммоль) и триметилсилилтрифлат (5,78 г; 26,0 ммоль). Эту реакционную смесь перемешивали под аргоном при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в дихлорметане (200 мл). Органический слой промывали насыщенным бикарбонатом натрия (200 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали с получением белой пены. Последующее разделение неочищенного продукта с помощью быстрой хроматографии на колонке с силикагелем с использованием Cl2Cl2-->EtOAc в качестве элюента дало два продукта. Выход второй фракции составил 4,5 г (42%). 1H-ЯМР (СDСl3) δ 2.28 (s, 3Н, СН3), 4.65-4.81 (m, 3Н, С4•Н and C5•H), 5.60 (m, 1H, С3•Н), 5.72 (s, 1H), 6.11 (m, 1H), 7.24-8.06 (m, 16H, PhH) and 9.40 (br s, 1H, NH). Первая фракция (4,20 г) по данным 1H-ЯMP не соответствовала желаемому продукту.
бензоил-L-рибофуранозу (10,12 г, 20 ммоль) и триметилсилилтрифлат (5,78 г; 26,0 ммоль). Эту реакционную смесь перемешивали под аргоном при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в дихлорметане (200 мл). Органический слой промывали насыщенным бикарбонатом натрия (200 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали с получением белой пены. Последующее разделение неочищенного продукта с помощью быстрой хроматографии на колонке с силикагелем с использованием Cl2Cl2-->EtOAc в качестве элюента дало два продукта. Выход второй фракции составил 4,5 г (42%). 1H-ЯМР (СDСl3) δ 2.28 (s, 3Н, СН3), 4.65-4.81 (m, 3Н, С4•Н and C5•H), 5.60 (m, 1H, С3•Н), 5.72 (s, 1H), 6.11 (m, 1H), 7.24-8.06 (m, 16H, PhH) and 9.40 (br s, 1H, NH). Первая фракция (4,20 г) по данным 1H-ЯMP не соответствовала желаемому продукту.
ПРИМЕР 24
1-β-Рибофуранозил-6-метилурацил (41)
Раствор (40) (4,50 г; 7,86 ммоль) растворяли в насыщенном растворе аммиака в метаноле (60 мл). Эту реакционную смесь 17 часов грели в стальном баллоне при 100oС. Реакционную смесь охлаждали до комнатной температуры и концентрировали с получением масла. Остаток чистили дальше с помощью быстрой хроматографии на колонке с силикагелем, используя дихлорметан и метанол (9: 1) в качестве элюента. Чистые фракции собирали и упаривали с получением белого твердого вещества. Далее это вещество перекристаллизовывали из 2-пропанола с получением 1,98 г (94%) чистого (41): т. пл. 175-177oС; 1H-ЯМР (Me2SO4-d6) δ 2.24 (s, 3Н, СН3), 3.42-3.57 (m, 2H, С5•Н), 3.68 (m, 1H, С4•Н), 4.0 (m. 1H, С3•Н), 4.53 (m, 1H, C2•H), 4.68, 4.94, 5.22 (m, 3Н, 3 ОН, D2O exchangeable), 5.43 (d, 1H, C1•H, J1',2'=3.85 Hz), 5.56 (s, 1H, C5H) and 11,25 (s, 1H, NH). Анализ. Рассчитано для С10Н14N2O8 (258,23): С 46,51; Н 5,46; N 10,85. Найдено: С 46,66; Н 5,26; N 10,66.
ПРИМЕР 25
1-(2',3',5'-Три бензоил-β-L-рибофуранозил)-5-азацитидин (42)
бензоил-β-L-рибофуранозил)-5-азацитидин (42)
5-Азацитозин (1,12 г; 10,0 ммоль) суспензировали в смеси гексаметилдисилазина (50 мл) и сульфата аммония (100 мг). Эту реакционную смесь 6 часов грели при 135oС с образованием флегмы. Затем удаляли растворители в вакууме и полученный таким образом остаток дважды упаривали с сухим толуолом (2•50 мл), чтобы удалить последние следы гексаметилдисилазина. Полученное таким образом твердое вещество сушили под вакуумом в течение 6 часов. К раствору 2,4-N, бис(триметилсисил)-5-азацитидина (10,0 ммоль) в сухом 1,2-дихлорэтане (150 мл) при 10oС добавляли  ацетил-2,3,5-три
ацетил-2,3,5-три бензоил-β-L-рибофуранозу (5,06 г; 10 ммоль) и тетрахлорид олова (1,68 мл; 14,16 ммоль). Эту реакционную смесь 2 часа перемешивали а атмосфере аргона при 10oС. Реакционную смесь проверяли с помощью ТСХ, используя гексан и этилацетат (7:3). ТСХ показала, что исходных материалов не осталось. Реакционную смесь разводили дихлорметаном (250 мл). Органический слой промывали насыщенным бикарбонатом натрия (200 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали до остатка. Этот остаток растворяли в толуоле и фильтровали через целит, чтобы удалить непрореагировавший 5-азацитозин. Фильтрат упаривали досуха и остаток (5,20 г) растворяли в этаноле и снова фильтровали через целит. Целевое соединение кристаллизовали из фильтрата в виде игл (4,45 г; 81%): т. пл. 186-187oС; 1H-ЯМР (СDСl3) δ 4.62-4.66 (m, 3Н. С4•Н and C5•H), 6,01 (m, 3H, C1•H, C2•H and C3•H), 7.26-8.06 (m, 17H, NH2 and РhН) and 8.48 (s, 1H, С6Н).
бензоил-β-L-рибофуранозу (5,06 г; 10 ммоль) и тетрахлорид олова (1,68 мл; 14,16 ммоль). Эту реакционную смесь 2 часа перемешивали а атмосфере аргона при 10oС. Реакционную смесь проверяли с помощью ТСХ, используя гексан и этилацетат (7:3). ТСХ показала, что исходных материалов не осталось. Реакционную смесь разводили дихлорметаном (250 мл). Органический слой промывали насыщенным бикарбонатом натрия (200 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали до остатка. Этот остаток растворяли в толуоле и фильтровали через целит, чтобы удалить непрореагировавший 5-азацитозин. Фильтрат упаривали досуха и остаток (5,20 г) растворяли в этаноле и снова фильтровали через целит. Целевое соединение кристаллизовали из фильтрата в виде игл (4,45 г; 81%): т. пл. 186-187oС; 1H-ЯМР (СDСl3) δ 4.62-4.66 (m, 3Н. С4•Н and C5•H), 6,01 (m, 3H, C1•H, C2•H and C3•H), 7.26-8.06 (m, 17H, NH2 and РhН) and 8.48 (s, 1H, С6Н).
ПРИМЕР 26
4-Амино-1-β-L-рибофуранозилтриазин-2(1Н)он (5-азацитидин, 43)
Соединение (42) (4,0 г; 7,19 ммоль) растворяли в абсолютном метаноле (60 мл), нагревали до кипения и обрабатывали 0,5 М раствором метоксида натрия (20 мл; 10,0 ммоль). Исходный материал быстро растворялся, и в растворе немедленно откладывался продукт. Эту смесь держали 4 часа при комнатной температуре и в течение ночи в холодильнике. Кристаллы собирали, промывали охлажденным во льду метанолом (10 мл) и сушили под пониженным давлением при комнатной температуре. Выход составил 1,50 г (86%). Образец для анализа получали перекристаллизацией из смеси воды и ацетона (1:1): т. пл. 220-222oС. 1H-ЯМР (D2О) δ 3.78-3.97 (m, 2H, С5•Н), 4.13 (m, 1Н, С4•Н), 4.20 (m, 1H, С3•Н), 4.33 (m, 1H, С2•Н), 6.31 (d, 1H, C1•H), J1',2'=2.5 Hz) and 8.24 (s, 1H, C6H). Анализ. Рассчитано для C8H12N4O5 (244,20): С 39,35; Н 4,95; N 22,94. Найдено: С 34.09; Н 4,28; N 22,98.
ПРИМЕР 27
1-(2',3',5'-Три бензоил-β-L-рибофуранозил)-6-азауридин (44)
бензоил-β-L-рибофуранозил)-6-азауридин (44)
6-Азаурацил (1,36 г; 12,0 ммоль) суспензировали в смеси гексаметилдисилазина (50 мл) и сульфата аммония (50 мг). Эту реакционную смесь 6 часов грели при 135oС с образованием флегмы. Затем удаляли растворители в вакууме и полученный таким образом остаток дважды упаривали с сухим толуолом (2•50 мл), чтобы удалить последние следы гексаметилдисилазина. Твердое вещество сушили в вакууме в течение 6 часов и использовали на следующей стадии синтеза без дальнейшей характеризации. К раствору 2,4-N,бис(триметилсисил)-5-азацитидина (12,0 ммоль) в сухом 1,2-дихлорэтане (60 мл) при 10oС добавляли  ацетил-2,3,5-три
ацетил-2,3,5-три бензоил-β-L-рибофуранозу (5,06 г, 10 ммоль) и тетрахлорид олова (1,68 мл; 14,16 ммоль). Эту реакционную смесь 6 часов перемешивали в атмосфере аргона при комнатной температуре. Реакционную смесь проверяли с помощью ТСХ, используя гексан и этилацетат (7:3). ТСХ показала, что исходных материалов не осталось. Реакционную смесь разводили дихлорметаном (250 мл). Органический слой промывали насыщенным бикарбонатом натрия (150 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали до белой пены. Этот остаток растворяли в дихлорметане (100 мл) и фильтровали через целит, чтобы удалить непрореагировавший 6-азаурацил. Фильтрат упаривали до остатка (4,50 г), который растворяли в минимальном количестве этанола и снова фильтровали через целит. Целевое соединение кристаллизовали из фильтрата в виде игл с получением 4,50 г (79%) чистого (44): т. пл. 193-195oС; 1H-ЯМР (Me2SO4-d6) δ 4.47-4.67 (m, 3H, С5•Н), 4,71 (m, 1Н, С4•Н), 5.85 (m, 1Н, С3•Н), 5.93 (m, 1H, С2•Н), 6.23 (d, 1H, J1',2'=2.56 Hz, C1•H), 7.26-8.06 (m, 16H, C5H and РhН) and 12.32 (s, 1H, NH).
бензоил-β-L-рибофуранозу (5,06 г, 10 ммоль) и тетрахлорид олова (1,68 мл; 14,16 ммоль). Эту реакционную смесь 6 часов перемешивали в атмосфере аргона при комнатной температуре. Реакционную смесь проверяли с помощью ТСХ, используя гексан и этилацетат (7:3). ТСХ показала, что исходных материалов не осталось. Реакционную смесь разводили дихлорметаном (250 мл). Органический слой промывали насыщенным бикарбонатом натрия (150 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали до белой пены. Этот остаток растворяли в дихлорметане (100 мл) и фильтровали через целит, чтобы удалить непрореагировавший 6-азаурацил. Фильтрат упаривали до остатка (4,50 г), который растворяли в минимальном количестве этанола и снова фильтровали через целит. Целевое соединение кристаллизовали из фильтрата в виде игл с получением 4,50 г (79%) чистого (44): т. пл. 193-195oС; 1H-ЯМР (Me2SO4-d6) δ 4.47-4.67 (m, 3H, С5•Н), 4,71 (m, 1Н, С4•Н), 5.85 (m, 1Н, С3•Н), 5.93 (m, 1H, С2•Н), 6.23 (d, 1H, J1',2'=2.56 Hz, C1•H), 7.26-8.06 (m, 16H, C5H and РhН) and 12.32 (s, 1H, NH).
ПРИМЕР 28
1-β-L-Рибофуранозил-6-азаурацил (6-азауридин 45)
Соединение 44 (4,5 г, 7,95 ммоль) растворяли в растворе аммиака в абсолютном метаноле (60 мл) и помещали в стальной баллон. Затем его 16 часов грели при 100oС. После этого реакционный сосуд охлаждали до комнатной температуры и растворитель удаляли под вакуумом. Полученный остаток перетирали с горячим толуолом (2•50 мл). Остаток растворяли в 95% этаноле и оставляли при комнатной температуре. Полученные белые твердые кристаллы собирали фильтрацией и сушили в вакууме. Выход составил 1,75 г (89%): т. пл. 151-153oС; 1H-ЯМР (Me2SO4-d6) δ 3.30-3.47 (m, 2H, С5•Н), 3.73 (m, 1H, С4•Н), 3.92 (m, 1H, С3•Н), 4.17 (m, 1H, С2•Н), 4.62, 4.98, 5.22 (3br s, 3H, 3 ОН, D2O exchangeable), 5.82 (d, 1H, С1•Н, J1',2'=3.85 Hz), 7.48 (s, 1H, C5H) and 11.20 (br s, 1H, NH). Анализ. Рассчитано для С8Н11N3О6 (245,19): С 39,19; Н 4,52; N 17,14. Найдено: С 38,81; Н 4,58; N 17,04.
ПРИМЕР 29
Диэтиловый эфир имидазол-4,5-дикарбоновой кислоты (46)
Имидазол-4,5-дикарбоновую кислоту (7,55 г; 50,0 ммоль) растворяли в абсолютном этиловом спирте (120 мл). Этот раствор охлаждали в ледяной бане до 0oС и в течение часа продували сухим газообразным HCl. Затем реакционную смесь грели с образованием флегмы при 80oС 7 часов, в течение которых весь исходный материал вступал в реакцию. Растворитель удаляли, полученный остаток растворяли в дихлорметане (200 мл) и органический слой нейтрализовали триэтиламином. Органический слой промывали холодной водой (100 мл) и соляным раствором (50 мл), сушили над безводным сульфатом натрия и концентрировали в вакууме с получением 5,50 г (52%) белого твердого вещества: т. пл. 175-177oС; 1H-ЯМР (CDCl3) δ 1.40 (t, 3H), 4.41 (m, 2H), 7.84 (1Н, С2Н) and 11.55 (br s, 1H, NH).
ПРИМЕР 30
Диэтиловый эфир 1-(2', 3', 5'-три бензоил-β-L-рибофуранозил)-имидазол-4,5-дикарбоновой кислоты (47)
бензоил-β-L-рибофуранозил)-имидазол-4,5-дикарбоновой кислоты (47)
Смесь диэтилового эфира имидазол-4,5-дикарбоновой кислоты (2,65 г; 12,50 ммоль) и сульфата аммония (50 мг) 6 часов грели при 135oС вместе с гексаметилдисилазином (50 мл) с образованием флегмы. Эту реакционную смесь упаривали досуха и остаток дважды упаривали с сухим толуолом (2•50 мл), чтобы удалить последние следы гексаметилдисилазина. Полученное таким образом твердое вещество сушили в вакууме в течение 6 часов и использовали для следующей стадии без дальнейшей характеризации. К раствору вышеуказанного остатка (12,5 ммоль) в 1,2-дихлорэтане (60 мл) добавляли при 10oС  ацетил-2,3,5-три
ацетил-2,3,5-три бензоил-L-рибофуранозу (5,06 г, 10 ммоль) и тетрахлорид олова (1,68 мл; 14,16 ммоль). Эту реакционную смесь перемешивали в атмосфере аргона при комнатной температуре в течение 6 часов. Реакционную смесь проверяли с помощью ТСХ, используя гексан и этилацетат (7:3). ТСХ показала, что исходных материалов не осталось. Реакционную смесь разбавляли дихлорметаном (200 мл). Органический слой промывали холодным насыщенным бикарбонатом натрия (200 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали с получением белой пены. Этот остаток растворяли в дихлорметане (100 мл) и фильтровали через целит для удаления солей олова. После упаривания в вакууме остаток (4,70 г) растворяли в этаноле и снова фильтровали через целит. Целевое соединение 47 кристаллизовали из фильтрата в виде игл. Выход составил 4,70 г (72%): т. пл. 134-146oС; 1H-ЯМР (СDСl3) δ 1.28 (t, 3Н, СН3), 1.37 (t, 3Н, СH3), 4.28-4.40 (m, 4H, 2 CH2), 4.65-4.88 (m, 3H, C4•H and С5•Н), 5.85 (m, 2H, C2•H and С3•Н), 6.68 (d, 1H, C1•H, J1',2'=3.90 Hz) and 7.26-8.08 (m, 16H,C2H and PhH).
бензоил-L-рибофуранозу (5,06 г, 10 ммоль) и тетрахлорид олова (1,68 мл; 14,16 ммоль). Эту реакционную смесь перемешивали в атмосфере аргона при комнатной температуре в течение 6 часов. Реакционную смесь проверяли с помощью ТСХ, используя гексан и этилацетат (7:3). ТСХ показала, что исходных материалов не осталось. Реакционную смесь разбавляли дихлорметаном (200 мл). Органический слой промывали холодным насыщенным бикарбонатом натрия (200 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали с получением белой пены. Этот остаток растворяли в дихлорметане (100 мл) и фильтровали через целит для удаления солей олова. После упаривания в вакууме остаток (4,70 г) растворяли в этаноле и снова фильтровали через целит. Целевое соединение 47 кристаллизовали из фильтрата в виде игл. Выход составил 4,70 г (72%): т. пл. 134-146oС; 1H-ЯМР (СDСl3) δ 1.28 (t, 3Н, СН3), 1.37 (t, 3Н, СH3), 4.28-4.40 (m, 4H, 2 CH2), 4.65-4.88 (m, 3H, C4•H and С5•Н), 5.85 (m, 2H, C2•H and С3•Н), 6.68 (d, 1H, C1•H, J1',2'=3.90 Hz) and 7.26-8.08 (m, 16H,C2H and PhH).
ПРИМЕР 31
1-β-L-Рибофуранозилимидазол-4,5-дикарбоксамид (48)
Соединение (47) (4,0 г; 6,09 ммоль) растворяли в растворе аммиака в абсолютном метаноле (60 мл) и 16 часов грели при 100oС в стальном баллоне. Затем реакционную смесь охлаждали до комнатной температуры. Продукт кристаллизовали из метанола. Преципитировавший продукт удаляли фильтрацией, а фильтрат концентрировали дальше с получением второй порции продукта. Объединенный продукт еще раз перекристаллизовывали из метанола с получением 1,68 г (100%) белого твердого вещества: т. пл. 224-226oС; 1H-ЯМР (Me2SO4-d6) δ 3.53-3.75 (m, 2H, С5•Н), 3.84 (m, 1H, C4•H), 3.96 (m, 2H, C2•H and С3•Н), 4.97, 5.16, 5.63 (3 br s, 3Н, 3 ОН, D2O exchangeable), 6.49 (d, 1H, C1•H, J1',2'=2.1 Hz), 7.60 (s, 1H, CONH2), 7.88 (s, 1H, CONH2), 7.99 (s, 1H, CONH2), 8.48 (s, 1H, C2H) and 10,59 (s, 1H, CONH2). Анализ. Рассчитано для C10H14N4O6 (286.24): С 41,96; Н 4,93; N 19,57. Найдено: С 41,89; Н 5,05; N 19,41.
ПРИМЕР 32
Этиловый эфир 1-(2', 3', 5'-три бензоил-β-L-рибофуранозил)-3-гидрокси-1,2-пиразол-4-карбоновой кислоты (49)
бензоил-β-L-рибофуранозил)-3-гидрокси-1,2-пиразол-4-карбоновой кислоты (49)
Смесь этилового эфира 3-гидрокси-1,2-пиразол-4-карбоновой кислоты и сульфата аммония (50 мг) в гексаметилдисилазине (50 мл) нагревали до образования флегмы в течение 6 часов. Реакционную смесь упаривали досуха и полученный остаток дважды упаривали с сухим толуолом (2•50 мл), чтобы удалить последние следы гексаметилдисилазина. Полученное твердое вещество сушили в вакууме в течение 6 часов и использовали как таковой в дальнейшей реакции. К раствору вышеуказанного триметилсилильного производного (12,5 ммоль) в сухом 1,2-дихлорэтане (60 мл) добавляли при 10oС  ацетил-2,3,5-три
ацетил-2,3,5-три бензоил-L-рибофуранозу (5,06 г; 10 ммоль) и тетрахлорид олова (1,68 мл; 14,16 ммоль). Эту реакционную смесь 6 часов перемешивали в атмосфере аргона при комнатной температуре. Реакционную смесь разводили дихлорметаном (200 мл). Органический слой промывали насыщенным бикарбонатом натрия (200 мл), водой (100 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали до пены. Этот остаток растворяли в дихлорметане (70 мл) и фильтровали через целит, чтобы удалить соли олова. Неочищенный продукт чистили быстрой хроматографией на колонке с силикагелем, используя CH2Cl2-->EtOAc в качестве элюента. Чистые фракции объединяли и упаривали с получением 3,50 г (57%) белой пены: 1H-ЯМР (CDCl3) δ 1.36 (t, 3H, СН3), 4.30 (m, 2H, СН2), 4.52-4.82 (m, 3H, C4•H and C5•H), 6.08-6.32 (m, 3H, C1•H, С2•Н and С3•Н) and 7.62-8.08 (m, 16H, C5H and PhH).
бензоил-L-рибофуранозу (5,06 г; 10 ммоль) и тетрахлорид олова (1,68 мл; 14,16 ммоль). Эту реакционную смесь 6 часов перемешивали в атмосфере аргона при комнатной температуре. Реакционную смесь разводили дихлорметаном (200 мл). Органический слой промывали насыщенным бикарбонатом натрия (200 мл), водой (100 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали до пены. Этот остаток растворяли в дихлорметане (70 мл) и фильтровали через целит, чтобы удалить соли олова. Неочищенный продукт чистили быстрой хроматографией на колонке с силикагелем, используя CH2Cl2-->EtOAc в качестве элюента. Чистые фракции объединяли и упаривали с получением 3,50 г (57%) белой пены: 1H-ЯМР (CDCl3) δ 1.36 (t, 3H, СН3), 4.30 (m, 2H, СН2), 4.52-4.82 (m, 3H, C4•H and C5•H), 6.08-6.32 (m, 3H, C1•H, С2•Н and С3•Н) and 7.62-8.08 (m, 16H, C5H and PhH).
ПРИМЕР 33
1-β-L-Рибофуранозил-3-гидрокси-1,2-пиразол-4-карбоксамид (50)
Раствор (49) (3,50 г; 5,71 ммоль) в насыщенном растворе аммиака в метаноле (60 мл) 16 часов грели при 100oС в стальном баллоне. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток перетирали с толуолом (2•50 мл), чтобы удалить бензамид. Этот остаток растворяли в минимальном количестве абсолютного этанола и оставляли при комнатной температуре на ночь. Полученные кристаллы удаляли фильтрацией, а фильтрат концентрировали дальше с получением второй порции продукта. Объединенный продукт еще раз перекристаллизовывали из этанола до твердого вещества, которое собирали фильтрацией и сушили в вакууме с получением 1 г (68%): т. пл. 178-180oС; 1H-ЯМР (Me2SO4-d6) δ 3.37-3.52 (m, 2H, С5•Н), 3.78 (m, 1Н, С4•Н), 3.98 (m, 1Н, С3•Н), 4.19 (m, 1H, С2•Н), 4.81, 5.05, 5.34 (3 br s, 3H, 3 ОН, D2О exchangeable), 5.38 (d, 1H, C1•H, J1',2'=4,2 Hz), 6.98 (bs, 1H, CONH2), 7.16 (bs, 1H, CONH2), 8.08 (s, 1H, C5H) and 10.98 (bs, 1H, C3OH). Анализ. Рассчитано для C9H13N3O6 (259,22): С, 41.70; Н, 5.05; N, 16.21. Найдено: С 41,52; Н 5.23; N 16.40.
ПРИМЕР 34
1-Азидо-2,3-изопропилидин-β-L-рибофураноза (51)
К раствору 2,3,5-три бензоил-1-азидо-β-L-рибофуранозы (9,0 г 18,48 ммоль) в абсолютном метаноле (60 мл) добавляли 0,5 М раствор метоксида натрия (10,0 мл; 5,0 ммоль). Эту реакционную смесь в течение ночи перемешивали при комнатной температуре. ТСХ реакционной смеси (гексан/этилацетат, 7: 3) свидетельствовала о полном превращении исходного материала в более полярное соединение. Реакционную смесь нейтрализовали сухой смолой Dowex 50 в протонированной форме и смолу удаляли фильтрацией. Фильтрат упаривали досуха и растворяли в воде (50 мл). Водный слой экстрагировали дихлорметаном (2•100 мл), чтобы удалить метилбензоат, а затем водный слой концентрировали в вакууме. Остаток сушили дальше над пятиокисью фосфора и использовали как таковой для следующей стадии синтеза без дальнейшей характеризации.
бензоил-1-азидо-β-L-рибофуранозы (9,0 г 18,48 ммоль) в абсолютном метаноле (60 мл) добавляли 0,5 М раствор метоксида натрия (10,0 мл; 5,0 ммоль). Эту реакционную смесь в течение ночи перемешивали при комнатной температуре. ТСХ реакционной смеси (гексан/этилацетат, 7: 3) свидетельствовала о полном превращении исходного материала в более полярное соединение. Реакционную смесь нейтрализовали сухой смолой Dowex 50 в протонированной форме и смолу удаляли фильтрацией. Фильтрат упаривали досуха и растворяли в воде (50 мл). Водный слой экстрагировали дихлорметаном (2•100 мл), чтобы удалить метилбензоат, а затем водный слой концентрировали в вакууме. Остаток сушили дальше над пятиокисью фосфора и использовали как таковой для следующей стадии синтеза без дальнейшей характеризации.
Вышеуказанный неочищенный продукт (3,0 г; 17,14 ммоль) суспензировали в сухом ацетоне (200 мл) и обрабатывали 1,1-диметоксипропаном (50 мл) и высушенной в вакууме смолой Dowex 50 в протонированной форме (5,0 г). Эту реакционную смесь 2 часа перемешивали при комнатной температуре и фильтровали, и смолу промывали сухим ацетоном (100 мл). Фильтрат упаривали досуха. Остаток чистили быстрой хроматографией на силикагеле, используя CH2Cl2-->EtOAc в качестве элюента. Чистые фракции объединяли и концентрировали с получением 3.60 г (97%) продукта в виде масла: 1H-ЯМР (CDCl3) δ 1.44 and 1.27 (2s, 6H, isopropylidene СН3), 2.70 (br s, 1H, C5•OH, exchangeable), 3.66 (m, 2Н, С5•Н), 4.34 (m, 1Н, С4•Н), 4.46 (d, 1H, С3•Н), 4.72 (d, 1H, C2•H) and 5.50 (s, 1H, C1•H).
ПРИМЕР 35
1-Азидо изопропилидин
изопропилидин трет-бутилдиметилсилил-β-L-рибофураноза (52)
трет-бутилдиметилсилил-β-L-рибофураноза (52)
К раствору 1-азидо изопропилидин-β-L-рибофуранозы (4,20 г; 20 ммоль) в сухом DMF (25 мл) добавляли имидазол (2,38 г, 35 ммоль) и трет-бутилдиметилсилилхлорид (4,5 г, 35 ммоль). Эту реакционную смесь в течение ночи перемешивали при комнатной температуре в атмосфере аргона. ТСХ реакционной смеси через 16 часов свидетельствовала о полном превращении исходного материала в продукт. Растворитель удаляли в вакууме и остаток растворяли в дихлорметане (200 мл). Органический слой промывали водой (100 мл), насыщенным бикарбонатом натрия (100 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали до маслянистого продукта. Дальнейшая очистка быстрой хроматографией на колонке с силикагелем с использованием гексана/этилацетата (9: 1) дала 6,22 г (94%) целевого соединения в виде масла: 1H-ЯМР (СDСl3) δ 0.07 (s, 6H), 0.9 (s, 9H), 1.27 and 1.47 (2s, 6H, isopropilidene СН3), 3.66 (m, 2H, C5•H), 4.34 (m, 1H, С4•Н), 4.46 (d, 1H, С3•Н), 4.72 (d, 1H, C2•H) and 5.50 (s, 1H, C1•H).
изопропилидин-β-L-рибофуранозы (4,20 г; 20 ммоль) в сухом DMF (25 мл) добавляли имидазол (2,38 г, 35 ммоль) и трет-бутилдиметилсилилхлорид (4,5 г, 35 ммоль). Эту реакционную смесь в течение ночи перемешивали при комнатной температуре в атмосфере аргона. ТСХ реакционной смеси через 16 часов свидетельствовала о полном превращении исходного материала в продукт. Растворитель удаляли в вакууме и остаток растворяли в дихлорметане (200 мл). Органический слой промывали водой (100 мл), насыщенным бикарбонатом натрия (100 мл) и соляным раствором (100 мл), сушили над сульфатом натрия и концентрировали до маслянистого продукта. Дальнейшая очистка быстрой хроматографией на колонке с силикагелем с использованием гексана/этилацетата (9: 1) дала 6,22 г (94%) целевого соединения в виде масла: 1H-ЯМР (СDСl3) δ 0.07 (s, 6H), 0.9 (s, 9H), 1.27 and 1.47 (2s, 6H, isopropilidene СН3), 3.66 (m, 2H, C5•H), 4.34 (m, 1H, С4•Н), 4.46 (d, 1H, С3•Н), 4.72 (d, 1H, C2•H) and 5.50 (s, 1H, C1•H).
ПРИМЕР 36
1-Амино  изопропилидин
изопропилидин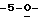 трет-бутилдиметилсилил-β-L-рибофураноза (53)
трет-бутилдиметилсилил-β-L-рибофураноза (53)
Смесь 1-азидо изопиридин-β-L-рибофуранозы (6,0 г; 18 ммоль) и Pd/C (0,25 г) в МеОН (50 мл) в течение ночи гидрогенировали при давлении 50 фунтов на квадратный дюйм в гидрогенаторе Парра. Реакционную смесь фильтровали и катализатор промывали метанолом (20 мл). Объединенный фильтрат упаривали досуха и в течение ночи сушили в вакууме над P2O5 и использовали как таковой для последующей реакции без характеризации. Выход составил 5,0 г (90%).
изопиридин-β-L-рибофуранозы (6,0 г; 18 ммоль) и Pd/C (0,25 г) в МеОН (50 мл) в течение ночи гидрогенировали при давлении 50 фунтов на квадратный дюйм в гидрогенаторе Парра. Реакционную смесь фильтровали и катализатор промывали метанолом (20 мл). Объединенный фильтрат упаривали досуха и в течение ночи сушили в вакууме над P2O5 и использовали как таковой для последующей реакции без характеризации. Выход составил 5,0 г (90%).
ПРИМЕР 37
Этиловый эфир 5-амино-(2', 3' изопропилидин-
изопропилидин- трет-бутилдиметилсилил-β-L-рибофуранозил)-имидазол-4-карбоновой кислоты (54)
трет-бутилдиметилсилил-β-L-рибофуранозил)-имидазол-4-карбоновой кислоты (54)
К перемешиваемому раствору 53 (5,0 г; 16,44 моль) в сухом Cl2Cl2 (60 мл) 15 минут добавляли раствор этилового эфира N-циано-N-(этоксикарбонилметил)-формимида (4,0 г; 22,18 ммоль; Robinson, D.H., et al., J. Chem. Soc., Perkin 1, 1715-1717, 1972). Эту реакционную смесь в течение ночи перемешивали при комнатной температуре в атмосфере аргона. Реакционную смесь разводили в CH2Cl2 (100 мл) и органический слой промывали насыщенным NаНСО3 (100 мл), водой (50 мл) и соляным раствором (50 мл). Органический экстракт сушили и концентрировали с получением неочищенного продукта. Этот неочищенный продукт чистили быстрой хроматографией на силикагеле, используя CH2Cl2-->EtOAc в качестве элюента. Чистые фракции объединяли и упаривали с получением 5,5 г (76%) белой пены: 1H-ЯМР (СDСl3) δ 0.28 (m, 6Н), 1.1 (m, 9Н), 1.55 (m, 9H), 4.00 (m, 2H, C5•H), 4.53 (m, 3H), 5.0 (m, 1H), 5.78 (m, 1Н), 6.06 (d, 1H, C1•H) and 7.44 (s, 1Н, С2Н).
ПРИМЕР 38
5-Амино-(2', 3' изопропилидин
изопропилидин  трет-бутилдиметилсилил-β-L-рибофуранозил)-имидазол-4-карбоксамид (55)
трет-бутилдиметилсилил-β-L-рибофуранозил)-имидазол-4-карбоксамид (55)
Раствор (54) (5,0 г; 11,33 ммоль) в растворе аммиака в метаноле (60 мл) 12 часов грели в стальном баллоне при 100oС. Стальной баллон охлаждали, осторожно открывали и реакционную смесь концентрировали. Неочищенный продукт чистили быстрой хроматографией на силикагеле, используя CH2Cl2-->EtOAc в качестве элюента. Чистые фракции объединяли и упаривали с получением 4,0 г (88%) белой пены.
ПРИМЕР 39
5-Амино-(2',3' изопропилидин-β-L-рибофуранозил)-имидазол-4-карбоксамид (56)
изопропилидин-β-L-рибофуранозил)-имидазол-4-карбоксамид (56)
К перемешиваемому раствору (55) (4,0 г, 9,97 ммоль) в дихлорметане (50 мл) при комнатной температуре добавляли Et3N•3HF (50 ммоль). Эту реакционную смесь перемешивали в течение ночи и упаривали досуха. Остаток чистили быстрой хроматографией на силикагеле, используя CH2Cl2-->EtOAc в качестве элюента. Чистые фракции объединяли и упаривали с получением 2,10 г (71%) белой пены.
ПРИМЕР 40
5-Амино-1-β-L-рибофуранозилимидазол-4-карбоксамид (57)
К перемешиваемому раствору (56) (2,0 г, 6,71 ммоль) в дихлорметане (20 мл) добавляли 90% СF3СООН (20 мл) при температуре 0oС. Эту реакционную смесь перемешивали при 0oС в течение 1 часа и упаривали досуха. Остаток упаривали с сухим метанолом (20 мл). Этот процесс повторяли три раза, чтобы удалить последние следы трифторуксусной кислоты. Остаток обрабатывали раствором NH4OH (10 мл) и упаривали досуха. Остаток упаривали с сухим этанолом (3•20 мл). Остаток кристаллизовали из этанола с получением 1,5 г (87%) чистого продукта.
ПРИМЕР 41
Метиловый эфир 1-β-L-(2', 3', 5'-три бензоил)-рибофуранозил-2-оксо-Δ4-имидазолин-4-карбоновой кислоты (59)
бензоил)-рибофуранозил-2-оксо-Δ4-имидазолин-4-карбоновой кислоты (59)
Смесь метилового эфира 2-oкco-Δ4-имидaзoлин-4-кaрбoнoвoй кислоты (58) (542 мг, 3,82 ммоль), гексаметилдисилазана (HMDS, 28 мл) и (NH4)2SO4 (75 мг 0,56 ммоль) грели при температуре образования флегмы. За 40 минут получался прозрачный раствор, и реакционную смесь держали при температуре образования флегмы еще 3,5 часа. Избыток HMDS выпаривали и продукт, коричневое масло, сушили дальше под вакуумом в течение 1 часа.
К вышеуказанному сухому силиловому основанию добавляли раствор  ацетил-2,3,5-три
ацетил-2,3,5-три бензоил-L-рибофуранозы (1,93 мг; 3,82 ммоль) в безводном дихлорэтане при комнатной температуре с последующим добавлением SnCl4 (1,39 г, 0,63 мл; 5,35 ммоль) по каплям. После добавления реакционную смесь оставляли при комнатной температуре на ночь (около 17 часов). Реакционную смесь фильтровали через слой силикагеля, через который пропускали ЕtOАс. Раствор в ЕtOАс промывали насыщенным NаНСО3, фильтровали и трижды промывали соляным раствором. Органическую фазу отделяли, сушили над Na2SO4, концентрировали и чистили быстрой хроматографией на силикагеле, используя (86% CH2Cl2, 14% ЕtOАс) с получением 797 мг (36%) продукта в виде беловатого твердого вещества: 1H-ЯМР (Me2SO4-d6) δ 3.70 (s, 3H), 4.60 (dd, 1H, J1',2'= 12.7, 6.6 Hz), 4.70 (m, 2H), 5.93 (dd, 1H), 5.98 (d, 1H), 6.05 (dd. 1H), 7.46 (m, 6H), 7.63 (m, 3H), 7.71 (s, 1H), 7.91 (m, 6H) and 11.15 (s, 1H).
бензоил-L-рибофуранозы (1,93 мг; 3,82 ммоль) в безводном дихлорэтане при комнатной температуре с последующим добавлением SnCl4 (1,39 г, 0,63 мл; 5,35 ммоль) по каплям. После добавления реакционную смесь оставляли при комнатной температуре на ночь (около 17 часов). Реакционную смесь фильтровали через слой силикагеля, через который пропускали ЕtOАс. Раствор в ЕtOАс промывали насыщенным NаНСО3, фильтровали и трижды промывали соляным раствором. Органическую фазу отделяли, сушили над Na2SO4, концентрировали и чистили быстрой хроматографией на силикагеле, используя (86% CH2Cl2, 14% ЕtOАс) с получением 797 мг (36%) продукта в виде беловатого твердого вещества: 1H-ЯМР (Me2SO4-d6) δ 3.70 (s, 3H), 4.60 (dd, 1H, J1',2'= 12.7, 6.6 Hz), 4.70 (m, 2H), 5.93 (dd, 1H), 5.98 (d, 1H), 6.05 (dd. 1H), 7.46 (m, 6H), 7.63 (m, 3H), 7.71 (s, 1H), 7.91 (m, 6H) and 11.15 (s, 1H).
ПРИМЕР 42
1-β-L-Рибофуранозил-2-оксо-Δ4-имидазолин-4-карбоксамид (60)
Соединение (59) (1,26 г, 2,15 ммоль) растворяли в растворе аммиака в метаноле (45 мл; предварительно насыщен аммиаком при 0oС). Раствор герметизировали в стальном баллоне и грели при 95oС в течение 15 часов. Эту реакционную смесь охлаждали до комнатной температуры, растворитель выпаривали и остаток трижды промывали с помощью СНСl3, чтобы удалить образованный в реакции бензамид. Затем осадок вносили в МеОН (15 мл) и грели при температуре образования флегмы. К прозрачному раствору при температуре образования флегмы медленно добавляли СНСl3, пока не начинали образовываться следы преципитата. Эту горячую смесь быстро
фильтровали отсасыванием и фильтрат упаривали досуха с получением светло-коричневого масла. Это масло смачивали безводным СН3СN с получением продукта в виде светло-коричневого твердого вещества: выход 322 мг (58%); т. пл. 174-178oС. 1H-ЯМР (Me2SO4-d6) δ 3.48 (m, 2H), 3.77 (m, 1H), 3.94 (m, 1H), 4.05 (m, 1H), 4.90 (m, 1H), 5.08 (d, 1H), 5.30 (d, 1H), 5.36 (d, 1H), 7.30 (s, 1Н), 7.31 (br s, 2H) and 10.47 (br s, 1Н).
ПРИМЕР 43
2,3,5-Три бензоил-β-L-рибофуранозил-1-карбонитрил (61)
бензоил-β-L-рибофуранозил-1-карбонитрил (61)
К перемешиваемой смеси  ацетил-2,3,5-три
ацетил-2,3,5-три бензоил-β-L-рибофуранозы (высушенной при 60oС, 1 мм, 12 часов; 12,6 г; 24,9 ммоль) в сухом дихлорметане (высушенном над сульфатом магния и хранившемся над молекулярным ситом, 125 мл) при 0-2oС в атмосфере аргона добавляли цианид триметилсилила (высушен над молекулярным ситом, 24 часа; 4,70 мл; 37,50 ммоль). Затем к этой реакционной смеси медленно добавляли хлорид олова (1,0 мл; 8,76 ммоль), поддерживая температуру реакции на уровне 0-2oС. Полученную смесь перемешивали при температуре -5oС-0oС еще 1,5 часа. Через 2 часа реакционную смесь медленно в течение 30 минут добавляли к интенсивно перемешиваемому холодному (5oС) раствору гидроксида натрия (1,5 л), поддерживая во время добавления температуру этой смеси на уровне 5-8oС. Слои разделяли и органический слой промывали водой (3•500 мл) до нейтральности, а затем сушили над безводным сульфатом магния. Органический экстракт фильтровали, а осушитель промывали дихлорметаном (3•50 мл). Фильтрат и промывную жидкость объединяли и этот раствор концентрировали (<30oС, 20 мм) до малого объема, и оставшийся раствор фильтровали через слой целита. Дальнейшую очистку осуществляли с помощью колонки для быстрой хроматографии на силикагеле, используя дихлорметан в качестве элюента. Растворы дихлорметана объединяли и упаривали (<30oС, 20 минут) с получением белой пены. Этот неочищенный продукт чистили быстрой хроматографией на силикагеле с использованием дихлорметана в качестве элюента. Чистые фракции объединяли и упаривали с получением сиропа. Этот сироп смешивали с сухим этанолом (100 мл) и смесь грели (примерно 60oС) с получением гомогенного раствора. Охлаждение этого раствора до комнатной температуры дало белый кристаллический продукт. Кристаллическое твердое вещество отфильтровывали и промывали холодным этанолом и сушили над P2O5 с получением 7,47 г (63%) (61): т. пл. 55-57oС; 1H-ЯМР (СDСl3) δ 4.61 (m, 1Н, С4•Н), 4.78 (m, 2Н, С5•Н), 5.00 (d, 1Н, С1•Н), 5.88 (t, 1Н, С3•Н), 6.05 (m, 1H, С2•Н), 7.45-8.07 (m, 15H, РhН).
бензоил-β-L-рибофуранозы (высушенной при 60oС, 1 мм, 12 часов; 12,6 г; 24,9 ммоль) в сухом дихлорметане (высушенном над сульфатом магния и хранившемся над молекулярным ситом, 125 мл) при 0-2oС в атмосфере аргона добавляли цианид триметилсилила (высушен над молекулярным ситом, 24 часа; 4,70 мл; 37,50 ммоль). Затем к этой реакционной смеси медленно добавляли хлорид олова (1,0 мл; 8,76 ммоль), поддерживая температуру реакции на уровне 0-2oС. Полученную смесь перемешивали при температуре -5oС-0oС еще 1,5 часа. Через 2 часа реакционную смесь медленно в течение 30 минут добавляли к интенсивно перемешиваемому холодному (5oС) раствору гидроксида натрия (1,5 л), поддерживая во время добавления температуру этой смеси на уровне 5-8oС. Слои разделяли и органический слой промывали водой (3•500 мл) до нейтральности, а затем сушили над безводным сульфатом магния. Органический экстракт фильтровали, а осушитель промывали дихлорметаном (3•50 мл). Фильтрат и промывную жидкость объединяли и этот раствор концентрировали (<30oС, 20 мм) до малого объема, и оставшийся раствор фильтровали через слой целита. Дальнейшую очистку осуществляли с помощью колонки для быстрой хроматографии на силикагеле, используя дихлорметан в качестве элюента. Растворы дихлорметана объединяли и упаривали (<30oС, 20 минут) с получением белой пены. Этот неочищенный продукт чистили быстрой хроматографией на силикагеле с использованием дихлорметана в качестве элюента. Чистые фракции объединяли и упаривали с получением сиропа. Этот сироп смешивали с сухим этанолом (100 мл) и смесь грели (примерно 60oС) с получением гомогенного раствора. Охлаждение этого раствора до комнатной температуры дало белый кристаллический продукт. Кристаллическое твердое вещество отфильтровывали и промывали холодным этанолом и сушили над P2O5 с получением 7,47 г (63%) (61): т. пл. 55-57oС; 1H-ЯМР (СDСl3) δ 4.61 (m, 1Н, С4•Н), 4.78 (m, 2Н, С5•Н), 5.00 (d, 1Н, С1•Н), 5.88 (t, 1Н, С3•Н), 6.05 (m, 1H, С2•Н), 7.45-8.07 (m, 15H, РhН).
ПРИМЕР 44
2,3,5-Три бензоил-β-L-(+)-рибофуранозилаллотиоамид (62)
бензоил-β-L-(+)-рибофуранозилаллотиоамид (62)
Через суспензию L-цианосахара (61) (6,10 г; 12,95 ммоль) в сухом этаноле (105 мл) 10 минут пропускали H2S. Затем к этому раствору добавляли N,N-диметиламинопиридин (DMAP, 158 мг; 1,3 ммоль). Реакционную смесь держали при 15-20oС и насыщали сероводородом в течение 2,5 ч. (Примечание: Исходный материал, который был суспензией, по ходу этой реакции растворялся). Через 2,5 часов пропускание Н2S прекращали, реакционную смесь закрывали пробкой и оставляли перемешиваться на ночь. На следующее утро реакцию проверяли с помощью ТСХ (гексан/ЕtOАс; 7:3). ТСХ свидетельствовала о полном превращении исходного материала в аллотиоамид. Реакционную смесь охлаждали на водяной бане и в течение 1 часа пропускали через нее аргон, чтобы удалить избыток H2S. Затем реакционную смесь концентрировали на роторном испарителе с получением 6,20 г (95%) пенистого материала: 1H-ЯМР (СDСl3) δ 4.78 (m, 3Н, С4•Н and С5•Н), 5.12 (d, 1H, С1•Н), 5.72 (t, 1H, С3•Н), 5.98 (m, 1H, С2•Н), 7.45-8.12 (m, 15H, PhH) and 8.50 (br s, 2H, NH2).
ПРИМЕР 45
Этиловый эфир 2-(2', 3',5-три бензоил-β-L(+)-рибофуранозил)-тиазол-4-карбоновой кислоты (63)
бензоил-β-L(+)-рибофуранозил)-тиазол-4-карбоновой кислоты (63)
К перемешиваемой суспензии аллотиоамида (62) (5,05 г; 10 ммоль) в сухом 1,2-диметоксиэтане (DМЕ, 100 мл) при 0oС добавляли безводный NaHCO3 (8,4 г; 100 ммоль). К этой суспензии под аргоном в течение 10 минут по каплям добавляли этиловый эфир бромпировиноградной килоты (3,75 мл; 30 ммоль). Реакционную смесь перемешивали при 0oС под аргоном в течение 5 часов. Реакцию проверяли с помощью ТСХ (гексан/ЕtOАс; 7:3). ТСХ свидетельствовала о наличии следов исходного материала. Реакционную смесь оставляли при 0-5oС еще на 1 час, в течение которого большая часть исходного материала превращалась в продукт. Затем реакционную смесь охлаждали до -15oС в бане из смеси сухого льда с ацетоном. Затем к реакционной смеси в течение 15 минут каплями через отливочную воронку добавляли раствор 2,6-лютидина (7,0 мл; 60 ммоль) и трифторуксусного ангидрида (4,16 мл; 30 ммоль) в сухом DMF (20 мл). Температуру реакционной смеси под аргоном поддерживали на уровне -15oС. Затем реакционную смесь фильтровали и концентрировали. Полученный остаток растворяли в CH2Cl2 (200 мл) и органический слой промывали 5% раствором NаНСО3 (100 мл), водой (100 мл) и соляным раствором (100 мл), сушили и концентрировали до масла темно-красного цвета. Этот неочищенный продукт чистили быстрой хроматографией на колонке с силикагелем, используя гексан/ЕtOАс в качестве элюента, с получением 5,96 г (99%) чистого продукта: 1H-ЯМР (CDCl3) δ 1.30 (t, 3Н, СН2СH3), 4.30 (t, 2Н, СH2СН3), 4.55-4.78 (m, 3Н, С4•Н and C5•H), 5.72 (d, 1H), 5.82 (m, 2Н), 7.25-8.04 (m, 15H, PhH) and 8.06 (s, 1H, C5H).
ПРИМЕР 46
Этиловый эфир β-L(+)-рибофуранозилтиазол-4-карбоновой кислоты (64)
Соединение (63) (6,0 г; 10 ммоль) растворяли в сухом этаноле (60 мл) (Примечание: соединение растворяли нагреванием с помощью нагнетателя горячего воздуха). К этому раствору под аргоном добавляли порошок NaOEt (200 мг, 3,0 ммоль). Реакционную смесь в течение ночи перемешивали под аргоном. Реакцию проверяли с помощью ТСХ, используя гексан/ЕtOАс 7:3 и ЕtOАс/МеОН 9:1. ТСХ свидетельствовала о полном превращении исходного материала в более полярный продукт. Затем реакционную смесь нейтрализовали сухой смолой Dowex 5x-8 в протонированной форме. Смолу удаляли фильтрацией и фильтрат концентрировали под вакуумом на роторном испарителе. Затем остаток коричневого цвета чистили быстрой хроматографией на колонке с силикагелем, используя ЕtOАс-->МеОН. Чистые фракции объединяли и концентрировали с получением 2,31 г (77%) чистого продукта. 1H-ЯМР (СDСl3) δ 1.30 (t, 3H, СН2СН3), 3.56 (m, 2H, C5•H), 3.86 (m, 2H), 4.0 (m, 1H), 4.26 (t, 2H, СН2СН3), 4.82-5.04 (3m, 3H, 3 ОН), 5.42 (d, 1H, C1•H) and 8.46 (s, 1H, C5H).
ПРИМЕР 47
β-L(+)-Рибофуранозилтиазол-4-карбоксамид (65)
Раствор (64) (1,0 г; 3,32 ммоль) в растворе аммиака в метаноле (50 мл) перемешивали в стальном баллоне при комнатной температуре. Через 17 часов баллон охлаждали и осторожно открывали и раствор упаривали до остатка. Остаток подвергали быстрой хроматографии на колонке с силикагелем, используя в качестве элюента этилацетат и метанол (9:1). Продукт перекристаллизовывали из абсолютного этанола. Выход 580 мг (85%): т. пл. 146-148oС; 1H-ЯМР (Ме2SО4-d6) δ 3.48 (m, 2H, C5•H), 3.85 (m, 2H), 4.03 (m, 1H), 4.80 (t, 1H, С5•ОН). 4.88 (d, 1H, С3•ОН), 5.32 (d, 1H, C2•OH), 5.02 (d, 1H, J1',2'=5.1 Hz), 7.52 (bs, 1H, CONH2), 7.64 (bs, 1H, CONH2) and 8.16 (s, 1H, C5H). Анализ. Рассчитано для C9H12N2SO5 (260,2): С 41,53; Н 4,65; N 10,76; S 12,32. Найдено: С 41,73; Н 4,60; N 10,55; S 12,25.
ПРИМЕР 48
Метиловый эфир β-L-рибофуранозил-1-карбоксимидной кислоты (66)
К перемешиваемой суспензии 2,3,5-три бензоил-β-L-рибофуранозилцианида (14,13 г; 30,0 ммоль) в сухом метаноле (60 мл) добавляли в атмосфере аргона метоксид натрия (0,358 г, 6.64 ммоль; 0,5 М раствор, Fluka). Этот раствор, ставший гомогенным через 5 минут, перемешивали 2,5 часа при комнатной температуре. Эту реакционную смесь нейтрализовали смолой Dowex 50W-X8 в протонированной форме (высушена в течение 16 часов при 100oС и 0,05 мм рт. ст.; 3,0 г; 5,1 молярных эквивалентов/г). Смолу отфильтровывали и растворитель удаляли при температуре ниже 40oС на роторном испарителе. Полученный остаток промывали метанолом. Метанольные смывы концентрировали с получением второй и третьей порций (66). Эти три порции объединяли и перекристаллизовывали из сухого метанола с получением 4,35 г (66%) продукта: т. пл. 140-142oС; 1H-ЯМР (СDСl3) δ 3.46 (s, 3H, ОСН3), 3.50-3.80 (m, 5Н), 3.98 (d, 1H), 4.98 (br s, 3H) and 8.27 (s, 1Н, NH).
бензоил-β-L-рибофуранозилцианида (14,13 г; 30,0 ммоль) в сухом метаноле (60 мл) добавляли в атмосфере аргона метоксид натрия (0,358 г, 6.64 ммоль; 0,5 М раствор, Fluka). Этот раствор, ставший гомогенным через 5 минут, перемешивали 2,5 часа при комнатной температуре. Эту реакционную смесь нейтрализовали смолой Dowex 50W-X8 в протонированной форме (высушена в течение 16 часов при 100oС и 0,05 мм рт. ст.; 3,0 г; 5,1 молярных эквивалентов/г). Смолу отфильтровывали и растворитель удаляли при температуре ниже 40oС на роторном испарителе. Полученный остаток промывали метанолом. Метанольные смывы концентрировали с получением второй и третьей порций (66). Эти три порции объединяли и перекристаллизовывали из сухого метанола с получением 4,35 г (66%) продукта: т. пл. 140-142oС; 1H-ЯМР (СDСl3) δ 3.46 (s, 3H, ОСН3), 3.50-3.80 (m, 5Н), 3.98 (d, 1H), 4.98 (br s, 3H) and 8.27 (s, 1Н, NH).
ПРИМЕР 49
2-[(Аминокарбонил)-карбонил] -1-(β-L-рибофуранозилиминометил)-гидразин (67)
Метиловый эфир имидной кислоты (66) (4,83 г, 25,26 моль) и оксамидогидразид (2,28 г, 26,00 ммоль) растворяли в сухом диметилсульфоксиде (100 мл). После перемешивания этой реакционной среды в течение 20 часов при комнатной температуре растворитель отгоняли в вакууме при 55oС. Оставшееся твердое вещество суспензировали в метаноле и растворимую часть собирали фильтрацией (нерастворимой частью оказался непрореагировавший гидразид) и концентрировали до примерно 25 мл. При добавлении этого раствора по каплям в ацетонитрил (500 мл) получали белый преципитат: выход 4,35 г (66%); 1H-ЯМР (Me2SO4-d6) δ 3.47-3.60 (m, 2H), 3.3.60-3.88 (m, 3H), 4.07 (d, 1H), 4.15 (d, 1H), 4.85-5.2 (br s, 2H), 7.70, 8.09 (2 br s, 2H) and 10.05 (br s, 1H, C= NH).
ПРИМЕР 50
3-β-L-Рибофуранозил-1,2,4-триазол-5-карбоксамид (С-рибавирин; 68)
Соединение (67) (4,0 г; 15,2 ммоль) 15 минут грели под вакуумом (0,1 мм) при 135oС. После охлаждения колбы стеклянистый материал обрабатывали метанолом и грели на паровой бане. Во время этого процесса твердое вещество начинало образовывать преципитат. Через 2 часа это твердое вещество выделяли и после концентрирования фильтрата получали вторую порцию. Общий выход продукта составил 2,65 г (71%): т. пл. 193-195oС; 1H-ЯМР (Me2SO4-d6) δ 3.43 (m, 2H, C5•H), 3.75 (m, 1H, С4•Н), 3.88 (m, 1H, С3•Н), 4.12 (m, 1Н, C2•H), 4.57 (d, 1H, C1•H, J1',2'=5.7 Hz), 7.62 (bs, 1H, CONH2), 7.86 (bs, 1H, CONH2) and 10.0 (bs, 1H, NH). Анализ. Рассчитано для С8Н12N4O5 (244,2): С 39,35; Н 4,95; N 22,94. Найдено: С 39,38; Н 4,73; N 22,43.
ПРИМЕР 51 Тритил
Тритил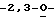 изопропилиден-β-L-рибофураноза (69)
изопропилиден-β-L-рибофураноза (69)
К раствору 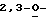 изопропилиден-β-L-рибофуранозы (10,5 г, 55,26 ммоль) в сухом пиридине (100 мл) под аргоном добавляли каталитическое количество DMAP (12,2 мг; 0,1 ммоль). Затем к этому перемешиваемому раствору добавляли тритилхлорид (15,56 г; 56 ммоль). Реакционную смесь в течение ночи перемешивали при комнатной температуре в атмосфере аргона. Пиридин удаляли под вакуумом, остаток растворяли в СН2Сl2 (250 мл) и органический слой промывали 10% раствором NаНСО3 (2•100 мл) и соляным раствором. Органический слой сушили над Na2SO4 и концентрировали в вакууме. Полученный остаток чистили быстрой хроматографией на колонке с силикагелем, используя гексан -->ЕtOАс в качестве элюента. Чистые фракции объединяли и концентрировали с получением 15,74 г (69%) продукта: 1H-ЯМР (CDCl3) δ 1.27 and 1.41 (2s, 6H, isopropylidene CH3), 3.25-3.56 (m, 2H, С5•Н), 3.86 (m, 2H), 4.0 (m, 1H), 4.70 (m, 1H), 5.24 (d, 1H, J1',2'=3.50 Hz, C1H) and 7.17-7.35 (m, 15H, PhH).
изопропилиден-β-L-рибофуранозы (10,5 г, 55,26 ммоль) в сухом пиридине (100 мл) под аргоном добавляли каталитическое количество DMAP (12,2 мг; 0,1 ммоль). Затем к этому перемешиваемому раствору добавляли тритилхлорид (15,56 г; 56 ммоль). Реакционную смесь в течение ночи перемешивали при комнатной температуре в атмосфере аргона. Пиридин удаляли под вакуумом, остаток растворяли в СН2Сl2 (250 мл) и органический слой промывали 10% раствором NаНСО3 (2•100 мл) и соляным раствором. Органический слой сушили над Na2SO4 и концентрировали в вакууме. Полученный остаток чистили быстрой хроматографией на колонке с силикагелем, используя гексан -->ЕtOАс в качестве элюента. Чистые фракции объединяли и концентрировали с получением 15,74 г (69%) продукта: 1H-ЯМР (CDCl3) δ 1.27 and 1.41 (2s, 6H, isopropylidene CH3), 3.25-3.56 (m, 2H, С5•Н), 3.86 (m, 2H), 4.0 (m, 1H), 4.70 (m, 1H), 5.24 (d, 1H, J1',2'=3.50 Hz, C1H) and 7.17-7.35 (m, 15H, PhH).
ПРИМЕР 52
3-Этоксикарбонил-2-оксопропилидентрифенилфосфоран (70)
Раствор хлорида { 3-(этоксикарбонил)-2-оксопропил} -трифенилфосфония (21,34 г; 500 ммоль) в воде (450 мл) в течение 10 минут добавляли к раствору карбоната натрия (3,1 г, 25,0 ммоль) (Примечание: немедленно после добавления получали белый преципитат). Эту реакционную смесь в течение ночи перемешивали при комнатной температуре. Полученный преципитат отфильтровывали через воронку с фильтром Шотта. Преципитат растворяли в дихлорметане (100 мл), сушили над сульфатом натрия и концентрировали с получением 18,13 г (93%) белого твердого вещества. Этот материал в течение ночи сушили над пятиокисью фосфора. 1H-ЯМР (СDСl3) δ 1/26 (t, 3Н), 3.34 (s, 2H), 3.36-3.84 (d, 1Н), 4.19 (m, 2H) and 7.48-7.68 (m, 15H, PhH).
ПРИМЕР 53
Этиловый эфир 4-(2',3' изопропилиден-5'
изопропилиден-5' тритил-α- и β-L-рибофуранозил)-3-оксобутановой кислоты (71)
тритил-α- и β-L-рибофуранозил)-3-оксобутановой кислоты (71)
Смесь (70) (10,9 г, 25,23 ммоль) и 3-этоксикарбонил-2-оксопропилидентрифенилфосфорана (11,8 г, 30 ммоль) в безводном ацетонитриле (30 мл) грели при температуре образования флегмы в течение 90 часов. Растворитель выпаривали под пониженным давлением и остаток подвергали быстрой хроматографии на колонке с силикагелем. Элюция гексаном-этилацетатом (9:1) дала продукт (α:β примерно 2:1) в виде пены (10,15 г; 74%).
ПРИМЕР 54
Этиловый эфир 2-диазо-4-(2',3' изопропилиден-5'
изопропилиден-5' тритил-α- и β-L-рибофуранозил)-3-оксобутановой кислоты (72)
тритил-α- и β-L-рибофуранозил)-3-оксобутановой кислоты (72)
В раствор (71) (9,85 г; 18,08 ммоль) в безводном ацетонитриле (50 мл) последовательно добавляли триэтиламин (1,83 г, 18,1 ммоль) и толуол-p-сульфонилазид (10 мл). Эту смесь 30 минут держали при комнатной температуре. Затем растворитель выпаривали под пониженным давлением и остаток подвергали быстрой хроматографии на колонке с силикагелем. Элюция гесаном-этилацетатом (9:1) дала 8,90 (86%) 72 (α:β примерно 1:1) в виде пены.
ПРИМЕР 55
Этиловый эфир 4-гидрокси-3-(2', 3' изопропилиден-5'
изопропилиден-5'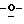 тритил-β-L-рибофуранозил)-пиразол-5-карбоновой кислоты (73)
тритил-β-L-рибофуранозил)-пиразол-5-карбоновой кислоты (73)
Раствор (72) (8,53 г; 14,92 моль) в сухом DME (60 мл) по каплям в течение 30 минут в атмосфере аргона добавляли к перемешиваемой охлажденной во льду суспензии гидрида натрия (NaH) (60%, в виде дисперсии; 1,80 г; 75,0 ммоль) в сухом DME (60 мл). Температура реакционной среды постепенно поднялась до 20oС, и смесь перемешивали еще 3 часа при комнатной температуре. Реакционную смесь анализировали с помощью ТСХ, используя гексан/ЕtOАс (3:1) или дихлорметан/ ЕtOАс (9:1). ТСХ свидетельствовала о завершении реакции. Затем к охлажденной во льду перемешиваемой реакционной смеси по каплям добавляли раствор уксусной кислоты (4,50 мл; 75 ммоль) в DME (10 мл). Растворитель выпаривали под пониженным давлением с получением остатка, к которому добавляли воду (50 мл) и диэтиловый эфир (100 мл). Эфирный слой отделяли, сушили над безводным сульфатом натрия и концентрировали. Остаток подвергали быстрой хроматографии на колонке с силикагелем, используя гексан-этилацетат (3: 1) в качестве элюента. Чистые фракции собирали и упаривали с получением (73) в виде смеси α и β (6,40 г; 73%): 1H-ЯМР (CDCl3) δ 1.31 (t, 3H), 1.42-1.65 (m, 6H), 3.19-3.27 (m, 2H), 4.44-4.75 (m, 3H), 4.75 (m, 1H), 5.19 (d, 1Н), 6.99 (br s, OH, exchangeable), 7.26-7.51 (m, 15H, PhH).
ПРИМЕР 56
4-Гидрокси-3-(2',3' изопропилиден-5'
изопропилиден-5' тритил-β-L-рибофуранозил)-пиразол-5-карбоксамид (74)
тритил-β-L-рибофуранозил)-пиразол-5-карбоксамид (74)
Раствор эфира (73) (6.30 г, 10,7 ммоль) в сухом растворе аммиака в метаноле (70 мл) 12 часов грели в стальном баллоне при 90-95oС. Растворитель выпаривали под пониженным давлением и остаток подвергали быстрой хроматографии на колонке с силикагелем, используя в качестве элюента гексан/этилацетат (3:2). Требуемые фракции объединяли и упаривали с получением 4,54 г (78%) продукта в виде стеклянистого вещества, содержащего смесь β:α (СDСl3) δ 1.40-1.62 (2s, 6H), 3.11-3.24 (m, 2H), 4.37 (m, 1Н), 4.65 (m, 1H), 5.11 (dd, 1H), 5.27 (d, 1H), 6.99 (br s, OH, exchangeable) and 7.23-7.50 (m, 17H).
ПРИМЕР 57
3-β-L-Рибофуранозил-4-гидроксипиразол-5-карбоксамид (L-пиразомицин; 75)
Раствор (74) (4,40 г; 8,13 ммоль) в 90% CF3CO2H (20 мл) перемешивали при комнатной температуре в течение 45 минут. Затем растворитель удаляли при 5oС под пониженным давлением с получением белого твердого вещества (1,90 г; 90,48%). Полученный остаток подвергали быстрой хроматографии на колонке с силикагелем, используя EtOAc-iPrOH-H2O (4:1:2) в качестве элюента. Фракции, содержавшие α и β изомеры чистого соединения, объединяли по отдельности и упаривали при <20oС. Перекристаллизация из воды дала 800 мг чистого β-изомера: т. пл. 11-113oС; 1H-ЯМР β-изомера (D2O) δ 3.73-3.78 (m, 2H), 4.0 (m, 1H), 4.19 (m, 1H). 4.35 (m, 1H) and 4.90-4.93 (d, 1H, J1',2'=7.42 Hz). Анализ. Рассчитано для C9H13N3O6 (259,22): С 41,70; Н 5,05; N 16,21. Найдено: С 41,88; Н 5,04; N 16,58. Выход смеси изолированных α:β составил 1,90 г (90%).
100 мг изомера выделяли в виде пены; 1H-ЯМР α-изомера (D2O) δ 3.65-3.85 (m, 2H), 4.06-4.11 (m, 1H), 4.32-4.41 (m, 2H) and 5.20 (d, 1H, J1'2'=3.30 Hz). Анализ. Рассчитано для С9Н13N3О6: С 41,70; Н 5,05; N 16,21. Найдено: С 41.91; Н 5.08; N 16.02.
Также выделили 1,0 г неразделенной смеси L-пиразомицина.
Чистоту изомеров α:β также установили с помощью обратнофазной ВЭЖХ, используя градиент ацетонитрила в воде 0-10%. Время удержания (Rt) у α-изомера было 5,716, а у β-изомера 7,135. По результатам ВЭЖХ, чистота смеси α- и β-изомеров L-пиразомицина была 99,0%.
ПРИМЕР 58
Приготовление гидрохлорида 2,5-ангидро-L-аллоамидина (76)
Метиловый эфир 2,5-ангидро-L-аллоимидиновой кислоты (3,82 г; 20,0 ммоль) и хлорида аммония (1,07 г; 20,0 ммоль) растворяли в растворе аммиака в метаноле (60 мл, насыщен при температуре смеси сухого льда с ацетоном в течение 1 часа). Затем этой смеси давали перемешиваться в стальном баллоне при комнатной температуре в течение 16 часов. Стальной баллон охлаждали и осторожно открывали и раствор упаривали досуха. Полученное твердое вещество сушили с получением 4,10 г целевого соединения с количественным выходом.
ПРИМЕР 59
2-(β-L-Рибофуранозил)-пиримидин-6(1Н)-оксо-4-карбоновая кислота (77)
К раствору гидрохлорида 2,5-ангидро-L-аллоимидина (4,0 г; 18,66 ммоль) в воде (60 мл) добавляли гидроксид натрия (1 н.; 20 мл; 20,0 ммоль) и этиловый эфир оксалоацетата натрия (4,20 г; 20,0 ммоль). Этой реакционной смеси давали перемешиваться 16 часов при комнатной температуре и затем нейтрализовали ее до рН 2 смолой Dowex 50W-X8 в протонированной форме. Реакционную смесь фильтровали и концентрировали до минимального объема. Добавляли силикагель и упаривали досуха. Полученный порошок наносили сверху на колонку для быстрой хроматографии и элюировали смесью этилацетат/ацетон/метанол/вода (3/1/1/1), пока не выходило самое подвижное соединение. Затем колонку элюировали метанолом и фракции, содержащие это соединение, объединяли. Метанол удаляли с получением гигроскопического соединения светло-коричневого цвета. Изолированный выход составил 4,50 г (89%). Это соединение использовали в следующей стадии как таковое без дальнейшей характеризации.
ПРИМЕР 60
Этиловый эфир 2-(β-L-рибофуранозил)-пиримидин-6(1Н)-оксо-4-карбоновой кислоты (78)
Тщательно высушенную суспензию кислоты (77) (4,50 г, 16,5 ммоль) в сухом этаноле (100 мл) охлаждали в ледяной бане и в течение 5 минут продували сухим хлористым водородом. К этой реакционной смеси добавляли триэтиловый эфир ортомуравьиной кислоты (20 мл) и смеси давали перемешиваться при комнатной температуре в течение 24 часов. Растворитель удаляли под вакуумом и полученное твердое вещество темной окраски чистили дальше быстрой хроматографией на колонке с силикагелем, используя смесь дихлорметан/метанол (9/1). Чистые фракции объединяли и концентрировали с получением 4,55 г (92%) твердого вещества. Поскольку это соединение оказалось не чистым, его превращали дальше в соответствующий тетраацетат с выходом 47%. Этот тетраацетат чистили колоночной хроматографией.
ПРИМЕР 61
2-(β-L-Рибофуранозил)-пиримидин-6(1 Н)-оксо-4-карбоксамид (79)
Раствор вышеуказанного тетраацетилированного эфира (1,80 г; 4,22 ммоль) в насыщенном растворе аммиака в метаноле (60 мл) 17 часов нагревали в стальном баллоне при 100oС. Реакционную смесь охлаждали и концентрировали с получением белого твердого вещества. Далее это твердое вещество перетирали с этилацетатом и фильтровали. Твердое вещество перекристаллизовывали из абсолютного этанола с получением 0,83 г (82%) чистого продукта в виде белого твердого вещества: т. пл. 200-202oС; 1H-ЯМР (Me2SO4-d6) δ 3.35-3.57 (m, 2H, С5•Н), 3.84 (m, 1H, С4•Н), 3.98 (m, 1H, С3•Н), 4.22 (m, 1H, C2•H), 4.75 (t, 1H, C5•OH, D2O exchangeable), 4.80 (d, 1H, C1•H, J1',2'=5.77 Hz), 4.89 (d, 1H, C3•OH, D2O exchangeable), 5.15 (d, 1H, C2•OH, D2O exchangeable), 7.85 (d, 1H), 7.98 (bs, 1H, CONH2), 8/19 (bs, 1H, CONH2) and 9.0 (d, 1H, NH). Анализ. Рассчитано для С10Н13N3O4 (239,23): С 44,28; Н 4,83; N 15,49. Найдено: С 44,58; Н 5,17; N 15,28.
Пример 62
Метил-β-L-арабинопиранозид (81)
К суспензии L-арабинозы (100 г; 667 ммоль) в безводном МеОН (450 мл)) при комнатной температуре в атмосфере аргона добавляли раствор HCl/MeOH (7,3 г сухого HCl в 50 мл МеОН). Эту смесь 2 часа грели при температуре образования флегмы и затем охлаждали до комнатной температуры. Раствор концентрировали до примерно 3/4 его объема с получением суспензии. Твердый преципитат отфильтровывали и промывали холодным МеОН (20 мл) с получением первой порции в виде кристаллического порошка (35,23 г). Фильтрат концентрировали (35oС) до 1/4 его объема. Твердый преципитат отфильтровывали, промывали и сушили как описано выше с получением второй порции (9,66 г) бесцветного кристаллического порошка. Концентрирование и фильтрование продолжали с получением дополнительных 28,31 г продукта (общий выход 73,2 г; 67%). 1H-ЯМР (D2O) δ 3.30 (s, ОСН3, 3Н), 3.56 (dd, 1H, H5), 3.73 (m, 1H, H4), 3.77 (dd, 1H, H5), 3.83 (bs, 1H, H2), 4.73 (m, 1H, H1).
ПРИМЕР 63
Метил-3,4-изопропилиден-β-L-арабинопиранозид (82)
К смеси метил-β-L-арабинопиранозида (81) (23,33 г; 142,26 ммоль) и диметоксипропана (55 мл; 448 ммоль) в сухом DMF (185 мл) добавляли Amberlyst 15 (протонированная форма; 1,42 г) и эту суспензию 18 часов перемешивали при комнатной температуре. Раствор упаривали с получением сиропа, который растворяли в ЕtOАс (200 мл) и промывали соляным раствором (50 мл), насыщенным раствором NаНСО3 и соляным раствором (20 мл). Водные смывы объединяли и экстрагировали, используя ЕtOАс (5•20 мл), который промывали раствором NaCl/H2O и объединяли с органическим
раствором. Раствор в EtOAc сушили над безводным Na2SO4 и упаривали досуха с получением сиропа (29,2 г, количественно). 1H-ЯМР (СDСl3) δ 1.36 and 1.53 (2s, 6H, isopropylidene-СН3), 2.43 (d, 1H, 2'-ОН), 3.44 (s, 2H, ОСН3), 3.78 (m, 1Н, Н2), 3.93 (s, 2H, H5), 4.15-4.25 (m, 2H, Н3 & Н4), 4.71 (d, 1H, H1).
ПРИМЕР 64
Метил-3,4-изопропилиден 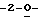 [(метилтио)-тиокарбонил] -β-L-арабинопиранозид (83)
[(метилтио)-тиокарбонил] -β-L-арабинопиранозид (83)
Вышеуказанный сироп (82) (29,2 г; 142,26 ммоль) растворяли в безводном THF (190 мл) и охлаждали до 0oС. К этому раствору медленно в атмосфере аргона добавляли NaH (55-65%, 6,9 г; 172,5 ммоль). Суспензию грели при температуре образования флегмы в течение 2 часов и охлаждали до 0oС. К смеси добавляли дисульфид углерода (21 мл; 349,14 ммоль) и полученную темную смесь 2 часа перемешивали при комнатной температуре. К этой смеси при 0oС добавляли метилйодид (12,4 мл; 160,64 ммоль) и смесь перемешивали 16 часов. Эту смесь вливали в ледяную воду (300 мл) и экстрагировали, используя EtOAc (3х50 мл). Раствор в EtOAc сушили и упаривали, пока не преципитировались кристаллы. Эту суспензию оставляли в холодильнике на 16 часов. Кристаллы отфильтровывали и промывали гексаном с получением первой порции продукта (21,61 г) в виде желтоватого порошка. Фильтрат концентрировали, держали в течение ночи при 0oС и фильтровали с получением второй порции продукта (16,51 г). Это повторяли еще два раза с получением дополнительных 1,44 г продукта (39,62 г; 94,7% для двух стадий от (81)). Т. пл. 127-130oС. 1H-ЯМР (СDСl3) δ 1.39 and 1.55 (2s, 6H, isopropylidene-СН3), 2.60 (s, 3Н, SСН3), 3.40 (s, 3Н, ОСН3), 4.01 (s, 2H, H5), 4.30 (m, 1H, H4), 4.50 (dd, 1H, H3), 4.98 (d, 1H, H1), 5.78 (dd, 1H, H2).
ПРИМЕР 65
Метил-2-дезокси-β-L-эритро-пентопиранозид (84)
Соединение (83) (40 г; 136 ммоль) и AIBN (24,61 г; 150 ммоль) растворяли в сухом диоксане (400 мл) нагреванием на масляной бане (100oС). Эту смесь 15 минут продували аргоном при 100oС с последующим добавлением дифенилсилана (51,4 мл; 272 ммоль). Температуру масляной бани поднимали до 130oС и смесь 16 часов грели с образованием флегмы. Дополнительно добавляли дифенилсилан (2 мл; 10,8 моль) и AIBN (1,27 г; 7,7 ммоль) и нагревание в условиях образования флегмы продолжали еще 5 часов. Дополнительно добавляли AIBN (0,2 г; 1,2 ммоль) и нагревание в условиях образования флегмы продолжали еще 1 час. Смесь охлаждали и упаривали с получением соединения (84) в виде сиропа, который смешивали с 80% НОАс (544 мл) и 16 часов перемешивали при комнатной температуре. Эту смесь упаривали с получением сиропа, который распределяли между водой и эфиром. Водный слой промывали эфиром и объединенный органический слой экстрагировали водой. Водный раствор упаривали с получением соединения (85) в виде сиропа (16,36 г; 81,4% для двух стадий после (83). 1H-ЯМР (CDCl3) δ 1.89 (dd, 2H, Н2), 2.30 (d, 1H, ОН), 2.47 (d, 1H, ОН), 3.35 (s, 3H, ОСН3), 3.88-3.69 (m, 3Н, Н4 & H5), 4.03 (m, 1Н, H3), 4.79 (t, 1H, H1).
ПРИМЕР 66
2'-дезокси-β-L-эритро-пентоза (86)
Соединение (85) (16,36 г; 110,5 ммоль) растворяли в 0,8 M водном растворе HCl (546 мл) и полученную смесь 72 часа перемешивали при комнатной температуре. Смесь нейтрализовали 1 н. водным раствором NaOH до рН 6-7 и упаривали с получением сиропа. Неочищенный продукт чистили на колонке с силикагелем (4•15 см), элюируемой смесью CH2Cl2/MeOH (от 1:0 до 95:5). Нужные фракции упаривали с получением (86) в виде сиропа (10,53 г; 71,1%).
ПРИМЕР 67
Метил-2'-дезокси-β-L-эритро-пентоза (87)
Соединение (86) (15,68 г; 117,0 ммоль) растворяли в сухом МеОН (342 мл) и к полученному раствору добавляли 1% HCl/MeOH (35 мл). Этот раствор держали при КОМНАТНОЙ ТЕМПЕРАТУРЕ в течение часа и нейтрализовали с помощью Ру (55 мл) при 5oС до рН ~6. Эту смесь упаривали с силикагелем и чистили на колонке с силикагелем (1•5 см), элюируемой смесью CH2Cl2/MeOH (от 98:2 к 96:4) с получением соединения (87) в виде сиропа (13,94 г; 80,5%). 1H-ЯМР (СDСl3) δ 2.22-2.44 (m, 2H, H2), 3.50 and 3.59 (2s, 3H, ОСH3), 3.75-3.88 (m, 2Н, H5), 4.26 (m, 1H, Н4), 4.66 (m, 1H, Н3), 5.25 (t, 1H, H1).
ПРИМЕР 68
Метил-2'-дезокси-3,5-ди- -пара-толуоил-L-эритро-пентоза (88)
-пара-толуоил-L-эритро-пентоза (88)
Соединение (87) (9,00 г; 60,8 ммоль) растворяли в пиридине (180 мл) и охлаждали в бане с тающим льдом. К этому холодному раствору в течение 30 минут добавляли толуоилхлорид (18 мл) и полученный раствор 16 часов перемешивали при комнатной температуре. Смесь упаривали досуха, экстрагировали, используя ЕtOАс, промывали соляным раствопром, сушили и упаривали. Неочищенный продукт чистили на колонке с силикагелем (3•15 см), используя в качестве элюента гексан/ЕtIАс (от 1:0 к 5:1). Упаривание надлежащих фракций дало соединение (88) в виде сиропа (22,63 г; 97%). 1H-ЯМР (СDСl3) δ 2.40 (2s, 6H, 2•СН3), 3.35 (s, 3H, ОСН3 of β-anomer), 5.21 (dd, 1H, H1 of β-anomer), 5.41 (m, 1H, H3 of α-anomer), 5.59 (m, 1H, H3 of β-anomer), 7.18-8.02 (m, 8H, Aromatic).
ПРИМЕР 68а
2'-Дезокси-3',5'-ди пара-толуоил-α-L-эритро-пентофуранозилхлорид (13)
пара-толуоил-α-L-эритро-пентофуранозилхлорид (13)
Соединение (88) (33 г; 57,3 ммоль) растворяли в сухом эфире (200 мл) и этот раствор охлаждали до 0oС в ледяной бане. Раствор в течение ~5 минут продували сухим НСl, пока смесь не кристаллизовалась. Затем реакционную смесь в течение ночи держали в холодильнике. Преципитированное твердое вещество отфильтровывали, промывали холодным эфиром и немедленно сушили под вакуумом над NaOH с получением соединения (13) в виде бесцветного кристаллического порошка (19,28 г). Фильтрат концентрировали и обрабатывали хлористым водородом и в течение ночи держали в холодильнике. Фильтрация, промывка и сушка дали дополнительные 1,2 г продукта (всего 20,48 г; 92%), т. пл. 118-121oС. 1H-ЯМР (CDCl3) δ 2.39 (2s, 6H, aromatic-СН3), 2.28 (m, 2H, Н2), 4.65 (m, 2H, H5), 4.86 (q, 1H, Н4), 5.57 (m, 1H, Н3), 6.48 (d, 1H, Н1), 7.25 (2d, 4H, aromatic-H), 7.95 (2d, 4H, aromatic-H).
ПРИМЕР 69
Метиловый эфир 1-(2'-дезокси-3', 5'-ди  пара-толуоил-β-L-эритро-пентофуранозил)-1,2,4-триазол-5-карбоновой кислоты (89), метиловый эфир 1-(2'-дезокси-3,5'-ди
пара-толуоил-β-L-эритро-пентофуранозил)-1,2,4-триазол-5-карбоновой кислоты (89), метиловый эфир 1-(2'-дезокси-3,5'-ди лара-толуоил-β-L-эритро-пентофуранозил)-1,2,4-триазол-2-карбоновой кислоты (90) и метиловый эфир 1-(2'-дезокси-3',5'-ди
лара-толуоил-β-L-эритро-пентофуранозил)-1,2,4-триазол-2-карбоновой кислоты (90) и метиловый эфир 1-(2'-дезокси-3',5'-ди пара-толуоил-β-L-эритро-пентофуранозил)-1,2,4-триазол-3-карбоновой кислоты (91)
пара-толуоил-β-L-эритро-пентофуранозил)-1,2,4-триазол-3-карбоновой кислоты (91)
К раствору метилового эфира 1,2,4-триазол-3-карбоновой кислоты (1,27 г; 10 ммоль) в сухом ацетонитриле (50 мл) добавляли гидрид натрия (60% в масле, 0,5 г, 12,5 ммоль). Эту смесь 30 минут перемешивали при комнатной температуре. К суспензии добавляли сухой порошок хлоруглевода (13) и перемешивали смесь при комнатной температуре в течение 16 часов. Смесь упаривали с получением остатка, который распределяли между водой и EtOAc и экстрагировали, используя EtOAc. Водный раствор экстрагировали, используя ЕtOАс. Объединенный раствор в EtOAc промывали соляным раствором и упаривали досуха. Смесь чистили на колонке с силикагелем (3•20 см), используя ЕtOН/гексан (1,2:1) в качестве элюента, с получением (98) (1,72 г), (90) (0,98 г) и (91) (0,45 г).
1H-ЯМР (СDСl3) (89): δ 2.52 (2s, 6H, СН3), 2.82 (m, 1H, H2'), 3.45 (m, 1H, H2'), 4.60 (dd, 1H, Н5'), 4.72 (dd, 1H, Н5'), 4.76 (m, 1H, H4'), 6.03 (m, 1H, H3'), 7.29-3.38 (m, 5H, aromatic-H and Н5'), 7.97-8.12 (m, 5H, aromatic-H and C5H). (90): δ 2.50 & 2.53 (2s, 6H, СН3), 2.95 (m, 1H, H2'), 3.20 (m, 1H, H2'), 4.09 (s, 3H, ОСН3), 4.72 (m, 3Н, Н4 & Н5), 5.83 (m, 1H, Н3'), 6.47 (t, 1H, Н1'), 7.36 (dd, 4H, aromatic-H), 8.02 (dd, 4Н, aromatic-H), 8.51 (s, 1H, C5H). (91): δ 2.53 (m, 7H, Н2' & 2хСН3), 3.16 (m, 1H, H2'), 4.13 (s, 3Н, ОСН3), 4.69-4.85 (m, 3H, H4' & Н5'), 5.73 (m, 1H, Н3'), 6.88 (q, 1H, H1'), 7.35 (dd, 4H, aromatic-H), 7.94 & 8.05 (dd, aromatic-H), 8.76 (s, 1H, C5H).
ПРИМЕР 70
1-(2'-Дезокси-β-L-эритро-пентофуранозил)-1,2,4-триазол-5-карбоксамид (92)
Смесь (89) (1,77 г; 3,70 моль) и насыщенного раствора аммиака в метаноле (40 мл) 16 часов грели в стальном баллоне при 55oС. После охлаждения этот раствор упаривали с силикагелем и чистили на колонке с силикагелем, элюируемой смесью CH2Cl2/MeOH (10:1) с получением соединения (92) в виде бесцветного порошка (297 мг, 35%). 1H-ЯМР (DMSO-d6): δ 2.27 (m, 1H, H2'), 2.60 (m, 1H, H2'), 3.32 (m, 1H, Н5'), 3.48 (m, 1H, Н5'), 3.80 (m, 1H, H4'), 4.41 (m, 1H, H3'), 7.12 (t, 1H, H1'), 8.06 (s, 1H, NH), 8.14 (s, 1H, C5H), 8.27 (s, 1H, NH).
ПРИМЕР 71
1-(2'-Дезокси-β-L-эритро-пентофуранозил)-1,2,4-триазол-3-карбоксамид (93)
Смесь (91) (0,45 г; 0,94 моль) и насыщенного раствора аммиака в метаноле (20 мл) 16 часов грели в стальном баллоне при 55oС. После охлаждения этот раствор упаривали досуха. Остаток чистили на колонке с силикагелем, элюируемой смесью CH2Cl2/MeOH (10:1), с получением соединения (93) (54 мг; 25%). 1H-ЯМР (DMSO-d6): δ 2.44 (m, 1H, H2'), 2.38 (m, 1H, H2'), 3.61 (m, 2H, H5'), 3.85 (m, 1H, H4'), 4.30 (m, 1H, H3'), 6.70 (t, 1H, H1'), 7.94 (s, 1H, NH), 8.33 (s, 1H, NH), 8.98(s, 1H, C5H).
ПРИМЕР 72
1-(2'-Дезокси-3', 5'-ди пара-толуоил-β-L-эритро-пентофуранозил)-2,4-дицианопиррол (94)
пара-толуоил-β-L-эритро-пентофуранозил)-2,4-дицианопиррол (94)
К раствору 2,4-дицианопиррола (302 мг; 2,58 ммоль) в сухом ацетонитриле (25 мл) добавляли гидрид натрия (60% в масле, 125 мг; 2,6 ммоль). Эту смесь 30 минут перемешивали при комнатной температуре. Добавляли хлоруглевод (13) (1 г, 2,58 ммоль) и перемешивали суспензию при комнатной температуре в течение 16 часов. Эту смесь упаривали с получением твердого остатка, который распределяли между водой и ЕtOАс. Водный раствор экстрагировали, используя ЕtOАс. Объединенный экстракт в ЕtOАс промывали водой и соляным раствором, сушили и упаривали досуха. Продукт чистили на колонке с силикагелем (3•20 см), используя гексан/ЕtOАс в качестве элюента, с получением (94) в виде масла (605 мг, 50%). 1H-ЯМР (СDСl3): δ 2.29 (s, 3Н, aromatic-СН3), 2.55 (s, 3Н, aromatic-СН3), 2.67 (m, 1H, H2'), 2.98 (m, 1H, H2'), 4.74-4.89 (m, 3Н, H4' & Н5'), 5.77 (m, 1H, Н3'), 6.36 (t, 1H, H1'), 7.18 (s. 1H, С5Н), 7.38 (m, 4H, aromatic-H), 7.68 (s, 1H, С3Н), 7.97 (d, 2H, aromatic-H), 8.05 (d, 2H, aromatic-Н).
ПРИМЕР 73
1-(2'-Дезокси-β-L-эритро-пентофуранозил)-2,4-дицианопиррол (95)
Смесь (94) (605 мг; 1,29 ммоль) и насыщенного раствора аммиака в метаноле (20 мл) 16 часов грели в стальном баллоне при 65oС. После охлаждения раствор упаривали досуха. Неочищенный продукт чистили на колонке с силикагелем, элюируемой смесью CH3Cl3/MeOH (10: 1), с получением (95) (158 мг; 52%). 1H-ЯМР (СD3ОD): δ 2.55 (m, 2H, Н2), 3.82 (m, 2H, Н5'), 4.08 (m, 1H, Н4'), 4.53 (m, 1H, Н3'), 6.38 (t, 1H, Н1'), 7.37 (s, 1H, С5Н), 8.19 (s, 1H, С3Н).
ПРИМЕР 74
1-(2'-Дезокси-β-L-эритро-пентофуранозил)-пиррол-2,4-дикарбоксамид (96)
К раствору (95) (90 мг; 0,385 ммоль) в водном растворе NH4OH (29,6%, 9 мл) добавляли H2O2. Этот раствор перемешивали при комнатной температуре в течение двух часов и упаривали досуха. Остаток чистили на колонке с силикагелем, элюируемой смесью CH2Cl2/MeOH (10:1), с получением соединения (96) (75 мг; 72%). 1H-ЯМР (CD3OD): δ 2.31 & 2.61 (m, 2H, H2'), 3.89 (m, 2H, Н5'), 4.04 (m, 1H, Н4'), 4.45 (m, 1H, Н3'), 6.93 (t, 1H, H1'), 7.24 (s, 1H, C5H), 8.09 (s, 1Н, С3Н).
ПРИМЕР 75
Метиловый эфир 1-(2'-дезокси-3', 5'-ди пара-толуоил-β-L-эритро-пентофуранозил)-пиразол-3,5-дикарбоновой кислоты (97)
пара-толуоил-β-L-эритро-пентофуранозил)-пиразол-3,5-дикарбоновой кислоты (97)
К раствору метилового эфира пиразол-3,5-дикарбоновой кислоты (458 г; 2,5 ммоль) в сухом ацетонитриле (25 мл) добавляли гидрид натрия (60% в масле; 144 мг; 3,0 ммоль). Эту смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли хлоруглевод (13) (970 мг, ~95%; 2,5 ммоль) и 4 часа перемешивали суспензию при комнатной температуре. Эту смесь упаривали досуха и остаток чистили на колонке с силикагелем (3•7 см), используя гексан/ЕtOАс (10:1) в качестве элюента, с получением (97) в виде масла (470 мг; 35%). 1H-ЯМР (CDCl): δ 2.40 (d, aromatic-СН3), 2.70 (m, 1H, H2'), 3.60 (m, 1H, H2'), 3.93 (d, 6H, ОСН3), 4.48-4.65 (m, 3H, H4' & Н5'), 5.89 (m, 1H, Н3'). 7.18-7.29 (m, 4H, aromatic-H & H1'), 7.38 (s, 1H, С4Н), 7.90 (m, 4H, aromatic-H).
ПРИМЕР 76
1-(2'-Дезокси-β-L-эритро-пентофуранозил)-пиразол-3,5-дикарбоксамид (98)
Смесь (97) (270 мг; 0,51 ммоль) и насыщенного раствора аммиака в метаноле (20 мл) 16 часов грели в стальном баллоне при 100oС. После охлаждения раствор упаривали и чистили на колонке с силикагелем, используя гексан/ЕtOАс (10:1) в качестве элюента, с получением (98) в виде бесцветного порошка (189 мг; 73%). 1H-ЯМР (DMSO-d6): δ 2.25 (m, 1H, H2'), 2.89 (m, 1H, H2'), 3.42 (m, 1H, Н5'), 3.39 (m, 1H, Н5'), 3.86 (q, 1H, Н4'), 4.55 (m, 1H, H3'), 4.77 (t, 1H, ОН), 5.27 (d, 1H, ОН), 7.17 (t, 1H, H1'), 7.29 (s, 1H, С4Н), 7.51 (s, 1H, NH), 7.66 (s, 1H, NH), 7.80 (s, 1H, NH), 8.19 (s, 1H, NH).
ПРИМЕР 77
Метиловый эфир 1-(2'-дезокси-3', 5'-ди пара-толуоил-β-L-эритро-пентофуранозил)-пиразол-4-карбоновой кислоты (99)
пара-толуоил-β-L-эритро-пентофуранозил)-пиразол-4-карбоновой кислоты (99)
К раствору метилового эфира пиразол-4-карбоновой кислоты (315 мг; 2,5 ммоль) в сухом ацетонитриле (30 мл) добавляли гидрид натрия (60% в масле; 144 мг; 3,0 ммоль). Эту смесь 15 минут перемешивали при 50oС. Добавляли хлоруглерод 13 (1г, ~95%; 2,5 моль) и 2 часа перемешивали суспензию при комнатной температуре. Смесь упаривали с получением остатка, который распределяли между водой и ЕtOАс. Водный раствор экстрагировали, используя ЕtOАс. Объединенный экстракт в ЕtOАс промывали водой и соляным раствором, сушили над Na2SO4 и упаривали досуха. Остаток чистили на колонке с силикагелем (3•12 см), используя гексан/ЕtOАс (4:1), с получением (99) в виде кристаллического порошка (549 мг, 46%). 1H-ЯМР (СDСl3): δ 2.40 (d, 6H, aromatic-СН3), 2.72 (m, 1H, Н2'), 3.16 (m, 1H, H2'), 3.79 (d, 3Н, ОСН3), 4.51-4.62 (m, 3Н, H4' & Н5'), 5.77 (m, 1H, Н3'), 6.20 (t, 1H, H1'), 7.24 (m, 4H, aromatic-H), 7.92 (m, 5H, aromatic-H & C5H), 8.10 (s, 1H, С3Н).
ПРИМЕР 78
1-(2'-Дезокси-β-L-эритро-пентофуранозил)-пиразол-4-карбоксамид (100)
Смесь (99) (500 мг; 1,046 ммоль) и насыщенного раствора аммиака в метаноле (30 мл) 16 часов грели в стальном баллоне при 100oС. После охлаждения раствор упаривали и остаток чистили на колонке с силикагелем, используя CH2Cl2/MeOH (10: 1), с получением (100) в виде желтой пены (50 мг; 20%). 1H-ЯМР (DMSO-d6): δ 2.30 (m, 1H, H2'), 2.56 (m, 1H, Н2'), 3.41-3.56 (m, 2H, Н5'), 3.84 (m, 1H, Н4'), 4.36 (m, 1H, Н3'), 4.88 (bs, 1H, ОН), 5.32 (bs, 1H, ОН), 6.11 (t, 1H, H1'), 7.08 (s, 1H, NH), 7.63 (s. 1H, NH), 7.90 (d, 1H, C5H), 8.36 (d, 1H, С3Н), 7.80 (s, 1H, NH), 8.19 (s, 1H, NH).
ПРИМЕР 79
Метил-α-L-ликсопиранозид (102)
К раствору HCl в метаноле [600 мл; 0,5% вес/объем; приготовлен in situ в реакции ацетилхлорида (6,0 мл)] добавляли L-ликсозу ((101); 118 г; 786 ммоль) и 5 часов грели его при температуре образования флегмы [реакция была завершена через 4 часа (по ТСХ с 30% MeOH/CH2Cl2), после чего ее продолжали еще 1 час (в сумме 5 часов)] в условиях исключения влаги (с предохранительной трубкой, содержащей CaCl2). Эту реакционную смесь в течение 10 минут при перемешивании нейтрализовали предварительно обработанной1 основной смолой Amberlite IRA 410 (100 г). Смолу отфильтровывали и промывали метанолом (3•50 мл). Объединенные смывы упаривали с получением бесцветного сиропа. Этот сироп упаривали с этилацетатом (2•50 мл) и под конец перекристаллизовывали (царапая стеки RB-колбы или действуя ультразвуком) из этилацетата (500 мл) с получением белого кристаллического продукта (102) (87 г, 67% в первой и второй порциях вместе). 1H-ЯМР (300 МГц, (СD3)2СО): δ 3.15 (bs, 3Н, ОН), 3.41 (s, 3H, ОСН3), 3.48 (m, 1H), 3.66-3.72 (m, 2H), 3.73-3.87 (m, 2H), 4.64 (d, 1H, Н5', J=2.7 Hz).
ПРИМЕР 80
Метил изопропилиден-α-L-ликсопиранозид (103)
изопропилиден-α-L-ликсопиранозид (103)
К суспензии (102) (69 г; 420 ммоль) в смеси 2,2-диметоксипропана (200,0 мл) и безводного ацетона (200,0 мл) добавляли 4М раствор HCl в диоксане (4,0 мл) и перемешивали эту реакционную смесь при 25oС в течение 16 часов. ТСХ (50% этилацетат/СН2Сl2) реакционной смеси свидетельствовала о полном превращении исходного материала. Реакционную смесь гасили твердым бикарбонатом натрия (500 мг) и фильтровали. Фильтрат упаривали и полученный маслянистый остаток (розоватого цвета) чистили быстрой хроматографией на силикагеле, используя СН2Сl2/этилацетат (от 100/0 к 80/20 с шагом 5%) в качестве элюента, с получением продукта (103) (80 г; 93,2%). 1H-ЯМР (300 МГц, (СDСl3: δ 1.31 (s, 3Н), 1.47 (s, 3H), 2.95 (bs, 1H), 3.41 (s, 3H), 3.66-3.77 (m, 3Н), 4.07 (dd, 1H, J=6.04 & 2.75 Hz), 4.16 (t, 1H, J=6.04 & 4.67 Hz), 4.60 (d, 1H, J=2.74 Hz).
ПРИМЕР 81
Метил-4-азидо-4-дезокси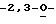 изопропилиден-β-D-рибопиранозид (104)
изопропилиден-β-D-рибопиранозид (104)
К смеси пиридина (6,4 мл; 79,65 ммоль) и диметиламинопиридина (100 мг; 0,72 ммоль) в безводном СН2Сl2 (400 мл) медленно при -20oС добавляли трифторметансульфоновый ангидрид (11,82 мл; 71,68 ммоль). Смесь 5 минут перемешивали при -20oС и затем к ней добавляли раствор (103) (5,0 г; 24,50 ммоль) в СН2Сl2 (50,0 мл). Эту реакционную смесь перемешивали при -20oС в течение 15 минут. Затем ее вливали в смесь воды со льдом (500 мл) и органический слой отделяли. Водную фазу экстрагировали, используя CH2Cl2(2•100 мл). Объединенный органический слой промывали соляным раствором (500 мл), сушили (Na2SO4) и упаривали с получением бледно-желтого смолистого твердого вещества (14 г).
К раствору вышеуказанного метил трифторметансульфонил
трифторметансульфонил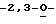 изопропилиден-β-L-ликсопиранозида в смеси DMF (350 мл) и тетраметилмочевины (50 мл) добавляли при 0-5oС (баня с тающим льдом) азид натрия (30,0 г; 461,53 ммоль; 18,83 экв.) и 3 часа перемешивали смесь при 23oС. Выпаривали летучие вещества и разводили остаток смесью CH2Cl2 (500 мл) и воды (200 мл). Органический слой отделяли и промывали водой (2•250 мл) и соляным раствором (300 мл), сушили (Na2SO4) и упаривали с получением маслянистого остатка, который чистили быстрой хроматографией на силикагеле, используя гексан/этилацетат (100/0; 97,5/2,2 и 95/5) в качестве элюента, с получением продукта (104) (2,8 г, 49,88% для двух стадий). %). 1H-ЯМР (300 МГц, (СDCСl3): δ 1.36 (s, 3Н, СН3), 1.54 (s, 3Н, СН3), 3.42 (s, 3H, ОСН3), 3.7-3.9 (m, 3Н), 4.01 (dd, 1H, J=6.05 & 3.85 Hz), 4.48 (d, 1H, J=3.85 Hz), 4.51 (m, 1H).
изопропилиден-β-L-ликсопиранозида в смеси DMF (350 мл) и тетраметилмочевины (50 мл) добавляли при 0-5oС (баня с тающим льдом) азид натрия (30,0 г; 461,53 ммоль; 18,83 экв.) и 3 часа перемешивали смесь при 23oС. Выпаривали летучие вещества и разводили остаток смесью CH2Cl2 (500 мл) и воды (200 мл). Органический слой отделяли и промывали водой (2•250 мл) и соляным раствором (300 мл), сушили (Na2SO4) и упаривали с получением маслянистого остатка, который чистили быстрой хроматографией на силикагеле, используя гексан/этилацетат (100/0; 97,5/2,2 и 95/5) в качестве элюента, с получением продукта (104) (2,8 г, 49,88% для двух стадий). %). 1H-ЯМР (300 МГц, (СDCСl3): δ 1.36 (s, 3Н, СН3), 1.54 (s, 3Н, СН3), 3.42 (s, 3H, ОСН3), 3.7-3.9 (m, 3Н), 4.01 (dd, 1H, J=6.05 & 3.85 Hz), 4.48 (d, 1H, J=3.85 Hz), 4.51 (m, 1H).
ПРИМЕР 82
N-Ацетил-4-амино-4-дезокси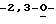 изопропилиден-β-D-метилрибофуранозид (105)
изопропилиден-β-D-метилрибофуранозид (105)
К раствору (104) (6,5 г; 28,38 ммоль) в МеОН (50 мл) добавляли NаНСО3 (2,38 г; 23,38 ммоль) и затем Pd/C (5% вес/вес, 650мг). Эту реакционную смесь в течение 1 часа хорошо встряхивали в атмосфере Н2 (40 фунтов на квадратный дюйм) при комнатной температуре. ТСХ (30% этилацетат/гексан) свидетельствовала о завершении реакции. Реакционную смесь фильтровали через слой целита и фильтрат упаривали досуха. Остаток упаривали с толуолом (2•20 мл) и пиридином (2•20 мл). Затем полученный остаток без дальнейшей очистки переводили в следующую реакцию.
К смеси вышеуказанного остатка (5,76 г; неочищенный продукт вышеуказанной реакции) и DMAP (0,059 г; 0,425 ммоль) в пиридине (6,8 мл; 84,5 ммоль) добавляли при 0oС уксусный ангидрид (4,01 мл; 42,57 ммоль). Эту реакционную смесь 2 часа перемешивали при комнатной температуре. ТСХ (100% этилацетат) свидетельствовала о завершении реакции. Добавляли МеОН (1,0 мл) и выпаривали летучие вещества. Остаток растворяли в СН2Сl2 (300 мл) и затем этот раствор промывали холодным перегнанным HCl (0,5 M; 3•200 мл), насыщенным NaHCO3 (200 мл) и соляным раствором (200 мл), сушили (Na2SO4) и упаривали. Полученный остаток чистили быстрой хроматографией на силикагеле, используя этилацетат/гексан (от 0/100 к 20/80, к 50/50 и к 100/0) в качестве элюента, с получением продукта (105) (с суммарным выходом 3,76 г; 54,11%). 1H-ЯМР (300 МГц, (CDCCl3): δ 1.45 (s, 3Н, СН3), 1.51 (s, 3Н, СН3), 2.0 (s, 3Н, СОСН3), 3.37 (t, 1H), 3.45 (s, 3Н, ОСН3), 3.85 (dd, 1H), 4.05 (dd, 1H), 4.38 (dd, 1H), 4.40 (d, 1H, J=4.5 Hz), 4.58 (m, 1H), 5.78 (bd, 1H).
ПРИМЕР 83
1,2,3,5-Тетра ацетил-4-дезокси-4-(ацетамидо)-D-рибофураноза (106)
ацетил-4-дезокси-4-(ацетамидо)-D-рибофураноза (106)
Раствор (105) (5,0 г; 20,40 ммоль) в смеси дистиллированной воды и АсОН (1: 1, 50 мл) грели при 70-75oС в течение 1,5 часов. ТСХ (100% этилацетат) свидетельствовала о завершении реакции. Добавляли абсолютный ЕtOН (2•30 мл) и вместе с ним выпаривали летучие вещества с получением сухого твердого остатка. К этому твердому веществу добавляли смесь ледяной уксусной кислоты и уксусного ангидрида (50 мл; 1:1), охлаждали полученную смесь до 0oС и обрабатывали ее концентрированной H2SO4 (1,5 мл). Реакционную смесь 30 минут перемешивали при 0oС и 2 дня держали при 4oС. Реакционную смесь обрабатывали безводным NaOAc (15 г) и 30 минут перемешивали при комнатной температуре. Затем реакционную смесь выливали в смесь воды со льдом (300 мл) и экстрагировали, используя CH2Cl2(2•250 мл). Объединенный органический слой промывали водой (2•250 мл) и соляным раствором (400 мл), сушили (Nа2SO4) и упаривали. Полученный неочищенный остаток чистили быстрой хроматографией на силикагеле, используя MeOH/CH2Cl2 (от 0/100 к 3/97) в качестве элюента, с получением чистого (106) (3,71 г, 50,82%). 1H-ЯМР (300 МГц, (CDCCl3): δ 1.99-2.11 (m, 15H, 5•СH3), 4.17-4.5 (m, 2Н), 5.27-5.52 (m, 2H), 6.35 (s, 0.75H), 6.53 (d, 0.25H, J=4.8 Hz).
ПРИМЕР 84
Метиловый эфир 1-(2', 3', 5'-три ацетил-4'-дезокси-4'-ацетамидо-β-D-рибофуранозил)-1,2,4-триазол-3-карбоновой кислоты (107)
ацетил-4'-дезокси-4'-ацетамидо-β-D-рибофуранозил)-1,2,4-триазол-3-карбоновой кислоты (107)
Суспензию метилового эфира 1,2,4-триазол-3-карбоновой кислоты (1,77 мг; 13,97 ммоль) и сульфата аммония (177 мг) в гексаметилдисилазане (40 мл) грели в течение 2,5 часов при температуре образования флегмы в атмосфере N2. Реакционную смесь упаривали досуха и остаток суспензировали в 1,2-дихлорэтане (50 мл). Затем ее обрабатывали раствором (106) (4,4 г; 12,25 ммоль) в 1,2 дихлорэтане (50 мл). Затем к реакционной смеси добавляли дымящийся SnCl4 (1,63 мл; 13,97 ммоль) при 0-5oС (баня с тающим льдом) и перемешивали ее при комнатной температуре в течение 1 часа. Реакционную смесь осторожно гасили насыщенным раствором NаНСО3 (50 мл) и затем разводили в CH2Cl2 (200 мл). Эту смесь фильтровали через слой целита (5 г) и промывали, используя CH2Cl2 (100 мл). Органический слой фильтрата отделяли и водный слой экстрагировали, используя CH2Cl2 (2•100 мл). Объединенный органический слой промывали водой (2•300 мл) и соляным раствором (500 мл), сушили (Na2SO4) и упаривали. Полученный неочищенный остаток перекристаллизовывали из этилацетата (40 мл) с получением чистого целевого продукта (107) (2,7 г; 51,71%). 1H-ЯМР (300 МГц, (CDCCl3): δ 2.03-2.15 (m, 12H, 4•СОСН3), 3.95 (s, 3Н, ОСН3), 4.2 (m, 1Н, Н5'), 4.43 (m, 2H, H4' & Н5'), 5.65 (dd, 1H, Н3', J=4.67 & 1.1 Hz), 6.17 (t, 1H, H2', J= 4.67 & 6.04 Hz), 6.28 (d, 1H, H1', J=6.04 Hz), 8.47 (s, 0.8H, major rotamer, C5H), 8.60 (s, 0.14H, minor rotamer, C5H). Анализ. Рассчитано для C17H22N4O9: С 47,89; Н 5,20; N 13,14. Найдено: С 47,94; Н 5.40; N 13,27.
ПРИМЕР 85
1-(4'-Дезокси-4'-ацетамидо-β-D-рибофуранозил)-1,2,4-триазол-3-карбоксамид (108)
Раствор (107) (2,7 г; 6.33 ммоль) в насыщенном растворе аммиака в метаноле (100 мл) 16 часов перемешивали в стальном баллоне при комнатной температуре. Реакционную смесь упаривали досуха и полученный остаток чистили быстрой хроматографией на окиси алюминия, используя смесь этилацетат/н-пропиловый спирт/вода (от 64/4/32 к 77/14/29%, нижний слой) в качестве элюента, с получением целевого соединения (108) (1,7 г). Продукт содержал ~5% этилового спирта, а также был немного загрязнен ацетамидом. 1H-ЯМР (300 МГц, CD3OD): δ 1.87 (s, 0.86H, СОСН3, минорный ротамер min), 2.14 (s, 2.14H, СОСН3, основной ротамер maj), 3.87 (d. 2H, H5', J=6.59 Hz), 4.1-4.01 (m, 1H, Н4'), 4.26 (d, 0.75H, J=4.12 Hz, Н3', maj), 4.31 (t, 0.25H, J=3.8 Hz, H3', min), 4.54 (t, 0.25H, J=4.1 Hz, Н2', min), 4.85 (dd, 0.75H, J=4.4 & 6.05 Hz, H2', maj), 6.03 (d, 0.75, J=6.04 Hz, H1', maj), 6.81 (d, 0.25H, J=4.13 Hz, Н1', min), 8.69 (s, 0.75H, J=6.04 Hz, H1', maj), 6.81 (d, 0.25H, J=4.13 Hz, H1', min), 8.69 (s, 0.75H, C5H, maj), 8.95 (s, 0.25H, C5H, min). Анализ. Рассчитано для C10H15N5O5: C42,10; Н 5,30; N 24,55. Найдено: С 42,21; Н 5,19; N 24,23.
ПРИМЕР 86
1-[(3', 5' (1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-D-рибофуранозил]-1,2,4-триазол-3-карбоксамид (109)
(1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-D-рибофуранозил]-1,2,4-триазол-3-карбоксамид (109)
Суспензию (108) (0,7 г; 2,45 ммоль) в пиридине (15 мл) обрабатывали 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксаном (1,06 мл; 3,31 ммоль) и 16 часов перемешивали при комнатной температуре. Реакционную смесь осторожно гасили насыщенным раствором NаНСО3 (5 мл) и разводили в СН2Сl2. Органический слой отделяли от водного слоя и экстрагировали, используя CH2Cl2 (2•25 мл). Объединенный органический слой промывали водой (2•100 мл) и соляным раствором (100 мл), сушили (Na2SO4) и упаривали. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/МеОН (100/0-98/2-95/5-90/10) в качестве элюента, с получением (109) (0,7 г; 54%). 1H-ЯМР (300 МГц, CDCl3): δ 0.93-1.18 (m, 24H), 1.38 (m, 2H), 2.02 (s, 0.84H, СОСН3, минорный ротамер min), 2.15 (s, 2.16H, СОСН3, основной ротамер maj), 3.83 (m, 1H, H5), 3.98-4.13 (m, 1H, H5), 4.33 (d, 0.34Н, J=3.85 Hz, Н2', min), 4.42 (d, 0.66H, J=4.67 Hz, H2', maj), 4.52 (dd, 0.34H, H4', min), 4.65 (dd, 0.66H, H4', min), 5.99 (s, 0.34H, H1', min), 6.40 (s, 0.66H, H1', maj), 6.91 (bs, 0.66H, maj), 7.01 (bs, 0.34H, min), 8.37 (s, 0.66H, C5H, maj), 8.53 (s, 0.34, C5H, min).
ПРИМЕР 87
1-[2' (пара-Толилтионоформил)-3', 5'
(пара-Толилтионоформил)-3', 5' (1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-D-рибофуранозил] -1,2,4-триазол-3-карбоксамид (110)
(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-D-рибофуранозил] -1,2,4-триазол-3-карбоксамид (110)
К раствору (109) (0,6 г; 1,138 ммоль) в смеси CH2Cl2 (9 мл) и пиридина (1 мл) добавляли  (пара-толил)-тионохлорформат (2,219 мл; 1,42 ммоль) и реакционную смесь 16 часов перемешивали при комнатной температуре. Реакционную смесь гасили насыщенным раствором NaHCО3 (5 мл) и разводили в CH2Cl2 (100 мл). Органический слой отделяли от водной фазы и экстрагировали, используя CH2Cl2 (2•25 мл). Объединенный органический слой промывали водой (2•100 мл) и соляным раствором (100 мл), сушили (Na2SO4) и упаривали. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/этилацетат (100/0-95/5-90/10) в качестве элюента, с получением чистого продукта (110) (0,35 г; 45%). 1H-ЯМР (300 МГц, CDCl3): δ 1.04-1.15 (m, 24H), 1.32 (m, 2H), 2.01 (s, 1H, СОСН3, минорный ротамер min), 2.19 (s, 2H, СОСH3, основной ротамер maj), 2.25 (s, 3Н, СН3), 3.92 (m, 1H, Н5'), 4.05 (m, 1H, Н5'), 4.68-4.81 (m, 1H, Н4'), 5.5 (t, 1H, J=6.05 & 5.22 Hz, Н3'), 5.75 (bs, 0.66H, maj), 5.88 (bs, 0.34H, min), 6.10 (d, 1H, J=4.67 Hz, H2'), 6.17 (s, 0.34H, H1', min), 6.54 (s, 0.66H, H1', maj), 6.87 (bs, 0.66H, maj), 6.96 (d, 2H, J=8.24 Hz, aromatic-H), 6.98 (bs, 0.34H, min), 7.20 (d. 2H, J=8.24 Hz), 8.40 (s, 0.66H, C5H, maj), 8.68 (s, 0.34H, C5H, min).
(пара-толил)-тионохлорформат (2,219 мл; 1,42 ммоль) и реакционную смесь 16 часов перемешивали при комнатной температуре. Реакционную смесь гасили насыщенным раствором NaHCО3 (5 мл) и разводили в CH2Cl2 (100 мл). Органический слой отделяли от водной фазы и экстрагировали, используя CH2Cl2 (2•25 мл). Объединенный органический слой промывали водой (2•100 мл) и соляным раствором (100 мл), сушили (Na2SO4) и упаривали. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/этилацетат (100/0-95/5-90/10) в качестве элюента, с получением чистого продукта (110) (0,35 г; 45%). 1H-ЯМР (300 МГц, CDCl3): δ 1.04-1.15 (m, 24H), 1.32 (m, 2H), 2.01 (s, 1H, СОСН3, минорный ротамер min), 2.19 (s, 2H, СОСH3, основной ротамер maj), 2.25 (s, 3Н, СН3), 3.92 (m, 1H, Н5'), 4.05 (m, 1H, Н5'), 4.68-4.81 (m, 1H, Н4'), 5.5 (t, 1H, J=6.05 & 5.22 Hz, Н3'), 5.75 (bs, 0.66H, maj), 5.88 (bs, 0.34H, min), 6.10 (d, 1H, J=4.67 Hz, H2'), 6.17 (s, 0.34H, H1', min), 6.54 (s, 0.66H, H1', maj), 6.87 (bs, 0.66H, maj), 6.96 (d, 2H, J=8.24 Hz, aromatic-H), 6.98 (bs, 0.34H, min), 7.20 (d. 2H, J=8.24 Hz), 8.40 (s, 0.66H, C5H, maj), 8.68 (s, 0.34H, C5H, min).
ПРИМЕР 88
1-[(3', 5' (1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-2'-4'-дидезокси-4'-ацетамидо-β-D-рибофуранозил]-1,2,4-триазол-3-карбоксамид (111)
(1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-2'-4'-дидезокси-4'-ацетамидо-β-D-рибофуранозил]-1,2,4-триазол-3-карбоксамид (111)
Раствор (110) (0,35 г; 0,516 ммоль) в толуоле (20 мл) 20 минут продували аргоном и затем обрабатывали 2,2'-азобисизобутиронитрилом (0,084 г; 0,516 ммоль) и трибутилингидридом (0,274 мл; 1,03 ммоль). Эту реакционную смесь 3 часа грели в токе аргона при температуре образования флегмы. Реакционную смесь упаривали досуха и неочищенный остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/этилацетат (100/0-90/10-70/30-40/60-20/80-0/100) в качестве элюента, с получением продукта (111) (0.23 г, 87%). 1H-ЯМР (300 МГц, CDCl3): δ 0.95-1.15 (m, 24H), 1.24 (m, 2H), 2.0 (s, 0.9H, СОСН3, минорный ротамер min), 2.13 (s, 2.1H, СОСН3, основной ротамер maj), 2.36-2.58 (m, 1H, H2'), 2.84 (m, 1H, H2'), 3.74-3.92 (m, 2H, Н5'), 4.11 (m, 0.66H, H4', maj), 4.49-4.67 (m, 0.34H, H4', min), 5.3 (m, 1H, H3'), 5.88 (bs, 0.66H, maj), 6.01 (bs, 0.34H, min), 6.14 (d, 0.34H, J=6.02 Hz, H1', min), 6.54 (d, 0.66H, J=7.97 Hz, Н1', maj), 6.89 (bs, 0.66H, maj), 7.03 (bs, 0.34H, min), 8.35 (s, 0.66H, C5H, maj), 8.55 (s, 0.44H, C5H, min).
ПРИМЕР 89
1-(2', 4'-Дидезокси-4'-ацетамидо-β-D-рибофуранозил)-1,2,4-триазол-3-карбоксамид (112)
Раствор (111) (0,23 г; 0,45 ммоль) в CH2Cl2 (5 мл) обрабатывали трисгидрофторидом триэтиламина (0,29 мл; 1,79 ммоль) при комнатной температуре. Реакционную смесь перемешивали 48 часов при комнатной температуре. Удаляли летучие вещества и чистили остаток быстрой хроматографией на силикагеле, используя СНСl3/МеОН (100/0-95/5-90/10) в качестве элюента, с получением чистого продукта (112) (0,08 г; 66%). 1H-ЯМР (300 МГц, CD3OD): δ 1.95 (s, 0.36H, СОСН3), минорный ротамер min), 2.16 (s, 2.64Н, СН3, основной ротамер maj), 2.51 (m, 1H, Н2'), 2.87 (m, 1H, Н2'), 3.82 (m, 2H, Н5'), 3.99 (t, 1H, J=6.87 Hz, H4'), 4.44 (d, 1H, J=4.12 Hz, Н3'), 6.54 (t, 1H, J=7.69 Hz, H1'), 8.63 (s, 0.88H, C5H, maj), 8.88 (s, 0>12 H, C5H, min). Анализ. Рассчитано для C10H15N5O2: С 44,60; H 5.62; N 26,01. Найдено: С 44,71; H 5,69; N 25.98.
ПРИМЕР 90
Метил 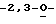 изопропилиден-α-D-ликсопиранозид (115)
изопропилиден-α-D-ликсопиранозид (115)
К метанольному раствору HCl [500 мл; 0,5% вес/объем, приготовлен in situ в реакции ацетилхлорида (5 мл; 70,37 ммоль) с МеОН (для ВЭЖХ по Fisher)] добавляли D-ликсозу ((113); 100 г; 666,66 ммоль) и 5 часов грели смесь при температуре образования флегмы в атмосфере N2. Реакционную смесь в течение 10 минут при перемешивании нейтрализовали предварительно обработанной1 смолой Amberlite IRA-410 в основной форме (100,0 г). Смолу отфильтровывали и промывали метанолом (3•125 мл). Объединенные смывы упаривали с получением бесцветного сиропа метил-β-D-ликсопиранозида (114; 110 г; количественный выход), которую направляли в следующую реакцию без дальнейшей очистки.
Примечание:
Получение предварительно обработанной смолы Amberlite IRA-410: Смолу (100 г) в течение 15 минут при перемешивании обрабатывали водным NaOH (0,5 М; 200 мл), фильтровали и промывали деионизированной водой (4•300 мл), пока рН смывов не становился нейтральным по индикаторной бумаге. Под конец смолу промывали безводным МеОН (3•30 мл) и использовали немедленно.
К суспензии (114) (110 г; 666,66 ммоль) в смеси 2,2-диметоксипропана (400,0 мл) и безводного ацетона (400,0 мл) добавляли раствор (4М) HCl в диоксане (8,0 мл) и эту реакционную смесь перемешивали 16 часов при 25oС. ТСХ (50% этилацетат/СН2Сl2) свидетельствовала о завершении реакции. Реакционную смесь гасили твердым бикарбонатом натрия (500 мг) и фильтровали. Фильтрат упаривали и маслянистый остаток (розоватого цвета) чистили быстрой хроматографией на силикагеле, используя СН2Сl2/этилацетат (от 100/0 к 80/20 с шагом 5%) в качестве элюента, с получением (115) (общий выход для обеих стадий: 63,97%; 87 г).
ПРИМЕР 91
Метил-4-азидо-4-дезокси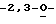 изопропилиден-β-L-рибопиранозид (116)
изопропилиден-β-L-рибопиранозид (116)
К смеси пиридина (6,432 мл; 79,9 моль) и диметиламинопиридина (105 мг; 0,75 ммоль) в безводном CH2Cl2 (600 мл) медленно при -20oС добавляли трифторметансульфоновый ангидрид (10,72 мл; 65 ммоль). Эту смесь 5 минут перемешивали при -20oС и затем добавляли к ней раствор (115) (10,2 г; 50 ммоль) в CH2Cl2 (100,0 мл). Эту реакционную смесь перемешивали в течение 15 минут при -20oС. ТСХ (15% этилацетат/гексан) свидетельствовала о завершении реакции. Реакционную смесь вливали в смесь воды и льда (500 мл) и отделяли органический слой. Водную фазу экстрагировали, используя СН2Сl2 (2•100 мл). Объединенный органический слой промывали водой (2•250 мл) и соляным раствором (500 мл), сушили (Na2SO4) и упаривали с получением промежуточного трифталата в виде бледно-желтого смолистого твердого вещества.
К раствору вышеуказанного метил трифторметансульфонил
трифторметансульфонил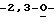 изопропилиден-β-D-ликсопиранозида (16 г) в DMF (300 мл), охлажденному до 0oС, добавляли азид лития (12,5 г; 255,6 моль) и перемешивали раствор при 23oС в течение 3 часов. Эту реакционную смесь разводили толуолом (200 мл) и летучие вещества выпаривали. Остаток растворяли в смеси СН2Сl2 (500 мл) и воды (200 мл). Органический слой отделяли и промывали водой (2•250 мл) и соляным раствором (300 мл), сушили (Na2SO4) и упаривали с получением маслянистого остатка, который при очистке быстрой хроматографией с использованием гексана/этилацетата (100/0; 97,5/2,5 и 95/5) в качестве элюента давал чистый азидо-продукт (116) (5,74 г; 50,2%). 1H-ЯМР (300 МГц, CDCl3): δ 1.38 (s, 3Н, СН3), 1.55 (s, 3Н, СН3), 3.44 (s, 3Н, ОСН3), 3.7-3.9 (m, 3Н), 4.03 (dd, 1H, J=6.32 & 3.85 Hz), 4.49 (d, 1H, J=3.84 Hz), 4.52 (m, 1H).
изопропилиден-β-D-ликсопиранозида (16 г) в DMF (300 мл), охлажденному до 0oС, добавляли азид лития (12,5 г; 255,6 моль) и перемешивали раствор при 23oС в течение 3 часов. Эту реакционную смесь разводили толуолом (200 мл) и летучие вещества выпаривали. Остаток растворяли в смеси СН2Сl2 (500 мл) и воды (200 мл). Органический слой отделяли и промывали водой (2•250 мл) и соляным раствором (300 мл), сушили (Na2SO4) и упаривали с получением маслянистого остатка, который при очистке быстрой хроматографией с использованием гексана/этилацетата (100/0; 97,5/2,5 и 95/5) в качестве элюента давал чистый азидо-продукт (116) (5,74 г; 50,2%). 1H-ЯМР (300 МГц, CDCl3): δ 1.38 (s, 3Н, СН3), 1.55 (s, 3Н, СН3), 3.44 (s, 3Н, ОСН3), 3.7-3.9 (m, 3Н), 4.03 (dd, 1H, J=6.32 & 3.85 Hz), 4.49 (d, 1H, J=3.84 Hz), 4.52 (m, 1H).
ПРИМЕР 92
N-Ацетил-4-амино-4-дезокси изопропилиден-β-L-метилрибопиранозид (117)
изопропилиден-β-L-метилрибопиранозид (117)
К раствору (116) (12,1 г; 52,83 ммоль) в МеОН (40,0 мл) добавляли Pd/C (5% вес/вес; 1,2 г) и эту реакционную смесь в течение 1 часа хорошо встряхивали в атмосфере Н2 (50 фунтов на квадратный дюйм) при комнатной температуре. ТСХ (30% этилацетат/гексан) свидетельствовала о завершении реакции. Реакционную смесь фильтровали через слой целита и фильтрат упаривали досуха и упаривали с толуолом (2•50 мл) и пиридином (2•25 мл). Затем остаток без дальнейшей очистки направляли в следующую реакцию.
К вышеуказанной неочищенной смеси добавляли DMAP (0,7 г; 5,0 ммоль) и пиридин (25,0 мл; 310,55 ммоль) в CH2Cl2 (250 мл), после чего при -5oС (баня из льда с ацетоном) добавляли уксусный ангидрид (25,0 мл; 265 ммоль). После добавления охлаждающую баню убирали и реакционную смесь перемешивали в течение 16 часов. ТСХ (100% этилацетат) свидетельствовала о завершении реакции. Добавляли МеОН и выпаривали летучие вещества. Остаток растворяли в CH2Cl2 (300 мл) и затем этот раствор промывали водой (2•200 мл) и соляным раствором (200 мл), сушили (Na2SO4) и упаривали. Остаток чистили быстрой хроматографией, используя этилацетат/гексан (от 5/95 к 20/80 к 60/40 к 80/20) в качестве элюента, с получением продукта (117) (общий выход 11,49 г, 88,83%). 1H-ЯМР (300 МГц, СDСl3): δ 1.34 (s, 3Н, СН3), 1.51 (s, 3H, СН3), 1.99 (s, 2Н, СОСH3), 3.37 (t, 1Н), 3.83 (dd, 1H, J=5.77 & 5.49 Hz), 4.01 (t, 1H, J= 5.77 & 4.67 Hz), 4.35 (t, 1H, J=5.5 & 4.67 Hz), 4.40 (d, 1H, J=4.4 Hz), 4.54 (m, 1H), 5.76(bd, 1H, J=7.97Hz).
ПРИМЕР 93
1,2,3,5-Тетра ацетил-4-дезокси-4-(ацетамидо)-L-рибофураноза (118)
ацетил-4-дезокси-4-(ацетамидо)-L-рибофураноза (118)
Раствор (117) (8,9 г ) в смеси дистиллированной воды и АсОН (1:1; 100 мл) в течение 1,5 часов грели при 70-75oС. ТСХ (100 этилацетат) свидетельствовала о завершении реакции. Добавляли абсолютный этанол (2х50 мл) и выпаривали с получением твердого остатка. К этому твердому веществу добавляли смесь ледяной уксусной кислоты и уксусного ангидрида (100,0 мл; 1:1) и охлаждали смесь до 0oС (баня с тающим льдом) и обрабатывали ее концентрированной серной кислотой H2SO4 (1,0 мл). Эту реакционную смесь 30 минут перемешивали при 0oС и затем 2 дня держали при 4oС. Реакционную смесь обрабатывали безводным NaOAc (10,0 г) и 30 минут перемешивали при комнатной температуре. Затем реакционную смесь вливали в смесь воды со льдом (400 мл) и экстрагировали, используя CH2Cl2 (2•250 мл). Объединенный органический слой промывали водой (2•500 мл) и соляным раствором (400 мл), сушили (Na2SO4) и упаривали. Полученный неочищенный продукт чистили быстрой хроматографией, используя этилацетат/гексан (от 25/75 к 50/50) в качестве элюента, с получением (118) (6,6 г; 50,67%). 1H-ЯМР (300 МГц, CDCl3): δ 2.0-2.12 (m, 15H, 5•СОСН3), 4.18-4.51 (m, 3H), 5.33-5.36 (m, 1Н), 5.45-5.55 (m. 1H), 6.36 (s, 0.75H), 6.55 (d, 0.25H, J=5.22 Hz).
ПРИМЕР 94
Метиловый эфир 1-(2',3',5'-триацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-1,2,4-триазол-3-карбоновой кислоты (119).
Суспензию метилового эфира 1,2,4-триазол-3-карбоновой кислоты (1,022 г, 8,05 ммоль) и сульфата аммония (100 мг) в гексаметилдисилазане (20 мл) 2,5 часа грели при температуре образования флегмы в атмосфере N2. Летучие вещества выпаривали и остаток суспензировали в 1,2-дихлорэтане (50 мл). Затем его обрабатывали раствором (118) (2,513 г, 7 моль) в 1,2 дихлорэтане (50 мл). Затем к реакционной смеси при 0-5oС (баня с тающим льдом) добавляли дымящийся SnCl4 (0,94 мл; 8,05 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь осторожно гасили насыщенным раствором NаНСО3 (50 мл) и разводили в CH2Cl2 (200 мл). Эту смесь фильтровали через слой целита (5 г) и промывали, используя CH2Cl2 (100 мл). Органический слой фильтрата отделяли и водный слой экстрагировали, используя CI2Cl2 (2•100 мл). Объединенный органический слой промывали водой (2•300 мл) и соляным раствором (500 мл), сушили (Na2SO4) и упаривали. Неочищенный остаток перекристаллизовывали из этилацетата (40 мл) с получением чистого целевого продукта (119) (1,8 г; 60,36%). 1H-ЯМР (300 МГц, СDСl3): δ 2.03-2.14 (m, 12Н, 4•СОСН3), 3.93 (s, 3H, ОСН3), 4.2 (m, 1H, Н5'), 5.64 (d, 1H, Н3', J= 4.67 Hz), 6.16 (t, 1H, H2', J=4.95 & 5.77), 6.27 (d, 1H, H1', J=6.06 Hz), 8.46 (s, 0.86H, major rotamer. C5H), 8.60 (s, 0.14H, minor rotamer, C5H). Анализ. Рассчитано для C17H22N4O9: С 47,89; Н 5,20; N 13,14. Найдено: С 47,77; Н 5.94; N 13.04.
ПРИМЕР 95
1-(4'-Дезокси-4'-ацетамидо-β-L-рибофуранозил)-1,2,4-триазол-3-карбоксамид (120)
Раствор (119) (3,26 г; 7,65 моль) в насыщенном растворе аммиака в метаноле (100 мл) 16 часов перемешивали при комнатной температуре. Реакционную смесь упаривали досуха и остаток чистили быстрой хроматографией на окиси алюминия, используя смесь растворителей этилацетат/н-пропиловый спирт/вода (нижний слой, 64/4/32 к 57/14/29%), с получением целевого продукта (120) (1,7 г, 77,98%). 1H-ЯМР (300 МГц, (DMSO-d6+D2O): δ 1.64 (s, 0.75H, СОСН3, минорный ротамер min), 2.0 (s, 2.25H, СОСН3 основной ротамер maj), 3.84-3.54 (m, 3H, Н-4' & Н-5'), 4.1 (m, 1H, Н3-'), 4.32 (t, 0.25Н, J=4.4 Hz, H-2', min), 4.56 (t, 0.75H, J=4.2 Hz, H-2', maj), 5.82 (d, 0.75H, J=6.32 Hz, H-1', maj), 6.01 (d, 0.25H, J=4.4 Hz, H-1', min), 8.74 (s, 0.75H, C5H, maj), 8.96 (s, 0.25H, C5H, min). Анализ. Рассчитано для C10H15N5O5: С 42,10; Н 5,30; N 24,55. Найднено: С 42,44; Н 5,49; N 24,69.
ПРИМЕР 96
1-[(3', 5' (1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил]-1,2,4-триазол-3-карбоксамид (121)
(1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил]-1,2,4-триазол-3-карбоксамид (121)
Суспензию (120) (0,75 г; 2,63 ммоль) в пиридине (15 мл) обрабатывали 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксаном (1,09 мл; 3,42 ммоль) и 16 часов перемешивали при комнатной температуре. Реакционную смесь осторожно гасили насыщенным раствором NaHCO3 (5 мл) и разводили в CH2Cl2 (100 мл). Органический слой отделяли и водный слой экстрагировали, используя СН2Сl2 (2•25 мл). Объединенный органический слой промывали водой (2•100 мл), соляным раствором (100 мл), сушили (Na2SO4) и упаривали. Полученный неочищенный остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/МеОН (100/0-98/2-95/5-90/10) в качестве элюента, с получением продукта (121) (0,75 г, 54%). 1H-ЯМР (300 МГц, СDСl3): δ 0.93-1.17 (m, 24Н), 1.37 (m, 2H). 2.01 (s, 1H, СОСН3, минорный ротамер min), 2.14 (s, 2H, СОСН3, основной ротамер maj), 2.98 (s, 0.34H, ОН, exchangeable, min), 3.34 (s, 0.66Н, ОН, exchangeable, maj), 3.78-3.84 (m, 1H, H5'), 3.97-4.14 (m, 2H, H4' & H5'), 4.33 (d, 0.34H, J=4.12 Hz, H2', min), 4.41 (d, 0.66H, J=4.95Hz, H2', maj), 4.52 (m, Н4', min), 4.65 (m, Н3', min), 5.28 (t, J=4.95 Hz, H3', maj), 5.80 (s, 0.66H, exchangeable, maj), 5.95 (s, 0.34H, exchangeable, min), 5.97 (s, 0.34H, H1', min), 6.39 (s, 0.66H, H1', maj), 6.89 (s, 0.66H, exchangeable, maj), 7.00 (s, 0.34, exchangeable, min), 8.37 (s, 0.66H, C5H, maj), 8.52 (s, 0.34H, C5H, min).
ПРИМЕР 97
1-[2' (пара-Толуоилтионоформил)-3'5'
(пара-Толуоилтионоформил)-3'5' (1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил] -1,2,4-триазол-3-карбоксамид (122)
(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил] -1,2,4-триазол-3-карбоксамид (122)
К раствору (121) (0,65 г, 1,23 ммоль) в смеси CH2Cl2 (9 мл) и пиридина (1 мл) добавляли  пара-толиловый эфир тионохлормуравьиной кислоты (0,285 мл; 1,85 ммоль) и эту реакционную смесь 16 часов перемешивали при комнатной температуре. Реакционную смесь гасили насыщенным раствором NаНСО3 (5 мл) и разводили в CH2Cl2 (100 мл). Органический слой отделяли и водный слой экстрагировали, используя CH2Cl2 (2•25 мл). Объединенный органический слой промывали водой (2•100 мл) и соляным раствором (100 мл), сушили (Na2SO4) и упаривали. Полученный неочищенный остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/этилацетат (100/0-95/5-90/10) в качестве элюента, с получением продукта (122) (0,33 г; 39,52%). 1H-ЯМР (300 МГц, CDCl3): δ 0.92-1.15 (m, 24H), 1.29 (m, 2H), 2.01 (s, 1H, СОСН3, минорный ротамер min), 2.19 (s, 2H, СОСН3, основной ротамер maj), 2.35 (s, 3H, СН3), 3.92 (m, 1H, Н5'), 4.07 (m, 2H, H4' & Н5'), 4.68-4.8 (m, Н3', min), 5.50 (m, Н3', maj), 5.7 (s, 0.66H, exchangeable, maj), 5.82 (s, 0.34, exchangeable, min), 6.10 (d, 1H, J=4.94 Hz, Н2'), 6.17 (s, 0.34H, H1', min), 6.54 (s, 0.66, H1', maj), 6.70 (s, 0.34H, exchangeable, min), 6.86 (s, 0.66, exchangeable, maj), 6.96 (d, 2H, J=8.52 Hz, aromatic-H), 7.21 (d, 2H, J=8.24 Hz, aromatic-H), 8.40 (s, 0.66H, C5H, maj), 8.69 (s, 0.34H, C5H, min).
пара-толиловый эфир тионохлормуравьиной кислоты (0,285 мл; 1,85 ммоль) и эту реакционную смесь 16 часов перемешивали при комнатной температуре. Реакционную смесь гасили насыщенным раствором NаНСО3 (5 мл) и разводили в CH2Cl2 (100 мл). Органический слой отделяли и водный слой экстрагировали, используя CH2Cl2 (2•25 мл). Объединенный органический слой промывали водой (2•100 мл) и соляным раствором (100 мл), сушили (Na2SO4) и упаривали. Полученный неочищенный остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/этилацетат (100/0-95/5-90/10) в качестве элюента, с получением продукта (122) (0,33 г; 39,52%). 1H-ЯМР (300 МГц, CDCl3): δ 0.92-1.15 (m, 24H), 1.29 (m, 2H), 2.01 (s, 1H, СОСН3, минорный ротамер min), 2.19 (s, 2H, СОСН3, основной ротамер maj), 2.35 (s, 3H, СН3), 3.92 (m, 1H, Н5'), 4.07 (m, 2H, H4' & Н5'), 4.68-4.8 (m, Н3', min), 5.50 (m, Н3', maj), 5.7 (s, 0.66H, exchangeable, maj), 5.82 (s, 0.34, exchangeable, min), 6.10 (d, 1H, J=4.94 Hz, Н2'), 6.17 (s, 0.34H, H1', min), 6.54 (s, 0.66, H1', maj), 6.70 (s, 0.34H, exchangeable, min), 6.86 (s, 0.66, exchangeable, maj), 6.96 (d, 2H, J=8.52 Hz, aromatic-H), 7.21 (d, 2H, J=8.24 Hz, aromatic-H), 8.40 (s, 0.66H, C5H, maj), 8.69 (s, 0.34H, C5H, min).
ПРИМЕР 98
1-[(3, '5' (1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-2', 4'-дидезокси-4'-ацетамидо-β-L-рибофуранозил]-1,2,4-триазол-3-карбоксамид (123)
(1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-2', 4'-дидезокси-4'-ацетамидо-β-L-рибофуранозил]-1,2,4-триазол-3-карбоксамид (123)
Раствор (122) (0,325 г; 0,48 ммоль) в толуоле (20 мл) 20 минут продували аргоном. К раствору добавляли 2,2'-азобисизобутиронитрил (0,078 г; 0,48 ммоль) и гидрид трибутилина (0,25 мл; 0,96 ммоль). Эту реакционную смесь 6 часов грели в токе аргона при температуре образования флегмы. Реакционную смесь упаривали досуха и неочищенный остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/этилацетат (100/0-90/10-70/30-40/60-20/80-0/100) в качестве элюента, с получением (123) (0,22 г, 89,68%). 1H-ЯМР (300 МГц, CDCl3): δ 0.92-1.15 (m, 24H), 1.28 (m, 2H), 2.0 (s, 0.66H, COCH3, минорный ротамер min), 2.13 (s, 2.34H, СОСН3, основной ротамер maj), 2.36-2.58 (m, 1H, H2'), 2.85 (dd, 1H, J=13.73 & 7.41 Hz, H2'), 3.74-4.09 (m, 3Н, Н4' & Н5'), 4.49-4.67 (m, Н3', min), 5.30 (m, Н3', maj), 5.83 (s, 0.8H, exchangeable, maj), 5.94 (s, 0.2H, exchangeable, min), 6.14 (d, 0.2H, J=6.05 Hz, H1', min), 6.54 (d, 0.8H, J=8.24 Hz, Н1', maj), 6.89 (s, 0.8H, exchangeable, maj), 7.04 (s, 0.2H, exchangeable, min), 8.35 (s, 0.8H, C5H, maj), 8.55 (s, 0.2H, C5H, min).
ПРИМЕР 99
1-(2', 4'-Дидезокси-4'-ацетамидо-β-L-рибофуранозил)1,2,4-триазол-3-карбоксамид (124)
Раствор (123) (0,2 г; 0,39 ммоль) в СН2Сl2 (5 мл) обрабатывали трис-гидрофторидом триэтиламина (0,25 мл; 1,56 ммоль) при комнатной температуре. Эту реакционную смесь перемешивали 48 часов и летучие вещества выпаривали досуха. Остаток чистили быстрой хроматографией на силикагеле, используя СН3Cl/МеОН (100/0-95/5-90/10-85/15) в качестве элюента, с получением (124) (0,08 г; 66%). 1H-ЯМР (300 МГц, СD3ОD): δ 1.95 (s, 0.36Н, СОСН3, минорный ротамер min), 2.16 (s, 2.64H, СОСН3, основной ротамер maj), 2.51 (m, 1H, H2'), 2.87 (m, 1H, Н2'), 3.82 (m, 2H, Н5'), 3.99 (t, 1H, J=6.87 Hz, H4'), 4.44 (d, 1H, J=4.12 Hz, Н3'), 6.54 (t, 1H, J=7.69 Hz, H1'), 8.63 (s, 0.88H, C5H, maj), 8.88 (s, 0.12H, С5Н, min). Анализ. Рассчитано для С10Н15N5О4: С 44,60; Н 5,62; N 26,01. Найдено: С 44,69; Н 5,71; N 26,10.
ПРИМЕР 100
1-(2', 3', 5'-Три  ацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-тимин (125)
ацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-тимин (125)
Суспензию тимина (1,26 г; 10 ммоль) и сульфата аммония (126 мг) в гексаметилдисилазане (25 мл) 5 часов грели при температуре образования флегмы в атмосфере N2. Эту реакционную смесь упаривали досуха и остаток суспензировали в 1,2-дихлорэтане (50 мл). Добавляли раствор 118 (2,513 г; 7 ммоль) в 1,2-дихлорэтане (50 мл), а затем при температуре 0-5oС (баня с тающим льдом) добавляли дымящийся SnCl4 (1,17 мл; 10 ммоль, 1,42 экв.). Эту реакционную смесь 1 час перемешивали при комнатной температуре. Реакционную смесь осторожно гасили насыщенным раствором NаНСО3 (50 мл) и разводили в Cl2Cl2 (200 мл). Смесь фильтровали через слой целита (5 г) и промывали, используя CH2Cl2 (100 мл). Органический слой фильтрата отделяли и водный слой экстрагировали, используя СН2Сl2 (2•100 мл). Объединенный органический слой промывали водой (2•300 мл) и соляным раствором (500 мл), сушили (Na2SO4) и упаривали. Полученный остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/ацетон (95/5-90/10-85/15-80/20) в качестве элюента, с получением чистого целевого продукта (125) (2,9 г, количественно). 1H-ЯМР (300 МГц, СDСl3): δ 1.89-2.2 (m, 15H, 4•СОСН3 & С5СН3), 4.08 (m, 0.5Н, Н5'), 4.37-4.56 (m, 2.5H, H4' & Н5'), 5.32 (m, 0.5H, Н3'), 5.47 (m, 1.5H, H2' & Н3'), 6.15 (m, 0.5H, Н1'), 6.37 (d, 0.5H, J=6.6 Hz, H1'), 7.16 (s, 0.5H, C3H), 7.44 (s, 0.5H, C3H), 9.02 (s, 0.5H, NH, exchangeable), 9.20 (s, 0.5H, NH, exchangeable).
ПРИМЕР 101
1-(4'-Дезокси-4'-ацетамидо-β-L-рибофуранозил)-тимин (126)
Раствор (125) (3,1 г, 7,29 ммоль) в насыщенном растворе аммиака в метаноле (100 мл) 16 часов перемешивали в стальном баллоне при комнатной температуре. Стальной баллон охлаждали до 0oС и открывали и содержимое упаривали досуха. Остаток чистили быстрой хроматографией на силикагеле, используя СНCl3/МеОН (95/5-90/10-85/15) в качестве элюента, с получением целевого продукта (126) (1,72 г: 78,86%). 1H-ЯМР (300 МГц, (DMSO-d6+D2O): δ 1.70 (s, 1.35H, СОСН3, минорный ротамер min), 1.73 (s, 1.35H, C5CH3), 1.77(s, 1.65H, С5СН3), 1.98 (s, 1.65H, СОСН3, основной ротамер maj), 3.95-3.57 (m, 4H, Н3', Н4' & Н5'), 4.13 (t, 0.55H, J=4.67 Hz, H2', maj), 4.20 (dd, 0.45H, J=4.4 Hz, H2', min), 5.72 (d, 0.45H, J=6.6 Hz, H1', min), 5.88 (d, 0.55H, J=5.77 Hz, H1', maj), 7.68 (s, 0.45H, C6H, min), 8.00 (s, 0.55H, С6Н, maj). Анализ. Рассчитано для C13H17N3O6: С 48,16; Н 5,73; N 14,04. Найдено: С 48,23; Н 5,81; N 14,29.
ПРИМЕР 102
1-[(3', 5' (1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил] -тимин (127) и 1-[(2',3'-O-(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-4'-дидезокси-4'-ацетамидо-β-1-рибофуранозил]-тимин (128)
(1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил] -тимин (127) и 1-[(2',3'-O-(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-4'-дидезокси-4'-ацетамидо-β-1-рибофуранозил]-тимин (128)
Суспензию (126) (1,72 г; 5,75 ммоль) в пиридине (25 мл) обрабатывали 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксаном (2,75 мл; 8,59 ммоль) и 16 часов перемешивали при комнатной температуре. Реакционную смесь осторожно гасили насыщенным раствором NаНСО3 (5 мл) и разводили в СН2Сl2 (100 мл). Органический слой отделяли и водный слой экстрагировали, используя СН2Сl2 (2•25 мл). Объединенный органический слой промывали водой (2•100 мл) и соляным раствором (100 мл), сушили (Na2SO4) и упаривали. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя гексан/этилацетат (90/10-80/20-60/40-20/80-0/100) с получением смеси неразделяемых региоизомерных продуктов (127) и (128) (2,15 г; 69%). 1H-ЯМР (300 МГц, CDCl3): δ 1.0 (m), 1.9-2.13 (m), 3.79-4.13 (m), 4.3-4.47 (m), 4.74 (d), 5.07-5.21 (m), 5.47 (s), 5.84 (s), 5.93 (d, J=3.3 Hz), 7.20 (s), 7.53 (s), 7.78 (s), 8.88 (s), 9.13(s), 9.67(s).
ПРИМЕР 103
1-[2' (пара-Толилтионоформил)-3', 5'-0-(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил] -тимин (129) и 1-[5'
(пара-Толилтионоформил)-3', 5'-0-(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил] -тимин (129) и 1-[5' (пара-толилтионоформил)-2', 3'
(пара-толилтионоформил)-2', 3' (1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил]-тимин (130)
(1,1,3,3-тетраизопропил-1,3-дисилоксандиил)-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил]-тимин (130)
К смеси (127) и (128) (2 г; 3,69 ммоль) в пиридине (20 мл) добавляли  пара-толуоиловый эфир тионохлормуравьиной кислоты (0,712 мл; 4,43 ммоль) и эту реакционную смесь 16 часов перемешивали при комнатной температуре. Реакционную смесь гасили насыщенным раствором NaHCO3 (5 мл) и разводили в CH2Cl2 (100 мл). Органический слой отделяли и водный слой экстрагировали, используя CH2Cl2 (2•25 мл). Объединенный органический слой промывали водой (2•100 мл) и соляным раствором (100 мл), сушили (Na2SO4) и упаривали. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя гексан/этилацетат (90/10-80/20-70/30-40/60) в качестве растворителя, с получением более быстрого продукта (0,9 г) и менее быстрого продукта (0,8 г). Общий выход обоих продуктов составил 1,7 г (66,54%). Анализ обоих продуктов с помощью 1H-ЯМР показал, что менее быстрым продуктом был (129), а более быстрым продуктом (130). (129): 1H-ЯМР (300 МГц, СDСl3): δ 0.98-1.06 (m, 24H), 1.89 (s, 3Н, СН3), 2.00 (s, 2H, СОСН3, основной ротамер maj), 2.25 (s, 1Н, СОСН3, минорный ротамер min), 2.34 (s, 3Н, СН3), 3.85-4.03 (m, 2H, Н5'), 4.38-4.80 (m, 2H, Н3' & Н4'), 5.3 (m, 0.24H, Н2', min), 5.7 (m, 0.76H, Н2', maj), 6.01 (s, 1H, H1'), 6.95 (d, 2H, J=8.52 Hz, aromatic-H), 7.20 (d, 2H, J= 8.52 Hz, aromatic-H), 7.35 (s, 0.24H, C6H), 7.57 (s, 0.76H, C6H), 8.23 (bs, 0.24H, NH, exchangeable), 8.64 (s, 0.76H, NH, exchangeable). (130): 1H-ЯМР (300 МГц, СDСl3): δ 1.00-1.06 (m, 26H), 1.87 (s, 3Н, СН3), 2.02 (s, 2.4H, COCH3, основной ротамер maj), 2.22 (s, 0.6H, СОСН3, минорный ротамер min), 4.22-4.5 (m, 3Н, H4' & Н5'), 4.88 (m, 1H, Н3'), 5.22 (m, 1H, H2'), 6.01 (d, 1H, J=3.02 Hz, H1'), 6.94 (d, 2H, J=8.24 Hz, aromatic-H), 7.21 (d, 2H, J= 8.52 Hz, aromatic-H), 7.78 (s, 1H, C6H), 8.38 (bs, 0.16H, NH, exchangeable, min), 8.44 (s, 0.84H, NH, exchangeable, maj).
пара-толуоиловый эфир тионохлормуравьиной кислоты (0,712 мл; 4,43 ммоль) и эту реакционную смесь 16 часов перемешивали при комнатной температуре. Реакционную смесь гасили насыщенным раствором NaHCO3 (5 мл) и разводили в CH2Cl2 (100 мл). Органический слой отделяли и водный слой экстрагировали, используя CH2Cl2 (2•25 мл). Объединенный органический слой промывали водой (2•100 мл) и соляным раствором (100 мл), сушили (Na2SO4) и упаривали. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя гексан/этилацетат (90/10-80/20-70/30-40/60) в качестве растворителя, с получением более быстрого продукта (0,9 г) и менее быстрого продукта (0,8 г). Общий выход обоих продуктов составил 1,7 г (66,54%). Анализ обоих продуктов с помощью 1H-ЯМР показал, что менее быстрым продуктом был (129), а более быстрым продуктом (130). (129): 1H-ЯМР (300 МГц, СDСl3): δ 0.98-1.06 (m, 24H), 1.89 (s, 3Н, СН3), 2.00 (s, 2H, СОСН3, основной ротамер maj), 2.25 (s, 1Н, СОСН3, минорный ротамер min), 2.34 (s, 3Н, СН3), 3.85-4.03 (m, 2H, Н5'), 4.38-4.80 (m, 2H, Н3' & Н4'), 5.3 (m, 0.24H, Н2', min), 5.7 (m, 0.76H, Н2', maj), 6.01 (s, 1H, H1'), 6.95 (d, 2H, J=8.52 Hz, aromatic-H), 7.20 (d, 2H, J= 8.52 Hz, aromatic-H), 7.35 (s, 0.24H, C6H), 7.57 (s, 0.76H, C6H), 8.23 (bs, 0.24H, NH, exchangeable), 8.64 (s, 0.76H, NH, exchangeable). (130): 1H-ЯМР (300 МГц, СDСl3): δ 1.00-1.06 (m, 26H), 1.87 (s, 3Н, СН3), 2.02 (s, 2.4H, COCH3, основной ротамер maj), 2.22 (s, 0.6H, СОСН3, минорный ротамер min), 4.22-4.5 (m, 3Н, H4' & Н5'), 4.88 (m, 1H, Н3'), 5.22 (m, 1H, H2'), 6.01 (d, 1H, J=3.02 Hz, H1'), 6.94 (d, 2H, J=8.24 Hz, aromatic-H), 7.21 (d, 2H, J= 8.52 Hz, aromatic-H), 7.78 (s, 1H, C6H), 8.38 (bs, 0.16H, NH, exchangeable, min), 8.44 (s, 0.84H, NH, exchangeable, maj).
ПРИМЕР 104
1-[(3', 5' (1,1,3,3-Тетраизопропил -1,3-дисилоксандиил)-2', 4'-дидезокси-4'-ацетамидо-β-L-рибофуранозил]-тимин (131)
(1,1,3,3-Тетраизопропил -1,3-дисилоксандиил)-2', 4'-дидезокси-4'-ацетамидо-β-L-рибофуранозил]-тимин (131)
Раствор (129) (0,74 г, 1,070 ммоль) в толуоле (25 мл) 20 минут продували аргоном. К этому раствору добавляли 2,2'-азобисизобутиронитрил (0,174 г; 1,074 ммоль) и затем трибутил гидрид олова (0,56 мл; 2,113 ммоль; 2 экв.). Эту реакционную смесь 6 часов грели при температуре образования флегмы в потоке аргона. Летучие вещества выпаривали и неочищенный остаток чистили быстрой хроматографией на силикагеле, используя гексан/этилацетат (100/0-90/10-80/20-70/30-60/40) в качестве элюента с получением (131) (0,45 г; 80,03%). 1H-ЯМР (300 МГц, CDCl3): δ 0.98-1.06 (m, 24H), 1.26 (m, 2H), 1.90 (s, 3H, СН3), 1.97 (s, 2.25H, СОСН3, основной ротамер maj), 2.17 (s, 0.75H, СОСН3, минорный ротамер min), 2.2-2.4 (m, 1.5H, Н2'), 2.65 (m, 0.5H, H2'), 3.67 (m, H, Н5'), 4.0 (m, 1H, Н5'), 4.32 (m), 4.64 (m, 1H, Н4'), 5.12 (m, 1H, Н3'), 5.71 (m, 0.15H, Н1', min), 6.05 (m, 0.85H, J=6.05 Hz, H1', maj), 7.33 (s, 0.15H, C6H, min), 7.53 (s, 0.85H, C6H, maj), 8.23 (bs, 0.15H, NH, exchangeable, min), 8.76 (s, 0.85H, NH, exchangeable, maj).
ПРИМЕР 105
1-(2',4'-Дидезокси-4'-ацетамидо-β-L-рибофуранозил)-тимин (132)
Раствор (131) (0,38 г, 0,72 моль) в CH2Cl2 (10 мл) обрабатывали трис-гидрофторидом триэтиамина (0,585 мл; 3,6 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 48 часов и упаривали досуха. Остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/МеОН (100/0-97/3-94/6-90/10) в качестве элюента, до получения целевого соединения (132) (0,19 г; 92,75%). 1H-ЯМР (300 МГц, СD3ОD): δ 1.83 (s, 1.35Н, С5СН3), 1.86 (s, 1.65H, C5CH3), 1.95 (s, 1.35H, СОСН3, минорный ротамер min), 2.18 (s, 1.65H, СОСН3, основной ротамер maj), 2.37 (m, 2H, H2'), 4.05-3.73 (m, 3H, H4' & Н5'), 4.32 (d, 0.55H, J=3.85 Hz, Н3', maj), 4.36 (bs, 0.45H, Н3', min), 6.26 (t, 0.45H, J=8.2 Hz, Н1', min), 6.49 (t, 0.55H, J=7.4 Hz, H1', maj), 7.70 (s, 0.55H, С6Н, maj), 8.18 (s, 0.45H, C6H, min). Анализ. Рассчитано для C12H17N3O5•1/2H2O: С 49,31; H 6,21; N 14,38. Найдено: С 49,49; H 6,43; N 14,51.
ПРИМЕР 106
1-[(2', 3' (1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-5', 4'-дидезокси-4'-ацетамидо-β-L-рибофуранозил]-тимин (133)
(1,1,3,3-Тетраизопропил-1,3-дисилоксандиил)-5', 4'-дидезокси-4'-ацетамидо-β-L-рибофуранозил]-тимин (133)
Раствор (130) (1,35 г; 1,954 ммоль) в толуоле (40 мл) 20 минут продували аргоном. К этому раствору добавляли 2,2'-азобисизобутиронитрил (0,32 г; 1,954 ммоль) и трибутилгидрид олова (1,035 мл; 3,905 ммоль). Эту реакционную смесь в течение 1,5 часов грели при температуре образования флегмы в токе аргона. Реакционную смесь упаривали досуха. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя гексан/этилацетат (100/0-90/10-80/20-70/30-60/40) в качестве элюента, до получения (133) (0,7 г, 68,24%). 1H-ЯМР (300 МГц, CDCl3): δ 0.98-1.05 (m, 24H), 1.24 (m, 2H), 1.46 (d, 1.2H, Н5', минорный ротамер min), 1.52 (d, 2.8H, J=6.9 Hz, Н5', основной ротамер maj), 1.89 (s, 1.2H, СОСН3, min), 1.93 (s, 3Н, СН3), 2.09 (s, 2.8H, СОСН3, maj), 3.86 (m, 0.6Н, Н4', maj), 3.98 (m, 0.4, Н4', min), 4.12-4.36 (m, 1H, Н3'), 5.18 (m, 1H, H2'), 5.30 (d, 0.6H, J=6.05 Hz, H1', maj), 5.98 (d, 0.4H, J= 3.57 Hz, H1', min), 6.99 (s, 0.4H, C6H, min), 7.08 (s, 0.6H, C6H, maj), 8.53 (bs, 0.6H, NH, exchangeable, maj), 8.66 (bs, 0.4H, NH, exchangeable, min).
ПРИМЕР 107
1-(5',4'-Дидезокси-4'-ацетамидо-β-L-рибофуранозил)-тимин (134)
Раствор (133) (0,6 г; 1,14 моль) в CH2Cl2 (20 мл) обрабатывали трис-гидрофторидом триэтиламина (0,558 г; 3,42 ммоль) при комнатной температуре. Эту реакционную смесь перемешивали при комнатной температуре в течение 20 часов и выпаривали летучие вещества досуха. Остаток чистили быстрой хроматографией на силикагеле. используя СН3Сl/МеОН (100/0-97/3-94/6-90/10) в качестве элюента, с получением (134) (0,2 г; 61,83%). 1H-ЯМР (300 МГц, CD3OD): δ 1.40 (d, 0.36H, J=6.87 Hz, Н5', минорный ротамер min), 1.48 (d, 0/64H, J= 6.87 Hz, Н5', основной ротамер maj), 1.87 (s, 1.08Н, С5СН3), 1.91 (s, 1.92H, С5СН3), 1.91 (s, 1.08H, СОСН3, min), 2.09 (s, 1.92H, СОСН3, maj), 3.84-4.09 (m, 2H, Н3' & Н4'), 4.33 (m, 0.36H, H2', min), 4.67 (m, 0.64H, H2', maj), 5.71 (d, 0.64H, J=6.87 Hz, H1', maj), 6.08 (d, 0.36H, J=5.77 Hz, Н1', min). 7.21 (s, 0.36H, C6H, min), 7.27 (s, 0.64H, C6H, maj). Анализ. Рассчитано для C12H17N3O5: С 50,88; Н 6,05; N 14,83. Найдено: С 50,91; Н 6,23; N 14.91.
ПРИМЕР 108
1-(2', 3', 5' Триацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-6-азаурацил (135)
Триацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-6-азаурацил (135)
Суспензию 6-азаурацила (0,909 г; 8,05 ммоль) и сульфата аммония (100 мг) в гексаметилдисилазане (20 мл) 2 часа грели при температуре образования флегмы в атмосфере N2. Эту реакционную смесь упаривали досуха и остаток суспензировали в 1,2-дихлорэтане (50 мл). К этому перемешиваемому раствору добавляли раствор (118) (2,513 г, 7 ммоль) в 1,2-дихлорэтане (50 мл), а затем добавляли дымящийся SnCl4 (0,94 мл; 8,05 ммоль; 1,15 экв.) при 0-5oС (баня с тающим льдом). Реакционную смесь 16 часов перемешивали при комнатной температуре. Реакционную смесь осторожно гасили насыщенным раствором NаНСО3 (50 мл) и разводили в CH2Cl2 (200 мл). Эту смесь фильтровали через слой целита (5 г) и промывали, используя CH2Cl2 (100 мл). Органический слой фильтрата отделяли и водный слой экстрагировали, используя CH2Cl2 (2•100 мл). Объединенный органический слой промывали водой (2•300 мл) и соляным раствором (500 мл), сушили (Na2SO4) и упаривали. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя гексан/этилацетат (85/15-70/30-50/50-30/70-0/100) в качестве элюента, с получением целевого продукта (135) (0,5 г; 17%). 1H-ЯМР (300 МГц, СDСl3): δ 2.01-2.15 (m, 12H), 4.12-4.48 (m, 3H, H4' & Н5'), 5.47 (m, 1H, Н3'), 5.57 (m, 0.2H, H2', минорный ротамер min), 5.64 (m, 0.8H, H2', основной ротамер maj), 6.41 (d, 0.2H, J=5.4 Hz, H1, min), 6.52 (d, 0.8H, J=7.2 Hz, Н1', maj), 7.38 (d, 0.6H, J=2.1 Hz, C5H, maj), 7.54 (s, 0.4H, C5H, min), 9.22 (bs, 0.6H, NH, exchangeable, maj), 9.46 (bs, 0.4, NH, exchangeable, min).
ПРИМЕР 109
1-(2', 3', 5' Триацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-6-карбометоксиурацил (136)
Триацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-6-карбометоксиурацил (136)
Суспензию 6-карбометоксиурацила (1,7 г; 10 ммоль) и сульфата аммония (170 мг) в гексаметилдисилазане (25 мл) 2 часа грели при температуре образования флегмы в атмосфере N2. Эту реакционную смесь упаривали досуха и остаток суспензировали в 1,2-дихлорэтане (50 мл). К этому перемешиваемому раствору добавляли раствор (118) (2,513 г, 7 ммоль) в 1,2-дихлорэтане (50 мл), а затем дымящийся SnCl4 (1,17 мл; 10 ммоль; 1,42 экв.) при 0-5oС (баня с тающим льдом). Реакционную смесь 16 часов перемешивали при комнатной температуре. Реакционную смесь осторожно гасили насыщенным раствором NаНСО3 (50 мл) и разводили в CH2Cl2 (200 мл). Эту смесь фильтровали через слой целита (5 г) и промывали, используя СН2Сl2 (100 мл). Органический слой фильтрата отделяли и водный слой экстрагировали, используя CH2Cl2 (2•100 мл). Объединенный органический слой промывали водой (2•300 мл) и соляным раствором (500 мл), сушили (Nа2SО4) и упаривали. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя гексан/этилацетат (95/5-80/20-70/30-50/50) в качестве элюента, с получением чистого целевого продукта (136) (2,2 г; 67%). 1H-ЯМР (300 МГц, СDСl3): δ 1.99-2.10 (m, 12H), 3.92 (s, 2H, ОСН3, основной ротамер maj), 3.98 (s, 1Н, ОСН3, минорный ротамер min), 4.00-4.07 (m, 1H, Н5'), 4.53 (m, 2H, H4' & Н5'), 5.48 (d, 0.8H, J=4.67 Hz, Н3', maj), 5.53 (d, 0.2H, J=4.94 Hz, Н3', min), 6.13 (m, 0.2H, H2', min), 6.20 (dd, 0.8H, J=4.67 & 7.96 Hz, H2', maj), 6.30 (s, 0.8H, H1', maj), 6.37 (s, 0.2H, H1', min), 6.58 (d, 0.2H, J=6.87 Hz, C5H, min), 6.68 (d, 0.8H, J= 7.99 Hz, C5H, maj), 8.70 (bs, 0.8H, NH, exchangeable, maj), 8.89 (bs, 0.2H, NH, exchangeable, min).
ПРИМЕР 110
1-(2', 3', 5' Триацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-5-фторурацил (137)
Триацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-5-фторурацил (137)
Суспензию 5-фторурацила (1,3 г; 10 ммоль) и сульфата аммония (130 мг) в гексаметилдисилазане (25 мл) 4 часа грели при температуре образования флегмы в атмосфере N2. Реакционную смесь упаривали досуха и остаток суспензировали в 1,2-дихлорэтане (50 мл). Затем этот раствор обрабатывали раствором (118) (2,513 г, 7 ммоль) в 1,2-дихлорэтане (50 мл) и затем дымящимся SnСl4 (1.17 г; 10 ммоль; 1,42 экв. ). Реакционную смесь осторожно гасили насыщенным раствором NаНСО3 (50 мл) и разводили в CH2Cl2 (200 мл). Эту смесь фильтровали через слой целита (5 г). Органический слой фильтрата отделяли и водный слой экстрагировали, используя СН2Сl2 (2•100 мл). Объединенный органический слой промывали водой (2•300 мл) и соляным раствором (500 мл), сушили (Na2SO4) и упаривали. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя СНCl3/ацетон (80/20) в качестве элюента, с получением чистого продукта (137) (1г, 33,30%). 1H-ЯМР (300 МГц, СDСl3): δ 2.02-2.21 (m, 12H), 4.10 (m, 0.5H, Н5'), 4.43-4.45 (m, 2.5H, H4' & Н5'), 5.30 (m, 0.5H, Н3'), 5.43-5.54 (m, 1.5H, H2' & Н3'), 6.09 (t, 0.5H, J=6.32 & 4.94 Hz, H1'), 6.28 (d, 0,5H, J=4.94 Hz, H1'), 7.48 (d, 0.5H, J=5.77 Hz, C6H), 7.95 (d, 0.5H, J= 5.77 Hz, С6Н), 9.19 (bs, 0.5H, NH, exchangeable), 9.34 (bs, 0.5H, NH, exchangeable).
ПРИМЕР 111
1-(2', 3', 5' Триацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-5-фторцитозин (138)
Триацетил-4'-дезокси-4'-ацетамидо-β-L-рибофуранозил)-5-фторцитозин (138)
Суспензию 5-фторцитозина (1,29 г; 10 ммоль; 1,42 экв.) и сульфата аммония (322 мг) в гексаметилдисилазане (40 мл) 4 часа грели при температуре образования флегмы в атмосфере N2. Летучие вещества выпаривали и остаток суспензировали в 1,2-дихлорэтане (50 мл). К этому перемешиваемому раствору добавляли раствор (118) (2,513 г; 7 ммоль) в дихлорэтане (50 мл) и затем дымящийся SnCl4 (1,17 мл; 10 ммоль; 1,42 экв.) при 0-5oС (баня с тающим льдом). Эту реакционную смесь 16 часов перемешивали при комнатной температуре. Реакционную смесь осторожно гасили насыщенным раствором Na2CO3 (50 мл) и разводили в CH2Cl2 (200 мл). Эту смесь фильтровали через слой целита (5 г). Органический слой фильтрата отделяли и водный слой экстрагировали, используя CH2Cl2 (2•100 мл). Объединенный органический слой промывали водой (2•300 мл) и соляным раствором (500 мл), сушили (Na2SO4) и упаривали. Неочищенный остаток чистили быстрой хроматографией на силикагеле, используя СНCl3/ацетон (80/20) в качестве элюента, с получением чистого продукта (137) (1г; 33,30%). 1H-ЯМР (300 МГц, СDСl3): δ 1.98-2.18 (m, 12H), 4.08 (m, 0.5H, Н5'), 4.37-4.61 (m, 2.5H, H4' & Н5'), 5.28 (m, 1H, Н3'), 5.44 (t, 0.8H, J=4.67 Hz, H2', основной ротамер maj), 5.55 (d, 0.2H, J=4.39 Hz, Н2', минорный ротамер min), 5.76 (bs, 1H, NH2, exchangeable), 6.27 (bt, 0.23H, H1', min), 6.37 (d, 0.77H, J=3.57 Hz, Н1', maj), 7.49 (d, 0.23H, J=5.77 Hz, C6H, min), 7.79 (bs, 1H, NH2, exchangeable), 7.94 (d, 0.77H, J=6.23 Hz, C6H, maj).
ПРИМЕР 112
1-(4'-Дезокси-4'-ацетамидо-β-L-рибофуранозил)-6-азаурацил (139)
Раствор (135) (0,45 г; 1,09 ммоль) в насыщенном растворе аммиака в метаноле (10 мл) 16 часов перемешивали в стальном баллоне при комнатной температуре. Стальной баллон охлаждали и открывали и содержимое упаривали досуха. Остаток чистили быстрой хроматографией на силикагеле, используя СНCl3/МеОН (95/5-90/10-85/15) в качестве элюента, с получением целевого продукта (139) (0,18 г, 57,62%). 1H-ЯМР (300 МГц, DMSO-d6): δ 1.84 (s, 1.35Н, СОСH3, минорный ротамер min), 1.95 (s, 1.65H, СОСH3, основной ротамер maj), 3.46-3.85 (m, 3Н, H4' & Н5'), 4.01 (m, 1H, Н-3'), 4.22 (m, 1H, Н2'), 4.89 (m, 0.45H, ОН, min, exchangeable), 5.01 (m, 0.55H, ОН, maj, exchangeable), 5.15 (s, 0.45H, ОН, min, exchangeable), 5.28 (s, 0.55H, ОН, maj, exchangeable), 5.39 (d, 0.45H, J= 6.86 Hz, OH, min, exchangeable), 5.50 (d, 0.55H, J= 5.7 Hz, OH, maj, exchangeable), 6.0 (d, 0.46H, J=7.15 Hz, H1', min), 6.05 (d, 0.55H, J=5.5 Hz, H1', maj), 7.50 (s, 0.45H, С5Н, min), 7.61 (s, 0.55H, C5H, maj), 12.2 (bs. 1H, NH, exchangeable). Анализ. Рассчитано для C10H14N4O6: С 41,96; Н 4,93; N 19,57. Найдено: С 42,03; Н 5,11; N 19,64.
ПРИМЕР 113
1-(4'-Дезокси-4'-ацетамидо-β-L-рибофуранозил)-урацил-6-карбоксамид (140)
Раствор (136) (2 г, 4,26 ммоль) в насыщенном растворе аммиака в метаноле (20 мл) 16 часов перемешивали в стальном баллоне при комнатной температуре. Стальной баллон охлаждали и открывали и содержимое упаривали досуха. Полученный остаток чистили быстрой хроматографией на силикагеле, используя СНCl3/МеОН (95/5-90/10-85/15) в качестве элюента, с получением целевого продукта (140) (1 г, 71,49%). 1H-ЯМР (300 МГц, DMSO-d6): δ 1.70 (s, 1H, СОСH3, минорный ротамер min), 1.89 (s, 2H, СОСН3, основной ротамер maj), 3.97-3.44 (m, 4H, Н3', H4' & Н5'), 4.73 (m, 1H, Н2'), 6.26-6.08 (m, 2H, С5Н & H1'). Анализ. Рассчитано для C12H16N4O7: С 43,90; Н 4.91; N 17,07. Найдено: С 43,99; Н 5,06; N 17,21.
ПРИМЕР 114
1-(4'-Дезокси-4'-ацетамидо-β-L-рибофуранозил)-5-фторурацил (141)
Раствор (137) (1 г; 2,33 ммоль) в насыщенном растворе аммиака в метаноле (20 мл) 16 часов перемешивали в стальном баллоне при комнатной температуре. Стальной баллон охлаждали до 0oС и открывали и содержимое упаривали досуха. Остаток чистили быстрой хроматографией на силикагеле, используя СНCl3/МеОН (95/5-90/10-85/15) в качестве элюента, с получением целевого продукта (141) (0,6 г, 84,95%). 1H-ЯМР (300 МГц, DMSO-d6): δ 1.72 (s, 1.35Н, СОСН3, минорный ротамер min), 1.83 (s, 1.65H, СОСН3, основной ротамер maj), 3.35-4.18 (m, 5H, Н2', Н3', Н4' & Н5'), 5.74 (d, 0.45H, J=5.77 Hz, H1', min), 5.84 (d, 0.55H, J=4.1 Hz, H1', maj), 8.25 (s, 0.45H, C6H, min), 8.53 (s, 0.55H, C6H, maj), 11.77 (bs, 1H, NH, exchangeable). Анализ. Рассчитано для С11Н14FN3О6: С 43,57; Н 4.65; N 13,86. Найдено: С43,40; Н 4,71; N 13,80.
ПРИМЕР 115
1-(4'-Дезокси-4'-ацетамидо-β-L-рибофуранозил)-5-фторцитозин (142)
Раствор (138) (1 г; 2,33 ммоль) в насыщенном растворе аммиака в метаноле (20 мл) 16 часов перемешивали в стальном баллоне при комнатной температуре. Стальной баллон охлаждали и открывали и содержимое упаривали досуха. Остаток чистили быстрой хроматографией на силикагеле, используя СНСl3/МеОН (95/5-90/10-85/15) в качестве элюента, с получением целевого продукта (142) (0,64 г, 90,70%). 1H-ЯМР (300 МГц, СD3ОD): δ 1.92 (s, 2H, COCH3, основной ротамер maj), 2.17 (s, 1H, СОСН3, минорный ротамер min), 3.75-3.93 (m, 2H, Н5'), 4.16-4.28 (m, 1.5H, Н3' & H4'), 4.49 (t, 0.5H, J=4.4 & 4.94 Hz, Н3'), 4.65 (s, 1H, H2'), 5.77 (d, 0.33H, J=5.22 Hz, Н1', min), 6.11 (dd, 0.66H, J=1.92 & 4.12 Hz, H1', maj), 8.19 (d, 0.33H, J=6.86 Hz, C6H, min), 8.66 (d, 0.66H, J=7.15 Hz, C6H, maj). Анализ. Рассчитано для C11H15FN4O5: С 43,71; Н 5,00; N 18,54. Найдено: С 43,77; Н 5,17; N18.79.
Понятно, что описанные выше воплощения всего лишь иллюстративны и что специалисты могут их модифицировать. Соответственно, это изобретение не должно считаться ограниченным раскрытыми здесь воплощениями, но должно быть ограничено только так, как это определено прилагаемой формулой изобретения.
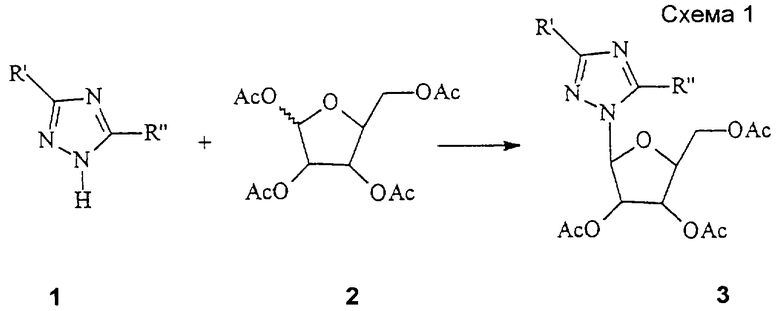
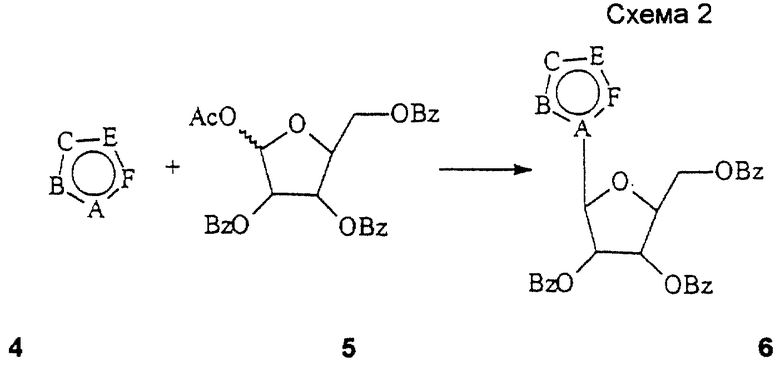
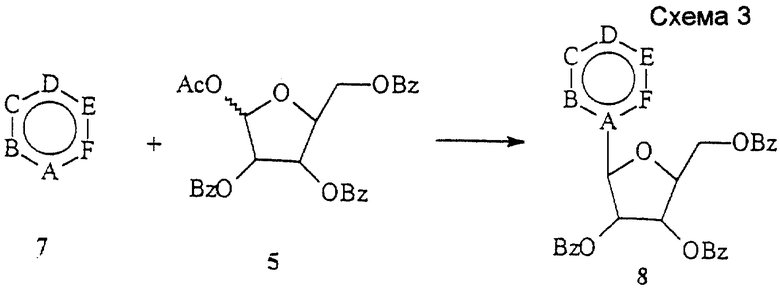

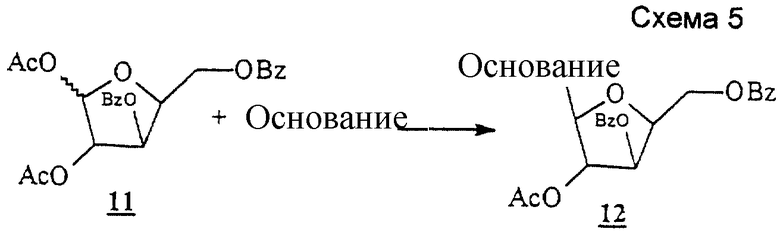
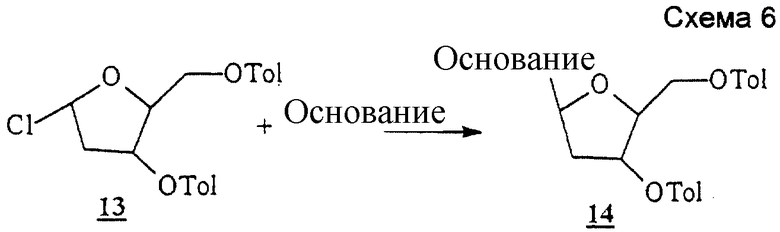
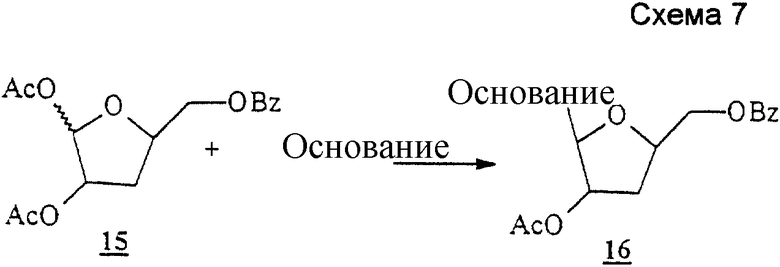
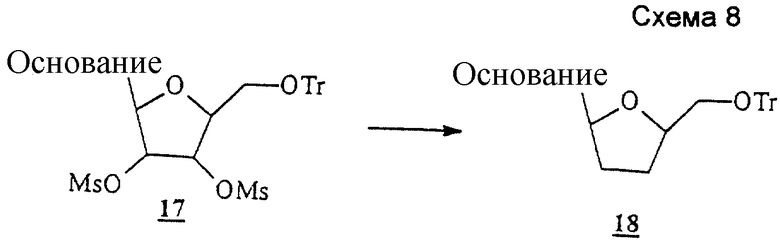
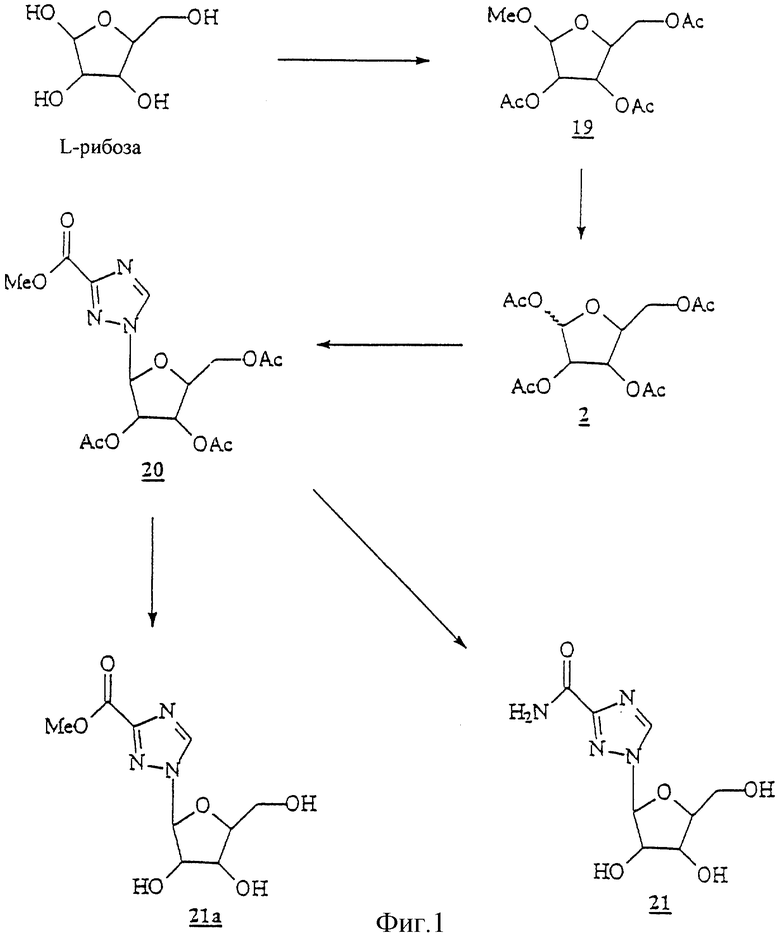
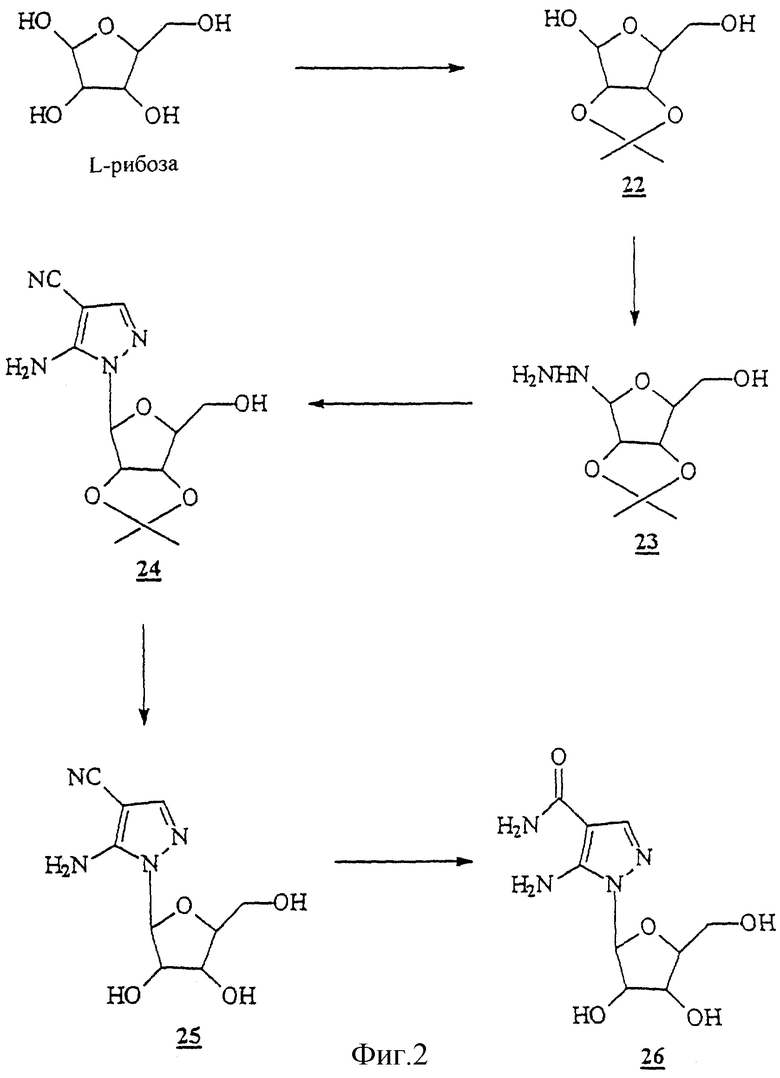
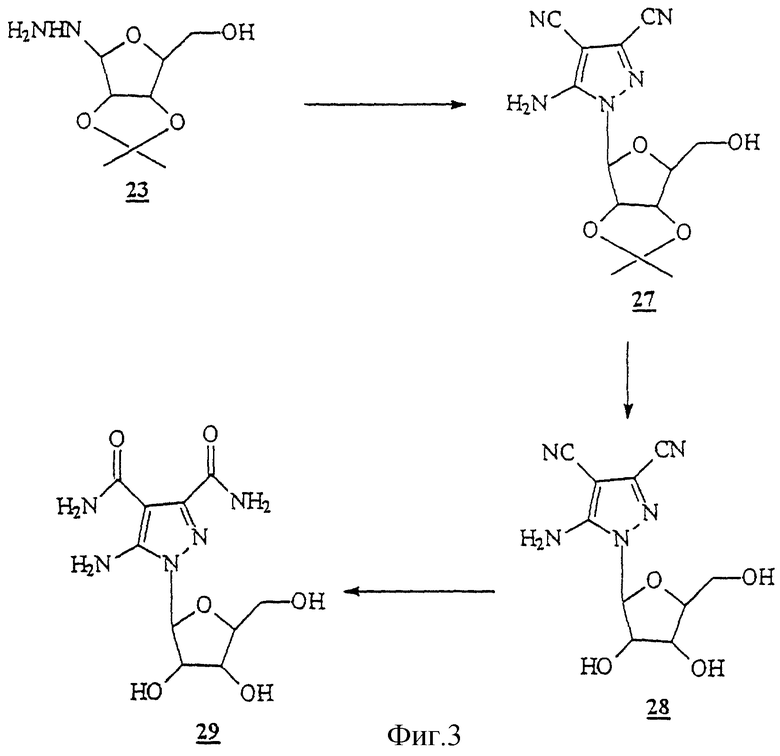
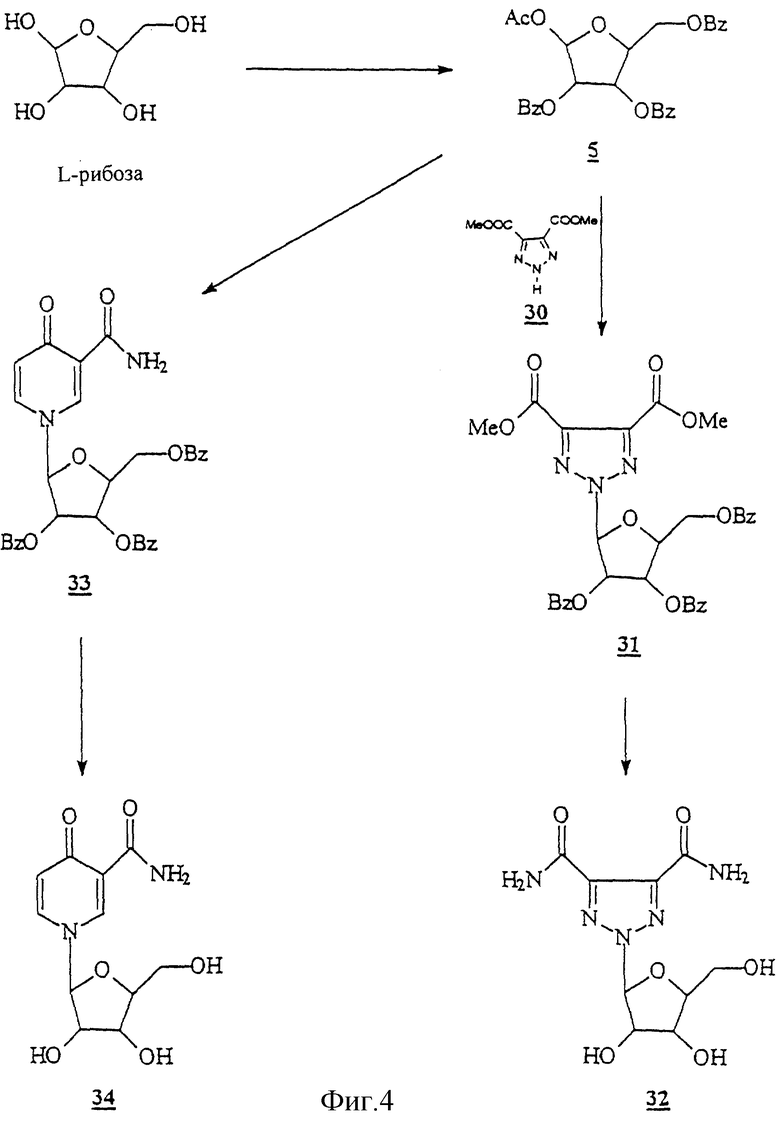
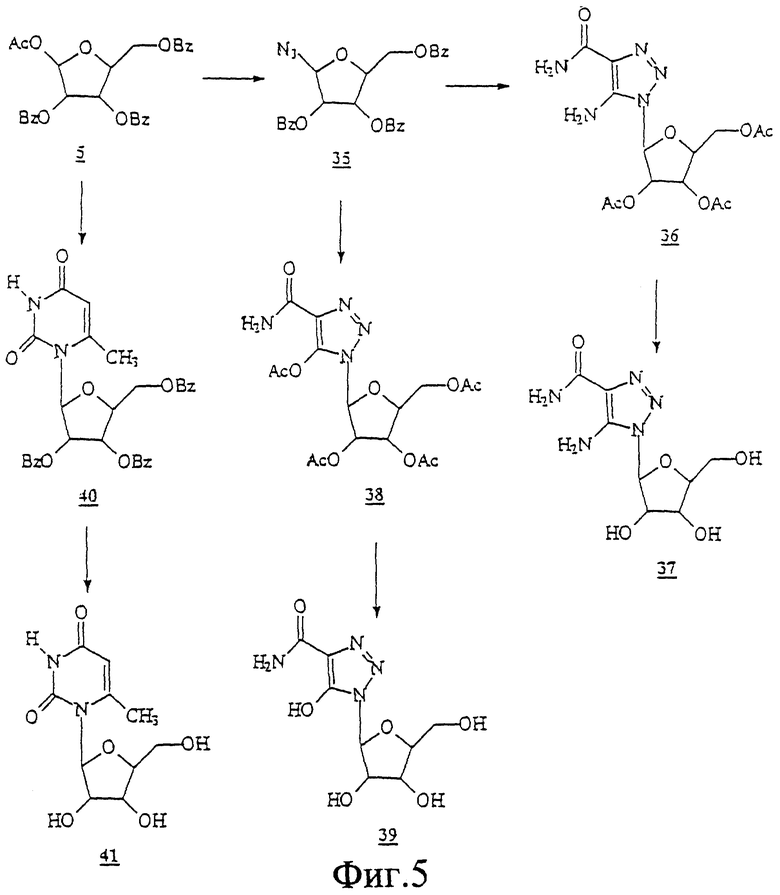
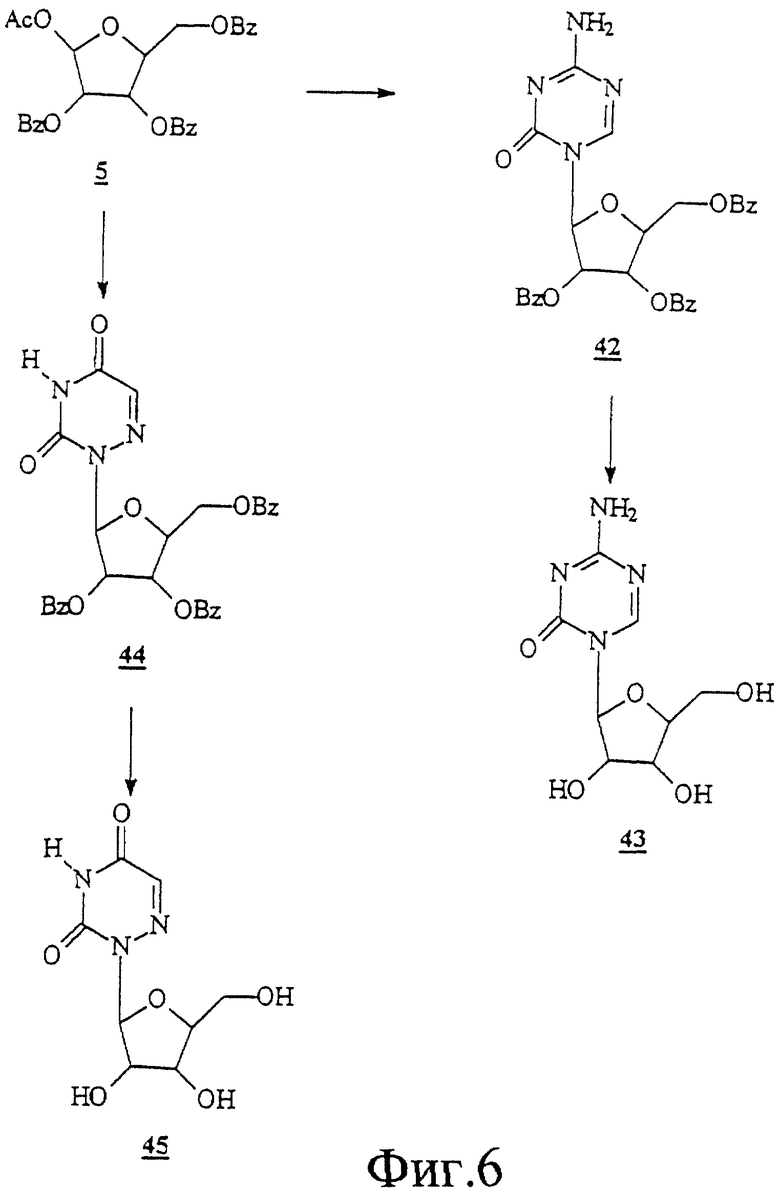
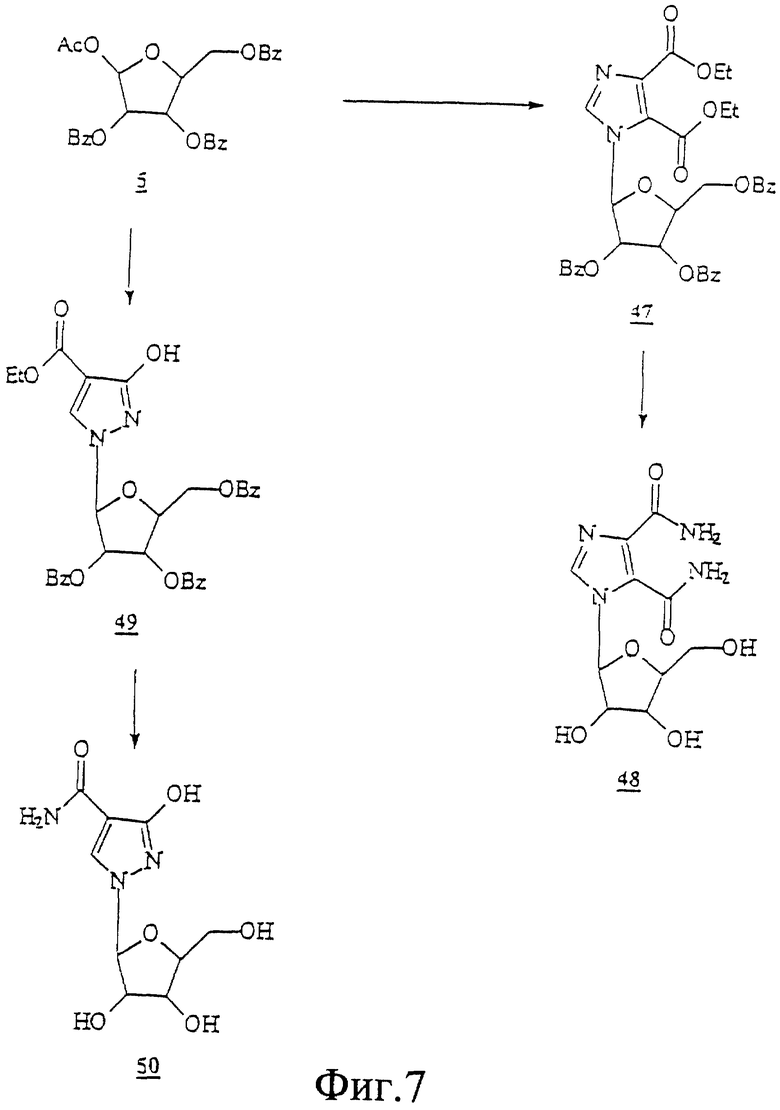
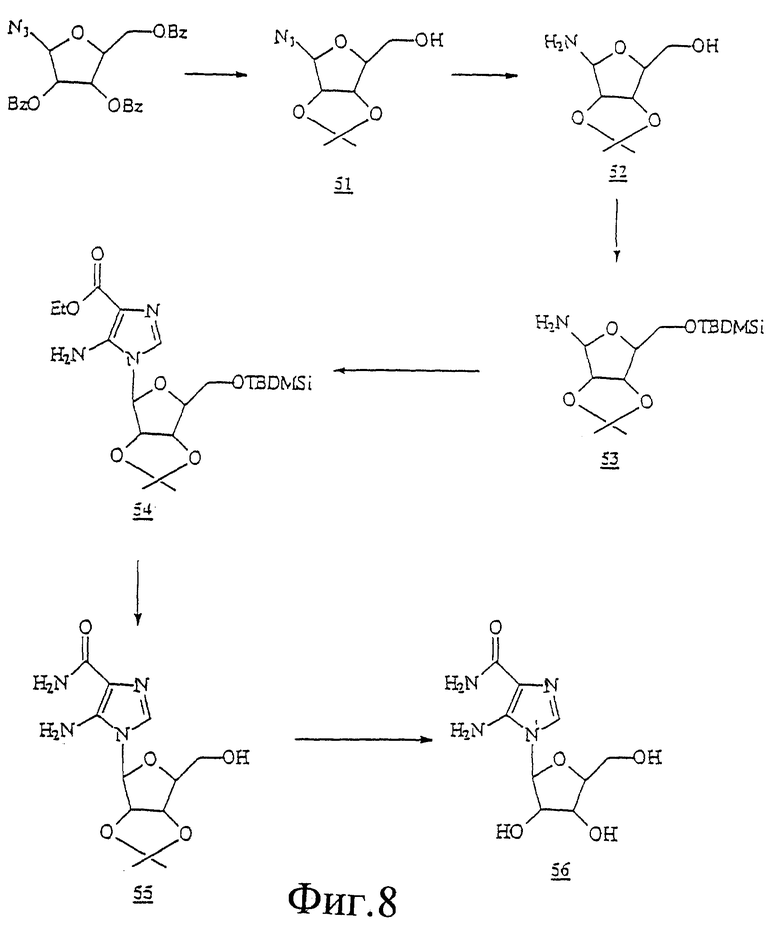
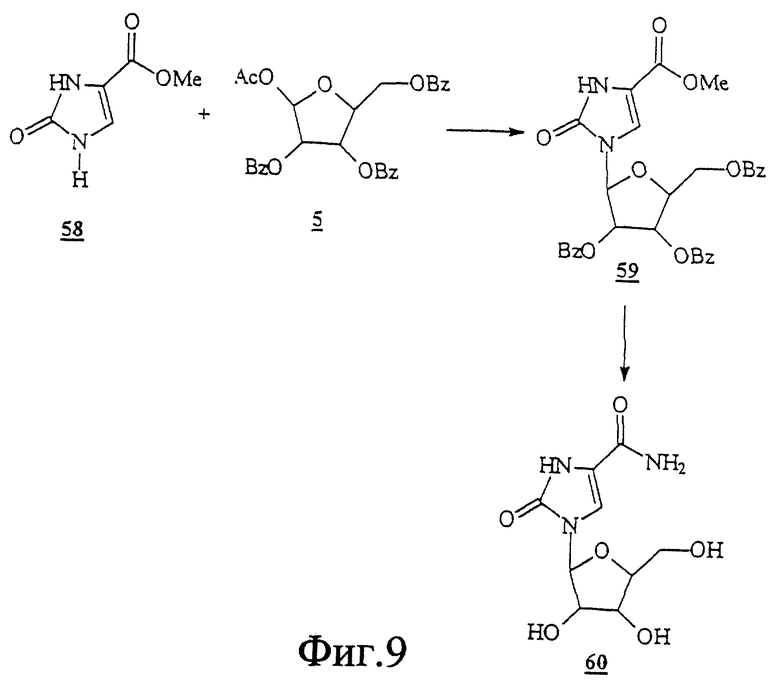
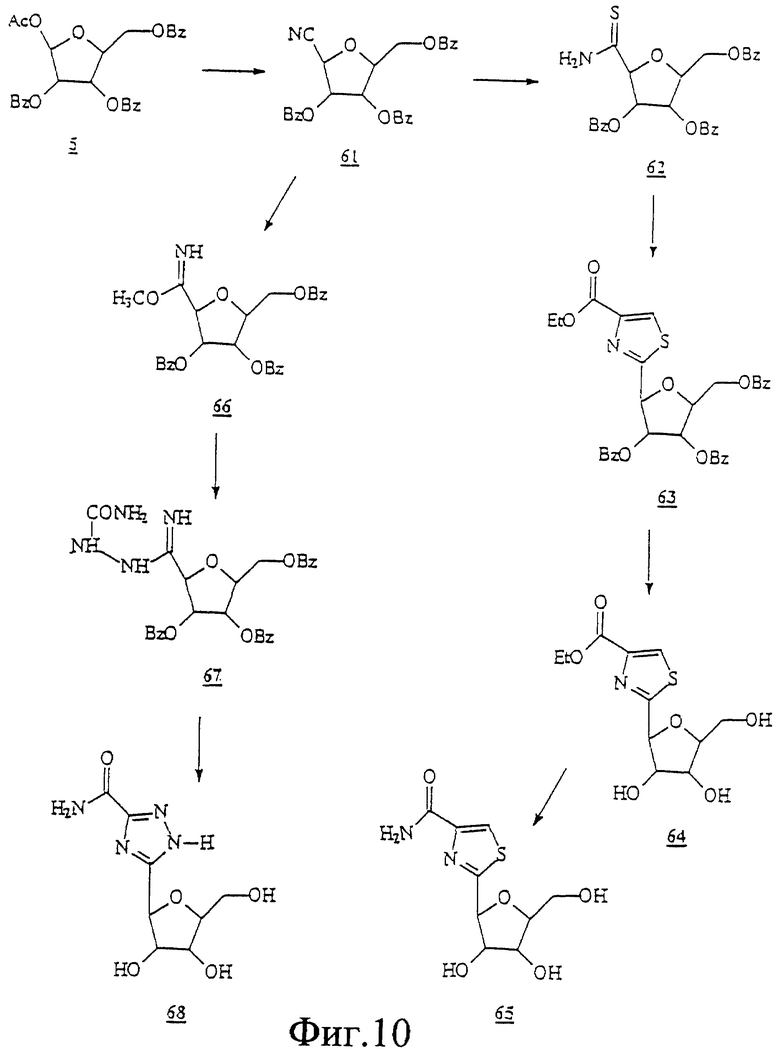
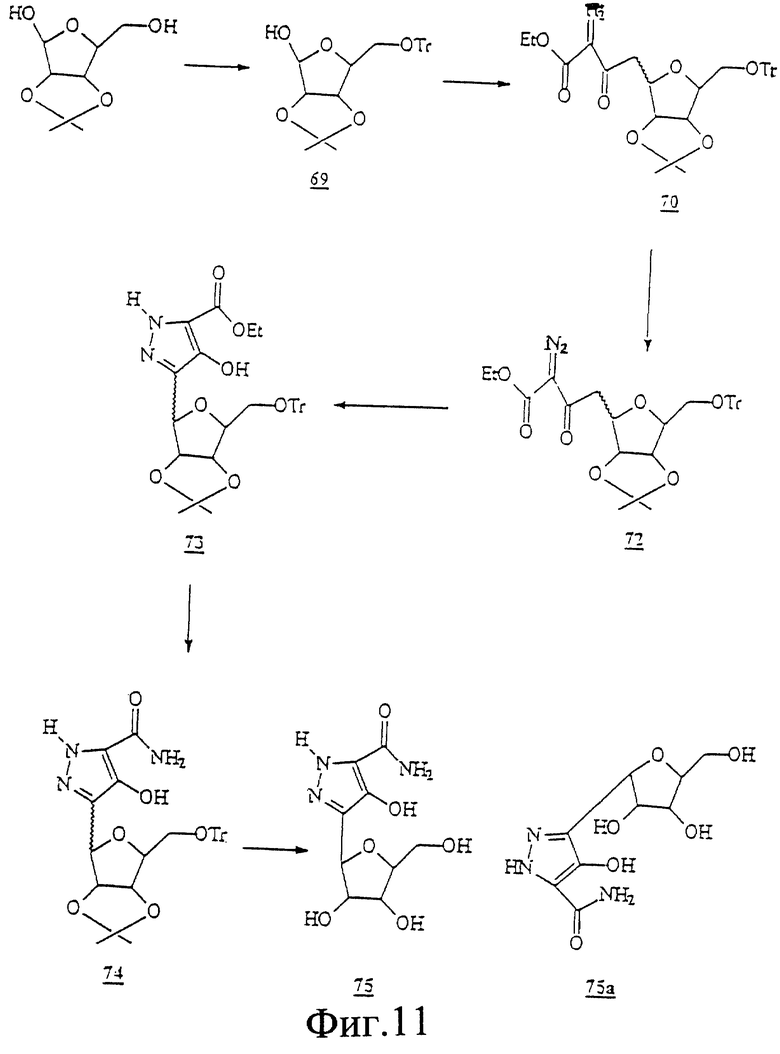
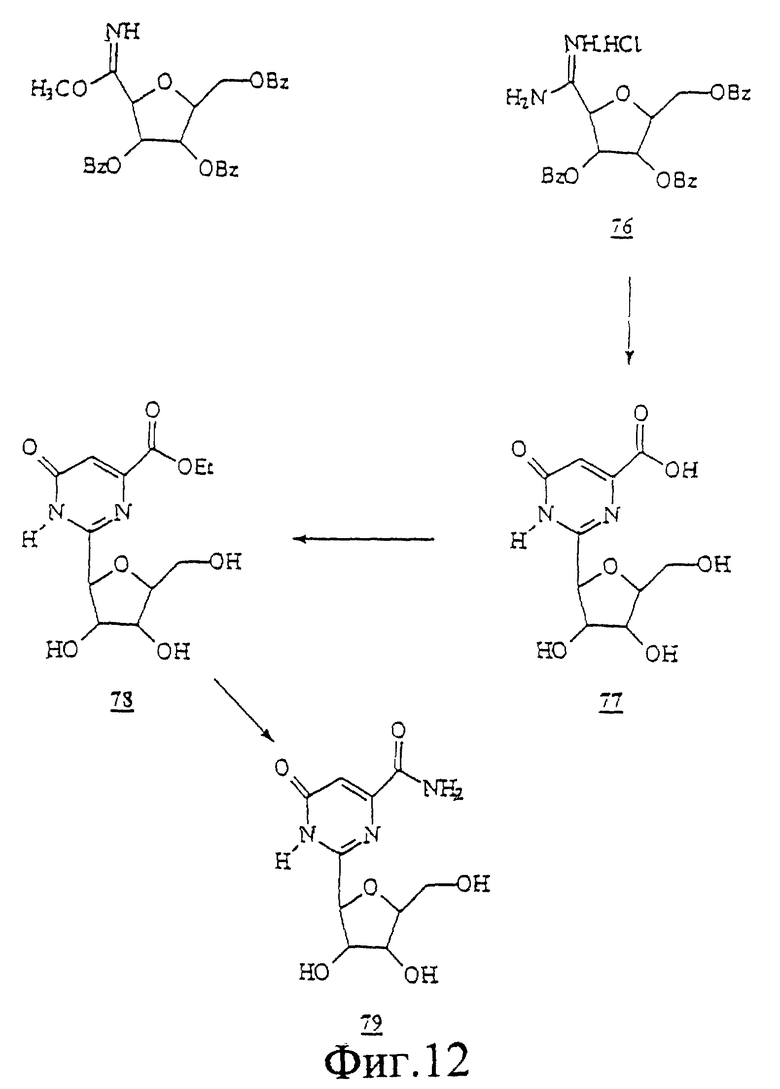
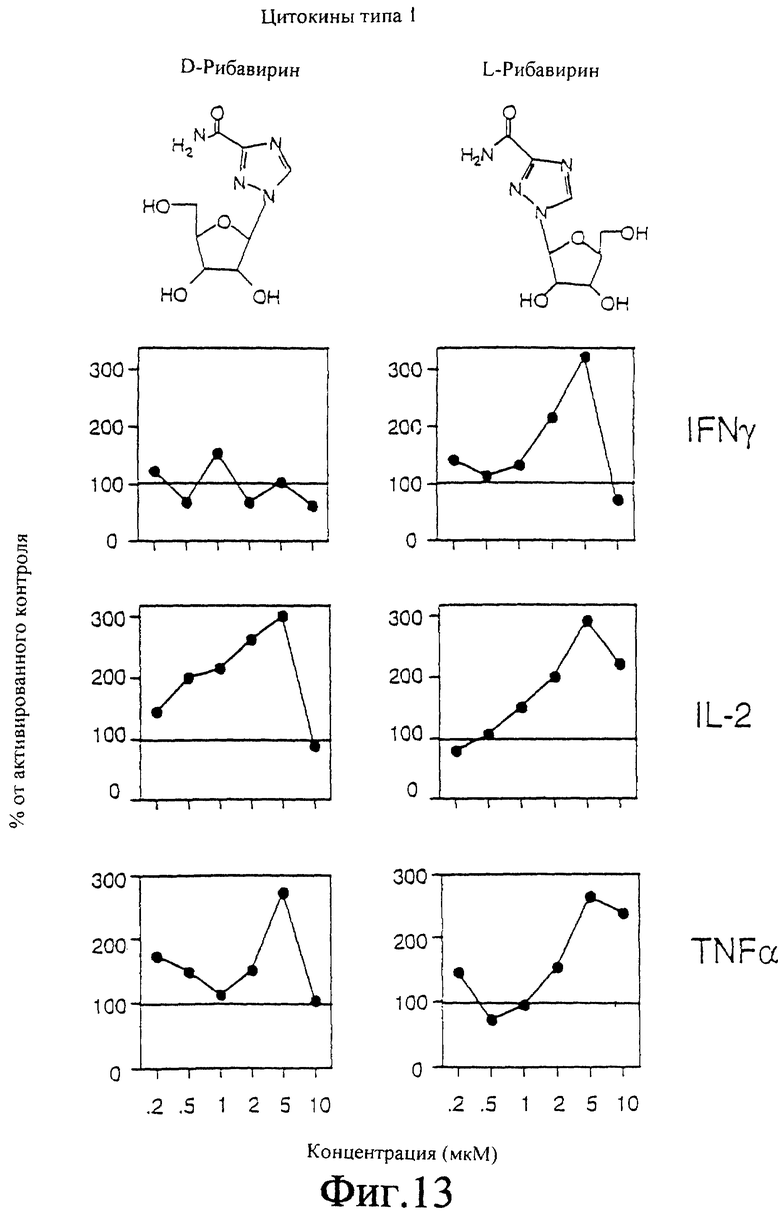
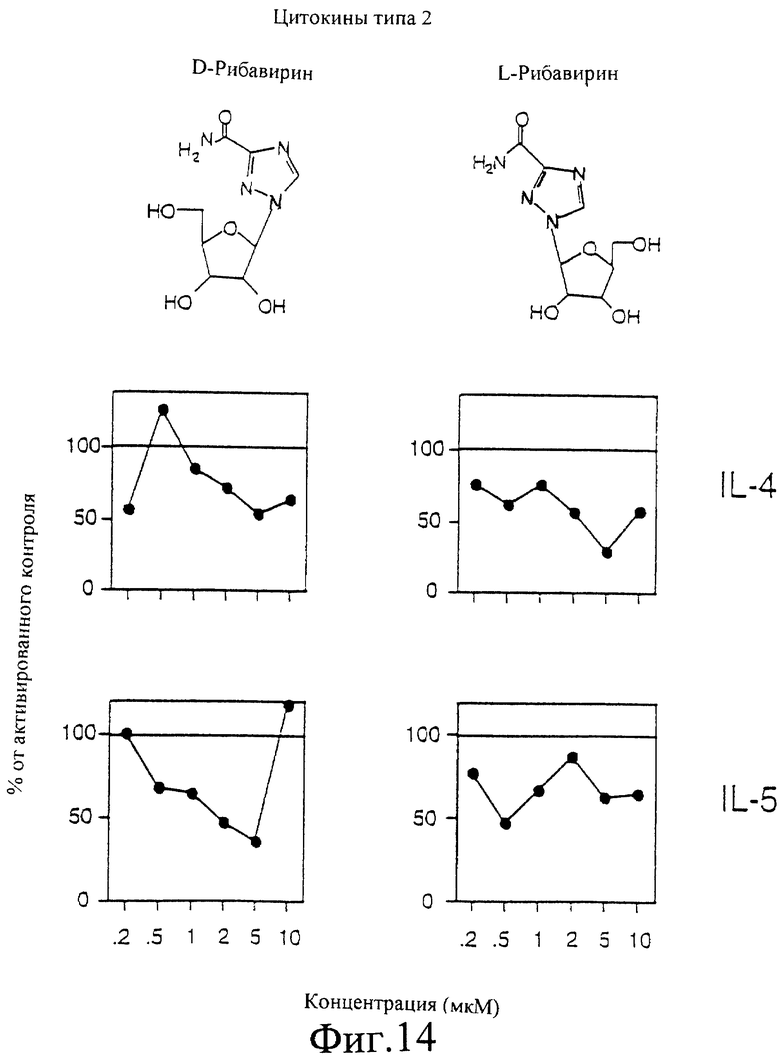
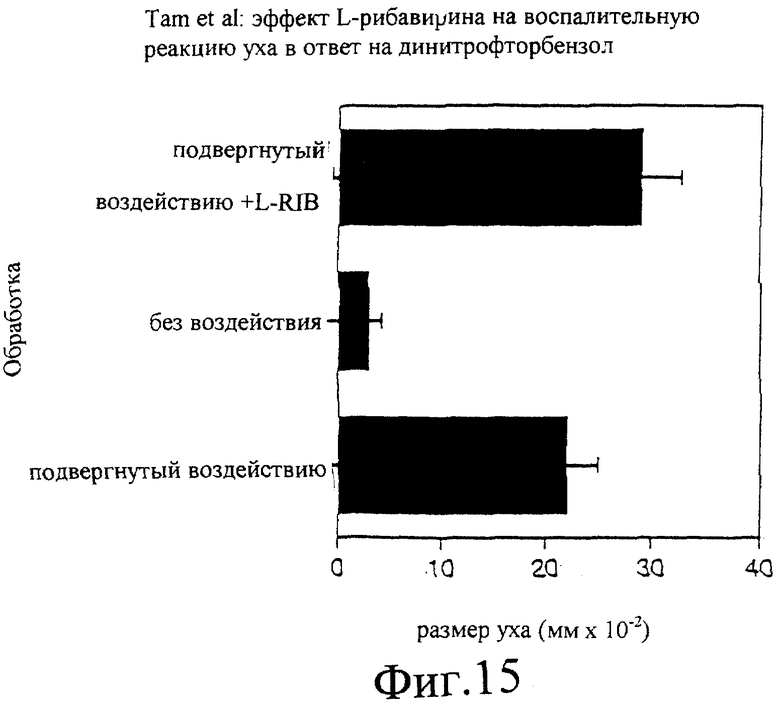

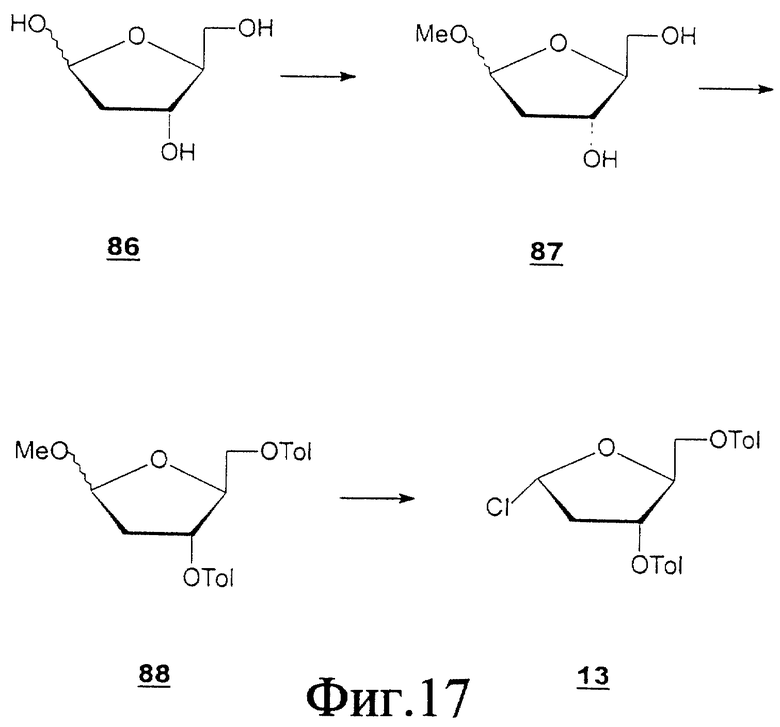
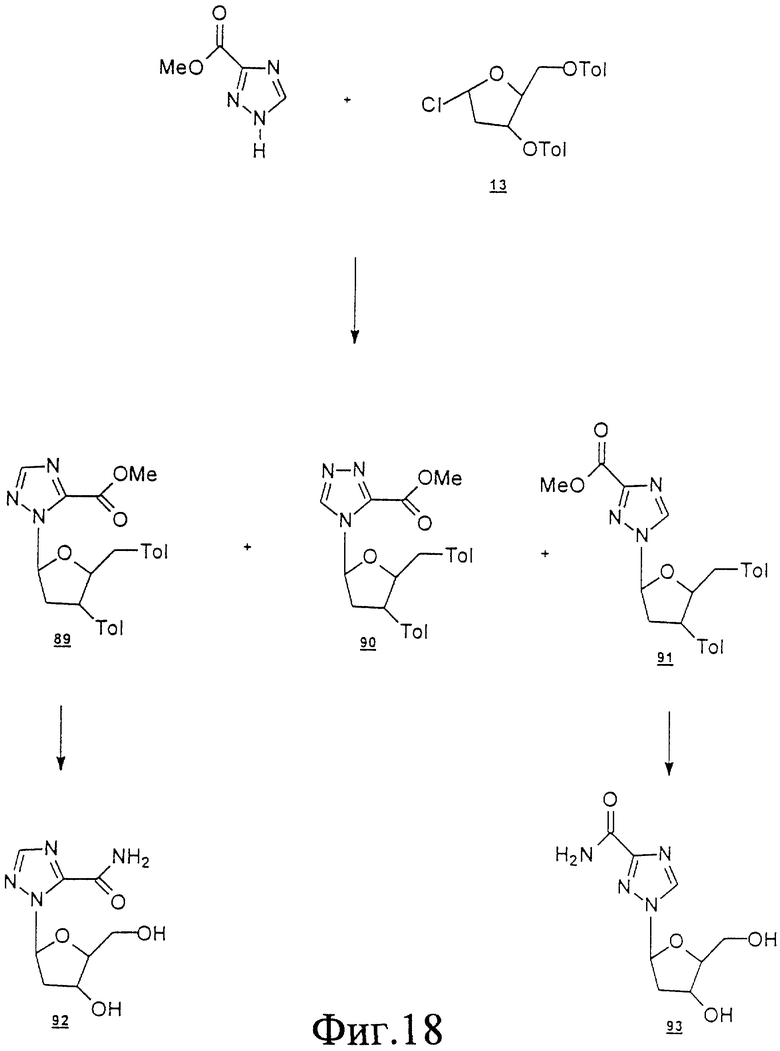
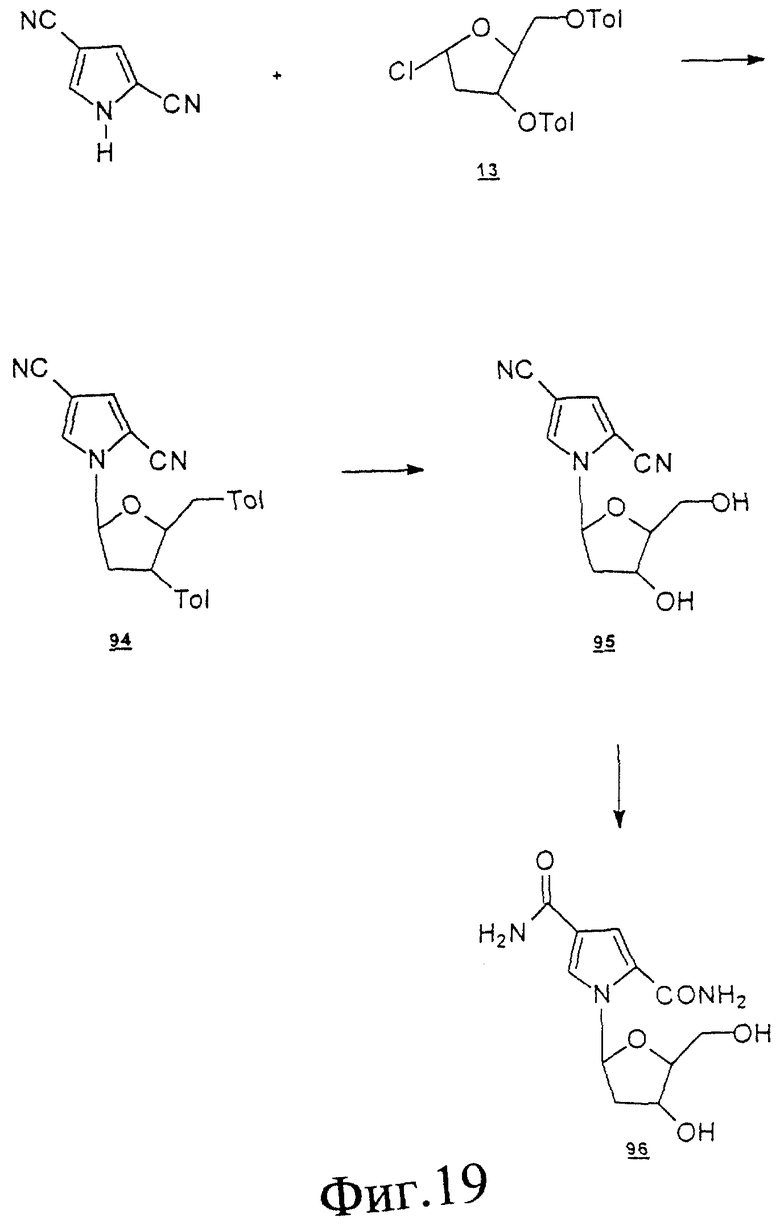
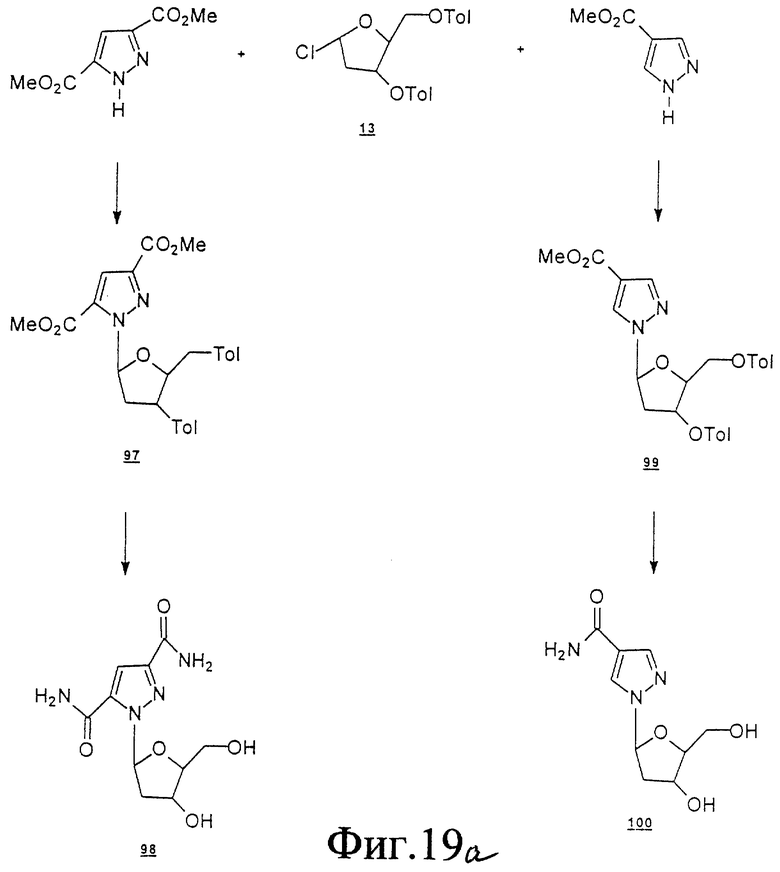
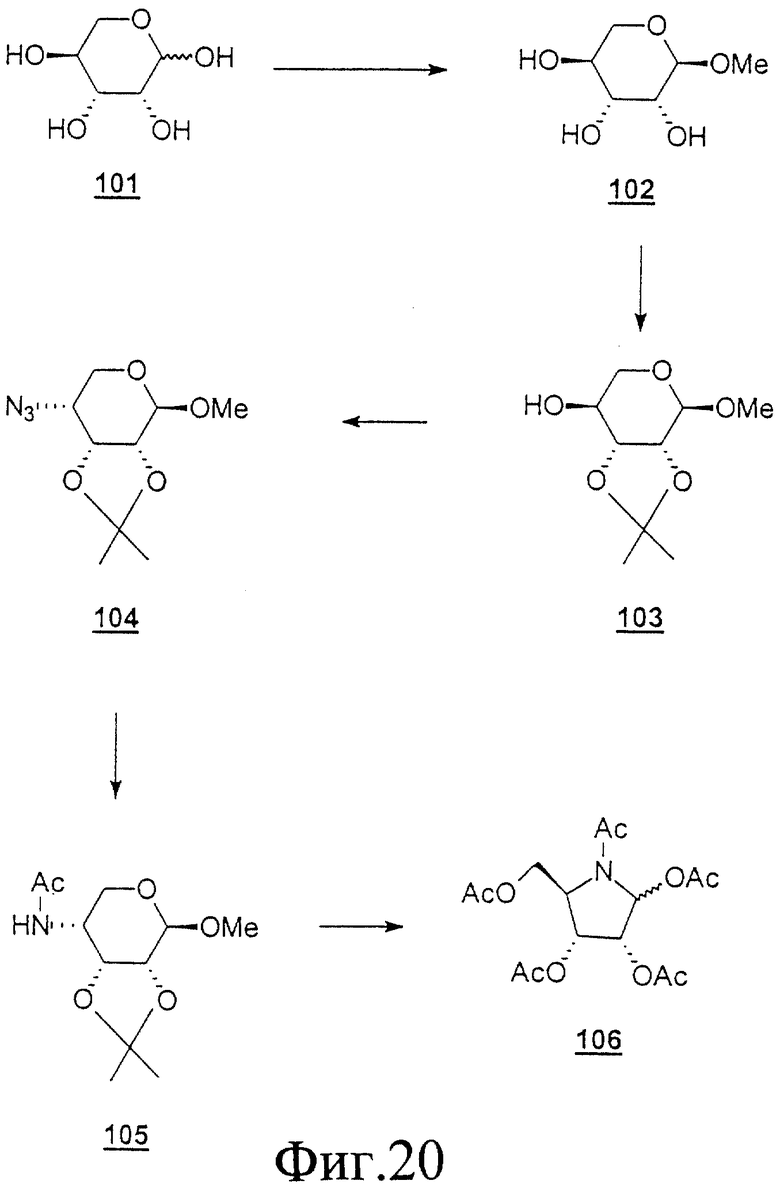
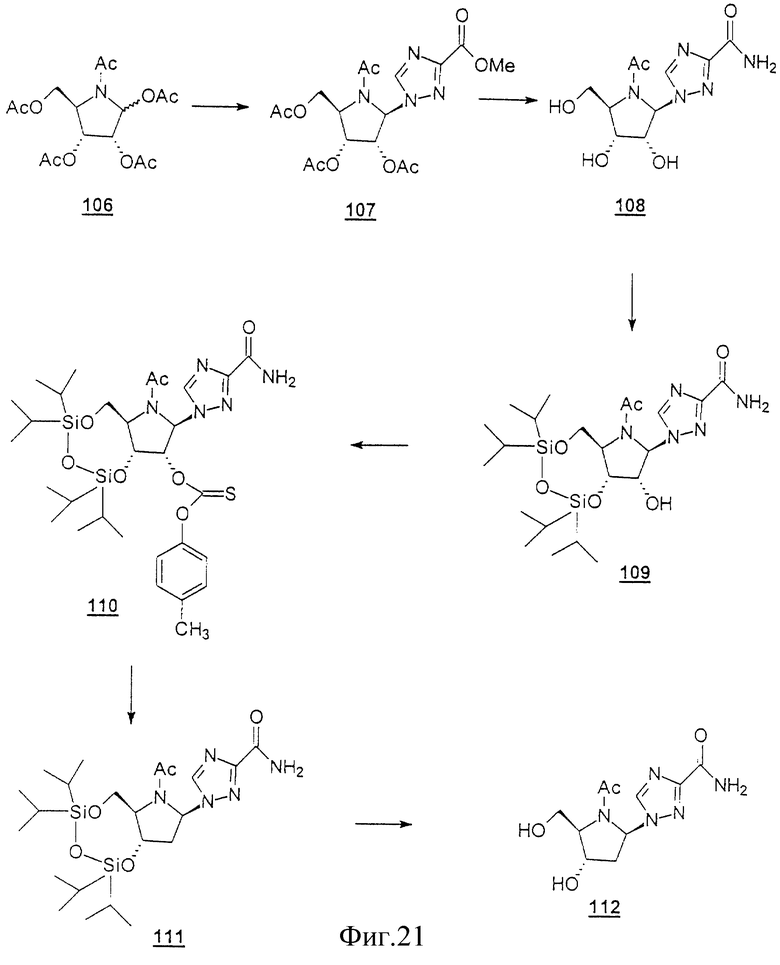
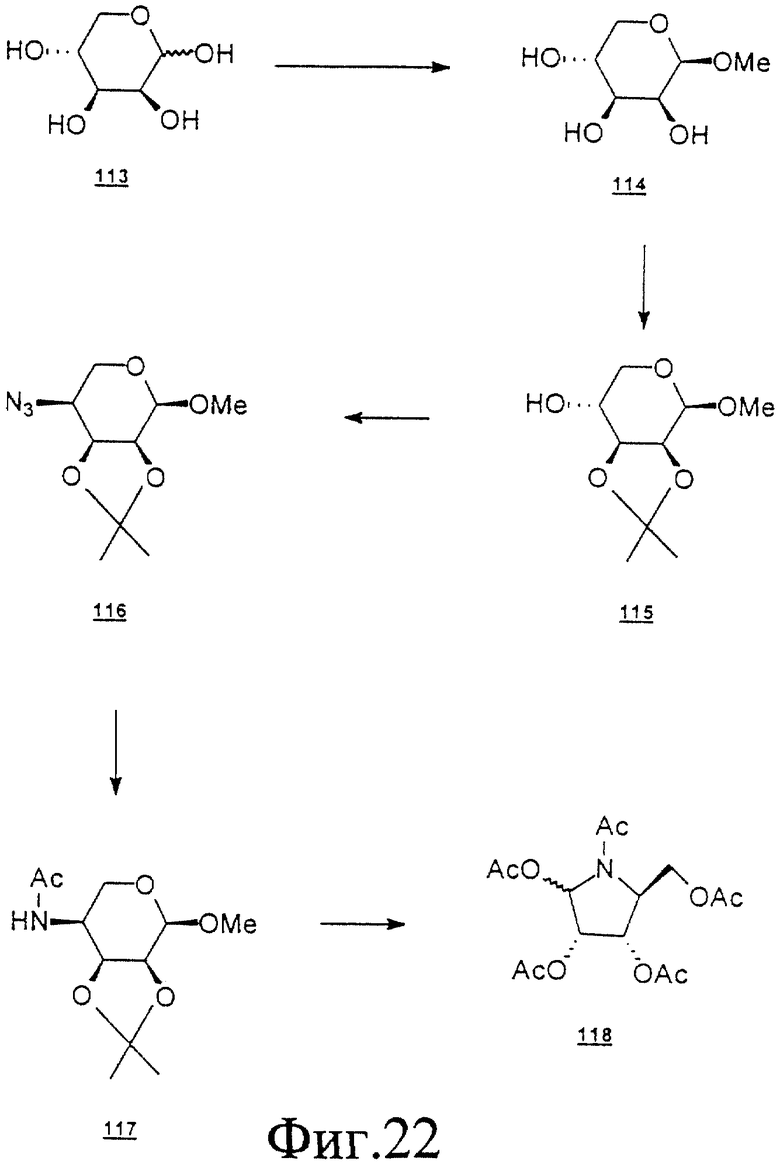
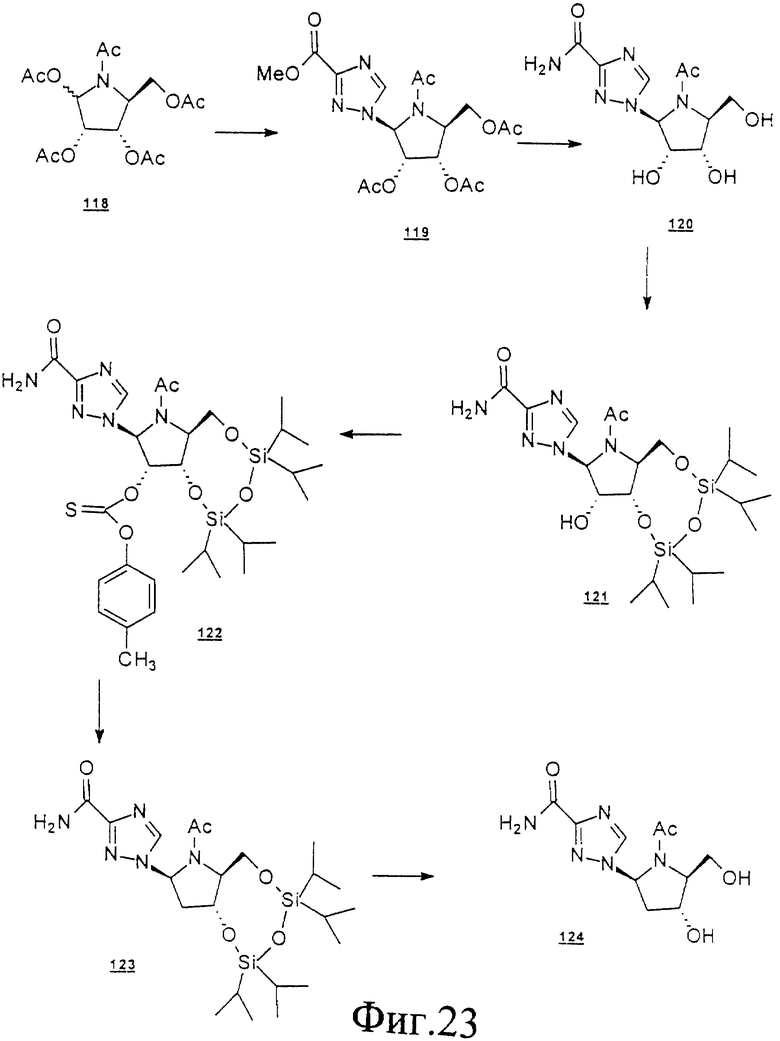
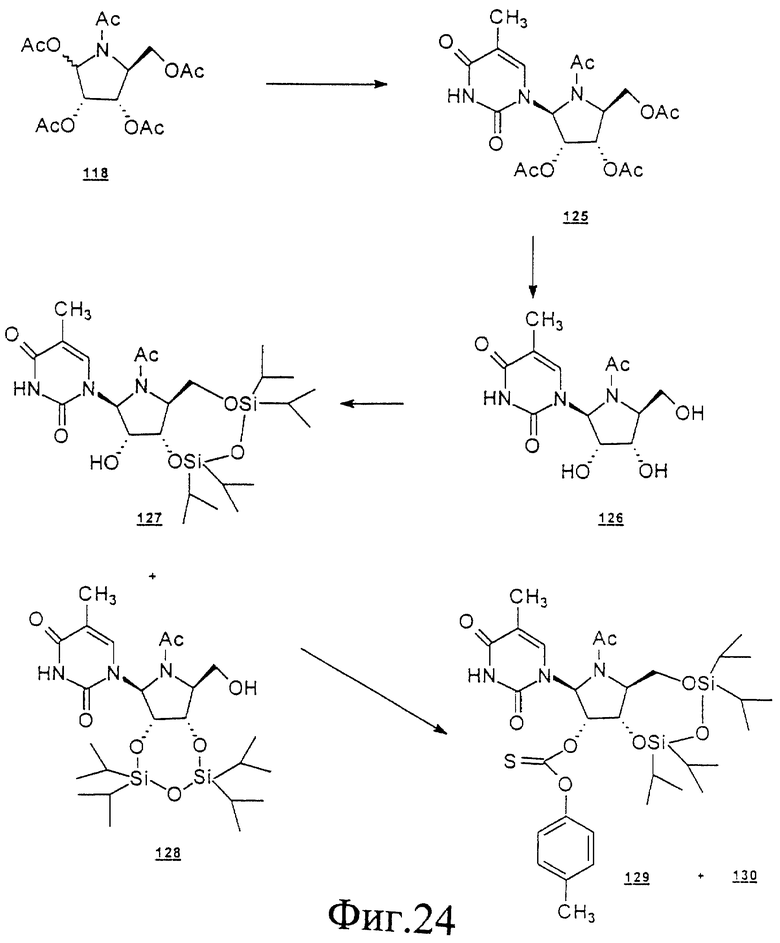
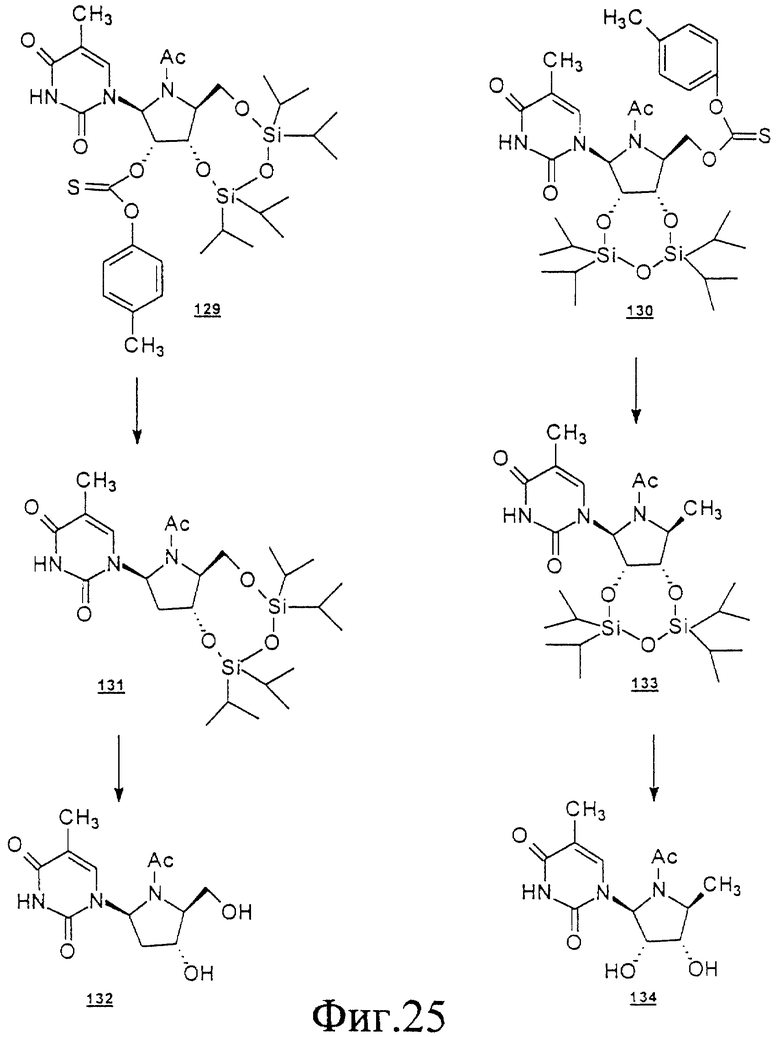
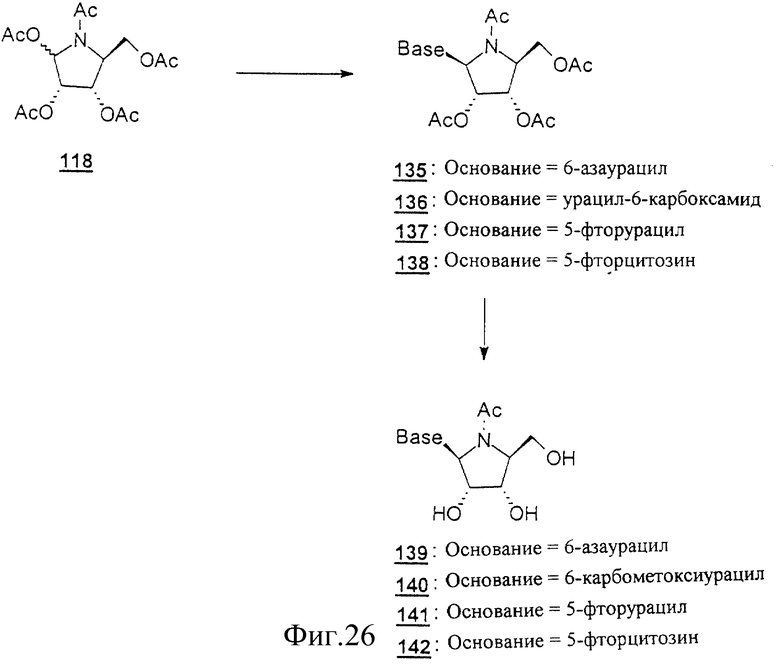
Изобретение относится к производному нуклеозида формулы I. Описана фармацевтическая композиция на его основе, обладающая иммуномодулирующей активностью. Также описаны способы лечения воспалительных состояний и противоположной модуляции Th1- и Th2-активностей у пациента путем введения ему соединения формулы I. 4 с. и 5 з.п.ф-лы, 27 ил.
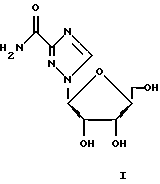
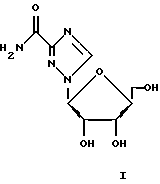
2. Фармацевтическая композиция, обладающая иммуномодулирующей активностью, включающая в себя соединение по п. 1 в смеси с по меньшей мере одним фармацевтически приемлемым носителем.
| US 5559101 А, 24.09.1996 | |||
| DE 19518216 А1, 11.04.1996 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| US 5041426 А, 20.10.1991 | |||
| US 5565438 А, 15.10.1996 | |||
| МИХАЙЛОВ С.Н | |||
| Достижения и перспективы направленного поиска антивирусных веществ в ряду нуклеозидов и их производных | |||
| Биоорг | |||
| химия, 1992, т | |||
| Способ использования делительного аппарата ровничных (чесальных) машин, предназначенных для мериносовой шерсти, с целью переработки на них грубых шерстей | 1921 |
|
SU18A1 |
| МЕХАНИЧЕСКАЯ ТРАМБОВКА ДЛЯ ИЗГОТОВЛЕНИЯ ИСКУССТВЕННЫХ ЖЕРНОВОВ | 1923 |
|
SU1033A1 |
