Область техники, к которой относится изобретение
Настоящее изобретение относится к новым производным морфина, фармацевтическим композициям, содержащим указанные новые производные морфина, и их применению для устранения боли и половой дисфункции.
Уровень техники
Морфин в настоящее время является лекарственным средством, выбранным для лечения острой боли любой интенсивности. Это опиоидное лекарственное средство применяют в примерно 80% случаев послеоперационной острой боли. Несмотря на высокую эффективность, применение морфина предполагает разнообразное побочное действие, специфическое для опиоидов, такое как угнетение дыхания, тошнота, рвота, задержка прохождения содержимого через кишечник, аддикция и допустимая доза (Minoru Nariata et al., 2001, Pharmacol et Ther., 89, 1-15).
Существуют три основных класса опиоидных рецепторов: мю (µ), каппа (κ) и дельта (δ), которые все принадлежат к семейству рецепторов, сопряженных с протеином G. В анализе вестерн-блоттингом, FRET (резонансный перенос энергии флуоресценции) и BRET (резонансный перенос энергии биолюминесценции) большое значение придается мономерам и димерам указанных трех типов рецепторов (George S.R. & al., J. Biol. Chem., 2000, 275: 26128-26135; Jordan B.A. & al., Nature, 1999, 399: 697-700; Gomes I. & al., J. Neurosci., 2000, 20: RC110). Димеры могут представлять собой или гомодимеры или гетеродимеры. Такая олигомеризация мешает физиологической роли опиоидных рецепторов и составляет новый терапевтический подход высокого потенциала.
На самом деле, опиоидные производные, синтезированные в димерной форме, показывают лучшую аффинность и более эффективны, чем мономеры в случае рецепторов µ, δ и κ (Hazum & al., Biochem. Biophys. Res. Comm., 1982, 104: 347-353). Их аналгетическая активность после внутривенного введения более высокая и продолжительность их действия больше (WO 2005/063263). Эта последняя характеристика позволяет снижать число введений при сохранении максимальной аналгетической активности по сравнению с мономерами и другими опиоидами, такими как морфин, которые обнаруживают меньшую продолжительность активности.
Более того, многочисленные исследования (Gomes I. & al., J. Neurosci., 2000, 20: RC110) показали солокализацию рецепторов µ и δ с одной стороны и рецепторов δ и κ с другой стороны, что делает физиологически возможным существование гетеродимерных рецепторов. Такая гипотеза подтверждена анализом методом FRET, иммунопреципитацией и профилями связывания (Rios & al., Pharm. & Ther., 2001, 92: 71-87).
С фармакологической точки зрения взаимодействие молекулы с опиоидными рецепторами приводит к более или менее значительной аналгетической активности, а также побочному действию.
Например, связывание рецепторов µ в значительной мере ответственно за аналгетическую активность молекулы. Существует два подтипа рецепторов µ: подтип µ1 с сильной аффинностью и плохой функциональной активностью и подтип µ2 с плохой аффинностью и сильной функциональной активностью (Pasternak & Wood, 1986, Life Sci., 38: 1869-1898). Взаимодействие с рецепторами µ1 приводит к аналгетической супраспинальной реакции, объединенной с уменьшением оборота ацетилхолина, в то время как взаимодействие рецепторов µ1 приводит к аналгезирующему спинальному действию, обычно известному в случае генерации угнетения дыхания и задержки прохождения содержимого через кишечник.
Рецепторы κ и δ играют особую роль в перильстатике кишечника и аналгетической активности. Более того, их ингибирование вносит вклад в ослабление явления зависимости и угнетения дыхания (Rothman et al., 2000, J. Subst. Abuse Treat, 19: 277-281; Shook et al., 1990, Am. Rev. Respir. Dis, 142: 895-909; J. Neurosci, 2005, Mar., 23; 25(12): 3229-33; J. Pharmacol. Exp.Ther., 2001, May, 297(2): 597-605; J. Pharmacol. Exp. Ther., 1998, Dec, 287(3): 815-23).
Указанные рецепторы присутствуют в центральной нервной системе, особенно на уровне заднего рога спинного мозга, а также на периферическом уровне (Stein et al., 1995, Ann. Med., 27(2): 219-21; Janso, Stein, Curr. Opin. Pharmacol., 2001, 1(1): 62-65).
Наиболее идентифицированные виды побочного действия, такие как угнетение дыхания, тошнота, зависимость и аддикция, являются основными видами побочного действия. Способом ограничения нежелательных эффектов при сохранении аналгетической активности является создание опиоидов, которые наряду с тем, что взаимодействуют с периферической системой, постепенно диффундируют в головной мозг (С.Stein, 1990, J. of Pain and Symptom Management, 6(3): 119-124; Zajaczowska R., Reg. Anesth. Pain Med., 2004, 29(5): 424-429; Likar et al., 1998, Pain, 76(1-2): 145-50; Tokuyama et al., Life Sci., 1998, 62(17-18): 1677-81; Junien, Aliment Pharmacol. Ther., 1995, 9(2): 117-26; Dehave-Hudkins et al., J. Pharmacol. Exp. Ther., 1999, 289(1): 494-502; Stein et al., N. Engl. J. Med., 1991, 325: 1123-1126). Действительно, представляется, что активация опиоидных рецепторов центральной нервной системы остается наиболее эффективным способом получения обезболивания, сравнимого с обезболиванием, получаемым от морфина.
Созданы и синтезированы некоторые двухвалентные лиганды для того, чтобы взаимодействовать с гетеродимерами µ/δ. В этом отношении Daniels et al. исследовали лиганды, которые состояли из агониста рецептора µ оксиморфона и антагониста рецептора δ налтриндола (Daniels et al., PNAS, 2005, 102(5): 19208, WO 2006/073396). Последний хорошо известен из литературы как специфический антагонист рецепторов δ. Авторы посредством интрацеребровентрикулярного введения получили аналгетическую активность для димера, сравнимую с оксиморфоном, и значительное снижение феноменов толерантности и физической зависимости. Более того, оказывается, что такие димеры действуют на центральном уровне. Действительно, оказывается, что их ED50 и ED50 морфина сравнимы после внутривенного и интрацеребровентрикулярного (i.c.v) введения.
Широко известно, что морфин претерпевает значительный метаболизм, что приводит к образованию морфин-6-глюкуронида (M6G). Такой метаболит плохо поступает в головной мозг из-за своей гидрофильной природы. Его аналгетическая активность при общем введении оказывается более сильной, чем активность, вызванная морфином, в то время как тошнота и рвота уменьшаются (Paul et al., 1989, J. Pharmacol. Exp. Ther., 251: 477-483; Frances et al., 1992, J. Pharmacol. Exp. Ther., 262: 25-31). Однако он может спровоцировать, подобно морфину, синдром аддикции. Более того, как и все метаболиты, он быстро удаляется из организма. M6G показывает более высокую аффинность к µ (или сравнимую, в зависимости от исследований), чем аффинность, показанная морфином, в то время как аффинность к κ более низкая (Current Topics in Medicinal Chemistry, 2005, 5, 585-594).
Поэтому представляется, что M6G является интересной основой для создания опиоида, такого же эффективного как морфин, но без его нежелательного побочного действия.
M6G получен синтетическим путем в 1968 (Hidetoshi et al., 1968), и его аналгезирующие свойства высоко оцениваются в ряде публикаций (например, в Frances et al., 1982; Kilpatrick et al.). В патентах ЕР 597915 B1 и US 6046313 описывается синтез M6G и производных M6G, в особенности, в положении 3, а ЕР 1086114 B1 относится к селективному способу синтеза β-аномера M6G. Заявка WO 95/05831 относится к применению M6G для лечения боли путем перорального введения.
Описаны производные M6G. Например, патент ЕР 975648 В1 относится к специфической защите для производного M6G, в котором связь между атомами углерода в положении 7-8 является насыщенной. В заявке WO 2005/063263 описываются производные M6G, сульфированные путем замещения благодаря группе, содержащей тиольную функциональную группу или атом серы. Такие серусодержащие производные показывают высокую аналгетическую активность и меньшее побочное действие.
Опиоидные производные, конъюгированные с углеводами, альтернативные M6G и его производным, описаны в заявке на патент ЕР 816375.
Однако потребность в разработке производных морфина, показывающих высокую аналгетическую активность и меньшее побочное действие, остается.
Раскрытие изобретения
Основной целью настоящего изобретения являются новые производные M6G, предпочтительно, димерные или двухвалентные лиганды, показывающие сильную аналгетическую активность, большую длительность действия и меньшее побочное действие, чем используемые в настоящее время опиоиды. Указанные производные предназначены:
- для постепенного распространения на основном уровне путем модуляции гидрофобности осуществленных химических модификаций,
- для взаимодействия с рецепторами или гомодимерных или гетеродимерных опиоидов,
- для демонстрации сильной аффиности к рецепторам µ с ингибированием в то же время рецепторов κ и/или δ для того, чтобы уменьшить какое-либо побочное действие.
Авторы установили, что присоединение группы, содержащей тиольную функциональную группу на карбоксигруппе глюкуронидной группы M6G и модификация гидроксила в положении 3 в особенности простой эфирной группой может дать возможность получить соединения, показывающие сильную аффиность как к µ, так и к κ вместе с сильной аналгетической активностью. Такие свойства являются особенно правильными с димеризованными соединениями или двухвалентными лигандами, полученными окислением тиольных функциональных групп и последующим образованием дисульфидного мостика.
В литературе показано, что дисульфидные связи могут улучшить свойства in vivo активных молекул: такие связи, относительно устойчивые в плазме, расщепляются в клетках (G. Saito et al., Advanced Drug Delivery Reviews, 2003, 55, 199-215).
Предлагаемым эффектом таких замещений является модификация фармакологического профиля по сравнению с уже известными производными M6G.
Группу в положении 3 выбирают с учетом возрастания липофильности M6G и улучшения его общей аналгетической активности путем благоприятствования его переходу гематоэнцефалического барьера (ГЭБ (ВВВ)). Однако в литературе показано, что некоторые модификации гидроксила в положении 3 морфиновой группы провоцируют утрату аналгетической активности и даже токсичность (Abdulghani A. Houdi et al., 1996, Pharm. Biochem. and Beh., 53(3), 665-671; Salvatella et al., 2004, Biorg. & Med. Chem. Lett., 14, 905-908). Авторы показали, что замещение в положении 3 метоксигруппой (производное 6-кодеинглюкуронида) приводит к неактивному и токсичному соединению (см. пример 1). Поэтому результат, полученный для соединений по настоящему изобретению, является неожиданным.
Другое применение таких производных M6G относится к половой дисфункции и особенно к ускоренной эякуляции. Действительно, эффективность введения аналгетиков для лечения ускоренной эякуляции описана в литературе (Eledjam et al., Safarinejad et al.), например, местное применение аналгетиков, таких как лидокаин, инъекция трамадола (Safarinejad et al., US 23186872 А), или даже применение фармацевтических препаратов, содержащих смесь виагра/агонист опиоидного рецептора дельта (US 06974839).
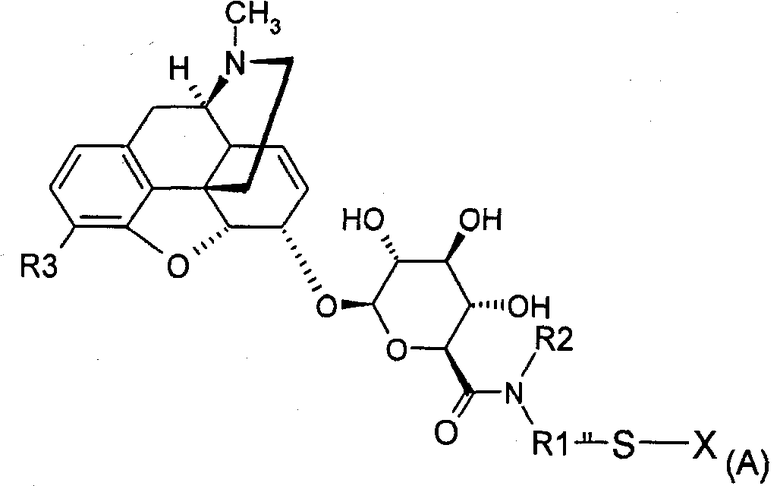
Поэтому настоящее изобретение относится к соединению формулы (А)
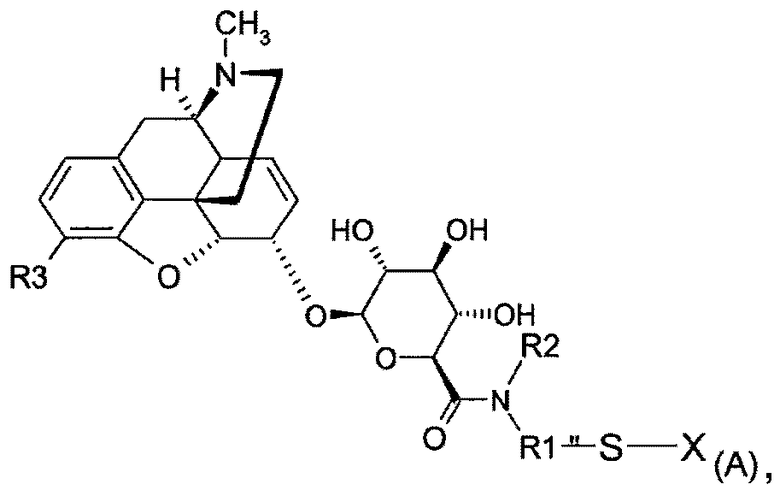
где
вся (А), за исключением заместителя X, обозначается как MR36G-NR1R2-S;
R1 представляет собой (С1-С10)-алкильную группу, причем алкильная цепь может прерываться одним или несколькими гетероатомами, выбранными среди атомов О, S и N;
R2 представляет собой водород, (С1-С5)-алкильную группу, насыщенную или ненасыщенную, линейную или разветвленную, или арил, гетероарил или (C1-C5)-алкиларильную группу;
R3 представляет собой Y(C=Z)R или группу YR, причем Y и Z представляют собой, независимо, кислород или серу, R представляет собой (С1-С6)-алкильную группу, линейную или разветвленную, насыщенную или ненасыщенную, при условии, что R3 является иным, чем O-СН3;
Х представляет собой водород, радикал -S-R4-W или радикал MR36G-NR1R2-S, причем R4 представляет собой (С1-С8)-алкильную группу, которая может содержать амидную, сложноэфирную или простую эфирную связи, и
W представляет собой или антагонист рецептора δ или антагонист рецептора κ,
а также к любой из его фармацевтически приемлемых солей.
Когда R1 представляет собой замещенный алкил, заместитель можно выбрать из группы, состоящей из (С1-С5)-алкильной группы, насыщенной или ненасыщенной, аминогруппы, группы COOR5; группы CONR5R6, причем R5 и R6 представляют собой, независимо, водород, насыщенную или ненасыщенную (С1-С20)-алкильную группу, необязательно замещенную, арил, гетероарил; из (С1-С20)-алкилов предпочтителен (C1-С10)-алкил, содержащий альдегидную функциональную группу и/или кетогрупу.
Когда R2 является замещенным, заместитель может представлять собой насыщенную или ненасыщенную (С1-С4)-алкильную группу.
Предпочтительно, когда W представляет собой антагонист рецептора δ, его выбирают из группы, состоящей из налтриндола, налтрибена, 7-бензилиденналтрексона (BNTX) и 7-(5',6'-бензо-2'-спироинданил)налтрексона (BSINTX).
Когда W представляет собой антагонист рецептора κ, его выбирают из группы, состоящей из 5'-гуанидиноналтриндола и норбиналтрофимина (nor-BNI). Радикал W соединяется с R4, так что его антагонистическая активность сохраняется.
Когда Х представляет собой радикал MR36G-NR1R2-S, радикалы R1, R2 и R3 в обоих радикалах MR36G-NR1R2-S могут быть или одинаковыми, или различными.
Алкил обозначает насыщенный или ненасыщенный углеводородный радикал, линейный или разветвленный, замещенный или незамещенный. Термин «(C1-С10)-алкильная группа» относится к радикалу, такому как описанный выше, содержащему 1-10 атомов углерода, предпочтительно выбранному из группы, состоящей из метила, этила, пропила, изопропила, бутила, изобутила, трет-бутила, пентила, гексила, гептила, октила, нонила и децила. Термин «(С1-С5)-алкильная группа» относится к радикалу, такому как описанный выше, содержащему 1-5 атомов углерода, например, выбранному из группы, состоящей из метила, этила, пропила, изопропила, бутила, изобутила, трет-бутила и пентила. Термин «(С1-С4)-алкильная группа» относится к радикалу, такому как описанный выше, содержащему 1-4 атома углерода, в особенности выбранному из группы, состоящей из метила, этила, пропила, изопропила, бутила, изобутила и трет-бутила. Термин «(С1-С6)-алкильная группа» относится к радикалу, такому как описанный выше, содержащему 1-6 атомов углерода, в особенности выбранному из группы, состоящей из метила, этила, пропила, изопропила, бутила, изобутила, трет-бутила, пентила и гексила. Термин «(С2-С6)-алкильная группа» относится к радикалу, такому как описанный выше, содержащему 2-6 атомов углерода, в особенности выбранному из группы, состоящей из этила, пропила, изопропила, бутила, изобутила, трет-бутила, пентила и гексила. Термин «(С1-С8)-алкильная группа» относится к радикалу, такому как описанный выше, содержащему 1-8 атомов углерода, предпочтительно выбранному из группы, состоящей из метила, этила, пропила, изопропила, бутила, изобутила, трет-бутила, пентила и гексила, гептила и октила. Термин «(С1-С3)-алкильная группа» относится к радикалу, такому как описанный выше, содержащему 1-3 атома углерода, в особенности выбранному из группы, состоящей из метила, этила, пропила и изопропила. Термин «(С2-С3)-алкильная группа» относится к радикалу, такому как описанный выше, содержащему 2-3 атома углерода, в особенности выбранному из группы, состоящей из этила, пропила и изопропила.
Термин «арил» обозначает ароматический углеводородный радикал, замещенный или незамещенный, содержащий, предпочтительно, 6-14 атомов углерода. Предпочтительно арильные радикалы по изобретению выбирают из фенила, нафтила (например, 1-нафтила или 2-нафтила), бифенила (например, 2-, 3- или 4-бифенила), антрила или флуоренила. Особенно предпочтительными являются замещенные или незамещенные фенильные группы.
Термин «гетероарил» обозначает ароматический углеводородный радикал, включающий один или несколько гетероатомов, таких как атомы азота, серы и кислорода, замещенный или незамещенный, содержащий, предпочтительно, 6-14 атомов углерода. Например, можно назвать такие группы, как пиридинил, пиридазинил, пиримидил, пиразил, триазинил, пирролил, пиразолил, имидазолил, (1, 2, 3)- и (1, 2, 4)-триазолил, пиразинил, пиримидинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, изоксазолил или оксазолил и т.д.
Термин «алкиларил» обозначает арильный радикал, замещенный алкильной группой. Термины «арил» и «алкил» соответствуют данным ранее подробным определениям. Примерами алкиларильных групп являются толил, мезитил и ксилил.
Точнее, настоящее изобретение относится к производным морфина, представленным формулами (В), (С) и (D):
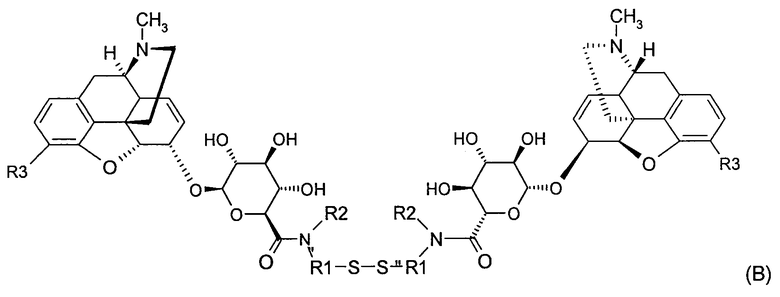
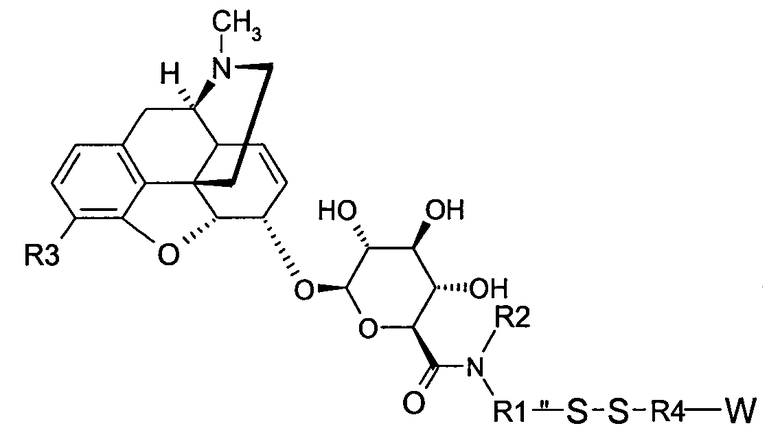
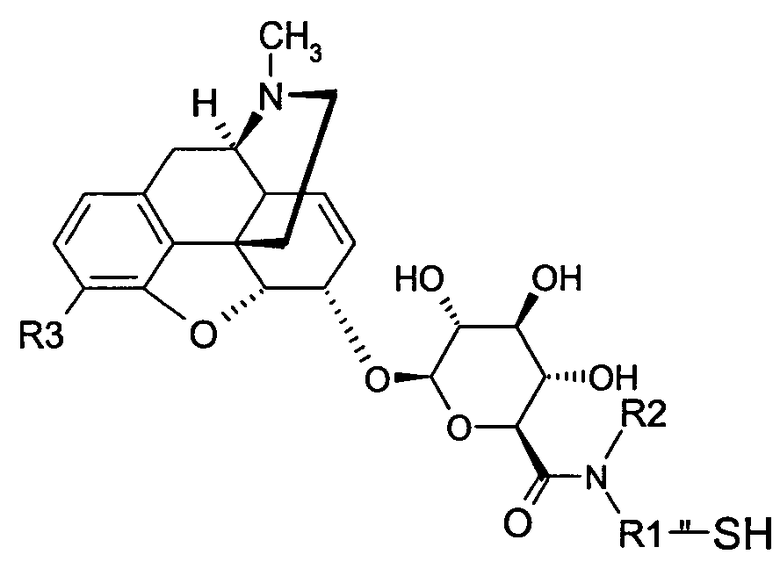
где R1, R2, R3, R4 и W имеют значения, указанные для формулы (А), причем радикалы R1, R2 и R3 в обоих радикалах MR36G-NR1R2-S являются или одинаковыми или различными,
а также к их любым фармацевтически приемлемым солям.
В предпочтительном воплощении Y и Z в радикале R3 представляют собой атомы кислорода. В другом предпочтительном воплощении R в радикале R3 представляет собой (С2-С6)-алкил, разветвленный или неразветвленный, насыщенный или ненасыщенный, предпочтительно, (С2-С3)-алкил, разветвленный или неразветвленный, насыщенный или ненасыщенный, предпочтительно, насыщенный. В отдельном воплощении R3 представляет собой Y(C=Z)R, причем R представляет собой (С1-С6)-алкильную группу, линейную или разветвленную, насыщенную или ненасыщенную, предпочтительно, (C1-С3)-алкил. В другом предпочтительном воплощении R3 представляет собой YR, причем R представляет собой (С2-С6)-алкильную группу, линейную или разветвленную, насыщенную или ненасыщенную, предпочтительно, (С2-С3)-алкил.
В предпочтительном воплощении R4 представляет собой группу -(CH2)n-C(O)-NH-или -(CH2)n-NH-C(O)-, где n равен целому числу от 1 до 8, предпочтительно, от 1 до 4.
Предпочтительные соединения по изобретению представляют собой соединения формул (A)-(D), обладающие одной или несколькими следующими характеристиками:
- группа R3 представляет собой -OR, где R представляет собой, в частности, этил или изопропил; и/или
- R2 представляет собой водород; и/или
- R1 представляет собой линейную алкильную группу -(CH2)2-; и/или
- R4 представляет собой группу -(CH2)2-C(O)-NH-; и/или
- W представляет собой налтриндол.
Также настоящее изобретение относится к соединениям формул (A)-(D), в которых
- R2 представляет собой водород, и R1 представляет собой линейную алкильную группу -(СН2)2-; и/или
- R2 представляет собой водород, и R3 представляет собой -OR, где R представляет собой, в частности, этил или изопропил; и/или
- R4 представляет собой группу -(CH2)2-C(O)-NH-, и W представляет собой налтриндол; и/или
- R1 представляет собой линейную алкильную группу -(СН2)2-, и R3 представляет собой -OR, где R представляет собой, в частности, этил или изопропил; и/или
- R1 представляет собой линейную алкильную группу -(СН2)2-, R2 представляет собой водород, и R3 представляет собой -OR, где R представляет собой, в частности, этил или изопропил.
В определенном воплощении соединение имеет формулу (В), и оба R2 представляют собой атомы водорода, и оба R1 представляют собой линейные алкилы -(СН2)2-. В другом определенном воплощении соединение имеет формулу (С), и R2 представляет собой водород, R1 представляет собой линейную алкильную группу -(СН2)2-, и R4 представляет собой группу -(CH2)2-C(O)-NH-.
Асимметричные атомы углерода, содержащиеся в соединениях формул (A)-(D), могут давать R- или S-конфигурацию.
Димерные предпочтительные соединения представлены в структурах (I), (II) или (III).
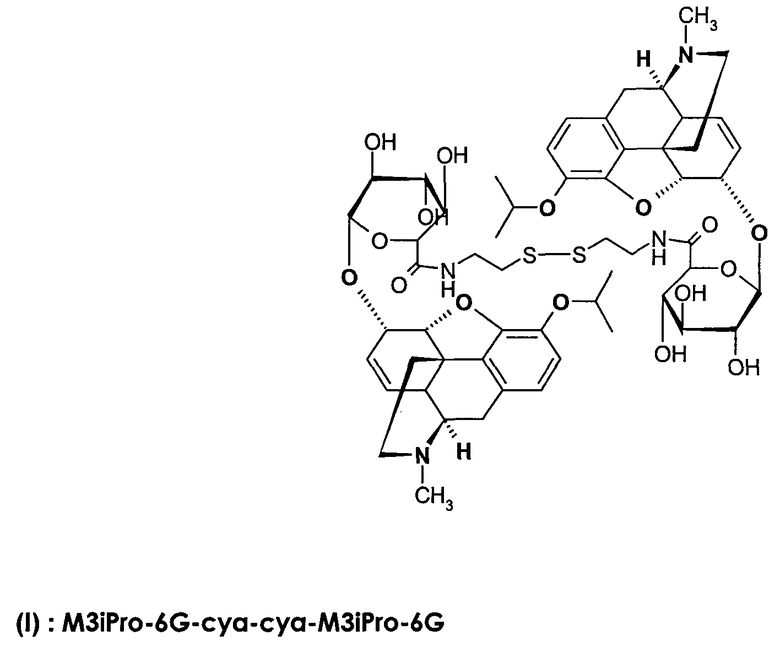
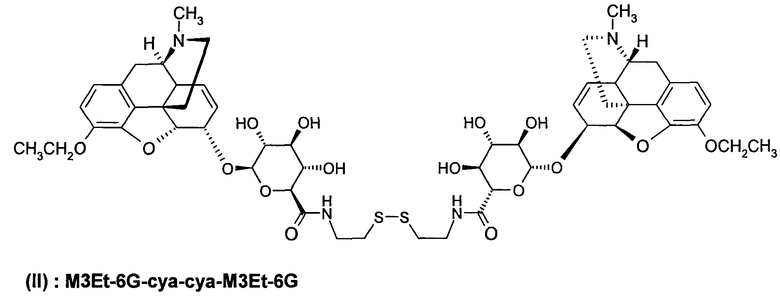
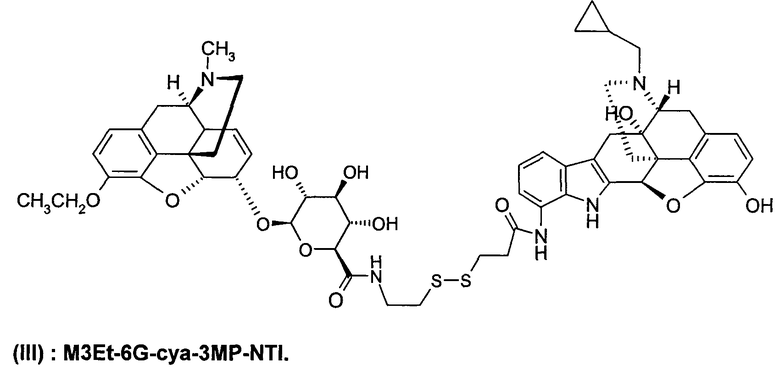
Этильная группа может быть заменена метилом с образованием соединения C6G-суа-3МР-NTI.
Соединения по настоящему изобретению можно получить с использованием способов, известных специалистам в данной области техники.
Изобретение относится к соединению по изобретению как лекарственному средству.
Изобретение также относится к фармацевтической композиции, содержащей в качестве активного компонента одно из вышеописанных соединений или одну из его фармацевтически приемлемых солей, такую как, например, но без ограничения, ацетат, сульфат или гидрохлорид.
Фармацевтическая композиция по настоящему изобретению может находиться в соответствующей форме, зависящей от способа введения:
- для парентерального введения, например, посредством препарата, инъецируемого подкожной инъекцией или внутривенной инъекцией или внутримышечной инъекцией;
- для перорального введения,например, посредством пилюль, капсул, порошков, гранул, пероральных растворов или суспензий или с немедленным высвобождением или пролонгированным высвобождением или даже отсроченным высвобождением лекарственного средства;
- для наружного введения и, в частности, чрескожного введения, например, посредством пэтча, мази или геля;
- для интраназального введения посредством аэрозолей и спреев;
- для ректального введения, например, посредством суппозиториев;
- для подъязычного введения;
- для внутриглазного введения.
Фармацевтически удобный носитель для каждого способа введения можно выбрать среди носителей, используемых в настоящее время.
В одном воплощении композиция может включать другой активный компонент.
Изобретение относится к применению соединения формулы, выбранной из числа формул (A)-(D) и (I)-(III), или одной из его фармацевтически приемлемых солей для получения лекарственного средства, предназначенного для лечения боли, в частности острой боли, хронической боли, невропатической боли, мышечной боли, костной боли, послеоперационной боли, мигрени, боли, связанной с раком, боли при люмбаго, боли при артрозе, боли, связанной с диабетом, или боли, ассоциированной со СПИДом. Настоящее изобретение также относится к применению соединения формулы, выбранной из числа формул (A)-(D) и (I)-(III), или одной из его фармацевтически приемлемых солей для получения лекарственного средства, предназначенного для лечения половых дисфункций, в частности ускоренной эякуляции.
Настоящее изобретение относится к способу лечения боли у субъекта, включающему введение терапевтически эффективной дозы соединения формулы, выбранной из числа формул (A)-(D) и (I)-(III), или одной из его фармацевтически приемлемых солей. В частности, боль представляет собой острую боль, хроническую боль, невропатическую боль, мышечную боль, костную боль, послеоперационную боль, мигрень, боль, связанную с раком, боль при люмбаго, боль при артрозе, боль, связанную с диабетом, или боль, ассоциированную со СПИДом. Кроме того, настоящее изобретение относится к способу лечения у субъекта половых дисфункций, в частности, ускоренной эякуляции, включающему введение терапевтически эффективной дозы соединения формулы, выбранной из числа формул (A)-(D) и (I)-(III), или одной из его фармацевтически приемлемых солей.
Настоящее изобретение также относится к соединению формулы, выбранной из числа формул (A)-(D) и (I)-(III), или одной из его фармацевтически приемлемых солей для лечения боли или лечения половых дисфункций.
Краткое описание фигур
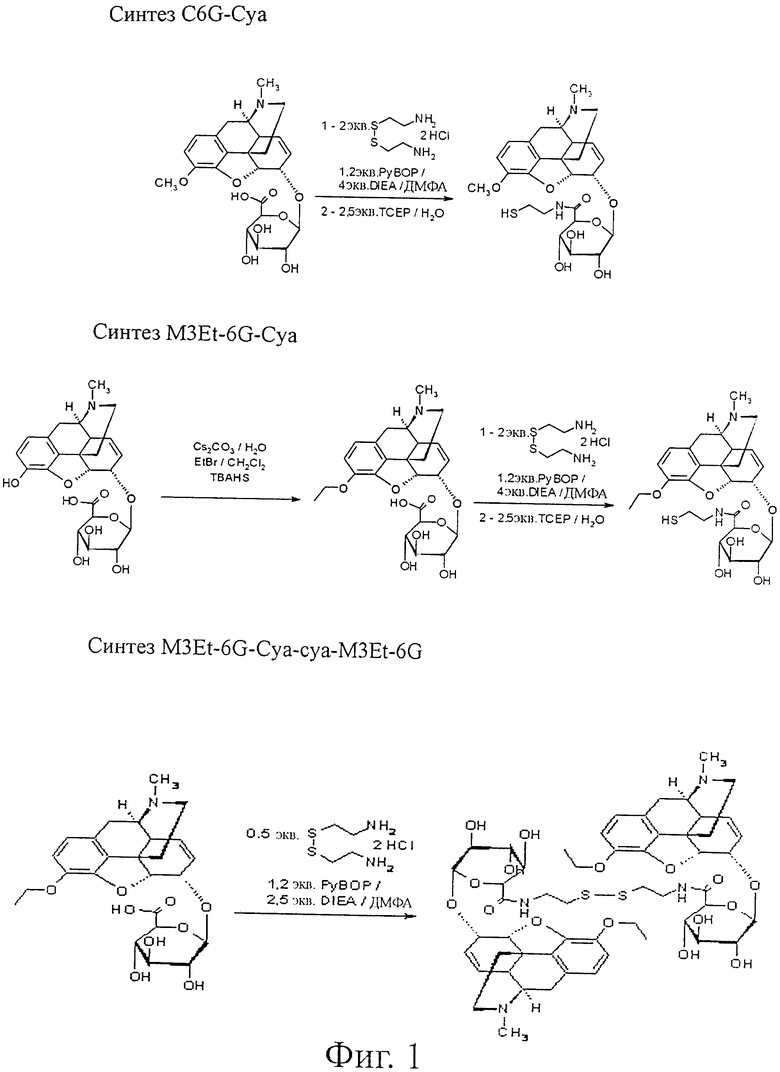
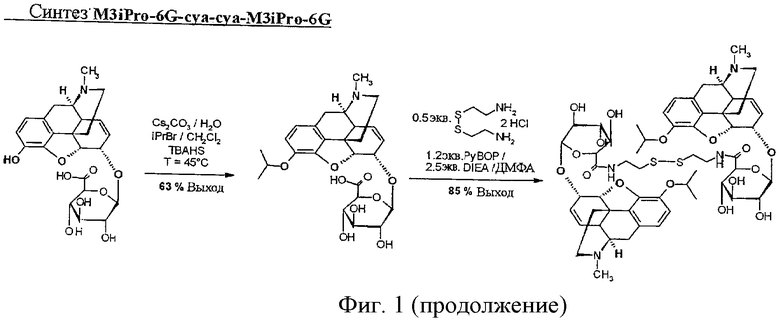
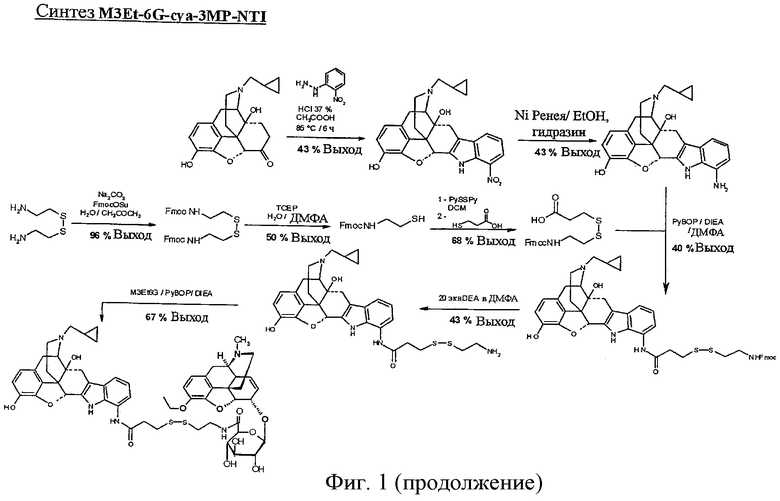
Фиг.1. Синтез C6G-cya, M3Et-6G-cya, M3Et-6G-cya-cya-M3Et-6G, М3-изо-Pr-6G-суа-суа-М3-изо-Pr-6G и M3Et-6G-cya-3MP-NTI.
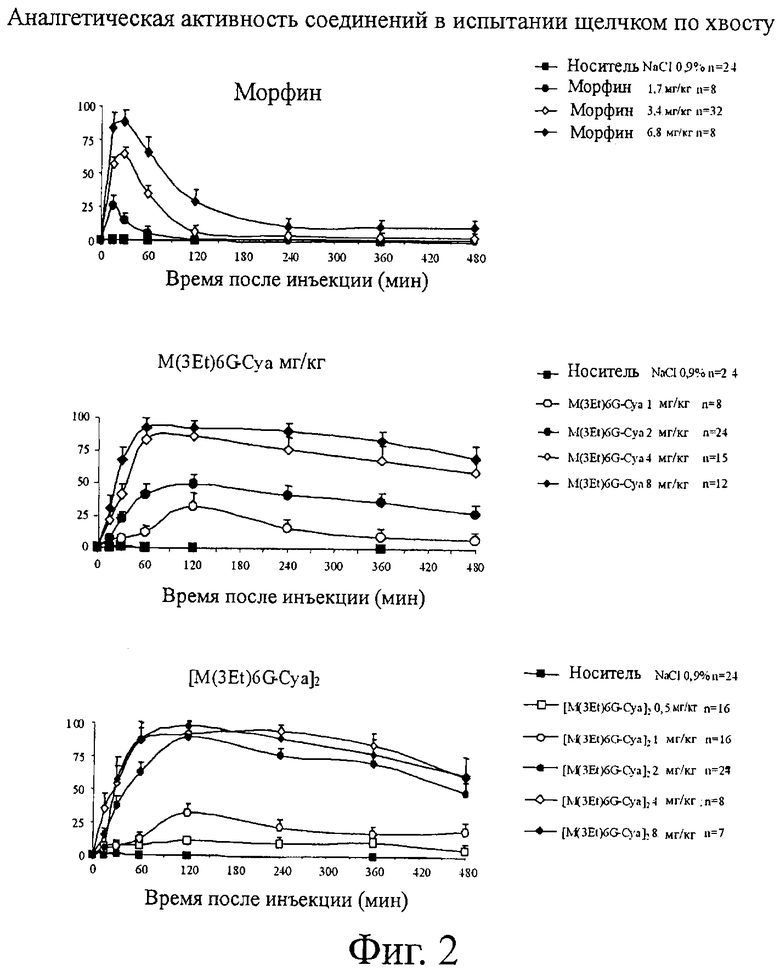
Фиг.2. Аналгетическая активность соединений при испытании щелчком по хвосту.
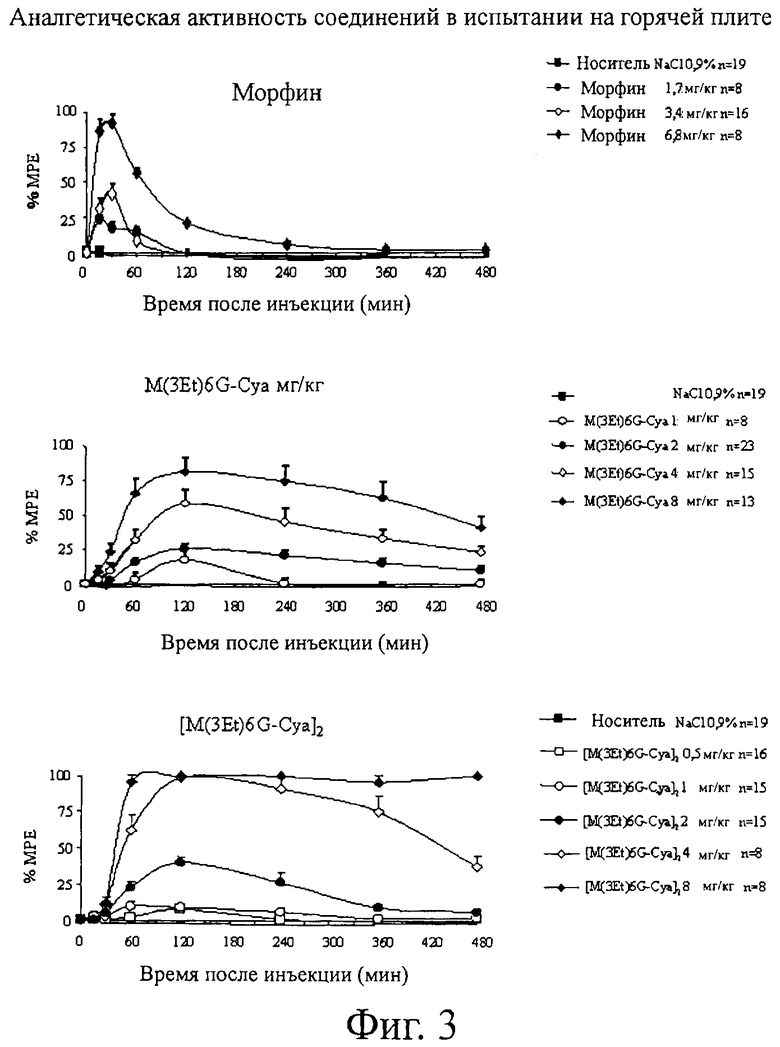
Фиг.3. Аналгетическая активность соединений при испытании на горячей плите.
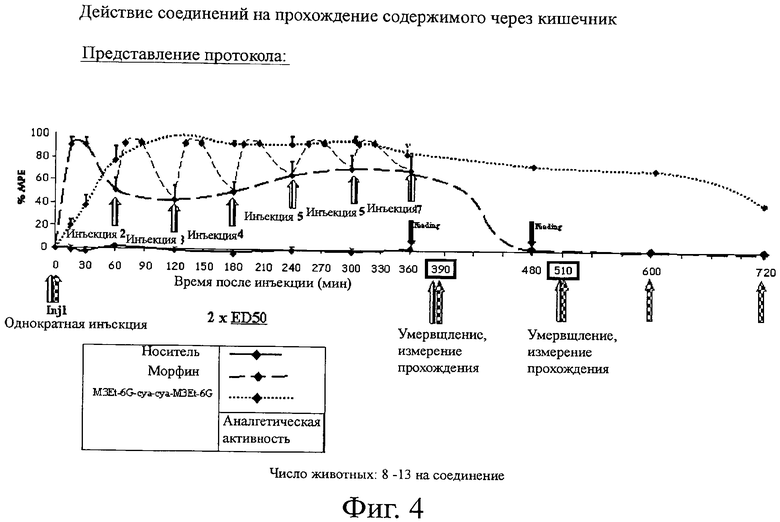
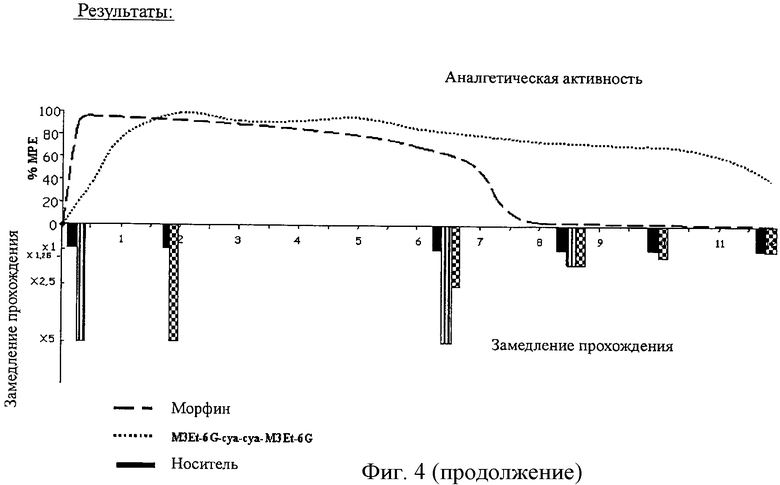
Фиг.4. Действие соединений на прохождение содержимого через кишечник.
Изобретение не ограничивается примерами, приведенными ниже для пояснения.
Осуществление изобретения
Примеры
А. Синтез
Ниже подробно описываются различные способы синтеза, осуществляемые для получения рассматриваемых продуктов. Постадийные схемы реакций приводятся на фиг.1.
После реакций осуществляют высокоэффективную жидкостную хроматографию с обращенной фазой (ВЭЖХ) и масс-спектрометрию MALDI-TOF. Продукты реакций идентифицируют, и определяют их чистоту методами высокоэффективной жидкостной хроматографии с обращенной фазой (ВЭЖХ) и масс-спектрометрии.
Пример 1. Синтез C6G-Cya
В реакторе 2 молярных эквивалента цистеамина растворяют в безводном диметилформамиде (ДМФА) в концентрации 138 г/л. Добавляют 1 молярный эквивалент коммерческого кодеин-6-глюкуронид · ТФК после растворения в безводном ДМФА в концентрации 47 г/л.
Добавляют 4 молярных эквивалента диизопропилэтиламина (DIEA), и реактор помещают на ледяную баню (0°С). В реакционную смесь прикапывают предварительно полученный раствор 1,2 молярных эквивалентов гексафторфосфата бензотриазол-1-илокситриспирролидинофосфония (РуВОР) в ДМФА в концентрации 225 г/л, и затем смесь перемешивают при комнатной температуре в течение 3 часов.
Затем восстанавливают дисульфидную связь, добавляя 2,5 молярных эквивалента трис(2-карбоксиэтил)фосфина (ТСЕР), предварительно растворенного в смеси вода/0,1% ТФК в концентрации 214 г/л. После 2-часового взаимодействия продукт очищают препаративной ВЭЖХ.
После лиофилизации получают 10,3 мг C6G-cya. [М+Н]+=535; МТФК=648; чистота 98%; выход = 93%.
Пример 2. Синтез M3Et-6G-cya
Синтез M3Et-6G
В реактор загружают 1 молярный эквивалент дигидрата морфин-6-глюкуронида (M6G) и растворяют в воде в концентрации 100 г/л. Добавляют 5 эквивалентов карбоната цезия, и смесь перемешивают в течение 5 минут при комнатной температуре.
К смеси добавляют объем дихлорметана, равный объему воды. Добавляют последовательно 5 эквивалентов бромэтана и 2 эквивалента гидросульфата тетрабутиламмония (TBAHS). Перемешивание продолжают при комнатной температуре в течение 72 часов. Продукт реакции очищают препаративной ВЭЖХ.
Эксперимент с ядерным эффектом Оверхаузера (NOE) методом протонного ядерного магнитного резонанса (ЯМР) показывает, что этильная группа присоединена к атому кислорода в положении 3 производного морфина. Действительно, регулируемое облучение этильной группы вызывает сигналы ароматических протонов фенола.
Получают 45,8 мг M3Et-6G. [М+Н]+=490; МТФК=603; чистота 93%; выход = 55%.
Синтез M3Et-6G-cya
В реакторе 2 молярных эквивалента дигидрохлорида цистеамина растворяют в ДМФА в концентрации 71 г/л. Добавляют 1 эквивалент M3Et-6G, предварительно растворенного в ДМФА в концентрации 96 г/л. Смесь разбавляют ДМФА и добавляют 4 молярных эквивалента DIEA. Реактор охлаждают до 0°С и прикапывают предварительно полученный раствор 1,2 молярных эквивалентов РуВОР в ДМФА в концентрации 23 г/л.
Перемешивание продолжают при комнатной температуре в течение 3 часов и затем добавляют 2,5 молярных эквивалента ТСЕР, растворенного в смеси вода/0,1% ТФК в концентрации 21 г/л. После 4-часовго перемешивания реакция завершается. Продукт очищают препаративной ВЭЖХ.
Получают 46,9 мг M3Et-6G-cya. [М+Н]+=549; МТФК=662; чистота 95%; выход = 89%.
Пример 3. Синтез M3Et-6G-cya-cya-M3Et-6G
В реакторе 1 молярный эквивалент дигидрохлорида цистеамина растворяют в ДМФА в концентрации 86 г/л. Добавляют 2 молярных эквивалента M3Et-6G-cya, предварительно растворенного в ДМФА в концентрации 97 г/л. Затем вводят 5 молярных эквивалентов DIEA и смесь охлаждают до 0°С на ледяной бане. Прикапывают раствор 2,4 молярных эквивалентов РуВОР в ДМФА концентрации 22,9 г/л. Перемешивание продолжают при комнатной температуре в течение 3 часов. Затем продукт реакции очищают препаративной ВЭЖХ.
Получают 40,5 мг димера M3Et-6G-cya-cya-M3Et-6G. [М+Н]+=1096; МТФК=1322; чистота 95%; выход = 81%.
Пример 4. Синтез М3-изо-Pr-6G-суа-суа-М3-изо-Pr-6G
Синтез М3-изо-Pr-6G
В 10-мл колбе 1 молярный эквивалент M6G растворяют в воде в концентрации 100 г/л. Добавляют 5 молярных эквивалентов карбоната цезия, 2 молярных эквивалента TBAHS, 1 мл дихлорметана и 5 молярных эквивалентов бромизопропана. Смесь перемешивают при 45°С - температуре кипения дихлорметана - в течение ночи.
Дихлорметан выпаривают и продукт реакции очищают препаративной ВЭЖХ.
Получают 61,7 мг М3-изо-Pr-6G. [М+Н]+=504; МТФК=617; чистота 95%; выход = 63%.
Синтез М3-изо-Pr-6G-суа-суа-М3-изо-Pr-6G
В пробирке Фалькона 1 молярный эквивалент дигидрохлорида цистеамина растворяют в ДМФА в концентрации 11,2 г/л. Добавляют 2 молярных эквивалента М3-изо-Pr-6G, предварительно растворенного в ДМФА в концентрации 100 г/л. Затем вводят 5 молярных эквивалентов чистого DIEA и смесь охлаждают до 0°С и перемешивают в течение 5 минут. Затем добавляют 2,4 молярных эквивалентов РуВОР, предварительно растворенного в ДМФА в концентрации 240 г/л, и смесь перемешивают при комнатной температуре в течение 1 часа.
Димер очищают препаративной ВЭЖХ.
Получают 46,4 мг М3-изо-Pr-6G-суа-суа-М3-изо-Pr-6G. [М+Н]+=1123; МТФК=1350; чистота 95%; выход = 85%.
Пример 5. Синтез M3Et-6G-cya-3MP-NTI
Синтез 7'-нитроналтриндола
В колбе в смесь 50/50 37% соляной кислоты и ледяной уксусной кислоты вводят 1 молярный эквивалент налтрексон · HCl · H2O и 1 молярный эквивалент (2-нитрофенил)гидразина в концентрации 57 г/л. Смесь перемешивают в течение 6 час 30 мин при 85°С и затем охлаждают до 0°С, нейтрализуют насыщенным раствором NaHCO3 и экстрагируют 3 раза этилацетатом. Органические фазы объединяют, сушат над сульфатом натрия, фильтруют и упаривают при пониженном давлении. Затем полученное твердое вещество очищают препаративной ВЭЖХ.
Получают 90 мг 7'-нитроналтриндола. [М+Н]+=460; МТФК=573; чистота 92%; выход = 43%.
Синтез 7'-аминоналтриндола
В 50-мл пробирку Фалькона загружают 1 молярный эквивалент 7'-нитроналтриндола, растворенного в этаноле в концентрации 34,6 г/л. Добавляют избыток никеля Ренея (50% суспензия в воде) и прикапывают 10 молярных эквивалентов водного гидразина. После 1-часового взаимодействия смесь центрифугируют, спиртовую фазу извлекают и выпаривают этанол при пониженном давлении. Полученный остаток снова растворяют в минимальном количестве смеси вода (0,1% ТФК) / ацетонитрил (0,1% ТФК), 50/50 и вводят в колонку для препаративной ВЭЖХ.
Получают 44,1 мг продукта реакции. [М+Н]+=430; МТФК=657; чистота 95%; выход = 43%.
Синтез Fmoc-цистеамин-3МР
В колбе 2 молярных эквивалента Fmoc-OSu растворяют в ацетоне в концентрации 320 г/л. Добавляют 1 молярный эквивалент цистамин · 2HCl и 1,6 молярных эквивалента карбоната натрия с объемом воды, равным объему ацетона. Смесь затвердевает. Добавляют смесь вода/ацетон, 50/50, для удвоения объема растворителя.
После перемешивания в течение 2 час 30 мин при комнатной температуре ацетон выпаривают на ротороном испарителе и полученную суспензию растворяют в дихлорметане. Водную фазу экстрагируют дихлорметаном, и органические фазы последовательно промывают раствором KHSO3 и насыщенным раствором NaCl.
Затем конечную органическую фазу сушат над сульфатом натрия, фильтруют и упаривают при пониженном давлении и получают 2,53 г Fmoc-цистамина. [М+Н]+=598; чистота 95%; выход = 96%.
Осуществляют восстановление дисульфидного мостика, растворяя 1 молярный эквивалент Fmoc-цистамина в ДМФА в концентрации 25 г/л и добавляя 2,5 молярных эквивалентов трис(2-карбоксиэтил)фосфина (ТСЕР), предварительно растворенного в воде (0,1% ТФК) в концентрации 400 г/л. После 1-часового перемешивания при комнатной температуре добавляют избыток воды и смесь экстрагируют дихлорметаном. Органические фазы объединяют, промывают насыщенным раствором NaCl, сушат над Na2SO4 и фильтруют через пористое стекло. Дихлорметан выпаривают. Затем продукт реакции высаживают, добавляя воду (0,1% ТФК), отфильтровывают на фильтре из пористого стекла, растворяют в смеси H2O (0,1% ТФК)/ACN (0,1% ТФК) (25/75) и затем лиофилизуют.
Получают 252 мг продукта реакции. [М+Н]+=300; чистота 95%; выход = 50%.
Раствор 3 молярных эквивалента 2,2'-дитиодипиридина в дихлорметане в концентрации 122 г/л охлаждают при 0°С.
Прикапывают предварительно полученный раствор 1 молярного эквивалента Fmoc-цистеамина в дихлорметане в концентрации 18 г/л в течение 10 минут.
После 3-часового взаимодействия при комнатной температуре смесь охлаждают при 0°С и прикапывают в течение 20 минут 5,5 молярных эквивалентов d3-меркаптопропионовой кислоты, разведенной в дихлорметане в концентрации 18 мкл/мл. После взаимодействия в течение ночи при комнатной температуре дихлорметан выпаривают при пониженном давлении. Полученный остаток очищают препаративной ВЭЖХ.
Получают 231 мг Fmoc-цистеамин-3МР. [М+Н]+=400; чистота 95%; выход = 68%.
Синтез цистеамин-3МР-7'-аминоналтриндола
Растворяют 1 молярный эквивалент 7'-аминоналтриндола в ДМФА в концентрации 70 г/л. Добавляют 2,1 молярных эквивалента Fmoc-цистеамин-3МР, растворенного в ДМФА в концентрации 66 г/л, и 6 молярных эквивалентов чистого DIEA. Смесь охлаждают при 0°С без перемешивания. Прикапывают предварительно полученный раствор 1,1 молярного эквивалента РуВОР в ДМФА в концентрации 200 г/л. После 60-минутного взаимодействия смесь очищают препаративной ВЭЖХ. Получают 22,5 мг Fmoc-цистеамин-3МР-7'-аминоналтриндола. [М+Н]+=815; МТФК=928; чистота 94%; выход = 36%.
Растворяют 1 молярный эквивалент Fmos-цистеамин-3МР-7'-аминоналтриндола в ДМФА в концентрации 15 г/л. Добавляют 20 молярных эквивалентов чистого DEA, и смесь перемешивают в течение 30 минут при комнатной температуре.
Смесь очищают препаративной ВЭЖХ.
Получают 9,3 мг цистеамин-3МР-7'-аминоналтриндола. М+Н]+=593; МТФК=820; чистота 81%; выход = 43%.
Синтез M3Et-6G-cya-3MP-NTI
Растворяют 1 молярный эквивалент Суа-3МР-7'-аминоналтриндола в ДМФА в концентрации 70 г/л. Добавляют 1 молярный эквивалент M3Et-6G, растворенного в ДМФА в концентрации 70 г/л, с 5 молярными эквивалентами DIEA. Смесь охлаждают до 0°С и к смеси прикапывают раствор 1,2 молярных эквивалентов РуВОР в ДМФА в концентрации 200 г/л. После 45-минутного перемешивания при комнатной температуре смесь очищают препаративной ВЭЖХ.
Получают 11 мг химерного продукта. [М+Н]+=1064; МТФК=1291; чистота 95%; выход = 67%.
В. Исследование аффинности к опиоидным рецепторам
B.1/ Экспериментальная процедура
Сравнивают аффинность производных M6G-C6G-cya, M3Et-6G-cya и M3Et-6G-cya-cya-M3Et-6G к каждому из трех подтипов опиоидных рецепторов µ, δ и κ.
Для того чтобы определить аффинность к рецепторам µ, гомогенаты клеточных мембран (рецепторы µ человека, трансфицированные в клетки НЕК-293) инкубируют при 22°С в течение 120 мин с 0,5 нМ [3H][D-Ala, N-MePhe, Cly(ol)] энкефалина (DAMGO) или в присутствии одного из соединений по изобретению или в его отсутствие в буфере, содержащем 50 мМ трис-HCl (рН 7,4) и 5 мМ MgCl2.
Для того чтобы определить аффинность к рецепторам κ, гомогенаты мозжечка морской свинки (250 мкг белка) инкубируют при 22°С в течение 80 мин с 0,7 нМ [3H]U-69593 или в присутствии одного из соединений по изобретению или в его отсутствие в буфере, содержащем 50 мМ трис-HCl (рН 7,4), 10 мМ MgCl2 и 1 мМ ЭДТК.
Для того чтобы определить аффинность к рецепторам S, гомогенаты клеточных мембран (рецепторы Мю человека, трансфицированные в клетки СНО) инкубируют при 22°С в течение 120 мин с 0,5 нМ [3H]DADLE или в присутствии одного из соединений по изобретению или в его отсутствие в буфере, содержащем 50 мМ трис-HCl (рН 7,4) и 5 мМ MgCl2.
Неспецифическое связывание определяют в присутствии 10 мкМ налтрексона. После инкубации образцы быстро фильтруют через стеклянные фильтры (GF/B, Packard), предварительно инкубируют с 0,3% полиэтиленимином и смывают несколько раз холодным 50 мМ трис-HCl с использованием «96-sample cell harvester» (Unifllter, Packard). Затем фильтры сушат и счетчиком измеряют радиоактивность.
Агонистическую/антагонистическую активность соединений по изобретению в отношении рецепторов κ оценивают с использованием семявыносящих протоков сегментов предстательной железы кролика, вытянутых и стимулированных электрическим током 0,1 Гц в течение 1 мс.
Для того чтобы измерить агонистическую активность, получают реакцию контроля (пик сокращений ткани), воздействуя на ткани высокой концентрацией (0,1 мкМ) U-69593, который является специфическим агонистом рецептора κ.
Затем ткани подвергают воздействию возрастающих концентраций соединения по изобретению или специфического агониста. Различные концентрации накапливают, и каждую концентрацию поддерживают в контакте с тканью до тех пор, пока не получат устойчивую реакцию, или максимум в течение 15 мин.
Если отмечают характерную реакцию агониста (т.е. подавление сокращений), ссылочный антагонист норбиналторфимин (nor-BNI, 0,01 нМ) испытывают при самой высокой концентрации соединения для подтверждения роли рецепторов κ при реакции.
Для того чтобы измерить антагонистическую активность, получают реакцию контроля (пик сокращений ткани), воздействуя на ткани высокой концентрацией (0,1 мкМ) специфического агониста U-69593.
Затем ткани подвергают воздействию возрастающих концентраций соединения по изобретению или специфического агониста. Различные концентрации накапливают, и каждую концентрацию поддерживают в контакте с тканью до тех пор, пока не получат устойчивую реакцию, или до 15 мин.
Антагонистическую реакцию наблюдают, когда амплитуда пиков сокращений, вызванных электрической стимуляцией, схожа с амплитудой, наблюдаемой с nor-BNI.
Измеренный параметр представляет собой максимальное изменение амплитуды пиков сокращений, вызванных электрической стимуляцией, с различными концентрациями соединения.
В.2/ Результаты
Результаты по аффинности к рецепторам µ, δ и κ подробно приводятся ниже в таблице 1.
NC: не учитывается, поскольку менее 25% от подавления при самых высоких концентрациях.
Результаты показывают:
- аффиность к рецепторам µ для соединений по изобретению схожа с аффиностью морфина;
- аффинность соединений по изобретению к рецепторам δ при сравнении с морфином теряется;
- аффиность мономерных соединений C6G-суа и M3Et-6G-cya к рецепторам k схожа с аффиностью морфина и аффинность улучшается (фактор 6) в случае димера M3Et-6G-cya-cya-M3Et-6G.
Агонистическую/антагонистическую активность соединения M3Et-6G-cya-cya-M3Et-6G измеряют так, как описано выше (§ В1): соединение по изобретению ведет себя как антагонист рецептора κ.
Результаты по определению агонистической/антагонистической активности приводятся ниже в таблице 2.
Оценка антагонистической активности
Реакции указаны в % от контроля реакции с U-69593 (уменьшение амплитуды сокращений).
С. Исследование аналгетической активности
C.1. Тип процедур
Аналгетическую активность определяют испытаниями как «щелчок по хвосту» ("Tail flick"), так и способом «горячая плита» ("Hot plate").
Испытание «щелчок по хвосту» (тест D'Amour et Smith, 1941, Pharmacol. Exp. Ther., 72, 74-79) состоит в помещении хвоста мыши, после введения продукта, перед источником инфракрасного излучения, для того чтобы получить ноцицептивный раздражитель (температура поверхности 55°С). Время реакции (RT) мыши (период задержки между включением светового луча и моментом, когда мышь отдергивает свой хвост) измеряют дважды при увеличении времени 8 раз от 15 мин до 480 мин после введения продукта. Максимальный 10-секундный период выбран как максимальное время реакции для того, чтобы избежать повреждения тканей животного.
Продукты вводят внутривенно в дозах в интервале от 0,5 до 8 мг/кг (8 мышей на группу).
Осуществляют два измерения времени реакции перед введением продукта для каждой мыши, с тем чтобы установить базисную линию.
Испытание «горячая плита» состоит в помещении мыши на горячую плиту при 54°С и измерении времени появления одной из следующих форм поведения:
- подергивание с участием, по меньшей мере, одной из четырех лап;
- облизывание передних или задних лап,
- превышение 30-секундного периода на горячей плите;
- активация скорости перемещения.
Время отдергивания измеряют дважды в различные сроки после инъекции продукта от 15 мин до 360 мин.
Перед введением испытываемых продуктов определяют базисную линию для каждой мыши.
Продукты вводят внутривенно в дозах в интервале от 0,5 до 8 мг/кг (8 мышей на группу).
С.2/ Результаты
Что касается испытания щелчком по хвосту, то результаты для морфина, M3Et-6G-суа, M3Et-6G-cya-cya-M3Et-6G, C6G-cya-cya-C6G, М3-изо-Pro-6G-суа-суа-М3-изо-Pro-6G, M3Et-6G-cya-3MP-NTI и cya-M3Et-6G указываются в виде средних величин для группы % МРЕ±ср.-кв. откл. (S.E.M.) и приводятся на фигуре 2.
МРЕ, %, представляет процент от возможного максимального действия и соответствует следующей формуле:
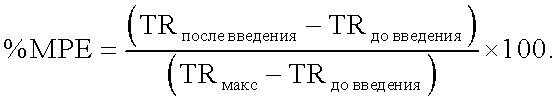
Для каждого продукта вычисляют ED50 (активная доза для 50% животных) и AUC (площадь под кривой) и представляют в приведенных ниже таблицах 3 (ED50) и 4 (AUC).
ND: не определена (отсутствие аналгезирующего ответа)
Что касается испытания на горячей плите, то результаты, полученные для морфина, M3Et-6G-cya и M3Et-6G-cya-cya-M3Et-6G, указываются в виде средних величин для группы % МРЕ±ср.-кв. откл. (S.E.M.) и приводятся на фиг.3.
Для каждого продукта вычисляют ED50 (активная доза для 50% животных) и AUC (площадь под кривой) и представляют в приведенных ниже таблицах 5 (ED50) и 6 (AUC).
Результаты показывают, что соединения по изобретению обладают аналгетической активностью, по меньшей мере, лучшей, чем активность, показанная морфином. Действительно, их ED50 составляет от 1,3 до 2 мг/кг по сравнению с 3,3 мг/кг для морфина.
Более того, длительность аналгетической активности значительно выше для соединений M3Et6G-Cya и M3Et-6G-cya-cya-M3Et-6G, чем для морфина. Действительно, соединения по изобретению остаются активными, по меньшей мере, в два раза дольше, чем морфин.
D. Исследование прохождения содержимого через кишечник у мышей
D.1/ Тип процедур
Мышей Awake кормят насильно путем введения в пищевод питательной трубки с 1-мл шприцем. Вводят постепенно 600 мкл пасты для принудительного кормления (состоящей, в основном, из активированного угля). Через 30 мин после насильного кормления животных умервщляют. После рассечения брюшной стенки кишки обнажают по всей их длине. Измеряют расстояние между кардией и прямой кишкой (общая длина), а также расстояние между кардией и отмеченным продвижением.
Действие соединения M3Et-6G-cya-cya-M3Et-6G на констипацию измеряют при максимальном действии после iv инъекции при ED50 и затем сравнивают с действием морфина.
Для того чтобы приблизиться к клиническому случаю послеоперационной боли, при которой пациент нуждается в аналгезирующем действии в течение нескольких часов, используют следующий протокол:
соединение M3Et-6G-cya-cya-M3Et-6G и морфин инъецируют при двукратной ED50.
Для того чтобы сохранить максимальную аналгезию в течение 6 час 30 мин, дозу морфина инъецируют каждый час, что, вероятно, соответствует морфинофому насосу в стационаре. Нет необходимости инъецировать дополнительно дозу M3Et-6G-cya-cya-M3Et-6G, так как активность является максимальной в течение, по меньшей мере, 6 часов.
Затем измеряют прохождение от 30 мин после насильного кормления мышей до 6 часов после введения.
Повторное измерение осуществляют через 2 часа 30 мин после последней инъекции морфина или 8 час 30 мин после инъекции M3Et-6G-cya-cya-M3Et-6G. Наконец, осуществляют измерения через 10 час и 12 час после инъекции M3Et-6G-cya-cya-M3Et-6G, для того чтобы оценить скорость возврата к нормальному прохождению.
D.2/ Результаты
Процент прохождения вычисляют следующим образом:
расстояние до маркера ×100 / общая длина.
Данные приводятся на фиг.4.
Не наблюдают существенного различия в прохождении между соединением M3Et-6G-cya-cya-M3Et-6G и морфином при максимальном аналгезирующем действии после инъекции при ED50 (подавление продвижения на примерно 90%).
В случае сохранения максимальной аналгезии на протяжении 6 час 30 мин подавление прохождения с соединением M3Et-6G-cya-cya-M3Et-6G (52-56%) меньше по сравнению с морфином (82%).
Когда аналгетическую активность поддерживают на максимуме морфином, прохождение полностью прекращается. Прохождение возобновляется, когда прекращаются инъекции морфина вместе с падением аналгетической активности.
В случае соединения M3Et-6G-cya-cya-M3Et-6G прохождение только замедляется, в то время как аналгетическая активность остается максимальной. Скорость прохождения становится нормальной между 10 и 12 часами с аналгезией 50%.
| название | год | авторы | номер документа |
|---|---|---|---|
| 3,4,4-ТРЕХЗАМЕЩЕННЫЕ ПИПЕРИДИНИЛ-N-АЛКИЛКАРБОКСИЛАТЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2145958C1 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ДЛЯ КУПИРОВАНИЯ ВИСЦЕРАЛЬНОЙ БОЛИ | 2010 |
|
RU2429874C1 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2009 |
|
RU2477281C2 |
| АНАЛОГИ БУПРЕНОРФИНА | 2009 |
|
RU2520222C2 |
| ПРИМЕНЕНИЕ ОПИОРФИНА В КАЧЕСТВЕ ПСИХОСТИМУЛИРУЮЩЕГО АГЕНТА | 2009 |
|
RU2526819C2 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ УЛУЧШЕНИЯ СВОЙСТВ КОЖИ, ВКЛЮЧАЮЩЕЕ В КАЧЕСТВЕ ДЕЙСТВУЮЩЕГО ИНГРЕДИЕНТА ПРОИЗВОДНОЕ МОРФИНАНА ИЛИ ЛЮБУЮ ИЗ ЕГО ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫХ КИСЛОТНО-АДДИТИВНЫХ СОЛЕЙ | 2008 |
|
RU2440117C1 |
| СИНТЕТИЧЕСКИЕ ПЕПТИДНЫЕ АМИДЫ | 2007 |
|
RU2500685C2 |
| БИЦИКЛО-3.1.1-ГЕПТАН-ЗАМЕЩЕННЫЕ БЕНЗИМИДАЗОЛОН- И ХИНАЗОЛИНОН-ПРОИЗВОДНЫЕ АГОНИСТЫ ORL1 РЕЦЕПТОРОВ ЧЕЛОВЕКА | 2004 |
|
RU2357964C2 |
| 4-ГИДРОКСИБЕНЗОМОРФАНЫ | 2005 |
|
RU2480455C2 |
| КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ТРАМАДОЛ И ЦЕЛЕКОКСИБ, ДЛЯ ЛЕЧЕНИЯ БОЛИ | 2010 |
|
RU2707752C2 |
Настоящее изобретение относится к соединениям формулы (А), где вся (А), за исключением заместителя X, обозначается как MR36G-NR1R2-S; R1 представляет собой линейный или разветвленный (С1-С10)-алкил; R2 - водород; R3 - группа YR, причем Y - кислород, R - линейный или разветвленный (С1-С6)-алкил при условии, что R3 не является O-СН3; Х представляет собой водород, радикал -S-R4-W или радикал MR36G-NR1R2-S, причем R4 представляет собой (C1-C8)-алкил, -(CH2)n-C(O)-NH-, или -(СН2)n-NH-C(O)-, где n равен целому числу от 1 до 8; и W - налтриндол, а также к их фармацевтически приемлемым солям. Кроме того, изобретение относится к фармацевтической композиции на основе указанных соединений для получения лекарственного средства для лечения боли, а также к применению данных соединений для получения лекарственного средства для лечения боли. 3 н. и 6 з.п. ф-лы, 6 табл., 4 ил.
1. Соединение формулы (А)
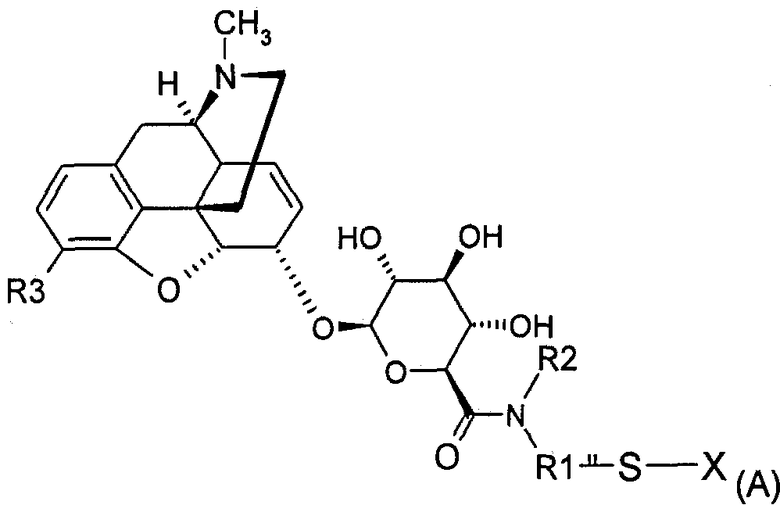
где вся (А), за исключением заместителя X, обозначается как MR36G-NR1R2-S;
R1 представляет собой линейную или разветвленную, насыщенную (C1-С10)-алкильную группу;
R2 представляет собой водород;
R3 представляет собой группу YR, причем Y представляет собой независимо кислород, R представляет собой линейную или разветвленную насыщенную (С1-С6)-алкильную группу, при условии, что R3 не является O-СН3;
Х представляет собой водород, радикал -S-R4-W или радикал MR36G-NR1R2-S,
причем R4 представляет собой насыщенную (С1-С8)-алкильную группу, группу -(CH2)n-C(O)-NH- или группу -(CH2)n-NH-C(O)-, где n равен целому числу от 1 до 8: и
W представляет собой налтриндол,
а также любая его фармацевтически приемлемая соль.
2. Соединение по п.1, где R3 представляет собой YR, причем R представляет собой линейную или разветвленную, насыщенную (С2-С3)-алкильную группу.
3. Соединение по п.1, где R4 представляет собой группу -(СН2)n-С(О)-NH-, где n равен целому числу от 1 до 4.
4. Соединение по п.1, где соединение имеет одну или несколько следующих характеристик:
- группа R3 представляет собой -OR, где R представляет собой, в частности, этил или изопропил; и/или
- R2 представляет собой водород; и/или
- R1 представляет собой линейную алкильную группу -(СН2)2-, и/или
- R4 представляет собой группу -(СН2)2-С(O)-NH-; и/или
- W представляет собой налтриндол.
5. Соединение по п.1, где Х представляет собой радикал -S-R4-W или радикал MR36G-NR1R2-S, причем радикалы R1, R2 и R3 в обоих радикалах MR36G-NR1R2-S являются или одинаковыми, или различными.
6. Соединение по п.5, которое представляет формула, выбранная из группы, состоящей из формул (I), (II) и (III)
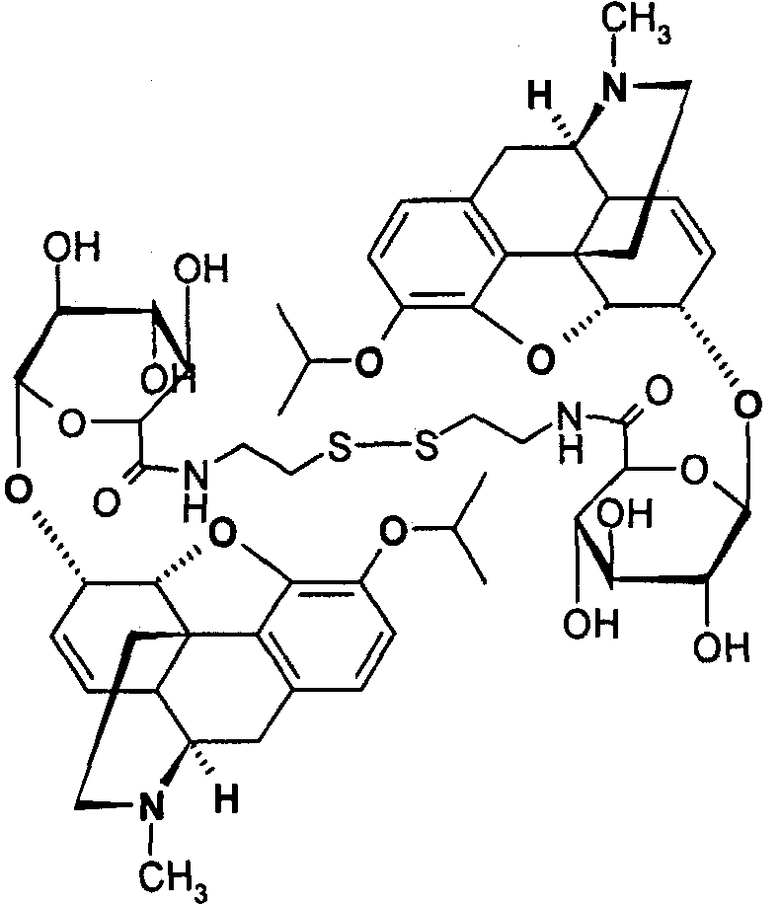
(I): M3iPro-6G-cya-cya-M3iPro-6G
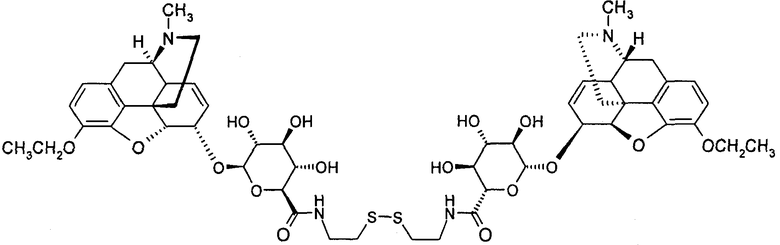
(II): M3Et-6G-cya-cya-M3Et-6G
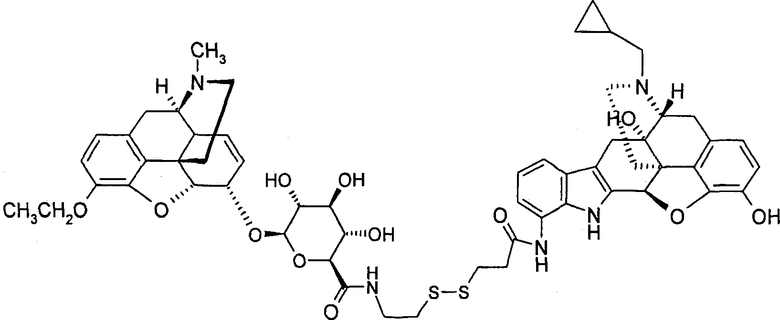
(III): M3Et-6G-cya-3MP-NTI.
7. Соединение по любому из пп.1-6 в форме лекарственного средства для лечения боли.
8. Фармацевтическая композиция для получения лекарственного средства для лечения боли, включающая соединение по любому из пп.1-6 и фармацевтически удобный носитель.
9. Применение соединения по любому из пп.1-6 для получения лекарственного средства для лечения боли.
| US 7365055 В2, 29.04.2008 | |||
| ЕР 0816375 A1, 07.01.1998 | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| RU 2005130166 A, 10.06.2006 | |||
| Salvatella M | |||
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |

