Область техники, к которой относится изобретение
Настоящее изобретение относится к способу эффективного отделения химического элемента от урана (VI), начиная с азотнокислой водной фазы, в цикле извлечения для урана, где данный химический элемент присутствует в данной водной фазе в концентрации ниже, чем концентрация урана (VI), или даже как следовый элемент, и в случае, когда он является менее извлекаемым с помощью экстрагента, применяемого в данном цикле извлечения, чем уран (VI).
Химический элемент может быть предпочтительно четырехвалентным актинидом, таким как нептуний или торий 228 (который является продуктом распада урана 232), но заявленный способ может быть применен к отделению любого другого химического элемента, например циркония, способного частично извлекаться совместно с ураном из кислотной водной фазы в цикле извлечения для урана.
Способ согласно настоящему изобретению может быть применен для переработки облученных ядерных топлив, и особенно в цикле очистки урана, известном как «второй урановый цикл» PUREX процесса или любого другого процесса, производного от последнего, или с целью облегчения очистки урана от загрязнителя, такого как нептуний, или с целью рекуперации химического элемента, который присутствует в качестве следового элемента в сконцентрированном по урану водном потоке, поступающем из первого цикла очистки данного процесса, и который эффективен также для тория 228; данные две цели, кроме того, не являются взаимно исключающими.
Предшествующий уровень техники
На современных заводах для повторной переработки отработанных ядерных топлив, таких как COGEMA UP3 и UP2-800 во Франции, Thorp в Великобритании или Rokkasho в Японии, повторная переработка отработанных ядерных топлив основана на PUREX процессе и включает несколько циклов очистки.
Первый цикл очистки имеет целью отделить совместно уран и плутоний от продуктов деления, затем выполнить разделение этих двух элементов на два потока.
Таким образом, этот первый цикл начинается с операции, состоящей в совместном извлечении урана и плутония, первого в степени окислении VI, второго в степени окисления IV, из азотнокислой водной фазы, которая получается при растворении отработанного ядерного топлива в азотной кислоте и которая соответственно упоминается как «жидкость после растворения» (dissolution liquor).
Данное совместное извлечение выполняют в не смешивающейся с водой органической фазе, которая составлена из экстрагента, имеющего высокое сродство к урану (VI) и плутонию (IV), в данном случае три-н-бутилфосфата (в дальнейшем TBP), обычно 30 об.% раствора в органическом разбавителе, обычно углеводороде, таком как гидрированный тетрапропилен (в дальнейшем ТРН), н-додекан или «керосин без запаха». Оно дополнительно включает стадию промывки органической фазы, выполняемую азотнокислым водным раствором, которая предназначена, чтобы улучшить очистку урана и плутония от продуктов деления.
Впоследствии выполняют операцию разделения урана и плутония. Она основана на легком изменении степени окисления плутония (IV), который переводят в трехвалентное состояние, в котором его экстрагируемость с помощью TBP является минимальной, чтобы способствовать его переходу в азотнокислый водный раствор и, таким образом, отделить его от урана (VI), который сам остается в органической фазе. Данное восстановление выполняют, применяя восстанавливающий агент, нитрат четырехвалентного урана, и агент, разлагающий соединения азотистой кислоты (anti-nitrous agent), гидразин, роль которого состоит в том, чтобы стабилизировать как нитрат четырехвалентного урана, так и плутоний (III) при помощи разрушения азотистой кислоты.
Нептуний, присутствующий в жидкости после растворения, извлекается главным образом в форме нептуния (VI), совместно с ураном и плутонием. Подобным образом, торий 228, который существует в водном растворе только в четырехвалентном состоянии, частично извлекается вместе с ураном и плутонием (Germain et al., Journal of Inorganic Nuclear Chemistry, 1970, vol.32, pp.245-253).
Во время операции разделения нептуний (VI) восстанавливается нитратом четырехвалентного урана в нептуний (IV), в каковом состоянии он является извлекаемым с помощью TBP, однако менее чем в степени окислении VI. Поэтому он остается в органической фазе вместе с ураном, как это делает и большая часть тория (IV).
После операции разделения уран (VI) отделяют от органической фазы посредством азотнокислого водного раствора, а четырехвалентные актиниды, которые следовали за ним во время предшествующих операций (нептуний (IV), плутоний (IV) и торий (IV), причем два последних элемента присутствовали в качестве следовых элементов), отделяются вместе с ним.
Исходящий водный поток, поступающий от операции отделения урана, после операции концентрации между циклами подвергается затем второму циклу очистки, называемому «второй урановый цикл», чтобы очистить уран от нептуния. Последний удаляют с помощью регулирования степени окисления до состояния V, в котором он является неспособным к экстракции при помощи TBP.
Данное регулирование является более трудным для выполнения, чем операция, которая состоит в регулировании степени окисления нептуния до состояния IV и в удалении последнего благодаря насыщению органической фазы ураном (VI), которое возможно, поскольку риск критичности будет устранен вследствие удаления главной части плутония на стадии разделения. Вычисления, подтвержденные экспериментальными испытаниями, показывают, что эта конфигурация, однако, подразумевает в части экстрактора, расположенной непосредственно вблизи фронта извлечения урана, накопление азотной кислоты таким образом, чтобы позволить образование пика нептуния (IV).
Данная конфигурация, с одной стороны, предоставляет систему, чрезвычайно чувствительную даже к наименьшим изменениям в насыщенности органической фазы ураном (VI), а с другой, делает остановку цикла без загрязнения фракции урана, переработанной в конце операции, очень трудной.
Более того, удаление тория 228 из фронта извлечения урана хотя и облегчено относительно удаления нептуния (IV) вследствие более низкой экстрагируемости четырехвалентного тория с помощью TBP, однако остается ограниченным по той же самой причине, а именно образования пика тория выше по течению относительно направления потока органической фазы в экстракторе.
Поэтому авторы настоящего изобретения предложили способ, который позволяет в цикле для извлечения урана во «втором урановом цикле» PUREX процесса очень эффективно отделять химический элемент от урана (VI), начиная от азотнокислой водной фазы, когда данный химический элемент присутствует в данной водной фазе в концентрации ниже, чем концентрация урана (VI), или даже в качестве следового элемента и когда он, кроме того, является менее экстрагируемым с помощью экстрагента, выбранного для проведения данного цикла извлечения, чем уран (VI).
Более того, авторы настоящего изобретения предложили способ, который позволяет извлекать химический элемент в концентрированной и очищенной форме в случае, когда этот элемент является ценным.
Краткое изложение сути настоящего изобретения
Данная и другие цели достигнуты в способе для отделения химического элемента от урана (VI), начиная с азотнокислой водной фазы А1, в цикле извлечения для урана, включающем:
a) стадию извлечения урана (VI), на которой водная фаза протекает при скорости потока D1 в первом экстракторе (10) в противотоке к органической фазе, не смешивающейся с водой и содержащей экстрагент, и
b) стадию для промывки органической фазы, полученной после стадии а), азотнокислой водной фазой A2, на которой органическая фаза протекает во второй экстрактор (11) в противотоке к фазе A2;
в котором первый и второй экстракторы связаны друг с другом таким способом, что водная фаза, протекающая в первом экстракторе, образована фазой A1 и фазой A2, полученной после стадии b); и
в котором поскольку химический элемент присутствует в фазе A1 в концентрации ниже, чем концентрация урана (VI), и является менее экстрагируемым указанным экстрагентом, чем уран (VI), он накапливается в органической фазе в ходе стадии a);
причем указанный способ характеризуется тем, что стадия a) включает отбор части водной фазы, протекающей в первом экстракторе, при пике накопления химического элемента в органической фазе, или, иначе, выше по течению (относительно направления потока органической фазы) от этого пика.
Согласно настоящему изобретению точное положение места отбора в первом экстракторе выбирают в зависимости от основной цели, которой стремятся добиться.
Таким образом, отбор выше по течению от пика накопления химического элемента в органической фазе позволит получать улучшенную очистку урана от этого химического элемента и наоборот и поэтому будет предпочтителен в случае, когда требуется, по существу, очистить уран от загрязнителя, тогда как отбор на пике накопления химического элемента в органической фазе позволит получать максимальную концентрацию химического элемента, которая должна быть получена, в отобранной части водной фазы, но за счет большего загрязнения этого элемента ураном, и будет поэтому предпочтителен в случае, когда требуется, по существу, извлечь этот элемент в целях его использования.
Где бы ни находилось положение места отбора, предпочтительно, чтобы пик накопления химического элемента в органической фазе был очень четким или, по меньшей мере, настолько явным, насколько это возможно.
Соответственно эксплуатационные режимы на стадиях a) и b) предпочтительно выбирают таким образом, чтобы получить:
- с одной стороны, очень высокое извлечение урана на стадии a) и также насыщенность ураном органической фазы до того, как она покинет первый экстрактор; и
- с другой стороны, коэффициент извлечения химического элемента больше чем 1 и предпочтительно много больше чем 1 в части первого экстрактора, расположенной выше по течению (относительно направления потока органической фазы) фронта извлечения урана, но меньше чем 1 и предпочтительно много меньше чем 1 во втором экстракторе, а также в части первого экстрактора, ближайшей к вводу в него водной фазы.
Таким образом, в случае, например, когда химический элемент, который должен быть отделен от урана, представляет собой торий 228 и когда в качестве органической фазы для применения выбран три-н-бутилфосфат концентрацией 30 об.% в органическом разбавителе, данные критерии будут удовлетворены, как показано в экспериментальных результатах, которые описаны в публикации автора Germain и др., при поддержании:
- в части первого экстрактора, расположенной выше по течению от фронта извлечения урана: концентрации азотной кислоты выше чем 2 моль/л при отношении скоростей потоков органической фазы/водной фазы около 3 в первом экстракторе;
- во втором экстракторе и в части первого экстрактора, ближайшей к вводу водной фазы в последний: концентрации азотной кислоты, равной или меньшей чем 2 моль/л при отношении скоростей потоков органической фазы/водной фазы около 6 во втором экстракторе.
Это является одинаково применимым и к другим химическим частицам, которые обладают поведением, аналогичным поведению тория 228, таким как, например, нептуний (IV).
Кроме того, желательно, чтобы скорость потока D2 отбора представляла долю скорости потока D1, такую, что продукт концентрирования химического элемента в водной фазе в момент отбора и скорость потока D2 равнялись потоку этого элемента, поступающему в первый реактор, таким образом, чтобы входной и выходной потоки указанного элемента были сбалансированы.
В случае когда желательно извлекать химический элемент в целях его использования, способ согласно настоящему изобретению дополнительно включает одну или более стадию для концентрирования и очистки химического элемента, присутствующего в отобранной части водной фазы.
Таким образом, в первом предпочтительном варианте воплощения способа согласно настоящему изобретению последнее включает стадию c1) для извлечения урана, присутствующего в отобранной части водной фазы, посредством органической фазы того же состава, как тот, который применялся на стадии a), сопряженную со стадией d1) для промывки данной органической фазы азотнокислой водной фазой A3.
Согласно настоящему изобретению стадию c1) преимущественно выполняют добавлением отобранной части водной фазы к фазе A3, полученной после стадии d1, и переработкой получающегося водного потока в третьем экстракторе в противотоке к органической фазе.
Таким образом, с помощью отбора части этого водного потока на стадии c1) на выходе из экстрактора и в то же время возвращая оставшуюся часть этого потока в первый экстрактор, данный химический элемент может быть извлечен в сконцентрированной и очищенной форме по отношению к формам, полученным на стадиях a) и b), без образования дополнительных исходящих потоков.
В другом предпочтительном варианте воплощения способа согласно настоящему изобретению, который особенно хорошо адаптирован для случая, когда химический элемент присутствует в качестве следового элемента в фазе A1, этот способ включает стадию c2), состоящую в подвергании отобранной части водной фазы одному или более хроматографированию на неподвижной твердой фазе для того, чтобы сконцентрировать и очистить химический элемент, присутствующий в этой фазе.
Предпочтительно неподвижная твердая фаза представляет собой ионообменную смолу.
Согласно настоящему изобретению экстрагент, присутствующий в органических фазах, используемых на данных стадиях, может быть любой молекулой, про которую известно, что она демонстрирует специфическое сродство по отношению к урану. Однако предпочтительно применяется триалкилфосфат и, в частности, три-н-бутилфосфат, который является экстрагентом в PUREX процессе. В этом случае последний предпочтительно применяется в виде 30%-ного раствора (по объему) в органическом разбавителе, преимущественно линейном или разветвленном додекане, таком как н-додекан или гидрированный тетрапропилен.
Фаза A1 предпочтительно представляет собой азотнокислую водную фазу высокой кислотности с концентрацией азотной кислоты примерно от 4 до 6 моль/л, тогда как азотнокислые водные фазы, используемые для операций промывки (фазы A2 и A3), имеют низкую кислотность, например, с концентрацией азотной кислоты менее чем или равной 2 моль/л.
Способ согласно настоящему изобретению предоставляет многочисленные преимущества, а именно:
- допущение возможности очень эффективной очистки урана (VI) от химического элемента и наоборот;
- допущение также возможности очень эффективной очистки химического элемента от загрязнителей, которые являются неизвлекаемыми из урана (VI);
- допущение возможности концентрирования и очистки химического элемента, если это является желательным для извлечения данного элемента.
Кроме того, при использовании в цикле извлечения урана, после остановки цикла способ согласно настоящему изобретению позволяет избежать происходящего загрязнения урана химическим элементом: чтобы достичь этого, требуется только выполнять стадию a), подавая в первый реактор очищенный уран, в то же время продолжая отбирать часть водной фазы, протекающей в этом реакторе, чтобы повторно сорбировать накапливание химического элемента, не загрязняя последнюю порцию урана.
Согласно настоящему изобретению химический элемент предпочтительно представляет собой четырехвалентный актинид, выбранный из тех, которые расположены между нептунием и торием 228.
Другой целью настоящего изобретения является операция повторной переработки отработанного ядерного топлива, которая характеризуется тем, что в ней осуществляется процесс разделения, такой, как определено ранее.
Еще одной целью настоящего изобретения является цикл очистки урана из PUREX процесса, который характеризуется тем, что он включает реализацию способа разделения, такого, как определено ранее, в котором химический элемент, который должен быть отделен от урана (VI), предпочтительно является четырехвалентным актинидом, выбранным из тех, которые расположены между нептунием и торием 228.
Настоящее изобретение будет лучше понято из приложенных чертежей.
Само собой разумеется, что чертежи приведены только для иллюстрации предмета настоящего изобретения и ограничивают этот предмет.
Краткое описание чертежей
Фигуры 1A и 1B демонстрируют схему цикла извлечения и профили концентрации, а фигуры 2 и 3 относятся к примерам вариантов воплощения способа согласно настоящему изобретению. Таким образом:
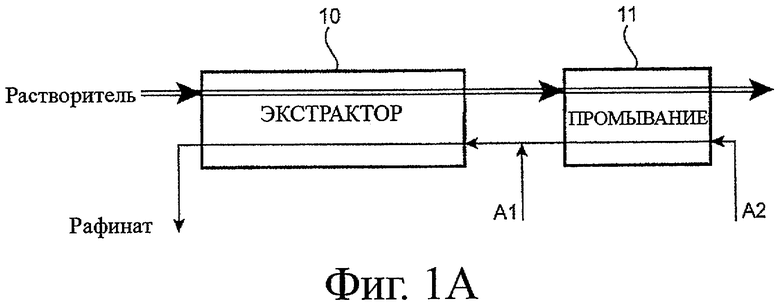
- фигура 1A соответствует принципиальной схеме цикла извлечения для очистки урана (VI), присутствующего в азотнокислой водной фазе, от химического элемента, менее экстрагируемого, чем уран, например, в структуре «второго уранового цикла» PUREX процесса;
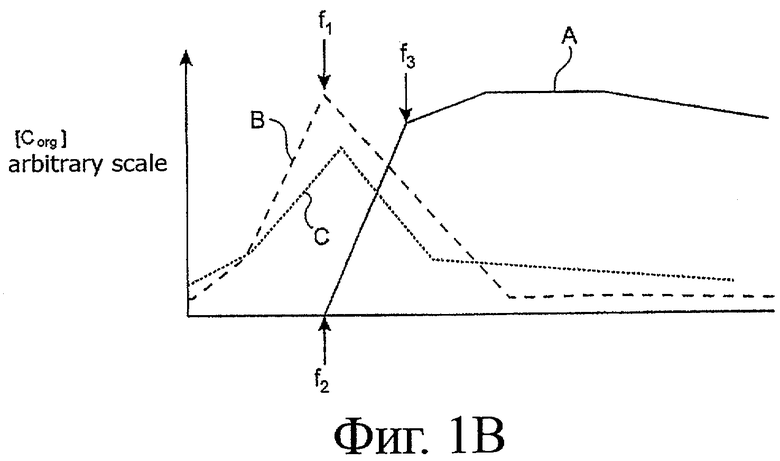
- фигура 1B показывает профили концентрации [Corg] урана (VI) (кривая A) и химического элемента (кривая B) в органической фазе вместе с профилем кислотности (кривая C) этой фазы в ходе цикла извлечения, показанного на фигуре 1A;
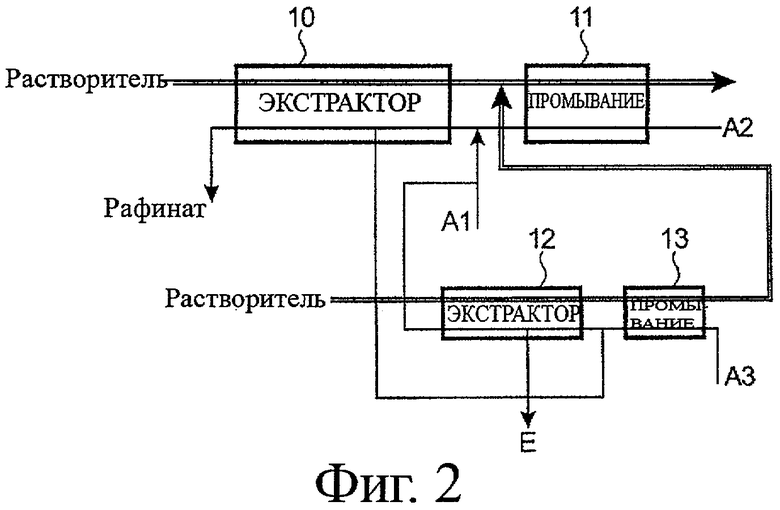
- фигура 2 иллюстрирует пример первого предпочтительного варианта воплощения способа согласно настоящему изобретению;
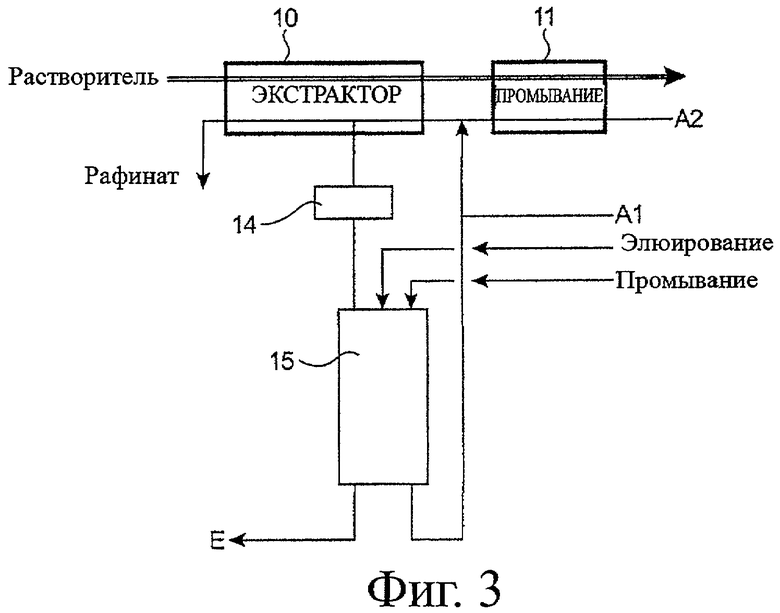
- фигура 3 иллюстрирует пример второго предпочтительного варианта воплощения способа согласно настоящему изобретению.
На фигурах 1A, 2 и 3 экстракторы обозначены прямоугольниками, потоки органической фазы обозначены двойной линией, и эта фаза упрощенно обозначена как «растворитель», тогда как водные потоки обозначены одинарной линией.
Операции промывки водных фаз с помощью органической фазы преднамеренно не показаны для упрощения чертежей.
Подробное описание настоящего изобретения
Прежде всего, касаясь фигуры 1A, она схематично показывает цикл извлечения, такой, который способен быть претворенным, для очистки урана (VI), присутствующего в азотнокислой водной фазе, обозначенной как фаза A1, от химического элемента, обозначенного как элемент Е, менее экстрагируемого, чем уран, например, в структуре «второго уранового цикла» PUREX процесса.
В этой фазе A1 уран (VI) присутствует в высокой концентрации, например около 400 г/л, тогда как элемент E, который является, например, нептунием (IV) или торием 228 (который также является четырехвалентным), присутствует при намного более низкой концентрации, например около 1% или менее, чем концентрация урана, или, иначе, в качестве следового элемента.
Касаясь концентрации азотной кислоты в фазе A1, она равна, например, от 4 до 6 моль/л.
В данном примере цикл извлечения включает:
- стадию извлечения, цель которой состоит в том, чтобы извлечь уран (VI) из фазы A1 посредством органической фазы, не смешивающейся с водой и содержащей экстрагент, способный к извлечению урана и, в меньшей степени, элемента E, и
- стадию промывки органической фазы азотнокислой водной фазой (далее обозначенной как фаза A2), цель которой состоит в том, чтобы отделить от органической фазы часть элемента E, которая была извлечена совместно с ураном (VI) на предшествующей стадии.
Органическая фаза состоит, например, из 30 об.% три-н-бутилфосфата в додекане, тогда как фаза A2 представляет собой, например, водный раствор с кислотностью менее чем 2 моль/л HNO3, например около 1 моль/л.
Как можно увидеть на фигуре 1A, эти стадии извлечения и промывки выполняют в двух различных экстракторах, 10 и 11 соответственно, которые предпочтительно являются многоступенчатыми экстракторами (типа смесителя-отстойника или пульсационной колонны), которые связаны вместе и в которых потоки растворителя и водной фазы установлены в противоположных направлениях.
Поэтому экстрактор 10 снабжают:
- с одного из его концов свежим потоком органической фазы,
- с другого из его концов водным потоком, образованным фазой A2, используемой, чтобы промывать органическую фазу в экстракторе 11, к которой добавляют фазу A1; тогда как экстрактор 11 снабжается:
- с одного из его концов органической фазой, используемой, чтобы экстрагировать уран в экстракторе 10, и
- с другого из его концов свежим потоком фазы A2.
Теперь будет рассмотрена фигура 1B, которая показывает профили концентрации [Corg] урана (кривая A) и элемента E (кривая B) в органической фазе вместе с профилем кислотности (кривая C) этой фазы на всем его пути в экстракторах 10 и 11.
Эта фигура показывает, что происходит в части экстрактора 10, расположенной выше по течению (в направлении циркуляции органической фазы) фронта извлечения урана, которая соответствует части кривой A, заключенной между стрелками f2 и f3, - накопление азотной кислоты в органической фазе, которое способствует поступлению элемента E в органическую фазу и образованию пика накопления данного элемента в данной органической фазе (и, неразделимым образом, также в водной фазе).
Существование этого пика накопления элемента E противодействует эффективному разделению урана и этого элемента и, следовательно, возможности удовлетворительной очистки урана от элемента E и, аналогично, элемента E от урана.
Поэтому часть водной фазы, протекающей в экстракторе 10, отбирают на пике накопления элемента E в органической фазе или до начала этого пика, другими словами, в конфигурации, показанной на фигуре 1B, в части реактора 10, расположенной по стрелке f1 или выше по течению (относительно направления потока органической фазы) от этой стрелки.
Как показано ранее, точное положение места отбора выбирают в зависимости от параметра, который желательно одобрить.
Таким образом, отбор на пике накопления химического элемента в органической фазе позволит получить максимальную концентрацию химического элемента в отобранной части водной фазы, но за счет относительно высокого загрязнения этого элемента ураном, тогда как отбор выше по течению от этого пика будет допускать улучшенную очистку урана от урана и наоборот, но за счет более низкой концентрации элемента E в отобранной части водной фазы.
Теперь будет рассмотрена фигура 2, которая иллюстрирует пример первого предпочтительного варианта воплощения способа, в котором часть водной фазы, отобранная в экстракторе 10, подвергнута дополнительному циклу извлечения, чтобы сконцентрировать и очистить элемент E.
Следовательно, заявленный способ включает два цикла извлечения, сопряженные друг с другом, а именно:
- первый цикл, который идентичен тому, который проиллюстрирован на фигуре 1A, с тем отличием, что часть водной фазы, протекающая в экстракторе 10, отобрана на пике накопления элемента E в органической фазе или выше по течению (относительно направления потока органической фазы) от этого пика, и
- второй цикл, который включает:
- по существу, стадию извлечения, цель которой состоит в том, чтобы извлечь уран (VI), присутствующий в отобранной части водной фазы, в экстракторе 10 из этой части водной фазы посредством органической фазы, которая преимущественно имеет тот же состав, который используется в первом цикле извлечения (TBP с концентрацией 30 об.% в органическом разбавителе), и
- стадию промывки органической фазы азотнокислой водной фазой (далее обозначенной как фаза A3), например водным раствором с кислотностью менее чем 2 моль/л HNO3, цель которой состоит в том, чтобы отделить от органической фазы часть элемента E, извлеченного совместно с ураном на предшествующей стадии.
Как показано на фигуре 2, стадии извлечения и промывки этого второго цикла выполняют подобно тем же стадиям первого цикла в двух различных экстракторах, 12 и 13 соответственно, и в которых потоки растворителя и водных фаз установлены в противоположных направлениях.
Таким образом, экстрактор 12 снабжают:
- с одного из его концов свежим потоком органической фазы, а
- с другого из его концов водным потоком, образованным фазой A3, используемой, чтобы промывать органическую фазу в экстракторе 13, к которой добавлена отобранная в экстракторе 10 часть водной фазы;
тогда как экстрактор 13 снабжается:
- с одного из его концов органической фазой, используемой для того, чтобы извлекать уран в экстракторе 12, а
- с другого из его концов свежим потоком фазы A3.
Отбирая часть водной фазы, протекающей в экстракторе 12, и в то же время направляя, с одной стороны, оставшуюся часть этой водной фазы с выхода из этого экстрактора, так, чтобы объединить с фазой A1 до того, как последняя будет добавлена к фазе A2, и, с другой стороны, органическую фазу, поступающую из экстрактора 13, так, чтобы объединить с органической фазой, поступающей из экстрактора 10 до того, как последняя входит в экстрактор 11, возможно сделать так, чтобы второй цикл извлечения работал под управлением обратной связи первого, и возвращать в части водной фазы, отобранной в экстракторе 12, элемент E, как сконцентрированный, так и очищенный, без образования дополнительных исходящих потоков по отношению к тем, которые образованы в первом цикле извлечения.
Теперь будет рассмотрена фигура 3, которая иллюстрирует пример второго предпочтительного варианта воплощения способа согласно настоящему изобретению, в котором отобранная часть водной фазы в экстракторе 10 подвергается дополнительным операциям хроматографирования на ионообменной смоле, чтобы сконцентрировать и очистить элемент E, который она содержит, и который особенно хорошо приспособлен к случаю, в котором элемент E появляется в качестве следового элемента в фазе A1.
В этом примере данный способ включает, прежде всего, цикл извлечения, который является идентичным показанному на фигуре 1A с тем отличием, что часть водной фазы, протекающую в экстракторе 10, отбирают на пике накопления элемента E в органической фазе или выше по течению (относительно направления потока органической фазы) от этого пика.
Затем часть отобранной водной фазы подают в буферный резервуар 14, где ее сохраняют прежде, чем направить к колонне 15, заполненной ионообменной смолой.
Элюирование, затем промывание смолы азотнокислыми водными фазами позволяют извлекать на выходе из колонны фракции, богатые элементом E, и фракции, богатые ураном (VI). Последние направляют, чтобы объединить с фазой A1 до того, как она добавлена к фазе A2.
| название | год | авторы | номер документа |
|---|---|---|---|
| УСОВЕРШЕНСТВОВАНИЕ СПОСОБА PUREX И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2400841C2 |
| СПОСОБ ПЕРЕРАБОТКИ ОТРАБОТАННОГО ЯДЕРНОГО ТОПЛИВА, ВКЛЮЧАЮЩИЙ СТАДИЮ ОЧИСТКИ УРАНА (VI) ОТ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО АКТИНИДА (IV) ПУТЕМ ПОЛУЧЕНИЯ КОМПЛЕКСА ДАННОГО АКТИНИДА (IV) | 2014 |
|
RU2663882C1 |
| СПОСОБ ПЕРЕРАБОТКИ ОТРАБОТАННОГО ЯДЕРНОГО ТОПЛИВА, НЕ ТРЕБУЮЩИЙ ВОССТАНОВИТЕЛЬНОЙ РЕЭКСТРАКЦИИ ПЛУТОНИЯ | 2011 |
|
RU2558332C9 |
| СПОСОБ ОТДЕЛЕНИЯ УРАНА ( VI ) ОТ АКТИНОИДОВ ( IV ) И/ИЛИ ( VI ) И ЕГО ИСПОЛЬЗОВАНИЕ | 2004 |
|
RU2352006C2 |
| УЛУЧШЕННЫЙ СПОСОБ ПЕРЕРАБОТКИ ОТРАБОТАННОГО ЯДЕРНОГО ТОПЛИВА | 2010 |
|
RU2537952C2 |
| СПОСОБ РЕГЕНЕРАЦИИ ОТРАБОТАННОГО ЯДЕРНОГО ТОПЛИВА И ПОЛУЧЕНИЯ СМЕШАННОГО УРАН-ПЛУТОНИЕВОГО ОКСИДА | 2007 |
|
RU2431896C2 |
| СПОСОБ ОБРАБОТКИ ВОДНОГО АЗОТНОКИСЛОГО РАСТВОРА, ПОЛУЧЕННОГО ПРИ РАСТВОРЕНИИ ОТРАБОТАВШЕГО ЯДЕРНОГО ТОПЛИВА, ВЫПОЛНЯЕМЫЙ В ОДНОМ ЦИКЛЕ И НЕ ТРЕБУЮЩИЙ КАКОЙ-ЛИБО ОПЕРАЦИИ, ВКЛЮЧАЮЩЕЙ ВОССТАНОВИТЕЛЬНУЮ РЕЭКСТРАКЦИЮ ПЛУТОНИЯ | 2016 |
|
RU2706954C2 |
| СПОСОБ ПЕРЕРАБОТКИ ОБЛУЧЕННОГО ТОПЛИВА АЭС | 2013 |
|
RU2535332C2 |
| СПОСОБ ВЫВЕДЕНИЯ НЕПТУНИЯ ПРИ ФРАКЦИОНИРОВАНИИ ДОЛГОЖИВУЩИХ РАДИОНУКЛИДОВ | 2010 |
|
RU2454740C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОВМЕСТНОГО РАСТВОРА U И Pu | 2014 |
|
RU2561065C1 |
Способ отделения химического элемента от урана (VI), начиная от азотнокислой водной фазы А1 в цикле извлечения для урана, включает несколько стадий. Первая стадия - извлечение урана (VI), в которой водная фаза протекает со скоростью потока D1 в первом экстракторе в противотоке к органической фазе, не смешивающейся с водой и содержащей экстрагент. Вторая стадия предназначена для промывания органической фазы, полученной после первой стадии, азотнокислой водной фазой А2, на которой органическая фаза протекает во втором экстракторе в противотоке к фазе А2. При этом первый и второй экстракторы связаны друг с другом таким способом, что водная фаза, протекающая в первом экстракторе, образована фазой А1 и фазой А2, полученной после второй стадии. Поскольку химический элемент присутствует в фазе А1 в концентрации ниже, чем концентрация урана (VI), и является менее экстрагируемым указанным экстрагентом, чем уран (VI), он накапливается в органической фазе в ходе первой стадии. Причем первая стадия включает отбор части водной фазы, протекающей в первом экстракторе, на пике накопления химического элемента в органической фазе или, иначе, выше по течению, относительно направления потока органической фазы, от этого пика. Предложены также способ переработки отработанного ядерного топлива и цикл очистки урана. Заявленные изобретения обеспечивают извлечение ценного химического элемента в концентрированной и очищенной форме. 3 н. и 13 з.п. ф-лы, 4 ил.
1. Способ отделения химического элемента от урана (VI), начиная от азотнокислой водной фазы А1, в цикле извлечения для урана, который включает следующие стадии:
a) стадию извлечения урана (VI), в которой водная фаза протекает со скоростью потока D1 в первом экстракторе (10) в противотоке к органической фазе, несмешивающейся с водой и содержащей экстрагент, и
b) стадию для промывания органической фазы, полученной после стадии а), азотнокислой водной фазой А2, на которой органическая фаза протекает во втором экстракторе (11) в противотоке к фазе А2;
в котором первый и второй экстракторы связаны друг с другом таким способом, что водная фаза, протекающая в первом экстракторе, образована фазой А1 и фазой А2, полученной после стадии b); и поскольку химический элемент присутствует в фазе А1 в концентрации ниже, чем концентрация урана (VI), и является менее экстрагируемым указанным экстрагентом, чем уран (VI), он накапливается в органической фазе в ходе стадии а);
причем стадия а) включает отбор части водной фазы, протекающей в первом экстракторе, на пике накопления химического элемента в органической фазе или, иначе, выше по течению, относительно направления потока органической фазы, от этого пика.
2. Способ по п.1, в котором скорость потока D2 отобранной части представляет собой долю скорости потока D1, такую, что продукт концентрирования химического элемента в водной фазе, протекающей в первом экстракторе, когда происходит отбор, и скорость потока D2, равны потоку этого элемента, входящему в первый экстрактор.
3. Способ по п.1, который дополнительно включает одну или более стадию для концентрирования и очистки химического элемента, присутствующего в отобранной части водной фазы.
4. Способ по п.3, который включает стадию с1) для извлечения урана, присутствующего в отобранной части водной фазы, посредством органической фазы того же состава, как та, которая применяется на стадии а), сопряженную со стадией d1) для промывки этой органической фазы азотнокислой водной фазой A3.
5. Способ по п.4, в котором стадию c1) выполняют, добавляя отобранную часть водной фазы к фазе A3, полученной после стадии d1), и заставляя получающийся водный поток протекать в третьем экстракторе (12) в противотоке к органической фазе.
6. Способ по п.5, в котором стадия c1) включает отбор части водного потока, протекающего в третьем экстракторе, и возвращение другой части к первому экстрактору.
7. Способ по п.3, который включает стадию c2), состоящую в подвергании отобранной части водной фазы одному или более хроматографированию на неподвижной твердой фазе.
8. Способ по п.7, в котором неподвижная твердая фаза является ионообменной смолой.
9. Способ по п.1, в котором экстрагент представляет собой три-н-бутилфосфат, предпочтительно применяемый, как 30 об.%-ный раствор в органическом разбавителе, предпочтительно в додекане.
10. Способ по п.9, в котором фаза А1 имеет молярность от 4 до 6 М.
11. Способ по п.9, характеризующийся тем, что фаза А2 демонстрирует молярность, меньшую чем или равную 2 М.
12. Способ по п.9, в котором фаза A3 имеет молярность, меньшую чем или равную 2 М.
13. Способ по п.1, в котором химический элемент представляет собой четырехвалентный актинид, выбранный из тех, которые расположены между нептунием и торием 228.
14. Способ переработки отработанного ядерного топлива, который осуществляют на основе способа разделения по п.1.
15. Цикл очистки урана из PUREX процесса, который осуществляют на основе способа разделения по п.1.
16. Цикл очистки по п.15, в котором отделяют четырехвалентный актинид, выбранный из тех элементов, которые расположены между нептунием и торием, от урана (VI).
| FR 2862804 A1, 27.05.2005 | |||
| WO 9962824 A1, 09.12.1999 | |||
| СПОСОБ СОВМЕСТНОГО СЕЛЕКТИВНОГО ВОССТАНОВЛЕНИЯ ПЛУТОНИЯ (IV) И НЕПТУНИЯ (VI) ДЛЯ ОТДЕЛЕНИЯ ИХ ОТ УРАНА (VI) | 1998 |
|
RU2155711C1 |
| СПОСОБ ПЕРЕРАБОТКИ ОБЛУЧЕННОГО ЯДЕРНОГО ТОПЛИВА (ВАРИАНТЫ) | 2003 |
|
RU2249267C2 |

