Техническая область
Настоящее изобретение в основном относится к области систем контролируемой доставки и, более конкретно, систем контролируемой доставки, содержащих активное вещество, способное обеспечивать местный анестетический эффект, где системы являются пригодными для применения в связи с хирургическим и медицинским лечением и в качестве лекарственных средств для применения в процедурах послеоперационного восстановления.
Предпосылки изобретения
Биоразлагаемые системы для контролируемой доставки активных веществ хорошо известны в данной области. Биоразлагаемые носители для доставки лекарственного средства являются применимыми, поскольку они устраняют необходимость удалять устройство с истощившимся лекарственным средством.
Наиболее распространенными материалами носителей, применяемыми для систем контролируемой доставки, являются полимеры. Область биоразлагаемых полимеров быстро развивалась с тех пор, как синтез и биоразлагаемость полимолочной кислоты были описаны Kulkarni et al. (1966). Arch. Surg. 93:839. Примеры других полимеров, которые были опубликованы как применимые в качестве связующего материала для систем контролируемой доставки, включают в себя полиангидриды, полиэфиры, такие как полигликолиды и сополимеры лактида и гликолида, полиаминокислоты, такие как полилизин, полимеры и coполимеры полиэтиленоксида, полиэтиленоксид с концевой акрильной группой, полиамиды, полиуретаны, полиортоэфиры, полиакрилонитрилы, и полифосфазены. См., например, патенты США №№ 4891225 и 4906474 (полиангидриды); 4767628 (полилактид, сополимер молочной и гликолевой кислот); 4530840 (полилактид, полигликолид и coполимеры) и 5234520 (биоразлагаемые полимеры для контролируемой доставки при лечении периодонтального заболевания).
Хорошо известны разлагаемые материалы биологического происхождения, включая, например, поперечно-сшитый желатин. Гиалуроновую кислоту сшивали и использовали в качестве разлагаемого набухающего полимера для биомедицинских применений (см., например, патент США 4957744 и Della Valle et al. (1991) Polym. Mater. Sci. Eng., 62:731-735).
Разработаны также биоразлагаемые гидрогели для использования в системах контролируемой доставки, и они служат носителями биологически активных материалов, таких как гормоны, ферменты, антибиотики, антинеопластические средства и суспензии клеток. См., например, патент США № 5149543.
Композиции гидрогеля также являются общеупотребительными в качестве субстратов для культуры клеток и тканей, слепочных материалов для протезирования, перевязочных материалов, или в качестве твердофазных материалов для применений в вытеснительной или аффинной хроматографии. Например, непористые, деформированные и/или дериватизированные композиции агарозного гидрогеля использовали в способах высокоэффективной жидкостной хроматографии и аффинной хроматографии (Li et al. (1990) Preparative Biochem. 20:107-121), а суперпористые шарики агарозного гидрогеля применяли в качестве подложки в хроматографии гидрофобного взаимодействия (Gustavsson et al. (1999) J. Сhromatography 830:275-284).
В настоящее время в качестве носителей веществ, в частности биологически активных соединений, используют многие дисперсионные системы. Дисперсионные системы, используемые для фармацевтических и косметических составов, можно классифицировать или как суспензии, или как эмульсии. Суспензии состоят из твердых частиц, размером в диапазоне от нескольких нанометров до сотен микрон, диспергированных в жидкой среде с использованием суспендирующих средств. Твердые частицы включают в себя микросферы, микрокапсулы и наносферы. Эмульсии в основном представляют собой дисперсии одной жидкости в другой, стабилизированные пленкой на границе раздела из эмульгаторов, таких как сурфактанты и липиды. Эмульсионные составы включают в себя эмульсии вода в масле и масло в воде, гетерогенные эмульсии, микроэмульсии, микрокапельки и липосомы. Микрокапельки представляют собой однослойные фосфолипидные везикулы, состоящие из сферического липидного слоя с масляной фазой внутри, например, описанные в патентах США №№ 4622219 и 4725442. Липосомы представляют собой фосфолипидные везикулы, полученные смешиванием не растворимых в воде полярных липидов с водным раствором. Неблагоприятная энтропия, обусловленная смешиванием нерастворимого липида с водой, приводит к высокоупорядоченному агрегату концентрических замкнутых мембран фосфолипида с заключенным водным раствором.
Описан ряд систем для образования имплантата in situ. Например, в патенте США № 4938763 описан способ образования имплантата путем растворения нереакционно-способного, нерастворимого в воде термопластического полимера в биологически совместимом, водорастворимом растворителе для получения жидкости, помещения жидкости внутрь организма и позволения растворителю рассеиваться для получения твердого имплантата. Раствор полимера можно поместить в организм посредством шприца. Имплантат может принимать форму окружающей полости. Альтернативно, имплантат можно образовать из реакционно-способных, жидких олигомерных полимеров, не содержащих растворителя и отверждаемых на месте для формирования твердых веществ, обычно добавлением отверждающего катализатора.
В данной области описан ряд полимерных систем контролируемой доставки для доставки местных анестетиков. Хотя такие полимерные системы доставки могут обеспечивать подходящие свойства контролируемого высвобождения анестетика и дополнительно преодолевать недостатки, связанные с инъекцией чистых анестетиков (например, рассеивание от участка-мишени, вход в кровоток и системную токсичность), трудно преодолеть конкретные недостатки, связанные с полимерными системами, такие как неспособность избежать начального системного выброса высвобождения анестетика или необходимость предоставлять усиливающие средства, чтобы преодолеть слишком низкое высвобождение анестетика из систем.
Сущность изобретения
Предоставлены неполимерные системы носителя для контролируемой доставки для введения интересующего анестетического средства. То есть, таким образом, целью настоящего изобретения является получение длительно действующей системы контролируемой доставки, высвобождающей анестетик в течение длительного периода времени, достаточного для обеспечения местного анестетического действия в участке введения по меньшей мере приблизительно 24 часа после введения, предпочтительно по меньшей мере приблизительно 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно 48-72 часа после введения. Целью настоящего изобретения является также то, что высвобождение активного анестетического средства из длительно действующей анестетической композиции происходит без начального выброса.
Более конкретно, целью настоящего изобретения является получение композиции, содержащей анестетик и фармацевтически приемлемый неполимерный носитель. Неполимерный носитель контролирует высвобождение анестетика с обеспечением анестетического действия, характеризующегося длительной местной анестезией после введения субъекту без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения, предпочтительно по меньшей мере приблизительно 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно 48-72 часа после введения.
В одном аспекте изобретения неполимерный носитель является достаточным для обеспечения либо контролируемого профиля высвобождения анестетика первого порядка, либо профиля высвобождения анестетика псевдонулевого порядка. В конкретных вариантах осуществления анестетик представляет собой местный анестетик, например местный анестетик амидного или сложноэфирного типа. В предпочтительном варианте осуществления анестетик представляет собой бупивакаин, который дополнительно может быть представлен в форме свободного основания. В других вариантах осуществления композиция способна обеспечивать длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно, предпочтительно по меньшей мере приблизительно 250 нг/мл, или по меньшей мере приблизительно 300 нг/мл, или по меньшей мере приблизительно 350 нг/мл.
В другом аспекте изобретения неполимерный носитель является по существу нерастворимым в воде или в водной биологической системе. В таких композициях фармацевтический препарат может дополнительно содержать растворитель, который является диспергируемым, растворимым в воде или водной системе или смешиваемым с ней. Растворитель, таким образом, может быть органическим растворителем, способным рассеиваться, диффундировать или вымываться из композиции при помещении в биологическую систему, в результате чего носитель затем может коагулировать или осаждаться с образованием твердого имплантата in situ.
В другом аспекте изобретения неполимерный носитель является жидким, предпочтительно жидким материалом носителя с высокой вязкостью («HVLCM»), обладающим вязкостью по меньшей мере приблизительно 5000 сП при 37°С, который без примесей не кристаллизуется в условиях окружающей среды или в физиологических условиях. Такие жидкие материалы носителя можно сочетать с растворителем, в котором материал носителя является растворимым. При применении HVLCM предпочтительно, чтобы растворитель являлся достаточным для снижения вязкости HVLCM. В конкретных вариантах осуществления растворителем может быть второе анестетическое средство, такое как бензиловый спирт. Композиции могут быть представлены в любой подходящей форме, например в виде эмульсии, пасты, геля, суспензии, крема, пленки, спрея, твердого вещества, частицы, микрочастицы, порошка, имплантата или жидкости. В конкретных вариантах осуществления композиция дополнительно включает материал, несмешиваемый с неполимерным носителем, например, где композиция представляет собой эмульсию. В данных композициях носитель может присутствовать либо в диспергированной, либо в дисперсионной фазе эмульсии.
Целью настоящего изобретения является также получение композиции, содержащей анестетик и фармацевтически приемлемый неполимерный носитель. Неполимерный носитель контролирует высвобождение анестетика с обеспечением анестетического действия, характеризующегося длительной местной анестезией после введения субъекту, где композиция в дальнейшем способна обеспечивать длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно, предпочтительно по меньшей мере приблизительно 250 нг/мл, или по меньшей мере приблизительно 300 нг/мл, или по меньшей мере приблизительно 350 нг/мл. В одном аспекте изобретения композиция способна обеспечивать длительную устойчивую среднюю концентрацию в плазме (Css) на период по меньшей мере приблизительно 48 часов. В другом аспекте композиция дополнительно характеризуется как не обладающая каким-либо существенным начальным выбросом. В других аспектах неполимерный носитель является достаточным для обеспечения либо контролируемого профиля высвобождения анестетика первого порядка, либо профиля высвобождения анестетика псевдонулевого порядка. В конкретных вариантах осуществления анестетик представляет собой местный анестетик, например местный анестетик амидного или сложноэфирного типа. В предпочтительном варианте осуществления анестетик представляет собой бупивакаин, который дополнительно может быть представлен в форме свободного основания.
В другом аспекте изобретения неполимерный носитель является по существу нерастворимым в воде или в водной биологической системе. В таких композициях фармацевтический препарат может дополнительно содержать растворитель, который является диспергируемым, растворимым в воде или водной системе или смешиваемым с ней. Растворитель, таким образом, может быть органическим растворителем, способным рассеиваться, диффундировать или вымываться из композиции при помещении в биологическую систему, в результате чего носитель затем может коагулировать или осаждаться с образованием твердого имплантата in situ.
В другом аспекте изобретения неполимерный носитель является жидким, предпочтительно жидким материалом носителя с высокой вязкостью («HVLCM»), обладающим вязкостью по меньшей мере приблизительно 5000 сП при 37°С, который без примесей не кристаллизуется в условиях окружающей среды или в физиологических условиях. Такие жидкие материалы носителя можно сочетать с растворителем, в котором материал носителя является растворимым. При применении HVLCM предпочтительно, чтобы растворитель являлся достаточным для снижения вязкости HVLCM. В конкретных вариантах осуществления растворителем может быть второе анестетическое средство, такое как бензиловый спирт. Композиции могут быть представлены в любой подходящей форме, например в виде эмульсии, пасты, геля, суспензии, крема, пленки, спрея, твердого вещества, частицы, микрочастицы, порошка, имплантата или жидкости. В конкретных вариантах осуществления композиция дополнительно включает материал, несмешиваемый с неполимерным носителем, например, где композиция представляет собой эмульсию. В данных композициях носитель может присутствовать либо в диспергированной, либо в дисперсионной фазе эмульсии.
Относящимся к цели изобретения является получение композиции, содержащей первый анестетик, второй анестетик и фармацевтически приемлемый неполимерный носитель. Второй анестетик в композиции является растворителем для первого анестетика и обеспечивает начальное анестетическое действие при введении субъекту. Неполимерный носитель контролирует высвобождение первого анестетика с обеспечением последующего анестетического действия, характеризующегося длительной местной анестезией с началом в течение приблизительно 2 часов от введения субъекту без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения, предпочтительно по меньшей мере приблизительно 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно 48-72 часа после введения.
В одном аспекте изобретения неполимерный носитель является достаточным для обеспечения либо контролируемого профиля высвобождения анестетика первого порядка, либо профиля высвобождения анестетика псевдонулевого порядка. В других вариантах осуществления композиция способна обеспечивать длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно, предпочтительно по меньшей мере приблизительно 250 нг/мл, или по меньшей мере приблизительно 300 нг/мл, или по меньшей мере приблизительно 350 нг/мл. В других конкретных вариантах осуществления первый анестетик представляет собой местный анестетик, например местный анестетик амидного или сложноэфирного типа. В других вариантах осуществления второй анестетик также представляет собой растворитель для неполимерного носителя. Второй анестетик может быть растворителем, таким как спирт, ароматический спирт, кислота или производное кислоты, или любой комбинацией таких растворителей. В предпочтительном варианте осуществления второй анестетик представляет собой бензиловый спирт. В другом предпочтительном варианте осуществления первый анестетик представляет собой бупивакаин, который дополнительно может быть представлен в форме свободного основания.
В другом аспекте изобретения неполимерный носитель является по существу нерастворимым в воде или в водной биологической системе. В таких композициях фармацевтический препарат может дополнительно содержать растворитель, который является диспергируемым, растворимым в воде или водной системе или смешиваемым с ней. Растворитель, таким образом, может быть органическим растворителем, способным рассеиваться, диффундировать или вымываться из композиции при помещении в биологическую систему, в результате чего носитель затем может коагулировать или осаждаться с образованием твердого имплантата in situ.
В другом аспекте изобретения неполимерный носитель является жидким, предпочтительно жидким материалом носителя с высокой вязкостью («HVLCM»), обладающим вязкостью по меньшей мере приблизительно 5000 сП при 37°С, который без примесей не кристаллизуется в условиях окружающей среды или в физиологических условиях. Такие жидкие материалы носителя можно сочетать с растворителем, в котором материал носителя является растворимым. При применении HVLCM предпочтительно, чтобы растворитель являлся достаточным для снижения вязкости HVLCM. В конкретных вариантах осуществления растворителем может быть второе анестетическое средство, такое как бензиловый спирт. Композиции могут быть представлены в любой подходящей форме, например в виде эмульсии, пасты, геля, суспензии, крема, пленки, спрея, твердого вещества, частицы, микрочастицы, порошка, имплантата или жидкости. В конкретных вариантах осуществления композиция дополнительно включает материал, несмешиваемый с неполимерным носителем, например, где композиция представляет собой эмульсию. В данных композициях носитель может присутствовать либо в диспергированной, либо в дисперсионной фазе эмульсии.
Относящимся к цели изобретения является также получение композиции, содержащей неполимерный, нерастворимый в воде жидкий материал носителя с высокой вязкостью («HVLCM»), обладающий вязкостью по меньшей мере приблизительно 5000 сП при 37°С, который без примесей не кристаллизуется в условиях окружающей среды или в физиологических условиях, первый анестетик и второй анестетик. Здесь снова второй анестетик является растворителем для первого анестетика и обеспечивает начальное анестетическое действие при введении субъекту. HVLCM контролирует высвобождение первого анестетика с обеспечением последующего анестетического действия, характеризуемого длительной местной анестезией с началом в течение приблизительно 2 часов от введения субъекту без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения, предпочтительно по меньшей мере приблизительно 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно 48-72 часа после введения. В конкретных вариантах осуществления композиция способна обеспечивать длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно, предпочтительно по меньшей мере приблизительно 250 нг/мл, или по меньшей мере приблизительно 300 нг/мл, или по меньшей мере приблизительно 350 нг/мл.
В одном аспекте изобретения первый анестетик представляет собой местный анестетик, например местный анестетик амидного или сложноэфирного типа. В других вариантах осуществления второй анестетик также является растворителем для HVLCM. Второй анестетик может быть растворителем, таким как спирт, ароматический спирт, кислота или производное кислоты, или любой комбинацией таких растворителей. В предпочтительном варианте осуществления второй анестетик представляет собой бензиловый спирт. В другом предпочтительном варианте осуществления первый анестетик представляет собой бупивакаин, который дополнительно может быть представлен в форме свободного основания. В других предпочтительных вариантах осуществления HVLCM представляет собой сложный эфир, такой как сложный эфир сахара, например изобутират ацетата сахарозы. В таких композициях может быть полезно предусматривать растворитель, в котором HVVLCM является растворимым.
Дополнительно относящимся к цели изобретения является получение композиции, содержащей неполимерный, нерастворимый в воде жидкий материал носителя с высокой вязкостью («HVLCM»), обладающий вязкостью по меньшей мере приблизительно 5000 сП при 37°С, который без примесей не кристаллизуется в условиях окружающей среды или в физиологических условиях, первый анестетик и второй анестетик. Здесь снова второй анестетик в композиции является растворителем для первого анестетика и обеспечивает начальное анестетическое действие при введении субъекту. HVLCM контролирует высвобождение первого анестетика с обеспечением последующего анестетического действия, характеризующегося длительной местной анестезией, где композиция далее способна обеспечивать длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно, предпочтительно по меньшей мере приблизительно 250 нг/мл, или по меньшей мере приблизительно 300 нг/мл, или по меньшей мере приблизительно 350 нг/мл.
В одном аспекте изобретения композиция способна обеспечивать длительную устойчивую среднюю концентрацию анестетика в плазме (Css) на период по меньшей мере приблизительно 48 часов. В другом аспекте композиция дополнительно характеризуется как не обладающая каким-либо существенным начальным выбросом.
В другом аспекте изобретения первый анестетик представляет собой местный анестетик, например местный анестетик амидного или сложноэфирного типа. В других вариантах осуществления второй анестетик является также растворителем для HVLCM. Второй анестетик может быть растворителем, таким как спирт, ароматический спирт, кислота или производное кислоты, или любой комбинацией таких растворителей. В предпочтительном варианте осуществления второй анестетик представляет собой бензиловый спирт. В другом предпочтительном варианте осуществления первый анестетик представляет собой бупивакаин, который дополнительно может быть представлен в форме свободного основания. В других предпочтительных вариантах осуществления HVLCM представляет собой сложный эфир, такой как сложный эфир сахара, например изобутират ацетата сахарозы. В таких композициях может быть полезно предусматривать растворитель, в котором HVVLCM является растворимым.
Дополнительной целью изобретения является предоставление способа обеспечения у субъекта анестетического действия в участке. Способ включает введение композиции на участок, около участка, в участок или по соседству с участком, где композиция содержит анестетик и фармацевтически приемлемый неполимерный носитель. Неполимерный носитель контролирует высвобождение анестетика с обеспечением анестетического действия, характеризующегося длительной местной анестезией после введения субъекту без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения.
В одном аспекте изобретения анестетик представляет собой местный анестетик, например местный анестетик амидного или сложноэфирного типа.
Относящимся к цели изобретения является предоставление способа обеспечения у субъекта анестетического действия в участке. Способ включает введение композиции на участок, около участка, в участок или по соседству с участком, где композиция содержит анестетик и фармацевтически приемлемый неполимерный носитель. Неполимерный носитель контролирует высвобождение анестетика с обеспечением анестетического действия, характеризующегося длительной местной анестезией после введения субъекту, где композиция далее способна обеспечивать длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно.
В одном аспекте изобретения анестетик представляет собой местный анестетик, например местный анестетик амидного или сложноэфирного типа.
Дополнительной целью изобретения является предоставление способа обеспечения у субъекта анестетического действия в участке. Способ включает введение композиции на участок, около участка, в участок или по соседству с участком, где композиция содержит первый анестетик, второй анестетик и фармацевтически приемлемый неполимерный носитель. Второй анестетик является растворителем для первого анестетика и обеспечивает начальное анестетическое действие в участке при введении. Неполимерный носитель контролирует высвобождение первого анестетика с обеспечением последующего анестетического действия, характеризуемого длительной местной анестезией в участке с началом в течение приблизительно 2 часов от введения без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения, предпочтительно по меньшей мере приблизительно 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно 48-72 часа после введения.
В одном из аспектов изобретения неполимерный носитель является жидким, предпочтительно жидким материалом носителя с высокой вязкостью («HVLCM»), который является нерастворимым в воде и обладает вязкостью по меньшей мере приблизительно 5000 сП при 37°С и, кроме того, не кристаллизуется без примесей в условиях окружающей среды или в физиологических условиях. Такие жидкие материалы носителя можно сочетать с растворителем, в котором материал носителя является растворимым. При применении HVLCM предпочтительно, чтобы растворитель являлся достаточным для снижения вязкости HVLCM. В конкретных вариантах осуществления растворителем может быть второе анестетическое средство, такое как бензиловый спирт.
В другом аспекте изобретения первый анестетик представляет собой местный анестетик, например местный анестетик амидного или сложноэфирного типа. В других вариантах осуществления второй анестетик является также растворителем для HVLCM. Второй анестетик может быть растворителем, таким как спирт, ароматический спирт, кислота или производное кислоты, или любой комбинацией таких растворителей. В предпочтительном варианте осуществления второй анестетик представляет собой бензиловый спирт. В другом предпочтительном варианте осуществления первый анестетик представляет собой бупивакаин, который дополнительно может быть представлен в форме свободного основания. В других предпочтительных вариантах осуществления HVLCM представляет собой сложный эфир, такой как сложный эфир сахара, например изобутират ацетата сахарозы. В таких композициях может быть полезно предусматривать растворитель, в котором HVVLCM является растворимым.
В другом аспекте изобретения композицию вводят в участок местным введением, чрескожным введением, инъекцией или в виде имплантата. В конкретных вариантах осуществления композицию вводят в участок, который представляет собой хирургическую рану, и композицию вводят в рану и/или по соседству с раной. Дополнительной целью изобретения является способ обеспечения у субъекта анестетического действия в участке. Способ включает введение композиции на участок, около участка, в участок или по соседству с участком, где композиция содержит первый анестетик, второй анестетик и фармацевтически приемлемый неполимерный носитель. Второй анестетик является растворителем для первого анестетика и обеспечивает начальное анестетическое действие в участке после введения. Неполимерный носитель контролирует высвобождение первого анестетика с обеспечением последующего анестетического действия, характеризуемого длительной местной анестезией в участке, и композиция дополнительно способна обеспечивать длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно.
В одном аспекте изобретения неполимерный носитель является жидким, предпочтительно жидким материалом носителя с высокой вязкостью («HVLCM»), который является нерастворимым в воде и обладает вязкостью по меньшей мере приблизительно 5000 сП при 37°С и который, кроме того, без примесей не кристаллизуется в условиях окружающей среды или в физиологических условиях. Такие жидкие материалы носителя можно сочетать с растворителем, в котором материал носителя является растворимым. При применении HVLCM предпочтительно, чтобы растворитель являлся достаточным для снижения вязкости HVLCM. В конкретных вариантах осуществления растворителем может быть второе анестетическое средство, такое как бензиловый спирт. В другом аспекте изобретения первый анестетик представляет собой местный анестетик, например местный анестетик амидного или сложноэфирного типа. В других вариантах осуществления второй анестетик является также растворителем для HVLCM. Второй анестетик может быть растворителем, таким как спирт, ароматический спирт, кислота или производное кислоты, или любой комбинацией таких растворителей. В предпочтительном варианте осуществления второй анестетик представляет собой бензиловый спирт. В другом предпочтительном варианте осуществления первый анестетик представляет собой бупивакаин, который дополнительно может быть представлен в форме свободного основания. В других предпочтительных вариантах осуществления HVLCM представляет собой сложный эфир, такой как сложный эфир сахара, например изобутират ацетата сахарозы. В таких композициях может быть полезно предусматривать растворитель, в котором HVVLCM является растворимым.
В другом аспекте изобретения композицию вводят в участок местным введением, чрескожным введением, инъекцией или в виде имплантата. В конкретных вариантах осуществления композицию вводят в участок, который представляет собой хирургическую рану, и композицию вводят в рану и/или по соседству с раной.
Преимуществом настоящего изобретения является то, что материал неполимерного носителя способен контролировать высвобождение анестетического средства как во избежание начального выброса высвобождения, так и для обеспечения длительного анестетического действия по меньшей мере приблизительно на 24 часа. Дополнительным преимуществом по изобретению является то, что композиции легко сконструировать для получения любого числа различных фармацевтических форм и, кроме того, для получения широкого ряда различных фармакологических характеристик высвобождения в зависимости от предназначенного участка введения и медицинского применения.
Эти и другие цели, аспекты и преимущества по настоящему изобретению будут легко понятны специалисту в данной области при чтении настоящего описания и спецификации.
Краткое описание чертежей
На фигуре 1 показаны средние уровни бупивакаина в плазме в течение 7 суток (фармакодинамические результаты) из примера 3, где данные когорты 1 представлены на нижней кривой, а данные когорты 2 представлены на верхней кривой.
На фигуре 2 показаны средние уровни бупивакаина в плазме в течение 0-144 часов (фармакодинамические результаты) из примера 4, когорта 1.
На фигуре 3 показаны средние уровни бупивакаина в плазме в течение 0-12 часов (фармакодинамические результаты) из примера 4, когорта 1.
На фигуре 4 показаны средние уровни бупивакаина в плазме в течение 0-300 часов (фармакодинамические результаты) из примера 4, когорта 2, где данные подгруппы 3 представлены на нижней кривой (◇) данные подгруппы 2 представлены на средней кривой  и данные подгруппы 1 представлены на верхней кривой (Δ).
и данные подгруппы 1 представлены на верхней кривой (Δ).
На фигуре 5 показаны средние уровни бупивакаина в плазме в течение 0-12 часов (фармакодинамические результаты) из примера 4, когорта 2, где данные подгруппы 3 представлены на нижней кривой (◇), данные подгруппы 2 представлены на средней кривой и данные подгруппы 1 представлены на верхней кривой (Δ).
На фигуре 6 показаны средние показатели боли в участке разреза «в покое», записанные с использованием 0-100 мм визуально-аналоговой шкалы (VAS) из примера 4, когорта 2, где данные подгруппы 3 представлены на верхней кривой (Δ), данные подгруппы 2 представлены на средней кривой и данные подгруппы 1 представлены на нижней кривой (◇).
Подробное описание конкретных вариантов осуществления
Перед подробным описанием настоящего изобретения следует понимать, что данное изобретение не ограничено приведенными в качестве конкретных примеров материалами носителя или параметрами способа, так как они, конечно, могут меняться. Следует понимать также, что применяемая здесь терминология предназначена для описания конкретных вариантов осуществления по изобретению и не предназначена, чтобы быть ограничивающей.
Полное содержание всех публикаций, патентов и патентных заявок, процитированных здесь, или выше, или ниже, приведено здесь в качестве ссылки.
Следует отметить, что, как используется в данном описании и прилагаемой формуле изобретения, формы единственного числа включают в себя объекты ссылки во множественном числе, если содержание ясно не требует иного. Таким образом, например, обозначение «неполимерный носитель» включает в себя смесь двух или более таких носителей, обозначение «растворитель» включает в себя смесь двух или более таких растворителей, обозначение «анестетик» включает в себя смесь двух или более таких средств и т.п.
Целью настоящего изобретения является получение длительно действующей системы контролируемой доставки, высвобождающей анестетик в течение длительного периода времени, достаточного для обеспечения местного анестетического действия в участке введения по меньшей мере приблизительно на 24 часа после введения, предпочтительно по меньшей мере приблизительно на 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно на 48-72 часа после введения. Целью настоящего изобретения является также то, что высвобождение активного анестетического средства из анестетической композиция длительного действия происходит без начального выброса. Дополнительной целью настоящего изобретения является то, что композиция высвобождает активное анестетическое средство из длительно действующей анестетической композиции для обеспечения длительной устойчивой средней концентрации анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно, предпочтительно по меньшей мере приблизительно 250 нг/мл, или по меньшей мере приблизительно 300 нг/мл, или по меньшей мере приблизительно 350 нг/мл.
Целью настоящего изобретения является также получение композиции, содержащей анестетик и фармацевтически приемлемый неполимерный носитель. Неполимерный носитель контролирует высвобождение анестетика с обеспечением анестетического действия, характеризуемого длительной местной анестезией после введения субъекту без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения, предпочтительно по меньшей мере приблизительно 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно 48-72 часа после введения.
Целью настоящего изобретения является также получение композиции, содержащей анестетик и фармацевтически приемлемый неполимерный носитель. Неполимерный носитель контролирует высвобождение анестетика с обеспечением анестетического действия, характеризуемого длительной местной анестезией после введения субъекту, где композиция обеспечивает длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно, предпочтительно по меньшей мере приблизительно 250 нг/мл, или по меньшей мере приблизительно 300 нг/мл, или по меньшей мере приблизительно 350 нг/мл.
Относящимся к цели изобретения является получение композиции, содержащей первый анестетик, второй анестетик и фармацевтически приемлемый неполимерный носитель. Второй анестетик в композиции является растворителем для первого анестетика и обеспечивает начальное анестетическое действие при введении субъекту. Неполимерный носитель контролирует высвобождение первого анестетика с обеспечением последующего анестетического действия, характеризующегося длительной местной анестезией с началом в течение приблизительно 2 часов от введения субъекту без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения, предпочтительно по меньшей мере приблизительно 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно 48-72 часа после введения.
Относящимся к цели изобретения является также получение композиции, содержащей неполимерный, не растворимый в воде жидкий материал носителя с высокой вязкостью («HVLCM»), обладающий вязкостью по меньшей мере приблизительно 5000 сП при 37°С, который без примесей не кристаллизуется в условиях окружающей среды или в физиологических условиях, первый анестетик и второй анестетик. Здесь снова второй анестетик является растворителем для первого анестетика и обеспечивает начальное анестетическое действие при введении субъекту. HVLCM контролирует высвобождение первого анестетика с обеспечением последующего анестетического действия, характеризующегося длительной местной анестезией с началом в течение приблизительно 2 часов от введения субъекту без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения, предпочтительно по меньшей мере приблизительно 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно 48-72 часа после введения.
Дополнительно относящимся к цели изобретения является получение композиции, содержащей неполимерный, не растворимый в воде жидкий материал носителя с высокой вязкостью («HVLCM»), обладающий вязкостью по меньшей мере приблизительно 5000 сП при 37°С, который без примесей не кристаллизуется в условиях окружающей среды или в физиологических условиях, первый анестетик и второй анестетик. Здесь снова второй анестетик является растворителем для первого анестетика и обеспечивает начальное анестетическое действие при введении субъекту. HVLCM контролирует высвобождение первого анестетика с обеспечением последующего анестетического действия, характеризующегося длительной местной анестезией, и композиция обеспечивает длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно, предпочтительно по меньшей мере приблизительно 250 нг/мл, или по меньшей мере приблизительно 300 нг/мл, или по меньшей мере приблизительно 350 нг/мл.
Дополнительной целью изобретения является предоставление способа обеспечения у субъекта анестетического действия в участке. Способ включает введение композиции на участок, около участка, в участок или по соседству с участком, где композиция содержит анестетик и фармацевтически приемлемый неполимерный носитель. Неполимерный носитель контролирует высвобождение анестетика с обеспечением анестетического действия, характеризующегося длительной местной анестезией после введения субъекту без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения.
Дополнительной целью изобретения является предоставление способа обеспечения у субъекта анестетического действия в участке. Способ включает введение композиции на участок, около участка, в участок или по соседству с участком, где композиция содержит анестетик и фармацевтически приемлемый неполимерный носитель. Неполимерный носитель контролирует высвобождение анестетика с обеспечением последующего анестетического действия, характеризуемого длительной местной анестезией после введения субъекту, и композиция обеспечивает длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно.
Дополнительной целью изобретения является предоставление способа обеспечения у субъекта анестетического действия в участке. Способ включает введение композиции на участок, около участка, в участок или по соседству с участком, где композиция содержит первый анестетик, второй анестетик и фармацевтически приемлемый неполимерный носитель. Второй анестетик является растворителем для первого анестетика и обеспечивает начальное анестетическое действие в участке при введении. Неполимерный носитель контролирует высвобождение первого анестетика с обеспечением последующего анестетического действия, характеризующегося длительной местной анестезией на месте с началом в течение приблизительно 2 часов от введения без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения, предпочтительно по меньшей мере приблизительно 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно 48-72 часа после введения.
Целью изобретения является также предоставление способа обеспечения у субъекта анестетического действия в участке. Способ включает введение композиции на участок, около участка, в участок или по соседству с участком, где композиция содержит первый анестетик, второй анестетик и фармацевтически приемлемый неполимерный носитель. Второй анестетик является растворителем для первого анестетика и обеспечивает начальное анестетическое действие в участке при введении. Неполимерный носитель контролирует высвобождение первого анестетика с обеспечением последующего анестетического действия, характеризуемого длительной местной анестезией в участке, и композиция обеспечивает длительную устойчивую среднюю концентрацию анестетика в плазме (Css) по меньшей мере приблизительно 200 нг/мл на период по меньшей мере приблизительно 24 часа, когда композицию вводят подкожно.
Фраза «без начального выброса», как используется здесь, означает, что конкретное упомянутое средство не высвобождается из композиции при нормальном введении и становится фармакологически доступным в существенном количестве в течение предопределенного начального периода. Специалист в данной области может легко определить наличие и уровень начального выброса средства из данной композиции с применением общепринятых способов фармакологического тестирования, хорошо известных в данной области. Подходящие способы характеризации выброса высвобождения in vitro включают способ лопастного USP II с использованием стандартного буфера, условий смешивания и нагревания. Характеристики выброса высвобождения данной композиции можно также легко определить с использованием общепринятого тестирования in vivo, такого как мониторинг концентрации интересующего средства в плазме субъекта-животного в течение данного периода времени. В композициях по настоящему изобретению предпочтительно менее чем приблизительно 40-60% анестетического средства высвобождается в течение первых 24 часов, более предпочтительно менее чем приблизительно 30-50% и даже более предпочтительно менее чем приблизительно 20-40% высвобождается в течение начального периода времени. В других конкретных предпочтительных вариантах осуществления менее чем приблизительно 5-10% анестетического средства высвобождается в течение первого часа, более предпочтительно менее чем приблизительно 3-7% высвобождается в течение данного начального периода времени.
Соответственно, композиции по настоящему изобретению будут содержать по меньшей мере одно анестетическое средство в системе контролируемого высвобождения, которая высвобождает анестетик в течение длительного периода времени. В конкретных вариантах осуществления анестетик присутствует в настоящей композиции в количестве от приблизительно 95 до приблизительно 1 процента по массе относительно общей массы композиции (% мас.), в количестве от приблизительно 30 до 1% мас., в количестве от приблизительно 25 до 5% мас. или в количестве приблизительно 20-10% мас., в зависимости от особенностей анестетика и его предполагаемого использования.
Как используется здесь, термин «анестетик» обозначает любое средство, которое обеспечивает обратимое местное онемение, обезболивание, блокирование проводимости импульса вдоль аксонов нерва и других возбудимых мембран, такое как местное блокирование ноцицептивных путей (афферентное и/или эфферентное), аналгезию и/или анестезию. См., например, Strichartz, G.R. (Ed.) Local Anesthetics, Handbook of Experimental Pharmacology, vol. 81, Springer, Berlin/New York, (1987). Термин включает также любое средство, которое при местном введении обеспечивает локализованное (местное) полное или частичное ингибирование сенсорного восприятия и/или моторной функции. Примеры общепринятых средств, пригодных для применения в качестве анестетиков при осуществлении изобретения, включают в качестве неограничивающих примеров амбукаин, амоланон, амилкаин, беноксинат, бензиловый спирт, бензокаин, бетоксикаин, бифенамин, бупивакаин, бутакаин, бутамбен, бутаниликаин, бутетамин, бутоксикаин, картикаин, хлорпрокаин, кокаэтилен, кокаин, циклометикаин, дибукаин, диметизохин, диметокаин, диперодон, диклонин, экогонидин, экогонин, этидокаин, эупроцин, феналкомин, формокаин, гексилкаин, гидрокситетракаин, изобуанин, изобутил п-аминобензоат, лейцинокаин, левобупивакаин, левоксадрол, лидокаин, мепивакаин, меприлкаин, метабутоксикаин, метилхлорид, миртекаин, наепаин, октакаин, ортокаин, оксетазаин, парентоксикаин, фенакаин, фенол, пиперокаин, пиридокаин, полидоканол, прамоксин, прилокаин, прокаин, пропанокаин, пропаракаин, пропипокаин, пропоксикаин, псевдококаин, пиррокаин, ропивакаин, салициловый спирт, тетракаин, толикаин, тримекаин, ксилокаин, золамин, их анестетически активные производные, аналоги, и любые фармацевтически приемлемые соли, и любую их смесь.
Предпочтительными являются местные анестетики амидного или сложноэфирного типа. Местные анестетики амидного типа характеризуются наличием амидной функциональной группы, тогда как местные анестетики сложноэфирного типа содержат сложноэфирную функциональную группу. Предпочтительные местные анестетики амидного типа включают лидокаин, бупивакаин, прилокаин, мепивакаин, этидокаин, ропивакаин и дибукаин. Предпочтительные местные анестетики сложноэфирного типа включают тетракаин, прокаин, бензокаин и хлорпрокаин. Наиболее предпочтительным местным анестетиком является бупивакаин.
Анестетическое средство предусмотрено в композиции в нейтральной форме, в форме свободного основания или в форме фармацевтически приемлемой соли. Термин «фармацевтически приемлемая соль», как используется здесь, обозначает такие соли, которые сохраняют биологическую эффективность и свойства нейтральных анестетиков и не являются в других отношениях неприемлемыми для фармацевтического применения. Фармацевтически приемлемые соли включают соли кислой или основной групп, которые могут присутствовать в анестетических средствах. Те анестетические средства, которые являются основными по природе, способны образовывать широкий ряд солей с различными неорганическими и органическими кислотами. Фармацевтически приемлемые кислотно-аддитивные соли основных анестетиков, пригодных для применения в данном изобретении, представляют собой такие, которые образуют нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинaт, ацетат, лактат, салицилат, цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкаронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат (т.е. 1,1'-метилен-бис(2-гидрокси-3-нафтоат)). Анестетические средства, содержащие аминогруппу, могут образовывать фармацевтически приемлемые соли с различными аминокислотами, в дополнение к кислотам, упомянутым выше. Приемлемые основные соли могут быть получены из оснований, которые образуют нетоксичные соли, например соли алюминия, кальция, лития, магния, калия, натрия, цинка и диэтаноламина. См., например, Berge et al. (1977) J. Pharm. Sci. 66:1-19.
Способность анестетического средства обеспечивать состояние длительной местной анестезии относится к способности указанного средства вызывать поддающееся оценке состояние локализованного (местного) полного или частичного ингибирования сенсорного восприятия и/или моторной функции. Различные способы и инструменты для осуществления такой оценки будут ясно очевидны специалисту в данной области. Что касается субъектов-животных, не относящихся к человеку, данные способы включают измерение спонтанной локомоции у тестируемых крыс (с использованием, например, коммерчески доступного оборудования и программного обеспечения от Med Associates Inc., St. Albans, VT), где для тестируемых субъектов могут быть собраны данные об общей пройденной дистанции, показателях ходьбы, стереотипии, подъему на задние лапы, времени, проведенному в различных движениях, и времени, проведенному на отдыхе; визуализацию реакции на укол булавкой у крыс; и модель отдергивания лап крыс от горячей пластины, например, согласно способу, подробно описанному в IACUC No. 9511- 2199.
Сенсорное исследование у субъектов-людей также является применимым способом оценки местного анестетического действия. Тестирование сфокусировано на трех основных областях, механическом тестировании (укол булавкой, волокна Фрея), термическом (теплое, горячее, холодное) и тактильном тестировании (прикосновение). Способы такого тестирования описаны в литературе. См., например, Dahl, et al. (1993) Pain 53:43-51; Moiniche, et al. (1993) Brit. J. of Anaesthesia 71:201-205; Moiniche, et al. (1993) Regional Anesthesia 18:300-303; Pedersen, et al. (1996) Anesthesiology 84(5): 1020-1026; Pedersen, et al. (1996) Brit. J. of Anaesthesia 76(6): 806-810; и Pedersen, et al. (1998) Pain 74:139-151. Например, местную анестетическую активность тестируемого средства можно исследовать по отношению к началу, пиковому значению и длительности действия с использованием конкретных способов: 1) механическое сенсорное тестирование (порог распознавания механической боли с использованием волокон Фрея); 2) сверхпороговое (механическое) тестирование с использованием отдельного волокна Фрея; 3) термическое сенсорное тестирование (порог распознавания теплого); 4) порог распознавания боли от горячего; 5) сверхпороговое (горячее) тестирование; 6) порог распознавания холодного; и 7) тактильное сенсорное тестирование (порог распознавания механического прикосновения). Эти данные являются показательными для ощущаемого субъектом местного облегчения боли, местного онемения и/или местной блокады нерва в ответ на введение тестируемого анестетического средства. Болевую реакцию можно характеризовать с использованием вербальной шкалы оценок 0-10 (например, где 0 = отсутствие боли, а 10 = наихудшая вообразимая боль) или визуальной аналоговой шкалы от 0 до 100 мм (например, где 0 = отсутствие боли, а 100 мм = наихудшая вообразимая боль).
Что касается выбора конкретного анестетического средства, специалистам в данной области будет известно также, что фармакологические свойства каждого средства-кандидата будут меняться, например, по отношению к началу и интенсивности анестетического действия, длительности и т.п. Конкретные средства могут обеспечивать умеренное анестетическое действие, с довольно быстрым началом активности, но короткой продолжительностью. Такие средства можно использовать в композициях по настоящему изобретению для обеспечения «начального анестетического действия», где их, как правило, объединяют с различными анестетическими средствами, обеспечивающими «длительную местную анестезию», характеризующуюся более постепенным началом активности, но более сильным действием и более длительной его продолжительностью. Примером анестетика, который можно применять для обеспечения начального анестетического действия, является бензиловый спирт. Примером анестетика, который можно применять для обеспечения длительной местной анестезии, является бупивакаин. Дополнительные средства, которые можно использовать для обеспечения начального анестетического действия, могут включать органические вещества, обычно применяемые в качестве растворителей и/или облегчающих проникновение средств, такие как этанол, диметилсульфоксид, N-метилпирролидон, полиэтиленгликоль и сложные эфиры конкретных жирных кислот. Эти и другие подобные средства могут обеспечивать очень умеренное начальное анестетическое действие, например, при применении они могут охлаждать или иначе десенсибилизировать/вызвать онемение участка ткани, таким образом, частично ингибируя сенсорное восприятие в данном участке. Каждый раз, когда средство применяют при осуществлении изобретения для обеспечения начального анестетического действия, его предусматривают в подходящей композиции в количестве, достаточном для обеспечения указанного действия, и таким образом, что средство можно быстро высвобождать из композиции для обеспечения предполагаемого действия. Составление таких подходящих композиций (содержащих средство для обеспечения начального анестетического действия) находится в компетенции специалиста в данной области в сочетании с руководством и объяснением, предоставленным настоящим описанием.
В конкретных вариантах осуществления изобретение относится к композиции, которая содержит два анестетических средства, первый анестетик и второй анестетик, где второе анестетическое средство является растворителем для первого анестетического средства. В этих конкретных композициях второе анестетическое средство обычно применяют для обеспечения начального анестетического действия, а первое анестетическое средство применяют для обеспечения последующего анестетического действия, характеризуемого длительной местной анестезией с началом в течение приблизительно 2 часов от введения субъекту без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения или даже дольше. В конкретных предпочтительных вариантах осуществления первое анестетическое средство обеспечивает длительную местную анестезию с началом в течение приблизительно 1-2 часа от введения, а в других предпочтительных вариантах осуществления первое анестетическое средство обеспечивает длительную местную анестезию с началом в течение приблизительно от 30 минут до 1 часа от введения. В других конкретных вариантах осуществления второй анестетик также является растворителем для системы носителя для контролируемой доставки.
Анестетическое средство будет служить растворителем для другого анестетического средства, когда одно средство является по меньшей мере частично растворенным в другом растворяющем средстве при изготовлении композиции. Кроме того, анестетический растворитель присутствует в композиции в количестве, достаточном как для обеспечения начального анестетического действия, так и для по меньшей мере частичного растворения другого анестетического средства. В конкретных вариантах осуществления второй анестетик, таким образом, присутствует в количестве от приблизительно 95 до приблизительно 1 процента по массе относительно общей массы композиции (% мас.), или в количестве от приблизительно 75 до 10% мас., или в количестве от приблизительно 50 до 15% мас.
При осуществлении изобретения можно применять ряд подходящих анестетических средств, которые служат также растворителями для других анестетических средств. Подходящие средства включают ароматические спирты, кислоты и производные кислот, и их комбинации. Особенно предпочтительным анестетическим средством, которое можно использовать в качестве растворителя для дополнительного анестетика, является бензиловый спирт.
Системы носителя для контролируемой доставки, применяемые в композициях по настоящему изобретению, классифицируют как неполимерные носители. Фармацевтически приемлемый неполимерный носитель является, как правило, биологически совместимым и предпочтительно биоразлагаемым, биоразрушаемым или биоадсорбируемым. Вещество является биологически совместимым, если оно и любые продукты его деградации не оказывают значительных, вредных или неблагоприятных действий, не вызывают существенного раздражения тканей или некроза, когда его вводят в живую ткань. «Биоразлагаемый» или «биоразрушаемый», применяемые здесь попеременно, означают указанный неполимерный материал, который будет разлагаться или разрушаться in vivo с образованием более мелких химических соединений, где такая деградация может происходить, например, в результате ферментативных, химических и физических процессов. «Биоадсорбируемый» означает, что данный неполимерный материал может разрушаться и адсорбироваться внутри организма субъекта-животного, например клетки, ткани или подобного.
Неполимерный материал носителя применяют для контроля высвобождения по меньшей мере одного анестетического средства из композиций по настоящему изобретению таким способом, чтобы обеспечивать длительную местную анестезию с началом в течение приблизительно 2 часов от введения и продолжительностью по меньшей мере приблизительно 24 часа или дольше. В некоторых композициях по настоящему изобретению неполимерный материал носителя является достаточным для обеспечения по меньшей мере для одного анестетика либо контролируемого профиля высвобождения первого порядка, либо профиля высвобождения псевдонулевого порядка. Соответственно, неполимерный носитель будет присутствовать в композиции в количестве от приблизительно 99,5 до приблизительно 1 процента по массе относительно общей массы композиции (% мас.), или в количестве приблизительно от 95 до 10% мас., или в количестве приблизительно от 75 до 25% мас.
Выбор подходящего неполимерного носителя находится в общей компетенции специалистов в данной области с использованием объяснения и руководства, предоставленных настоящим описанием и спецификацией. Например, специалисту в данной области доступны многочисленные системы фармацевтически приемлемых неполимерных носителей для получения жидкости, спрея, крема, лосьона, мази, геля, взвеси, масла, эмульсии, микроэмульсии, твердого вещества, пластыря, пленки, частицы, микрочастицы, порошка или другой приемлемой формы фармацевтической композиции. Эти и другие системы носителя описаны, например, в Remington's Pharmaceutical Sciences, 16th Edition, 1980 и 17th Edition, 1985, оба опубликованы Mack Publishing Company, Easton, PA.
Композиции по настоящему изобретению могут дополнительно содержать один или несколько дополнительных компонентов, например фармацевтически приемлемые эксципиенты, такие как диспергирующие средства, наполнители, связующие вещества, носители, стабилизаторы, вещества, способствующие скольжению, антиоксиданты, регуляторы pH, вещества, препятствующие раздражению, и т.п. Опытный специалист в данной области будет понимать, что конкретные эксципиенты могут выполнять несколько вышеупомянутых функций в любом конкретном составе. Таким образом, любое число подходящих эксципиентов можно смешивать с композициями по настоящему изобретению или вводить в них для обеспечения объемных свойств, изменения скоростей высвобождения активного вещества, увеличения или затруднения поглощения воды, контроля pH, предоставления структурной поддержки, облегчения процесса изготовления и других применений, известных специалистам в данной области. Термин «эксципиент», как правило, относится к по существу инертному материалу, который является нетоксичным и не взаимодействует с другими компонентами композиции вредным образом. Соотношение, при котором конкретный эксципиент может присутствовать в композиции, зависит от цели, для которой предусмотрен наполнитель, и вида эксципиента.
Например, приемлемые эксципиенты, которые могут действовать также в качестве стабилизаторов для активных веществ, включают глюкозу, сахарозу, лактозу, трегалозу, маннит, сорбит, инозит, декстран и т.п. фармацевтической чистоты. Такие стабилизаторы, таким образом, могут представлять собой сахарид, такой как моносахарид, дисахарид, полисахарид или сахарный спирт. Другие приемлемые эксципиенты включают крахмал, целлюлозу, фосфаты натрия или кальция, сульфат кальция, лимонную кислоту, винную кислоту, глицин и их комбинации. Примеры гидрофобных эксципиентов, которые могут быть добавлены для замедления гидратации и кинетики растворения, включают жирные кислоты и их фармацевтически приемлемые соли (например, стеарат магния, стеариновую кислоту, стеарат цинка, пальмитиновую кислоту и пальмитат натрия).
Может также быть полезным применение в композициях по настоящему изобретению в качестве эксципиента заряженного липида и/или детергента. Подходящие заряженные липиды включают, без ограничения, фосфатидилхолины (лецитин) и т.п. Детергенты обычно представляют собой неионный, анионный, катионный или амфотерный сурфактант. Примеры подходящих сурфактантов включают, например, сурфактанты Tergitol® и Triton® (Union Carbide Chemicals and Plastics); полиоксиэтиленсорбитаны, например сурфактанты TWEEN® (Atlas Chemical Industries); полисорбаты; простые эфиры полиоксиэтилена, например Brij; фармацевтически приемлемые сложные эфиры жирных кислот, например лаурилсульфат и его соли; амфифильные сурфактанты (глицериды и т.д.); и подобные материалы.
Другие эксципиенты могут быть добавлены для изменения пористости, например, такие вещества, как сахароза, глюкоза, хлорид натрия, сорбит, лактоза, полиэтиленгликоль, маннит, фруктоза, поливинилпирролидон или их подходящие комбинации. Кроме того, анестетическое средство или средства могут быть диспергированы в маслах (например, кунжутном масле, кукурузном масле, растительном), или их смеси с фосфолипидом (например, лецитином), или триглицеридах жирных кислот со средней цепью (например, Миглиол 812) для получения масляной суспензии.
В композиции по настоящему изобретению можно вводить дополнительные эксципиенты, включая разбавители различной буферной емкости (например, Трис-HCl, ацетат); средства, изменяющие pH и ионную силу; добавки, такие как антиоксиданты (например, аскорбиновая кислота, глутатион, метабисульфит натрия); консерванты (например, Thimersol, бензиловый спирт, метилпарабен, пропилпарабен); и диспергирующие средства, такие как водорастворимые полисахариды (например, маннит, лактоза, глюкоза, крахмалы), гиалуроновая кислота, глицин, фибрин, коллаген и неорганические соли (например, хлорид натрия).
В конкретных вариантах осуществления изобретения неполимерный носитель является по существу нерастворимым в воде или в водной биологической системе. Такие неполимерные материалы носителя включают, но не ограничиваются ими: стерины, такие как холестерин, стигмастерин, β-ситостерин и эстрадиол; сложные эфиры холестерина, такие как холестерин стеарат; C12-C24 жирные кислоты, такие как лауриновая кислота, миристиновая кислота, пальмитиновая кислота, стеариновая кислота, арахидиновая кислота, бегеновая кислота и лигноцериновая кислота; C18-C36 моно-, ди- и триацилглицериды, такие как глицерилмоноолеат, глицерилмонолинолеат, глицерилмонолаурат, глицерилмонодокозаноат, глицерилмономиристат, глицерилмонодеценоат, глицерилдипальмитат, глицерилдидокозаноат, глицерилдимиристат, глицерилдидеценоат, глицерилтридокозаноат, глицерилтримиристат, глицерилтридеценоат, глицеринтристеарат и их смеси; сложные эфиры сахарозы и жирных кислот, такие как дистеарат сахарозы и пальмитат сахарозы; сложные эфиры сорбитана и жирных кислот, такие как моностеарат сорбитана, монопальмитат сорбитана и тристеарат сорбитана; C16-C18 жирные спирты, такие как цетиловый спирт, миристиловый спирт, стеариловый спирт и цетостеариловый спирт; сложные эфиры жирных спиртов и жирных кислот, такие как цетилпальмитат и цетеарилпальмитат; ангидриды жирных кислот, такие как стеариновый ангидрид; фосфолипиды, включая фосфатидилхолин (лецитин), фосфатидилсерин, фосфатидилэтаноламин, фосфатидилинозит и их лизопроизводные; сфингозин и его производные; сфингомиелины, такие как стеарил, пальмитоил и трикозанил сфингомиелины; керамиды, такие как стеарил и пальмитоил керамиды; гликосфинголипиды; ланолин и ланолиновые спирты; и их комбинации и смеси. Конкретные предпочтительные неполимерные носители включают холестерин, глицерилмоностеарат, глицеринтристеарат, стеариновую кислоту, стеариновый ангидрид, глицерилмоноолеат, глицерилмонолинолеат и ацетилированные моноглицериды.
Если один из вышеупомянутых неполимерных материалов носителя выбирают для использования в композиции по настоящему изобретению, его, как правило, будут сочетать с совместимым и подходящим органическим растворителем для материала носителя с получением композиции, обладающей консистенцией, изменяющейся от водянистой до вязкой до пастообразной замазки или пасты. Консистенция композиции будет меняться в зависимости от таких факторов, как растворимость неполимерного носителя в растворителе, концентрации неполимерного носителя, концентрации анестетического средства и/или присутствия дополнительных анестетических средств, добавок и эксципиентов. Растворимость неполимерного носителя в конкретном растворителе будет меняться в зависимости от таких факторов, как его кристалличность, гидрофильность, ионный характер и липофильность. Соответственно, ионный характер и концентрацию неполимерного носителя в растворителе можно регулировать для достижения желаемой растворимости. Предпочтительными неполимерными материалами носителя являются такие, которые обладают низкой кристалличностью, неполярными свойствами и являются более гидрофобными.
Подходящие органические растворители для использования в композициях, как правило, представляют собой такие, которые являются биологически совместимыми, фармацевтически приемлемыми и будут по меньшей мере частично растворять неполимерный носитель. Органический растворитель, кроме того, будет обладать растворимостью в воде в диапазоне от смешиваемого до растворимого до диспергируемого. В конкретных вариантах осуществления растворитель выбирают так, что он способен диффундировать, рассеиваться или вымываться из композиции in situ в водной системе и в жидкостях, обнаруженных в участке введения, таким образом, образуя твердый имплантат. Предпочтительно растворитель обладает параметром растворимости Гильдебранда (HLB) от приблизительно 9-13 (кал/см3)1/2. Предпочтительно степень полярности растворителя способна обеспечивать по меньшей мере приблизительно 5% растворимость в воде.
Подходящие органические растворители, таким образом, включают, но не ограничиваются ими: замещенные гетероциклические соединения, такие как N-метил-2-пирролидон (NMP) и 2-пирролидон (2-пирол); сложные эфиры угольной кислоты и алкиловых спиртов, такие как пропиленкарбонат, этиленкарбонат и диметилкарбонат; жирные кислоты, такие как уксусная кислота, молочная кислота и гептановая кислота; сложные эфиры алкила и моно-, ди- и трикарбоновых кислот, такие как 2-этоксиэтилацетат, этилацетат, метилацетат, этиллактат, этилбутират, диэтилмалонат, диэтилглюконат, трибутилцитрат, диэтилсукцинат, трибутирин, изопропилмиристат, диметиладипат, диметилсукцинат, диметилоксалат, диметилцитрат, триэтилцитрат, ацетилтрибутилцитрат, глицерилтриацетат; алкилкетоны, такие как ацетон и метилэтилкетон; эфирные спирты, такие как 2-этоксиэтанол, диметиловый эфир этиленгликоля, гликофурол и глицеринформаль; спирты, такие как этанол и пропанол; полигидроксиспирты, такие как пропиленгликоль, полиэтиленгликоль (PEG), глицерин (глицерол), 1,3-бутиленгликоль и изопропилиденгликоль (2,2-диметил-1,3-диоксолон-4-метанол); Солкеталь; диалкиламиды, такие как диметилформамид, диметилацетамид; диметилсульфоксид (ДМСО) и диметилсульфон; тетрагидрофуран; лактоны, такие как ε-капролактон и бутиролактон; циклические алкиламиды, такие как капролактам; ароматические амиды, такие как N,N-диметил-м-толуамид и 1-додецилазациклогептан-2-он; и тому подобное; и их смеси и комбинации. Предпочтительные растворители включают N-метил-2-пирролидон, 2-пирролидон, диметилсульфоксид, этиллактат, пропиленкарбонат, гликофурол, глицеринформаль и изопропилиденгликоль.
Органические растворители будут представлены в композиции в количестве от приблизительно 99,5 до приблизительно 1 процента по массе относительно общей массы композиции (% мас.), в количестве от приблизительно 95 до 10% мас., в количестве от приблизительно 75 до 25% мас. или в количестве от приблизительно 60 до 40% мас., в зависимости от выбранных неполимерного носителя, органического растворителя, анестетического средства, добавки и/или эксципиента, применяемых в композиции. В конкретных вариантах осуществления органический растворитель диффундирует или вымывается из композиции в водной среде при помещении в биологическую систему, где материал неполимерного носителя коагулирует с образованием твердого вещества. Предпочтительно неполимерный носитель отверждается in situ с образованием твердого вещества в течение приблизительно 1-5 суток после введения (имплантации), предпочтительно в течение приблизительно 1-3 суток, предпочтительно в течение приблизительно 2 часов.
Ряд подходящих добавок можно включать в композицию для придания композиции выбранных характеристик. Например, можно включать незначительное количество биоразлагаемого термопластичного полимера, такого как полилактид, поликапролактон, полигликолид или их coполимер, для получения более когерентного твердого имплантата или композиции с большей вязкостью, чтобы удерживать ее на месте, в то время как она отверждается. Такие термопластичные полимеры описаны в патенте США № 4938763 Dunn et al.
Необязательно в композицию можно включать порообразующее средство. Порообразующее средство может представлять собой любое органическое или неорганическое, фармацевтически приемлемое вещество, которое является по существу растворимым в воде или жидкостях организма и будет рассеиваться из материала неполимерного носителя и/или твердого вещества имплантата в окружающей жидкости организма в участке имплантата. Порообразующее средство предпочтительно может быть нерастворимым в органическом растворителе для образования однородной смеси с материалом неполимерного носителя. Порообразующее средство может также быть не смешиваемым с водой веществом, которое быстро распадается до водорастворимого вещества. В конкретных композициях порообразующее средство объединяют в смеси с неполимерным носителем и органическим растворителем. Подходящие порообразующие средства, которые можно использовать в композиции, включают, например, сахара, такие как сахароза и глюкоза, соли, такие как хлорид натрия и карбонат натрия, полимеры, такие как гидроксипропилцеллюлоза, карбоксиметилцеллюлоза, полиэтиленгликоль и поливинилпирролидон, и т.п. Предпочтительными являются твердые кристаллы, обеспечивающие определенный размер пор, такие как соль или сахар.
Другие варианты осуществления настоящего изобретения относятся к композициям, где неполимерный носитель представляет собой жидкость. Жидкий неполимерный носитель предпочтительно представляет собой жидкий материал носителя с высокой вязкостью («HVLCM»), являющийся нерастворимым в воде и обладающий вязкостью по меньшей мере 5000 сП (и, необязательно, по меньшей мере 10000, 15000; 20000; 25000 или даже 50000 сП) при 37°С, который без примесей не кристаллизуется в условиях окружающей среды или в физиологических условиях. Термин «нерастворимый в воде» относится к материалу, который растворим в воде в соотношении менее одного процента по массе в условиях окружающей среды. Термин «неполимерный» относится к сложным эфирам или смешанным сложным эфирам, по существу не обладающим повторяющимися звеньями в кислой группе эфира, так же как к сложным эфирам или смешанным сложным эфирам, обладающим кислыми группами, где функциональные звенья в кислой группе повторяются малое число раз (т.е. к олигомерам). Как правило, вещества, обладающие более чем пятью идентичными и смежными повторяющимися звеньями или мерами в кислой группе сложного эфира, исключены из термина «неполимерный», как используется здесь, однако вещества, содержащие димеры, тримеры, тетрамеры или пентамеры, входят в объем данного термина. Когда сложный эфир образован из групп гидроксисодержащих карбоновых кислот, которые можно далее этерифицировать, таких как молочная кислота или гликолевая кислота, число повторяющихся звеньев вычисляют на основании числа групп лактида или гликолида, вместо числа групп молочной кислоты или гликолевой кислоты, где повторяющееся звено лактида содержит две группы молочной кислоты, этерифицированные по их соответствующим гидрокси- и карбоксигруппам, и где повторяющееся звено гликолида содержит две группы гликолевой кислоты, этерифицированные по их соответствующим гидрокси- и карбоксигруппам. Сложные эфиры, имеющие от 1 до приблизительно 20 этерифицированных полиолов в их спиртовой группе или от 1 до приблизительно 10 глицериновых групп в их спиртовой группе, считаются неполимерными, как этот термин используется в данном описании.
В конкретном варианте осуществления вязкость HVLCM уменьшают, в некоторых случаях значительно, когда смешивают с растворителем для получения жидкого материала носителя с низкой вязкостью («LVLCM»), который можно вводить с использованием общепринятых медицинских устройств. Композицию с LVLCM, как правило, легче поместить в организм, чем композицию с HVLCM, так как она легче протекает внутрь шприца и вытекает из шприца или других устройств для имплантации. Ее также можно легко составлять в виде эмульсии. LVLCM может обладать любой желательной вязкостью, но его вязкость, как правило, ниже, чем у соответствующего HVLCM. В качестве примера, области значений вязкости для LVLCM менее чем приблизительно 6000 сП, менее чем приблизительно 4000 сП, менее чем приблизительно 1000 сП или менее чем 200 сП, как правило, пригодны для применений in vivo.
Конкретным HVLCM, используемым в композициях по изобретению, может быть один или несколько из множества материалов. Подходящие материалы включают неполимерные сложные эфиры или смешанные сложные эфиры одной или нескольких карбоновых кислот. В конкретном варианте осуществления сложный эфир образован из карбоновых кислот, которые этерифицированы полиолом, имеющим от приблизительно 2 до приблизительно 20 гидроксигрупп, и которые могут содержать от 1 до приблизительно 20 этерифицированных полиолов. Особенно подходящие карбоновые кислоты для образования кислой части сложного эфира HVLCM включают карбоновые кислоты, имеющие одну или несколько гидроксигрупп, например, полученные раскрывающим кольцо алкоголизом лактонов или циклических карбонатов или алкоголизом ангидридов карбоновых кислот. Аминокислоты также пригодны для образования сложных эфиров с полиолом. В конкретном варианте осуществления сложный эфир или смешанный сложный эфир содержит спиртовую группу, имеющую одну или несколько концевых гидроксигрупп, которые этерифицированы одной или несколькими карбоновыми кислотами, полученными алкоголизом ангидрида карбоновой кислоты, такого как циклический ангидрид.
Неограниченные примеры подходящих карбоновых кислот, которые можно этерифицировать для получения HVLCM, включают гликолевую кислоту, молочную кислоту, ε-гидроксикапроновую кислоту, серин и любые соответствующие лактоны или лактамы, триметиленкарбонат и диоксанон. Гидроксисодержащие кислоты сами по себе можно дополнительно этерифицировать посредством реакции их гидроксигрупп с дополнительной карбоновой кислотой, которая может быть такой же или отличной от других групп карбоновой кислоты в материале. Подходящие лактоны включают, но не ограничиваются ими, гликолид, лактид, ε-капролактон, бутиролактон и валеролактон. Подходящие карбонаты включают в качестве, но не ограничиваются ими, триметиленкарбонат и пропиленкарбонат.
Спиртовую группу сложного эфира или смешанного сложного эфира можно получить из полигидроксиспирта, имеющего от приблизительно 2 до приблизительно 20 гидроксигрупп, и, как указано выше, можно получить этерификацией 1-20 молекул полиола. Подходящие спиртовые группы включают группы, полученные удалением одного или нескольких атомов водорода из: монофункциональных C1-C20 спиртов, дифункциональных C1-C20 спиртов, трифункциональных спиртов, гидроксисодержащих карбоновых кислот, гидроксисодержащих аминокислот, фосфатсодержащих спиртов, тетрафункциональных спиртов, сахарных спиртов, моносахаридов и дисахаридов, сахарных кислот и полиэфирных полиолов. Более конкретно, спиртовые группы могут включать одно или несколько из: додеканола, гександиола, более конкретно 1,6-гександиола, глицерина, гликолевой кислоты, молочной кислоты, гидроксимасляной кислоты, гидроксивалериановой кислоты, гидроксикапроновой кислоты, серина, ATP, пентаэритрита, маннита, сорбита, глюкозы, фруктозы, сахарозы, глюкуроновой кислоты, простых эфиров полиглицерина, содержащих от 1 до приблизительно 10 звеньев глицерина, полиэтиленгликолей, содержащих от 1 до приблизительно 20 звеньев этиленгликоля.
В конкретных вариантах осуществления изобретения по меньшей мере одна из групп карбоновой кислоты сложных эфиров или смешанных сложных эфиров HVLCM содержит по меньшей мере одну оксигруппу. В более конкретном варианте осуществления каждая из групп карбоновой кислоты содержит по меньшей мере одну оксигруппу.
В другом конкретном варианте осуществления по меньшей мере одна из групп карбоновой кислоты сложных эфиров или смешанных сложных эфиров по изобретению содержит 2-4 атома углерода. В более конкретном варианте осуществления каждая из групп карбоновой кислоты сложных эфиров или смешанных сложных эфиров по изобретению содержит 2-4 атома углерода.
В другом более конкретном варианте осуществления изобретения по меньшей мере одна из групп карбоновой кислоты сложного эфира или смешанного сложного эфира по изобретению имеет 2-4 атома углерода и содержит по меньшей мере одну оксигруппу. В другом более конкретном варианте осуществления по изобретению каждая из групп карбоновой кислоты сложного эфира или смешанного сложного эфира по изобретению имеет 2-4 атома углерода и содержит по меньшей мере одну оксигруппу.
В конкретном варианте осуществления HVLCM может представлять собой изобутират ацетата сахарозы (SAIB) или какой-либо другой сложный эфир группы сахарного спирта с одной или несколькими группами алкановой кислоты.
В конкретном варианте осуществления изобретение относится к композициям, где HVLCM имеет структуру, выбранную из группы, состоящей из:
I:
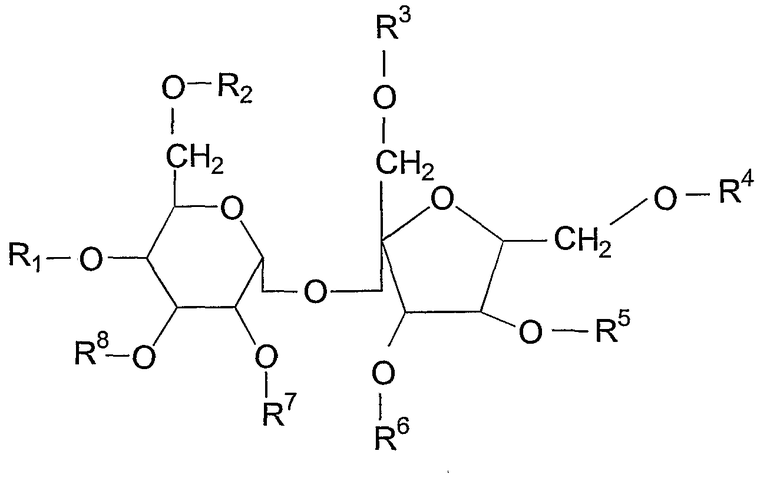
где R1, R2, R3, R4, R5, R6, R7 и R8 независимо выбирают из группы, состоящей из водорода, алканоила, гидрокси-замещенного алканоила и ацилокси-замещенного алканоила;
где по меньшей мере три из R1, R2, R3, R4, R5, R6, R7 и R8 являются отличными от водорода;
и
где, когда R1, R2, R3, R4, R5, R6, R7 и R8 выбирают из группы, состоящей из ацетила и изобутирила, по меньшей мере три из R1, R2, R3, R4, R5, R6, R7 и R8 представляют собой ацетил;
II:

где R1, R2 и R3 независимо выбирают из группы, состоящей из водорода, алканоила, гидрокси-замещенного алканоила и ацилокси-замещенного алканоила, и где n означает число от 1 до 20;
III:
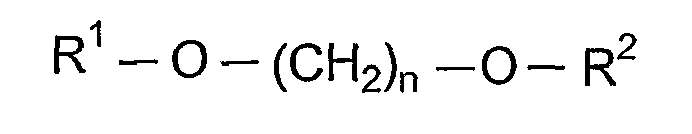
где n представляет собой целое число от 4 до 8, и R1 и R2 независимо выбирают из группы, состоящей из водорода, алканоила, гидрокси-замещенного алканоила и ацилокси-замещенного алканоила;
IV:
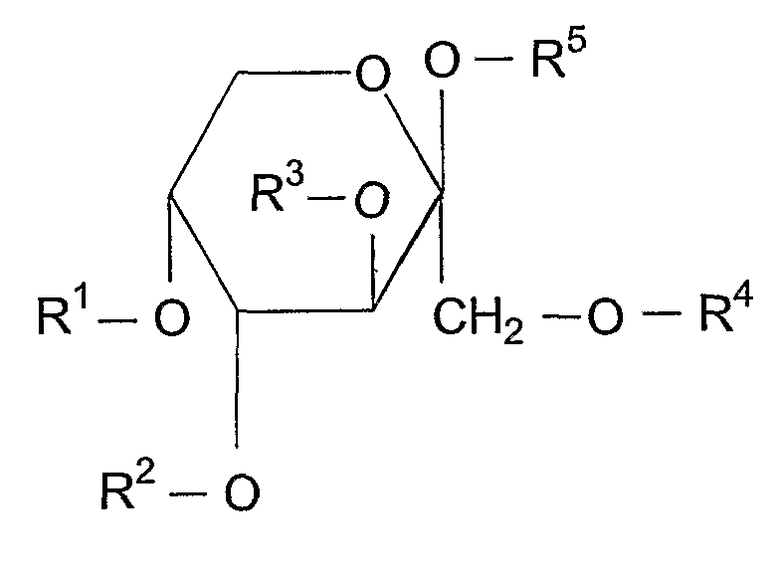
V:
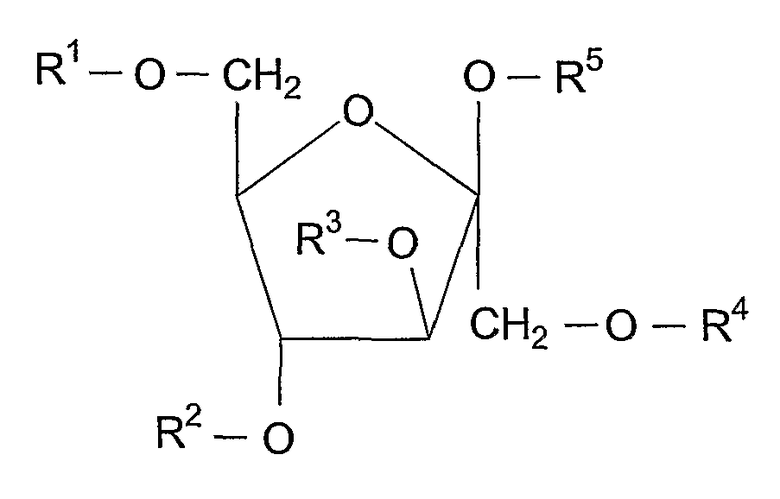
где в формулах IV и V R1, R2, R3, R4 и R5 независимо выбирают из группы, состоящей из водорода, алканоила, гидрокси-замещенного алканоила и ацилокси-замещенного алканоила;
VI:
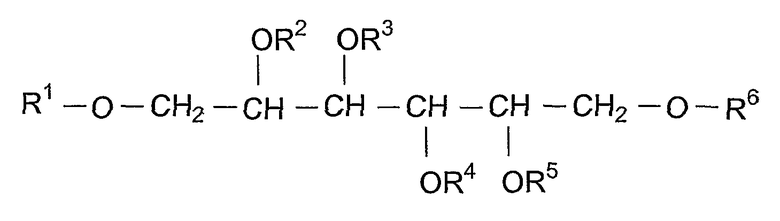
VII:
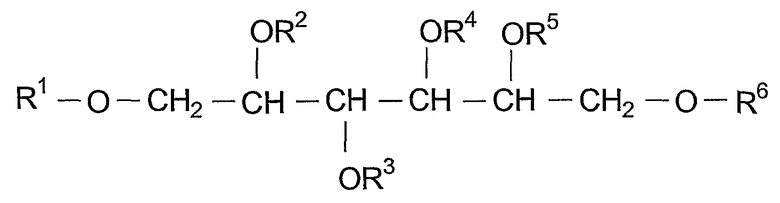
где в формулах VI и VII R1, R2, R3, R4, R5 и R6 независимо выбирают из группы, состоящей из водорода, алканоила, гидрокси-замещенного алканоила и ацилокси-замещенного алканоила;
VIII:
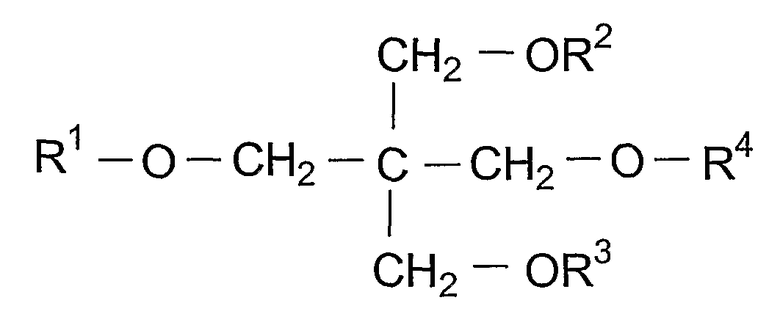
где R1, R2, R3 и R4 независимо выбирают из группы, состоящей из водорода, алканоила, гидрокси-замещенного алканоила и ацилокси-замещенного алканоила.
В каждой из формул I-VIII одна или несколько из групп алканоила, гидрокси-замещенного алканоила и ацилокси-замещенного алканоила может быть группой алканоила, имеющей 2-6 атомов углерода, включая углерод карбонила. Более того, в другом более конкретном варианте осуществления изобретения каждая из формул I-VIII содержит по меньшей мере одну гидрокси-замещенную или ацилокси-замещенную алканоильную группу. В более конкретном варианте осуществления по меньшей мере одна из этих гидрокси-замещенных или ацилокси-замещенных алканоильных групп является алканоильной группой, имеющей 2-6 атомов углерода, включая углерод карбонила.
Ацильные группы, образующие ацилокси-заместители HVLCM, могут быть любой группой, полученной из карбоновой кислоты согласно общепринятому определению термина «ацил». Более конкретно, ацильные группы в композиции по изобретению могут быть в форме R9CO-, где R9 необязательно представляет собой окси-замещенный алкил с 2-6 атомами углерода. Данное окси-замещение может принимать форму гидрокси-замещения, или замещения дополнительными ацильными группами. Например, R9 может представлять собой олигомер окси-замещенных карбоновых кислот, соединенных сложноэфирной связью между гидроксигруппой одной кислоты и карбоксигруппой другой кислоты. В более конкретном примере R9 может содержать 1-5 лактидных или гликолидных звеньев, где лактидное звено содержит две группы молочной кислоты, этерифицированные вместе, и гликолидное звено содержит две группы гликолевой кислоты, этерифицированные вместе. Альтернативно, R9 может содержать смешанные лактидные и гликолидные звенья или может содержать смешанные молочную кислоту и гликолевую кислоту, без присутствия лактидных или гликолидных звеньев.
Конкретные материалы HVLCM включают компоненты формул II или III, где R1, R2 и R3 независимо представляют собой лактоил, полилактоил, ε-капроил, гидроксиацетил или полигидроксиацетил, в частности полилактоил и ε-капроил или полилактоил и полигидроксиацетил.
Применение окси-замещенных групп карбоновой кислоты с относительно небольшой цепью (2-6 атомов углерода) в сложном эфире или в смешанном сложном эфире по изобретению является предпочтительным. Когда эти кислые группы присутствуют в форме олигомерных сложных эфиров (т.е. последующая группа кислоты связана с предыдущей группой кислоты посредством этерификации последующей карбоксигруппы предыдущей оксигруппой), гидролиз материала значительно проще, чем для олигомеров, полученных более чем с 6 атомами углерода, поскольку материал является более гидрофильным. Как правило, для доставки лекарственного средства желательно, чтобы HVLCM был нерастворимым в воде, однако он может быть несколько гидрофильным. Как правило, ожидают, что HVLCM, синтезированные с более гидрофильными звеньями (как определяют по более высокому соотношению O:C), будут быстрее поглощать воду и быстрее деградировать. Например, ожидают, что HVLCM, полученный ковалентным связыванием 4 молей гликолида с одним молем глицерина, будет быстрее поглощать воду и быстрее деградировать, чем HVLCM, полученный ковалентным связыванием 2 молей гликолида и 2 молей лактида с одним молем глицерина. Похожего увеличения можно ожидать для более гибких молекул и для более разветвленных, сферических молекул на основании аргументов свободного объема. Использование гибких и разветвленных молекул может также обладать преимуществом снижения вязкости LVLCM. Использование карбоновых кислот и/или полиолов с различной длиной цепи и использование карбоновых кислот, имеющих окси-замещение, позволяет точный контроль степени гидрофильности и растворимости полученного сложного эфира. Эти материалы значительно устойчивы к растворению in vivo, так что они способны обеспечивать контролируемое высвобождение переносимого анестетического средства в организме, сопровождающееся гидролизом или с последующим гидролизом окси-связей in vivo.
В более конкретном варианте осуществления из HVLCM исключают сложный эфир - изобутират ацетата сахарозы, имеющий соотношение ацетата к кислым группам изобутирата 2:6. Тем не менее, сложный эфир - изобутират ацетата сахарозы, имеющий соотношение ацетата к группам изобутирата 2:6, включают в объем изобретения для применения в аэрозольных составах. Данный материал можно получить согласно способам, описанным в патенте США № 2931802.
Как правило, подходящие сложные эфиры HVLCM могут быть получены взаимодействием одного или нескольких спиртов, в частности одного или нескольких полиолов, которые будут образовывать спиртовую группу получающихся сложных эфиров, с одной или несколькими карбоновыми кислотами, лактонами, лактамами, карбонатами или ангидридами карбоновых кислот, которые будут образовывать кислые группы получающихся сложных эфиров. Реакцию этерификации можно проводить просто нагреванием, хотя в некоторых случаях можно применять добавление катализатора этерификации - сильной кислоты или сильного основания. Альтернативно, можно применять такой катализатор этерификации, как 2-этилгексаноат олова. Нагретую реакционную смесь, с катализатором или без него, нагревают при перемешивании, затем высушивают, например, в вакууме, для удаления любых непрореагировавших исходных веществ и получения жидкого продукта. Изобутираты ацетата сахарозы можно получить, следуя методикам, описанным в патенте США № 2931802.
В этом отношении полиол можно рассматривать как инициатор олигомеризации, в том смысле, что он предоставляет субстрат для этерификации карбоновых кислот, в частности олигомеров лактида, гликолида или других этерифицированных гидрокси-замещенных карбоновых кислот.
В конкретных вариантах осуществления HVLCM можно смешивать с понижающим вязкость растворителем для получения жидкого материала носителя с пониженной вязкостью (LVLCM), который затем можно смешивать с одним или несколькими анестетическими средствами, подлежащими доставке, перед введением. Эти растворители могут быть водорастворимыми, нерастворимыми в воде или смешиваемыми с водой и могут включать ацетон, бензиловый спирт, бензилбензоат, N-(бетагидроксиэтил)лактамидбутиленгликоль, капролактам, капролактон, кукурузное масло, децилметилсульфоксид, диметиловый эфир, диметилсульфоксид, 1-додецилазациклогептан-2-он, этанол, этилацетат, этиллактат, этилолеат, глицерин, гликофурол (тетрагликоль), изопропилмиристат, метилацетат, метилэтилкетон, N-метил-2-пирролидон, MIGLYOL® (сложные эфиры каприловой и/или каприновой кислот с глицерином или алкиленгликолями, например MIGLYOL® 810 или 812 (каприловые/каприновые триглицериды), MIGLYOL® 818 (каприловый/каприновый/линолевый триглицерид), MIGLYOL® 829 (каприловый/каприновый/янтарный триглицерид), MIGLYOL® 840 (дикаприлат/капрат пропиленгликоля)), олеиновую кислоту, арахисовое масло, полиэтиленгликоль, пропиленкарбонат, 2-пирролидон, кунжутное масло, солкеталь ([±]-2,2-диметил-1,3-диоксолан-4-метанол), тетрагидрофуран, TRANSCUTOL® (моноэтиловый эфир диэтиленгликоля, карбитол), триацетин, триэтилцитрат, дифенилфталат и их комбинации. Кроме того, если композицию используют в виде аэрозоля, например, для местного применения, растворитель может представлять собой или содержать один или несколько пропеллентов, таких как CFC-пропелленты, подобные трихлорфторметану и дихлорфторметану, не CFC-пропелленты, подобные тетрафторэтану (R-134a), 1,1,1,2,3,3,3-гептафторпропану (R-227), диметиловому эфиру, пропану и бутану. В частности, подходящие растворители и/или пропелленты включают бензилбензоат, бензиловый спирт, триацетин, триэтилцитрат, диметилсульфоксид, этанол, этиллактат, глицерин, гликофурол (тетрагликоль), N-метил-2-пирролидон, MIGLYOL® 810, полиэтиленгликоль, пропиленкарбонат, 2-пирролидон и тетрафторэтан.
Другие возможные растворители включают перфтордекалин, перфтортрибутиламин, метоксифлуран, глицеринформаль, тетрагидрофурфуриловый спирт, диглим и диметилизосорбид.
Когда композицию применяют как LVLCM для введения анестетического средства, она должна содержать растворитель, в котором растворим HVLCM. В конкретных случаях анестетическое средство также является растворимым в растворителе. В дополнительных случаях растворитель представляет собой второе анестетическое средство, в котором первое анестетическое средство является растворимым. Растворитель предпочтительно является нетоксичным и в остальном биологически совместимым.
В конкретных вариантах осуществления растворитель является по меньшей мере водорастворимым, так что он после введения будет быстро диффундировать в жидкости организма или другую водную среду, в результате чего композиция коагулирует и/или становится более вязкой. В других вариантах осуществления растворитель не является полностью смешиваемым с водой или жидкостями организма, так что диффузия растворителя из композиции и соответствующее увеличение вязкости композиции являются замедленными. Подходящие растворители, обладающие этим свойством, по меньшей мере в некоторой степени, включают бензилбензоат, MIGLYOL® 810, бензиловый спирт и триэтилцитрат. Бензиловый спирт может быть особенно подходящим, так как он также является анестетическим средством.
Когда в качестве HVLCM используют сложные эфиры 1,6-гександиола или глицерина, некоторыми из возможных растворителей являются этанол, N-метилпирролидон, пропиленкарбонат и PEG 400.
Растворитель обычно добавляют к композиции в количестве в диапазоне от приблизительно 99,7 процента до приблизительно 0,5 процента по массе относительно общей массы композиции (% мас.), от приблизительно 95 процентов до приблизительно 1% мас., от приблизительно 75 до приблизительно 10% мас. или от приблизительно 50 до 15% мас. Растворитель обычно присутствует в композиции в количестве в диапазоне от приблизительно 55 процентов до 10% мас.
В других вариантах осуществления изобретения композиция содержит материал, который не является смешиваемым с HVLCM, такой, что, когда его объединяют с HVLCM, отдельно или в комбинации с растворителем для HVLCM, полученная композиция образует эмульсию. Такие эмульсии могут содержать HVLCM в дисперсной фазе, как в случае смесей SAIB/MIGLYOL®, эмульгированных в воде или глицерине, или могут содержать HVLCM в качестве компонента дисперсионной фазы, как в случае водного раствора, эмульгированного в HVLCM, или раствора HVLCM в несмешиваемом с водой растворителе.
Любая из вышеописанных неполимерных систем контролируемой доставки может быть составлена в виде жидкости, спрея, крема, лосьона, мази, геля, взвеси, масла, эмульсии, микроэмульсии, твердого вещества, пластыря, пленки, частицы, микрочастицы, порошка или другой подходящей формы фармацевтической композиции, пригодной для использования в способах по настоящему изобретению. В такие композиции анестетическое средство (например, первое анестетическое средство) включают в количестве, достаточном для доставки субъекту, подлежащему лечению, эффективного количества для достижения желаемого эффекта. Количество анестетического средства, включенного в композицию, зависит от конечных желаемых продолжительности и профиля высвобождения и концентрации анестетика, требуемых для предназначенного эффекта.
Концентрация анестетика в композиции зависит также от скоростей всасывания, инактивации и выделения данного конкретного агента, так же как других факторов, известных специалистам в данной области. Следует заметить, что значения дозы будут также изменяться с тяжестью состояния, подлежащего облегчению. Кроме того, следует понимать, что для любого конкретного субъекта конкретные режимы дозирования следует регулировать с течением времени согласно индивидуальной необходимости и профессионального суждения лица, управляющего введением композиции или контролирующего его, и что диапазоны концентраций, описанные здесь, являются только примерными и не предназначены для ограничения объема или осуществления заявленной в формуле изобретения композиции. Композицию можно вводить в одной дозе или можно разделить на ряд меньших доз для введения в различные интервалы времени, или последовательно или одновременно. Анестетическое средство или средства обычно будут присутствовать в композиции в диапазоне от приблизительно 0,1 до приблизительно 99,5 процента по массе относительно общей массы композиции (% мас.), от приблизительно 0,5 до приблизительно 70% мас. или от приблизительно 1 процента до приблизительно 50% мас. Однако можно использовать диапазоны с настолько низкими верхними пределами, как приблизительно 40%, 30%, 20% или 10%, и диапазоны с настолько высокими нижними пределами, как приблизительно 5%, 3% или 2%. Для очень активных анестетических средств диапазоны могут составлять менее чем 1% по массе и, возможно, менее чем 0,0001%.
Как растворимые, так и нерастворимые анестетические средства можно распределять с использованием неполимерных материалов носителя для контролируемой доставки. Более того, дополнительно можно составлять композиции с полимерными эксципиентами для получения связующего для доставки с модифицированными свойствами, например большей или меньшей скоростью деградации. Из полученной композиции можно образовать микросферы, или макроскопический имплантат, или другие формы и размеры согласно способам, известным в данной области. Альтернативно, предварительно образованную микросферу, имплантат или полимерную частицу с включенным туда анестетическим средством или средствами можно сочетать с неполимерным носителем.
Микросферы могут быть получены рядом способов, известных в данной области, так же как способами, описанными в патентах США № 6291013 и 6440493. Полимерную частицу можно получить с использованием экструзии расплава, грануляции, смешивания с растворителем, адсорбции или подобных способов, или можно адсорбировать анестетическое средство на полимерную матрицу, такую как ионообменная смола. Полученный материал при объединении с подходящим неполимерным носителем можно вводить парентерально. В других вариантах осуществления анестетическое средство можно объединять с неполимерным материалом, таким как фосфат кальция или сахароза, для обеспечения свойств покрытия/барьера, удлиняющих деградацию. Затем неполимерный носитель будет образовывать вторичный барьер для обеспечения улучшенных характеристик доставки. Фаза неполимерного носителя может содержать или не содержать другие биологически активные вещества, согласно конкретным требованиям выбранного применения. Этими другими биологически активными средствами могут быть любые подходящие терапевтические и/или профилактические лекарственные средства, при условии, что добавляемое вещество пригодно для введения в микросферы или имплантаты согласно способам, известным в данной области.
Как обсуждалось выше, множество добавок можно, необязательно, добавлять к композициям по настоящему изобретению для модификации его свойств и, в частности, для модификации свойств высвобождения из композиции в отношении содержащихся там анестетических средств. Добавки могут присутствовать в любом количестве, достаточном, чтобы придавать композиции желательные свойства. Количество используемой добавки, как правило, будет зависеть от природы добавки и эффекта, которого необходимо достичь, и его легко может определить исполнитель. Подходящие добавки описаны в патенте США № 5747058, полное содержание которого приведено здесь в качестве ссылки. Более конкретно, подходящие добавки включают воду, биоразлагаемые полимеры, небиоразлагаемые полимеры, природные масла, синтетические масла, углеводороды или производные углеводорода, неорганические соли, BSA (бычий сывороточный альбумин), сурфактанты, органические соединения, такие как сахара, и органические соли, такие как цитрат натрия. Как правило, чем менее водорастворимой, т.е. более липофильной, является добавка, тем больше она будет уменьшать скорость высвобождения анестетического средства по сравнению с такой же композицией без добавки. Кроме того, может быть желательным включать добавки, увеличивающие такие свойства, как эффективность или пористость композиции.
Добавление добавок можно использовать также для удлинения времени доставки анестетического средства, что делает композицию пригодной для медицинских применений, требующих долговременного введения или чувствительных к нему. Подходящие в этом отношении добавки включают описанные в патентах США №№ 5747058 и 5736152. В частности, подходящие добавки для этой цели включают полимерные добавки, такие как целлюлозные полимеры и биоразлагаемые полимеры. Подходящие целлюлозные полимеры включают ацетаты целлюлозы, простые эфиры целлюлозы и бутираты ацетата целлюлозы. Подходящие биоразлагаемые полимеры включают полилактоны, полиангидриды и полиортоэфиры, в частности полимолочную кислоту, полигликолевую кислоту, поликапролактон и их coполимеры.
Добавка, когда присутствует в композиции, как правило, присутствует в количестве в диапазоне от приблизительно 0,01 процента до приблизительно 20 процентов по массе, более конкретно от приблизительно 0,1 процента до приблизительно 20 процентов по массе, относительно общей массы композиции, и более конкретно присутствует в композиции в количестве в диапазоне от приблизительно 1, 2 или 5 процентов до приблизительно 10 процентов по массе. Конкретные добавки, такие как буферы, присутствуют в композиции только в небольших количествах.
Следующие категории являются неограничивающими примерами классов добавок, которые можно использовать в композициях по настоящему изобретению.
Одной категорией добавок являются биоразлагаемые полимеры и олигомеры. Полимеры можно применять для изменения профиля высвобождения подлежащего доставке анестетического средства, для придания прочности композиции или для модификации свойств композиции другим способом. Неограничивающие примеры подходящих биоразлагаемых полимеров и олигомеров включают: поли(лактид), поли(лактид-ко-гликолид), поли(гликолид), поли(капролактон), полиамиды, полиангидриды, полиаминокислоты, полиортоэфиры, полицианоакрилаты, поли(фосфазины), поли(фосфоэфиры), полиэфирамиды, полидиоксаноны, полиацетали, поликетали, поликарбонаты, полиортокарбонаты, разлагаемые полиуретаны, полигидроксибутираты, полигидроксивалераты, полиалкиленоксалаты, полиалкиленсукцинаты, поли(яблочная кислота), хитин, хитозан и coполимеры, терполимеры, оксидированная целлюлоза или комбинации или смеси вышеуказанных материалов.
Примеры поли(α-гидроксикислот) включают поли(гликолевую кислоту), поли(DL-молочную кислоту) и поли(L-молочную кислоту) и их coполимеры. Примеры полилактонов включают поли(ε-капролактон), поли(δ-валеролактон) и поли(γ-бутиролактон).
Без намерения быть связанными какой-либо теорией полагают, что, когда композиция содержит биоразлагаемый полимер, часть полимера может осаждаться или коагулировать на поверхности композиции, по мере того как любой включенный в состав растворитель диффундирует из материала после введения субъекту. Полимер можно, таким образом, добавлять в качестве модифицирующего высвобождение средства, чтобы воздействовать на высвобождение анестетического средства или средств, или можно добавлять в качестве части композиции, содержащей предварительно образованные микросферы, имплантаты или молотые частицы полимера. Осаждением или коагуляцией полимера образуют оболочку, по меньшей мере частично окружающую жидкую сердцевину такой композиции. Эта оболочка является пористой и позволяет растворителю продолжать диффундировать через нее в окружающую ткань. Скорость высвобождения растворителя и объем образования оболочки, так же как ее пористость, можно контролировать количеством и типом растворителя и полимера, использованных в композиции.
Другими добавками для применения в настоящих композициях являются не биоразлагаемые полимеры. Неэродируемые полимеры, которые можно применять в качестве добавок, включают в качестве неограничивающих примеров: полиакрилаты, полимеры этилен-винилацетата, целлюлозу и производные целлюлозы, ацил-замещенные ацетаты целлюлозы и их производные, неэродируемые полиуретаны, полистиролы, поливинилхлорид, поливинилфторид, поливинил (имидазол), хлорсульфонированные полиолeфины, полиэтиленоксид и полиэтилен.
Предпочтительные не биоразлагаемые полимеры включают поливинилпирролидон, этиленвинилацетат, полиэтиленгликоль, ацетобутират целлюлозы («CAB») и ацетопропионат целлюлозы («CAP»).
Дополнительным классом добавок, которые можно применять в настоящих композициях, являются природные и синтетические масла и жиры. Масла животного происхождения или полученные из семян или орехов растений обычно включают глицериды жирных кислот, главным образом олеиновой, пальмитиновой, стеариновой и линолевой. Как правило, чем больше водорода содержит молекула, тем более вязким становится масло.
Подходящие природные и синтетические масла включают в качестве неограничивающих примеров растительное масло, арахисовое масло, триглицериды со средней цепью, соевое масло, миндальное масло, оливковое масло, кунжутное масло, фенхелевое масло, камелиевое масло, кукурузное масло, касторовое масло, хлопковое масло и соевое масло, или сырое, или рафинированное, и триглицериды жирных кислот со средней цепью.
Жиры обычно представляют собой сложные эфиры глицерина и высших жирных кислот, таких как стеариновая и пальмитиновая. Такие сложные эфиры и их смеси являются твердыми при комнатной температуре и обладают кристаллической структурой. Примерами являются сало и животный жир. Как правило, масла и жиры увеличивают гидрофобность системы неполимерного носителя, замедляют деградацию и поглощение воды. Все описанные выше композиции можно применять в способах по настоящему изобретению для обеспечения длительной местной анестезии в участке-мишени. В частности, композиции могут быть составлены в виде жидкости, спрея, крема, лосьона, мази, геля, взвеси, масла, эмульсии, микроэмульсии, твердого вещества, пластыря, пленки, частицы, микрочастицы, порошка или любой другой подходящей формы фармацевтической композиции и затем введены субъекту посредством способов местной, чрескожной, парентеральной (например, инъекция, имплантат и т.д.) доставки или подобных. Композиции, содержащие анестетик и фармацевтически приемлемый неполимерный носитель, используют для обеспечения анестетического действия, характеризуемого длительной местной анестезией после введения субъекту без начального выброса и продолжительностью по меньшей мере приблизительно 24 часа после введения, предпочтительно по меньшей мере приблизительно 36-48 часов после введения и более предпочтительно по меньшей мере приблизительно 48-72 часа после введения. В конкретных вариантах осуществления начало местной анестезии происходит в течение приблизительно 2 часов от введения субъекту, предпочтительно в течение приблизительно 1 часа от введения и в некоторых случаях в течение приблизительно 30 минут от введения субъекту.
Термин «субъект», как используется здесь, относится к любому позвоночному, у которого желательно обеспечить состояние местной анестезии. Термин, таким образом, в широком смысле относится к любому животному, подлежащему лечению композициями по настоящему изобретению, такому как птицы, рыбы и млекопитающие, включая человека. В конкретных вариантах осуществления способы по настоящему изобретению пригодны для обеспечения длительной анестезии в ветеринарной практике и животноводстве, например для птиц и млекопитающих, где долговременное состояние местной анестезии является удобным или желательным. В конкретных случаях композиции, в частности, подходят для использования домашними животными, такими как собаки или кошки, и, кроме того, их можно использовать для лошадей. В предпочтительных вариантах осуществления термин «субъект» обозначает относящегося к человеку субъекта. Кроме того, термин «субъект» не обозначает конкретного возраста, и композиции, таким образом, пригодны для использования субъектами любого возраста, такими как субъекты младенческого, подросткового, взрослого и пожилого возраста.
В предпочтительных вариантах осуществления композиции по настоящему изобретению подходят, в частности, для лечения ран. Системы неполимерного носителя позволяют легко применять анестетическое средство или средства для раны, или непосредственно внутри раны, и/или рядом с раной, с использованием очень простых способов применения, таких как закапывание, распыление, штрихование, намазывание, заливка или другая ручная обработка раны композицией в виде спрея, крема, лосьона, мази, геля, взвеси, масла, эмульсии, микроэмульсии, гибкого твердого вещества или пластыря, пленки, частицы, микрочастицы или порошка. Таким образом, композиции можно применять для раны любого размера или формы и обеспечивать равномерное распределение анестетического средства или средств по всей области раны для лучшего удержания и эффективности. Раны, которые можно обрабатывать с использованием таких способов, могут меняться от самых неглубоких до глубоких, от поверхностных до надрезов и от хирургических (или в другом случае преднамеренных) до случайных. Если композиция предназначена для инъекции, ее можно наносить в подкожное пространство с использованием смещаемых инъекций рядом с раной по всем сторонам или внешним границам. Можно применять также комбинированные способы, такие где композицию помещают непосредственно в рану, например, перед хирургическим закрытием просвета, и, кроме того, вдоль раны. В особенно предпочтительном варианте осуществления способы по изобретению включают применение настоящих композиций в качестве местного анестетика для лечения послеоперационной инцизионной боли. Использование настоящих композиций этим способом может устранять или по меньшей мере уменьшать необходимость предоставления дополнительного лечения, такого как введение системных наркотических аналгетиков для лечения такой послеоперационной боли. Соответственно, композиции можно применять для лечения послеоперационной боли, сопровождающей все типы медицинских процедур, такие как большие операции (например, торакотомия, восстановление аорты, резекция кишки), промежуточные операции (например, кесарево сечение, гистерэктомия и аппендэктомия) и малые операции (лапароскопия, артроскопия и процедуры биопсии), которая в ином случае может быть изнуряющей и может требовать лечение боли в течение 3-5 дней после операции.
Таким образом, описанные здесь композиции можно вводить при практическом осуществлении настоящих способов с использованием широкого разнообразия способов. Например, композиции можно вводить местно, системно (например, на слизистую (перорально, ректально, вагинально или назально), парентерально (внутривенно, подкожно, внутримышечно или внутрибрюшинно) или т.п. Композиции можно вводить посредством инъекции, заливки, разбрызгивания, аэрозоля или покрытого аппликатора. Аэрозоли или туманы из композиции можно вводить с использованием аэрозольного пропеллента, например, для местного введения или с использованием подходящего распылителя, например, для назального или перорального введения на слизистые.
Предпочтительно композиции вводят в виде жидкости посредством инъекции или в виде аэрозоля, пасты или эмульсии. При применении в аэрозоле любой растворитель, присутствующий в растворе аэрозоля, будет, как правило, испаряться после нанесения, позволяя композиции создавать пленку. Альтернативно, аэрозоль или эмульсию можно получать без растворителя. В данной ситуации аэрозольный пропеллент может также функционировать как растворитель. Получение аэрозолей и эмульсий можно осуществлять с использованием способов, известных специалистам в данной области. См., например, Ansel, H.C. et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, Sixth Edition (1995).
Кроме применений, описанных выше, настоящие композиции можно вводить посредством осмотических насосов. В одном из вариантов осуществления разработано устройство для имплантации в ткань субъекта и для осуществления длительного высвобождения в течение времени.
Возможно также вводить композиции по изобретению с использованием пористой или непористой трубки, желательно сделанной из экструдированного биоразлагаемого полимера. Можно получить трубку с различными степенями пористости в зависимости от композиции и желаемых характеристик высвобождения. Композицию по изобретению вставляют в трубку, и концы трубки можно оставлять открытыми, позволяя биологически активному соединению диффундировать из концов трубки, или можно закрыть дополнительным пористым или непористым полимером. Пористые крышки и пористые трубки позволяют соединению со временем диффундировать через поры. Непористые крышки, так же как непористые трубки, позволяют анестетическим средствам, которые являются растворимыми в полимере, диффундировать через него в окружающие ткани. Непористые материалы, которые не являются растворителями для анестетика, но являются биоразлагаемыми, будут высвобождать анестетик, когда они достаточно деградируют. Композиции по изобретению можно получать и хранить как многокомпонентные системы, пока они не готовы для введения. Число различных компонентов будет отчасти зависеть от характеристик композиции. Перед введением компоненты объединяют и смешивают, например, для получения однородной композиции, которую можно затем вводить субъекту. Растворители или добавки можно добавлять к одному или всем компонентам, или они могут составлять отдельный компонент, который также смешивают с другими перед введением. Разделение композиции на многокомпонентную смесь позволяет оптимизировать условия хранения для каждого компонента и минимизировать любые вредные взаимодействия между компонентами с течением времени. В результате увеличивается стабильность при хранении.
ПРИМЕРЫ
Ниже приведены примеры конкретных вариантов осуществления для выполнения настоящего изобретения. Примеры представлены только для иллюстративных целей и не предназначены для ограничения объема настоящего изобретения каким-либо образом.
Общие способы
Эффективность in vivo композиций и способов по изобретению можно оценить у крыс с использованием модели горячей пластины, например, способом, подробно описанным в IACUC № 9511-2199. Критериями эффективности, принятыми для композиций по изобретению, являются среднее запаздывание больше чем приблизительно на 2 секунды, с прерыванием при 12 секундах (данное прерывание устанавливают для предупреждения любого возможного повреждения животного). Запаздывания на 2 секунды являются доказательными для статистически значимого действия местного анестетика. Предпочтительно среднее запаздывание для крыс в модели горячей пластины превышает 7 секунд. Предпочтительно процент отвечающих составляет 50% или более. Предпочтительно композиции по изобретению обеспечивают среднее запаздывание для крыс в модели горячей пластины от более чем приблизительно 7 секунд до приблизительно 12 секунд, с процентом крыс, для которых показан эффект, составляющим по меньшей мере приблизительно 50% из тестированных.
Сущность способа горячей пластины для крыс следующая. Используют самцов крыс Sprague Dawley (Harlan Laboratories, Indianapolis, Ind.) со средней массой 275 г. Исследование на горячей пластине заключается в осторожной поддержке тела животного, в то время как подошвенную поверхность задней лапы помещают на горячую пластину при 56°С. Исходное запаздывание определяют перед односторонней инъекцией анестетической композиции около седалищного нерва крысы.
Сенсорное тестирование на моделях для человека также применимо для тестирования композиций по настоящему изобретению. Местную анестетическую активность можно оценивать по отношению к началу, пиковому значению и длительности действия с использованием семи конкретных способов: (a) механическое сенсорное тестирование (порог распознавания механической боли с использованием волокон Фрея); (b) сверхпороговое (механическое) тестирование с использованием отдельного волокна Фрея; (c) термическое сенсорное тестирование (порог распознавания теплого); (d) порог распознавания боли от горячего; (e) сверхпороговое (горячее) тестирование и (g) тактильное сенсорное тестирование (порог распознавания механического прикосновения). Различные степени или уровни результатов могут быть доказательными для ощущаемого субъектом местного обезболивания, местного онемения и/или местной блокады нерва. Анестетическую активность композиций и способов по изобретению можно дополнительно характеризовать в отношении безопасности различными показателями активности, такими как системные уровни в плазме крови, полученные после введения в локализованный участок.
Порог распознавания механической боли определяют как наименьшую силу или число волокон Фрея, дающие определенное ощущение боли или дискомфорта, и порог распознавания механического прикосновения определяют как наименьшую силу или число волокон Фрея, дающие определенное ощущение прикосновения или давления. Пороги распознавания механического прикосновения и пороги распознавания механической боли можно определять одновременно с использованием волокон Фрея возрастающей жесткости (VFH) (доступны от Somedic A/B, Stockholm, Sweden). Ранее было определено, что каждое VFH, нажатое против равновесия до небольшого сгиба, обеспечивает силу, которая увеличивается для каждого волокна, покрывая полный диапазон от 3 до 402 миллиньютон (мН) (VFH No. 7=3 мН; VFH No. 8=13 мН; VFH No. 9=20 мН; VFH No. 10=39 мН; VFH No. 11=59 мН; VFH No. 12=98 мН; VFH No. 13=128 мН; VFH No. 14=133 мН; VFH No. 15=314 мН; VFH No. 16=350 мН; VFH No. 17=402 мН).
Соответственно, у субъекта-человека область после инъекции композиции, полученной по настоящему изобретению, можно стимулировать 8 раз каждым VFH со скоростью приблизительно 2 стимула в секунду, начиная с VFH No. 7 и двигаясь к VFH No. 17. Наименьший номер VFH, ощущаемых как прикосновение или давление (порог распознавания механического прикосновения), и наименьший номер волокна, половина из восьми стимуляций которым являются болезненными или неприятными (порог распознавания механической боли), записывают. Процедуру повторяют еще два раза и записывают медиану из трех измерений. Если для VFH No. 17 не получают ощущения прикосновения или давления, значение порога распознавания механического прикосновения оценивают как 18. Если для VFH No. 17 не получают какой-либо боли или дискомфорта, величину порога распознавания механической боли оценивают как 18. Сверхпороговый болевой ответ - механический для волокна Фрея - определяют стимулированием области инъекции пять раз VFH No. 17 (402 мН). Субъект оценивает боль с использованием шкалы VRS 0-10, где ноль (0) = отсутствие боли, а десять = (10) боль настолько интенсивна, насколько можно вообразить.
Как обсуждалось выше, этот тест проводят с отдельным жестким волокном Фрея, которое определяют как вызывающее болевую реакцию у субъектов. Болевую реакцию определяют стимуляцией области после инъекции или другой обработки 5 раз VFH No. 17. Субъекты оценивали боль по вербальной шкале оценок (VRS) от 0 до 10, как выше.
Термическое тестирование (сверхпороговую болевую реакцию на горячее) для обработанной области определяют по стимулированию 45°С, продолжающемуся 5 секунд, с использованием компьютеризированного термодатчика (доступного от Thermostest, Somedic A/B, Stockholm, Sweden) на обработанных областях. Субъект оценивает боль по вербальной шкале оценок (VRS) 0-10.
Порог распознавания теплого определяют как наименьшее воспринимаемое увеличение температуры от 32°С, порог распознавания боли от горячего определяют как наименьшую температуру, воспринимаемую как болезненную, и порог распознавания холодного определяют как наименьшее воспринимаемое снижение температуры от 32°С. Порог распознавания теплого, порог распознавания боли от горячего и порог распознавания холодного определяют компьютеризированным термодатчиком (доступным от Somedic A/B, Stockholm, Sweden) на обработанных областях. Субъекты получают инструкции нажимать на кнопку, как только достигают конкретного ощущения. Термические пороги определяют от исходных 32°С и увеличивают (порог распознавания теплого и порог распознавания боли от горячего) или уменьшают (порог распознавания холодного) со скоростью изменения 1°С в секунду. Верхний предел прекращения составляет 52°С для порога распознавания теплого и порога распознавания боли от горячего. Нижний предел прекращения составляет 25°С для порога распознавания холодного.
Порог распознавания теплого, порог распознавания боли от горячего и порог распознавания холодного вычисляют как медиану трех измерений с интервалами по 10 секунд между каждыми стимулами. Если субъект не воспринимал тепло или боль при 52°С, величину 53°С записывали как порог распознавания теплого; если субъект не воспринимал боль при 52°С, величину 53°С записывали как порог распознавания боли от горячего; и если субъект не воспринимал холод или боль при 25°С, величину 24°С записывали как порог распознавания холодного.
Пример 1
Систему неполимерного жидкого носителя, содержащую анестетическое средство, получали следующим образом. Изобутират ацетата сахарозы (SAIB) объединяли с растворителем для носителя SAIB N-метилпирролидоном (NMP) с получением смеси 70:30. К этой смеси добавляли или 2,5% (мас./об.), или 5% бупивакаин (свободное основание) для получения двух тестовых композиций.
Самцов крыс Sprague Dawley (275-300 г) разделяли на две тестовые группы по 8 животных каждая. Тестовые составы вводили четвероногим с использованием иглы и шприца для доставки либо 25, либо 50 мг доз бупивакаина. Затем использовали тест реакции вздрагивания кожи для определения наличия местного анестетического действия, где использовали непроизвольное вздрагивание при кожной стимуляции с использованием булавки для 10 случайных областей внутри 1 см относительно участка инъекции. Затем вычисляли процент ингибирования распознавания булавки взятием исходного ответа минус тестовый ответ, деленного на исходный ответ и умноженного на 100.
Полученные результаты указывают на то, что обе тестовые композиции обеспечивают местное анестетическое действие продолжительностью до приблизительно 60-72 часов, с началом активности в течение 1 часа от введения.
Пример 2
Субъекты. Для исследования использовали самцов крыс Фишера 344, (Charles River Laboratories) (N=96). Животных содержали при обращенном цикле свет:темнота (темнота 5:00-17:00) в виварии с контролем температуры и влажности. В периоды экспериментов крысам предоставляли доступ к пище и воде по желанию. Все эксперименты проводили во время фазы темноты цикла свет:темнота. Все процедуры были одобрены индустриальным комитетом по уходу за животными и их использованию центра медико-санитарных дисциплин университета Wake Forest.
Хирургическая процедура. После проведения анестезии 5% парами изофлурана в кислороде животным выбривали левый нижний квадрант брюха. Во время хирургической процедуры поддерживали анестезию с использованием 2,0-2,5% паров изофлурана в кислороде. На 0,5 см ниже самого нижнего ребра на левой стороне и параллельно ему располагали 3 см разрез, проникающий в брюшную полость. Внутренними органами и мускулатурой энергично манипулировали, вставляя 5-7 см указательного пальца в брюшную полость и растягивая мускулатуру. Приблизительно 10 см тонкого кишечника выводили на поверхность и осторожно манипулировали с ними. Кишечник помещали внутрь брюшной полости и рану зашивали в 3 слоя, состоящих из перитонеальной выстилки, мышц брюшного пресса и кожи, с использованием хромированного кетгута 4.0. Внешнюю поверхность ран покрывали порошком антибиотика (Polysporin®, Glaxo-Wellcome, Research Triangle Park, NC) и животным i.m. вводили 75000 ед. прокаина пенициллина G. Ложно-обработанных животных анестезировали, брили, выдерживали под изофлурановой анестезией в течение 20 мин и вводили им i.m. прокаин пенициллин G.
Введение композиции бупивакаина с контролируемым высвобождением. Тестовым животным вводили или носитель (70:30, SAIB:NMP), 1,25% (мас./об.), 2,5% или 5% бупивакаин согласно таблице 1. Композицию вводили после сшивания кожи с использованием способа смещаемой инъекции 1,0 и 1,5 мл шприцом иглой 22 ga, так что 0,25 мл композиции равномерно распределяли на протяжении области разреза приблизительно на 0,5 см выше участка раны. Второе введение проводили таким же образом на 0,5 см ниже участка раны. Для групп K и L проводили дополнительное введение 0,1 мл или 1,25% (мас./об.), или 5% бупивакаина поверх перитонеальной выстилки с использованием способа смещаемой инъекции перед сшиванием внешнего мышечного слоя. Композиции с бупивакаином позволяют оставаться на перитонеальной выстилке 1-2 мин перед сшиванием внешней мышцы для достижения достаточной вязкости и, таким образом, предотвращения вымывания композиции через внешнюю мышцу в течение процесса зашивания.
Группы обрабатывали, как описано ниже. Все группы, за исключением группы B, подвергали хирургической лапаротомии, как описано выше.
Измерение спонтанной локомоции. Исследовательское поведение оценивали, начиная с 24 часов после лапаротомии, с использованием коммерчески доступного оборудования и программного обеспечения (Med Associates Inc., St. Albans, VT). Камеры для активности состояли из акриловых замкнутых пространств размером 17” × 17”, высотой 15” с открытым верхом. Двойные наборы из 16 инфракрасных излучателей, разделенных 1”, помещали в направлениях X и Y, на 1” выше поверхности пола, с выровненными по одной линии инфракрасными детекторами на противоположных сторонах камеры. Третий набор инфракрасных излучателей и детекторов помещали в направлении X, на 7 см выше поверхности пола, так что крысам, используемым в этих исследованиях, требовалось подниматься на задние лапы, чтобы прерывать эти лучи. Каждую камеру для активности помещали в пространство с ослабленными светом и звуком. Сеансы проводили через 16, 24, 40, 48 и 72 часа после операции, и они длились 60 мин. Собранные измерения включали общую пройденную дистанцию, общее число прерываний лучей как в X, так и Y направлении (показатели ходьбы), повторяющиеся прерывания лучей в пределах 3 см от животного в отсутствие локомоции (стереотипия), общее число прерываний в верхнем направлении X (подъем на задние лапы), время, проведенное в движении, время, проведенное в стереотипии, время, проведенное в подъемах на задние лапы, и время, проведенное на отдыхе. Все измерения собирали отрезками по 6 мин в течение сеанса, а также суммировали для целого сеанса.
Измерение возвращения анестезии с использованием теста укола булавкой. Через 96 часов после операции животных быстро помещали в прозрачную акриловую приемную камеру, которая давала доступ к брюшной области. Немного затупленной иглой 20 ga надавливали в области в пределах 0,5 см от участка брюшной раны с достаточной силой до заметной вмятины в брюшной ткани. Иглу оставляли на месте на 10 секунд или пока животное не отреагирует вздрагиванием или движением от иглы. Каждое животное тестировали 2-3 раза в участке раны. Участок, по меньшей мере на 10 см отдаленный от участка раны, тестировали таким же образом в качестве измерения положительного контроля.
Анализ данных. Данные анализировали с использованием 2-факторного ANOVA для повторяющихся измерений с индивидуальными индексами поведения, служащими независимыми измерениями, и группой лечения и временем после операции в качестве зависимых переменных. После этого проводили анализы с использованием t-теста Бонферрони-Данна для множественных сравнений с группой ложной операции, служащей контролем, и с использованием защищенных LSD по Фишеру для всех попарных сравнений.
Результаты (пройденное расстояние). Эффекты хирургии и околооперативного лечения либо носителем, либо бупивакаином показаны для каждой временной точки фигур 1-5. Имело место значительное среднее действие группы лечения и времени после операции на пройденную дистанцию без существенного взаимодействия между этими переменными (таблица 1).
Результаты 2-факторного ANOVA для пройденной дистанции (см)
Последующим анализом с использованием t-теста Бонферрони-Данна для множественных сравнений с контрольной группой выявили, что все группы с введенным бупивакаином достоверно не отличались от ложной обработки, в то время как животные с разрезом без лечения или после лечения носителем оставались статистически отличными от ложно-обработанных контролей в течение исследования (таблица 2). Последующим анализом всех попарных сравнений с использованием защищенными LSD по Фишеру обнаружили, что только группы с введением 5% бупивакаина или 1,25% бупивакаина как в участке перитонеальной выстилки, так и на внешнюю мышцу достоверно не отличались от ложной обработки (таблица 3).
Защищенные LSD по Фишеру для всех попарных сравнений, пройденная дистанция (см)
Показатели ходьбы. Статистические результаты для данных показателей ходьбы являются качественно сходными с результатами, описанными для пройденной дистанции выше. Данные показателей ходьбы оценивали для каждой отдельной временной точки после операции. Как и для пройденной дистанции в течение сеанса, имело место значительное основное действие и группы лечения, и времени после операции на показатели ходьбы, без существенного взаимодействия между этими переменными (таблица 4).
Результаты 2-факторного ANOVA для показателей ходьбы
Последующим анализом t-тестом Бонферрони-Данна с использованием показателей ходьбы в качестве независимого измерения получали результаты, сходные с данными для пройденной дистанции (таблица 5).
t-тест Бонферрони-Данна для множественных сравнений, показатели ходьбы
Анализом всех попарных сравнений защищенными LSD по Фишеру обнаружили, что группа с ложной обработкой достоверно отличалась только от контрольной группы с разрезом, обработанной носителем группы и группы, обработанной наименьшей дозой бупивакаина (1,25%) (таблица 6).
Защищенные LSD по Фишеру для всех попарных сравнений, показатели ходьбы
Показатели стереотипии. Отсутствовали достоверные различия между группами для близких движений (стереотипии), однако имело место основное достоверное действие времени после операции и достоверное взаимодействие между группой лечения и временем после операции (таблица 7, фигуры 11-15). Последующим анализом с использованием t-теста Бонферрони-Данна выявили, что временная точка 40 часов достоверно отличалась от других временных точек по данному измерению (таблица 8). Исключением временной точки 40 часов и повторным анализом оставшихся данных 2-факторным ANOVA выявили более или менее достоверное действие группы лечения на стереотипическое поведение, достоверное основное действие времени после операции и достоверное взаимодействие между группой лечения и временем после операции (таблица 9). Последующим анализом t-тестом Бонферрони-Данна не выявили достоверных различий между группами лечения, однако в t-тесте защищенных LSD по Фишеру обнаружили, что все группы значительно отличались от ложно-обработанных контролей, за исключением наивысшей дозы бупивакаина (5%), так же как группы, обработанной 1,25% бупивакаином, и на уровне перитонеальной выстилки, и на слое мышц брюшного пресса (таблица 10).
2-факторный ANOVA, показатели стереотипии
Бонферрони-Данн для множественных сравнений с контролем, время после операции
2-факторный ANOVA с исключением временной точки 40 ч, показатели стереотипии
Защищенные LSD по Фишеру для всех попарных сравнений, за исключением временной точки 40 ч, показатели стереотипиии
Вертикальные показатели (подъем на задние лапы). Имело место основное достоверное действие на подъем на задние лапы как группы лечения, так и времени после операции, так же как достоверное взаимодействие между этими переменными (таблица 11). Последующим анализом с использованием t-теста Бонферрони-Данна выявили достоверные различия между ложно-обработанным контролем и всеми группами лечения (таблица 12). По результатам теста Фишера для защищенных LSD получили согласующиеся данные с отсутствием достоверных различий, за исключением отличия группы с ложной обработкой от всех остальных обработок (таблица 13).
2-факторный ANOVA, вертикальные показатели
Бонферрони-Данн для множественных сравнений с контролем, вертикальные показатели
Защищенные LSD по Фишеру для всех попарных сравнений, вертикальные показатели
Стимул укола булавкой. Животных подвергали стимулу укола булавкой в области внутри 0,5 см от разреза через 96 часов после операции. Все животные отвечали на этот стимул, за исключением одного животного в группе HH (1,25% бупивакаин, введенный как на слой мышц брюшного пресса, так и на перитонеальную выстилку). Это животное нормально отвечало на стимул укола булавкой, когда его применяли в участке приблизительно в 3 см от участка разреза.
Результаты. Послеоперационное лечение носителем, применяемым для композиции с длительным высвобождением (контроль), не оказывало значительного действия на измерения активности по сравнению с лапаротомией без другого лечения. Имело место явное действие носителя на стереотипическое поведение в некоторых из поздних временных точек. Эти данные согласованно подтверждают достоверное аналгетическое действие 5% бупивакаина в композиции с длительным высвобождением, по-видимому, имеющее место в поздних исследуемых временных точках. Композиция с контролируемым высвобождением 5% бупивакаина постоянно обеспечивала существенную аналгезию и изменяла действие лапаротомии на все измерения поведения, за исключением вертикальной активности. Некоторые аналгетические действия были обнаружены после введения 1,25% бупивакаина как на уровне перитонеальной выстилки, так и на внешнем слое мышц брюшного пресса, однако действия не были такими стойкими, как обнаруженные после введения 5% бупивакаина только на внешний слой мышц брюшного пресса. Действие 5% бупивакаина является более сильным во временных точках 48 и 72 часа. Причина(причины), по которым данные животных показали меньшую активность в более ранних временных точках, не является(не являются) совершенно очевидными, так как эти животные демонстрировали нормальное поведение при кормлении и уходе за поверхностью тела в их домашних клетках и, по-видимому, отсутствие стресса. Эти данные доказывают, что местного анестетического действия с использованием композиций по изобретению с контролируемым высвобождением, содержащих анестетик бупивакаин, достаточно, чтобы контролировать боль после операции.
Пример 3
Следующую оценку повышения дозы, безопасности и фармакокинетики проводили для здоровых человеческих субъектов-добровольцев, чтобы оценить безопасность/переносимость и предварительное фармакокинетическое действие композиции с контролируемым высвобождением бупивакаина, содержащей неполимерный носитель изобутират ацетата сахарозы.
Композиции составляли с использованием свободного основания бупивакаина, составленного в неполимерном носителе изобутирате ацетата сахарозы (SAIB), дополнительно содержащем N-метил-2-пирролидон (NMP), действующий в качестве растворителя для бупивакаина и носителя SAIB. Композицию получали смешиванием носителя SAIB и растворителя NMP (70:30 носителя) с 5% мас. бупивакаина для получения отдельных доз, содержащих 137,5 мг бупивакаина в объеме инъекции 2,5 мл. Композицию предоставляли в виде жидкости для инъекций.
В исследование входило две когорты. Когорта 1 состояла из 6 здоровых субъектов-мужчин, в возрасте от 22 до 38. Для когорты 1 все субъекты получали инъекции общим объемом 2,5 мл композиции SAIB/NMP/бупивакаин (содержащей 137,5 мг бупивакаина) в первый участок введения, введенные в абдоминальную подкожную область в форме 5 см смещаемой инъекции; и 2,5 мл инъекции плацебо, содержащей только носитель (SAIB/NMP), во второй участок введения, также введенные в абдоминальную подкожную область в форме 5 см смещаемой инъекции. После введений субъектов наблюдали вплоть до восьми часов для мониторинга местного состояния тканей в участке введения и собирали образцы плазмы. Дополнительные образцы плазмы отбирали на сутки 1, 2, 3, 4 и 28. Во всех образцах плазмы тестировали концентрацию бупивакаина в крови с использованием общепринятых способов.
Когорта 2 также состояла из 6 здоровых субъектов-мужчин в возрасте от 22 до 38. Когорту 2 разделяли на две подгруппы, где первой подгруппе (n = 3) вводили инъекции общим объемом 5 мл композиции SAIB/NMP/бупивакаин (содержащей 275 мг бупивакаина), введенные в абдоминальную подкожную область в форме двух 5 см смещаемых инъекций по 2,5 мл каждая. Второй подгруппе (n = 3) вводили инъекции плацебо общим объемом 2,5 мл, содержащие только носитель (SAIB/NMP), также введенные в абдоминальную подкожную область в форме двух 5 см смещаемых инъекций по 1,25 мл каждая. Здесь снова, после введения, субъектов наблюдали вплоть до восьми часов для мониторинга местного состояния тканей в участке введения и собирали образцы плазмы. Дополнительные образцы плазмы собирали на сутки 1, 2, 3, 4 и 28.
В качестве результата исследования обнаружили, что носитель SAIB и композиции SAIB/NMP/бупивакаина были хорошо переносимыми, где инъекции не приводили к какому-либо заметному покраснению, отеку, зуду, изменению окраски или любому другому неблагоприятному симптому в участке инъекции, или любой нежелательной реакции ткани в процессе исследования. Кроме того, оценкой фармакокинетики бупивакаина показали достаточно медленное (пролонгированное) высвобождение активного бупивакаина из носителя SAIB, с высвобождением активного бупивакаина в течение периода 3-4 суток. Эти фармакокинетические результаты представлены на фигуре 1. Как можно видеть, концентрации бупивакаина в плазме Cmax после инъекций в когорте 1 (84 нг/мл) и когорте 2 (174 нг/мл) были ниже опубликованных установленных диапазонов токсичной концентрации в плазме (приблизительно 1-4 мкг/мл). Кроме того, сравнениями между кривыми когорты 1 и когорты 2 показали, что уровни бупивакаина в плазме увеличиваются по существу линейным образом с повышением дозы. Более того, оценкой AUC показали 100% биологическую доступность бупивакаина.
Пример 4
Следующую оценку повышения дозы, безопасности, фармакокинетики и фармакодинамики (эффективности) проводили для пациентов-людей, подвергавшихся процедурам хирургической пластики грыжи, чтобы оценить эффективность и фармакокинетическое действие композиции с контролируемым высвобождением бупивакаина, содержащей неполимерный носитель изобутират ацетата сахарозы и полученной в соответствии с настоящим изобретением. В исследовании сравнивали эффективность настоящих композиций SAIB/бупивакаин, введенных подкожно в сочетании с инфильтратом в рану солевого раствора (плацебо) или бупивакаина гидрохлорида (Marcain®), с эффективностью коммерчески доступного раствора бупивакаина (Marcain®, бупивакаина гидрохлорид BP, 5,28 мг/мл, эквивалентно 5 мг/мл безводного бупивакаина гидрохлорида), введенного подкожно и в виде инфильтрата в открытую паховую область пациентов с пластикой грыжи.
Тестовую композицию составляли/составляют с использованием свободного основания бупивакаина, составленного в неполимерном носителе изобутирате ацетата сахарозы (SAIB), дополнительно содержащем бензиловый спирт (BA), который действует в качестве растворителя для бупивакаина и носителя SAIB. Бензиловый спирт также является анестетическим средством. Композицию получали/получают сочетанием приблизительно 66% мас. носителя SAIB, 22% мас. растворителя/анестетика бензилового спирта и 12% мас. бупивакаина для получения отдельных доз, содержащих 159,5 мг бупивакаина в объеме инъекции 1,25 мл (319 мг в 2,5 мл общего объема). Композицию предоставляли/предоставляют в виде пригодной для инъекций прозрачной жидкости.
Дизайн исследования включает 3 когорты с вплоть до 91 пациентом (6 пациентов для когорты 1; 15 пациентов для когорты 2; и вплоть до 70 пациентов для когорты 3). В частности, когорта 1 состояла из 6 здоровых субъектов-мужчин в возрасте от 23 до 52. В когорте 1 всем пациентам вводили инъекции общим объемом 2,5 мл композиции SAIB/BA/бупивакаин (содержащей 319 мг бупивакаина), введенные в виде двух смещаемых подкожных инъекций вдоль каждой стороны хирургической раны (0,5 мл/см вдоль предполагаемых 5 см общей длины разрезанной раны) с 10 мл солевого раствора, инфильтрованного в разрезанную рану (включая субфасциальную) перед закрытием раны. Смещаемые инъекции вводили в 0,5-1,0 см от границ разреза раны и параллельно им, и их проводили продвижением иглы подкожно, параллельно и вдоль длины разреза, вливая непрерывно с выниманием иглы. Анестетическое/аналгетическое действие оценивали с использованием тестов времени до первого дополнительного аналгетического средства и общего употребления дополнительного аналгетического лекарственного средства (в течение 4 суток). Концентрацию бупивакаина в плазме измеряли периодически на всем протяжении курса исследования, в частности в течение первых 24 часов для оценки амплитуды раннего высвобождения бупивакаина из композиции SAIB с контролируемым высвобождением.
Результаты теста времени до первого дополнительного аналгетического средства представлены ниже в таблице 14.
Время до первого дополнительного аналгетического средства
Результаты общего употребления дополнительного аналгетического лекарственного средства (в течение 4 суток) приведены ниже в таблице 15.
Общее употребление дополнительного аналгетического лекарственного средства
Как и в исследовании примера 3, снова обнаружили, что композиция SAIB/BA/бупивакаин была хорошо переносимой, где инъекции не приводили к какому-либо заметному покраснению, отеку, зуду, изменению окраски или любому другому неблагоприятному симптому в участке инъекции, или любой нежелательной реакции ткани в процессе исследования. Кроме того, оценкой фармакокинетики бупивакаина показали пролонгированное высвобождение активного бупивакаина из носителя SAIB с высвобождением активного бупивакаина в течение периода 4 суток. Фармакокинетические результаты представлены на фигурах 2 и 3. Как можно видеть, композиция SAIB/BA/бупивакаин быстро высвобождала активный бупивакаин (в течение приблизительно 1 часа от введения) без начального выброса, и для нее показали по существу постоянное высвобождение с установившимся режимом в течение по меньшей мере первых 3 суток лечения. Наблюдаемая средняя Cmax составляла 277 нг/мл ± 109; Tmax составляло 23 часа ± 21; и Css составляла 191 нг/мл ± 13.
Когорта 2 состояла из 15 здоровых субъектов-мужчин, в возрасте от 26 до 54. Когорту 2 разделяли на три подгруппы, где первой подгруппе (n = 5) вводили инъекции композиции SAIB/BA/бупивакаин (содержащей 638 мг бупивакаина) общим объемом 5,0 мл, введенной как две смещаемые подкожные инъекции вдоль каждой стороны хирургической раны (0,5 мл/см вдоль предполагаемых 5 см общей длины разрезанной раны) с 10 мл солевого раствора, инфильтрованного в разрезанную рану (включая субфасциальную) перед закрытием раны. Второй подгруппе (n = 5) вводили инъекции композиции SAIB/BA/бупивакаин (содержащей 638 мг бупивакаина) общим объемом 5 мл, введенные как две смещаемые подкожные инъекции вдоль каждой стороны хирургической раны (0,5 мл/см вдоль предполагаемых 5 см общей длины разрезанной раны) с 10 мл Marcain® (0,5% бупивакаин-HCl), инфильтрованного в разрезанную рану (включая субфасциальную) перед закрытием раны для получения всего 688 мг бупивакаина, введенного каждому пациенту. Третьей подгруппе (n = 5) вводили инъекции композиции Marcain® (0,5% бупивакаин-HCl) общим объемом 5 мл, введенные как две смещаемые подкожные инъекции вдоль каждой стороны хирургической раны (0,5 мл/см вдоль предполагаемых 5 см общей длины разрезанной раны) вместе с 10 мл Marcain®, инфильтрованного в разрезанную рану (включая субфасциальную) перед закрытием раны для получения всего 75 мг бупивакаина, введенного каждому пациенту.
Анестетическое/аналгетическое действие оценивали с использованием тестов времени до первого дополнительного аналгетического лекарственного средства, показателей боли в области разреза «в покое» и общего употребления дополнительного аналгетического лекарственного средства (в течение 4 суток). Концентрацию бупивакаина в плазме измеряли периодически в течение курса исследования, в частности в течение первых 24 часов для оценки амплитуды раннего высвобождения бупивакаина из композиции SAIB с контролируемым высвобождением.
Результаты теста времени до первого дополнительного аналгетического лекарственного средства и теста общего употребления дополнительного аналгетического лекарственного средства (в течение 4 суток) для всех трех подгрупп когорты 2 приведены ниже в таблице 16.
Среднее время до первого дополнительного аналгетического лекарственного средства и общего употребления дополнительного аналгетического лекарственного средства (в течение 4 дней)
Композиция SAIB/BA/бупивакаин снова была хорошо переносимой (подгруппа пациентов 1 и 2), где инъекции не приводили к какому-либо заметному покраснению, отеку, зуду, изменению окраски или любому другому неблагоприятному симптому в участке инъекции, или любой нежелательной реакции ткани в процессе исследования. Кроме того, оценкой фармакокинетики бупивакаина показали пролонгированное высвобождение активного бупивакаина из носителя SAIB, высвобождающего активный бупивакаин в течение периода 4 суток. Фармакокинетические результаты представлены на фигурах 4 и 5. Как можно видеть, композиция SAIB/BA/бупивакаин быстро высвобождала активный бупивакаин (в течение приблизительно 1 часа от введения) без начального выброса и демонстрировала по существу постоянное высвобождение с установившимся режимом в течение по меньшей мере первых 3 суток лечения.
Фармакодинамика для всех трех подгрупп когорты 2 приведена ниже в таблице 17.
Фармакодинамика для когорты 2
Как можно видеть из результатов исследования когорты 2, настоящие композиции с контролируемым высвобождением обеспечивают эффективное местное анестетическое действие в течение периода по меньшей мере 4 суток после операции, значительно уменьшая необходимость дополнительных аналгетических лекарственных средств. Фактически 50% пациентов, получающих композиции SAIB/BA/бупивакаин по настоящему изобретению (5 из 10 пациентов в подгруппах 1 и 2), не требовали дополнительных обезболивающих лекарственных средств в течение всего периода 4 суток. Те пациенты в подгруппах 1 и 2, которым требовались дополнительные аналгетические лекарственные средства, все еще были способны ожидать первых дополнительных обезболивающих лекарственных средств в течение приблизительно 2-3 суток, что указывает на эффективное местное анестетическое действие в течение периода по меньшей мере 2 суток после операции. Кроме того, количества доз дополнительных аналгетических лекарственных средств в подгруппах 1 и 2 были существенно уменьшены по сравнению с контрольными пациентами (подгруппа 3), кому требовалось в среднем 11 доз в течение 4 суток тестового периода по сравнению с 2,4-2,6 дозами в течение того же периода.
Более того, обзор фармакокинетических данных для когорты 2 позволяет предполагать, что эффективную подкожную дозу 638-688 мг бупивакаина можно воспроизводимо вводить с использованием композиций с контролируемым высвобождением по настоящему изобретению для обеспечения эффективной устойчивой концентрации бупивакаина в плазме приблизительно 300 нг/мл.
Результаты теста показателей боли в области разреза «в покое» для всех трех подгрупп когорты 2 представлены на фигуре 6. Данные подгруппы 3 представлены на верхней кривой (Δ), данные подгруппы 2 представлены на средней кривой и данные подгруппы 1 представлены на нижней кривой (◇). Для удобства среднее время до первого дополнительного аналгетика показано на каждой кривой. Интенсивность боли в области разреза записывали с использованием 0-100 мм визуальной аналоговой шкалы (VAS) с показателями в диапазоне от 0 (отсутствие боли) до 100 (наихудшая вообразимая боль). Каждый показатель VAS записывали в виде отдельной вертикальной линии на шкале. Тест проводили следующим образом. На сутки операции (сутки 0) показатели боли в области разреза сначала записывали через 60 минут после введения тестируемой композиции (как обсуждалось выше, подгруппе 1 вводили SAIB/BA/бупивакаин и солевой раствор; подгруппе 2 вводили SAIB/BA/бупивакаин и Marcain®; и подгруппе 3 вводили Marcain® и Marcain®). В дальнейшем показатели боли в области разреза записывали каждые 30 минут до оцениваемой временной точки 4 часа, а затем каждый час до оцениваемой временной точки 8 часов и, наконец, в оцениваемой временной точке 12 часов. В последующие сутки от 1 до 3 показатели боли в области разреза записывали утром, исходя из времени, когда вводили тестируемую композицию на сутки 0. Эти последующие измерения проводили с 4-часовыми интервалами в течение 12-часового периода оценки (4 измерения). Время применения любых сопутствующих (дополнительных) лекарственных средств также отмечали в течение этой 4-суточной оценки.
Как можно видеть из обзора результатов теста показателей боли в области разреза, показанного на фигуре 6, обе подгруппы, которым вводили тестируемые композиции SAIB/BA/бупивакаин (подгруппы 1 и 2), показали меньшие средние показатели VAS в любое время во время теста по сравнению с группой, которой вводили тестируемую композицию Marcain® (подгруппа 3). Эти результаты показывают, что композиции по настоящему изобретению обеспечивают длительную местную анестезию в участке разрезанной раны продолжительностью по меньшей мере приблизительно 36-48 часов после введения субъекту.
Пациенты когорты 3 будут разделены на 2 подгруппы лечения. Первой подгруппе будут вводить инъекции композиции SAIB/BA/бупивакаин (содержащей 958 мг бупивакаина) общим объемом 7,5 мл, введенные как две смещаемые подкожные инъекции вдоль каждой стороны хирургической раны (0,75 мл/см вдоль предполагаемых 5 см общей длины разрезанной раны) вместе с 10 мл Marcain® (0,5% бупивакаин-HCl), инфильтрованного в разрезанную рану (включая субфасциальную), перед закрытием раны для получения всего 1,008 мг бупивакаина, введенного каждому пациенту. Второй подгруппе будут вводить инъекции композиции Marcain® (0,5% бупивакаин-HCl) общим объемом 7,5 мл, введенные как две смещаемые подкожные инъекции вдоль каждой стороны хирургической раны (0,75 мл/см вдоль предполагаемых 5 см общей длины разрезанной раны) вместе с 10 мл Marcain®, инфильтрованных в разрезанную рану (включая субфасциальную) перед закрытием раны для получения всего 87,5 мг бупивакаина, введенного каждому пациенту.
Анестетическое/аналгетическое действие оценивали с использованием тестов времени до первого дополнительного аналгетического лекарственного средства и общего употребления дополнительного аналгетического лекарственного средства (в течение 4 суток). Концентрацию бупивакаина в плазме измеряли периодически в течение курса исследования, в частности в течение первых 24 часов для оценки амплитуды раннего высвобождения бупивакаина из композиции SAIB с контролируемым высвобождением. Ожидают, что более высокая доза композиции SAIB/BA/бупивакаин с контролируемым высвобождением, полученной по настоящему изобретению, будет обеспечивать такие же или даже более высокие результаты эффективности по сравнению с результатами когорты 2 тестируемых субъектов.
Понятно, что настоящее изобретение, описанное таким образом, его варианты и модификации, как будет очевидно специалистам в данной области, будет входить в объем прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНЕСТЕТИЧЕСКИЕ КОМПОЗИЦИИ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2018 |
|
RU2791481C2 |
| АНЕСТЕТИЧЕСКИЕ КОМПОЗИЦИИ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2820649C2 |
| ДОЗИРОВАННЫЕ ФОРМЫ АНЕСТЕЗИРУЮЩИХ СРЕДСТВ С ДЛИТЕЛЬНЫМ ВЫСВОБОЖДЕНИЕМ ДЛЯ ОБЕЗБОЛИВАНИЯ | 2003 |
|
RU2332985C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ БОЛИ | 2020 |
|
RU2812902C2 |
| СТАБИЛЬНЫЕ НЕВОДНЫЕ ОДНОФАЗНЫЕ ГЕЛИ И КОМПОЗИЦИИ НА ИХ ОСНОВЕ ДЛЯ ДОСТАВКИ ИЗ ИМПЛАНТИРУЕМОГО УСТРОЙСТВА | 2003 |
|
RU2342118C2 |
| ДЕПО-СОСТАВЫ МЕСТНОГО АНЕСТЕТИКА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2013 |
|
RU2664700C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ДОСТАВКИ С РЕГУЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ | 2005 |
|
RU2390355C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2008 |
|
RU2470907C2 |
| КОМПОЗИЦИЯ СО СШИТОЙ ГИАЛУРОНОВОЙ КИСЛОТОЙ ДЛЯ НАРАЩИВАНИЯ ТКАНЕЙ | 2003 |
|
RU2351367C9 |
| КОМПОЗИЦИИ ПРОЛОНГИРОВАННОГО ДЕЙСТВИЯ С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2003 |
|
RU2355385C2 |
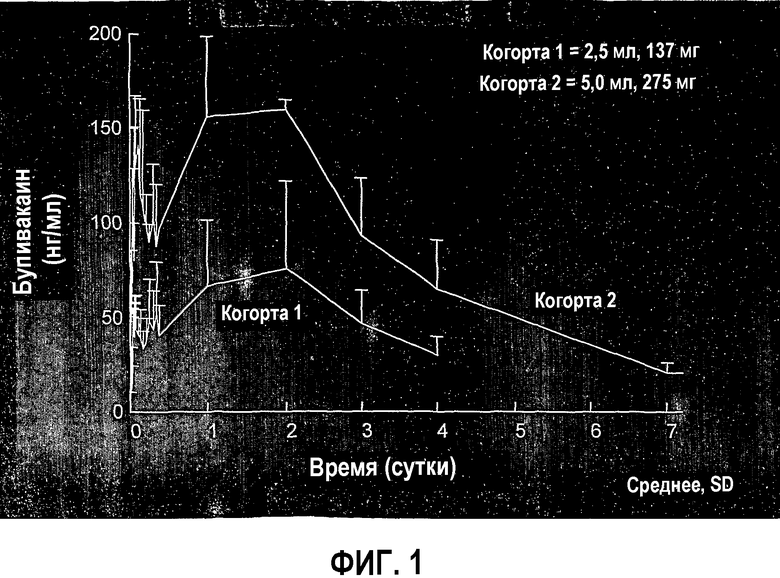
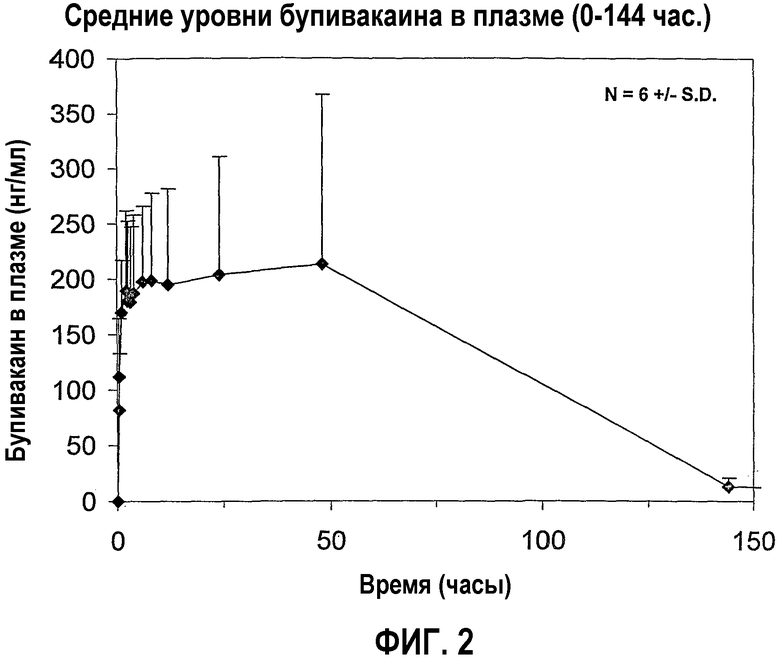
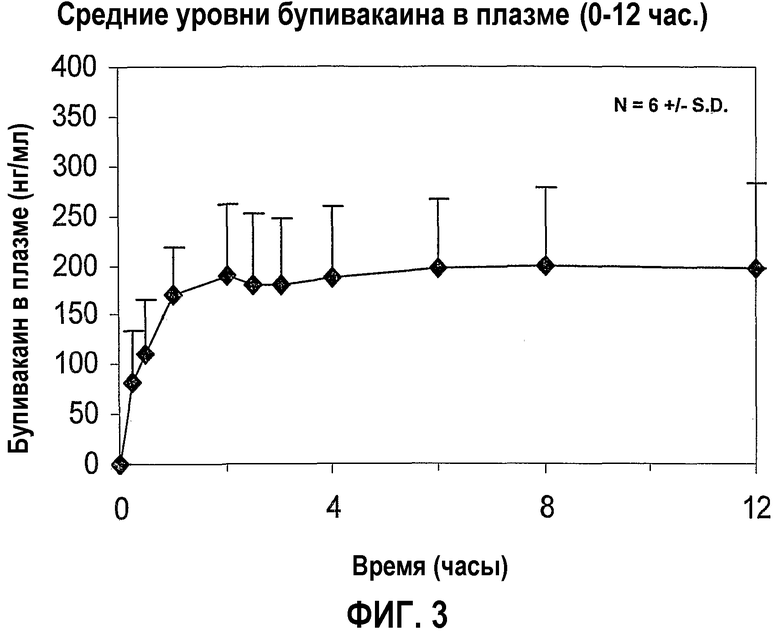
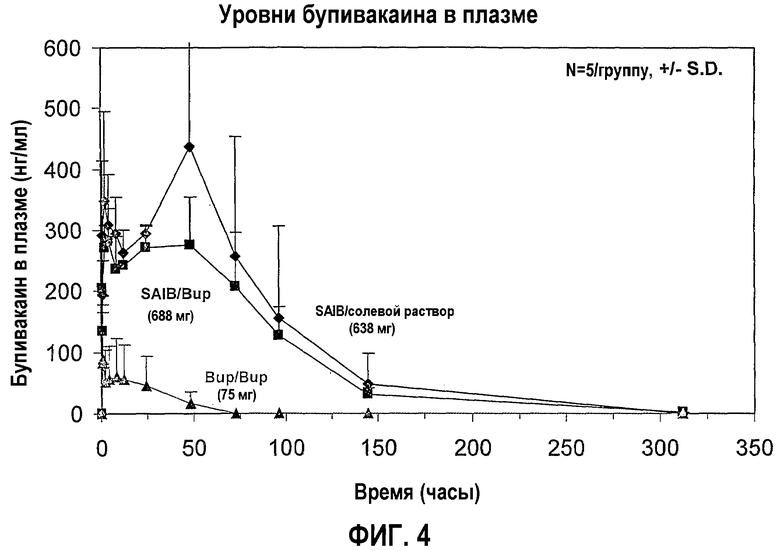
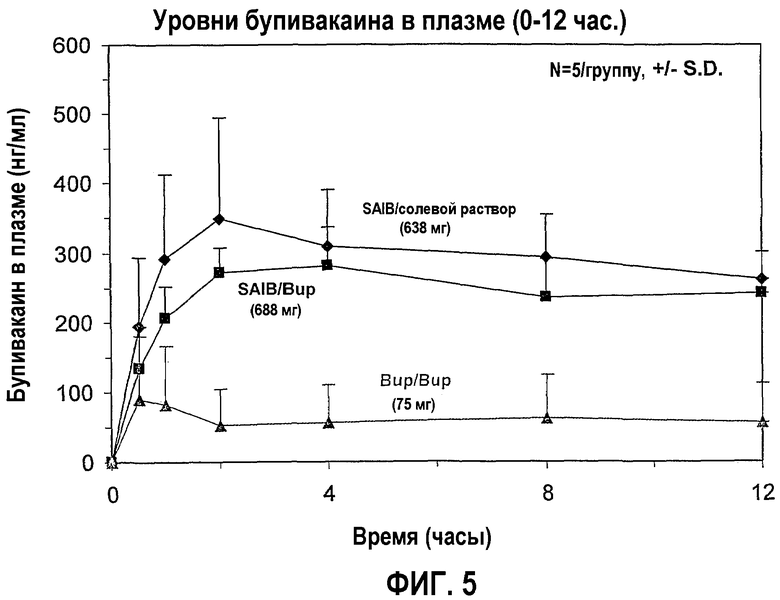
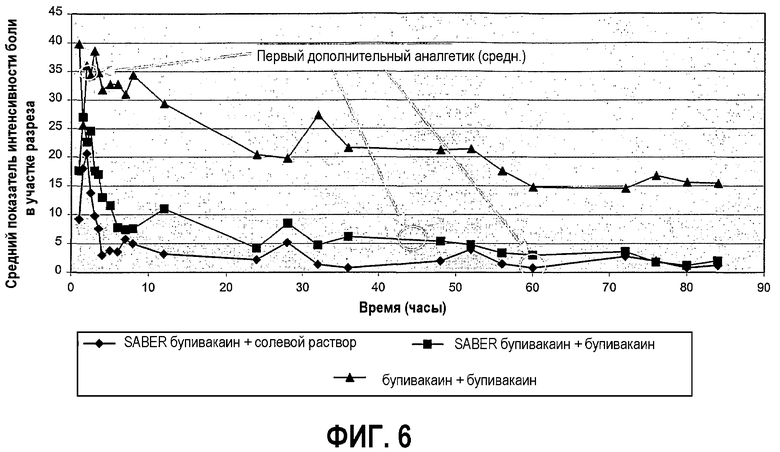
Изобретение относится к лекарственным средствам и касается жидкой композиции для обеспечения длительной местной анестезии после введения субъекту, содержащей бупивакаин в количестве от 30 до 5% мас. от общей массы композиции, изобутират ацетат сахарозы в качестве фармацевтически приемлемого носителя и бензиловый спирт в качестве растворителя для указанного носителя. Также раскрыт способ обеспечения длительной местной анестезии у субъекта, включающий введение субъекту жидкой композиции. Композиция по изобретению высвобождает бупивакаин в течение длительного периода времени без начального выброса. 2 н. и 15 з.п. ф-лы, 6 ил., 17 табл.
1. Жидкая композиция для обеспечения длительной местной анестезии после введения субъекту, содержащая бупивакаин в количестве от 30 до 5 мас.% от общей массы композиции, изобутират ацетат сахарозы в качестве фармацевтически приемлемого носителя и бензиловый спирт в качестве растворителя для указанного носителя.
2. Композиция по п.1, где бупивакаин присутствует в форме свободного основания.
3. Композиция по п.1, где бупивакаин присутствует в количестве от 25 до 5 мас.% от общей массы композиции.
4. Композиция по п.3, где указанное количество составляет от 20 до 10 мас.% от общей массы композиции.
5. Композиция по любому из предшествующих пунктов, где указанный носитель присутствует в количестве от 75 до 25 мас.% относительно общей массы композиции.
6. Композиция по любому из пп.1-4, состоящая из 66 мас.% изобутирата ацетата сахарозы, 22 мас.% бензилового спирта и 12 мас.% бупивакаина.
7. Способ обеспечения длительной местной анестезии у субъекта, включающий введение субъекту жидкой композиции, включающей бупивакаин в количестве от 30 до 5 мас.% от общей массы композиции, изобутират ацетат сахарозы в качестве фармацевтически приемлемого носителя и бензиловый спирт в качестве растворителя для указанного носителя.
8. Способ по п.7, где участком введения является хирургическая рана.
9. Способ по п.8, где указанную композицию вводят в рану и/или по соседству с раной.
10. Способ по любому из пп.7-9, где указанную композицию вводят посредством заливки.
11. Способ по любому из пп.7-9, где указанным субъектом является человек, перенесший хирургическую пластику грыжи или аппендэктомию.
12. Способ по любому из пп.7-9, где указанную композицию используют для лечения послеоперационной боли.
13. Способ по любому из пп.7-9, где бупивакаин присутствует в указанной композиции в форме свободного основания.
14. Способ по любому из пп.7-9, где бупивакаин присутствует в указанной композиции в количестве от 25 до 5 мас.% от общей массы композиции.
15. Способ по любому из пп.7-9, где бупивакаин присутствует в указанной композиции в количестве от 20 до 10 мас.% от общей массы композиции.
16. Способ по любому из пп.7-9, где указанный носитель присутствует в указанной композицици в количестве от 75 до 25 мас.% от общей массы композиции.
17. Способ по любому из пп.7-9, где указанная композиция состоит из 66 мас.% изобутирата ацетата сахарозы, 22 мас.% бензилового спирта и 12 мас.% бупивакаина.
| US 2004101557 A1, 27.05.2004 | |||
| JOHNSON RM "Applications of continuous site-directed drug delivery" Proceedings of the Western Pharmacology Society, 2002; 45:219-22 | |||
| US 2003152637 A1, 14.08.2003 | |||
| US 5747058 A, 05.05.1998 | |||
| Okumu FW "Sustained delivery of human growth hormone from a novel gel system: SABER" Biomaterials, 2002 Nov;23(22):4353-8, реферат |

