ОБЛАСТЬ ТЕХНИКИ
Изобретение касается использования улучшенного катализатора при гидрокрекинге углеводородных масел.
УРОВЕНЬ ТЕХНИКИ
Гидрокрекинг - это универсальный метод переработки нефти, который популярен и широко используется в нефтеперерабатывающей промышленности. Гидрокрекинг дает возможность переработать широкий спектр разветвленных исходных продуктов в разнообразные желаемые продукты. Исходные продукты, которые можно перерабатывать этим методом, включают тяжелые нефти, керосин, каталитически крекированные продукты рециркуляции и высококипящий газойль коксования, оливковое масло первого отжима. Жестким гидрокрекингом может превратить эти материалы в бензин и низкокипящие парафины; менее жесткий дает возможность превратить высококипящие исходные продукты в более легкие нефтепродукты, например дизельное топливо и авиационный керосин.
Гидрокрекинг обычно проводят при умеренных температурах от 350°С до 450°С (650°F и 850°F) и высоких давлениях свыше 7,000 кПа (избыточное давление 1,000), так как исходя из термодинамики процесса гидрокрекинга более высокие температуры нежелательны. Кроме того, требуется высокое парциальное давление водорода, обычно избыточное давление как минимум 800, чтобы препятствовать старению катализатора и таким образом поддерживать достаточную его активность, чтобы иметь возможность регулировать процесс, проходящий в стационарном слое катализатора в течение периода от одного до двух лет без необходимости его замены.
Экологические факторы в процессах гидродесульфурации и гидрокрекинга, особенно в случае выделения оксидов серы и оксидов азота, играют для нефтепереработчиков большую роль, чем в прошлом. Наличие побочных продуктов водорода при реформинге нефти, несомненно, также содействовало повышению этой роли. Другие факторы также способствовали повышению важности гидрообработки. Среди этих факторов то, что сырая нефть становится дефицитным продуктом. Также нефтеперерабатывающие заводы, которые проводят каталитический крекинг со взвешенным катализатором (ККВК), производят в больших объемах деалкилированные, ароматические трудноочищаемые сточные воды, известные как циклические масла ККВК. Снижение спроса на мазутную продукцию, в которую эти циклические масла ККВК добавляли раньше, способствовало разработке их переработки объединением с гидрокрекированными исходными продуктами. Побочные продукты гидрокрекинга, в отличие от каталитического крекирования, можно эффективно улучшить.
Гидрокрекинг - это признанный процесс очистки нефти. Исходные продукты гидрокрекинга обязательно поддаются гидрообработке перед подачей на установку для гидрокрекинга, чтобы избавиться от соединений серы и азота и металлов и, кроме того, чтобы насытить олефины и частично провести насыщение ароматики. Серу, соединения азота и кислорода можно удалить в виде неорганической серы, азота и воды до гидрокрекинга, также можно проводить промежуточное разделение, как в методе Unicracking-JHC. Хотя присутствие больших количеств аммиака может приводить к уменьшению эффективности крекинга на последующих стадиях гидрокрекинга, этого можно избежать, повышая жесткость условий ведения процесса гидрокрекинга.
При гидрообработке имеет место целый ряд различных реакций гидрогенирования в том числе насыщение олефинов и ароматических колец, но жесткость условий процесса ограничена, чтобы минимизировать крекинг. Продукты гидрообработки затем подаются на установку для гидрокрекинга, в которой проходят различные реакции крекинга и гидрогенирования.
В установке для гидрокрекинга реакции крекинга включают прохождение гидрогенирования олефинов, а гидрогенирование в свою очередь обеспечивает тепло для крекинга, так как реакции гидрогенирования экзотермические, в то время как реакции крекинга эндотермические; реакция в общем проходит с образованием большого количества тепла, поскольку количество тепла, выделенного при экзотермических реакциях гидрогенирования обычно, намного большее, чем количество тепла, поглощенного эндотермическими реакциями крекинга. Этот излишек тепла повышает температуру в реакторе и ускоряет скорость реакции, что контролируется при помощи охлаждения водородом.
Традиционные катализаторы гидрокрекинга играют роль кислоты и также катализируют гидрогенирование. Кислотная функция катализатора обеспечивается пористым носителем, как, например, глиноземом, глинозем-кремнеземом или смесью кристаллического цеолита, как, например, фожазита, Х цеолита, Y цеолита или морденита аморфных носителей, как, например, глинозем-кремнезема. В принципе, желательно использование пористого тела с относительно большим размером пор более 7А, потому что объемным полициклическим ароматическим соединениям, которые составляют большую часть обычных исходных продуктов, необходима такая величина пор, чтобы пройти во внутреннюю структуру катализатора, где проходит большая часть реакций крекинга.
Функция гидрогенирования в катализаторе гидрокрекинга осуществляется благодаря переходным металлам или комбинациям металлов. Могут использоваться благородные металлы VIUA группы Периодической системы элементов Менделеева, особенно платина и палладий, но в общем предпочитают обычные металлы IVA, VIA и VIIIA групп из-за их низкой стоимости и относительно большой устойчивости к отравлению загрязняющими примесями. Предпочитаемые обычные металлы для использования как компонентов гидрогенирования - это хром, молибден, вольфрам, кобальт и никель; и комбинации металлов, как, например, молибден-никель, молибден-кобальт, никель-кобальт, вольфрам-никель, молибден-никель-кобальт и титан-вольфрам-никель, которые являются достаточно эффективными и пригодными.
Пористые материалы, которые обеспечивает кислотную функциональность катализатора, могут включать или аморфный, или кристаллический материал, или оба сразу. Аморфные материалы имеют существенные преимущества при обработке самых высококипящих исходных продуктов, которые содержат существенные количества объемных полициклических соединений (ароматические соединения такие, как полинафталины), так как аморфные материалы обычно обладают порами разных размеров и большими порами с размерами порядка 100-400 ангстрем (Å), которые являются достаточно большими, чтобы обеспечить вход объемных исходных соединений во внутреннюю структуру материала, где проходят катализируемые кислотой реакции. Типичные аморфные материалы этого типа включают глинозем и глинозем-кремнезем, а также их смеси, возможно вместе с другими неорганическими оксидами, например кремния, магия или титана.
Кристаллические материалы, особенно цеолиты с большим размером пор, как, например, цеолит Х и Y, оказались эффективными в ряде реакций гидрокрекинга, так как они имеют преимущество, по сравнению с аморфными материалами, обладая большей степенью активности, что дает возможность проводить гидрокрекинг при более низких температурах, которые желательны в реакциях гидрогенирования с точки зрения термодинамики. Кроме того, кристаллические катализаторы более устойчивые, чем аморфные материалы, как, например, глинозем. Кристаллические материалы, однако, не подходят для всех реакций, так как самый большой размер поры у них обычно около 7.4 ангстрем в Х и Y цеолите, слишком маленькие, чтобы могли пройти некоторые объемные исходные соединения. Поэтому гидрокрекинг остаточных фракций и высококипящих исходных соединений требует менее активный аморфный катализатор.
Гидрокрекингом с кристаллическим катализатором обычно производят значительные количества продуктов в интервале температур кипения бензина (приблизительно 330°F, 165°С). Так как гидрокрекингованный бензин стремятся делать с относительно низким октановым числом, он нуждается в дальнейшей обработке, например в риформинге, перед тем как его можно будет отправить в парк смешения нефтеперегонного завода, поэтому гидрокрекинг не очень привлекательный способ изготовления бензина. С другой стороны, этот метод хорош для получения дистиллятной фракции, особенно реактивного топлива, топочного мазута и дизельного топлива, так как при гидрокрекинге достаточно небольшое количество гетероатомных примесей, что желательно для этой продукции. При гидрокрекинге топлива требуется такое большое количество цеолитного катализатора, как только возможно для достижения желаемой кислотности; с другой стороны количество матрицы, на которой находится металлическая составляющая, ограничено, и при увеличении количества цеолитного катализатора количество необходимого для металла носителя уменьшается, в результате чего активность гидрогенирования становится ограниченной при высоких загрузках цеолита, необходимых при гидрокрекинге топлив.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с этим изобретением гидрообработка и/или гидрокрекинг проводят при наличии катализатора на основе кристаллического цеолита, имеющего макропористую структуру, где макропоры структуры по сути окружены или покрыты кристаллами цеолита.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
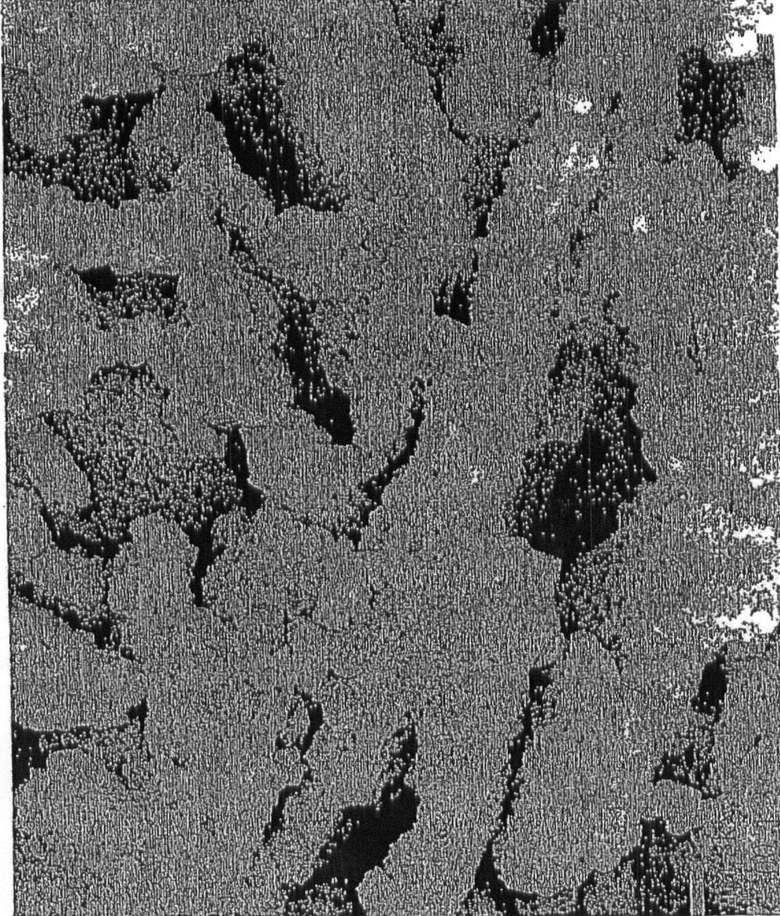
На чертеже показана фотография SEM катализатора цеолита, представленного в данном изобретении, как показано в Примере 6.
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Исходные продукты
Данный процесс может использоваться для гидрокрекинга различных исходных продуктов, например сырой нефти, отбензиненной нефти, отгонов вакуумной перегонки, газойля коксования, рециклового газойля, гудронов ККВК, вакуумного газойля, деасфальтированных остатков и других густых смазочных масел. Эти исходные продукты могут необязательно поддаваться гидрообработке до введения в данный процесс гидрокрекинга. Исходные продукты, особенно не прошедшие гидрообработку, будут содержать существенное количество веществ, кипящих при температуре выше 260°С (500°F), и обычно имеют точку кипения около 290°С (550°F), чаще около 340°С (650°F). Обычно диапазон температур кипения составляет от 340°С до 565°С (650°F до 1050°F) или от 340°С до 510°С (650°F до 950°F), но нефти с более узким интервалом кипения также могут обрабатываться, например, кипящие при температуре от около 340°С до 455°С (650°F до 850°F). Тяжелый газойль, являющийся рецикловым газойлем и другие немазутные продукты также можно поддавать гидрокрекингу. Вещества, полученные из каменного угля, сланцевого или битуминозный песка, также можно поддавать гидрокрекингу. Можно использовать совместно обрабатываемые материалы, кипящие при температуре ниже 260°С (500°F), но они большей частью не прореагируют. Исходные продукты, содержащие более легкие фракции, обычно имеют точку кипения выше 150°С (около 300°F). Составляющие исходных продуктов, кипящие в диапазоне от 290° до 340°С (от около 550° до 650°F), могут быть переработаны в продукты, кипящие при температуре от 230°С до 290°С (от около 450° до 550°F), но более тяжелые фракции предпочтительней превращать в летучие соединения, и, таким образом, более легкие фракции могут не прореагировать, если не повысится жесткость условий ведения процесса, что приведет к вступлению в реакцию всех соединений исходных продуктов. Отдельные углеводородные исходные продукты, которые можно вводить в процесс, - это масло рецикла ККВК, имеющее точку кипения как минимум около 343°С (650°F). Другими примерами исходных продуктов могут быть материалы с относительно большим содержанием неароматических углеводородов, например парафиновые исходные продукты, например исходные продукты, имеющие как минимум 20 массовых процентов, или как минимум 50 массовых процентов, или как минимум 60 массовых процентов парафина. Исходные продукты, в том числе прошедшие гидрообработку, которые можно использовать в данном процессе, включают материалы, имеющие как минимум 70 массовых процентов углеводородов с точкой кипения как минимум 204°С (400°F).
Условия ведения процесса
Исходные продукты нагревают и передают на гидрообработку над катализаторами в присутствии водорода. Поскольку с точки зрения термодинамики гидрокрекинг нежелательно проводить при температурах выше 450°С (около 850°F), то ее обычно не поднимают выше этого значения. Кроме того, поскольку гидрообработка и реакции гидрокрекинга экзотермические, исходные продукты не нужно нагревать до температуры, желательной для реакции в слое катализатора, которая составляет около 290°, обычно от 360°С до 440°С (около 550°, обычно от 675°F до 825°F). В начале циклического процесса используемая температура будет находиться внизу этого температурного ряда, но по мере старения катализатора температура может увеличиваться для поддерживания желаемой степени его активности. Обычно, сырая нефть проходит над катализатором в присутствии водорода. Объемная скорость нефти составляет обычно около от 0.1 до 10 LHSV (часовая объемная скорость жидкости), лучше от 0.2 до 2.0 LHSV, и скорость циркуляции водорода - от около 1400 до 5600 ст. куб.фт./барр.н и чаще от 300 до 800 (около от 1685 до 4500 ст. куб.фт./барр.н). Парциальное давление водорода составляет обычно как минимум 75 процентов общего давления в системе, а давление на входе в реактор обычно составляет от 400 до 1500 избыточного давления (абсолютное от около 2860 до 10445 кПа), чаще от 800 до 1500 избыточного давления (абсолютное около 5620 до 10445 кПа) с изменением давления от малого до умеренного, что предпочтительно в случае данного катализатора, также можно вести процесс при избыточном давлении 1500 (абсолютное около 10445 кПа) с такими же преимуществами, особенно в случае гидрокрекинга топлива. Характерным для данного процесса является ведение его при высоком давлении от около 1500 до 5000 избыточного давления (абсолютное около 10445 до 34575 кПа), хотя можно вести его и при более высоких давлениях, ограниченных возможностями оборудования. При низких конверсиях, например меньше 50 массовых процентов конверсии продуктов, кипящих при 345°С (650°F), давление может быть значительно ниже нормального, что обусловлено методом. Авторы обнаружили, что общее давление в системе около 700-1200 избыточного давления (абсолютное около 4930-8375 кПа) является эффективнее, по сравнению с давлением как минимум 1500 избыточного давления (около 10445 кПа), обычно используемого при коммерческом гидрокрекинге. Низкую конверсию можно получить варьированием других параметров реакции, например температуры, объемной скорости, выбором катализатора и даже понижением давления. Более низкие давления желательны с точки зрения эксплуатации оборудования, так как в этом случае оно менее массивное и потому более дешевое. Также при более низких давлениях обычно меньше проходит насыщение ароматических соединений, благодаря чему уменьшается общее количество водорода, поглощенного в течение процесса. Полная конверсия может достигаться изменением различных условий ведения в зависимости от природы исходных соединений и от желаемых свойств продуктов. Можно вести процесс с низким уровнем конверсии, меньше 50 массовых процентов, получая продукты с низкой температурой кипения 340°С (650°F) из исходных продуктов тяжелой нефти, пока качество продукта является удовлетворительным. Конверсия может, конечно, быть еще меньше, например 30 или 40 массовых процентов. Степень крекинга, чтобы выделить (С4) газолин, который получается при этих низких конверсиях, соответственно низкая, равно как конверсия первой широкой фракции дистиллята (200°С, 400°F); селективность процесса соответственно высокая и чрезмерно глубокий крекинг до более легких и менее желаемых продуктов минимизирован. Верится, что при каскадном процессе этот эффект обеспечен, в частности, благодаря влиянию аммиака, пролонгированного с самой начальной стадии. Контроль конверсии можно проводить разными методами, например контролем температуры, давления, объемной скорости и других параметров реакции. Подачу лучше начинать через катализатор гидрообработки перед катализатором гидрокрекинга для того, чтобы превратить азот и серу в газообразный аммиак и сероводород. На этой стадии гидрокрекинг минимизирован, но проходит частичное гидрогенирование полициклических ароматических соединений, степень конверсии низкокипящих продуктов (345°С, 650°F) ограничена. Катализатор, используемый на этой стадии, может являться соответствующим катализатором денитрогенации (денитрификации). Катализаторы этого типа относительно стабильные к отравлению азотными и сернистыми примесями исходных продуктов, включают неблагородные металлы на аморфных, пористых носителях, например кремнеземе, глиноземе, глинозем-кремнеземе или магнезия-кремнеземе. Поскольку экстенсивный крекинг не желателен на этой стадии процесса, кислотная функция носителя может быть относительно низкой по сравнению с последующими катализаторами гидрокрекинга. Металлическая составляющая может являться одним металлом VIA и VINA групп Периодической системы элементов Менделеева, например никелем, кобальтом, хромом, ванадием, молибденом, вольфрамом, или комбинацией металлов, как, например, молибден-никелем, молибден-никель-кобальтом, молибден-кобальтом, вольфрам-никелем или титан-вольфрам-никелем. В общем, металлическая составляющая будет выбираться так, чтобы обеспечить эффективное гидрогенирование; в общем катализатор должен иметь хорошие гидрогенизирующие и минимальные крекингирующие свойства. Катализатор должен быть предварительно сульфурирован, чтобы превратить металлическую составляющую (обычно импрегнированный в носитель и превращенный в оксид) в соответствующий сульфид. На стадии гидрообработки (денитрогенации) азот и примеси серы превращаются в аммиак и сероводород. В то же время полициклические ароматические соединения частично гидрируются, образуя нафтены и гидроароматические соединения, которые хорошо поддаются крекингу на второй стадии. Поток с первой стадии можно передавать непосредственно на вторую или на стадию гидрокрекинга без отделения промежуточного аммиака или сероводорода. Квенчингом водорода можно регулировать температуру потока и катализатора на второй стадии. Возможно отделение промежуточного аммиака и сероводорода и легких фракций благодаря катализаторам гидрокрекинга на основе благородных металлов, которые чувствительнее к примесям. Относительные пропорции катализаторов гидрокрекинга и гидрообработки могут меняться в зависимости от исходного продукта, чтобы превратить азот исходных продуктов в аммиак перед тем, как смесь пойдет на следующую стадию гидрокрекинга; цель - избавиться от азота, чтобы в определенной точке достичь желаемую степень конверсии каталитического гидрокрекинга при оптимальном сочетании объемной скорости и температуры реакции. Чем больше количество азота в потоке, тем больше будет соотношение катализатора гидрообработки (денитрогенации) к катализатору гидрокрекинга. Если количество азота в потоке низкое, соотношение катализаторов может тоже быть низким, например 10:90 (по объему, денитрогенация: гидрокрекинг). Часто соотношения являются 25:75 или 75:25. Во многих случаях будет эффективным приблизительно равное соотношение объемов, например 40:60, 50:50 или 60:40.
Поток со стадии денитрогенации/десульфуризации подается на стадию гидрокрекинга для крекинга частично прогидрированных ароматических соединений и для прохождения других характерных реакций, которые имеют место над катализатором гидрокрекинга. Гидрокрекинг осуществляется при наличии катализатора, который содержит три основных компонента. Первый компонент - металл, который обеспечивает желаемое гидрогенирование, этот компонент находится на одном или двух пористых носителях, а именно материал-основе катализатора с макропорами (которая может частично обеспечивать кислотную функцию катализатора), и цеолит, который является Y цеолитом, который окружает или покрывает макропоры матрицы.
Металлический компонент катализатора гидрокрекинга
Гидрогенирование-дегидрогенирование обеспечено компонентом, включающим металл или комбинацию металлов. Могут использоваться благородные металлы VINA группы, особенно палладий, платина, или неблагородные металлы IVA, VIA и VIIIA групп, особенно хром, молибден, вольфрам, кобальт и никель. Комбинация металлов включает как минимум один металл VIA группы, например вольфрам, и как минимум один металл VIIA группы, например никель, особенно предпочитаемая для многих воплощений комбинация, например, молибден-никеля, никель-кобальта, вольфрам-никеля, молибден-никель-кобальта и титан-вольфрам-никеля. В некоторых случаях предпочтительными являются палладий или платина.
Содержание металлического компонента меняется в зависимости от его каталитической активности. Поэтому чрезвычайно активные благородные металлы можно использовать в меньших количествах, чем менее активные неблагородные металлы. Например, около 1 массового процента или меньше палладия или платины будут эффективными, а в предпочитаемой комбинации неблагородных металлов содержится около 7 мас.% никеля и от около 2 до около 21 мас.% вольфрама. Металлический компонент может составлять больше около 30 процентов в монослое, содержание металла может составлять вплоть до около 40% или даже больше. Компонент, отвечающий за гидрогенирование, можно заменить на материал-основу, где металл находится в форме катиона, куда его можно импрегнировать или смешать. Соединения палладия или платины, в которых металл находится в форме катионного комплекса, например Pd(NH3)4Cl2 или Pt(NH3)4Cl2, особенно эффективны при замене этих металлов на основу. Анионные комплексы, например молибдат, ванадат и метавольфрамат ионы, могут использоваться в случаях, когда металлическая составляющая должна быть импрегнирована в основу.
Цеолитный компонент катализатора гидрокрекинга
Катализатор гидрокрекинга, представленный в этом изобретении, является цеолитом Y, который образовывается in-situ и находится в макропорах матрицы. Катализатор гидрокрекинга, содержащий цеолит, соответственно используется в матричной форме и может быть в виде экструдата, таблеток или других форм, чтобы обеспечивать прохождение газов над катализатором с минимальным падением давления. Кристаллический компонент цеолита может быть матричным или средним между активными и инертными материалами, как, например, глины, кремнезем и/или оксиды металлов, как, например, глинозем, титан и/или цирконий. Недисперсные глиноземы особенно эффективны. Оксиды металлов могут находится в обычном состоянии или в форме студенистых осадков или гелей, смесей кремнезема и оксидов металлов. Инертные материалы удобно подавать как разжижители для регулировки степени конверсии процесса таким образом, чтобы получить продукты экономично и в нужной последовательности без использования других средств для контроля скорости реакции. Желательно использовать как минимум часть материалов описанной выше матрицы в коллоидной форме, чтобы облегчить выведение связанных компонентов катализатора. Относительные пропорции тонко диспергированного материала цеолита и матрицы находятся в широком диапазоне, где количество кристаллической составляющей от около 1 до около 90 массовых процентов, чаще, особенно, когда смесь находится в виде дроби, в границах от 2 до около 80 массовых процентов смеси. Катализатор нужно соответственно насытить серой, например нагреванием в среде сероводорода, чтобы превратить оксиды металлов, например Со или NiO, в соответствующие сульфиды. Катализатор гидрокрекинга, представленный в этом изобретении, можно получать из микрогранул, например, полученных стандартным методом, как описано в U.S. Pat. Nos. 6,656,347; 6,942,784 и 6,943,132, полная информация по которому приводится в ссылке. Микрогранулы используют, чтобы подготовить катализатор гидрокрекинга, образованный in-situ, полученный из цеолита Y и нецеолитной матрицы с избытком глинозема. Нецеолитную матрицу с избытком глинозема, представленную в данном изобретении, лучше получать из исходного материала, такого как водный каолин в форме ультрамелкого порошка, в котором размер как минимум 90 мас.% частиц составляет меньше 2.0 микрон, лучше чтобы как минимум 90 мас.% меньше 1 микрона. Водный ультрамелкий каолин размельчают и кальцинируют экзотермически. Известный цеолит микрогранул для ККВК крекинга был образован из матрицы с избытком глинозема, полученной из каолина, имеющего больший размер частиц, чем тот, который используется в этом изобретении и который большей частью кальцинируют исходя из характерного для него экзотермического эффекта. Satintone® No. 1 (коммерчески доступный каолин, который кальцинирован исходя из характерного для него экзотермического эффекта без значительного образования муллит) - это материал, используемый изначально для коммерческого получения матриц с избытком глинозема. Satintone® No. 1 получают из водного каолина, в котором размер 70% частиц составляет меньше 2 микрон. Другими исходными материалами, которые используют, чтобы получить матрицу с избытком глинозема, являются мелко диспергированный водный каолин (например, ASP® 600, коммерчески доступный водный каолин, описанный в Engelhard Technical Bulletin No. TI-1004, под названием "Aluminum Silicate Pigments" (EC-1167)), в большей степени кальцинированный исходя из характерного для него экзотермического эффекта. Рекламная глина нашла самое широкое распространение в коммерческом использовании и имела огромный успех во всем мире. Эти большие частицы глины уже являются общеизвестными в области образования матрицы с избытком глинозема для микрогранул катализатора ККВК и не являются дефицитом.
Под "ультрамелким" порошком подразумевают, что размер как минимум 90 мас.% частиц водного каолина составляет меньше 2 микрон в диаметре, лучше меньше 1 микрона, как установлено Sedigraph™ (или осаждение). В частности, использование водных каолиновых наполнителей с такими размерами частиц при пульверизации и кальцинирование исходя из характерного экзотермического эффекта дают большую макропористость даже в микрогранулированном катализаторе после кристаллизации цеолита. Рыхлую упаковку кальцинированного ультрамелкого каолина можно сравнить с "карточным домиком", в котором отдельные частицы находятся в беспорядочных положениях относительно друг друга. Кроме того, кальцинированный ультрамелкий каолин находится в виде крупнопористой массы со строением "карточного домика" с не только пористыми частицами, но и пористой областью между ними. Пульверизация водного ультрамелкого каолина необходима, чтобы обеспечить случайное расположение частиц каолина.
Каолиновые глины или наполнители и природные гидрированные силикаты алюминия приблизительной формулы Al2O3 2SiO2 X Н2О, где Х обычно является каолтинитом, накритом, диккитом и галлозитом, - разновидность минералов ряда каолина. Известно, что когда каолин нагревают на воздухе, первое изменение происходит при около 550°С и связано с эндотермической реакцией дегидроксилирования. Полученный материал обычно является мета-каолином. Мета-каолин устойчив до температуры около 975°С и подвергается экзотермической реакции. Этот материал обычно является каолином, который подвергся характерным экзотермическим реакциям. Некоторые называют его неполным кремний-алюминиевым шпинелем или гамма-глиноземом (смотрите Donald W. Breck, Zeolite Molecular Sieves, published by John Wiley and Sons, 1974, p.314-315). При дальнейшем нагревании до около 1,050°С начинают образовываться высокотемпературные фазы, а также муллит. Конверсия муллита зависит от взаимоотношения температуры и времени и наличия минерализатора исходя их известной в данной области литературы. В предпочитаемых воплощениях этого изобретения пылевидный ультрамелкий водный каолин, используемый для получения матрицы с избытком глинозема, кальцинируют исходя из характерного экзотермического эффекта с или без образования муллита. Особенно предпочтительный исходный материал матрицы, который используется в этом изобретении, для образования микрогранул цеолита с макропорами - это Ansilex® 93. Ansilex® 93 состоит из мелких частиц твердого сырого каолина, которые сушат распылительной сушкой, размельчают и кальцинируют, чтобы получить мелкоабразивные наполнители, как описано в U.S. Pat. No. 3,586,523, Fanselow, et. al., полная информация по которому приводится в ссылке. Водный исходный материал ультрамелкой матрицы сушат распылительной сушкой, размельчают и кальцинируют исходя из характерного экзотермического эффекта, необязательно получая муллит. Вышеупомянутый U.S. Pat. No. 4,493,902 представляет кальцинирование каолиновой глины до муллита, пока интенсивность дифракции рентгеновского луча не станет равна справочному значению для кристаллической структуры.
В то время как исходя из данного изобретения, чтобы кальцинировать каолин согласно его экзотермическому эффекту такой, чтобы интенсивность дифракции рентгеновских лучей соответствовала справочной постоянной для кристаллической структуры, желательно кальцинировать каолин исходя из сверхэкзотермического эффекта, чтобы превратить каолин в мелкий кристаллический муллит. У мелкокристаллического муллита соответствующие дифракционные линии и выщелоченный химический состав, характерный для полностью кристаллического муллита, но дифракционные линии менее выражены, так как кристаллиты более мелкие. Соотношение ширина/интенсивность дифракционных линий и размера кристаллита известно. Желательно кальцинировать каолин при более высоких температурах, чем его экзотермический эффект, получая мелкокристаллическую муллитовую матрицу, так как полное кальцинирование каолина до муллита на практике требует чрезмерно высоких температур и времени. К тому же кальцинирование каолина при более высоких температурах, чем его экзотермический эффект, до полностью кристаллического муллита может приводить к потере макроскопичности за счет спекания. Кроме того, размер пор может уменьшаться и объемная плотность существенно увеличивается после кальцинирования каолина до полностью кристаллического муллита. Поэтому желательно, чтобы ультрамелкий каолин, кальцинированный при температурах его экзотермического эффекта, имел 20-80% интегрированных площадей пика дифракции рентгеновских лучей относительно образца каолина, содержащего кристаллический муллит. Лучше, чтобы ультрамелкий каолин кальцинировали при температурах его экзотермического эффекта так, чтобы 50-70% интегрированных площадей его пика дифракции рентгеновских лучей соответствовали полностью кристаллическому муллиту. Необыкновенным в использовании продукта Ansilex® является то, что он получен из твердой каолиновой глины. Твердые каолиновые глины обычно имеют легкий серый окрас или цвет и поэтому называются "серыми глинами". Этим твердым каолиновым глинам, кроме того, характерно разложение на несимметричные куски, имеющие шероховатую поверхность. Твердые каолиновые глины также содержат значительное количество железа, обычно около от 0.6 до 1 мас.% Fe2O3. Твердые каолиновые глины описаны в Grim's "Applied Clay Mineralology", 1962, MaGraw Hill Book Company, p.394-398, полная информация по которому приводится в ссылке. Случайно открыли, что твердые каолиновые глины также можно использовать в качестве исходных материалов для получения микрогранул мета-каолина in-situ, но не особо эффективно. Матрицу с избытком глинозема можно получить из глиноземсодержащих материалов с характерной пористостью, которая играет роль при распределении кальцинированного материала. Разработана методика определения объема пор кальцинированного глиноземсодержащего материала, которые в конечном счете формируют матрицу катализатора, представленного в изобретении. Метод определения объема пор водного кальцинированного глиноземсодержащего материала состоит в определении минимального количества воды, необходимой, чтобы сделать из твердого образца глинистую суспензию. При тесте твердый образец в колбе смешивают с водой, содержащей дисперсант, например Colloid 211, Viking Industries, Atlanta, Ga., используя мешалку или шпатель. Воду к сухому образцу добавляют только в количестве, необходимом, чтобы превратить сухое вещество в единую массу жидкой глины, которая начинает вытекать только под своим собственным весом. Начальная точка появления жидкой глины (ISP) вычисляется по весу образца и используемой воды. Начальная точка появления жидкой глины может быть вычислена, как указано ниже: ISP=[(граммы сухого образца)/(граммы сухого образца плюс граммы добавленной воды)]×100. Единица измерения безразмерная и составляет процентное содержание твердого вещества. Это количество воды больше, чем (внутренний) водный объем пор образца, но четко с ним коррелируется. Низкая начальная точка появления жидкой глины из твердого вещества означает большую возможность абсорбировать воду или большой объем пор в образце. Кальцинированные глиноземсодержащие материалы, из которых получают матрицу с избытком глинозема в соответствии с этим изобретением, будут иметь начальную точку появления жидкой глины при меньше 57% твердого вещества, лучше от 48 до 52% твердого вещества. Такие характеристики имеет Satintone® No. 1, у которого начальная точка появления жидкой глины при свыше 58% твердого вещества. Соответственно не только водный ультрамелкий каолин можно использовать при получении глиноземсодержащих материалов, из которых получают матрицу для микрогранулированного катализатора, но и неслоистый каолин, пластинчатый глинозем и преципитат алюминия. Средства дерасщепления блоков каолина известны в данной области науки и техники. Предпочитаемым является метод, в котором используют абразивный материал, как песок или стеклянные микрошарики. Последующее расслоение и размельчение проводят, чтобы получить беспорядочную упаковку или "карточный домик".
Также исходными глиноземными макропористыми материалами, которые могут использоваться для получения матрицы катализатора, могут быть, например, некаолиновые вещества, например тригидрат алюминия, например байерит и гиббсит, переходные глиноземы, моногидраты алюминия, как, например, бомит и псевдобомит, вспененные глиноземы, глиноземсодержащие аэрогели, гидроталькит, глинозем-кремнеземные когели и бокситы.
Желательно, чтобы получить исходный материал матрицы, представленной в изобретении, обрабатывать водный каолин методом пульверизации-кальцинирования-пульверизации, так как при использовании водного каолина как исходного материала для получения мета-каолина для образования реакционноспособных предшественников микрогранул возникает повышенная износостойкось в большом объеме порового пространства. Менее привлекательным является способ получения предшественников матрицы путем мокрого дробления с последующей деагломерацией. Перемалывание уменьшает объем пор микрогранул, все остальные параметры не изменяются. Метод, при котором уменьшается объем пор микрогранул, эффективен, когда заранее кальцинированный порошок мета-каолина используют для получения реакционноспособных предшественников микрогранул. Деагломерация эффективна при поддержании требуемого содержимого цеолита, а также для улучшения дисперсии перемолотой матрицы в микрогранулах, за счет того что происходит разрыв сильных связей, которые иначе могли бы упрочнить полученный катализатор, что нежелательно.
Также в пределах данного изобретения получение матрицы из химически синтезированного шпинеля и/или муллита (см. Okata, et al., "Characterization of spinel phase from SiO2-Al2O3 xerogels and the formation process of mullite", Journal of the American Ceramic Society, 69 [9] 652-656 (1986), полная информация по которому приводится в ссылке) и исходя из нее два вида ксерогелей можно получить медленным и быстрым гидролизом тетраэтокси силана и нонагидрата нитрата алюминия, растворенного в этаноле. Медленный метод гидролиза включает гелеобразование упомянутой выше смеси в сушилке при 60°С от одной до двух недель, тогда как быстрый метод гидролиза включает добавление раствора гидроокиси аммония к смеси и сушку на воздухе. В ксерогеле, полученном медленным методом гидролиза, муллит кристаллизуется непосредственно из аморфной смеси при сильном нагревании, тогда как из ксерогелей, полученных быстрым гидролизом, кристаллизуется сначала полученный шпинель и только потом муллит. Длительность процесса зависит от водного объема пор кальцинированного синтетического материала в пределах этого изобретения, такие материалы могут использоваться для получения матрицы для катализатора, представленного в изобретении.
Также матрицу можно получить из других исходных глиноземных некаолиновых материалов. Например, предшественником глиноземной матрицы могут быть производные гидратов алюминия, например моногидраты и тригидраты. Тригидраты, например байерит и гиббсит, могут кристаллизоваться с образованием больших частиц с соотношением геометрических размеров около 1. Соотношение геометрических размеров, стремящееся к 1, указывает на квазисферическое строение, которое подразумевает неплотную упаковку и малый объем пор, компенсировать это можно, например, соответствующей пульверизацией и кальцинированием, увеличивающими пористость материала. Предшественники с высоким соотношением геометрических размеров от 1 обладают необходимой макроскопичностью, и поэтому практически не возникает необходимость в повторной обработке. Известны методы получения материалов с пластинчатым и игольчатым строением, например гиббсит, и такое строение сохраняется в кальцинированном продукте. Например, гиббсит может быть кальцинирован до альфа глинозема. Во время кальцинирования образуется большая внутренняя пористость, которая является метастабильной и не сохраняется в полученном альфа глиноземе, но оригинальное строение кристаллита и макроскопичность сохраняются в кальцинированной продукции, если спекание не проводить в особо жестких условиях. Байерит и Si-стабилизированный байерит используют в качестве модернизированных глиноземов. Соответственно, такой байерит, в том числе байерит, кальцинированный до эта-глинозема, после соответствующей обработки или синтеза может быть получен с необходимой макроскопичностью и использован как материал для получения матрицы, представленной в данном изобретении, и может воспроизводить строение цеолитной матрицы катализатора, получаемого in-situ. Примеры байерита, используемого в катализаторах, описаны в US Pat. Nos. 6,165,351; 5,547,564; 5,306,417; 5,304,526, полная информация по которым приводится в ссылке.
Также известно кальцинирование гиббсита электрическим разрядом, продукт которого чрезвычайно реакционноспособный и может быть повторно гидратирован до высшей формы - бемита. Получение кальцинированного гиббсита электрическим разрядом обычно начинается дроблением на шаровой мельнице гиббсита от 100 микрон. Материал имеет небольшой объем макропор, так как соотношение геометрических размеров после дробления на шаровой мельнице составляет приблизительно 1. Смешиванием с добавками, например зернистым глиноземом, фосфатами или глинами можно получить пористые бемиты из кальцинированного гиббсита. В U.S. Pat. Nos. 6,403,526 и 6,451,200 приводят примеры такой обработки, полная информация по патентам приводится в ссылке. Глиноземные гели можно получить с содержанием как моногидрата оксида алюминия, так и тригидрата оксида алюминия. В гелевых системах гидраты оксида алюминия являются основной твердой фазой, а вторая фаза может быть водой, смесью воды и органического растворителя или воздухом (в случае аэрогелей). В зависимости от метода получения твердое вещество может находиться в виде как дискретных частиц с размером от нескольких нанометров до микрометров, так и в форме трехмерных полимерных сетей, в которых частицы твердого вещества связаны химическими связями. Такие трехмерные полимерные сети могут быть макропорами и подходят в качестве исходного соединения для получения матрицы катализатора согласно данному изобретению. Полимерную сеть и, в частности, гели можно получить направленным гидролизом алкоксида алюминия в органических растворителях. Такие гели используют для получения пористых керамических материалов. Свойства гелей, полученных из алкоксида алюминия, зависят от температуры, количества воды, состава алкоксида, природы растворителя и стабильности его смеси с водой, присутствия электролитов и рН раствора. Все эти факторы оказывают влияние на конечный продукт.
Гидраты алюминия амфотерные и, поэтому, растворимы в сильных кислотах и сильных основаниях. В водных растворах среднее рН и растворимость этих соединений самая низкая. За счет крутости кривой растворимости маленькие значения рН могут вызвать значительное перенасыщение раствора и, следовательно, быстрое выпадение осадка. Соответственно, гидраты алюминия лучше получить в виде коллоидного размера, после чего их легко коагулировать в гель. Факторы, которые определяют физические свойства гелей, - например размер частиц, химический состав, температура, скорость осаждения, конечное рН, ионный состав, ПАВ, концентрация начальных растворов, перемешивание и время выдержки. Быстрая нейтрализация раствора соли алюминия с основаниями дает гель с избыточной водой, который содержит непостоянное количество остаточных кислотных анионов. При удалении лишней воды и ионов можно получить макропористые материалы, которые можно использовать как предшественников матрицы, представленной в этом изобретении. Большие и маленькие объемы пор моногидратов глинозема общеизвестны. Большая макроскопичность как минимум в случае псевдобемита может объясняться процессами, включающими осаждение солей алюминия. Кристаллиты этих веществ могут быть волокнистыми. Эти вещества обычно используются при экструзии макропористого глинозема, которая обычно проходит вместе с, например, гидрообработкой и т.п. Макропористость этих материалов полезна в случае катализатора, представленного в изобретении, когда структура макропор воспроизводится в катализаторе. Помол и диспергирование проводят для получения частиц определенного размера и пористости. Такие материалы описаны в U.S. Pat. Nos. 2,656,250 и 2,915,475, полная информация по патентам приводится в ссылке.
Существует большой раздел науки, описывающий Si-Al когели для ККВК и других процессов. Эти методы не практикуются сейчас, так как полученные продукты имеют большой объем пор, небольшую плотность ККВК (гель-катализаторы), но очень маленькую износостойкость. Катализаторы полученные in-situ и связанные с растворителем демонстрируют намного большую износостойкость, и поэтому когели заменили технологией получения коллоидного связующего вещества. Свойства, неудовлетворительные в случае когелей для ККВК, являются превосходными в случае материалов-предшественников матрицы катализатора гидрокрекинга, полученных in-situ, представленных в этом изобретении. При кальцинировании в насыщенных глиноземом фазах геля формируются безкремневые переходные глиноземы, Si-Al шпинели или Si-модифицированные глиноземы, альфа глиноземы и/или муллиты. Вероятно, в богатых на кремнезем регионах формируется большое количество аморфного глинозем-кремнезема. Метод должен быть одинаково эффективным как в случае получения каолиновых активных матриц, так и в случае получения питательной среды для выращивания цеолита. Примеры получения когеля, которые описывают, но не ограничивают изобретение, описаны в U.S. Pat. Nos. 4,310,441 и 4,499,197, полная информация по патентам приводится в ссылке. Матрицы с большим объемом пор могут также быть получены из вспененного глинозема и пористого боксита. Процесс гидротермического старения геля и/или повторная гидратация, в автоклаве или при повышенном давлении, может привести к взаимопереходу между фазами предшественника гидроокиси алюминия, как описано выше.
Водный объем пор кристаллизированных цеолитных микрогранул согласно изобретению, полученных из глиноземсодержащих материалов, определяется тестом ISP, с целью получения матрицы катализатора со значительной макроскопичностью, т.е. с порами диаметром как минимум 600 ангстрем. Лучше, чтобы объем пор цеолитных микрогранул был больше 0.27 см3/г, желательно больше 0.30 см3/г Hg для пор диаметром 40-20,000 ангстрем. Предпочтительным является катализатор с объем макропор, диаметр которых от 600 до 20,000 ангстрем, как минимум 0.07 см3/г Hg и лучше как минимум 0.10 см3/г Hg. Хотя обычный цеолитный катализатор имеет макропористость, сравнимую с катализатором, представленным в этом изобретении, обычный катализатор не содержит цеолитную матрицу нового строения и принцип действия катализатора, представленного в этом изобретении. Катализаторы, представленные в этом изобретении, будут иметь площадь поверхности БЭТ меньше 500 м2/г, лучше меньше 475 м2/г и лучше всего в пределах около 300-450 м2/г. Умеренная площадь катализатора, представленного в этом изобретении в комбинации с макроскопичностью дает желаемую активность и выборочность относительно газолина при уменьшении выхода газа и кокса. Квалифицированный в этой области специалист сразу оценит площадь поверхности и значительно увеличенную активность, которая зависит от доступного объема поры. Предпочтительные площади поверхности конечного катализатора (свежего) выбраны так, что площадь поверхности при 1500°F через четыре часа и пропаривания при давлении 1 атм составляет меньше 300 м2/г. Кроме того, макроскопичность катализатора сохраняется, даже если часть матрицы получена из твердых глиноземсодержащих материалов, водный объем пор которых не соответствует желаемому исходя из изобретения, как определили тестом ISP. Было найдено, что при смешивании каолиновой глины и ультрамелкой каолиновой глины, которую кальцинировали исходя из экзотермического эффекта, получая катализатор с большим объемом пор, широкими макропорами, но с низким содержимым цеолита. Такие катализаторы могут быть ценны при крекинге с исключительно строгими условиями.
Общая методика производства цеолитных микрогранул согласно этому изобретению известна в науке и технике, ее прототипом может быть способ, раскрытый в U.S. Pat. No. 4,493,902. Согласно патенту готовят водную реакционноспособную глинистую суспензию тонко диспергированной водной каолиновой глины и/или мета-каолина и глиноземсодержащего материала, который формирует матрицу, например ультрамелкого каолина, который кальцинирован исходя из его характерного экзотермического эффекта. Водную жидкую глину затем сушат распылением, получают микрогранулы, включающие смесь водного каолина и/или мета-каолина и каолина, который как минимум по большей части кальцинирован исходя из его характерного экзотермического эффекта, с целью получения матрицы с избытком глинозема. Желательно к водной жидкой глине перед сушкой распылением добавить умеренное количество силиката натрия. Во время и после сушки силикат натрия играет связующую роль между частицами каолина.
Реакционноспособную жидкую каолиновую глину, образующую микрогранулы, можно получить из гидратированного каолина или кальцинированного водного каолина (мета-каолина) или их смесей. Исходные соединения для получения жидкой каолиновой глины выбирают из ASP® 600 или ASP® 400 каолина или их смесей, полученных из твердого необработанного белого каолина. Более соответствующий размер частиц водного каолина у производных серых глин, например наполнитель LHT. Успешно можно использовать гидроочистку каолиновой глины из Средней Грузии. Кальцинированные продукты этого водного каолина можно использовать в качестве исходного соединения для получения мета-каолина. Самый большой объем пор был получен при использовании мета-каолина влажным размалыванием исходных соедиений матрицы. Связывающий силикат лучше использовать в виде смеси силиката натрия с SiO2 в соотношении Na2O от 1.5 до 3.5, предпочтительно в соотношении от 2.88 до 3.22. Инициатор цеолита (например, 3 30 мас.% каолина) также можно добавить к водной жидкой глине перед сушкой. Под "инициатором цеолита" подразумевается любой материал, содержащий кремнезем и глинозем, так как оба они инициируют процесс кристаллизации цеолита, который не сможет пройти в отсутствие инициатора, или существенно сокращают процесс кристаллизации цеолита, по сравнению с происходящим в отсутствие инициатора. Такие соединения также известны как "зародыши цеолита". Инициатор цеолита может не фиксироваться дифракцией рентгеновского луча. Добавление инициатора цеолита в водную жидкую каолиновую глину, который сохнет в микрогрануле, в данном изобретении называют "внутренним зародышем". Также инициатор цеолита может добавляться в каолиновые микрогранулы после того, как они сформированы, и перед началом процесса кристаллизации, что здесь именуется "внешним зародышем". Инициатор цеолита, используемый в настоящем изобретении, можно получить из целого ряда исходных материалов. Например, инициатор цеолита может состоять из рециклированных мелких фракций, производимых непосредственно в течение процесса кристаллизации. Другие возможные инициаторы цеолита включают мелкие фракции, производимые в течение процесса кристаллизации другого цеолитного продукта или аморфного инициатора цеолита в растворе силиката натрия. Как используется здесь, "аморфный инициатор" цеолита подразумевает инициатор цеолита, при дифракции рентгеновского луча которого не выявляется кристалличность.
Зародыши можно получать, как представлено в U.S. Pat. No. 4,493,902, a желательно следуя U.S. Pat. No. 4,631,262. После сушки распылением микрогранулы можно сразу кальцинировать или предварительно провести нейтрализацию кислотой, чтобы, кроме того, увеличить обмен ионов катализатора после кристаллизации. Процесс нейтрализации кислотой включает смешивание некальцинированных соединений, сушку распылением микрогранул и добавление неорганической кислоты к жидкой глине при перемешивании и контролируемом рН. Скорость добавления твердого вещества и кислоты поддерживают такую, чтобы рН составлял около 2.7, лучше всего от около 2.5 до 4.5 (держать рН около 3). Связующий силикат натрия превращают в гель, пока не получат кремнезем и растворимую натриевую соль, которую фильтруют и промывают отдельно от микрогранул. Кремнезем-связанные микрогранулы кальцинируют. Кальцинирование проводят при температуре и в течение определенного времени (например, в течение двух часов в муфельной печи с температурой в камере около 1,350°F), что является достаточным, чтобы превратить какое-либо вещество каолиновой гидратированной микрогранулы в мета-каолин, не изменяя структуры предварительно кальцинированных компонентов каолиновых микрогранул. Полученные кальцинированные пористые микрогранулы включают смесь мета-каолина и каолиновой глины, кальцинированные исходя из характерного экзотермического эффекта, причем в микрогранулах присутствуют два вида кальцинированного каолина одновременно. Также каким-либо подходящим кальцинированным глиноземом можно заменить каолин, кальцинированный исходя из его экзотермического эффекта, как описано выше. Вообще, соотношение масс мета-каолина к кальцинированному глинозему должно составлять от около 1:0.66 до 1:4, лучше от 1:1.5 до 1:3. Поэтому кальцинированные микрогранулы в общем должны включать около 25-60 мас.% мета-каолина и около 40-75 мас.% каолина, который кальцинирован исходя из его характерного экзотермического эффекта. Желательно, чтобы обеспечивалось 30-40 мас.% мета-каолина и 60-70 мас.% каолина, кальцинированного исходя из его характерного экзотермического эффекта. Обязательным является наличие Na2O и Si2O, полученных из силиката натрия.
Y-фожазит можно кристаллизировать, смешивая микрогранулы кальцинированного каолина с соответсвующим количеством других компонентов (в том числе раствором силиката натрия в воде), как будет подробно описано ниже, с последующим нагреванием полученной жидкой глины при определенной температуре и определенное время (например, при 200°-215°F в течение 10-24 часов), достаточное для кристаллизации Y-фожазита в микрогранулах. Можно следовать U.S. Pat. No. 4,493,902.
Способы кристаллизации, которые использовали, основаны на ряде допущений и определенных исходных материалах. Зародыши описаны в U.S. Pat. No. 4,631,262, и лучше использовать внешние зародыши. Предполагается, что компоненты мета-каолина SiO2, Al2O3 и Na2O, зародыши, раствор силиката натрия, кальцинированное связующее силиката натрия и силикагель 100% реакционноспособны. Предполагается, что реакционная способность глинозема и кремнезема в каолине, кальцинированном исходя из его экзотермического эффекта до шпинеля, составляет 1% и 90% соответственно. Хотя и используют эти два значения, но они не являются точными. Предполагается, что реакционная способность глинозема и кремнезема в каолине, кальцинированном исходя из его экзотермического эффекта до муллита, составляет 0% и 67% соответственно. Эти два значения относительно точные, конкретнее, инертность 3:2 муллита при кристаллизации и полном растворении свободной фазы кремнезема. Так как количество мета-каолинового глинозема в синтезе является ограниченным и объем цеолита намного больший, чем соответственный объем мета-каолина, важно ограничить выход цеолита соответственно необходимому объему пор микрогранул. Иначе после кристаллизации поры будут недостаточного объема. Так представлено в известной литературе. С другой стороны, если недостающий реагент находится в микрогрануле для выращивания цеолита и соответственно делает твердым катализатор, можно добавить дополнительную питательную среду - глинозем, например микрогранулы мета-каолина, исходя из известных в данной области знаний. Поэтому жесткий контроль процесса стоит проводить относительно объема пор и трения. Исходя из всего вышеизложенного представлены следующие соотношения массы реагентов, используемых в способах кристаллизации. Инертные компоненты не учитываются только в случае определения количества зародыша, которое определяется как соотношение массы зародыша Al2O3 к полной массе микрогранул.
Силикат натрия и гидроокись натрия можно добавлять в кристаллизатор разными способами. Например, реагенты можно вводить в виде водной смеси силиката натрия N® Brand и гидроокиси натрия. Или как минимум часть силиката натрия можно вводить в виде маточного раствора, полученного во время кристаллизации другого цеолитсодержащего продукта.
После окончания процесса кристаллизации микрогранулы, содержащие Y-фожазит, отделяют как минимум от основной части маточного раствора, например, фильтрованием. Желательно промыть микрогранулы водой во время или после фильтрования. Цель промывки - удалить маточный раствор, который входит в состав микрогранулы. Можно практиковать "удерживание кремнезема". U.S. Pat. No. 4,493,902 at column 12, lines 3-31, касается удерживания кремнезема, полная информация по патенту приводится в ссылке. Микрогранулы, которые отфильтровывают, содержат цеолит Y-фожазит в натриевой форме. Обычно микрогранулы содержат больше чем около 8 мас.% Na2O. Y цеолит обычно синтезируют в формах, имеющих соотношения кремнезем: глинозем около 5:1. Эти синтезированные формы Y цеолита можно обрабатывать различными методами, которые дают возможность исключить из них структурированный алюминий. Многие подобные методы заключаются в выведении алюминия из структурной решетки цеолита при помощи соответствующих химических веществ. Большой объем работ по получению алюминий-дефицитного фожазита приводится в Advances in Chemistry Series No. 121, Molecular Sieves, G.T.Kerr, American Chemical Society, 1973. Определенные методы получения деалюминированного цеолита описаны в: Catalysis by Zeolites (International Symposium on Zeolites, Lyon, Sept. 9-11, 1980), Elsevier Scientific Publishing Co., Amsterdam, 1980 (деалюминирование Y цеолита под действием тетрахлорида кремния); U.S. Pat. No. 3,442,795 и G.B. No. 1,058,188 (гидролиз и удаление алюминия комплексообразованием); G.B. No. 1,061,847 (кислотная экстракция алюминия); U.S. Pat. No. 3,493,519 (удаление алюминия выпариванием и комплексообразованием); U.S. Pat. No. 3,591,488 (удаление алюминия выпариванием); U.S. Pat. No. 4,273,753 (деалюминирование галогенидами и галоидоокисями кремния); U.S. Pat. No. 3,691,099 (кислотная экстракция алюминия); U.S. Pat. No. 4,093,560 (деалюминирование обработкой солями); U.S. Pat. No. 3,937,791 (удаление алюминия растворами Cr(III)); U.S. Pat. No. 3,506,400 (выпаривание после комплексообразования); U.S. Pat. No. 3,640,681 (экстракция алюминия ацетилацетонатами после бидегидроксилирования); U.S. Pat. No. 3,836,561 (удаление алюминия кислотой); DE-OS No. 2,510,740 (обработка цеолита хлором или не содержащими хлор газами при высоких температурах), NL No. 7,604,264 (кислотная экстракция), JA No. 53,101,003 (обработка ЭДТА или другими веществами для удаления алюминия) и J. Catalysis 54 295 (1978) (гидротермическая обработка после кислотной экстракции).
Высококремнистые формы Y цеолита можно получать выпариванием или кислотной экстракцией структурированного алюминия (или двумя этими способами), но из-за того что Y цеолит, в нормальном состоянии, неустойчивый к действию кислоты, поэтому должен быть сразу преобразованным в кислотостойкую форму. Методы получения этой известной и одной из самых популярных форм кислотостойкого Y цеолита известен как "Ультрастабильный Y" (USY); как описано в U.S. Pat. Nos. 3,293,192 и 3,402,996 and the publication, Society of Chemical Engineering (London) Monograph Molecular Sieves, page 186 (1968) by С.V.McDaniel and P.K.Maher, в ссылке приведена подробная информация по получению цеолита.
В принципе, "ультрастабильный" означает Y цеолит, который чрезвычайно устойчив к разрушению кристаллической решетки при высокотемпературной обработке и выпаривании и характеризуется содержанием R2O (где R - это Na, К или любой другой ион щелочного металла) меньше 4 массовых процентов, лучше меньше 1 массового процента, размером элементарной ячейки меньше 24.5 ангстрем и молярным соотношением кремнезема к глинозему около от 3.5 до 7 и выше. Ультрастабильную форму Y цеолита получают прежде всего благодаря существенному уменьшению ионов щелочных металлов и размера элементарной ячейки. Ультрастабильный цеолит характеризуется как меньшим размером элементарной ячейки, так и низким содержанием щелочного металла в кристаллической структуре. Ультрастабильную форму Y цеолита можно получить основным обменом между Y цеолитом и водным раствором соли аммония, например нитрата аммония, пока в цеолите не уменьшиться содержание до 4 массовых процентов. Полученный цеолит после обмена кальцинируют при температуре от 540°С до 800°С в течение нескольких часов, охлаждают и снова поддают основному обмену с водным раствором соли аммония, пока количество щелочного металла не уменьшиться до меньше чем 1 массового процента, после чего продукт промывают и кальцинируют опять при температуре от 540°С до 800°С, получая на выходе ультрастабильный Y цеолит. Последовательный ионный обмен и термическая обработка приводят к существенному уменьшению содержания щелочного металла в исходном цеолите и уменьшению размера элементарной ячейки, что характерно для особо высокостабильного цеолита Y-типа. Затем ультрастабильный Y цеолит можно поддавать кислотной экстракции с образованием высококремнистой формы цеолита.
Другие методы увеличения соотношения кремнезема к глинозему в Y цеолите путем кислотной экстракции описаны в U.S. Pat. Nos. 4,218,307; 3,591,488 и 3,691,099, ссылка на которые дается для более подробного ознакомления с этими методами. Катализатор согласно изобретению включает микрогранулы, содержащие как минимум 40 мас.%, лучше от 50 мас.% до 65 мас.% Y-фожазита, являющего собой кристаллический натрийсодержащий цеолит типа фожазита. Термин Y-фожазит означает синтетический цеолит типа фожазита в натриевой форме, дифракция рентгеновских лучей образца которого описана в Breck, Zeolite Molecular Sieves, p.369, Table 4.90 (1974), с размером элементарной кристаллической ячейки натриевой формы (после промывки любого кристаллизационного маточного раствор цеолита) меньше около 24.75 Å, что можно определить методом, описанным в стандартной методике ASTM "Determination of the Unit Cell Size Dimension of a Faujasite Type Zeolite" (Designation D3942-80) или любым аналогичным методом. Термин Y-фожазит означает цеолит в натриевой форме, равно как в любой известной модифицированной форме, в том числе, например, редкой земли, аммониевой, обменной и стабилизированной форме. Процент цеолита типа Y-фожазит в микрогранулах катализатора определяют, когда цеолит находится в натриевой форме (после промывки, чтобы удалить кристаллизационный маточный раствор, содержащийся в микрогранулах), методом, описанным в стандартной методике ASTM "Relative Zeolite Diffraction Intensities" (Designation D3906-80) или любым аналогичным методом. Необходимо тщательно уравновесить микрогранулы перед измерением дифракции рентгеновских лучей, иначе результаты могут быть существенно измененными.
Микрогранулы, содержащие макропористую матрицу и in-situ Y цеолит, могут быть образованы в катализаторе, используемом для гидрокрекинга, любым известным методом. Обычно микрогранулы смешивают с матричным материалом, описанным выше, и экструдируют или другим способом получают необходимую форму. Псевдобемитовые глиноземы, которые можно полностью и частично пептизировать, являются особенно эффективными связующими веществами. Они также помогают при экструзии. Эффективным количеством бемитовых глиноземов является 5-20 мас.% от общей массы катализатора. Диаметр типичного экструдата составляет от 1/16 до 1/8 дюймов. Экструдаты могут находиться в любой известной форме, в том числе цилиндрической, многолепестковой или клевероподобной. Микрогранулы могут быть насыщены металлическим составляющим до соединения с матричным материалом или экструзии, или насыщать ими можно уже собственно катализатор.
Далее можно сформировать цеолит in-situ из матричного материала, который может быть экструдированным или сформированным другим способом. Водный каолин, мета-каолин и матричный материал, образующий макропоры, могут перемешиваться, а затем формируются до реакции, чтобы сформировался цеолит. Цеолитобразующая реакция подобна, если не идентична, реакции получения цеолитных микрогранул, описанной выше. Полагается, что так же как в случае кристаллизации микрогранул, кристаллизация маточного раствора должна проходить при движении жидкости. Это должно препятствовать образованию внешнего зародыша фожазита и мелких фракций цеолита. Кристаллизация слоев экструдата и маточного раствора в щадящем восходящем потоке была успешной. Для улучшения экструдируемости или образования смеси предцеолит/матрица и ее целостности при кристаллизации могут быть добавлены небольшие количества связующих веществ, например кремнезема.
Чертеж иллюстрирует уникальное строение катализаторов гидрокрекинга согласно этому изобретению, полученных из уникального глиноземсодержащего материала, используемого для получения матрицы катализатора. Микрогранулы катализатора согласно изобретению имеют совершенно другое строение, чем известные микрогранулы катализатора, особенно относительно увеличенного объема пор, структуры цеолитной матрицы и средней площади поверхности. Как видно из чертежа, катализатор, представленный в изобретении, включает макропористую матрицу, в которой макропоры образованы из пористых плоских матричных структур случайной конфигурации, которые покрывают поверхность кристаллами цеолита. Поэтому, макропоры катализатора покрыты активными кристаллами цеолита. В чертеже поверхность мезопористой матрицы состоит из частиц муллита.
Макроскопичность катализатора позволяет углеводородам входить в катализатор свободно, а увеличенная поверхность макропор позволяет этим углеводородам контактировать с каталитической поверхностью. Важно, что углеводороды могут беспрепятственно взаимодействовать с цеолитом, что делает катализатор настолько активным.
Пример 1
Были получены микрогранулы с содержанием 30 частей мета-каолина (МК), 20 частей влажного Ansilex 93™ наполнителя среднего помола, 20 частей HiOpaque™ наполнителя среднего помола, кальцинированного при температуре выше 1,050°С, и 30 частей NuSurf™ наполнителя, который кальцинировали при температуре выше 1,050°С, пульверизировали и дробили гидроэнергетически. К этой смеси кальцинированного каолина добавляли 15 частей SiO2, полученного из силиката натрия N-brand®. Эти микрогранулы не нейтрализовали кислотой. Исходным источником мета-каолина был Metamax™, пульверизированный порошок, который превращали в материал с содержанием твердого вещества 55% смешиванием с водопроводной водой и 3 мл ПАВ Colloid 211 (Viking Industries, Atlanta, GA) на кг кальцинированного каолина. В периодическом процессе, во время которого сухой каолин добавляли в воду, которая уже содержала ПАВ, использовали воздушный смеситель Cowles. По мере загустения смеси каолин добавляли медленней. Получалась дилатантная суспензия, но продолжить смешивание мешала вязкость. Постепенно добавляли каолин и продолжали перемешивание в течение 45 минут или дольше, пока зрительно прекращалась дилатантность суспензии.
Влажный Ansilex 93™ среднего помола получали из коммерчески производимых жидких глин с содержанием твердого вещества 50% и 60%. Около 7 галлонов этих жидких глин помещали в расходный бак четырехлитровой мельницы со средним помолом и перемешиванием (Premier Mill Corp., Reading PA). После трехкратного прохождения мельницы по 4 минуты каждое получали 51% твердого вещества с размерами 90% частиц меньше 1.66 мкм исходя из лазерного рассеивания (Horiba).
HiOpaque™ - кальцинированный наполнитель, полученный из непластинчатого каолина. Коммерчески производимый наполнитель был дополнительно кальцинирован на кордиеритовых поддонах в заранее нагретой высокотемпературной электрической печи при 2350°F в течение четырех часов, чтобы получить хорошо кристаллизированный муллит с максимальным выходом. Затем полученный материал дробили, чтобы получить пудру, демонтировали и дробили в мельнице для мокрого измельчения, как описано выше. Конечный продукт содержал 37% твердых веществ, а размер частиц в 90% составил <3.24 мкм исходя из параметров, полученных лазерным рассеиванием. У каждой из этих измельченных жидких глин была очень низкая вязкость.
NuSurf™ - это грубая фракция непластинчатого водного каолина. Этот продукт кальцинировали при 2350°F в течение четырех часов на кордиеритовых поддонах, чтобы получить хорошо кристаллизированный муллит с максимальным выходом, дробили, затем дробили в гидроэнергетической воздушной мельнице (Micron Master Jet Pulverizer, Jet Pulverizer Co., Palmyra, NY), пока 90% частиц не достигало размера <8.51 мкм. Этот материал превращали в жидкую недилатантную глину, содержащую 50% твердого вещества, водопроводную воду и 3 мл Colloid 211 на кг сухого каолина в смесителе Cowles.
Недостаточно суспензированная жидкая глина свидетельствует о том, что водный наполнитель был недостаточно пульверизирован перед кальцинированием или дробление в гидроэнергетической воздушной мельнице уплотнило частицы. Каждая из четырех составляющих жидких глин до смешивания поддерживалась в суспензированном состоянии в герметичных сейсмоприемниках.
Жидкую глину для распылительной сушки получали смешиванием четырех жидких глин в соотношении 30:20:20:30 на 4 кг сухих основных компонентов, как описано выше, в смесителе Cowles. Непосредственно к этой жидкой глине добавляли 2.14 кг N-brand® силиката натрия, образовывая смесь, включающую 45.7% твердых веществ, которая была достаточно жидкой для накачивания и распылительной сушки. Материал сушили распылительной сушкой, используя 0.6-миллиметровую насадку для однокомпонентной жидкости при 700 фунт/кв.дюйм.
ABD (кажущаяся объемная плотность) материала после сушки составляла 0.71 г/мл и анализировалась с 3.9% Na2O или 13.1% связующих веществ, таких как SiO2. Этот продукт был непосредственно кальцинирован при 1500°F в течение двух часов в заранее нагретой печи с использованием кордиеритовых поддонов. Продукт содержал поры объемом 0.356 м2/г и диаметром 40-20,000 Å, который определяли ртутной порометрией, 76 мкм APS и 14.2 мас.% кислоторастворимые компоненты (U.S. Patent No. 5,023,220; column 18, line 59) и 0.71 г/см3 ABD.
Пример 2
Были получены микрогранулы с содержанием 30 частей МК, 20 частей влажного Ansilex 93™ наполнителя среднего помола, 20 частей HiOpaque™ наполнителя среднего помола, кальцинированного при температуре выше 1,050°С, и 30 частей NuSurf™ муллита, полученного пульверизацией водного наполнителя перед кальцинированием при температуре выше 1,050°С, 15 частей SiO2, полученного из N-brand® силиката натрия. Микрогранулы нейтрализовали кислотой.
Исходным веществом для МК была вторая партия Metamax™, полученная из 55% твердого вещества и дисперсанта С211. Ansilex 93™ наполнитель был таким же, как в Примере 1. Гранулированный NuSurf™ муллит готовили, кальцинируя непластинчатый водный наполнитель при 2350°F в течение четырех часов на кордиеритовых поддонах в заранее нагретой высокотемпературной электрической печи. Затем полученный материал дробили, чтобы получить пудру, демонтировали и дробили в мельнице для мокрого измельчения, как описано выше. Конечный продукт содержал 46% твердых веществ, а размер частиц в 90% составил <2 мкм исходя из параметров, полученных лазерным рассеиванием.
В этом примере NuSurf™ муллит получили с большим объемом пор и со структурой карточного домика. Непластинчатый водный наполнитель NuSurf™ суспензировали с водой и дисперсантом, сушили распылительной сушкой с получением плотно упакованных микрогранул, затем дробили, чтобы получить порошок с низкой плотностью. Этот водный порошок кальцинировали при 2350°F на кордиеритовых поддонах в течение четырех часов, чтобы образовался кристаллизованный муллит. Продукт дробили, получали порошок, который затем смешивали в смесителе Cowles, получая смесь, содержащую 50% твердых веществ, 4 мл С211 на кг кальцинированного каолина. Полученная жидкая глина была очень дилатантной. Перемешивание длилось до тех пор, пока дилатантность не исчезала. Размер частицы составлял 90% <14.75 мкм. Каждая из четырех составляющих жидких глин до смешивания поддерживалась в суспензированном состоянии в герметичных сейсмоприемниках. Жидкую глину для распылительной сушки получали смешиванием четырех жидких глин в соотношении 30:20:20:30 на 3.93 кг сухих основных компонентов, как описано выше, в смесителе Cowles. Непосредственно к 2.11 кг жидкой глины добавляли N-brand® силикат натрия, получая смесь, содержащую 48% твердых веществ, которая была достаточно жидкой для накачивания и распылительной сушки. Материал сушили на распылительной сушке, используя 0.6-миллиметровую насадку для однокомпонентной жидкости при 700 фунт/кв.дюйм.
ABD материала после сушки составляла 0.72 г/мл и анализировалась с 4.01% Na2O или 13.5% связующих веществ, таких как SiO2. Этот продукт был нейтрализован кислотой. Сухие микрогранулы добавляли в реактор с холодной водой при перемешивании и добавляли необходимое количество 40 мас.% H2SO4, поддерживая рН жидкой глины между около 2.5 и 4. После загрузки микрогранул смесь перемешивали в течение 10 минут, контролируя рН, фильтровали, промывали приблизительно 2 галлонами воды на кг микрогранул, а затем сушили 12 часов при температуре около 350°F.
Нейтрализованные кислотой микрогранулы непосредственно кальцинировали при 1500°F в заранее нагретой печи на кордиеритовых поддонах в течение трех часов. Продукт содержал поры объемом 0.402 м2/г с диаметром 40-20,000 Å, определенным ртутной порометрией, 77 мкм APS и 14.4 мас.% кислоторастворимые компоненты и 0.66 г/см3 ABD.
Пример 3
Микрогранулы с исключительно большим объем и с необыкновенно широкими макропорами готовили, используя 30 частей МК и 70 частей NuSurf™ муллита, который получали, размельчая перед кальцинированием при температуре выше 1,050°С. Кальцинированная смесь каолина сушилась распылительной сушкой с 15 частями SiO2, полученного из N-brand® силиката натрия. Микрогранулы нейтрализовали кислотой.
Исходным материалом для МК был такой же Мetаmax™, как в примере 2. NuSurf™ муллит был таким же, как в примере 2. Обе жидкие глины до смешивания поддерживались в суспензированном состоянии в герметичных сейсмоприемниках.
Жидкую глину для распылительной сушки получали смешиванием двух жидких глин в соотношении 30:70 на 4.00 кг сухих основных компонентов, как описано выше, в смесителе Cowles. К 2.14 кг жидкой глины добавляли силиката натрия N-brand®, получая смесь, содержащую 48% твердых веществ, которая была достаточно жидкой для накачивания и распылительной сушки. Материал сушили распылительной сушкой, используя 0.6-миллиметровую насадку для однокомпонентной жидкости при 700 фунт/кв.дюйм.
ABD материала после сушки составляла 0.56 г/мл и анализировалась с 3.92% Na2O или 13.1% связующих веществ, таких как SiO2. Этот продукт был нейтрализован кислотой, как указано в примере 2.
Нейтрализованные кислотой микрогранулы непосредственно кальцинировали при 1500°F в заранее нагретой печи на кордиеритовых поддонах в течение трех часов. Продукт содержал поры объемом 0.407 м2/г с диаметром 40-20,000 Å, и 0.156 м2/г с диаметром 20,000 - 40,000 при повторном измерении методом ртутной порометрии, 86 мкм APS и 10.46 мас.% кислоторастворимых компонентов с 0.53 г/см3 ABD.
Примеры 4-6
Микрогранулы, полученные в примерах 1-3, кристаллизировали в течение 23 часов, получая У цеолит согласно методу 4,493,902 и 5,395,809 со следующими результатами. Зародыши описываются в 4,631,262.
Ртутный объем пор этих материалов значительно выше, чем у известных до этого материалов. Натриевые формы катализатора затем поддавались ионному обмену с получением конечного продукта, как указано ниже. Катализатор в натриевой форме добавляли при перемешивании к 27 мас.% раствору нитрата аммония при 180° F и рН 2.8-3.2 и прикапывали 50% HNO3, чтобы поддерживать рН на нужном уровне. После добавления всего количества катализатора жидкая глина перемешивалась в течение 15 минут, фильтровалась, дважды промывалась деионизированной водой.
Провели два таких обмена при соотношении массы катализатора к 27 мас.% нитрата аммония 1:2. Образцы редкой земли поддавали обмену при 180° F и рН 4, получая около 3% редкоземельных элементов на катализаторе. Содержание Na2O в этот момент составляло от 1.8 до 1.9 массы, значительно меньше, чем в известных формах. Частично подвергшиеся обмену материалы сохли, а затем кальцинировались при 1150°F в заранее нагретой печи на кордиеритовых поддонах в течение двух часов, пока влажность не составила 25 мас.%. После кальцинирования процесс обмена аммония повторяли пять (Пример 4) или три раза, затем продукт опять кальцинировали до содержания 25 мас.% влажности при 1150°F, чтобы сформировался конечный продукт. Результаты представлены ниже.
Видно, что использование большего количества определенным образом размельченного водного дерасслоенного каолина приводит к получению большего объема пор катализатора и более широких макропор. На чертеже показана SEM фотография катализатора. Темные области - это макропоры, которые находятся в случайном порядке или в структуре "карточного домика", слоев матрицы, полученной из непластинчатого наполнителя. Более маленькие зернышки, находящиеся между большими кристаллами цеолита, можно идентифицировать как кристаллы муллита. Большие кристаллы с оболочкой (покрытые) или другим образом покрытую муллит-матрицу можно идентифицировать как Y цеолит.
Включение как муллита, так и шпинеля ведет к трехмерной картинке размера поры. Пористость, свойственная шпинелю и муллиту, видимая при ртутном измерении пор, указывает на то, что в мезопористых матрицах не закупоривается цеолит, который в них образуется.
Настоящее изобретение относится к катализатору гидрокрекинга, который включает образованный in situ Y-фожазит, и к процессу гидрокрекинга. Описан катализатор гидрокрекинга, который состоит из сформированной матрицы, полученной экструзией металлов, катализирующих гидрогенирование, и слоя цеолита, кристаллизованного на поверхности пористой глиноземсодержащей матрицы, где матрица со слоем цеолита имеет такое строение, чтобы обеспечить макропоры, на стенках которых образуется слой цеолита, и цеолит образуется in situ в глиноземсодержащей матрице. Описан также процесс гидрокрекинга и восстановления углеводородов, включающий взаимодействие фракции нефти с водородом и описанным выше катализатором при необходимых для гидрокрекинга условиях и включающий также восстановление фракций нефти, кипящих при температуре ниже около 200°С. Технический эффект - повышение активности катализатора. 2 н. и 7 з.п. ф-лы, 3 табл., 1 ил.
1. Процесс гидрокрекинга и восстановления углеводородов, который включает взаимодействие фракции нефти с катализатором и водородом при необходимых для гидрокрекинга условиях, где катализатор состоит из комбинации металлов, катализирующих гидрогенирование, и слоя цеолита, кристаллизованного на поверхности пористой глиноземсодержащей матрицы, где матрица со слоем цеолита имеет такое строение, чтобы обеспечить макропоры, на стенках которых образуется слой цеолита, где сказанный процесс также включает восстановление фракций нефти, кипящих при температуре ниже около 200°С.
2. Процесс по п.1, где металл, катализирующий гидрогенирование, выбирают из группы, состоящей из металлов групп VA, VIA, и VIIIA Периодической системы элементов Менделеева.
3. Процесс по п.1, где фракция нефти - это фракция нефти после гидрообработки.
4. Процесс по п.1, где матрица со слоем цеолита объединена со связующим веществом с образованием связанного цеолита, где связанному цеолиту придают форму экструзией.
5. Процесс по п.1, где цеолит - это Y цеолит.
6. Процесс по п.1, где пористой глиноземсодержащей матрице придают форму, например, экструзией, а цеолит образуется in-situ в данной матрице.
7. Процесс по п.1, где матрица со слоем цеолита имеет пористость, полученную методами ртутной порометрии, как минимум 0,07 см3/г для пор 600-20,000 Å диаметром.
8. Катализатор гидрокрекинга и восстановления углеводородов, который состоит из сформированной матрицы, полученной экструзией, металлов, катализирующих гидрогенирование, и слоя цеолита, кристаллизованного на поверхности пористой глиноземсодержащей матрицы, где матрица со слоем цеолита имеет такое строение, чтобы обеспечить макропоры, на стенках которых образуется слой цеолита, и цеолит образуется in-situ в глиноземсодержащей матрице.
9. Катализатор по п.8, где катализатор находится в форме от 1/16 до 1/8 дюймового экструдата.
| Сплав на основе магния для протекторов | 1987 |
|
SU1502648A1 |
| US 6174430 B1, 16.01.2001 | |||
| US 5393409 A, 28.02.1995 | |||
| RU 2004137797/15 A1, 10.09.2005 | |||
| RU 2004100239/04 A1, 20.06.2005. | |||

