ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым катализаторам каталитического крекинга с псевдоожиженным слоем катализатора, содержащим микросферы, которые содержат цеолит типа Y-фожазит и обладают исключительно высокой активностью и другими желательными характеристиками, способу получения таких катализаторов и применению таких катализаторов для крекинга нефтяного сырья, в частности, в рамках процессов с малой продолжительностью пребывания.
С 1960-х годов большинство коммерческих катализаторов каталитического крекинга с псевдоожиженным слоем катализатора содержали цеолиты в качестве активного компонента. Такие катализаторы принимают форму мелких частиц, называемых микросферами, которые содержат в качестве активного компонента как цеолитный, так и нецеолитный компонент. Зачастую нецеолитный компонент известен под названием матрица для цеолитного компонента катализатора. Нецеолитный компонент, как известно, выполняет ряд важных функций, которые относятся и к каталитическим и к физическим свойствам катализатора. Облад описал эти функции следующим образом: "Считается, что матрица выступает в качестве поглотителя натрия в сите, добавляя, таким образом, устойчивости цеолитным частицам в матричном катализаторе. Матрица служит дополнительной функцией: разбавления цеолита; стабилизации его в отношении нагревания и выпаривания и механического истирания; обеспечивая высокую пористость таким образом, что цеолит может быть применен с его максимальной способностью и регенерация может быть произведена без проблем; и, в конце концов, она обеспечивает объемные свойства, которые являются важными для передачи тепла в процессе регенерации и крекинга и сохранения тепла в крупномасштабных установках каталитического крекинга. "A.G. Oblad Molecular Sieve Cracking Catalysts, The Oil And Gas Journal, 70, 84 (Mar. 27, 1972).
В существующем уровне техники катализаторов каталитического крекинга с псевдоожиженным слоем катализатора, активный цеолитный компонент встроен в микросферах катализатора с помощью одного из двух общих способов. В одном из способов, цеолитный компонет кристаллизуют, а затем вводят в микросферы в отдельной стадии. Во втором способе, in-situ способе, сначала образуют микросферы и цеолитный компонент затем кристаллизуется в самих микросферах, чтобы обеспечить микросферы, которые содержат как цеолитный, так и нецеолитный компоненты.
Уже давно является признанным тот факт, что для катализатора каталитического крекинга с псевдоожиженным слоем катализатора, чтобы быть коммерчески успешным, он должен иметь коммерчески приемлемые характеристики активности, селективности и стабильности. Он должен быть достаточно активным, чтобы дать экономически привлекательные показатели выработки, он должен иметь хорошую избирательность к производству продуктов, которые являются желательными, и не производить продукты, которые являются нежелательными, и он должен быть достаточно стабильным гидротермально и устойчивым к истиранию, чтобы иметь коммерчески полезный срок годности.
Как правило, ККП (каталитический крекинг с псевдоожиженным слоем катализатора) коммерчески практикуется в циклическом режиме. В ходе таких операций углеводородное сырье контактирует с катализатором из горячих, активных, твердых частиц без добавленного водорода, к примеру, при давлении до около 50 фунтов на квадратный дюйм и температурах до около 650°C. Катализатор представляет собой порошок с размером частиц около 20-200 микрон в диаметре и со средним размером примерно 60-100 микрон. Порошок проталкивается вверх через зону лифт-реактора, псевдоожижается и тщательно смешивается с углеводородным сырьем. Углеводородное сырье подвергают крекингу при указанных выше высоких температурах с помощью катализатора и разделяют на различные углеводородные продукты. По мере того, как углеводородное сырье поддается крекингу в присутствии крекирующего катализатора с образованием горючего и олефинов, на катализаторе оседает нежелательный углеродистый осадок, известный как «кокс». Отработанный катализатор содержит кокс, а также металлы, которые присутствуют в исходном сырье. Катализаторы для ККП представляют собой, как правило, алюмосиликатные композиции с большими порами, включая, в том числе, фожазит или цеолит Y.
Частицы закоксованного катализатора отделяют от углеводородных продуктов крекинга, и после зачистки перемещают в регенератор, где кокс сжигается для регенерации катализатора. Регенерированный катализатор затем стекается вниз из регенератора к основанию воронки.
Эти циклы крекинга и регенерации при высоких скоростях потока и температурах имеют тенденцию к физическому разрушению катализатора на еще более мелкие частицы, называемые "пылью". Эта пыль имеет диаметр до 20 микрон по сравнению со средним диаметром частиц катализатора от около 60 до около 100 микрон. В определении сохранения целостности катализаторов, и соответственно, их экономической эффективности, устойчивость к истиранию является ключевым параметром. В то время, как начальный размер частиц можно контролировать путем регулирования первоначальной сушки распылением катализатора, если сопротивление истиранию плохое, каталитическая крекинг-установка может производить большое количество пыли размером 0-20 микрон, которая не должна высвобождаться в атмосферу. Коммерческие каталитические крекинг-установки включают циклонные и электростатические фильтры для предотвращения переноса пыли по воздуху. Квалифицированному специалисту в данной области техники также понятно, что чрезмерное образование пыли катализатора увеличивает стоимость катализатора для нефтепереработчика. Избыток пыли может привести к увеличенному добавлению катализатора и разбавлению каталитически жизнеспособных частиц.
Патент США №4,493,902, содержание которого включено в данное описание посредством ссылки, раскрывает новые катализаторы флюидизированного крекинга, которые содержат устойчивые к истиранию, с высоким содержанием цеолита, каталитически активные микросферы, содержащие более чем около 40%, предпочтительно 50-70 мас. % Y фожазита и способы получения таких катализаторов путем кристаллизации более, чем около 40% натрий Y цеолита в пористых микросферах, состоящих из смеси двух различных форм химически реактивной кальцинированной глины, именуемые, метакаолином (каолин, кальцинированный с подвержением сильной эндотермической реакции, связанной с дигидроксилированием) и каолиновой глиной, кальцинированная в более тяжелых условиях, чем те, которые используются для преобразования каолина в метакаолин, то есть, каолиновую глину кальцинируют, чтобы каолину пройти характерную экзотермическую реакцию каолина, иногда также называемую шпинельная форма кальцинированного каолина. В предпочтительном варианте осуществления, микросферы, содержащие две формы кальцинированной каолиновой глины, погружают в щелочной раствор силиката натрия, который нагревают, предпочтительно до тех пор, пока максимально возможное количество Y фожазита не кристаллизуется в микросферы.
На практике '902 технологии, пористые микросферы, в которых кристаллизуется цеолит предпочтительно получают путем образования водой суспензии порошкообразной сырой (гидратированной) каолиновой глины (Al2O3:2SiO2:2H2O) и порошкообразной кальцинированной каолиновой глины, которая подверглась экзотермической реакции вместе с небольшим количеством силиката натрия, который действует в качестве псевдоожижающего агента для суспензии, которую загружают в распылительную сушилку для образования микросфер, а затем действуют таким образом, чтобы обеспечить физическую целостность с компонентами высушенных распылением микросфер. Высушенные распылением микросферы, содержащие смесь гидратированной каолиновой глины и каолина, кальцинированного с подвержением экзотермической реакции затем кальцинируют в контролируемых условиях, менее серьезных, чем те, которые требуются для того, чтобы подвергнуть каолин прохождению экзотермического эффекта, с целью обезводить гидратированную часть каолина микросферы и повлиять на его превращение в метакаолин, и это дает в результате микросферы, содержащие желаемую смесь метакаолина, каолина, кальцинированного с прохождением экзотермической реакции и натрий силикатного связующего вещества. В иллюстративных примерах '902 патента приблизительно равные массы гидратированной глины и шпинели присутствуют в подающем устройстве распылительной сушилки и полученные в результате кальцинированные микросферы содержат немного больше глины, которая подверглась экзотермическому эффекту, чем метакаолин. В '902 патенте сообщается, что кальцинированные микросферы содержат около 30-60 мас. % метакаолина и около 40-70 мас. % каолина, который характеризуется его характерным экзотермическим эффектом. Менее предпочтительный способ, описанный в патенте, предполагает сушку распылением суспензии, которая содержит смесь каолиновой глины, предварительно кальцинированной до состояния метакаолина и каолин, кальцинированный с прохождением экзотермического эффекта, но без включения какого-либо гидратированного каолина в суспензию, таким образом, обеспечивая микросферы, содержащие и метакаолин и каолин, кальцинированный с подвержением экзотермическому эффекту, непосредственно, без кальцинирования с превращением гидратированного каолина в метакаолин.
При осуществлении изобретения, описанного в '902 патенте, микросферы, состоящие из каолина, кальцинированного с подвержением экзотермическому эффекту и метакаолина, реагируют с едким обогащенным раствором силиката натрия в присутствии инициатора кристаллизации (затравки) для превращения диоксида кремния и оксида алюминия в микросферах в синтетический фожазит натрия (цеолит Y). Эти микросферы отделяют от маточного раствора силиката натрия, с ионным обменом с редкоземельными металлами, ионами аммония или ими обоими для образования редкоземельных элементов или различных известных стабилизированных форм катализаторов. Технология '902 патента обеспечивает средства для достижения желаемого и уникального сочетания высокого содержания цеолита в сочетании с высокой активностью, хорошей селективностью и термической стабильностью, а также сопротивлением к истиранию.
Упомянутая выше технология имела широкий коммерческий успех. Ввиду доступности микросфер с высоким содержанием цеолита, которые также являются устойчивыми к истиранию, теперь для нефтеперерабатывающих заводов доступны катализаторы, разработанные индивидуально на заказ с конкретными целями производительности, такими как повышенная активность и/или селективность, не подвергаясь дорогостоящим механическим модернизациям. Значительная часть ККП катализаторов, поставляемая в настоящее время отечественным и зарубежным нефтепереработчикам, основана на этой технологии. Нефтеперерабатывающие заводы, у которых ККП системы ограничены максимально допустимыми температурами регенератора или мощностью воздуходувки стремятся к повышению селективности, приводящему к сокращению выработки кокса, тогда как нормирование газовых компрессоров дает катализаторы, которые сокращение газа делает весьма желательным. Казалось бы, небольшое сокращение кокса может представлять собой значительную экономическую выгоду для работы ККП установки с воздуходувкой или ограничениями температуры генератора.
Характеристика активности и селективности катализаторов, образованных с помощью способов по '902 патенту достигаются, в общем случае, даже при том, что катализаторы имеют относительно низкую общую пористость по сравнению с катализатором каталитического крекинга с псевдоожиженным слоем катализатора, полученного путем введения цеолита в матрицу. В частности, микросферы таких катализаторов, в некоторых случаях, имеют общую пористость менее, чем около 0.15 см3/г или даже менее, чем около 0.10 см3/г. В основном, микросферы '902 патента имеют общую пористость менее, чем 0.30 см3/г. Используемый здесь термин "общая пористость" означает объем пор, которые имеют диаметры в диапазоне 35-20,000 Å, как определено методом ртутной порометрии. К удивлению, '902 патент отмечает, что микросферы, имеющие общую пористость менее, чем около 0.15 см3/г проявляют активности и обнаружены характеристики селективности. К примеру, такой результат противоречит ранее раскрытому в данной области техники, что малые объемы пор «могут привести к потере селективности в виду диффузионных ограничений».
Считается, что относительно низкая пористость образующейся микросферы катализатора, как в '902 патенте, не оказывает негативного влияния на активность и характеристики селективности, так как микросферы '902 патента не ограничивают диффузию в сравнении с типичными условиями обработки ККП, которые применялись во время патента. В частности, время контакта катализатора с исходным сырьем, которое должно быть подвержено крекингу, обычно составляло не менее 5 секунд или больше. Таким образом, в то время как типичные ККП катализаторы, образованные механическим введением цеолита в пределы матрицы, могут быть более пористыми, время реакции ККП лифт-реакторов из существующего уровня техники не дает никаких преимуществ в активности или селективности. Этот результат стал причиной вывода о том, что процессы переноса были вовсе не ограничивающими в ККП катализаторах, по крайне мере, за пределами цеолитной структуры. Утверждения, сделанные наоборот, были несовместимы с фактами и были легко отклонены как корыстные. Важно отметить, сопротивление истиранию микросфер, приготовленных в соответствии с '902 патентом, превосходило обычные ККП катализаторы, в которых кристаллизованный цеолитный каталитический компонент был физически включен в нецеолитную матрицу.
В последнее время, тем не менее, были разработаны ККП аппараты, которые значительно сокращают время контакта между катализатором и исходным сырьем, которое должно быть подвержено крекингу. Обычно, реактор представляет собой вертикальный трубопровод, в котором катализатор и углеводородное сырье входит в нижнюю часть вертикального трубопровода и проходит через вертикальный трубопровод. Горячий катализатор воздействует на крекинг углеводорода во время прохождения через вертикальный трубопровод и при высвобождении из вертикального трубопровода, при этом крекированные продукты отделяются от катализатора. Катализатор затем подают в регенератор, где удаляется кокс, таким образом, очищая катализатор, и в то же время, обеспечивая необходимое тепло для катализатора в реакторе, представляющего собой вертикальный трубопровод. Новейшие реакторы, представляющие собой вертикальный трубопровод, работают при меньшем времени пребывания сырья и более высоких рабочих температурах, чтобы минимизировать селективность кокса и удельный кокс. Некоторые из конструкций даже не используют вертикальный трубопровод, дополнительно сокращая время контакта меньше одной секунды. Селективность бензина и сухого газа можно улучшить в результате аппаратных изменений. Эти модификации ККП установок позиционируются, как ценные вне зависимости от типа купленного катализатора, что предполагает отсутствие систематических проблем в состоянии уровня техники каталитических технологий.
Обработка все более и более тяжелого исходного сырья в процессах ККП типа и тенденции такого сырья к увеличению выработки кокса и выхода нежелательных продуктов также привели к новым способам контактирования исходного сырья с катализатором. Способы контактирования ККП катализатора с очень короткими периодами контакта имели особый интерес. Таким образом, короткие временные интервалы контакта менее чем 3 секунды в вертикальном трубопроводе, и ультра короткие временные интервалы контакта 1 секунда или менее показали улучшение селективности бензина при одновременном уменьшении выработки кокса и сухого газа.
Для того, чтобы компенсировать продолжающийся спад в катализаторе по времени контакта масла в ККП обработке, "равновесные" катализаторы в применении имеют тенденцию становиться более активными. Таким образом, должно быть достигнуто увеличение полной площади поверхности катализатора, также как и увеличивается уровень ускорителей катализа из оксидов редкоземельных металлов, добавляемых в катализаторы. Кроме того, температура крекинга растет, чтобы компенсировать уменьшение конверсии. К сожалению, было обнаружено, что плотность в градусах АНИ нижней части, образующейся в процессе короткого времени контакта ("КВК") часто возрастает после частичного переоборудования, в результате чего некоторые предполагают, что самая тяжелая доля углеводородного сырья занимает больше времени для крекирования. Кроме того, в то время как высокая полная площадь поверхности катализатора оценена, ККП процесс все еще оценивает сопротивление истиранию. Соответственно, хоть и не очевидно для тех, кто специализируется в данной области техники, становится все более и более вероятно, что требуется оптимизация ККП катализаторов, которые используются в настоящее время, для обработки с новым коротким временем контакта и ультра коротким временем контакта.
Сейчас создается теория, что при обработке углеводородов с коротким временем контакта, дополнительное усовершенствование может быть получено путем устранения ограничений диффузии, которые все еще могут существовать в данных катализаторах. Такое решение принимается, даже когда эти материалы превосходят при применении. Создается теория, что улучшение этих катализаторов может быть произведено путем оптимизации пористости катализатора и устранением заграждения активного участка и диффузионных ограничений связующих фаз, присутствующих в катализаторах, полученных так называемым методом внедрения.
В принадлежащей тому же правообладателю США 6,943,132 было обнаружено, что если нецеолитная, богатая на алюминий матрица катализатора получена из источника сверхтонкого водного каолина, имеющего такой размер микрочастицы, что 90 мас. % частиц водного каолина составляет менее чем 2 микрона, и которые измельчают и кальцинируют посредством экзотермической реакции, могут быть приготовлены микропористые цеолитные микросферы.
Ультратонкий водный каолин сушат в распылительной сушилке, или подходящем для данного процесса агрегате, затем дезагломерируют с применением высокоэнергетических измельчателей, или процедур сухого помола. Эти процессы агрегатов практикуются для сокращения агломератов и возвращения кальцинированного сырья до размера частиц, подобного тому, как было измерено в суспензии, как отмечалось выше. Присутствие агломерированных структур изменяют размер частиц и свойства объемной плоскости кальцинированного каолина. Во время изменения фазы из водного каолина, происходит агломерация и спекание. Измеренный размер частицы грубеет на протяжении диапазонов размеров частиц. Большие агломерированные структуры имеют более высокую плотность, таким образом, меньшую пористость. Структурирование перед кальцинированием расширяет объем пор путем цементирования контактных точек частиц, которые в кальцинированных в полной мере каолинах теоретически поддерживаются путем исключения аморфного диоксида кремния. Тепловой переход к шпинели вытесняет один моль диоксида кремния на один моль образованной шпинели. Переход муллита от шпинели удаляет четыре дополнительных моля.
В более общем смысле, матрица ККП катализатора, применяемая для достижения макропористости ККП катализатора, получается из источника оксида алюминия, такого как каолин, кальцинированный с помощью экзотермического эффекта, который имеет заданный объем водяной поры, что выгодно отличается от кальцинированного каолина из существующего уровня техники, используемого для формирования матрицы катализатора. Объем водяной поры получают из теста Точки Зарождения Суспензии ("ТЗС"), который описан в патенте.
Морфология микросферических катализаторов, которые получают в соответствии с патентом США 6,943,132, является уникальной в сравнении с in-situ микросферическими катализаторами, полученными ранее. Применение распыленного, ультратонкого водного каолина, кальцинированного через экзотермический эффект дает in-situ цеолитные микросферы, которые имеют макропористую структуру, в которой макропоры структуры, по сути, являются покрытыми или выстланы цеолитом после кристаллизации. Макропористость, как это определено в документе, означает, что катализатор имеет объем микропор в диапазоне пор 600-20,000 Å не менее 0.07 см3/г внедренной ртути. Катализаторы также имеют площадь поверхности БЭТ менее, чем 500 м2/г. Новый катализатор является оптимальным для ККП обработки, в том числе обработки с короткими временными интервалами контакта, в которой углеводородное сырье контактирует с катализатором в течение времени около 3 секунд или менее.
Высокая пористость внутри микросферы важна, чтобы максимизировать каталитическую активность путем устранения типичных снижений скоростей за счет диффузии молекул сырой нефти в рамках структуры микросферы. Поскольку пористость микросферы увеличивается, тем не менее, скорость, с которой микросферы разрушаются и размельчаются на более мелкие частицы в рамках усилений условий эксплуатации ККП блока; что приводит к увеличению скорости присоединения свежего катализатора и увеличению выбросов твердых частиц из блока. Обработка или композиционные механизмы для снижения скорости, при которой ККП катализатор размельчается до общего объема пор, имеют принципиальное значение для повышения эффективности и соответствующей стоимости катализатора.
Как описано в находящейся одновременно на рассмотрении патентного ведомства принадлежащей тому же правообладателю заявки США номер №13/042,808, поданной 8 марта 2011, было обнаружено, что оксиды металлов, такие как каолин, которые использовались в качестве матричных материалов для ККП катализаторов, при обработке небольших количеств полифосфата с последующим нагреванием, неожиданно обеспечивают внутреннюю структуризацию таким образом, чтобы увеличилась внутренняя пористость оксида. Оксиды металлов, которые могут изменять кристаллические фазы при нагревании при таком же состоянии, могут поддерживать внутренний объем пор во время изменения фазы, как раз увеличивается твердость оксида метала в процессе термообработки.
Обработка полифосфатом аммония (АПФ) предшествующих порошков матриц обеспечивает улучшенную внутреннюю структуру, что дает ККП катализаторы с большим объемом пор, полезные, в частности, в ВГО крекинга. Объем пор в микро (20-30 ангстрем) диапазоне, как известно, приводит к увеличенному коксу. Мезопоры (30-100 ангстрем), как известно, производят улучшенную коксовую селективность и макропоры (100-10,000 ангстрем) дают возможность иметь место диффузии, позволяющую краткое реакционное время контакта. Размеры пор выше представляют собой классификацию микро, мезо и макро размеры пор в соответствии с настоящим изобретением. Отработанный продукт АПФ имеет мало микропор и повышенный объем мезо и макропор. При заданном объеме пор прекурсор матрицы, обработанный АПФ генерирует истирание несколько лучше, чем аналогичный катализатор с большим объемом пор, как описано в патенте США 6,943,132. Таким образом, продукты с большим объемом пор могут быть сгенерированы с аналогичными свойствами истирания.
В находящейся одновременно на рассмотрении патентного ведомства принадлежащей тому же правообладателю заявки США номер №13/919,225, поданной 17 июня 2013 г., описано, что обработка водного каолина катионными полиэлектролитами, перед переработкой в субстрат микросферы для последующего роста цеолита, дает ККП катализаторы с улучшенным сопротивлением истиранию, даже не смотря на то, что в структуре катализатора поддерживают хороший объем пор.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Улучшенное сопротивление истиранию, а также контролируемый объем пор теперь обеспечены для ККП катализатора из микросферы, образованной путем объединения прекурсора матрицы, обработанного полифосфатом и суспензии водного каолина, обработанной катионным полиэлектролитом. Комбинированная обработка полифосфатом и катионным полиэлектролитом неожиданно приводит к улучшениям в сопротивлении истиранию, сохраняя при этом высокий общий объем пор, несмотря на то, что соотношение объема мезопор к объему макропор полученного ККП катализатора увеличивается.
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ
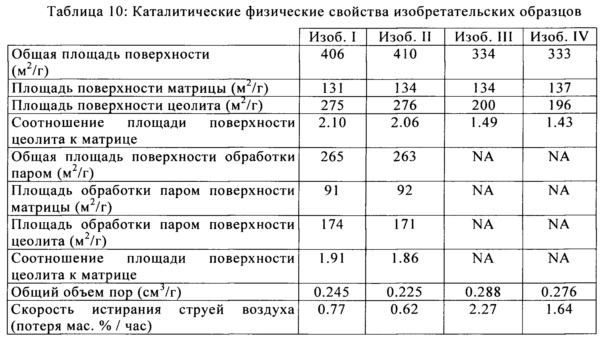
На Фигуре 1 представлен график устойчивости к истиранию в сопоставлении с высоким объемом пор коммерческого катализатора, образованного путем добавления полиамина.
На Фигуре 2 представлен график сравнения объема мезо/макро ртутных пор против сопротивления истиранию сравнительных образцов катализатора и образованных с помощью настоящего изобретения.
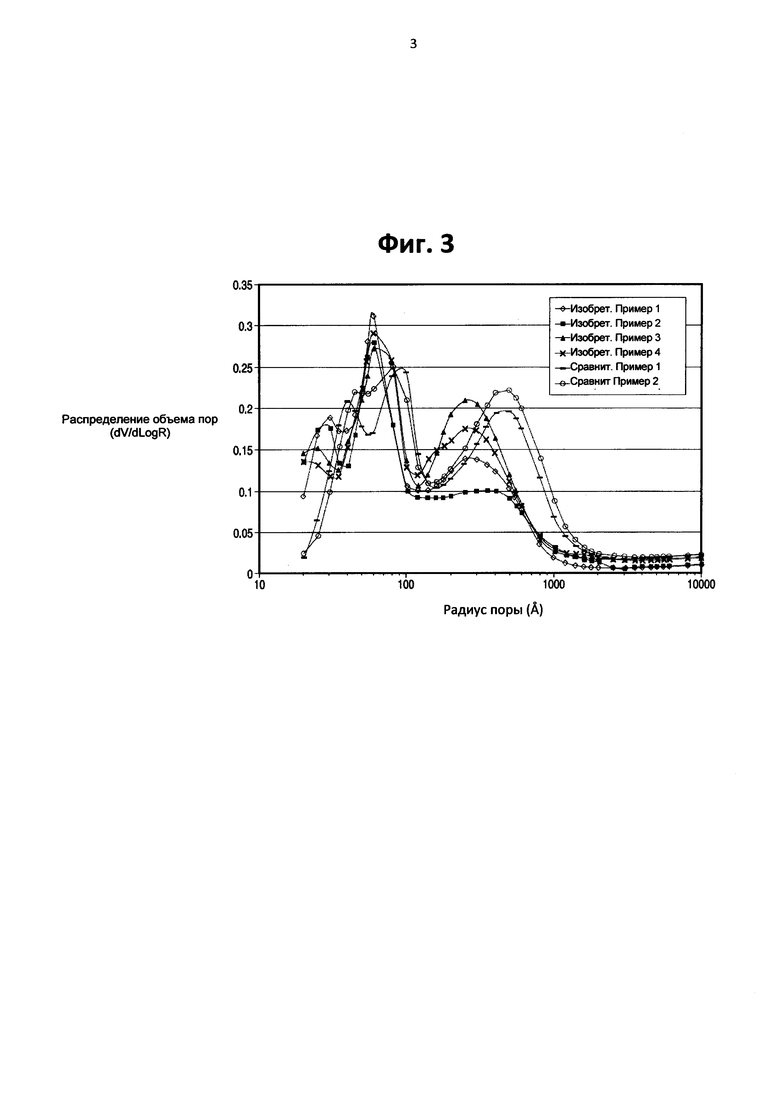
На Фигуре 3 представлен график, показывающий распределение размера пор в пределах сравнительных катализаторов, и катализаторов, образованных с помощью настоящего изобретения, в которых отличия в образовании катализатора были в части количества метакаолина, содержащегося в микросферах, применяемых для образования катализатора.
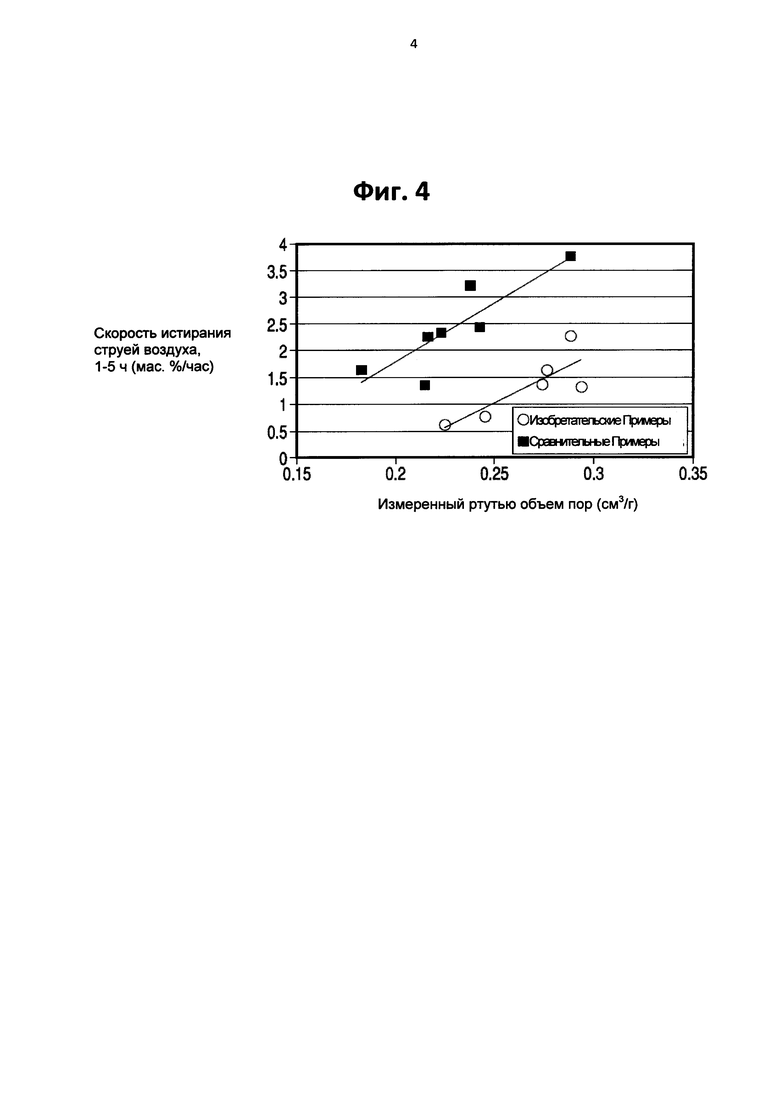
На Фигуре 4 представлен график сравнения скорости истирания сравнительных образцов катализатора, и катализаторов, образованных с помощью способа настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Каталитические микросферы настоящего изобретения получают общим способом, как описано в принадлежащем одному и тому же правообладателю патента США №4,493,902. Предпочтительно, хотя и не обязательно, что нецеолитная, богатая на оксид алюминия матрица катализаторов настоящего изобретения будет получена из водного каолинового источника, который находится в форме ультратонкого порошка, в котором, по меньшей мере, 90 мас. % частиц имеют размер менее, чем 2.0 микрон, предпочтительно, по меньшей мере, 70 мас. % менее, чем 1 микрон, как описано в упомянутом выше патенте США №6,943,132. Ультратонкий водный каолин измельчают и кальцинируют с помощью экзотермического эффекта. Кроме того, в пределах объема настоящего изобретения, когда цеолитные микросферы будут выполнены с богатой оксидом алюминия матрицей, полученной из каолина, который имеет больший размер, и кальцинируют, по меньшей мере, главным образом, через его характеристический экзотермический эффект. Satintone®No.1, (коммерчески доступный каолин, который был кальцинирован через его характеристический экзотермический эффект без какого-либо образования муллита) представляет собой материал, применяемый на коммерческой основе для образования богатой оксидом алюминия матрицы. Satintone®No.1 является производным от водного каолина, в котором 70% частиц имеют размер менее, чем 2 микрон. Другие источники, применяемые для образования богатой оксидом алюминия матрицы, включают тонкоизмельченный водный каолин (к примеру, ASP® 600, коммерчески доступный водный каолин, описанный в Engelhard Technical Bulletin No. TI-1004, в главе "Aluminum Silicate Pigments" (EC-1167)) кальцинированный, по меньшей мере, главным образом, через его характеристический экзотермический эффект. Опробованная глина нашла самое широко распространенное коммерческое применение и встретила огромный успех во всем мире.
Общая процедура изготовления ККП микросфер настоящего изобретения хорошо известна в данной области техники и может следовать из процедуры, описанной в патенте США №4,493,902. Как описано в документе, изготавливают водную суспензию реактивного тонко измельченного водного каолина и/или метакаолина и содержащего оксид алюминия материала, который образует матрицу, такую как ультратонкий каолин, который кальцинирован через его характеристический экзотермический эффект. Водную суспензию затем высушивают распылением до получения микросферы, содержащей смесь водного каолина и/или метакаолина и каолина, который был кальцинирован по меньшей мере, главным образом, через его характеристический экзотермический эффект с образованием высокоглиноземистой матрицы. Предпочтительно, умеренное количество силиката натрия добавляют к водной суспензии, прежде чем она высушивается распылением, чтобы функционировать в качестве связующего вещества между частицами каолина.
Суспензия реактивного каолина, которая образует микросферы, может быть образована из гидратированного каолина или кальцинированного водного каолина (метакаолина) или их смеси. Водный каолин сырьевой суспензии может соответствующим образом быть либо сам по себе, либо как смесь ASP® 600 или ASP® 400 каолина, полученных из сырой руды крупнозернистого белого каолина. Водные каолины с мелким размером частиц также могут быть применены, включая те, которые получены из серых глинистых отложений, таких как LHT R пигмент. Очищенные обработанные водой каолиновые глины из Средней Джорджии были успешно применены. Кальцинированные продукты этих водных каолинов могут быть применены в качестве метакаолинового компонента сырьевой суспензии.
Новые микросферы, демонстрирующие пониженные темпы износа, главным образом производятся из смеси частиц водного каолина и частиц кальцинированного каолина. Композиция смеси состоит, как правило, из от 25 частей до 75 частей водного каолина и от 75 до 25 частей кальцинированного каолина. Частицы водного каолина диаметром приблизительно от 0.20 до 10 микрон, как измерено с помощью Sedigraph, были суспендированы в воде в диапазоне твердых частиц от 30 до 80 мас. %, как ограниченные вязкостью процесса с добавлением соответствующего диспергатора. Предпочтительно, 90 мас. % или более частиц имеют размер менее, чем 2 микрона. Кальцинированный каолин состоит из каолинита, который был нагрет мимо экзотермического преобразования кристаллической фазы, чтобы сформировать шпинель (то, что некоторые авторитетные источники именуют как дефектная алюминий-кремниевая шпинель или гамма-глинозем фаза) или муллит. В предпочтительном варианте осуществления, кальцинированный каолин или водный каолин присутствуют в количестве от 30 мас. % до 70 мас. % указанного катализатора.
Силикат для связующего вещества предпочтительно обеспеченный силикатом натрия с соотношением SiO2 к Na2O от 1.5 до 3.5, и в особенности предпочтительно соотношением от 2.88 до 3.22.
Связующее вещество затем добавляют до уровня от 0 до 20 мас. % (при измерении с SiO2) перед сушкой распылением суспензии с образованием керамических пористых бусин, которые имеют средний размер частиц от 20 до 200 мкм. Высушенные распылением бусины нагревают за эндотермическим переходом каолинита, который начинается при 550°C с образованием метакаолина. Полученные в результате микросферы затем кристаллизуют, обменивают основаниями, кальцинируют, и, как правило, но не всегда обменивают основаниями и кальцинируют второй раз.
Некоторое количество (к примеру, от 1 до 30 мас. % каолина) цеолитного инициатора также может быть добавлено к водной суспензии перед ее сушкой распылением. Используемый в данном документе термин "цеолитный инициатор" должен включать в себя любой материал, содержащий диоксид кремния и оксид алюминия, который позволяет проходить процессу кристаллизации цеолита, который произошел бы в отсутствие инициатора, или значительно сокращает процесс кристаллизации цеолита, который будет происходить в отсутствие инициатора. Такие материалы также известны как "затравка цеолита". Цеолитный инициатор или может или не может обладать обнаруживаемой с помощью рентгеновской дифракции кристалличностью.
Добавление цеолитного инициатора в водную суспензию каолина прежде чем она будет высушена распылением в микросферы упоминается в данном документе, как "внутреннее затравливание". Альтернативно, цеолитный инициатор может быть смешан с каолиновыми микросферами после того, как они образуются и до начала инициирования процесса кристаллизации, методика, которая упоминается в данном документа как "внешнее затравливание".
Цеолитный инициатор, используемый в настоящем изобретении, может быть получен из нескольких источников. К примеру, цеолитный инициатор может содержать переработанную пыль, произведенную в процессе его кристаллизации. Другие цеолитные инициаторы, которые могут быть применены, включают пыль, произведенную во время процесса кристаллизации других цеолитных продуктов, или аморфный цеолитный инициатор в растворе силиката натрия. Используемый в данном документе термин "аморфный цеолитный инициатор" означает цеолитный инициатор, который обладает не обнаруживаемой с помощью рентгеновской дифракции кристалличностью.
Затравки могут быть получены в соответствии с тем, как описано в патенте США №4,493,902. В особенности предпочтительные затравки описаны в патенте США №4,631,262.
После сушки распылением, микросферы могут быть кальцинированы непосредственно, или в качестве альтернативы промыты кислотой для дополнительного повышения ионного обмена катализаторов после кристаллизации. Процесс промывки кислотой включает совместную подачу некальцинированных, высушенных распылением микросфер и минеральных кислот в перемешанную суспензию при контролируемом рН. Скорость добавления твердых веществ и кислот подгоняют для поддержания рН от около 2 до 7, более предпочтительно от около 2.5 до 4.5 с планируемым показателем около 3 рН. Связующее вещество из силиката натрия загущают до оксида кремния и растворимой натриевой соли, которую затем фильтруют и вымывают из микросфер. Связанные силикагелем микросферы затем кальцинируют. В любом случае, кальцинирование проводят при температуре и в течение времени (к примеру, в течение двух часов в муфельной печи при температуре камеры около 1,350°F), достаточных для превращения любого гидратированного каолинового компонента микросферы в метакаолин, оставляя ранее кальцинированные каолиновые компоненты микросферы без существенных изменений. Полученные в результате кальцинированные пористые микросферы включают смесь метакаолина и каолиновой глины, кальцинированной через ее характеристический экзотермический эффект, в котором два вида кальцинированного каолина присутствуют в тех же микросферах. В качестве альтернативы, любой подходящий кальцинированный оксид алюминия может заменить каолин, кальцинированный с помощью экзотермического эффекта, как описано выше.
Y-фожазит подвергается кристаллизации путем смешивания микросфер из кальцинированного каолина с соответствующими количествами других компонентов (включая, по меньшей мере, силикат натрия и воду), как описано в патенте США 4,493,902, а затем нагревания полученной в результате суспензии до температуры и в течение времени (к примеру, до 200°-215°F в течение 10-24 часов), достаточного, чтобы кристаллизовать Y-фожазит в микросферы. Можно следовать предписаниям патента США №4,493,902, как описано.
После того, как процесс кристаллизации завершается, микросферы, содержащие Y-фожазит отделяют от, по меньшей мере, значительной части их маточного раствора, к примеру, путем фильтрации. Может быть желательным промывка микросфер путем контактирования их с водой или во время или после стадии фильтрации. Нераспределенный кремнезем поддерживается в синтетическом продукте на разных уровнях. Кремнезем образует силикагель, который придает функциональные возможности для конкретных применений конечного продукта.
Микросферы, которые фильтруются, содержат цеолит Y-фожазит в натриевой форме. Как правило, микросферы содержат более, чем около 8 мас. % Na2O. Для приготовления микросфер в качестве активных катализаторов, значительная часть ионов натрия в микросферах замещаются ионами аммония и редкоземельными ионами, или обоими.
Ионный обмен может проводиться с помощью нескольких различных методов ионного обмена. Предпочтительно, микросферы сначала обмениваются один или несколько раз с солью аммония, такой как нитрат аммония или сульфатный раствор при рН около 3. Типичный технологичный процесс замещения обменного комплекса будет иметь несколько ленточных фильтров, которые обрабатывают противоток продукта, чтобы поменять поток раствора. Количество равновесных стадий определяется с помощью общего натрия, который удаляется и оптимизацией химической стоимости. Типичный процесс включает от 3 до 6 равновесных стадий в каждом процессе замещения обменного комплекса. Ионный обмен(ы) с ионами аммония предпочтительно следует за одним или более ионных обменов с редкоземельными ионами при рН около 3. Редкоземельные могут быть представлены в виде одного редкоземельного материала или в виде смеси редкоземельных материалов. Предпочтительно, редкоземельные представлены в виде нитратов или хлоридов. Предпочтительные микросферы изобретения подвергаются ионному обмену до содержания между 0% и 12 мас. % ОРЭ, более предпочтительно от 1% до 5 мас. % ОРЭ (оксид редкоземельного элемента) и менее, чем около 0.5, более предпочтительно всего 0.1 мас. % Na2O. Как хорошо известно, промежуточное кальцинирование потребуется для достижения этих уровней карбоната натрия.
После того, как ионный обмен завершен, микросферы сушат. Могут быть применены многие сушащие конструкции, включая барабанную, аэрофонтанную и сушку распылением. Процедура, описанная выше для ионообменных микросферных ККП катализаторов настоящего изобретения является хорошо известной и, как таковой, такой процесс не образует основу настоящего изобретения.
Настоящее изобретение направлено на улучшение объема пор и сопротивление истиранию цеолит-содержащего ККП катализатора, образованного способом, описанным выше. С этой целью, суспензию водного каолина, применяемую в обеспечении реактивного питательного вещества для цеолитного образования, обрабатывают катионным полиэлектролитом и второй оксид металла, к примеру, кальцинированный каолиновый порошок, применяемый в обеспечении каталитической матрицы, обрабатывают полифосфатом перед обработкой в субстрат микросферы. Добавление катионного полиэлектролита снижает скорость истирания получаемого в результате ККП катализатора, как измерено с помощью воздушной струи (ASTM метод D5757-00) и роликовых фрикционных испытаний по сравнению с контрольными образцами, полученными без добавления полиэлектролита в том же общем объеме пор катализатора. Точный механизм, приводящий к улучшенному истиранию находится под исследованием, но полиэлектролиты известны и используются в покрытых и заполненных бумагой изделиях, требующих флокуляции и смешивания водных и кальцинированных каолиновых частиц. Считается, что добавление полиэлектролита в смесь суспензии водного каолина придает локализованную структуру через образование мелких наполнителей, которые поддерживаются с помощью этапов распылительной сушки и кальцинирования процесса образования микросферы.
Несмотря на то, что структурирование водного каолина связывается с соответствующим улучшением износа катализатора, полиэлектролиты, такие как полиамин, могут быть добавлены в любой момент в течении процесса формирования прекурсора микросфер. Порядок добавления полиэлектролита не ограничивается конкретной точкой во время процесса формирования суспензии прекурсора микросфер. К примеру, полиэлектролит можно было бы добавить к водной суспензии перед смешиванием кальцинированного каолина или он может быть добавлен после смешивания водной и кальцинированной суспензий. Конкретное описание процесса относится к применению полиэлектролита с водной и кальцинированной каолиновой суспензиями, применяемых для формирования микросферы, но не ограничивается этими материалами. Другие прекурсоры оксидов металлов могут быть применены в составе микросфер в качестве либо матричного компонента, либо матричного компонента или цеолитного питательного вещества. Эти компоненты могут потребовать введения альтернативного полиэлектролита с отличающейся ММ и катионным зарядом от тех, которые выбраны специально для составов, содержащих один водный и/или кальцинированный каолин, чтобы достичь желаемой структуры в высушенной распылением микросфере. Не ограничивающие примеры включают глинозем, гидрат оксида алюминия, оксид алюминия, кремнезем и глиноземно-кремнеземные материалы, такие как глина.
Количество катионного полиэлектролита, добавленного к каолиновой суспензии, является минимальным и, тем не менее, было обнаружено существенное улучшение в сопротивлении истиранию в готовом катализаторе. Таким образом, было обнаружено, что количества от около 0.1 до 5 фунтов или от 0.005 до 0.25 мас. % полиэлектролита на тонну сухого каолина (некальцинированный и кальцинированный) приводят к желаемым результатам. Более предпочтительно, от 0.5 до 2 фунтов на тонну или от 0.025 до 0.1 мас. % полиэлектролита относительно общего содержания каолина в пересчете на сухое вещество эффективно добавлены. Увеличение дозировок полиэлектролита более, чем 5 фунтов полиэлектролита на тонну может быть возможным, но только с соответствующим уменьшением уровня содержания твердых веществ, с тем, чтобы иметь возможность дальнейшей переработки и высушивания распылением. Следует понимать, что процент применяемого полиэлектролита основан на всех каолиновых твердых веществах, присутствующих в суспензии, которая применяется для формирования микросфер, до кристаллизации цеолитов.
Катионные полиэлектролиты, применяемые в этом изобретении известны в уровне техники как объемообразующие агенты для флокуляции водного каолина в наполнении бумаг и нанесении покрытий. Многие такие агенты также известны как флокулянты для увеличения скорости, при которой фильтруются глиняные суспензии. Смотри, к примеру, патенты США №4,174,279 и 4,767,466. Полезные катионные полиэлектролитные флокулянты включают полиамины, четвертичные аммониевые соли, диаллил аммониевые полимерные соли, диметил диаллил аммоний хлорид (полидиаллилдиметиламмония хлориды). Катионные полиэлектролитные флокулянты отличаются высокой плотностью положительного заряда, как определено путем деления общего числа положительных зарядов на молекулу молекулярной массы (ММ). ММ химических составов находится между 10,000 и 1,000,000 (к примеру, между 50,000 и 250,000) с плотностью положительного заряда, превышающего 1X10-3. Такие материалы не содержат анионных групп, таких как карбоксильные или карбонильные группы. Положительно заряженные центры, расположенные близко к концу полимерной цепи реагируют, и образуется мост с анионными участками, такими как сайты оксида алюминия, подверженного воздействию вдоль грани пластины близлежащих водных частиц, пока доступные заряженные центры глины или заряженные центры полимера не истощатся. Мостообразование усиливает связь между частицами, обеспечивая таким образом высокое сопротивление к сдвигу набухшей глинистой минеральной композиции. Присутствие ионов хлора в фильтрате в случае диметилдиаллил аммоний хлорида может быть признаком того, что, по меньшей мере, одна стадия реакции между частицами глины и соли четвертичного полимера происходит с помощью механизма ионного обмена.
Полиамины серии Kemira Superfloc С-500 представляют собой жидкости, катионные полимеры различных молекулярных масс. Они эффективно работают как первичные коагулянты и агенты, нейтрализующие заряд в процессах разделения жидкость - твердое тело в самых различных отраслях промышленности. Доступный диапазон химии гарантирует, что есть продукт, подходящий для каждого отдельного применения. У многих, если не у всех упомянутых продуктов есть цепи разветвленного полимера. MAGNAFLOC LT7989, LT7990 и LT7991 от BASF также представляют собой полиамины, которые содержатся в около 50% растворе, и применяемые в настоящем изобретении.
В дополнение к алкилдиаллил четвертичным аммониевым солям, другие катионные четвертичные аммониевые флокулянты получают путем сополимеризации алифатических вторичных аминов с эпихлоргидрином. Тем не менее, другие водорастворимые катионные полиэлектролиты являются поли(четвертичный аммоний) полиэфирными солями, которые содержат четвертичный азот в полимерной цепи и являются цепями, которые расширены эфирными группами. Их получают из водорастворимых поли(четвертичных аммониевых солей), содержащих боковые гидроксильные группы и бифункциональные реактивные агенты с продлевающими цепями; такие полиэлектролиты получают обработкой N,N,N(1),N(1)тетраалкилгидроксиалкилендиамина и органического дигалогенида, такого как дигидроалкана или дигалогенэфир с эпоксидным галогеналканом. Такие полиэлектролиты и их применение в флокулировании глины описано в патенте США №3,663,461.
В дополнение к данному изобретению, оксиды металлов, такие как каолин, которые образуют матрицу ККП катализатора, подвергают взаимодействию со структурирующим агентом в форме полифосфата, будь то твердая полифосфатная соль или жидкий полифосфат, такой как полифосфат аммония, перед образованием микросфер, а именно в соответствии с принципами США 4,493,902, описанными выше. Не наблюдается какая-либо критичность в длине полифосфатной цепи и, соответственно, ди-полифосфаты, три-полифосфатны, и более высокие полифосфатные цепи до тысячи или более полезны в данном изобретении. Смеси полифосфата и ортофосфатов, таких как фосфорная кислота, являются возможными до тех пор, пока количество компонента ортофосфорной кислоты не будет чрезмерным. Предпочтительно, содержание ортофосфорной кислоты не должно быть больше, чем 50 мас. % любой структурирующей смеси с одним или более полифосфатами. Количество ортофосфата или ортофосфорной кислоты должно быть сведено к минимуму, поскольку многие, если не все, анионно диспергированные матрицы из оксидов металлов будут флоккулировать в присутствии фосфорной кислоты, и не обеспечивает найденный эффект структурирования. Флокуляция также в значительной степени затрудняет обработку этих материалов. Особенно предпочтительный класс структурирующих агентов представляет собой аммониевые полифосфаты, которые являются часто растворимыми в воде и являются жидкими, чтобы быть легко обработанными жидкими растворами оксидов металлов, которые будут обрабатывать. Особую полезность представляет собой жидкий фосфат аммония, к примеру, жидкие удобрения, такие как 11-37-0 и 10-34-0 и тому подобные, которые имеют доступное содержание фосфата от около 33% до 37%, содержание ортофосфата около 27% и содержание полифосфата около 65-75%. Этот материал 100% растворимый в воде.
Количество полифосфатного структурирующего агента, добавленного к прекурсору матрицы из оксида металла, такого как водный каолин, который подлежит обработке, чтобы обеспечить внутреннее структурирование, является минимальным. Таким образом, количество добавленного полифосфата относительно твердых веществ из оксидов металлов (каолин) может находиться в диапазоне всего лишь от 0.01 до 5 мас. %. Более конкретно, количество полифосфата будет в диапазоне от около 0.05 до 2 мас. % и, более конкретно, от около 0.05 до 0.5 мас. %. Было обнаружено, что даже эти небольшие количества полифосфата могут обеспечить значительные изменения в площади поверхности и внутреннем объеме пор оксида металла относительно необработанных материалов. В общем, процесс обработки оксида металла с целью добавить к нему внутреннее структурирование включает суспендирование оксида в воде и смешивание полифосфатного структурирующего агента в жидкой форме с водной суспензией оксида металла. Может быть включена суспензия диспергатора, такая как гидроксид натрия, карбонат натрия, полиакрилат натрия, силикат натрия, тетра-пирофосфат натрия, метасиликат натрия, гексаметафосфат натрия, и/или натрия три-полифосфат. Как было отмечено выше, конкретные полифосфаты, такие как полифосфат аммония, находятся в жидкой форме и могут быть просто добавлены к суспензии. Другие соли полифосфатов могут требовать растворения в растворителе. Поскольку смесь подвергают распылительной сушке под вакуумом или нагреванию и, как правило, требуется дополнительный этап нагрева для обеспечения структурирования, растворитель, вместе с тем представляющий собой предпочтительно воду, может быть органическим растворителем, который будет испаряться либо в процессе распылительной сушки и будет полностью удален во время любого последующего процесса нагревания.
Распылительная сушка суспензионной смеси дает в результате смесь частиц оксида металла и агента структурирования полифосфата. Содержание влаги снижается ниже 5.0 мас. %, как правило, ниже 2.0 мас. %. Вслед за распылительной сушкой, может быть применен этап размельчения для разрушения высушенных распылением частиц в обеспечение однородной смеси оксида металла и структурирующего агента. Последующее нагревание приводит к реакции полифосфата с оксидом металла и формированию новой внутренней структуры в оксиде, обеспечивая дополнительный объем пор и более высокую площадь поверхности. Дальнейшее нагревание для закрепления материала приводит к дополнительной твердости и к новому дополнительному объему пор, площадь поверхности обработанного оксида не так резко снижается, как у необработанных материалов. Следует понимать, что в то время, как сушку распылением предпочтительно применяют для получения смеси металлического оксида, подлежащего обработке и полифосфатного структурирующего агента, могут быть использованы другие способы смешивания, как хорошо известно в области техники смешивания. Могут быть использованы другие методы сушки, к примеру, распылительный гранулятор, ультразвуковой распылитель, распылитель Lamrot (laminar operating rotary atomizer) и т.д.
Катионный полиэлектролит и полифосфат могут быть добавлены к отдельным суспензиям оксидов металлов или, в случае полиэлектролита, к смешанной водной и кальцинированной суспензии. Затем суспензии могут быть высушены с применением различных доступных способов обработки, таких как распылительная сушка, для формирования микросфер, которые затем могут быть обработаны так, чтобы инициировать рост цеолита. Катионные обработанные полиэлектролитом суспензии формируют питательную основу для роста цеолита, в то время как обработанная полифосфатом суспензия представляет собой матрицу полученного катализатора ККП.
Способ изготовления
Способ согласно настоящему изобретению может теперь быть более конкретно описан для обработки отдельных партий водного каолина, который применяется для формирования микросфер, которые применяют для кристаллизации цеолита. Следует понимать, что процесс обработки оксидов металлов, в то время как по существу одинаков для всех типов оксидов металла, может иметь конкретные детали, которые отличаются от обработки каолина. Частицы водного каолина диаметром приблизительно от 0.20 до 10 микрон суспендируют отдельными партиями в воде в диапазоне твердых частиц от 30 до 80 мас. %, как ограниченные вязкостью процесса. Более типично, суспензия будет содержать 40-70 мас. % твердых частиц водного каолина и, еще более, от 50 до 65 мас. % твердых частиц каолина в воде. Могут быть приготовлены суспензии комнатной температуры, несмотря на то, что суспензия может быть нагрета до 150°F, если необходимо, до введения в распылительную сушилку. Смешанным с одной из водных каолиновых суспензий является полифосфат, к примеру, жидкий полифосфат аммония, к примеру, сорт удобрения полифосфат аммония, такой как 11-37-0, 10-34-0 и тому подобные. Приблизительно, от 0.01 до 5 мас. % полифосфата аммония в качестве доступного содержания фосфата может быть смешано с водной суспензией каолина относительно твердых частиц каолина. Предпочтительно, количество полифосфатного структурирующего агента будет в нижней части указанного диапазона, обычно от около 0.01 до 0.2 мас. % в качестве доступного содержания фосфата и, более предпочтительно, около 0.15 мас. % в качестве доступного содержания фосфата, относительно твердых частиц каолина. Смесь водной суспензии водного каолина и жидкого полифосфата аммония подвергают распылительной сушке в обычном оборудовании распылительной сушки. Распылительная сушка может быть выполнена в вакууме или атмосферном давлении при температурах от около 70°F до 550°F для удаления воды.
Размер полученных распылительной сушкой частиц содержит смесь оксидов металла, таких как водный каолин, глинозем, и т.д. или их смесь с полифосфатом аммония, как правило, будет находиться в диапазоне от около 20 до 200 микрон. Перед нагреванием смесь частиц, высушенных распылением подвергают реакциям структурирования, что может быть полезным для измельчения частиц распылительной сушкой в порошок, чтобы обеспечить более однородную смесь полифосфата аммония и оксида металла. Затем смесь может быть нагрета в воздухе в любой обжиговой печи. При повышении температуры, полифосфат аммония разлагается при или выше 350°F. Продукты разложения являются преимущественно полифосфорной и ортофосфорной кислотой. Так как нагрев продолжается, оксид металла будет преобразован в другую кристаллическую форму. К примеру, в отношении каолина, водный каолин преобразуется в метакаолин, на котором оксид алюминия в каолиновой решетке становится химически активен. Фосфатные материалы реагируют с алюминиевыми участками в каолине для формирования нового структурирования внутри частицы каолина. Считается, что постепенно нарастающая структура внутри частицы каолина, скорее всего, создается из-за полифосфорной кислоты, реагирующей с присутствующим химически активным оксидом алюминия, формируя алюминий/фосфатные мосты, в то время как продукты находятся в метакаолиновой фазе.
Разложение полифосфатов при низкой температуре дает реакцию структурирования, которая имеет место, когда каолин переходит в метакаолиновую фазу. Есть неожиданные преимущества для процесса, который эффективно расширяет уровень техники кальцинирования каолина. Первое преимущество заключается в создании дополнительной площади поверхности и объема пор в решетке каолина. Эту степень структурирования можно контролировать, делая решетку более абсорбирующей. Второе, реакция полифосфата позволяет проводить структурирование при низкой температуре, что значительно ниже пороговой температуры, где метакаолин подвергается решетчатой реконфигурации в шпинель и муллит. Вновь созданная структура может служить в качестве поглотителя для сбора диоксида кремния, удаленного при переходах метакаолина в шпинель и муллит с постепенно нарастающей температурной обработкой. Это преимущество значительно расширяет возможности управления процессом кальцинирования и может воздействовать на применение потоков (т.е. силикат натрия, борат натрия или тому подобные), которые понижают температуру, при которой переходы шпинели и муллита имеют место для создания уникальных решетчатых структур.
Затем каолин нагревают до температуры выше характерного экзотермического эффекта, т.е., 2000-2200°F, для формирования фаз шпинели и муллита. Предпочтительно, кальцинирование продолжается до содержания муллита около 40%. Таким образом, твердость по Моосу продукта кальцинированного каолина может быть увеличена с обычного диапазона 4.5 до 6.5 без образования агломератов. Важно отметить, что объем пор в пределах каолина не столь резко снижается по сравнению с необработанным каолином, который превращается в значительной степени в муллитовую фазу. Кальцинированный продукт измельчали снова, чтобы сделать порошок и хранить.
Отдельную суспензию водной глины, эквивалентную той, которая описана выше, обрабатывают примерно 1 фунтом полиамина/сухая тонна каолина (сухого) при сдержанном перемешивании. Если вязкость по Брукфильду высока, то вязкость может быть уменьшена с помощью полиакрилатного диспергирующего вещества средней молекулярной массы. Альтернативный вариант осуществления заключается в обработке перемешанной суспензии, содержащей оба, как обработанную водосодержащую глину, так и ПФА, кальцинированные промежуточные компоненты.
Обработанную полиамином суспензию (от 30% до 70%, в пересчете на общую массу твердых частиц каолина, предпочтительно от 48 до 60%) смешивают с порошком полифосфата аммония АПФ (от 30% до 70%, предпочтительно от 40 до 52%) с образованием суспензии. Объединенную суспензию подвергают распылительной сушке, при этом линейно впрыскивают с 8 мас. % силиката натрия (3.22 модуля = SiO2/Na2O в качестве связующего вещества). Высушенный распылением шарик сформирован непосредственным впрыском в обжиговую печь при температуре продукта между 1400-1600°F для преобразования водного каолина в реакционный метакаолин. Температура кальцинирования контролирует уровень сформированного метакаолина. Метакаолин представляет собой реактивный агент для роста цеолита. Содержание метакаолина предназначено для получения желаемого содержания цеолита, обычно между 40 до 60 мас. %, в перерасчете на общую массу микросфер.
После того, как подходящая микросфера приготовлена, ее помещают в реактор для кристаллизации и суспендируют с раствором силиката натрия, раствором гидроксида натрия, инициатором кристаллизации (затравка) и количеством воды. Количество реагирующих веществ определяется серией соотношений реактивных компонентов, выраженных как Si2O/Na2O (обычно 2.0-3.0), H2O/Na2O (обычно 5.0-9.0), SiO2/Al2O3 (обычно 5.0-8.0) и массовым соотношением затравки к реактивному оксиду алюминия в системе (обычно 0.002-0.003). Перемешиваемую смесь затем нагревают до 210°F в течение периода обычно 10-20 часов до тех пор, пока кристаллизация цеолита не прекращается, как определено с помощью химического теста реактивного раствора, индицирующего содержание Al2O3<0.05%. В качестве альтернативы, испытание твердых частиц дифракцией рентгеновских лучей ("ДРЛ") для определения содержания цеолита Y ("ZY") в образцах, взятых на один час из реакционной смеси не показывает увеличение содержания ZY, что указывало бы на завершение реакции. Полностью кристаллизованный продукт Na-Y затем отделяют фильтрованием от жидкой фазы и промывают для удаления остаточного силиката натрия. Затем промытые твердые вещества подвергают ионному обмену для удаления натрия, применяя ряд ионных обменов с раствором соли аммония в комбинации с кальцинированием с получением конечного продукта почти свободного натрия. Часто, уровень редкоземельных элементов водится во время процесса ионного обмена (~1-5% в качестве ОРЭ на катализатор) для улучшения гидротермальной стабильности. Типичная характеристика продукта состоит из измерения площади поверхности путем многоточечного анализа БЭТ, общего химического состава с помощью РФА, а также анализа объема пор под высоким давлением проникновения ртутного столба, с применением инструмента порозиметра для того, чтобы определить общий объем пор в см3/г и распределение пор по размерам по сравнению с радиусом пор. Каталитическое тестирование может быть сделано путем крекинга стандартного минерального масла над катализатором для определения активности и селективности к нефтепродуктам, таким как бензин.
Хотя приведенное выше описание относится к катализаторам ККП, содержащим цеолит, образованный in situ из микросфер, содержащих метакаолин, полагают, что улучшения в сопротивлении истиранию может быть достигнуто с помощью данного изобретения, в котором цеолит включен в матрицу. В таком случае водный каолин, обработанный катионным полиэлектролитом может быть смешан с кальцинированным каолином, образованным из каолина, обработанного полифосфатом, но любое нагревание комбинированных материалов было бы таким, что не преобразовывает водный каолин в метакаолин. В таком случае, водный каолин теперь будет являться частью матрицы. Кристаллы цеолита могут быть добавлены в один или оба суспензию водного каолина или порошок кальцинированного каолина. Связующее вещество, известное в уровне техники для введения цеолита в матрицу материалов, может также быть добавлено. Катализатор согласно изобретению предпочтительно имеет мезо/макро соотношение от 0.65 до 1.2, более предпочтительно от 0.675 до 1.1. Катализатор согласно изобретению предпочтительно имеет интенсивность расходования воздушной струи, по меньшей мере, 0.5 и менее, чем 2.5, более предпочтительно по меньшей мере 0.5 и менее, чем 1.5.
Пример 1
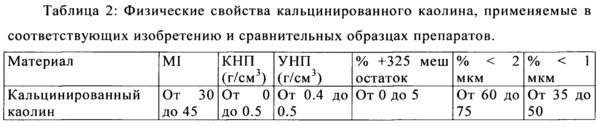
Этот пример и соответствующие данные были получены и описаны в принадлежащий одному и тому же правообладателю заявки США с серийным номером №13/042,790. Способы и результаты, которые упоминаются здесь, приведены для сравнительных целей. Использовали суспензию водного каолина, состоящую из частиц с более, чем 70% имеющими эквивалентный сферический диаметр менее, чем 2 мкм как измерено с помощью Sedigraph 5200 и менее, чем 0.5% частиц, захваченных на 325-меш экране. Водный каолин в количестве 37.5 сухой мас. % смешивали с кальцинированным каолином в количестве 62.5 сухой мас. % для получения пяти образцов согласно изобретению, каждый из которых имел общий уровень твердых частиц суспензии ~50 мас. %. Физические свойства каолинов показаны в Таблицах 1 и 2.
Внедренный кальцинированный каолин состоял из материала, который был нагрет за пределами характерного экзотермического преобразования при ~950°C с образованием шпинели, муллита или комбинации шпинели и муллита. Индекс муллита (МИ) представляет собой соотношение пика муллита в образце каолина до 100% муллитового эталонного образца с указанием степени термической обработки для кальцинированного каолина как измерено РДА на инструменте Panalytical Cubix Pro. Кажущаяся насыпная плотность (КНП) представляет собой вес на единицу объема материала, в том числе истинное объемное паросодержание. Утрамбованная насыпная плотность (УНП) является мерой объемной плотности следующей входной работы в целях стимулирования более плотной упаковки частиц, измеренной с помощью Волюметра TAP-PACK Volumeter (ISO 787-11).

Superfloc C577 (катионный полиамин) разводили до содержания 1% твердых веществ в воде и добавляли в глинистые суспензии изобретения при дозировке 1.0 сухой фунт полимера на тонну сухой глины. Стандартный пневматический миксер был применен во время добавления. Силикат натрия сорт #40 (3.22 модулей или 3.22 частей SiO2 на 1.0 часть Na2O) затем добавляют в качестве связующего вещества к смеси при дозировке 4 мас. % на основе SiO2. Альтернативно, количество связующего вещества может варьироваться от 0 мас. % до 20 мас. % на основе SiO2 добавления силиката натрия. Суспензию изобретения затем высушивают распылением до образования микросфер со средним размером частиц (СРЧ) от 80 до 90 микрон, как измерено с помощью лазерного анализа размера частиц (Microtrac SRA 150). Другие способы сушки будут одинаково эффективны для снижения влажности продукта до уровня ниже 2 мас. % (как измерено СЕМ Labwave 9000 анализатором влажности). Полученные в результате микросферы были кальцинированы в лабораторной печи при 815°C (1500°F) в течение 1 часа.
Пять сравнительных образцов также были получены из таких же исходных каолиновых компонентов, как и изобретательные образцы, применяя ту же самую процедуру, за исключением того, что катионный полиамин был опущен и концентрация связующего вещества из силиката натрия была увеличена с целью поддержания целостности микросферы перед кристаллизацией (8 мас. % на основе SiO2 к каолину, который был добавлен к материалу). В случаях Сравнительных образцов IV и V, соотношение водного к кальцинированному использованному каолину было изменено до 40:60 соответственно. Каждый из изобретательского и сравнительного образцов были получены с различными количествами питательных метакаолиновых микросфер для создания различного общего объема пор микросферы, с целью продемонстрировать улучшение наблюдаемого истирания по отношению к переменному общему объему пор.
После образования микросферы, кристаллизацию цеолита проводили на изобретательских образцах и сравнительных образцах, как это показано в Таблицах 3 и 4, где затравками были мелкие алюмосиликатные частицы, применяемые для инициирования кристаллизации цеолита и роста. Силикат натрия с составом 21.6 мас. % SiO2 и 11.6 мас. % Na2O (1.87 модулей, как определено в виде частей SiO2 на части Na2O) был переработан и получен из коммерческого производства микросфер. Питательные микросферы, состоящие в основном из метакаолина, который является растворимым в основной кристаллизационной среде, служат в качестве источников питательных веществ для продолжения роста цеолита Y. Затравки, применяемые для инициирования кристаллизации цеолита описаны в Патенте США №4,493,902, и Патенте США №4,631,262, которые включены в настоящую заявку посредством ссылки.
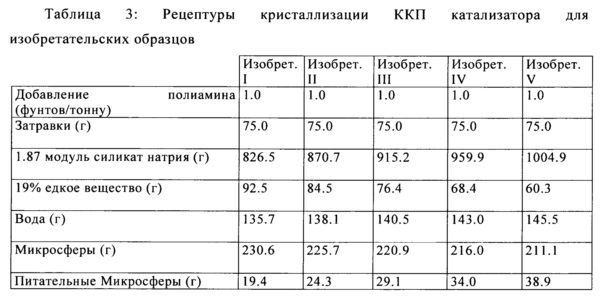
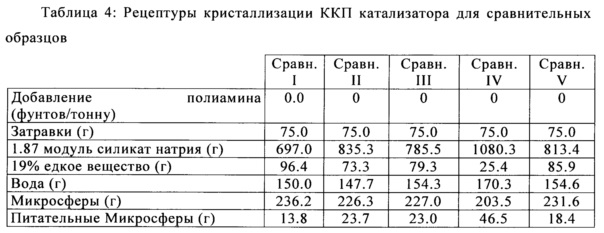
Y-фожазиту позволяли кристаллизоваться путем смешивания кальцинированного каолина микросфер с соответствующими количествами других составляющих (включая, по меньшей мере, силикат натрия и воду), как описано в Патенте США №5,395,809, идеи которого включены в настоящее изобретение посредством ссылки, и затем полученную в результате суспензию нагревали до температуры от 200° до 215°F в течение 10-24 часов, в достаточной мере, чтобы кристаллизовать Y-фожазит в микросферы. Микросферы кристаллизуют до требуемого содержания цеолита (как правило, приблизительно до 50-65), фильтруют, промывают, проводят аммониевый ионный обмен, проводят аммониевый ионный обмен с редкоземельными катионами, кальцинируют, проводят ионный обмен второй раз с ионами аммония, и кальцинируют второй раз. В публикации США №2012/0228194 изложена эта процедура, и она включена в настоящую заявку посредством ссылки.
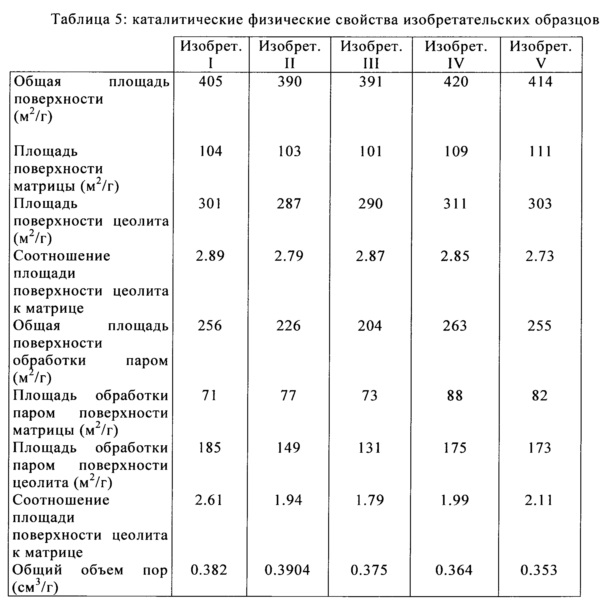
В Таблице 5 и Таблице 6 приведен список физических свойств полученных в результате изобретательских и сравнительных образцов после кристаллизации и последующих циклов ионного обмена и кальцинации. Общая площадь поверхности (ОПП), площадь поверхности матрицы (ППМ), и площадь поверхности цеолита (ППЦ) были определены с помощью BET анализа изотермы абсорбции азота, с применением приборов Micromeritics TriStar или TriStar 2. В то время как образцы, образованные в этом примере, дают катализаторы повышенной активности /повышенной площадью поверхности, изобретение в данном описании не предназначено для ограничения по площади поверхности или каталитической активности образованного катализатора. Это изобретение осуществляет улучшение в сопротивлении истиранию независимо от активности катализатора.
После первоначального тестирования производимых катализаторов, осуществляли выпаривание для имитации неактивных или равновесных физических свойств катализатора из нефтеперерабатывающего завода. Процесс состоит из обработки паром катализатора при 1500°F в течение 4 или более часов. Пористость катализатора определяли методом ртутной порометрии с применением Micromeritics Autopore 4. Общий объем пор представляет собой суммарный объем пор, имеющих диаметр в диапазоне от 35 до 20,000 Å.
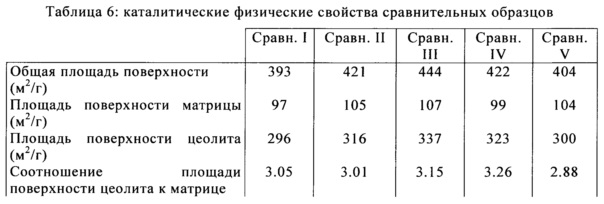
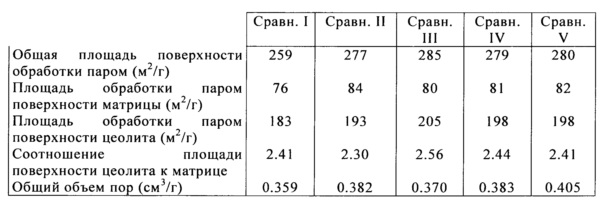
Истирание струей воздуха было измерено для изобретательских и сравнительных образцов против объема пор. Значения скорости истирания струей воздуха измеряли с применением прибора внутреннего пользования в соответствии с ASTM стандартным методом D5757. Как правило, для данного способа производства катализатора и композиции, скорость истирания возрастает, как повышается пористость частиц данного катализатора. Добавление полиэлектролитного флокулянта до образования микросферы изменяет укладку получающихся в результате частиц до формирования микросферы, приводящей к керамической структуре, которая является более стойкой к истиранию. В частности, получающаяся в результате новая каталитическая структура обладает повышенной стойкостью к истиранию, получающегося в результате воздействия механизма разрушения абразивного типа (истирание мелких частиц по отношению к общему размеру исходной частицы). На Фигуре 1 показано наблюдаемое снижение скорости истирания для изобретательских образцов (показаны в виде ромбов) при эквивалентных или повышенных общих объемах пор, чем у сравнительных образцов (показаны в виде квадратов).
Пример 2
Суспензия водного каолина, состоящая из частиц, более чем 79% которых имеют эквивалентный сферический диаметр менее, чем 2 мкм, как измерено Sedigraph 5200 и менее, чем 0.5% частиц захваченных на используемом 325 ячеечном экране. Водный каолин в количестве от 46 до 52 сухой мас. % смешивают с кальцинированным каолином в количестве от 48 до 54 сухой мас. % для получения шести изобретательских образцов, каждый из которых имеет общий уровень содержания твердых веществ в суспензии ~50 мас. %.
Компоненты кальцинированного каолина для обоих и изобретательских и сравнительных образцов были образованы из того же источника суспензии водного каолина. Тем не менее, для изобретательских образцов 37.0 мас. % раствор полифосфата аммония был добавлен в суспензию при 0.15 сухой мас. % как доступный фосфат водного каолина перед кальцинированием. Для получения сравнительных образцов кальцинированный каолин предварительно не обрабатывали фосфатом. Физические свойства указанных компонентов, связанных с водным и кальцинированным каолином детализированы в Таблице 1 Примера 1 и Таблице 7 ниже.
Введенный кальцинированный каолин состоял из материала, который был нагрет без характерного экзотермического перехода при ~950°C с образованием шпинели, фазы муллита или комбинации шпинели и муллита.

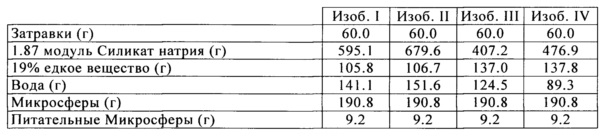
Superfloc С577 (катионный полиамин) был разбавлен до 1% твердых веществ в воде, и затем добавлен в изобретательские образцы I-IV при дозах 1.0 сухой фунт полимера на тонну сухой глины с применением стандартного пневматического миксера. Силикат натрия сорта номер 40 (3.22 модуль или 3.22 частей SiO2 на 1.0 часть Na2O) затем добавили в качестве связующего вещества в смесь при дозировке 4 мас. % на основе SiO2. Альтернативно, количество связующего вещества может варьироваться от 0 мас. % до 20 мас. % на основе SiO2 добавления силиката натрия. Изобретательскую суспензию затем высушивают распылением до образования микросфер со средним размером частиц (СРЧ) от 80 до 90 микрон, как измерено с помощью лазерного анализа размера частиц (Microtrac SRA 150). Другие способы сушки будут одинаково эффективны для снижения влажности продукта до уровня ниже 2 мас. % (как измерено с помощью анализатора влажности СЕМ Labwave 9000). Полученные в результате микросферы были кальцинированны в лабораторной печи при 815°C (1500°F) в течение 1 часа.
Два сравнительных образца были получены из тех же каолиновых исходных компонентов, с применением той же процедуры, за исключением того, что были опущены компоненты катионного полиамина и полифосфата. Концентрация связующего вещества из силиката натрия была одинаковой для обоих и изобретательского и сравнительного образцов (8 мас. % на основе SiO2 к каолину). Оба и изобретательский и сравнительный образцы были произведены с различными количествами питательных метакаолиновых микросфер для создания финальных каталитических образцов с различными значениями общего объема пор, с целью продемонстрировать улучшение истирания, наблюдаемое в диапазоне общего объема пор.
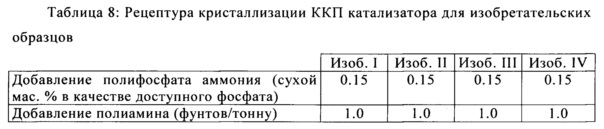
После образования микросфер, кристаллизацию цеолита проводили с применением следующих композиций для изобретательских образцов, показанных в Таблице 8, где затравками были мелкие алюмосиликатные частицы, применяемые для инициирования кристаллизации цеолита и роста. Силикат натрия с композицией 21.6 мас. % SiO2 и 11.6 мас. % Na2O (1.87 модуль, как приведено в виде частей SiO2 на части Na2O) были переработаны и выработаны из коммерческого производства микросфер. Питательные микросферы, состоящие в основном из метакаолина, которые являются растворимыми в основной кристаллизационной среде, служат в качестве питательных веществ для продолжения роста цеолита Y. Затравки, применяемые для инициирования кристаллизации цеолита, описаны в патенте США №4,493,902, и патенте США №4,631,262, которые включены в данную заявку посредством ссылки.

Y-фожазиту позволяли кристаллизоваться путем смешивания микросфер кальцинированного каолина с соответствующими количествами других компонентов (включая, по меньшей, мере силикат натрия и воду), как описано в Патенте США №5,395,809, учения которого включены в настоящую заявку посредством ссылки, и затем нагревания полученной в результате суспензии до температуры от 200° до 215°F в течение 10-24 часов, достаточных для кристаллизации Y-фожазита в микросферы. Микросферы кристаллизовали до требуемого содержания цеолита (как правило, приблизительно 50-65), фильтровали, промывали, подвергали ионному обмену с ионами аммония, подвергали ионному обмену с редкоземельными катионами, кальцинировали, подвергали второй раз ионному обмену с ионами аммония, и кальцинировали второй раз. В публикации США №2012/0228194 изложена эта процедура, и она включена в настоящую заявку посредством ссылки.
В Таблице 10 и Таблице 11 приведен список физических свойств полученных в результате изобретательских и сравнительных образцов после кристаллизации и последующих циклов ионного обмена и кальцинации. Общая площадь поверхности (ОПП), площадь поверхности матрицы (ППМ), и площадь поверхности цеолита (ППЦ) были определены с помощью БЭТ анализа изотермы абсорбции азота, с применением приборов Micromeritics TriStar или TriStar 2. В то время как образцы, образованные в этом примере, дают катализаторы повышенной активности /повышенной площади поверхности, изобретение в данном описании не предназначено для ограничения по площади поверхности или каталитической активности образованного катализатора. Это изобретение осуществляет улучшение в сопротивлении истиранию независимо от активности катализатора.
После первоначального тестирования производимых катализаторов, осуществляли выпаривание для имитации неактивных или равновесных физических свойств катализатора из нефтеперерабатывающего завода. Процесс состоит из обработки паром катализатора при 1500°F в течение 4 или более часов. Пористость катализатора определяли методом ртутной порометрии с применением Micromeritics Autopore 4. Общий объем пор представляет собой суммарный объем пор, имеющих диаметр в диапазоне от 30 до 10,000 Å.
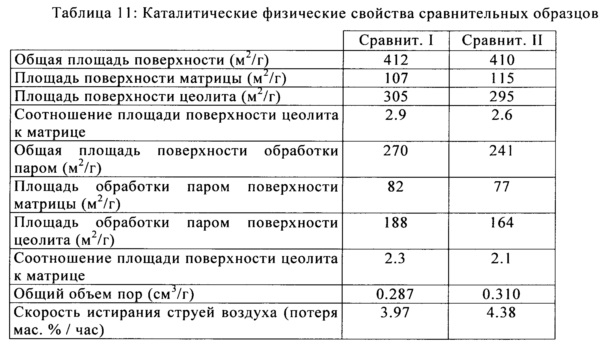
Фигура 2 представляет собой сравнение объема мезопор с объемом макропор против истирания для сравнительных каталитических образцов, показанных в квадратах и изобретательских катализаторов, образованных с помощью способа настоящего изобретения, показанных в виду ромбов. Соотношение мезо/макро описывается как суммарный объем пор (см3/г), измеренный с помощью ртутной порометрии для пор, имеющих радиус 30-100 Å, деленный на суммарный объем пор (см3/г) для пор, имеющих радиус 100-10000 Å.
Как можно видеть, катализаторы, образованные в соответствии с настоящим изобретением имеют более высокий объем мезопор, чем сравнительные каталитические образцы, и существенно улучшенное сопротивление истиранию. Обратившись к Фигуре 4 можно заметить, что улучшенное сопротивление истиранию не является результатом снижения объема пор, так как катализаторы настоящего изобретения имели более высокий объем пор, чем коммерческие катализаторы. Фигура 3 иллюстрирует распределение объема пор катализаторов, образованных в соответствии с настоящим изобретением, при этом изобретательские образцы с I по IV показаны в сравнении со сравнительными образцами с I по II. Как было отмечено на Фигуре 2, изобретательские катализаторы демонстрируют повышенную мезопористость по сравнению с макропористостью. Распределение объема пор на Фигуре 3 отражает этот сдвиг в распределении с каждым из изобретательских примеров, имеющих увеличенную общую мезопористость и уменьшение в размере пор на пике макропористости. Эти изменения в размере пор наблюдаются в каждом из катализаторов, образованных в соответствии с настоящим изобретением. Распределение объема пор в области макропор может быть подогнано с помощью других средств, таких как изменение содержания метакаолина и/или содержания силиката/затравки, но распределение объема пор в области мезопор остается по существу таким же, независимо от вариаций в распределении объема макропор. Показанные преимущества изобретательской технологии состоят в улучшенных показателях производительности катализаторов при истирании, в то же время, имея достаточный общий объем пор, каталитическая производительность не ухудшается в виду ограничений массовой диффузии. Кроме того, также было обнаружено, что путем изменения соотношения полиамина к полифосфату аммония, пики радиуса пор в областях мезо- и макропор могут быть сдвинуты, предлагая дополнительную гибкость в физических каталитических свойствах, которые влияют на истирание и параметры каталитической производительности.
Пример 3
Фигура 4 сравнивает скорости истирания струей воздуха сравнительных ККП катализаторов, представленных в виде квадратов, в том числе коммерческого катализатора, полученного в соответствии с учениями Патента США №6,943,132, по сравнению с изобретательскими образцами, отмеченные данными, представленными в виде точек. Продемонстрированные сравнительные и изобретательские образцы были получены в соответствии с процедурами, описанными в Примере 2, но не из образцов, как описано в Примере 2. В частности, на Фигуре 4, для изобретательских образцов полифосфат аммония добавляли при дозировке 0.15 мас. % в качестве доступного фосфата к суспензии водного каолина перед образованием кальцинированного каолина, применяемого в последней стадии смешивания. Полиамин добавляли к смеси водного и кальцинированного каолина при дозировке 1 фунт/тонну. В каждом изобретательском примере, количество обработанного полиамином каолина в микросфере составило от 48 до 52 мас. % и обработанного полифосфатом каолина составило от 52 до 48 мас. % от общего содержания каолина микросферы. Как показано на Фигуре 4, изобретательский катализатор имеет больший общий ртутный объем пор, чем сравнительный катализатор, и, тем не менее, сопротивление истиранию значительно улучшается по сравнению с коммерческим катализатором и поддерживается на значительно более высоком уровне объема пор по отношению к катализатору, обработанному только полифосфатом. Кроме того, сравнивая Фигуру 4 с Фигурой 1, можно видеть, что катализатор, обработанный только полиамином, дает меньшие улучшения по сопротивлению истиранию в сравнении с комбинированной обработкой по настоящему изобретению.
Настоящее изобретение относится к новым катализаторам каталитического крекинга с псевдоожиженным слоем катализатора, содержащим микросферы, и к способу каталитического крекинга с псевдоожиженным слоем катализатора. Мезопористый катализатор включает: 1) смесь кальцинированного каолина, модифицированного с помощью полифосфата путем нагревания, с водным каолином, обработанную катионным полиэлектролитом; и 2) активный цеолитный крекирующий компонент, содержащий цеолит Y, при этом указанный катализатор находится в форме частиц, которые имеют средний размер от 20 до 200 микрон в диаметре. Комбинированная обработка полифосфатом и катионным полиэлектролитом дает неожиданные улучшения в сопротивлении истиранию, сохраняя при этом высокий общий объем пор, даже когда соотношение объема мезопор к объему макропор образованного катализатора увеличивается. 2 н. и 5 з.п. ф-лы, 4 ил., 11 табл., 3 пр.
1. Мезопористый катализатор для каталитического крекинга углеводородов с псевдоожиженным слоем катализатора, включающий: 1) смесь кальцинированного каолина, модифицированного с помощью полифосфата путем нагревания, с водным каолином, обработанную катионным полиэлектролитом; и 2) активный цеолитный крекирующий компонент, содержащий цеолит Y, при этом указанный катализатор находится в форме частиц, которые имеют средний размер от 20 до 200 микрон в диаметре.
2. Мезопористый катализатор по п. 1, отличающийся тем, что указанный катионный полиэлектролит выбран из группы, которая состоит из полиаминов, четвертичных аммониевых солей, солей диаллил аммониевого полимера, диметил диаллил хлорида аммония и их смесей.
3. Мезопористый катализатор по п. 1, отличающийся тем, что указанный кальцинированный каолин или указанный водный каолин присутствует в количестве от 30 до 70 мас. % указанного катализатора.
4. Мезопористый катализатор по п. 1, отличающийся тем, что указанный кальцинированный каолин структурирован с помощью указанного полифосфата в количестве от 0,01 до 5 мас. % указанного кальцинированного каолина.
5. Мезопористый катализатор по п. 1, отличающийся тем, что указанный водный каолин обработан указанным катионным полиэлектролитом в количестве от 0,005 до 0,25 мас. % кальцинированного каолина и водного каолина.
6. Мезопористый катализатор по п. 1, отличающийся тем, что указанный катализатор имеет мезо/макро соотношение от 0,65 до 1,2.
7. Способ крекинга углеводорода в условиях каталитического крекинга с псевдоожиженным слоем катализатора (ККП) с указанным мезопористым катализатором по п. 1.
| КАТАЛИЗАТОР КРЕКИНГА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2367518C2 |
| МИКРОСФЕРИЧЕСКИЙ КАТАЛИЗАТОР ДЛЯ КРЕКИНГА НЕФТЯНЫХ ФРАКЦИЙ И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2011 |
|
RU2473385C1 |
| US 4493902 A1, 15.01.1985 | |||
| US 6943132 B2, 13.09.2005 | |||
| US 20130098804 A1, 25.04.2013 | |||
| US 20030199386 A1, 23.10.2003 | |||
| US 20120228194 A1, 13.09.2012. | |||

