Родственные заявки
Данная заявка является заявкой в частичное продолжение патентной заявки Соединенных Штатов № 11/023971, поданной 28 декабря 2004 г., которая представляет собой заявку - частичное продолжение патентной заявки Соединенных Штатов № 10/696389, зарегистрированной 29 октября 2003 г., которая заявляет преимущество патентной заявки Соединенных Штатов № 60/421923, поданной 29 октября 2002 г.
Введение
Некоторые технологии замедленного высвобождения, подходящие для введения ингаляцией, используют липосомы и липидные комплексы для обеспечения пролонгированного терапевтического эффекта лекарственного средства в легких и системно посредством замедленного высвобождения и способности нацеливать лекарственное средство и повышать поглощения лекарственного средства в патологических очагах. Настоящее изобретение включает в себя липосомальный противоинфекционный агент и способы лечения легочных инфекций с использованием липосомального или представляющего собой липидные комплексы противоинфекционного агента.
Как сообщается в Goodman and Gilman, The Pharmaceutical Basis of Therapeutics, восьмое издание, «Поскольку частота нефротоксичности и ототоксичности связана с концентрацией, до которой накапливается аминогликозид, критически важным является уменьшение поддерживающей дозы указанных лекарственных средств у пациентов с нарушенной функцией почек». Поскольку аминогликозиды могут вызывать дисфункцию вестибулярного аппарата или слуха и нефротоксичность, независимо от нарушенных функций пациента, важным является, главным образом, уменьшение поддерживающих доз. Настоящее изобретение обеспечивает резкое уменьшение токсичности, что позволяет использовать более высокие дозы, чем обычно.
У пациентов, страдающих муковисцидозом (CF), наблюдается секреция густой слизи и/или мокроты в легких, частые, как следствие, инфекции, и биопленки, образующиеся в результате бактериальной колонизации. Все указанные жидкости и материалы создают барьеры для эффективного воздействия противоинфекционных агентов на инфекции. Настоящее изобретение преодолевает указанные барьеры и даже позволяет уменьшать дозирование (по количеству или по частоте), что уменьшает лекарственную нагрузку на пациентов. В случае, главным образом, легочных инфекций, схема дозирования по настоящему изобретению обеспечивает средство уменьшения лекарственной нагрузки.
В случае липосомальной системы доставки лекарственного средства зачастую необходимо как можно больше понизить соотношение липид-лекарственное средство (L/D) для минимизации липидной нагрузки, чтобы избежать сатурационных эффектов в организме. В случае ингаляционной доставки в легкие это может быть особенно актуальным из-за постоянного применения, введение липосом может опережать клиренс, что ограничивает введение и, таким образом, эффективность лекарственного продукта. Более низкое соотношение L/D позволит вводить больше лекарственного средства, прежде чем будет достигнут порог дозирование/клиренс.
Сущность изобретения
Посредством способов инфузии, описанных в настоящем документе, были созданы липосомы, практически не содержащие анионных липидов среднего размера (<1 мкм), которые удерживают противоинфекционные агенты в массовом соотношении липид-противоинфекционный агент обычно приблизительно от 4:1 до 0,5:1. Были измерены удерживаемые липосомами объемы, и на основе указанных чисел можно рассчитать, какое теоретическое удержание будет иметь место, если противоинфекционный агент ведет себя как идеальное растворенное вещество (т.е. не взаимодействует с мембраной липосом, но идеально удерживается вместе с водой). На основе данного сравнения наблюдаются величины удержания, которые в 3-5 раз превышают ожидаемые, что указывает на наличие особого взаимодействия, которое обеспечивает удержание, превышающее ожидаемое, и более низкие, чем ожидаемые, соотношения липид-противоинфекционный агент. Раствор, в котором содержится липосомальная форма, будет иметь приблизительно ту же концентрацию противоинфекционного агента, которая имеет место внутри липосом. Однако, по расчетам, внутренняя концентрация противоинфекционного агента приблизительно в 3 раза выше.
Частично, настоящее изобретение относится к липосомальной противоинфекционной преп. форме, включающей липидную преп. форму и противоинфекционный агент, в которой липидная композиция практически не содержит анионных липидов, и в которой массовое соотношение липида и противоинфекционного агента составляет приблизительно от 4:1 до 1:1. В некоторых вариантах осуществления настоящего изобретения массовое соотношение липида и противоинфекционного агента составляет приблизительно от 3:1 до 1:1, приблизительно от 2:1 до 1:1 или приблизительно 1:1.
В другом варианте осуществления настоящее изобретение относится к липидной композиции, включающей в себя противоинфекционный агент, в которой соотношение липида и противоинфекционного агента составляет приблизительно 1:1 или менее, приблизительно 0,75:1 или менее, или приблизительно 0,5:1 или менее.
В некоторых вариантах осуществления настоящего изобретения липидная противоинфекционная композиция включает в себя липосому, имеющую средний диаметр приблизительно от 0,2 мкм до 1,0 мкм. В некоторых других вариантах осуществления настоящего изобретения средний диаметр составляет приблизительно от 0,2 мкм до 0,5 мкм. В некоторых других вариантах осуществления настоящего изобретения средний диаметр составляет приблизительно от 0,2 мкм до 0,3 мкм.
В некоторых вариантах осуществления настоящего изобретения противоинфекционный агент может представлять собой любой противоинфекционный агент, хорошо известный специалистам. В некоторых вариантах осуществления настоящего изобретения противоинфекционный агент может представлять собой аминогликозид, включая, без ограничения, амикацин, тобрамицин или гентамицин, или их фармацевтически приемлемые соли.
В некоторых вариантах осуществления настоящего изобретения липидная композиция включает в себя нейтральный липид. В некоторых вариантах осуществления настоящего изобретения липидная композиция не содержит анионных липидов. В некоторых других вариантах осуществления настоящего изобретения липид представляет собой фосфолипид, включая, без ограничения, фосфатидилхолин, такой как дипальмитоилфосфатидилхолин или диолеилфосфатидилхолин; или липид может представлять собой стероид, такой как стерол, включая, без ограничения, холестерин; или липид может представлять собой их комбинацию.
Частично, настоящее изобретение относится к способу изготовления липидной противоинфекционной композиции, описанной выше, включающему в себя инфузию в водный или спиртовой раствор, или в смесь противоинфекционного агента липидно-спиртового раствора, или смеси при температуре ниже температуры фазового перехода по меньшей мере одного из липидных компонентов нейтрального липида, в котором инфузия производится сверху. В некоторых вариантах осуществления настоящего изобретения спирт представляет собой этанол.
В некоторых вариантах осуществления настоящего изобретения концентрация липидно-спиртового раствора или смеси составляет приблизительно от 10 до 30 мг/мл. В некоторых вариантах осуществления настоящего изобретения концентрация противоинфекционного водного или спиртового раствора или смеси составляет приблизительно от 20 до 70 мг/мл. В некоторых вариантах осуществления настоящего изобретения концентрация нейтрального липидно-спиртового раствора или смеси составляет приблизительно от 10 до 30 мг/мл, а концентрация противоинфекционного водного или спиртового раствора, или смеси составляет приблизительно от 20 до 70 мг/мл. Однако специалисту будет понятно, что концентрации могут варьировать или могут быть каким-либо другим образом оптимизированы, в зависимости от используемого липида и/или противоинфекционного агента.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутой ранее липидной композиции, в которой противоинфекционный агент выбран из следующих агентов: аминогликозида, тетрациклина, сульфонамида, п-аминобензойной кислоты, диаминопиримидина, хинолона, β-лактама, β-лактама и ингибитора β-лактамазы, хлорапфеникола, макролида, линомицина, клиндамицина, спектиномицина, полимиксина В, колистина, ванкомицина, бацитрацина, изониазида, рифампина, этамбутола, этионамида, аминосалициловой кислоты, циклосерина, капреомицина, сульфона, клофазимина, талидомида, полиенового противогрибкового агента, флуцитозина, имидазола, триазола, гризеофульвина, терконазола, бутоконазола циклопиракса, циклопирокса оламина, галопрогина, толнафтата, нафтифина, тербинафина или их комбинации. В некоторых вариантах осуществления настоящее изобретение относится к упомянутой ранее липидной композиции, в которой противоинфекционный агент представляет собой аминогликозид. В еще одном варианте осуществления настоящего изобретения аминогликозид выбран из следующих агентов: амикацина, гентамицина или тобрамицина. В еще одном варианте осуществления настоящего изобретения противоинфекционный агент представляет собой амикацин. В еще одном варианте осуществления настоящего изобретения противоинфекционный агент представляет собой гентамицин. В еще одном варианте осуществления настоящего изобретения противоинфекционный агент представляет собой тобрамицин.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутой ранее липидной композиции, в которой липидная композиция представляет собой липосому.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутой ранее липидной композиции, в которой липидная композиция включает в себя фосфолипид. В некоторых вариантах осуществления настоящего изобретения липидная композиция включает в себя стероид. В некоторых вариантах осуществления настоящего изобретения липидная композиция включает в себя стерол. В некоторых вариантах осуществления настоящего изобретения липидная композиция включает в себя дипальмитоилфосфатидилхолин (DPPC). В некоторых вариантах осуществления настоящего изобретения липидная композиция включает в себя холестерин. В некоторых вариантах осуществления настоящего изобретения липидная композиция включает в себя фосфолипид и стероид. В некоторых вариантах осуществления настоящего изобретения липидная композиция включает в себя фосфолипид и стерол. В некоторых вариантах осуществления настоящего изобретения липидная композиция включает в себя DPPC и холестерин. В некоторых вариантах осуществления настоящее изобретение относится к упомянутой ранее композиции, в которой липидная композиция включает в себя DPPC, диолеилфосфатидилхолин (DOPC) и холестерин.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутой ранее композиции, в которой липидная композиция включает в себя DPPC и холестерин в молярном соотношении приблизительно 20:1, 10:1, 5:1, 2:1 или 1:1.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутой ранее композиции, в которой липидная композиция включает в себя DPPC, DOPC и холестерин в молярном соотношении приблизительно 5-20:1-20:0,5-1.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутой ранее композиции, в которой липидная композиция представляет собой липосому, а противоинфекционный агент представляет собой амикацин.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутой ранее композиции, в которой липидная композиция представляет собой липосому, противоинфекционный агент представляет собой амикацин и липидная композиция включает в себя фосфолипид и стерол.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутой ранее композиции, в которой липидная композиция представляет собой липосому, противоинфекционный агент представляет собой амикацин и липидная композиция включает в себя DPPC и холестерин.
В другом варианте осуществления настоящее изобретение относится к способу изготовления липидной композиции, включающей в себя противоинфекционный агент, включающему в себя: смешивание потока липидного раствора или смеси с потоком противоинфекционного раствора или смеси, в котором два потока смешиваются на одной линии. В некоторых вариантах осуществления настоящего изобретения два потока входят в Y-коннектор до смешивания на одной линии.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором поток липидного раствора или смеси и поток противоинфекционного раствора или смеси смешивают при общей скорости потока приблизительно от 700 до 900 мл/мин. В некоторых вариантах осуществления настоящего изобретения поток липидного раствора или смеси и поток противоинфекционного раствора или смеси смешивают при общей скорости потока приблизительно 800 мл/мин. В некоторых вариантах осуществления настоящего изобретения поток липидного раствора или смеси добавляют при скорости потока приблизительно от 200 до 400 мл/мин. В некоторых вариантах осуществления настоящего изобретения поток липидного раствора или смеси добавляют при скорости потока приблизительно 300 мл/мин. В некоторых вариантах осуществления настоящего изобретения поток противоинфекционного раствора или смеси добавляют при скорости потока приблизительно от 400 до 600 мл/мин. В некоторых вариантах осуществления настоящего изобретения поток противоинфекционного раствора или смеси добавляют при скорости потока приблизительно 500 мл/мин. В некоторых вариантах осуществления настоящего изобретения поток липидного раствора или смеси добавляют при скорости потока приблизительно 300 мл/мин, а поток противоинфекционного раствора или смеси добавляют при скорости потока приблизительно 500 мл/мин.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором температура объединенных потоков составляет приблизительно 30-40°С. В некоторых вариантах осуществления настоящего изобретения температура липидного раствора или смеси составляет приблизительно 30°С, и температура противоинфекционного раствора или смеси составляет приблизительно 30°С. В некоторых вариантах осуществления настоящего изобретения температура липидного раствора или смеси составляет приблизительно 50°С, а температура противоинфекционного раствора или смеси является комнатной.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором способ изготовления липидной композиции, включающей в себя противоинфекционный агент, дополнительно включает в себя стадию разбавления объединенных потоков водой по меньшей мере приблизительно через 20 секунд после смешивания.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором концентрация противоинфекционного раствора или смеси составляет приблизительно от 30 до 50 мг/мл. В некоторых вариантах осуществления настоящего изобретения концентрация противоинфекционного раствора или смеси составляет приблизительно от 40 до 50 мг/мл.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором поток липидного раствора или смеси добавляют при скорости потока приблизительно 300 мл/мин, а поток противоинфекционного раствора или смеси добавляют при скорости потока приблизительно 500 мл/мин; температура объединенных потоков составляет приблизительно 30-40°С; объединенные потоки разбавляют водой по меньшей мере приблизительно через 20 секунд после смешивания; и концентрация противоинфекционного раствора или смеси составляет приблизительно от 40 до 50 мг/мл.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором растворы или смеси являются водными или спиртовыми. В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором липидная композиция представляет собой липосому.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором противоинфекционный агент выбран из следующих агентов: аминогликозида, тетрациклина, сульфонамида, п-аминобензойной кислоты, диаминопиримидина, хинолона, β-лактама, β-лактама и ингибитора β-лактамазы, хлорапфеникола, макролида, линомицина, клиндамицина, спектиномицина, полимиксина В, колистина, ванкомицина, бацитрацина, изониазида, рифампина, этамбутола, этионамида, аминосалициловой кислоты, циклосерина, капреомицина, сульфона, клофазимина, талидомида, полиенового противогрибкового агента, флуцитозина, имидазола, триазола, гризеофульвина, терконазола, бутоконазола циклопиракса, циклопирокса оламина, галопрогина, толнафтата, нафтифина, тербинафина или их комбинации. В некоторых вариантах осуществления противоинфекционный агент представляет собой аминогликозид. В некоторых вариантах осуществления настоящего изобретения противоинфекционный агент представляет собой аминогликозид, выбранный из следующих агентов: амикацина, гентамицина или тобрамицина. В некоторых вариантах осуществления настоящего изобретения противоинфекционный агент представляет собой амикацин. В некоторых вариантах осуществления настоящего изобретения противоинфекционный агент представляет собой гентамицин. В некоторых вариантах осуществления настоящего изобретения противоинфекционный агент представляет собой тобрамицин.
В некоторых вариантах осуществления настоящее изобретение относится упомянутому ранее способу, в котором липид включает в себя фосфолипид. В некоторых вариантах осуществления настоящего изобретения липид включает в себя стероид. В некоторых вариантах осуществления настоящего изобретения липид включает в себя стерол. В некоторых вариантах осуществления настоящего изобретения липид включает в себя DPPC. В некоторых вариантах осуществления настоящего изобретения липид включает в себя холестерин. В некоторых вариантах осуществления настоящего изобретения липид включает в себя фосфолипид и стерол. В некоторых вариантах осуществления настоящего изобретения липид включает в себя DPPC и холестерин.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором липидная композиция представляет собой липосому, а противоинфекционный агент представляет собой амикацин.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором липидная композиция представляет собой липосому, противоинфекционный агент представляет собой амикацин и липид включает в себя фосфолипид и стерол.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором липидная композиция представляет собой липосому, противоинфекционный агент представляет собой амикацин и липид включает в себя DPPC и холестерин.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором липидная композиция имеет соотношение липида и противоинфекционного агента приблизительно 1:1 или менее.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором липидная композиция имеет соотношение липида и противоинфекционного агента приблизительно 0,75:1 или менее.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором липидная композиция имеет соотношение липида и противоинфекционного агента приблизительно 0,5:1 или менее.
В некоторых вариантах осуществления настоящее изобретение относится к упомянутому ранее способу, в котором липидная композиция представляет собой липосому, противоинфекционный агент представляет собой амикацин, липид включает в себя DPPC и холестерин, а соотношение липида и противоинфекционного агента составляет приблизительно 1:1 или менее.
В другом варианте осуществления настоящее изобретение относится к способу лечения легочных инфекций у пациента, который в этом нуждается, включающему в себя введение пациенту терапевтически эффективного количества липосомальной противоинфекционной композиции, включающей в себя липидную композицию и противоинфекционный агент, в котором доза противоинфекционного агента составляет приблизительно 100 мг в день или менее. В еще одном варианте осуществления настоящего изобретения доза противоинфекционного агента составляет приблизительно от 30 мг до 50 мг через день. В еще одном варианте осуществления настоящего изобретения доза противоинфекционного агента составляет приблизительно от 30 мг до 50 мг каждый третий день.
В другом варианте осуществления настоящее изобретение относится к упомянутому ранее способу лечения, в котором липосома имеет средний диаметр приблизительно от 0,2 мкм до 1,0 мкм. В еще одном варианте осуществления настоящего изобретения липосома имеет средний диаметр приблизительно от 0,2 мкм до 0,5 мкм или приблизительно от 0,2 мкм до 0,3 мкм.
В другом варианте осуществления настоящее изобретение относится к упомянутому ранее способу лечения, в котором легочная инфекция является результатом муковисцидоза.
В другом варианте осуществления настоящее изобретение относится к упомянутому ранее способу лечения, в котором массовое соотношение липида и противоинфекционного агента составляет приблизительно от 4:1 до 0,5:1, приблизительно от 3:1 до 0,5:1, приблизительно от 2:1 до 0,5:1 или приблизительно от 1:1 до 0,5:1.
В другом варианте осуществления настоящее изобретение относится к упомянутому ранее способу лечения, в котором противоинфекционный агент выбран из следующих агентов: аминогликозида, тетрациклина, сульфонамида, п-аминобензойной кислоты, диаминопиримидина, хинолона, β-лактама, β-лактама и ингибитора β-лактамазы, хлорапфеникола, пенициллинов, цефалоспоринов, макролида, линомицина, клиндамицина, кортикостероидов, простагландина, спектиномицина, полимиксина В, колистина, ванкомицина, бацитрацина, изониазида, рифампина, этамбутола, этионамида, аминосалициловой кислоты, циклосерина, капреомицина, сульфона, клофазимина, талидомида, полиенового противогрибкового агента, флуцитозина, имидазола, триазола, гризеофульвина, терконазола, бутоконазола циклопиракса, циклопирокса оламина, галопрогина, толнафтата, нафтифина, тербинафина или их комбинации. В другом варианте осуществления настоящего изобретения противоинфекционный агент представляет собой аминогликозид. В другом варианте осуществления настоящего изобретения противоинфекционный агент представляет собой амикацин.
В другом варианте осуществления настоящее изобретение относится к упомянутому ранее способу лечения, в котором липидная композиция включает в себя нейтральные липиды. В другом варианте осуществления настоящего изобретения все липиды, которые составляют липидную композицию, представляют собой нейтральные липиды. В другом варианте осуществления настоящего изобретения липосома не содержит анионных липидов. В другом варианте осуществления настоящего изобретения липидная композиция включает в себя фосфолипид. В другом варианте осуществления настоящего изобретения липидная композиция включает в себя стерол. В другом варианте осуществления настоящего изобретения липидная композиция включает в себя DPPC и холестерин.
В другом варианте осуществления настоящее изобретение относится к упомянутому ранее способу лечения, в котором противоинфекционный агент представляет собой амикацин, а липидная композиция включает в себя DPPC и холестерин.
В другом варианте осуществления настоящее изобретение относится к упомянутому ранее способу лечения, в котором противоинфекционный агент представляет собой амикацин, массовое соотношение липида и противоинфекционного агента составляет приблизительно от 4:1 до 1:1 и липидная композиция включает в себя DPPC и холестерин. В другом варианте осуществления настоящего изобретения массовое соотношение составляет приблизительно от 3:1 до 1:1, приблизительно от 2:1 до 1:1 или приблизительно 1:1.
В другом варианте осуществления настоящее изобретение относится к упомянутому ранее способу лечения, в котором противоинфекционный агент представляет собой амикацин, массовое соотношение липида и противоинфекционного агента составляет приблизительно от 4:1 до 1:1, липидная композиция включает в себя DPPC и холестерин, а легочная инфекция является результатом муковисцидоза. В другом варианте осуществления настоящего изобретения массовое соотношение составляет приблизительно от 3:1 до 1:1, приблизительно от 2:1 до 1:1 или приблизительно 1:1.
В другом варианте осуществления настоящее изобретение относится к упомянутому ранее способу лечения, в котором противоинфекционный агент представляет собой амикацин, массовое соотношение липида и противоинфекционного агента составляет приблизительно от 4:1 до 0,5:1, липидная композиция включает в себя DPPC и холестерин, а липосома имеет средний диаметр приблизительно от 0,1 мкм до 0,5 мкм. В другом варианте осуществления настоящего изобретения средний диаметр составляет приблизительно от 0,2 мкм до 0,4 мкм или приблизительно от 0,2 мкм до 0,3 мкм.
В другом варианте осуществления настоящее изобретение относится к упомянутому ранее способу лечения, в котором противоинфекционный агент представляет собой амикацин, массовое соотношение липида и противоинфекционного агента составляет приблизительно от 4:1 до 0,5:1, липидная композиция включает в себя DPPC и холестерин, легочная инфекция является результатом муковисцидоза, а липосома имеет средний диаметр приблизительно от 0,1 мкм до 1,0 мкм. В другом варианте осуществления настоящего изобретения средний диаметр составляет приблизительно от 0,2 мкм до 0,5 мкм или приблизительно от 0,2 мкм до 0,3 мкм.
Указанные варианты осуществления настоящего изобретения, другие варианты осуществления и их признаки и характеристики будут понятны из описания, чертежей и формулы изобретения, представленных ниже.
Краткое описание чертежей
Фиг. 1 изображает схему поперечного сечения мокроты/биопленки у пациентов с муковисцидозом.
Фиг. 2 изображает графическую картину нацеливающего и депонирующего эффекта лекарственного средства по настоящему изобретению.
Фиг. 3 и 4 изображают графические картины бактериологии амикацина в различных формах.
Фиг. 5 изображает графическую картину замедленного высвобождения для липосомального/комплексированного амикацина и тобрамицина.
Фиг. 6 изображает данные по свободному или комлексированному ципрофлоксацину.
Фиг. 7 изображает графическую картину местонахождения лекарственного средства в легком при использовании различных схем дозирования.
Фиг. 8 изображает графически двухпоточный процесс поточной инфузии для изготовления липосомальных противоинфекционных композиций.
Фиг. 9 изображает смешиваемость амикацина сульфата с этанолом/водой. Линии представляют максимальную концентрацию амикацина (базисные), способную смешиваться с раствором этанола при комнатной температуре (КТ) и при 40°. При более высоких концентрациях амикацин образует отдельную жидкую фазу (коацерваты), которая позже выпадает в осадок в виде кристаллов. Вертикальные линии показывают концентрацию этанола в инфузионной смеси липид/амикацин (300/500 частей) и после добавления 200 частей воды.
Подробное описание
Настоящее изобретение описывает липидную композицию, включающую в себя противоинфекционный агент, в которой размер и соотношения липида и лекарственного средства меньше, чем было известно ранее. Настоящее изобретение описывает также способ изготовления указанных липидных композиций.
1. Определения
Для удобства, перед описанием настоящего изобретения приводятся некоторые термины, использованные в описании, примерах и прилагаемой формуле изобретения. Указанные описания следует читать в свете представленного описания и понимать, как это делает специалист в данной области. Если не указано иное, все технические и научные термины, использованные в настоящем документе, имеют то значение, которое является общепринятым для специалистов в данной области.
Артикли «а» и «an» используются в настоящем документе для обозначения одного или более одного (т.е. по меньшей мере одного) грамматического объекта. Например, «an element» означает один элемент или более, чем один элемент.
Термин «биодоступный» известен специалистам и относится к форме предмета изобретения, которая позволяет ей, или части введенного количества, всасываться, инкорпорироваться или каким-либо другим образом быть физиологически доступным для субъекта или пациента, которому ее вводят.
Термины «включает» или «включающий» используются во включающем, открытом смысле, означая, что могут быть включены другие элементы.
Термины «инкапсулированный» и «инкапсулирующий» относятся к адсорбции противоинфекционных агентов на поверхности композиции на липидной основе, ассоциации противоинфекционных агентов в интерстициальной области двойных слоев или между двумя одинарными слоями, к удержанию противоинфекционных агентов в пространстве между двумя двойными слоями или удержанию противоинфекционных агентов в пространстве, окруженном внутренним наибольшим двойным слоем или одинарным слоем.
Термин «включающий», используемый в настоящем документе, означает «включающий, без ограничения». «Включающий» и «включающий, без ограничения» используются взаимозаменяемым образом.
Термины «липидная противоинфекционная композиция» или «Лип-противоинфекционный агент», или «Lip-An», обсуждаемые в настоящем документе, представляют собой любую форму противоинфекционной композиции, в которой по меньшей мере около 1% масс. противоинфекционного агента ассоциировано с липидом, или в виде части комплекса с липидом, или в виде липосомы, в которой антибиотик может быть в водной фазе или в гидрофобной двухслойной фазе, или в межфазной области головных групп липосомального двойного слоя. Предпочтительно, по меньшей мере приблизительно 5% или по меньшей мере приблизительно 10%, или по меньшей мере приблизительно 20%, или по меньшей мере приблизительно 25% может быть ассоциировано. Ассоциацию можно измерить сепарацией через фильтр, на котором задерживается липид и ассоциированный с липидом противоинфекционный агент, а свободный противоинфекционный агент проходит в фильтрат. «Липосомальная противоинфекционная композиция» представляет собой липидную противоинфекционную композицию, в которой липидная композиция представляет собой форму липосомы.
Термин «млекопитающее» известен специалистам, и примеры млекопитающих включают людей, приматов, крупный рогатый скот, свиней, собак, кошек и грызунов (например, мышей и крыс).
«Пациент», «субъект» или «хозяин», которых следует лечить описываемым способом, могут означать как человека, так и животное, не являющееся человеком.
Термин «фармацевтически приемлемые соли» известен специалистам и относится к относительно нетоксичным, неорганическим или органическим кислотно-аддитивным солям соединений, включая, например, соли, содержащиеся в композициям по настоящему изобретению.
Термин «инфузия растворителя» представляет собой процесс, который включает в себя растворение одного или более липидов в малом, предпочтительно минимальном, количестве совместимого с процессом растворителя, с образованием липидной суспензии или раствора (предпочтительно, раствора), а затем добавление раствора к водной среде, содержащей биологически активные агенты. Обычно совместимый с процессом растворитель представляет собой растворитель, который можно отмыть в ходе водного процесса, такого как диализ. Композицию после циклической смены охлаждение/нагревание предпочтительно изготавливают путем инфузии растворителя; предпочтительной является инфузия этанола. В качестве растворителей предпочтительными являются спирты. «Инфузия этанола», тип инфузии растворителя, представляет собой процесс, который включает в себя растворение одного или более липидов в малом, предпочтительно минимальном, количестве этанола, с образованием липидного раствора, а затем добавление раствора к водной среде, содержащей биологически активные агенты. «Малое» количество растворителя представляет собой количество, совместимое с формированием липосом или липидных комплексов в процессе инфузии. Термин «инфузия растворителя» может также включать в себя процесс поточной инфузии, когда два потока компонентов композиции сначала смешиваются на одной линии.
Термин «практически не содержит» известен специалистам и относится к незначительным количествам или менее.
Термин «терапевтический агент» известен специалистам и относится к любому химическому компоненту, который представляет собой биологически, физиологически или фармакологически активное вещество, которое действует локально или системно на субъекта. Примеры терапевтических агентов, также называемых «лекарственными средствами», описаны в хорошо известной специалистам литературе, такой как Merck Index, the Physicians Desk Reference и The Pharmacological Basis of Therapeutics, и они включают в себя, без ограничения, лекарства, витамины, минеральные добавки, вещества, используемые для лечения, профилактики, диагностики, излечения или облегчения болезни или заболевания, вещества, которые действуют на структуру или функцию организма, или пролекарства, которые становятся биологически активными или более активными, после того, как они были помещены в физиологическое окружение.
Фраза «терапевтически эффективное количество», используемая в настоящем документе, означает, что количество соединения, материала или композиции, включающей в себя липидную противоинфекционную композицию по настоящему изобретению, является эффективным для получения некоторого желательного терапевтического эффекта путем ингибирования легочных инфекций.
Термин «лечение» известен специалистам и относится к излечению, а также к облегчению по меньшей мере одного симптома любого состояния или заболевания. Термин «лечение» относится также к профилактическому лечению, которое защищает или предотвращает состояние или заболевание.
2. Противоинфекционные агенты
Противоинфекционными агентами являются агенты, которые действуют против инфекций, такие как бактериальные, микобактериальные, грибковые, вирусные или протозойные инфекции. Противоинфекционные агенты, охватываемые настоящим изобретением, включают в себя, без ограничения, аминогликозиды (например, стрептомицин, гентамицин, тобрамицин, амикацин, нетилмицин, канамицин и т.п.), тетрациклины (такие как хлортетрациклин, окситетрациклин, метациклин, доксициклин, микоциклин и т.п.), сульфонамиды (например, сульфаниламид, сульфадиазин, сульфаметаоксазол, сульфизоксазол, сульфацетамид и т.п.), парааминобензойную кислоту, диаминопиримидины (такие как триметоприм, часто используемый в сочетании с сульфаметоксазолом, пиразинамидом и т.п.), хинолоны (такие как налидиксовая кислота, циноксацин, ципрофлоксацин и норфлоксацин и т.п.), пенициллины (такие как пенициллин G, пенициллин V, ампициллин, амоксициллин, бакампициллин, карбенициллин, карбенициллина инданил, тикарциллин, азлоциллин, мезлоциллин, пиперациллин и т.п.), пенициллин, резистентный к пенициллиназе (такой как метициллин, оксациллин, клоксациллин, диклоксациллин, нафциллин и т.п.), цефалоспорины первого поколения (такие как цефадроксил, цефалексин, цефрадин, цефалотин, цефапирин, цефазолин и т.п.), цефалоспорины второго поколения (такие как цефаклор, цефамандол, цефоницид, цефокситин, цефотетан, цефуроксим, цефуроксима аксетил, цефметазол, цефпрозил, лоракарбеф, цефоранид и т.п.), цефалоспорины третьего поколения (такие как цефепим, цефоперазон, цефотаксим, цефтизоксим, цефтриаксон, цефтазидим, цефиксим, цефподоксим, цефтибутен и т.п.), другие бета-лактамы (такие как имипенем, меропенем, азтреонам, клавулановая кислота, сульбактам, тазобактам и т.п.), ингибиторы бета-лактамазы (такие как клавулановая кислота), хлорапфеникол, макролиды (такие как эритромицин, азитромицин, кларитромицин и т.п.), линкомицин, клиндамицин, спектиномицин, полимиксин В, полимиксины (такие как полимиксин A, B, C, D, E1 (колистин А) или Е2, колистин В или С и т.п.), ванкомицин, бацитрацин, изониазид, рифампин, этамбутол, этионамид, аминосалициловую кислоту, циклосерин, капреомицин, сульфоны (такие как дапсон, сульфоксон-натрий и т.п.), клофазимин, талидомид или любой другой антибактериальный агент, который может быть инкапсулирован в липиды. Противоинфекционные агенты могут включать в себя противогрибковые агенты, включая полиеновые противогрибковые агенты (такие как амфотерицин В, нистатин, натамицин и т.п.), флуцитозин, имидазолы (такие как н-тиконазол, клотримазол, эконазол, кетоконазол и т.п.), триазолы (такие как итраконазол, флуконазол и т.п.), гризеофульвин, терконазол, бутоконазол циклопиракс, циклопирокс оламин, галопрогин, толнафтат, нафтифин, тербинафин или любой другой противогрибковый агент, который может быть инкапсулирован в липиды или образовывать комплексы с липидами. Обсуждение и примеры направлены, главным образом, на амикацин, однако объем заявки не ограничивается указанным противоинфекционным агентом. Могут быть использованы комбинации лекарственных средств.
Особенно предпочтительные противоинфекционные агенты включают в себя аминогликозиды, хинолоны, полиеновые противогрибковые агенты и полимиксины.
Также в качестве подходящих противоинфекционных агентов для использования в липидных противоинфекционных композициях по настоящему изобретению включены фармацевтически приемлемые аддитивные соли и комплексы противоинфекционных агентов. В случаях, когда соединения могут иметь один или более хиральных центров, за исключением описанных, настоящее изобретение включает в себя каждое уникальное рацемическое соединение, а также каждое уникальное нерацемическое соединение.
В случаях, когда противоинфекционные агенты имеют ненасыщенные углерод-углеродные связи, как цис (Z), так и транс (Е) изомеры входят в объем настоящего изобретения. В случаях, когда противоинфекционные агенты могут существовать в таутомерных формах, таких как кето-енольные таутомеры, такие как 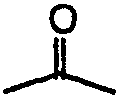 и
и 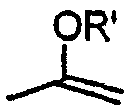 , каждая таутомерная форма рассматривается как включенная в объем изобретения, существует ли она в равновесной форме или в одной форме путем соответствующего замещения R'. Значение любого заместителя в любом появлении не зависит от его значения или от значения любого другого заместителя, при любом появлении.
, каждая таутомерная форма рассматривается как включенная в объем изобретения, существует ли она в равновесной форме или в одной форме путем соответствующего замещения R'. Значение любого заместителя в любом появлении не зависит от его значения или от значения любого другого заместителя, при любом появлении.
Также в качестве подходящих противоинфекционных агентов для использования в липидных противоинфекционных композициях по настоящему изобретению включены пролекарства соединений платины. Пролекарствами считают любые ковалентно связанные носители, которые высвобождают активное родительское соединение in vivo.
3. Легочные инфекции
Среди легочных инфекций (таких как муковисцидоз), которые можно лечить с использованием способов по настоящему изобретению, находятся инфекции, вызываемые Pseudomonas (например, P.aeruginosa, P.paucimobilis, P.putida, P.fluorescens и P.acidovorans), стафилококками, резистентным к метициллину Staphylococcus aureus (MRSA), стрептококками (включая Streptococcus pneumoniae), Escherichia coli, Klebsiella, Enterobacter, Serratia, Haemophilus, Yersinia pesos, Burkholderia pseudomallei, B.cepacia, B.gladioli, B.multivorans, B.vietnamiensis, Mycobacterium tuberculosis, комплексом M.avium (MAC) (M.avium и M.intracellulare), M.kansasii, M.xenopi, M.marinum, M.ulcerans или комплексом M.fortuitum (M.fortuitum и M.chelonei).
4. Способы лечения
В одном варианте осуществления настоящее изобретение включает в себя способ лечения, включающий в себя введение терапевтически эффективного количества липидной противоинфекционной композиции.
В случае, когда конкретные дозы не приводятся ниже, предпочтительная доза по настоящему изобретению составляет 50% или менее, 35% или менее, 20% или менее, или 10%, или менее, от минимального количества свободного лекарственного средства (которое, разумеется, может представлять собой соль), которое является эффективным, если доставляется в легкие посредством небулайзера, в отношении уменьшения количества КОЕ в легких на один порядок после 14-дневного курса лечения. Сравнительное количество свободного лекарственного средства представляет собой кумулятивное количество, которое следует использовать в период дозирования, проводимого с использованием введения лекарственного средства по настоящему изобретению. Сравнительный минимум свободного лекарственного средства, определенный в данном абзаце, представляет собой «сравнительное количество свободного лекарственного средства».
Варианты осуществления настоящего изобретения, не относящиеся к лечению CF, можно использовать для любого животного, хотя, предпочтительно, для человека. Относительные количества для данного животного измеряются по отношению к указанному животному.
Схема дозирования предпочтительно - один раз в день или менее. В предпочтительных вариантах осуществления настоящего изобретения схема дозирования - один раз в два дня, один раз в три дня, один раз в неделю или менее. Например, схема дозирования может быть один раз в два дня или менее, с использованием 50% или менее сравнительного количества свободного лекарственного средства. Или, например, схема дозирования может быть один раз в день, с использованием 35% или менее сравнительного количества свободного лекарственного средства. См. фиг. 3 и 4, данные по животным, показывающие, что липидные противоинфекционные композиции по настоящему изобретению являются более эффективными, чем свободное лекарственное средство.
Для лечения инфекций эффективное количество противоинфекционного агента будет определяться клиницистами, но оно включает в себя количество, эффективное для лечения, уменьшения, улучшения, устранения или профилактики одного или более симптомов заболевания, которое пытаются лечить, или состояния, которого пытаются избежать или лечить, или для какого-либо другого клинически распознаваемого изменения в патологии заболевания или состояния. Улучшение включает в себя уменьшение частоты или тяжести инфекций у животных, которых лечили с профилактической целью. В некоторых вариантах осуществления настоящего изобретения эффективное количество представляет собой количество, эффективное для лечения или улучшения после того, как симптомы легочной инфекции уже появились. В некоторых других вариантах осуществления настоящего изобретения эффективное количество представляет собой количество, эффективное для лечения или улучшения средней частоты или тяжести инфекций у животных, которых лечили с профилактической целью (по статистическим данным).
Липосому или другие липидные системы доставки можно вводить путем ингаляции с использованием распыленного спрея, порошка или аэрозоля, или путем интратекального введения. Введение путем ингаляции является предпочтительным. Конечным результатом является менее частое введение и увеличенный терапевтический индекс по сравнению со свободным лекарственным средством или парентеральной формой лекарственного средства. Липосомы или другие липидные композиции являются особенно выгодными в силу их способности защищать лекарственное средство, при одновременной совместимости с выстилкой легких или сурфактантом легких.
Настоящее изобретение включает в себя способы лечения легочных грамотрицательных инфекций. Инфекцией, подходящей для лечения, является хроническая инфекция, вызываемая Pseudomonas, у пациентов с CF. Известные способы лечения легочных инфекций (таких как у пациентов с CF) аминогликозидом включают в себя введение приблизительно 200-600 мг амикацина или тобрамицина в день путем ингаляции. Настоящее изобретение позволяет осуществлять лечение введением, в одном предпочтительном варианте осуществления, 100 мг или менее амикацина в день (или введением, нормализованным до 100 мг в день или менее, если дозирование производится реже). В еще одном другом варианте осуществления настоящего изобретения производится введение 60 мг или менее амикацина каждый день. И в еще одном другом варианте осуществления настоящего изобретения производится введение приблизительно от 30 до 50 мг, не чаще, чем один раз в два дня. Наиболее предпочтительный вариант осуществления настоящего изобретения включает в себя введение приблизительно от 30 до 50 мг один раз в два дня или один раз в три дня.
5. Липиды и липосомы
Липиды, используемые в композициях по настоящему изобретению, могут быть синтетическими, полусинтетическими или натуральными, включая фосфолипиды, токоферолы, стероиды, жирные кислоты, гликопротеины, такие как альбумин, анионные липиды и катионные липиды. Липиды могут быть анионными, катионными или нейтральными. В одном варианте осуществления настоящего изобретения липидная композиция практически не содержит анионных липидов. В одном варианте осуществления настоящего изобретения липидная композиция содержит только нейтральные липиды. В другом варианте осуществления настоящего изобретения липидная композиция не содержит анионных липидов. В другом варианте осуществления настоящего изобретения липид представляет собой фосфолипид. Фосфолипиды включают в себя яичный фосфатидилхолин (EPC), яичный фосфатидилглицерин (EPG), яичный фосфатидилинозит (EPI), яичный фосфатидилсерин (EPS), фосфатидилэтаноламин (ЕРЕ) и яичную фосфатидную кислоту (ЕРА); соевые аналоги, соевый фосфатидилхолин (SPC); SPG, SPS, SPI, SPE и SPA; гидрированные яичные и соевые аналоги (например, HEPC, HSPC), другие фосфолипиды, построенные сложноэфирными связями жирных кислот по 2 и 3 позициям глицерина, содержащими цепочки от 12 до 26 атомов углерода, и различных главных групп по 1 позиции глицерина, которые включают в себя холин, глицерин, инозит, серин, этаноламин, а также соответствующие фосфатидные кислоты. Цепочки указанных жирных кислот могут быть насыщенными или ненасыщенными, и фосфолипид может быть составлен из жирных кислот с различными длинами цепей и различными степенями ненасыщенности. В частности, в состав композиций может входить дипальмитоилфосфатидилхолин (DPPC), главный составной компонент натурального легочного сурфактанта, а также диолеоилфосфатидилхолин (DOPC). Другие примеры включают в себя димиристоилфосфатидилхолин (DMPC) и димиристоилфосфатидилглицерин (DMPG), дипальмитоилфосфатидхолин (DРPC) и дипальмитоилфосфатидилглицерин (DРPG), дистеароилфосфатидилхолин (DSPC) и дистеароилфосфатидилглицерин (DSPG), диолеилфосфатидилэтаноламин (DOPE) и смешанные фосфолипиды, такие как пальмитоилстеароилфосфатидилхолин (PSPC) и пальмитоилстеароилфосфатидилглицерин (PSPG), триацилглицерин, диацилглицерин, серанид, сфингозин, сфингомиелин и одинарно ацилированные фосфолипиды, такие как моноолеоилфосфатидилэтаноламин (МОРЕ).
Используемые липиды могут включать в себя аммониевые соли жирных кислот, фосфолипиды и глицериды, стероиды, фосфатидилглицерины (PG), фосфатидные кислоты (РА), фосфатидилхолины (РС), фосфатидилинозиты (PI) и фосфатидилсерины (PS). Жирные кислоты включают в себя жирные кислоты с длинами углеродной цепи от 12 до 26 атомов углерода, которые являются насыщенными или ненасыщенными. Некоторые конкретные примеры включают в себя: миристиламин, пальмитиламин, лауриламин и стеариламин, дилауроилэтилфосфохолин (DLEP), димиристоилэтилфосфохолин (DMEP), дипальмитоилэтилфосфохолин (DPEP) и дистеароилэтилфосфохолин (DSEP), хлорид N-(2,3-ди-(9(Z)октадеценилокси)проп-1-ил-N,N,N-триметиламмония (DOTMA) и 1,2-бис(олеоилокси)-3-(триметиламмоний)пропан (DOTAP). Примеры стероидов включают в себя холестерин и эргостерол. Примеры PG, PA, PI, PC и PS включают в себя DMPG, DPPG, DSPG, DMPA, DPPA, DSPA, DMPI, DPPI, DSPI, DMPS, DPPS и DSPS, DSPC, DPPG, DMPC, DOPC, яичный РС.
Липосомы или липидные противоинфекционные композиции, составленные из фосфатидилхолинов, таких как DPPC, помогают захвату клетками в легких, такими как альвеолярные макрофаги, и помогают замедлить высвобождение противоинфекционного агента в легких (Gonzales-Rothi et al. (1991)). Отрицательно заряженные липиды, такие как PG, PA, PS и PI, помимо уменьшения агрегации частиц, могут играть роль в характеристиках замедленного высвобождения ингаляционной композиции, а также в транспорте композиции через легкие (трансцитоз) для системного поглощения. Стероловые соединения, как полагают, влияют на характеристики высвобождения и просачивания композиции.
Липосомы представляют собой полностью закрытые липидные двухслойные мембраны, содержащие в себе удерживаемый водный объем. Липосомы могут представлять собой однослойные пузырьки (имеющие один мембранный двойной слой) или многослойные пузырьки (напоминающие лук структуры, характеризующиеся множеством мембранных двойных слоев, каждый из которых отделен от следующего водным слоем). Двойной слой состоит из двух липидных монослоев, имеющих гидрофобную «хвостовую» область и гидрофильную «головную» область. Структура мембранного двойного слоя такова, что гидрофобные (неполярные) «хвосты» липидных монослоев ориентированы по направлению к центру двойного слоя, в то время как гидрофильные «головы» ориентированы по направлению к водной фазе. Липидные противоинфекционные композиции представляют собой ассоциации липида и противоинфекционного агента. Указанная связь может быть ковалентной, ионной, электростатической, нековалентной или стерической. Указанные комплексы являются нелипосомальными и не способны захватывать дополнительные водорастворимые растворенные вещества. Примеры указанных комплексов включают в себя липидные комплексы амфотенцина В (Janoff et al., Proc. Nat.Acad.Sci., 85:6122-6126, 1988) и кардиолипины, комплексированные с доксорубицином.
Липидный клатрат представляет собой пространственную, напоминающую клетку структуру, в которой содержится один или более липидов; указанная структура удерживает в себе биологически активный агент. Указанные клатраты включены в объем настоящего изобретения.
Пролипосомы представляют собой композиции, которые могут стать липосомами или липидными комплексами при контакте с водной жидкостью. Может быть необходимо перемешивание или какое-либо другое смешивание. Указанные пролипосомы включены в объем настоящего изобретения.
Липосомы можно изготавливать различными способами (см., например, Bally, Cullis et al., Biotechnol. Adv. 5(1):194, 1987). С помощью процедуры Bangham (J.Mol.Biol., J.Mol.Biol. 13(1):238-52, 1965) получают обычные многослойные пузырьки (MLV). Lenk et al. (патенты США №№ 4552803, 5030453 и 5169637), Fountain et al. (патент США № 4588578) и Cullis et al. (патент США № 4975282) описывают способы изготовления многослойных липосом, имеющих практически равное межслойное распределение растворенного вещества в каждом из их водных компартментов. Paphadjopoulos et al., патент США № 4235871, описывает изготовление липосом, имеющих небольшое количество слоев, путем выпаривания обратной фазы.
Однослойные везикулы можно изготавливать из MLV с использованием ряда методик, например, экструзией по Cullis et al. (патент США № 5008050) и Loughrey et al. (патент США № 5059421). Ультразвуковую обработку и гомогенизацию можно использовать для изготовления однослойных липосом меньшего размера из липосом большего размера (см., например, Paphadjopoulos et al., Biochim. Biophys. Acta., 135:624-638, 1967; Deamer, патент США № 4515736 и Chapman et al., Liposome Technol., 1984, стр. 1-18).
Оригинальное изготовление липосом по Bangham et al. (J.Mol.Biol., 1965, 13:238-252) включает в себя суспендирование фосфолипидов в органическом растворителе, который затем выпаривают до сухости, в результате чего на реакционном сосуде остается фосфолипидная пленка. Затем добавляют необходимое количество водной фазы, смесь оставляют «набухать» и полученные липосомы, которые состоят из многослойных везикул (MLV) диспергируют механическими средствами. Указанное изготовление обеспечивает основу для изготовления малых, обработанных ультразвуком, однослойных везикул, описанных Papаhadjopoulos et al. (Biochim. Biophys. Acta., 1967, 135:624-638), и больших однослойных везикул.
Методики изготовления больших однослойных везикул (LUV), такие как выпаривание обратной фазы, процедуры инфузии и дилюция с использованием детергента, можно использовать для изготовления липосом. Обзор указанных и других способов изготовления липосом можно найти в тексте Liposomes, Marc Ostro, изд., Marcel Dekker, Inc., New York, 1983, глава 1, подходящие части которого включены в настоящий документ в качестве ссылки. См. также Szoka, Jr. et al., (1980, Ann. Rev. Biophys. Bioeng., 9:467), подходящие части которого также включены в настоящий документ в качестве ссылки.
Другие методики, которые используются для изготовления везикул, включают методики, при которых образуются везикулы выпаривания обратной фазы (REV), Papаhadjopoulos et al., патент США № 4235871. Другой класс липосом, которые можно использовать, представляют собой липосомы, характеризующиеся как имеющие практически равное межслойное распределение растворенного вещества. Указанный класс липосом называется стабильными многослойными везикулами (SPLV), как определено в патенте США № 4522803, выданном Lenk et al., и включает в себя монофазные везикулы, как описано в патенте США № 4588578, выданном Fountain, et al., и замороженные и оттаявшие многослойные везикулы (FATMLV), как описано выше.
Ряд стеролов и их водорастворимых производных, таких как гемисукцинат холестерина, использовались для изготовления липосом; см., в частности, Janoff et al., патент США № 4721612, выданный 26 января 1988 г., озаглавленный «Стероидные липосомы». Mayhew et al. описали способ уменьшения токсичности антибактериальных агентов и противовирусных агентов путем инкапсулирования их в липосомы, содержащие альфа-токоферол и некоторые его производные. Также ряд токоферолов и их водорастворимых производных использовались для изготовления липосом, см. Janoff et al., патент США № 5041278.
6. Способы изготовления
Процесс изготовления липосом или липидных противоинфекционных композиций включает процесс «инфузии растворителя». Последний представляет собой процесс, который включает в себя растворение одного или более липидов в малом, предпочтительно минимальном, количестве совместимого с процессом растворителя, с образованием липидной суспензии или раствора (предпочтительно, раствора), а затем инфузию раствора в водную среду, содержащую противоинфекционный агент. Обычно совместимый с процессом растворитель представляет собой растворитель, который можно отмыть в ходе водного процесса, такого как диализ или диафильтрация. «Инфузия этанола», тип инфузии растворителя, представляет собой процесс, который включает в себя растворение одного или более липидов в малом, предпочтительно минимальном, количестве этанола, с образованием липидного раствора, а затем инфузию раствора в водную среду, содержащую противоинфекционный агент. «Малое» количество растворителя представляет собой количество, совместимое с формированием липосом или липидных комплексов в процессе инфузии. Указанные процессы описаны в Lee et al., патентная заявка США № 10/634144, зарегистрирована 4 августа 2003 г., Pilkiewicz et al., патентная заявка США № 10/383173, зарегистрирована 5 марта 2003 г., и Boni et al., патентная заявка США № 10/383004, зарегистрирована 5 марта 2003 г.; указанные заявки целиком включены в настоящий документ в качестве ссылок.
Стадия инфузии липидно-спиртового раствора в водный или спиртовой раствор или смесь, содержащие противоинфекционный агент, можно осуществлять выше или ниже поверхности водного или спиртового раствора или смеси, содержащих противоинфекционный агент. Предпочтительно, стадию осуществляют над поверхностью раствора или смеси.
Липосомы также можно изготавливать способами, описанными в одновременно рассматриваемых патентных заявках США: 10/383004, поданной 5 марта 2003 г.; 10/634144, поданной 4 августа 2003 г.; 10/224293, поданной 20 августа 2002 г.; и 10/696389, поданной 29 октября 2003 г., описания которых целиком включены в настоящий документ в качестве ссылок.
Задание размеров липосом или липидной композиции можно осуществлять с использованием ряда способов, таких как экструзия, обработка ультразвуком и гомогенизация, которые хорошо известны специалистам и легко осуществляются на практике. Экструзия включает в себя пропускание липосом под давлением один раз или более через фильтры, имеющие определенные размеры пор. Фильтры обычно изготавливают из поликарбоната, но фильтры могут быть изготовлены из любого стойкого материала, который не взаимодействует с липосомами и который является достаточно прочным, чтобы осуществлять экструзию под достаточным давлением. Предпочтительные фильтры включают в себя «прямые транзитные фильтры», поскольку они обычно могут выдерживать более высокое давление предпочтительных способов экструзии по настоящему изобретению. «Извилистые» фильтры также можно использовать. Для экструзии также можно использовать асимметричные фильтры, такие как фильтры Anopore™, при использовании которых липосомы экструдируют через пористый фильтр из оксида алюминия с разветвленными порами.
Размеры липосом или липидной композиции можно уменьшать также с использованием ультразвуковой обработки, при которой ультразвуковая энергия используется для разрушения или расщепления липосом, которые затем спонтанно формируют липосомы меньших размеров. Обработку ультразвуком осуществляют погружением стеклянной пробирки, содержащей суспензию липосом, в эпицентр ультразвука ультразвукового аппарата типа бани. Альтернативно, можно использовать ультразвуковой аппарат зондового типа, в котором ультразвуковая энергия генерируется вибрацией титанового зонда в прямом контакте с суспензией липосом. Аппараты для гомогенизации и размалывания, такие как гомогенизатор Gifford Wood, Polytron™ или Microfluidizer, также можно использовать для дробления липосом или липидных композиций большего размера на липосомы или липидные композиции меньшего размера.
Полученные липосомальные композиции можно разделять на гомогенные популяции с использованием способов, известных специалистам, таких как тангенциальная фильтрация потока. В данной процедуре популяции липосом или липидных композиций, разнородные в том, что касается размеров, пропускают через тангенциальная фильтры потока, в результате чего получают популяцию липосом с более высоким и/или более низким пределом размеров. В случае, когда используются два фильтра различных размеров, т.е. имеющие различные диаметры пор, липосомы, имеющие меньший размер, чем первый диаметр пор, проходят через фильтр. Указанный фильтрат можно подвергать тангенциальной фильтрации потока через второй фильтр, имеющий меньший размер пор по сравнению с первым фильтром. То, что задержано указанным фильтром, представляет собой липосомальную/комплексированную популяцию, имеющую верхний и нижний пределы размеров, определенные размерами пор первого и второго фильтров, соответственно.
Mayer et al. установили, что проблемы, связанные с эффективным удержанием липофильных ионизируемых биологически активных агентов, таких как антинеопластические агенты, например, антрациклинов или алкалоидов барвинка, могут быть смягчены путем использования трансмембранных ионных ингредиентов. Помимо индуцирования большего поглощения указанные трансмембранные градиенты могут также увеличивать задержку противоинфекционного агента в липосомальной композиции.
Липидные противоинфекционные композиции оказывают замедленное противоинфекционное действие и обладают более низкой токсичностью, что позволяет осуществлять менее частое введение и обеспечивает повышение терапевтического индекса. В доклинических испытаниях на животных и при сравнении с ингаляционным тобрамицином (нелипосомальным и не на основе липидов) на эквивалентном уровне доз, липосомальный амикацин, как было установлено, показывает, во время периода времени, вскоре после введения и через 24 часа, уровни лекарственного средства в легких, которые в два - несколько сот раз превышали уровни тобрамицина. Помимо этого липосомальный амикацин поддерживал указанные уровни свыше 24 часов. На животных, которые служили моделями, имитирующими инфекцию Pseudomonas, наблюдающуюся у пациентов с CF, липосомальный амикацин, как было показано, значимо элиминировал инфекцию в легких животных по сравнению со свободными аминогликозидами.
Легочный сурфактант вносит поправку в увеличение объема и компрессию легких во время дыхания. Это осуществляется путем покрытия легких комбинацией липида и белка. Липид представлен в виде монослоя с гидрофобными цепями, направленными вовне. Липид представляет 80% легочного сурфактанта; наибольшую часть липида представляет фосфатидилхолин, 50% которого представляет дипальмитоилфосфатидилхолин (DPPC) (Veldhuizen et al., 1998). Белки сурфактанта (SP), которые присутствуют, выполняют функцию поддержания структуры и облегчают как расширение, так и компрессию легочного сурфактанта, как это наблюдается во время дыхания. Из них SP-B и SP-C, в частности, показывают литическое поведение и могут лизировать липосомы (Hagwood et al., 1998; Johansson, 1998). Указанное литическое поведение может облегчать постепенное разрушение липосом. Липосомы также могут напрямую перевариваться макрофагами посредством фагоцитоза (Couveur et al., 1991; Gonzales-Roth et al., 1991; Swenson et al., 1991). Захват липосом альвеолярными макрофагами представляет собой другое средство, посредством которого лекарственные средства могут доставляться в область патологии.
Липиды, предпочтительно используемые для формирования липосомальных или липидных композиций для ингаляции, имеют общее происхождение с эндогенными липидами, обнаруженными в легочном сурфактанте. Липосомы состоят из двойных слоев, которые удерживают желательное лекарственное средство. Они могут иметь конфигурацию многослойных везикул из концентрических двойных слоев, с лекарственным средством, удерживаемым в липиде различных слоев или в водном пространстве между слоями. В настоящем изобретении используются уникальные процессы для создания уникальных липосомальных или липидных противоинфекционных композиций. Как процессы, так и продукт указанных процессов являются частью настоящего изобретения.
6.1 Способ поточной инфузии
В одном особенно предпочтительном варианте осуществления настоящего изобретения липосомальные противоинфекционные композиции по настоящему изобретению изготавливают с использованием способа поточной инфузии, в котором поток липидного раствора смешивают с потоком противоинфекционного раствора на одной линии. Например, два раствора можно смешивать на одной линии внутри трубы для смешивания, которой предшествует Y-коннектор, как изображено на фиг. 8. В данном случае способ поточной инфузии отличается от способа инфузии, описанного выше, в котором липидный раствор вливают в виде потока в массу противоинфекционного раствора. Неожиданно было установлено, что указанный способ инфузии обеспечивает более низкие соотношения липида и лекарственного средства и более высокую эффективность инкапсуляции. Способ можно дополнительно улучшить путем оптимизации параметров, таких как скорость потока, температура, концентрация противоинфекционного агента, и путем добавления соли после стадии инфузии.
6.1.а Эффект скоростей потока
Скорости отдельных потоков изменяли при сохранении общей скорости потока 800 мл/мин. Чтобы этого добиться, использовали два отдельных насоса, с различными скоростями нагнетания. Смешанные растворы инфузировали в течение 10 с в химический стакан, содержавший раствор NaCl, таким образом, что конечная концентрация NaCl составляла 1,5%, а конечная концентрация этанола не превышала 30%. После смешивания 1 мл аликвоту пропускали через фильтрационную колонку Sephadex G-75 для отделения свободного амикацина от инкапсулированного. 1 мл фракцию с наибольшей плотностью (определенной по видимой мутности) собирали для дальнейшего анализа. Результаты представлены в таблице 1. В результате увеличения соотношения скоростей потока липид/амикацин получали почти постоянное L/D до 300/500 мл/мин. При дальнейшем увеличении скорости липида L/D начало увеличиваться, и размер частиц также начал увеличиваться. В то же время более высокие скорости потока липида обеспечивали лучший возврат амикацина (эффективность инкапсуляции), поскольку было добавлено больше массы липида.
мл/мин
АМК,
мг/мл
АМК,
%
мг/мл
частиц
(по
объемной массе)
%
Серия 3 со скоростями потока липид/амикацин 300/500 мл/мин показала лучшие L/D и размер частиц, в сочетании с достаточно высоким возвратом амикацина. Таким образом, было принято решение использовать указанные скорости потоков для всех дальнейших экспериментов.
Для того, чтобы воспроизвести результаты при выбранных условиях, изготавливали полностью промытую серию (серия 6) с использованием диафильтрации, как показано в таблице 2. 10% раствор NaCl добавляли в химический стакан перед инфузией, с получением конечной концентрации 2% (по сравнению с 1,5% в сериях, указанных в таблице 1). Полученное L/D (1,71) не было таким хорошим, как в серии 3 в таблице 1, а размер частиц был больше. Возможно, это произошло из-за неблагоприятного эффекта высокой концентрации NaCl, которая контактировала с липосомами на ранних стадиях их формирования. Образцы, разделенные (промытые) с использованием колонок для гель-фильтрации, имели тенденцию к лучшим L/D, чем у образцов, промытых с использованием диафильтрации. Это может иметь отношение к различным степеням напряжения, которому подвергались липосомы, или просто образцы, разделенные с использованием колонки для гель-фильтрации, содержали фракцию липосом с лучшим L/D, которая не представляет популяцию целиком.
%
%
6.1.b Эффекты температуры процесса
Параметры сохраняли те же, что в серии 3, за исключением того, что количество добавленного раствора NaCl было меньше, с конечной концентрацией 1,0%. Раствор добавляли еще раз до начала инфузии, поскольку при коротком времени инфузии было трудно осуществлять добавление во время инфузии. Также во время инфузии поточный миксер сдвигался к концу трубки для смешивания под давлением потока. Положение миксера было 5 см от переднего конца трубки, вместо 0 см для серии 3. Это может быть важным, поскольку соотношение L/D, полученное при тех же температурных условиях 40/40°С в серии 20, составляло 0,55, почти половину от соотношения в серии 3. При сравнении инкапсуляции амикацина при различных температурах инфузии неожиданно оказалось, что более низкие температуры давали лучшие L/D. Из испытанных температур, температуры липид/амикацин 30/30°С и 50/RT давали сходные соотношения 0,32 и 0,37. И снова, как в сериях 1-5, числа от указанных промытых с использованием гель-фильтрации образцов были низкими, возможно, меньше, чем если бы серии промывали с использованием диафильтрации.
%
В отдельных экспериментах было установлено, что смешивание 90% этанола и воды при 30°С и 30°С или 50°С и 22°С, соответственно, дает сходную конечную температуру приблизительно 36°С. Это наводит на мысль о том, что скорее температура конечной смеси, чем температура отдельных компонентов, является важной для инкапсуляции амикацина. Температуры 50°С/RT использовались в примерах 6-15. В примерах 16-18 температуры 30°С и 30°С для двух потоков использовались со сравнимыми результатами, хотя наблюдалась несколько меньшая инкапсуляция амикацина.
6.1.с. Эффект постинфузионного добавления водного объема
Внимание затем было сфокусировано на стадиях добавления раствора NaCl и процессе промывания. Параметры процесса меняли по различным направлениям. Непосредственно после стадии инфузии при скоростях потоков 300/500 концентрация этанола в смеси достигает 34%. При указанной концентрации амикацин имеет ограниченную растворимость (см. фиг. 9).
Если начинать с маточного раствора амикацина 50 мг/мл, то после смешивания с липидным раствором общая концентрация амикацина будет более 30 мг/мл, в которой по меньшей мере половина (15 мг/мл) приходится на свободный амикацин, что предполагает 50% эффективность инкапсуляции. Это выше предела растворимости в 34% этаноле. Одним возможным решением указанной проблемы является добавление большего количества воды в сосуд со смесью липид/амикацин, что уменьшает концентрацию как этанола, так и амикацина. Например, добавление 200 частей воды (или раствора NaCl) к 800 частям липида/амикацина уменьшает концентрацию этанола до 27% (фиг. 9). Это делает амикацин растворимым при 15 мг/мл или даже выше, в зависимости от температуры.
Помимо этого добавление NaCl может стабилизировать осмотические условия. Когда формируются липосомы, и амикацин инкапсулируется с внутренней концентрацией 200-300 мг/мл, остается только приблизительно 15 мг/мл или около того не инкапсулированного амикацина. В отсутствии солевого раствора это будет создавать осмотический дисбаланс, который, в свою очередь, может приводить к утечке амикацина. Добавление 150 частей 10% NaCl к 800 частям липида/амикацина даст приблизительно 1,5% конечную концентрацию NaCl (вне липосом).
Был изготовлен ряд серий, в которые добавляли различные количества раствора NaCl (или воды в некоторых сериях) в разное время относительно инфузии (см. таблицу 5, составленную из таблиц 2 и 3). В таблице можно увидеть общую тенденцию, позволяющую сделать следующие выводы:
- некоторый интервал времени между инфузией и добавлением водного объема требуется для получения более низкого L/D (если используется короткая трубка для смешивания). Из серий 6-15, серии с интервалом 20 с или более имели более низкий L/D. Одно возможное объяснение заключается в том, что липосомы не образуются полностью немедленно после перемешивания потоков. Если используется более длинная трубка для смешивания (серии 16-18), что обеспечивает более продолжительное время смешивания, интервал времени не требуется.
- Добавление раствора NaCl высокой концентрации для балансировки осомоляльности на самом деле не способствует удержанию амикацина. Фактически, добавление чистой воды с подходящим интервалом времени дает самое низкое L/D и общую концентрацию амикацина.
- Добавление 100 частей 10% NaCl (серия 9) через 5 мин после инфузии дало конкурентное соотношение L/D, но не дало настолько же хорошую общую концентрацию амикацина. Возможно, NaCl, когда он присутствует на ранних стадиях при относительно высоких концентрациях этанола, вызывает повышение агрегации и вязкости.
после
после
после
после
6.1.d. Эффект маточного раствора противоинфекционного агента
Ранее было установлено, что использование маточного раствора амикацина 50 мг/мл обеспечивает наилучшее удержание. Уменьшение концентрации маточного раствора амикацина 40 мг/мл увеличивало L/D, когда его использовали в обычных процессах. В двухпоточном процессе поточной инфузии концентрация этанола достигает более высоких уровней, поэтому современный 50 мг/мл амикацин может не быть оптимальной концентрацией.
В таблице 6 суммированы эффекты от использования различных концентраций маточного раствора амикацина. Концентрация 40 мг/мл давала сравнимые или лучшие величины L/D, и даже улучшение возврата амикацина. Использование меньших концентраций амикацина относительно постоянного количества липида и обеспечение сходного L/D давали более высокий процент инкапсуляции (серия 12). Дальнейшее уменьшение концентрации маточного раствора амикацина до 30 мг/мл давало незначительное увеличение L/D, хотя возврат был все еще впечатляющим (серия 13).
%
%
Уменьшение концентрации маточного раствора амикацина имеет другие последствия. Оно уменьшает концентрацию свободного амикацина в смеси липид/амикацин после инфузии, что позволяет ему оставаться растворимым при более высокой концентрации этанола. Предполагая, что липид и амикацин смешивают в соотношении 300/500, концентрация маточного раствора амикацина составляет 50 мг/мл, а эффективность инкапсуляции составляет 37%, то исходный свободный амикацин должен составлять приблизительно 20 мг/мл. Подобно этому концентрация маточного раствора амикацина 40 мг/мл, с 52% инкапсуляцией должны обеспечивать приблизительно 12 мг/мл свободного амикацина. Концентрация маточного раствора амикацина 30 мг/мл, с 46% инкапсуляцией должны обеспечивать приблизительно 10 мг/мл свободного амикацина.
7. Соотношение липида и лекарственного средства
Имеется несколько путей увеличения удержания липосомами противоинфекционных агентов (например, аминогликозидов, таких как амикацин, тобрамицин, гентамицин). Один путь заключается в изготовлении очень больших липосом (>1 мкм), в которых удерживаемый объем на количество липида является большим. Указанный подход для достижения более малого соотношения L/D является непрактичным для ингаляции (распыления) липосом, поскольку 1) напряжение сдвига во время распыления имеет тенденцию разрушать липосомы в зависимости от размеров, когда более крупные липосомы (>0,5 мкм) демонстрируют большее высвобождение, и 2) более мелкие размеры капель, необходимые для хорошего депонирования в легких, составляют менее приблизительно 3 мкм. Таким образом, для ингаляции желательно поддерживать самый маленький, насколько это возможно, размер липосом, чтобы избежать слишком большого высвобождения. В настоящее время средний диаметр липосом, описанных в настоящем документе, составляет менее приблизительно 0,4 мкм (см. таблицу 4).
Другим подходом для уменьшения L/D является использование отрицательно заряженных липидов. Аминогликозиды, перечисленные выше, имеют большой положительный заряд, с 4-5 аминами на соединение. В терапевтических композициях обычно используются сульфатные соли указанных аминогликозидов. Вместе с многокатионным характером проявляется сильное связывание с отрицательно заряженными липосомами. Это обеспечивает более сильное удержание во время формирования липосом. Целью противоинфекционных композиций является обеспечение замедленного высвобождения в среду легких. Быстрый клиренс липосом в силу поглощения макрофагами будет действовать противоположным образом. Хорошо документально зафиксирован тот факт, что отрицательно заряженные липосомы в значительно более высокой степени захватываются макрофагами, чем нейтральные липосомы. Таким образом, желательно использовать нейтральные липосомы.
Одна группа технологий, которые обеспечивают высокую степень удержания в маленьких липосомах, основана на градиентной загрузке, когда градиент рН, градиент сульфата аммония или градиент Mg-сульфата используются для загрузки аминовых лекарственных средств в липосомы: см. патенты США №№ 5578320, 5736155, 5837279, 5922350 (градиент рН); 5837282, 5785987 (градиент Mg-сульфата); и 5316771 (градиент сульфата аммония). Указанные технологии работают только в случае проникающих через мембрану аминов (моноаминов, нейтральная форма которых является проникающей, таких как доксорубицин и даунорубицин). Градиентная загрузка не будет работать в случае определенных противоинфекционных агентов, таких как аминогликозиды, поскольку они не проникают (слишком большие и имеют слишком высокий заряд).
Все процессы, описанные в настоящем документе, могут быть легко адаптированы для асептического производства в промышленных масштабах. Конечный размер липосом может быть скорригирован посредством изменений липидной композиции, концентрации, наполнителей и параметров обработки.
Соотношение липида и лекарственного средства при использовании способов по настоящему изобретению составляет приблизительно от 4:1 до 1:1. В другом варианте осуществления настоящего изобретения соотношение липида и лекарственного средства составляет приблизительно от 3:1 до 1:1, от 2:1 до 1:1, приблизительно 1:1 или менее, приблизительно 0,75:1 или менее или приблизительно 0,5:1 или менее. В дальнейшем процентная доля свободного противоинфекционного агента, присутствующая после того, как продукт прошел диализ определенной продолжительности, уменьшается.
8. Результаты
8.1 Биопленочные барьеры при легочных инфекциях
Препятствием для лечения инфекционных заболеваний, таких как Pseudomonas aeruginosa, ведущая причина хронического заболевания у пациентов с муковисцидозом, является проникновение лекарственного средства в барьер на эпителиальных клетках, состоящий из мокроты/биопленки (фиг. 1). На фиг. 1 кольцевидные формы изображают липосомальную противоинфекционную композицию, символ «+» представляет свободный противоинфекционный агент, символ «-» представляет муцин, альгинат и ДНК, а закрашенные прямоугольники - Pseudomonas aeruginosa. Указанный барьер состоит из колонизированной и планктонной P.аeruginosa, вкрапленной в альгинат или в экополисахариды из бактерий, а также ДНК из разрушенных лейкоцитов и муцин из легочных эпителиальных клеток, которые обладают результирующим отрицательным зарядом (Costerton, et al., 1999). Указанный отрицательный заряд связывает и предотвращает проникновение положительно заряженных лекарственных средств, таких как аминогликозиды, что делает их биологически неэффективными (Mendelman et al., 1985). Удержание противоинфекционных агентов внутри липосомальных или липидных композиций может защитить или частично защитить противоинфекционные агенты от неспецифичного связывания с мокротой/биопленкой, позволяя липосомальным или липидным композициям (с удерживаемым внутри аминогликозидом) проникать (фиг. 1).
Амикацин, как было показано, обладает в высокой степени резистентностью к бактериальным ферментам, обеспечивая больший процент чувствительных клинических изолятов, чем обнаруживают в случае других аминогликозидов, включая тобрамицин и гентамицин (Price et al., 1976). В частности, изоляты P.аeruginosa являются значительно более чувствительными к амикацину, чем другие аминогликозиды, в то же время не демонстрируя перекрестной резистентности (Damaso et al., 1976).
Замедленное высвобождение и депо-эффект липосомальных композиций амикацина отчетливо виден на фиг. 2. В данном исследовании крысам давали тобрамицин посредством интратрахеального и внутривенного введения. Крысам давали также липосомальные композиции амикацина интратрахеально в той же дозе (4 мг/крыса). Данные показывают, что только в случае липосомальной композиции амикацина достигается замедленное высвобождение и депо-эффект. Фактически, через 24 часа после введения, только липосомальные композиции амикацина демонстрируют значимые уровни лекарственного средства в легких животных, в то время как композиции тобрамицина обнаруживали ничтожно малые уровни, прежде всего, вследствие, как полагают, быстрого системного всасывания. Это более чем стократное повышение аминогликозида в легких, в случае липосомальных противоинфекционных композиций, поддерживает идею липосомальных композиций противоинфекционных агентов с замедленным высвобождением, которые можно вводить значительно реже, чем разрешенную в настоящее время композицию TOBI® (раствор тобрамицина для ингаляций, выпускаемый компанией Chiron Corporation, Ameryville, CA).
Помимо этого присутствие мокроты/биопленки предотвращает проникновение свободных аминогликозидов из-за связывания противоинфекционных агентов с ее поверхностью (фиг. 1). Таким образом, дозы выше 1000 г тобрамицина/грамм легочной ткани требуются для получения терапевтического эффекта у пациентов с CF. Это преодолевается с использованием липосомальных композиций амикацина. Так, терапевтический уровень лекарственного средства сохраняется в течение более длительного периода времени при использовании липосомальных композиций амикацина по сравнению со свободным тобрамицином. Указанное облегчение связывания и проникновения может стать средством, благодаря которому липосомальные композиции амикацина могут значительно уменьшать бактериальную резистентность, обнаруживаемую повсеместно, когда антибактериальные агенты присутствуют in vivo на уровнях ниже минимальной ингибирующей концентрации.
8.2 Фармакокинетика
Фармакокинетика амикацина изучалась на крысах после интратрахеального (ИТ) введения свободного тобрамицина или липосомальных композиций амикацина. Указанные данные сравнивали с распределением, полученным в легких после инъекции свободного тобрамицина в хвостовую вену. Во всех случаях каждой крысе вводили дозу 4 мг. Как можно видеть на фиг. 2, гораздо большее депонирование аминогликозида можно обеспечить посредством ИТ по сравнению с инъекцией. Депо-эффект липосомальной противоинфекционной технологии также продемонстрирован в указанном случае, по сравнению с тобрамицином, введенным посредством как ИТ, так и ВВ; более чем стократное увеличение лекарственного средства в случае липосомальных композиций амикацина все еще остается в легких спустя двадцать четыре часа после введения. Так, терапевтический уровень лекарственного средства сохраняется в течение более длительного периода времени при использовании липосомальных композиций амикацина по сравнению со свободным тобрамицином.
Связывание аминогликозида с мокротой у пациентов с CF представляет собой проблему, особенно, если указанное связывание уменьшает биологическую активность противоинфекционного агента (Hunt et al., 1995). Для того, чтобы определить, могут ли липосомальные композиции амикацина сохранять биологическую активность в течение длительного периода времени, здоровым крысам вводили липосомальные композиции амикацина посредством закапывания в трахею. Затем следовало их удаление через 2 или 24 часа посредством бронхиального альвеолярного лаважа (BAL), для определения биологической активности. Образцы концентрировали с использованием ультрафильтрации с последующим фильтрованием (0,2 микрона) для удаления контаминирующих легочных микробов. Концентрацию амикацина определяли с использованием инструмента TDX, а биологическую активность определяли с использованием анализа с разведением на бульоне Mueller Hinton (Pseudomonas aeruginosa). Результаты показаны в таблице 7.
(мкг/мл)
(мкг/мл)
Как видно из представленной выше таблицы, восстановленная отфильтрованная липосомальная композиция амикацина была способна убивать P.aeruginosa в анализе с разведением на бульоне Mueller Hinton даже после 24 часов при МИК 4. В случае 2 часов была получена МИК 2, которая аналогична МИК, полученной в случае профильтрованного липосомального/комлексированного маточного раствора амикацина. Таким образом, липосомальная композиция амикацина все еще была активной после 24-часового пребывания в легком. Через 24 часа после введения свободный тобрамицин в той же дозе не поддавался выявлению в BAL. Это показывает, что липосомальная противоинфекционная композиция не только задерживается в легком, но также является свободно доступной для проникновения в мокроту/биопленку с течением времени. Указанные данные, объединенные с теми фактами, как очевидно следует из фиг. 2 и таблицы 9 (ниже), что липосомальные композиции амикацина высвобождают свободный противоинфекционный агент в течением времени, в то же время сохраняя высокие уровни противоинфекционного агента в легких, подтверждают разумное обоснование, что указанная система может обеспечивать замедленный противоинфекционный эффект по прошествии длительного времени. Указанный эффект должен оказаться значимым для уменьшения как биологической нагрузки P.aeruginosa, так и развития резистентности из-за недостаточных уровней противоинфекционного агента.
В качестве in vitro демонстрации медленного высвобождения липосомальной композиции амикацина и ее замедленного противоинфекционного действия композицию инкубировали в мокроте пациентов с хронической обструктивной болезнью легких (COPD), содержащей PAOI мукоидную Pseudomonas. Липосомальную композицию амикацина инкубировали также в альгинате, содержащем PAO1 мукоидную Pseudomonas. В обоих случаях наблюдалось продленное и усиленное устранение Pseudomonas в течение продолжительного периода времени, как показано в таблице 8.
амикацина
(мкг/мл)
Классические кривые гибели микроорганизмов не применимы к технологии липосомальных противоинфекционных композиций, поскольку липосомальные композиции демонстрируют замедленное высвобождение противоинфекционного агента с усиленным противоинфекционным эффектом. Липосомальная композиция защищает амикацин от мокроты и/или альгината вплоть до его высвобождения. Со временем наблюдается полная гибель, согласующаяся с моделью замедленного противоинфекционного эффекта медленного высвобождения, без помех или инактивации противоинфекционного агента.
Эффективность липосомальных композиций амикацина изучали с использованием модели хронической легочной инфекции (Cash et al., 1979), в которой P.aeruginosa, залитую в матрикс из гранул агарозы, закапывали в трахею крысам. Указанная модель мукоидной Pseudomonas на животных была разработана таким образом, чтобы напоминать инфекции Pseudomonas, наблюдающиеся у больных с CF. Некоторые из клинических коррелятов CF включают: сходную легочную патологию, развитие иммунных комплексных расстройств и превращение в штаммы P.aeruginosa мукоидного фенотипа (Cantin and Woods, 1999). Легкие крыс инфицировали с использованием свыше 107 КОЕ мукоидной Pseudomonas (штамм РАО1), полученной из изолята от пациента с CF, а затем лечили их (а) свободным аминогликозидом, (b) липидным носителем отдельно, в качестве нелекарственного контроля, и (с) липосомальной композицией амикацина. Помимо этого композиции сначала подвергали скринингу на способность убивать P.aeruginosa in vitro на модифицированных чашках Kirby-Bauer.
Различные липосомальные композиции амикацина испытывали на основе различных липидных составов или параметров изготовления, в результате чего получали различные зоны гибели в экспериментах in vitro. Данный эксперимент был разработан для того, чтобы определить возрастание эффективности, полученной при использовании липосомальных композиций аминогликозида, по сравнению со свободным аминогликозидом. Сравнивали пустые контрольные липидные композиции, две различных липосомальных композиции амикацина и свободный амикацин, и свободный тобрамицин в тех концентрациях аминогликозида, что и липосомальных противоинфекционных композициях. Помимо этого также вводили в 10 раз более высокую дозу свободного амикацина и в 10 раз более высокую дозу свободного тобрамицина. Введение осуществляли ИТ один раз в день в течение семи дней. Результаты (фиг. 3) показывают, что липосомальный амикацин в двух композициях (различающихся по липидному составу) продемонстрировал значимое уменьшение уровней КОЕ и действовал лучше в том, что касается уменьшения КОЕ, чем свободный амикацин или свободный тобрамицин в 10 раз более высоких дозах. На фиг. 5 Lip-An-14 представляет собой DPPC/Chol/DOPC/DOPG (42:45:4:9) и 10 мг/мл амикацина, Lip-An-15 представляет собой DPPC/Chol (1:1) также при 10 мг/мл. Все липид-липидные и липид-лекарственные соотношения представлены как массовые соотношения.
Следующий эксперимент (фиг. 14) был разработан для того, чтобы продемонстрировать медленное высвобождение и замедленные противоинфекционные способности липосомальных композиций амикацина. Введение осуществляли через день в течение 14 дней, в противоположность ежедневному в течение семи дней введению, как в предыдущих экспериментах. Результаты показывают, что липосомальный амикацин в двух композициях (различающихся по липидному составу) обладал в 10-100 раз большей силой (большей способностью уменьшать уровни КОЕ), чем свободный амикацин или свободный тобрамицин. Суточная доза для человека 600 мг TOBI® (раствор тобрамицина для ингаляций, выпускаемый компанией Chiron Corporation, Ameryville, CA) или приблизительно 375 мг/м2, соответствует суточной дозе для крысы 9,4 мг. Таким образом, данные можно напрямую коррелировать с 10-100-кратным улучшением в отношении эффективности для человека. Следует отметить, что уменьшение на два log является самым лучшим, которое можно наблюдать на указанной модели. 100-кратное уменьшение P.aeruginosa в исследованиях мокроты коррелировали с улучшением функции легких (Ramsey et al., 1993). Замедленное высвобождение липосомальных композиций амикацина показывает, что можно использовать более низкую дозу и/или менее частое введение, чтобы получить большее уменьшение бактериального роста, чем можно получить при использовании свободного аминогликозида.
Эффективность липосомальной композиции амикацина изучали с использованием модели хронической легочной инфекции, в которой P.aeruginosa заливали в матрикс из гранул агарозы, который закапывали в трахею крысам Sprague-Dawley. Спустя три дня свободный амикацин или липосомальный амикацин вводили каждый день (фиг. 3) или через день (фиг. 4) в дозе 1 мг, или 10 мг свободного амикацина, или 1 мг липосомального амикацина, а также пустые липосомы (липидный носитель) в качестве контроля; группы состояли из пяти крыс.
Гомогенизированные легкие крыс (замороженные) после 14-дневного эксперимента изучали на предмет содержания и активности аминогликозида. Клиническое химическое исследование осуществляли с использованием инструмента TDX, в то время как биологическое исследование осуществляли путем измерения зон ингибирования на пластинах из агара, в который была залита Bacillus subtilis. Результаты показаны в таблице 9:
Массы лекарственных средств представлены для лекарственного средства, стандартизированного к отсутствию любой солевой формы.
Результаты, представленные в таблице 10, показывают, что аминогликозид присутствует и является активным в случае обеих липосомальных противоинфекционных композиций, в то время как мало свободного аминогликозида можно было выявить даже при использовании в 10 раз более высокой дозы. Указанные дополнительные результаты устанавливают характеристики замедленного высвобождения липосомальных противоинфекционных композиций, а также подтверждают, что противоинфекционный агент, который остается, все еще является активным. Из представленных композиций только свободный тобрамицин (0,1 микрограмм/мл) демонстрировал какие-либо поддающиеся обнаружению уровни аминогликозида в почках.
Замедленное высвобождение и депо-эффект липосомальной композиции амикацина дополнительно показаны на фиг. 5. У крыс моделировали хроническую легочную инфекцию, закапывая в трахею P.aeruginosa, залитую в матрикс из гранул агарозы, с использованием тех же гранул, которые использовали при изучении эффективности. Затем крысам давали свободный тобрамицин или липосомальный амикацин (композиция Lip-An-14) посредством интратрахеального введения в одной и той же дозе (2 мг). Данные, измеренные в микрограммах противоинфекционного агента на грамм легочной ткани с течением времени, показали, что липосомальный противоинфекционный агент демонстрирует замедленное высвобождение и депо-эффект, в то время как свободный тобрамицин обнаруживал ничтожно малые уровни в легких через 24 часа, прежде всего, вследствие, как полагают, быстрого системного всасывания. Это более чем стократное повышение противоинфекционного агента в легких у зараженной крысы, в случае липосомальных композиций амикацина, поддерживает идею липосомального противоинфекционного агента с замедленным высвобождением, который можно вводить значительно реже, чем разрешенную в настоящее время композицию TOBI® (раствор тобрамицина для ингаляций, выпускаемый компанией Chiron Corporation, Ameryville, CA).
Фармакокинетику амикацина изучали на крысах после интратрахеального (ИТ) введения свободного тобрамицина или липосомального амикацина. Каждой крысе вводили дозу 2 мг. Депо-эффект липосомальной противоинфекционной технологии продемонстрирован в том, что по сравнению со свободным тобрамицином, введенным ИТ, более чем стократное увеличение лекарственного средства в случае липосомального амикацина все еще сохраняется в инфицированных легких через двадцать четыре часа после введения. Таким образом, терапевтический уровень лекарственного средства поддерживается в течение более длительного периода времени при использовании липосомальных композиций, по сравнению со свободным тобрамицином.
Фиг. 7 показывает замечательное время пребывания и аккумуляцию эффективных количеств противоинфекционного агента в легких, результат, который устанавливает, что можно использовать относительно нечастое введение. Каждая доза представляет собой 4 часа ингаляции (у крысы, 3 крысы в группе, как выше) распыленных липосомальных композиций амикацина (DPPC/Chol., 1:1) при концентрации амикацина 15 мг/мл. Введение осуществлялось на первый день; на первый, третий и пятый день; или на первый, второй, третий, четвертый и пятый день. Крыс, от которых получили указанные данные, умерщвляли после получения соответствующей указанной дозы. Композицию изготавливали, как описано в примере.
Сходные противоинфекционные агенты можно использовать для лечения внутриклеточных инфекций, таких как легочная форма сибирской язвы и туляремии. В случае легочной формы сибирской язвы споры сибирской язвы достигают альвеол в форме аэрозоля. Ингалированные споры перевариваются макрофагами легких в альвеолах и переносятся в регионарные трахеобронхиальные лимфоузлы или средостенные лимфоузлы через лимфатическую систему (Pile et al., 1998; Gleiser et al., 1968). Макрофаг играет главную роль в обоих инфицирующих путях и вносит основной вклад в саморазрушение хозяина при системной (ингаляционной) сибирской язве. Помимо своего вклада и замедленное высвобождение, и нацеливание, технология липосомальных противоинфекционных композиций может усиливать клеточный захват и может использовать альвеолярные макрофаги и эпителиальные клетки легких в нацеливании и доставке лекарственного средства. Наличие указанных свойств, как полагают, облегчает лечение указанных внутриклеточных инфекций, которые наблюдаются в легких и переносятся макрофагами. Более важно, что указанные свойства должны делать противоинфекционный агент более эффективным в том, что липосомальный противоинфекционный агент будет подвергаться фагоцитозу именно теми клетками, которые несут в себе болезнь. Противоинфекционный агент будет направленно высвобождаться внутри клетки, атакуя инфекцию до того, как она будет диссеминирована. Инкапсулированное лекарственное средство может представлять собой уже разрешенное к применению лекарственное средство, такое как ципрофлоксацин, тетрациклин, эритромицин или амикацин. Разработаны липосомальные композиции ципрофлоксацина.
В данном исследовании указанное соединение вводили мышам и сравнивали свободный ципрофлоксацин при интратрахеальном введении и свободный ципрофлоксацин при пероральном введении со всеми тремя соединениями в одной и той же дозе (фиг. 6). Доза для каждой мыши составляла 15 мкг; в каждой группе по три мыши. Липосомальный ципрофлоксацин был в DPPC/холестерине (9:1), 3 мг/мл ципрофлоксацина; композицию изготавливали как описано в примере. Соотношение липида и лекарственного средства составляло 12,5:1 по массе. При сравнении с введенным перорально ципрофлоксацином липосомальный ципрофлоксацин присутствовал в легких мышей в количествах, которые на два порядка превышали количества свободного ципрофлоксацина. Кроме того, только липосомальный ципрофлоксацин показал уровни лекарственного средства в легких спустя 24 часа, в то время как перорально введенное лекарственное средство не поддавалось обнаружению менее чем через два часа. Указанные данные свидетельствуют в пользу применения липосомальных композиций ципрофлоксацина и других противоинфекционных агентов, таких как аминогликозиды, тетрациклины и макролиды, для лечения и профилактики внутриклеточных болезней, используемых биотеррористами.
8.3. Параметры липосом
Липиды, которые выбраны для использования, растворяют в этаноле для получения липидно-спиртового раствора. Липидно-спиртовой раствор инфузируют в водный или спиртовой раствор, содержащий молекулу биологически активного агента, предназначенного для удержания в липосомах. Липиды спонтанно образуют везикулы.
Таблица 10 описывает параметры липосомальных противоинфекционных композиций, в которых липиды представляют собой DPPC и холестерин.
** Только удерживаемое количество амикацина учитывалось при расчете L/D.
Дополнительную информацию по изготовлению липосомальных противоинфекционных композиций можно найти в PCT/US03/06847, зарегистрированной 5 марта 2003 г., которая целиком включена в настоящий документ в качестве ссылки.
Удерживаемый объем является основной характеристикой липосомальной композиции и определяется как объем внутрилипосомальной водной фазы на единицу липида. Обычно он выражается в единицах мкл/мкмоль. Часто полагают, что когда образуются липосомы, концентрация растворенного вещества внутри липосом равна концентрации вне липосом, в массе раствора. Более высокий удерживаемый объем тогда должен вести к более высокому соотношению лекарственное средство/липид, т.е. к более высокой общей концентрации лекарственного средства для конечной композиции.
При изготовлении липосомального амикацина, однако, было установлено, что фактическое соотношение лекарственное средство/липид, которое можно обеспечить, было в 3 раза выше, чем можно было ожидать на основании удерживаемого объема. Таблица 11 показывает результаты для 4 различных образцов изготовления липидных противоинфекционных композиций (см. пример 1 в разделе примеры).
параметр
Образцы 1 и 2 были изготовлены с использованием процедуры инфузии этанола, описанной в настоящем документе, а образцы 3 и 4 были изготовлены с использованием технологии получения липосом, известной специалистам.
Концентрации амикацина определяли с помощью иммунофлюоресцентного анализа, с использованием реагента INNOFLUO Seradyn и анализатора TDx. Липиды определяли с помощью ВЭЖХ с обращенной фазой, с использованием колонки С-8 и детектора рассеянного света.
Липосомальный объем (объем, занимаемый липосомами, на единицу всего объема) в образцах № 1-3 определяли измерением концентрации флюоресцентной пробы (Sulforhodamine 101 или Carboxyfluorescein) в общем объеме и в объеме фильтрата композиции, полученном центрифугированием на фильтрующем устройстве CentriSart. Концентрация пробы в фильтрате выше, чем средняя концентрация из-за исключения пробы из объема, занимаемого липосомами.
В образце № 4 липосомальный объем определяли измерением концентрация ионов калия в образце после добавления его фиксированного количества, 250 мкл KCl (Vadd) в 10 мл липосомальной суспензии (V0). Образцы затем центрифугировали в течение 30 мин на 4000 об/мин и надосадочную жидкость собирали для измерения ионов калия (K) с использованием чувствительного к калию электрода Cole-Parmer. Измеренная концентрация калия была всегда выше ожидаемой из-за исключения ионов калия из объема, занятого липосомами. В контроле равное количество KCl добавляли в 10 мл солевого раствора. Измеряли концентрацию калия в контроле Кс. Водный и липосомальные объемы затем устанавливали следующим образом:
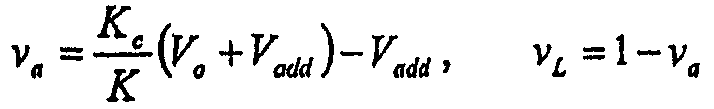
Зная липосомальный объем и концентрацию липида, можно определить удерживаемый объем:
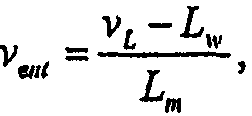
где Lw и Lm представляют собой массовую и молярную концентрации липида, соответственно. Плотность липида допускают близкой к 1 мг/мл. Соответственно, можно определить ожидаемое соотношение липид/лекарственное средство, которое будет иметь образец, если лекарственное средство распределяется идеально в водных пространствах внутри и снаружи липосом:
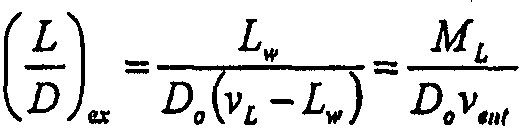
где D0 представляет собой объемную концентрацию лекарственного средства во время формирования липосом, ML представляет собой среднюю молекулярную массу липидов.
Как можно видеть, фактические соотношения L/D для образцов № 1 и № 2 (1,8 и 1,9) устойчиво ниже, чем можно было ожидать от равномерного распределения амикацина (5,6 и 6,0), в то время как L/D для образцов № 3 и № 4 ближе к теоретическим величинам.
Аналогичное сравнение проводили для 2 образцов изготовления липидных противоинфекционных композиций, в которых противоинфекционным агентом был гентамицина сульфат (см. пример 3 в разделе примеры) данные в таблице 12 показывают, что способ, описанный в настоящем документе, обеспечивает неожиданно высокое удержание гентамицина. В обоих образцах № 5 и № 6 фактические соотношения L/D почти вдвое превышали теоретически ожидаемые величины.
8.4. Высвобождение лекарственного средства, опосредованное инфекцией P.aeruginosa
Высвобождение лекарственного средства в активной форме вблизи от инфекций является важным аспектом действия липосомальных композиций лекарственных средств по настоящему изобретению. Потенциал для указанного нацеленного высвобождения испытывали путем мониторинга высвобождения лекарственного средства после инкубации с мокротой от пациента с CF, высвобождения в легких крыс, которым предварительно инокулировали P.aeruginosa, а также активности против культур P.aeruginosa.
Высвобождение амикацина прямой инкубацией культуры P.aeruginosa с липосомальной композицией амикацина по настоящему изобретению обсуждалось ранее. Для дальнейшего изучения данного феномена липосомальную композицию амикацина инкубировали с препаратом мокроты от пациента, страдающего муковисцидозом, с инфекцией P.aeruginosa. Выделенную мокроту разжижали бычьей ДНКазой I и альгинатной лиазой в течение 2 ч при 37°С. Липосомальную композицию амикацина или растворимый амикацин (1 мг/мл амикацина) смешивали 1:1 с разжиженной мокротой или контролем и инкубировали при 37°С при осторожном встряхивании. Аликвоты анализировали на концентрацию амикацина с использованием анализатора Abbott TDx. Интактные липосомы лизировали в отдельной аликвоте каждого образца с использованием детергента, 1% Triton X-100. Для анализа использовали надосадочные жидкости от каждого образца. В течение 48 часов (80-90%) амикацина высвобождалось зависимым от времени образом из липидной композиции при указанных условиях, что показывает на то, что высвобождение лекарственного средства может наблюдаться в участках инфекции в легких при CF.
Высвобождение лекарственного средства из липосом in vivo сравнивали у крыс, которым закапывали агаровые гранулы, содержащие P.aeruginosa (3,5×104 КОЕ каждой крысе), и у контрольных крыс. Через три дня после инстилляции крысам давали вдыхать липосомальные композиции амикацина по настоящему изобретению (приблизительно 6 мг/кг суточная доза) каждый день (группа, которой не вводили бактерии) или через день в течение 14 дней (группа, которой вводили гранулы). Через 24 часа после последнего лечения общий амикацин и свободный амикацин измеряли, как описано выше. У крыс, которые получали бактерии, в среднем приблизительно 50-70% обнаруженного амикацина составляла свободная форма, т.е. высвобожденная из липосом. У крыс, которые не получали бактерии, приблизительно 20-25% лекарственного средства составляла свободная форма. Указанные данные убедительно предполагают, что высвобождение свободного амикацина из липосомы может быть опосредовано присутствием P.aeruginosa in vivo.
Тест in vitro на высвобождение и активность осуществляли в условиях, аналогичных условиям при изучении фармакокинетики в легких, когда было предварительно показано, что свободный антибиотик исчезает в течение нескольких часов. Свободный амикацин или липосомальную композицию амикацина инкубировали с P.aeruginosa РА01 (приблизительно 108/мл) в стерильных 0,5 мл картриджах Slide-A-Lyzer при различных концентрациях лекарственного средства. Свободное лекарственное средство диализируется из картриджей при данных условиях в течение часов. Спустя 24 часа образцы изымали из картриджей и высевали на чашки, чтобы подсчитать КОЕ. В предварительных экспериментах свободный амикацин лишь слегка уменьшал КОЕ в указанных образцах, в то время как два log уменьшение КОЕ наблюдалось в случае липидных композиций, содержавших амикацин, при той же концентрации амикацина (50 мкг/мл). Указанные данные наводят на мысль о том, что амикацин в самом деле высвобождается в активной форме в присутствии бактерий, и что медленное высвобождение, обеспечиваемое композицией, делает применение лекарственного средства более эффективным.
Взаимодействие липосомальных композиций амикацина по настоящему изобретению с P.aeruginosa или ее факторами вирулентности приводит к высвобождению амикацина, возможно, направленному высвобождению, в участке инфекции. Когда амикацин высвобожден, он обладает активностью против P.aeruginosa, а медленное высвобождение вблизи от бактерий может иметь преимущество перед неспецифическим распределением и быстрым клиренсом ингалированного свободного лекарственного средства.
8.5. Влияние ингалированных липосомальных лекарственных композиций на функцию альвеол
Макрофаги
Липосомальные композиции амикацина по настоящему изобретению, в одном варианте осуществления настоящего изобретения, представляют собой инкапсулированную в липосомах в наномасштабе (200-300 нм) форму амикацина, которая изготовлена для лечения хронических инфекций P.aeruginosa у пациентов, страдающих муковисцидозом. Она разработана для ингаляции с замедленным высвобождением в легком. Поскольку альвеолярные макрофаги, как известно, жадно поглощают частицы в указанных пределах размеров, влияние липосомальных композиций на указанные клетки представляет особый интерес. Основные и стимулированные функции крысиных альвеолярных макрофагов, полученных посредством лаважа, изучали с введением и без введения липосомальных композиций амикацина и сравнивали с различными контролями.
Аэрозоли липосомальных композиций амикацина, амикацина, липосом-плацебо и физиологического раствора генерировали с использованием небулайзера PARI LC Star, и давали их вдыхать самкам крысы CD®IGS в камере для ингаляций только носом. Ингаляционную терапию осуществляли в течение 4 часов в течение 14 дней подряд, таким образом, чтобы суточная легочная доза общего липида составляла приблизительно 12 мг/кг для группы липосомального амикацина и 11 мг/кг для группы плацебо-липосом. Половину крыс умерщвляли на 15 день. Остальных крыс умерщвляли на 43 день. Жидкость после бронхиального альвеолярного лаважа (BALF) собирали от каждой крысы и хранили при -80°С для последующего анализа на оксид азота (представленного общими нитратами) и фактор некроза опухолей альфа (TNF-α). Клетки из BALF собирали центрифугированием, подсчитывали и культивировали в среде с липополисахаридом (LPS) и без него в течение 24 часов. Надосадочные жидкости из указанных культур собирали центрифугированием и анализировали на оксид азота TNF-α. Фагоцитарную функцию BAL макрофагов ((106)/мл) определяли измерением по захвату опсонизированных флюоресцентных микросфер (0,2 мкм, 2(106)/мл) за ночь.
Ингаляция липосомальной композиции амикацина, пустых липосом, растворимого амикацина или физиологического раствора в течение 14 последовательных дней не вызывала значительной острой или отсроченной воспалительной реакции в легких крыс, как следует из уровней оксида азота (нитратов) и TNF-α в BALF, которые незначимо отличались от контролей, хотя имелась ранняя тенденция к более высоким уровням NO во всех группах, получавших ингалянты, включая контроли. Общее восстановление клеток незначимо отличались во всех группах с ранней тенденцией к большему количеству полиморфно-ядерных лейкоцитов во всех группах, получавших ингалянты. Крысиные альвеолярные макрофаги имели нормальные функции после воздействия аэрозолей указанных выше испытуемых веществ, несмотря на тот факт, что они выглядели увеличенными на 15 день в группах с ингаляцией липосом. Концентрации нитратов и TNF-α, выявленные после культивирования альвеолярных макрофагов в среде на 15 или 43 день исследования, незначимо отличались от контролей. Макрофаги реагировали нормально после стимуляции с использованием LPS, вырабатывая значительные концентрации оксида азота (20-40 нмоль/106 клеток) и TNF-α (5-20 нмоль/106 клеток). Указанные макрофаги обладали также нормальной фагоцитарной функцией, как показано идентичным захватом флюоресцентных гранул по сравнению с нелеченными контролями.
Ингаляция липосомальных композиций амикацина в течение 14 последовательных дней не оказывала значимого влияния на функцию альвеолярных макрофагов в том, что касается фагоцитоза опсонизированных гранул, выработки медиаторов воспаления TNF и NO.
9. Дозы
Дозировка любых композиций по настоящему изобретению будет меняться в зависимости от симптомов, возраста и массы тела пациента, природы и тяжести заболевания, которое лечат или предотвращают, пути введения и формы композиции, являющейся предметом изобретения. Любые из указанных композиций можно вводить в виде однократной дозы или разделенных доз. Дозировки для композиций по настоящему изобретению можно легко определить способами, известными специалистам, или способом, описанным в настоящем документе.
В некоторых вариантах осуществления настоящего изобретения доза рассматриваемых соединений будет составлять, главным образом, приблизительно от 0,01 нг до 10 г на кг массы тела, особенно, приблизительно от 1 нг до 0,1 г на кг, и, более конкретно, приблизительно от 100 нг до 50 мг на кг.
Эффективную дозу или количество и любые возможные влияния на время введения композиции, возможно, будет необходимо идентифицировать для каждой конкретной композиции по настоящему изобретению. Это можно осуществить обычным экспериментом, как описано в настоящем документе, с использованием одной или более групп животных (предпочтительно, по меньшей мере 5 животных на группу), или в ходе испытаний на людях, если это уместно. Эффективность любой рассматриваемой композиции и способа лечения или профилактики можно оценивать введением композиции и оценкой эффекта введения, путем измерения одного или более подходящих показателей, и сравнением величин указанных показателей после лечения с величинами тех же показателей до лечения.
Точное время введения и количество любой конкретной рассматриваемой композиции, которое будет обеспечивать наиболее эффективное лечение данного пациента, будет зависеть от активности, фармакокинетики и биодоступности рассматриваемой композиции, физиологического состояния пациента (включая возраст, пол, тип и продолжительность заболевания, общее состояние здоровья, чувствительность к данной дозе и типу лекарственного средства), пути введения и т.п. Указания, представленные в настоящем документе, можно использовать для оптимизации лечения, например, для определения оптимального времени и/или количества введения, что потребует не более чем рутинного экспериментирования, состоящего из мониторинга субъекта и корректировки дозы и/или времени введения.
Во время лечения здоровье пациента можно мониторировать измерением одного или более подходящих показателей в заранее определенные моменты времени во время периода лечения. Лечение, включая композицию, количества, время введения и состав, можно оптимизировать в соответствии с результатами указанного мониторинга. Можно периодически производить повторную оценку пациента для определения степени улучшения путем измерения тех же параметров. Корректировку количества (количеств) рассматриваемой композиции и, возможно, времени введения, можно осуществлять, руководствуясь указанными повторными оценками.
Лечение можно начинать с более низких доз, которые меньше оптимальной дозы соединения. Затем дозу можно понемногу увеличивать, до достижения оптимального терапевтического эффекта.
Использование рассматриваемых композиций может уменьшить требующуюся дозу для любого отдельного агента, содержащегося в композициях (например, противоинфекционного агента), поскольку начало и продолжительность эффекта различных агентов могут быть взаимодополнящими.
Токсичность и терапевтическую эффективность рассматриваемых композиций можно определить с использованием стандартных фармацевтических процедур, на клеточных культурах или экспериментальных животных, например, для определения LD50 и ED50.
Данные, полученные исследованиями с использованием клеточных культур и животных, можно использовать для определения пределов доз для человека. Доза любой рассматриваемой композиции предпочтительно находится в пределах циркулирующих концентраций, которые включают в себя ED50 с малой токсичностью или с отсутствием токсичности. Доза может варьировать в указанных пределах, в зависимости от используемой лекарственной формы и пути введения. Для композиций по настоящему изобретению терапевтически эффективная доза может быть установлена первоначально в исследованиях на клеточных культурах.
10. Препаративная форма
Липидные противоинфекционные препаративные формы по настоящему изобретению могут включать в себя водную дисперсию липосом. Препаративная форма может содержать липидные наполнители для формирования липосом, и соли/буферы для обеспечения соответствующей осмолярности и рН. Препаративная форма может включать в себя фармацевтический наполнитель. Фармацевтический наполнитель может представлять собой жидкость, разбавитель, растворитель или инкапсулирующий материал, участвующие в носительстве или транспортировании любой рассматриваемой композиции или ее компонента из одного органа или части тела в другой орган или часть тела. Каждый носитель должен быть «приемлемым» в том смысле, что он должен быть совместимым с рассматриваемой композицией и ее компонентами и не быть вредным для пациента. Подходящие наполнители включают в себя трегалозу, раффинозу, маннит, сахарозу, лейцин, трилейцин и хлорид кальция. Примеры других подходящих наполнителей включают в себя (1) сахара, такие как лактоза и глюкоза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как натрий-карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) наполнители, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) физиологический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) растворы фосфатного буфера и (21) другие нетоксичные совместимые вещества, применяемые в фармацевтических композициях.
ПРИМЕРЫ
Пример 1
Далее представлено подробное описание изготовления 150 мл липосомального/комплексированного амикацина.
Общий исходный объем = 1,5 л
Содержание этанола = 23,5% (об./об.)
Липидный состав: DPPC/Chol (молярное соотношение 1:1)
Исходный [липид] = 7,6 мг/мл
Исходный [амикацина сульфат] = 57,3 мг/мл
Объем конечного продукта = 150 мл
I) Cоставление смеси и инфузия:
7,47 г DPPC и 3,93 г холестерина растворяли непосредственно в 352,5 мл этанола на водяной бане с температурой 50°С. 85,95 г амикацина сульфата растворяли непосредственно в 1147,5 мл буфера PBS. Раствор затем титровали с использованием ионов NaOH или КОН для доведения рН приблизительно до 6,8.
352,5 мл этанола/липида добавляли или инфузировали в 1147,5 мл амикацина/буфера, с получением общего исходного объема 1,5 л. Этанол/липид нагнетали @30 mUмин (также называется скоростью инфузии) с использованием перистальтического насоса в раствор амикацин/буфер, который быстро перемешивали при 150 об/мин в реакционном сосуде на диске для перемешивания при комнатной температуре.
Продукт перемешивали при комнатной температуре в течение 20-30 минут.
II) Диафильтрация или стадия «Промывания»:
сосуд для смешивания подключали к перистальтическому насосу и диафильтрационному картриджу. Диафильтрационный картридж представляет собой мембрану из полого волокна с отсечкой молекулярной массы 500 килодальтон. Продукт прокачивали из реакционного сосуда через диафильтрационный картридж, а затем назад в сосуд для смешивания при комнатной температуре. Противодавление приблизительно 7 фунтов/кв.дюйм создавали во всем картридже. Свободный амикацин и этанол принудительно нагнетали через мембрану из полого волокна посредством противодавления, оставляя позади липосомальный амикацин (продукт). Продукт промывали 8 раз при комнатной температуре. Добавляли свежий буфер PBS (посредством другого перистальтического насоса) в реакционный сосуд, чтобы компенсировать удаление растворенного вещества и сохранения постоянного объема продукта.
Продукт концентрировали.
ПРИМЕР 2
Высокое удержание амикацина липосомами. Четыре образца липидных противоинфекционных композиций изготавливали при различных концентрациях липида и противоинфекционного агента, согласно следующим процедурам.
Образец № 1. 1,72 кг амикацина сульфата растворяли в 23 литрах физиологического раствора (0,9% NaCl) и рН доводили до 6,5 добавлением необходимого количества NaOH. Липиды - 98,2 г DРPC и 51,8 г холестерина растворяли в 7 литрах этанола. Липосомы формировали инфузией липидного раствора в раствор амикацина со скоростью приблизительно 600 мл/мин при постоянном перемешивании. Полученную суспензию затем промывали для удаления этанола и незахваченного липосомами амикацина посредством диафильтрации с использованием картриджа из полого волокна Amersham с размером пор 500 кДа. Суспензию концентрировали до конечного объема приблизительно 3,5 л.
Образец № 2. Процедура была аналогична той, которая использовалась для образца № 1, с уменьшением количеств всех материалов в 100 раз. 17,2 г амикацина сульфата растворяли в 230 мл физиологического раствора (0,9% NaCl) и рН доводили до 6,6 добавлением необходимого количества NaOH. Липиды - 0,982 г DРPC и 0,518 г холестерина растворяли в 70 мл этанола. Липосомы формировали инфузией липидного раствора в раствор амикацина со скоростью приблизительно 300 мл/мин при постоянном перемешивании. Полученную суспензию затем промывали для удаления этанола и незахваченного липосомами амикацина посредством диафильтрации с использованием картриджа из полого волокна Amersham. Суспензию концентрировали до конечного объема приблизительно 35 мл.
Образец № 3. Липосомы изготавливали с использованием процедуры, известной как SPLV. 1,4 г амикацина сульфата растворяли в 20 мл физиологического раствора (0,9% NaCl) и рН доводили до 3,3. Липиды, 0,666 г DРPC и 0,333 г холестерина растворяли в 40 мл дихлорметана. Растворы амикацина и липидов смешивали в 500 мл колбе с круглым дном и короткое время обрабатывали ультразвуком для формирования эмульсии. Колбу затем соединяли с системой BUCHI Rotavapor для удаления дихлорметана при низком вакууме (-5 дюймов Hg) и температуре 50° и постоянном вращении, до тех пор, пока амикацин-липидная смесь не образует гель. Когда гель в конечном итоге оседал, вакуум постепенно повышали до -20 дюймов и сушку продолжали в течение еще 30 минут. Конечный объем сформированной суспензии липосом составлял 22 мл.
Образец № 4. Процедура была аналогична той, которая использовалась для образца № 3. 1,3 г амикацина сульфата растворяли в 20 мл физиологического раствора и рН доводили до 6,5 добавлением NaOH. Липиды, 0,583 г DРPC и 0,291 г холестерина растворяли в 35 мл дихлорметана. Процедуру ультразвуковой обработки пропускали. Стадию удаления растворителя с использованием системы Rotavapor осуществляли при температуре 40° в течение 2 ч. Конечный объем составлял 20 мл.
ПРИМЕР 3
Высокое удержание гентамицина липосомами.
Образец № 5. 20,0 г гентамицина сульфата растворяли в 230 мл физиологического раствора (0,9% NaCl) и рН доводили до 6,5 добавлением необходимого количества серной кислоты. Липиды - 0,982 г DРPC и 0,518 г холестерина растворяли в 70 мл этанола. Липосомы формировали инфузией липидного раствора в раствор гентамицина со скоростью приблизительно 500 мл/мин при постоянном перемешивании. Незахваченный липосомами гентамицин и этанол удаляли посредством диафильтрации с использованием картриджа из полого волокна Amersham. Суспензию концентрировали до конечного объема приблизительно 35 мл.
Образец № 6. Процедура была аналогична той, которая использовалась для образца № 5, за исключением: 17,0 г гентамицина сульфата растворяли в 230 мл 100 мМ раствора Na2SO4 и рН доводили до 6,5 добавлением необходимого количества H2SO4. Липиды - 0,982 г DРPC и 0,518 г холестерина растворяли в 75 мл этанола.
ПРИМЕР 4
Удержание липосомами других форм амикацина.
Образец № 7. Процедура была аналогична той, которая использовалась для образца № 2 примера 2. 10,7 г основания амикацина и 4,2 г лимонной кислоты растворяли в 230 мл физиологического раствора (0,9% NaCl). рН полученного раствора амикацина/цитрата составляла 6,2. Липиды - 0,982 г DРPC и 0,518 г холестерина растворяли в 70 мл этанола. Липосомы формировали инфузией липидного раствора в раствор амикацина со скоростью приблизительно 500 мл/мин при постоянном перемешивании. Незахваченный липосомами амикацин и этанол удаляли посредством диафильтрации с использованием картриджа из полого волокна Amersham. Суспензию концентрировали до конечного объема приблизительно 35 мл.
Фактическое соотношение липид/лекарственное средство было аналогичным таковому для образца № 2 и вновь ниже ожидавшегося (удержание лекарственного средства выше ожидавшегося). С учетом того факта, что удерживаемый объем в образце № 7 составлял лишь 1,5 (по сравнению с 2,5 для образца № 2), соотношение ожидаемое/фактическое L/D составляло целых 5,2. Таким образом, липосомальный амикацина цитрат, подобно амикацина сульфату, также может в высокой степени удерживаться липосомами.
ПРИМЕР 5
Биодоступность амикацина из ингалированных липосомальных композиций амикацина у крыс
Скорость высвобождения амикацина из липосом измеряли после ингаляции крысами и сравнивали с ингалированным растворимым амикацином.
Испытуемые агенты распыляли с использованием небулайзера Pari LC Star, присоединенного к камере для ингаляций только носом. Крысы CD®IGS получали установленную легочную депонированную дозу 6 мг/кг амикацина в форме липосомальной композиции или 5 мг/кг растворимого амикацина в виде однократной дозы или ежедневных доз в течение 14 дней подряд. Легкое или другую ткань гомогенизировали на аппарате Polytron. Кинетику клиренса амикацина из легкого изучали по анализам легочных гомогенатов в различные моменты времени после лечения однократной дозой или через 1 день и 28 дней после введения множества доз. Уровни амикацина измеряли с использованием иммунофлюоресцентной поляризации на анализаторе Abbott TDx® в отсутствии или в присутствии 1% Triton X-100, который высвобождает амикацин из липосом. В цельные образцы легких были добавлены липосомы перед гомогенизацией, чтобы изучить высвобождение амикацина при указанных условиях. Свободный и общий амикацин измеряли в присутствии и в отсутствии 1% Triton X-100, чтобы оценить утечку лекарственного средства.
Липосомальный амикацин, добавленный в цельные образцы легких, не показал значимого высвобождения амикацина в результате гомогенизации тканей на аппарате Polytron в отсутствие указанного детергента. Однако добавление 1% Triton X-100 приводило к возврату всего ожидавшегося лекарственного средства. Таким образом, можно было провести прямое сравнение общего уровня амикацина (с детергентом) и свободно доступных уровней в легочной ткани.
Высокая общая концентрация амикацина (при 500-600 мкг/г легочной ткани) наблюдалась непосредственно после 6 мг/кг однократной дозы липосомального амикацина, которая медленно уменьшалась приблизительно на 50% в течение 7-дневного периода времени. Временной профиль для высвобождения свободного амикацина из указанных липосом показал первоначальную высокую концентрацию свободного лекарственного средства, возможно, в результате высвобождения амикацина после распыления. За указанной фазой последовал самый низкий уровень предпочтительно, через 24 часа, с последующим увеличением, достигшим максимума 279 мкг/г через 96 часов после введения. К концу 7-дневного эксперимента значительная часть лекарственного средства, остававшегося в легком, находилась в свободной форме (приблизительно 50-70%). Представлялось, что малая часть растворимого лекарственного средства, введенного ингаляцией, также оставалась в легком в течение длительного периода времени. Однако наибольшая часть амикацина, введенного в растворимой форме, исчезала в течение нескольких часов, а видимая AUC свободного амикацина свыше 7 дней была в 2 раза выше для животных, получавших липосомальный амикацин, чем у животных, получавших растворимый амикацин. Некоторые аспекты указанного поведения можно качественно смоделировать с подходящими константами скорости для клиренса и медленного высвобождения лекарственного средства из липосом.
Спустя 14 дней подряд введения (24 часа после последней дозы) более 20% общего амикацина в легких крыс, которые получали липосомальный амикацин, присутствовало в форме свободного лекарственного средства (приблизительно 650 мкг/г). Общий уровень свободного лекарственного средства был даже выше, чем количество у крыс, которые ингалировали растворимый амикацин (при 500 мкг/г).
Свободный амикацин медленно высвобождается из липосом липосомальных композиций амикацина в легких здоровых животных во временном масштабе за дни. Свободное лекарственное средство, которое высвободилось, демонстрирует относительно длительное пребывание в легком, что видно по значительному депонированию свободного лекарственного средства в легких.
ПРИМЕР 6
Процесс поточной инфузии
Суть процесса поточной инфузии заключается в том, что поток липидного раствора смешивается с потоком противоинфекционного раствора «на одной линии» через, например, Y-коннектор, который соединяется с длиной трубки, называемой трубкой для смешивания, где может наблюдаться дальнейшее смешивание. С этой точки зрения указанный новый процесс отличается от «обычного» процесса инфузии этанола, когда липидный раствор инфузируют в виде потока в массу раствора амикацина.
Изготовление растворов амикацина и липидов
12,0 г амикацина сульфата растворяли в 200 мл воды и рН доводили до 6,5 добавлением необходимых количеств 25% раствора NaOH. Липиды, 1,480 г DРPC и 0,520 г холестерина растворяли в смеси 60 мл этанола и 10 мл воды. Указанные количества дали 300 мл серии после инфузии при скорости потока липид/амикацин 300/500 мл/мин, соответственно. Объемы можно пропорционально корректировать для больших масштабов или если желательными являются разные скорости потоков.
Раствор амикацина изготавливали в соответствии с указанными выше результатами, с концентрацией приблизительно 40 мг/мл (по основанию). Липидный раствор представлял собой DPPC/Chol (молярное соотношение 60/40), с общим содержанием липида приблизительно 20 мг/мл раствора (90% этанол). Липиды нагревали приблизительно до 40° для более быстрого растворения.
Точные количества, необходимые для 300 мл серии: амикацин 150 мл, липид 90 мл и 60 мл дополнительного физиологического раствора (или воды), которые добавляли после или во время инфузии для корректировки конечной концентрации этанола.
Процедура изготовления
Один вариант осуществления инфузионной системы показан на фиг. 8.
Растворы липида и амикацина смешивали на одной линии с использованием Y-коннектора (ID 3,2 мм, OD 6,4 мм) при скоростях потоков приблизительно 300/500 мл/мин (т.е. объемное соотношение приблизительно 1/1,67 вместо приблизительно 1/3,35 при обычном процессе). Трубку MasterFlex L/S 25 (ID 4,8 мм) использовали для доставки липидного раствора и трубку L/S 17 (ID 6,4 мм) использовали для доставки раствора амикацина. Для получения синхронных скоростей потока использовали два напора насоса с одним приводом MasterFlex.
Согласно площадям поперечного сечения трубок теоретическое соотношение скоростей потоков должно составлять 4,82/6,42=0,562=1/1,78. Когда привод насоса устанавливали на 500 мл/мин для трубки для амикацина L/S 17, измеренные скорости потоков составляли приблизительно 300/500=1/1,67.
Поскольку липидный раствор содержит 90% этанола, поточная смесь содержала приблизительно 34% этанола. Для предотвращения осаждения амикацина можно добавлять раствор NaCl после или во время инфузии, при скорости потока 100-200 мл/мин (допускают, что липосомы уже сформированы на указанный момент времени). Таким образом, конечная смесь будет содержать приблизительно 27% этанола, в которой весь свободный амикацин, как ожидается, будет растворимым.
Общая скорость потока жидкой инфузии, 800-1000 мл/мин, является сравнимой со скоростью проникновения потока, когда используются два больших диафильтрационных картриджа. Это делает возможной одновременную инфузию и концентрацию посредством диафильтрации.
Полученную суспензию липосом промывали для удаления свободного амикацина посредством диафильтрации с использованием картриджа из полого волокна Amersham UFP-500-C-3MA (площадь мембраны 140 см2, ID волокна 0,5 мм). На первой стадии суспензию концентрировали почти до половины исходного объема (150 мл). Затем, во время диафильтрации для промывки, суспензию отправляли на рециркуляцию и в смесь подавали свежий физиологический раствор со скоростью приблизительно 6 мл/мин, чтобы добиться соответствия со скоростью проникновения и, таким образом, поддерживать постоянный объем. Диафильтрацию продолжали до тех пор, пока не был перекачен 4-кратный суспензионный объем поданного физиологического раствора (т.е. 4×150 мл = 600 мл). Указанная процедура диафильтрации/промывания будет называться 4 «промываниями». В конечном итоге, суспензию концентрировали (диафильтрацией без подачи физиологического раствора), с получением конечного продукта с желаемой концентрацией амикацина и липида. Скорость рециркуляционного потока во время стадии диафильтрации составляла приблизительно 350 мл/мин, а во время стадии окончательного концентрирования она постепенно уменьшалась приблизительно до 150 мл/мин, чтобы поддерживать давление на входе ниже 10 фунтов/кв. дюйм.
Ссылки



Включение в качестве ссылок
Публикации и ссылки, включая, без ограничения, патенты и патентные заявки, процитированные в данном описании, целиком включены в настоящий документ в качестве ссылок, в целой процитированной части, как если бы каждая отдельная публикация или ссылка была конкретно и индивидуально упомянута как включенная в настоящий документ в качестве ссылки. Любая патентная заявка, по отношению к которой настоящая заявка заявляет приоритет, также включена в настоящий документ в качестве ссылки, как описано выше для публикаций и ссылок.
Эквиваленты
В то время как настоящее изобретение было описано с акцентом на предпочтительные варианты осуществления, специалисту будет понятно, что можно использовать вариации в предпочтительных устройствах и способах, и что изобретение можно осуществлять на практике иначе, чем конкретно описано в настоящем документе. Соответственно, настоящее изобретение включает в себя все модификации, охватываемые идеей и объемом настоящего изобретения, как определено формулой изобретения, ниже.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИСТЕМЫ ДЛЯ ЛЕЧЕНИЯ ЛЕГОЧНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2657510C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЛЕГОЧНЫХ НАРУШЕНИЙ СОСТАВАМИ ЛИПОСОМАЛЬНОГО АМИКАЦИНА | 2009 |
|
RU2537238C2 |
| СТАБИЛИЗИРОВАННЫЕ СОСТАВЫ ВАНКОМИЦИНА | 2013 |
|
RU2675859C2 |
| СТАБИЛИЗИРОВАННЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2732567C2 |
| СТАБИЛИЗИРУЮЩИЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2833053C2 |
| СПЕЦИАЛЬНО РАЗРАБОТАННЫЕ ЛИПОСОМЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2672106C2 |
| ЛИПОСОМАЛЬНЫЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ СЛАБОКИСЛОТНЫЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА, И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2778886C2 |
| ЛИОФИЛИЗИРОВАННЫЕ ЛИПОСОМЫ | 2012 |
|
RU2780489C2 |
| СПЕЦИАЛЬНО РАЗРАБОТАННЫЕ ЛИПОСОМЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2780026C2 |
| ЛИОФИЛИЗИРОВАННЫЕ ЛИПОСОМЫ | 2012 |
|
RU2648753C2 |
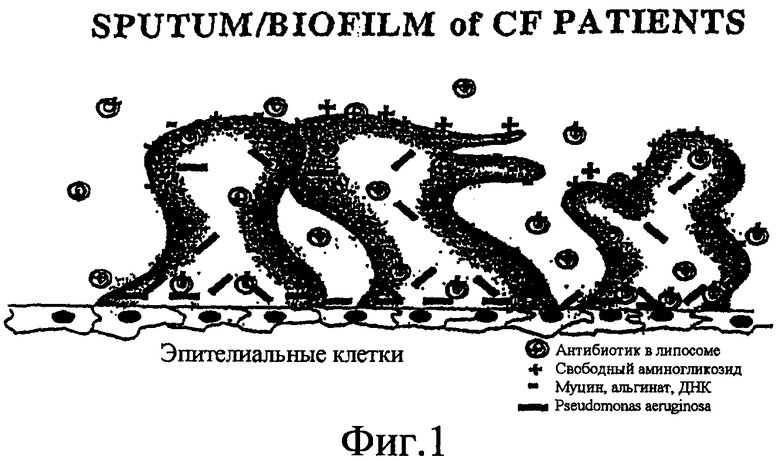
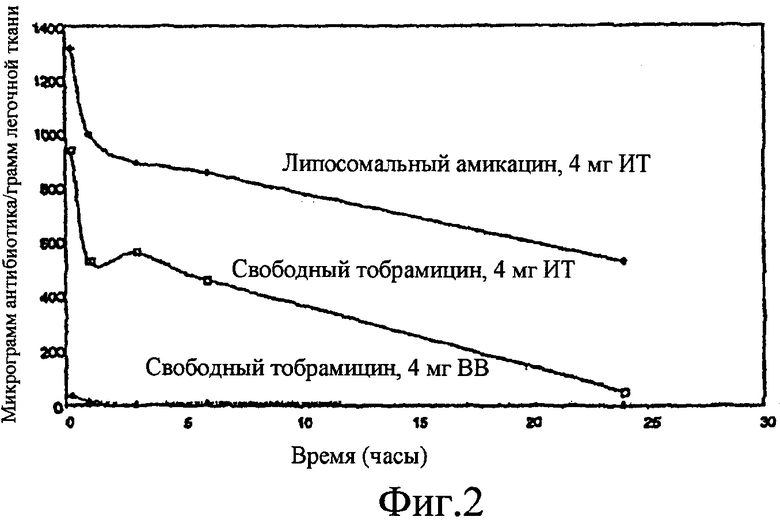

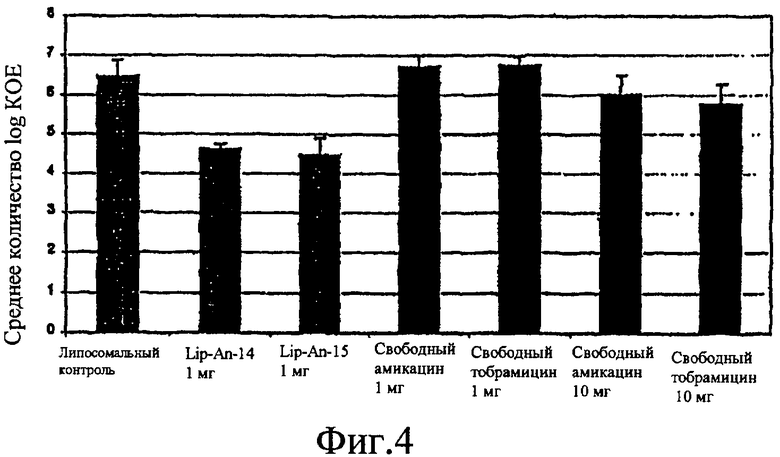
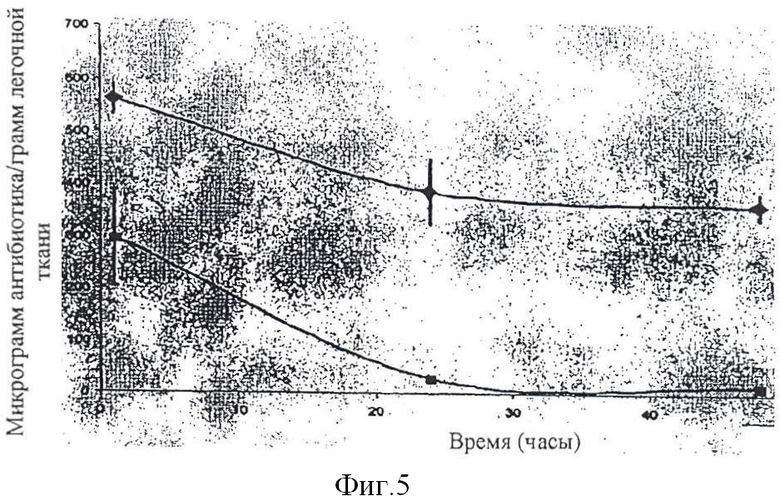
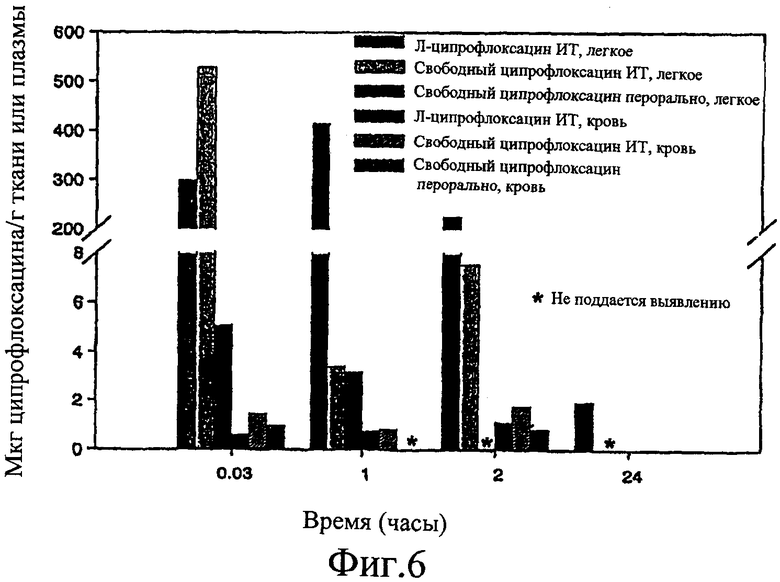
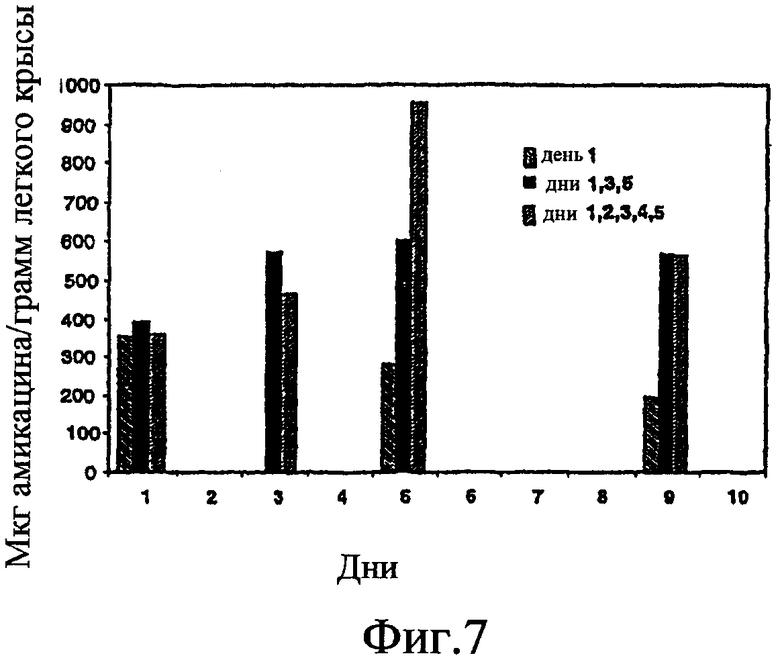
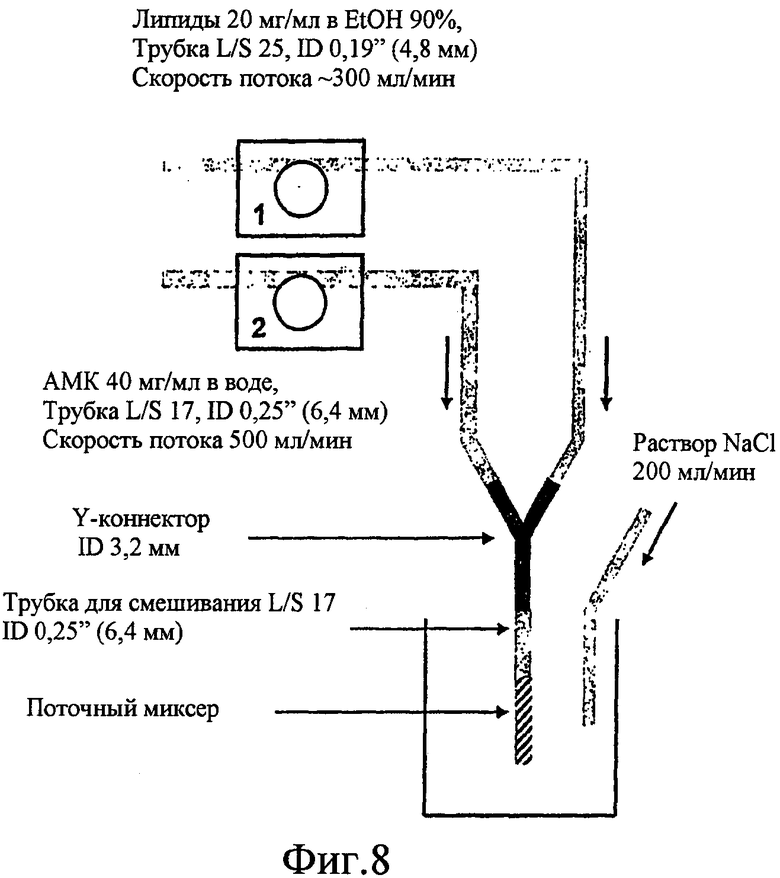
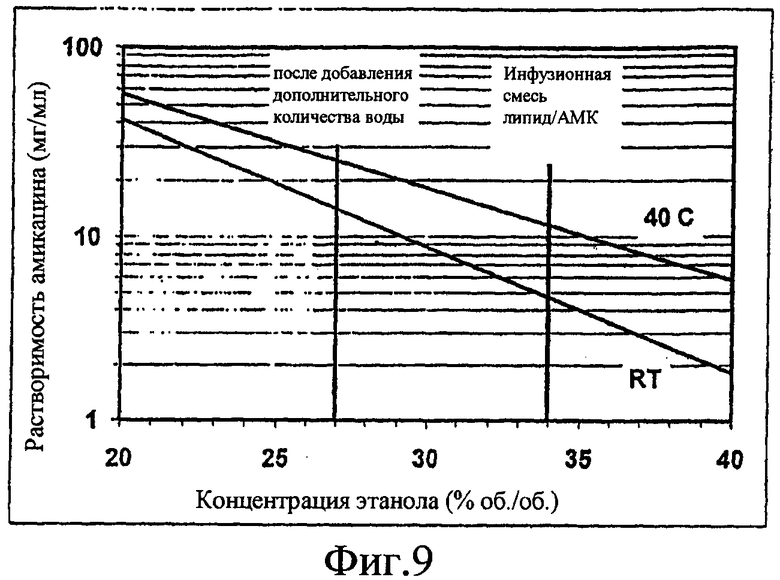
Изобретение относится к области медицины, а именно фармацевтике. Создана липосомная препаративная форма аминогликозида, имеющая липидный двойной слой и инкапсулированный в него аминогликозид, в которой массовое количество липида составляет менее чем или равное количеству аминогликозида, причем липидный двойной слой включает нейтральный фосфолипид и стерол, а липосома имеет средний диаметр от 0,1 до 0,5 микрон. Также настоящее изобретение относится к способу лечения пациента с легочной инфекцией, которое включает в себя введение пациенту терапевтически эффективного количества липидной противоинфекционной препаративной формы и к способу лечения пациента, страдающего муковисцидозом, который основан на введении пациенту терапевтически эффективного количества липидной противоинфекционной препаративной формы по настоящему изобретению. Использование заявленного липосомального противоинфекционного агента обеспечивает пролонгированный терапевтический эффект системно, путем замедленного высвобождения. 3 н. и 76 з.п. ф-лы, 13 табл., 9 ил.
1. Липосомная препаративная форма аминогликозида, включающая липосому, имеющую липидный двойной слой и инкапсулиованный в него аминогликозид, в которой массовое количество липида составляет менее чем или равное количеству аминогликозида, причем липидный двойной слой включает нейтральный фосфолипид и стерол и липосома имеет средний диаметр от 0,1 до 0,5 мкм.
2. Липосомная препаративная форма аминогликозида по п.1, в которой массовое количество липида составляет менее чем или равное 75% от массового количества аминогликозида.
3. Липосомная препаративная форма аминогликозида по п.1, в которой массовое количество липида составляет менее чем или равное 50% от массового количества аминогликозида.
4. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозид выбран из группы, состоящей из амикацина, гентамицина и тобрамицина.
5. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является амикацин.
6. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является гентамицин.
7. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является тобрамицин.
8. Липосомная препаративная форма аминогликозида по п.1, в которой нейтральным фосфолипидом является дипальмитоилфосфатидилхолин (DPPC).
9. Липосомная препаративная форма аминогликозида по п.1, в которой стеролом является холестерин.
10. Липосомная препаративная форма аминогликозида по п.1, в которой нейтральным липидом является DPPC и стеролом является холестерин.
11. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является амикацин, нейтральным липидом является DPPC и стеролом является холестерин.
12. Способ лечения легочной инфекции у пациента, включающий введение пациенту терапевтически эффективного количества липосомной препаративной формы аминогликозида по п.1.
13. Способ по п.12, в котором легочной инфекцией является инфекция Pseudomonas staphylococcal, Streptococcal, Escherichia, Kiebsiella, Enterobacter, Serratia, Haemophilus, Yersinia, Burkholderia или Mycobacterium.
14. Способ лечения легочной инфекции у пациента с муковисцидозом, включающий введение пациенту терапевтически эффективного количества липосомной препаративной формы аминогликозида по п.1.
15. Липосомная препаративная форма аминогликозида по п.1, которая, по существу, не содержит анионных липидов.
16. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является аминогликозида сульфат.
17. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является амикацина сульфат.
18. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является тобрамицина сульфат.
19. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является гентамицина сульфат.
20. Способ по п.13, в котором легочной инфекцией является инфекция Pseudomonas.
21. Способ по п.20, в котором инфекцию Pseudomonas выбирают из группы, состоящей из P.aeruginosa, P.paucimobilis, P.putida, P.fluorescens и P.acidovarans.
22. Способ по п.21, в котором инфекцией Pseudomonas является инфекция P.aeruginosa.
23. Способ по п.13, в котором инфекцией является стафилококковая инфекция.
24. Способ по п.23, в котором стафилококковая инфекция является инфекция Staphylococcus aureus, резистентная к метициллину (MRSA).
25. Способ по п.13, в котором инфекцией является стрептококковая инфекция.
26. Способ по п.25, в котором стафилококковая инфекция является инфекцией Staphylococcus pneumonia.
27. Способ по п.13, в котором инфекцией является инфекция Escherichia.
28. Способ по п.27, в котором инфекцией Escherichia является инфекция Escherichia coli.
29. Способ по п.13, в котором инфекцией является инфекция Yersinia.
30. Способ по п.29, в котором инфекцией Yersinia является инфекция Yersinia pesos.
31. Способ по п.13, в котором инфекцией является инфекция Burkholderia.
32. Способ по п.31, в котором инфекцией Burkholderia является инфекция, выбранная из группы, состоящей из Burkholderia pseudomallei, B.cepacia, В.gladioli, B.multivorans, B.vietnamiensis.
33. Способ по п.13, в котором инфекцией является инфекция Mycobacterium.
34. Способ по п.33, в котором инфекцию Mycobacterium выбирают из группы, состоящей из Mycobacterium tuberculosis, комплекса M.avium (MAC), M.avium, M.intracellulare, M.kansasii, M.xenopi, M.marinum, M.ulcerans, комплексом M.fortuitum, M.fortuitum или M.chelonei.
35. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозид выбран из группы, состоящей из амикацина, гентамицина и тобрамицина.
36. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является амикацин.
37. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является гентамицин.
38. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является тобрамицин.
39. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является аминогликозида сульфат.
40. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является амикацина сульфат.
41. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является тобрамицина сульфат.
42. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является гентамицина сульфат.
43. Липосомная препаративная форма аминогликозида по п.2, в которой нейтральным фосфолипидом является фосфатидилхолин.
44. Липосомная препаративная форма аминогликозида по п.2, в которой нейтральным фосфолипидом является дипальмитоилфосфатидилхолин (DPPC).
45. Липосомная препаративная форма аминогликозида по п.2, в которой стеролом является холестерин.
46. Липосомная препаративная форма аминогликозида по п.2, в которой нейтральным липидом является DPPC и стеролом является холестерин.
47. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является амикацин и нейтральным липидом является DPPC.
48. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является амикацин и стеролом является холестерин.
49. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является амикацин, нейтральным липидом является DPPC и стеролом является холестерин.
50. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является амикацина сульфат и нейтральным липидом является DPPC.
51. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является амикацина сульфат и стеролом является холестерин.
52. Липосомная препаративная форма аминогликозида по п.2, в которой аминогликозидом является амикацина сульфат, нейтральным липидом является DPPC и стеролом является холестерин.
53. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозид выбран из группы, состоящей из амикацина, гентамицина и тобрамицина.
54. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является амикацин.
55. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является гентамицин.
56. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является тобрамицин.
57. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является аминогликозида сульфат.
58. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является амикацина сульфат.
59. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является тобрамицина сульфат.
60. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является гентамицина сульфат.
61. Липосомная препаративная форма аминогликозида по п.3, в которой нейтральным фосфолипидом является фосфатидилхолин.
62. Липосомная препаративная форма аминогликозида по п.3, в которой нейтральным фосфолипидом является дипальмитоилфосфатидилхолин (DPPC).
63. Липосомная препаративная форма аминогликозида по п.3, в которой стеролом является холестерин.
64. Липосомная препаративная форма аминогликозида по п.3, в которой нейтральным липидом является DPPC и стеролом является холестерин.
65. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является амикацин, нейтральным липидом является DPPC.
66. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является амикацин и стеролом является холестерин.
67. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является амикацин, нейтральным липидом является DPPC и стеролом является холестерин.
68. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является амикацина сульфат и нейтральным липидом является DPPC.
69. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является амикацина сульфат и стеролом является холестерин.
70. Липосомная препаративная форма аминогликозида по п.3, в которой аминогликозидом является амикацина сульфат, нейтральным липидом является DPPC и стеролом является холестерин.
71. Липосомная препаративная форма аминогликозида по п.1, в которой массовое количество липида составляет менее чем или равное 91% от массового количества аминогликозида.
72. Липосомная препаративная форма аминогликозида по п.1, в которой массовое количество липида составляет менее чем или равное 83% от массового количества аминогликозида.
73. Липосомная препаративная форма аминогликозида по п.1, в которой массовое количество липида составляет менее чем или равное 60% от массового количества аминогликозида.
74. Липосомная препаративная форма аминогликозида по п.1, в которой нейтральным фосфолипидом является фосфатидилхолин.
75. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является амикацин и нейтральным липидом является DPPC.
76. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является амикацин и стеролом является холестерин.
77. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является амикацина сульфат и нейтральным фосфолипидом является DPPC.
78. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является амикацина сульфат и стеролом является холестерин.
79. Липосомная препаративная форма аминогликозида по п.1, в которой аминогликозидом является амикацина сульфат, нейтральным фосфолипидом является DPPC и стеролом является холестерин.
| US 5759571 А, 02.06.1998 | |||
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ЛЕГОЧНЫХ ИНФЕКЦИЙ | 2001 |
|
RU2185818C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРОРАЛЬНОЙ ПРЕПАРАТИВНОЙ ФОРМЫ ПРОЛОНГИРОВАННОГО ДЕЙСТВИЯ С РЕГУЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ АКТИВНОГО ВЕЩЕСТВА В ЗАВИСИМОСТИ ОТ ВИДА И КОЛИЧЕСТВА НАПОЛНЕНИЯ ЖЕЛУДКА И ПИЩЕВАРИТЕЛЬНОГО ТРАКТА | 1999 |
|
RU2235540C2 |

