Настоящее изобретение относится к способу асимметрического каталитического восстановления окскарбазепина (10,11-дигидро-10-оксо-5H-дибенз/b,f/азепин-5-карбоксамида).
Карбамазепин (I) и окскарбазепин (II) установлены в качестве лекарств первой линии, применяемых для лечения эпилепсии
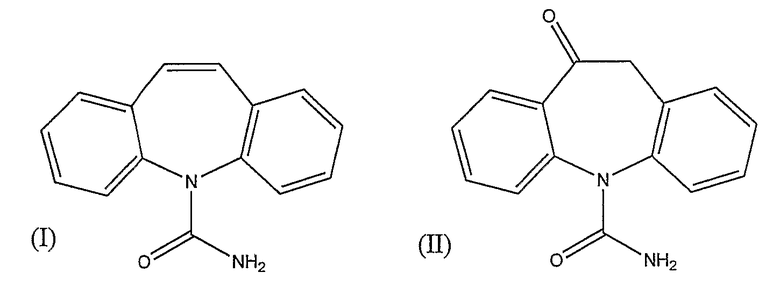
После перорального введения человеку окскарбазепин (II) быстро метаболизирует до фармакологически активной смеси 4:1 (S)- и (R)-энантиомеров 10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида (III)
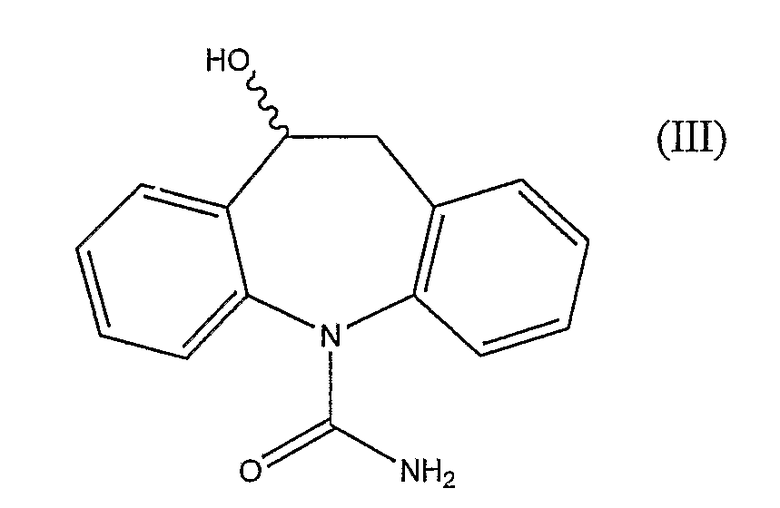
WO 02/096881 описывает двухстадийный способ получения рацемической смеси (III) карбамазепина. WO 02/092572 описывает способ получения рацемической смеси (III) окскарбазепина и, кроме того, описывает способ выделения (S)- и (R)-энантиомеров (III) из рацемической смеси. Энантиомеры могут быть использованы в качестве промежуточных соединений при получении (S)-(-)-10-ацетокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамида и (R)-(+)-10-ацетокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамида, двух лекарственных средств на основе изолированных изомеров, которые могут быть применены для лечения эпилепсии и других нарушений центральной нервной системы (Benes et al, США 5.753.646). WO 2004/031155 описывает способ энантиоселективного получения (S)- и (R)-энантиомеров (III) путем асимметрического восстановления окскарбазепина. Асимметрическое восстановление осуществляют в присутствии рутениевого катализатора и гидридного источника. Подходящий катализатор может быть получен из [RuCl2(п-цимол)]2 и (S,S)-N-(4-толуолсульфонил)дифенилэтилендиамина (здесь далее обозначаемого (S,S)-TsDPEN). Смесь муравьиной кислоты и триэтиламина (в молярном соотношении 5:2) используют в качестве гидридного источника. Описанный способ требует использования очень низкого соотношения субстрат:катализатор, т.е., высокого количества катализатора (например, соотношение 86:1 по примеру 1). Первым основным недостатком применения такого высокого количества катализатора является то, что остаточный уровень металла рутения, наиболее нежелательной загрязняющей примеси в продукте, является высоким и указанная примесь трудно извлекаема, и, кроме того, продукт непригоден для применения в качестве активного фармацевтического ингредиента (API) или в качестве промежуточного соединения, приводящего к API на последней стадии. Существует нормативное руководство по остаточным металлам, возникающим из катализаторов, и пероральные концентрационные пределы для остаточного рутения контролируются особенно строго. Второй основной недостаток состоит в том, что рутениевый катализатор является дорогостоящим. Каталитическая система, описанная в WO 2004/031155, весьма неэффективна, и одни только затраты на каталитическую систему делают способ экономически нерентабельным для крупносерийного производства.
Способ, описанный в WO 2004/031155, также основан на использовании гидридного источника в больших количествах (7 эквивалентов муравьиной кислоты и 2,7 эквивалентов триэтиламина). Покупные источники смеси муравьиная кислота/триэтиламин (триэтиламмонийформиат) доступны, но смесь является дорогостоящей. Значительный избыток используемой в способе муравьиной кислоты потенциально опасен, поскольку муравьиная кислота может разлагаться в присутствии катализатора, вызывая постепенное или спонтанное выделение диоксида углерода и легко воспламеняющегося газообразного водорода, а также вызывая нарастание давления в корпусе реактора. Преждевременное разложение гидридного источника также представляет собой, что реакция восстановления значительно замедляется, и полная конверсия не достигается даже при длительных временах взаимодействия, что делает взаимодействие еще менее эффективным и, в конечном счете, приводит к загрязненному продукту.
Согласно примерам WO 2004/031155, сырой продукт, полученный асимметрическим восстановлением окскарбазепина, очищают колоночной хроматографией на силикагеле. Масштабная очистка методом хроматографии является медленной, дорогостоящей и, в большинстве случаев, практически нецелесообразной по причине низкой пропускной способности. Способ, описанный в WO 2004/031155, непригоден для применения в крупном масштабе, в плане эффективности, и не может рассматриваться как промышленно конкурентный в экономическом отношении способ производства.
Итак, настоящее изобретение ставит задачу разработки улучшенного способа получения (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида и (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида, где способ полностью пригоден для промышленного серийного производства. К удивлению, оказалось, что разработанный способ обеспечивает высокие выходы оптически чистого (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида и (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида при использовании сильно сниженного количества катализатора (т.е. высокого соотношения субстрат/катализатор).
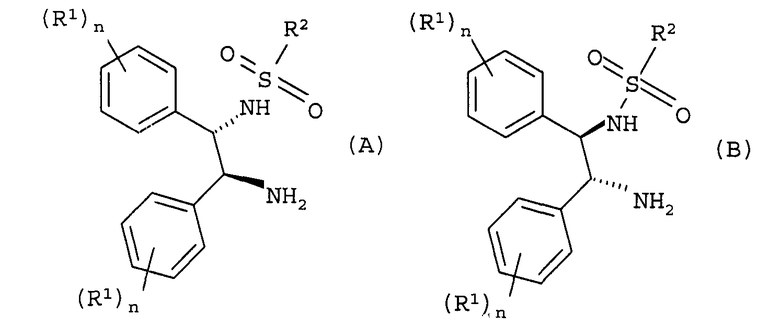
Следовательно, настоящее изобретение представляет способ получения (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида или (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида путем восстановления окскарбазепина в присутствии катализатора и гидридного источника, в указанном способе катализатор основан на комбинации [RuX2(L)]2, где X представляет собой хлор, бром или иод и L представляет собой ариловый или арилалифатический лиганд, и лиганда формулы (A) или формулы (B)
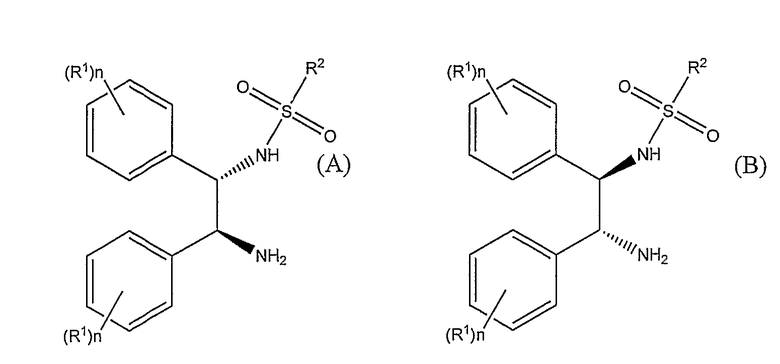
где R1 выбирают из группы, включающей C1-6-алкокси и C1-6-алкил, n представляет собой число от 0 до 5, и когда n представляет собой число от 2 до 5, то R1 может быть одинаковым или различным, и R2 представляет собой алкил, замещенный алкил, арил, замещенный арил, алкарил или замещенный алкарил;
где гидридным источником являются либо NR3R4R5 и муравьиная кислота, либо [R3R4R5NH][OOCH] и, необязательно, муравьиная кислота, либо [M][OOCH]x и муравьиная кислота, где R3, R4 и R5 представляют собой C1-6-алкил, M представляет собой щелочной металл или щелочноземельный металл и x равно 1 или 2, и где в ходе процесса pH поддерживают от 6,5 до 8.
Настоящее изобретение дает возможность получения оптически чистого (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида или (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида. Используемое выражение "оптически чистый" включает соединения, обладающие оптической чистотой 75-100%, предпочтительно, 92-99,5%, более предпочтительно, 96-99,5%.
Заявителями также установлено, что путем контроля pH взаимодействия теперь возможно достигать высоких выходов при выделении оптически чистого (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида или (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида, используя приемлемо малые количества катализатора и реагентов гидридного источника. Количество остаточного рутения в образующемся продукте очень низкое, что делает указанный продукт приемлемым для применения в качестве промежуточного API. Теперь способ удобен для крупномасштабных работ и экономически эффективен по причине меньших затрат на катализатор и характеризуется упрощенным процессом выделения и улучшенными выходами.
Активный катализатор получают из [RuX2(L)]2 и лиганда формулы (A) или формулы (B)
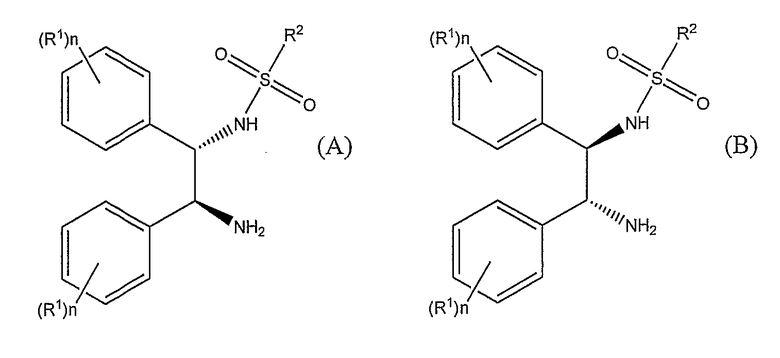
где X представляет собой хлор, бром или иод, предпочтительно, хлор; L представляет собой ариловый или арилалифатический лиганд, такой как п-цимол (изопропилметилбензол), бензол, гексаметилбензол или мезитилен, и, предпочтительно, п-цимол, R1 выбирают из группы, включающей C1-6-алкокси и C1-6-алкил, и n представляет собой число от 0 до 5. Когда n представляет собой число от 2 до 5, то R1 может быть одинаковым или различным. Предпочтительно, n равно либо 0, либо 1, и R1, предпочтительно, представляет собой либо метокси-, либо метильную группу. Наиболее предпочтительно, R1 представляет собой метоксигруппу в пара-положении.
R2 представляет собой такую группу, как алкил, замещенный алкил, арил, замещенный арил, алкарил или замещенный алкарил; где алкил может быть линейным, разветвленным, циклическим или мостиковым, и алкильные, арильные или алкарильные группы могут быть замещены алкильными, алкокси-, галогеновыми или кето- группами. Когда R2 группа представляет собой алкильную группу, целесообразно, чтобы указанная группа содержала 1-9 атомов углерода. Когда алкильная группа является замещенной на R2 группе, целесообразно, чтобы заместитель алкильной группы содержал 1-9 атомов углерода. Целесообразно, чтобы заместитель алкокси- или кето-группы содержал 1-9 атомов углерода. Предпочтительно, чтобы заместитель алкоксигруппы означал метокси.
Предпочтительные R2 группы приведены ниже:
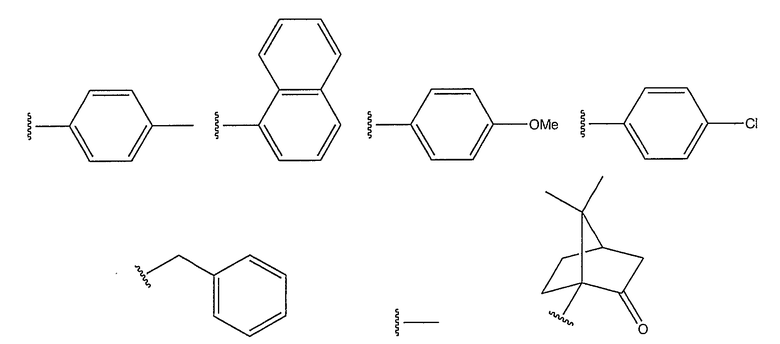
R2 предпочтительно представляет собой фенильную группу, замещенную метилом, наиболее предпочтительно, фенильную группу, замещенную метилом в пара-положении. Предпочтительные лиганды формул (A) и (B) приведены ниже:
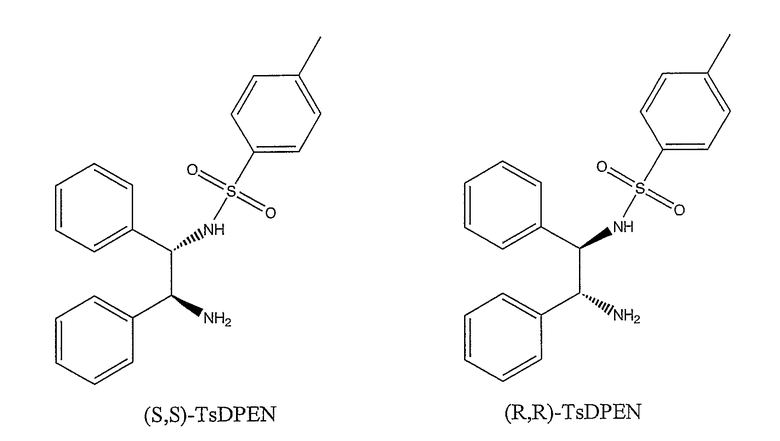
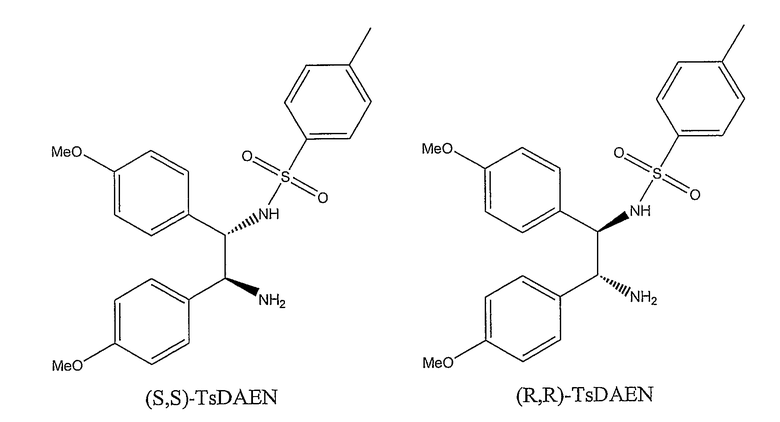
Наиболее предпочтительными лигандами являются (S,S)-TsDAEN и (R5R)-TsDAEN. Неожиданно было установлено, что замещение фенильных колец, в частности, метокси-заместителем приводит к катализатору, обладающему большей эффективностью при асимметрическом восстановлении окскарбазепина. Следовательно, значительно меньшие количества такого катализатора требуются для получения (S)- и (R)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида из окскарбазепина по сравнению с другими лигандами.
Способы с применением катализаторов, основанных на лигандах формулы (A), приводят к получению (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида и способы с применением катализаторов, основанных на лигандах формулы (B), приводят к получению (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида.
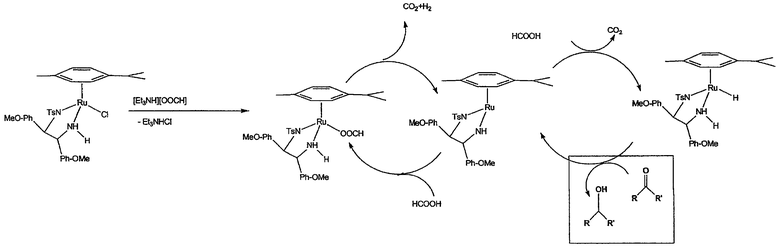
Катализатор, предпочтительно, получают на месте, например, объединением [RuX2L]2 и лиганда формулы (A) или (B) в инертной атмосфере, в растворителе, таком как диметилформамид (ДМФА). На фиг.1 представлен пример каталитического цикла, который, как предполагается, имеет место (гидридным источником в данном примере является [Et3NH][OOCH]).
Целесообразно, чтобы молярное соотношение окскарбазепина и рутениевого катализатора (которое эквивалентно молярному соотношению окскарбазепина и рутения) составляло, по меньшей мере, 500:1, предпочтительно, по меньшей мере, 1000:1, более предпочтительно, по меньшей мере, 1500:1. Способ может быть успешно осуществлен при молярном соотношении окскарбазепина и рутениевого катализатора, равном 2700:1, таким образом, предусматривается, что применение способа может давать преимущество при соотношениях окскарбазепина и рутениевого катализатора не менее 2000:1, более предпочтительно, не менее 2500:1. Можно ожидать, что осуществление способа возможно при соотношениях окскарбазепина и рутениевого катализатора не менее 3000:1. Молярные соотношения ниже 500:1 нежелательны, поскольку катализатор на основе драгоценного металла обходится дорого и может приводить к неприемлемо высоким остаточным уровням рутения в выделенном продукте. Способ по изобретению, согласно которому регулируют pH реакционной смеси, обеспечивает существенно улучшенные соотношения субстрат:катализатор по сравнению с известными из уровня техники способами, где pH не регулируют, например в способе по примеру 1 согласно WO 2004/03115 используют соотношение окскарбазепин и рутения 86:1.
Гидридным источником являются либо NR3R4R5 и муравьиная кислота, либо [R3R4R5NH][OOCH] и, необязательно, муравьиная кислота, либо [M][OOCH]x и муравьиная кислота, где R3, R4 и R5 представляют собой C1-6-алкил, M представляет собой щелочной металл или щелочноземельный металл и x равно 1 или 2. R3, R4 и R5 могут быть одинаковыми или различными, но, предпочтительно, являются все одинаковыми. C1-6-алкильные группы могут быть линейными, разветвленными или циклическими. Предпочтительно, R3, R4 и R5 представляют собой этил, пропил или бутил, наиболее предпочтительно, этил. M предпочтительно представляет собой Na, Li или K, наиболее предпочтительно, Na. Когда M представляет собой щелочной метал, x равен 1, и когда M представляет собой щелочноземельный металл, x равен 2.
Реагенты [R3R4R5NH][OOCH], например [Et3NH][OOCH], являются коммерчески доступными. [Et3NH][OOCH], обычно используемый в реакциях асимметрического восстановления, синтезируют из H2, CO2 и NEt3 в присутствии рутениевого катализатора, и поэтому указанный реагент является дорогостоящим, по этой причине желательно свести к минимуму применение реагента такого типа. Когда муравьиную кислоту и NR3R4R5 смешивают в стехиометрическом количестве, устанавливается следующее кислотно-основное равновесие:
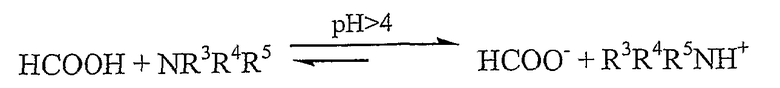
При низком pH гидридный источник существует в кислотной форме, тогда как при больших pH гидридный источник существует в форме сопряженного основания, той форме, что участвует в каталитическом цикле.
Согласно первому варианту осуществления изобретения гидридным источником являются NR3R4R5 и муравьиная кислота, предпочтительно, триэтиламин и муравьиная кислота. Указанный вариант осуществления позволяет избежать применения является дорогостоящих реагентов [R3R4R5NH][OOCH]. Целесообразно, NR3R4R5 добавлять к реакционной смеси в начале технологического процесса. Предпочтительно, добавляют менее двух эквивалентов NR3R4R5, наиболее предпочтительно, порядка одного эквивалента. Количество муравьиной кислоты, добавляемое к реакционной смеси в начале технологического процесса, может быть сведено к минимуму. Такое сведение к минимуму выгодно, поскольку муравьиная кислота в ходе процесса разлагается на монооксид углерода и водород, что потенциально опасно в случае использования больших количеств муравьиной кислоты. Целесообразно добавлять к реакционной смеси в начале процесса менее 1,5 эквивалентов муравьиной кислоты, предпочтительно, менее 1 эквивалента, наиболее предпочтительно, менее 0,2 эквивалента. Если в начале процесса добавляют менее 1 эквивалента муравьиной кислоты, в ходе взаимодействия следует добавлять дополнительное количество муравьиной кислоты, обеспечивая, в целом, около 1-3 эквивалента муравьиной кислоты.
По второму варианту осуществления изобретения гидридным источником является [R3R4R5NH][OOCH], предпочтительно, [Et3NH][OOCH], с добавлением или без добавления муравьиной кислоты. Целесообразно, [R3R4R5NH][OOCH] добавлять к реакционной смеси в начале технологического процесса. Предпочтительно, используют менее двух эквивалентов [R3R4R5NH][OOCH], желательно, порядка одного эквивалента. Целесообразно добавлять к реакционной смеси в начале процесса менее 0,5 эквивалентов муравьиной кислоты, наиболее предпочтительно, менее 0,2 эквивалента. В ходе протекания взаимодействия к реакционной смеси может быть добавлено дополнительное количество муравьиной кислоты.
Согласно третьему варианту осуществления изобретения гидридным источником являются [M][OOCH]x и муравьиная кислота. Целесообразно, [M][OOCH]x добавлять к реакционной смеси в начале технологического процесса. Предпочтительно, используют менее двух эквивалентов [M][OOCH]x, желательно, порядка одного эквивалента. Целесообразно добавлять к реакционной смеси в начале процесса менее 1,5 эквивалентов муравьиной кислоты, предпочтительно, менее 1 эквивалента, наиболее предпочтительно, менее 0,2 эквивалента. Если в начале процесса добавляют менее 1 эквивалента муравьиной кислоты, в ходе взаимодействия следует добавлять дополнительное количество муравьиной кислоты, обеспечивая, в целом, около 1-3 эквивалента муравьиной кислоты.
Способ по изобретению допускает сниженные количества реагентов гидридного источника по сравнению с известными из уровня техники способами, например в способе WO 2004/03115, где pH не регулируют, используют семь эквивалентов муравьиной кислоты и 2,7 эквивалента триэтиламина. В частности, способ по настоящему изобретению сводит к минимуму опасность, связанную с добавлением больших количеств муравьиной кислоты к реакционной смеси в начале взаимодействия.
pH реакционной смеси поддерживают в ходе взаимодействия в пределах от 6,5 до 8. Контроль pH имеет важное значение для обеспечения хороших конверсий и приемлемо высоких выходов продукта, предпочтительно, свыше 85%, при этом используются приемлемо низкие количества катализатора (например, соотношение субстрат:катализатор равно 500 или более). Контроль за pH можно осуществлять известными специалистам из уровня техники способами, но предпочтительный способ состоит в применении электрода Гамильтона, гелевого наполнения, как описано в примерах по настоящему изобретению.
Предпочтительный способ регулирования pH состоит в добавлении в ходе взаимодействия муравьиной кислоты управляемым способом, например титрованием. Наиболее предпочтительно, pH поддерживают в пределах от 7,0 до 7,8 контролируемым добавлением муравьиной кислоты. По первому варианту осуществления изобретения, где гидридным источником являются NR3R4R5 и муравьиная кислота, до 1,5 эквивалентов муравьиной кислоты может быть добавлено в начале технологического процесса и затем дополнительное количество муравьиной кислоты может быть добавлено, по мере необходимости, для поддержания pH. Однако, предпочтительно, не добавлять муравьиную кислоту к реакционной смеси, когда добавляется NR3R4R5, и всю муравьиную кислоту добавлять постепенно, управляемым способом, например, по каплям при титровании, поддерживая, таким образом, pH в пределах от 6,5 до 8. Предпочтительно добавлять всю муравьиную кислоту постепенно, управляемым способом, поскольку такой способ сводит к минимуму опасность, связанную с разложением муравьиной кислоты. По второму варианту осуществления изобретения, где гидридный источник включает [R3R4R5NH][OOCH], предпочтительно не добавлять муравьиную кислоту в начале процесса, но добавлять впоследствии муравьиную кислоту для поддержания pH. Муравьиную кислоту добавлять постепенно, управляемым способом, например, по каплям при титровании, поддерживая pH в пределах от 6,5 до 8. По третьему варианту осуществления изобретения, где гидридным источником являются [M][OOCH]x и муравьиная кислота, до 1,5 эквивалентов муравьиной кислоты может быть добавлено в начале технологического процесса, и затем дополнительное количество муравьиной кислоты может быть добавлено, по мере необходимости, для поддержания pH. Однако, предпочтительно, не добавлять муравьиную кислоту к реакционной смеси, когда добавляется [M][OOCH]x, и всю муравьиную кислоту добавлять постепенно, управляемым способом, например, по каплям при титровании, поддерживая, таким образом, pH в пределах от 6,5 до 8.
Растворимость окскарбазепина является весьма низкой в большинстве фармацевтически приемлемых технологических растворителях, даже при повышенных температурах. Подходящие растворители включают диметилформамид (ДМФА), этилацетат (EtOAc), ацетонитрил, изопропилацетат, тетрагидрофуран, 1,2-дихлорэтан, диметоксиэтан и/или воду. Предпочтительно, чтобы растворитель включал, по меньшей мере, один полярный апротонный растворитель, такой как ДМФА или ацетонитрил, поскольку указанные растворители смешиваются как с органической, так и неорганической фазами. Неожиданно оказалось, что стандартные дезоксигенированные растворители марки чистый для анализа пригодны для использования в способе по настоящему изобретению. Предпочтительной системой растворителей для способа по настоящему изобретению является смесь двух или более растворителей, выбираемых из группы, включающей ДМФА, EtOAc, ацетонитрил и воду. В первом варианте осуществления изобретения, где гидридным источником являются NR3R4R5 и муравьиная кислота, подходящий растворитель включает 0-25% ДМФА, 0-25% воды и 75-95% EtOAc или 0-25% ацетонитрила, 0-25% воды и 75-95% EtOAc, предпочтительно, 0-20% ДМФА, 5-20% воды и 80-90% EtOAc. Наиболее предпочтительным растворителем является смесь 10% ДМФА, 10% воды и 80% EtOAc. По второму варианту осуществления изобретения, где гидридным источником является [R3R4R5NH][OOCH], с добавлением или без добавления муравьиной кислоты, подходящий растворитель включает 5-25% ДМФА и 75-95% EtOAc, 5-25% ацетонитрила и 75-95% EtOAc, 5-25% ДМФА и 75-95% воды или 5-25% ацетонитрила и 75-95% воды. Согласно третьему варианту осуществления изобретения, где гидридным источником являются [M][OOCH]x и муравьиная кислота, подходящий растворитель включает 0-25% ДМФА, 0-25% воды и 75-95% EtOAc или 0-25% ацетонитрила, 0-25% воды и 75-95% EtOAc, предпочтительно, 0-20% ДМФА, 5-20% воды и 80-90% EtOAc.
В заслуживающем особого внимания варианте осуществления способа восстановление происходит в присутствии катализатора межфазного переноса. Подходящие катализаторы межфазного переноса включают галоидные соли четвертичного алкиламмония, такие как, например, Bu4NBr. Целесообразно, чтобы соотношение катализатора межфазного переноса и катализатора составляло от 0,01 до 0,5, предпочтительно, около 0,1.
Взаимодействие может быть проведено при различных температурах и давлениях. Целесообразно осуществлять взаимодействие при атмосферном давлении и температуре кипения предпочтительной системы растворителей. Наружная температура 100-120°C, более предпочтительно, 105-110°C, подходит для большинства предпочтительных систем растворителей.
Реакционное время зависит от основных факторов, таких как соотношение окскарбазепина и катализатора. Предпочтительно, взаимодействие должно быть завершено менее чем за 36 часов, более предпочтительно, менее чем за 24 часа, и высокие выходы достигаются, с применением способа по настоящему изобретению, за реакционное время менее чем 24 часа, при соотношениях окскарбазепина и катализатора даже больших, чем 2000:1.
В первом варианте осуществления изобретения, где гидридным источником являются NR3R4R5 и муравьиная кислота, продукт может самопроизвольно осаждаться из реакционной смеси при охлаждении ниже температуры кипения указанной смеси. Перед фильтрованием к реакционной смеси добавляют подходящий растворитель, предпочтительно метилтретбутиловый эфир (MTBE). По второму варианту осуществления изобретения, где гидридным источником является [R3R4R5NH][OOCH], с добавлением или без добавления муравьиной кислоты, продукт может быть выделен осаждением сырого продукта из подходящей смеси растворителей, предпочтительно, либо из смеси метанол/вода, либо из смеси метанол/MTBE, при 0-5°C. Технология обработки чрезвычайно простая по сравнению с известными из уровня техники способами, такими как описаны в WO 2004/031155, где технология обработки требует нейтрализации избытка муравьиной кислоты, экстракции, сушки, выпаривания растворителя и флэш хроматографии. Указанные процедуры неприемлемы для крупномасштабного производства. По данному изобретению, простая технология обработки приводит к низкому содержанию остаточного рутения в выделенном продукте, в соответствии с предполагаемым использованием указанного продукта в качестве конечного промежуточного продукта для получении APIs.
По альтернативному варианту осуществления, (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамид или (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамид может быть осажден путем удалении реакционного растворителя при добавлении воды в целях поддержания реакционного объема, по существу, на постоянном уровне. Реакционный растворитель, которым предпочтительно является этилацетат, может быть удален путем перегонки. Температура перегонки, предпочтительно, составляет, по меньшей мере, 60°C. Как правило, масса воды, заменяющей реакционный растворитель, может быть в пределах 80-120%, более предпочтительно, 90-110% от массы удаленного растворителя. По данному варианту осуществления, осажденный (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамид или (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамид может быть впоследствии выделен фильтрованием и дополнительно очищен, предпочтительно, путем ресуспендирования в растворителе, которым, предпочтительно, является этилацетат, и повторного фильтрования.
Другое преимущество настоящего изобретения состоит в возможности протекания взаимодействия при высокой концентрации субстрата, например, 0,5-1,5 M, так что объемная производительность реакции очень хорошая. Это в особенности важно, когда касается крупномасштабного производства.
(S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамид или (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамид, полученный по способу настоящего изобретения, может быть использован в качестве API и формулирован в конечную фармацевтическую продукцию, либо может быть преобразован, путем дополнительного химического превращения, в другой API, например (S)-(-)-10-ацетокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид может быть получен этерификацией (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида. Настоящее изобретение также касается способа получения соединения формулы (C) или (D)
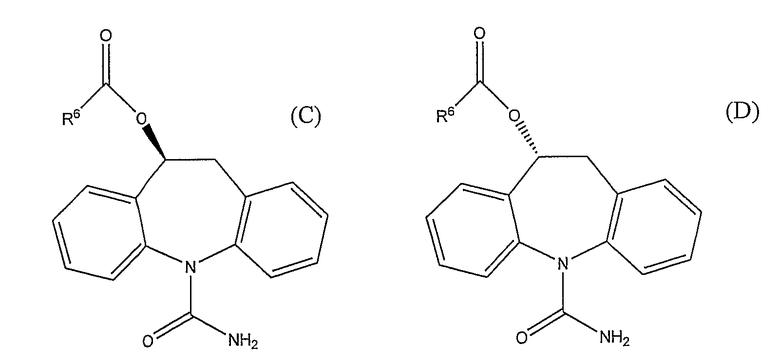
где R6 представляет собой водород, алкил, галогеналкил, аралкил, циклоалкил, циклоалкиалкил, алкокси, арил или пиридил; включающего первую стадию, представляющую собой способ получения (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида или (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида по изобретению, и вторую стадию, где (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамид или (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамид ацилируют. Соединения, полученные указанным способом, могут быть оптически чистыми, где понятие оптически чистые представляет собой соединения, обладающие оптической чистотой 75-100%, предпочтительно от 92-99,5%, более предпочтительно, от 96-99,5%.
R6 может означать линейный или разветвленный C1-18-алкил, который может быть замещен галогеном (F, Cl, Br или I). R6 может также означать циклоалкил (циклическую C3-C6-насыщенную группу) или арил (незамещенный фенил или фенил, замещенный алкокси, галогеном или нитрогруппой). Предпочтительно, R6 представляет собой CH3. Соединения формул (C) и (D) дополнительно описаны в США 5,753,646. Подходящие способы ацилирования описаны в США 5,753,646 и WO 02/092572, например (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамид может быть подвергнут взаимодействию с ацетилхлоридом или уксусным ангидридом в дихлорметане, что дает (S)-(-)-10-ацетокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид. Соответствующие стереоизомеры следующих дополнительных соединений также могут быть получены преобразованием (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида или (R)-(-)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида с применением подходящих способов, как описано в США 5,753,646:
(1) 10-бензоилокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(2) 10-(4-метоксибензоилокси)-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(3) 10-(3-метоксибензоилокси)-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(4) 10-(2-метоксибензоилокси)-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(5) 10-(4-нитробензоилокси)-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(6) 10-(3-нитробензоилокси)-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(7) 10-(2-нитробензоилокси)-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(8) 10-(4-хлорбензоилокси)-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(9) 10-(3-хлорбензоилокси)-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(10) 10-(2-ацетоксибензоилокси)-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(11) 10-пропионилокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(12) 10-бутирилокси-10,1-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(13) 10-пивалоилокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(14) 10-[(2-пропил)пентаноилокси]-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(15) 10-[(2-этил)гексаноил]-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(16) 10-стеароилокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(17) 10-циклопентаноилокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(18) 10-циклогексаноил-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(19) 10-фенилацетокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(20) 10-(4-метоксифенил)ацетокси-10,11-дигидро-5H-дибенз/b,f/-азепин-5-карбоксамид
(21) 10-(3-метоксифенил)ацетокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(22) 10-(4-нитрофенил)ацетокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(23) 10-(3-нитрофенил)ацетокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(24) 10-никотиноилокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(25) 10-изоникотиноилокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(26) 10-хлорацетокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(27) 10-бромацетокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(28) 10-формилокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(29) 10-этоксикарбонилокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
(30) 10-(2-хлорпропионилокси-10,11-дигидро-5H-дибенз/b,f/азепин-5-карбоксамид
Далее изобретение раскрыто с помощью примеров, которые не рассматриваются как ограничивающие.
Пример 1: Асимметрическое восстановление окскарбазепина с использованием NEt 3 и pH-контролируемого добавления HCOOH
В 5-горлую, круглодонную колбу на 2 л загружают окскарбазепин (635 ммоль, 169 г), колбу снабжают двумя водяными обратными конденсаторами, подключенных к линии Шленка (два обратных конденсатора используют для обеспечения двух путей выхода для газообразных CO2 и H2, выделяющихся при взаимодействии), бюреткой для титрования и электродом Гамильтона, гелевого наполнения, входящим без зазора в навинчивающуюся пробку GL25 с выемкой, снабженную уплотнительным кольцом из PTFE/силикон. К исходному материалу, в токе N2, добавляют с помощью мерного цилиндра EtOAc (480 мл, недегазированный, чистый для ВЭЖХ), H2O (48 мл, недегазированная, чистая для ВЭЖХ) и Et3N (1,1 экв., 699 ммоль, 97,5 мл, недегазированный, Fluka, 99,9% чистота). Впрыскивают катализатор (полученный отдельно, на месте, в трубке Шленка на 50 мл, в токе N2, путем перемешивания [RuCl2(п-цимол)]2 (0,1588 ммоль, 97,2 мг) и (S,S)-TsDAEN (2,2 экв. в расчете на металлический димерный предшественник, 0,3493 ммоль, 159 мг) в ДМФА (13 мл, дегазированный, безводный), при комнатной температуре, в течение 10-15 мин). Трубку Шленка споласкивают небольшими порциями оставшегося ДМФА (5×7 мл), которые впрыскивают в реакционную смесь. Комбинация растворителей на данный момент: 10% ДМФА-10% H2O-80% EtOAc (б./б./б.); и концентрация субстрата перед титрованием равна 1,1 M. Круглодонную колбу помещают на масляную баню, предварительно нагретую до 105°C, и реакционную смесь перемешивают с помощью магнитной мешалки при нагревании до температуры кипения с обратным холодильником (Tмасляной бани=105°C, внутренняя T=72-77°C). Когда реакционная смесь начинает стекать по дефлегматору, pH реакционной смеси составляет около 8,8. С этого момента начинают титрование/медленное добавление 12,5 M раствора HCOOH в смеси 20% ДМФА/EtOAc. pH медленно доводят до 7,4 и затем поддерживают постоянным на указанной величине в течение 12 час путем медленного добавления раствора HCOOH. Конверсия по ВЭЖХ спустя 15 час равна 99%. Дальнейшее перемешивание до 20 час приводит к образованию белого осадка, и оставшийся 1% окскарбазепина не расходуется. В целом на реакцию расходуется приблизительно 3,7 экв. HCOOH в расчете на исходный материал. Суммарная концентрация субстрат/продукт в конце взаимодействия равна 0,86 M. Спустя 20 час нагревание прекращают и реакционную смесь перемешивают и оставляют медленно охлаждаться. Когда температура масляной бани достигает примерно 80°C, к реакционной смеси добавляют 500 мл MTBE и оставляют охлаждаться до комнатной температуры при перемешивании. Реакционную смесь перемешивают при 0-5°C около 30 мин, фильтруют и осадок повторно промывают охлажденными порциями MTBE до тех пор, пока фильтрат не обесцветится. Полученный белый осадок сушат на воздухе, затем при высоком вакууме, что дает белый порошок: 95% выход изолированного продукта (152 г). ВЭЖХ: продукт 99,6%, 97,8% e.e., окскарбазепин 0,4%.
В связи с тем, что в указанных реакционных условиях (10% ДМФА-10% H2O-80% EtOAc (об./об./об.); 1,1 экв. Et3N и HCOOH) продукт выкристаллизовывается при температуре кипения флегмы, растворитель во время обработки не выпаривают. Очень низкая растворимость продукта в MTBE допускает не только дальнейшее осаждение исходного материала, но также способствует очистке/удалению остаточного рутения, ДМФА и реагентов путем промывки фильтрата в больших количествах, без потери в выходе изолированного продукта. Уровень рутения в продукте составляет в пределах 5-50 ч./млн.
Пример 2: Асимметрическое восстановление окскарбазепина с использованием [Et 3 NH][OOCH] и pH-контролируемого добавления HCOOH
В 4-горлую, круглодонную колбу на 500 мл загружают окскарбазепин (159 ммоль, 40 г), колбу снабжают водяным обратным конденсатором, подключенным к линии Шленка, бюреткой для титрования и электродом Гамильтона, гелевого наполнения, входящим без зазора в навинчивающуюся пробку GL25 с выемкой, снабженную уплотнительным кольцом из PTFE/силикон. Колбу продувают N2 около 30 мин. К исходному материалу, в токе N2, добавляют с помощью шприца EtOAc (78 мл, дегазированный, безводный), [Et3NH][OOCH] промышленно выпускаемый Fluka (1,07 экв., 170 ммоль, 25 мл, недегазированный, Fluka). Впрыскивают катализатор (полученный отдельно, на месте, в трубке Шленка на 20 мл, путем перемешивания [RuCl2(п-цимол)]2 (0,0265 ммоль, 16,2 мг и (S,S)-TsDAEN (2,2 экв. в расчете на металлический димерный предшественник, 0,0582 ммоль, 25 мг) в ДМФА (5 мл, дегазированный, безводный), при комнатной температуре, в течение 10-15 мин). Трубку Шленка споласкивают небольшими порциями оставшегося ДМФА (5×3 мл), которые впрыскивают в реакционную смесь. Комбинация растворителей на данный момент: 20% ДМФА-80% EtOAc (об./об.); и концентрация окскарбазепина перед титрованием равна 1,3 M. Круглодонную колбу помещают на масляную баню, предварительно нагретую до 105°C, и реакционную смесь перемешивают с помощью магнитной мешалки при нагревании до температуры кипения с обратным холодильником (Tмасляной бани=105°C). Когда реакционная смесь начинает стекать по дефлегматору, pH реакционной смеси составляет около 6,8. Реакционная смесь медленно приобретает багровый оттенок, и pH начинает повышаться по мере расходования HCOOH из триэтиламмонийформиата. Когда pH достигает 7,4-7,45, начинают титрование/медленное добавление 12,5 M раствора HCOOH в смеси 20% ДМФА/EtOAc. pH поддерживают на уровне pH=7,4 в течение 12 час путем медленного добавления раствора HCOOH. Спустя 17 час реакционная смесь становится ярко-пурпуровой, при некотором разложении катализатора, обнаруживаемого на стенках колбы. Конверсия по ВЭЖХ спустя 17 час равна 98%. К этому моменту pH раствора равняется 7,8, и добавление дополнительного количества раствора HCOOH продолжают при 7,7. Дальнейшее перемешивание до 23 час приводит к 99% конверсии. На реакцию расходуется приблизительно 4,7 экв. HCOOH в расчете на исходный материал. Спустя 23 час нагревание прекращают и реакционную смесь перемешивают и оставляют медленно охлаждаться. Реакционную смесь концентрируют, добавляют 100 мл MTBE и растворитель вновь удаляют. Добавляют 15 мл MeOH и белую пастообразную массу нагревают до температуры кипения с обратным холодильником около 5 мин и затем медленно добавляют 250 мл MTBE к указанной, стекающей по дефлегматору, смеси. Образующуюся смесь перемешивают при нагревании до температуры кипения с обратным холодильником в течение 30 мин, охлаждают до RT, затем до 0-5°C, и перемешивают в течение 30 мин. Смесь фильтруют охлажденной и промывают охлажденными порциями MTBE до обесцвечивания фильтрата (8×50 мл). Полученный белый осадок сушат на воздухе, затем при высоком вакууме, что дает белый порошок: 94% выход изолированного продукта (37,9 г). ВЭЖХ: продукт 99,5%, 97,8% e.e., окскарбазепин 0,5%. Уровень рутения в продукте составляет в пределах 5-50 ч./млн.
Сопоставление 1a: Асимметрическое восстановление с контролем и без контроля pH (гидридным источником являются NEt 3 и HCOOH)
Взаимодействия осуществляют, используя способ, аналогичный способу примера 1. Соотношение субстрат/катализатор равно 2000 и растворителем является смесь 20% H2O/EtOAc. Лигандом служит (S,S)-Ts-DAEN. В примере 3 1 экв. NEt3 и 1 экв. HCOOH добавляют к реакционной смеси в начале взаимодействия. Дополнительное количество HCOOH добавляют в ходе взаимодействия, поддерживая pH равным 7,4. В примере сравнения 1 4,4 экв. NEt3 и 4 экв. HCOOH предварительно смешивают в H2O и добавляют к реакционной смеси в EtOAc в начале взаимодействия. В таблице 1 представлены результаты примера 3 и примера сравнения 1:
Выход pH-контролируемой реакции (пример 3) намного больше, чем выход реакции с неконтролируемым pH (пример сравнения 1), несмотря на тот факт, что используют большие количества реагентов гидридного источника.
Сопоставление 1b: Асимметрическое восстановление с контролем и без контроля pH (гидридным источником являются [Et 3 NH][OOCH] и HCOOH)
Взаимодействия осуществляют, используя способ, аналогичный способу примера 2. Соотношение субстрат/катализатор равно 1000 и растворителем является смесь 10% ДМФА/EtOAc. Лигандом служит Ts-DPEN вместо Ts-DAEN. Используют 20 г окскарбазепина вместо 40 г. [Et3NH][OOCH] добавляют к реакционной смеси в начале взаимодействия, без дополнительного добавления HCOOH. В таблице 2 представлены результаты двух примеров сравнения с использованием различных количеств [Et3NH][OOCH]:
(%)
5 эквивалентов дорогостоящего реагента [Et3NH][OOCH] обеспечивают 100% конверсию после 22 часов, тогда как при использовании лишь 2 эквивалентов реагента достигается конверсия менее 50%. pH реакционной смеси возрастает в ходе взаимодействия.
Взаимодействие повторяют в масштабе 10 г, используя соотношение субстрат/катализатор, равное 1500:1, и растворитель 20% ДМФА/EtOAc. В примерах сравнения 4 и 5 используют 5 эквивалентов [Et3NH][OOCH] и без добавления дополнительного количества HCOOH. В примере 4 используют только один эквивалент [Et3NH][OOCH], но добавляют дополнительно HCOOH в ходе взаимодействия, поддерживая pH равным 7,4. В таблице 3 представлены результаты примеров сравнения 4 и 5 и примера 4:
Сопоставление примеров сравнения 4 и 5 и примера 4 показывает, что, контролируя pH путем добавления HCOOH, можно использовать значительно меньшее количество дорогостоящего реагента [Et3NH][OOCH], при этом взаимодействие достигает почти полной конверсии за 7 часов, вместо 20-25 часов.
Сопоставление 2: Асимметрическое восстановление с использованием различных лигандов
Активность катализаторов, включающих лиганды (S,S)-TsDAEN и (S,S)-TsDPEN, сравнивают при масштабе 20-40 г. Катализаторы получают на месте путем перемешивания [RuCl2(п-цимол)]2 и либо (S,S)-TsDAEN, либо (S,S)-TsDPEN, в течение 5-10 минут в ДМФА, перед добавлением окскарбазепина и 1,07 эквивалентов [Et3NH][OOCH]. 12,5 M раствор HCOOH в смеси 20% ДМФА/EtOAc медленно впрыскивают при pH=7,4. Соотношение окскарбазепина и катализатора равно 3000:1. В таблице 4 представлены результаты примеров 5 и 6 (применение (S,S)-TsDAEN) и примера 7 (применение (S,S)-TsDPEN):
Примеры с (S,S)-TsDAEN демонстрируют значительно большую конверсию, чем пример с (S,S)-TsDPEN, и дают близкую энантиоселективность.
Сопоставление 3: Асимметрическое восстановление окскарбазепина с использованием катализатора межфазного переноса
В таблице 5 представлены результаты трех реакций асимметрического восстановления, где в дополнение к рутениевому катализатору используют катализатор межфазного переноса. Для каждого взаимодействия катализатор получают на месте путем добавления [RuCl2(п-цимол)]2 и лиганда к EtOAc и перемешивания. (NB (R,R)-TsDTEN имеет ту же структуру, что и (R,R)-TsDPEN, за тем исключением, что фенильные группы заменены на толильные группы). Катализатор межфазного переноса присутствует в количестве 0,1 эквивалента от Bu4NBr. Гидридный источник представлен 2 эквивалентами [Et3NH][OOCH], и в ходе взаимодействия к реакционной смеси медленно добавляют дополнительное количество муравьиной кислоты. Соотношение окскарбазепина и катализатора равно 2000:1, и наружная температура реакции равна 110°C. Конверсия в примере 9, где лигандом является (R,R)-TsDAEN, значительно больше, чем конверсия в примерах 8 и 10, где лигандами служат (S,S)-TsDPEN и (R,R)-DTEN.
Пример 3: Ацетилирование (S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамида
(S)-(+)-10,11-дигидро-10-гидрокси-5H-дибенз/b,f/азепин-5-карбоксамид (500 г), полученный асимметрическим гидрированием с переносом, как описано выше, и 4-(N,N-диметиламино)пиридин (4 г) суспендируют в дихлорметане (5,07 л). К суспензии добавляют пиридин (210 мл). Реакционную смесь нагревают до температуры кипения с обратным холодильником, после чего добавляют по каплям уксусный ангидрид (240 мл). Полученный желтовато-коричневый раствор перемешивают в течение 2 часов и затем охлаждают до 30°C.
После чего реакционную смесь гасят добавлением серной кислоты. После перемешивания в течение 10 мин слои разделяют. Органический слой промывают дважды насыщенным водным раствором бикарбоната натрия и затем водой. Приблизительно половину дихлорметана затем удаляют путем выпаривания, и изопропанол (5 л) добавляют к смеси, которую потом оставляют стоять в течение ночи. Упаривают дополнительное количество растворителя (приблизительно, 1,5 л) и полученную суспензию охлаждают примерно до 3°C. Спустя 3 часа твердое вещество отделяют фильтрованием, промывают охлажденным изопропанолом и затем сушат в вакууме в течение ночи. Высушенное твердое вещество суспендируют в изопропаноле (6,5 л) и образовавшуюся белую суспензию нагревают до температуры кипения с обратным холодильником. Сразу после образования раствора нагревание прекращают и реакционную смесь перемешивают в течение ~ 1 час при 1-5°C. Твердые вещества отделяют фильтрованием, промывают охлажденным изопропанолом и сушат в вакууме, получая 524,2 г белого твердого вещества, выход 90%, химическая чистота 99,96%, (R)-изомер - ниже предела обнаружения.
Обнаружено, что остаточное содержание рутения составляет менее 2 ч./млн. Согласно нормативному руководству, пероральный концентрационный предел равен 5 ч./млн.
Пример 4: Асимметрическое восстановление окскарбазепин с использованием большего соотношения окскарбазепин:катализатор
Указанное восстановление, описанное в примере 1, осуществляют на окскарбазепине (357 ммоль, 90 г), используя [RuCl2(п-цимол)]2 (0,066 ммоль, 40,4 мг) и (S,S)-TsDAEN (0,145 ммоль, 61,9 мг) и четырехкратное количество воды. Взаимодействие завершается за 27 часов.
Этилацетат отгоняют из опытной партии, поддерживая исходный объем партии добавлением воды (по каплям). Во время перегонки температуру поддерживают выше 60°C. После отгонки приблизительно 1/3, продукт начинает осаждаться.
Смесь охлаждают до 5°C, выдерживают при указанной температуре в течение одного часа и затем фильтруют. Плотный осадок на фильтре промывают водой. Затем влажный осадок с фильтра ресуспендируют в этилацетате (350 мл) и нагревают до температуры кипения с обратным холодильником в течение 0,5 часа. После чего охлаждают до 5°C и выдерживают при указанной температуре в течение 1 часа. Затем смесь фильтруют и извлекают твердые вещества с помощью этилацетата (120 мл). Сушка при высоком вакууме дает не совсем белый порошок: выход изолированного продукта 88% (79,8 г): ВЭЖХ: продукт 99,8%, 98,4% e.e., окскарбазепин 0,09%.
Следует понимать, что раскрытое выше изобретение может быть видоизменено.
Описан способ получения (S)-(+)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамида или (R)-(-)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамида путем восстановления окскарбазепина в присутствии катализатора и гидридного источника. Катализатор основан на комбинации [RuX2(L)]2, где Х представляет собой хлор, бром или иод, и L представляет собой ариловый или арилалифатический лиганд, и лиганда формулы (А) или формулы (В)
где значения радикалов раскрыты в формуле изобретения. Гидридным источником являются либо NR3R4R5 и муравьиная кислота, либо [R3R4R5NH][OOCH] и, необязательно, муравьиная кислота, либо [М][ООСН]х и муравьиная кислота, где R3, R4 и R5 представляют собой C1-6-алкил, М представляет собой щелочной металл или щелочноземельный металл и х равно 1 или 2. рН в ходе процесса поддерживают от 6,5 до 8. Растворитель, в котором проходит восстановление, при этом включает, по меньшей мере, один полярный апротонный растворитель. 4 н. и 26 з.п. ф-лы, 1 ил., 5 табл.
1. Способ получения соединения, выбранного из (S)-(+)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамида или (R)-(-)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамида, путем восстановления окскарбазепина в присутствии катализатора и гидридного источника, где катализатор основан на комбинации [RuX2(L)]2, где Х представляет собой хлор, бром или йод, и L представляет собой ариловый или арилалифатический лиганд, и лиганда формулы (А) или формулы (В):
где R1 выбирают из группы, включающей С1-6-алкокси и С1-6-алкил, n представляет собой число от 0 до 5, и когда n представляет собой число от 2 до 5, то R1 может быть одинаковым или различным, и R2 представляет собой алкил, замещенный алкил, арил, замещенный арил, алкарил или замещенный алкарил;
где гидридный источник выбирают из группы, в которую входят NR3R4R5 и муравьиная кислота, либо [R3R4R5NH][OOCH] и, необязательно, муравьиная кислота, либо [М][ООСН]х и муравьиная кислота, где R3, R4 и R5 представляют собой С1-6-алкил, М представляет собой щелочной металл или щелочноземельный металл и х равно 1 или 2,
где в ходе процесса рН поддерживают от 6,5 до 8,
в котором восстановление происходит в растворителе, и где растворитель включает, по меньшей мере, один полярный апротонный растворитель.
2. Способ по п.1, в котором Х представляет собой хлор.
3. Способ по п.1 или 2, где L представляет собой п-цимол.
4. Способ по п.1 или 2, в котором n равно 1, и R1 представляет собой метоксигруппу или метильную группу.
5. Способ по п.4, в котором n равно 1, и R1 представляет собой метоксигруппу или метильную группу в пара-положении.
6. Способ по любому из пп.1 и 2, в котором лигандом формулы (А) или формулы (В) является (S,S)-N-(4-толуолсульфонил)ди(метоксифенил)этилендиамин, ((S,S)-TsDAEN) или (R,R)-N-(4-толуолсульфонил)ди(метоксифенил)этилендиамин, ((R,R)-TsDAEN).
7. Способ по любому из пп.1, 2 и 5, в котором молярное соотношение окскарбазепина и рутениевого катализатора равно, по меньшей мере, 500:1.
8. Способ по п.7, в котором молярное соотношение окскарбазепина и рутениевого катализатора равно, по меньшей мере, 1500:1.
9. Способ по п.7, в котором молярное соотношение окскарбазепина и рутениевого катализатора равно, по меньшей мере, 2500:1.
10. Способ по любому из пп.1, 2, 5, 8 и 9, в котором гидридным источником являются NR3R4R5 и муравьиная кислота.
11. Способ по п.10, в котором менее двух эквивалентов NR3R4R5 и менее 1 эквивалента муравьиной кислоты добавляют к реакционной смеси в начале процесса.
12. Способ по любому из пп.1, 2, 5, 8, 9 и 11, в котором гидридным источником являются [R3R4R5NH][OOCH] и, необязательно, муравьиная кислота.
13. Способ по п.10, в котором менее двух эквивалентов [R3R4R5H][ООСН] и менее 0,5 эквивалентов муравьиной кислоты добавляют к реакционной смеси в начале процесса.
14. Способ по любому из пп.1, 2, 5, 8 и 9, в котором гидридным источником являются [М][ООСН]х и муравьиная кислота.
15. Способ по п.14, в котором менее двух эквивалентов [М][ООСН]х и менее 1 эквивалента муравьиной кислоты добавляют к реакционной смеси в начале процесса.
16. Способ по любому из пп.1, 2, 5, 8, 9, 11, 13 и 15, в котором R3, R4 и R5 представляют собой этил, пропил или бутил.
17. Способ по любому из пп.1, 2, 5, 8, 9, 11, 13 и 15, в котором рН поддерживают в пределах от 6,5 до 8 путем добавления, управляемым способом, в ходе взаимодействия муравьиной кислоты.
18. Способ по п.17, в котором рН поддерживают в пределах от 7,0 до 7,8 путем добавления, управляемым способом, в ходе взаимодействия муравьиной кислоты.
19. Способ по п.1, в котором растворитель включает диметилформамид или ацетонитрил.
20. Способ по п.1, в котором восстановление происходит в растворителе, включающем 0-25% ДМФА, 0-25% воды и 75-95% EtOAc или 0-25% ацетонитрила, 0-25% воды и 75-95% EtOAc.
21. Способ по п.1, в котором восстановление происходит в растворителе, включающем 5-25% ДМФА и 75-95% EtOAc, 5-25% ацетонитрила и 75-95% EtOAc, 5-25% ДМФА и 75-95% воды или 5-25% ацетонитрила и 75-95% воды.
22. Способ по любому из пп.1, 2, 5, 8, 9, 11, 13, 15, 18 и 19, в котором процесс осуществляют в условиях нагревания до температуры кипения с обратным холодильником.
23. Способ по любому из пп.1, 2, 5, 8, 9, 11, 13, 15, 18 и 19, в котором восстановление происходит в присутствии катализатора межфазного переноса и катализатором межфазного переноса является галоидная соль четвертичного алкиламмония.
24. Способ по любому из пп.1, 2, 5, 8, 9, 11, 13, 15, 18 и 19, в котором (S)-(+)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамид или (R)-(-)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамид выделяют либо добавлением метилтретбутилового эфира (МТВЕ) и фильтрованием, либо осаждением из смеси метанол/вода или смеси метанол/МТВЕ при 0-5°С.
25. Способ по любому из пп.1, 2, 5, 8, 9, 11, 13, 15, 18 и 19, в котором (S)-(+)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамид или (R)-(-)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамид осаждают путем удаления реакционного растворителя с добавлением воды для поддерживания реакционного объема, по существу, на постоянном уровне.
26. Способ по п.25, в котором осажденный (S)-(+)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-3-карбоксамид или (R)-(-)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамид выделяют фильтрованием с последующим ресуспендированием в этилацетате и фильтрованием.
27. Способ по любому из пп.1, 2, 5, 8, 9, 11, 13, 15, 18, 19 и 26, в котором (S)-(+)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамид или (R)-(-)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамид имеет оптическую чистоту в пределах 92-100%.
28. Способ получения соединения формулы (С) или (D)
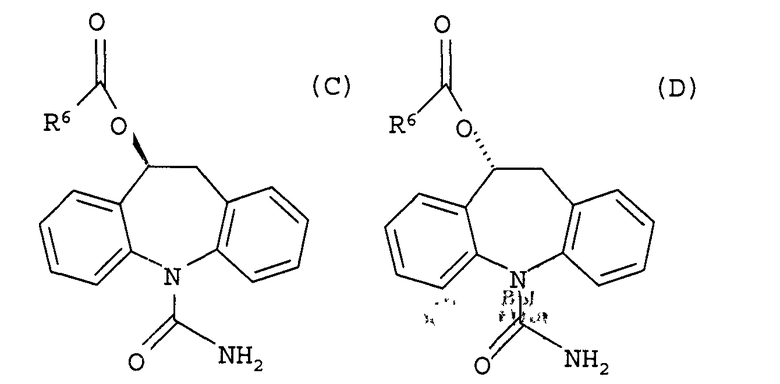
где R6 представляет собой водород, алкил, галогеналкил, аралкил, циклоалкил, циклоалкиалкил, алкокси, арил или пиридил;
включающий первую стадию, представляющую собой способ получения (S)-(+)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-6-карбоксамида или (R)-(-)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамида по любому из предшествующих пунктов, и вторую стадию, где (S)-(+)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамид или (R)-(-)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамид ацилируют.
29. Способ получения (S)-(-)-10-ацетокси-10,11-дигидро-5Н-дибенз/b,f/азепин-6-карбоксамида, включающий первую стадию, представляющую собой способ получения (S)-(+)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамида по любому из предшествующих пп.1-28, и вторую стадию, где (S)-(+)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамид ацилируют.
30. Способ получения (R)-(+)-10-ацетокси-10,11-дигидро-5Н-дибенз/b,f/азепин-5-карбоксамида, включающий первую стадию, представляющую собой способ получения (R)-(-)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-5-карбоксамида по любому из предшествующих пп.1-28, и вторую стадию, где (R)-(-)-10,11-дигидро-10-гидрокси-5Н-дибенз/b,f/азепин-6-карбоксамид ацилируют.
| WO 2004031155 A1, 15.04.2004 | |||
| WU, XIAOFENG et al | |||
| ANGEWANTE CHEMIE, INTERNATIONAL EDITION, 44 (22), 3407-3411, 25.04.2005 | |||
| US 5753646 A, 19.05.1998 | |||
| MA Y et al | |||
| Organic Letters, ACS, v.5, no.12, 2003, pp.2103-2106 | |||
| ЗАМЕЩЕННЫЕ ДИГИДРОДИБЕНЗ/B, F/АЗЕПИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИЭПИЛЕПТИЧЕСКОЙ АКТИВНОСТЬЮ | 1996 |
|
RU2168502C2 |

