Изобретение относится области химико-фармацевтической промышленности, в частности к способам получения лекарственных форм парацетамола, имеющего широкое применение в медицине.
Парацетамол (N-(4-гидроксифенил)ацетамид) - широко используемый ненаркотический анальгетик, оказывающий также противовоспалительное и жаропонижающее действие. Для него известно несколько кристаллических полиморфных модификаций, одна из которых (моноклинная Форма I, Р21/n) термодинамически устойчива, может быть легко получена кристаллизацией из различных растворителей (Nichols G.; Frampton C.S. // J. Pharm. Sci. 1998, 87, 684-693.) [l]. Коммерчески используемая Форма I плохо прессуется, в отличие от другой кристаллической полиморфной модификации (ромбическая Форма II, Pcab), которая легко прессуется в таблетки без наполнителей (Di Martino P., Guyot-Hermann A.-M., Conflant Р., Drache М., Guyot J.-C. // Int. J. Pharm. 1996, 128, 1-8.) [2], (Joris E., Di Martino P., Berneron C., Guyot-Hermann A.-M., Guyot J.-C. // Pharm. Res. 1998, 15(7), 1122-1130.) [3], лучше растворяется, но невоспроизводимо получается в фазово чистом виде и, что хуже всего, метастабильна и самопроизвольно превращается в моноклинную форму при хранении (Di Martino Р., Guyot-Hermann A.-M., Conflant Р., Drache М., Guyot J.-C. // Int. J. Pharm. 1996, 128, 1-8.) [2].
Известен способ получения таблеток парацетамола (Патент РФ №2068689 «Способ получения таблеток парацетамола) [4], включающий смешение и увлажнение компонентов таблеточной массы, влажное гранулирование, сушку гранулята, сухое гранулирование и опудривание гранулята. При смешении и увлажнении компонентов таблеточной массы проводят последовательно: сначала смешение парацетамола с крахмалом 2-3% влажности в соотношении 1:(0,066-0,068), затем увлажнение полученной смеси крахмальным клейстером в соотношении 1:(0,27-0,29), сушку гранулята проводят при температуре 40-50°C, опудривание проводят смесью крахмала 2-3% влажности со стеариновой кислотой при соотношении 1:(0,97-0,99) и при соотношении массы гранулята к массе опудривающей смеси 1:(0,015-0,017). Видно, что для производства таблеточной массы необходимо использовать целую серию различного оборудования, что приводит к дополнительным затратам материалов и времени.
Попытка получения чистого моноклинного парацетамола, пригодного для прямого прессования, была предпринята в работе (Fachaux J.-M., Guyot-Hermann A.-M., Guyot J.-С, Conflant P., Drache М., Veesler S., Boistelle R. // Powder Technology, 1995, 82, 123-128.) [5] и заключалась в перекристаллизации парацетамола из раствора или суспензии в 1,4-диоксане, с образованием сольвата парацетамола с диоксаном (1:½), с последующим удалением сольватированого диоксана. При этом образовывались высокопористые частицы парацетамола. При этом авторы отмечали, что существенным неудобством такого способа получения парацетамола для таблетирования является высокая термическая устойчивость сольвата, а также существенно низкий выход продукта при использовании насыщенных растворов (~21 г парацетамола из 1000 мл насыщенного раствора при 50°C. Использование более высоких температур (80-90°C) позволяло повысить выход продукта до 20-30% (при высоких температурах возможно частичное окисление парацетамола), и несколько больший выход при использовании суспензии (при 50°C выход составлял около 164 г из 1000 мл суспензии, содержащей 200 г парацетамола). Несмотря на то, что авторами было показано некоторое улучшение прессуемости и скорости растворения полученных ими образцов парацетамола по сравнению с исходным реактивом, дальнейшие испытания проходили с использованием стандартных наполнителей, использующихся для приготовления таблеток парацетамола.
Возможность получения прессуемых форм парацетамола, которые были бы устойчивы при хранении, привлекает большой интерес как научного сообщества, так и фармацевтических компаний. Известен ряд работ зарубежных исследователей, посвященных поиску новых способов получения парацетамола, пригодного для непосредственного таблетирования без использования наполнителей, а также приготовления парацетамолсодержащих смесей с замедленной или увеличенной скоростью высвобождения лекарственного препарата. В качестве методов решения предлагалось использовать вместо чистого парацетамола его смеси с поливинилпирролидоном (Garekani H.A., Ford J.L., Rubinstein M.H., Rajabi-Siahboomi A.R. // Int. J. Pharm. 2000, 208, 87-99.) [6], углеводами (Gonnissen Y., Remon J.P., Vervaet C. // Eur. J. Pharm. Biopharm. 2007, 67, 220-226.) [7], (Boateng, J.S., Auffret, A.D., Matthews, K.H., Humphrey, M.J., Stevens, H.N., Eccleston, G.M. Characterization of freeze-dryied wafers and solvent evaporated films as potential drug delivery systems to mucosal surfaces. Int. J. Pharm. 2010, 389, 24-31) [8], (Wlosnewski, J.C., Kumpugdee-Vollrath, M., Sriamornsak, P. Effect of drying technique and desintegrant on physical properties and drug release behavior of microcrystalline cellulose-based pellets prepared by extrusion/spheronization. // Chem. Eng. Res. Design. 2010, 88, 100-108.) [9], хитозаном и алгинатом натрия (Lai H.L., Abu'Khalil A., Craig D. Q. М. // Int. J. Pharm. 2003, 251, 175-181.) [10], либо смешанные кристаллы на основе щавелевой кислоты, нафталина и др. Karki S., (Friscic Т., Fabian L., Laity P.R., Day C.M., Jones W. // Adv. Mater. 2009, 21, 3905-3909.) [11] или соединения включения с гидроксипропил-β-циклодекстрином Talegaonkar S., Khan A.Y., Khar R.K., Ahmad F.J., Khan Z.I. // Iranian J. Pharm. Res. 2007, 6, 95-99.) [12]. В качестве инструментальных методов в этих работах преимущественно используются методы распылительной и лиофильной сушки водных растворов парацетамола.
Существенными недостатками данных методик являются необходимость использования и испарения в ходе процесса больших объемов растворителей (обусловленная плохой растворимостью парацетамола), либо сложность удаления большого количества льда сублимацией (приводящая к существенному увеличению времени эксперимента для наработки сравнительно малых количеств препарата), что приводит к большим энергозатратам на единицу массы продукта.
В литературе описаны инструментальные методы получения высокодисперсных образцов лекарственных препаратов, основанные на распылительной либо лиофильной сушке (Hu J., Johnston K.R, Williams R.O. // Drug Dev. Ind. Pharm. 2004, 30 (3), 233-245.) [13]. При этом использование больших скоростей охлаждения с образованием аморфных фаз с последующим удалением растворителей сублимацией позволяет избежать укрупнения образующихся кристаллитов вследствие отсутствия контакта с жидкой фазой.
Данный способ по техническим приемам наиболее близок к предлагаемому изобретению.
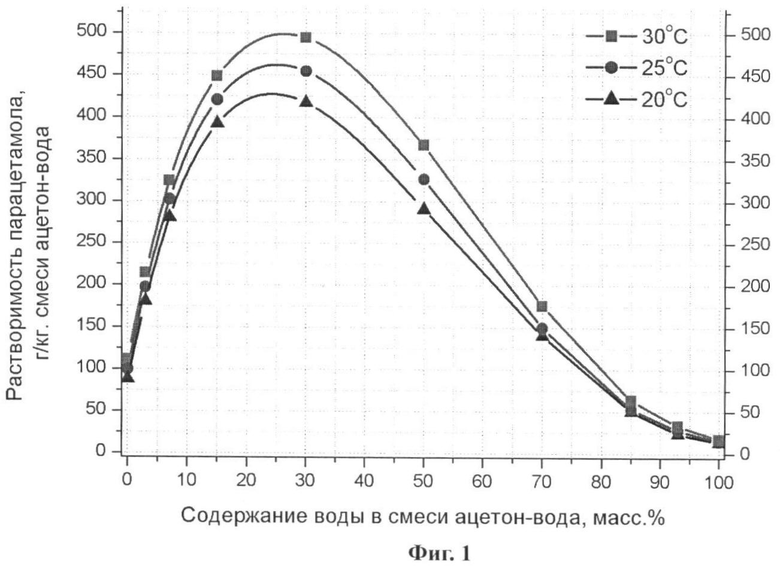
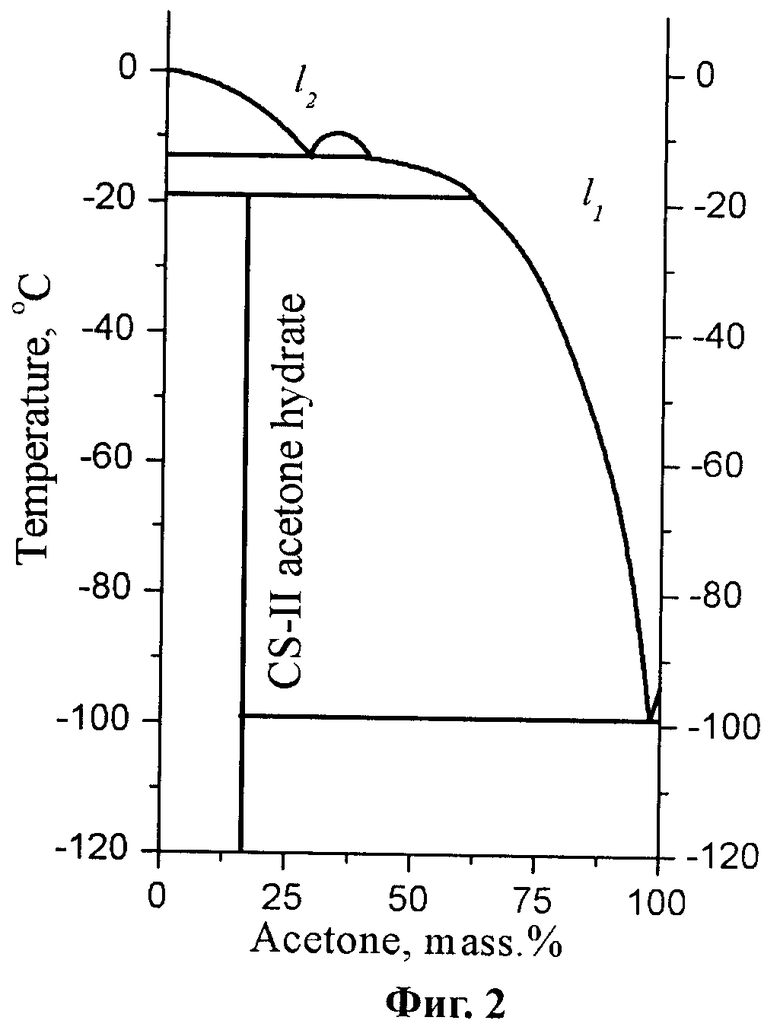
Известно, что существенное увеличение растворимости парацетамола достигается при использовании в качестве растворителя двухкомпонентной смеси - легкокипящая жидкость (диоксан, этанол, ацетон) - вода (Фиг.1) (Granberg, R.A., Rasmuson, A.C. Solubility of Paracetamol in Binary and Ternary Mixtures of Water + Acetone + Toluene. // J. Chem. Eng. Data. 2000, 45, 478-483.) [14]. Дополнительным аргументом в пользу использования систем с диоксаном и ацетоном является образование клатратных гидратов (молярное отношение легкокипящая жидкость - вода 1:17) в этих системах при низких температурах (Фиг.2), вследствие чего имеется возможность проводить эксперимент таким образом, чтобы в условия пониженного давления в широком интервале концентраций и температур сосуществовали только твердые фазы. (Dyadin Yu. A., Bondaryuk I.V., Zhurko F.V. Clathrate hydrates at high pressures. Inclusion compounds, V.5, Eds. J.L. Atwood, J.E.D. Davies and D.D. MacNicol, Oxford University Press, Oxford. 1991, 214-275.) [15] При этом несмотря на то благоприятное условие, что температура плавления диоксана позволяет удалять растворитель сублимацией при температуре, лимитируемой скоростью удаления льда, как более труднолетучего компонента в этой системе в твердом состоянии, данная смесь растворителей была отвергнута вследствие установленного нами факта образования в данной системе устойчивого сольвата парацетамола.
Задачей настоящего изобретения является разработка более простого и эффективного способа получения высокодисперсной формы моноклинной полиморфной модификации парацетамола, обладающей лучшей динамикой растворения и пригодной для непосредственного таблетирования без использования наполнителей.
Поставленная задача была решена использованием в качестве растворителя системы ацетон - вода, быстрым охлаждением растворов распылением раствора в емкость с жидким азотом либо охлаждением раствора на охлажденной в жидком азоте медной пластине с последующим удалением растворителей нагревом (ступенчатым повышением температуры от -196°C до +30°C) получившейся твердой смеси в вакуумированном термостатируемом сосуде при давлении <5·10-2 мм рт.ст. В таком случае при указанных условиях эксперимента происходит удаление компонентов используемой смеси растворителей сублимацией и разложение образующихся при отжиге замороженных растворов промежуточных фаз.
В работе использовали порошок моноклинной полиморфной модификации парацетамола (Merck), ацетон квалификации «хроматографически чистый» и дистиллированную воду.
Были проведены следующие предварительные эксперименты:
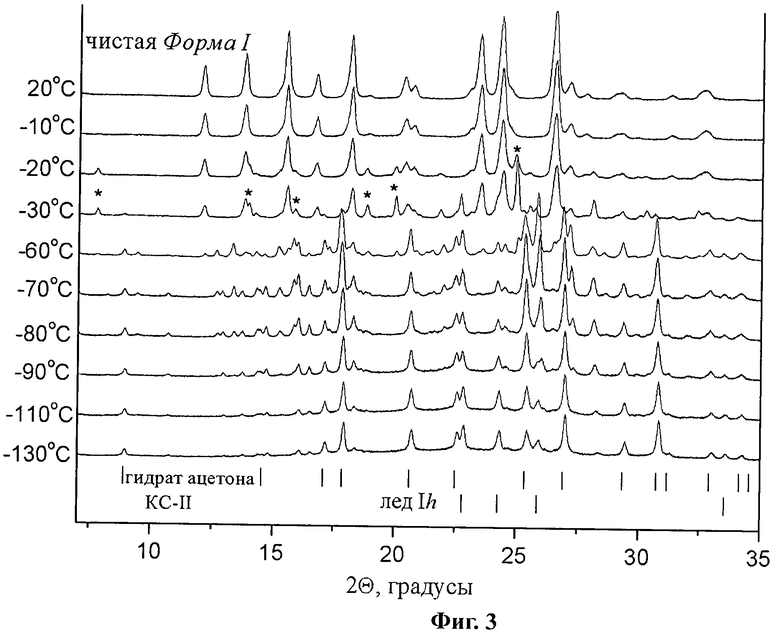
К началу нашего исследования полностью отсутствовали литературные данные как о составе фаз, образующихся при быстром замораживании растворов парацетамола в используемой смеси растворителей, так и о процессах, протекающих при отжиге образующихся в данной системе аморфных фазах. Эти сведения были абсолютно необходимы как для ответа на вопрос, возможно ли использование данной смеси растворителей для поставленной нами задачи, так и подбора температурных режимов сублимационной сушки. С целью идентификации фаз, образующихся при быстром замораживании растворов и регистрации фазовых превращений, происходящих при отжиге замороженных растворов в системах ацетон - вода и парацетамол - ацетон - вода, были записаны порошковые дифрактограммы (порошковый дифрактометр Bruker D8 Advance, оборудованный низкотемпературной приставкой ТТК 450 Anton Paar, с ячейкой для исследований в условиях вакуума до 10-3 мм рт.ст.) в температурном интервале от -140 до 20°C при атмосферном и при пониженном давлении (Р<5·10-2 мм рт.ст.). Результаты дифракционного эксперимента, проведенного с образцами замороженного раствора парацетамола в смеси ацетон - вода (30 масс.% воды) при различных температурах в условиях пониженного давления (Р<5·10-2 мм рт.ст.), представлены на Фиг.3, где * обозначены наиболее сильные рефлексы тригидрата парацетамола. На дифрактограмме, записанной при -130°С, нами были надежно идентифицированы только рефлексы, отвечающие фазам гидрата ацетона КС-II и льда Ih. По-видимому, при добавлении парацетамола происходит образование аморфной фазы ацетона при быстром охлаждении раствора, который вследствие проведения эксперимента в области пара на фазовой диаграмме ацетона удаляется сублимацией из ячейки для дифракционных исследований.
Дифракционный эксперимент, проведенный с образцом замороженного раствора парацетамол-ацетон-вода при атмосферном давлении, показал, что при температурах ниже температуры плавления эвтектики в двойной системе ацетон - вода, также наблюдались только рефлексы, отвечающие фазам гидрата ацетона КС-II и льда Ih, несмотря на то, что исходя из состава используемой нами смеси ацетон - вода, в данных условиях эксперимента должны были наблюдаться рефлексы гидрата ацетона КС-II и низкотемпературной полиморфной модификации ацетона. Низкое качество полученных в данном эксперименте дифрактограмм (не приводятся) обусловлено присутствием аморфной фазы (наличие характерного «горба»), подтверждает наше предположение об образовании аморфной фазы ацетона. На дифрактограммах, записанных при повышении температуры от -130°C до -70°C, мы наблюдали появление и увеличение интенсивности рефлексов новой фазы, не относящейся ни к известным полиморфным модификациям парацетамола, ни его известным гидратам. По-видимому, неизвестная фаза, наблюдавшаяся при температурах -130°C до -70°C, представляет собой полигидрат парацетамола либо упаковочный комплекс парацетамола с ацетоном, либо тройное соединение, которое можно представить как гидрат-сольват парацетамола. При дальнейшем повышении температуры наблюдалось появление и рост рефлексов, соответствующих тригидрату парацетамола и моноклинной полиморфной модификации парацетамола; исчезновение рефлексов тригидрата парацетамола при температурах выше -20°C свидетельствует о его разложении. На дифрактограммах, записанных при -10°C и +20°C (последняя стадия сушки), присутствуют только рефлексы, отвечающие чистой Форме I парацетамола. Появление на дифрактограммах рефлексов моноклинной полиморфной модификации парацетамола только при сравнительно высоких температурах можно объяснить превышением температуры эксперимента некоторой температуры стеклования (glass transition temperature) (Liu, J. Physical Characterization of Pharmaceutical Formulations in Frozen and Freeze-Dried Solid States: Techniques and Applications in Freeze-Drying Development. Pharmaceutical Development and Technology. 2006, 11, 3-28.) [16] парацетамола в данной системе, выше которой происходит кристаллизация парацетамола в твердой фазе. При этом поскольку увеличение интенсивности рефлексов моноклинной полиморфной модификации парацетамола происходит даже при температурах, при которых уже в условиях нашего эксперимента отсутствуют другие соединения, это подтверждает наше предположение об образовании парацетамола Формы I при кристаллизации из аморфной фазы.
Температурный режим и давление для предлагаемого способа получения высокодисперсного парацетамола были подобраны на основании полученных нами экспериментальных данных о процессах, происходящих при отжиге замороженных растворов в системе парацетамол - ацетон - вода в температурном интервале от -196°C до +30°C, а также известных литературных данных по диаграмме плавкости системы ацетон - вода и положению кривых сублимаций льда и ацетона на соответствующих фазовых диаграммах.
Таким образом экспериментально было установлено:
1. Лимитирующей стадией сушки становится удаление аморфного ацетона сублимацией при температурах ниже температуры плавления эвтектики в двойной системе ацетон - вода, поскольку неполное удаление ацетона сублимацией при этих температурах приводит к образованию жидкой фазы ацетон - вода с содержанием до 5% масс. воды при температуре -80°C, что приводит к повышению растворимости парацетамола, его последующей кристаллизации при испарении смеси растворителей и, как следствие, появлению хорошо ограненных кристаллов. Следующая стадия сушки, выполняемая при температуре порядка -35°C, имеет целью разложение неизвестной фазы, являющейся, по-видимому, полигидратом парацетамола или его упаковочным комплексом с ацетоном, частичное удаление льда Ih сублимацией и частичное разложение тригидрата парацетамола. Сушка при температуре -7°C имеет целью удаление оставшегося количества льда Ih и разложение тригидрата парацетамола. Заключительная стадия сушки проводится при температуре +30°C и имеет целью удаление следовых количеств воды за счет большего давления паров воды при этой температуре согласно фазовой диаграмме воды.
2. Оптимальный для данного метода диапазон концентраций используемой смеси ацетон - вода - от 30 до 15 масс.% воды. При большем содержании воды в используемых растворах существенно уменьшается растворимость парацетамола в данной смеси растворителей и увеличивается относительная доля льда, который необходимо удалять сублимацией, а при концентрации воды в исходном растворе <15% резко уменьшается растворимость парацетамола в данной смеси и сильно увеличивается относительное количество жидкой фазы, находящееся в равновесии с твердым клатратным гидратом ацетона, либо только жидкой фазы при температурах выше -60°C, что в случае небольших колебаний заданных параметров (продолжительность выдержки при данной температуре, давление) приводит к перекристаллизации парацетамола с образованием хорошо ограненных кристаллов, что недопустимо в рамках поставленной задачи.
3. Оптимальный для данного метода диапазон концентраций парацетамола в исходной смеси - 80-95% от известной на основании литературных данных равновесной растворимости парацетамола в данной смеси растворителей при заданной температуре. При концентрациях парацетамола >95% необходимо либо подогревание раствора до температуры порядка +50°C либо длительное перемешивание, что приводит к испарению ацетона с соответствующим понижением растворимости парацетамола в получившейся смеси и увеличению времени подготовки раствора, также необходимости подогревать форсунку (в случае, если применяемый для охлаждения раствора способ заключается в распылении раствора в емкость с жидким азотом) для предотвращения кристаллизации парацетамола из раствора в подводящем капилляре. При концентрациях парацетамола <80% увеличивается относительная доля растворителей, которые необходимо удалить, что приводит к увеличению времени эксперимента и энергозатрат на единицу массы продукта.
Характеристики получаемого продукта.
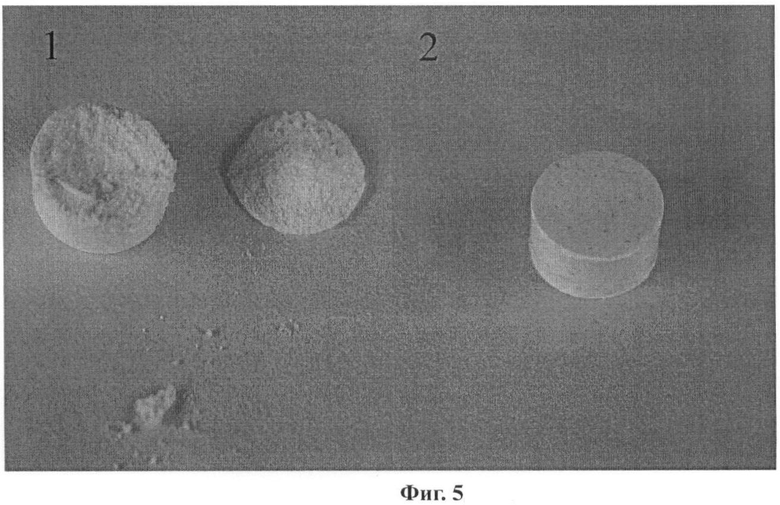
По данным сканирующей электронной микроскопии (настольный сканирующий электронный микроскоп Hitachi ТМ-1000, напыление Pt), полученные при различном увеличении образцы представляют собой плоские частицы с линейными размерами 1-10 мкм, толщиной 60-150 нм, объединенные в агломераты размерами 30-200 мкм (в зависимости от способа охлаждения исходного раствора) (Фиг.4). Методом рентгенофазового анализа было установлено, что все полученные образцы представляют собой чистую моноклинную полиморфную модификацию парацетамола. Образцы полученного нами высокодисперсного парацетамола продемонстрировали существенно лучшую способность к прессованию (измеритель прочности Zwick/Roell Z010, диаметр поршня 6.0 мм, масса образца ~150 мг) по сравнению с обычными поликристаллическими образцами коммерческого препарата Формы I. Следует также отметить, что при изготовлении таблеток прессованием поликристаллических образцов товарного реактива происходит прилипание таблеток к поршню, для предотвращения чего в промышленности используют различные добавки, облегчающие скольжение (например, стеарат магния); при прессовании образцов парацетамола, полученных в данной работе, таблетка легко отходит от поршня. На Фиг.5 продемонстрирована различная способность к прессованию образцов парацетамола, полученного в данной работе, и чистой Формы I. Давление прессования 345 МПа. (1 - таблетка, приготовленная прессованием товарного реактива, прилипает к поршню и легко разрушается при попытке снятия с поршня; 2 - таблетка, полученная прессованием парацетамола, приготовленного предложенным нами способом).
Пористость изготовленных таблеток (6,0% при 345 МПа) Фиг.6 оказалась сравнимой с пористостью таблеток, полученных прессованием образцов чистой Формы II (5,2% при 335 МПа) (Joris E., Di Martino P., Berneron С., Guyot-Hermann A.-M., Guyot J.-C. // Pharm. Res. 1998, 15 (7), 1122-1130.) [3].
В то же время дифракционным экспериментом было установлено, что при таблетировании не происходит образования Формы II. Улучшение прессуемости происходит, по-видимому, вследствие высокой поверхностной энергии, получающихся при использовании данного метода частиц парацетамола (Huttenrauch R., Friecke S., Zieike P. // Pharm. Res., 1985, 6, 302-306.) [17], обусловленной большой площадью поверхности и формой частиц, что способствует их лучшей агрегации.
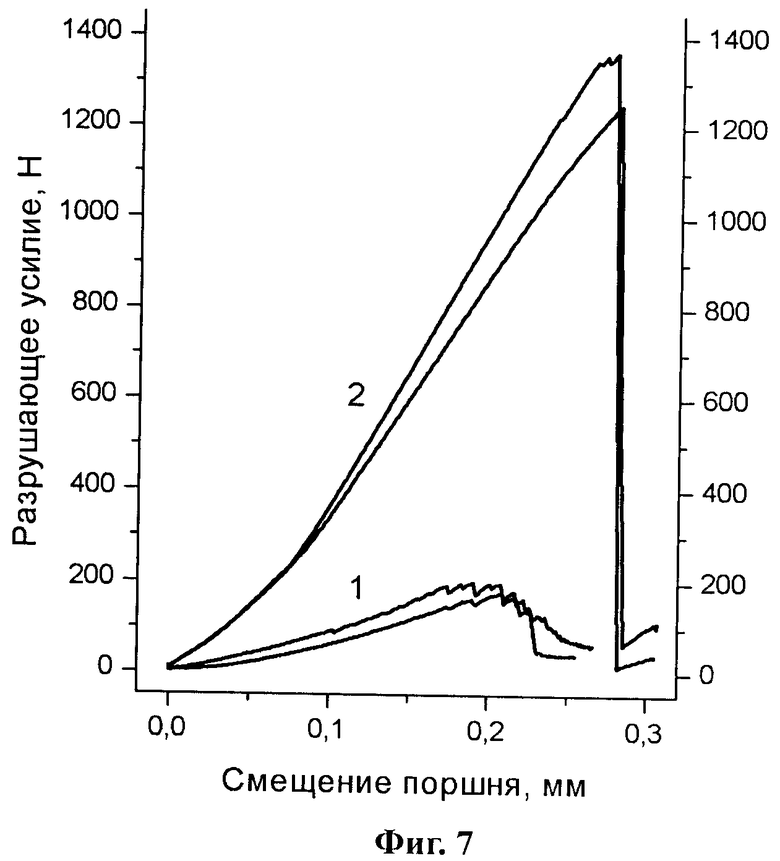
Эксперименты по определению разрушающего усилия (разрушающее усилие направлено: а) вдоль оси цилиндра, б) вдоль диаметра цилиндра) приготовленных таблеток показали существенно более высокую прочность таблеток, приготовленных из образцов парацетамола, полученных в данной работе (способ «а»: 1118±148Н, б: 45,5±5 Н), по сравнению с таблетками, приготовленными из чистого коммерческого моноклинного парацетамола (способ «а»: 184±18 Н, б: 8±1 Н). На Фиг.7 представлены результаты эксперимента по определению разрушающего усилия таблеток парацетамола (разрушающее усилие приложено вдоль оси цилиндра), приготовленных из: 1 - товарного реактива (Merck); 2 - образца, полученного в данной работе.
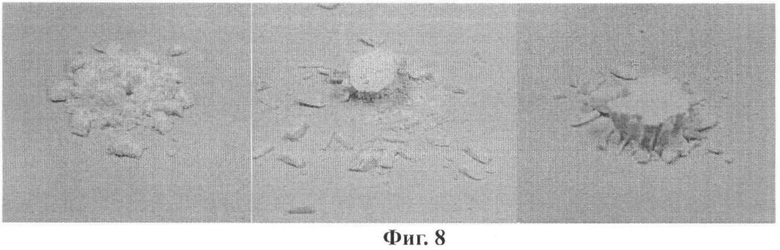
Разрушающие усилие таблеток, приготовленных прессованием образцов парацетамола, полученных в данной работе, определенное по способу б), используемому в фарм. промышленности (1,27±0,27 МПа), оказалось несколько больше разрушающего усилия таблеток, приготовленных прессованием чистой Формы II (~0,95 МПа для таблеток, полученных при усилии прессования 335 МПа) [3] и существенно больше усилия, требуемого для разрушения таблеток, приготовленных прессованием коммерческого моноклинного парацетамола, определенного как в данной работе (0,225±0,03 МПа), так и другими авторами (~0,2 МПа [3] (поликристаллический образец); ~0,38 МПа [7] (получен распылительной сушкой)) и несколько меньше или на уровне усилия разрушения таблеток, приготовленных прессованием смешанных кристаллов (1,15-2,79 МПа) [11] или смесей с углеводами, полученных распылительной сушкой (0,45-2,39 МПа) [7]. При этом, в отличие от таблеток, приготовленных из поликристаллического парацетамола (реактив), при разрушении таблетки, приготовленной из препарата, полученного в данной работе, образец после эксперимента не крошился, но представлял собой совокупность осколков, сохраняющих твердость исходной таблетки. На Фиг.8 представлен вид таблеток после эксперимента по определению разрушающего усилия (разрушающее усилие приложено вдоль оси цилиндра): слева - таблетка, приготовленная из товарного реактива (Merck); две таблетки справа - из образца, полученного в данной работе.
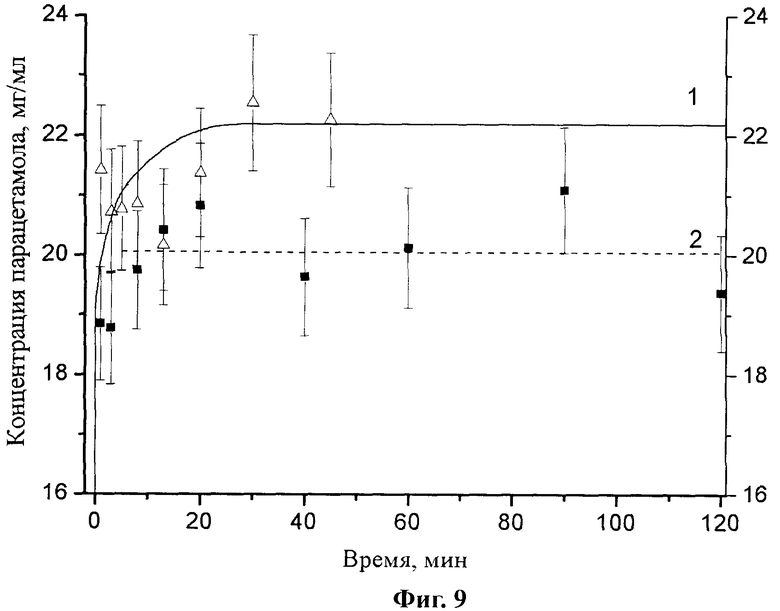
Образцы моноклинного парацетамола, полученного по нашей методике, хорошо растворяются в воде. Растворение проводили с использованием тестера растворимости 705 DS (Varian), через определенные промежутки времени производили отбор проб, которые подвергались фильтрованию в термостатируемые пробирки. Концентрацию парацетамола определяли фотометрически (спектрофотометр Сагу 50 (Varian), l=10 мм, λ=243 нм) после необходимого разбавления фильтрата. Сравнение динамики растворения образцов парацетамола (тестер растворимости 705 DS (Varian)), полученных по предложенной нами методике и традиционным способом (Фиг.9, где 1 - полученный в данной работе, 2 - товарный реактив моноклинного парацетамола (Merck)), показало, что в течение длительного времени (несколько суток) концентрация парацетамола в растворе превышает равновесную (Grant D. J. W., Mehdizadeh M., Chow A. H.-L., Fail-brother J. E. // Int. J. Pharm. 1984, 18, 25-33.) [18]. Достижение на начальных этапах растворения значений концентраций, превышающих равновесные, является типичным для лекарственных веществ со значительной степенью аморфизации, нанодисперсных образцов или метастабильных кристаллических модификаций, например для образцов, полученных при механической активации (Shakhtshneider Т.Р., Boldyrev V.V. Mechanochemical synthesis and mechanical activation of drugs in Reactivity of Molecular Solids / Ed. by E. Boldyreva, V. Boldyrev. John Wiley & Sons, LTD, England, 1999, p.271-312.) [19].
Таким образом, предложенный метод впервые позволил решить задачу получения устойчивой формы парацетамола, пригодной для прямого прессования без наполнителей в таблетки, сочетающие устойчивость при хранении, высокую механическую прочность и высокую скорость растворения с образованием пересыщенных растворов, сохраняющихся в течение длительного времени, что важно для биодоступности препарата. Предлагаемый способ прост в осуществлении, сравнительно легко может быть преобразован в технологический процесс промышленного масштаба.
Примеры реализации способа
Пример 1
В 26.9 г смеси ацетон - вода (29.9 масс.% воды) при перемешивании и небольшом подогревании раствора (до +30°C) растворили 10.8 г моноклинной полиморфной модификации парацетамола. Раствор распыляли через пульверизатор (диаметр подводящего капилляра 0,4 мм, избыточное давление распыляющего газа 1 атм) в емкость с жидким азотом. Образующуюся смесь твердых фаз (лед, твердый ацетон, клатратный гидрат ацетона, тригидрат парацетамола) помещали в охлажденный до температуры жидкого азота массивный держатель (масса держателя и подставки>массы смеси примерно в 3 раза), который помещали при температуре жидкого азота в вакуумную камеру, камеру закрывали и затем понижали давление в до Р<5·10-2 мм рт.ст. Затем вакуумную камеру помещали в термостат с температурой теплоносителя -35°C, с выдерживанием в течение 5 часов при этой температуре. Затем температуру теплоносителя повышали до -7°C и +30°C, с выдерживанием в течение 4 часов при каждой температуре. Затем давление в камере повышали до Р=1 атм заполнением камеры сухим воздухом, камеру открывали, доставали держатель с образцом, образец помещали в предварительно взвешенный бюкс, взвешивали и помещали в эксикатор с сухой атмосферой для дальнейшего хранения. Выход ~8 г(75%).
Аналогичные примеру 1 эксперименты были проведены с растворами парацетамола в смеси ацетон - вода (25 масс.% воды) с концентрациями парацетамола, равными 85 и 90% от равновесной растворимости в данной смеси и с вариациями использования подводящих капилляров различного диаметра (от 0.4 до 1.0 мм) и избыточного давления распыляющего газа (0.7-1.5 атм.).
Рентгенофазовый анализ показал, что полученные во всех экспериментах образцы представляет собой чистую моноклинную полиморфную модификацию парацетамола
По данным сканирующей электронной микроскопии, полученные образцы представляют собой плоские частицы с линейными размерами 2-5 мкм, толщиной 60-150 нм, объединенные в ассоциаты размерами 30-100 мкм.
Аналогичные эксперименты проводили со смесью растворителей с концентрациями 15 и 20 масс.% воды.
Пример 2
В 31.9 г смеси ацетон - вода (30.1 масс.% воды) при перемешивании и небольшом подогревании раствора (до +30°C) растворили 13.0 г. моноклинной полиморфной модификации парацетамола. Раствор распрыскивали из шприца на лежащую в жидком азоте медную пластину с постоянным сбиванием образующейся твердой фазы в емкость с жидким азотом. Образующуюся смесь твердых фаз (лед, твердый ацетон, клатратный гидрат ацетона, тригидрат парацетамола) помещали в охлажденный до температуры жидкого азота массивный держатель (масса держателя и подставки > массы смеси примерно в 3 раза), который помещали при температуре жидкого азота в вакуумную камеру, камеру закрывали и затем понижали давление в до Р<5·10-2 мм рт.ст. Затем вакуумную камеру помещали в термостат с температурой теплоносителя -35°C, с выдерживанием в течение 5 часов при этой температуре. Затем температуру теплоносителя повышали до -7°C и +30°C, с выдерживанием в течение 4 часов при каждой температуре. Затем давление в камере повышали до Р=1 атм заполнением камеры сухим воздухом, камеру открывали, доставали держатель с образцом, образец помещали в предварительно взвешенный бюкс, взвешивали и помещали в эксикатор с сухой атмосферой для дальнейшего хранения. Выход - 11 г. (85%).
Аналогичные примеру 2 эксперименты были проведены с растворами парацетамола в смеси ацетон - вода (25 масс.% воды) с концентрациями парацетамола, равными 85 и 90% от равновесной растворимости в данной смеси.
Рентгенофазовый анализ показал, что полученные во всех экспериментах образцы представляет собой чистую моноклинную полиморфную модификацию парацетамола.
По данным сканирующей электронной микроскопии, полученные образцы представляют собой плоские частицы с линейными размерами 2-10 мкм, толщиной 60-150 нм, объединенные в ассоциаты размерами 200-300 мкм.
Аналогичные эксперименты поводили со смесью растворителей с концентрациями 15 и 20 масс.% воды.
Источники информации


| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОДИСПЕРСНОГО ИБУПРОФЕНА | 2012 |
|
RU2491919C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОДИСПЕРСНЫХ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ САЛЬБУТАМОЛА | 2012 |
|
RU2504370C1 |
| СПОСОБ ПОЛУЧЕНИЯ БЕТА ГЛИЦИНА | 2010 |
|
RU2425025C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОДИСПЕРСНОГО МЕЛОКСИКАМА | 2011 |
|
RU2465892C1 |
| ВЫСОКОДИСПЕРСНАЯ КОМБИНИРОВАННАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ БЕКЛОМЕТАЗОНА И САЛЬБУТАМОЛА С БЕТА-ГЛИЦИНОМ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2013 |
|
RU2539374C1 |
| ВЫСОКОДИСПЕРСНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ БУДЕСОНИДА С БЕТА-ГЛИЦИНОМ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2013 |
|
RU2539376C1 |
| Способ получения аморфных форм соединений, обладающих противоопухолевой активностью | 2023 |
|
RU2841137C2 |
| КРИСТАЛЛИЧЕСКАЯ МОДИФИКАЦИЯ 3β-ГИДРОКСИ-5-АНДРОСТЕН-17-ОНА FVII И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2013 |
|
RU2528990C1 |
| ИЗОЛИРОВАННАЯ ОРТОРОМБИЧЕСКАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА 4-[6-АЦЕТИЛ-3-[3-(4-АЦЕТИЛ-3-ГИДРОКСИ-2-ПРОПИЛФЕНИЛТИО)ПРОПОКСИ]-2-ПРОПИЛ-ФЕНОКСИ]МАСЛЯНОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕЕ ОСНОВЕ | 2004 |
|
RU2317979C2 |
| Водорастворимая композиция, обладающая противоопухолевой активностью и способ ее получения | 2015 |
|
RU2611362C1 |


Изобретение относится к области химико-фармацевтической промышленности и касается способа получения высокодисперсного парацетамола путем быстрого охлаждении раствора парацетамола в системе ацетон - вода при содержании воды 15-30% и концентрации парацетамола 80-95% от равновесной растворимости в данной смеси при заданной температуре распылением его в емкость с жидким азотом либо охлаждением раствора на охлажденной в жидком азоте медной пластине с последующим удалением растворителей сублимацией из получившейся твердой смеси ступенчатым повышением температуры от -196°С до -80°С до полного удаления ацетона, затем до -35°С для разложения сольвата парацетамола, затем до -7°С для разложения тригидрата парацетамола и полного удаления льда в вакуумированном термостатируемом сосуде при давлении <5·10-2 мм рт.ст. Изобретение обеспечивает простой и эффективный способ получения высокодисперсного парацетамола. 1 з.п. ф-лы, 9 ил., 2 пр.
1. Способ получения высокодисперсного парацетамола путем быстрого охлаждения раствора парацетамола и удаления растворителя сублимацией, отличающийся тем, что в качестве растворителя используют систему ацетон-вода при содержании воды 15-30%, при этом концентрация парацетамола составляет 80-95% от равновесной растворимости в данной смеси при заданной температуре, быстрое охлаждение раствора осуществляют распылением раствора в емкость с жидким азотом, либо охлаждением раствора на охлажденной в жидком азоте медной пластине с последующим удалением растворителей сублимацией из получившейся твердой смеси ступенчатым повышением температуры от -196 до -80°С до полного удаления ацетона, затем до -35°С до полного разложения сольвата парацетамола, затем до -7°С до полного разложения тригидрата парацетамола и удаления льда в вакуумированном термостатируемом сосуде при давлении <5·10-2 мм рт.ст.
2. Способ по п.1, отличающийся тем, что извлечение образца производят в сухой атмосфере после нагревания термостатируемого сосуда до температуры окружающей среды.
| RU 2142792, C1, 20.12.1999 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПОРОШКООБРАЗНОЙ ВОДОРАСТВОРИМОЙ ШИПУЧЕЙ КОМПОЗИЦИИ (ВАРИАНТЫ) | 2005 |
|
RU2288594C2 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ АГЛОМЕРАЦИИ ТРУДНОРАСТВОРИМЫХ И ЧУВСТВИТЕЛЬНЫХ К ГИДРОЛИЗУ ВЕЩЕСТВ | 1997 |
|
RU2191065C2 |
| ГРАНУЛЫ, СОДЕРЖАЩИЕ ПАРАЦЕТАМОЛ, НПВС И САХАРОСПИРТ, ПОЛУЧЕННЫЕ ЭКСТРУЗИЕЙ РАСПЛАВА | 2005 |
|
RU2389478C2 |

