Область техники, к которой относится изобретение
Полиморфная форма А, установленная методом дифракции рентгеновских лучей в порошке 4-[6-ацетил-3-[3-(4-ацетил-3-гидрокси-2-пропилфенилтио)пропокси]-2-пропил-фенокси]масляной кислоты, обладает высокой растворимостью и биодоступностью по сравнению с другими кристаллическими формами.
Уровень техники
Лейкотриены - это метаболиты арахидоновой кислоты через 5′-липоксигеназный путь и являются важными медиаторами аллергических реакций типа реакции, связанной с бронхиальной астмой. Лекарственные средства, оказывающие антагонистическое действие на лейкотриены, полезны для лечения аллергических заболеваний.
Синтез и биологическая активность многих производных феноксиалкилкарбоновых кислот, являющихся антагонистами лейкотриенов, описаны в Ohashi et al., US 4985585. Эти соединения получены в лабораторных масштабах методом колоночной хроматографии на силикагеле неочищенных смесей продукта. Растворитель выпаривали, получая бледно-желтое масло либо бесцветные кристаллы, и не прилагали никаких сознательных усилий, чтобы контролировать морфологию кристаллов.
Мы заметили, что 4-[6-ацетил-3-[3-(4-ацетил-3-гидрокси-2-пропилфенилтио)-пропокси]-2-пропилфенокси]масляная кислота (1), представленная в примере 33 у Ohashi et al., обладает пероральной активностью при лечении астмы и аллергических заболеваний, а твердое вещество может образовывать кристаллы в нескольких особых полиморфных формах при получении его в большом объеме. Оказалось, что условия кристаллизации, в особенности температура, имеют критическое значение при получении различных полиморфных форм.
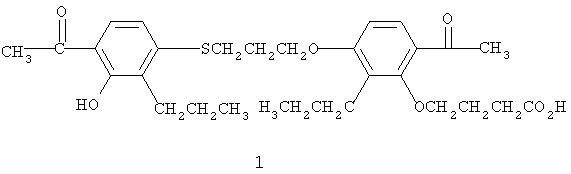
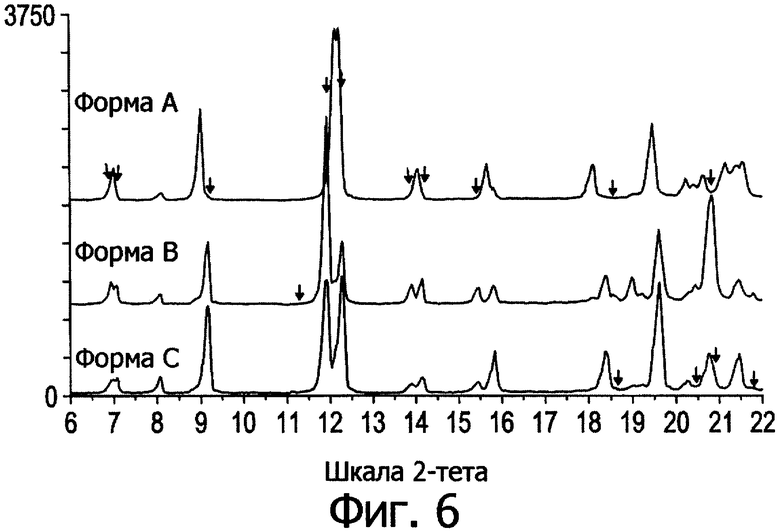
Мы также заметили, что по растворимости и биодоступности одна из этих полиморфных форм, идентифицированная как орторомбические кристаллы (форма V в табл.1 и форма А на фиг.6), превосходит другие полиморфные формы, и поэтому форма А обеспечивает улучшенные твердые составы.
Раскрытие изобретения
Настоящее изобретение предусматривает фармацевтическую композицию, содержащую соединение формулы (1) в особой кристаллической форме:
вместе с фармацевтически приемлемым носителем или наполнителем, причем особая кристаллическая форма состоит из полиморфной формы А, по существу, свободной от нежелательных полиморфов. Под "по существу свободной" понимается то, что обнаруживается мало или совсем не обнаруживаются нежелательные полиморфы методом дифрактометрии рентгеновских лучей в порошке (PXRD). Как правило, чистота полиморфной формы превышает 90% (определяется по высоте пика на записи дифракции рентгеновских лучей). Предпочтительно искомая кристаллическая форма по изобретению, по меньшей, мере на 95% представлена полиморфной формой А (фиг.6) при измерении по относительной высоте пика в районе 9° 2-тета.
Настоящее изобретение также предусматривает способ получения формы А соединения формулы (1) с чистотой, по меньшей, мере 90% относительно других полиморфов. Типичный способ кристаллизации включает стадии растворения соединения (1) в 5-10 весовых частях теплого этанола и 1-10 частях воды, перемешивания полученной суспензии при 20-25°С в течение 15-60 минут, а затем охлаждения до 5-10°С в течение следующих 1-4 часов, добавления 5-15 частей воды, перемешивания смеси при 5-10°С в течение следующих 1-4 часов и выделения кристаллов соединения (1), содержащих, по меньшей мере, около 90 вес.% формы А (фиг.6).
Соответственно, предусматривается фармацевтическая композиция, содержащая соединение формулы (1) в твердом виде:
вместе с фармацевтически приемлемым носителем или наполнителем, при условии, что соединение формулы (1) находится в полиморфной форме А и практически свободно от других полиморфных форм. В предпочтительном воплощении изобретения соединение формулы (1) находится в виде орторомбических кристаллов. Также изобретение может осуществляться в виде таблеток или капсул. Предпочтительно композиция по изобретению дает, по существу, такой профиль PXRD, как представлено для полиморфной формы А на фиг.6. Кроме того, предпочтительно, по меньшей мере, 90% соединения формулы (1) представлено полиморфной формой А при определении по высоте пика PXRD в районе 9° 2-тета. Композиция может дополнительно включать лактозу и микрокристаллическую целлюлозу. Таблетки могут иметь различный вес, к примеру, от 250 до 500 мг.
Настоящее изобретение также касается композиций, содержащих изолированные кристаллы соединения формулы (1):
в которых изолированные кристаллы соединения формулы (1) находятся в полиморфной форме А и, по существу, свободны от других полиморфов. В предпочтительном воплощении изолированные кристаллы соединения формулы (1) находятся в виде орторомбических кристаллов. Изолированные кристаллы соединения формулы (1) по настоящему изобретению предпочтительно дают, по существу, такой же профиль PXRD, как представлено для полиморфной формы А на фиг.6. Более предпочтительно изолированные кристаллы соединения формулы (1), по меньшей мере, на 90% представлены полиморфной формой А при определении по высоте пика PXRD в районе 9° 2-тета. Изобретение также предусматривает композиции с изолированными кристаллами соединения формулы (1), которые содержат, по меньшей мере, 90% полиморфной формы А по отношению к другим полиморфным формам.
Краткое описание чертежей
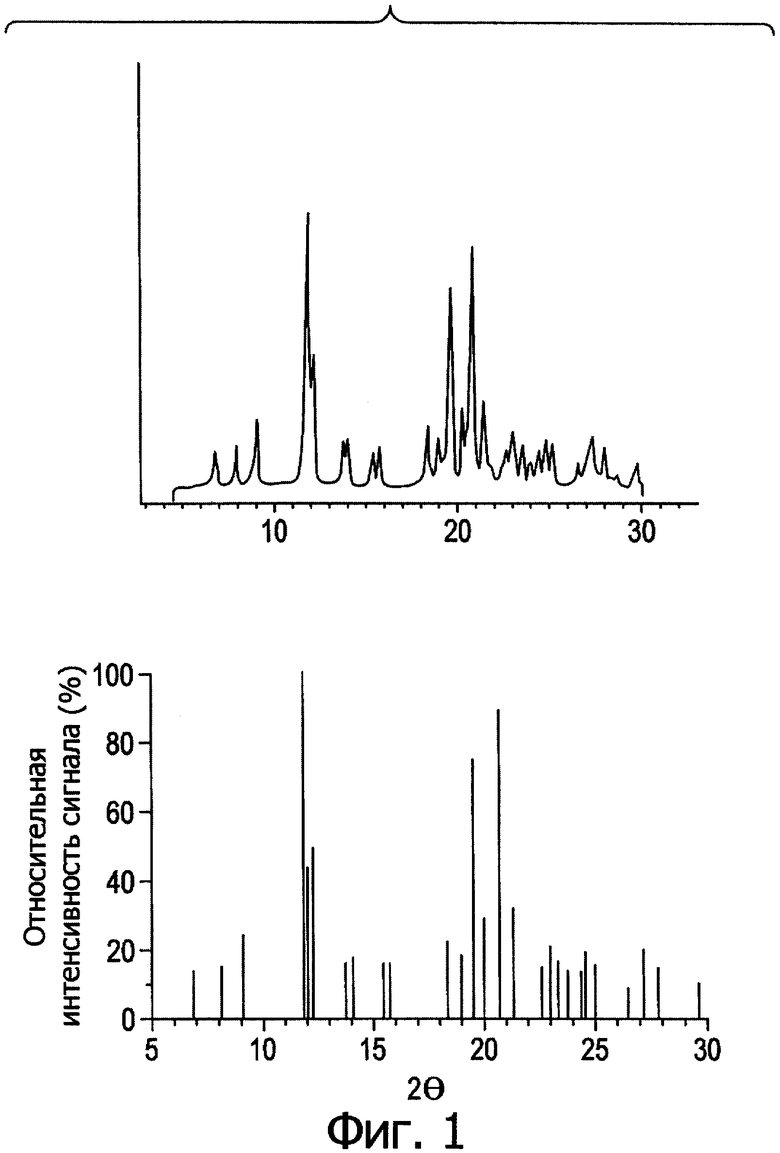
Фиг.1. Профиль дифракции рентгеновских лучей в порошке DSC для формы I.
Фиг.1а. Кривая дифференциальной сканирующей калориметрии (DSC) для формы I. Скорость повышения температуры: 10 град./мин, количество образца: 5,58 мг.
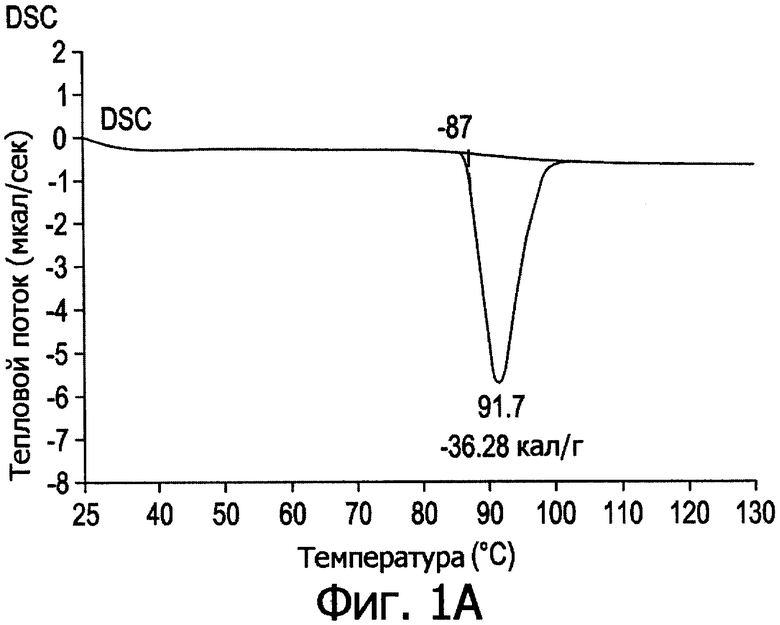
Фиг.2. Профиль дифракции рентгеновских лучей в порошке и кривая DSC для формы II.
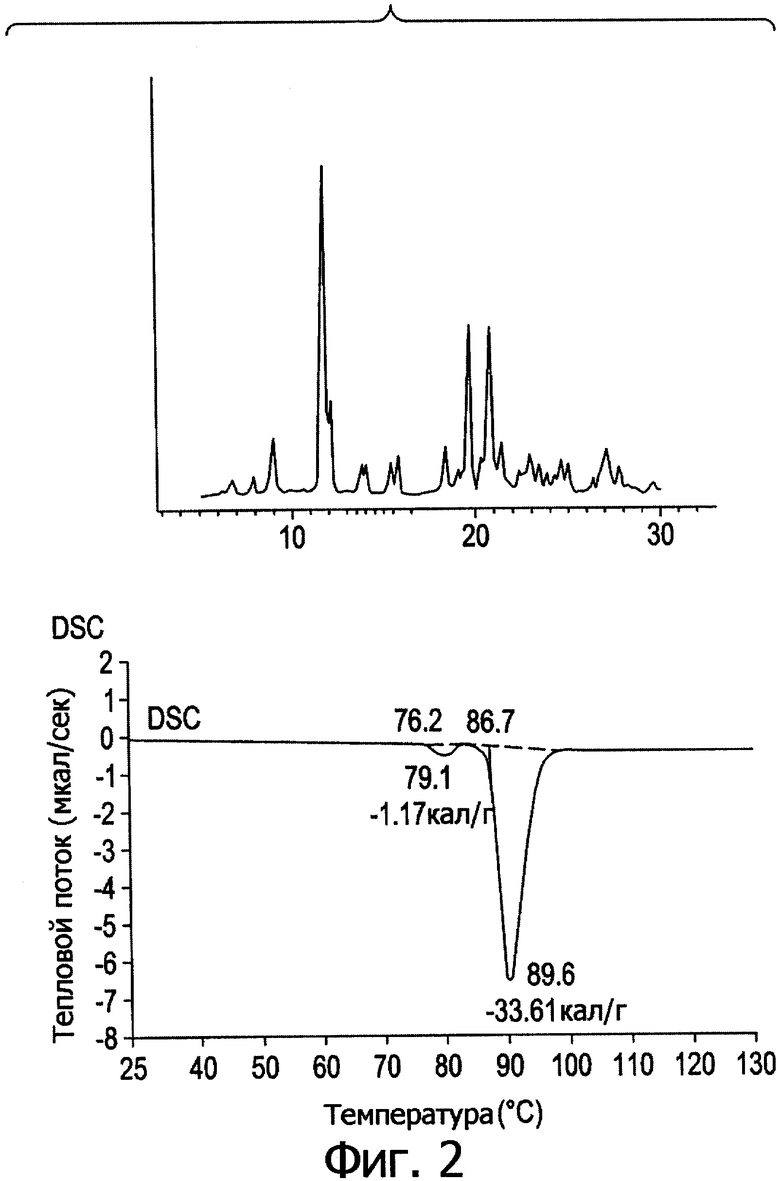
Фиг.3. Профиль дифракции рентгеновских лучей в порошке и кривая DSC для формы III.
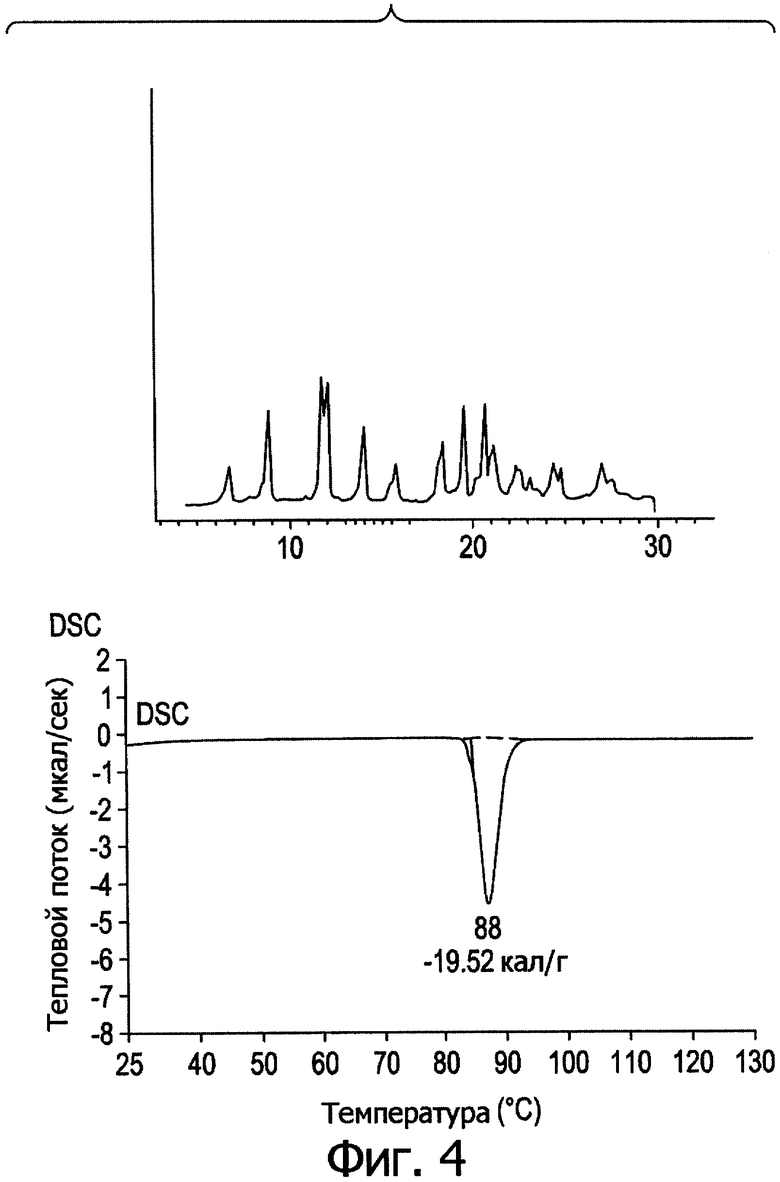
Фиг.4. Профиль дифракции рентгеновских лучей в порошке и кривая DSC для формы IV.
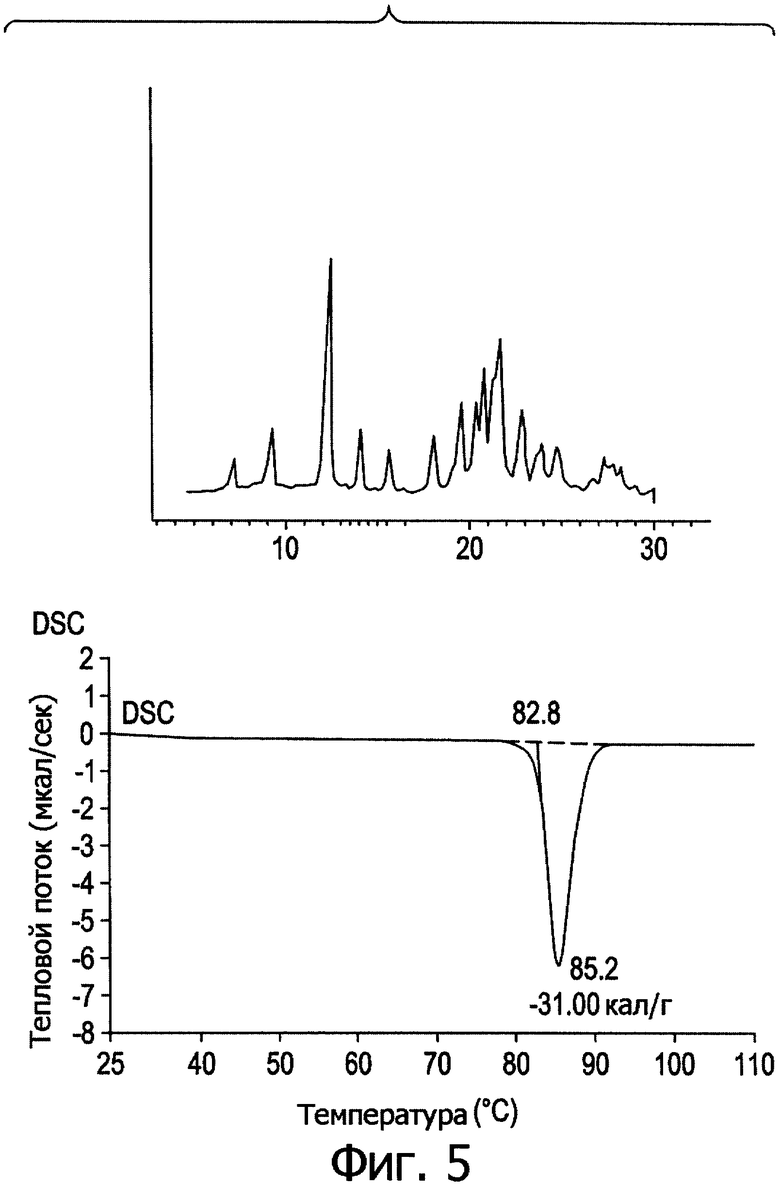
Фиг.5. Профиль дифракции рентгеновских лучей в порошке и кривая DSC для формы V.
Фиг.6. Профили дифракции рентгеновских лучей для трех полиморфных форм.
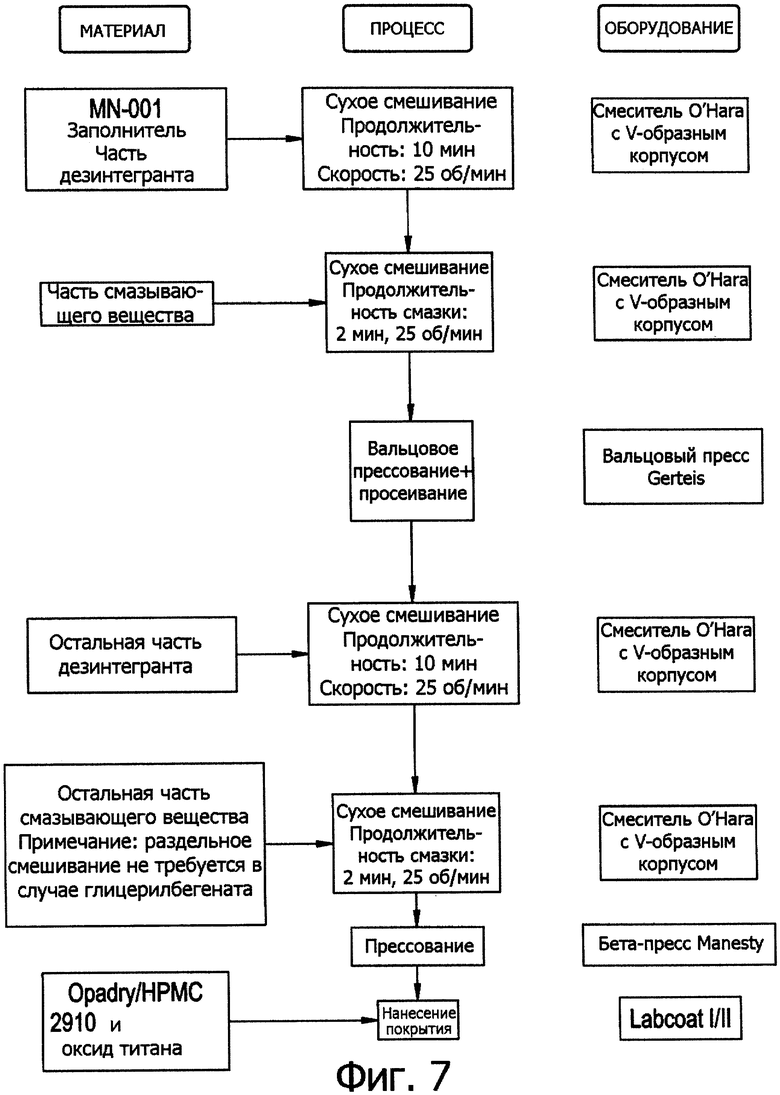
Фиг.7. Схема процесса сухой грануляции.
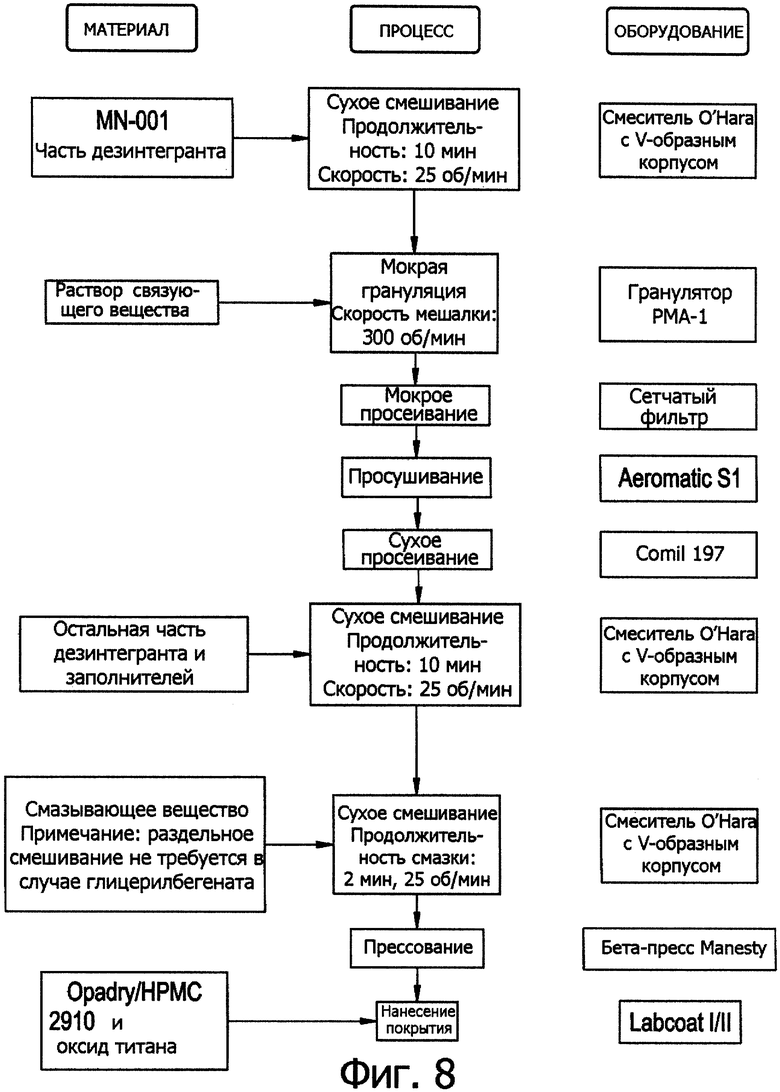
Фиг.8. Схема процесса мокрой грануляции.
Осуществление изобретения
Сложный эфир (4) можно синтезировать при реакции фенола формулы (2):
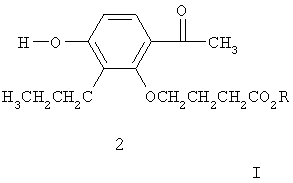
где R означает защитную группу для кислоты типа метила или этила, с соединением брома формулы (3):
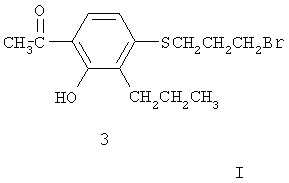
в органическом растворителе, к примеру, в ацетоне, метилэтилкетоне, диэтилкетоне или диметилформамиде. Реакцию можно проводить при температуре от ниже комнатной до температуры кипения растворителя в присутствии неорганического основания, например, карбоната калия или карбоната натрия. Также рекомендуется добавление иодида калия. Можно использовать аналоги соединения (3) с альтернативными уходящими группами, такими как хлор и тозилат, для осуществления реакции конденсации.
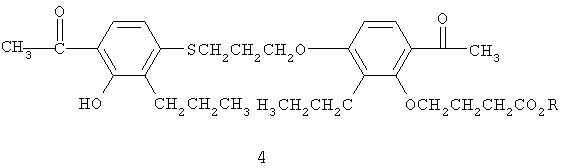
После удаления защитной группы для кислоты путем щелочного гидролиза сложного эфира и экстракционной обработки получают соединение (1) в виде белого твердого вещества.
Перекристаллизацию белого твердого вещества с получением практически чистых кристаллов формы А (фиг.6) (например, 90% и более, предпочтительно не менее 95%) можно осуществить при растворении соединения (1) в 5-10 весовых частях этанола при 25-40°С, получая желтый или оранжевый раствор. В этанольный раствор вносят 1-10 частей воды и перемешивают при 20-25°С в течение 15-60 минут, а затем еще при 5-10°С в течение 1-4 часов, предпочтительно 2,0-3,0 часов, при этом образуется беловатая суспензия.
В эту суспензию добавляют 5-15 частей воды и перемешивают смесь при 5-10°С в течение следующих 1-4 часов, предпочтительно 1,5-2,0 часов. Твердый белый или беловатый продукт выделяют фильтрованием под вакуумом, при этом осадок на фильтре промывают водой и сушат под вакуумом при 25-40°С в течение 12-24 часов.
При других условиях перекристаллизации также можно получить форму А, к примеру, растворяя соединение (1) в низшем спирте (изопропаноле) и охлаждая раствор с образованием кристаллов.
Терапевтические составы
Фармацевтические композиции, содержащие орторомбическую форму соединения (1), могут быть составлены для перорального применения с инертными наполнителями типа крахмала как вяжущего наполнителя, одного или в сочетании с микрокристаллической целлюлозой и подходящим смазывающим веществом. К другим подходящим наполнителям относятся поливинилпирролидон, желатин, гидроксицеллюлоза, гуммиарабик, полиэтиленгликоль, маннит, хлорид натрия, цитрат натрия и любые другие наполнители, известные специалистам в области фармацевтических композиций.
Наполнители в таблетках обычно классифицируют в соответствии с их функцией, как-то: разбавители (также называемые заполнителями объема и просто заполнителями), связующие вещества, удерживающие все ингредиенты вместе в готовой таблетке, дезинтегранты, способствующие распадению таблетки при попадании в жидкую среду с высвобождением активного ингредиента, и смазывающие вещества, облегчающие отделение готовых таблеток из пресс-формы. Кроме того, таблетки могут содержать и другие вещества, предназначенные для улучшения процесса таблетирования, такие как добавки, повышающие текучесть, ароматизаторы, подслащивающие вещества и антиоксиданты.
Таблетирование и некоторые операции заполнения капсул основываются на способности определенных порошков к цементированию при сжатии (прессовании). Прессованные таблетки могут быть получены путем мокрой грануляции, сухой грануляции или прямого прессования. Процесс мокрой грануляции включает смешивание компонентов в виде порошка, приготовление гранулирующего вяжущего раствора связующего вещества, тщательное перемешивание компонентов вместе с гранулирующим раствором связующего вещества до образования теста, пропускание этой массы через сито, высушивание, измельчение, добавление смазывающего вещества и прессование таблеток из полученной смеси.
Предпочтительным способом получения таблеток является мокрая грануляция, содержащая полиморфную форму А соединения формулы (1), стандартную лактозу, микрокристаллическую целлюлозу 101, кросскармелозу, стеарат магния и очищенную воду, покрытая слоем белого Opadry II. Таблетки должны весить от 100 до 1000 мг, предпочтительно от 250 до 500 мг.
Сухая грануляция включает стадии смешивания компонентов в виде порошка, прессование смеси в твердые куски, размельчение кусков до требуемого размера частиц, просеивание, добавление других компонентов при необходимости и прессование таблеток из этой смеси. Наиболее экономичный способ таблетирования - прямое прессование, оно требует всего двух стадий: смешивания сухих компонентов и прессования таблеток из смеси.
К подходящим связующим веществам для прямого прессования относятся микрокристаллическая целлюлоза, поддающиеся прессованию сахара, определенные соли кальция, лактоза и декстроза. Из них предпочтительна микрокристаллическая целлюлоза. Этот наполнитель также проявляет хорошие показатели распадаемости. К другим хорошим связующим относятся фосфаты кальция и поддающиеся прессованию сахара. Связующие из солей кальция обычно требуют применения дезинтегрантов. Маннит и сорбит обладают определенными преимуществами в смысле вкуса, но они не имеют связывающей способности и требуют дезинтегранта.
Как правило, таблетки проявляют твердость свыше 2 тыс. фунтов (kp)/см2, более предпочтительно твердость свыше 5, наиболее предпочтительно от 10 до 20 kp/см, и время распадения менее 30 минут, более предпочтительно менее 15 минут при измерении стандартным тестом в соответствии с фармакопеей США (USP) на распадение в воде.
Полиморфная форма А соединения формулы (1) также может быть составлена в виде капсул. К твердым носителям относятся крахмал, лактоза, сульфат кальция дигидрат, teffa alba, стеарат магния или стеариновая кислота, тальк, пектин, гуммиарабик, агар или желатин. Носитель может также включать материал для продолжительного выделения типа моностеарата глицерина или дистеарата глицерина, один или вместе с воском. Содержание твердого носителя варьирует, но предпочтительно оно составляет от 20 мг до 1 г на единичную дозу.
Инкапсулирование может осуществляться любым удобным способом, как правило, с применением полимерного покрытия, используемого для микроинкапсуляции, энтеро-солюбильного покрытия, многослойного покрытия и т.п. Полимерное покрытие может быть устойчивым к распадению при контакте со слюной, но немедленно высвобождает соединение при контакте с желудочным соком в желудке с тем, чтобы контролировать вкус композиции. С другой стороны, полимерное покрытие может быть устойчивым к быстрому распаду в присутствии желудочного сока. К подходящим полимерам для покрытия относятся биодеградируемые полимеры, такие как полимолочная кислота, полигликолевая кислота, сополимеры молочной и гликолевой кислоты, их полиортоэфиры и полиангидриды. Соединение также можно заключить в капсулы нанесением покрытия из полимера типа полисахарида (например, метил- или этилцеллюлозы) либо заключением внутри липосомной системы доставки. Подходящие методы получения композиций, содержащих микроинкапсулированные активные ингредиенты, описаны, к примеру, в US Patents 4462982, 4710384, 5178878 и 5709886. Предпочтительно микроинкапсулированные соединения имеют средний размер частиц от около 50 микрон до около 120 микрон (например, от около 70 микрон до около 100 микрон).
Типичные дозы соединения (1) в таблетках и капсулах составляют от 1,0 до 100 мг/кг. Промежутки между введением зависят от возраста, веса и общего состояния пациента. В общем случае препарат вводится от одного до четырех раз в день.
Примеры
Обычно таблетки формируют с использованием такого носителя, как модифицированный крахмал, один или в сочетании с 10 вес.% карбоксиметилцеллюлозы (Avicel). В процессе таблетирования составы подвергают прессованию при давлении от 1000 до 3000 фунтов. Таблетки предпочтительно проявляют среднюю твердость от 1,5 до 8,0 kp/см2, предпочтительно от 5,0 до 7,5 kp/см2. Время распадения колеблется от 30 секунд до около 15 или 20 минут. В следующих примерах изложены конкретные воплощения изобретения, но их не следует трактовать как ограничивающие его область действия.
Пример 1
Синтез этил-4-[6-ацетил-3-[3-(4-ацетил-3-гидрокси-2-пропилфенилтио)-пропокси]-2-пропилфенокси]бутирата
При перемешивании в смесь из этилового эфира 4-(6-ацетил-3-гидрокси-2-пропилфенокси) масляной кислоты (1,6 г), иодида калия (0,5 г) и карбоната калия (1,45 г) в ацетоне (30 мл) по каплям добавляли раствор 4-(3-бромпропилтио)-2-гидрокси-3-пропилфенилэтанона (1,9 г) в ацетоне (10 мл) при кипячении с обратным холодильником. После кипячения с обратным холодильником в течение 6 часов смесь охлаждали до комнатной температуры, и неорганические материалы отделяли фильтрованием. Фильтрат концентрировали, а осадок отделяли и очищали колоночной хроматографией на силикагеле (элюировали смесью бензол:этилацетат=9:1), получая указанное в заголовке соединение в виде неочищенных кристаллов (2,1 г, 72,4%), которые перекристаллизовывали из этанола, получая бесцветные кристаллы с т.пл. 65-66°С.
Пример 2
Синтез 4-[6-ацетил-3-[3-(4-ацетил-3-гидрокси-2-пропилфенилтио)пропокси]-2-пропилфенокси] масляной кислоты
В смесь из этил-4-[6-ацетил-3-[3-(4-ацетил-3-гидрокси-2-пропилфенилтио)-пропокси]-2-пропилфенокси]бутирата (2,1 г) в этаноле (10 мл) добавляли раствор гидроксида натрия (0,26 г), растворенного в воде (10 мл). После нагревания на горячей водяной бане в течение 5 минут смесь охлаждали добавлением ледяной воды и подкисляли добавлением соляной кислоты, а затем экстрагировали этилацетатом. Органический слой промывали водой, сушили над сульфатом натрия и концентрировали. Образовавшийся осадок отделяли и очищали колоночной хроматографией на силикагеле (элюировали смесью этанол:метиленхлорид = 3:100), получая указанное в заголовке соединение (1,3 г, 65,2%) в виде бесцветных кристаллов с т. пл. 79-81°С.
Пример 3
Кристаллический полиморфизм
После перекристаллизации в индивидуальных растворителях соединение (1) подвергали дифрактометрии рентгеновских лучей в порошке, термическому анализу и определяли растворимость в эфире; таким образом проводили предварительную оценку кристаллического полиморфизма. Результаты свидетельствуют, что соединение (1) находится в 5 различных кристаллических полиморфных модификациях.
На фиг.1-5 представлены профили дифракции рентгеновских лучей в порошке и дифференциальной сканирующей калориметрии (DSC) в метастабильных типах кристаллов I-V. В табл.1 представлены способы получения типов I-V и их растворимость в эфире.
Получение кристаллических полиморфов и их растворимость в эфире
Из табл.1 видно, что температура кристаллизации имеет решающее значение при получении различных кристаллических полиморфов. При получении ингредиента в большом масштабе кристаллизация происходит в больших размерах, и при отсутствии контроля за температурой может образоваться смесь из стабильных и метастабильных кристаллов, что приведет к большому разбросу в физико-химических свойствах и биодоступности между различными партиями продукции, против чего следует принимать меры.
Пример 4
Методика кристаллизации в большом масштабе для получения орторомбического полиморфа - кристаллов типа V (формы А)
Растворяли 34 г беловатого твердого соединения (1) в 204 мл (6 частей относительно массы сухого осадка на фильтре) этанола (40°С), получая желтый или оранжевый раствор. При умеренном перемешивании в этанольный раствор вносили 43 мл (1,3 часть) воды. Реакционную смесь охлаждали до 20-25°С и помешивали при 20-25°С примерно 15 минут, а затем еще при 10-15°С в течение 1-2 часов, так что она приобретала вид беловатой суспензии.
Затем в полученную суспензию вносили 364 мл (10,7 частей) воды и помешивали смесь при 5-10°С в течение 1-2 часов. Твердый белый или беловатый продукт выделяли фильтрованием под вакуумом. Осадок на фильтре промывали 2×30 мл воды. Беловатое твердое вещество сушили под вакуумом при 35-40°С в течение 24 часов.
Пример 5
Данные по растворимости соединения (1) в смеси этанол/вода (2:1)
Образцы соединения (1) (5 г) суспендировали в смеси этанол/вода (2:1, 100 мл) и помешивали в течение 1 часа при температурах 22, 30 и 40°С соответственно. Суспензии фильтровали и твердые вещества высушивали под вакуумом при комнатной температуре в течение ночи, получая нерастворимый материал. Растворимости рассчитывали методом вычитания, исходя из полученного материала.
Пример 6
В общем, таблетки методом мокрой грануляции получали с использованием вяжущего раствора, состоящего из водного раствора гидроксипропилцеллюлозы. Гранулирование осуществляли в грануляторе с большим срезывающим усилием, а полученную влажную массу сушили в псевдоожиженном слое, измельчали, перемешивали с внешними наполнителями, улучшающими распадаемость, текучесть и прессуемость, после чего подвергали таблетированию на таблетировочном прессе. На эти ядра таблеток наносили слой покрытия для придания стандартного вида и улучшения комплаентности (т.е. легкости проглатывания). К наполнителям относились, не ограничиваясь этим, кросскармелоза натриевая, стеарат магния, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, лактоза, глицерилбегенат, поливинилпирролидон, маннит, диоксид титана и микрокристаллическая целлюлоза.
Пример 7
В общем, состав для сухой грануляции получали путем сухого смешивания (в бункерном смесителе или миксере с большим срезывающим усилием) порции из порошков связующего вещества, дезинтегранта и смазывающего вещества. Из этой сухой смеси порошков формировали гранулы при помощи вальцового пресса, снабженного возвратно-поступательным гранулятором. Размеры отверстий сита, ширину зазора, усилие, скорость вальцов и скорость гранулятора устанавливали таким образом, чтобы оптимизировать физические параметры гранулирования, известные специалистам в области фармацевтической технологии. К наполнителям относились, не ограничиваясь этим, кросскармелоза натриевая, стеарат магния, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, лактоза, глицерилбегенат, поливинилпирролидон, маннит, диоксид титана и микрокристаллическая целлюлоза.
Пример 8
Конкретные составы для сухой грануляции
Предлагаемые прототипы исходного состава композиций для сухой грануляции
Процесс сухой грануляции изложен на схеме на фиг.7.
Пример 9
Конкретные составы для мокрой грануляции
Предлагаемые прототипы исходного состава композиций для мокрой грануляции
Процесс мокрой грануляции изложен на схеме на фиг.8.
Выше были подробно описаны предпочтительные воплощения изобретения. Специалистам в этой области будет нетрудно установить разнообразные модификации и улучшения его. Предыдущие примеры следует рассматривать как неограничивающие и типичные для изобретения, описанного в вышеприведенном описании и заявленного в нижеприведенной формуле изобретения.
Пример 10
Анализ методом PXRD
Образцы готовили стандартным методом фронтального уплотнения и прогоняли на дифрактометре D5000 Diffractometer System фирмы Siemens. Использовали источник CuKα с высокой разрешающей способностью, работающий при 50 кВ/35 мА. Вторичный луч подвергали монохроматизации при помощи твердотельного детектора Kevex. Для сбора данных использовали режим пошагового сканирования в диапазоне 2,5-35° (2-тета). Полученные данные обрабатывали с помощью программы Diffrac Plus™.
На фиг.6 представлены отдельные части профилей дифракции трех различных полиморфов, определенных как форма А (вероятно, это орторомбическая структура, обозначаемая как тип V), а также форма В (I) и форма С (II) (обе представляют собой моноклинные решетки).
Видно, что верхний профиль сильно отличается от двух остальных. Различия отмечены стрелками над верхней кривой. Большинство одиночных пиков на верхнем профиле стали двойными на двух остальных. Это явно свидетельствует о структурном переходе с понижением общей симметрии. Для того чтобы найти критерии для лучшего различения этих полиморфов, была предпринята попытка индексировать неизвестные решетки. Результаты свидетельствуют об орторомбической решетке (верхняя кривая, форма А) и моноклинной решетке (средняя кривая, форма В). Нижняя кривая (форма С) также имеет моноклинную решетку, аналогично форме В, но при отсутствии некоторых рефлексов (отмечены стрелками), что могло произойти вследствие каких-то структурных отличий.
Структура формы А очень близка форме V в табл.1 и на фиг.5, хотя и есть некоторые отличия в диапазоне 19-25° 2-тета. С другой стороны, профили дифракции полиморфных форм I и II хорошо совпадают с формами В и С, поскольку все они, как видно, проявляют расщепление основных рефлексов вследствие понижения общей симметрии из орторомбической до моноклинной.
Поскольку кристаллографические характеристики всех пяти полиморфных форм, описанных в табл.1, трудны для воспроизведения, мы будем характеризировать структурное состояние соединения (1) в фармацевтических образцах только по его появлению в виде формы А в соответствии с методом PXRD.
Настоящее изобретение относится к изолированной орторомбической кристаллической форме 4-[6-ацетил-3-[3-(4-ацетил-3-гидрокси-2-пропилфенилтио)пропокси]-2-пропил-фенокси]масляной кислоты, обеспечивающей превосходную растворимость, которая (i) по существу свободна от моноклинных кристаллических форм, о чем свидетельствует анализ дифракции рентгеновских лучей на порошке (XRPD), показывающий отсутствие дублетных пиков между 11,5 и 16 (по шкале 2-Тета), и (ii) проявляет по меньшей мере в два раза большую растворимость при 30°С в водном этаноле, чем моноклинная кристаллическая форма. Соединение обладает пероральной активностью при лечении астмы и аллергических заболеваний, описана также фармацевтическая композиция на основе указанного выше соединения. 2 н. и 4 з.п. ф-лы, 8 ил., 1 табл.
| US 4985585 A, 15.01.1991. |

