Область техники
Настоящее изобретение относится к способам лечения повреждения суставов у индивидов, страдающих этим нарушением.
Предпосылки создания изобретения
Воспаление суставов является широко известным клиническим проявлением при различных аутоиммунных нарушениях, в том числе ревматоидном артрите (RA), псориатическом артрите (PsA), системной красной волчанке (SLE), синдроме Шегрена и полимиозите. У большинства больных при физическом обследовании обнаруживаются деформации суставов, но обычно только у больных RA и PsA обнаруживаются эрозии кости при обследовании с получением изображений.
RA представляет собой хроническое воспалительное заболевание, которое поражает приблизительно 0,5-1% взрослой популяции северной Европы и Северной Америки и немного меньшую часть в других частях света (Alamonosa and Drosos, Autoimmun. Rev., 4: 130-136 (2005)). Он является системным воспалительным заболеванием, характеризующимся хроническим воспалением синовиальной оболочки пораженных суставов, которое, в конечном счете, приводит к утрате ежедневной функции вследствие хронических боли и усталости. У большинства больных также имеет место прогрессирующее разрушение хряща и кости в пораженных суставах, которое может, в конечном счете, привести к необратимой нетрудоспособности. Долгосрочный прогноз RA является плохим, при этом у приблизительно 50% больных развивается значительная функциональная неполноценность в пределах 10 лет от момента диагноза (Keystone, Rheumatology, 44 (Suppl. 2): ii8-ii12 (2005)). Вероятная продолжительность жизни уменьшается в среднем на 3-10 лет (Alamanos and Rosos, выше). Больные с высоким титром ревматоидного фактора (RF) (приблизительно 80% больных) имеют более агрессивное заболевание (Bukhari et al., Arthritis Rheum. 46: 906-912 (2002)), с худшим долгосрочным прогнозом и повышенной смертностью по сравнению с больными, отрицательными по RF (Heliovaara et al., Ann. Rheum. Dis. 54: 811-814 (1995)).
Патогенез хронических воспалительных заболеваний кости, таких как RA, не полностью понятен. Такие заболевания сопровождаются утратой кости вокруг поврежденных суставов вследствие повышенной резорбции остеокластов. Этот процесс опосредуется, по большей части, увеличенной местной продукцией провоспалительных цитокинов (Teitelbaum Science, 289: 1504-1508 (2000); Goldring and Gravallese Arthritis Res. 2(1): 33-37 (2000)). Эти цитокины могут действовать непосредственно на клеточные линии остеокластов или опосредованно, влияя на продукцию остеобластными/стромальными клетками важного фактора дифференциации остеокластов, рецепторного активатора лиганда NF<B (RANKL), I и/или его растворимого ложного рецептора, остеопротегерина (OPG), (Hossbauer et al. J. Bone Miner. Res. 15(1): 2-12 (2000)). TNF-альфа является главным медиатором воспаления, значение которого в патогенезе различных форм утраты кости поддерживается несколькими линиями экспериментальных и клинических доказательств (Feldmann et al. Cell 85(3): 307-310 (1996). Однако TNF-альфа не является существенным для остеокластогенеза (Douni et al. J. Inflamm. 47: 27-38 (1996)), эрозийного артрита (Campbell et al. J. Clin. Invest. 107(12): 1519-1527 (2001)) или остеолизиса (Childs et al. J. Bon. Min. Res. 16: 338-347 (2001)), поскольку они могут наблюдаться в отсутствие TNF-альфа.
В частности, при RA иммунный ответ, как полагают, инициируется/поддерживается одним или несколькими антигенами, присутствующими в синовиальном компартменте, что приводит к притоку клеток острого воспаления и лимфоцитов в сустав. Последующие атакующие цепи воспаления приводят к образованию инвазивной и эрозивной ткани, называемой паннусом. Он содержит пролиферирующие подобные фибробластам синовиоциты и макрофаги, продуцирующие провоспалительные цитокины, такие как фактор-альфа некроза опухолей (TNF-альфа) и интерлейкин-1 (IL-1). Большой вклад в повреждение ткани вносят местное высвобождение протеолитических ферментов, различные медиаторы воспаления и активация остеокластов. Имеет место утрата суставного хряща и образование эрозий кости. Воспалительный процесс может затронуть окружающие сухожилия и синовиальную сумку. В конечном счете, нарушается целостность структуры сустава, что приводит к нетрудоспособности.
Точное влияние В-клеток в иммунопатогенезе RA не полностью охарактеризовано. Однако существует несколько возможных механизмов, с помощью которых В-клетки могут принимать участие в патологическом процессе (Silverman and Carson, Arthritis Res. Ther., 5 Suppl. 4: S1-6 (2003)).
Исторически считают, что В-клетки вносят вклад в патологический процесс при RA, являясь, главным образом, предшественниками продуцирующих аутоантитела клеток. Был идентифицирован ряд специфичностей аутоантител, в том числе аутоантитела к коллагену типа II и протеогликанам, а также к ревматоидным факторам. Образование больших количеств антител приводит к образованию иммунных комплексов и активации системы комплемента. Это в свою очередь усиливает иммунный ответ и может привести к местныму лизису клеток. Повышенный синтез RF и истощение комплемента коррелировало с активностью заболевания. Присутствие самого RF связано с более тяжелой формой RА и наличием дополнительных признаков, кроме суставных.
Недавно полученные данные (Janeway and Katz, J. Immunol., 138: 1051 (1998); Rivera et al., Int. Immunol., 13: 1583-1593 (2001)) указывают на то, что В-клетки являются в высокой степени эффективными антигенпрезентирующими клетками (АРС). В-клетки RF-положительных больных могут быть особенно сильными АРС, поскольку их поверхностный иммуноглобулин делает возможным легкий захват любых иммунных комплексов, не обращая внимание на антигены, присутствующие внутри них. Многие антигены могут, таким образом, процессироваться для презентации Т-клеткам. Кроме того, недавно было сделано предположение, что это может также сделать возможной самоподдержку В-клеток RF-положительных больных (Edwards et al., Immunology, 97: 188-196 (1999)).
Для активации Т-клеток в клетку должны быть доставлены два сигнала; один - через Т-клеточный рецептор (TCR), который распознает процессированный пептид в присутствии антигена главного комплекса гистосовместимости (MHC), а второй - через костимулирующие молекулы. При активации В-клетки экспрессируют костимулирующие молекулы на своей поверхности и могут, таким образом, обеспечить второй сигнал активации Т-клеток и образования эффекторных клеток.
В-клетки могут усиливать свою собственную функцию, а также функцию других клеток благодаря продукции цитокинов (Harris et al., Nat. Immunol., 1: 475-482 (2000)). Среди некоторых цитокинов, которые могут продуцировать В-клетки в синовиальной оболочке при RA, находятся TNF-альфа и IL-1, лимфотоксин-альфа, IL-6 и IL-10.
Хотя считают, что активация Т-клеток является решающим компонентом патогенеза RA, в недавней работе, в которой использовались эксплантаты синовиальной оболочки человека для мышей с тяжелыми комбинированными иммунодефицитными нарушениями (SCID), было показано, что активация Т-клеток и ретенция в суставе критическим образом зависят от присутствия В-клеток (Takemura et al., J. Immunol., 167: 4710-4718 (2001). Точная роль в этом В-клеток не ясна, поскольку другие АРС, по-видимому, не имели такого же эффекта на Т-клетки.
Повреждение структуры суставов является важным последствием хронического синовиального воспаления. У 60-95% больных ревматоидным артритом (RA) обнаруживается по крайней мере одна рентгенографическая эрозия в пределах 3-8 лет от начала заболевания (Paulus et al., J. Rheumatol., 23: 801-805 (1996); Hulsmans et al., Arthritis Rheum. 43: 1927-1940 (2000)). При RA на ранней стадии корреляция между рентгенографическими показателями повреждения и функциональной способностью является слабой, но после 8 лет болезни коэффициенты корреляции могут достигать настолько больших значений, как 0,68 (Scott et al., Rheumatology, 39: 122-132 (2000)). У 1007 больных, моложе 60 лет, имеющих RA на протяжении по крайней мере четырех лет, Wolfe и др. (Arthritis Rheum, 43 Suppl. 9: S403 (2000)) обнаружили значительную связь между скоростью прогрессирования рентгенографического показателя повреждения Larsen (Larsen et al., Acta Radiol. Diagn. 18: 481-491 (1977)), увеличением степени нетрудоспособности, оплачиваемой из средств социального обеспечения, и уменьшением дохода семьи.
Профилактика или замедление рентгенографического повреждения является одной из целей лечения RA (Edmonds et al., Arthritis Rheum. 36: 336-340 (1993)). При контролируемом клиническом испытании на протяжении 6 или 12 месяцев документировали, что прогрессирование рентгенографических показателей повреждения было более быстрым в группе плацебо, чем в группе, которая получала метотрексат (МТХ) (Sharp et al., Arthritis Rheum. 43: 495-505 (2000), лефлуномид (Sharp et al., выше), сульфасалазин (SSZ) (Sharp et al., выше), преднизолон (Kirwan et al., N. Engl. J. Med., 333: 142-146 (1995); Wassenburg et al., Arthritis Rheum, 42: Suppl 9: S243 (1999)), антагонист рецептора интерлейкина-1 (Bresnihan et al., Arthritis Rheum, 41: 2196-2204 (1998)) или комбинацию инфликсимаб/МТХ (Lipsky et al., N. Eng. J. Med., 343: 1594-1604 (2000)), и что рентгенографическое прогрессирование после лечения этанерцептом было менее быстрым, чем после лечения МТХ (Bathon et al., N. Engl. J. Med., 343: 1586-1593 (2000)). В других исследованиях оценивалось рентгенографическое прогрессирование у больных, которые получали лечение кортикостероидами (Joint Committee of the Medical Research Council and Nuffield Foundation, Ann. Rheum. Dis. 19: 331-337 (1960); Van Everdingen et al., Ann. Intern. Med., 136: 1-12 (2002)), циклоспорином А (Pasero et al., J. Rheumatol., 24: 2113-2118 (1997); Forre, Arthritis Rheum., 37: 1506-1512 (1994)), MTX в сравнении с азатиоприном (Jeurissen et al., Ann. Intern. Med., 114: 999-1004 (1991)), MTX в сравнении с ауранофином (Weinblatt et al., Arthritis Rheum., 36: 613-619 (1993)), МТХ (мета-анализ) (Alarcon et al., J. Rheumatol., 19: 1868-1873 (1992)), гидроксихлорохином (HCQ) в сравнении с SSZ (Van der Heijde et al., Lancet, 1: 1036-10380), SSZ (Hannonen et al., Arthritis Rheum. 36: 1501-1509 (1993)), комбинацией COBRA (Combinatietherapei Bij Reumatoide Artritis) преднизолона, MTХ и SSL (Boers et al., Lancet, 350: 309-318 (1997); Landewe et al., Arthritis Rhreum., 46: 347-356 (2002)), комбинацией MTX, SSZ и HCQ (O'Dell et al., N. Engl. J. Med., 334: 1287-1291 (1996); Mottonen et al., Lancet, 353: 1568-1573 (1999)); комбинацией циклофосфамида, азатиоприна и HCQ (Csuka et al., JAMA, 255: 2115-2119 (1986)) и комбинацией адалимумаба с MTX (Keystone et al., Arthritis Rheum., 46 Suppl. 9: S205 (2002)).
Управление по контролю за продуктами и медикаментами (FDA) недавно разрешило утверждение на этикетке, что некоторые лекарственные средства, например лефлуномид, этанерцепт и инфликсимаб, замедляют прогрессирование рентгенографического повреждения сустава. Эти утверждения основаны на статистически значимых различиях в скоростях прогрессирования, наблюдаемых между случайно заданными группами, получающими лечение, и контрольными группами. Однако скорость прогрессирования у индивидуумов в пределах групп, получающих лечение, и контрольных групп в значительной степени перекрываются; следовательно, несмотря на значительную разницу между группами, получающими лечение, эти данные нельзя использовать для оценки вероятности того, что у больного, на начальных этапах лечения, будет наблюдаться благоприятный исход в отношении прогрессирования рентгенографического повреждения. Были предложены различные способы определения категории спаренных рентгенограмм индивидуальных больных как не прогрессирующей, например, показатели повреждения, составляющие 0, в обоих моментах времени, нет увеличения показателей повреждения, нет новых суставов с эрозиями, и изменение показателя, не превышающее наименьшего определяемого различия (т.е. 95% доверительный интервал для различия между повторными считываниями одной и той же рентгенограммы) (Lassere et al., J. Rheumatol., 26: 731-739 (1999)).
Определение того, увеличилось ли структурное повреждение у конкретного больного во время интервала между моментами времени получения спаренных рентгенограмм, которые приходятся на начало и конец 6- или 12-месячного клинического испытания, было трудным по нескольким причинам. Скорость рентгенографического повреждения не является постоянной внутри популяции больных RA; маленькая часть больных может иметь быстро прогрессирующее повреждение, но у многих больных может наблюдаться небольшое прогрессирование или его может не быть; особенно если интервал является относительно коротким. Способы определения показателей рентгенографического повреждения, например Sharp (Sharp et al., Arthritis Rheum., 14: 706-720 (1971); Sharp et al., Arthritis Rheum., 28: 1326-13335 (1985)), Larsen (Larsen et al., Acta Radial. Diagn., 18: 481-491 (1977)) и модификации этих способов (Van der Heijde, J. Rheumatol., 27: 261-263 (2000)) зависят от оценки и интерпретации специалистом, анализирующим рентгенограмму, того факта, является реальным кажущееся нарушение субхрящевой кортикальной пластины или является ли реальным уменьшение расстояния между поверхностями на противоположных сторонах сустава, или оно обусловлено незначительным изменением положения сустава относительно пленки и рентгеновского луча, изменением рентгеновской экспозиции или другим техническим фактором.
Следовательно, определенный показатель является приближением действительного повреждения и для многих индивидов наименьшее определяемое различие между повторными показателями одних и тех же рентгенограмм является больше действительного изменения, которое может иметь место во время интервала между моментами времени базовой линии и конечных рентгенограмм. Если специалист не различает временного порядка следования пленок, неизбежные ошибки в показателях могут быть в одном из двух направлений, приводя к кажущемуся «излечиванию» при снижении показателей или кажущемуся быстрому прогрессированию, если ошибка специалиста увеличивает различие между пленками. Когда в исследование вовлечена достаточно большая популяция больных, которым случайным образом назначено получение эффективного лечения, по сравнению с плацебо, погрешности с положительным и отрицательным знаком специалиста, проводящего анализ, компенсируют друг друга и можно определить небольшое, но реальное различие между подвергаемыми лечению группами.
Погрешность клинических показателей, используемых для количественной оценки активности заболевания RA, вызывала сходные трудности; статистически значимые различия между определенными результативными показателями из клинических испытаний не подходили для оценки вероятности улучшения индивидуума, на ранних этапах лечения (Paulus et al., Arthritis Rheum., 33: 477-484 (1990)). Атрибуция индивидуального улучшения стала возможной на практике с созданием American College of Rheumatology (ACR) составных критериев 20% улучшения (ACR20), с помощью которого пациента относили в категорию пациентов с улучшениями, если существовало 20% снижении числа болезненных и опухших суставов, и 20% улучшение в по крайней мере 3 из 5 дополнительных показателях (боль, физическая функция, общая оценка здоровья пациентом, общая оценка здоровья врачом и уровни реагентов острой фазы) (Felson et al., Arthritis Rheum., 38: 727-735 (1995)). Все эти показатели имеют большую ценность для наименьшего определяемого различия, зато в результате требования одновременного улучшения в 5 из 7 аспектов одного и того же процесса (активности заболевания) случайность 7 ошибок измерений ограничена и легко приписать индивидууму реальное улучшение.
При RA повреждение сустава является широко известным признаком. Рентгенологические параметры нарушения суставов рассматриваются в качестве ключевых результативных показателей при характеристике исхода заболевания. На недавнем заседании по согласованию OMERACT (результативных показателей при ревматологических клинических испытаниях) рентгенология была выбрана частью основного набора результативных показателей для продольных исследований по данным наблюдений (Wolfe et al., Arthritis Rheum., 41 Supp 9: S204 (1998) abstract). Рентгенология также является частью требуемого WHO/ILAR (World Health Organization/International League of Associations for Rheumatology) основного набора показателей для продолжительных клинических испытаний (Tugwell and Boers, J. Rheumatol., 20: 528-530 (1993)).
Доступные данные в отношении результата рентгенологического повреждения при RA были получены как при кратковременных, так и при продолжительных исследованиях. При кратковременных исследованиях больных RA с недавно возникшим заболеванием рентгенограммы, получаемые каждые 6 месяцев, показали, что после первоначального быстрого прогрессирования происходит снижения скорости прогрессирования рентгенологического повреждения рук и ног после 2-3 лет (Van der Heijde et al., Arthritis Rheum., 35: 26-34 (1992); Fex et al., Br. J. Rheumatol., 35: 1106-1055 (1996)). При длительных исследованиях с получением рентгенограмм менее часто была обнаружена постоянная скорость прогрессирования с неумолимым ухудшением повреждения вплоть до 25 лет продолжительности заболевания (Wolfe and Sharp, Arthritis Rheum., 41: 1571-1582 (1998); Gradual et al., Arthritis Rheum., 41: 1470-1480 (1998); plant et al., J. Rheumatol., 25: 417-426 (1998); Kaarela and Kautiainen, J. Rheumatol., 24: 1285-1287 (1997)). Обусловлены ли эти различия в рентгенографической картине прогрессирования различиями в способах определения показателей не ясно.
Используемые системы определения показателей отличаются по числу подвергаемых определению суставов, наличию независимых показателей для эрозий (ERO) и сужения суставного пространства (JSN), максимальному показателю на сустав и взвешиванию рентгенологической аномалии. До сих пор нет консенсуса в отношении предпочтительного способа определения показателей. В течение первых трех лет наблюдения при исследовании группы больных артритом на ранней стадии обнаружено, что JSN и ERO отличаются по их вкладу в измеряемое прогрессирование рентгенологического повреждения рук и ног (Van der Heijde et al., Arthritis Rheum., 35: 26-34 (1992)). Кроме того, обнаружено, что способы, в которых независимо определяются показатели для JSN и ERO, такие как показатели Sharp и Kellgren, являются более чувствительными к изменению при RA на ранней стадии, чем способы, в которых используются общие показатели, такие как показатель Larsen (Plant et al., J. Rheumatol., 21: 1808-1813 (1994); Cuchacovich et al., Arthritis Rheum., 35: 736-739 (1992). Способ определения показателя Sharp является очень трудоемким способом (Van der Heijde, Baillieres Clin. Rheumatol., 10: 435-533 (1996)). При RA на поздней или деструктивной стадии обнаружено, что способы Sharp и Larsen обеспечивают сходную информацию. Однако чувствительность к смене различных способов определения показателей при заболевании на поздней стадии еще не была исследована, и это может служить доводом в пользу того, что способы определения показателей, в которых независимо измеряются ERO и JSN, обеспечивают пригодную информацию (Pincus et al., J. Rheumatol., 24: 2106-2122 (1997)). Смотри также работу Drossaers-Bakker et al., Arthritis Rheum., 43: 1465-1472 (2000), в которой сравниваются три системы определения рентгенологических показателей для продолжительной оценки RA.
Paulus и др. (Arthritis Rheum., 50: 1083-1096 (2004)) определяли рентгенографическое повреждение суставов у индивидуумов с RA, участвующих в клинических испытаниях, как прогрессирующее или непрогрессирующее, и заключили, что повреждение устава при RA в наблюдаемой группе можно определить как прогрессирующее или непрогрессирующее с использованием составного определения, которое включает ряд неточных и родственных, но различных измерений структурного повреждения сустава. Представляется, что при повседневном наблюдении больных RA должно иметь место изменение интервала между парой рентгенограмм, составляющее по крайней мере пять единиц рентгенографического показателя повреждения по Sharp, прежде чем рассматривать структурное изменение реальным и использовать его в качестве основы для решения проводить лечение.
На протяжении последних 10 лет был достигнут основной прогресс в лечение RA. Комбинированное использование модифицирующих существующее заболевание противоревматических средств (DMARD) вместе с новыми биологическими агентами обеспечило более высокие уровни эффективности у большей доли больных, в то время как ранняя диагностика и лечение заболевания также улучшило исходы.
Этанерцепт является полностью человеческим слитым белком, ингибирующим фактор некроза опухолей (TNF) и последующий каскад воспалительных цитокинов. Продемонстрировано, что этанерцепт является безопасным и эффективным для быстрого снижения активности заболевания у взрослых с RA и для поддержки этого улучшения (Bathon et al., N. Eng. J. Med., 343: 1586-1593 (2000); Moreland et al., N. Engl. J. Med., 337: 141-147 (1997); Moreland et al., Ann. Intern. Med., 130: 478-486 (1999); Weinblatt et al., N. Engl. J. Med., 340: 253-259 (1999); Moreland et al., J. Rheumatol., 28: 1238-1244 (2001)). Он является одинаково эффективным для детей с ювенильным ревматоидным полиартритом (Lovell et al., N. Engl. J. Med., 342: 763-769 (2000)). Этанерцепт разрешен для использования в виде монотерапии, а также в виде комбинированной терапии с МТХ, для лечения RA.
Утрата функции и рентгенографическое изменение имеют место на ранней стадии в течение болезни. Эти изменения можно отсрочить или предупредить с помощью использования некоторых DMARD. Хотя некоторые DMARD были первоначально клинически эффективными и хорошо переносились, с течением времени многие из этих лекарственных средств стали менее эффективными или проявляют увеличенную токсичность. На основе своей эффективности и переносимости МТХ стал стандартной терапией, с которой сравнивают другие лечения (Bathon et al., N. Eng. J. Med., 343: 1586-1593 (2000); Albert et al., J. Rheumatol., 27: 644-652 (2000)).
В недавних исследованиях изучалось рентгенографическое прогрессирование у больных RA на поздней стадии, которые принимали лефлуномид, МТХ или плацебо (Stand et al., Arch. Intern. Med., 159: 2542-2550 (1999)), а также у больных, которые принимали лефлуномид плюс МТХ или плацебо плюс МТХ, с последующим частичным реагированием на МТХ (Lipsky et al., N. Engl. J. Med., 343: 1594-1602 (2000); Maini et al., Lancet, 354: 1932-1939 (1999)). В первый год Enbrel-испытания RA на ранней стадии обнаружено, что этанерцепт является значительно более эффективным, чем МТХ в улучшении признаков и симптомов заболевания и в ингибировании рентгенографического прогрессирования (Bathon et al., N. Eng. J. Med., 343: 1586-1593 (2000)). Genovese и др. (Arthritis Rheum. 46: 1443-1450 (2002) сообщают результаты второго года исследования, делая заключение, что этанерцепт в качестве монотерапии является безопасным и превосходит МТХ в уменьшении активности заболевания, в остановке структурного повреждения и в снижении нетрудоспособности на протяжении 2 лет у больных агрессивным RA на ранней стадии.
Кроме того, снижение рентгенографического прогрессирования в руках и ногах наблюдалось у больных ревматоидным артритом ранней стадии после получения инфликсимаба в комбинации с метотрексатом (Van der Heijde et al., Annnals Rhermatic Diseases 64: 418-419 (2005)). У больных ревматоидным артритом на ранней стадии наблюдалось клинически значимое и устойчивое улучшение физической функции после лечения инфликсимабом (Smolen et al., Annals Rheumatic Diseases 64: 418 (2005)). Об эффекте инфликсимаба и метотрексата на рентгенографическое прогрессирование у больных ревматоидным артритом на ранней стадии сообщается Van der и др. (Annals Rheumatic Diseases 64: 417 (2005)). Лечение инфликсимабом больных алкилозирующим спондилоартритом приводит к изменению маркеров воспаления и сменяемости кости, связанных с его клинической эффективностью (Visvanathan et al., Annals Rheumatic Diseases 64: 319 (2005)).
Об эффекте лечения инфликсимабом на плотность минеральных веществ в кости у больных алкилозирующим спондилоартритом (AS), наблюдаемым при рандомизированном, контролируемым плацебо испытании, названным ASSERT, сообщается Van der Heijde et al., Annals Rheumatic Diseases 64: 319 (2005). Обнаружено, что инфликсимаб уменьшает усталость и боль у больных AS в результате ASSERT (Van der Heijde et al., Annals Rheumatic Diseases 64: 318-319 (2005). Кроме того, эффективность и безопасность инфликсимаба для больных AS в результате ASSERT описывается Van der Heijde и др. (Arthritis Rheum. 5: 582-591 (2005). Авторы делают заключение, что инфликсимаб хорошо переносится и эффективен для большой группы больных AS на протяжении периода исследования, составляющего 24 недели. Кроме того, эффект лечения инфликсимабом воспаления позвоночника был оценен при получении изображения с помощью магнитного резонанса в рандомизированном, контролируемым плацебо, испытании 279 больных AS (Van der Heijde et al., Annals Rheumatic Diseases 64: 317 (2005)). Вопрос о способе, с помощью которого следует определять эффект лечения на рентгенографическое прогрессирование в позвоночнике больных AS, рассматривается Heijde и др. (Arthritis Rheum. 52: 1979-1985 (2005).
Результаты анализов рентгенограмм при международном, контролируемом испытании инфликсимаба для лечения псориатического артрита (IMPACT) спустя год сообщаются Antoni и др. (Annals Rheumatic Diseases 64: 107 (2005)). Доказательство рентгенографической пользы лечения инфликсимабом плюс МТХ больных ревматоидным артритом без клинического улучшения с детальным субанализом данных испытания антител против фактора некроза опухолей для лечения ревматоидного артрита с исследованием сопутствующей терапии приводится Smolen и др. (Arthritis Rheum. 52: 1020-1030 (2005)). Рентгенографическое прогрессирование, измеренное с помощью среднего изменения модифицированного van der Heijde показателя Sharp, было намного больше у больных, получающих МТХ плюс плацебо, чем у больных, получающих инфликсимам плюс МТХ. Авторы делают заключение, что даже у больных без клинического улучшения лечение инфликсимабом плюс МТХ приносило значительную пользу, что касается деструктивного процесса, что говорит о том, что у таких больных эти два показателя заболевания разобщены. Связь между рентгенографическим повреждением на базовой линии и улучшением физической функции после лечения больных, страдающих ревматоидным артритом, инфликсимабом описывается Breedveld et al., Annals Rheumatic Diseases 64: 52-55 (2005). Структурное повреждение оценивается с использованием модифицированного van der Heijde показателя Sharp. Авторы делают заключение, что более сильное повреждение сустава на базовой линии ассоциируется с более плохой физической функцией на базовой линии и меньшим увеличением физической функции после лечения, что подчеркивает важность раннего вмешательства для замедления прогрессирования разрушения сустава.
Антитела против CD20 и лечение ими
Лимфоциты являются одним из многих типов лейкоцитов, продуцируемых в костном мозге во время процесса гемопоэза. Существует две основных популяции лимфоцитов: В-лимфоциты (В-клетки) и Т-лимфоциты (Т-клетки). Представляющими здесь особый интерес лимфоцитами являются В-клетки.
В-клетки созревают в костном мозге и покидают костный мозг, экспрессируя на своих клеточных поверхностях антигенсвязывающее антитело. Когда не обученная В-клетка впервые встречает антиген, в отношении которого является специфичным ее связанное с мембраной антитело, клетка начинает быстро делиться, и ее потомство дифференцируется в В-клетки памяти и эффекторные клетки, называемые «плазмацитами». В-клетки памяти имеют более продолжительное время жизни и продолжают экспрессировать связанное с мембраной антитело с той же самой специфичностью, что и первоначальная родительская клетка. Плазмациты не продуцируют связанное с мембраной антитело, но вместо этого продуцируют антитело в форме, которая может секретироваться. Секретируемые антитела являются главными эффекторными молекулами гуморального иммунитета.
Антиген CD20 (также называемый ограниченный В-лимфоцитами человека антиген дифференциации, Вр35 или В1) представляет собой четырехпроходной, гликозилированный интегральный мембранный белок с молекулярной массой, составляющей 35 кДа, находящийся на пре-В-клетках и зрелых В-лимфоцитах (Valentine et al., J. Biol. Chem. 264(19): 11282-11287 (1989) и Einfeld et al., EMBO J. 7(3): 711-717 (1988)). Антиген также экспрессируется на более чем 90% В-клеточных неходжкинских лимфом (NHL) (Anderson et al. Blood 63(6): 1424-1433 (1984)), но он не обнаружен на гемопоэтических стволовых клетках, про-В-клетках, нормальных плазмацитах и в других нормальных тканях (Tedder et al. J. Immunol. 135(2): 973-979 (1985)). CD20 регулирует раннюю стадию(и) активационного процесса инициации клеточного цикла и дифференциации (Tedder et al., выше) и, возможно, функции кальциевых ионных каналов (Tedder et al., J. Cell Biochem. 14D: 195 (1990). В активированных В-клетках CD20 подвергается фосфорилированию (Riley and Sliwkowski Semin Oncol, 27(12), 17-24 (2000)). CD20 появляется на поверхности В-лимфоцитов на стадии пре-В-клеток и обнаруживается на зрелых В-клетках и В-клетках памяти, но не на плазмацитах (Stashenko et al. J. Immunol. 125: 1678-1685 (1980)); Clark and Ledbetter Adv. Cancer Res. 52, 81-149 (1989)). CD20 имеет активность в отношении кальциевых каналов и может играть роль в развитии В-клеток. Связь между лизисом периферических В-клеток CD20+ in vitro и активностью ритуксимаба in vivo не ясна. Ритуксимаб проявляет антителозависимую клеточноопосредованную цитотоксичность (ADCC) in vitro (Reff et al. Blood 83: 435-445 (1994)). Сильная комплементзависимая цитотоксическая (CDC) активность ритуксимаба также наблюдалась в отношении клеток и клеточных линий лимфомы (Reff et al., выше, 1994) и в некоторых моделях ксенотрансплантатов на мышах (Di Gaetano et al. J. Immunol. 171: 1581-1587 (2003)). Показано, что несколько антител против CD20, в том числе ритуксимаб, когда сшиты с помощью вторичного антитела или другим способом, индуцируют апоптоз in vitro (Ghetie et al. Proc. Natl. Acad. Sci. 94, 7509-7514 (1997)).
При условии, что CD20 экспрессируется в В-клеточных лимфомах, этот антиген может служить кандидатом для «нацеливания» на такие лимфомы. В сущности, такое нацеливание можно обобщенно представить следующим образом: больному вводят антитела, специфичные в отношении поверхностного антигена CD20 В-клеток. Эти антитела против CD20 специфично связываются с антигеном CD20 (по-видимому) как нормальных, так и злокачественных В-клеток; связанное с поверхностным антигеном CD20 антитело может привести к разрушению и истощению неопластических В-клеток. Кроме того, антитело против CD20 можно конъюгировать с химическими агентами или радиоактивными метками, имеющими активность уничтожать опухоль, так что агент специфически «доставляется» к неопластическим В-клеткам. Независимо от подхода главная цель - уничтожить опухоль; специфический подход может определяться конкретным используемым антителом против CD20, и, следовательно, доступные подходы к нацеливанию на антиген CD20 могут значительно варьировать.
Антителом ритуксимаб (RITUXAN®) является полученное методами генной инженерии химерное крысиное/человеческое моноклональное антитело, направленное против антигена CD20. Ритуксимаб является антителом, называемым «С2В8» в патенте США № 5736137, выданным 7 апреля 1998 (Anderson et al.). Ритуксимаб показан для лечения больных рецидивирующими или резистентными недоброкачественными или фолликулярными, CD20-положительными, В-клеточными неходжкинскими лимфомами. Исследования in vitro механизма действия показали, что ритуксимаб связывается с комплементом человека и лизирует лимфоидные В-клеточные линии через CDC (Reff et al., Blood 83(2): 435-445 (1994)). Кроме того, он обладает значительной активностью в анализах ADCC. Более недавно показано, что ритуксимаб оказывает антипролиферативные действия в анализах включения меченного тритием тимидина и непосредственно индуцируют апоптоз, в то время как другие антитела против CD19 и CD20 не оказывают таких действий (Maloney et al. Blood 88(10): 637a (1996)). Экспериментально также наблюдали синергизм между ритуксимабом и химиотерапевтическими агентами и токсинами. В частности, ритуксимаб сенсибилизирует устойчивые к лекарственным средствам линии клеток В-клеточных лимфом человека к цитотоксическим действиям доксорубицина, CDDDP, VP-16, дифтерийного токсина и рицина (Demidem et al., Cancer Chemotherapy & Radiopharmaceuticals 12(3): 177-186 (1997)). In vivo преклинические исследования показали, что ритуксимаб истощает В-клетки из периферической крови, лимфатических узлов и костного мозга яванских макак, предположительно через опосредуемыми комплементом и клетками процессы (Reff et al., Blood 83: 435-445 (1994)).
В Соединенных Штатах в ноябре 1997 г. ритуксимаб был разрешен для лечения больных рецидивирующими или резистентными недоброкачественными или фолликулярными, CD20+ В-клеточными неходжкинскими лимфомами в дозе 375 мг/м2 раз в неделю на протяжении четырех доз. В апреле 2001 Управление по контролю за продуктами и медикаментами (FDA) разрешило дополнительные показания для лечения недоброкачественных неходжкинских лимфом: повторное лечение (раз в неделю на протяжении четырех доз) и дополнительную схему дозирования (раз в неделю на протяжении восьми доз). Более 300000 больных подверглись воздействию ритуксимаба или в виде монотерапии, или в комбинации с иммунодепрессантами или химиотерапевтическими агентами. Больных подвергали лечению ритуксимабом в виде профилактической терапии в течение вплоть до 2 лет (Hainsworth et al., J. Clin. Oncol. 21: 1746-1751 (2003); Hainsworth et al., J. Clin. Oncol. 20: 4261-4267 (2002)). Также ритуксимаб использовали для лечения злокачественных и не злокачественных плазмаклеточных нарушений (Treon and Anderson, Semin. Oncol. 27: 79-85 (2000)).
Ритуксимаб также исследовали при различных не злокачественных аутоиммунных нарушениях, при которых В-клетки и аутоантитела, по-видимому, играют роль в патофизиологии заболевания (Edwards et al., Biochem. Soc. Trans. 30: 824-828 (2002)). Сообщалось, что ритуксимаб сильно уменьшает признаки и симптомы, например, ревматоидного артрита (RA) (Leandro et al., Ann. Rheum. Dis. 61: 883-888 (2002); Edwards et al., Arthritis Rheum., 46 (Suppl. 9): S46 (2002); Stahl et al., Ann. Rheum. Dis., 62 (Suppl. 1): OP004 (2003); Emery et al., Arthritis Rheum. 48(9): S439 (2003)), обыкновенной волчанки (Eisenberg, Arthritis. Res. Ther. 5: 157-159 (2003); Leandro et al. Arthritis Rheum. 46: 2673-2677 (2002); Gorman et al., Lupus, 13: 312-316 (2004)), иммунной тромботической пурпуры (D'Arena et al., Leuk. Lymphoma 44: 561-562 (2003); Stasi et al., Blood, 98: 952-957 (2001); Saleh et al., Semin. Oncol., 27 (Supp 12): 99-103 (2000); Zaia et al., Haematolgica, 87: 189-195 (2002); Ratanatharathorn et al., Ann. Int. Med., 133: 275-279 (2000)), истинной эритроцитарной аплазии (Auner et al., Br. J. Haematol., 116: 725-728 (2002)); аутоиммунной анемии (Zaja et al., Haematologica 87: 189-195 (2002) (опечатка появляется в Haematologica 87: 336 (2002))), болезни холодового агглютинина (Layios et al., Leukemia, 15: 187-8 (2001); Berentsen et al., Blood, 103: 2925-2928 (2004); Berentsen et al., Br. J. Haematol., 115: 79-83 (2001); Bauduer, Br. J. Haematol., 112: 1083-1090 (2001); Damiani et al., Br. J. Haematol., 114: 229-234 (2001)); синдрома типа В тяжелой инсулинорезистентности (Coll et al., N. Engl. J. Med., 350: 310-311 (2004)); смешанной криоглобулинемии (DeVita et al., Arthritis Rheum. 46 Suppl. 9: S206/S469 (2002)); тяжелой миастении (Zaja et al., Neurology, 55: 1062-63 (2000); Wylam et al., J. Pediatr., 143: 674-677 (2003)); грануломатоза Вегенера (Specks et al., Arthritis & Rheumatism 44: 2836-2840 (2001)); рефракторной обыкновенной пузырчатки (Dupuy et al., Arch Dermatol., 140: 91-96 (2004)); дерматомиозита (Levine, Arthritis Rheum., 46 (Suppl. 9): S1299 (2002)), синдрома Шегрена (Somer et al., Arthritis & Rheumatism, 49: 394-398 (2003)), активной криоглобулинемии смешанного типа II (Zaja et al., Blood, 101: 3827-3834 (2003)); обыкновенной пузырчатки (Dupay et al., Arch. Dermatol., 140: 91-95 (2004)); аутоиммунной невропатии (Pestronk et al., J. Neurol. Neurosurg. Psychiatry 74: 485-489 (2003)); паранеопластического синдрома пляшущих глаз и судорожного подергивания мышц (Pranzatelli et al. Neurology 60 (Suppl. 1) PO5.128: A395 (2003)) и возвращающегося-прекращающегося рассеянного склероза (RRMS) (Cross et al. (abstract) “Preliminary Results from a Phase II Trial of Rituximab in MS” Eighth Annual Meeting of the Americas Committes for Research and Treatment in Multiple Sclerosis, 20-21 (2003)).
Исследование фазы II (WA16291) проводилось на больных ревматоидным артритом (RA) с предоставлением данных контроля на протяжении 48 недель в отношении безопасности и эффективности ритуксимаба (Emery et al., Arthritis Rheum 48(9): S439 (2003); Szczepanski et al. Arthritis Rheum 48(9): S121 (2003)). В целом 161 больных равномерно рандомизировали в четыре подвергаемые лечению группы: метотрексат, только ритуксимаб, ритуксимаб плюс метотрексат и ритуксимаб плюс циклофосфамид (СТХ). Схема лечения ритуксимабом была один грамм, вводимый внутривенно в дни 1 и 15. Большинство больных RA хорошо переносили инфузии ритуксимаба, при этом у 36% больных имел место по крайней мере один побочный эффект во время первой инфузии (по сравнению с 30% больных, получающих плацебо). В общем, большинство побочных эффектов считались по тяжести от слабых до умеренных и были сбалансированы по всем подвергаемых лечению группам. Во всех четырех группах на протяжении 48 недель было в общем 19 серьезных побочных эффектов, которые были немного более частыми в группе ритуксимаб/CTX. Инфекционные заболевания были хорошо сбалансированы по всем группам. Средняя частота серьезных инфекций в этой популяции больных RA была 4,66 на 100 пациентов в год, что ниже частоты инфекций, требующей госпитализации, у больных RA (9,57 на 100 пациентов в год), о которой сообщалось в общественном эпидемиологическом исследовании (Doran et al., Arthritis Rheum. 46: 2287-2293 (2002)).
Сообщаемый профиль безопасности ритуксимаба при использовании небольшого количества больных неврологическими нарушениями, в том числе аутоиммунной невропатией (Pestronk et al., выше); синдромом пляшущих глаз и судорожного подергивания мышц (Pranzatelli et al., выше) и RRRMS (Cross et al., выше), был схож с профилем, о котором сообщалось при онкологии или RA. В проводящемся, финансируемом исследователями испытании (IST) ритуксимаба в комбинации с интерфероном-бета (IFN-β) или глатирамера ацетатом на больных RRRMS (Cross et al., выше) 1 из 10 подвергаемых лечению больных был госпитализирован для наблюдения в течение ночи после того, как у него имели место лихорадка и озноб средней степени после первой инфузии ритуксимаба, в то время как для остальных 9 больных полностью выполнили схему из четырех инфузий без каких-либо сообщенных побочных эффектов.
Патенты и патентные публикации, касающиеся антител против CD20 и связывающих CD20 молекул, включают патенты США № 5776456, 5736137, 5843439, 6399061 и 6682734, а также US 2002/0197255, US 2003/0021781, US 2003/0082172, US 2003/0095963, US 2003/0147885 (Anderson et al.); патент США № 6455043 и WO 2000/09160 (Grillo-Lopez, A.); WO 2000/27428 (Grillo-Lopez and White); WO 2000/27433 (Grillo-Lopez and Leonard); WO 2000/44788 (Braslawsky et al.); WO 2001/10462 (Rastetter, W.); WO 2001/10461 (Rastetter and White); WO 2001/10460 (White and Grillo-Lopez); US 2001/0018041, US 2003/0180292, WO 2001/34194 (Hanna and Hariharan); US 2002/0006404 и WO 2002/04021 (Hanna and Hariharan); US 2002/0012665, WO 2001/74388 и 6896885В5 (Hanna, N.); US 2002/0058029 (Hanna, N.); US 2003/0103971 (Hariharan and Hanna); US 2005/0123540 (Hanna et al); US 2002/0009444 и WO 2001/80884 (Grillo-Lopes, A.); WO 2001/97858; US 2005/0112060 и патент США № 6846476 (White, C.); US 2002/0128488 и WO 2002/34790 (Reff, M.); WO 2002/060955 (Braslawsky et al.); WO 2002/096948 (Braslawsky et al.); WO 2002/079255 (Reff and Davies); патент США № 6171586 и WO 1998/56418 (Lam et al.); WO 1998/58964 (Raju, S.); WO 1999/22764 (Raju, S.); WO 1999/51642, патент США № 6194551, патент США № 6242195, патент США № 6528624 и патент США № 6538124 (Idusogie et al.); WO 2000/42072 (Presta, L.); WO 2000/67796 (Curd et al.); WO 2001/03734 (Grillo-Lopez et al.): US 2002/0004587 и WO 2001/77342 (Miller and Presta); US 2002/0197256 (Grewal, I.); US 2003/0157108 (Presta, L.); патенты США № 6565827, 6090365, 6287537, 6015542, 5843398 и 5595721 (Kaminski et al.); патенты США № 5500362, 5677180, 5721108, 6120767, 6652852 и 6893625 (Robinson et al.); патент США № 6410391 (Raubitschek et al.); патент США № 6224866 и WO 00/20864 (Barbera-Guillem, E.); WO 2001/13945 (Barbera-Guillem, E.); WO 2000/67795 (Goldenberg); US 2003/0133930; WO 200/74718 и US 2005/091300A1 (Goldenberg and Hansen); US 2003/0219433 и WO 2003/68821 (Hansen et al.); WO 2004/058298 (Goldenberg and Hansen); WO 2000/76542 (Golay et al.); WO 2001/72333 (Wolin and Rosenblatt); патент США № 6368596 (Ghetie et al.); патент США № 6306393 и US 2002/0041847 (Goldenberg, D.); US 2003/0026801 (Weiner and Hartmann); WO 2002/102312 (Engleman, E.); US 2003/0068664 (Albitar et al.); WO 2003/002607 (Leung, S.); WO 2003/049694, US 2002/0009427 и US 2003/0185796 (Wolin et al.); WO 2003/061694 (Sing and Siegall); US 2003/0219818 (Bohen et al.); US 2003/0219433 и WO 2003/068821 (Hansen et al.); US 2003/0219818 (Bohen et al.); US 2002/0136719 (Shenoy et al.); WO 2004/032828 и US 2005/0180972 (Wahl et al.); и WO 2002/56910 (Hayden-Ledbetter). Смотри также патент США № 5849898 и ЕР 330191 (Seed et al); EP 332865A2 (Meyer and Weiss); патент США № 4861579 (Meyer et al.); US 2001/0056066 (Bugelski et al.); WO 1995/03770 (Bhat et al.); US 2003/0219433 A1 (Hansen et al.); WO 2004/035607 (Teeling et al.); WO 2005/103081 (Teeling et al.); WO 2004/056312 (Lowman et al); US 2004/0093621 (Shitara et al.); WO 2004/103404 (Watkins et al.); WO 2005/000901 (Tedder et al.): US 2005/0025764 (Watkins et al.); WO 2005/016969 (Carr et al); US 2005/0069545 (Carr et al.); WO 2005/014618 (Chang et al.); US 2005/0079174 (Barbera-Guillem and Nelson); US 2005/0106108 (Leung and Hansen); WO 2005/044859 и US 2005/0123546 (Umana et al.); WO 2005/070963 (Allan et al.); US 2005/0186216 (Ledbetter and Hayden-Ledbetter); US 2005/0202534 (Hayden-Ledbetter and Ledbetter); US 2005/0202028 (Hayden-Ledbetter and Ledbetter); US 2005/0202023 (Hayden-Ledbetter and Ledbetter); патент США № 6183744 (Goldenberg) и патент США № 6897044 (Braslawski et al.).
Публикации, относящиеся к лечению ритуксимабом, включают: Perotta and Abuel, “Response of chronic relapsing ITP of 10 years duration to rituximab” Abstract #3360 Blood 10(1) (part 1-2): p. 88B (1998); Perotta et al., “Rituxan in the treatment of chronic idiopathic thrombocytopenic purpura (ITP)”, Blood 94: 49 (abstract) (1999); Matthews, R., “Medical Heretics” New Scientist (7 April, 2001); Leando et al., “Clinical outcome in 22 patients with rheumatoid arthritis treated with B lymphocyte depletion” Ann. Rheum. Dis, выше; Leandro et al., “Lymphocyte depletion in rheumatoid arthritis: early evidence for safety, efficacy and dose response” Arthritis and Rheumatism 44(9): S370 (2001); Leandro et al., “An open study of B lymphocyte depletion in systemic lupus erythematosus”, Arthritis and Rheumatism, 46: 2673-2677 (2002), где в течение 2-недельного периода каждый больной получал две инфузии по 500 мг ритуксимаба, две инфузии по 750 мг циклофосфамида и большую дозу пероральных кортистероидов, и где двое подвергаемых лечению больных перенесли рецидив на 7 и 8 месяцах, соответственно, и их лечили повторно, хотя с использованием отличных протоколов; “Successful long-term treatment of systemic lupus erythematosus with rituximab maintenance therapy” Weide et al., Lupus, 12: 779-782 (2003), где больного лечили ритуксимабом (375 мг/м2 × 4 повтора с интервалами в одну неделю) и, кроме того, для больного использовался ритуксимаб каждые 5-6 месяцев и затем использовалось профилактическое лечение ритуксимабом 375 мг/м2 каждые три месяца, а второго больного рефрактерным SLE успешно лечили ритуксимабом, и он получал профилактическое лечение каждые три месяца, при этом оба больных хорошо реагировали на лечение ритуксимабом; Edwards and Cambridge, “Sustained improvement in rheumatoid arthritis following a protocol designed to deplete B lymphocytes” Rheumatology 40: 205-211 (2001); Cambridge et al., “B lymphocyte depletion in patients with rheumatoid arthritis: serial studies of immunological parameter” Arthritis Rheum., 46 (Suppl. 9): S1350 (2002); Cambridge et al., “Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis” Arthritis Rheum., 48: 2146-2154 (2003); Edwards et al., “B-lymphocyte depletion therapy in rheumatoid arthritis and other autoimmune disorder” Biochem Soc. Trans., выше; Edwards et al., “Efficacy and safety of rituximab, a B-cell targeted chimeric monoclonal antibody: A randomized, placebo controlled trial in patients with rheumatoid arthritis. Arthritis and Rheumatism 46(9): S197 (2002); Edwards et al., “Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis” N. Engl. J. Med. 350: 2572-2582 (2004); Pavelka et al., Ann. Rheum. Dis. 63: (S1): 289-290 (2004); Emery et al., Arthritis Rheum. 50 (S9): S659 (2004); Levine and Pestronk, “IgM antibody-related polyneuropathies: B-cell depletion chemotherapy using rituximab” Neurology 52: 1701-1704 (1999), Uchida et al., “The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy” J. Exp. Med. 1999: 1659-1669 (2004); Gong et al., “Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy” J. Immunol. 174: 817-826 (2005); Hamaguchi et al., “The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice” J. Immunol. 174: 4389-4399 (2005); Cragg et al. “The biology of CD20 and its potential as a target for mAb therapy” Curr. Dir. Autoimmune. 8: 140-174 (2005); Eisenberg, “Mechanisms of autoimmunity” Immunol. Res. 27: 203-218 (2003); DeVita et al., “Efficacy of selective B cell blockade in the treatment of rheumatoid arthritis” Arthritis & Rheum. 46: 2029-2033 (2002); Hidashida et al. “Treatment of DMARD-refractory rheumatoid arthritis with rituximab”, работа, представленная на Annual Scientific Meeting of the American College of Rheumatology; Oct. 24-29; New Orleans, LA 2002; Tuscano, J. “Successful treatment of infliximab-refractory rheumatoid arthritis with rituximab” работа, представленная на Annual Scientific Meeting of the American College of Rheumatology; Oct. 24-29; New Orleans, LA 2002 и опубликованная Tuscano, Arthritis Rheum. 46: 3420 (2002); “Pathogenic roles of B cells in human autoimmunity; insights from the clinic” Martin and Chan, Immunity 20: 517-527 (2004); Silverman and Weisman, “Rituximab therapy and autoimmune disorder, prospects for anti-B cell therapy”, Arthritis and Rheumatism, 48: 1484-1492 (2003); Kazkaz and Isenberg, “Anti B cell therapy (rituximab) in the treatment of autoimmune diseases”, Current opinion in pharmacology, 4: 398-402 (2004); Virgolini and Vanda, “Rituximab in autoimmune diseases”, Biomedicine & pharmacotherapy, 58: 299-309 (2004); Klemmer et al., “Treatment of antibody mediated autoimmune disorder with a AntiCD20 monoclonal antibody Rituximab”, Arthritis and Rheumatism, 48: (9) 9,S (SEP), page S624-S624 (2003); Kneitz et al., “Effective B cell depletion with rituximab in the treatment of autoimmune disease”, Immunobiology, 206: 519-527 (2002); Arzoo et al., “Treatment of refractory antibody mediated autoimmune disorder with an anti-CD20 monoclonal antibody (rituximab)” Annals of the Rheumatic Diseases, 61 (10), p. 922-924 (2002) Comment in Ann Rheum. Dis. 61: 863-866 (2002); “Future strategies in immunotherapy” by Lake and Dionne, in Burger's Medicinal Chemistry and Drug Discovery (2003 by John Wiley & Sons, Inc.) дата поступления статьи онлайн: 15 января 2003 (Chapter 2 “Antibody-Directed Immunotherapy”); Liang and Tedder, Wiley Encyclopedia of Molecular Medicine, Section: CD20 as an Immunotherapy Target, дата поступления статьи онлайн: 15 января 2002, озаглавленной “CD20”; приложение 4А, озаглавленное “Monoclonal Antibodies to Human Cell Surface Antigens” by Stockinger et al., eds: Coligan et al., in Current Protocols in Immunology (2003 John Wiley & Sons, Inc) дата поступления онлайн: май 2003; дата печатной публикации: февраль 2003; Penichet and Morrison, “CD Antibodies/molecules: Definition; Antibody Engineering” in Wiley Encyclopedia of Molecular Medicine Section: Chimeric, Humanized and Human Antibodies, дата поступления онлайн: 15 января 2002.
Кроме того, смотри Looney “B cells as a therapeutic target in autoimmune diseases other than rheumatoid arthritis” Rheumatology, 44 Suppl. 2: ii13-ii17 (2005); Chambers and Isenberg, “Anti-B cell therapy (rituximab) in the treatment of autoimmune diseases” Lupus 14(3): 210-214 (2005); Looney et al., “B-cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalating trial of rituximab” Arthritis Rheum. 50: 2580-2589 (2004); Looney, “Treating human autoimmune disease by depleting B cells” Ann. Rheum. Dis. 61: 863-866 (2002); Edelbauer et al., “Rituximab in childhood systemic lupus erythematosus refractory to conventional immunosuppresssion case report” Pediatr. Nephrol. 20(6): 811-813 (2005); D'Cruz and Hughes, “The treatment of lupus nephritis” BMJ 330(7488): 377-378 (2005); Looney, “B cell-targeted therapy in diseases other than rheumatoid arthritis” J. Rheumatol. Suppl. 73: 25-28; discussion 29-30 (2005); Sfikakis et al., “Remission of proliferative lupus nephritis following B cell depletion therapy is preceded by down-regulation of the T cell costimulatory molecule CD40 ligand: an open-label trial” Arthritis Rheum. 52(2): 501-513 (2005); Rastetter et al., “Rituximab: expanding role in therapy for lymphomas and autoimmune diseases” Annu. Rev. Med. 55: 477-503 (2004); Silverman, “Anti-CD20 therapy in systemic lupus erythematosus: a step closer to the clinic” Arthritis Rheum. 52(2): 371-7 (2005), опечатка в: Arthritis Rheum. 52(4): 1342 (2005); Ahn et al., “Long-term remission from life-threatening hypercoagulable state associated with lupus anticoagulant (LA) following rituximab therapy” Am. J. Hematol. 78(2): 127-129 (2005); Tahir et al., “Humanized anti-CD20 monoclonal antibody in the treatment of severe resistant systemic lupus erythematosus in a patient with antibodies against rituximab” Rheumatology, 44(4): 561-562 (2005), Epub 2005 Jan 11: Looney et al., “Treatment of SLE with anti-CD20 monoclonal antibody” Curr. Dir. Autoimmun. 8: 193-205 (2005); Cragg et al., “The biology of CD20 and its potential as a target for mAb therapy” Curr. Dir. Autoimmun. 8: 140-174 (2005); Gottenberg et al., “Tolerance and short term efficacy of rituximab in 43 patients with systemic autoimmune diseases” Ann. Rheum. Dis. 64(6): 913-920 (2005) Epub 2004 Nov 18: Tokunaga et al., “Down-regulation of CD40 and CD80 on B cells in patients with life-threatening systemic lupus erythematosus after successful treatment with rituximab” Rheumatology 44(2): 176-182 (2005), Epub 2004 Oct. 19. Смотри также Leandro et al., “B cell repopulation occurs mainly from naïve B cells in patient with rheumatoid arthritis and systemic lupus erythematosus” Arthritis Rheum., 48 (Suppl 9): S1160 (2003).
Specks и др. в “Response of Wegener's granulomatosis to anti-CD20 chimeric monoclonal antibody therapy” Arthritis & Rheumatism 44(12): 2836-2840 (2001) описывают успешное применение четырех инфузий по 375 мг/м2 ритуксимаба и большой дозы кортикостероидов для лечения грануломатоза Вегенера. Лечение повторяли через 11 месяцев, когда происходил рецидив cANCA, но лечение было без глюкокортикоидов. На 8 месяце после второго курса лечения ритуксимабом заболевание пациентов оставалось в полной ремиссии. Кроме того, в другом исследовании обнаружено, что ритуксимаб является хорошо переносимым, эффективным индуцирующим ремиссию агентом для тяжелого сопровождающего ANCA васкулита при использовании в дозе по 375 мг/м2 × 4 вместе с пероральным преднизоном в количестве 1 мг/кг/день, которое снижали еженедельно на 4-40 мг/день и до полного прекращения на протяжении последующих 16 недель. Четырех больных лечили повторно только ритуксимабом в отношении титров рецидива/нарастания ANCA. Для индукции ремиссии и сохранения устойчивой ремиссии (6 месяцев или дольше), помимо глюкокортикоидов, дополнительных иммунодепрессантов, по-видимому, не требуется. Смотри поступления онлайн реферата и заявление Keogh и др., “Rituximab for Remission Induction in Severe ANCA-Associated Vasculitis: Report of a Prospective Open-Label Pilot Trial in 10 Patients”, American College of Rheumatology, Session Number: 28-100, Session Title: Vasculitis, Session Type: ACR Concurrent Session, Primary Category: 28 Vasculitis, Session 10/18/2004 (www.abstractsonline.com/viewer/SearchResults.asp). Смотри также Keogh et al., Kidney Blood Press. Res. 26: 293 (2003), где сообщается, что одиннадцать больных с рефрактерным сопровождающим ANCA васкулитом подвергались лечению четырьмя еженедельными дозами 375 мг/м2 ритуксимаба и большой дозы глюкокортикоидов, что приводило к ремиссии.
Больным рефрактерным сопровождающим ANCA васкулитом вводили ритуксимаб вместе с иммунодепрессантами, такими как внутривенный циклофосфамид, микофенолат мофетил, азатиоприн или лефлуномид, с видимой эффективностью (Eriksson, “Short-term outcome and safety in 5 patients with ANCA-positive vasculitis treated with rituximab”, Kidney and Blood Pressure Research, 26: 294 (2003) (пять больных сопровождающим ANCA васкулитом, подвергнутых лечению 375 мг/м2 ритуксимаба раз в неделю на протяжении 4 недель, реагировали на лечение); Jayne et al., “B-cell depletion with rituximab for refractory vasculitis” Kidney and Blood Pressure Research, 26: 294-295 (2003) (у шести больных рефрактерным васкулитом, получивших четыре еженедельных инфузии по 375 мг/м2 ритуксимаба с циклофосфамидом вместе с фоновой иммунодепрессией и преднизолоном, имели место серьезные снижения активности васкулита). Еще одно сообщение об использовании ритуксимаба вместе с внутривенным циклофосфамидом при 375 мг/м2 на дозу в 4 дозах для введения больным рефрактерным системным васкулитом, предоставлено Jayne, poster 88 (11th International Vasculitis and ANCA workshop), 2003 American Society of Nephrology. Смотри также Stone and Specks, “Rituximab Therapy for the Induction of Remission and Tolerance in ANCA-associated Vasculitis”, in the Clinical Trial Research Summary of the 2002-2003 Immune Tolerance Network, http://www.immunetolerance.org/research/autoimmune/trials/stone.html, в которой предлагается испытание ритуксимаба для лечения сопровождающего ANCA васкулита на протяжении в целом 18 месяцев. Смотри также Eriksson, J. Internal Med., 257: 540-548 (2005) в отношении девяти больных с положительным в отношении ANCA васкулитом, которых успешно лечили двумя или четырьмя еженедельными дозами по 500 мг ритуксимаба, а также Keogh и др. al., Arthritis and Rheumatism, 52: 262-268 (2005), которые сообщили, что у 11 больных рефрактерным сопровождающим ANCA васкулитом лечение или повторное лечение четырьмя еженедельными дозами по 375 мг/м2 ритуксимаба индуцировало ремиссию путем истощения В-лимфоцитов, при этом исследование проводилось с января 2000 по сентябрь 2002.
В отношении активности гуманизированного антитела против CD20 смотри, например, Vugmeyster и др., “Depletion of B cells by humanized anti-CD20 antibody PRO70769 in Macaca fascicularis” J. Immunother. 28: 212-219 (2005). В отношении обсуждения моноклонального антитела человека смотри Baker и др., “Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator” Arthritis Rheum. 48: 3253-3265 (2003).
Обнаружения исследования WA17043, рандомизированного, двойного слепого исследования фазы IIb с использованием диапазона доз больных ревматоидным артритом, которые неадекватно реагировали на DMARD (в том числе анти-TNF агенты) (Emery et al., European League against Rheumatism (EULAR) (June 2005) OP0007; Van Vollenhoven et al., EULAR (June 2005) SAT0072), указывают на то, что комбинация ритуксимаба с МТХ сопровождается клинически и статистически значимым улучшением симптомов заболевания. В этом исследовании определены дозы ритуксимаба в комбинации с МТХ, которые необходимо далее исследовать и подтвердить в пределах установки клинического исследования фазы III. Смотри также World Pharmaceutical Nres, www.scrippharma.com свидетельство о праве - статья, датированная 13 июня 2005, озаглавленная “Rituximab a future challenge for anti-TNFs?”, в которой описываются исследования EULAR и приводится предположение о том, смогут ли данные рентгенологического анализа при исследовании REFLEX фазы III отразить замедление ритуксимабом повреждения сустава. Кроме того, в заявке WO 2004/091657, опубликованной 28 октября 2004, описывается лечение больных ревматоидным артритом, которые неадекватно реагируют на TNFα-ингибиторное лечение антителами против CD20, причем рентгенографически доказано, что у больных имеется по крайней мере один сустав с определенной эрозией, свойственной ревматоидному артриту, как определено с использованием центрального места считывания (любой сустав рук, запястий или ног за исключением DIP-уставов рук). Возможное вторичное конечное положение включает изменение модифицированного рентгенографического общего показателя Sharp, показателя эрозии и показателя сужения суставного пространства, которые можно проанализировать с использованием континуальной или категоричной методологии, что подходит. Исследовательские конечные положения и анализы могут включать рентгенографические анализы, включающие долю больных без прогрессирования эрозий, которые можно оценить на 24 неделе или позже. Смотри также заявку US 2005/00001862, опубликованную 25 августа 2005, эквивалентную заявке WO 2005/060999, опубликованной 7 июля 2005, касающейся лечения больных ревматоидным артритом ритуксимабом, причем возможное вторичное концевое положение и исследовательские концевые положения и анализы включают те, которые описаны в WO 2004/091657, выше.
Несмотря на прогресс в лечении заболевания сустава значительному числу больных противопоказаны имеющиеся в настоящее время лечения, они не переносятся больными или значительное число больных недостаточно отвечает на имеющиеся в настоящее время лечения. Следовательно, требуются новые варианты лечения, в частности такие, которые могут быть нацелены на различные аспекты патологии заболевания и приводить к схожим или лучшим степеням клинической пользы.
Краткое изложение сущности изобретения
Настоящее изобретение включает введение антагониста CD20, обеспечивающее схему безопасного и активного лечения индивидов с повреждением сустава, которая включает выбор схемы эффективного введения лекарственного средства и запланированное или не запланированное повторное лечение. Этот антагонист является эффективным как при первоначальном лечении, так и для контроля рефрактерного заболевания.
Соответственно заявлено изобретение. В первом аспекте настоящее изобретение относится к способу лечения повреждения сустава у индивида, включающему введение индивиду антитела против CD20 и рентгенографическую проверку индивида, спустя по крайней мере приблизительно один месяц от момента введения, с помощью которой определяют уменьшение повреждения сустава по сравнению с базовой линией до введения, причем количество вводимого антитела против CD20 является эффективным для получения уменьшения повреждения сустава.
В другом аспекте настоящее изобретение относится к способу контролирования лечения повреждения сустава у индивида, включающему введение индивиду эффективного количества антитела против CD20 и определение с помощью рентгенографии спустя по крайней мере приблизительно один месяц от момента введения, произошло ли уменьшение повреждения сустава по сравнению с базовой линией до введения, причем уменьшение в сравнении с базовой линией у индивида после лечения указывает на то, что антитело против CD20 оказывает эффект на повреждение сустава. В предпочтительном варианте осуществления настоящего изобретения степень уменьшения в сравнение с базовой линией определяют второй раз после введения антитела против CD20.
В другом аспекте настоящим изобретением обеспечивается способ определения, нужно ли продолжать введение индивиду с повреждением сустава антитела против CD20, включающий определение с помощью рентгенографии уменьшения повреждения сустава у индивида после введения антитела против CD20 первый раз, определение с помощью рентгенографии уменьшения повреждения сустава у индивида после введения антитела против CD20 второй раз, сравнение рентгенографических показателей у индивида в первый раз и во второй раз и, если показатель меньше во второй раз, чем в первый раз, продолжение введения антитела против CD20.
В еще одном аспекте настоящее изобретение направлено на изделие производства, включающее
(а) контейнер, содержащий антитело против CD20; и
(b) листовку-вкладыш с инструкциями в отношении лечения повреждения сустава у индивида, причем в инструкциях указывается на то, что индивиду вводится антитело против CD20, и затем он проходит рентгенографическую проверку, спустя по крайней мере приблизительно один месяц от момента введения, с помощью которой определяют уменьшение повреждения сустава по сравнению с базовой линией до введения, причем количество вводимого антитела против CD20 является эффективным для получения уменьшения повреждения сустава.
В предпочтительном аспекте изделие дополнительно включает контейнер, содержащий второе лекарственное средство, причем антитело против CD20 представляет собой первое лекарственное средство, дополнительно включающий инструкции в листовке-вкладыше в отношении лечения индивида эффективным количеством второго лекарственного средства.
В другом варианте осуществления настоящего изобретения обеспечивается способ лечения повреждения сустава у индивида, включающий введение индивиду антитела против CD20 и рентгенографическую проверку индивида, спустя по крайней мере приблизительно 52 недели от момента введения, с помощью которой определяют уменьшение повреждения сустава по сравнению с базовой линией до введения, причем количество вводимого антитела против CD20 является эффективным для получения уменьшения повреждения сустава.
В еще одном варианте осуществления настоящим изобретением обеспечивается способ контролирования лечения повреждения сустава у индивида, включающий введение индивиду эффективного количества антитела против CD20 и определение с помощью рентгенографии спустя по крайней мере приблизительно 52 недели от момента введения, произошло ли уменьшение повреждения сустава по сравнению с базовой линией до введения, причем уменьшение в сравнении с базовой линией у индивида после лечения указывает на то, что антитело против CD20 оказывает эффект на повреждение сустава.
Кроме того, настоящим изобретением обеспечивается изделие производства, включающее
(а) контейнер, содержащий антитело против CD20; и
(b) листовку-вкладыш с инструкциями в отношении лечения повреждения сустава у индивида, причем в инструкциях указывается на то, что индивиду вводится антитело против CD20, и затем он проходит рентгенографическую проверку, спустя по крайней мере приблизительно 52 недели от момента введения, с помощью которой определяют уменьшение повреждения сустава по сравнению с базовой линией до введения, причем количество вводимого антитела против CD20 является эффективным для получения уменьшения повреждения сустава.
В другом аспекте настоящим изобретением обеспечивается способ лечения повреждения сустава у индивида, причем (а) индивиды неадекватно реагировали на один или несколько ингибиторов в виде антител против фактора некроза опухолей (TNF); (b) индивиды получили по крайней мере один предшествующий курс лечения антителом против CD20, и (с) лечение включает назначение по крайней мере одного дополнительного курса лечения антителом против CD20.
Эти и другие аспекты будут более понятны из остального описания, включающего примеры и прилагаемую формулу изобретения.
Краткое описание чертежей
На фиг.1 демонстрируется проект исследования лечения больных RA контролем (плацебо плюс МТХ) или ритуксимабом (1000 мг × 2) плюс МТХ (здесь пример 1).
На фиг.2 демонстрируются ответы ACR на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.3 демонстрируются ответы ACR на протяжении шести месяцев лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.4 демонстрируются изменения DAS28 на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.5 демонстрируются ответы EULAR на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.6 демонстрируются ремиссия и заболевание низкой активности на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.7 демонстрируется медианный С-реактивный белок (CRP) на протяжении шести месяцев лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.8 демонстрируется доля больных RA с клинически релевантным улучшением функции на шестом месяце лечения, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.9 демонстрируется изменение в процентах набора показателей ACR на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.10 демонстрируется изменение FACIT-F на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.11 демонстрируются изменения в классах SF-36 (психическое и физическое здоровье) на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.12 демонстрируется ревматоидный фактор в целом на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.13 демонстрируется среднее изменение общего показателя Sharp-Genant на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.14 демонстрируется среднее изменение показателя эрозии Sharp-Genant на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.15 демонстрируется среднее изменение показателя сужения суставного пространства (JSN) Sharp-Genant на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.16 демонстрируется доля больных RA без изменения показателя эрозии на шестом месяце лечения, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.17 демонстрируется изменение рентгенографических конечных положений на 24 неделе (исследовательское конечное положение) лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.18 демонстрируется среднее изменение в процентах параметров набора показателей ACR на 24 неделе лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.19 демонстрируется медианный CD19 на шестом месяце лечения больных RA контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.20 демонстрируются наиболее часто сообщаемые побочные эффекты при исследовании больных RA на протяжении шести месяцев, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.21 демонстрируются эффекты, приводящие к исключению, при исследовании больных RA на протяжении шести месяцев, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.22 демонстрируются эффекты, происходящие во время/в пределах 24 часов от инфузий, при исследовании больных RA на протяжении шести месяцев, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.23 демонстрируются острые реакции на инфузии при исследовании больных RA на протяжении шести месяцев, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.24 демонстрируются серьезные побочные эффекты, происходящие во время/в пределах 24 часов от инфузий, при исследовании больных RA на протяжении шести месяцев, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.25 демонстрируется класс системных органов - инфекции и инвазии при исследовании больных RA на протяжении шести месяцев, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.26 демонстрируются серьезные инфекции при исследовании больных RA на протяжении шести месяцев, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.27 демонстрируется частота инфекций при исследовании больных RA на протяжении шести месяцев, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.28 демонстрируется НАСА при исследовании больных RA на протяжении шести месяцев, причем больных подвергают лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.29 демонстрируются уровни IgG на протяжении шести месяцев у больных RA, подвергнутых лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.30 демонстрируются уровни IgА на протяжении шести месяцев у больных RA, подвергнутых лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.31 демонстрируются уровни IgМ на протяжении шести месяцев у больных RA, подвергнутых лечению контролем или ритуксимабом (1000 мг × 2) плюс МТХ.
На фиг.32А демонстрируется выравнивание последовательностей, позволяющее сравнивать аминокислотные последовательности вариабельного домена легкой цепи (VL) крысиного антитела 2Н7 (SEQ ID NO: 1), гуманизированного варианта 2H7.v16 (SEQ ID NO: 2) и легкой цепи каппа человека подгруппы I (SEQ ID NO: 3). CDR VL 2Н7 и hu2H7.v16 являются следующими: CDR1 (SEQ ID NO: 4), CDR2 (SEQ ID NO: 5) и CDR3 (SEQ ID NO: 6).
На фиг.32В демонстрируется выравнивание последовательностей, позволяющее сравнивать аминокислотные последовательности вариабельного домена тяжелой цепи (VН) крысиного антитела 2Н7 (SEQ ID NO: 7), гуманизированного варианта 2H7.v16 (SEQ ID NO: 8) и консенсусной последовательности тяжелой цепи человека подгруппы III (SEQ ID NO: 9). CDR VН 2Н7 и hu2H7.v16 являются следующими: CDR1 (SEQ ID NO: 10), CDR2 (SEQ ID NO: 11) и CDR3 (SEQ ID NO: 12).
На фиг.32А и 32В в скобки заключены CDR1, CDR2 и CDR3, фланкированные каркасными областями FR1-FR4, как указано. 2Н7 относится к крысиному антителу 2Н7. Звездочки между двумя рядами последовательностей указывают положения, которые являются различными между двумя последовательностями. Нумерация остатков находится в соответствии с Kabat et al. Sequences of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991), со вставками в виде a, b, c, d и e.
На фиг.33 демонстрируется аминокислотная последовательность зрелой легкой цепи 2H7.v16 (SEQ ID NO: 13).
На фиг.34 демонстрируется аминокислотная последовательность зрелой тяжелой цепи 2H7.v16 (SEQ ID NO: 14).
На фиг.35 демонстрируется аминокислотная последовательность зрелой тяжелой цепи 2H7.v31 (SEQ ID NO: 15). Легкая цепь 2H7.v31 является такой же, как в 2H7.v16.
На фиг.36 демонстрируется выравнивание последовательностей, позволяющее сравнивать аминокислотные последовательности легкой цепи гуманизированного варианта 2H7.v16 (SEQ ID NO: 2) и гуманизированного варианта 2H7.v138 (SEQ ID NO: 28).
На фиг.37 демонстрируется выравнивание последовательностей, позволяющее сравнивать аминокислотные последовательности тяжелой цепи гуманизированного варианта 2H7.v16 (SEQ ID NO: 8) и гуманизированного варианта 2H7.v138 (SEQ ID NO: 29).
На фиг.38 демонстрируется выравнивание зрелых легких цепей 2H7.v16 и 2H7.v511 (SEQ ID NO: 13 и 30, соответственно), с нумерацией по Kabat остатков вариабельных доменов и Eu-нумерацией остатков константных доменов.
На фиг.39 демонстрируется выравнивание зрелых тяжелых цепей 2H7.v16 и 2H7.v511 (SEQ ID NO: 14 и 31, соответственно), с нумерацией по Kabat остатков вариабельных доменов и Eu-нумерацией остатков константных доменов.
На фиг.40А демонстрируется последовательность вариабельного домена легкой цепи гуманизированного 2H7.v114 (SEQ ID NO: 32); на фиг.40В демонстрируется последовательность вариабельного домена тяжелой цепи гуманизированного 2H7.v114 (SEQ ID NO: 33); и на фиг.40С демонстрируется последовательность полноразмерной тяжелой цепи гуманизированного 2H7.v114 (SEQ ID NO: 34), с нумерацией по Kabat остатков вариабельных доменов и Eu-нумерацией остатков константных доменов.
На фиг.41 демонстрируется распределение пациентов клинического испытания REFLEX на 56 неделе, в том числе проводящееся лечение подгрупп пациентов, выбираемых из групп лечения и плацебо клинического испытания фазы III REXLEX.
На фиг.42 демонстрируется изменение рентгенографических конечных положений на 56 неделе.
На фиг.43 демонстрируется среднее изменение общего показателя Sharp-Genant на протяжении времени.
На фиг.44 демонстрируется кумулятивное распределение изменения общего показателя Sharp-Genant.
На фиг.45 демонстрируются анализы восприимчивости: изменение общего показателя Sharp-Genant.
На фиг.46 демонстрируются пациенты без рентгенографических изменений на 56 неделе.
Подробное описание предпочтительных вариантов осуществления настоящего изобретения
I. Определения
«В-клетка» представляет собой лимфоцит, который созревает в костном мозге и включает не обученную В-клетку, В-клетку памяти или эффекторную В-клетку (плазмациты). Здесь В-клетка представляет собой нормальную или не злокачественную В-клетку.
«Маркер поверхности В-клетки» или «поверхностный антиген В-клетки» здесь представляет собой антиген, экспрессируемый на поверхности В-клетки, на который может быть нацелен связывающийся с ним антагонист. Примеры маркеров поверхности В-клетки включают маркеры поверхности лейкоцитов CD10, CD19, CD20, CD21, CD22, CD23, CD24, CD37, CD40, CD53, CD72, CD73, CD74, CDw75, CDw76, CD77, CDw78, CD79a, CD79b, CD80, CD81, CD82, CD83, CDw84, CD85 и CD86 (в отношении описаний смотри The Leukocyte Antigen Facts Book, 2nd Edition. 1997, ed. Barclay et al. Academic Press, Harcourt Brace & Co., New York). Другие маркеры поверхности В-клетки включают RP105, FcRH2, CR2 B-клетки, CCR6, P2X5, HLA-DOB, CXCR5, FCER2, BR3, Btig, NAG14, SLGC16270, FcRH1, IRTA2, ATWD578, FcRH3, IRTA1, FcRH6, BCMA и 239287. Представляющий особый интерес маркер поверхности В-клетки предпочтительно экспрессируется на В-клетках по сравнению с другими не-В-клеточными тканями млекопитающего и может экспрессироваться как на предшественниках В-клеток, так и на зрелых В-клетках. Здесь предпочтительными маркерами поверхности В-клетки являются CD20 и CD22.
Антиген «CD20», или «CD20», представляет собой не гликозилированный фосфопротеин с молекулярной массой приблизительно 35 кДа, обнаруживаемый на поверхности свыше 90% В-клеток периферической крови или лимфоидных органов. CD20 присутствует как на нормальных В-клетках, так и на злокачественных В-клетках, но не экспрессируется на стволовых клетках. Другие встречающиеся в литературе названия CD20 включают «ограниченный В-лимфоцитами антиген» и «Вр35». Антиген CD20 описывается, например, Clark и др. (Proc. Natl. Acad. Sci. (USA) 82: 1766 (1985)).
Антиген «CD22», или «CD22», также известный как BL-CAM или Lyb8, представляет собой интегральный мембранный гликопротеин типа 1 с молекулярной массой приблизительно 130 (восстановленный)-140 кДа (не восстановленный). Он экспрессируется и в цитоплазме, и в клеточной мембране В-лимфоцитов. Антиген CD22 появляется рано при дифференциации В-лимфоцитов приблизительно в тот же период, что и антиген CD19. В отличие от других В-клеточных маркеров экспрессия CD20 в мембране ограничена поздними периодами дифференциации, заключенными между зрелыми В-клетками (CD22+) и плазмацитами (CD22-). Антиген CD22 описывается, например, Wilson и др. (J. Exp. Med. 173: 137 (1991)) и Wilson и др. (J. Immunol. 150: 5013 (1993)).
«Антагонист» представляет собой молекулу, которая при связывании с CD20 на В-клетках уничтожает или истощает В-клетки у млекопитающего и/или мешает одной или нескольким функциям В-клеток, например, путем уменьшения или предотвращения гуморального ответа, индицируемого В-клеткой. Предпочтительно антагонист способен истощать В-клетки (т.е. снижать уровни циркулирующих В-клеток) у млекопитающего, обработанного им. Такое истощение можно получить через различные механизмы, такие как ADCC (антителозависимая клеточноопосредованная цитотоксичность) и/или CDC (комплементзависимая цитотоксичность), ингибирование пролиферации В-клеток и/или индукция гибели В-клеток (например, через апоптоз). Антагонисты, включаемые в пределы объема настоящего изобретения, включают антитела, синтетические пептиды и пептиды с природными последовательностями, иммуноадгезины и антагонисты в виде небольших молекул, которые связываются с CD20, необязательно конъюгированные или слитые с другой молекулой. Предпочтительный антагонист включает антитело.
Здесь «антагонист в виде антитела» представляет собой антитело, которое при связывании с маркером поверхности В-клетки на В-клетках уничтожает или истощает В-клетки у млекопитающего и/или мешает одной или нескольким функциям В-клеток, например, путем уменьшения или предотвращения гуморального ответа, индуцируемого В-клеткой. Предпочтительно антагонист в виде антитела способен истощать В-клетки (т.е. снижать уровни циркулирующих В-клеток) у млекопитающего, обработанного им. Такое истощение можно получить через различные механизмы, такие как ADCC и/или CDC, ингибирование пролиферации В-клеток и/или индукция гибели В-клеток (например, через апоптоз).
Здесь термин «антитело» используется в самом широком значении и, в частности, включает моноклональные антитела, поликлональные антитела, мультиспецифические антитела (например, биспецифические антитела), образованные из по крайней мере двух интактных антител, и фрагменты антител при условии, что они проявляют желаемую биологическую активность.
«Фрагменты антител» включают часть интактного антитела, предпочтительно включающую его антигенсвязывающий район. Примеры фрагментов антител включают фрагменты Fab, Fab', F(ab')2 и Fv, диатела, линейные антитела, одноцепочечные молекулы антител и мультиспецифические антитела, образованные из фрагментов антител.
Для целей настоящего изобретения «интактное антитело» - это антитело, включающее вариабельные домены тяжелой и легкой цепи, а также Fc-фрагмент.
«Антитело, которое связывается с маркером поверхности В-клетки» представляет собой молекулу, которая при связывании с маркером поверхности В-клетки уничтожает или истощает В-клетки у млекопитающего и/или мешает одной или нескольким функциям В-клеток, например, путем уменьшения или предотвращения гуморального ответа, индицируемого В-клеткой. Предпочтительно антитело способно истощать В-клетки (т.е. снижать уровни циркулирующих В-клеток) у млекопитающего, обработанного им. Такое истощение можно получить через различные механизмы, такие как ADCC и/или CDC, ингибирование пролиферации В-клеток и/или индукция гибели В-клеток (например, через апоптоз). В одном предпочтительном варианте осуществления настоящего изобретения маркером поверхности В-клетки является CD20 или CD22, так что антителом, которое связывается с маркером поверхности В-клетки, является антитело, которое связывается с CD20 или CD22, соответственно, или «антитело против CD20» или «антитело против CD22», соответственно. Примеры антител против CD22 включают антитела, описанные в ЕР 1476120 (Tedder and Tuscano), EP 1485130 (Tedder) и EP 1504035 (Popplewell et all.), а также антитела, описанные в US 2004/0258682 (Leung et al.). В еще более предпочтительном варианте осуществления настоящего изобретения антителом является антитело против CD20. Особенно предпочтительный вариант - это антитело против CD20 или CD22, предпочтительно антитело против CD20.
Примеры антител против CD20 включают «С2В8», которое в настоящее время называют «ритуксимаб» (“RITUXAN®/MABTHERA®”) (патент США № 5736137); иттрий-[90]-меченное 2В8 крысиное антитело, обозначенное “Y2B8” или “Ibritumomab Tiuxetan” (ZEVALIN®), коммерчески доступное от Biogen Idec, Inc. (например, патент США № 5736137; 2В8, депонированное в АТСС (Американскую коллекцию типовых культур) под номером доступа НВ 11388 22 июня 1993); крысиный IgG2a “B1”, также называемый ”Tositumomab”, необязательно меченный 131I с образованием антитела “131I-B1” или «иод-131-тоситумомаб» (BEXXARTM), коммерчески доступные от Corixa (смотри также, например, патент США № 5595721); крысиное моноклональное антитело “1F5” (например, Press et al. Blood 69(2): 584-591 (1987)) и его варианты, включающие «заплатанное каркасными областями» или гуманизированное антитело 1F5 (например, WO 2003/002607, Leung, S., депонирование в АТСС HB 96450); крысиное антитело 2Н7 и химерное антитело 2Н7 (например, патент США № 5677189); гуманизированное антитело 2Н7 (например, WO 2004/056312 (Lowman et al) и как изложено ниже); полностью человеческое антитело с высокой аффинностью, нацеленное на молекулу CD20 в клеточной мембране В-клеток, HUMAX-CD20TM (Genmab, Дания; смотри, например, Glennie and van de Winkel, Drug Discovery Today 8: 503-510 (2003) и Cragg et al., Blood 101: 1045-1052 (2003)); моноклональные антитела человека, описанные в WO 2004/035607 и WO 2005/103081 (Teeling et al., GenMab/Medarex); антитела, имеющие сложные N-гликозид-связанные сахарные цепи, связанные с Fc-фрагментом, описанные в US 2004/0093621 (Shitara et al.); моноклональные антитела и антигенсвязывающие фрагменты, связывающиеся с CD20 (например, WO 2005/000901, Tedder et al.), такие как НВ20-3, НВ20-4, НВ20-25 и МВ20-11; одноцепочечные белки, связывающиеся с CD20 (например, US 2005/0186216 (Ledbetter and Hayden-Ledbetter); US 2005/0202534 (Hayden-Ledbetter and Ledbetter); US 2005/0202028 (Hayden-Ledbetter and Ledbetter); US 2005/0202023 (Hayden-Ledbetter and Ledbetter) - Trubion Pharm. Inc.); CD20-связывающие молекулы, такие как серии антител АМЕ, например антитела АМЕ-33ТМ, описанные, например, в WO 2004/103404 и US 2005/0025764 (Watkins et al., Applied Molecular Evolution, Inc.) и антитела против CD20 с мутациями в Fc, описанные, например, в WO 2005/070963 (Allan et al., Applied Molecular Evolution, Inc.); CD20-связывающие молекулы, такие как молекулы, описанные в WO 2005/016969 и US 2005/0069545 (Carr et al.); биспецифические антитела, описанные, например, в WO 2005/014618 (Chang et al.); гуманизированные моноклональные антитела LL2, описанные, например, в US 2005/0106108 (Leung and Hansen; Immunomedics); химерные или гуманизированные антитела B-Ly1 против CD20, описанные, например, в WO 2005/044859 и US 2005/0123546 (Umana et al., GlycArt Biotechnology AG); антитело А20 или его варианты, такие как химерное или гуманизированное антитело А20 (сА20, hА20, соответственно) и IMMUN-106 (например, US 2003/0219433, Immunomedics); и моноклональные антитела L27, G28-2, 93-1B3, B-C1 или NU-B2, доступные от International Leukocyte Typing Workshop (например, Valentine et al., In: Leukocyte Typing III (McMichael, Ed., p. 440, Oxford University Press (1987)). Предпочтительными здесь антителами против CD20 являются химерные, гуманизированные антитела или антитела человека против CD20, более предпочтительно ритуксимаб, гуманизированное антитело 2Н7, химерное или гуманизированное антитело А20 (Immunomedics), антитело человека против CD20 HUMAX-CD20TM (Genmab) и иммуноглобулины/белки, связывающиеся с CD20, (Trubion Pharm. Inc.).
Термины «ритуксимаб» или «RITUXAN®» здесь относится к полученному с помощью методов генной инженерии химерному крысиному/человеческому моноклональному антителу, направленному против антигена CD20 и обозначаемому “C2B8” в патенте США № 5736137, в том числе его фрагментам, сохраняющим способность связывать CD20.
Исключительно для целей настоящего изобретения и если не указано иное, «гуманизированное антитело 2Н7» относится к гуманизированному антителу против CD20, или его антигенсвязывающему фрагменту, включающему одну, две, три, четыре, пять или шесть следующих последовательностей CDR:
последовательность CDR L1 RASSSVSYXH, где Х представляет собой M или L (SEQ ID NO: 35), например, SEQ ID NO: 4 (фиг.32А),
последовательность CDR L2 SEQ ID NO: 5 (фиг.32А),
последовательность CDR L3 QQWXFNPPT, где Х представляет собой S или A (SEQ ID NO: 36), например, SEQ ID NO: 6 (фиг.32А),
последовательность CDR H1 SEQ ID NO: 10 (фиг.32B),
последовательность CDR H2 AIYPGNGXTSYNQKFKG, где Х представляет собой D или A (SEQ ID NO: 37), например, SEQ ID NO: 11 (фиг.32B), и
последовательность CDR H3 VVYYSXXYWYFDV, где Х в положении 6 представляет собой N, A, Y, W или D, а Х в положении 7 представляет собой S или R (SEQ ID NO: 38), например, SEQ ID NO: 12 (фиг.32B).
Здесь гуманизированные антитела 2Н7 включают антитела с аминокислотными последовательностями тяжелой цепи, содержащими С-концевой лизин, и с аминокислотными последовательностями тяжелой цепи, не содержащими С-концевой лизин. Вышеуказанные последовательности CDR обычно присутствуют в пределах последовательностей каркасных областей вариабельных областей легкой и тяжелой цепей, таких как по существу консенсусные остатки FR человека подгруппы I легкой цепи каппа человека (VL6I) и по существу консенсусные остатки FR человека подгруппы III тяжелой цепи (VHIII). Смотри также WO 2004/056312 (Lowman et al.).
Вариабельная область тяжелой цепи может быть соединена с константной областью цепи IgG человека, которая может быть, например, областью IgG1 или IgG3, в том числе константными областями природной последовательности и не природной последовательности.
В предпочтительном варианте осуществления настоящего изобретения такое антитело включает последовательность вариабельного домена тяжелой цепи SEQ ID NO: 8 (v16, как показано на фиг.32В), при этом оно необязательно также включает последовательность вариабельного домена легкой цепи SEQ ID NO: 2 (v16, как показано на фиг.32А), которое необязательно включает одну или несколько аминокислотных замен в положениях 56, 100 и/или 100а, например, D56A, N100A или N100Y и/или S100aR в вариабельном домене тяжелой цепи и одну или несколько аминокислотных замен в положениях 32 и/или 92, например M32L и/или S92A в вариабельном домене легкой цепи. Предпочтительно антитело является интактным антителом, включающим аминокислотную последовательность легкой цепи SEQ ID NO: 13 или 30 и аминокислотную последовательность тяжелой цепи SEQ ID NO: 14, 15, 29, 31, 34 или 39, при этом последовательность SEQ ID NO: 39 приведена ниже.
Предпочтительным гуманизированным антителом 2Н7 является окрелизумаб (Genentech, Inc.).
Антитело здесь может дополнительно включать по крайней мере одну аминокислотную замену в Fc-фрагменте, которая увеличивает активность ADCC, например антитело, в котором аминокислотные замены находятся в положениях 298, 333 и 334, предпочтительно S298A, E333A и K334A, используя Eu-нумерацию остатков тяжелой цепи. Смотри также патент США № 6737056 (L. Presta).
Любое из этих антител может включать по крайней мере одну замену в Fc-фрагменте, которая увеличивает связывание с FcRn или время полужизни в сыворотке, например замену в положении 434 тяжелой цепи, такую как N434W. Смотри также патент США № 6737056 (L. Presta).
Любое из этих антител может, кроме того, включать по крайней мере одну замену в Fc-фрагменте, которая увеличивает активность CDC, например, антитело, включающее по крайней мере замену в положении 326, предпочтительно K326A или K326W. Смотри также патент США № 6528624 (Idusogie et al.).
Некоторые предпочтительные гуманизированные варианты 2Н7 являются такими, которые включают вариабельный домен легкой цепи SEQ ID NO: 2 и вариабельный домен тяжелой цепи SEQ ID NO: 8, в том числе варианты с заменами в Fc-фрагменте (если присутствует) или без них, и такими, которые включают вариабельный домен тяжелой цепи с заменами в SEQ ID NO: 8: N100A, или D56А и N100A, или D56A, N100Y и S100aR; и вариабельный домен тяжелой цепи с заменами в SEQ ID NO: 2: M32L, или S92A, или M32L и S92A.
М34 в вариабельном домене тяжелой цепи 2Н7.v16 был идентифицирован в качестве возможного источника стабильности антитела и является другим возможным кандидатом для замены.
Резюмируя некоторые различные предпочтительные варианты осуществления настоящего изобретения, вариабельные области вариантов, основанных на 2Н7.v16, включают аминокислотные последовательности v16 за исключением положений аминокислотных замен, указанных в таблице ниже. Если не указано иное, варианты 2Н7 будут иметь такую же легкую цепь, как и v16.
Примеры вариантов гуманизированного антитела 2Н7
Одно предпочтительное гуманизированное антитело 2Н7 включает последовательность вариабельного домена легкой цепи 2H7.v16:

и последовательность вариабельного домена тяжелой цепи 2H7.v16:

Когда гуманизированное антитело 2H7.v16 является интактным антителом, оно может включать аминокислотную последовательность легкой цепи:

и аминокислотную последовательность тяжелой цепи SEQ ID NO: 14 или:

Другое предпочтительное гуманизированное антитело 2Н7 включает последовательность вариабельного домена легкой цепи 2H7.v511:

и последовательность вариабельного домена тяжелой цепи 2H7.v511:

Когда гуманизированное антитело 2H7.v511 является интактным антителом, оно может включать аминокислотную последовательность легкой цепи:

и аминокислотную последовательность тяжелой цепи SEQ ID NO: 31 или:

Смотри фиг.38 и 39, на которых выровнены зрелые легкая и тяжелая цепи, соответственно, гуманизированного антитела 2H7.v511 с гуманизированным антителом 2H7.v16, используя последовательность с С-концевым лизином для тяжелой цепи.
Когда гуманизированное антитело 2H7.v31 является интактным антителом, оно может включать аминокислотную последовательность легкой цепи:

и аминокислотную последовательность тяжелой цепи SEQ ID NO: 15 или:

или

Предпочтительным вариантом осуществления здесь является случай, когда антитело является гуманизированным антителом 2Н7, включающим последовательности вариабельных доменов SEQ ID NO: 2 и 8 (вариант 16). Другим предпочтительным вариантом осуществления здесь является случай, когда антитело является гуманизированным антителом 2Н7, включающим последовательности вариабельных доменов SEQ ID NO: 39 и 40 (вариант 511). Кроме того, предпочтительным является случай, когда антитело является гуманизированным антителом 2Н7, включающим последовательности вариабельных доменов SEQ ID NO: 32 и 33 (смотри фиг.40, вариант 114), таким как антитело, включающее вариабельный домен легкой цепи SEQ ID NO: 32 и аминокислотную последовательность тяжелой цепи SEQ ID NO: 34. Кроме того, предпочтительным является случай, когда антитело является гуманизированным антителом 2Н7, включающим вариабельный домен тяжелой цепи с заменами: N100A, или D56А и N100A, или D56A, N100Y и S100aR - в SEQ ID NO: 8 и вариабельный домен тяжелой цепи с заменами: M32L, или S92A, или M32L и S92A - в SEQ ID NO: 2.
«Антителозависимая клеточно-опосредованная цитотоксичность» или «ADCC» относится к клеточно-опосредованной реакции, в которой неспецифические цитотоксические клетки, экспрессирующие рецепторы для Fc (FcR) (например, природные киллеры (NK), нейтрофилы и макрофаги) узнают связанное антитело на клетке-мишени и впоследствии вызывают лизис клетки-мишени. Основные клетки, опосредующие ADCC, NK, экспрессируют только FcγRIII, в то время как моноциты экспрессируют FcγRI, FcγRII и FcγRIII. Резюме экспрессии FcR на гемопоэтических клетках - это таблица 3 на странице 464 Ravetch and Kinet, Annu. Rev. Immunol. 9: 477-492 (1991). Для оценки активности ADCC представляющей интерес молекулы можно провести анализ ADCC, например, такой, который описан в патенте США № 5500362 или 5821337. Применимые для таких анализов эффекторные клетки включают мононуклеарные клетки периферической крови (PBMC) или природные киллеры (NK). Альтернативно или дополнительно, активность ADCC представляющей интерес молекулы можно оценить in vivo, например, в модели на животном, такой как модель, описанная Clynes et al. PNAS (USA) 95: 652-656 (1998).
«Эффекторными клетками человека» являются лейкоциты, которые экспрессируют один или несколько FcR и осуществляют эффекторные функции. Предпочтительно клетки экспрессируют FcγRIII и осуществляют эффекторную функцию ADCC. Примеры лейкоцитов человека, опосредующих ADCC, включают мононуклеарные клетки периферической крови (PBMC), природные киллеры (NK), моноциты, цитотоксические Т-клетки и нейтрофилы, при этом предпочтительными являются PBMC и NK.
Термины «рецептор для Fc» или «FcR» используются для описания рецептора, который связывается с Fc-фрагментом антитела. Предпочтительным FcR является FcR человека с природной последовательностью. Кроме того, предпочтительным FcR является FcR, который связывает антитело IgG (гамма-рецептор) и включает рецепторы подклассов FcγRI, FcγRII и FcγRIII, в том числе аллельные варианты и подвергнутые альтернативному сплайсингу формы этих рецепторов. Рецепторы FcγRII включают FcγRIIА (активирующий рецептор) и FcγRIIВ (ингибирующий рецептор), имеющие сходные аминокислотные последовательности, которые различаются, главным образом, в их цитоплазматических доменах. Активирующий рецептор FcγRIIА содержит мотив активации на основе тирозина иммунорецептора (ITAM) в своем цитоплазматическом домене. Ингибирующий рецептор FcγRIIВ содержит мотив ингибирования на основе тирозина иммунорецептора (ITIM) в своем цитоплазматическом домене (смотри Daëron, Annu. Rev. Immunol. 15: 203-234 (1997)). Обзор FcR приведен Ravetch и Kinet (Annu. Rev. Immunol. 9: 457-492 (1991)); Capel и др. (Immunomethods 4: 25-34 (1994)) и de Haas и др. (J. Lab. Clin. Med. 126: 330-341 (1995)). Другие FcR, в том числе FcR, которые будут идентифицированы в будущем, охватываются термином «FcR» здесь. Этот термин также включает неонатальный рецептор, FcRn, который ответственен за перенос IgG матери плоду (Guyer et al., J. Immunol. 117: 587 (1976) и Kim et al., J. Immunol. 24: 249 (1994)).
«Комплементзависимая цитотоксичность» или «CDC» относится к способности молекулы лизировать мишень в присутствии комплемента. Путь активации комплемента инициируется связыванием первого компонента системы комплемента (C1q) с молекулой (например, антителом), образующей комплекс с родственным антигеном. Для оценки активации комплемента можно выполнить анализ CDC, например, как описано Gazzano-Santoro и др. (J. Immunol. Methods 202: 163 (1996)).
«Ингибиторные в отношении роста» антитела представляют собой антитела, предотвращающие или уменьшающие пролиферацию клетки, экспресирующей антиген, с которым связывается антитело. Например, антитело может предотвращать или уменьшать пролиферацию В-клеток in vitro и/или in vivo.
Антитела, «индуцирующие апоптоз», представляют собой антитела, индуцирующие запрограммированную гибель клетки, например В-клетки, определяемую с помощью стандартных анализов апоптоза, таких как связывание аннексина V, фрагментация ДНК, сморщивание клетки, дилатация эндоплазматического ретикулума, фрагментация клетки и/или образование мембранных везикул (называемых апоптозными телами).
«Природные антитела» обычно представляют собой гетеротетрамерные гликопротеины с молекулярной массой приблизительно 150000 дальтон, состоящие из двух идентичных легких (L) цепей и двух идентичных тяжелых (H) цепей. Каждая легкая цепь связана с тяжелой цепью одной ковалентной дисульфидной связью, в то время как число дисульфидных связей варьирует среди тяжелых цепей различных изотипов иммуноглобулинов. Каждая из тяжелой и легкой цепи также имеет регулярно расположенные внутрицепочечные дисульфидные мостики. Каждая тяжелая цепь имеет на одном конце вариабельный домен (VH), за которым следует ряд константных доменов. Каждая легкая цепь имеет вариабельный домен (VL) на одном конце и константный домен на другом ее конце; константный домен легкой цепи находится напротив первого константного домена тяжелой цепи, а вариабельный домен легкой цепи находится напротив вариабельного домена тяжелой цепи. Полагают, что специфические аминокислотные остатки образуют границу контакта между вариабельными доменами легкой и тяжелой цепей.
Термин «вариабельный» относится к тому факту, что последовательности определенных частей вариабельных доменов сильно отличаются среди антител и используются для связывания и определения специфичности каждого конкретного антитела в отношении его конкретного антигена. Однако вариабельность не распределена равномерно по вариабельным доменам антитела. Она сконцентрирована в трех сегментах, называемых гипервариабельными участками вариабельных доменов как легкой цепи, так и тяжелой цепи. Более высококонсервативные части вариабельных доменов называются каркасными областями (FR). Вариабельные домены природных тяжелых и легких цепей включают, каждая, четыре FR, в значительной степени принимающие β-складчатую конформацию, соединенные тремя гипервариабельными участками, которые образуют петли, соединяющие и в некоторых случаях отчасти образующие β-складчатую структуру. Гипервариабельные участки каждой цепи удерживаются вместе на близком расстоянии с помощью FR и вместе с гипервариабельными участками другой цепи вносят вклад в образование антигенсвязывающего сайта антител (смотри, Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991)). Константные домены не вовлечены непосредственно в связывание антитела с антигеном, но проявляют различные эффекторные функции, такие как участие антитела в ADCC.
В результате расщепления антител папаином получают два идентичных антигенсвязывающих фрагмента, называемых фрагменты “Fab”, каждый с единственным антигенсвязывающим сайтом, и остаточный фрагмент “Fc”, название которого отражает его способность легко кристаллизоваться. Обработка пепсином дает фрагмент “F(ab)2”, имеющий два антигенсвязывающих сайта и способный образовывать поперечные связи с антигеном.
“Fv” является минимальным фрагментом антитела, который содержит полный узнающий антиген и антигенсвязывающий сайт. Этот фрагмент состоит из димера одного вариабельного домена тяжелой цепи и одного вариабельного домена легкой цепи в тесной, нековалентной связи. Фрагмент находится в такой конфигурации, что три гипервариабельных участка каждого вариабельного домена взаимодействуют с определением антигенсвязывающего сайта на поверхности димера VH-VL. Вместе шесть гипервариабельных участков придают антителу специфичность в отношении связывания антигена. Однако даже единственный вариабельный домен (или половина Fv, включающая только три гипервариабельных участка, специфичных в отношении антигена) обладает способностью узнавать и связывать антиген, хотя с более низкой аффинностью, чем полный связывающий сайт.
Фрагмент Fab также содержит константный домен легкой цепи и первый константный домен (СН1) тяжелой цепи. Фрагменты Fab' отличаются от фрагментов Fab добавочным содержанием небольшого числа остатков с С-конца СН1-домена тяжелой цепи, в том числе одного или нескольких цистеинов от шарнирной области антитела. Здесь Fab'-SH является обозначением Fab', в котором остаток (остатки) цистеина константных доменов несут по крайней мере одну свободную тиольную группу. Фрагменты антитела F(ab)2 первоначально были получены в виде пары фрагментов Fab', имеющих шарнирные цистеины между собой. Также известны другие химические взаимодействия фрагментов антител.
«Легкие цепи» антител (иммуноглобулинов) любого вида позвоночных можно отнести к одному из двух четко отличимых типов, называемых каппа (κ) и лямбда (λ) на основе аминокислотных последовательностей их константных доменов.
В зависимости от аминокислотной последовательности константного домена тяжелых цепей антитела можно отнести к различным классам. Существует пять основных классов интактных антител: IgA, IgD, IgE, IgG и IgM - и некоторые из них можно, кроме того, подразделить на подклассы (изотипы), например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2. Константные домены тяжелой цепи, соответствующие различным классам антител, называют α, δ, ε, γ и µ, соответственно. Субъединичные структуры и пространственные конфигурации различных классов иммуноглобулинов хорошо известны.
Фрагменты антител «одноцепочечные Fv» или «scFv» включают домены VH и VL антитела, где эти домены присутствуют в одной полипептидной цепи. Предпочтительно полипептид Fv, кроме того, включает полипептидный линкер между доменами VH и VL, который предоставляет возможность scFv образовывать требуемую для связывания антигена структура. В отношении обзора scFv смотри Plückthun in The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., Springer-Verlag, New York, pp. 269-315 (1994).
Термин «диатела» относится к небольшим фрагментам антител с двумя антигенсвязывающими сайтами, включающим вариабельный домен тяжелой цепи (VH), соединенный с вариабельным доменом легкой цепи (VL) в одной и той же полипептидной цепи (VH-VL). В результате использования линкера, являющегося слишком коротким для того, чтобы сделать возможным спаривание двух доменов одной и той же цепи, домены вынуждены спариваться с комплементарными областями другой цепи и образовывать два антигенсвязывающих сайта. Более полно диатела описываются, например, в ЕР 404097, WO 93/11161 и Hollinger et al., Proc. Natl. Acad. Sci. USA, 90: 6444-6448 (1993).
Используемый здесь термин «моноклональное антитело» относится к антителу, получаемому из популяции по существу гомогенных антител, т.е. индивидуальные антитела, образующие популяцию, являются идентичными и/или связываются с одним и тем же эпитопом, за исключением возможных вариантов, которые могут возникнуть во время продукции моноклонального антитела, при этом такие варианты, как правило, присутствуют в небольших количествах. В противоположность препаратам поликлональных антител, которые обычно включают различные антитела, направленные против различных детерминант (эпитопов), каждое моноклональное антитело направлено против единственной детерминанты на антигене. Помимо их специфичности моноклональные антитела выгодны тем, что они не содержат примесей других иммуноглобулинов. Атрибут «моноклональное» показывает, что свойством антитела является то, что его получают из по-существу гомогенной популяции антител и его не следует рассматривать в качестве требования продукции антитела каким-либо конкретным способом. Например, моноклональные антитела, получаемые в соответствии с настоящим изобретением, можно получить с помощью гибридомной технологии, впервые описанной Kohler и др. (Nature, 256: 495 (1975)), или методов рекомбинантных ДНК (смотри, например, патент США № 4816567). «Моноклональные антитела» можно также выделить из фаговых библиотек антител, используя, например, методы, описанные Clackson и др. (Nature, 352: 624-628 (1991)) и Marks и др. (J. Mol. Biol., 222: 581-597 (1991)).
Моноклональные антитела здесь, в частности, включают «химерные» антитела (иммуноглобулины), в которых часть тяжелой и/или легкой цепи идентична или гомологична соответствующим последовательностям в антителах, происходящих из конкретных видов или принадлежащих конкретному классу или подклассу антител, в то время как остальная часть цепи (ей) идентична или гомологична соответствующим последовательностям в антителах, происходящих из других видов или принадлежащих другому классу или подклассу антител, а также фрагменты таких антител при условии, что проявляют желаемую биологическую активность (патент США № 4816537; Morrison et al., Proc. Natl. Acad. Sci. USA, 81: 6851-6855 (1984)). Представляющие здесь интерес химерные антитела включают «приматизированные» антитела, включающие антигенсвязывающие последовательности вариабельных доменов, происходящие из не являющегося человеком примата (например, обезьяны Нового Света, такой как бабуин, макак резус или яванский макак), и последовательности константных областей человека (патент США № 5693780).
«Гуманизированные» формы нечеловеческих (например, крысиных) антител являются химерными антителами, которые содержат минимальную последовательность, происходящую из нечеловеческого иммуноглобулина. В большинстве случаев гуманизированные антитела представляют собой иммуноглобулины человека (реципиентное антитело), в котором остатки из гипервариабельного участка реципиента заменены остатками из гипервариабельного участка иммуноглобулинов не являющихся человеком видов (донорского антитела), таких как мышь, крыса, кролик или не являющийся человеком примат, имеющих желаемую специфичность, аффинность и качество. В некоторых случаях остатки каркасных областей (FR) иммуноглобулина человека заменяют соответствующими нечеловеческими остатками. Кроме того, гуманизированные антитела могут включать остатки, не обнаруживаемые в реципиентом антителе или донорском антителе. Такие модификации осуществляют для дальнейшего улучшения характеристики антитела. Как правило, гуманизированное антитело будет включать по существу все из по крайней мере одного и обычно двух вариабельных доменов, в которых все или по существу все из гипервариабельных петлевых фрагментов соответствуют таковым из нечеловеческого иммуноглобулина, и все или по существу все из FR являются FR последовательности иммуноглобулина человека, за исключением замены (замен) в FR, отмеченных выше. Гуманизированное антитело необязательно также будет включать по крайней мере часть константной области иммуноглобулина, обычно иммуноглобулина человека. В отношении дальнейших подробностей смотри Jones et al., Nature 321: 522-525 (1986); Riechmann et al., Nature 332: 323-329 (1988); и Presta, Curr. Op. Struct. Biol. 2: 593-586 (1992)).
Используемый здесь термин «гипервариабельный участок» относится к аминокислотным остаткам антитела, которые ответственны за связывания антигена. Гипервариабельный участок включает аминокислотные остатки из «определяющего комплементарность района» или «CDR» (например, остатки 24-34 (L1), 50-56 (L2) и 89-97 (L3) в вариабельном домене легкой цепи и 31-34 (Н1), 50-65 (Н2) и 95-102 (Н3) в вариабельном домене тяжелой цепи (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD. (1991)) и/или остатки из «гипервариабельного петлевого фрагмента» (например, остатки 26-32 (L1), 50-52 (L2) и 91-96 (L3) в вариабельном домене легкой цепи и 26-32 (Н1), 53-55 (Н2) и 96-101 (Н3) в вариабельном домене тяжелой цепи (Clothia and Lesk J. Mol. Biol. 196: 901-917 (1987)). Остатки «каркасной области» или «FR» представляют собой остатки вариабельного домена, отличные от остатков гипервариабельного участка, определенного здесь.
«Не конъюгированное антитело» является антителом (как здесь определено), которое не конъюгировано с гетерологичной молекулой, такой как цитотоксическая составляющая, полимер или радиоактивная метка.
«Изолированное» антитело представляет собой антитело, которое было идентифицировано и/или отделено от компонента его природного окружения. Компоненты-примеси его природного окружения являются веществами, которые мешают диагностическим или терапевтическим применениям антитела, и могут включать ферменты, гормоны и другие белковоподобные или небелковоподобные растворенные вещества. В предпочтительных вариантах осуществления настоящего изобретения антитело будет очищено (1) до более чем 95% по весу антитела, как определено с помощью метода Лоури, и наиболее предпочтительно до более чем 99% по весу, (2) до степени, достаточной для получения по крайней мере 15 остатков N-концевой или внутренней аминокислотной последовательности с помощью использования секвенатора, рабочим элементом которого является вращающийся сосуд, или (3) до гомогенности при электрофоре в SDS-ПААГ в восстанавливающих или невостанавливающих условиях, используя кумасси голубой или предпочтительно окрашивание серебром. Выделенное антитело включает антитело in situ внутри рекомбинантных клеток, поскольку по крайней мере один компонент природного окружения антитела не присутствует. Обычно, однако, выделенное антитело получают с помощью по крайней одной стадии очистки.
Термин «повреждение сустава» используется в самом широком значении и относится к повреждению или частичному или полному разрушению любой части одного или нескольких суставов, в том числе соединительной ткани и хряща, причем повреждение включает структурное и/или функциональное повреждение любого происхождения и может вызывать или может не вызывать боль в суставе/артралгию. Оно включает, без ограничения, повреждение сустава, сопровождаемое или являющееся результатом воспалительного заболевания суставов, а также невоспалительного заболевания суставов. Это повреждение может быть вызвано любым состоянием, таким как аутоиммунное заболевание, такое как волчанка (например, системная красная волчанка), артрит (например, острый и хронический артрит, ревматоидный артрит, в том числе начинающийся в юношестве ревматоидный артрит, ювенильный идиопатический артрит (JIA) или ювенильный RA (JRA), и стадиями, такими как ревматоидный синовит, подагрический артрит, острый иммунологический артрит, хроническое воспаление суставов, дегенеративный артрит, индуцированный коллагеном типа II артрит, инфекционный артрит, септический артрит, артрит Лима, пролиферативный артрит, псориатический артрит, болезнь Стилла, вертебральный артрит, остеоартрит, артрит хронический прогрессирующий, артрит деформирующий, полиартрит хронический первичный, реактивный артрит, климактерический артрит, эстроген-обедненный артрит и алкилозирующий спондилоартрит/ревматоидный спондилит, ревматическим аутоиммунным заболеванием, отличным от RA, значительным системным поражением, вторичным по отношению к RA, (включающим, но без ограничения, васкулит, фиброз легкого или синдром Фелти), синдромом Шегрена, в частности вторичным таким синдромом, вторичным ограниченным кожей васкулитом с RA, серонегативной спондилоартропатией, болезнью Лима, болезнью воспаленного кишечника, склеродермией, воспалительной миопатией, смешанным заболеванием соединительных тканей, любым синдромом наложения, бурситом, тендинитом, остеомиелитом, инфекционными заболеваниями, включающими грипп, корь, ревматическую атаку, вирусный синдром Эпштейна-Барра, гепатит, эпидемический паротит, краснуху и ветряную оспу, хондромацией надколенника, коллагеновым колитом, аутоиммунными заболеваниями, сопровождаемыми коллагенозом, воспалением сустава, необычайным напряжением или перегрузкой, такими как растяжение или напряжение, повреждением, включающим перелом, подагру, особенно обнаруживаемую в большом пальце стопы, а также может быть вызвано неврологическими заболеваниями, гемофилическими нарушениями (например, гемофилической артропатией), мышечными нарушениями, прогрессирующими нарушениями, заболеваниями кости, заболевания хряща и сосудистыми нарушениями. Для целей настоящего изобретения суставы - это точки контакта между элементами скелета (позвоночных, таких как животное) с частями, которые окружают и поддерживают его, и включают, но без ограничения, например, тазобедренные суставы, суставы между позвонками позвоночника, суставы между позвоночником и тазом (крестцово-подвздошные сочленения), суставы, где сухожилия и связки присоединяются к костям, суставы между ребрами и позвоночником, плечевые суставы, коленные суставы, суставы ног, локтевые суставы, суставы рук, пальцев, голеностопные суставы и суставы пальцев стопы, но особенно суставы рук и ног.
Здесь «индивид» является являющимся человеком индивидом, в том числе пациентом, подходящим для лечения, у которого наблюдается или наблюдался один или несколько признаков, симптомов или других индикаторов повреждения сустава, у которого было диагностировано, или, например, только что диагностировано, или ранее диагностировано, повреждение сустава и теперь наблюдается рецидив, или который подвержен риску развития повреждения сустава, независимо от причины. Индивида могли ранее лечить антителом против CD20 или не лечить. Индивид, подходящий для лечения повреждения сустава, может быть необязательно идентифицирован в качестве индивида, которого подвергли скринингу, например, крови, в отношении увеличения уровней инфильтрующих клеток CD20 или подвергают скринингу с использованием анализа для обнаружения аутоантител, причем продукция аутоантител оценивается качественно, и предпочтительно количественно. Индивида могли ранее не лечить вторым лекарственным средством, используемым, когда начинают лечение, т.е. индивида ранее не лечили, например, иммунодепрессантом, таким как метотрексат, на «базовой линии» (т.е. в контрольный момент времени до введения первой дозы антитела против CD20 в способе лечения настоящего изобретения, такой как день скрининга индивида до того, как начинается лечение). Таких индивидов, как правило, считают кандидатами для лечения таким вторым лекарственным средством.
«Лечение» индивида здесь относится как к терапевтическому лечению, так и к профилактическим или предупредительным мерам. Нуждающиеся в лечении индивиды включают индивидов, уже имеющих повреждение сустава, а также индивидов, у которых повреждение сустава или прогрессирование повреждения сустава должно быть предупреждено. Следовательно, индивид может быть диагностирован в качестве индивида, имеющего повреждение сустава, или он может быть предрасположен или восприимчив к повреждению сустава, или он может иметь ограниченное повреждение сустава, которое, вероятно, будет прогрессировать при отсутствии лечения. Лечение является здесь успешным, если повреждение сустава уменьшается или излечивается, или прогрессирование структурного повреждения сустава останавливается или замедляется по сравнению с тем, что было до введения. Успешное лечение, кроме того, включает полное или частичное предупреждение развития повреждения сустава. Для целей настоящего изобретения замедление или уменьшение повреждения сустава или прогрессирования повреждения сустава эквивалентно остановке, уменьшению или реверсированию повреждения сустава.
«Клиническое улучшение» относится к предупреждению дальнейшего прогрессирования повреждения сустава или любому улучшению в повреждении сустава в результате лечения, как определяется с помощью проверки, отличной от рентгенографической проверки. Таким образом, клиническое улучшение можно, например, определить с помощью оценки числа болезненных или опухших суставов, показателя тяжести оценки псориаза, общей клинической оценки индивида, оценки скорости осаждения эритроцитов или оценки величины уровня С-реактивных белков.
Для целей настоящего изобретения индивид находится в стадии «ремиссии», если он/она не имеют симптомов активного повреждения сустава, таких как симптомы, определяемые с помощью раскрытых здесь способов, и не имеют прогрессирования повреждения сустава, оцененного на базовой линии или в определенный момент времени во время лечения. Индивиды, не находящиеся в ремиссии, включают, например, индивидов, у которых имеет место ухудшение или прогрессирование повреждения сустава. Такие индивиды, у которых имеет место возвращение симптомов, в том числе активное повреждение сустава, представляют собой индивидов, которые перенесли «рецидив» или имели «рецидив».
«Симптом» повреждения сустава - это любой болезненный признак или отклонение от нормальной структуры, функции или ощущения, которые имеются у индивида и указывают на повреждение сустава, такие как те, которые отмечены выше, в том числе болезненные или опухшие суставы.
Выражение «эффективное количество» относится к количеству антитела или антагониста, являющемуся эффективным для лечения повреждения сустава, в том числе количеству, являющемуся эффективным для получения уменьшения повреждения сустава по сравнению с базовой линией до введения такого количества, что определяется с помощью рентгенографической проверки. Эффективное количество других лекарственных средств, таких как вторых лекарственных средств, представляет собой количество такого лекарственного средства, которое является эффективным для лечения повреждения сустава или других нежелательных эффектов, в том числе побочных эффектов или симптомов или других состояний, сопровождающих повреждение сустава, в том числе лежащего в основе заболевания или нарушения.
«Общий модифицированный показатель Sharp» означает показатель, полученный для оценки рентгенограмм с использованием способа согласно Sharp, модифицированного Genant (Am. J. Med., 30: 35-47 (1983)). Первичная оценка будет представлять собой изменение общего показателя Sharp-Genant от скрининга. Показатель Sharp-Genant объединяет показатель эрозии и показатель сужения суставного пространства как рук, так и ног. Повреждение сустава измеряют при этой проверке, определяя показатель по среднему изменению по соотношению «меньше» показателя на базовой линии (когда пациент подвергается скринингу или проверке до первого введения антагониста CD20 здесь).
Используемый здесь термин «ревматоидный артрит» относится к распознаваемому болезненному состоянию, которое можно диагностировать в соответствии с пересмотренными в 2000 г. American Rheumatoid Association критериями для классификации ревматоидного артрита или любыми схожими критериями. Физиологические индикаторы RA включают опухание симметричных суставов, являющееся, как полагают, неизменной характеристикой ревматоидного артрита. Обычно имеет место веретенообразное опухание проксимальных межфаланговых (PIP) суставов рук, а также пястно-фаланговых (MCP) суставов, запястий, локтей, коленок, лодыжек и плюснефаланговых (MTP) суставов, и опухание легко определяется. Боль при пассивном движении является самым чувствительным тестом воспаления сустава, и воспаление и структурная деформация часто ограничивают диапазон движения пораженного сустава. Типичные видимые изменения включают локтевую девиацию пальцев в МСР суставах, гиперэкстезию или гиперфлексию МСР и PIP суставов, сгибательные контрактуры локтевых суставов и подвывихи костей запястья и пальцев стопы. Индивиды с ревматоидным артритом могут быть устойчивы к DMARD, так что DMARD являются не эффективными или не полностью эффективными для лечения симптомов. Кроме того, кандидаты для лечения в соответствии с настоящим изобретением включают тех, которые неадекватно реагировали на предшествующее или текущее лечение ингибиторами TNF, такими как этанерцепт, инфликсимаб и/или адалимумаб, из-за токсичности или неадекватной эффективности (например, этанерцепт в течение 3 месяцев в количестве 25 мг дважды в неделю или по крайней мере 4 инфузии инфликсимаба в количестве 3 мг/кг). Под больным «активным ревматоидным артритом» подразумевается больной с активными, а не латентными симптомами ревматоидного артрита. Индивидами с «активным ревматоидным артритом на ранней стадии» являются индивиды с активным ревматоидным артритом, протекающим, как диагностировано, в течение по крайней мере 8 недель, но не дольше четырех лет, в соответствии с пересмотренными в 1987 г. ACR критериями для классификации RA.
Псориатический артрит (PsA) является воспалительным заболеванием сустава, характеризующимся экстенсивной резорбцией кости. Также как здесь раскрыто, образцы крови больных PsA, в частности больных с эрозиями кости на рентгенограммах простого класса, демонстрируют заметное увеличение предшественников остеокластов (ОСР) по сравнению со здоровыми контролями.
«Воздействие антителом» относится к приведению в контакт или воздействию антителом настоящего изобретения в одной или нескольких дозах на протяжении периода времени, составляющего от приблизительно 1 дня до приблизительно 5 недель. Дозы можно назначать один раз или через постоянные или нерегулярные интервалы на протяжении этого периода воздействия, такие как, например, одна доза еженедельно на протяжении четырех недель или две дозы, разделенные интервалом времени, составляющим приблизительно 13-17 дней. Первоначальное и более позднее воздействие антителом разделены во времени друг от друга, как подробно здесь описывается.
Воздействие, не назначаемое или не предоставляемое до определенного момента времени «от первоначального воздействия» или от любого предшествующего воздействия, означает, что момент времени для второго или более позднего воздействия определяют от момента времени, в который были назначены дозы предшествующего воздействия, если более одной дозы назначалось при этом воздействии. Например, если при первоначальном воздействии назначают две дозы, второе воздействие не назначают до момента времени, составляющего приблизительно 16-54 недель, определяемого от момента времени, когда была назначена первая или вторая доза в пределах этого предшествующего воздействия. Аналогично, если назначают три дозы, второе воздействие можно определить от момента времени назначения первой, второй или третьей дозы в пределах предшествующего воздействия. Предпочтительно «от первоначально воздействия» или от любого предшествующего воздействия определяют от момента времени назначения первой дозы.
Термин «иммунодепрессант», используемый здесь для вспомогательной терапии, относится к веществам, действующим с подавлением иммунной системы млекопитающего, которого здесь подвергают лечению. Они включают вещества, которые подавляют продукцию цитокинов, супрессируют экспрессию аутоантигенов или маскируют антигены МНС. Примеры таких агентов включают 2-амино-6-арил-5-замещенные пиримидины (смотри патент США № 4665077); нестероидные противовоспалительные лекарственные средства (NSAID); ганцикловир, такролимус, глюкокортикоиды, такие как кортизол или альдостерон, противовоспалительные агенты, такие как ингибитор циклооксигеназы, ингибитор 5-липоксигеназы или антагонист рецептора для лейкотриенов; антагонисты пуринов, такие как азатиоприн или микофенолат мофетил (MMF); алкилирующие агенты, такие как циклофосфамид; бромокриптин; даназол; дапсон; глютаральдегид (который маскирует антигены МНС, как описано в патенте США № 4120649); антиидиотипические антитела против антигенов МНС или фрагментов МНС; циклоспорин А; стероиды, такие как кортикостероиды или глюкокортикостероиды или аналоги глюкокортикоидов, например преднизон, метилпреднизолон, в том числе метилпреднизолон натрия сукцинат SOLU-MEDROL® и дексаметазон; ингибиторы дигидрофолатредуктазы, такие как метотрексат (пероральный или подкожный); антималярийные агенты, такие как хлорохин и гидроксихлорохин; сульфасалазин; лефлуномид; антагонисты цитокинов, такие как антитела против цитокинов или антитела против рецепторов для цитокинов, в том числе антитела против интерферона-альфа, -бета или -гамма, антитела против фактора некроза опухолей (TFA)-альфа (инфликсимаб (REMICADE®) или адалимумаб); анти-TFA-альфа иммуноадгезин (этанерцепт); антитела против TFA-бета, антитела против интерлейкина-2 (IL-2) и антитела против рецептора для IL-2, антитела против рецептора для интерлейкина-6 (IL-6) и антагонисты этого рецептора; антитела против LFA-1, в том числе антитела против CD11a и CD18, антитела против L3T4; гетерологичный глобулин против лимфоцитов; пан-Т антитела, предпочтительно антитела против CD3 или CD4/CD4a; растворимый пептид, содержащий связывающий LFF-3 домен (заявка WO 90/08187, опубликованная 26/7/90); стрептокиназа; трансформирующий фактор-бета роста (TGF-бета); стептодорназа; РНК или ДНК хозяина; FK506; RS-61443; хлорамбуцил; дезоксиспергуалин; рапамицин; рецептор Т-клетки (Cohen et al., патент США № 5114721); фрагменты рецептора Т-клетки (Offner et al., Science, 251: 430-432 (1991); WO 90/11294; Ianeway, Nature, 341: 482 (1989); и WO 91/01133); антагонисты BAFF, такие как антитела против BAFF и антитела против BR3 и антагонисты zTNF4 (для обзора смотри Mackay and Mackay, Trends Immunol., 23: 113-5 (2002)); биологические агенты, мешающие передаче сигналов от Т-хелперов, такие как антитела против рецептора для CD40 или антитела против лиганда CD40 (CD154), в том числе блокирующие антитела против лиганда CD40-CD40 (например, Durie et al., Science, 261: 1328-30 (1993); Mohan et al., J. Immunol., 154: 1470-89 (1995)) и CTLA4-Ig (Finck et al., Science, 265: 12225-7 (1994)); и антитела против рецепторов Т-клеток (ЕР 340109), такие как Т10В9. Некоторые иммунодепрессанты здесь также представляют собой DMARD, такие как метотрексат. Примеры предпочтительных иммунодепрессантов здесь включают циклофосфамид, хлорамбуцил, азатиоприн, лефлуномид, MMF или метотрексат.
Термин «цитокин» является общим термином для белков, высвобождаемых одной клеточной популяцией, которые действуют на другую клетку в качестве межклеточных медиаторов. Примерами таких цитокинов являются лимфокины, монокины, интерлейкины (IL), такие как IL-1, IL-1α, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-11, IL-12, IL-15, в том числе rIL-2 PROLEUKIN®; фактор некроза опухолей, такой как TNF-α или TNF-β; и другие полипептидные факторы, включающие LIF и сборный лиганд (KL). Используемый здесь термин «цитокин» включает белки из природных источников или из культуры рекомбинантных клеток и биологически активные эквиваленты цитокинов с природными последовательностями, в том числе синтетически получаемые небольшие молекулярные частицы и их фармацевтически приемлемые производные и соли. «Антагонист цитокина» представляет собой молекулу, которая ингибирует или оказывает антагонистическое действие в отношении таких цитокинов с помощью любого механизма, в том числе, например, антитела против цитокина, антитела против рецептора цитокина и иммуноадгезины.
Термин «гормон» относится к гормонам в виде полипептидов, которые, как правило, секретируются железистыми органами с протоками. Гормоны включают, например, гормоны роста, такие как гормон роста человека, N-метионил-гормон роста человека и гормон роста быка; гормон паращитовидной железы; тироксин; инсулин; проинсулин; релаксин; эстрадиол; гормонозаместительную терапию; андрогены, такие как калюстерон, дромостанолона пропионат, эпитиостанол, мепитиостан или тестолактон; прорелаксин; гормоны в виде гликопротеинов, такие как фолликулостимулирующий гормон (FSH), стимулирующий гормон щитовидной железы (TSH) и лютеинизирующий гормон (LH); пролактин, плацентарный лактоген, связанный с гонадотропином пептид мыши, высвобождающий гонадотропин гормон; ингибин; активин; ингибирующее вещество Миллера; и тромбопоэтин. Используемый здесь термин «гормон» включает белки из природных источников или из культуры рекомбинантных клеток и биологически активные эквиваленты гормонов с природными последовательностями, в том числе синтетически получаемые небольшие молекулярные частицы и их фармацевтически приемлемые производные и соли.
Термин «фактор роста» относится к белкам, которые усиливают рост и включают, например, фактор роста печени; фактор роста фибробластов; фактор роста эндотелия сосудов; факторы роста нервов, такие как NGF-β; происходящий из тромбоцитов фактор роста; трансформирующие факторы роста (TGF), такие как TGF-α и TGF-β; инсулиноподобный фактор I и II роста; эритропоэтин (ЕРО); остеоиндуктивные факторы; интерфероны, такие как интерферон-α, -β и -γ, и колониестимулирующие факторы (CSF), такие как макрофагальный CSF (M-CSF); гранулоцитарно-макрофагальный CSF (GM-CSF); и гранулоцитарный CSF (G-CSF). Используемый здесь термин «фактор роста» включает белки из природных источников или из культуры рекомбинантных клеток и биологически активные эквиваленты фактора роста с природной последовательностью, в том числе синтетически получаемые небольшие молекулярные частицы и их фармацевтически приемлемые производные и соли.
Термин «интегрин» относится к белку-рецептору, который позволяет клеткам связываться с экстраклеточным матриксом и реагировать с ним и вовлечен в ряд клеточных функций, таких как заживление ран, дифференциация клеток, хоминг опухолевых клеток и апоптоз. Они являются частью большого семейства рецепторов адгезии клеток, которые вовлечены во взаимодействия клетка-экстраклеточный матрикс и клетка-клетка. Функциональные интегрины состоят из двух трансмембранных гликопротеиновых субъединиц, называемых альфа и бета, которые нековалентно связаны. Все альфа-субъединицы, как и бета-субъединицы, гомологичны в некоторой степени друг другу. Рецепторы всегда содержат одну альфа-цепь и одну бета-цепь. Примеры включают альфа6бета1, альфа3бета1, альфа7бета1, LFA-1 и т.п. Используемый здесь термин «интегрин» включает белки из природных источников или из культуры рекомбинантных клеток и биологически активные эквиваленты интегрина с природной последовательностью, в том числе синтетически получаемые небольшие молекулярные частицы и их фармацевтически приемлемые производные и соли.
Для целей настоящего изобретения «фактор-альфа некроза опухолей (TNF-альфа)» относится к молекуле TNF-альфа, включающей аминокислотную последовательность, описанную Pennica и др. (Nature, 312: 721 (1984)) или Aggarwal и др. (JBC, 260: 2345 (1985)). «Ингибитор TNF-альфа» здесь представляет собой агент, который ингибирует, до некоторой степени, биологическую функцию TNF-альфа, как правило, благодаря связыванию TNF-альфа и нейтрализации его активности. Примерами ингибиторов TNF, специально здесь предусмотренными, являются этанерцепт (ENBREL®), инфликсимаб (REMICADE®) и адалимумаб (HUMIRATM).
Примеры «модифицирующих заболевание противоревматических лекарственных средств» или «DMARD» включают гидроксихлорохин, сульфасалазин, метотрексат, лефлуномид, этанерцепт, инфликсимаб (плюс пероральный или подкожный метотрексат), азатиоприн, D-пеницилламин, соли золота (пероральные), соли золота (внутримышечные), миноциклин, циклоспорин, в том числе циклоспорин А и циклоспорин для местного применения, стафилококковый белок А (Goodyears and Silverman, J. Exp. Med., 197, (9), p.1125-39 (2003)), в том числе их соли и производные и т.п. Предпочтительным DMARD здесь является метотрексат.
Примеры «нестероидных противовоспалительных лекарственных средств» или «NSAID» включают аспирин, ацетилсалициловую кислоту, ибупрофен, флурбипрофен, напроксен, индометацин, сулиндак, толметин, фенилбумазон, диклофенак, кетопрофен, бенорилат, мефенамовую кислоту, метотрексат, фенбуфен, азапропазон; ингибиторы COX-2, такие как целекоксиб (CELEBREX®; 4-(5-(4-метилфенил)-3-(трифторметил)-1Н-пиразол-1-ил)бензолсульфонамид, валдекоксиб (BEXTRA®), мелоксикам (MOBIC®), GR253035 (Glaxo Wellcome) и MK966 (Merck Sharp & Dohme), в том числе их соли и производные, и т.п. Предпочтительно они являются аспирином, напроксеном, ибупрофеном, индометацином или толметином.
Примеры «антагонистов интегрина или антител против интегрина» здесь включают антитело против LFA-1, такое как эфализумаб (RAPTIVA®), коммерчески доступное от Genentech, или антитело против интегрина альфа4, такое как натализумаб (ANTEGREN®), доступное от Biogen, или диазациклические производные фенилаланина (WO 2003/89410), производные фенилаланина (WO 2003/70709, WO 2002/28830, WO 2002/16329 и WO 2003/53926), производные фенилпропионовой кислоты (WO 2003/10135), производные энамина (WO 2001/79173), производные пропановой кислоты (WO 2000/37444), производные алкановой кислоты (WO 2000/32575), замещенные производные фенила (патенты США № 6677339 и 6348463), ароматические производные амина (патент США № 6369229), полипептиды домена дезинтегрина ADAM (US 2002/0042368), антитела против интегрина альфаVбета3 (ЕР 633845), аза-соединенные бициклические производные аминокислот (WO 2002/02556) и т.п.
«Кортикостероиды» относятся к любому одному из нескольких синтетических или природных веществ с общей химической структурой стероидов, которые имитируют или увеличивают эффекты природных кортикостероидов. Примеры синтетических кортикостероидов включают преднизон, преднизолон (в том числе метилпреднизолон, такой как метилпреднизолон натрия сукцинат SOLU-MEDROL®), дексаметазон или дексаметазон и триамцинолон, гидрокортизон и бетаметазон. Предпочтительными кортикостероидами здесь являются преднизон, метилпреднизолон, гидрокортизон или дексаметазон.
Термины «BAFF» или «полипептид BAFF», «TALL-1» или «полипептид TALL-1» и «BlyS», при использовании здесь, включают «полипептиды BAFF с природными последовательностями» и «варианты BAFF». «BAFF» является названием, данным таким полипептидам, которые имеют любую из аминокислотных последовательностей, представленных ниже:
Последовательность BAFF человека (SEQ ID NO: 16):

Последовательность BAFF мыши (SEQ ID NO: 17):

и их гомологам, фрагментам и вариантам, имеющим биологическую активность природного BAFF. Биологическую активность BAFF можно выбрать из группы, состоящей из увеличения выживаемости В-клеток, стимулирования созревания В-клеток и связывания с BR3. Аминокислотные последовательности вариантов BAFF предпочтительно идентичны на по крайней мере 80% или любое последующее целое число вплоть до 100%, в том числе более предпочтительно на по крайней мере 90% и даже более предпочтительно на по крайней мере 95%, природной последовательности полипептида BAFF.
Полипептид BAFF с «природной последовательностью» включает полипептид, имеющий ту же самую аминокислотную последовательность, что и соответствующий полипептид BAFF, полученный из природного источника. Например, BAFF существует в растворимой форме после отщепления от поверхности клетки с помощью протеаз типа фурина. Такие полипептиды BAFF со природными последовательностями можно выделить из природных источников, или их можно получить с помощью рекомбинантного и/или синтетического способа.
Термин «полипептид BAFF с природной последовательностью» или «природный BAFF», в частности, включает природные усеченные или секретируемые формы (например, последовательность экстраклеточного домена), природные вариантные формы (например, полученные в результате альтернативного сплайсинга формы) и природные аллельные варианты полипептида. Термин «BAFF» включает такие полипептиды, которые описаны Shu и др. (J. Leukocyte Biol., 65: 680 (1999); номер доступа в GenBank - AF136293); в заявке WO 1998/18921, опубликованной 7 мая 1998; ЕР 869180, опубликованной 7 октября 1998; в заявке WO 1998/27114, опубликованной 25 июня 1998; в заявке WO 1999/12964, опубликованной 18 марта 1999; в заявке WO 1999/33980, опубликованной 8 июля 1999; Moore и др. (Science, 285: 260-263 (1999)); Schneider и др. (J. Exp. Med., 189: 1747-1756 (1999)) и Mukhopadhyay и др. (J. Biol. Chem., 274: 15978-15981 (1999)).
Используемый здесь термин «антагонист BAFF» используется в самом широком значении и включает любую молекулу, которая (1) связывает полипептид BAFF с природной последовательностью или связывает природную последовательность BAFF с частичным или полным блокированием взаимодействия BR3 с полипептидом BAFF и (2) частично и полностью блокирует, ингибирует или нейтрализует активность BAFF с природной последовательностью. В одном предпочтительном варианте осуществления настоящего изобретения блокируемым рецептором для BAFF является рецептор BR3. Активность природного BAFF приводит, среди прочих вещей, к увеличению выживаемости В-клеток и/или стимуляции созревания В-клеток. В одном варианте осуществления настоящего изобретения ингибирование, блокирование или нейтрализация активности BAFF приводит к уменьшению числа В-клеток. Антагонист BAFF в соответствии с настоящим изобретением будет частично или полностью блокировать, ингибировать или нейтрализовать одну или несколько биологических активностей полипептида BAFF in vitro и/или in vivo. В одном варианте осуществления настоящего изобретения биологически активный BAFF усиливает какое-либо одно или комбинацию следующих явлений in vitro и/или in vivo: увеличенную выживаемость В-клеток, увеличенный уровень IgG и/или IgM, увеличенное число плазмацитов и процессирование NF-κb2/100 в p52 NF-κb в В-клетках селезенки (например, Batten et al., J. Exp. Med 192: 1453-1465 (2000); Moore et al., Science 285: 260-263 (1999); Kayagaki et al. Immunity 17: 515-524 (2002)).
Как отмечено выше, антагонист BAFF может действовать напрямую или опосредованно с частичным или полным блокированием, ингибированием или нейтрализацией передачи сигнала от BAFF in vitro и/или in vivo. Например, антагонист BAFF может непосредственно связывать BAFF. Например, предусматриваются антитела против BAFF, которые связываются в пределах района BAFF человека, включающего остатки 162-275 и/или остаток, близлежащий от остатка, выбираемого из группы, состоящей из 162, 163, 206, 211, 231, 233, 264 и 264 BAFF человека, так что антитело стерически затрудняет связывание BAFF с BR3, где такие номера остатков относятся к SEQ ID NO: 16. В другом примере непосредственным связующим веществом является полипептид, включающий любую часть рецептора для BAFF, которая связывает BAFF, такую как экстраклеточный домен рецептора для BAFF или его фрагменты и варианты, которые связывают природный BAFF. В другом примере антагонист BAFF включает полипептиды, имеющие последовательность полипептида, включающую последовательность формулы I: X1-C-X3-D-X5-L-X7-X8-X9-X10-X11-X12-C-X14-X15-X16-X17 (формула I) (SEQ ID NO:18), где X1, X3, X5, X7, X8, X9, X10, X11, X12, X14, X15 и X17 представляют собой любую аминокислоту за исключением цистеина, а X16 представляет собой аминокислоту, выбираемую из группу, состоящей из L, F, I и V; и где полипептид не включает цистеин в пределах семи аминокислотных остатков в направлении N-конца от самого N-концевого цистеина С и в направлении С-конца от самого С-концевого цистеина С формулы I.
В одном варианте осуществления полипептид, включающий последовательность формулы I, имеет два С, соединенных дисульфидной связью, при этом X5LX7X8 образуют конформацию структуры бета-оборота типа I с центром оборота между L и X7; и имеет положительное значение двугранного угла фи X8. В одном варианте осуществления настоящего изобретения X10 выбирают из группы, состоящей из W, F, V, L, I, Y, M и неполярной аминокислоты. В другом варианте осуществления настоящего изобретения X10 представляет собой W. В другом варианте осуществления X3 представляет собой аминокислоту, выбираемую из группы, состоящей из M, V, L, I, Y, F, W и неполярной аминокислоты. В другом варианте осуществления настоящего изобретения X5 выбирают из группы, состоящей из V, L, P, S, I, A и R. В другом варианте осуществления X7 выбирают из группы, состоящей из V, T, I и L. В другом варианте осуществления X8 выбирают из группы, состоящей из R, K, G, N, H и D-аминокислоты. В другом варианте осуществления X9 выбирают из группы, состоящей из H, K, A, R и Q. В другом варианте осуществления X11 представляет собой I или V. В другом варианте осуществления X12 выбирают из группы, состоящей из P, A, D, E и S. В другом варианте осуществления X16 представляет собой L. В одном конкретном варианте осуществления последовательность формулы I представляет собой последовательность, выбираемую из группы, состоящей из ECFDLLVRAWVPCSVLK (SEQ ID NO: 19), ECFDLLVRHWVPCGLLR (SEQ ID NO: 20), ECFDLLVRRWVPCEMLG (SEQ ID NO: 21), ECFDLLVRSWVPCHMLR (SEQ ID NO: 22), ECFDLLVRHWVACGLLR (SEQ ID NO: 23) и QCFDRLNAWVPCSVLK (SEQ ID NO: 24). В предпочтительном варианте осуществления антагонист BAFF включает любую аминокислотную последовательность, выбираемую из группы, состоящей из SEQ ID NO: 19, 20, 21, 22 и 23.
В еще одном примере антагонисты BAFF включают полипептиды, имеющие последовательность полипептида, включающую последовательность формулы II: X1-C-X3-D-X5-L-V-X8-X9-W-V-P-C-X14-X15-L-X17 (формула II) (SEQ ID NO:25), где X1, X3, X5, X8, X9, X14, X15 и X17 представляют собой любую аминокислоту за исключением цистеина, и где полипептид не включает цистеин в пределах семи аминокислотных остатков в направлении N-конца от самого N-концевого цистеина С и в направлении С-конца от самого С-концевого цистеина С формулы II.
В одном варианте осуществления полипептид, включающий последовательность формулы II, имеет дисульфидную связь между двумя С и имеет конформацию структуры бета-оборота типа I, образуемую X5LX7X8, с центром оборота между L и X7; и имеет положительное значение двугранного угла фи X8. В другом варианте формулы II X3 представляет собой аминокислоту, выбираемую из группы, состоящей из M, А, V, L, I, Y, F, W и неполярной аминокислоты. В другом варианте формулы II X5 выбирают из группы, состоящей из V, L, P, S, I, A и R. В другом варианте формулы II X8 выбирают из группы, состоящей из R, K, G, N, H и D-аминокислоты. В другом варианте формулы II X9 выбирают из группы, состоящей из H, K, A, R и Q.
В дополнительном варианте осуществления рецептор для BAFF, из которого происходит экстраклеточный домен или его связывающий BAFF фрагмент или его связывающий BAFF вариант, является TACI, BR3 или BCMA. Альтернативно, антагонист BAFF может связываться с экстраклеточным доменом BR3 с природной последовательностью в его связывающим BAFF районе с частичным или полным блокированием, ингибированием или нейтрализацией связывания BAFF с BR3 in vitro, in situ или in vitro. Например, такой непрямой антагонист представляет собой антитело против BR3, которое связывается в районе BR3, включающем остатки 23-38 BR3 человека, определенного ниже (SEQ ID NO: 26), или районе, близлежащем к таким остаткам, так что связывание BR3 человека с ВAFF стерически затрудняется.
В некоторых вариантах осуществления антагонист ВAFF в соответствии с настоящим изобретением включает антитела против ВAFF и иммуноадгезины, включающие экстраклеточный домен рецептора для ВAFF, или их фрагменты или варианты, которые связывают природный ВAFF. В дополнительном варианте осуществления рецептор для BAFF, из которого происходит экстраклеточный домен или его связывающий BAFF фрагмент, или его связывающий BAFF вариант, является TACI, BR3 или BCMA. В еще одном варианте осуществления иммуноадгезин включает аминокислотную последовательность формулы I или формулы II, определенную выше, в том числе аминокислотную последовательность, выбираемую из группы, состоящей из SEQ ID NO: 19, 20, 21, 22, 23 и 24.
В соответствии с одним вариантом осуществления антагонист BAFF связывается с полипептидом BAFF или полипептидом BR3 с аффинностью связывания, составляющей 100 нМ или меньше. В соответствии с другим вариантом осуществления антагонист BAFF связывается с полипептидом BAFF или полипептидом BR3 с аффинностью связывания, составляющей 10 нМ или меньше. В соответствии с еще одним вариантом осуществления антагонист BAFF связывается с полипептидом BAFF или полипептидом BR3 с аффинностью связывания, составляющей 1 нМ или меньше.
Используемые здесь термины «BR3», «полипептид BR3» или «рецептор BR3» включают «полипептиды BR3 с природными последовательностями» и «варианты BR3» (которые здесь дополнительно определяются). «BR3» является названием, данным таким полипептидам, которые включают следующую аминокислотную последовательность и ее гомологи, и их вариантам или фрагментам, которые связывают природный BAFF:
Последовательность BR3 человека (SEQ ID NO: 26):

Полипептид BR3 настоящего изобретения можно выделить из множества источников, например, из типов тканей человека или из другого источника, или получить с помощью рекомбинантных и/или синтетических способов. Термин «BR3» включает полипептиды BR3, описанные в WO 2002/24909 и WO 2003/14294.
Термин «полипептид BR3 с природной последовательностью» или «природный BAFF» включает полипептид, имеющий такую же аминокислотную последовательность, что и соответствующий полипептид BR3, получаемый из природного источника. Такие полипептиды BR3 с природными последовательностями можно выделить из природного источника или их можно получить с помощью рекомбинантного и/или синтетического способа. Термин «полипептид BR3 с природной последовательностью», в частности, охватывает природные усеченные, растворимые или секретируемые формы (например, последовательность экстраклеточного домена), природные вариантные формы (например, полученные в результате альтернативного сплайсинга формы) и природные аллельные варианты полипептида. Полипептид BR3 настоящего изобретения включает полипептид BR3, включающий или состоящий из непрерывной последовательности аминокислотных остатков 1-184 BR3 человека (SEQ ID NO: 26).
«Экстраклеточный домен» BR3 или «ECD» относится к форме полипептида BR3, которая по существу не содержит трансмембранного и цитоплазматического доменов. ECD-формы BR3 включают полипептид, включающий любую аминокислотную последовательность, выбираемую из группы, состоящей из аминокислот 1-77, 2-62, 2-71, 1-61, 7-71, 23-38 и 2-63 BR3 человека. В настоящем изобретении предусмотрены антагонисты BAFF, которые являются полипептидами, включающими любую из отмеченных выше ECD-форм BR3 человека и их вариантов и фрагментов, которые связывают природный BAFF.
Мини-BR3 представляет собой основный район из 26 остатков связывающего BAFF домена BR3, т.е. аминокислотную последовательность: TPCVPAECFDLLVRHCVACGLLRTPR (SEQ ID NO: 27).
«Вариант BR3» означает полипептид BR3, аминокислотная последовательность которого идентична по крайней мере на 80% аминокислотной последовательности полноразмерного BR3 или ECR BR3 с природными последовательностями, и связывается с полипептидом BAFF с природной последовательностью. Необязательно вариант BR3 включает один богатый цистеином домен. Такие полипептиды-варианты BR3 включают, например, полипептиды BR3, в которых один или несколько аминокислотных остатков добавлены или делетированы с N- и/или С-концов, а также в пределах одного или нескольких внутренних доменов полноразмерной аминокислотной последовательности. Также предусмотрены фрагменты ECD BR3, которые связываются с полипептидом BAFF с природной последовательностью. В соответствии с одним вариантом осуществления аминокислотная последовательность вариантного полипептида BR3 идентична на по крайней мере приблизительно 80%, на по крайней мере приблизительно 81%, на по крайней мере приблизительно 82%, на по крайней мере приблизительно 83%, на по крайней мере приблизительно 84%, на по крайней мере приблизительно 85%, на по крайней мере приблизительно 86%, на по крайней мере приблизительно 87%, на по крайней мере приблизительно 88%, на по крайней мере приблизительно 89%, на по крайней мере приблизительно 90%, на по крайней мере приблизительно 91%, на по крайней мере приблизительно 92%, на по крайней мере приблизительно 93%, на по крайней мере приблизительно 94%, на по крайней мере приблизительно 95%, на по крайней мере приблизительно 96%, на по крайней мере приблизительно 97%, на по крайней мере приблизительно 98% или на по крайней мере приблизительно 99% аминокислотной последовательности полипептида BR3 или его точно определенного фрагмента (например, ECD). Вариантные полипептиды BR3 не включают природную последовательность полипептида BR3. В соответствии с другим вариантом осуществления размер вариантных полипептидов BR3 составляет по крайней мере приблизительно 10 аминокислот, по крайней мере приблизительно 20 аминокислот, по крайней мере приблизительно 30 аминокислот, по крайней мере приблизительно 40 аминокислот, по крайней мере приблизительно 50 аминокислот, по крайней мере приблизительно 60 аминокислот или по крайней мере приблизительно 70 аминокислот.
В одном предпочтительном варианте осуществления здесь антагонисты BAFF представляют собой иммуноадгезины, включающие часть BR3, TACI или BCMA, которая связывается с BAFF, или их варианты, которые связываются с BAFF. В других вариантах осуществления антагонист BAFF является антителом против BAFF. «Антитело против BAFF» представляет собой антитело, которое связывает BAFF и предпочтительно связывает BAFF в районе BAFF человека, включающем остатки 162-275 последовательности BAFF человека, раскрытой здесь под названием «BAFF» (SEQ ID NO: 16). В другом варианте осуществления антагонистом BAFF является антитело против BR3. «Антитело против BR3» является антителом, которое связывает BR3, и предпочтительно является антителом, которое связывается с BR3 в районе BR3 человека, включающем остатки 23-38 последовательности BR3 человека, раскрытой здесь под названием «BR3» (SEQ ID NO: 26). В общем, положения аминокислот BAFF человека и BR3 человека, на которые здесь ссылаются, находятся в соответствии с нумерацией положений в SEQ ID NO: 16 и 26, соответственно, раскрытых здесь под названиями «BAFF» и «BR3».
Другие примеры связывающих BAFF полипептидов или антител против BAFF можно найти, например, в WO 2002/092620, WO 2003/014294, Gordon et al., Biochemistry 42(20): 5977-5983 (2003), Kelley et al., J. Biol. Chem. 279 (16): 16727-16735 (2004), WO 1998/18921, WO 2001/12812, WO 2000/68378 и WO 2000/40716.
Используемый здесь термин «листовка-вкладыш» относится к инструкциям, обычно включаемым в торговые упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозе, введении, противопоказаниях, других терапевтических продуктах, объединяемых с упакованным продуктом, и/или предостережениях, касающихся использования таких терапевтических продуктов, и т.п.
«Лекарственное средство» представляет собой активное лекарственное средства для лечения повреждения сустава или его симптомов или побочных эффектов.
II. Лечение
В одном аспекте настоящим изобретением обеспечивается способ лечения повреждения сустава у индивида, такого как пациент, включающий введение индивиду антагониста, предпочтительно антитела, который связывается с маркером поверхности В-клетки, (более предпочтительно антитела против CD20) и рентгенографическую оценку того, демонстрируется ли у индивида, подвергаемого лечению, уменьшение повреждения сустава.
Таким образом, изобретением предусматривается способ лечения повреждения сустава у индивида, включающий введение индивиду антагониста, который связывается с маркером поверхности В-клетки, и рентгенографическую проверку индивида, по крайней мере спустя приблизительно один месяц от момента введения, с помощью которой определяют уменьшение повреждения сустава по сравнению с базовой линией до введения, причем количество вводимого антагониста является эффективным для получения уменьшения повреждения сустава, что является показателем успешного лечения индивида в отношении повреждения сустава.
Изобретением также предусматривается способ лечения повреждения сустава у индивида, включающий введение индивиду антитела, которое связывается с маркером поверхности В-клетки, и рентгенографическую проверку индивида, по крайней мере спустя приблизительно один месяц от момента введения, с помощью которой определяют уменьшение повреждения сустава по сравнению с базовой линией до введения, причем количество вводимого антитела является эффективным для получения уменьшения повреждения сустава, что является показателем успешного лечения индивида в отношении повреждения сустава.
Изобретением, кроме того, предусматривается способ лечения повреждения сустава у индивида, включающий введение индивиду антитела против CD20 и рентгенографическую проверку индивида, по крайней мере спустя приблизительно один месяц от момента введения, с помощью которой определяют уменьшение повреждения сустава по сравнению с базовой линией до введения, причем количество вводимого антитела против CD20 является эффективным для получения уменьшения повреждения сустава, что является показателем успешного лечения индивида в отношении повреждения сустава.
В предпочтительном варианте осуществления настоящего изобретения рентгенографическая проверка после введения антагониста или антитела, такого как антитело против CD20, имеет место спустя по крайней мере приблизительно два месяца, более предпочтительно по крайней мере приблизительно 10 недель, еще более предпочтительно по крайней мере приблизительно три месяца, еще предпочтительнее по крайней мере приблизительно четыре месяца, еще более предпочтительно по крайней мере приблизительно пять месяцев, еще предпочтительнее по крайней мере приблизительно 24 недели или по крайней мере приблизительно шесть месяцев, и наиболее предпочтительно по крайней мере приблизительно 52 недели от момента введения антагониста или антитела. В другом предпочтительном варианте осуществления настоящего изобретения с помощью проверки определяют общий модифицированный показатель Sharp.
В другом предпочтительном варианте осуществления настоящего изобретения индивида подвергают повторному лечению, при котором способ дополнительно включает введение индивиду антагониста или антитела, такого как CD20, в количестве, эффективном для получения непрерывного или сохраняемого уменьшения повреждения сустава по сравнению с эффектом предшествующего введения антагониста или антитела, такого как антитело против CD20. Таким образом, индивиду может вводиться вторая доза антагониста или антитела, и он подвергается оценке с помощью рентгенографической проверки через по крайней мере приблизительно один месяц (или предпочтительно более чем приблизительно два месяца, более предпочтительно приблизительно 24 недели или приблизительно 6 месяцев) после такого введения второй дозы для определения того, является ли введение второй дозы эффективным (т.е. вводится ли эффективное количество антагониста или антитела) для сохранения эффектов введения первой дозы или увеличения уменьшения повреждения сустава по сравнению с эффектом введения первой дозы. Эту схему повторного лечения можно повторить, если желательно или необходимо получение или сохранение уменьшения повреждения сустава, которое является индикатором успешного лечения повреждения сустава.
В другом варианте осуществления настоящего изобретения антагонист или антитело, такое как антитело против CD20, дополнительно вводят (продолжают вводить) индивиду, даже если у индивида нет клинического улучшения во время рентгенографической проверки после предшествующего введения, такого как первое введение антагониста или антитела. В последнем из двух названных вариантов осуществления клиническое улучшение предпочтительно определяют с помощью оценки числа болезненных или опухших суставов, показателя тяжести оценки псориаза, общей клинической оценки индивида, оценки скорости осаждения эритроцитов или оценки величины уровня С-реактивных белков.
Для целей настоящего изобретения второе воздействие антагонистом или антителом для повторного лечения представляет собой то, что в следующий раз индивида подвергают лечению антагонистом или, например, антителом против CD20 после первоначального воздействия антителом, при этом нет промежуточного лечения или воздействия антагонистом или, например, антителом против CD20 между первоначальным и вторым воздействиями. Такое повторное лечение может быть запланированным или не запланированным, но предпочтительно оно является запланированным повторным введением дозы, в частности, для защиты органов, таких как почки, от повреждения. При использовании антитела, особенно антитела против CD20, второе воздействие антителом предпочтительно составляет приблизительно 0,5-4 грамма, более предпочтительно приблизительно 1,5-3,5 грамма, еще более предпочтительно приблизительно 1,5-2,5 грамма, при этом второе воздействие предоставляется не раньше приблизительно 20-35 недель (предпочтительно приблизительно 23-30, более предпочтительно приблизительно 23-28 недель) от первоначального воздействия.
В способе предусматривается введение индивиду эффективного количества антагониста или, например, антитела против CD20 с предоставлением третьего воздействия антагонистом или антителом (если антителом, более предпочтительно антителом против CD20) предпочтительно в количестве приблизительно 0,5-4 грамма, более предпочтительно приблизительно 1,5-3,5 грамма, еще более предпочтительно приблизительно 1,5-2,5 грамма, при этом третье воздействие предоставляется не раньше приблизительно 46-60 недель (предпочтительно приблизительно 46-55, более предпочтительно приблизительно 46-52 недели) от первоначального воздействия. Предпочтительно, чтобы дополнительное воздействие антагонистом или антителом не предоставлялось раньше, чем приблизительно 70-75 недель от первоначального воздействия, и еще более предпочтительно, чтобы дополнительное воздействие антагонистом или антителом не предоставлялось раньше, чем приблизительно 74-80 недель от первоначального воздействия.
При использовании антитела индивиду здесь может быть предоставлено одно или несколько воздействий антителом в виде единственной дозы антитела или в виде отдельных доз, например, приблизительно 1-4 отдельных доз антитела (например, образующих первую и вторую дозу, или первую, вторую и третью дозу, или первую, вторую, третью и четвертую дозу, и т.п.). Конкретное число доз (или одна, две или три или более), используемых для каждого воздействия антителом, зависит, например, от типа подвергаемого лечению повреждения сустава, типа используемого антитела, от того, использовалось ли второе лекарственное средство, какого оно было типа, в каком количестве и сколько раз оно использовалось, как отмечено ниже, и от способа и частоты введения. При введении отдельных доз более позднюю дозу (например, вторую или третью дозу) предпочтительно вводят спустя приблизительно 1-20 дней, более предпочтительно спустя приблизительно 6-16 дней и наиболее предпочтительно спустя приблизительно 14-16 дней от момента времени введения предшествующей дозы. Отдельные дозы предпочтительно вводят в пределах общего периода, составляющего приблизительно 1 день - 4 недели, более предпочтительно приблизительно 1-20 дней (например, в пределах периода, составляющего 6-18 дней). В одном таком аспекте отдельные дозы вводят приблизительно один раз в неделю, при этом вторую дозу вводят спустя приблизительно одну неделю от момента введения первой дозы, третью или последующую дозу вводят спустя приблизительно одну неделю от момента введения второй дозы. Каждая такая отдельная доза антитела предпочтительно составляет приблизительно 0,5-1,5 грамма, более предпочтительно приблизительно 0,75-1,3 грамма.
В наиболее предпочтительном варианте осуществления изобретения предоставляется способ лечения повреждения сустава у индивида, включающий введение индивиду эффективного количества антитела, которое связывается с маркером поверхности В-клетки, (например, антитела против CD20) с предоставлением первоначального воздействия антителом с последующим вторым воздействием антителом, причем второе воздействие не предоставляется раньше, чем приблизительно 16-54 недель от первоначального воздействия, и каждое воздействие антителом предоставляется индивиду в виде единственной дозы или в виде двух или трех отдельных доз антитела. Предпочтительно в таком способе каждое воздействие антителом составляет приблизительно 0,5-4 грамма, и наиболее предпочтительно количества, установленные выше.
В одном варианте осуществления настоящего изобретения индивиду предоставляются по крайней мере три воздействия антителом, в частности, приблизительно 3-60 воздействий и, в более частном случае, приблизительно 3-40, в самом частном случае, приблизительно 3-20 воздействий. Предпочтительно такие воздействия назначают с интервалами, составляющими каждый 24 недели. В одном варианте осуществления настоящего изобретения каждое воздействие антителом предоставляется в виде единственной дозы антитела. В альтернативном варианте осуществления настоящего изобретения каждое воздействие антителом предоставляется в виде отдельных доз антитела. Однако не каждое воздействие антителом требуется предоставлять в виде единственной дозы или в виде отдельных доз.
В одном предпочтительном варианте осуществления настоящего изобретения в качестве первоначального воздействия вводится приблизительно 2-3 грамма антитела против CD20. При введении в качестве первоначального воздействия приблизительно 3 грамм вводят приблизительно 1 грамм антитела против CD20 раз в неделю на протяжении приблизительно трех недель. При введении в качестве первоначального воздействия 2 грамм антитела против CD20 вводят приблизительно 1 грамм антитела против CD20, а затем через приблизительно две недели еще приблизительно 1 грамм антитела. В предпочтительном аспекте второе воздействие осуществляют через приблизительно 24 недели или шести месяцев от первоначального воздействия, и его назначают в количестве приблизительно 2 граммов. В альтернативном предпочтительном аспекте второе воздействие осуществляют через приблизительно 24 недели или шесть месяцев от первоначального воздействия, и его назначают в количестве приблизительно 1 грамм антитела, а затем через приблизительно две недели еще приблизительно 1 грамм антитела.
В предпочтительном варианте осуществления метода многократного воздействия настоящего изобретения индивид находится в стадии ремиссии после первоначального или любого более позднего воздействия антагонистом или антителом. Более предпочтительно способ многократного воздействия здесь включает запланированное повторное введение дозы или повторное лечение, так чтобы индивид находился в стадии ремиссии при обеспечении второго и предпочтительно всех воздействий антагонистом или антителом. Такое повторное введение дозы планируется для предупреждения любого рецидива или повреждения органа, а не для терапевтического лечения. Наиболее предпочтительно, когда индивид находится в стадии ремиссии на протяжении по крайней мере приблизительно 24 недель или шести месяцев, и еще наиболее предпочтительно на протяжении по крайней мере приблизительно девяти месяцев, и даже еще наиболее предпочтительно на протяжении по крайней мере приблизительно 52 недель или одного года с момента последнего воздействия антагонистом или антителом, используемого в способе повторного лечения.
В еще одном варианте осуществления настоящего изобретения индивида подвергают лечению одним и тем же антагонистом или антителом, таким как антитело против CD20, на протяжении по крайней мере двух воздействий антагонистом или антителом и предпочтительно на протяжении каждого воздействия антагонистом или антителом. Таким образом, первоначальное и второе воздействия антагонистом или антителом предпочтительно осуществляют с использованием одного и того же антагониста или антитела, и более предпочтительно все воздействия антагонистом или антителом осуществляют с использованием одного и того же антагониста или антитела, т.е. лечение на протяжении первых двух воздействий и предпочтительно всех воздействий осуществляют с использованием одного типа антагониста или антитела, которое связывается с маркером поверхности В-клетки, такого как антитело против CD20, например, все с использованием ритуксимаба или все с использованием одного и того же гуманизированного антитела 2Н7.
Во всех способах настоящего изобретения, изложенных здесь, антагонист (такой как антитело против CD20 и маркера поверхности В-клетки) может быть не конъюгированным, такой как не конъюгированное антитело, или может быть конъюгированным с другой молекулой для дополнительной эффективности, например, для увеличения времени полужизни. Предпочтительным антителом против CD20 здесь является химерное, гуманизированное антитело против CD20 или антитело человека против CD20, более предпочтительно ритуксимаб, гуманизированное антитело 2Н7 (например, включающее последовательности вариабельных доменов SEQ ID NO: 2 и 8, или гуманизированное антитело 2Н7, включающее последовательности вариабельных доменов SEQ ID NO: 39 и 40, или включающее последовательности вариабельных доменов SEQ ID NO: 32 и 33, или включающее вариабельный домен тяжелой цепи с заменами N100A, или D56A и N100A, или D56A, N100Y и S100aR в SEQ ID NO: 8 и вариабельный домен легкой цепи с заменами M32L, или S92A, или M32L и S92A в SEQ ID NO: 2), химерное или гуманизированное антитело А20 (Immunomedics), антитело человека против CD20 HUMAX-CD20TM (Genmab) или одноцепочечные белки, связывающиеся с CD20 (Trubion Pharm. Inc.). Еще более предпочтительным является ритуксимаб или гуманизированное антитело 2Н7.
В дополнительном варианте осуществления всех способов настоящего изобретения индивид никогда ранее не подвергался лечению одним или несколькими лекарственными средствами, такими как анти-TNF-альфа ингибитор, например антитела против TNF-альфа или антитела против рецептора для TNF-альфа, для лечения, например, артрита или иммунодепрессантом(ами) для лечения повреждения сустава и лежащей в основе причины, такой как аутоиммунное нарушение, и/или никогда ранее не подвергался лечению антагонистом (например, антителом против маркера поверхности В-клетки) (например, никогда ранее не подвергался лечению антителом против CD20). В одном таком варианте осуществления индивид никогда ранее не подвергался лечению антителом против альфа-4-интегрина и костимулирующим модулятором, биологическим агентом, DMARD, отличным от МТХ, за исключением азатиоприна и/или лефлуномида, истощающему клетки лечению, включающему агенты, проходящие клинические испытания (например, CAMPATH, анти-CD4, анти-CD5, анти-CD3, анти-CD19, анти-CD11a, анти-CD2 или BLys/BAFF), живой/ослабленной вакциной в пределах 28 дней до базовой линии или внутрисуставными или парентеральными глюкокортикоидами в пределах 4 недель до базовой линии.
В еще одном дополнительном аспекте у индивида мог быть рецидив повреждения сустава, или он мог страдать повреждением органа, таким как повреждение почки, до начала лечения с помощью любого из вышеуказанных способов, в том числе после первоначального или более позднего воздействия антагонистом или антителом. Однако, предпочтительно, чтобы у индивида не было рецидива повреждения сустава, и более предпочтительно, чтобы у него не было такого рецидива до по крайней мере первоначального лечения.
В другом варианте осуществления настоящего изобретения антагонист (например, антитело против CD20) является единственным лекарственным средством, вводимым индивиду для лечения повреждения сустава. В другом варианте осуществления изобретения антагонист (например, антитело против CD20) является одним из лекарственных средств, используемых для лечения повреждения сустава. В дополнительном варианте осуществления настоящего изобретения у индивида нет злокачественной опухоли, в том числе солидных опухолей, опухолей кроветворной системы или карциномы in situ (за исключением базальноклеточной и чешуйчатоклеточной карциномы кожи, которую подвергли иссечению и вылечили). В еще одном дополнительном варианте осуществления настоящего изобретения у индивида нет ревматоидного артрита (RA). В другом аспекте у индивида нет ревматического аутоиммунного заболевания, отличного от RA, или значительного системного поражения, вторичного по отношению к RA, (включающего, но без ограничения, васкулит, фиброз легкого или синдром Фелти). В другом варианте осуществления изобретения у индивида нет вторичного синдрома Шегрена или вторичного ограниченного кожей васкулита. В другом варианте осуществления настоящего изобретения у индивида нет функционального состояния класса IV, определяемого в соответствии с классификацией ACR функциональных состояний при RA. В дополнительном варианте осуществления настоящего изобретения у индивида нет воспалительного заболевания сустава, отличного от RA, (включающего, но без ограничения, подагрический, реактивный артрит, псориатический артрит, серонегативную спондилоартропатию, болезнь Лима) или другого системного аутоиммунного заболевания (включающего, но без ограничения, системную красную волчанку, болезнь воспаленного кишечника, склеродермию, воспалительную миопатию, смешанное заболевание соединительных тканей или любой синдром наложения). В другом варианте осуществления настоящего изобретения у индивида нет ювенильного идиопатического артрита (JIA) или ювенильного RA (JRA) и/или RA до возраста 16 лет. В другом варианте осуществления настоящего изобретения у индивида нет значимого и/или неконтролируемого заболевания сердца или легких (в том числе обструктивного заболевания легких) или значимого сопутствующего заболевания, включающего, но без ограничения, нарушения нервной системы, почек, печени, эндокринной системы или желудочно-кишечного тракта, ни первичного, ни вторичного иммунодефицита (в анамнезе или активного в текущий момент), в том числе известной истории ВИЧ-инфекции. В другом аспекте у индивида нет какого-либо неврологического (врожденного или приобретенного), сосудистого или системного нарушения, которое могло бы оказать влияние на какую-либо оценку эффективности, в частности боли в суставе и опухания, (например, болезни Паркинсона, церебрального паралича, невропатии при сахарном диабете). В еще дополнительном варианте осуществления настоящего изобретения у индивида нет рассеянного склероза. В еще дополнительном варианте осуществления настоящего изобретения у индивида нет обыкновенной волчанки или синдрома Шегрена. В еще другом варианте осуществления настоящего изобретения у индивида нет аутоиммунного заболевания. В еще одном аспекте настоящего изобретения повреждение сустава не сопровождается аутоиммунным заболеванием или аутоиммунным заболеванием, отличным от артрита, или риском развития аутоиммунного заболевания или аутоиммунного заболевания, отличного от артрита. Для целей самых последних формулировок «аутоиммунное заболевание» здесь представляет собой заболевание или нарушение, направленное против собственных тканей или органов индивидуума и возникающее из таких тканей и органов, или их сокомпонент или проявление или являющееся их результатом состояние. Не ограничиваясь какой-либо одной теорией, при аутоиммунных заболеваниях человека В-клетки проявляют патогенное действии через множество механистических путей, включающих продукцию аутоантител, образование иммунных комплексов, активацию дендритных клеток и Т-клеток, синтез цитокинов, прямое высвобождение цитокинов и формирующих очаг для эктопического неолимфогенеза.
В предпочтительном варианте осуществления настоящего изобретения повреждение сустава вызвано артритом, асептическим разрыхлением ортопедических имплантатов сустава, не соединением перелома, спондилоартропатиями, псориазом иди болезнью Крона. Более предпочтительно повреждение сустава вызвано артритом, который более предпочтительно является ревматоидным артритом, остеоартритом, алкилозирующим спондилоартритом или псориатическим артритом.
В еще дополнительных вариантах осуществления настоящего изобретения, если антагонистом является антитело, антитело вводят внутривенно или подкожно.
В дополнительных предпочтительных аспектах, если антагонистом является антитело, антитело вводят в дозе, составляющей приблизительно 0,4-4 грамма, более предпочтительно приблизительно 1,5-3,5 грамма, еще более предпочтительно приблизительно 1,5-2,5 грамма. В другом аспекте антитело предпочтительно вводят в дозе, составляющей приблизительно 0,4-1,3 грамма, с частотой одна-четыре дозы в пределах периода, составляющего приблизительно один месяц, более предпочтительно приблизительно 500 мг-1 грамм, еще более предпочтительно от приблизительно 500 мг или приблизительно 750 мг до приблизительно 1,1 грамма, и более предпочтительно антитело вводят в виде двух-трех доз. Еще более предпочтительно антитело вводят в пределах периода, составляющего приблизительно 2-3 недели.
В другом предпочтительном аспекте индивид является отрицательным в отношении ревматоидного фактора. В другом аспекте индивид является положительным в отношении ревматоидного фактора.
В другом предпочтительном аспекте описанного выше способа индивиду вводили метотрексат до базовой линии или начала лечения. Более предпочтительно метотрексат вводили в дозе, составляющей приблизительно 10-25 мг/неделю. Также предпочтительно, когда метотрексат вводили в течение по крайней мере приблизительно 12 недель до базовой линии, и еще более предпочтительно, когда метотрексат вводили в постоянной дозе в течение последних четырех недель до базовой линии. В других вариантах осуществления метотрексат вводили перорально или парентерально.
В особенно предпочтительных вариантах осуществления определенного выше способа повреждение сустава вызвано ревматоидным артритом, и индивид неадекватно реагировал на один или несколько ингибиторов в виде антител против фактора некроза опухолей (TNF), и/или индивиду вводят метотрексат вместе с антагонистом, например антителом против CD20, и/или антагонистом является антитело против CD20, которое вводят внутривенно в дозе приблизительно 1000 мг × 2 в дни 1 и 15 от начала лечения.
В еще дополнительных вариантах осуществления настоящим изобретением обеспечивается способ контролирования лечения повреждения сустава у индивида, включающий введение индивиду эффективного количества антагониста в отношении маркера поверхности В-клетки (такого как антитело против такого маркера, в том числе антитело против CD20) и определение с помощью рентгенографии спустя по крайней мере приблизительно один месяц от момента введения, произошло ли уменьшение повреждения сустава по сравнению с базовой линией до введения, причем уменьшение в сравнении с базовой линией у индивида после лечения указывает на то, что антагонист или антитело, такое как антитело против CD20, оказывает эффект на повреждение сустава. Предпочтительно степень уменьшения в сравнение с базовой линией определяют второй раз после введения антагониста или антитела, такого как антитело против CD20. Также предпочтительно, когда опредедение проводят спустя по крайней мере приблизительно 24 недели от момента введения.
Также сюда включен способ контролирования лечения повреждения сустава у индивида, включающий введение индивиду эффективного количества антагониста в отношении маркера поверхности В-клетки (такого как антитело против такого маркера, в том числе антитело против CD20) и определение с помощью рентгенографии спустя по крайней мере приблизительно 52 недели от момента введения, произошло ли уменьшение повреждения сустава по сравнению с базовой линией до введения, причем уменьшение в сравнении с базовой линией у индивида после лечения указывает на то, что антагонист или антитело, такое как антитело против CD20, оказывает эффект на повреждение сустава. Предпочтительно степень уменьшения в сравнение с базовой линией определяют второй раз после введения антагониста или антитела, такого как антитело против CD20.
В еще одном аспекте настоящим изобретением обеспечивается способ определения того, нужно ли продолжать введение индивиду с повреждением сустава антагониста в отношении маркера поверхности В-клетки (такого как антитело против такого маркера, в том числе антитело против CD20), включающий определение с помощью рентгенографии уменьшения повреждения сустава у индивида после введения антагониста, такого как антитело против CD20, первый раз, определение с помощью рентгенографии уменьшения повреждения сустава у индивида после введения антагониста, такого как антитело против CD20, второй раз, сравнение рентгенографических показателей у индивида в первый раз и во второй раз и, если показатель меньше во второй раз, чем в первый раз, продолжение введения антагониста или антитела, такого как антитело против CD20.
В еще дополнительном варианте осуществления настоящим изобретением способ лечения включает стадию проверки ответа индивида на лечение после стадии введения для определения того, является уровень ответа эффективным для лечения повреждения сустава. Например, включается стадия проверки рентгенографического показателя после введения и сравнения его с рентгенографическим показателем базовой линии, полученным до введения, для определения того, является ли лечение эффективным с помощью определения, изменился ли рентгенографический показатель и насколько он изменился. Эту проверку можно повторять через различные запланированные или не запланированные интервалы времени после введения для определения сохранности частичной или полной ремиссии. Альтернативно, способы настоящего изобретения включают стадию проверки, до введения, у индивида того, имеется ли у него один или несколько биомаркеров или симптомов повреждения сустава, как изложено выше. В другой способ может быть включена стадия проверки клинической истории индивида, как детализировано выше, например, чтобы исключить инфекции или злокачественную опухоль в качестве причин, например первичных причин, состояния индивида, до введения индивиду антитела или антагониста. Предпочтительно повреждение сустава является первичным (т.е. основным заболеванием), а не вторичным, таким как вторичное по отношению к инфекции или злокачественной опухоли, или солидным опухолям, или опухолям жидкостей организма.
В одном варианте осуществления всех способов настоящего изобретения для лечения повреждения сустава у индивида не вводится никакого другого лекарственного средства, кроме антагониста, такого как антитело против CD20.
В любом из способов настоящего изобретения предпочтительно, когда вместе с антагонистом или антителом, которое связывается с маркером поверхности В-клетки, индивиду вводится эффективное количество второго лекарственного средства (где антагонист или антитело, которое связывается с маркером поверхности В-клетки, (например, антитело против CD20) является первым лекарственным средством). Вторым лекарственным средством может быть одно или несколько лекарственных средств, и оно включает, например, иммунодепрессант, антагонист цитокина, такой как антитело против цитокина, фактор роста, гормон, интегрин, антагонист или антитело против интегрина или любую их комбинацию. Тип такого второго лекарственного средства зависит от различных факторов, включающих тип повреждения сустава, тяжесть повреждения сустава, состояние и возраст индивида, тип и дозу применяемого первого лекарственного средства и т.п.
Примеры таких дополнительных лекарственных средств включают лекарственное средство класса интерферонов, такое как интерферон-альфа (например, от Amarillo Biosciences, Inc.), IFN-бета-1а (REBIF® и AVONEX®) или IFN-бета-1b (BETASERON®), олигопептид, такой как глатирамера ацетат (COPAXONE®), агент, блокирующий лиганд CD40-CD40, иммунодепрессант (такой как метоксантрон (NOVANTRONE®), метотрексат, циклофосфамид, хлорамбуцил, лефлуномид и азатиоприн), внутривенно вводимый иммуноглобулин (гамма-глобулин), истощающую лимфоциты терапию (например, митоксантрон, циклофосфамид, антитела CAMPATHTM, анти-CD4, кладрибин, полипептидная конструкция с по крайней мере двумя доменами, включающая аутореактивный антиген с исключенной иммуногенностью или его фрагмент, который специфически узнается Ig-рецепторами аутореактивных В-клеток (WO 2003/68822), облучение всего организма, трансплантация костного мозга), антагонист или антитело против интегрина (например, антитело против LFA-1, такое как эфализумаб/RAPTIVA®, коммерчески доступное от Genentech, или антитело против альфа-4-интегрина, такое как натализумаб/ANTEGREN®, доступное от Biogen, или другие, отмеченные выше), лекарственные средства, с помощью которых лечат симптомы, вторичные по отношению к повреждению сустава или связанные с таким повреждением, такие как те, которые здесь отмечены, стероид, такой как кортикостероид (например, преднизолон, метилпреднизолон, такой как метилпреднизолон натрия сукцинат SOLU-MEDROL® для инъекции, преднизон, такой как преднизон в низкой дозе, дексаметазон или глюкокортикоид, например, через инъекцию в сустав, в том числе системную кортикостероидную терапию), имуносупрессирующую терапию без истощения лимфоцитов (например, MMF или циклоспорин), понижающее уровень холестерина лекарственное средство класса «статины» (которое включает церивастатин (BAYCOLTM), флувастатин (LESCOLTM)), аторвастатин (LIPITORTM), ловастатин (MEVACORTM), правастатин (PRAVACHOLTM) и симвастатин (ZOCORTM), эстрадиол, тестостерон (необязательно в повышенных дозах; Stuve et al. Neurology 8: 290-301 (2002)), андроген, гормонозаместительную терапию, ингибитор TNF, такой как антитело против TNF-альфа, DMARD, NSAID, плазмаферез или замещение плазмы, триметопримсульфаметоксазол (BACTRIMTM, SEPTRATM), микофенолат мофетил, Н2-блокаторы или ингибиторы протоновых насосов (во время использования необязательно ульцерогенной иммуносупрессирующей терапии), левотироксин, циклоспорин А (например, SANDIMMUNE®), аналог соматастатина, DMARD или NSAID, антагонист цитокина, такой как антитело, антиметаболит, иммунодепрессант, операцию с целью реабилитации, радиоактивный йод, тиреоидэктомию, антагонист BAFF, такой как антитела против BAFF или BR3 или иммуноадгезины, антитело против рецептора для CD40 или анти-CD40 лиганд (CD154), антагонист/антитело против рецептора для IL-6, другой антагонист или антитело против маркера поверхности В-клетки, такие как гуманизированное антитело 2Н7 или другое гуманизированное антитело, или антитело человека против CD20 с ритуксимабом; блокаторы IL-1, такие как rHUIL-1Ra (Amgen-Synergen) и ингибитор тиапрофеновой кислоты I-1B (Hoechst); и костимулирующие модификаторы, такие как ORENCIA® (абатацепт) (Bristol-Myers Squibb); энлимомаб (моноклональное антитело против ICAM-1); CDO-855 (гуманизированное антитело, которое специфически связывается с районом комплекса МНС класса II, Celltech); CH-3298 (Chiroscience); ацеметацин (Merck); GW353430 (моноклональное антитело против CD23, Glaxo Wellcome); GR 252025 (ингибитор СОХ02, Glaxo Wellcome); 4162W94 (гуманизированное антитело против CD4, Glaxo Wellcome); азатиоприн (DMARD, Glaxo Wellcome); пеницилламин и фенопрофен (Eli Lilly); и т.п.
Предпочтительными такими лекарственными средствами являются антибиотик, антитело против интегрина, гамма-глобулин, контролирующий боль агент, антагонист интегрина, антитело против CD4, кладрибин, триметопримсульфаметоксазол, Н2-блокатор, ингибитор протонового насоса, циклоспорин, понижающее уровень холестерина лекарственное средство класса «статины», эстрадиол, тестостерон, андроген, гормонозаместительная терапия, ингибитор TNF, такой как ингибитор TNF-альфа, DMARD, NSAID (для лечения, например, симптомов опорно-двигательного аппарата), левотироксин, циклоспорин А, аналог соматастатина, антагонист цитокина (в том числе антагонист рецептора цитокина), антиметаболит, антагонист BAFF, такой как антитело против BAFF или антитело против BR3, особенно антитело против BAFF, иммунодепрессант, такой как метотрексат или кортикостероид, бисфосфонат, гормон и другое антитело против маркера поверхности В-клетки, такое как комбинация ритуксимаба и гуманизированное антитело 2Н7 или другое гуманизированное антитело против CD20.
Более предпочтительными такими лекарственными средствами являются антибиотик, иммунодепрессант, такой как метотрексат или кортикостероид, DMARD, контролирующий боль агент, антагонист интегрина, NSAID, антагонист цитокина, бисфосфонат или гормон, или их комбинация.
В одном особенно предпочтительном варианте осуществления настоящего изобретения второе лекарственное средство представляет собой DMARD, который предпочтительно выбирают из группы, состоящей из ауранофина, хлорохина, D-пеницилламина, инъецируемого золота, перорального золота, гидроксихлорохина, сульфасалазина, миокризина и метотрексата.
В другом таком варианте осуществления настоящего изобретения второе лекарственное средство представляет собой NSAID, который предпочтительно выбирают из группы, состоящей из пентазы, месалазина, асакола, кодеина фосфата, бенорилата, фенбуфена, напросина, диклофенака, этодолака и индометацина, аспирина и ибупрофена.
В другом таком варианте осуществления настоящего изобретения второе лекарственное средство представляет собой контролирующий боль агент, который предпочтительно выбирают из группы, состоящей из парацетамола и декстропропоксифена.
В дополнительном таком варианте осуществления настоящего изобретения второе лекарственное средство представляет иммунодепрессант, который предпочтительно выбирают из группы, состоящей из этанерцепта, инфликсимаба, адалимумаба, лефлуномида, анакинры, азатиоприна, метотрексата и циклофосфамида.
В другом предпочтительном варианте осуществления настоящего изобретения второе лекарственное средство выбирают из группы, состоящей из остеопротегерина (OPG), этанерцепта, инфликсимаба, адалимумаба, кинарета, раптивы, RANKFc, анти-RANKL, памидроната, алендроната, актонеля, золендроната, клодроната, метотрексата, азулфидина, гидроксихлорохина, доксициклина, лефлуномида, сульфасалазина (SSZ), преднизолона, антагониста рецептора интерлейкина-1, преднизона и метилпреднизолона.
В еще предпочтительных вариантах осуществления настоящего изобретения второе лекарственное средство выбирают из группы, состоящей из инфликсимаба, комбинации инфликсимаб/метотрексат (МТХ), этанерцепта, кортикостероида, циклоспорина А, азатиоприна, ауранофина, гидроксихлорохина (HCQ), комбинации преднизолона, МТХ и SSZ, комбинаций МТХ, SSL и HCQ, комбинации циклофосфамида, азатиоприна и HCQ и комбинации адалимумаба с МТХ. Если второе лекарственное средство является кортикостероидом, предпочтительно оно является преднизоном, преднизолоном, метилпреднизолоном, гидрокортизоном или дексаметазоном. Также предпочтительно вводить кортикостероид в более низких количествах, чем количества, используемые, если антитело против CD20 не вводится индивиду, подвергаемому лечению кортикостероидом. Наиболее предпочтительно, когда вторым лекарственным средством является метотрексат.
Все эти вторые лекарственные средства можно использовать в комбинации друг с другом или сами по себе с первым лекарственным средством, так что выражение «второе лекарственное средство», используемое здесь, не означает, что это единственное лекарственное средство помимо первого лекарственного средства, соответственно. Таким образом, второе лекарственное средство необязательно является одним лекарственным средством, и оно может включать или состоять из более чем одного такого лекарственного средства.
Эти вторые лекарственные средства, изложенные здесь, используются, как правило, в тех же самых дозах и с помощью способов введения, используемых здесь ранее, или в дозах, составляющих приблизительно 1-99% используемых ранее доз. Если такие вторые лекарственные средства, тем не менее, используются, предпочтительно их использовать в более низких количествах, чем в том случае, когда первое лекарственное средство не присутствует, особенно при введениях доз, следующих за первоначальным введении дозы, с первым лекарственным средством, для того, чтобы устранить или уменьшить побочные эффекты, вызванные в связи с этим.
Для способов повторного лечения, описанных здесь, когда второе лекарственное средство вводится в эффективном количестве вместе с воздействием антагонистом или антителом, его можно вводить с использованием любого числа воздействий, например, только одного воздействия или более одного воздействия. В одном варианте осуществления второе лекарственное средство вводят с использованием первоначального воздействия. В другом варианте осуществления второе лекарственное средство вводят с использованием первоначального и второго воздействий. В еще дополнительном варианте осуществления второе лекарственное средство вводят с использованием всех воздействий. Предпочтительно, чтобы после первоначального воздействия, например, стероидом, количество такого второго лекарственного средства уменьшалось или исключалось для того, чтобы уменьшить воздействие на индивида агента с побочными эффектами, такого как преднизон, преднизолон, метилпреднизолон и циклофосфамид.
Комбинированное введение второго лекарственного средства включает совместное введение (сопутствующее введение), используя отдельные композиции или единственную фармацевтическую композицию, и последующее введение в том или ином порядке, причем предпочтительно, чтобы существовал период времени, пока оба (или все) активные агенты (лекарственные средства) одновременно проявят свои биологические активности.
Антитело или антагонист настоящего изобретения вводят любым подходящим способом, в том числе с помощью парентерального, местного, подкожного, внутрибрюшинного, внутрилегочного, интраназального введения и/или введения внутрь повреждения. Парентеральные инфузии включают внутримышечное, внутривенное, внутриартериальное, внутрибрюшинное или подкожное введение. Также предусмотрено интратекальное введение (смотри, например заявку US 2002/0009444, Grillo-Lopez, A, касающуюся интратекальной доставки антитела против CD20). Кроме того, антитело или антагонист можно подходящим образом ввести с помощью импульсной инфузии, например, с использованием уменьшающихся доз антитела или антагониста. Предпочтительно, дозы вводят внутривенно или подкожно, и более предпочтительно с помощью внутривенной инфузии(й).
Если предоставляются множественные воздействия антителом, каждое воздействие может быть предоставлено с использованием одного того же или различных способов введения. В одном варианте осуществления настоящего изобретения каждое воздействие осуществляют с помощью внутривенного введения. В другом варианте осуществления настоящего изобретения каждое воздействие осуществляют с помощью подкожного введения. В еще одном варианте осуществления настоящего изобретения воздействия осуществляют с помощью внутривенного и подкожного введения.
В одном варианте осуществления настоящего изобретения антитело против CD20 вводят в виде медленного внутривенного вливания, а не внутривенного проталкивания или болюса. Например, стероид, такой как преднизолон или метилпреднизолон (например, приблизительно 80-120 мг внутривенно, более предпочтительно приблизительно 100 мг внутривенно) вводят за приблизительно 30 минут до какой-либо инфузии антитела против CD20. Антитело против CD20 вводят, например, через выделенную линию.
Для первоначальной дозы многодозового воздействия антителом против CD20 или для единственной дозы, если в воздействие включена только одна доза, такую инфузию предпочтительно начинают со скоростью приблизительно 50 мг/час. Ее можно усилить, например, при скорости приблизительно 50 мг/час приращения каждые приблизительно 30 минут до максимума, составляющего приблизительно 400 мг/час. Однако, если у индивида имеет место связанная с инфузией реакция, скорость инфузии предпочтительно уменьшают, например, до половины текущей скорости, например, со 100 мг/час до 50 мг/час. Предпочтительно инфузию такой дозы антитела против CD20 (например, общая доза приблизительно 1000 мг) завершают через приблизительно 255 минут (4 часа 15 минут). Необязательно индивиды получают профилактическое лечение с использованием ацетаминофена/парацетамола (например, приблизительно 1 г) и дифенгидрамина HCl (например, приблизительно 50 мг или эквивалентная доза схожего агента) перорально за приблизительно 30-60 минут до начала инфузии.
Если для достижения общего воздействия назначается более одной инфузии (дозы) антитела против CD20, вторую или последующую инфузию антитела против CD20 в этом варианте инфузий предпочтительно начинают при более высокой скорости, чем первоначальную инфузию, например, при приблизительно 100 мг/час. Эту скорость можно увеличить, например, при скорости приблизительно 100 мг/час приращения каждые приблизительно 30 минут до максимума, составляющего приблизительно 400 мг/час. Для индивидов, у которых место связанная с инфузией реакция, скорость инфузии предпочтительно уменьшают до половины этой скорости, например, со 100 мг/час до 50 мг/час. Предпочтительно инфузию такой второй или последующей дозы антитела против CD20 (например, общая доза приблизительно 1000 мг) завершают через приблизительно 195 минут (3 часа 15 минут).
В другом варианте осуществления настоящего изобретения обеспечивается способ лечения повреждения сустава у индивида, включающий введение индивиду антагониста в отношении маркера поверхности В-клетки, такого как антитело против такого маркера, например, антитело против CD20, и рентгенографическую проверку индивида, по крайней мере спустя приблизительно 52 недели от момента введения, с помощью которой определяют уменьшение повреждения сустава по сравнению с базовой линией до введения, причем количество вводимого антагониста или антитела, такого как антитело против CD20, является эффективным для получения уменьшения повреждения сустава, что является указанием эффективного лечения индивида.
В этом способе предпочтительно проверкой определяется общий модифицированный показатель Sharp. В другом предпочтительном варианте осуществления изобретения антагонистом является антитело против CD20. Более предпочтительно антитело против CD20 является ритуксимабом или гуманизированным антителом 2Н7, включающим последовательности вариабельных доменов SEQ ID NO: 2 и 8, или гуманизированным антителом 2Н7, включающим последовательности вариабельных доменов SEQ ID NO: 39 и 40, или гуманизированным антителом 2Н7, включающим последовательности вариабельных областей SEQ ID NO: 32 и 33, или гуманизированным антителом 2Н7, включающим вариабельный домен тяжелой цепи с заменами N100A, или D56A и N100A, или D56A, N100Y и S100aR в SEQ ID NO: 8 и вариабельный домен легкой цепи с заменами M32L, или S92A, или M32L и S92A в SEQ ID NO: 2.
В другом предпочтительном варианте осуществления настоящего изобретения повреждение сустава вызвано артритом, предпочтительно RA и более предпочтительно активным RA на ранней стадии. В другом предпочтительном варианте осуществления настоящего изобретения индивида ранее не подвергали лечению иммунодепрессантом до введения первой дозы антагониста или антитела, такого как антитело против CD20, в способе лечения. В предпочтительном варианте осуществления настоящего изобретения антагонист или антитело вводят в дозе, составляющей приблизительно 0,4-4 грамма, и более предпочтительно антагонист или антитело вводят в дозе, составляющей приблизительно 0,4-1,3, с частотой одна-четыре дозы в пределах периода, составляющего приблизительно один месяц. Еще более предпочтительно доза составляет приблизительно 500 мг-1,2 грамма, а в других вариантах осуществления она составляет приблизительно 750 мг-1,1 грамма. В таких аспектах антагонист или антитело предпочтительно вводят в виде двух-трех доз и/или вводят в пределах периода, составляющего приблизительно 2-3 недели.
В другом аспекте такой способ дополнительно включает повторное лечение индивида путем предоставления дополнительного введения индивиду антагониста, такого как антитело против CD20, в количестве, эффективном для получения непрерывного или сохраняемого уменьшения повреждения сустава по сравнению с эффектом предшествующего введения антагониста или антитела, такого как антитело против CD20. В одном аспекте этого варианта осуществления антагонист или антитело, такое как антитело против CD20, дополнительно вводят индивиду даже, если у индивида нет клинического улучшения во время рентгенографической проверки после предшествующего введения. Повторное лечение можно начинать через по крайней мере приблизительно 24 недели после первого введения антагониста, такого как антитело против CD20, и необязательно начинают одно или несколько дополнительных повторных лечений. В другом варианте осуществления дополнительное повторное лечение начинают через по крайней мере приблизительно 24 недели после второго введения антагониста, такого как антитело против CD20. В дополнительном предпочтительном аспекте повреждение сустава уменьшалось после повторного лечения по сравнению со степенью повреждения сустава после первой рентгенографической оценки.
Предпочтительно в этом способе, относящемся к оценке спустя приблизительно 52 недели, вводят в эффективном количестве второе лекарственное средство, причем антагонист или антитело, такое как антитело против CD20, является первым лекарственным средством. В одном аспекте второе лекарственное средство представляет собой более одного лекарственного средства. В другом аспекте второе лекарственное средство представляет собой антибиотик, иммунодепрессант, модифицирующее заболевание противоревматическое лекарственное средство (DMARD), контролирующий боль агент, антагонист интегрина, нестероидное противовоспалительное лекарственное средство (NSAID), антагонист цитокина, бисфосфонат или гормон, или их комбинацию, более предпочтительно метотрексат. Индивид может быть отрицательным или положительным в отношении ревматоидного фактора. Также предпочтительно вводить антагонист, такой как антитело против CD20, внутривенно или подкожно, наиболее предпочтительно внутривенно.
Обсуждение способов продуцирования, модифицирования и приготовления таких антител приводится ниже.
III. Продуцирование антител
В способах и изделиях производства настоящего изобретение используется, или включено, антитело, которое связывается с маркером поверхности В-клетки, особенно антитело, которое связывается с CD20. В соответствии с этим здесь описываются способы получения таких антител.
Антиген CD20, используемый для продуцирования, или скринирования, антитела (антител) может представлять собой, например, растворимую форму CD20 или его часть, содержащую желаемый эпитоп. Альтернативно или дополнительно, для получения, или скринирования, антитела (антител) можно использовать клетки, экспрессирующие CD20 на своей клеточной поверхности. Для квалифицированных в данной области техники специалистов очевидны другие формы CD20, применимые для получения антител.
Ниже приведено описание в виде примеров способов продуцирования антител, применимых в соответствии с настоящим изобретением.
(i) Поликлональные антитела
Предпочтительно поликлональные антитела индуцируют у животных с помощью многократных подкожных или внутрибрюшинных инъекций релевантного антигена и адъюванта. Может быть полезным конъюгирование релевантного антигена с белком, являющимся иммуногенным для иммунизируемых видов, например, с гемоцианином лимфы улитки, сывороточным альбумином, бычьим тироглобулином или соевым ингибитором трипсина, используя бифункциональный или дериватизирующий агент, например, малеимидобензоилсульфосукцинимидовый эфир (конъюгация через остатки цистеина), N-гидроксисукцинимид (через остатки лизина), глютаральдегид, янтарный ангидрид, SOCl2 или R1N=C=NR, где R и R1 представляют собой различные алкильные группы.
Животных иммунизируют с использованием антигена, иммуногенных конъюгатов или производных с помощью объединения, например, 100 мкг или 5 мкг белка или конъюгата (для кроликов или мышей, соответственно) с 3 объемами полного адъюванта Фрейнда и введения раствора внутрикожно во множество мест. Спустя один месяц животных подвергают повторной иммунизации с использованием 1/5-1/10 первоначального количества пептида или конъюгата в полном адъюванте Фрейнда путем подкожной инъекции в множество мест. Спустя 7-14 дней у животных берут кровь и сыворотку анализируют в отношении титра антител. Животных подвергают повторной иммунизации до достижения титром плата. Предпочтительно животного повторно иммунизируют конъюгатом того же самого антигена, но конъюгированного с отличным белком и/или через отличный сшивающий агент. Конъюгаты также можно получить в культуре рекомбинантных клеток в виде слитых белков. Также агрегирующие агенты, такие как квасцы, подходящим образом используют для усиления иммунного ответа.
(ii) Моноклональные антитела
Моноклональные антитела получают из популяции по существу гомогенных антител, т.е. индивидуальные антитела, образующие популяцию, являются идентичными и/или связываются с одним и тем же эпитопом, за исключением возможных вариантов, которые возникают во время продукции моноклонального антитела, при этом такие варианты, как правило, присутствуют в малых количествах. Таким образом, атрибут «моноклональное» показывает, что свойством антитела является то, что оно не является смесью дискретных или поликлональных антител.
Например, моноклональные антитела можно получить с помощью гибридомной технологии, впервые описанной Kohler и др. (Nature, 256: 495 (1975)), или методов рекомбинантных ДНК (смотри, например, патент США № 4816567).
При гибридомной технологии мышь или другое подходящее животное-хозяин, такое как хомяк, иммунизируют, как описано здесь выше, для индуцирования лимфоцитов, которые продуцируют или способны продуцировать антитела, которые специфически связываются с используемым для иммунизации белком. Альтернативно, лимфоциты можно подвергнуть иммунизации in vitro. Лимфоциты затем сливают с миеломными клетками, используя подходящий агент слияния, такой как полиэтиленгликоль, с образованием гибридомной клетки (Goding, Monoclonal Antibodies: Principles and Practice, pp. 59-103 (Academic Press, 1986)).
Полученные таким образом гибридомные клетки засевают и выращивают в подходящей для культивирования среде, которая предпочтительно содержит одно и несколько веществ, которые ингибируют рост или выживание не слившихся, родительских миеломных клеток. Например, если в исходных миеломных клетках отсутствует фермент гипоксантин-гуанин-фосфорибозилтрансфераза (HGPRT или HPRT), среда для культивирования гибридом обычно включает вещества: гипоксантин, аминоптерин и тимидин (среда НАТ) - которые предотвращают рост дефицитных в отношении HGPRT клеток.
Предпочтительными миеломными клетками являются такие клетки, которые эффективно сливаются, поддерживают стабильную продукцию на высоком уровне антител отобранными продуцирующими антитело клетками и чувствительны к среде, такой как среда НАТ. Среди них предпочтительными линиями миеломных клеток являются линии крысиных миеломных клеток, такие как линии, происходящие из опухолей мыши МОРС-21 и МРС-11, доступные от Salk Institute Cell Distribution Center, San Diego, California США, и клетки SP-2 или X63-Ag8-653, доступные из Американской коллекции типовых культур, Rockville, Maryland, США. Для продукции моноклональных антител человека также описаны линии миеломных клеток человека и линии гетеромиеломных мышиных-человеческих клеток (Kozbor, J. Immunol., 133: 3001 (1984); Brodeur et al., Monoclonal Antibody Production Techniques and Aplications, pp. 51-63 (Marcel Dekker, Inc., New York, 1987)).
Среду для культивирования, в которой выращиваются гибридомные клетки, анализируют на продукцию моноклональных антител, направленных против антигена. Предпочтительно специфичность связывания моноклональных антител, продуцируемых гибридомными клетками, определяют с помощью иммунопреципитации или с помощью анализа связывания in vitro, такого как радиоиммуноанализ (RIA) или твердофазный иммуноферментный анализ (ELISA).
Аффинность связывания моноклонального антитела можно, например, определить с помощью анализа Scatchard (Munson et al., Anal. Biochem., 107: 220 (1980)).
После идентификации гибридомных клеток, продуцирующих антитела желаемой специфичности, аффинности и/или активности, клоны можно субклонировать с помощью методов серийных разведений и выращивать с использованием стандартных способов (Goding, Monoclonal Antibodies: Pripciples and Practice, pp. 59-103 (Academic Press, 1986)). Подходящие для этой цели среды для культивирования включают, например, среду D-MEM или RPMI-1640. Кроме того, гибридомные клетки можно выращивать in vivo в виде асцитной жидкости опухолей у животных.
Моноклональные антитела, секретируемые субклонами, подходящим образом отделяют от среды для культивирования, асцитной жидкости или сыворотки с помощью традиционных методик очистки иммуноглобулинов, таких как, например, хроматография на протеин-А-SEPHAROSETM, гидроксилапатите, гель-электрофорез, диализ или аффинная хроматография.
ДНК, кодирующую моноклональные антитела, легко выделить и секвенировать с использованием традиционных методик (например, с использованием олигонуклеотидных проб, которые способны специфически связываться с генами, кодирующими тяжелую и легкую цепи крысиных антител). Гибридомные клетки служат в качестве предпочтительного источника такой ДНК. После выделения ДНК можно встроить в экспрессирующие векторы, которыми затем трансфицируют клетки-хозяев, такие как клетки E.coli, обезьяньи клетки COS, клетки яичника китайского хомячка (СНО) или миеломные клетки, которые в противном случае не продуцируют белок в виде иммуноглобулина, для получения синтеза моноклональных антител в рекомбинантных клетках-хозяевах. Обзорные статьи, касающиеся рекомбинантной экспрессии в бактериях ДНК, кодирующей антитело, включают Skerra et al., Curr. Opinion in Immunol., 5: 256-262 (1993) и Plückthum, Immunol. Revs., 130: 151-188 (1992).
В дополнительном варианте осуществления антитела или фрагменты антител можно выделить из фаговых библиотек антител, используя методы, описанные McCafferty и др.(Nature, 348: 552-554 (1990)). Clackson и др. (Nature, 352: 624-628 (1991)) и Marks и др. (J. Mol. Biol., 222: 581-597 (1991)) описывают выделение крысиных и человеческих антител, соответственно, используя фаговые библиотеки. В последующих публикациях описывается получение антител человека с высокой аффинностью с помощью перетасовки цепей (Marks et al., Bio/Technology, 10: 779-783 (1992)), а также комбинаторного инфицирования и in vivo рекомбинации в качестве стратегии конструирования больших фаговых библиотек (Waterhouse et al., Nuc. Acids. Res., 21: 2265-2266 (1993)). Таким образом, эти методы являются жизнеспособными альтернативами традиционным гибридомным технологиям для выделения моноклональных антител.
ДНК также можно модифицировать, например, путем замены последовательностями, кодирующими константные домены тяжелой и легкой цепи человека, гомологичных крысиных последовательностей (патент США № 4816567; Morrison et al., Proc. Natl Acad. Sci. USA, 81: 6851 (1984)), и путем ковалентного соединения с кодирующей иммуноглобулин последовательностью всей или части последовательности, кодирующей не являющийся иммуноглобулином полипептид.
Обычно такими не являющимися иммуноглобулином полипептидами замещают константные домены антитела или ими замещают вариабельные домены одного антигенсвязывающего сайта антитела с созданием химерного двухвалентного антитела, включающего один антигенсвязывающий сайт, имеющий специфичность в отношении антигена, и другой антигенсвязывающий сайт, имеющий специфичность в отношении отличного антигена.
Кроме того, антитела, включающие вариантный фрагмент Fc с высокой аффинностью к FcγR, применимы для лечения заболеваний, при которых желательна увеличенная эффективность функции эффекторных клеток, таких как аутоиммунные заболевания, изложенные, например, в US 2005/0037000 и WO 2004/63351 (Macrogenics, Inc. STAVENHAGEN et al).
(iii) Гуманизированные антитела
Способы гуманизации антител не являющихся человеком видов описаны в данной области техники. Предпочтительно гуманизированное антитело имеет один или несколько аминокислотных остатков, введенных в него из источника, не являющегося человеком. На эти нечеловеческие аминокислотные остатки часто приводится ссылка в виде «импортированные» остатки, которые обычно берут из «импортируемого» вариабельного домена. Гуманизацию можно по существу выполнить согласно способу Winter и соавторов (Jones et al., Nature, 321: 522-525 (1986); Riechmann et al., Nature, 332: 323-327 (1988); Verhoeyen et al., Science, 239: 1534-1536 (1988)) путем замещения последовательностями гипервариабельных участков соответствующих последовательностей антитела человека. Соответственно, такие «гуманизированные» антитела являются химерными антителами (патент США № 4816567), в которых заметно менее чем интактный вариабельный домен человека был замещен соответствующей последовательностью из не являющихся человеком видов. На практике гуманизированные антитела обычно представляют собой антитела человека, в которых некоторые остатки гипервариабельных участков и возможно некоторые остатки FR замещены остатками из аналогичных сайтов антител грызунов.
Выбор вариабельных доменов человека как легкой, так и тяжелой цепи, используемых при получении гуманизированных антител, очень важен для снижения антигенности. В соответствии с так называемым способом «оптимальной подгонки» последовательность вариабельного домена антитела грызунов используют для скрининга всей библиотеки известных последовательностей вариабельных доменов человека. Последовательность человека, являющуюся самой близкой к последовательности грызуна, затем берут в качестве каркасной области (FR) человека для гуманизированного антитела (Sims et al., J. Immunol., 151: 2296 (1993); Chothia et al., J. Mol. Biol., 196: 901 (1987)). В другом способе используется конкретная каркасная область, происходящая из консенсусной последовательности всех антител человека конкретной подгруппы вариабельных областей тяжелой или легкой цепи. Одну и ту же каркасную область можно использовать для нескольких различных гуманизированных антител (Carter et al.,Proc. Natl. Acad. Sci. USA, 89: 4285 (1992); Presta et al., J. Immunol., 151: 2623 (1993)).
Кроме того, важно, чтобы антитела подвергались гуманизации с сохранением высокой аффинности к антигену и других благоприятных биологических свойств. Для достижения этой цели, в соответствии с предпочтительным способом, гуманизированные антитела готовят с использованием процесса анализа исходных последовательностей и различных концептуальных гуманизированных продуктов, используя трехмерные модели исходной и гуманизированных последовательностей. Трехмерные модели иммуноглобулинов обычно доступны и знакомы квалифицированным в данной области техники специалистам. Имеются компьютерные программы, которые иллюстрируют и отображают возможные трехмерные конформационные структуры отобранных последовательностей иммуноглобулинов-кандидатов. Обследование этих отображений делает возможным анализ вероятной роли остатков в функционировании последовательности иммуноглобулина-кандидата, т.е. анализ остатков, которые оказывают влияние на способность иммуноглобулина-кандидата связываться со своим антигеном. Таким образом остатки FR можно отобрать и объединить из реципиентных и импортируемых последовательностей так, чтобы достигалась желаемая характеристика антитела, например, увеличенная аффинность к антигену(ам)-мишени. Как правило, остатки гипервариабельных участков непосредственно и наиболее заметно оказывают влияние на связывание с антигеном.
(iv) Антитела человека
В качестве альтернативы гуманизации можно получить антитела человека. Например, в настоящее время можно получить трансгенных животных (например, мышей), которые способны, при иммунизации, продуцировать полный набор антител человека в отсутствие эндогенной продукции иммуноглобулинов. Например, описано, что гомозиготная делеция гена области соединения (JH) тяжелой цепи антитела у химерных и зародышевых мутантных мышей приводит к полному ингибированию эндогенной продукции антитела. Перенос набора генов зародышевых иммуноглобулинов человека в таких зародышевых мутантных мышей приведет к продукции антител человека при введении антигена (смотри, например, Jakobovits et al., Proc. Natl. Acad. Sci. USA, 90: 2551 (1993); Jakobovits et al., Nature, 362: 255-258 (1993); Bruggermann et al., Year in Immuno., 7: 33 (1993) и патенты США № 5591669, 5589369 и 5545807).
Альтернативно для получения антител человека и фрагментов антител in vitro, из наборов генов вариабельных (V) доменов иммуноглобулинов от не иммунизированных доноров, можно использовать технологию фагового дисплея (McCafferty et al., Nature 348: 552-553 (1990)). В соответствии с этой технологией гены V-доменов антитела клонируют с сохранением рамки считывания в ген покровного белка, основного или неосновного, нитевидного фага, такого как М13 или fd, и представляют в виде функциональных фрагментов антитела на поверхности фаговой частицы. Поскольку нитевидная частица содержит копию одноцепочечной ДНК фагового генома, отборы на основе функциональных свойств антитела также приводят к отбору гена, кодирующего антитело, проявляющее эти свойства. Таким образом, фаг имитирует некоторые свойства В-клетки. Фаговый дисплей можно выполнить в различных форматах; для обзора смотри, например, Johnson et al., Current Opinion in Structural Biology 3: 564-571 (1993). Для фагового дисплея можно использовать несколько источников сегментов V-генов. Clackson и др. (Nature, 352: 624-628 (1991)) выделили набор разнообразных антител против оксазолона из небольшой произвольной комбинаторной библиотеки V-генов, происходящих из селезенок иммунизированных мышей. Можно сконструировать набор V-генов от неиммунизированных доноров, являющихся людьми, и антитела против набора разнообразных антигенов (в том числе аутоантигенов) можно выделить, следуя по существу методам, описанным Marks и др. (J. Mol. Biol. 222: 581-597 (1991)) или Griffith и др. (EMBO J. 12: 725-734 (1993)). Смотри также патенты США № 5565332 и 5573905.
Антитела человека могут также продуцироваться in vitro активированными В-клетками (смотри патенты США № 5567610 и 5229275).
(v) Фрагменты антител
Для получения фрагментов антител были разработаны различные методы. Традиционно такие фрагменты получали через протеолитическое расщепление интактных антител (смотри, например, Morimoto et al., Journal of Biochemical and Biophysical Methods 24: 107-117 (1992); Brennan et al., Science 229: 81 (1985)). Однако в настоящее время эти фрагменты могут продуцироваться непосредственно рекомбинантными клетками-хозяевами. Например, фрагменты антител можно выделить из фаговых библиотек антител, описанных выше. Альтернативно, Fab'-SH-фрагменты можно непосредственно выделить из E.coli и химически связать с образованием фрагментов F(ab')2 (Carter et al., Bio/Technology 10: 163-167 (1992)). В соответствии с другим подходом фрагменты F(ab')2 можно выделить непосредственно из культуры рекомбинантных клеток-хозяев. Для квалифицированного специалиста-практика очевидны другие методы получения фрагментов антител. В других вариантах осуществления настоящего изобретения выбранным антителом является одноцепочечный Fv-фрагмент (scFv). Смотри WO 93/16185, патенты США № 5571894 и 5587458. Фрагмент антитела может также быть «линейным антителом», например, описанным в патенте США № 5641870. Такие фрагменты - линейные антитела могут быть моноспецифическими или биспецифическими.
(vi) Биспецифические антитела
Биспецифические антитела представляют собой антитела, обладающие специфичностями связывания с по крайней мере двумя различными эпитопами. Например, биспецифические антитела могут связываться с двумя различными эпитопами антигена CD20. Другие такие антитела могут связываться с CD20 и, кроме того, со вторым маркером поверхности В-клетки. Альтернативно, связывающееся с CD20 плечо можно объединить с плечом, которое связывается со стимулирующей молекулой на лейкоците, такой как молекула рецептора Т-клетки (например, CD2 или CD3), или Fc-рецепторами для IgG (FcγR), такими как FcγRI (CD64), FcγRII (CD32) и FcγRIII (CD16), для того, чтобы сфокусировать клеточные защитные механизмы на В-клетке. Биспецифические антитела можно также использовать для локализации определенных агентов на В-клетке. Эти антитела обладают связывающим CD20 плечом и плечом, которое связывает агент (например, метотрексат). Биспецифические антитела можно приготовить в виде полноразмерных антител или фрагментов антител (например, биспецифические антитела F(ab')2).
Способы получения биспецифических антител известны в данной области техники. Традиционное получение полноразмерных биспецифических антител основано на коэкспрессии пар тяжелая цепь-легкая цепь двух иммуноглобулинов, где две цепи имеют различные специфичности (Millstein et al., Nature, 305: 537-539 (1983)). Из-за случайного расхождения тяжелой и легкой цепей иммуноглобулинов эти гибридомы (квадромы) продуцируют возможную смесь 10 различных молекул антител, из которых только одна имеет правильную биспецифическую структуру. Очистка правильной молекулы, которую обычно осуществляют с помощью стадий аффинной хроматографии, является довольно громоздкой, а выходы продуктов низкими. Схожие методики раскрыты в WO 93/08829 и Traunecker и др. (EMBO J., 10: 3655-3659 (1991)).
В соответствии с отличным подходом вариабельные домены антитела с желаемыми специфичностями связывания (сайтами соединения антитела с антигеном) сливают с последовательностями константных доменов иммуноглобулинов. Предпочтительно слияние осуществляют с константным доменом тяжелой цепи иммуноглобулина, включающим по крайней мере часть шарнирной, СН2 и СН3 областей. Предпочтительно, чтобы первая константная область тяжелой цепи (СН1), содержащая сайт, необходимый для связывания легкой цепи, присутствовала в по крайней мере одном из слияний. ДНК, кодирующие слияния с тяжелой цепью иммуноглобулина и, если желательно, легкую цепь иммуноглобулина, встраивают в отдельные экспрессирующие векторы и котрансфицируют в подходящий организм-хозяина. Это обеспечивает большую гибкость в регулировании соотношений трех полипептидных фрагментов в тех вариантах осуществления, при которых неравное соотношение трех полипептидных цепей, используемых в конструкции, обеспечивает оптимальные выходы. Однако можно встроить последовательности, кодирующие две или все три полипептидные цепи, в один экспрессирующий вектор, когда экспрессия по крайней мере двух полипептидных цепей в равных соотношениях приводит к высоким выходам, или когда соотношения не имеют особого значения.
В предпочтительном варианте этого подхода биспецифические антитела состоят из гибридной тяжелой цепи иммуноглобулина с первой специфичностью связывания в одном плече и гибридной пары тяжелая цепь-легкая цепь иммуноглобулина (с обеспечением второй специфичности связывания) в другом плече. Обнаружено, что эта несимметричная структура облегчает отделение желаемого биспецифического соединения от нежелательных комбинаций цепей иммуноглобулина, поскольку присутствие легкой цепи иммуноглобулина только в одной половине биспецифической молекулы обеспечивает легкость способа отделения. Этот подход описан в WO 94/04690. В отношении дополнительных подробностей получения биспецифических антител смотри, например, Suresh et al., Methods in Enzymology, 121: 210 (1986).
В соответствии с другим подходом, описанным в патенте США № 5731168, область контакта между парой молекул антитела можно сконструировать с помощью генно-инженерных методов с максимизацией процента гетеродимеров, извлекаемых из культуры рекомбинантных клеток. Предпочтительная область контакта включает по крайней мере часть СН3-домена константного домена антитела. В этом способе одну или несколько небольших боковых цепей аминокислот с поверхности контакта первой молекулы антитела замещают более большими боковыми цепями (например, тирозином или триптофаном). Компенсирующие «углубления» размером, идентичным или схожим с размером более больших боковых цепей (цепи), создают на поверхности контакта второй молекулы антитела путем замены больших боковых цепей аминокислот более маленькими (например, аланином или треонином). Это обеспечивает механизм увеличения выхода гетеродимера в сравнении с другими нежелательными конечными продуктами, такими как гомодимеры.
Биспецифические антитела включают сшитые антитела или «гетероконъюгаты». Например, одно из антител в гетероконъюгате может быть связано с авидином, другое - с биотином. Такие антитела предложили, например, для нацеливания клеток иммунной системы на нежелательные клетки (патент США № 4676980) и для лечения ВИЧ-инфекции (WO 91/00360, WO 92/200373 и ЕР 03089). Антитела в виде гетероконъюгатов можно получить, используя любые подходящие способы сшивания. Подходящие сшивающие агенты хорошо известны в данной области техники и раскрыты в патенте США № 4676980 вместе с рядом методов сшивания.
В литературе также описаны методы получения биспецифических антител из фрагментов антител. Например, биспецифические антитела можно приготовить с помощью химической связи. Brennan и др. (Science, 229: 81 (1985)) описывают методики, в которой интактные антитела протеолитически расщепляют с образованием фрагментов F(ab')2. Эти фрагменты восстанавливают в присутствии дитиольного комплексообразующего агента натрия арсенита для стабилизации вицинальных дитиолов и предотвращения межмолекулярного дисульфидного образования. Образованные фрагменты Fab' затем превращают в производные тионитробензоата (TNB). Одно из производных Fab'-TNB затем вновь превращают в Fab'-тиол с помощью восстановления с использованием меркаптоэтиламина и смешивают с эквимолярным количеством другого производного Fab'-TNB с образованием биспецифического антитела. Полученные биспецифические антитела можно использовать в качестве агентов для избирательной иммобилизации ферментов.
Также описаны различные методы получения и выделения фрагментов биспецифических антител непосредственно из культуры рекомбинантных клеток. Например, биспецифические антитела получали, используя лейциновые молнии (Kostelny et al., J. Immunol., 148(5): 1547-1553 (1992). Пептиды в виде лейциновых молний белков Fos и Jun связывали с частями Fab' двух различных антител с помощью слияния генов. Антитела в виде гомодимеров восстанавливали в шарнирной области с образованием мономеров и затем вновь окисляли с образованием антител в виде гетеродимеров. Этот способ можно также использовать для получения антител в виде гомодимеров. Технология «диател», описанная Hollinger и др. (Proc. Natl. Acad. Sci. USA, 90: 6444-6448 (1993)) обеспечила альтернативный механизм получения фрагментов биспецифических антител. Фрагменты включают вариабельный домен тяжелой цепи (VH), соединенный с вариабельным доменом легкой цепи (VL) с помощью линкера, являющегося слишком коротким для того, чтобы сделать возможным спаривание двух доменов одной и той же цепи. Соответственно, домены VH и VL одного фрагмента вынуждены спариваться с комплементарными доменами VH и VL другого фрагмента с образованием, таким образом, двух антигенсвязывающих сайтов. Также сообщалось о другой стратегии получения фрагментов биспецифических антител с помощью использования димеров одноцепочечных Fv-фрагментов (sFv) (смотри Gruber et al., J. Immunol., 152: 5368 (1994)).
Предусмотрены антитела с более чем двумя валентностями. Например, можно приготовить триспецифические антитела (Tutt et al., J. Immunol. 147: 60 (1991)).
IV. Конъюгаты и другие модификации антитела
Здесь предусмотрены модификации антитела. Таким образом, в одном варианте осуществления антитело можно конъюгировать с другой молекулой, например, для увеличения времени полужизни или стабильности или в противном случае для улучшения фармакокинетики антитела. Например, антитело можно связать с одним из разнообразных небелковоподобных полимеров, например полиэтиленгликолем (PEG), полипропиленгликолем, полиоксиалкиленами или сополимерами полиэтиленгликоля и полипропиленгликоля. Фрагменты антитела, такие как Fab', связанные с одной или несколькими молекулами PEG, являются особенно предпочтительным вариантом осуществления настоящего изобретения.
Описываемые здесь антитела также можно приготовить в виде липосом. Содержащие антитело липосомы готовят с помощью способов, известных в данной области техники, таких как способы, описанные Epstein и др. (Proc. Natl. Acad. Sci. USA, 82: 3688 (1985)); Hwang и др. (Proc. Natl. Acad. Sci. USA, 77: 4030 (1980)); в патентах США № 4485045 и 4544545 и заявке WO 97/38731, опубликованной 23 октября 1997. Липосомы с увеличенным временем циркуляции раскрыты в патенте США № 5013556.
Особенно полезные липосомы можно получить с помощью обращенно-фазового метода напыления с использованием липидной композиции, включающей фосфатидилхолин, холестерин и PEG-дериватизированный фосфатидилэтаноламин (PEG-PE). Липосомы пропускают через фильтры с определенным размером пор с получением липосом с желаемым диаметром. Фрагменты Fab' антитела настоящего изобретения можно конъюгировать с липосомами, как описано Martin и др. (J. Biol. Chem. 257: 286-288 (1982)) посредством реакции дисульфидного обмена.
Предусмотрена модификация(и) аминокислотной последовательности белковых или пептидных антител, описанных здесь. Например, может быть желательным увеличение аффинности связывания и/или улучшение других биологических свойств антитела. Варианты аминокислотной последовательности антитела готовят путем введения соответствующих нуклеотидных замен в нуклеиновую кислоту антитела или путем пептидного синтеза. Такие модификации включают, например, делеции и/или вставки и/или замены остатков в пределах аминокислотных последовательностей антитела. В конечной конструкции может присутствовать любая комбинация делеции, вставки и замены при условии, что конечная конструкция обладает желаемыми свойствами. Аминокислотные замены могут также изменять посттрансляционную модификацию антитела, такую как изменение числа или положения сайтов гликозилирования.
Пригодный способ идентификации определенных остатков или районов антитела, имеющих предпочтительное для мутагенеза месторасположение, называют «сканирующим аланином мутагенезом», как описано Cunningham и Wells (Science, 244: 1081-1085 (1989)). В этом способе остаток или группу остатков-мишеней идентифицируют (например, заряженные остатки, такие как arg, asp, his, lys и glu) и замещают нейтральной или отрицательно заряженной аминокислотой (наиболее предпочтительно аланином или полиаланином) для оказания влияния на взаимодействие аминокислот с антигеном. Месторасположения аминокислот, демонстрирующие функциональную чувствительность к заменам, затем уточняют путем введения дополнительных или других вариантов в места замены. Таким образом, несмотря на то, что место введения вариации в аминокислотную последовательность заранее определено, природа самой мутации необязательно должна быть заранее определена. Например, для анализа функционирования мутации в данном месте осуществляют сканирующий аланином мутагенез или произвольный мутагенез в кодоне-мишени или районе-мишени и экспрессированные варианты антитела скринируют на желаемую активность.
Вставки в аминокислотную последовательность включают амино- и/или карбоксил-концевые слияния, длина которых находится в диапазоне от одного остатка до полипептидов, содержащих сотню или более остатков, а также вставки внутрь последовательности одного или множества аминокислотных остатков. Примеры концевых вставок включают антитело с метионильным остатком на N-конце или антитело, слитое с полипептидом или полимером. Другие вставочные варианты молекулы антитела включают слияние фермента или полипептида, который увеличивает полужизнь антитела в сыворотке, с N- или С-концом антитела.
Другим типом варианта является вариант аминокислотной замены. В этих вариантах по крайней мере один аминокислотный остаток в молекуле антитела заменен отличным остатком. Представляющие наибольший интерес места для мутагенеза антитела с заменой включают гипервариабельные участки, но также предусмотрены изменения в FR. Консервативные замены представлены в таблице ниже под заголовком «предпочтительные замены». Если такие замены приводят к изменению биологической активности, то можно ввести более существенные замены, обозначенные в таблице ниже «примеры замен» или дополнительно описанные ниже со ссылкой на классы аминокислот, и продукты скринируют.
Существенные модификации биологических свойств антитела осуществляют путем отбора замен, которые значительно отличаются по их эффекту на сохранение (а) структуры полипептидного остова в области замены, например, в виде складчатой или спиральной конформации, (б) заряда или гидрофобности молекулы в сайте-мишени, или (в) массы боковых цепей. Аминокислоты можно сгруппировать в соответствии со схожестью свойств их боковых цепей (A.L. Lehninger, in Biochemistry, second ed., pp. 73-75, Worth Publishers, New York (1975)):
неполярные: Ala(A), Val(V), Leu(L), Ile(I), Pro(P), Phe(F), Trp(W), Met(M);
незаряженные полярные: Gly(G), Ser(S), Thr(T), Cys(C), Tyr(Y), Asn(N), Gln(Q);
кислые: Asp(D), Glu(E);
основные: Lys(K), Arg(R), His(H).
Альтернативно, природные остатки можно разделить на группы на основе общих свойств боковых цепей:
гидрофобные: норлейцин, Met, Ala, Val, Leu, Ile;
нейтральные гидрофильные: Cys, Ser, Thr, Asn, Gln;
кислые: Asp, Glu;
основные: His, Lys, Arg;
остатки, оказывающие влияние на ориентацию цепи: Gly, Pro;
ароматические: Trp, Tyr, Phe.
Неконсервативные замены влекут за собой замену члена одного из этих классов на другой класс.
Любой остаток цистеина, не вовлеченный в сохранение правильной конфирмации антитела, также может быть замещен, как правило, серином для увеличения стабильности молекулы к окислению и для предотвращения аберрантного сшивания. Обратно, связь(и) между цистеинами может быть добавлена в антитело для увеличения ее стабильности (в частности, когда антителом является фрагмент антитела, такой как фрагмент Fv).
Особенно предпочтительный тип варианта замены включает замену одного или нескольких остатков гипервариабельных участков исходного антитела. Как правило, получаемый в результате вариант(ы), отбираемый для дальнейшей разработки, имеет улучшенные биологические свойства относительно исходного антитела, из которого они получены. Традиционный способ получения таких вариантов замены представляет собой созревание аффинности с использованием фагового дисплея. Кратко, несколько сайтов гипервариабельных участков (например, 6-7 сайтов) мутируют с получением всех возможных аминокислотных замен в каждом сайте. Варианты антитела, полученные таким образом, представляют в одновалентной форме в частицах нитеобразных фагов в виде слияний с продуктом гена III М13, упакованных внутри каждой частицы. Представляемые фагами варианты затем скринируют на их биологическую активность (т.е. аффинность связывания), как здесь раскрыто. Для идентификации сайтов гипервариабельных участков - кандидатов на модификацию можно осуществить сканирующий аланином мутагенез с идентификацией остатков гипервариабельных участков, вносящих значительный вклад в связывание с антигеном. Альтернативно или дополнительно, может быть полезным проанализировать кристаллическую структуру комплекса антиген-антитело для идентификации точек контакта между антителом и антигеном. Такие остатки контакта и близлежащие остатки являются кандидатами на замену в соответствии с методами, разработанными здесь. После получения таких вариантов панель вариантов подвергают скринингу, как здесь описано, и антитела с лучшими свойствами в одном или нескольких релевантных исследованиях могут быть отобраны для дальнейшей разработки.
Другой тип аминокислотного варианта антитела изменяет первоначальную картину гликозилирования антитела. Такое изменение включают делецию одной или нескольких углеводных составляющих, обнаруживаемых в антителе, и/или добавление одного или нескольких сайтов гликозилирования, которые присутствуют в антителе.
Гликозилирование полипептидов обычно является или N-связанным, или О-связанным. N-связанное относится к присоединению углеводной составляющей к боковой цепи остатка аспарагина. Для ферментативного присоединения углеводной составляющей к боковой цепи остатка аспарагина узнаваемыми последовательностями являются трипептидные последовательности аспарагин-Х-серин и аспарагин-Х-треонин, где Х является любой аминокислотой, за исключением пролина. Таким образом, присутствие одной из двух этих трипептидных последовательностей в полипептиде создает потенциальный сайт гликозилирования. О-связанное относится к присоединению одного из сахаров N-ацетилгалактозамина, галактозы или ксилозы к гидроксиаминокислоте, наиболее часто серину или треонину, хотя можно также использовать 5-гидроксипролин или 5-гидроксилизин.
Добавление к антителу сайтов гликозилирования традиционно осуществляют путем изменения аминокислотной последовательности так, чтобы она содержала одну или несколько описанных выше трипептидных последовательностей (для сайтов N-связанного гликозилирования). Изменение может быть также осуществлено путем добавления, или замены, одного или нескольких остатков серина или треонина в последовательность исходного антитела (для сайтов О-связанного гликозилирования).
Если антитело включает фрагмент Fc, углевод, присоединяемый к нему, может быть изменен. Например, антитела со зрелой углеводной структурой, в которой отсутствует фукоза, присоединенной к фрагменту Fc антитела, описаны в заявке US 2003/0157108 (Presta, L.). Смотри также US 2004/0093621 (Kyowa Hakko Kogyo Co., Ltd). На антитела с биссекторным N-ацетилглюкозамином (GlcNAc) в углеводе, присоединенном к фрагменту Fc антитела, ссылаются в WO 2003/011878, Jean-Mairet et al. и патенте США № 6602684, Umana et al. Об антителах с по крайней мере одним остатком галактозы в олигосахариде, присоединенном к фрагменту Fc антитела, сообщается в WO 1997/30087, Patel et al. Смотри также WO 1998/58964 (Raju, S.) и WO 1999/22764 (Raju, S.), касающиеся антител с измененным углеводом, присоединенным к фрагменту Fc антитела. Смотри также US 2005/0123546 (Umana et al.) в отношении антигенсвязывающих молекул с модифицированным гликозилированием.
Предпочтительный вариант гликозилирования здесь включает фрагмент Fc, к которому присоединяется углеводная структура, в которой отсутствует фукоза. Такие варианты имеют увеличенную ADCC-функцию. Необязательно фрагмент Fc, кроме того, включает одну или несколько аминокислотных замен, которые дополнительно увеличивает ADCC, например, замены в положениях 298, 333 и/или 334 фрагмента Fc (Eu-нумерация остатков). Примеры публикаций, относящихся к «дефукозилированным» или «дефицитным в отношении фукозы» антителам, включают US 2003/0157108; WO 2000/61739; WO 2001/29246; US 2003/0115614; US 2002/0164328; US 2004/0093621; US 2004/0132140; US 2004/0110704; US 2004/0110282; US 2004/0109865; WO 2003/085119; WO 2003/084570; WO 2005/035586; WO 2005/035778; WO 2005/053742; Okazaki et al., Mol. Biol. 336: 1239-1249 (2004); Yamane-Ohnuki et al. Biotech. Bioeng. 87: 614 (2004). Примеры линий клеток, продуцирующих дефукозилированные антитела, включают клетки Lec13 CHO, дефицитные в отношении фукозилирования белков (Ripka et al. Arch. Biochem. Biophys. 249: 533-545 (1986); заявка US 2003/0157108 A1, Presta, L.; и WO 2004/056312 A1, Adams et al., особенно в примере 11), и дефицитные линии клеток, такие как дефицитные в отношении гена альфа-1,6-фукозилтрансферазы, FUT8, клетки СНО (Yamane-Ohnuki et al. Biotech. Bioeng. 87: 614 (2004)).
Молекулы нуклеиновых кислот, кодирующие варианты аминокислотных последовательностей антитела, готовят с использованием множества способов, известных в данной области техники. Эти способы включают, но без ограничения, выделение из природного источника (в случае природных вариантов аминокислотных последовательностей) или приготовление с помощью опосредуемого олигонуклеотидами (или сайт-направленного) мутагенеза, мутагенеза с использованием ПЦР и кассетного мутагенеза ранее приготовленного варианта или не являющейся вариантом версии антитела.
Может быть желательной модификация антитела настоящего изобретения относительно эффекторной функции, например, для того, чтобы увеличить ADCC и/или CDC антитела. Этого можно достичь путем введения одной или нескольких аминокислотных замен во фрагмент Fc антитела. Альтернативно или дополнительно, во фрагмент Fc можно ввести остаток (остатки) цистеина, что делает, таким образом, возможным образование дисульфидных связей между цепями в этом районе. Гомодимерное антитело, полученное таким образом, может иметь улучшенную способность к интернализации и/или увеличенную способность к комплемент-опосредуемому уничтожению клеток и ADCC. Смотри Caron et al., J. Exp. Med. 176: 1191-1195 (1992) и Shopes, J. Immunol. 148: 2918-2922 (1992). Гомодимерные антитела можно также приготовить с использованием гетеробифункциональных сшивающих агентов, как описано Wolff и др. (Cancer Research 53: 2560-2565 (1993)). Альтернативно, можно получить антитело, которое имеет два фрагмента Fc и может, таким образом, обладать увеличенными способностями к лизису с участием комплемента и ADCC. Смотри Stevenson et al., Anti-Cancer Drug Design 3: 219-230 (1989).
В WO 00/42072 (Presta, L.) описываются антитела с увеличенной ADCC-функцией в присутствии эффекторных клеток человека, причем антитела включают аминокислотные замены в их фрагменте Fc. Предпочтительно антитела с увеличенной ADCC включают замены в положениях 298, 333 и/или 334 фрагмента Fc. Предпочтительно измененный фрагмент Fc представляет собой фрагмент Fc IgG1, включающий или состоящий из замен в одном, двух или трех из этих положений.
Антитела с измененным связыванием C1q и/или CDC описываются в WO 99/51642, патенте США № 6194551B1, патенте США № 6242195B1, патенте США № 6528624B1 и патенте США № 6538124 (Idusogie et al.). Антитела включают аминокислотную замену в одном или нескольких положениях аминокислот 270, 322, 326, 327, 329, 313, 333 и/или 334 их фрагмента Fc.
Для увеличения полужизни антитела в сыворотке можно встроить связывающий эпитоп рецептора выживаемости в антитело (особенно фрагмент антитела), как описано, например, в патенте США № 5739277. Используемый здесь термин «связывающий эпитоп рецептора выживаемости» относится к эпитопу фрагмента Fc молекулы IgG (например, IgG1, IgG2, IgG3 или IgG4), который ответственен за увеличение in vivo полужизни молекулы IgG в сыворотке. Антитела с заменами в их фрагменте Fc и увеличенными полужизнями в сыворотке также описываются в WO 00/42072 (Presta, L.).
Также предусмотрены полученные с помощью генно-инженерных методов антитела с тремя или более (предпочтительно четырьмя) функциональными антигенсвязывающими сайтами (заявка US 2002/0004587 A1, Miller et al.).
V. Фармацевтические композиции
Терапевтические композиции антител, используемые в соответствии с настоящим изобретением, готовят для хранения смешиванием антитела, имеющего желаемую степень чистоты, с необязательными фармацевтически приемлемыми носителями, наполнителями или стабилизаторами (Remington's Pharmaceutical Sciences 16th edition, Osol, A, Ed. (1980)) в форме лиофилизированных композиций или водных растворов. Приемлемые носители, наполнители или стабилизаторы являются нетоксичными для реципиентов в используемых дозах и концентрациях и включают буферы, такие как фосфатный, цитратный и другие органические кислоты; антиоксиданты, включающие аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензиламмония хлорид; гексаметония хлорид; бензалкония хлорид, бензетония хлорид, фенол, бутиловый или бензиловый спирт, алкилпарабены, такие как метилпарабен или пропилпарабен; катехин; резорцин; циклогексанол; 3-пентанол и м-крезол); низкомолекулярные (менее чем приблизительно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углероды, в том числе глюкозу, маннозу или декстрины; хелатообразующие агенты, такие как EDTA; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противойоны, такие как натрий; металлокомплексы (например, комплексы Zn-белок); и/или неионные поверхностно-активные вещества, такие как TWEENTM, PLURONICSTM или полиэтиленгликоль (PEG).
Примеры содержащих антитело против CD20 композиций описаны в WO 98/56418. В этой публикации описывается жидкая многодозовая композиция, включающая 40 мг/мл ритуксимаба, 25 мМ ацетат, 150 мМ трегалозу, 0,9% бензилового спирта, 0,02% полисорбата 20 при рН 5,0, которая имеет минимальный срок годности 2 года при хранении при 2-8ºС. Другая представляющая интерес, содержащая антитело против CD20 композиция включает 10 мг/мл ритуксимаба в 9,0 мг/мл хлорида натрия, 7,35 мг/мл цитрата натрия дигидрата, 0,7 мг/мл полисорбата 80 и стерильной воде для инъекции, рН 6,5.
Лиофилизированные композиции, адаптированные для подкожного введения, описаны в патенте США № 6267958 (Andya et al.). Такие лиофилизированные композиции можно воссоздать с использованием подходящего разбавителя до высокой концентрации белка, и восстановленную композицию можно вводить подкожно млекопитающему, подвергаемому здесь лечению.
Также предусмотрены кристаллизованные формы антитела. Смотри, например, US 2002/0136719A1 (Shenoy et al.).
Композиция настоящего изобретения может также содержать более одного активного компонента (второе лекарственное средство, как отмечено выше), если необходимо, предпочтительно компоненты с дополнительными активностями, которые не оказывают неблагоприятный эффект друг на друга. Тип и эффективные количества таких лекарственных средств зависят, например, от количества антитела, присутствующего в композиции, и клинических параметров индивида. Предпочтительные такие лекарственные средства отмечены выше.
Активные ингредиенты можно также поместить в микрокапсулы, приготовленные, например, с помощью методов коацервации или межфазной полимеризации, например, гидроксиметилцеллюлозные или желатиновые микрокапсулы и микрокапсулы из поли-(метилметакрилата), соответственно, в коллоидные системы доставки лекарственных средств (например, липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие способы описаны в Remington's Pharmaceutical Sciences 16th edition, Osol, A, Ed. (1980).
Можно приготовить препараты с непрерывным высвобождением. Подходящие примеры препаратов с непрерывным высвобождением включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие антитело, которые находятся в форме фасонных изделий, например, пленок, или микрокапсул. Примеры матриц с непрерывным высвобождением включают полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт), полилактиды (патент США № 3773919), сополимеры L-глютаминовой кислоты и γ-этил-L-глютамата, неразлагаемый этилен-винилацетат, разлагаемые сополимеры молочной кислоты и гликолевой кислоты, такие как LUPRON DEPOTTM (инъецируемые микросферы, состоящие из сополимера молочной кислоты и гликолевой кислоты и лейпролида ацетата) и поли-D-(-)-3-гидроксибутировая кислота.
Композиции, используемые для введения, должны быть стерильными. Это легко осуществить с помощью фильтрации через мембраны для стерильной фильтрации.
VI. Изделия производства
В другом варианте осуществления настоящего изобретения предоставляются изделия производства, содержащие материал, используемый для лечения повреждения сустава, описанный выше. Настоящим изобретение, в частности, предоставляется изделие производства, включающее (а) контейнер, содержащий антагонист, такой как антитело, которое связывается с маркером поверхности В-клетки (например, антитело против CD20) (предпочтительно контейнер включает антитело и фармацевтически приемлемый носитель или разбавитель); и (b) листовку-вкладыш с инструкциями в отношении лечения повреждения сустава у индивида, причем в инструкциях указывается на то, что индивиду вводится антагонист или антитело (например, антитело против CD20), и затем он проходит рентгенографическую проверку спустя по крайней мере приблизительно один месяц от момента введения, с помощью которой определяют уменьшение повреждения сустава по сравнению с базовой линией до введения, причем количество вводимого антагониста или антитела, такого как антитело против CD20, является эффективным для получения уменьшения повреждения сустава, что является указанием на успешное лечение индивида.
В предпочтительном варианте осуществления этого аспекта изобретения изделие производства настоящего изобретения дополнительно включает контейнер, содержащий второе лекарственное средство, причем антагонист или антитело представляет собой первое лекарственное средство, дополнительно включающий инструкции в листовке-вкладыше в отношении лечения индивида эффективным количеством второго лекарственного средства. Вторым лекарственным средством может быть любое второе лекарственное средство, изложенное выше, при этом примеры второго лекарственного средства являются изложенными выше вторыми лекарственными средствами, включающими антибиотик, иммунодепрессант, модифицирующее заболевание противоревматическое лекарственное средство (DMARD), контролирующий боль агент, антагонист интегрина, нестероидное противовоспалительное лекарственное средство (NSAID), антагонист цитокина, бисфосфонат или гормон, или их комбинацию, более предпочтительно DMARD, NSAID, контролирующий боль агент или иммунодепрессант. Более предпочтительно вторым лекарственным средством является метотрексат.
В этом аспекте листовка-вкладыш находится на контейнере или связана с контейнером. Подходящие контейнеры включают, например, пузырьки, флаконы, шприцы и т.п. Контейнеры могут состоять из множества материалов, таких как стекло или пластик. Контейнер удерживает или содержит композицию, которая является эффективной для лечения повреждения сустава, и может иметь стерильное входное отверстие (например, контейнер может быть мешком с раствором для внутривенного введения или флаконом с пробкой, прокалываемой иглой для подкожных инъекций). По крайней мере один активный агент в композиции является антагонистом или антителом. Этикетка или листовка-вкладыш указывают на то, что композиция применима для лечения повреждения сустава у индивида, подходящего для лечения с использованием специфического руководства, касающегося вводимых количеств антагониста или антитела и интервалов между их введениями и любого другого предоставляемого лекарственного средства. Изделие производства может, кроме того, включать дополнительный контейнер, включающий фармацевтически приемлемый буфер для разбавления, такой как бактериостатическая вода для инъекции (BWFI), забуференный фосфатом солевой раствор, раствор Рингера и/или раствор декстрозы. Изделие производства может, кроме того, включать другие материалы, желаемые с коммерческой точки зрения и точки зрения пользователя, в том числе другие буферы, разбавители, фильтры, иголки и шприцы.
В более конкретном варианте осуществления настоящего изобретения изделие производства включает (а) контейнер, содержащий антагонист, такой как антитело, которое связывается с маркером поверхности В-клетки (например, антитело против CD20) (предпочтительно контейнер включает антитело и фармацевтически приемлемый носитель или разбавитель); и (b) листовку-вкладыш с инструкциями в отношении лечения повреждения сустава у индивида, причем в инструкциях указывается на то, что индивиду вводится антагонист или антитело (например, антитело против CD20), и затем он проходит рентгенографическую проверку, спустя по крайней мере приблизительно 52 недели от момента введения, с помощью которой определяют уменьшение повреждения сустава по сравнению с базовой линией до введения, причем количество вводимого антитела против CD20 является эффективным для получения уменьшения повреждения сустава, что является указанием на успешное лечение индивида. В предпочтительном варианте осуществления изделие включает контейнер, содержащий второе лекарственное средство, причем антагонист или антитело (такое как антитело против CD20) представляет собой первое лекарственное средство, дополнительно включающий инструкции в листовке-вкладыше в отношении лечения индивида эффективным количеством второго лекарственного средства. Предпочтительно вторым лекарственным средством является метотрексат.
Дальнейшие детали настоящего изобретения иллюстрируются следующими не ограничивающими примерами. Содержание всех ссылок в настоящем описании прямо включено сюда посредством ссылки.
V
Пример 1:
Эффективность и безопасность ритуксимаба у пациентов с неадекватным ответом на предшествующую анти-TNF терапию или непереносимостью такой терапии
Это является рандомизированным, двойным слепым клиническим испытанием фазы III с использованием параллельных групп и множества центров, названным рандомизированной оценкой длительной эффективности ритуксимаба при RA (REFLEX). На фиг.1 демонстрируется проект исследования.
Целями этого исследования были:
• определение эффективности и безопасности ритуксимаба при использовании в комбинации с метотрексатом (МТХ) у 517 больных активным ревматоидным артритом, которые неадекватно реагировали на одну или несколько анти-TNF терапий;
• изучение фармакокинетических и фармакодинамических параметров ритуксимаба в этой популяции больных (например, продолжительности истощения В-клеток и эффектов на иммуноглобулины и ревматоидный фактор).
Результатами этого исследования были:
первичное конечное положение
• доля пациентов с ответом ACR20 на 24 неделе;
вторичные конечные положения
• доля пациентов с ответами ACR50 и ACR70 на 24 неделе,
• изменение от базовой линии в DAS28 на 24 неделе,
• ответ EULAR на 24 неделе,
• изменение от базовой линии в основном наборе ACR;
• изменение от базовой линии в SF-36,
• изменение модифицированного Genant рентгенографического общего показателя Sharp на 56 неделе,
• изменение модифицированного Genant рентгенографического показателя Sharp на 24 неделе (исследовательское положение),
• изменение показателя эрозии и показателя сужения суставного пространства.
Ключевыми критериями включения в это исследование были:
• неадекватное реагирование на предшествующее или текущее лечение этанерцептом, инфликсимабом или адалимумабом из-за токсичности или неадекватной эффективности:
этанерцептом в течение ≥3 месяцев в количестве 25 мг дважды в неделю,
инфликсимабом с помощью по крайней мере 4 инфузией в расчете ≥3 мг/кг,
адалимумабом в течение ≥3 месяцев в количестве 40 мг через неделю,
• получение МТХ в дозе 10-25 мг/неделю (перорально или парентерально) на протяжении по крайней мере 12 недель, при этом на протяжении последних 4 недель до скрининга назначалась постоянная доза;
• отмена всех других DMARD/модификаторов биологических ответов за по крайней мере 4 недели до рандомизации (за 8 недель отмена инфликсимаба, лефлуномида и адалимумаба),
• количество преднизона, равное ≤10 мг/день,
• подсчет опухших суставов (SJC) ≥8 (оценка 66 суставов) и подсчет болезненных суставов (TJC)≥8 (оценка 68 суставов),
• или CRP≥1,5 мг/дл (15 мг/л), или ESR≥28 мм/ч,
• рентгенографическое доказательство наличия по крайней мере одного сустава с определенной эрозией, свойственной ревматоидному артриту.
Лечение данного исследования было следующим:
• группы А:
ритуксимаб две внутривенных инфузии по 1000 мг в дни 1 и 15,
• группы В:
плацебо внутривенные инфузии в дни 1 и 15,
• обеих групп:
100 мг внутривенно метилпреднизолона до каждой инфузии ритуксимаба/плацебо,
60 мг/д преднизона в дни 2-7 и 30 мг/д в дни 8-14.
Результаты через 6 месяцев:
Популяции пациентов:
ITT
• рандомизированные
• получившие часть инфузии
• проанализированные как рандомизированные
На протокол
• как указано выше, но соблюдая протокол
Безопасность
• рандомизированные
• получившие часть инфузии
• проанализированные как получившие
Демографии пациентов
Результаты исследования, касающиеся эффективности, продемонстрированы на фиг.1-12.
Рентгенографические результаты исследования продемонстрированы на фиг.13-17.
Среднее изменение параметров основного набора ACR продемонстрированы на фиг.18.
Анализ исследования, касающийся безопасности, продемонстрирован на фиг.19-31.
Из исследования REFLEX были сделаны следующие выводы.
• Введение ритуксимаба сопровождалось значительным увеличением частоты ответа ACR20 по сравнению с плацебо (первичное конечное положение).
• Все вторичные и исследовательские конечные положения (DAS, EULAR, основной набор ACR) подтверждали первичный анализ.
• Рентгенографические результаты показывают, что для подвергнутой лечению группы демонстрируется более низкий общий показатель Sharp-Genant на протяжении 24 недель по сравнению с группой-плацебо (фиг.13), более низкий показатель эрозии Sharp-Genant на протяжении 24 недель по сравнению с группой-плацебо (фиг.14), более низкий JSN-показатель Sharp-Genant на протяжении 24 недель по сравнению с группой-плацебо (фиг.15), намного большая доля пациентов без изменения показателя эрозии на 24 неделе лечения по сравнению с группой-плацебо (фиг.16), при этом сводка рентгенографических конечных положений на 24 неделе для группы плацебо и подвергнутой лечению группы представлена на фиг.17.
• Ритуксимаб, как правило, хорошо переносился.
Связанные с инфузиями эффекты
Частота всех инфекций в сравнении с плацебо
Незначительное увеличение частоты серьезных инфекций
Отсутствие значительного влияния на иммуноглобулины
Низкий НАСА
Результаты через 56 недель:
(n=186)
(n=277)
На фиг.41, кроме того, демонстрируется распределение пациентов в клиническом испытании REFLEX на 56 неделе, в том числе проводящиеся лечения подгрупп пациентов, выбираемых из подвергнутой лечению группы и группы-плацебо, клинического испытания фазы III REFLEX.
Как показано на фиг.42, на 56 неделе среднее изменение общего модифицированного Genant показателя Sharp (Genant, Am. J. Med., 30: 35-47 (1983)), сужения суставного пространства (JSN) и показателя эрозии продемонстрировали статистически значимое улучшение по сравнению с плацебо.
Эффективность лечения ритуксимабом, кроме того, иллюстрируются с помощью среднего изменения общего показателя Genant-Sharp с течением времени. Как показано на фиг.43, улучшение продолжалось с 24 недели по 56 неделю.
На фиг.44 демонстрируется кумулятивное распределение изменения общего показателя Genant-Sharp.
На фиг.45 демонстрируются результаты анализов восприимчивости, выраженной с помощью изменения общего показателя Genant-Sharp. Подвергнутая лечению ритуксимабом + МТХ группа постоянно превосходила группу, подвергнутую лечению плацебо + МТХ.
На фиг.46 демонстрируется процент пациентов без рентгенографических изменений в точке наблюдения, равняющейся 56 неделям, в состоянии их суставов, как измерено с помощью показателя эрозии и модифицированного Genant показателя Sharp, соответственно.
В заключение, результаты этого клинического испытания показывают, что ритуксимаб значительно ингибирует рентгенографическое прогрессирование у больных ревматоидным артритом (RA), которые неадекватно реагировали на один или несколько ингибиторов TNF или не переносили их. Кроме того, это исследование впервые показывает, что нацеленная на В-клетки терапия может ингибировать рентгенографическое прогрессирование.
Пример 2
Ритуксимаб для RA: программа фазы III
Важный результат для оценки RA включает ингибирование прогрессирования структурного повреждения сустава и улучшение физической функции. Этот результат особенно важен для пациентов, у которых RA диагностирован недавно, поскольку раннее воздействие на таких пациентов имеет потенциально более длительную пользу (Genovese et al., Arthritis Rheum. 46: 1443-1450 (2002)). Поэтому популяция больных RA на ранней стадии является соответствующей популяцией для исследования этих важных результатов.
Что касается подходящего контрольного лечения таких пациентов, используемой в настоящее время терапией золотого стандарта для RA на ранней стадии является МТХ. В результате, отбор популяции не подвергавшихся лечению МТХ пациентов и сравнение ритуксимаба плюс МТХ с только МТХ обеспечивает сравнение этого нового подхода к лечению с имеющимся в настоящем времени золотым стандартом для этих пациентов.
Это исследование является рандомизированным, контролируемым, разрешимым слепым исследованием с использованием параллельных групп и множества центров для оценки безопасности и эффективности ритуксимаба в комбинации с МТХ по сравнению с только МТХ для не подвергавшихся лечению МТХ больных активным RA.
Первичные цели:
1) определение эффективности ритуксимаба в предотвращении прогрессирования структурного повреждения сустава и оценка безопасности ритуксимаба для больных активным RA, начавших лечение МТХ;
2) оценка эффективности ритуксимаба в улучшении физической функции пациентов и признаков и симптомов RA;
3) изучение с помощью подхода анализа популяции фармакокинетических (РК) параметров ритуксимаба в целевой популяции больных RA и влияния ковариантов на РК-параметры;
4) исследование длительной эффективности и безопасности дальнейших курсов лечения ритуксимабом.
Первый курс лечения - проект исследования:
Группа А: ритуксимаб 500 мг внутривенно × 2 плюс МТХ (7,5 мг с увеличением до 20 мг перорально)
Группа В: ритуксимаб 1000 мг внутривенно × 2 плюс МТХ (7,5 мг с увеличением до 20 мг перорально)
Группа С: плацебо внутривенно × 2 плюс МТХ (7,5 мг с увеличением до 20 мг перорально)
Для подходящих пациентов группы С со 104 недели будет доступно повторное лечение с использованием курсов ритуксимаба 500 мг внутривенно × 2 плюс МТХ.
Следующие правила применимы для повторного лечения в соответствии с вышеуказанными схемами:
Второй курс лечения
Второй курс лечения ритуксимабом плюс МТХ или плацебо плюс МТХ будут назначать как можно быстрее после достижения следующего:
1. Прошло как минимум 24 недели с момента первой инфузии последнего курса лечения исследуемым лекарственным средством.
2. DAS28-ESR >2,6.
3. Удовлетворены критерии желательности для абсолютного числа нейтрофилов (не ниже 1,5 × 103 мкл), IgG (не ниже 5,0 мг/мл) и IgM (не ниже 0,4 мг/мл) в последнем анализе образца крови.
Кроме того, пациенты должны удовлетворять критериям исключения из годности 8, 10 и 11, описанные ниже:
1. Значимое заболевание сердца или легких (в том числе обструктивное заболевание легких).
2. Первичный или вторичный иммунодефицит (в анамнезе или активный в текущий момент), в том числе известная история ВИЧ-инфекции.
3. Известная активная инфекция любого вида (за исключением микоза ногтевых лож) или любой крупный эпизод инфекции, требующей госпитализации или лечения вводимыми внутривенно антиинфекционными агентами, в пределах 4 недель инфузии или завершение приема пероральных антиинфекционных агентов в пределах 2 недель до инфузии.
Пациентов, которые не удовлетворяют критериям для второго курса лечения ритуксимабом/плацебо на 24 неделе, будут обследовать каждые 4-8 недель, и они впоследствии получат второй курс лечения в то время, когда они станут подходящими на основе вышеуказанных критериев.
Дальнейшие курсы лечения:
С 48 недели пациенты могут стать подходящими для получения третьего курса лечения ритуксимабом/плацебо. Третий курс и после может только быть назначен пациентам, которые удовлетворили вышеуказанным критериям для второго курса лечения и которые, как полагает исследователь, имели релевантную клиническую реакцию после или первого, или второго курса лечения ритуксимабом. Пациенты, которые никогда не имели клиническую реакцию, должны быть исключены из испытания с последующим врачебным наблюдением в целях безопасности (SFU).
Пациентов, которые, как полагают, имели клиническую реакцию, но в текущий момент времени не удовлетворяют критериям для дополнительного курса лечения ритуксимабом, будут обследовать каждые 4 недели, с 48 недели по 56 неделю, и затем каждые 8 недель, и они впоследствии получат дополнительные курсы лечения в то время, когда они станут подходящими на основе вышеуказанных критериев.
Все пациенты будут получать премедикацию 100 мг внутривенно метилпреднизолона до каждой инфузии. Все пациенты также будут получать постоянную дозу фолата (по крайней мере 5 мг/неделю) в виде одной дозы или в виде дробной еженедельной дозы.
Все пациенты должны продолжать получать любой фоновый кортикостероид (по крайней мере 10 мг/мл преднизона или эквивалента) или вводимые перорально нестероидные противовоспалительные лекарственные средства (NSAID) в постоянной дозе. Рандомизацию будут стратифицировать по зонам (США или остальной мир) и ревматоидному фактору (положительные или отрицательные) для того, чтобы гарантировать сбалансированное распределение пациентов по зоне или RF-статусу между подвергаемыми лечению группами.
Пациенты будут посещать клинику один раз каждые 4 недели на протяжении первых 24 недель и каждые 8 недель впоследствии (за исключением недель 48-56, когда визиты будут происходить каждые 4 недели) для оценок эффективности, безопасности, иммунологии и качества жизни. Рентгенографические оценки будут проводить при скрининге, на неделе 24 и неделе 52. Рентгенографические оценки будут также проводить через 2 и 3 года от введения первой дозы исследуемого лекарственного средства.
Оценка первичного конечного положения (изменения общего модифицированного показателя Sharp) будет иметь место на 52 неделе. Вторичные и исследовательские конечные положения будут включать, кроме того, рентгенографические конечные положения и признаки и симптомы, физическую функцию и конечные положения в виде ремиссии.
В любое время после рентгенографической оценки на 52 неделе пациенты, у которых не наблюдается 20% уменьшения как числа опухших суставов, так и числа болезненных суставов, могут получить спасительное лечение увеличенной дозой МТХ или одного небиологического DMARD.
Все пациенты, которые были исключены из исследования в любой момент или завершили полный цикл лечения, должны вернуться для оценок SFU на 4, 12, 24, 36 и 48 неделе после исключения или завершения. Результативно наблюдать пациента на протяжении одного года после исключения пациента из исследования/завершения пациентом лечения. Если число периферических В-клеток (CD19) пациента не вернулось до уровня базовой линии или до нормального диапазона, какой ниже, после одного года, визиты для наблюдения в целях безопасности должны продолжаться с 12-недельными интервалами до тех пор, пока не произойдет пополнение В-клеток.
При всех сообщаемых серьезных инфекционных неблагоприятных эффектах должен проводиться клинический анализ крови с определением лейкоцитарной формулы, подсчетом тромбоцитов, количественным определением Ig и подсчетом CD19 в пределах одной недели от момента, когда инфекционный неблагоприятный эффект стал серьезным.
Пациентов, исключенных из испытания с SFU, необходимо убеждать вернуться для проведения всех запланированных рентгенографических оценок (на неделях 24, 52, 104 и 152) независимо от момента их исключения из исследования. Рентгенограммы таких пациентов следует получать в соответствии с первоначальной графиком оценок, относительно исходного дня рандомизации.
В это исследование будет привлечено приблизительно 852 пациента и их будут рандомизировать равно на три подвергаемые лечению группы. Пациентов будут стратифицировать по зоне (США или остальной мир) и RF-статусу (RF-положительные - по крайней мере 20 МЕ/мл или RF-отрицательные - менее чем 20 МЕ/мл). Общую долю RF-отрицательных пациентов будут ограничивать до 20% от общего размера популяции. Комплектование будет конкурсным, при этом не более 70% и не менее 30% будет зарегистрировано в любой из двух зон. Замены пациентов не будет, если лечение пациента прекратят по какой-либо причине.
Целевая популяция
Целевая популяция для этого исследования представляет собой больных активным RA на ранней стадии, которые не подвергались лечению МТХ.
Критерии исключения:
Пациенты должны удовлетворять следующим критериям для того, что они подходили для вхождения в исследование:
1. Способны и желать дать письменное информированное согласие и выполнять требования протока исследования.
2. Пациенты, у которых RA диагностирован в течение по крайней мере 8 недель, но не более 4 лет, в соответствии с пересмотренными в 1987 г. критериями ACR для классификации RA.
3. Пациенты, не подвергавшиеся лечению МТХ и считающиеся кандидатами на лечение МТХ.
4. Число опухших суставов (SJC) по крайней мере 8 (максимум 66) и число болезненных суставов (TJC) по крайней мере 8 (максимум 68) при скрининге и на базовой линии.
5. При скрининге CRP по крайней мере 1,2 мг/дл (12 мг/л).
6. Возраст 18-80 лет.
7. Глюкокортикоиды по крайней мере 10 мг/день преднизолона или эквивалента разрешаются при постоянстве в течение по крайней мере в течение четырех недель до базовой линии.
8. Использование NSAID разрешается при постоянстве в течение по крайней мере двух недель до базовой линии.
9. Для пациентов с репродуктивной способностью (мужчины и женщины) использование надежных средств контрацепции (например, гормональных контрацептивов, накладок, внутриматочного противозачаточного средства, физического барьера) во время всего участия в исследовании.
10. Должны быть согласны принимать орально фолат.
11. Только для RF-отрицательных пациентов рентгенографическое доказательство наличия по крайней мере одного сустава с определенной эрозией, свойственной ревматоидному артриту.
12. Пациенты, которые должны получить лечение или которые в текущий момент получают амбулаторное лечение.
Критерии исключения:
Связанные с RA исключения
1. Ревматическое аутоиммунное заболевание, отличное от RA, или значительное системное поражение, вторичное по отношению к RA (включающее, но без ограничения, васкулит, фиброз легкого или синдром Фелти). Разрешается вторичный синдромом Шегрена или вторичный ограниченный кожей васкулит с RA.
2. Функциональное состояние класса IV, определяемое в соответствии с классификацией ACR функциональных состояний при RA.
3. История и наличие в текущий момент воспалительного заболевания сустава, отличного от RA, (включающего, без ограничения, подагрический, реактивный артрит, псориатический артрит, серонегативную спондилоартропатию, болезнь Лима), или другого системного аутоиммунного заболевания (включающего, но без ограничения, системную красную волчанку, болезнь воспаленного кишечника, склеродермию, воспалительную миопатию, смешанное заболевание соединительных тканей или любой синдром наложения).
4. Диагноз ювенильного идиопатического артрита (JIA) или ювенильного RA (JAR) и/или RA до возраста 16 лет.
Связанные с общим состоянием здоровья исключения
5. Любая хирургическая операция, в том числе операция на кости/суставе/синовэктомия (в том числе соединение или замещение сустава), в пределах 12 недель до базовой линии или запланированная во время исследования.
6. Отсутствие доступа к периферическим венам.
7. Беременность или кормление грудью.
8. Значимое и/или неконтролируемое заболевание сердца или легких (в том числе обструктивное заболевание легких).
9. Доказательство значимого сопутствующего заболевания, включающего, но без ограничения, нарушения нервной системы, почек, печени, эндокринной системы или желудочно-кишечного тракта, которое, по мнению исследователя, будет препятствовать участию пациента.
10. Первичный или вторичный иммунодефицит (в анамнезе или активный в текущий момент), в том числе известная история ВИЧ-инфекции.
11. Известная активная инфекция любого вида (за исключением микоза ногтевых лож) или любой крупный эпизод инфекции, требующий госпитализации или лечения вводимыми внутривенно антиинфекционными агентами, в пределах 4 недель базовой линии или завершение приема пероральных антиинфекционных агентов в пределах 2 недель до базовой линии.
12. История глубокой тканевой инфекции (например, фасцит, абсцесс, остеомиелит) в пределах 52 недель до базовой линии.
13. История серьезной рецидивирующей или хронической инфекции (для скрининга инфекции грудной клетки рентгенограмму грудной клетки будут получать при скрининге, если она не была получена в пределах 12 недель до скрининга).
14. История злокачественной опухоли, в том числе солидных опухолей, опухолей кроветворной системы или карциномы in situ (за исключением базальноклеточной и чешуйчатоклеточной карциномы кожи, которую подвергли иссечению и вылечили).
15. Любое неврологическое (врожденное или приобретенное), сосудистое или системное нарушение, которое могло бы оказать влияние на какую-либо оценку эффективности, в частности боли в суставе и опухлости, (например, болезнь Паркинсона, церебральный паралич, невропатия при сахарном диабете).
16. Активные на текущий момент злоупотребление алкоголем или лекарственная зависимость или история злоупотребления алкоголем или лекарственной зависимости в пределах 24 недель до базовой линии.
Связанные с лекарственными средствами исключения
17. История тяжелой аллергической или анафилактической реакции на биологический агент или известная гиперчувствительность к любому компоненту ритуксимаба или к крысиным белкам.
18. Предшествующее лечение любым разрешенным или проходящим клиническое испытание биологическим агентом для RA.
19. Предшествующее лечение антителом против альфа-4-интегрина или костимулирующим модулятором.
20. Сопутствующее лечение любым биологическим агентом или DMARD, отличным от МТХ. Лечение должно быть прекращено за 14 дней до базовой линии, за исключением следующих: азатиоприн - за по крайней мере 28 дней; лефлуномид - за по крайней мере 8 недель (или по крайней мере 14 дней после выведения в течение 11 дней с помощью стандартного холестирамина или активированного древесного угля).
21. Предшествующее лечение с помощью любых истощающих клетки терапий, в том числе проходящих клинические испытания агентов (например, CAMPATH, анти-CD4, анти-CD5, анти-CD3, анти-CD19, анти-CD11a, анти-CD22, BLys/BAFF и анти-CD20).
22. Лечение любым проходящим клиническое испытание агентом в пределах 28 дней от базовой линии или пяти полужизней проходящего клиническое испытание агента (что длиннее).
23. Введение живой/ослабленной вакциной в пределах 28 дней до базовой лини (рекомендуется тщательное изучение записи о вакцинации пациента и необходимости иммунизации до введения ритуксимаба/плацебо).
24. Внутрисуставные или парентеральные глюкокортикоиды в пределах 4 недель до базовой линии.
25. Непереносимость глюкокортикоидов или противопоказания к внутривенному введению глюкокортикоидов.
Исключения, связанные с лабораторными показателями
26. Положительный тест на хорионический гонадотропин человека в сыворотке, проведенный до первой инфузии ритуксимаба.
27. Положительные тесты на поверхностный антиген вируса гепатита В (HBsAg), основное антитело против вируса гепатита В (HBsAb) или серология гепатита С.
28. Гемоглобин менее чем 8,0 г/дл.
29. Концентрации IgG и/или IgM в сыворотке ниже 5,0 и 0,40 мг/мл, соответственно.
30. Абсолютное число нейтрофилов (ANS) ниже 1,5 × 103/мкл.
31. AST или ALT выше чем в 2,5 раза верхней границы нормы.
Окончание лечение определяют как временную точку, равную 3 годам, после чего следует дополнительный период, равный по крайней мере году SFU.
Ритуксимаб (500 мг или 1000 мг) плюс МТХ или плацебо плюс МТХ будут вводить с помощью внутривенных инфузий в дни 1 и 15. Пациенты могут подходить для повторного лечения (две дозы с интервалом в 14 дней) с максимальной частотой, составляющей одно повторное лечение каждые 24 недели. Пациенты, первоначально рандомизированные для получения ритуксимаба плюс МТХ, будут получать повторное лечение в одной и той же дозе на протяжении всего исследования. Со 104 недели пациенты, первоначально рандомизированные для получения плацебо плюс МТХ, могут подходить для получения ритуксимаба (500 мг) плюс МТХ. Перед каждой инфузией должна назначаться премедикация 100 мг внутривенно метилпреднизолона.
Всем группам будут назначать орально таблетки метотрексата (7,5 мг/неделю с увеличением до 20 мг/неделю).
Рекомендуется, чтобы все пациенты были подвергнуты премедикации парацетамолом/ацетаминофеном (1 г перорально) и дифенгидрамина HCl (100 мг внутривенно или вводимым орально эквивалентным антигистамином) за 30-60 минут до начала инфузии для снижения возможности реакций на инфузию.
Пациенты будут получать фолат или эквивалент (по крайней мере 5 мг/неделю) в виде одной дозы или в виде дробной еженедельной дозы.
Пациенты могут продолжать получать любой фоновый кортикостероид (по крайней мере 10 мг/мл преднизона или эквивалента). Для уменьшения боли, если требуется, могут использоваться аналгетики.
Первичное конечное положение представляет собой изменение от скрининга общего модифицированного показателя Sharp на 52 неделе, используя модифицированную намеревающуюся лечиться (ITT) популяцию.
Рентгенографические вторичные конечные положения представляют собой:
1) изменение модифицированного общего показателя Sharp на 24 и 104 неделе,
2) изменение модифицированного Sharp показателя эрозии на 52 неделе,
3) изменение модифицированного показателя сужения суставного пространства на 52 неделе,
4) долю пациентов без рентгенографического прогрессирования на 52 неделе (определенного в виде изменения общего модифицированного показателя Sharp, составляющего менее 0 или равного 0). Кроме того, будут анализировать долю пациентов без рентгенографического прогрессирования на 24 и 104 неделе.
Рентгенографические исследовательские конечные положения представляют собой:
1) изменение модифицированных показателей Sharp с течением времени,
2) долю пациентов без рентгенографического прогрессирования на 24, 52 и 104 неделе, дополнительно представленная в следующих подкатегориях:
а) доля пациентов без изменения модифицированного Sharp показателя эрозии от скрининга,
b) доля пациентов без изменения модифицированного показателя сужения суставного пространства от скрининга,
с) доля пациентов без вновь подвергшихся эрозии суставов.
Рентгенографические оценки будут осуществлять следующим образом: отдельные рентгенограммы каждой руки сзади-спереди и каждой ноги спереди-сзади будут получать согласно расписанию оценок. При посещении с целью скринирования читаемость и качество рентгенограмм (которые можно найти в инструкции для рентгенографических оценок рук, запястий и ног) следует подтверждать, прежде чем пациент покинет место. Рентгенограммы RF-отрицательных пациентов при посещении с целью скринирования будут проверяться на рентгенографическое доказательство наличия по крайней мере одного сустава с определенной эрозией, свойственной RA, с использованием центрального места считывания. Все рентгенограммы будут оценивать с использованием способа Sharp, модифицированного Genant (Am. J. Med., 30: 35-47 (1983)). Первичная оценка будет представлять собой изменение от скрининга общего модифицированного показателя Sharp на 52 неделе. Общий модифицированный показатель Sharp объединяет показатель эрозии и показатель сужения суставного пространства как рук, так и ног. Максимальный общий показатель эрозии в руках составляет 100, в ногах - 42, максимальный показатель сужения суставного пространства в руках составляет 100, в ногах - 48. Максимально достижимый общий модифицированный показатель Sharp составляет 290. Изменение показателя на 52 неделе следует рассчитывать следующим образом:
Изменение = показатель на 52 неделе - показатель при скринировании.
Для расчета общего модифицированного показателя Sharp будут получать рентгенограммы рук и ног. До начала испытания всем рентгенологическим отделениям будут участвовать в учебных семинарах для стандартизации рентгенограмм. Это будет включать стандартизацию для процедур проверки оборудования, пленок, кассет, размещения рук/ног и процедур получения подходящих рентгенограмм.
Ожидается, что изменение общего модифицированного показателя Sharp будет ассиметричным и, следовательно, не распределенным нормально. Поэтому, изменение общего модифицированного показателя Sharp на 52 неделе будут проверять между подвергаемыми лечению группами с использованием непараметрического статистического испытания со стратификацией по зоне и RF-статусу. Однако если обнаружится, что данные распределяются приблизительно нормально, их будут анализировать с помощью модели дисперсионного анализа (ANOVA) с использованием зоны, RF-статуса и подвергаемых лечению групп в качестве эксплоративных термов в этой модели.
Будет использоваться принцип замыкания для корректирования множества сравнений в первичном конечном положении. Первое сравнение будет проводиться между каждой из трех подвергаемых лечению групп с использованием статистического испытания Kruskal-Wallis.
Три подвергаемые лечению группы будут считаться отличными, если существует достаточное статистическое доказательство отклонения следующей нулевой гипотезы:
Н0: µ1=µ2=µ3, т.е. нет доказательства того, что существует какое-либо отличие в изменении общего модифицированного показателя Sharp в какой-либо из подвергаемых лечению групп,
и будет приниматься альтернативная гипотеза:
Н1: µ1 не равен µ2, или µ1 не равен µ3, или µ2 не равен µ3, т.е существует отличие в изменении общего модифицированного показателя Sharp в по крайней мере одного из попарных сравнений подвергаемых лечению групп.
Если результаты проверки являются статистически значимыми на уровне ∀=0,05, делают заключение, что существует отличие в изменении от базовой линии общего модифицированного показателя Sharp на 52 неделе между подвергаемыми лечению группами.
Впоследствии каждую из групп, подвергаемых лечению ритуксимабом, сравнивают с группой плацебо на уровне ∀=0,05, как описано выше. Первичными сравнениями будут считать сравнения отдельной группы, подвергаемой лечению ритуксимабом, относительно группы плацебо, используя статистическое испытание Van Elteren.
Каждую из подвергнутых лечению ритуксимабом групп будут считать превосходящей группу плацебо, если существует достаточное статистическое доказательство отклонения следующей нулевой гипотезы:
Н0: µ1=µ2, т.е. нет доказательства того, что изменение общего модифицированного показателя Sharp в группе, подвергаемой лечению ритуксимабом, превосходит такое изменение в группе плацебо,
и будет приниматься альтернативная гипотеза:
Н1: µ1 не равен µ2, т.е. изменение общего модифицированного показателя Sharp в группе, подвергаемой лечению ритуксимабом, превосходит такое изменение в группе плацебо.
Если результаты проверки являются статистически значимыми на уровне ∀=0,05, будут делать заключение, что в группе, подвергаемой лечению ритуксимабом, демонстрируются большее изменение от базовой линии общего модифицированного показателя Sharp на 52 неделе по сравнению с группой плацебо.
Будут предприниматься все усилия для обеспечения, когда возможно, возвращения пациентов для рентгенографических проверок, даже если пациент был исключен из испытания лекарственного средства. Отсутствующие данные 52 недели будут приписываться, используя следующие способы.
При отсутствии рентгенографических данных 52 недели рентгенографические данные 24 недели будут использовать для того, чтобы линейно экстраполировать результаты 52 недели от такого пациента. Пациентов, которых преждевременно исключили из испытания до 52 недели, будут включать в качестве части анализа рентгенографических данных 52 недели. Любого пациента, для которого нет рентгенографических данных после скрининга, будут исключать из модифицированной ITT-популяции и, следовательно, из анализа первичного конечного положения.
Будут исследовать влияние возможного дисбаланса в предоставлениях лечения в пределах конкретных мест. Будут проводиться анализы восприимчивости для оценки влияния сильно несбалансированных мест на первичный анализ.
Что касается рентгенографических вторичных конечных положений, изменение модифицированного общего показателя Sharp на 24 и 104 неделе будет анализироваться таким же образом, как определено для первичного конечного положения. Изменение модифицированного Sharp показателя эрозии на 52 неделе будет анализироваться таким же образом, как определено для первичного конечного положения. Кроме того, будет анализироваться изменение модифицированного Sharp показателя эрозии на 24 и 104 неделе. Изменение модифицированного показателя сужения суставного пространства на 52 неделе будет анализироваться таким же образом, как определено для первичного конечного положения. Кроме того, будет анализироваться изменение модифицированного показателя сужения суставного пространства на 24 и 104 неделе. Долю пациентов без рентгенографического прогрессирования на 52 неделе (определенного в виде изменения общего модифицированного показателя Sharp, составляющего менее 0 или равного 0) будут оценивать следующим образом. Различие в долях будут проверять с использованием статистического испытания Cochran-Mantel-Haenszel (CMH) со стратификацией по зонам и RF-статусу. Кроме того, будут анализировать долю пациентов без рентгенографического прогрессирования на 24 и 104 неделе.
Что касается рентгенографических исследовательских конечных положений, будет представлено изменение модифицированных показателей Sharp с течением времени. Доля пациентов без рентгенографического прогрессирования на 24, 52 и 104 неделе, кроме того, будет представлена в следующих подкатегориях:
• доля пациентов без изменения модифицированного показателя эрозии от скрининга,
• доля пациентов без изменения модифицированного показателя сужения суставного пространства от скрининга,
• доля пациентов без вновь подвергшихся эрозии суставов.
Детали рентгенографической оценки суставов и шкал степеней (Genant, Am. J. Med., 30: 35-47 (1983)) являются следующими:
Шкалы степеней:
1. Сужение суставного пространства (JSN)
Степень 0 - норма
Степень 0,5 - стертое JSN или сомнительные обнаружения
Степень 1 - легкое JSN (фокальное или незначительное)
Степень 1,5 - от легкого до умеренного JSN
Степень 2,0 - умеренное JSN
Степень 2,5 - от умеренного до сильного JSN
Степень 3,0 - сильное JSN
Степень 3,5 - сильное JSN, близкое к анкилозу
Степень 4,0 - определенный анкилоз.
2. Эрозии (дискретные нарушения кортикальной поверхности)
Степень 0 - норма
Степень 0,5 - стертая потеря непрерывности кортикальной поверхности или сомнительные обнаружения эрозии кости
Степень 1 - Легкая. Определенные, но небольшие эрозии одной или обоих формирующих сустав костей, обычно в областях без покрытия, в которые вовлечено менее 25% суставной поверхности.
Степень 1,5 - От легкой до умеренной. Эрозии от небольших до средних, в которые вовлечено менее 25% суставной поверхности одной или обоих формирующих сустав костей.
Степень 2,0 - Умеренная. Эрозии от средних до больших, в которые вовлечено 25-50% суставной поверхности обоих формирующих сустав костей.
Степень 2,5 - От умеренной до тяжелой. Эрозии, в которые вовлечено 51-75% суставной поверхности.
Степень 3,0 - Тяжелая. Эрозии, в которые вовлечено 76-90% суставной поверхности.
Степень 3,5 - Очень тяжелая. Эрозии, в которые вовлечено 100% суставной поверхности (полное разрушение суставных поверхностей).
Оценка суставов
1. Сужение суставного пространства в руке (13 суставов на руку)
а. Все проксимальные межфаланговые (PIP) суставы на пальцах II-V.
b. Межфаланговый сустав на пальце I.
с. Все пястно-фаланговые (МРС) суставы.
d. Капрометакапральные (СМС) суставы пальцев III-V в виде одной единицы.
e. Примыкающее к головчатой кости (объединенные ладьевидная кость и головчатая кость и объединенные ладьевидная и головчатая кость) пространство.
f. Лучезапястный сустав.
2. Показатели эрозии в руке (14 суставов на руку)
а. Все проксимальные межфаланговые (PIP) суставы на пальцах II-V.
b. Межфаланговый сустав на пальце I.
с. Все пястно-фаланговые (МРС) суставы.
d. Капрометакапральные (СМС) суставы пальца I.
e. Ладьевидная кость.
f. Дистальная лучевая кость.
g. Дистальный локтевой.
Показатели будут суммироваться раздельно (14 × 3,5 максимум на сустав × 2 = 98 для эрозий и 13 × 4 максимум на сустав × 2 для JSN). Каждую сумму будут нормализовать по шкале 0-100. Оба показателя будут сложены для получения общего показателя (шкала 0-200).
3. Сужение суставного пространства и эрозии (6 суставов на ногу)
а. Все плюснефаланговые (МТР) суставы.
b. Все межфаланговые суставы на пальце I.
Показатели будут суммироваться раздельно (6 × 3,5 максимум на сустав × 2 = 42 для эрозий и 6 × 4 максимум на сустав × 2 = 48 для JSN). Оба показателя будут сложены для получения общего показателя со шкалой 0-90.
Первоначальные рентгенограммы будут отсылаться в центральное место прочтения, где будет проверяться их качество. Если первоначальная рентгенограмма имеет неприемлемое качество, центр посоветует в отношении того, какие требуются изменения, и даст указание получить вторую рентгенограмму.
Для всех рандомизированных в этом исследовании пациентов, получивших по крайней мере часть дозы ритуксимаба (в том числе исключенных из испытания с последующим врачебным наблюдением в целях безопасности), будут получать рентгенограммы рук/запястий и ног на неделях 24, 52, 104 и 152. Пациентов необходимо убеждать вернуться для проведения всех рентгенографических оценок независимо от момента их исключения из исследования. Рентгенограммы таких пациентов следует получать в соответствии с первоначальным графиком оценок, относительно исходного дня рандомизации.
Дополнительными вторичными и исследовательскими конечными положениями будут являться признаки и симптомы, физическая функция, ремиссия и сообщаемые пациентами результаты.
Изменение общего модифицированного показателя Sharp на 52 неделе будут проверять между подвергаемыми лечению группами с использованием непараметрического статистического испытания со стратификацией по зонам и RF-статусу. Однако если обнаружится, что данные распределяются приблизительно нормально, их будут анализировать с помощью модели дисперсионного анализа (ANOVA) с использованием зоны, RF-статуса и подвергаемых лечению групп в качестве эксплоративных термов в этой модели.
Очевидно, что желательным является сохранение пациентов в состоянии низкой активности заболевания путем ограничения вспышек заболевания и возможно ограничения прогрессирования структурного повреждения. В исследовании DANCER, на которое выше приводится ссылка (Emery et al., EULAR, выше, и Van Vollenhoven et al., EULAR, выше), на 24 недели приблизительно 90% пациентов, подвергнутых лечению ритуксимабом, не достигали ремиссии EULAR (DAS28). В исследовании, являющимся предметом этого примера, такие пациенты будут подходить для получения ими первого повторного лечения ритуксимабом на 24 неделе. Обязательное повторное лечение на основе DAS28-ESR предоставляет объективную информацию, касающуюся принципа повторного лечения на основе активности заболевания. Минимальный период, составляющий 24 недели, между курсами рекомендуется на основе фармакокинетических и фармакодинамических параметров ритуксимаба. Не ограничиваясь какой-либо одной теорией, фармакокинетические и фармакодинамические параметры ритуксимаба, как представляется, демонстрируют, что в это время (на 24 неделе) уровни лекарственного средства ниже уровня обнаружения, и существует доказательство возвращения
периферических клеток CD19+. Это происходит параллельно явному увеличению активности заболевания после этого момента времени, который, следовательно, представляет целесообразную точку, с которой можно назначать дополнительные курсы лечения.
Ожидается, что ритуксимаб (или гуманизированное антитело 2Н7 вместо ритуксимаба) в комбинации с МТХ будут эффективными для достижения первичного конечного положения в предупреждении прогрессирования структурного повреждения сустава, как изложено в этом примере. Также ожидается, что схема этого исследования достигнет одного или нескольких вторичных рентгенографических конечных положений. Таким образом, ожидается, что введение первой дозы ритуксимаба или гуманизированного антитела 2Н7 (в количестве 500 мг × 2 или 100 мг × 2) вместе с метотрексатом уменьшит повреждение сустава от базовой линии (до первого введения антитела против CD20), что измеряется с помощью модифицированного общего показателя Sharp, через по крайней мере приблизительно один месяц от базовой линии или начала лечения, предпочтительно на по крайней приблизительно 24 неделе от базовой линии, более предпочтительно на по крайней приблизительно 52 неделе от базовой линии и в дальнейших временных точках вплоть до 104 недели от базовой линии. Также ожидается, что пациентов можно будет эффективно подвергать повторному лечению на 24 или 52 неделе от базовой линии для сохранения этого предупреждения прогрессирования повреждения сустава.
Предсказывается и ожидается, что введение индивиду ритуксимаба или гуманизированного антитела 2Н7 согласно протоколу запланированного введения повторных доз, изложенному выше, будет эффективным для предупреждения прогрессирования структурного повреждения сустава на 52 неделе или позже. Ожидается, что эти результаты будут значительно лучше результатов контроля.
Также ожидается, что на приблизительно 48-54 неделе еще одна 1 г - или 2 г - доза антитела против CD20 (например, ритуксимаба или гуманизированного антитела 2Н7), назначенная в виде одного введения или распределенная на протяжении приблизительно 14-16 дней в количестве 0,5 - или 1-грамм, будет эффективной для лечения повреждения сустава в течение всего второго года, с или без одного или нескольких вторых лекарственных средств, таких как иммунодепрессанты. Таким образом, антитело против CD20 следует вводить первоначально в пределах приблизительно 2-недельного периода времени с последующим другим лечением на приблизительно 4-8 месяце, с последующим другим лечением через приблизительно один год от первоначального лечения (определяемого от момента времени, когда вводится любая из доз), с последующим лечением через приблизительно два года от первоначального лечения, с ожидаемым успехом, в количестве приблизительно 0,5 грамм или 1 грамм × 2-4 введения для каждого лечения, вводимом вместе, приблизительно раз в неделю или приблизительно через неделю на протяжении приблизительно 2-4 недель. Ожидается, что результаты этого лечения будут намного лучше результатов контроля с плацебо. Ожидается, что этот протокол повторного лечения будет успешно использоваться на протяжении нескольких лет с небольшими побочными эффектами или без них.
Также предполагается, что к первичному конечному положению приведет использование ритуксимаба или другого антитела против CD20 в виде монотерапии с использованием той же схемы при той же дозе или более высокой дозе без второго лекарственного средства, такого как МТХ, и что повторное лечение в виде монотерапии с использование той же схемы при той же дозе или более высокой дозе будет успешным.
Пример 3:
Исследование эффективности повторного лечения ритуксимабом больных ревматоидным артритом
В этом примере описывается рандомизированное, двойное слепое, контролируемое плацебо исследование фазы III с использованием множества центров повторного лечения ритуксимабом индивидов с RA, получающих фоновый метотрексат.
Первичной целью этого исследования является оценка эффективности повторного лечения ритуксимабом индивидов с активным RA, которые получают МТХ и которые неадекватно реагировали на ингибиторы TNF.
Вторичными целями этого исследования являются следующие:
• оценить безопасность повторного лечения ритуксимабом индивидов с активным RA, которые получают МТХ и которые неадекватно реагировали на ингибиторы TNF.
• оценить безопасность ритуксимаба для индивидов с активным RA, которые получают МТХ и которые неадекватно реагировали на ингибиторы TNF.
Это является рандомизированным, двойным слепым, контролируемым плацебо исследованием фазы III с использованием множества центров, оценивающим эффективность повторного лечения ритуксимабом индивидов с RA, получающих МТХ. Исследование состоит из четырех частей: скринирование, период лечения (ритуксимаб с доступной маркировкой в течение первого курса и для подходящих индивидов двойное слепое, рандомизированное повторное лечение), последующее врачебное наблюдению в целях безопасности (SFU) и контроль В-клеток. Индивиды должны были неадекватно реагировать на лечение одним или несколькими ингибиторами TNF из-за токсичности или неадекватной эффективности. Приблизительно 555 индивидов вступят в период лечения в приблизительно 150 местах исследования в Соединенных Штатах. RF-положительные и RF-отрицательные индивиды будут регистрироваться и равно распределяться между подвергаемыми лечению группами, при этом общая доля RF-отрицательных индивидов будет ограничена до 20% общего размера популяции.
До дня 1 индивидам прекратят назначать все DMARD за исключением МТХ (лефлуномид, адалимумаб и инфликсимаб за ≥8 недель и этанерцепт за ≥4 недели). Все индивиды будут продолжать получать МТХ 10-25 мг/кг в постоянной дозе на протяжении исследования. Посещение с целью скринирования может происходить вплоть до 56 дней до получения первой дозы исследуемого лекарственного средства в зависимости от требований для выведения.
Все индивиды, удовлетворяющие критериям годности и зарегистрированные для испытания, будут получать ритуксимаб в течение первого курса лечения. Курс лечения ритуксимабом определяется как две 1000 мг внутривенные дозы, введенные с интервалом в 14 дней, с премедикацией 100 мг внутривенно метилпреднизолона до каждой дозы ритуксимаба. Все пациенты будут также получать постоянную дозу фолата (≥5 мг/неделю). Все пациенты должны продолжать получать любой фоновый кортикостероид (≤10 мг/мл преднизона или эквивалента) или вводимые орально нестероидные противовоспалительные лекарственные средства (NSAID) в постоянной дозе.
Первую дозу ритуксимаба следует вводить в пределах 24 часов после оценок базовой линии. Однако если требуется, разрешается вплоть до 72 часов между оценками базовой линии и первой дозой исследуемого лекарственного средства.
Во время 28-40 недели индивидов с активным заболеванием на основе показателя активности заболевания в 28 суставах (DAS28 - скорость осаждения эритроцитов [ESR]), составляющего ≥2,6, считают подходящими для повторного лечения, и их подвергнут рандомизации в соотношении 2:1 для получения повторного лечения с использованием одного дополнительного курса лечения ритуксимабом (группа А) или плацебо (группа В). Индивидов, не удовлетворяющих критериям для второго курса лечения ритуксимабом во время недель 28-40, будут продолжать наблюдать с целью безопасности и эффективности. Индивидов, удовлетворяющих критериям для повторного лечения во время недель 28-40, но отказавшихся от повторного лечения по какой-либо причине, исключат из периода лечения и подвергнут последующему врачебному наблюдению с целью безопасности.
Будут осуществляться стандартные оценки артрита и безопасности. Фармакодинамические измерения будут включать B-клетки CD19+, иммуноглобулины и аутоантитела. Показатели DAS28- ESR будут рассчитываться исследователем при регулярно запланированных посещениях (Prevoo et al. Arthritis Rheum 38: 44-48 (1995); DAS-score.nl 2005 DAS-score.nl 2005: home of the DAS. Department of Rheumatology University Medical Center Nijmegan - the Netherlands [процитированный 1 сентября 2005], который имеется на сайте: http://www.das-score.nl/www.das-score.nl/index.html).
Период лечения продолжается 72 недели (с дня 1 по 72 неделю). Все индивиды, исключенные из периода лечения в любое время или получавшие повторное лечение ритуксимабом/плацебо между неделя 24-40 и завершившие период лечения, должны вернуться для оценок SFU на 4, 12, 24, 36 и 48 недели после исключения или завершения. За индивидами будут наблюдать в течение по крайней мере 48 недель после введения им последней дозы ритуксимаба. Индивидов, получивших только один курс лечения ритуксимабом (т.е. тех, которые не отвечали требованиям для повторного лечения во время исследования) и завершили период лечения, не будут возвращать для SFU.
Индивидов, у которых число периферических В-клеток не восстановилось к концу периода лечения или периода SFU, следует продолжать наблюдать с целью лабораторных оценок и проявления серьезных побочных эффектов каждые 12 недель до восстановления В-клеток. Восстановление В-клеток определяют как возвращение числа периферических В-клеток к величинам базовой линии или нижней границы норы (LLN), что ниже.
На 16 неделе после повторного лечения или позже индивиды, у которых не наблюдается 20% уменьшения как числа болезненных суставов (TJC), так и числа опухших суставов (SJC) по сравнению с базой линией, могут получить спасительное лечение одним небиологическим DMARD, выбор которого - по усмотрению лечащего врача.
В исследовании повторного лечения ритуксимабом RA у индивидов достигались устойчивые ответы Американской коллегии ревматологии (ACR) 20, 50 и 70 (Pavelka et al. “Efficacy and safety following repeated courses of rituximab in patients with active rheumatoid arthritis”, Abstract presented at ELULAR 2005). Однако поскольку это исследование является исследованием с доступной маркировкой, нет сравниваемых с контролем данных, с помощью которых оценивается эффективность и безопасность повторного лечения ритуксимабом RA. Настоящее исследование разрабатывается для оценки эффективности повторного лечения в контролируемом плацебо испытании с одним дополнительным курсом лечения ритуксимабом индивидов с активным RA. Индивиды с активным заболеванием, как определено с помощью DAS 28-ESR ≥2,6, будут подвергаться лечению с использованием второго курса лечения исследуемым лекарственным средством (ритуксимабом или плацебо) на протяжении 24-40 недель.
Целью повторного лечения ритуксимабом является предупреждение вспышек, повышение постоянного контроля над заболеванием и возможно предупреждение прогрессирования заболевания. Критерий DAS 28-ESR ≥2,6 для повторного лечения будет служить гарантией того, что повторному лечению подвергаются индивиды с клинически значимой активностью заболевания. DAS 28-ESR ≥2,6 будут рассчитывать с использованием числа опухших и болезненных суставов, ESR и общей оценки активности заболевания у пациента (по 100 мм визуальной аналоговой шкале [VAS]).
Первичным конечным положением этого исследования является доля подвергшихся повторному лечению индивидов с ACR20 на 48 неделе относительно базовой линии (день 1), не относительно времени повторного лечения. Базовая линия (день 1), не время повторного лечения, была выбран для оценки суммарной пользы повторного лечения ритуксимабом индивидов с RA от умеренной до тяжелой. Не ограничиваясь какой-либо одной теорией, предполагают, что повторное лечение RTX приводит к сохранению эффекта небольшого улучшения на 48 неделе относительно 24 недели, в сравнении с ухудшением у подвергнутых лечению плацебо индивидов. Хотя может существовать очевидная польза от лечения для подвергшихся повторному лечению индивидов, ответы ACR20 на 48 неделе относительно недели 24 в качестве базовой линии для обеих групп могут быть незначительными. Таким образом, это исследование не разрабатывается для оценки улучшения со времени повторного лечения.
Основываясь на данных 24 недели в исследовании фазы III (REFLEX, WA17042/U2646s/IDEC102-20), приблизительно 91% индивидов, подвергнутых лечению ритуксимабом, удовлетворяют критерию повторного лечения (DAS 28-ESR≥2,6) на 24 неделе этого протокола. Обязательное повторное лечение на основе DAS 28-ESR предоставит объективную информацию об эффективности с использованием основанного на активности заболевания принципа повторного лечения. Минимальный период, составляющий 24 недели, основан на фармакокинетических и фармакокинетических параметрах ритуксимаба. Это происходит параллельно очевидному увеличению активности заболевания после этого момента времени, который, следовательно, представляет целесообразное время для повторного лечения.
Первичным измерением результатов является доля подвергшихся повторному лечению индивидов с ответом ACR20 на 48 неделе относительно базовой линии (день 1).
Вторичными измерениями результатов являются следующие измерения:
• доля подвергшихся повторному лечению индивидов с ответами ACR50 и ACR70 на 48 неделе относительно базовой линии (день 1);
• изменение DAS 28-ESR на 48 неделе относительно базовой линии (день 1) у подвергшихся повторному лечению индивидов;
• доля подвергшихся повторному лечению индивидов, достигших ответа Европейской Лиги против ревматизма (EULAR) (хорошего или среднего) на 48 неделе относительно базовой линии (день 1);
• изменение в основном наборе ACR на 48 неделе относительно базовой линии (день 1) у подвергшихся повторному лечению индивидов (SJC, TJC, анкета оценки здоровья [HAQ], общие оценки пациентом и врачом, оценка боли пациентом, С-реактивный белок [CRP] и ESR);
• ACR-N на 48 неделе у подвергшихся повторному лечению индивидов;
• изменение в уменьшенной шкале SF-36 и суммарных показателях от базовой линии (день 1) на 48 неделе у подвергшихся повторному лечению индивидов;
• изменение функциональной оценки усталости при лечении хронического заболевания (FACIT-F)-оценки от базовой линии (день 1) на 48 неделе у подвергшихся повторному лечению индивидов;
• доля ответов ACR20, ACR50 и ACR70 на 48 неделе у всех индивидов.
Исследовательскими измерениями результатов являются следующие измерения:
• доля всех индивидов, достигших ответа ACR20, ACR50 и ACR70 на 72 неделе в сравнении с базовой линией;
• доля индивидов с ремиссией DAS 28-ESR (DAS 28-ESR <2,6) на 48 неделе;
• доля индивидов с заболеванием с низкой активностью DAS 28-ESR (DAS 28-ESR ≤3,2) на 48 неделе;
• доля индивидов с ремиссией DAS 28-ESR (DAS 28-ESR <2,6) на 72 неделе;
• доля индивидов с заболеванием с низкой активностью DAS 28-ESR (DAS 28-ESR≤2,2) на 72 неделе.
Подходящие индивиды с RF-положительным (≥20 МЕ/мл) или RF-отрицательным RA, а также индивиды, которые получают МТХ и ранее или в текущий момент неадекватно реагируют на один или несколько ингибиторов TNF, будут подвергнуты скринингу для участия в исследовании. RF-отрицательные индивиды будут ограничены до 20% от общей зарегистрированной популяции.
Индивиды должны удовлетворять следующим критериям для того, что они подходили для вхождения в исследование:
• Подписать форму информированного согласия;
• Способность и желание выполнять требования протока исследования;
• Возраст 18-80 лет;
• Диагноз RA в течение по крайней мере 6 месяцев, в соответствии с пересмотренными в 1987 г. критериями ACR для классификации RA (Hochberg et al. Arthritis Rheum. 35: 498-502 (1992)):
Класс I
Полный функциональный объем со способностью выполнять все обычные работы без затруднений.
Класс II
Функциональный объем, соответствующий выполнению нормальных работ, несмотря на затруднение вследствие неудобства или ограниченной подвижности одного или нескольких суставов.
Класс III
Функциональный объем, соответствующий выполнению только малого числа работ обычного рода или самообслуживания или их невыполнению.
Класс IV
В большой степени или полностью нетрудоспособный индивид, при этом индивид прикован к крови или коляске, обслуживая себя немного или не обслуживая.
• Получать амбулаторно лечение для RA;
• Следующая документированная активность RA от умеренной до средней при скрининге:
TJC≥8 (максимальный показатель - 68)
SJC≥8 (максимальный показатель - 66) и
Анормальный CRP≥0,6 мг/дл или ESR≥28 мм/ч.
Документированный неадекватный ответ на предшествующее или проводящиеся в текущий момент лечение одним или несколькими из следующих средств: этанерцептом, инфликсимабом и/или адалимумабом - из-за токсичности или несоответствующего эффективности.
Несоответствующая эффективность состоит из лечения этанерцептом на протяжении ≥3 месяцев в дозах 25 мг дважды в неделю или 50 мг раз в неделю, по крайней мере четырех инфузий ≥3 мг/кг инфликсимаба или 40 мг адалимумаба через неделю на протяжении ≥3 месяцев.
• Применение МТХ 10-25 мг/неделю в течение ≥12 недель до дня 1 в постоянной дозе на протяжении ≥4 недель.
• Готовность получать перорально фолиевую кислоту.
• При приеме фоновых кортикостероидов (≤10 мг/день преднизолона или эквивалента) применение кортикостероидов должно быть в постоянной дозе на протяжении 4 недель до дня 1.
• Применение NSAID разрешается при постоянстве дозы в течение ≥2 недель до дня 1.
• Для мужчин и женщин с репродуктивной способностью готовность использовать надежные средства контрацепции (например, гормональный контрацептив, внутриматочное противозачаточное средство, физический барьер) в течение ≥30 дней до дня 1 и во время исследования или на протяжении периода времени, когда у индивида истощены периферические В-клетки CD19+, что дольше.
Индивиды, удовлетворяющие любому из следующих критериев, будут исключаться из вхождения в исследование:
А. Общие
• Ревматическое аутоиммунное заболевание, отличное от RA, или значительное системное поражение, вторичное по отношению к RA, (например, васкулит, фиброз легкого или синдром Фелти). Вторичный синдром Шегрена с RA разрешается.
• История и наличие в текущий момент, воспалительного заболевания сустава, отличного от RA, (например, подагрического, реактивного артрита, псориатического артрита, серонегативной спондилоартропатии или болезни Лима) или другого системного аутоиммунного заболевания (например, системной красной волчанки, болезни воспаленного кишечника, склеродермии, воспалительной миопатии или синдрома наложения).
• Функциональное состояние класса IV, определяемое в соответствии с классификацией ACR функциональных состояний при RA.
• Любая хирургическая операция, в том числе операция на кости/суставе/синовэктомия (в том числе соединение или замещение сустава), в пределах 12 недель до дня 1 или запланированная в пределах 48 недель после дня 1.
• Известная гиперчувствительность к любому компоненту гуманизированного или крысиного моноклонального антитела.
• Введение живой вакцины в пределах 4 недель до дня 1.
• Значимое заболевание сердца или легких (в том числе обструктивное заболевание легких).
• Доказательство значимого неконтролируемого сопутствующего заболевания, включающего, но без ограничения, нарушения нервной системы, почек, печени, эндокринной системы или желудочно-кишечного тракта.
• Известная активная бактериальная, вирусная, грибковая, микобактериальная инфекция или другая инфекция (в том числе туберкулез или атипичное микобактериальное заболевание, за исключением микозов ногтевых лож) или любой крупный эпизод инфекции, требующий госпитализации или лечения вводимыми внутривенно антибиотиками в пределах 4 недель от дня 1 или пероральными антибиотиками в пределах 2 недель от дня 1.
• История серьезной рецидивирующей или хронической инфекции (для скрининга инфекции грудной клетки рентгенограмму грудной клетки будут получать при скрининге, если она не была получена в пределах 12 недель до скрининга).
• История или активный в текущий момент первичный или вторичный иммунодефицит, в том числе ВИЧ-инфекция.
• История злокачественной опухоли, в том числе солидных опухолей и опухолей кроветворной системы (за исключением базальноклеточной и чешуйчатоклеточной карциномы кожи, которую подвергли иссечению и вылечили).
• История значимой цитопении или других заболеваний костного мозга.
• История злоупотребления алкоголем или лекарственной или химической зависимости в пределах 24 недель до дня 1.
• Беременность или лактация.
• Невропатии и невроваскулопатии, которые могли бы оказать влияние на оценку боли.
• Плохой доступ к периферическим венам.
• Непереносимость глюкокортикоидов или противопоказания к внутривенному введению глюкокортикоидов.
b. Лабораторные критерии исключения
• Гемоглобин <8,0 г/дл.
• Абсолютное число нейтрофилов <1,5 × 103/мкл.
• IgM<0,40 мг/мл
• IgG<5,0 мг/мл.
• Аспартатаминотрансфераза (AST) или аланинаминотрансфераза (ALT) > в 2,5 раза верхней границы нормы.
• Положительный тест на поверхностный антиген вируса гепатита В или антитело против вируса гепатита С в сыворотке.
• Для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб) положительный тест на беременность с использованием сыворотки при скрининге.
• Запрещенные принимаемые ранее или сопутствующие лекарственные средства.
• Применение на текущий момент любого DMARD, отличного от МТХ
• Сопутствующее лечение любым биологическим агентом.
Лечение должно быть прекращено за по крайней мере 14 дней до дня 1, за исключением следующих: лефлуномид - за ≥8 недель (или ≥14 дней после выведения в течение 11 дней с помощью стандартного холестирамина), инфликсимаб - за ≥8 недель и адалимумаб за ≥8 недель.
• Любое предшествующее лечение ритуксимабом или с помощью других истощающих клетки терапий, в том CAMPATH, анти-CD4, анти-CD5, анти-CD3, анти-CD19, анти-CD11a, анти-CD22, BLys/BAFF и других анти-CD20 агентов.
• Предшествующее лечение анти-α4-интегрин агентом, включающим натализумаб.
• Предшествующее лечение в пределах 6 месяцев от скринирования γ-глобулином IV или Prosorba® Column.
После завершения всех скринирующих оценок и проверки того, что все индивиды удовлетворяют всем критериям включения и исключения, персонал на местах войдет в контакт с интерактивной системой речевого ответа (ivrs) для получения номера индивида и подтверждения регистрации индивида для лечения с учетом исследуемого лекарственного средства. Все зарегистрированные индивиды будут получать ритуксимаб в качестве первоначального курса лечения.
Во время недель 24-40 индивидов, подходящих для повторного лечения, подвергнут рандомизации в соотношении 2:1 в или группу А (повторное лечение ритуксимабом), или группу В (плацебо; смотри таблицу 1). Если индивиды удовлетворяют критериям годности для повторного лечения во время недель 24-40, персонал на местах войдет в контакт с IVRS для начала рандомизации индивидов и получения номера набора исследуемого лекарственного средства.
Независимый провайдер IVRS осуществит рандомизацию и будет владеть кодами к назначенному лечению. Рандомизацию стратифицируют в исследовательском центре по RF-статусу (RF-положительные, RF-отрицательные) на базовой линии и уменьшениям на 24 неделе числа опухших и болезненных суставов (≥20% уменьшение или <20% уменьшение). Во время прекращения работы вслепую у провайдера IVRS будут запрошены коды к назначенному лечению и коды к используемым для лечения наборам. Переданную документацию и данные, включенные в переданную документацию, будут сохранять зарегистрированными.
Исследуемые подвергаемые лечению группы
1000 мг внутривенно в дни 1 и 15 для курса повторного лечения для подходящих индивидов (DAS28-ESR≥2,6 и удовлетворяют критериям для повторного лечения) во время недель 24-40
Плацебо внутривенно в дни 1 и 15 для курса повторного лечения для подходящих индивидов (DAS28-ESR≥2,6 и удовлетворяют критериям для повторного лечения) во время недель 24-40
Во время повторного лечения от персонала на местах и Genentech будет скрыто назначенное группам лечение. Для предотвращения возможного раскрытия из-за наблюдаемой эффективности и лабораторных изменений во время повторного лечения будет использоваться подход с использованием двух экспертов для оценки эффективности и безопасности.
От персонала на местах и Genentech будет скрыто число периферических В-клеток CD19 до момента разблокирования базы данных для первичного конечного положения на 48 неделе для всех индивидов, зарегистрированных в испытании. Следовательно, все индивиды, завершившие период лечения или период SFU до разблокирования базы данных, будут считаться истощенными в отношении периферических В-клеток и будут оставаться под наблюдением в отношении их В-клеток.
Эксперт по оценке эффективности (или назначенное должное лицо) должен быть ревматологом или квалифицированным экспертом по оценке артрита. Эксперт по оценке эффективности не должен быть научным руководителем. Эксперт по оценке эффективности будет иметь только доступ к данным по эффективности и будет отвечать за обеспечение показателей суставов и выполнение общей оценки врачом активности заболевания VAS только. Для обеспечения согласованной оценки на протяжении всего испытания одному и тому же эксперту следует проводить оценку отдельных индивидов на протяжении всех посещений во время исследования. Во время посещения в течение исследования все сообщаемые индивидом результаты должны завершаться до всех других оценок.
Эксперт по оценке безопасности (или назначенное должное лицо) должен быть ревматологом и будет иметь доступ как к данным по безопасности, так и данным по эффективности. Эксперт по оценке безопасности не должен быть научным руководителем. Эксперт по оценке безопасности будет иметь доступ к исходным документам, лабораторным результатам и формам историй болезней (CRF) и будет отвечать за расчет DAS28-ESR и принятие решения в отношении лечения на основе клинического ответа и лабораторных показателей индивида. Эксперт по оценке безопасности будет выполнять все оценки эффективности и регистрировать результаты любых оценок эффективности по поручению эксперта по оценке эффективности.
Используемый в этом исследовании ритуксимаб представляет собой стерильный, прозрачный, бесцветный, свободный от консервантов жидкий концентрат для внутривенного введения. Ритуксимаб поставляется в концентрации 10 мг/мл в 500 мг (50 мл) одноразовых флаконах. Продукт приготовлен для внутривенного введения в 9,0 мг/мл хлорида натрия, 7,35 мг/мл цитрата натрия дигидрата, 0,7 мг/мл полисорбата 80 и стерильной воде для инъекции. рН доводят до 6,5.
Все зарегистрированные индивиды будут получать курс лечения ритуксимабом (1000 мг ритуксимаба с помощью внутривенного вливания в дни 1 и 15) в качестве первичного лечения. Во время недель 24-40 индивидов, подходящих для повторного лечения, подвергнут рандомизации в группу А для получения дополнительного курса лечения (две дозы) ритуксимабом 1000 мг внутривенно с интервалом в 14 дней или в группу В для получения курса (две дозы) плацебо 1000 мг внутривенно с интервалом в 14 дней.
Для всех индивидов рекомендуется премедикация 1000 мг перорального ацетаминофена и 50 мг перорального дифенгидрамина, и необходимо, чтобы введение 100 мг внутривенно метилпреднизолона было завершено в пределах 30-60 минут до каждой инфузии ритуксимаба/плацебо.
Все пациенты будут продолжать получать МТХ в постоянной дозе 10-25 мг/неделю и получать постоянную дозу фолата (≥5 мг/неделю) в виде одной дозы или в виде дробной еженедельной дозы.
Инфузии ритуксимаба/плацебо следует проводить индивидам под тщательным наблюдением исследователя или назначенного должностного лица в больнице или клинике, в которых немедленно доступны все средства реанимации. Хотя ритуксимаб можно вводить амбулаторно, по усмотрению исследователя индивидов можно госпитализировать для наблюдения. Ритуксимаб/плацебо следует вводить в виде медленных внутривенных вливаний. Нельзя вводить в виде внутривенного проталкивания или болюса. В конце каждой инфузии внутривенное соединение следует оставлять на месте в течение по крайней мере 1 часа для того, чтобы иметь возможность ввести внутривенно лекарственные средства, если потребуется. Если на протяжении этого времени побочных эффектов не наблюдаются, внутривенное соединение удаляют.
Используя соответствующую асептическую технику, следует извлечь необходимое количество ритуксимаба/плацебо и развести до конечной концентрации 4 мг/мл в мешке для инфузий, содержащем или 0,9% хлорида натрия, USP, или 5% декстрозы в воде, USP. Следует осторожно перевернуть мешок, чтобы перемешать раствор. Флаконы с ритуксимабом являются биологически и химически стабильными при 2ºС-8ºС (36ºF-46ºF). Флаконы не должны использоваться после срока годности лекарственного средства. Ритуксимаб следует защищать от воздействия прямых солнечных лучей.
После воссоздания в мешке для внутривенных вливаний раствор ритуксимаба для вливаний можно хранить при 2ºС-8ºС (36ºF-46ºF) в течение 24 часов. Показано, что растворы ритуксимаба являются стабильными в течение дополнительных 24 часов при комнатной температуре (23ºС или 73ºF). Однако поскольку растворы ритуксимаба не содержат консерванта, разведенные растворы следует подвергнуть охлаждению (2ºС-8ºС). Несовместимости между ритуксимабом и мешками из поливинилхлорида или полиэтилена не наблюдалось.
Что касается повторного лечения, DAS28-ESR будут рассчитывать с использованием числа суставов и общей оценки пациентом активности заболевания (VAS) от посещения в текущий момент и ESR от посещения в текущий момент или предшествующего посещения только, если нет ESR от посещения в текущий момент. Для получения повторного лечения исследуемым лекарственным средством (ритуксимабом или плацебо) подходят пациенты, удовлетворяющие следующим критериям при любом посещении на протяжении недель 24-40):
• DAS28-ESR≥2,6
• Для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб) отрицательный тест на беременность при использовании их мочи.
Кроме того, индивиды, отвечающие требованиям повторного лечения по DAS28-ESR≥2,6 на протяжении недель 24-40, должны также удовлетворять следующим критериям для того, чтобы подвергнуться повторному лечению на основе результатов от предшествующего посещения, если нет текущих результатов:
• Гемоглобин ≥8,0 г/дл.
• Абсолютное число нейтрофилов ≥1,5 × 103/мкл.
• IgM≥0,40 мг/мл
• IgG≥5,0 мг/мл.
• Нет значимого заболевания сердца или легких (в том числе обструктивного заболевания легких).
• Нет первичного или вторичного иммунодефицита, в том числе известной истории ВИЧ-инфекции.
• Нет доказательства значимого неконтролируемого сопутствующего заболевания, включающего, но без ограничения, нарушения нервной системы, почек, печени, эндокринной системы или желудочно-кишечного тракта.
• Нет активной инфекции любого вида (за исключением микозов ногтевых лож) или любого крупного эпизода инфекции, требующего госпитализации или лечения вводимыми внутривенно антибиотиками в пределах 4 недель от инфузии, или завершения приема вводимых орально антибиотиков в пределах 2 недель до инфузии.
Во время этого исследования изменять дозу ритуксимаба не разрешается. В случае связанной с инфузией реакции скорость инфузии можно регулировать. Если у индивида имеет место связанная с инфузией реакция, требующая прерывания инфузии, и исследователь принимает решение не возобновлять инфузию, индивид должен быть исключен из периода исследуемого лечения и подвергнут SFU.
Все индивиды будут получать сопутствующие 10-25 мг/неделю МТХ, как предписано их лечащим врачом. Индивиды должны подвергаться лечению МТХ в течение ≥12 недель до вхождения в исследование и должны оставаться на постоянной дозе МТХ в течение ≥4 недель до дня 1 и во время всего исследования, если только не требуется изменение вследствие токсичности.
Могут иметь место некоторые побочные эффекты, обычно сопровождающие лечение МТХ. Для минимизации токсичности МТХ все индивиды, подвергаемые лечению МТХ, будут получать постоянную дозу фолиевой кислоты (≥5 мг/неделю) в виде в виде одной дозы раз в неделю или в виде дробной ежедневной дозы. Схема приема назначается по усмотрению исследователя.
Разрешается ежедневное лечение ≤10 мг преднизона или эквивалента, если доза является постоянной в течение ≥4 недель до дня 1. Доза должна оставаться постоянной на протяжении всего исследования, если только не требуется изменение вследствие токсичности.
Разрешается применение NSAID, если доза является постоянной в течение ≥2 недель до дня 1. Доза должна оставаться постоянной на протяжении всего исследования, если только не требуется изменение вследствие токсичности.
Кроме того, разрешается ежедневное лечение ≤325 мг ацетилсалициловой кислоты для профилактики сердечно-сосудистых заболеваний.
Можно использовать, если требуется, дополнительные аналгетики. Однако индивиды не должны принимать аналгетики в пределах 12 часов до посещения, при котором выполняются оценки эффективности. Могут быть осуществлены корректировки схемы приема аналгетиков, но изменения должны быть документированы в соответствующих CRF.
Внутрисуставные введения кортикостероидов не рекомендуются, особенно во время первых 48 недель; однако их можно использовать ограниченно для регулирования активности RA у индивидов во время исследования. Не более одного сустава на протяжении 24-недельного периода должно подвергаться инъекции, и один и тот же сустав не должен подвергаться инъекции более одного раза на протяжении любого 48-недельного периода. Одна инъекция не должна превышать 40 мг триамцинолона (или эквивалента), и суммарная доза кортикостероида не должна превышать 80 мг триамцинолона (или эквивалента) во время любого 48-недельного периода.
Во время исследования индивиды могут продолжать получать фоновый кортикостероид в дозе ≤10 мг/день преднизона (или эквивалента). В случае вспышки заболевания или не являющего RA состояния, такого как астма, требующих лечения пероральными кортикостероидами, следует назначать соответствующие дозы в течение максимум 2 недель и снижать до предшествующего уровня настолько быстро, насколько это возможно терапевтически.
Увеличение дозы кортикостероидов следует считать ухудшением состояния индивида по сравнению с базовой линией и должно быть отмечено в качестве побочного эффекта в CRF.
В этом исследовании не разрешаются вводимые внутривенно или внутримышечно кортикостероиды, отличные от тех, которые определены в протокольных лечениях.
Увеличение дозы кортикостероидов следует считать ухудшением состояния индивида по сравнению с базовой линией и должно быть отмечено в качестве побочного эффекта в CRF.
Индивиды должны оставаться на постоянной дозе МТХ в течение ≥4 недель до дня 1 и во время всего исследования, если только не требуется изменение вследствие токсичности.
На 16 неделе после повторного лечения или позже индивиды, у которых не наблюдается 20% уменьшения как числа болезненных суставов (TJC), так и числа опухших суставов (SJC) по сравнению с базовой линией, могут получить спасительное лечение одним небиологическим DMARD, выбор которого - по усмотрению лечащего врача. Индивиды, получившие спасительное лечение, не будут исключаться из исследования.
До внутривенных вливаний метилпреднизолона и ритуксимаба/плацебо должны быть взяты лабораторные образцы.
Сообщаемые индивидом данные (например, общая оценка пациентом активности заболевания, оценка пациентом боли) могут быть только документированы включенной в исследование медсестрой/исследователем по поручению индивида, если индивиду трудно писать во время посещения, или он не может читать. Это должно быть четко документировано в записях индивида.
Для предотвращения возможного прекращения работы вслепую для оценки эффективности и безопасности будет использоваться подход с двумя экспертами по оценке (экспертом по оценке эффективности и экспертом по оценке безопасности). Важно, чтобы оценки, выполняемые индивидом и экспертом по оценки эффективности, были сделаны до оценок экспертом по безопасности.
Для общей оценки индивидом его или ее активности заболевания во время последних 24 часов должна использоваться 100-мм горизонтальная VAS, где левый край линии представляет отсутствие активности заболевания (без симптомов или без симптомов артрита), а правый край представляет максимальную активность заболевания (максимальная активность заболевания артрита).
Для оценки индивидом его или ее степени боли во время последних 24 часов должна использоваться 100-мм горизонтальная VAS, где левый край линии представляет отсутствие боли, а правый край представляет невыносимую боль.
Показатель нетрудоспособности Stanford HAQ представляет собой заполняемую индивидом анкету, специфичную в отношении RA. Она состоит из 20 вопросов, относящихся к наборам из восьми компонентов: одевание/уход за одеждой, подъем, прием пищи, хождение, гигиена, досягаемость, сжатие и активности. Анкету будут оценивать на основе инструкций от Stanford University Medical Center (Fries et al. Arthritis Rheum 23: 137-145 (1980)).
Для оценки усталости будет использоваться FACIT-F. FACIT-F представляет собой анкету из 13 элементов, в которой индивиду необходимо оценить каждый вопрос по шкале 0-4. Эта оценка была первоначально разработана для хронических заболеваний и в настоящее время ее правильность подтверждается для индивидов с RA (Cella et al. J. Rheumatology 32(5): 811-819 (2005)).
SF36 представляет собой средство определения общего связанного со здоровьем качества жизни, которое было в большой степени проверено в отношении его психометрических свойств и широко используется в клинических и эпидемиологических исследованиях (Ware et al. How to Score Version Two of the SF-36 Health Survey. Lincoln, RI: Qualitymetric Incorporated, 2000). SF36 (версия 2) предоставляется Medical Outcomes Trust (Boston, MA, США).
Оценка с максимальным числом 66 для опухших суставов и максимальным числом 68 для болезненных суставов должна осуществляться ревматологом или квалифицированным экспертом по оценке суставов. Суставы будут оценивать и классифицировать как опухшие или не опухшие и болезненные или не болезненные на основе манипулирования давлением и суставами при физической проверке. Не должны подвергаться оценке полностью протезированные суставы или суставы с артродезом; однако все прочие суставы должны подвергаться оценке. Суставы, подвергаемые оценке в отношении опухлости и болезненности, предоставлены ниже:
Височно-нижнечелюстной сустав
Грудиноключичный сустав
Акромиально-ключичный сустав
Плечиа
Локтевые суставы∗
Запястья∗
Межфаланговый сустав на пальце Iа
Дистальные межфаланговые суставы на пальцах 2-5
Проксимальные межфаланговые суставы на пальцах 2-5а
Пястно-фаланговые (МРС) суставы на пальцах 1-5а
Тазобедренные суставы (только в отношении болезненности)
Коленные суставы а
Голеностопные суставы
Плюсневые суставы
Межфаланговые суставы пальцев стопы 1-5
Плюснефаланговые суставы пальцев стопы 1-5
а Включает 28 суставов, используемых для расчета показателя активности заболевания (DAS28).
Суставы, подвергнутые операции или манипуляции, должны оцениваться следующим образом:
• Операции: Любой сустав, замещенный или соединенный в любое время до или во время исследования, следует документировать как не поддающийся оценке на протяжении исследования.
Любой сустав, подвергшийся синовэктомии (в том числе химической и радиологической синовэктомии), следует документировать как «не сделано (ND)» в течение 24 недель после синовэктомии. После этого времени сустав можно оценивать снова.
• Внутрисуставные инъекции: Любой сосуд, подвергнутый внутрисуставной инъекции кортикостероида, следует документировать как «ND» в течение последующих 12недель. После этого времени сустав можно оценивать снова.
• Артроцентез: Любой сустав, который подвергается пункции синовиальной жидкости, не будет оцениваться при следующем запланированном посещении и будет оценен как ND. После этого времени сустав можно оценивать снова.
Для оценки врачом активности заболевания у индивида во время последних 24 часов должна использоваться 100-мм горизонтальная VAS, где левый край линии представляет отсутствие активности заболевания (без симптомов или без симптомов артрита), а правый край представляет максимальную активность заболевания. Это должно осуществляться экспертом по оценке эффективности, который может быть или может не быть врачом.
CRP будут анализировать в центральной лаборатории. ESR будет определяться с использованием способа Westergren в местной лаборатории.
DAS 28-ESR≥2,6 был выбран в качестве пороговой величины для повторного лечения в этом испытании. Индивиды с DAS 28-ESR≥2,6 во время недель 24-40 могут подходить для получения второго курса лечения исследуемым лекарственным средством (ритуксимабом или плацебо).
Общая физическая оценка (в том числе сердечно-сосудистой, дыхательной, желудочно-кишечной и нервной систем) должна проводиться в моменты времени, указанные в графике оценок. Любые сохраняющиеся отклонения должны отмечаться каждый раз, когда проводится оценка. Диагноз новых отклонений должен быть документирован как побочный эффект, если уместно.
Признаки жизни (частоту сердечных сокращений, систолическое и диастолическое кровяное давление и температуру) следует определять в моменты времени, указанные в графике оценок. Оценки должны проводиться после того, как индивид находился в полулежащем состоянии в течение по крайней мере 5 минут.
Электрокардиограммы (ECG) двенадцати отведений должны сниматься в моменты времени, указанные в графике оценок.
Рентгенограммы грудной клетки сзади-спереди и сбоку следует получать при скрининге, и они должны анализироваться исследователем и назначенным должностным лицом. Если при скрининге рентгенограммы грудной клетки, полученные в пределах последних 12 недель, не демонстрируют клинически значимых отклонений, и нет признаков или симптомов, говорящих о заболевании легких, что должно было бы привести к исключению индивида из испытания, повторного получения рентгенограммы грудной клетки не требуется.
Центральной лабораторией будут проводиться гематология, серология, биохимия, анализ мочи, тест на беременность с использованием сыворотки, проточная цитометрия, иммунология и CRP-анализ; Genentech будет проводить фармакокинетические анализы и НАСА-анализ; а тест на беременность с использованием мочи и ESR-оценки будут осуществляться локально в местах исследования. Для всех лабораторных оценок будут предоставлены инструкции о порядке работы и наборы - снабжение, в том числе снабжение для фармакокинетических анализов и НАСА-анализа. Лабораторные оценки будут включать следующее:
• Гематология/CBC (клинический анализ крови): гемоглобин, гематокритное число, эритроциты (RBC), лейкоциты (WBC) с определением лейкоцитарной формулы и подсчет тромбоцитов.
• Серология: поверхностный антиген вируса гепатита В (HBsAg) и антитело против вируса гепатита С (HCV).
• Биохимия сыворотки: AST/SGOT, ALT/SGPT, щелочная фосфатаза, общий белок, альбумин, общий билирубин, азот мочевины в крови (BUN), мочевая кислота, креатинин случайная глюкоза, калий, натрий, хлорид, кальций и фосфор.
• Анализ мочи: кровь, белок и глюкоза (микроскопическая проверка при аномалии и применимости).
• Тест на беременность: При скрининге для всех женщин с репродуктивной способностью (в том числе с перевязкой маточных труб) будет проводиться тест на беременность с использованием их сыворотки. Кроме того, при всех остальных посещениях будут назначаться обычные тесты на беременность с использованием мочи. Если тест на беременность с использованием мочи является положительным, его необходимо подтвердить с помощью тест на беременность с использованием сыворотки.
• Уровни компонентов комплемента С3 и С4 в сыворотке.
• Иммунологические оценки: количественные оценки иммуноглобулинов (общий Ig, IgG, IgA и IgМ), RF (общие концентрации и концентрации изотипов) и антитело (IgG) против циклического пептида с цитруллином (ССР).
• Расширенный анализ с использованием клеточного сортера с активацией флуоресценции (FACS): оцениваемые популяции клеток будут включать моноциты (CD14 и CD16); NK-клетки (CD56); субпопуляции Т-клеток (CD3, CD4, CD8, CD45RO и CD45RA); и субпопуляции В-клеток (CD19, CD27, CD38 и IgD). Могут также оцениваться маркеры активации (CD25, CD69, CD40L и CD80).
• FACS-анализ В-клеток: абсолютное число В-клеток (CD19) только.
• Анализ НАСА-ответа будет проводиться с использованием ELISA для всех зарегистрированных индивидов.
• Фармакокинетические анализы: Образцы сыворотки будут получать для фармакокинетического анализа во время посещений, указанных в SOA, а также в те же моменты времени, что и НАСА. Для концентраций ритуксимаба в сыворотке требуется правильная интерпретация результатов НАСА.
• Необязательные биомаркерные образцы:
Для индивидов, давших отдельное согласие, необязательные исследовательские образцы (цельная кровь, сыворотка) будут взяты во время курса исследования для исследовательских оценок биомаркеров. Образцы цельной крови, помещенные в РНК-пробирки PAXgeneTM, будут использоваться для получения профиля экспрессии генов. Оценки маркеров, связанных с RA или ритуксимабом, в образце сыворотки могут включать, но без ограничения, определение цитокинов/хемокинов и количественное определение маркеров обновления кости и хряща. Все образцы будут брать в одни и те же временные точки для оптимальной совместимости данных.
Ожидается, что информация от этого анализа стимулирует и облегчит индивидуализированную медико-санитарную помощь посредством лучшего понимания механизма действия ритуксимаба, связанных эффективности и безопасности, прогностических факторов для хорошего ответа и возможно решающих факторов прогрессирования RA (или других аутоиммунных заболеваний). Эти данные будут храниться вплоть до 15 лет после закрытия базы данных.
Все индивиды должны дать письменное информированное согласие до начала выполнения каких-либо специфических для исследования процедур или оценок, в том числе изменений используемой в текущий момент схемы лечения. Скринирующие оценки могут быть выполнены в течение 56 дней до первой инфузии ритуксимаба в день 1. Сообщаемые индивидом оценки следует завершать до начала других клинических оценок.
Если индивид не удовлетворяет лабораторному критерию исключения при скрининге, исследователь может повторить тест до двух раз в пределах периода скринирования. Если индивид не удовлетворяет лабораторному критерию в третий раз, его считают вышедшим из исследования при скринировании. Образец крови или лабораторную проверку не будут считать подвергнутыми повторной проверке, если образец был подвергнут повторной проверке из-за проблем обращения с образцом, разрушения или целостности образца. Индивид, не прошедший скринирование, может быть подвергнут повторному скринированию.
Индивиды могут быть подвергнуты повторному скринированию, если они не удовлетворяли всем критериям годности в пределах 56 дней от первоначального посещения в целях скринирования. Индивид, подвергаемый повторному скринированию, должен повторно пройти весь процесс скринирования и повторно дать согласие до начала любых специфичных для исследования процедур. Индивиды могут быть подвергнуты повторному скринированию только раз.
а. Посещение с целью скринирования (день 56)
• Письменное информированное согласие
• Анализ критериев включения и исключения
• Контакт с IVRS для получения присвоения номера скрининга индивида
• Демографические данные (например, пол, возраст, раса/этничность)
• Полная история болезни (в том числе история вакцинаций)
• Сопутствующие лекарственные средства, принимаемые в пределах 12 недель до скрининга, в том числе вакцины, все предшествующие DMARD и биологические агенты
• Признаки жизни (частота сердечных сокращений, кровяное давление и температура)
• Полная физическая проверка, в том числе измерения роста и веса
• Оценка суставов
• ECG 12 отведений
• Рентгеновский анализ грудной клетки
Если рентгеновский анализ грудной клетки проводился в пределах 12 недель до скринирования и не продемонстрировал клинически значимых отклонений, при скринировании рентгеновский анализ грудной клетки не требуется.
• Оценки центральной лаборатории
Гематология/CBC
Поверхностный антиген вируса гепатита В или антитело против вируса гепатита С
Тест на беременность с использованием сыворотки для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
Биохимия сыворотки
Анализ мочи
Тест на беременность с использованием сыворотки для женщин
CRP
IgG и IgМ
Общий RF
• ESR (местная лаборатория; метод Westergren)
Все оценки во время периода лечения следует проводить в пределах точно определенного временного окна для каждого посещения. Оценки и процедуры, запланированные на тот день, когда вводится ритуксимаб, должны проводиться до инфузии ритуксимаба, если только не указано иное. Для этого исследования день 1 является днем первоначальной инфузии ритуксимаба. Посещение в день 1 должно быть в такой день, который делает возможным последующие посещения (например, посещение в день 15) без задержки. Сообщаемые индивидом оценки следует проводить до начала других клинических оценок. Для индивидов, подходящих для повторного лечения, посещение, во время которого индивид удовлетворит критериям годности для повторного лечения, считается квалифицирующим посещением.
а. День 1
Все оценки будут проводиться за 30 минут до инфузии, если только не определено иное.
• Анализ критериев включения и исключения
• Контакт с IVRS для получения номера регистрации индивида и учета исследуемого лекарственного вещества
• Общая оценка пациентом активности заболевания (VAS)
• Оценка пациентом боли (VAS)
• Показатель нетрудоспособности Stanford HAQ
• FASIT-F
• SF-36
• Признаки жизни (частота сердечных сокращений, кровяное давление и температура): до инфузии, во время инфузии (каждые 15 минут в течение 1 часа и затем каждые 30 минут до конца инфузии) и после инфузии (каждые 30 минут в течение 1 часа после инфузии)
• Физическая проверка, в том числе измерение веса тела
• Оценка суставов
• Общая оценка врачом активности заболевания (VAS)
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC (в пределах 30 минут до и после инфузии)
Биохимия сыворотки
Анализ мочи
Расширенный FACS (в пределах 30 минут до и после инфузии)
В-клетки CD19 (в пределах 30 минут до и после инфузии)
CRP
Иммуноглобулины
RF
Антитело против ССР
С3, С4
Фармакокинетический образец (в пределах 30 минут до и после инфузии)
Образец для НАСА
• ESR (местная лаборатория; метод Westergren)
• Необязательные исследовательские образцы для оценки биомаркеров (цельная кровь и образцы сыворотки)
• Назначение метилпреднизолона
• Введение ритуксимаба
• Побочные эффекты
• Сопутствующие лекарственные средства
b. День 15 (±1 день)
Все оценки будут проводиться за 30 минут до инфузии, если только не определено иное.
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Признаки жизни (частота сердечных сокращений, кровяное давление и температура): до инфузии, во время инфузии (каждые 15 минут в течение 1 часа и затем каждые 30 минут до конца инфузии) и после инфузии (каждые 30 минут в течение 1 часа после инфузии)
• Оценки центральной лаборатории
Гематология/CBC (в пределах 30 минут до и после инфузии)
Биохимия сыворотки
Анализ мочи
С3, С4
Фармакокинетический образец (в пределах 30 минут до и после инфузии)
• Назначение метилпреднизолона
• Введение ритуксимаба
• Побочные эффекты
• Сопутствующие лекарственные средства
с. Недели 4, 12 и 20 (дни 28, 84 и 140, соответственно; ±3 дня)
• Общая оценка пациентом активности заболевания (VAS)
• Оценка пациентом боли (VAS)
• HAQ
• FASIT-F (неделя 12 только)
• Оценка суставов
• Общая оценка врачом активности заболевания (VAS)
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC
Биохимия сыворотки
Анализ мочи
Расширенный FACS (неделя 12 только)
В-клетки CD19 (недели 4 и 20 только)
CRP
С3, С4 (неделя 4 только)
Фармакокинетический образец
Иммуноглобулины
• ESR (местная лаборатория; метод Westergren)
• Необязательные исследовательские образцы для оценки биомаркеров (цельная кровь и образцы сыворотки) (неделя 12 только)
• Побочные эффекты
• Сопутствующие лекарственные средства
d. Неделя 24 (день 168±3 дня)
• Общая оценка пациентом активности заболевания (VAS)
• Оценка пациентом боли (VAS)
• HAQ
• FASIT-F
• SF-36
• Оценка суставов
• Общая оценка врачом активности заболевания (VAS)
• Расчет DAS 28-ESR c использование подсчета суставов и общей оценки пациентом активности заболевания (VAS) от этого посещения и ESR от предшествующего посещения, если нет результата ESR.
Оценивается годность индивида для повторного лечения на основе рассчитанной DAS 28-ESR и лабораторных результатов от предшествующего посещения.
Если индивид подходит для повторного лечения, осуществляют переход к дню 1 повторного лечения и выполняют оценки, как определено.
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC
Биохимия сыворотки
Анализ мочи
Расширенный FACS
CRP
Иммуноглобулины
RF
Антитело против ССР
Фармакокинетический образец
Образец для НАСА
• ESR (местная лаборатория; метод Westergren)
• Необязательные исследовательские образцы для оценки биомаркеров (цельная кровь и образцы сыворотки)
• Побочные эффекты
• Сопутствующие лекарственные средства
е. Неделя 28 (день 196±3 дня)
• Общая оценка пациентом активности заболевания (VAS)
• Оценка пациентом боли (VAS)
• HAQ
• Оценка суставов
• Общая оценка врачом активности заболевания (VAS)
• Выполняется только, если индивид не подвергся повторному лечению, расчет DAS 28-ESR c использование подсчета суставов и общей оценки пациентом активности заболевания (VAS) от этого посещения и ESR от предшествующего посещения, если нет результата ESR.
Оценивается годность индивида для повторного лечения на основе рассчитанной DAS 28-ESR и лабораторных результатов от предшествующего посещения.
Если индивид подходит для повторного лечения, осуществляют переход к дню 1 повторного лечения и выполняют оценки, как определено.
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC
Биохимия сыворотки
Анализ мочи
В-клетки CD19
CRP
Иммуноглобулины
Фармакокинетический образец
• ESR (местная лаборатория; метод Westergren)
• Побочные эффекты
• Сопутствующие лекарственные средства
f. Недели 32, 40 и 44 (дни 224, 280 и 308, соответственно; ±3 дня)
• Общая оценка пациентом активности заболевания (VAS)
• Оценка пациентом боли (VAS)
• HAQ
• FASIT-F (только неделя 32)
• Оценка суставов
• Общая оценка врачом активности заболевания (VAS)
• Выполняется только, если индивид не подвергся повторному лечению, расчет DAS 28-ESR c использование подсчета суставов и общей оценки пациентом активности заболевания (VAS) от этого посещения и ESR от предшествующего посещения, если нет результата ESR (только недели 32 и 40).
Оценивается годность индивида для повторного лечения на основе рассчитанной DAS 28-ESR и лабораторных результатов от предшествующего посещения.
Если индивид подходит для повторного лечения, осуществляют переход к дню 1 повторного лечения и выполняют оценки, как определено.
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC
Биохимия сыворотки
Анализ мочи
Расширенный FACS (выполняется только, если индивид не подвергся повторному лечению; только неделя 44)
В-клетки CD19
Иммуноглобулины
CRP
Фармакокинетический образец
• ESR (местная лаборатория; метод Westergren)
• Побочные эффекты
• Сопутствующие лекарственные средства
g. Неделя 48 (день 336±3 дня)
• Общая оценка пациентом активности заболевания (VAS)
• Оценка пациентом боли (VAS)
• HAQ
• FASIT-F
• SF-36
• Оценка суставов
• Общая оценка врачом активности заболевания (VAS)
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC
Биохимия сыворотки
Анализ мочи
Расширенный FACS
CRP
Иммуноглобулины
RF
Антитело против ССР
Фармакокинетический образец
Образец для НАСА
• ESR (местная лаборатория; метод Westergren)
• Необязательные исследовательские образцы для оценки биомаркеров (цельная кровь и образцы сыворотки)
• Побочные эффекты
• Сопутствующие лекарственные средства
h. Неделя 60 (день 420±7 дней)
• Общая оценка пациентом активности заболевания (VAS)
• Оценка пациентом боли (VAS)
• HAQ
• FASIT-F
• Оценка суставов
• Общая оценка врачом активности заболевания (VAS)
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC
Биохимия сыворотки
Анализ мочи
Расширенный FACS
CRP
Иммуноглобулины
Необязательные исследовательские образцы для оценки биомаркеров (цельная кровь и образцы сыворотки)
• ESR (местная лаборатория; метод Westergren)
• Побочные эффекты
• Сопутствующие лекарственные средства
i. Неделя 72 (день 504±7 дней)
Индивиды, у которых число периферических В-клеток (CD19+) не восстановилось, после завершения этого посещения войдут в период слежения за В-клетками. Восстановление В-клеток определяют как возвращение числа периферических В-клеток к величинам базовой линии или нижней границы норы (LLN), что ниже.
• Общая оценка пациентом активности заболевания (VAS)
• Оценка пациентом боли (VAS)
• HAQ
• FASIT-F
• SF-36
• Признаки жизни (частота сердечных сокращений, кровяное давление и температура)
• Физическая проверка, в том числе веса
• Оценка суставов
• Общая оценка врачом активности заболевания (VAS)
• ECG 12 отведений
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC
Биохимия сыворотки
Анализ мочи
Расширенный FACS
CRP
Иммуноглобулины
RF
Антитело против ССР
• ESR (местная лаборатория; метод Westergren)
• Необязательные исследовательские образцы для оценки биомаркеров (цельная кровь и образцы сыворотки)
• Побочные эффекты
• Сопутствующие лекарственные средства
Индивиды, подходящие для повторного лечения во время недель 24-30, будут подвергнуты рандомизации для получения дополнительного курса лечения исследуемым лекарственным средством. Для индивидов должны быть завершены все оценки дня 1 повторного лечения, которые не были проведены во время квалифицирующего посещения. Оценки, проведенные при квалифицирующем посещении, повторять не требуется.
Если инфузии не могут быть проведены в тот же день, что и квалифицирующее посещение, индивиды должны вернуться для проведения инфузии в пределах 72 часов квалифицирующего посещения.
После курса повторного лечения (дни 1 и 15 повторного лечения) следующее посещение индивида планируется на основе графика оценок от момента времени, в который индивид был квалифицирован для повторного лечения. Например, если индивид был квалифицирован для повторного лечения на 32 неделе, после курса повторного лечения (дни 1 и 15 повторного лечения) следующим запланированным посещением индивида должно быть посещение на 40 неделе.
а. День 1 повторного лечения (R1)
Все оценки будут проводиться за 30 минут до инфузии, если только не определено иное.
• Рандомизация и назначение исследуемого лекарственного средства через IVRS
• Общая оценка пациентом активности заболевания (VAS)
• Оценка пациентом боли (VAS)
• Показатель нетрудоспособности Stanford HAQ
• FASIT-F
• SF-36
• Признаки жизни (частота сердечных сокращений, кровяное давление и температура): до инфузии, во время инфузии (каждые 15 минут в течение 1 часа и затем каждые 30 минут до конца инфузии) и после инфузии (каждые 30 минут в течение 1 часа после инфузии)
• Физическая проверка, в том числе измерения веса тела
• Оценка суставов
• Общая оценка врачом активности заболевания (VAS)
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC (в пределах 30 минут до и после инфузии)
Биохимия сыворотки
Анализ мочи
CRP
Расширенный FACS (в пределах 30 минут до и после инфузии)
В-клетки CD19 (в пределах 30 минут до и после инфузии)
Иммуноглобулины
RF
Антитело против ССР
С3, С4
Фармакокинетический образец (в пределах 30 минут до и после инфузии)
Образец для НАСА
• ESR (местная лаборатория; метод Westergren)
• Необязательные исследовательские образцы для оценки биомаркеров (цельная кровь и образцы сыворотки)
• Назначение метилпреднизолона
• Введение исследуемого лекарственного вещества
• Побочные эффекты
• Сопутствующие лекарственные средства
b. День 15 повторного лечения (R15; ±1 день)
Все оценки будут проводиться за 30 минут до инфузии, если только не определено иное.
• Признаки жизни (частота сердечных сокращений, кровяное давление и температура): до инфузии, во время инфузии (каждые 15 минут в течение 1 часа и затем каждые 30 минут до конца инфузии) и после инфузии (каждые 30 минут в течение 1 часа после инфузии)
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC (в пределах 30 минут до и после инфузии)
Биохимия сыворотки
Анализ мочи
C3, С4
Фармакокинетический образец (в пределах 30 минут до и после инфузии)
• Назначение метилпреднизолона
• Введение исследуемого лекарственного вещества
• Побочные эффекты
• Сопутствующие лекарственные средства
Индивидов, которые были исключены ранее из периода лечения по какой-либо причине, попросят вернуться в период до 14 дней после исключения (посещение в связи ранним исключением из лечения) и затем вернуться на 4 неделе после посещения в связи с исключением и затем каждые 12 недель на протяжении по крайней мере 48 недель от момента времени исключения индивидов. Следующие оценки будут выполняться при посещении в связи с ранним исключением из лечения:
• Общая оценка пациентом активности заболевания (VAS)
• Оценка пациентом боли (VAS)
• HAQ
• FASIT-F
• SF-36
• Признаки жизни (частота сердечных сокращений, кровяное давление и температура)
• Физическая проверка, в том числе веса
• Оценка суставов
• Общая оценка врачом активности заболевания (VAS)
• ECG 12 отведений
• Тест на беременность с использованием мочи для женщин с репродуктивной способностью (в том числе с перевязкой маточных труб)
• Оценки центральной лаборатории
Гематология/CBC
Биохимия сыворотки
Анализ мочи
CRP
В-клетки CD19
Иммуноглобулины
RF
Антитело против ССР
Фармакокинетический образец
Образец для НАСА
Необязательные исследовательские образцы для оценки биомаркеров (цельная кровь и образцы сыворотки)
• ESR (местная лаборатория; метод Westergren)
• Побочные эффекты
• Сопутствующие лекарственные средства
Оценки при врачебном наблюдении с целью безопасности (SFU) будут проводиться на неделях 4, 12, 24, 36 и 48 SFU после раннего исключения или завершения периода лечения, как определено для следующих индивидов:
• Индивиды, подвергшиеся повторному лечению во время недель 24-40 и завершившие период лечения (т.е. посещение на 72 неделе)
• Индивиды, исключенные рано из периода лечения.
Следующие оценки будут проводиться при каждом посещении с целью SFU:
• Все побочные эффекты вплоть до недели 4
• Серьезные побочные эффекты и инфекционные неблагоприятные эффекты после недели 4
• Сопутствующие лекарственные средства, используемые для лечения этих эффектов
• Оценки центральной лаборатории
Гематология/CBC
Иммуноглобулины
Фармакокинетический образец
Образец для НАСА
В-клетки CD19
При последнем посещении с целью SFU индивиды, у которых число периферических В-клеток (CD19+) не восстановилось, войдут в период слежения за В-клетками. Восстановление В-клеток определяют как возвращение числа периферических клеток CD19+ к величинам базовой линии или нижней границы норы (LLN), что ниже. При всех сообщаемых серьезных инфекционных неблагоприятных эффектах CBC c определением лейкоцитарной формулы, количественным определением Ig и подсчетом CD19 будут определяться в пределах 1 недели от возникновения.
Индивиды, у которых число периферических В-клеток (CD19+) не восстановилось, при последнем определенном протоком посещении (неделя 72 или конец SFU) войдут в период слежения за В-клетками, и они будут возвращаться каждые 12 недель от последнего посещения до восстановления В-клеток для посещений с целью исследования. Восстановление В-клеток определяют как возвращение числа периферических клеток CD19+ к величинам базовой линии или нижней границы норы (LLN), что ниже.
При каждом посещении с целью контроля В-клеток будут проводиться следующие оценки и процедуры:
• Серьезные побочные эффекты и инфекционные неблагоприятные эффекты
• Сопутствующие лекарственные средства, используемые для лечения этих эффектов
• Оценки центральной лаборатории
Гематология/CBC
Иммуноглобулины
Проточная цитометрия: подсчет В-клеток (CD19+)
При всех сообщаемых серьезных инфекционных неблагоприятных эффектах CBC c определением лейкоцитарной формулы, количественным определением Ig и подсчетом CD19 будут определяться в пределах 1 недели от возникновения.
Образцы крови, сыворотки и мочи будут получать в точно определенные моменты времени. В лабораторных руководствах будут предоставлены инструкции в отношении обработки, хранения и транспортировки образцов в Genentech и центральную клиническую лабораторию. Будут использоваться стандартные анализы для гематологии, серологии, биохимии, сывороточного β-hCG, проточной цитометрии, иммунологии и анализа мочи.
С помощью ELISA для фармакокинетических параметров ритуксимаба определяются уровни ритуксимаба в образцах сыворотки человека. В ELISA используется аффинно-очищенное поликлональное козье антитело против ритуксимаба в качестве захватывающего реагента и козье антитело против F(ab)2 IgG мыши, конъюгированное с пероксидазой хрена (HRP), в качестве реагента для обнаружения.
ELISA для антитела против ритуксимаба НАСА является анализом сопряжения, в котором используется ритуксимаб в качестве захватывающего реагента и биотинилированный ритуксимаб и стрептавидин-HRP для обнаружения. Для этого анализа используется калибровочная кривая, полученная с использованием аффинно-очищенных поликлональных козьих антител против ритуксимаба, и оно подтверждается с помощью иммуноистощения с использованием ритуксимаба. Результаты этого анализа будут сообщаться относительно этого поликлонального антитела в относительных единицах (ОЕ)/мл.
Индивиды могут быть исключены из периода лечения или для них может быть прерван период лечения в какое-либо время, но они должны вернуться в исследовательский центр в пределах 14 дней для посещения при раннем исключении из лечения, с целью SFU и возможно слежения за В-клетками. За индивидами, для которых был прерван период лечения, будут наблюдать с целью оценок безопасности. Должны быть приложены все усилия для получения этих оценок для индивидов, для которых был рано прерван период лечения. Основная причина раннего прерывания должна быть документирована на соответствующей странице CRF.
Причины прерывания лечения индивида исследователем включают, но без ограничения, следующие:
• Сознательный отзыв согласия
• Любое медицинское состояние, которое может подвергнуть риску безопасность, если он или она продолжат участие в исследовании, как определено исследователем.
• Определение исследованием, что продолжение участия в исследовании не в интересах индивида.
Индивиды с положительным тестом на беременность с использованием мочи или сыворотки в любое время во время исследования.
Индивиды, которых рано исключили, не будут замещаться.
Genentech имеет право прекратить это исследование в любое время. Причины прекращения этого исследования могут включать, но без ограничения, следующие причины:
• Частота или тяжесть побочных эффектов в этом или других исследованиях указывает на возможную опасность здоровью индивидов.
• Регистрация индивидов не является удовлетворительной.
• Документирование дат является неточным или неполным.
Это исследование предназначено для оценки эффективности одного курса повторного лечения ритуксимабом или плацебо подходящих индивидов и безопасности лечения ритуксимабом индивидов с активным RA, которые получают МТХ и которые ранее или в текущей момент неадекватно реагируют на одну или несколько анти-TNF терапий. Годность индивида для повторного лечения основывается на критерии ремиссии DSA 28-ESR (DSA 28-ESR ≥ 2,6). Будет зарегистрировано приблизительно 555 индивидов. Исследование является исследованием с доступной маркировкой для первого курса лечения (ритуксимабом) и слепым для индивидов, подходящих для курса повторного лечения (исследуемым лекарственным средством). Во время недель 24-40 индивиды, удовлетворяющие критериям повторного лечения, будут подвергнуты рандомизации в соотношении 2:1 или в группу ритуксимаба, или в группу плацебо.
Если не указано иное, все статистические испытания являются двусторонними, и их будут проводить при уровне значимости □=0,5.
Первичным конечным положением является доля индивидов с ответом ACR20 на 48 неделе относительно базовой линии (день 1). Анализ раскрытых данных начнут после того, как результаты оценок всех индивидов на 48 неделе будут находиться в базе данных, и база данных будет очищена и заморожена.
Для сегмента лечения ритуксимабом с доступной маркировкой (первые 24 недели исследования) число зарегистрированных индивидов будет подвергаться табулированию по центру. Для индивидов, для которых сегмент лечения с доступной маркировкой был прерван, будут суммироваться причины прерывания. Будут суммироваться нарушения основных критериев годности, другие отклонения от основного протокола и число индивидов, получивших все запланированные дозы.
Для сегмента слепого повторного лечения исследуемым лекарственным средством число рандомизированных индивидов будет табулироваться по центру или группе лечения. Распределение индивидов будет суммироваться по группе лечения относительно рандомизации индивидов, лечения и завершения исследования. Будут суммироваться по группе лечения нарушения основных критериев годности, другие отклонения от основного протокола и число индивидов, получивших все запланированные дозы. Для индивидов, для которых контролируемый плацебо сегмент повторного лечения был прерван, причины прерывания будут перечисляться и суммироваться по группе лечения.
Базовую линию подвергаемых лечению групп будут оценивать на сопоставимость относительно демографий (т.е. возраста, пола, расы/этничности) и характеристик базовой линии (т.е. веса тела, продолжительности RA, RF-статуса базовой линии, SJC/TJC базовой линии, DAS 28-ESR базовой линии и числа предшествующих TNF-терапий). Базовая линия какой-либо переменной величины будет определяться как последняя имеющаяся величина до первого введения ритуксимаба (день 1).
Безопасность будет оцениваться по сводке побочных эффектов, смертей, результатов лабораторных испытаний, признаков жизни и НАСА. Эти сводки будут получать в виде общей сводки для сегмента лечения с доступной маркировкой и по группе лечения для контролируемого плацебо сегмента повторного лечения. Анализы безопасности будут основываться на индивидах, получивших любое количество исследуемого лекарственного средства. Индивиды будут анализироваться в соответствии с фактически полученным лечением.
Следующие касающиеся безопасности сводки будут включены в анализы безопасности.
Дословные описания возникающих при лечении побочных эффектов будут отображены в терминах толкового словаря MedDRA. Побочные эффекты будут табулироваться в соответствии с классом системных органов, группы лечения и степени NCI CTCAE. Табуляции для серьезных побочных эффектов, связанных с инфекциями неблагоприятных эффектов, связанных с инфузией ритуксимаба реакций и побочных эффектов, приводящих к прекращению лечения исследуемым лекарственным средством, будут основываться на всех подвергнутых лечению индивидах и будут табулироваться после первой инфузии, входящей в первоначальный курс лечения ритуксимабом и курс повторного лечения.
Кроме того, серьезные побочные эффекты будут суммироваться в виде встречаемости на 100 пациентов в год после первой инфузии, входящей в первоначальный курс лечения ритуксимабом и курс повторного лечения, а также на протяжении всего периода наблюдения.
Смерти индивидов и основная причина смерти будут перечислены и/или суммированы.
Будут предоставлены сводки с использованием количественного показателя распределения лабораторных величин и изменений от базовой линии на протяжении всего исследования. Будет суммироваться доля индивидов, у которых имеют место возникающие при лечении лабораторные отклонения.
Будут предоставлены сводки по группе лечения с использованием количественного показателя распределения признаков жизни и изменений от базовой линии на протяжении всего исследования.
Будет суммироваться доля индивидов с измеряемым антительным ответом на ритуксимаб.
Намеревающаяся лечиться (ITT) популяция состоит из всех индивидов, которые подвергаются рандомизации в контролируемом плацебо сегменте повторного лечения и получают какое-либо исследуемое лекарственное средство. ITT-популяция представляет собой популяцию для первичного анализа первичного и вторичных конечных положений. Индивиды будут анализироваться в соответствии с рандомизированным для них лечением.
Первичное конечное положение представляет собой долю подвергшихся повторному лечению индивидов с ответом АСR20 на 48 неделе по сравнению с базовой линией (день 1).
Для достижения АСR20 требуется ≥20% снижения как TJC, так и SJC по сравнению с базовой линией, а также ≥20% улучшения трех из пяти следующих дополнительных измерений:
• Общей оценки врачом активности заболевания
• Общей оценки пациентом активности заболевания (VAS)
• Оценки пациентом боли (VAS)
• HAQ
• Реагента острой фазы (CRP или ESR)
При расчете АСR20 будет использоваться CRP. Если CRP отсутствует или его определение не проведено, будет использоваться ESR.
Первичный анализ различий в частоте ответа АСR20 между подвергаемой повторному лечению плацебо группой и группой, подвергаемой повторному лечению ритуксимабом, будет представлен с использованием статистического испытания Cochran-Mantel-Haenszel со стратификацией по RF-статусу на базовой линии. Результаты будут суммироваться с использованием количественного показателя распределения по подвергаемой лечению группе и выражаться в виде долей, с соответствующими корректируемыми 95% доверительными интервалами (CI) между частотами ответов и р-величинами.
Частоты ответов АСR20 будут также анализировать с использованием логистической регрессии и проверки связи между ответом АСR20 на 48 неделе и подвергаемой лечению группой при контролировании RF-статуса на базовой линии с использованием модели логистической регрессии. Оценки параметров от этой модели будут проверяться с помощью табулирования стандартных ошибок, вальдовских статистик и вероятностей успешных исходов с использованием соответствующих 95% CI и р-величин.
Анализы восприимчивости также будут выполняться с использованием установки возможных эксплоративных термов для ответов ACR20 (например, SJC на базовой линии).
Вторичные конечные положения будут анализироваться следующим образом.
• Долю индивидов с ответами ACR50 и ACR70 на 48 неделе будут анализировать таким же образом, как определено для первичного конечного положения.
• Изменение от базовой линии в DAS28-ESR на 48 неделе будут анализировать с помощью модели дисперсионного анализа (ANOVA) с использованием группы повторного лечения плацебо/ритуксимабом и RF-статуса в качестве эксплоративных термов в этой модели.
• Упорядоченный класс ответа ACR (ответы ACR70, ответы ACR50-70, ответы ACR20-50 и ответы, не являющиеся ответами ACR20) на 48 неделе будут анализироваться с использованием модели кумулятивных логит-преобразований со стратификацией по RF-статусу базовой линии. Оценки параметров от этой модели будут проверяться с помощью табулирования стандартных ошибок, вальдовских статистик и вероятностей успешных исходов с использованием соответствующих 95% CI и р-величин.
• Степени ответов (хороший или умеренный) EULAR на 48 неделе будут анализировать таким же образом, как определено для первичного конечного положения.
• Изменение от базовой линии в основном наборе ACR (SJC, TJC, HAQ, общие оценки пациентом и врачом, боль, CRP и ESR) на 48 неделе будут анализировать с помощью модели дисперсионного анализа (ANOVA) с использованием группы повторного лечения плацебо/ритуксимабом и RF-статуса на базовой линии в качестве эксплоративных термов в этой модели.
• ACRn на 48 неделе и площадь под кривой концентрация-время (AUC) ACR на 48 неделе будут анализировать с помощью модели дисперсионного анализа (ANOVA) с использованием группы повторного лечения плацебо/ритуксимабом и RF-статуса на базовой линии в качестве эксплоративных термов в этой модели.
• Будет даваться отчет об изменение в уменьшенной шкале SF-36 и суммарных показателях от базовой линии на 48 неделе для показателей восьми предметных областей и показателей психического и физического компонента, используя модель дисперсионного анализа (ANOVA), с использованием группы повторного лечения плацебо/ритуксимабом и RF-статуса на базовой линии в качестве эксплоративных термов в этой модели. Кроме того, показатели психического и физического компонента будут дополнительно классифицироваться и анализироваться.
• Изменение функциональной оценки усталости при лечении хронического заболевания (FACIT-F)-оценки от базовой линии на 48 неделе будут анализировать с помощью модели дисперсионного анализа (ANOVA) с использованием группы повторного лечения плацебо/ритуксимабом и RF-статуса на базовой линии в качестве эксплоративных термов в этой модели.
• Долю индивидов, достигающих DAS28-ESR ремиссии (DAS28-ESR>2,6) на 48 неделе, будут анализировать таким же образом, как определено для первичного конечного положения.
• Долю индивидов, достигающих DAS28-ESR заболевания с низкой активностью (DAS28-ESR≤3,2) на 48 неделе, будут анализировать таким же образом, как определено для первичного конечного положения.
Будут определять фармакокинетические (РК) параметры, происходящие из концентраций в сыворотке ритуксимаба, в том числе максимальная концентрация в сыворотке (Cmax), время достижения максимальной концентрации в сыворотке (Tmax), площадь под кривой концентрация-время (AUC), системный клиренс (CL), объем распределения (V) и время полужизни (t1/2), для всех индивидов с использованием популяционной РК-модели. Из-за нечастых отборов образцов фаза распределения может быть не очень хорошо охарактеризована. РК-данные от этого исследования будут объединять с данными от других исследований для популяционного РК-анализа.
Для популяционного РК-анализа будут определяться РК-параметры в виде общих средних для всей исследуемой популяции вместе с оценками внутри- и межиндивидных вариаций и оценки случайной ошибки. Оценки параметров отдельных индивидов будут рассчитываться с использованием процедур анализа ошибки предположения, что временная последовательность является доказательством причинной связи. Будет приготовлен план будущего анализа, и популяционный РК-анализ будет представляться в отчете, отдельном от отчета о клиническом испытании.
Популяционный РК-анализ будет включать исследовательский анализ для определения ковариацией базовой линии, которые оказывают влияние на фармакокинетические параметры ритуксимаба в этой популяции пациентов. Проверяемые ковариации базовой линии будут включать демографические и другие характеристики индивида, такие как тяжесть заболевания и избранные лабораторные показатели.
Данные будут суммироваться с использованием количественного показателя распределения, в том числе среднего, стандартного отклонения, среднего геометрического, коэффициента вариации, медианы и диапазона.
Возможные фармакодинамические маркеры образцов крови, в том числе число В-клеток, количественные уровни Ig, субпопуляции лимфоцитов и концентрация НАСА, будут суммироваться с использованием количественного показателя. Для этих анализов величины на базовой линии, измеренные в образце до введения дозы в день 1, будут использовать для расчета изменений от базовой линии в каждый момент времени взятия образца.
Исследовательские анализы будут проводиться для определения возможной связи между фармакодинамическими маркерами, РК-измерениями и клиническим ответом и будут точно определены в плане статистического анализа.
Приблизительно 555 индивидов будет зарегистрировано. Допустив процент отсева вплоть до 25% во время сегмента лечения с доступной маркировкой и вплоть до 10% индивидов, достигающих DAS 28-ESR ремиссии на 24 неделе и не подвергающихся повторному лечению, с соотношением рандомизации 2:1, этот размер популяции будет обладать 80% способностью к обнаружению различий в 16% в частотах ответа ACR20 от базовой линии на 48 неделе между группой повторного лечения ритуксимабом (50% ACR20-ответчиков) и группой плацебо (34% ACR20-ответчиков) с использованием точного критерия Fisher.
Оценки безопасности будут состоять из контроля и документирования определяемых протоколом побочных эффектов (АЕ) и серьезных побочных эффектов (SAE); измерения определяемых протоколом лабораторных (гематология, клиническая биохимия и анализ мочи) переменных показателей; измерения определяемых протоколом признаков жизни и других определяемых протоколом проверок, которые считаются критическими для оценки безопасности исследуемого лекарственного средства.
Для контролирования связанных с инфузией реакций признаки жизни будут определяться непосредственно перед инфузией, каждые 15 минут во время первого часа инфузии, затем каждые 30 минут в оставшееся время инфузии и каждые 30 минут в течение одного часа после инфузии, в дни введения исследуемого лекарственного средства. Дополнительные показатели могут определяться в случае связанной с инфузией реакции (например, гипотензии и/или лихорадки).
АЕ представляет собой неблагоприятный и непредусмотренный признак, симптом или заболевание, временно связанные с использованием лекарственного препарата, проходящего клиническое испытание, или другим предписываемым протоколом вмешательством, независимо от атрибуции.
Это включает следующие АЕ:
• АЕ, ранее не наблюдаемые у индивида, которые возникают во время определяемого протоколом периода сообщения о АЕ, в том числе признаки или симптомы, связанные с RA, которые не присутствовали до периода сообщения о АЕ;
• Осложнения, возникающие в результате предписываемых протоколом вмешательств (например, инвазивных процедур, таких как биопсии);
• Если применимо, АЕ, которые возникают до назначения исследуемого лечения, связанные с выведением лекарственного средства, не вхождением в период лечения или другим предписываемым протоколом вмешательством;
• Предсуществующие медицинские состояния (отличные от исследуемого состояния), которое по оценку исследователя ухудшилось по тяжести или частоте или изменило характер во время определяемого протоколом периода сообщения о АЕ.
АЕ следует классифицироваться как SAE, если он удовлетворяет следующим критериям:
• Он приводит к смерти (т.е. АЕ фактически вызывает смерть или приводит к ней).
• Он представляет собой угрозу жизни (т.е. АЕ, по мнению исследователя, подвергает индивида непосредственному риску смерти. Он не включает АЕ, который, если бы происходил в более тяжелой форме, мог вызвать смерть).
• Он вызывает необходимость или удлиняет срок госпитализации.
• Он приводит к постоянной или значительной нетрудоспособности (т.е. АЕ приводит к существенному нарушению способности индивида выполнять нормальные жизненные функции).
• Он приводит к врожденной аномалии/врожденному дефекту у новорожденного, рожденного матерью, подвергшейся воздействию лекарственного продукта, находящегося на стадии клинического испытания.
• Он считается исследователем значимым медицинским случаем по медицинской оценке (например, может подвергнуть индивида риску или может потребовать медицинского/хирургического вмешательства для предотвращения одного из результатов, перечисленных выше).
Все АЕ, которые не удовлетворяют какому-либо из критериев серьезности, должны считаться несерьезными АЕ.
Термины «тяжелый» и «серьезный» не являются синонимами. Тяжесть относится к степени конкретного АЕ, например, слабая (степень 1), умеренная (степень 2) или тяжелая (степень 3) инфаркта миокарда. «Серьезный» относится к регуляторному определению и основан на исходе индивида или результате эффекта или критериях для действий, обычно связанных с эффектами, которые вызывают угрозу жизни или функционирования индивида. Серьезность служит ориентиром для определения регуляторных обязательств спонсора перед соответствующими регуляторными органами в отношении сообщения.
Тяжесть и серьезность должны оцениваться независимо при документировании АЕ и SAE в CRF.
Исследователи будут оценивать случаи АЕ и SAE во всех временных точках оценки индивида на протяжении исследования. Все АЕ и SAE, которые были или добровольно сообщены индивидом, обнаружены включенным в исследовании персоналом во время расспросов, или определены при физической проверке, лабораторном испытании, или другим образом, будут вноситься в медицинскую документацию и на соответствующую АЕ- или SAE-страницу CRF.
Для каждого зарегистрированного АЕ или SAE будет описываться его продолжительность (т.е. даты начала и окончания), тяжесть (смотри таблицу 2), критерии регуляторной серьезности, если применимы, заподозренная связь с лекарственным препаратом, проходящим клиническое испытание (смотри следующее руководство) и предпринятые действия.
Шкала степеней (тяжести) АЕ, которую можно найти в NCI CTCAE, будет использоваться для сообщаемых АЕ.
Шкала степеней (тяжести) побочных эффектов
Замечание: Независимо от тяжести некоторые эффекты могут также удовлетворять регуляторным критериям серьезности.
Дается ссылка на определения SAE.
Использовать эти альтернативные определения для эффектов степеней 1, 2, 3 и 4, когда наблюдаемый или сообщаемый АЕ не перечислен в NCI CTCAE.
Для обеспечения совместимости причинной связи АЕ и SAE исследователи должны применять следующее общее руководство:
• Да
Существует вероятная временная связь между возникновением АЕ и введением лекарственного препарата, проходящего клиническое испытание, и АЕ нельзя точно объяснить клиническим состоянием индивида, интеркуррентным заболеванием или сопутствующими терапиями; и/или АЕ соответствует известному примеру ответа на лекарственный препарат, проходящий клиническое испытание; и/или АЕ прекращается при прекращении введения лекарственного препарата, проходящего клиническое испытание, или при уменьшении его дозы и, если применимо, вновь появляется при повторном введении лекарственного препарата.
• Нет
Существует доказательство, что этиология АЕ отлична от лекарственного препарата, проходящего клиническое испытание (например, предсуществующее медицинское состояние, лежащее в основе заболевание, интеркуррентное заболевание или сопутствующее лекарственное средство); и/или нет вероятной временной связи между АЕ и введением лекарственного препарата, проходящего клиническое испытание, (например, рак, диагностированный спустя 2 дня после первой дозы исследуемого лекарственного средства).
Замечание: Оценка исследователем причинной связи для отдельных случаев АЕ является частью процесса документирования исследования. Независимо от оценки причинной связи «Да» или «Нет» для отдельных случаев АЕ спонсор будет своевременно оценивать все сообщаемые SAE против кумулятивного испытания продукта для идентификации и быстрого сообщения возможно новых, касающихся безопасности решений исследователям и соответствующим регуляторным органам.
RA следует документировать как АЕ или SAE только, если по оценке исследователя, неожиданно произошло ухудшение его тяжести и/или частоты или изменение его природы в любое время во время исследования. При документировании неожиданного ухудшения ревматоидного артрита на странице АЕ или SAE CRF важно передать концепцию изменения состояния путем включения соответствующих дескрипторов (например, «ускоренный ревматоидный артрит»).
Ожидается, что повторное лечение в соответствии с протоколом настоящего изобретения (или отличным антителом против CD20) будет эффективным для предотвращения или замедления прогрессирования структурного повреждения сустава и эрозии, вызванной RA. Предполагается, что эффективные результаты будут иметь место, если МТХ не будет применяться, т.е. будет использоваться монотерапия с использованием антитела против CD20.
Пример 4
Исследование эффективности ритуксимаба для больных псориатическим артритом
Следуют протоколу примера 2 за исключением того, пациентов подвергают лечению в отношении повреждения сустава, вызванного псориатическим артритом. Ожидается, что будут получены схожие с результатами при ревматоидном артрите результаты (при использовании ритуксимаба или гуманизированного антитела 2Н7), т.е. прогрессирование структурного повреждения сустава будет предотвращено после первой дозы антитела против CD20 (с метотрексатом или без него в зависимости, например, от доз, что может быть отрегулировано соответствующим образом) спустя по крайней мере приблизительно один месяц от базовой линии или начала лечения, предпочтительно по крайней мере 24 недели от базовой линии, более предпочтительно по крайней мере 52 недели от базовой линии и в других временных точках вплоть до 104 недели от базовой линии.
Эти основанные на ритуксимабе схемы лечения бросают вызов используемому в настоящее время стандарту лечения и, как ожидается, продемонстрируют увеличение фактической клинической пользы, при этом первичной целью является предупреждение прогрессирования повреждения сустава. Ожидается, что эти результаты будут значительно лучше результатов для контрольной группы.
Предсказывается и ожидается, что введение индивиду ритуксимаба или гуманизированного антитела 2Н7 в соответствии с протоколом запланированного введения повторных доз, изложенном в примере 2, будет эффективным для предупреждения прогрессирования структурного повреждения сустава на 52 неделе или позже. Ожидается, что эти результаты будут значительно лучше результатов контроля.
Также ожидается, что на приблизительно 48-54 неделе еще одна 1 г - или 2 г - доза антитела против CD20 (например, ритуксимаба или гуманизированного антитела 2Н7), назначенная в виде одного введения или распределенная на протяжении приблизительно 14-16 дней по 0,5 - или 1-грамм по количеству, будет эффективной для лечения повреждения сустава в течение всего второго года, с или без одного или нескольких вторых лекарственных средств, таких как иммунодепрессанты. Таким образом, антитело против CD20 следует вводить первоначально в пределах приблизительно 2-недельного периода времени с последующим другим лечением через приблизительно 4-8 месяцев, с последующим другим лечением через приблизительно один год от первоначального лечения (определяемого от момента времени, когда вводится любая из доз), с последующим лечением через приблизительно два года от первоначального лечения, с ожидаемым успехом, в количестве приблизительно 0,5 грамм или 1 грамм × 2-4 введения для каждого лечения, вводимом вместе, приблизительно раз в неделю или приблизительно через неделю на протяжении приблизительно 2-4 недель. Ожидается, что результаты этого лечения будут намного лучше результатов контроля с плацебо. Ожидается, что этот протокол повторного лечения будет успешно использоваться на протяжении нескольких лет с небольшими побочными эффектами или без них.
Пример 5
Исследование эффективности ритуксимаба при лечении больных остеоартритом и болезнью Крона
Ожидается, что результаты примера 2 будут успешно получены при использовании пациентов, имеющих повреждение сустава в результате остеоартрита или болезни Крона, независимо от того, используется ли ритуксимаб или другое антитело против CD20, такое как гуманизированное антитело 2Н7, и от того, используется или нет иммунодепрессант, с регулированием доз, что хорошо известно квалифицированным в данной области техники специалистам. Кроме того, если пациенты первоначально подвергаются лечению антителом против CD20 и затем повторному лечению таким антителом спустя приблизительно шесть месяцев или приблизительно один год после того момента, когда они впервые подверглись лечению, в других отношениях с использованием одних и тех доз и других протоколов примера 2, ожидается, что у них будет продолжаться предотвращение прогрессирования повреждения сустава в течение длительного периода времени.
Ожидается, что ритуксимаб или другое антитело против CD20 будут по крайней мере также эффективны, как и традиционная схема лечения для индукции и сохранения ремиссии повреждения сустава, представляя значительные преимущества по сравнению со стандартной терапией в силу их лучшего профиля побочных эффектов, например, намного меньшую, чем у стероидов токсичностью и лучшую устойчивость к восстановлению.
Ожидается, что пациенты в группе лечения будут хорошо переносить ритуксимаб, а В-клетки будут быстро истощаться. Ожидается, что для индукции ремиссии и сохранения длительной ремиссии (6 месяцев или дольше) у пациентов, подвергаемых лечению антителом против CD20, кроме метотрексата, если потребуется, дополнительные иммунодепрессанты не потребуются.









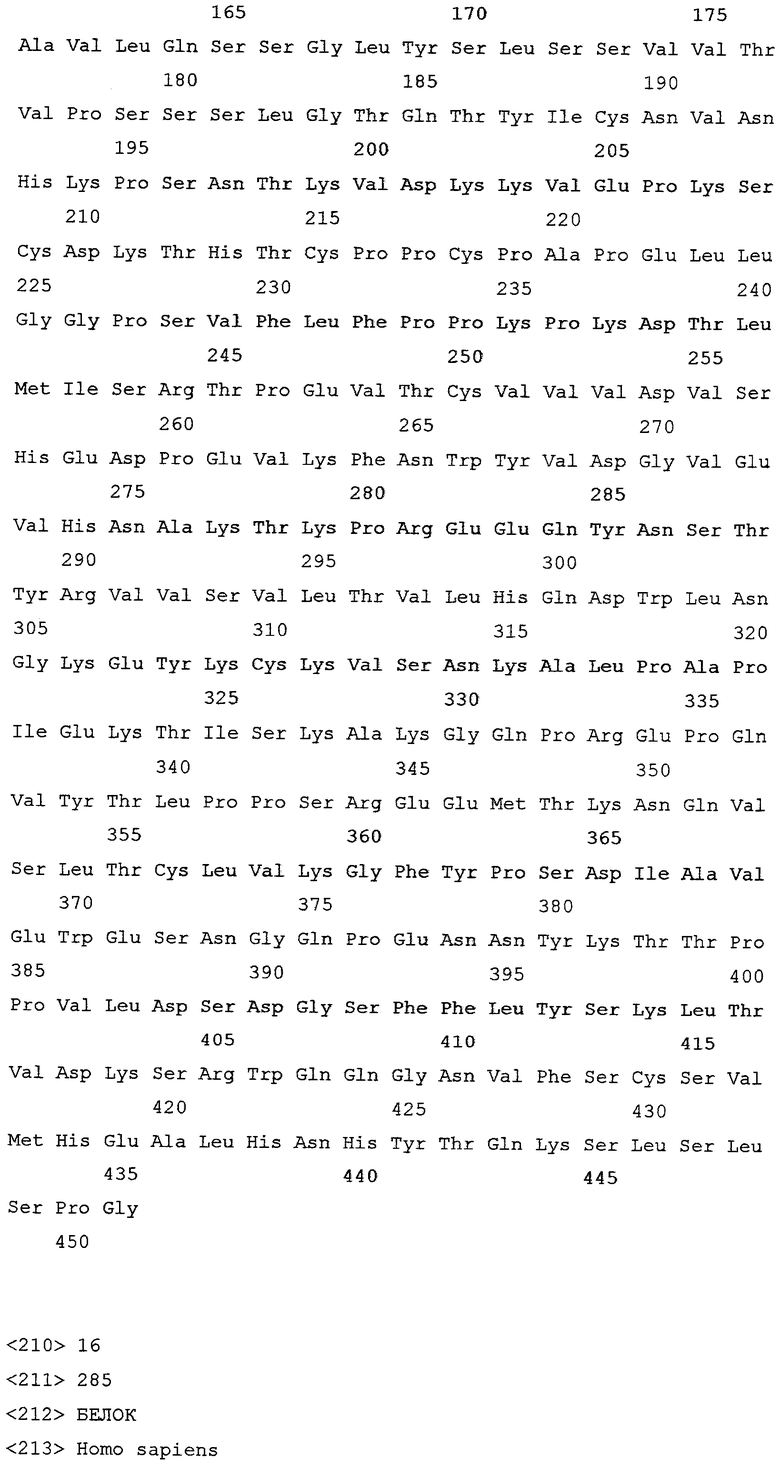






























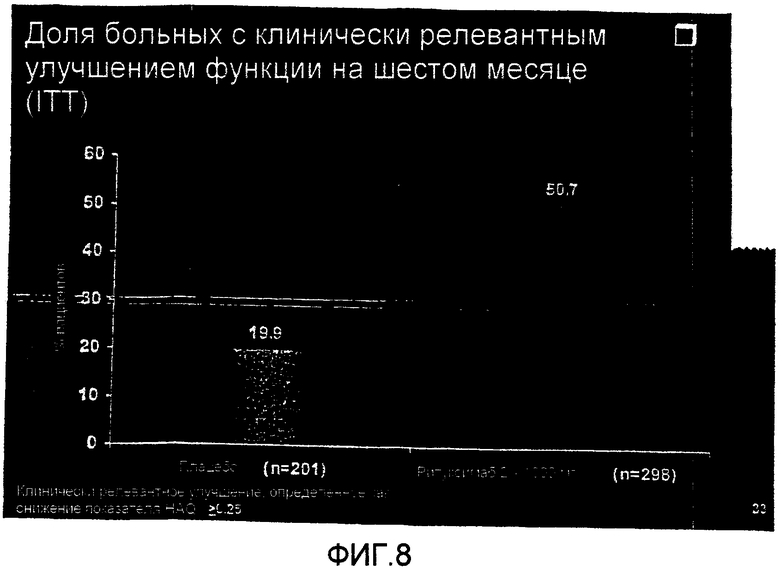

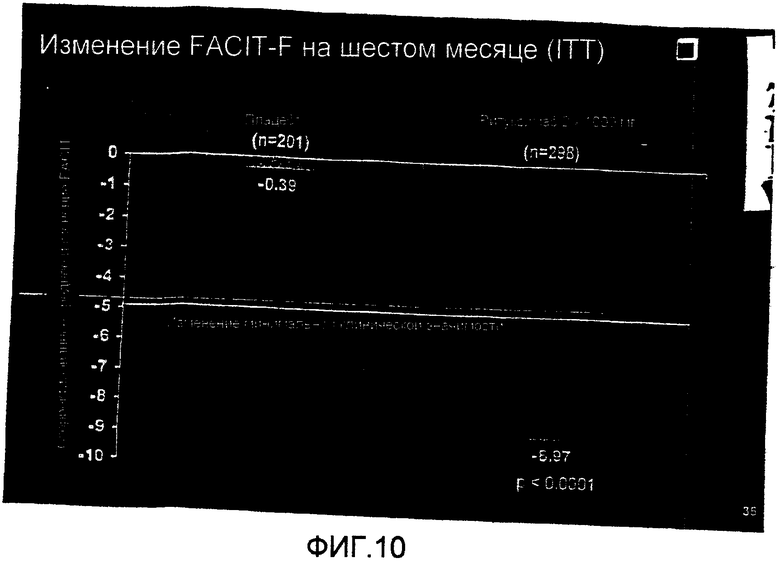




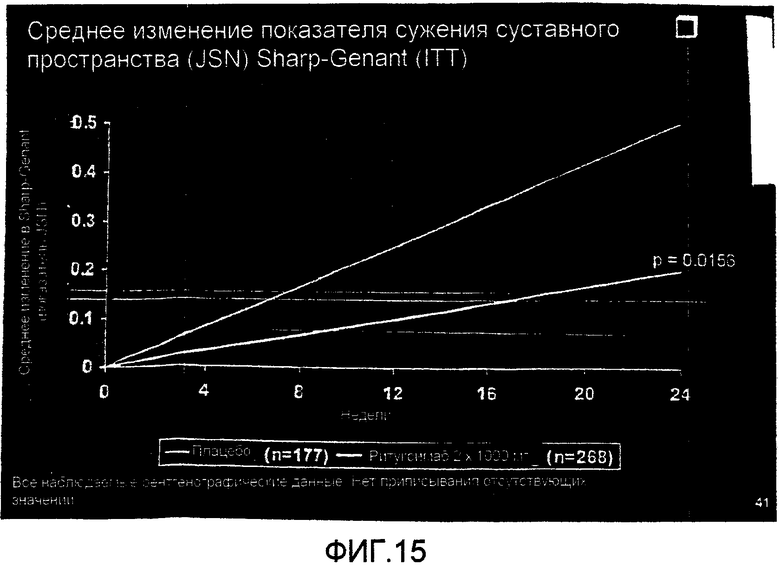







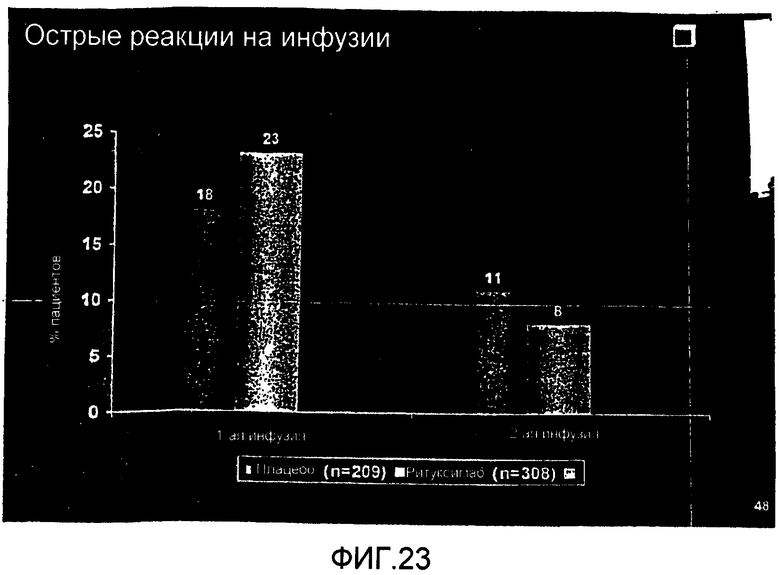








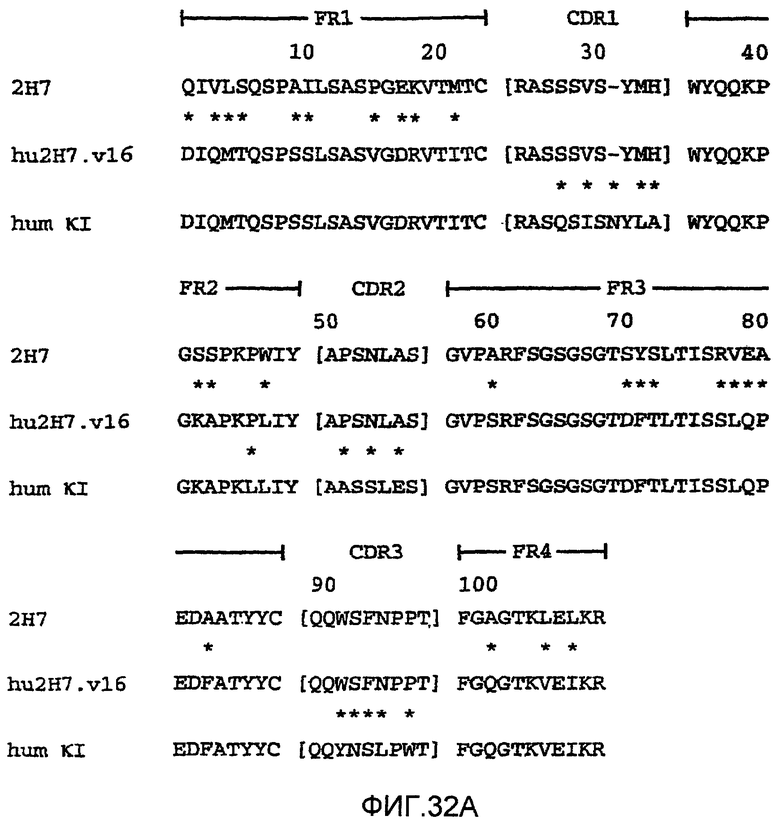















Группа изобретений относится к медицине, а именно к ревматологии, и может быть использована для лечения повреждения сустава у индивида, страдающего ревматоидным артритом (RA). Для этого пациенту, который проявляет неадекватный ответ или имеет чрезмерную чувствительность к одному или нескольким ингибиторам фактора некроза опухоли (TNF), вводят ритуксимаб. При этом пациенту был проведен, по меньшей мере, один курс лечения ритуксимабом, и лечение включает проведение, по крайней мере, еще одного курса лечения ритуксимабом. Дополнительный курс лечения проводят через 24 недели после начала предшествующего курса лечения ритуксимабом. Каждый курс включает введение двух внутривенных доз по 1000 мг с интервалом 14 дней. Также способ включает проведения через 52 недели после начала введения ретуксимаба радиографического анализа. Группа изобретений обеспечивает уменьшение повреждения сустава у указанной группы пациентов. 2 н. и 7 з.п. ф-лы, 46 ил., 2 табл., 5 пр.
1. Способ лечения повреждения сустава у индивида, страдающего ревматоидным артритом (RA), где способ включает введение указанному индивиду ритуксимаба, где (а) индивид проявляет неадекватный ответ или имеет чрезмерную чувствительность к одному или нескольким ингибиторам фактора некроза опухоли (TNF); (b) у индивида был проведен по меньшей мере один курс лечения ритуксимабом, и (с) лечение включает проведение по крайней мере еще одного курса лечения ритуксимабом, где дополнительный курс лечения проводят по крайней мере через 24 недели после начала предшествующего курса лечения ритуксимабом, и каждый курс лечения включает введение двух внутривенных доз по 1000 мг с интервалом 14 дней.
2. Способ по п.1, где индивид отвечал по меньшей мере на один ранее проводимый курс лечения ритуксимабом.
3. Способ по п.1, где RA является активным RA.
4. Способ по п.1, где дополнительный курс лечения проводят по меньшей мере через 24-48 недель после начала предыдущего курса лечения риткусимабом.
5. Способ по п.1, где возраст индивида составляет от 18 до 80 лет.
6. Способ по п.1, где указанное лечение эффективно для остановки или замедления прогресса структурного поражения сустава, вызванного ревматоидным артритом или для профилактики развития поражения сустава, вызванного ревматоидным артритом.
7. Способ по любому из пп.1-6, где лечение включает введение ритуксимаба и метотрексата.
8. Способ лечения структурного повреждения сустава у индивида, страдающего ревматоидным артритом (RA), где метод включает проведение курса лечения ритуксимабом у индивида и проведение у пациента через по крайней мере 52 недели после начала введения радиографического анализа для измерения уменьшения повреждения суставов по сравнению с базовым уровнем до введения, где курс лечения эффективно замедляет прогрессирование указанного структурного повреждения суставов по данным радиографического анализа, где (а) индивид проявляет неадекватный ответ или имеет чрезмерную чувствительность к одному или нескольким ингибиторам фактора некроза опухоли (TNF); (b) у индивида был проведен по меньшей мере один курс лечения ритуксимабом, и (с) лечение включает проведение по крайней мере еще одного курса лечения ритуксимабом, где дополнительный курс лечения проводят по крайней мере через 24 недели после начала предшествующего курса лечения ритуксимабом, и каждый курс лечения включает введение двух внутривенных доз по 1000 мг с интервалом 14 дней.
9. Способ по п.8, где лечение дополнительно включает введение метотрексата.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| RU 2005105307 A, 27.08.2005 | |||
| ИСПОЛЬЗОВАНИЕ ФОСФОНАТОВ И НЕСТЕРОИДНЫХ ПРОТИВОВОСПАЛИТЕЛЬНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ АРТРИТА, СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2119794C1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| НАСОНОВ Е.Л | |||
| Новые направления в лечении ревматоидного артрита | |||
| Фарматека, 2003, №5(68), с.3-7 | |||
| KIRWAN J.R | |||
| et al | |||
| The effect of glucocorticoids on joint destruction in | |||

