Область техники
Изобретение относится к молекулярной биологии и медицине и рассматривает создание и применение тест-системы для определения наличия точечных мутаций в гене фумарилацетоацетат гидролазы, вызывающих поражение печени у новорожденных детей.
Одним из наиболее частых нарушений метаболизма, выявляемых в период новорожденности, является повышение концентрации билирубина в сыворотке крови. Известно, что причинами гипербилирубинемии являются: повышенное образование билирубина при распаде гемоглобина в клетках ретикулоэндотелиальной системы, снижение конъюгации билирубина, нарушение его экскреции или сочетание некоторых из вышеперечисленных факторов. Частота гипербилирубинемии составляет около 55% у доношенных новорожденных. В большинстве случаев гипербилирубинемия новороженных является функциональной (связана с незрелостью ферментативной системы) и проходит в течение первых недель жизни. Тем не менее, повышение уровня билирубина может быть связано с патологией экскреторной функции гепатобилиарной системы и носить злокачественный характер, в частности приводить к необратимому поражению центральной нервной системы.
За последние десятилетия углубилось понимание основ и патофизиологии многих болезней печени, был установлен ряд новых нозологических форм, появились возможности эффективного лечения многих заболеваний, в частности стала применяться диетотерапия (при галактоземии и тирозинемии), стала возможной трансплантация печени. На сегодняшний день число распознанных наследственных болезней печени у новорожденных составляет около 20 различных форм. Гены всех этих заболеваний картированы, и во многих случаях определен спектр основных патогенных мутаций. Для некоторых генов (кодирующих в основном ферменты) также охарактеризованы полиморфные варианты, представленные преимущественно однонуклеотидными заменами, которые могут влиять на остаточную активность белка. Однако дифференциальная диагностика большинства из них крайне затруднительна ввиду схожести клинических проявлений. В нашей стране число пациентов, которым быстро и правильно устанавливается диагноз, сравнительно невелико. Большинство детей погибают в раннем возрасте, не получая адекватного лечения, а отягощенные семьи не имеют информации о наследственном заболевании.
Поскольку в ряде случаев причины злокачественной гипербилирубинемии новорожденных остаются не выясненными, изучение роли генетических факторов в формировании данного патологического состояния - актуальная научная задача, имеющая большое практическое значение.
Уже на первом месяце жизни могут выявляться первые клинические признаки, свидетельствующие о наследственной патологии печени и желчевыводящих протоков и проявляющиеся в виде синдрома холестаза. Одной из причин нарушения экскреторной функции гепатобилиарной системы в неонатальный период (неонатального холестаза) являются наследственные заболевания. По приблизительным подсчетам распространенность врожденных или наследственных заболеваний печени у детей составляет 1 случай на 2500 новорожденных детей. Именно у таких детей развиваются тяжелые формы печеночной патологии. Правильно и быстро установленный диагноз в данном случае является очень важным, т.к. позволяет прогнозировать течение болезни и проводить адекватную терапию. Однако дифференциальная диагностика этой группы заболеваний чрезвычайно сложна.
Объектом исследования являются гены фумарилацетоацетат гидролазы и альфа-1-антитрипсина человека.
Цель работы - разработка системы, обеспечивающей диагностику и генетический анализ нескольких наследственных болезней из группы врожденных нарушений обмена веществ, приводящих к развитию неонатальной гипербилирубинемии: тирозинемии типа 1 и недостаточности альфа-1-антитрипсина.
Разработана тест-система, которая представляет собой биочип для детекции мутаций и полиморфизмов генов фумарилацетоацетат гидролазы и альфа-1-антитрипсина на микрочипе. В процессе работы проводились дизайн и оптимизация последовательностей олигонуклеотидных последовательностей для иммобилизации на микрочипе. Подобраны условия гибридизации на микрочипе. В результате исследования подобраны ДНК детей, больных тирозинемией типа 1 и недостаточностью альфа-1-антитрипсина, несущие нуклеотидные замены в областях исследуемых мутаций и полиморфизмов генов фумарилацетоацетат гидролазы и альфа-1-антитрипсина. Проведен анализ фрагментов этих ДНК методами полиморфизма длин рестрикционных фрагментов (ПДРФ), секвенирования, гибридизации на микрочипе. Проведен сравнительный анализ результатов, полученных этими методами.
Целью нашей работы является разработка системы, обеспечивающей одновременную диагностику и генетический анализ двух наследственных болезней из группы врожденных нарушений обмена веществ, приводящих к развитию неонатальной гипербилирубинемии: тирозинемии типа 1, недостаточности альфа-1-антитрипсина. Для решения этой задачи должна быть создана тест-система на основе одного микрочипа для выявления мутаций в генах галактозо-1-фосфат уридил трансферазы; фумарилацетоацетат гидролазы и в гене альфа-1 антитрипсина. Разрабатываемый метод обеспечивает точный генетический анализ и позволяет максимально сократить сроки проведения диагностики.
Наследственная тирозинемия тип 1.
Одним из факторов, вызывающих тяжелейшие нарушения функционирования печени, является нарушение метаболизма тирозина, вызываемое мутациями в генах, кодирующих белки-ферменты.
На сегодняшний день известно три типа тирозинемии: тирозинемия типа 1 или гепаторенальная тирозинемия (недостаточность фумарилацетоацетат гидролазы), тирозинемия тип 2 (недостаточность тирозин трансаминазы) и тирозинемия тип 3 (хавкинсурия или недостаточность 4-гидроксифенилпируват диоксигеназы).
Среди этих заболеваний тирозинемия 1 типа является наиболее тяжелой, требующей неотложной коррекции патологии. Массовый скрининг новорожденных, проводимый во многих странах, направлен на выявление именно этой формы тирозинемии. Своевременная диагностика тирозинемии позволяет вовремя начать диетотерапию и избежать серьезных последствий для здоровья ребенка, а также снизить экономические издержки, связанные с медицинской реабилитацией при поздней диагностике.
Тирозинемия тип 1 описана U.Baber et al. в 1956 году. Тип наследования - аутосомно-рецессивный. Заболевание связано с недостаточностью фумарилацетоацетат гидролазы, катализирующей последнюю ступень деградации тирозина. Вследствие недостаточности фермента нарушается обмен тирозина, в тканях накапливаются его метаболиты: фумарилацетоацетат, малеилацетоацетат, сукцинилацетон, сукцинилацетоацетат, которые оказывают токсическое действие на клетки печени и проксимальных почечных канальцев, в результате чего страдают процессы канальцевой реабсорбции, в первую очередь, фосфатов. Происходит вторичное ингибирование активности ряда ферментов: 4-гидроксифенилпируват диоксигеназы, порфобилиноген синтазы, метионинаденозил трансферазы, дегидратазы дельта-ами-нолевулиновой кислоты, ферментов глюконеогенеза, что влечет за собой значительные биохимические расстройства. Нарушается антиоксидантная защита, падает общая антиокислительная активность плазмы.
Основными методами подтверждения диагноза тирозинемия типа 1 являются биохимические тесты, основанные на определении концентрации тирозина в крови и сукцинилацетона в моче. Для подтверждения диагноза осуществляется исследование фермента фумарилацетоацетат гидролазы в лейкоцитах или фибробластах. Разработаны способы пренатальной диагностики заболевания путем исследования сукцинилацетона в амниотической жидкости и активности фумарилацетоацетат гидролазы в культуре амниоцитов или клетках хориона. Трудности интерпретации метаболических изменений связаны с тем, что при поражении печени другой этиологии (как наследственной, так и ненаследственной) возможно повышение уровня тирозина в крови. Поэтому молекулярно-генетические методы играют важную роль в диагностике этого заболевания. Проведение мутационного анализа в случае тирозинемии типа 1 может быть полезно для выявления ложно-положительных результатов, полученных при проведении биохимических тестов.
Ген фумарилацетоацетат гидроксилазы (FAH) картирован на 15 хромосоме (локус q23-q25) и состоит из 14 экзонов, разделенных 13 интронами. На сегодняшний день в гене описано более 70 мутаций, которые преимущественно представлены миссенс-мутациями. В таблице 1 приведены данные по всем описанным в литературе мутациям гена FAH.
Наиболее распространенной мутацией у пациентов из Европы, Пакистана, Турции и США является внутриинтронная замена IVS12+5 G>A [Rootwelt H, Høie K, Berger R, Kvittingen EA. Fumarylacetoacetase mutations in tyrosinaemia type I. Hum Mutat. 1996; 7 (3):239-43; Rootwelt H, Kristensen T, Berger R, Høie K, Kvittingen EA. Tyrosinemia type I-complex splicing defects and a missense mutation in the fumarylacetoacetase gene. Hum Genet. 1994 Sep; 94(3):235-9]. Мутация IVS 6-1 G>T превалирует в Центральной и Восточной Европе [Bergman AJ, van den Berg IE, Brink W, Poll-The ВТ, Ploos van Amstel JK, Berger R. Spectrum of mutations in the fumarylacetoacetate hydrolase gene of tyrosinemia type I patients in northwestern Europe and Mediterranean countries. Hum Mutat. 1998; 12(1): 19-26], а в Испании составляет около 70% всех мутантных аллелей [Arranz JA, Piñol F, Kozak L, Pérez-Cerdá C, Cormand B, Ugarte M, Riudor E. Splicing mutations, mainly IVS6-1(G>T), account for 70% of fumarylacetoacetate hydrolase (FAH) gene alterations, including 7 novel mutations, in a survey of 29 tyrosinemia type I patients. Hum Mutat. 2002 Sep; 20(3):180-8]. Для некоторых популяций характерны свои особенности профиля мутаций, так в популяции евреев-ашкенази мутация Pro261Leu составляет почти 100% мутантных аллелей [Elpeleg ON, Shaag A, Holme E, Zughayar G, Ronen S, Fisher D, Hurvitz H. Mutation analysis of the FAH gene in Israeli patients with tyrosinemia type I. Hum Mutat. 2002 Jan; 19(l):80-l]. В Финляндии мутация Trp262Term составляет 80% мутантных аллелей, в то время как у пациентов из других частей Европы данная мутация не детектировалась [St-Louis М, Leclerc В, Laine J, Salo MK, Holmberg С, Tanguay RM. Identification of a stop mutation in five Finnish patients suffering from hereditary tyrosinemia type I. Hum Mol Genet. 1994 Jan; 3(l):69-72].
С целью разработки биочипа нами были проанализированы данные литературы и частоты мутаций в гене FAH у пациентов с тирозинемией типа 1 из Российской Федерации.
Наиболее частые мутации в гене FAH у пациентов с тирозинемией типа 1 из Российской Федерации: Pro342Leu; Argl74Term; IVS 12+5 G->A; IVS6-1 G->T.
Учитывая вышеизложенное, для создания биочипа были выбраны следующие мутации в гене FAH: Pro342Leu, Argl74Term, IVS 12+5 G->A, IVS6-1 G->T, IVS7-1 G->A.
Альфа-1-антитрипсина недостаточность.
Альфа-1-антитрипсина недостаточность - аутосомно-рецессивное наследственное заболевание, обусловленное мутациями в гене SERPINA1. Альфа-1-антитрипсин является ингибитором широкого спектра сериновых протеаз, играя важную роль в защите тканей от протеолиза. В число ингибируемых им сериновых протеаз входят протеазы системы свертывания крови и фибринолиза и протеазы иммунного ответа. Нарушение ингибиции этих групп протеаз объясняет симптомы повышенной кровоточивости у больных и частую гибель детей от интеркурретных заболеваний и сепсиса. К раннему летальному исходу приводят те формы недостаточности альфа-1-анитрипсина, которые манифестируют заболеванием печени в неонатальный период. Более легкие формы заболевания манифестируют позже. Течение заболевания хроническое. Оно приводит к хронической печеночной недостаточности или менее часто протекает как асимптоматическая гепатоспленомегалия или холанокарцинома. У взрослых отмечается высокая частота цирроза печени. Также отмечен высокий риск развития эмфиземы легких.
Основными методами подтверждения диагноза альфа-1-антитрипсина недостаточности являются биохимические тесты, основанные на определении концентрации афльфа-1-антитрипсина в плазме крови. Также исследуется электрофоретическая подвижность (изоэлектрофокусирование в полиакриламидном геле). У гомозиготных носителей мутаций она изменена по сравнению с контролем. Проведение мутационного анализа в случае альфа-1-антитрипсина недостатоточности может быть полезно для выявления ложно-положительных результатов, полученных при проведении биохимических тестов.
Ген SERPINA1 картирован на 14 хромосоме (локус q32.1) и состоит из 5 экзонов, разделенных 4 интронами. На сегодняшний день в гене описано более 40 мутаций, которые преимущественно представлены миссенс-мутациями. Наиболее распространенной мутацией, обуславливающей развитие ранней формы заболевания, является мутация Е342К (аллель Z). Второй частой мутацией является мутация E264V, которая в сочетании с аллелем может вызывать развитие эмфиземы легких.
ДНК-анализ, как диагностико-подтверждающая процедура, основан на определении мутаций с помощью ПЦР и последующего рестрикционнго анализа.
Для создания биочипа были выбраны частые мутации в гене SERPINA1: Е342К, E264V.
Для обнаружения точечных мутаций в различных генах применяются следующие методы.
1. Экстенция цепи меченным дидезоксирибонуклеозидтрифосфатом (SNP-single nucleotide polymorphism);
2. Аллель - специфичная ПЦР (Allele specific PCR);
3. Изучение полиморфизма фрагментов после рестрикции (RFLP-restriction fragment length polymorphism);
4. Анализ конформационного полиморфизма одно- и двухцепочечной ДНК (SSCP и DSCP-single-(double)-strand conformation polymorphism);
5. Гибридизацией на микроматрицах (Hybridization on microai Tay);
6. Секвенирование (sequencing);
7. Дидезоксифингерпринтинг (ddF-method);
8. ПЦР-гетеродуплексный анализ (PCR-heteroduplex analysis);
9. Метод несовершенного дуплекса с РНК (RNA mismatch analysis);
10. Метод структурно-специфичного расщепления (Structure-specific endonuclease cleavage);
11. Метод зондов (Line Probe assay (LiPA));
12. Метод PhaB.
Однако все перечисленные выше методы имеют определенные недостатки, которые на практике могут существенно усложнить массовый скрининг пациентов. Так, методы экстенции цепи и аллель - специфичная ПЦР (1, 2) требуют постановки независимых реакций по числу изучаемых мутаций (т.е. несколько десятков проб для одного пациента в случае гена галактоза-1-фосфат уридилтрансферазы) и, соответственно, большого количества изучаемого образца.
Методы изучения полиморфизма (3, 4), ПЦР - гетеродуплексный анализ (8) и метод несовершенного дуплекса с РНК (9) трудоемки, занимают большое количество времени, дают косвенное заключение о типе мутации и требуют типовых стандартов (на каждую мутацию); кроме того, для метода несовершенного дуплекса предъявляются повышенные требования к отсутствию РНК-азы. Также данный метод трудоемкий и не приемлем для детекции дуплекса G-U.
Гибридизация на микроматрицах (5) требует сложного компьютерного обсчета полученных результатов и дорогостоящих расходных материалов (собственно микроматрица).
Прямое секвенирование (6) требует выделения последовательности гена, отличается высокой себестоимостью и требует секвенирующее устройство.
Методы структурно-специфического расщепления (10) и дидезоксифингерпринтинг (7) требуют предварительной стандартизации (подбора условий), что увеличивает трудоемкость, а также наличия типовых стандартов. Кроме того, дидезоксифингерпринтинг выполняется с радиоактивно-меченным зондом.
Метод зондов (11) отличается высокой себестоимостью и работает в случае небольшого количества мутаций.
Осуществление метода PhaB (12) занимает большое количество времени, поскольку связано с репликацией фагов и последующей регистрацией лизирования на культуре М. smegmatis, а также он очень трудоемок.
Данные недостатки устраняются в настоящем изобретении.
Метод гибридизации на биочипах выгодно отличается от всех вышеперечисленных методик возможностью определения нескольких присутствующих мутаций одновременно, низкой себестоимостью, малым временем получения результата. Также он не требует дорогостоящего оборудования и высококвалифицированного персонала.
При проведении патентного поиска нами не было обнаружено аналогов заявленного изобретения. Представленное изобретение обладает рядом преимуществ по сравнению с другими методами медико-генетической диагностики тирозинемии типа 1.
1. При помощи заявленного биочипа можно детектировать одновременно несколько различных точечных мутаций в гене FAH и SERPINA1, что позволит с большей вероятностью исключить ложно-положительные результаты при постановке диагноза пациенту.
2. Заявленный биочип будет более приемлем, чем более дорогостоящие и трудоемкие методы для использования в рутинной медико-генетической диагностике тирозинемии 1 типа в клинических лабораториях.
Таким образом, заявленное нами изобретение позволит более просто и точно определять наличие мутаций в генах РАН и SERPINA1, что позволит в значительной мере улучшить диагностику тирозинемии I типа и недостаточности альфа-1-антитрипсина.
Раскрытие изобретения.
Заявленные изобретения относятся к биочипу, который позволяет детектировать наличие точечных мутаций у пациента-человека; применению указанного биочипа для постановки диагноза пациенту-человеку, а также способу получения и использования указанного биочипа.
Краткое описание фигур.
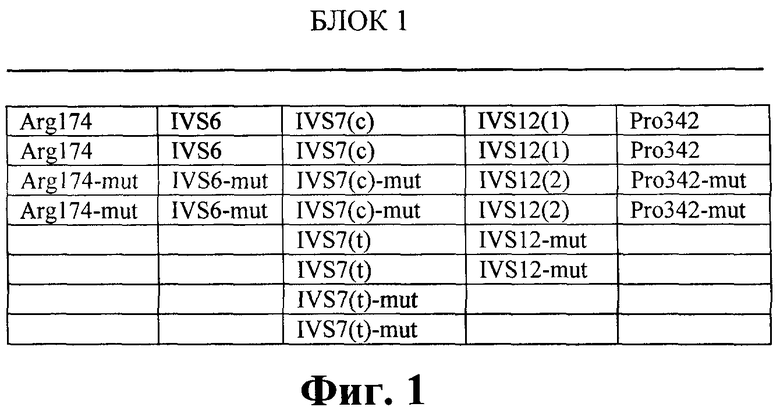
Фигура 1. Нанесение олигонуклеотидов, комплементарных различным мутациям в гене FAH, на биочип (блок 1).
Фигура 2. Нанесение олигонуклеотидов, комплементарных различным мутациям в гене SERPINA1, на биочип (блок 2).
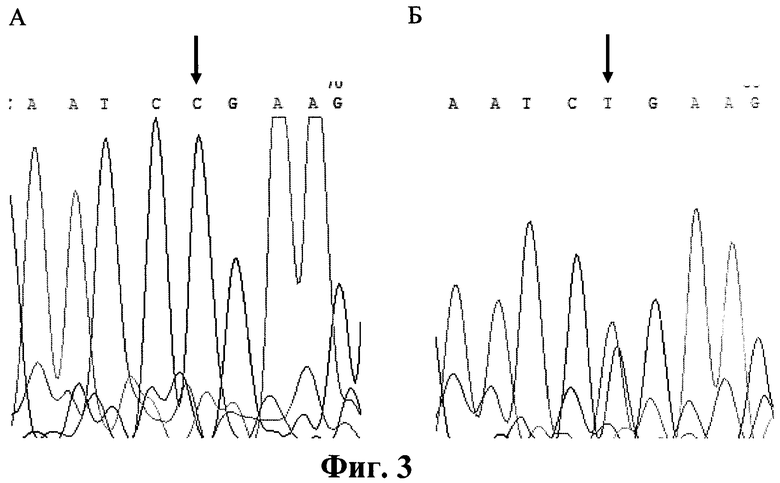
Фигура 3. Детекция мутации Argl74 (CGA->TGA). Прямое ДНК-секвенирование. Стрелками указана локализация мутации. А. Фрагмент экзона 6 гена FAH. Норма. Б. Фрагмент экзона 6 гена FAH. Мутация Argl74Term в гетерозиготном состоянии.
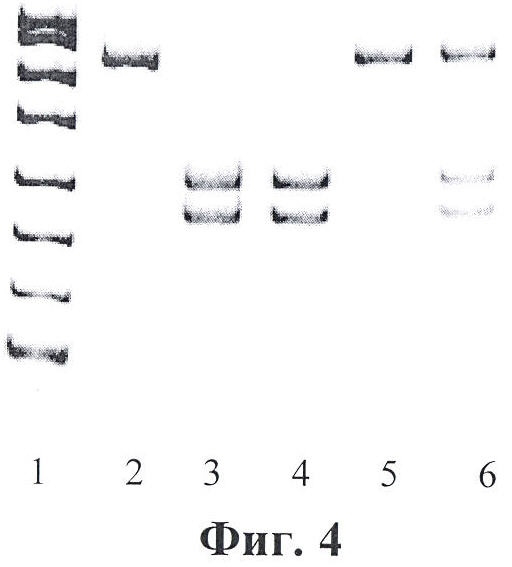
Фигура 4. Детекция мутации IVS6-1 g->t. ПДРФ анализ. При возникновении мутации IVS 6-1 G->T исчезает сайт рестрикции для эндонуклеазы рестрикции Bsp 1720 I (GC^TNAGC). Дорожки: 1 - маркеры молекулярного веса; 2 - фрагмент экзона 7 гена FAH до рестрикции; 3, 4 - фрагмент экзона 7 гена FAH после обработки эндонуклеазой рестрикции Bsp 1720 I, норма; 5 - фрагмент экзона 7 гена FAH после обработки эндонуклеазой рестрикции Bsp 1720 I, мутация IVS 6-1 G->T в гомозиготном состоянии; 6 - фрагмент экзона 7 гена FAH после обработки эндонуклеазой рестрикции Bsp 1720 I, мутация IVS 6-1 G->T в гетерозиготном состоянии.
Фигура 5. Детекция мутации IVS7-1 g->a. ПДРФ анализ. При возникновении мутации IVS7-1 G->A появляется сайт рестрикции для эндонуклеазы рестрикции Alu I (AG^CT). Дорожки: 1 - маркеры молекулярного веса; 2 - фрагмент экзона 8 гена FAH до рестрикции; 3 - фрагмент экзона 8 гена FAH после обработки эндонуклеазой рестрикции Alu I, норма.
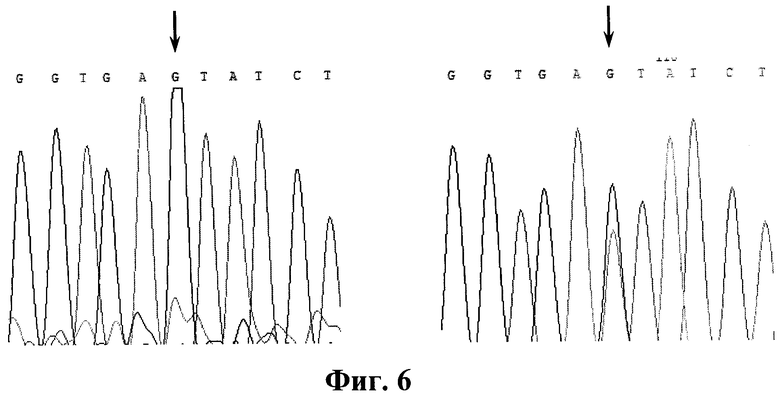
Фигура 6. Детекция мутации IVS 12+5 g->a. Прямое ДНК-секвенирование. Стрелками указана локализация мутации. А. Фрагмент интрона 6 - экзона 7 гена FAH. Норма. Б. Фрагмент интрона 6 - экзона 7 гена РАН, мутация IVS 12+5 G->A в гетерозиготном состоянии.
Фигура 7. Детекция мутации Pro342Leu (CCG->CTG). Прямое ДНК-секвенирование. Стрелками указана локализация мутации. А. фрагмент экзона 12 гена FAH. Норма. Б. Фрагмент экзона 12 гена FAH, мутация Pro342Leu в гомозиготном состоянии.
Фигура 8. Детекция мутаций E264V (GAA->GTA) и Е342К (GAG->AAG). Дорожки: 1 - маркер молекулярного веса 100bp DNA ladder; 2 - ПЦР-продукт экзона 5 гена альфа-1-антитрипсина до рестрикции (мутация Е342К); 3-ПЦР-продукт экзона 5 гена альфа-1-антитрипсина образца с мутацией Е342К после рестрикции с эндонуклеазой Taq I; 4-ПЦР-продукт экзона 5 гена альфа-1-антитрипсина контрольного образца (норма) после рестрикции с эндонуклеазой Taq I; 5-ПЦР-продукт экзона 3 гена альфа-1-антитрипсина до рестрикции (мутация E264V); 6, 7-ПЦР-продукты экзона 3 гена альфа-1-антитрипсина контрольных образцов (норма) после рестрикции с эндонуклеазой Taq I.
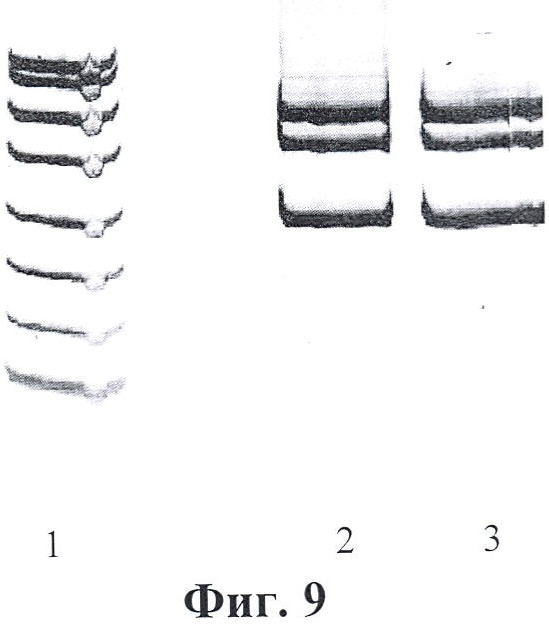
Фигура 9. Электрофореграмма мультиплексных ПЦР для первого раунда (ген FAH). Дорожки: 1 - маркер молекулярного веса; 2-3 - первый раунд ПЦР: фрагмент экзонов 6-7 гена FAH (мутации Argl74Term, IVS6-1 g-t) - 463 пн + фрагмент экзона 12 гена FAH (мутации IVS 12+5 g-a, Pro342Leu) - 382 пн + фрагмент экзона 8 гена FAH (мутация IVS7-1 g-a) - 252 пн.
Фигура 10. Электрофореграмма мультиплексных ПЦР для второго раунда (ген FAH). Дорожки: 1 - маркер молекулярного веса; 2 - второй раунд ПЦР: фрагмент экзона 12 гена FAH (мутация IVS 12+5 g-a) - 149 пн+фрагмент экзона 7 гена FAH (мутация IVS6-1 g-t) - 129 пн; 3 - второй раунд ПЦР: фрагмент экзона 12 гена FAH (мутация Pro342Leu) - 140 пн+фрагмент экзона 8 гена FAH (мутация IVS37-1 g-a) - 121 пн+фрагмент экзона 6 гена FAH (мутация Argl74Term) - 111 пн.
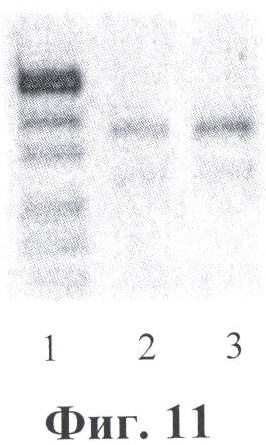
Фигура 11. Электрофореграмма мультиплексных ПЦР для первого раунда (ген SERPINA1). Дорожки: 1 - маркер молекулярного веса; 2-3 - второй раунд ПЦР: фрагмент экзона 3 гена SERPINA1 (мутация E264V) - 374 пн+фрагмент экзона 5 гена SERPINA1 (мутация Е342К) - 289 пн.
Фигура 12. Электрофореграмма мультиплексных ПЦР для второго раунда (ген SERPINA1). Дорожки: 1 - маркер молекулярного веса; 2-3 - второй раунд ПЦР: фрагмент экзона 3 гена SERPINA1 (мутация E264V) - 139 пн+фрагмент экзона 5 гена SERPINA1 (мутация Е342К) - 92 пн.
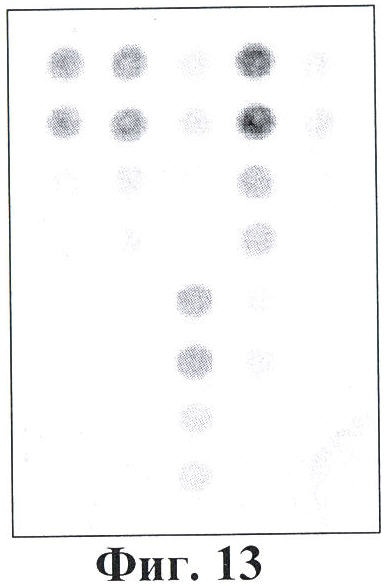
Фигура 13. Гибридизационная картина образца ДНК, полученного из человека, не несущего мутации в гене FAH с биочипом. Расположение точек соответствует фиг.1.
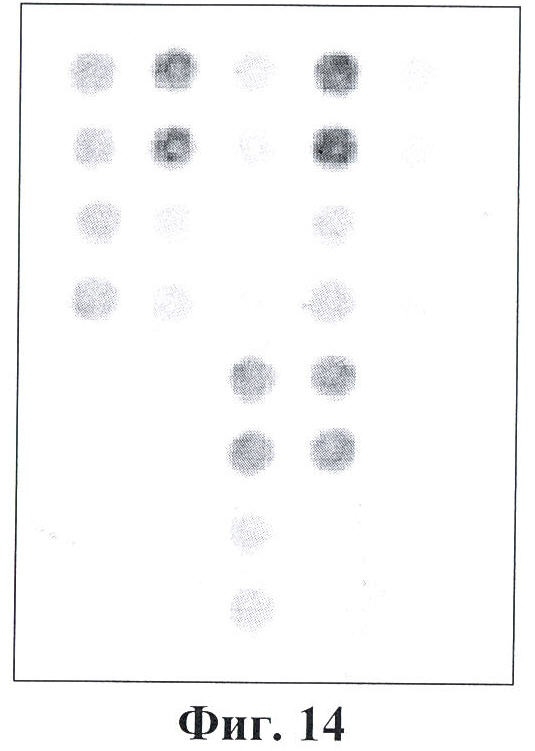
Фигура 14. Гибридизационная картина образца ДНК, полученного из человека, несущего мутации Argl74Term и IVS12+5 g->a в гетерозиготном состоянии в гене FAH с биочипом. Расположение точек соответствует фиг.1.
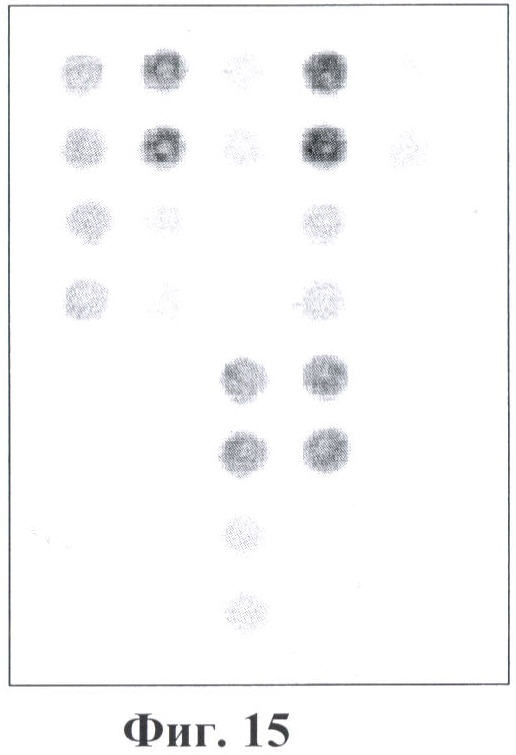
Фигура 15. Гибридизационная картина образца ДНК, полученного из человека, несущего мутацию IVS12+5 a->g в гетерозиготном состоянии в гене FAH с биочипом. Расположение точек соответствует фиг.1.
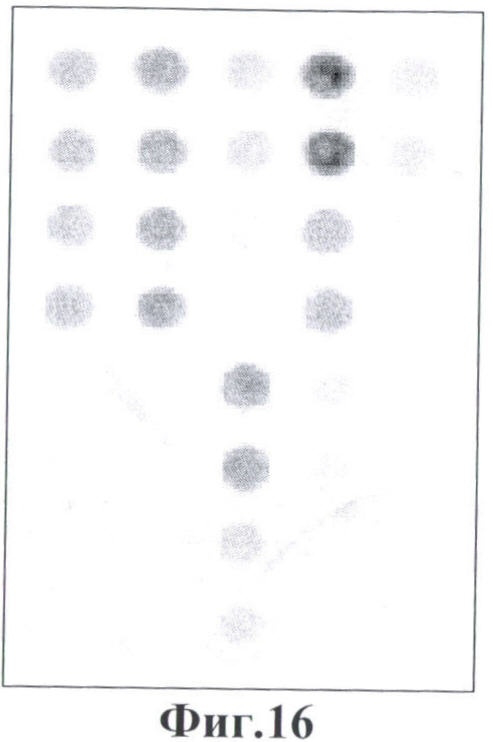
Фигура 16. Гибридизационная картина образца ДНК, полученного из человека, несущего мутации Argl74Term и IVS6-1 g->t в гетерозиготном состоянии в гене FAH с биочипом. Расположение точек соответствует фиг.1.
Фигура 17. Гибридизационная картина образца ДНК, полученного из человека, несущего мутацию Pro342Leu в гомозиготном состоянии в гене FAH с биочипом. Расположение точек соответствует фиг.1.
Фигура 18. Гибридизационная картина образца ДНК, полученного из человека, не несущего мутации в гене SERPINA1 с биочипом. Расположение точек соответствует фиг.2.
Фигура 19. Гибридизационная картина образца ДНК, полученного из человека, несущего мутацию Е342К в гомозиготном состоянии в гене SERPINA1 с биочипом. Расположение точек соответствует фиг.2.
Осуществление изобретения
Осуществление изобретения проиллюстрировано следующими примерами.
Пример 1. Установление мутаций, использованных для создания биочипа.
С целью разработки блока-1 биочипа нами были проанализированы данные литературы и частоты мутаций в гене FAH у пациентов с тирозинемией тип 1 из Российской Федерации.
Частоты мутаций Pro342Leu, Argl74Term, IVS 12+5 G->A, IVS6-1 G->T составляют на данный момент исследования 44%, 18%, 18% и 9% соответственно. При анализе литературных данных также были выбраны мутации и полиморфизмы, которые с наибольшей частотой встречались в других популяциях.
Учитывая вышеизложенное, для создания биочипа были выбраны следующие мутации в гене FAH: Pro342Leu, Argl74Term, IVS 12+5 G->A, IVS6-1 G->T, IVS7-1 G->A.
С целью разработки блока-2 биочипа нами были проанализированы данные литературы и частоты мутаций в гене SERPINA1 у пациентов с альфа-1-антитрипсина недостаточностью из Российской Федерации. Наиболее распространенной мутацией, обуславливающей развитие ранней формы заболевания, является мутация Е342К (аллель Z). Второй частой мутацией является мутация E264V, которая в сочетании с аллелем может вызывать развитие эмфиземы легких.
Учитывая вышеизложенное, для создания биочипа были выбраны две мутации в гене SERPINA1: Е342К, E264V.
Пример 2. Дизайн и оптимизация последовательностей олигонуклеотидов для иммобилизации на микрочипе.
Дизайн олигонуклеотидных последовательностей для иммобилизации на микрочипе проводили с учетом анализа нуклеотидной последовательности генов фумарилацетоацетат гидролазы (FAH) и альфа-1-антитрипсина (SERPINA1) в областях клинически значимых мутаций. Для этой цели использовали программу Oligo 6 (США).
Пример 3. Конструирование микрочипа для детекции полиморфизма в гене галактоза-1-фосфат уридилтрансферазы.
При выборе полиморфных локусов, анализ которых будет проводиться при помощи биочипа по изобретению, руководствовались следующими подходами: 1) в генах выбирались функционально значимые аллели, встречающиеся с наибольшей частотой; 2) включали только те нуклеотидные замены, полиморфизм которых уже изучен в ряде популяций России.
Последовательности нуклеотидов, использованных для создания биочипа по изобретению, приведены в таблицах 1; 2; 3 и 4.

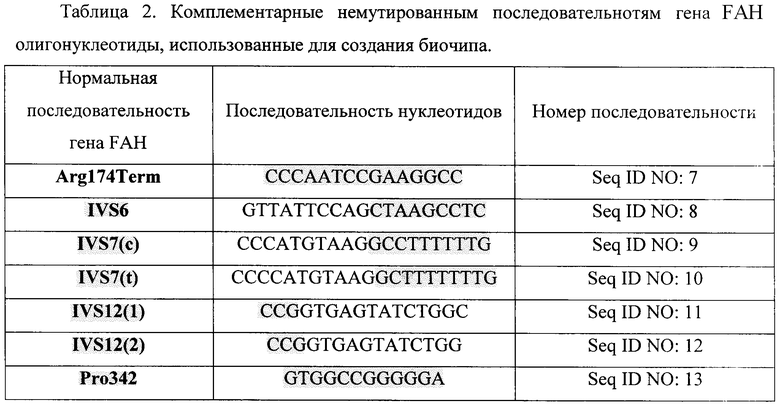


Олигонуклеотиды, приведенные в таблицах 1, 2, 3, 4, синтезированы с использованием стандартной фосфоамитидной процедуры на автоматическом синтезаторе 394 DNA/RNA Synthesizer ("Applied Biosystems", США) На 3'-конце олигонуклеотидов находится спейсер со свободной аминогруппой, который вводили при синтезе с помощью 3'-Amino-Modifier C7 CPG 500 ("Glen Reseach", США) с целью проведения фотоиндуцируемой совместной полимеризации олигонуклеотидов и компонентов полиакриламидного геля.
С целью повышения надежности определения сигнала каждую олигонуклеотидную пробу продублировали. Для подбора последовательности олигонуклетидов, обеспечивающих максимальную специфичность, в биочипе использовались несколько типов олигонуклеотидов, комплементарных одной и той же мутации или немутантной последовательности.
Факт наличия немутантных и мутантных аллелей в геноме пациента установлен при анализе интенсивности флюоресценции соответствующих ячеек на чипе. Нанесение олигонуклетидов, комплементарных нормальной последовательности и различным мутациям гена FAH, на биочип показано на фигуре 1, а комплементарных нормальной последовательности и различным мутациям гена SERPINA1 - показано на фигуре 2 (обозначение олигонуклеотидов такое же, как и в таблицах 1, 2, 3, 4).
Таким образом, сконструированный биочип позволяет определять наличие мутаций в генах фумарилацетоацетат гидролазы (FAH) и альфа-1-антитрипсина (SERPINA1), приводящих к возникновению заболевания.
Пример 4. Подготовка образцов ДНК от здоровых доноров и больных галактоземией 1 типа для тестирования биочипа.
В результате работы были отобраны образцы ДНК больных галактоземией детей для каждой из десяти исследуемых мутаций. Эти образцы ДНК подготовлены для тестирования полученного ДНК-чипа.
Все выбранные нами для создания биочипа мутации были подтверждены методом прямого нерадиоактивного секвенирования и/или анализом полиморфизма длины рестрикционных фрагментов. В качестве примеров на фигурах 2-8 приведены результаты части экспериментов. Как видно на приведенных фигурах мутации хорошо детектируются с помощью данных методов. Таким образом, в результате данной работы были отобраны образцы ДНК, которые можно применить для тестирования полученного биочипа.
Пример 5. Гибридизация биочипа с ДНК-зондами с известной структурой и с ДНК-зондами, полученными от пациентов.
В результате исследования подобраны олигонуклеотидные праймеры и условия проведения реакции для проведения второго раунда ПЦР на последовательностях генов FAH и SERPINA1, полученной в первом раунде ПЦР, для дальнейшей детекции патогенно-значимых нуклеотидных замен генов FAH и SERPINA1 на микрочипе. Для проведения первого и второго раундов мультиплексной ПЦР генов FAH и SERPINA1 были подобраны следующие олигонуклеотидные праймеры (таблицы 5 и 6 соответственно).
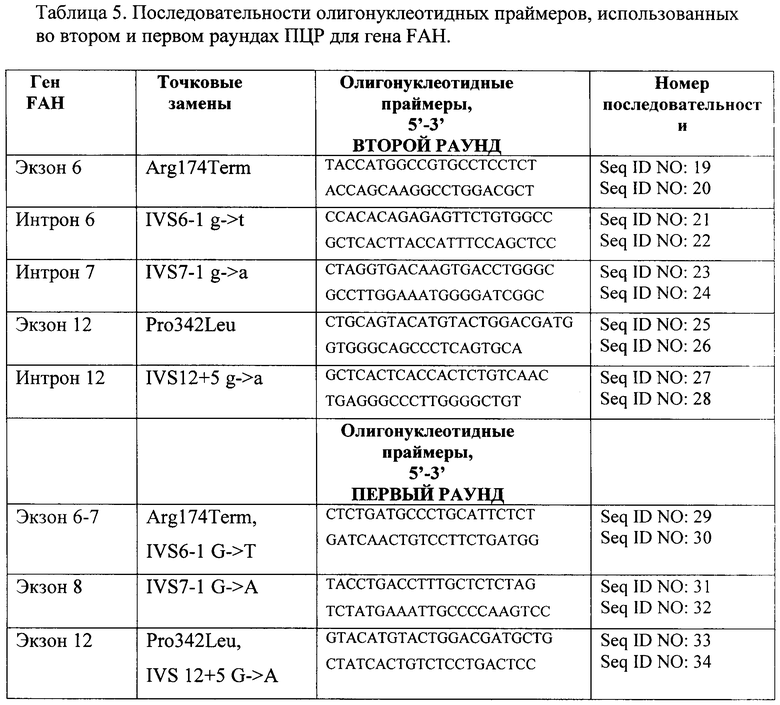
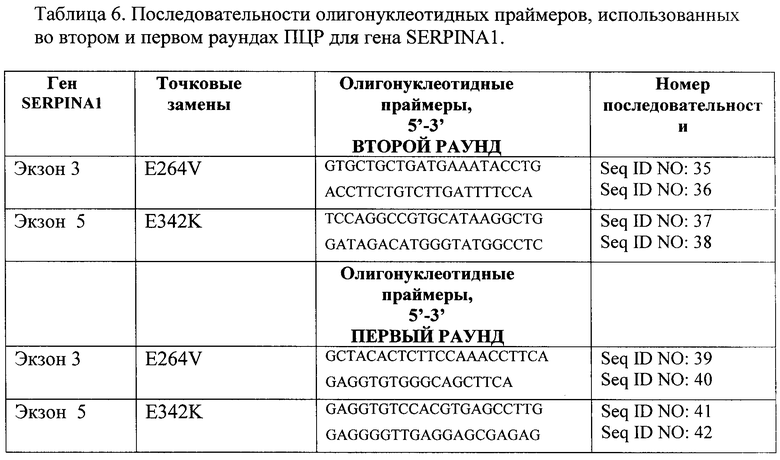
Так как экзоны гена РАН располагаются плотным кластером, то, чтобы избежать перекрестной ПЦР, второй раунд проводили следующим образом: аликвота от первого раунда ПЦР объемом 2 мкл с последующей мультиплексной ПЦР.
1. Фрагмент экзона 12 гена FAH (мутация Pro342Leu) + фрагмент экзона 8 гена FAH (мутация IVS37-1 g-a)+фрагмент экзона 6 гена FAH (мутация Argl74Term).
2. Фрагмент экзона 12 гена РАН (мутация IVS12+5 g-a) + фрагмент экзона 7 гена FAH (мутация IVS6-1 g-t).
Электрофореграмма мультиплексных ПЦР для гена РАН представлена на фигурах 9 и 10.
Для гена SERPINA1 второй раунд ПЦР проводили следующим образом: аликвота от первого раунда ПЦР объемом 2 мкл с последующей мультиплексной ПЦР: фрагмент экзона 3 гена SERPINA1 (мутация E264V)+фрагмент экзона 5 гена SERPINA1 (мутация Е342К).
Электрофореграмма мультиплексных ПЦР для гена SERPINA1 представлена на фигурах 11 и 12.
Реакционная смесь (25 мкл) на втором этапе ПЦР содержала 0,5 пмоль каждого праймера, 2,5 мкл буфера для ПЦР с 2,5 мМ MgCl2 («Силекс», Россия), 6,25 нМ каждого из dNTP («Силекс», Россия), 0,2 нМ флюоресцентно-меченного dUTP-Cy5 и 1 ед. акт. Taq-полимеразы («Силекс», Россия). Для амплификации использовали программируемый ДНК-амплификатор фирмы «ДНК-технология» (Россия). После предварительной денатурации ДНК (3 минуты при 94°C) проводили амплификацию в режиме: 33 цикла амплификации: 94°С - 30 сек, 59°С - 30 сек, 72°С - 1 мин. Заключительный синтез 72°С - 3 минуты.
Гибридизацию на микрочипе проводили с использованием флюоресцентно-меченных образцов, полученных на второй стадии реакции ПЦР, в 32 мкл смеси следующего состава: 8 мкл формамида ("Serva", США), 8 мкл 20XSSPE ("Promega", США), 4 мкл амплификата и 12 мкл Н2О. Гибридизационную смесь денатурировали при 95°С (5 мин), охлаждали на льду (3 мин), наносили на биочип и оставляли на ночь при температуре 37°С. Далее чип отмывали в 1XSSPE в течение 5 мин при 20°С и высушивали.
Флюоресцентный сигнал от ячеек регистрировали с помощью широкопольного люминисцентного микроскопа, снабженного камерой ПЗС и программным обеспечением Imageware («Биочип-ИМБ», Россия). Использовали нормированные сигналы флюоресценции Jm=(Im-I0)/(Bm-I0), где Im - интенсивность сигнала на единицу площади внутренней части составляющей ячейки, Bm - фоновый сигнал, отражающий распределение освещенности микрочипа, I0 - темновой ток ПЗС матрицы, a m - номер ячейки чипа. При анализе интенсивности флюоресценции использовали среднее значение между двумя дублирующими ячейками. Сигналы Jm сортировали по возрастанию. Ячейкой сравнения считали ячейку, после которой наблюдали резкое увеличение нормированного сигнала. Считали, что ячейка имеет положительный сигнал, если ее сигнал превышал сигнал ячейки сравнения. Затем сравнивали между собой ячейки, содержащие нуклеотидную последовательность дикого типа и соответствующую мутантную последовательность, выявляя доминирующий сигнал, который превышал пороговое значение. Пороговое значение сигнала определяли статистически, обработав более 20 гибридизационных картин. Если в паре положительных сигналов «проба дикого типа - мутантная проба» ни один из сигналов не доминировал, значимыми считали сигналы с обеих ячеек, что соответствовало гетерозиготному состоянию анализируемого локуса.
Пример 6. Анализ данных, полученных при гибридизации на биочипе по изобретению.
6.1. Результат гибридизации продуктов ПЦР-реакции, полученных с использованием геномной ДНК доноров, не болеющих тирозинемией типа 1, с биочипом по изобретению приведен на фигуре 13 и в таблице 7.
Согласно сведениям, приведенным в таблице 7 и на фигуре 13, уровень нормированных сигналов флюоресценции (Jm) в точках, соответствующих нормальной последовательности гена FAH, в несколько раз выше, чем в точках, соответствующих мутированным последовательностям (в соответствующих парах олигонуклеотидов). Таким образом, заявленный биочип не дает ложноположительных результатов в случае пациента, не несущего указанных мутаций в последовательности гена FAH.
6.2. Результат трех различных гибридизаций продуктов ПЦР-реакции, полученных с использованием геномной ДНК доноров, несущих мутации Argl74Term IVS12+5 g->a, с биочипом по изобретению приведен на фигуре 14 и в таблице 8.
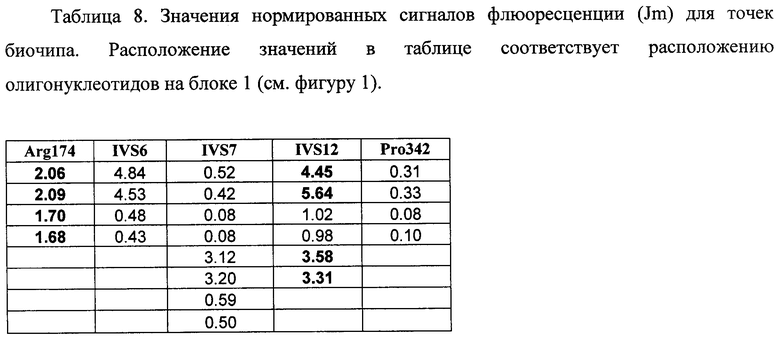
Согласно сведениям, приведенным в таблице 8 и на фигуре 14, уровень сигналов флюоресценции в точках, соответствующих мутациям Argl74Term и IVS12+5 g->a в гене FAH, такой же, как и в точках, соответствующих последовательности, не содержащей данную замену в интроне 3. Данный факт говорит о том, что мутация находится в гетерозиготном состоянии. Во всех остальных точках уровень сигнала, соответствующий нормальным последовательностям, в несколько раз сильнее сигнала, соответствующего мутированным последовательностям. Таким образом, заявленный биочип позволяет установить наличие мутаций Argl74Term и IVS12+5 g->a в гене FAH с высокой специфичностью (даже в гетерозиготном состоянии). При этом не возникает ложноположительных сигналов, соответствующих другим мутациям.
6.3. В результате гибридизации продуктов ПЦР-реакции, полученных с использованием геномной ДНК пациента, несущего мутацию IVS12+5 g->a в гене FAH в гетерозиготном состоянии, был получен результат, приведенный на фигуре 15 и в таблице 9.
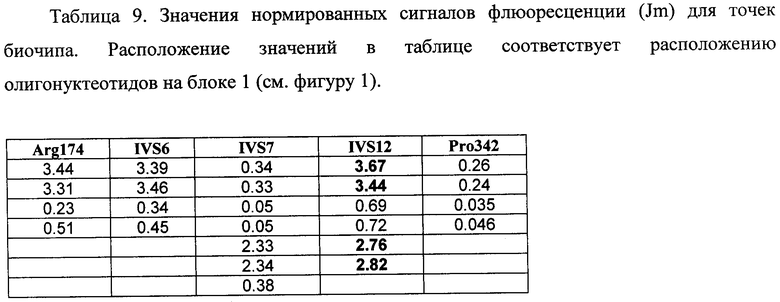
Согласно сведениям, приведенным в таблице 9 и на фигуре 15, уровень сигналов флюоресценции в точках, соответствующих мутации IVS12+5 g->a в гене FAH, такой же, как и в точках, соответствующих последовательностям, не содержащим данные замены. Данный факт говорит о том, что мутация находятся в гетерозиготном состоянии. Во всех остальных точках уровень сигнала, соответствующий нормальным последовательностям, в несколько раз сильнее сигнала, соответствующего мутированным последовательностям. Таким образом, заявленный биочип позволяет установить наличие мутации IVS12+5 g->a в гене FAH с высокой специфичностью (даже в гетерозиготном состоянии). При этом не возникает ложноположительных сигналов, соответствующих другим мутациям.
6.4. В результате гибридизации продуктов ПЦР-реакции, полученных с использованием геномной ДНК пациента, несущего мутации Argl74Term и IVS6 g->t в гене FAH в гетерозиготном состоянии, был получен результат, приведенный на фигуре 16 и в таблице 10.
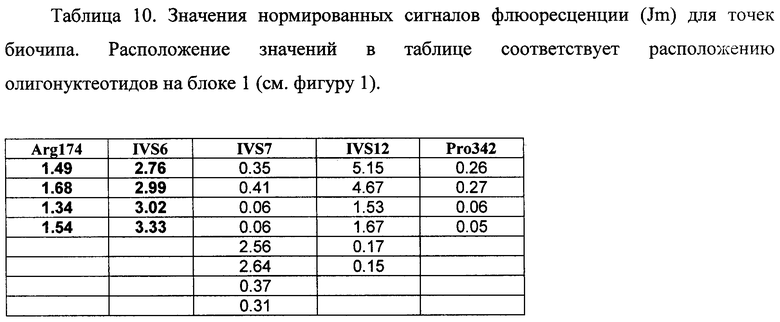
Согласно сведениям, приведенным в таблице 10 и на фигуре 16 уровень сигналов флюоресценции в точках, соответствующих мутациям Argl74Term и IVS6 g->t в гене FAH, такой же, как и в точках, соответствующих последовательностям, не содержащим данные замены. Данный факт говорит о том, что мутация находится в гетерозиготном состоянии. Во всех остальных точках уровень сигнала, соответствующий нормальным последовательностям, в несколько раз сильнее сигнала, соответствующего мутированным последовательностям. Данный факт показывает, что заявленный биочип позволяет установить наличие мутаций Argl74Term и IVS6 g->t в гене FAH с высокой специфичностью даже в гетерозиготном состоянии. При этом не возникает ложноположительных сигналов, соответствующих другим мутациям.
6.5. В результате гибридизации продуктов ПЦР-реакции, полученных с использованием геномной ДНК пациента, несущего мутацию Pro342Leu в гене FAH в гомозиготном состоянии, был получен результат, приведенный на фигуре 17 и в таблице 11.
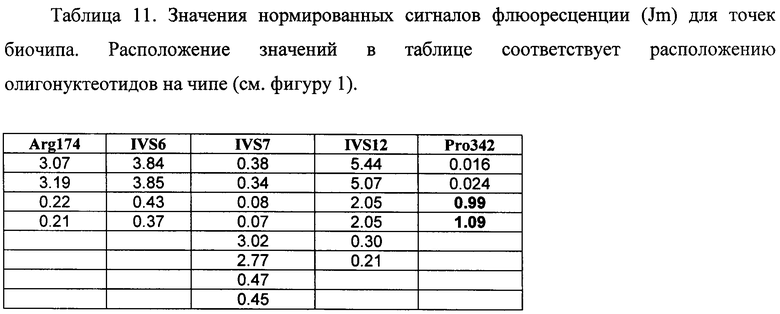
Согласно сведениям, приведенным в таблице 11 и на фигуре 17, уровень сигналов флюоресценции в точках, соответствующих мутации Pro342Leu в гене FAH, в несколько десятков раз сильнее, чем в точках, соответствующих последовательностям, не содержащим данные замены. Данный факт говорит о том, что мутация находится в гомозиготном состоянии. Во всех остальных точках уровень сигнала, соответствующий нормальным последовательностям, в несколько раз сильнее сигнала, соответствующего мутированным последовательностям. Таким образом, биочип позволяет установить наличие мутации Pro342Leu в гене FAH с высокой специфичностью (в гомозиготном состоянии). При этом не возникает ложноположительных сигналов, соответствующих другим мутациям.
6.6. В результате гибридизации продуктов ПЦР-реакции, полученных с использованием геномной ДНК пациента, не несущего мутации в гене SERPINA1, был получен результат, приведенный на фигуре 18 и в таблице 12.

Согласно сведениям, приведенным в таблице 12 и на фигуре 18, уровень нормированных сигналов флюоресценции (Jm) в точках, соответствующих нормальной последовательности гена SERPINA1, в несколько раз выше, чем в точках соответствующих мутированным последовательностям (в соответствующих парах олигонуклеотидов). Таким образом, заявленный биочип не дает ложноположительных результатов в случае пациента, не несущего указанных мутаций в последовательности гена SERPINA1.
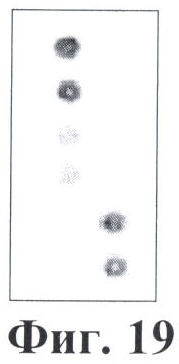
6.7 В результате гибридизации продуктов ПЦР-реакции, полученных с использованием геномной ДНК пациента, несущего мутацию Е342К в гене SERPINA1 в гомозиготном состоянии, был получен результат, приведенный на фигуре 19 и в таблице 13.

Согласно сведениям, приведенным в таблице 13 и на фигуре 19, уровень сигналов флюоресценции в точках, соответствующих мутации Е342К в гене SERPINA1, в несколько раз сильнее, чем в точках, соответствующих последовательностям, не содержащим данные замены. Данный факт говорит о том, что мутация находится в гомозиготном состоянии. Во всех остальных точках уровень сигнала, соответствующий нормальным последовательностям, в несколько раз сильнее сигнала, соответствующего мутированным последовательностям. Данный факт показывает, что заявленный биочип позволяет установить наличие мутации Е342К в гене SERPINA1 с высокой специфичностью (в гомозиготном состоянии). При этом не возникает ложноположительных сигналов, соответствующих другим мутациям.




















Изобретение относится к области молекулярной биологии и может быть использовано в медицине, в частности при диагностике наследственных нарушений обмена, приводящих к развитию неонатальной гипербилирубинемии. В данном изобретении предлагается тест-система, представленная набором олигонуклеотидов, позволяющих выявлять точковые мутации в генах FAN SERPINA1, которые вызывают соответственно тирозинемию 1 типа и недостаточность альфа-1-антитрипсина. Описан способ детекции указанных точковых мутаций, предусматривающий проведение двухраундной мультиплексной ПЦР с использованием соответствующих наборов специфических праймеров и гибридизацию полученных ПЦР-продуктов с биочипом, содержащим тест-систему по изобретению. Предложенный метод обеспечивает возможность проведения в короткие сроки точного генетического анализа. 3 н.п. ф-лы, 13 табл., 19 ил., 6 пр.
1. Тест-система, которая представляет собой биочип для детекции точковых мутаций в генах РАН и SERPINA1, вызывающих тирозинемию 1 типа человека и недостаточность альфа-1-антитрипсина человека, содержащая олигонуклеотиды, имеющие последовательности Seq ID NO: 1, Seq ID NO: 2, Seq ID NO: 3, Seq ID NO: 4, Seq ID NO: 5, Seq ID NO: 6, Seq ID NO: 7, Seq ID NO: 8, Seq ID NO: 9, Seq ID NO: 10, Seq ID NO: 11, Seq ID NO: 13, Seq ID NO: 14. Seq ID NO: 15, Seq ID NO: 16, Seq ID NO: 17.
2. Применение тест-системы по п.1 для детекции точковых мутаций в генах FAH и SERPINA1, вызывающих тирозинемию 1 типа человека и недостаточность альфа-1-антитрипсина человека.
3. Способ детекции точковых мутаций в генах FAH и SERPINA1, вызывающих тирозинемию 1 типа человека и недостаточность альфа-1-антитрипсина человека, состоящий в
1) постановке двухраундной мультиплексной ПЦР на геномной ДНК пациента с использованием праймеров, имеющих последовательности Seq ID NO: 19, Seq ID NO: 20, Seq ID NO: 21, Seq ID NO: 22, Seq ID NO: 23, Seq ID NO: 24, Seq ID NO: 25, Seq ID NO: 26, Seq ID NO: 27, Seq ID NO: 28, ID NO: 29, Seq ID NO: 30, Seq ID NO: 31, Seq ID NO: 32, Seq ID NO: 33, Seq ID NO: 34, Seq ID NO: 35, Seq ID NO: 36, Seq ID NO: 37, Seq ID NO: 38, Seq ID NO: 39, Seq ID NO: 40, Seq ID NO: 41, Seq ID NO: 42 и применяемых согласно таблицам 5 и 6.
2) гибридизации полученных продуктов ПЦР с биочипом по п.1,
3) анализе результатов гибридизации с установлением наличия мутации и ее гомозиготного или гетерозиготного состояния.
| LIU С | |||
| ЕТ AL., Gastroenterology, 132(1), 119-126, 2007 | |||
| BARTELS С | |||
| ЕТ AL., Am | |||
| Transl | |||
| Res., 1(4), 406-411, 2009 | |||
| FANEUF D | |||
| ЕТ AL., J | |||
| Clin | |||
| Invest., 90 (4), 1185-1192, 1992 | |||
| ROOTWELT H | |||
| ET AL., Hum | |||
| Mutat., 7(3), 239-243, 1996 | |||
| ARRANZ J | |||
| ЕТ AL., Hum | |||
| Mutat., 20(3), 180-188, 2002. |

