Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения ароматических углеводородов из метана, в частности из природного газа.
Уровень техники
Ароматические углеводороды, в частности бензол, толуол, этилбензол и ксилолы, являются важными химическими продуктами массового производства в нефтехимической промышленности. В настоящее время ароматические соединения наиболее часто получают по разнообразным методам из исходных материалов на основе сырой нефти, включая каталитический реформинг и каталитический крекинг. Однако по мере того как мировые поставки исходных материалов на основе сырой нефти уменьшаются, потребность в нахождении альтернативных источников ароматических углеводородов возрастает.
Одним возможным альтернативным источником ароматических углеводородов служит метан, который является основным компонентом природного газа и биогаза. Мировые запасы природного газа постоянно пополняются, и в настоящее время месторождений природного газа открывается больше, чем нефтяных месторождений. Из-за проблем, связанных с транспортировкой больших объемов природного газа, большую часть природного газа, добываемого вместе с нефтью, в частности в отдаленных местах, сжигают в факеле. Следовательно, привлекательным методом переработки природного газа является превращение содержащихся в нем алканов непосредственно в более высокомолекулярные углеводороды, такие как ароматические углеводороды, при условии, что могут быть преодолены сопутствующие этому технические трудности.
Значительная часть способов, предложенных в настоящее время для превращения метана в жидкие углеводороды, включает в себя вначале превращение, или конверсию, метана в синтез-газ, смесь Н2 и СО. Однако производство синтез-газа связано с большими капитальными затратами и является энергоемким, вследствие чего предпочтительны пути, которые не требуют получения синтез-газа.
Был предложен ряд альтернативных способов прямого превращения метана в более высокомолекулярные углеводороды. Один такой способ предусматривает каталитическое окислительное взаимодействие метана до олефинов с последующим каталитическим превращением этих олефинов в жидкие углеводороды, включая ароматические углеводороды. Так, например, в патенте US 5336825 описан двухстадийный способ окислительного превращения метана в углеводороды, находящиеся в температурном интервале кипения бензиновой фракции и включающие в себя ароматические углеводороды. На первой стадии в присутствии свободного кислорода с использованием промотированного редкоземельным металлом катализатора из оксида щелочноземельного металла при температуре от 500 до 1000°С метан превращают в этилен и небольшие количества олефинов С3 и C4. Затем этилен и более высокомолекулярные олефины, образовавшиеся на первой стадии, над кислотным твердым катализатором, включающим пентасиловый цеолит с высоким содержанием диоксида кремния, превращают в жидкие углеводороды, находящиеся в температурном интервале кипения бензиновой фракции.
Однако этим способам окислительного взаимодействия присущи проблемы, заключающиеся в том, что их осуществление предполагает проведение высокоэкзотермических и потенциально опасных реакций горения метана и сопровождается выбросом влияющих на экологию оксидов углерода в больших количествах.
Потенциально привлекательным направлением переработки метана непосредственно в более высокомолекулярные углеводороды, в частности этилен, бензол и нафталин, является дегидроароматизация или восстановительное взаимодействие. Этот метод, как правило, предусматривает контактирование метана с катализатором, включающим такой металл, как рений, вольфрам и молибден, нанесенный на цеолит, такой как ZSM-5, при высокой температуре, в частности от 600 до 1000°С. Каталитически активной формой металла часто является элементная форма с нулевой валентностью, карбид или оксикарбид.
Например, в патенте US 4727206 описан способ получения жидкостей, богатых ароматическими углеводородами, введением метана при температуре от 600 до 800°С в отсутствие кислорода в контакт с каталитической композицией, включающей в себя алюмосиликат с молярным отношением диоксида кремния к оксиду алюминия по меньшей мере 5:1, причем в алюмосиликат добавлен галлий или его соединение и металл группы VIIB периодической таблицы элементов или его соединение.
Кроме того, в патенте US 5026937 описан способ ароматизации метана, включающий подачу потока исходных материалов, содержащего водород (мольное содержание 0,5%) и метан (мольное содержание 50%), в реакционную зону, содержащую по меньшей мере один слой твердого катализатора, включающего ZSM-5, галлий и фосфорсодержащий оксид алюминия, в условиях превращения, включающих температуру от 550 до 750°С, абсолютное давление ниже 10 атм (1000 кПа) и объемную скорость подачи газа от 400 до 7500 ч-1.
Далее, в патентах US 6239057 и US 6426442 описан способ получения углеводородов с более высоким числом углеродных атомов, например бензола, из углеводородов с низким числом углеродных атомов, таких как метан, введением этого последнего в контакт с катализатором, содержащим пористый носитель, такой как ZSM-5, на котором диспергирован рений и металл-промотор, такой как железо, кобальт, ванадий, марганец, молибден, вольфрам, или их смесь. После пропитки носителя рением и металлом-промотором катализатор активируют обработкой водородом и/или метаном при температуре примерно от 100 до 800°С в течение времени примерно от 0,5 до 100 ч. При этом утверждается, что добавление СО или CO2 в метановый исходный материал повышает выход бензола и стабильность катализатора.
Однако успешное применение восстановительного взаимодействия для получения ароматических соединений в промышленном масштабе требует решения ряда серьезных технически сложных задач. Например, процесс восстановительного взаимодействия и эндотермичен, и ограничен в термодинамическом отношении. Таким образом, если при ведении процесса не предусмотреть существенного дополнительного подогрева, эффект охлаждения в ходе реакции приводит к понижению температуры реакции, достаточному для значительного уменьшения скорости реакции и общего термодинамического превращения.
Более того, осуществление процесса сопровождается образованием углерода и других нелетучих материалов, которые накапливаются на поверхности катализатора, вызывая снижение его активности и потенциально нежелательные сдвиги селективности. Кроме того, при свойственных процессу высоких температурах активные металлические соединения (МоСх, WCx и т.д.) на поверхности катализатора могут мигрировать, образовывать агломераты или изменять фазу, что опять же приводит к нежелательному ухудшению степени превращения и селективности действия. Поэтому приходится проводить частую окислительную регенерацию катализатора для того, чтобы удалить углерод и другие нелетучие материалы, накопившиеся на его поверхности, и по возможности перераспределить активные металлические соединения. Однако в зависимости от состава катализатора регенерация в окислительной среде может иметь определенные нежелательные побочные эффекты. Например, металл на поверхности катализатора может переходить из каталитически активного элементного или обогащенного углеродом состояния в менее активное окисленное состояние. Также катализатор после регенерации может проявлять повышенную активность в плане отложения кокса (коксообразования) и связанного с ним образования водорода. Таким образом, существует заинтересованность в разработке процессов восстановительного взаимодействия, в которых регенерация катализатора осуществляется в неокислительных условиях.
Например, в японской патентной публикации Kokai 2003-26613 от 29.01.2003 описан способ производства ароматических углеводородов и водорода из низшего углеводорода, содержащего по меньшей мере 60 мол.% метана, в присутствии катализатора, такого как молибден, вольфрам или рений на носителе ZSM-5, характеризующийся тем, что катализатор периодически и попеременно переводят из рабочего цикла, в котором катализатор контактируют с низшим углеводородом, в цикл регенерации, в котором катализатор контактируют с водородом. Обычно рабочий цикл составляет от 1 до 20 минут, предпочтительно от 1 до 10 минут, а цикл регенерации - от 1 до 30 минут, предпочтительно примерно от 5 до 20 минут.
Кроме того, в международной публикации WO 2006/011568 от 02.02.2006 описан способ производства ароматических углеводородов и водорода из сырого газа, содержащего низший углеводород, характеризующийся тем, что смесь сырьевого газа и водородсодержащего газа вводят в контакт с катализатором, таким как металлосиликат на основе молибдена и/или родия, при высокой температуре, такой как 750°С, и периодически, во время циклов регенерации, подачу вышеупомянутого сырьевого газа прекращают, а подачу водородсодержащего газа продолжают. Как утверждается, непрерывная подача водорода как во время рабочего цикла, так и во время цикла регенерации уменьшает отношение длительности регенерации к длительности рабочего цикла.
В публикации заявки US 2003/0083535 раскрыт способ ароматизации метансодержащего исходного материала, характеризующийся тем, что между реакторной системой и регенерационной системой осуществляют циркуляцию катализатора дегидроароматизации, причем для регенерации различных частей катализатора его в разное время вводят в контакт с разными регенераторными газами, включающими O2, H2 и Н2О. Процентную долю катализатора, контактирующую с каждым регенераторным газом, регулируют для сохранения в реакторной системе и регенерационной системе режима теплового баланса. Эта реакторная система включает в себя псевдоожиженный слой катализатора в лифт-реакторе, а регенерационная система включает в себя второй псевдоожиженный слой катализатора, содержащийся в реакторе со стационарным кипящим слоем. После прохождения через регенерационную систему горячий регенерированный катализатор рециркулируют в реакторную систему по транспортировочной системе, которая может включать восстановительный сосуд для повышения активности регенерированного катализатора введением катализатора в псевдоожиженном слое в контакт с потоком восстанавливающего газа, содержащего водород и/или метан.
Раскрытие изобретения
В основу изобретения положена задача разработки усовершенствованного способа ароматизации метана, в котором регенерация катализатора осуществляется водородсодержащим газом.
Одним объектом настоящего изобретения является способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий:
(а) подачу в реакционную зону метансодержащего исходного материала и зернистого каталитического материала,
(б) обеспечение работы реакционной зоны в условиях, эффективных для превращения по меньшей мере части метана в более высокомолекулярный(-ые) углеводород(-ы) с сопутствующим отложением углеродистого материала на зернистом каталитическом материале, вызывающим дезактивацию каталитического материала,
(в) удаление из реакционной зоны по меньшей мере части дезактивированного зернистого каталитического материала,
(г) нагревание по меньшей мере части удаленного из реакционной зоны зернистого каталитического материала до температуры примерно от 700°С примерно до 1200°С, осуществляемое прямым или косвенным контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива,
(д) регенерирование нагретой части зернистого каталитического материала водородсодержащим газом в регенерационной зоне в условиях, эффективных для превращения по меньшей мере части отложившегося углеродистого материала в метан, и
(е) возврат по меньшей мере части зернистого каталитического материала со стадии (д) в реакционную зону.
Еще одним объектом настоящего изобретения является способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий:
(а) подачу в реакционную зону метансодержащего исходного материала и зернистого каталитического материала,
(б) обеспечение работы реакционной зоны в условиях, эффективных для превращения по меньшей мере части метана в более высокомолекулярный(-ые) углеводород(-ы) с отложением углеродистых материалов на катализаторе, вызывающем дезактивацию катализатора,
(в) удаление из реакционной зоны по меньшей мере части дезактивированного зернистого каталитического материала,
(г) нагревание первой части удаленного из реакционной зоны зернистого каталитического материала до температуры примерно от 700°С примерно до 1200°С, осуществляемое прямым или косвенным контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива,
(д) регенерирование нагретой первой части зернистого каталитического материала водородсодержащим газом в первой регенерационной зоне в условиях, включающих первое давление и эффективных для превращения по меньшей мере части отложившихся на зернистом каталитическом материале углеродистых материалов в метан,
(е) перемещение второй части удаленного из реакционной зоны зернистого каталитического материала со стадии (в) или стадии (д) во вторую регенерационную зону,
(ж) регенерирование второй части зернистого каталитического материала водородсодержащим газом во второй регенерационной зоне в условиях, включающих второе давление, отличающееся от первого давления, и эффективных для превращения по меньшей мере части отложившихся на зернистом каталитическом материале углеродистых материалов в метан, и
(з) возврат по меньшей мере части регенерированного зернистого каталитического материала со стадии (д) и по меньшей мере части зернистого каталитического материала со стадии (ж) в реакционную зону.
В одном варианте осуществления изобретения реакционная зона представляет собой реакционную зону с подвижным (движущимся) слоем катализатора и может работать с обратным температурным профилем. Исходный материал также может содержать по меньшей мере одно из следующих веществ: СО, СО2, Н2, Н2О и/или О2. Удаленный зернистый каталитический материал можно нагревать на стадии (г) до температуры примерно от 800°С примерно до 1000°С, в частности примерно от 850°С примерно до 950°С. Условия на стадии (д) регенерации могут включать в себя абсолютное давление, составляющее по меньшей мере 100 кПа, в частности примерно от 150 до 700 кПа. Условия на стадии (ж) регенерации могут включать в себя абсолютное давление, составляющее по меньшей мере 500 кПа, в частности примерно от 1000 кПа примерно до 5000 кПа. Стадии (д) и (ж) регенерации могут проводиться в отдельных регенерационных зонах.
В одном варианте осуществления изобретения условия реакции, протекающей в реакционной зоне на стадии (б), являются неокислительными. На стадии (б) условия реакции в реакционной зоне могут включать в себя температуру примерно от 400°С примерно до 1200°С, абсолютное давление примерно от 1 кПа примерно до 1000 кПа и объемную скорость подачи газа примерно от 0,01 ч-1 примерно до 1000 ч-1.
В одном варианте осуществления изобретения, зернистый каталитический материал является катализатором дегидроциклизации, содержащим металл или его соединение на неорганическом носителе. Зернистый каталитический материал может включать в себя по меньшей мере одно из следующих веществ: молибден, вольфрам, рений, соединение молибдена, соединение вольфрама, соединение цинка и соединение рения, на ZSM-5, диоксиде кремния или оксиде алюминия.
Краткое описание чертежей
На фиг.1 представлена схема осуществления способа превращения метана в более высокомолекулярные углеводороды в соответствии с первым вариантом выполнения изобретения.
На фиг.2 представлена схема осуществления способа превращения метана в более высокомолекулярные углеводороды в соответствии со вторым вариантом выполнения изобретения.
На фиг.3 представлен график зависимости выхода метана от температуры при термопрограммированной обработке водородом закоксованного Mo/ZSM-5 катализатора в примере 6, осуществлявшейся в реакторе идеального вытеснения при динамике повышения температуры, составлявшей 5°С/мин.
На фиг.4 представлен график зависимости содержания кокса (мас.%) от времени регенерации при регенерировании водородом закоксованного Mo/ZSM-5 катализатора в примере 7 при 850°С и различных парциальных давлениях водорода.
На фиг.5А и 5Б показано изменение соответственно степени превращения метана (%) и селективности по бензолу и толуолу при повышении температуры регенерирования с 875°С до 925°С.
Осуществление изобретения
В контексте настоящего описания понятие "более высокомолекулярный(-ые) углеводород(-ы)" означает углеводород(-ы), содержащий больше одного углеродного атома на молекулу; оксигенат, содержащий по меньшей мере один углеродный атом на молекулу, например этан, этилен, пропан, пропилен, бензол, толуол, ксилолы, нафталин и/или метилнафталин; и/или органическое(-ие) соединение(-ия), содержащее по меньшей мере один углеродный атом и по меньшей мере один неводородный атом, например метанол, этанол, метиламин и/или этиламин.
В контексте настоящего описания понятие "ароматический углеводород (углеводороды)" означает молекулы, содержащие одно или несколько ароматических колец. Примерами ароматических углеводородов являются бензол, толуол, ксилолы, нафталин и метилнафталины.
Понятия "кокс" и "углеродистый материал" используются в настоящем описании как взаимозаменяемые для обозначения углеродсодержащих материалов, которые в реакционных условиях представляют собой по существу нелетучие твердые вещества с низким содержанием водорода относительно содержания углерода (в частности, со значением молярного соотношения Н:С менее 0,8, наиболее вероятно менее 0,5). Они могут включать в себя кристаллический графит, графитовые листовые или пластинчатые материалы, графитовые фрагменты, аморфный углерод или другие углеродсодержащие структуры, которые в реакционных условиях представляют собой по существу нелетучие твердые материалы. Когда речь идет о твердом коксе, более твердом коксе или огнеупорном или трудно поддающемся термической переработке коксе, имеются в виду типы кокса, которые вследствие либо его структуры, либо расположения тяжелее удалять с помощью реагента (как правило, кислорода или водорода), используемого для превращения кокса в газообразные материалы.
В контексте настоящего описания понятие "дезактивация" катализатора означает потерю с течением времени каталитической активности и/или селективности. Катализатор оказывается дезактивированным, если его каталитическая активность по меньшей мере на 1% ниже, в другом случае по меньшей мере на 5% ниже, в другом случае по меньшей мере на 10% ниже, в другом случае по меньшей мере на 15% ниже, в другом случае по меньшей мере на 20% ниже, в другом случае по меньшей мере на 25% ниже, в другом случае по меньшей мере на 30% ниже, в другом случае по меньшей мере на 35% ниже, в другом случае по меньшей мере на 40% ниже, в другом случае по меньшей мере на 45% ниже, в другом случае по меньшей мере на 50% ниже, в другом случае по меньшей мере на 55% ниже, в другом случае по меньшей мере на 60% ниже, в другом случае по меньшей мере на 65% ниже, в другом случае по меньшей мере на 70% ниже, в другом случае по меньшей мере на 75% ниже, в другом случае по меньшей мере на 80% ниже, в другом случае по меньшей мере на 85% ниже, в другом случае по меньшей мере на 90% ниже, в другом случае по меньшей мере на 95% ниже или в другом случае по меньшей мере на 100% ниже, чем каталитическая активность свежего катализатора или регенерированного катализатора. Не ограничивая себя какой-либо теорией, дезактивацию катализатора можно рассматривать как явление, в котором изменяются структура и/или состояние катализатора, что приводит к потере активных участков на поверхности катализатора и, таким образом, вызывает ухудшение эффективности катализатора. Так, например, дезактивация катализатора может происходить вследствие коксообразования (закоксовывания), блокирования активных участков или деалюминирования алюмосиликатного молекулярного сита вследствие обработки водяным паром.
В контексте настоящего описания понятие реактор "с подвижным (движущимся) слоем" означает зону или сосуд с контактированием твердых частиц и газообразных потоков таким образом, что расходная скорость (U) газа ниже скорости, необходимой для пневмотранспортного уноса твердых частиц в виде разжиженной фазы, с целью сохранения слоя твердых частиц с порозностью менее 95%. Реактор с подвижным слоем может работать в нескольких режимах течения, включая режим оседающего движения (оседания) или режим движения уплотненного слоя (U<Umf), режим с барботажем пузырей (Umf<U<Umb), режим с канало- и поршнеобразованием (Umb<U<Uc), режим, переходный к турбулентному, и собственно турбулентный режим псевдоожижения (Uc<U<Utr) и режим с высокой скоростью потока (U>Utr). Эти разные режимы псевдоожижения описаны, например, в работах Kunii, D., Levenspiel, О., глава 3, Fluidization Engineering, издание 2-е, Butterworth-Heinemann, Boston, 1991, и Walas, S.M., глава 6, Chemical Process Equipment, Butterworth-Heinemann, Boston, 1990.
В контексте настоящего описания понятие "оседающий слой" служит для обозначения зоны или сосуда, где частицы (зерна) контактируют с газообразными потоками таким образом, что расходная скорость (U) газа меньше минимальной скорости, необходимой для псевдоожижения твердых частиц, минимальной скорости псевдоожижения (Umf), U<Umf, в по меньшей мере части реакционной зоны, и/или зоны или сосуда, работающего со скоростью, которая выше минимальной скорости псевдоожижения при одновременном поддержании градиента свойства газа и/или твердого вещества (таком как температура, состав газа или твердого вещества и т.д.) вдоль оси снизу вверх в реакторном слое при применении внутрикорпусных устройств реактора с целью свести к минимуму обратное перемешивание газа и твердого вещества. Описание минимальной скорости псевдоожижения приведено, например, в главе 3 работы "Fluidization Engineering", D.Kunii и O.Levenspiel, издание 2-е, Butterworth-Heinemann, Boston, 1991 и главе 6 работы "Chemical Process Equipment" S.M. Walas, Butterworth-Heinemann, Boston, 1990, содержание которых в полном объеме включено в описание путем ссылки.
В контексте настоящего описания понятие "реактор с псевдоожиженным слоем" служит для обозначения зоны или сосуда, где твердые частицы контактируют с газообразными потоками таким образом, что расходная скорость (U) газа достаточна для псевдоожижения твердых частиц (т.е. выше минимальной скорости псевдоожижения Umf) и ниже скорости, необходимой для пневмотранспортного уноса твердых частиц в виде разжиженной фазы, с целью сохранения слоя твердых частиц с порозностью менее 95%. Используемое в настоящем описании понятие "каскадные псевдоожиженные слои" служит для обозначения последовательного расположения отдельных псевдоожиженных слоев таким образом, что при этом может иметь место градиент свойства газа и/или твердого вещества (такого как температура, состав газа или твердого вещества, давление и т.д.) по мере перехода твердого вещества или газа от одного псевдоожиженного слоя со сменой уровня каскада к другому слою. График изменения минимальной скорости псевдоожижения дан, например, в приведенных выше опубликованных работах Kunii и Walas.
В контексте настоящего описания понятие "лифт-реактор" обозначает зону или сосуд (такой как вертикальная цилиндрическая труба), применяемый для чисто вертикальной транспортировки твердых частиц в режимах псевдоожижения с высокой скоростью потока (быстрого псевдоожижения) или псевдоожижения с пневмотранспортом. Режимы псевдоожижения с высокой скоростью потока и псевдоожижения с пневмотранспортом характеризуются расходными скоростями (U) газа, превышающими скорость транспортировки (Utr). Режимы псевдоожижения с высокой скоростью потока и псевдоожижения с пневмотранспортом описаны также в приведенных выше опубликованных работах Kunii и Walas.
В контексте настоящего описания указания на нагрев "косвенным контактированием" с газообразными продуктами горения включают в себя теплоперенос через поверхность теплопередачи и/или применение теплоносителя (газ, жидкость или твердое вещество), который нагревают газообразными продуктами горения и который отдает свое тепло зернистому каталитическому материалу.
В настоящем изобретении предлагается способ получения ароматических углеводородов путем контактирования метансодержащего исходного материала, обычно вместе с H2, H2O, О2, СО и/или СО2, с зернистым катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения метана в ароматические углеводороды и водород. По мере протекания реакции на катализаторе откладывается кокс, понижая тем самым активность или селективность катализатора, и, следовательно, из реакционной зоны непрерывно или периодически отводят часть закоксованного катализатора и направляют ее в отдельную регенерационную зону, где закоксованный катализатор вводят в контакт с водородсодержащим регенерирующим газом. Поскольку реакция дегидроциклизации является эндотермической, к закоксованному катализатору, отведенному из реакционной зоны, прямым или косвенным контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива, подводят тепло для повышения его температуры до требуемой температуры регенерирования, обычно составляющей примерно от 700°С примерно до 1200°С. Затем часть нагретого закоксованного катализатора может быть возвращена в реакционную зону с обеспечением тепла для реакции дегидроциклизации, а остаток нагретого катализатора вводят в контакт с водородсодержащим регенерирующим газом в регенерационной зоне в таких условиях, в которых по меньшей мере часть кокса, отложившегося на катализаторе, превращается в метан. Далее регенерированный катализатор возвращают в реакционную зону.
В одном варианте осуществления изобретения регенерирование проводят отводом из реакционной зоны двух или более порций закоксованного катализатора, подводом тепла к этим порциям катализатора и контактированием нагретых порций катализатора с водородсодержащим газом в отдельных регенерационных зонах, работающих в таких условиях, в которых парциальные давления водорода по меньшей мере в двух из регенерационных зон разнятся.
Кроме того, изобретение дает способ утилизации водорода, получаемого в качестве побочного продукта реакции дегидроциклизации и, в частности, способ превращения по меньшей мере части этого водорода в более ценную продукцию.
Исходный материал
В предлагаемом в изобретении способе можно использовать любой метансодержащий исходный материал, но в общем предлагаемый способ предусмотрен для использования в качестве исходного материала природного газа. К другим подходящим метансодержащим исходным материалам относятся материалы, получаемые из таких источников, как угольные пласты, места захоронения отходов, ферментация сельскохозяйственных или муниципальных отходов и/или газообразные потоки нефтеперерабабатывающих предприятий.
Содержащие метан исходные материалы, такие как природный газ, как правило, помимо метана содержат диоксид углерода и этан. Этан и другие алифатические углеводороды, которые могут содержаться в исходном материале, на стадии дегидроциклизации могут быть, разумеется, превращены в целевые ароматические продукты. Кроме того, как это обсуждается ниже, диоксид углерода также может быть превращен в полезные ароматические продукты либо непосредственно на стадии дегидроциклизации, либо опосредованно путем превращения в метан и/или этан на стадии снижения содержания водорода.
В используемых в предлагаемом в изобретении способе содержащих метан потоках также обычно присутствуют азот- и/или серосодержащие примеси, которые перед использованием этих потоков могут быть удалены или их количество может быть уменьшено до низких концентраций. В одном из вариантов осуществления изобретения исходный материал, подаваемый на стадию дегидроциклизации, содержит менее 100 част./млн, например менее 10 част./млн, в частности менее 1 част./млн, каждого из соединений азота и серы.
Для содействия уменьшению коксообразования в дополнение к метану в исходный материал, подаваемый на стадию дегидроциклизации, можно добавлять по меньшей мере одно из следующих веществ: водород, вода, моноксид углерода и диоксид углерода. Эти добавки можно вводить в виде отдельных совместно подаваемых потоков исходных материалов или они могут присутствовать в метановом потоке, например, в случае, когда метановый поток получают как производный от природного газа, содержащего диоксид углерода. К другим источникам диоксида углерода можно отнести дымовые газы, установки СПГ, водородные установки, аммиачные установки, гликольные установки и фталевоангидридные установки.
В одном варианте осуществления изобретения исходный материал, подаваемый на стадию дегидроциклизации, содержит диоксид углерода и включает в себя примерно от 90 мол.% примерно до 99,9 мол.%, в частности примерно от 97 мол.% примерно до 99 мол.% метана и примерно от 0,1 мол.% примерно до 10 мол.%, в частности примерно от 1 мол.% примерно до 3 мол.% СО2. В другом варианте осуществления изобретения исходный материал, подаваемый на стадию дегидроциклизации, содержит моноксид углерода и включает в себя примерно от 80 мол.% примерно до 99,9 мол.%, в частности примерно от 94 мол.% примерно до 99 мол.%, метана и примерно от 0,1 мол.% примерно до 20 мол.%, в частности примерно от 1 мол.% примерно до 6 мол.% СО. В еще одном варианте осуществления изобретения исходный материал, подаваемый на стадию дегидроциклизации, содержит водяной пар и включает в себя примерно от 90 мол.% примерно до 99,9 мол.%, в частности примерно от 97 мол.% примерно до 99 мол.% метана и примерно от 0,1 мол.% примерно до 10 мол.%, в частности примерно от 1 мол.% примерно до 5 мол.% водяного пара. И в еще одном варианте осуществления изобретения исходный материал, подаваемый на стадию дегидроциклизации, содержит водород и включает в себя примерно от 80 мол.% примерно до 99,9 мол.%, в частности примерно от 95 мол.% примерно до 99 мол.% метана и примерно от 0,1 мол.% примерно до 20 мол.%, в частности примерно от 1 мол.% примерно до 5 мол.% водорода.
Исходный материал, подаваемый на стадию дегидроциклизации, может также содержать более высокомолекулярные углеводороды, чем метан, включая ароматические углеводороды. Такие более высокомолекулярные углеводороды могут возвращаться в процесс со стадии снижения содержания водорода, добавляться в виде отдельных совместно подаваемых исходных материалов или могут присутствовать в метановом потоке, например в случае, когда в исходном материале в виде природного газа содержится этан. К более высокомолекулярным углеводородам, возвращаемым в процесс со стадии снижения содержания водорода, как правило, относятся моноциклические ароматические соединения и/или парафины и олефины, содержащие преимущественно 6 или менее, в частности 5 или менее, например 4 или менее, как правило, 3 или менее атома углерода. Обычно исходный материал, подаваемый на стадию дегидроциклизации, содержит менее 5 мас.%, в частности менее 3 мас.% углеводородов С3+.
Дегидроциклизация
На стадии дегидроциклизации предлагаемого в изобретении способа метансодержащий исходный материал вводят в контакт с зернистым катализатором дегидроциклизации в условиях, обычно - неокислительных, а как правило - восстановительных, эффективных для превращения метана в более высокомолекулярные углеводороды, включая бензол и нафталин. При этом основными результирующими реакциями являются:


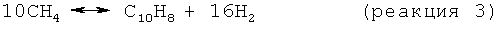
Моноксид и/или диоксид углерода, который может присутствовать в исходном материале, повышает активность и стабильность катализатора, содействуя протеканию реакций, таких как:

но негативно влияет на равновесие, позволяя протекать параллельным результирующим реакциям, таким как:
 .
.
В предлагаемом в изобретении способе может использоваться любой катализатор дегидроциклизации, эффективный для превращения метана в ароматические соединения, хотя обычно катализатор включает в себя металлический компонент, в частности переходный металл или его соединение, на неорганическом носителе. В подходящем варианте осуществления изобретения металлический компонент присутствует в количестве примерно от 0,1 мас.% примерно до 20 мас.%, в частности примерно от 1 мас.% примерно до 10 мас.%, по массе всего катализатора. Обычно металл присутствует в катализаторе в форме свободных элементов или в форме карбида.
К подходящим металлическим компонентам для катализатора относятся кальций, магний, барий, иттрий, лантан, скандий, церий, титан, цирконий, гафний, ванадий, ниобий, тантал, хром, молибден, вольфрам, марганец, рений, железо, рутений, кобальт, родий, иридий, никель, палладий, медь, серебро, золото, цинк, алюминий, галлий, кремний, германий, индий, олово, свинец, висмут и трансурановые металлы. Такие металлические компоненты могут присутствовать в форме свободных элементов или в виде соединений металлов, таких как оксиды, карбиды, нитриды и/или фосфиды, и их можно использовать самостоятельно или в сочетании. В качестве одного из металлических компонентов также могут использоваться платина и осмий, но обычно они не являются предпочтительными.
Неорганический носитель может быть либо аморфным, либо кристаллическим и может представлять собой, в частности, оксид, карбид или нитрид бора, алюминия, кремния, фосфора, титана, скандия, хрома, ванадия, магния, марганца, железа, цинка, галлия, германия, иттрия, циркония, ниобия, молибдена, индия, олова, бария, лантана, гафния, церия, тантала, вольфрама или других трансурановых элементов. Кроме того, носителем может быть пористый материал, такой как микропористый кристаллический материал или мезопористый материал. В контексте настоящего описания понятие "микропористый" относится к порам диаметром менее 2 нм, тогда как понятие "мезопористый" относится к порам диаметром от 2 до 50 нм.
К подходящим микропористым кристаллическим материалам относятся силикаты, алюмосиликаты, титаносиликаты, алюмофосфаты, металлофосфаты, кремнеалюмофосфаты или их смеси. Такие микропористые кристаллические материалы включают в себя материалы с каркасами типов MFI (например, ZSM-5 и силикалит), MEL (например, ZSM-11), MTW (например, ZSM-12), TON (например, ZSM-22), МТТ (например, ZSM-23), FER (например, ZSM-35), MFS (например, ZSM-57), MWW (например, МСМ-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, МСМ-36, МСМ-49 и МСМ-56), IWR (например, ITQ-24), KFI (например, ZK-5), ВЕА (например, цеолит бета), ITH (например, ITQ-13), MOR (например, морденит), FAU (например, цеолиты X, Y, ультрастабилизированный Y и деалюминированный Y), LTL (например, цеолит L), IWW (например, ITQ-22), VFI (например, VPI-5), AEL (например, SAPO-11), AFI (например, ALPO-5) и AFO (SAPO-41), а также такие материалы, как МСМ-68, EMM-1, EMM-2, ITQ-23, ITQ-24, ITQ-25, ITQ-26, ETS-2, ETS-10, SAPO-17, SAPO-34 и SAPO-35. К подходящим мезопористым материалам относятся МСМ-41, МСМ-48, МСМ-50, FSM-16 и SBA-15.
Примеры предпочтительных катализаторов включают в себя молибден, вольфрам, цинк, рений, а также их соединения и сочетания на ZSM-5, диоксиде кремния или оксиде алюминия.
Металлический компонент может быть диспергирован на неорганическом носителе любыми средствами, хорошо известными в данной области техники, такими как соосаждение, пропитка по влагоемкости, выпаривание, обычная пропитка, распылительная сушка, золь-гелевое, ионообменное, химическое парофазное осаждение, диффузионное и физическое смешение. Кроме того, неорганический носитель может быть модифицирован по известным методам, таким, например, как обработка водяным паром, кислотная промывка, промывка каустической содой и/или обработка кремнийсодержащими соединениями, фосфорсодержащими соединениями и/или элементами или соединениями элементов групп 1, 2, 3 и 13 Периодической таблицы элементов. Такие модификации можно использовать для изменения поверхностной активности носителя, а также для затруднения или улучшения доступа к любой внутренней пористой структуре носителя.
В некоторых вариантах осуществления изобретения в реакцию дегидроциклизации, в дополнение к каталитическому зернистому материалу, может вводиться некаталитический зернистый материал. Некаталитический зернистый материал можно использовать в качестве материала для транспорта энергии (тепла) в систему и/или для заполнения пространства, в зависимости от потребности обеспечения требуемых гидродинамических условий. Некаталитический зернистый материал может образовывать макрочастицы без связующего вещества или частицы могут быть связаны неорганическим связующим веществом, таким как глина, диоксид кремния, оксид алюминия, диоксид циркония или другой оксид металла, используемый для содействия сохранению физической целостности частиц. В предпочтительном варианте частицы обладают по существу сферической формой. Примерами приемлемого некаталитического зернистого материала служат диоксид кремния, оксид алюминия, керамика и карбид кремния с малой удельной площадью поверхности.
Стадию дегидроциклизации осуществляют введением метансодержащего исходного материала в контакт с катализатором дегидроциклизации в реакционных зонах с одним или несколькими неподвижными слоями, подвижными слоями или псевдоожиженными слоями. Обычно исходный материал в реакционной зоне или каждой реакционной зоне вводят в контакт с подвижным слоем катализатора дегидроциклизации, где исходный материал движется в противоток направлению движения катализатора дегидроциклизации. В одном варианте осуществления изобретения реакционная зона включает в себя реактор с оседающим слоем, под которым подразумевают вертикально расположенный реактор, в котором зернистый катализатор поступает в верхней части реактора или вблизи нее и движется под действием собственного веса с образованием слоя катализатора, в то время как исходный материал поступает в реактор в основании реактора или вблизи него и движется вверх через слой катализатора.
В варианте осуществления изобретения с использованием оседающего слоя движение катализатора дегидроциклизации в реакционной зоне практически свободно от псевдоожижения. В контексте настоящего описания понятие "практически свободно от псевдоожижения" означает, что средняя скорость движения потока газа в реакторе ниже минимальной скорости псевдоожижения. Понятие "практически свободно от псевдоожижения" в контексте настоящего описания также означает, что средняя скорость движения потока газа в реакторе меньше 99%, в частности меньше 95%, как правило, меньше 90%, даже меньше 80%, минимальной скорости псевдоожижения. Когда реакционная зона или каждая реакционная зона работает с оседающим слоем, зернистый каталитический материал и/или любой зернистый некаталитический материал характеризуется средним размером частиц, составляющим примерно от 0,1 мм примерно до 100 мм, в частности примерно от 1 мм примерно до 5 мм, например примерно от 2 мм примерно до 4 мм. В некоторых вариантах осуществления изобретения по меньшей мере 90 мас.% зернистого каталитического материала и/или по меньшей мере 90 мас.% зернистого некаталитического материала имеет размер частиц, составляющий примерно от 0,1 мм примерно до 100 мм, в частности примерно от 1 мм примерно до 5 мм, например примерно от 2 мм примерно до 4 мм.
В альтернативном варианте реакцию дегидроциклизации проводят в нескольких последовательно соединенных реакторах с псевдоожиженным слоем, в которых зернистый катализатор каскадирует в одном направлении из одного реактора в следующий смежный реактор в этом ряду, в то время как исходный материал пропускают в противоположном направлении через реакторы и между ними. Когда каждая реакционная зона работает с псевдоожиженным слоем, каталитический зернистый материал и/или любой некаталитический зернистый материал характеризуется средним размером частиц, составляющим примерно от 0,01 мм примерно до 10 мм, в частности примерно от 0,05 мм примерно до 1 мм и в частности примерно от 0,1 мм примерно до 0,6 мм. В некоторых вариантах осуществления изобретения по меньшей мере 90 мас.% каталитического зернистого материала и/или по меньшей мере 90 мас.% некаталитического зернистого материала имеет размер частиц, составляющий примерно от 0,01 мм примерно до 10 мм, в частности примерно от 0,05 мм примерно до 1 мм, например примерно от 0,1 мм примерно до 0,6 мм.
Как правило, отношение массового расхода каталитического зернистого материала плюс любой некаталитический зернистый материал к массовому расходу углеводородного исходного материала в зоне реакции дегидроциклизации или в каждой зоне реакции дегидроциклизации составляет примерно от 1:1 примерно до 100:1, в частности примерно от 1:1 примерно до 40:1, в частности примерно от 5:1 до 20:1.
Реакция дегидроциклизации является эндотермической, а значит по мере протекания этой реакции температура в каждой зоне реакции дегидроциклизации обычно проявляет тенденцию к понижению от максимальной температуры до минимальной температуры. Приемлемые условия для стадии дегидроциклизации включают в себя максимальную температуру примерно от 700°С примерно до 1200°С, в частности примерно от 800°С примерно до 950°С, и минимальную температуру примерно от 400°С примерно до 800°С, в частности примерно от 500°С примерно до 700°С. Однако, как обсуждается ниже, для уменьшения падения температуры во время реакции дегидроциклизации в эту реакцию подводят тепло, а значит в некоторых конфигурациях может оказаться возможным уменьшение разницы между максимальной и минимальной температурами по существу до нуля. В другом варианте осуществления изобретения, вводя в реакцию дегидроциклизации нагретый катализатор, можно создавать обратный температурный профиль, т.е. температура реакции на выходе для технологического газа будет выше температуры на входе для технологического газа.
В одном варианте осуществления изобретения для создания обратного температурного профиля в системе для проведения реакции дегидроциклизации движение исходного материала и зернистого катализатора дегидроциклизации организуют в противотоке, благодаря чему, несмотря на эндотермическую природу реакции дегидроциклизации, разница между температурой реакции для газообразного отходящего потока на выходе из системы для проведения реакции дегидроциклизации и температурой реакции для метансодержащего исходного материала на входе в систему для проведения реакции дегидроциклизации составляет по меньшей мере +10°С, в частности по меньшей мере +50°С, например по меньшей мере +100°С и даже по меньшей мере +150°С.
В любом случае, поскольку реакция дегидроциклизации является эндотермической, зернистый каталитический материал поступает в систему для проведения реакции дегидроциклизации при первой, высокой температуре, как правило, примерно от 800°С примерно до 1200°С, в частности примерно от 900°С примерно до 1100°С, а выходит из реакционной системы при второй, более низкой температуре, как правило, примерно от 500°С примерно до 800°С, в частности примерно от 600°С примерно до 700°С.Общая разность температур зернистого каталитического материала после прохождения через реакционные зоны составляет по меньшей мере 100°С.
Другие условия, создаваемые в реакции дегидроциклизации, обычно включают в себя давление, составляющее примерно от 1 кПа примерно до 1000 кПа, в частности примерно от 10 кПа примерно до 500 кПа, например примерно от 50 кПа примерно до 200 кПа, и объемную скорость подачи газа, составляющую примерно от 0,01 ч-1 примерно до 1000 ч-1, в частности примерно от 0,1 ч-1 примерно до 500 ч-1, в частности примерно от 1 ч-1 примерно до 20 ч-1. В подходящем варианте осуществления изобретения стадию дегидроциклизации осуществляют в отсутствие О2.
Основными компонентами потока, отходящего со стадии дегидроциклизации, являются водород, бензол, нафталин, моноксид углерода, этилен и непрореагировавший метан. Этот отходящий поток, как правило, содержит по меньшей мере на 5 мас.%, в частности по меньшей мере на 10 мас.%, например по меньшей мере на 20 мас.%, предпочтительно по меньшей мере на 30 мас.%, больше ароматических колец, чем исходный материал.
Бензол и нафталин выделяют из потока, отходящего со стадии дегидроциклизации, например, экстракцией растворителем с последующим разделением на фракции, и эти вещества могут получить в потоке продукции. Однако, как это обсуждается ниже, до или после извлечения продукции по меньшей мере часть этих ароматических компонентов может быть подвергнута алкилированию с получением более ценных материалов, таких как ксилолы. Более того, как обсуждается ниже, предлагаемый в изобретении способ предусматривает утилизацию водорода, образующегося в качестве побочного продукта реакции дегидроциклизации, в частности превращение по меньшей мере части водорода в более ценную продукцию.
Регенерирование катализатора
Реакция дегидроциклизации характеризуется тенденцией к отложению кокса на катализаторе, а значит для сохранения активности катализатора дегидроциклизации по меньшей мере часть катализатора необходимо регенерировать непрерывно или периодически. Обычно этого достигают отводом из реакционной зоны или каждой реакционной зоны части катализатора, осуществляемым на периодической или на непрерывной основе, и перемещением этой части катализатора в отдельную регенерационную зону. В регенерационной зоне закоксованный катализатор дегидроциклизации вводят в контакт с водородсодержащим газом в условиях, эффективных для превращения по меньшей мере части отложившегося на нем углеродистого материала в метан. Обычно водородсодержащий газ не содержит значительных количеств метана или других углеводородов; содержание углеводородов, как правило, составляет меньше 20 мол.%, в частности менее 18 мол.%, менее 15 мол.%, менее 10 мол.%, менее 8 мол.%, менее 6 мол.%, менее 4 мол.% или менее 2 мол.%. В одном варианте осуществления изобретения водород, необходимый для регенерирования, получают, по меньшей мере частично, из водородсодержащего потока, отходящего из реакции дегидроциклизации.
В подходящем варианте осуществления изобретения условия регенерирования включают в себя температуру, составляющую примерно от 700°С примерно до 1200°С, в частности примерно от 800°С примерно до 1000°С, в частности примерно от 850°С примерно до 950°С, и абсолютное давление, составляющее по меньшей мере 100 кПа, в частности примерно от 150 кПа примерно до 5000 кПа. Обычно же закоксованный катализатор дегидроциклизации, удаляемый из реакционной зоны или из каждой реакционной зоны, характеризуется более низкой температурой, чем та, что оптимальна для регенерирования, а значит удаляемый катализатор вначале нагревают до требуемой температуры регенерирования прямым или косвенным (опосредованным) контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива. Нагревание проводят в нагревательной зоне, которая может находиться в том же сосуде, что и регенерационная зона, или которая может находиться в сосуде, отдельном от регенерационной зоны.
Под "дополнительным источником топлива" имеется в виду то, что источник топлива физически отделен от катализатора, а значит не является коксом, образующимся на катализаторе в качестве побочного продукта реакции дегидроциклизации. Как правило, дополнительный источник топлива включает в себя углеводород, такой как метан, в частности, приемлемый источник топлива представляет собой природный газ, используемый в качестве исходного материала, направляемого в процесс. В подходящем варианте осуществления изобретения в нагревательной зоне поддерживают бедную кислородом атмосферу, благодаря чему при сжигании углеводородного топлива для нагрева первой части катализатора образуется синтез-газ, который затем может быть использован для получения дополнительного углеводородного продукта и/или топлива. Кроме того, в случае прямого теплопереноса к катализатору дегидроциклизации применение бедной кислородом атмосферы препятствует окислению карбидов металлов, присутствующих в катализаторе, и сводит к минимуму среднее парциальное давление водяного пара, тем самым уменьшая гидротермическое старение катализатора.
В другом варианте осуществления изобретения подходящим дополнительным источником топлива является водород, в частности часть водорода, образующегося в качестве побочного продукта реакции ароматизации.
В случае если катализатор дегидроциклизации нагревают прямым путем, или непосредственно, закоксованный катализатор, отводимый из реакционной зоны, целесообразно вводить в контакт прямо с горящим источником топлива в нагревательной зоне. В другом варианте осуществления изобретения источник топлива сжигают в отдельной зоне горения, и газообразные продукты горения, образующиеся в зоне горения, направляют в нагревательную зону для нагрева катализатора. В еще одном варианте осуществления изобретения катализатор дегидроциклизации может нагреваться непрямым теплообменом, например, с использованием газообразных продуктов горения для нагрева инертной среды (газа, жидкости или твердого вещества) или поверхности теплопередачи и последующим контактированием закоксованного катализатора с нагретой инертной средой или поверхностью теплопередачи.
В одном практическом варианте осуществления изобретения нагревательная зона является удлиненной, а закоксованный катализатор пропускают через нагревательную зону от входа, расположенного у одного конца нагревательной зоны или вблизи него, к выходу, расположенному у другого конца нагревательной зоны или вблизи него, причем подвод тепла к первой части катализатора осуществляется во множестве мест, распределенных по длине нагревательной зоны. Таким образом можно распределить подвод тепла к катализатору по длине нагревательной зоны, сведя тем самым к минимуму температуры на поверхности катализатора и внутренние градиенты.
В случае если первую часть катализатора нагревают прямым контактированием с горящим источником топлива в нагревательной зоне, постепенного нагрева катализатора можно достичь, подавая по существу все дополнительное топливо во входной конец нагревательной зоны и затем подавая в эту нагревательную зону кислородсодержащий газ с определенным шагом в указанном множестве мест, распределенных по длине нагревательной зоны. В другом варианте осуществления изобретения по существу весь кислородсодержащий газ, необходимый для сжигания дополнительного топлива, можно подавать во входной конец нагревательной зоны, а дополнительное топливо - с определенным шагом в указанном множестве мест, распределенных по длине нагревательной зоны.
В случае если первую часть катализатора нагревают прямым контактированием с горячими газообразными продуктами горения, образующимися в отдельной зоне горения, постепенного нагрева катализатора можно достичь подачей горячих газообразных продуктов горения в указанное множество мест, распределенных по длине нагревательной зоны.
В одном варианте осуществления изобретения нагревательная зона представляет собой вертикальную трубу, и во время стадии повторного нагревания первую часть катализатора пропускают вверх по вертикальной трубе. На практике нагревательная зона может включать несколько вертикальных труб, соединенных параллельно. В другом варианте осуществления изобретения нагревательная зона может включать в себя подвижный слой указанного катализатора.
В одном варианте осуществления изобретения закоксованный катализатор дегидроциклизации, удаляемый из реакционной зоны, разделяют по меньшей мере на две части, которые нагревают так, как изложено выше, и затем направляют в отдельные регенерационные зоны, работающие под разными давлениями. Так, например, одна регенерационная зона работает при абсолютном давлении, составляющем по меньшей мере 100 кПа, в частности примерно от 150 кПа примерно до 700 кПа, как изложено выше, а другая регенерационная зона работает при абсолютном давлении, составляющем по меньшей мере 500 кПа, в частности примерно от 1000 кПа примерно до 5000 кПа. Так, было установлено, как продемонстрировано в примерах, что регенерирование при более высоких давлениях обеспечивает более быстрое удаление кокса, а также удаление более огнеупорного кокса. Вместе с тем, также было установлено, что для удаления кокса при более высоких давлениях требуется более дорогостоящее оборудование. По этой причине может быть целесообразным часть кокса удалять при более низком парциальном давлении водорода на менее дорогостоящем оборудовании, а еще одну часть кокса - при более высоком парциальном давлении водорода на более дорогостоящем оборудовании.
Регенерационная зона или каждая регенерационная зона может представлять собой реактор, работающий как реактор с псевдоожиженным слоем, кипящим слоем, оседающим слоем, лифт-реактор или их сочетание. На практике каждая регенерационная зона может включать в себя несколько реакторов, в частности несколько лифт-реакторов, соединенных параллельно, или несколько реакторов, соединенных последовательно, в частности лифт-реактор и следующий за ним реактор с оседающим слоем. После регенерирования катализатор возвращают в реакционную зону.
В другом варианте осуществления изобретения, в частности, если реакцию дегидроциклизации проводят в реакторе с неподвижным слоем, регенерирование можно проводить без извлечения катализатора из реакционной зоны, временно прерывая подачу метансодержащего исходного материала в реакционную зону, нагревая реакционную зону до температуры регенерирования, составляющей примерно от 700°С примерно до 1200°С, прямым и/или непрямым контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива, регенерируя зернистый каталитический материал водородсодержащим газом и затем возобновляя подачу метансодержащего исходного материала в реакционную зону. Разумеется, что нагревание реакционной зоны до температуры регенерирования можно осуществлять перед прерыванием подачи метансодержащего исходного материала.
Повторный нагрев катализатора
Поскольку реакция дегидроциклизации является эндотермической, в реакцию необходимо подводить тепло. В предлагаемом в изобретении способе это удобно достигается отводом части катализатора из реакционной зоны либо на периодической, либо на непрерывной основе, подводом тепла к катализатору и последующим возвратом нагретого катализатора назад в реакционную зону. Поскольку стадия регенерирования водородом, описанная выше, также предполагает нагревание катализатора и последующий возврат нагретого регенерированного катализатора обратно в реакционную зону, один возможный путь подвода тепла в реакцию дегидроциклизации заключается в процессе регенерирования.
В другом варианте осуществления изобретения часть тепла или все тепло, необходимое для поддержания реакции дегидроциклизации, может быть обеспечено отдельной стадией повторного нагрева катализатора. В этом варианте часть катализатора, отводимую для реакционной зоны, перемещают в отдельную нагревательную зону, где катализатор вновь нагревают прямым или непрямым контактированием с горячими газообразными продуктами горения, образующимися при сжигании дополнительного источника топлива. Затем нагретый катализатор возвращают в реакционную зону с регенерированием водородом или без него.
Повторное науглероживание катализатора
Необходимо иметь в виду, что нагревание катализатора дегидроциклизации для целей регенерирования и/или для переноса тепла обратно в реакцию дегидроциклизации может подвергнуть катализатор воздействию высокотемпературных окислительных условий, особенно если нагрев катализатора предполагает прямой контакт с горячими газообразными продуктами горения. В результате присутствующие в катализаторе дегидроциклизации металлы, такие как рений, вольфрам или молибден, во время стадии нагрева могут перейти из их каталитически активной элементной или карбидной формы в оксидные вещества. Таким образом, перед возвратом в реакционную зону регенерированный и/или повторно нагретый катализатор может быть перемещен в зону обработки катализатора, отдельную от регенерационной зоны, нагревательной зоны и реакционной зоны, где катализатор входит в контакт с науглероживающим газом, содержащим по меньшей мере один углеводород, выбранный из метана, этана, пропана, бутана, изобутена, гексана, бензола и нафталина. В некоторых случаях науглероживающий газ может также содержать по меньшей мере одно из следующих веществ: СО2, СО, H2, Н2О и инертные разбавители. В другом варианте осуществления изобретения науглероживающий газ может представлять собой смесь водорода и по меньшей мере одного из СО и СО2. Более того, может возникнуть необходимость введения катализатора в контакт последовательно с множеством разных науглероживающих газов, каждый из которых включает в себя углеводород, выбранный из метана, этана, пропана, бутана, изобутена, гексана, бензола и нафталина, или смесь водорода и по меньшей мере одного из СО и СО2.
Обычно максимальная температура в зоне обработки катализатора составляет примерно от 400°С примерно до 1100°С, в частности примерно от 500°С примерно до 900°С, причем минимальная температура находится в пределах от 300°С до 500°С. Как правило, зона обработки катализатора работает при абсолютных давлениях, составляющих от 10 до 100 фунтов/кв.дюйм (от 69 до 690 кПа), в частности в пределах от 15 до 60 фунтов/кв.дюйм (от 103 до 414 кПа). Обычно средняя продолжительность пребывания каталитических частиц в зоне обработки катализатора находится в пределах от 0,1 до 100 мин, например в пределах от 1 до 20 мин. В этих условиях науглероживающий газ взаимодействует с металлоксидными материалами на поверхности катализатора, возвращая металл в его каталитически активную элементную или карбидную форму. Кроме того, науглероживающий газ способен взаимодействовать с активными центрами поверхности на носителе катализатора, уменьшая их склонность к образованию кокса в зоне реакции дегидроароматизации.
Для поддержания температуры, необходимой для науглероживания регенерированного катализатора, тепло можно подводить к катализатору и/или науглероживающему газу перед стадией науглероживания или во время ее проведения. Так, например, тепло к катализатору может подводиться косвенным нагревом, введением в контакт с горячими газами, отходящими из реакционной зоны или нагревательной зоны, введением в контакт с горячим газообразным потоком, отходящим из процесса науглероживания, или смешением с нагретым катализатором из нагревательной зоны. Тепло удобно подводить к науглероживающему газу с помощью наружной топки или теплообменника либо посредством нагретого катализатора из нагревательной зоны.
Зона обработки катализатора может работать как реактор с псевдоожиженным слоем, реактор с кипящим слоем, реактор с оседающим слоем, лифт-реактор или циркуляционный лифт-реактор. В одном варианте зона обработки катализатора включает в себя реактор с оседающим слоем. В другом варианте осуществления изобретения зона обработки катализатора включает в себя единственный реактор с псевдоожиженным слоем, снабженный внутренними отбойными заслонками для предотвращения обратного перемешивания, или несколько реакторов с псевдоожиженным слоем, соединенных последовательно, причем регенерированный катализатор каскадирует между смежными реакторами. В любом случае контакту катализатора с газом в зоне обработки катализатора способствуют путем организации движения регенерированного катализатора и науглероживающего газа в зоне обработки катализатора в противоположных направлениях. С применением такого противоточного движения в зоне обработки катализатора может сформироваться температурный профиль, благодаря которому науглероживание регенерированного катализатора первоначально происходит при низкой температуре, но по мере продвижения катализатора через слой температура наглероживания повышается.
В некоторых случаях может быть желательно до стадии науглероживания сначала вводить нагретый нерегенерированный катализатор в контакт с богатым Н2 потоком для частичного или полного восстановления металлического компонента катализатора. Также может быть желательным подвергать науглероженный катализатор последующей обработке водородом и/или углекислым газом для удаления любого избытка углерода, который мог отложиться на катализаторе на стадии науглероживания.
На практике, по мере протекания реакции дегидроциклизации в процесс добавляют свежий катализатор дегидроциклизации для возмещения потери катализатора вследствие механического износа или дезактивации, и несмотря на существование множества средств добавления свежего катализатора, во избежание разрушения катализатора свежий катализатор обычно желательно добавлять в область процесса, работающую при температуре ниже максимальной температуры в каждой зоне реакции дегидроциклизации. В одном варианте осуществления изобретения свежий катализатор дегидроциклизации добавляют в процесс введением в зону обработки катализатора, благодаря чему свежий катализатор вводят в контакт с науглероживающим газом перед перемещением в реакционную зону для введения в контакт с метансодержащим исходным материалом. В другом варианте осуществления изобретения катализатор можно добавлять в области более низкой температуры реакторной системы с обратным температурным профилем.
Управление содержанием водорода
Поскольку водород является одним из основных компонентов отходящего со стадии дегидроциклизации потока, этот отходящий поток после извлечения ароматических продуктов может подвергаться обработке для снижения содержания водорода в отходящем потоке перед возвратом непрореагировавшего метана на стадию дегидроциклизации для достижения максимальной утилизации исходного материала. Стадия снижения содержания водорода, как правило, включает в себя взаимодействие по меньшей мере части водорода, содержащегося в отходящем со стадии дегидроциклизации потоке, с кислородсодержащими материалами, такими как СО и/или СО2, с получением воды и второго отходящего потока, обладающего более низким содержанием водорода в сравнении с первым отходящим (со стадии дегидроциклизации) потоком. Подходящие способы снижения содержания водорода описаны ниже и в международной заявке PCT/US2005/044042 (публикация WO/2006/068814), поданной авторами настоящего изобретения 2 декабря 2005 г.
В подходящем варианте стадия снижения содержания водорода включает в себя (I) метанирование и/или этанирование, (II) процесс Фишера-Тропша, (III) синтез спиртов С1-С3, в частности метанола, и других оксигенатов, (IV) синтез легких олефинов, парафинов и/или ароматических соединений посредством метанола или диметилового эфира как промежуточного продукта и/или (V) селективное сжигание водорода. Для получения наибольшего выигрыша эти стадии можно осуществлять последовательно; например, вначале может быть проведен процесс Фишера-Тропша с получением потока, обогащенного углеводородами С2+, с последующим метанированием для достижения высокой степени превращения Н2.
Обычно на стадии снижения содержания водорода образуются углеводороды, как это описано ниже, причем в этом случае после выделения одновременно получаемой воды по меньшей мере часть углеводородов целесообразно возвращать на стадию дегидроциклизации. Например, если углеводороды, получаемые на стадии снижения содержания водорода, включают в себя парафины и олефины, часть углеводородов, возвращаемая на стадию дегидроциклизации, обычно включает в себя парафины или олефины с шестью или менее атомами углерода, в частности с пятью или менее атомами углерода, например с четырьмя или менее атомами углерода или с тремя или менее атомами углерода. Если углеводороды, получаемые на стадии снижения содержания водорода, включают в себя ароматические соединения, целесообразно, чтобы часть углеводородов, возвращаемая на стадию дегидроциклизации, включала в себя моноциклические ароматические вещества.
Метанирование/этанирование
В одном варианте осуществления изобретения стадия снижения содержания водорода включает в себя реакцию по меньшей мере части водорода, присутствующего в отходящем со стадии дегидроциклизации потоке, с диоксидом углерода с образованием метана и/или этана в соответствии со следующими результирующими реакциями:
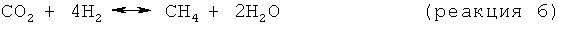
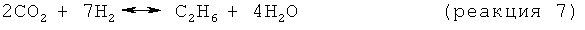
В целесообразном варианте осуществления изобретения используемый диоксид углерода является частью потока природного газа, как правило, того же потока природного газа, что используется в качестве исходного материала, подаваемого на стадию дегидроциклизации. Когда диоксид углерода является частью метансодержащего потока, отношение CO2:СН4 в этом потоке целесообразно поддерживать в пределах примерно от 1:1 примерно до 0,1:1. Смешение содержащего диоксид углерода потока и отходящего со стадии дегидроциклизации потока целесообразно обеспечивать подачей газообразных исходных материалов на вход струйного насоса.
На стадии снижения содержания водорода с получением метана или этана, как правило, используется молярное отношение Н2:СО2, близкое к стехиометрическим пропорциям, требуемым для целевой реакции 6 или реакции 7, хотя, если необходимо получить содержащий СО2 или содержащий Н2 второй отходящий поток, в это стехиометрическое отношение могут быть внесены небольшие изменения. Стадию снижения содержания водорода с получением метана или этана целесообразно осуществлять в присутствии бифункционального катализатора, содержащего металлический компонент, в частности переходный металл или его соединение, на неорганическом носителе. К подходящим металлическим компонентам относятся медь, железо, ванадий, хром, цинк, галлий, никель, кобальт, молибден, рутений, родий, палладий, серебро, рений, вольфрам, иридий, платина, золото, галлий и их сочетания и соединения. Неорганическим носителем может быть аморфный материал, такой как диоксид кремния, оксид алюминия или диоксид кремния/оксид алюминия, или подобный тем, которые перечислены для катализатора дегидроароматизации. Кроме того, неорганическим носителем может быть кристаллический материал, такой как микропористый или мезопористый кристаллический материал. К подходящим пористым кристаллическим материалам относятся алюмосиликаты, алюмофосфаты и кремнеалюмофосфаты, перечисленные выше для катализатора дегидроциклизации.
Стадия снижения содержания водорода с получением метана и/или этана может проводиться в широком диапазоне условий, включая температуру примерно от 100°С примерно до 900°С, в частности примерно от 150°С примерно до 500°С, например примерно от 200°С примерно до 400°С, давление примерно от 200 кПа примерно до 20000 кПа, в частности примерно от 500 кПа примерно до 5000 кПа, и объемную скорость подачи газа примерно от 0,1 ч-1 примерно до 10000 ч-1, в частности примерно от 1 ч-1 примерно до 1000 ч-1. Значения степени превращения СО2, как правило, находятся в пределах от 20 до 100%, а предпочтительно более 90%, в частности более 99%. Эту экзотермическую реакцию можно проводить в нескольких слоях катализатора с отводом тепла между слоями. Кроме того, для получения максимально возможных кинетических скоростей, процесс в первом (первых) по потоку слое (слоях) можно проводить при более высоких температурах, а для получения максимально возможного термодинамического превращения, процесс в последнем (последних) по потоку слое (слоях) можно проводить при более низких температурах.
Основными продуктами такой реакции являются вода и, в зависимости от молярного отношения Н2:CO2, метан, этан и высшие алканы совместно с некоторыми ненасыщенными углеводородами с двумя и более атомами углерода. Кроме того, предпочтительна некоторая частичная гидрогенизация диоксида углерода до моноксида углерода. После удаления воды метан, моноксид углерода, любой непрореагировавший диоксид углерода и более высокомолекулярные углеводороды можно направлять непосредственно на стадию дегидроциклизации для получения дополнительных ароматических продуктов.
Процесс Фишера-Тропша
В еще одном варианте осуществления изобретения стадия снижения содержания водорода включает в себя реакцию по меньшей мере части водорода, присутствующего в отходящем со стадии дегидроциклизации потоке, с моноксидом углерода по методу Фишера-Тропша с получением парафинов и олефинов С2-С5.
Процесс Фишера-Тропша хорошо известен в данной области техники (см., например, патенты US 5348982 и US 5545674, содержание которых включено в настоящее описание путем ссылки). Этот процесс, как правило, включает в себя реакцию водорода и моноксида углерода в молярном отношении примерно от 0,5:1 примерно до 4:1, в частности примерно от 1,5:1 примерно до 2,5:1, при температуре примерно от 175°С примерно до 400°С, в частности примерно от 180°С примерно до 240°С, и при давлении примерно от 1 бар примерно до 100 бар (от 100 до 10000 кПа), в частности примерно от 10 бар примерно до 40 бар (от 1000 до 4000 кПа), в присутствии катализатора Фишера-Тропша, обычно нанесенного на носитель или используемого без подложки элемента группы VIII, неблагородного металла, например Fe, Ni, Ru, Co, с промотором или без него, например с рутением, рением, гафнием, цирконием, титаном. Носителями, когда их используют, могут служить огнеупорные оксиды металлов, таких как металлы группы IVB, т.е. диоксид титана, диоксид циркония или диоксид кремния, оксид алюминия или диоксид кремния/оксид алюминия. В одном варианте осуществления изобретения катализатор включает в себя не вызывающий конверсии катализатор, например кобальт или рутений, в частности кобальт, с рением или цирконием в качестве промотора, в частности кобальт и рений, нанесенные на диоксид кремния или диоксид титана, обычно на диоксид титана.
В еще одном варианте осуществления изобретения катализатор синтеза углеводородов включает в себя металл, такой как Сu, Cu/Zn и Cr/Zn, на носителе ZSM-5, и процесс ведут с получением значительных количеств моноциклических ароматических углеводородов. Пример такого процесса описан в работе Jose Erena Study of Physical Mixtures of Cr2O3-ZnO and ZSM-5 Catalysts for the Transformation of Syngas into Liquid Hydrocarbons; Ind. Eng. Chem Res. 1998, 37, 1211-1219, содержание которой включено в настоящее описание путем ссылки.
Выделяют жидкости Фишера-Тропша, т.е. С5+, и от более тяжелых углеводородов отделяют легкие газы, например непрореагировавшие водород и СО, углеводороды С1-С3 или С4 и воду. Затем более тяжелые углеводороды могут быть выделены как продукты или направлены на стадию дегидроциклизации для получения дополнительных ароматических продуктов.
Присутствие моноксида углерода, необходимого для реакции Фишера-Тропша, можно полностью или частично обеспечить за счет моноксида углерода, присутствующего в метансодержащем исходном материале или подаваемого совместно с ним и образовавшегося в качестве побочного продукта на стадии дегидроциклизации. При необходимости можно генерировать дополнительное количество моноксида углерода, подавая диоксид углерода, содержащийся, например, в природном газе, к катализатору конверсии, в результате чего при обратной реакции конверсии водяного газа получают моноксид углерода:

и следующей реакцией:
СН4+H2O↔СО+3Н2
В еще одном варианте осуществления изобретения стадия снижения содержания водорода включает в себя реакцию по меньшей мере части водорода, присутствующего в отходящем со стадии дегидроциклизации потоке, с моноксидом углерода с получением спиртов С1-С3, в частности метанола. Получение метанола и других оксигенатов из синтез-газа также хорошо известно и описано, например, в патентах US 6114279, US 6054497, US 5767039, US 5045520, US 5254520, US 5610202, US 4666945, US 4455394. US 4565803, US 5385949, содержание которых включено в настоящее описание путем ссылки. Используемый синтез-газ, как правило, обладает молярным отношением водорода (Н2) к оксидам углерода (СО+СО2) в интервале примерно от 0,5:1 примерно до 20:1, в частности в интервале примерно от 2:1 примерно до 10:1, причем диоксид углерода необязательно присутствует в количестве не более 50 мас.% по общей массе синтез-газа.
Катализатор, используемый в процессе синтеза метанола, обычно включает в себя оксид по меньшей мере одного элемента, выбранного из группы, включающей медь, серебро, цинк, бор, магний, алюминий, ванадий, хром, марганец, галлий, палладий, осмий и цирконий. В целесообразном варианте осуществления изобретения катализатор представляет собой катализатор на основе меди, например в форме оксида меди, необязательно в присутствии оксида по меньшей мере одного элемента, выбранного из серебра, цинка, бора, магния, алюминия, ванадия, хрома, марганца, галлия, палладия, осмия и циркония. В целесообразном варианте осуществления изобретения катализатор содержит оксид меди и оксид по меньшей мере одного элемента, выбранного из цинка, магния, алюминия, хрома и циркония. В одном варианте осуществления изобретения катализатор синтеза метанола выбирают из группы, состоящей из оксидов меди, оксидов цинка и оксидов алюминия. В более предпочтительном варианте осуществления изобретения катализатор содержит оксиды меди и цинка.
Процесс синтеза метанола можно проводить в широком интервале температур и давлений. Приемлемые температуры находятся в интервале примерно от 150°С примерно до 450°С, в частности примерно от 175°С примерно до 350°С, например примерно от 200°С примерно до 300°С. Приемлемые давления находятся в интервале примерно от 1500 кПа примерно до 12500 кПа, в частности примерно от 2000 кПа примерно до 10000 кПа, в частности примерно от 2500 кПа примерно до 7500 кПа. Объемные скорости подачи газа колеблются в зависимости от типа проводимого процесса, но обычно объемная скорость подачи газа при пропускании газа через слой катализатора находится в интервале примерно от 50 ч-1 примерно до 50000 ч-1, в частности примерно от 250 ч-1 примерно до 25000 ч-1, в частности примерно от 500 ч-1 примерно до 10000 ч-1. Эту экзотермическую реакцию можно проводить либо в неподвижных, либо в псевдоожиженных слоях, включающих несколько слоев катализатора, с отводом тепла между слоями. Кроме того, для получения максимально возможных кинетических скоростей, процесс в первом (первых) по потоку слое (слоях) можно проводить при более высоких температурах, а для получения максимально возможного термодинамического превращения, процесс в последнем (последних) по потоку слое (слоях) можно проводить при более низких температурах.
Получаемый метанол и/или другие оксигенаты могут направляться в продажу как самостоятельный продукт, могут использоваться для алкилирования ароматических соединений, образующихся на стадии дегидроциклизации, до более ценных продуктов, таких как ксилолы, или могут использоваться в качестве исходного материала для получения более низкомолекулярных олефинов, в частности этилена и пропилена. Превращение метанола в олефины является хорошо известным процессом, который описан, например, в патенте US 4499327, содержание которого включено в настоящее описание путем ссылки.
Селективное сжигание водорода
В еще одном варианте осуществления изобретения стадия снижения содержания водорода включает в себя селективное сжигание водорода, которое представляет собой процесс, в котором водород в смешанном потоке взаимодействует с кислородом с образованием воды или водяного пара без существенного взаимодействия в потоке углеводородов с кислородом с образованием моноксида углерода, диоксида углерода и/или оксигенированных углеводородов. Обычно селективное сжигание водорода проводят в присутствии кислородсодержащего твердого материала, такого как смешанный оксид металла, высвобождающего часть связанного кислорода для водорода.
Один подходящий способ селективного сжигания водорода описан в патенте US 5430210, содержание которого включено в настоящее описание путем ссылки, и включает в себя контактирование в реакционных условиях первого потока, включающего углеводород и водород, и второго потока, содержащего кислород, с раздельными поверхностями мембраны, непроницаемой для не содержащих кислорода газов и содержащей оксид металла, селективный в отношении сжигания водорода, и выделение продукта селективного сжигания водорода. Этот оксид металла, как правило, представляет собой смешанный оксид висмута, индия, сурьмы, таллия и/или цинка.
В патенте US 5527979, содержание которого включено в настоящее описание путем ссылки, описан способ чистой каталитической окислительной дегидрогенизации алканов с получением алкенов. Этот способ включает в себя одновременную равновесную дегидрогенизацию алканов до алкенов и селективное сжигание образующегося водорода для проведения равновесной реакции дегидрогенизации с дальнейшим образованием алкенов. Так, в частности, исходный алкановый материал дегидрируют над катализатором равновесной дегидрогенизации в первом реакторе, а затем отходящий из первого реактора поток совместно с кислородом направляют во второй реактор, содержащий катализатор из оксида металла, который служит для катализа селективного сжигания водорода. Катализатор равновесной дегидрогенизации может включать в себя платину, а катализатор селективного сжигания из оксида металла может включать в себя висмут, сурьму, индий, цинк, таллий, свинец и теллур или их смесь.
В патентной заявке US 2004/0152586, которая была опубликована 5 августа 2004 г. и содержание которой включено в настоящее описание путем ссылки, описан способ снижения содержания водорода в отходящем из крекинг-установки потоке. В этом способе используют каталитическую систему, включающую в себя (1) по меньшей мере один твердый кислотный компонент крекинга и (2) по меньшей мере один компонент селективного сжигания водорода на металлической основе, состоящий по существу из (а) комбинации металлов, выбранных из группы, включающей: I) по меньшей мере один металл из группы 3 и по меньшей мере один металл из групп 4-15 Периодической таблицы элементов; II) по меньшей мере один металл из групп 5-15 Периодической таблицы элементов и по меньшей мере один металл из по меньшей мере одной из групп 1, 2 и 4 Периодической таблицы элементов; III) по меньшей мере один металл из групп 1 и 2, по меньшей мере один металл из группы 3 и по меньшей мере один металл из групп 4-15 Периодической таблицы элементов; и IV) два или более металлов из групп 4-15 Периодической таблицы элементов; и (б) кислорода и/или серы, причем кислород и/или сера химически связан(-а) как внутри, так и между металлами.
Реакцию селективного сжигания водорода в соответствии с настоящим изобретением обычно проводят при температуре, находящейся в интервале примерно от 300°С примерно до 850°С, и давлении, находящемся в интервале примерно от 1 атм примерно до 20 атм (от 100 до 2000 кПа).
Выделение/обработка ароматических продуктов
Помимо водорода, другими продуктами стадии дегидроциклизации являются бензол и нафталин. Эти продукты, как правило, можно выделять из потока, отходящего со стадии дегидроциклизации, экстракцией растворителем с последующим разделением на фракции, а затем сразу отправлять в продажу в качестве химической продукции массового производства. В другом варианте осуществления изобретения часть бензола и/или нафталина или весь бензол и/или нафталин можно алкилировать с получением, например, толуола, ксилолов и алкилнафталинов, и/или можно подвергнуть гидрогенизации с получением, например, циклогексана, циклогексена, дигидронафталина (бензилциклогексена), тетрагидронафталина (тетралина), гексагидронафталина (дициклогексена), октагидронафталина и/или декагидронафталина (декалина). Подходящие способы алкилирования и гидрогенизации описаны ниже, а подробнее описаны в заявках, поданных авторами настоящего изобретения, а именно в международных заявках PCT/US 2005/043523 (публикация WO/2006/068800), поданной 2 декабря 2005 г., и PCT/US 2005/044038, поданной 2 декабря 2005 г.
Алкилирование ароматических соединений
Алкилирование ароматических соединений, таких как бензол и нафталин, хорошо известно в данной области техники и, как правило, включает в себя реакцию олефина, спирта или алкилгалогенида с ароматическими веществами в газообразной или жидкой фазе в присутствии кислотного катализатора. К подходящим кислотным катализаторам относятся цеолиты со средними порами (т.е. обладающие индексом проницаемости (Constraint Index), определенным в патенте US 4016218 и составляющим от 2 до 12), включая материалы, обладающие каркасами типов MFI (например, ZSM-5 и силикалит), MEL (например, ZSM-11), MTW (например, ZSM-12), TON (например, ZSM-22), МТТ (например, ZSM-23), MFS (например, ZSM-57) и FER (например, ZSM-35), и ZSM-48, а также цеолиты с крупными порами (т.е. обладающими индексом проницаемости, меньшим 2), такие как материалы с каркасами типов ВЕА (например, цеолит бета), FAU (например, ZSM-3, ZSM-20, цеолиты X, Y, ультрастабилизированный Y и деалюминированный Y), MOR (например, морденит), MAZ (например, ZSM-4), MEI (например, ZSM-18) и MWW (например, МСМ-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, МСМ-36, МСМ-49 и МСМ-56).
В одном варианте осуществления рассматриваемого способа бензол выделяют из потока, отходящего со стадии дегидроциклизации, и затем алкилируют олефином, таким как этилен, получаемый в качестве побочного продукта на стадии снижения содержания водорода с применением этанирования/метанирования. Типичные условия проведения парофазного алкилирования бензола этиленом включают в себя температуру, составляющую примерно от 650°F примерно до 900°F (от 343 до 482°С), манометрическое давление примерно от атмосферного примерно до 3000 фунтов/кв.дюйм (от 100 до 20800 кПа), объемную скорость подачи газа (ОС) по этилену, составляющую примерно от 0,5 ч-1 примерно до 2,0 ч-1, и мольное отношение бензола к этилену, составляющее от 1:1 до 30:1. Жидкофазное алкилирование бензола этиленом можно проводить при температуре от 300°F до 650°F (от 150 до 340°С), при манометрическом давлении примерно до 3000 фунтов/кв.дюйм (20800 кПа), при ОС по этилену, составляющей примерно от 0,1 ч-1 примерно до 20 ч-1, и при мольном отношении бензола к этилену, составляющем от 1:1 до 30:1.
Этилирование бензола целесообразно проводить в по меньшей мере частично жидкофазных условиях с использованием катализатора, включающего в себя по меньшей мере один из цеолита бета, цеолита Y, MCM-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, ITQ-13, ZSM-5 МСМ-36, МСМ-49 и МСМ-56.
Этилирование бензола можно проводить на месте проведения процесса дегидроциклизации/снижения содержания водорода, или же бензол для превращения в этилбензол можно транспортировать в другой регион. Затем полученный этилбензол можно отправлять в продажу, использовать в качестве предшественника, например при получении стирола, или изомеризовать по методам, хорошо известным в данной области техники, с получением смешанных ксилолов.
В другом варианте осуществления рассматриваемого способа алкилирующий агент представляет собой метанол или диметиловый эфир (ДМЭ) и используется для алкилирования бензола и/или нафталина, выделяемого из отходящего со стадии дегидроциклизации потока, с получением толуола, ксилолов, метилнафталинов и/или диметилнафталинов. Если метанол или ДМЭ используется для алкилирования бензола, это целесообразно осуществлять в присутствии катализатора, включающего в себя цеолит, такой как ZSM-5, цеолит бета, ITQ-13, MCM-22, МСМ-49, ZSM-11, ZSM-12, ZSM-22, ZSM-23, ZSM-35 и ZSM-48, модифицированный обработкой водяным паром таким образом, чтобы он обладал диффузионным параметром для 2,2-диметилбутана, составляющим примерно от 0,1 до 15 с-1 при его измерении при температуре 120°С и давлении 2,2-диметилбутана 60 торр (8 кПа). Такой способ селективен в отношении получения пара-ксилола и описан, например, в патенте US 6504272, содержание которого включено в настоящее описание путем ссылки. Если метанол используется для алкилирования нафталина, это целесообразно осуществлять в присутствии катализатора, включающего в себя ZSM-5, MCM-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, ITQ-13, МСМ-36, МСМ-49 или МСМ-56. Такой способ может применяться для селективного получения 2,6-диметилнафталина, и он описан, например, в патентах US 4795847 и US 5001295, содержание которых включено в настоящее описание путем ссылки.
Если в предлагаемом в изобретении способе в качестве алкилирующего агента используется метанол или ДМЭ, этот агент можно вводить в процесс как отдельный исходный материал или он может, по меньшей мере частично, образовываться in situ путем добавления содержащего диоксид углерода газообразного исходного материала, такого как поток природного газа, в часть потока, отходящего со стадии дегидроциклизации, или весь этот поток. В частности, отходящий со стадии дегидроциклизации поток перед любым выделением ароматических компонентов можно направлять в реактор обратной конверсии и проводить реакцию с содержащим диоксид углерода исходным материалом в условиях повышения содержания моноксида углерода в этом отходящем потоке, т.е. такими реакциями, как вышеприведенные реакции 5 и 8.
Кроме того, в реактор обратной конверсии можно направлять метан и СО2 и/или водяной пар с получением синтез-газа, который затем может быть смешан с частью отходящего со стадии дегидроциклизации потока для регулирования отношений Н2/СО/СО2 в зависимости от потребности для стадии алкилирования.
Как правило, реактор обратной конверсии содержит катализатор, включающий в себя переходный металл на носителе, такой как Fe, Ni, Cr, Zn на оксиде алюминия, диоксиде кремния или диоксиде титана, и работает в условиях, включающих в себя температуру, составляющую примерно от 500°С примерно до 1200°С, в частности примерно от 600°С примерно до 1000°С, например примерно от 700°С примерно до 950°С, и давление, составляющее примерно от 1 кПа примерно до 10000 кПа, в частности примерно от 2000 кПа примерно до 10000 кПа, например примерно от 3000 кПа примерно до 5000 кПа. Объемные скорости подачи газа могут варьироваться в зависимости от типа используемого процесса способа, но обычно объемная скорость подачи газа через каталитический слой находится в интервале примерно от 50 ч-1 примерно до 50000 ч-1, в частности примерно от 250 ч-1 примерно до 25000 ч-1, более предпочтительно примерно от 500 ч-1 примерно до 10000 ч-1.
Затем отходящий из реактора обратной конверсии поток может быть направлен в реактор алкилирования, работающий в условиях, обеспечивающих протекание таких реакций, как следующие:

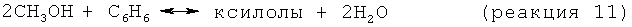
Подходящие для такого реактора условия алкилирования включают в себя температуру, составляющую примерно от 100 примерно до 700°С, давление, составляющее примерно от 1 примерно до 300 атм (от 100 до 30000 кПа) и объемную скорость подачи сырья, составляющую для ароматического углеводорода примерно от 0,01 ч-1 примерно до 100 ч-1. Подходящий катализатор включает в себя молекулярное сито, обладающее индексом проницаемости от 1 до 12, такое как ZSM-5, как правило, совместно с одним из металлов или оксидов металлов, таких как медь, хром и/или оксид цинка.
Если катализатор алкилирования включает в себя молекулярное сито, последнее целесообразно модифицировать для изменения его диффузионных характеристик таким образом, чтобы превалирующим изомером ксилола, получаемого реакцией 11, был пара-ксилоп. К приемлемым средствам диффузионной модификации относятся обработка водяным паром и осаждение ex-situ или in situ соединений кремния, кокса, оксидов металлов, таких как MgO, и/или Р на поверхности или в устьях пор молекулярного сита. Также предпочтительным является введение в молекулярное сито активного металла таким образом, чтобы обеспечить насыщение более высокореакционноспособных веществ, таких как олефины, которые могут образовываться в качестве побочных продуктов и которые в противном случае могли бы вызвать дезактивацию катализатора.
Затем отходящий из реактора алкилирования поток можно направлять в секцию разделения, в которой ароматические продукты вначале отделялись бы от водорода и других низкомолекулярных материалов, целесообразно экстракцией растворителем. Далее ароматические продукты можно разделять на бензольную фракцию, толуольную фракцию, фракцию C8 и тяжелую фракцию, включающую в себя нафталин и алкилированные нафталины. Затем ароматическая фракция C8 направлена в процесс кристаллизации или сорбции для отделения ценного п-ксилольного компонента, а оставшиеся смешанные ксилолы могут либо отправляться в продажу в качестве готовой продукции, либо направляться в контур изомеризации для получения дополнительного количества п-ксилола. Толуольная фракция может либо удаляться в качестве готовой к сбыту продукции, либо возвращаться в реактор алкилирования, либо направляться в установку диспропорционирования толуола, в частности в установку селективного диспропорционирования толуола, для получения дополнительного количества п-ксилола.
Гидрогенизация ароматических соединений
В дополнение к стадии алкилирования или вместо нее по меньшей мере часть ароматических компонентов в отходящем со стадии дегидроциклизации потоке можно гидрировать с получением полезных продуктов, таких как циклогексан, циклогексен, дигидронафталин (бензилциклогексен), тетрагидронафталин (тетралин), гексагидронафталин (дициклогексен), октагидронафталин и/или декагидронафталин (декалин). Эти продукты можно использовать в качестве топлив и химических промежуточных продуктов, а в случае тетралина и декалина - и в качестве растворителя для экстракции ароматических компонентов из потока, отходящего со стадии дегидроциклизации.
Гидрогенизацию целесообразно, но необязательно, проводить после выделения ароматических компонентов из потока, отходящего со стадии дегидроциклизации, и целесообразно использовать часть водорода, образующегося в результате реакции дегидроциклизации. Подходящие способы гидрогенизации ароматических соединений хорошо известны в данной области техники, и в них, как правило, используют катализатор, включающий Ni, Pd, Pt, Ni/Mo или сульфидированные Ni/Мо, нанесенные на носитель из оксида алюминия или диоксида кремния. Подходящие для процесса гидрогенизации рабочие условия включают в себя температуру, составляющую примерно от 300°F примерно до 1000°F (от 150 до 540°С), в частности примерно от 500°F примерно до 700°F (от 260 до 370°С), манометрическое давление, составляющее примерно от 50 примерно до 2000 фунтов/кв.дюйм (от 445 до 13890 кПа), в частности примерно от 100 примерно до 500 фунтов/кв.дюйм (от 790 до 3550 кПа), и объемную скорость подачи сырья, составляющую примерно от 0,5 ч-1 примерно до 50 ч-1, в частности примерно от 2 ч-1 примерно до 10 ч-1.
Для получения материалов, подходящих для полимеризации или другого последующего химического превращения, также может быть желательна частичная гидрогенизация, позволяющая оставить в продукте одну или несколько олефиновых углерод-углеродных связей. Подходящие способы частичной гидрогенизации хорошо известны в данной области техники, и в них, как правило, используют катализатор, включающий в себя благородные металлы, причем предпочтителен рутений, нанесенный на оксиды металлов, такие как La2O3-ZnO. Также могут использоваться гомогенные каталитические системы с благородными металлами. Примеры способов частичной гидрогенизации описаны в патентах US 4678861, US 4734536, US 5457251, US 5656761, US 5969202 и US 5973218, содержание которых в полном объеме включено в настоящее описание путем ссылки.
Альтернативный способ гидрогенизации предусматривает гидрокрекинг нафталинового компонента при низком давлении с получением алкилбензолов, осуществляемый над таким катализатором, как сульфидированные Ni/W или сульфидированный Ni, нанесенный на аморфный алюмосиликат или цеолит, такой как цеолит X, цеолит Y или цеолит бета. Подходящие для гидрокрекинга низкого давления рабочие условия включают в себя температуру, составляющую примерно от 300°F примерно до 1000°F (от 150 до 540°С), в частности примерно от 500°F примерно до 700°F (от 260 до 370°С), манометрическое давление, составляющее примерно от 50 примерно до 2000 фунтов/кв.дюйм (от 445 до 13890 кПа), в частности примерно от 100 примерно до 500 фунтов/кв.дюйм (от 790 до 3550 кПа), и объемную скорость подачи сырья, составляющую примерно от 0,5 ч-1 примерно до 50 ч-1, в частности примерно от 2 ч-1 примерно до 10 ч-1.
Далее изобретение рассматривается более конкретно со ссылкой на прилагаемые чертежи и следующие неограничивающие примеры.
На фиг.1 представлена упрощенная схема реактора дегидроциклизации с подогревателем катализатора и одним регенератором катализатора для превращения метана в ароматические углеводороды в первом варианте осуществления изобретения. В этом варианте осуществления изобретения реактор дегидроциклизации содержит вертикально расположенный реактор 11 с оседающим слоем, в который через вход 12, расположенный вблизи верхней части реактора 11, поступает поток нагретого и регенерированного зернистого катализатора, и из которого через выход 13, расположенный вблизи основания реактора 11, выходит поток охлажденного закоксованного катализатора. Метановый исходный материал (сырье) 15 вводится в реактор 11 вблизи его основания. Как правило, нагретый катализатор поступает в реактор 11 при температуре примерно 850°С, а охлажденный закоксованный катализатор выходит из реактора при температуре примерно 650°С.
Охлажденный закоксованный катализатор движется под действием силы тяжести (самотеком) от выхода 13 к основанию вертикального трубного подогревателя 16, где катализатор увлекается смесью воздуха и метанового топлива, проходящей в вертикальную трубу через коллектор 17. Катализатор транспортируется вверх по вертикальному трубному подогревателю 16 воздушно-метановой смесью и во время его прохождения по вертикальному трубному подогревателю 16 нагревается за счет горения метана. Смесь, поступающая в вертикальный трубный подогреватель 16 через коллектор 17, содержит достаточно топлива, необходимого для нагрева катализатора до требуемой температуры реакции, но недостаточно кислорода. Поэтому в вертикальный трубный подогреватель 16 через множество впускных отверстий 18, распределенных по длине вертикальной трубы (для упрощения на фиг.1 обозначено только одно впускное отверстие 18, но в действительности их число может быть гораздо большим), вводят дополнительный воздух, благодаря чему нагрев катализатора происходит постепенно, по мере движения катализатора вверх по вертикальному трубному подогревателю 16. Обычно по достижении верхней части вертикального трубного подогревателя 16 температура катализатора снова составляет примерно 850°С.
Выйдя из верхней части вертикального трубного подогревателя 16, нагретый катализатор проходит в сепаратор 19, где твердый зернистый катализатор отделяется от газообразных продуктов горения и затем направляется в бункер 20, а затем в верхний конец вертикально расположенного стояка 21 катализатора. Газообразные продукты горения выпускают из сепаратора через выходной патрубок 22 и затем, прежде чем они пойдут на рекуперацию тепла, их направляют в циклоны 22 (не показаны) для удаления катализаторной пыли. При использовании воздуха в качестве среды горения в подогревателе 16 выходящие газообразные продукты горения, как правило, содержат 40-70 мас.% N2, <1 мас.% O2, 1-30 мас.% H2, 2-20 мас.% СО, 1-20 мас.% CO2 и 5-25 мас.% Н2О.
Скорость движения катализатора через систему и внутренний диаметр стояка 21 таковы, что нагретый катализатор накапливается в стояке 21, образуя плотно упакованный слой, движущийся под действием силы тяжести через стояк 21, и поступая в регенератор 23, присоединенный к нижнему концу стояка 21. В зависимости от высоты стояка 21 давление в регенераторе может поддерживаться на уровне, необходимом для эффективной регенерации катализатора водород. Например, расчеты показали, что для достижения абсолютного давления в регенераторе примерно от 700 до 1000 кПа при использовании катализатора Mo/ZSM-5, циркулирующего таким образом, что абсолютное давление в сепараторе 19 примерно равно 140 кПа, высота стояка должна составлять примерно от 100 до 300 футов (от 30 до 91 м).
Катализатор из стояка 21 поступает в регенератор 23 через вход 24, расположенный вблизи верхней части регенератора 23 и движется вниз через регенератор 23 навстречу потоку водорода 25, нагнетаемого в регенератор 23 вблизи его основания. Регенерированный катализатор выходит из регенератора 23 через выход 26, также расположенный вблизи основания регенератора 23, и затем через еще одну вертикальную трубу 28 движется в отделительный сосуд 31, прежде чем снова поступить в реактор 11 через вход 12. Эта вертикальная труба 28 и отделительный сосуд 31 предназначены для выделения водорода, захваченного катализатором, а при необходимости, для содействия движению катализатора по вертикальной трубе 28, в основание вертикальной трубы 28 может нагнетаться метан 28.
Фиг.2 иллюстрирует осуществление способа превращения метана в ароматические углеводороды во втором варианте осуществления изобретения, в котором реактор дегидроциклизации снабжен подогревателем катализатора, а также первым и вторым регенераторами катализатора, включенными последовательно и работающими при различных давлениях. В этом варианте осуществления изобретения реактор дегидроциклизации также содержит вертикально расположенный реактор 111 с оседающим слоем, в который через входы 112, 113, расположенные вблизи верхней части реактора 111, поступает поток нагретого и регенерированного зернистого катализатора, и из которого через выход 114, расположенный вблизи основания реактора 111, выходит поток охлажденного закоксованного катализатора. Метановый исходный материал 115 вводится в реактор 111 вблизи его основания. Как правило, нагретый катализатор поступает в реактор 111 при температуре примерно 850°С, а охлажденный закоксованный катализатор выходит из реактора при температуре примерно 650°С.
Охлажденный закоксованный катализатор движется под действием силы тяжести от выхода 114 к основанию вертикального трубного подогревателя 116, где катализатор увлекается смесью воздуха и метанового топлива, проходящей в вертикальную трубу через коллектор 117. Катализатор транспортируется вверх по вертикальному трубному подогревателю 116 воздушно-метановой смесью и во время его прохождения по вертикальному трубному подогревателю 116 нагревается за счет горения метана. Смесь, поступающая в вертикальный трубный подогреватель 116 через коллектор 117, содержит достаточно топлива, необходимого для нагрева катализатора до требуемой температуры реакции, но недостаточно кислорода. Поэтому в вертикальный трубный подогреватель 116 через множество впускных отверстий 118, распределенных по длине вертикальной трубы (для упрощения на фиг.2 обозначено только одно впускное отверстие 118, но в действительности их число может быть гораздо большим), вводят дополнительный воздух, благодаря чему нагрев катализатора происходит постепенно, по мере движения катализатора вверх по вертикальному трубному подогревателю 116. Обычно по достижении верхней части вертикального трубного подогревателя 116 температура катализатора снова составляет примерно 850°С.
Выйдя из верхней части вертикального трубного подогревателя 116, нагретый катализатор проходит в циклонный сепаратор 119, где твердый зернистый катализатор отделяется от газообразных продуктов горения и затем подается в верхнюю часть первого регенератора 121, работающего при относительно низком давлении. Первый регенератор 121 расположен по вертикали над вторым регенератором 122, работающим при относительно высоком давлении, и соединен со вторым регенератором 122 посредством вертикально расположенного стояка 123 для катализатора. В первый регенератор 121 вблизи его основания нагнетается поток водорода 125, который движется вверх через первый регенератор 121. Высота стояка 123 определяет разность давлений между первым и вторым регенераторами 121, 122.
Во второй регенератор 122 вблизи его основания нагнетается поток водорода 124, который движется вверх через второй регенератор 122, стояк 123 для катализатора и первый регенератор 121 навстречу потоку катализатора, движущемуся под действием силы тяжести из первого регенератора 121 во второй регенератор 122. Водород регенерирует закоксованный катализатор по мере движения последнего через регенераторы, и часть регенерированного катализатора возвращается в реактор 111 через вход 112 из первого регенератора 121, а остальная часть регенерированного катализатора возвращается в реактор 111 через вход 113 из второго регенератора 122.
Ниже следует более подробное описание изобретения со ссылкой на следующие неограничивающие примеры.
Пример 1
Пример 1 демонстрирует применение совместно подаваемых исходных материалов (таких как Н2, СО2 и Н2О) для достижения пониженной скорости закоксовывания катализатора Mo/ZSM-5 во время дегидроциклизации метана с получением преимущественно бензола. Пример 2 продемонстрирует, что благодаря уменьшению количества кокса, отложившегося на катализаторе во время контакта с углеводородом, можно поддерживать высокую эффективность после нескольких циклов контакта с углеводородом и регенерирования и что более низкая скорость закоксовывания за один цикл преобразуется в более низкую результирующую скорость отложения кокса за несколько циклов.
Катализаторы Mo/ZSM-5 готовили двумя методами: (1) пропиткой по влагоемкости носителя NH4 +-ZSM-5 (имеющего отношение Si/Al2, равное 25) требуемым количеством раствора гептамолибдата аммония с последующей сушкой при 120°С в течение 2 часов и завершающим прокаливанием при 500°С в течение 6 часов в токе воздуха, и (2) размалыванием в шаровой мельнице оксида молибдена с носителем NH4 +-ZSM-5 (имеющим отношение Si/Al2, равное 25) в течение 2 часов с последующим прокаливанием при 500°С в течение 5 ч в токе воздуха. Содержание молибдена (в пересчете на металл в мас.%) варьировали изменением концентрации гептамолибдата аммония в пропиточном растворе или количества оксида молибдена, добавленного в измельчаемую смесь.
Каталитическое испытание приготовленных катализаторов Mo/ZSM-5 осуществляли с помощью вибрационных микровесов с сужающимся элементом (англ. Tapered-Element Oscillating Microbalance, сокр. ТЕОМ), позволяющих точно и с быстрым временем отклика определять изменения массы катализатора во время реакции. Катализатор (после прокаливания) таблетировали, измельчили и просеяли для выделения частиц крупностью 20-40 меш. Приблизительно 0,10 г просеянных частиц катализатора загрузили в держатель образца микровесов ТЕОМ (общий объем образца: 0,20 см3) и с использованием подкладок из кварцевого волокна уплотнили с получением неподвижного слоя катализатора. Функциональные свойства катализатора при дегидроциклизации метана до бензола определяли при 800°С, а также при абсолютном давлении 20 фунтов/кв.дюйм (138 кПа) в экспериментах А-Б и при абсолютном давлении 14,7 фунтов/кв.дюйм (101 кПа) в экспериментах В-Д, указанных в таблице 1, с использованием исходного материала, содержавшего указанные совместно подаваемые материалы (CO2, Н2О, Н2), Ar (Ar используют в качестве внутреннего стандарта), остальное - СН4, при заданной объемной скорости (по метану). Отходивший из реакции поток анализировали с помощью масс-спектрометра для определения концентраций метана, бензола, нафталина, водорода и аргона. Скорость отложения кокса на катализаторе (т.е. образования тяжелых углеродистых отложений, не улетучивавшихся с поверхности катализатора) определяли непосредственно по изменениям массы, наблюдавшимся с помощью микровесов. Приведенные значения функциональных свойств катализатора (например, производительность по бензолу, превращение метана, селективность по бензолу) являются совокупными или средними значениями за период времени, начинающийся с момента подачи метана и заканчивающийся моментом, когда мгновенный выход бензола уменьшился до <2%. Результаты приведены в таблице 1.
Как показано в табл.1, при содержании Мо 4,6% для экспериментов А и Б средняя скорость отложения кокса уменьшалась с 5,2 мас.% в час при отсутствии материалов, совместно подаваемых с метаном (т.е. при составе подаваемого потока: 95% СН4 и 5% Ar) до 2,2 мас.% в час при подаче совместно с метаном 2% СО2 и 10% Н2 (т.е. при составе подаваемого потока: 2% CO2, 10% H2, 83,6% СН4 и 4,4% Ar). Производительность по бензолу увеличилась с 0,5 г бензола, полученного на грамм катализатора при отсутствии совместно подаваемых материалов, до 1,3 г бензола на грамм катализатора. Это показывает, что присутствие СО2 и H2 как подаваемых совместно с метаном материалов способно значительно улучшить производительность катализаторов Mo/ZSM-5 по бензолу за счет снижения скоростей коксообразования. Эксперименты В, Г и Д демонстрируют влияние совместной подачи Н2О и Н2 на катализатор, содержащий 2,7% Мо на ZSM-5. Средняя скорость закоксовывания, как было установлено, уменьшилась с 8,6 мас.%/ч при отсутствии совместно подаваемых материалов до 6,0 мас.%/ч при совместной с метаном подаче 3% водяного пара и до 3,8 мас.%/ч при совместной с метаном подаче 20% Н2. В обоих случаях было установлено, что с добавлением совместно подаваемого материала производительность и селективность по бензолу повышались.
Пример 2
Катализаторы Mo/ZSM-5 готовили методами, описанными выше в примере 1. Катализаторы в синтезированном состоянии подвергли циклическому старению, которое состояло из (1) периода контакта с углеводородом, когда на катализатор воздействовали метаном СН4, подаваемым при 800°С и при абсолютном давлении 14,7 фунта/кв.дюйм (101 кПа) совместно с указанными материалами в течение 5 мин при указанной объемной скорости по СН4, и (2) последующего периода регенерирования, когда катализатор нагревали до 850°С и при абсолютном давлении 14,7 фунта/кв.дюйм (101 кПа) в газообразном H2 со скоростью 10°С/мин и выдерживали при 850°С в течение заданного времени при объемной скорости подачи газа 9000 см [в нормальных условиях]/г катализатора/ч. После завершения стадии регенерирования катализатор перед повторной подачей метанового исходного материала охлаждали до 800°С. Перед циклическим старением катализатор в синтезированном состоянии предварительно науглероживали нагреванием катализатора в газообразной смеси 15% CH4-H2 со 150°С до 800°С со скоростью 5°С/мин и выдерживали при 800°С в течение 1 часа. После того как катализаторы пропустили через указанное число циклов контакта с углеводородом и регенерирования, отработавшие катализаторы удаляли и испытывали на функциональные свойства на микровесах ТЕОМ.
Приблизительно 0,10 г отработавшего катализатора загрузили в держатель образца микровесов ТЕОМ и с использованием подкладок из кварцевого волокна уплотнили с получением неподвижного слоя катализатора. Функциональные свойства катализатора при дегидроциклизации метана до бензола определяли при 800°С, а также при абсолютном давлении 14,7 фунта/кв.дюйм (101 кПа) для экспериментов А-Г и при абсолютном давлении 20 фунтов/кв.дюйм (138 кПа) для экспериментов Д-Ж в таблице 2 с использованием смеси исходного материала из 95% СН4 и 5% Ar (Ar используют в качестве внутреннего стандарта) и при объемной скорости подачи сырья, соответствующей подаче 4 г СН4 на грамм катализатора в час. Отходивший из реакции поток анализировали с применением масс-спектрометра для определения концентраций метана, бензола, нафталина, водорода и аргона. Определение кокса на катализаторе осуществляли с использованием термопрограммированного окисления отработавшего катализатора в термогравиметрическом анализаторе (ТГА).
В таблице 2 сравниваются накопившийся на катализаторе кокс после нескольких циклов контакта катализатора с углеводородом и регенерирования катализатора и остаточная производительность отработавших катализаторов по бензолу. Эксперименты А-В демонстрируют функциональные свойства катализатора 2,7% Mo/ZSM-5 после 0, 20 и 40 циклов. Эксперимент Г демонстрирует функциональные свойства катализатора 2,7% Mo/ZSM-5 после 40 циклов. Эксперименты Д-Ж демонстрируют функциональные свойства катализатора 4,6% Mo/ZSM-5 после 0, 50 и 389 циклов. При сопоставлении экспериментов В и Г видно, что добавление 20% H2 в качестве материала, подаваемого совместно с метаном, уменьшило результирующее отложение кокса на катализаторе примерно на 55%, с 2,0 мас.% (в случае без совместно подаваемого материала) до 0,9 мас.% (при совместной с метаном подаче 20% Н2). Было установлено, что при совместной с метаном подаче 20% Н2 помимо снижения коксообразования производительность по бензолу после 40 циклов значительно, примерно на 25%, повысилась (0,25 г бензола/г катализатора), если сравнивать со случаем отсутствия совместно подаваемого материала (0,20 г бензола/г катализатора). Эксперименты Д-Ж показывают, что результирующее накопление кокса на катализаторе может быть сведено к минимуму, если в качестве совместно подаваемого материала добавлять 2% CO2. Было установлено, что после 50 циклов суммарное отложение кокса на катализаторе составило всего 0,15 мас.%, что значительно меньше (примерно на 93%) отложения кокса на катализаторе, наблюдаемого после 40 циклов в случае отсутствия совместно подаваемого материала (т.е. 2,0 мас.%), даже несмотря на то, что объемная скорость подачи исходного материала во время контакта с углеводородом увеличилась с 1 до 4. После 50 циклов не наблюдалось никакого ухудшения производительности по бензолу. После 389 циклов измеренное отложение кокса на катализаторе составило примерно 0,5 мас.%, указывая на то, что суммарное накопление кокса на катализаторе в течение долговременного циклического старения может быть сведено к минимуму.
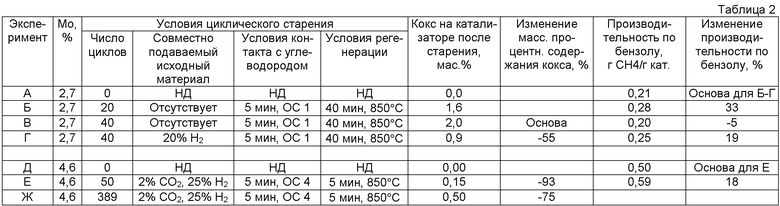
Пример 3
Примеры 3 и 4 призваны показать влияние температуры регенерирования на выход бензола при дегидроциклизации метана за несколько циклов.
Катализатор, содержащий примерно 4 мас.% Мо на ZSM-5, использовали для ароматизации исходного материала, содержащего 86,65 мол.% СН4, 1,8% С2Н6, 0,9% CO2, 0,45% Н2 и 10 мол.% Ar. Для расчета значений выхода бензола отходящий поток анализировали методом газовой хроматографии. Катализатор попеременно использовали в циклах реакции и регенерирования, причем отношение времени реакции к времени регенерации было 1:1. Условия и реакции, и регенерации включали в себя температуру 800°С и манометрическое давление примерно 7 фунтов/кв.дюйм (абс.давление 149 кПа). Значения выхода бензола по прошествии приблизительно 15 минут с начала реакции и в несколько моментов времени после начала эксперимента перечислены ниже:
Пример 4
Процесс, описанный в примере 3, повторили, но с проведением каждого цикла регенерации при температуре 850°С и манометрическом давлении примерно 7 фунтов/кв.дюйм (абс. давление 149 кПа). Условия реакции и отношение времени реакции к времени регенерации остались теми же, что в примере 3. Значения выхода бензола по прошествии приблизительно 15 минут с начала реакции и в несколько моментов времени после начала эксперимента перечислены ниже:
Отсюда видно, что значения выхода бензола в примере 4 (где температура регенерирования составляла 850°С) выше, чем в примере 3 (где температура регенерирования составляла 800°С).
Пример 5
Этот пример призван показать, что регенерация при более высоком давлении выгодна с точки зрения улучшения селективности катализатора, т.е. повышения селективности по бензолу и уменьшения селективности в отношении образования кокса.
Катализатор, содержащий примерно 4 мас.% Мо на ZSM-5, использовали для ароматизации исходного материала, содержащего 86,65 мол.% СН4, 1,8% С2Н6, 0,9% CO2, 0,45% Н2 и 10 мол.% Ar. Для расчета значений выхода бензола отходящий поток анализировали методом газовой хроматографии. Катализатор попеременно использовали в циклах реакции и регенерирования, причем метансодержащий исходный материал подавали к катализатору в течение каждого 20-минутного цикла реакции, а водород подавали к катализатору в течение каждого 40-минутного цикла регенерации. В течение первых 90% каждого цикла реакции в метансодержащий исходный материал в качестве совместно подаваемого материала добавляли 20% H2, но на завершающих 10% каждого цикла реакции это добавление прекращали. Манометрическое давление реакции составляло примерно 7 фунтов/кв.дюйм (абс. давление 149 кПа), а температуру реакции повышали в течение каждого цикла реакции от начального значения примерно 700°С до конечного значения примерно 800°С.Средняя температура в течение каждого цикла регенерации равнялась примерно 850°С, максимальная же температура составляла 860°С. В начале испытательного эксперимента манометрическое давление в течение каждого цикла регенерации составляло 34 фунта/кв.дюйм (абс. давление 335 кПа), а стабилизировавшиеся значения селективности и выхода бензола перечислены ниже. По прошествии заданного времени манометрическое давление в течение каждого цикла регенерации понижали примерно до 7 фунтов/кв.дюйм (абс. давление 149 кПа) и спустя примерно 11 ч после изменения давления регенерации вновь измеряли значения селективности и выхода бензола, которые представлены ниже. Отсюда видно, что при более высоком давлении регенерации селективность по бензолу и выход бензола были выше, а селективность в отношении кокса - ниже.
Пример 6
Этот пример призван показать, что при использовании содержащего кокс катализатора Mo/HZSM-5 дегидроциклизации метана при высоком давлении H2 можно удалить больше кокса, чем при низком давлении Н2.
Катализатор 5 мас.% Mo/ZSM-5 приготовили размалыванием в шаровой мельнице оксида молибдена с носителем NH4 +-ZSM-5 (имеющим отношение Si/Al2, равное 25) в течение 2 часов с последующим прокаливанием при 500°С в течение 5 часов в токе воздуха. Полученный катализатор предварительно науглероживали в газообразной смеси, содержавшей 15% СН4 и 85% Н2, при 800°С и объемной скорости 1,2 в течение 1 часа в кварцевом реакторе идеального вытеснения. Превращение метана выполняли при температуре 800°С, объемной скорости 1,2 и абсолютном давлении 20 фунтов/кв.дюйм (138 кПа) с целью дегидроциклизации метана до бензола с использованием исходного материала, содержавшего 95 об.% СН4 и 5 об.% Ar (аргон использовали в качестве внутреннего стандарта). Реакцию превращения метана остановили спустя 1 час нахождения катализатора в контакте с углеводородом. Затем, после охлаждения до температуры окружающей среды из реактора взяли образец катализатора и на этом образце катализатора измерили количество кокса, которое примерно составило 8,9 мас.% [(масса кокса)/(суммарная масса кокса и Mo/ZSM-5)].
Приблизительно 100 мг содержащего кокс образца катализатора поместили в насадку трубчатого реактора диаметром ¼'' (6,35 мм). Температуру реактора подняли от температуры окружающей среды примерно до 1000°С, повышая ее со скоростью 5°С/мин в потоке Н2, подававшемся с расходом 50 мл/мин. Концентрации метана и водорода в отходящем потоке реактора анализировали методом ГХ-МС в режиме реального времени. Количество кокса, газифицированного водородом, вычисляли по концентрации метана в отходящем потоке и расходу водорода; других углеродсодержащих веществ, кроме метана, не наблюдалось.
На фиг.3 показано изменение количества метана (ммоль), выделяемого из реактора при линейном повышении температуры реактора, начинающемся от температуры окружающей среды, при двух различных парциальных давлениях водорода. Из графика видно, что при абсолютном парциальном давлении водорода 20 фунтов/кв.дюйм (138 кПа), максимум концентрации метана достигается при температуре около 850°С. При более высоком абсолютном парциальном давлении водорода (75 фунтов/кв.дюйм [517 кПа]), температура максимальной концентрации уменьшилась примерно до 750°С. Величина пика концентрации выделяющегося метана при абсолютном давлении 75 фунтов/кв.дюйм гораздо выше, чем при 20 фунтов/кв.дюйм, что означает гораздо более высокую интенсивность удаления кокса при высоких давлениях. Что более важно, значительное уменьшение температуры максимальной концентрации метана при высоком давлении по сравнению с низким давлением означает то, что при данной температуре регенерации водородом не весь кокс, удаленный при высоком давлении, может быть удален при низком давлении.
В таблице 3 сведены значения количества кокса (массовая доля в процентах) на образце катализатора в зависимости от температуры реактора во время термопрограммированной обработки водородом (ТО-Н2). Из приведенных значений видно, что при любой данной температуре количество кокса, удаленное при более высоком давлении, гораздо выше, чем количество кокса, удаленное при низких давлениях. Например, после TO-H2 при абсолютном давлении 20 фунтов/кв.дюйм осталось примерно 2,6 мас.% кокса, тогда как при абсолютном давлении 75 фунтов/кв.дюйм осталось только 0,3 мас.% кокса.
Пример 7
Катализатор 2,7 мас.% Mo/ZSM-5 приготовили пропиткой по влагоемкости носителя NH4 +-ZSM-5 (имеющего отношение Si/Al2, равное 25) раствором гептамолибдата аммония с последующей сушкой при 120°С в течение 2 часов и завершающим прокаливанием при 500°С в течение 6 часов в токе воздуха. Полученный катализатор поместили в насадку реактора в виде неподвижного слоя. Температуру реактора подняли до 800°С.При этой температуре 800°С в реактор подавали метан, поддерживая контакт метана с катализатором в течение 20 мин при объемной скорости подачи 1,2 и абсолютном парциальном давлении метана, составлявшем примерно 15 фунтов/кв.дюйм (103 кПа). После 20 минут контакта катализатора с метаном подачу метанового исходного материала отключили и для газификации кокса на катализаторе включили подачу водорода. Продолжительность газификации водородом составила 40 мин при 800°С.Эти две стадии повторили еще 39 раз, после чего по окончании стадии газификации водородом образец катализатора извлекли из реактора. Измерили количество кокса на образце катализаторе, которое составило примерно 8,7 мас.% [(масса кокса)/(суммарная масса кокса и Mo/ZSM-5)].
Приблизительно 100 мг содержащего кокс образца катализатора поместили в насадку трубчатого реактора диаметром ¼'' (6,35 мм). Температуру реактора подняли от температуры окружающей среды примерно до 850°С, повышая ее со скоростью 5°С/мин в токе гелия, подававшегося с расходом 50 мл/мин. После того как температура реактора стабилизировалась на 850°С, через реактор пропускали водород, подававшийся с расходом 50 мл/мин. Концентрации метана и водорода в отходящем потоке реактора анализировали методом ГХ-МС в режиме реального времени. Количество кокса, газифицированного водородом, вычисляли по концентрации метана в отходящем потоке и расходу водорода.
На фиг.4 представлено изменение содержания кокса на катализаторе (массовая доля в процентах) в зависимости от времени регенерирования водородом при 850°С и различных парциальных давлениях водорода. Из приведенного графика видно, что первоначальная (до истечения 40 мин) скорость газификации кокса была гораздо выше при более высоких давлениях, что согласуется с результатами, приведенными в примере 6. В промышленных применениях желательно, чтобы время регенерирования водородом было как можно меньшим. В результате преимущество значительно более быстрой регенерации для удаления кокса при высоких давлениях становится исключительно заметным.
В таблице 4 приведено содержание кокса (мас.%) при различных временах регенерирования. Из приведенных данных видно, что количество кокса, оставшегося на катализаторе после 36 мин регенерирования водородом при абсолютном парциальном давлении водорода 21 фунт/кв.дюйм (145 кПа) и температуре 850°С, составило 7,7 мас.%, что примерно на 31% выше, чем количество кокса, оставшегося на катализаторе (5,9 мас.%) после 36 мин регенерирования водородом при той же температуре и абсолютном парциальном давлении водорода 105 фунтов/кв.дюйм (724 кПа).
Также важно отметить, что при постоянной температуре (850°С) после начального более быстрого уменьшения содержания кокса газификация кокса идет очень медленно. Кривые содержания кокса начали выравниваться спустя примерно 100 мин нахождения в водороде. Обратим внимание на значительные различия между количеством кокса, остающимся на катализаторе при различных давлениях регенерации в конце процесса регенерирования водородом продолжительностью 2 часа. Например, количество кокса, оставшееся спустя примерно 2 часа регенерирования в водороде при абсолютном парциальном давлении водорода 21 фунт/кв.дюйм (145 кПа), составило 6,6 мас.%, что на 35% выше количества кокса, оставшегося при абсолютном парциальном давлении водорода 105 фунтов/кв.дюйм (724 кПа). При абсолютном давлении 21 фунт/кв.дюйм для достижения уровня кокса, аналогичного полученному при 105 фунтов/кв.дюйм, может потребоваться много часов дополнительного времени регенерации. На практике эти результаты означают, что не весь кокс, удаляемый при высоких давлениях, можно удалить при низких давлениях в тех же температурных условиях регенерирования водородом.
Пример 8
Катализаторы Mo/ZSM-5 приготовили пропиткой по влагоемкости носителя NH4ZSM-5 (с отношением Si/Al2, равным 28) требуемым количеством раствора гептамолибдата аммония с последующей сушкой при 120°С в течение 2 часов и завершающим прокаливанием при 500°С в течение 6 часов в токе воздуха. Номинальное целевое содержание молибдена (массовая процентная доля металла в общей массе катализатора) составляло 2,7 мас.%; небольшие колебания содержания молибдена на сделанные выводы не влияют. Каждый образец катализатора Mo/ZSM-5 (после прокаливания) таблетировали, измельчили и просеяли для выделения частиц крупностью 30-60 меш. Каталитическое испытание катализаторов Mo/ZSM-5 осуществляли в кварцевом реакторе, содержавшем насадку для получения неподвижного слоя с использованием подкладок из кварцевого волокна.
Функциональные свойства катализатора при дегидроциклизации метана до бензола определяли при различных температурах с использованием в качестве исходного материала смеси 95 мас.% СН4 и 5 мас.% аргона (аргон использовали в качестве внутреннего стандарта) при объемной скорости подачи сырья (по метану) 1,2 ч-1. Все экспериментальные данные были получены при абсолютном давлении 138 кПа (20 фунтов/кв.дюйм) и при том же давлении осуществляли также все моделирование. Отходивший из реакции поток анализировали с помощью масс-спектрометра и газового хроматографа для определения концентраций метана, бензола, толуола, этилена, нафталина, водорода и аргона. Скорость отложения кокса на катализаторе (т.е. образования тяжелых углеродистых отложений, не улетучивавшихся с поверхности катализатора) определяли посредством баланса углерода. Добавляя Н2 в исходный материал при двух температурах (750 и 800°С) и в концентрации соответственно 6 и 20 мол.%, получили дополнительные данные.
Для целей эксперимента в примере 8 полученные экспериментальные данные были сведены к двум величинам: Сел.БТН и Сел.Кокс. Величина Сел.БТН представляет собой среднюю селективность на основе молярной концентрации углерода, определяемую суммой молей углерода в продукции: бензоле, толуоле и нафталине, деленной на моли углерода, содержавшегося в метане, который прореагировал. Сел.Кокс представляет собой среднюю селективность на основе молярной концентрации углерода, определяемой суммой молей углерода, оставшегося в реакторе, деленной на моли углерода, содержавшегося в метане, который прореагировал. Сумма Сел.БТН и Сел.Кокс не равна 100% из-за образования других небольших продуктов, преимущественно этилена. Поскольку часто затруднительно получить точные экспериментальные данные термодинамического превращения, для нахождения значения Превр.Бл использовалось распространяемое на коммерческой основе программное обеспечение (PROII/6,0 Copyright 2003 Invensys Systems Inc.) для моделирования. Превр.Бл определяется как максимально термодинамически достижимое превращение метана в бензол и водород (т.е. отсутствие модельного ограничения, благодаря которому отсутствуют другие продукты, такие как кокс, нафталин, этилен и т.д.) при данной температуре и абсолютном давлении 138 кПа (20 фунтов/кв.дюйм). Результаты эксперимента и моделирования представлены в таблице 5.
Разумеется, что количественные показатели селективности и превращения могут изменяться ввиду использования различных каталитических композиций, применения совместно подаваемых материалов (СО2, СО, H2O, H2, О2, этан, пропан и т.д.), различных рабочих давлений и/или различных объемных скоростей подачи сырья, но если точный уровень рассмотренного в описании улучшения может меняться, то общее направление рассмотренных в описании улучшений все же будет достигаться. Кроме того, необходимо иметь в виду, что при выполнении рассмотренных ниже моделирующих вычислений исходили из того, что направляемый в реактор метановый исходный материал всегда предварительно нагревался до одной и той же температуры (600°С), и во всех случаях использовалась номинальная скорость подачи метана 100 кг/ч. Также исходили из того, что температуру катализатора, направляемого в реакторные системы с подвижным слоем, поддерживали на одном и том же значении (850°С). Количество катализатора, необходимое для поддержания этой температуры, рассчитывали для каждой конфигурации реактора. Для простоты было принято допущение, что теплопроводность, коэффициент температуропроводности и излучательная способность поверхности катализатора оставались постоянными. В приведенной ниже таблице 6 перечислены физические постоянные и свойства катализатора, использованные в расчетах.
Для обеспечения возможности моделирования различных конфигураций реактора для Сел.БТН, Сел.Кокс и Превр.Бл вывели уравнения путем получения для приведенного выше набора экспериментальных точек многочленных уравнений по критерию наилучшей подгонки; экспериментальные точки, где в исходный материал включался Н2, для вычисления уравнений не привлекались. Полученные уравнения и значения R2 представлены ниже:
Сел.БТН=(1,81818181818345Е-10)Т4-(5,41010101010501Е-07)Т3+(5,88000000000377Е-04)Т2-(2,78591414141575Е-01)Т+4,97583333333585Е+01
R2 БTH=9,99810335105254Е-01
Сел.Кокс=(-1,85878787878687Е-10)Т4+(5,62280808080511Е-07)Т3-(6,21721666666349Е-04)Т2+(2,99664027416883Е-01)Т-5,33408809523590Е+01
R2 Кокс=9,99958406639717Е-01
Превр.Бл=(1,91428571428569Е-06)Т2-(1,81714285714283Е-03)Т+4,53357142857135Е-01
R2 Бл=9,99955208049633Е-01,
где Т - температура в °С.
Во всех примерах R2 - это коэффициент детерминации, который сопоставляет расчетные и фактические значения y и находится в интервале значений от 0 до 1. Если он равен 1, то в выборке отличная корреляция - различие между расчетным значением y и фактическим значением y отсутствует. Если коэффициент детерминации имеет другое крайнее значение - 0, уравнение регрессии не поможет при прогнозировании значения y. Вариант, используемый в настоящем описании, основан на анализе разложения дисперсий:
В приведенном выше определении
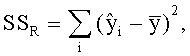
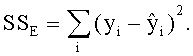
Другими словами, SST - это полная сумма квадратов, SSR - регрессионная сумма квадратов, a SSE - сумма квадратов погрешностей.
R2 БТН - коэффициент детерминации для корреляции Сел.БТН,
R2 Кокс - коэффициент детерминации для корреляции Сел.Кокс, и
R2 Бл - коэффициент детерминации для корреляции Превр.Бл.
Этот набор уравнений использовался для вычисления значений выхода, которые достигались бы для различных конфигураций реактора, где величина ВыходБТН была определена как произведение Сел.БТН×Превр.Бл, проинтегрированное по профилю распределения температур в реакторной системе, а величина ВыходКокс была определена как произведение Сел.Кокс×Превр.Бл, проинтегрированное по профилю распределения температур в реакторной системе. Хотя было установлено и показано в таблице 5, что побочный продукт H2 улучшает селективность реакции, эти уравнения такое улучшение селективности не учитывают, обеспечивая консервативную оценку в отношении уровня улучшения, обеспечиваемого при осуществлении предлагаемого способа.
Реактор с транспортом катализатора или лифт-реактор
Используя приведенные выше уравнения для реактора с транспортом катализатора, или лифт-реактора, с адиабатическим понижением температуры при температуре на входе 850°С, требуемую скорость циркуляции катализатора для поддержания на выходе температуры 800°С определили как 3211 кг в час (кг/ч) исходя из номинальной скорости подачи метана 100 кг/ч при 600°С.При этом были вычислены следующие значения выхода и селективности:
Сел.БТН=51%
Сел.Кокс=40%
выходБТН=12%
выходКокс=8,9%
ΔТреакции=-50°С (отрицательная температура 50°С),
где ΔТреакции определяется как температура продукта на выходе реакции (т.е. последняя температура, при которой каталитическая реакция протекает до того как углеводородный продукт выходит из реакторной системы) минус температура углеводородного сырья на входе в реакцию (т.е. первая температура, при которой протекает каталитическая реакция, когда углеводородное сырье поступает в реакторную систему).
Реактор с неподвижным слоем катализатора
Моделирование сопоставимых процессов в потенциальных системах с неподвижным слоем катализатора показало еще худшие характеристики, чем в случае реактора с транспортом катализатора или лифт-реактора, потому что в конфигурации с неподвижным слоем все тепло реакции должно было подводиться с метансодержащим потоком (поскольку для подвода тепла в реакционную зону движущийся катализатор не использовали). Следовательно, для работы реактора с неподвижным слоем требовалось нагревать метансодержащий поток до температуры, намного превышавшей требуемую температуру на выходе 800°С, что, таким образом приводило к увеличению модуля показателя ΔТреакции, составившего -60°С или еще больше по модулю со знаком минус.
Реактор с оседающим слоем катализатора
В случае моделирования оседающего слоя катализатора с обратным температурным профилем и перепадом между температурой подаваемого катализатора и температурой на выходе процесса, составлявшим 50°С, реактор работал на входе при 620°С, а на выходе - при 800°С, скорость циркуляции катализатора была уменьшена до 717 кг/ч, а результаты реакции улучшились:
Сел.БТН=89%
Сел.Кокс=7%
ВыходБТН=20%
ВыходКокс=1,5%
ΔТреакции=+180°C
Пример 9
Основываясь на предсказанных при моделировании преимуществах обратного температурного профиля, для подтверждения результатов моделирования была сконструирована установка лабораторного масштаба. Хотя модель была ориентирована на работу реакционной системы как системы с оседающим слоем катализатора, лабораторный реактор обладал неподвижным слоем катализатора с обратным температурным профилем, обусловленным применением внешних нагревателей. Во всех случаях экспериментально наблюдаемые степени превращения оказывались ниже степеней превращения, спрогнозированных с помощью модели. Это может быть обусловлено наличием связанных с экспериментом помех лабораторного масштаба, таких как обход слоя катализатора и/или обратное смешение, обусловленное гидродинамическим режимом, в котором работают реакторы лабораторного масштаба.
Катализатор Mo/ZSM-5 приготовили измельчением в шаровой мельнице 7,5 мас.% Мо (массовая процентная доля металла в общей массе катализатора) в виде МоО3 с NH4ZSM-5 носителем (имеющим отношение Si/Al2, равное 25) в течение 2 часов с последующим прокаливанием при 500°С в течение 5 часов на воздухе. Катализатор таблетировали, измельчили и просеяли для выделения частиц крупностью 20-40 меш. Каталитическое испытание Mo/ZSM-5 катализатора осуществляли в кварцевом реакторе внутренним диаметром 7 мм с неподвижным слоем при длине слоя примерно 18 см. Для того чтобы все слои были одинаковой длины, в качестве разбавителя слоя катализатора использовали инертные частицы кварца (крупностью 20-50 меш).
Функциональные свойства катализатора при дегидроциклизации метана до бензола определяли с использованием в качестве исходного материала смеси 95 мас.% СН4 и 5 мас.% аргона (аргон использовали в качестве внутреннего стандарта). Все экспериментальные данные реакции получали при абсолютном давлении 20 фунтов/кв.дюйм (138 кПа). Для определения концентраций продуктов отходивший из реакции поток анализировали с помощью масс-спектрометра.
Для сравнения было проведено десять отдельных экспериментов в отношении функциональных свойств катализатора. Во всех экспериментах катализатор активировали нагреванием в среде 15 об.% СН4, 80 об.% H2, 5 об.% Ar со скоростью нагрева 5°С/мин до 800°С и выдержкой в течение 30 мин. За этим следовало старение катализатора с пятью циклами реакции и регенерирования (также одинаковыми для всех десяти экспериментов). Каждая реакционная часть продолжалась 20 мин при 800°С в среде 95 об.% СН4 и 5 об.% Ar, использовавшейся в качестве исходного материала, при объемной скорости подачи исходного материала 1,4 ч-1, определявшейся по СН4. Каждая регенерационная часть состояла из подключения к источнику Н2, нагрева до 850°С с 10-минутной выдержкой и последующего охлаждения вновь до 800°С (с общим временем нахождения в среде Н2 14 мин). Эти десять экспериментов различались только их шестым реакционным циклом, который осуществляли в среде 95 об.% СН4 и 5 об.% Ar, использовавшейся в качестве исходного материала, в течение 4 ч. Условия для шестого цикла выбирали для сравнения влияния температурного профиля слоя катализатора при разных объемных скоростях подачи исходного материала. В частности, эксперименты с 1-5 проводили, варьируя объемной скоростью подачи исходного материала в пределах от 0,25 до 8 ч-1 при поддержании слоя катализатора в изотермических условиях при 800°С. В противоположность этому эксперименты 6-10 проводили в том же интервале значений объемной скорости, но с линейным градиентом температуры в слое катализатора, которая повышалась от 650°С на входе для исходного материала до 800°С на выходе для продуктов (обратный температурный профиль). Результаты определения функциональных свойств катализатора для десяти экспериментов во время реакционного цикла №6 сведены в таблице 7.
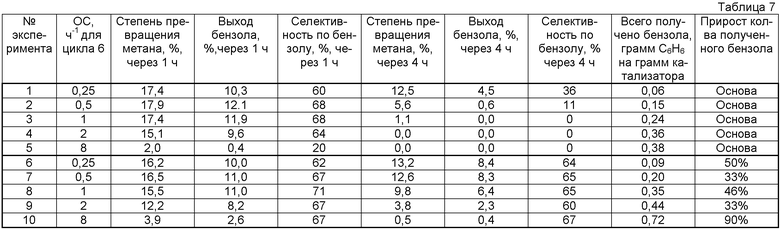
Результаты в таблице 7 показывают очевидное преимущество ведения процесса с обратным температурным профилем, который при большинстве значений объемной скорости подачи исходного материала улучшал мгновенную селективность в течение более коротких промежутков времени процесса и неизменно обеспечивал пролонгирование селективности по бензолу в течение более продолжительных периодов времени процесса. Это позволило увеличить суммарное количество продукции по сравнению с изотермическим слоем при всех объемных скоростях подачи исходного материала.
Хотя примеры 8 и 9 относятся к реакторам с оседающим слоем катализатора, аналогичное улучшение селективности было бы продемонстрировано и для других реакторных систем с эквивалентным обратным температурным профилем.
Как показало моделирование, улучшились значения выхода, селективности и скорости циркуляции катализатора. Кроме того, реакцию в целом можно осуществлять в одной реакционной зоне, таким образом сводя к минимуму потребность в оборудовании. Как возможный вариант, можно использовать и две или более реакционные зоны.
Как это иллюстрируется примерами 8 и 9, конфигурация реактора с оседающим слоем или иные реакторые системы с аналогичным обратным температурным профилем делают возможным превращение метана в более высокомолекулярные углеводороды, например ароматические соединения, при уменьшенных потерях из-за старения/механического истирания катализатора, улучшенной действенности и более высокой селективности, т.е. при меньшем образовании кокса, чем в конфигурациях с неподвижным слоем и/или с транспортом слоя или восходящим движением слоя в вертикальных трубных конфигурациях.
Пример 10
Этот пример призван показать влияние температуры регенерирования на стабильность степени превращения метана и селективности по бензолу и толуолу при дегидроциклизации метана за несколько циклов.
Катализатор 7,5 мас.% Mo/ZSM-5 приготовили размалыванием в шаровой мельнице оксида молибдена с носителем NH4 +-ZSM-5 (имеющим отношение Si/Al2, равное 25) в течение 2 часов с последующим прокаливанием при 500°С в течение 5 часов в токе воздуха. Катализатор таблетировали, измельчили и просеяли для выделения частиц крупностью 20-40 меш. Каталитическое испытание катализатора Mo/ZSM-5 проводили в кварцевом реакторе с неподвижным слоем. Образец катализатора массой приблизительно 300 мг поместили в трубчатый реактор диаметром 1/4'' (6,35 мм). В качестве разбавителя слоя катализатора использовали инертные частицы кварца (крупностью 20-40 меш).
Катализатор в синтезированном состоянии предварительно науглероживали нагреванием катализатора в газообразной смеси 15% СН4-Н2 со 150°С до 800°С со скоростью 5°С/мин и выдерживали при 800°С в течение 0,5 часа. Затем катализатор подвергли циклическому старению, которое состояло из: (1) периода контакта с углеводородом, когда на катализатор воздействовали углеводородным исходным материалом (молярный состав: 13,4% H2, 78,2% СН4, 1,8% СО, 1,5% С6Н6, 0,2% С2Н4 и 4,9% Ar) при 785°С, абсолютном давлении 23 фунтов/кв.дюйм (159 кПа) и объемной скорости 5 ч-1 по CH4, и (2) последующего периода регенерирования, где катализатор нагревали примерно до 850-925°С в среде газообразного H2 при абсолютном давлении 100 фунтов/кв.дюйм (689 кПа) в течение заданного времени при объемной скорости подачи газа 34000 см [в нормальных условиях]/г катализатора/ч. После завершения стадии регенерирования катализатор перед повторной подачей углеводородного исходного материала охлаждали до 785°С.Отходящий поток реактора анализировали методами ГХ и МС в режиме реального времени.
В течение первых 3670 циклов катализатор подвергали действию углеводородного исходного материала в течение 4-6 минут при 785°С и регенерировали в среде Н2 в течение 6-12 минут при 850°С или 875°С для каждого цикла. По мере увеличения числа циклов катализатор проявлял все более быстрое ухудшение активности в отношении степени превращения метана и селективности по бензолу и толуолу. Для восстановления активности катализатора его регенерировали во время этих циклов 8 раз в течение 12-23 часов. Продолжительные регенерации могли временно восстановить катализатор, но его функциональные свойства все равно быстро ухудшались.
На фиг.5А и 5Б показана характеристика изменения выраженной в процентах степени превращения метана и селективности по бензолу и толуолу при повышении температур регенерации с 875°С до 925°С.После цикла 3610 катализатор регенерировали в среде H2 при 900°С в течение 20 часов, затем катализатор вернули в циклический процесс и в каждом цикле между циклами 3610 и 3670 его подвергали действию углеводородного исходного материала в течение 4 минут при 785°С и регенерировали в среде Н2 в течение 12 минут при 875°С. Катализаторы проявили быстрое ухудшение активности в отношении превращения метана и селективности по бензолу и толуолу. После цикла 3670 катализатор регенерировали в среде Н2 при 925°С в течение 20 часов, затем катализатор вернули в циклический процесс и в каждом цикле между циклами 3680 и 3800 его подвергали действию углеводородного исходного материала в течение 4 минут при 785°С и регенерировали в среде Н2 в течение 12 минут при 925°С. В случае регенерирования при температуре 925°С катализатор показал стабильность степени превращения метана и селективности по бензолу и толуолу, демонстрируя преимущество более высокой температуры регенерирования для более старого катализатора.
Другие варианты осуществления изобретения
1. Способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий:
(а) подачу в реакционную зону метансодержащего исходного материала и зернистого каталитического материала,
(б) обеспечение работы реакционной зоны в условиях, эффективных для превращения по меньшей мере части метана в более высокомолекулярный(-ые) углеводород(-ы) с сопутствующим отложением углеродистого материала на зернистом каталитическом материале, вызывающим дезактивацию каталитического материала,
(в) удаление из реакционной зоны по меньшей мере части дезактивированного зернистого каталитического материала,
(г) нагревание по меньшей мере части удаленного из реакционной зоны зернистого каталитического материала до температуры примерно от 700°С примерно до 1200°С, осуществляемое прямым и/или косвенным контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива,
(д) регенерирование нагретой части зернистого каталитического материала водородсодержащим газом в первой регенерационной зоне в условиях, эффективных для превращения по меньшей мере части отложившегося углеродистого материала в метан, и
(е) возврат по меньшей мере части зернистого каталитического материала со стадии (д) в реакционную зону.
2. Способ по п.1, в котором реакционная зона представляет собой реакционную зону с подвижным слоем.
3. Способ по п.2, в котором реакционная зона с подвижным слоем работает с обратным температурным профилем.
4. Способ по любому из предыдущих пунктов, в котором исходный материал содержит также по меньшей мере одно из следующих веществ: СО, CO2, Н2, H2O и/или O2.
5. Способ по любому из предыдущих пунктов, в котором удаленный из реакционной зоны зернистый каталитический материал нагревают на стадии (г) до температуры примерно от 800°С примерно до 1000°С, предпочтительно примерно от 850°С примерно до 950°С.
6. Способ по любому из предыдущих пунктов, в котором при нагревании на стадии (г) каталитический материал вводят в прямой контакт с дополнительным источником топлива или газообразными продуктами его горения.
7. Способ по любому из предыдущих пунктов, в котором каталитический материал нагревают путем прямого контакта с инертной средой или поверхностью теплообмена, нагреваемой газообразными продуктами горения.
8. Способ по любому из предыдущих пунктов, в котором дополнительный источник топлива содержит углеводород и/или водород.
9. Способ по любому из предыдущих пунктов, в котором дополнительный источник топлива содержит метан.
10. Способ по любому из предыдущих пунктов, в котором дополнительный источник топлива содержит углеводород, и этот углеводород сжигают в бедной кислородом атмосфере с получением синтез-газа.
11. Способ по любому из предыдущих пунктов, в котором дополнительный источник топлива содержит водород.
12. Способ по любому из предыдущих пунктов, в котором дополнительный источник топлива содержит водород, образующийся в качестве побочного продукта процесса превращения метана.
13. Способ по любому из предыдущих пунктов, включающий также:
(ж) удаление из реакционной зоны дополнительной части зернистого каталитического материала,
(з) нагревание дополнительной части зернистого каталитического материала путем прямого и/или косвенного контакта с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива, и
(и) возврат по меньшей мере части зернистого каталитического материала со стадии (з) в реакционную зону.
14. Способ по любому из предыдущих пунктов, включающий также:
(i) перемещение второй части удаленного из реакционной зоны зернистого каталитического материала со стадии (в) или стадии (д) во вторую регенерационную зону,
(ii) регенерирование второй части зернистого каталитического материала водородсодержащим газом во второй регенерационной зоне в условиях, включающих в себя давление, отличающееся от давления в первой регенерационной зоне, и эффективных для превращения по меньшей мере части отложившихся на зернистом каталитическом материале углеродистых материалов в метан, и
(iii) возврат по меньшей мере части регенерированного зернистого каталитического материала со стадии (ж) в реакционную зону.
15. Способ по п.14, в котором условия регенерирования (ii) включают в себя абсолютное давление, составляющее по меньшей мере 500 кПа, предпочтительно примерно от 1000 кПа примерно до 5000 кПа.
16. Способ по п.14 или 15, в котором давление во второй регенерационной зоне создают посредством вертикального столба зернистого каталитического материала.
17. Способ по любому из предыдущих пунктов, в котором условия регенерирования на стадии (д) включают в себя абсолютное давление, составляющее по меньшей мере 100 кПа, предпочтительно примерно от 150 кПа примерно до 5000 кПа.
18. Способ по любому из предыдущих пунктов, в котором давление в первой регенерационной зоне создают посредством вертикального столба зернистого каталитического материала.
19. Способ по любому из предыдущих пунктов, в котором условия реакции, протекающей в реакционной зоне на стадии (б), включают в себя температуру примерно от 400°С примерно до 1200°С, абсолютное давление примерно от 1 кПа примерно до 1000 кПа и объемную скорость подачи исходного материала примерно от 0,01 ч-1 примерно до 1000 ч-1.
20. Способ по любому из предыдущих пунктов, в котором зернистый каталитический материал является катализатором дегидроциклизации, содержащим металл или его соединение на неорганическом носителе.
21. Способ по любому из предыдущих пунктов, в котором зернистый каталитический материал включает в себя по меньшей мере одно из следующих веществ: молибден, вольфрам, рений, соединение молибдена, соединение вольфрама, соединение цинка и соединение рения, на ZSM-5, диоксиде кремния или оксиде алюминия.
22. Способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий:
(а) подачу метансодержащего исходного материала в реакционную зону, содержащую зернистый каталитический материал,
(б) обеспечение работы реакционной зоны в условиях, эффективных для превращения по меньшей мере части метана в более высокомолекулярный(-ые) углеводород(-ы) с сопутствующим отложением углеродистого материала на зернистом каталитическом материале, вызывающим дезактивацию каталитического материала,
(в) прерывание подачи метансодержащего исходного материала в реакционную зону,
(г) нагревание реакционной зоны до температуры регенерирования, составляющей от 700 до 1200°С, осуществляемое прямым и/или косвенным контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива,
(д) регенерирование находящегося в нагретой реакционной зоне зернистого каталитического материала водородсодержащим газом в условиях, эффективных для превращения по меньшей мере части отложившегося углеродистого материала в метан, и
(е) возобновление подачи метансодержащего исходного материала в реакционную зону.
23. Способ по п.22, в котором подачу метансодержащего исходного материала прерывают после нагревания на стадии (г).
24. Способ по п.22 или 23, в котором нагревание на стадии (г) проводят в присутствии водородсодержащего газа.
25. Способ по любому из п.п.22-24, в котором реакционная зона является реакционной зоной с неподвижным слоем.
26. Способ по любому из п.п.22-25, в котором условия регенерирования на стадии (д) включают в себя абсолютное давление, составляющее по меньшей мере 100 кПа.
Хотя настоящее изобретение описано и проиллюстрировано со ссылкой на конкретные варианты его выполнения, для специалиста очевидна возможность осуществления изобретения и в вариантах, отличных от рассмотренных выше. По этой причине для определения истинного объема охраны настоящего изобретения следует обращаться только к его формуле.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2007 |
|
RU2459789C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ АЛИФАТИЧЕСКИХ | 2008 |
|
RU2461537C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ МЕТАНА | 2007 |
|
RU2454389C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2007 |
|
RU2454390C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2006 |
|
RU2408565C2 |
| ПОЛУЧЕНИЕ АЛКИЛИРОВАННЫХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2005 |
|
RU2417974C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ МЕТАНА | 2007 |
|
RU2448079C2 |
| ПОЛУЧЕНИЕ ЖИДКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2005 |
|
RU2405764C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2005 |
|
RU2418780C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2009 |
|
RU2514915C2 |
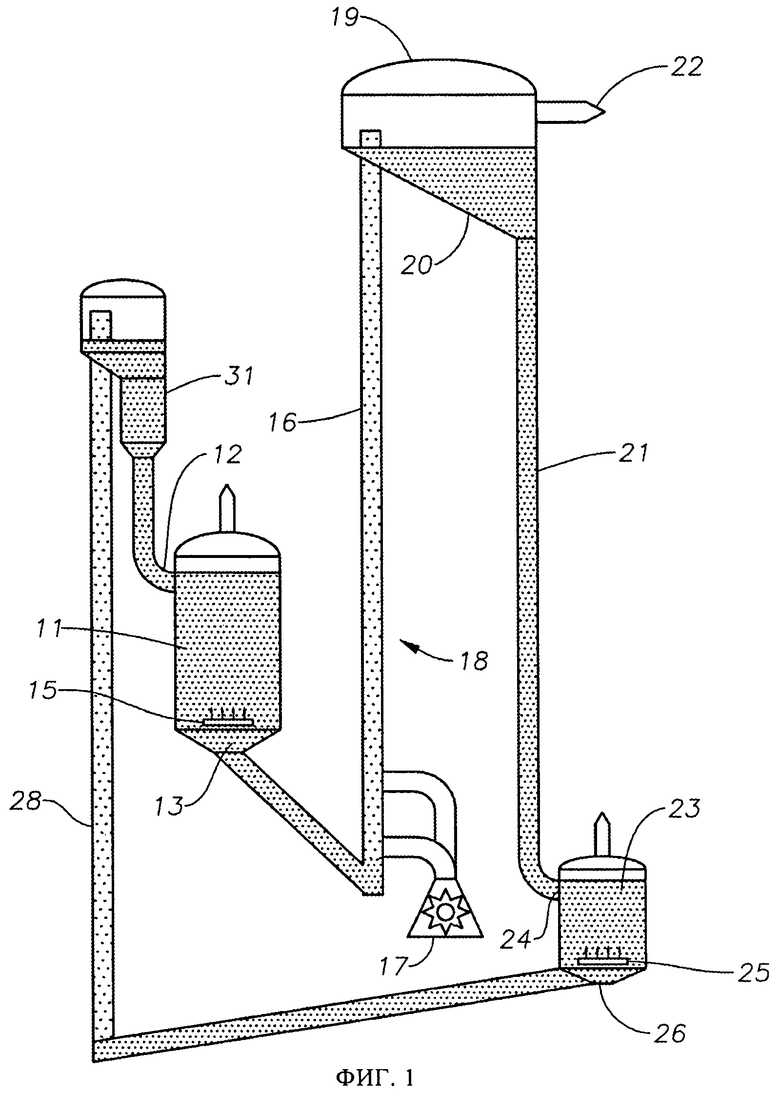
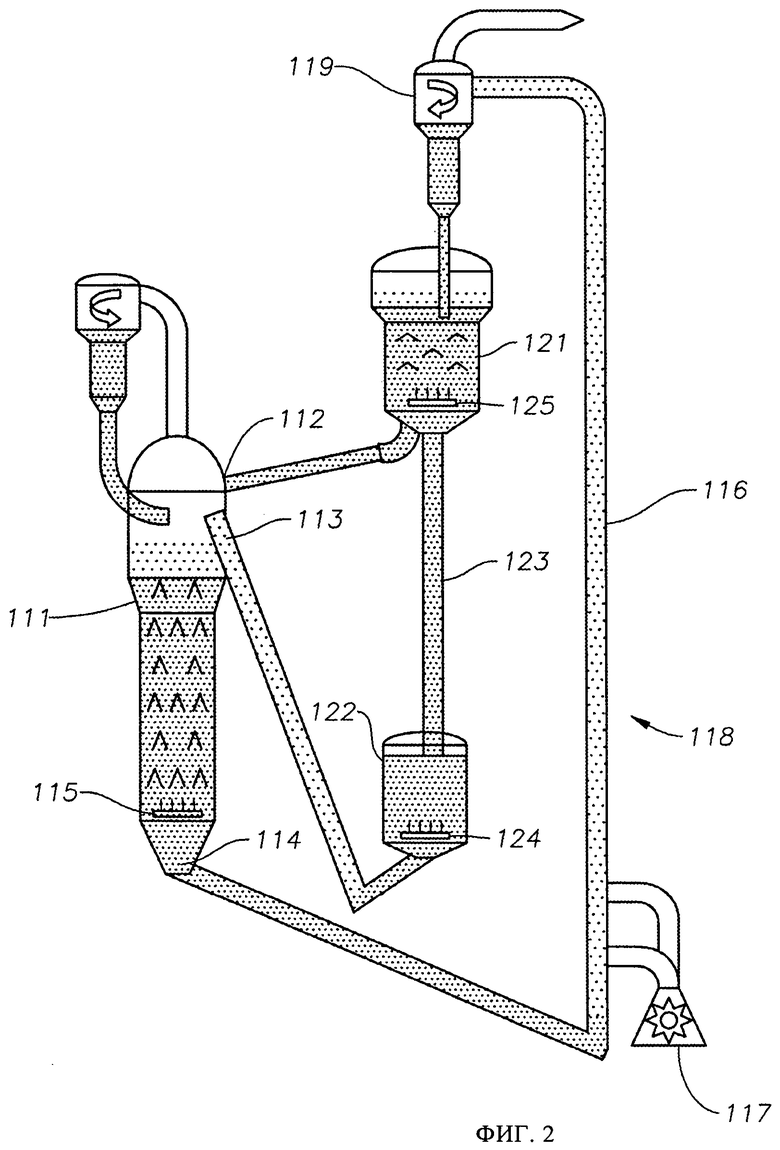
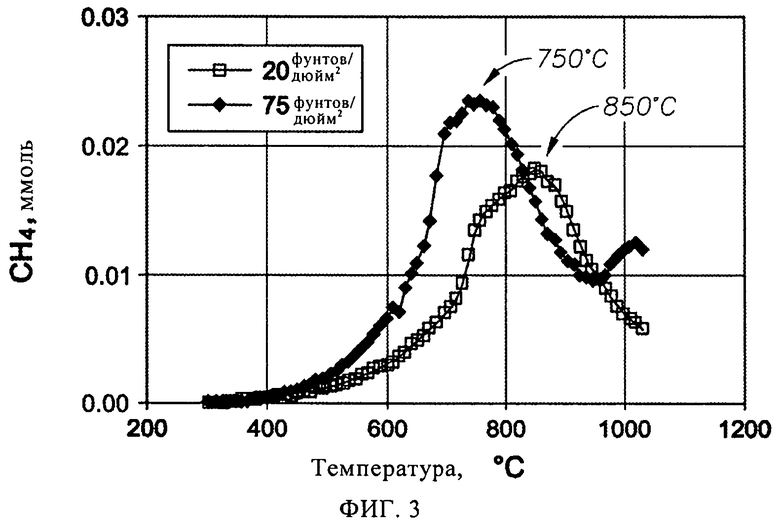
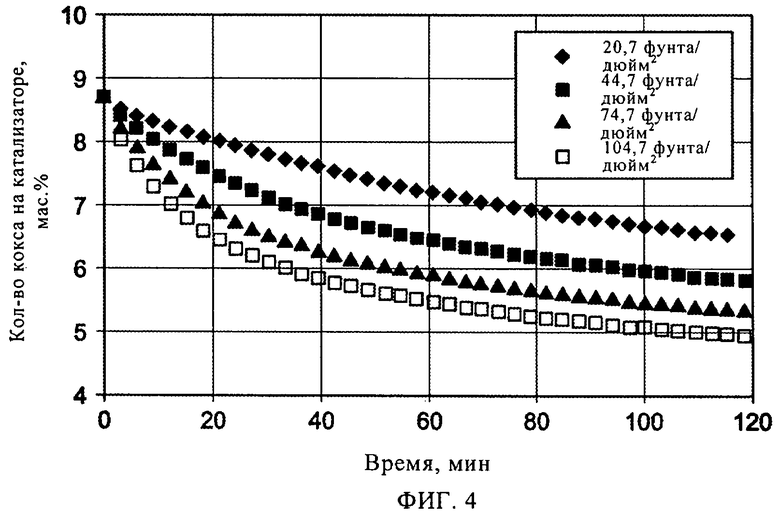
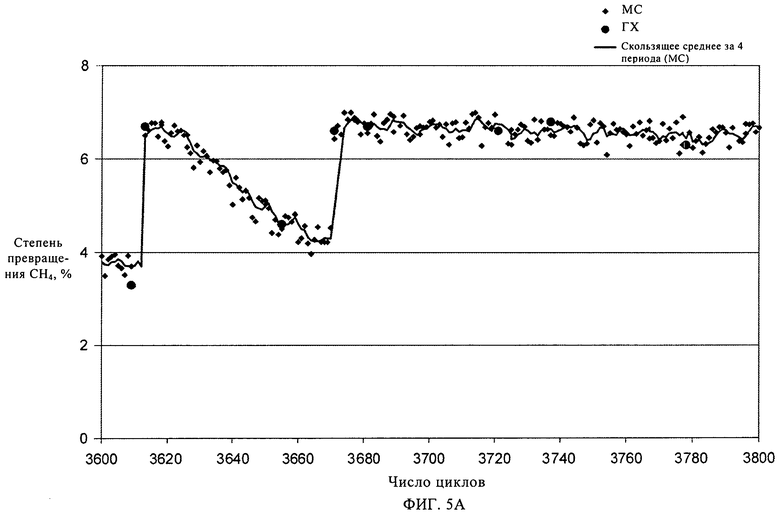
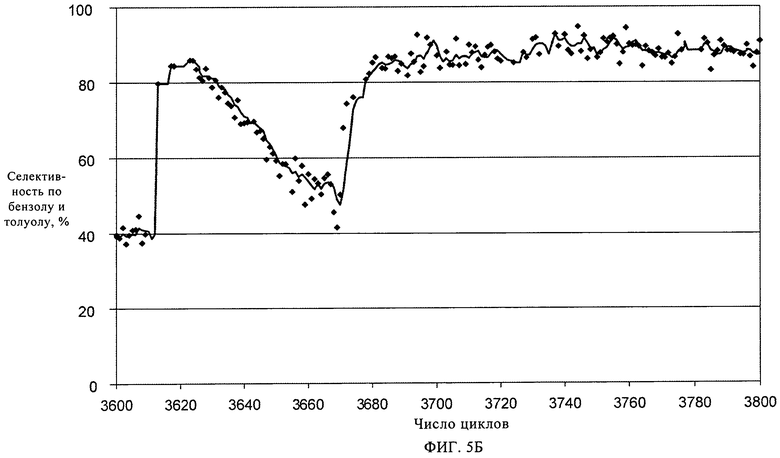
Изобретение относится к способу получения ароматических углеводородов из метана, в частности природного газа. Изобретение касается способа превращения метана в ароматические углеводороды, в котором в реакционную зону подают метансодержащий исходный материал и зернистый каталитический материал, обеспечивают работу реакционной зоны в условиях реакции, эффективных для превращения по меньшей мере части метана в ароматические углеводороды с сопутствующим отложением углеродистого материала на зернистом каталитическом материале, вызывающего его (катализатора) дезактивацию. По меньшей мере часть дезактивированного зернистого каталитического материала удаляют из реакционной зоны и нагревают до температуры примерно от 700°С примерно до 1200°С прямым или косвенным контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива. Затем нагретый зернистый каталитический материал регенерируют водородсодержащим газом в условиях, эффективных для превращения по меньшей мере части отложившегося углеродистого материала в метан, и возвращают регенерированный зернистый каталитический материал обратно в реакционную зону. Технический результат - усовершенствование способа ароматизации метана. 2 н. и 23 з.п. ф-лы, 6 ил., 7 табл., 10 пр.
1. Способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий:
(а) подачу в реакционную зону метансодержащего исходного материала и зернистого каталитического материала,
(б) обеспечение работы реакционной зоны в условиях, эффективных для превращения по меньшей мере части метана в более высокомолекулярный(-ые) углеводород(-ы) с сопутствующим отложением углеродистого материала на зернистом каталитическом материале, вызывающим дезактивацию каталитического материала,
(в) удаление из реакционной зоны по меньшей мере части дезактивированного зернистого каталитического материала,
(г) нагревание по меньшей мере части удаленного из реакционной зоны зернистого каталитического материала до температуры примерно от 700°С примерно до 1200°С, осуществляемое прямым или косвенным контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива,
(д) регенерирование нагретой части зернистого каталитического материала водородсодержащим газом в первой регенерационной зоне в условиях, эффективных для превращения по меньшей мере части отложившегося углеродистого материала в метан, и
(е) возврат по меньшей мере части зернистого каталитического материала со стадии (д) в реакционную зону.
2. Способ по п.1, в котором реакционная зона представляет собой реакционную зону с подвижным слоем.
3. Способ по п.2, в котором реакционная зона с подвижным слоем работает с обратным температурным профилем.
4. Способ по одному из пп.1-3, в котором исходный материал содержит также по меньшей мере одно из следующих веществ: СО, СО2, Н2, Н2О и/или О2.
5. Способ по одному из пп.1-3, в котором удаленный из реакционной зоны зернистый каталитический материал нагревают на стадии (г) до температуры примерно от 800°С примерно до 1000°С.
6. Способ по одному из пп.1-3, в котором при нагревании на стадии (г) каталитический материал вводят в прямой контакт с дополнительным источником топлива или газообразными продуктами его горения.
7. Способ по одному из пп.1-3, в котором каталитический материал нагревают путем прямого контакта с инертной средой или поверхностью теплообмена, нагреваемой газообразными продуктами горения.
8. Способ по одному из пп.1-3, в котором дополнительный источник топлива содержит углеводород и/или водород.
9. Способ по одному из пп.1-3, в котором дополнительный источник топлива содержит метан.
10. Способ по одному из пп.1-3, в котором дополнительный источник топлива содержит углеводород, и этот углеводород сжигают в бедной кислородом атмосфере с получением синтез-газа.
11. Способ по одному из пп.1-3, в котором дополнительный источник топлива содержит водород.
12. Способ по одному из пп.1-3, в котором дополнительный источник топлива содержит водород, образующийся в качестве побочного продукта процесса превращения метана.
13. Способ по одному из пп.1-3, включающий также:
(ж) удаление из реакционной зоны дополнительной части зернистого каталитического материала,
(з) нагревание дополнительной части зернистого каталитического материала путем прямого или косвенного контакта с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива, и
(и) возврат по меньшей мере части зернистого каталитического материала со стадии (з) в реакционную зону.
14. Способ по одному из пп.1-3, включающий также:
(i) перемещение второй части удаленного из реакционной зоны зернистого каталитического материала со стадии (в) или стадии (д) во вторую регенерационную зону,
(ii) регенерирование второй части зернистого каталитического материала водородсодержащим газом во второй регенерационной зоне в условиях, включающих в себя давление, отличающееся от давления в первой регенерационной зоне, и эффективных для превращения по меньшей мере части отложившихся на зернистом каталитическом материале углеродистых материалов в метан, и
(iii) возврат по меньшей мере части регенерированного зернистого каталитического материала со стадии (ж) в реакционную зону.
15. Способ по п.14, в котором давление во второй регенерационной зоне создают посредством вертикального столба зернистого каталитического материала.
16. Способ по одному из пп.1-3, в котором условия регенерирования на стадии (д) включают в себя абсолютное давление, составляющее по меньшей мере 100 кПа, предпочтительно примерно от 150 кПа примерно до 5000 кПа.
17. Способ по одному из пп.1-3, в котором давление в первой регенерационной зоне создают посредством вертикального столба зернистого каталитического материала.
18. Способ по одному из пп.1-3, в котором условия реакции, протекающей в реакционной зоне на стадии (б), включают в себя температуру примерно от 400°С примерно до 1200°С, абсолютное давление примерно от 1 кПа примерно до 1000 кПа и объемную скорость подачи исходного материала примерно от 0,01 ч-1 примерно до 1000 ч-1.
19. Способ по одному из пп.1-3, в котором зернистый каталитический материал является катализатором дегидроциклизации, содержащим металл или его соединение на неорганическом носителе.
20. Способ по одному из пп.1-3, в котором зернистый каталитический материал включает в себя по меньшей мере одно из следующих веществ: молибден, вольфрам, рений, соединение молибдена, соединение вольфрама, соединение цинка и соединение рения, на ZSM-5, диоксиде кремния или оксиде алюминия.
21. Способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий:
(а) подачу метансодержащего исходного материала в реакционную зону, содержащую зернистый каталитический материал,
(б) обеспечение работы реакционной зоны в условиях, эффективных для превращения по меньшей мере части метана в более высокомолекулярный(-ые) углеводород(-ы) с сопутствующим отложением углеродистого материала на зернистом каталитическом материале, вызывающим дезактивацию каталитического материала,
(в) прерывание подачи метансодержащего исходного материала в реакционную зону,
(г) нагревание реакционной зоны до температуры регенерирования, составляющей от 700 до 1200°С, осуществляемое прямым или косвенным контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива,
(д) регенерирование находящегося в нагретой реакционной зоне зернистого каталитического материала водородсодержащим газом в условиях, эффективных для превращения по меньшей мере части отложившегося углеродистого материала в метан, и
(е) возобновление подачи метансодержащего исходного материала в реакционную зону.
22. Способ по п.21, в котором подачу метансодержащего исходного материала прерывают после нагревания на стадии (г).
23. Способ по п.21 или 22, в котором нагревание на стадии (г) проводят в присутствии водородсодержащего газа.
24. Способ по п.21 или 22, в котором реакционная зона является реакционной зоной с неподвижным слоем.
25. Способ по п.21 или 22, в котором условия регенерирования на стадии (д) включают в себя абсолютное давление, составляющее по меньшей мере 100 кПа.
| WO 03000826 А2, 03.01.2003 | |||
| Устройство для резки чайного листа и т.п. | 1938 |
|
SU57278A1 |
| WO 2006068814 А2, 29.06.2006 | |||
| СПОСОБ ПЕРЕРАБОТКИ УГЛЕВОДОРОДНОГО СЫРЬЯ НА ОСНОВЕ АЛИФАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 1998 |
|
RU2152977C1 |

