ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет для заявки на патент US №60/794280, поданной 21 апреля 2006 г., содержание которой в полном объеме включено в настоящее описание в качестве ссылки. По настоящей заявке испрашивается также приоритет для заявки на патент US №60/794058, поданной 21 апреля 2006 г., содержание которой в полном объеме включено в настоящее описание в качестве ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способу получения ароматических углеводородов из метана и, в частности, из природного газа.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Ароматические углеводороды, в особенности бензол, толуол, этилбензол и ксилолы, являются важными химическими продуктами массового производства в нефтехимической промышленности. В настоящее время ароматические соединения наиболее часто получают по разнообразным методам из исходных материалов на основе сырой нефти, включая каталитический реформинг и каталитический крекинг. Однако по мере того как мировые поставки исходных материалов на основе сырой нефти уменьшаются, возрастает потребность найти альтернативные источники ароматических углеводородов.
Одним возможным альтернативным источником ароматических углеводородов служит метан, который является основным компонентом природного газа и биогаза. Объем разведанных мировых запасов природного газа постоянно увеличивается, и в настоящее время открывают больше месторождений природного газа, чем нефти. Из-за проблем, связанных с транспортировкой больших объемов природного газа, большую часть природного газа, добываемого вместе с нефтью, в особенности в отдаленных местах, сжигают в факеле и направляют в отход. Следовательно, привлекательным методом повышения сортности природного газа является превращение алканов, содержащихся в природном газе, непосредственно в более высокомолекулярные углеводороды, такие как ароматические соединения, при условии, что могут быть преодолены сопутствующие этому технические трудности.
Значительная часть предлагаемых в настоящее время способов превращения метана в жидкие углеводороды включает вначале превращение метана в синтез-газ, смесь Н2 и СО. Однако получение синтез-газа связано с большими капитальными затратами и является энергоемким; следовательно, предпочтительны пути, которые не требуют генерирования синтез-газа.
Предложен ряд альтернативных способов прямого превращения метана в более высокомолекулярные углеводороды. Один такой способ включает каталитическое окислительное сочетание метана до олефинов с последующим каталитическим превращением олефинов в жидкие углеводороды, включающие ароматические углеводороды. Так, например, в US №5336825 описан двухстадийный способ окислительного превращения метана в углеводороды с пределами кипения бензиновой фракции, включающие ароматические углеводороды. На первой стадии в присутствии свободного кислорода с использованием промотированного редкоземельным металлом катализатора из оксида щелочноземельного металла при температуре в пределах от 500 до 1000°С метан превращают в этилен и небольшие количества С3- и С4олефинов. Затем этилен и более высокомолекулярные олефины, образовавшиеся на первой стадии, над кислотным твердым катализатором, включающим пентасиловый цеолит с высоким содержанием диоксида кремния, превращают в жидкие углеводороды с пределами кипения бензиновой фракции.
Однако эти способы окислительного сочетания страдают проблемами, заключающимися в том, что они включают высокоэкзотермические и потенциально опасные реакции сжигания метана, и в том, что при их осуществлении образуются большие количества воздействующих на окружающую среду оксидов углерода.
Потенциально привлекательный путь повышения сортности метана непосредственно до более высокомолекулярных углеводородов, в особенности до этилена, бензола и нафталина, заключается в дегидроароматизации или восстановительном сочетании. Этот метод, как правило, включает контактирование метана с катализатором, включающим такой металл, как рений, вольфрам и молибден, нанесенный на цеолит, такой как ZSM-5, при высокой температуре, в частности от 600 до 1000°С. Часто каталитически активные материалы в виде металлов находятся в форме элемента с нулевой валентностью, карбида или оксикарбида.
Например, в US №4727206 описан способ получения жидкостей, богатых ароматическими углеводородами, введением метана при температуре в пределах от 600 до 800°С в отсутствие кислорода в контакт с каталитической композицией, включающей алюмосиликат, обладающий молярным отношением диоксида кремния к оксиду алюминия по меньшей мере 5:1, причем упомянутый алюмосиликат вводят с (I) галлием или его соединением и (II) металлом группы VIIB Периодической таблицы элементов или его соединением.
Кроме того, в US №5026937 описан способ ароматизации метана, который включает стадии подачи потока исходных материалов, который включает больше 0,5 мольного % водорода и 50 мольных % метана, в реакционную зону, содержащую по меньшей мере один слой твердого катализатора, включающего ZSM-5, галлий и фосфорсодержащий оксид алюминия, в условиях превращения, которые включают температуру от 550 до 750°С, абсолютное давление ниже 10 ат (1000 кПа) и среднечасовую скорость подачи газа от 400 до 7500 ч-1.
В US №№6239057 и 6426442 описан способ получения углеводородов с более высоким числом углеродных атомов, например бензола, из углеводородов с низким числом углеродных атомов, таких как метан, введением этого последнего в контакт с катализатором, включающим пористый носитель, такой как ZSM-5, который содержит диспергированный на нем рений и промоторный металл, такой как железо, кобальт, ванадий, марганец, молибден, вольфрам, или их смесь. После пропитки носителя рением и промоторным металлом катализатор активируют обработкой водородом и/или метаном при температуре от примерно 100 до примерно 800°С в течение времени от примерно 0,5 до примерно 100 ч. Добавление СО или СО2 в метановый исходный материал повышает выход бензола и стабильность катализатора.
Соответственно, существует потребность в разработке способа превращения метана в более высокомолекулярный углеводород (углеводороды), который обеспечивает высокую эффективность теплопереноса, адекватное контактирование углеводорода/катализатора и/или улучшенные технологические условия для достижения максимальной селективности в отношении целевых более высокомолекулярных углеводородов, например ароматического соединения (соединений), при одновременном сведении к минимуму коксообразования.
Объектом изобретения, представленного в настоящем описании, является разработка усовершенствованного способа ароматизации метана, в котором эксплуатационные свойства улучшают путем регулирования температуры и композиционного профиля в реакторной системе.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном варианте объектом настоящего изобретения является способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий следующие стадии:
а) подача в реакционную зону углеводородного исходного материала, содержащего метан;
б) подача внутрь реакционной зоны некоторого количества каталитического материала;
в) поддержание в реакционной зоне обратного температурного профиля и
г) работа реакционной зоны в реакционных условиях, достаточных для превращения по меньшей мере части метана в первый отходящий поток, содержащий более высокомолекулярный углеводород (углеводороды).
В другом варианте объектом настоящего изобретения является способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, в двух или большем числе реакционных зон, работающих последовательно, включающий следующие стадии:
а) подача внутрь каждой реакционной зоны некоторого количества каталитического материала;
б) подача в первую реакционную зону углеводородного исходного материала, содержащего метан;
в) перенос по меньшей мере части отходящего потока первой реакционной зоны во вторую реакционную зону;
г) поддержание в первой реакционной зоне более низкой средней температуры, чем во второй реакционной зоне; и
д) работа реакционных зон в реакционных условиях, достаточных для превращения по меньшей мере части метана в первый отходящий поток, содержащий более высокомолекулярный углеводород (углеводороды).
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 представлена схема осуществления способа превращения метана в более высокомолекулярные углеводороды в соответствии с первым вариантом выполнения изобретения.
На фиг.2 представлена схема осуществления способа превращения метана в более высокомолекулярные углеводороды в соответствии со вторым вариантом выполнения изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ВЫПОЛНЕНИЯ ИЗОБРЕТЕНИЯ
Используемое в настоящем описании понятие "более высокомолекулярный углеводород (углеводороды)" служит для обозначения углеводорода (углеводородов), содержащего больше одного углеродного атома на молекулу, оксигената, содержащего по меньшей мере один углеродный атом на молекулу, например этана, этилена, пропана, пропилена, бензола, толуола, ксилолов, нафталина и/или метилнафталина; и/или органического соединения (соединений), включающего по меньшей мере один углеродный атом и по меньшей мере один неводородный атом, например метанола, этанола, метиламина и/или этиламина.
Используемое в настоящем описании понятие "ароматический углеводород (углеводороды)" служит для обозначения вещества, содержащего одно или несколько ароматических колец. Примерами ароматических углеводородов являются бензол, толуол, ксилолы, нафталин и метилнафталины.
Понятия "кокс" и "углеродистый материал" используют в настоящем описании как взаимозаменяемые для обозначения углеродсодержащих материалов, которые в реакционных условиях представляют собой, по существу, нелетучие твердые материалы с низким содержанием водорода относительно содержания углерода (в частности значение молярного соотношения Н/С меньше 0,8, наиболее вероятно меньше 0,5). Они могут включать кристаллический графит, графитовые листовые материалы, графитовые фрагменты, аморфный углерод или другие углеродсодержащие структуры, которые в реакционных условиях представляют собой, по существу, нелетучие твердые материалы. Когда ссылаются на твердый кокс, более твердый кокс или огнеупорный кокс, это означает ссылку на типы кокса либо благодаря структуре, либо вследствие участка, который труднее удалить с помощью реагента (как правило, кислорода или водорода), используемого для превращения кокса в газообразные материалы.
Используемое в настоящем описании понятие "дезактивация" катализатора означает потерю с течением времени каталитической активности и/или селективности. Катализатор оказывается дезактивированным, если его каталитическая активность по меньшей мере на 1% ниже, по другому варианту по меньшей мере на 5% ниже, по другому варианту по меньшей мере на 10% ниже, по другому варианту по меньшей мере на 15% ниже, по другому варианту по меньшей мере на 20% ниже, по другому варианту по меньшей мере на 25% ниже, по другому варианту по меньшей мере на 30% ниже, по другому варианту по меньшей мере на 35% ниже, по другому варианту по меньшей мере на 40% ниже, по другому варианту по меньшей мере на 45% ниже, по другому варианту по меньшей мере на 50% ниже, по другому варианту по меньшей мере на 55% ниже, по другому варианту по меньшей мере на 60% ниже, по другому варианту по меньшей мере на 65% ниже, по другому варианту по меньшей мере на 70% ниже, по другому варианту по меньшей мере на 75% ниже, по другому варианту по меньшей мере на 80% ниже, по другому варианту по меньшей мере на 85% ниже, по другому варианту по меньшей мере на 90% ниже, по другому варианту по меньшей мере на 95% ниже или по другому варианту по меньшей мере на 100% ниже, чем каталитическая активность свежего катализатора или регенерированного катализатора. Не основываясь на какой-либо теории, полагают, что дезактивация катализатора может быть явлением, в котором структура и/или состояние катализатора меняются, что приводит к потере активных участков на поверхности катализатора и, таким образом, вызывает ухудшение эксплуатационных свойств катализатора. Так, например, дезактивация катализатора может происходить вследствие коксообразования, блокирования активных участков или деалюминирования алюмосиликатного молекулярного сита вследствие обработки водяным паром.
Используемое в настоящем описании понятие "реактор с подвижным слоем" означает зону или сосуд с контактированием твердых частиц и газообразных потоков таким образом, что расход газа на единицу сечения потока (U) ниже скорости, необходимой для пневмотранспортировки с разбавленной фазой твердых частиц с целью сохранения слоя твердых частиц с долей свободного пространства меньше 95%. Реактор с подвижным слоем может работать в нескольких режимах потоков, включая отстойный или режим движения уплотненного слоя (U<Umf), псевдоожиженный режим (Umf<U<Umb), режим с комкованием частиц (Umb<U<Uc), переходный к турбулентному и собственно турбулентный режим псевдоожижения (Uc<U<Utr) и режим с высокой скоростью потока (U>Utr). Эти разные режимы псевдоожижения описаны, например, в работах Kunii, D., Levenspiel, О., глава 3, Fluidization Engineering, издание 2-е, Butterworth-Heinemann, Boston, 1991, и Walas, S.M., глава 6, Chemical Process Equipment, Butterworth-Heinemann, Boston, 1990.
Используемое в настоящем описании понятие "отстойный слой" служит для обозначения зоны или сосуда, в котором частицы контактируют с газообразными потоками таким образом, что расход газа на единицу сечения потока (U) меньше минимальной скорости, необходимой для псевдоожижения твердых частиц, минимальной скорости псевдоожижения (Umf), U<Umf, в по меньшей мере части реакционной зоны, и/или работающего со скоростью, которая выше минимальной скорости псевдоожижения при одновременном сохранении градиента в свойстве газа и/или твердого вещества (таком как температура, состав газа или твердого вещества и т.д.) вдоль оси снизу вверх в реакторном слое при применении внутрикорпусных устройств реактора с целью свести к минимуму обратное перемешивание газа и твердого вещества. Описание минимальной скорости псевдоожижения приведено, например, в главе 3 работы "Fluidization Engineering", D.Kunii и O.Levenspiel, издание 2-е, Butterworth-Heinemann, Boston, 1991 и главе 6 работы "Chemical Process Equipment" S.M.Walas, Butterworth-Heinemann, Boston, 1990, которые в полном объеме включены в качестве ссылок.
Используемое в настоящем описании понятие "реактор с псевдоожиженным слоем" означает зону или сосуд с контактированием твердых частиц и газообразных потоков таким образом, что расход газа на единицу сечения потока (U) достаточен для псевдоожижения твердых частиц (т.е. выше минимальной скорости псевдоожижения Umf) и ниже скорости, необходимой для пневмотранспортировки с разбавленной фазой твердых частиц с целью сохранить слой твердых частиц с долей свободного пространства меньше 95%. Используемое в настоящем описании понятие "каскадированные псевдоожиженные слои" служит для обозначения последовательного расположения отдельных псевдоожиженных слоев таким образом, что при этом может иметь место градиент свойства газа и/или твердого вещества (такого как температура, состав газа или твердого вещества, давление и т.д.), поскольку твердое вещество или газ каскадирует от одного псевдоожиженного слоя к другому. Описание минимальной скорости псевдоожижения находится, например, в приведенных выше опубликованных работах Kunii и Walas.
Используемое в настоящем описании понятие "вертикальный трубный реактор" обозначает зону или сосуд (такой как вертикальный цилиндрический патрубок), применяемый для вертикальной в принципе транспортировки твердых частиц в режимах псевдоожижения с высокой скоростью потока или псевдоожижения с пневмотранспортировкой. Режимы псевдоожижения с высокой скоростью потока и псевдоожижения с пневмотранспортировкой характеризуются скоростями газа на единицу сечения потока (U), которые больше, чем скорость транспортировки (Utr). Режимы псевдоожижения с высокой скоростью потока и псевдоожижения с пневмотранспортировкой описаны также в приведенных выше опубликованных работах Kunii и Walas.
Используемые в настоящем описании ссылки на нагрев "косвенным контактированием" с газообразными продуктами горения служат как охватывающие теплоперенос через поверхность теплопередачи и/или применение теплоносителя (газ, жидкость или твердое вещество), который нагревают газообразными продуктами горения и который отдает свое тепло каталитическому порошкообразному материалу.
Необходимо иметь в виду, что реакционная зона, обладающая обратным температурным профилем, представляет собой реакционную зону, в которой реакционная температура на входе в реакционную зону ниже, чем реакционная температура при выпускном приспособлении для технологического газа, что является противоположностью температурному профилю, достигаемому естественным образом для эндотермической реакции, такой как ароматизация метана. По-другому обратный температурный профиль может означать температурный профиль ряда катализаторных зон, в котором первая (как она определяется впускным приспособлением для исходного материала) реакционная зона работает при более низкой реакционной температуре, чем работают или работает (как она определяется выпускным приспособлением для технологического газа) последующая реакционная зона (зоны), а именно обратно температурному профилю, достигаемому естественным образом для эндотермической реакции. Этот обратный температурный профиль может возникнуть при противоточных движениях исходного материала и порошкообразного катализатора дегидроциклизации, достигаемых путем, например, введения горячего катализатора в верхнюю часть реакционной зоны таким образом, чтобы катализатор двигался вниз через реакционную зону, причем из основания реакционной зоны удаляют катализатор с пониженной температурой. Исходный материал вводят вблизи основания реакционной зоны, и он движется в противоток к катализатору вверх по реакционной зоне таким образом, что входит в контакт с самой горячей частью катализатора при выпускном приспособлении для технологического газа. По-другому обратный температурный профиль может быть достигнут подводом тепла вдоль реакторной зоны (зон).
В одном варианте объектом настоящего изобретения является способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий следующие стадии:
а) подача в реакционную зону углеводородного исходного материала, содержащего метан;
б) подача внутрь реакционной зоны некоторого количества каталитического материала;
в) поддержание в реакционной зоне обратного температурного профиля и
в) работа реакционной зоны в реакционных условиях, достаточных для превращения по меньшей мере части метана в первый отходящий поток, содержащий более высокомолекулярный углеводород (углеводороды).
В еще одном варианте объектом настоящего изобретения является способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, в двух или большем числе реакционных зон, работающих последовательно, включающий следующие стадии:
а) подача внутрь каждой реакционной зоны некоторого количества каталитического материала;
б) подача в первую реакционную зону углеводородного исходного материала, содержащего метан;
в) перенос по меньшей мере части отходящего потока первой реакционной зоны во вторую реакционную зону;
г) поддержание в первой реакционной зоне более низкой средней температуры, чем во второй реакционной зоне; и
д) работа реакционных зон в реакционных условиях, достаточных для превращения по меньшей мере части метана в первый отходящий поток, содержащий более высокомолекулярный углеводород (углеводороды).
В подходящем варианте реакционная зона (зоны) может представлять собой реакционную зону (зоны) с подвижным слоем или неподвижным слоем.
В подходящем варианте исходный материал далее включает по меньшей мере один из СО, СО2, H2, Н2О и/или О2.
В подходящем варианте каталитический материал может характеризоваться коксом, удаленным реакцией кокса с водородом или кислородом; в предпочтительном варианте это удаление водородом осуществляют под давлениями по меньшей мере 100 кПа, в частности в пределах от примерно 150 до примерно 5000 кПа. Когда в подходящем варианте реакционная зона (зоны) представляет собой подвижный слой (слои), часть катализатора отводят из реакционной зоны, по меньшей мере часть кокса, отложившегося на катализаторе, удаляют окислением и катализатор с пониженным содержанием кокса возвращают в реакционную зону или часть катализатора отводят из реакционной зоны, по меньшей мере часть кокса, отложившегося на катализаторе, удаляют реакцией с водородом с получением метана и катализатор с пониженным содержанием кокса возвращают в реакционную зону. По-другому, когда реакционная зона (зоны) представляет собой неподвижный слой (слои), подачу углеводородов циклически останавливают и вместо этого в реакционную зону направляют кислородсодержащий поток, благодаря чему по меньшей мере часть кокса, отложившегося на катализаторе, удаляют окислением, затем кислородсодержащий поток останавливают и вновь начинают подачу углеводородов или, по-другому, подачу углеводородов циклически останавливают и вместо этого в реакционную зону направляют водородсодержащий поток, благодаря чему по меньшей мере часть кокса, отложившегося на катализаторе, удаляют превращением в метан, затем водородсодержащий поток останавливают и вновь начинают подачу углеводородов.
В одном варианте реакционные условия в реакционной зоне в (б) являются неокислительными условиями. В подходящем варианте реакционные условия в реакционной зоне в (б) включают температуру от примерно 400 до примерно 1200°С, абсолютное давление от примерно 1 до примерно 1000 кПа и среднечасовую скорость подачи сырья от примерно 0,01 до примерно 1000 ч-1.
В одном варианте каталитический материал представляет собой катализатор дегидроциклизации, включающий металл или его соединение на неорганическом носителе. В подходящем варианте каталитический порошкообразный материал включает по меньшей мере один из молибдена, вольфрама, рения, соединения молибдена, соединения вольфрама, соединения цинка и соединения рения на ZSM-5, диоксиде кремния или оксиде алюминия.
В подходящем варианте начальная температура катализируемой реакции ниже примерно 750°С, предпочтительно ниже примерно 700°С, по другому варианту ниже примерно 650°С, а конечная температура катализируемой реакции превышает примерно 700°С, предпочтительно выше примерно 800°С, по другому варианту выше примерно 850°С.
В подходящем варианте подают достаточное количество катализатора, благодаря которому степень приближения в % к термодинамическому равновесному превращению метана в бензол в средней части реакционной зоны (в смысле количества катализатора, контактировавшего с углеводородом) превышает примерно 25%, предпочтительно больше примерно 50%, наиболее предпочтительно больше примерно 75%. По-другому варианту подают достаточное количество катализатора, благодаря которому степень приближения в % к термодинамическому равновесному превращению метана в бензол в средней части реакционной зоны (в смысле половины общего перепада температур на пути через реакционную зону) превышает примерно 25%, предпочтительно больше примерно 50%, наиболее предпочтительно больше примерно 75%.
В подходящем варианте обратного температурного профиля в неподвижном слое катализатора добиваются прерывистым прямым контактным нагреванием газообразными продуктами горения или инертной средой, нагретой газообразными продуктами горения. По другому варианту обратного температурного профиля в неподвижном слое катализатора добиваются теплопереносом через поверхность теплопередачи, где поверхность теплопередачи нагревают благодаря теплопереносу от горения за счет излучения и/или теплопроводности. В подходящем варианте поверхность теплопередачи является металлической или керамической. В подходящем варианте катализатор размещен в одной или нескольких параллельных трубках, а эти трубки размещены внутри печи, обеспечивающей тепло для сохранения обратного температурного профиля. По-другому варианту катализатор размещен в сосуде с одной или несколькими трубками, проходящими через слой, по этим трубкам транспортируют газообразные продукты горения для сохранения обратного температурного профиля. По другому варианту, когда используют две или большее число реакционных зон, ступенчатого изменения температуры в неподвижных слоях катализатора добиваются нагреванием потока углеводородов теплопереносом через поверхность теплопередачи; нагревание потока углеводородов производят между реакционными зонами.
В одном варианте противоточное движение исходного материала и порошкообразного катализатора дегидроциклизации осуществляют с учетом создания обратного температурного профиля в реакционной зоне или в каждой реакционной зоне, благодаря чему, несмотря на эндотермическую природу реакции дегидроциклизации, разница между реакционной температурой при выпускном приспособлении для технологического газа из реакционной зоны и реакционной температурой на входе в реакционную зону составляет по меньшей мере +10°С, в частности по меньшей мере +50°С, например по меньшей мере +100°С и даже по меньшей мере +150°С.
В одном варианте объектом настоящего изобретения является способ получения ароматических углеводородов введением исходного материала, содержащего метан, в некоторых вариантах совместно с Н2, Н2О, О2, СО и/или CO2, в контакт с порошкообразным катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения метана в ароматические углеводороды и водород. По мере того как протекает эта реакция, на катализаторе накапливается кокс, понижая тем самым активность катализатора, и, следовательно, часть закоксованного катализатора может быть непрерывно или периодически отведена из реакционной зоны и направлена через отдельную регенерационную зону, где закоксованный катализатор вводят в контакт с водородсодержащим регенераторным газом. Поскольку реакция дегидроциклизации является эндотермической, к закоксованному катализатору, отводимому из реакционной зоны, прямым и/или косвенным контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива, подводят тепло для повышения его температуры до целевой температуры регенерирования, которая в некоторых вариантах составляет от примерно 700 до примерно 1200°С. Затем часть нагретого закоксованного катализатора может быть возвращена в реакционную зону с получением тепла для реакции дегидроциклизации, тогда как остаток нагретого катализатора в регенерационной зоне вводят в контакт с водородсодержащим регенераторным газом в таких условиях, в которых по меньшей мере часть кокса на катализаторе превращают в метан. Далее регенерированный катализатор возвращают в реакционную зону.
В одном варианте регенерирование проводят отводом двух или большего числа частей закоксованного катализатора из реакционной зоны, подводом тепла к этим частям катализатора и контактированием нагретых частей катализатора с водородсодержащим газом в отдельных регенерационных зонах, работающих в таких условиях, в которых парциальные давления водорода в по меньшей мере двух из регенерационных зон разнятся.
ИСХОДНЫЙ МАТЕРИАЛ
В способе по изобретению можно использовать любой метансодержащий исходный материал, но в общем предлагаемый способ предусмотрен для применения с исходным природным газом. Другие приемлемые метансодержащие исходные материалы включают те, которые получают из таких источников, как угольные пласты, захоронения отходов, ферментация сельскохозяйственных или муниципальных отходов и/или газообразные потоки нефтепереработки.
Метансодержащие исходные материалы, такие как природный газ, как правило, содержат, в дополнение к метану, диоксид углерода и этан. Этан и другие алифатические углеводороды, которые могут содержаться в исходном материале, на стадии дегидроциклизации могут быть, разумеется, превращены в целевые ароматические продукты. Кроме того, как это обсуждается ниже, диоксид углерода также может быть превращен в полезные ароматические продукты либо непосредственно на стадии дегидроциклизации, либо косвенным путем, посредством превращения в метан и/или этан на стадии снижения содержания водорода.
Перед применением метансодержащих потоков в способе по изобретению азот- и/или серусодержащие примеси, которые также, как правило, находятся в этих потоках, могут быть удалены или их количество может быть уменьшено до низких концентраций. В одном из вариантов исходный материал, подаваемый на стадию дегидроциклизации, содержит меньше 100 ч./млн, например меньше 10 ч./млн, в частности меньше 1 ч./млн, каждого из соединений азота и серы.
В дополнение к метану, с целью содействовать уменьшению коксообразования в исходный материал, подаваемый на стадию дегидроциклизации, можно добавлять по меньшей мере один из водорода, воды, моноксида углерода и диоксида углерода. Эти добавки могут быть введены в виде отдельных совместно подаваемых исходных материалов или могут находиться в метановом потоке, например таком, как в случае, когда метановый поток дериватизируют из природного газа, включающего диоксид углерода. Другие источники диоксида углерода могут включать отходящие газы, установки СПГ, водородные установки, аммиачные установки, гликольные установки и фталевоангидридные установки.
В одном варианте исходный материал, подаваемый на стадию дегидроциклизации, содержит диоксид углерода и включает от примерно 90 до примерно 99,9 мольного %, в частности от примерно 97 до примерно 99 мольных %, метана и от примерно 0,1 до примерно 10 мольных %, в частности от примерно 1 до примерно 3 мольных %, СО2. В другом варианте исходный материал, подаваемый на стадию дегидроциклизации, содержит моноксид углерода и включает от примерно 80 до примерно 99,9 мольного %, в частности от примерно 94 до примерно 99 мольных %, метана и от примерно 0,1 до примерно 20 мольных %, в частности от примерно 1 до примерно 6 мольных %, СО. В еще одном варианте исходный материал, подаваемый на стадию дегидроциклизации, содержит водяной пар и включает от примерно 90 до примерно 99,9 мольного %, в частности от примерно 97 до примерно 99 мольных %, метана и от примерно 0,1 до примерно 10 мольных %, в частности от примерно 1 до примерно 5 мольных %, водяного пара. Однако в еще одном варианте исходный материал, подаваемый на стадию дегидроциклизации, содержит водород и включает от примерно 80 до примерно 99,9 мольного %, в частности от примерно 95 до примерно 99 мольных %, метана и от примерно 0,1 до примерно 20 мольных %, в частности от примерно 1 до примерно 5 мольных %, водорода.
Исходный материал, подаваемый на стадию дегидроциклизации, может также содержать более высокомолекулярные углеводороды, чем метан, включая ароматические углеводороды. Такие более высокомолекулярные углеводороды могут быть возвращены в процесс со стадии снижения содержания водорода, добавлены в виде отдельных совместно подаваемых исходных материалов или могут находиться в метановом потоке, таком как, например, в случае, когда в исходном природном газе содержится этан. Более высокомолекулярные углеводороды, возвращаемые в процесс со стадии снижения содержания водорода, как правило, включают моноциклические ароматические соединения и/или парафины и олефины, содержащие преимущественно 6 или меньше, в частности 5 или меньше, например 4 или меньше, как правило 3 или меньше углеродных атомов. Обычно исходный материал, подаваемый на стадию дегидроциклизации, содержит меньше 5 мас.%, в частности меньше 3 мас.%, углеводородов С3+.
ДЕГИДРОЦИКЛИЗАЦИЯ
На стадии дегидроциклизации предлагаемого способа метансодержащий исходный материал вводят в контакт с порошкообразным катализатором дегидроциклизации в условиях, обычно в неокислительных условиях, а, как правило, в восстановительных условиях, эффективных для превращения метана в более высокомолекулярные углеводороды, включая бензол и нафталин. В принципе, проводят следующие результирующие реакции:
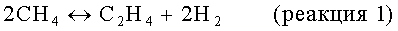
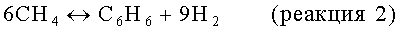

Моноксид и/или диоксид углерода, который может находиться в исходном материале, повышает активность и стабильность катализатора содействием протеканию реакций, таких как:
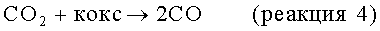
но негативно влияет на равновесие, позволяя протекать параллельным результирующим реакциям, таким как:
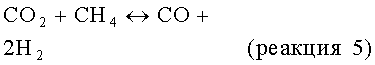
В способе по изобретению можно использовать любой катализатор дегидроциклизации, эффективный для превращения метана в ароматические соединения, хотя обычно катализатор включает металлический компонент, в особенности переходный металл или его соединение, на неорганическом носителе. В подходящем варианте металлический компонент содержится в количестве в пределах от примерно 0,1 до примерно 20%, в частности в пределах от примерно 1 до примерно 10 мас.%, в пересчете на массу всего катализатора. Обычно металл содержится в катализаторе в форме карбида.
Приемлемые для катализатора металлические компоненты включают кальций, магний, барий, иттрий, лантан, скандий, церий, титан, цирконий, гафний, ванадий, ниобий, тантал, хром, молибден, вольфрам, марганец, рений, железо, рутений, кобальт, родий, иридий, никель, палладий, медь, серебро, золото, цинк, алюминий, галлий, кремний, германий, индий, олово, свинец, висмут и трансурановые металлы. Такие металлические компоненты могут содержаться в форме свободных элементов или в виде соединений металлов, таких как оксиды, карбиды, нитриды и/или фосфиды, и их можно использовать самостоятельно или в сочетании. В качестве одного из металлических компонентов могут быть также использованы платина и осмий, но обычно они не предпочтительны.
Неорганический носитель может быть либо аморфным, либо кристаллическим и, в частности, может представлять собой оксид, карбид или нитрид бора, алюминия, кремния, фосфора, титана, скандия, хрома, ванадия, магния, марганца, железа, цинка, галлия, германия, иттрия, циркония, ниобия, молибдена, индия, олова, бария, лантана, гафния, церия, тантала, вольфрама или других трансурановых элементов. Кроме того, носителем может быть пористый материал, такой как микропористый кристаллический материал и мезопористый материал. Используемое в настоящем описании понятие "микропористый" относится к порам, обладающим диаметром меньше 2 нм, тогда как понятие "мезопористый" относится к порам, обладающим диаметром от 2 до 50 нм.
Приемлемые микропористые кристаллические материалы включают силикаты, алюмосиликаты, титаносиликаты, алюмофосфаты, металлофосфаты, кремнеалюмофосфаты и их смеси. Такие микропористые кристаллические материалы включают материалы, обладающие каркасами типов MFI (например, ZSM-5 и силикалит), MEL (например, ZSM-11), MTW (например, ZSM-12), TON (например, ZSM-22), МТТ (например, ZSM-23), FER (например, ZSM-35), MFS (например, ZSM-57), MWW (например, МСМ-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, МСМ-36, МСМ-49 и МСМ-56), IWR (например, ITQ-24), KFI (например, ZK-5), ВЕА (например, бета-цеолит), ITH (например, ITQ-13), MOR (например, морденит), FAU (например, цеолиты X, Y, ультрастабилизированный Y и деалюминированный Y), LTL (например, цеолит L), IWW (например, ITQ-22), VFI (например, VPI-5), AEL (например, SAPO-11), AFI (например, ALPO-5) и AFO (SAPO-41), а также такие материалы, как МСМ-68, EMM-1, EMM-2, ITQ-23, ITQ-24, ITQ-25, ITQ-26, ETS-2, ETS-10, SAPO-17, SAPO-34 и SAPO-35. Приемлемые мезопористые материалы включают МСМ-41, МСМ-48, МСМ-50, FSM-16 и SBA-15.
Примеры предпочтительных катализаторов включают молибден, вольфрам, цинк, рений и их соединения и сочетания на ZSM-5, диоксиде кремния или оксиде алюминия.
Металлический компонент может быть диспергирован на неорганическом носителе с помощью любого средства, хорошо известного в данной области техники, такого как соосаждение, пропитка до начальной влажности, выпаривание, обычная пропитка, распылительная сушка, золь-гелевое, ионообменное, химическое паровое осаждение, диффузионное и физическое смешение. Кроме того, неорганический носитель может быть модифицирован по известным методам, таким как, например, обработка водяным паром, кислотная промывка, промывка каустической содой и/или обработка кремнийсодержащими соединениями, фосфорсодержащими соединениями и/или элементами или соединениями элементов групп 1, 2, 3 и 13 Периодической таблицы элементов. Такие модификации можно использовать для изменения поверхностной активности носителя и препятствия или улучшения доступа к любой внутренней пористой структуре носителя.
В некоторых вариантах в реакцию дегидроциклизации, в дополнение к каталитическому порошкообразному материалу, может быть направлен некаталитический порошкообразный материал. Некаталитический порошкообразный материал можно использовать в качестве материала для передачи энергии (тепла) в систему и/или для заполнения пространства, в зависимости от потребности обеспечения требуемых гидродинамических условий. Некаталитический порошкообразный материал может образовывать макрочастицы без связующего вещества или частицы могут быть связаны неорганическим связующим веществом, таким как глина, диоксид кремния, оксид алюминия, диоксид циркония или другой оксид металла, используемый для содействия сохранению физической целостности частиц. В предпочтительном варианте частицы обладают, по существу, сферической формой. Примерами приемлемого некаталитического порошкообразного материала служат диоксид кремния, оксид алюминия, керамика и карбид кремния с малой удельной площадью поверхности.
Стадию дегидроциклизации осуществляют введением метансодержащего исходного материала в контакт с катализатором дегидроциклизации в реакционных зонах с одним или несколькими неподвижными слоями, подвижными слоями или псевдоожиженными слоями. Обычно исходный материал в реакционной зоне или каждой реакционной зоне вводят в контакт с подвижным слоем катализатора дегидроциклизации, где исходный материал движется в противоток направлению движения катализатора дегидроциклизации. В одном варианте реакционная зона включает реактор с отстойным слоем, под которым подразумевают вертикально расположенный реактор, в котором порошкообразный катализатор поступает по месту или вблизи верхней части реактора и движется под действием собственного веса с образованием каталитического слоя, в то время как исходный материал поступает в реактор по месту или вблизи основания реактора и движется вверх через каталитический слой.
Движение катализатора дегидроциклизации в реакционной зоне в варианте отстойного слоя практически свободно от псевдоожижения. Понятие "практически свободно от псевдоожижения", используемое в настоящем описании, означает, что средняя скорость движения газа в реакторе ниже минимальной скорости псевдоожижения. Понятие "практически свободно от псевдоожижения", используемое в настоящем описании, также означает, что средняя скорость движения газа в реакторе меньше 99%, в частности меньше 95%, как правило меньше 90%, даже меньше 80%, минимальной скорости псевдоожижения. Когда реакционная зона или каждая реакционная зона работает как отстойный слой, порошкообразный каталитический материал и/или любой порошкообразный некаталитический материал обладают средним размером частиц от примерно 0,1 до примерно 100 мм, в частности от примерно 1 до примерно 5 мм, например от примерно 2 до примерно 4 мм. В некоторых вариантах по меньшей мере 90 мас.% порошкообразного каталитического материала и/или по меньшей мере 90 мас.% порошкообразного некаталитического материала обладают размером частиц от примерно 0,1 до примерно 100 мм, в частности от примерно 1 до примерно 5 мм, в частности от примерно 2 до примерно 4 мм.
В альтернативном варианте реакцию дегидроциклизации проводят в нескольких последовательно соединенных реакторах с псевдоожиженным слоем, в которых порошкообразный катализатор каскадирует в одном направлении из одного реактора в следующий смежный реактор в этом ряду, в то время как исходный материал пропускают через и между реакторами в противоположном направлении. Когда каждая реакционная зона работает как псевдоожиженный слой, каталитический порошкообразный материал и/или любой некаталитический порошкообразный материал обладает средним размером частиц от примерно 0,01 до примерно 10 мм, в частности от примерно 0,05 до примерно 1 мм и в частности от примерно 0,1 до примерно 0,6 мм. В некоторых вариантах по меньшей мере 90 мас.% каталитического порошкообразного материала и/или по меньшей мере 90 мас.% некаталитического порошкообразного материала обладают размером частиц от примерно 0,01 до примерно 10 мм, в частности от примерно 0,05 до примерно 1 мм и в частности от примерно 0,1 до примерно 0,6 мм.
Как правило, массовое отношение расхода каталитического порошкообразного материала плюс любой некаталитический порошкообразный материал к расходу углеводородного исходного материала в зоне реакции или в каждой зоне реакции дегидроциклизации составляет от примерно 1:1 до примерно 100:1, в частности от примерно 1:1 до примерно 40:1, в частности от примерно 5:1 до 20:1.
Реакция дегидроциклизации является эндотермической, и, следовательно, по мере протекания этой реакции температура в каждой зоне реакции дегидроциклизации обычно проявляет тенденцию к понижению от максимальной температуры до минимальной температуры. Приемлемые условия для стадии дегидроциклизации включают максимальную температуру от примерно 700 до примерно 1200°С, в частности от примерно 800 до примерно 950°С, и минимальную температуру от примерно 400 до примерно 800°С, в частности от примерно 500 до примерно 700°С. Однако, как обсуждается ниже, для того чтобы уменьшить падение температуры во время реакции дегидроциклизации в эту реакцию подводят тепло и, следовательно, в некоторых конфигурациях может оказаться возможным уменьшить разницу между максимальной и минимальной температурами, по существу, до нуля. По другому варианту может оказаться возможным достижение обратного температурного градиента подачей нагретого катализатора в реакцию дегидроциклизации, т.е. реакционная температура при выпускном приспособлении для технологического газа выше, чем реакционная температура при впускном приспособлении для технологического газа.
В одном варианте для создания обратного температурного профиля через систему реакции дегидроциклизации осуществляют противоточное движение исходного материала и порошкообразного катализатора дегидроциклизации, благодаря чему, несмотря на эндотермическую природу реакции дегидроциклизации, разница между реакционной температурой газообразного отходящего потока при выпускном приспособлении из системы реакции дегидроциклизации и реакционной температурой метансодержащего исходного материала при впускном приспособлении в систему реакции дегидроциклизации составляет по меньшей мере +10°С, в частности по меньшей мере +50°С, например по меньшей мере +100°С и даже по меньшей мере +150°С.
В любом случае поскольку реакция дегидроциклизации является эндотермической, каталитический порошкообразный материал поступает в систему реакции дегидроциклизации при первой, высокой температуре, как правило от примерно 800 до примерно 1200°С, в частности от примерно 900 до примерно 1100°С, а выходит из реакционной системы при второй, более низкой температуре, как правило от примерно 500 до примерно 800°С, в частности от примерно 600 до примерно 700°С. Общая разница температур каталитического порошкообразного материала на пути через реакционные зоны составляет по меньшей мере 100°С.
Другие условия, создаваемые в реакции дегидроциклизации, обычно включают давление от примерно 1 до примерно 1000 кПа, в частности от примерно 10 до примерно 500 кПа, например от примерно 50 до примерно 200 кПа, и среднечасовую скорость подачи сырья от примерно 0,01 до примерно 1000 ч-1, в частности от примерно 0,1 до примерно 500 ч-1, в частности от примерно 1 до примерно 20 ч-1. В подходящем варианте стадию дегидроциклизации осуществляют в отсутствии О2.
Основными компонентами отходящего со стадии дегидроциклизации потока являются водород, бензол, нафталин, моноксид углерода, этилен, кокс и непрореагировавший метан. Этот отходящий поток, как правило, включает по меньшей мере на 5 мас.%, в частности по меньшей мере на 10 мас.%, например по меньшей мере на 20 мас.%, предпочтительно по меньшей мере на 30 мас.%, ароматических колец больше, чем исходный материал.
Из отходящего из дегидроциклизации потока выделяют бензол и нафталин, например экстракцией растворителем с последующим разделением на фракции, и они могут быть выделены как поток продуктов. Однако, как это обсуждается ниже, перед или после извлечения продуктов по меньшей мере часть этих ароматических компонентов может быть обработана на стадии алкилирования с получением более ценных материалов, таких как ксилолы. Более того, как обсуждается ниже, в предлагаемом способе используют образование водорода в качестве побочного продукта реакции дегидроциклизации и, в частности, по меньшей мере часть водорода превращают в более ценные продукты.
РЕГЕНЕРИРОВАНИЕ КАТАЛИЗАТОРА
Реакция дегидроциклизации характеризуется тенденцией к отложению кокса на катализаторе и, следовательно, для того чтобы сохранить активность катализатора дегидроциклизации по меньшей мере часть катализатора может быть непрерывно или периодически регенерирована. Этого, как правило, добиваются отводом части катализатора из реакционной зоны или каждой реакционной зоны либо на прерывистой, либо на непрерывной основе и ее переносом в отдельную регенерационную зону. В регенерационной зоне закоксованный катализатор дегидроциклизации вводят в контакт с водородсодержащим газом в условиях, эффективных для превращения по меньшей мере части содержащегося на нем углеродистого материала в метан. Обычно водородсодержащий газ не включает значительных количеств метана или других углеводородов, причем содержание углеводородов, как правило, составляет меньше 20 мольных %, в частности меньше 10 мольных %, например меньше 2 мольных %. В одном варианте водород, необходимый для регенерирования, по меньшей мере отчасти получают из водородсодержащего потока, отходящего из реакции дегидроциклизации.
В подходящем варианте условия регенерирования включают температуру от примерно 700 до примерно 1200°С, в частности от примерно 800 до примерно 1000°С, в частности от примерно 850 до примерно 950°С, и давление по меньшей мере 100 кПа, в частности в пределах от примерно 150 до примерно 5000 кПа. Обычно, однако, закоксованный катализатор дегидроциклизации, удаляемый из реакционной зоны или каждой реакционной зоны, характеризуется более низкой температурой, чем оптимальная для регенерирования, и, следовательно, удаляемый катализатор вначале нагревают до целевой температуры регенерирования прямым и/или косвенным контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива. Нагревание проводят в нагревательной зоне, которая может находиться в том же сосуде, что и регенерационная зона, или которая может находиться в сосуде, отделенном от регенерационной зоны.
Под понятием "дополнительный источник топлива" имеют в виду то, что источник топлива физически отделен от катализатора и, следовательно, на катализаторе в качестве побочного продукта реакции дегидроциклизации кокс не образуется. Как правило, дополнительный источник топлива включает углеводород, такой как метан, и, в частности, приемлемый источник топлива представляет собой природный газ, используемый в качестве исходного материала, направляемого в процесс. В подходящем варианте в нагревательной зоне поддерживают бедную кислородом атмосферу, благодаря чему при сжигании углеводородного топлива для нагрева первой части катализатора образуется синтез-газ, который затем может быть использован для получения дополнительного углеводородного продукта и/или топлива. Кроме того, в случае прямого теплопереноса к катализатору дегидроциклизации применение бедной кислородом атмосферы ингибирует окисление карбидов металлов, содержащихся в катализаторе, и сводит к минимуму среднее парциальное давление водяного пара, тем самым уменьшая гидротермическое старение катализатора.
По другому варианту приемлемый дополнительный источник топлива представляет собой водород, и в частности часть водорода, образующегося в качестве побочного продукта реакции ароматизации.
Когда катализатор дегидроциклизации нагревают прямым путем, закоксованный катализатор, отводимый из реакционной зоны, целесообразно вводить в контакт непосредственно с горящим источником топлива в нагревательной зоне. По другому варианту источник топлива сжигают в отдельной зоне горения, и газообразные продукты горения, образующиеся в зоне горения, направляют в нагревательную зону для нагрева катализатора. По другому варианту катализатор дегидроциклизации может быть нагрет непрямым теплообменом, таким как, например, с использованием газообразных продуктов горения для нагрева инертной среды (газа, жидкости или твердого вещества) или поверхности теплопередачи и последующим контактированием закоксованного катализатора с нагретой инертной средой или поверхностью теплопередачи.
В одном практическом варианте нагревательная зона является удлиненной, и закоксованный катализатор пропускают по нагревательной зоне из впускного приспособления по месту или вблизи одного конца нагревательной зоны до выпускного приспособления по месту или вблизи другого конца зоны нагрева, причем тепло передают первой части катализатора на множестве участков, отделенных промежутками по длине зоны нагрева. Таким путем поступление тепла к катализатору может быть распределено по длине нагревательной зоны, сведением тем самым к минимуму температур поверхности катализатора и внутренних градиентов.
Когда первую часть катализатора нагревают прямым контактированием с горящим источником топлива в нагревательной зоне, постепенный нагрев катализатора может быть достигнут направлением, по существу, всего дополнительного топлива во впускной конец нагревательной зоны и затем подачей кислородсодержащего газа по нарастающей в упомянутую нагревательную зону на упомянутом множестве отделенных промежутками участках по длине нагревательной зоны. По другому варианту, по существу, весь кислородсодержащий газ, необходимый для сжигания дополнительного топлива, может быть направлен во впускной конец нагревательной зоны, а дополнительное топливо направлено по нарастающей в нагревательную зону на упомянутом множестве отделенных промежутками участков.
Когда первую часть катализатора нагревают прямым контактированием с горячими газообразными продуктами горения, образующимися в отдельной зоне горения, постепенный нагрев катализатора может быть достигнут подачей горячих газообразных продуктов горения к упомянутому множеству отделенных промежутками участков по длине нагревательной зоны.
В одном варианте нагревательная зона представляет собой вертикальную трубу, и во время стадии повторного нагревания первую часть катализатора пропускают вверх по вертикальной трубе. На практике нагревательная зона может включать несколько вертикальных труб, соединенных параллельно. По другому варианту нагревательная зона может включать подвижный слой упомянутого катализатора.
В одном варианте закоксованный катализатор дегидроциклизации, удаляемый из реакционной зоны, разделяют на по меньшей мере две части, которые нагревают так, как изложено выше, и затем направляют в отдельные регенерационные зоны, работающие под разными давлениями. Так, например, одна регенерационная зона работает под давлением по меньшей мере 100 кПа, в частности в пределах от примерно 150 до примерно 700 кПа, как изложено выше, тогда как другая регенерационная зона работает под давлением по меньшей мере 500 кПа, в частности в пределах от примерно 1000 до примерно 5000 кПа. Так, например, установлено, как продемонстрировано в примерах, что регенерирование под более высокими давлениями обеспечивает более быстрое удаление кокса, а также удаление более огнеупорного кокса. Однако установлено также, что для возможности удаления кокса под более высокими давлениями потребуется более дорогостоящее оборудование. По этой причине могут оказаться целесообразными удаление части кокса под более низким парциальным давлением водорода в менее дорогостоящем оборудовании и удаление дополнительной части кокса под более высоким парциальным давлением водорода в более дорогостоящем оборудовании.
Регенерационная зона или каждая регенерационная зона может представлять собой реактор, работающий как псевдоожиженный слой, кипящий слой, отстойный слой, вертикальный трубный реактор или их сочетание. На практике каждая регенерационная зона может включать несколько реакторов, в частности несколько соединенных параллельно вертикальных трубных реакторов или несколько реакторов, соединенных последовательно, в частности вертикальный трубный реактор с последующим отстойным слоем. После регенерирования катализатор возвращают в реакционную зону.
В альтернативном варианте и, в особенности, когда реакцию дегидроциклизации проводят в реакторе с неподвижным слоем, регенерирование может быть осуществлено без удаления катализатора из реакционной зоны, временным прерыванием подачи метансодержащего исходного материала в реакционную зону, нагреванием реакционной зоны до температуры регенерирования от примерно 700 до примерно 1200°С прямым и/или непрямым контактированием с газообразными продуктами горения, получаемыми сжиганием дополнительного топлива, регенерированием порошкообразного каталитического материала водородсодержащим газом, а затем возобновлением подачи метансодержащего исходного материала в реакционную зону. Необходимо иметь в виду, что нагревание реакционной зоны до температуры регенерирования можно осуществлять перед прерыванием подачи метансодержащего исходного материала.
ПОВТОРНЫЙ НАГРЕВ КАТАЛИЗАТОРА
Поскольку реакция дегидроциклизации является эндотермической, в реакцию необходимо подводить тепло. В предлагаемом способе этого целесообразно добиваться отводом части катализатора из реакционной зоны либо на периодической, либо на непрерывной основе, подводом тепла к катализатору и затем возвратом нагретого катализатора назад в реакционную зону. Поскольку стадия регенерирования водородом, описанная выше, также включает нагревание катализатора и затем возврат нагретого регенерированного катализатора назад в реакционную зону, один возможный путь подвода тепла в реакцию дегидроциклизации заключается в процессе регенерирования.
По другому варианту некоторое количество или все тепло, потребное для поддержания реакции дегидроциклизации, может быть обеспечено отдельной стадией повторного нагрева катализатора. В этом варианте часть катализатора, отводимую для реакционной зоны, переносят в отдельную нагревательную зону, где катализатор вновь нагревают прямым или непрямым контактированием с горячими газообразными продуктами горения, генерируемыми сжиганием дополнительного источника топлива. Затем нагретый катализатор возвращают в реакционную зону с регенерированием водородом или без него.
По-другому варианту для применений в неподвижном слое катализатор может быть нагрет циклическим прерыванием потока углеводородного сырья и пропусканием горячих газов над каталитическим слоем. Эти горячие газы могут быть продуктами горения или инертным газом. Такой горячий газ пропускают преимущественно через каталитический слой в направлении, противоположном направлению потока углеводородного сырья, благодаря чему в каталитическом слое формируется обратный температурный профиль.
ПОВТОРНОЕ ЗАКОКСОВЫВАНИЕ КАТАЛИЗАТОРА
Необходимо иметь в виду, что нагревание катализатора дегидроциклизации с целью регенерирования и/или для теплопереноса вновь в реакцию дегидроциклизации способно подвергать катализатор воздействию высокотемпературных окислительных условий, преимущественно когда нагрев катализатора включает прямое контактирование с горячими газообразными продуктами горения. В результате металлы, такие как рений, вольфрам и молибден, содержащиеся в катализаторе дегидроциклизации, во время стадии нагрева могут быть превращены из их каталитически активной элементной или карбидной формы в оксидные материалы. Таким образом, перед возвратом в реакционную зону регенерированный катализатор и/или повторно нагретый катализатор может быть перенесен в зону обработки катализатора отдельно от регенерационной зоны, нагревательной зоны и реакционной зоны, где регенерированный катализатор вводят в контакт с закоксовывающим газом, содержащим по меньшей мере один углеводород, выбранный из метана, этана, пропана, бутана, изобутена, гексана, бензола и нафталина. В некоторых случаях закоксовывающий газ может также включать по меньшей мере один из СО2, СО, Н2, Н2О и инертных разбавителей. По другому варианту закоксовывающий газ может представлять собой смесь водорода и по меньшей мере одного из СО и СО2. Более того может возникнуть необходимость введения катализатора в контакт последовательно со множеством разных закоксовывающих газов, каждый из которых включает углеводород, выбранный из метана, этана, пропана, бутана, изобутена, гексана, бензола и нафталина, или смесь водорода и по меньшей мере одного из СО и CO2.
Для того чтобы избежать повреждения катализатора, процесс закоксовывания регулируют таким образом, чтобы максимальная температура в зоне обработки катализатора была ниже максимальной температуры в зоне реакции дегидроциклизации, хотя, как правило, максимальная температура закоксовывания выше, чем максимальная температура, достигаемая в зоне окислительной регенерации. Обычно максимальная температура в зоне обработки катализатора составляет от 400 до 1100°С, в частности от 500 до 900°С, тогда как минимальная температура находится в пределах от 300 до 500°С. Как правило, зона обработки катализатора работает под давлениями в пределах от 10 до 100 фунтов/кв.дюйм (от 69 до 690 кПа), в частности в пределах от 15 до 60 фунтов/кв.дюйм (от 103 до 414 кПа). Обычно средняя продолжительность пребывания каталитических частиц в зоне обработки катализатора находится в пределах от 0,1 до 100 мин, например в пределах от 1 до 20 мин. В этих условиях закоксовывающий газ взаимодействует с металлоксидными материалами на катализаторе с возвратом металла в его каталитически активную элементную или карбидную форму. Кроме того, закоксовывающий газ способен взаимодействовать с активными поверхностными участками на носителе катализатора с ослаблением их тенденции к образованию кокса в зоне реакции дегидроароматизации.
Для того чтобы поддержать температуру, необходимую для закоксовывания регенерированного катализатора, тепло можно подводить к катализатору и/или закоксовывающему газу перед или во время стадии закоксовывания. Так, например, тепло может быть подведено к катализатору косвенным нагревом, введением в контакт с горячим отходящим из реакционной зоны или нагревательной зоны газом, введением в контакт с горячим газообразным отходящим из процесса закоксовывания потоком или смешением с нагретым катализатором из нагревательной зоны. Тепло удобно подводить к закоксовывающему газу посредством внешней печи или теплообменника или посредством нагретого катализатора из нагревательной зоны.
Зона обработки катализатора может работать как реактор с псевдоожиженным слоем, реактор с кипящим слоем, реактор с отстойным слоем, вертикальный трубный реактор или циркуляционный вертикальный трубный реактор. В одном варианте зона обработки катализатора включает реактор с отстойным слоем. По другому варианту зона обработки катализатора включает единственный реактор с псевдоожиженным слоем с внутренними отбойными заслонками с целью предотвратить обратное перемешивание или несколько реакторов с псевдоожиженным слоем, соединенных последовательно, причем регенерированный катализатор каскадирует между смежными реакторами. В любом случае контактированию в зоне обработки катализатора содействуют с помощью устройства, в котором в зоне обработки катализатора регенерированный катализатор и закоксовывающий газ движутся в противоположных направлениях. С применением такого противоточного движения в зоне обработки катализатора может быть сформирован температурный профиль, благодаря которому закоксовывание регенерированного катализатора первоначально происходит при низкой температуре, но, по мере того как катализатор продвигается через слой, температура закоксовывания повышается.
В некоторых случаях может возникнуть необходимость того, чтобы нагретый нерегенерированный катализатор вначале контактировал с богатым Н2 потоком для частичного или полного восстановления металлического компонента катализатора перед стадией закоксовывания. Может также возникнуть потребность подвергнуть закоксованный катализатор постобработке Н2 и/или СО2 для десорбции всего избытка углерода, который может предварительно отложиться на катализаторе на стадии закоксовывания.
На практике, по мере того как протекает реакция дегидроциклизации, в процесс обычно добавляют свежего катализатора дегидроциклизации для возмещения потери катализатора за счет механического износа или дезактивации, и несмотря на существование множества средств добавления свежего катализатора, для того чтобы избежать повреждения катализатора, свежий катализатор обычно необходимо добавлять в зону процесса, который протекает при температуре ниже максимальной температуры в каждой зоне реакции дегидроциклизации. В одном варианте свежий катализатор дегидроциклизации добавляют в процесс введением в зону обработки катализатора, благодаря чему свежий катализатор вводят в контакт с закоксовывающим газом перед переносом в реакционную зону для введения в контакт с метансодержащим исходным материалом. В другом варианте катализатор можно добавлять в зоны более низкой температуры реакторной системы с обратным температурным профилем.
КОНСТРУКЦИОННЫЕ МАТЕРИАЛЫ ДЛЯ РЕАКЦИОННЫХ СОСУДОВ, ВНУТРИКОРПУСНЫХ УСТРОЙСТВ И ПОВЕРХНОСТЕЙ ТЕПЛОПЕРЕДАЧИ
В подходящем варианте в процессе превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, предусмотрено контактирование исходного материала, содержащего метан, с катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения метана в ароматические углеводороды, где реакционная зона содержится внутри реактора и где реактор или внутренний компонент реактора обладает по меньшей мере одной поверхностью, которая подвергается химическому воздействию исходного материала и которую изготавливают из огнеупорного материала, который проявляет поглощение углерода (масса абсорбированного углерода на единицу подвергаемой воздействию площади металлической поверхности) меньше 25 г/м2, предпочтительно 15 г/м2, а наиболее предпочтительно 10 г/м2, когда воздействует смесь 50 об.% метана и 50 об.% H2 при 900°С в течение 168 ч.
В другом варианте в процессе превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, предусмотрено контактирование исходного материала, содержащего метан, с катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения метана в ароматические углеводороды, где реакционная зона содержится внутри реактора и где реактор или внутренний компонент реактора обладает по меньшей мере одной поверхностью, которая подвергается химическому воздействию исходного материала и которую изготавливают из огнеупорного металла или сплава, который включает или способен к образованию и/или сохранению в данных условиях непрерывного оксидного или карбидного слоя, стойкого в данных условиях.
В подходящем варианте по меньшей мере одну поверхность изготавливают из огнеупорного сплава, содержащего по меньшей мере 2 мас.% по меньшей мере одного из алюминия, магния и церия.
В подходящем варианте по меньшей мере одну поверхность выполняют из молибдена, вольфрама, хрома и/или ниобия.
В подходящем варианте по меньшей мере одна поверхность подвергается химическому воздействию исходного материала через огнеупорное износостойкое покрытие, как правило, включающее по меньшей мере один из керамики, фосфида, нитрида, карбида и оксида.
Тем не менее, в еще одном варианте в процессе превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, этот процесс включает контактирование исходного материала, содержащего метан, с катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения метана в ароматические углеводороды, где реакционная зона содержится внутри реактора и где реактор или внутренний компонент реактора обладает по меньшей мере одной поверхностью, которая подвергается химическому воздействию исходного материала и которую изготавливают из огнеупорного сплава, содержащего по меньшей мере 2 мас.% по меньшей мере одного из алюминия, магния и церия.
В подходящем варианте по меньшей мере одну поверхность изготавливают из сплава на железной основе. В подходящем варианте сплав на железной основе включает, в дополнение к железу, хром, в частности в пределах от примерно 15 до примерно 25 мас.% хрома, и по меньшей мере 2 мас.%, в частности в пределах от примерно 4 до примерно 6 мас.%, алюминия.
В подходящем варианте по меньшей мере одну поверхность изготавливают из сплава на никелевой основе. В подходящем варианте сплав на никелевой основе включает в дополнение к никелю, хром, в частности в пределах от примерно 15 до примерно 30 мас.% хрома, и по меньшей мере 2 мас.%, в частности в пределах от примерно 3 до примерно 5 мас.%, алюминия.
Используемые в настоящем описании понятия "сплав на железной основе" и "сплав на никелевой основе" служат для обозначения сплава, содержащего больше 50 мас.% соответственно железа и никеля.
Объектом настоящего изобретения является способ получения ароматических углеводородов введением исходного материала, содержащего метан, в контакт, как правило совместно с Н2, СО и/или CO2, с катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения метана в ароматические углеводороды и водород. Реакционная зона может находиться внутри реактора, и реактор или внутренний компонент реактора может обладать по меньшей мере одной поверхностью, которая подвергается химическому воздействию исходного материала и которую изготавливают из огнеупорного материала, который проявляет поглощение углерода (масса абсорбированного углерода на единицу подвергаемой воздействию площади металлической поверхности) меньше 25 г/м2, предпочтительно 15 г/м2, а наиболее предпочтительно 10 г/м2, когда воздействует смесь 50 об.% метана и 50 об.% Н2 при 900°С в течение 168 ч. Как правило, по меньшей мере одну поверхность изготавливают из огнеупорного сплава, содержащего металлический компонент, обладающий или способный к образованию и/или сохранению в данных условиях стойкого непрерывного оксидного слоя, и/или из огнеупорного металла или сплава, способного к образованию в данных условиях стойкого непрерывного карбидного слоя.
Огнеупорные материалы, которые стойки к закоксовыванию, когда контактируют с исходный материалом, содержащим метан, для получения ароматических углеводородов при высоких температурах, идентифицированы. Эти огнеупорные материалы, которые могут быть непосредственно подвергнуты воздействию технологических газов и условий, можно использовать в виде сплошных металлических сплавов или в виде плакировки на обычных сплавах, когда стойкость к закоксовыванию обычных сплавов улучшают благодаря высокой стойкости к закоксовыванию их поверхностной плакировки.
Когда поверхность металлического сплава при высоких температурах подвергается воздействию углеводородных газов, такая металлическая поверхность способна катализировать превращение углеводородов в кокс, что приводит к значительному накоплению кокса. Образующие карбиды металлы (такие как молибден, вольфрам и т.д.), которые в условиях восстановительного сочетания стойки к закоксовыванию поверхности благодаря образованию поверхностного слоя из карбида металла, идентифицированы. Эти образующие карбиды металлы можно использовать в металлургии сплошного изделия или в виде поверхностных покрытий, или в виде плакировки на обычных сплавах с достижением улучшенного закоксовывания/стойкости к закоксовыванию.
Когда поверхность подвергается воздействию вызывающей эрозию окружающей среды, такой как высокоскоростные газы и/или движение частиц катализаторов, может возникнуть необходимость обеспечить поверхность огнеупорным износостойким покрытием, как правило, включающим по меньшей мере одно из керамики, фосфида, нитрида, карбида и оксида. Это объясняется тем, что воздействие на поверхность сплава высокоскоростных газов и/или движущихся каталитических частиц может вызвать эрозию защитного слоя оксида или карбида металла, находящегося на его поверхности, что может привести к повышенной скорости закоксовывания сплошного сплава. Более того, поверхностная эрозия способна обеднять преимущественно металлический сплав структурной составляющей его металла, которая образует защитный слой (такой как алюминий), тем самым вызывая в технологических условиях образование более трудно устранимых дефектов в поверхностном защитном оксидном/карбидном слое. В дополнение к уменьшению поверхностной эрозии, эти износостойкие покрытия могут служить в качестве термоизоляторов, которые в некоторых случаях применения способны понижать поверхностные температуры сплава, когда их используют в сочетании с охлаждающими системами. Поскольку нижняя поверхность обладает стойкостью к закоксовыванию, отсутствует потребность в том, чтобы износостойкое покрытие обладало также стойкостью к проникновению углерода. Примеры реакторных поверхностей, которые целесообразно обеспечивать износостойкими покрытиями, охватывают рабочие поверхности реакторных внутрикорпусных устройств, таких как газораспределители, заслонки и циклоны.
СНИЖЕНИЕ СОДЕРЖАНИЯ ВОДОРОДА
Поскольку водород является основным компонентом отходящего из дегидроциклизации потока, после извлечения ароматических продуктов отходящий поток может быть подвергнут обработке на стадии снижения содержания водорода с целью понизить содержание водорода в отходящем потоке перед возвратом непрореагировавшего метана на стадию дегидроциклизации и максимизировать утилизацию исходного материала. Стадия снижения содержания водорода, как правило, включает реакцию по меньшей мере части водорода в отходящем из дегидроциклизации потоке с кислородсодержащими материалами, такими как СО и/или СО2, с получением воды и второго отходящего потока, обладающего более низким содержанием водорода в сравнении с первым отходящим (из дегидроциклизации) потоком. Приемлемые способы снижения содержания водорода описаны ниже и в совместно рассматриваемой заявке, серийный номер РСТ № PCT/US 2005/044042, поданной авторами настоящего изобретения 2 декабря 2005 г.
В подходящем варианте стадия снижения содержания водорода включает (I) метанирование и/или этанирование, (II) процесс Фишера-Тропша, (III) синтез спиртов с C1 по С3, в особенности метанола, и других оксигенатов, (IV) синтез легких олефинов, парафинов и/или ароматических соединений посредством метанола или диметилового эфира как промежуточного продукта и/или (V) селективное сжигание водорода. Для достижения наибольшей эффективности эти стадии можно осуществлять последовательно; например, вначале может быть проведен процесс Фишера-Тропша с получением обогащенного C2+ потока с последующим метанированием для достижения высокой степени превращения Н2.
На стадии снижения содержания водорода обычно, как правило, так, как изложено ниже, образуются углеводороды, причем в этом случае после выделения одновременно получаемой воды по меньшей мере часть углеводородов целесообразно возвращать на стадию дегидроциклизации. Так, например, когда углеводороды, получаемые на стадии снижения содержания водорода, включают парафины и олефины, часть, возвращаемая на стадию дегидроциклизации, обычно включает парафины или олефины с 6 или меньшим числом углеродных атомов, в частности с 5 или меньшим числом углеродных атомов, например с 4 или меньшим числом углеродных атомов или с 3 или меньшим числом углеродных атомов. Когда углеводороды, получаемые на стадии снижения содержания водорода, включают ароматические соединения, часть, возвращаемая на стадию дегидроциклизации, обычно включает моноциклические ароматические материалы.
По другому варианту водород можно выделять из потока углеводородов с применением физических методов разделения, таких как криогенная дистилляция, адсорбция с колебанием давления, адсорбция с колебанием температуры и/или с применением мембранных систем. Может оказаться необходимым выделить водород физическим путем, когда существует возможность размещения для обогащенного водородом потока.
Метанирование/этанирование
В одном варианте стадия снижения содержания водорода включает реакцию по меньшей мере части водорода в отходящем из дегидроциклизации потоке с диоксидом углерода с образованием метана и/или этана в соответствии со следующими результирующими реакциями:
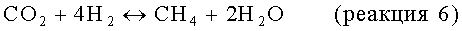

В целесообразном варианте используемый диоксид углерода является частью потока природного газа, а как правило того же потока природного газа, который используют как исходный материал, подаваемый на стадию дегидроциклизации. Когда диоксид углерода является частью метансодержащего потока, CO2/СН4 этого потока в целесообразном варианте сохраняют в пределах от примерно 1/1 до примерно 0,1/1. Смешения содержащего диоксид углерода потока и отходящего из дегидроциклизации потока в целесообразном варианте добиваются подачей газообразных исходных материалов во впускное приспособление струйного насоса.
На стадии снижения содержания водорода с получением метана или этана как правило используют молярное соотношение Н2/СО2, близкое к стехиометрическим пропорциям, требуемым для целевой реакции 6 или реакции 7, хотя, если необходимо получить содержащий CO2 или содержащий Н2 второй отходящий поток, в стехиометрическое соотношение могут быть внесены небольшие изменения. Стадию снижения содержания водорода с получением метана или этана в целесообразном варианте осуществляют в присутствии бифункционального катализатора, включающего металлический компонент, в особенности переходный металл или его соединение, на неорганическом носителе. Приемлемые металлические компоненты включают медь, железо, ванадий, хром, цинк, галлий, никель, кобальт, молибден, рутений, родий, палладий, серебро, рений, вольфрам, иридий, платину, золото, галлий и их сочетания и соединения. Неорганическим носителем может быть аморфный материал, такой как диоксид кремния, оксид алюминия и диоксид кремния/оксид алюминия, или подобный тем, которые перечислены для катализатора дегидроароматизации. Кроме того, неорганическим носителем может быть кристаллический материал, такой как микропористый или мезопористый кристаллический материал. Приемлемые пористые кристаллические материалы включают алюмосиликаты, алюмофосфаты и кремнеалюмофосфаты, перечисленные выше для катализатора дегидроциклизации.
Стадия снижения содержания водорода с получением метана и/или этана может быть осуществлена в широком диапазоне условий, включая температуру от примерно 100 до примерно 900°С, в частности от примерно 150 до примерно 500°С, например от примерно 200 до примерно 400°С, давление от примерно 200 до примерно 20000 кПа, в частности от примерно 500 до примерно 5000 кПа, и среднечасовую скорость подачи сырья от примерно 0,1 до примерно 10000 ч-1, в частности от примерно 1 до примерно 1000 ч-1. Значения степени превращения СО2, как правило, находятся в пределах от 20 до 100%, а предпочтительно больше 90%, в частности больше 99%. Эту экзотермическую реакцию можно проводить во множестве каталитических слоев с отводом тепла между слоями. Кроме того, для того чтобы максимизировать кинетические скорости, процесс в переднем слое (слоях) можно проводить при более высоких температурах, а для того чтобы максимизировать термодинамическое превращение, в последнем слое (слоях) его можно проводить при более низких температурах.
Основными продуктами такой реакции являются вода и, в зависимости от молярного соотношения Н2/СО2, метан, этан и более высокомолекулярные алканы совместно с некоторыми ненасыщенными С2- и более высокомолекулярными углеводородами. Кроме того, предпочтительна некоторая частичная гидрогенизация диоксида углерода до моноксида углерода. После удаления воды метан, моноксид углерода, весь непрореагировавший диоксид углерода и более высокомолекулярные углеводороды можно направлять непосредственно на стадию дегидроциклизации для получения дополнительных ароматических продуктов.
Процесс Фишера-Тропша
В другом варианте стадия снижения содержания водорода включает реакцию по меньшей мере части водорода в отходящем из дегидроциклизации потоке с моноксидом углерода в соответствии с процессом Фишера-Тропша с получением парафинов и олефинов с С2 по С5.
Процесс Фишера-Тропша в данной области техники известен хорошо (см., например, патенты US №№5348982 и 5545674, включенные в настоящее описание в качестве ссылок). Этот процесс, как правило, включает реакцию водорода и моноксида углерода в молярном соотношении от примерно 0,5/1 до примерно 4/1, в частности от примерно 1,5/1 до примерно 2,5/1, при температуре от примерно 175 до примерно 400°С, в частности от примерно 180 до примерно 240, и под давлением от примерно 1 до примерно 100 бар (от 100 до 10000 кПа), в частности от примерно 10 до примерно 40 бар (от 1000 до 4000 кПа), в присутствии катализатора Фишера-Тропша, обычно нанесенного или не нанесенного на носитель элемента группы VIII, неблагородного металла, например Fe, Ni, Ru, Co, с промотором или без него, например с рутением, рением, гафнием, цирконием, титаном. Носителями, когда их используют, могут служить огнеупорные оксиды металлов, таких как группы IVB, т.е. диоксид титана, диоксид циркония или диоксид кремния, оксид алюминия или диоксид кремния/оксид алюминия. В одном варианте катализатор включает не вызывающий конверсии катализатор, например кобальт или рутений, предпочтительно кобальт, с рением или цирконием в качестве промотора, предпочтительно с кобальтом и рением, нанесенными на диоксид кремния или диоксид титана, обычно на диоксид титана.
В другом варианте катализатор синтеза углеводородов включает металл, такой как Cu, Cu/Zn и Cr/Zn, на ZSM-5, и процесс проводят до получения значительных количеств моноциклических ароматических углеводородов. Пример такого процесса описан в работе Jose Erena Study of Physical Mixtures of Cr2O3-ZnO and ZSM-5 Catalysts for the Transformation of Syngas into Liquid Hydrocarbons; Ind. Eng. Chem. Res. 1998, 37, 1211-1219, включенной в настоящее описание в качестве ссылки.
Выделяют жидкости Фишера-Тропша, т.е. С5+, и от более тяжелых углеводородов отделяют легкие газы, например непрореагировавшие водород и СО, с C1 по С3 или С4 и воду. Затем более тяжелые углеводороды могут быть выделены как продукты или направлены на стадию дегидроциклизации для получения дополнительных ароматических продуктов.
Наличие моноксида углерода, требующегося для реакции Фишера-Тропша, может быть полностью или частично обеспечено благодаря имеющемуся или совместно подаваемому с метансодержащим исходным материалом и полученному в качестве побочного продукта на стадии дегидроциклизации моноксиду углерода. Если необходимо, дополнительный моноксид углерода может быть генерирован за счет подачи диоксида углерода, содержащегося, например, в природном газе, к катализатору конверсии, благодаря чему моноксид углерода получают обратной реакцией конверсии водяного газа:

и следующей реакцией:

Синтез спиртов
В еще одном варианте стадия снижения содержания водорода включает реакцию по меньшей мере части водорода в отходящем из дегидроциклизации потоке с моноксидом углерода с получением спиртов с C1 по С3, в особенности метанола. Получение метанола и других оксигенатов из синтез-газа также хорошо известно и представлено, например, в патентах US №№6114279, 6054497, 5767039, 5045520, 5254520, 5610202, 4666945, 4455394, 4565803, 5385949, описания к которым включены в настоящее описание в качестве ссылок. Используемый синтез-газ, как правило, обладает молярным отношением водорода (Н2) к оксидам углерода (СО+СО2) в интервале от примерно 0,5/1 до примерно 20/1, в частности в интервале от примерно 2/1 до примерно 10/1, причем диоксид углерода необязательно содержится в количестве не больше 50 мас.% в пересчете на общую массу синтез-газа.
Катализатор, используемый в процессе синтеза метанола, обычно включает оксид по меньшей мере одного элемента, выбранного из группы, включающей медь, серебро, цинк, бор, магний, алюминий, ванадий, хром, марганец, галлий, палладий, осмий и цирконий. В подходящем варианте катализатор представляет собой катализатор на основе меди, в частности в форме оксида меди, необязательно в присутствии оксида по меньшей мере одного элемента, выбранного из серебра, цинка, бора, магния, алюминия, ванадия, хрома, марганца, галлия, палладия, осмия и циркония. В подходящем варианте катализатор содержит оксид меди и оксид по меньшей мере одного элемента, выбранного из цинка, магния, алюминия, хрома и циркония. В одном варианте катализатор синтеза метанола выбирают из группы, включающей оксиды меди, оксиды цинка и оксиды алюминия. В более предпочтительном варианте катализатор содержит оксиды меди и цинка.
Процесс синтеза метанола может быть осуществлен в широком интервале температур и давлений. Приемлемые температуры находятся в интервале от примерно 150 до примерно 450°С, в частности от примерно 175 до примерно 350°С, в частности от примерно 200 до примерно 300°С. Приемлемые давления находятся в интервале от примерно 1500 до примерно 12500 кПа, в частности от примерно 2000 до примерно 10000 кПа, в частности от примерно 2500 до примерно 7500 кПа. Среднечасовые скорости подачи газа варьируют в зависимости от типа процесса, который проводят, но обычно среднечасовая скорость подачи газа в потоке газа через каталитический слой находится в интервале от примерно 50 до примерно 50000 ч-1, в частности от примерно 250 до примерно 25000 ч-1, в частности от примерно 500 до примерно 10000 ч-1. Эту экзотермическую реакцию можно проводить либо в неподвижных, либо в псевдоожиженных слоях, включающих множество каталитических слоев, с отводом тепла между слоями. Кроме того, для того чтобы максимизировать кинетические скорости, процесс в переднем слое (слоях) можно проводить при более высоких температурах, а для того чтобы максимизировать термодинамическое превращение, в последнем слое (слоях) его можно проводить при более низких температурах.
Получаемые метанол и/или другие оксигенаты могут быть направлены на продажу как самостоятельный продукт, могут быть использованы для алкилирования ароматических соединений, образующихся на стадии дегидроциклизации, до более высокоценных продуктов, таких как ксилолы, или могут быть использованы в качестве исходного материала для получения более низкомолекулярных олефинов, в особенности этилена и пропилена. Превращение метанола в олефины является хорошо известным процессом, который описан, например, в патенте US №4499327, включенном в настоящее описание в качестве ссылки.
Селективное сжигание водорода
Тем не менее, в еще одном варианте стадия снижения содержания водорода включает селективное сжигание водорода, которое представляет собой процесс, в котором водород в смешанном потоке взаимодействует с кислородом с образованием воды или водяного пара без существенного взаимодействия в потоке углеводородов с кислородом с получением моноксида углерода, диоксида углерода и/или оксигенированных углеводородов. Обычно селективное сжигание водорода проводят в присутствии кислородсодержащего твердого материала, такого как смешанный оксид металла, который обычно высвобождает часть связанного кислорода для водорода.
Один приемлемый способ селективного сжигания водорода описан в патенте US №5430210, включенном в настоящее описание в качестве ссылки, он включает контактирование в реакционных условиях первого потока, включающего углеводород и водород, и второго потока, включающего кислород, с раздельными поверхностями мембраны, непроницаемой для не содержащих кислорода газов, где упомянутая мембрана включает оксид металла, селективный в отношении сжигания водорода, и выделение продукта селективного сжигания водорода. Этот оксид металла, как правило, представляет собой смешанный оксид металла висмута, индия, сурьмы, таллия и/или цинка.
В патенте US №5527979, включенном в настоящее описание в качестве ссылки, описан способ чистой каталитической окислительной дегидрогенизации алканов с получением алкенов. Этот способ включает одновременную равновесную дегидрогенизацию алканов до алкенов и селективное сжигание образующегося водорода для проведения равновесной реакции дегидрогенизации с дальнейшим образованием алкенов. Так, в частности, алкановый исходный материал дегидрируют над катализатором равновесной дегидрогенизации в первом реакторе, а затем отходящий из первого реактора поток совместно с кислородом направляют во второй реактор, содержащий катализатор из оксида металла, который служит для катализа селективного сжигания водорода. Катализатор равновесной дегидрогенизации может включать платину, а катализатор селективного сжигания из оксида металла может включать висмут, сурьму, индий, цинк, таллий, свинец и теллур или их смесь.
В заявке на патент US №2004/0152586, опубликованной 5 августа 2004 г. и включенной в настоящее описание в качестве ссылки, описан способ снижения содержания водорода в отходящем из крекинг-установки потоке. В этом способе используют каталитическую систему, включающую (1) по меньшей мере один твердый кислотный компонент крекинга и (2) по меньшей мере один компонент селективного сжигания водорода на металлической основе, состоящий, по существу, из (а) сочетания металлов, выбранных из группы, включающей: I) по меньшей мере один металл из группы 3 и по меньшей мере один металл из групп с 4 по 15 Периодической таблицы элементов; II) по меньшей мере один металл из групп с 5 по 15 Периодической таблицы элементов и по меньшей мере один металл из по меньшей мере одной из групп 1, 2 и 4 Периодической таблицы элементов; III) по меньшей мере один металл из групп 1 и 2, по меньшей мере один металл из группы 3 и по меньшей мере один металл из групп с 4 по 15 Периодической таблицы элементов; и IV) два или большее число металлов из групп с 4 по 15 Периодической таблицы элементов; и (б) по меньшей мере одного из кислорода и серы, где этот по меньшей мере один из кислорода и серы химически связан как внутри, так и между металлами.
Реакцию селективного сжигания водорода по настоящему изобретению обычно проводят при температуре в интервале от примерно 300 до примерно 850°С и под давлением в интервале от примерно 1 до примерно 20 ат (от 100 до 2000 кПа).
ВЫДЕЛЕНИЕ/ОБРАБОТКА АРОМАТИЧЕСКИХ ПРОДУКТОВ
В дополнение к водороду, другими продуктами стадии дегидроциклизации являются бензол и нафталин. Эти продукты могут быть выделены из отходящего из дегидроциклизации потока, как правило, экстракцией растворителем с последующим разделением на фракции, а затем поставлены для продажи непосредственно как химические продукты массового производства. По другому варианту некоторое количество или весь бензол и/или нафталин может быть алкилирован с получением, например, толуола, ксилолов и алкилнафталинов, и/или может быть подвергнут гидрогенизации с получением, например, циклогексана, циклогексена, дигидронафталина (бензилциклогексена), тетрагидронафталина (тетралина), гексагидронафталина (дициклогексена), октагидронафталина и/или декагидронафталина (декалина). Приемлемые способы алкилирования и гидрогенизации описаны ниже и более подробно в совместно рассматриваемых заявках авторов настоящего изобретения РСТ №№ PCT/US 2005/043523 (реестр патентного поверенного №2004В156), поданной 2 декабря 2005 г., и PCT/US 2005/044038 (реестр патентного поверенного №2004В155), поданной 2 декабря 2005 г.
Алкилирование ароматических соединений
Алкилирование ароматических соединений, таких как бензол и нафталин, в данной области техники хорошо известно и, как правило, включает реакцию олефина, спирта или алкилгалогенида с ароматическими материалами в газообразной или жидкой фазе в присутствии кислотного катализатора. Приемлемые кислотные катализаторы включают цеолиты со средними порами (т.е. те, которые обладают ограничивающим показателем от 2 до 12, как определено в US №4016218), включая материалы, обладающие каркасами типов MFI (например, ZSM-5 и силикалит), MEL (например, ZSM-11), MTW (например, ZSM-12), TON (например, ZSM-22), МТТ (например, ZSM-23), MFS (например, ZSM-57) и FER (например, ZSM-35), и ZSM-48, а также цеолиты с крупными порами (т.е. те, которые обладают ограничивающим показателем меньше 2), такие как материалы, обладающие каркасами типов ВЕА (например, бета-цеолит), FAU (например, ZSM-3, ZSM-20, цеолиты X, Y, ультрастабилизированный Y и деалюминированный Y), MOR (например, морденит), MAZ (например, ZSM-4), MEI (например, ZSM-18) и MWW (например, МСМ-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, МСМ-36, МСМ-49 и МСМ-56).
В одном варианте предлагаемого способа бензол выделяют из отходящего из дегидроциклизации потока и затем алкилируют олефином, таким как этилен, получаемый в качестве побочного продукта на стадии снижения содержания водорода с применением этанирования/метанирования. Типичные условия проведения парофазного алкилирования бензола этиленом включают температуру от примерно 650 до примерно 900°F (от 343 до 482°С), манометрическое давление от примерно атмосферного до примерно 3000 фунтов/кв.дюйм (от 100 до 20800 кПа), ССПС в пересчете на этилен от примерно 0,5 до примерно 2,0 ч-1 и мольное отношение бензола к этилену от 1/1 до 30/1. Жидкофазное алкилирование бензола этиленом можно проводить при температуре в пределах от 300 до 650°F (от 150 до 340°С), под манометрическим давлением до примерно 3000 фунтов/кв.дюйм (20800 кПа), ССПС в пересчете на этилен от примерно 0,1 до примерно 20 ч-1 и при мольном отношении бензола к этилену от 1/1 до 30/1.
В целесообразном варианте этилирование бензола проводят в условиях по меньшей мере частично жидкой фазы с использованием катализатора, включающего по меньшей мере один из бета-цеолита, цеолита Y, МСМ-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, ITQ-13, ZSM-5 МСМ-36, МСМ-49 и МСМ-56.
Этилирование бензола может быть осуществлено по месту процесса дегидроциклизации/снижения содержания водорода или бензол может быть транспортирован в другой регион для превращения в этилбензол. Затем полученный этилбензол может быть поставлен для продажи, использован как предшественник, например при получении стирола, или изомеризован по методам, хорошо известным в данной области техники, в смешанные ксилолы.
В другом варианте предлагаемого способа алкилирующий агент представляет собой метанол или диметиловый эфир (ДМЭ), его используют для алкилирования бензола и/или нафталина, выделяемого из отходящего из дегидроциклизации потока, с получением толуола, ксилолов, метилнафталинов и/или диметилнафталинов. Когда метанол или ДМЭ используют для алкилирования бензола, в целесообразном варианте это осуществляют в присутствии катализатора, включающего цеолит, такой как ZSM-5, бета-цеолит, ITQ-13, MCM-22, MCM-49, ZSM-11, ZSM-12, ZSM-22, ZSM-23, ZSM-35 и ZSM-48, который предварительно модифицируют обработкой водяным паром таким образом, чтобы он обладал диффузионным параметром для 2,2-диметилбутана примерно от 0,1 до 15 с-1, когда его определяют при температуре 120°С и давлении 2,2-диметилбутана 60 торр (8 кПа). Такой способ селективен в отношении получения пара-ксилола, он изложен, например, в патенте US №6504272, включенном в настоящее описание в качестве ссылки. Когда метанол используют для алкилирования нафталина, в целесообразном варианте это осуществляют в присутствии катализатора, включающего ZSM-5, MCM-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, ITQ-13, МСМ-36, MCM-49 или МСМ-56. Такой способ можно применять для селективного получения 2,6-диметилнафталина, он изложен, например, в патентах US №№4795847 и 5001295, включенных в настоящее описание в качестве ссылок.
Когда в способе по изобретению в качестве алкилирующего агента используют метанол или ДМЭ, его можно вводить в процесс как отдельный исходный материал или он может быть по меньшей мере частично получен in situ добавлением содержащего диоксид углерода газообразного исходного материала, такого как поток природного газа, в часть или весь отходящий со стадии дегидроциклизации поток. Так, в частности, отходящий из дегидроциклизации поток перед каким-либо выделением ароматических компонентов можно направлять в реактор обратной конверсии и проводить реакцию с содержащим диоксид углерода исходным материалом в условиях повышения содержания моноксида углерода в этом отходящем потоке, т.е. такими реакциями, как вышеприведенные реакции 5 и 8.
Кроме того, в реактор обратной конверсии можно направлять метан и CO2 и/или водяной пар с получением синтез-газа, который затем может быть смешан с частью отходящего из дегидроциклизации потока для регулирования соотношений Н2/СО/СО2 в зависимости от потребности для стадии алкилирования.
Как правило, реактор обратной конверсии содержит катализатор, включающий переходный металл на носителе, такой как Fe, Ni, Cr, Zn на оксиде алюминия, диоксиде кремния или диоксиде титана, и работает в условиях, включающих температуру от примерно 500 до примерно 1200°С, в частности от примерно 600 до примерно 1000°С, например от примерно 700 до примерно 950°С, и давление от примерно 1 до примерно 10000 кПа, в частности от примерно 2000 до примерно 10000 кПа, например от примерно 3000 до примерно 5000 кПа. Среднечасовые скорости подачи газа можно варьировать в зависимости от типа применяемого способа, но обычно среднечасовая скорость подачи газа в потоке газа через каталитический слой находится в интервале от примерно 50 до примерно 50000 ч-1, в частности от примерно 250 до примерно 25000 ч-1, более предпочтительно от примерно 500 до примерно 10000 ч-1.
Затем отходящий из реактора обратной конверсии поток может быть направлен в реактор алкилирования, работающий в условиях, обеспечивающих протекание таких реакций, как следующие:
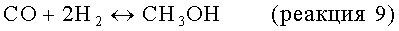


Приемлемые для такого реактора условия алкилирования включают, по-видимому, температуру от примерно 100 примерно до примерно 700°С, давление от примерно 1 до примерно 300 ат (от 100 до 30000 кПа) и ССПС для ароматического углеводорода от примерно 0,01 до примерно 100 ч-1. Приемлемый катализатор включает, по-видимому, молекулярное сито, обладающее ограничивающим показателем от 1 до 12, такое как ZSM-5, как правило, совместно с одним из металлов или оксидов металлов, таких как медь, хром и/или оксид цинка.
Когда в подходящем варианте катализатор алкилирования включает молекулярное сито, это последнее модифицируют для изменения его диффузионных характеристик таким образом, чтобы превалирующим изомером ксилола, получаемого реакцией 11, был пара-ксилол. Приемлемое средство диффузионной модификации включает обработку водяным паром и осаждение ex-situ или in situ соединений кремния, кокса, оксидов металлов, таких как MgO, и/или Р на поверхности или на входах в поры молекулярного сита. Предпочтительно также то, что активный металл вводят в молекулярное сито таким образом, чтобы обеспечить насыщение более высокореакционноспособных материалов, таких как олефины, которые способны образовываться в качестве побочных продуктов и которые в противном случае могли бы вызвать дезактивацию катализатора.
Затем отходящий из реактора алкилирования поток можно было бы направить в секцию разделения, в которой ароматические продукты вначале отделяли бы от водорода и других низкомолекулярных материалов, целесообразно экстракцией растворителем. Далее ароматические продукты можно было бы разделить на бензольную фракцию, толуольную фракцию, С8фракцию и тяжелую фракцию, включающую нафталин и алкилированные нафталины. Затем ароматическая С8фракция могла бы быть направлена в процесс кристаллизации или сорбции для отделения ценного п-ксилольного компонента, а оставшиеся смешанные ксилолы либо поставлены для продажи как продукт, либо направлены в контур изомеризации для получения дополнительного количества п-ксилола. Толуольная фракция могла бы быть либо удалена как находящий сбыт продукт, либо возвращена в реактор алкилирования, либо направлена в установку диспропорционирования толуола, в частности в установку селективного диспропорционирования толуола, для получения дополнительного количества п-ксилола.
Гидрогенизация ароматических соединений
В дополнение к стадии алкилирования или вместо нее по меньшей мере часть ароматических компонентов в отходящем из дегидроциклизации потоке может быть гидрирована с получением полезных продуктов, таких как циклогексан, циклогексен, дигидронафталин (бензилциклогексен), тетрагидронафталин (тетралин), гексагидронафталин (дициклогексен), октагидронафталин и/или декагидронафталин (декалин). Эти продукты можно использовать в качестве топлив и химических промежуточных продуктов, а в случае тетралина и декалина эти последние можно использовать в качестве растворителя для экстракции из отходящего из дегидроциклизации потока ароматических компонентов.
Гидрогенизацию целесообразно, но необязательно, проводить после выделения из отходящего из дегидроциклизации потока ароматических компонентов и целесообразно использовать часть водорода, образуемого реакцией дегидроциклизации. Приемлемые способы гидрогенизации ароматических соединений в данной области техники известны хорошо, и в них как правило используют катализатор, включающий Ni, Pd, Pt, Ni/Mo или сульфидированные Ni/Mo, нанесенные на оксид алюминия или диоксид кремния как носитель. Приемлемые для процесса гидрогенизации рабочие условия включают температуру от примерно 300 до примерно 1000°F (от 150 до 540°С), в частности от примерно 500 до примерно 700°F (от 260 до 370°С), манометрическое давление от примерно 50 до примерно 2000 фунтов/кв.дюйм (от 445 до 13890 кПа), в частности от примерно 100 до примерно 500 фунтов/кв.дюйм (от 790 до 3550 кПа), и ССПС от примерно 0,5 до примерно 50 ч-1, в частности от примерно 2 до примерно 10 ч-1.
Для того чтобы получить материалы, приемлемые для полимеризации или другого последующего химического превращения, может оказаться также необходимой частичная гидрогенизация с целью оставить в продукте одну или несколько олефиновых углерод-углеродных связей. Приемлемые способы частичной гидрогенизации в данной области техники известны хорошо, и в них, как правило, используют катализатор, включающий благородные металлы, причем рутений в предпочтительном варианте наносят на оксиды металлов, такие как La2O3/ZnO. Могут быть также использованы гомогенные каталитические системы с благородными металлами. Примеры способов частичной гидрогенизации описаны в патентах US №№4678861, 4734536, 5457251, 5656761, 5969202 и 5973218, содержания которых в полном объеме включены в настоящее описание в качестве ссылок.
Альтернативный способ гидрогенизации включает гидрокрекинг низкого давления нафталинового компонента с получением алкилбензолов над таким катализатором, как сульфидированные Ni/W или сульфидированный Ni, нанесенный на аморфный алюмосиликат или цеолит, такой как цеолит X, цеолит Y и бета-цеолит. Приемлемые для гидрокрекинга низкого давления рабочие условия включают температуру от примерно 300 до примерно 1000°F (от 150 до 540°С), в частности от примерно 500 до примерно 700°F (от 260 до 370°С), манометрическое давление от примерно 50 до примерно 2000 фунтов/кв.дюйм (от 445 до 13890 кПа), в частности от примерно 100 до примерно 500 фунтов/кв.дюйм (от 790 до 3550 кПа), и ССПС от примерно 0,5 до примерно 50 ч-1, в частности от примерно 2 до примерно 10 ч-1.
Изобретение далее более конкретно описано со ссылкой на прилагаемые чертежи и следующие неограничивающие примеры.
Что касается фиг.1, то на чертеже проиллюстрирована упрощенная схема дегидроциклизационной реакторной системы для превращения метана в ароматические соединения в соответствии с одним из вариантов выполнения изобретения. В этом варианте дегидроциклизационный реактор включает реакционную зону 11 с отстойным слоем, в которой каталитический порошкообразный материал движется из верхней части этой реакционной зоны к основанию реакционной зоны, в то время как исходный материал пропускают через реакционную зону в противоположном направлении. Нагретый каталитический порошкообразный материал истекает через впускное приспособление, расположенное возле верхней части реактора 11, по линии 12. Охлажденный каталитический порошкообразный материал движется из реактора 11 посредством выпускных приспособлений, расположенных вблизи основания реактора 11, и его отводят по линиям 13 и 14. Метановый исходный материал вводят в реактор 11 вблизи его основания по линии 15. Продукт и непрореагировавший метан движутся из реактора 11 посредством выпускного приспособления 16, смежного с верхней частью реактора 11. Как правило, нагретый катализатор поступает в реактор 11 при температуре примерно 850°С, а охлажденный катализатор выходит из реактора при температуре примерно 600°С. На фиг.1 изображен реактор 11, включающий одну реакционную зону. Однако для того, кто является обычным специалистом в данной области техники, очевидно, что реакторная система может включать больше одной зоны.
Что касается фиг.2, то на чертеже проиллюстрирована упрощенная схема дегидроциклизационной реакторной системы для превращения метана в ароматические соединения в соответствии с одним вариантом выполнения изобретения. В этом варианте дегидроциклизационный реактор включает два последовательно соединенных реактора 21 и 22 с подвижными слоями, в которых порошкообразный катализатор каскадирует из одного реактора в следующий смежный реактор в этом ряду в одном направлении, в то время как исходный материал пропускают через и между реакторами в противоположном направлении. Нагретый катализатор продвигается по линии 23 через впускное приспособление, расположенное вблизи верхней части реактора 21, из которой охлажденный катализатор продвигается посредством выпускного приспособления 24, расположенного вблизи основания реактора 21, в реактор 22 посредством впускного приспособления 25. Этот катализатор продвигается из реактора 22 посредством выпускного приспособления вблизи основания реактора 22 по линии 26. Метановый исходный материал 27 вводят в реактор 22 вблизи его основания по линии 27. Продукт и непрореагировавший метан движутся из реактора 22 через выпускное приспособление вблизи верхней части реактора 22 посредством выпускного приспособления 29 и поступают в реактор 21 посредством впускного приспособления вблизи его основания по линии 28. Конечный продукт удаляют из реактора 21 через выпускное приспособление вблизи верхней части реактора 21 по линии 20. Как правило, нагретый катализатор поступает в реактор 21 при температуре примерно 850°С, а охлажденный катализатор выходит из реактора 22 при температуре примерно 600°С. На фиг.2 изображены зоны, находящиеся в отдельных сосудах, однако обе зоны могут быть расположены в одном сосуде с соответствующими внутрикорпусными устройствами для поддержания работы обеих зон. Для того, кто является обычным специалистом в данной области техники, очевидно, что реакторная система может включать больше двух каскадирующих подвижных слоев, например 3, 4 или 5 последовательно соединенных реакторов или зон с подвижными слоями.
Для того, кто является обычным специалистом в данной области техники, очевидно, что варианты, рассмотренные в данном применении, не представляют все возможные варианты устройства или способа, приведенные в настоящем описании. Кроме того, многие элементы оборудования и устройство, а также некоторые стадии обработки могут оказаться необходимыми для промышленных, технических или даже экспериментальных целей. Примеры таких оборудования, устройства и технологических стадий представляют собой, хотя ими их список не ограничен, дистилляционные колонны, ректификационные колонны, теплообменники, насосы, клапаны, манометры, термометры, сепараторы жидкости/пара, сушильные и/или очистительные аппараты для исходного материала и продуктов, очистительные аппараты для глины, оборудование для хранения исходного материала и/или продуктов и средства технологического контроля и регулирования процессов и стадий. Хотя такие оборудование, устройство и стадии, потребность в которых для понимания сущности настоящего описания отсутствует, на чертежах не представлены, для иллюстрации различных объектов изобретения некоторые из них время от времени могут быть упомянуты. Необходимо также отметить, что некоторые из единиц оборудования в процессе могут быть, в зависимости от условий проведения процессов, размещены в других местах.
Изобретение далее более конкретно описано со ссылкой на следующие неограничивающие примеры.
Пример 1
Пример 1 демонстрирует применение совместно подаваемых материалов (таких как Н2, СО2 и Н2О) с целью добиться пониженной скорости коксообразования на Mo/ZSM-5 катализаторе во время дегидроциклизации метана с получением, главным образом, бензола. Пример 2 показывает, что благодаря уменьшению количества кокса, который образует отложения на катализаторе во время "углеводородного периода", существует возможность сохранить высокую эффективность после многочисленных циклов "углеводорода" и регенерирования и что более низкая скорость закоксовывания за один цикл преобразуется в более низкую результирующую скорость отложения кокса в течение многочисленных циклов.
Mo/ZSM-5 катализаторы готовили с применением двух методов: (1) посредством пропитки требуемым количеством раствора гептамолибдата аммония NH4 +-ZSM-5 носителя (обладавшего значением соотношения Si/Al2 25) посредством начальной влажности с последующими сушкой при 120°С в течение 2 ч и конечным кальцинированием при 500°С в течение 6 ч в токе воздуха, и (2) измельчением в шаровой мельнице оксида молибдена с NH4 +-ZSM-5 носителем (обладавшем значением соотношения Si/Al2 25) в течение 2 ч с последующим кальцинированием при 500°С в течение 5 ч в токе воздуха. Содержание молибдена (в пересчете на металл в мас.%) варьировали изменением концентрации гептамолибдата аммония в пропиточном растворе или количества оксида молибдена, добавленного в измельчаемую смесь.
Каталитическое испытание приготовленных Mo/ZSM-5 катализаторов осуществляли с применением прибора Tapered-Element Oscillating Microbalance (ТЕОМ) с короткими периодами срабатывания, позволявшего точно определять изменения массы катализатора во время реакции. Катализатор (после кальцинирования) таблетировали, измельчали и просеивали до размеров частиц от 20 до 40 меш. Приблизительно 0,10 г просеянных каталитических частиц помещали в держатель для образца прибора ТЕОМ (общий объем образца: 0,20 куб.см) и с использованием подкладок из кварцевого волокна укладывали с получением неподвижного слоя. Эксплуатационные свойства катализатора при дегидроциклизации метана до бензола определяли при 800°С и под абсолютным давлением 20 фунтов/кв.дюйм (138 кПа) в экспериментах А и Б или 14,7 фунтов/кв.дюйм (101 кПа) в экспериментах с В по Д в таблице 1 с использованием исходного материала, содержавшего указанные конкретные совместно подаваемые материалы (СО2, Н2О, Н2), Ar (где Ar применяют в качестве внутреннего стандарта), а остальное - СН4, при указанной среднечасовой скорости подачи сырья (в пересчете на метан). Отходивший из реакции поток анализировали с помощью масс-спектрометра для определения концентраций метана, бензола, нафталина, водорода и аргона. Скорость отложения кокса на катализаторе (т.е. тяжелое углеродистое отложение, которое с поверхности катализатора не улетучивается) определяли непосредственно по изменениям массы, отмечаемым с помощью микровесов. Значения, приведенные для эксплуатационных свойств катализатора (например, производительность по бензолу, превращенный метан, селективность в отношении бензола), являются совокупными или средними значениями за период времени, начиная с момента инжекции метана и заканчивая, когда текущая селективность в отношении бензола уменьшается до <2%.
Как показано в таблице 1, при содержании Мо 4,6% в экспериментах А и Б средняя скорость отложения кокса уменьшалась с 5,2 мас.% в час в случае отсутствия совместно подаваемых материалов (т.е. исходная композиция из 95% СН4/5% Ar) до 2,2 мас.% в час в случае с 2% СО2 и 10% Н2 как совместно подаваемого материала (т.е. исходная композиция из 2% CO2, 10% Н2, 83,6% СН4 и 4,4% Ar). Производительность по бензолу увеличивалась с 0,5 г бензола, полученного на грамм катализатора для случая без совместно подаваемого материала, до 1,3 г бензола на грамм катализатора. Это показывает, что присутствие СО2 и Н2 как совместно подаваемых материалов может значительно улучшить производительность по бензолу Mo/ZSM-5 катализаторов за счет снижения скоростей коксообразования. Эксперименты В, Г и Д демонстрируют влияние совместно подаваемых Н2О и Н2 на катализатор, включающий 2,7% Мо на ZSM-5. Средняя скорость закоксовывания, как было установлено, уменьшалась с 8,6 мас.%/ч без совместно подаваемого материала до 6,0 мас.%/ч с 3% водяного пара как совместно подаваемого материала и до 3,8 мас.%/ч с 20% Н2 как совместно подаваемого материала. В обоих случаях производительность и селективность в отношении бензола, как было установлено, с добавлением совместно подаваемого материала повышались.
Пример 2
Mo/ZSM-5 катализаторы готовили с применением методов, описанных ранее в примере 1. Катализаторы в синтезированном виде подвергали циклическому старению, которое состояло из (1) "углеводородного" периода, когда на катализатор воздействовали подачей СН4 при 800°С и под абсолютным давлением 14,7 фунта/кв.дюйм (101 кПа) с указанными конкретными совместно подаваемыми материалами в течение 5 мин при указанной ССПС в пересчете на СН4, с последующим (2) периодом регенерирования, когда катализатор нагревали до 850°С и под абсолютным давлением 14,7 фунта/кв.дюйм (101 кПа) в газообразном Н2 со скоростью 10°С/мин и выдерживали при 850°С в течение указанного времени при ССПГ 9000 куб.см [в нормальных условиях]/г·катализатора/ч. После завершения стадии регенерирования катализатор охлаждали до 800°С перед повторной инжекцией метанового исходного материала. Перед циклическим старением в синтезированном виде катализатор предварительно закоксовывали нагреванием катализатора в газообразной смеси 15% СН4/Н2 со 150 до 800°С со скоростью 5°С/мин и выдерживали при 800°С в течение 1 ч. После того как катализаторы подвергали указанному числу циклов "углеводорода" и регенерирования, отработавшие катализаторы удаляли и испытывали на эксплуатационные свойства в ТЕОМ.
Приблизительно 0,10 г отработавшего катализатора помещали в держатель для образца прибора ТЕОМ и с использованием подкладок из кварцевого волокна укладывали с получением неподвижного слоя. Эксплуатационные свойства катализатора при дегидроциклизации метана до бензола определяли при 800°С и под абсолютным давлением 14,7 фунта/кв.дюйм (101 кПа) для экспериментов с А по Г и под абсолютным давлением 20 фунтов/кв.дюйм (138 кПа) для экспериментов с Д по Ж в таблице 2 с использованием 95% СН4/5% Ar как исходного материала (где Ar применяют в качестве внутреннего стандарта) и при среднечасовой скорости подачи сырья 4 г СН4, подаваемого на грамм катализатора в час. Отходивший из реакции поток анализировали с применением масс-спектрометра для определения концентраций метана, бензола, нафталина, водорода и аргона. Определение кокса на катализаторе осуществляли с использованием программированного по температуре окисления отработавшего катализатора в термо-гравиметрическом анализаторе (ТГА).
В таблице 2 сопоставлены аккумулированный кокс на катализаторе после многочисленных циклов "углеводорода" и регенерирование и остаточная производительность по бензолу отработавших катализаторов. Эксперименты с А по В демонстрируют эксплуатационные свойства 2,7% Mo/ZSM-5 катализатора после 0, 20 и 40 циклов. Эксперимент Г демонстрирует эксплуатационные свойства 2,7% Mo/ZSM-5 катализатора после 40 циклов. Эксперименты с Д по Ж демонстрируют эксплуатационные свойства 4,6% Mo/ZSM-5 катализатора после 0, 50 и 389 циклов. При сопоставлении экспериментов В и Г можно видеть, что добавление 20% Н2 как совместно подаваемого материала уменьшало результирующее отложение кокса на катализаторе примерно на 55%, с 2,0 мас.% (в случае без совместно подаваемого материала) до 0,9 мас.% (для 20% Н2 как совместно подаваемого материала). В дополнение к снижению коксообразования, производительность по бензолу после 40 циклов, как было установлено, была значительно выше, примерно на 25%, с 20% Н2 как совместно подаваемого материала (0,25 г бензола/г катализатора), если сравнивать со случаем без совместно подаваемого материала (0,20 г бензола/г катализатора). Эксперименты с Д по Ж показывают, что результирующее накопление кокса на катализаторе может быть дополнительно сведено к минимальному посредством добавления 2% СО2 как совместно подаваемого материала. После 50 циклов совокупный кокс на катализаторе, как было установлено, составлял всего 0,15 мас.%, что значительно меньше (примерно на 93%), чем кокс на катализаторе, отмечаемый после 40 циклов для случая без совместно подаваемого материала (т.е. 2,0 мас.%), даже несмотря на то, что ССПС исходного материала во время "углеводородного периода" увеличивали с 1 до 4. После 50 циклов никакого ухудшения производительности по бензолу не отмечали. После 389 циклов кокс на катализаторе определяли как составлявший примерно 0,5 мас.%, а это указывало на то, что результирующее накопление кокса на катализаторе в течение долговременного циклического старения может быть сведено к минимуму.
мент
ность по бензолу (г СН4/г кат.)
ности по бензолу, %
Пример 3
Этот пример предназначен для того, чтобы показать, что регенерирование под более высоким давлением обладает преимуществом улучшения селективности катализатора, т.е. улучшения селективности в отношении бензола и понижения селективности в отношении коксообразования.
Катализатор, включавший примерно 4 мас.% Мо на ZSM-5, использовали для ароматизации исходного материала, включавшего 86,65 мольных % СН4, 1,8% С2Н6, 0,9% CO2, 0,45% Н2 и 10 мольных % Ar. Для расчета значений выхода бензола отходивший поток анализировали газовой хроматографией. Катализатор использовали в эксперименте с чередующимися циклами реакции и регенерирования, причем метансодержащий исходный материал подавали к катализатору в течение каждого 20-минутного реакционного цикла, а водород подавали к катализатору в течение каждого 40-минутного регенерационного цикла. Для первых 90% каждого реакционного цикла в метансодержащий исходный материал в качестве совместно подаваемого материала добавляли 20% H2, но завершали во время конечных 10% каждого реакционного цикла. Реакционное манометрическое давление составляло примерно 7 фунтов/кв.дюйм (149 кПа), тогда как реакционную температуру повышали в течение каждого реакционного цикла от начального значения примерно 700°С до конечного значения примерно 800°С. Средняя температура в течение каждого регенерационного цикла была равной примерно 850°С, причем максимальная температура составляла 860°С. В начале испытательного эксперимента манометрическое давление в течение каждого регенерационного цикла составляло 34 фунта/кв.дюйм (335 кПа), а стабилизированные значения селективности и выхода бензола были такими, как перечисленные ниже. По прошествии предопределенного времени манометрическое давление в течение каждого регенерационного цикла понижали до примерно 7 фунтов/кв.дюйм (149 кПа) и, спустя примерно 11 ч после изменения регенерационного давления, вновь определяли значения селективности и выхода бензола, они представлены ниже. Можно видеть, что под более высоким регенерационным давлением селективность в отношении и выход бензола оказывались выше, а селективность в отношении кокса ниже.
Манометрическое давление: 34 фунта/кв.дюйм 7 фунтов/кв.дюйм
Выход бензола, % от
Значения селективности, превращенный углерод в % от исходного:
Пример 4
Mo/ZSM-5 катализаторы готовили посредством пропитки требуемым количеством раствора гептамолибдата аммония NH4ZSM-5 носителя (обладавшего значением соотношения Si/Al2 28) посредством начальной влажности с последующими сушкой при 120°С в течение 2 ч и конечным кальцинированием при 500°С в течение 6 ч в токе воздуха. Номинальное целевое содержание молибдена (мас.% металла в пересчете на общую массу катализатора) составляло 2,7 мас.%; небольшие варьирования содержания молибдена на сделанные заключения не влияли. Каждый образец Mo/ZSM-5 катализатора (после кальцинирования) таблетировали, измельчали и просеивали до размеров частиц от 30 до 60 меш. Каталитическое испытание Mo/ZSM-5 катализаторов осуществляли в кварцевом реакторе, содержавшем насадку для получения неподвижного слоя с использованием подкладок из кварцевого волокна.
Эксплуатационные свойства катализатора при дегидроциклизации метана до бензола определяли при различных температурах с использованием в качестве исходного материала 95 мас.% СН4/5 мас.% аргона (аргон используют в качестве внутреннего стандарта) при среднечасовой скорости подачи сырья (в пересчете на метан) 1,2 ч-1. Все экспериментальные данные получали под абсолютным давлением 138 кПа (20 фунтов/кв.дюйм) и под идентичным давлением осуществляли также все моделирование. Отходивший из реакции поток анализировали с помощью масс-спектрометра и газового хроматографа для определения концентраций метана, бензола, толуола, этилена, нафталина, водорода и аргона. Скорость отложения кокса на катализаторе (т.е. тяжелого углеродистого отложения, которое с поверхности катализатора не улетучивается) определяли посредством углеродного баланса. Дополнительные данные получали при двух температурах (750 и 800°С) с Н2, который добавляли в исходный материал при соответственно 6 и 20 мольных %.
В целях эксперимента в примере 4 экспериментальные данные объединяли в две величины: Сел.БТН и Сел.Кокс. Сел.БТН обозначает среднюю селективность на углеродной молярной основе, как ее определяют по сумме молей углерода в продукте, содержавшемся в бензоле, толуоле и нафталине, деленной на моли углерода, содержавшегося в метане, который прореагировал. Сел.Кокс обозначает среднюю селективность на углеродной молярной основе, как ее определяют по сумме молей углерода, который оставался в реакторе, деленной на моли углерода, содержавшегося в метане, который прореагировал. Сумма Сел.БТН и Сел.Кокс не равна 100% благодаря образованию других небольших продуктов, по преимуществу этилена. Поскольку часто затруднительно получить точные экспериментальные данные термодинамического превращения, для установления значения Превр.Бл. применяют технически доступное программное обеспечение (PROII/6,0 Copyright 2003 Invensys Systems Inc.) для моделирования. Превр.Бл. определяют как максимально термодинамически достижимое превращение метана в бензол и водород (т.е. отсутствие модельного ограничения, благодаря которому отсутствуют другие продукты, такие как кокс, нафталин, этилен и т.д.) при данных температуре и абсолютном давлении 138 кПа (20 фунтов/кв.дюйм). Результаты эксперимента и моделирования представлены в таблице 3.
Понятно, что разные каталитические композиции, применение совместно подаваемых материалов (СО2, СО, Н2О, Н2, О2, этан, пропан и т.д.), разные рабочие давления и/или разные объемные скорости способны изменять значения селективности и превращения, но в то время как точная степень улучшения, представленная в настоящем описании, может меняться, направленные улучшения, приведенные в настоящем описании, все-таки достижимы. Кроме того, необходимо иметь в виду, что в качестве базиса для моделирования расчетов, обсуждаемых ниже, допускают, что направляемый в реактор метановый исходный материал всегда предварительно нагревали до той же температуры (600°С) и во всех случаях использовали номинальную скорость подачи метана 100 кг/ч. В качестве базиса предусматривали также то, что катализатор, направляемый в реакторные системы с подвижным слоем, выдерживали при той же температуре (850°С). Количество катализатора, необходимое для поддержания этой температуры, рассчитывали для каждой реакторной конфигурации. Для простоты допускали, что теплопроводность, коэффициент температуропроводности и излучательная способность поверхности катализатора оставались постоянными. В следующей таблице 4 перечислены физические постоянные и свойства катализатора, использованные в расчетах.
Для возможности моделирования различных реакторных конфигураций для Сел.БТН, Сел.Кокс и Превр.Бл. уравнения выводили путем выведения наилучшим образом соответствующих многочленных уравнений для приведенных выше базовых координат; эти базовые координаты, где в исходный материал включали Н2, в расчеты уравнений не включали. Полученные уравнения и значения R2 представлены ниже:
Сел.БТН=(1,81818181818345Е-10)Т4-(5,41010101010501Е-07)Т3+(5,88000000000377Е-04)Т2-(2,78591414141575Е-01)Т+4,97583333333585Е+01 R2 БTH=9,99810335105254Е-01
Сел.Кокс=(-1,85878787878687Е-10)Т4+(5,62280808080511Е-07)Т3-(6,21721666666349Е-04)Т2+(2,9966402741б883Е-01)Т-5,33408809523590Е+01 R2 Кокс=9,99958406639717Е-01
Превр.Бл.=(1,914285714285б9Е-06)Т2-(1,81714285714283Е-03)Т+4,53357142857135Е-01 R2 Бл.=9,99955208049бЗЗЕ-01, где Т обозначает температуру в градусах С.
Во всех примерах R2 обозначает коэффициент определения, который сопоставляет расчетные и фактические значения у и находится в интервале значений от 0 до 1. Если он обозначает 1, существует абсолютная корреляция в образце - отсутствует разница между расчетным значением y и фактическим значением y. Если коэффициент определения при другом экстремуме обозначает 0, уравнение регрессии не может принести пользу в прогнозировании значения y. Вариант, используемый в настоящем описании, основан на анализе несоответствия разложения следующим образом:
 .
.
В приведенном выше определении
 ,
, 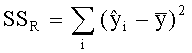 ,
, 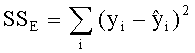 .
.
Другими словами, SST обозначает общую сумму квадратов, SSR обозначает регрессивную сумму квадратов, а SSE обозначает сумму возведенных в квадрат погрешностей.
 обозначает коэффициент определения для корреляции Сел.БТН,
обозначает коэффициент определения для корреляции Сел.БТН,
 обозначает коэффициент определения для корреляции Сел.Кокс, а
обозначает коэффициент определения для корреляции Сел.Кокс, а
 обозначает коэффициент определения для корреляции Превр.Бл..
обозначает коэффициент определения для корреляции Превр.Бл..
Этот ряд уравнений использовали для расчета значений выхода, которых достигают, по-видимому, для различных реакторных конфигураций, где ВыходБТН определяли как произведение Сел.БТН×Превр.Бл., интегрированное по температурному профилю в реакторной системе, а ВыходКокс определяли как произведение Сел.Кокс×Превр.Бл., интегрированное по температурному профилю в реакторной системе. Хотя признано и в таблице 3 продемонстрировано, что селективность реакции в отношении Н2 как побочного продукта улучшена, в этих уравнениях улучшением селективности пренебрегали, благодаря чему они обеспечивали консервативную оценку уровня улучшения, которое обеспечивало бы осуществление предлагаемого способа.
Транспортировка или вертикальный трубный реактор - сравнительный
С использованием приведенных выше уравнений для транспортировки или вертикального трубного реактора с адиабатическим понижением температуры при температуре на входе 850°С требуемая скорость циркуляции катализатора для сохранения температуры на выходе 800°С составляла 3211 кг в час (кг/ч) в пересчете на номинальную скорость подачи метана 100 кг/ч при 600°С. Были вычислены следующие значения выхода и селективности:
Сел.БТН = 51%
Сел.Кокс = 40% ВыходБТН = 12%
ВыходКокс = 8,9%
ΔТреакции = -50°С (отрицательные 50°С);
где ΔТреакции определяют как реакционную температуру продукта на выходе (т.е. последнюю температуру, при которой протекает каталитическая реакция перед тем, как углеводородный продукт выходит из реакторной системы) минус реакционная температура углеводородного сырья на входе (т.е. первая температура, при которой протекает каталитическая реакция, когда углеводородное сырье поступает в реакторную систему).
Адиабатический реактор с неподвижным слоем - сравнительный
Осуществление моделирования потенциально сравнительных неподвижных слоев приводило к еще более худшим эксплуатационным свойствам, чем с транспортировкой или вертикальным трубным реактором, потому что в конфигурации с неподвижным слоем все тепло реакции следовало подводить с помощью метансодержащего потока (поскольку для подвода тепла в реакционную зону движущийся катализатор не использовали). Следовательно, для работы реактора с неподвижным слоем требовалось, чтобы метансодержащий поток был нагрет до температуры, намного превышавшей целевую температуру на выходе 800°С, что приводило тем самым к большей величине ΔТреакции, значение которой составляет -60 или более отрицательное.
Реактор с отстойным слоем
В случае, имитировавшем отстойный слой катализатора с обратным температурным профилем и приближением в 50°С температуры между температурой подаваемого катализатора и технологической температурой на выходе, впускное приспособление работало при 620°С, а выпускное приспособление работало при 800°С, скорость циркуляции катализатора уменьшали до 717 кг/ч, а результаты реакции были улучшенными:
Сел.БТН = 89%
Сел.Кокс = 7%
ВыходБТН = 20%
ВыходКокс = 1,5%
ΔТреакции = +180°C.
Каскадировавшие псевдоожиженные слои (2 псевдоожиженных слоя)
Аналогично тому, что показано на фиг.2; этот пример предусмотрен для 2 каскадировавших псевдоожиженных слоев. Под каскадировавшими псевдоожиженными слоями подразумевают, что существуют 2 или большее число реакционных стадий или зон, работающих при разных температурах с порошкообразным катализатором, движущимся из одной стадии в следующую, и углеводородным газом, движущимся из одной стадии в следующую в направлении, противоположном направлению движения катализатора. В случае, имитировавшем два каскадировавших псевдоожиженных слоя катализатора, первый слой работал при 731°С, а второй слой работал при 800°С; требуемую скорость циркуляции катализатора уменьшали до 1367 кг/ч, и результаты реакции были улучшенными:
Сел.БТН = 81%
Сел.Кокс = 13%
ВыходБТН = 18%
ВыходКокс = 2,8%
ΔТреакции = +69°C
ΔТкатализатора = -119°С (отрицательные 119°С).
Каскадировавшие псевдоожиженные слои (3 псевдоожиженных слоя)
В случае, имитировавшем три каскадировавших псевдоожиженных слоя катализатора, первый слой работал при 690°С, второй слой работал при 753°С, а третий слой работал при 800°С, скорость циркуляции катализатора уменьшали до 1020 кг/ч, и результаты реакции были улучшенными: Сел.БТН = 85% Сел.Кокс = 10% ВыходБТН = 19% ВыходКокс = 2,2% ΔТреакции = +110°С
ΔТкатализатора = -160°С (отрицательные 160°С).
Каскадировавшие псевдоожиженные слои (4 псевдоожиженных слоя)
В случае, имитировавшем четыре каскадировавших псевдоожиженных слоя катализатора, первый слой работал при 669°С, второй слой работал при 723°С, третий слой работал при 762°С, а четвертый слой работал при 800°С, скорость циркуляции катализатора уменьшали до 900 кг/ч и результаты реакции были улучшенными: Сел.БТН = 86% Сел.Кокс = 9% ВыходБТН = 19% ВыходКокс = 2,0%
ΔТреакции = +131°С
ΔТкатализатора = -181°С (отрицательный 181°С).
Каскадировавшие псевдоожиженные слои (5 псевдоожиженных слоя)
В случае, имитировавшем пять каскадировавших псевдоожиженных слоев катализатора, первый слой работал при 655°С, второй слой работал при 703°С, третий слой работал при 737°С, четвертый слой работал при 767°С, а пятый слой работал при 800°С, скорость циркуляции катализатора уменьшали до 838 кг/ч и результаты реакции были улучшенными:
Сел.БТН = 87%
Сел.Кокс = 8%
ВыходБТН = 20%
ВыходКокс = 1,8%
ΔТ = +145°С
ΔТкатализатора = -195°С (отрицательные 195°С).
Как проиллюстрировано приведенными выше примерами с каскадировавшими псевдоожиженными слоями, улучшенных результатов достигали в большем числе реакционных зон, хотя необходимо иметь в виду, что капитальные затраты в эту реакционную систему возрастали с увеличением числа зон (или стадий). Существует оптимальное число зон (или стадий), которое зависит от экономичности процесса.
Изотермический неподвижный слой - сравнительный
В случае, имитировавшем единственный неподвижный слой катализатора, этот слой работал при 800°С и результатами являлись:
Сел.БТН = 68%
Сел.Кокс = 24%
ВыходБТН = 15%
ВыходКокс = 5,4%
ΔТреакции = 0°C
ΔTкaтaлизaтopa: величина не применима, неподвижные слои катализатора
Изотермические неподвижные слои с температурными ступенями (2 слоя)
В случае, имитировавшем два неподвижных слоя катализатора с температурными ступенями, первый слой работал при 700°С, второй слой работал при 800°С и результаты реакции были улучшенными:
Сел.БТН = 83%
Сел.Кокс = 12%
ВыходБТН = 19%
ВыходКокс = 2,7%
ΔТреакции = +100°C
ΔTкaтaлизaтopa: величина не применима, неподвижные слои катализатора
Изотермические неподвижные слои с температурными ступенями (4 слоя)
В случае, имитировавшем четыре неподвижных слоя катализатора с температурными ступенями, первый слой работал при 650°С, второй слой работал при 700°С, третий слой работал при 750°С, а четвертый слой работал при 800°С и результаты реакции были улучшенными:
Сел.БТН = 87%
Сел.Кокс = 8%
ВыходБТН = 20%
ВыходКокс = 1,9%
ΔТреакции = +150°С
ΔТкатализатора: величина не применима, неподвижные слои катализатора
Пример 5
Основываясь на прогнозированных на моделях преимуществах обратного температурного профиля, для подтверждения моделированных результатов конструировали установку лабораторного масштаба. Хотя эту модель ориентировали на работу реакционной системы как отстойного слоя, лабораторный реактор обладал неподвижным слоем катализатора с обратным температурным профилем, обусловленным применением внешних нагревателей. Во всех случаях экспериментально наблюдаемые превращения оказывались ниже прогнозированных по моделям превращений. Это могло происходить вследствие экспериментальных артефактов лабораторного масштаба, таких как обход слоев и/или обратное смешение благодаря гидродинамическому режиму, в котором работают реакторы лабораторного масштаба.
Mo/ZSM-5 катализатор готовили посредством измельчения в шаровой мельнице 7,5 мас.% Мо (мас.% металла в пересчете на общую массу катализатора) в виде МоО3 с NH4ZSM-5 носителем (обладавшим значением соотношения Si/Al2 25) в течение 2 ч с последующим кальцинированием при 500°С в течение 5 ч на воздухе. Катализатор таблетировали, измельчали и просеивали до размеров частиц от 20 до 40 меш. Каталитическое испытание Mo/ZSM-5 катализатора осуществляли в кварцевом реакторе с неподвижным слоем, внутренним диаметром 7 мм и с длиной слоя примерно 18 см. В качестве разбавителя в слое использовали инертные кварцевые частицы (от 20 до 50 меш), благодаря чему все слои обладали одинаковой длиной.
Эксплуатационные свойства катализатора при дегидроциклизации метана до бензола определяли с использованием 95 об.% CH4/5 об.% Ar как исходного материала (аргон использовали как внутренний стандарт). Все экспериментальные реакционные данные получали под абсолютным давлением 20 фунтов/кв.дюйм (138 кПа). Для определения концентраций продуктов отходивший из реакции поток анализировали с помощью масс-спектрометра.
Для сравнения проводили десять отдельных экспериментов на эксплуатационные свойства катализатора. Во всех экспериментах катализатор активировали нагреванием в 15 об.% СН4/80 об.% Н2/5 об.% Ar со скоростью нагрева 5°С/мин до 800°С и выдержкой в течение 30 мин. За этим следовало старение катализатора с 5 циклами реакции и регенерирования (также идентичными для всех десяти экспериментов). Каждая реакционная часть продолжалась 20 мин при 800°С в 95 об.% СН4/5 об.% Ar как исходного материала при среднечасовой скорости подачи сырья (ССПС) 1,4 ч-1 в пересчете на СН4. Каждая регенерационная часть состояла из подключения к Н2, нагрева до 850°С с 10-минутным временем выдержки, затем охлаждения вновь до 800°С (с общим временем с Н2 14 мин). Эти десять экспериментов различались только их шестым реакционным циклом, который осуществляли в 95 об.% СН4/5 об.% Ar как исходном материале в течение 4 ч. Условия для шестого цикла выбирали для сравнения эффектов температурного профиля слоя катализатора при разных объемных скоростях. Так, в частности, эксперименты с 1 по 5 проводили при варьировании значений ССПС в пределах от 0,25 до 8 ч-1 с выдержкой слоя в изотермальных условиях при 800°С. В противоположность этому эксперименты с 6 по 10 проводили в таком же интервале значений ССПС, но с линейным градиентом температуры в слое от 650°С при впускном приспособлении для исходного материала до 800°С при выпускном приспособлении для продуктов (обратный температурный профиль). Результаты определения эксплуатационных свойств катализатора для десяти экспериментов во время реакционного цикла №6 сведены в таблицу 5.
Результаты в таблице 5 показывают, что наблюдалось очевидное преимущество проведения процесса с обратным температурным профилем, который улучшал текущую селективность при самых высоких объемных скоростях в течение более коротких промежутков времени процесса и, следовательно, обуславливал пролонгированную селективность в отношении бензола, в течение более продолжительных периодов времени процесса. Это обуславливало более значительное совокупное получение, если сравнивать с изотермическим слоем при всех объемных скоростях.
Хотя примеры 4 и 5 предусмотрены для реакторов особых типов, аналогичное улучшение селективности проявлялось, по-видимому, в других реакторных системах с эквивалентным обратным температурным профилем или ступенчатым изменением температуры.
Как продемонстрировано моделированием, предлагаемыми по изобретению концепциями были улучшены значения выхода, селективности и скорости циркуляции катализатора. Кроме того, реакция в целом может быть осуществлена в единственной реакционной зоне, тем самым сводя к минимальному требуемое оборудование. Можно использовать, что необязательно, две или большее число реакционных зон.
Как проиллюстрировано примерами 4 и 5, обратный температурный профиль или ступенчатое изменение температуры создает возможность превращения метана в более высокомолекулярные углеводороды, например ароматические соединения, при уменьшенных потерях из-за старения/механического истирания катализатора, улучшенной действенности и более высокой селективности; т.е. меньшее образование кокса, чем в обычном неподвижном слое и/или транспортировке иди вертикальных трубных конфигурациях.
В другом варианте объектом настоящего изобретения являются:
1. Способ превращения метана в более высокомолекулярный углеводород (углеводороды), содержащий ароматический углеводород (углеводороды), в реакционной зоне, включающий следующие стадии:
(а) подача в упомянутую реакционную зону углеводородного исходного материала, содержащего метан;
(б) подача внутрь упомянутой реакционной зоны некоторого количества каталитического материала;
(в) поддержание в упомянутой реакционной зоне обратного температурного профиля и
(г) работа упомянутой реакционной зоны в реакционных условиях, достаточных для превращения по меньшей мере части упомянутого метана в первый отходящий поток, содержащий упомянутый более высокомолекулярный углеводород (углеводороды).
2. Способ по п.1, в котором упомянутая реакционная зона представляет собой реакционную зону с подвижным слоем.
3. Способ по п.1, в котором упомянутая реакционная зона представляет собой реакционную зону с неподвижным слоем.
4. Способ по одному из пп.1-3, в котором упомянутый исходный материал далее включает по меньшей мере один из СО, СО2, Н2, Н2О и/или О2.
5. Способ по одному из пп.1-4, в котором начальная температура катализируемой реакции ниже примерно 750°С, предпочтительно ниже примерно 700°С, по другому варианту ниже примерно 650°С.
6. Способ по одному из пп.1-5, в котором конечная температура катализируемой реакции превышает примерно 700°С, предпочтительно выше примерно 800°С, по другому варианту выше примерно 850°С.
7. Способ по одному из пп.1-6, в котором подают достаточное количество каталитического материала, благодаря которому степень приближения в % к термодинамическому равновесному превращению метана в бензол в средней части реакционной зоны (в смысле количества каталитического материала, контактировавшего с углеводородом) превышает примерно 25%, предпочтительно больше примерно 50%, наиболее предпочтительно больше примерно 75%.
8. Способ по одному из пп.1-6, в котором подают достаточное количество каталитического материала, благодаря которому степень приближения в % к термодинамическому равновесному превращению метана в бензол в средней части реакционной зоны (в смысле половины общего перепада температур на пути через реакционную зону) превышает примерно 25%, предпочтительно больше примерно 50%, наиболее предпочтительно больше примерно 75%.
9. Способ по одному из пп.1-8, в котором упомянутого обратного температурного профиля в упомянутом каталитическом материале добиваются прерывистым прямым контактным нагреванием газообразными продуктами горения или инертной средой, нагретой упомянутыми газообразными продуктами горения.
10. Способ по одному из пп.1-9, в котором упомянутого обратного температурного профиля в упомянутом каталитическом материале добиваются теплопереносом через поверхность теплопередачи.
11. Способ по п.10, в котором поверхность теплопередачи нагревают благодаря теплопереносу от горения за счет излучения и/или теплопроводности.
12. Способ по п.10, в котором упомянутая поверхность теплопередачи является металлической или керамической.
13. Способ по п.10, в котором каталитический материал размещен в одной или нескольких параллельных трубках, а эти трубки размещены внутри печи, обеспечивающей тепло для сохранения упомянутого обратного температурного профиля.
14. Способ по п.10, в котором каталитический материал размещен в сосуде с одной или несколькими трубками, проходящими через слой; по упомянутым трубкам транспортируют газообразные продукты горения для сохранения упомянутого обратного температурного профиля.
15. Способ по п.2, в котором из реакционной зоны отводят часть каталитического материала, по меньшей мере часть кокса, отложившегося на каталитическом материале, удаляют окислением и каталитический материал с пониженным содержанием кокса возвращают в реакционную зону.
16. Способ по п.2, в котором из реакционной зоны отводят часть каталитического материала, по меньшей мере часть кокса, отложившегося на каталитическом материале, удаляют реакцией с водородом с получением метана и каталитический материал с пониженным содержанием кокса возвращают в реакционную зону.
17. Способ по п.1, в котором подачу углеводородов циклически останавливают и вместо этого в реакционную зону направляют кислородсодержащий поток, благодаря чему по меньшей мере часть кокса, отложившегося на каталитическом материале, удаляют окислением, затем кислородсодержащий поток останавливают и вновь начинают подачу углеводородов.
18. Способ по п.1, в котором подачу углеводородов циклически останавливают и вместо этого в реакционную зону направляют водородсодержащий поток, благодаря чему по меньшей мере часть кокса, отложившегося на каталитическом материале, удаляют превращением в метан, затем водородсодержащий поток останавливают и вновь начинают подачу углеводородов.
19. Способ по одному из пп.1-18, в котором упомянутый каталитический материал представляет собой катализатор дегидроциклизации, включающий металл или его соединение на неорганическом носителе.
20. Способ по одному из пп.1-18, в котором упомянутый каталитический материал включает по меньшей мере один из молибдена, вольфрама, рения, соединения молибдена, соединения вольфрама, соединения цинка и соединения рения на ZSM-5, диоксиде кремния или оксиде алюминия.
21. Способ превращения метана в более высокомолекулярный углеводород (углеводороды), содержащий ароматический углеводород (углеводороды), в двух или большем числе реакционных зон, работающих последовательно, включающий следующие стадии:
(а) подача внутрь каждой реакционной зоны некоторого количества каталитического материала;
(б) подача в первую реакционную зону углеводородного исходного материала, содержащего метан;
(в) перенос по меньшей мере части отходящего потока упомянутой первой реакционной зоны во вторую реакционную зону;
(г) поддержание в упомянутой первой реакционной зоне более низкой средней температуры, чем в упомянутой второй реакционной зоне, и
(д) проведение процесса в упомянутых реакционных зонах в реакционных условиях, достаточных для превращения по меньшей мере части упомянутого метана в первый отходящий поток, содержащий упомянутый более высокомолекулярный углеводород (углеводороды).
22. Способ по п.21, в котором упомянутые реакционные зоны представляют собой реакционные зоны с подвижным слоем.
23. Способ по п.21, в котором упомянутые реакционные зоны представляют собой реакционные зоны с неподвижным слоем.
24. Способ по одному из пп.21-23, в котором упомянутый исходный материал далее включает по меньшей мере один из СО, СО2, Н2, Н2О и/или O2.
25. Способ по одному из пп.21-24, в котором упомянутая начальная температура катализируемой реакции ниже примерно 750°С, предпочтительно ниже примерно 700°С, по другому варианту ниже примерно 650°С.
26. Способ по одному из пп.21-25, в котором упомянутая конечная температура катализируемой реакции превышает примерно 700°С, предпочтительно выше примерно 800°С, по другому варианту выше примерно 850°С.
27. Способ по одному из пп.21-26, в котором подают достаточное количество каталитического материала, благодаря которому степень приближения в % к термодинамическому равновесному превращению метана в бензол в средней части реакционной зоны (в смысле количества каталитического материала, контактировавшего с углеводородом) превышает примерно 25%, предпочтительно больше примерно 50%, наиболее предпочтительно больше примерно 75%.
28. Способ по одному из пп.21-26, в котором подают достаточное количество каталитического материала, благодаря которому степень приближения в % к термодинамическому равновесному превращению метана в бензол в средней части реакционной зоны (в смысле половины общего перепада температур на пути через реакционную зону) превышает примерно 25%, предпочтительно больше примерно 50%, наиболее предпочтительно больше примерно 75%.
29. Способ по п.21, в котором поддержания в упомянутой первой реакционной зоне более низкой средней температуры, чем в упомянутой второй реакционной зоне, добиваются теплопереносом через поверхность теплопередачи в контакте с каталитическим материалом в реакционных зонах.
30. Способ по п.29, в котором поверхность теплопередачи нагревают благодаря теплопереносу от горения за счет излучения и/или теплопроводности.
31. Способ по п.29, в котором упомянутая поверхность теплопередачи является металлической или керамической.
32. Способ по п.29, в котором каталитический материал размещен в одной или нескольких параллельных трубках, а эти трубки размещены внутри печи, обеспечивающей тепло для сохранения упомянутого обратного температурного профиля.
33. Способ по п.29, в котором каталитический материал размещен в сосуде с одной или несколькими трубками, проходящими через слой, причем по упомянутым трубкам транспортируют газообразные продукты горения для сохранения упомянутого обратного температурного профиля.
34. Способ по п.21, в котором поддержания в упомянутой первой реакционной зоне более низкой средней температуры, чем в упомянутой второй реакционной зоне, добиваются нагреванием потока углеводородов благодаря теплопереносу через поверхность теплопередачи, причем упомянутый нагрев потока углеводородов осуществляют между упомянутыми реакционными зонами.
35. Способ по п.22, в котором из реакционной зоны отводят часть каталитического материала, по меньшей мере часть кокса, отложившегося на каталитическом материале, удаляют окислением и каталитический материал с пониженным содержанием кокса возвращают в реакционную зону.
36. Способ по п.22, в котором из реакционной зоны отводят часть каталитического материала, по меньшей мере часть кокса, отложившегося на каталитическом материале, удаляют реакцией с водородом с получением метана и каталитический материал с пониженным содержанием кокса возвращают в реакционную зону.
37. Способ по п.21, в котором подачу углеводородов циклически останавливают и вместо этого в реакционную зону направляют кислородсодержащий поток, благодаря чему по меньшей мере часть кокса, отложившегося на каталитическом материале, удаляют окислением, затем кислородсодержащий поток останавливают и вновь начинают подачу углеводородов.
38. Способ по п.21, в котором подачу углеводородов циклически останавливают и вместо этого в реакционную зону направляют водородсодержащий поток, благодаря чему по меньшей мере часть кокса, отложившегося на каталитическом материале, удаляют превращением в метан, затем водородсодержащий поток останавливают и вновь начинают подачу углеводородов.
39. Способ по одному из пп.21-38, в котором упомянутый каталитический материал представляет собой катализатор дегидроциклизации, включающий металл или его соединение на неорганическом носителе.
40. Способ по одному из пп.21-38, в котором упомянутый каталитический материал включает по меньшей мере один из молибдена, вольфрама, рения, соединения молибдена, соединения вольфрама, соединения цинка и соединения рения на ZSM-5, диоксиде кремния или оксиде алюминия.
41. Способ по п.16 или п.36, в котором реакцию с водородом проводят под давлением, превышающем давление, под которым метансодержащий исходный материал вступает в реакцию с образованием ароматических соединений.
42. Способ по п.16 или п.36, в котором реакцию с водородом проводят под давлением в пределах от примерно 150 до примерно 5000 кПа.
43. Способ по п.1 или п.21, в котором из потока углеводородных продуктов выделяют ароматический продукт с получением обедненного ароматическими соединениями остаточного потока метана и Н2.
44. Способ по п.43, в котором упомянутый остаточный поток метана и Н2 в дальнейшем обрабатывают таким образом, что по меньшей мере часть Н2 может быть выделена из потока углеводородов реакцией с кислородсодержащими материалами или с применением физических методов разделения (таких как криогенная дистилляция, адсорбция с колебанием давления, адсорбция с колебанием температуры и/или с применением мембранных систем) с получением богатого метаном остаточного потока.
45. Способ по п.44, в котором по меньшей мере часть упомянутого богатого метаном остаточного потока возвращают в упомянутую реакционную зону.
46. Способ по п.43, в котором упомянутые выделенные ароматические продукты далее обрабатывают реакцией с алкилирующим агентом с получением алкилированных ароматических соединений.
47. Способ по п.43, в котором упомянутые выделенные ароматические продукты далее обрабатывают реакцией с содержащим Н2 потоком с получением гидрированных материалов или алкилароматических соединений.
Хотя настоящее изобретение описано и проиллюстрировано со ссылкой на конкретные варианты его выполнения, для обычных специалистов в данной области техники вполне очевидно, что сама сущность изобретения приводит к вариантам, которые нет необходимости иллюстрировать в настоящем описании. По этой причине с целью определить фактический объем настоящего изобретения следует обращаться только к прилагаемой формуле изобретения.
Некоторые варианты и особенности представлены с использованием ряда выраженных числами верхних пределов и и ряда выраженных числами нижних пределов. Необходимо иметь в виду, что интервалы от любого нижнего предела до любого верхнего предела во всех случаях, если не указано иное, являются предполагаемыми. Некоторые нижние пределы, верхние пределы и интервалы приведены в одном или нескольких пунктах формулы изобретения ниже. Все числовые значения являются "примерными" или "приблизительными" указываемыми значениями, и необходимо учитывать экспериментальную погрешность и варианты, которые, по-видимому, предусматривает специалист в данной области техники.
Выше определено значение различных понятий. В той степени, в которой понятие, использованное в пункте формулы изобретения, выше не определено, имеющие к этому отношение специалисты в данной области техники его должны рассматривать в самом широком смысле, который приведен в по меньшей мере одной печатной публикации или изданном патенте. Более того, все упомянутые в настоящем описании патенты, методы испытаний и другие документы, считая приоритетные документы, упомянутые в данной заявке, включены в него в полном объеме в качестве ссылок в той степени, в которой они не соответствуют настоящей заявке согласно всем юрисдикциям, которые такое включение допускают.
Хотя изложенное выше относится к вариантам выполнения настоящего изобретения, можно рекомендовать другие и дополнительные варианты выполнения изобретения, не выходя при этом из основного его объема, который определяет следующая формула изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2007 |
|
RU2454390C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ МЕТАНА | 2007 |
|
RU2448079C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ МЕТАНА | 2007 |
|
RU2454389C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2006 |
|
RU2408565C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ АЛИФАТИЧЕСКИХ | 2008 |
|
RU2461537C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2008 |
|
RU2460581C2 |
| ПОЛУЧЕНИЕ АЛКИЛИРОВАННЫХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2005 |
|
RU2417974C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2007 |
|
RU2462444C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ И СИНТЕЗ-ГАЗА ИЗ МЕТАНА | 2007 |
|
RU2458899C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2009 |
|
RU2514915C2 |
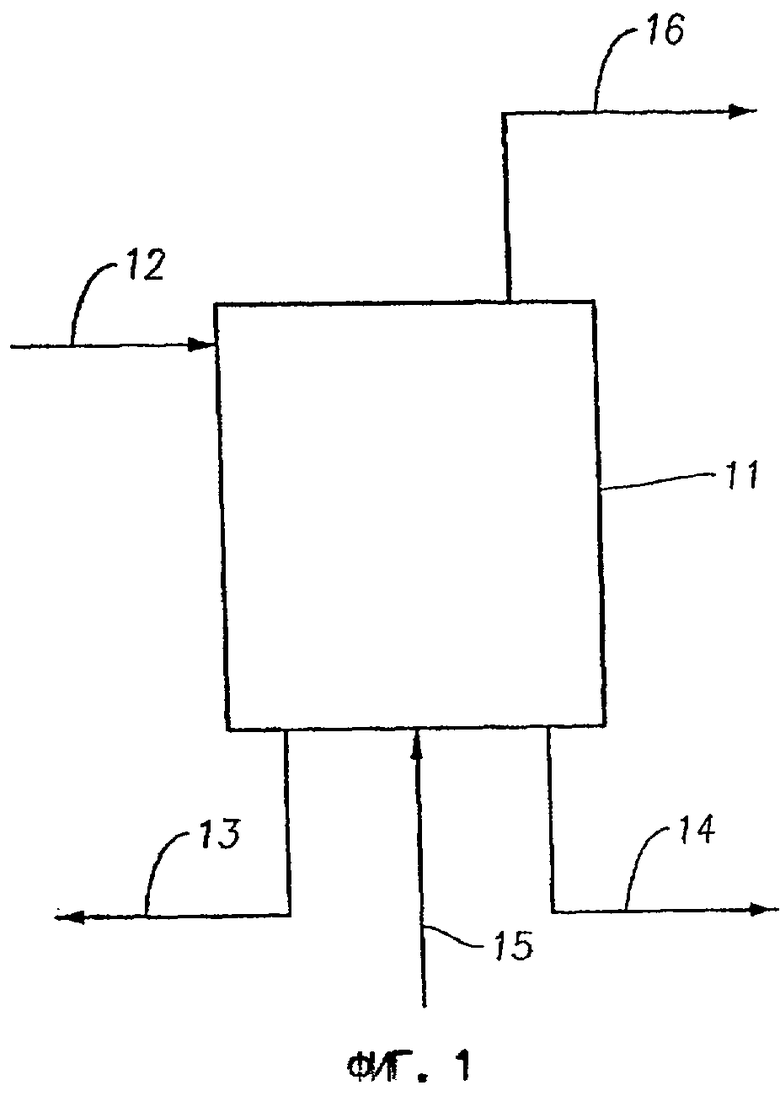
Изобретение относится к способу превращения метана в более высокомолекулярный углеводород (углеводороды), содержащий ароматический углеводород (углеводороды), в реакционной зоне. Способ включает следующие стадии: (а) подача в упомянутую реакционную зону углеводородного исходного материала, содержащего метан; (б) подача внутрь упомянутой реакционной зоны некоторого количества каталитического материала; (в) поддержание в упомянутой реакционной зоне обратного температурного профиля и (г) работа упомянутой реакционной зоны в реакционных условиях, достаточных для превращения по меньшей мере части упомянутого метана в первый отходящий поток, содержащий упомянутый более высокомолекулярный углеводород (углеводороды). Использование настоящего способа позволяет обеспечить высокую эффективность теплопереноса и достичь максимальной селективности в отношении целевых углеводородов при одновременном сведении к минимуму коксообразования. 14 з.п. ф-лы, 5 пр., 5 табл., 2 ил.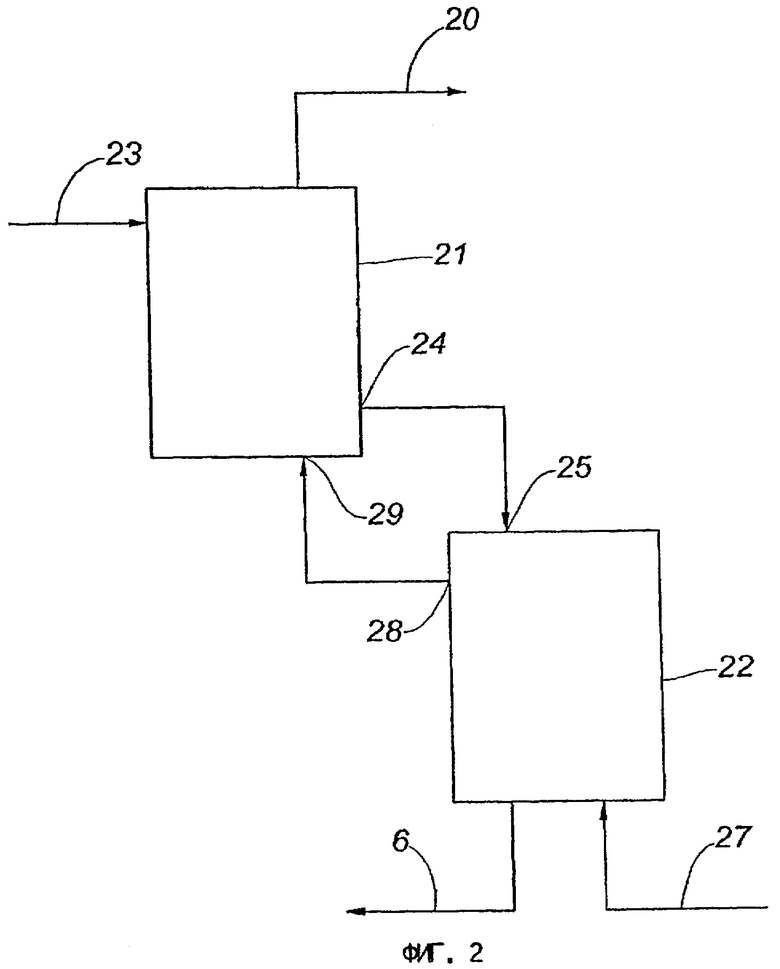
1. Способ превращения метана в более высокомолекулярный углеводород (углеводороды), содержащий ароматический углеводород (углеводороды), в реакционной зоне, включающий следующие стадии:
(а) подача в упомянутую реакционную зону углеводородного исходного материала, содержащего метан;
(б) подача внутрь упомянутой реакционной зоны некоторого количества каталитического материала;
(в) поддержание в упомянутой реакционной зоне обратного температурного профиля и
(г) работа упомянутой реакционной зоны в реакционных условиях, достаточных для превращения по меньшей мере части упомянутого метана в первый отходящий поток, содержащий упомянутый более высокомолекулярный углеводород (углеводороды).
2. Способ по п.1, в котором упомянутый исходный материал далее включает по меньшей мере один из СО, СО2, Н2, Н2О и/или О2.
3. Способ по п.1, в котором начальная температура катализируемой реакции ниже примерно 750°С.
4. Способ по п.1, в котором конечная температура катализируемой реакции превышает примерно 700°С.
5. Способ по п.1, в котором подают достаточное количество каталитического материала, благодаря которому степень приближения в % к термодинамическому равновесному превращению метана в бензол в средней части реакционной зоны (в смысле количества каталитического материала, контактировавшего с углеводородом) превышает примерно 25%, предпочтительно больше примерно 50%, наиболее предпочтительно больше примерно 75%.
6. Способ по п.1, в котором подают достаточное количество каталитического материала, благодаря которому степень приближения в % к термодинамическому равновесному превращению метана в бензол в средней части реакционной зоны (в смысле половины общего перепада температур на пути через реакционную зону) превышает примерно 25%, предпочтительно больше примерно 50%, наиболее предпочтительно больше примерно 75%.
7. Способ по п.1, в котором упомянутого обратного температурного профиля в упомянутом каталитическом материале добиваются прерывистым прямым контактным нагреванием газообразными продуктами горения или инертной средой, нагретой упомянутыми газообразными продуктами горения.
8. Способ по п.1, в котором упомянутого обратного температурного профиля в упомянутом каталитическом материале добиваются теплопереносом через поверхность теплопередачи.
9. Способ по п.8, в котором поверхность теплопередачи нагревают благодаря теплопереносу от горения за счет излучения и/или теплопроводности.
10. Способ по п.8, в котором упомянутая поверхность теплопередачи является металлической или керамической.
11. Способ по п.8, в котором каталитический материал размещен в одной или нескольких параллельных трубках, а эти трубки размещены внутри печи, обеспечивающей тепло для сохранения упомянутого обратного температурного профиля.
12. Способ по п.8, в котором каталитический материал размещен в сосуде с одной или несколькими трубками, проходящими через слой; по упомянутым трубкам транспортируют газообразные продукты горения для сохранения упомянутого обратного температурного профиля.
13. Способ по п.1, в котором указанная реакционная зона представляет собой реакционную зону с подвижным слоем.
14. Способ по п.1, в котором указанная реакционная зона представляет собой реакционную зону с неподвижным слоем.
15. Способ по п.1, в котором
(i) способ включает две или более реакционные зоны, работающие последовательно;
(ii) по меньшей мере часть эффлюента из первой реакционной зоны переносится во вторую реакционную зону, и
(iii) указанная первая реакционная зона поддерживается при более низкой средней температуре, чем в упомянутой второй реакционной зоне.
| US 2003083535 A1, 01.05.2003 | |||
| US 5026937 A, 25.06.1991 | |||
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 1995 |
|
RU2118634C1 |
| US 2002072642 A1, 13.06.2002 | |||
| Способ получения бензола | 1981 |
|
SU1004335A1 |
| SU 1811153 A1, 27.03.1996. | |||

