ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способу получения ароматических углеводородов из метана и, в частности, из природного газа.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Ароматические углеводороды, в особенности бензол, толуол, этилбензол и ксилолы являются важными химическими продуктами массового производства в нефтехимической промышленности. В настоящее время ароматические соединения наиболее часто получают по разнообразным методам из исходных материалов на основе сырой нефти, включая каталитический реформинг и каталитический крекинг. Однако по мере того как мировые поставки исходных материалов на основе сырой нефти уменьшаются, возрастает потребность найти альтернативные источники жидких углеводородов.
Одним возможным альтернативным источником ароматических углеводородов служит метан, который является основным компонентом природного газа и биогаза. Объем разведанных мировых запасов природного газа постоянно увеличивается, и в настоящее время открывают больше месторождений природного газа, чем нефти. Из-за проблем, связанных с транспортировкой больших объемов природного газа, большую часть природного газа, добываемого вместе с нефтью, в особенности в отдаленных местах, сжигают в факеле и направляют в отход. Следовательно, особенно привлекательным методом повышения сортности природного газа является превращение алканов, содержащихся в природном газе, непосредственно в более высокомолекулярные углеводороды, такие как ароматические соединения, при условии, что могут быть преодолены сопутствующие этому технические трудности.
Значительная часть способов превращения метана в жидкие углеводороды включает вначале превращение метана в синтез-газ, смесь H2 и СО. Получение синтез-газа связано с большими капитальными затратами и является энергоемким, следовательно, предпочтительны пути, которые не требуют генерирования синтез-газа.
Предложен ряд альтернативных способов прямого превращения метана в более высокомолекулярные углеводороды. Один такой способ включает каталитическое окислительное сочетание метана до олефинов с последующим каталитическим превращением олефинов в жидкие углеводороды, включающие ароматические углеводороды. Так, например, в US №5336825 описан двухстадийный способ окислительного превращения метана в углеводороды с пределами кипения бензиновой фракции, включающие ароматические углеводороды. На первой стадии в присутствии свободного кислорода с использованием промотированного редкоземельным металлом катализатора из оксида щелочно-земельного металла при температуре в пределах от 500 до 1000°С метан превращают в этилен и небольшие количества С3- и С4-олефинов. Затем этилен и более высокомолекулярные олефины, образовавшиеся на первой стадии, над кислым твердым катализатором, включающим пентасиловый цеолит с высоким содержанием диоксида кремния, превращают в жидкие углеводороды с пределами кипения бензиновой фракции.
Однако эти методы окислительного сочетания страдают проблемами, заключающимися в том, что они включают высокоэкзотермические и потенциально опасные реакции сжигания метана, и в том, что при их осуществлении образуются большие количества воздействующих на окружающую среду оксидов углерода.
В качестве пути повышения сортности метана до более высокомолекулярных углеводородов, в особенности этилена, бензола и нафталина предложена также дегидроароматизация метана посредством высокотемпературного восстановительного сочетания. Так, например, в US №4727206 описан способ получения жидких продуктов, богатых ароматическими углеводородами, введением метана при температуре в пределах от 600 до 800°С в отсутствии кислорода в контакт с каталитической композицией, включающей алюмосиликат, обладающий молярным отношением диоксида кремния к оксиду алюминия по меньшей мере 5:1, причем в упомянутый алюмосиликат вводят (I) галлий или его соединение и (II) металл группы VII B Периодической таблицы элементов или его соединение.
Кроме того, в US №5026937 описан способ ароматизации метана, который включает стадии подачи потока исходных материалов, который включает больше 0,5 мольного % водорода и 50 мольных % метана, в реакционную зону, содержащую по меньшей мере один слой твердого катализатора, включающего ZSM-5 и фосфорсодержащий оксид алюминия, в условиях превращения, которые включают температуру от 550 до 750°С, абсолютное давление ниже 10 ат (1000 кПа) и среднечасовую скорость подачи газа от 400 до 7500 ч-1. Отходящий поток продуктов включает, как сказано, метан, водород, по меньшей мере 3 мольных % С2-углеводородов и по меньшей мере 5 мольных % ароматических С6-С8-углеводородов. После конденсации для удаления фракции С4+ с целью выделить водород и легкие углеводороды (метан, этан, этилен и т.д.), содержащиеся в отходящем потоке продуктов, предложены криогенные методы.
В US №№6239057 и 6426442 описан способ получения углеводородов с более высоким числом углеродных атомов, например бензола, из углеводородов с низким числом углеродных атомов, таких как метан, введением этого последнего в контакт с катализатором, включающим пористый носитель, такой как ZSM-5, который содержит диспергированный на нем рений и промоторный металл, такой как железо, кобальт, ванадий, марганец, молибден, вольфрам или их смесь. Добавление СО или СО2 в исходный материал повышает, как сказано, выход бензола и стабильность катализатора.
Однако существующие технические решения по дегидроароматизации метана часто характеризуются низкой селективностью в отношении ароматических соединений и могут потребовать совместной подачи дорогостоящих добавок для повышения селективности в отношении ароматических соединений. Более того в любом процессе восстановительного сочетания получают большие количества водорода, вследствие чего для экономической жизнеспособности необходим путь для эффективной утилизации водорода как побочного продукта. Поскольку месторождения природного газа часто находятся в отдаленных районах, эффективная утилизация водорода может оказаться довольно сложной задачей.
Другая проблема, свойственная применению восстановительного сочетания для повышения сортности метана до более высокомолекулярных углеводородов, состоит в том, что в реакцию должно быть направлено значительное количество тепла. Так, например, этот процесс является не только высокоэндотермическим, но реакция также ограничена термодинамически. Таким образом, охлаждающий эффект, вызываемый реакцией, в достаточной мере понижает реакционную температуру для существенного уменьшения скорости реакции и общего термодинамического превращения, если каким-либо образом не подводить дополнительного тепла. Были предложены различные методы подвода тепла для ароматизации метана, но до сих пор ни один из предложенных методов не был признан полностью удовлетворительным.
Например, один известный метод обеспечения реакционным теплом процесса ароматизации метана заключается в применении теплообменной текучей среды, движущейся по реакционной зоне, которая снабжает катализатор в реакционной зоне непрямым теплом. Однако этот метод теплообмена проявляет тенденцию оказываться неэффективным и вызывает нарушение истечения катализатора в реакторах с ненеподвижными слоями.
Известна также подача реакционного тепла для процесса ароматизации метана с использованием больше одной реакционной зоны в виде их последовательности в сочетании с повторным нагреванием реагентов между реакционными зонами. При этом межстадийном повторном нагревании отходящий из реактора поток из первого слоя катализатора нагревают до целевой температуры на входе во второй, последующий слой катализатора.
Один метод межстадийного повторного нагревания включает применение непрямого теплообмена, при котором отходящий из предыдущей реакционной зоны поток перед направлением в последующую реакционную зону пропускают через теплообменник. Для этого метода непрямого теплообмена используемая высокотемпературная текучая среда может представлять собой высокотемпературный водяной пар, газообразные продукты горения, высокотемпературный технологический поток или любую другую легкодоступную высокотемпературную текучую среду. Этот метод межстадийного нагрева не приводит к разбавлению реагентов, но обуславливает некоторое падение давления в системе и может вызвать воздействие на реагенты нецелесообразно высоких температур.
Так, например, в российском патенте №2135441 описан способ превращения метана в более тяжелые углеводороды, в котором метан смешивают с по меньшей мере 5 мас.% углеводорода С3+, такого как бензол, а затем вводят в контакт с катализатором, включающим металлическую платину, обладающую под парциальным давлением метана по меньшей мере 0,05 МПа и при температуре по меньшей мере 440°С степенью окисления выше нуля. Этот способ осуществляют в мультистадийной реакторной системе с использованием межстадийного повторного нагрева непрямым теплообменом. Водород, образующийся в процессе, может быть введен в контакт с оксидами углерода с получением дополнительного метана, который после удаления одновременно получаемой воды можно добавлять в метановый исходный материал. Продукты превращения метана представляют собой газообразную фазу С2-С4 и жидкую фазу С5+, но в соответствии с примерами при этом мало (меньше 5 мас.%) или отсутствует фактическое увеличение количества ароматических колец в сравнении с исходным материалом.
Другой метод межстадийного нагрева представляет собой метод окислительного повторного нагрева, который включает подмешивание в реагенты регулируемого количества кислорода и селективное окисление водорода, образующегося в процессе ароматизации. Окисление осуществляют в присутствии катализатора, который селективно содействует окислению водорода в сравнении с разрушительным горением или окислением более ценного исходного материала и получаемых углеводородов. Однако такая реакция генерирует водяной пар, который может быть вредным для катализатора ароматизации и может взаимодействовать с метаном с образованием водорода и моноксида углерода. Более того при применении второго катализатора селективного окисления этот метод страдает дополнительной сложностью и дороговизной.
Альтернативный технический прием для снабжения реакционным теплом процесса восстановительного сочетания состоит в использовании того факта, что по мере того как протекает реакция ароматизации, катализатор образует кокс. Этот кокс постепенно дезактивирует катализатор и, следовательно, для удаления кокса и повторной активации катализатора этот последний должен быть повторно регенерирован. Регенерирование, которое включает контактирование катализатора с кислородсодержащим газом, является высокоэкзотермическим процессом и, следовательно, его можно использовать в качестве источника существенного количества тепла для процесса в целом. Такой способ описан в международной публикации №WO 03/000826, в которой катализатор дегидроароматизации циркулирует между реакторной системой и регенераторной системой, где катализатор вводят в контакт с разными регенерирующими газами, включающими O2, Н2 и H2O, в разные моменты времени для регенерирования разных частей катализатора. Для сохранения сбалансированного теплового режима реакторной системы и регенерационной системы регулируют процент катализатора, контактирующего с каждым регенерирующим газом. Реакторная система включает псевдоожиженный слой катализатора в вертикальном трубном реакторе, а регенерационная система включает второй псевдоожиженный слой катализатора, поддерживаемый в реакторе с псевдоожиженным слоем.
Однако способы, в которых осуществляют стадию регенерирования катализатора для снабжения реакционным теплом, страдают проблемой, состоящей в том, что катализатор нуждается в нагреве до температуры, которая намного превышает целевую реакционную температуру в процессе регенерирования, что приводит к ускоренной деструкции катализатора и, следовательно, к сокращенному сроку службы катализатора. Более того для сохранения теплового баланса процесс требует высокой селективности в отношении скорее кокса, чем целевых ароматических продуктов.
Следовательно, все еще существует потребность в усовершенствованном способе подачи реакционного тепла для ароматизации метана.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном варианте объектом настоящего изобретения является способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий:
(а) контактирование исходного материала, содержащего метан, с катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения упомянутого метана в ароматические углеводороды;
(б) перенос первой части упомянутого катализатора из реакционной зоны в зону нагрева;
(в) нагрев первой части катализатора в зоне нагрева введением катализатора в контакт с горячими газообразными продуктами горения, образующимися при сжигании дополнительного источника топлива; и
(г) возврат нагретой первой части катализатора в реакционную зону.
В подходящем варианте при упомянутом нагреве (в) упомянутую первую часть катализатора вводят в контакт непосредственно с упомянутым источником топлива. По другому варианту упомянутый источник топлива сжигают в зоне горения отдельно от упомянутой зоны нагрева, и газообразные продукты горения, образующиеся в зоне горения, направляют в зону нагрева.
В подходящем варианте упомянутый дополнительный источник топлива включает углеводород и/или водород.
Когда дополнительный источник топлива включает углеводород, этот углеводород в предпочтительном варианте представляет собой метан, а упомянутый дополнительный источник топлива в предпочтительном варианте включает часть исходного материала, контактировавшего на стадии (а). В подходящем варианте углеводородное топливо сжигают в бедной кислородом атмосфере с получением синтез-газа, и синтез-газ эффективно используют для получения дополнительного углеводородного продукта и/или топлива.
Когда дополнительный источник топлива включает водород, упомянутое топливо включает водород, образовавшийся в качестве побочного продукта упомянутого контактирования (а).
В подходящем варианте упомянутая зона нагрева является удлиненной, и тепло передают упомянутой первой части катализатора на множестве участков, размещенных с промежутками по длине зоны нагрева. В одном варианте, по существу, все дополнительное топливо подают в один конец зоны нагрева, а кислородсодержащий газ подают по нарастающей в упомянутую зону нагрева на упомянутом множестве отделенных промежутками участках. В другом варианте, по существу, весь кислородсодержащий газ подают в один конец зоны нагрева, а дополнительное топливо подают по нарастающей в упомянутую зону нагрева на упомянутом множестве отделенных промежутками участков. В еще одном варианте упомянутые горячие газообразные продукты горения генерируют в зоне горения отдельно от упомянутой зоны нагрева и подают к упомянутому множеству отделенных промежутками участков.
В предпочтительном варианте способ далее включает перенос второй части катализатора в зону регенерирования отдельно от упомянутой зоны нагрева, и контактирование упомянутой второй части катализатора с регенерирующим газом в упомянутой зоне регенерирования для удаления по меньшей мере части кокса, образуемого упомянутым контактированием (а).
В подходящем варианте температура зоны регенерирования равна или ниже температуры реакционной зоны.
В еще одном варианте объектом изобретения является способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий:
(а) контактирование исходного материала, содержащего метан, с катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения упомянутого метана в ароматические углеводороды;
(б) перенос первой части упомянутого катализатора из реакционной зоны в зону нагрева;
(в) нагрев первой части катализатора в зоне нагрева прямым контактированием катализатора с горячими газообразными продуктами горения, образующимися при сжигании дополнительного источника топлива;
(г) возврат нагретой первой части катализатора в реакционную зону;
(д) перенос второй части упомянутого катализатора из реакционной зоны в зону регенерирования отдельно от упомянутой зоны нагрева;
(е) контактирование упомянутой второй части катализатора с регенерирующим газом в упомянутой зоне регенерирования в условиях, эффективных для по меньшей мере частичного удаления кокса из упомянутой второй части катализатора; и
(ж) возврат регенерированной второй части катализатора в реакционную зону.
В подходящем варианте упомянутый перенос (б) и (д) и упомянутый возврат (г) и (ж) проводят непрерывно.
В подходящем варианте упомянутая реакционная зона включает вертикально расположенный реактор с отстойным слоем, в котором упомянутый исходный материал поступает по месту или вблизи основания реактора, а нагретую первую часть катализатора и регенерированную вторую часть катализатора возвращают в реактор по месту или вблизи верхней части реактора. В подходящем варианте упомянутые первую и вторую части катализатора удаляют на стадии (б) по месту или вблизи основания реактора. В подходящем варианте упомянутые ароматические углеводороды извлекают из реактора по месту или вблизи верхней части реактора.
Используемое в настоящем описании понятие "катализатор дегидроциклизации" служит как охватывающее не только активный компонент (компоненты) катализатора, но также любые инертные твердые частицы, которые могут содержаться, в дополнение к активному компоненту (компонентам), для улучшения физических свойств катализатора и/или для содействия теплопереносу.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 представлена схема дегидроциклизационного реактора и подогревателя катализатора в соответствии с первым вариантом выполнения изобретения.
На фиг.2 представлена схема дегидроциклизационного реактора с подогревателем катализатора и регенератором катализатора в соответствии со вторым вариантом выполнения изобретения.
На фиг.3 представлена схема дегидроциклизационного реактора с несколькими псевдоожиженными слоями в соответствии с третьим вариантом выполнения изобретения.
На фиг.4 представлена схема аппарата с подъемом катализатора для использования с вертикальными трубами, применяемыми в первом, втором и третьем вариантах выполнения изобретения.
На фиг.5 сопоставлен температурный профиль отходящего газа с температурными профилями на разных радиальных участках внутри катализаторной частицы, обладающей диаметром 3650 мкм, когда она нагрета в вертикальной трубе, причем все тепло поступает от горения топлива в основании вертикальной трубы.
На фиг.6 сопоставлен температурный профиль отходящего газа с температурными профилями на разных радиальных участках внутри катализаторной частицы, обладающей диаметром 3650 мкм, когда она нагрета в вертикальной трубе, причем тепло поступает от сжигания топлива на разделенных промежутками участках по длине вертикальной трубы.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ВЫПОЛНЕНИЯ ИЗОБРЕТЕНИЯ
Используемое в настоящем описании понятие "высший углеводород (углеводороды)" служит для обозначения углеводорода (углеводородов), содержащего больше одного углеродного атома на молекулу, оксигената, содержащего по меньшей мере один углеродный атом на молекулу, например этана, этилена, пропана, пропилена, бензола, толуола, ксилолов, нафталина и/или метилнафталина; и/или органического соединения (соединений), включающего по меньшей мере один углеродный атом и по меньшей мере один неводородный атом, например метанола, этанола, метиламина и/или этиламина.
Используемое в настоящем описании понятие "ароматический углеводород (углеводороды)" служит для обозначения вещества, содержащего одно или несколько ароматических колец. Примерами ароматических углеводородов являются бензол, толуол, ксилолы, нафталин и метилнафталины.
Используемое в настоящем описании понятие "реактор с подвижным слоем" означает зону или сосуд с контактированием твердых частиц и газообразных потоков таким образом, что расход газа на единицу сечения потока (U) ниже скорости, необходимой для пневмотранспортировки с разбавленной фазой твердых частиц с целью сохранения слоя твердых частиц с долей свободного пространства меньше 95%. Реактор с подвижным слоем может работать в нескольких режимах потоков, включая отстойный или режим движения уплотненного слоя (U<Umf), псевдоожиженный режим (Umf<U<Umb), режим с комкованием частиц (Umb<U<Uc), переходный к турбулентному и собственно турбулентный режим псевдоожижения (Uc<U<Utr) и режим с высокой скоростью потока (U>Utr). Эти разные режимы псевдоожижения описаны, например, в работах Kunii, D., Levenspiel, О., глава 3, Fluidization Engineering, издание 2-е, Butterworth-Heinemann, Boston, 1991 и Walas, S.M., глава 6, Chemical Process Equipment, Butterworth-Heinemann, Boston, 1990.
Используемое в настоящем описании понятие "отстойный слой" служит для обозначения зоны или сосуда, в котором частицы контактируют с газообразными потоками таким образом, что расход газа на единицу сечения потока (U) меньше минимальной скорости, необходимой для псевдоожижения твердых частиц, минимальной скорости псевдоожижения (Umf), U<Umf, в по меньшей мере части реакционной зоны, и/или работающего со скоростью, которая выше минимальной скорости псевдоожижения при одновременном сохранении градиента в свойстве газа и/или твердого вещества (таком как температура, состав газа или твердого вещества и т.д.) вдоль оси снизу вверх в реакторном слое при применении внутрикорпусных устройств реактора с целью свести к минимуму обратное перемешивание газа и твердого вещества. Описание минимальной скорости псевдоожижения приведено, например, в главе 3 работы "Fluidization Engineering", D.Kunii и O.Levenspiel, издание 2-е, Butterworth-Heinemann, Boston, 1991 и главе 6 работы "Chemical Process Equipment" S.M.Walas, Butterworth-Heinemann, Boston, 1990, которые в полном объеме включены в качестве ссылок.
Используемое в настоящем описании понятие "реактор с псевдоожиженным слоем" означает зону или сосуд с контактированием твердых частиц и газообразных потоков таким образом, что расход газа на единицу сечения потока (U) достаточен для псевдоожижения твердых частиц (т.е. выше минимальной скорости псевдоожижения Umf) и ниже скорости, необходимой для пневмотранспортировки с разбавленной фазой твердых частиц с целью сохранить слой твердых частиц с долей свободного пространства меньше 95%. Используемое в настоящем описании понятие "каскадированные псевдоожиженные слои" служит для обозначения последовательного расположения отдельных псевдоожиженных слоев таким образом, что при этом может иметь место градиент свойства газа и/или твердого вещества (такого как температура, состав газа или твердого вещества, давление и т.д.), поскольку твердое вещество или газ каскадирует от одного псевдоожиженного слоя к другому. Описание минимальной скорости псевдоожижения находится, например, в приведенных выше опубликованных работах Kunii и Walas.
Используемое в настоящем описании понятие "вертикальный трубный реактор" обозначает зону или сосуд (такой как вертикальный цилиндрический патрубок), применяемый для вертикальной в принципе транспортировки твердых частиц в режимах псевдоожижения с высокой скоростью потока или псевдоожижения с пневмотранспортировкой. Режимы псевдоожижения с высокой скоростью потока и псевдоожижения с пневмотранспортировкой характеризуются скоростями газа на единицу сечения потока (U), которые больше, чем скорость транспортировки (Utr). Режимы псевдоожижения с высокой скоростью потока и псевдоожижения с пневмотранспортировкой описаны также в приведенных выше опубликованных работах Kunii и Walas.
Объектом настоящего изобретения является способ получения ароматических углеводородов введением исходного материала, содержащего метан, как правило, совместно с H2, CO и/или СО3, в контакт с катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения метана в ароматические углеводороды и водород. Как обсуждалось выше, реакция дегидроциклизации является эндотермической, и объектом настоящего изобретения является способ подачи тепла в реакцию удалением из реакционной зоны части катализатора, нагреванием этой части катализатора в зоне нагрева горячими газообразными продуктами горения, образующимися при сжигании дополнительного источника топлива, и затем возвратом нагретой части катализатора в реакционную зону.
Кроме того, объектом изобретения является способ утилизации водорода, образующегося в качестве побочного продукта реакции дегидроциклизации, и, в частности, способ превращения по меньшей мере части водорода в более ценные продукты.
Исходный материал
В способе по изобретению можно использовать любой метансодержащий исходный материал, но в общем предлагаемый способ предусмотрен для применения с исходным природным газом. Другие приемлемые метансодержащие исходные материалы включают те, которые получают из таких источников, как угольные пласты, захоронения отходов, ферментация сельскохозяйственных или муниципальных отходов и/или газообразные потоки нефтепереработки.
Метансодержащие исходные материалы, такие как природный газ, как правило, содержат, в дополнение к метану диоксид углерода и этан. Этан и другие алифатические углеводороды, которые могут содержаться в исходном материале, на стадии дегидроциклизации могут быть, разумеется, превращены в целевые ароматические продукты. Кроме того, как это обсуждается ниже, диоксид углерода также может быть превращен в полезные ароматические продукты либо непосредственно на стадии дегидроциклизации, либо косвенным путем, посредством превращения в метан и/или этан на стадии снижения содержания водорода.
Перед применением метансодержащих потоков в способе по изобретению азот- и/или серусодержащие примеси, которые также, как правило, находятся в этих потоках, могут быть удалены или их количество может быть уменьшено до низких концентраций. В одном из вариантов исходный материал, подаваемый на стадию дегидроциклизации, содержит меньше 100 ч./млн, например меньше 10 ч./млн, в частности меньше 1 ч./млн каждого из соединений азота и серы.
В дополнение к метану, с целью содействовать уменьшению коксообразования в исходный материал, подаваемый на стадию дегидроциклизации, можно добавлять по меньшей мере один из водорода, воды, моноксида углерода и диоксида углерода. Эти добавки могут быть введены в виде отдельных совместно подаваемых исходных материалов или могут находиться в метановом потоке, например, таком как в случае, когда метановый поток дериватизируют из природного газа, включающего диоксид углерода. Другие источники диоксида углерода могут включать отходящие газы, установки СПГ, водородные установки, аммиачные установки, гликольные установки и фталевоангидридные установки.
В одном варианте исходный материал, подаваемый на стадию дегидроциклизации, содержит диоксид углерода и включает от примерно 90 до примерно 99,9 мольного %, в частности от примерно 97 до примерно 99 мольных % метана и от примерно 0,1 до примерно 10 мольных %, в частности от примерно 1 до примерно 3 мольных % СO2. В другом варианте исходный материал, подаваемый на стадию дегидроциклизации, содержит моноксид углерода и включает от примерно 80 до примерно 99,9 мольного %, в частности от примерно 94 до примерно 99 мольных % метана и от примерно 0,1 до примерно 20 мольных %, в частности от примерно 1 до примерно 6 мольных % СО. В еще одном варианте исходный материал, подаваемый на стадию дегидроциклизации, содержит водяной пар и включает от примерно 90 до примерно 99,9 мольного %, в частности от примерно 97 до примерно 99 мольных %, метана и от примерно 0,1 до примерно 10 мольных %, в частности от примерно 1 до примерно 5 мольных % водяного пара. Однако в еще одном варианте исходный материал, подаваемый на стадию дегидроциклизации, содержит водород и включает от примерно 80 до примерно 99,9 мольного %, в частности от примерно 95 до примерно 99 мольных % метана и от примерно 0,1 до примерно 20 мольных %, в частности от примерно 1 до примерно 5 мольных % водорода.
Исходный материал, подаваемый на стадию дегидроциклизации, может также содержать более высокомолекулярные углеводороды, чем метан, включая ароматические углеводороды. Такие более высокомолекулярные углеводороды могут быть возвращены в процесс со стадии снижения содержания водорода, добавлены в виде отдельных совместно подаваемых исходных материалов или могут находиться в метановом потоке, таком как, например, в случае, когда в исходном природном газе содержится этан. Более высокомолекулярные углеводороды, возвращаемые в процесс со стадии снижения содержания водорода, как правило, включают моноциклические ароматические соединения и/или парафины и олефины, содержащие преимущественно 6 или меньше, в частности 5 или меньше, например 4 или меньше, как правило, 3 или меньше углеродных атомов. Обычно исходный материал, подаваемый на стадию дегидроциклизации, содержит меньше 5 мас.%, в частности меньше 3 мас.% углеводородов С3+.
Дегидроциклизация
На стадии дегидроциклизации предлагаемого способа метансодержащий исходный материал вводят в контакт с катализатором дегидроциклизации в условиях, как правило, в неокислительных условиях, а предпочтительно в восстановительных условиях, эффективных для превращения метана в более высокомолекулярные углеводороды, включая бензол и нафталин. В принципе проводят следующие результирующие реакции:
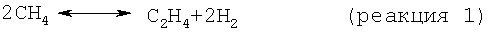
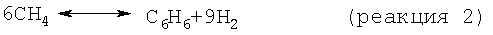
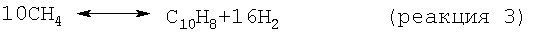
Моноксид и/или диоксид углерода, который может находиться в исходном материале, повышает активность и стабильность катализатора содействием протеканию реакций, таких как:
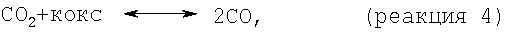
но негативно влияет на равновесие, позволяя протекать параллельным результирующим реакциям, таким как:
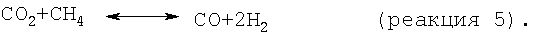
Приемлемые для стадии дегидроциклизации условия включают температуру от примерно 400 до примерно 1200°С, в частности от примерно 500 до примерно 975°С, например от примерно 600 до примерно 950°С, давление от примерно 1 до примерно 1000 кПа, в частности от примерно 10 до примерно 500 кПа, например от примерно 50 до примерно 200 кПа, и среднечасовую скорость подачи сырья от примерно 0,01 до примерно 1000 ч-1, в частности от примерно 0,1 до примерно 500 ч-1, например от примерно 1 до примерно 20 ч-1. В подходящем варианте стадию дегидроциклизации осуществляют в отсутствии O2.
В способе по изобретению можно использовать любой катализатор дегидроциклизации, эффективный для превращения метана в ароматические соединения, хотя обычно катализатор включает металлический компонент, в особенности переходный металл или его соединение на неорганическом носителе. В подходящем варианте металлический компонент содержится в количестве в пределах от примерно 0,1 до примерно 20%, в частности в пределах от примерно 1 до примерно 10 мас.%, в пересчете на массу всего катализатора. Обычно металл содержится в катализаторе в форме карбида.
Приемлемые для катализатора металлические компоненты включают кальций, магний, барий, иттрий, лантан, скандий, церий, титан, цирконий, гафний, ванадий, ниобий, тантал, хром, молибден, вольфрам, марганец, рений, железо, рутений, кобальт, родий, иридий, никель, палладий, медь, серебро, золото, цинк, алюминий, галлий, кремний, германий, индий, олово, свинец, висмут и трансурановые металлы. Такие металлические компоненты могут содержаться в форме свободных элементов или в виде соединений металлов, таких как оксиды, карбиды, нитриды и/или фосфиды, и их можно использовать самостоятельно или в сочетании. В качестве одного из металлических компонентов могут быть также использованы платина и осмий, но обычно они не предпочтительны.
Неорганический носитель может быть либо аморфным, либо кристаллическим и, в частности, может представлять собой оксид, карбид или нитрид бора, алюминия, кремния, фосфора, титана, скандия, хрома, ванадия, магния, марганца, железа, цинка, галлия, германия, иттрия, циркония, ниобия, молибдена, индия, олова, бария, лантана, гафния, церия, тантала, вольфрама или других трансурановых элементов. Кроме того, носителем может быть пористый материал, такой как микропористый кристаллический материал и мезопористый материал. Используемое в настоящем описании понятие "микропористый" относится к порам, обладающим диаметром меньше 2 нм, тогда как понятие "мезопористый" относится к порам, обладающим диаметром от 2 до 50 нм.
Приемлемые микропористые кристаллические материалы включают силикаты, алюмосиликаты, титаносиликаты, титаноалюмосиликаты, алюмофосфаты, металлофосфаты, металлоалюмофосфаты, кремнеалюмофосфаты и их смеси. Такие микропористые кристаллические материалы включают материалы, обладающие каркасами типов MFI (например, ZSM-5, TX-1, TS-2 и силикалит), MEL (например, ZSM-11), MTW (например, ZSM-12), TON (например, ZSM-22), МТТ (например, ZSM-23), FPER (например, ZSM-35), MFS (например, ZSM-57), MWW (например, МСМ-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, МСМ-36, МСМ-49 и МСМ-56), IWR (например, ITQ-24), KFI (например, ZK-5), *ВЕА (например, бета-цеолит), IТН (например, ITQ-13), MOR (например, морденит), FAU (например, цеолиты X, Y, ультрастабилизированный Y и деалюминированный Y), LTL (например, цеолит L), IWW (например, ITQ-22), VFI (например, VPI-5), AEL (например, SAPO-11), AFI (например, ALPO-5) и AFO (SAPO-41), а также такие материалы, как МСМ-68, EMM-1, EMM-2, ITQ-23, ITQ-24, ITQ-25, ITQ-26, ETS-2, ETS-10, ETAS-10, ETGS-10, SAPO-17, SAPO-34 и SAPO-35. Приемлемые мезопористые материалы включают МСМ-41, МСМ-48, МСМ-50, FSM-16 и SBA-15.
Примеры предпочтительных катализаторов включают молибден, вольфрам, рений и их соединения и сочетания на ZSM-5, диоксиде кремния или оксиде алюминия.
Металлический компонент может быть диспергирован на неорганическом носителе с помощью любого средства, хорошо известного в данной области техники, такого как соосаждение, пропитка до начальной влажности, выпаривание, обычная пропитка, распылительная сушка, золь-гелевое, ионообменное, химическое паровое осаждение, диффузионное и физическое смешение. Кроме того, неорганический носитель может быть модифицирован по известным методам, таким как, например, обработка водяным паром, кислотная промывка, промывка каустической содой и/или обработка кремнийсодержащими соединениями, фосфорсодержащими соединениями и/или элементами или соединениями элементов групп 1, 2, 3 и 13 Периодической таблицы элементов. Такие модификации можно использовать для изменения поверхностной активности носителя и препятствия или улучшения доступа к любой внутренней пористой структуре носителя.
Стадию дегидроциклизации осуществляют введением метансодержащего исходного материала в контакт с катализатором дегидроциклизации в реакционных зонах с одним или несколькими неподвижными слоями, подвижными слоями или псевдоожиженными слоями. Обычно исходный материал в реакционной зоне или каждой реакционной зоне вводят в контакт с подвижным слоем катализатора дегидроциклизации, где потоки исходных материалов движутся в противоток с направлением движения катализатора дегидроциклизации. В одном варианте реакционная зона включает реактор с отстойным слоем, под которым подразумевают вертикально расположенный реактор, в котором порошкообразный катализатор поступает по месту или вблизи верхней части реактора и движется под действием гравитации с образованием каталитического слоя, в то время как исходный материал поступает в реактор по месту или вблизи основания реактора и движется вверх через каталитический слой. В альтернативном варианте реакционная зона включает несколько последовательно соединенных реакторов с псевдоожиженными слоями, в которых порошкообразный катализатор последовательно каскадно перемещают в одном направлении из одного реактора в следующий, смежный с ним реактор, в то время как исходный материал пропускают через и между реакторами в противоположном направлении.
Реакция дегидроциклизации является эндотермической и для того чтобы подводить в реакцию тепло, первую часть катализатора отводят из реакционной зоны либо на прерывистой, либо в более предпочтительном варианте на непрерывной основе и переносят в отдельную зону нагрева, где первую часть катализатора нагревают прямым контактированием с горячими газообразными продуктами горения, получаемыми сжиганием дополнительного источника топлива. Затем нагретую первую часть катализатора возвращают в реакционную зону.
Под понятием "дополнительный источник топлива" имеют в виду то, что источник топлива физически отделен от катализатора и, следовательно, на катализаторе в качестве побочного продукта реакции дегидроциклизации кокс не образуется. Как правило, дополнительный источник топлива включает углеводород, такой как метан, и, в частности, приемлемый источник топлива представляет собой природный газ, используемый в качестве исходного материала, направляемого в процесс. В подходящем варианте в зоне нагрева поддерживают бедную кислородом атмосферу, благодаря чему при сжигании углеводородного топлива для нагрева первой части катализатора образуется синтез-газ, который затем может быть использован для получения дополнительного углеводородного продукта и/или топлива. Кроме того, применение бедной кислородом атмосферы ингибирует окисление карбидов металлов, содержащихся в катализаторе дегидроциклизации, и сводит к минимуму среднее парциальное давление водяного пара, тем самым уменьшая гидротермическое старение катализатора.
По другому варианту приемлемый дополнительный источник топлива представляет собой водород и, в частности, часть водорода, образующегося в качестве побочного продукта реакции ароматизации.
В подходящем варианте упомянутую первую часть катализатора в зоне нагрева вводят в контакт непосредственно с горящим источником топлива. По другому варианту источник топлива сжигают в зоне горения отдельно от упомянутой зоны нагрева, и газообразные продукты горения, образующиеся в зоне горения, направляют в зону нагрева для нагрева первой части катализатора.
В одном практическом варианте зона нагрева является удлиненной, и первую часть катализатора пропускают через зону нагрева из впускного приспособления по месту или вблизи одного конца зоны нагрева до выпускного приспособления по месту или вблизи другого конца зоны нагрева, причем тепло передают первой части катализатора на множество участков, отделенных промежутками по длине зоны нагрева. Таким путем поступление тепла к первой части катализатора может быть распределено по длине зоны нагрева, сведением тем самым к минимуму температур поверхности катализатора и внутренних градиентов.
Когда первую часть катализатора нагревают прямым контактированием с горящим источником топлива в зоне нагрева, постепенный нагрев катализатора может быть достигнут направлением, по существу, всего дополнительного топлива во впускной конец зоны нагрева и затем подачей кислородсодержащего газа по нарастающей в упомянутую зону нагрева на упомянутом множестве отделенных промежутками участках по длине зоны нагрева. По другому варианту, по существу, весь кислородсодержащий газ, необходимый для сжигания упомянутого дополнительного топлива, может быть направлен во впускной конец зоны нагрева, а дополнительное топливо направлено по нарастающей в зону нагрева на упомянутом множестве отделенных промежутками участков.
Когда первую часть катализатора нагревают прямым контактированием с горячими газообразными продуктами горения, образующимися в отдельной зоне горения, постепенный нагрев катализатора может быть достигнут подачей горячих газообразных продуктов горения к упомянутому множеству отделенных промежутками участков по длине зоны нагрева.
В одном варианте зона нагрева представляет собой вертикальную трубу, и во время стадии повторного нагревания упомянутую первую часть катализатора пропускают вверх по вертикальной трубе. На практике зона нагрева может включать несколько вертикальных труб, соединенных параллельно. По другому варианту упомянутая зона нагрева может включать подвижный слой упомянутого катализатора.
Как правило, первая часть катализатора при поступлении в зону нагрева находится при температуре от примерно 500 до примерно 900°С, а при покидании зоны нагрева находится при температуре от примерно 800 до примерно 1000°С. Горячие газообразные продукты горения, как правило, находятся при температуре ниже 1300°С, предпочтительно ниже 1100°С, более предпочтительно ниже 1000°С, например при температуре в интервале от примерно 800°С до ниже 1000°С. Как правило, зона нагрева работает под абсолютными давлениями в пределах от 10 до 100 фунтов/кв. дюйм (от 69 до 690 кПа), более предпочтительно в пределах от 15 до 60 фунтов/кв. дюйм (от 103 до 414 кПа). Как правило, средняя продолжительность пребывания каталитических частиц в зоне нагрева находится в пределах от 0,1 до 100 с, более предпочтительно в пределах от 1 до 10 с.
Перед повторным введением в реакционную зону, а предпочтительно после прохождения через зону нагрева первая часть катализатора может быть подвергнута обработке на одной или нескольких стадиях десорбции для по меньшей мере частичного удаления (а) кокса или тяжелых углеводородов, которые могут предварительно образовываться на поверхности катализатора, и/или (б) воды или кислорода, который может предварительно адсорбироваться катализатором. Десорбцию для удаления кокса или тяжелых углеводородов удобно проводить введением первой части катализатора в контакт с водяным паром, водородом и/или СО2, тогда как десорбцию для удаления воды или кислорода удобно проводить введением первой части катализатора в контакт с метаном, СО2 или водородом.
Кроме того, поскольку стадии повторного нагрева может быть свойственна тенденция к окислению каталитически активных металлических материалов, в особенности карбидов металлов, содержащихся в первой части катализатора, перед повторным введением в реакционную зону повторно нагретый катализатор в предпочтительном варианте подвергают обработке на стадии закоксовывания. В подходящем варианте стадию закоксовывания осуществляют введением первой части катализатора в контакт с Н2, СО, СО2 и/или углеводородом, таким как метан, этан и пропан, она может быть осуществлена одновременно со стадией десорбции воды/кислорода или отдельно от нее.
Являющейся также эндотермической реакции дегидроциклизации свойственна тенденция к отложению кокса на катализаторе и, следовательно, для сохранения активности катализатора дегидроциклизации вторую часть катализатора отводят из реакционной зоны либо на прерывистой либо (в более предпочтительном варианте) на непрерывной основе и переносят в отдельную зону регенерирования. Газ, используемый для транспортировки второй части катализатора в зону регенерирования, может включать О2, но в предпочтительном варианте содержит меньше О2, чем воздух, в частности меньше 10 мас.% О2, наиболее предпочтительно меньше 5% О2. Транспортирующий газ может включать СО2 и/или Н2 для газификации части кокса из второй части катализатора, но в предпочтительном варианте практически свободен от Н2О и находится при низкой температуре (как правило, ниже 200°С), вследствие чего поток катализатора не окисляется и нагревается выше целевой температуры зоны регенерирования.
В зоне регенерирования вторую часть катализатора вводят в контакт с кислородсодержащим газом в условиях для по меньшей мере частичного удаления имеющегося на катализаторе кокса и регенерирования тем самым катализатора. Регенерирующий газ в предпочтительном варианте содержит меньше О2, чем воздух, в частности меньше 10 мас.%, более предпочтительно меньше 5 мас.% О2, и в предпочтительном варианте, по существу, свободен от Н2О. Регенерирующий газ может также включать СО2 для газификации части кокса во второй части катализатора. Удобными источниками регенерирующего газа служат обедненный О2, обогащенный N2 поток из установки разделения воздуха и поток после удаления большого количества СО2 в результате переработки промышленного или природного газа, в который предварительно добавляют воздух или О2 для достижения целевой концентрации О2. Как правило, регенерирующий газ циркулирует между зоной регенерирования и зоной обработки, где использованный регенерирующий газ охлаждают для конденсации избытка воды, добавляют свежего кислородсодержащего газа (предпочтительно воздуха) для поддержания целевой концентрации О2 и часть удалят продувкой для сохранения постоянного давления. Как правило, зона регенерирования работает под абсолютными давлениями в пределах от 10 до 100 фунтов/кв.дюйм (от 69 до 690 кПа), более предпочтительно в пределах от 15 до 60 фунтов/кв.дюйм (от 103 до 414 кПа).
Зона регенерирования может представлять собой реактор, работающий как псевдоожиженный слой, кипящий слой, отстойный слой, вертикальный трубный реактор или их сочетание. На практике зона регенерирования может включать несколько реакторов, в частности несколько соединенных параллельно вертикальных трубных реакторов. Зона регенерирования должна работать при минимальной температуре, необходимой для удаления требуемого количества кокса при предполагаемой продолжительности пребывания, и, в частности, температура не должна превышать точки, при которой происходит улетучивание оксида металла или носитель катализатора подвергается быстрому ухудшению. Обычно температура в зоне регенерирования ниже температуры реакционной зоны и температура в зоне регенерирования, как правило, составляет от примерно 400 до примерно 700°С, в частности от примерно 550 до примерно 650°С. Продолжительность пребывания катализатора в зоне регенерирования также должна быть минимизирована для того, чтобы уменьшить скорость старения катализатора и довести до максимума процент времени, которое катализатор проводит в реакторе, выполняя полезную работу. Как правило, средняя продолжительность пребывания каталитических частиц в зоне регенерирования находится в пределах от 0,1 до 100 мин, более предпочтительно в пределах от 1 до 20 мин.
После покидания зоны регенерирования вторую часть катализатора возвращают в реакционную зону, хотя перед тем как регенерированный катализатор повторно вводят в реакционную зону, может возникнуть необходимость ввести регенерированную вторую часть катализатора в контакт с метаном для по меньшей мере частичного удаления из нее адсорбированной воды и/или кислорода. Кроме того, перед повторным введением регенерированного катализатора в реакционную зону может возникнуть необходимость подвергнуть регенерированную вторую часть катализатора обработке на стадии закоксовывания введением в контакт с Н2, СО, СО2 и/или углеводородом, таким как метан, этан и пропан. Десорбция воды/кислорода и закоксовывание регенерированного катализатора могут быть осуществлены в одну стадию или на отдельных стадиях.
В подходящем варианте отношение массы первой части катализатора, переносимого в данное время в зону нагрева, к массе второй части катализатора, переносимого в то же время в зону регенерирования, находится в интервале от примерно 5:1 до примерно 100:1, предпочтительно от примерно 10:1 до примерно 20:1.
В одном практическом варианте стадию дегидроциклизации осуществляют в вертикально расположенном реакторе с отстойным слоем с исходным материалом, поступающим в реактор по месту или вблизи его основания, и нагретой первой частью катализатора и регенерированной второй частью катализатора, возвращаемыми в реактор по месту или вблизи верхней части реактора. В подходящем варианте упомянутые первую и вторую части катализатора удаляют по месту или вблизи основания реактора, а отходящий технологический поток выделяют по месту или вблизи верхней части реактора.
В альтернативном варианте стадию дегидроциклизации осуществляют в нескольких реакторах с псевдоожиженным слоем, соединенных в последовательный ряд, причем исходный материал поступает в первый реактор в этом ряду, а нагретую первую часть катализатора и регенерированную вторую часть катализатора возвращают в последний реактор в этом ряду. В подходящем варианте упомянутые первую и вторую части катализатора удаляют из первого реактора.
Основными компонентами отходящего со стадии дегидроциклизации потока являются водород, бензол, нафталин, моноксид углерода, этилен и непрореагировавший метан. Этот отходящий поток, как правило, включает по меньшей мере на 5 мас.%, в частности по меньшей мере на 10 мас.%, например по меньшей мере на 20 мас.%, предпочтительно по меньшей мере на 30 мас.%, ароматических соединений больше, чем исходный материал.
Затем из отходящего из дегидроциклизации потока выделяют бензол и нафталин, например, экстракцией растворителем с последующим разделением на фракции. Однако, как это обсуждается ниже, перед или после извлечения продуктов по меньшей мере часть этих ароматических компонентов может быть обработана на стадии алкилирования с получением более ценных материалов, таких как ксилолы.
Снижение содержания водорода
Поскольку водород является основным компонентом отходящего из дегидроциклизации потока, после извлечения ароматических продуктов отходящий поток подвергают обработке на стадии снижения содержания водорода с целью понизить содержание водорода в отходящем потоке перед возвратом непрореагировавшего метана на стадию дегидроциклизации и максимизировать утилизацию исходного материала. Стадия снижения содержания водорода, как правило, включает реакцию по меньшей мере части водорода в отходящем из дегидроциклизации потоке с кислородсодержащими материалами, предпочтительно СО и/или СО2, с получением воды и второго отходящего потока, обладающего более низким содержанием водорода в сравнении с первым отходящим (из дегидроциклизации) потоком. Приемлемые способы снижения содержания водорода описаны ниже и в совместно рассматриваемой заявке РСТ №PCT/US2005/044042 (реестр патентного поверенного №2004В154), поданной 2 декабря 2005 г.
В подходящем варианте стадия снижения содержания водорода включает (I) метанирование и/или этанирование, (II) процесс Фишера-Тропша, (III) синтез спиртов с C1 по С3, в особенности метанола, и других оксигенатов, (IV) синтез легких олефинов, парафинов и/или ароматических соединений посредством метанола или диметилового эфира как промежуточного продукта и/или (V) селективное сжигание водорода. Для достижения наибольшей эффективности эти стадии можно осуществлять последовательно; например, вначале может быть проведен процесс Фишера-Тропша с получением обогащенного С2+ потока с последующим метанированием для достижения высокой степени превращения Н2.
На стадии снижения содержания водорода обычно, как правило, так, как изложено ниже, образуются углеводороды, причем в этом случае после выделения одновременно получаемой воды по меньшей мере часть углеводородов целесообразно возвращать на стадию дегидроциклизации. Так, например, когда углеводороды, получаемые на стадии снижения содержания водорода, включают парафины и олефины, часть, возвращаемая на стадию дегидроциклизации, обычно включает парафины или олефины с 6 или меньшим числом углеродных атомов, в частности, с 5 или меньшим числом углеродных атомов, например, с 4 или меньшим числом углеродных атомов или с 3 или меньшим числом углеродных атомов. Когда углеводороды, получаемые на стадии снижения содержания водорода, включают ароматические соединения, часть, возвращаемая на стадию дегидроциклизации, обычно включает моноциклические ароматические материалы.
Метанирование/этанирование
В одном варианте стадия снижения содержания водорода включает реакцию по меньшей мере части водорода в отходящем из дегидроциклизации потоке с диоксидом углерода с получением метана и/или этана в соответствии со следующими результирующими реакциями:
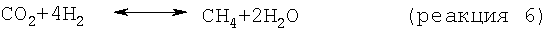
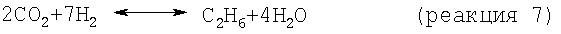
В целесообразном варианте используемый диоксид углерода является частью потока природного газа, а предпочтительно того же потока природного газа, который используют как исходный материал, подаваемый на стадию дегидроциклизации. Когда диоксид углерода является частью метансодержащего потока, CO2/СН4 этого потока в целесообразном варианте сохраняют в пределах от примерно 1/1 до примерно 0,1/1. Смешения содержащего диоксид углерода потока и отходящего из дегидроциклизации потока в целесообразном варианте добиваются подачей газообразных исходных материалов во впускное приспособление струйного насоса.
На стадии снижения содержания водорода с получением метана или этана, как правило, используют молярное соотношение Н2/CO2, близкое к стехиометрическим пропорциям, требуемым для целевой реакции 6 или реакции 7, хотя если необходимо получить содержащий CO2 или содержащий Н2 второй отходящий поток, в стехиометрическое соотношение могут быть внесены небольшие изменения. Стадию снижения содержания водорода с получением метана или этана в целесообразном варианте осуществляют в присутствии бифункционального катализатора, включающего металлический компонент, в особенности переходный металл или его соединение, на неорганическом носителе. Приемлемые металлические компоненты включают медь, железо, ванадий, хром, цинк, галлий, никель, кобальт, молибден, рутений, родий, палладий, серебро, рений, вольфрам, иридий, платину, золото, галлий и их сочетания и соединения. Неорганическим носителем может быть аморфный материал, такой как диоксид кремния, оксид алюминия и диоксид кремния/оксид алюминия, или подобный тем, которые перечислены для катализатора дегидроароматизации. Кроме того, неорганическим носителем может быть кристаллический материал, такой как микропористый или мезопористый кристаллический материал. Приемлемые пористые кристаллические материалы включают алюмосиликаты, алюмофосфаты и кремнеалюмофосфаты, перечисленные выше для катализатора дегидроциклизации.
Стадия снижения содержания водорода с получением метана и/или этана может быть осуществлена в широком диапазоне условий, включая температуру от примерно 100 до примерно 900°С, в частности, от примерно 150 до примерно 500°С, например, от примерно 200 до примерно 400°С, давление от примерно 200 до примерно 20000 кПа, в частности, от примерно 500 до примерно 5000 кПа, и среднечасовую скорость подачи сырья от примерно 0,1 до примерно 10000 ч-1, в частности, от примерно 1 до примерно 1000 ч-1. Значения степени превращения СО2, как правило, находятся в пределах от 20 до 100%, а предпочтительно больше 90%, в частности больше 99%. Эту экзотермическую реакцию можно проводить во множестве каталитических слоев с отводом тепла между слоями. Кроме того, для того чтобы максимизировать кинетические скорости, процесс в переднем слое (слоях) можно проводить при более высоких температурах, а для того чтобы максимизировать термодинамическое превращение, в последнем слое (слоях) его можно проводить при более низких температурах.
Основными продуктами такой реакции являются вода и в зависимости от молярного соотношения Н2/СО2 метан, этан и более высокомолекулярные алканы совместно с некоторыми ненасыщенными С2- и более высокомолекулярными углеводородами. Кроме того, предпочтительна некоторая частичная гидрогенизация диоксида углерода до моноксида углерода. После удаления воды метан, моноксид углерода, весь непрореагировавший диоксид углерода и более высокомолекулярные углеводороды можно направлять непосредственно на стадию дегидроциклизации для получения дополнительных ароматических продуктов.
Процесс Фишера-Тропша
В другом варианте стадия снижения содержания водорода включает реакцию по меньшей мере части водорода в отходящем из дегидроциклизации потоке с моноксидом углерода в соответствии с процессом Фишера-Тропша с получением парафинов и олефинов с С2 по С5.
Процесс Фишера-Тропша в данной области техники известен хорошо (см., например, патенты US №№5348982 и 5545674, включенные в настоящее описание в качестве ссылок). Этот процесс, как правило, включает реакцию водорода и моноксида углерода в молярном соотношении от примерно 0,5/1 до примерно 4/1, предпочтительно от примерно 1,5/1 до примерно 2,5/1, при температуре от примерно 175 до примерно 400°С, предпочтительно от примерно 180 до примерно 240, и под давлением от примерно 1 до примерно 100 бар (от 100 до 10000 кПа), предпочтительно от примерно 10 до примерно 40 бар (от 1000 до 4000 кПа), в присутствии катализатора Фишера-Тропша, обычно нанесенного или ненанесенного на носитель элемента группы VIII, неблагородного металла, например Fe, Ni, Ru, Co, с промотором или без него, например с рутением, рением, гафнием, цирконием, титаном. Носителями, когда их используют, могут служить огнеупорные оксиды металлов, таких как группы IV B, т.е. диоксид титана, диоксид циркония или диоксид кремния, оксид алюминия или диоксид кремния/оксид алюминия. В одном варианте катализатор включает не вызывающий конверсии катализатор, например кобальт или рутений, предпочтительно кобальт, с рением или цирконием в качестве промотора, предпочтительно с кобальтом и рением, нанесенными на диоксид кремния или диоксид титана, предпочтительно на диоксид титана.
В другом варианте катализатор синтеза углеводородов включает металл, такой как Сu, Cu/Zn и Cr/Zn, на ZSM-5, и процесс проводят до получения значительных количеств моноциклических ароматических углеводородов. Пример такого процесса описан в работе Jose Erena Study of Physical Mixtures of Cr2O3-ZnO and ZSM-5 Catalysts for the Transformation of Syngas into Liquid Hydrocarbons; Ind. Eng. Chem. Res. 1998, 37, 1211-1219, включенной в настоящее описание в качестве ссылки.
Выделяют жидкости Фишера-Тропша, т.е. С5+, и от более тяжелых углеводородов отделяют легкие газы, например непрореагировавшие водород и СО, с C1 по С3 или С4 и воду. Затем более тяжелые углеводороды могут быть выделены как продукты или направлены на стадию дегидроциклизации для получения дополнительных ароматических продуктов.
Наличие моноксида углерода, требующегося для реакции Фишера-Тропша, может быть полностью или частично обеспечено благодаря имеющемуся или совместно подаваемому с метансодержащим исходным материалом и полученному в качестве побочного продукта на стадии дегидроциклизации моноксиду углерода. Если необходимо, дополнительный моноксид углерода может быть генерирован за счет подачи диоксида углерода, содержащегося, например, в природном газе, к катализатору конверсии, благодаря чему моноксид углерода получают обратной реакцией конверсии водяного газа:
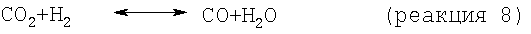
и следующей реакцией:
СН4+H2O↔СО+3Н2
Синтез спиртов
В еще одном варианте стадия снижения содержания водорода включает реакцию по меньшей мере части водорода в отходящем из дегидроциклизации потоке с моноксидом углерода с получением спиртов с C1 по С3, в особенности метанола. Получение метанола и других оксигенатов из синтез-газа также хорошо известно и представлено, например, в патентах US №№6114279, 6054497, 5767039, 5045520, 5254520, 5610202, 4666945, 4455394, 4565803, 5385949, описания к которым включены в настоящее описание в качестве ссылок. Используемый синтез-газ, как правило, обладает молярным отношением водорода (Н2) к оксидам углерода (СО+СО2) в интервале от примерно 0,5/1 до примерно 20/1, предпочтительно в интервале от примерно 2/1 до примерно 10/1, причем диоксид углерода необязательно содержится в количестве не больше 50 мас.% в пересчете на общую массу синтез-газа.
Катализатор, используемый в процессе синтеза метанола, обычно включает оксид по меньшей мере одного элемента, выбранного из группы, включающей медь, серебро, цинк, бор, магний, алюминий, ванадий, хром, марганец, галлий, палладий, осмий и цирконий. В подходящем варианте катализатор представляет собой катализатор на основе меди, в частности в форме оксида меди, необязательно в присутствии оксида по меньшей мере одного элемента, выбранного из серебра, цинка, бора, магния, алюминия, ванадия, хрома, марганца, галлия, палладия, осмия и циркония. В подходящем варианте катализатор содержит оксид меди и оксид по меньшей мере одного элемента, выбранного из цинка, магния, алюминия, хрома и циркония. В одном варианте катализатор синтеза метанола выбирают из группы, включающей оксиды меди, оксиды цинка и оксиды алюминия. В более предпочтительном варианте катализатор содержит оксиды меди и цинка.
Процесс синтеза метанола может быть осуществлен в широком интервале температур и давлений. Приемлемые температуры находятся в интервале от примерно 150 до примерно 450°С, в частности от примерно 175°С до примерно 350°С, например от примерно 200 до примерно 300°С. Приемлемые давления находятся в интервале от примерно 1500 до примерно 12500 кПа, в частности от примерно 2000 до примерно 10000 кПа, например от примерно 2500 до примерно 7500 кПа. Среднечасовые скорости подачи газа варьируют в зависимости от типа процесса, который проводят, но обычно среднечасовая скорость подачи газа в потоке газа через каталитический слой находится в интервале от примерно 50 до примерно 50000 ч-1, в частности от примерно 250 до примерно 25000 ч-1, более предпочтительно от примерно 500 до примерно 10000 ч-1. Эту экзотермическую реакцию можно проводить либо в неподвижных либо в псевдоожиженных слоях, включающих множество каталитических слоев, с отводом тепла между слоями. Кроме того, для того чтобы максимизировать кинетические скорости, процесс в переднем слое (слоях) можно проводить при более высоких температурах, а для того чтобы максимизировать термодинамическое превращение, в последнем слое (слоях) его можно проводить при более низких температурах.
Получаемые метанол и/или другие оксигенаты могут быть направлены на продажу как самостоятельный продукт, их можно использовать для алкилирования ароматических соединений, образующихся на стадии дегидроциклизации, до более высокоценных продуктов, таких как ксилолы, или можно использовать в качестве исходного материала для получения более низкомолекулярных олефинов, в особенности этилена и пропилена. Превращение метанола в олефины является хорошо известным процессом, который описан, например, в патенте US №4499327, включенном в настоящее описание в качестве ссылки.
Селективное сжигание водорода
Тем не менее в еще одном варианте стадия снижения содержания водорода включает селективное сжигание водорода, которое представляет собой процесс, в котором водород в смешанном потоке взаимодействует с кислородом с образованием воды или водяного пара без существенного взаимодействия в потоке углеводородов с кислородом с получением моноксида углерода, диоксида углерода и/или оксигенированных углеводородов. Обычно селективное сжигание водорода проводят в присутствии кислородсодержащего твердого материала, такого как смешанный оксид металла, который обычно высвобождает часть связанного кислорода для водорода.
Один приемлемый способ селективного сжигания водорода описан в патенте US №5430210, включенном в настоящее описание в качестве ссылки, он включает контактирование в реакционных условиях первого потока, включающего углеводород и водород, и второго потока, включающего кислород, с раздельными поверхностями мембраны, непроницаемой для не содержащих кислорода газов, где упомянутая мембрана включает оксид металла, селективный в отношении сжигания водорода, и выделение продукта селективного сжигания водорода. Этот оксид металла, как правило представляет собой смешанный оксид металла висмута, индия, сурьмы, таллия и/или цинка.
В патенте US №5527979, включенном в настоящее описание в качестве ссылки, описан способ чистой каталитической окислительной дегидрогенизации алканов с получением алкенов. Этот способ включает одновременную равновесную дегидрогенизацию алканов до алкенов и селективное сжигание образующегося водорода для проведения равновесной реакции дегидрогенизации с получением алкенов. Так, в частности, алкановый исходный материал дегидрируют над катализатором равновесной дегидрогенизации в первом реакторе, а затем отходящий из первого реактора поток совместно с кислородом направляют во второй реактор, содержащий катализатор из оксида металла, который служит для катализа селективного сжигания водорода. Катализатор равновесной дегидрогенизации может включать платину, а катализатор селективного сжигания из оксида металла может включать висмут, сурьму, индий, цинк, таллий, свинец и теллур или их смесь.
В заявке на патент US №2004/0152586, опубликованной 5 августа 2004 г. и включенной в настоящее описание в качестве ссылки, описан способ снижения содержания водорода в отходящем из крекинг-установки потоке. В этом способе используют каталитическую систему, включающую (1) по меньшей мере один твердый кислотный компонент крекинга и (2) по меньшей мере один компонент селективного сжигания водорода на металлической основе, состоящий, по существу, из (а) сочетания металлов, выбранных из группы, включающей:
I) по меньшей мере один металл из группы 3 и по меньшей мере один металл из групп с 4 по 15 Периодической таблицы элементов;
II) по меньшей мере один металл из групп с 5 по 15 Периодической таблицы элементов и по меньшей мере один металл из по меньшей мере одной из групп 1, 2 и 4 Периодической таблицы элементов;
III) по меньшей мере один металл из групп 1 и 2, по меньшей мере один металл из группы 3 и по меньшей мере один металл из групп с 4 по 15 Периодической таблицы элементов; и
IV) два или большее число металлов из групп с 4 по 15 Периодической таблицы элементов;
и (б) по меньшей мере одного из кислорода и серы, где этот по меньшей мере один из кислорода и серы химически связан как внутри, так и между металлами.
Реакцию селективного сжигания водорода по настоящему изобретению обычно проводят при температуре в интервале от примерно 300 до примерно 850°С и под давлением в интервале от примерно 1 до примерно 20 ат (от 100 до 2000 кПа).
Выделение/обработка ароматических продуктов
Основными продуктами стадии дегидроциклизации являются бензол и нафталин. Эти продукты могут быть выделены из отходящего из дегидроциклизации потока, как правило, экстракцией растворителем с последующим разделением на фракции, а затем поставлены для продажи непосредственно как химические продукты массового производства. По другому варианту некоторое количество или весь бензол и/или нафталин может быть алкилирован с получением, например, толуола, ксилолов и алкилнафталинов, и/или может быть подвергнут гидрогенизации с получением, например, циклогексана, циклогексена, дигидронафталина (бензилциклогексена), тетрагидронафталина (тетралина), гексагидронафталина (дициклогексена), октагидронафталина и/или декагидронафталина (декалина). Приемлемые способы алкилирования и гидрогенизации описаны ниже и более подробно в совместно рассматриваемых заявках РСТ №№PCT/US 2005/043523 (реестр патентного поверенного №2004В156), поданной 2 декабря 2005 г., и PCT/US 2005/044038 (реестр патентного поверенного №2004В155), поданной 2 декабря 2005 г.
Алкилирование ароматических соединений
Алкилирование ароматических соединений, таких как бензол и нафталин, в данной области техники хорошо известно и, как правило, включает реакцию олефина, спирта или алкилгалогенида с ароматическими материалами в газообразной или жидкой фазе в присутствии кислотного катализатора. Приемлемые кислотные катализаторы включают цеолиты со средними порами (т.е. те, которые обладают ограничивающим показателем от 2 до 12, как определено в US №4016218), включая материалы, обладающие каркасами типов MFI (например, ZSM-5 и силикалит), MEL (например, ZSM-11), MTW (например, ZSM-12), TON (например, ZSM-22), МТТ (например, ZSM-23), MFS (например, ZSM-57) и FER (например, ZSM-35), и ZSM-48, а также цеолиты с крупными порами (т.е. те, которые обладают ограничивающим показателем меньше 2), такие как материалы, обладающие каркасами типов ВЕА (например, бета-цеолит), FAU (например, ZSM-3, ZSM-20, цеолиты X, Y, ультрастабилизированный Y и деалюминированный Y), MOR (например, морденит), MAZ (например, ZSM-4), MEI (например, ZSM-18) и MWW (например, МСМ-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, МСМ-36, МСМ-49 и МСМ-56).
В одном варианте предлагаемого способа бензол выделяют из отходящего из дегидроциклизации потока и затем алкилируют олефином, таким как этилен, получаемый в качестве побочного продукта на стадии снижения содержания водорода с применением этанирования/метанирования. Типичные условия проведения парофазного алкилирования бензола этиленом включают температуру от примерно 650 до примерно 900°F (от 343 до 482°С), манометрическое давление от атмосферного до примерно 3000 фунтов/кв.дюйм (от 100 до 20800 кПа), ССПС в пересчете на этилен от примерно 0,5 до примерно 2,0 ч-1 и мольное отношение бензола к этилену от 1/1 до 30/1. Жидкофазное алкилирование бензола этиленом можно проводить при температуре в пределах от 300 до 650°F (от 150 до 340°С), под манометрическим давлением до примерно 3000 фунтов/кв.дюйм (20800 кПа), ССПС в пересчете на этилен от примерно 0,1 до примерно 20 ч-1 и при мольном отношении бензола к этилену от 1/1 до 30/1.
В предпочтительном варианте этилирование бензола проводят в условиях по меньшей мере частично жидкой фазы с использованием катализатора, включающего по меньшей мере один из бета-цеолита, цеолита Y, MCM-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, ITQ-13, ZSM-5 МСМ-36, МСМ-49 и МСМ-56.
Этилирование бензола может быть осуществлено по месту процесса дегидроциклизации/снижения содержания водорода или бензол может быть транспортирован в другой регион для превращения в этилбензол. Затем полученный этилбензол может быть поставлен для продажи, использован как предшественник, например, при получении стирола, или изомеризован по методам, хорошо известным в данной области техники, в смешанные ксилолы.
В другом варианте предлагаемого способа алкилирующий агент представляет собой метанол или диметиловый эфир (ДМЭ), его используют для алкилирования бензола и/или нафталина, выделяемого из отходящего из дегидроциклизации потока, с получением толуола, ксилолов, метилнафталинов и/или диметилнафталинов. Когда метанол или ДМЭ используют для алкилирования бензола, в целесообразном варианте это осуществляют в присутствии катализатора, включающего цеолит, такой как ZSM-5, бета-цеолит, ITQ-13, MCM-22, МСМ-49, ZSM-11, ZSM-12, ZSM-22, ZSM-23, ZSM-35 и ZSM-48, который предварительно модифицируют обработкой водяным паром таким образом, чтобы он обладал диффузионным параметром для 2,2-диметилбутана примерно от 0,1 до 15 с-1, когда его определяют при температуре 120°С и давлении 2,2-диметилбутана 60 Торр (8 кПа). Такой способ селективен в отношении получения пара-ксилола, он изложен, например, в патенте US №6504272, включенном в настоящее описание в качестве ссылки. Когда метанол используют для алкилирования нафталина, в целесообразном варианте это осуществляют в присутствии катализатора, включающего ZSM-5, MCM-22, PSH-3, SSZ-25, ERB-1, ITQ-1, ITQ-2, ITQ-13, МСМ-36, МСМ-49 или МСМ-56. Такой способ можно применять для селективного получения 2,6-диметилнафталина, он изложен, например, в патентах US №№4795847 и 5001295, включенных в настоящее описание в качестве ссылок.
Когда в способе по изобретению в качестве алкилирующего агента используют метанол или ДМЭ, его можно вводить в процесс как отдельный исходный материал или он может быть по меньшей мере частично получен in situ добавлением содержащего диоксид углерода газообразного исходного материала, такого как поток природного газа, в часть или весь отходящий со стадии дегидроциклизации поток. Так, в частности, отходящий из дегидроциклизации поток перед каким-либо выделением ароматических компонентов можно направлять в реактор обратной конверсии и проводить реакцию с содержащим диоксид углерода исходным материалом в условиях повышения содержания моноксида углерода в этом отходящем потоке, т.е. такими реакциями, как вышеприведенные реакции 5 и 8.
Кроме того, в реактор обратной конверсии можно направлять метан и СО2 и/или водяной пар с получением синтез-газа, который затем может быть смешан с частью отходящего из дегидроциклизации потока для регулирования соотношений Н2/СО/СО2 в зависимости от потребности для стадии алкилирования.
Как правило, реактор обратной конверсии содержит катализатор, включающий переходный металл на носителе, такой как Fe, Ni, Cr, Zn на оксиде алюминия, диоксиде кремния или диоксиде титана, и работает в условиях, включающих температуру от примерно 500 до примерно 1200°С, в частности от примерно 600 до примерно 1000°С, например от примерно 700 до примерно 950°С, и давление от примерно 1 до примерно 10000 кПа, в частности от примерно 2000 до примерно 10000 кПа, например от примерно 3000 до примерно 5000 кПа. Среднечасовые скорости подачи газа можно варьировать в зависимости от типа применяемого способа, но обычно среднечасовая скорость подачи газа в потоке газа через каталитический слой находится в интервале от примерно 50 до примерно 50000 ч-1, в частности от примерно 250 до примерно 25000 ч-1, более предпочтительно от примерно 500 до примерно 10000 ч-1.
Затем отходящий из реактора обратной конверсии поток может быть направлен в реактор алкилирования, работающий в условиях, обеспечивающих протекание таких реакций, как следующие:
Приемлемые для такого реактора условия алкилирования включают, по-видимому, температуру от 100 примерно до примерно 700°С, давление от примерно 1 до примерно 300 ат (от 100 до 30000 кПа) и ССПС для ароматического углеводорода от примерно 0,01 до примерно 100 ч-1. Приемлемый катализатор включает, по-видимому, молекулярное сито, обладающее ограничивающим показателем от 1 до 12, такое как ZSM-5, как правило, совместно с одним из металлов или оксидов металлов, таких как медь, хром и/или оксид цинка.
Когда в предпочтительном варианте катализатор алкилирования включает молекулярное сито, это последнее модифицируют для изменения его диффузионных характеристик таким образом, чтобы превалирующим изомером ксилола, получаемого реакцией 11, был пара-ксилол. Приемлемое средство диффузионной модификации включает обработку водяным паром и осаждение ex-situ или in situ соединений кремния, кокса, оксидов металлов, таких как MgO, и/или Р на поверхности или на входах в поры молекулярного сита. Предпочтительно также то, что активный металл вводят в молекулярное сито таким образом, чтобы обеспечить насыщение более высокореакционноспособных материалов, таких как олефины, которые могут быть образованы в качестве побочных продуктов и которые в противном случае могли бы вызвать дезактивацию катализатора.
Затем отходящий из реактора алкилирования поток можно было бы направить в секцию разделения, в которой ароматические продукты вначале отделяли бы от водорода и других низкомолекулярных материалов, целесообразно экстракцией растворителем. Далее ароматические продукты можно было бы разделить на бензольную фракцию, толуольную фракцию, С8-фракцию и тяжелую фракцию, включающую нафталин и алкилированные нафталины. Затем ароматическая С8фракция могла бы быть направлена в процесс кристаллизации или сорбции для отделения ценного п-ксилольного компонента, а оставшиеся смешанные ксилолы либо поставлены для продажи как продукт либо направлены в контур изомеризации для получения дополнительного количества п-ксилола. Толуольная фракция могла бы быть либо удалена как находящий сбыт продукт либо возвращена в реактор алкилирования, либо направлена в установку диспропорционирования толуола, а предпочтительно в установку селективного диспропорционирования толуола для получения дополнительного количества п-ксилола.
Гидрогенизация ароматических соединений
В дополнение к стадии алкилирования или вместо нее по меньшей мере часть ароматических компонентов в отходящем из дегидроциклизации потоке может быть гидрирована с получением полезных продуктов, таких как циклогексан, циклогексен, дигидронафталин (бензилциклогексен), тетрагидронафталин (тетралин), гексагидронафталин (дициклогексен), октагидронафталин и/или декагидронафталин (декалин). Эти продукты можно использовать в качестве топлив и химических промежуточных продуктов, а в случае тетралина и декалина эти последние можно использовать в качестве растворителя для экстракции из отходящего из дегидроциклизации потока ароматических компонентов.
Гидрогенизацию целесообразно, но необязательно, проводить после выделения из отходящего из дегидроциклизации потока ароматических компонентов и целесообразно использовать часть водорода, образуемого реакцией дегидроциклизации. Приемлемые способы гидрогенизации ароматических соединений в данной области техники известны хорошо и в них, как правило, используют катализатор, включающий Ni, Pd, Pt, Ni/Mo или сульфидированные Ni/Mo, нанесенные на оксид алюминия или диоксид кремния как носитель. Приемлемые для процесса гидрогенизации рабочие условия включают температуру от примерно 300 до примерно 1000°F (от 150 до 540°С), в частности от примерно 500 до примерно 700°F (от 260 до 370°С), манометрическое давление от примерно 50 до примерно 2000 фунтов/кв.дюйм (от 445 до 13890 кПа), в частности от примерно 100 до примерно 500 фунтов/кв.дюйм (от 790 до 3550 кПа), и ССПС от примерно 0,5 до примерно 50 ч-1, в частности от примерно 2 до примерно 10 ч-1.
Для того чтобы получить материалы, приемлемые для полимеризации или другого последующего химического превращения, может оказаться также необходимой частичная гидрогенизация с целью оставить в продукте одну или несколько олефиновых углерод-углеродных связей. Приемлемые способы частичной гидрогенизации в данной области техники известны хорошо и в них, как правило, используют катализатор, включающий благородные металлы, причем рутений в предпочтительном варианте наносят на оксиды металлов, такие как La2O3/ZnO. Могут быть также использованы гомогенные каталитические системы с благородными металлами. Примеры способов частичной гидрогенизации описаны в патентах US №№4678861, 4734536, 5457251, 5656761, 5969202 и 5973218, содержания которых в полном объеме включены в настоящее описание в качестве ссылок.
Альтернативный способ гидрогенизации включает гидрокрекинг низкого давления нафталинового компонента с получением алкилбензолов над таким катализатором, как сульфидированные Ni/W или сульфидированный Ni, нанесенный на аморфный алюмосиликат или цеолит, такой как цеолит X, цеолит Y и бета-цеолит. Приемлемые для гидрокрекинга низкого давления рабочие условия включают температуру от примерно 300 до примерно 1000°F (от 150 до 540°С), в частности от примерно 500 до примерно 700°F (от 260 до 370°С), манометрическое давление от примерно 50 до примерно 2000 фунтов/кв.дюйм (от 445 до 13890 кПа), в частности от примерно 100 до примерно 500 фунтов/кв.дюйм (от 790 до 3550 кПа), и ССПС от примерно 0,5 до примерно 50 ч-1, в частности от примерно 2 до примерно 10 ч-1.
Различные неограничивающие варианты выполнения изобретения далее более конкретно описаны со ссылкой на прилагаемые чертежи и примеры.
На фиг.1 проиллюстрирована упрощенная схема дегидроциклизационного реактора и подогревателя катализатора в соответствии с первым вариантом выполнения изобретения. В этом варианте дегидроциклизационный реактор включает вертикально расположенный реактор 11 с отстойным слоем, в который нагретый катализатор истекает через впускное приспособление 12, расположенное возле верхней части реактора 11, из которой охлажденный поток катализатора истекает посредством снабженного клапаном выпускного приспособления 13, расположенного возле основания реактора 11. Как правило, нагретый катализатор поступает в реактор 11 при температуре примерно 900°С, а охлажденный катализатор выходит из реактора при температуре примерно 650°С.
Метановый исходный материал 14 вводят в реактор 11 вблизи основания реактора, а дополнительный метан или метан плюс воздух 15 в качестве дополнительного топлива используют для транспортировки охлажденного катализатора в подогреватель 16 катализатора. Как правило, количество метана, используемого в качестве дополнительного топлива, составляет примерно 120 мас.% от количества метана, используемого в качестве исходного материала 14 для процесса дегидроциклизации, когда целью является совместное получение синтез-газа. Как правило, количество метана, используемого в качестве дополнительного топлива, составляет примерно 65 мас.% от количества метана, используемого в качестве исходного материала 14 для процесса дегидроциклизации, когда целью не является совместное получение синтез-газа.
Подогреватель 16 катализатора выполнен в форме вертикально расположенной трубы, причем охлажденный катализатор транспортируют вверх по этой вертикальной трубе с помощью метанового топлива, а кислород инжектируют в вертикальную трубу через множество впускных приспособлений 17, размещенных через промежутки по длине вертикальной трубы. Кислород, инжектируемый в подогреватель 16, вызывает горение метанового топлива и тем самым повышает температуру катализатора по мере его передвижения по этой вертикальной трубе. Количество кислорода, вводимого через каждое впускное приспособление 17, регулируют таким образом, чтобы сохранить в вертикальной трубе бедную кислородом атмосферу, благодаря чему горение дополнительного топлива и, следовательно, нагревание катализатора происходят постепенно, по мере того как катализатор передвигается вверх по этой вертикальной трубе. Это сводит к минимуму воздействие на катализатор чрезмерно высоких температур, а также приводит к неполному окислению топлива, вследствие чего отходящий из процесса горения поток богат моноксидом углерода. Отходящий из процесса горения поток выходит из реактора 16 через выпускное приспособление 18 и, как правило, включает 59 мас.% Н2, 30 мас.% СО, 8 мас.% Н2О и 3 мас.% СО2. Таким образом, отходящий из процесса горения поток включает удобный источник синтез-газа, который можно использовать для получения дополнительного углеводородного продукта и/или топлива.
Перед тем как нагретый катализатор возвращают в реактор 11 через впускное приспособление 12 по месту или вблизи верхней части вертикальной трубы 16, этот нагретый катализатор проходит в секцию 19 десорбции для катализатора, где катализатор вводят в контакт с добавляемым метаном для удаления воды или кислорода, которые могут быть предварительно адсорбированы катализатором. Отходящий технологический поток, содержащий ароматические углеводороды, удаляют из реактора 11 посредством выпускного приспособления (не показано) по месту или вблизи верхней части реактора 11.
Второй вариант выполнения изобретения продемонстрирован на фиг.2, согласно которой дегидроциклизационный реактор снабжен как подогревателем катализатора, так и отдельным регенератором катализатора. Как и в первом примере, дегидроциклизационный реактор включает вертикально расположенный реактор 21 с отстойным слоем, в который нагретый катализатор поступает через впускное приспособление 22, расположенное возле верхней части реактора 21, из которого охлажденный катализатор перемещается посредством первого и второго выпускных приспособлений соответственно 23, 24, расположенных возле основания реактора 21. Метановый исходный материал 25 вводят в реактор 21 вблизи его основания.
Первая часть охлажденного катализатора движется под действием гравитации из первого выпускного приспособления 23 в основание вертикального трубного подогревателя 26, где катализатор захватывается смесью воздуха и метанового топлива, проходящей в вертикальную трубу через коллектор 27. Катализатор транспортируют вверх по вертикальной трубе 26 смесью воздуха/метана и нагревают во время его прохождения по этой вертикальной трубе 26 сжиганием метана. Смесь, поступающая в вертикальную трубу 26 через коллектор 27, содержит все топливо, необходимое для нагрева катализатора до требуемой реакционной температуры, но бедна кислородом. Следовательно, в вертикальную трубу 26 через множество впускных приспособлений 28, размещенных через промежутки по длине вертикальной трубы (для простоты на фиг.2 показаны только два впускных приспособления 28, но в действительности их число может быть гораздо большим), вводят дополнительный воздух, благодаря чему нагрев катализатора происходит постепенно, по мере того как катализатор перемещается вверх по этой вертикальной трубе 26.
При выходе из верхней части вертикальной трубы 26 нагретый катализатор проходит в сепаратор 29, где твердый порошкообразный катализатор отделяют от газообразных продуктов горения и затем направляют в активаторную/десорбционную колонну 32. Далее газообразные продукты горения перед тем как их подвергают обработке для рекуперации тепла, направляют в циклоны 31 для удаления катализаторной мелочи. При применении воздуха в качестве среды горения в подогревателе газообразные продукты горения, как правило, включают 67,9 мас.% N2, 0,2 мас.% O2, 1,3 мас.% Н2, 3,6 мас.% СО, 7,9 мас.% СО2 и 17,3 мас.% H2O.
Вторая часть охлажденного катализатора движется под действием гравитации из второго выпускного приспособления 24 в основание вертикального трубного регенератора 33, где катализатор захватывается потоком кислородсодержащего газа и транспортируется вверх по вертикальному трубному регенератору. По мере того как вторая часть катализатора проходит через регенератор 33, кокс, образовавшийся на катализаторе в дегидроциклизационном реакторе 21, с катализатора выжигают, тем самым нагревая катализатор. Однако работу регенератора 33 в предпочтительном варианте регулируют, например уменьшением температуры кислородсодержащего газа, направляемого в регенератор 33, таким образом, чтобы температура второй части катализатора, выходящей из регенератора, была ниже температуры второй части катализатора, выходящей из реактора 21. Температура второй части катализатора, выходящей из регенератора 33, как правило, составляет примерно 550°С, тогда как при выходе из реактора 21 вторая часть катализатора находится при температуре примерно 650°С.
При выходе из верхней части регенератора 33 вторая часть катализатора проходит в сепаратор 34, в котором твердый порошкообразный катализатор отделяют от газообразных продуктов горения, которые затем направляют в циклоны 31 для удаления катализаторной мелочи. Далее выделенные каталитические частицы движутся в активаторную/десорбционную колонну 32.
В активаторной/десорбционной колонне 32 регенерированный катализатор вначале контактирует с потоком 35 углеводорода, такого как метан, этан и пропан, а также H2 и/или СО, движущимся вверх из нижних частей сосуда, для повторного закоксовывания металла на катализаторе, поскольку на стадии регенерирования не только удаляют из катализатора поверхностный кокс, но этой стадии также свойственна тенденция к окислению каталитически активных карбидных материалов на металлическом компоненте катализатора. Затем катализатор вводят в контакт с потоком 36 метана для удаления воды или кислорода, которые могут быть предварительно адсорбированными катализатором. Далее регенерированный катализатор объединяют с повторно нагретым катализатором из вертикальной трубы 26 и объединенный катализатор вводят в контакт с водородом и/или потоком 37 CO2 для удаления всего остаточного кокса или тяжелых углеводородов. После десорбционной обработки катализатора горячие газы движутся вверх, содействуя нагреву регенерированной части катализатора в секции повторного закоксовывания. Затем через впускное приспособление 22 объединенный катализатор возвращают в реактор 21. В предпочтительном варианте все газообразные потоки, направленные в сосуд, предварительно нагревают с целью свести к минимуму температурные потери. Хотя на чертеже все действия показаны в единственном сосуде, совершенно очевидно, что для того чтобы упростить конструкцию или проведение процесса, эти действия можно осуществлять в нескольких сосудах.
Третий вариант выполнения изобретения продемонстрирован на фиг.3, согласно которой дегидроциклизационный реактор включает несколько, в данном случае 3 вертикально размещенных через промежутки последовательно соединенных реакторов 41 с псевдоожиженным слоем, в которых горячий катализатор из секции 42 десорбции подогревателя катализатора (не показана) поступает в самый верхний реактор 41 и движется вниз по противоточному принципу относительно метана, который вводят через впускное приспособление 43 в самый нижний реактор 41. Охлажденный катализатор удаляют из самого нижнего реактора 41 и направляют в подогреватель катализатора. Подогреватель и регенератор, применяемые в конструкции с псевдоожиженным слоем, показанной на фиг.3, как правило, могли бы быть аналогичными тем, которые продемонстрированы на фиг.2. Однако следует иметь в виду, что размер частиц катализатора, используемого в конструкции с псевдоожиженным слоем, показанной на фиг.3, как правило, оказывается меньше, например, в интервале от примерно 50 до примерно 500 мкм, чем размер частиц катализатора, используемого в конструкции с отстойным слоем, показанной на фиг.1 и 2, как правило, в интервале от примерно 1000 до примерно 10000 мкм.
На фиг.4 проиллюстрировано одно приемлемое устройство для регулирования скорости потока катализатора в вертикальных трубах, применяемых в подогревателе катализатора на фиг.1 и 2 и/или регенераторе катализатора на фиг.2. Это устройство включает сборную емкость 51 для катализатора, которая приспособлена для слоя охлажденного/отработавшего катализатора, получаемого из дегидроциклизационного реактора (не показан) посредством патрубков 52. Подъемный газ либо в форме бедного кислородом воздуха для регенерирования либо воздушно-топливной смеси для повторного нагрева катализатора подают в емкость 51 посредством патрубка 53 и разделяют клапанами 54, 55 на первичный газообразный поток и вторичный газообразный поток. Первичный газообразный поток направляют по патрубку 56 в зону емкости 51 непосредственно ниже нижнего конца вертикальной трубы 57, вследствие чего первичные подъемные газообразные потоки поднимаются по этой вертикальной трубе 57 без прохождения на сколько-нибудь существенную толщину через каталитический слой.
Вторичный подъемный газ направляют по патрубку 58 в зону емкости 51, отделенную существенным расстоянием от нижнего конца вертикальной трубы 57 таким образом, чтобы для достижения вертикальной трубы вторичный подъемный газ должен был двигаться через значительную толщину каталитического слоя. Этот вторичный подъемный газ толкает каталитические частицы в поток первичного подъемного газа, которым они захватываются и поднимаются по вертикальной трубе 57. Путем регулирования клапанов 54, 55 для варьирования относительных скоростей потоков первичного и вторичного подъемных газов можно варьировать скорость потока каталитических частиц по этой вертикальной трубе 57 и, следовательно, скорость потока катализатора внутри вертикальной трубы 57. Обычно скорость потока вторичного подъемного газа варьируют в пределах от 5 до 15% от общего газообразного потока.
В вариантах, продемонстрированных на чертежах, вертикальные трубы, применяемые в подогревателе катализатора и регенераторе катализатора, характеризуются в общем постоянным внутренним диаметром по длине вертикальной трубы. Однако в определенном случае может возникнуть необходимость в конструкции, у которой внутренний диаметр вертикальной трубы увеличивается от основания к верхней части вертикальной трубы.
Хотя на этих чертежах не показано, "низкопотенциальную теплоту" из других частей процесса необходимо использовать для предварительного нагрева метана (как в качестве исходного материала, который должен быть превращен в ароматические соединения, так и в качестве топлива для подогревателя катализатора), предпочтительно до примерно 600°С, и для предварительного нагрева кислородсодержащего исходного материала, направляемого в подогреватель катализатора, до максимально доступной температуры.
Пример 1.Зависящий от пространства и времени температурный профиль катализатора в подогревателе
Внутренний (пространственный) температурный профиль внутри катализаторной частицы и его зависимость от времени аксиально вверх вдоль вертикальной трубы рассчитывали для двух случаев: (А) когда весь воздух, необходимый для окисления метана, добавляют вблизи основания вертикальной трубы (т.е. при нулевой продолжительности пребывания катализатора в вертикальной трубе), и (Б) когда подаваемый воздух распределяют аксиально вдоль вертикальной трубы (либо за отдельные интервалы продолжительности пребывания катализатора, как было бы практически возможно, либо в точках "непрерывной" инжекции воздуха при бесконечно малых интервалах (или длинах вертикальной трубы) для имитации теоретических эксплуатационных свойств в лучших случаях). Эта методология для расчета зависящего от времени температурного профиля внутри катализаторной частицы (по предположению, сферической) T(r, t) включала решение в числовом виде упрощенного уравнения теплопереноса для установившегося режима, 1-мерного теплопереноса с негомогенной реакцией в сочетании с граничным условием конвективного и за счет излучения теплообмена на поверхности частиц:
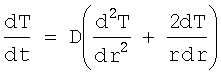

где r обозначает радиальное расстояние от центра сферы, t обозначает продолжительность пребывания катализатора в вертикальной трубе, D обозначает коэффициент температуропроводимости, k обозначает теплопроводность катализатора, h обозначает коэффициент конвективной теплопередачи, Тповерхности и Тотходящ обозначают соответственно температуру поверхности катализатора (при r=R) и температуру массы отходящего газа (при r>>R), σ обозначает постоянную Стефана-Больцмана, а ε обозначает коэффициент тепловых потерь с поверхности катализатора. Для простоты предполагают, что теплопроводность катализатора, коэффициент температуропроводимости и коэффициент тепловых потерь с поверхности остаются постоянными. Коэффициент конвективной теплопередачи рассчитывали с использованием свойств отходящего газа возле верхней части и условий в основании вертикальной трубы с помощью корреляции Дженкоплиса (Geankoplis) для выделенной сферы в газообразном потоке. Поскольку разница коэффициентов теплопередачи составляла <20%, по всей вертикальной трубе использовали среднее значение. В следующей таблице 1 перечислены физические постоянные и свойства катализатора, использованного в модели.
В дополнение к расчетам теплопередачи общий энергетический баланс между катализаторной частицей и отходящим газом должен удовлетворять следующему:
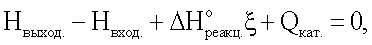
где Нвыход и Нвход обозначают энтальпии отходящего газа в условиях при впускном приспособлении и выпускном приспособлении (для каждого приращения времени), 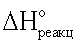 обозначает изменение энтальпии вследствие реакции ξ молей метана, а Окат обозначает тепло, переданное катализатору за приращение времени. Энтальпию газовой смеси рассчитывали с использованием:
обозначает изменение энтальпии вследствие реакции ξ молей метана, а Окат обозначает тепло, переданное катализатору за приращение времени. Энтальпию газовой смеси рассчитывали с использованием:
H-H298 (кДж/моль) = At+Bt2/2+Ct3/3+Dt4/4-Et+F-H
(где t обозначает Т(К)/1000),
где с А по Н обозначают постоянные теплоемкости для конкретных материалов. Уравнение энергетического баланса позволяет рассчитывать температуру отходящего газа аксиально вверх вдоль вертикальной трубы.
Частное дифференциальное уравнение теплопередачи решали в числовом виде с использованием явного метода конечных разностей:
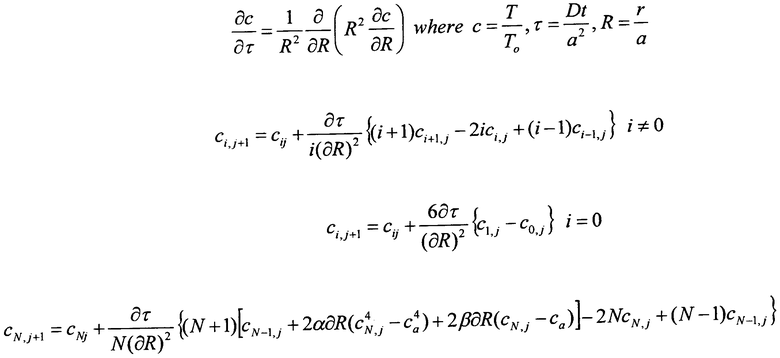
где подстрочный символ i обозначает радиальный показатель от 0 до N, подстрочный символ j обозначает временной показатель, подстрочный символ а обозначает свойство отходящего газа, а α и β обозначают безразмерные постоянные соответственно излучения и конвекции.
Случай А: завершающее добавление воздуха вблизи основания вертикальной трубы
На фиг.5 показан температурный профиль катализатора для разных радиальных координат внутри катализаторной частицы в зависимости от продолжительности пребывания катализатора в вертикальной трубе. Эти профили получали для диаметра катализатора (а) 3650 мкм, который, по-видимому, является типичным для реактора с отстойным (непсевдоожиженным) слоем. Начальная температура отходящего газа в нулевой момент времени является приростом адиабатической температуры 2254°С. Катализаторная частица после 0,95 с достигает среднемассовой температуры 850°С, хотя в этот момент времени все еще существуют значительные градиенты внутренней температуры. После 0,95 с катализаторная частица выходит из вертикальной трубы и отделяется от отходящего газа с последующим уравновешиванием этих внутренних градиентов, что приводит к предположению об отсутствии дальнейшего теплообмена с технологическими газами. После примерно 3,5 с катализаторная частица достигает равновесного состояния при 850°С. Благодаря высоким первоначальным температурам отходящего газа температура поверхности катализатора значительно превышает целевые 850°С, достигая после 0,05 с максимума в 1073°С. Необходимо отметить, что временная ступень, использованная в этой числовой имитации, для аккуратного сбора данных о ситуациях в кратковременных масштабах составляла 0,0004 с.
Случай Б: аксиальное распределение воздуха вдоль вертикальной трубы
На фиг.6 показан температурный профиль катализатора для разных радиальных координат внутри катализаторной частицы в зависимости от продолжительности пребывания катализатора в вертикальной трубе. Эти профили получали для такого же диаметра катализатора, 3650 мкм (как на фиг.5). Воздух распределяют вдоль вертикальной трубы (примерно 3,5 фута на точку инжекции), и количество воздуха, добавляемого в каждой точке инжекции, определяют по количеству энергии, необходимой для повышения температуры отходящего газа вновь до 1000°С. После использования всего доступного воздуха дальнейшая инжекция кислорода отсутствует, и вследствие теплообмена с твердыми частицами отходящий газ охлаждается. Катализаторная частица достигает среднемассовой температуры 850°С после 1,96 с, по прошествии примерно удвоенного количества времени, необходимого в случае А (когда весь воздух инжектировали вблизи основания вертикальной трубы), что согласуется с уменьшенной термальной движущей силой. Однако максимальная температура поверхности 904°С существенно ниже, чем 1073°С в случае А. Этот технический прием позволяет сводить к минимуму воздействие на катализатор очень высоких температур, содействуя тем самым уменьшению дезактивации катализатора в результате спекания, улетучивания или изменений активного участка и улучшению его механической целостности благодаря уменьшенным термическим градиентам.
Пример 2.Регулирование максимальной температуры поверхности катализатора
Сведение к минимальной температуре поверхности катализатора имеет важное значение для ограничения гидротермической дезактивации катализатора в вертикальной трубе. Двумя факторами, которые могут оказать значительное влияние на температурный профиль поверхности катализатора, являются (1) максимальная температура отходящего газа (регулируемая варьированием профиля инжекции воздуха вдоль вертикальной трубы) и (2) размер частиц катализатора. Другие свойства катализатора и текучей среды (такие как, теплопроводность, коэффициент тепловых потерь с поверхности, коэффициент конвективного теплообмена, теплоемкость и т.д.) могут повлиять на температурные профили катализатора, но являются более трудными для регулирования из-за перспективы катализатора и/или конструкции реактора. Этот пример показывает, что манипулирование температурой отходящего газа (для конкретного размера частицы катализатора) дает возможность сводить к минимальным температуры поверхности катализатора. В следующих имитациях пользовались точками "непрерывной" инжекции воздуха при бесконечно малых интервалах времени пребывания катализатора (или длинах вертикальных труб) для сохранения фиксированной температуры отходящего газа (до тех пор, пока не использовали весь воздух) для имитации теоретических эксплуатационных свойств в лучших случаях (т.е. минимально необходимая продолжительность пребывания).
В таблице 2 приведены результаты имитации для частиц двух размеров: (1) 3650 мкм, представляющих случай реактора с отстойным слоем, и (2) 250 мкм, представляющих случай реактора с псевдоожиженным слоем. Для фиксированного размера частиц и выбора начальной температуры отходящего газа в таблице 2 приведена максимальная температура поверхности, которая воздействует, по-видимому, на частицу во время нагрева и минимальной продолжительности пребывания, необходимой для повышения средней температуры частиц до 850°С. В экстремальном случае, когда весь воздух инжектируют вблизи основания вертикальной трубы, максимальная температура поверхности для 250-микрометровых частиц 871°С благодаря их более высокому отношению поверхности к объему и более короткому размеру транспортировки существенно ниже, чем 1073°С для 3650-микрометровых частиц. Более того, для частиц меньших размеров значительно уменьшается также требуемая продолжительность пребывания. При фиксированном размере частиц уменьшение начальной температуры отходящего газа в изотермальной зоне уменьшает максимальную температуру поверхности катализатора и увеличивает необходимую продолжительность пребывания. В экстремальном случае, когда температура отходящего газа постоянна по всей вертикальной трубе (900°С), максимальная температура поверхности для 250-микрометровых частиц только на 5°С выше конечной средней температуры и на 22°С выше для 3650-микрометровых частиц. Требуемая продолжительность пребывания для частиц этих размеров и температур отходящего газа могут быть достигнуты с использованием вертикальных труб целесообразных высот и скоростей частиц.
Хотя настоящее изобретение описано и проиллюстрировано со ссылкой на конкретные варианты его выполнения, для обычных специалистов в данной области техники вполне очевидно, что сама сущность изобретения приводит к вариантам, которые нет необходимости иллюстрировать в настоящем описании. По этой причине с целью определить фактический объем настоящего изобретения следует обращаться только к прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2007 |
|
RU2454390C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ МЕТАНА | 2007 |
|
RU2448079C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ МЕТАНА | 2007 |
|
RU2454389C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ МЕТАНА | 2007 |
|
RU2459789C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ИЗ АЛИФАТИЧЕСКИХ | 2008 |
|
RU2461537C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2008 |
|
RU2460581C2 |
| ПОЛУЧЕНИЕ АЛКИЛИРОВАННЫХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2005 |
|
RU2417974C2 |
| ПОЛУЧЕНИЕ ЖИДКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2005 |
|
RU2405764C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ И СИНТЕЗ-ГАЗА ИЗ МЕТАНА | 2007 |
|
RU2458899C2 |
| ПОЛУЧЕНИЕ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ ИЗ МЕТАНА | 2005 |
|
RU2418780C2 |
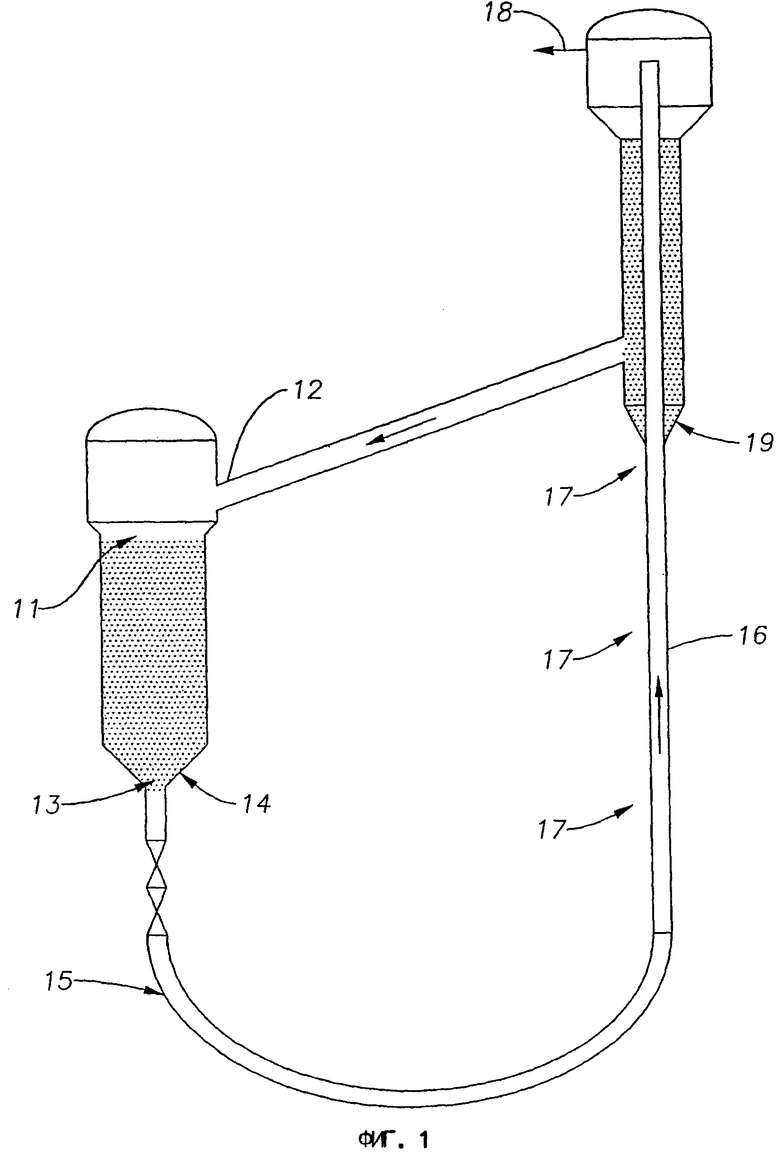
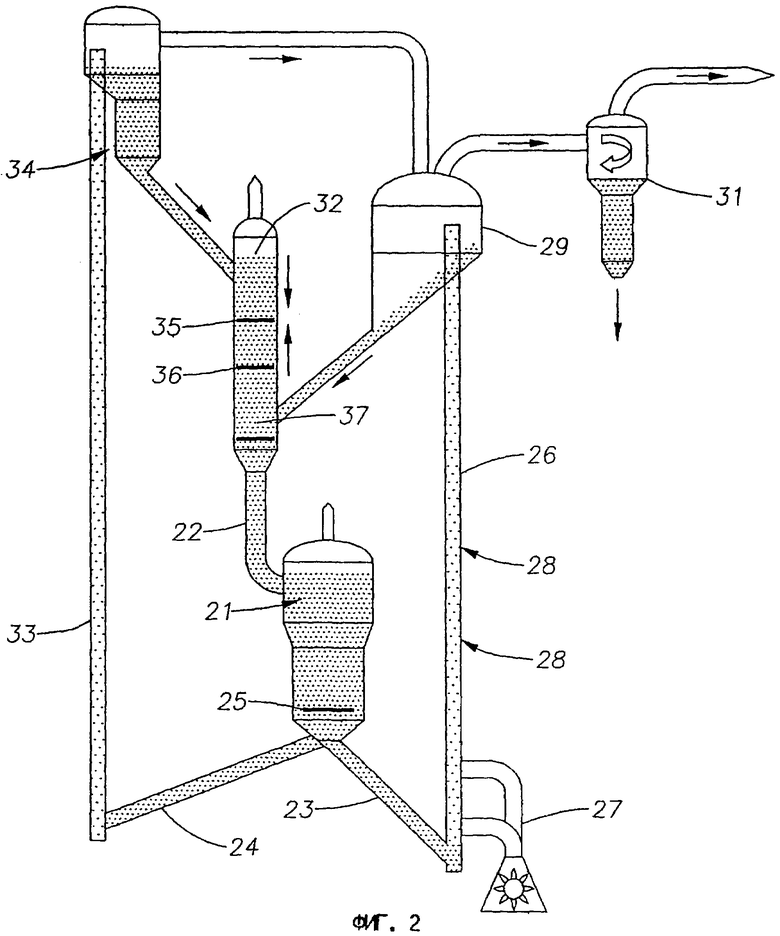
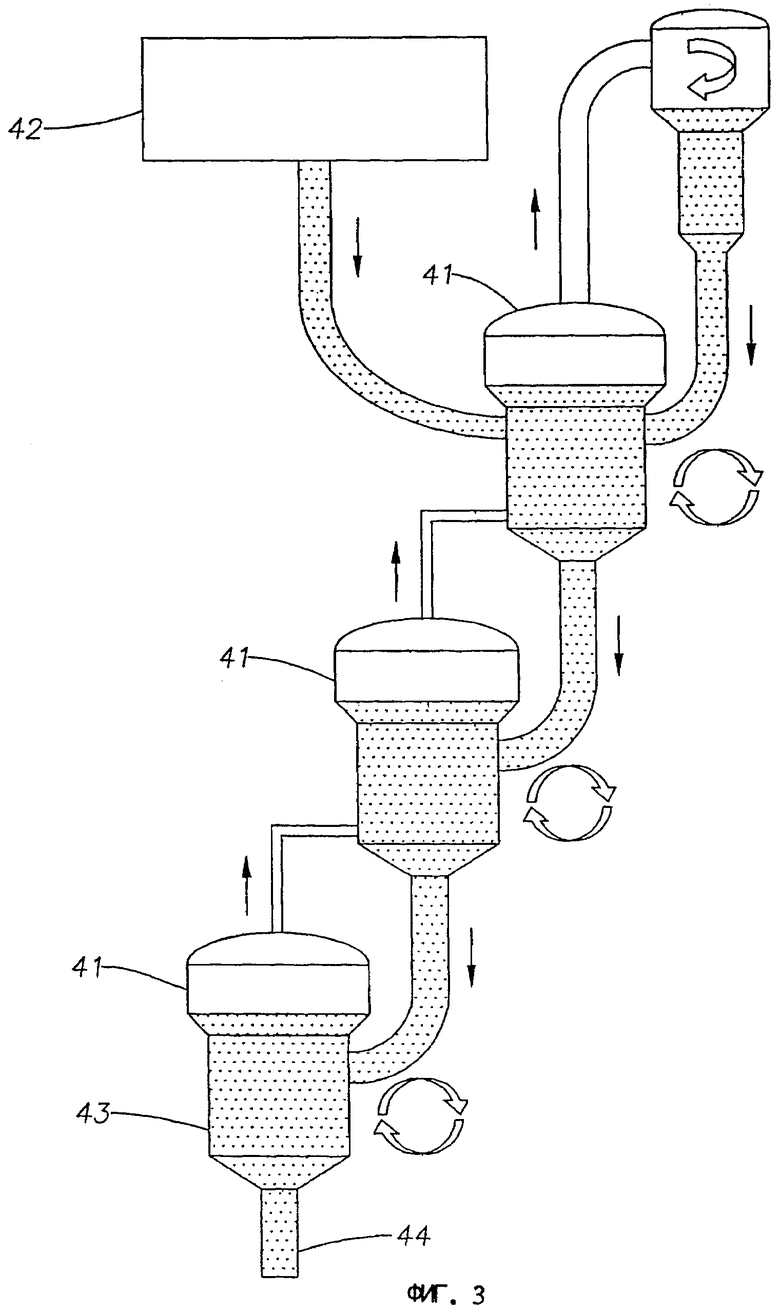
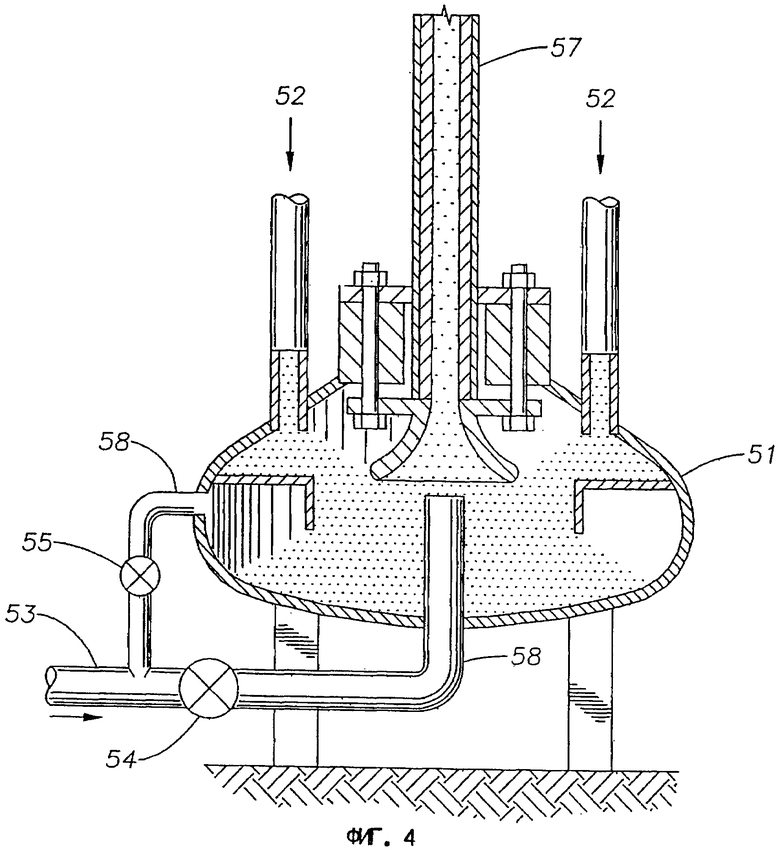
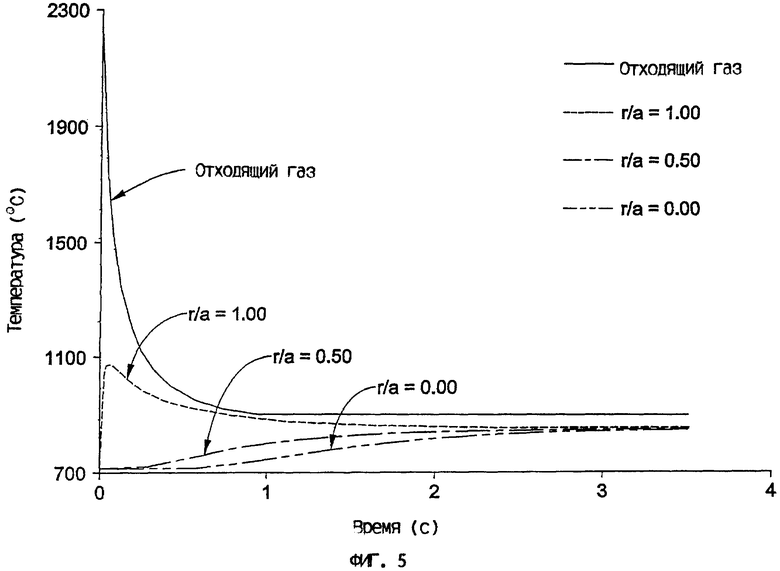
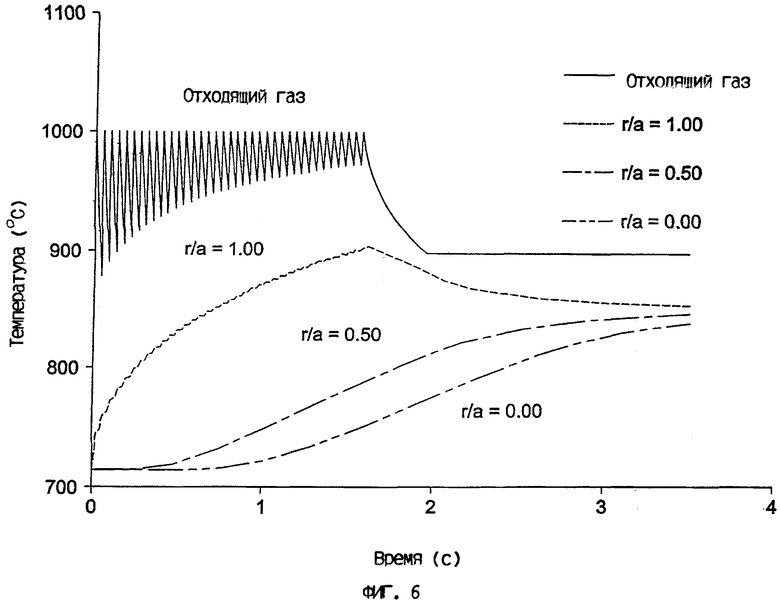
Изобретение относится к способу получения ароматических углеводородов из метана. Предложен способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, при осуществлении которого исходный материал, содержащий метан, вводят в контакт с катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения упомянутого метана в ароматические углеводороды. Первую часть этого катализатора переносят из реакционной зоны в зону нагрева, где первую часть катализатора нагревают введением катализатора в контакт с горячими газообразными продуктами горения, получаемыми сжиганием дополнительного источника топлива, находящегося отдельно от катализатора. Затем нагретую первую часть катализатора возвращают в реакционную зону. Технический результат - повышение срока службы катализатора. 25 з.п. ф-лы, 6 ил., 2 табл.
1. Способ превращения метана в более высокомолекулярные углеводороды, содержащие ароматические углеводороды, включающий:
(а) контактирование исходного материала, содержащего метан, с катализатором дегидроциклизации в реакционной зоне в условиях, эффективных для превращения упомянутого метана в ароматические углеводороды;
(б) перенос первой части упомянутого катализатора из реакционной зоны в зону нагрева;
(в) нагрев первой части катализатора в зоне нагрева введением катализатора в контакт с горячими газообразными продуктами горения, образующимися при сжигании дополнительного источника топлива, причем источник топлива находится отдельно от катализатора; и
(г) возврат нагретой первой части катализатора в реакционную зону.
2. Способ по п.1, в котором при упомянутом нагреве (в) упомянутую первую часть катализатора вводят в контакт непосредственно с упомянутым источником топлива.
3. Способ по п.1, в котором упомянутый источник топлива сжигают в зоне горения отдельно от упомянутой зоны нагрева и газообразные продукты горения, образующиеся в зоне горения, направляют в зону нагрева.
4. Способ по п.1, в котором упомянутый дополнительный источник топлива включает углеводород и/или водород.
5. Способ по п.4, в котором упомянутый дополнительный источник топлива включает углеводород, и упомянутый углеводород сжигают в бедной кислородом атмосфере с получением синтез-газа.
6. Способ по п.1, в котором упомянутая зона нагрева является удлиненной, и тепло упомянутой первой части катализатора передают на множестве участков, отделенных промежутками по длине зоны нагрева.
7. Способ по п.6, в котором, по существу, все дополнительное топливо подают в один конец зоны нагрева, а кислородсодержащий газ подают по нарастающей в упомянутую зону нагрева на упомянутом множестве отделенных промежутками участков.
8. Способ по п.6, в котором горячие газообразные продукты горения, образующиеся в зоне горения отдельно от упомянутой зоны нагрева, подают к упомянутому множеству отделенных промежутками участков.
9. Способ по п.1, в котором упомянутая зона нагрева представляет собой вертикальную трубу, и упомянутую первую часть катализатора направляют вверх по этой вертикальной трубе.
10. Способ по п.1, в котором упомянутая первая часть катализатора при поступлении в упомянутую зону нагрева находится при температуре от примерно 500 до примерно 900°С, а при покидании упомянутой зоны нагрева находится при температуре от примерно 800 до примерно 1000°С.
11. Способ по п.1, в котором упомянутые горячие газообразные продукты горения находятся при температуре ниже 1300°С.
12. Способ по п.1, дополнительно включающий обработку упомянутой первой части катализатора на стадии отделения для по меньшей мере частичного удаления из нее кокса и/или тяжелых углеводородов.
13. Способ по п.12, в котором упомянутая стадия отделения включает контактирование упомянутой первой части катализатора с водяным паром, водородом и/или CO2.
14. Способ по п.12 или 13, в котором упомянутое отделение осуществляют после упомянутого нагрева (в).
15. Способ по п.1, дополнительно включающий контактирование нагретой первой части катализатора с метаном для по меньшей мере частичного удаления из нее адсорбированных воды и/или кислорода.
16. Способ по п.1, в котором упомянутый катализатор включает металл, и нагретую первую часть катализатора подвергают обработке на стадии закоксовывания - перед повторным введением в реакционную зону.
17. Способ по п.1, дополнительно включающий перенос второй части катализатора в зону регенерирования отдельно от упомянутой зоны нагрева и контактирование упомянутой второй части катализатора с регенерирующим газом в упомянутой зоне регенерирования для удаления кокса, образуемого упомянутым контактированием (а).
18. Способ по п.17, в котором температура в упомянутой зоне регенерирования составляет от примерно 400 до примерно 700°С.
19. Способ по п.17 или 18, в котором отношение массы катализатора, переносимого в данное время в зону нагрева, к массе катализатора, переносимого в то же время в зону регенерирования, находится в интервале от примерно 5:1 до примерно 100:1.
20. Способ по п.17, в котором упомянутый регенерирующий газ содержит кислород.
21. Способ по п.17, в котором упомянутая зона регенерирования представляет собой вертикальную трубу или подвижный слой.
22. Способ по п.17, в котором регенерированную вторую часть катализатора подвергают обработке на стадии закоксовывания перед повторным введением регенерированного катализатора в реакционную зону.
23. Способ по п.1, в котором упомянутый исходный материал в упомянутой реакционной зоне вводят в контакт с подвижным слоем упомянутого катализатора дегидроциклизации.
24. Способ по п.17, в котором упомянутая реакционная зона включает вертикально расположенный реактор с отстойным слоем, где упомянутый исходный материал поступает в реактор по месту или вблизи основания реактора, а нагретую первую часть катализатора и регенерированную вторую часть катализатора возвращают в реактор по месту или вблизи верхней части реактора.
25. Способ по п.1, в котором упомянутый исходный материал в упомянутой реакционной зоне вводят в контакт с одним или несколькими псевдоожиженными слоями упомянутого катализатора дегидроциклизации.
26. Способ по п.25, в котором упомянутая реакционная зона включает несколько соединенных в последовательный ряд реакторов с псевдоожиженным слоем, в которых нагретую первую часть катализатора направляют в первый реактор в упомянутом ряду и она движется по принципу противотока относительно упомянутого исходного материала, который вводят в конечный реактор в упомянутом ряду.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 2003144565 A1, 31.07.2003 | |||
| US 5026937 A, 25.06.1991 | |||
| СПОСОБ ПОЛУЧЕНИЯ СМЕСИ НАФТАЛИНОВЫХ УГЛЕВОДОРОДОВ ИЗ ЛЕГКИХ УГЛЕВОДОРОДНЫХ ГАЗОВ (ВАРИАНТЫ) | 2003 |
|
RU2227793C1 |

