ОБЛАСТЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Данное изобретение относится к способу получения органического соединения, которое соответствующим образом может быть использовано для получения органического соединения, помеченного радиоактивным галогеном. Указанное соединение представляет собой действующее вещество агентов, используемых для позитронно-эмиссионной томографии (PET).
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Анализ с помощью медицинской радиологии дает возможность диагностирования путем внутривенного введения в человеческое тело агента, содержащего в качестве действующего вещества соединение, помеченное специфическим радиоизотопом в качестве действующего вещества (в дальнейшем называемое радиофармацевтическим препаратом), и регистрации излучения, испускаемого соединением, с последующим отображением информации, получаемой от излучения. Анализ при помощи медицинской радиологии эффективен при диагностировании спектра заболеваний, включающих заболевания сердца и рак, и характеризуется не только высокой специфичностью и чувствительностью к заболеваниям, но также преимуществом в получении информации о характере повреждений, связанных с другими способами анализа.
Кроме того, радиофармацевтические препараты получают при помощи введения метки в непомеченное соединение (в дальнейшем называемого "предшественник для введения метки"), при необходимости, и осуществления подготовительных процессов, таких как очистка.
Следовательно, предшественник для введения метки очень важен как сырье для синтеза действующего начала, и способ получения недорогого предшественника для введения метки в большом количестве необходим в целях разработки радиофармацевтических препаратов.
В последнее время (18F)1-амино-3-фторциклобутанкарбоновая кислота (в дальнейшем называемая (18F)-FACBC) исследуется и разрабатывается как новейший препарат для радио. Ожидается разработка (18F)-FACBC как агента для диагностирования опухолей, так как ее изучают на предмет способности поглощения ее клеткой через аминокислотный переносчик, в особенности высокопролиферативными опухолевыми клетками, в которых активно идут процессы синтеза.
Известно, что (18F)-FACBC существует в форме двух разных стереоизомеров, называемых син-формой и анти-формой (в дальнейшем называемые син-(18F)-FACBC и анти-(18F)-FACBC). Так как стереоизомеры, как правило, различны по физическим и химическим свойствам, син-(18F)-FACBC и анти-(18F)-FACBC определяются как разные соединения при разработке медицинских препаратов. Следовательно, когда (18F)-FACBC используется как действующее вещество медицинского препарата, в состав этого препарата должен входить практически только один из стереоизомеров.
С другой стороны, для (18F)-FACBC важно найти способ получения большого количества недорогого помечаемого соединения.
Как правило, в качестве предшественников для введения метки радиоактивных соединений, помеченных 18F, используются вещества, в которых на помечаемые участки вводится уходящая группа, такая как трифлат, и такие соединения обычно получают следующим образом. Сначала синтезируется вещество, замещаемое OH-группой в позиции 18F. Затем уходящая группа, такая как трифлат, присоединяют к OH-группе, после чего при необходимости вводится защитная группа. Такой же способ синтеза описан для предшественника (18F)-FACBC для введения метки (патентный документ 1).
Кстати, в случае (18F)-FACBC на стадии введения 18F происходит инверсия конфигурации. Следовательно, к примеру, при синтезе предшественника для введения метки, используемого для синтеза анти-(18F)-FACBC, должно быть синтезировано соединение, в котором OH-группа введена в син-положение, положение 18F. Аналогично, при синтезе предшественника для введения метки, используемого в синтезе син-(18F)-FACBC, должно быть синтезировано соединение, в котором OH-группа введена в анти-положение.
Для удобства названия соединений в дальнейшем сокращаются и обозначаются следующим образом. В соответствии с требованиями перед названиями ставятся приставки "анти-" или "син-", которые указывают на стереоизомер, а случай, когда они не ставятся, означает смесь стереоизомеров.
Защищенная форма: FACBC, в которой аминогруппа и карбоксильная группа защищены.
OH-форма: защищенная форма, в которую введена OH-группа в положение, в которое должен быть введен 18F.
Аддукт уходящей группы: OH-форма, в которую ее OH-группа соединена с уходящей группой.
Международная публикация № WO97/017092 описывает способ синтеза метилового эфира 1-третбутилкарбамат-3-трифторметансульфонокси-циклобутан-1-карбоновой кислоты в качестве предшественника для введения метки (18F)-FACBC (патентный документ 1).
Международная публикация № WO04/056725 описывает способ синтеза метилового эфира син-1-трет-бутоксикарбониламино-3-(1,2,3,4-тетрафтор-2-(1,1,2,2-тетрафторо-2-йодоэтокси)этансульфонилокси)-циклобутановой карбоновой кислоты как предшественника для введения метки анти-(18F)-FACBC. Как уже упоминалось выше, это способ синтеза син-OH-формы и затем синтеза аддукта уходящей син-группы.
Патентный документ 1: Международная публикация № WO97/017092, брошюра
Патентный документ 2: Международная публикация № WO04/056725, брошюра
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
ЗАДАЧА, РЕШАЕМАЯ ИЗОБРЕТЕНИЕМ
Как упоминалось выше, Международная публикация No. WO97/017092 (брошюра) описывает способ синтеза помечаемого предшественника (18F)-FACBC. Однако публикация не проводит различий между стереоизомерами на всех стадиях.
Международная публикация № WO 04/056725 (брошюра) описывает способ синтеза син-1-трет-бутоксикарбониламино-3-(1,2,3,4-тетрафторо-2-(1,1,2,2-тетрафтор-2-йодэтокси)этансульфонилокси)-циклобутановой карбоновой кислоты как помечаемого предшественника анти-(18F)-FACBC. Однако этот способ усложнен тем, что каждая стадия до синтеза анти-OH-формы требует очистки от стереоизомера.
Описанные выше обстоятельства обусловили создание настоящего изобретения и помогло разработке простого способа получения аддукта уходящей син-группы как предшественника анти-(18F)-FACBC.
СРЕДСТВА РЕШЕНИЯ ЗАДАЧИ
В результате кропотливых исследований авторы настоящего изобретения обнаружили, что аддукт уходящей син-группы и аддукт уходящей анти-группы обладают различной способностью вступать в реакцию с основанием. Когда специальное основание добавляется в реакционную систему на стадии введения уходящей группы, аддукт уходящей син-группы, который нерастворим в воде, не демонстрирует способности вступать в реакцию с основанием и является стабильным. С другой стороны, аддукт уходящей анти-группы легко реагирует с основанием и превращается в водорастворимое соединение. Путем выбора способа очистки, в котором используется различие в растворимости аддуктов уходящих групп, аддукт уходящей син-группы может быть избирательно получен без сложной очистки даже из смеси син-OH-формы и анти-OH-формы при добавлении к ним специального основания, чтобы повлиять на реакцию на стадии введения уходящей группы.
Данный способ получения соединения-предшественника анти-(18F)-FACBC отличается наличием:
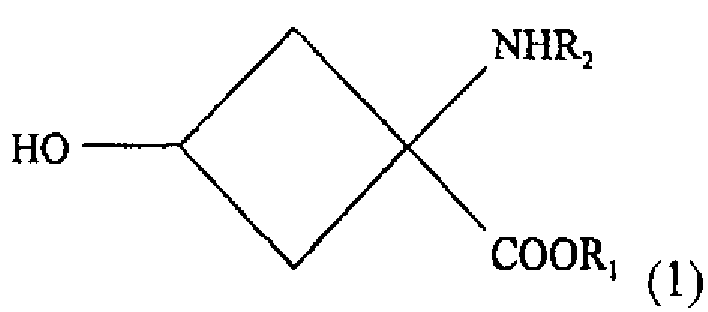
- стадии взаимодействия, обеспечивающей условия для взаимодействия раствора, содержащего соединение со следующей химической формулой (1):

вместе с соединением, реагирующим с OH-группой соединения, представленного формулой (1) с превращением в уходящую группу, и основанием, и
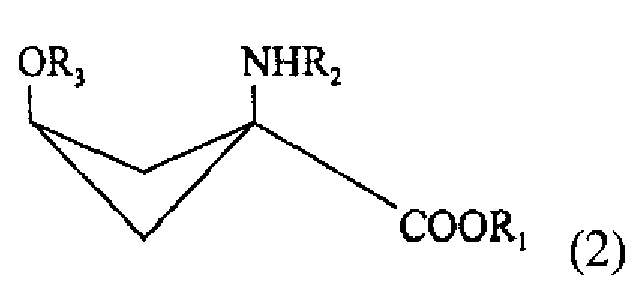
- стадии очистки реакционного раствора, для получения практически индивидуального стереоизомера соединения со следующей химической формулой (2):

R1 обозначает защитную группу карбоксильной группы, и ее выбор специально не ограничен при условии, что это защитная группа, используемая для карбоксильной группы. Желательно использовать линейную или разветвленную алкильную группу с 1-5 атомами углерода или ароматический заместитель. Более желательно использовать использовать метильную, этильную, трет-бутильную или фенильную группу.
R2 обозначает защитную группу аминогруппы, и ее выбор не ограничен специально при условии, что это защищающая группа, используемая для аминогруппы. Желательно использовать линейный или разветвленный алкилоксикарбонильный заместитель с 2-7 атомами углерода, линейный или разветвленный алкенилоксикарбонильный заместитель с 3-7 атомами углерода, насыщенную или ненасыщенную алкилкарбонильную группу с 2-7 атомами углерода, бензилоксикарбонильный заместитель с 7-12 атомами углерода, алкилдитиооксикарбонильный заместитель с 2-7 атомами углерода, линейный или разветвленный алкиламидный заместитель с 1-6 атомами углерода, линейный или разветвленный алкениламидный заместитель с 2-6 атомами углерода, бензамидный заместитель с 6-11 атомами углерода, ароматический иминовый заместитель с 6-11 атомами углерода, линейный или разветвленный алкиламиновый заместитель с 1-6 атомами углерода, линейный или разветвленный алкиленаминовый заместитель с 2-6 атомами углерода, или бензиламиновый заместитель с 6-11 атомами углерода. Более желательно использовать трет-бутоксикарбонильную, арилоксикарбонильную группу или н-бензальаминовый заместитель.
Примеры соединения, реагирующего с OH-группой, представленного химической формулой (1) с получением уходящей группы на стадии взаимодействия, включают линейную или разветвленную алкилсульфоновую кислоту с 1-10 атомами углерода, линейную или разветвленную галоалкилсульфоновую кислоту с 1-9 атомами углерода, ароматическую сульфоновую кислоту и хлорид ароматической сульфоновой кислоты. В качестве линейной или разветвленной галоалкилсульфоновой кислоты с 1-10 атомами углерода желательно использовать метансульфоновую кислоту. В качестве линейной или разветвленной галоалкилсульфоновой кислоты с 1-9 атомами углерода желательно исползовать трифторметансульфоновую кислоту. В качестве ароматической сульфоновой кислоты желательно использовать соединение, выбранное из группы, состоящей из толуолсульфоновой кислоты, нитробензолсульфоновой кислоты и бензолсульфоновой кислоты. В качестве хлорида ароматической сульфоновой кислоты желательно использовать соединение, выбранное из группы, состоящей из хлорида толуолсульфоновой кислоты, хлорида бензолсульфоновой кислоты и хлорида нитробензолсульфоновой кислоты.
Примерами основания, добавляемого на стадии реакции, служат одно или более отобраных из группы, состоящей из линейного или разветвленного алкиламина, от первичного до третичного, с 1-10 атомами углерода; азотсодержащего гетероциклического соединения с 2-20 атомами углерода и азотсодержащего гетероароматического соединения с 2-20 атомами углерода. В качестве алкиламина, от первичного до четвертичного, желательно выбрать этиламин, пропиламин, изопропиламин, диметиламин, диэтиламин, дипропиламин, диизопроиламин, триметиламин, триэтиламин или трипропиламин. В качестве азотсодержащего гетероциклического соединения желательно выбрать пирролидин, имидазолин, пиразолидин, пиперидин, пиперазин, 1,4-диазабицикло(2.2.2.)октан, пирролин, азиридин или 1,8-диазабицикло(5.4.0)ундека-7-ен. В качестве азотсодержащего гетероароматического соединения желательно выбрать пиррол, пиридин, пиримидин, пиразин, триазин, имидазол или пиразол. Более желательно использовать пиридин.
Выбор растворителя, используемого на стадии реакции, не ограничен специально при условии, что это растворитель, который может растворить соединение, представленное формулой (1), так же, как и соединение, реагирующее с OH-группой этого соединения с последующим превращением в уходящую группу и основание, и не реагирует с этими соединениями. Желательно использовать полярный растворитель, более желательно использовать хлороформ и пиридин.
Желательно, чтобы температура реакции была ниже комнатной, более желательно, не более 4°C, а лучше всего - около 0°C. В данном случае следует отметить, что если температура реакции слишком высокая, это часто приводит к образованию побочных продуктов. При этом температура реакции должна быть не ниже температуры замерзания растворителя. Однако следует отметить, что при слишком низкой температуре реакции ее протекание часто становится слишком медленным. Желаемый вариант осуществления включает в себя способ проведения реакции при регулируемой температуре на ледяной бане или в охлажденном растворителе. Время реакции не ограничено специально при условии, что это достаточное время для проведения реакции.
В качестве процесса очистки на стадии очистки могут быть применены различные процессы, использующие разницу в растворимости. Желательно применить процесс жидкость-жидкостной экстракции, процесс разделения на колонке или процесс очистки суспензий. Более желательно применить процесс жидкость-жидкостной экстракции.
ДЕЙСТВИЕ ИЗОБРЕТЕНИЯ
В соответствии с настоящим изобретением стало возможным легко и эффективно получать аддукт уходящей син-группы, представляющий собой помечаемый предшественник, удобный для получения анти-(18F)-FACBC.
ЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее будет описан процесс получения соединения данного изобретения, причем в качестве примера будет взято получение этилового эфира син-1-(N-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)-циклобутан-1-карбоновой кислоты.
Сначала раствор 5-(3-бензилоксициклобутан)гидантоина нагревают с обратным холодильником в насыщенном растворе гидроксида бария, и в нагретый раствор добавляют серную кислоту, чтобы привести значение его pH примерно к 7. Затем раствор фильтруют и фильтрат концентрируют, чтобы позволить 1-амино-3-бензилоксициклобутан-1-карбоновой кислоте выпасть в осадок в виде белых кристаллов (стадия 1). Кислота, используемая для регулировки pH, может быть другой кислотой, а не только серной, но она должна образовывать нерастворимую в воде неорганическую соль с барием. При этом в настоящем изобретении стереоизомер 5-(3-бензилоксициклобутан)гидантоина, используемый на данной стадии, не требует очистки, и можно использовать смесь син-формы и анти-формы как таковую.
Далее получаемую 1-амино-3-бензилоксициклобутан-1-карбоновую кислоту полностью высушивают, чтобы удалить воду и затем растворяют в этаноле. Далее в этаноловый раствор добавляют основание и хлористый тионил в указанном порядке, размешивают при комнатной температуре и затем нагревают с обратным холодильником при 95°С. После того как реакция проходит полностью, раствор концентрируют при пониженном давлении, чтобы получить этиловый эфир 1-амино-3-бензилоксициклобутан-1-карбоновой кислоты в виде белых кристаллов (стадия 2).
Основание, добавляемое в реакционный раствор на данной стадии, может быть любым основанием, способным связать соляную кислоту, образующуюся в процессе реакции. Желательно использовать триэтиламин. Необходимое количество основания такое же, как, или больше, чем количество хлористого тионила.
Количество хлористого тионила должно быть таким же, как, или больше, чем количество исходного вещества в реакции, а именно 1-амино-3-бензилоксициклобутан-1-карбоновой кислоты. Если количество хлористого тионила слишком мало, этерификация этиловым спиртом может проходить недостаточно, что невыгодно. Если количество хлористого тионила слишком велико, образуется избыточная соляная кислота и, таким образом, возникает необходимость в большем количестве основания, что невыгодно. В желательных вариантах осуществления количество хлористого тионила равно или меньше 5 эквивалентам 1-амино-3-бензилоксициклобутан-1-карбоновой кислоты.
Затем этиловый эфир 1-амино-3-бензилоксициклобутан-1-карбоновой кислоты добавляют к раствору малого количества основания в спиртовом растворителе, таком как этанол. Получаемую смесь размешивают при охлаждении и к смеси добавляют трет-бутиловый бикарбонат, для того чтобы позволить им прореагировать при комнатной температуре (Фиг.1, стадия 3). В качестве спиртового растворителя желательно использовать этанол, хотя можно использовать различные спирты.
Количество основания должно быть достаточно малым относительно количества спирта, но если оно слишком мало, протекание реакции становится невыгодно медленным. В желательном варианте осуществления используют отношение спирта к основанию, равное 9:1. Количество трет-бутилового бикарбоната должно быть равно одному эквиваленту 1-амино-3-бензилоксициклобутан-1-карбоновой кислоты или более, желательно 1,5 эквивалентов 1-амино-3-бензилоксициклобутан-1-карбоновой кислоты.
Эта процедура дает возможность получить этиловый эфир 1-(н-(трет-бутоксикарбонил)амино)-3-бензилоксициклобутан-1-карбоновой кислоты.
Этиловый эфир 1-(н-(трет-бутоксикарбонил)амино)-3-бензилоксициклобутан-1-карбоновой кислоты, синтезированный согласно описанному выше, растворяют в спиртовом растворителе, таком как этанол, или уксусно-эфирном растворителе, таком как этиловый эфир уксусной кислоты, и в раствор добавляют палладий на активированном угле (в количестве 10% или более по весу относительно субстрата) в атмосфере водорода, чтобы позволить им прореагировать при размешивании при комнатной температуре. Затем реакционный раствор фильтруют через селит, фильтрат концентрируют и очищают, чтобы получить этиловый эфир 1-(н-(трет-бутоксикарбонил)амино)-3-гидроксициклобутан-1-карбоновой кислоты (стадия 4).
Полученный этиловый эфир 1-(н-(трет-бутоксикарбонил)амино)-3-гидроксициклобутан-1-карбоновой кислоты растворяют в органическом растворителе, таком как диэтиловый эфир. Затем путем добавления основания и трифторметансульфонового ангидрида к получаемому раствору и обеспечения реакционных условий получают этиловый эфир син-1-(н-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)циклобутан-1-карбоновой кислоты (стадия 5).
В настоящем изобретении используемый на данной стадии этиловый эфир 1-(н-(трет-бутоксикарбонил)амино)-3-гидроксициклобутан-1-карбоновой кислоты не требует очистки, и смесь син-форма/анти-форма может быть использована как таковая, в отличие от способов, описанных ранее (например согласно способу, описанному в статье (международная публикация № WO04/056725, брошюра)).
В качестве основания может быть использован алкиламин, азотсодержащее гетероциклическое соединение и азотсодержащее гетероароматическое соединение. В желательном варианте осуществления в качестве растворителя используется пиридин, чтобы, кроме того, растворять основание.
И наконец, путем очистки реакционного раствора требуемое соединение этиловый эфир син-1-(н-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)-циклобутан-1-карбоновой кислоты может быть получено как практически единственный стереоизомер. В качестве желательного способа очистки может быть применен процесс экстракции жидкость-жидкость, в котором воду, органический растворитель, такой как эфир, и кислоту добавляют в реакционный раствор для очистки органического слоя.
В настоящем изобретении могут быть синтезированы соединения, отличные от описанных выше, по стадиям, сходным с вышеописанными. К примеру, когда синтезируется соединение, в котором заместитель в виде эфира галоалкилсульфоновой кислоты, отличный от трифлата, заместитель в виде алкилсульфоновой кислоты или заместитель в виде эфира ароматической сульфоновой кислоты связан с атомом углерода в положении 3 циклобутанового кольца, реакция на стадии 5 может быть проведена таким же образом, как описано выше, за исключением того, что вместо трифторметансульфонового ангидрида используется другой галогенсульфонил или сульфоновый ангидрид.
При синтезе соединения, в котором алкилоксикарбонильный заместитель, отличный от трет-бутоксикарбонильного, алкенилоксикарбонильный заместитель или бензилоксикарбонильный заместитель связаны с аминогруппой в положении 1, реакция на вышеописанном стадии 3 может быть проведена с применением различных алкилхлороформиатов, алкенилхлороформиатов или бензилхлороформиатов соответственно, вместо трет-бутильного бикарбоната. Схожим образом соединение, в котором ароматический иминовый заместитель связан с аминогруппой, может быть синтезировано путем проведения реакции бензальдегида, имеющего заместитель, с аминогруппой на стадии 3.
Когда синтезируются 1-пропилэфировая форма и изопропилэфировая форма, в качестве спирта для реакции на вышеописанной стадии 2 могут быть использованы соответственно 1-пропанол и изопропанол.
Далее в качестве примера использования новейших аминокислотных органических соединений в соответствии настоящим изобретением, будет описан способ, по которому можно синтезировать анти-(18F)-FACBC с использованием синтезированного ранее этилового эфира син-1-(н-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)-циклобутан-1-карбоновой кислоты.
Синтез анти-(18F)-FACBC проводится в две стадии: стадия присоединения радиоактивного фтора к предшественнику и стадия снятия защитных групп с соединения, к которому был присоединен радиоактивный фтор.
Радиоактивный фтор может быть получен каким-либо известным способом, например способом, в котором воду, обогащенную H2 18O используют как мишень и подвергают протонной бомбардировке. В этом случае радиоактивный фтор присутствует в воде, обогащенной H2 18O, использованной в качестве мишени. Воду, обогащенную H2 18O и содержащую радиоактивный фтор пропускают, к примеру, через хроматографическую колонку с анионообменной смолой, так что радиоактивный фтор адсорбируется и собирается на колонке, таким образом отделяясь от воды, обогащенной H2 18O. Далее через колонку пропускают раствор карбоната калия, для того чтобы элюировать радиоактивный фтор, и к элюату добавляют катализатор межфазного переноса, после чего элюат выпаривают досуха, таким образом активируя радиоактивный фтор.
Затем высушенный радиоактивный фтор растворяют в ацетонитриле и к ацетонитриловому раствору добавляют этиловый эфир син-1-(н-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)-циклобутан-1-карбоновой кислоты в качестве предшественника, чтобы позволить им прореагировать при нагревании. В результате радиоактивный фтор присоединяется к предшественнику, и, таким образом, синтезируется этиловый эфир анти-(18F)-1-(N-(трет-бутоксикарбонил)амино)-3-фторциклобутан-1-карбоновой кислоты.
Получаемый этиловый эфир син-1-(н-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)-циклобутан-1-карбоновой кислоты деэтерифицируют и лишают защитных групп для того, чтобы получить анти-(18F)-FACBC как требуемое соединение. Дезэтерификация и снятие защитных групп могут быть выполнены, к примеру, способом, описанным в статье (патентный документ; международная публикация No. WO97/017092, брошюра). Количество добавляемой кислоты не требует ограничений, при условии, что это количество обеспечивает кислотность, достаточную для снятия защитных групп.
ПРИМЕР
Далее настоящее изобретение будет описано более подробно со ссылкой на Примеры; однако следует понимать, что особенности Примеров не накладывают ограничений на настоящее изобретение.
Пример 1. Синтез этилового эфира син-1-(N-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)-циклобутан-1-карбоновой кислоты.
Гидролиз син-гидантоина (Фиг.1, стадия 1)
250 мл насыщенного раствора гидроксида бария добавили к 6,15 г (что соответствует 25 ммоль) 5-(3-бензилоксициклобутан)гидантоина и нагрели с обратным холодильником на масляной бане при 114°С в течение 24 часов или более, для того чтобы выделить 1-амино-3-бензилоксициклобутан-1-карбоновую кислоту. Затем применили анализ ТСХ с использованием в качестве подвижных растворителей двух видов систем: хлороформ/метанол = 5/1 (значение R f син-гидантоина = около 0,6) и хлороформ/метанол = 95/1 (значение R f син-гидантоина = около 0,3), и завершение реакции было подтверждено (путем окрашивания фосфомолибденовой кислотой в УФ). При этом соотношение син/анти-форм гидантоина, использованного в реакции, было около 65/35.
После того как завершение реакции было подтверждено, реакционный раствор охладили до комнатной температуры и добавили около 27 мл серной кислоты (1 моль/мл), чтобы нейтрализовать реакционный раствор. После нейтрализации реакционный раствор размешивали при комнатной температуре в течение 5 минут и образовавшийся осадок удалили путем фильтрации. Фильтрат сконцентрировали, получив 19,07 г твердого белого вещества - смеси 1-амино-3-бензилоксициклобутан-1-карбоновой кислоты и неорганической соли.
Этерификация этиловым спиртом (Фиг.1, стадия 2)
19,07 г 1-амино-3-бензилоксициклобутан-1-карбоновой кислоты, полностью высушенной для удаления воды, растворили в 250 мл этанола. К этому раствору добавили 9,5 мл триэтиламина (что соответствует 75 ммоль) и охлаждали при 78°С в течение 20 минут, затем добавили 4,6 мл хлористого тионила (что соответствует 62,5 ммоль). Реакционный раствор размешивали при 0°С в течение 1 часа, затем при комнатной температуре в течение 1 часа, после чего нагревали с обратным холодильником на масляной бане при температуре 95°С в течение ночи. Завершение реакции подтвердили с помощью анализа методом ТСХ, используя в качестве подвижного растворителя хлороформ/метанол = 95/1 (значение R f искомого соединения = около 0,6) (подтвердили путем окрашивания фосфомолибденовой кислотой в УФ). После того, как завершение реакции было подтвержено, реакционный раствор сконцентрировали при пониженном давлении для того, чтобы выделить этиловый эфир 1-амино-3-бензилоксициклобутан-1-карбоновой кислоты.
Добавление Boc (Фиг.1, стадия 3)
7,64 г этилового эфира 1-амино-3-бензилоксициклобутан-1-карбоновой кислоты растворили в 250 мл смеси этанол/триэтиламин = 9/1. После того, как раствор охладили в ледяной бане в течение 15 минут, в раствор добавили 8,6 мл (что соответствует 37,5 ммоль) трет-бутилового бикарбоната и размешивали при комнатной температуре в течение ночи. Завершение реакции было подтверждено с помощью анализа методом ТСХ с использованием подвижного растворителя гексан/этилацетата = 1:1 (значение R f требуемого продукта реакции = около 0,6) (подтвердили путем окрашивания фосфомолибденовой кислотой в УФ). После того, как завершение реакции было подтверждено, реакционный раствор сконцентрировали при пониженном давлении для того, чтобы получить осадок в виде белых кристаллов. К осадку прилили 150 мл охлажденного этилацетата и 150 мл 0,5 моль/л охлажденной соляной кислоты, размешивали в ледяной бане в течение 5 минут и оставили стоять при комнатной температуре до полного разделения. Органический слой извлекли и промыли дважды 150 мл воды, 150 мл насыщенного водного раствора гидрокарбоната натрия, дважды 150 мл воды и 150 мл насыщенного раствора соли в указанном порядке, высушили безводным сульфатом натрия и сконцентрировали при пониженном давлении, чтобы извлечь малое количество желтого маслянистого вещества. С помощью серии операций получили 8,82 г желтого маслянистого вещества. После того, как с помощью анализа методом ТСХ было подтверждено, что в осадке остались реагенты, осадок в течение небольшого времени очистили с помощью метода колоночной хроматографии с силикагелем (гексан/этилацетат = 1/1), получив 4,9282 г (что соответствует 14 ммоль) этилового эфира 1-(н-(трет-бутоксикарбонил)амино)-3-бензилоксициклобутан-1-карбоновой кислоты в виде белых кристаллов.
Дебензилирование (Фиг.1, стадия 4)
К 4,9282 г (что соответствует 14 ммоль) этилового эфира 1-(н-(трет-бутоксикарбонил)амино)-3-бензилоксициклобутан-1-карбоновой кислоты добавили 150 мл этанола, а затем 900 мг палладия на активированном угле (10% палладия) и смесь очистили водородом, а затем размешивали при комнатной температуре в течение ночи. После окончания реакции палладий удалили путем фильтрации с использованием селита, а фильтрат сконцентрировали при пониженном давлении, получив осадок в виде 5,74 г белых кристаллов. Реакцию контролировали с помощью анализа методом ТСХ, используя в качестве подвижной фазы гексан/этилацетат = 1/1(значение R f искомого продукта = около 0,2) (подтвердили путем окрашивания нингидрином в УФ), для того чтобы подтвердить окончание реакции. Затем осадок отделили и очистили с помощью колоночной хроматографии с силикагелем (гексан/этилацетат = 1/1, гексан/этилацетат = 4/1), получив 1,61 г (что соответствует 6,2 ммоль) этилового эфира 1-(н-(трет-бутоксикарбонил)амино)-3-гидроксициклобутан-1-карбоновой кислоты в виде белых кристаллов.
На основе интегрирования Н-ЯМР транс-протонов, связанных с атомом углерода в третьем положении в син-форме, и цис-протонов, связанных с атомом углерода в третьем положении в анти-форме, подсчитали, что молярное соотношение син-формы и анти-формы равно 14/46 для полученного этилового эфира 1-(н-(трет-бутоксикарбонил)амино)-3-гидроксициклобутан-1-карбоновой кислоты.
Присоединение трифлата (Фиг.1, стадия 5)
259 мг (1 ммоль; отношение син/анти в начале реакции было 14/46) этилового эфира 1-(н-(трет-бутоксикарбонил)амино)-3-гидроксициклобутан-1-карбоновой кислоты растворили в 4 мл пиридина и перемешивали в ледяной бане 20 минут. Потом к раствору добавили 0,26 мл (что соответствует 1,5 ммоль) трифторметансульфонового ангидрида, а затем смесь в неизменном виде размешивали 30 минут. Реакцию контролировали с помощью анализа методом ТСХ, используя в качестве подвижной фазы растворителя гексан/диэтиловый эфир = 1/1 (значение R f искомого продукта = около 0,6) (подтвердили путем окрашивания нингидрином в УФ), чтобы подтвердить окончание реакции. Анализ методом ТСХ показал не только пятно при значении R f=0,4, означающее этиловый эфир син-1-(н-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)-циклобутан-1-карбоновой кислоты, но также пятно в исходной точке (гексан/диэтиловый эфир = 1/1). После того как завершение реакции было подтверждено, в реакционный раствор добавили 50 мл воды и 50 мл эфира, а потом провели экстакцию и промывание - дважды с 50 мл 1 моль/л соляной кислоты, дважды с 50 мл воды и дважды с насыщенным солевым раствором в указанном порядке. После высушивания безводным сульфатом натрия провели концентрацию при пониженном давлении, получив 297,2 мг светло-желтых кристаллов. TLC показал, что пятно в области исходной точки исчезло и, таким образом, не осталось других пятен, кроме пятна при значении R f=0,4 (гексан/диэтиловый эфир = 1/1). Выпавший в результате реакции осадок отделили и очистили с помощью колоночной хроматографии с силикагелем (гексан/диэтиловый эфир = 3:1), получив 222,8 мг белых кристаллов, представляющих собой этиловый эфир син-1-(н-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)-циклобутан-1-карбоновой кислоты, что было подтверждено с помощью анализа методом ЯМР.
Результаты измерений методом ЯМР (внутренний стандарт: тетраметилсилан) полученного этилового эфира син-1-(н-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)-циклобутан-1-карбоновой кислоты были следующими.
Для анализа ЯМР использовали: JNM-ECP-500 (произведено JEOL, Ltd.)
1H-ЯМР (растворитель: CDCl3, резонансная частота: 500 МГц): δ 5,41-5,35 (м, 1H), 5,32 (б, 1H), 4,26 (кв, 2H, J=7 Гц), 3,10-3,02 (м, б, 4Н), 1,45 (с, 9Н), 1,31 (т, 3Н, J=7,0 Гц).
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение может быть применено в области синтеза радиоактивных, помеченных фтором аминокислотных соединений, пригодных для средств ПЭТ, и полезно для применения в области радиофармацевтических препаратов.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 показывает схему синтеза этилового эфира син-1-(н-(трет-бутоксикарбонил)амино)-3-(((трифторметил)сульфонил)окси)-циклобутан-1-карбоновой кислоты в случае, когда в качестве исходного материала используется син/анти гидантоин.
Фиг.2 показывает спектр 1H-ЯМР этилового эфира син-1-(н-(трет-бутоксикарбонил)амино)-3-гидрокси-циклобутан-1-карбоновой кислоты.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ-ПРЕДШЕСТВЕННИК ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ, МЕЧЕННОГО РАДИОАКТИВНЫМ ГАЛОГЕНОМ | 2006 |
|
RU2428415C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАДИОАКТИВНОГО, МЕЧЕННОГО ФТОРОМ ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ | 2008 |
|
RU2476423C2 |
| ОЧИСТКА СОЕДИНЕНИЯ-ПРЕДШЕСТВЕННИКА КРИСТАЛЛИЗАЦИЕЙ | 2011 |
|
RU2586881C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАДИОАКТИВНОГО ФТОР-МЕЧЕННОГО ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ | 2007 |
|
RU2434846C2 |
| УПРОЩЕНИЕ СПОСОБА ПОЛУЧЕНИЯ СОЕДИНЕНИЯ-ПРЕДШЕСТВЕННИКА | 2011 |
|
RU2593372C2 |
| ПОЛУЧЕНИЕ ПРЕДШЕСТВЕННИКА ДЛЯ ПЭТ | 2011 |
|
RU2587308C2 |
| СТЕРЕОСЕЛЕКТИВНЫЙ СИНТЕЗ АМИНОКИСЛОТ ДЛЯ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЯ ОПУХОЛИ | 2006 |
|
RU2376282C2 |
| ПОЛУЧЕНИЕ ПРОИЗВОДНОГО 1-АМИНО-3-ГИДРОКСИЦИКЛОБУТАН-1-КАРБОНОВОЙ КИСЛОТЫ | 2012 |
|
RU2596829C2 |
| Получение 18F-флуцикловина | 2013 |
|
RU2640805C2 |
| КОМПОЗИЦИЯ 18F- ФЛУЦИКЛОВИНА В ЦИТРАТНЫХ БУФЕРАХ | 2012 |
|
RU2623163C2 |


Изобретение относится к способу получения соединения -предшественника радиоактивного соединения, помеченного фтором, формулы (2), который включает: стадию взаимодействия, обеспечивающего условия для взаимодействия раствора, содержащего вещество со следующей химической формулой (I): где R1 обозначает защитную группу карбоксильной группы, a R2 - защитную группу аминогруппы вместе с основанием, выбранным из группы, состоящей из алкиламинов, от первичных до четвертичных, с неразветвленной или разветвленной цепью, с 1-10 атомами углерода, азотсодержащих гетероциклических веществ с 2-20 атомами углерода и азотсодержащих гетероароматических веществ с 2-20 атомами углерода, и соединением, реагирующим с ОН-группой соединения с химической формулой (1), с превращением в уходящую группу, выбранным из группы, состоящей из алкилсульфоновой кислоты с неразветвленной или разветвленной цепью из 1-10 атомов углерода, галоалкилсульфоновой кислоты с неразветвленной или разветвленной цепью из 1-9 атомов углерода, ароматической сульфоновой кислоты и хлорида ароматической сульфоновой кислоты; а также - стадию очистки реакционного раствора, получаемого на стадии взаимодействия, для получения практически индивидуального стереоизомера вещества со следующей химической формулой (2), где R1 обозначает защитную группу карбоксильной группы, R2 - защитную группу аминогруппы, a R3 - уходящую группу. Технический результат - разработан способ получения помеченного предшественника, удобного для получения аминокислоты, помеченной радиоактивным фтором, используемой для позитронно-эмиссионной топографии. 8 з.п. ф-лы, 2 ил., 1пр.
1. Способ получения соединения-предшественнника радиоактивного соединения, помеченного фтором, формулы (2)

который включает:
- стадию взаимодействия, обеспечивающего условия для взаимодействия раствора, содержащего вещество со следующей химической формулой (I):

где R1 обозначает защитную группу карбоксильной группы, a R2 - защитную группу аминогруппы вместе с основанием, выбранным из группы, состоящей из алкиламинов, от первичных до четвертичных, с неразветвленной или разветвленной цепью, с 1-10 атомами углерода, азотсодержащих гетероциклических веществ с 2-20 атомами углерода и азотсодержащих гетероароматических веществ с 2-20 атомами углерода, и соединением, реагирующим с ОН-группой соединения с химической формулой (1), с превращением в уходящую группу, выбранным из группы, состоящей из алкилсульфоновой кислоты с неразветвленной или разветвленной цепью из 1-10 атомов углерода, галоалкилсульфоновой кислоты с неразветвленной или разветвленной цепью из 1-9 атомов углерода, ароматической сульфоновой кислоты и хлорида ароматической сульфоновой кислоты; а также
- стадию очистки реакционного раствора, получаемого на стадии взаимодействия, для получения практически индивидуального стереоизомера вещества со следующей химической формулой (2);

где R1 обозначает защитную группу карбоксильной группы, R2 -защитную группу аминогруппы, a R3 - уходящую группу.
2. Способ получения по п.1, в котором алкилсульфоновая кислота является метансульфоновой кислотой.
3. Способ получения по п.1, в котором галоалкилсульфоновая кислота является трифторметансульфоновой кислотой.
4. Способ получения по п.1, в котором ароматическая сульфоновая кислота является толуолсульфоновой кислотой, нитробензолсульфоновой кислотой или бензолсульфоновой кислотой.
5. Способ получения по п.1, в котором хлорид ароматической сульфоновой кислоты представляет собой хлорид толуолсульфоновой кислоты, хлорид бензолсульфоновой кислоты или хлорид нитробензолсульфоновой кислоты.
6. Способ получения по п.1, в котором алкиламин представляет собой этиламин, пропиламин, изопропиламин, диметиламин, диэтиламин, дипропиламин, диизопропиламин, триметиламин, триэтиламин или трипропиламин.
7. Способ получения по п.1, в котором азотсодержащее гетероциклическое вещество представляет собой пирролидин, имидазолин, пиразолидин, пиперидин, пиперазин, 1,4-диазабицикло(2.2.2)октан, пирролин, азиридин, или 1,8-диазабицикло(5.4.0)ундека-7-ен.
8. Способ получения по п.1, в котором азотсодержащее гетероароматическое соединение представляет собой пиррол, пиридин, пиримидин, пиразин, триазин, имидазол или пиразол.
9. Способ получения по пп.1-8, в котором стадия очистки осуществляется с помощью экстракции жидкость-жидкость, разделения с помощью разделительной колонки, очистки суспензии или процесса рекристаллизации.
| WO 9717092 А1, 15.05.1997 | |||
| WO 2004056725 А1, 08.07.2004 | |||
| WO 2006126410 А1, 30.11.2006 | |||
| RU 2003137589 А, 20.05.2005. |

