Область техники
Настоящее изобретение относится к соединению-предшественнику, использование которого может быть подходящим для производства органических соединений, меченных радиоактивным галогеном, или активных ингредиентов для диагностических средств, используемых в позитронно-эмиссионной томографии и однофотонной эмиссионной компьютерной томографии.
Уровень техники
Радиологическое исследование, представленное позитронно-эмиссионной томографией (называемой далее в описании ПЭТ) и однофотонной эмиссионной компьютерной томографией (называемой далее в описании ОЭКТ), является эффективным при диагностике ряда заболеваний, включая сердечную недостаточность и рак. Данные технологии включают введение пациенту агента, меченного специфическим радиоизотопом (называемого далее в описании радиофармпрепарат), с последующим детектированием γ-излучения, испускаемого агентом непосредственно или опосредованно. Радиологическое исследование отличается тем, что оно обладает не только высокой специфичностью и чувствительностью к заболеваниям, а также по сравнению с другими способами исследования обладает тем преимуществом, что обеспечивает информацию о развитии лезий.
Например, [18F]2-фтор-2-дезокси-D-глюкоза (называемая далее в описании “18F-ФДГ”), один из радиофармпрепаратов, используемых для исследования ПЭТ, обладает тенденцией к концентрированию в областях, где метаболизм глюкозы повышен, поэтому возможным становится специфическое детектирование опухолей, в которых повышен метаболизм глюкозы.
Радиологическое исследование проводится путем слежения за распределением введенного радиофармпрепарата, и полученные таким образом данные варьируются в зависимости от природы радиофармпрепарата. Поэтому для различных заболеваний были разработаны различные радиофармпрепараты, и некоторые из них нашли клиническое применение. Были разработаны, например, диагностические средства для различных опухолей, диагностические средства для исследования кровотока и агенты для картирования рецепторов.
В последние годы в качестве новых радиофармпрепаратов была разработана группа соединений аминокислот, меченных радиоактивным галогеном, включающая [18F]1-амино-3-фторциклобутанкарбоновую кислоту (называемая далее в описании [18F]-ФАЦБК), и их клиническое применение находится в стадии разработки (патентный документ 1, и непатентные документы 1 и 2). [18F]-ФАЦБК рассматривается как эффективное диагностическое средство для интенсивно пролиферирующих опухолей, так как она обладает тем свойством, что специфически переносится аминокислотным транспортером.
Из способов получения [18F]-ФАЦБК раскрыты способы, которые включают: предложение метилового эфира 1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты в качестве предшественника для мечения, замещение трифлатной группы в положении 3 предшественника радиоактивным фтором и снятие защитных групп полученного соединения в кислых условиях (патентный документ 1 и непатентные документы 1 и 2).
Патентный документ 1: выложенный патент Японии №2000-500442.
Непатентный документ 1: Jonathan McConathy et al. Improved synthesis of anti-[18F]FACBC: improved preparation of labeling precursor and automated radiosynthesis. Applied Radiation and Isotopes, (Netherlands), 2003, 58, p. 657-666.
Непатентный документ 2: Timothy M. Shoup et al. Synthesis and Evaluation of [18F]1-Amino-3-fluorocyclobutane-1-carboxylic Acid to Image Brain Tumors. The Journal of Nuclear Medicine, 1999, 40. P. 331-338.
Раскрытие изобретения
Проблемы, решаемые при помощи изобретения
Однако исследования, предпринятые авторами настоящего изобретения, показали, что согласно способу получения [18F]-ФАЦБК, раскрытому к настоящему моменту, в полученной [18F]-ФАЦБК остается метанол в качестве удерживаемого растворителя. Метанол определяется как растворитель класса 2 согласно руководству МКГ: “Примеси: Руководство для удерживаемых растворителей”, и рассматривается как растворитель, остаточный уровень которого в лекарственном средстве должен регулироваться.
Настоящее изобретение было разработано с учетом вышеописанных обстоятельств. Соответственно объектом настоящего изобретения является предложение нового органического соединения аминокислоты, которое может быть использовано в качестве соединения-предшественника для введения метки для соединений аминокислоты, меченных радиоактивным галогеном, имеющих циклобутановый циклический остов, включающие [18F]-ФАЦБК, и которое предотвращает удерживание метанола соединениями аминокислоты, меченных радиоактивным галогеном, получаемых таким образом.
Средства для решения проблем
В результате исследования авторы настоящего изобретения обнаружили, что при образовании эфирной связи между атомом углерода в положении 1 циклобутанового кольца и алкильной группой с 2 или 3 атомами углерода возможно предотвращение удерживания метанола синтезированным соединением. Таким образом, достигается осуществление настоящего изобретения.
Согласно настоящему изобретению предлагается соединение-предшественник для органических соединений, меченных радиоактивным галогеном, которое представлено следующей формулой (1):
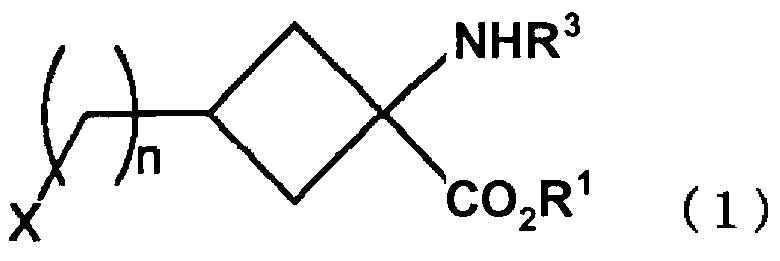
В формуле (1), приведенной выше, n представляет собой целое число 0 или от 1 до 4, приемлемая величина которого может варьироваться в зависимости от типа соединений аминокислоты, меченных радиоактивным галогеном, которые в результате получают. Например, если соединение, получаемое в результате, представляет собой соединение, в котором галоген образует связь непосредственно в положении 3 циклобутанового кольца (например, [18F]-ФАЦБК), то n равно 0, в то время как если соединение, получаемое в результате, представляет собой соединение, в котором галоген образует связь с положением 3 циклобутанового кольца через метиленовую цепь, такое как [18F]1-амино-3-фторметилциклобутанкарбоновая кислота, то n равно 1.
В формуле (1), приведенной выше, R1 представляет собой этильный, 1-пропильный или изопропильный заместитель, и предпочтительно этильный заместитель.
В формуле (1), приведенной выше, X представляет собой галоген или группу, представленную OR2. R2 выбран из группы, состоящей из галогеналкилсульфоновой кислоты с неразветвленной или разветвленной цепью с от одного до 10 атомами углерода, триалкилстаннила с от 3 до 12 атомами углерода, фторсульфоновой кислоты и сульфоновой кислоты, содержащей ароматические заместители, и предпочтительным является заместитель, выбранный из группы, состоящей из толуолсульфоновой кислоты, нитробензолсульфоновой кислоты, бензолсульфоновой кислоты, трифторметансульфоновой кислоты, фторсульфоновой кислоты, перфторалкилсульфоновой кислоты, триметилстанила и триэтилстанила. В качестве заместителя галогена предпочтительно могут быть использованы заместители бром или хлор.
R3 предпочтительно выбран из группы, состоящей из алкилоксикарбонила с неразветвленной или разветвленной цепью с от 2 до 7 атомами углерода, алкенилоксикарбонила с неразветвленной или разветвленной цепью с от 3 до 7 атомами углерода, бензилоксикарбонила, содержащего от 7 до 12 атомов углерода, который может содержать заместитель, алкилдитиооксикарбонила с от 2 до 7 атомами углерода, алкиламида с неразветвленной или разветвленной цепью с от одного до 6 атомами углерода, алкениламида с неразветвленной или разветвленной цепью с от 2 до 6 атомами углерода, бензамида с от 6 до 11 атомами углерода, который может содержать заместитель, циклического имида с от 4 до 10 атомами углерода, ароматического имина с от 6 до 11 атомами углерода, который может содержать заместитель, алкиламина с неразветвленной или разветвленной цепью с от одного до 6 атомами углерода, алкениламина с неразветвленной или разветвленной цепью с от 2 до 6 атомами углерода, и бензиламина с от 6 до 11 атомами углерода, который может содержать заместитель. Предпочтительно R3 представляет собой заместитель, выбранный из группы, состоящей из т-бутоксикарбонильной группы, аллилоксикарбонильной группы, фталимидной группы и N-бензилиденамина, более предпочтительно R3 представляет собой т-бутоксикарбонильную группу или фталимидную группу.
Действие изобретения
Соединение согласно настоящему изобретению может быть использовано в качестве соединения-предшественника для введения метки для соединений аминокислот, меченных радиоактивным галогеном, имеющего циклобутановый остов. При использовании соединения согласно настоящему изобретению в качестве предшественника для введения метки стало возможным предотвратить удерживание метанола получаемыми соединениями аминокислоты, меченными радиоактивным галогеном.
Лучшее воплощение изобретения
Далее, способ получения соединения согласно настоящему изобретению будет описан, с использованием в качестве примера синтеза этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]циклобутан-1-карбоновой кислоты, показанного на фиг. с 1 по 3.
Во-первых, раствор син-5-(3-бензилоксициклобутан)гидантоина в насыщенном растворе гидроксида бария нагревали с обратным холодильником и к раствору, нагреваемому с обратным холодильником, добавляли серную кислоту для доведения его рН до приблизительно 7. Затем раствор фильтровали, и фильтрат концентрировали для осаждения син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты в виде белых кристаллов. Кислота, используемая для доведения рН, может быть кислотой, отличной от серной кислоты, но необходимо, чтобы кислота образовывала неорганическую соль с барием, нерастворимую в воде (фиг. 1, стадия 1).
Син-1-амино-3-бензилоксициклобутан-1-карбоновую кислоту сушили до полного удаления воды и затем растворяли в этаноле. Затем к этанольному раствору добавляли основание и тионилхлорид в указанном порядке, перемешивали при комнатной температуре и затем нагревали с обратным холодильником при приблизительно 95°С. После полного завершения взаимодействия раствор концентрировали при пониженном давлении с получением этилового эфира син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты в виде белых кристаллов (фиг. 1, стадия 2).
Основание, добавляемое к реакционному раствору, на вышеупомянутой стадии может представлять собой любое основание, при условии, что оно нейтрализует хлорводородную кислоту, образующуюся во время взаимодействия. Предпочтительно может быть использован триэтиламин. Количество используемого основания такое же или превышает таковое для тионилхлорида.
Необходимо, чтобы количество тионилхлорида было такое же или превышало таковое для исходного вещества взаимодействия, а именно син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты. Если количество тионилхлорида слишком мало, этерификация с получением этилового эфира происходит с недостаточной эффективностью, что является нежелательным. Если количество тионилхлорида слишком велико, образуется избыток хлорводородной кислоты, и поэтому необходимо большее количество основания, что является нежелательным. В предпочтительных воплощениях количество тионилхлорида равно или менее 5 эквивалентов син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты.
Затем этиловый эфир син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты добавляли к раствору небольшого количества основания в спиртовом растворителе, таком как этанол. Полученную суспензию перемешивали при охлаждении и к суспензии добавляли т-бутил дикарбонат для взаимодействия данных соединений при комнатной температуре (фиг. 1, стадия 3). В качестве спиртового растворителя предпочтительно может быть использован этанол, хотя возможно использование спиртов различных типов. Необходимо, чтобы количество основания было достаточно небольшим по отношению к таковому для спирта, а если количество слишком мало, то скорость реакции понижается, что является нежелательным. В предпочтительных воплощениях используется раствор, в котором соотношения спирта и основания составляет 9:1. Необходимо, чтобы количество т-бутил дикарбоната было равно одному эквиваленту или более син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты, и предпочтительно равно 1,5 эквивалентам син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты.
Данная операция делает возможным получение этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-бензилокси-циклобутан-1-карбоновой кислоты.
Этиловый эфир син-1-(N-(т-бутоксикарбонил)амино)-3-бензилокси-циклобутан-1-карбоновой кислоты, синтезированный как описано выше, растворяли в спиртовом растворителе, таком как этанол, или растворителе эфире уксусной кислоты, таком как этиловый эфир уксусной кислоты, и к раствору добавляли палладий на активированном угле (количество: 10% мас./мас. или более по отношению к количеству субстрата) в атмосфере водорода для взаимодействия данных соединений при перемешивании при комнатной температуре. Затем реакционный раствор фильтровали через Celite и фильтрат концентрировали и очищали с получением этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-гидрокси-циклобутан-1-карбоновой кислоты (фиг. 2, стадия 4).
Полученный этиловый эфир син-1-(N-(т-бутоксикарбонил)амино)-3-гидрокси-циклобутан-1-карбоновой кислоты растворяли в веществе, обладающем основными свойствами, таком как пиридин, с последующим добавлением трифторметансульфонового ангидрида. Целевое соединение этиловый эфир син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]циклобутан-1-карбоновой кислоты получали добавлением воды, органического растворителя, такого как эфир, и кислоты к полученному раствору и очисткой органического слоя (фиг. 3, стадия 5).
Соединения согласно настоящему изобретению, отличные от описанных выше, так же могут быть синтезированы на стадиях, подобных описанным выше. Например, если синтезируют соединение, в котором заместитель эфир галогеналкилсульфоновой кислоты отличен от заместителя трифлата, заместитель эфир алкилсульфоновой кислоты или заместитель эфир сульфоновой кислоты, несущей ароматические заместители, образует связь с атомом углерода в положении 3 циклобутанового кольца, взаимодействие на стадии 5 может быть проведено таким же образом, как описано выше, за исключением того, что вместо трифторметансульфонового ангидрида используют различные галогенангидриды сульфоновой кислоты или сульфоновый ангидрид.
Если синтезируют соединение, в котором заместитель триалкилстаннил образует связь с атомом углерода в положении 3 циклобутанового кольца, соединение, являющееся спиртом, этиловый эфир син-1-(N-(т-бутоксикарбонил)амино)-3-гидрокси-циклобутан-1-карбоновой кислоты или подобное окисляют до кетона или альдегида, и кетон или альдегид участвуют в реакции Виттига с использованием соли фосфония, такой как метилфосфонил йодид, с образованием винилгалогенида в 3 положении, с последующим взаимодействием с гидридом триалкилолова. Соединение, в котором галоген образует связь с атомом углерода в 3 положении, может быть получено путем взаимодействия вышеописанного соединения, являющегося спиртом, с гидрогалогенидом или подобными.
Если синтезируют соединение, в котором заместитель алкилоксикарбонил отличен от заместителя т-бутоксикарбонила, заместитель алкенилоксикарбонил или заместитель бензилоксикарбонил образует связь с аминогруппой в положении 1, взаимодействие на вышеописанной стадии 3 может быть проведено с использолванием алкилхлорформиатов, алкенилхлорформиатов или бензилхлорформиатов соответственно, вместо т-бутил дикарбоната. Подобным образом, если синтезируют соединение, в котором заместитель циклический имид образует связь с аминогруппой, для взаимодействия с аминогруппой на вышеописанной стадии 3 могут быть использованы различные циклические ангидриды кислот, такие как фталиевый ангидрид. Соединение, в котором заместитель ароматический имин образует связь с аминогруппой, может быть синтезировано при взаимодействии бензальдегида, несущего заместитель, с аминогруппой на стадии 3. Соединения, имеющие другие функциональные группы, так же могут быть синтезированы с использованием сочетаний известных способов (Theodora W. Greene. Protective groups in organic synthesis. 3rd edition. USA, Jon Wiley & Sons, Inc., 1999, pp. 531, 550-561, and 573-586).
Если синтезируют форму 1-пропилового эфира и форму изопропилового эфира, возможно использование 1-пропанола и изопропанола соответственно в качестве спирта, вступающего во взаимодействие на вышеописанной стадии 2.
Затем в качестве примера использования новых органических соединений аминокислот согласно настоящему изобретению будет описан способ, в котором анти-[18F]-ФАЦБК синтезируют с использованием синтезированного ранее этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты.
Синтез анти-[18F]-ФАЦБК проводят в две стадии: стадия введения радиоактивного фтора в предшественник и стадия снятия защитных групп соединения, в которое был введен радиоактивный фтор.
Радиоактивный фтор может быть получен известным способом, например способом, в котором воду, обогащенную H2 18O, используют в качестве мишени и подвергают бомбардировке протонами. В данном случае, радиоактивный фтор присутствует в воде, обогащенной H2 18O, используемой в качестве мишени. Воду, обогащенную H2 18O, содержащую радиоактивный фтор, пропускают через, например, анионобменную колонку так, что радиоактивный фтор адсорбируется и накапливается в колонке, таким образом, его отделяют от воды, обогащенной H2 18O. После этого, через колонку пропускают раствор карбоната калия для элюирования радиоактивного фтора и к элюату добавляют катализатор межфазного переноса и упаривают досуха, активируя, таким образом, радиоактивный фтор.
Затем высушенный радиоактивный фтор растворяют в ацетонитриле, и к раствору ацетонитрила, в качестве предшественника, добавляют этиловый эфир син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты, взаимодействие данных соединений происходит при нагревании. В результате, радиоактивный фтор вводят в предшественник, посредством чего синтезируют этиловый эфир анти-[18F]-1-(N-(т-бутоксикарбонил)амино)-3-фторциклобутан-1-карбоновой кислоты.
Для полученного этилового эфира анти-[18F]-1-(N-(т-бутоксикарбонил)амино)-3-фторциклобутан-1-карбоновой кислоты проводят снятие защитных групп с получением анти-[18F]-ФАЦБК в качестве целевого соединения. Снятие защитных групп может быть проведено, например, в кислых условиях. Кислые условия могут быть созданы различными способами, например способом, в котором к раствору, который содержит этиловый эфир анти-[18F]-1-(N-(т-бутоксикарбонил)амино)-3-фторциклобутан-1-карбоновой кислоты, добавляют кислоту. Количество добавляемой кислоты не ограничивается, и кислоту добавляют до тех пор, пока кислых условий не будет достаточно для снятия защитных групп.
Другие соединения согласно настоящему изобретению, отличные от описанного выше соединения, так же могут быть использованы в качестве предшественников для введения метки для соединений, меченных радиоактивным галогеном, таким же образом, как описано выше.
Например, соединения, в которых заместитель триалкилстаннил образует связь с атомом углерода в положении 3 циклобутанового кольца, могут быть смешаны и приведены во взаимодействие с различными радиоактивными галогенами и окислителями в зависимости от цели таким образом, чтобы получить соединения, меченные радиоактивным галогеном. В соединения, в которых заместитель галоген образует связь с атомом углерода в положении 3, метка - радиоактивный галоген может быть введена с использованием реакции нуклеофильного замещения или реакции изотопного обмена. Если введение метки - радиоактивного галогена проводят с использованием реакции нуклеофильного замещения, может быть проведена следующая реакция замещения. Например, галоген, образующий связь с атомом углерода в положении 3, представляет собой йод, йод может быть замещен фтором, хлором или бромом, если галоген, образующий связь с атомом углерода в положении 3, представляет собой бром, то бром может быть замещен хлором или фтором, и если галоген, образующий связь с атомом углерода в положении 3, представляет собой хлор, хлор может быть замещен фтором.
ПРИМЕРЫ
Здесь настоящее изобретение будет описано более подробно со ссылками к примерам, однако должно быть понятно, что особенности примеров не ограничивают настоящее изобретение.
Условия анализа, в которых проводили газовую хроматографию, в каждом примере и сравнительном примере следующие.
Оборудование: GC-1700AF/aoc (произведенный Shimadzu Corporation).
Колонка: SPB-1 (произведена SUPELCO, 30 м × 0,53 мм в. д., размер частиц для заполнения: 3 мкм).
Температура колонки: 40°С (3,3 минуты)→90°С (0,5 минут) (скорость увеличения температуры: 20°С/мин).
Температура на входе: 250°С.
Температура детектора: 220°С.
Газ-носитель: гелий.
Скорость расплескивания: 1:10.
Линейная скорость: 30 см/сек.
Пример 1
Синтез этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты
Гидролиз син-гидантоина (фиг. 1, стадия 1)
Син-5-(3-бензилоксициклобутан)гидантоин синтезировали согласно способу, описанному в литературе (Jonathan McConathy et al. Applied Radiation and Isotopes. 2003, 58, p. 657-666).
К раствору 72,8 г (соответствует 0,418 моль) 3-бензилоксициклобутан-1-она в 2,86 л этанола по каплям добавляли раствор, полученный растворением 397 г (соответствует 4,13 моль) карбоната аммония и 88,4 г (соответствует 1,65 моль) хлорида аммония в 2,86 л воды, и перемешивали при комнатной температуре в течение 30 минут. Затем 121,0 г (соответствует 1,86 моль) цианида калия добавляли к смеси и перемешивали при 60°С в течение ночи. Реакционный раствор концентрировали и полученное желтое твердое вещество промывали 1,06 л воды для удаления солей. К твердому веществу добавляли 927 млм метанола, проводили азеотропную дистилляцию и очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/метанол = 98/2) с получением 55,3 г син-5-(3-бензилоксициклобутан)гидантоина.
250 мл насыщенного раствора хлорида бария добавляли к 6,15 г (соответствует 25 ммоль) син-5-(3-бензилоксициклобутан)гидантоина и нагревали с обратным холодильником на масляной бане при 114°С в течение 24 часов или дольше. Затем проводили анализ ТСХ с использованием в качестве подвижной фазы систем двух видов: хлороформ/метанол = 5/1 (величина Rf син-гидантоина = приблизительно 0,6) и хлороформ/метанол = 95/1 (величина Rf син-гидантоина = приблизительно 0,3), и завершение реакции было подтверждено (свечение в УФ и окрашивание с фосфорномолибденовой кислотой).
После подтверждения завершения реакции реакционный раствор охлаждали до комнатной температуры и добавляли приблизительно 24 мл 1 моль/мл серной кислоты для нейтрализации реакционного раствора. После нейтрилизации реакционный раствор далее перемешивали при комнатной температуре в течение 5 минут и образующийся осадок удаляли фильтрованием. Фильтрат концентрировали с получением 5,67 г син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты в виде белых кристаллов.
Этерификация с получением этилового эфира (фиг. 1, стадия 2)
5,67 г син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты, которую сушили до полного удаления воды, растворяли в 200 мл этанола. К данному раствору добавляли 9,5 мл (соответствует 75 ммоль) триэтиламина и охлаждали до -78°С в течение 20 минут, и затем добавляли 4,6 мл (соответствует 62,5 ммоль) тионилхлорида. Реакционный раствор перемешивали при 0°С в течение 1 часа и при комнатной температуре в течение 1 часа с последующим нагреванием с обратным холодильником на масляной бане при 95°С в течение ночи. Завершение реакции подтверждали анализом ТСХ с использованием подвижной фазы хлороформ/метанол = 95/1 (величина Rf целевого соединения = приблизительно 0,6 (подтверждено свечением в УФ и окрашиванием с фосфорномолибденовой кислотой). После подтверждения завершения реакции реакционный раствор концентрировали при пониженном давлении с получением 7,64 г этилового эфира син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты в виде белых кристаллов.
Введение Вос (фиг. 1, стадия 3)
7,64 г этилового эфира син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты растворяли в 250 мл смешанного раствора этанол/триэтиламин = 9/1. Затем раствор охлаждали на ледяной бане в течение 15 минут, к раствору добавляли 8,6 мл (соответствует 37,5 ммоль) т-бутил дикарбоната и перемешивали при комнатной температуре в течение ночи. Завершение реакции подтверждали анализом ТСХ с использованием подвижной фазы гексан/этилацетат = 1:1 (величина Rf целевого соединения = приблизительно 0,6) (подтверждено свечением в УФ и окрашиванием с фосфорномолибденовой кислотой). После подтверждения завершения реакции реакционный раствор концентрировали при пониженном давлении с получением белых кристаллов в остатке. К остатку добавляли 150 мл охлажденного этилацетата и 150 мл 0,5 моль/л охлажденной хлорводородной кислоты, перемешивали на ледяной бане в течение 5 минут, оставляли для разделения фаз. Органический слой экстрагировали и дважды промывали 150 мл воды, 150 мл насыщенного водного раствора гидрокарбоната натрия, дважды 150 мл воды и дважды 150 мл насыщенного солевого раствора в указанном порядке, сушили безводным сульфатом натрия и концентрировали при пониженном давлении с получением желтой масляной субстанции. Отдельно водный слой экстрагировали и промывали дважды 150 мл этилацетата, дважды 150 мл воды и 150 мл насыщенного солевого раствора в указанном порядке, сушили безводным сульфатом натрия и концентрировали при пониженном давлении с получением небольшого количества желтой масляной субстанции. При осуществлении данных операций было получено 8,82 г светло-желтой масляной субстанции. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат = 1/1) с получением 8,04 г (соответствует 23 ммоль) этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-бензилокси-циклобутан-1-карбоновой кислоты в виде белых кристаллов.
Дебензилирование (фиг. 2, стадия 4)
К 8,04 г (соответствует 23 ммоль) этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-бензилокси-циклобутан-1-карбоновой кислоты добавляли 150 мл этанола и затем 960 мг палладия на активированном угле (палладий 10%) для проведения замещения на водород при перемешивании при комнатной температуре в течение ночи. По окончании взаимодействия, палладий на активированном угле удаляли фильтрованием с использованием Celite и фильтрат концентрировали при пониженном давлении с получением 5,74 г белых кристаллов в остатке. За взаимодействием следили по средствам анализа ТСХ с использованием в качестве подвижной фазы гексан/этилацетат = 1:1 (величина Rf целевого соединения = приблизительно 0,2) (подтверждено свечением в УФ и окрашиванием с фосфорномолибденовой кислотой) для подтверждения завершения взаимодействия. Затем остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат = 1/1, гексан/этилацетат = 4/1) с получением 5,36 г (соответствует 20,7 ммоль) этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-гидрокси-циклобутан-1-карбоновой кислоты в виде белых кристаллов.
Введение трифлата (фиг. 3, стадия 5)
2,07 г (8 ммоль) этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-гидрокси-циклобутан-1-карбоновой кислоты растворяли в 26 мл пиридина и перемешивали на ледяной бане в течение 20 минут. Затем добавляли 2,0 мл (соответствует 12 ммоль) ангидрида трифторметансульфоновой кислоты и перемешивали в течение 30 минут. За взаимодействием следили по средствам анализа ТСХ с использованием подвижной фазы гексан/диэтиловый эфир = 1/1 (величина Rf целевого соединения взаимодействия = приблизительно 0,6) (подтверждено свечением в УФ и окрашиванием с нингидрином) для подтверждения завершения взаимодействия. После подтверждения завершения взаимодействия к реакционному раствору добавляли 100 мл воды и 100 мл эфира, экстракцию и промывание проводили дважды 100 мл 1 моль/л хлорводородной кислоты, дважды 100 мл воды и дважды 100 мл насыщенного солевого раствора в указанном порядке. После сушки безводным сульфатом натрия проводили концентрирование при пониженном давлении с получением 2,78 г светло-желтых кристаллов. Реакционную смесь очищали хроматографией на силикагеле (гексан/диэтиловый эфир = 3/1) с получением белых кристаллов и полученные белые кристаллы снова перекристаллизовывали с использованием пентан/диэтиловый эфир с получением 1,84 г (соответствует 4,7 ммоль) этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]циклобутан-1-карбоновой кислоты.
Результаты исследования ЯМР (внутренний стандарт: тетраметилсилан) для полученного этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты приведены далее.
Использовали ЯМР спектрометр: JNM-ECP-500 (произведенный JEOL, Ltd.)
1Н-ЯМР (растворитель: CDCl3, резонансная частота: 500 МГц): δ 5,41-5,35 (м, 1Н), 5,32 (ш, 1Н), 4,26 (кв, 2Н, J=7 Гц), 3,10-3,02 (м, ш, 4Н), 1,45 (с, 9Н), 1,31 (т, 3Н, J=7,0 Гц).
13С-ЯМР (растворитель: CDCl3, резонансная частота: 125 МГц): δ 172,60, 154,46, 118,48, 75,88, 51,97, 40,87, 28,29, 14,11.
Сравнительный пример 1
Анти-[18F]ФАЦБК синтезировали с использованием метилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]циклобутан-1-карбоновой кислоты в качестве предшественника для введения метки и измерение проводили для удерживаемого растворителя при синтезе анти-[18F]ФАЦБК.
Метиловый эфир син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]циклобутан-1-карбоновой кислоты синтезировали согласно способу, описанному в литературе (Jonathan McConathy et al. Applied Radiation and Isotopes. 2003, 58, pp. 657-666).
H2 18O, содержащую фторид-ион [18F] (активность: 3,27 ГВк, исправленное значение в момент начала синтеза), пропускали через анионобменную колонку для адсорбции и накопления на колонке [18F] фторид-иона. Затем смесь водного раствора карбоната калия (133 ммоль/л, 0,3 мл) и раствора 40 мг Kryptfix 222 (под товарным знаком, произведенный Merck & Co., Inc.) в 1,5 мл ацетонитрила пропускали через ту же колонку для элюирования [18F] фторид-иона.
Элюат нагревали до 110°С для упаривания воды и добавляли ацетонитрил (0,5 мл ×2) и подвергали азеотропной дистилляции с последующим упариванием досуха. К сухому [18F] фториду добавляли раствор 30 мг метилового эфира 1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты в 1 мл ацетонитрила и нагревали до 85°С в течение 3 минут. Затем к раствору добавляли 4 мл диэтилового эфира и затем дважды добавляли 3 мл того же, и смесь пропускали через Sep-PakSilica (под товарным знаком, произведенный Japan Waters) с получением раствора [18F]фтор-меченного соединения в ацетонитрил/диэтиловый эфир.
К полученному раствору [18F]фтор-меченного соединения в ацетонитрил/диэтиловый эфир добавляли 1,5 мл 4 моль/л хлорводородной кислоты и нагревали при 120°С в течение 15 минут для снятия защитных групп с получением анти-[18F] ФАЦБК. Для полученного анти-[18F]ФАЦБК проводили газовую хроматографию в условиях, описанных выше, для количественного определения метанола и этанола. Как показано в таблице 1, метанол детектировали в концентрациях 17,4±0,6 чнм.
Пример 2
H2 18O, содержащую фторид-ион [18F] (активность: 36,63 ГВк, исправленное значение в момет начала синтеза), пропускали через анион-обменную колонку для адсорбции и накопления на колонке [18F] фторид-иона. Затем смесь водного раствора карбоната калия (133 ммоль/л, 0,3 мл) и раствора 40 мг Kryptfix 222 (под товарным знаком, произведенный Merck & Co., Inc.) в 1,5 млм ацетонитрила пропускали через ту же колонку для элюирования [18F] фторид-иона.
Элюат нагревали до 110°С для упаривания воды и добавляли ацетонитрил (0,5 мл ×2) и подвергали азеотропной дистилляции с последующим упариванием досуха. К сухому [18F] фториду добавляли раствор 32 мг этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты, полученного в примере 1, в 1 мл ацетонитрила и нагревали до 85°С в течение 3 минут. Затем к раствору добавляли 4 мл диэтилового эфира и затем дважды добавляли 3 мл того же, и смесь пропускали через Sep-PakSilica (под товарным знаком, произведенный Japan Waters) с получением раствора [18F]фтор-меченного соединения в ацетонитрил/диэтиловый эфир.
К полученному раствору [18F]фтор-меченного соединения в ацетонитрил/диэтиловый эфир добавляли 1,5 мл 4 моль/л хлорводородной кислоты и нагревали при 120°С в течение 15 минут для снятия защитных групп с получением анти-[18F] ФАЦБК. Для полученного анти-[18F]ФАЦБК проводили газовую хроматографию в условиях, описанных выше, для количественного определения метанола и этанола. Как показано в таблице 2, метанол не был детектирован, в то время как этанол был детектирован в концентрациях 24,1±0,8 чнм.
Результаты подтвердили, таким образом, что использование соединения согласно настоящему изобретению в качестве предшественника для введения метки делает возможным предотвращение удерживания метанола синтезированным анти-[18F] ФАЦБК.
Применение в промышленности
В качестве соединения согласно настоящему изобретению предлагаются органические соединения, меченные радиоактивным галогеном, которые используются в качестве радиофармпрепаратов в радиологическом исследовании с использованием ПЭТ или ОЭКТ и являются полезными в области радиофармпрепаратов.
Краткое описание чертежей
На фиг. 1 показана схема синтеза этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-бензилокси-циклобутан-1-карбоновой кислоты;
на фиг. 2 показана схема синтеза этилового эфира син-1-(N-(т-бутоксикарбонил)амино)-3-гидрокси-циклобутан-1-карбоновой кислоты; и
на фиг. 3 показана схема синтеза эфира син-1-(N-(т-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты.



Изобретение относится к новым соединениям-предшественникам формулы (1), которые могут найти применение для получения органических соединений, меченных радиоактивным галогеном, используемых в позитронно-эмиссионной томографии и однофотонной эмиссионной компьютерной томографии. В формуле (1)

n представляет собой 0; R1 представляет собой этильный, 1-пропильный или изопропильный заместитель; Х представляет собой группу, представленную OR2; R2 представляет собой перфторалкилсульфонильный заместитель с неразветвленной или разветвленной цепью, содержащий от 1 до 7 атомов углерода; и R3 представляет собой алкилоксикарбонил с неразветвленной или разветвленной цепью, содержащий от 2 до 7 атомов углерода. 2 з.п. ф-лы, 2 табл., 3 ил.
1. Соединение-предшественник для органических соединений, меченных радиоактивным галогеном, представленное следующей формулой (I):
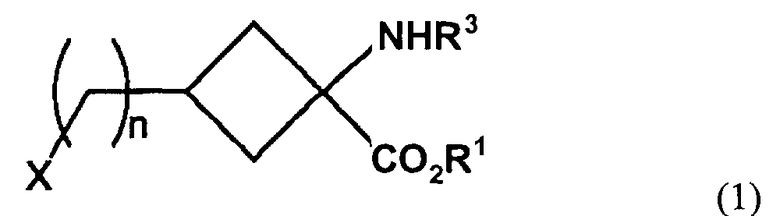
где n представляет собой 0; R1 представляет собой этильный, 1-пропильный или изопропильный заместитель; Х представляет собой группу, представленную OR2; R2 представляет собой перфторалкилсульфонильный заместитель с неразветвленной или разветвленной цепью, содержащий от 1 до 7 атомов углерода; и R3 представляет собой алкилоксикарбонил с неразветвленной или разветвленной цепью, содержащий от 2 до 7 атомов углерода.
2. Соединение по п.1, в котором R1 представляет собой этильный заместитель.
3. Соединение по п.1 или 2, где R3 представляет собой т-бутоксикарбонильную группу.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| L.MARTARELLO et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| MED | |||
| CHEM., 2002, 45(11), 2250-2259 | |||
| L.J.WANG et al, Syntheses of new conformationally constrained S-[2-[(1-iminoethyl)amino]ethyl]homocysteine | |||

