Область изобретения
Настоящее изобретение относится к способу получения радиофармацевтических предшественников и, в частности, к защищенным производным аминокислот, которые используют в качестве предшественников для получения аминокислот, меченных радиоактивным изотопом, для применения в позитрон-эмиссионной томографии (PET). Кроме того, изобретение включает способ получения указанных меченных радиоактивным изотопом аминокислот.
Предшествующий уровень техники
В последние годы в качестве новых радиофармацевтических средств был разработан ряд соединений, представляющих собой меченные радиоактивным галогеном аминокислоты, включая [18F]1-амино-3-фторциклобутанкарбоновую кислоту ([18F]-FACBC). [18F]-FACBC считается эффективной в качестве диагностического агента в отношении высокопролиферативных опухолей, поскольку обладает свойством специфично переноситься переносчиками аминокислот.
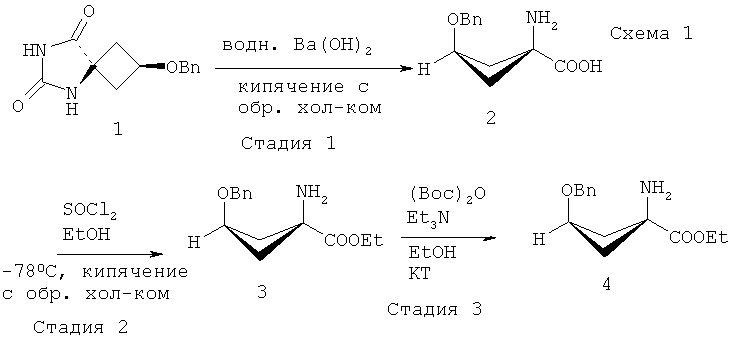
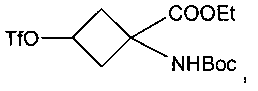
В ЕР 1978015 (А1) предложены предшественники соединения [18F]-FACBC и способы получения указанных предшественников. В ЕР 1978015 (А1) конкретно описан способ получения предшественника - этилового эфира син-1-(N-(трет-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты, включающий следующие стадии:
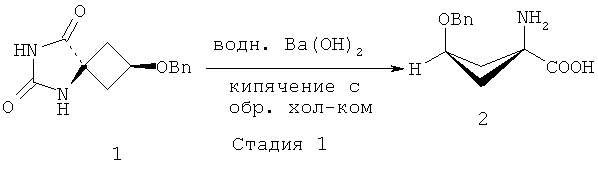
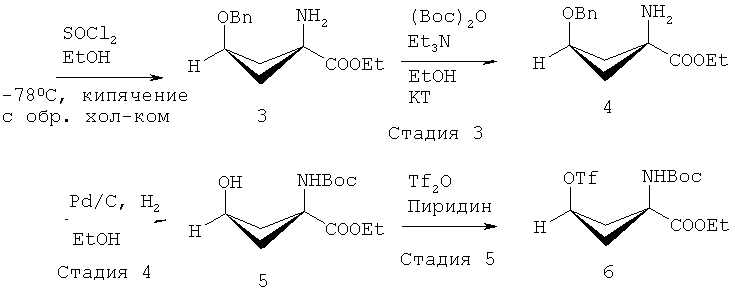
В ЕР 1978015 (А1) описано, что стадия 1 на приведенной выше реакционной схеме включает гидролиз син-5-(3-бензилоксициклобутан)-гидантоина 1 путем добавления гидроксида бария Ва(ОН)2 к раствору и кипячения с обратным холодильником этой смеси при 114°C в течение 24 часов или больше. син-1-Амино-3-бензилоксициклобутан-1-карбоновую кислоту 2 на стадии 2 образования сложного этилового эфира растворяют в этаноле (EtOH) и подвергают взаимодействию с тионилхлоридом (SOCl2), получая этиловый эфир син-1-амино-3-бензилоксициклобутан-1-карбоновой кислоты 3. Стадия 3 включает присоединение трет-бутоксикарбонила (Boc) к аминной функциональной группе путем взаимодействия соединения 3 с трет-бутилдикарбонатом (Вос)2O, и полученное вещество очищают хроматографией, получая этиловый эфир син-1-(N-(трет-бутоксикарбонил)амино)-3-бензилокси-циклобутан-1-карбоновой кислоты 4. Затем на стадии 4 с бензил-защищенного промежуточного соединения 4 снимают защиту путем растворения соединения 4 в этаноле (EtOH), добавления палладия на активированном угле (Pd/C) и приложения к реакционной смеси небольшого положительного давления H2. Полученное вещество очищают хроматографией, получая этиловый эфир син-1-(N-(трет-бутоксикарбонил)амино)-3-бензилокси-циклобутан-1-карбоновой кислоты 5 для использования на стадии 5, которая включает взаимодействие соединения 5 с трифторметансульфоновым ангидридом (Tf2O), за которым следует хроматографическая очистка с последующей перекристаллизацией вещества с получением этилового эфира син-1-(N-(трет-бутоксикарбонил)амино)-3-[((трифторметил)сульфонил)окси]-циклобутан-1-карбоновой кислоты 6.
Вышеописанный известный способ является относительно сложным, дорогостоящим и трудоемким, особенно когда применяется для крупномасштабного производства соединения-предшественника. Было бы желательно иметь способ, который является более простым для осуществления, более экономически эффективным и более пригодным для крупномасштабного промышленного производства.
Краткое изложение сущности изобретения
Настоящее изобретение относится к способу, полезному в получении соединений-предшественников для [18F]-FACBC и подобных соединений, который является более пригодным для крупномасштабного промышленного производства, чем способы, известные ранее. По сравнению с известным способом способ по настоящему изобретению позволяет получать такие соединения в промышленном масштабе без необходимости использования в работе больших количеств растворителей, а также приводит к улучшенным выходам.
Подробное описание изобретения
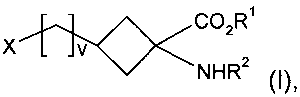
В одном аспекте настоящее изобретение относится к способу получения соединения формулы I:
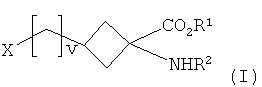 ,
,
где:
R1 представляет собой C1-5алкильную группу с прямой или разветвленной цепью;
R2 представляет собой амино-защитную группу;
v является целым числом от 0 до 4; и
Х представляет собой уходящую группу, выбранную из галогена или группы -O-SO2-R3, где R3 представляет собой галоген, C1-10алкил с прямой цепью или разветвленной цепью, C1-10галогеналкил с прямой цепью или разветвленной цепью и C6-10арил,
включающему:
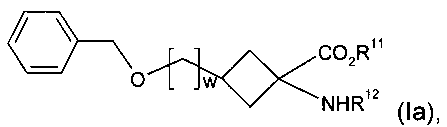
(а) дебензилирование соединения формулы Ia:
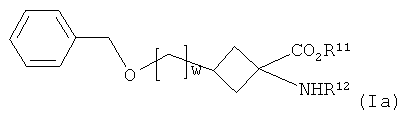 ,
,
где R11, R12 и w являются такими, как определено для R1, R2 и v в формуле I соответственно;
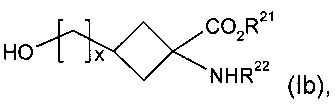
(б) кристаллизацию реакционной смеси со стадии (а) с получением очищенного соединения формулы Ib:
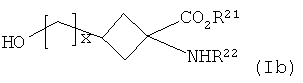
где R21, R22 и х являются такими, как определено для R1, R2 и v в формуле I соответственно;
(в) превращение очищенного соединения формулы I, полученного на стадии (б), в соединение формулы I путем взаимодействия с подходящей формой X, где Х является таким, как определено для формулы I.
Термин "алкил", используемый отдельно или в комбинации, означает группу с прямой цепью или разветвленной цепью, имеющую общую формулу CnH2n+1. Величина n в этой общей формуле указана в конкретных случаях. Примеры некоторых предпочтительных алкильных групп включают метильную, этильную, 1-пропильную или изопропильную группы.
Под термином "защитная группа" понимают группу, которая ингибирует или подавляет нежелательные химические реакции, но которая предназначена быть достаточно реакционноспособной, чтобы она могла отщепляться от рассматриваемой функциональной группы в достаточно мягких условиях, при которых остальная часть молекулы не изменяется, с получением целевого продукта. Защитные группы хорошо известны специалистам в данной области и описаны в "Protective Groups in Organic Synthesis", Theorodora W. Greene and Peter G.M. Wuts (Fourth Edition, John Wiley & Sons, 2007). Подходящие амино-защитные группы хорошо известны в данной области. Подходящей амино-защитной группой R2 является карбамат. Предпочтительно, R2 выбран из: трет-бутилкарбамата (ВОС), 9-флуоренилметилкарбамата (Fmoc), метилкарбамата, этилкарбамата, 2-хлор-3-инденилметилкарбамата (Climoc), бенз[f]инден-3-илметилкарбамата (Bimoc), 2,2,2-трихлорэтилкарбамата (Troc), 2-хлорэтилкарбамата, 1,1-диметил-2,2-дибромэтилкарбамата (DB-f-BOC), 1,1-диметил-2,2,2-трихлорэтилкарбамата (ТСВОС), бензилкарбамата (Cbz) и дифенилметилкарбамата. Для получения N-трет-бутоксикарбонила R2 наиболее предпочтительно представляет собой трет-бутилкарбамат.
Термин "уходящая группа" относится к группировке, подходящей для нуклеофильного замещения, и означает молекулярный фрагмент, уходящий с электронной парой при гетеролитическом разрыве связи.
Термин "галоген" или "галогено-", используемый отдельно или в комбинации, относится к заместителю, выбранному из фтора, хлора, брома или йода.
Термин "C1-10галогеналкил" относится к алкильной группе, которая определена выше, содержащей 1-10 атомов углерода, где по меньшей мере один водород заменен галогеном, при этом галоген является таким, как определено выше.
Термин "C6-10арил" относится к моновалентному ароматическому углеводороду, имеющему единственное кольцо (то есть фенилу) или конденсированные кольца (то есть нафталину). Если не указано иное, такие арильные группы обычно содержат от 6 до 10 кольцевых атомов углерода.
Термин "дебензилирование" относится к отщеплению от соединения бензильного заместителя. Термин "бензид" относится к группе с химической структурой С6Н5СН2-. Дебензилирование является способом, хорошо известным в данной области, и его обычно осуществляют посредством "каталитического гидрирования", представляющего собой реакцию, в результате которой углерод-углеродная связь расщепляется или подвергается "разрушению" под действием водорода. Гидрогенолиз обычно осуществляют каталитическим методом, например, с использованием палладия на угле (Pd/C) в качестве катализатора. Если на стадии дебензилирования используют такой катализатор, как Pd/C, то удаление катализатора из реакционной смеси осуществляют путем фильтрования перед началом следующей стадии. Термин "фильтрование" относится к механическому отделению твердых веществ от жидкостей. Неограничивающие примеры подходящих средств для фильтрования для применения в настоящем изобретении включают воронку с фильтром из пористого стекла или воронку для фильтрования, дополненную фильтром из стекловолокна, хотя также подходят и другие более специализированные методы фильтрования. В общем случае, после стадии дебензилирования (а) и перед стадией кристаллизации (б) реакционный растворитель удаляют путем сушки. Сушка может быть осуществлена методами, хорошо известными специалисту в данной области, например путем упаривания в токе азота и/или вакуумной сушки.
Термин "кристаллизация" обычно относится к процессу образования кристаллов твердого вещества, выпадающих в осадок из раствора. Кристаллизацию можно использовать в качестве способа очистки вследствие того, что правильно образованные кристаллы, как ожидается, будут чистыми, поскольку каждая молекула или ион должна(ен) идеально встраиваться в решетку, покидая раствор. Чтобы произошла кристаллизация из раствора, он должен быть перенасыщенным. Это означает, что раствор должен содержать больше частиц растворенного вещества, чем он мог бы содержать в равновесии (насыщенный раствор). Этого можно достичь разными методами, включая выпаривание растворителя, охлаждение раствора, добавление второго растворителя для уменьшения растворимости растворенного вещества (метод, известный как добавление антирастворителя или "глушение" (drown-out)), химическую реакцию и изменение pH. В способе по изобретению после стадии (а) готовят раствор реагентов. Этот раствор готовят, используя первый растворитель, в котором эти реагенты легко растворимы.
Термин "подходящая форма X" означает X, как он определен в данном описании, в форме, которая может заместить гидроксильную функциональную группу в реакции замещения.
Соединения формулы Ia можно получить, следуя описанным в ЕР 1978015 (А1) способам или адаптируя их. Например, соединение 4, конкретно описанное в ЕР 1978015 (А1), является соединением формулы Ia, подходящим для применения в способе по настоящему изобретению. Способ получения указанного соединения 4, описанный в ЕР 1978015 (А1), проиллюстрирован ниже на Схеме 1:
Способы получения соединения формулы Ia также описаны McConathy et al (AppI Rad Isotop 2003; 58: 657-666). Соединение 6 на Фиг.2 из McConathy et al является соединением формулы Ia. Способ получения указанного соединения 6, описанный в McConathy et al, проиллюстрирован ниже на Схеме 2:
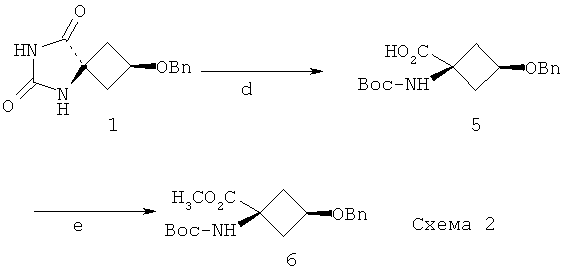
Гидантоин 1 обрабатывали 3 н. водным гидроксидом натрия при 180°C, затем ди-трет-бутилдикарбонатом, получая N-Вос-кислоту 5. Метиловый эфир 6 получали с высоким выходом путем взаимодействия соединения 5 с триметилсилилдиазометаном.
В компетенции специалиста в данной области адаптировать описанные выше способы предшествующего уровня техники для получения других соединений формулы Ia, которые подпадают под определение настоящего изобретения. Если исходное гидантоиновое соединение включает смесь син- и анти-энантиомеров, то это приемлемо. Нет необходимости в активном разделении энантиомеров на какой-либо стадии способа. В действительности, как описано здесь в примере 2, в кристаллическом продукте было достигнуто небольшое обогащение по син-изомеру. Во время предварительных экспериментов было обнаружено, что такое обогащение является более выраженным на ранних стадиях кристаллизации. При несколько более низком суммарном выходе было зарегистрировано соотношение син/(син + анти) выше 90%. Таким образом, способ по изобретению имеет дополнительное преимущество, заключающееся в том, что с его помощью можно разделять изомеры.
Предпочтительно, R1 представляет собой метил или этил; наиболее предпочтительно представляет собой этил. Такое предпочтительное определение для R1 равно применимо к R11 и R21.
R2 предпочтительно представляет собой карбонатную сложноэфирную защитную группу, где термин "карбонатный сложный эфир" относится к функциональной группе, состоящей из карбонильной группы с двумя алкоксигруппами, расположенными по обеим ее сторонам, имеющей общую структуру RxO=(C=O)ORy. Наиболее предпочтительно R2 представляет собой трет-бутоксикарбонильную группу. Такое предпочтительное определение для R2 равно применимо к R12 и R22.
Предпочтительно, v равно 0 или 1; наиболее предпочтительно равно 0. Такое предпочтительное определение для v равно применимо к w и х.
Особенно предпочтительное соединение формулы I представляет собой:
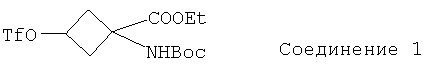 .
.
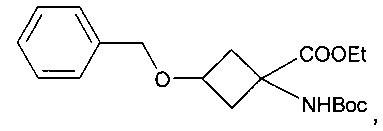
Особенно предпочтительное соединение формулы Ia представляет собой:
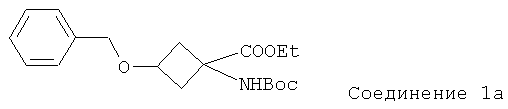 .
.
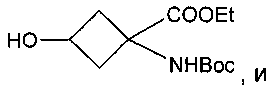
Особенно предпочтительное соединение формулы Ib представляет собой:
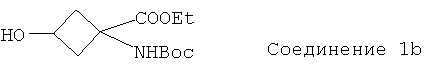 .
.
В приведенных выше соединениях 1, 1а и 1b Et означает этил, OTf - трифторметансульфоновую кислоту, а Boc - трет-бутилоксикарбонил.
Способ по настоящему изобретению уменьшает продолжительность процесса и снижает стоимость товара по сравнению со способами предшествующего уровня техники. В частности, для изготовления партий соединений формулы I в промышленном масштабе способом предшествующего уровня техники, в котором для очистки соединения формулы Ib используют стадию флэш-хроматографии, потребуется большая колонка с диоксидом кремния и большие количества растворителя. Благодаря использованию кристаллизации вместо флэш-хроматографии удается избежать применения больших количеств растворителей, что оказывается полезным как с точки зрения стоимости, так и безопасности обслуживающего персонала.
В предпочтительном воплощении Х представляет собой группу -O-SO2-R3. Когда Х представляет собой -O-SO2-R3, то Х наиболее предпочтительно выбран из группы, состоящей из толуолсульфоновой кислоты, нитробензолсульфоновой кислоты, бензолсульфоновой кислоты, трифторметансульфоновой кислоты, фторсульфоновой кислоты и перфторалкилсульфоновой кислоты. В особенно предпочтительном воплощении -O-SO2-R3 представляет собой трифторметансульфоновую кислоту. Группа -O-SO2-R3 может быть присоединена на стадии (в) способа по изобретению путем взаимодействия соединения формулы I с электрофильным производным желаемой группы -O-SO2-R3, которое является примером "подходящей формы X". Например, если желательно присоединить трифторметансульфоновую кислоту, то соединение формулы Ib можно подвергнуть взаимодействию с трифторметансульфоновым ангидридом.
В альтернативном предпочтительном воплощении Х представляет собой галоген. Если Х представляет собой галоген, то наиболее предпочтительно представляет собой бром или хлор. Стадия (в), где Х представляет собой галоген, может быть осуществлена способами, хорошо известными специалистам в данной области. Например, соединение формулы Ib, где Х представляет собой хлор, может быть получено путем взаимодействия соединения формулы I с хлоридсодержащим реагентом, таким как тионилхлорид, пентахлорид фосфора (PCl5), трихлорид фосфора (PCl3), каждый из которых является примером "подходящей формы X". Соединение формулы Ib, где Х представляет собой бром, может быть получено путем взаимодействия соединения формулы I с бромсодержащим реагентом, таким как бромистоводородная кислота (HBr) или трибромид фосфора (PBr3), каждый из которых опять является примером "подходящей формы X".
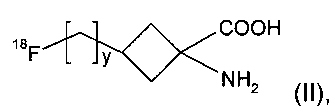
Соединение формулы I представляет собой соединение-предшественник, полезное в радиохимическом синтезе некоторых 18F-меченных соединений. Таким образом, согласно настоящему изобретению также предложен способ получения соединения формулы II:
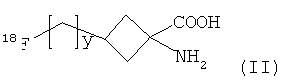 ,
,
где y является таким, как определено для v в формуле I, радиохимическим синтезом, включающий:
(1) получение соединения формулы I способом, определенным в данном описании;
(2) взаимодействие указанного соединения формулы I с подходящим источником 18F-фторида с получением соединения формулы IIa:
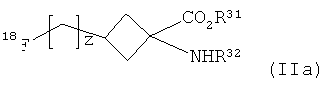 ,
,
где R31, R32 и z являются такими, как определено для R1, R2 и v в формуле I соответственно; и
(3) снятие защиты с соединения формулы IIa, полученного на стадии (2), для удаления R31 и R32.
В типичном случае [18F]-фторид-ион получают в виде водного раствора, который представляет собой продукт облучения мишени - [18О]-воды. Обычно для превращения [18F]-фторида в реакционноспособный нуклеофильный реагент перед его использованием в реакциях нуклеофильного мечения радиоактивным изотопом, проводят определенные стадии. Как и в случае нерадиоактивного фторирования, эти стадии включают удаление воды от [18F]-фторид-иона и предоставление подходящего противоиона (Handbook of Radiopharmaceuticals 2003, Welch & Redvanly eds., Chapter 6, pp.195-227). Затем проводят реакцию радиофторирования с использованием безводных растворителей (Aigbirhio et al., 1995, J FluorChem, 70: pp.279-87).
Для улучшения реакционной способности [18F]-фторид-иона в отношении реакций фторирования добавляют катионный противоион, после чего удаляют воду. Чтобы растворимость [18F]-фторид-иона сохранилась, противоион должен обладать достаточной растворимостью в безводном реакционном растворителе. Таким образом, используемые противоионы включают большие, но "мягкие" ионы металлов, таких как рубидий или цезий, калий в комплексе с криптандом, таким как Kryptofix™, или соли тетраалкиламмония. Предпочтительным противоионом в отношении реакций фторирования является калий в комплексе с криптандом, таким как Kryptofix™, вследствие его хорошей растворимости в безводных растворителях и улучшенной реакционной способности по отношению к фторидам.
Стадию снятия защиты (3) осуществляют способами, которые хорошо известны специалистам в данной области. Большое разнообразие защитных групп, а также способы их удаления описаны в "Protective Groups in Organic Synthesis", Theorodora W. Greene and Peter G.M. Wuts (Fourth Edition, John Wiley & Sons, 2007). В предпочтительном воплощении перед удалением амино-защитной группы R32 удаляют карбокси-защитную группу R31. Например, если R31 представляет собой Et, то ее можно удалить основным гидролизом, и если R32 представляет собой Boc, то ее можно удалить потом кислотным гидролизом.
Дипазон подходящих и предпочтительных определений для v, как он предложен выше для формулы I, равно применим к у и z в формулах II и IIa соответственно.
Дипазон подходящих и предпочтительных определений R1 и R2, как они предложены выше для формулы I, равно применим к R31 и R32 соответственно в формулах II и IIa.
В предпочтительном воплощении указанное соединение формулы II представляет собой:
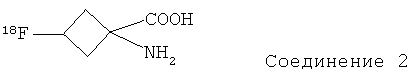 ,
,
а указанное соединение формулы На представляет собой:
 ,
,
где Et представляет собой этил, и Boc представляет собой трет-бутилоксикарбонил.
В предпочтительном воплощении стадии (2) и (3) осуществляют на автоматическом синтезаторе. Радиоактивные [18F]-индикаторы в настоящее время часто в целях удобства получают на автоматическом приборе для радиохимического синтеза. В продаже имеется несколько вариантов такого прибора, включая Tracerlab™ и Fastlab™ (оба от GE Healthcare Ltd.). Такой прибор обычно содержит "кассету", часто одноразовую, в которой проводят радиохимические превращения, которая вставляется в данный прибор для осуществления радиохимического синтеза. Кассета обычно содержит каналы для жидкостей, реакционный сосуд и отверстия для присоединения содержащих реагенты флаконов, а также любых картриджей для твердофазной экстракции, используемых на стадиях очистки после радиохимического синтеза.
Типичная кассета для автоматизированного синтеза соединения формулы II включает:
(1) сосуд, содержащий соединение формулы I, которое определено в данном описании; и
(2) средство для элюирования содержимого сосуда подходящим источником [18F]-фторида, который определен в данном описании;
(3) ионообменный картридж для удаления избытка [18F]-фторида и
(4) картридж для снятия защиты с соединения формулы IIa для образования соединения формулы II.
Далее изобретение будет описано с использованием нижеследующих экспериментальных примеров.
Краткое описание примеров
Пример 1 представляет собой пример сравнения, описывающий способ получения соединения формулы I из предшествующего уровня техники.
В примере 2 описан способ получения соединения формулы I согласно настоящему изобретению.
Список сокращений, использованных в примерах
Примеры
Пример 1. Способ получения соединения 1 из предшествующего уровня техники
1(а). Синтез и очистка соединения 1а
3-Бензилоксициклобутан-1-он получают согласно способу, описанному McConathy et al (AppI Radiat Isotop 2003, 58: 657-666). 3-Бензилоксициклобутан-1-он подвергают взаимодействию с цианидом калия, карбонатом аммония и хлоридом аммония. 5-(3-Бензилоксициклобутан)гидантоин выделяют из реакционной смеси путем кристаллизации и проводят реакцию раскрытия кольца в Ва(ОН)2 (насыщ.) при кипячении с обратным холодильником. Реакционную смесь нейтрализуют с использованием H2SO4, выпавший в осадок BaSO4 отфильтровывают и путем упаривания фильтрата выделяют аминокислоту. 1-Амино-3-бензилокси-циклобутанкарбоновую кислоту превращают в этиловый эфир 1-амино-3-бензилокси-циклобутанкарбоновой кислоты с помощью SOCl2 и Et3N в этаноле. После концентрирования реакционной смеси в вакууме получают этиловый эфир 1-амино-3-бензилокси-циклобутанкарбоновой кислоты, выделенный в виде солевой смеси. Аминогруппу защищают группой Boc, используя boc-ангидрид в Et3N и этаноле. Этиловый эфир 3-бензилокси-1-трет-бутоксикарбониламино-циклобутанкарбоновой кислоты (соединение 1а) выделяют экстрактивной обработкой с последующей флэш-хроматографией.
1(b). Синтез и очистка соединения 1b
Соединение 1а (полученное согласно Примеру 1(а); 31,83 г, 91 ммоль) растворяли в этаноле (600 мл) и уксусной кислоте (8 мл, 139 ммоль) в атмосфере N2 в реакционной колбе, присоединенной к источнику Н2. К полученной смеси добавляли увлажненный Pd на угле (6,28 г, 10% масс./масс.). Подачу N2 заканчивали и реакционную колбу осторожно вакуумировали и заполняли Н2; этот процесс повторяли дважды. При необходимости в реакционную смесь дополнительно добавляли H2. Реакционную смесь перемешивали при температуре окружающей среды в течение 2 суток до полной конверсии (за протеканием реакции следили посредством TLC). Реакционную смесь фильтровали через стекловолоконный фильтр, осадок на фильтре промывали этанолом (160 мл), затем фильтрат упаривали в вакууме при температуре ниже 40°C, что позволило получить неочищенное соединение 1b (24,64 г). Неочищенное соединение 1b перерастворяли в дихлорметане (500 мл), добавляли SiO2 (65 г) и упаривали в вакууме при температуре ниже 40°C, что позволило получить адсорбат для хроматографической очистки.
Система для флэш-хроматографии: SiO2 (360 г) загружали в стеклянную колонку ⌀=13 см до высоты приблизительно 5 см и обрабатывали гептаном, а затем гептаном с добавлением 30% этилацетата. В качестве адсорбата на верх колонки наносили неочищенное соединение. На верх колонки осторожно добавляли морской песок (88 г). Затем колонку элюировали: гептаном с добавлением 30% этилацетата (3 фракции, в сумме 2000 мл), гептаном с добавлением 50% этилацетата (8 фракций, в сумме 2750 мл) и гептаном с добавлением 70% этилацетата (8 фракций, в сумме 4000 мл). Продукт выделяли во фракциях 8-19, эти фракции объединяли и упаривали в вакууме при 38°C, что позволило получить Соединение 1b (20,1 г, 86%). Чистота по ГХ (газовая хроматография) 99,8%.
1(с). Синтез и очистка соединения 1
Соединение 1b (20,1 г, 78 ммоль) растворяли в дихлорметане (500 мл) и добавляли пиридин (19 мл, 235 ммоль), полученный раствор охлаждали до температуры ниже 5°C и порциями в течение 30 мин добавляли трифторметансульфоновый ангидрид (19,5 мл, 115 ммоль). Во время добавления температуру реакционной смеси поддерживали ниже 5°C, по завершении добавления реакционную смесь перемешивали на ледяной бане в течение 1 ч (за протеканием реакции следили посредством TLC), реакцию гасили путем добавления воды (500 мл). Смесь экстрагировали, используя Et2O (950 мл), водную фазу отбрасывали, органическую фазу промывали HCl (500 мл, 1М), рассолом (500 мл, насыщ. водн.) и сушили над Na2SO4 (56 г). Неочищенную смесь фильтровали через воронку с фильтром из пористого стекла, осадок на фильтре промывали Et2O (100 мл), объединенный фильтрат упаривали в вакууме при температуре ниже 30°C, что позволило получить неочищенное соединение 1 (28,11 г). Неочищенное соединение 1 перерастворяли в дихлорметане (400 мл), добавляли SiO2 (80 г) и упаривали в вакууме при температуре ниже 30°C, что позволило получить адсорбат для хроматографической очистки.
Система для флэш-хроматографии: SiO2 (330 г) загружали в стеклянную колонку ⌀=7 см до высоты приблизительно 19 см и обрабатывали смесью пентан : диэтиловый эфир (3:1). В качестве адсорбата на верх колонки наносили неочищенное соединение. На верх колонки осторожно добавляли морской песок (50 г). Затем колонку элюировали смесью пентан : диэтиловый эфир (3:1), размер фракции 250 мл; продукт выделяли во фракциях 5-12, которые объединяли и упаривали в вакууме при температуре ниже 30°C, что позволило получить соединение 1 (21,94 г). К этому веществу в колбе для упаривания добавляли диэтиловый эфир (50 мл) и медленно перемешивали на испарителе при температуре ниже 35°C до тех пор, пока не растворились все твердые вещества. Нагревание отключали и смесь медленно охлаждали до 25°C в течение 1 ч 5 мин; раствор медленно перемешивали при температуре окружающей среды в течение 1 ч 20 мин. Затем смесь охлаждали до температуры ниже 5°C и выдерживали при этой температуре в течение 20 мин, после чего смесь охлаждали до температуры ниже -20°C в течение 15 мин и перемешивали при этой температуре в течение 1 ч 30 мин. К раствору добавляли гептан (110 мл) и перемешивали в течение 1 ч 20 мин. Кристаллы собирали фильтрованием на предварительно охлажденной воронке с фильтром из пористого стекла и промывали ледяным гептаном (110 мл, ниже -5°C). В результате реакции получали соединение 1 (19,47 г; 64%), чистота по ЯМР (ядерный магнитный резонанс) +99%.
Пример 2. Способ по изобретению для получения соединения 1
0,5300 г неочищенного соединения 1b, полученного согласно способу, описанному в примере 1(b) (то есть включающему гидрирование, фильтрование и упаривание, но без флэш-хроматографии), растворяли в 5 мл абсолютного этанола при температуре окружающей среды. Раствор медленно концентрировали путем продувки азотом. Во время этого процесса образовывались зародыши кристаллов и росли кристаллы. Примерно через один час упаривание останавливали. Количество оставшегося этанола составило 0,3500 г (0,43 мл), и смесь содержала значительное количество кристаллов. Добавляли 1 мл н-гептана и продолжали упаривание путем продувки. Когда смесь растворителей была почти выпарена (осталось примерно 0,2 мл растворителя), упаривание останавливали и добавляли 1 мл н-гептана. Через 15 мин кристаллы отфильтровывали и промывали примерно 3 мл н-гептана. Кристаллы сушили в вакууме, фильтрат упаривали путем продувки азотом, а затем сушили в вакууме. Выход после выделения составил 0,4873 г кристаллов (91,9%) при степени извлечения 92,9%.
Кристаллы хорошо фильтруются, размер можно контролировать скоростью упаривания, а также скоростью добавления н-гептана.
Пример 2. Способ крупномасштабного получения очищенного соединения 1b
Неочищенное вещество: неочищенная реакционная смесь после гидрирования соединения 1а до соединения 1b в виде этанольного раствора после отфильтровывания катализатора и промывки. Этанол: 2,5-3,8 литра.
Оборудование: вакуумный испаритель, колба для упаривания, оборудование для фильтрования. Операцию сначала можно провести в большой колбе для упаривания, а после уменьшения объема осуществить перенос в небольшую колбу. Альтернативно, в небольшой колбе объемом 500 или 1000 мл, пополняя содержимое непрерывно или малыми порциями.
1. Прозрачный раствор концентрируют путем упаривания в колбе для вакуумирования до суммарного объема 100-200 мл. В растворе образуются зародыши кристаллов, и продукт кристаллизуется с образованием густой суспензии.
2. Добавляют 200 мл н-гексана и через 10 мин перемешивания (путем вращения) суспензию концентрируют до объема примерно 150 мл.
3. Добавляют новую порцию н-гептана (200 мл) и повторяют стадию 2.
4. Через 30 мин вращения (при температуре окружающей среды или ниже) фильтруют суспензию и н-гептаном промывают кристаллы.
5. Кристаллы сушат в вакууме.
| название | год | авторы | номер документа |
|---|---|---|---|
| УПРОЩЕНИЕ СПОСОБА ПОЛУЧЕНИЯ СОЕДИНЕНИЯ-ПРЕДШЕСТВЕННИКА | 2011 |
|
RU2593372C2 |
| ПОЛУЧЕНИЕ ПРЕДШЕСТВЕННИКА ДЛЯ ПЭТ | 2011 |
|
RU2587308C2 |
| СОЕДИНЕНИЕ-ПРЕДШЕСТВЕННИК ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ, МЕЧЕННОГО РАДИОАКТИВНЫМ ГАЛОГЕНОМ | 2006 |
|
RU2428415C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАДИОАКТИВНОГО, МЕЧЕННОГО ФТОРОМ ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ | 2008 |
|
RU2476423C2 |
| ПРОЦЕСС ПОЛУЧЕНИЯ СОЕДИНЕНИЙ-ПРЕДШЕСТВЕННИКОВ ДЛЯ РАДИОАКТИВНЫХ ГАЛОГЕНПОМЕЧЕННЫХ СОЕДИНЕНИЙ | 2007 |
|
RU2466984C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАДИОАКТИВНОГО ФТОР-МЕЧЕННОГО ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ | 2007 |
|
RU2434846C2 |
| Катализируемый галогенидом цинка способ циклизации, приводящий к образованию трициклических индолов | 2013 |
|
RU2647867C2 |
| СПОСОБ ФТОРИРОВАНИЯ АНИЛИДНЫХ ПРОИЗВОДНЫХ И ФТОРИРОВАННЫЕ ПРОИЗВОДНЫЕ БЕНЗОТИАЗОЛА В КАЧЕСТВЕ IN VIVO ВИЗУАЛИЗИРУЮЩИХ АГЕНТОВ | 2006 |
|
RU2419601C2 |
| СТЕРЕОСЕЛЕКТИВНЫЙ СИНТЕЗ АМИНОКИСЛОТ ДЛЯ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЯ ОПУХОЛИ | 2006 |
|
RU2376282C2 |
| Получение 18F-флуцикловина | 2013 |
|
RU2640805C2 |
Изобретение относится к способу получения предшественников для радиофармацевтических препаратов, применяемых в позитрон-эмиссионной томографии. Конкретно изобретение относится к способу получения соединения формулы I, в которой R1 представляет собой С1-5алкильную группу с прямой или разветвленной цепью; R2 представляет собой амино-защитную группу; v является целым числом от 0 до 4; X представляет собой уходящую группу, выбранную из галогена или группы -O-SO2-R3, где R3 представляет собой галоген, С1-10алкил с прямой цепью или разветвленной цепью, С1-10галогеналкил с прямой цепью или разветвленной цепью и С6-10арил. Способ включает (а) дебензилирование соединения формулы Ia, где R11, R12 и w являются такими, как определено для R1, R2 и v в формуле I соответственно; (б) кристаллизацию реакционной смеси со стадии (а) с получением очищенного соединения формулы Ib, где R21, R22 и х являются такими, как определено для R1, R2 и v в формуле I соответственно; (в) превращение очищенного соединения формулы Ib, полученного на стадии (б), в соединение формулы I путем взаимодействия с подходящей формой X, где X является таким, как определено для формулы I. Способ позволяет повысить выход целевого соединения, а также достигнуть некоторого обогащения по син-изомеру при кристаллизации. Изобретение относится также к способу получения соединения формулы II с использованием способа получения соединения формулы I. 2 н. и 17 з.п. ф-лы, 2 пр.
1. Способ получения соединения формулы I
где
R1 представляет собой С1-5алкильную группу с прямой или разветвленной цепью;
R2 представляет собой амино-защитную группу;
v является целым числом от 0 до 4; и
X представляет собой уходящую группу, выбранную из галогена или группы -O-SO2-R3, где R3 представляет собой галоген, С1-10алкил с прямой цепью или разветвленной цепью, С1-10галогеналкил с прямой цепью или разветвленной цепью и С6-10арил;
включающий:
(а) дебензилирование соединения формулы Ia
где R11, R12 и w являются такими, как определено для R1, R2 и v в формуле I соответственно;
(б) кристаллизацию реакционной смеси со стадии (а) с получением очищенного соединения формулы Ib
где R21, R22 и х являются такими, как определено для R1, R2 и v в формуле I соответственно;
(в) превращение очищенного соединения формулы Ib, полученного на стадии (б), в соединение формулы I путем взаимодействия с подходящей формой X, где X является таким, как определено для формулы I.
2. Способ по п.1, где R1, R11 и R21 представляют собой этил.
3. Способ по п.1, где R2, R12 и R22 выбраны из группы, состоящей из трет-бутоксикарбонильной группы, аллилоксикарбонильной группы, фталимидной группы и N-бензилиденаминного заместителя.
4. Способ по п.1, где v, w и х равны 0 или 1.
5. Способ по п.1, где X представляет собой группу -O-SO2-R3.
6. Способ по п.5, где X выбран из толуолсульфоновой группы, нитробензолсульфоновой группы, бензолсульфоновой группы, трифторметансульфоновой группы, фторсульфоновой группы, перфторалкилсульфоновой группы, триметилстаннилсульфоновой группы и триэтилстаннилсульфоновой группы.
7. Способ по п.6, где X представляет собой трифторметансульфоновую группу.
8. Способ по любому из пп.1-7, где указанное соединение формулы I представляет собой
указанное соединение формулы Ib представляет собой
 и
и
указанное соединение формулы Ia представляет собой
где Et представляет собой этил, OTf представляет собой трифторметансульфоновую группу и Boc представляет собой трет-бутилоксикарбонил.
9. Способ по п.1, где X представляет собой галоген.
10. Способ по п.9, где указанный галоген представляет собой бром или хлор.
11. Способ получения соединения формулы II
где у является таким, как определено для v в п.1, радиохимическим синтезом, включающий:
(1) получение соединения формулы I способом по п.1;
(2) взаимодействие указанного соединения формулы I с подходящим источником 18F-фторида с получением соединения формулы IIa
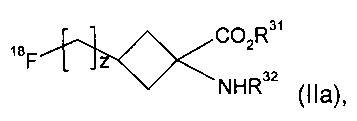
где R31, R32 и z являются такими, как определено в п.1 для R1, R2 и v соответственно; и
(3) снятие защиты с соединения формулы IIa, полученного на стадии (2), для удаления R31 и R32.
12. Способ по п.11, где указанный источник 18F-фторида представляет собой 18F-фторид в присутствии противоиона, где указанный противоион выбран из рубидия, цезия, калия в комплексе с криптандом или тетраалкиламмониевой соли.
13. Способ по п.11, где указанное снятие защиты включает удаление R31 с последующим удалением R32.
14. Способ по п.11, где у и z являются одинаковыми и равны 0 или 1.
15. Способ по п.11, где R31 представляет собой этил.
16. Способ по п.11, где R32 представляет собой трет-бутоксикарбонильную группу.
17. Способ по п.11, где указанное соединение формулы II представляет собой
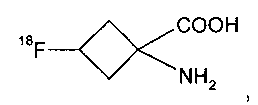
и указанное соединение формулы IIa представляет собой
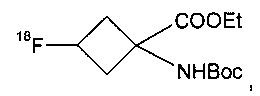
где Et представляет собой этил и Boc представляет собой трет-бутилоксикарбонил.
18. Способ по п.17, где указанная стадия снятия защиты включает удаление Et основным гидролизом и удаление Boc кислотным гидролизом.
19. Способ по любому из пп.11-18, где стадии (2) и (3) осуществляют на автоматическом синтезаторе.
| EP 1978015 A1, 08.10.2008 | |||
| СПОСОБ ДИАГНОСТИРОВАНИЯ УРАВНОВЕШЕННОСТИ СТАНКОВ-КАЧАЛОК ШТАНГОВЫХ НАСОСНЫХ УСТАНОВОК | 2002 |
|
RU2230229C1 |
| L | |||
| J | |||
| WANG et al., Syntheses of New Conformationally Constrained S-[2-[(1-Iminoethyl)amino]ethyl]homocysteine Derivatives as Potential Nitric Oxide Synthase Inhibitors, HETEROATOM CHEMISTRY, 2002, Vol | |||
| Насос | 1917 |
|
SU13A1 |
| RU 2008148851 A, 20.06.2010. | |||

