ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям, которые обладают ингибирующим действием по отношению к связыванию сфингозин-1-фосфата, имеющим различные физиологические эффекты (действия), с его рецептором Edg-1 (рецептор 1 типа гена дифференциации эндотелиальных клеток, S1P1), а также относится к фармацевтическим препаратам, включающим эти соединения в качестве действующих ингредиентов.
УРОВЕНЬ ТЕХНИКИ
Сфингозин-1-фосфат (далее обозначаемый как "S1P") является физиологически активным липидом, который образуется при метаболизме сфинголипидов (например, сфингомиелина) в клетках. Известно, что S1P обладает широким спектром действия, таким как индуцирование клеточной дифференцировки, стимулирование роста клеток, регулирование подвижности клетки и ингибирование апоптоза, и также известно, что он проявляет физиологическое действие, такое как ангиогенез, индуцирование брадикардии, активация клеток воспаления и активация тромбоцитов (Непатентный документ 1).
Что касается S1P рецепторов, то известны следующие 5 подтипов: Edg-1(S1P1), Edg-3(S1P3), Edg-5(S1P2), Edg-6(S1P4) и Edg-8(S1P5) (Непатентный документ 2).
Среди этих подтипов, Edg-1(S1P1) экспрессирует исключительно в иммуноцитах (например, T-клетках, дендритных клетках) и васкулярных эндотелиальных клетках, что указывает на то, что Edg-1(S1P1) активно способствует S1P-стимулированной миграции T-клеток (Непатентный документ 3), миграции тучных клеток (Непатентный документ 4) и выходу T- и B-клеток из лимфоидных органов (Непатентный документ 5) и ангиогенезу (Непатентный документ 6) и что он связан с аутоиммунными заболеваниями, такими как болезнь Крона, синдром раздраженной толстой кишки, синдром Шегрена, рассеянный склероз и системная красная волчанка, а также другими заболеваниями, такими как ревматоидный артрит, астма, атопический дерматит, отторжение после трансплантации органа, рак, ретинопатия, псориаз, остеоартрит, возрастная дегенерация желтого пятна и т.д.
Следовательно, лиганды, которые могут связывать Edg-1(S1P1), должны быть эффективными при лечении и профилактике этих заболеваний.
Известные на настоящий момент Edg-1(S1P1) лиганды включают конкретные типы производных тиофена (Непатентный документ 7), производных фосфорной кислоты (Патентные документы 1 и 2, Непатентные документы 8 и 9) и производных тиазолидина (Патентный документ 3), производных карбоновой кислоты (Патентные документы 4, 5, 6 и 8, Непатентные документы 10 и 11), производных, содержащих аминогруппу (Патентный документ 7), производных пиррола (Патентный документ 9) и производных триазола (Патентные документы 10 и 11).
Патентный документ 1: WO2002/18395
Патентный документ 2: JP 2003-137894 A
Патентный документ 3: JP 2002-332278 A
Патентный документ 4: WO2002/092068
Патентный документ 5: WO2003/105771
Патентный документ 6: WO2004/058149
Патентный документ 7: WO2004/103279
Патентный документ 8: WO2005/058848
Патентный документ 9: WO2005/123677
Патентный документ 10: WO2006/013948
Патентный документ 11: WO2007/083089
Непатентный документ 1: J Biol Chem. 2004, 279:20555, FASEB J 2002, 16:625, Proceedings of the Japanese Society for Immunology 2003, 33:2-J-W30-20-P
Непатентный документ 2: Pharmacol Res 2003, 47:401
Непатентный документ 3: FASEB J 2002, 16:1874
Непатентный документ 4: J Exp Med 2004, 199:959
Непатентный документ 5: Nature 2004, 427:355
Непатентный документ 6: J Clin Invest 2000, 106:951, Biocchim Biophys Acta 2002, 1582:222
Непатентный документ 7: J Biol Chem 2004, 279:13839
Непатентный документ 8: Bioorg Med Chem Lett 2003, 13:3401
Непатентный документ 9: J Biol Chem. 2005; 280:9833
Непатентный документ 10: J Med Chem. 2004, 47:6662
Непатентный документ 11: J Med Chem. 2005, 48:6169
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ЗАДАЧИ, РЕШАЕМЫЕ ИЗОБРЕТЕНИЕМ
Задачей настоящего изобретения является разработка соединений с новой структурой, которые обладают ингибирующим действием по отношению к связыванию S1P с его рецептором Edg-1(S1P1) и которые применяют для фармацевтических целей.
СРЕДСТВА РЕШЕНИЯ ЗАДАЧ
В результате обширных и интенсивных поисков соединений, которые могут служить лигандами для Edg-1(S1P1), авторы настоящего изобретения обнаружили, что эта задача может быть решена путем применения соединения следующей формулы (I) или ее фармацевтически приемлемой соли. Это открытие и привело к созданию настоящего изобретения.
Далее будут приведены варианты осуществления соединения формулы (I) (далее называемого как "соединение настоящего изобретения").
(1)
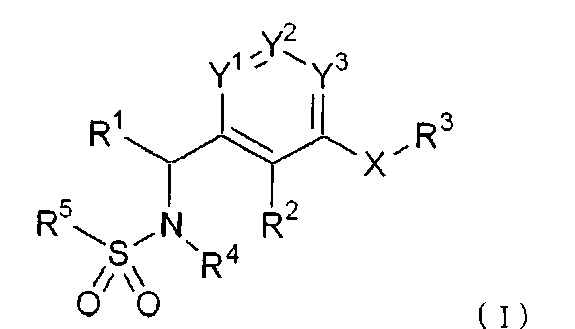
Соединение, представленное формулой (I), или его фармацевтически приемлемая соль
Формула 1
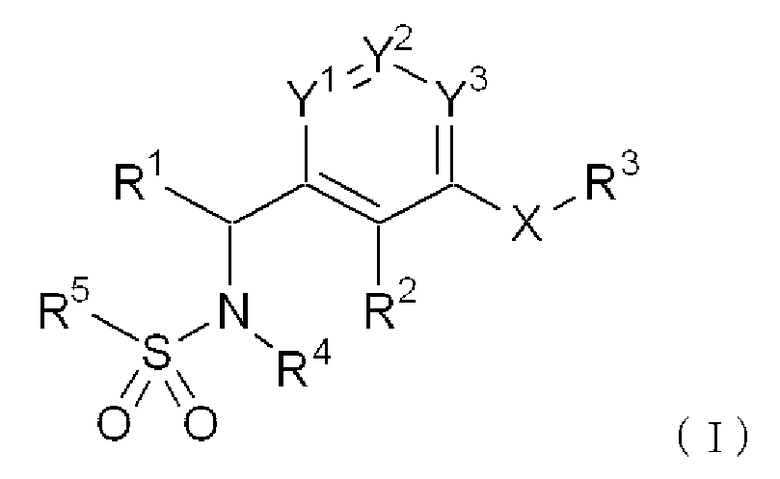
{где Y1 представляет атом азота или группу, представленную CRA, Y2 представляет атом азота или группу, представленную CRB, Y3 представляет атом азота или группу, представленную CRC, где
RA, RB и RC, которые могут быть одинаковыми или различными, и каждая представляет атом водорода или C1-C6алкильную группу (за исключением случая, когда Y1 является CRA, Y2 является CRB и Y3 является CRC),
X представляет атом кислорода, атом серы, группу, представленную формулой -SO-, группу, представленную формулой -SO2-, или группу, представленную формулой -NR6- (где R6 представляет атом водорода или C1-C6алкильную группу),
R1 представляет атом водорода, C1-C6 алкильную группу или бензильную группу,
R2 представляет атом водорода, C1-C6алкильную группу или C3-C8циклоалкильную группу,
R3 представляет
(i) C1-C6алкильную группу, которая может быть замещена от 1 до 3 заместителями, выбранными из группы A [где группа A состоит из атома галогена, фенильной группы, C1-C6алкоксигруппы, аминогруппы, которая может быть замещена одной или двумя C1-C6алкильными группами, морфолиногруппы, и пиперазиногруппы, которая может быть замещена C1-C6алкильной группой (алкильными группами)],
(ii) C3-C8циклоалкильную группу, или
(iii) фенильную группу, нафтильную группу или изохинолинильную группу, каждая из которых может быть замещена от 1 до 3 заместителями, выбранными из группы B [где группа B состоит из атома галогена, C1-C6алкильной группы, C1-C6алкоксигруппы, аминогруппы, которая может быть замещена одной или двумя C1-C6алкильными группами, морфолиногруппы, пиперазиногруппы, которая может быть замещена C1-C6алкильной группой (алкильными группами), C1-C6алканоиламиногруппой и C1-C6алкилсульфониламиногруппой],
R4 представляет атом водорода или C1-C6алкильную группу, и
R5 представляет фенильную группу, тиенильную группу, тиазолильную группу, пиридильную группу, нафтильную группу, инданильную группу, дигидробензофуранильную группу, бензодиоксолильную группу, бензотиадиазолильную группу, бензотиенильную группу или хинолинильную группу, каждая из которых может быть замещена от 1 до 5 заместителями, выбранными из группы C [где группа C состоит из C1-C6алкильной группы, атома галогена, трифторметильной группы, C1-C6алкоксигруппы, трифторметоксигруппы, нитрогруппы, цианогруппы и C2-C7алканоильной группы], либо C2-C8алкенильную группу, которая может быть замещена фенильной группой (фенильными группами)}.
(2)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) Y1 является атомом азота или CH, Y2 является CRB и Y3 является атомом азота или CH.
(3)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) Y1 и Y2 каждая является CH и Y3 является атомом азота.
(4)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) X является атомом кислорода.
(5)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) R1 является атомом водорода или C1-C6алкильной группой.
(6)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) R1 является атомом водорода, метильной группой или этильной группой.
(7)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) R2 является атомом водорода или C1-C6алкильной группой.
(8)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) R2 является метильной группой или этильной группой.
(9)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) R4 является атомом водорода.
(10)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) R3 является фенильной группой, нафтильной группой или изохинолинильной группой, каждая из которых может быть замещена от 1 до 3 заместителями, выбранными из группы D [где группа D состоит из атома галогена, C1-C6алкильной группы, C1-C6алкоксигруппы, аминогруппы, которая может быть замещена одной или двумя C1-C6алкильными группами, морфолиногруппы и пиперазиногруппы, которая может быть замещена C1-C6алкильной группой (алкильными группами)].
(11)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) R3 является фенильной группой, в которой мета-положение замещено одним заместителем, выбранным из группы E [где группа E состоит из аминогруппы, которая может быть замещена одной или двумя C1-C6алкильными группами, морфолиногруппы и пиперазиногруппы, которая может быть замещена C1-C6алкильной группой (алкильными группами)].
(12)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) R5 является фенильной группой, тиенильной группой, нафтильной группой, дигидробензофуранильной группой, бензодиоксолильной группой, бензотиадиазолильной группой, бензотиенильной группой или хинолинильной группой, каждая из которых может быть замещена от 1 до 3 заместителями, выбранными из группы F [где группа F состоит из C1-C6алкильной группы, атома галогена, трифторметильной группы, C1-C6алкоксигруппы, нитрогруппы, цианогруппы и C2-C7алканоильной группы], или C2-C8алкенильной группы, которая может быть замещена фенильной группой (фенильными группами).
(13)
Соединение или его фармацевтически приемлемая соль, согласно приведенному выше пункту (1), где в формуле (I) R5 является фенильной группой, замещенной двумя или тремя атомами галогена, или нафтильной группой, замещенной одним или двумя атомами галогена.
(14)
Фармацевтический препарат, включающий соединение или его фармацевтически приемлемую соль согласно любому одному из приведенных выше пунктов (1)-(13).
(15)
Фармацевтический препарат согласно приведенному выше пункту (14), который является лечебным средством при аутоиммунном заболевании, таком как болезнь Крона, синдром раздраженной толстой кишки, синдром Шегрена, рассеянный склероз или системная красная волчанка, ревматоидный артрит, астма, атопический дерматит, отторжение после трансплантации органа, рак, ретинопатия, псориаз, остеоартрит или возрастная дегенерация желтого пятна.
ПРЕИМУЩЕСТВА ИЗОБРЕТЕНИЯ
Было обнаружено, что соединения настоящего изобретения являются лигандами, которые прочно связывают Edg-1(S1P1), как это показано в описанном далее примере испытания.
НАИЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение будет описано более подробно.
Предполагается, что используемая здесь фраза "за исключением случая, когда Y1 является CRA, Y2 является CRB и Y3 является CRC" означает, что ароматическое кольцо, содержащее Y1, Y2 и Y3 в качестве его конституентных атомов, не является бензольным кольцом. То есть это означает, что, по меньшей мере, один из Y1, Y2 и Y3 является атомом азота.
Термин "атом галогена" относится к атому фтора, атому хлора, атому брома или атому йода.
Термин "C1-C6алкильная группа" относится к линейной или разветвленной алкильной группе, содержащей от 1 до 6 углеродных атомов. Примеры включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, трет-бутильную группу, втор-бутильную группу, н-пентильную группу, изопентильную группу, неопентильную группу, трет-пентильную группу, и н-гексильную группу.
Термин "C3-C8циклоалкильная группа" относится к циклоалкильной группе, содержащей от 3 до 8 углеродных атомов. Примеры включают циклопропильную группу, циклобутильную группу, циклопентильную группу, и циклогексильную группу.
Термин "C2-C8алкенильная группа" относится к линейной или разветвленной алкенильной группе, содержащей от 2 до 8 углеродных атомов. Примеры включают винильную группу, аллильную группу, 1-пропенильную группу, изопропенильную группу, 1-бутенильную группу, 2-бутенильную группу, 3-бутенильную группу, 1,3-бутадиенильную группу, 2-метилаллильную группу, 2-метилпропенильную группу, 2-пентенильную группу и 3-метилбут-2-енильную группу.
Термин "C1-C6алкоксигруппа" относится к линейной или разветвленной алкоксигруппе, содержащей от 1 до 6 углеродных атомов. Примеры включают метоксигруппу, этоксигруппу, пропоксигруппу, изопропоксигруппу, бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу, трет-бутоксигруппу, пентилоксигруппу и гексилоксигруппу.
Термин "C1-C6алкилсульфонильная группа" относится к линейной или разветвленной алкилсульфонильной группе, содержащей от 1 до 6 углеродных атомов. Примеры включают метансульфонильную группу, этансульфонильную группу, пропан-2-сульфонильную группу и гексансульфонильную группу.
Термин "C1-C6алкилсульфониламиногруппа" относится к группе, состоящей из определенной выше C1-C6алкилсульфонильной группы и присоединенной к ней аминогруппы. Примеры включают метансульфониламиногруппу, этансульфониламиногруппу, пропан-2-сульфониламиногруппу и гексансульфониламиногруппу.
Термин "C2-C7алканоильная группа" относится к линейной или разветвленной алканоильной группе, содержащей от 2 до 7 углеродных атомов. Примеры включают ацетильную группу, пропаноильную группу, бутаноильную группу и гексаноильную группу.
Термин "C1-C6алканоильная группа" относится к линейной или разветвленной алканоильной группе, содержащей от 1 до 6 углеродных атомов. Примеры включают формильную группу, ацетильную группу, пропаноильную группу и бутаноильную группу.
Термин "C1-C6алканоиламиногруппа" относится к группе, состоящей из определенной выше C1-C6алканоильной группы и присоединенной к ней аминогруппы. Примеры включают формиламиногруппу, ацетиламиногруппу, пропаноиламиногруппу и бутаноиламиногруппу.
Предполагается, что фраза "аминогруппа, которая может быть замещена одной или двумя C1-C6алкильными группами", включает, например, аминогруппу, метиламиногруппу, этиламиногруппу, изопропиламиногруппу, гексиламиногруппу, диметиламиногруппу, диэтиламиногруппу, диизопропиламиногруппу и дигексиламиногруппу.
Фраза "пиперазиногруппа, которая может быть замещена C1-C6алкильной группой (алкильными группами)", относится к пиперазиногруппе, которая может быть замещена линейной или разветвленной алкильной группой (алкильными группами), содержащей от 1 до 6 углеродных атомов. Примеры включают пиперазиногруппу, метилпиперазиногруппу и изопропил-пиперазиногруппу.
Термин "фармацевтически приемлемая соль" относится к соли щелочного металла, щелочноземельного металла, аммония или алкиламмония, или соли минеральной кислоты, или органической кислоты. Примеры включают соль натрия, соль калия, соль кальция, соль аммония, соль алюминия, соль триэтиламмония, ацетатную соль, пропионатную соль, бутиратную соль, формиатную соль, трифторацетатную соль, малеатную соль, тартратную соль, цитратную соль, стеаратную соль, сукцинатную соль, этилсукцинатную соль, лактобионатную соль, глюконатную соль, глюкогептатную соль, бензоатную соль, метансульфонатную соль, этансульфонатную соль, 2-гидроксиэтансульфонатную соль, бензолсульфонатную соль, паратолуолсульфонатную соль, лаурилсульфатную соль, малатную соль, аспартатную соль, глутаматную соль, адипатную соль, соль цистеина, соль N-ацетилцистеина, гидрохлоридную соль, гидробромидную соль, фосфатную соль, сульфатную соль, гидройодидную соль, никотинатную соль, оксалатную соль, пикратную соль, тиоцианатную соль, ундеканоатную соль, соль с акрилатным полимером и соль с карбоксивинильным полимером.
Соединения настоящего изобретения могут иметь стереоизомеры, включающие оптические изомеры, диастереоизомеры и геометрические изомеры. Все эти стереоизомеры и их смеси также входят в объем настоящего изобретения. Могут также существовать некоторые соединения и промежуточные соединения, например, такие как кетоенольные таутомеры.
Далее будут приведены предпочтительные варианты соединения настоящего изобретения.
Предпочтительно, чтобы Y1 являлся атомом азота или CH, предпочтительно, чтобы Y2 являлся CRB, и предпочтительно, чтобы Y3 являлся атомом азота или CH. Более предпочтительно, чтобы Y1 и Y2 каждый являлся CH, и Y3 являлся атомом азота.
Предпочтительным примером X является атом кислорода.
Предпочтительным примером R1 является атом водорода или C1-C6алкильная группа. Более предпочтительными являются атом водорода, метильная группа или этильная группа, и еще более предпочтительной является метильная группа.
Предпочтительным примером R2 является атом водорода или C1-C6алкильная группа. Более предпочтительными являются метильная группа или этильная группа, и еще более предпочтительной является этильная группа.
Предпочтительным примером R4 является атом водорода.
Предпочтительным примером R3 является фенильная группа, у которой мета-положение замещено одним заместителем, выбранным из группы E [где группа E состоит из аминогруппы, которая может быть замещена одной или двумя C1-C6алкильными группами, морфолиногруппы и пиперазиногруппы, которая может быть замещена C1-C6алкильной группой (алкильными группами)]. Более предпочтительными являются 3-(4-метилпиперазино)фенильная группа или 3-морфолинофенильная группа.
Предпочтительным примером R5 является фенильная группа, замещенная двумя или тремя атомами галогена, или нафтильная группа, замещенная одним или двумя атомами галогена. Более предпочтительными являются 3,4-дихлорфенильная группа, 2,3,4-трихлорфенильная группа или 5-хлор-2-нафтильная группа.
Предпочтительными оптически активными формами соединений настоящего изобретения являются соединения, имеющие следующую структуру:
Формула 2
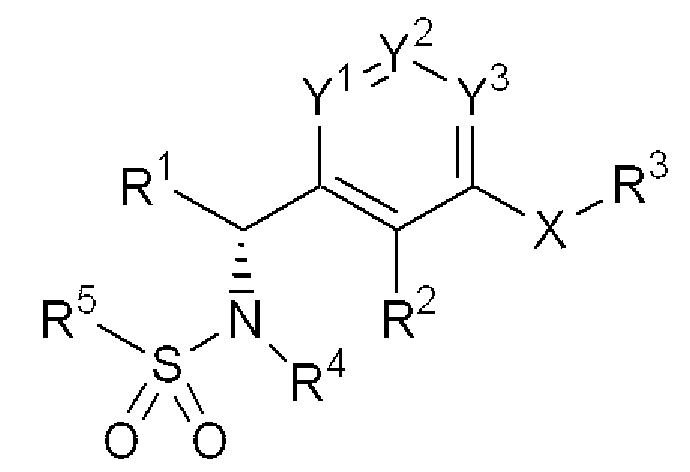
Соединения настоящего изобретения могут быть синтезированы при помощи приведенных далее в качестве примера методик. Следует отметить, что следующие методики приводятся в целях иллюстрации, и методики синтеза соединений настоящего изобретения не являются ограничениями для изобретения.
Формула 3
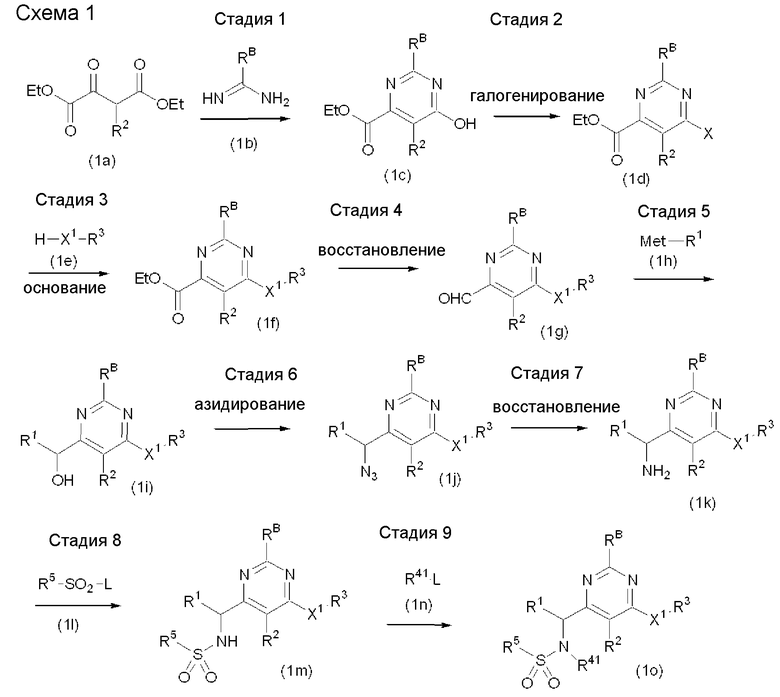
Формула 4
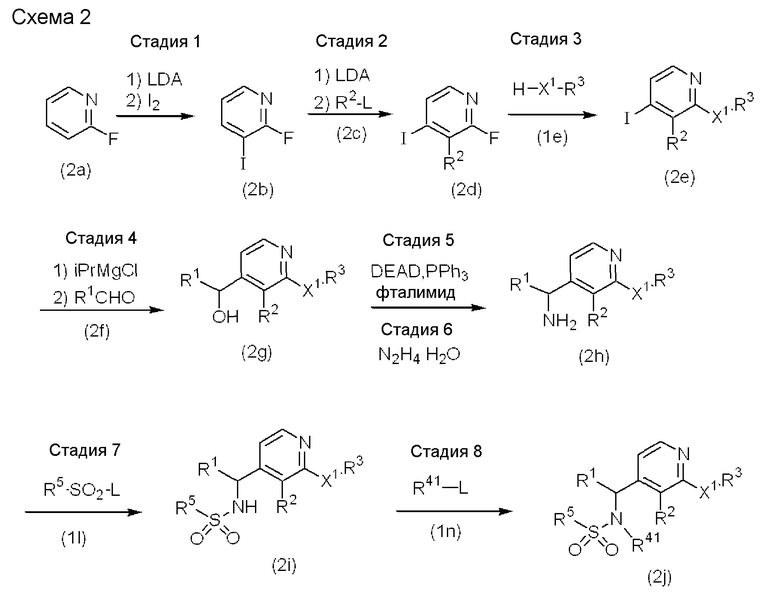
Формула 5
Формула 6
В приведенных выше стадиях, Y1, Y2, Y3, RB, R1, R2, R3, R4, R5 и X определены выше, R41 определяется так же, как R4, но за исключением атома водорода, X1 представляет атом кислорода, атом серы или группу, представленную формулой -NR6- (где R6 представляет атом водорода или C1-C6алкильную группу), Met представляет типичный металл или комплекс типичного металла с лигандом, таким как атом галогена и т.д. (например, Li, Na, MgCl2, MgBr2), и L представляет уходящую группу (где уходящей группой может являться, например, атом галогена, такой как атом хлора, атом брома или атом йода, ацетилоксигруппа, метансульфонилоксигруппа или п-толуолсульфонилоксигруппа).
Схема 1
Стадия 1: Соединение, представленное формулой (1a), может быть приведено во взаимодействие с соединением, представленным формулой (1b), с получением соединения, представленного формулой (1c).
Стадия 2: Соединение, представленное формулой (1c), может быть приведено во взаимодействие с галогенирующим реагентом с получением соединения, представленного формулой (1d). Примеры галогенирующего реагента включают POCl3, PCl5 и SOCl2. Количество используемого галогенирующего реагента обычно составляет от 1 до 10 эквивалентов, предпочтительно от 5 до 10 эквивалентов соединения, представленного формулой (1c). Если необходим растворитель, то может быть использован любой растворитель при условии, что он является инертным в отношении данной реакции, включая галогенированные растворители (например, CCl4, CHCl3, CH2Cl2), DMF, DMA, NMP, DMPU, HMPA, DMSO или их смеси. Температура реакции изменяется в пределах от 0°C до температуры кипения растворителя, предпочтительно от комнатной температуры до температуры кипения растворителя. Хотя время реакции может меняться в зависимости от температуры реакции и/или исходного реагента, обычно оно составляет от 1 часа до 12 часов.
Стадия 3: Соединение, представленное формулой (1d), может быть приведено во взаимодействие с соединением, представленным формулой (1e), в присутствии основания с использованием или без использования растворителя с получением соединения, представленного формулой (1f). Количество используемого соединения (1e) обычно составляет от 1 до 5 эквивалентов, предпочтительно от 1 до 3 эквивалентов соединения, представленного формулой (1d). Примеры основания включают соли щелочных металлов (например, Na2CO3, K2CO3, Cs2CO3, NaHCO3, KHCO3, NaOH, NaH, NaNH2, трет-BuOK, трет-BuONa), амины (например, Et3N, iPr2NEt, iPr2NH, пирролидин, пиперидин), AcONa и AcOK. Количество используемого основания обычно составляет от 1 до 10 эквивалентов, предпочтительно от 1 до 3 эквивалентов соединения, представленного формулой (1d). Температура реакции изменяется в пределах от 0°C до 300°C, и реакцию можно проводить, например, при нормальном давлении, при повышенном давлении или при микроволновом излучении. Примеры реакционного растворителя, который может быть использован, включают эфиры (например, диоксан, THF, Et2O), DMF, DMA, NMP, DMPU, HMPA, DMSO или их смеси. В случае необходимости вводят добавки. Примеры добавки включают соли металлов (например, CuI, CuCl) или порошок меди. Хотя время реакции может меняться в зависимости от температуры реакции и/или исходного реагента, оно обычно составляет от 1 часа до 12 часов.
Стадия 4: Соединение, представленное формулой (1f), может быть приведено во взаимодействие с восстановителем с получением соединения, представленного формулой (1g). Примеры восстановителя включают NaBH4, KBH4, LiB(H)Et3, LiB(втор-Bu)3H, (i-Bu)2AlH, AlH(O-трет-Bu)3, LiAlH4, LiHAl(O-трет-Bu)3 и NaH2Al(OCH2CH2OCH3). Количество восстановителя составляет от 0,5 до 5 эквивалентов, предпочтительно от 0,5 до 1,2 эквивалентов соединения, представленного формулой (1f). Примеры растворителя, который может быть использован, включают эфиры (например, диоксан, THF, диэтиловый эфир), гексан, бензол, толуол или их смеси. Температура реакции изменяется в пределах от -78°C до комнатной температуры, предпочтительно от -78°C до 0°C. Хотя время реакции может меняться в зависимости от температуры реакции и/или исходного реагента, оно обычно составляет от 30 минут до 4 часов.
Стадия 5: Соединение, представленное формулой (1g), может быть приведено во взаимодействие с соединением, представленным формулой (1h), с получением соединения, представленного формулой (1i). Количество используемого соединения, представленного формулой (1h), составляет от 1 до 10 эквивалентов, предпочтительно от 1,1 до 1,5 эквивалентов соединения, представленного формулой (1g). Примеры растворителя, который может быть использован, включают эфиры (например, диоксан, THF, Et2O) или их смеси. Температура реакции изменяется в пределах от -78°C до комнатной температуры, предпочтительно от -30°C до 0°C. Хотя время реакции может меняться в зависимости от температуры реакции и/или исходного реагента, оно обычно составляет от 30 минут до 24 часов.
Стадия 6: Соединение, представленное формулой (1i), может быть приведено во взаимодействие с метансульфонилхлоридом, п-толуолсульфонилхлоридом, трифлатным ангидридом или другим подобным реагентом в растворителе и, в случае необходимости, в присутствии основания, такого как пиридин или триэтиламин, с последующим взаимодействием с азидирующим реагентом (например, NaN3, LiN3, Zn(N3)2), или, в качестве варианта, может быть непосредственно обработано смесью диэтил азодикарбоксилат (DEAD)/PPh3/NH3, смесью дифенилфосфорилазид (DPPA)/1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), смесью Zn(N3)2/2 пиридин или другими подобными реагентами с получением соединения, представленного формулой (1j). Примеры растворителя включают эфиры (например, диоксан, THF), галогенированные растворители (например, CH3CN, CCl4, CHCl3, CH2Cl2), бензол и толуол.
Стадия 7: Соединение, представленное формулой (1j), может быть приведено во взаимодействие с восстановителем в растворителе и, в случае необходимости, в присутствии катализатора (например, Pd/C, Pd(OH)2/C, PtO2) с получением соединения, представленного формулой (1k). Примеры восстановителя включают водород, формиат аммония, гидразин, PPh3, и Mg. Примеры растворителя включают эфиры (например, диоксан, THF, Et2O), спирты (например, MeOH, EtOH) и AcOEt.
Стадия 8: Соединение, представленное формулой (1k), может быть приведено во взаимодействие с соединением, представленным формулой (1l), в присутствии основания с использованием или без использования растворителя, с последующим образованием при необходимости соли с получением соединения, представленного формулой (1m), или его фармацевтически приемлемой соли. Количество используемого соединения, представленного формулой (1l), составляет обычно от 1 до 5 эквивалентов, предпочтительно от 1 до 1,2 эквивалентов соединения, представленного формулой (1k). Примеры основания, которое может быть использовано, включают гидроксиды щелочных металлов (например, NaOH, KOH), соли щелочных металлов (например, NaHCO3, K2CO3) и амины (например, Et3N, iPr2NEt, iPr2NH). Количество основания обычно составляет от 1 до 10 эквивалентов, предпочтительно от 1,0 до 3,0 эквивалентов соединения, представленного формулой (1k). Температура реакции изменяется в пределах от 0°C до температуры кипения растворителя, предпочтительно от 0°C до комнатной температуры. Если необходим растворитель, то любой растворитель может быть использован при условии, что он должен быть инертным в отношении реакции, включая галогенированные углеводороды (например, CHCl3, CH2Cl2), эфиры (например, диоксан, THF, Et2O) или их смеси. Хотя время реакции может меняться в зависимости от температуры реакции и/или исходного реагента, оно обычно составляет от 30 минут до 24 часов.
Стадия 9: Соединение, представленное формулой (1m), может быть приведено во взаимодействие с соединением, представленным формулой (1n), в присутствии основания с использованием или без использования растворителя, с последующим образованием при необходимости соли с получением соединения, представленного формулой (1o), или его фармацевтически приемлемой соли. Количество используемого соединения, представленного формулой (1n), составляет от 1 до 10 эквивалентов, предпочтительно от 1,1 до 1,5 эквивалентов соединения, представленного формулой (1m). Примеры основания, которое может быть использовано, включают гидроксиды щелочных металлов (например, NaOH, KOH), соли щелочных металлов (например, NaHCO3, K2CO3) и амины (например, Et3N, iPr2NEt, iPr2NH). Количество основания обычно составляет от 1 до 10 эквивалентов, предпочтительно от 1,0 до 3,0 эквивалентов соединения, представленного формулой (1m). Температура реакции изменяется в пределах от 0°C до температуры кипения растворителя, предпочтительно от 0°C до комнатной температуры. Если необходим растворитель, то может быть использован любой растворитель при условии, что он является инертным в отношении реакции, включая воду, эфиры (например, диоксан, THF, Et2O), DMF, DMA, NMP, DMPU, HMPA, DMSO или их смеси. Хотя время реакции может меняться в зависимости от температуры реакции и/или исходного реагента, обычно оно составляет от 30 минут до 24 часов.
Схема 2
Стадии 1 и 2: 2-фторпиридин может быть приведен во взаимодействие с LDA и затем с йодом, и полученный 2-фтор-3-йодпиридин может быть приведен во взаимодействие с LDA и затем с соединением, представленным формулой (2c) с получением соединения, представленного формулой (2d) (J. Org. Chem., 1993, 58, 7832-7838).
Стадия 3: Исходя из соединения, представленного формулой (2d), и соединения, представленного формулой (1e), для получения соединения, представленного формулой (2e), может быть использована такая же методика, как приведенная на стадии 3 схемы 1.
Стадия 4: Соединение, представленное формулой (2e), может быть приведено во взаимодействие с основанием и затем с соединением, представленным формулой (2f), с получением соединения, представленного формулой (2g). Примеры основания включают iPrMgCl, н-BuLi и LDA. Количество используемого основания составляет от 1 до 10 эквивалентов, предпочтительно от 1,1 до 1,5 эквивалентов соединения, представленного формулой (2e). Количество используемого соединения, представленного формулой (2f), составляет от 1 до 10 эквивалентов, предпочтительно от 2 до 3 эквивалентов соединения, представленного формулой (2e). Примеры растворителя, который может быть использован, включают эфиры (например, диоксан, THF, Et2O) и их смеси. Температура реакции изменяется в пределах от -78°C до комнатной температуры, предпочтительно от -78°C до -30°C. Хотя время реакции может меняться в зависимости от температуры реакции и/или исходного реагента, оно обычно составляет от 30 минут до 24 часов.
Стадии 5 и 6: Соединение, представленное формулой (2g), может быть приведено во взаимодействие с фталимидом в присутствии DEAD и PPh3, и полученное соединение может быть приведено во взаимодействие с гидразином с получением соединения, представленного формулой (2h). В качестве варианта может быть использована такая же методика, как приведенная на стадии 6 и 7 схемы 1, с получением соединения, представленного формулой (2h), из соединения, представленного формулой (2g).
Стадия 7: Исходя из соединения, представленного формулой (2h), и соединения, представленного формулой (1l), для получения соединения, представленного формулой (2i), может быть использована такая же методика, как приведенная на стадии 8 схемы 1.
Стадия 8: Исходя из соединения, представленного формулой (2i), и соединения, представленного формулой (1n), для получения соединения, представленного формулой (2j), может быть использована такая же методика, как приведенная на стадии 9 схемы 1.
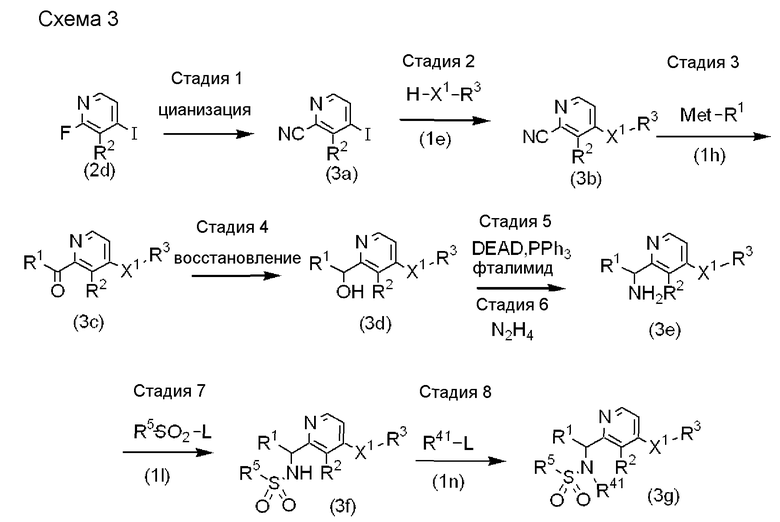
Схема 3
Стадия 1: Соединение, представленное формулой (2d), полученное на стадиях 1 и 2 схемы 3, может быть подвергнуто цианизации с получением соединения, представленного формулой (3a). Примеры цианизирующего реагента включают NaCN, KCN и CuCN. Примеры растворителя, который может быть использован, включают DMF, DMA, NMP, DMPU, HMPA, DMSO или их смеси. В случае необходимости, вводится добавка. Примеры добавки включают краун-эфиры (например, 15-краун-5 эфир, 18-краун-6 эфир) и катализаторы межфазного переноса (например, н-Bu4NOH).
Стадия 2: Исходя из соединения, представленного формулой (3a), и соединения, представленного формулой (1e), для получения соединения, представленного формулой (3b), может быть использована такая же методика, как приведенная на стадии 3 схемы 1.
Стадия 3: Исходя из соединения, представленного формулой (3b), и соединения, представленного формулой (1h), может быть получено соединение, представленное формулой (3c). Количество используемого соединения, представленного формулой (1h), составляет от 1 до 10 эквивалентов, предпочтительно от 3 до 5 эквивалентов соединения, представленного формулой (3b). Примеры растворителя, который может быть использован, включают эфиры (например, диоксан, THF, Et2O) или их смеси. Температура реакции изменяется в пределах от -78°C до температуры кипения растворителя, предпочтительно от 0°C до комнатной температуры. Хотя время реакции может меняться в зависимости от температуры реакции и/или исходного реагента, обычно оно составляет от 30 минут до 24 часов.
Стадия 4: Соединение, представленное формулой (3c), может быть приведено во взаимодействие с восстановителем в растворителе с получением соединения, представленного формулой (3d). Примеры восстановителя включают NaBH4, KBH4, LiB(втор-Bu)3H, (i-Bu)2AlH и LiAlH4. Количество восстановителя составляет от 0,5 до 5 эквивалентов, предпочтительно от 0,5 до 1,2 эквивалентов соединения, представленного формулой (3c). Примеры растворителя включают эфиры (например, диоксан, THF, Et2O) и спирты (например, MeOH, EtOH). Температура реакции изменяется в пределах от -78°C до температуры кипения растворителя, предпочтительно от 0°C до комнатной температуры. Хотя время реакции может меняться в зависимости от температуры реакции и/или исходного реагента, оно обычно составляет от 30 минут до 2 часов.
Стадии 5 и 6: Исходя из соединения, представленного формулой (3d), для получения соединения, представленного формулой (3e), может быть использована такая же методика, как приведенная на стадиях 5 и 6 схемы 2.
Стадия 7: Исходя из соединения, представленного формулой (3e), и соединения, представленного формулой (1l), для получения соединения, представленного формулой (3f), может быть использована такая же методика, как приведенная на стадии 8 схемы 1.
Стадия 8: Исходя из соединения, представленного формулой (3f), и соединения, представленного формулой (1n), для получения соединения, представленного формулой (3g), может быть использована такая же методика, как приведенная на стадии 9 схемы 1.
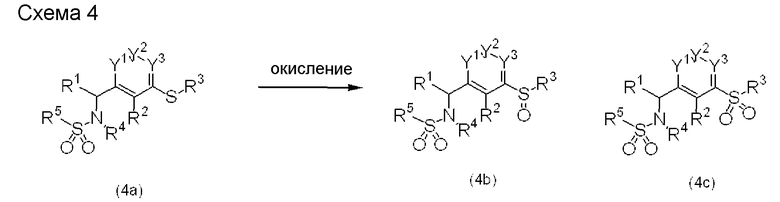
Схема 4
Соединение, полученное так, как показано на схемах 1-3, где X1 является атомом серы, то есть соединение, представленное формулой (4a), может быть приведено во взаимодействие с окислителем, с последующим образованием при необходимости соли с получением соединений, представленных формулами (4b) и (4c), или их фармацевтически приемлемых солей. Примеры окислителя, который может быть использован, включают органические перкислоты (например, м-хлорпербензойная кислота, гексагидрат моноперфталата магния, перуксусная кислота, надмуравьиная кислота), неорганические и органические пероксиды (например, пероксид водорода, аддукт мочевины с пероксидом водорода/фталевый ангидрид, гидропероксид трет-бутила, гидропероксид кумола), периодат натрия, Oxone®, N-бром-сукцинимид, N-хлорсукцинимид, хлорамин-T, гипохлорит трет-бутила, диацетат йодбензола и аддукт бром-1,4-диазабицикло[2,2,2]октана. Количество используемого окислителя составляет от 1 до 10 эквивалентов, предпочтительно от 1 до 3 эквивалентов соединения, представленного формулой (4a). Если необходим растворитель, то может быть использован любой растворитель при условии, что он является инертным в отношении реакции, включая галогенированные углеводороды, такие как метиленхлорид и хлороформ. Температура реакции изменяется в пределах от -78°C до температуры кипения растворителя, предпочтительно от 0°C до 40°C. Хотя время реакции может меняться в зависимости от температуры реакции и/или исходного реагента, оно обычно составляет от 30 минут до 24 часов.
В методиках синтезов, использованных в приведенных выше схемах, для синтеза соединений настоящего изобретения может также быть соответствующим образом изменена последовательность стадий.
Для применения в качестве фармацевтических препаратов соединения настоящего изобретения могут быть дополнены обычно используемыми вспомогательными веществами, расширителями, регуляторами pH, солюбилизаторами и т.д., и затем сформированы при помощи стандартных методов в таблетки, гранулы, пилюли, капсулы, порошки, растворы, суспензии, инъекции и т.д. Полученные таким образом фармацевтические препараты могут быть введены в виде пероральных или парентеральных лекарственных форм.
Соединения настоящего изобретения могут быть введены взрослым пациентам в количестве от 1 до 1000 мг в день в виде разовой дозы или дробных доз. Эта доза может быть увеличена или уменьшена соответствующим образом, исходя из вида заболевания, возраста, массы тела и симптомов у пациента, и т.д.
Далее настоящее изобретение будет описано более подробно с помощью следующих примеров и примера испытания.
Пример 1
3,4-Дихлор-N-[1-(5-этил-2-метил-6-п-толуилоксипиримидин-4-ил)этил]бензолсульфонамид (Соединение 1)
Формула 7

Этиловый эфир 5-этил-6-гидрокси-2-метилпиримидин-4-карбоновой кислоты
Формула 8
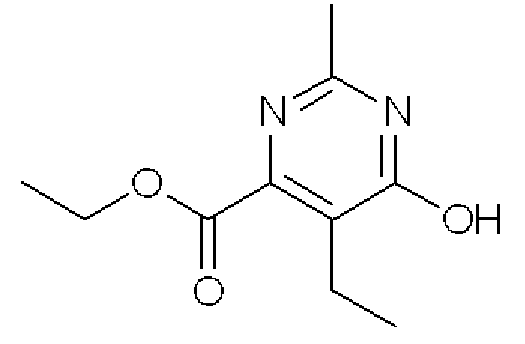
(1) Раствор NaOEt, полученный из Na (1,63 г) и EtOH (30 мл), добавляли по каплям в атмосфере азота к гидрохлориду ацетамидина (6,69 г) в EtOH (50 мл). Нерастворимые материалы отфильтровывали, и фильтрат добавляли к диэтиловому эфиру 2-этил-3-оксоянтарной кислоты (15,3 г). Реакционную смесь кипятили с обратным холодильником в течение 21 часов и затем испаряли для удаления растворителя, и полученный остаток очищали с помощью колоночной хроматографии (OH-тип SiO2, AcOEt/гексан = от 0% до 25%) с получением названного соединения (3,71 г, твердое вещество оранжевого цвета).
1H ЯМР (600 МГц, ДМСО-D6) δ м.д.: 1,02 (т, J=7,3 Гц, 3H), 1,28 (т, J=7,2 Гц, 3H), 2,26 (с, 3H), 2,39 (кв, J=7,3 Гц, 2H), 4,29 (кв, J=7,2 Гц, 2H)
Этиловый эфир 6-хлор-5-этил-2-метилпиримидин-4-карбоновой кислоты
Формула 9
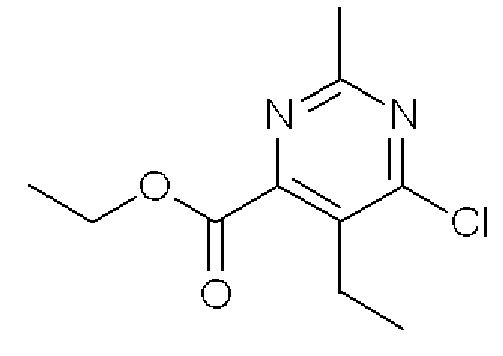
(2) К раствору соединения (1,42 г), полученного в примере 1-(1), в 1,2-дихлорэтане (3 мл) добавляли в атмосфере азота POCl3 (3,2 мл) и кипятили с обратным холодильником в течение 1,5 часов. Реакционную смесь охлаждали до 0°C, нейтрализовывали с помощью NaOH (2,0 M в воде) и экстрагировали CHCl3. Органический слой сушили над MgSO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (OH-тип SiO2, AcOEt/гексан = от 0% до 25%) с получением названного соединения (1,11 г, бесцветное масло).
1H ЯМР (600 МГц, ДМСО-D6) δ м.д.: 1,16 (т, J=7,5 Гц, 3H), 1,33 (т, J=7,2 Гц, 3H), 2,61 (с, 3H), 2,72 (кв, J=7,5 Гц, 2H), 4,41 (кв, J=7,2 Гц, 2H)
Этиловый эфир 5-этил-2-метил-6-п-толуилоксипиримидин-4-карбоновой кислоты
Формула 10

(3) К раствору 4-крезола (1,02 мл) в DMF (20 мл) добавляли при комнатной температуре NaH (388 мг, 60% в минеральном масле) и перемешивали при комнатной температуре в течение 10 минут, затем добавляли соединение, полученное в примере 1-(2), (1,11 г) и перемешивали при комнатной температуре в течение еще 10 минут. Реакционную смесь разбавляли насыщенным водным раствором хлорида аммония и экстрагировали AcOEt. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, AcOEt/гексан = от 0% до 25%) с получением названного соединения (1,06 г, бесцветное масло).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,28 (т, J=7,5 Гц, 3H), 1,44 (т, J=7,2 Гц, 3H), 2,38 (с, 3H), 2,51 (с, 3H), 2,83 (кв, J=7,5 Гц, 2H), 4,47 (кв, J=7,2 Гц, 2H), 6,99-7,04 (м, 2H), 7,19-7,24 (м, 2H)
5-Этил-2-метил-6-п-толуилоксипиримидин-4-карбальдегид
Формула 11
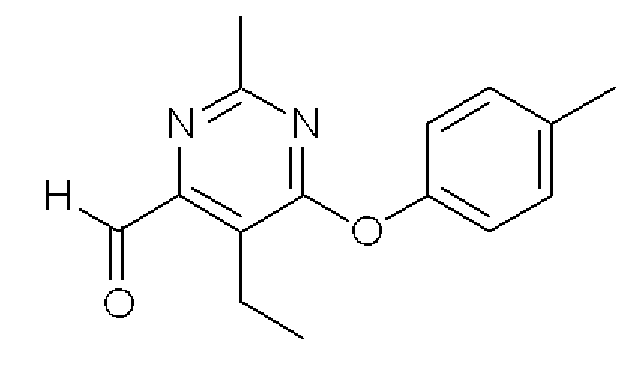
(4) К раствору соединения, полученного в примере 1-(3), (552 мг) в толуоле (10 мл) добавляли в атмосфере азота при -78°C DiBAL-H (2,76 мл, 1,0M в толуоле) и перемешивали при этой температуре в течение 2,5 часов, затем добавляли DiBAL-H (2,76 мл, 1,0M в толуоле) и перемешивали при этой температуре в течение еще 1,0 часа. Реакционную смесь разбавляли с помощью HCl (2,0M в воде) и экстрагировали CHCl3. Органический слой сушили над MgSO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (OH-тип SiO2, AcOEt/гексан = от 0% до 25%) с получением названного соединения (443 мг, бесцветное твердое вещество).
1H ЯМР (600 МГц, CDCl3) δ м.д,: 1,26 (т, J=7,5 Гц, 3H), 2,39 (с, 3H), 2,55 (с, 3H), 3,11 (кв, J=7,5 Гц, 2H), 7,01-7,05 (м, 2H), 7,21-7,25 (м, 2H), 10,10 (с, 1H)
1-(5-Этил-2-метил-6-п-толуилоксипиримидин-4-ил)этанол
Формула 12
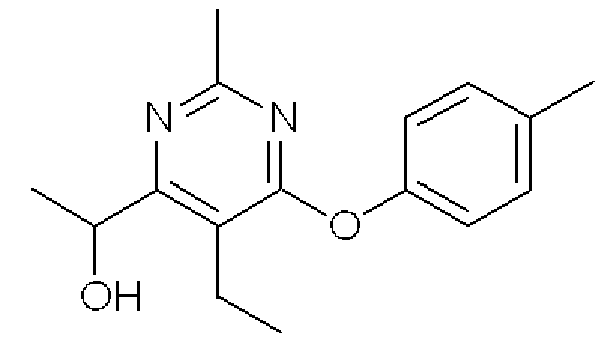
(5) К раствору соединения, полученного в примере 1-(4), (443 мг) в Et2O (10 мл) добавляли при -30°C в атмосфере азота MeMgBr (0,75 мл, 3,0 ммоль, в Et2O). После перемешивания при 0°C в течение 1 часа реакционную смесь разбавляли насыщенным водным раствором хлорида аммония и экстрагировали AcOEt. Органический слой сушили над Na2SO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (OH-тип SiO2, AcOEt/гексан = от 0% до 25%) с получением названного соединения (422 мг, бесцветное твердое вещество).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,24 (т, J=7,3 Гц, 3H), 1,43 (д, J=6,4 Гц, 3H), 2,38 (с, 3H), 2,46 (с, 3H), 2,58-2,73 (м, 2H), 4,55-4,62 (м, 1H), 4,95-5,03 (м, 1H), 6,99-7,04 (м, 2H), 7,18-7,22 (м, 2H)
4-(1-Азидоэтил)-5-этил-2-метил-6-п-толуилоксипиримидин
Формула 13
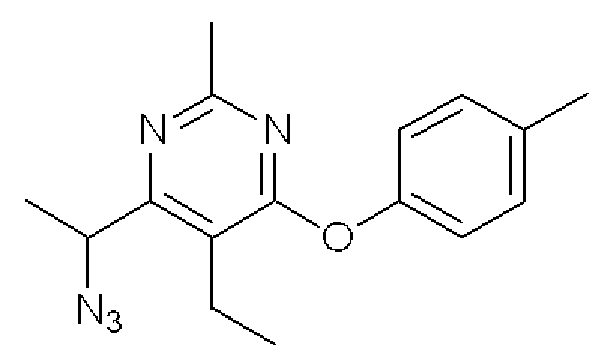
(6) К раствору соединения, полученного в примере 1-(5), (422 мг) в толуоле (10 мл) добавляли дифенилфосфорилазид (DPPA) (0,50 мл) и 1,8-диазабицикло[5,4,0]ундец-7-ен (DBU) (0,70 мл) и перемешивали при 50°C в течение 7,5 часов. Затем добавляли к этой смеси DPPA (0,50 мл) и DBU (0,35 мл) и перемешивали в течение ночи при 60°C. Реакционную смесь разбавляли водой и экстрагировали AcOEt. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и затем испаряли для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип силикагель, AcOEt/гексан = от 0% до 10%) с получением названного соединения (370 мг, бесцветное масло).
1H ЯМР (600 МГц, CDCl3) δ м.д,: 1,23 (м, 3H), 1,63 (д, J=6,8 Гц, 3H), 2,38 (с, 3H), 2,47 (с, 3H), 2,68-2,81 (м, 2H), 4,67 (кв, J=6,8 Гц, 1H), 6,99-7,03 (м, 2H), 7,18-7,22 (м, 2H)
1-(5-Этил-2-метил-6-п-толуилоксипиримидин-4-ил)этиламин
Формула 14
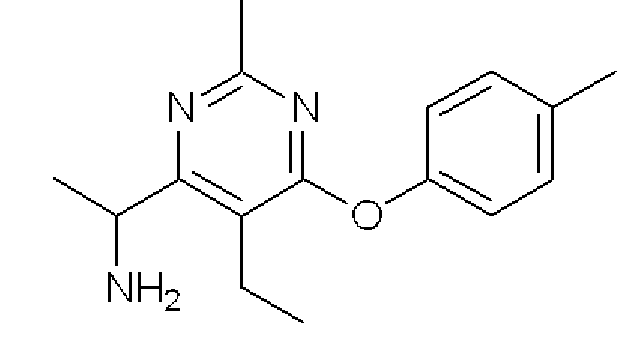
(7) Смесь соединения, полученного в примере 1-(6), (370 мг) и палладия на активированном угле (100 мг, Pd 10 масс.%) в AcOEt (10 мл) перемешивали в атмосфере водорода (около 1 атм) при комнатной температуре в течение 1,5 часов. Реакционную смесь фильтровали через целит, и фильтрат концентрировали с получением названного соединения (330 мг, бесцветное твердое вещество).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,22 (т, J=7,6 Гц, 3H), 1,39 (д, J=6,7 Гц, 3H), 2,37 (с, 3H), 2,45 (с, 3H), 2,65-2,79 (м, 2H), 4,32 (кв, J=6,7 Гц, 1H), 6,98-7,03 (м, 2H), 7,16-7,21 (м, 2H)
3,4-Дихлор-N-[1-(5-этил-2-метил-6-п-толуилоксипиримидин-4-ил)этил]бензолсульфонамид (Соединение 1)
Формула 15
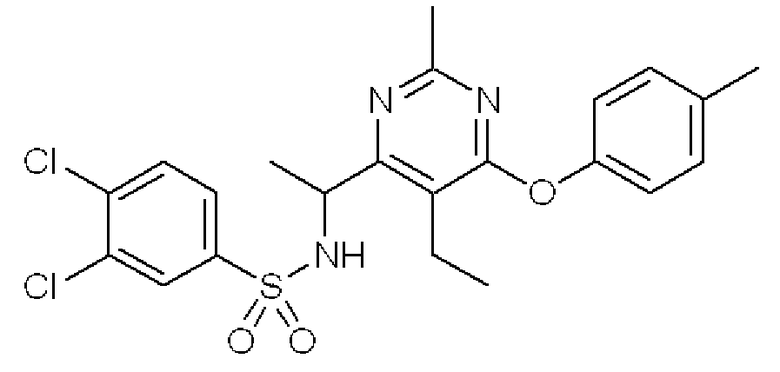
(8) К раствору соединения, полученного в примере 1-(7), (126 мг) в THF (3,0 мл) добавляли при комнатной температуре Et3N (0,20 мл) и 3,4-дихлорбензолсульфонилхлорид (171 мг) и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали при помощи колоночной хроматографии на силикагеле (NH-тип SiO2, AcOEt/гексан = 50%), затем перекристаллизовывали (AcOEt/гексан) с получением названного соединения (соединение 1) (177 мг, бесцветный порошок).
1H ЯМР (600 МГц, ДМСО-D6) δ м.д,: 1,16 (т, J=7,6 Гц, 3H), 1,33 (д, J=6,9 Гц, 3H), 2,12 (с, 3H), 2,33 (с, 3H), 2,53-2,70 (м, 2H), 4,72-4,81 (м, 1H), 6,93-7,00 (м, 2H), 7,20-7,26 (м, 2H), 7,48-7,54 (м, 1H), 7,64-7,69 (м, 1H), 7,70-7,74 (м, 1H), 8,55 (ушир.с, 1H)
Пример 2
3,4-Дихлор-N-(1-{3-этил-2-[3-(4-метилпиперазин-1-ил)-фенокси]пиридин-4-ил}этил)бензолсульфонамид (Соединение 3)
Формула 16
2-Фтор-3-йодпиридин
Формула 17
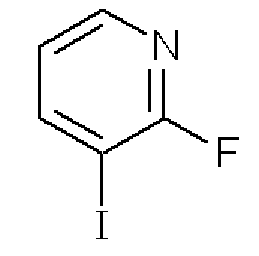
(1) К раствору, приготовленному путем добавления диизопропиламида лития (54,1 мл, 2M в смеси гептан/THF/этилбензол) в THF (100 мл), добавляли по каплям в атмосфере азота раствор 2-фторпиридина (10,503 г) в THF (10 мл) при -78°C и перемешивали при этой температуре в течение 4 часов. К реакционной смеси добавляли по каплям раствор йода (13,728 г) в THF (10 мл) и перемешивали при этой же температуре в течение 2 часов. После добавления воды (5 мл) реакционную смесь нагревали до комнатной температуры, разбавляли насыщенным солевым раствором и затем экстрагировали эфиром. Органический слой сушили над Na2SO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (кислотный OH-тип SiO2, AcOEt/гексан = 9%) с получением названного соединения (8,94 г, светло-желтое масло).
1H ЯМР (200 МГц, CDCl3) δ м.д,: 6,93-7,00 (м, 1H), 8,13-8,22 (м, 2H)
3-Этил-2-фтор-4-йодпиридин
Формула 18
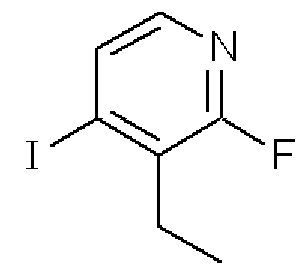
(2) К раствору, приготовленному путем добавления диизопропиламида лития (15,0 мл, 2M в смеси гептан/THF/этилбензол) к THF (30 мл), добавляли по каплям в атмосфере азота при -78°C раствор соединения, синтезированного в примере 2-(1), (6,68 г) в THF (10 мл) и перемешивали при этой температуре в течение 1 часа. К реакционной смеси добавляли по каплям раствор этилйодида (4,67 г) в THF (10 мл) и перемешивали при этой же температуре в течение 4 часов. После добавления воды (5 мл) реакционную смесь нагревали до комнатной температуры, разбавляли насыщенным солевым раствором и затем экстрагировали эфиром. Органический слой сушили над Na2SO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (кислотный OH-тип SiO2, AcOEt/гексан = от 9% до 18%) с получением названного соединения (2,554 г, желтое масло).
1H ЯМР (200 МГц, CDCl3) δ м.д.: 1,17 (т, J=7,5 Гц, 3H), 2,83 (кв, J=7,5 Гц, 2H), 7,59 (д, J=5,3 Гц, 1H), 7,67 (д, J=5,3 Гц, 1H)
1-[3-(3-Этил-4-йодпиридин-2-илокси)фенил]-4-метилпиперазин
Формула 19
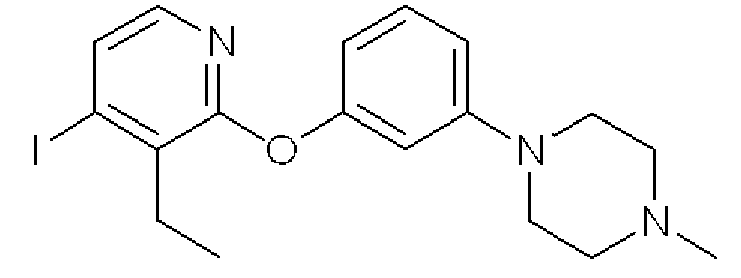
(3) К раствору 3-(4-метилпиперазин-1-ил)фенола (505 мг) в DMF (5 мл) добавляли в атмосфере азота гидрид натрия (115 мг) и перемешивали при комнатной температуре в течение 15 минут. К реакционной смеси добавляли соединение, полученное в примере 2-(2), (549 мг) и перемешивали при 130°C в течение 1 часа. Реакционную смесь фильтровали и затем концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, AcOEt/гексан = 50%) с получением названного соединения (882 мг, твердое вещество светло-желтого цвета).
1H ЯМР (200 МГц, CDCl3) δ м.д.: 1,21 (т, J=7,5 Гц, 3H), 2,34 (с, 3H), 2,52-2,57 (м, 4H), 2,87-2,98 (м, 2H), 3,20-3,25 (м, 4H), 6,53 (дд, J=7,9, 2,2 Гц, 1H), 6,64 (т, J=2,2 Гц, 1H), 6,77 (дд, J=7,9, 2,2 Гц, 1H), 7,25 (т, J=7,9 Гц, 1H), 7,39 (д, J=5,3 Гц, 1H), 7,60 (д, J=5,3 Гц, 1H)
1-{3-Этил-2-[3-(4-метилпиперазин-1-ил)фенокси]пиридин-4-ил}этанол
Формула 20
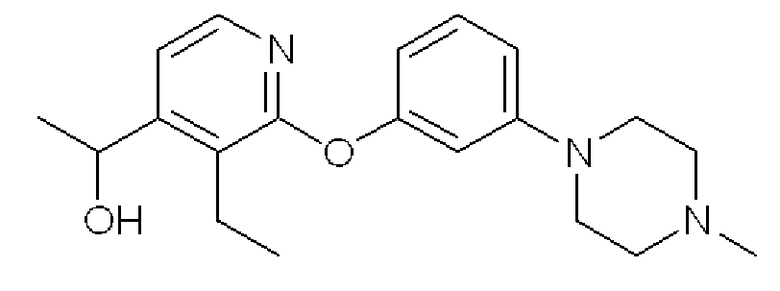
(4) К раствору соединения, полученного в примере 2-(3), (665 мг) в THF (2 мл) добавляли в атмосфере азота хлорид изопропилмагния (2,4 мл, 1M в THF) при -50°C и перемешивали при этой температуре в течение 2 часов. К реакционной смеси добавляли ацетальдегид (208 мг) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли насыщенным солевым раствором и экстрагировали смесью THF-эфир. После сушки над Na2SO4 и фильтрации растворитель отгоняли при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, MeOH/CHCl3 = от 0% до 5%) с получением названного соединения (308 мг, светло-желтое масло).
1H ЯМР (200 МГц, CDCl3) δ м.д.: 1,23 (т, J=7,5 Гц, 3H), 1,49 (д, J=6,6 Гц, 3H), 2,32 (с, 3H), 2,52-2,57 (м, 4H), 2,68-2,82 (м, 2H), 3,19-3,24 (м, 4H), 5,18 (кв, J=6,6 Гц, 1H), 6,53 (дд, J=8,0, 2,2 Гц, 1H), 6,65 (т, J=2,2 Гц, 1H), 6,72 (дд, J=8,0, 2,2 Гц, 1H), 7,18 (д, 1H), 7,24 (т, J=8,0 Гц, 1H), 8,02 (д, J=5,3 Гц, 1H)
2-(1-{3-Этил-2-[3-(4-метилпиперазин-1-ил)фенокси]пиридин-4-ил}этил)изоиндол-1,3-дион
Формула 21
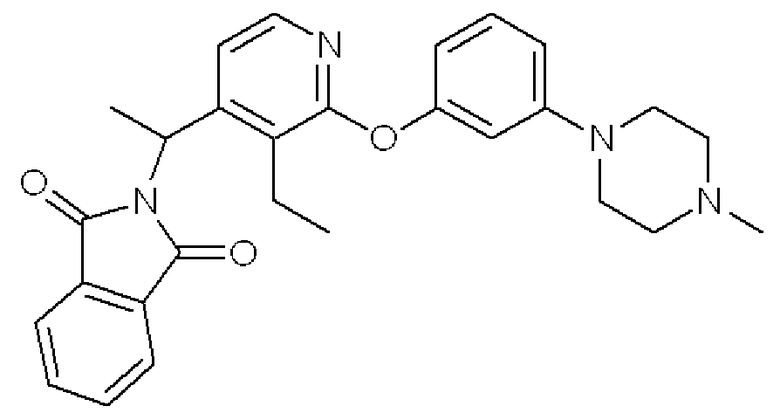
(5) К раствору соединения, полученного в примере 2-(4), (201 мг), трифенилфосфина (232 мг) и фталимида (113 мг) в THF (10 мл) добавляли в атмосфере азота при 0°C диэтил азодикарбоксилат (DEAD, 113 мг, 40% в толуоле) и перемешивали при этой температуре в течение 2 часов. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии (кислотный OH-тип SiO2, MeOH/CHCl3 = от 0% до 9%) с получением названного соединения (227 мг, твердое вещество светло-желтого цвета).
1H ЯМР (200 МГц, CDCl3) δ м.д.: 1,19 (т, J=7,5 Гц, 3H), 1,92 (д, J=7,0 Гц, 3H), 2,33 (с, 3H), 2,51-2,56 (м, 4H), 3,18-3,23 (м, 4H), 5,77 (кв, J=7,0 Гц, 1H), 6,52 (дд, J=7,9, 2,2 Гц, 1H), 6,63 (т, J=2,2 Гц, 1H), 6,72 (дд, J=7,9, 2,2 Гц, 1H), 7,20 (д, J=8,3 Гц, 1H), 7,39 (д, J=5,3 Гц, 1H), 7,70-7,76 (м, 4H), 7,79-7,86 (м, 4H), 8,01 (д, J=5,3 Гц, 1H)
1-{3-Этил-2-[3-(4-метилпиперазин-1-ил)фенокси]пиридин-4-ил}этиламин
Формула 22
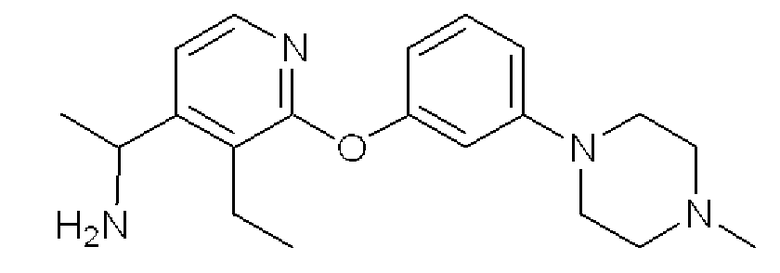
(6) К раствору соединения, полученного в примере 2-(5), (227 мг), в этаноле (5 мл) добавляли моногидрат гидразина (72 мг) и кипятили с обратным холодильником в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и затем концентрировали. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, MeOH/CHCl3 = от 0% до 9%) с получением названного соединения (146 мг, светло-желтое масло).
1H ЯМР (200 МГц, CDCl3) δ м.д.: 1,24 (т, J=7,5 Гц, 3H), 1,38 (д, J=6,6 Гц, 3H), 2,34 (с, 3H), 2,52-2,57 (м, 4H), 2,70-2,89 (м, 2H), 3,20-3,24 (м, 4H), 4,44 (кв, J=6,6 Гц, 1H), 6,53 (дд, J=8,0, 2,2 Гц, 1H), 6,66 (т, J=2,2 Гц, 1H), 6,72 (дд, J=8,0, 2,2 Гц, 1H), 7,12 (д, J=5,3 Гц, 1H), 7,23 (т, J=8,0 Гц, 1H), 8,01 (д, J=5,3 Гц, 1H)
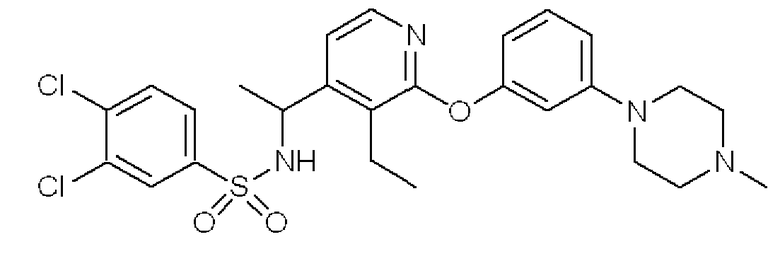
3,4-Дихлор-N-(1-{3-этил-2-[3-(4-метилпиперазин-1-ил)-фенокси]пиридин-4-ил}этил)бензолсульфонамид (Соединение 3)
Формула 23
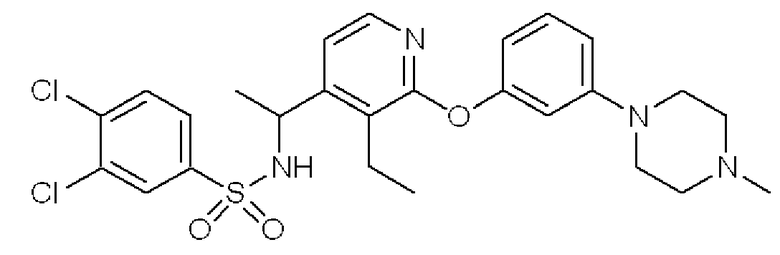
(7) К раствору соединения, полученного в примере 2-(6), (146 мг) в THF (3 мл) добавляли в атмосфере азота при 0°C 3,4-дихлорбензолсульфонилхлорид (116 мг) и триэтиламин (65 мг) и перемешивали при этой температуре в течение 1 часа. Реакционную смесь фильтровали и затем концентрировали. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, MeOH/CHCl3 = от 0% до 5%), затем перекристаллизовывали (AcOEt-гексан) с получением названного соединения (соединение 3) (122 мг, бесцветный порошок).
1H ЯМР (200 МГц, CDCl3) δ м.д.: 1,21 (т, J=7,5 Гц, 3H), 1,46 (д, J=6,59 Гц, 3H), 2,35 (с, 3H), 2,53-2,59 (м, 4H), 2,66-2,79 (м, 2H), 3,21-3,26 (м, 4H), 4,90 (квинтет, J=7,5 Гц, 1H), 5,04 (ушир.д, J=7,5 Гц, 1H), 6,50 (дд, J=8,1, 2,2 Гц, 1H), 6,63 (д, J=5,3 Гц, 1H), 6,65 (т, J=2,2 Гц, 1H), 6,75 (дд, J=7,9, 2,2 Гц, 1H), 7,25 (дд, J=8,1, 7,9 Гц, 1H), 7,44 (д, J=8,3 Гц, 1H), 7,50 (дд, J=8,3, 2,2 Гц, 1H), 7,73 (д, J=2,2 Гц, 1H), 7,79 (д, J=5,3 Гц, 1H)
Пример 3
*3,4-Дихлор-N-(1-{3-этил-2-[3-(4-метилпиперазин-1-ил)-фенокси]пиридин-4-ил}этил)бензолсульфонамид (Соединения 28 и 29)
Формула 24
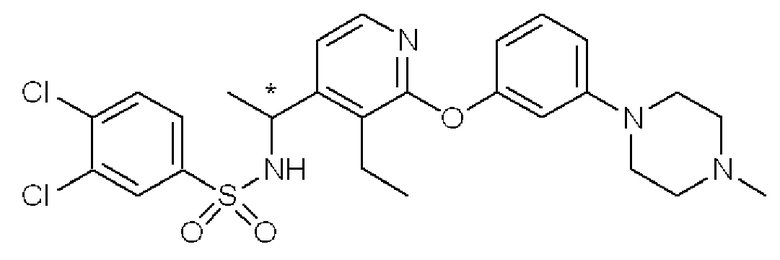
Соединение, полученное в примере 2, (100 мг) было разделено на оптические изомеры с помощью колонки для оптического разделения (колонка: CHIRALPAK AD [Daicel Chemical Industries, Ltd., Japan], диаметр 2 см × длина 25 см; элюент: i-PrOH/гексан = 10%, расход: 6,0 мл/мин) с получением названного соединения (соединение 28) [(R)-форма, 11 мг, бесцветный порошок, строение которого было определено с помощью рентгеноструктурного анализа] и другого названного соединения (соединение 29) [(S)-форма, 12 мг, бесцветный порошок, строение которого было определено с помощью рентгеноструктурного анализа].
(R)-3,4-Дихлор-N-(1-{3-этил-2-[3-(4-метилпиперазин-1-ил)-фенокси]пиридин-4-ил}этил)бензолсульфонамид (Соединение 28)
Формула 25
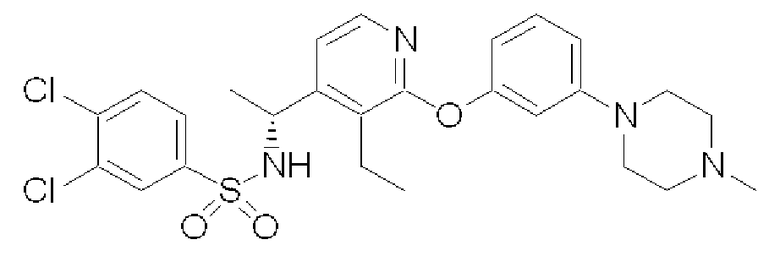
Время удерживания: 23,0 мин (колонка: CHIRALPAK AD [Daicel Chemical Industries, Ltd., Japan], диаметр 4,6 мм × длина 250 мм; элюент: i-PrOH/гексан = 20%; расход: 0,5 мл/мин; детектирование: УФ 254 нм)
(S)-3,4-Дихлор-N-(1-{3-этил-2-[3-(4-метилпиперазин-1-ил)-фенокси]пиридин-4-ил}этил)бензолсульфонамид (Соединение 29)
Формула 26
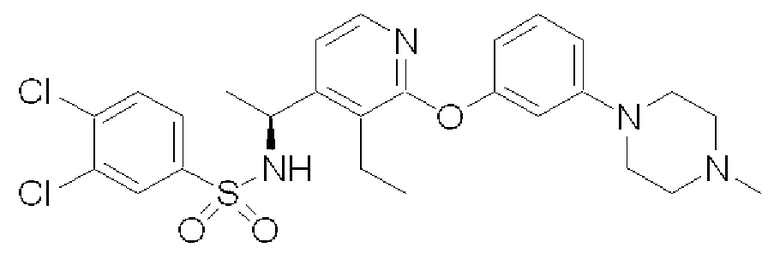
Время удерживания: 28,9 мин (колонка: CHIRALPAK AD [Daicel Chemical Industries, Ltd., Japan], диаметр 4,6 мм × длина 250 мм; элюент: i-PrOH/гексан = 20%; расход: 0,5 мл/мин; детектирование: УФ 254 нм)
Пример 4
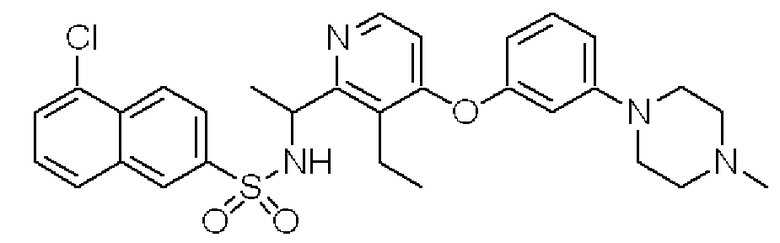
(1-{3-Этил-4-[3-(4-метилпиперазин-1-ил)фенокси]пиридин-2-ил}этил)амид 5-хлорнафталин-2-сульфоновой кислоты (Соединение 4)
Формула 27
3-Этил-4-йодпиридин-2-карбонитрил
Формула 28
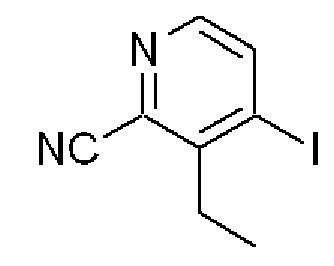
(1) К раствору 3-этил-2-фтор-4-йодпиридина, полученного в примере 2-(2), (3,425 г) в диметилсульфоксиде (5 мл) добавляли в атмосфере азота цианид натрия (668 мг) и перемешивали при 150°C в течение 3 часов. Реакционную смесь разбавляли раствором гидроксида калия и экстрагировали эфиром. Органический слой сушили над MgSO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (кислотный OH-тип SiO2, AcOEt/гексан = от 9% до 11%) с получением названного соединения (250 мг, желтое масло) и исходного реагента (1,61 г). К раствору собранного исходного реагента (1,61 г) в диметилсульфоксиде (15 мл) добавляли цианид калия (626 мг) и перемешивали при 150°C в течение 3 часов. Реакционную смесь обрабатывали аналогичным образом с получением названного соединения (349 мг, суммарный выход: 599 мг).
1H ЯМР (200 МГц, CDCl3) δ м.д. 1,28 (т, J=7,7 Гц, 3H), 3,06 (кв, J=7,7 Гц, 2H), 7,96 (д, J=4,8 Гц, 1H), 8,09 (д, J=4,8 Гц, 1H)
3-Этил-4-[3-(4-метилпиперазин-1-ил)фенокси]пиридин-2-карбонитрил
Формула 29

(2) К раствору соединения, полученного в примере 4-(1), (599 мг) в диметилформамиде (20 мл) добавляли в атмосфере азота 3-(4-метилпиперазин-1-ил)фенол (894 мг), порошок меди (74 мг), йодид меди (222 мг) и карбонат цезия (2,273 г) и перемешивали при 150°C в течение 1 часа. Реакционную смесь разбавляли с помощью THF и фильтровали для удаления твердых веществ, и фильтрат испаряли при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (OH-тип SiO2, MeOH/CHCl3 = от 0% до 11%) с получением названного соединения (682 мг, желтое масло).
1H ЯМР (200 МГц, CDCl3) δ м.д. 1,32 (т, J=7,5 Гц, 3H), 2,36 (с, 3H), 2,54-2,59 (м, 4H), 3,02 (кв, J=7,5 Гц, 2H), 3,21-3,26 (м, 4H), 6,50 (дд, J=8,4, 2,2 Гц, 1H), 6,59 (т, J=2,2 Гц, 1H), 6,76 (д, J=5,7 Гц, 1H), 6,82 (дд, J=8,4, 2,2 Гц, 1H), 7,7 (т, J=8,4 Гц, 1H), 8,30 (д, J=5,7 Гц, 1H)
1-{3-Этил-4-[3-(4-метилпиперазин-1-ил)фенокси]пиридин-2-ил}этанон
Формула 30
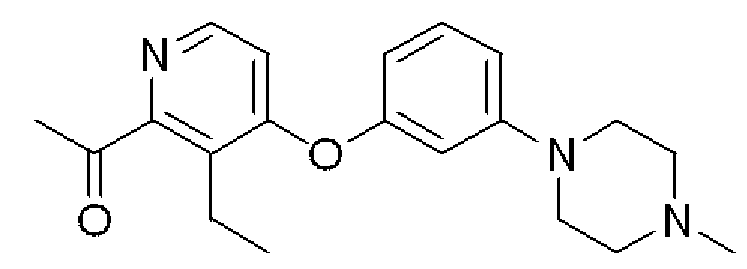
(3) К раствору соединения, полученного в примере 4-(2), (570 мг) в THF (10 мл) добавляли в атмосфере азота при комнатной температуре йодид метилмагния (2,65 мг, 2 M в эфире) и перемешивали при этой температуре в течение 2 часов. К этой смеси добавляли водный раствор хлористоводородной кислоты (10 мл, 1M) и перемешивали в течение 30 минут при комнатной температуре. Реакционную смесь разбавляли водным раствором гидроксида калия и экстрагировали смесью THF/эфир. Органический слой сушили над Na2SO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (кислотный OH-тип SiO2, гексан/CHCl3 = 50%) с получением названного соединения (415 мг, желтое масло).
1H ЯМР (200 МГц, CDCl3) δ м.д. 1,27 (т, J=7,5 Гц, 3H), 2,35 (с, 3H), 2,54-2,59 (м, 4H), 2,70 (с, 3H), 3,00 (кв, J=7,5 Гц, 2H), 3,20-3,25 (м, 4H), 6,51 (дд, J=8,4, 2,2 Гц, 1H), 6,60 (т, J=2,2 Гц, 1H), 6,73 (д, J=5,3 Гц, 1H), 6,78 (дд, J=8,4, 2,2 Гц, 1H), 7,27 (т, J=8,4 Гц, 1H), 8,27 (д, J=5,3 Гц, 1H)
1-{3-Этил-4-[3-(4-метилпиперазин-1-ил)фенокси]пиридин-2-ил}этанол
Формула 31

(4) К раствору соединения, полученного в примере 4-(3), (415 мг) в этаноле (10 мл) добавляли при комнатной температуре тетрагидроборат натрия (93 мг) и перемешивали при этой температуре в течение 1 часа. Реакционную смесь разбавляли раствором гидроксида калия и экстрагировали смесью THF/эфир. Органический слой сушили над Na2SO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, гексан/CHCl3 = 33%) с получением названного соединения (371 мг, светло-желтое масло).
1H ЯМР (200 МГц, CDCl3) δ м.д. 1,23 (т, J=7,5 Гц, 3H), 1,46 (д, J=6,2 Гц, 3H), 2,35 (с, 3H), 2,54-2,59 (м, 4H), 2,62-2,85 (м, 2H), 3,20-3,25 (м, 4H), 5,05 (кв, J=6,2 Гц, 1H), 6,51 (дд, J=8,4, 2,2 Гц, 1H), 6,56 (д, J=5,7 Гц, 1H), 6,60 (т, J=2,2 Гц, 1H), 6,77 (дд, J=8,4, 2,2 Гц, 1H), 7,27 (т, J=8,4 Гц, 1H), 8,21 (д, J=5,7 Гц, 1H)
2-(1-{3-Этил-4-[3-(4-метилпиперазин-1-ил)фенокси]пиридин-2-ил}этил)изоиндол-1,3-дион
Формула 32
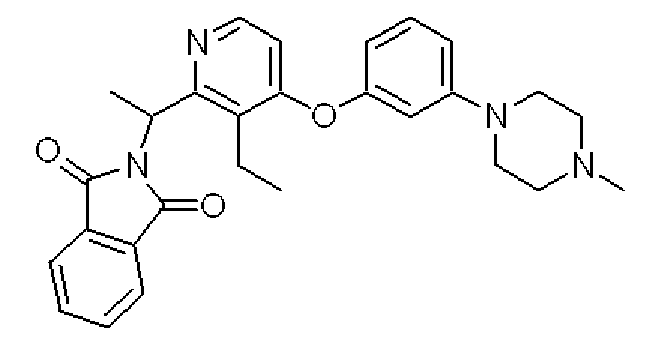
(5) К раствору соединения, полученного в примере 4-(4), (371 мг), трифенилфосфина (428 мг) и фталимида (208 мг) в THF (5 мл) добавляли в атмосфере азота при 0°C диэтил азодикарбоксилат (DEAD, 710 мг, 40% в толуоле) и перемешивали при этой температуре в течение 1 часа. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, CHCl3) с получением названного соединения (467 мг, желтое масло).
1H ЯМР (200 МГц, CDCl3) δ м.д. 1,19 (т, J=7,5 Гц, 3H), 1,96 (д, J=7,5 Гц, 3H), 2,34 (с, 3H), 2,53-2,58 (м, 4H), 2,65-2,97 (м, 2H), 3,18-3,23 (м, 2H), 5,81 (кв, J=7,5 Гц, 1H), 6,49 (дд, J=8,4, 2,2 Гц, 1H), 6,55 (д, J=5,3 Гц, 1H), 6,57 (т, J=2,2 Гц, 1H), 6,75 (дд, J=8,4, 2,2 Гц, 1H), 7,25 (т, J=8,4 Гц, 1H), 7,67-7,72 (м, 2H), 7,81-7,85 (м, 2H), 8,25 (д, J=5,3 Гц, 1H)
1-{3-Этил-4-[3-(4-метилпиперазин-1-ил)фенокси]пиридин-2-ил}этиламин
Формула 33

(6) К раствору соединения, полученного в примере 4-(5), (467 мг) в этаноле (5 мл) добавляли при комнатной температуре моногидрат гидразина (149 мг) и кипятили с обратным холодильником в течение 1 часа. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, MeOH/CHCl3 = от 0% до 11%) с получением названного соединения (249 мг, желтое масло).
1H ЯМР (200 МГц, CDCl3) δ м.д. 1,23 (т, J=7,0 Гц, 3H), 1,42 (д, J=6,6 Гц, 3H), 2,34-2,35 (м, 3H), 2,54-2,58 (м, 4H), 2,69-2,90 (м, 2H), 3,19-3,25 (м, 4H), 4,37 (кв, J=6,6 Гц, 1H), 6,48 (дд, J=8,4, 2,2 Гц, 1H), 6,51 (д, J=5,7 Гц, 1H), 6,60 (т, J=2,2 Гц, 1H), 6,76 (дд, J=8,4, 2,2 Гц, 1H), 7,25 (т, J=8,4 Гц, 1H), 8,25 (д, J=5,7 Гц, 1H)
(1-{3-Этил-4-[3-(4-метилпиперазин-1-ил)фенокси]пиридин-2-ил}этил)амид 5-хлорнафталин-2-сульфоновой кислоты (Соединение 4)
Формула 34
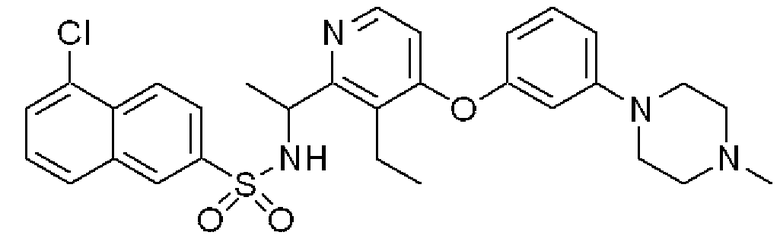
(7) К раствору соединения, полученного в примере 4-(6), (100 мг) в THF (5 мл) добавляли при комнатной температуре 5-хлор-2-нафталинсульфонилхлорид (84 мг) и триэтиламин (45 мг) и перемешивали при этой температуре в течение 1 часа. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии (кислотный OH-тип SiO2, CHCl3), затем перекристаллизовывали (эфир-гексан) с получением названного соединения (54 мг, бесцветный порошок).
1H ЯМР (200 МГц, CDCl3) δ м.д. 0,97 (т, J=7,3 Гц, 3H), 1,44 (д, J=6,6 Гц, 3H), 2,36 (с, 3H), 2,55-2,60 (м, 4H), 3,18-3,23 (м, 4H), 4,74-4,88 (м, 1H), 6,10 (дд, J=8,4, 1,0 Гц, 1H), 6,20 (д, J=5,7 Гц, 1H), 6,42-6,44 (м, 1H), 6,55 (д, J=8,8 Гц, 1H), 6,73 (дд, J=8,4, 1,0 Гц, 1H), 7,18 (т, J=8,4 Гц, 1H), 7,46 (т, J=8,4 Гц, 1H), 7,65-7,79 (м, 3H), 7,95 (д, J=5,7 Гц, 1H), 8,12 (д, J=9,2 Гц, 1H), 8,28 (с, 1H)
Пример 5
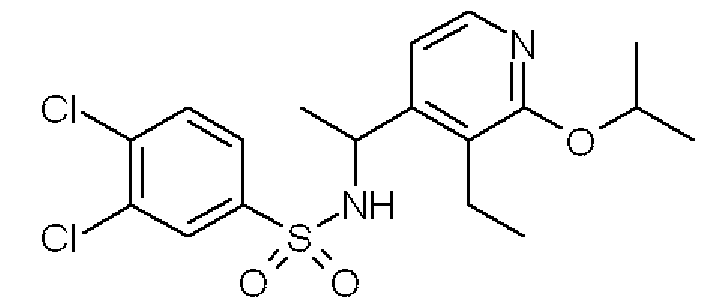
3,4-Дихлор-N-[1-(2-изопропокси-3-этилпиридин-4-ил)этил]-бензолсульфонамид (Соединение 26)
Формула 35
3-Этил-2-фторпиридин-4-карбальдегид
Формула 36
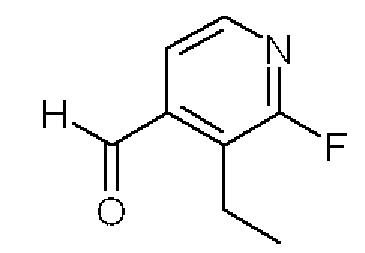
(1) К раствору 3-этил-2-фтор-4-йодпиридина, полученного в примере 2-(2), (3,45 г) в THF (35 мл) добавляли в атмосфере азота при комнатной температуре хлорид изопроприлмагния (7,6 мл, 2,0M в THF) и перемешивали при 45°C в течение 2 часов. К реакционной смеси добавляли диметилформамид (2,1 мл) и перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли водой и экстрагировали AcOEt. Полученный органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (OH-тип SiO2, AcOEt/гексан = от 0% до 50%) с получением названного соединения (1,71 г, бесцветное масло).
1H ЯМР (600 МГц CDCl3) δ м.д.: 1,29 (т, J=7,7 Гц, 3H), 3,06 (кв, J=7,7 Гц, 2H), 7,53-7,57 (м, 1H), 8,25-8,28 (м, 1H), 10,36 (с, 1H)
1-(3-Этил-2-фторпиридин-4-ил)этанол
Формула 37
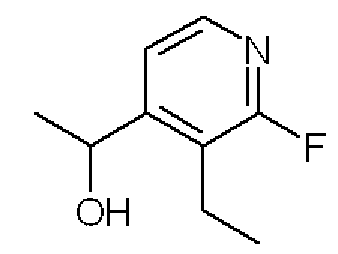
(2) К раствору соединения, полученного в примере 5-(1), (1,00 г) в THF (10 мл) добавляли при 0°C в атмосфере азота MeMgBr (0,75 мл, 3,0 ммоль, в Et2O) и перемешивали при 0°C в течение 2 часов. Реакционную смесь разбавляли водой и экстрагировали AcOEt. Полученный органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (OH-тип SiO2, AcOEt/гексан = от 0% до 50%) с получением названного соединения (1,05 г, бесцветное масло).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,21 (т, J=7,6 Гц, 3H), 1,49 (д, J=6,4 Гц, 3H), 2,58-2,74 (м, 2H), 5,17 (кв, J=6,4 Гц, 1H), 7,34-7,38 (м, 1H), 8,03-8,08 (м, 1H)
2-[1-(3-Этил-2-фторпиридин-4-ил)этил]изоиндол-1,3-дион
Формула 38
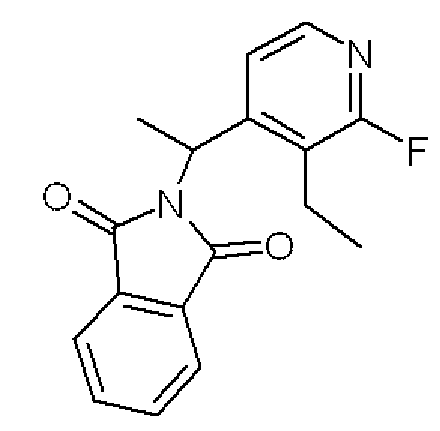
(3) К раствору соединения, полученного в примере 5-(2), (1,05 г) в THF (5 мл) добавляли в атмосфере азота при комнатной температуре трифенилфосфин (2,30 г), фталимид (1,10 г) и диэтил азодикарбоксилат (DEAD, 3,76 мл, 2 M в толуоле) и перемешивали при этой температуре в течение 2 часов. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии (кислотный SiO2, AcOEt/гексан = от 0% до 40%) с получением названного соединения (1,43 г, твердое вещество желтого цвета).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,15 (т, J=7,6 Гц, 3H), 1,89 (д, J=7,1 Гц, 3H), 2,67-2,88 (м, 2H), 5,73 (кв, J=7,1 Гц, 1H), 7,58-7,63 (м, 1H), 7,71-7,75 (м, 2H), 7,81-7,86 (м, 2H), 8,03-8,07 (м, 1H)
1-(3-Этил-2-фторпиридин-4-ил)этиламин
Формула 39
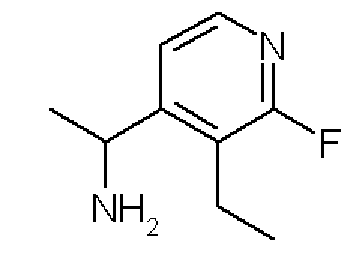
(4) К раствору соединения, полученного в примере 5-(3), (1,43 г) в этаноле (20 мл) добавляли моногидрат гидразина (0,7 мл) и перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, AcOEt/гексан = от 0% до 99%) с получением названного соединения (535 мг, бесцветное масло).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,20 (т, J=7,6 Гц, 3H), 1,36 (д, J=6,4 Гц, 3H), 2,64-2,78 (м, 2H), 4,42 (кв, J=6,4 Гц, 1H), 7,30-7,36 (м, 1H), 8,00-8,05 (м, 1H)
1-(3-Этил-2-изопропоксипиридин-4-ил)этиламин
Формула 40
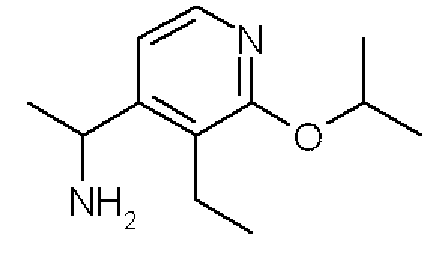
(5) Смесь соединения, полученного в примере 5-(4), (200 мг), изопропанола (0,5 мл), Cs2CO3 (775 мг) и N,N'-диметилпропиленмочевины (DMPU) (1,0 мл) перемешивали при 200°C в течение 3 часов. После охлаждения до комнатной температуры добавляли CHCl3 и хлористоводородную кислоту (1,0 н.), и водный слой экстрагировали CHCl3, подщелачивали с помощью NaOH и дополнительно экстрагировали CHCl3. Органический слой сушили над MgSO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя, получая в результате названное соединение (213 мг, бесцветное масло).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,12 (т, J=7,6 Гц, 3H), 1,32-1,38 (м, 9H), 2,59-2,69 (м, 2H), 4,35 (кв, J=6,4 Гц, 1H), 5,28-5,36 (м, 1H), 6,92-6,95 (м, 1H), 7,96-8,00 (м, 1H)
3,4-Дихлор-N-[1-(2-изопропокси-3-этилпиридин-4-ил)этил]-бензолсульфонамид (Соединение 26)
Формула 41

(6) К раствору соединения, полученного в примере 5-(5), (150 мг) в THF (2 мл) добавляли при 0°C в атмосфере азота 3,4-дихлорбензолсульфонилхлорид (212 мг) и триэтиламин (0,2 мл) и перемешивали при этой температуре в течение 2 часов. Реакционную смесь фильтровали и затем концентрировали. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, AcOEt/гексан = от 0% до 50%), затем перекристаллизовывали (AcOEt-гексан) с получением названного соединения (62 мг, бесцветный порошок).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,03-1,11 (м, 3H), 1,29-1,36 (м, 6H), 1,42 (д, J=6,4 Гц, 3H), 2,47-2,60 (м, 2H), 4,77-4,85 (м, 1H), 4,85-4,90 (м, 1H), 5,20-5,29 (м, 1H), 6,40-6,43 (м, 1H), 7,37-7,40 (м, 1H), 7,42-7,46 (м, 1H), 7,64-7,67 (м, 1H), 7,73-7,77 (м, 1H)
Пример 6
[1-(3-Этил-2-п-толуилсульфанилпиридин-4-ил)этил]амид 5-хлорнафталин-2-сульфоновой кислоты (Соединение 58)
Формула 42
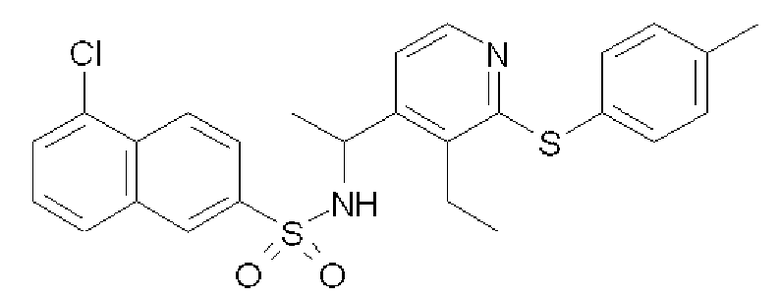
1-(3-Этил-2-п-толуилсульфанилпиридин-4-ил)этиламин
Формула 43
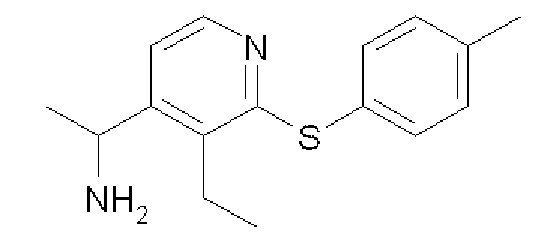
(1) В устойчивую к повышенному давлению пробирку с завинчивающейся крышкой вводили соединение, полученное в примере 5-(4), (500 мг), DMPU (1,0 мл), 4-метилбензолтиол (443 мг) и карбонат цезия (1,94 г) и перемешивали при 200°C в течение 2 часов. После охлаждения до комнатной температуры добавляли CHCl3 и хлористоводородную кислоту (1,0 н.), и водный слой экстрагировали CHCl3, подщелачивали с помощью NaOH и затем экстрагировали CHCl3, Органический слой сушили над MgSO4, фильтровали и затем испаряли при пониженном давлении для удаления растворителя, получая в результате названное соединение (601 мг, твердое вещество светло-желтого цвета).
1H ЯМР (600 МГц, CDCl3) δ м.д. 1,27 (т, J=7,6 Гц, 3H), 1,36 (д, J=6,8 Гц, 3H), 2,36 (с, 3H), 2,79-2,94 (м, 2H), 4,39 (кв, J=6,8 Гц, 1H), 7,15-7,21 (м, 3H), 7,37-7,42 (м, 2H), 8,18-8,22 (м, 1H)
[1-(3-Этил-2-п-толуилсульфанилпиридин-4-ил)этил]амид 5-хлорнафталин-2-сульфоновой кислоты (Соединение 58)
Формула 44
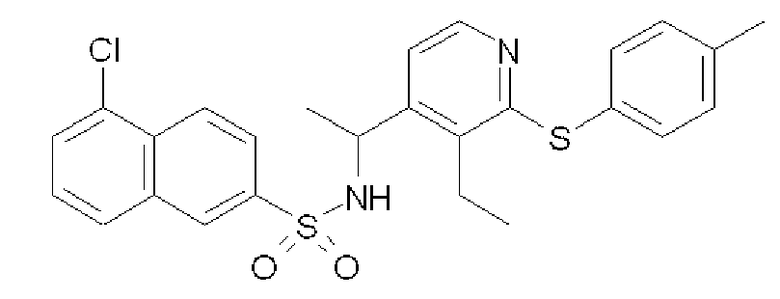
(2) К раствору соединения, полученного в примере 6-(1), (302 мг) в THF (5,0 мл) добавляли при комнатной температуре 5-хлор-2-нафталинсульфонилхлорид (284 мг) и триэтиламин (310 мкл) и перемешивали при этой температуре в течение 1 часа. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, AcOEt/гексан = 10%), затем перекристаллизовывали (эфир-гексан) с получением названного соединения (385 мг, бесцветный порошок).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,20 (т, J=7,6 Гц, 3H), 1,41 (д, J=6,9 Гц, 3H), 2,36 (с, 3H), 2,63-2,89 (м, 2H), 4,82-4,91 (м, 1H), 4,95-5,02 (м, 1H), 6,64-6,68 (м, 1H), 7,14-7,19 (м, 2H), 7,22-7,25 (м, 2H), 7,48-7,53 (м, 1H), 7,70-7,82 (м, 4H), 8,18-8,22 (м, 1H), 8,24-8,28 (м, 1H)
Пример 7
{1-[3-Этил-2-(толуол-4-сульфонил)пиридин-4-ил]этил}амид 5-хлорнафталин-2-сульфоновой кислоты (Соединение 59)
Формула 45
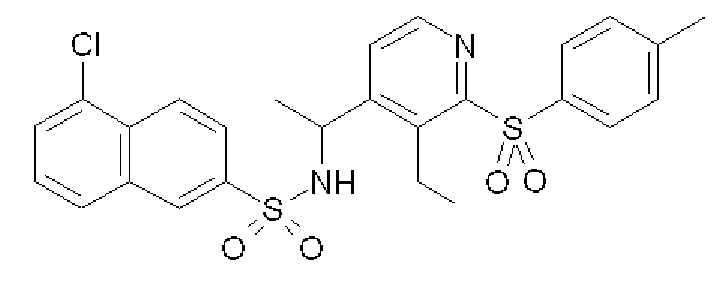
К раствору соединения, полученного в примере 6-(2), (150 мг) в хлороформе (6,0 мл) добавляли м-хлорпербензойную кислоту (535 мг) и перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли 5% водным раствором Na2S2O3 и экстрагировали хлороформом. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. После фильтрации, растворитель отгоняли при пониженном давлении, и полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, AcOEt/гексан = от 0% до 100%, MeOH/CHCl3 = от 0% до 10%) с получением названного соединения (соединение 59) (бесцветное порошкообразное соединение, 116 мг).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,33 (т, J=7,6 Гц, 3H), 1,41 (д, J=6,9 Гц, 3H), 2,46 (с, 3H), 3,06-3,40 (м, 2H), 4,93-4,99 (м, 1H), 5,00-5,05 (м, 1H), 7,06-7,09 (м, 1H), 7,31-7,36 (м, 2H), 7,50-7,56 (м, 1H), 7,66-7,84 (м, 6H), 8,12-8,16 (м, 1H), 8,24-8,29 (м, 1H)
Пример 8
{1-[3-Этил-2-(3-морфолин-4-илфенокси)пиридин-4-ил]этил}-метиламид 5-хлорнафталин-2-сульфоновой кислоты (Соединение 60)
Формула 46
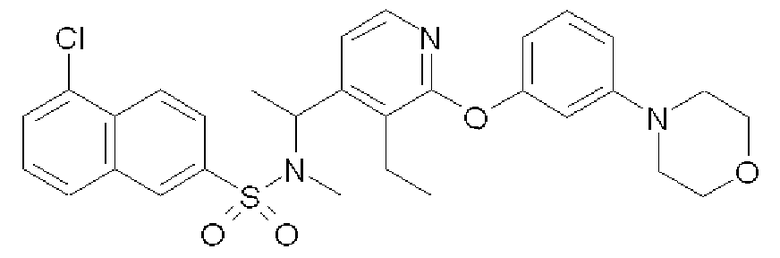
{1-[3-Этил-2-(3-морфолин-4-илфенокси)пиридин-4-ил]этил}- амид 5-хлорнафталин-2-сульфоновой кислоты (Соединение 27)
Формула 47
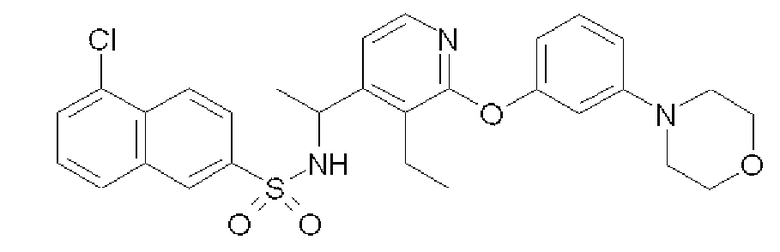
(1) 1-[3-Этил-2-(3-морфолин-4-илфенокси)пиридин-4-ил]-этиламин (168 мг), который был получен таким же образом, как в примере 5-(4), за исключением того, что изопропанол в примере 5-(4) был заменен 3-морфолинофенолом, растворяли в THF (2 мл). К этому раствору добавляли при комнатной температуре 5-хлор-2-нафталинсульфонилхлорид (156 мг) и триэтиламин (140 мкл) и перемешивали при этой температуре в течение ночи. Реакционную смесь концентрировали, и полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, AcOEt/гексан = от 10% до 99%), затем перекристаллизовывали (EtOH) с получением названного соединения (127 мг, бесцветный порошок).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,16 (т, J=7,6 Гц, 3H), 1,43 (д, J=6,9 Гц, 3H), 2,58-2,77 (м, 2H), 3,12-3,18 (м, 4H), 3,82-3,86 (м, 4H), 4,87-4,94 (м, 1H), 4,94-4,99 (м, 1H), 6,32-6,36 (м, 1H), 6,53-6,57 (м, 1H), 6,66-6,73 (м, 2H), 7,20-7,24 (м, 1H), 7,48-7,53 (м, 1H), 7,67-7,73 (м, 2H), 7,78-7,82 (м, 2H), 8,25-8,32 (м, 2H)
{1-[3-Этил-2-(3-морфолин-4-илфенокси)пиридин-4-ил]этил}-метиламид 5-хлорнафталин-2-сульфоновой кислоты (Соединение 60)
Формула 48
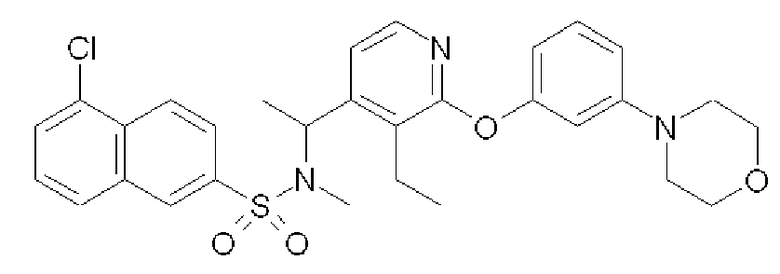
(2) К раствору соединения, полученного в примере 8-(1), (50 мг) в DMF (0,5 мл) добавляли K2CO3 (25 мг) и MeI (7 мкл) и перемешивали при комнатной температуре в течение 3 дней. Реакционную смесь разбавляли водой и экстрагировали хлороформом. Полученный органический слой промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и затем концентрировали. Полученный неочищенный продукт очищали с помощью колоночной хроматографии (NH-тип SiO2, AcOEt/гексан = от 0% до 50%), затем перекристаллизовывали (Et2O-гексан) с получением названного соединения (соединение 60) (29 мг, бесцветный порошок).
1H ЯМР (600 МГц, CDCl3) δ м.д.: 1,30 (т, J=7,6 Гц, 3H), 1,33 (д, J=7,3 Гц, 3H), 2,75 (с, 3H), 2,95-3,07 (м, 2H), 3,14-3,21 (м, 4H), 3,80-3,87 (м, 4H), 5,61-5,69 (м, 1H), 6,54-6,58 (м, 1H), 6,64-6,66 (м, 1H), 6,71-6,75 (м, 1H), 6,81-6,84 (м, 1H), 7,24-7,29 (м, 1H), 7,52-7,57 (м, 1H), 7,72-7,77 (м, 1H), 7,87-7,95 (м, 3H), 8,38-8,43 (м, 2H)
Для получения соединений, приведенных ниже в таблице, исходя из соответствующих исходных реагентов, могут быть использованы такие же методики, как в примерах 1-8, с последующим образованием при необходимости соли.
Соединения, полученные в приведенных выше примерах, наряду с другими соединениями также показаны в таблице.
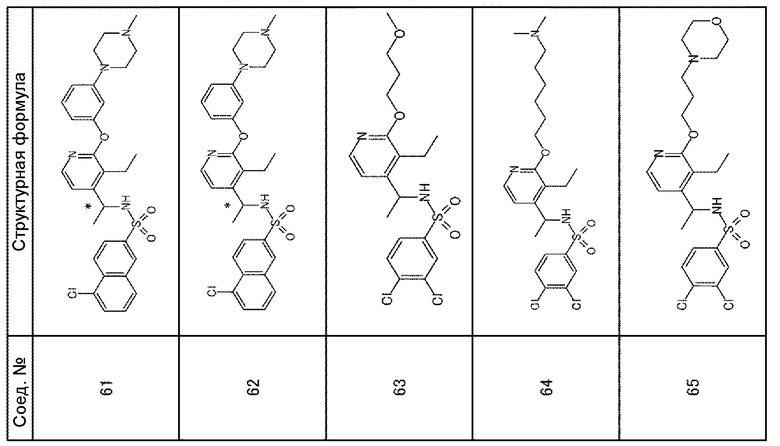
Следующие соединения были получены из соединения 31 путем разделения на оптические изомеры таким же образом, как показано в примере 3. Данные по их временам удерживания на хиральной колонке приведены ниже.
Соединение 61: время удерживания: 12,1 мин (колонка: CHIRALPAK AD [Daicel Chemical Industries, Ltd,, Japan], диаметр 4,6 мм × длина 250 мм; элюент: i-PrOH/гексан = 30%; расход: 0,5 мл/мин; детектирование: УФ 254 нм)
Соединение 62: время удерживания: 17,7 мин (колонка: CHIRALPAK AD [Daicel Chemical Industries, Ltd,, Japan], диаметр 4,6 мм × длина 250 мм; элюент: i-PrOH/гексан = 30%; расход: 0,5 мл/мин; детектирование: УФ 254 нм)
Для следующих соединений приведены данные 1H-ЯМР.
Соединение 63: (600 МГц, CDCl3) δ м.д.: 1,09 (т, J=7,6 Гц, 3H), 1,42 (д, J=6,9 Гц, 3H), 2,01-2,09 (м, 2H), 2,50-2,70 (м, 2H), 3,37 (с, 3H), 3,56 (т, J=6,4 Гц, 2H), 4,30-4,39 (м, 2H), 4,78-4,86 (м, 1H), 4,90-5,08 (м, 1H), 6,44-6,48 (м, 1H), 7,37-7,42 (м, 1H), 7,42-7,47 (м, 1H), 7,63-7,65 (м, 1H), 7,74-7,78 (м, 1H)
Соединение 64: (600 МГц, CDCl3) δ м.д.: 1,09 (т, J=7,6 Гц, 3H), 1,36-1,43 (м, 5H), 1,44-1,55 (м, 4H), 1,74-1,82 (м, 2H), 2,23 (с, 6H), 2,25-2,30 (м, 2H), 2,51-2,63 (м, 2H), 4,24 (т, J=6,7 Гц, 2H), 4,81 (кв, J=6,9 Гц, 1H), 6,48-6,52 (м, 1H), 7,37-7,40 (м, 1H), 7,44-7,47 (м, 1H), 7,64-7,66 (м, 1H), 7,73-7,76 (м, 1H)
Соединение 65: (600 МГц, CDCl3) δ м.д.: 1,09 (т, J=7,6 Гц, 3H), 1,42 (д, J=6,9 Гц, 3H), 1,89-2,06 (м, 2H), 2,35-2,71 (м, 8H), 3,70-3,79 (м, 4H), 4,26-4,34 (м, 2H), 4,74-4,86 (м, 1H), 5,53 (ушир.с, 1H), 6,48-6,56 (м, 1H), 7,36-7,42 (м, 1H), 7,44-7,47 (м, 1H), 7,63-7,65 (м, 1H), 7,72-7,76 (м, 1H)
Таблица 1-1
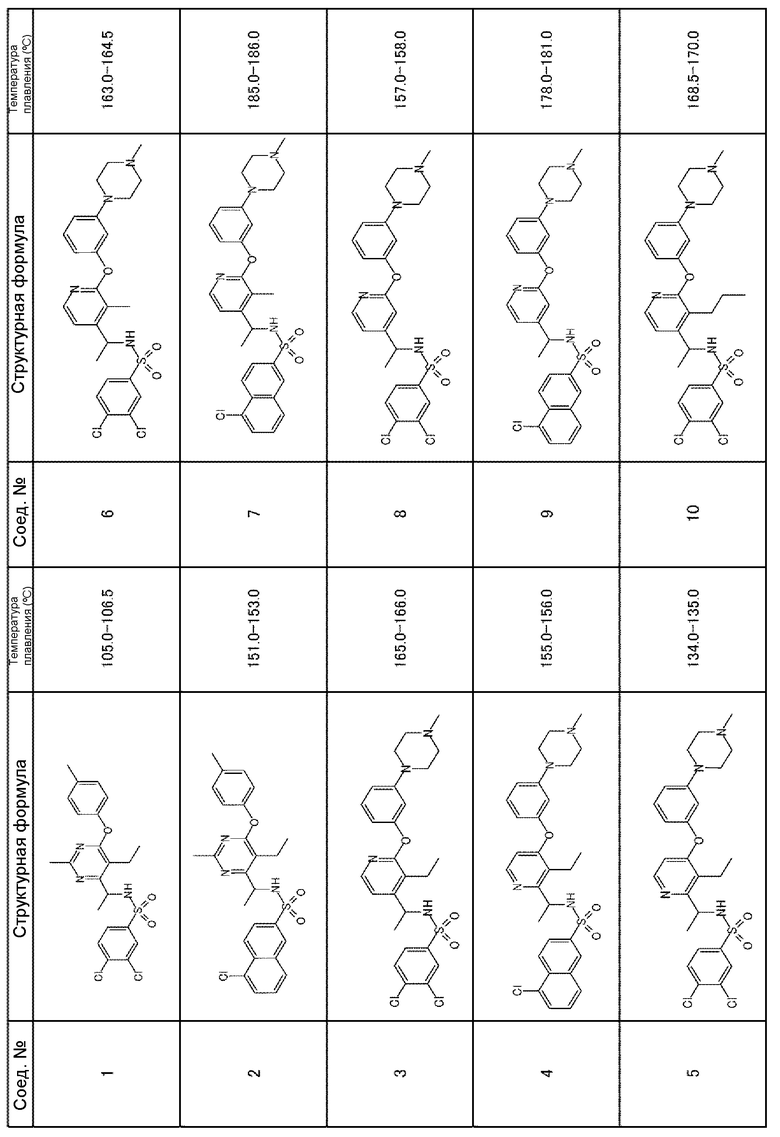
Таблица 1-2
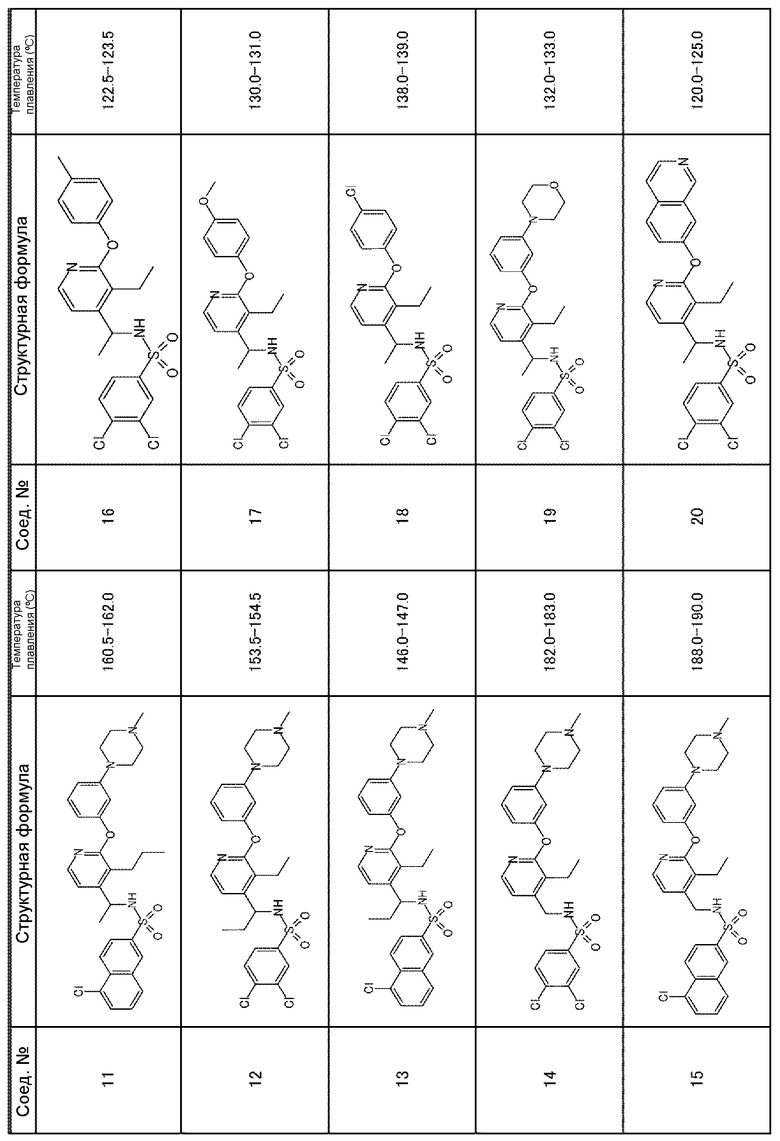
Таблица 1-3
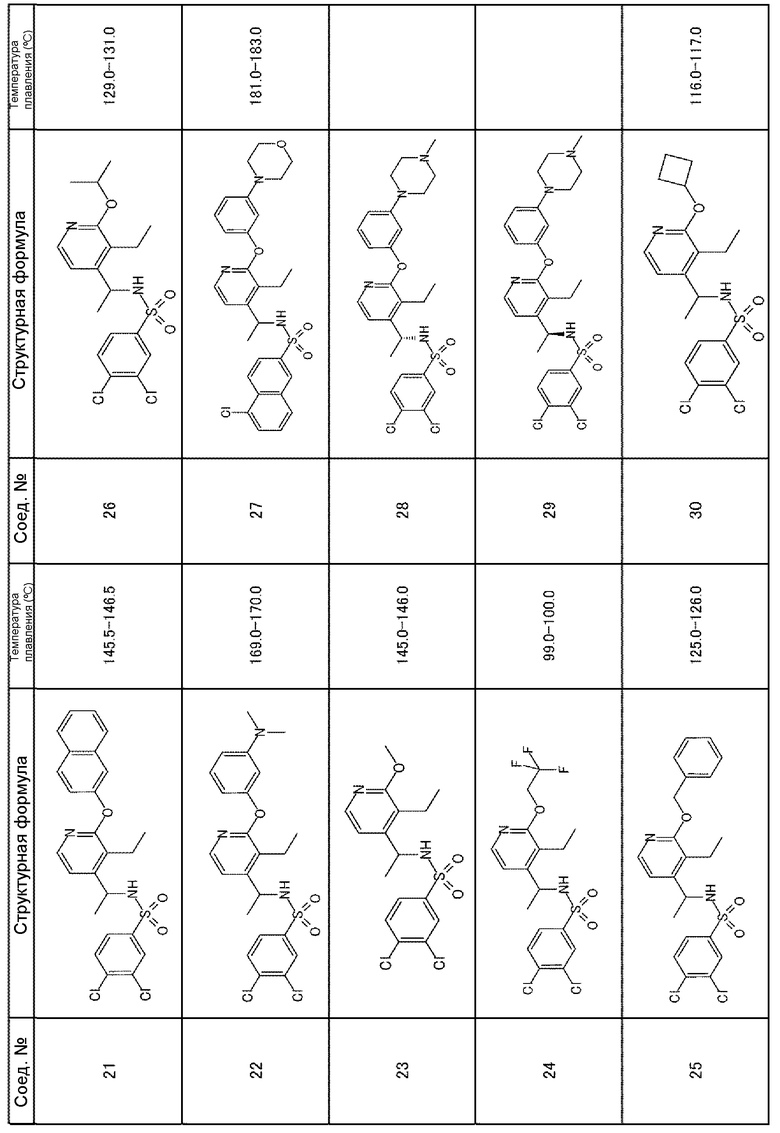
Таблица 1-4
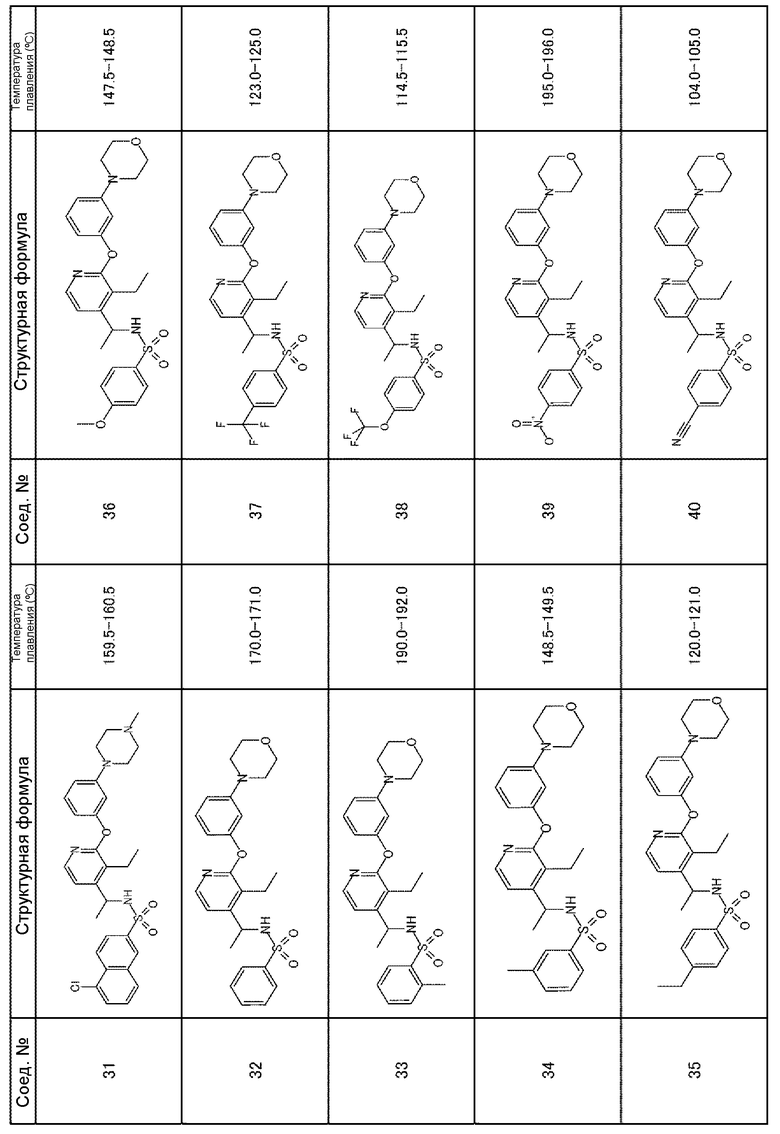
Таблица 1-5
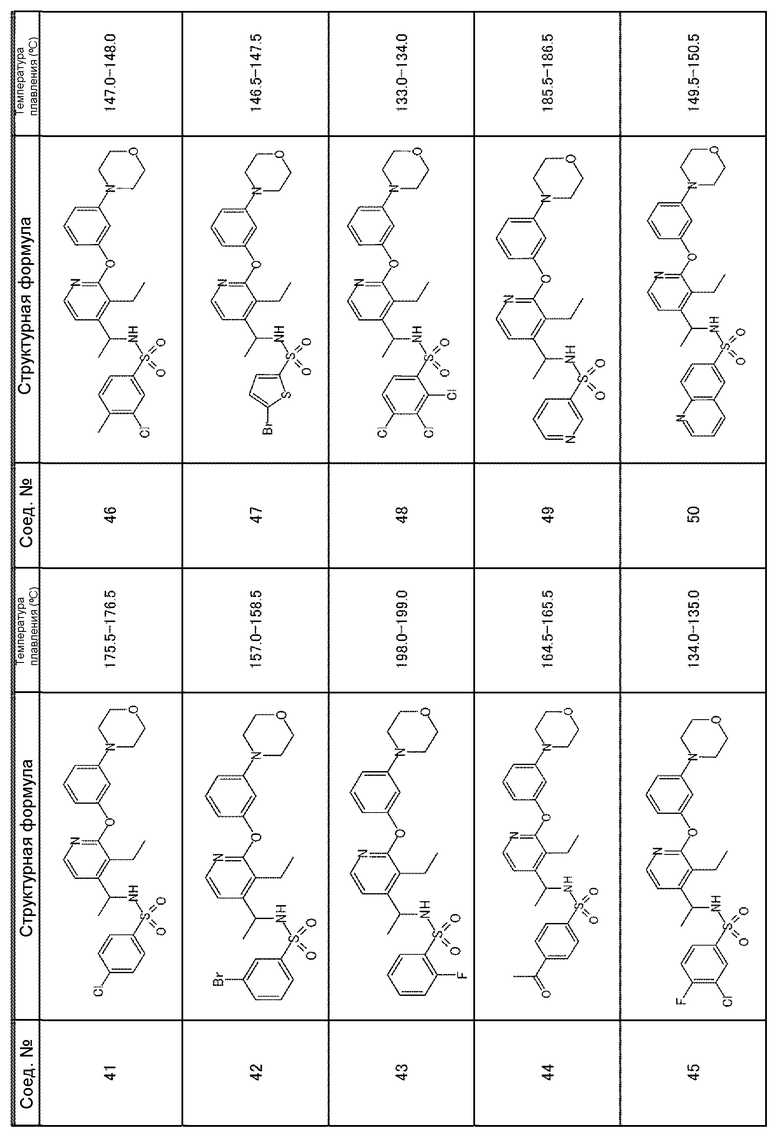
Таблица 1-6
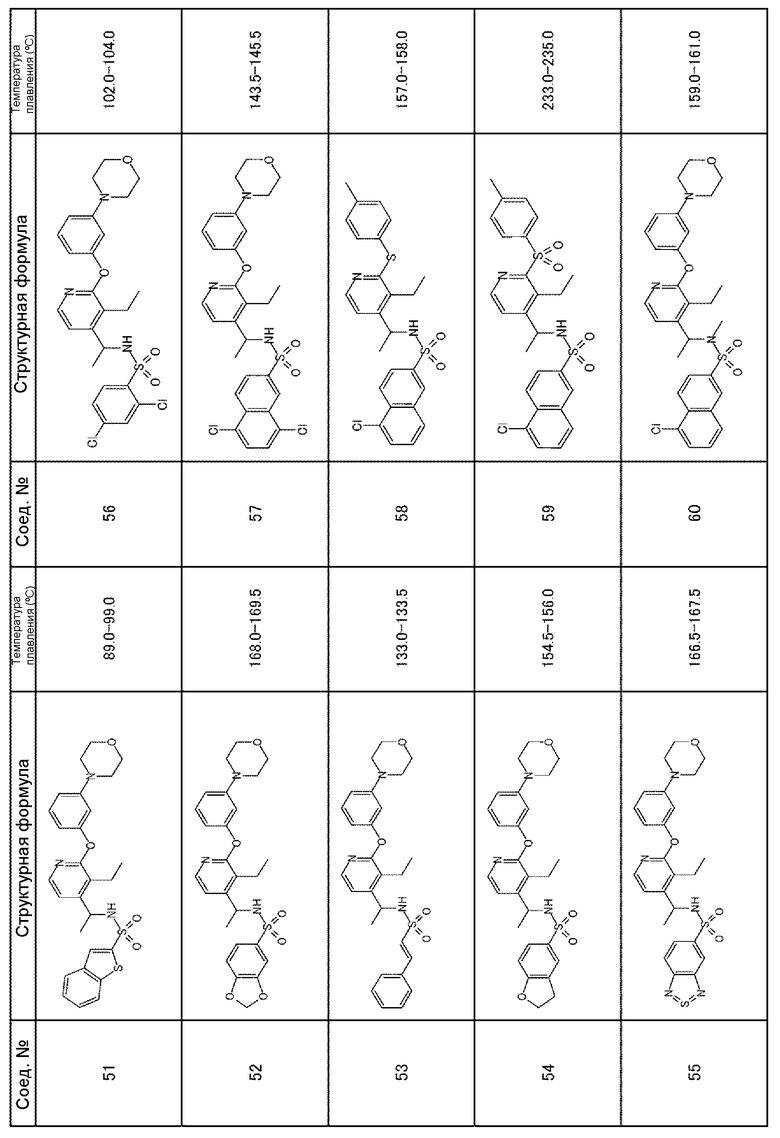
Таблица 1-7
Пример испытания 1 (испытание на связывание S1P 1 )
Мембранную фракцию клеток линии HEK-293, несущую Edg-1(S1P1) ген человека, использовали для исследования соединений настоящего изобретения на их ингибирующее действие по отношению к Edg-1(S1P1) связыванию, как описано в публикации Science, 2002, 296:346 (Kd=0,15 нМ и Bmax=2,5 фмоль/мкг для связывания с [33P]-S1P). Мембранную фракцию получали путем обработки солюбилизирующим буфером (1 мМ Tris/HCl, pH 7,2) в течение 10 минут на льду с последующим центрифугированием при 1000×g в течение 5 минут для удаления нерастворимой фракции и затем при 40000×g в течение 30 минут при 4°C. Полученную мембранную фракцию растворяли в связывающем буфере (20 мМ Tris-HCl, pH 7,4, 100 мМ NaCl, 15 мМ NaF, 2 мМ деоксипиридоксин, 4 мг/мл не содержащий жирных кислот BSA), затем добавляли связывающий буфер, содержащий [33P]-S1P (ARC, конечная концентрация: 0,1 нМ), и раствор испытуемого соединения в ДМСО (конечная концентрация соединения: 10-5M, конечная концентрация ДМСО: 0,1%). После перемешивания реакционный раствор инкубировали при 30°C в течение 1 часа. Мембранную фракцию собирали на фильтре Unifilter-96 GF/C (Perkin Elmer) при помощи коллектора и промывали четыре раза связывающим буфером, затем сушили на фильтре. После добавления 25 мкл Microscint 0 (Perkin Elmer) измеряли радиоактивность при помощи Top Count NXT (Packard) для расчета количества (A), то есть количества [33P]-S1P, связывающегося с мембранной фракцией в присутствии соединения.
Такую же методику использовали в отсутствие какого-либо испытуемого соединения для расчета величины (B), то есть величины [33P]-S1P связывания. Кроме того, эту же методику также использовали для HEK-293 клеток, не несущих Edg-1(S1P1) ген в отсутствие какого-либо испытуемого соединения для расчета величины (C), то есть фоновой величины [33P]-S1P связывания.
Степень ингибирования Edg-1(S1P1) связывания рассчитывали для каждого соединения с помощью следующего уравнения.
Степень ингибирования (%)=[1-(A-C)/(B-C)]×100
Соединения № 1-62 подвергали приведенному выше испытанию. Для каждого из испытываемых соединений степень ингибирования Edg-1(S1P1) связывания составляла 3% или более при добавлении соединения в количестве 10 мкМ.
Кроме того, рассчитывали активность каждого испытуемого соединения как значение (IC50 значение), характеризующееся как 50% радиоактивности, получаемой в отсутствие какого-либо испытуемого соединения. А именно проводили испытание связывания мембранной системы в присутствии испытуемого соединения при различных концентрациях для расчета величины IС50 на основании зависимой от дозы кривой ингибирования, анализируемой при помощи программы для анализа данных Origin (Lightstone Corp., Japan).
Результаты показали, что перечисленные ниже соединения имеют величины IС50 200 нМ или менее и характеризовались высокими степенями ингибирования связывания: соединения 7, 11, 22 и 57.
Кроме того, ниже приведены более подробные IС50 данные для индивидуальных соединений (единица: нМ):
соединение 3:107; соединение 4:121; соединение 13:47; соединение 19:66; соединение 27:51; соединение 28:49; соединение 31:35; соединение 48:63; соединение 55:126; и соединение 61:33.
ПРИМЕР А
Таблетки, содержащие 100 мг активного вещества - соединения по изобретению
Способ приготовления:
Активное вещество - соединение(я) по изобретению, смешивают с моногидратом лактозы, микрокристаллической целлюлозой, кальций карбоксиметилцеллюлозой, и гидроксипропилцеллюлозой, и смесь размалывают в мельнице. Смесь после перемешивания загружают в высокоскоростной гранулятор, перемешивают 1 мин и гранулируют в течение 4-8 минут в присутствии воды. Полученный гранулят сушат 4 0 минут при 70°С. Высушенный гранулят пропускают через сито 500 мкм. Полученный гранулят смешивают со стеаратом магния, в V-образном смесителе в течение 3 минут при 30 об/мин. Полученную смесь прессуют в таблетки с помощью роторной таблетирующей машины.
Вес таблетки: 200 мг, диаметр 8 мм, круглая.
ПРИМЕР В
Таблетки, содержащие 25 мг активного вещества - соединения по изобретению
Способ приготовления:
Активное вещество - соединение(я) по изобретению, смешивают с D-маннитом, микрокристаллической целлюлозой, кальций карбоксиметилцеллюлозой и гидроксипропилцеллюлозой, и смесь размалывают в мельнице. Смесь после перемешивания загружают в высокоскоростной гранулятор, перемешивают 1 мин, и гранулируют в течение 4-8 минут в присутствии воды. Полученный гранулят сушат при 70°С, пока отходящий воздух не достигнет температуры 40°С. Высушенный гранулят пропускают через сито 500 мкм. Полученный гранулят смешивают со стеаратом магния, в V-образном смесителе в течение 3 минут при 30 об/мин. Полученную смесь прессуют в таблетки с помощью роторной таблетирующей машины.
Вес таблетки: 230 мг, диаметр 8 мм, круглая.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ Так как соединения настоящего изобретения являются отличными Edg-l(SlP1) лигандами, они могут применяться в качестве лечебных и/или профилактических средств при аутоиммунных заболеваниях, таких как болезнь Крона, синдром раздраженной толстой кишки, синдром Шегрена, рассеянный склероз и системная красная волчанка, а также других заболеваниях, таких как ревматоидный артрит, астма, атопический дерматит, отторжение после трансплантации органа, рак, ретинопатия, псориаз, остеоартрит, возрастная дегенерация желтого пятна и т.д.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР СВЯЗЫВАНИЯ СФИНГОЗИН-1-ФОСФАТА | 2007 |
|
RU2395499C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА | 2007 |
|
RU2409570C2 |
| ПРОИЗВОДНОЕ ТРИАЗОЛА | 2005 |
|
RU2383536C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2013 |
|
RU2640416C2 |
| ПРОИЗВОДНОЕ АМИНОКАРБОНОВОЙ КИСЛОТЫ И ПРИМЕНЕНИЕ УКАЗАННОГО ВЕЩЕСТВА В МЕДИЦИНСКИХ ЦЕЛЯХ | 2005 |
|
RU2433121C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ СИГМА РЕЦЕПТОРОВ | 2011 |
|
RU2582338C2 |
| ПРОИЗВОДНОЕ 1,2,4-ТРИАЗОЛОНА | 2011 |
|
RU2566754C2 |
| 3-АЛКИЛИДЕНГИДРАЗИНО-ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВАТОРОВ РЕЦЕПТОРА ТРОМБОПОЭТИНА | 2004 |
|
RU2358970C2 |
| ЦИННАМИДНОЕ СОЕДИНЕНИЕ | 2005 |
|
RU2361872C2 |
| ЗАМЕЩЕННОЕ АКРИЛАМИДНОЕ ПРОИЗВОДНОЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2006 |
|
RU2404966C2 |
Соединение, представленное формулой (I), или его фармацевтически приемлемая соль, где Y1 представляет атом азота или группу, представленную CRA, Y2 представляет атом азота или группу, представленную CRB, Y3 представляет атом азота или группу, представленную CRC, RA, RB и RC, которые могут быть одинаковыми или различными, каждая представляет атом водорода и т.д. (за исключением случая, когда Y1 является CRA, Y2 является CRB и Y3 является CRC), X представляет атом кислорода и т.д., R1 представляет С1-С6алкильную группу и т.д., R2 представляет С1-С6алкильную группу и т.д., R3 представляет необязательно замещенную фенильную группу и т.д., R4 представляет атом водорода и т.д., и R5 представляет необязательно замещенную фенильную группу и т.д.), обладает ингибирующим действием по отношению к связыванию S1P с его рецептором Edg-1(S1P1) и может применяться в качестве лечебного средства при аутоиммунных заболеваниях, ревматоидном артрите, астме, атопическом дерматите, отторжении после трансплантации органа, раке, ретинопатии, псориазе, остеоартрите или возрастной дегенерации желтого пятна, и т.д. 2 н. и 11 з.п. ф-лы, 9 пр., 1 табл., 4 сх.
1. Соединение формулы (I) или его фармацевтически приемлемая соль:

{где Y1 представляет атом азота или группу, представленную CRA; Y2 представляет атом азота или группу, представленную CRB; Y3 представляет атом азота или группу, представленную CRC, где
RA, RB и RC могут быть одинаковыми или различными и каждая представляет атом водорода или С1-С6алкильную группу (за исключением случая, когда Y1 является CRA, Y2 является CRB и Y3 является CRC);
X представляет атом кислорода, атом серы, группу, представленную формулой -SO2-;
R1 представляет атом водорода или С1-С6алкильную группу;
R2 представляет атом водорода или С1-С6алкильную группу;
R3 представляет
(i) С1-С6алкильную группу, которая может быть замещена от 1 до 3 заместителями, выбранными из группы А [где группа А состоит из атома галогена, фенильной группы, С1-С6алкоксигруппы, аминогруппы, которая может быть замещена одной или двумя С1-С6алкильными группами, и морфолиногруппы],
(ii) С3-С8циклоалкильную группу или
(iii) фенильную группу, нафтильную группу или изохинолинильную группу, каждая из которых может быть замещена от 1 до 3 заместителями, выбранными из группы В [где группа В состоит из атом галогена, С1-С6алкильной группы, С1-С6алкоксигруппы, аминогруппы, которая может быть замещена одной или двумя С1-С6алкильными группами, морфолиногруппы и пиперазиногруппы, которая может быть замещена С1-С6алкильной группой (алкильными группами)];
R4 представляет атом водорода или С1-С6алкильную группу и
R5 представляет фенильную группу, тиенильную группу, пиридильную группу, нафтильную группу, дигидробензофуранильную группу, бензодиоксолильную группу, бензотиадиазолильную группу, бензотиенильную группу или хинолинильную группу, каждая из которых может быть замещена от 1 до 5 заместителями, выбранными из группы С [где группа С состоит из С1-С6алкильной группы, атома галогена, трифторметильной группы, С1-С6алкоксигруппы, трифторметоксигруппы, нитрогруппы, цианогруппы и С2-С7алканоильной группы], либо С2-С8алкенильную группу, которая может быть замещена фенильной группой (фенильными группами), при условии, что исключается следующее соединение:
2. Соединение или его фармацевтически приемлемая соль по п.1, где в формуле (I) Y1 является атомом азота или СН; Y2 является CRB и Y3 является атомом азота или СН.
3. Соединение или его фармацевтически приемлемая соль по п.1, где в формуле (I) Y1 и Y2 каждая является СН и Y3 является атомом азота.
4. Соединение или его фармацевтически приемлемая соль по п.1, где в формуле (I) X является атомом кислорода.
5. Соединение или его фармацевтически приемлемая соль по п.1, где в формуле (I) R1 является атомом водорода, метальной группой или этильной группой.
6. Соединение или его фармацевтически приемлемая. соль по п.1, где в формуле (I) R2 является метальной группой или этильной группой.
7. Соединение или его фармацевтически приемлемая соль по п.1, где в формуле (I) R4 является атомом водорода.
8. Соединение или его фармацевтически приемлемая соль по п.1, где в формуле (I) R3 является фенильной группой, нафтильной группой или изохинолинильной группой, каждая из которых может быть замещена от 1 до 3 заместителями, выбранными из группы D [где группа D состоит из атома галогена, С1-С6алкильной группы, С1-С6алкоксигруппы, аминогруппы, которая может быть замещена одной или двумя С1-С6алкильными группами, морфолиногруппы и пиперазиногруппы, которая может быть замещена С1-С6алкильной группой (алкильными группами)].
9. Соединение или его фармацевтически приемлемая соль по п.1, где в формуле (I) R3 является фенильной группой, в которой метаположение замещено одним заместителем, выбранным из группы Е [где группа Е состоит из аминогруппы, которая может быть замещена одной или двумя С1-С6алкильными группами, морфолиногруппы и пиперазиногруппы, которая может быть замещена С1-С6алкильной группой (алкильными группами)].
10. Соединение или его фармацевтически приемлемая соль по п.1, где в формуле (I) R5 является фенильной группой, тиенильной группой, нафтильной группой, дигидробензофуранильной группой, бензодиоксолильной группой, бензотиадиазолильной группой, бензотиенильной группой или хинолинильной группой, каждая из которых может быть замещена от 1 до 3 заместителями, выбранными из группы F [где группа F состоит из С1-С6алкильной группы, атома галогена, трифторметильной группы, С1-С6алкоксигруппы, нитрогруппы, цианогруппы и С2-С7алканоильной группы], или С2-С8алкенильной группы, которая может быть замещена фенильной группой (фенильными группами).
11. Соединение или его фармацевтически приемлемая соль по п.1, где в формуле (I) R5 является фенильной группой, замещенной двумя или тремя атомами галогена, или нафтильной группой, замещенной одним или двумя атомами галогена.
12. Фармацевтическая композиция, которая обладает ингибирующим действием по отношению к связыванию сфингозин-1-фосфата (S1P) с его рецептором Edg-l(SlP1) (рецептор 1 типа гена дифференциации эндотелиальных клеток, SlP1), включающая соединение согласно любому из пп.1-11 или его фармацевтически приемлемую соль и вспомогательные вещества.
13. Фармацевтическая композиция по п.12, которая является лечебным средством при аутоиммунном заболевании, таком как болезнь Крона, синдром раздраженной толстой кишки, синдром Шегрена, рассеянный склероз или системная красная волчанка, ревматоидный артрит, астма, атопический дерматит, отторжение после трансплантации органа, рак, ретинопатия, псориаз, остеоартрит или возрастная дегенерация желтого пятна.
| WO 2007083089 А1, 26.07.2007 | |||
| WO 2006013948 А1, 09.02.2006 | |||
| ВОДОРАСТВОРИМАЯ КОМПОЗИЦИЯ ДЛЯ ДОБЫЧИ И ТРАНСПОРТА НЕФТИ | 1985 |
|
RU1391199C |
| RU 2003127677 А1, 11.03.2002 | |||
| WO 2007024922 А1, 01.03.2007 | |||
| WO 2006097487 A1, 21.09.2006 | |||
| RU 2004112562 A, 19.09.2002 | |||
| RU 2003134010 A, 20.05.2005. | |||

