Настоящее изобретение относится к спироциклическим производным циклогексана, к способу их получения, к лекарственным средствам, содержащим эти соединения, и к применению спироциклических производных циклогексана для получения лекарственных средств.
Гептадекапептид ноцицептин является эндогенным лигандом рецептора ORL1 (Opioid-Receptor-Like) (Meunier et al., Nature 377, 1995, S.532-535), который относится к семейству опиоидных рецепторов и может быть обнаружен в большинстве участках головного мозга и спинного мозга и обладает высоким сродством к рецептору ORL1. Рецептор ORL1 является гомологом µ, κ и δ опиоидных рецепторов и аминокислотная последовательность пептида ноцицептина обладает сильным сходством с последовательностями известных опиоидных пептидов. Индуцированная ноцицептином активность рецептора путем сопряжения с Gi/o-белками приводит к ингибированию аденилатциклазы (Meunier et al., Nature 377, 1995, с.532-535).
Пептид ноцицептин после интрацеребровентрикулярного применения показывает проноцицептивную и гиперальгетическую активность в различных экспериментальных моделях (животные) (Reinscheid et al., Science 270,1995, с.792-794). Эти данные могут быть объяснены как ингибирование стресс-индуцированной анальгезии (Mogil et al., Neuroscience 75, 1996, с.333-337). В связи с этим также могла быть доказана анксиолитическая активность ноцицептина (Jenck et al., Proc. Natl. Acad. Sci. USA 94, 1997, 14854-14858).
С другой стороны, в различных экспериментальных моделях, в особенности после интратекального применения, можно было также продемонстрировать антиноцицептивный эффект ноцицептина. Ноцицептин действует антиноцицептивно в различных моделях боли, например в тесте «отдергивание хвоста» (tail flick) на мышах (King et al., Neurosci. Lett., 223, 1997, 113-116). В моделях для невропатических болей равным образом можно было доказать антиноцицептивное действие ноцицептина, которое в этом отношении является особенно интересным, что эффективность ноцицептина увеличивается после аксотомии спинно-мозговых нервов. Что в противоположность к классическим опиоидам, уменьшает их эффективность при этих условиях (Abdulla и Smith, J. Neurosci., 18, 1998, с.9685-9694).
Кроме того, ORL1-рецептор участвует еще в регуляции других физиологических и патофизиологических процессов. В частности сюда относятся обучение и формирование памяти (Manabe etal., Nature, 394,1997, с.577-581), слуховая способность (Nishi et al., EMBO J., 16,1997, с.1858-1864), а также многочисленные другие процессы. В обзорной статье Calo et al. (Br. J. Pharmacol., 129, 2000, 1261-1283) предоставлен обзор показаний или биологических процессов, для которых ORL1-рецептор имеет большое значение или мог бы иметь с большой вероятностью. В частности упоминаются: анальгезия, стимуляция и регуляция приема пищи, влияние на µ-агонисты, такие как морфин, лечение явлений синдрома отмены, снижение наркотического потенциала опиоидов, анксиолизис, модуляция двигательной активности, нарушения памяти, эпилепсия; модуляция нейромедиаторного распределения, в особенности глутамата, серотонина и допамина, и вместе с этим нейродегенеративные заболевания; воздействие на сердечно-сосудистую систему, возникновение эрекции, диурез, антинатрийурез, электролитный баланс, артериальное кровяное давление, заболевания, связанные с задержкой воды, желудочно-кишечная подвижность (диарея), расслабляющие эффекты на дыхательные пути, мочеиспускательный рефлекс (недержание мочи). Далее обсуждается применение агонистов и антагонистов в качестве аноретиков, анальгетиков (также при совместном приеме с опиоидами) или ноотропов.
Соответственно являются разносторонними возможности применения соединений, которые привязаны к ORL1-рецептору и его активируют или ингибируют. Однако наряду с этим именно в области болевой терапии, а также и другие из приведенных показаний, имеют большое значение опиоидные рецепторы, такие как µ-рецептор, но и другие подтипы этих опиоидных рецепторов, а именно δ и κ. Соответственно является благоприятным, если соединение также оказывает действие на эти опиоидные рецепторы.
В WO 2004043967 раскрываются спироциклические производные циклогексана, которые обладают высоким сродством к ORL1-рецептору, а также к µ-опиоидному рецептору. WO 2004043967 также, в общем, описывает группу, в которой R3 означает алкил или циклоалкил. Однако не раскрыты примерные соединения, которые являются составной частью этой подгруппы.
Растворимость является важным свойством для биодоступности и решающим фактором в отношении активности и вместе с тем также для успеха разработки лекарственных средств. Для того, чтобы повысить растворимость применяют дорогостоящие процессы, такие как, например, получение микрочастиц или наночастиц (например, Exp. Op. Dug Disc. 2007, 2, 145), однако более простой и спланированной является разработка соединений, которые при одновременной эффективности обладают высокой растворимостью.
Недостатком раскрытых в WO 2004043967 примерных соединений является незначительная растворимость соединений.
Задача настоящего изобретения состояла в том, чтобы предоставить в распоряжение лекарственные средства, которые оказывают действие на систему ноцицептин/ORL1-рецептор и имеют более высокую растворимость, чем соединения, известные из WO 2004043967.
Неожиданно теперь было обнаружено, что определенные соединения, которые хотя, в общем, и описаны в WO 2004043967, однако не раскрыты при помощи примерных соединений, обладают более высокой растворимостью, чем приведенные там примерные соединения.
Поэтому объектом изобретения являются производные циклогексана общей формулы I
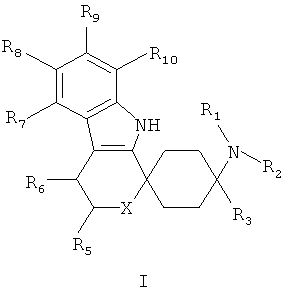
в которой
R1 и R2, независимо друг от друга означают Н; С1-5-алкил каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный или полизамещенный или незамещенный; C3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, монозамещенный или полизамещенный или незамещенный; арил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный;
или остатки R1 и R2 вместе означают CH2CH2OCH2CH2, CH2CH2NR11CH2CH2 или (CH2)3-6.
причем R11 означает Н; С1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный или полизамещенный или незамещенный; С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, монозамещенный или полизамещенный или незамещенный; арил-, или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный;
R3 означает С1-8-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный или полизамещенный или незамещенный; C3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, монозамещенный или полизамещенный или незамещенный;
R5 означает =O; Н; С1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный или полизамещенный; COOR13, CONR13, OR13; C3-8-циклоалкил, насыщенный или ненасыщенный, незамещенный или монозамещенный или полизамещенный; арил-, или гетероарил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный или монозамещенный или полизамещенный;
R6 означает H; F, Cl, NO2, CF3, OR13, SR13, SO2R13, SO2OR13, CN, COOR13, NR14R15; С1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный или полизамещенный; С3-8-циклоалкил, насыщенный или ненасыщенный, незамещенный или монозамещенный или полизамещенный; арил-, или гетероарил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный или монозамещенный или полизамещенный;
или R5 и R6 совместно означают (CH2)n с n=2, 3, 4, 5 или 6, причем отдельные атомы водорода также могут быть замещены посредством F, Cl, Br, I, NO2, CF3, OR13, CN или С1-5-алкил;
R7, R8, R9 и R10 независимо друг от друга означают
Н, F, Cl, Br, I, NO2, CF3, OR13, SR13, SO2R13 SO2OR13, NHC(=O)NR14R15, SO2NR14R15, CN, COOR13, NR14R15; С1-5-алкил, C3-8-циклоалкил, незамещенный или монозамещенный или полизамещенный; арил-, или гетероарил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный или монозамещенный или полизамещенный;
причем R13 означает Н; С1-5-алкил каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный или полизамещенный; С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, незамещенный или монозамещенный или полизамещенный; арил-, или гетероарил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, C3-8-циклоалкил или гетероарил, незамещенный или монозамещенный или полизамещенный;
R14 и R15 независимо друг от друга означают Н; С1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный или полизамещенный; или С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, незамещенный или монозамещенный или полизамещенный; арил-, или гетероарил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный или монозамещенный или полизамещенный;
или R14 и R15 вместе образуют CH2CH2OCH2CH2, CH2CH2NR16CH2CH2 или (CH2)3-6,
причем R16 означает Н; С1-5-алкил насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный или полизамещенный;
X означает О, S, SO, SO2 или NR17;
R17 означает Н; С1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный; COR12 или SO2R12,
причем R12 означает H; С1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный или полизамещенный или незамещенный; С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, монозамещенный или полизамещенный или незамещенный; арил-, или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный; OR13; NR14R15;
в виде рацемата; энантиомеров, диастереомеров, смесей энантиомеров или диастереомеров или отдельного энантиомера или диастереомера; оснований и/или солей физиологически совместимых кислот или катионов.
При комбинации различных остатков, например R7, R8, R9 и R10, а также комбинации остатков с их заместителями, как, например, OR13, SR13, SO2R13 или COOR13, один заместитель, например, R13, в пределах одного вещества может принимать различные значения для двух или нескольких остатков, например R7, R8, R9 и R10.
Соединения согласно изобретению показывают хорошее связывание с ORL1-рецептором, а также с другими опиоидными рецепторами.
Выражения "С1-8-алкил" "С1-5-алкил" и "С1-3-алкил" в смысле данного изобретения включают ациклические насыщенные или ненасыщенные углеводородные остатки, которые могут быть разветвленными или с прямой цепью, а также незамещенными или моно- или полизамещенными, с 1, 2, 3, 4, 5, 6, 7 или 8 С-атомами соответственно с 1, 2, 3, 4 или 5 С-атомами соответственно 1, 2 или 3 С-атомами, т.е. С1-8-алканилы, С2-8-алкенилы и С2-8-алкинилы соответственно С1-5-алканилы, С2-5-алкенилы и С2-5-алкинилы соответственно С1-3-алканилы, С2-3-алкенилы и С2-3-алкинилы. При этом алкенилы имеют по меньшей мере одну двойную углерод-углеродную связь и алкинилы по меньшей мере одну тройную углерод-углеродную связь. Преимущественно алкил выбран из группы, которая включает метил, этил, n-пропил, 2-пропил, n-бутил, изо-бутил, втор.-бутил, трет.-бутил, n-пентил, изо-пентил, нео-пентил, n-гексил, 2-гексил; этиленил (винил), этинил, пропенил (-CH2CH=CH2, -СН=СН-CH3, -С(=CH2)-CH3), пропинил (-СН-С≡СН, -С≡С-CH3), 1,1-диметилэтил, 1,1-диметилпропил, бутенил, бутинил, пентенил, пентинил, гексил, гексенил, гексинил, гептил, гептенил, гептинил, октил, октенил или октинил. Особенно предпочтительны в смысле данного изобретения метил, этил, n-пропил и n-бутил.
Выражение "циклоалкил" или "С3-8-циклоалкил" для целей этого изобретения означает циклические углеводороды с 3, 4, 5, 6, 7 или 8 атомами углерода, причем углеводороды могут быть насыщенными или ненасыщенными (но не ароматическими), незамещенными или моно- или полизамещенными. Выгодно С3-8-циклоалкил выбран из группы, содержащей циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклопентенил, циклогексенил, циклогептенил и циклооктенил. Особенно предпочтительны в смысле данного изобретения циклобутил, циклопентил и циклогексил.
Под обозначением (СН2)3-6 следует понимать -CH2-CH2-CH2-, -CH2-CH2-CH2-CH2-, -CH2-CH2-CH2-CH2-CH2- и CH2-CH2-CH2-CH2-CH2-CH2-.
Выражение "арил" в смысле данного изобретения означает карбоциклические кольцевые системы с по меньшей мере одним ароматическим циклом, но без гетероатомов только в одном из колец, в частности фенилы, нафтилы и фенантренилы, фторантренилы, фторенилы, индалилы и тетралинилы. Арильные остатки могут также быть конденсированы с другими насыщенными, (частично) ненасыщенными или ароматическими кольцевыми системами. Каждый арильный остаток может быть незамещенным или монозамещенным или полизамещенным, причем арильные заместители могут быть одинаковыми или различными и в любом и возможном положении арила. Особенно предпочтительными являются остатки фенила или нафтила.
Выражение "гетероарил" означает 5-, 6- или 7-членный циклический ароматический остаток, который содержит, по меньшей мере, 1, при необходимости также 2, 3, 4 или 5 гетероатомов, причем гетероатомы являются одинаковыми или различными и гетероцикл может быть незамещенныйм или моно- или полизамещенным; в случае замещения в гетероцикле заместители могут быть одинаковыми или различными и находиться в любом и возможном положении гетероарила. Гетероцикл также может быть частью бициклической или полициклической системы. Предпочтительными гетероатомами являются азот, кислород и сера. Предпочтительно, чтобы гетероарильный остаток был выбран из группы, которая включает пирролил, индолил, фурил (фуранил), бензофуранил, тиенил (тиофенил), бензотиенил, бензотиадиазолил, бензотиазолил, бензотриазолил, бензодиоксоланил, бензодиоксанил, фталазинил, пиразолил, имидазолил, тиазолил, оксазолил, изоксазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, пиранил, индазолил, пуринил, индолизинил, хинолинил, изохинолинил, хиназолинил, карбазолил, феназинил, фенотиазинил или оксадиазолил, причем связь к соединениям общей структуры I может осуществляться через любой и возможный кольцевой член гетероарильного остатка.
В связи с определениями заместителей „алкил" означает „C1-5-алкил", если „алкил" не обозначен подробнее отдельно.
В связи с "алкил" и „циклоалкил" под понятием "замещенный" в смысле данного изобретения понимают замещение одного или нескольких водородных остатков посредством F, Cl, Br, I, -CN, NH2, NH-алкил, NH-арил, NH-гетероарил, NH-циклоалкил, NH-алкил-арил, NH-алкил-гетероарил, NH-алкил-ОН, N(алкил)2, N(алкил-арил)2, N(алкил-гетероарил)2, N(циклоалкил)2, N(алкил-ОН)2, NO2, SH, S-алкил, S-арил, S-гетероарил, S-алкил-арил, S-алкил-гетероарил, S-циклоалкил, S-алкил-ОН, S-алкил-SH, ОН, О-алкил, О-арил, О-гетероарил, О-алкил-арил, О-алкил-гетероарил, О-циклоалкил, О-алкил-ОН, СНО, C(=O)C1-6-алкил, С(=S)С1-6-алкил, С(=O)-арил, С(=S)арил, С(=O)С1-6-алкил-арил, С(=S)С1-6-алкил-арил, С(=O)-гетероарил, С(=S)-гетероарил, С(=O)-циклоалкил, С(=S)-циклоалкил, СО2Н, СО2-алкил, CO2-алкил-арил, C(=O)NH2, C(=O)NH-алкил, С(=O)NHарил, С(=O)NH-циклоалкил, С(=O)N(алкил)2, С(=O)N(алкил-арил)2, С(=O)N(алкил-гетероарил)2, С(=O)N(циклоалкил)2, SO-алкил, SO2-алкил, SO2NH2, SO3H, РО(O-С1-6-алкил)2 =O, =S, причем под полизамещенными остатками следует понимать те остатки, которые или в различных, или в одинаковых атомах замещены многократно, например, дважды или трижды, например, трижды в одинаковом С-атоме, как в случае CF3 или -CH2CF3 или в различных местах, как в случае -СН(OH)-СН=СН-CHCl2. Многократное замещение может происходить с одинаковыми или с различными заместителями. При необходимости один заместитель со своей стороны также может быть замещенным, так что -О-алкил в частности включает также -О-CH2-CH2-O-CH2-CH2-ОН. В смысле данного изобретения является предпочтительным, если алкил или циклоалкил замещены с помощью F, Cl, Br, I, CN, CH3, С2Н5, NH2, NO2, SH, CF3 ОН, OCH3, циклопентил, циклогексил, ОС2Н5 или N(CH3)2, предпочтительно F, Cl, Br, I, CN, CH3, С2Н5, NH2, NO2, SH, CF3, ОН, OCH3, ОС2Н5 или N(CH3)2. В высшей степени является предпочтительным, если алкил или циклоалкил замещены посредством ОН, OCH3 или ОС2Н5.
Что касается "арил" или "гетероарил", то в смысле данного изобретения под "моно- или полизамещенный" понимают однократное или многократное, например, двух-, трех- четырех- или пятикратное замещение одного или нескольких атомов водорода кольцевой системы посредством F, Cl, Br, I, CN, NH2, NH-алкил, NH-арил, NH-гетероарил, NH-алкил-арил, NH-алкил-гетероарил, NH-циклоалкил, NH-алкил-ОН, NH(алкил)2, NH(алкил-арил)2, N(алкил-гетероарил)2, N(циклоалкил)2, N(алкил-OH)2, NO2, SH, S-алкил, S-циклоалкил, S-арил, S-гетероарил, S-алкил-арил, S-алкил-гетероарил, S-циклоалкил, S-алкил-ОН, S-алкил-SH, ОН, О-алкил, О-циклоалкил, О-арил, О-гетероарил, О-алкил-арил, О-алкил-гетероарил, О-циклоалкил, O-алкил-ОН, СНО, C(=O)C1-6-алкил, С(=S)С1-6-алкил, С(=O)арил, С(=S)арил, С(=O)-С1-6-алкил-арил, С(=S)С1-6-алкил-арил, С(=O)-гетероарил, С(=S)-гетероарил, С(=O)-циклоалкил, С(=S)-циклоалкил, CO2H, CO2-алкил, CO2-алкил-арил, C(=O)NH2, C(=O)NH-алкил, С(=O)NН-арил, С(=O)NH-циклоалкил, С(=O)N(алкил)2, С(=O)N(алкил-арил)2, С(=O)N(алкил-гетероарил)2, С(=O)N(циклоалкил)2, S(O)-алкил, S(O)-арил, SO2-алкил, SO2-арил, SO2NH2, SO3H, CF3; алкил, циклоалкил, арил и/или гетероарил; в одном или при необходимости различных атомах (причем один заместитель при необходимости в свою очередь может быть замещен). При этом многократное замещение происходит с одинаковым или с различными заместителями. Особенно является предпочтительным в смысле данного изобретения, если арил или гетероарил замещены посредством F, Cl, Br, I, CN, СН3, С2Н5, NH2, NO2, SH, CF3, ОН, OCH3, ОС2Н5 или N(CH3)2.
Под понятием соль следует понимать всякую форму действующего вещества согласно изобретению, в котором оно принимает ионную форму соответственно заряжено и спарено с противоионом (катионом или анионом) соответственно находится в растворе. Под эти также следует понимать комплексы действующего вещества с другими молекулами и ионами, в особенности комплексы, которые комплексованы ионными взаимодействиями. В особенности под этим понимают (и это также является предпочтительной формой осуществления этого изобретения) физиологически совместимые соли, в особенности физиологически совместимые соли с катионами или основаниями и физиологически совместимые соли с анионами или кислотами, или также соль, образованную с физиологически совместимой кислотой или физиологически совместимым катионом.
Под понятием физиологически совместимой соли с анионами или кислотами в смысле данного изобретения понимают соли, по меньшей мере, одного из соединений согласно изобретению - в большинстве случаев, например в азоте, протонированном - как катион с, по меньшей мере, одним анионом, которые являются совместимыми физиологически - в особенности при применении у человека и/или млекопитающего животного. В особенности под этим в смысле данного изобретения понимают соль, образованную с помощью физиологически совместимой кислоты, то есть соли соответствующего действующего вещества с неорганическими или органическими кислотами, которые физиологически - в особенности при применении человеком и/или млекопитающим животным - являются совместимыми. Примерами физиологически совместимых солей определенных кислот являются соли: соляной кислоты, бромистоводородной кислоты, серной кислоты, метансульфокислоты, муравьиной кислоты, уксусной кислоты, щавелевой кислоты, янтарной кислоты, яблочной кислоты, винной кислоты, миндальной кислоты, фумаровой кислоты, молочной кислоты, лимонной кислоты, глутаминовой кислоты, сахариновой кислоты, монометилсебациновой кислоты, 5-оксо-пролин, гексан-1-сульфокислоты, никотиновой кислоты, 2-, 3- или 4-аминобензойной кислоты, 2,4,6-триметил-бензойной кислоты, α-липоновой кислоты, ацетилглицин, ацетилсалициловой кислоты, гиппуровой кислоты и/или аспарагиновой кислоты. Особенно предпочтительны хлористоводородная соль, цитрат и полуцитрат.
Под понятием соли, образованной физиологически совместимой кислотой, в смысле данного изобретения понимают соли соответствующего действующего вещества с неорганическими соответственно органическими кислотами, которые совместимы физиологически - в особенности при применении у человека и/или млекопитающего животного. Особенно предпочтителен гидрохлорид и цитрат. Примерами физиологически совместимых кислот являются: соляная кислота, бромистоводородная кислота, серная кислота, метансульфокислота, муравьиная кислота, уксусная кислота, щавелевая кислота, янтарная кислота, винная кислота, миндальная кислота, фумаровая кислота, молочная кислота, лимонная кислота, глутаминовая кислота, сахариновая кислота, монометилсебациновая кислота, 5-оксо-пролин, гексан-1-сульфокислота, никотиновая кислота, 2-, 3- или 4-аминобензойная кислота, 2,4,6-триметил-бензойная кислота, α-липоновая кислота, ацетилглицин, ацетилсалициловая кислота, гиппуровая кислота и/или аспарагиновая кислота.
Под понятием физиологически совместимой соли с катионами или основаниями в смысле данного изобретения понимают соли, по меньшей мере, одного из соединений согласно изобретению - большей частью одной (депротонированной) кислоты - как анион с, по меньшей мере, одним, предпочтительно неорганическим, катионом, которые совместимы физиологически - в особенности при применении у человека и/или млекопитающего животного. Особенно предпочтительны соли щелочных и щелочноземельных металлов, а также аммониевые соли, и в особенности (моно-) или (ди-) натриевые-, (моно-) или (ди-) калиевые, магниевые или кальциевые соли.
Под понятием соли, образованной с физиологически совместимым катионом, в смысле данного изобретения понимают соли, по меньшей мере, одного из соответствующих соединений как анион с по меньшей мере одним неорганическим катионом, который совместим физиологически - в особенности при применении у человека и/или млекопитающего животного. Особенно предпочтительны соли щелочных и щелочноземельных металлов, а также соли аммония, но в особенности (моно-) или (ди-) натриевые, (моно-) или (ди-) калиевые, магниевые или кальциевые соли.
Предпочтительны соединения общей формулы I,
в которой
R1 и R2, независимо друг от друга означают Н; С1-5-алкил каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный или полизамещенный или незамещенный; С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, монозамещенный или полизамещенный или незамещенный; арил, незамещенный или монозамещенный или полизамещенный; или С1-3-алкилом связанный арил, С3-8-циклоалкил или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный;
или остатки R1 и R2 вместе означают CH2CH2OCH2CH2, CH2CH2NR11CH2CH2 или (CH2)3-6,
причем R11 означает Н; С1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный или полизамещенный или незамещенный; С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, монозамещенный или полизамещенный или незамещенный; арил-, или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный;
R3 означает С1-8-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный или полизамещенный или незамещенный; С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, монозамещенный или полизамещенный или незамещенный;
R5 означает =O; Н; С1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный или полизамещенный; COOR13, CONR13, OR13; С3-8-циклоалкил, насыщенный или ненасыщенный, незамещенный или монозамещенный или полизамещенный; арил-, или гетероарил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный или монозамещенный или полизамещенный;
R6 означает Н; F, Cl, NO2, CF3, OR13, SR13, SO2R13, SO2OR13, CN, COOR13, NR14R15; С1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный или полизамещенный; С3-8-циклоалкил, насыщенный или ненасыщенный, незамещенный или монозамещенный или полизамещенный; арил-, или гетероарил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный или монозамещенный или полизамещенный;
или R5 и R6 совместно означают (CH2)n с n=2, 3, 4, 5 или 6, причем отдельные атомы водорода также могут быть замещены посредством F, Cl, Br, I, NO2, CF3, OR13, CN или С1-5-алкил;
R7, R8, R9 и R10 независимо друг от друга означают
Н, F, Cl, Br, I, NO2, CF3, OR13, SR13, SO2R13, SO2OR13, NHC(=O)NR14R15, SO2NR14R15, CN, COOR13, NR14R15; С1-5-алкил, С3-8-циклоалкил, незамещенный или монозамещенный или полизамещенный; арил-, или гетероарил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный или монозамещенный или полизамещенный;
причем R13 означает Н; С1-5-алкил каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный или полизамещенный; С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, незамещенный или монозамещенный или полизамещенный; арил-, или гетероарил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный или монозамещенный или полизамещенный;
R14 и R15 независимо друг от друга означают Н; С1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный или полизамещенный; или С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, незамещенный или монозамещенный или полизамещенный; арил-, или гетероарил, незамещенный или монозамещенный или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный или монозамещенный или полизамещенный;
или R14 и R15 вместе образуют CH2CH2OCH2CH2, CH2CH2NR16CH2CH2 или (CH2)3-6,
причем R16 означает Н; С1-5-алкил насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный или полизамещенный;
X означает О, S, SO, SO2 или NR17;
R17 означает Н; С1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный; COR12 или SO2R12,
причем R12 означает Н; С1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный или полизамещенный или незамещенный; С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, монозамещенный или полизамещенный или незамещенный; арил-, или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный; или связанный С1-3-алкилом арил, C3-8-циклоалкил или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный; OR13; NR14R15.
причем „алкил замещенный" или „циклоалкил замещенный" означает алкил или циклоалкил, замещенный с помощью F, Cl, Br, I, CN, CH3, С2Н5, NH2, NO2, SH, CF3 ОН, ОСН3, циклопентил, циклогексил, ОС2Н5 или N(CH3)2 и „арил замещенный или „гетероарил замещенный" означает арил или гетероарил, замещенный с помощью F, Cl, Br, I, CN, CH3, С2Н5, NH2, NO2, SH, CF3 ОН, ОСН3, ОС2Н5 или N(CH3)2,
в виде рацемата; энантиомеров, диастереомеров, смесей энантиомеров или диастереомеров или отдельного энантиомера или диастереомера; оснований и/или солей физиологически совместимых кислот или катионов.
Для предпочтительной формы осуществления изобретения спироциклических производных циклогексана согласно изобретению относится, что
R1 и R2 независимо друг от друга означают Н, С1-5-алкил, разветвленный или неразветвленный, насыщенный или ненасыщенный, незамещенный или монозамещенный или полизамещенный, или фенил или бензил, незамещенный или монозамещенный или полизамещенный,
или совместно означают кольцо и означают (CH2)3-6,
в особенности
R1 и R2 независимо друг от друга означают Н, метил, этил, n-пропил, или совместно -CH2CH2CH2- или -CH2CH2CH2CH2-, причем предпочтительно только один из остатков R1 и R2 означает Н.
Особенно предпочтительно R1 и R2 независимо друг от друга означают Н, CH3 или С2Н5, причем не оба остатка R1 и R2 означают Н,
или R1 и R2 образуют кольцо и означают -CH2CH2CH2- или -CH2CH2CH2CH2-.
В высшей степени предпочтительно R1 и R2 означают Н или CH3, причем R1 и R2 не одновременно означают CH3, в особенности CH3.
Также предпочтительны спироциклические производные циклогексана общей формулы I, в которой
R3 означает этил, n-пропил, 2-пропил, аллил, n-бутил, изо-бутил, втор.-бутил, трет.-бутил, n-пентил, изо-пентил, нео-пентил, n-гексил, метилциклопентил, метилциклогексил, циклопентил или циклогексил, каждый раз незамещенный или монозамещенный или полизамещенный с помощью ОН, ОСН3 или ОС2Н5. в особенности
R3 означает этил, n-пропил, 2-пропил, аллил, n-бутил, изо-бутил, втор.-бутил, трет.-бутил, n-пентил, изо-пентил, нео-пентил, n-гексил циклопентил или циклогексил, каждый раз незамещенный или монозамещенный или полизамещенный с помощью ОН, ОСН3 или ОС2Н5.
Особенно предпочтительны замещенные производные циклогексана общей формулы I, в которой R3 означает этил, n-пропил или n-бутил, незамещенный или монозамещенный или полизамещенный с помощью ОСН3, ОН или ОС2Н5, в особенности посредством OCH3.
К предпочтительной форме осуществления спироциклических производных циклогексана согласно изобретению относится, что
остаток R5 означает Н, CH3, СООН, COOCH3, CH2Офенил, причем фенильный остаток может быть замещен посредством F, Cl, Br, I, CN, CH3, С2Н5, NH2, NO2, SH, CF3 ОН, ОСН3, ОС2Н5или N(CH3)2, или CH2OH.
Особенно предпочтительны замещенные производные циклогексана, в которых R5 означает Н.
Предпочтительны также замещенные производные циклогексана общей формулы I, в которой R6 может означать Н; метил, этил, CF3, бензил или фенил, причем бензильный или фенильный остаток может быть замещен посредством F, Cl, Br, I, CN, CH3, С2Н5, NH2, NO2, SH, CF3 ОН, OCH3, ОС2Н5 или N(CH3)2.
Особенно предпочтительны спироциклические производные циклогексана, в которых R6 означает Н.
Далее предпочтительны спироциклические производные циклогексана, в которых R7, R8, R9 и R10 независимо друг от друга означают Н; С1-5-алкил, разветвленный или неразветвленный, незамещенный или моно- или полизамещенный; F, Cl, Br, I, CF3, ОН, OCH3, NH2, СООН, COOCH3, NHCH3 тиенил, пиримидинил, пиридил, N(CH3)2 или NO2
предпочтительно
один из остатков R7 R8, R9 и R10 означает Н; С1-5-алкил, разветвленный или неразветвленный, незамещенный или моно- или полизамещенный; F, Cl, Br, I, ОН, OCH3, СООН, COOCH3, NH2, NHCH3 или N(CH3)2 или NO2, в то время как остальные остатки представляют собой Н,
или
два из остатков R7 R8 R9 и R10 независимо друг от друга означают Н; С1-5
алкил, разветвленный или неразветвленный, незамещенный или моно- или полизамещенный; F, Cl, Br, I, ОН, OCH3, СООН, COOCH3, NH2, NHCH3 или
N(CH3)2 или NO2, в то время как остальные остатки представляют собой Н.
Особенно предпочтительны спироциклические производные циклогексана, в которых R7 R8, R9 и R10 означают Н, F, ОН, Cl или OCH3.
Соединения, в которых X означает О, являются совершенно особенно предпочтительными. Далее в высшей степени особенно предпочтительны соединения общей формулы I, где X означает NR17.
При этом предпочтительны спироциклические производные циклогексана, в которых R17 означает COR12 и R12 означает Н; С1-5-алкил; С3-8-циклоалкил;
или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, каждый раз монозамещенный или полизамещенный или незамещенный; NR14R15;
в особенности R12 означает Н; бензил, фенэтил, фенэтенил; каждый раз незамещенный или замещенный с помощью ОСН3; CH3, 2,2-диметилпропил или циклопентил.
В высшей степени предпочтительны соединения из группы:
4',9'-дигидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(3'H)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(1'H)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-гидрокси-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2,2,2-трифторацетат
6'-гидрокси-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-метилкарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-циклопентилкарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-(2,2)-диметилпропанкарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-(3,4-диметоксибензилкарбонил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-этиламинокарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-4-метоксибензиламинокарбонил-спиро[циклогексан-1,1,(1'Н)-пиридо[3,4-b]индол]-4-амин
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-метил-спиро[циклогексан-1,1,(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N-этил-N-метил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N-бензил-N-метил-4-бутил-спиро[циклогексан-1,1'(3,Н)-пирано[3,4-b]индол]-4-амин
6'-фтор-4',9'-дигидро-N-бензил-N-метил-4-бутил-спиро[циклогексан-1,1'(3'H)-пирано[3,4-b]индол]-4-амин
4-бутил-6'-фтор-4-(N-морфолино)-1',3',4',9'-тетрагидроспиро[циклогексан-1,1'-пирано[3,4-b]индол]
4-бутил-6'-фтор-4-(N-морфолино)-1',3',4',9'-тетрагидроспиро[циклогексан-1,1'-пирано[3,4-b]индол]
4',9'-дигидро-N,N-диметил-4-метоксипропил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N,N-диметил-4-метоксипропил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-(3-метоксипропил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
4',9'-дигидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
4',9'-дигидро-N,N-диметил-4-циклопентил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-циклопентил-спиро[циклогексан-1,1'(3'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
4',9'-дигидро-N,N-диметил-4-циклогексил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N,N-диметил-4-циклогексил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-циклогексил-спиро[циклогексан-1,1'(3'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-(3-метоксипропил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N,N-диметил-4-метоксипропил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-этиламинокарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин
4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N-бензил-4-аллил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N-фенил-4-аллил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин
6'-фтор-4',9'-дигидро-N-(4-метоксибензил)-4-аллил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин
N-{6'-фтор-4',9'-дигидро-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-ил}-пирролидин, 2-гидрокси-1,2,3-пропантрикарбоксилат
N-{6'-фтор-4',9'-дигидро-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-ил}-пиперидин
N-{6'-фтор-4',9'-дигидро-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-ил}-пиперидин, 2-гидрокси-1,2,3-пропантрикарбоксилат
N-{6'-фтор-4',9'-дигидро-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-ил}-n-метилпиперазин, 2-гидрокси-1,2,3-пропантрикарбоксилат
4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-гидрокси-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'H)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
6'-фтор-4',9'-дигидро-N,N-диметил-4-циклопентилметил-спиро[циклогексан 1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-(2-фенилэтенкарбонил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат
при необходимости также как смесь.
Вещества согласно изобретению оказывают влияние, например, на релевантный в связи с различными заболеваниями ORL1-рецептор, так что они пригодны в качестве фармацевтического действующего вещества в лекарственном средстве. Поэтому другим объектом изобретения являются лекарственные средства, содержащие, по меньшей мере, одно спироциклическое производное циклогексана согласно изобретению, а также при необходимости пригодные добавки и/или вспомогательные вещества и/или при необходимости другие действующие вещества.
Лекарственные средства согласно изобретению наряду с по меньшей мере одним спироциклическим производным циклогексана согласно изобретению при необходимости содержат пригодные добавки и/или вспомогательные вещества, а также носители, наполнители, растворители, разбавители, красители и/или связующие вещества, и могут применяться как жидкие лекарственные формы в виде инъекционных растворов, капель или сиропов, как полутвердые лекарственные формы в виде гранулятов, таблеток, гранул, пэтчей, капсул, пластырей/распыляемых пластырей или аэрозолей. Выбор вспомогательных веществ и т.п., а также их используемые количества зависят от того, как должно применяться лекарственное средство - орально, перорально, парентерально, внутривенно, внутрибрюшинно, внутрикожно, внутримышечно, интраназально, буккально, ректально или местно, например, наноситься на кожу, слизистые оболочки или в глаза. Для орального применения пригодны препараты в виде таблеток, драже, капсул, гранулятов, капель, соков и сиропов, для парентерального, местного и ингаляционного применения пригодны растворы, суспензии, легко восстанавливаемые сухие препараты, а также спреи. Спироциклические производные циклогексана согласно изобретению являются пригодными для чрескожных препаратов, в препаратах пролонгированного действия, в растворенной виде или в пластыре, при необходимости с добавлением средств, способствующих кожному проникновению. Препаративные формы, применяемые перорально или чрескожно, могут высвобождать замедленно спироциклические производные циклогексана согласно изобретению. Согласно изобретению спироциклические производные циклогексана могут также применяться в парентеральных формах длительного действия, таких как, например, имплантаты или имплантационные насосы. В принципе, к лекарственным средствам согласно изобретению могут добавляться другие действующие вещества, известные специалисту в данной области техники.
Вводимые пациентам количества действующего вещества варьируются в зависимости от веса пациента, от вида применения, показания и степени тяжести заболевания. Обычно применяют от 0,00005 до 50 мг/кг, предпочтительно от 0,001 до 0,5 мг/кг по меньшей мере одного спироциклического производного циклогексана согласно изобретению.
В предпочтительной форме лекарственного средства содержащееся согласно изобретению спироциклическое производное циклогексана находится в виде чистого диастереомера и/или энантиомера, в виде рацемата или как неэквимолярная или эквимолярная смесь диастереомеров и/или энантиомеров.
Как определено в вводной части из уровня техники, ORL1-рецептор был в особенности идентифицирован в проявлении боли. Соответственно спироциклические производные циклогексана согласно изобретению могут применяться для получения лекарственного средства для лечения боли, особенно острой, невропатической или хронической боли.
Поэтому другим объектом изобретения является применение спироциклического производного циклогексана согласно изобретению для получения лекарственного средства для лечения боли, в особенности острой, висцеральной, невропатической или хронической боли.
Другим объектом изобретения является применение спироциклического производного циклогексана согласно изобретению для получения лекарственного средства для лечения состояний страха, стресса и связанных со стрессом синдромов, депрессий, эпилепсии, болезни Альцгеймера, старческого слабоумия, общих познавательных дисфункций, нарушений обучения и памяти (как ноотроп), синдромов отмены, злоупотребления и/или алкогольной, и/или наркотической, и/или медикаментозной зависимости, сексуальных дисфункций, сердечно-сосудистых заболеваний, гипотонии, гипертензии, тиннитуса, зуда, мигрени, тугоухости, недостаточной подвижности кишечника, нарушенного приема пищи, анорексии, ожирения, локомоторных расстройств, диареи, кахексии, недержания мочи соответственно как мышечный релаксант, противосудорожное средство или анестетик соответственно для совместного приема при лечении с опиоидным анальгетиком или с анестетиком, для диуреза или антинатрийуреза, анксиолизиса, для модуляции двигательной активности, для модуляции распределения нейромедиаторов и лечения связанных с этим нейродегенеративных заболеваний, для лечения синдромов отмены и/или для снижения наркотического потенциала опиоидов.
При этом в одном из указанных выше применений может быть предпочтительным, если применяемое спироцикличесоке производное циклогексана находится в виде чистого диастереомера и/или энантиомера, в виде рацемата или как не эквимолярная или эквимолярная смесь диастереомеров и/или энантиомеров.
Другим объектом изобретения является способ лечения, в особенности при одном из указанных выше показаний, нечеловеческого млекопитающего или человека, которое или который нуждается в лечении болей, в особенности хронических болей, путем введения терапевтически действующей дозы спироциклического производного циклогексана согласно изобретению, или лекарственного средства согласно изобретению.
Другим объектом изобретения является способ получения спироциклических производных циклогексана согласно изобретению как приведено в нижеследующем описании и примерах. В особенности при этом пригоден способ получения спироциклического производного циклогексана согласно изобретению, при котором производное циклогексана общей формулы Е подвергается взаимодействию с производным индола общей формулы F или Н.
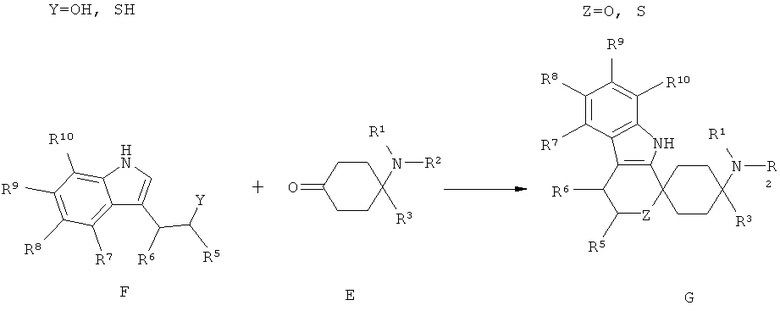
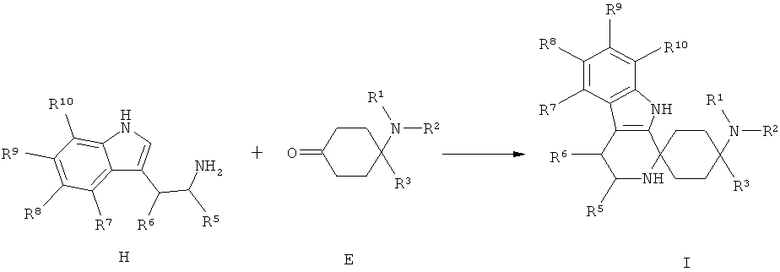
Триптофолы типа F (Y=О) могут реагировать в реакциях типа Окса-Пиктет-Шпенглера (Oxa-Pictet-Spengler), и триптамины типа Н в реакциях типа Пиктет-Шпенглера (Pictet-Spengler), с кетонами при добавлении, по меньшей мере, одного приемлемого реагента из группы кислот, кислотных ангидридов, сложных эфиров или слабо кисло реагирующих солей или кислот Льюиса при образовании продуктов формулы I. Для X=SH реакция осуществляется аналогично.
При этом предпочтительно используют, по меньшей мере, один реагент из группы карбоновых кислот, фосфорных кислот или сульфокислот или их соответствующих ангидридов, триалкилсилилового эфира карбоновой кислоты, кисло реагирующих солей, минеральных кислот или кислот Льюиса, выбранных из группы, состоящей из бортрифторида, хлорида индия(III), тетрахлорида титана, хлорида алюминия(III), или при добавлении, по меньшей мере, одной соли переходного металла, предпочтительно при добавлении, по меньшей мере, одного трифлата переходного металла (трифторметансульфоната переходного металла), особенно предпочтительно при добавлении, по меньшей мере, одного трифторметансульфоната переходного металла, выбранного из группы, состоящей из трифторметансульфоната скандия(III), трифторметансульфоната иттербия(III) и трифторметансульфоната индия(III), при необходимости при добавлении целита, с твердофазно связанными реактандами или реагентами, при повышенной или сниженной температуре, с или без микроволнового облучения, при необходимости в пригодном растворителе или смеси растворителей, как, например, хлорированных или нехлорированных, предпочтительно ароматических, углеводородов, ацетонитриле; в простоэфирных растворителях, предпочтительно в диэтиловом эфире или ТГФ; или в нитрометане, в пригодных случаях также в спиртах или воде.
Особенно предпочтительно при этом используют пара-толуолсульфонат пиридиния, пентоксид фосфора в присутствии целита, эфират бортрифторида, трифторуксусную кислоту, тетраизопропиловый эфир ортотитановой кислоты вместе с трифторуксусной кислотой, триметилсилиловый эфир трифторметансульфокислоты, трифторметансульфокислоту, метансульфокислоту, трифторуксусную кислоту, уксусную кислоту, фосфорную кислоту, полифосфорную кислоту, полифосфатный эфир, п-толуолсульфокислоту, соляную кислоту HCl-газ, серную кислоту вместе с ацетатным буфером, тетрахлорид олова.
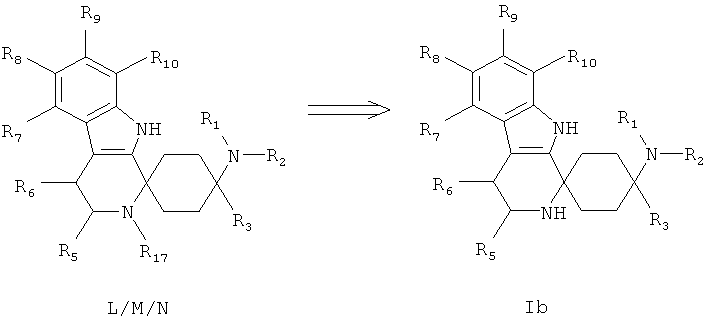
Вторичные амины -I могут быть ацилированы, сульфонированы или карбамоилированы согласно известным для специалиста в данной области техники способам до получения соединений типа L/M/N. Предпочтительно эти реакции проводят при повышенной температуре, особенно предпочтительно при облучении микроволнами.
Один такой метод, известный специалисту в данной области техники, может представлять собой взаимодействие с ангидридом или хлорангидридом кислоты при добавлении основания, например триэтиламина.
Синтез кетоновых структурных звеньев
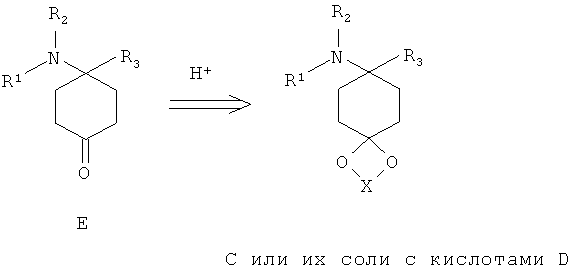
Соединения формулы Е могут быть выделены из соответствующих ацеталей С, или из их солей D, согласно известным для специалиста в данной области техники методам путем снятия защиты с помощью кислот. При этом X выбран из группы алкил, алкил/ алкилиден/ с арил или алкил (насыщенный/ненасыщенный) замещенным алкилиденом
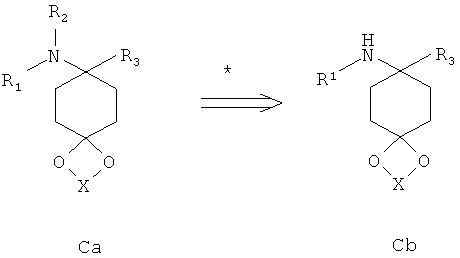
Аминоацетали Cb максимально с одним заместителем у атома азота могут быть переведены в соответствии со способом, известным специалисту в данной области техники, например, путем восстановительного аминирования, в соответствующие аминоацетали Са с одним или двумя другими заместителями у азота.
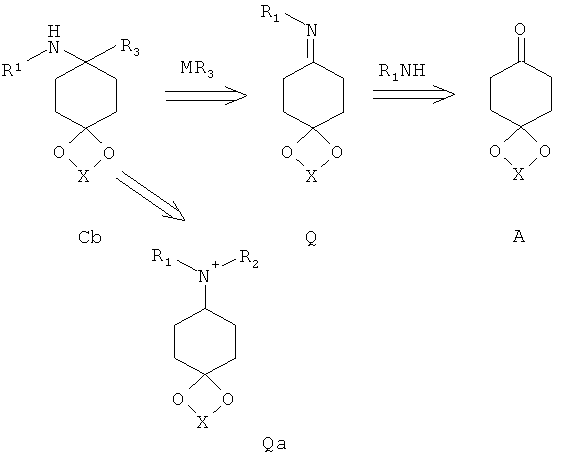
Аминоацетали Cb с максимально одним заместителем у атома азота могут быть получены в соответствии со способом, известным специалисту в данной области техники, путем присоединения углеродных нуклеофилов к иминам Q, предпочтительно с металлоорганическими соединениями в инертных растворителях, особенно предпочтительно с реактивами Гриньяра или литийорганическими соединениями, предпочтительно в простых эфирах, предпочтительно при температурах от -100° до комнатной температуры.
Аминоацетали C с двумя заместителями у атома азота также могут быть получены в соответствии со способом, известным специалисту в данной области техники, путем присоединения углеродных нуклеофилов к солям енаминов Qa, предпочтительно с металлоорганическими соединениями в инертных растворителях.
Получение иминов известно из литературных источников.
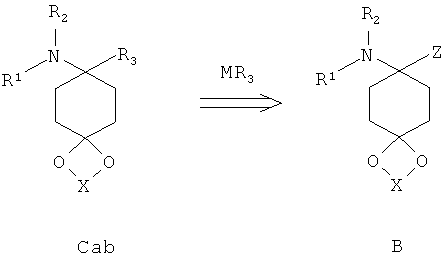
Ацетали С также могут быть получены путем замещения пригодных уходящих групп Z в структурах формулы В. Пригодными уходящими группами являются предпочтительно цианогруппы; 1,2,3-триазол-1-ил-группы. Другими пригодными уходящими группами являются 1Н-бензо[d][1,2,3]триазол-1-ил-группы и пиразол-1-ил-группы (Katritzky et al., Synthesis 1989, 66-69). Особенно предпочтительным путем до получения соединений структуры С является взаимодействие аминонитрилов В с соответствующими металлоорганическими соединениями, предпочтительно соединениями Гриньяра, предпочтительно в простых эфирах, предпочтительно при комнатной температуре. Металлоорганические соединения или могут быть приобретены в продаже, или могут быть получены согласно известным способам. Другой особенно предпочтительный путь до получения соединений структуры С представляет собой взаимодействие аминотриазолов В с соответствующими металлоорганическими соединениями, предпочтительно соединениями Гриньяра, предпочтительно в простых эфирах, предпочтительно при комнатной температуре.
Металлоорганические соединения или могут быть приобретены в продаже, или могут быть получены согласно методам, известным из литературных источников.
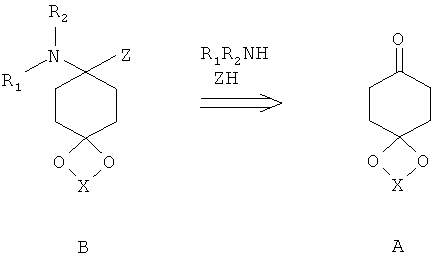
Структуры формулы В возможно получить путем реакции кетонов А с аминами и кислотными реактантами Z-H. Пригодными реактантами Z-H являются, например, цианистый водород, 1,2,3-триазол, бензотриазол или пиразол.
Особенно предпочтительным путем до получения соединений структуры В является взаимодействие кетонов с цианидами металлов и соответствующим амином в присутствии кислоты, предпочтительно в спирте, при температурах от - 40 до 60°С, предпочтительно при комнатной температуре с цианидами щелочных металлов в метаноле.
Другой особенно предпочтительный путь до получения соединений структуры В представляет собой взаимодействие кетонов с 1,2,3-триазолом и соответствующим амином при обезвоживающих условиях, предпочтительно с применением водоотделителя при повышенной температуре в инертном растворителе или при применении молекулярного сита или другого сушильного агента. Аналогично возможно ввести В подобные структуры с бензотриазоловыми или пиразоловыми вместо триазоловых групп.
Соединения общих формул F и Н или могут быть приобретены в продаже, или их получение известно из уровня техники, или для специалиста в данной области техники очевидным образом вытекает из уровня техники. В особенности релевантными для этого являются нижеследующие процитированные источники: Jirkovsky et al., J. Heterocycl. Chem., 12, 1975, 937-940; Beck et al., J. Chem. Soc. Perkin 1, 1992, 813-822; Shinada et al., Tetrahedron Lett., 39, 1996, 7099-7102; Garden et al., Tetrahedron, 58, 2002, 8399-8412; Lednicer et al., J. Med. Chem., 23, 1980, 424-430; Bandini et al. J. Org. Chem. 67, 15; 2002, 5386-5389; Davis et al., J. Med. Chem. 35, 1, 1992, 177-184; Yamagishi et al., J. Med. Chem. 35, 11,1992, 2085-2094; Gleave et al.; Bioorg. Med. Chem. Lett. 8, 10, 1998, 1231-1236; Sandmeyer, Helv. Chim. Acta; 2; 1919; 239; Katz et al.; J. Med. Chem. 31, 6, 1988; 1244-1250; Вас et al. Tetrahedron Lett. 1988, 29, 2819; Ma et al. J. Org. Chem. 2001, 66, 4525; Kato et al. J. Fluorine Chem. 99, 1, 1999, 5-8.
Исследования растворимости
Исследования растворимости были проведены на основании пяти соединений согласно изобретению и пяти примерных соединений. Данные были собраны при помощи серии соединений, которые, не считая остаток у R3, имеют много общего, чем гарантируется сопоставимость.
Выявилось, что соединения, которые в R3 имеют алкильный остаток, являются явно лучше растворимыми, чем соединения, которые в R3 имеют фенильный или тиенильный остаток. Неожиданно именно эта структурная вариация вызывает повышение растворимости. Введение ОН-группы в R8, типичная дериватизация (метаболизация), которая у живого организма проводится для повышения растворимости, чтобы вывести соединение через почки, не приводило к сравнимому повышению растворимости (соединения V-4 и V-5).
Примеры
Нижеследующие примеры служат для детального объяснения изобретения, однако не ограничивают общие изобретательские идеи.
Выходы полученных соединений не являются оптимизированными.
Все температуры не корригируются.
Указание „простой эфир" означает диэтиловый эфир, „ЕЕ" этилацетат и „ДХМ" дихлорметан. Указание „эквиваленты" означает эквиваленты количества веществ, „Тпл." точка плавления соответственно область плавления, „разл." разложение, „КТ" комнатная температура, „абс." абсолютный (безводный), "рац." рацемический, „конц." концентрированный, „мин" минуты, „ч" часы, „дн." дни, „объемн.%" объемный процент, „m%" массовый процент и „М" представляет собой указание концентрации в моль/л.
В качестве неподвижной фазы для колоночной хроматографии использовали силикагель 60 (0.040-0.063 мм) фирмы Е.Merck, Дармштадт.
Исследования тонкослойной хроматографией проводили с помощью ВЭТСХ - готовых пластин, силикагель 60 F 254, фирмы Е.Merck, Дармштадт.
Соотношение компонентов смеси растворителей для хроматографических исследований всегда указаны в объемн./объемн.
Кетоны
Структурное звено В-1:
8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил (В-1)
К смеси из 4N соляной кислоты (50 мл) и метанола (30 мл) при охлаждении льдом добавляли 40-проц. водный раствор диметиламина (116 мл, 0.92 моль), циклогексан-1,4-дион-моноэтиленкеталь (30.0 г, 0.192 моль) и цианид калия (30.0 г, 0.46 моль). Смесь 72 ч при комнатной температуре перемешивали и затем после добавления воды (80 мл) экстрагировали простым эфиром (4×100 мл). После концентрирования раствора осадок ресуспендировали в дихлорметане (200 мл) и высушивали сульфатам магния в течение ночи. Органическую фазу концентрировали и получали кеталь В-1 в виде белого твердого вещества.
Выход: 38.9 г (96%) Точка плавления: 86-88°С
1Н-ЯМР (ДМСО-d6): 1.57 (2H, m); 1.72 (2H; m); 1.85 (2H, m); 1.99 (2H, m); 2.25 (6H, s); 3.87 (4H, m).
13С-ЯМР (ДМСО-d6): 30.02; 31.32; 60.66; 63.77; 106.31; 118.40.
Структурное звено В-2:
8-(этилметиламино)-1,4-диоксаспиро[4.5]декан-8-карбонитрил (В-2)
К смеси из 4 N соляной кислоты (15 мл, 60 ммоль) и метанола (10 мл) при охлаждении льдом сначала добавляли этилметиламин (16.0 г, 23 мл, 262 ммоль) и воду (10 мл), после этого 1,4-диоксаспиро[4.5]дека-8-он (9.40 г, 60 ммоль), а также цианид калия (9.20 г, 141 ммоль). Реакционную смесь перемешивали 5 дн. при комнатной температуре. Затем добавляли воду (100 мл) и раствор экстрагировали диэтиловым эфиром (5×50 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме.
Выход: 10.8 г (80%), желтое масло
1Н-ЯМР (ДМСО-d6): 1.04 (t, 3H, J=7.1 Hz); 1.50-1.59 (m, 2Н); 1.68-1.77 (m, 2Н); 1.89-1.95 (m, 2Н); 1.98-2.06 (m, 2Н); 2.23 (s, 3H); 2.42-2.48 (m, 2Н, наложенный от ДМСО-сигнала); 3.87 (s, 4Н).
Структурное звено Е-1:
Это структурное звено было получено вместо желаемого целевого продукта. Является очевидным, что D-1 также может быть целенаправленно получен из бромистого этилмагния и В-1.
(8-этил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин (D-1)
Смесь этилбромида (30.0 г, 0.3 моль) и 3-бромпиридина (16.0 г, 0.1 моль) добавляли по каплям к магниевому порошку (10.0 г) в диэтиловый эфир (50 мл). После того как было завершено образование Гриньяра, серый раствор при 0°С перемешивали в течение 15 мин с аминонитрилом В-1 (10.5 г, 47.6 ммоль) в ТГФ (80 мл) и реакционный раствор перемешивали в течение ночи при комнатной температуре. Затем реакционный раствор при охлаждении льдом смешивали с 20%-ным раствором хлорида аммония (50 мл) и водой (50 мл). Реакционный раствор разбавляли с диэтиловым эфиром (100 мл), органическую фазу отделяли и водную фазу экстрагировали 2 × с помощью Et2O (100 мл). Объединенные органические фазы промывали водой (50 мл) и раствором NaCl (50 мл), высушивали над Na2SO4, фильтровали и растворитель удаляли в вакууме. Осадок ресуспендировали в 2-бутаноне (200 мл) и при 0°С смешивали с Me3SiCl (10 мл). Реакционный раствор при влажности воздуха перемешивали 5 ч и выпавшее в осадок твердое вещество отсасывали.
Выход: 6.8 г (64%), светло-коричневое твердое вещество
1Н-ЯМР (ДМСО-d6): 0.94 (3H, t); 1.51-1.60 (2H, m); 1.77-1.86 (8H, m); 2.64 (6H, 2s); 3.83-3.89 (4H, m).
4-диметиламино-4-этил-циклогексанон (Е-1)
Гидрохлорид D-1 (6.67 г, 0.026 ммоль) растворяли в 6N HCl (40 мл) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь дважды экстрагировали диэтиловым эфиром (100 мл). Затем при охлаждении льдом подщелачивали посредством 5N NaOH и снова трижды экстрагировали с помощью Et2O (100 мл). Объединенные органические фазы высушивали над NaSO4, фильтровали и растворитель удаляли в вакууме.
Выход: 4.16 г (92%), коричневое масло
1Н-ЯМР (ДМСО-d6): 0.81 (3H, t); 1.43-1.50 (2H, q); 1.67-1.89 (2H, m); 1.83-1.89 (2Н, m); 1.99-2.06 (2H, m); 2.22 (6H, 2 s); 2.39-2.43 (4H, m).
13С-ЯМР (ДМСО-d6): 8.71; 21.99; 30.41; 36.17; 37.07; 38.66; 55.53; 210.57.
Структурное звено Е-2:
Вариант 1:
(8-бутил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид (D-2)
8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил В-1 (10.5 г, 50 ммоль) помещали в ТГФ (150 мл) при охлаждении льдом и аргон. В течение 15 мин добавляли по каплям 2М хлорид бутилмагния в ТГФ (62.5 мл, 125 ммоль) и перемешивали 16 ч при комнатной температуре. Состав смешивали при охлаждении льдом с 20%-ным раствором хлорида аммония (37 мл) и водой (50 мл) и экстрагировали простым эфиром (3×50 мл). Органическую фазу промывали водой (1×50 мл) и насыщенным раствором хлорида натрия (1×50 мл), органическую фазу высушивали посредством Na2SO4 и концентрировали в вакууме.
Сырой продукт (2.05 г) растворяли в этилметилкетоне (75 мл), при охлаждении льдом смешивали с ClSiMe3 (9.5 мл, 75 ммоль) и 6 ч перемешивали при комнатной температуре. Выпавший в осадок белый остаток отсасывали и высушивали в вакууме.
Выход: 3.1 г (22%)
1Н-ЯМР (ДМСО-d6): 0.91 (3H, t); 1.31 (4H, m); 1.56 (2H, m); 1.75 (8H, m); 2.64 (6H, s); 3.87 (4H, s); 9.87 (1H, s).
Вариант 1:
4-бутил-4-диметиламино-циклогексанон (Е-2)
8-бутил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид D-2 (3.10 г, 11.1 ммоль) помещали в Н2О (4.7 мл) и конц. HCl (7 мл) и 24 ч перемешивали при комнатной температуре. Состав экстрагировали простым эфиром (1×15 мл), водную фазу при охлаждении льдом подщелачивали посредством 5N NaOH и экстрагировали дихлорметаном (3×20 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вакууме.
Выход: 1.96 г (89%), масло
1Н-ЯМР (ДМСО-d6): 0.88 (3H, t); 1.23 (4H, m); 1.40 (2H, m); 1.68 (2H, m); 1.91 (2H, m); 2.31 (2H, m); 2.22 (6H, s); 2.42 (2H, m).
13С-ЯМР (ДМСО-d6): 13.91; 23.21; 26.06; 29.53; 31.07; 37.04; 38.88; 55.36; 210.37.
Вариант 2:
(8-бутил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид (D-2)
К раствору аминонитрила В-1 (38,3 г, 0,182 моль) в абс. тетрагидрофуране (420 мл) при охлаждении смесью льда и поваренной соли медленно добавляли 2М раствор хлорида n-бутилмагния в ТГФ (228 мл, 0,456 моль) под аргоном. При этом температура реакции не должна превышать выше 10°С. Затем перемешивали 16 ч при комнатной температуре. Образовался коричневый прозрачный раствор. Для переработки реакционной смеси при охлаждении льдом (от 0 до 10°С) добавляли по каплям насыщенный раствор хлорида аммония (150 мл). При этом образовалось белое твердое вещество, которое было растворено путем добавления воды (приблизительно 250 мл). Реакционную смесь экстрагировали диэтиловым эфиром (4×100 мл). Органическую фазу промывали водой (100 мл) и насыщенным раствором NaCl (100 мл), высушивали и концентрировали. Оставалось желтое масло (44,5 г), которое кроме желаемого соединения бутила содержало еще исходный продукт нитрил. Сырой продукт растворяли в этилметилкетоне (275 мл), при охлаждении льдом смешивали с ClSiMe3 (32 мл, 0,245 моль) и перемешивали в открытой колбе при комнатной температуре. Гидрохлорид D-2 отделяли путем многократной фильтрации с интервалом в 2 ч. После продолжения реакции в 6-8 ч можно было выделить гидрохлорид D-2 с выходом в 82% (41,8 г) в виде белого твердого вещества.
Вариант 2:
4-бутил-4-диметиламино-циклогексанон (Е-2)
Гидрохлорид D-2 (41,8 г, 0,15 ммоль) растворяли в воде (78 мл) и при перемешивании и охлаждении льдом смешивали с 37-проц. соляной кислотой (100 мл, 1,2 моль). Прозрачную реакционную смесь перемешивали 7 дней при комнатной температуре. После оконченного гидролиза реакционную смесь экстрагировали диэтиловым эфиром (2×70 мл). Органические экстракты отбрасывали. Водную фазу при охлаждении льдом подщелачивали с помощью 5N раствора едкого натра (приблизительно 250 мл) и сильно перемешивали. Раствор экстрагировали диэтиловым эфиром (3×100 мл). Объединенные органические экстракты промывали водой (2×70 мл), высушивали и концентрировали. Кетон Е-2 выделили в виде светло-коричневого масла с выходом в 96% (28,4 г). Выход кетона Е-2 - в пересчете на использованный в первой стадии кеталь - составил 75%.
Структурное звено Е-3:
1-хлор-4-метокси-бутан
Гидрид натрия (24.0 г, 1.0 моль) и йодметан (142 г, 1.0 моль) помещали в абс. ТГФ (350 мл). Под аргоном и при охлаждении льдом добавляли по каплям в течение 1.5 ч раствор 4-хлорбутан-1-ол (54 г, 0.5 моль) в абс. ТГФ (50 мл), при этом возникало легкое газообразование. Состав перемешивали 24 ч при комнатной температуре.
К реакционному раствору прикапывали 20%-ный раствор NH4Cl (130 мл). Органическую фазу отделяли, высушивали над Na2SO4 и сушильный агент отфильтровывали.
Органическую фазу дистиллировали под нормальным давлением.
Точка кипения: 150-162°С
Выход: 10.4 г (17%)
1Н-ЯМР (ДМСО-d6): 1.93 (2H, m); 3.23 (3H; s); 3.44 (2H, t); 3.66 (2H, t).
[8-(4-метокси-бутил)-1,4-диокса-спиро[4.5]дец-8-ил]-диметил-амин (С-3)
Магний (1.62 г, 66.8 ммоль) и I2 в абс. диэтиловом эфире (25 мл) под атмосферой аргона и временном нагревании смешивали с раствором 1-хлор-4-метокси-бутана (8.19 г, 66.8 ммоль) в абс. простом эфире (12 мл). Смесь перемешивали 1 ч при кипячении с обратным холодильником, до тех пор пока магний не растворился далее.
При охлаждении льдом добавляли по каплям раствор 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрила В-1 (10.5 г, 50.1 ммоль) в абс. ТГФ (40 мл). При этом в осадок выпадал вязкий остаток, для лучшего смешивания добавляли остальной абс. ТГФ (20 мл). Смесь перемешивали при комнатной температуре 24 ч.
Состав при охлаждении льдом смешивали с раствором NH4Cl (20%-ный, 80 мл) и водой (100 мл), органическую фазу отделяли и водную фазу экстрагировали простым эфиром (3×100 мл).
Объединенные органические фазы промывали насыщенным раствором NaCl (80 мл) и водой (80 мл), высушивали над Na2SO4 и концентрировали в вакууме. Сырой продукт очищали флеш-хроматографией с хлороформ/метанолом (50:1⇒20:1⇒9:1).
Выход: 6.44 г (59%), желтое масло
13С-ЯМР (ДМСО-d6): 19.81; 27.10; 29.26; 30.34; 35.79; 37.48; 57.76: 63.72; 64.07; 71.35; 106.46.
4-диметиламино-4-(4-метокси-бутил)-циклогексанон (Е-3)
[8-(4-метокси-бутил)-1,4-диокса-спиро[4.5]дец-8-ил]-диметил-амин (С-3) (6.44 г, 23.7 ммоль) растворяли в воде (9.3 мл), смешивали с конц. HCl (14.6 мл) и перемешивали при комнатной температуре 4 дн.
Реакционную смесь промывали простым эфиром (2×50 мл). Затем раствор подщелачивали с помощью 5N NaOH и экстрагировали дихлорметаном (3×50 мл). Объединенные органические фазы промывали водой (50 мл), высушивали над Na2SO4, фильтровали и растворитель удаляли в вакууме.
Выход: 4.91 г (91%), желтое масло
13С-ЯМР (ДМСО-d6): 20.56; 29.75; 29.83; 30.98; 36.92; 37.06; 55.40: 57.73; 71.76; 210.39.
Структурное звено Е-4:
1-хлор-3-метокси-пропан
3-метоксипропан-1-ол (47.1 г, 50 мл, 0.523 моль) растворяли в пиридине (41.3 г, 42.6 мл, 0.523 моль), охлаждали до 10°С и при 10-30°С при сильном помешивании смешивали по каплям с тионилхлоридом (93.3 г, 56.9 мл, 0.784 моль). При этом в осадок выпадал твердый остаток, смесь потом еще перемешивали 3 ч при 65°С.
Состав выливали в смесь изо льда (130 г) и конц. HCl (26 мл). Водный раствор экстрагировали простым эфиром (2×20 мл) и объединенные органические фазы промывали раствором K2CO3. При добавлении сушильного агента К2СО3 наблюдалось сильное газообразование, поэтому раствор отстаивался в течение ночи.
Сушильный агент отфильтровывали и органическую фазу промывали раствором К2СО3 до щелочной реакции. Органическую фазу отделяли, промывали водой и высушивали через K2CO3, фильтровали и дистиллировали при нормальном давлении.
Точка кипения: 113°С
Выход: 41.2 г (72%), бесцветная жидкость
1Н-ЯМР (ДМСО-d6): 1.93 (2H, m); 3.23 (3H; s); 3.44 (2H, t); 3.66 (2H, t).
[8-(3-метокси-пропил)-1,4-диокса-спиро[4.5]дец-8-ил]-диметил-амин (С-4)
Магний (10.0 г, 92 ммоль) и J2 в абс. диэтиловом эфире (30 мл) под атмосферой аргона и временном нагревании по каплям смешивали с раствором 1-хлор-3-метокси-пропана (10.0 г, 92 ммоль) в абс. простом эфире (15 мл). Затем состав при кипячении с обратным холодильником перемешивали еще 60 мин, магний после этого не был полностью растворен.
При охлаждении льдом добавляли по каплям раствор 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрила В-1 (9.68 г, 46 ммоль) в абс. ТГФ (30 мл). При этом в осадок выпадал вязкий остаток, для лучшего смешивания добавляли еще 100 мл абс. ТГФ. Смесь перемешивали при комнатной температуре 24 ч.
Состав при охлаждении льдом смешивали с 20%-ным раствором NH4Cl (100 мл) и водой (120 мл), органическую фазу отделяли и водную фазу экстрагировали простым эфиром (3×120 мл).
Объединенные органические фазы промывали насыщенным раствором NaCl (120 мл) и воды (120 мл), высушивали над Na2SO4 и концентрировали в вакууме. Выход сырого продукта составил 10.8 г коричневого масла. 9.8 г сырого продукта очищали флеш-хроматографией с CHCl3/МеОН (50:1⇒20:1⇒9:1).
Выход: 8.11 г (75%), желтое масло
1Н-ЯМР (ДМСО-d6): 1.44 (8H, m); 1.62 (4H; m); 2.25 (6H, s); 3.21 (3H, s); 3.31 (2H, m); 3.82 (4H, s).
13С-ЯМР (ДМСО-d6): 23.99; 26.52; 28.87; 29.88; 36.97; 55.24: 57.67; 63.40; 72.62; 108.07.
4-диметиламино-4-(3-метокси-пропил)-циклогексанон (Е-4)
Амин С-4 (8.11 г, 31.5 ммоль) растворяли в воде (12 мл), при охлаждении льдом смешивали с конц. HCl (19.5 мл) и 3 дн. перемешивали при комнатной температуре. Реакционную смесь промывали простым эфиром (2×75 мл). Затем раствор подщелачивали с помощью 5N NaOH и экстрагировали дихлорметаном (3×75 мл). Объединенные органические фазы промывали водой (75 мл), высушивали над Na2SO4, фильтровали и растворитель удаляли в вакууме.
Выход: 6.03 г (90%), желтое масло
1Н-ЯМР (ДМСО-d6): 1.44 (4H, m); 1.68 (2H; m); 1.88 (2H, m); 2.00 (1H, m); 2.05 (1H, m); 2.20 (6H, s); 2.41 (2H, m); 3.22 (3H, s); 3.28 (2H, m).
13С-ЯМР (ДМСО-d6): 24.01; 26.34; 30.88; 36.15; 37.06; 55.26: 57.70; 72.55; 210.39.
Структурное звено Е-5:
(8-циклогексил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин (С-5)
К раствору аминонитрила В-1 (10.5 г, 50 ммоль) в абс. ТГФ (150 мл) под аргоном и охлаждении льдом в течение 15 мин прикапывали раствор 2М циклогексил-хлорид магния в простом эфире (62.5 мл, 125 ммоль) при 5-10°С и затем перемешивали при комнатной температуре в течение ночи. Для переработки реакционной смеси добавляли при охлаждении льдом 20%-ный раствор хлорида аммония (50 мл) и воду (50 мл) и экстрагировали простым эфиром (3×100 мл). Органическую фазу промывали водой и насыщенным раствором хлорида натрия, высушивали над сульфатом натрия и концентрировали в вакууме. Оставшийся осадок очищали флеш-хроматографией с CHCl3/МеОН (20:1).
Выход: 1.18 г (9%), бесцветное масло
1Н-ЯМР (ДМСО-d6): 1.05 (6H, m); 1.43 (5H; m); 1.61 (8H, m), 2.35 (6H, s); 3.86 (4H, s).
4-циклогексил-4-диметиламино-циклогексанон (Е-5)
(8-циклогексил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин (С-5) (1.18 г, 4.41 ммоль) смешивали с 6N соляной кислотой (7 мл) и перемешивали при комнатной температуре в течение ночи. После оконченного гидролиза реакционную смесь экстрагировали простым эфиром (2×10 мл), водный раствор при охлаждении льдом подщелачивали посредством 5N раствора едкого натра, реакционную смесь экстрагировали дихлорметаном (3×20 мл), органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме. Сырой продукт (637 мг) очищали флеш-хроматографией с CHCl3/МеОН (9:1).
Выход: 366 мг (37%), бесцветное масло
1Н-ЯМР (ДМСО-d6): 1.08 (5H, m); 1.68 (8H; m); 1.99 (4H, m); 2.29 (2H, s), 2.41 (6H, s).
Структурное звено Е-6:
(8-циклопентил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин (С-6)
К раствору аминонитрила В-1 (10.5 г, 50 ммоль) в абс. ТГФ (150 мл) под аргоном и охлаждении льдом в течение 15 мин прикапывали 2М раствор циклопентил-бромид магния в простом эфире (62.5 мл, 125 ммоль) при 5-10°С и затем 72 ч. перемешивали при комнатной температуре. Для переработки реакционной смеси при охлаждении льдом добавляли 20%-ный раствор хлорида аммония (50 мл) и воду (50 мл) и экстрагировали простым эфиром (3×100 мл). Органическую фазу промывали водой и насыщенным раствором хлорида натрия, высушивали над сульфатом натрия и концентрировали в вакууме. Оставшийся осадок отделяли флеш-хроматографией с CHCl3/МеОН (40⇒20:1). Так как желаемый продукт еще не был чистым, то применяли последующую колоночную хроматографию с CHCl3/МеОН (40:1).
Выход: 692 мг (5%), бесцветное масло
1Н-ЯМР (ДМСО-d6): 1.23 (2H, m); 1.46 (9 Н; m); 1.69 (4H, m); 2.04 (1H, m); 2.23 (6H, s); 3.86 (4H, s).
4-циклопентил-4-диметиламино-циклогексанон (Е-6)
Кеталь С-6 (0.68 г, 2.68 ммоль) смешивали с 6N соляной кислотой (5 мл) и при комнатной температуре в течение ночи перемешивали. После оконченного гидролиза реакционную смесь экстрагировали простым эфиром (2×20 мл), водный раствор при охлаждении льдом подщелачивали с помощью 5N раствора едкого натра, экстрагировали дихлорметаном (3×10 мл), органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме.
Выход: 424 мг (76%), бесцветное масло
1Н-ЯМР (ДМСО-d6): 1.28 (2H, m); 1.54 (8H; m); 1.99 (4H, m); 2.14 (1H, m); 2.29 (6H, s).
13С-ЯМР (ДМСО-d6): 24.58; 28.13; 29.24; 36.07; 37.79; 42.97; 57.07; 210.67.
Структурное звено Е-7:
(8-бутил-1,4-диоксаспиро[4.5]дец-8-ил)этилметиламин (С-7)
К 2 М раствору хлорида бутилмагния в тетрагидрофуране (20 мл, 40 ммоль) при 0°С под аргоном прикапывали раствор В-2 (3.50 г, 15.6 ммоль) в тетрагидрофуране (50 мл) и смесь перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь при охлаждении льдом осторожно смешивали с насыщенным раствором хлорида аммония (60 мл), значение pH подправляли с помощью раствора едкого натра до 10 и экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт без очистки подвергали дальнейшему взаимодействию.
4-бутил-4-(этилметиламино)циклогексанон (Е-7)
Раствор С-7 (4.43 г, 17.3 ммоль) в ацетоне (15 мл) сначала смешивали с водой (2.5 мл) и потом с концентрированной соляной кислотой (2.5 мл) и в течение выходных перемешивали при комнатной температуре. Затем реакционную смесь подщелачивали с помощью 2 М раствора карбоната калия (pH 10), экстрагировали диэтиловым эфиром (3×40 мл), объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали с помощью флеш-хроматографии (200 г, 20×5.7 см) с циклогексан / этилацетат (2:1).
Выход: 2.08 г (57%), желтое масло
1Н-ЯМР (ДМСО-d6): 0.87 (t, 3H, J=7.0 Hz); 1.00 (t, 3H, J=7.0 Hz); 1.20-1.29 (m, 4H); 1.38-1.42 (m, 2H); 1.63-1.71 (m, 2H); 1.92-2.00 (m, 4H); 2.20 (s, 3H); 2.36-2.47 (m, 4H).
Структурное звено E-8
Бензилметил-[8-(4Н-[1,2,3]триазин-1-ил)-1,4-диоксаспиро[4.5]дец-8-ил]амин
Раствор 1,4-диоксаспиро[4,5]декан-8-она (3.9 г, 25 ммоль), N-бензилметиламин (3.32 г, 3.54 мл, 27.5 ммоль) и 1,2,3 триазол (2.07 г, 30 ммоль) в толуоле (40 мл) 8 ч нагревали в водоотделителе (Dean-Stark) с обратным холодильником. Реакционный раствор после охлаждения до комнатной температуры использовали непосредственно в дальнейшем.
Бензил-(8-бутил-1,4-диоксаспиро[4.5]дец-8-ил)метиламин (D-8)
К 2 М раствору хлорида n-бутилмагния в тетрагидрофуране (50 мл, 100 ммоль) при 0°С под потоком аргона прикапывали реакционный раствор бензилметил-[8-(4H-[1,2,3]триазин-1-ил)-1,4-диоксаспиро[4.5]дец-8-ил]амина (20 мл, прибл. 25 ммоль). Смесь нагревали до комнатной температуры и 2 ч перемешивали и затем выливали в насыщенный раствор хлорида аммония (60 мл). Фазы отделяли, водную фазу экстрагировали диэтиловым эфиром (3×30 мл), объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт ресуспендировали в дихлорметане, нерастворимые компоненты отфильтровывали, фильтрат снова концентрировали в вакууме и осадок (6.31 г) очищали путем флеш-хроматографии (400 г, 20×7.6 см) с циклогексан / этилацетатом (9:1).
Выход: 3.4 г (43% за две стадии), бесцветное масло
1Н-ЯМР (ДМСО-d6): 0.90 (t, 3H, J=6.8 Hz); 1.18-1.33 (m, 4Н); 1.36-1.47 (m, 4Н); 1.51-1.59 (m, 2Н); 1.70-1.93 (m, 4Н); 2.03 (3H, s); 3.57 (s, 2Н); 3.85 (s, 4Н); 7.15-7.25 (m, 1Н); 7.27-7.36 (m, 4H).
4-(бензилметиламино)-4-бутилциклогексанон (Е-8)
Раствор D-8 (3.40 г, 10.7 ммоль) в ацетоне (70 мл) смешивали с водой (10 мл) и 37% соляной кислотой (14.1 мл) и 5.5 ч перемешивали при комнатной температуре. Затем к смеси медленно прикапывали насыщенный раствор карбоната калия, пока не достигли pH 10. Смесь экстрагировали диэтиловым эфиром (4×40 мл), объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме.
Выход: 2.3 г (74%), желтоватое масло
1Н-ЯМР (ДМСО-d6): 0.91 (t, 3H, J=6.74Hz); 1.20-1.37 (m, 5Н); 1.48-1.59 (m, 2Н); 1.78 (dt, 2Н, J=13.7 и 5.5 Hz); 2.00-2.17 (m, 4H); 2.09 (s, 3H); 2.50-2.60 (m, 1H); 3.66 (s, 2H); 7.12-7.26 (m, 1H); 7.26-7.38 (m, 4H).
Структурное звено Е-9:
1,4-диоксаспиро[4.5]дец-8-илиден)-(4-метоксибензил)амин
К раствору 1,4-диоксаспиро[4.5]декан-8-она (10.0 г, 64 ммоль) в дихлорметане (100 мл) добавляли молекулярное сито 4 Ǻ (20 г) и 4-метоксибензиламин (11.8 г, 83 ммоль). Суспензию перемешивали 16 ч при комнатной температуре, потом фильтровали и фильтрат без дальнейшей переработки использовали в дальнейшей стадии.
1Н-ЯМР (300 MHz, CDCl3): 1.81 (t, J=6.3 Hz, 2H); 1.89 (t, J=6.3 Hz, 2H); 2.50 (t, J=6.3 Hz, 4H); 3.74 (s, 3H); 3.95 (s, 4H); 4.45 (s, 2H); 6.83 (d, J=8.6 Hz, 2H); 7.18 (d, J=8.6 Hz, 2H).
13С-ЯМР (100 MHz, CDCl3): 25.0; 34.0; 34.8; 36.2; 53.8; 55.1; 64.3; 108.3; 113.7; 128.0; 128.6; 158.2; 171.2.
(8-аллил-1,4-диоксаспиро[4.5]дец-8-ил)-(4-метоксибензил)амин (C-9)
К раствору 1,4-диоксаспиро[4.5]дец-8-илиден)-(4-метоксибензил)амина (17.6 г, 64 ммоль) в дихлорметане (120 мл) прикапывали 1 М раствор бромид аллилмагния (100 мл, 100 ммоль) в диэтиловый эфир и реакционную смесь 4 ч перемешивали при комнатной температуре. Затем смесь при охлаждении льдом выливали в насыщенный раствор хлорида аммония (100 мл) и экстрагировали дихлорметаном (3×40 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Осадок очищали путем флеш-хроматографии (400 г, 20×7.6 см) с хлороформ / метанолом (10:0.1).
Выход: 10.8 г (53%), коричневое масло
1Н-ЯМР (300 MHz, d6-ДМСО): 1.30 (br s, 1Н); 1.42 (t, J=11.5 Hz, 4H); 1.51-1.64 (m, 2H); 1.72-1.86 (m, 2H); 2.18 (d, J=7.3 Hz, 2H); 3.51 (s, 2H); 3.72 (s, 3H), 3.83 (s, 4H); 4.99-5.16 (m, 2H); 5.76-5.93 (m, 1H); 6.82-6.89 (m, 2H); 7.24 (m, 2H).
13С-ЯМР (100 MHz, d6-ДМСО): 29.9; 32.1; 41.8; 44.3; 52.6; 54.9; 63.4; 108.3; 113.4; 117.2; 128.9; 133.6; 134.8; 157.9.
4-аллил-4-(4-метоксибензиламино)циклогексанон (E-9)
К раствору (8-аллил-1,4-диоксаспиро[4.5]дец-8-ил)-(4-метоксибензил)амина (С-9) (1.0 г, 3.15 ммоль) в ацетоне (10 мл) и воде (0.5 L) добавляли концентрированную соляную кислоту (0.5 мл) и смесь перемешивали 16 ч при комнатной температуре. Затем раствор смешивали с насыщенным раствором гидрокарбоната натрия (40 мл) и экстрагировали дихлорметаном (3×40 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме.
Выход: 864 мг (100%), коричневое масло
1Н-ЯМР (400 MHz, d6-ДМСО): 1.64 (dt, J=13.2, 4.6 Hz, 2H); 1.89 (d, J=13.0 Hz, 2H); 2.04 (d, J=14.9 Hz, 2H); 2.30 (d, J=7.2 Hz, 2H); 2.45-2.63 (m, 2H); 3.61 (s, 2H); 3.72 (s, 3H); 5.12 (dd, J=13.1, 11.2 Hz, 2H); 5.90 (dt, J=17.1, 7.3 Hz, 1H); 6.86 (d, J=8.3 Hz, 2H); 7.28 (d, J=8.2 Hz, 2H). NH-сигнал было не возможно идентифицировать.
13С-ЯМР (100 MHz, d6-ДМСО): -3.1; -0.9; 4.4; 7.4; 15.7; 17.9; 76.4; 80.5; 92.1; 96.4; 97.5; 120.9; 174.1.
Структурное звено Е-10:
фенил-(1,4-диоксаспиро[4.5]дец-8-илиден)амин
По аналогии с синтезом кетона Е-13 синтезировали соответствующий N-фенил-замещенный кетон Е-10. По аналогии с синтезом бензил-(1,4-диоксаспиро[4.5]дец-8-илиден)амина (сравн. структурное звено Е-13) 1,4-диоксаспиро[4.5]декан-8-он количественно при отщеплении воды подвергали взаимодействию с анилином до получения имина фенил-(1,4-диоксаспиро[4.5]дец-8-илиден)амин.
(8-аллил-1,4-диоксаспиро[4.5]дец-8-ил)-фенил-амин (С-10)
При последующей реакции фенил-(1,4-диоксаспиро[4.5]дец-8-илиден)амина с бромидом аллилмагния (по аналогии с С-13) можно было выделить желаемый (8-аллил-1,4-диоксаспиро[4.5]дец-8-ил)-фенил-амин (С-10) с хорошим выходом.
4-аллил-4-фениламиноциклогексанон (Е-10)
К раствору С-10 (333 мг, 1.22 ммоль) в ацетоне (10 мл) и воде (0.5 мл) добавляли концентрированную соляную кислоту (0.5 мл) и смесь 2 дн. перемешивали при комнатной температуре. Реакционную смесь затем смешивали с насыщенным раствором гидрокарбоната натрия (40 мл) и экстрагировали дихлорметаном (3×40 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме.
Выход: 285 мг (100%), бесцветные кристаллы
Точка плавления: 76-78°С
1Н-ЯМР (400 MHz, d6-ДМСО): 1.78 (dt, J=13.0, 4.6Hz, 2H); 2.06-2.29 (m, 4H); 2.49 (m, 4H); 5.00 (dd, J=10.1, 1.9 Hz, 2H); 5.30 (s, 1H); 5.65-5.87 (m, 1H); 6.58 (t, J=7.2 Hz, 1H); 6.82 (dd, J=8.5 Hz, 2H); 7.07 (m, 2H).
13С-ЯМР (100 MHz, d6-ДМСО): 34.5; 36.3; 41.0; 53.8; 115.3; 116.4; 117.5; 128.7; 134.3; 147.2; 210.4.
Структурное звено E-11:
Вариант 1:
(1,4-диоксаспиро[4.5]дец-8-илиден)фенилимин
Раствор 1,4-диоксаспиро[4.5]дека-8-она (5.46 г, 35 ммоль) и анилина (3.35 г, 3.28 мл, 36 ммоль) в толуоле (100 мл) нагревали 15 ч в водоотделителе, который дополнительно был загружен безводным сульфатом натрия (2 г). Для контроля взаимодействия брали пробу, концентрировали в вакууме и немедленно измерили 1Н-ЯМР-спектр в ДМСО.
После полного взаимодействия реакционный раствор концентрировали в вакууме и осадок растворяли в безводном тетрагидрофуране.
1Н-ЯМР (ДМСО-d6): 1.70 (t, 2Н, J=6.7 Hz); 1.86-1.94 (m, 2Н); 2.21 (t, 2Н, J=6.8 Hz); 2.35 (t, 2H, J=7.0 Hz); 3.91-3.94 (m, 4H); 6.67-6.71 (m, 2H); 6.96-7.04 (m, 1H); 7.24-7.31 (m, 2H).
Вариант 2:
(1,4-диоксаспиро[4.5]дец-8-илиден)фенилимин
Раствор 1,4-диоксаспиро[4.5]декан-8-она (6.20 г, 39.6 ммоль) в дихлорметане (65 мл) смешивали с молекулярным ситом 4 Ǻ (12.5 г) и анилином (3.80 г, 3.73 мл, 40.8 ммоль) и перемешивали в течение выходных при комнатной температуре. Для контроля взаимодействия брали пробу, концентрировали в вакууме и немедленно 1Н-ЯМР-спектр ресуспендировали в CDCl3. После полного взаимодействия реакционную смесь фильтровали и фильтрат концентрировали в вакууме.
1Н-ЯМР (CDCl3): 1.76 (t, 2Н, J=6.6Hz); 1.93-2.05 (m, 2Н); 2.35 (t, 2Н, J=6.7 Hz); 2.64 (t, 2H, J=6.6Hz); 3.96-4.02 (m, 4H); 6.71 (d, 2H, J=7.8 Hz); 7.05 (t, 1H, J=7.2 Hz); 7.29 (t, 2H, J=7.9 Hz).
Вариант 1:
(8-бутил-1,4-диоксаспиро[4.5]дец-8-ил)фениламин (C-11) и 4-бутил-4-фениламиноциклогексанон (Е-11)
К 1.6 М раствор n-бутиллития в n-гексане (27 мл, 42 ммоль) при 0°С под аргоном прикапывали раствор (1,4-диоксаспиро[4.5]дец-8-илиден)фенилимина (17 ммоль) в безводном тетрагидрофуране. Затем реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи. После этого реакционную смесь при охлаждении льдом смешивали с водой (40 мл) и экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические фазы концентрировали в вакууме и осадок очищали с помощью флеш-хроматографии (100 г, 20×4.0 см) с циклогексан / этилацетатом (9:1) и 1% триэтиламином.
С-11:
Выход: 645 мг (13%), коричневое масло
1Н-ЯМР (ДМСО-d6): 0.78 (t, 3H, J=6.8 Hz); 1.17-1.22 (m, 4Н); 1.42-1.71 (m, 8Н); 1.94-2.03 (m, 2Н); 3.83 (s, 4Н); 4.93 (s, 1Н); 6.49 (t, 1Н, J=7.3 Hz); 6.71 (d, 2H, J=8.0 Hz); 7.01 (t, 2H, J=7.8 Hz).
E-11:
Выход: 1.01 г (24%), коричневое масло
1Н-ЯМР (ДМСО-d6): 0.78 (t, 3H, J=7.0 Hz); 1.12-1.30 (m, 4H); 1.57-1.87 (m, 4H); 2.04-2.15 (m, 2H); 2.19-2.31 (m, 2H); 2.40-2.60 (m, 2H, наложенный от ДМСО-сигнала); 5.25 (s, 1H); 6.55 (t, 1H, J=7.2 Hz); 6.77 (d, 2H, J=8.6 Hz); 7.00-7.09 (m, 2H).
Сверх этого была еще получена смесь С-11 и Е-11 (816 мг, прибл. 20%).
Вариант 2:
(8-бутил-1,4-диоксаспиро[4.5]дец-8-ил)фениламин (С-11)
К 1.6 М раствору n-бутиллития в n-гексане (63 мл, 98 ммоль) при 0°С под аргоном прикапывали раствор (1,4-диоксаспиро[4.5]дец-8-илиден)фенилимина (39.6 ммоль) в безводном тетрагидрофуране. Затем реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи. После этого реакционную смесь при охлаждении льдом смешивали с водой (40 мл) и экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические фазы высушивали сульфатом натрия, концентрировали в вакууме и осадок очищали с помощью флеш-хроматографии (100 г, 20×4.0 см) с циклогексан / этилацетат (9:1) и 1% триэтиламин.
Выход: 2.83 г (25%), коричневое масло 4-бутил-4-фениламиноциклогексанон (Е-11)
Раствор С-11 (645 мг, 2.23 ммоль) в ацетоне (15 мл) смешивали с водой (2.5 мл) и концентрированной соляной кислотой (2.5 мл) и в течение выходных перемешивали при комнатной температуре. Затем реакционную смесь подщелачивали раствором карбоната калия (pH 10), экстрагировали диэтиловым эфиром (3×30 мл), объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме.
Выход: 547 мг (100%); коричневое масло
1Н-ЯМР (ДМСО-d6): 0.78 (t, 3H, J=7.0 Hz); 1.16-1.26 (m, 4H); 1.65-1.78 (m, 4H); 2.04-2.13 (m, 2H); 2.21-2.29 (m, 2H); 2.42-2.58 (2H, наложенный от ДМСО-сигнала); 5.25 (s, 1Н); 6.55 (t, 1Н, J=7.2 Hz); 6.77 (d, 2H, J=7.7 Hz); 7.03 (d, 2H, J=7.3 Hz); 7.07 (d, 2H, J=7.3 Hz).
Структурное звено E-12:
4-(8-бутил-1,4-диоксаспиро[4.5]дец-8-ил)морфолин (С-12)
В прокаленной колбе подогревали раствор морфолина (4.79 г, 4.8 мл, 55 ммоль), 1,4-диоксаспиро[4.5]дец-8-она (7.8 г, 50 ммоль) и 1,2,3-триазола (4.14 г, 60 ммоль) в толуоле (50 мл) 7 ч при кипячении с обратным холодильником в водоотделителе. Затем под аргоном к охлажденному до 0°С раствору прикапывали 2 М раствор хлорида n-бутилмагния в тетрагидрофуран (100 мл, 200 ммоль) таким образом, что внутренняя температура оставалась ниже 30°С. Реакционную смесь 2 ч перемешивали при комнатной температуре и после этого при охлаждении ледяной водой до 20% прикапывали раствор хлорида аммония (120 мл). Органическую фазу отделяли и водную фазу экстрагировали этилацетатом (3×100 мл). Объединенные органические фазы промывали с помощью 2 N раствора натрового щелока (100 мл) и воды (100 мл), высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт (7.67 г) очищали путем флеш-хроматографии (400 г, 20×7.5 см) с этилацетат / циклогексаном (1:2).
Выход: 3.86 г (27%), бесцветное масло
1Н-ЯМР (CDCl3): 0.88 (t, J=6.9 Hz, 3H); 1.14-1.73 (m, 12H); 1.88 (dt, J=12.6, 3.4Hz, 2H); 2.44-2.61 (m, 4H); 3.56-3.73 (m, 4H); 3.93 (m, 4H).
4-бутил-4-морфолин-4-илциклогексанон (E-12)
К раствору С-12 (3.40 г, 12 ммоль) в ацетоне (20 мл) добавляли 6 М соляную кислоту (5 мл). Через 24 ч реакционный раствор смешивали с дополнительной 6 М соляной кислотой (2.5 мл), перемешивали последующие 20 ч. при комнатной температуре, затем подщелачивали раствором 25% карбоната калия (pH~10) и экстрагировали диэтиловым эфиром (3×25 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт (2.7 г) очищали путем флеш-хроматографии (200 г, 20×5.6 см) с этилацетат / циклогексаном (1:4).
Выход: 2.18 г (76%), бесцветное масло
1Н-ЯМР (CDCl3): 0.90 (t, 3H, J=7.0 Hz); 1.09- 2.23 (m, 12H); 2.55 (dd, 2H, J=14.3, 5.8 Hz); 2.59-2.65 (m, 4H); 3.67-3.73 (m, 4H).
Структурное звено E-13:
бензил-(1,4-диоксаспиро[4.5]дец-8-илиден)амин
К раствору 1,4-диоксаспиро[4.5]декан-8-она (10.0 г, 64 ммоль) в дихлорметане (100 мл) добавляли молекулярное сито 4 Å (20 г) и бензиламин (8.90 г, 83 ммоль) и реакционную смесь перемешивали 16 ч при комнатной температуре. Затем суспензию фильтровали и фильтрат концентрировали в вакууме.
Выход: 15.6 г (99%), желтоватое масло
1Н-ЯМР (300 MHz, CDCl3): 1.83 (t, J=6.3 Hz, 2H); 1.92 (t, J=6.6Hz, 2H); 2.53 (s, 4H); 3.98 (s, 4H); 4.54 (s, 2H); 7.25 (m, 5H).
13С-ЯМР (100 MHz, CDCl3): 25.6; 34.2; 34.9; 36.3; 54.6; 64.4; 108.0; 126.6; 127.9; 128.4; 140.2; 171.7.
(8-аллил-1,4-диоксаспиро[4.5]дец-8-ил)-бензил-амин (C-13)
К раствору бензил-(1,4-диоксаспиро[4.5]дец-8-илиден)амина (15.6 г, 63.7 ммоль) в дихлорметане (120 мл) прикапывали 1 М раствор бромида аллилмагния (127 мл, 127 ммоль) и реакционную смесь перемешивали 72 ч. при комнатной температуре. Затем смесь при охлаждении льдом осторожно выливали в насыщенный раствор хлорида аммония (100 мл) и экстрагировали дихлорметаном (3×40 мл). Объединенные органические фазы высушивали сульфатом натрия, концентрировали в вакууме и осадок очищали путем флеш-хроматографии (400 г, 20×7.6 см) с хлороформ / метанолом (10: 0.2).
Выход: 5.92 г (32%), коричневое масло
1Н-ЯМР (300 MHz, d6-ДМСО): 1.25-1.52 (m, 4H); 1.53-1.66 (m, 2H); 1.74-1.87 (m, 2H); 2.20 (d, J=7.4 Hz, 2H); 3.59 (s, 2H); 3.83 (s, 4H); 4.89-5.19 (m, 2H); 5.86 (tdd, J=14.9, 10.4, 7.3 Hz, 1H); 7.20 (t, J=7.0 Hz, 1H); 7.20-7.35 (m, 4H).
13С-ЯМР (100 MHz, d6-ДМСО): 29.9; 32.0; 41.9; 44.9; 52.7; 63.4; 63.4; 108.3; 117.2; 126.3; 127.8; 127.9; 134.8; 141.8.
4-аллил-4-бензиламиноциклогексанон (E-13)
К раствору C-13 (500 мг, 1.74 ммоль) в ацетоне (20 мл) и воде (2 мл) добавляли концентрированную соляную кислоту (2 мл) и смесь 16 ч перемешивали при комнатной температуре. Затем реакционную смесь смешивали с раствором гидрокарбоната натрия (40 мл) и экстрагировали этилацетатом (3×40 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме.
Выход: 423 мг (100%), коричневое масло
1Н-ЯМР (300 MHz, d6-ДМСО): 1.64 (dt, J=13.2, 4.9 Hz, 2H); 1.75-1.97 (m, 2H); 2.04 (dd, J=14.8, 3.4Hz, 2H); 2.31 (d, J=7.3 Hz, 2H); 2.46-2.65 (m, 2H); 3.68 (s, 2H); 5.03-5.20 (m, 2H); 5.81-6.00 (m, 1H); 7.14-7.26 (m, 1H); 7.26-7.35 (m, 2H); 7.39 (m, 2H). NH-сигнал невозможно было определить.
13С-ЯМР (100.4 MHz, d6-ДМСО): 33.8; 36.4; 41.4; 45.0; 52.8; 117.2; 117.5; 126.4; 127.9; 134.5; 141.5; 211.1.
Структурное звено Е-14:
1-(8-пирролидин-1-ил-1,4-диоксаспиро[4.5]дец-8-ил)-1 Н-[1,2,3]триазол
К раствору 1,4 диоксаспиро[4,5]декан-8-она (3.9 г, 25 ммоль) в толуоле (40 мл) добавляли пирролидин (1.95 г, 2.29 мл, 27.5 ммоль), 1,2,3-триазол (2.07 г, 30 ммоль) и молекулярное сито 4 Å (7.14 г). Смесь перемешивали 7 ч при 90°С. Затем раствор декантировали и немедленно подвергали дальнейшему взаимодействию.
1-(8-бутил-1,4-диоксаспиро[4.5]дец-8-ил)пирролидин (С-14)
К 2 М раствору хлорида n-бутилмагния (25 мл, 50 ммоль) в тетрагидрофуране при охлаждении льдом и аргоном прикапывали реакционный раствор только что полученного триазолового производного (прибл. 6.9 г, 25 ммоль) в толуоле (38 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре и затем выливали в насыщенный раствор хлорида аммония (60 мл). Фазы отделяли и водную экстрагировали диэтиловым эфиром (3×70 мл). Объединенные органические фазы высушивали сульфатом натрия, концентрировали в вакууме и осадок (12 г) очищали путем флеш-хроматографии (400 г, 20×7.6 см) с этилацетат / метанолом (9:1).
Выход: 2.70 г (40% за две стадии), коричневое масло (С-14)
1Н-ЯМР (ДМСО-d6): 0.87 (t, 3H, J=7.1 Hz); 1.12-1-29 (m, 4Н); 1.30-1.45 (m, 4Н); 1.46-1.60 (m, 4Н); 1.61-1.75 (m, 6Н); 1.93 (t, 1Н, J=7.1 Hz); 2.36 (t, 1H, J=7.0 Hz), 2.58 (br s, 2H), 3.83 (s, 4H).
4-бутил-4-пирролидин-1-ил-циклогексанон (E-14)
Раствор C-14 (2.70 г, 10.1 ммоль) в ацетоне (100 мл) смешивали с водой (10.0 мл) и 37% соляной кислотой (14.0 мл) и перемешивали в течение ночи при комнатной температуре. Затем к смеси медленно прикапывали 4 М раствор едкого натра, пока не достигло pH 10. Смесь экстрагировали диэтиловым эфиром (4×40 мл), объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт (2.6 г) очищали путем флеш-хроматографии (260 г, 30×5.6 см) с этилацетат / метанолом (9:1).
Выход: 1.06 г (47%), коричневое масло (Е-14)
1Н-ЯМР (ДМСО-d6): 0.88 (t, 3H, J=6.7 Hz); 1.14-1.34 (m, 4Н); 1.40-1.50 (m, 2Н); 1.62-1.88 (m, 8Н); 2.04 (dt, 2Н, J=15.0, 3.9 Hz); 2.42 (ddd, 2H, J=6.3, 11.8, 15.5 Hz); 2.63 (t, 4H, J=6.0 Hz).
Структурное звено E-15:
4-(8-[1,2,3]триазол-1-ил-1,4-диоксаспиро[4.5]дец-8-ил)пиперидин
В прокаленной колбе смешивали раствор пиперидина (1.87 г, 2.17 мл, 22 ммоль), 1,4-диоксаспиро[4.5]дец-8-он (3.12 г, 20 ммоль) и 1,2,3-триазол (1.66 г, 24 ммоль) в толуоле (20 мл) с молекулярным ситом 4 Å и перемешивали 7 ч при 104°С при кипячении с обратным холодильником. Затем этот раствор отдекантировали от молекулярного сита. Молекулярное сито промывали толуолом и отфильтровывали. Объединенные жидкие фазы подвергали дальнейшему взаимодействию как 0.6 М раствор.
4-(8-бутил-1,4-диоксаспиро[4.5]дец-8-ил)пиперидин (С-15)
В прокаленной колбе 2 М раствор хлорид н-бутилмагния в тетрагидрофуране (22 мл, 44 ммоль) под аргоном при 0°С по каплям смешивали с 0.6 М раствором только что полученного триазолового производного в толуоле (18 мл, 11 ммоль) в течение 1 ч. Смесь перемешивали 2 ч. при комнатной температуре и после этого при охлаждении ледяной водой до 20% прикапывали раствор хлорида аммония (24 мл). Органическую фазу отделяли и водную фазу экстрагировали диэтиловым эфиром (4×20 мл). Объединенные органические фазы промывали 2 N раствором едкого натра (30 мл) и водой (20 мл), высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт (1.9 г) очищали путем флеш-хроматографии (100 г, 22×4 см) с этилацетат / циклогексаном (1:2).
Выход: 1.03 г (33%), бесцветное масло (С-15)
1Н-ЯМР (ДМСО-d6): 0.86 (t, 3H, J=6.9 Hz); 1.09-1.52 (m, 16Н); 1.60-1.79 (m, 4H); 2.44 (brs,4H), 3.82 (s, 4H).
4-бутил-4-пиперидин-4-илциклогексанон (E-15)
К раствору С-15 (1.0 г, 3.6 ммоль) в ацетоне (15 мл) добавляли 6 М соляную кислоту (5 мл). Реакционный раствор перемешивали 6 дн. при комнатной температуре, затем подщелачивали с помощью 25% раствора карбоната калия (pH~9) и экстрагировали диэтиловым эфиром (3×20 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме.
Выход: 860 мг (100%), бесцветное масло (Е-15)
1Н-ЯМР (ДМСО-d6): 0.87 (t, 3H, J=6.9 Hz); 1.06-1.54 (m, 14Н); 1.54-1.74 (m, 3H); 1.88-2.07 (m, 4H); 2.21-2.46 (m, 3H).
Структурное звено Е-16:
1-метил-4-(8-[1,2,3]триазол-1-ил-1,4-диоксаспиро[4.5]дец-8-ил)пиперазин
В прокаленной колбе нагревали раствор N-метилпиперазина (2.60 г, 2.88 мл, 26 ммоль), 1,4-диоксаспиро[4.5]декан-8-он (3.90 г, 25 ммоль) и 1,2,3-триазол (1.87 г.27 ммоль) в толуоле (25 мл) 6 ч в водоотделителе с обратным холодильником.
Затем реакционный раствор переводили в запираемый измерительный цилиндр и сырой продукт подвергали дальнейшему взаимодействию.
1-(8-бутил-1,4-диоксаспиро[4.5]дец-8-ил)-4-метилпиперазин (С-16)
К раствору только что полученного триазолового производного (12.5 ммоль) в толуоле (12 мл) под аргоном прикапывали раствор 2 М хлорида н-бутилмагния в тетрагидрофуране (15 мл, 30 ммоль) так, что внутренняя температура оставалась ниже 24°С. После оконченного добавления реакционную смесь перемешивали 2 ч при комнатной температуре и потом охлаждали до 0°С и прикапывали к 20% раствору хлорида аммония (50 мл), водную фазу экстрагировали диэтиловым эфиром (3×40 мл), объединенные органические фазы промывали 2 N раствором едкого натра (70 мл) и водой (70 мл), высушивали сульфатом натрия и концентрировали в вакууме.
Сырой продукт С-16 (3.57 г) подвергали дальнейшему взаимодействию.
4-бутил-4-(4-метилпиперазин-1-ил)циклогексанон (Е-16)
Раствор С-16 (3.57 г, 12.0 ммоль) в ацетоне (15 мл) сначала смешивали с водой (2.5 мл) и потом с концентрированной соляной кислотой (2.5 мл) и в течение выходных перемешивали при комнатной температуре. Затем реакционную смесь подщелачивали с помощью 2 М раствора карбоната калия (pH 10), экстрагировали диэтиловым эфиром (3×40 мл), объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали с помощью флеш-хроматографии (200 г, 20×5.7 см) с метанолом.
Выход: 2.04 г (67%), желтое масло (Е-16)
1Н-ЯМР(ДМСО-d6): 0.87 (t, 3H, J=7.0 Hz); 1.16-1.28 (m, 4H); 1.37-1.43 (m, 2H); 1.66 (dt, 2H, J=13.5, 4.5 Hz); 1.90-2.02 (m, 4H); 2.15 (s, 3H); 2.28-2.43 (m, 6H); 2.53-2.57 (m, 4H).
13С-ЯМР: 13.9; 23.2; 26.3; 31.0 (2C); 31.9; 36.1 (2C); 44.0 (2C); 45.7; 55.5; 55.8; 210.4.
Структурное звено E-17:
8-(циклопентилметил)-N,N-диметил-1,4-диоксаспиро[4.5]декан-8-амин
К смеси магния (3.64 г, 150 ммоль) в абс. простом эфире (30 мл) под аргоном добавляли по каплям раствор йодметилциклопентана (31.5 г, 150 ммоль) в абс. простом эфире (150 мл) так, что простой эфир слегка кипел. Затем реакционный раствор 30 мин кипятили с обратным холодильником, охлаждали до КТ и по каплям смешивали с раствором 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрила В-1 (10.5 г, 50 ммоль) в абс. ТГФ (100 мл). Реакционный раствор начинал вскипать, и белое твердое вещество выпадало в осадок. Кипятили 6 ч с обратным холодильником и затем перемешивали при КТ в течение ночи. Для переработки реакционной смеси при охлаждении льдом добавляли 20%-ный раствор NH4Cl (200 мл) и экстрагировали простым эфиром (3×100 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вакууме. Оставшийся осадок очищали флеш-хроматографией с ЕЕ/EtOH (20:1).
Выход: 13.4 г (100%)
1Н-ЯМР (ДМСО-d6): 1.04 (2H, m); 1.37 (4H, m); 1.45-1.78 (17 Н, m); 2.13 (6H, s); 3.62 (4H, s).
4-циклопентилметил-4-диметиламино-циклогексанон (Е-17)
(8-циклопентилметил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин (13.4 г, 50 ммоль) при комнатной температуре смешивали с 5%-ной серной кислотой (600 мл) и 3 дн. перемешивали при КТ. Реакционный состав экстрагировали простым эфиром (2×50 мл). Затем водную фазу при охлаждении ледяной ванной подщелачивали посредством 5N NaOH и экстрагировали дихлорметаном (3×50 мл). Органическую фазу высушивали над Na2SO4 и в вакууме концентрировали до сухости.
Выход: 8.46 г (76%), бесцветные кристаллы.
1Н-ЯМР (ДМСО-d6): 1.04 (2H, m); 1.48 (6H, m); 1.83 (5H, m); 1.93 (4H, m); 2.20 (6H, s); 2.44 (2H, m).
13С-ЯМР (ДМСО-d6): 24.7; 31.7; 34.6; 35.4; 36.1; 36.2; 36.9; 55.9; 210.4.
Индольные структурные звенья - F&Н
Структурное звено F-1:
Триптофол (F-1) (CAS.: 526-55-6), коммерчески доступный
Структурное звено F-2:
Сложный этиловый эфир (5-фтор-3-гидрокси-2-охо-2,3-дигидро-1Н-индол-3-ил)уксусной кислоты1
(S.J.Garden, R.В. da Silva, А.С.Pinto, Tetrahedron 2002, 58, 8399-8412 (особенно стр.8406).
5-фторизатин (10 ммоль) растворяли в смеси из этанол/пиридин/уксусной кислоты (50 мл, 15:5:2), смешивали с этил-манолатом калия (1,87 г, 11 ммоль) и в течение 14 ч кипятили с обратным холодильником. Протекание реакции контролировали с помощью ТХ (элюент: этилацетат/гексан 1:1). Для переработки смесь растворителей отгоняли в вакууме. Осадок ресуспендировали в этилацетате (50 мл) и встряхивали с водой (50 мл). После разделения фаз водную фазу дважды экстрагировали этилацетатом (по 30 мл). Комбинированные органические фазы промывали с помощью 2N HCl (50 мл), высушивали над Na2SO4 и в вакууме концентрировали до 20 мл. К раствору добавляли гексан пока использовалась кристаллизация желаемого продукта. Для дополнения кристаллизации состав охлаждали 12 ч до 10°С. Твердое вещество отсасывали и высушивали в вакууме. Выход: 89%
2-(5-фторо-1H-индол-3-ил)этанол (F-2)2
2S.J.Garden, R.B. da Silva, S.C.Pinto, Tetrahedron 2002, 58, 8399-8412.
Только что полученный альдольный продукт (10 ммоль) под Ar-атмосферой растворяли в абсолютном ТГФ (20 мл). Затем состав при охлаждении в водяной бане смешивали с ВН3×ТГФ (40 мл, 1 М раствор, 40 ммоль) и перемешивали при комнатной температуре в течение 14 ч. За протеканием реакции наблюдали с помощью ТХ. После окончания реакции реакционный раствор добавляли к смеси из этилацетата (50 мл) и H2O (50 мл). После разделения фаз водную фазу дважды экстрагировали этилацетатом (по 30 мл). Комбинированные органические фазы высушивали над Na2SO4 и концентрировали в вакууме. Осадок фильтровали через силикагель с этилацетатом. Полученный после удаления растворителя продукт (F-2) в большинстве случаев находился в виде исключительно чистого масла и кристаллизовался спонтанно. При необходимости осуществляли очистку колоночной хроматографией на силикагеле. Выход 95%
Структурное звено F-3:
3-(2-гидрокси-этил)-1H-индол (F-3)
LiAlH4 (1.99 г, 52.3 ммоль) помещали в абс. ТГФ (60 мл) под аргоном и (5-гидрокси-1Н-индол-3-ил)-уксусную кислоту (5.00 г, 26.2 ммоль) в абс. ТГФ (100 мл) добавляли в течение 30 мин. Состав кипятили 3 ч с обратным холодильником. Для переработки при охлаждении льдом к составу добавляли ТГФ (10 мл) и H2O (4 мл) и 30 мин. дополнительно перемешивали. Состав фильтровали через целит, подвергали последующей промывке дихлорметаном (150 мл) и фильтрат концентрировали в вакууме.
Выход: 2.17 г (47%)
1Н-ЯМР (ДМСО-d6): 2.74 (2H, m); 3.60 (3H, m); 6.58 (1H, m); 6.78 (1H, s); 6.99 (1H, s); 7.08 (1H, d); 10.5 (1H, bs).
Пример АА-1:
4',9'-дигидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(3'H)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (2:1) (один из двух возможных диастереомеров)
Триптофол F-1 (484 мг, 3.00 ммоль) и кетон Е-1 (507 мг, 3.00 ммоль) растворяли в дихлорметане (25 мл) и смешивали с метансульфокислотой (316 мг, 3.30 ммоль). Реакционный раствор в течение ночи перемешивали при комнатной температуре. К нему снова добавляли метансульфокислоту (316 мг, 3.30 ммоль) и перемешивали последующие 3 ч. Реакционный раствор подщелачивали с помощью 1N NaOH, органическую фазу отделяли и водную фазу трижды экстрагировали дихлорметаном (15 мл). Объединенные органические фазы высушивали над Na2SO4, фильтровали и растворитель удаляли под сниженным давлением. Сырой продукт очищали колоночной хроматографией с CHCl3/EtOH (10:1).
Выход: 672 мг (72%); белое твердое вещество
1Н-ЯМР (ДМСО-d6): 0.85 (3H, t); 1.23-1.75 (8H, br. m); 2.14 (2H, br. m); 2.28 (6H, br. s); 2.01 (6H, s); 2.66 (2H, t); 3.89 (2H, t); 6.93 (1H, t); 7.01 (1H, t); 7.29-7.37 (2H, 2d); 10.80 (br, 1H).
Образование соответствующего цитрата происходило с помощью только что полученного простого спироэфира (0.66 г, 2.11 ммоль) в горячем EtOH (10 мл) и лимонной кислоте (405 мг, 2.11 ммоль), растворенной в горячем EtOH (2 мл). Перемешивали 2 ч при комнатной температуре. Выпавшее в осадок твердое вещество АА-1 отсасывали и высушивали.
Выход: 889 мг (82%), белое твердое вещество (АА-1) Точка плавления: 240-242°С
1Н-ЯМР (ДМСО-d6): 0.89 (3H, t); 1.53 (2H, m); 1.62 (4H, br. t); 1.67 (2H, br. t); 2.12 (2H, br. t); 2.55 (6H, s); 2.57-2.70 (4 H, m); 3.90 (2H, t); 6.97 (1H, t); 7.05 (1H, t); 7.35-7.39 (2H, 2 d); 10.73 (1H, br).
13С-ЯМР (ДМСО-d6): 8.86; 22.15; 23.47; 25.22; 37.17; 44.19; 59.08; 71.23; 72.07; 99.65; 105.28; 111.35; 117.62; 118.39; 120.65; 126.38; 135.61; 139.04; 171.84.
Пример АА-2:
6,-фтор-4,,9,-дигидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (неполярный диастереомер)
4-диметиламино-4-этил-циклогексанон Е-1 (600 мг, 3.55 ммоль) и 5-фтор-триптофол F-2 (852 мг, 3.55 ммоль) помещали под аргоном в абс. CH2Cl2 (15 мл) и затем смешивали с метансульфокислотой (250 µл, 3.89 ммоль). Состав перемешивали 72 ч при комнатной температуре, до щелочной реакции смешивали с 1N NaOH и экстрагировали с помощью CH2Cl2 (3×20 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вакууме. Осадок очищали путем флеш-хроматографии с CHCl3/МеОН (20:1, 4:1, 1:1+1% TEA).
Полученный при этом неполярный продукт циклизации (164 мг, 0.496 ммоль) растворяли в горячем этаноле (5 мл) и добавляли растворенную в горячем этаноле лимонную кислоту (90 мг, 0.496 ммоль). Состав охлаждали до комнатной температуры и выпавший в осадок остаток АА-2 отсасывали и высушивали в вакууме.
Выход: 124 мг (7%) (АА-2)
Точка плавления: 233-236°С
1Н-ЯМР (ДМСО-d6): 0.88 (3H, t); 1.47 (2H, m); 1.53-1.87 (8H, m); 2.05 (2H, t); 2.48 (6H, m); 2.60 (4H, m); 3.91 ((2H, t); 6.83 (1H, m); 7.12 (1H, m); 7.35 (1H, m); 10.74 (1H, s).
Пример АА-3:
2',3',4',9'-тетрагидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (один из двух возможных диастереомеров)
Триптамин Н-1 (528 мг, 3.30 ммоль) и кетон Е-1 (507 мг, 3.30 ммоль) растворяли в метаноле (15 мл) и перемешивали в течение ночи. Метанол удаляли под сниженным давлением, осадок ресуспендировали в дихлорэтане (15 мл) и смешивали с трифторуксусной кислотой (494 мг, 3.30 ммоль). Реакционный раствор перемешивали 72 ч при комнатной температуре, подщелачивали с помощью 1N NaOH, органическую фазу отделяли и водную фазу трижды экстрагировали дихлорметаном (15 мл). Объединенные органические фазы высушивали над NaSO4, фильтровали и растворитель удаляли под сниженным давлением. Сырой продукт очищали колоночной хроматографией с CHCl3/МеОН (1:4).
Выход: 265 мг (26%), белое твердое вещество
1Н-ЯМР (ДМСО-d6): 0.85 (3H, t); 1.32-1.48 (6H, br. m); 1.82 (2H, br. t); 2.11 (2H, br. t); 2.25 (6H, s); 2.53 (2H, t); 2.96 (2H, t); 6.78 (2H, dt); 6.95 (1H, dt); 7.29 (2H, d); 10.49 (br, 1H).
13С-ЯМР (ДМСО-d6): 9.25; 22.87; 23.47; 26.21; 30.66; 38.66; 51.31; 55.49; 106.23; 110.85; 116.94; 117.64; 119.76; 126.77; 135.35; 144.85.
Образование соответствующего цитрата происходило с помощью только что полученного спироамина (0.25 г, 0.80 ммоль) в горячем EtOH (10 мл) и лимонной кислоте (0.15 г, 0.80 ммоль), растворенной в горячем EtOH (1 мл). 2 ч перемешивали при комнатной температуре. Выпавшее в осадок твердое вещество АА-3 отсасывали и высушивали.
Выход: 347 мг (86%), белое твердое вещество
Точка плавления: 228-230°С
1Н-ЯМР (ДМСО-d6): чистые, но очень широкие сигналы, поэтому нет распределения.
13С-ЯМР (ДМСО-d6): 9.01; 23.77; 25.62; 28.83; 37.09; 44.23; 55.47; 71.23; 105.42; 111.22; 117.64; 118.52; 121.22; 125.82; 135.76; 171.01.
Пример АА-4:
4',9,-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (один из двух возможных диастереомеров)
Циклогексанон Е-2 (394 мг, 2 ммоль) и триптофол F-1 (322 мг, 2 ммоль) под аргоном помещали в абс. CH2Cl2 (15 мл). Затем добавляли метансульфокислоту (142 µл, 2.2 ммоль) и 24 ч перемешивали при комнатной температуре.
Для переработки состав смешивали с 1N NaOH и экстрагировали с помощью CH2Cl2 (3×15 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вакууме. Осадок очищали путем флеш-хроматографии с CHCl3/МеОН (9:1) и затем перекристаллизировали из этанола.
Выход: 330 мг (49%)
Анализировали ЯМР-спектры свободного основания, так как спектры цитрата были плохо растворимы.
1Н-ЯМР (ДМСО-d6): 0.91 (3H, t); 1.25 (6H, m); 1.55 (4H, m); 1.73 (2H, m); 2.11 (2H, m); 2.26 (6H, s); 2.66 (2H, t); 3.91 (2H, t); 6.98 (2H, m); 7.32 (2H, m); 10.72 (1H,s).
13С-ЯМР (ДМСО-d6): 14.04; 18.50; 22.18; 23.37; 26.61; 26.99; 29.93; 30.79; 37.24; 55.14; 55.97; 58.75; 71.99; 104.90; 111.21; 117.41; 118.19; 120.33; 126.42; 135.81; 139.74.
Образование соответствующего цитрата происходило с помощью только что полученного простого спироэфира (150 мг, 0.44 ммоль), который растворяли в горячем этаноле (5 мл) и к которому добавляли растворенную в горячем этаноле (1 мл) лимонную кислоту (84 мг, 0.44 ммоль). Затем раствор охлаждали до комнатной температуры и 2 ч перемешивали. Выпавший в осадок белый остаток АА-4 отсасывали и высушивали в вакууме.
Выход: 180 мг (77%) (АА-4) Точка плавления: 210-214°С
Пример АА-5:
6-фтор-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (2:1) (один из двух возможных диастереомеров)
4-бутил-4-диметиламино-циклогексанон Е-2 (394 мг, 2 ммоль) и 5-фтор-триптофол F-2 (482 мг, 2 ммоль) помещали под аргоном в абс. CH2Cl2 (15 мл) и затем смешивали с трифторметансульфокислотой (194 µл, 2.2 ммоль).
Состав перемешивали 72 ч при комнатной температуре. Для переработки раствор смешивали с 1N NaOH и экстрагировали с помощью CH2Cl2 (3×15 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вакууме. Осадок очищали путем флеш-хроматографии с CHCl3/МеОН (9:1 до 1:1). Для дальнейшей очистки продукт перекристаллизировали из этанола.
Выход: 119 мг (16%)
Оценивали ЯМР-спектры свободного основания, так как спектры цитрата были плохо растворены.
1Н-ЯМР (ДМСО-d6): 0.90 (3H, t); 1.19 (6H, m); 1.54 (4H, m); 1.67 (2H, m); 2.12 (2H, m); 2.24 (6H, s); 2.59 (2H, t); 3.88 (2H, t); 6.83 (1H, m); 7.12 (1H, m); 7.28 (1H, m); 10.85 (1H, s).
13С-ЯМР (ДМСО-d6): 14.03; 18.49; 22.10; 23.36; 26.59; 26.93; 29.87; 30.74; 37.22; 55.12; 55.97; 58.69; 72.01; 102.16; 102.39; 105.39; 108.00; 108.26; 111.90; 111.99; 126.55; 126.65; 132.43; 141.93; 155.56; 157.85.
Образование соответствующего цитрата происходило с помощью только что полученного простого спироэфира. Это спиросоединение (119 мг, 0.33 ммоль) растворяли в горячем этаноле (5 мл) и добавляли растворенную в горячем этаноле лимонную кислоту (63 мг, 0.33 ммоль). Состав охлаждали до комнатной температуры, выпавший в осадок белый остаток (АА-5) отсасывали и высушивали в вакууме.
Выход: 106 мг (58%) (АА-5)
Точка плавления: 217-220°С
Пример АА-6:
6'-фтор-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (один из двух возможных диастереомеров)
Пример АА-6 получали аналогично примеру АА-5. Однако при осаждении цитрата вместо полуцитрата выделили цитрат.
Е-2 (4.0 г/ 20.3 ммоль) и фтортриптофол F-2 (4.89 г/ 20.3 ммоль) помещали под аргоном в абс. CH2Cl2 (50 мл). Затем добавляли метансульфокислоту (1.44 мл / 22.33 ммоль) и перемешивали 48 ч. при комнатной температуре. Для переработки состав смешивали с 1N NaOH и 10 мин сильно перемешивали. Фазы отделяли, водную фазу экстрагировали с помощью CH2Cl2 (1×30 мл), при этом в осадок выпадало твердое вещество, которое отсасывали и перекристаллизировали из этанола. Органическую фазу высушивали над Na2SO4 и концентрировали в вакууме. Осадок равным образом перекристаллизировали из этанола. Оба твердых вещества представляли собой целевой продукт.
Выход: 1.9 г (26%)
Только что полученный продукт циклизации (1.0 г, 2.77 ммоль) растворяли в горячем этаноле (5 мл). Добавляли растворенную в горячем этаноле лимонную кислоту (0.528 г, 2.77 ммоль). Состав охлаждали до комнатной температуры, при этом в осадок выпадал белый остаток. Осадок (АА-6) отсасывали и высушивали в вакууме.
Выход: 1.5 г (98%) (АА-6)
Пример АА-7:
6'-гидрокси-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2,2,2-трифторацетат (1:1) (один из двух возможных диастереомеров)
3-(2-гидрокси-этил)-1H-индол F-3 (620 мг, 3.49 ммоль) и кетон Е-2 (680 мг, 3.49 ммоль) помещали в абс. CH2Cl2 (100 мл) под аргоном, при охлаждении льдом смешивали с ТМС-трифлатом (686 µл, 3.55 ммоль) в CH2Cl2 (2 мл) и 30 мин перемешивали при комнатной температуре. Состав перемешивали последующие 16 ч при комнатной температуре. Для переработки добавляли H2O (22 мл) и K2CO3 (490 мг, 3.55 ммоль) и перемешивали при комнатной температуре 20 мин. Фазы отделяли. Водную фазу экстрагировали дихлорметаном (2×20 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вакууме.
Только что полученный продукт циклизации (100 мг, 0.273 ммоль) растворяли в горячем этаноле (5 мл). Добавляли растворенную в горячем этаноле лимонную кислоту (52 мг, 0.273 ммоль). Состав охлаждали до комнатной температуры, при этом в осадок выпадал белый остаток. Осадок АА-7 высушивали в вакууме.
Выход: 48 мг (31%), согласно ЯМР сигналы цитрата отсутствовали.
Примечание: вероятно по ошибке была внесена трифторуксусная кислота, так что был получен не желаемый цитрат, а соль трифторуксусной кислоты.
Выход: 109 мг (9%) (АА-7)
Точка плавления: 265-269°С
1Н-ЯМР (ДМСО-d6): 0.94 (3H, t); 1.29 (4H, m); 1.60 (2H, m); 1.81 (4H, t); 1.96 (2H, t); 2.40 (4H, m); 2.59 (6H, m); 3.87 (2H; t); 6.55 (1H, d); 6.70 (1H, s); 7.04 (1H, d); 8.54 (1H, s); 9.45 (1H, bs): 10.98 (1H, bs).
13С-ЯМР (ДМСО-d6): 13.77; 22.14; 22.70; 24.86; 26.07; 29.10; 30.94; 37.12; 59.36; 66.16; 70.51; 101.97; 104.52; 110.81; 111.05; 126.74; 129.56; 138.88; 150.28 (свободное основание).
Пример АА-8:
6'-гидрокси-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (2:3) (один из двух возможных диастереомеров)
3-(2-гидрокси-этил)-1Н-индол F-3 (2.68 г, 15.09 ммоль) и кетон Е-2 (2.94 г, 15.09 ммоль) помещали в абс. CH2Cl2 (100 мл) под аргоном и при охлаждении льдом смешивали с трифлатом (2.96 мл, 15.34 ммоль) в CH2Cl2 (5 мл) и 30 мин перемешивали при комнатной температуре. Состав перемешивали последующие 16 ч при комнатной температуре. Для переработки добавляли H2O (110 мл) и K2CO3 (2.45 г) и перемешивали при комнатной температуре 20 мин. Фазы отделяли. Водную фазу экстрагировали дихлорметан (2×20 мл).
Органическую фазу высушивали над Na2SO4 и концентрировали в вакууме. Осадок очищали путем флеш-хроматографии с CHCl3/МеОН (9:1) и перекристаллизовывали в этиловом эфире уксусной кислоты. Выход: 476 мг (9%)
Только что полученный простой спироэфир (471 мг, 1.32 ммоль) растворяли в горячем этаноле (5 мл). Добавляли растворенную в горячем этаноле лимонную кислоту (245 мг, 1.32 ммоль). Состав охлаждали до комнатной температуры, при этом в осадок не выпадал белый остаток. Состав концентрировали в вакууме до сухости.
Выход: 524 мг (72%) (АА-8)
Пример АА-9:
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (неполярный диастереомер)
Триптамин Н-1 (2,43 г, 15,2 ммоль) и кетон Е-2 (3,0 г, 15,2 ммоль) растворяли в абс. метаноле (90 мл) и 25 ч перемешивали при комнатной температуре под аргоном. Реакционную смесь затем концентрировали. Осадок растворяли в абс. 1,2-дихлорэтане (150 мл), быстро смешивали с трифторуксусной кислотой (10,4 мл, 15,5 г, 136 ммоль) и перемешивали 3 дн. при комнатной температуре. Коричневый раствор при охлаждении льдом смешивали с 1N раствором едкого натра (130 мл) и 20 мин перемешивали при комнатной температуре. Фазы раствора отделяли. Водную фазу экстрагировали с помощью 1,2-дихлорэтана (2×70 мл). Органические фазы объединяли, промывали водой (50 мл), высушивали и концентрировали. Коричневый маслянистый остаток смешивали с метанолом (60 мл), вследствие чего начиналась кристаллизация. Суспензию перемешивали еще 10 мин. Бесцветные кристаллы отсасывали и промывали метанолом (60 мл) (1,28 г). При этом речь шла о чистом неполярном спироамине. Фильтрат концентрировали и полученное коричневое твердое вещество снова смешивали с метанолом (50 мл) и перемешивали 1 ч в ледяной ванне. После отсасывания и промывания холодным метанолом (20 мл) можно было выделить 673 мг неполярного спироамина. Фильтрат концентрировали и осадок (2,4 г) отделяли хроматографически [силикагель 60 (130 г); метанол (500 мл), метанол/триэтиламин (100:1, 1,5 I)]. Неполярный спироамин получали вместе с примесями (1,02 г). Эту фракцию смешивали с холодным метанолом (10 мл) и отсасывали. Полученное твердое вещество (332 мг) представляло собой чистый неполярный продукт. Неполярный спироамин получали с общим выходом в 44% (2,28 г) с точкой плавления в 180-182°С. Полярный спироамин выделяли в дополнительной фракции с выходом в 12% (622 мг) с точкой плавления в 93-96°С.
Только что полученный неполярный спироамин (92 мг, 0.27 ммоль) растворяли в горячем этаноле (5 мл). Добавляли растворенную в горячем этаноле лимонную кислоту (51 мг, 0.27 ммоль). Состав охлаждали до комнатной температуры, при этом в осадок выпадал белый остаток. Осадок АА-9 отфильтровывали и высушивали в вакууме.
Выход: 58 мг (40%) (АА-9)
Пример АА-10:
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-метилкарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (неполярный диастереомер)
Ацетилхлорид (0,126 мл, 139 мг, 1,77 ммоль) растворяли под аргоном в абс. дихлорметане (5 мл) и при комнатной температуре смешивали со свободным основанием неполярного спироамина АА-9 (200 мг, 0,59 ммоль), растворенного в дихлорметане (15 мл), в течение 30 мин. Через 15 мин было очевидным осаждение, которое к концу добавления снова растворилось. После продолжения реакции в 30 мин снова возникало осаждение. Перемешивали еще последующий 21 ч при комнатной температуре. Для переработки бесцветный состав смешивали с водой (10 мл) и 1N раствором едкого натра (5 мл) и 1 ч. перемешивали. Фазы отделяли. Водную фазу экстрагировали дихлорметаном (20 мл). Объединенные органические фазы промывали водой (20 мл), высушивали и концентрировали. При этом получали масло (277 мг) бежевого цвета, которое отделяли хроматографически [силикагель 60 (35 г); этилацетат/метанол (20:1, 300 мл)].
Выход: 56% (125 мг)
Точка плавления: 163-166°С
Только что полученный неполярный амид (125 мг, 0,327 ммоль) растворяли при 50°С в этаноле (5 мл) и смешивали с этанольным раствором (3 мл) лимонной кислоты (70 мг, 0,36 ммоль). После продолжения реакции в 3 ч при комнатной температуре при помощи фильтрации отделяли бесцветный цитрат АА-10 и промывали этанолом (2×3 мл). Неполярный амид получали как цитрат с выходом в 63% (118 мг) с точкой плавления в 220-222°С.
Пример АА-11:
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-циклопентилкарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (2:1) неполярный диастереомер
Хлорид циклопентанкарбоновой кислоты (0,215 мл, 234 мг, 1,77 ммоль) под аргоном растворяли в абс. дихлорметане (5 мл) и при комнатной температуре в течение 45 мин смешивали с неполярным спироамином (неполярное свободное основание из АА-9, 200 мг, 0,59 ммоль), растворенным в дихлорметане (15 мл). Перемешивали еще последующие 1,5 ч при комнатной температуре. Для переработки бесцветный состав смешивали с водой (10 мл) и 1N раствором едкого натра (5 мл) и 1 ч перемешивали. Фазы отделяли. Водную фазу экстрагировали дихлорметаном (20 мл). Объединенные органические фазы промывали водой (20 мл), высушивали и концентрировали. При этом получали масло (325 мг) бежевого цвета, которое отделяли хроматографически [силикагель 60 (40 г); этилацетат, (350 мл)]. Амид выделяли как бесцветное гигроскопическое твердое вещество с выходом в 87% (222 мг).
Только что полученный амид (186 мг, 0,427 ммоль) при 60°С растворяли в этаноле (8 мл) и смешивали с этанольным раствором (3 мл) лимонной кислоты (90 мг, 0,47 ммоль). Немедленно начиналось осаждение. После продолжения реакции в 2 ч при комнатной температуре отделяли бесцветный цитрат АА-11 при помощи фильтрации и промывали этанолом (2×3 мл). Неполярный амид получали как цитрат с выходом в 69% (183 мг) с точкой плавления в 228-230°С.
Пример АА-12:
2',3',4',9,-тетрагидро-N,N-диметил-4-бутил-2'-(2,2)-диметилпропанкарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (неполярный диастереомер)
Хлорид 3,3-диметилмасляной кислоты (0,246 мл, 238 мг, 1,77 ммоль) растворяли под аргоном в абс. дихлорметане (5 мл) и при комнатной температуре в течение 30 мин смешивали со свободным основанием неполярного спироамина АА-9 (200 мг, 0,59 ммоль), растворенного в дихлорметане (15 мл). После продолжения реакции в 24 ч желтый реакционный раствор смешивали с водой (10 мл) и 1N раствором едкого натра (5 мл) и 1 ч перемешивали. Фазы отделяли. Водную фазу экстрагировали дихлорметаном (20 мл). Объединенные органические фазы промывали водой (20 мл), высушивали и концентрировали. При этом получали масло бежевого цвета (322 мг), которое отделяли хроматографически [силикагель 60 (40 г); этилацетат (250 мл), этилацетат/метанол (4:1, 400 мл), метанол (300 мл)]. Амид выделяли как бесцветное масло только с выходом в 7% (40 мг).
Ацилирование повторяли как описано ранее. Реакционный раствор оставался бесцветным. Но прерывание реакции происходило уже через 1,5 ч. После хроматографического отделения реакционной смеси [силикагель 60 (40 г); этилацетат (250 мл)] выделяли амид теперь с выходом в 78% (200 мг) как бесцветное твердое вещество с точкой плавления в 220-222°С.
Выделенный неполярный амид (230 мг, 0,525 ммоль) растворяли при 50°С в этаноле (8 мл) и смешивали с этанольным раствором (4 мл) лимонной кислоты (111 мг, 0,578 ммоль). После продолжения реакции в 16 ч при комнатной температуре при помощи фильтрации отделяли бесцветный цитрат и промывали этанолом (2×3 мл). Неполярный спироамин АА-12 получали как цитрат с выходом в 66% (219 мг) с точкой плавления в 216-218°С.
Пример АА-13:
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-(3,4-диметоксибензилкарбонил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (неполярный диастереомер)
3,4-диметоксифенилацетилхлорид (380 мг, 1,77 ммоль) под аргоном растворяли в абс. дихлорметане (5 мл) и при комнатной температуре в течение 50 мин. смешивали со свободным основанием неполярного спироамина АА-9 (200 мг, 0,59 ммоль), растворенного в дихлорметане (15 мл). Немедленно возникало осаждение. Перемешивали еще последующие 1,5 ч при комнатной температуре. Для переработки состав смешивали с водой (10 мл) и 1N раствором едкого натра (5 мл) и 1 ч перемешивали. Фазы отделяли. Водную фазу экстрагировали дихлорметаном (20 мл). Объединенные органические фазы промывали водой (20 мл), высушивали и концентрировали. При этом получали масло бежевого цвета (357 мг), которое отделяли хроматографически [силикагель 60 (40 г); этилацетат, (250 мл), этилацетат/метанол (8:1, 200 мл)]. Амид выделяли как бесцветное твердое вещество с выходом в 75% (230 мг) с точкой плавления в 135-140°С.
Только что выделенный неполярный амид (216 мг, 0,417 ммоль) растворяли при 60°С в этаноле (11 мл) и смешивали с этанольным раствором (3 мл) лимонной кислоты (89 мг, 0,46 ммоль). После продолжения реакции в 5 ч при комнатной температуре при помощи фильтрации отделяли бесцветный цитрат и промывали этанолом (2×3 мл). Неполярный амид получали в виде цитрата АА-13 с выходом в 92% (270 мг) с точкой плавления в 188-190°С.
Пример АА-14:
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2-этиламинокарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (неполярный диастереомер, сумма ротамеров прибл. 95%)
Свободное основание неполярного спироамина АА-9 (204 мг, 0,6 ммоль) суспендировали в абс. ацетонитриле (30 мл) и смешивали с этилизоцианатом (0,052 мл, 47 мг, 0,66 ммоль). Реакционную смесь 6 ч кипятили с обратным холодильником. Прозрачный раствор концентрировали. Маслянистый остаток ресуспендировали в диэтиловом эфире (20 мл) и промывали водой (5 мл). После высушивания и концентрирования получали неполярную мочевину в виде бесцветного твердого вещества с выходом в 57% (139 мг) с точкой плавления 154-158°С.
Только что выделенную неполярную мочевину (139 мг, 0,4 ммоль) растворяли в этаноле (10 мл) и смешивали с этанольным раствором (5 мл) лимонной кислоты (85 мг, 0,44 ммоль). После продолжения реакции в 20 ч при комнатной температуре при помощи фильтрации отделяли бесцветный цитрат. Так как продукт имел вязкую консистенцию, то промывали диэтиловым эфиром (2×3 мл). Дополнительный продукт невозможно было выделить из фильтрата. Неполярную мочевину получали в виде цитрата АА-14 с выходом в 38% (90 мг) с точкой плавления в 215-231°С.
Пример АА-15:
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-4-метоксибензиламинокарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (полярный диастереоизомер)
4-метоксибензилизоцианат (0,75 ммоль) растворяли в абс. ацетонитриле (30 мл), смешивали с триэтиламином (0,07 мл, 511 мг, 5 ммоль) и свободным основанием неполярного спироамина АА-9 (170 мг, 0,5 ммоль). Реакционную смесь нагревали 6 ч до кипения, причем реакционный раствор становился прозрачным. Так как в ТХ не наблюдалось никакого взаимодействия, то кипятили с обратным холодильником последующие 7 ч. Состав концентрировали. Твердый бесцветный остаток смешивали с диэтиловым эфир, суспензию перемешивали 15 мин и потом отсасывали. Получали неполярную мочевину АА-15 с выходом в 92% (200 мг).
Пример АА-16:
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-метил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (неполярный диастереомер)
Свободное основание неполярного спироамина АА-9 (200 мг, 0,59 ммоль) смешивали с водой (0,04 мл) и при 0°С растворяли в 95-проц. муравьиной кислоте (0,6 мл, 732 мг, 15,9 ммоль). При этой температуре добавляли 37-проц. водный раствор формальдегида (0,46 мл, 178 мг, 5,9 ммоль), 10 мин перемешивали в ледяной ванне и состав нагревали 1 ч до 100°С. При охлаждении льдом раствор бежевого цвета смешивали с водой (5 мл) и 1N раствором едкого натра (15 мл). Мутную смесь перемешивали 30 мин при комнатной температуре, смешивали с дихлорметаном (20 мл) и еще раз 30 мин перемешивали. Фазы отделяли. Водную фазу экстрагировали дихлорметаном (15 мл). Объединенные органические фазы промывали водой (15 мл), высушивали и концентрировали. Осадок (225 мг) представлял собой масло бежевого цвета, которое отделяли хроматографически [силикагель 60 (40 г); этилацетат (250 мл)]. Спироамин выделяли в виде бесцветного твердого вещества с выходом в 25% (51 мг).
Только что выделенный неполярный спироамин (51 мг, 0,144 ммоль) растворяли при 60°С в этаноле (2 мл) и смешивали с этанольным раствором (2 мл) лимонной кислоты (64 мг, 0,316 ммоль). После продолжения реакции в 6 ч отсасывали цитрат 5/6 в виде бесцветного твердого вещества, промывали этанолом (2×2 мл) и диэтиловым эфиром (2×5 мл). Неполярный спироамин получали в виде гигроскопического цитрата АА-16 с выходом в 47% (37 мг).
Пример АА-17:
6'-фтор-4',9'-дигидро-N-этил-N-метил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (один из двух возможных диастереомеров)
Раствор Е-7 (500 мг, 2 ммоль) и 5-фтортриптофол F-2 (430 мг, 2.4 ммоль) в безводном дихлорметане (25 мл) при охлаждении льдом смешивали с трифторметансульфокислотой (450 мг, 265 µл, 3 ммоль) и в течение ночи перемешивали при комнатной температуре. Затем реакционную смесь смешивали с 0.5 N раствор едкого натра (10 мл), 2 ч перемешивали при комнатной температуре, органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (2×20 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали с помощью флеш-хроматографии (100 г, 20×4.0 см) с циклогексан / этилацетат (9:1) и 1% триэтиламин.
Выход: 469 мг (53%), белое твердое вещество
Точка плавления: 112-121°С
1Н-ЯМР (ДМСО-d6): 0.89 (t, 3H, J=6.8 Hz); 1.13 (t, 3H, J=6.9 Hz); 1.18-1.33 (m, 4H); 1.51-1.58 (m, 4H); 1.65-1.73 (m, 2H); 1.65-1.73 (m, 2H); 2.04-2.13 (m, 2H); 2.22(s, 3H); 2.40-2.48 (m, 2H); 2.62 (t, 2H, J=5.3 Hz); 3.88 (t, 2H, J=5.3 Hz); 6.80-6.88 (m, 1H); 7.11 (dd, 1H, J=9.8, 2.3 Hz); 7.31 (dd, 1H, J=8.8, 4.6 Hz); 10.67 (s, 1H).
13С-ЯМР: 14.0; 14.9; 20.0; 22.1, 23.4; 26.6; 27.3 (2C); 29.9 (2C); 32.7; 42.5; 56.1; 58.6; 72.1; 102.3 (d, J=23 Hz); 105.4 (d, J=4 Hz); 108.2 (d, J=26 Hz); 111.9 (d, J=10 Hz); 126.1 (d, J=10 Hz); 132.4; 141.9; 156.7 (d, J=231).
Полученный простой спироэфир (366 мг, 0.98 ммоль) смешивали в горячем изопропаноле (60 мл) с лимонной кислотой (232 мг, 1.21 ммоль) в изопропаноле (5 мл). Выпавший в осадок остаток АА-17 отфильтровывали и высушивали.
Выход: 203 мг (37%), белое твердое вещество АА-17
Точка плавления: 206-209°С
1Н-ЯМР (ДМСО-d6): 0.86 (t, 3H, J=6.9 Hz); 1.12-1.29 (m, 7Н); 1.31-1.81 (m, 6Н); 1.98-2.09 (m, 2Н); 2.36 (s, 3H); 2.46-2.69 (m, 10H), 3.85 (t, 2Н, J=5.4 Hz); 6.82 (dt, 1H, J=9.3, 2.6 Hz); 7.09 (dd, 1H, J=9.8, 2.4 Hz); 7.30 (dd, 1H, J=8.7, 4.6 Hz); 10.55 (s, 1H).
Пример AA-18:
6'-фтор-4',9'-дигидро-N-бензил-N-метил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин (один из двух возможных диастереомеров)
Раствор Е-8 (500 мг, 1.73 ммоль) и 2-(5-фтор-1Н-индол-3-ил)этанола F-2 (311 мг, 1.73 ммоль) в безводном дихлорметане (30 мл) при охлаждении льдом смешивали с трифторметансульфокислотой (346 мг, 204 µл, 2.30 ммоль) и в течение ночи перемешивали при комнатной температуре. Затем реакционную смесь смешивали с 0.5 М раствором едкого натра (17 мл) и 1 ч перемешивали при комнатной температуре. Фазы отделяли, водную фазу экстрагировали дихлорметаном (3×20 мл), объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт (954 мг) очищали путем флеш-хроматографии (100 г, 20×3.6 см) с циклогексан / этилацетатом (9:1).
Выход: 424 мг (56%), аморфное, белое твердое вещество АА-18
Точка плавления: 58-62°С
1Н-ЯМР (ДМСО-d6): 0.97 (t, 3H, J=6.79 Hz); 1.38-1.49 (m, 6Н); 1.77-1.87 (m, 4Н); 1.88-1.96 (m, 4Н); 2.10 (s, 3H); 2.63 (t, 2Н, J=5.2 Hz); 3.62 (s, 2H); 3.89 (t, 2H, J=5.2 Hz); 6.87 (dt, 1H, J=9.1 и 2.5 Hz); 7.13 (dd, 2H, J=9.8 и 2.4 Hz); 7.24-7.35 (m, 5H); 11.03 (s, 1H).
13С-ЯМР (ДМСО-d6): 14.3; 22.1; 23.1; 25.1; 25.4; 26.3; 30.4; 31.5; 34.1; 53.4; 56.5; 58.8; 71.7; 102.4 (d, J=23 Hz); 105.6 (d, J=5 Hz); 108.3 (d, J=26 Hz); 111.6 (d, J=11 Hz); 126.2; 126.6 (d, J=10 Hz); 127.8; 128.=; 132.2; 141.7; 141.9; 156.7 (d, J=231 Hz).
Пример AA-19:
6'-фтор-4',9'-дигидро-N-фенил-N-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин (один из двух возможных диастереомеров)
Раствор Е-11 (368 мг, 1.5 ммоль) и 2-(5-фтор-1Н-индол-3-ил)этанол F-2 (269 мг, 1.5 ммоль) в безводном дихлорметане при 10°С смешивали как можно быстрее с трифторметансульфокислотой (300 мг, 177 µл, 2.0 ммоль) и в течение ночи перемешивали при комнатной температуре. Для контроля взаимодействия брали пробу (0.5 мл), которую промывали 0.5 N раствором едкого натра, и органическую фазу высушивали сульфатом натрия. После полного взаимодействия реакционную смесь смешивали с 0.5 N раствором едкого натра (10 мл), перемешивали 2 ч при комнатной температуре, органическую фазу отделяли, водную фазу экстрагировали дихлорметаном (2×20 мл), объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Затем сырой продукт очищали с помощью флеш-хроматографии (18 г, 20×2.0 см) с циклогексан / этилацетат (9:1) и 1% триэтиламин.
Выход: 327 мг (54%), белое твердое вещество АА-19
Точка плавления: 150-162°С
1Н-ЯМР (ДМСО-d6): 0.87 (t, 3H, J=6.9 Hz); 1.25-1.35 (m, 4Н); 1.77-1.97 (m, 10H); 2.64 (t, 2Н, J=5.2 Hz); 3.90 (t, 2H, J=5.3 Hz); 4.92 (s, 1H); 6.50 (t, 1H, J=7.1 Hz); 6.75 (d, 2H, J=7.9 Hz); 6.83-6.90 (m, 1H); 7.02 (t, 2H, J=7.8 Hz); 7.14 (dd, 1H, J=9.8, 2.5 Hz); 7.30 (dd, 1H, J=8.7, 4.6 Hz); 11.03 (s, 1H).
13С-ЯМР: 14.2; 22.0; 22.7; 25.1; 30.7; 30.9; 31.1; 54.0; 58.8; 71.6; 102.5 (d, J=23 Hz); 105.6 (d, J=5 Hz); 108.3 (d, J=26 Hz); 111.6 (d, J=11 Hz); 115.2; 126.6 (d, J=10 Hz); 128.5; 132.2; 141.6; 147.5; 156.7 (d, J=237 Hz).
Пример AA-20:
4-бутил-6'-фтор-4-(N-морфолино)-1',3',4',9'-тетрагидроспиро[циклогексан-1,1'-пирано[3,4-b]индол] (неполярный диастереомер)
К раствору Е-12 (479 мг, 2 ммоль) и 2-(5-фтор-1Н-индол-3ил)этанол F-2 (358 мг, 2 ммоль) дихлорметан (50 мл) при охлаждении ледяной водой прикапывали трифторметансульфокислоту (400 мг, 236 µл, 2.66 ммоль). Реакционную смесь 20 ч перемешивали при комнатной температуре, затем смешивали с 0.5 М раствором едкого натра (20 мл) и после этого 3 ч перемешивали при комнатной температуре. Органическую фазу отделяли, водную экстрагировали дихлорметаном (3×20 мл), объединенные органические фазы промывали раствором хлорида натрия (50 мл), высушивали сульфатом натрия и концентрировали в вакууме. Смесь изомеров (815 мг) отделяли путем флеш-хроматографии (100 г, 22×4 см) с этилацетат / циклогексан (1:3).
Фракция 1: неполярный диастереоизомер, АА-20
Выход: 259 мг (32%), белое твердое вещество
Точка плавления: >250°С
1Н-ЯМР (CDCl3): 0.92 (t, 3H, J=6.5 Hz); 1.19-2.10 (m, 14H); 2.58-2.65 (m, 4H); 2.75 (t, 2H, J=5.3 Hz); 3.75-3.81 (m, 4H); 3.99 (t, 2H, J=5.4 Hz); 6.91 (dt, 1H, J=8.8, 1.8 Hz); 7.12 (dd, 1H, J=9.5, 2.5 Hz); 7.30-7.26 (m, 1H), 7.55 (s, 1H).
13С-ЯМР (CDCl3): 14.1; 22.5; 23.8; 26.7 (2 C); 26.9; 30.3 (2 C); 33.4; 45.1(2 C); 56.1; 59.6; 68.5 (2 C); 72.3; 103.3 (d, J=23 Hz); 107.5 (d, J=4 Hz); 109.7 (d, J=26 Hz); 111.3 (d, J=10 Hz); 127.6 (d, J=10 Hz); 132.1; 141.2; 157.9 (d, J=235 Hz).
Фракция 2: полярный диастереоизомер, сравн. пример АА-21
Выход: 335 мг (42%), белое твердое вещество
Точка плавления: 238-241°С
1Н-ЯМР (CDCl3): 0.98 (t, 3H, J=6.4 Hz); 1.30-2.05 (m, 14Н); 2.63-2.68 (m, 4H); 2.75 (t, 2H, J=5.3 Hz); 3.68-3.72 (m, 4H); 3.99 (t, 2H, J=5.4 Hz); 6.90 (dt, 1H, J=9.3, 2.4 Hz); 7.12 (dd, 1H, J=9.4, 2.0 Hz); 7.24 (dd, 1H, J=8.8, 4.3 Hz); 7.63 (s, 1H).
13С-ЯМР (CDCl3): 14.4; 22.4; 23.6; 25.3; 25.6 (2 C); 30.7; 32.4 (2 C); 45.7 (2 C); 56.4; 59.6; 68.2 (2 C); 71.9; 103.4 (d, J=24 Hz); 107.8; 109.8 (d, J=27 Hz); 111.3 (d, J=9 Hz); 127.5; 132.1; 140.7; 158.0 (d, J=234 Hz).
Пример AA-21:
4-бутил-6'-фтор-4-(N-морфолино)-1',3',4',9'-тетрагидроспиро[циклогексан-1,1'-пирано[3,4-b]индол] (полярный диастереомер)
Полученный под примером АА-20 полярный диастереомер приведен далее как пример АА-21.
АА-21 (полярный диастереоизомер)
Выход: 335 мг (42%), белое твердое вещество
Точка плавления: 238-241°С
1Н-ЯМР (CDCl3): 0.98 (t, 3H, J=6.4 Hz); 1.30-2.05 (m, 14Н); 2.63-2.68 (m, 4H); 2.75 (t, 2H, J=5.3 Hz); 3.68-3.72 (m, 4H); 3.99 (t, 2H, J=5.4 Hz); 6.90 (dt, 1H, J=9.3, 2.4 Hz); 7.12 (dd, 1H, J=9.4, 2.0 Hz); 7.24 (dd, 1H, J=8.8, 4.3 Hz); 7.63 (s, 1H).
13С-ЯМР (CDCl3): 14.4; 22.4; 23.6; 25.3; 25.6 (2 С); 30.7; 32.4 (2 С); 45.7 (2 С); 56.4; 59.6; 68.2 (2 С); 71.9; 103.4 (d, J=24 Hz); 107.8; 109.8 (d, J=27 Hz); 111.3 (d, J=9 Hz); 127.5; 132.1; 140.7; 158.0 (d, J=234 Hz).
Пример AA-22:
4',9'-дигидро-N,N-диметил-4-метоксипропил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (один из двух возможных диастереомеров)
Кетон Е-4 (275 мг, 1.26 ммоль) и триптофол F-1 (206 мг, 1.26 ммоль) растворяли в абс. дихлорметане (10 мл), под аргоном смешивали с метансульфокислотой (0.13 мл, 2.05 ммоль) и 20 ч перемешивали при комнатной температуре. После добавления 1N NaOH (10 мл) и CH2Cl2 (20 мл) перемешивали еще 10 мин, фазы отделяли, водную фазу дважды экстрагировали с помощью CH2Cl2, объединенные органические фазы промывали водой, высушивали над Na2SO4 и раствор концентрировали в вакууме. Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/МеОН (20:1).
Выход: 327 мг (73%)
При взаимодействии с молярным количеством лимонной кислоты в этаноле в осадок выпал цитрат АА-22 в виде твердого вещества.
Выход: 281 мг (АА-22)
Точка плавления: 207-208°С
1Н-ЯМР (ДМСО-d6): 1.35-1.56 (8H, m); 1.71 (2H; t); 2.14 (2H, t); 2.26 (6H, s); 2.64 (2H, t); 3.25 (3H, s); 3.36 (2H s); 3.89 (2H, t); 6.95 (2H, m); 7.32 (2H, m); 10.72 (1H, bs), свободное основание.
13С-ЯМР (ДМСО-d6): 22.13; 24.27; 25.80; 27.78; 29.26; 37.16; 44.12; 57.81; 59.09: 71.16; 72.18; 105.25; 111.38; 117.58; 118.35; 120.62; 126.36; 135.63; 138.96; 171.95; 177.09, цитрат.
Пример АА-23:
6'-фтор-4',9'-дигидро-N,N-диметил-4-метоксипропил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (неполярный диастереомер)
Кетон Е-4 (426 мг, 2 ммоль) и 5-фтор-триптофол F-2 (362 мг, 2 ммоль) растворяли в абс. дихлорметане (10 мл), под аргоном смешивали с метансульфокислотой (0.14 мл, 2.2 ммоль) и 24 ч перемешивали при комнатной температуре. После добавления 1N NaOH (10 мл) фазы отделяли, водную фазу экстрагировали с помощью CH2Cl2 (3×10 мл), объединенные органические фазы промывали водой (10 мл), высушивали над Na2SO4 и раствор концентрировали в вакууме. Оставшийся осадок отделяли путем флеш-хроматографии с CHCl3/МеОН (20:1 чистый метанол).
Выход: 408 мг (54%) неполярное соединение, 218 мг (29%) полярное соединение
При взаимодействии неполярного соединения с молярным количеством лимонной кислоты в этаноле в осадок выпал цитрат в виде бесцветного твердого вещества.
Выход: 384 мг, неполярное соединение АА-23
Точка плавления: 210-213°С
1Н-ЯМР (ДМСО-d6): 1.52 (4H, m); 1.70 (4H, m); 1.83 (2H; m); 2.14 (2H, m); 2.60-2.73 (12H, m); 3.25 (3H, s); 3.35 (2H m); 3.89 (2H, t); 6.83 (1H, m); 7.13 (1H, m); 7.36(1H, m); 10.91 (1H, bs).
Пример АА-24:
2',3',4',9'-тетрагидро-N,N-диметил-4-(3-метоксипропил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (неполярный диастереомер)
Кетон Е-4 (426 мг, 2 ммоль) и триптамин Н-1 (320 мг, 2 ммоль) растворяли в абс. метаноле (10 мл) и 20 ч перемешивали при комнатной температуре. Затем растворитель удаляли в вакууме, осадок растворяли в DCE (20 мл), смешивали с трифторуксусной кислотой (2 мл) и перемешивали 5 ч при комнатной температуре. После добавления 1N NaOH (10 мл) и CH2Cl2 (10 мл) перемешивали еще 20 мин., фазы отделяли, водную фазу экстрагировали с помощью CH2Cl2 (2×10 мл), объединенные органические фазы промывали водой (10 мл), высушивали над Na2SO4 и раствор концентрировали в вакууме. Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/МеОН (9:1 без триэтиламин ⇒4:1+1% триэтиламин).
Выход: 350 мг (49%) неполярное соединение, загрязненное исходным кетоном, 321 мг (45%) полярное соединение, загрязненное
При взаимодействии неполярного соединения с молярным количеством лимонной кислоты в этаноле в осадок выпал цитрат АА-24 в виде бесцветного твердого вещества.
Выход: 264 мг, неполярный диастереомер АА-24 (чистый)
Точка плавления: 247-248°С
1Н-ЯМР (ДМСО-d6): 1.44-1.55 (4H, m); 1.79 (6H; m); 2.33-2.63 (12H, m); 2.86 (2H, m); 3.25 (3H, s); 3.38 (4H m); 7.00 (1H, m); 7.07 (1H, m); 7.39 (2H, m); 11.04 (1H, bs).
Пример АА-25:
4',9'-дигидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (один из двух возможных диастереомеров)
Кетон Е-3 (455 мг, 2 ммоль) и триптофол F-1 (326 мг, 2 ммоль) растворяли в абс. дихлорметане (10 мл), под аргоном смешивали с метансульфокислотой (0.14 мл, 2.2 ммоль) и 24 ч перемешивали при комнатной температуре. После добавления 1N NaOH (15 мл) и CH2Cl2 (25 мл) перемешивали еще 10 мин, потом фазы отделяли, водную фазу дважды экстрагировали посредством CH2Cl2 (10 мл), объединенные органические фазы промывали водой (10 мл), высушивали над Na2SO4 и раствор концентрировали в вакууме. Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/МеОН (20:1).
Выход: 687 мг (93%)
При взаимодействии с молярным количеством лимонной кислоты в этаноле в осадок выпадал цитрат АА-25 в виде бесцветного твердого вещества.
Выход: 152 мг, белое твердое вещество
Точка плавления: 214-215°С
1Н-ЯМР (ДМСО-d6): 1.33 (2H, m); 1.51 (4H; m); 1.75 (4H, m) 1.95 (2H, t); 2.14 (2H, t); 2.66 (10H, m); 3.31 (3H, s); 3.36 (2H t); 3.90 (2H, s); 6.98 (2H, m); 7.38 (2H, m); 10.88 (1H, bs), цитрат.
13С-ЯМР (ДМСО-d6): 21.04; 22.16; 26.93; 29.90; 30.23; 30.91; 37.19; 55.17; 57.74; 58.75; 71.85; 104.91; 111.18; 117.41; 118.18; 120.33; 126.40; 135.81; 139.71, свободное основание.
Пример АА-26:
6'-фтор-4',9'-дигидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (полярный диастереоизомер)
Кетон Е-3 (426 мг, 2 ммоль) и 5-фтор-триптофол F-2 (362 мг, 2 ммоль) растворяли в абс. дихлорметане (10 мл), под аргоном смешивали с метансульфокислотой (0.14 мл, 2.2 ммоль) и перемешивали при комнатной температуре 24 ч. После добавления 1N NaOH (10 мл) реакция раствора) фазы отделяли, водную фазу экстрагировали с помощью CH2Cl2 (3×10 мл), объединенные органические фазы промывали водой (10 мл), высушивали над Na2SO4 и раствор концентрировали в вакууме. Оставшийся осадок отделяли путем флеш-хроматографии с CHCl3/МеОН (20:1).
Выход: 613 мг (79%)
При взаимодействии с молярным количеством лимонной кислоты в этаноле в осадок выпадал цитрат АА-26 в виде бесцветного твердого вещества.
Точка плавления: 216-218°С
1Н-ЯМР (ДМСО-d6): 1.12 (2H, m); 1.50 (4H; m); 1.68 (4H, m) 1.86 (2H, t); 2.06 (2H, t); 2.56 (10H, m); 3.22 (3H, s); 3.34 (5H m); 3.87 (2H, s); 4.34 (1H, bs); 6.81 (1H, t); 7.11 (1H, m); 7.34 (1H, m); 10.81 (1H, bs), цитрат.
13С-ЯМР (ДМСО-d6): 21.04; 22.08; 26.88; 29.84; 30.23; 30.86; 37.10; 55.16; 57.74; 58.69; 71.84; 72.00; 102.16; 102.38; 105.37; 108.00; 111.89; 111.98; 126.65; 132.44; 141.91; 155.56; 157.85, свободное основание.
Пример АА-27:
2',3',4',9'-тетрагидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (неполярный диастереомер)
Кетон Е-3 (455 мг, 2 ммоль) и триптамин Н-1 (320 мг, 2 ммоль) растворяли в абс. метаноле (10 мл) и перемешивали при комнатной температуре 20 ч. Затем растворитель удаляли в вакууме, осадок растворяли в DCE (20 мл), смешивали с трифторуксусной кислотой (2 мл) и 5 ч перемешивали при комнатной температуре. После добавления 1N NaOH (10 мл) и CH2Cl2 (10 мл) перемешивали еще 30 мин, фазы отделяли, водную фазу экстрагировали посредством CH2Cl2 (2×10 мл), объединенные органические фазы промывали водой (10 мл), высушивали над Na2SO4 и раствор концентрировали в вакууме. Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/МеОН (9:1 без триэтиламин ⇒4:1+1% триэтиламин).
Выход: 273 мг (37%), неполярное соединение
335 мг (48%), полярное соединение, загрязненное
При взаимодействии неполярного диастереомера с молярным количеством лимонной кислоты в этаноле в осадок выпадал цитрат АА-27 в виде бесцветного твердого вещества.
Выход: 204 мг, неполярный диастереомер АА-27
Точка плавления: 236-240°С
1Н-ЯМР (ДМСО-d6): 1.40 (2H, m); 1.63 (4H, m); 1.80-2.08 (8H; m); 2.52-266 (10H, m); 3.12 (2H, t); 3.26 (3H s); 3.41 (2H, m); 6.94 (1H, m); 7.06 (1H, m); 7.37 (2H, m); 10.86 (1H, bs).
Пример АА-28:
4',9'-дигидро-N,N-диметил-4-циклопентил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (2:1) (один из двух возможных диастереомеров)
Кетон Е-6 (235 мг,1.1 ммоль) и триптофол F-1 (180 мг, 1.12 ммоль) растворяли в абс. дихлорметане (5 мл) и под аргоном смешивали с метансульфокислотой (0.1 мл, 1.5 ммоль) и 20 ч перемешивали при комнатной температуре. После добавления 1N NaOH (5 мл) и CH2Cl2 (10 мл) перемешивали еще 10 мин, фазы отделяли, водную фазу дважды экстрагировали посредством CH2Cl2, объединенные органические фазы промывали водой, высушивали (Na2SO4) и раствор концентрировали в вакууме. Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/МеОН (20:1).
Выход: 361 мг, получали смесь веществ, при взаимодействии с молярным количеством лимонной кислоты в этаноле в осадок выпадал цитрат АА-28 в виде бесцветного твердого вещества.
Выход: 302 мг (50%), 1 диастереомер АА-28
Точка плавления: 200-202°С
1Н-ЯМР (ДМСО-d6): 1.35 (6H, m); 1.61 (8H, m); 1.98 (3H, m); 2.36 (8H; m); 2.84 (1H, m); 2.59 (2H; s); 3.74 (2H, m); 6.83 (2H, m); 7.23 (2H, m); 10.63 (1H, s).
13С-ЯМР (ДМСО-d6): 22.10; 23.87; 24.67; 28.15; 29.42; 38.26; 42.72; 43.46; 59.14; 71.29; 72.08; 105.32; 111.24; 117.64; 118.41; 120.71; 126.36; 135.51; 138.91; 171.40; 175.86.
Пример АА-29:
2',3',4',9'-тетрагидро-N,N-диметил-4-циклопентил-спиро[циклогексан-1,1'(1,Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (один из двух возможных диастереомеров)
Кетон Е-6 (209 мг, 1.0 ммоль) и триптамин Н-1 (160 мг, 1.0 ммоль) растворяли в абс. метаноле (10 мл) и 20 ч перемешивали при комнатной температуре. Затем растворитель удаляли в вакууме, осадок растворяли в дихлорэтане (10 мл), смешивали с трифторуксусной кислотой (1.0 мл) и 5 дн. перемешивали при комнатной температуре. После добавления 1N NaOH (10 мл) и CH2Cl2 (10 мл) перемешивали еще 20 мин, фазы отделяли, водную фазу дважды экстрагировали с помощью CH2Cl2, объединенные органические фазы промывали водой, высушивали (Na2SO4) и раствор концентрировали в вакууме. Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/МеОН (20:1). При взаимодействии с молярным количеством лимонной кислоты в этаноле в осадок выпадал цитрат АА-29 в виде бесцветного твердого вещества.
Выход: 226 мг (64%) 1 диастереомер АА-29
Цитрат: Точка плавления: 229-230°С
Так как ЯМР-спектры цитрата были плохо растворены, то добавляли ЯМР-спектры свободных оснований.
1Н-ЯМР (ДМСО-d6): 1.43 (12H, m); 1.80 (2H, t); 2.07 (3H, m); 2.35 (6H, s); 2.55 (2H, m); 3.00 (2H, t); 3.37 (1H, bs); 6.96 (2H, m); 7.30 (2H, m); 10. 55 (1H, s).
13С-ЯМР (ДМСО-d6): 22.53; 24.57; 24.81; 28.04; 30.72; 37.85; 38.66; 43.97; 52.07; 57.12; 106.26; 111.00; 117.20; 117.90; 120.09; 126.89; 135.59; 141.62.
Пример АА-30:
4',9',-дигидро-N,N-диметил-4-циклогексил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (2:1) (один из двух возможных диастереомеров)
Кетон Е-5 (175 мг, 0.78 ммоль) и триптофол F-1 (126 мг, 0.78 ммоль) растворяли в абс. дихлорметане (5 мл) и под аргоном смешивали с метансульфокислотой (0.07 мл, 1.1 ммоль) и 72 ч перемешивали при комнатной температуре. После добавления 1N NaOH (5 мл) и CH2Cl2 (10 мл) перемешивали еще 10 мин, фазы отделяли, водную фазу дважды экстрагировали с помощью CH2Cl2, объединенные органические фазы промывали водой, высушивали (Na2SO4) и раствор концентрировали в вакууме. Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/МеОН (20:1). При взаимодействии с молярным количеством лимонной кислоты в этаноле в осадок выпадал цитрат АА-30 в виде твердого вещества.
Выход: 110 мг (39%) 1 диастереомер АА-30 цитрат: Точка плавления: 230-231°С
1Н-ЯМР (ДМСО-d6): 1.10 (6H, m); 1.77 (12H, m); 2.07 (2H, m); 2.66 (10H; m); 3.88 (2H, m); 6.97 (2H, m); 7.36 (2H, m); 10.72 (1H, s).
13С-ЯМР (ДМСО-d6): 22.16; 24.64; 26.00; 28.77; 30.09; 43.01; 43.62; 59.01; 71.52; 72.16; 105.20; 111.24; 117.59; 118.35; 120.61; 126.43; 135.64; 139.26; 171.56; 176.14.
Пример АА-31:
6'-фтор-4',9'-дигидро-N,N-диметил-4-циклогексил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (один из двух возможных диастереомеров)
Кетон Е-5 (137 мг, 0.61 ммоль) и 5-фтор-триптофол F-2 (109 мг, 0.61 ммоль) растворяли в абс. дихлорметане (4 мл), смешивали под аргоном с метансульфокислотой (0.065 мл, 1.0 ммоль) и перемешивали при комнатной температуре 48 ч. После добавления 1N NaOH (5 мл) и CH2Cl2 (10 мл) перемешивали еще 20 мин, фазы отделяли, водную фазу дважды экстрагировали с помощь. CH2Cl2, объединенные органические фазы промывали водой, высушивали (Na2SO4) и раствор концентрировали в вакууме. Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/MeOH (20:1). При взаимодействии с молярным количеством лимонной кислоты в этаноле в осадок выпадал цитрат АА-31 в виде бесцветного твердого вещества.
Выход: 172 мг (73%), 1 диастереомер АА-31
Цитрат: Точка плавления: 204-205°С
1Н-ЯМР (ДМСО-d6): 1.11 (6H, m); 1.43 (2H, m); 1.56 (4H, m); 1.77 (6H, m); 2.06 (2H, m); 2.57 (7 Н; m); 3.00 (2H, m); 6.90 (1H, m); 6.98 (1H, m); 7.30 (2H, m); 10.51 (1H, s).
13С-ЯМР (ДМСО-d6): 22.04; 24.57; 25.97; 26.58; 28.72; 30.04; 38.38; 43.25; 58.93; 71.52; 72.11; 102.35; 102.58; 105.64; 108.52; 112.03; 126.56; 132.21; 171.32; 175.49.
Пример АА-32:
2',3',4',9'-тетрагидро-N,N-диметил-4-циклогексил-спиро[циклогексан-1,1'(1,Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (один из двух возможных диастереомеров)
Кетон Е-5 (175 мг, 0.78 ммоль) и триптамин Н-1 (125 мг, 0.78 ммоль) растворяли в абс. метаноле (8 мл) и 20 ч перемешивали при комнатной температуре. Затем растворитель удаляли в вакууме, осадок растворяли в дихлорэтане (10 мл), смешивали с трифторуксусной кислотой (0.8 мл) и 4 ч перемешивали при комнатной температуре. После добавления 1N NaOH (5 мл) и CH2Cl2 (10 мл) перемешивали еще 20 мин, фазы отделяли, водную фазу дважды экстрагировали с помощью CH2Cl2, объединенные органические фазы промывали водой, высушивали (Na2SO4) и раствор концентрировали в вакууме. Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/МеОН (9:1).
Выход: 160 мг (56%) 1 диастереомер
Цитрат: Точка плавления: 228-229°С
ЯМР-спектры свободного основания:
1Н-ЯМР (ДМСО-d6): 1.13 (6H, m); 1.72 (10H, m); 1.97 (2H, m); 2.59 (10H; m); 3.88 (2H, m); 6.86 (1H, t); 7.14 (1H, m); 7.32 (1H, m); 10.74 (1H, s).
13С-ЯМР (ДМСО-d6): 22.58; 25.06; 26.32; 26.81; 28.85; 31.26; 38.22; 45.32; 51.91; 57.69; 72.11; 106.30; 110.97; 117.22; 117.91; 120.10; 126.94; 135.58; 141.69
Только что полученный простой спироэфир (140 мг, 0.38 ммоль) растворяли в горячем этаноле (4 мл) и смешивали с раствором лимонной кислоты (73 мг, 0.38 ммоль) в этаноле (2 мл). После 2-часового отстаивания в холодильнике отсасывали образовавшееся твердое вещество АА-32 и высушивали в вакууме.
Выход: 160 мг (75%) (АА-32)
Точка плавления: 228-229°С
Пример АА-33:
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(1'H)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (полярный диастереомер)
Полученный под примером АА-9 полярный простой спироэфир (90 мг, 0.26 ммоль) растворяли в горячем этаноле (5 мл). Добавляли растворенную в горячем этаноле лимонную кислоту (48 мг, 0.26 ммоль). Состав охлаждали до комнатной температуры, при этом в осадок выпадал белый остаток. Осадок отфильтровывали и высушивали в вакууме.
Выход: 89 мг (75%) (АА-33)
Пример АА-34:
6'-фтор-4',9'-дигидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (полярный диастереомер)
Свободные основания полярного простого спироэфира из примера АА-2 (142 мг, 0.429 ммоль) растворяли в горячем этаноле (5 мл) и добавляли растворенную в горячем этаноле лимонную кислоту (78 мг, 0.429 ммоль). Состав охлаждали до комнатной температуры и концентрировали в вакууме.
Выход: 212 мг (11%) (АА-34)
Точка плавления: 72-75°С
1Н-ЯМР (ДМСО-d6): 1.05 (3H, t); 1.64 (2H, m); 1.94 (6H, m); 2.48 (2H, m); 2.55 (6H, s); 3.89 (2H, t); 6.87 (1H, m); 7.14 (1H, m); 7.29 (1H, m); 11.04 (1H, s).
Пример АА-35:
2',3',4',9'-тетрагидро-N,N-диметил-4-(3-метоксипропил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (полярный диастереомер, чистота <95%)
Кетон Е-4 (426 мг, 2 ммоль) и триптамин Н-1 (320 мг, 2 ммоль) растворяли в абс. метаноле (10 мл) и 20 ч перемешивали при комнатной температуре. Затем растворитель удаляли в вакууме, осадок растворяли в DCE (20 мл), смешивали с трифторуксусной кислотой (2 мл) и 5 ч перемешивали при комнатной температуре. После добавления 1N NaOH (10 мл) и CH2Cl2 (10 мл) перемешивали еще 20 мин, фазы отделяли, водную фазу экстрагировали с помощью CH2Cl2 (2×10 мл), объединенные органические фазы промывали водой (10 мл), высушивали над Na2SO4 и раствор концентрировали в вакууме. Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/МеОН (9:1 без триэтиламин ⇒4:1+1% триэтиламин).
Выход: 350 мг (49%) неполярное соединение, загрязненное исходным кетоном 321 мг (45%) полярное соединение, загрязненное.
При взаимодействии полярного соединения с молярным количеством лимонной кислоты в этаноле в осадок выпадал цитрат АА-35 в виде бесцветного твердого вещества.
Выход: 267 мг, полярный диастереомер АА-35
Точка плавления: 228-229°С
1Н-ЯМР (ДМСО-d6): 1.65 (4H, m); 1.88 (4H; m); 2.05 (4H, m); 2.47-2.59 (10H, m); 2.69 (2H, t); 3.18 (2H, t); 3.30 (3H s); 3.43 (2H, m); 6.97 (1H, m); 7.07 (1H, m); 7.33 (2H, m); 10.95(1H, bs).
Пример АА-36:
6'-фтор-4',9'-дигидро-N,N-диметил-4-метоксипропил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (полярный диастереомер)
Кетон Е-4 (426 мг, 2 ммоль) и 5-фтор-триптофол F-2 (362 мг, 2 ммоль) растворяли в абс. дихлорметане (10 мл), смешивали под аргоном с метансульфокислотой (0.14 мл, 2.2 ммоль) и перемешивали при комнатной температуре 24 ч. После добавления 1N NaOH (10 мл) фазы отделяли, водную фазу экстрагировали с помощью CH2Cl2 (3×10 мл), объединенные органические фазы промывали водой (10 мл), высушивали над Na2SO4 и раствор концентрировали в вакууме. Оставшийся осадок отделяли путем флеш-хроматографии с CHCl3/МеОН (20:1 чистый метанол).
Выход: 408 мг (54%) неполярное соединение
218 мг (29%) полярное соединение
При взаимодействии полярного соединения с молярным количеством лимонной кислоты в этаноле осадок не выпадал, поэтому раствор концентрировали и получали белое, аморфное твердое вещество АА-36.
Выход: 239 мг, полярное соединение, АА-36
1Н-ЯМР (ДМСО-d6): 1.65 (4H, m); 1.97 (8H; m); 2.56-2.68 (12H, m); 3.31 (3H, s); 3.45 (2H m); 3.89 (2H, t); 6.88 (1H, m); 7.17 (1H, m); 7.32 (1H, m); 11.06 (1H, bs).
Пример АА-37
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-этиламинокарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (полярный диастереомер)
Полярный спироамин (свободное основание из АА-9, 133 мг, 0,39 ммоль) суспендировали в абс. ацетонитриле (30 мл) и смешивали с этилизоцианатом (0,034 мл, 31 мг, 0,43 ммоль). Реакционную смесь 1,5 ч кипятили с обратным холодильником. После охлаждения до комнатной температуры выкристаллизовывалось бесцветное твердое вещество. После отсасывания получали полярную мочевину АА-37 с выходом в 46% (74 мг) с точкой плавления в 182-184°С.
Пример АА-38:
4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (полярный диастереомер)
Кетон Е-2 (2.0 г/10.15 ммоль) и триптофол F-1 (1.63 г/10.15 ммоль) под аргоном помещали в абс. дихлорметан (70 мл) и затем смешивали с метансульфокислотой (720 µл/11.16 ммоль). Состав 24 ч перемешивали при комнатной температуре. Для переработки состав смешивали с 1N NaOH и экстрагировали посредством дихлорметана (3×15 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вакууме. Осадок очищали путем флеш-хроматографии с CHCl3/МеОН (9:1,1:1).
Выход: Фракция 1: неполярный диастереомер 2.18 г (загрязненный триптофолом)
Фракция 2: полярный диастереомер 862 мг (25%)
Фракцию 2 (862 мг, 2.52 ммоль) растворяли в горячем этаноле (5 мл). Добавляли растворенную в горячем этаноле лимонную кислоту (480 мг, 2.52 ммоль). Состав охлаждали до комнатной температуры, при этом в осадок выпадал белый остаток. Осадок АА-38 отфильтровывали и высушивали в вакууме.
Выход: 476 мг (35%), полярный АА-38
Пример АА-39:
2',3',4',9'-тетрагидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (полярный диастереомер)
Кетон Е-3 (455 мг, 2 ммоль) и триптамин Н-1 (320 мг, 2 ммоль) растворяли в абс. метаноле (10 мл) и при комнатной температуре 20 ч перемешивали. Затем растворитель удаляли в вакууме, осадок растворяли в DCE (20 мл), смешивали с трифторуксусной кислотой (2 мл) и 5 ч перемешивали при комнатной температуре. После добавления 1N NaOH (10 мл) и CH2Cl2 (10 мл) перемешивали еще 30 мин, фазы отделяли, водную фазу экстрагировали с помощью CH2Cl2 (2×10 мл), объединенные органические фазы промывали водой (10 мл), высушивали над Na2SO4 и раствор концентрировали в вакууме.
Оставшийся осадок очищали путем флеш-хроматографии с CHCl3/МеОН (9:1 без триэтиламин → 4:1+1% триэтиламин).
Выход: 273 мг (37%), неполярное соединение
335 мг (48%), полярное соединение, загрязненное
При взаимодействии полярного диастереомера с молярным количеством лимонной кислоты в этаноле в осадок выпадал цитрат АА-39 в виде бесцветного твердого вещества.
Выход: 223 мг, полярный диастереомер АА-39
Точка плавления: 202-204°С
1Н-ЯМР (ДМСО-d6): 1.41 (4H, m); 1.53 (2H, m); 1.73 (6H; m); 2.31-2.61 (10H, m); 2.84 (2H, m); 3.35 (7 Н, m); 7.01 (1H, m); 7.09 (1H, m); 7.41 (2H, m); 10.95 (1H, bs).
Пример АА-40
6'-фтор-4',9'-дигидро-N-бензил-4-аллил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (2:1) (один из двух возможных диастереомеров)
Раствор Е-13 (398 мг, 1.64 ммоль) и 2-(5-фтор-1Н-индол-3-ил)этанол F-2 (293 мг, 1.64 ммоль) в абсолютном дихлорметане (20 мл) при комнатной температуре смешивали с трифторметансульфокислотой (328 мг, 556 µл, 2.18 ммоль) и 16 ч перемешивали при комнатной температуре. Реакционный раствор затем смешивали с 0.5 М раствором едкого натра (10 мл) и перемешивали 2 ч. при комнатной температуре. Фазы отделяли и водную фазу экстрагировали дихлорметаном (3×30 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме.
Выход: 649 мг (98%), слегка желтоватое твердое вещество
Точка плавления: 45-48°С
1Н-ЯМР (300 MHz, d6-ДМСО): 1.49-1.73 (m, 6Н); 1.84 (t, J=6.8 Hz, 1H); 2.08 (dd, J=15.5, 11.7 Hz, 2H); 2.21 (d, J=7.0 Hz, 2H); 2.63 (t, J=5.3 Hz, 2H); 3.67 (d, J=6.4 Hz, 2H); 3.88 (t, J=5.2 Hz, 2H); 4.95-5.16 (m, 2H); 5.94 (m, 1H); 6.79-6.90 (m, 1H); 7.13 (dd, J=9.9, 2.5 Hz, 1H); 7.19-7.41 (m, 4H); 7.49 (d, J=7.0 Hz, 2H); 10.86 (s, 1H).
13С-ЯМР (100 MHz, d6-ДМСО): 22.1; 29.3; 29.7; 38.9; 43.7; 45.1; 52.4; 58.8; 71.8; 102.4 (d, J=23 Hz); 105.4; 108.2 (d, J 26 Hz); 111.7; 116.8; 126.4; 126.7; 127.9; 128.1; 132.1; 135.1; 141.8; 142.0; 156.4 (d, J=231 Hz).
Один из полученных простых спироэфиров (300 мг, 0.74 ммоль) в изопропаноле смешивали с раствором лимонной кислоты (142 мг, 0.74 ммоль) в изопропаноле (1.2 мл) при 70°С. В холоде продукт выпадал в осадок в виде полуцитрата АА-40.
Выход: 389 мг (100%), бесцветные кристаллы АА-40
Точка плавления: 133°С
1Н-ЯМР (300 MHz, d6-ДМСО): 1.58-1.80 (m, 6Н); 1.96-2.18 (m, 2Н); 2.32 (d, J=7.2 Hz, 2H); 2.57 (d, J=15.2 Hz, 1H); 2.65 (dd, J=12.8, 9.5 Hz, 3H); 3.84 (s, 2H); 3.88 (t, J=5.2 Hz, 2H); 4.34 (s, 1H); 5.09-5.21 (m, 2H); 5.95 (tdd, J=17.3, 10.0, 7.2 Hz, 1H); 6.80-6.92 (m, 1H); 7.14 (dd, J=9.9, 2.5 Hz, 1H); 7.24-7.45 (m, 4H); 7.49-7.57 (m, 2H); 10.72 (s, 1H).
13С-ЯМР (100 MHz, d6-ДМСО): 22.0; 28.6; 29.5; 38.8; 42.9; 43.5; 45.0; 54.5; 58.9; 71.5; 71.8; 102.4 (d, J=23 Hz); 105.6; 108.3 (d, J=23 Hz); 111.8; 117.8; 126.7; 127.0; 128.2; 128.8; 132.0; 134.2; 141.7; 156.3 (d, J=231 Hz); 171.3; 175.8.
Пример AA-41
6'-фтор-4',9'-дигидро-N-фенил-4-аллил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин (один из двух возможных диастереомеров)
Раствор Е-10 (277 мг, 1.14 ммоль) и 2-(5-фтор-1Н-индол-3-ил)этанол F-2 (170 мг, 1.14 ммоль) в абсолютном дихлорметан (20 мл) при комнатной температуре смешивали с трифторметансульфокислотой (342 мг, 580 µл, 2.28 ммоль) и 16 ч перемешивали при комнатной температуре. Реакционный раствор смешивали затем с 0.5 М раствором едкого натра (10 мл) и 2 ч перемешивали при комнатной температуре. Фазы отделяли и водную фазу экстрагировали с помощью дихлорметана (3×30 мл). Объединенные органические фазы высушивали сульфатом натрия, концентрировали в вакууме и осадок очищали путем флеш-хроматографии (200 г, 20×5.6 см) с циклогексан / этилацетат (5:1→3:2).
АА-41:
Выход: 296 мг (66%), бесцветное твердое вещество
Точка плавления: 52-54°С
1Н-ЯМР (300 MHz, d6-ДМСО): 1.60-2.14 (m, 8Н); 2.64 (t, J=5.1 Hz, 2H); 2.77 (d, J=6.8 Hz, 2H); 3.90 (t, J=5.1 Hz, 2H); 4.98 (s, 1H); 5.11 (dd, J=13.7, 2.6 Hz, 2H); 5.73-5.91 (m, 1H); 6.53 (t, J=7.2 Hz, 1H); 6.80 (d, J=7.7 Hz, 2H); 6.87 (dd, J=9.6, 2.6 Hz, 1H); 7.04 (t, J=7.9 Hz, 2H); 7.15 (dd, J=9.9, 2.6 Hz, 1H); 7.30 (dd, J=8.7 Hz, 1H); 11.06 (s, 1H).
13С-ЯМР (100 MHz, d6-ДМСО): 22.0; 30.4; 30.9; 35.4; 54.0; 58.9; 71.4; 102.5 (d, J=23 Hz); 105.6; 108.3 (d, J=26 Hz); 111.6; 115.5; 115.9; 117.1; 126.6; 128.5; 132.1; 135.1; 141.5; 147.1; 155.7 (d, J=230 Hz).
Пример AA-42
6'-фтор-4',9'-дигидро-N-(4-метоксибензил)-4-аллил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин (один из двух возможных диастереомеров)
Раствор Е-9 (843 мг, 3.08 ммоль) и 2-(5-фтор-1Н-индол-3-ил)этанол F-2 (552 мг, 3.08 ммоль) в абсолютном дихлорметане (30 мл) при комнатной температуре смешивали с трифторметансульфокислотой (600 мг, 4.0 ммоль) и перемешивали 72 ч при комнатной температуре. Затем добавляли дополнительную трифторметансульфокислоту (300 мг, 2.0 ммоль) и снова 16 ч перемешивали. Реакционный раствор после этого смешивали с 0.5 М раствором едкого натра (10 мл) и 2 ч перемешивали при комнатной температуре. Фазы отделяли и водную фазу экстрагировали дихлорметаном (3×30 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме.
Выход: 1.32 г (99%), желтоватое твердое вещество АА-42
Точка плавления: 54-56°С
1Н-ЯМР (400 MHz, d6-ДМСО): 1.50 (d, J=11.9 Hz, 2H); 1.72 (m, 4H); 1.91 (d, J=14.4 Hz, 2H); 2.55 (d, J=5.0 Hz, 2H); 2.64 (t, J=5.0 Hz, 2H); 3.63 (d, J=2.9 Hz, 2H); 3.72 (s, 3H); 3.88 (dd, J=5.2, 4.8 Hz, 2H); 5.18 (m, 3H); 5.88-6.04 (m, 1H); 6.80-6.93 (m, 4H); 7.08-7.17 (m, 1H); 7.27 (m, 2H); 11.01 (s, 1H).
13С-ЯМР (100 MHz, d6-ДМСО): 22.0; 30.4; 31.1; 35.3; 44.1; 53.2; 54.9; 58.8; 71.8; 102.4 (d, J=23 Hz); 105.6; 108.2 (d, J=25 Hz); 108.5; 111.6; 113.4; 116.9; 126.6; 129.1; 132.1; 133.9; 135.5; 141.7; 156.3 (d, J=233 Hz).
Пример AA-43
N-{6'-фтор-4',9'-дигидро-4-бутил-спиро[циклогексан-1,1, (3,Н)-пирано[3,4-b]индол]-4-ил}-пирролидин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (один из двух возможных диастереомеров)
Раствор Е-14 (1.06 г, 4.7 ммоль) и 2-(5-фтор-1Н-3-ил)этанол F-2 (854 мг, 4.7 ммоль) в безводном дихлорметане (60 мл) при охлаждении льдом и аргоном смешивали с трифторметансульфокислотой (949 мг, 552 µл, 6.3 ммоль) и перемешивали 1 дн. при комнатной температуре. После этого добавляли дополнительно трифторметансульфокислоту (300 мг, 174 µл, 2 ммоль) и снова перемешивали 1 дн. при комнатной температуре. Затем реакционную смесь смешивали с 0.5 М раствором едкого натра (48 мл) и 20 мин перемешивали. Фазы отделяли, водную фазу экстрагировали дихлорметаном (2×20 мл) и объединенные органические фазы высушивали сульфатом натрия. Сырой продукт (1.8 г) очищали путем флеш-хроматографии (180 г, 20×5.6 см) с хлороформ / метанол (95:5).
Выход: 370 мг (19%), желтоватое твердое вещество (фракция 1)
Продукт представлял собой гидрохлорид. Предположительно хлористый водород возникал из применяемого для хроматографии хлороформа.
1Н-ЯМР (CDCl3): 0.97 (t, 3H, J=6.8 Hz), 1.35-1.41 (m, 4Н); 1.46-1.52 (m, 2Н); 1.57 (d, 2Н, J=14.6 Hz), 1.89-1.98 (m, 4H); 2.22 (dt, 2H, J=14.6, 6.0 Hz), 2.35-2.45 (m, 2H); 2.72 (t, 2H, J=5.3 Hz), 2.78 (dt, 2H, J=14.6, 3.5 Hz); 3.10 (dt, 2H, J=13.0, 6.9 Hz), 3.63 (dt, 2H, J=12.2 и 6.6 Hz), 3.92 (t, 2H, J=5.3 Hz), 6.81 (dt, 1H, J=9.2 и 2.5 Hz), 7.06 (dd, 1H, J=9.7, 2.4 Hz), 7.37 (dd, 1H, J=8.8, 4.5 Hz); 10.36 (br s, 1H); 11.04 (s, 1H).
13С-ЯМР (CDCl3): 13.9; 22.6; 23.4; 25.1; 26.6; 27.0; 29.5; 32.6; 48.2; 60.3; 66.5; 71.0; 102.4 (d, J=23 Hz); 106.1 (d, J=4 Hz); 109.2 (d, J=10 Hz); 112.4 (d, J=10 Hz); 126.3 (d, J=10 Hz); 132.4; 139.8; 157.5 (d, J=233 Hz).
Кроме того, получали еще неочищенный продукт (фракция 2, 322 мг, 17%) и не взаимодействующий кетон (Фракция 3, 227 мг, 23%).
1Н-ЯМР-спектр сырой смеси продуктов показал, что образовались только один диастереоизомер и алкен, причем однако последний не был выделен.
Раствор фракция 1 (350 мг, 0.83 ммоль) в хлороформе (20 мл) промывали раствором гидрокарбоната натрия, органическую фазу высушивали сульфатом натрия и концентрировали в вакууме.
Выход: 204 мг (70%), аморфное, желтоватое твердое вещество
Точка плавления: 70°С
1Н-ЯМР (CDCl3): 0.93 (t, 3H, J=6.7 Hz), 1.21-1.38 (m, 4Н); 1.38-1.42 (m, 2Н); 1.48 (d, 2Н, J=12.8 Hz); 1.74 (d, 2H, J=12.8 Hz); 1.74-1.84 (m, 4H); 1.88 (dt, 2H, J=13.5, 2.9 Hz); 2.04 (dt, 2H, J=13.2, 3.2 Hz); 2.69 (t, 4 H, J=5.8 Hz); 2.74 (t, 2H, J=5.4 Hz); 3.99 (t, 2H, J=5.4 Hz); 6.87 (dt, 1H, J=9.1, 2.5 Hz); 7.11 (dd, 1H, J=9.5, 2.4 Hz); 7.23 (dd, 1H, J=8.7, 4.3 Hz); 7.90 (s, 1H).
13С-ЯМР (CDCl3): 14.2; 22.5; 24.0; 24.1; 24.8; 27.0; 28.6; 30.8; 31.1; 44.1; 54.7; 59.7; 72.4; 103.2 (d, J=24 Hz); 107.1 (d, J=5 Hz); 109.4 (d, J=26 Hz); 111.2 (d, J=10 Hz); 127.6 (d, J=10 Hz); 132.0; 141.7; 157.8 (d, J=234 Hz).
Раствор только что полученного желтого твердого вещества (свободное основание фракции 1) (180 мг, 0.46 ммоль) в горячем этаноле (15 мл) смешивали с горячим раствором лимонной кислоты (90 мг, 0.46 ммоль) в этаноле (1.2 мл). При этом в осадок выпадал белый остаток, который после охлаждения отфильтровывали.
Выход: 137 мг (50%), белое твердое вещество (АА-43)
Точка плавления:198-199°С
1Н-ЯМР (ДМСО-d6): 0.92 (t, 3H, J=6.7 Hz); 1.20-1.40 (m, 4H); 1.44-1.64 (m, 4H); 1.71 (br d, 2H, J=12.7 Hz); 1.90 (br s, 6H); 2.12 (br t, 2H, J=12.7 Hz); 2.57 (d, 2H, J=15.0 Hz); 2.63 (t, 2H, J=4 Hz); 2.66 (d, 2H, J=15.0 Hz); 3.07 (br s, 4H); 3.89 (t, 2H, J=5.1 Hz); 6.87 (dt, 1H, J=9.1, 2.4 Hz); 7.15 (dd, 1H, J=9.9, 2.3 Hz); 7.37 (dd, 1H, J=8.5,4.4 Hz); 10.64 (s, 1H); прибл. 11-12 (очень br s, 2-3H).
Пример AA-44
N-{6'-фтор-4',9'-дигидро-4-бутил-спиро[циклогексан-1,1'(3'H)-пирпно[3,4-b]индол]-4-ил}-пиперидин (неполярный диастереомер)
К раствору Е-15 (860 мг, 3.6 ммоль) и 2-(5-фтор-1Н-индол-3ил)этанола F-2 (645 мг, 3.6 ммоль) в дихлорметане (70 мл) при охлаждении ледяной водой прикапывали трифторметансульфокислоту (702 мг, 408 µл, 4.68 ммоль). Реакционную смесь перемешивали 20 ч при комнатной температуре, затем смешивали с 0.5 М раствором едкого натра (36 мл) и после этого перемешивали 2.5 ч при комнатной температуре. Органическую фазу отделяли и водную экстрагировали дихлорметаном (3×20 мл). Объединенные органические фазы промывали раствором хлорида натрия (40 мл), высушивали сульфатом натрия и концентрировали в вакууме. Смесь изомеров (1.4 г) отделяли путем флеш-хроматографии (140 г, 23×5.4 см) с этилацетат / циклогексан (1:3→1:2) и после этого с этилацетатом.
Фракция 1 (неполярный диастереоизомер)
Выход: 98 мг (7%), белое твердое вещество
Точка плавления: 126-130°С
1Н-ЯМР (CDCl3): 0.92 (t, 3H, J=6.8 Hz); 1.20-1.83 (m, 18H); 1.99-2.10 (m, 2H); 2.56 (m, 4H); 2.74 (t, 2H, J=5.4 Hz); 3.99 (t, 2H, J=5.4 Hz); 6.89 (dt, 1H, J=9.0, 2.5 Hz); 7.11 (dd, 1H, J=9.5, 2.5 Hz); 7.29-7.25 (m, 1H), 7.62 (s, 1H).
13С-ЯМР (CDCl3):14.2; 22.5; 23.9; 25.4; 27.0; 27.6 (2); 28.0 (2); 30.5 (2); 33.6; 45.7; 56.4; 59.6; 72.6; 103.2 (d, J=23 Hz); 107.3 (d, J=4 Hz); 109.5 (d, J=26 Hz); 111.3 (d, J=10 Hz); 127.6 (d, J=10 Hz); 132.0; 141.6; 157.9 (d, J=234 Hz).
Фракция 2 (полярный диастереоизомер)
Выход: 360 мг (25%), бесцветное масло
1Н-ЯМР (CDCl3): 0.97 (t, 3H, J=6.4 Hz); 1.29-1.80 (m, 18H); 2.63-2.68 (m, 4H); 1.99 (t, 2H, J=11.2 Hz); 2.54-2.63 (m, 4H); 2.74 (t, 2H, J=5.4 Hz); 3.99 (t, 2H, J=5.4 Hz); 6.89 (dt, 1H, J=9.0, 2.4 Hz); 7.12 (dd, 1H, J=9.4, 2.2 Hz); 7.21-7.25 (m, 1H);7.63 (s, 1H).
Пример AA-45
N-{6'-фтор-4',9,-дигидро-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-ил}-пиперидин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) (полярный диастереомер)
К горячему раствору полученного под примером АА-44 полярного диастереомера (фракция 2, 220 мг, 0.55 ммоль) в 2-пропаноле (25 мл) добавляли 0.5 М раствор лимонной кислоты в 2-пропаноле (1.38 мл, 0.69 ммоль). Выпавший в осадок остаток отфильтровывали и высушивали в вакууме.
Выход: 160 мг (49%), белое твердое вещество (АА-45)
Точка плавления: 236-238°С
1Н-ЯМР (ДМСО-d6): 0.98 (t, 3H, J=6.9 Hz); 1.21-2.06 (m, 20Н); 2.56 (d, 2H, J=15.1 Hz); 2.47 (d, 2H, J=15.1 Hz); 2.65 (t, 2H, J=5.1 Hz), 2.90 (br s, 4H), 3.90 (t, 2H, J=5.1 Hz, 2H), 6.89 (ddd, 1H, J=9.6, 8.9, 2.6 Hz); 7.16 (dd, 1H, J=9.9, 2.5 Hz); 7.29-7.35 (m, 1H); 11.03 (s, 1H).
Пример AA-46
N-{6'-фтор-4',9,-дигидро-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-ил}-n-метилпиперазин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:2) (один из двух возможных диастереомеров)
Раствор Е-16 (631 мг, 2.5 ммоль) и 5-фтортриптофол F-2 (449 мг, 2.5 ммоль) в безводном дихлорметане (25 мл) при охлаждении льдом смешивали с трифторметансульфокислотой (900 мг, 530 µл, 6 ммоль) и в течение выходных перемешивали при комнатной температуре. Для контроля взаимодействия отбирали пробу (0.5 мл), которую промывали 0.5 N раствором едкого натра и органическую фазу высушивали сульфатом натрия. После полного взаимодействия реакционную смесь смешивали с 0.5 N раствором едкого натра (10 мл), 2 ч перемешивали при комнатной температуре, водную фазу экстрагировали дихлорметаном (2×20 мл), объединенные органические фазы высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт очищали с помощью флеш-хроматографии (200 г, 20×5.7 см) с метанолом.
Фракция 1:
Выход: 144 мг (14.0%), белое твердое вещество
Точка плавления: 74-81°С
1Н-ЯМР (ДМСО-d6): 0.88 (t, 3H, J=6.7 Hz); 1.14-1.36 (m, 7Н); 1.55 (t, 4Н, J=12.2 Hz); 1.68 (t, 2H, J=12.2 Hz); 2.04 (t, 2H, J=13.0 Hz); 2.23 (s, 3H); 2.42-2.48 (m, 4H); 2.52-2.57 (m, 3H); 2.62 (t, 2H, J=5.4 Hz); 3.88 (t, 2H, J=5.4 Hz); 6.86 (dt, 1H, J=9.3, 2.6 Hz); 7.12 (dd, 1H, J=9.9, 2.5 Hz); 7.37 (dd, 1H, J=8.7, 4.6 Hz); 10.57 (s, 1H).
В добавление к этому получали еще две смешанные фракции 2 & 3 (652 и 213 мг, 84%) в виде желтого масла, которые содержали простой спироэфир и побочный продукт в соотношении в прибл. 9:1.
Раствор фракции 2 & 3 (796 мг, 1.93 ммоль) в кипящем этаноле (15 мл) смешивали с раствором лимонной кислоты (928 мг, 4.8 ммоль) в горячем этаноле (8 мл). Через некоторое время в осадок выпадал белый остаток, который после охлаждения отфильтровывали.
Выход: 675 мг (85%), белое твердое вещество (АА-46)
Точка плавления: 213-220°С
1Н-ЯМР (ДМСО-d6): 0.90 (t, 3H, J=6.9 Hz); 1.15-1.37 (m, 7H); 1.51-1.63 (m, 4H); 1.71 (t, 2H, J=12.8 Hz); 1.99 (t, 2H, J=13.0 Hz); 2.46-2.80 (m, 16H, наложенный от ДМСО-сигнала); 3.12 (br s, 4H); 3.89 (t, 2H, J=5.4 Hz); 6.89 (dt, 1H, J=9.4, 2.6 Hz); 7.15 (dd, 1H, J=9.8, 2.4 Hz); 7.35 (dd, 1H, J=8.7, 4.5 Hz); 10.49 (s, 1H).
13С-ЯМР (ДМСО-d6): 14.0; 22.1; 23.2; 26.5; 26.7 (2C); 29.7 (2C); 34.1; 42.0; 42.8; 44.2 (2C); 54.3; 55.8; 58.7; 71.5; 72.0; 102.5 (d, J=24 Hz); 105.8 (d, J=5 Hz); 108.4 (d, J=26 Hz); 111.8 (d, J=11 Hz); 126.9 (d, J=10 Hz); 132.3; 141.8; 156.8 (d, J=231 Hz); 171.4 (2C); 176.8.
Пример АА-47
4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1), неполярный диастереомер
В примере АА-47 речь идет о цитрате получившегося под примером АА-38 неполярного диастереомера (Фракция 1). Этот цитрат был осажден в соответствии со стандартным способом.
Пример АА-48
6'-гидрокси-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1), неполярный диастереомер
Индол F-3 (2.17 г, 12.2 ммоль) и кетон Е-2 (2.37 г, 12.2 ммоль) помещали в абс. дихлорметан (100 мл) под аргоном, при охлаждении льдом смешивали с ТМС-трифлатом (2.37 мл, 14.4 ммоль) в дихлорметане (5 мл) и 30 мин перемешивали при КТ. Состав перемешивали последующие 16 ч при КТ. Для переработки добавляли H2O (85 мл) и К2СО3 (1.90 г) и перемешивали при КТ 20 мин. Фазы отделяли. Водную фазу экстрагировали дихлорметаном (2×40 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вакууме. Осадок очищали флеш-хроматографией с CHCl3/МеОН (9:1, 1:1, МеОН).
Выход: неполярный диастереомер 1.12 г (26%)
полярный диастереомер 0.911 г (21%)
Только что полученный неполярный диастереомер (991 мг, 2.78 ммоль) растворяли в горячем этаноле и смешивали с лимонной кислотой (529 мг, 2.78 ммоль), растворенной в этаноле (5 мл). Выпавший в осадок остаток отсасывали и высушивали в вакууме.
Выход: 567 мг (38%)
Точка плавления: 240-241°С
1Н-ЯМР (ДМСО-d6): 0.92 (3H, t); 1.29 (4H, m); 1.46 (2H, m); 1.75 (4H, t); 1.85 (2H, t); 2.10 (2H, m); 2.54-2.69 (10H, m); 3.87 (2H; t); 6.54 (1H, d); 6.68 (1H, s); 7.16 (1H, d); 8.51 (1H, s, ОН); 10.53 (1H, s).
13С-ЯМР (ДМСО-d6): 13.91; 22.20; 23.05; 25.90; 26.29; 29.29; 30.65; 37.20; 44.44; 59.06; 60.52; 71.28; 72.08; 101.80; 104.34; 110.52; 111.58; 127.02; 130.11; 139.56; 150.27; 172.05; 177.47.
Пример АА-49
6'-гидрокси-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1), полярный диастереомер
Полученный под примером АА-48 полярный диастереомер (900 мг, 2.52 ммоль) растворяли в горячем этаноле/диоксан (5 мл, 30 мл) (плохорастворимый). Затем лимонную кислоту (480 мг, 2.52 ммоль) растворяли в горячем этаноле (5 мл) и добавляли. Состав охлаждали до КТ, при этом в осадок выпадало мало остатка, поэтому добавляли простой эфир. Выпавший в осадок остаток отсасывали и высушивали в вакууме.
Выход: 874 мг (63%)
Точка плавления: 160-170°С
1Н-ЯМР (ДМСО-d6): 0.97 (3H, t); 1.43 (4H, m); 1.65 (2H, m); 1.92 (9 Н, m); 2.51-2.67 (10H, m); 3.88 (2H; t); 6.58 (1H, d); 6.70 (1H, s); 7.12 (1H, d); 8.56 (1H, s, ОН); 10.63 (1H, s).
13С-ЯМР (ДМСО-d6): 14.00; 22.09; 22.68; 24.48; 24.74; 28.32; 30.91; 37.46; 44.27; 59.13; 64.84; 70.64; 71.1608; 101.96; 104.69; 110.80; 111.17; 127.03; 129.96; 138.62; 150.36; 171.21; 176.86.
Пример АА-50
6'-фтор-4',9'-дигидро-N,N-диметил-4-циклопентилметил-спиро[циклогексан 1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (2:1) один из двух возможных диастереомеров
Кетон Е-17 (223 мг, 1.0 ммоль) и 5-фтор-триптофол (179 мг, 1.0 ммоль) растворяли в абс. дихлорметане (10 мл), под аргоном смешивали с метансульфокислотой (0.1 мл, 1.5 ммоль) и перемешивали при КТ 3 дн. После добавления 1N NaOH (10 мл) и CH2Cl2 (20 мл) перемешивали еще 10 мин, фазы отделяли, водную фазу дважды экстрагировали с помощью CH2Cl2, объединенные органические фазы промывали водой, высушивали (Na2SO4) и концентрировали в вакууме. Оставшийся осадок отделяли флеш-хроматографией с CHCl3/МеОН (20:1).
Выделяли 388 мг твердого вещества, которое согласно ЯМР имелось в виде соли, его растворяли в CH2Cl2, промывали раствором 1N NaOH, высушивали над Na2SO4 и концентрировали в вакууме.
Выход: 310 мг (81%), образовался только 1 диастереомер
1Н-ЯМР (ДМСО-d6): 1.16 (2H, m); 1.51-1.84 (14H, m); 2.05 (2H, m); 2.45 (5H, m); 2.74 (6H, s); 3.90 (2H, m); 6.87 (1H, t); 7.17 (1H, m); 7.26 (1H, m); 8.44 (1H, bs); 11.5(1H, bs). соль?
Только что полученный амин (310 мг, 0.81 ммоль) растворяли в горячем этаноле (10 мл) и смешивали с раствором лимонной кислоты (155 мг, 0.81 ммоль) в горячем этаноле (5 мл). После 2-часового отстаивания в холодильнике образовавшееся твердое вещество отсасывали и высушивали в вакууме.
Выход: 316 мг (81%), образовался полуцитрат.
Точка плавления: 222-223°С
1Н-ЯМР (ДМСО-d6): 1.11 (2H, m); 1.48-1.98 (15H, m); 2.15 (2H, m); 2.58 (6H, s); 2.65 (11H, m); 3.89 (2H, m); 6.83 (1H, m); 7.15 (1H, m); 7.37 (1H, m); 10. (1H, bs); 11.01 (1H, s), полуцитрат.
13С-ЯМР (ДМСО-d6): 20.1; 24.6; 26.3; 29.2; 34.4; 35.6; 36.9; 44.2; 59.1; 61.6; 71.2; 72.5; 102.5; 105.6;108.2; 112.2; 126.5; 132.3; 141.1; 155.6; 157.9; 172.1; 177.3.
Пример АА-51
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-(2-фенилэтенкарбонил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (2:1) неполярный диастереомер
Хлорид коричной кислоты (441 мг, 2,65 ммоль) растворяли под аргоном в абс. тетрагидрофуране (30 мл) и при комнатной температуре смешивали со свободным основанием полученного под примером АА-9 неполярного спироамина (300 мг, 0,88 ммоль), растворенного в абс. тетрагидрофуране (15 мл), в течение 20 мин. При этом крепкого осаждения не образовывалось. После продолжения реакции в 1,5 ч реакционную смесь разбавляли с водой (10 мл), при охлаждении льдом смешивали с 1N раствором едкого натра (10 мл) и потом перемешивали 2 ч. Тетрагидрофуран удаляли в вакууме. При этом в осадок выпадало твердое вещество, которое отделяли при помощи фильтрации и промывали водой (3×10 мл). Сырой продукт (408 мг) отделяли хроматографически [силикагель 60 (50 г); этилацетат (500 мл)]. Неполярный амид выделяли в виде бесцветного твердого вещества с выходом в 76% (314 мг).
Только что полученный неполярный амид (296 мг, 0,63 ммоль) при 80°С суспендировали в этаноле (14 мл) и смешивали с этанольным раствором (3 мл) лимонной кислоты (133 мг, 0,69 ммоль). Из прозрачного раствора при охлаждении до комнатной температуры в осадок выпадало твердое вещество. Перемешивали 16 ч при комнатной температуре. Смесь 2 ч выдерживали при 5°С. Бесцветное твердое вещество отделяли при помощи фильтрации и промывали диэтиловым эфиром (3×3 мл).
Неполярный цитрат АА-51 получали таким образом с выходом в 85% (302 мг) в виде полуцитрата с точкой плавления в 154-157°С.
13С-ЯМР (101 MHz, ДМСО-D6) d част. на млн: (неполярный диастереоизомер) 13.9, 22.2, 23.0, 26.2, 27.5, 29.5, 30.6, 37.3, 42.4, 44.0, 59.0, 71.6, 105.9, 111.3, 117.5, 118.4, 120.6, 123.1, 126.2, 127.8, 128.7, 129.3, 135.1, 135.5, 139.9, 140.2, 169.4, 171.4, 176.6
Пример АА-52
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-(2-фенилэтенкарбонил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин, 2-гидрокси-1,2,3-пропантрикарбоксилат (1:1) полярный диастереомер
Хлорид коричной кислоты (С, 441 мг, 2,65 ммоль) растворяли под аргоном в абс. тетрагидрофуране (30 мл) и при комнатной температуре смешивали со свободным основанием полученного под примером АА-9 полярного спироамина (300 мг, 0,88 ммоль, растворенный в абс. тетрагидрофуране (15 мл), в течение 20 мин. При этом образовалось легкое осаждение. После продолжения реакции в 1,5 ч реакционную смесь разбавляли с водой (10 мл), при охлаждении льдом смешивали с 1N раствором едкого натра (10 мл) и 1 ч перемешивали. Тетрагидрофуран удаляли в вакууме. При этом в осадок выпадало твердое вещество, которое при помощи фильтрации отделяли и промывали водой (3×10 мл). Сырой продукт (384 мг) отделяли хроматографически [силикагель 60 (50 г); этилацетат/метанол 1: 4 (750 мл)]. Полярный амид выделяли в виде твердого вещества бежевого цвета с выходом в 43% (177 мг).
13С-ЯМР (101 MHz, CDCl3) d част. на млн: (полярный диастереоизомер) 14.0, 22.4, 23.5, 25.9, 27.3, 31.5, 31.6, 37.8, 43.3, 58.8, 106.7, 111.8, 117.6, 119.3, 121.6, 122.1, 126.6, 127.7, 128.8, 129.6, 135.0, 136.0, 138.8, 141.9, 170.9
Только что полученный полярный амид (157 мг, 0,334 ммоль) растворяли в этаноле (5 мл) и смешивали с этанольным раствором (2 мл) лимонной кислоты (72 мг, 0,37 ммоль). Перемешивали 16 ч при комнатной температуре, причем не наблюдалось осаждения. Смесь концентрировали, ресуспендировали в этаноле (2 мл) и медленно смешивали с диэтиловым эфиром (30 мл). Через 1,5 ч отделяли бесцветное твердое вещество при помощи фильтрации и промывали диэтиловым эфиром (3×3 мл). Полярный цитрат АА-52 таким образом получали с выходом в 73% (161 мг).
Сравнительные исследования растворимости:
Для определения растворимости соединений использовали серию опытов посредством разбавления раствора 20 мг/мл в ДМСО с водным буферным раствором. При прохождении лекарственных средств через пищеварительный тракт лекарственные средства принимают различные значения pH. В желудке ожидаются значения pH в 1-3, и по окончании прохождения желудка в кишечнике значения pH ожидаются в пределах 6-8. Так как растворимости могут быть pH-зависимыми, то применяют водные буферы при различных значениях pH (pH 1, 100 mM HCl; pH 2, 10 тМ HCl; pH 4, 50 mM лимонной кислоты, титрованной с 1 N NaOH; pH 6, 50 mM цитрат натрия, титрованный с 1 N HCl; pH 7, 50 mM трис.HCl; pH 8, трис.HCl), которые при комнатной температуре удерживают установленные значения pH в окончательном растворе.
Так как ДМСО при возрастающей концентрации способствует образованию метастабильных перенасыщенных водных растворов, то концентрированные растворы разбавляли 1:100 в водном буфере. Растворы взбалтывали в закрытых сосудах, по меньшей мере, 15 часов. При этом разбавляли 10 µл ДМСО концентрированного раствора в 990 µл водном буфере и в пригодных сосудах (например, Эппендорф-сосудах), так что концентрация была постоянной при 1% v/v. Затем растворы центрифугировали и пробы прозрачной жидкой фракции переводили в пробоотборные сосуды, два эквивалента содержали 50% ацетонитрил в 0,1 N HCl.
Калибровочные растворы получали путем разбавления ДМСО-концентрированных растворов в метаноле (1:100). Из этих разбавлений получали другие разбавленные растворы в метаноле (1:100, 1:200, 1:400 и 1:800). Пробы анализировали посредством ОФ ВЭЖХ с УФ-детектированием. Посредством регрессионного анализа выводили линейные калибровочные уравнения, в общем, с коэффициентом корреляции в более 0,95. С помощью экспериментально установленных калибровочных проверок определяли концентрации соединений в буферных растворах. Посредством описанного эксперимента можно было определить максимальные концентрации в 200 µг/мл вещества в буфере. Более высокая растворимость не смогла быть квантифицирована.
Корреляция между растворимостью и вариацией R3 была показана при помощи нижеследующих соединений.
R1, R2=СН3; R5, R6, R7, R9, R10=Н, R8=F, X=О
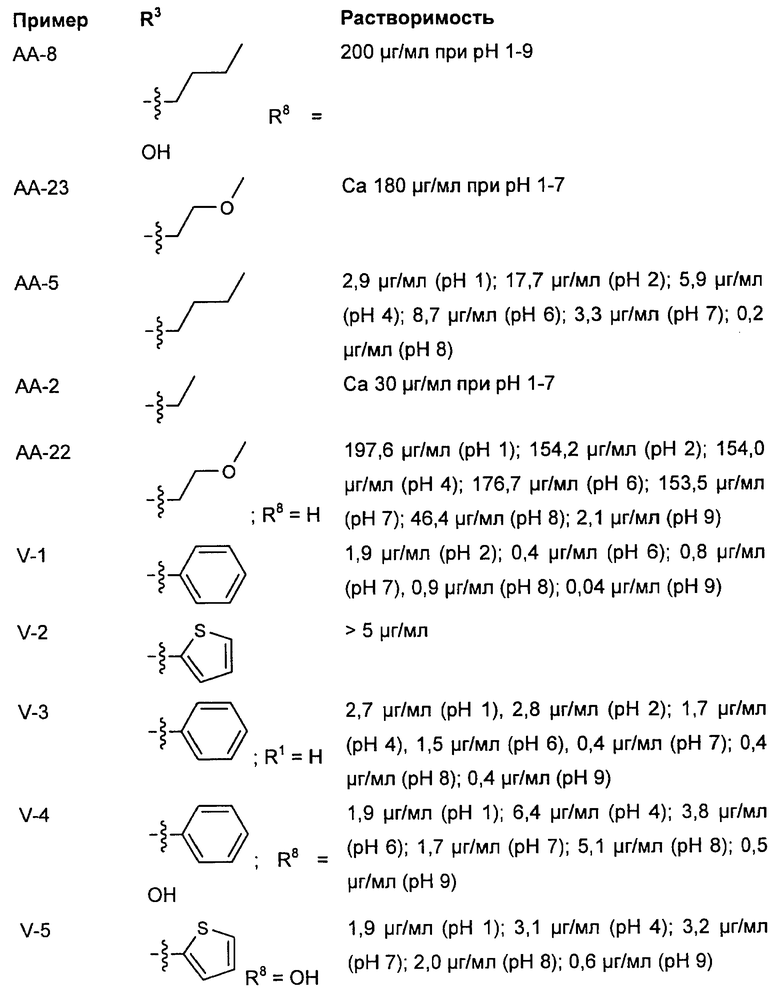
Соединения согласно изобретению обладают чрезвычайно высоким сродством к ORL1 соответственно к µ-опиоидному рецептору. Сродство находится в подобном порядке величины как оба сравнительных соединения. Однако они не обладают повышенной растворимостью.
Между pH 1 и pH 8 наблюдалась незначительная pH-зависимость растворимости. Если pH ниже 8, то растворимость соединений снижается.
Исследования эффективности соединений согласно изобретению:
Собранные в нижеследующих анализах и моделях данные приведены в таблице 1.
Измерение ORL1-связывания
Производные циклогексана общей формулы I были исследованы в анализе связывания рецепторов с 3Н-ноцицептин/орфанин FQ с мембранами рекомбинантных CHO-ORL1 клеток. Эта тестовая система была проведена согласно методу, представленному в Ardati et al. (Mol. Pharmacol., 51, 1997, с.816-824). Концентрация 3Н-ноцицептин/орфанин FQ при этом исследовании составила 0.5 нМ. Анализы связывания были проведены с 20 µг мембранного протеина по 200 µл состава в 50 мМ Hepes, pH 7,4, 10 мМ MgCl2 и 1 mM EDTA. Связывание к ORL1-рецептору определяли с применением по 1 мг WGA-SPA Beads (Amersham-Pharmacia, Фрайбург), путем одночасовой инкубации состава при КТ и последующего измерения в сцинтилляционном счетчике Trilux (Wallac, Финляндия). Сродство указано в таблице 1 как наномолярное Ki-значение в или % ингибирования при с=1 µМ.
Измерение µ-связывания
Сродство рецепторов к человеческому µ-опийному рецептору определяли в гомогенном составе в микротитрационных планшетах. Для этого инкубировали разбавленные серии соответствующих проверяемых соединений с рецепторным мембранным препаратом (15-40 µг протеина на 250 µл инкубационного состава) СНО-К1-клеток, которые выражают человеческий µ-опийный рецептор (RB-HOM-рецепторный мембранный препарат фирмы NEN, Zaventem, Бельгия) в присутствии 1 нмоль/л радиоактивного лиганда [3Н]-налоксон (NET719, фирма NEN, Zaventem, Бельгия), а также 1 мг WGA-SPA-Beads (Wheat germ agglutinin SPA Beads фирмы Amersham/Pharmacia, Фрайбург, Германия) в общем объееме 250 µл в течение 90 минут при комнатной температуре. В качестве инкубационного буфера применяли 50 ммоль/л трис-HCl дополненный с 0,05 мас.% азида натрия и с 0,06 мас.% альбумина сыворотки крупного рогатого скота. Для определения неспецифического связывания дополнительно добавляли 25 µмоль/л налоксон. После окончания девяностоминутного инкубационного периода микротитрационные планшеты отделяли центрифугированием и в течение 20 минут при 1000 г и радиоактивность измеряли в β-счетчике (Microbeta-Trilux, фирма PerkinElmer Wallac, Фрайбург, Германия). Определяли процентное вытеснение радиоактивного лиганда из его связывания к человеческому µ-опийному рецептору при концентрации контрольного вещества в 1 µмоль/л и указывали как процентное ингибирование (% ингибирования) специфического связывания. Частично исходя из процентного вытеснения, благодаря различным концентрациям контрольных соединений общей формулы I IC50, рассчитывали ингибирующие концентрации, которые вызывали 50-процентное вытеснение радиоактивного лиганда. Путем пересчета с помощью соотношения Чена-Прусоффа (Cheng-Prusoff) получали Ki-значения для контрольных веществ.
Проверка аналгезии в тесте «отдергивание хвоста» (Tail-Flick) на мышах
Мышей каждый раз по одной помещали в клетку для проведения испытаний и основание хвоста подвергали фокусирующему тепловому излучению электрической лампы (Tail-flick-Typ 50/08/1.bc, Labtec, Dr. Hess). Интенсивность лампы устанавливали таким образом, что время от включения лампы до внезапного отдергивания хвоста (болевая латентность) у неподвергшихся обработке мышей составило от 3 до 5 секунд. Перед введением растворов, содержащих соединение согласно изобретению, или соответствующих стандартных растворов мыши в течение пяти минут дважды подвергались предварительному тесту, и среднее значение этих измерений подсчитывали как среднее значение предварительного теста.
Растворы соединения общей формулы I согласно изобретению, а также стандартные растворы потом вводили внутривенно. Измерение боли проводили каждые 10, 20, 40 и 60 минут после внутривенного введения.
Анальгезирующее действие определяли как возрастание болевой латентности (% максимально возможного антиноцицептивного эффекта) согласно нижеследующей формуле:
[(Т1-Т0)/(Т2-Т0)]×100.
При этом время Т0 представляет собой латентный период перед введением, время T1 является латентным периодом после применения комбинации действующих веществ и время Т2 максимальную выдержку (12 секунд).
В двух случаях тест проводили аналогичным образом на крысах.
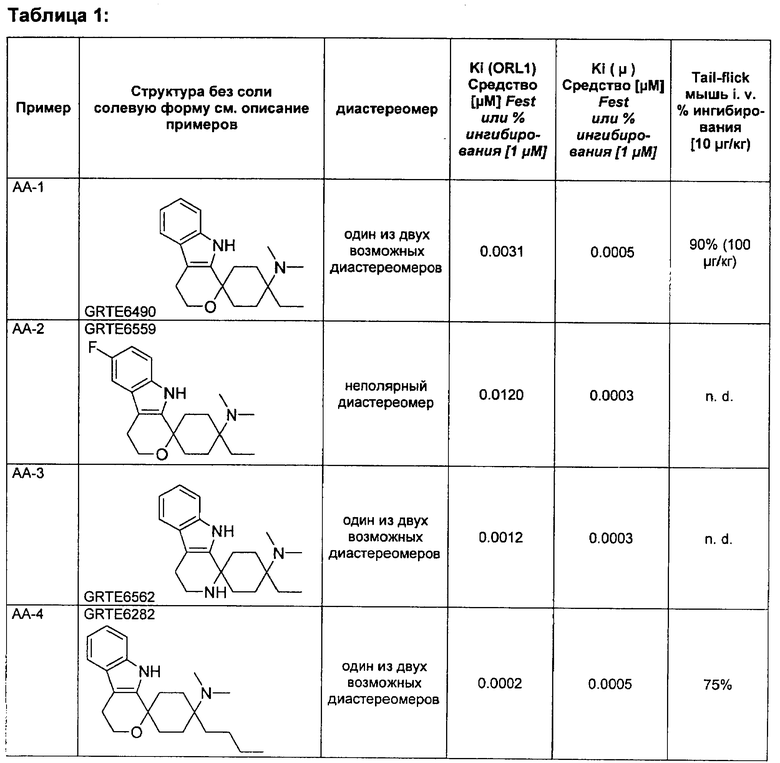
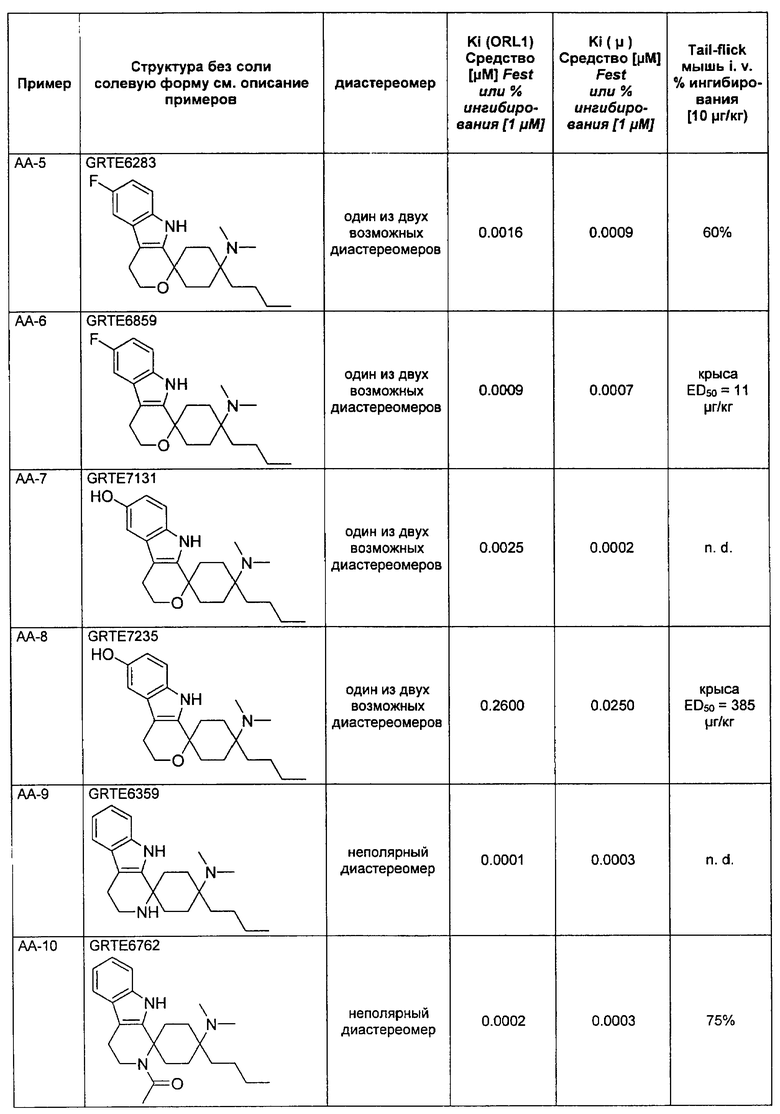
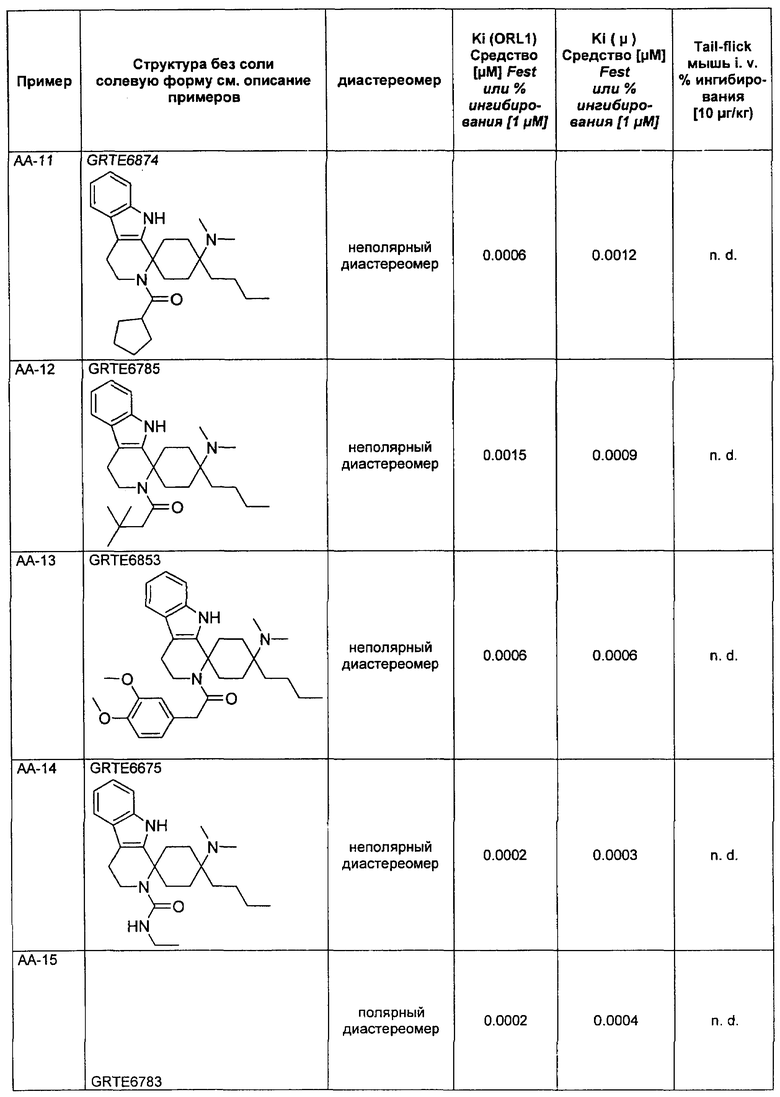
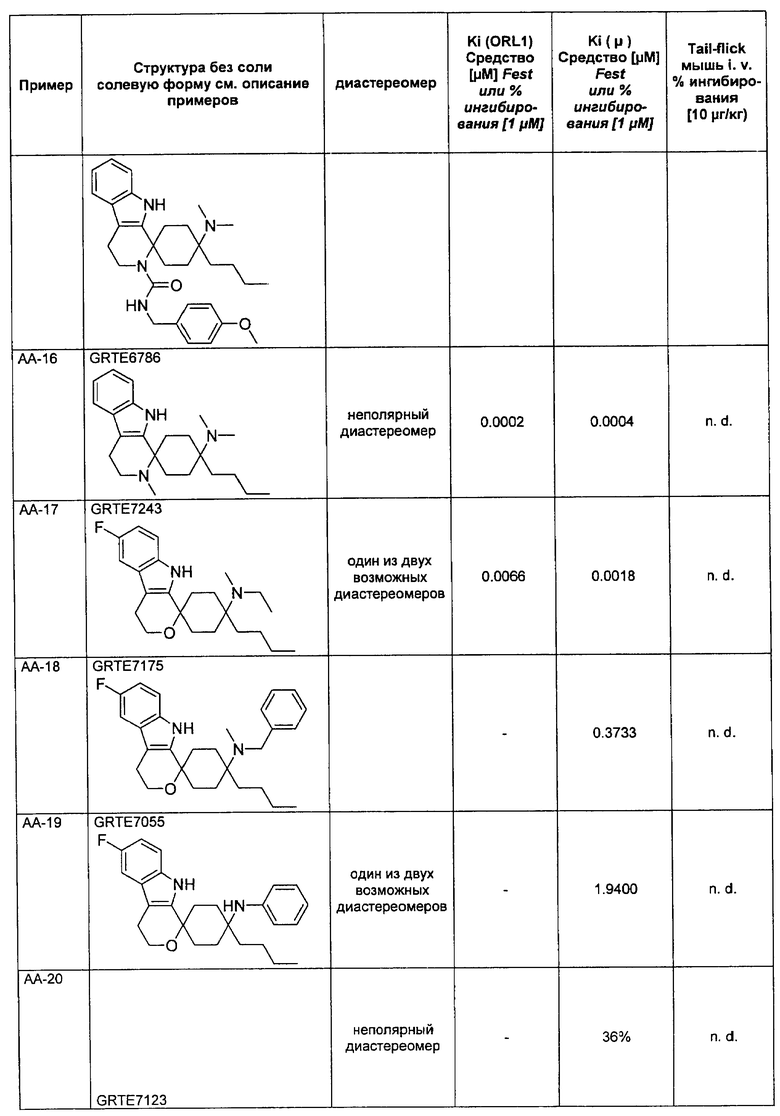
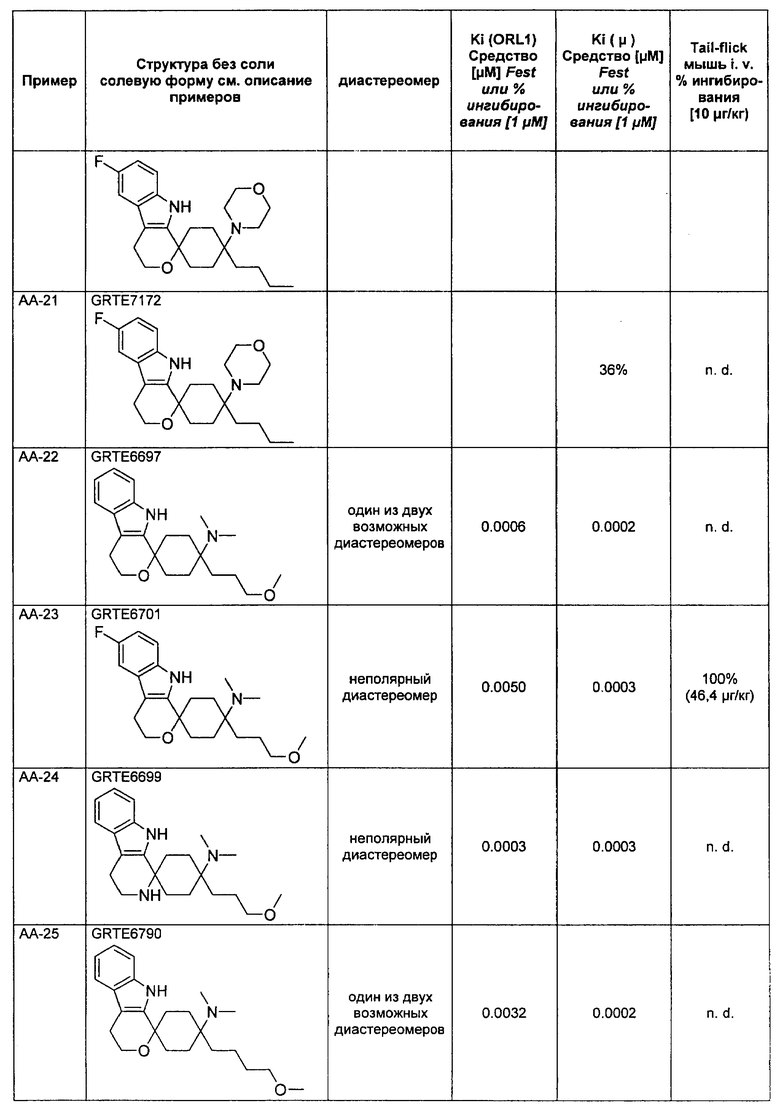
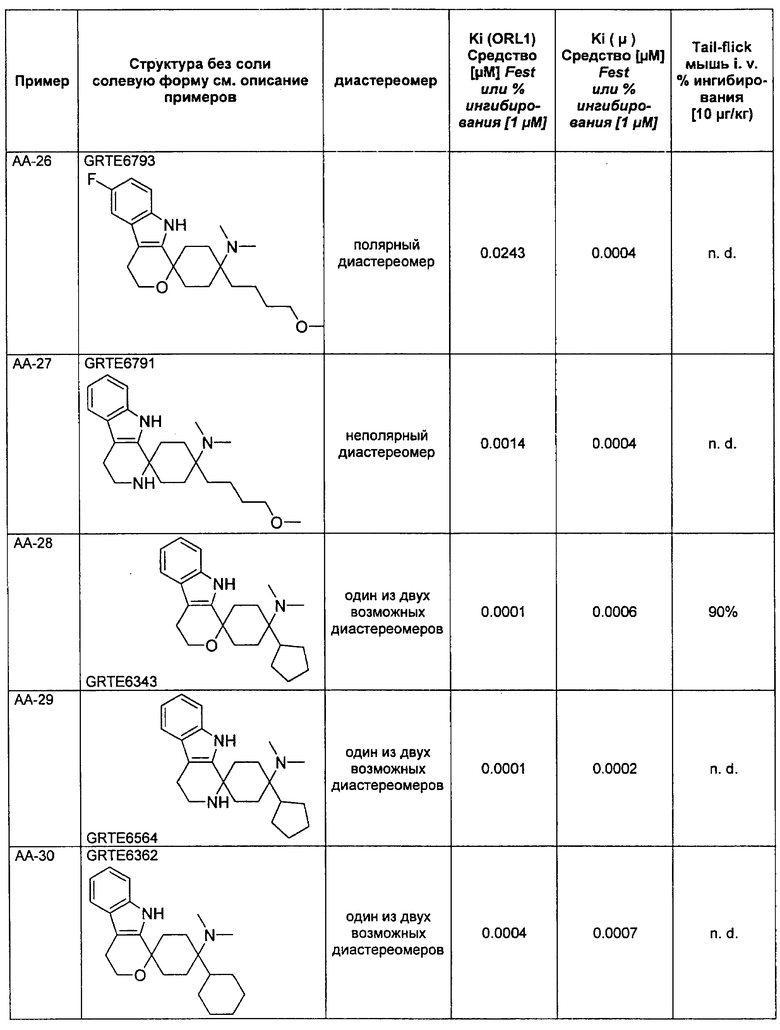
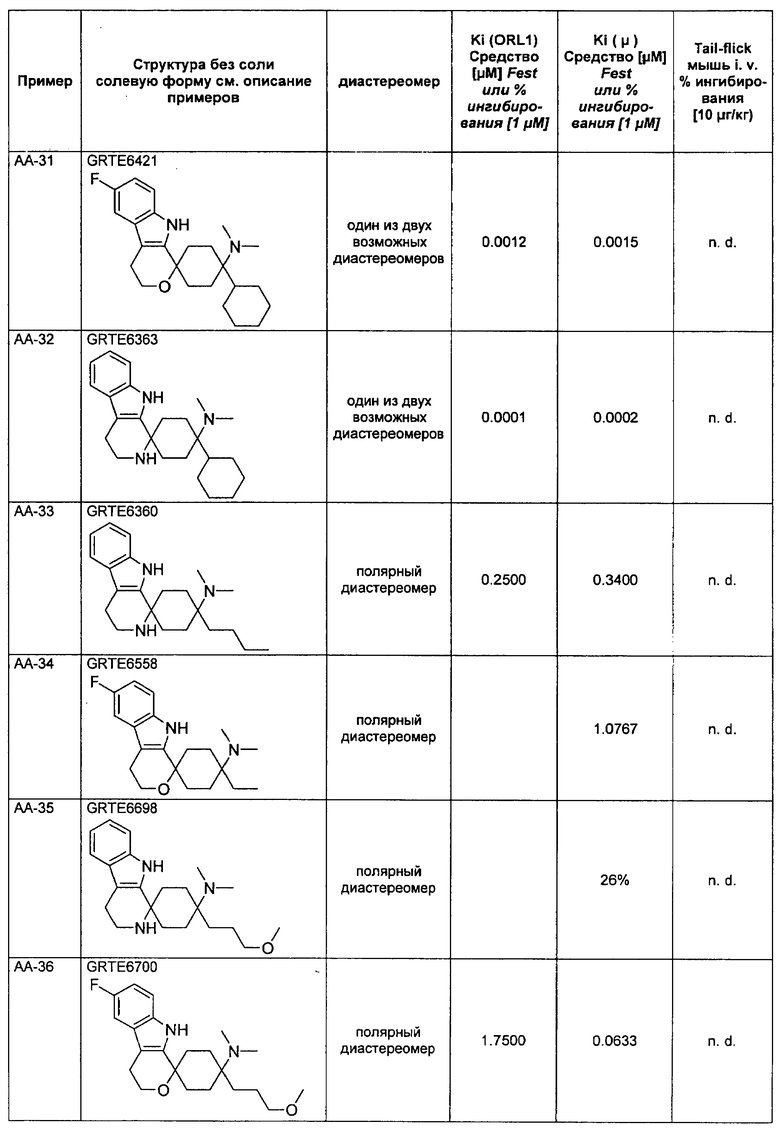
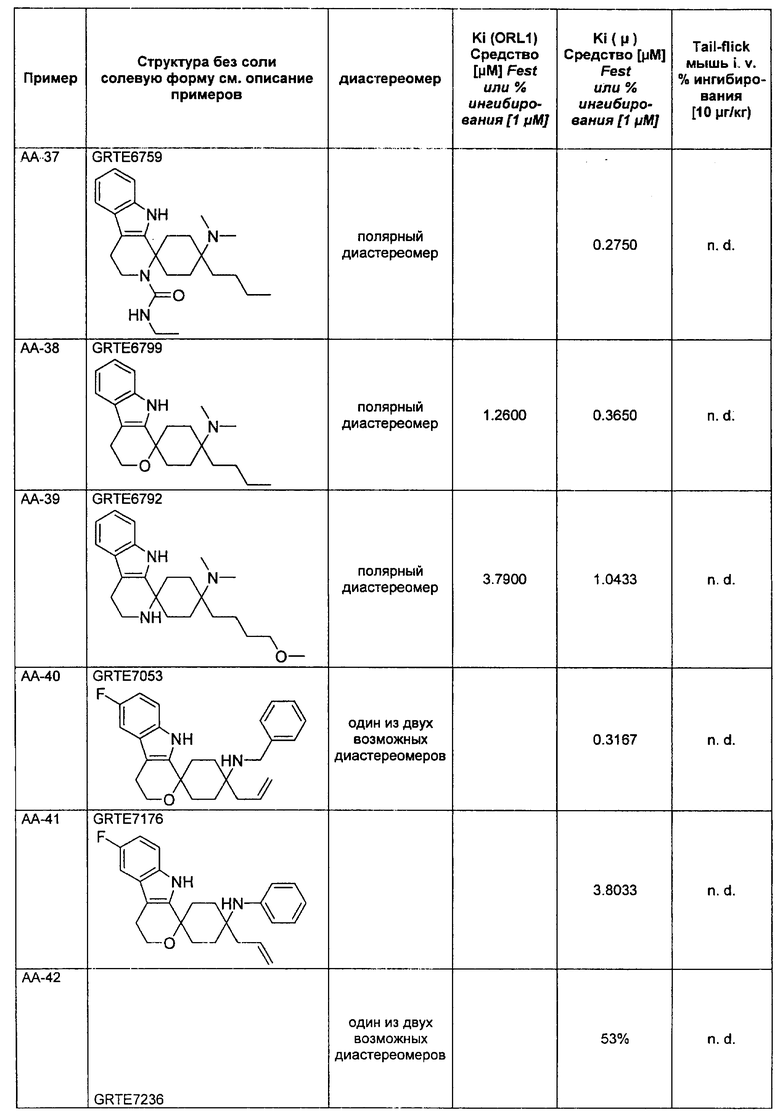
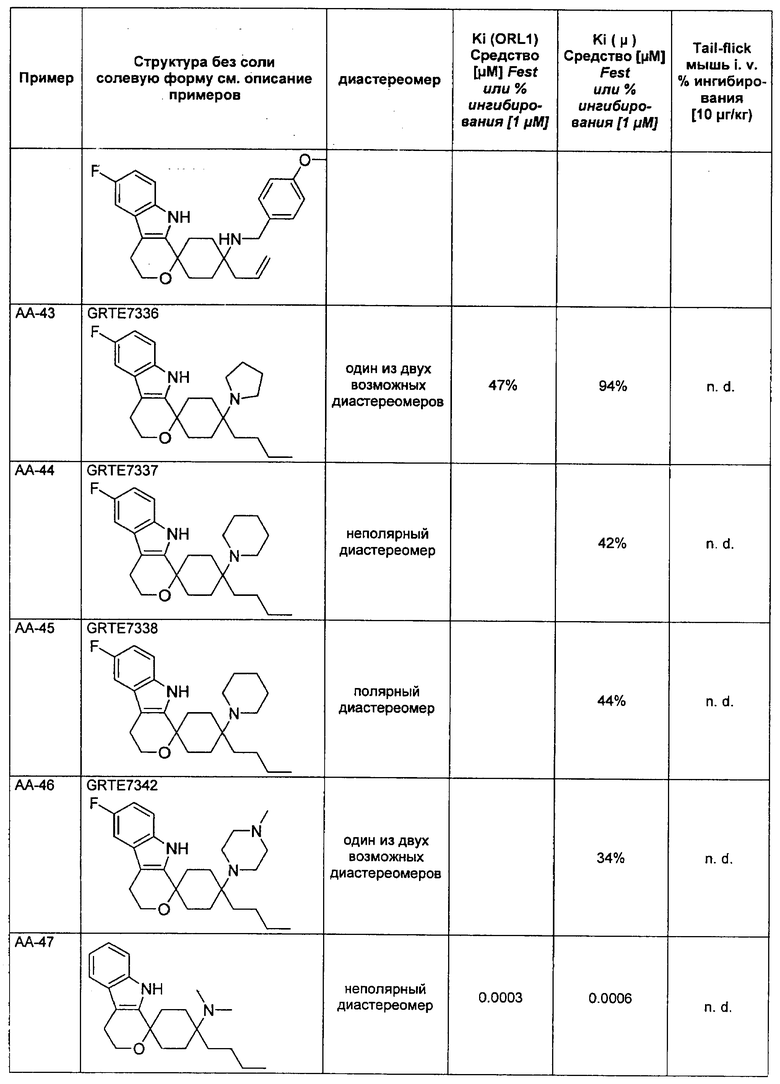
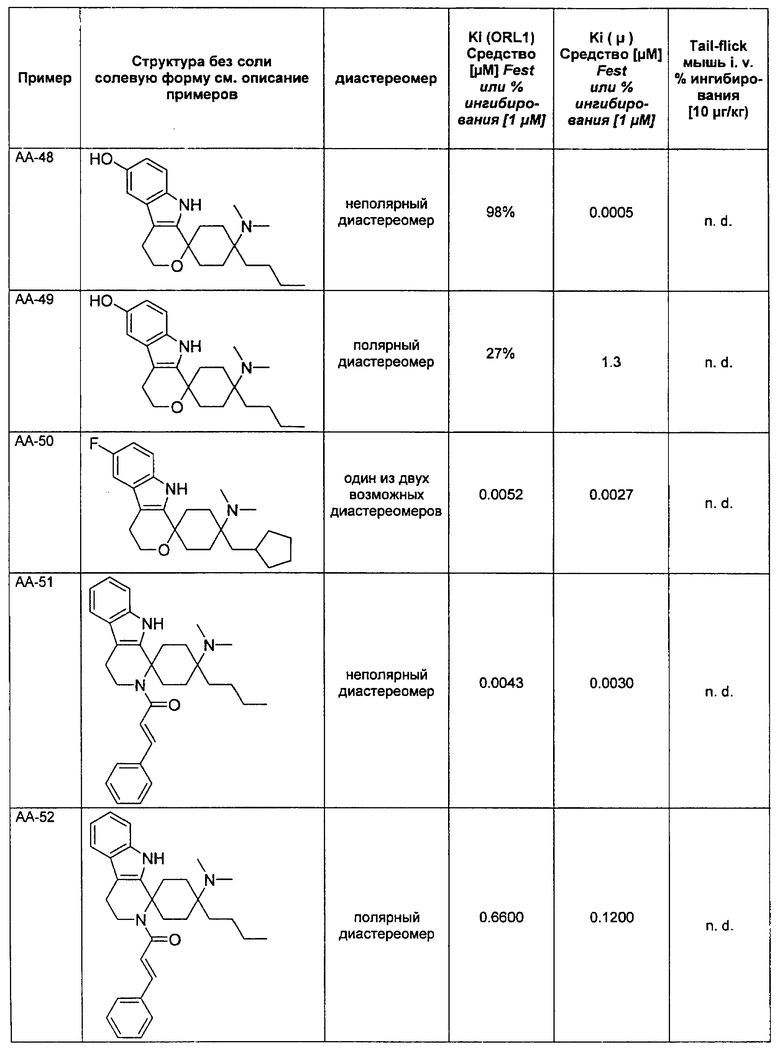
Парентеральный раствор спироциклического производного циклогексана согласно изобретению
38 г одного из спироциклического производного циклогексана согласно изобретению, здесь пример 3, при комнатной температуре растворяли в 1 л воды для инъекционных целей и затем путем добавления безводной глюкозы для инъекционных целей устанавливали до изотонных условий.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПИРОЦИКЛИЧЕСКИЕ АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ | 2007 |
|
RU2468028C2 |
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 2008 |
|
RU2470933C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2532545C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2503660C2 |
| ПРОИЗВОДНЫЕ СПИРО(5.5)УНДЕКАНА | 2009 |
|
RU2515895C2 |
| ДИСПИРОПИРРОЛИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2012 |
|
RU2612534C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ГИДРОКСИМЕТИЛЦИКЛОГЕКСИЛАМИНЫ | 2009 |
|
RU2514192C2 |
| 5-ЗАМЕЩЕННЫЕ ИНДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2008 |
|
RU2487873C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ C-KIT | 2016 |
|
RU2754858C2 |
Изобретение относится к спироциклическим производным циклогексана формулы I
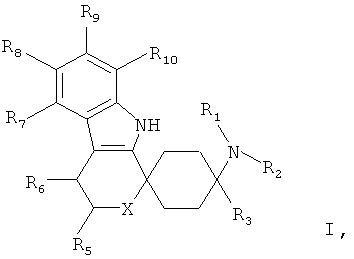
где значения R1-10 приведены в пункте 1 формулы. Соединения обладают сродством к ORL1 рецептору, что позволяет использовать их для получения лекарственного средства для лечения боли, в особенности острой, невропатической или хронической боли. Описан способ получения соединений I. 5 н. и 7 з.п. ф-лы, 1 табл, 52 пр.
1. Спироциклические производные циклогексана общей формулы I
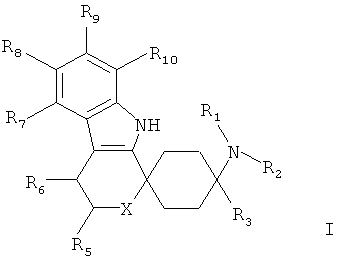
в которой R1 и R2 независимо друг от друга означают Н; C1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный; С3-8-циклоалкил, каждый раз насыщенный; фенил, незамещенный или монозамещенный ОСН3; или связанный С1-3-алкилом фенил, незамещенный или монозамещенный ОСН3; или связанный С1-3-алкилом С3-8-циклоалкил, каждый раз насыщенный и незамещенный; или остатки R1 и R2 вместе означают СН2СН2ОСН2СН2, CH2CH2NR11CH2CH2 или (CH2)3-6; причем R11 означает С1-5-алкил, каждый раз насыщенный, разветвленный или неразветвленный и незамещенный;
R3 означает C1-8-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный ОСН3 или незамещенный; С3-8-циклоалкил, каждый раз насыщенный и незамещенный; или связанный C1-3-алкилом С3-8-циклоалкил, каждый раз насыщенный и незамещенный;
R5 означает Н;
R6 означает Н; F, Cl или OR13;
R7, R8, R9 и R10 независимо друг от друга означают Н, F, Cl, Br, I или OR13; причем R13 означает Н;
R14 и R15 независимо друг от друга означают Н, C1-5-алкил или связанный C1-3-алкилом фенил, монозамещенный ОСН3;
Х означает О или NR17;
R17 означает Н; С1-5алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный; или COR12;
причем R12 означает Н; C1-5-алкил, каждый раз насыщенный, разветвленный или неразветвленный; С3-8-циклоалкил, каждый раз насыщенный и незамещенный; фенилэтенил; связанный С1-3-алкилом фенил, где фенил может быть монозамещенным или полизамещенным ОСН3; или NR14R15;
за исключением соединения 2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амина (неполярный диастереомер);
в виде рацемата; энантиомеров, диастереомеров, смесей энантиомеров или диастереомеров или отдельного энантиомера или диастереомера; оснований и/или солей физиологически совместимых кислот.
2. Спироциклические производные циклогексана по п.1, где R1 и R2 независимо друг от друга означают Н; С1-5алкил, разветвленный или неразветвленный, насыщенный или ненасыщенный; или фенил или бензил, незамещенный или монозамещенный ОСН3; или остатки R1 и R2 совместно означают (СН3)3-6.
3. Спироциклические производные циклогексана по п.2, где R1 и R2 означают Н или СН3, причем R1 и R2 не одновременно означают СН3, в особенности СН3.
4. Спироциклические производные циклогексана по п.1, где R3 означает этил, n-пропил, 2-пропил, аллил, n-бутил, изобутил, втор-бутил, трет-бутил, n-пентил, изопентил, нео-пентил, n-гексил, каждый раз монозамещенные ОСН3; циклопентил или циклогексил.
5. Спироциклические производные циклогексана по п.1, где R6 может означать Н.
6. Спироциклические производные циклогексана по п.1, где Х означает О.
7. Спироциклические производные циклогексана по п.1, где Х означает NR17.
8. Спироциклические производные циклогексана по п.1, в которых R17 означает
COR12 и R12 означает Н; С1-5-алкил; С3-8-циклоалкил; или связанный С1-3-алкилом фенил, где фенил может быть монозамещенным или полизамещенным ОСН3; NR14R15.
9. Спироциклические производные циклогексана по п.1, выбранные из группы, включающей 4',9'-дигидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-этил-спиро[циклогексан-1, 1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-гидрокси-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2,2,2-трифторацетат,
6'-гидрокси-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b] индол]-4-амин-2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-метилкарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-циклопентилкарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-(2,2)-диметилпропанкарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-(3,4-диметоксибензилкарбонил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-этиламинокарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-4-метоксибензиламинокарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин,
2',3',4',9',-тетрагидро-N,N-диметил-4-бутил-2'-метил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N-этил-N-метил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N-бензил-N-метил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин,
6'-фтор-4',9'-дигидро-N-фенил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин,
4-бутил-6'-фтор-4-(N-морфолино)-1',3',4',9'-тетрагидроспиро[циклогексан-1,1'-пирано [3,4-b]индол],
4-бутил-6'-фтор-4-(N-морфолино)-1',3',4',9'-тетрагидроспиро[циклогексан-1,1'-[3,4-b]индол],
4',9'-дигидро-N,N-диметил-4-метоксипропил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N,N-диметил-4-метоксипропил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-(3-метоксипропил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
4',9'-дигидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
4',9'-дигидро-N,N-диметил-4-циклопентил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-циклопентил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
4',9'-дигидро-N,N-диметил-4-циклогексил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N,N-диметил-4-циклогексил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-циклогексил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N,N-диметил-4-этил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-(3-метоксипропил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N,N-диметил-4-метоксипропил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3,'4,',9'-тетрагидро-N,N-диметил-4-бутил-2'-этиламинокарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин,
4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-(4-метоксибутил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N-бензил-4-аллил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N-фенил-4-аллил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин,
6'-фтор-4',9'-дигидро-N-(4-метоксибензил)-4-аллил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин,
N-{6'-фтор-4',9'-дигидро-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-ил}-пирролидин 2-гидрокси-1,2,3-пропантрикарбоксилат,
N-{6'-фтор-4',9'-дигидро-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-ил}-пиперидин,
N-{6'-фтор-4',9'-дигидро-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-ил}-пиперидин 2-гидрокси-1,2,3-пропантрикарбоксилат,
N-{6'-фтор-4',9'-дигидро-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-ил}-n-метилпиперазин 2-гидрокси-1,2,3-пропантрикарбоксилат,
4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-гидрокси-4',9'-дигидро-N,N-диметил-4-бутил-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
6'-фтор-4',9'-дигидро-N,N-диметил-4-циклопентилметил-спиро[циклогексан 1,1'(3'Н)-пирано[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат,
2',3',4',9'-тетрагидро-N,N-диметил-4-бутил-2'-(2-фенилэтенкарбонил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин 2-гидрокси-1,2,3-пропантрикарбоксилат, при необходимости также их смеси.
10. Способ получения спироциклических производных циклогексана по одному из пп.1-9, отличающийся тем, что исходный продукт общей формулы Е
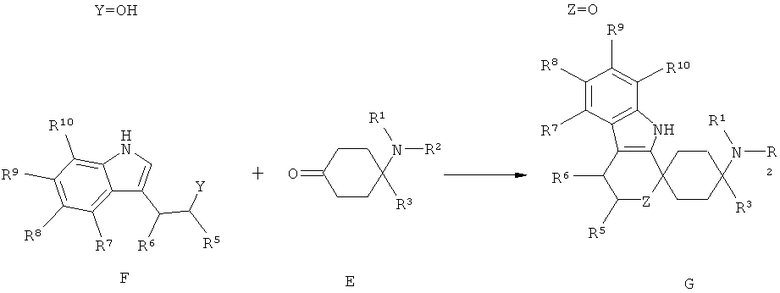
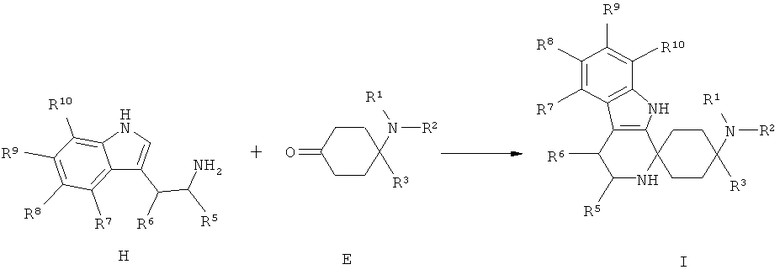
при добавлении кислоты, или ее триметилсилилового эфира, например триметилсилилового эфира трифторметансульфокислоты, трифторметансульфокислоты, уксусной кислоты, фосфорной кислоты, метансульфокислоты или трифторуксусной кислоты, в пригодном растворителе, например дихлорэтане, дихлорметане, хлороформе, ацетонитриле, диэтиловом эфире или нитрометане, подвергают взаимодействию с исходным продуктом общей формулы F или Н, причем остатки R1-R3 и R5-R10 имеют значения, указанные в п.1.
11. Лекарственное средство, обладающее сродством к ORL1-рецептору, содержащее по меньшей мере одно спироциклическое производное циклогексана по одному из пп.1-9, а также при необходимости содержащее пригодные добавки и/или вспомогательные вещества.
12. Применение спироциклического производного циклогексана по одному из пп.1-9 для получения лекарственного средства для лечения боли, в особенности острой, невропатической или хронической боли.
13. Применение спироциклического производного циклогексана по одному из пп.1-9 для получения лекарственного средства для лечения состояний страха, стресса и связанных со стрессом синдромов, депрессий, эпилепсии, болезни Альцгеймера, старческого слабоумия, общих познавательных дисфункций, нарушений обучения и памяти (как ноотроп), синдромов отмены, злоупотребления и/или алкогольной, и/или наркотической, и/или медикаментозной зависимости, сексуальных дисфункций, сердечно-сосудистых заболеваний, гипотонии, гипертензии, тиннитуса, зуда, мигрени, тугоухости, недостаточной подвижности кишечника, нарушенного приема пищи, анорексии, ожирения, локомоторных расстройств, диареи, кахексии, недержания мочи соответственно как мышечный релаксант, противосудорожное средство или анестетик соответственно для совместного приема при лечении с опиоидным анальгетиком или с анестетиком, для диуреза или антинатрийуреза, анксиолизиса, для модуляции двигательной активности, для модуляции распределения нейромедиаторов и лечения связанных с этим нейродегенеративных заболеваний, для лечения синдромов отмены и/или для снижения наркотического потенциала опиоидов.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| RU 2006110519 A, 27.07.2006. | |||

