Настоящее изобретение относится к замещенным спироциклическим азаиндольным производным, к способу их получения, лекарственным средствам, содержащим эти соединения, и к применению замещенных спироциклических азаиндольных производных для получения лекарственных средств.
Лечение хронических и нехронических болевых состояний для медицины имеет большое значение. Во всем мире существует потребность в хорошо действующей болевой терапии. Неотложная потребность действий для удовлетворяющего пациентов и целенаправленного лечения хронических и нехронических болевых состояний, причем под этим следует понимать успешное и удовлетворительное лечение болей пациентов, документируется в большом количестве научных работ, которые в последнее время появились в области применяемых анальгетиков или фундаментального исследования ноцицепции.
Классические µ-опиоиды, такие как морфин, при лечении сильных и сильнейших болей являются хорошо действующими и для болевой терапии имеют огромное значение, если наряду с µ-опиоидным рецептором также оказывают влияние другие опиоидные рецепторы, в особенности ORL-1-рецептор, так как чистые µ-опиоиды также обладают нежелательными побочными действиями, такими как запор и дыхательная депрессия, а также могут приводить к зависимости. Опиоидные рецепторы δ, κ и ORL-1 также участвуют в болевой чувствительности (Opioids: Introduction, cc.127-150, Further Opioid Receptors, 455-476, в: Analgesics - From Chemistry and Pharmacology to Clinical Application, Wiley VCH, 2002).
Кроме того, ORL1-рецептор еще участвует в регуляции других физиологических и патофизиологических процессов. Сюда же среди прочего относятся обучение и формирование памяти (Manabe et al., Nature, 394, 1997, S.577-581), способность слышать (Nishi et al., EMBO J., 16, 1997, S.1858-1864), а также другие многочисленные процессы. В обзорной статье Calo et al. (Br. J. Pharmacol., 129, 2000, 1261-1283) предоставлен обзор показаний или биологических процессов, для которых ORL1-рецептор имеет значение или мог бы иметь с большой вероятностью. В частности, упоминаются: аналгезия, стимулирование и регулирование приема пищи, влияние на µ-агонистов, таких как морфин, лечение явлений синдрома отмены, снижение наркотического потенциала опиоидов, анксиолизис, модуляция двигательной активности, нарушения памяти, эпилепсия; модуляция нейромедиаторного распределения, в особенности глутамата, серотонина и допамина, и вместе с этим нейродегенеративные заболевания; влияние сердечно-сосудистой системы, возникновение эрекции, диурез, антинатрийурез, электролитный баланс, артериальное кровяное давление, заболевания, связанные с задержкой воды, желудочно-кишечная подвижность (диарея), расслабляющие эффекты на дыхательные пути, мочеиспускательный рефлекс (недержание мочи). Далее обсуждается применение агонистов и антагонистов в качестве аноретиков, анальгетиков (также при совместном приеме с опиоидами) или ноотропов.
Из уровня техники (WO 04043967) известны сходные по структуре соединения, которые обладают сродством к ORL-1-рецептору и к µ-опиоидному рецептору. Однако в этих соединениях ароматический гетероцикл представляет собой индольное кольцо, в котором ни один атом углерода не может быть замещен атомом азота.
Задача настоящего изобретения состояла в том, чтобы предоставить в распоряжение другие лекарственные средства, которые оказывают влияние на систему опиоидного рецептора и тем самым пригодны для лекарственных средств, в особенности для лечения различных связанных с этой системой заболеваний, соответственно для применения при непосредственно связанных с этим показаниях.
Поэтому объектом изобретения являются замещенные спироциклические азаиндольные производные общей формулы I,
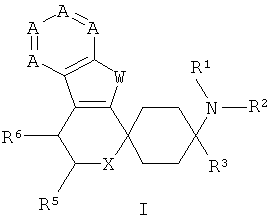
в которой
А означает N или CR7-10, причем А по меньшей мере один раз и самое большее дважды означает N,
W означает NR4,
Х означает NR17, О или S,
R1 и R2 независимо друг от друга означают Н, C1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный, или полизамещенный, или незамещенный; С3-8-циклоалкил, каждый раз монозамещенный, или полизамещенный, или незамещенный; или связанный C1-3-алкилом арил, С3-8-циклоалкил или гетероарил, каждый раз монозамещенный, или полизамещенный, или незамещенный;
или остатки R1 и R2 вместе означают CH2CH2OCH2CH2, CH2CH2NR11CH2CH2 или (СН2)3-6,
причем R11 означает Н; C1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный, или полизамещенный, или незамещенный; С3-8-циклоалкил, каждый раз монозамещенный, или полизамещенный, или незамещенный; арил или гетероарил, каждый раз монозамещенный, или полизамещенный, или незамещенный; или связанный C1-3-алкилом арил, С3-8-циклоалкил или гетероарил, каждый раз монозамещенный, или полизамещенный, или незамещенный; С(O)фенил, С(O)гетероарил, С(O)С1-5-алкил, каждый раз замещенный или незамещенный;
R3 означает C1-8-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный, или полизамещенный, или незамещенный; С3-8-циклоалкил, каждый раз монозамещенный, или полизамещенный, или незамещенный; арил или гетероарил, каждый раз незамещенный, или монозамещенный, или полизамещенный; связанный C1-3-алкильной группой арил, гетероарил или С3-8-циклоалкил, каждый раз незамещенный, или монозамещенный, или полизамещенный;
R4 означает Н; С1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный, или монозамещенный, или полизамещенный; арил или гетероарил, каждый раз замещенный или незамещенный; связанный C1-3-алкильной группой арил, гетероарил или циклоалкил, каждый раз монозамещенный, или полизамещенный, или незамещенный; COR12; SO2R12,
причем R12 означает Н; C1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный, или полизамещенный, или незамещенный; С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, монозамещенный, или полизамещенный, или незамещенный; арил или гетероарил, каждый раз монозамещенный, или полизамещенный, или незамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, каждый раз монозамещенный, или полизамещенный, или незамещенный; OR13; NR14R15;
R5 означает =O; Н; COOR13, CONR13, OR13; C1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный, или монозамещенный, или полизамещенный; С3-8-циклоалкил, насыщенный или ненасыщенный, незамещенный, или монозамещенный, или полизамещенный; арил или гетероарил, незамещенный, или монозамещенный, или полизамещенный; или связанный C1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный, или монозамещенный, или полизамещенный;
R6 означает Н; F, Cl, NO2, CF3, OR13, SR13, SO2R13, SO2OR13, CN, COOR13, NR14R15; C1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный, или монозамещенный, или полизамещенный; С3-8-циклоалкил, насыщенный или ненасыщенный, незамещенный, или монозамещенный, или полизамещенный; арил или гетероарил, незамещенный, или монозамещенный, или полизамещенный; или связанный C1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный, или монозамещенный, или полизамещенный;
или R5 и R6 совместно означают (СН2)n с n = 2, 3, 4, 5 или 6, причем отдельные атомы водорода также могут быть замещены посредством F, Cl, Br, I, NO2, CF3, OR13, CN или C1-5-алкила;
R7, R8, R9 и R10 независимо друг от друга означают
Н, F, Cl, Br, I, NO2, CF3, OR13, SR13, SO2R13, SO2OR13, CN, COOR13, NR14R15; C1-5-алкил, С3-8-циклоалкил, незамещенный, или монозамещенный, или полизамещенный; арил или гетероарил, незамещенный, или монозамещенный, или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный, или монозамещенный, или полизамещенный;
причем R13 означает Н; C1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный, или монозамещенный, или полизамещенный; С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, незамещенный, или монозамещенный, или полизамещенный; арил или гетероарил, незамещенный, или монозамещенный, или полизамещенный; или связанный С1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный, или монозамещенный, или полизамещенный;
R14 и R15 независимо друг от друга означают Н; C1-5-алкил, каждый раз насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный, или монозамещенный, или полизамещенный; или С3-8-циклоалкил, каждый раз насыщенный или ненасыщенный, незамещенный, или монозамещенный, или полизамещенный; арил или гетероарил, незамещенный, или монозамещенный, или полизамещенный; или связанный C1-3-алкилом арил, С3-8-циклоалкил или гетероарил, незамещенный, или монозамещенный, или полизамещенный;
или R14 и R15 вместе образуют CH2CH2OCH2CH2, CH2CH2NR16CH2CH2 или (СН2)3-6,
причем R16 означает Н, С1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный, незамещенный, или монозамещенный, или полизамещенный;
R17 означает Н; C1-8-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный; COR12 или SO2R12;
в виде рацемата; энантиомеров, диастереомеров, смесей энантиомеров или диастереомеров или отдельного энантиомера или диастереомера; оснований и/или солей физиологически совместимых кислот.
Соединения согласно изобретению показывают хорошее связывание к µ-опиоидному рецептору и ORL-1-рецептору.
Понятия "C1-8-алкил", "C1-3-алкил" и "С1-5-алкил" в смысле этого изобретения охватывают ациклические насыщенные или ненасыщенные углеводородные остатки, которые могут быть разветвленными или с прямой цепью, а также незамещенными или моно- или полизамещенными, с от 1 до 8 или от 1 до 3 С-атомами или 1-5 С-атомами, т.е. C1-8-алканилы, С2-8-алкенилы и С2-8-алкинилы или С1-3-алканилы, С2-3-алкенилы и С2-3-алкинилы или C1-5-алканилы, С2-5-алкенилы и С2-5-алкинилы. При этом алкенилы имеют, по меньшей мере, одну C-C-двойную связь и алкинилы по меньшей мере одну C-C-тройную связь. Преимущественно алкил выбран из группы, которая включает метил, этил, n-пропил, 2-пропил, n-бутил, изо-бутил, втор-бутил, трет-бутил, n-пентил, изо-пентил, нео-пентил, n-гексил, 2-гексил, n-гептил, n-октил, 1,1,3,3-тетраметилбутил; этиленил (винил), этинил, пропенил (-CH2CH=CH2, -СН=СН-СН3, -С(=СН2)-СН3), пропинил (-СН-С≡СН, -C≡C-СН3), бутенил, бутинил, пентенил, пентинил, гексенил, гексинил, гептенил, гептинил, октенил и октинил. Особенно выгодны метил, этил, n-пропил, n-бутил, втор-бутил, изо-бутил.
Выражение "циклоалкил" или "С3-8-циклоалкил" означает для целей этого изобретения циклические углеводороды с 3, 4, 5, 6, 7 или 8 атомами углерода, причем углеводороды могут быть насыщенными или ненасыщенными (но не ароматическими), незамещенными или моно- или полизамещенными. Что касается циклоалкила, понятие также включает насыщенные или ненасыщенные (но не ароматические) циклоалкилы, в которых один или два атома углерода посредством гетероатома S, N или О. Предпочтителен С3-8-циклоалкил из группы, которая содержит циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклопентенил, циклогексенил, циклогептенил и циклооктенил, а также тетрагидропиранил, диоксанил, диоксоланил, морфолинил, пиперидинил, пиперазинил, пиразолинонил и пирролидинил. Особенно предпочтителен циклопропил, циклобутил, циклопентил и циклогексил.
Выражение "арил" означает в смысле этого изобретения ароматические углеводороды, в частности фенилы и нафтилы. Ариловые остатки могут также быть конденсированными с другими насыщенными, (частично) ненасыщенными или ароматическими циклическими системами, так что ариловый остаток образует ароматическую циклическую систему самое большее в 20 С-атомов. Каждый из этих С6-20-ариловых остатков может быть незамещенным, или монозамещенным, или полизамещенным, причем ариловые заместители могут быть одинаковыми или различными и находиться в любом и возможном положении арила. Предпочтительно арил выбран из группы, которая содержит фенил, 1-нафтил, 2-нафтил, которые каждый раз могут быть незамещенными или моно- или полизамещенными. Особенно предпочтителен фениловый остаток.
Выражение "гетероарил" означает 5-, 6- или 7-членный циклический ароматический остаток, который содержит, по меньшей мере, 1, при необходимости также 2, 3, 4 или 5 гетероатомов, причем гетероатомы являются одинаковыми или различными и гетероцикл может быть незамещенным или моно- или полизамещенным; в случае замещения у гетероцикла заместители могут быть одинаковыми или различными и находиться в любом и возможном положении гетероарила. Гетероцикл также может быть частью бициклической или полициклической системы в совокупности с до 20 кольцевыми членами. Предпочтительными гетероатомами являются азот, кислород и сера. Является предпочтительным, когда гетероариловый остаток выбран из группы, которая содержит пирролил, индолил, фурил (фуранил), бензофуранил, тиенил (тиофенил), бензотиенил, бензотиадиазолил, бензотиазолил, бензотриазолил, бензодиоксоланил, бензодиоксанил, фталазинил, пиразолил, имидазолил, тиазолил, оксазолил, изоксазолил, пиридил, пиридазинил, пиримидинил, пиразинил, пиранил, индазолил, пуринил, индолизинил, хинолинил, изохинолинил, хиназолинил, карбазолил, феназинил, фенотиазинил, триазолил или оксадиазолил, причем связывание к соединениям общей структуры I может осуществляться любым и возможным кольцевым членом гетероарилового остатка. Предпочтительны пиридил, фурил, тиенил, индолил, бензотиенил, пирролил, триазолил и изоксазолил, особенно предпочтительно пиридил, тиенил, бензотиенил и триазолил.
Выражение "связанный C1-3-алкилом арил или гетероарил" в целях настоящего изобретения означает, что C1-3-алкил и арил или гетероарил имеют определенные выше значения и ариловый или гетероариловый остаток привязан С1-3-алкильной группой к соединению общей структуры I. Особенно предпочтительны в смысле этого изобретения бензил и фенэтил.
В связи с "алкилом" или "циклоалкилом" под понятием "замещенный" в смысле этого изобретения понимают замещение водородного остатка посредством F, Cl, Br, I, -CN, NH2, NH-C1-6-алкила, NH-С1-6-алкил-ОН, N(С1-6-алкил)2, N(C1-6-алкил-OH)2, NO2, SH, S-C1-6-алкила, S-бензила, OCF3, O-C1-6-алкила, ОН, О-С1-6-алкил-ОН, =O, C1-6-алкила, бензила, O-бензила, O-фенила, С(=O)С1-6-алкила, CO2H, CO2-C1-6-алкила, причем под полизамещенными остатками следует понимать те остатки, которые или в различных или в одинаковых атомах замещены многократно, например дважды или трижды, к примеру трижды в одинаковом С-атоме, как в случае CF3 или -CH2CF3, или в различных местах, как в случае -CH(OH)-CH=CH-CHCl2. Многократное замещение может происходить с одинаковым или с различным заместителем. Предпочтительно для целей настоящего изобретения "монозамещенный или полизамещенный" в связи с алкилом означает замещение с помощью СООСН3, ОСН3, ОН, COOC2H5, F или Cl.
Что касается "арила" и "гетероарила", в смысле этого изобретения под "моно- или полизамещенным" понимают моно- или поли-, например дважды, трижды или четырехкратное, замещение одного или нескольких атомов водорода циклической системы посредством F, Cl, Br, I, CN, NH2, NH-C1-6-алкила, NH-C1-6-алкил-ОН, N(C1-6-алкил), N(С1-6-алкил-ОН)2, NO2, SH, S-C1-6-алкила, ОН, O-С1-6-алкила, О-С1-6-алкил-ОН, С(=O)С1-6-алкила,  ;
;  СО2Н, CO2-С1-6-алкила, CF3, OCF3, C1-6-алкила; в одном или при необходимости различных атомах (причем заместитель при необходимости со своей стороны может быть замещенным). Многократное замещение при этом происходит с одинаковым или различным заместителем. Для "арила" и "гетероарила" при этом предпочтительными заместителями являются -F, -Cl, -CF3, -О-СН3, ОН, метил, этил, n-пропил, нитро, трет-бутил, и -CN. Особенно предпочтительны -F и -Cl.
СО2Н, CO2-С1-6-алкила, CF3, OCF3, C1-6-алкила; в одном или при необходимости различных атомах (причем заместитель при необходимости со своей стороны может быть замещенным). Многократное замещение при этом происходит с одинаковым или различным заместителем. Для "арила" и "гетероарила" при этом предпочтительными заместителями являются -F, -Cl, -CF3, -О-СН3, ОН, метил, этил, n-пропил, нитро, трет-бутил, и -CN. Особенно предпочтительны -F и -Cl.
Под понятием соли, образованной с помощью физиологически совместимой кислоты, в смысле этого изобретения понимают соли соответствующего действующего вещества с неорганическими или органическими кислотами, которые являются физиологически совместимыми - в особенности при применении у человека и/или млекопитающего. Предпочтительным является гидрохлорид, цитрат, полуцитрат и метансульфанат. Особенно предпочтителен метансульфанат. Примерами физиологически совместимых кислот являются: соляная кислота, бромистоводородная кислота, серная кислота, метансульфоновая кислота, муравьиная кислота, уксусная кислота, щавелевая кислота, янтарная кислота, винная кислота, миндальная кислота, фумаровая кислота, молочная кислота, яблочная кислота, малеиновая кислота, лимонная кислота, глутаминовая кислота, 1,1-диоксо-1,2-дигидроλ6-бензо[d]изотиазол-3-он (сахариновая кислота), монометилсебациновая кислота, 5-оксо-пролин, гексан-1-сульфоновая кислота, никотиновая кислота, 2-, 3- или 4- аминобензойная кислота, 2,4,6-триметил-бензойная кислота, α-липоновая кислота, ацетилглицин, гиппуровая кислота, фосфорная кислота и/или аспарагиновая кислота. Предпочтительны лимонная кислота, метансульфоновая кислота и соляная кислота. Особенно предпочтительна метансульфоновая кислота.
Под понятием (СН2)3-6 или (СН2)4-5 следует понимать -СН2-СН2-СН2-, -CH2-CH2-CH2-CH2-, -CH2-CH2-CH2-CH2-CH2- и CH2-CH2-CH2-CH2-CH2-CH2- или -CH2-CH2-CH2-CH2- и -CH2-СН2-СН2-СН2-СН2-.
Предпочтительны соединения общей формулы I,
в которой остатки A, W, Х и R1-17 имеют приведенные выше значения,
причем
указанные выше С1-8-алкилы, С1-5-алкилы, С1-3-алкилы или C1-3-алкилены или С3-8-циклоалкильные остатки каждый раз могут быть моно- или полизамещенными с помощью F, Cl, Br, I, CN, NH2, NH-C1-6-алкила, NH-C1-6-алкил-ОН, N(С1-6-алкил)2, N(С1-6-алкил-ОН)2, NO2, SH, S-C1-6-алкила, ОН, O-C1-6-алкила, O-С1-6-алкил-ОН, С(=O)С1-6-алкила, CO2H, СО2-С1-6-алкила, CF3, OCF3, C1-6-алкила,
приведенные выше ариловые или гетероариловые остатки каждый раз могут быть моно- или полизамещенными с помощью F, Cl, Br, I, CN, NH2, NH-C1-6-алкила, NH-C1-6-алкил-ОН, N(С1-6-алкил)2, N(С1-6-алкил-ОН)2, NO2, SH, S-C1-6-алкила, ОН, O-C1-6-алкила, O-С1-6-алкил-ОН, С(=O)С1-6-алкила, CO2H, CO2-C1-6-алкила, CF3, OCF3, С1-6-алкила, ; или фенокси,
в виде рацемата; энантиомеров, диастереомеров, смесей энантиомеров или диастереомеров или отдельного энантиомера или диастереомера; оснований и/или солей физиологически совместимых кислот.
В дальнейшем описанные как предпочтительные остатки и группы соответственно заместители в соединениях согласно изобретению могут комбинироваться с широчайшим значением остальных остатков, а также с предпочтительными значениями других остатков и групп соответственно заместителей.
К предпочтительной форме осуществления спироциклических азаиндольных производных согласно изобретению относится, что
R1 и R2 независимо друг от друга означают Н; C1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный, или полизамещенный, или незамещенный;
или остатки R1 и R2 вместе образуют цикл и означают СН2СН2ОСН2СН2, CH2CH2NR11CH2CH2 или (СН2)3-6,
причем R11 означает Н; С1-5-алкил, насыщенный или ненасыщенный, разветвленный или неразветвленный, монозамещенный, или полизамещенный, или незамещенный.
Особенно предпочтительны спироциклические азаиндольные производные, в которых R1 и R2 независимо друг от друга означают СН3 или Н, причем R1 и R2 не одновременно означают Н,
или R1 и R2 означают (CH2)3.
Совершенно особенно предпочтительны спироциклические азаиндольные производные, в которых R1 и R2 означают СН3
Далее предпочтительны спироциклические азаиндольные производные, в которых
R3 означает этил, пропил, бутил, пентил, гексил, гептил, циклопентил, циклогексил, фенил, бензил, нафтил, антраценил, тиофенил, бензотиофенил, фурил, бензофуранил, бензодиоксоланил, индолил, инданил, бензодиоксанил, пирролил, пиридил, пиримидил или пиразинил, каждый раз незамещенный, или монозамещенный, или полизамещенный; связанный насыщенной, неразветвленной С1-3-алкильной группой С5- или С6-циклоалкил, фенил, нафтил, антраценил, тиофенил, бензотиофенил, пиридил, фурил, бензофуранил, бензодиоксоланил, индолил, инданил, бензодиоксанил, пирролил, пиримидил, триазолил или пиразинил, каждый раз незамещенный, или монозамещенный, или полизамещенный;
в особенности
R3 означает пропил, бутил, пентил, гексил, фенил, фурил, тиофенил, нафтил, бензил, бензофуранил, индолил, инданил, бензодиоксанил, бензодиоксоланил, пиридил, пиримидил, пиразинил, триазолил или бензотиофенил, каждый раз незамещенный, или монозамещенный, или полизамещенный; связанный насыщенной, неразветвленной C1-3-алкильной группой фенил, фурил или тиофенил, каждый раз незамещенный, или монозамещенный, или полизамещенный.
Особенно предпочтительны спироциклические азаиндольные производные, в которых R3 означает пропил, бутил, пентил, гексил, фенил, фенэтил, тиофенил, пиридил, триазолил, бензотиофенил или бензил, каждый раз замещенный или незамещенный, особенно предпочтительно пропил, 3-метоксипропил, бутил, пентил, гексил, фенил, 3-метилфенил, 3-фторфенил, бензо[1,3]-диоксолил, тиенил, бензотиофенил, 4-хлорбензил, бензил, 3-хлорбензил, 4-метилбензил, 2-хлорбензил, 4-фторбензил, 3-метилбензил, 2-метилбензил, 3-фторбензил, 2-фторбензил, 1-метил-1,2,4-триазолил или фенэтил.
Совершенно особенно предпочтительны спироциклические азаиндольные производные, в которых R3 означает бутил, этил, 3-метоксипропил, бензотиофенил, фенил, 3-метилфенил, 3-фторфенил, бензо[1,3]-диоксолил, бензил, 1-метил-1,2,4-триазолил, тиенил или фенэтил.
R4 преимущественно означает Н.
Кроме того, предпочтительны спироциклические азаиндольные производные, в которых R5 означает Н, C1-5-алкил, разветвленный или неразветвленный, незамещенный, или монозамещенный, или полизамещенный, или COOR13.
Особенно предпочтительны спироциклические азаиндольные производные, в которых R5 означает СН3, CH2OH, COOH или СООСН3.
Совершенно особенно предпочтительны спироциклические азаиндольные производные, в которых R5 означает Н.
Предпочтительны также спироциклические азаиндольные производные, в которых R6 означает Н, C1-5-алкил, арил или соединенный С1-3-алкильной группой арил.
Особенно предпочтительны спироциклические азаиндольные производные, в которых R6 означает Н, СН3, фенил или бензил.
Совершенно особенно предпочтительны спироциклические азаиндольные производные, в которых R6 означает Н.
Сверх этого также предпочтительны спироциклические азаиндольные производные, в которых R7, R8, R9 и R10 независимо друг от друга означают Н; метил, этил, пропил, бутил; пиридил, O-бензил, F, Cl, Br, I, CF3, ОН, ОСН3, NH2, COOH, СООСН3, NHCH3 или N(СН3)2 или NO2.
Особенно предпочтительны спироциклические азаиндольные производные, в которых R7, R8, R9 и R10 независимо друг от друга означают Н, F, ОН, СН3, Cl, ОСН3, Br или NO2.
Совершенно особенно предпочтительны спироциклические азаиндольные производные, в которых R7, R8, R9 и R10 означают Н.
Предпочтительны также замещенные азаиндольные производные общей формулы I, в которой А один раз означает N и остальные остатки А принимают значение CR7-9 или CR8-10 или CR7 и CR9-10 или CR7-8 и CR10.
Особенно предпочтительны соединения общих формул Ia и Ib:
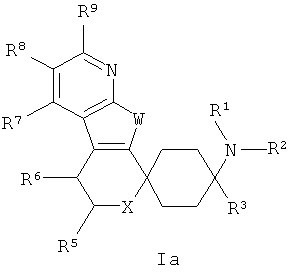
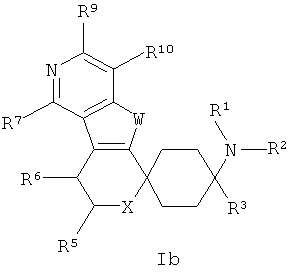
в которых X, W и остатки R1-R10, которые в широчайшем определении приведенных, а также которые в обозначенных как предпочтительные определениях могут принимать описанные значения.
Совершенно особенно предпочтительны замещенные азаиндольные производные из группы
(1) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]
(2) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; метансульфонат
(3) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)]
(4) 4-(диметиламино)-4-тиофен-2-ил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; метансульфонат
(5) 4-(диметиламино)-4-тиофен-2-ил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)]
(6) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (4:3)
(7) 4-(метиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(8) 4-(метиламино)-4-тиофен-2-ил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (4:3)
(9) 4-(диметиламино)-4-бензо[1,3-диоксол]-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1)
(10) 4-(диметиламино)-4-(бензотиофен-2-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(12) 4-(диметиламино)-4-(3-фторфенил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (4:3)
(13) 4-(диметиламино)-4-(3-метилфенил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(14) 4-(диметиламино)-4-(бут-1-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1)
(15) 4-(диметиламино)-4-фенилэтил-спиро[циклогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(17) 4-(диметиламино)-4-этил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (2:3)
(18) 4-(диметиламино)-4-фенилэтил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (2:3)
(19) 4-бензил-4-морфолино-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(20) 4-(диметиламино)-4-(3-метоксипропил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(21) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(22) 4-(диметиламино)-4-тиофен-2-ил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(23) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)], цитрат (1:1)
(24) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)], цитрат (1:1)
(25) 4-бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(26) 4-бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1)
(27) 4-бензил-4-морфолино-спиро[циклогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(28) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1)
(29) 4-(ацетидин-1-ил)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1)
(30) 4-бутил-4-(пирролидин-1-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (2:1)
(31) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-тиопирано[3,4-b]-7-аза-индол)], цитрат (1:1)
(32) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-тиопирано[3,4-b]-5-аза-индол)], цитрат (1:1)
(33) 4-(1-метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1)
(34) 4-(1-метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1)
в виде рацемата; энантиомеров, диастереомеров, смесей энантиомеров или диастереомеров или отдельного энантиомера или диастереомера; основания и/или солей физиологически совместимых кислот.
Вещества согласно изобретению действуют, например, на связанный с различными заболеваниями релевантный µ-опиоидный рецептор, так что они пригодны в качестве фармацевтического действующего вещества в лекарственном средстве. Поэтому другим объектом изобретения являются лекарственные средства, содержащие, по меньшей мере, одно спироциклическое азаиндольное производное согласно изобретению, а также при необходимости пригодные добавки, и/или вспомогательные вещества, и/или при необходимости другие действующие вещества.
Лекарственные средства согласно изобретению наряду с, по меньшей мере, одним спироциклическим азаиндольным производным согласно изобретению содержат при необходимости пригодные добавки и/или вспомогательные вещества, такие как носители, наполнители, растворители, разбавители, красители, и/или связующие вещества и могут применяться в качестве жидких лекарственных форм в виде инъекционных растворов, капель или сиропов, в качестве полутвердых лекарственных форм в виде гранулятов, таблеток, гранул, пэтчей, капсул, пластырей/распылительных пластырей или аэрозолей. Выбор вспомогательных веществ и т.д., а также их используемое количество зависят от того, должно ли лекарственное средство применяться орально, перорально, парентерально, внутривенно, внутрибрюшинно, внутрикожно, внутримышечно, интраназально, буккально, ректально или местно, например на кожу, слизистые оболочки или в глаза. Для орального применения пригодны препараты в виде таблеток, драже, капсул, гранул, капель, соков и сиропов, для парентерального, местного и ингаляционного применения пригодны растворы, суспензии, легко восстанавливаемые сухие препараты, а также спреи. Спироциклические азаиндольные производные согласно изобретению являются пригодными чрескожными композициями для применения в препарате пролонгированного действия, в растворенном виде или в пластыре, при необходимости с добавлением средств, способствующих проникновению сквозь кожу. Применяемые орально или чрескожно препаративные формы могут высвобождать замедленно спироциклические азаиндольные производные согласно изобретению. Спироциклические азаиндольные производные согласно изобретению могут также применяться в парентеральных формах пролонгированного действия, таких как, например, имплантаты или имплантационные насосы. В принципе к лекарственным средствам согласно изобретению могут добавляться другие действующие вещества, которые известны специалисту в данной области техники.
Количество вводимого пациентам действующего вещества варьируется в зависимости от веса пациента, от способа применения, показания и степени тяжести заболевания. Обычно применяют от 0,00005 до 50 мг/кг, предпочтительно от 0,01 до 5 мг/кг по меньшей мере одного спироциклического азаиндольного производного согласно изобретению.
Для всех указанных выше форм лекарственных средств согласно изобретению является особенно предпочтительным, если лекарственное средство наряду с, по меньшей мере, одним спироциклическим азаиндольным производным содержит еще одно дополнительное действующее вещество, в особенности опиоид, преимущественно сильный опиоид, в особенности морфин, или анестетик, преимущественно гексобарбитал или галотан.
В предпочтительной форме лекарственного средства содержащееся спироциклическое азаиндольное производное согласно изобретению находится в виде чистого диастереомера и/или энантиомера, в виде рацемата или в виде неэквимолярной или эквимолярной смеси диастереомеров и/или энантиомеров.
ORL-1-рецептор и µ-опиоидный рецептор в особенности были идентифицированы в болевой чувствительности. Соответственно спироциклические азаиндольные производные согласно изобретению могут применяться для получения лекарственного средства для лечения боли, в особенности острой, невропатической, хронической боли или воспалительной боли.
Поэтому другим объектом изобретения является применение спироциклического азаиндольного производного согласно изобретению для получения лекарственного средства для лечения боли, в особенности острой, внутренней, невропатической или хронической боли.
Другим объектом изобретения является применение спироциклического азаиндольного производного согласно изобретению для получения лекарственного средства для лечения состояний страха, стресса и связанных со стрессом синдромов, депрессий, эпилепсии, болезни Альцгеймера, старческого слабоумия, каталепсии, общих познавательных дисфункций, нарушений обучения и памяти (как ноотроп), синдромов отмены, злоупотребления и/или зависимости алкогольной, и/или наркотической, и/или медикаментозной, сексуальных дисфункций, сердечно-сосудистых заболеваний, гипотонии, гипертензии, тиннитуса, зуда, мигрени, глухоты, недостаточной подвижности кишечника, нарушенного приема пищи, анорексии, ожирения, двигательных нарушений, диареи, кахексии, недержания мочи или как мышечный релаксант, противосудорожное средство или анестетик или для совместного приема при лечении с опиоидным анальгетиком или с анестетиком, для диуреза или антинатрийуреза, анксиолизиса, для модуляции двигательной активности, для модуляции распределения нейромедиаторов и лечения связанных с этим нейродегенеративных заболеваний, для лечения синдромов отмены и/или для снижения наркотического потенциала опиоидов.
При этом в одном из указанных выше применений может быть предпочтительным, если применяемое спироциклическое азаиндольное производное находится в виде чистого диастереомера и/или энантиомера, рацемата или неэквимолярной или эквимолярной смеси диастереомеров и/или энантиомеров.
Другим объектом изобретения является способ лечения, в особенности при одном из вышеуказанных показаний, нечеловеческого млекопитающего или людей, которому или которым необходимо лечение болей, в особенности хронических болей, путем введения терапевтически эффективной дозы спироциклического азаиндольного производного согласно изобретению или лекарственного средства согласно изобретению.
Другим объектом изобретения является способ получения согласно изобретению спироциклического азаиндольного производного, как приведено в нижеследующем описании и примерах.
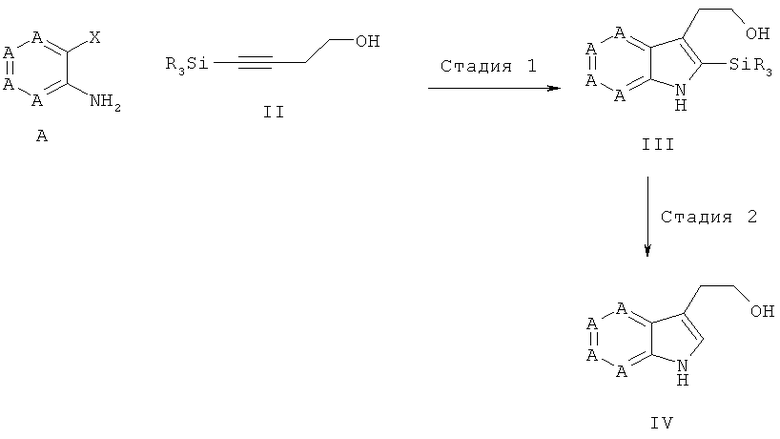
На стадии 1 соединения приведенной выше общей формулы А, в которой Х означает галогенный остаток или сложный эфир сульфоновой кислоты, особенно предпочтительно йод, бром или трифторметансульфонат, в значении индольного синтеза согласно Лароку подвергают взаимодействию с алкинами общей формулы II в реакционной среде, преимущественно выбранной из группы, состоящей из метанола, этилацетата, этанола, изопропанола, n-бутанола, диоксана, хлороформа, дихлорметана, пиридина, диметилсульфоксида, толуола, тетрагидрофурана, диметилформамида, ацетонитрила, диэтилового эфира, воды и соответствующих смесей, особенно предпочтительно выбранной из группы, состоящей из диметилформамида, этилацетата, тетрагидрофурана, воды и соответствующих смесей, преимущественно при добавлении, по меньшей мере, одного палладиевого катализатора, преимущественно выбранного из группы, состоящей из дихлорида палладия(II) [PdCl2], ацетата бис(трифенилфосфин)-палладия(II) [Pd(PPh3)2(OAc)2], хлорида бис(трифенилфосфин)-палладия(II) [PdCl2(PPh3)2], ацетата палладия(II) [Pd(OAc)2; Ac = ацетат], хлорида бис(ацетонитрил)-палладия(II) [(CH3CN2)PdCl2], хлорида бис(бензонитрил)-палладия(II) [(PhCN)2PdCl2] и тетракис(трифенилфосфин)палладия [(PPh3)4Pd], особенно предпочтительно выбранного из группы, состоящей из Pd(PPh3)2(OAc)2, (PPh3)4Pd и PdCl2(PPh3)2, при необходимости в присутствии по меньшей мере одного фосфина, преимущественно одного фосфина, выбранного из группы, состоящей из трифенилфосфина, три-(трет-бутил)-фосфина, трифениларсина и три-(орто-толуил)-фосфина, особенно предпочтительно в присутствии трифенилфосфина, при необходимости с добавлением по меньшей мере одной неорганической соли, предпочтительно с добавлением хлорида лития или хлорида тетрабутиламмония, при необходимости с добавлением по меньшей мере одного неорганического основания, преимущественно выбранного из группы, состоящей из карбоната калия, карбоната натрия, ацетата калия, гидрокарбоната натрия и карбоната цезия, и/или при добавлении, по меньшей мере, одного органического основания, выбранного из группы, состоящей из триэтиламина, диизопропиламина, диизопропилэтиламина и [1,4]-диазабицикло-[2.2.2]октана, при температурах преимущественно от -70°С до 300°С, особенно предпочтительно от -70°С до 150°С, до получения соединений общей формулы III.
Соединения общей формулы А имеются в продаже или известны из литературных источников. Для примера синтезы получения соединений общей формулы А описаны в части примеров.
На стадии 2 соединения общей формулы III подвергают взаимодействию в реакционной среде, преимущественно выбранной из группы, состоящей из метанола, этилацетата, этанола, изопропанола, n-бутанола, диоксана, хлороформа, дихлорметана, пиридина, диметилсульфоксида, толуола, тетрагидрофурана, диметилформамида, ацетонитрила, диэтилового эфира, воды и соответствующих смесей, особенно предпочтительно выбранной из группы, состоящей из ацетонитрила, тетрагидрофурана, метанола, этанола, этилацетата, пиридина, воды и соответствующих смесей, в присутствии фторида, выбранного из группы, состоящей из фторида тетра-n-бутиламмония, плавиковой кислоты (HF, HF-пиридин), фторида калия и/или фторида натрия, фторида цезия, или в присутствии органической или неорганической кислоты, преимущественно HCl, уксусной кислоты, трифторуксусной кислоты, бортрифторида, при температурах преимущественно от -70°С до 300°С, особенно предпочтительно от -70°С до 150°С, до получения соединений общей формулы IV.
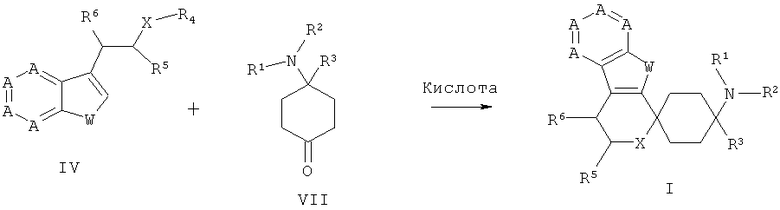
Для получения спироциклических соединений общей формулы VIII кетоны общей формулы VII подвергают взаимодействию с гетероароматами общей формулы IV с добавлением по меньшей мере одной органической кислоты или ее сложного триметилсилилового эфира, преимущественно выбранного из группы, состоящей из сложного триметилсилилового эфира трифторметансульфоновой кислоты, трифторметансульфоновой кислоты, метансульфоновой кислоты, трифторуксусной кислоты, уксусной кислоты, фосфорной кислоты, p-толуолсульфоновой кислоты или одной неорганической кислоты, выбранной из группы, состоящей из бортрифторида, хлорида индия(III), тетрахлорида титана, хлорида алюминия(III), или с добавлением по меньшей мере одной соли переходного металла, преимущественно с добавлением по меньшей мере одного трифлата переходного металла (трифторметансульфонат переходного металла), особенно предпочтительно с добавлением по меньшей мере одного трифторметансульфоната переходного металла, выбранного из группы, состоящей из трифторметансульфоната скандия(III), трифторметансульфоната иттербия(III) и трифторметансульфоната индия(III), в пригодном растворителе или смеси растворителей, таком как, к примеру, дихлорметан, дихлорэтан, хлороформ, ацетонитрил, диэтиловый эфир или нитроэтан, при температурах от 0 до 150°С при необходимости с применением микроволн.
Синтезы производных циклогексанона с общей формулой VII известны из литературных источников (WO 04043967, WO 0290317, US 4065573, Lednicer et al., J. Med. Chem., 23, 1980, 424-430).
Примеры
Следующие примеры служат для подробного пояснения изобретения, однако не ограничивают общие изобретательские идеи.
Выходы полученных соединений не являются оптимизированными.
Все температуры являются неизменяемыми.
Сокращения
Микроволна: Biotage Initiator, 2,45 ГГц.
Кетоновые элементарные звенья
Общие элементарные звенья
8-Диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил:
Вариант 1: 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил
К смеси из 4N соляной кислоты (50 мл) и метанола (30 мл) прибавляли при охлаждении льдом 40-проц. водный раствор диметиламина (116 мл, 0,92 моль), циклогексан-1,4-дион-моноэтиленкеталь (30 г, 0,192 моль) и цианид калия (30 г, 0,46 моль). Смесь перемешивали 74 ч при комнатной температуре и затем после добавления воды (80 мл) экстрагировали диэтиловым эфиром (4×100 мл). После концентрирования остаток ресуспендировали в дихлорметане (200 мл) и высушивали сульфатом магния в течение ночи. Органическую фазу концентрировали и кеталь получали в виде белого твердого вещества с точкой плавления в 86-88°C с выходом в 97% (40 г).
Вариант 2: 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил
К смеси из 4N соляной кислоты (50 мл) и метанола (30 мл) при охлаждении льдом прибавляли 40-проц. водный раствор диметиламина (116 мл, 0,92 моль), циклогексан-1,4-дион-моноэтиленкеталь (30,0 г, 0,192 моль) и цианид калия (300 г, 0,46 моль). Смесь перемешивали 72 ч при комнатной температуре и затем после добавления воды (80 мл) экстрагировали простым эфиром (4×100 мл). После концентрирования раствора остаток ресуспендировали в дихлорметане (200 мл) и сульфатом магния в течение ночи высушивали. Органическую фазу концентрировали и получали кеталь в виде белого твердого вещества.
Выход: 38,9 г (96%).
Точка плавления: 86-88°С.
1H-ЯМР (ДМСО-d6): 1.57 (2Н, m); 1.72 (2H; m); 1.85 (2H, m); 1.99 (2H, m); 2.25 (6H, s); 3.87 (4H, m).
13С-ЯМР (ДМСО-d6): 30.02; 31.32; 60.66; 63.77; 106.31; 118.40.
8-Метиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил
К смеси из 4N соляной кислоты (12,5 мл) и метанола (7,5 мл) прибавляли при охлаждении льдом 40%-ный водный раствор метиламина (29,0 мл, 0,23 моль), циклогексан-1,4-дион-моноэтиленкеталь (7,50 г, 0,048 моль) и цианид калия (7,50 г). Смесь перемешивали 7 дн. при комнатной температуре. После добавления воды (20 мл) экстрагировали простым эфиром (4×25 мл). После концентрирования раствора остаток ресуспендировали в дихлорметане (50 мл) и высушивали с помощью MgSO4 в течение ночи. Органическую фазу концентрировали и получали кеталь в виде масла, которое кристаллизировалось.
Выход: 7,05 г (80%).
1H-ЯМР (ДМСО-d6): 1.54 (2H, m); 1.71 (4H, m); 1.95 (2H, m); 2.30 (3H, d); 2.72 (1H, q); 3.86 (4H, s).
Элементарное звено Ket-1
Диметил-(8-фенил-1,4-диоксаспиро[4.5]дец-8-ил)амин-гидрохлорид
К 1,82 M раствору хлорида фенилмагния в ТГФ (109 мл, 0,198 моль) под аргоном и охлаждением льдом в течение 15 мин прибавляли 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил (21 г, 0,1 моль), растворенный в ТГФ (210 мл), и затем перемешивали 16 ч при комнатной температуре. Для переработки реакционной смеси при охлаждении льдом добавляли насыщенный раствор хлорида аммония (150 мл) и экстрагировали диэтиловым эфиром (3×100 мл). Органическую фазу встряхивали с помощью воды (100 мл) и насыщенного раствора NaCl (100 мл) и концентрировали. Оставалось желтое масло (25,2 г). Исходный продукт растворяли в этилметилкетоне (280 мл) и при охлаждении льдом смешивали с ClSiMe3 (18,8 мл, 0,15 моль). После продолжительности реакции в 6 ч можно было выделить диметил-(8-фенил-1,4-диоксаспиро[4.5]дец-8-ил)амин-гидрохлорид с выходом в 35% (10,5 г) в виде белого твердого вещества.
4-Диметиламино-4-фенилциклогексанон (Ket-1)
Диметил-(8-фенил-1,4-диоксаспиро[4.5]дец-8-ил)амин-гидрохлорид (10,5 г, 35,2 ммоль) растворяли в 7,5N соляной кислоте (36 мл) и перемешивали 96 ч при комнатной температуре. После оконченного гидролиза реакционную смесь экстрагировали диэтиловым эфиром (2×50 мл). Водную фазу при охлаждении льдом подщелачивали с помощью 5N натрового щелока, экстрагировали дихлорметаном (3×50 мл) и концентрировали. Таким образом, 4-диметиламино-4-фенилциклогексанон (Ket-1) можно было выделить в виде желтого твердого вещества с точкой плавления в 104-108°C с выходом в 97% (7,4 г).
Элементарное звено Ket-2
Диметил-(8-тиофен-2-ил-1,4-диоксаспиро[4.5]дец-8-ил)амин-гидрохлорид
2-Йодтиофен (22,9 г, 109 ммоль) растворяли под аргоном в ТГФ (80 мл) и в течение 30 мин при 0°С смешивали с 2М хлоридом изопропилмагния (35,7 мл, 72 ммоль) в ТГФ. После продолжительности реакции в 1 ч при 3-5°С прибавляли 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил (10 г, 47,6 ммоль), растворенный в тетрагидрофуране (20 мл), и 20 ч перемешивали при комнатной температуре. Переработка состава осуществлялась путем добавления насыщенного раствора NH4Cl (85 мл) и экстракции диэтиловым эфиром (3×100 мл). Органическую фазу встряхивали с помощью воды (50 мл) и насыщенного раствора NaCl (50 мл) и концентрировали. Можно было получить темно-коричневое масло (21,3 г). Исходный продукт растворяли в этилметилкетоне (140 мл) и смешивали с ClSiMe3 (9,1 мл, 71,4 ммоль). После продолжительности реакции в 6 ч выделяли диметил-(8-тиофен-2-ил-1,4-диоксаспиро[4.5]дец-8-ил)амин-гидрохлорид в виде белого кристаллического соединения с выходом в 60% (8,74 г).
4-Диметиламино-4-тиофен-2-илциклогексанон (Ket-2)
Диметил-(8-тиофен-2-ил-1,4-диоксаспиро[4.5]дец-8-ил)амин-гидрохлорид (8,68 г, 28,6 ч оконченного гидролиза реакционную смесь экстрагировали диэтиловым эфиром (2×50 мл). Водную фазу подщелачивали при охлаждении льдом посредством 5N натрового щелока, экстрагировали дихлорметаном (3×50 мл) и концентрировали. Ket-2 получали, таким образом, в виде желтого твердого вещества с точкой плавления в 108-110°C с выходом в 89% (5,66 г).
Элементарное звено Ket-3
Сложный диметиловый эфир 4-циано-4-фенилгептандионовой кислоты
Фенилацетонитрил (11,7 г, 0,1 моль) и метилакрилат (47 мл, 0,5 моль) помещали в трет-бутанол (60 мл) и нагревали до кипения. Затем источник нагревания удаляли. Растворенный в трет-бутаноле (23 мл) тритон В (гидроксид бензилтриметиламмония, 40-проц. в метаноле, 15,2 мл) прибавляли по каплям сначала медленно, затем быстро. После прикапывания состав кипятили 4 ч в колбе с обратным холодильником. В течение ночи реакционную смесь охлаждали до комнатной температуры. Для переработки состав смешивали с толуолом (100 мл) и водой (70 мл). Органическую фазу отделяли и промывали водой (1×70 мл) и насыщенным раствором хлорида натрия (1×50 мл). После высушивания с помощью Na2SO4 растворитель из-за сильной загазованности дистиллировали в вытяжке. Очистку осуществляли путем дистилляции в трубке с шаровым расширением при давлении в 7,8×10-2 мбар и температуре 235°С. Целевой сложный диметиловый эфир 4-циано-4-фенилгептандионовой кислоты можно было выделить с выходом в 21,45 г (72%) в виде бесцветного вязкотекучего вещества.
Сложный метиловый эфир 5-циано-2-оксо-5-фенилциклогексанкарбоновой кислоты
Сложный диметиловый эфир 4-циано-4-фенилгептандионовой кислоты (14,45 г, 0,05 моль) растворяли в сухом тетрагидрофуране (350 мл). Затем добавляли порционно трет-бутилат натрия (9,6 г, 0,1 моль). При этом добавлении реакционная смесь окрасилась в оранжевый цвет. После этого состав кипятили 5 ч в колбе с обратным холодильником. Во время кипячения образовалась тягучая суспензия бежевого цвета. В течение ночи реакционную смесь охлаждали до комнатной температуры. При охлаждении льдом к реакционной смеси медленно прибавляли по каплям 2,5N уксусную кислоту (170 мл). Затем состав смешивали с толуолом (100 мл). Органическую фазу отделяли и промывали насыщенным раствором гидрокарбоната натрия (3×70 мл), водой (3×50 мл) и раствором хлорида натрия (1×70 мл). После высушивания с помощью Na2SO4 растворитель в ротационном выпарном аппарате отгоняли и остаток перекристаллизовывали из метанола. Целевой продукт можно было выделить с выходом в 10,7 г (83%) в виде желтого твердого вещества с точкой плавления в 75-80°С.
4-Циано-4-фенилциклогексанон
Сложный метиловый эфир 5-циано-2-оксо-5-фенилциклогексанкарбоновой кислоты (7,71 г, 0,03 моль) растворяли в 10-проц. H2SO4 и концентрированной уксусной кислоте (240 мл). Реакционную смесь перемешивали 24 ч при 100°С. За протеканием реакции наблюдали с помощью DC. Для переработки состав при охлаждении льдом разбавляли с водой (400 мл) и экстрагировали этилацетатом (3×100 мл). Затем органическую фазу тщательно промывали водой (6×100 мл), насыщенным раствором гидрокарбоната натрия (10×100 мл) и насыщенным раствором хлорида натрия (1×100 мл). После высушивания с помощью Na2SO4 растворитель отгоняли в ротационном выпарном аппарате. Целевой продукт можно было выделить с выходом в 5,46 г (92%) с точкой плавления в 106-107°С.
8-Циано-8-фенил-1,4-диоксаспиро[4.5]декан
4-Циано-4-фенилциклогексанон (5,97 г, 30 ммоль) ресуспендировали в толуоле (200 мл) и смешивали с этиленгликолем (4 мл, 71,6 ммоль). После добавления p-толуолсульфоновой кислоты (86 мг, 0,5 ммоль) состав нагревали до кипения в водоотделителе. Протекание реакции наблюдали с помощью DC хроматографии. Через 20 ч в DC исходный продукт больше не обнаруживался. После охлаждения раствор толуола встряхивали с водой (5×30 мл) и насыщенным водным раствором NaC (3×20 мл) и высушивали над Na2SO4. После удаления растворителя в ротационном выпарном аппарате получался целевой кеталь с выходом в 6,8 г (94%) в виде белого твердого вещества с точкой плавления в 108-110°С.
8-Фенил-1,4-диоксаспиро[4.5]декан-8-карбоновая кислота
(Schneider, Woldemar; Krombholz, Gottfried; ARPMAS; Arch. Pharm. (Weinheim Ger.); 313; 6; 1980; 487-498)
8-Циано-8-фенил-1,4-диоксаспиро[4.5]декан (4,86 г, 20 ммоль) растворяли в этиленгликоле (40 мл), смешивали с NaOH (4 г, 100 ммоль) и затем в колбе с обратным холодильником нагревали до кипения. Протекание реакции наблюдали с помощью DC. Через 20 ч нитрил больше не обнаруживался. Для переработки состав смешивали со льдом (прибл. 100 г), наслаивали простым эфиром (40 мл) и подкисляли путем медленного добавления полуконцентрированной HCl (50 мл). Водную фазу экстрагировали простым эфиром (3×30 мл). Объединенные органические экстракты промывали насыщенным раствором NH4C- (2×30 мл), высушивали над Na2SO4 и в ротационном выпарном аппарате осушали. Путем перекристаллизации полученного твердого вещества из толуола выделяли целевую карбоновую кислоту в виде кристаллического твердого вещества с точкой плавления в 134-139°C с выходом в 3,1 г (59%).
8-Изоцианато-8-фенил-1,4-диоксаспиро[4.5]декан
8-Фенил-1,4-диоксаспиро[4.5]декан-8-карбоновую кислоту (3 г, 11,5 ммоль) помещали в анизол (30 мл). Полученную суспензию в ванной из льда и поваренной соли охлаждали до температуры в 0°С и смешивали с триэтиламином (2,25 мл, 16 ммоль). Образовался светлый раствор, который перемешивали в течение последующих 15 мин при 0°С. Затем смесь в течение 5 мин смешивали с азидом дифенилового эфира фосфорной кислоты (2,5 мл, 11,5 ммоль). Реакционную смесь перемешивали 20 мин при 0°С, в течение последующих 20 мин доводили до КТ и затем нагревали в масляной бане в течение 2 ч до 100°С (температура ванны). Для переработки анизол отгоняли в вакууме масляного насоса. Хроматографическую очистку проводили на силикагеле с толуолом. Целевой продукт получали в виде кристаллического твердого вещества с точкой плавления в 38-41°C с выходом в 2,7 г (91%).
Метил-(8-фенил-1,4-диоксаспиро[4.5]дец-8-ил)амин
LiAlH4 (535 мг, 14,08 ммоль) суспендировали при исключении влажности воздуха в сухом ТГФ (4 мл). 8-Изоцианато-8-фенил-1,4-диоксаспиро[4.5]декан (2,29 г, 8,8 ммоль, растворенный в 40 мл сухого ТГФ) прибавляли по каплям в течение 20 мин. После полного добавления реакционную смесь 4 ч нагревали в колбе с обратным холодильником до кипения. После охлаждения реакционную смесь при охлаждении льдом смешивали сначала осторожно с водным ТГФ (1 мл H2O в 3 мл), затем с 1,7 мл 15-проц. натрового щелока и в заключение с 5 мл H2O. Состав перемешивали 20 мин и затем фильтровали через силикагель. Полученную после многократной промывки фильтровального осадка этилацетатом смесь растворителей концентрировали в ротационном выпарном аппарате до сухости. Целевой продукт получали с выходом в 2,1 г (97%) в виде вязкого масла.
4-Метиламино-4-фенилциклогексанон (Ket-3)
(Upjohn Lednicer, US 4065573 A1, 1977)
Метил-(8-фенил-1,4-диоксаспиро[4.5]дец-8-ил)амин (2,1 г, 8,4 ммоль) переливали со смесью из конц. HCl (15 мл) и воды (8 мл) и перемешивали 5 дней при КТ. Для переработки реакционную смесь разбавляли с водой (20 мл) и экстрагировали простым эфиром (3×30 мл). Эфирную фазу отводили. Водную фазу затем подщелачивали с помощью 2N NaOH и экстрагировали дихлорметаном (3×30 мл). Полученную таким образом органическую фазу высушивали с помощью Na2SO4 и затем концентрировали в ротационном выпарном аппарате. Кетон Ket-3 можно было получить путем хроматографической очистки на силикагеле с этилацетат/этанол (4:1) с выходом в 1,38 г (81%) в виде твердого вещества с точкой плавления в 32-38°С.
Элементарное звено Ket-4: 2-(хлорметил)тиофен
Тиофен (50 г) смешивали с раствором конц. HCl (25 мл) и смесь охлаждали до 0-5°С. Под постоянным HCl-газовым потоком теперь прибавляли по каплям водный раствор формальдегида (54,8 мл, 40%) в течение промежутка времени в 4 ч при 0-15°С. Реакционную смесь перемешивали в течение 10 мин при КТ и затем смешивали с этилацетатом (500 мл). Органическую фазу экстрагировали насыщенным раствором NaHCO3 (3×250 мл) и водой (1×250 мл) и высушивали над Na2SO4. Дистилляция под вакуумом при 100°С-110°С (температура масляной бани) дала (60°С температура верха) целевой продукт (8 мм ртутного столба).
Выход: 24 г (30%), бесцветное масло.
2-(Тиофен-2-ил)ацетонитрил
Раствор 2-(хлорметил)тиофена (22 г) в ДХМ (60 мл) смешивали со смесью из воды (90 мл) и NaCN (12,2 г). Реакционную смесь при 35-40°С в течение 18 ч нагревали с обратным холодильником. Смесь охлаждали до КТ и фазу ДХМ отделяли. Экстрагировали с помощью ДХМ (2×100 мл) и объединенные органические фазы промывали насыщенным раствором NaCl и высушивали над Na2SO4. Растворители удаляли при сниженном давлении и остаток при 140-150°С (температура масляной бани) дистиллировали (температура верха: 115-120°С). Последующая хроматографическая очистка (SiO2, 5% EtOAc/n-гексан) дала целевой продукт.
Выход: 9,2 г (45%), светло-коричневое масло.
Этил 3-бромпропионат
Раствор этилакрилата (200 г) в простом эфире (400 мл) охлаждали до 0-5°С. В отдельном реакционном сосуде в течение промежутка времени в 3 ч добавляли по каплям бром (278 мл) ктетралину (213 мл) и выделившийся HBr-газ направляли к раствору этилакрилата.
Реакционную смесь перемешивали в течение 12 ч. Простой эфир удаляли при сниженном давлении и остаток при 70°С дистиллировали (9 мм ртутного столба).
Выход: 360 г (99%), бесцветное масло.
Этил 5-циано-2-оксо-5-(тиофен-2-ил)циклогексанкарбоксилат
Раствор 2-(тиофен-2-ил)ацетонитрила (10 г) в толуоле (300 мл) смешивали с этил-3-бромпропионатом (33,8 г) и смесь охлаждали до -10°С. NaNH2 (27 г) прибавляли порционно в течение промежутка времени в 1 ч (температуру удерживали ниже 0°С). Реакционную смесь нагревали до КТ и в течение 1 ч кипятили с обратным холодильником (111°С). Затем смесь охлаждали до 0-5°С и смешивали с АсОН/вода (50 мл/100 мл). Толуоловую фазу отделяли и водную фазу экстрагировали толуолом (3×200 мл). Объединенные органические экстракты промывали с помощью 5% раствора Na2CO3 (1×150 мл) и воды (1×00 мл) и высушивали над Na2SO4.
Растворитель удаляли при сниженном давлении.
Выход: 12 г (55%), темно-коричневое масло.
4-Циано-4-(тиофен-2-ил)циклогексанон
Этил 5-циано-2-оксо-5-(тиофен-2-ил)циклогексанкарбоксилат (12 г) в уксусной кислоте (120 мл) смешивали с конц. HCl (60 мл). Реакционную смесь в течение 3 ч нагревали в колбе с обратным холодильником (110°С-120°С). Смесь охлаждали до 0°С и с помощью раствора 2N NaOH (200 мл) нейтрализовали (рН-7). Водную фазу экстрагировали этилацетатом (3×150 мл). Объединенные органические фазы промывали водой (1×300 мл) и насыщенным раствором NaHCO3 (1×300 мл) и высушивали над Na2SO4. Растворители удаляли при сниженном давлении и остаток очищали с помощью колоночной хроматографии (SiO2, 15% EtOAc/n-гексан).
Выход: 37 г (43%), светло-желтое твердое вещество.
8-Циано-8-(тиофен-2-ил)-1,4-диоксаспиро[4.5]декан
Раствор 4-циано-4-(тиофен-2-ил)циклогексанона (15 г) в бензоле (120 мл) смешивали с этиленгликолем (9,08 г) и п-толуолсульфоновой кислотой (0,0139 г) и реакционную смесь в течение 4 ч при 110°С (аппаратура Dean-Stark) кипятили с обратным холодильником в водоотделителе. Реакционную смесь охлаждали до КТ и органическую фазу промывали водным раствор бикарбоната натрия (1×150 мл), водой (1×150 мл) и насыщенным раствором NaCl (1×150 мл). После высушивания над Na2SO4 растворители удаляли при сниженном давлении.
Выход: 16,5 г (90%), светло-желтое твердое вещество.
8-(Тиофен-2-ил)-1,4-диоксаспиро[4.5]декан-8-карбоновая кислота
8-Циано-8-(тиофен-2-ил)-1,4-диоксаспиро[4.5]декан (20 г) в этиленгликоле (240 мл) смешивали с KОН (22,48 г) и реакционную смесь кипятили с обратным холодильником в течение 16 ч при 140-150°С. Смесь охлаждали до КТ и затем при 0-5°С наслаивали простым эфиром (500 мл). Прибавляли ледяную воду (250 мл) и HCl (30 мл) и значение pH водной фазы устанавливали до pH~2. Органическую фазу отделяли, промывали водой (1×300 мл) и насыщенным раствором NaCl (1×300 мл) и высушивали над Na2SO4. Растворитель удаляли при сниженном давлении.
Выход: 20,5 г (95%), желтое твердое вещество.
8-Изоцианато-8-(тиофен-2-ил)-1,4-диоксаспиро[4.5]декан
К раствору 8-(тиофен-2-ил)-1,4-диоксаспиро[4.5]декан-8-карбоновой кислоты (26 г) в толуоле (221 мл) добавляли TEA (14,1 мл) и DPPA (32,38 г) и реакционную смесь нагревали в течение 30 мин до 60-70°С. Затем толуол удаляли при сниженном давлении и остаток очищали с помощью колоночной хроматографии (сырой SiO2, 1. течение: 10% EtOAc/n-гексан, 2. течение: 10% EtOAc/гексан).
Выход: 14 г (54%), светло-зеленое масло.
N-метил-8-(тиофен-2-ил)-1,4-диоксаспиро[4.5]декан-8-амин
8-Изоцианато-8-(тиофен-2-ил)-1,4-диоксаспиро[4.5]декан (4 г) растворяли в сухом ТГФ (140 мл) и раствор охлаждали до 0-10°С. LAH (4 г) прибавляли порционно в течение промежутка времени в 30 мин и реакционную смесь в течение еще 30 мин нагревали до 60°С. Смесь охлаждали до 0-10°С и смешивали с насыщенным раствором хлорида аммония (100 мл). Теперь смесь фильтровали через целит и промывали этилацетатом (3×150 мл). После удаления растворителей сырой продукт ресуспендировали в этилацетате (200 мл) и перемешивали в течение 3 мин при 0-10°С. Этилацетатную фазу отделяли, водную фазу подщелачивали насыщенным раствором NaOH (pH~10-14) и экстрагировали этилацетатом (3×200 мл). Объединенные органические фазы высушивали над Na2SO4. Растворитель удаляли при сниженном давлении.
Выход: 7,5 г (56%), бесцветное твердое вещество.
4-Метиламино-4-тиофен-2-ндциклогексанон (Ket-4)
N-метил-8-(тиофен-2-ил)-1,4-диоксаспиро[4.5]декан-8-амин (2 г, 7,9 ммоль) смешивали со смесью из конц. HCl (15 мл) и воды (8 мл) и 5 дней перемешивали при КТ. Для переработки реакционную смесь разбавляли с водой (30 мл) и экстрагировали простым эфиром. Эфирную фазу отводили. Затем водную фазу подщелачивали с помощью 2N NaOH и экстрагировали дихлорметаном (3×30 мл). Полученную таким образом органическую фазу высушивали посредством Na2SO4 и затем концентрировали в ротационном выпарном аппарате. Остаток очищали путем колоночной хроматографии [силикагель 60 (50 г); 500 мл этилацетат/этанол (5:1)] и Ket-4 получали с выходом в 1,4 г (85%) в виде твердого вещества с точкой плавления в 72-74°С.
Элементарное звено Ket-5
(8-Бензо[1,3]диоксол-5-ил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид
К раствору 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрила (5,25 г, 25 ммоль) в абс. ТГФ (75 мл) под аргоном и охлаждением льдом в течение 15 мин добавляли по каплям раствор бромида 1М 3,4-(метилендиокси)фенилмагния в толуол/ТГФ (1:1) (62,5 мл, 62,5 ммоль) при 5-10°С и затем при КТ перемешивали 20 ч. Для переработки реакционной смеси при охлаждении льдом добавляли 20%-ный раствор хлорида аммония (20 мл) и воду (25 мл) и смесь экстрагировали простым эфиром (3×50 мл). Органическую фазу промывали водой и насыщенным раствором хлорида натрия, высушивали над сульфатом натрия и концентрировали в вакууме. Оставалось бесцветное масло (11,26 г), которое растворяли в этилметилкетоне (35 мл) и при охлаждении льдом смешивали с триметилхлорсиланом (4,75 мл, 37,5 ммоль). Перемешивали в открытой колбе 5 ч при КТ. При этом в осадок выпадало бесцветное твердое вещество, которое отсасывали и на воздухе высушивали.
Выход: 2,7 г (32%).
1Н-ЯМР (ДМСО-d6): 1.71 (2Н, t); 1.72 (2H; d); 2.09 (2H, t); 2.43 (6H, s); 2.84 (2H, d); 3.82 (4H, m); 6.11 (2H, s); 7.07 (1H, d); 7.15 (1H, d); 7.32 (1H, s); 10.74 (1H, bs).
4-Бензо[1,3]диоксол-5-ил-4-диметиламино-циклогексанон (Ket-5)
(8-Бензо[1,3]диоксол-5-ил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид (2,70 г, 7,91 ммоль) смешивали с 6N соляной кислотой (10 мл) и перемешивали при КТ в течение ночи. После оконченного гидролиза реакционную смесь экстрагировали простым эфиром (2×20 мл), водный раствор при охлаждении льдом подщелачивали с помощью 5N натрового щелока, реакционную смесь экстрагировали дихлорметаном (3×50 мл), органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме.
Выход (Ket-5): 1,99 г (96%), бесцветные кристаллы.
Точка плавления: 122-124°С.
1Н-ЯМР (ДМСО-d6): 2.01 (6H, s); 2.10 (4H, m); 2.43 (6H, m); 6.01 (2H, s); 6.88 (2H, m); 7.02 (1H, s).
13С-ЯМР (ДМСО-d6): 32.39; 36.68; 38.88; 58.82; 100.76; 107.12; 107.67; 120.46; 131.34; 145.69; 147.03; 210.27.
Элементарное звено Ket-6
2-Йод-бензо[b]тиофен
В 500 мл трехгорлой колбе помещали под атмосферой аргона бутиллитий 1,6М в гексан (112,5 мл, 180 ммоль) и абс. простой эфир (70 мл) и охлаждали в ледяной ванне до 0°С. Затем бензотиофен (20,1 г, 150 ммоль), растворенный в абс. простом эфире (40 мл), и при охлаждении льдом в течение 30 минут прибавляли по каплям и перемешивали 2,5 ч в ледяной ванне. Реакционный состав стоял в течение ночи в холодильнике. В 500 мл трехгорлой колбе под атмосферой аргона помещали йод (75,0 г) и абс. простой эфир (50 мл) и прибавляли по каплям раствор соединения лития при охлаждении льдом. Состав медленно нагревали до комнатной температуры, гидролизировали водой, промывали раствором тиосульфата натрия и органическую фазу высушивали над сульфатом натрия. Затем реакционный раствор концентрировали в вакууме и очищали с помощью флеш-хроматографии с циклогексаном.
Выход: 24,1 г (62%), полутвердые, светло-коричневые кристаллы.
1Н-ЯМР (ДМСО-d6): 7.32 (2Н, m); 7.75 (1H, s); 7.81 (1H, m); 7.93 (1H, m).
(8-Бензо[b]тиофен-2-ил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид
IB 100 мл в трехгорлой колбе помещали Mg (238 мг) в абс. простом эфире (2 мл) под аргоном, туда медленно прибавляли по каплям 2-йод-бензо[b]тиофен (2,51 г, 9,6 ммоль) в абс. простом эфире (8 мл). После добавления абс. простого эфира (10 мл) состав 5 ч кипятили в колбе с обратным холодильником. Реакционный раствор охлаждали в ледяной ванне и при 10°С по каплям добавляли 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил (1,03 г, 4,9 ммоль) в ТГФ (10 мл). Состав перемешивали при комнатной температуре в течение ночи, реакционную смесь смешивали при охлаждении льдом с раствором NH4Cl (5 мл) и водой (7 мл) и экстрагировали простым эфиром (3×30 мл). Органич. фазу промывали водой (30 мл) и затем насыщенным раствором NaCl (20 мл), высушивали над Na2SO4 и концентрировали в вакууме.
Выход: 1,99 г (66%).
Исходный продукт растворяли в этилметилкетоне (19 мл), при охлаждении льдом смешивали с триметилхлорсиланом (1,63 мл, 12,8 ммоль) и перемешивали при комнатной температуре 5 ч. Образовавшийся осадок отсасывали и высушивали в вакууме.
Выход: 600 мг (35%).
1H-ЯМР (ДМСО-d6): 1.46 (2H, m); 1.79 (2H, m); 2.37 (2H, m); 2.63 (6H, s); 2.75 (2H, m); 7.47 (2H, m); 7.91 (1H, s); 7.95 (1H, m); 8 06 (1H, m); 11.40 (1H, s).
13С-ЯМР (ДМСО-d6): 30.43; 31.13; 37.84; 63.88; 66.42; 105.84; 122.48; 124.55; 124.89; 125.71; 128.99; 135.00; 138 91; 139 58.
4-Бензо[b]тиофен-2-ил-4-диметиламино-циклогексанон (Ket-6)
(8-Бензо[b]тиофен-2-ил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид (0,60 г, 1,7 ммоль) растворяли в воде (0,8 мл), смешивали с концентрированной соляной кислотой (1,04 мл, 151 ммоль) и перемешивали 3 дн. при комнатной температуре. После оконченного гидролиза реакционную смесь экстрагировали диэтиловым эфиром (2×25 мл) и водную фазу подщелачивали с помощью 5N натрового щелока, экстрагировали дихлорметаном (3×25 мл), высушивали над сульфатом натрия и концентрировали в вакууме.
Выход (Ket-6): 0,44 г (95%).
1Н-ЯМР (ДМСО-d6): 2.19 (10Н, m); 2.52 (4H, m); 7.35 (3H, m); 7.84 (1H, m); 7.91 (1H, m).
13С-ЯМР (ДМСО-d6): 33.74; 36.51; 38.05; 58.60; 121.87; 121.94; 123.35; 124.02; 124.16; 138.19; 139.17; 144.28; 209.50.
Элементарное звено Ket-7
Вариант 1: [8(3-фтор-фенил)-1,4-диоксаспиро[4.5]дец-8-ил]-диметил-амин гидрохлорид
К раствору 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрила (19,8 г, 94 ммоль) в ТГФ (100 мл) под аргоном и охлаждением льдом прибавляли в течение 15 мин раствор бромида 0,5М 3-фторфенилмагния в ТГФ (3,750 мл, 375 ммоль) и затем перемешивали 16 ч при комнатной температуре. Для переработки реакционной смеси при охлаждении льдом добавляли насыщенный раствор хлорида аммония (150 мл) и воду (60 мл) и экстрагировали диэтиловым эфиром (3×100 мл). Органическую фазу встряхивали водой (50 мл) и насыщенным раствором NaCl (50 мл) и концентрировали. Оставалось коричневое масло (26,5 г), которое кроме соединения фенила 4 содержало еще кеталь 2. Исходный продукт растворяли в этилметилкетоне (156 мл) и при охлаждении льдом смешивали с ClSiMe3 (17,8 мл, 141 ммоль). После продолжительности реакции в 6 ч можно было выделить гидрохлорид с выходом в 55% (16,3 г) в виде белого твердого вещества с точкой плавления в 275-278°С.
Вариант 2: [8-(3-фтор-фенил)-1,4-диокса-спиро[4.5]дец-8-ил]-диметил-амин гидрохлорид
Раствор 1-бром-3-фторбензола (5,00 г, 28,6 ммоль) в абс. простом эфире (15 мл) добавляли по каплям к суспензии магния (694 мг, 28,6 ммоль) в абс. простом эфире (10 мл) таким образом, что простой эфир вскипал. После окончательного добавления перемешивали 10 мин при КТ, после этого магний был полностью растворен. Реакционный раствор охлаждали в ледяной ванне и при 10°С по каплям добавляли 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил (3,00 г, 14,3 ммоль) в абс. ТГФ (30 мл). Состав перемешивали при комнатной температуре в течение ночи, реакционную смесь при охлаждении льдом смешивали с 20%-ным раствором NH4Cl (20 мл) и водой (30 мл) и экстрагировали простым эфиром (3×50 мл). Органич. фазу промывали водой (50 мл) и затем насыщенным раствором NaCl (50 мл), высушивали над Na2SO4 и концентрировали в вакууме.
Исходный продукт растворяли в этилметилкетоне (25 мл), при охлаждении льдом смешивали с ClSiMe3 (3,2 мл, 25 ммоль) и перемешивали при комнатной температуре 5 ч. Образовавшийся осадок отфильтровывали и высушивали в вакууме.
Выход: 2,8 г (62%).
1H-ЯМР (ДМСО-d6): 1.91 (8Н, m); 2.54 (6H, s); 3.91 (4H, d); 7.37 (1H, m); 7.61 (3H, m).
Вариант 1: 4-диметиламино-4-(3-фтор-фенил)-циклогексанон (Ket-7)
[8-(3-Фтор-фенил)-1,4-диокса-спиро[4.5]дец-8-ил]-диметил-амин гидрохлорид (7,2 г, 22,75 ммоль) растворяли в воде (9,6 мл), смешивали с концентрированной соляной кислотой (14 мл, 455 ммоль) и 4 дн. перемешивали при комнатной температуре. После оконченного гидролиза реакционную смесь экстрагировали диэтиловым эфиром (2×50 мл), водную фазу при охлаждении льдом подщелачивали с помощью 5N натрового щелока, причем продукт выпадал в осадок. Кетон Ket-7 можно было выделить в виде желтого твердого вещества с точкой плавления в 83-88°С и выходом в 50% (6,05 г).
Вариант 2: 4-диметиламино-4-(3-фтор-фенил)-циклогексанон (Ket-7)
[8-(3-Фтор-фенил)-1,4-диокса-спиро[4.5]дец-8-ил]-диметил-амин гидрохлорид (2,80 г, 8,86 ммоль) растворяли в воде (3,7 мл), смешивали с концентрированной соляной кислотой (5,5 мл) и перемешивали при КТ 4 дн. После оконченного гидролиза реакционную смесь экстрагировали простым эфиром (2×10 мл), водный раствор при охлаждении льдом подщелачивали с помощью 5N натрового щелока, реакционную смесь экстрагировали дихлорметаном (3×50 мл), органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме. Исходный продукт очищали путем флеш-хроматографии с CHCl3/МеОН (20:1).
Выход (Ket-7): 676 мг (32%), бесцветное твердое вещество.
Точка плавления: 62-67°С.
1H-ЯМР (ДМСО-d6): 2.02 (6H, s); 2.12 (5H, m); 2.45 (3H, m); 7.24 (3H, m); 7.43 (1H, m).
Элементарное звено Ket-8
Диметил-(8-m-толил-1,4-диокса-спиро[4.5]дец-8-ил)-амин гидрохлорид
В 500 мл трехгорлой колбе помещали 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил (8,4 г, 40 ммоль) в абс. ТГФ (150 мл) под аргоном и охлаждением льдом. При 0°С в течение 15 мин прибавляли по каплям бромид m-толилмагния, 1М раствор в ТГФ (100 мл, 100 ммоль). Реакционный состав затем перемешивали 16 ч при комнатной температуре.
Состав при охлаждении льдом смешивали с раствором хлорида аммония (20%-ный, 37 мл) и водой (50 мл) и простым эфиром (3×50 мл) экстрагировали.
Органическую фазу промывали водой (50 мл) и насыщенным раствором NaCl, высушивали над сульфатом натрия и концентрировали в вакууме. Выход неочищенного продукта составил 11,25 г (коричневое масло).
Исходный продукт растворяли в этилметилкетоне (60 мл) и при 0°С смешивали с триметилхлорсиланом (7,6 мл, 60 ммоль). После 5 ч помешивания при комнатной температуре выпавший в осадок остаток отсасывали и повторно промывали небольшим количеством холодного этилметилкетона.
Выход: 5,64 г (45%), белое твердое вещество.
Точка плавления: 230-234°С.
1Н-ЯМР (ДМСО-d6): 1.19 (2Н, t); 1.67 (2H; d); 2.13 (2H, t); 2.44 (9H, m); 2.89 (2H, d); 3.87 (4H, m); 7.43 (4H, m); 10.82 (1H, bs).
4-Диметиламино-4-m-толил-циклогексанон (Ket-8)
Диметил-(8-m-толил-1,4-диокса-спиро[4.5]дец-8-ил)-амин гидрохлорид (2,76 г, 10 ммоль) растворяли в воде (4,2 мл), смешивали с концентрированной соляной кислотой (6,15 мл) и 76 ч перемешивали при КТ.
Раствор экстрагировали простым эфиром (2×25 мл), эфирную фазу отводили. К водному раствору по каплям добавляли 5N NaOH, пока он не был щелочным. Затем экстрагировали дихлорметан (3×25 мл), органическую фазу повторно промывали водой (25 мл), высушивали над Na2SO4 и концентрировали.
Выход Ket-8: 1,69 г (73%), желтое масло.
1Н-ЯМР (ДМСО-d6): 2.05 (10H, m); 2.35 (3H, s); 2.52 (2H, m); 2.62 (2H, m); 7.12 (1H, m); 7.23 (3H, m).
Элементарное звено Ket-9
Вариант 1: (8-бутил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид
8-Диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил (10,5 г, 50 ммоль) помещали в ТГФ (150 мл) при охлаждении льдом и аргоном. В течение 15 мин по каплям прибавляли хлорид 2М бутилмагния в ТГФ (62,5 мл, 125 ммоль) и перемешивали 16 ч при КТ.
Состав при охлаждении льдом смешивали с 20%-ным раствором хлорида аммония (37 мл) и водой (50 мл) и экстрагировали простым эфиром (3×50 мл). Органич. фазу промывали водой (1×50 мл) и насыщенным раствором хлорида натрия (1×50 мл), органическую фазу высушивали с помощью Na2SO4 и концентрировали в вакууме. Исходный продукт (2,05 г) растворяли в этилметилкетоне (75 мл), при охлаждении льдом смешивали с ClSiMe3 (9,5 мл, 75 ммоль) и 6 ч перемешивали при КТ. Выпавший в осадок белый остаток отсасывали и высушивали в вакууме.
Выход: 3,1 г (22%).
1H-ЯМР (ДМСО-d6): 0.91 (3Н, t); 1.31 (4H, m); 1.56 (2H, m); 1.75 (8H, m); 2.64 (6H, s); 3.87 (4H, s); 9.87 (1H, s).
Вариант 1: 4-бутил-4-диметиламино-циклогексанон (Ket-9)
8-Бутил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид (3,10 г, 11,1 ммоль) помещали в H2O (4,7 мл) и конц. HCl (7 мл) и 24 ч перемешивали при КТ. Состав экстрагировали простым эфиром (1×15 мл), водную фазу при охлаждении льдом подщелачивали с помощью 5N NaOH и экстрагировали дихлорметаном (3×20 мл). Органич. фазу высушивали над Na2SO4 и концентрировали в вакууме.
Выход: 1,96 г (89%), масло.
1H-ЯМР (ДМСО-d6): 0.88 (3H, t); 1.23 (4H, m); 1.40 (2H, m); 1.68 (2H, m); 1.91 (2H, m); 2.31 (2H, m); 2.22 (6H, s); 2.42 (2H, m).
13С-ЯМР (ДМСО-d6): 13.91; 23.21; 26.06; 29.53; 31.07; 37.04; 38.88; 55.36; 210.37.
Вариант 2: (8-бутил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид
К раствору 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрила (38,3 г, 0,182 моль) в абс. тетрагидрофуране (420 мл) при охлаждении смесью изо льда и поваренной соли медленно прибавляли под аргоном раствор хлорида 2М n-бутилмагния в ТГФ (228 мл, 0,456 моль). При этом температура реакции не должна превышать 10°С. Затем перемешивали 16 ч при комнатной температуре. Образовался коричневый прозрачный раствор. Для переработки реакционной смеси прибавляли по каплям при охлаждении льдом (от 0 до 10°С) насыщенный раствор хлорида аммония (150 мл). При этом образовалось белое твердое вещество, которое растворяли путем добавления воды (приблизительно 250 мл). Реакционную смесь экстрагировали диэтиловым эфиром (4×100 мл). Органическую фазу промывали водой (100 мл) и насыщенным раствором NaCl (100 мл), высушивали и концентрировали. Оставалось желтое масло (44,5 г), которое кроме целевого бутилового соединения содержало еще исходный продукт нитрил. Исходный продукт растворяли в этилметилкетоне (275 мл), при охлаждении льдом смешивали с ClSiMe3 (32 мл, 0,245 моль) и перемешивали в открытой колбе при комнатной температуре. Гидрохлорид отделяли путем многократной фильтрации с интервалом в 2 ч. После продолжительности реакции в 6-8 ч можно было выделить (8-бутил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид с выходом в 82% (41,8 г) в виде белого твердого вещества.
Вариант 2: 4-бутил-4-диметиламино-циклогексанон (Ket-9)
(8-Бутил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид (41,8 г, 0,15 ммоль) растворяли воде (78 мл) и при помешивании и охлаждении льдом смешивали с 37-проц. соляной кислотой (100 мл, 1,2 моль). Прозрачный реакционный раствор перемешивали 7 дней при комнатной температуре. После оконченного гидролиза реакционную смесь экстрагировали диэтиловым эфиром (2×70 мл). Органические экстракты отводили. Водную фазу при охлаждении льдом подщелачивали посредством 5N натрового щелока (приблизительно 250 мл) и сильно перемешивали. Раствор экстрагировали диэтиловым эфиром (3×100 мл). Объединенные органические экстракты промывали водой (2×70 мл), высушивали и концентрировали. 4-Бутил-4-диметиламино-циклогексанон (Ket-9) выделяли в виде светло-коричневого масла с выходом в 96% (28,4 г). Выход кетона - в пересчете на использованный в первой стадии кеталь - составил 75%.
Элементарное звено Ket-10
Диметил-(8-фенэтил-1,4-диокса-спиро[4.5]дец-8-ил)амин-гидрохлорид
К раствору 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрила (39 г, 186 ммоль) в ТГФ (300 мл) прибавляли под аргоном и охлаждением льдом в течение 15 мин раствор 1М хлорида 2-фенилэтилмагния в ТГФ (550 мл, 550 ммоль) и затем перемешивали 16 ч при комнатной температуре. Для переработки реакционной смеси при охлаждении льдом добавляли насыщенный раствор хлорида аммония (295 мл) и воду (120 мл) и экстрагировали диэтиловым эфиром (3×150 мл). Органическую фазу промывали водой (100 мл) и насыщенным раствором NaCl (100 мл) и затем концентрировали. Оставалось коричневое масло (60,4 г). Исходный продукт растворяли в этилметилкетоне (310 мл) и при охлаждении льдом смешивали с ClSiMe3 (35,6 мл, 282 ммоль). Через 16 ч при КТ образовавшееся твердое вещество отсасывали и промывали этилметилкетоном.
Выход: 50 г (83%).
Точка плавления: 275-278°С.
Диметиламино-4-фенэтилциклогексанон (Ket-10)
Диметил-(8-фенэтил-1,4-диокса-спиро[4.5]дец-8-ил)амин-гидрохлорид (50 г, 154 ммоль) растворяли в воде (60 мл), смешивали с концентрированной соляной кислотой (97,2 мл, 3,16 моль) и 4 дн. перемешивали при комнатной температуре. После оконченного гидролиза реакционную смесь экстрагировали диэтиловым эфиром (2×100 мл) и водную фазу при охлаждении льдом подщелачивали с помощью 5N натрового щелока, причем в осадок выпадало твердое вещество, которое отсасывали, промывали с помощью H2O (3×20 мл) и затем высушивали.
Выход Ket-10: 25,3 г (67%), желтое твердое вещество.
Точка плавления: 60°С.
Элементарное звено Ket-12
Это элементарное звено получали при указанных условиях реакции вместо желаемого целевого продукта. Является очевидным, что (8-этил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид также целенаправленно может быть получен из бромида этилмагния и 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрила.
(8-Этил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид
Смесь этилбромида (30,0 г, 0,3 моль) и 3-бромпиридин (16,0 г, 0,1 моль) по каплям подавали к магнию в порошке (10,0 г) в диэтиловом эфире (50 мл). После того как было окончено образование Гриньяра, серый раствор при 0°С в течение 15 мин смешивали с 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрилом (10,5 г, 47,6 ммоль) в ТГФ (80 мл) и реакционный раствор перемешивали в течение ночи при КТ. Затем реакционный раствор при охлаждении льдом смешивали с 20%-ным раствором хлорида аммония (50 мл) и водой (50 мл). Реакционный раствор разбавляли диэтиловым эфиром (100 мл), органич. фазу отделяли и водн. фазу 2× экстрагировали с помощью EtaO (100 мл). Объединенные органич. фазы промывали водой (50 мл) и раствором NaCl (50 мл), высушивали над Na2SO4, фильтровали и растворитель удаляли в вакууме. Остаток ресуспендировали в 2-бутаноне (200 мл) и при 0°С смешивали с Me3SiCl (10 мл). Реакционный раствор 5 ч перемешивали при влажности воздуха и выпавшее в осадок твердое вещество отсасывали.
Выход: 6,8 г (64%), светло-коричневое твердое вещество.
1Н-ЯМР (ДМСО-d6): 0.94 (3Н, t); 1.51-1.60 (2H, m); 1.77-1.86 (8H, m); 2.64 (6H, 2 s); 3.83-3.89 (4H, m).
4-Диметиламино-4-этил-циклогексанон (Ket-12)
(8-Этил-1,4-диокса-спиро[4.5]дец-8-ил)-диметил-амин гидрохлорид (6,67 г, 0,026 ммоль) растворяли в 6N HCl (40 мл) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь дважды экстрагировали диэтиловым эфиром (100 мл). Затем при охлаждении льдом подщелачивали с помощью 5N NaOH и вновь три раза экстрагировали с помощью Et20 (100 мл). Объединенные органич. фазы высушивали над NaSO4, фильтровали и растворитель удаляли в вакууме.
Выход: 4,16 г (92%), коричневое масло.
1Н-ЯМР (ДМСО-d6): 0.81 (3Н, t); 1.43-1.50 (2H, q); 1.67-1.89 (2H, m); 1.83-1.89 (2H, m); 1.99-2.06 (2H, m); 2.22 (6H, 2 s); 2.39-2.43 (4H, m).
13С-ЯМР (ДМСО-d6): 8.71; 21.99; 30.41; 36.17; 37.07; 38.66; 55.53; 210.57.
Элементарное звено Ket-13
4-(8-Бензил-1,4-диоксаспиро[4.5]дец-8-ил)морфолин
В нагретой колбе нагревали раствор морфолина (958 мг, 0,96 мл, 11 ммоль), 1,4-диоксаспиро[4.5]дец-8-он (1,56 г, 10 ммоль) и 1,2,3-триазол (829 мг, 12 ммоль) в толуоле (10 мл) 6 ч кипятили в водоотделителе с обратным холодильником. Затем этот раствор под аргоном добавляли по каплям к 2М раствору хлорида бензилмагния в тетрагидрофуране (20 мл, 40 ммоль) таким образом, что внутренняя температура оставалась ниже 24°С. Смесь перемешивали 2 ч при комнатной температуре и после этого при охлаждении воды со льдом добавляли по каплям к 20% раствору хлорида аммония (25 мл). Органическую фазу отделяли и водную фазу экстрагировали диэтиловым эфиром (3×20 мл). Объединенные органические фазы промывали с помощью 2N натрового щелока (40 мл) и воды (40 мл), высушивали с помощью сульфата натрия и концентрировали в вакууме. Исходный продукт (4 г) очищали путем флеш-хроматографии (400 г, 20×7,5 см) с этилацетат/циклогексан (1:3).
Выход: 2,87 г (90%), белые кристаллы.
Точка плавления: 97-101°C.
1H-ЯМР (CDCl3): 1.35-1.52 (m, 4Н); 1.72-1.96 (m, 4H); 2.61-2.66 (m, 4H); 2.67 (s, 2H); 3.68-3.75 (m, 4H); 3.78-3.92 (m, 4H); 7.08-7.34 (m, 5H).
4-Бензил-4-морфолин-4-илциклогексанон (Ket-13)
К раствору 4-(8-бензил-1,4-диоксаспиро[4.5]дец-8-ил)морфолина (1,00 г, 3,15 ммоль) в ацетоне (5 мл) добавляли 6М соляную кислоту (5 мл). Через 24 ч реакционный раствор смешивали с дополнительной 6М соляной кислотой (2,5 мл), перемешивали последующие 3 дн. при комнатной температуре, затем подщелачивали (pH~9) с помощью 25% раствора карбоната калия и экстрагировали диэтиловым эфиром (3×20 мл). Объединенные органические фазы высушивали с помощью сульфата натрия и концентрировали в вакууме.
Выход: 753 мг (87%), белое твердое вещество (Ket-13).
Точка плавления: 124-127°С.
1H-ЯМР (CDCl3): 1.47-1.72 (m, 2H); 1.98-2.14 (m, 4H); 2.48-2.68 (m, 2H); 2.70-2.77 (m, 2H); 2.78 (s, 4H); 3.72-3.81 (s, 4H); 7.12-7.36 (m, 5H).
Элементарное звено Ket-14
1-Хлор-3-метокси-пропан
(Letsinger; Schnizer; J. Org. Chem.; 16; 1951; 704, 706)
3-Метоксипропан-1-ол (47,1 г, 50 мл, 0,523 моль) растворяли в пиридине (41,3 г, 42,6 мл, 0,523 моль), охлаждали до 10°С и при 10-30°С при сильном помешивании по каплям смешивали с тионилхлоридом (93,3 г, 56,9 мл, 0,784 моль). При этом в осадок выпадал твердый остаток, затем смесь перемешивали 3 ч при 65°С.
Состав выливали на смесь из льда (130 г) и конц. HCl (26 мл). Водный раствор экстрагировали простым эфиром (2×20 мл) и объединенные органические фазы промывали с помощью раствора K2CO3. При добавлении сушильного агента K2CO3 наблюдалось сильное газообразование, поэтому раствор оставляли отстаиваться в течение ночи.
Сушильный агент отфильтровывали и органическую фазу промывали раствором K2CO3 до щелочной реакции. Органическую фазу отделяли, промывали водой и высушивали над K2CO3, фильтровали и дистиллировали при нормальном давлении.
Точка кипения: 113°С.
Выход: 41,2 г (72%), бесцветная жидкость.
1Н-ЯМР (ДМСО-d6): 1.93 (2H, m); 3.23 (3H, s); 3.44 (2H, t); 3.66 (2H, t).
[8-(3-Метокси-пропил)-1,4-диокса-спиро[4.5]дец-8-ил]-диметил-амин
Магний (10,0 г, 92 ммоль) и I2 в абс. диэтиловом эфире (30 мл) под атмосферой аргона и при временном нагревании смешивали по каплям с раствором 1-хлор-3-метокси-пропана (10,0 г, 92 ммоль) в абс. простом эфире (15 мл). Затем состав при кипячении с обратным холодильником 60 мин перемешивали, после этого магний был не полностью растворенным.
При охлаждении льдом по каплям прибавляли раствор 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрила (9,68 г, 46 ммоль) в абс. ТГФ (30 мл). При этом в осадок выпадал вязкий остаток, для лучшего перемешивания добавляли еще 100 мл абс. ТГФ. Смесь перемешивали при комнатной температуре 24 ч.
Состав при охлаждении льдом смешивали с 20%-ным раствором NH4Cl (100 мл) и водой (120 мл), органическую фазу отделяли и водную фазу экстрагировали простым эфиром (3×120 мл).
Объединенные органические фазы промывали насыщенным раствором NaCl (120 мл) и водой (120 мл), высушивали над Na2SO4 и концентрировали в вакууме. Выход неочищенного продукта составил 10,8 г коричневого масла.
9,8 г неочищенного продукта очищали с помощью флеш-хроматографии с CHCl3/МеОН 
Выход: 8,11 г (75%), желтое масло.
1Н-ЯМР (ДМСО-d6): 1.44 (8Н. m); 1.62 (4H; m); 2.25 (6H, s); 3.21 (3H, s); 3.31 (2H, m); 3.82 (4H, s).
13С-ЯМР (ДМСО-d6): 23.99; 26.52; 28.87; 29.88; 36.97; 55.24; 57.67; 63.40; 72.62; 108.07.
4-Диметиламино-4-(3-метокси-пропил)-циклогексанон (Ket-14)
[8-(3-Метокси-пропил)-1,4-диокса-спиро[4.5]дец-8-ил]-диметил-амин (8,11 г, 31,5 ммоль) растворяли в воде (12 мл), при охлаждении льдом смешивали с конц. HCl (19,5 мл) и 3 дн. перемешивали при комнатной температуре. Реакционную смесь промывали простым эфиром (2×75 мл). Затем раствор подщелачивали с помощью 5N NaOH и экстрагировали дихлорметаном (3×75 мл). Объединенные органические фазы промывали водой (75 мл), высушивали над Na2SO4, фильтровали и растворитель удаляли в вакууме.
Выход: 6,03 г (90%), желтое масло.
1Н-ЯМР (ДМСО-d6): 1.44 (4H, m); 1.68 (2H; m); 1.88 (2H, m); 2.00 (1H, m); 2.05 (1H, m); 2.20 (6H, s); 2.41 (2H, m); 3.22 (3H, s); 3.28 (2H, m).
13С-ЯМР (ДМСО-d6): 24.01; 26.34; 30.88; 36.15; 37.06; 55.26; 57.70; 72.55; 210.39.
Элементарное звено Ket-15
8-Ацетидин-1-ил-1,4-диоксаспиро[4.5]декан-8-карбонитрил
К смеси 4N соляной кислоты (8,1 мл), метанола (4,9 мл) и ацетидина (8,5 г, 10 мл, 149 ммоль) подавали при охлаждении льдом сначала 1,4-диоксаспиро[4.5]декан-8-он (4,84 г, 31 ммоль) и после этого цианид калия (4,85 г, 74,4 ммоль) в воде (15 мл). Смесь перемешивали 5 дн. при комнатной температуре, затем смешивали с водой (50 мл) и экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические фазы высушивали с помощью сульфата натрия и концентрировали в вакууме.
Выход: 6,77 г (98%), масло.
1H-ЯМР (ДМСО-d6): 1.45-1.63 (m, 4H); 1.67-1.82 (m, 4H); 1.99 (q, 2H, J=7.1 Hz); 3.21 (t, 4H, J=7.1 Hz); 3.86 (s, 4H).
1-(8-Фенил-1,4-диоксаспиро[4.5]дец-8-ил)ацетидин
2M раствор хлорида фенилмагния в тетрагидрофуране (12 мл, 24 ммоль) под аргоном и охлаждением льдом по каплям смешивали с раствором только что полученного нитрила (2,20 г, 9,9 ммоль) в безводном тетрагидрофуране (25 мл) и после этого перемешивали при комнатной температуре в течение ночи. После добавления насыщенного раствора хлорида аммония (5 мл) и воды (5 мл) фазы разделяли и водную экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические фазы высушивали с помощью сульфата натрия и концентрировали в вакууме. Исходный продукт очищали путем флеш-хроматографии (100 г, 20×4,0 см) с этилацетат/циклогексан (1:1).
Выход: 670 мг (25%), бесцветное масло.
1H-ЯМР (ДМСО-d6): 1.27-1.40 (m, 2H); 1.55-2.00 (m, 8Н); 2.86 (t, 4H, J=6.8 Hz); 3.76-3.89 (m, 4H); 7.24-7.45 (m, 5H).
4-Ацетидин-1-ил-4-фенилциклогексанон (Ket-15)
Раствор только что полученного ацеталя (370 мг, 1,3 ммоль) в ацетоне (30 мл) смешивали с 6N соляной кислотой (2 мл) и в течение ночи перемешивали при комнатной температуре. Путем добавления 5N натрового щелока устанавливали pH 10 и водную фазу экстрагировали дихлорметаном (3×20 мл). Объединенные органические фазы высушивали с помощью сульфата натрия и концентрировали в вакууме.
Выход: 274 мг (92%), белое твердое вещество (Ket-15).
Точка плавления: не определяется.
1Н-ЯМР (ДМСО-d6): 1.67 (td, 2H, J=13.8, 6.9 Hz); 1.95-2.13 (m, 4H); 2.20-2.33 (m, 2H); 2.40-2.47 (m, 1H); 2.52-2.57 (m, 1H); 2.94 (t, 4H; J=6.9 Hz); 7.28-7.47 (m, 5H).
Элементарное звено Ket-16
1-(8-Пирролидин-1-ил-1,4-диоксаспиро[4.5]дец-8-ил)-1H-[1,2,3]триазол
К раствору 1,4 диоксаспиро[4.5]декан-8-она (3,9 г, 25 ммоль) в толуоле (40 мл) подавали пирролидин (1,95 г, 2,29 мл, 27,5 ммоль), 1,2,3-триазол (2,07 г, 30 ммоль) и молекулярное сито 4 Å (7,14 г). Смесь перемешивали 7 ч при 90°С. Затем раствор декантировали и немедленно подвергали дальнейшему взаимодействию.
1-(8-Бутил-1,4-диоксаспиро[4.5]дец-8-ил)пирролидин
К 2М раствору хлорида n-бутилмагния (25 мл, 50 ммоль) в тетрагидрофуране при охлаждении льдом и аргоне добавляли по каплям реакционный раствор только что полученного производного триазола (прибл. 6,9 г, 25 ммоль) в толуоле (38 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре и затем выливали в насыщенный раствор хлорида аммония (60 мл). Фазы разделяли и водную экстрагировали диэтиловым эфиром (3×70 мл). Объединенные органические фазы высушивали с помощью сульфата натрия, концентрировали в вакууме и остаток (12 г) очищали путем флеш-хроматографии (400 г, 20×7,6 см) с этилацетат/метанол (9,1).
Выход: 2,70 г (40% за две стадии), коричневое масло.
1H-ЯМР (ДМСО-d6): 0.87 (t, 3Н, J=7.1 Hz); 1.12-1.29 (m, 4H); 1.30-1.45 (m, 4H); 1.46-1.60 (m, 4H); 1.61-1.75 (m, 6Н); 1.93 (t, 1Н, J=7.1 Hz); 2.36 (t, 1H, J=7.0 Hz), 2.58 (br s, 2H), 3.83 (s, 4H).
4-Бутил-4-пирролидин-1-ил-циклогексанон (Ket-16)
Раствор только что полученного ацеталя (2,70 г, 10,1 ммоль) в ацетоне (100 мл) смешивали с водой (10,0 мл) и 37% соляной кислотой (14,0 мл) и перемешивали в течение ночи при комнатной температуре. Затем к смеси медленно добавляли по каплям 4М натровый щелок, пока pH не достиг 10. Смесь экстрагировали диэтиловым эфиром (4×40 мл), объединенные органические фазы высушивали с помощью сульфата натрия и концентрировали в вакууме. Исходный продукт (2,6 г) очищали путем флеш-хроматографии (260 г, 30×5,6 см) с этилацетат/метанол (9,1).
Выход: 1,06 г (47%), коричневое масло (Ket-16).
1H-ЯМР (ДМСО-d6): 0,88 (t, 3Н, J=6.7 Hz); 1.14-1.34 (m, 4H); 1.40-1.50 (m, 2H); 1.62-1.88 (m, 8H); 2.04 (dt, 2H, J=15.0, 3.9 Hz); 2.42 (ddd, 2H, J=6.3, 11.8, 15.5 Hz); 2.63 (t, 4H, J=6.0 Hz).
Элементарное звено Ket-17
Метил-[8-(2-метил-2Н-[1,2,4]триазол-3-ил)-1,4-диокса-спиро[4.5]дец-8-ил]-амин
Под атмосферой аргона помещали бутиллитий (2,5М в гексане, 9,2 мл, 23,0 ммоль) и охлаждали до -78°С. 1-Метил-1,2,4-триазол (1,30 мл, 23 ммоль) растворяли в абс. тетрагидрофуране (60,0 мл) и добавляли по каплям при охлаждении льдом при -78°С. Затем при этой температуре 10 мин перемешивали. К данному раствору быстро прибавляли по каплям 8-метиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил (2,12 г, 10,8 ммоль) в абс. тетрагидрофуране (15 мл) в ванной из охлаждающей смеси при -78°С. После добавления реакционный раствор 1 ч перемешивали в ванной из охлаждающей смеси и затем медленно нагревали до 0°С. При комнатной температуре реакционную смесь перемешивали в течение ночи. Затем гидролизировали при 0°С водой (10 мл), водную фазу экстрагировали хлороформом (3×50 мл), органическую фазу промывали водой (50 мл) и насыщенным раствором NaCl (50 мл), высушивали над Na2SO4 и концентрировали в вакууме.
Продукт очищали с помощью флеш-хроматографии с хлороформ/метанолом (15:1).
Выход: 1,93 г (71%).
1H-ЯМР (ДМСО-d6): 1.54 (2Н, m); 1.72 (2H, m); 1.91 (5H, m); 2.10 (2H, m); 3.84 (4H, m); 4.01 (3H, s); 7.74 (1H, s).
4-Метиламино-4-(2-метил-2H-[1,2,4]триазол-3-ил)-циклогексанон (Ket-17)
Метил-[8-(2-метил-2Н-[1,2,4]триазол-3-ил)-1,4-диоксаспиро[4.5]дец-8-ил]-амин (4,2 г, 16,646 ммоль) при комнатной температуре смешивали с 5-проц. серной кислотой (330 мл) и 3 дн. перемешивали при комнатной температуре. Реакционный состав смешивали с простым эфиром (120 мл). Фазы разделяли. Водную фазу подщелачивали с помощью 5N NaOH и экстрагировали дихлорметаном (4×50 мл). Органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме до сухости. Продукт можно было получить в виде бесцветного кристаллического твердого вещества с выходом в 83% (2,89 г, 13,862 ммоль).
Индольные элементарные звенья
Синтезы элементарных звеньев
Синтезы йодопиридинаминов известны из литературных источников и представлены посредством ортометаллирования соответствующих защищенных пивалоилом аминопиридинов в трехступенчатой последовательности: J.A.Turner, J.Org. Chem. 1983, 48, 3401; L.Estel, F.Marsais, G.Quéguiner, J.Org. Chem. 1988, 53, 2740; J. Malm, В. Rehn, A.-B.Hörnfeldt, S.Gronowitz, J.Het. Chem. 1994, 31, 11.
Синтез 4-(триэтилсилил)бут-3-ин-1-ола описан в литературных источниках и осуществляется аналогично следующим инструкциям: В.С.Bishop, I.F.Cottrell, D.Hands Synthesis, 1997, 1315; С.Cheng, D.R.Lieberman, R.D.Larsen, R.A.Reamer, T.R.Verhoeven, P.J.Reider Tet. Lett., 1994, 6981.
Азатриптофолы получают путем гетероаннелирования Ларока (Larock) с палладием в качестве посредника, триэтилсилилом, защищенные предшественники известны из литературных источников: F.Ujjainwalla, D.Warner Tet. Lett., 1998, 5355.
Общие элементарные звенья
Триэтил(4-(триэтилсилил)бут-3-инилокси)силан
3-Бутин-1-ол (34,99 г, 0,50 моль) растворяли в ТГФ (1,2 I) под азотом и охлаждали до -30°С. К этому раствору добавляли по каплям в течение 15 мин n-BuLi (640 мл, 1,02 моль, 1,6М раствор в n-гексане) таким образом, что температура не превышала -20°С. Через 1 ч при -20°С добавляли по каплям раствор триэтилхлорсилана (171,4 мл, 1,02 моль) в ТГФ (300 мл) в течение 30 мин. Ванну из охлаждающей смеси удаляли и реакционный раствор перемешивали в течение 15 ч при КТ. При охлаждении в ледяной ванной реакционную смесь резко охлаждали водным раствором Na2CO3 (1%-ным) и экстрагировали гексаном (2×500 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (300 мл) и высушивали над MgSO4. После фильтрации сушильного агента растворители удаляли в ротационном выпарном аппарате и остаток очищали дистилляцией (р=0,05 мбар, температура верха = 115-110°С) и получали триэтил(4-(триэтилсилил)бут-3-инилокси)силан (88,9 г, 60%).
4-(Триэтилсилил)бут-3-ин-1-ол
Триэтил(4-(триэтилсилил)бут-3-инилокси)силан (32,99 г, 110,66 ммоль) растворяли в МеОН (336 мл), смешивали с 2N HCl (62 мл) и смесь 4 ч перемешивали при КТ. Затем к раствору прибавляли гексан (250 мл) и H2O (200 мл) и водную фазу экстрагировали гексаном (3×100 мл). Объединенные органические фазы промывали посредством H2O (100 мл) и затем насыщенным раствором NaCl (50 мл) и высушивали над MgSO4. После фильтрации сушильного агента растворители удаляли в ротационном выпарном аппарате. Колоночная хроматография остатка (гексан/простой эфир = 4:1, затем простой эфир) дала 4-(триэтилсилил)бут-3-ин-1-ол (19,6 г, 96%) в виде бесцветного масла.
Элементарное звено Ind-1
N-(пиридин-2-ил)пиваламид
Под азотом ресуспендировали 2-аминопиридин (25,00 г, 265,6 ммоль) в ДХМ (425 мл) и смешивали с NEt3 (46,00 мл, 332 ммоль) Раствор охлаждали до -5°С, по каплям смешивали с раствором триметилацетилахлорида (35,95 мл, 292,20 ммоль) в ДХМ (50 мл) и перемешивали последующие 15 мин при -5°С. Затем смесь перемешивали 2 ч при КТ. Суспензию промывали с помощью H2O (200 мл), затем разбавленным раствором NaHCO3 и органическую фазу высушивали над MgSO4. После фильтрации сушильного агента и удаления растворителя в ротационном выпарном аппарате остаток (48,30 г) перекристаллизовывали из гексана (100 мл) при кипении. Получали N-(пиридин-2-ил)пиваламид (42,80 г, 91%) в виде бесцветных кристаллов.
N-(3-йодпиридин-2-ил)пиваламид
N-(пиридин-2-ил)пиваламид (14,25 г, 80 ммоль) и ТМЭДА (29,80 мл, 200 ммоль) растворяли в ТГФ (400 мл) под азотом и при -75°С по каплям смешивали с n-BuLi (125 мл, 200 ммоль; 1,6М раствор в n-гексане). Смесь перемешивали 15 мин при -75°С, затем 2 ч при -10°С. После вторичного охлаждения до -75°С прибавляли по каплям раствор йода (50,76 г, 200 ммоль) в ТГФ (200 мл) и реакционную смесь перемешивали в течение 2 ч. Нагревали до 0°С и резко охлаждали насыщенным водным раствором тиосульфата натрия. Водную фазу экстрагировали с помощью ДХМ (2×150 мл) и объединенные органические фазы высушивали над MgSO4. После фильтрации сушильного агента и удаления растворителя в ротационном выпарном аппарате остаток очищали с помощью колоночной хроматографии (простой эфир: циклогексан = 3:1) и таким образом получали N-(3-йодпиридин-2-ил)пиваламид (14,80 г, 61%).
3-Йодпиридин-2-амин
N-(3-йодпиридин-2-ил)пиваламид (13,80 г, 45,36 ммоль) ресуспендировали в H2SO4 (24 мас.%, 394 мл) и смесь 60 мин перемешивали в колбе с обратным холодильником. После охлаждения до КТ нейтрализовали с помощью 4N NaOH и твердым NaHCO3, водную фазу экстрагировали посредством ДХМ (3×200 мл) и объединенные органические фазы высушивали над MgSO4. После фильтрации растворитель удаляли в ротационном выпарном аппарате. Получали 3-йодпиридин-2-амин (9,70 г, 97%) в виде твердого вещества кремового цвета.
2-(2-(Триэтилсилил)-1Н-пирроло[2,3-b]пиридин-3-ил)этанол
Под азотом смесь из 3-йодпиридин-2-амина (0,46 г, 2,09 ммоль), 4-(триэтилсилил)бут-3-ин-1-ола (0,58 г, 3,14 ммоль), 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорид-дихлорометана (0,083 г, 0,105 ммоль), хлорида лития (0,086 г, 2,09 ммоль) и карбоната натрия (0,44 г, 4,18 ммоль) в ДМФ (21 мл) перемешивали 15 ч при 100°С. Реакционную смесь охлаждали до КТ, смешивали с EtOAc/простой эфир (1:1) и подавали в Н2О. Двухфазную суспензию фильтровали через фильтрующую землю. После разделения фаз водную фазу экстрагировали с помощью EtOAc (2x). Органические фазы объединяли и промывали посредством H2O и насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворители удаляли в ротационном выпарном аппарате. Колоночная хроматография остатка (n-гексан/EtOAc = 2:1, затем n-гексан/EtOAc = 1:1) дала 2-(2-(триэтилсилил)-1Н-пирроло[2,3-b]пиридин-3-ил)этанол (0,46 г, 80%).
2-(1Н-пирроло[2,3-b]пиридин-3-ил)этанол (Ind-1)
2-(2-(Триэтилсилил)-1Н-пирроло[2,3-b]пиридин-3-ил)этанол (1,00 г, 3,62 ммоль) перемешивали с ТБАФ (10,86 мл, 10,86 ммоль; 1М раствор в ТГФ) 6 ч при 50°С и затем 10 ч при КТ. Растворитель удаляли в ротационном выпарном аппарате и остаток очищали колоночной хроматографией (ДХМ/МеОН = 9:1, затем 1:1). Получали 2-(1Н-пирроло[2,3-b]пиридин-3-ил)этанол (0,46 г, 79%) в виде белого твердого вещества.
Элементарное звено Ind-2
2-(1Н-пирроло[2,3-с]пиридин-3-ил)этанол (Ind-2)
2-(2-(Триэтилсилил)-1Н-пирроло[2,3-с]пиридин-3-ил)этанол (1,99 г, 7,24 ммоль) растворяли в ТГФ (17 мл) и при 0°С смешивали с ТБАФ (7,96 мл, 7,96 ммоль; 1М раствор в ТГФ). Смесь перемешивали в течение 1 ч при 0°С и затем нагревали до КТ. Следовало вторичное добавление ТБАФ (7,96 мл, 7,96 ммоль, 1М раствор в ТГФ) и H2O (5 капель). Через 2 дн. при КТ добавляли 20 мл H2O и водную фазу экстрагировали посредством ДХМ (3×60 мл). Органические фазы объединяли и промывали насыщенным раствором NaCl (2×20 мл) и высушивали над MgSO4. После фильтрации сушильного агента растворители удаляли в ротационном выпарном аппарате и остаток очищали колоночной хроматографией (ДХМ/МеОН = 4:1). Получали 2-(1Н-пирроло[2,3-с]пиридин-3-ил)этанол (0,60 г, 51%) в виде бесцветного масла, которое со временем затвердевало.
Элементарное звено Ind-3
N-(5-(трифторометил)пиридин-2-ил)пиваламид
Под азотом ресуспендировали 5-(трифторометил)пиридин-2-амин (15 г, 92,5 ммоль) в ДХМ (190 мл) и смешивали с Net3 (16 мл, 115,7 ммоль). Раствор охлаждали до -5°С, по каплям смешивали с раствором триметилацетилхлорида (12,5 мл, 101,8 ммоль) в ДХМ (65 мл) и перемешивали последующие 15 мин в ледяной ванне. Затем смесь перемешивали 2 ч при КТ. Суспензию промывали посредством H2O (150 мл), затем разбавленным раствором NaHCO3 и органическую фазу высушивали над MgSO4. После фильтрации сушильного агента и удаления растворителя в ротационном выпарном аппарате остаток кристаллизировался. Получали N-(5-(трифторметил)пиридин-2-ил)пиваламид (20,8 г, 91).
N-(3-йодо-5-(трифторметил)пиридин-2-ил)пиваламид
N-(5-(трифторметил)пиридин-2-ил)пиваламид (22 г, 89,4 ммоль) и ТМЭДА (33,3 мл, 223,4 ммоль) растворяли в ТГФ (400 мл) под азотом и при -75°С по каплям смешивали с n-BuLi (139,6 мл, 223,4 ммоль; 1,6М раствор в n-гексане). Смесь 2 ч перемешивали при -75°С. Сохраняя температуру, прибавляли по каплям раствор йода (56,7 г, 223,4 ммоль) в ТГФ (280 мл) и реакционную смесь перемешивали в течение 2 ч. Нагревали до 0°С и резко охлаждали насыщенным водным раствором тиосульфата натрия. Водную фазу экстрагировали посредством ДХМ (2×150 мл) и объединенные органические фазы высушивали над MgSO4. После фильтрации сушильного агента и удаления растворителя в ротационном выпарном аппарате остаток очищали с помощью колоночной хроматографии (простой эфир/циклогексан = 1:1) и таким образом получали N-(3-йодо-5-(трифторметил)пиридин-2-ил)пиваламид (13 г, 39,1%).
3-Йод-5-(трифторметил)пиридин-2-амин
N-(3-йод-5-(трифторметил)пиридин-2-ил)пиваламид (16,8 г, 45,1 ммоль) ресуспендировали в H2SO4 (24 мас.%, 392 мл) и смесь 60 мин перемешивали в колбе с обратным холодильником. После охлаждения до КТ нейтрализовали с помощью 4N NaOH и твердого NaHCO3, водную фазу экстрагировали посредством ДХМ (3×200 мл) и объединенные органические фазы высушивали над MgSO4 После фильтрации растворитель удаляли в ротационном выпарном аппарате. Получали 3-йодо-5-(трифторметил)пиридин-2-амин (13 г, 100%) в виде твердого вещества кремового цвета.
2-(2-(Триэтилсилил)-5-(трифторметил)-1Н-пирроло[2,3-b]пиридин-3-ил)этанол
Под азотом перемешивали смесь из 3-йодо-5-(трифторметил)пиридин-2-амина (13 г, 45,1 ммоль), 4-(триэтилсилил)бут-3-ин-1-ола (12,4 г, 67,7 ммоль), 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорид-дихлорометана (1,78 г, 2,26 ммоль), хлорида лития (1,86 г, 45,1 ммоль) и карбоната натрия (9,54 г, 90,3 ммоль) в ДМФ (460 мл) 15 ч при 100°С. Реакционную смесь охлаждали до КТ, смешивали с 2 L EtOAc/простой эфир (1:1) и подавали в 2 L H2O. Двухфазную суспензию фильтровали через фильтрующую землю. После разделения фаз водную фазу экстрагировали с помощью EtOAc (2×1 L). Органические фазы объединяли и промывали посредством H2O и насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворители удаляли в ротационном выпарном аппарате. Колоночная хроматография остатка (простой трет-бутилметиловый эфир/n-гексан = 2:3). Затем полученную смешанную фракцию ресуспендировали 4× каждый раз в 20 мл дихлорметана и отсасывали от белого кристаллического твердого вещества. Первые 3 фракционных остатка объединяли 2-(2-(триэтилсилил)-5-(трифторметил)-1Н-пирроло[2,3-b]пиридин-3-ил)этанол (3,8 г, 24,4%).
2-(5-(Трифторметил)-1Н-пирроло[2,3-b]пиридин-3-ил)этанол (Ind-3)
2-(2-(Триэтилсилил)-5-(трифторметил)-1Н-пирроло[2,3-b]пиридин-3-ил)этанол (3,8 г, 11,0 ммоль) перемешивали с ТБАФ (33,1 мл, 33,1 ммоль; 1М раствор в ТГФ) 6 ч при 50°С и затем 10 ч при КТ. Растворитель удаляли в ротационном выпарном аппарате. Получали 2-(5-(трифторометил)-1Н-пирроло[2,3-b]пиридин-3-ил)этанол (2,4 г, 94,5%).
Элементарное звено Ind-4
S-2-(1Н-пирроло[2,3-b]пиридин-3-ил)этил этантиоат
Под защитным газом растворяли трифенилфосфин (3,56 г, 13,57 ммоль) в абсолютном ТГФ (20 мл). Светлый раствор охлаждали до -5°С. При помешивании прибавляли по каплям растворенный в ТГФ (20 мл) диизопропил-азодикарбоксилат (2,75 г, 13,6 ммоль) в течение 15 мин. При этом в осадок выпадал белый остаток. Суспензию перемешивали 30 мин при -5°С. После этого в течение 30 мин прибавляли по каплям смесь из Ind-1 (1100 мг, 6,78 ммоль) и тиоуксусной кислоты (965 мкл, 13,57 ммоль), растворенной в ТГФ (20 мл). Реакция была слегка экзотермической. Затем температуру удерживали еще один час при -5°С. При медленном нагревании до комнатной температуры из суспензии раствор становился прозрачным. После 18 ч помешивания при 23°С отгоняли ТГФ в значительной мере. Полученную смесь веществ (8,5 г, коричневое масло) разбавляли с этилацетатом (30 мл) и экстрагировали с помощью 1N соляной кислоты (1×10 мл, 3×5 мл). К объединенным водным фазам осторожно добавляли насыщенный раствор гидрокарбоната натрия (25 мл). Смесь экстрагировали дихлорметаном (3×10 мл). Объединенные органические фазы высушивали над сульфатом натрия, фильтровали и растворитель отгоняли в вакууме. S-2-(1Н-пирроло[2,3-b]пиридин-3-ил)этил этантиоат получали в виде почти белого твердого вещества (1,12 г, 75%).
2-(1H-пирроло[2,3-b]пиридин-3-ил)этантиол-гидрохлорид (Ind-4)
Под аргоном охлаждали метанол (20 мл) до ниже 0°С. Затем медленно прибавляли по каплям ацетилхлорид (3 мл, 42 ммоль). Реакция была экзотермической. Во время прикапывания температуру удерживали ниже 15°С. Реакционную смесь перемешивали 1 ч при комнатной температуре. S-2-(1Н-пирроло[2,3-b]пиридин-3-ил)этил этантиоат (600 мг, 2,724 ммоль) растворяли в метанол/дихлорметане 1:1 (10 мл) и затем прибавляли по каплям в течение 10 мин. Реакционную смесь перемешивали 18 ч при 23°С. Растворитель отгоняли. Остаток оранжевого цвета суспендировали в циклогексане (5 мл), фильтровали и промывали циклогексаном (3×1 мл). Ind-4 бежевого цвета, вероятно, получали как гидрохлорид (578 мг, 99%, точка плавления 138-150°С).
Элементарное звено Ind-5
S-2-(1Н-пирроло[3,2-с]пиридин-3-ил)этил этантиоат
Под защитным газом растворяли трифенилфосфин (3,83 г, 14,6 ммоль) в абсолютном ТГФ (20 мл). Светлый раствор охлаждали до -5°С. При помешивании в течение 15 мин прибавляли по каплям растворенный в ТГФ (20 мл) диизопропил-азодикарбоксилат (2,18 г, 10,78 ммоль). При этом в осадок выпадал белый остаток. Суспензию перемешивали 30 мин при -5°С. После этого в течение 30 мин прибавляли по каплям смесь из Ind-2 (1700 мг, 5,38 ммоль, чистота прибл. 51% (г/г)) и тиоуксусной кислоты (765 мкл, 10,75 ммоль), растворенной в ТГФ (20 мл). Реакция была слегка экзотермической. Затем температуру еще один час удерживали при -5°С. При медленном нагревании до комнатной температуры из суспензии становился светлый раствор. После 65 ч помешивания при 23°С отгоняли ТГФ в значительном количестве. Полученную смесь веществ (7,3 г, коричневое масло) разбавляли с этилацетатом (30 мл) и экстрагировали с помощью 1N соляной кислоты (1×10 мл, 3×5 мл). К объединенным водным фазам осторожно добавляли насыщенный раствор гидрокарбоната натрия (60 мл). Смесь экстрагировали дихлорметаном (3×10 мл). Объединенные органические фазы высушивали над сульфатом натрия. После фильтрации растворитель отгоняли в вакууме. S-2-(1Н-пирроло[3,2-с]пиридин-3-ил)этил этантиоат получали в виде коричневого масла (1,05 г, 82%).
2-(1Н-пирроло[3,2-с]пиридин-3-ил)этантиол-гидрохлорид (Ind-5)
Под аргоном охлаждали метанол (30 мл) до ниже 0°С. Затем медленно прибавляли по каплям ацетилхлорид (3 мл, 42 ммоль). Реакция была экзотермической. Во время прикапывания температуру удерживали ниже 15°С. Реакционную смесь перемешивали 1 ч при комнатной температуре. S-2-(1Н-пирроло[3,2-с]пиридин-3-ил)этил этантиоат (1,05 г, 4,77 ммоль) растворяли в метаноле (10 мл) и затем в течение 10 мин прибавляли по каплям. Реакционную смесь перемешивали 18 ч при 23°С. Растворитель отгоняли. Остаток оранжевого цвета суспендировали в диэтиловом эфире (10 мл), фильтровали и промывали диэтиловым эфиром (3×1 мл). Ind-5 бежевого цвета получали, вероятно, как гидрохлорид (975 мг, 95%, точка плавления 182-195°С).
Примеры
Пример 1
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], 1 диастереомер
Кетон Ket-1 (0,13 г, 0,62 ммоль) помещали вместе с Ind-1 (0,10 г, 0,62 ммоль) в дихлорметан ультрасухой (4 мл). Микроволновый сосуд был закрыт с помощью перегородки и промыт азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,47 мл, 2,47 ммоль). Смесь нагревали 2×20 мин при 120°С в микроволне. Затем реакционную смесь смешивали с 2N NaOH и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3x). Объединенные органические экстракты промывали водой и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество (0,71 г) смешивали с метанолом (7 мл) и смесь перемешивали в течение 2 ч при комнатной температуре. Твердое вещество отсасывали, повторно промывали небольшим количеством метанола и высушивали.
Выход (Прим. 1): 0,17 г (78%), твердое вещество кремового цвета.
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.65-2.8 (m, 16H), 3.17 (s, 1H), 3.96 (m, 2H), 6.93-6.8 (m, 1H), 7.40-7.70 (bm, 4H), 7.76 (d, 1H), 8.06 (d, 1H), 11.40 (s, 1H).
Пример 2
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; метансульфонат, смесь диастереомеров
Кетон Ket-1 (0,13 г, 0,62 ммоль) помещали вместе с Ind-1 (0,10 г, 0,62 ммоль) в дихлорметан (4 мл, ультрасухой). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,26 мл, 1,36 ммоль). Смесь нагревали 2×10 мин при 90°С в микроволне. Затем вновь прибавляли триметилсилилтрифторметансульфонат (0,26 мл, 1,36 ммоль) и смесь 1×10 мин, затем 1×20 мин при 120°С в микроволне нагревали.
Реакционную смесь в заключение смешивали с 2N NaOH и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3x). Объединенные органические экстракты промывали водой и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (7 мл) и смесь перемешивали в течение 2 ч при комнатной температуре. Твердое вещество отсасывали, промывали небольшим количеством метанола, высушивали (0,14 г, 63%). Для осаждения метансульфоната твердое вещество (0,14 г, 0,39 ммоль) взмучивали в дихлорметане (2 мл) и смешивали с метансульфоновой кислотой (0,028 мл). Теперь к светлому раствору прибавляли через 1 мин ацетон (0,5 мл) и по каплям простой эфир, пока не получили перемешиваемую смесь. Перемешивали 30 мин при комнатной температуре. Затем осадок при исключении воздуха отсасывали, порционно промывали простым эфиром и высушивали в высоком вакууме при 50°С. Выход (Прим. 2): 0,16 г (90%), смесь диастереомеров (прибл. 6:1).
Пример 3
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)], смесь диастереомеров
Кетон Ket-1 (0,13 г, 0,62 ммоль) помещали вместе с Ind-2 (0,10 г, 0,62 ммоль) в ультрасухом дихлорметане (4 мл). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,47 мл, 2,47 ммоль) Смесь 40 мин при 120°С нагревали в микроволне. Затем реакционную смесь смешивали с 2N NaOH и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали с помощью дихлорметана (3x). Объединенные органические экстракты промывали водой и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (7 мл) и смесь перемешивали в течение 48 ч при комнатной температуре. Твердое вещество отсасывали, повторно промывали небольшим количеством метанола и высушивали.
Выход (Прим. 3): 0,06 г (28%), твердое вещество кремового цвета (смесь диастереомеров: прибл. 5:1).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: диастереомер 1: 1.63 (t, 2H), 2.13 (bd, 2H), 2.16 (t, 2H), 2.27 (s, 6H), 2.11 (bd, 2H), 2.77 (m, 2H), 3.98 (m, 2H), 7.26 (d, 1H), 7.37 (d, 1H), 7.5 (m, 1H), 7.56 (t, 2H), 7.63 (m, 2H), 8.12 (d, 1H), 8.76 (s, 1H), 11.33 (bs, 1H), дополнительные пики, диастереомер 2: 1.74-1.80 (bt, 4H), 2.06 (s, 6H), 3.91 (m, 2H), 7.43-7.48 (m, 1H), 8,19 (d, 1H), 8.84 (s, 1H), 11.8 (bs, 1H).
Пример 4
4-(Диметиламино)-4-тиофен-2-ил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; метансульфонат, 1 диастереомер
Кетон Ket-2 (0,13 г, 0,62 ммоль) и 2-(1Н-пирроло[2,3-b]пиридин-3-ил)этанол Ind-1 (0,10 г, 0,62 ммоль) помещали под азотом в дихлорметане (4 мл, ультрасухой). Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,48 мл, 2,47 ммоль). Смесь 5 дней перемешивали при комнатной температуре. После добавления тетрагидрофурана смесь подщелачивали с помощью 1М водного раствора Na2CO3 (pH 11) и 20 мин перемешивали при комнатной температуре. Органическую фазу отделяли и водную фазу экстрагировали тетрагидрофураном (2х). Объединенные органические фазы промывали насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворители удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (3 мл) и 18 ч перемешивали при комнатной температуре. Твердое вещество отсасывали, промывали небольшим количеством метанола и высушивали в вакууме масляного насоса при 50°С (0,09 г, 43%). Для осаждения метансульфоната твердое вещество (0,07 г, 0,19 ммоль) взмучивали в дихлорметане (2 мл) и при комнатной температуре смешивали с метансульфоновой кислотой (0,014 мл). Суспензию через 5 мин смешивали с простым эфиром (10 мл). После 30 мин помешивания при комнатной температуре твердое вещество отсасывали при исключении воздуха, промывали простым эфиром и при 50°С в вакууме масляного насоса высушивали.
Выход (Прим. 4): 0,07 г (79%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.87-1.98 (m, 4H), 2.26-2.35 (m, 5H), 2.59-2.65 (m, 8H), 2.71 (t, 2H), 3.97 (t, 2H), 6.98-7.04 (m, 1H), 7.32 (t, 1H), 7.55 (d, 1H), 7.83 (d, 1H), 7.92 (d, 1H), 8.11 (d, 1H), 9.55 (bs, 1H), 11.60 (s, 1H).
Пример 5
4-(Диметиламино)-4-тиофен-2-ил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)], 1 диастереомер
Кетон Ket-2 (0,13 г, 0,62 ммоль) и Ind-2 (0,10 г, 0,62 ммоль) помещали под азотом в дихлорметане (4 мл, ультрасухой). Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,48 мл, 2,47 ммоль). Смесь перемешивали 6 дней при комнатной температуре. После добавления тетрагидрофурана смесь подщелачивали с помощью 1М водного раствора Na2CO3 (pH 11) и 20 мин перемешивали при комнатной температуре. Органическую фазу отделяли и водную фазу экстрагировали этиловым эфиром уксусной кислоты (2х). Объединенные органические фазы промывали насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворители удаляли в ротационном выпарном аппарате. Очистка колоночной хроматографией остатка (дихлорметан:метанол = 19:1) дала Прим. 5 (0,05 г, 22%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.84 (bt, 2H), 2.01 (bd, 2H), 2.29 (bt, 2H), 2.5 (s, 6H напластованный ДМСО), 2.60 (bd, 2H), 2.85 (t, 2H), 4.01 (t, 2H), 7.30-7.34 (m, 1H), 7.48-7.53 (m, 1H), 7.63 (d, 1H), 7.86-7.91 (m, 1H), 8.29 (d, 1H), 9.09 (s, 1H), 12.37 (bs, 1H).
Пример 6
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (4:3), 1 диастереомер
Ket-1 (0,27 г, 1,23 ммоль) помещали вместе с Ind-1 (0,20 г, 1,23 ммоль) в дихлорметане (4 мл, ультрасухой). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (1,1 мл, 4,9 ммоль). Смесь нагревали 40 мин при 120°С в микроволне. Реакционную смесь в заключение смешивали с 1М раствором Na2CO3 (pH 11) и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3х). Объединенные органические экстракты промывали водой и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (7 мл) и смесь перемешивали в течение 2 ч при комнатной температуре. Твердое вещество отсасывали, промывали небольшим количеством метанола, высушивали (0,257 г, 57%).
Для осаждения цитрата твердое вещество (0,1 г, 0,27 ммоль) взмучивали в горячем этаноле (6 мл) и смешивали с также горячим раствором лимонной кислоты (0,053 г) в этаноле (1,4 мл). Раствор перемешивали 3 дня при комнатной температуре. Затем осадок отсасывали, порционно промывали простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 6): 0,109 г (71%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.68-1.89 (m, 4H), 2.1-2.3 (m, 8H), 2.50-2.58 (q, 3Н), 2.68 (m, 4H), 3.96 (m, 2H), 6.93-6.99 (m, 1H), 7.43-7.64 (m, 5H), 7.75 (d, 1H), 8.06 (d, 1H), 11.40 (s, 1H).
Пример 7
4-(Метиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Кетон Ket-3 (0,125 г, 0,62 ммоль) помещали вместе с Ind-1 (0,10 г, 0,62 ммоль) в дихлорметане (4 мл, ультрасухой). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,48 мл, 2,5 ммоль). Смесь нагревали 30 мин при 120°С в микроволне. Реакционную смесь в заключение смешивали с раствором 2N NaOH и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3x). Объединенные органические экстракты промывали водой и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Очистка колоночной хроматографией остатка (дихлорметан:метанол = 4:1) дала 4-(метиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (0,06 г, 28%). Для осаждения цитрата взмучивали 4-(метиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (0,053 г, 0,15 ммоль) в горячем этаноле (4 мл) и смешивали с также горячим раствором лимонной кислоты (0,032 г) в этаноле (1 мл). Раствор перемешивали 3 ч при комнатной температуре. Затем осадок отсасывали, порционно промывали простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 7): 0,07 г (85%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.76-1.83 (bt, 2H), 1.87-1.93 (bd, 2H), 2.08 (s, 3H), 2.16-2.25 (bt, 2H), 2.47 (d, 2H). 2.54 (d, 2H), 2.55-2.60 (m, 2H), 2.70 (bt, 2H), 3.98 (bt, 2H), 6.95-6.99 (m, 1H), 7.49 (t, 1H), 7.56 (t, 2H), 7.66 (d, 2H), 7.77 (d, 1H), 8.08 (d, 1H), 11.46 (s, 1H), Breites Signal при 8.5-12.0.
Пример 8
4-(Метиламино)-4-тиофен-2-ил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (4:3), 1 диастереомер
Кетон Ket-4 (0,13 г, 0,62 ммоль) помещали вместе c Ind-1 (0,10 г, 0,62 ммоль) под азотом в дихлорметане (4 мл, ультрасухой). Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,48 мл, 2,5 ммоль). Смесь перемешивали 15 дней при комнатной температуре. После добавления тетрагидрофурана смесь подщелачивали с помощью 1М раствора Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали тетрагидрофураном (2х). Объединенные органические экстракты промывали насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (5 мл) и смесь перемешивали в течение 18 ч при комнатной температуре. Твердое вещество отсасывали, промывали небольшим количеством метанола, высушивали в вакууме масляного насоса (0,118 г, 54%). Для осаждения цитрата взмучивали твердое вещество (0,112 г, 0,32 ммоль) в горячем этаноле (4 мл) и смешивали с также горячим раствором лимонной кислоты (0,066 г) в этаноле (1,5 мл). Раствор перемешивали 3 ч при комнатной температуре. Затем осадок отсасывали, порционно промывали простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 8): 0,117 г (68%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.86-1.99 (m, 4H), 2.14 (s, 3H), 2.18 (bd, 2H), 2.31 (bd, 2H), 2.47 (d, 1.4H), 2.54 (d, 1.4H), 2.69 (t, 2H), 3.95 (t, 2H), 6.96-6.99 (m, 1H), 7.16 (m, 1H), 7.30 (m, 1H), 7.66 (d, 1H), 7.77 (d, 1H), 8.09 (d, 1H), 11.51 (s, 1H), шир. сигнал при 8.0-12.0.
Пример 9
4-(Диметиламино)-4-бензо[1,3-диоксол]-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
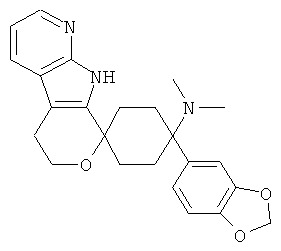
Кетон Ket-5 (0,16 г, 0,62 ммоль) помещали вместе с Ind-1 (0,10 г, 0,62 ммоль) под азотом в ультрасухом дихлорметане (4 мл). Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,48 мл, 2,5 ммоль). Смесь перемешивали 15 дней при комнатной температуре. После добавления тетрагидрофурана смесь подщелачивали с помощью 1М раствора Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали тетрагидрофураном (2х). Объединенные органические экстракты промывали насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (5 мл) и смесь в течение 2 ч перемешивали при комнатной температуре. Твердое вещество отсасывали, промывали небольшим количеством метанола и высушивали в вакууме масляного насоса при 50°С (0,052 г, 21%).
Для осаждения цитрата твердое вещество (0,047 г, 0,11 ммоль) взмучивали в горячем этаноле (4 мл) и смешивали с также горячим раствором лимонной кислоты (0,024 г) в этаноле (1 мл). Раствор 3 ч перемешивали при комнатной температуре. Затем осадок отсасывали, порционно промывали простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 9): 0,061 г (88%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.7-1.78 (t, 2H), 1.83-1.90 (d, 2H), 2.09-2.17 (m, 2H), 2,32-2.41 (m, 6H), 2.53 (d, 2H), 2.58 (d, 2H), 2.68 (t, 4H), 3.96 (t, 2H), 6.13 (s, 2H), 6.95-6.99 (m, 1H), 7.05-7.10 (m, 1H), 7.10-7.17 (m, 1H), 7.25 (m, 1H), 7.76 (d, 1H), 8.08 (d, 1H), 11.42 (bs, 1H).
Пример 10
4-(Диметиламино)-4-(бензотиофен-2-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Кетон Ket-6 (0,17 г, 0,62 ммоль) помещали вместе с Ind-1 (0,10 г, 0,62 ммоль) под азотом в дихлорметане (4 мл, ультрасухой). Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,48 мл, 2,5 ммоль). Смесь перемешивали 7 дней при комнатной температуре. После добавления тетрагидрофурана смесь подщелачивали с помощью 1М раствора Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3x). Объединенные органические экстракты промывали насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (5 мл) и смесь перемешивали в течение 2 ч при комнатной температуре. Твердое вещество отсасывали, промывали небольшим количеством метанола и высушивали в вакууме масляного насоса при 50°С (0,158 г, 61%).
Для осаждения цитрата твердое вещество (0,154 г, 0,37 ммоль) взмучивали в горячем этаноле (4 мл) и смешивали с также горячим раствором лимонной кислоты (0,071 г) в этаноле (2,5 мл). Раствор 3 ч перемешивали при комнатной температуре. Затем осадок отсасывали, порционно промывали простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 10): 0,193 г (85%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.89-2.03 (m, 4H), 2.19-2.28 (m, 2H), 2.39 (bs, 6H), 2.56-2.62 (m, 3H), 2.65-2.71 (m, 5H); 3.96 (m, 2H), 6.94-6.98 (m, 1H), 7.38-7.46 (m, 2H), 7.63 (bs, 1H), 7.76 (d, 1H), 7.93 (d, 1H), 8.02 (d, 1H), 8.05 (d, 1H), 11.43 (bs, 1H).
Пример 12
4-(Диметиламино)-4-(3-фторфенил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (4:3), 1 диастереомер
Кетон Ket-7 (0,145 г, 0,62 ммоль) помещали вместе с Ind-1 (0,10 г, 0,62 ммоль) под азотом в ультрасухом дихлорметане (4 мл). Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,48 мл, 2,5 ммоль). Смесь 7 дней перемешивали при комнатной температуре. После добавления дихлорметана смесь подщелачивали 1М раствором Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3x). Объединенные органические экстракты промывали насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (5 мл) и смесь перемешивали в течение 2 ч при комнатной температуре. Твердое вещество отсасывали, промывали небольшим количеством метанола и высушивали в вакууме масляного насоса при 50°С (0,054 г, 23%).
Для осаждения цитрата твердое вещество (0,054 г, 0,14 ммоль) взмучивали в горячем этаноле (3 мл) и смешивали с также горячим раствором лимонной кислоты (0,027 г) в этаноле (1 мл). Раствор 3 ч перемешивали при комнатной температуре. Затем осадок отсасывали, порционно промывали простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 12): 0,048 г (59%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.69 (bt, 2H), 1.85 (bd, 2H), 2.09 (bt, 2H), 2.20 (bs, 6H), 2.51-2.55 (d, 1.5H), 2.56-2.62 (d, 1.5H), 2.63 (m, 2H), 2.67 (t, 2H), 3.95 (t, 2H), 6.94-6.97 (m, 1H), 7.25-7.31 (m, 1H), 7.35-7.42 (m, 2H), 7.52-7.58 (m, 1H), 7.76 (d, 1H), 8.07 (d, 1H), 11.37 (s, 1H).
Пример 13
4-(Диметиламино)-4-(3-метилфенил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Кетон Ket-8 (0,142 г, 0,62 ммоль) помещали вместе с Ind-1 (0,10 г, 0,62 ммоль) под азотом в ультрасухом дихлорметане (4 мл). Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,48 мл, 2,5 ммоль). Смесь 7 дней перемешивали при комнатной температуре. После добавления дихлорметана смесь подщелачивали с помощью 1М раствора Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3х). Объединенные органические экстракты промывали насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (5 мл) и смесь перемешивали в течение 2 ч при комнатной температуре. Твердое вещество отсасывали, промывали небольшим количеством метанола и высушивали в вакууме масляного насоса при 50°С (0,117 г, 50%).
Для осаждения цитрата твердое вещество (0,112 г, 0,30 ммоль) взмучивали в горячем этаноле (4 мл) и смешивали с также горячим раствором лимонной кислоты (0,057 г) в этаноле (2 мл). Раствор 3 ч перемешивали при комнатной температуре. Затем осадок отсасывали, порционно промывали простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 13): 0,118 г (70%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.73 (m, 2H), 2.15 (m, 2H), 2.3 (bs, 6H), 2.43 (s, 3H), 2.56-2.58 (m, 3H), 2.67-2.69 (m, 4H), 3.96 (m, 2H), 6.95-6.97 (m, 1H), 7.32 (bm, 1), 7.42-7.44 (m, 3H), 7.77 (d, 1H), 8.07 (d, 1H), 11.41 (s, 1H).
Пример 14
4-(Диметиламино)-4-(бут-1-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1), 1 диастереомер
Кетон Ket-9 (0,3 г, 0,185 ммоль) помещали вместе с Ind-1 (0,10 г, 0,62 ммоль) под азотом в ультрасухом дихлорметане (4 мл). Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,48 мл, 2,5 ммоль). Смесь перемешивали 5 дней при комнатной температуре. После добавления дихлорметана смесь подщелачивали с помощью раствора 1М Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3х). Объединенные органические экстракты промывали насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Очистка колоночной хроматографией остатка (дихлорметан:метанол = 4:1) дала 4-(диметиламино)-4-(бут-1-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (0,026 г, 12%).
Для осаждения цитрата твердое вещество (0,02 г, 0,06 ммоль) взмучивали в горячем этаноле (2 мл) и смешивали с также горячим раствором лимонной кислоты (0,012 г) в этаноле (1 мл). Раствор 3 ч при комнатной температуре перемешивали. Затем осадок отсасывали, порционно промывали простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 14): 0,021 г (67%).
Пример 15
4-(Диметиламино)-4-фенилэтил-спиро[циклогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Кетон Ket-10 (0,319 г, 1,30 ммоль) помещали вместе c Ind-3 (0,3 г, 1,30 ммоль) в ультрасухом дихлорметане (3,4 мл). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (1,01 мл, 5,2 ммоль). Состав нагревали 10 мин при 120°С в микроволне. Затем реакционную смесь смешивали с 1М водным раствором Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3х). Объединенные органические экстракты промывали водой и насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное пенистое твердое вещество очищали с помощью колоночной хроматографии (силикагель KG 60, дихлорметан:метанол 9:1). Получали 4-(диметиламино)-4-фенилэтил-спиро[циклогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (0,14 г, 23,5%).
Для осаждения цитрата твердое вещество (0,140 г, 0,306 ммоль) растворяли в горячем этаноле (2 мл) и смешивали с лимонной кислотой (0,058 г) и с диэтиловым эфиром (10 мл). Раствор перемешивали при комнатной температуре. Твердое вещество отсасывали и высушивали в высоком вакууме при 60°С.
Выход (Прим. 15): 0,173 г (87%).
Пример 17
4-(Диметиламино)-4-этил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (2:3), смесь диастереомеров
Ket-12 (0,25 г, 1,48 ммоль) помещали вместе с Ind-1 (0,24 г, 1,48 ммоль) в ультрасухом дихлорметане (3,3 мл). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (1,14 мл, 5,92 ммоль). Состав нагревали при 120°С 20 мин в микроволне. Затем реакционную смесь смешивали с 1М водным раствором Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3х). Объединенные органические экстракты промывали водой и нас. раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (5 мл) и смесь в течение 2 ч перемешивали при комнатной температуре. Твердое вещество отсасывали, повторно промывали небольшим количеством метанола и высушивали в высоком вакууме при 50°С. Получали твердое вещество кремового цвета (0,07 г, 15,1%).
Твердое вещество (0,07 г, 0,22 ммоль) взмучивали в горячем этаноле (2 мл) и смешивали с также горячим раствором лимонной кислоты (0,043 г) в этаноле (1 мл). Раствор перемешивали 3 ч при комнатной температуре. Светлый раствор концентрировали до сухости и высушивали в высоком вакууме при 60°С.
Выход (Прим. 17): 0,083 г (62%).
Пример 18
4-(Диметиламино)-4-фенилэтил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (2:3), 1 диастереомер
Ket-10 (0,227 г, 0,925 ммоль) помещали вместе с Ind-1 (0,15 г, 0,925 ммоль) в ультрасухом дихлорметане (3,6 мл). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,715 мл, 3,70 ммоль). Состав перемешивали 10 дн. при КТ. Из-за неполного взаимодействия смесь нагревали 2× 15 мин при 120°С в микроволне. Затем реакционную смесь смешивали с 1М водным раствором Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3х). Объединенные органические экстракты промывали водой и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (7 мл) и смесь перемешивали в течение 1 ч при комнатной температуре. Твердое вещество отсасывали, повторно промывали небольшим количеством метанола и высушивали. Получали твердое вещество кремового цвета (0,15 г, 41,4%).
Твердое вещество (0,144 г, 0,37 ммоль) взмучивали в горячем этаноле (3 мл) и смешивали с также горячим раствором лимонной кислоты (0,071 г) в этаноле (2 мл). Раствор 3 ч при комнатной температуре перемешивали. Затем осадок отсасывали, промывали небольшим количеством этанола и 2х простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 18): 0,184 г (73%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.74-1.83 (m, 2H), 1.90-1.96 (m, 2H), 2.02-2.10 (m, 4H), 2.22-2.30 (m, 2H), 2.52-2.58 (d, 3Н), 2.58-2.65 (d, 3H), 2.65-2.75 (m, 5H), 2.79 (bs, 5H), 3.94 (t, 2H), 6.99-7.04 (m, 1H), 7.18-7.32 (m. 2H), 7.34-7.43 (m, 3Н), 7.82 (d, 1H), 8.14 (d, 1H), 11.70 (s, 1H).
Пример 19
4-Бензил-4-морфолино-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Ket-13 (0,252 г, 0,925 ммоль) помещали вместе с Ind-1 (0,15 г, 0,925 ммоль) в ультрасухом дихлорметане (3,6 мл). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,715 мл, 3,70 ммоль). Состав перемешивали 10 дн. при КТ. Затем реакционную смесь смешивали с 1М водным раствором Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3х). Объединенные органические экстракты промывали водой и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное твердое вещество смешивали с метанолом (7 мл) и смесь перемешивали 2 ч при комнатной температуре. Твердое вещество отсасывали, повторно промывали небольшим количеством метанола и высушивали в высоком вакууме при 50°С (0,248 г, 64,2%, твердое вещество кремового цвета).
Твердое вещество (0,234 г, 0,56 ммоль) взмучивали в горячем этаноле (5 мл) и смешивали с также горячим раствором лимонной кислоты (0,108 г) в этаноле (3 мл). Раствор в течение ночи перемешивали при комнатной температуре. Затем осадок отсасывали, промывали небольшим количеством этанола и 2х простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 19): 0,280 г (82%).
1H-ЯМР (600 МГц, D6-ДМСО) δ част. на млн: 1.35-1.55 (bs, 2H), 1.87 (bm, 4H), 2.20 (m, 2H), 2.62-2.74 (m, 10H), 3.13 (bm, 2H), 3.58 (bm, 4H), 3.92 (m, 2H), 7.00 (m, 1H), 7.17 (m, 1H), 7.32 (m, 1H), 7.46 (d, 1H), 7.80 (d, 1H), 8.16 (d, 1H), 11.66 (s, 1H).
Пример 20
4-(Диметиламино)-4-(3-метоксипропил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Ket-14 (0,197 г, 0,925 ммоль) помещали вместе с Ind-1 (0,15 г, 0,925 ммоль) в ультрасухом дихлорметане (3,6 мл). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (0,715 мл, 3,70 ммоль). Состав перемешивали 13 дн. при КТ. Состав нагревали 2× 15 мин при 120°С в микроволне. Затем реакционную смесь смешивали с 1М водным раствором Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3х). Объединенные органические экстракты промывали водой и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное масло очищали с помощью колоночной хроматографии (силикагель KG 60, дихлорметан:метанол 4:1) (0,1 г, 30,2%, твердое вещество кремового цвета).
Для осаждения цитрата твердое вещество (0,095 г, 0,27 ммоль) суспендировали в горячем этаноле (2 мл) и смешивали с также горячим раствором лимонной кислоты (0,051 г) в этаноле (2 мл). Раствор перемешивали в течение ночи при комнатной температуре. Затем осадок отсасывали, промывали небольшим количеством этанола и 2х простым эфиром и высушивали в высоком вакууме при 60°С.
Выход (Прим. 20): 0,111 г (76%).
Пример 21
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Кетон Ket-1 (247 мг, 1,1 ммоль) растворяли вместе с Ind-1 (184 мг, 1,1 ммоль) в дихлорметане (20 мл). Затем осуществляли добавление трифторметансульфоновой кислоты (363 мг, 0,215 мл, 2,42 ммоль), причем состав окрашивался в темный цвет. Перемешивали 20 ч при КТ. Для переработки реакционную смесь смешивали с 1N NaOH (8 мл) и 10 мин перемешивали. При этом цвет изменился с темно-красного на светло-коричневый. В осадок выпадало бесцветное твердое вещество. Твердое вещество (126 мг) отсасывали и промывали дихлорметаном (5 мл). Фазы фильтрата разделяли. Водную фазу экстрагировали дихлорметаном (2×10 мл). Объединенные органические фазы высушивали с помощью сульфата натрия, фильтровали и концентрировали в вакууме. Получали светло-коричневое твердое вещество (491 мг). Исходный продукт смешивали с этилацетатом (10 мл), при этом в осадок выпадало бесцветное твердое вещество, которое отсасывали (119 мг) и промывали этилацетатом (10 мл). Оба твердых вещества были идентичными и получали совместно (245 мг, 62%) с точкой плавления в 266-274°С.
Твердое вещество (215 мг, 0,59 ммоль) нагревали в этаноле (70 мл) до кипения. К мутному раствору добавляли лимонную кислоту (274 мг, 1,43 ммоль), растворенную в горячем этаноле (5 мл). Раствор был светлым. После 20-часового помешивания при КТ выпавший в осадок бесцветный остаток отсасывали и промывали этанолом (10 мл).
Прим. 21, таким образом, получали с выходом в 88% (288 мг).
1H-ЯМР (400 МГц, CD3OD) δ част. на млн: 1.01 (t, J=7.01 Hz, 1.5H), 1.53-1.78 (m, 2H), 1.78-1.93 (m, 2H), 2.00-2.41 (m, 8H), 2.42-2.61 (m, 5H), 2.61-2.82 (m, 3Н), 3.40 (d, J=6.83 Hz, 1H), 3.85-4.02 (m, 2H), 6.87-7.02 (m, 1H), 7.41-7.58 (m, 3Н), 7.58-7.69 (m, 2H), 7.73 (d, J=7.74 Hz, 1H), 8.03 (d, J=4.57 Hz, 1H), 11.40 (s, 1H).
13С-ЯМР (101 МГц, CD3OD) δ част. на млн: 18.5, 21.6, 26.2, 31.1, 37.4, 44.0, 56.0, 59.0, 70.8, 71.3, 104.3, 114.9, 118.6, 125.5, 128.6, 128.8, 129.1, 138.7, 141.8, 148.5, 171.1, 176.4.
Пример 22
4-(Диметиламино)-4-тиофен-2-ил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Кетон Ket-2 (268 мг, 1,2 ммоль) растворяли вместе с азатриптофолом Ind-1 (195 мг, 1,2 ммоль) в 1,2-дихлорэтане (20 мл). Затем осуществляли добавление трифторметансульфоновой кислоты (397 мг, 0,235 мл, 2,64 ммоль), причем состав окрасился в темно-лиловый цвет. Все вместе перемешивали 40 ч при КТ. Для переработки реакционную смесь с помощью 1N NaOH (8 мл) устанавливали до pH 11 и 15 мин перемешивали. При этом цвет изменился на светло-коричневый и в осадок выпало светло-коричневое твердое вещество. Твердое вещество (140 мг) отсасывали и промывали метанолом (15 мл). При этом продукт обесцветился. Фазы фильтрата разделяли. Водную фазу экстрагировали дихлорметаном (2×10 мл). Объединенные органические фазы высушивали с помощью сульфата натрия, фильтровали и концентрировали в вакууме. Твердый коричневый остаток перемешивали с метанолом (10 мл) 10 мин, затем отсасывали (58 мг) и промывали метанолом (10 мл). Оба твердых вещества были идентичными и объединены (198 мг, 45%).
Твердое вещество (123 мг, 0,33 ммоль) нагревали в этаноле (90 мл) до кипения. К мутному раствору подавали лимонную кислоту (152 мг, 0,79 ммоль), растворенную в горячем этаноле (5 мл). Раствор был светлым. Его кипятили дополнительно в течение 5 мин, затем охлаждали до комнатной температуры и концентрировали в вакууме до прибл. половины его объема. Затем раствор перемешивали 20 ч при комнатной температуре, причем в осадок выпадал бесцветный остаток. Его отсасывали и промывали этанолом (10 мл). Прим. 22 выделяли с выходом в 54% (100 мг) сточкой плавления в 278-283°С.
1H-ЯМР (400 МГц, CD3OD) δ част. на млн: 1.75-1.99 (m, 4H), 2.03-2.36 (m, 8H), 2.36-2.78 (m, 8H), 3.85-4.05 (m, 2H), 6.91-7.04 (m, 1H), 7.16-7.34 (m, 2H), 7.63-7.73 (m, 1H), 7.73-7.84 (m, 1H), 8.04-8.15 (m, 1H), 11.51 (s, 1H).
Пример 23
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)], цитрат (1:1), неполярный диастереомер
Пример 24
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)], цитрат (1:1), полярный диастереомер
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)] (неполярный и полярный диастереомер)
Кетон Ket-1 (267 мг, 1,23 ммоль) и 5-азатриптофол (Ind-2) (200 мг, 1,23 ммоль) растворяли в абс. 1,2-дихлорэтане (40 мл), смешивали с трифторметансульфоновой кислотой (0,12 мл, 204 мг, 1,36 ммоль) и 16 ч перемешивали при комнатной температуре. В реакционной колбе осаждалось светлое масло. Повторно прибавляли трифторметансульфоновую кислоту (0,12 мл, 204 мг, 1,36 ммоль) и 5 ч нагревали в колбе с обратным холодильником. Состав при комнатной температуре смешивали с водой (30 мл) и 1N натровым щелоком (10 мл) и 30 мин перемешивали. Фазы разделяли. Водную фазу экстрагировали 1,2-дихлорэтаном (3×30 мл). Органические фазы объединяли, высушивали сульфатом натрия и концентрировали. Остаток представлял собой твердое вещество бежевого цвета (455 мг), которое отделяли с помощью хроматографии [силикагель 60 (120 г); этилацетат/метанол 4:1 (500 мл), этилацетат/метанол 1:1 (500 мл), метанол (300 мл)]. 4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)] (неполярный диастереомер) выделяли в виде бесцветного твердого вещества с выходом в 30% (133 мг). Полярный диастереомер был загрязненным (158 мг, 35%).
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)], цитрат (1:1), неполярный диастереомер (Прим. 23)
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)] (неполярный диастереомер) (120 мг, 0,332 ммоль) растворяли при 60°С в этаноле (15 мл) и смешивали с этанольным раствором (2 мл) лимонной кислоты (140 мг, 0,73 ммоль). Через 22 ч прибавляли диэтиловый эфир (20 мл). Через 30 мин отделяли бесцветное твердое вещество путем фильтрации и промывали этанол/этилацетатом 1:1 (3 мл) и диэтиловым эфиром (2 мл). Прим. 23 получали с выходом в 56% (102 мг) с точкой плавления в 273-277°С.
1H-ЯМР (300 МГц, ДМСО-d6) δ част. на млн: 1.80-2.04 (m, 2H), 2.18-2.4 (m, 4H), 2.48 (s, 6H), 2.57 (dd, 4H), 2.79 (t, 2H), 3.91 (t, 2H), 7.15-7.3 (m, 6H), 8.24 (d, 1H), 8.94 (s, 1H), 12.17 (s, 1H).
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)], цитрат (1:1), полярный диастереомер (Прим. 24)
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)] (полярный диастереомер, загрязненный) (140 мг, 0,38 ммоль) растворяли в этаноле (7 мл) и смешивали с лимонной кислотой (163 мг, 0,85 ммоль), растворенной в этаноле (2 мл). Тот час же начиналось осаждение. Через 2 ч путем фильтрации отделяли бесцветное твердое вещество (103 мг) и промывали этанолом (2×2 мл) и диэтиловым эфиром (2 мл). Фильтрат смешивали с диэтиловым эфиром (10 мл), образовавшийся осадок отделяли и промывали диэтиловым эфиром (2×2 мл) (38 мг). Две фракции твердого вещества объединяли и смешивали с натровым щелоком и трихлорметаном (107 мг). Полученное твердое вещество в этаноле (30 мл) смешивали с лимонной кислотой (124 мг, 0,65 ммоль), растворенной в этаноле (3 мл). Через 20 мин отделяли твердое вещество и промывали этанолом (2×3 мл) и диэтиловым эфиром (2×2 мл) (59 мг). Фильтрат смешивали с диэтиловым эфиром (30 мл) и 16 ч перемешивали при комнатной температуре. Путем фильтрации и промывания диэтиловым эфиром (2×2 мл) выделяли бесцветное твердое вещество.
Выход (Прим. 24): 0,040 г (25%), точка плавления в 122-127°С.
1H-ЯМР (300 МГц, ДМСО-d6) δ част. на млн: 1.63 (t, 2H), 1.92 (d, 2H), 2.13-2.27 (m, 2H), 2.36 (s, 6H), 2.57 (dd, 4H), 2.68-2.83 (m, 4H), 3.99 (t, 2H), 7.32 (d, 1H), 7.49-7.63 (m, 3Н), 7.66-7.73 (m, 2H), 8.15 (d, 1H), 8.83 (s, 1H), 11.50 (s, 1H).
Пример 25
4-Бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), неполярный диастереомер
Пример 26
4-Бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), полярный диастереомер
4-Бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (неполярный и полярный диастереомер)
Кетон Ket-9 (462 мг, 2,34 ммоль) вместе с азатриптофолом Ind-1 (380 мг, 2,34 ммоль) растворяли в 1,2-дихлорэтане (40 мл). Затем осуществляли добавление трифторметансульфоновой кислоты (773 мг, 0,457 мл, 5,15 ммоль), причем состав обесцвечивался. Все вместе перемешивали 4 дн. при КТ. Для переработки реакционную смесь смешивали водой (10 мл), с помощью 1N NaOH (10 мл) устанавливали до pH 11 и 15 мин перемешивали. Затем фазы разделяли. Водную фазу экстрагировали дихлорметаном (2×10 мл). Объединенные органические экстракты высушивали с помощью сульфата натрия, фильтровали и в вакууме концентрировали. Коричневый масляный остаток смешивали с метанолом (10 мл), причем в осадок выпадало бесцветное твердое вещество, которое отсасывали и промывали метанолом (10 мл). При этом оно представляло собой 4-бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (неполярный диастереоизомер) (39 мг, 5%, незначительно загрязненный). Маточный раствор концентрировали и отделяли с помощью хроматографии [силикагель 60 (50 г); этилацетат (1200 мл), этилацетат/метанол 4:1 (500 мл), 1:1 (500 мл)]. Получали желтое твердое вещество (239 мг), которое 30 мин сильно перемешивали с хлороформом (25 мл), 1N NaOH (5 мл) и водой (10 мл). Затем фазы разделяли. Водную фазу экстрагировали хлороформом (2×10 мл). Объединенные органические экстракты высушивали с помощью сульфата натрия, фильтровали и в вакууме концентрировали. Желтый полутвердый остаток (230 мг, 30%) представлял собой полярный диастереоизомер, который содержал прибл. 24% неполярного продукта. Разделение должно было происходить над образованием цитрата.
4-Бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), неполярный диастереомер (Прим. 25)
4-Бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (неполярный диастереоизомер) (27 мг, 0,08 ммоль) растворяли в этаноле (20 мл) при небольшом нагревании и смешивали с лимонной кислотой (37 мг, 0,192 ммоль), растворенной в горячем этаноле (5 мл). После 20-часового помешивания при КТ светлый раствор почти полностью концентрировали и смешивали с диэтиловым эфиром (5 мл). В осадок начало выпадать бесцветное твердое вещество, которое отсасывали.
Выход (Прим. 25): 38 мг (88%).
Точка плавления: 86-92°С.
4-Бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), полярный диастереомер (Прим. 26)
4-Бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (неполярный диастереоизомер) (загрязненный неполярным диастереомером, 230 мг, 0,67 ммоль) растворяли в этаноле (20 мл) при небольшом нагревании и смешивали с лимонной кислотой (310 мг, 1,62 ммоль), растворенной в горячем этаноле (5 мл). После одночасового помешивания при КТ светлый раствор концентрировали прибл. до третьей части его объема и смешивали с диэтиловым эфиром (10 мл). В осадок начало выпадать бесцветное твердое вещество, которое отсасывали.
Выход (Прим. 26): 91 мг, (25%).
Точка плавления: 95-103°С.
Пример 27
4-Бензил-4-морфолино-спиро[циклогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Ket-13 (0,593 г, 2,17 ммоль) помещали вместе c Ind-3 (0,5 г, 2,17 ммоль) в ультрасухом дихлорметане (11 мл). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (1,68 мл, 8,69 ммоль). Состав нагревали 10 мин при 120°С в микроволне. Затем реакционную смесь смешивали с 1М водным раствором Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3х). Объединенные органические экстракты промывали водой и насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное пенистое твердое вещество очищали с помощью колоночной хроматографии (силикагель KG 60; дихлорметан:сложный эфир уксусной кислоты 9:1). Получали 4-бензил-4-морфолино-спиро[цикпогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (0,12 г, 8,2%) в виде твердого вещества кремового цвета.
Твердое вещество (0,120 г, 0,25 ммоль) растворяли в горячем этаноле (15 мл) и смешивали с лимонной кислотой (0,051 г) и диэтиловым эфиром (10 мл). Раствор перемешивали в течение ночи при комнатной температуре. Растворитель удаляли в ротационном выпарном аппарате. Остаток кристаллизовали с помощью n-гексан/этанола (10 мл; 9,5:0,5). Твердое вещество отсасывали и высушивали в высоком вакууме при 60°С.
Выход (Прим. 27): 0,173 г (100%).
Пример 28
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Ket-1 (0,283 г, 1,30 ммоль) помещали вместе с Ind-3 (0,3 г, 1,30 ммоль) в ультрасухом дихлорметане (3,4 мл). Микроволновый сосуд закрывали с помощью перегородки и промывали азотом. Затем осуществляли быстрое добавление триметилсилилтрифторметансульфоната (1,01 мл, 5,2 ммоль). Состав нагревали 10 мин при 120°С в микроволне. Затем реакционную смесь смешивали с 1М водным раствором Na2CO3 и 20 мин перемешивали. Органическую фазу отделяли и водную фазу экстрагировали дихлорметаном (3х). Объединенные органические экстракты промывали водой и насыщенным раствором NaCl и высушивали над MgSO4. После фильтрации сушильного агента растворитель удаляли в ротационном выпарном аппарате. Полученное пенистое твердое вещество очищали с помощью колоночной хроматографии (силикагель KG 60, дихлорметан:метанол 9:1) (0,116 г, 20,7%). Для осаждения цитрата твердое вещество (0,100 г, 0,23 ммоль) растворяли в горячем этаноле (2 мл) и смешивали с лимонной кислотой (0,058 г) и диэтиловым эфиром (45 мл). Перемешивали при комнатной температуре 1. Твердое вещество отсасывали и 5 ч высушивали в высоком вакууме при 60°С.
Выход (Прим. 28): 0,108 г (75%).
Пример 29
4-(Ацетидин-1-ил)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1), 1 диастереомер
Кетон Ket-15 (275 мг, 1,2 ммоль) растворяли вместе с азатриптофолом Ind-1 (195 мг, 1,2 ммоль) в дихлорметане (30 мл). Затем осуществляли добавление трифторметансульфоновой кислоты (397 мг, 0,235 мл, 2,64 ммоль), причем состав окрашивался в темный цвет. Перемешивали 20 ч при КТ. Для переработки реакционную смесь смешивали с 1N NaOH (10 мл) и водой (10 мл) и 10 мин перемешивали. При этом цвет изменился с темно-красного на светло-коричневый. Фазы фильтрата разделяли. Водную фазу экстрагировали дихлорметаном (2×10 мл). Объединенные органические фазы высушивали с помощью сульфата натрия, фильтровали и концентрировали в вакууме. Получали желтое твердое вещество, которое 10 мин перемешивали с дихлорметаном (5 мл), затем отсасывали и промывали дихлорметаном (5 мл) (116 мг, 26%, диастереоизомерно чистое, точка плавления: 269-274°С).
Твердое вещество (101 мг, 0,27 ммоль) нагревали в этаноле (60 мл) до кипения. К мутному раствору добавляли лимонную кислоту (125 мг, 0,65 ммоль), растворенную в горячем этаноле (5 мл). Раствор был прозрачным. После 20-часового помешивания при КТ раствор концентрировали до прибл. 3 мл и смешивали с диэтиловым эфиром (5 мл) до кристаллизации. Выпавший в осадок бесцветный остаток отсасывали и промывали диэтиловым эфиром (5 мл).
Выход (Прим. 29): 80 мг (53%).
Точка плавления: 208-212°С.
1H-ЯМР (300 МГц, ДМСО-d6) δ част. на млн: 1.50-1.81 (m, 2H), 1.81-2.04 (m, 4H), 2.04-2.25 (m, 2H), 2.30-2.50 (m, 2H), 2.56 (dd, J=26.01, 15.16 Hz, 4H), 2.64-2.77 (m, 2H), 3.47-3.77 (m, 4H), 3.97 (t, J=5.08 Hz, 2H), 6.89-7.07 (m, 1H), 7.46-7.89 (m, 6H), 8.00-8.16 (m, 1H), 11.44 (s, 1H).
13С-ЯМР (101 МГц, ДМСО-d6) δ част. на млн: 15.3, 21.6, 24.2, 30.6, 44.0, 47.6, 59.0, 62.8, 71.1, 71.3, 104.2, 114.9, 118.6, 125.5, 128.5, 128.9, 132.3, 138.8, 141.8, 148.5, 171.1, 176.5.
Пример 30
4-Бутил-4-(пирролидин-1-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (2:1), 1 диастереомер
Кетон Ket-16 (268 мг, 1,2 ммоль) и 7-азатриптофол (Ind-1, 195 мг, 1,2 ммоль) растворяли в абс. 1,2-дихлорэтане (40 мл) и смешивали с трифторметансульфоновой кислотой (0,117 мл, 198 мг, 1,32 ммоль) и перемешивали 3 дн. при комнатной температуре. 7 ч нагревали до 80°С (температура ванны), затем прибавляли трифторметансульфоновую кислоту (0,117 мл, 198 мг, 1,32 ммоль) и 8 ч перемешивали при 80°С. Смесь перемешивали еще 5 дней при комнатной температуре. Состав смешивали с водой (15 мл) и 1N натровым щелоком (5 мл) и 15 мин перемешивали. Фазы разделяли. Водную фазу экстрагировали с помощью 1,2-дихлорэтана (2×20 мл). Органические фазы объединяли, высушивали сульфатом натрия и концентрировали. Остаток представлял собой твердое вещество бежевого цвета (424 мг), которое ресуспендировали в этилацетат/метаноле 4:1 (3 мл). При этом в осадок начало выпадать бесцветное твердое вещество. Добавляли этилацетат (5 мл) и твердое вещество отделяли путем фильтрации. Получали 4-бутил-4-(пирролидин-1-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (диастереоизомерно чистый) с выходом в 12% (52 мг) сточкой плавления в 246-250°С.
Фильтрат концентрировали и отделяли с помощью хроматографии [силикагель 60 (50 г); этилацетат/метанол 4:1 (500 мл), метанол (200 мл)] и таким образом выделяли дополнительный продукт (107 мг, 24%).
Твердое вещество (91 мг, 0,247 ммоль) при 30°С растворяли в этаноле (7 мл) и смешивали с этанольным раствором лимонной кислоты (105 мг, 0,545 ммоль, 3 мл). Через 2 ч раствор смешивали с простым этиловым эфиром (50 мл) и перемешивали 2 дн. при комнатной температуре. При этом образовывался светло-коричневый осадок, который отделяли путем фильтрации (50 мг). Фильтрат концентрировали, ресуспендировали в этаноле (1 мл), смешивали с диэтиловым эфиром (20 мл) и 30 мин перемешивали. При этом в осадок выпадало бесцветное твердое вещество, которое фильтровали и промывали диэтиловым эфиром (2×2 мл) (50 мг). Оба твердых вещества были идентичными.
Выход (Прим. 30): 0,100 г (54%).
1H-ЯМР (300 МГц, ДМСО-d6) δ част. на млн: 1.02 (t, 3Н), 1.25-1.53 (m, 5H), 1.63-1.75 (m, 2H), 1.79-2.08 (m, 15H), 2.58-2.74 (m, 6H), 3.93 (t, 2H), 6.99-7.07 (m, 1H), 7.79-7.86 (m, 1H), 8.13-8.18 (m, 1H).
Пример 31
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-тиопирано[3,4-b]-7-аза-индол)]; цитрат (1:1), 1 диастереомер
Ind-4 (258 мг, 1,2 ммоль) под аргоном растворяли вместе с кетоном Ket-1 (261 мг, 1,2 ммоль) в абс. 1,2-дихлорэтане (24 мл) и смешивали с трифторметансульфоновой кислотой (0,44 мл, 4,96 ммоль). Состав перемешивали 1 ч при 150°С в микроволне. Для переработки реакционный раствор смешивали с насыщенным раствором гидрокарбоната натрия (10 мл). Смесь перемешивали в течение последующих 15 мин. После разделения фаз водную фазу экстрагировали дихлорметаном (3×10 мл). Объединенные органические экстракты высушивали над Na2SO4 и затем концентрировали. Полученный неочищенный продукт (346 мг, желтая пена) очищали колоночной хроматографией [силикагель 60 (20 г); циклогексан/этилацетат 4:1 (500 мл), циклогексан/этилацетат 1:1 (1000 мл)].
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-тиопирано[3,4-b]-7-аза-индол)] получали в виде твердого вещества бежевого цвета (87 мг, 19%, т.пл.: 246-253°С, диастереоизомерно чистое). Из водной фазы в течение ночи выпадал в осадок дополнительный продукт. Твердое вещество фильтровали и промывали водой (3×0,5 мл) и метанолом (2×0,5 мл) (51 мг, 11% твердое вещество бежевого цвета, точка плавления 262-267°С).
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-тиопирано[3,4-b]-7-аза-индол)] (135 мг, 0,358 ммоль, диастереоизомерно чистый) растворяли в дихлорметане (6 мл) и смешивали с лимонной кислотой (78 мг, 0,406 ммоль), растворенной в этилацетате (12 мл). Во время добавления кислоты остаток выпадал в осадок. Затем смесь перемешивали 17 ч при 23°С, потом фильтровали и осадок промывали этилацетатом (3×0,5 мл).
Выход (Прим. 31): 188 мг (92%), белое твердое вещество.
Точка плавления: 130-136°С.
1H-ЯМР (400 МГц, CD3OD) δ част. на млн: 2.01-2.15 (m, 4H), 2.52-2.71 (m, 7H), 2.71-2.90 (m, 5H), 2.90-3.05 (m, 6H), 7.02 (dd, J=7.79, 4.86 Hz, 1H), 7.59-7.72 (m, 3H), 7.74-7.81 (m, 2H), 7.81-7.88 (m, 1H), 8.08 (d, J=4.40 Hz, 1H).
13С-ЯМР (101 МГц, CD3OD) δ част. на млн: 24.4, 25.2, 28.4, 34.9, 38.4, 44.7, 45.4, 70.5, 74.2, 110.0, 116.4, 121.9, 127.9, 130.7, 130.8, 130.9, 131.5, 138.8, 143.1, 148.6, 174.7, 179.0.
Пример 32
4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-тиопирано[3,4-b]-5-аза-индол)], цитрат (1:1), 1 диастереомер
Ind-5 (430 мг, 2,0 ммоль) под аргоном растворяли вместе с кетоном Ket-1 (435 мг, 2,0 ммоль) в абс. 1,2-дихлорэтане (25 мл) и смешивали с трифторметансульфоновой кислотой (0,533 мл, 6,0 ммоль). Состав перемешивали 1 ч при 150°С в микроволне. Для переработки реакционный раствор смешивали с 1N раствором гидроксида натрия (20 мл). Смесь перемешивали последующие 15 мин. После разделения фаз водную фазу экстрагировали дихлорметаном (3×20 мл). Объединенные органические экстракты высушивали над Na2SO4 и затем концентрировали. Полученный неочищенный продукт (715 мг, коричневое твердое вещество) очищали колоночной хроматографией [силикагель 60 (30 г); циклогексан/этилацетат 4:1 (500 мл), циклогексан/этилацетат 2:1 (600 мл), этилацетат/метанол 100:1 (1000 мл), этилацетат/метанол 5:1 (600 мл)]. 4-(Диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-тиопирано[3,4-b]-5-аза-индол)] получали в виде почти белого твердого вещества (320 мг, 42%, т.пл.: 163-168°С, диастереоизомерно чистый).
Твердое вещество (233 мг, 0,617 ммоль) растворяли в горячем изопропаноле (10 мл) и смешивали с лимонной кислотой (130 мг, 0,677 ммоль), растворенной в горячем изопропаноле (10 мл). Во время добавления кислоты выпадал осадок. Затем смесь перемешивали 2 ч при 5°С, потом фильтровали и осадок промывали изопропанолом (2×0,5 мл) и ацетоном (3×0,5 мл).
Выход (Прим. 32): 350 мг (97%), белое твердое вещество.
Точка плавления: 132-140°С.
1H-ЯМР (400 МГц, ДМСО-d6) δ част. на млн: 1.04 (d, J=5.87 Hz, 1.5H), 1.82-2.03 (m, 4H), 2.16-2.39 (m, 7H), 2.49-2.70 (m, 5H), 2.75-2.91 (m, 2H), 2.91-3.14 (m, 4H), 3.68-3.86 (m, 0.25H), 7.24-7.44 (m, 1H), 7.44-7.84 (m, 5H), 8.07-8.27 (m, 1H), 8.74-8.94 (m, 1H), 11.47 (s, broad, 1H).
13С-ЯМР (101 МГц, ДМСО-d6) δ част. на млн: 23.3, 23.5, 25.4, 27.4, 33.4, 37.5, 44.0, 44.6, 62.0. 65.3, 71.5, 107.1, 109.8, 123.8, 126.6, 127.5, 128.6, 128.7, 131.9, 136.7, 138.1, 139.3, 140.4, 171.3, 176.5.
Пример 33
4-(1-Метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1), полярный диастереомер
Пример 34
4-(1-Метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1), неполярный диастереомер
4-(1-Метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (неполярный и полярный диастереомер)
Кетон Ket-17 (250 мг, 1,2 ммоль) и 7-азатриптофол (Ind-1,195 мг, 1,2 ммоль) растворяли в абс. 1,2-дихлорэтане (30 мл) и под аргоном смешивали с метансульфоновой кислотой (0,467 мл, 692 мг, 7,2 ммоль). Через 17 ч состав 7 ч перемешивали при температуре ванны в 90°С и 15 ч при 75°С. Реакционную смесь смешивали с 1N натровым щелоком (15 мл) и водой (10 мл) и 15 мин перемешивали. Фазы разделяли. Водную фазу экстрагировали 1,2-дихлорэтаном (2×20 мл). Органические фазы объединяли, высушивали сульфатом натрия и концентрировали. Остаток представлял собой твердое вещество бежевого цвета, которое ресуспендировали в этилацетате (5 мл). При этом начало выпадать в осадок бесцветное твердое вещество, которое отделяли путем фильтрации и промывали этилацетатом (2 мл).
Получали 4-(1-метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] в виде смеси диастереоизомеров (130 мг). Фильтрат концентрировали и отделяли с помощью хроматографии [силикагель 60 (30 г); этилацетат/метанол 6:1 (700 мл)] (220 мг, бесцветное твердое вещество, смесь диастереоизомеров).
Обе фракции вновь хроматографировали [силикагель 60 (20 г); хлороформ/метанол 30:1 (500 мл)] и при этом выделяли 4-(1-метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (неполярный диастереоизомер) с выходом в 40% (164 мг) в виде бесцветного твердого вещества с точкой плавления в 282-287°С. Полярный диастереоизомер получали с выходом в 11% (48 мг) с точкой плавления в 285-289°С.
4-(1-Метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1), полярный диастереомер (Прим. 33)
4-(1-Метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (полярный диастереомер) (48 мг, 0,136 ммоль) растворяли при нагревании в этаноле (20 мл) и смешивали с лимонной кислотой (58 мг, 0,3 ммоль), растворенной в этаноле (2 мл). После 3 дней помешивания при комнатной температуре прозрачный раствор почти полностью концентрировали и остаток смешивали с диэтиловым эфиром (5 мл) до кристаллизации.
Выход (Прим. 33): 71 мг (96%), бесцветное твердое вещество.
Точка плавления: 90-93°С.
1H-ЯМР (300 МГц, ДМСО-d6) δ част. на млн: 1.72-1.92 (m, 2H), 1.92-2.02 (m, 2H), 2.04 (s, 3Н), 2.07-2.19 (m, 4H), 2.56-2.83 (m, 10Н), 3.91 (t, J=5.17 Hz, 2H), 4.10 (s, 3Н), 6.94-7.09 (m, 1Н), 7.78 (s, 1 H), 7.79-7.86 (m, 1Н), 8.10-8.22 (m, 1 Н), 11.35 (s, 1Н).
13С-ЯМР (101 МГц, ДМСО-d6) δ част. на млн: 21.7, 27.0, 28.9, 29.0, 37.2, 42.8, 55.4, 59.1, 70.7, 72.3, 104.1, 115.0, 118.7, 125.7, 139.8, 141.8, 148.2, 148.9, 158.1, 171.2, 174.7.
4-(1-Метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], неполярный диастереомер (Прим. 34)
4-(1-Метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)] (неполярный диастереомер) (149 мг, 0,423 ммоль) растворяли в этаноле (20 мл) и смешивали с лимонной кислотой (179 мг, 0,93 ммоль), растворенной в этаноле (2 мл). После 3 дн. помешивания при комнатной температуре можно было отсасывать цитрат 8K/9K в виде бесцветного твердого вещества.
Выход (Прим. 34): 74 мг, (91%).
Точка плавления: 192-193°С.
1H-ЯМР (300 МГц, ДМСО-d6) δ част. на млн: 1.80-2.10 (m, 6H), 2.11 (s, 3Н), 2.52-2.77 (m, 8Н), 3.95 (t, J=5.27 Hz, 2H), 4.08 (s, 3Н), 6.93-7.04 (m, 1H), 7.73-7.83 (m, 1H), 7.99 (s, 1H), 8.06-8.16 (m, 1H), 11.41 (s, 1H).
13С-ЯМР (101 МГц, ДМСО-d6) δ част. на млн: 21.7, 27.7, 29.0, 31.2, 38.0, 38.9, 43.4, 57.0, 59.0, 71.2, 71.8, 104.1, 114.9, 118.6, 125.5, 139.2, 141.8, 148.5, 149.3, 152.4, 171.2, 175.7.
Исследования эффективности соединении согласно изобретению
Измерение связывания ORL1
Циклогексановые производные общей формулы I были исследованы в анализе связывания с 3H-ноцицептин/орфанин FQ с мембранами рекомбинантных CHO-ORL1 клеток. Эта тестовая система была проведена согласно методу, представленному Ardati et al. (Mol. Pharmacol., 51, 1997, cc.816-824). Концентрация 3H-ноцицептин/орфанина FQ в этих опытах составила 0,5 нМ. Анализы связывания были проведены с по 20 мкг мембранного протеина по 200 мкл состава в 50 мM Hepes, pH 7,4, 10 мM MgCl2 и 1 мM EDTA. Связывание к ORL1-рецептору определяли с применением по 1 мг WGA-SPA Beads (Amersham-Pharmacia, Фрайбург) путем одночасовой инкубации состава при КТ и последующего измерения в сцинтилляционном счетчике Trilux (Wallac, Финляндия). Сродство указано в таблице 1 как наномолярное Ki-значение в или % ингибирования при с=1 мкM.
Измерение µ-связывания
Сродство рецепторов к человеческому µ-опийному рецептору определяли в гомогенном составе в микротитрационных планшетах. Для этого инкубировали разбавленные серии соответствующих проверяемых соединений с рецепторным мембранным препаратом (15-40 мкг протеина на 250 мкл инкубационного состава) СНО-K1-клеток, которые выражают человеческий µ-опийный рецептор (RB-HOM-рецепторный мембранный препарат фирмы NEN, Zaventem, Бельгия) в присутствии 1 нмоль/л радиоактивного лиганда [3H]-налоксон (NET719, фирма NEN, Zaventem, Бельгия), а также 1 мг WGA-SPA-Beads (Wheat germ agglutinin SPA Beads фирмы Amersham/Pharmacia, Фрайбург, Германия) в общем объеме 250 мкл в течение 90 минут при комнатной температуре. В качестве инкубационного буфера применяли 50 ммоль/л трис-HCl, дополненный с 0,05 мас.% азида натрия и с 0,06 мас.% альбумина сыворотки крупного рогатого скота. Для определения неспецифического связывания дополнительно добавляли 25 мкмоль/л налоксона. После окончания девяностоминутного инкубационного периода микротитрационные планшеты отделяли центрифугированием и в течение 20 минут при 1000 г и радиоактивность измеряли в В-счетчике (Microbeta-Trilux, фирма PerkinElmer Wallac, Фрайбург, Германия). Определяли процентное вытеснение радиоактивного лиганда из его связывания к человеческому µ-опийному рецептору при концентрации контрольного вещества в 1 мкмоль/л и указывали как процентное ингибирование (% ингибирования) специфического связывания. Частично, исходя из процентного вытеснения, благодаря различным концентрациям контрольных соединений общей формулы I IC50 рассчитывали ингибирующие концентрации, которые вызывали 50-процентное вытеснение радиоактивного лиганда. Путем пересчета с помощью соотношения Чена-Прусоффа (Cheng-Prusoff) получали Ki-значения для контрольных веществ.
Проверка аналгезии в тесте «отдергивание хвоста» (Tail-Flick) на мышах
Мышей каждый раз по одной помещали в клетку для проведения испытаний и основание хвоста подвергали фокусирующему тепловому излучению электрической лампы (Tail-flick-Typ 50/08/1. bc, Labtec, Dr. Hess). Интенсивность лампы устанавливали таким образом, что время от включения лампы до внезапного отдергивания хвоста (болевая латентность) у неподвергшихся обработке мышей составило от 3 до 5 секунд. Перед введением растворов, содержащих соединение согласно изобретению, или соответствующих стандартных растворов мыши в течение пяти минут дважды подвергались предварительному тесту, и среднее значение этих измерений подсчитывали как среднее значение предварительного теста.
Растворы соединения общей формулы I согласно изобретению, а также стандартные растворы вводили потом внутривенно. Измерение боли проводили каждые 10, 20, 40 и 60 минут после внутривенного введения. Анальгезирующее действие определяли как возрастание болевой латентности (% максимально возможного антиноцицептивного эффекта) согласно нижеследующей формуле:
[(T1-T0)/(T2-T0)]×100
При этом время Т0 представляет собой латентный период перед введением, время T1 является латентным периодом после применения комбинации действующих веществ и время T2 - максимальной выдержкой (12 секунд).
Проверка аналгезии в тесте «отдергивание хвоста» (Tail-Flick) на крысах
Анальгезирующее действие испытуемых соединений исследовали фокальным лучом с помощью теста «отдергивания хвоста» (Tail-flick) на крысах в соответствии с методом Д'Амура и Смита (D'Amour and Smith) (J.Pharm. Exp. Ther. 72, 74 79 (1941)). Для этого использовали самок крыс Sprague Dawley с массой от 134 до 189 г. Животных по отдельности помещали в специальную клетку для проведения испытаний и основание хвоста подвергали фокусирующему тепловому излучению лампы (Tail-flick Тур 50/08/1.bc, Labtec, Dr. Hess). Интенсивность лампы устанавливали таким образом, что время от включения лампы до внезапного отдергивания хвоста (болевая латентность) у неподвергшихся обработке животных составило 2,5-5 секунд. Перед приемом испытуемых соединений животных дважды предварительно тестировали в течение 30 минут и среднее значение этих измерений подсчитывали как среднее значение предварительного теста. Измерение боли проводили через 20, 40 и 60 мин после внутривенной дозы. Анальгезирующее действие определяли как возрастание болевой латентности (% МРЕ) согласно следующей формуле:
[(T1-T0)/(T2-T0)]×100
При этом Т0 представляет собой латентный период перед и T1 латентный период после введения вещества, T2 является максимальной выдержкой (12 сек).
Для определения дозы зависимости применяли соответствующее испытуемое соединение в 3-5 логарифмически возрастающих доз, которые каждый раз включали предельную и максимальную действующую дозу, и значения ED50 определяли с помощью регрессионного анализа. Расчет ED50 осуществлялся по максимуму действия, через 20 минут после внутривенного введения вещества.
Нижеследующая таблица представляет примерные соединения в виде оснований без указания того, о каком диастереомере идет речь; в случаях, в которых примерное соединение было представлено как соль или выделено полярным и неполярным диастереомером, оно может быть взято из соответствующего описания синтеза.
Определенные Ki-значения приведены в следующей таблице. Ki-значения были определены не для всех соединений. Для примера были исследованы два соединения в тесте «отдергивания» хвоста (Tail-flick).
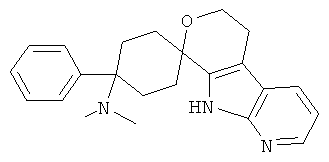
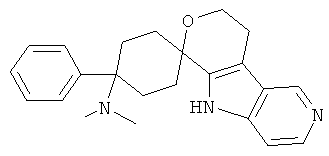
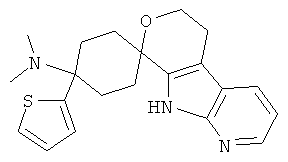
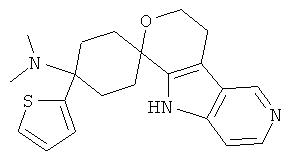
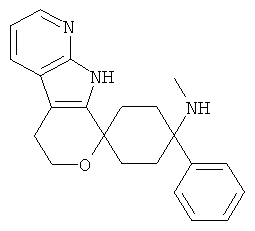
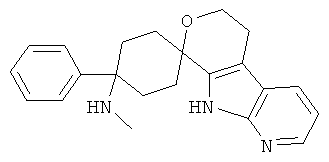
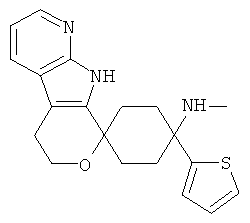
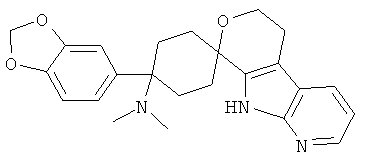
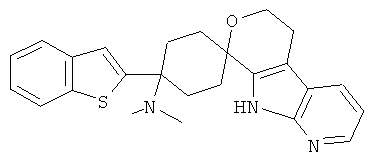
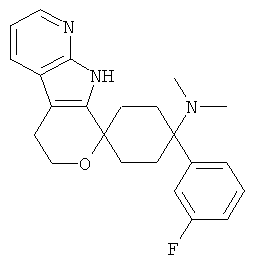
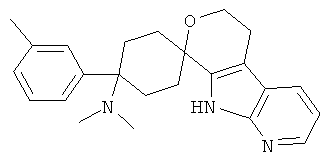
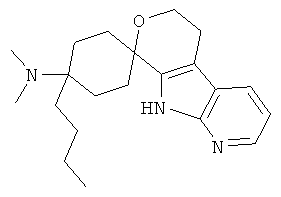
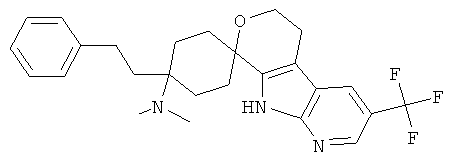
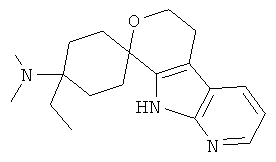
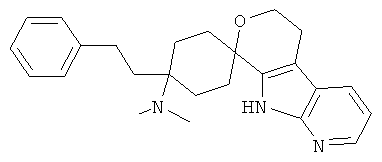
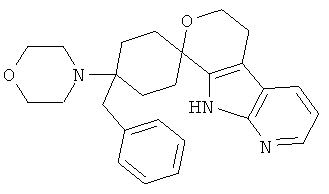
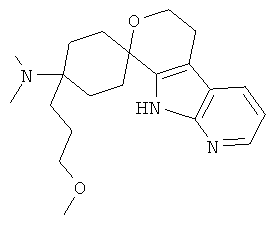
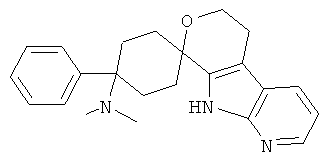
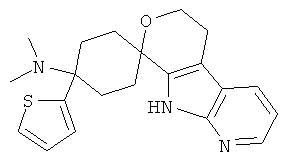
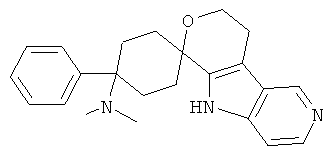
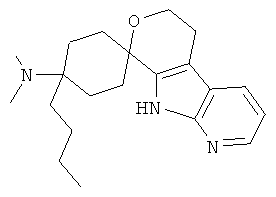
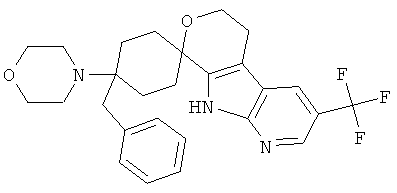
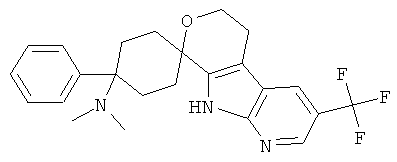
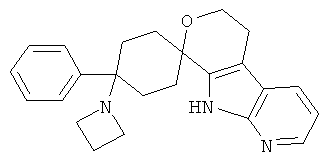
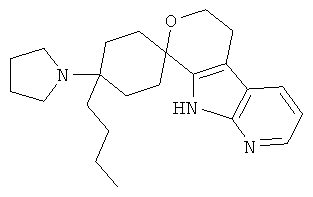
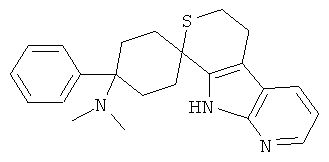
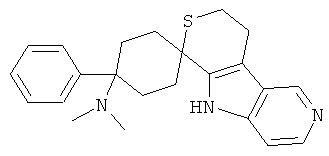
Парентеральный раствор спироциклического производного согласно изобретению
3 г одного из спироциклических азаиндольных производных согласно изобретению, в данном случае Пример 1, при комнатной температуре растворяли в 1 л воды для инъекционных целей и затем путем добавления безводной глюкозы для инъекционных целей устанавливали до изотонических условий.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 2008 |
|
RU2484092C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2532545C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2503660C2 |
| ПРОИЗВОДНЫЕ СПИРО(5.5)УНДЕКАНА | 2009 |
|
RU2515895C2 |
| ГИДРОКСИМЕТИЛЦИКЛОГЕКСИЛАМИНЫ | 2009 |
|
RU2514192C2 |
| ПРОИЗВОДНЫЕ ЦИС-ТЕТРАГИДРО-СПИРО(ЦИКЛОГЕСАН-1,1'-ПИРИДО[3,4-в]ИНДОЛ)-4-АМИНА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ НЕВРОПАТИЧЕСКОЙ И/ИЛИ ХРОНИЧЕСКОЙ БОЛИ | 2011 |
|
RU2592283C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2525236C2 |
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 2008 |
|
RU2470933C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОГЕКСИЛДИАМИНЫ | 2009 |
|
RU2526251C2 |
| ТИОФЕНИЛЬНЫЕ И ПИРРОЛИЛЬНЫЕ АЗЕПИНЫ В КАЧЕСТВЕ ЛИГАНДОВ СЕРОТОНИНОВОГО 5-HT РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2434872C2 |
Изобретение относится к новым спироциклическим азаиндольным производным формулы I:
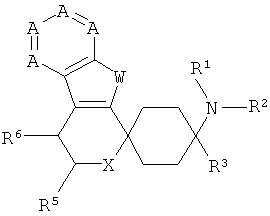
где один из А означает N, а другие означают CR7-10; W означает NR4; Х означает О, S; R1 и R2 независимо друг от друга означают Н; С1-5-алкил, каждый раз насыщенный, разветвленный или неразветвленный, или остатки R1 и R2 вместе означают CH2CH2OCH2CH2, (CH2)3-6; R3 означает C1-8-алкил, каждый раз насыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный -ОС1-6-алкилом; фенил, тиенил, морфолинил, бензотиофенил или бензодиоксолил, каждый раз незамещенный или монозамещенный F, C1-6-алкилом; или 5-членный гетероарил, содержащий три атома азота в качестве гетероатомов, замещенный С1-3-алкилом; связанный C1-3-алкильной группой фенил, незамещенный или монозамещенный F или C1-6-алкилом; R4 означает Н; R5 означает Н; R6 означает Н; R7, R8, R9 и R10 означают Н или CF3; в виде диастереомеров, смесей диастереомеров или отдельного диастереомера; оснований и/или солей физиологически совместимых кислот. Соединения пригодны для лечения ряда заболеваний, например боли, стресса, депрессий и других. Описан способ их получения. 5 н. и 6 з.п. ф-лы, 1 табл., 34 пр.
1. Спироциклические азаиндольные производные общей формулы I
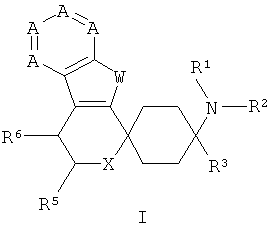
в которой один из А означает N, а другие означают CR7-10,
W означает NR4,
Х означает О или S,
R1 и R2 независимо друг от друга означают Н; C1-5-алкил, каждый раз насыщенный, разветвленный или неразветвленный,
или остатки R1 и R2 вместе означают СН2СН2ОСН2СН2 или (СН2)3-6,
R3 означает C1-8-алкил, каждый раз насыщенный, разветвленный или неразветвленный, незамещенный или монозамещенный -OC1-6-алкилом; фенил, тиенил, морфолинил, бензотиофенил или бензодиоксолил, каждый раз незамещенный или монозамещенный F или C1-6-алкилом; или 5-членный гетероарил, содержащий три атома азота в качестве гетероатомов, замещенный C1-3 алкилом; связанный С1-3-алкильной группой фенил, незамещенный или монозамещенный F или C1-6 алкилом;
R4 означает Н;
R5 означает Н;
R6 означает Н;
R7, R8, R9 и R10 означают Н или CF3;
в виде диастереомеров, смесей диастереомеров или отдельного диастереомера; оснований и/или солей физиологически совместимых кислот.
2. Спироциклические азаиндольные производные по п.1, в которых R1 и R2 означают СН3.
3. Спироциклические азаиндольные производные по п.1 или 2, в которых R3 означает пропил, бутил, пентил, гексил, фенил, тиофенил, бензил, бензодиоксоланил или бензотиофенил, каждый раз незамещенный или монозамещенный F, C1-6 алкилом; связанный насыщенной неразветвленной С1-3-алкильной группой фенил.
4. Спироциклические азаиндольные производные по п.3, в которых R3 означает бутил, этил, 3-метоксипропил, бензотиофенил, фенил, 3-метилфенил, 3-фторфенил, бензо[1,3]-диоксолил, бензил, 1-метил-1,2,4-триазолил, тиенил или фенэтил.
5. Спироциклические азаиндольные производные по п.1 или 2, в которых R7, R8, R9 и R10 означают Н.
6. Соединения по п.1 или 2, в которых общая формула I принимает значение одной из общих формул Ia или Ib
и группы Х и W, а также остатки R1-R10 имеют значения, приведенные в пп.1-5.
7. Спироциклические азаиндольные производные по п.1, выбранные из группы
(1) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]
(2) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; метансульфонат
(3) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано [3,4-b]-5-аза-индол)]
(4) 4-(диметиламино)-4-тиофен-2-ил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; метансульфонат
(5) 4-(диметиламино)-4-тиофен-2-ил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)]
(6) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (4:3)
(7) 4-(метиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(8) 4-(метиламино)-4-тиофен-2-ил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (4:3)
(9) 4-(диметиламино)-4-бензо[1,3-диоксол]-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(10) 4-(диметиламино)-4-(бензотиофен-2-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(12) 4-(диметиламино)-4-(3-фторфенил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (4:3)
(13) 4-(диметиламино)-4-(3-метилфенил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(14) 4-(диметиламино)-4-(бут-1-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1)
(15) 4-(диметиламино)-4-фенилэтил-спиро[циклогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(17) 4-(диметиламино)-4-этил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (2:3)
(18) 4-(диметиламино)-4-фенилэтил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (2:3)
(19) 4-бензил-4-морфолино-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(20) 4-(диметиламино)-4-(3-метоксипропил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(21) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(22) 4-(диметиламино)-4-тиофен-2-ил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(23) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)], цитрат (1:1), неполярный диастереомер,
(24) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-пирано[3,4-b]-5-аза-индол)], цитрат (1:1), полярный диастереомер,
(25) 4-бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), неполярный диастереомер,
(26) 4-бутил-4-(диметиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1), полярный диастереомер,
(27) 4-бензил-4-морфолино-спиро[циклогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(28) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(3-трифторметил-5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(29) 4-(ацетидин-1-ил)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1)
(30) 4-бутил-4-(пирролидин-1-ил)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)]; цитрат (2:1)
(31) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-тиопирано[3,4-b]-7-аза-индол)]; цитрат (1:1)
(32) 4-(диметиламино)-4-фенил-спиро[циклогексан-1,6'-(5,6,8,9-тетрагидро-тиопирано[3,4-b]-5-аза-индол)], цитрат (1:1)
(33) 4-(1-метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1), полярный диастереомер,
(34) 4-(1-метил-1Н-1,2,4-триазол-5-ил)-4-(метиламино)-спиро[циклогексан-1,8'-(5,6,8,9-тетрагидро-пирано[3,4-b]-7-аза-индол)], цитрат (1:1), неполярный диастереомер,
в виде диастереомеров, смесей диастереомеров или отдельного диастереомера; оснований и/или солей физиологически совместимых кислот.
8. Способ получения спироциклических азаиндольных производных общей формулы I по п.1, где Х означает О, при котором соединения общей формулы III подвергают превращению в реакционной среде в присутствии фторида, выбранного из группы, состоящей из фторида тетра-п-бутиламмония, плавиковой кислоты (HF, HF-пиридин), фторида калия и/или фторида натрия, фторида цезия или в присутствии органической или неорганической кислоты, преимущественно HCl, уксусной кислоты, трифторуксусной кислоты, бортрифторида, при температурах преимущественно от -70 до 300°С с получением соединений общей формулы IV
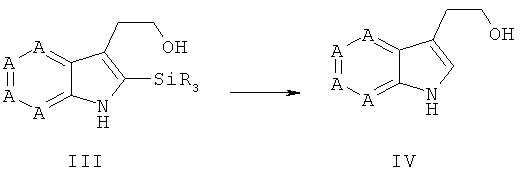
которые затем при добавлении по меньшей мере одной органической кислоты или ее сложного триметилсилилового эфира или одной неорганической кислоты или при добавлении по меньшей мере одной соли переходного металла подвергают взаимодействию с кетонами общей формулы VII
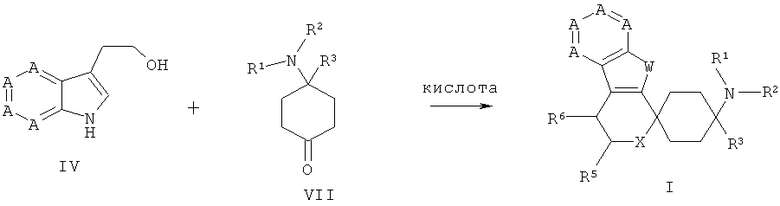
в пригодном растворителе или смеси растворителей, таких как дихлорметан, дихлорэтан, хлороформ, ацетонитрил, диэтиловый эфир или нитроэтан, при температурах от 0 до 150°С при необходимости с применением микроволн.
9. Лекарственное средство для лечения боли, содержащее, по меньшей мере, одно спироциклическое азаиндольное производное по п.1, при необходимости в виде его чистых диастереомеров в любом соотношении компонентов смеси; в виде его оснований или в виде его физиологически совместимых солей; а также при необходимости содержащее пригодные добавки и/или вспомогательные вещества.
10. Применение одного спироциклического азаиндольного производного по п.1 при необходимости в виде его диастереомеров в любом соотношении компонентов смеси; в виде его оснований или в виде его физиологически совместимых солей; для получения лекарственного средства для лечения боли, в особенности острой, невропатической или хронической боли.
11. Применение одного спироциклического азаиндольного производного по п.1 для получения лекарственного средства для лечения состояний страха, стресса и связанных со стрессом синдромов, депрессий, эпилепсии, болезни Альцгеймера, старческого слабоумия, каталепсии, общих познавательных дисфункций, нарушений обучения и памяти (как ноотроп), синдромов отмены, злоупотребления и/или зависимости алкогольной, и/или наркотической, и/или медикаментозной, сексуальных дисфункций, сердечно-сосудистых заболеваний, гипотонии, гипертензии, тиннитуса, зуда, мигрени, глухоты, недостаточной подвижности кишечника, нарушенного приема пищи, анорексии, ожирения, двигательных нарушений, диареи, кахексии, недержания мочи, или как мышечный релаксант, противосудорожное средство или анестетик, или для совместного приема при лечении с опиоидным анальгетиком или с анестетиком, для диуреза или антинатрийуреза, анксиолизиса, для модуляции двигательной активности, для модуляции распределения нейромедиаторов и лечения связанных с этим нейродегенеративных заболеваний, для лечения синдромов отмены и/или для снижения наркотического потенциала опиоидов.
| WO 2004043967 A1, 27.05.2004 | |||
| WO 2005063769 A1, 14.07.2005 | |||
| WO 2005066183 A1, 21.07.2005 | |||
| Рыбозащитное устройство водозаборного сооружения | 1983 |
|
SU1142587A1 |
| СПОСОБ ОЦЕНКИ ТЕЧЕНИЯ ХРОНИЧЕСКОГО ВОСПАЛИТЕЛЬНОГО ПРОЦЕССА В БРОНХОЛЕГОЧНОЙ СИСТЕМЕ БОЛЬНОГО МУКОВИСЦИДОЗОМ | 2002 |
|
RU2216737C1 |

