Область изобретения
Настоящее изобретение относится к способам лечения туберкулеза, включая разновидности с множественной лекарственной устойчивостью и латентный туберкулез. Более конкретно, настоящее изобретение относится к способу лечения туберкулеза у млекопитающего, при котором указанному млекопитающему, нуждающемуся в этом, вводят эффективное количество соединения формулы (I), (S)-N-[[3-[3-фтор-4-(4-тиоморфолинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамида, или его фармацевтически приемлемой соли в комбинации с по меньшей мере двумя агентами, полезными при лечении туберкулеза. Настоящее изобретение также относится к фармацевтической композиции, содержащей: (1) терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, (2) терапевтически эффективное количество по меньшей мере одного агента, полезного при лечении туберкулеза, и (3) один или более чем один фармацевтически приемлемый носитель или наполнитель.
Предшествующий уровень техники
Туберкулез (ТБ) приводит к смерти приблизительно 1,6 миллиона человек во всем мире каждый год, что делает его второй основной причиной смерти взрослых после ВИЧ. Каждый год происходит почти 500000 новых случаев ТБ с множественной лекарственной устойчивостью (multidrug-resistant, МЛУ), и недавнее появление ТБ с расширенной лекарственной устойчивостью (extensively drug-resistant, РЛУ-ТБ) предвещает новую эпидемию неизлечимого ТБ. (Смотри Dorman, S.Е. et al., Nat. Med 13:295-298 2007, Zignol, M. et al., J Infect. Dis 194:479-485, 2006.) Необходимы новые лекарственные средства с мощной противотуберкулезной активностью, особенно против неразмножающихся устойчивых организмов, для сокращения продолжительности лечения ТБ и, посредством этого, облегчения глобального внедрения лечения под непосредственным наблюдением. (Смотри O'Brien, R.J. et al., Am. J.Respir. Crit Care Med. 163:1055-1058, 2001.)
В настоящее время лечение туберкулеза, чувствительного к лекарственным средствам, заключается во введении комбинации по меньшей мере следующих лекарственных средств: изониазида, рифампина и пиразинамида. Для эффективного лечения вышеупомянутые лекарственные средства дают пациенту в начальной фазе лечения в течение 8 недель, на протяжении которых данные лекарственные средства используют в комбинации для уничтожения быстроразмножающейся популяции Mycobacterium tuberculosis, а также для предупреждения появления устойчивости к лекарственному средству. После начальной фазы лечения следует продолжение или фаза стерилизации в течение 18 недель, на протяжении которых дают два или более чем два стерилизующих лекарственных средства (например, изониазид и рифампин) для уничтожения периодически делящейся популяции (неразмножающихся устойчивых организмов) Mycobacterium tuberculosis.
В то время как вышеупомянутая комбинация лекарственных средств совместно обеспечивает лечение против чувствительной инфекции Mycobacterium tuberculosis за время от 4 до 6 месяцев, такая комбинированная терапия не всегда является успешной, особенно у пациентов, имеющих штаммы, устойчивые к лекарственным средствам. Большая продолжительность лечения, продолжающегося шесть месяцев, также может приводить к неприятным побочным эффектам. Кроме того, соблюдение схемы и режима лечения при относительно длинном курсе лечения обычно является плохим. Такое несоблюдение схемы и режима лечения может приводить к провалу лечения, приводя к развитию устойчивости к лекарственному средству.
Оксазолидиноны включают класс ингибиторов синтеза белка, которые блокируют трансляцию путем предотвращения образования инициирующего комплекса (относительно механизма, смотри K.Leach et al., Molecular Cell, 26:4, 460-462, 2007). Линезолид (LZD, Zyvox®), единственный продающийся на рынке оксазолидинон, обладает активностью в отношении грамположительных бактерий и в настоящее время одобрен для применения при осложненных инфекциях кожи и кожных структур и внутрибольничной пневмонии (Zyvox® листовка-вкладыш в упаковке). Однако он также является активным против многих видов микобактерий, включая Mycobacterium tuberculosis, для которой его МИК (минимальная ингибирующая концентрация) варьирует от 0,125 до 1 мкг/мл с МИК50 0,5 мкг/мл и МИК90 1 мкг/мл. (Смотри Alcala, L. et al., Antimicrob Agents Chemother 47: 416-417, 2003; Cynamon, M.H., et al., Antimicrob Agents Chemother 43: 1189-1191, 1999; Fattorini, L., et al., Antimicrob Agents Chemother 47: 360-362, 2003). В результате LZD используют в случае отличных от указанных на этикетке показаний для лечения не поддающихся воздействию случаев МЛУ- и РЛУ-ТБ. Несмотря на то что несколько серий случаев говорит о том, что LZD может способствовать успешному превращению культуры мокроты в таких случаях, его отдельная активность у пациентов с ТБ и его точный вклад в комбинированные схемы лечения остается неясным. Эти исследования также демонстрируют то, что продолжительность введения LZD может быть ограничена гематологической и нейрологической токсичностью, которая может возникать при долговременном введении. (Смотри Fortun, J., et al., 56: 180-185, 2005; Park, I.N., et al., Antimicrob Chemother 58: 701-704, 2006; Zignol, M., et al., J Infect. Dis 194: 479-485, 2006.) Следовательно, желательными являются новые оксазолидиноны с более мощной активностью in vivo против Mycobacterium tuberculosis и меньшим риском токсичности при длительном введении.
Ранее были описаны оксазолидиноны с более мощной активностью против Mycobacterium tuberculosis. (Смотри Barbachyn, M.R. et al., J Med Chem. 39: 680-685, 1996; Sood, R., et al., Antimicrob Agents Chemother 49: 4351-4353, 2005.) Противотуберкулезная активность соединения формулы (I), (S)-N-[[3-[3-фтор-4-(4-тиоморфолинил)фенил]-2-оксо-5-оксазолидинил]метил] ацетамида, была впервые описана в 1996. (Смотри Barbachyn, M.R., et al., J Med Chem. 39: 680-685, 1996.) Последующие эксперименты на мышиной модели выявили то, что соединение формулы (I) является более активным, чем LZD, при введении обоих лекарственных средств в дозе 100 мг/кг, но клиническая релевантность этой дозы LZD не была установлена, и активности соединения формулы (I) и LZD не были явно отличными при сравнении в более низких дозах. (Смотри Cynamon, M.H., et al., Antimicrob Agents Chemother 43: 1189-1191, 1999.) Кроме того, несмотря на то что соединение формулы (I), по-видимому, имеет умеренную активность при объединении с рифампином (RIF) в модели острой (ранней) инфекции, соединение формулы (I) не имеет дополнительной активности при объединении с изониазидом (INH). (Cynamon, M.H., et al., Antimicrob Agents Chemother 43: 1189-1191, 1999.) Важнее всего то, что активность соединения формулы (I), одного или в комбинации с RIF или INH, оценивали только на протяжении исходных 4 недель лечения, что является недостаточным для оценки активности соединения или комбинации агентов против неразмножающихся сохраняющихся клеток, которая в свою очередь в конечном счете определяет продолжительность лечения, необходимого для излечения (т.е. предупреждения рецидива после завершения лечения).
Следовательно, существует насущная потребность в разработке более новых схем лечения, которые могут быть использованы для предупреждения, лечения и/или снижения туберкулеза и/или для устранения угрозы появления туберкулеза с множественной лекарственной устойчивостью, или сокращения продолжительности лечения.
Краткое изложение сущности изобретения
Настоящее изобретение относится к способу лечения туберкулеза у млекопитающего, при котором указанному млекопитающему, нуждающемуся в этом, вводят эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли:

в комбинации с по меньшей мере двумя агентами, полезными при лечении туберкулеза.
В одном воплощении указанные по меньшей мере два агента выбраны из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, этамбутола, стрептомицина, канамицина, амикацина, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина, капреомицина, этионамида, циклосерина, пара-аминосалициловой кислоты, тиацетазона, кларитромицина, амоксициллина-клавулановой кислоты, имипенема, меропенема, виомицина, теризидона, клофазимина, ТМС-207, РА-824, ОРС-7683, LL-3858 и SQ-109.
В другом воплощении один из указанных по меньшей мере двух агентов выбран из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина, клофазимина и этамбутола.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой пиразинамид.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой рифампин.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой рифапентин.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой РА-824.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой ОРС-67683.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой ТМС-207.
В еще одном воплощении один из указанных по меньшей мере двух агентов выбран из группы, состоящей из моксифлоксацина, гатифлоксацина, левофлоксацина и офлоксацина.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой моксифлоксацин.
В конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид и рифампин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид, рифампин и изониазид.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид и рифапентин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид, рифапентин и изониазид.
В конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид и моксифлоксацин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид, моксифлоксацин и рифампин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид, моксифлоксацин и рифапентин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой РА-824 и пиразинамид.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой РА-824, пиразинамид и агент, выбранный из группы, состоящей из стрептомицина, канамицина, амикацина и капреомицина.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой РА-824 и моксифлоксацин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой РА-824, моксифлоксацин и агент, выбранный из группы, состоящей из стрептомицина, канамицина, амикацина и капреомицина.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ОРС-67683 и пиразинамид.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ОРС-67683, пиразинамид и агент, выбранный из группы, состоящей из стрептомицина, канамицина, амикацина и капреомицина.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ОРС-67683 и моксифлоксацин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ОРС-67683, моксифлоксацин и агент, выбранный из группы, состоящей из стрептомицина, канамицина, амикацина и капреомицина.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ТМС-207 и пиразинамид.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ТМС-207, пиразинамид и моксифлоксацин или изониазид.
В еще одном воплощении указанный туберкулез включает активный туберкулез или латентный туберкулез.
В еще одном воплощении указанный активный туберкулез включает чувствительный к лекарственным средствам, устойчивый к одному лекарственному средству, туберкулез с множественной лекарственной устойчивостью (МЛУ) или туберкулез с расширенной лекарственной устойчивостью (РЛУ).
В еще одном воплощении способ по настоящему изобретению позволяет полностью устранить туберкулез, чувствительный к лекарственным средствам, устойчивый к одному лекарственному средству, туберкулез с множественной лекарственной устойчивостью или туберкулез с расширенной лекарственной устойчивостью (РЛУ) по завершении лечения.
В еще одном воплощении указанный туберкулез вызван инфекцией Mycobacterium, выбранной из группы, состоящей из Mycobacterium tuberculosis, Mycobacterium bovis или другого родственного вида микобактерий.
В еще одном воплощении способ по настоящему изобретению предупреждает рецидив инфекции Mycobacterium после завершения лечения.
В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли вводят перорально.
В еще одном воплощении каждый из указанных по меньшей мере двух агентов вводят перорально.
В еще одном воплощении указанные по меньшей мере два агента вводят совместно в композиции.
В еще одном воплощении указанные по меньшей мере два агента вводят раздельно.
В еще одном воплощении указанные по меньшей мере два агента вводят совместно, и другой агент, полезный для лечения туберкулеза, вводят отдельно.
В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли вводят один раз в сутки (QD) или два раза в сутки (BID).
В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли вводят один раз в неделю, дважды в неделю, трижды в неделю или через день.
В одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве от примерно 10 мг до примерно 2000 мг.
В еще одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве от примерно 250 мг до примерно 1000 мг.
В еще одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве от примерно 600 мг до примерно 1000 мг.
В еще одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве от примерно 25 мг до примерно 1000 мг.
В еще одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве от примерно 50 мг до примерно 500 мг.
В еще одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве от примерно 100 мг до примерно 500 мг.
Настоящее изобретение также относится к способу лечения туберкулеза у млекопитающего после того, как указанное млекопитающее прошло начальную фазу лечения, при котором указанному млекопитающему, нуждающемуся в этом, вводят эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли:
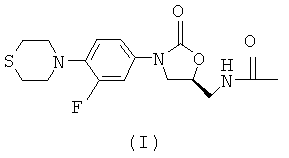
в комбинации с по меньшей мере одним агентом, полезным при лечении туберкулеза.
В одном воплощении указанный по меньшей мере один агент выбран из группы, состоящей из рифампина, рифапентина, рифабутина, пиразинамида, этамбутола, стрептомицина, канамицина, амикацина, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина, капреомицина, этионамида, циклосерина, пара-аминосалициловой кислоты, тиацетазона, кларитромицина, амоксициллина-клавулановой кислоты, имипенема, меропенема, клофазимина, виомицина, теризидона, ТМС-207, РА-824, ОРС-7683, LL-3858 и SQ-109.
В еще одном воплощении по меньшей мере один указанный агент выбран из группы, состоящей из рифампина, рифапентина, рифабутина, пиразинамида, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина и этамбутола.
В конкретном воплощении указанный по меньшей мере один агент представляет собой пиразинамид.
В конкретном воплощении указанный по меньшей мере один агент представляет собой рифампин.
В конкретном воплощении указанный по меньшей мере один агент представляет собой рифапентин.
В конкретном воплощении указанный по меньшей мере один агент представляет собой РА-824.
В конкретном воплощении указанный по меньшей мере один агент представляет собой ОРС-7683.
В конкретном воплощении указанный по меньшей мере один агент представляет собой ТМС-207.
В конкретном воплощении указанный по меньшей мере один агент выбран из группы, состоящей из моксифлоксацина, гатифлоксацина, левофлоксацина и офлоксацина.
В конкретном воплощении указанный по меньшей мере один агент представляет собой моксифлоксацин.
В еще одном воплощении указанный туберкулез включает активный туберкулез или латентный туберкулез.
В еще одном воплощении указанный активный туберкулез включает туберкулез, чувствительный к лекарственным средствам, устойчивый к одному лекарственному средству, туберкулез с множественной лекарственной устойчивостью (МЛУ) или туберкулез с расширенной лекарственной устойчивостью (РЛУ).
В еще одном воплощении способ по настоящему изобретению полностью устраняет туберкулез, чувствительный к лекарственным средствам, устойчивый к одному лекарственному средству, туберкулез с множественной лекарственной устойчивостью (МЛУ) и туберкулез с расширенной лекарственной устойчивостью (РЛУ) по завершении лечения.
В еще одном воплощении указанный туберкулез вызван инфекцией Mycobacterium, выбранной из группы, состоящей из Mycobacterium tuberculosis, Mycobacterium bovis или другого родственного вида микобактерий.
В еще одном воплощении способ по настоящему изобретению предупреждает рецидив инфекции Mycobacterium после завершения лечения.
В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли вводят перорально.
В еще одном воплощении указанный по меньшей мере один агент вводят перорально.
В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли и указанный по меньшей мере один агент вводят совместно в композиции.
В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли и указанный по меньшей мере один агент вводят раздельно.
В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли вводят один раз в сутки (QD) или два раза в сутки (BID).
В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли вводят один раз в неделю, дважды в неделю, трижды в неделю или через день.
В одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве от примерно 10 мг до примерно 2000 мг.
В еще одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве от примерно 25 мг до примерно 1000 мг.
В еще одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве от примерно 50 мг до примерно 500 мг.
В еще одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль вводят в количестве от примерно 100 мг до примерно 500 мг.
Настоящее изобретение также относится к фармацевтической композиции, содержащей:
1) терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли:

2) терапевтически эффективное количество по меньшей мере одного агента, полезного при лечении туберкулеза, и
3) один или более чем один фармацевтически приемлемый носитель или наполнитель.
В еще одном воплощении указанный по меньшей мере один агент выбран из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, этамбутола, стрептомицина, канамицина, амикацина, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина, капреомицина, этионамида, циклосерина, пара-аминосалициловой кислоты, тиацетазона, кларитромицина, клофазимина, амоксициллина-клавулановой кислоты, имипенема, меропенема, виомицина, теризидона, клофазимина, ТМС-207, РА-824, ОРС-7683, LL-3858 и SQ-109.
В еще одном воплощении указанный по меньшей мере один агент выбран из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина и этамбутола.
В конкретном воплощении указанный по меньшей мере один агент представляет собой пиразинамид.
В конкретном воплощении указанный по меньшей мере один агент представляет собой рифампин.
В конкретном воплощении указанный по меньшей мере один агент представляет собой рифапентин.
В конкретном воплощении указанный по меньшей мере один агент представляет собой изониазид.
В конкретном воплощении указанный по меньшей мере один агент представляет собой РА-824.
В конкретном воплощении указанный по меньшей мере один агент представляет собой ОРС-7683.
В конкретном воплощении указанный по меньшей мере один агент представляет собой ТМС-207.
В конкретном воплощении указанный по меньшей мере один агент выбран из группы, состоящей из моксифлоксацина, гатифлоксацина, левофлоксацина и офлоксацина.
В конкретном воплощении указанный по меньшей мере один агент представляет собой моксифлоксацин.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 10 мг до примерно 2000 мг соединения формулы (I) или его фармацевтически приемлемой соли.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 250 мг до примерно 1000 мг соединения формулы (I) или его фармацевтически приемлемой соли.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 600 мг до примерно 1000 мг соединения формулы (I) или его фармацевтически приемлемой соли.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 25 мг до примерно 1000 мг соединения формулы (I) или его фармацевтически приемлемой соли.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 50 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли.
Кроме того, любой препарат, включая комбинации, приведенные ниже, может содержать от 250 мг до 1000 мг соединения формулы (I) или его фармацевтически приемлемой соли или от 600 мг до 1000 мг соединения формулы (I) или его фармацевтически приемлемой соли.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли и примерно 600 мг рифампина.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли и примерно 300 мг изониазида.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли, примерно 300 мг изониазида и примерно 600 мг рифампина.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли, примерно 300 мг изониазида, примерно 600 мг рифампина и от примерно 20-25 мг/кг до примерно 50-70 мг/кг пиразинамида.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли и от примерно 10-15 мг/кг до примерно 20-30 мг/кг изониазида.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли, от примерно 10-15 мг/кг до примерно 20-30 мг/кг изониазида и от примерно 10 мг/кг до примерно 20 мг/кг рифампина.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли, от примерно 10-15 мг/кг до примерно 20-30 мг/кг изониазида, от 10 мг/кг до примерно 20 мг/кг рифампина и от примерно 15-30 мг/кг до примерно 50 мг/кг пиразинамида.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 50 мг до примерно 250 мг соединения формулы (I) или его фармацевтически приемлемой соли, примерно 75 мг изониазида, примерно 150 мг рифампина и примерно 400 мг пиразинамида.
В еще одном воплощении данная фармацевтическая композиция содержит от примерно 25 мг до примерно 250 мг соединения формулы (I) или его фармацевтически приемлемой соли и примерно 300 мг рифампина.
Настоящее изобретение также относится к производственному изделию, содержащему упакованную композицию, содержащую:
(а) (1) терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли:

(2) терапевтически эффективное количество по меньшей мере одного агента, полезного при лечении туберкулеза, и
(3) один или более чем один фармацевтически приемлемый носитель или наполнитель;
(б) вкладыш с инструкциями для введения упакованной композиции (а) для лечения туберкулеза; и
(в) контейнер для (а) и (б).
Настоящее изобретение также относится к фармацевтической упаковке для лечения туберкулеза у млекопитающего, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль:

и вкладыш с инструкциями для введения указанной композиции в комбинации с по меньшей мере одним агентом, полезным при лечении туберкулеза.
Эти и другие аспекты, преимущества и отличительные признаки данного изобретения станут очевидными из следующего подробного описания данного изобретения.
Подробное описание изобретения
Настоящее изобретение относится к способу лечения туберкулеза у млекопитающего, при котором указанному млекопитающему вводят эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли:

в комбинации с по меньшей мере двумя агентами, полезными при лечении туберкулеза.
Соединение формулы (I) по данному изобретению, (S)-N-[[3-[3-фтор-4-(4-тиоморфолинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамида, раскрыто в патенте США №5880118 (включенном сюда во всей его полноте посредством ссылки) в Примере 1. Как описано более подробно ниже, соединение формулы (I) по данному изобретению можно вводить в виде свободного основания или в форме его соли.
Фраза «фармацевтически приемлемая соль(ли)» при использовании здесь, если не указано иначе, включает соли кислотных или основных групп, которые могут присутствовать в соединениях формулы (I).
Например, соединения формулы (I), которые являются основными по природе, способны к образованию целого ряда солей" с разными неорганическими и органическими кислотами. Кислоты, которые можно использовать для получения фармацевтически приемлемых солей присоединения кислоты таких основных соединений, являются кислотами, которые образуют нетоксичные соли присоединения кислоты, т.е. соли, содержащие фармакологически приемлемые анионы, такие как соли гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, цитрат, кислый цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, пара-толуолсульфонат и памоат [т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)].
Примеры солей включают соли ацетат, акрилат, бензолсульфонат, бензоат (такой как хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат и метоксибензоат), бикарбонат, бисульфат, бисульфит, битартрат, борат, бромид, бутин-1,4-диоат, кальция эдетат, камсилат, хлорид, капроат, каприлат, цитрат, деканоат, дигидрофосфат, эдетат, эдисилат, эстолат, эзилат, этилсукцинат, формиат, фумарат, глюцептат, глюконат, глутамат, гликолят, гликолиларсанилат, гептаноат, гексин-1,6-диоат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, γ-гидроксибутират, йодид, изобутират, изотионат, лактат, лактобионат, лаурат, малат, малеат, малонат, манделат, мезилат, метафосфат, метансульфонат, метилсульфат, моногидрофосфат, мукат, напсилат, нафталин-1-сульфонат, нафталин-2-сульфонат, нитрат, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фенилацетаты, фенилбутират, фенилпропионат, фталат, фосфат/дифосфат, полигалактуронат, пропансульфонат, пропионат, пропиолат, пирофосфат, пиросульфат, салицилат, стеарат, субацетат, суберат, сукцинат, сульфат, сульфонат, сульфит, таннат, тартрат, теоклат, тозилат, триэтиодод и валерат, но не ограничиваются ими.
Данное изобретение также относится к солям присоединения основания соединений формулы (I). Химические основания, которые можно использовать в качестве реагентов для получения фармацевтически приемлемых основных солей соединений формулы (I), которые являются кислотными по природе, представляют собой основания, которые образуют нетоксичные основные соли с такими соединениями. Такие нетоксичные основные соли включают соли, полученные из таких фармакологически приемлемых катионов, как катионы щелочных металлов (например, калия и натрия), катионы щелочноземельных металлов (например, кальция и магния), соли присоединения аммония или водорастворимого амина, такого как N-метилглюкамин (меглумин), и соли низшего алканоламмония и другие основные соли фармацевтически приемлемых органических аминов, но не ограничиваются ими.
Также могут быть образованы гемисоли кислот и оснований, например, гемисульфат и гемикальциевые соли.
Для обзора подходящих солей, смотри Stahl and Wermuth, Handbook of Pharmaceutical Salts: Properties, Selection, and Use (Wiley-VCH, 2002). Способы получения фармацевтически приемлемых солей соединений формулы (I) по данному изобретению известны специалисту в данной области.
Термины «формула (I)» и «формулы (I) или их фармацевтически приемлемые соли» при использовании здесь определены так, что они включают все формы соединения формулы (I), включая изомеры, кристаллические и некристаллические формы, изоморфы, полиморфы, метаболиты, сольваты, гидраты и их пролекарства.
Термин «сольват» используется здесь для описания нековалентной или легко обратимой комбинации между растворителем и растворенным веществом или диспергентами и дисперсной фазой. Будет понятно, что данный сольват может находиться в форме твердого вещества, суспензии (например, суспензии или дисперсии) или раствора. Неограничивающие примеры растворителей включают этанол, метанол, пропанол, ацетонитрил, диметиловый эфир, диэтиловый эфир, тетрагидрофуран, хлористый метилен и воду. Термин «гидрат» используется, когда указанным растворителем является вода.
Принятой в настоящее время системой классификации органических гидратов является система, которая определяет гидраты с изолированным сайтом или канальные гидраты - смотри K.R.Morris, Polymorphism in Pharmaceutical Solids (Ed. H.G.Brittain, Marcel Dekker, 1995). Гидраты с изолированным сайтом представляют собой гидраты, в которых молекулы воды изолированы от непосредственного контакта друг с другом органическими молекулами, находящимися между ними. В канальных гидратах молекулы воды лежат в каналах кристаллической решетки, где они находятся рядом с другими молекулами воды.
Когда растворитель или вода являются прочно связанными, данный комплекс будет иметь хорошо определенную стехиометрию, независимо от влажности. Однако когда растворитель или вода являются слабо связанными, как в канальных сольватах и гигроскопических соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях нестехиометрические количества будут нормой.
Данное изобретение также относится к пролекарствам соединений формулы (I). Таким образом, определенные производные соединений формулы (I), которые сами по себе могут не иметь или иметь слабую фармакологическую активность, при введении в или на организм могут превращаться в соединения формулы (I), имеющие желательную активность, например, путем гидролитического расщепления. Такие производные именуются «пролекарствами». Дополнительную информацию по применению пролекарств можно найти в Pro-drugs as Novel Delivery Systems, Vol.14, ACS Symposium Series (T.Higuchi and W.Stella) и в Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (Ed. E.B.Roche, American Pharmaceutical Association).
Пролекарства согласно данному изобретению можно получить, например, заменой подходящих функциональных групп, присутствующих в соединениях формулы (I), определенными группировками, известными специалистам в данной области как «прогруппировки», как описано, например, у H.Bundgaard в Design of Prodrugs (Elsevier, 1985).
Некоторые неограничивающие примеры пролекарств согласно данному изобретению включают:
(1) где соединение формулы (I) содержит функциональную группу карбоновой кислоты, которая преобразована в подходящую метаболически лабильную группу (сложные эфиры, карбаматы и т.д.) соединения формулы (I);
(2) где соединение формулы (I) содержит спиртовую функциональную группу, которая преобразована в подходящую метаболически лабильную группу (простые эфиры, сложные эфиры, карбаматы, ацетали, кетали и т.д.) соединения формулы (I); и
(3) где соединение формулы (I) содержит первичную или вторичную функциональную аминогруппу или амид, которые преобразованы в подходящую метаболически лабильную группу, например, гидролизуемую группу (амиды, карбаматы, мочевины, фосфонаты, сульфонаты и т.д.) соединения формулы (I).
Дополнительные примеры заменяющих групп согласно вышеупомянутым примерам и примерам других типов пролекарств можно найти в вышеупомянутых ссылках.
Соединения формулы (I) по данному изобретению могут демонстрировать явления таутомерии и структурной изомерии. Например, соединения формулы (I) по данному изобретению могут существовать в нескольких таутомерных формах, включая енольную и имино форму, кето и енамино форму и их геометрические изомеры и смеси. Все такие таутомерные формы включены в пределы объема соединений формулы (I) по данному изобретению. Таутомеры существуют в виде смесей таутомерных наборов в растворе. В твердой форме обычно преобладает один таутомер. Даже несмотря на то, что может быть описан один таутомер, настоящее изобретение включает все таутомеры соединений формулы (I) по данному изобретению.
Настоящее изобретение также включает изотопно-меченые соединения, которые являются идентичными соединениям, перечисленным в формуле (I), приведенной выше, если не считать того, что один или более чем один атом заменен атомом, имеющим отличную атомную массу или массовое число от атомной массы или массового числа, обычно обнаруживаемых в природе. Примеры изотопов, которые могут быть включены в соединения формулы (I), включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 13C, 14C, 15N, 18O, 17O, 35S и 18F, но не ограничиваются ими. Некоторые изотопно-меченые соединения формулы (I) по данному изобретению, например, соединения, в которые включены радиоактивные изотопы, такие как 3H и 14C, являются полезными в анализах распределения в ткани лекарственного средства и/или субстрата. Тритированные, т.е. 3H, и углерод-14, т.е. 14C, изотопы являются особенно предпочтительными из-за простоты их получения и детектируемости. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может дать определенные терапевтические преимущества, происходящие из-за большей метаболической стабильности, например, увеличенного периода полувыведения in vivo или пониженных требований по дозировке, и, следовательно, может быть предпочтительным в некоторых обстоятельствах. Изотопно-меченые соединения формулы (I) по данному изобретению обычно можно получить путем проведения процедур, раскрытых в Схемах и/или Примерах и Получениях, приведенных ниже, путем замены реагента, не меченого изотопом, изотопно-меченым реагентом.
Соединения формулы (I) по данному изобретению могут демонстрировать полиморфизм. Полиморфы соединений формулы (I) по данному изобретению можно получить кристаллизацией соединения формулы (I) по изобретению при разных условиях. Например, можно использовать разные растворители (включая воду) или разные смеси растворителей для перекристаллизации; кристаллизацию при разных температурах; разные способы охлаждения, варьирующие от очень быстрого до очень медленного охлаждения на протяжении кристаллизации. Полиморфы также можно получать нагреванием или плавлением соединения формулы (I) по данному изобретению с последующим постепенным или быстрым охлаждением. Присутствие полиморфов можно определить спектроскопией ядерного магнитного резонанса (ЯМР) в твердом состоянии, ИК(инфра-красной)-спектроскопией, дифференциальной сканирующей калориметрией, дифракцией рентгеновских лучей на порошке или другими такими методиками.
Минимальным количеством соединения формулы (I) по данному изобретению, подлежащим введению, является эффективное количество. Термин «эффективное количество» означает количество соединения формулы (I) по данному изобретению, которое предупреждает начало, облегчает симптомы, останавливает развитие и/или устраняет ТБ инфекцию у млекопитающего, например человека.
Обнаружили, что терапевтически эффективное количество соединения формулы (I) по данному изобретению обладает желательными противотуберкулезными свойствами, описанными ниже. Однако наблюдается синергический эффект при введении соединения формулы (I) по данному изобретению в комбинации с по меньшей мере двумя агентами, полезными при лечении туберкулеза.
Под синергическим эффектом подразумевается то, что терапевтический эффект введения соединения формулы (I) по данному изобретению и по меньшей мере двух агентов, полезных при лечении туберкулеза, является большим, чем терапевтический эффект, полученный при введении эффективного количества либо одного соединения формулы (I) по данному изобретению, либо терапевтически эффективного количества по меньшей мере двух агентов, полезных при лечении туберкулеза, введенных индивидуально или в комбинации.
Такой синергизм является полезным в том, что он может обеспечить введение каждого из компонентов в данной комбинации в меньшем количестве, чем требовалось бы при индивидуальном введении, что может снизить вероятность вредных событий или неприятных побочных эффектов. В качестве альтернативы такой синергизм может сократить продолжительность лечения ТБ.
Таким образом, было обнаружено, что введение как соединения формулы (I) по данному изобретению, так и по меньшей мере двух агентов, полезных при лечении туберкулеза, например, по меньшей мере двух соединений, выбранных из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина и этамбутола, оказывает эффект, который приводит к улучшенному лечению туберкулеза по сравнению с эффектом, когда соединение формулы (I) по данному изобретению вводят само по себе, когда по меньшей мере два агента, полезных при лечении туберкулеза, вводят индивидуально, или когда меньшей мере два агента, полезных при лечении туберкулеза, вводят в комбинации друг с другом.
В одном воплощении данного изобретения было обнаружено, что введение как соединения формулы (I) по данному изобретению или его фармацевтически приемлемой соли, так и по меньшей мере двух соединений, выбранных из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина и этамбутола, оказывает эффект, который приводит к полному устранению туберкулеза по сравнению с неполным устранением, когда вводят соединение формулы (I) по данному изобретению или по меньшей мере два агента, полезных при лечении туберкулеза, индивидуально или в комбинации друг с другом.
Термин «полное устранение» означает то, что после схемы лечения комбинацией по настоящему изобретению в органе-мишени, т.е. в легких инфицированного млекопитающего, не наблюдали способных к размножению микобактерий. Отмечают, что в конце лечения инфицированных млекопитающих существующей схемой лечения лекарственным средством, т.е. комбинацией по меньшей мере двух агентов, полезных при лечении туберкулеза, изониазида, пиразинамида и рифампина, из органа-мишени, т.е. из легких, выделяют значительное количество способных к размножению туберкулезных бацилл. Это очевидно из данных в Таблице 3, приведенной ниже. Дополнительно отмечается, что даже после лечения в течение 4 месяцев посредством стандартной схемы лечения: 2 месяцев изониазида, пиразинамида и рифампина с последующими 2 месяцами изониазида и рифампина, 90% мышей снова заболевали после окончания лечения, что означает то, что живые бациллы сохраняются в момент завершения лечения, даже если они не могут быть культивированы во время завершения лечения, что очевидно из данных в Таблице 4.
В одном воплощении настоящего изобретения по меньшей мере два агента, полезных при лечении туберкулеза, используются в сочетании с соединением формулы (I), и описанные здесь фармацевтические композиции по данному изобретению являются следующими: изониазид, рифампин, рифапентин, рифабутин, пиразинамид, этамбутол, стрептомицин, канамицин, амикацин, моксифлоксацин, гатифлоксацин, левофлоксацин, офлоксацин, ципрофлоксацин, капреомицин, этионамид, циклосерин, пара-аминосалициловая кислота, тиацетазон, кларитромицин, амоксициллин-клавулановая кислота, имипенем, меропенем, клофазимин, виомицин, теризидон, ТМС-207, РА-824, ОРС-7683, LL-3858 и SQ-109.
В другом воплощении один из указанных по меньшей мере двух агентов выбран из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина и этамбутола.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой пиразинамид.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой рифампин.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой рифапентин.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой РА-824.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой ОРС-67683.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой ТМС-207.
В еще одном воплощении один из указанных по меньшей мере двух агентов выбран из группы, состоящей из моксифлоксацина, гатифлоксацина, левофлоксацина и офлоксацина.
В еще одном воплощении один из указанных по меньшей мере двух агентов представляет собой моксифлоксацин.
В конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид и рифампин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид, рифампин и изониазид.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид и рифапентин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид, рифапентин и изониазид.
В конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид и моксифлоксацин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид, моксифлоксацин и рифампин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой пиразинамид, моксифлоксацин и рифапентин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой РА-824 и пиразинамид.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой РА-824, пиразинамид и агент, выбранный из группы, состоящей из стрептомицина, канамицина, амикацина и капреомицина.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой РА-824 и моксифлоксацин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой РА-824, моксифлоксацин и агент, выбранный из группы, состоящей из стрептомицина, канамицина, амикацина и капреомицина.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ОРС-67683 и пиразинамид.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ОРС-67683, пиразинамид и агент, выбранный из группы, состоящей из стрептомицина, канамицина, амикацина и капреомицина.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ОРС-67683 и моксифлоксацин.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ОРС-67683, моксифлоксацин и агент, выбранный из группы, состоящей из стрептомицина, канамицина, амикацина и капреомицина.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ТМС-207 и пиразинамид.
В еще одном конкретном воплощении указанные по меньшей мере два агента представляют собой ТМС-207, пиразинамид и моксифлоксацин или изониазид.
Дополнительно обнаружили, что синергический эффект наблюдается при введении соединения формулы (I) по данному изобретению в комбинации с по меньшей мере одним агентом, полезным при лечении туберкулеза, например, с рифампином, во время фазы продолжения (стерилизации) лечения (после исходной фазы лечения), когда превалируют неразмножающиеся сохраняющиеся бактерии. Такой синергизм полезен тем, что может сокращать продолжительность лечения ТБ. Более важно то, что данные, приведенные ниже, показывают, что введение соединения формулы (I) в комбинации с по меньшей мере двумя агентами, полезными при лечении туберкулеза, может сокращать продолжительность терапии чувствительного к лекарственным средствам ТБ, ТБ, устойчивого к одному лекарственному средству, и ТБ с множественной лекарственной устойчивостью.
В одном воплощении настоящего изобретения способы по настоящему изобретению обеспечивают лечение млекопитающего после того, как данное млекопитающее подверглось исходной фазе лечения, включающей по меньшей мере один агент, полезный при лечении туберкулеза, используемый в сочетании с соединением формулы (I).
В другом воплощении по меньшей мере один агент, полезный при лечении туберкулеза, выбран из группы, состоящей из рифампина, рифапентина, рифабутина, пиразинамида, этамбутола, стрептомицина, канамицина, амикацина, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина, капреомицина, этионамида, циклосерина, пара-аминосалициловой кислоты, триацетазона, кларитромицина, амоксициллина-клавулановой кислоты, имипенема, меропенема, клофазимина, виомицина, теризидона, ТМС-207, РА-824, ОРС-7683, LL-3858 и SQ-109.
В еще одном воплощении по меньшей мере один указанный агент выбран из группы, состоящей из рифампина, рифапентина, рифабутина, пиразинамида, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина и этамбутола.
В конкретном воплощении указанный по меньшей мере один агент представляет собой пиразинамид.
В конкретном воплощении указанный по меньшей мере один агент представляет собой рифампин.
В конкретном воплощении указанный по меньшей мере один агент представляет собой рифапентин.
В конкретном воплощении указанный по меньшей мере один агент представляет собой РА-824.
В конкретном воплощении указанный по меньшей мере один агент представляет собой ОРС-7683.
В конкретном воплощении указанный по меньшей мере один агент представляет собой ТМС-207.
В конкретном воплощении указанный по меньшей мере один агент выбран из группы, состоящей из моксифлоксацина, гатифлоксацина, левофлоксацина и офлоксацина.
В конкретном воплощении указанный по меньшей мере один агент представляет собой моксифлоксацин.
Способы и композиции по данному изобретению являются особенно эффективными против туберкулеза, включая активный туберкулез и латентный туберкулез. В одном примере данный активный туберкулез включает чувствительный к лекарственным средствам туберкулез, туберкулез, устойчивый к одному лекарственному средству, туберкулез с множественной лекарственной устойчивостью и туберкулез с расширенной лекарственной устойчивостью. В другом примере согласно настоящему изобретению предложен способ полного устранения чувствительного к лекарственным средствам туберкулеза, устойчивого к одному лекарственному средству туберкулеза, туберкулеза с множественной лекарственной устойчивостью и туберкулеза с расширенной лекарственной устойчивостью (РЛУ) по завершении лечения.
Кроме того, способы и композиции по данному изобретению можно использовать в сочетании с диагностическими тестами для идентификации туберкулеза у млекопитающего, которые известны специалистам в данной области. Например, способы и композиции по данному изобретению можно использовать в сочетании с так называемым линейным анализом проб (line-probe assay, Hain Life-science GmbH), который можно использовать для идентификации генов, связанных с устойчивостью к рифампину и изониазиду для выявления туберкулеза с множественной лекарственной устойчивостью и/или туберкулеза с расширенной лекарственной устойчивостью. В сочетании со способами и композициями по данному изобретению также можно использовать другие анализы, которые известны специалистам в данной области.
В другом воплощении способы и композиции по данному изобретению являются особенно эффективными против туберкулеза, вызванного инфекцией Mycobacterium, выбранной из группы, состоящей из Mycobacterium tuberculosis, Mycobacterium bovis или другого родственного вида микобактерии, который известен специалисту в данной области. В одном примере согласно настоящему изобретению предложены способы и композиции для предупреждения рецидива инфекции Mycobacterium после завершения лечения.
Такая комбинация может быть предназначена для одновременного, раздельного или последовательного применения. В одном воплощении по меньшей мере два агента (или по меньшей мере один агент), полезных для лечения туберкулеза, вводят до введения соединения формулы (I) по данному изобретению. В другом воплощении по меньшей мере два агента (или по меньшей мере один агент), полезных для лечения туберкулеза, вводят после введения соединения формулы (I) по данному изобретению. В другом воплощении по меньшей мере два агента (или по меньшей мере один агент), полезных для лечения туберкулеза, вводят примерно в то же самое время, что и соединение формулы (I) по данному изобретению.
В некоторых случаях было бы полезным раздельное введение каждого соединения в разное время и разными путями. Таким образом, компоненты в данной комбинации, т.е. соединение формулы (I) по данному изобретению и по меньшей мере два агента (или по меньшей мере один агент) при лечении туберкулеза, не обязательно вводить по существу в то же самое время или в любом порядке. Данное введение может быть спланировано во времени таким образом, что пиковый фармакокинетический эффект одного соединения будет совпадать с пиковым фармакокинетическим эффектом другого.
Все активные ингредиенты могут быть приготовлены в виде препарата в отдельных или индивидуальных лекарственных формах, которые можно вводить совместно друг за другом. Другой возможностью является то, что если путь введения является тем же самым (например, пероральным), два или более чем два активных соединения могут быть приготовлены в виде препарата в единой форме для совместного введения, при этом, однако, оба способа совместного введения являются частью того же самого терапевтического лечения или схемы.
Предпочтительными агентами, полезными для лечения туберкулеза, могут быть следующие: изониазид, рифампин, рифапентин, рифабутин, пиразинамид, этамбутол, стрептомицин, канамицин, амикацин, моксифлоксацин, гатифлоксацин, левофлоксацин, офлоксацин, ципрофлоксацин, капреомицин, этионамид, циклосерин, пара-аминосалициловая кислота, тиацетазон, кларитромицин, амоксициллин-клавулановая кислота, имипенем, меропенем, клофазимин, виомицин, теризидон, ТМС-207, РА-824, ОРС-7683, LL-3858 и SQ-109. Данные агенты, полезные для лечения туберкулеза, можно использовать в настоящем изобретении в целом ряде форм, включая кислотную форму, солевую форму, рацематы, энантиомеры, сольваты и таутомеры. Данные агенты, полезные для лечения туберкулеза, можно вводить любым путем, полезным для введения указанных агентов, которые известны специалистам в данной области.
Данное изобретение также относится к композициям по изобретению, которые содержат: (1) терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, (2) по меньшей мере один агент, полезный при лечении туберкулеза, и (3) фармацевтически приемлемые носители или наполнители (далее: "композиции по данному изобретению").
Композиции по данному изобретению, которые являются подходящими для введения пациенту, нуждающемуся в этом (например, человеку), также именуются здесь "фармацевтические композиции по данному изобретению".
Фармацевтические композиции по данному изобретению могут находиться в любой форме, подходящей для введения пациенту. Например, фармацевтические композиции по данному изобретению могут находиться в форме, подходящей для перорального введения, такой как таблетка, капсула, пилюля, порошок, препараты замедленного высвобождения, раствор и суспензия; для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии; для местного введения в виде мази или крема; или для ректального введения в виде суппозитория. Фармацевтические композиции по данному изобретению могут находиться в стандартных лекарственных формах, подходящих для однократного введения точных дозировок.
Типичные формы для парентерального введения включают растворы или суспензии активных соединений в стерильных водных растворах, например, в водных растворах пропиленгликоля или декстрозы. Такие лекарственные формы, если желательно, могут быть подходящим образом забуференными.
В одном воплощении фармацевтические композиции по данному изобретению могут находиться в виде пероральной лекарственной формы. Неограничивающие примеры пероральных лекарственных форм включают такие, как, например, жевательные таблетки, капсулы, пилюли, лепешки, пастилки, саше, порошки, сиропы, эликсиры, растворы и суспензии и тому подобное, согласно стандартной фармацевтической практике. В другом воплощении фармацевтические композиции по данному изобретению также можно непосредственно доставлять в желудочно-кишечный тракт пациента через назогастральный зонд.
Соединение формулы (I) по данному изобретению будет присутствовать в фармацевтической композиции по данному изобретению в достаточном количестве для обеспечения желательного количества дозировки в описанном здесь интервале. Пропорциональное отношение соединения формулы (I) по данному изобретению к эксципиентам, естественно, будет зависеть от химической природы, растворимости и стабильности активных ингредиентов, а также от рассматриваемой лекарственной формы. Типично фармацевтические композиции по настоящему изобретению могут содержать от примерно 20% до примерно 99% соединения формулы (I) по данному изобретению по массе.
В одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли вводят в количестве от примерно 10 мг до примерно 2000 мг. В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли вводят в количестве от примерно 25 мг до примерно 1000 мг.
В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли вводят в количестве от примерно 50 мг до примерно 500 мг.
В еще одном воплощении соединение формулы (I) и его фармацевтически приемлемые соли вводят (в количестве) от примерно 100 мг до примерно 500 мг.
Методики приготовления в виде препарата и введения соединения формулы (I) по настоящему изобретению и композиций по данному изобретению можно найти в Remington: the Science and Practice of Pharmacy, 19th ed., Mack Pub. Co., Easton, Pa. (1995).
Термин «эксципиент» означает инертное вещество, которое объединяют с соединением формулы (I) для получения фармацевтической композиции или пероральной лекарственной формы.
Термин «фармацевтически приемлемый эксципиент» означает то, что данный эксципиент должен быть совместимым с другими ингредиентами данной композиции и не вредным для ее реципиента. Фармацевтически приемлемые эксципиенты выбирают в зависимости от планируемой лекарственной формы.
Таблетки, пилюли, капсулы и тому подобное могут содержать эксципиенты, выбранные из связующих веществ, таких как поливинилпирролидон, гидроксипропилметилцеллюлоза (НРМС), гидроксипропилцеллюлоза (НПЦ), сахароза, желатин, аравийская камедь, трагакантовая камедь или кукурузный крахмал; наполнителей, таких как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, двухосновный фосфат кальция, глицин и крахмал; разрыхлителей, таких как кукурузный крахмал, картофельный крахмал, альгиновая кислота, натрий крахмал гликолят, кроскармеллоза натрия и определенные комплексные силикаты; смазывающих веществ, таких как стеарат магния, лаурилсульфат натрия и тальк; и подсластителей, таких как сахароза, лактоза или сахарин. Когда стандартная лекарственная форма представляет собой капсулу, она может содержать, помимо веществ приведенного выше типа, жидкий носитель, такой как нелетучее жидкое масло. Могут присутствовать другие вещества в качестве покрытий или для модификации физической формы данной лекарственной формы. Например, таблетки могут быть покрыты шеллаком, сахаром или тем и другим.
В случае педиатрических пероральных суспензий и саше эти эксципиенты могут содержать суспендирующие вспомогательные вещества, такие как ксантановая камедь или гидроксипропилметилцеллюлоза, скользящие вещества, такие как коллоидный кремнезем, разбавители и наполнители, такие как диоксид кремния, корригенты, такие как корригенты, используемые в жевательных резинках, с ароматом апельсина, банана, малины и золотого сиропа или их смеси, подсластители, такие как аспартам или сахар, и стабилизаторы, такие как янтарная кислота. Препараты в виде порошка или гранул, такие как препараты педиатрических суспензий и саше, могут быть изготовлены с использованием методик, которые, в общем, являются традиционными в области изготовления фармацевтических препаратов и при изготовлении сухих препаратов для разведения до таких суспензий. Например, подходящей методикой является методика смешивания сухих порошковых или гранулированных ингредиентов.
Типично эффективная суточная доза (т.е. общая дозировка в течение примерно 24 часов) соединения формулы (I) по данному изобретению для взрослых составляет от примерно 10 мг до примерно 2000 мг; от примерно 25 мг до примерно 1000 мг; от примерно 50 мг до примерно 500 мг; и от 100 мг до примерно 500 мг с пищей или без нее. В некоторых случаях может быть необходимо применение дозировок вне этих границ.
Суточную дозировку соединения формулы (I) по данному изобретению обычно вводят от 1 до 4 раз ежесуточно равными дозами.
В одном воплощении за сутки вводят однократную дозу соединения формулы (I) по данному изобретению (т.е. примерно с 24-часовыми интервалами) (т.е. QD); в еще одном воплощении за сутки вводят две дозы соединения формулы (I) по данному изобретению (т.е. BID); в еще одном воплощении за сутки вводят три дозы соединения формулы (I) по данному изобретению (т.е. TID); и в еще одном воплощении за сутки вводят четыре дозы соединения формулы (I) по данному изобретению (т.е. QID); в еще одном воплощении однократную дозу соединения формулы (I) по данному изобретению вводят через сутки (т.е. примерно с 48-часовыми интервалами), в еще одном воплощении однократную дозу соединения формулы (I) по данному изобретению вводят дважды в неделю; в еще одном воплощении однократную дозу соединения формулы (I) по данному изобретению вводят трижды в неделю.
В одном воплощении эффективную дозу соединения формулы (I) по данному изобретению вводят BID примерно с 12-часовыми интервалами.
В другом воплощении эффективную дозу соединения формулы (I) по данному изобретению вводят TID примерно с 8-часовыми интервалами.
В другом воплощении эффективную дозу соединения формулы (I) по данному изобретению вводят QID примерно с 6-часовыми интервалами.
В одном воплощении эффективная доза соединения формулы (I) по данному изобретению составляет от примерно 25 мг до примерно 1000 мг, которые вводят BID примерно с 12-часовыми интервалами.
Пероральное введение является предпочтительным.
Фармацевтическую композицию по данному изобретению с фиксированной комбинацией доз, содержащей соединение формулы (I) по данному изобретению и по меньшей мере один агент, полезный для лечения туберкулеза, и фармацевтически приемлемые носители, можно изготовить традиционными в данной области способами. Например, любой специалист в данной области может изготовить данную комбинацию в форме таблетки.
Некоторые примеры по настоящему изобретению представляют собой комбинации и фармацевтические композиции, которые включают следующие неограничивающие смеси:
а) соединение формулы (I) или его фармацевтически приемлемая соль и пиразинамид;
б) соединение формулы (I) или его фармацевтически приемлемая соль и рифампин;
в) соединение формулы (I) или его фармацевтически приемлемая соль и рифапентин;
г) соединение формулы (I) или его фармацевтически приемлемая соль и РА-824;
д) соединение формулы (I) или его фармацевтически приемлемая соль и ТМС-207;
е) соединение формулы (I) или его фармацевтически приемлемая соль и агент, выбранный из группы, состоящей из моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина и моксифлоксацина;
ж) соединение формулы (I) или его фармацевтически приемлемая соль, изониазид и рифампин;
з) соединение формулы (I) или его фармацевтически приемлемая соль, изониазид, рифампин и пиразинамид; и
и) соединение формулы (I) или его фармацевтически приемлемая соль, рифампин и пиразинамид.
Другие примеры по настоящему изобретению представляют собой комбинации и фармацевтические композиции, которые включают следующие неограничивающие смеси:
к) от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли и примерно 600 мг рифампина;
л) от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли и примерно 300 мг изониазида;
м) от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли и примерно 300 мг изониазида и примерно 600 мг рифампина;
н) от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли, примерно 300 мг изониазида, примерно 600 мг рифампина и от примерно 20-25 мг/кг до примерно 50-70 мг/кг пиразинамида;
о) от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли и от примерно 10 мг/кг до примерно 20 мг/кг рифампина;
п) от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли и от примерно 10-15 мг/кг до примерно 20-30 мг/кг изониазида;
р) от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли, от примерно 10-15 мг/кг до примерно 20-30 мг/кг изониазида и от примерно 10 мг/кг до примерно 20 мг/кг рифампина;
с) от примерно 100 мг до примерно 500 мг соединения формулы (I) или его фармацевтически приемлемой соли, от примерно 10-15 мг/кг до примерно 20-30 мг/кг изониазида, от примерно 10 мг/кг до примерно 20 мг/кг рифампина и от примерно 15-30 мг/кг до примерно 50 мг/кг пиразинамида;
т) от примерно 50 мг до примерно 250 мг соединения формулы (I) или его фармацевтически приемлемой соли, примерно 75 мг изониазида, примерно 150 мг рифампина и примерно 400 мг пиразинамида; и
у) от примерно 25 мг до примерно 250 мг соединения формулы (I) или его фармацевтически приемлемой соли и примерно 300 мг рифампина.
Соединения формулы (I) по настоящему изобретению легко получить согласно способам синтеза, знакомым специалистам в данной области. Соединение формулы (I) по данному изобретению можно получить способом, аналогичным способу, описанному для получения из Примера 1, описанного в разделе Примеров патента США №5880118. Кроме того, соединение формулы (I) можно получить способами, изложенными в международных публикациях WO97/37980 и W099/24393, каждая из которых включена сюда посредством ссылки.
Другим примером получения (S)-N-[[3-[3-фтор-4-(4-тиоморфолинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамида является следующий пример.
Схема 1 иллюстрирует общую последовательность синтеза для получения соединений по настоящему изобретению.
СХЕМА I

Схема 1 иллюстрирует способ проведения синтеза соединений формулы (I) по данному изобретению в многоступенчатом синтезе через соединение формулы 6. Ссылаясь на Схему I, данный синтез начинается с образования промежуточного соединения (3) при взаимодействии (S)-эпихлоргидрина (1) со смесью подходящим образом замещенного бензальдегидного производного (2) (предпочтительно от 0,5 до 2 экв., наиболее предпочтительно - 1 экв.) и водного аммиака (предпочтительно от 0,5 до 3 экв., наиболее предпочтительно 1,5 экв.). Данную реакцию лучше всего проводить как в протонных, так и в непротонных ненуклеофильных и инертных растворителях, таких как спирты (включая C1-C6 разветвленные и линейные спирты и полиолы), простые эфиры (включая метил-трет-бутиловый эфир (МТВЕ), тетрагидрофуран (THF) и другие C1-C6 линейные, разветвленные и циклические эфиры), а также в хлорированных растворителях, таких как хлористый метилен. Предпочтительным растворителем является МТВЕ. Предпочтительными являются температуры в диапазоне от примерно 15 до примерно 60°С, наиболее предпочтительными от 30 до 50°С. После экстрактивного выделения и концентрирования получают имино-группировку (3). Ее затем кристаллизуют из второй жидкой фазы в присутствии неполярных углеводородных растворителей, таких как, без ограничения, алканы, смеси алканов (гексан, гептан, октан, изо-октан и имеющиеся в продаже смеси алканов), возможно в присутствии апротонных полярных растворителей, предпочтительно эфирных растворителей, таких как МТВЕ, или ароматических растворителей, таких как толуол, или хлорированных растворителей, таких как хлористый метилен или их смеси. Предпочтительными растворителями являются смесь МТВЕ и гептана или смесь толуола и гептана. Процесс кристаллизации можно проводить при температуре в интервале от температуры окружающей среды (примерно 18-25°С) до примерно 55°C, предпочтительно в интервале 30-50°С, более предпочтительно в интервале 38-45°C. Эта кристаллизация обеспечивает неожиданно высокий выход и дает значительно улучшенную энантиомерную чистоту после выделения посредством фильтрования. (S)-эпихлоргидрин (1) и бензальдегидное производное (2) имеются в продаже или могут быть получены способами, хорошо известными специалистам в данной области.
Замещенную имино-группировку (3) подвергают сочетанию с карбаматом (4) (что известно специалистам в данной области, смотри, например, J. Med. Chem., 1996, 39, (3), 680-685 и также Пример 2, приведенный ниже (предпочтительно от 1 до 3 экв., наиболее предпочтительно от 1,5 до 2 экв.)) с получением соответствующего (S)-оксазолидинонимина (5). Данную реакцию предпочтительно проводят при температуре в интервале от температуры окружающей среды до примерно 65°C в присутствии основания с рКа больше, чем 12, предпочтительно третичного алкоксидного основания, наиболее предпочтительно трет-бутоксида лития и апротонного ненуклеофильного растворителя (предпочтительно DMF, DMAc, ацетонитрил, C1-C6 линейные, разветвленные и циклические эфиры и/или хлорированные растворители, и/или смеси этих растворителей, наиболее предпочтительно МТВЕ или хлористый метилен). Наиболее предпочтительно температура составляет от примерно 30 до примерно 60°C, и время реакции составляет от 2 до 24 часов. Предпочтительно (S)-оксазолидинонимин (5) после водной экстрактивной обработки выделяют фильтрованием из смеси 1:1 простого эфира (включая МТВЕ, THF и другие C1-C6 линейные, разветвленные и циклические эфиры) и воды, наиболее предпочтительно МТВЕ. В качестве альтернативы, (5) выделяют после водного экстрактивного выделения путем фильтрования или кристаллизации из спирта (включая C1-C6 линейные и разветвленные спирты и полиолы); наиболее предпочтительно изопропанола. Гидролиз соединения (5) водным кислотным раствором дает соединение (6), и последующее ацилирование дает неочищенное соединение (7). Соединение (5) лучше всего гидролизуется смесью воды и сильной кислоты, такой как соляная кислота, и замещенный бензальдегидный побочный продукт удаляют экстракцией органическим растворителем, несмешивающимся с водой (предпочтительно толуолом, МТВЕ, хлористым метиленом или этилацетатом), наиболее предпочтительно этилацетатом. Образующийся водный раствор амингидрохлорида (6) предпочтительно ацилируют уксусным ангидридом, предпочтительно в присутствии воды и органического растворителя, несмешивающегося с водой (наиболее предпочтительно хлористого метилена). Превращение амингидрохлорида (6) в соединение (7) хорошо известно в литературе. (Смотри Brickner, S.J. et al. J. Med. Chem. 1996 39 (3) 673-679, патент США 5837870, патент США 5688792).
Приведенные ниже примеры дополнительно иллюстрируют и представляют соединения формулы (I) по данному изобретению, композиции по данному изобретению и способы применения соединения по данному изобретению. Следует понимать, что объем настоящего изобретения не ограничен каким-либо образом объемом следующих примеров и способов получения.
Пример 1
Получение (S)-1-хлор-3-[(4-хлор-Е-бензилиден)-амино]-пропан-2-ола
Способ А
В 5-л трехгорлую круглодонную колбу, оснащенную механической мешалкой, термопарой, обратным холодильником и греющим кожухом, загружают 4-хлорбензальдегид (351,0 г, 2,5 моль, 1,0 экв.). Затем загружают на круглое дно МТВЕ (1,5 л) с получением гомогенного раствора. Добавляют одной порцией водный аммиак (28% масс., 252,98 мл, 3,75 моль, 1,5 экв.), что приводит к образованию белого осадка, который превращается в жидкую суспензию в пределах 15 минут перемешивания. Затем в сосуд медленно загружают (S)-(+)-эпихлоргидрин (>99% э.и. (энантиомерный избыток), 196,0 мл, 2,5 моль, 1,0 экв.). Затем, через 40 минут, содержимое медленно нагревают до 43°C. Данную реакционную смесь перемешивают при 40°C в течение 18 часов, причем в этот момент времени сохранялось 8,4% площади эпихлоргидрина при определении ГХ (газовая хроматография). При охлаждении данную реакционную смесь переносят в делительную воронку, и разделяют слои. Нижний водный слой отбрасывают. Органический слой переносят в 3-л круглодонную колбу, концентрируют в вакууме до примерно половины объема (800-900 мл), одновременно медленно добавляют изо-октан (~750 мл) из подпитывающей трубки, пока наблюдается мутность. В двухфазную смесь вносят затравку ~4 мг соединения, указанного в заголовке. Данную реакционную смесь охлаждают в бане со льдом в течение 45 минут с перемешиванием. Осадок собирают и промывают холодным изо-октаном (500 мл). Данное твердое вещество сушат в течение 18 часов при 50°С в вакууме с получением 345,19 г (59% выход) соединения, указанного в заголовке, в виде белого твердого вещества. Анализ ГХ: 100%, 99,7% э.и., определенный хиральной СКФ (сверхкритической флюидной хроматографией, SFC). ГХ (условия: колонка 30-метровая НР-1, внутренний диаметр 0,25 мм; 0,25 мкм пленка и давление на входе в колонку 15 фунт-сила/кв.дюйм (103,421 кПа); инъекционный объем 1,0 мкл; начальная температура Tini=70°С, скорость изменения температуры 20°С/мин); время удерживания TR (эпихлоргидрин)=2,4 мин, TR (4-хлорбензальдегид)=4,8 мин и TR (соединение, указанное в заголовке)=9,7 мин; условия ВЭЖХ (высокоэффективная жидкостная хроматография): колонка Chiralpak AD-H 250 мм ×4,6 мм, элюирование 70% CO2/30% MeOH при скорости 3,0 мл/мин, детектирование при 255 нм. TR [соединение, указанное в заголовке]=3,9 мин; TR (энантиомер соединения, указанного в заголовке)=2,8 мин; 1Н ЯМР (400 МГц, CDCl3) δ 3.69 (bs, 2 Н), 3.80 (m, 2 Н), 4.15 (s, 1 Н), 7.41 (d, J=8 Гц, 2 Н), 7.69 (d, J=8 Гц, 2 Н), 8.33 (s, 1 Н); 13С ЯМР (CDCl3) δ 47.05, 63.09, 70.82, 128.93, 129.39, 134.08, 137.07, 162.30; ИК (KBr Pellet) 1630 см-1.
Способ Б
В 5-л трехгорлую круглодонную колбу, оснащенную механической мешалкой, термопарой, обратным холодильником и греющим кожухом, загружают 4-хлорбензальдегид (375 г, 2,67 моль, 1,0 экв.). Добавляют метанол (0,75 л) с получением гомогенного раствора после нагревания от 10 до 23°C. Добавляют одной порцией водный аммиак (28,4% масс., 264 мл, 3,95 моль, 1,5 экв.), что приводит к образованию двухфазного раствора после перемешивания в течение 15 минут при 23-26°C. Затем одной порцией добавляют (S)-(+)-эпихлоргидрин (99,3% э.и., 207 мл, 2,64 моль, 1,0 экв.). Данную реакционную смесь перемешивают при 23-24°C в течение 18 часов, затем нагревают до 40-45°С и перемешивают в течение 2,5 часов, причем в данный момент времени остается 0,26% площади (S)-эпихлоргидрина при определении ГХ (условия ГХ: 0,050 мл реакционной смеси в 1 мл ацетонитрила, инъецирование 1 микролитра; колонка 15 М DB-1, внутренний диаметр 0,25 мм; 0,25 мкм пленка и давление на входе в колонку 15 фунт-сила/кв.дюйм (103,421 кПа); инъекционный объем 1,0 мкл; Tini=38°C, скорость изменения температуры 10°C/мин); TR (эпихлоргидрин)=1,1 мин, TR (4-хлорбензальдегид)=6,9 мин и TR (соединение, указанное в заголовке)=16,0 мин). Данную смесь концентрируют в вакууме до общего объема 1250 мл. Добавляют толуол (250 мл), и данную смесь концентрируют в вакууме до общего объема 1250 мл. Добавляют толуол (250 мл), и данную смесь концентрируют в вакууме до общего объема 1145 мл. Добавляют толуол (355 мл), и данную смесь концентрируют в вакууме до общего объема 900 мл. Добавляют толуол (600 мл), и данную смесь концентрируют в вакууме до общего объема 1120 мл. Поддерживая температуру 45-50°C, добавляют гептан (1500 мл). Образующийся двухфазный раствор охлаждают до 45°C и вносят затравку. Данную смесь затем дополнительно охлаждают до 38°C в течение 1/2 часа, внося затравку после каждого 1 градуса охлаждения. Затем позволяют данной смеси дополнительно медленно охладиться до 23°C за 16 часов. Белые кристаллы затем собирают вакуум-фильтрацией и промывают гептаном комнатной температуры (180 мл). Продукт сушат в струе азота с получением соединения, указанного в заголовке (431,57 г, 70,4%). ВЭЖХ 95% площади [колонка Kromasil 150 мм ×4,6 мм, 254 нм, скорость тока 1,5 мл/мин; А=1000 мл воды +0,52 мл трифторуксусной кислоты +1,20 мл триэтиламина; Б=ацетонитрил; изократический 47:53 А:Б в течение 5 мин, затем градиент до 100% Б за 5 мин, TR [соединение, указанное в заголовке]=2,1 мин; TR (4-хлорбензальдегид)=2,3 мин]; 99,72% э.и., определенный посредством хиральной СФХ. Условия хиральной ВЭЖХ: колонка Chiralpak AD-H 250 мм ×4,6 мм, элюирование 70% CO2/30% MeOH при скорости 3,0 мл/мин, детектирование при 255 нм. TR [соединение, указанное в заголовке]=3,9 мин; TR (энантиомер соединения, указанного в заголовке)=2,8 мин; 1Н ЯМР (400 МГц, CDCl3) δ 3.69 (bs, 2 Н), 3.80 (m, 2 Н), 4.15 (s, 1 Н), 7.41 (d, J=8 Гц, 2 Н), 7.69 (d, J=8 Гц, 2 Н), 8.33 (s, 1 Н); 13С ЯМР (CDCl3) δ 47.05, 63.09, 70.82, 128.93, 129.39, 134.08, 137.07, 162.30.
Способ В
В 5-л трехгорлую круглодонную колбу, оснащенную механической мешалкой, термопарой, обратным холодильником и греющим кожухом, загружают 4-хлорбензальдегид (375 г, 2,67 моль, 1,0 экв.). Затем загружают МТВЕ (1,5 л) с получением гомогенного раствора после нагревания от 9 до 24°C. Добавляют одной порцией водный аммиак (28,4% масс., 265 мл, 3,97 моль, 1,5 экв.), что приводит к образованию двухфазного раствора после перемешивания в течение 15 минут при 23-26°C. Затем добавляют одной порцией (S)-(+)-эпихлоргидрин (99,3% э.и., 209 мл, 2,67 моль, 1,0 экв.). Данную реакционную смесь перемешивают при 23-24°C в течение 3 суток. Фазы разделяют, и верхнюю фазу концентрируют при атмосферном давлении от 2000 до 1000 мл общего объема (температура кипения от 58 до 67°C). Добавляют гептан (1700 мл), поддерживая температуру 45-50°С. Образующийся двухфазный раствор охлаждают до 45°C и вносят затравку. Данную смесь затем дополнительно охлаждают до 38°C в течение 1/2 часа, внося затравку после каждого 1 градуса охлаждения. Затем позволяют данной смеси дополнительно медленно охладиться до 23°C в течение 1 часа. Затем собирают снежно-белые тяжелые кристаллы вакуум-фильтрацией и промывают гептаном комнатной температуры (180 мл). Данный продукт сушат в струе азота с получением соединения, указанного в заголовке (462,43 г, 74,7%). ВЭЖХ 94% площади [колонка Kromasil 150 мм ×4,6 мм, 254 нм, скорость тока 1,5 мл/мин; А=1000 мл воды +0,52 мл трифторуксусной кислоты +1,20 мл триэтиламина; Б=ацетонитрил; изократический 47:53 А:Б в течение 5 мин, затем градиент до 100% Б в течение 5 мин. TR [соединение, указанное в заголовке]=2,1 мин; TR (4-хлорбензальдегид)=2,3 мин]; 99,92% э.и., определенный посредством хиральной СФХ. Условия хиральной ВЭЖХ: колонка Chiralpak AD-H 250 мм ×4,6 мм, элюирование 70% CO2/30% MeOH при скорости 3,0 мл/мин, детектирование при 255 нм. TR [соединение, указанное в заголовке]=3,9 мин; TR (энантиомер соединения, указанного в заголовке)=2,8 мин; 1Н ЯМР (400 МГц, CDCl3) δ 3.69 (bs, 2 Н), 3.80 (m, 2 Н), 4.15 (s, 1 Н), 7.41 (d, J=8 Гц, 2 Н), 7.69 (d, J=8 Гц, 2 Н), 8.33 (s, 1 Н); 13С ЯМР (CDCl3) δ 47.05, 63.09, 70.82, 128.93, 129.39, 134.08, 137.07, 162.30.
Пример 2
Получение бензилового эфира (3-фтор-4-морфолин-4-ил-фенил)-карбаминовой кислоты
Соединение, указанное в заголовке, можно получить согласно способу, описанному в J. Med. Chem., 1996, 39, (3), 680-685 и показанному на СХЕМЕ II.
СХЕМА II

Предложены дополнительные способы превращения Промежуточного соединения А в 3-фтор-4-тиоморфолин-4-иланилин (В).
Способ А
4-(2-Фтор-4-нитрофенил)тиоморфолин (А, 250 г, 1,03 моль) загружали в смесь диоксана (1400 мл), EtOH (1000 мл) и воды (600 мл) в 5000-мл трехгорлой круглодонной колбе, оснащенной механической мешалкой. В данную перемешиваемую смесь загружали хлорид аммония (166 г, 3,1 моль) с последующей загрузкой железного порошка (247 г, 4,25 моль), причем каждый загружали одиночными порциями. Данную реакционную смесь нагревали до температуры флегмообразования с энергичным перемешиванием. Данную реакционную смесь нагревали при температуре флегмообразования всего в течение 16 часов и затем давали ей охладиться до комнатной температуры. Темную смесь разводили EtOAc (800 мл), фильтровали через слой целита и концентрировали в вакууме до пастообразного остатка. Данный остаток распределяли между рассолом (1000 мл) и дихлорметаном (750 мл). Однократное фильтрование через целит удаляло частицы, которые препятствовали разделению фаз. Водный слой затем экстрагировали дополнительным количеством дихлорметана (750 мл). Объединенные органические слои сушили над безводным карбонатом калия и концентрировали в вакууме с получением 225 г темного твердого вещества. Это неочищенное вещество растворяли в дихлорметане (1000 мл), обрабатывали 200 г силикагеля (230-400 меш), и данную смесь концентрировали досуха. Данную пробку фильтровали через 500 г силикагеля (230-400 меш, упакованного в виде суспензии с 20% EtOAc/гексан), элюируя 20-30% EtOAc/гексаном с отбором 1000 мл фракций. Фракции 3-11 объединяли и концентрировали с получением 3-фтор-4-тиоморфолин-4-иланилина (В, 232 г, 106% выход) в виде беловатого твердого вещества. 1Н ЯМР показал желательное вещество наряду со следами остаточных растворителей для объяснения большего выхода, чем теоретический. 1Н ЯМР (400 МГц, CDCl3): δ 2.8 (m, 4Н), 3.2 (m, 4Н), 3.6 (s, 2Н), 6.4 (m, 2Н), 6.8 (m, 1Н).
Способ Б
В 2000-мл колбу с мешалкой Парра загружали 5%-ный сульфидированный палладий на углероде (Johnson Matthey, тип А103038-5, 18 г) и 4-(2-фтор-4-нитрофенил)тиоморфолин (А, 60 г, 0,25 моль). Данную смесь суспендировали в MeOH (1050 мл), и реакционную смесь гидрогенизировали при 50 фунт-сила/кв.дюйм (344,7 кПа) в течение 7 ч. Катализатор удаляли фильтрованием через целит, и осадок на фильтре хорошо промывали свежим MeOH. Прозрачный серый фильтрат концентрировали в вакууме с получением 3-фтор-4-тиоморфолин-4-иланилина (В, 51,3 г, 98% выход) в виде серого твердого вещества. 1Н ЯМР (400 МГц, CDCl3): δ 2.8 (m, 4Н), 3.2 (m, 4Н), 3.6 (s, 2Н), 6.4 (m, 2Н), 6.8 (m, 1Н).
Пример 3
Получение (5S)-5-{[(4-хлорбензилиден)амино]метил}-3-(3-фтор-4-тиоморфолин-4-илфенил)-1,3-оксазолидин-2-она
Соединение, указанное в заголовке в Примере 2 (194 г, 0,56 моль), и соединение, указанное в заголовке Примера 1 (195 г, 0,84 моль), и трет-бутоксид лития (116 г, 1,4 моль) загружали в 3000-мл трехгорлую круглодонную колбу в атмосфере азота. Реагенты суспендировали с метил-трет-бутиловым эфиром (1200 мл), и данную смесь нагревали до 56°C и перемешивали в течение 2 ч, по мере того, как постепенно образовывалось желтое твердое вещество. Данную реакционную смесь охлаждали до комнатной температуры и разбавляли 1200 мл воды. Затем данную смесь энергично перемешивали в течение 60 мин, по мере того, как твердое вещество менялось от темно-желтого до более бледного желтого твердого вещества. Данную смесь охлаждали до 10°C, фильтровали, и осадок на фильтре промывали ледяным метил-трет-бутиловым эфиром (450 мл). Образующееся светло-желтое твердое вещество сушили на воздухе в течение 30 мин, затем помещали в вакуумную печь и сушили при 40°C в течение ночи с получение соединения, указанного в заголовке (243 г, выход 99%). 1Н ЯМР (400 МГц, CDCl3): δ 2.8 (m, 4Н), 3.2 (m, 4Н), 3.9 (m, 2Н), 4.1 (m, 2Н), 5.0 (m, 1Н), 6.9 (m, 1Н), 7.2 (m, 1Н), 7.4 (m, 3Н), 7.6 m, 2Н), 8.4 (s, 1Н).
Пример 4
Получение N-{[(5S)-3-(3-фтор-4-тиоморфолин-4-илфенил)-2-оксо-1,3-оксазолидин-5-ил]метил}ацетамида
Соединение, указанное в заголовке в Примере 3 (243 г, 0,56 моль), объединяли с EtOAc (1300 мл) и водой (1300 мл) в 5000-мл трехгорлой круглодонной колбе, оснащенной механической мешалкой. Данную смесь обрабатывали каплями 12 н. HCl (140 мл, 1,68 моль), и смесь энергично перемешивали в течение 1 часа при комнатной температуре. Слои разделяли, и водный слой промывали EtOAc (1×500 мл). Образующийся водный раствор, содержащий гидрохлорид (S)-5-(аминометил)-3-(3-фтор-4-тиоморфолинофенил)оксазолидин-2-она, объединяли со смесью дихлорметана (1800 мл) и MeOH (120 мл), и в энергично перемешиваемую смесь загружали одной порцией уксусный ангидрид (132 мл, 1,4 моль) и затем обрабатывали 10 н. NaOH (200 мл, 2,0 моль) по каплям в течение 15 мин. В результате добавления основания получалась крайне густая реакционная смесь, которая постепенно разжижалась по мере роста pH и быстрого хода ацилирования. Данную реакционную смесь энергично перемешивали в течение 1 часа после того, как данная смесь разделялась на две фазы. В это время к данной смеси добавляли по каплям 10 М NaOH (160 мл, 1,6 моль), пока не достигали стабильного pH 7. Слои разделяли, водный слой экстрагировали дихлорметаном (250 мл), и объединенные органические слои сушили над безводным карбонатом калия. Летучие вещества удаляли в вакууме с получением беловатого твердого вещества, которое титровали с метил-трет-бутиловым эфиром (250 мл), собирали и сушили в вакууме с получением соединения, указанного в заголовке (5) (186,1 г, 94% выход), в виде мелкодисперсного белого твердого вещества с чистотой больше 98%, определенной ВЭЖХ (время удерживания =3,93 минуты, условия ВЭЖХ изложены ниже).
Данное неочищенное твердое вещество растворяли в теплом 6% метаноле в дихлорметане (1250 мл) в 5000-мл трехгорлой круглодонной колбе, оснащенной магнитной мешалкой. Данный раствор нагревали до температуры дефлегмации, разбавляли порционным (500 мл) добавлением 2500 мл изопропанола (IPA) и, для того чтобы поддерживать дефлегмацию, температуру увеличивали до 50-70°С. По завершении этого добавления IPA обратный холодильник заменяли насадкой для молекулярной перегонки, и продолжали перегонку в охлажденную колбу. Во время перегонки после сбора 500 мл дистиллята добавляли 500 мл порцию свежего IPA для поддержания постоянного присутствия от 2000 до 2500 мл IPA. После этого добавления (внутренняя температура в колбе падала до 60°С) данная смесь становилась слегка мутной и оставалось такой в течение оставшегося времени перегонки, становясь особенно мутной, когда температура дистиллята превышала 70°С; вещество в виде частиц появлялось, когда температура дистиллята превышала 75°С. Регулятор температуры устанавливали на 85°С и оставляли на этом уровне до завершения перегонки. Когда дистиллят очевидно представлял собой один изопропанол (82-83°С), объем уменьшали до 2500 мл горячего IPA, греющий кожух удаляли, перемешивание прерывали и удаляли лопастную мешалку из колбы. Давали данной смеси продолжать кристаллизоваться по мере того, как колба охлаждалась. Затем белое кристаллическое твердое вещество собирали фильтрованием, промывали метил-трет-бутиловым эфиром (250 мл) и сушили в вакууме при 40°С с получением 180 г (выход 91%) соединения, указанного в заголовке, с чистотой больше 99%, определенной ВЭЖХ (время удерживания=3,93 минуты, условия ВЭЖХ приведены ниже). 1Н ЯМР (400 МГц, DMSO-d6): δ 1.8 (s, 3Н), 2.7 (m, 4Н), 3.2 (m, 4Н), 3.4 (m, 2Н), 3.7 (m, 1Н), 4.7 (m, 1Н), 7.1 (m, 1Н), 7.15 (m, 1Н), 7.2 (m, 1Н), 8.2 (m, 1Н). Масс-спектр C16H20FN3O3S: m/z 354,1 (М+1).
Условия ВЭЖХ для анализов, упомянутых в тексте: HP Series 1100; колонка: Symmetry С8 5 мкм, 4,6×50 мм; скорость потока 1,2 мл/мин; растворитель А: вода с 0,1% муравьиной кислотой, растворитель Б: ацетонитрил с 0,1% муравьиной кислотой; инъекционный объем =10 мкл 1 мг/мл (ацетонитрил); градиент: растворитель Б 0-100% за 7 минут, затем 100% Б в течение 1 минуты; длина волны=254 нм.
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
В следующих примерах используются следующие сокращения, которые имеют указанные определения, если не отмечено иное: КОЕ представляет собой колониеобразующую единицу; INH представляет собой изониазид; RIF представляет собой рифампин; PZA представляет собой пиразинамид; MXF представляет собой моксифлоксацин, и LZD представляет собой линезолид.
Бактериальный штамм Mycobacterium tuberculosis H37Rv пассировали в мышах, замораживали в 1 мл аликвотах и хранили при -80°С до применения. Для каждой инъекции аликвоту размораживали и субкультивировали в бульоне Middlebrook 7Н9, дополненном смесью 10% олеиновая кислота-альбумин-декстроза-каталаза (OADC) (Difco, Detroit, MI) и 0,05% Tween 80 (Sigma, St. Louis MO). Самок мышей BALB/c (Charles River, Wilmington, MA), в возрасте от четырех до шести недель инфицировали посредством аэрозоля с использованием Inhalation Exposure System (Glascol Inc, Terre Haute, IN) и культуры логарифмической фазы в бульоне с оптической плотностью примерно 1,0 при 600 нм. После аэрозольной инфекции мышей рандомизировали в группы обработки (5 мышей на группу на момент времени). Необработанных мышей умерщвляли обычным способом (1) в сутки после инфекции для определения числа КОЕ, имплантированных в легкие, и (2) в сутки начала обработки для определения числа КОЕ до обработки.
Соединение формулы (I) по данному изобретению и LZD (приобретенный у Pfizer Inc. Ann Arbor, MI and Groton, CT.), используемые в тестах, описанных ниже, суспендировали в растворе, состоящем из 5% полиэтиленгликоля-200 (PEG-200) и 95% метилцеллюлозы (0,5%) в дистиллированной воде. MXF приобретали у Bayer (Rolling Meadows, IL), PZA приобретали у Fisher, и INH и RIF приобретали у Sigma. Маточные растворы готовили еженедельно, используя дистиллированную воду. Все растворы антибиотиков хранили при 4°C.
За исключением тех случаев, где указано иное, антибиотики вводили один раз в сутки, пять суток в неделю по 0,2 мл посредством кормления через желудочный зонд. Обе суспензии оксазолидинона кратко обрабатывали ультразвуком до применения и встряхивали между дозированиями. RIF давали за 1 час до введения других лекарственных средств для того, чтобы избежать вредного фармакокинетического взаимодействия.
Пример 5
Активность соединения формулы (I) in vitro
МИК (минимальная ингибирующая концентрация) соединения формулы (I) и LZD определяли способом разведений в агаре на агаре Middlebrook 7Н11, дополненном 10% OADC (Becton-Dickinson, Sparks, MD). Планшеты, содержащие последовательные двукратные концентрации соединения формулы (I) и LZD, варьирующие от 0,125 до 4 мкг/мл, инокулировали приблизительно 5×105 КОЕ Mycobacterium tuberculosis H37Rv. КОЕ подсчитывали после 28 суток инкубирования при 37°C с 5% CO2 в окружающей среде. МИК определяли как наименьшую концентрацию, ингибирующую рост бактерий по меньшей мере на 99%.
МИК соединения формулы (I) и LZD против Mycobacterium tuberculosis H37Rv составляла 0,25 мкг/мл.
Варьирующая в зависимости от дозы активность соединения формулы (I) и LZD в модели установившейся инфекции
Мышей инфицировали аэрозольным путем 4,44±0,04 log10 КОЕ. Начиная с 13 суток после аэрозольной инфекции, когда среднее число КОЕ в легком составляло 7,49±0,11 log10 и средняя масса селезенки составляла 105±11 мг, контрольные мыши получали одну из следующих обработок: дозы 25 мг/кг INH, от 25 мг/кг до 260 мг/кг LZD (однократная доза и 130 мг/кг дважды в сутки) и от 25 мг/кг до 100 мг/кг соединения формулы (I). В конце периода обработки, через 28 суток, только 2 из 5 необработанных мышей выживали. Средняя масса селезенки у этих живых мышей возрастала до 237±16 мг.
В отличие от этого монотерапия 25 мг/кг INH предотвращала увеличение селезенки (средняя масса селезенки =100±1 мг) и смерть. Дозозависимое уменьшение массы селезенки по отношению к необработанным контролям наблюдали с увеличением доз LZD от 25 мг/кг (200±47 мг) до 130 мг/кг (102±5 мг). Однако селезенки от мышей, обработанных 260 мг/кг LZD (как в виде одиночной дозы, так и по 130 мг/кг дважды в сутки), были больше, чем ожидалось (190±52 мг), и аналогичными по размеру селезенкам от мышей, обработанных 25 мг/кг/сутки LZD. Поскольку число и размер поражений легких уменьшались с увеличением суточных доз LZD, включая 260 мг/кг, не предполагалось, что увеличение селезенки, наблюдавшееся у мышей, получающих 260 мг/кг/сутки, было обусловлено пониженной противотуберкулезной активностью. Обработка соединением формулы (I) предотвращала увеличение селезенки при всех введенных дозах, включая дозу 25 мг/кг (средняя масса селезенки =113±17 мг).
Выживающие необработанные мыши претерпевали увеличение числа КОЕ почти до 8 log10. Обработка INH снижала среднее число КОЕ в легком до 5,62 log10, для log умерщвления 1,87 по сравнению с исходным значением. Все схемы, включающие соединение формулы (I), приводили к значительному снижению среднего числа КОЕ относительно исходного (p<0,01), начиная со снижения 0,78 log10 в присутствии 25 мг/кг. В дозе 50 мг/кг соединение формулы (I) демонстрировало бактерицидную активность (определенную по критерию снижения на 2-log) путем снижения среднего числа КОЕ до 5,28 log10, для log умерщвления 2,21, большего, чем наблюдаемый в присутствии INH. Несмотря на то что LZD также демонстрировал дозозависимую активность, его активность была более ограниченной, чем активность соединения формулы (I). Только при дозах LZD≥100 мг/кг было продемонстрировано значимое снижение исходного числа КОЕ (p<0,01). Даже при наивысшей протестированной дозе, 260 мг/кг, LZD не удовлетворял критерию снижения на 2-log, определяющему бактерицидную активность, продуцируя log умерщвления лишь 1,46. Соединение формулы (I) было значительно более активным, чем LZD, при каждой протестированной дозе.
Обработка соединением формулы (I) в дозе 25 мг/кг приводила к меньшему числу КОЕ, чем показатель КОЕ, наблюдающийся после обработки LZD в дозе 25 или 50 мг/кг (p<0,01), и не являющемуся значимо отличным от числа КОЕ, которое наблюдали после обработки LZD в дозе 100 или 130 мг/кг. В дозе 50 и 100 мг/кг соединение формулы (I) было более активным, чем любая доза LZD (p<0,001). Деление общей суточной дозы соединения формулы (I) и LZD на две суточные дозы не имело ясного эффекта (т.е. положительно или отрицательного) на активность (Таблица 1).
Следовательно, соединение формулы (I) демонстрировало, что оно является значительно более активным, чем эквивалентная человеческой доза LZD (т.е. 100 мг/кг), единственного клинически доступного оксазолидинона, который применяется по неодобренным показаниям для лечения МЛУ- и РЛУ-ТБ.
Фармакокинетика соединения формулы (I)
Для определения единичной дозы и профилей фармакокинетики в стационарном состоянии соединения формулы (I) и LZD в мышиной модели в приведенное выше исследование по интервалу дозы было вставлено дополнительное исследование с использованием мышей, обработанных 100 мг/кг соединения формулы (I) один раз в сутки или 130 мг/кг LZD один раз или дважды в сутки. Трех мышей на группу умерщвляли в 0,5, 1, 2, 4, 8 и 24 часа после обработки первой дозой (D1) и в вышеупомянутые моменты времени с добавлением момента времени 9 часов (т.е. через 1 час после второй суточной дозы) в сутки 24 (D24) обработки. Мышей анестезировали хлороформом и обескровливали проколом сердца. Цельную кровь собирали на льду и затем центрифугировали с получением сыворотки. К образцам добавляли ацетонитрил перед хранением в герметизированных пробирках с завинчивающимися крышками при -20°С. Образцы D24 отбирали и перерабатывали тем же самым способом и хранили при -20°С перед отсылкой совместно с образцами D1 в Pfizer (Groton, CT) для определения концентраций лекарственного средства. Супернатант впрыскивали (инъекционный объем 10 мкл) в ЖХ (Shimadzu SCL-10A, Kyoto, Япония) - МС/МС (Sciex API 3000, Applied Biosystems Group, Foster City, CA), используя колонку Hypersil C18 (5 мкм, 50×2,1 мм, Thermo Electron Corp, Waltham, MA) и ступенчатый градиент, состоящий из подвижной фазы А: вода с 0,05% 5 мМ формиатом аммония и Б: ацетонитрил/вода/5 мМ формиат аммония (80:20:0,05%). Электрораспылительная ионизация осуществлялась в режиме положительных ионов. Биоаналитические данные собирали с использованием Analyst (Version 1.4.1, Applied Biosystems Group, Foster City, CA). Фармакокинетические расчеты были основаны на средних сывороточных концентрациях и проводились с использованием некомпартментного подхода (линейное трапециевидное правило для расчета AUC (площадь под кривой) с помощью Watson 7.2 Bioanalytical LIMS [Thermo Electron Corp, Waltham, MA)). При расчетах AUC для D24 уровень аналитов в плазме в нулевой момент времени был установлен для 24-ч концентрации. Концентрации 0 использовали для кинетических расчетов для всех результатов, меньших нижнего предела количественной оценки (5 нг/мл).
Значения выбранного фармакокинетического параметра для соединения формулы (I) и LZD представлены в Таблице 2. Суточная доза LZD 130 мг/кг давала AUC в равновесном состоянии 379 мкг-ч/мл, приблизительно на 50% большую, чем предполагаемая AUC в равновесном состоянии, 215-294 мкг-ч/мл, наблюдающаяся у людей, которым перорально вводили 600-625 мг дважды в сутки. (Смотри Gee, T. et al.; Antimicrob Agents Chemother 45: 1843-1846, 2001; Stalker DJ et al., J Antimicrob Chemother 51: 1239-46, 2003.) На основе линейной кинетики в этом интервале дозы доза LZD 100 мг/кг у мышей представляет собой верхнюю границу интервала AUC в равновесном состоянии у человека, хотя и с Cmax, которая приблизительно в 3 раза выше, чем Cmax, полученная у людей. Как и ожидалось, имел место значительный метаболизм первого прохождения соединения формулы (I) до главного сульфоксидного метаболита и минорного сульфонового метаболита (Barbachyn et al. J Med Chem. 39: 680-685, 1996). Поскольку каждый из этих метаболитов имеет МИК90 0,5 мкг/мл против М. tuberculosis, близкую к МИК90 0,25 мкг/мл для соединения формулы (I), для фармакокинетического анализа концентрацию каждого метаболита добавляли к концентрации родительского соединения. AUC в равновесном состоянии для соединения формулы (I) и его метаболитов, наблюдаемая с суточной дозой 100 мг/кг, была приблизительно в 3 раза ниже, чем AUC в равновесном состоянии, наблюдаемая с LZD при 130 мг/кг. Тот факт, что доза соединения формулы (I) 25 мг/кг была такой же активной, как и данная доза LZD, говорит о том, что соединение формулы (I) является в 12 раз более эффективным in vivo, чем LZD, несмотря на аналогичные МИК для обоих соединений.
Пример 6
Активность комбинации in vivo
Мышей инфицировали аэрозолем 3,89±0,19 log10 КОЕ. Начиная с 14 суток после аэрозольной инфекции, когда среднее число КОЕ в легком составляло 7,37±0,05 log10, контрольные мыши получали одну из следующих обработок: один INH (25 мг/кг), один RIF (10 мг/кг), RIF+PZA (150 мг/кг), RIF+INH+PZA, MXF (100 мг/кг)+PZA или RIF+MXF+PZA. Тестируемые мыши получали одно соединение формулы (I) (100 мг/кг) или добавленное к каждой из контрольных схем. Дополнительные мыши оставались необработанными для того, чтобы служить в качестве негативных контролей. Мышей умерщвляли после 4 и 8 недель обработки для оценки массы селезенки и числа КОЕ в легком. Необработанные контрольные мыши умирали во время второго месяца данного эксперимента.
Синергический эффект добавления соединения формулы (I) к целому ряду схем с 1, 2 и 3 лекарственными средствами на число КОЕ в легком является очевидным из данных, приведенных в Таблице 3.
Монотерапия соединением формулы (I) по данному изобретению приводила к снижению числа КОЕ на 2,65 log10 от исходного уровня до 4,72 log10 за первые 28 суток, тогда как монотерапия INH и RIF снижала число КОЕ в легком на 1,58 и 1,63 log10 до 5,79 и 5,74 log10 соответственно. Примечательно то, что соединение формулы (I) по данному изобретению продолжало оказывать бактерицидную активность на протяжении второго месяца обработки, снижая число КОЕ до 2,70 log10 КОЕ больше, чем активность одного INH и RIF и аналогично числу КОЕ у мышей, обработанных по стандартной схеме первоочередной комбинацией RIF-INH-PZA, которая снижала число КОЕ до 2,47 log10. Комбинация INH и соединения формулы (I) по данному изобретению была более активной, чем одно соединение формулы (I), тогда как объединение RIF с соединением формулы (I) имело мультипликативный эффект, приводя к среднему числу КОЕ в легком почти в 30 раз меньшему, чем КОЕ, которые наблюдали с одним соединением формулы (I). Комбинация RIF-PZA имела активность, которая снижала среднее число КОЕ в легком до 1,05.
В этом примере добавление INH к RIF-PZA имело значительный антагонистический эффект, приводя к среднему числу КОЕ в легком 1,05±0,44 и 2,47±0,18 после 2 месяцев обработки RIF-PZA и RIF-INH-PZA соответственно. Этот эффект мог бы ложно свидетельствовать о том, что замена нового лекарственного средства на INH в схеме RIF-INH-PZA имеет значительный полезный эффект, даже если само новое лекарственное средство является полностью неактивным, просто потому, что данный антагонистический эффект INH был удален. Однако в этом примере соединение формулы (I) по данному изобретению имело аналогично сильные эффекты, было ли оно добавлено к RIF-INH-PZA или использовано вместо INH (т.е. в виде RIF-PZA-соединение формулы (I)). После 2 месяцев обработки среднее число КОЕ в легком составляло 0,47±0,20 и 0,50±0,33 у мышей, обработанных RIF-INH-PZA-соединение формулы (I) и RIF-PZA-соединение формулы (I) соответственно, приводя к среднему числу КОЕ в легком приблизительно в 3,5 раза меньшему, чем среднее число КОЕ в легком у мышей, получающих только RIF-PZA (p<0,05), что дает дополнительное доказательство того, что польза замены INH соединением формулы (I) по данному изобретению происходит не в результате удаления антагонистического влияния INH. Соединение формулы (I) по данному изобретению также улучшало активность схемы RIF-MXF-PZA. Обработка RIF-MXF-PZA в течение 2 месяцев приводила к среднему числу КОЕ в легком 0,61±0,38, причем 1 из 5 мышей не давала культуру, тогда как обработка RIF-MXF-PZA и соединением формулы (I) приводила к среднему числу КОЕ 0,12±0,27, причем 4 из 5 мышей не давали культуру (полное устранение (р=0,05 для различия средних чисел КОЕ)). Данное различие имело пограничную статистическую значимость, вероятно из-за малых чисел КОЕ, но оно подразумевает то, что соединение формулы (I) по данному изобретению может дополнительно улучшить стерилизующую активность данной схемы для предупреждения рецидива. Наконец, обработка MXF-PZA-соединение формулы (I) в течение 2 месяцев приводила к среднему числу КОЕ в легком 0,93±0,34, приводя к среднему числу КОЕ в легком почти в 175 раз меньшему, чем КОЕ, наблюдавшееся без соединения формулы (I), демонстрируя то, что соединение формулы (I) по данному изобретению способно заместить RIF в схеме сокращения лечения RIF-MXF-PZA без уменьшения активности данной схемы.
Пример 7
Активность in vivo комбинации после длительного введения и последующего врачебного наблюдения
Мышей инфицировали аэрозолем с 4,45±0,05 log10 КОЕ. Начиная с 14 суток после аэрозольной инфекции, когда среднее число КОЕ в легком составляло 7,92±0,15 log10, контрольные мыши получали RIF-INH-PZA в течение 8 недель с последующим RIF-INH в течение 8 недель. Одна когорта тестируемых мышей получала ту же самую схему, к которой было добавлено соединение формулы (I) (160 мг/кг) на протяжении всех 16 недель, добавлено на протяжении лишь первых 8 недель или добавлено на протяжении всех 16 недель с удалением INH после первых 8 недель. Другая когорта тестируемых мышей получала контрольную схему, в которую было добавлено LZD на протяжении всех 16 недель или добавлено на протяжении лишь первых 8 недель. Дополнительные мыши оставались необработанными для того, чтобы служить в качестве негативных контролей. Мышей умерщвляли после 8, 12 и 16 недель обработки для оценки числа КОЕ в легком. Дополнительных мышей оставляли без обработки в течение 12 недель после завершения 12 или 16 недель обработки. Необработанные контрольные мыши умирали на протяжении второго месяца данного эксперимента.
Синергический эффект добавления соединения формулы (I) (сокращенного в виде U) на число КОЕ в легком относительно контрольной схемы на протяжении первых 8 недель очевиден из данных, приведенных в Таблице 4. Бактерицидный эффект контрольной схемы возрастал на 2 порядка. Этот эффект является количественно и качественно отличным от антагонистического эффекта добавления LZD.
Обработка по схеме, включающей соединение формулы (I) (сокращено как U в Таблице 4), была связана с меньшей вероятностью рецидива (золотой стандарт полного устранения) после завершения 12 и 16 недель обработки по сравнению с мышами, получающими другие схемы. После 12 недель обработки по контрольной схеме все мыши оставались позитивными в отношении культуры, и ожидается, что они продемонстрировали бы 100%-ную частоту рецидивов. Это дополнительно поддерживается 90%-ной частотой рецидивов после 16 недель обработки.
Напротив, группы, получающие схему, включающую соединение формулы (I) в течение всех 16 недель, имели среднюю частоту рецидивов 40% после 12 недель и 5% после 16 недель. Когда соединение формулы (I) добавляли к контрольной схеме в течение лишь первых 8 недель, 85% и 35% мышей демонстрировали рецидивы после общих продолжительностей обработок 12 и 16 недель соответственно. С другой стороны, добавление LZD было антагонистическим и предотвращало полное устранение у любой мыши. В целом эти данные демонстрируют то, что соединение формулы (I) имеет синергический эффект при объединении со стандартной схемой первой линии и способно укорачивать продолжительность лечения на 1-2 месяца без потери эффективности.
Следовательно, соединение формулы (I) имеет синергический эффект при комбинировании по меньшей мере с двумя агентами, полезными для лечения туберкулеза. Важнее всего то, что комбинирование соединения формулы (I) по меньшей мере с двумя агентами для лечения туберкулеза значительно увеличивает бактерицидную активность, что свидетельствует о том, что оно может сокращать продолжительность химиотерапии чувствительного к лекарственным средствам ТБ, а также МЛУ-ТБ.
Пример 8
Пример 8А
Исследование по варьированию дозы в течение четырех недель в комбинации со стандартными агентами первой линии, рифампином, изониазидом и пиразинамидом
В Примере 8 описаны результаты исследования, в котором мышей инфицировали Mycobacterium tuberculosis аналогично способу, описанному в Примере 6, приведенном выше. За исключением контрольных вариантов всех мышей обрабатывали следующими тремя лекарственными средствами:
1) рифампин, 10 мг/кг (R),
2) изониазид, 25 мг/кг (Н), и
3) пиразинамид, 150 мг/кг (Z).
Мышей делили на восемь групп (8), и шесть из данных групп получали варьирующие дозы соединения Формулы I ("U"). Дозы варьировали от 12,5 мг/кг до 160 мг/кг, и они перечислены в Таблице 5, приведенной ниже.
Данное исследование проводили следующим образом. Шестинедельных самок мышей BALB/c инфицировали аэрозольным путем Mycobacterium tuberculosis H37Rv. В эти сутки (D-17) 5 мышей умерщвляли для определения числа КОЕ, имплантированных в легкие. Через семнадцать суток (D0) умерщвляли 5 дополнительных мышей для определения исходного числа КОЕ в легких. Остальных мышей рандомизировали в группы обработки, как показано в Таблице 5, и начинали проводить на них определенную схему обработки лекарственным средством. Обработку вводили один раз в сутки, 5 суток в неделю, за исключением группы 8, которая на всем протяжении получала обработку трижды в неделю (3/7). В момент завершения 4-недельного тестового периода всех животных умерщвляли для подсчета числа КОЕ в легком, за исключением 80 животных из Группы 4, описанной ниже. Этих животных использовали в 8-недельном протоколе, который описан ниже в Примере 8Б.
Как показано выше, соединение формулы I продемонстрировало дозозависимый эффект на число КОЕ. Добавление этого соединения к стандартной схеме RHZ продуцировало дополнительное уменьшение числа ТБ колоний, содержащихся в легком.
Пример 8Б
Исследование по варьированию дозировки в течение восьми недель
Восемь мышей из Группы 4 (в Примере 8А), которые получали 25 мг/кг соединения формулы I (U), наряду с рифампином (R), изониазидом (H) и пиразинамидом (Z), оценивали во второй фазе данного исследования. Их приписывали к одной из групп, описанных ниже в Таблице 6, и данное исследование продолжали в течение дополнительных 4 недель (т.е. результаты 8 недель).
Как описано выше, 5 животных служили в качестве контроля, и им не давали дополнительных лекарственных средств. Пяти давали комбинацию рифампина (R) и изониазида (Н), пяти давали один изониазид (H) или рифампин (R). Из двенадцати (12) остальных групп всем давали соединение формулы (I) в дозах, варьирующих от 12,5 мг/кг до 160 мг/кг. Шести из этих групп также дозировали рифампин (R). Рифампин и изониазид дозировали тем же самым способом и в том же самом количестве, что и в Примере 8А.
В момент завершения восьми(8)-недельного эксперимента всех животных умерщвляли и получали число КОЕ в легком. Были получены следующие результаты:
В начале этой второй фазы данные восемь животных имели среднее КОЕ 4,19±0,26 (на основе других мышей из Группы 4, которые были умерщвлены). Таблица 7 показывает результаты, полученные с контрольной группой, только рифампином (R), только изониазидом (H) и комбинацией как изониазида (H), так и рифампина (R).
Таблица 8 показывает результаты, полученные с мышами, получающими одно соединение формулы (I) (U) или в комбинации с рифампином (R).
Соединение формулы I (U) демонстрировало дозозависимую бактерицидную активность против сохраняющихся туберкулезных бацилл, которые оставались жизнеспособными после начальных 4 недель обработки RHZU25. Следующие эффекты наблюдали под влиянием монотерапии U: ингибирующий рост эффект при 12,5 мг/кг, бактериостатический эффект при 25 мг/кг, >0,5 log снижение при 50 мг/кг, >1 log снижение при 100 мг/кг и почти 2 log снижение при 160 мг/кг. Комбинация рифампина (R) и соединения формулы (I) U была синергической, и оказалось, что она имеет сильную бактерицидную активность против сохраняющихся бактерий. Когда соединение формулы (I) U вводили в дозах ≥25 мг/кг, его комбинация с рифампином R была более эффективной, чем стандартная схема (обработки) рифампином (R) и изониазидом (H). Результаты с одним U подтверждают заключение о том, что схемы, включающие U, могут иметь способность значительно сокращать продолжительность лечения как туберкулеза с множественной лекарственной устойчивостью, так и туберкулеза с расширенной лекарственной устойчивостью.
Пример 8В
Дополнительное исследование по варьированию дозы
Протокол, описанный выше в Примере 8А и 8Б, проводили при альтернативном отборе проб. Единственными существенными изменениями были число животных на группу и исходный период инкубации. В таблице 9 приведены полученные результаты.
Тестирование продолжали в течение дополнительных 4 недель с 60 мышами из тестовой группы 2, показанной тут же выше. Данные мыши получали либо одно U в варьирующих дозах, либо комбинацию U и R. Были получены следующие результаты.
Как описано выше, соединение формулы I демонстрировало дозозависимый эффект на число КОЕ. Добавление этого соединения к стандартной схеме рифампина (R) и изониазида (H) и одного рифампина (R) давало дополнительное уменьшение числа ТБ колоний, присутствующих в легком как в 4, так и в 8 недель.
Пример 9
В Примере 9 оценивали влияние соединения формулы I на существующие противотуберкулезные лекарственные средства первой и второй линии. Оценивали две дозы данного соединения, 25 мг/кг и 100 мг/кг. Оценивали следующие стандартные противотуберкулезные агенты в оговоренных дозах:
1) изониазид 10 мг/кг (Н)
2) рифампин 10 мг/кг (R)
3) пиразинамид 150 мг/кг (Z)
4) этамбутол 100 мг/кг (Eb)
5) моксифлоксацин 100 мг (M)
6) этионамид 50 мг/кг (Et)
7) амикацин 150 мг/кг подкожно (A)
8) циклосерин 250 мг/кг дважды в сутки (Cs)
9) пара-аминосалициловая кислота 750 мг/кг (Ps)
10) капреомицин 125 мг/кг подкожно (Cap)
11) клофазимин 20 мг/кг (C)
Данное исследование проводили следующим образом.
Способы
Мыши: самки BALB/c 6-недельного возраста
Инфекция: аэрозольная инфекция ~104 КОЕ M. tuberculosis H37Rv с последующей рандомизацией в группы обработки.
Обработка: начата на сутки 14 после инфекции (D0), когда число КОЕ в легких мышей составляло ~108 КОЕ.
Все лекарственные средства давали один раз в сутки (за исключением циклосерина - дважды в сутки) пероральным путем (за исключением амикацина и капреомицина - подкожная инъекция) 5 суток в неделю.
Мышей умерщвляют для определения числа КОЕ в легких. Согласно экспериментальной схеме 4 мышей умерщвляли через сутки после инфекции (D-13) и 2 неделями позднее (D0) для оценки числа имплантированных КОЕ и исходного числа КОЕ при инициации обработки соответственно. Активность каждой схемы оценивали по числу КОЕ в легком после 4 недель обработки.
Результаты
Мышей инфицировали ~4,5 log10 КОЕ. Через четырнадцать суток число КОЕ в легком в момент начала обработки на сутки ноль составляло приблизительно 8,3 log10. Необработанные мыши умирали в пределах первых 3 недель инфекции (Таблица 11). Выживание мышей, обработанных пиразинамидом (Z), циклосерином (Cs) и капреомицином (Cap), не было значимо отличным от необработанных контролей. Обработка пара-аминосалициловой кислотой (Ps) задерживала, но не предотвращала смертность. Обработка этамбутолом (Eb) и клофазимином (C) предотвращала смертность частично, но не полностью. Обработка изониазидом (H), рифампином (R), моксифлоксацином (M), амикацином (A) и этионамидом (Et) предотвращала смерть, как и обработка соединением формулы (I) в любой дозе одним или в комбинации с другими лекарственными средствами.
Помимо измерения выживания число КОЕ в легком также определяли, как описано выше и доложено ниже в Таблице 12.
Обзор данных показывает то, что добавление соединения Формулы I (U) значительно снижало бактериальную нагрузку дозозависимым образом при добавлении к стандартным противотуберкулезным лекарственным средствам. Эти результаты говорят о том, что соединение формулы I можно использовать для улучшения эффективности схем лечения устойчивого к лекарственным средствам туберкулеза (либо (туберкулеза) с множественной лекарственной устойчивостью, либо с расширенной лекарственной устойчивостью).
Все патенты, заявки, публикации, способы тестирования, литература и другие материалы, процитированные здесь, являются тем самым включенными сюда посредством ссылки во всей их полноте.
Группа изобретений относится к области медицины и предназначена для терапии туберкулеза. Применяют соединение формулы (I), (S)-N-[[3-[3-фтор-4-(4-тиоморфолинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамида, или его фармацевтически приемлемой соли в комбинации с по меньшей мере двумя агентами для изготовления лекарственного средства для лечения туберкулеза после того как субъект прошел начальную фазу лечения туберкулеза. Использование заявленной группы изобретений обеспечивает высокий терапевтический эффект при лечении туберкулеза. 3 н. и 7 з.п. ф-лы, 12 табл., 8 пр.
1. Применение соединения формулы:
или его фармацевтически приемлемой соли в комбинации по меньшей мере с двумя противотуберкулезными агентами в изготовлении лекарственного средства для лечения туберкулеза.
2. Применение по п.1, где указанные по меньшей мере два агента выбраны из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, этамбутола, стрептомицина, канамицина, амикацина, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина, капреомицина, этионамида, циклосерина, пара-аминосалициловой кислоты, тиацетазона, кларитромицина, амоксициллина-клавулановой кислоты, имипенема, меропенема, виомицина, теризидона, ТМС207, РА-824, ОРС-7683, LL-3858 и SQ-109.
3. Применение по п.1, где один из указанных по меньшей мере двух агентов выбран из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина и этамбутола.
4. Применение по п.1, где один из указанных по меньшей мере двух агентов выбран из группы, состоящей из пиразинамида, рифампина, рифапентина и изониазида.
5. Применение соединения формулы (I) или его фармацевтически приемлемой соли:
в комбинации по меньшей мере с двумя противотуберкулезными агентами в изготовлении лекарственного средства для лечения туберкулеза после того, как субъект прошел начальную фазу лечения туберкулеза.
6. Применение по п.5, где указанный по меньшей мере один агент выбран из группы, состоящей из рифампина, рифапентина, рифабутина, пиразинамида, этамбутола, стрептомицина, канамицина, амикацина, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина, капреомицина, этионамида, циклосерина, пара-аминосалициловой кислоты, тиацетазона, кларитромицина, амоксициллина-клавулановой кислоты, имипенема, меропенема, виомицина, теризидона, ТМС207, РА-824, ОРС-7683, LL-3858 и SQ-109.
7. Применение по п.5 или 6, где указанный активный туберкулез выбран из группы, состоящей из туберкулеза, чувствительного к лекарственным средствам, туберкулеза, устойчивого к одному лекарственному средству, туберкулеза с множественной лекарственной устойчивостью (multi-drug resistant, МЛУ) и туберкулеза с расширенной лекарственной устойчивостью (extensively drug-resistant, РЛУ).
8. Фармацевтическая композиция для лечения туберкулеза, включающая:
1) терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли:
2) терапевтически эффективные количества двух и более противотуберкулезных агентов, и
3) один или более чем один фармацевтически приемлемый носитель или наполнитель.
9. Фармацевтическая композиция по п.8, где указанные противотуберкулезные агенты выбраны из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, этамбутола, стрептомицина, канамицина, амикацина, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина, ципрофлоксацина, капреомицина, этионамида, циклосерина, пара-аминосалициловой кислоты, тиацетазона, кларитромицина, амоксициллина-клавулановой кислоты, имипенема, меропенема, виомицина, теризидона, ТМС207, РА-824, ОРС-7683, LL-3858 и SQ-109.
10. Фармацевтическая композиция по п.8 или 9, где указанные противотуберкулезные агенты выбраны из группы, состоящей из изониазида, рифампина, рифапентина, рифабутина, пиразинамида, моксифлоксацина, гатифлоксацина, левофлоксацина, офлоксацина и этамбутола.
| US 5880118 A, 09.03.1999 | |||
| CYNAMON M.H | |||
| Activities of several novel oxazolidinones against Mycobacterium tuberculosis in a Murine // Antimicrobal Agents and Chemotherapy, 1999, May, 43(5), 1189-1191 | |||
| LLUIS BALLELL | |||
| ET AL | |||
| New small-molecule synthetic antimycobacterials // Antimicrobal Agents and Chemotherapy, 2005, June, 49(6), 2153-2163. |

