ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к трициклическим производным фенотиазинов, феноксазинов, феназинов, акридинов, оксазепинов, диазепинов, ксантенов, тиоксантенов, а также к их применению.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Фенотиазиновые антипсихотические и антигистаминные препараты прохлорперазин, хлорпромазин и промазин продемонстрировали синергетическое взаимодействие с широким спектром антимикробных агентов. Минимальные ингибирующие концентрации антибиотиков в присутствии фенотиазинов в некоторых случаях снижались до 8000 раз.
Кроме того, известно, что фенотиазины повышают отношение NADH/NAD в Mycobacterium tuberculosis за счет ингибирования NADH:менахинон-оксидоредуктазы типа II. Таким путем фенотиазины способны усилить восстановленное состояние клетки, действуя синергетически с лекарственными средствами, которым для активации необходим микотиол, например, изониазидом и этионамидом. Такая комбинированная терапия помогает ограничить появление устойчивости к антимикробным агентам. Было доказано, что фенотиазины, например тиоридазин, проявляют активность против мульти- и экстремально резистентных к лекарственным средствам форм Mycobacterium tuberculosis.
Фенотиазины продемонстрировали широкий спектр биологической активности, хотя изначально они применялись в основном в качестве нейролептических препаратов. Эти нейролептические свойства ограничивают антимикробное применение данных препаратов при таких заболеваниях, как туберкулез, поскольку они могут вызывать нежелательные побочные эффекты в центральной нервной системе в тех дозировках, которые применяются для борьбы с микобактериями.
Таким образом, существует необходимость в модифицированных производных фенотиазина, которые не демонстрируют или демонстрируют минимальное нежелательное побочное воздействие на ЦНС.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению, разработано трициклическое производное формулы (1)
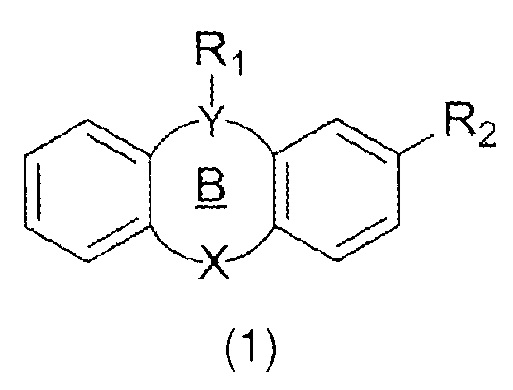 ,
,
где R1 представляет собой алкилсульфонатную или сульфонамидную группу; R2 представляет собой водород, галоген, замещенную алкильную группу, тиоэфирную или ацетильную группу; Y представляет собой N или C; X представляет собой S, SO, SO2, N, O, CH2, C(O), CO2, NHCO; и цикл B представляет собой 6, 7 или 8-членный циклоалкил. Если производное (1) является производным фенотиазина, R2 не является H.
В предпочтительном варианте осуществления изобретения R1 представляет собой (CH2)nSO3M, где n=1, 2, 3 или 4, и M = Na, K или H, или R1 представляет собой SO2Ar, где Ar необязательно является замещенным; R2 представляет собой H, Cl, Br, SMe, C(O)CH3 или CF3; Y представляет собой N или C; и X представляет собой S, SO, SO2, N, O, CH2, C(O), CO2, NHCO, и цикл B представляет собой 6, 7 или 8-членный циклоалкил. Заместители фрагмента Ar, если R1 представляет собой SO2Ar, включают орто- и пара-заместители, выбранные из NO2, NH2, SO3M, CO2R, где M = Na, K или H, и R = алкил.
Другие производные по настоящему изобретению, относящиеся к трициклическим производным, могут быть выбраны из:
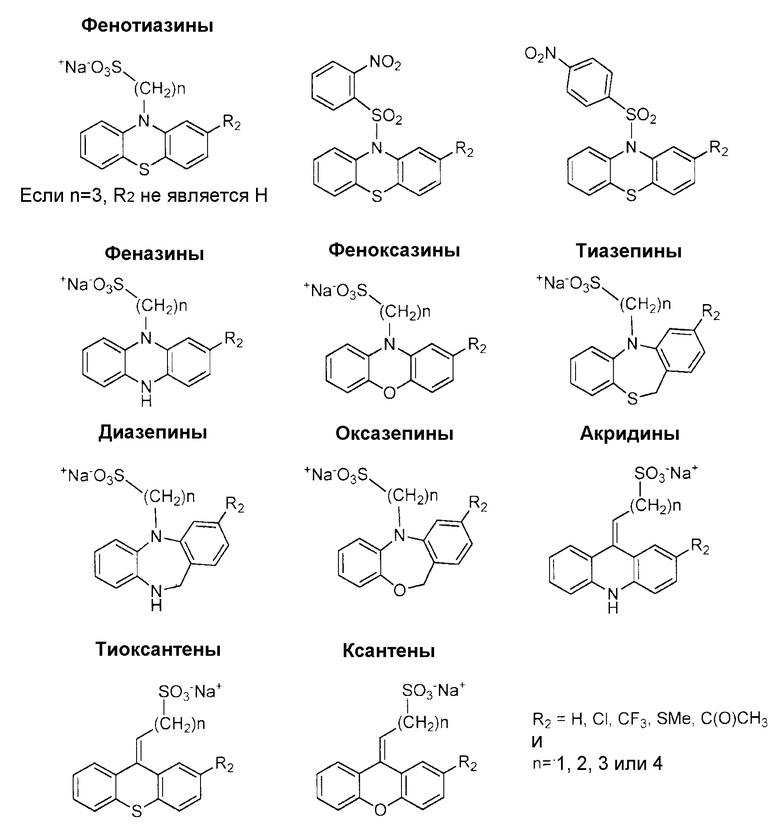 .
.
Дополнительные производные по настоящему изобретению, относящиеся к трициклическим производным, могут быть выбраны из:
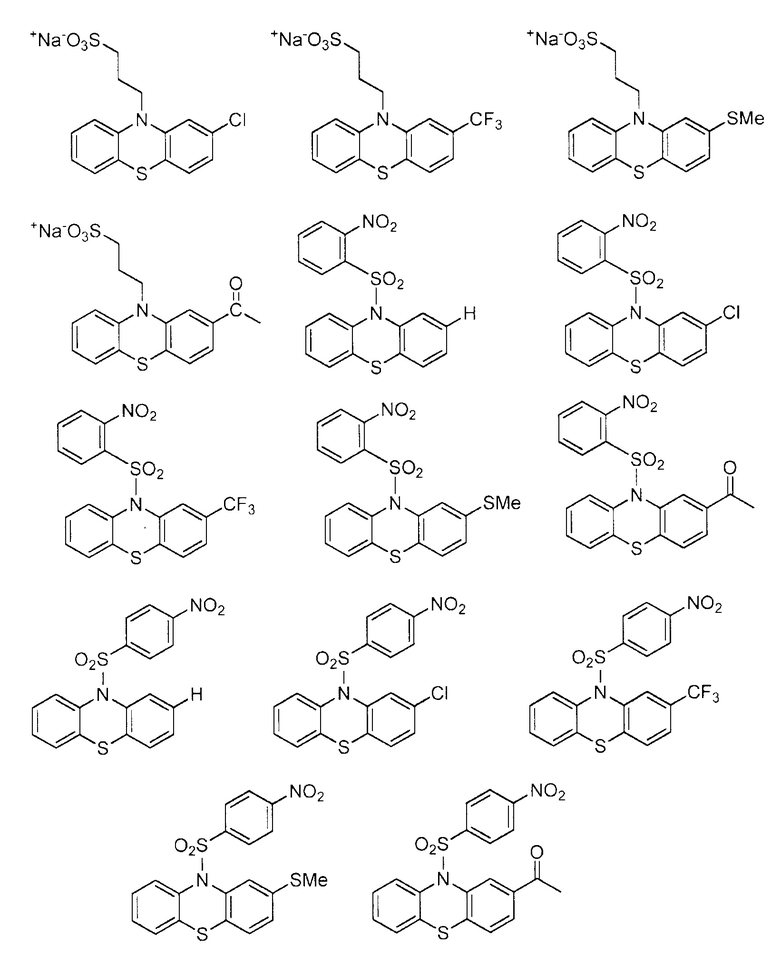 .
.
Настоящее изобретение относится к трициклическим производным формулы (1), которые определены выше, в т.ч. производным фенотиазина формулы (1), где n=3 и R2=H, для применения в лечении туберкулеза, предпочтительно чувствительных и резистентных к лекарственным средствам форм туберкулеза, к указанным производным для применения в лечении туберкулеза, где эти производные применяются индивидуально или в комбинации с известными препаратами против туберкулеза.
Настоящее изобретение относится также к способу лечения туберкулеза, где указанный способ включает введение терапевтически эффективного количества трициклического производного формулы (1), которое определено выше, в т.ч. производного фенотиазина формулы (1), где n=3 и R2=H, пациенту, которому это необходимо, где введение этого производного осуществляется в комбинации со вторым лекарственным средством против туберкулеза, причем оба препарата могут входить в состав одной дозированной формы или двух отдельных дозированных форм.
Другие аспекты настоящего изобретения относятся к противотуберкулезным лекарственным средствам, выбранным из изониазида, этионамида, этамбутола, пиразинамида, рифампицина, амикацина, канамицина, капреомицина, виомицина, энвиомицина, ципрофлоксацина, левофлоксацина, моксифлоксацина, рифабутина, кларитромицина, линезолида, тиоацетазона, протионамида.
Далее, настоящее изобретение относится к применению трициклического производного формулы (1), которое определено выше, в т.ч. производного фенотиазина формулы (1), где n=3 и R2=H, в производстве лекарственного средства для лечения туберкулеза.
Далее, настоящее изобретение относится к лекарственному средству в виде стандартной дозированной формы, отличающемуся тем, что каждая единица дозированной формы содержит трициклическое производное формулы (1), которое определено выше, в т.ч. производное фенотиазина формулы (1), где n=3 и R2=H, а также второе противотуберкулезное средство.
Другие аспекты настоящего изобретения относятся к противотуберкулезным лекарственным средствам, выбранным из изониазида, этионамида, этамбутола, пиразинамида, рифампицина, амикацина, канамицина, капреомицина, виомицина, энвиомицина, ципрофлоксацина, левофлоксацина, моксифлоксацина, рифабутина, кларитромицина, линезолида, тиоацетазона, протионамида.
КРАТКОЕ ОПИСАНИЕ ИЛЛЮСТРАТИВНОГО МАТЕРИАЛА
Ниже по тексту настоящее описание будет описано с привлечением примеров, в которых осуществляются ссылки на сопроводительный иллюстративный материал, в котором:
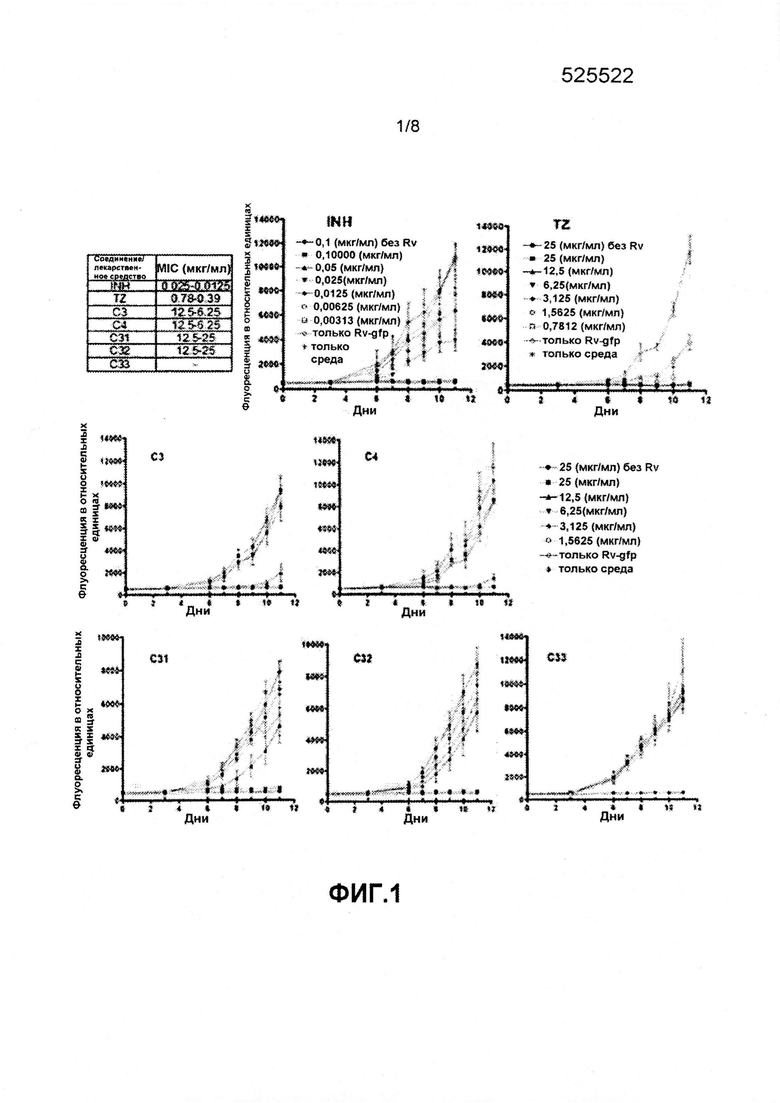
на фиг. 1 показана антимикобактериальная активность изониазида (INH), тиоридазина (TZ) и синтезированных в настоящем изобретении производных фенотиазина C3, C4, C31, C32 и C33 против M.tb H37Rv.gfp, которую определяли с использованием анализа GFAP на микропланшете, а также таблица, в которой приведены полученные значения MIC50;
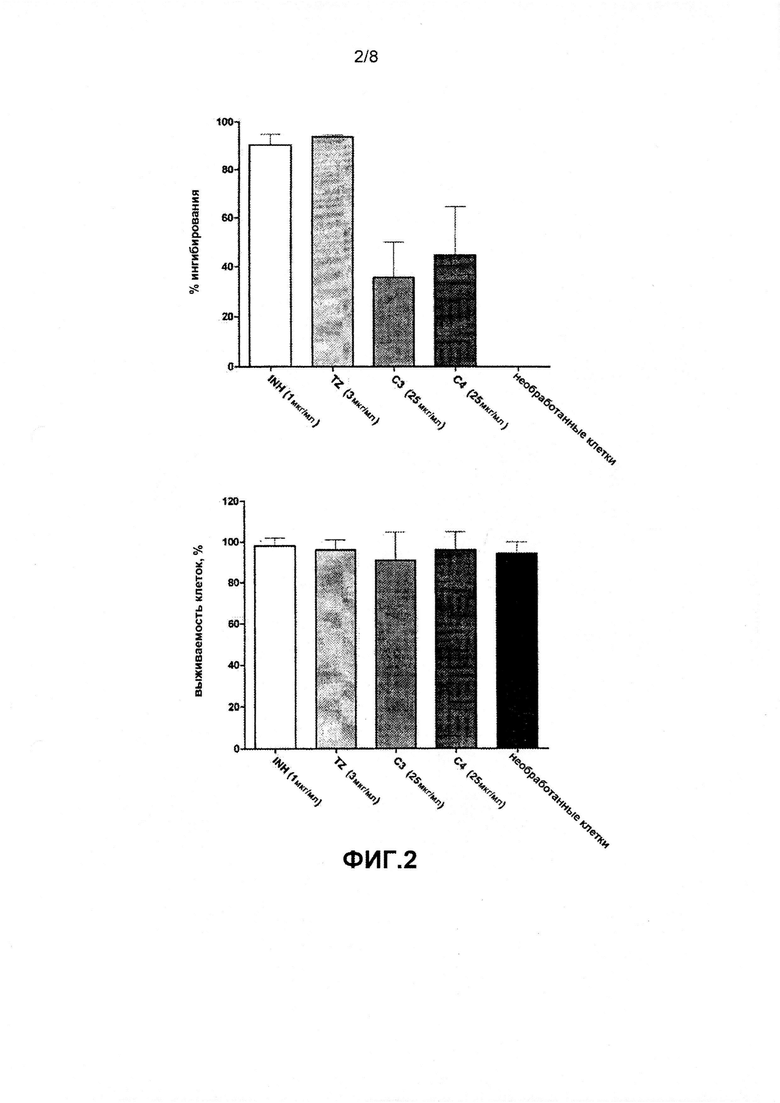
на фиг. 2 показана активность тестируемых соединений против внутриклеточных M.tb H37Rv при обработке инфицированных макрофагов INH, TZ и тестируемыми соединениями, соответственно, в указанных концентрациях в течение пяти дней;
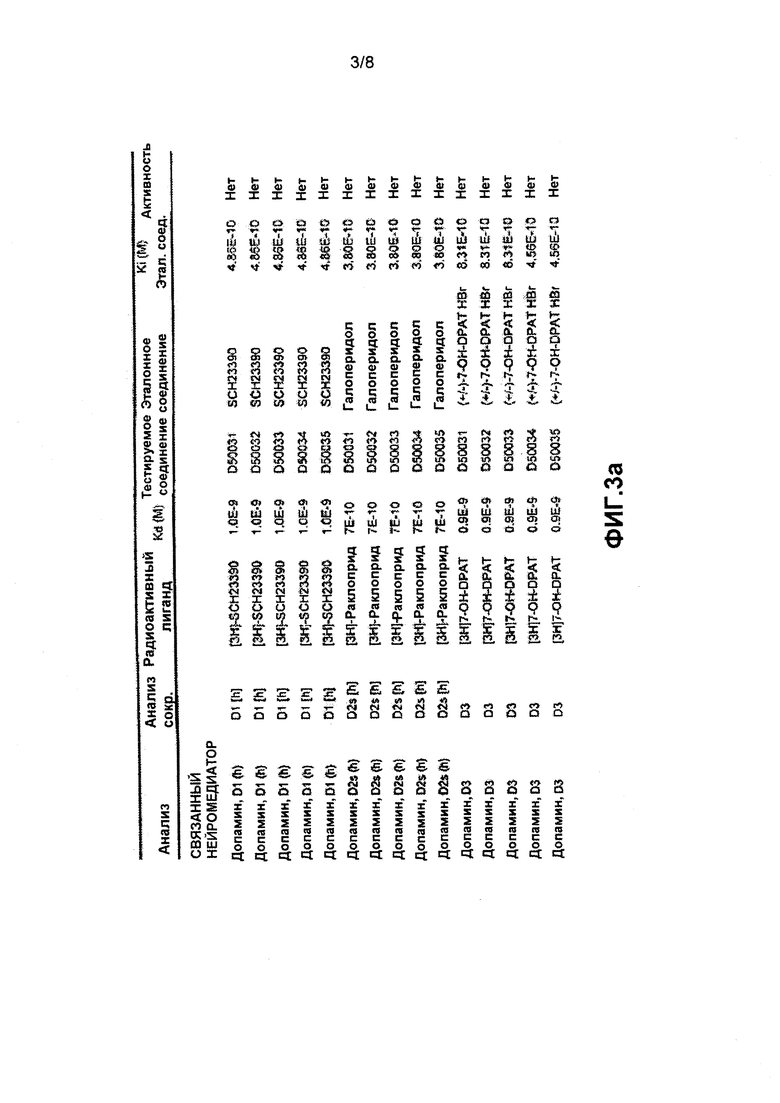
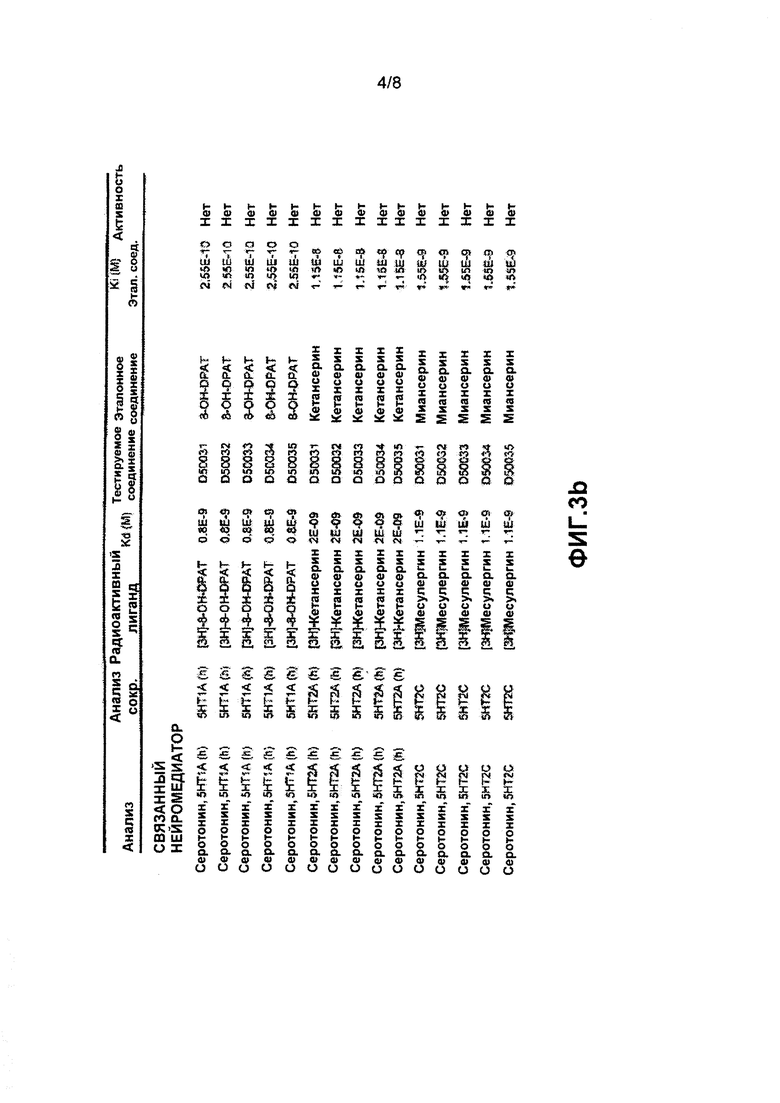
на фиг. 3a и 3b показаны результаты анализа связывания радиоактивного лиганда, указывающие на отсутствие активности, направленной на связывание с дофаминергическими рецепторами подтипов D1, D2, D3 и серотонинергическими рецепторами подтипов 5-HT1A, 5-HT2A и 5-HT2C;
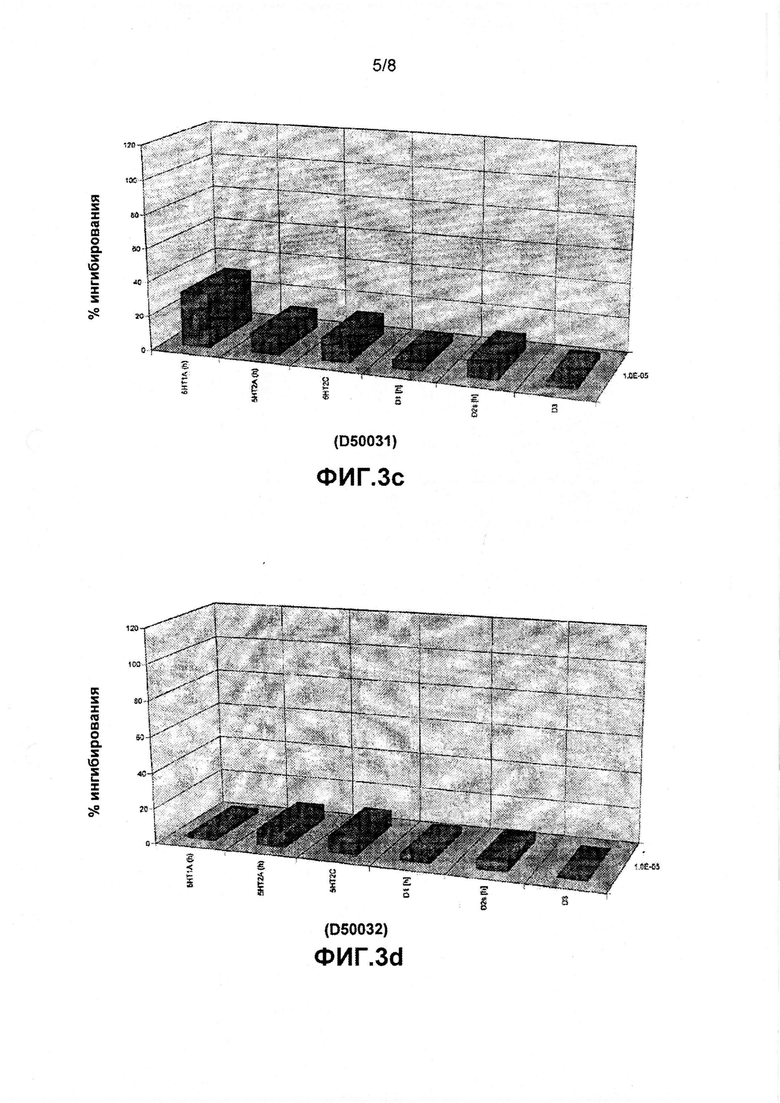
фиг. 3c является графическим представлением ингибирования C3 (D50031) в процентах относительно результатов для ингибиторов дофамина и серотонина;
фиг. 3d является графическим представлением ингибирования C4 (D50032) в процентах относительно результатов для ингибиторов дофамина и серотонина;
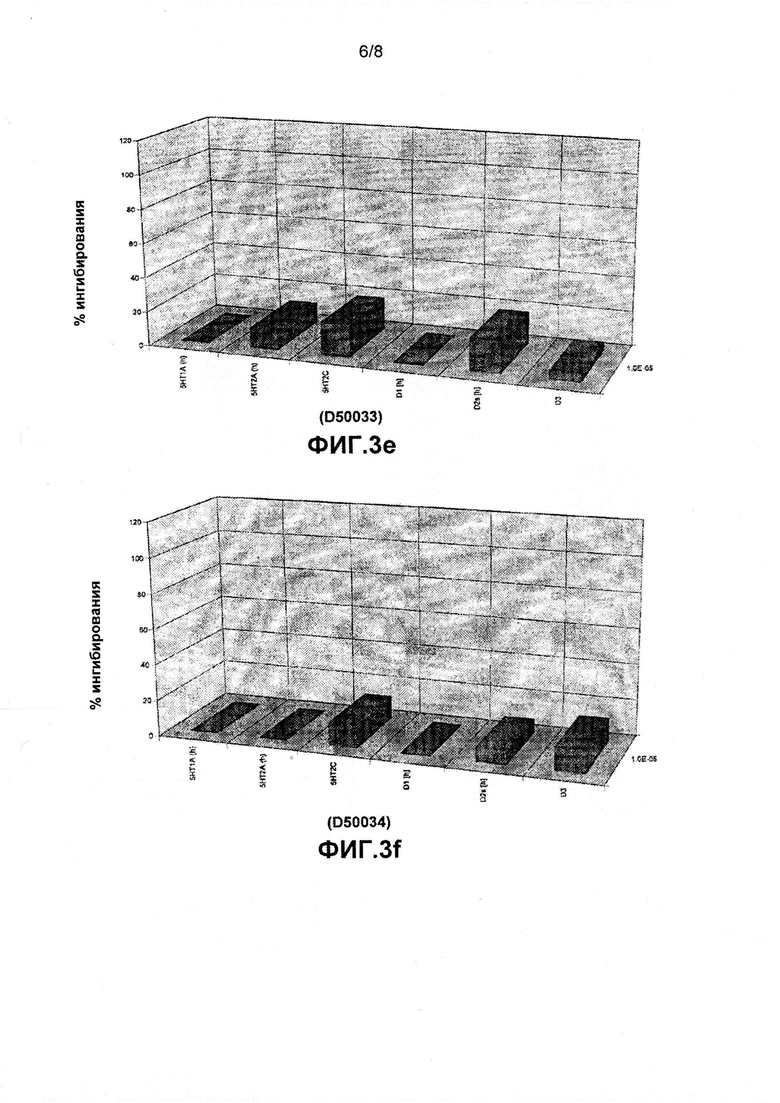
фиг. 3e является графическим представлением ингибирования C31 (D50033) в процентах относительно результатов для ингибиторов дофамина и серотонина;
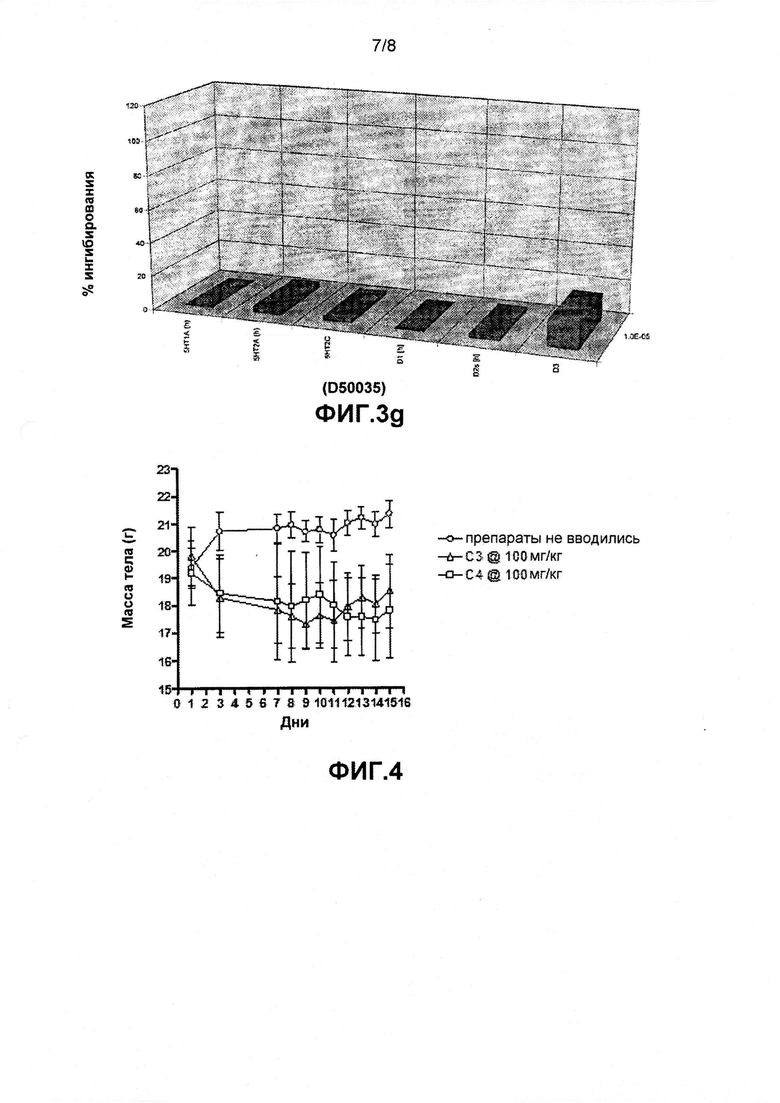
фиг. 3f является графическим представлением ингибирования C32 (D50034) в процентах относительно результатов для ингибиторов дофамина и серотонина; и
фиг. 3g является графическим представлением ингибирования C33 (D50035) в процентах относительно результатов для ингибиторов дофамина и серотонина;
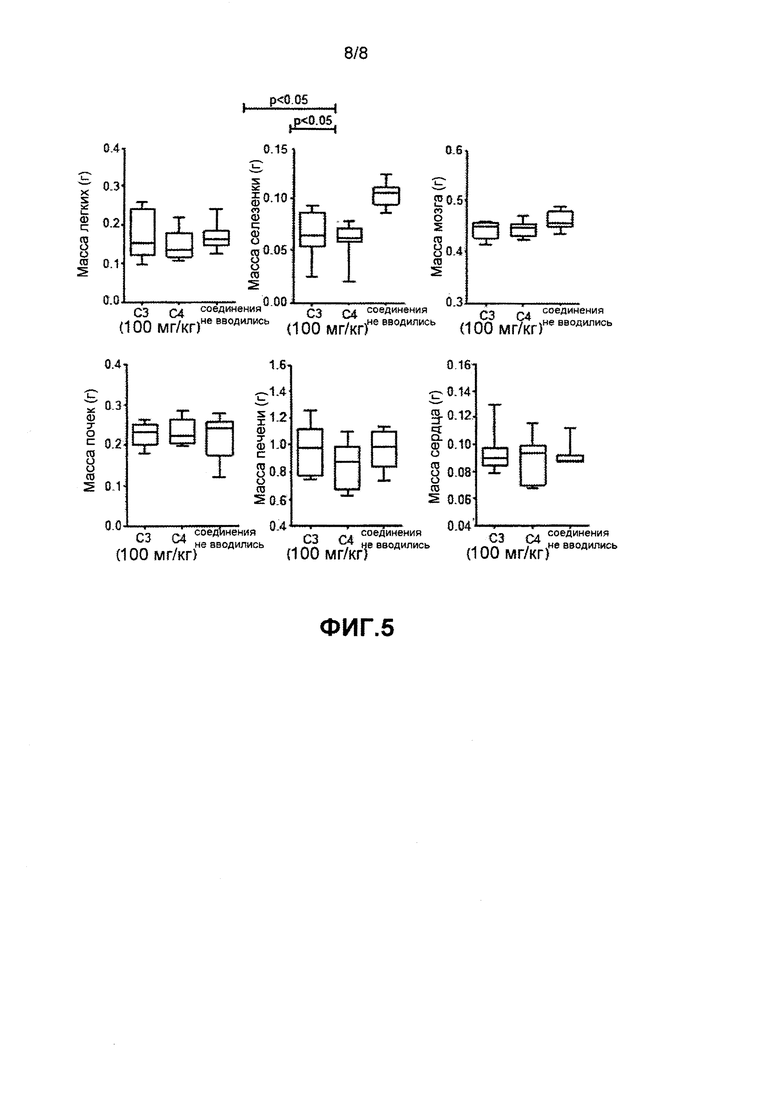
фиг. 4 представляет собой график, демонстрирующий дневные изменения массы тела мышей, получающих ежедневные дозы 100 мг/кг либо C3, либо C4, а также мышей, не получавших препаратов, в течение 14-дневного периода; и
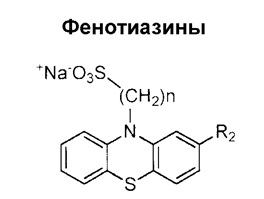
на фиг. 5 изображены графики, демонстрирующие массы органов мышей в конце 14-дневного эксперимента, в течение которого мышам вводили ежедневные дозы 100 мг/кг C3 либо C4 или не вводили препаратов.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ СО ССЫЛКАМИ НА ИЛЛЮСТРАТИВНЫЙ МАТЕРИАЛ
В изобретении предложены трициклические производные приведенной ниже общей формулы (1)
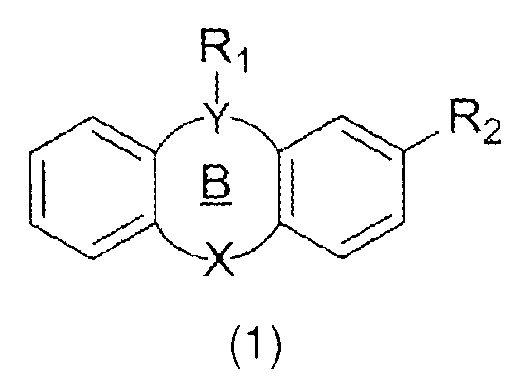 ,
,
где R1 представляет собой алкилсульфонатную или сульфонамидную группу; R2 представляет собой водород, галоген, замещенную алкильную группу, тиоэфирную или ацетильную группу; Y представляет собой N или C; X представляет собой S, SO, SO2, N, O, CH2, C(O), CO2, NHCO; и цикл B представляет собой 6-, 7- или 8-членный циклоалкил. Если производное (1) является производным фенотиазина, R2 не является H.
В предпочтительном варианте осуществления изобретения R1 представляет собой (CH2)nSO3M, где n=1, 2, 3 или 4, и M = Na, K или H, или R1 представляет собой SO2Ar, где Ar необязательно является замещенным; R2 представляет собой H, Cl, Br, SMe, C(O)CH3 или CF3; Y представляет собой N или C; X представляет собой S, SO, SO2, N, O, CH2, C(O), CO2, NHCO; и цикл B представляет собой 6, 7 или 8-членный циклоалкил.
Заместители фрагмента Ar, если R1 представляет собой SO2Ar, включают орто- и паразаместители, выбранные из NO2, NH2, SO3M, CO2R, где M = Na, K или H, и R = алкил.
Приведенные ниже соединения представляют собой предпочтительные варианты осуществления трициклических производных общей формулы (1):
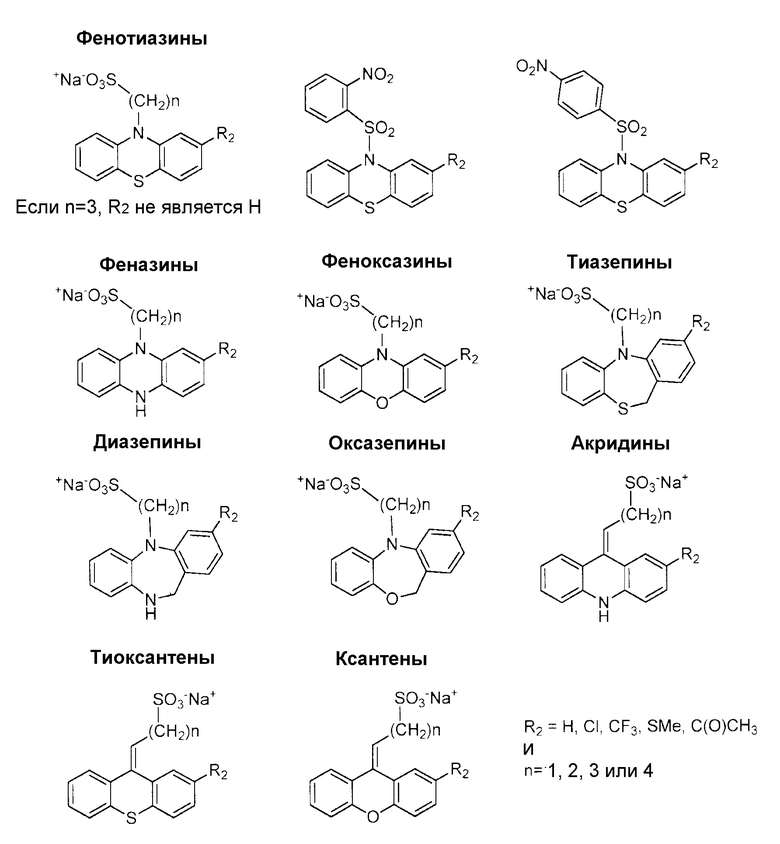 .
.
В более предпочтительных вариантах осуществления трициклические производные общей формулы (1) могут быть выбраны из:
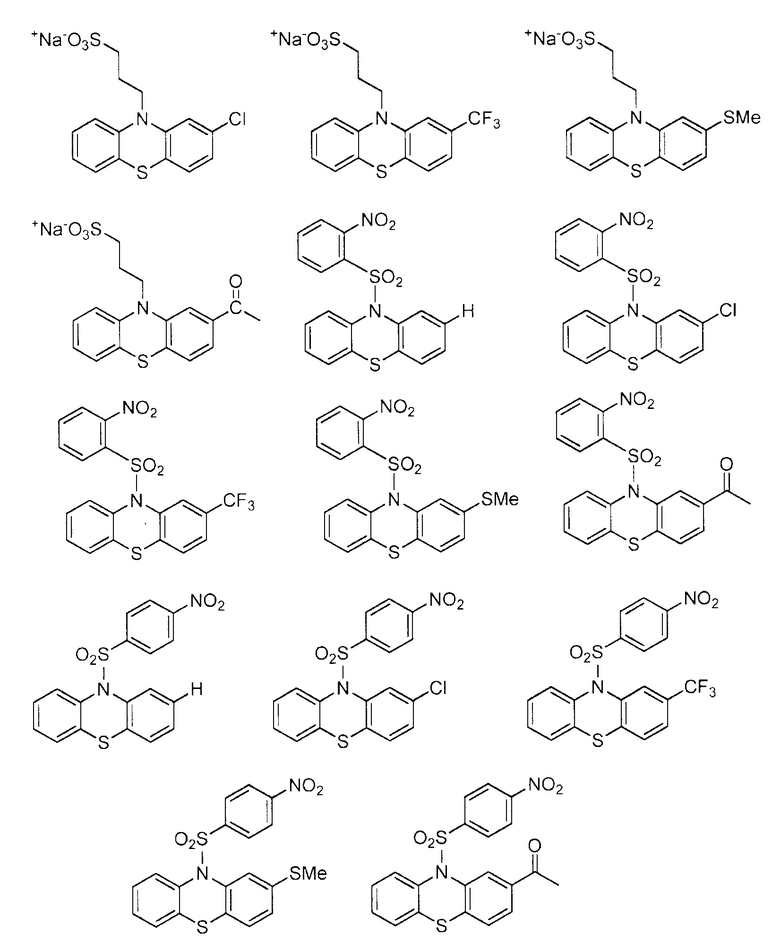 .
.
Способ получения N-алкилсульфонатов фенотиазина включает:
(a) получение аниона фенотиазина и
(b) взаимодействие указанного аниона с циклическими алкилсульфонатами, например, 1,3-пропан сультоном или 1,4-бутан сультоном, которое в общих чертах описано в патенте Соединенных Штатов №7855287.
Менее эффективный многостадийный способ синтеза был опубликован ранее в Journal of Physical Chemistry, 1986, 90, 2469-2415.
Было показано, что производные фенотиазина по настоящему изобретению имеют ограниченную токсичность против культур основных макрофагов, а также ограниченную психотропную активность.
Считается, что антишизофреническое действие фенотиазиновых препаратов включает блокирование синаптических дофаминовых рецепторов в мозге. С помощью молекулярных моделей заполнения пространства было показано, что предпочтительные Ван-дер-Вальсовы взаимодействия между аминогруппой боковой цепи фенотиазинов и заместителем в положении 2 цикла A могут способствовать образованию конформации, имитирующей структуру дофамина. Таким образом, модификации, которые вызывают отклонение от этой предпочтительной ориентации, могли бы уменьшить связывание с дофаминовым рецептором и уменьшить воздействие на ЦНС.
Стандартные исследования ингибирования дофамина и серотонина соединениями по настоящему изобретению выявили полную инактивацию связывания, что позволяет сделать предположение о полном устранении воздействия на ЦНС.
Соединения по настоящему изобретению продемонстрировали статистически значимое ингибирующее действие против вирулентных бактерий Mycobacterium tuberculosis, показав минимальную ингибирующую концентрацию (MIC50) ~6-12 мкг/мл в прямом анализе уничтожения бактерий.
В исследованиях in vitro было убедительно показано, что алкилсульфонаты производных фенотиазина, которые представлены выше, являются очень эффективными при уничтожении вирулентных бактерий M. tuberculosis, если они находятся в непосредственном контакте с бактериями, указывая на то, что соединения по настоящему изобретению обходят механизмы, которые используются M. tuberculosis для проявления устойчивости к лекарственным средствам, например, эффлюксный насос. Кроме того, было надежно установлено, что M. tuberculosis выживают в фагосомах макрофагов, т.е. первичных клеток-хозяев бактерий, где определенные стратегии уклонения позволяют им избежать слияния с лизосомами и за счет этого предотвратить разрушение лизосомальными ферментами. Одна из ключевых задач для потенциальных кандидатов в новые лекарственные средства заключается в преодолении нескольких подобных мембранных систем для попадания в целевой микроорганизм и инициирования его уничтожения. Полученные данные явным образом демонстрируют, что соединения по настоящему изобретению обладают способностью преодолевать все мембранные системы и подавлять размножение M.tuberculosis. По сравнению с тиоридазином, который проявляет нейролептическое и цитотоксическое действие, соединения по настоящему изобретению обладают более благоприятными свойствами в плане цитотоксичности и не обладают нейролептическими свойствами.
Известно, что фенотиазины улучшают эффективность изониазида в моделях латентности in vivo и in vitro. Однако фармакокинетика и фармакодинамика, которые способствуют проявлению их психотических свойств, препятствуют их применимости в качестве антимикробных препаратов in vivo.
Производные фенотиазина по настоящему изобретению могут применяться независимо или в комбинации с известными противотуберкулезными препаратами, например, средствами 1-й линии: этамбутолом, изониазидом, пиразинамидом, рифампицином; средствами 2-й линии: аминогликозидами (например, амикацином, канамицином), полипептидами (например, капреомицином, виомицином, энвиомицином); фторхинолонами (например, ципрофлоксацином, левофлоксацином, моксифлоксацином), тиоамидами (например, этионамидом, протионамидом); средствами 3-й линии: рифабутином, макролидами (например, кларитромицином), линезолидом, тиоацетазоном, тиоридазином, аргинином, витамином D и R207910.
Считается, что потенциальное синергетическое действие комбинированной терапии подобного типа особенно подходит для чувствительных и резистентных к лекарственным средствам форм M. tuberculosis.
Настоящее изобретение относится к способу лечения туберкулеза, где указанный способ включает введение пациенту, которому это необходимо, эффективного количества трициклического производного по настоящему изобретению. В одном из вариантов осуществления настоящего изобретения введение соединения по настоящему изобретению осуществляют в комбинации со вторым противотуберкулезным лекарственным средством. Применение этого комбинированного лечения осуществляют путем введения обоих препаратов, т.е. трициклического производного по настоящему изобретению и второго противотуберкулезного средства, в составе одной дозированной формы или последовательного введения двух отдельных дозированных форм.
Далее, настоящее изобретение относится к применению трициклического производного по настоящему изобретению для производства лекарственного средства для лечения туберкулеза. Это лекарственное средство производят в виде твердой дозированной формы для перорального введения, которая представляет собой таблетку, пилюлю или капсулу, или в виде жидкой дозированной формы для перорального введения, или в виде порошка в форме аэрозоля для легочной доставки, которые включают как трициклическое производное по настоящему изобретению, так и второе противотуберкулезное средство, или каждый из действующих ингредиентов отдельно для последовательного введения.
Лекарственное средство, включающее комбинацию действующих ингредиентов, которые являются трициклическим производным по настоящему изобретению и вторым противотуберкулезным средством, предпочтительно должно представлять собой стандартную дозированную форму. Т.е. оба действующих ингредиента предпочтительно будут содержаться в одной таблетке, пилюле, капсуле и т.п. Присутствие обоих действующих ингредиентов в одной дозированной форме упрощает введение препаратов пациентам, особенно, если пациенту необходимо самостоятельно вводить препараты. Это приводит также к улучшению приверженности пациента лечению и меньшей вероятности развития микробных форм, резистентных к лекарственным препаратам, в результате частичного невыполнения схемы лечения.
Химически модифицированные фенотиазины по настоящему изобретению сохраняют антимикробную активность и не обладают токсичностью по отношению к макрофагам. Специалист в данной области техники поймет, что новые фенотиазины по настоящему изобретению будут иметь измененные фармакокинетику и фармакодинамику проникновения через гематоэнцефалический барьер, что приводит к минимизации или исчезновению психотического действия. Кроме того, существуют многочисленные модификации и видоизменения вариантов осуществления настоящего изобретения, которые могли бы быть очевидны специалисту в данной области техники, которые, как считается, входят в объем настоящего изобретения, сущность которого определяется приведенным выше описанием и примерами.
ПРИМЕРЫ
Производные фенотиазина демонстрируют прямую антибактериальную активность против M. tuberculosis в культуре
Для скрининга бактерицидной/бактериостатической активности производных фенотиазина исследовали действие пяти соединений, а именно C3, C4, C31, C32 и C33 (показаны ниже), против M. tuberculosis, используя анализ GFP (зеленого флуоресцентного белка) в микропланшетах (GFPMA).
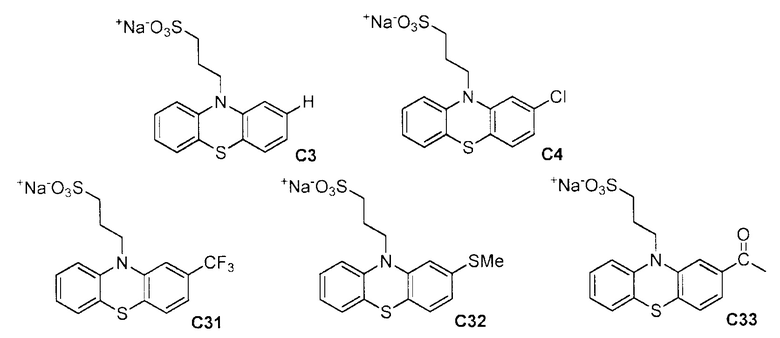
Результаты сравнивали с результатами для производного фенотиазина тиоридазина (TZ) и для лекарственного препарата первой линии изониазида (INH), которые использовались в качестве положительного контроля для оценки эффективности против микобактерий. Полученные результаты показали, что все тестируемые соединения, за исключением C33, продемонстрировали дозозависимое ингибирующее действие на рост M.tb-H37Rv.gfp, и полученные значения MIC50 показаны на фиг. 1. Полученные значения MIC50 для INH и TZ соответствовали опубликованным данным. Статистически значимая антибактериальная активность была продемонстрирована для C3 и C4, которые имели наиболее низкие значения MIC, а также для C31 и C32.
Производные фенотиазина являются нетоксичными для культуры макрофагов, выделенных из костного мозга
Исследовали токсическое действие C3, C4, C31, C32 и C33 на макрофаги, выделенные из костного мозга (BMDM), in vitro в концентрациях от 0,1953 до 25 мкг/мл. Костный мозг изолировали из бедренных костей интактных мышей C57BI/6 и культивировали макрофаги при 37°C в атмосфере 5% CO2 до зрелости. Полученные данные показали, что соединения не оказывали воздействия на жизнеспособность клеток при всех концентрациях после 5 дней инкубирования (таблица 1 ниже). В противоположность этому, TZ оказался токсичным и вызывал смерть клеток после 3 дней инкубирования (данные не показаны). Через 5 дней инкубирования с TZ наблюдалась смерть 100% клеток при концентрациях от 25 мкг/мл до 6,25 мкг/мл. Проведенный анализ включал также исследование INH в диапазоне концентраций от 0,00078 мкг/мл до 0,1 мкг/мл, и было обнаружено, что INH не является токсичным (данные не показаны).
Цитотоксичность тестируемых соединений в отношении макрофагов. Макрофаги обрабатывали соединениями в течение 5 дней и оценивали выживаемость клеток
Внутриклеточное ингибирование воспроизведения M. tuberculosis в макрофагах производными фенотиазина по настоящему изобретению
Для выяснения того, способны ли соединения по настоящему изобретению проникать сквозь систему клеточных мембран и попадать внутрь бактериальных клеток, ингибируя их размножение, BMDM инфицировали M. tuberculosis и культуру инфицированных клеток в течение 5 дней обрабатывали производными фенотиазина по настоящему изобретению, а именно C3 и C4. Рассчитывали показатели ингибирования внутриклеточного размножения бактерий соединениями C3 или C4 и выражали полученные величины в процентах от значений, полученных для необработанных клеточных культур. Одновременно определяли также токсическое действие препаратов на инфицированные макрофаги, используя анализ выживаемости клеток CellTiter-Blue Cell Viability Assay. Процентную долю выживших клеток вычисляли, исходя из значений для инфицированных клеток (обработанных или необработанных) относительно значений для неинфицированных клеток. Как видно на фиг. 2, как INH, так и TZ продемонстрировали более чем 90% ингибирование роста внутриклеточных M. tuberculosis в концентрациях 1 мкг/мл и 3 мкг/мл соответственно. Интересно отметить, что оба тестируемых соединения C3 и C4 при концентрации 25 мкг/мл продемонстрировали статистически значимую антибактериальную активность в диапазоне 40-50%, хотя и оказались менее эффективными по сравнению с INH и TZ. Заслуживает упоминания, что все инфицированные макрофаги выжили почти полностью после обработки лекарственными средствами. После этого макрофаги подвергали лизису и супернатант распределяли по планшетам для определения CFU.
Исследование связывания серотонина и дофамина
Результаты анализа связывания радиоактивных лигандов показали отсутствие способности связываться с дофаминергическими рецепторами подтипов D1, D2, D3 и серотонинергическими рецепторами подтипов 5-HT1A, 5-HT2A и 5-HT2C (фиг. 3a-g). В качестве эталонов для сравнения были выбраны и одновременно протестированы немодифицированные фенотиазины, а именно тиоридазин и хлорпромазин. Для структурных гомологов, т.е. C3, C4, C31, C32 и C33 (обозначенных как DS0031, DS0032, DS0033, DS0034, DS0035), удалось добиться почти полного устранения связывания с рецепторами дофамина и серотонина.
Токсический потенциал соединений C3 и C4 в модели мелких животных (мыши C57BI/6)
Оценивали токсичность производных фенотиазина C3 и C4 в дозировке 100 мг/кг, которая являлась максимальной исходной дозой, и сравнивали результаты с данными для известных, одобренных для клинического применения препаратов, а именно фенотиазина и тиоридазина (TZ), и с результатами для животных, которым не вводили препаратов.
Первое исследование проводили для оценки максимально переносимой дозы TZ у мышей, поскольку в литературе имелся широкий разброс данных в диапазоне от 160 мг/кг до 20 мг/кг. В этом исследовании самкам мышей C57BI/6 в возрасте 6-8 недель (n=10 особей/группу) вводили 100 мг/кг, 40 мг/кг, 20 мг/кг и 10 мг/кг препарата путем принудительного кормления через рот, при этом наблюдали за выживанием животных и регистрировали изменение массы их тела. В начальном исследовании введения сублетальных концентраций TZ вводили один раз в начале эксперимента в объеме 200 мкл и наблюдали за выживанием и изменением массы тела животных. Было обнаружено, что тестируемые дозы 40 мг/кг и 100 мг/кг оказывали временное токсическое действие и животные находились в параличе в течение 6 ч. Однако в течение 24-48 ч животные восстанавливались. Животные временно теряли массу тела, но восстанавливались через 3-4 дня. Введение одной дозы TZ не вызывало смертности в любой из исследованных дозировок в течение 2-недельного периода.
Для оценки токсичности введения одиночной дозы (100 мг/кг) C3 и C4 производные фенотиазина по настоящему изобретению вводили путем принудительного кормления через рот в объеме 200 мкл. Так же, как и в случае животных, не получавших препаратов, ни одно из животных не продемонстрировало статистически значимой потери массы на протяжении 14-дневного периода. Аналогично, у животных не наблюдалось потери массы тела при введении одиночной дозы TZ 20 мг/кг. Мыши демонстрировали временную летаргию, но восстанавливались через 24 ч. Исследования введения одиночной дозы повторяли с аналогичными результатами.
Затем оценивали токсическое действие у животных, которые получали препараты в ежедневном режиме. Говоря вкратце, мыши (n=10-15 особей/группу) получали либо TZ (10 мг/кг или 20 мг/кг); C3 (100 мг/кг) или C4 (100 мг/кг) в 200 мкл воды путем принудительного кормления через рот в ежедневном режиме. Изменения массы тела определяли и фиксировали также в ежедневном режиме (фиг. 4). Эксперимент завершали через 14 дней, всех животных умерщвляли гуманным способом, после чего извлекали и взвешивали органы (сердце, легкие, почки, печень и селезенку) (фиг. 5). Собирали кровь и сохраняли сыворотку для дальнейшего анализа.
Животные, не получавшие никаких препаратов, вначале набирали массу тела, а затем происходила ее стабилизация. Животные, которые ежедневно получали 100 мг/кг, продемонстрировали быструю потерю примерно 10% массы тела в первую неделю, после чего масса тела стабилизировалась с возможностью восстановления к концу недели 2.
Измерение массы органов животных, получавших C3 и C4, продемонстрировало статистически значимое снижение массы селезенки в обеих группах, по сравнению с животными, не получавшими препаратов. Масса всех остальных органов не зависела от введения препаратов.
Сравнительные профили токсичности соединений C3 и C4 оказались значимо лучше, чем профиль одобренного для клинического применения фенотиазина, т.е. TZ, даже при условии, что последний тестировался в концентрации, которая была в 10 раз ниже концентрации C3 или C4. При ежедневном введении любой из доз 100 мг/кг или 40 мг/кг TZ приводил к 100% смертности животных в течение 48 ч, тогда как все животные выжили после введения 100 мг/кг C3 или C4. Хотя мыши выжили после инфекции, животные, получавшие C3 и C4, тем не менее, потеряли приблизительно 10% массы тела. Что касается взвешенных органов, мозг, печень, легкие, почки и сердце продемонстрировали нормальное распределение массы, тогда как масса селезенки статистически значимо снизилась. В целом животные не продемонстрировали аномальных изменений и перенесли введение C3 в течение 14-дневного периода.
Следует иметь в виду, что существуют многочисленные модификации и видоизменения вариантов осуществления настоящего изобретения, которые могли бы быть очевидны специалисту в данной области техники и которые, как считается, входят в объем настоящего изобретения, сущность которого определяется приведенным выше описанием и примерами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ФЕНИЛ ФОРМАМИДИНА, ОБЛАДАЮЩИЕ АНТИМИКОБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ | 2018 |
|
RU2702224C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ТУБЕРКУЛЕЗА | 2009 |
|
RU2484819C2 |
| НОВЫЙ АНТИБАКТЕРИАЛЬНЫЙ АГЕНТ 3-(КАРБОКСИМЕТИЛ)-1-(ИМИДАЗО[1,2-b][1,2,4,5]ТЕТРАЗИН-3-ИЛ)-1Н-БЕНЗО[d]ИМИДАЗОЛ-3-ИУМ ХЛОРИД, ОБЛАДАЮЩИЙ АКТИВНОСТЬЮ ОТНОСИТЕЛЬНО NEISSERIA GONORRHOEAE И MYCOBACTERIUM TUBERCULOSIS | 2023 |
|
RU2828876C1 |
| ФУРИЛИДЕНФУРАНОНОВЫЕ ПРОИЗВОДНЫЕ УСНИНОВОЙ КИСЛОТЫ КАК НОВЫЕ ПРОТИВОТУБЕРКУЛЕЗНЫЕ АГЕНТЫ | 2013 |
|
RU2533707C1 |
| 5-метил-7-(3-нитро-[1,2,4]триазол-1-ил)-[1,2,4]триазоло[1,5-а]пиримидина, обладающий противотуберкулезной активностью в отношении возбудителя с множественной лекарственной устойчивостью, и способ его получения | 2018 |
|
RU2705591C1 |
| ИЗОНИКОТИНОИЛГИДРАЗОН ДИМЕФОСФОНА, ОБЛАДАЮЩИЙ ПРОТИВОТУБЕРКУЛЕЗНОЙ АКТИВНОСТЬЮ | 2011 |
|
RU2457212C1 |
| ПИРИДИНОИЛГИДРАЗОНЫ ДИАЛКИЛ(2-МЕТИЛ-4-ОКСОПЕНТ-2-ИЛ) ФОСФИНОКСИДОВ, ОБЛАДАЮЩИЕ ПРОТИВОТУБЕРКУЛЕЗНОЙ АКТИВНОСТЬЮ | 2012 |
|
RU2498990C1 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО ПРОЛОНГИРОВАННОГО ДЕЙСТВИЯ С ДОЗИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ В ОРГАНЫ-МИШЕНИ НА ОСНОВЕ D-ЦИКЛОСЕРИНА ДЛЯ ЛЕЧЕНИЯ РЕЗИСТЕНТНЫХ ФОРМ ТУБЕРКУЛЕЗА | 2008 |
|
RU2403041C2 |
| Соединение, фармацевтическая композиция, лекарственное средство, применение соединения, фармацевтической композиции, лекарственного средства | 2015 |
|
RU2723545C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОТИАЗИНОНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2006 |
|
RU2395508C1 |
Изобретение относится к производным
Технический результат: получены новые соединения – производные фенотиазина, которые могут быть использованы для лечения микробных инфекций, таких как туберкулез. 5 н. и 2 з.п. ф-лы, 1 табл., 11 ил.
1. Трициклическое производное формулы (1)
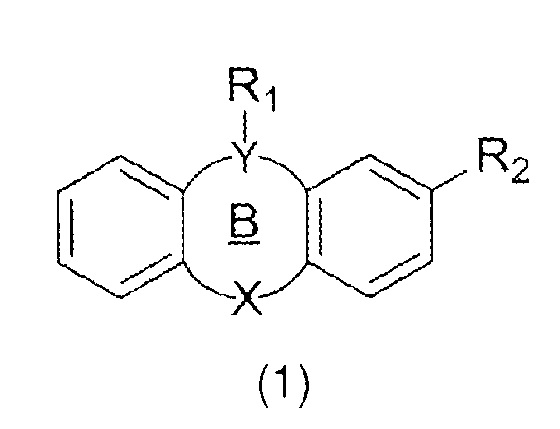 ,
,
где R1 представляет собой (CH2)nSO3M, где n=3, и M = Na, K;
R2 представляет собой Cl, Br, SMe, C(O)CH3 или CF3;
Y представляет собой N, X представляет собой S и цикл B представляет собой 6-членный циклоалкил.
2. Трициклическое производное по п. 1, где указанное трициклическое производное выбрано из:
 .
.
3. Трициклическое производное по п. 1 или 2, где указанное трициклическое производное выбрано из:
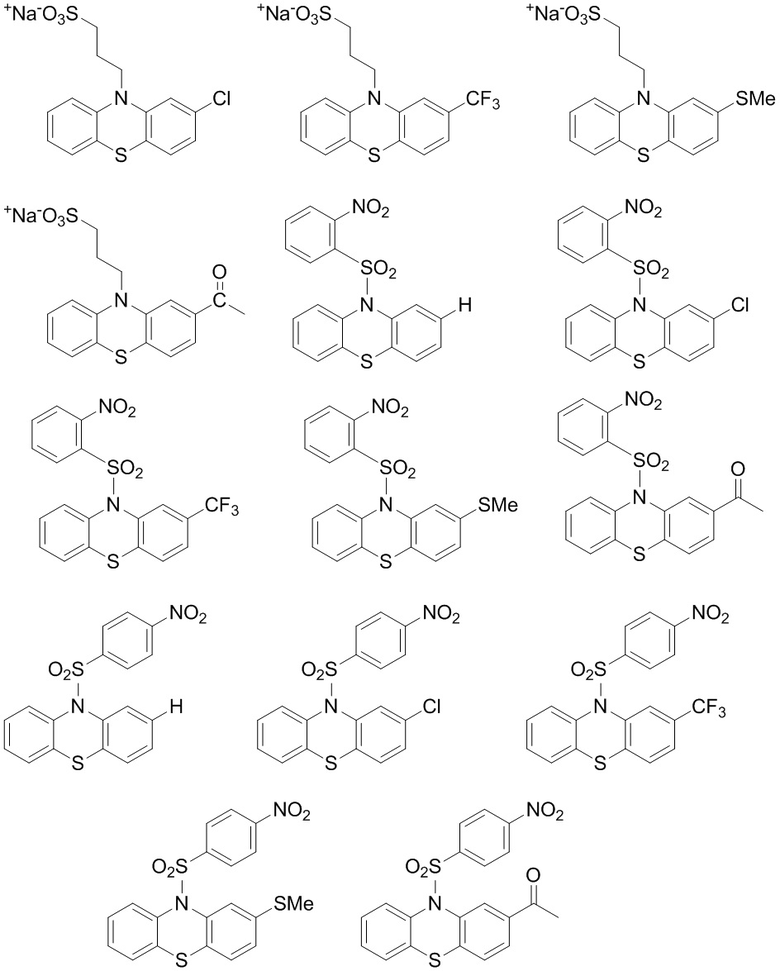
.
4. Применение трициклического производного формулы (1)
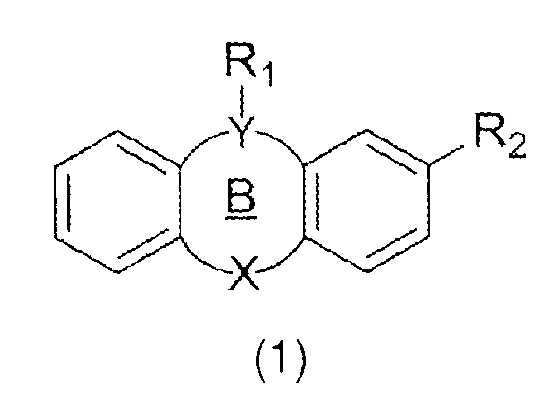 ,
,
где R1 представляет собой (CH2)nSO3M, где n=3, и M = Na, K;
R2 представляет собой H, Cl, Br, SMe, C(O)CH3 или CF3;
Y представляет собой N, X представляет собой S и цикл B представляет собой 6-членный циклоалкил, для лечения микробных инфекций.
5. Применение трициклического производного по п. 4 для лечения туберкулеза.
6. Способ лечения туберкулеза, который включает введение пациенту, которому это необходимо, терапевтически эффективного количества трициклического производного формулы (1)
,
где R1 представляет собой (CH2)nSO3M, где n=3, и M = Na, K;
R2 представляет собой H, Cl, Br, SMe, C(O)CH3 или CF3;
Y представляет собой N, X представляет собой S и цикл B представляет собой 6-членный циклоалкил.
7. Применение трициклического производного формулы (1)
,
где R1 представляет собой (CH2)nSO3M, где n=3, и M = Na, K;
R2 представляет собой H, Cl, Br, SMe, C(O)CH3 или CF3;
Y представляет собой N, X представляет собой S и цикл B представляет собой 6-членный циклоалкил, в производстве лекарственного средства для лечения туберкулеза.
| EP 1950207 A1, 30.07.2008 | |||
| AMARAL L | |||
| et al | |||
| Activity of phenothiazines against antibiotic-resistant mycobacterium tuberculosis: a rewiew supporting further studies that may elucidate the potential use of thioridazine as anti-tuberculosis therapy, JOURNAL OF ANTIMICROBIAL CHEMOTHER, 47, No:5, стр.: 505-511, 2001 | |||
| KAATZ G W | |||
| et al | |||
| PHENOTHIAZINES AND THIOXANTHENES INHIBIT MULTIDRUG EFFLUX PUMP ACTIVITY IN STAPHYLOCOCCUS AUREUS, ANTIMICROBIAL AGENTS AND CHEMOTHE, 47, Nor:2, стр.: 719 - 726, 2003 | |||
| WO 2005105145 A1, 10.11.2005 | |||
| Гидрохлорид 7-бром-2-амино-3Н-3-оксофенотиазина обладающий противотуберкулезной активностью | 1986 |
|
SU1340077A1 |

