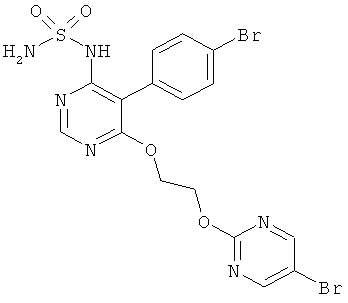
Настоящее изобретение относится к {5-(4-бромфенил)-6-[2-(5-бромпиримидин-2-илокси)этокси]пиримидин-4-ил}сульфамиду и его солям, способу получения этого соединения и его применению в медицине.
{5-(4-Бромфенил)-6-[2-(5-бромпиримидин-2-илокси)этокси]пиримидин-4-ил}сульфамид имеет формулу (I)
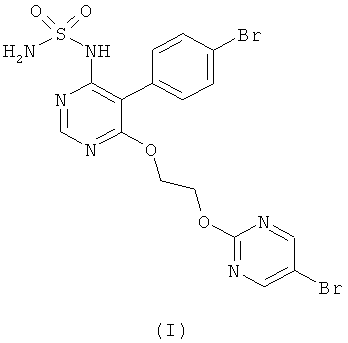
Соединение формулы (I) является ингибитором эндотелин рецептора и применяется в качестве антагониста эндотелин рецептора. Соединение формулы (I) является новым членом структурного семейства, которое в основном было открыто ранее в публикации WO 02/053557.
Неожиданно было установлено, что соединение формулы (I) обладает улучшенными свойствами по сравнению со структурно близкими соединениями, конкретно раскрытыми в WO 02/053557. В частности, соединение формулы (I) проявляет антагонистическую активность в отношении эндотелин рецептора в условиях in vivo со значительно более длительным периодом полураспада и значительно более коротким временем выведения по сравнению с соответствующими алкилированными производными. Это делает соединение формулы (I) особенно подходящим для применения в пролонгированных фармацевтических композициях.
Вследствие способности ингибировать связывание эндотелина соединение формулы (I) может быть использовано для лечения болезней, обусловленных повышенным сжатием сосудов, пролиферацией или воспалением, индуциируемых эндотелином. Примерами таких болезней являются гипертензия, легочная гипертензия, коронарные болезни, сердечная недостаточность, ишемия почек и миокарда, почечная недостаточность, церебральная ишемия, деменция, мигрень, субарахноидальное кровотечение, синдром Рейнольда, пальцевидная язва и портальная гипертензия. Эти соединения могут быть также использованы при лечении или профилактике атеросклероза, рестеноза после баллонной или стентовой ангиопластики, воспаления, язвы желудка и двенадцатиперстной кишки, рака, меланомы, рака простаты, гипертрофии простаты, эректильной дисфункции, потери слуха, слепоты, хронического бронхита, астмы, легочного фиброза, грамотрицательной септицемии, шока, серповидно-клеточной анемии, гломерулонефрита, почечной колики, глаукомы, болезней соединительных тканей, при лечении и профилактике диабетических осложнений, осложнений при сосудистой или сердечной хирургии или после трансплантации органов, осложнений, связанных с лечением циклоспорином, боли, гиперлипидемии, а также других болезней, связь которых с эндотелином в настоящее время установлена.
Соединение формулы (I) и его фармацевтически приемлемые соли могут быть, таким образом, использованы в качестве лекарственных средств, например, в форме фармацевтических композиций для энтерального или парентерального введения.
Термин "фармацевтически приемлемые соли" относится к нетоксичным, аддитивным солям неорганической или органической кислоты и/или основания. Ссылка может быть сделана на "Salt selection for basic drugs". Int. J. Pharm. (1986), 33, 201-217.
Изобретение поэтому, в первую очередь, относится к соединению формулы (I) или его соли (в частности, к фармацевтически приемлемой соли).
Изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли в качестве лекарственного средства.
Изобретение далее относится к фармацевтическим композициям, содержащим в качестве активного компонента соединение формулы (I) или его фармацевтически приемлемую соль и, по крайней мере, один фармацевтически инертный наполнитель.
Кроме того, соединение формулы (I) и его фармацевтически приемлемые соли могут быть использованы для получения лекарственного средства и являются подходящими для лечения гипертензии, легочной гипертензии, коронарных болезней, сердечной недостаточности, ишемии почек и миокарда, почечной недостаточности, церебральной ишемии, деменции, мигрени, субарахноидального кровотечения, синдрома Рейнольда, пальцевидной язвы и портальной гипертензии. Эти соединения могут быть также использованы при лечении или профилактике атеросклероза, рестеноза после баллонной или стентовой ангиопластики, воспаления, язвы желудка и двенадцатиперстной кишки, рака, меланомы, рака простаты, гипертрофии простаты, эректильной дисфункции, потери слуха, слепоты, хронического бронхита, астмы, легочного фиброза, грамотрицательной септицемии, шока, серповидно-клеточной анемии, гломерулонефрита, почечной колики, глаукомы, болезней соединительных тканей, при лечении и профилактике диабетических осложнений, осложнений при сосудистой или сердечной хирургии или после трансплантации органов, осложнений, связанных с лечением циклоспорином, боли, гиперлипидемии.
Более предпочтительно, соединение формулы (I) и его фармацевтически приемлемые соли могут быть использованы для получения лекарственного средства, и являются подходящими для лечения болезни, выбранной из группы, включающей гипертензию, легочную гипертензию (включая легочную артериальную гипертензию), диабетическую артериопатию, сердечную недостаточность, эректильную дисфункцию и стенокардию.
Согласно особенно предпочтительному варианту осуществления настоящего изобретения соединение формулы (I) и его фармацевтически приемлемые соли могут быть использованы для получения лекарственного средства и являются подходящими для лечения гипертензии (особенно артериальной гипертензии).
Согласно другому особенно предпочтительному варианту осуществления изобретения соединение формулы (I) и его фармацевтически приемлемые соли могут быть использованы для получения лекарственного средства, и являются подходящими для лечения легочной гипертензии (особенно легочной артериальной гипертензии).
Соединение формулы (I) может быть получено, как описано в WO 02/053557 или как приведено далее в описании (в частности, в примере).
Получение фармацевтических композиций может быть проведено методом, 20 известным любому специалисту из области техники (см., например, Remington, The Science and Practice of Pharmacy, 21st Edition (2005), Part 5, "Pharmaceutical Manufacturing" [published by Lippincott Williams & Wilkins]) посредством внесения описанного соединения формулы (I) или его фармацевтически приемлемых солей, необязательно в комбинации с другими подходящими субстанциями, в лекарственную форму для введения вместе с подходящим, нетоксичным, инертным, терапевтически совместимым твердым или жидким носителем и, при необходимости, обычными фармацевтическими добавками.
Соединение формулы (I) может быть получено в соответствии с настоящим изобретением при использовании методов, описанных далее.
Получение соединения формулы (I)
Аббревиатуры
Аббревиатуры, используемые в данном описании:
Ас - ацетил. Бок - трет-бутоксикарбонил, ДБУ - 1,8-диазабицикло[5.4.0]индец-7-ен, ДХМ - дихлорметан, ДАД - диодный сетчатый детектор, ДХМ - дихлорметан, ДМАП - 4-диметиламинопиридин, ДМФ - N,N-диметилформамид, ДМЭ - 1,2-диметоксиэтан, ЭА - этилацетат, ЭТ - эндотелин, эфир - диэтиловый эфир, ЖХ - жидкостная хроматография, МеОН - метанол, МС - масс-спектроскопия, ЯМР - ядерный магнитный резонанс, ТЭА - триэтиламин, ТГФ - тетрагидрофуран, ТСХ - тонкослойная хроматография, tR - время удерживания.
Общие препаративные методы
Соединение формулы (I) может быть получено в соответствии с общей последовательностью реакций, приведенных ниже, методами, приведенными в примере, или аналогичными методами. Оптимальные реакционные условия могут варьироваться в зависимости от особенностей используемых реагентов или растворителей, при этом такие условия могут быть выбраны специалистом в области техники путем проведения ряда оптимизированных опытов. В данном тексте описано только несколько синтетических возможностей, приводящих к соединению формулы (I).
Полученное таким образом соединение формулы (I), если необходимо, может быть превращено в его соли, а именно в его фармацевтически приемлемые соли, стандартными методами.
Соединение формулы (I) получают из соединения формулы (I)-1
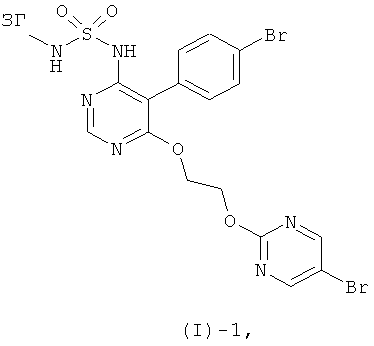
где ЗГ обозначает подходящую защитную группу, посредством отщепления защитной группы ЗГ. Подходящими защитными группами являются, например, бензильная группа, которая может быть отщеплена при помощи BCl3, или BBr3 (например, в растворителе, таком как хлороформ), или 4-метокси- или 2,4-диметоксибензильной группы, которые могут быть удалены посредством окислительного расщепления, например, с помощью аммонийнитрата цезия (например, в растворителе, таком как смесь ацетонитрила с водой) или 2,3-дихлор-5,6-дицианобензохинон (например, в растворителе, таком как ДХМ, 1,2-дихлорпентан, ацетон или толуол, в присутствии или отсутствие воды).
Соединения формулы (1)-1 могут быть получены посредством реакции соединения формулы (I)-2
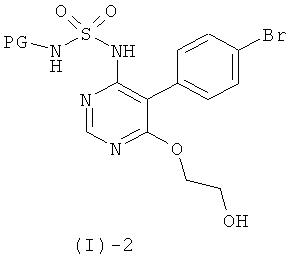
с соединением формулы (I)-3
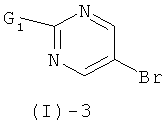
где G1 обозначает реакционную группу, такую как хлор или бром, или метилсульфонильную или этилсульфонильную группу, в присутствии сильного основания, такого как LiH, NaH, СаН2 и им подобных, в растворителе таком, как ТГФ, ДМФ, диоксан и им подобные, или в их смесях. Некоторые соединения формулы (I)-3 являются коммерчески доступными; другие могут быть получены с помощью стандартных методов, известных специалистам из области техники.
Соединения формулы (I)-2 могут быть получены посредством реакции соединения формулы (I)-4
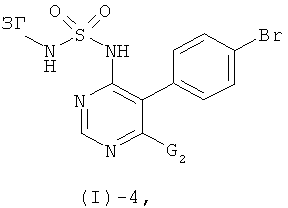
где G2 обозначает реакционную группу такую, как атом галогена, предпочтительно хлор, с этиленгликолем в присутствии основания такого, как трет-бутилат калия, NaH, LiH, и им подобных, в присутствии или отсутствие дополнительного растворителя такого, как 1,2-диметоксиэтан, ТГФ, диоксан (и особенно в присутствии 1,2-диметоксиэтана), предпочтительно при повышенных температурах (например, в интервале от 50 до 100°С, в частности при температурах от 80 до 100°С).
Соединения формулы (I)-4 могут быть получены посредством реакции соединения формулы (I)-5
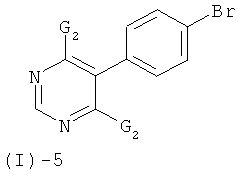
с соединением формулы (I)-6
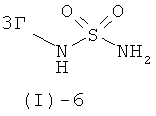
в присутствии основания такого, как трет-бутилат калия, ТЭА, этилдиизопропиламин и им подобные, или, предпочтительно, с солью соединения формулы (I)-6, предпочтительно с калиевой солью, в растворителе таком, как ДМСО, ДМФ, ТГФ и им подобных, или в их смесях, в присутствии или отсутствие дополнительного основания при температурах от 20 до 80°С и предпочтительно от 20 до 40°С.
Соединения формулы (I)-5 получают, например, обработкой соединения формулы (I)-7
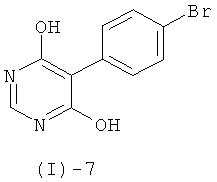
с помощью POCl3, PCl3, PCl5 или их смесей, или POBr3, в присутствии или отсутствие тетраэтиламмонийхлорида, триэтиламина или диметил- или диэтиланилина, и в присутствии или отсутствие дополнительного растворителя такого, как хлороформ, 1,2-дихлорэтан, толуол, ксилол или ацетонитрил, при повышенных температурах (например, в интервале от 60 до 120°С).
Соединение формулы (I)-7 получают посредством реакции соединения формулы (I)-8
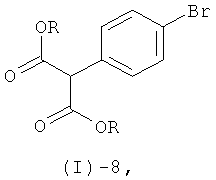
где R обозначает алкильную группу, предпочтительно метильную или этильную группу, с формамидином или его солью, аналогично методам, описанным в литературе (например, A.Gomtsyan et al., J. Med. Chem., (2002), 45, 3639-3648; W. Neidhart et al., Chimia, (1996), 50, 519-524).
Сложный эфир 2-(4-бромфенил)малоновой кислоты формулы (1)-8 может быть получен из коммерчески доступной 4-бромфенилуксусной кислоты по аналогии с приведенными в литературе методиками (например, J.Lee, J.-H.Lee, S.Y.Kim, N.A.Perry, N.E.Lev/in, J.A.Ayres, P.M.Blumberg, Bioorg. Med. n., 14 (2006), 2022-2031).
Сульфамиды формулы (I)-6 могут быть получены трехстадийным методом из хлорсульфонилизоцианата аналогично литературным методам (например, G.Dewynter et al., Tetrahedron, (1993), 49, 65-76; S.Ghassemi, K.Fuchs, Molecular Diversity, (2005), 9, 295-299; J.-Y.Winum et al., Organic Letters, (2001), 3, 2241-2243). На первой стадии хлорсульфонилизоцианат вводят в реакцию с трет-бутанолом и затем, на второй стадии, с соответствующим амином ЗГ-NH2 с получением Бок - защищенного промежуточного соединения формулы (I)-6. На третьей стадии Бок-группу отщепляют в кислых условиях, получая соединение формулы (I)-6. Альтернативно, соединение формулы (I)-6 может быть получено аналогично литературным методам (например, R.Е.Olson, et al., J.Med. Chem., (1999), 42, 1178-1192, и литературе, цитированной в этой публикации), посредством реакции соответствующего промежуточного сульфамоилхлорида формулы (I)-9
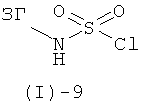
с аммиаком.
Способ по изобретению
Изобретение, следовательно, также относится к способу получения соединения формулы (I) по определению выше, заключающемуся в проведении следующих стадий:
а) реакции соединения формулы (I)-2B
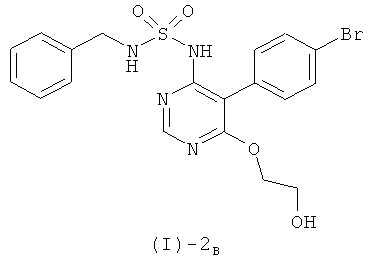
с соединением формулы (I)-3
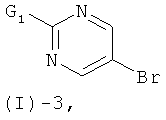
где G1 обозначает атомы хлора или брома или метилсульфонильную, или этилсульфонильную группу (предпочтительно, атом хлора), в присутствии сильного основания; и
б) отщеплении бензильной группы соединения формулы (I)-1B, полученного на стадии а)
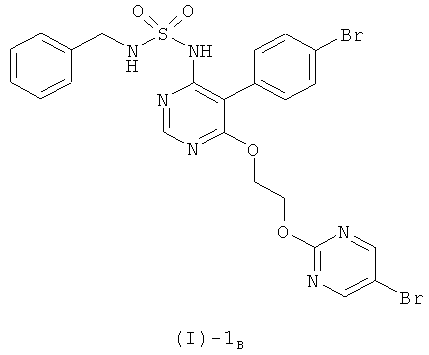
при помощи BCl3 или BBr3.
Предпочтительно, стадию а) приведенного выше процесса проводят в растворителе, выбранном из группы, включающей ТГФ, ДМФ и диоксан, или в смеси растворителей, выбранных из группы, включающей ТГФ, ДМФ и диоксан (например, в смеси ТГФ и ДМФ). Сильное основание для стадии а) описанного выше процесса предпочтительно выбирают из группы, включающей LiH, NaH и СаН2. На стадии а) реакцию предпочтительно проводят при температурах от 20°С до температуры кипения растворителя и, в частности, при температурах от 20°С до 70°С.
Предпочтительно, стадию б) приведенного выше процесса проводят в растворителе, выбранном из группы, включающей хлороформ и ДХМ, или в смеси хлороформа и ДХМ (например, в хлороформе), предпочтительно при температурах от 20°С до 40°С и, в частности, при температурах от 20°С до 30°С.
Согласно предпочтительному варианту проведения описанного выше способа соединение формулы (I)-2B получают из соединения формулы (I)-4, как указано в разделе "Общие препаративные методы" (ЗГ является бензилом), благодаря дополнительной стадии, описанной в этом разделе. Предпочтительно, согласно названному варианту, само соединение формулы (I)-4 получают из соединения формулы (I)-5 и соединения формулы (I)-6 (где ЗГ обозначает бензил), оба из которых описаны в разделе "Общие препаративные методы", благодаря дополнительной стадии, описанной в этом разделе (при этом соединение формулы (I)-5 и соединение формулы (I)-6 могут быть сами по себе получены благодаря дополнительным стадиям, как описано в разделе "Общие препаративные методы").
Описанный выше способ может быть также направлен на получение соли (в частности, фармацевтически приемлемой соли) соединения формулы (I). В этом случае способ включает дополнительную стадию превращения соединения формулы (I), полученного на стадии б), в его соль (в частности, в его фармацевтически приемлемую соль).
Предпочтительные варианты осуществления настоящего изобретения описаны в следующем примере, который более детально иллюстрирует изобретение, никоим образом не ограничивая его объема.
Пример
Соединения следующего примера получены согласно методикам, приведенным ниже. Все соединения охарактеризованы с помощью 1Н-ЯМР (300 МГц) и иногда с помощью 13С-ЯМР (75 МГц) (Varian Oxford, 300 МГц; химические сдвиги приведены в м.д. относительно используемого растворителя; мультиплеты: s=синглет, d=дублет, t=триплет; m=мультиплет), с помощью ЖХ-МС (Finnigan Navigator with HP 1100 Binary Pump и DAD, колонка: 4,6×50 мм, Develosil RP Aqueous, 5 мкм, 120 А, градиент: 5-95% ацетонитрила в воде, 1 мин, с 0,04% трифторуксусной кислоты, истечение: 4,5 мл/мин), tR дано в мин; с помощью ТСХ (ТСХ-пластины от фирмы Merck, силикагель 60 F254); и иногда с помощью температур плавления.
Препаративный метод А: калиевая соль бензилсульфамида:
A.i. Бензилсульфамид:
Хлорсульфонилизоцианат (14,14 г) растворяют в ДХМ (50 мл), охлаждают до температуры 0°С и в течение 30 мин добавляют раствор трет-BuOH (9,6 мл) в ДХМ (50 мл). Перемешивание продолжают дополнительно в течение 30 мин при комнатной температуре. Полученный таким образом раствор затем добавляют при температуре 0°С в течение 1 ч к раствору бензиламина (10,7 г) и ТФК (15,32 мл) в ДХМ (200 мл). Перемешивание продолжают в течение 10 ч при комнатной температуре. Смесь концентрируют в вакууме, переносят в этилацетат (500 мл) и промывают водой (дважды по 40 мл) и рассолом (30 мл), высушивают над MgSO4 и фильтруют. Фильтрат концентрируют в вакууме и сырое вещество кристаллизуют из этилацетата и высушивают в высоком вакууме, получая N-бензил-N′-трет-бутоксикарбонилсульфамид (13,68 г).
1Н ЯМР (CDCl3):(1,46 (s, 9H); 4,25 (s, 2H); 5,42 (s расширенный, 1Н); 7,30-7,40 (m, 5H).
ЖХ-МС:tR=0,90 мин, [M+H]+=287,09.
Это вещество растворяют в диоксане (20 мл) и добавляют к нему 4-молярный раствор HCl в диоксане (120 мл) в течение 1 ч при комнатной температуре. Смесь затем перемешивают в течение 8 ч, после чего растворитель выпаривают и остаток высушивают в высоком вакууме, получая бензилсульфамид в виде грязно-белого порошка (9,47 г).
1Н ЯМР (D6-ДМСО):δ (4,05 (d, J=6,4 Гц, 2H); 6,60 (s, 2H); 7,04 (s, J - 6,4 Гц, 1Н); 7,20-7,36 (m, 5H).
ЖХ-МС: tR=0,60 мин, [M+H+CH3CN]+=228,17.
A.ii. Калиевая соль бензилсульфамида:
К раствору бензилсульфамида (17,98 г) в МеОН (300 мл) осторожно добавляют трет-бутилат калия (10,8 г). Смесь перемешивают при комнатной температуре в течение 15 мин, после чего растворитель выпаривают.Остаток высушивают в высоком вакууме, получая калиевую соль бензилсульфамида в виде грязно-белого порошка (21,73 г).
Препаративный метод Б: 5-(4-бромфенил)-4,6-дихлорпиримидин:
Б.i. Метиловый эфир 4-бромфенилуксусной кислоты:
К раствору 4-бромфенилуксусной кислоты (50 г) в метаноле (250 мл) добавляют по каплям тионилхлорид (34,2 мл), поддерживая при этом температуру реакционной смеси в интервале от 0 до 5°С. После завершения прикапывания охлаждение прекращают и смесь оставляют самопроизвольно нагреваться до комнатной температуры. Затем перемешивание продолжают в течение 75 мин, после чего растворитель удаляют в вакууме. Полученное желтое масло растворяют в бензоле и снова концентрируют. Остаток растворяют в этилацетате, промывают водой, рассолом, 2-нормальным водным раствором Na2CO3 и снова рассолом. Органический экстракт высушивают над MgSO4, фильтруют, концентрируют и высушивают в высоком вакууме при температуре 85°С в течение 30 мин, получая ожидаемый продукт в виде желтого масла (52,4 г).
1Н-ЯМР (D6-ДМСО): δ 3,60 (s, 3Н); 3,67 (s, 2Н); 7,22 (d, 8,5, 2H); 7,50 (d, J -8,5 Гц, 2H).
Б.ii. Диметиловый эфир 2-(4-бромфенил)малоновой кислоты:
При температуре 40°С раствор промежуточного соединения (Б.i) (52 г) в ТГФ (100 мл) осторожно прибавляют в течение 40 мин к суспензии NaH (15,6 г) в сухом ТГФ (450 мл). Перемешивание продолжают в течение 70 мин без нагревания, при этом температура опускается до 27°С. Выделение газа при этом прекращается, после чего к реакционной смеси по каплям прибавляют диметилкарбонат (76,42 мл), поддерживая температуру смеси в пределах от 29 до 31°С. Затем перемешивание продолжают в течение 22 ч при комнатной температуре, смесь охлаждают до температуры -10°С и осторожно нейтрализуют ее до рН 6-7 с помощью водного раствора HCl, после чего ТГФ удаляют в вакууме. Остаток растворяют в этилацетате (700 мл), трижды промывают 1-нормальным водным раствором НС1 и однократно рассолом, а затем высушивают над MgSO4. Большую часть этилацетата выпаривают, после чего добавляют гексан. Полученный продукт кристаллизуется в течение ночи при температуре 4°С. Кристаллы отделяют, промывают гексаном и высушивают, получая ожидаемый продукт в виде бледно-желтых кристаллов (45,9 г).
1Н-ЯМР (D6-ДМСО): 5 3,66 (s, 6H); 5,07 (s, 1H); 7,30-7,34 (m, 2H); 7,55-7,59 (m, 2H).
E.iii. 5-(4-Бромфенил)пиримидин-4,6-диол:
Раствор промежуточного соединения (Б.ii) (11,73 г) в МеОН (100 мл) добавляют при температуре 0°С к раствору натрия (2,83 г) в МеОН (100 мл). Смесь перемешивают в течение 18 ч при комнатной температуре, после чего добавляют гидрохлорид формамидина (4,10 г). Образовавшуюся суспензию перемешивают при комнатной температуре в течение 4 ч. Растворитель удаляют и остаток суспендируют в 10%-ном водном растворе лимонной кислоты (100 мл), а затем перемешивают в течение 10 мин. Белый осадок отделяют, промывают 10%-ным водным раствором лимонной кислоты, водой, трижды выпаривают из циклогексана и высушивают в высоком вакууме при температуре 40°С, получая 5-(4-бромфенил)пиримидин-4,6-диол в виде бледно-бежевого порошка (9,90 г).
1Н-ЯМР(D6-ДМСО):δ 7,43-7,48 (m, 2Н), 7,50-7,55 (m, 2H), 8,13 (s, 1H), 12,1 (s расширенный, 2H).
ЖХ-МС: tR=0,62 мин, [М+Н]+=266,89/268,89 (Br-изотопы).
Б.iv. 5-(4-Бромфенил)-4,6-дихлорпиримидин:
К суспезии 5-(4-бромфенил)пиримидин-4,6-диола (9,90 г) в POCl3 (130 мл) осторожно прибавляют N,N-диметиланилин (13,5 мл). Смесь нагревают затем при температуре 130°С в течение 2 ч. Темно-коричневый раствор концентрируют в вакууме и остаток переносят в смесь воды со льдом. Суспензию затем разбавляют 2-нормальным водным раствором HCl и водой и перемешивают в течение 20 мин. Образовавшийся осадок отделяют и промывают водой. Твердое вещество растворяют в этилацетате, промывают 1-нормальным водным раствором HCl и рассолом. Органическую фазу высушивают над MgSO4 и выпаривают. Полученное вещество затем очищают с помощью колоночной хроматографии на силикагеле, элюируя смесью гексан: ЭА в соотношении от 95:5 до 1:1, затем кристаллизуют из смеси гексан: ЭА при температуре -20°С, получая при этом 4,6-дихлор-5-(4-бромфенил)пиримидин в виде бледно-желтых кристаллов (8,3 г).
1Н-ЯМР(D6-ДМСО):δ 7,39-7,44 (m, 2H), 7,72-7,76 (m, 2H), 8,94 (s, 1H), ЖХ-МС: tR=1,02 мин.
Пример 1: {5-(4-бромфенил)-6-[2-(5-бромпиримидин-2-илокси)этокси]пиримидин-4-ил}сульфамид:
1.i. [6-Хлор-5-(4-бромфенил)пиримидин-4-ил]амид бензилсульфаминовой кислоты
Раствор 5-(4-бромфенил)-4,6-дихлорпиримидина (4,00 г, 13,2 ммоля) и калиевой соли бензилсульфамида (7,38 г, 32,9 ммоля) в ДМСО (30 мл) перемешивают при комнатной температуре в течение 24 ч, после чего разбавляют 10%-ным водным раствором лимонной кислоты (200 мл). Образовавшуюся суспензию фильтруют, отделяют твердую часть и тщательно промывают ее водой и высушивают в высоком вакууме при температуре 40°С в течение 48 ч, получая ожидаемый продукт в виде белого порошка (6,15 г).
1Н ЯМР (CDCl3):δ 4,23 (d, J=5,9 Гц, 2Н); 5,94 (t расширенный, J=6 Гц, 1Н); 7,05 (d, J=8,2 Гц, 2Н); 7,20-7,35 (m, 5H); 7,68 (d, J=8,2 Гц, 2Н); 8,61 (s, 1H).
ЖХ-МС: tR=1,02 мин, [М+Н]+=452,95.
1.ii. [5-(4-Бромфенил)-6-(2-гидроксиэтокси)пиримидин-4-ил]амид бензилсульфаминовой кислоты
трет-Бутилат калия (18,5 г, 164,5 ммоля) прибавляют по частям к суспензии промежуточного соединения (1.i) (7,46 г, 16,4 ммоля) в этиленгликоле (50 мл). Смесь при этом становится теплой и вязкой, после чего ее разбавляют ДМЭ (75 мл). Затем смесь перемешивают при температуре 95°С в течение 24 ч, охлаждают до комнатной температуры, разбавляют водой (50 мл) и 10%-ным водным раствором лимонной кислоты (250 мл). Молочную суспензию экстрагируют этилацетатом (дважды по 300 мл). Объединенные органические экстракты высушивают над MgSO4, фильтруют и фильтрат концентрируют. Оставшееся кристаллическое вещество суспендируют в МеОН, отделяют, тщательно промывают МеОН и высушивают в высоком вакууме, получая ожидаемый продукт в виде белого кристаллического порошка (6,49 г).
1Н ЯМР (CDCl3):δ 2,50 (t расширенный, J=6 Гц, 1H); 3,80-3,88 (m, 2Н); 4,20 (d, J=5,9 Гц, 2Н); 4,46-4,50 (m, 2Н); 5,99 (t расширенный, J=6,4 Гц. 1H); 6,85 (s расширенный, 1H); 7,12 (d, J=8,2 Гц, 2Н); 7,23-7,34 (m, 5H); 7,64 (d, J=8,2 Гц, 2Н); 8,44 (s, 1H).
ЖХ-МС: tR=0,93 мин, [M+H]+=479,08.
1.iii. [5-(4-Бромфенил)-6-{2-(5-бромпиримидин-2-илокси)этокси}пиримидин-4-ил]амид бензилсульфаминовой кислоты
К раствору промежуточного соединения (1.ii) (6,49 г, 13,5 ммоля) в ТГФ (120 мл) осторожно прибавляют NaH (1,77 г, 40,6 ммоля, 55%-ная дисперсия в минеральном масле). Смесь перемешивают в течение 10 мин, после чего добавляют 2-хлор-5-бромпиримидин (3,93 г, 20,3 ммоля). Затем смесь разбавляют ДМФ (15 мл) и перемешивают при комнатной температуре в течение 20 мин, потом смесь нагревают до температуры 60°С и перемешивают в течение 3 ч, после чего снова охлаждают до комнатной температуры. Реакцию останавливают водой и 10%-ным водным раствором лимонной кислоты (250 мл) и далее смесь экстрагируют этилацетатом (дважды по 300 мл). Органические экстракты промывают водой, объединяют, высушивают над MgSO4, фильтруют и растворитель из фильтрата выпаривают. Сырой продукт кристаллизуют из смеси МеОН с эфиром. Кристаллическое вещество отделяют, промывают дополнительным количеством смеси МеОН с эфиром и высушивают в высоком вакууме, получая ожидаемый продукт в виде белого порошка (6,47 г).
1Н ЯМР (CDCl3): δ 4,20 (d, J=6,4 Гц, 2Н); 4,59-4,64 (m, 2H); 4,69-4,74 (m, 2H); 5,98 (t расширенный, J=6,4 Гц, 1Н); 6,83 (s расширенный, 1Н); 7,06-7,10 (m, 2H); 7,24-7,34 (m, 5H); 7,54-7,58 (m, 2H); 8,44 (s, 1Н); 8,50 (s, 2H).
ЖХ-МС: tR=1,06 мин, [М+H]+=634,98.
1.iv. {5-(4-Бромфенил)-6-[2-(5-бромпиримидин-2-илокси)этокси1пиримидин-4-ил} сульфамид:
Раствор трибромида бора (25,5 мл, 1-молярный в ДХМ) медленно прибавляют к раствору промежуточного соединения (1.iii) (6,50 г, 10,2 ммоля) в хлороформе (250 мл). Смесь становится мутной, при этом отделяется масляный слой. Смесь перемешивают при комнатной температуре и прибавляют следующие порции раствора трибромида бора (5 мл) с промежутками в 6, 24 и 33 ч. После добавления последней порции BBr3 образовавшуюся бежевую суспензию энергично перемешивают дополнительно в течение 2 ч, а затем осторожно гасят реакционную смесь добавлением МеОН. Смесь становится слегка теплой и прозрачной, после чего раствор промывают холодной водой (0°С, дважды по 150 мл). Промывные воды экстрагируют ДХМ. Объединенные органические экстракты промывают водой, высушивают над MgSO4, фильтруют и концентрируют. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле, элюируя смесью гептан: ЭА в соотношении 1:1, с последующей кристаллизацией из ДХМ. Очищенный кристаллический продукт высушивают в высоком вакууме при температуре 45°С в течение 48 ч, получая ожидаемый продукт в виде белого кристаллического порошка (1,62 г).
1Н ЯМР (CDCl3): δ 4,60-4,65 (m, 2H), 4,71-4,74 (m, 2H), 5,50 (s расширенный, 2H), 7,10 (s расширенный, 1Н), 7,13-7,17 (m, 2H), 7,55-7,59 (m, 2H), 8,49 (s, 2H), 8,50 (s, 1Н).
ЖХ-МС: tR=0.93 мин, [М+H]+=544,70.
Фармакологические свойства соединения по изобретению
1) Ингибирование связывания эндотелина с мембранами СНО клеток, несущих человеческие ЕТ рецепторы
Экспериментальные методы:
Для исследования конкурентного связывания мембран были использованы СНО клетки, экспрессирующие человеческие рекомбинантные ETA или ETB рецепторы. Были получены микросомальные мембраны из рекомбинантных СНО клеток, после чего анализ связывания был проведен, как было описано ранее (Breu V., et al, FEBS Lett. (1993), 334, 210).
Анализ проводили в 200 мкл 50 мМ Трис/HCl буфере, рН 7,4, включающем 25 мМ MnCl2, 1 мМ ЭДТА и 0,5% БСА в полипропиленовых планшетах для микротитрования. Мембраны, содержащие 0,5 мкг белка, были инкубированы в течение 2 ч при температуре 20°С с 8 пМ [125I]ЕТ-1 (4000 импульсов/мин) и возрастающих концентрациях немаркированных антагонистов. Максимальное и минимальное связывание устанавливалось в образцах без и с наличием 100 нМ ЕТ-1 соответственно. Спустя 2 ч мембраны были отфильтрованы на фильтровальных планшетах, содержащих GF/C фильтры (Unifilterplates от фирмы Canberra Packard S.A.ZÜrich, Switzerland). В каждую ячейку добавляли 50 мкл сцинтилляционного коктейля (MicroScint 20, фирма Canberra Packard S.A.Zurich, Switzerland), и планшеты для фильтрования считывались в микропланшетном счетчике (TopCount, фирма Canberra Packard S.A.ZÜrich, Switzerland).
Все тестируемые соединения растворяли, разбавляли и добавляли в ДМСО. Анализ проводился в присутствии 2,5% ДМСО, который, как было установлено, существенно не влияет на связывание. IC50 вычисляли, как 50%-ную концентрацию антагонистического ингибирования специфического связывания ЕТ-1. Для контрольных соединений были найдены следующие IC50 величины: ETA клетки: 0,075 нМ (н = 8) для ЕТ-1 и 118 нМ (н = 8) для ЕТ-3; ETВ клетки: 0,067 нМ (н = 8) для ЕТ-1 и 0,092 нМ (н = 3) для ЕТ-3.
Результаты:
IC50 величины, полученные для соединения формулы (I), приведены ниже в таблице 1.
[соединение формулы (I)]
2) Ингибирование эндотелин-индуцируемых мышечных сокращений на изолированных аортальных кольцах крыс (ETA рецепторы) и трахеальных кольцах крыс (ETB рецепторы)
Экспериментальные методы:
Функциональная ингибиторная потенция антагонистов эндотелина была проанализирована посредством ингибирования мышечного сокращения, индуцируемого эндотелином-1 на аортальных кольцах крыс (ETA рецерторы), и мышечного сокращения, индуцируемого сарафотоксином S6c на трахеальных кольцах крыс (ETB рецерторы). Взрослые особи Wistar крыс подвергались анастезии и обескровливанию. Торокальная (грудная) аорта или трахея вскрывались, препарировались и разрезались на кольца размером 3-5 мм. Эндотелий/эпителий удаляли посредством мягкого стирания интимальной поверхности. Каждое кольцо суспендировали в 10 мл бане для изолированного органа, наполненной раствором Кребса-Хензелайта (в мМ; NaCl 115, KCl 4,7, MgSO4 1,2, KH2PO4 1,5, NaHCO3 25, CaCl2 2,5, глюкоза 10), поддерживая температуру 37°С, и продуваемую газообразной смесью, состоящей из 95% О2 и 5% СО2. Кольца связывали с силовыми преобразователями, после чего регистрировали изометрическое растяжение (ЕМКА Technologies SA, Paris, France). Затем кольца вытягивались до остаточного растяжения 3 г (аорта) или 2 г (трахея). Совокупные дозы ЕТ-1 (аорта) или сарафотоксин S6c (трахея) были добавлены после 10-минутного инкубирования с тестируемым соединением или его растворителем. Функциональная ингибиторная потенция тестируемого соединения была проанализирована посредством подсчета концентрационного соотношения, то есть сдвигом вправо ЕС50, индуцируемого различными концентрациями тестируемого соединения. ЕС50 представляет собой концентрацию эндотелина, необходимого для получения полумаксимального мышечного сокращения, рА2 представляет собой отрицательный логарифм антагонистической концентрации, которая индуцирует двукратный сдвиг ЕС50 величины.
Результаты:
Величины рА2, полученные для соединения формулы (I) (n = 3), приведены ниже в таблице 2.
 [соединение формулы (I)]
[соединение формулы (I)]
Фармакокинетика после одноразового орального дозирования у крыс
Экспериментальные методы:
Животные, используемые для проведения исследования
Мужские особи крыс Wistar с массой тела 200-250 г были использованы для фармакокинетических экспериментов после ассимиляционного периода, по крайней мере, в течение 7 дней. Все животные содержались в условиях в соответствии с NIH-руководством. За два дня до эксперимента крысы были анестезированы смесью кетамина (90 мг/кг) и ксилазина 2% (10 мг/кг). Катетер был имплантирован в асептических условиях в яремную вену с целью многократного отбора проб крови. После отхода от основной анестезии животные индивидуально содержались в стандартных лабораторных условиях в клетках с проволочными сетчатыми крышами типа-3 Makrolon со стандартизованной мягкой деревянной подстилкой. Животные имели свободный доступ к воде и пище в оздоровительный период и во время проведения эксперимента.
Экспериментальная методика
Фармакокинетические эксперименты осуществлялись на Wistar крысах (m=2-3) после каннюлирования яремной вены для проведения серии отбора образцов крови. Тестируемые соединения вводились орально через зонд в дозах 10 мг/кг. Кровь затем отбиралась в соответствии с заранее установленными временными точками в течение 24 ч, после чего плазму отделяли центрифугированием. Лекарственную концентрацию в плазме количественно определяли с помощью жидкостной хроматографии совместно с масс-спектроскопией (лимит квантификации: 4,6 нг/мл). Фармакокинетический анализ осуществлялся при помощи некомпарментального анализа.
Результаты:
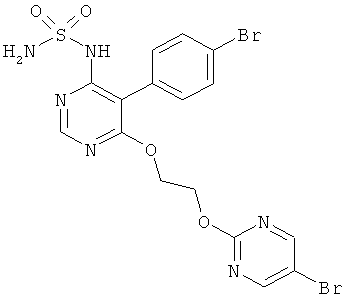
Время полураспада t1/2 и скорость клиренса, измеренные с помощью жидкостной хроматографии у крыс для соединения формулы (I) и для контрольного соединения из WO 02/053557, представлены ниже в таблице 3.
[соединение, раскрытое в WO 02/053557]
[соединение формулы (I)]
Фармакокинетика после введения мультиплетной оральной дозы человеку
Экспериментальные методы:
Это исследование проводилось как двойное-слепое, плацебо-контролируемое, рандомизированное, восходяще дозированное (фаза I) исследование.
Темы, зарегистрированные в исследовании
Многолетний уровень тестируемых доз, выбор 8 здоровых субъектов мужского пола для клинических испытаний после проведенного контроля за их право быть избранными для проведения массового обследования.
Избранные субъекты должны отвечать всем из следующих включенных критериев:
- Мужчины в возрасте от 20 до 50 лет (включительно).
- Здоровье на основе медицинской истории болезни и оценки, сделанной при скрининге.
- Индекс массы тела от 18 до 28 кг/м2.
- Нормальное кровяное давление (КД) и скорость пульса (СП), например, СКД: 100-140 мм Hg, ДКД: 50-90 мм Hg и СП: 45-90 бпм после 10 мин в положении лежа (ограничения включены).
- 12-Канальная ЭКГ без клинических соответствующих отклонений.
- Гематологические, биохимические и урологические тесты без отклонений от нормальных, принятых клинических пределов.
- Отрицательные результаты при лекарственном скрининге (кокаин, марихуана, опиаты, бензодиазепины, барбитураты, трициклические антидепрессанты, метадон и амфетамины).
- Способность к общению с исследователем на местном языке и к пониманию и исполнению требований исследования.
У выбранных субъектов не должен быть зарегистрирован ни один из следующих исключительных критериев:
- В течение трехлетнего периода, предшествующего сканированию, историческое или клиническое доказательство алкоголизма или лекарственной зависимости.
- В течение трехлетнего периода, предшествующего сканированию, историческое или клиническое доказательство любой болезни и/или наличия любого хирургического или медицинского условия, которое может препятствовать абсорбции, распределению, метаболизму или выделению изучаемого лекарства (например, печеночная или почечная недостаточность, сахарный диабет, сердечно-сосудистые нарушения, болезни поджелудочной железы, хронические симптомы ясно выраженных запора или диарреи или другие острые симптомы, связанные с желудочно-кишечным трактом, за исключением допускаемых аппендеэктомии или герниотомии).
- Наличие гепатитов В или С и/или положительных результатов, полученных из серологии гепатита, которые указывают на острый или хронический гепатит В или С (искючая вакцинированных субъектов).
- Положительные результаты на ВИЧ серологию.
- Курение.
- История клинической релевантной гиперчувствительности или серьезные неблагоприятные реакции на любое лекарство.
- Участие в другом клиническом исследовании в течение трехмесячного периода, предшествующего скрининговым испытаниям.
- Предварительное или сопутствующее лечение любым лекарственным препаратом (выписанным по рецепту или принимаемому без рецепта) за две недели до первого приема лекарства.
- Потеря 250 мл или более крови в течение трех месяцев перед испытанием.
- Симптомы клинического релевантного заболевания в течение 4-недельного периода, предшествующего скринингу (например, острая бактериальная, вирусная или грибковая инфекция).
Сопутствующее медикаментозное лечение, диетические аспекты, спирт, курение, физическая активность.
Никакое сопутствующее лечение не разрешается, за исключением лечения при ухудшающемся состоянии.
Субъекты голодают от 10 до 4 час после введения лекарства на 1 и 10 день проведения исследования. Во время проведения исследования субъекты получают следующую стандартизованную пищу:
- завтрак (1 раз в день) во 2-11 дни (никакого завтрака в 1 и 10 дни);
- ланч (1 раз в день) в 1 и 10 дни около полудня или приблизительно спустя 4 часа после принятия лекарства;
- легкая закуска (1 раз в день) в 1 и 10 дни приблизительно через 8 часов после введения лекарства и
- обед (1 раз в день) в 1 и 10 дни приблизительно около 19.00 ч или приблизительно через 11 часов после введения лекарства.
Субъекты в группах с различным дозированием получают одинаковую еду в соответствующие проведению эксперимента дни. Пища в 1 и 10 дни одинаковая. Потребление воды - по желанию.
От начала скрининга до конца проведения исследования, субъекты должны воздерживаться от усиленных физических упражнений и напряженных спортивных занятий (спортивные нагрузки) и не употреблять любые алкогольсодержащие напитки, грейпфруты или грейпфрутовый сок. Потребление ксантинсодержащих напитков не разрешается в клинике.
Экспериментальная методика
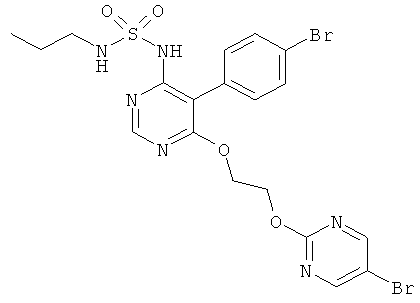
[5-(4-Бромфенил)-6-[2-(5-бромпиримидин-2-илокси)этокси]пиримидин-4-ил]амид пропилсульфаминовой кислоты (см. WO 02/053557 или WO 2007/031933; называемый в дальнейшем «Контрольное соединение») является пригодным в виде свободного основания для клинического испытания в твердых желатиновых капсулах для орального введения с дозировкой, составляющей 1 мг и 10 мг. Сопоставимые плацебо капсулы содержат те же самые вспомогательные вещества без «контрольного соединения».
Контрольное соединение вводят в виде восходящих мультиплетных доз в количестве 1, 3, 10 и 30 мг (соответственно в виде 1 капсулы по 1 мг, 3 капсул по 1 мг, 1 капсулы по 10 мг и 3 капсул по 10 мг). Субъекты подвергаются лечению в течение 10 дней. Присвоение номера и кода для идентификации субъекта основано на обязательстве анонимности. Только номер субъекта и дата рождения идентифицируют субъектов.
Это исследование осуществляется двойным-слепым методом. 6 субъектов рандомизируют по уровню дозы для лечения «контрольным соединением» и 2 субъекта лечатся путем введения плацебо.
Субъекты принимают лекарство, находясь в положении стоя, запивая его 150 мл воды, утром, после чего они голодают, по крайней мере, в течение 10 часов в 1 и 10 дни. В другие дни проведения исследования лекарство вводится за 30 мин до завтрака. Для каждого субъекта интервал между каждым приемом лекарства составляет 24±0,5 часа, но прием лекарства всегда проводится между 7.00 ч и 9.00 ч.
Все введения лекарств осуществляются под прямым медицинским наблюдением. Проверка ротовой полости проводится немедленно после каждого приема лекарства. Измерение в плазме уровня контрольного соединения и/или его метаболита, например, соединения формулы (I), во время аналитической фазы служит в качестве дальнейшего контроля соблюдения режима и схемы лечения.
Концентрация контрольного соединения и его метаболита, например, соединения формулы (I), в плазме и моче определяют с помощью ЖХ-МС. Предельное значение количественного анализа установлено на уровне 1 нг/мл для обоих анализов.
Результаты
Видимое элиминирование полураспада (t1/2), измеряемое у человека на 10-й день исследования, для соединения формулы (I) и для контрольного соединения в отношении каждого из различных доз суточного введения (1, 3, 10 и 30 мг соответственно) представлено ниже в таблице 4 (данные найдены геометрическим способом).
Краткое изложение фармакологических свойств соединения по изобретению
Как видно из полученных результатов, соединение формулы (I) является антагонистом рецептора эндотелина (см. "Ингибирование связывания эндотелина с мембранами из СНО клеток, несущих ЕТ рецерторы", таблица 1, и "Ингибирование эндотелин-индуцируемых мышечных сокращений на изолированных аортальных кольцах крыс (ETA рецепторы) и трахеальных кольцах крыс (ETB рецепторы), таблица 2), которое имеет значительно более высокий период полураспада (у крыс и людей) и значительно более низкий коэффициент выведения (у крыс), чем контрольное соединение WO 02/053557 (см. "Фармакокинетика после одноразового орального дозирования у крыс", таблица 3, и "Фармакокинетика после введения мультиплетной оральной дозы человеку", таблица 4).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИМИДИНСУЛЬФАМИДЫ И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ЭНДОТЕЛИАЛЬНЫХ РЕЦЕПТОРОВ | 2003 |
|
RU2329255C2 |
| БОРСОДЕРЖАЩИЕ ДИАЦИЛГИДРАЗИНЫ | 2014 |
|
RU2637946C2 |
| Замещенные производные бисфенилового эфира масляной кислоты в качестве ингибиторов NEP | 2019 |
|
RU2784522C2 |
| СТИМУЛЯТОРЫ sGC | 2011 |
|
RU2582679C2 |
| ФЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИММУНОМОДУЛЯТОРОВ | 2007 |
|
RU2442780C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 1,2,3,4-ТЕТРАГИДРОИЗОХИНОЛИНА | 2005 |
|
RU2378257C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНМОЧЕВИНЫ | 2014 |
|
RU2666894C2 |
| ГИДРОКСИПИРИДОКСАЗЕПИНЫ В КАЧЕСТВЕ АКТИВАТОРОВ NRF2 | 2020 |
|
RU2812931C2 |
| ПРОИЗВОДНЫЕ КАРБАМИНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1999 |
|
RU2179969C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ТИОФЕНА | 2006 |
|
RU2420523C2 |
Изобретение относится к соединению формулы (I)
, или к его соли. Изобретение также относится к способу получения указанного соединения, фармацевтической композиции, предназначенной для лечения болезней, обусловленных повышенным сжатием сосудов, пролиферацией или воспалением, вызванных эндотелином, на основе указанного соединения. Технический результат - получены новое соединение и его соли, которые могут найти применение в медицине для лечения гипертензии, легочной гипертензии, диабетической артериопатии, сердечной недостаточности, эректильной дисфункции и стенокардии. 4 н. и 10 з.п. ф-лы, 4 табл., 1 пр.
1. Соединение формулы (I)
,
или его соль.
2. Соединение формулы (I) по п.1 или его фармацевтически приемлемая соль в качестве лекарственного средства для лечения болезней, обусловленных повышенным сжатием сосудов, пролиферацией или воспалением, вызванных эндотелином.
3. Фармацевтическая композиция для лечения болезней, обусловленных повышенным сжатием сосудов, пролиферацией или воспалением, вызванных эндотелином, содержащая в качестве активного компонента соединение формулы (I) по п.1 или его фармацевтически приемлемую соль и по крайней мере одно терапевтически инертное вспомогательное вещество.
4. Применение соединения формулы (I) по п.1 или его фармацевтически приемлемой соли для получения лекарственного средства, предназначенного для лечения болезней, обусловленных повышенным сжатием сосудов, пролиферацией или воспалением, вызванных эндотелином.
5. Применение по п.4, где полученное лекарственное средство предназначено для лечения болезни, выбранной из группы, включающей гипертензию, легочную гипертензию, диабетическую артериопатию, сердечную недостаточность, эректильную дисфункцию и стенокардию.
6. Применение по п.5, где полученное лекарственное средство предназначено для лечения гипертензии.
7. Применение по п.5, где полученное лекарственное средство предназначено для лечения легочной гипертензии.
8. Применение по п.7, где полученное лекарственное средство предназначено для лечения легочной артериальной гипертензии.
9. Соединение формулы (I) по п.1 или его фармацевтически приемлемая соль для лечения болезней, обусловленных повышенным сжатием сосудов, пролиферацией или воспалением, вызванных эндотелином.
10. Соединение или фармацевтически приемлемая соль по п.9 для лечения болезни, выбранной из группы, включающей гипертензию, легочную гипертензию, диабетическую артериопатию, сердечную недостаточность, эректильную дисфункцию и стенокардию.
11. Соединение или фармацевтически приемлемая соль по п.9 для лечения гипертензии.
12. Соединение или фармацевтически приемлемая соль по п.9 для лечения легочной гипертензии.
13. Соединение или фармацевтически приемлемая соль по п.9 для лечения легочной артериальной гипертензии.
14. Способ получения соединения формулы (I) по п.1, включающий следующие стадии:
а) взаимодействия в растворителе, выбранном из группы, включающей тетрагидрофуран, N,N-диметилформамид и диоксан, или в смеси растворителей, выбранных из группы, включающей тетрагидрофуран, N,N-диметилформамид и диоксан, при температурах от 20°С до температуры кипения растворителя, соединения формулы (I)-2В
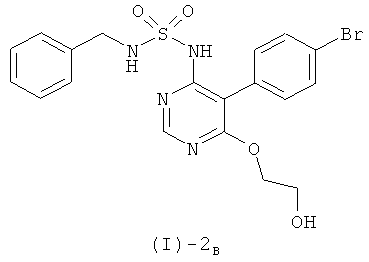
с соединением формулы (I)-3
где G1 обозначает атом хлора или брома, или метилсульфонильную или этилсульфонильную группу, в присутствии сильного основания, выбранного из группы, включающей LiH, NaH и СаН2; и
б) отщепления бензильной группы от соединения формулы (I)-1B, полученного на стадии (а),
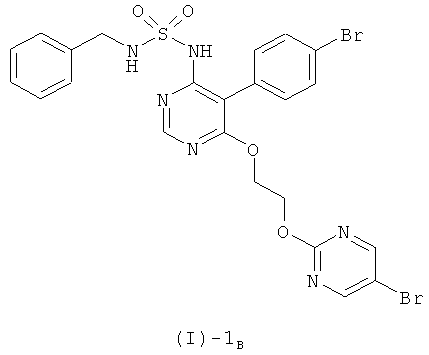
при температурах от 20°С до 40°С в растворителе, выбранном из группы, включающей хлороформ и дихлорметан, или в смеси хлороформа и дихлорметана, с использованием BCl3 или BBr3.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| RU 2005120769 A, 27.02.2006. | |||

