Настоящее изобретение относится к нуклеиновой кислоте общей формулы (I): GlXmGn или (II): ClXmCn, необязательно модифицированной липидом, которую предпочтительно применяют как таковую в качестве иммуностимулятора или в другом варианте в сочетании с другими биологически активными агентами, при этом иммуностимулятор, предлагаемый в изобретении, выполняет в композиции функцию адъюванта, который необязательно можно объединять с другими адъювантами. Таким образом, изобретение относится также к фармацевтической композиции или вакцине, каждая из которых содержит нуклеиновые кислоты формул (I) и/или (II) в качестве иммуностимулятора. Если фармацевтическая композиция содержит иммуностимулятор, предлагаемый в изобретении, в качестве адъюванта, то фармацевтическая композиция содержит по меньшей мере один дополнительное фармацевтически активный компонент, например, антигенный агент. Фармацевтическая композиция, предлагаемая в изобретении, как правило, может содержать фармацевтически приемлемый носитель и необязательно дополнительные вспомогательные вещества, добавки и/или дополнительные адъюванты. Настоящее изобретение относится также к применению фармацевтической композиции или вакцины, предлагаемой в изобретении, для лечения инфекционных болезней, раковых заболеваний, аллергий и аутоиммунных заболеваний. Настоящее изобретение относится также к применению иммуностимулирующего адъюванта, предлагаемого в изобретении, для приготовления фармацевтической композиции, предназначенной для лечения раковых заболеваний, инфекционных болезней, аллергий и аутоиммунных заболеваний.
При применении как обычных, так и генетических вакцин, часто возникает проблема, связанная с тем, что в обработанном или инокулированном организме часто возникает лишь невысокий и поэтому часто недостаточный иммунный ответ. По этой причине в вакцины или к фармацевтически активным компонентам часто добавляют так называемые адъюванты, представляющие собой субстанции или композиции, которые обладают способностью повышать и/или влиять целенаправленным образом на иммунный ответ, например на антиген. Например, известно, что эффективность некоторых применяемых для инъекций медицинских действующих веществ можно значительно повышать путем объединения действующего вещества с адъювантом, который обладает способностью высвобождать действующее вещество в клеточной системе хозяина и необязательно влиять на его поглощение клетками хозяина. Таким путем можно достигать действия, сопоставимого с периодическим введением нескольких небольших доз с регулярными интервалами. Понятие «адъювант» в контексте настоящего описания обычно относится к соединению или композиции, которое(ая) служит в качестве связующего вещества, носителя или вспомогательной субстанции для иммуногенов и/или других фармацевтических действующих веществ.
В данной области предложен в качестве адъювантов целый ряд соединений и композиций, например, адъювант Фрейнда, оксиды металлов (гидроксиды алюминия и т.д.), квасцы, неорганические хелаты или их соли, различные масла типа парафина, синтетические смолы, альгинаты, мукоиды, полисахариды, казеинаты, а также соединения, выделенные из крови и/или сгустков крови, такие, например, как фибриновые производные и т.д. Однако указанные адъюванты во многих случаях вызывают нежелательные побочные действия, например, раздражение и воспаление кожи в области введения. Кроме того, могут иметь место случаи токсических побочных действий, в частности некроз тканей. И, наконец, известные адъюванты в большинстве случаев вызывают неполную стимуляцию клеточного иммунного ответа, поскольку активируют только В-клетки.
Например, известно, что квасцы, оксиды металлов и хелаты солей ассоциированы с образованием стерильных абсцессов. Кроме того, у ученых существуют сомнения в том, что такие соединения полностью выводятся из организма. Скорее они приводят к образованию нежелательных неорганических остатков в организме. Хотя такие соединения, как правило, обладают низкой токсичностью, они могут в результате фагоцитоза проникать в клетки эндоплазматического ретикулума (литоральные и синусоидальные клетки печени и селезенки) в качестве части нежелательного дебриса. Кроме того, есть данные о том, что указанный дебрис может оказывать вредное воздействие на различные механизмы фильтрации в организме, например, в почках, печени или селезенке. Поэтому такие остатки представляют собой скрытый постоянный источник риска в организме, и, как правило, источник риска для иммунной системы.
Синтетические масла и продукты переработки нефти, применяемые в качестве адъювантов в известных прототипах, также обладают побочными действиями. Однако эти соединения являются нежелательными, в частности из-за их быстрого метаболизма в организме и расщепления на ароматические углеводороды. Однако известно, что такие ароматические углеводороды могут обладать очень высоким канцерогенным действием и/или могут вызывать непоправимое повреждение ДНК другими путями, например, в результате интеркаляции в ДНК. Кроме того, продемонстрировано, что указанные соединения связаны также с образованием стерильных абсцессов и редко могут быть удалены полностью из организма.
Соединения, выделенные из животных, например, желатин, также часто являются нежелательными в качестве адъювантов, применяемых для иммунной стимуляции. Хотя указанные соединения, как правило, не обладают деструктивным действием на организм-хозяин или представляющие интерес клетки-хозяева, они обычно слишком быстро мигрируют от места инъекции в организм-хозяин или в клетки-хозяева, в результате чего редко достигаются требования, обычно применяемые к адъюванту, такие, например, как замедленное высвобождение действующего вещества, необязательно инъецируемого вместе с адъювантом, и т.д. Такое быстрое распределение может в некоторых случаях противодействовать танинам или другим (неорганическим) соединениям. Однако метаболизм указанных дополнительных соединений и их приблизительное местонахождение в организме пока не полностью изучены. Однако в этом случае также можно предположить, что эти соединения накаливаются в дебрисе и поэтому могут оказывать воздействие на механизмы фильтрации, например, клеток почки, печени и/или селезенки. Кроме того, желатин, набухающий при парентеральном введении, может приводить в условиях in vivo к нежелательным побочным действиям, таким, например, как опухание, прежде всего в месте введения, и болезненное ощущение.
Соединения, выделенные из крови и/или сгустков крови, такие, например, как фибриновые производные и т.д., как правило, обладают иммуностимулирующими действиями. Однако большинство этих соединений, когда они присутствуют в качестве адъювантов, являются неприемлемыми из-за их побочных действий на иммунную систему (которые имеют место наряду с их требуемыми иммуногенными свойствами). Например, многие такие соединения рассматриваются как аллергенные и в некоторых обстоятельствах приводят к избыточной реакции иммунной системы, существенно превышающей требуемый уровень. Таким образом, эти соединения также являются непригодными в качестве адъювантов для иммунной стимуляции по вышеуказанным причинам.
Таким образом, первым объектом настоящего изобретения являются иммуностимуляторы, которые действуют в качестве адъювантов, при их введении в сочетании с другими биологическим активными соединениями, в частности, при их введении с иммуномодуляторами, предпочтительно в сочетании с соединениями, которые специфически стимулируют иммунную систему, такими как антигены.
Однако (неспецифические) иммуностимулирующие действия можно получать также путем непосредственного применения нуклеиновых кислот для инициации неспецифического иммунного ответа (врожденный иммунный ответ). Последовательности бактериальных CpG-ДНК не только служат в качестве носителей генетической информации. Например, известно, что ДНК играет основную роль в производстве неспецифических иммунных ответов. Бактериальная ДНК, например, действует в качестве сигнала «опасности», предназначенного для изменения иммунных клеток, таких как макрофаги и дендритные клетки, для усиления защитного Th1-поляризованного (специфического) T-клеточного иммунного ответа. Иммуностимулирующее действие, вероятно, обусловлено присутствием неметилированных CG-мотивов, и поэтому такие CpG-ДНК, предложено применять как таковые в качестве иммуностимуляторов (см.US 5663153). CpG-ДНК непосредственно вызывает активацию компонентов врожденный иммунной системы, приводят к повышающей регуляции костимулирующих молекул и провоспалительных цитокинов. Для достижения указанных иммуностимулирующих свойств ДНК можно применять также олигонуклеотиды ДНК, стабилизированные путем фосфоротиоатной модификации (US 6239116). И, наконец, в US 6406705 описаны иммуностимулирующие композиции, которые содержат синергетическую комбинацию олигодекосирибонуклеотида CpG и соединения, не относящегося к нуклеиновым кислотам, для усиления стимулирующего действия на врожденную иммунную систему.
Однако применение ДНК для усиления неспецифического иммунного ответа может быть менее предпочтительным с нескольких позиций. Расщепление ДНК происходит относительно медленно in vivo, поэтому, когда применяют иммуностимулирующую (чужеродную) ДНК, то может происходить образование антител к ДНК, что было подтверждено при создании моделей на животных, таких как мыши (Gilkeson и др., J. Clin. Invest. 95, 1995, cc.1398-1402). Персистентность (чужеродной) ДНК в организме может в результате приводить к сверх активации иммунной системы, что, как известно, вызывает у мышей спленомегалию (Montheith и др., Anticancer Drug Res., 12(5), 1997, cc. 421-432). Кроме того (чужеродная) ДНК может взаимодействовать с геномом хозяина и вызывать мутации, в частности при интеграции в геном хозяина. Например, может иметь место инсерция интродуцированной (чужеродной) ДНК в интактный ген, что представляет собой мутацию, которая может мешать или даже полностью элиминировать функцию эндогенного гена. В результате случаев такой интеграции, с одной стороны, может происходить нарушение ферментных систем, которые являются жизненно важными для клетки, а, с другой стороны, существует риск того, что измененная таким образом клетка может быть трансформирована в состояние дегенерации, если при интеграции (чужеродной) ДНК изменен ген, имеющий решающее значение для регуляции клеточного роста. Таким образом, в известных к настоящему времени процессах нельзя исключать возможный риск образования рака при применении (чужеродной) ДНК в качестве иммуностимулятора.
Таким образом, существенно более целесообразным является применение специфических молекул РНК в качестве соединения, предназначенного для вызывания неспецифического иммунного ответа с помощью врожденной иммунной системы. Известно, что олигонуклеотиды РНК связываются с рецепторами TLR-7/-8, тем самым, усиливая иммуностимулирующее действие. РНК, применяемая в качестве иммуностимулятора, как правило, имеет существенно более короткое время полужизни in vivo, чем ДНК. Несмотря на это, применение даже указанных специфических молекул РНК, известных в данной области в качестве иммуностимуляторов, имеет ограничение. Например, известные к настоящему времени в данной области специфические последовательности РНК обладают лишь ограниченной способностью проникать в клетки in vivo. В результате для иммунной стимуляции могут требоваться увеличенные количества РНК, что, даже без учета повышенной стоимости, связанной с необходимостью вводить повышенные количества РНК, включают риск значительного усиления описанных в целом выше нежелательных побочных действий, например, раздражение и воспаление в области введения. Кроме того, нельзя исключать токсические побочные действия при введении больших количеств иммуностимулятора.
Несмотря на продемонстрированные к настоящему времени успехи, сохраняется потребность и значительный интерес к улучшенным иммуностимуляторам, которые могут усиливать иммунный ответ врожденной иммунной систему пациента, благодаря иммунному ответу на них самих. Таким образом, вторым объектом изобретения являются иммуностимуляторы, которые усиливают неспецифический иммунный ответ путем активации врожденной иммунной системы пациента.
В качестве обоих объектов настоящего изобретения предложены молекулы нуклеиновых кислот следующих формул (I) и (II). Эти предлагаемые в изобретении молекулы нуклеиновых кислот активируют врожденную иммунную систему, вызывая тем самым неспецифический иммунный ответ, и действуют в качестве адъювантов (например, компонентов вакцины), которые поддерживают иммуностимулирующую активность второго соединения, специфически активирующего приобретенную иммунную систему.
Настоящее изобретение относится к нуклеиновой кислоте формулы (I):
GlXmGn,
в которой:
G обозначает гуанозин, урацил или аналог гуанозина или урацила;
Х обозначает гуанозин, урацил, аденозин, тимидин, цитозин или аналог указанных выше нуклеотидов;
l обозначает целое число от 1 до 40,
где, если l обозначает 1, то G обозначает гуанозин или его аналог,
если l>1, то по меньшей мере 50% нуклеотидов представляют собой гуанозин или его аналог;
m обозначает целое число, и оно равно по меньшей мере 3;
где, если m равно 3, то Х обозначает урацил или его аналог,
если m>3, то встречаются по меньшей мере 3 последовательных остатка урацила или аналога урацила;
n обозначает целое число от 1 до 40,
где, если n равно 1, то G обозначает гуанозин или его аналог,
если n>1, то по меньшей мере 50% нуклеотидов представляют собой гуанозин или его аналог.
Кроме того, настоящее изобретение относится к нуклеиновой кислоте формулы (II):
ClXmCn
в которой:
С обозначает цитозин, урацил или аналог цитозина или урацила;
Х обозначает гуанозин, урацил, аденозин, тимидин, цитозин или аналог указанных выше нуклеотидов;
l обозначает целое число от 1 до 40,
где, если l обозначает 1, то С обозначает цитозин или его аналог,
если l>1, то по меньшей мере 50% нуклеотидов представляют собой цитозин или его аналог;
m обозначает целое число, и оно равно по меньшей мере 3;
где, если m равно 3, то Х обозначает урацил или его аналог,
если m>3, то встречаются по меньшей мере 3 последовательных остатка урацила или аналога урацила;
n обозначает целое число от 1 до 40,
где, если n равно 1, то С обозначает цитозин или его аналог,
если n>1, то по меньшей мере 50% нуклеотидов представляют собой цитозин или его аналог.
Нуклеиновые кислоты формулы (I) или (II), предлагаемые в изобретении, как правило, представляют собой сравнительно короткие молекулы нуклеиновой кислоты. Так, нуклеиновая кислота либо формулы (I), либо формулы (II), предлагаемая в изобретении, как правило, включает примерно от 5 до 100 (но в конкретных вариантах осуществления изобретения она может состоять также из более чем 100 нуклеотидов, например, может включать вплоть до 200 нуклеотидов), от 5 до 90 или от 5 до 80 нуклеотидов, предпочтительно от 5 до 70, более предпочтительно примерно от 8 до 60, и более предпочтительно примерно от 15 до 60 нуклеотидов, более предпочтительно от 20 до 60, наиболее предпочтительно от 30 до 60 нуклеотидов. Если нуклеиновая кислота, предлагаемая в изобретении, включает, например, максимум 100 нуклеотидов, то, как правило, m должно быть ≤98.
Нуклеиновая кислота либо формулы (I), либо формулы (II), предлагаемая в изобретении, может представлять собой РНК или ДНК (например, кДНК), она может быть одноцепочечной или двухцепочечной, иметь форму гомо- или гетеродуплекса и быть линейной или кольцевой. Наиболее предпочтительно нуклеиновая кислота либо формулы (I), либо формулы (II), предлагаемая в изобретении, находится в форме одноцепочечной РНК.
G в нуклеиновой кислоте формулы (I), предлагаемой в изобретении, обозначает гуанозин или урацил, или его аналог. В этой связи аналоги гуанозинового или урацилового нуклеотида определяют как не встречающиеся в естественных условиях варианты встречающихся в естественных условиях нуклеотидов. В соответствии с этим аналоги гуанозина или урацила представляют собой дериватизированные химическим путем нуклеотиды, несущие не встречающиеся в нативных нуклеотидах функциональные группы, которые предпочтительно добавлены или удалены путем делении из встречающегося в естественных условиях гуанозинового или урацилового нуклеотида, или которыми заменены встречающиеся в естественных условиях функциональные группы гуанозинового или урацилового нуклеотида. Следовательно, можно модифицировать каждый компонент встречающегося в естественных условиях гуанозинового или урацилового нуклеотида, а именно, компонент, представляющий собой основание, сахарный (рибозный) компонент и/или фосфатный компонент, формирующие остов олигонуклеотида. Фосфатные фрагменты можно заменять, например, фосфорамидатами, фосфоротиоатами, пептидными нуклеотидами, метилфосфонатами и т.д.
Таким образом, аналоги гуанозина или урацила включают, но, не ограничиваясь только ими, любой встречающийся в естественных условиях или не встречающийся в естественных условиях гуанозин или урацил, который изменен химическим путем, например, путем ацетилирования, метилирования, гидроксилирования и т.д., включая, например, 1-метилгуанозин, 2-метилгуанозин, 2,2-диметилгуанозин, 7-метилгуанозин, дигидроурацил, 4-тиоурацил, 5-карбоксиметиламинометил-2-тиоурацил, 5-(карбоксигидроксилметил)урацил, 5-фторурацил, 5-бромурацил, 5-карбоксиметиламинометилурацил, 5-метил-2-тиоурацил, метиловый эфир N-урацил-5-оксиуксусной кислоты, 5-метиламинометилурацил, 5- метоксиаминометил-2-тиоурацил, 5'-метоксикарбонилметилурацил, 5-метоксиурацил, метиловый эфир урацил-5-оксиуксусной кислоты, урацил-5-оксиуксусную кислоту (v). Получение таких аналогов известно специалисту в данной области, оно описано, например, в US 4373071, US 4401796, US 4415732, US 4458066, US 4500707, US 4668777, US 4973679, US 5047524, US 5132418, US 5153319, US 5262530 и 5700642, содержание которых полностью включено в настоящее описание в качестве ссылки. В случае применения указанных выше аналогов предпочтительными согласно изобретению являются те аналоги, которые повышают иммуногенность нуклеиновой кислоты формулы (I), предлагаемой в изобретении, и/или не взаимодействуют с другими интродуцированными модификациями. По меньшей мере один аналог может присутствовать во фланкирующей последовательности Gl и/или Gn, необязательно по меньшей мере 10, 20, 30, 50, 60, 70, 80, 90% нуклеотидов во фланкирующих последовательностях Gl и/или Gn обладают свойствами указанных в настоящем описании аналогов, если фланкирующая последовательность вообще содержит по меньшей мере один аналог. Наиболее предпочтительно все нуклеотиды во фланкирующей последовательности являются аналогами, которые могут (что является наиболее предпочтительным) быть идентичными аналогами нуклеотидов одного и того же типа (например, все гуанозиновые нуклеотиды присутствуют в виде 1-метилгуанозина) или они могут быть различными (например, по меньшей мере два различных гуанозиновых аналога присутствуют вместо встречающегося в естественных условиях гуанозинового нуклеотида). Предпочтительно l и n представляют собой целые числа от 1 до 20, более предпочтительно от 1 до 10 и еще более предпочтительно от 2 до 8.
Количество нуклеотидов G в нуклеиновой кислоте формулы (I), предлагаемой в изобретении, определяется величиной l или n. l и n каждый независимо друг от друга представляют собой целые числа от 1 до 40, причем, если l или n равно 1, то G представляет собой гуанозин или его аналог, и если l или n>1, то по меньшей мере 50% нуклеотидов представляют собой гуанозин или его аналог. Например, если 1 или n=4, то Gl или Gn может представлять собой, но, не ограничиваясь только ими, например, GUGU, GGUU, UGUG, UUGG, GUUG, GGGU, GGUG, GUGG, UGGG или GGGG и т.д.; если l или n равно 5, то Gl или Gn могут представлять собой, например, GGGUU, GGUGU, GUGGU, UGGGU, UGGUG, UGUGG, UUGGG, GUGUG, GGGGU, GGGUG, GGUGG, GUGGG, UGGGG или GGGGG и т.д. Нуклеотид, смежный с Xm в нуклеиновой кислоте формулы (I), предлагаемой в изобретении, предпочтительно не представляет собой урацил.
С в нуклеиновой кислоте формулы (II), предлагаемой в изобретении, обозначает цитозин или урацил, или его аналог. В этой связи аналоги цитозинового или урацилового нуклеотида определяют как не встречающиеся в естественных условиях варианты встречающихся в естественных условиях цитозиновых или урациловых нуклеотидов. В соответствии с этим аналоги цитозина или урацила представляют собой дериватизированные химическим путем нуклеотиды, несущие не встречающиеся в нативных нуклеотидах функциональные группы, которые предпочтительно добавлены или удалены путем делеции из встречающегося в естественных условиях цитозинового или урацилового нуклеотида, или которыми заменены встречающиеся в естественных условиях функциональные группы цитозинового или урацилового нуклеотида. Следовательно, можно модифицировать каждый компонент встречающегося в естественных условиях цитозинового или урацилового нуклеотида, а именно, компонент, представляющий собой основание, сахарный (рибозный) компонент и/или фосфатный компонент, формирующий остов олигонуклеотида. Фосфатные фрагменты можно заменять, например, фосфорамидатами, фосфоротиоатами, пептидными нуклеотидами, метилфосфонатами и т.д.
Таким образом, аналоги цитозина или урацила включают, но, не ограничиваясь только ими, любой встречающийся в естественных условиях или не встречающийся в естественных условиях цитозин или урацил, который изменен химическим путем, например, путем ацетилирования, метилирования, гидроксилирования и т.д., включая, например, 2-тиоцитозин, 3-метилцитозин, 4-ацетилцитозин, дигидроурацил, 4-тиоурацил, 5-карбоксиметиламинометил-2-тиоурацил, 5-(карбоксигидроксилметил)урацил, 5-фторурацил, 5-бромурацил, 5-карбоксиметиламинометилурацил, 5-метил-2-тиоурацил, метиловый эфир N-урацил-5-оксиуксусной кислоты, 5-метиламинометилурацил, 5-метоксиаминометил-2-тиоурацил, 5'-метоксикарбонилметилурацил, 5- метоксиурацил, метиловый эфир урацил-5-оксиуксусной кислоты, урацил-5-оксиуксусную кислоту (v). Получение таких аналогов известно специалисту в данной области, оно описано, например, в US 4373071, US 4401796, US 4415732, US 4458066, US 4500707, US 4668777, US 4973679, US 5047524, US 5132418, US 5153319, US 5262530 и 5700642, содержание которых полностью включено в настоящее описание в качестве ссылки. В случае применения указанных выше аналогов предпочтительными согласно изобретению являются те аналоги, которые повышают иммуногенность нуклеиновой кислоты формулы (II), предлагаемой в изобретении, и/или не взаимодействуют с другими интродуцированными модификациями. По меньшей мере один аналог может присутствовать во фланкирующей последовательности Cl и/или Cn, необязательно по меньшей мере 10, 20, 30, 50, 60, 70, 80, 90% нуклеотидов во фланкирующих последовательностях Cl и/или Cn обладают свойствами указанных в настоящем описании аналогов, если фланкирующая последовательность вообще содержит по меньшей мере один аналог. Наиболее предпочтительно все нуклеотиды во фланкирующей последовательности являются аналогами, которые могут (что является наиболее предпочтительным) быть идентичными аналогами нуклеотидов одного и того же типа (например, все цитозиновые нуклеотиды во фланкирующей(их) последовательности(ях) присутствуют в виде 2-тиоцитозина) или они могут быть различными (например, по меньшей мере два различных аналога цитозина присутствуют вместо встречающихся в естественных условиях цитозиновых нуклеотидов во фланкирую щей(их) последовательности(ях)). Предпочтительно l и n представляют собой целые числа от 1 до 20, более предпочтительно от 1 до 10 и еще более предпочтительно от 2 до 8.
Аналогичным образом количество нуклеотидов С в нуклеиновой кислоте формулы (II), предлагаемой в изобретении, определяется величиной l или n. l и n каждый независимо друг от друга представляют собой целые числа от 1 до 40, причем, если l или n равно 1, то С представляет собой цитозин или его аналог, и если l или n>1, то по меньшей мере 50% нуклеотидов представляют собой цитозин или его аналог. Например, если l или n равно 4, то Cl или Cn может представлять собой, но, не ограничиваясь только ими, например, CUCU, CCUU, UCUC, UUCC, CUUC, CCCU, CCUC, CUCC, UCCC или СССС и т.д.; если l или n равно 5, то Cl или Cn может представлять собой, например, CCCUU, CCUCU, CUCCU, UCCCU, UCCUC, UCUCC, UUCCC, CUCUC, CCCCU, CCCUC, CCUCC, CUCCC, UCCCC или ССССС и т.д. Нуклеотид, смежный с Xm в нуклеиновой кислоте формулы (II), предлагаемой в изобретении, предпочтительно не представляет собой урацил.
В контексте настоящего описания понятие «идентичность» относится к сравнению последовательностей с референс-последовательностью, при этом сравнении определяют процент идентичности. Например, для того, чтобы определить процент идентичности двух нуклеотидных последовательностей, прежде всего следует расположить определенным образом последовательности относительно друг друга (выравнивание) для того, чтобы иметь возможность осуществлять последующее сравнение последовательностей. Для этой цели можно, например, в первую нуклеотидную последовательность вводить бреши и можно сравнивать нуклеотиды с нуклеотидами в соответствующем положении второй нуклеотидной последовательности. Если рассматриваемое положение в первой нуклеотидной последовательности занято таким же нуклеотидом, что и в соответствующем положении второй последовательности, то две последовательности являются идентичными в этом положении. Процент идентичности двух последовательностей является функцией количества идентичных положений, распределенных по последовательностям. Если, например, для конкретной нуклеиновой кислоты установлена определенная идентичность последовательности при сравнении с референс-нуклеиновой кислотой, имеющей определенную длину, то этот процент идентичности указывают по отношению к референс-нуклеиновой кислоте. Следовательно, если исходить, например, из нуклеиновой кислоты, которая идентична на 50% референс-нуклеиновой кислоте, состоящей из 100 нуклеотидов, то нуклеиновая кислота может представлять собой, например, нуклеиновую кислоту, состоящую из 50 нуклеотидов, которая полностью идентична участку референс-нуклеиновой кислоты, состоящему из 50 нуклеотидов. Однако, она может представлять собой также нуклеиновую кислоту, состоящую из 100 нуклеотидов, которая является идентичной на 50%, это означает, что в этом случае она имеет 50% идентичных нуклеиновых кислот с референс-нуклеиновой кислотой по всей ее длине. В альтернативном варианте нуклеиновая кислота может представлять собой нуклеиновую кислоту, состоящую из 200 нуклеотидов, которая на участке нуклеиновой кислоты, состоящем из 100 нуклеотидов, полностью идентична референс-нуклеиновой кислоте, состоящей из 100 нуклеотидов. Естественно, существуют другие нуклеиновые кислоты, также удовлетворяющие этим критериям.
Определение процента идентичности двух последовательностей можно осуществлять с помощью математического алгоритма. Примером предпочтительного математического алгоритма, который можно применять для сравнения двух последовательностей, является, но, не ограничиваясь только им, алгоритм, описанный у Karlin и др., PNAS USA, 90, 1993, cc.5873-5877. Этот алгоритм используется в программе NBLAST, с помощью которой можно идентифицировать последовательности, обладающие требуемой идентичностью с последовательностями, предлагаемыми в настоящем изобретении. Для осуществления сравнения последовательностей с использованием брешей можно применять программу «Gapped BLAST», описанную у Altschul и др., Nucleic Acids Res, 25, 1997, cc.3389-3402. При использовании программ BLAST и Gapped BLAST можно применять задаваемые по умолчанию параметры конкретной программы (например, NBLAST). Кроме того, последовательности можно сравнивать с помощью версии 9 программы GAP (программа глобального сравнительного анализа), разработанной «Genetic Computing Group» с использованием задаваемой по умолчанию матрицы (BLOSUM62) (значения от -4 до +11), где штраф за открытие бреши равен -12 (для первого нуля в бреши) и штраф за расширение бреши составляет -4 (для каждого дополнительного последующего нуля в бреши). После сравнения рассчитывают процент идентичности в виде процента относительно количества нуклеиновых кислот в заявляемой последовательности. Описанные методы определения процента идентичности двух нуклеотидных последовательностей можно применять соответственно также и для аминокислотных последовательностей с использованием соответствующих программ.
Когда в формуле (I) l или n>1, то предпочтительно также, чтобы по меньшей мере 60, 70, 80, 90 или даже 100% нуклеотидов представляли собой гуанозин или его аналог, как он определен выше в настоящем описании. Остальные нуклеотиды, дополняющие до 100% (если на долю гуанозина приходится менее 100% нуклеотидов) фланкирующие последовательности Gl и/или Gn, представляют собой урацил или его аналог, как он определен выше в настоящем описании. Предпочтительно также l и n каждый независимо друг от друга обозначают целое число от 2 до 30, более предпочтительно целое число от 2 до 20, и еще более предпочтительно целое число от 2 до 15. При необходимости нижний предел значений l или n можно варьировать, он составляет по меньшей мере 1, предпочтительно по меньшей мере 2, более предпочтительно по меньшей мере 3, 4, 5, 6, 7, 8, 9 или 10. Это определение применимо соответственно также и к формуле (II).
Х в нуклеиновой кислоте либо формулы (I), либо формулы (II), предлагаемой в изобретении, обозначает гуанозин, урацил, аденозин, тимидин, цитозин или его аналог. В этом контексте аналоги нуклеотидов определяют как не встречающиеся в естественных условиях варианты встречающихся в естественных условиях нуклеотидов. В соответствии с этим аналоги представляют собой дериватизированные химическим путем нуклеотиды, несущие не встречающиеся в нативных нуклеотидах функциональные группы, которые предпочтительно добавлены или удалены путем делеции из встречающегося в естественных условиях нуклеотида, или которыми заменены встречающиеся в естественных условиях функциональные группы нуклеотида. Следовательно, можно модифицировать каждый компонент встречающегося в естественных условиях нуклеотида, а именно, компонент, представляющий собой основание, сахарный (рибозный) компонент и/или фосфатный компонент, формирующий остов олигонуклеотида. Фосфатные фрагменты можно заменять, например, фосфорамидатами, фосфоротиоатами, пептидными нуклеотидами, метилфосфонатами и т.д. Предпочтительно по меньшей мере 10%, более предпочтительно по меньшей мере 20%, более предпочтительно по меньшей мере 30%, более предпочтительно по меньшей мере 50%, более предпочтительно по меньшей мере 70% и еще более предпочтительно по меньшей мере 90% всех обозначенных символом «X» нуклеотидов обладают свойствами аналога, как он определен в настоящем описании, если нуклеиновая кислота, предлагаемая в изобретении, вообще содержит по меньшей мере один аналог. Аналоги, замещающие конкретный тип нуклеотида в коровой последовательности, образованной «Xm», могут быть идентичными, например, все цитозиновые нуклеотиды, встречающиеся в коровых последовательностях, могут представлять собой 2-тиоцитозин, или они могут представлять собой различные аналоги конкретного нуклеотида, например, в коровой последовательности могут содержаться по меньшей мере два различных аналога цитозина.
Аналоги гуанозина, урацила, аденозина, тимидина, цитозина включают, но, не ограничиваясь только ими, любой встречающийся в естественных условиях или не встречающийся в естественных условиях гуанозин, урацил, аденозин, тимидин или цитозин, который изменен химическим путем, например, путем ацетилирования, метилирования, гидроксилирования и т.д., включая, 1-метиладенозин, 2-метиладенозин, 2-метилтио-N6-изопентиниладенозин, N6-метиладенозин, N6-изопентениладенозин, 2-тиоцитозин, 3-метилцитозин, 4-ацетилцитозин, 2,6-диаминопурин, 1-метилгуанозин, 2-метилгуанозин, 2,2-диметилгуанозин, 7-метилгуанозин, инозин, 1-метилинозин, дигидроурацил, 4-тиоурацил, 5-карбоксиметиламинометил-2-тиоурацил, 5-(карбоксигидроксилметил)урацил, 5-фторурацил, 5-бромурацил, 5-карбоксиметиламинометилурацил, 5-метил-2-тиоурацил, метиловый эфир N-урацил-5-оксиуксусной кислоты, 5-метиламинометилурацил, 5-метоксиаминометил-2-тиоурацил, 5'-метоксикарбонилметилурацил, 5-метоксиурацил, метиловый эфир урацил-5-оксиуксусной кислоты, урацил-5-оксиуксусную кислоту (v), квеозин, β-D-маннозилквеозин, вибутоксизин и инозин. Получение таких аналогов известно специалисту в данной области, оно описано, например, в US 4373071, US 4401796, US 4415732, US 4458066, US 4500707, US 4668777, US 4973679, US 5047524, US 5132418, US 5153319, US 5262530 и 5700642, содержание которых полностью включено в настоящее описание в качестве ссылки. В случае применения указанных выше аналогов предпочтительными согласно изобретению являются те аналоги, которые повышают иммуногенность нуклеиновой кислоты либо формулы (II), либо формулы (II), предлагаемой в изобретении, и/или не взаимодействуют с другими интродуцированными модификациями.
Количество Х в нуклеиновой кислоте либо формулы (II), либо формулы (II), предлагаемой в изобретении, определяется величиной m. m представляет собой целое число и, как правило, оно равно по меньшей мере 3, 4, 5, 6, 7, 8, 9 или 10, при этом, когда m равно 3, то X представляет собой урацил или его аналог, и когда m>3, то присутствуют по меньшей мере 3 расположенных непосредственно друг за другом остатка урацила или его аналогов. Такую последовательность, включающую по меньшей мере 3 расположенных непосредственно друг за другом остатка урацила, обозначают в контексте настоящего описания как «монотонная урациловая последовательность». Монотонная урациловая последовательность, как правило, содержит по меньшей мере 3, 4, 5, 6, 7, 8, 9 or 10, 10-15, 15-20, 20-25, 25-30, 30-50 или 50-90 остатков урацила или необязательно аналогов урацила, как они определены выше. Такая монотонная урациловая последовательность встречается по меньшей мере один раз в нуклеиновой кислоте либо формулы (I), либо формулы (II), предлагаемой в изобретении. Таким образом, может встречаться, например, 1, 2, 3, 4, 5 или большее количество монотонных урациловых последовательностей, содержащих по меньшей мере 3 остатка урацила или его аналогов, причем монотонные урациловые последовательности могут прерываться по меньшей мере одним остатком гуанозина, аденозина, тимидина, цитозина или его аналога, предпочтительно 2, 3, 4, 5 или большим количеством остатков. Например, если m равно 3, то Xm представляет собой UUU. Если m равно 4, то Xm может представлять собой, но, не ограничиваясь только ими, например, UUUA, UUUG, UUUC, UUUU, AUUU, GUUU или CUUU и т.д. Если n равно 10, то Xn, может представлять собой, но, не ограничиваясь только ими, например, UUUAAUUUUC, UUUUGUUUUA, UUUGUUUGUU, UUGUUUUGUU, UUUUUUUUUU и т.д. Нуклеотиды, смежные с Gl или Gn в нуклеиновой кислоте формулы (I), предлагаемой в изобретении, предпочтительно представляют собой урацил или его аналоги. Аналогично этому нуклеотиды, смежные с Cl или Cn в нуклеиновой кислоте формулы (II), предлагаемой в изобретении, предпочтительно представляют собой урацил или его аналоги.
Когда m>3, то, как правило, по меньшей мере 50%, предпочтительно по меньшей мере 60, 70, 80, 90 или даже 100% нуклеотидов представляют собой урацил или его аналог, как он определен выше. Тогда остальные нуклеотиды, дополняющие последовательность до 100% (если на долю урацила приходится менее 100% нуклеотидов в последовательности Xm) представляют собой гуанозин, урацил, аденозин, тимидин, цитозин или их аналоги, как они определены выше. Предпочтительно также m представляет собой целое число и оно равно по меньшей мере 4, 5, 6, 7, 8, 9 или 10, 10-15, 15-20, 20-25, 25-30, 30-50 или 50-90.
Нуклеиновая кислота формулы (I), предлагаемая в изобретении, наиболее предпочтительно содержит по меньшей мере одну из следующих последовательностей, представленных в SEQ ID NO:1-80:
GGUUUUUUUUUUUUUUUGGG (SEQ ID NO:1);
GGGGGUUUUUUUUUUGGGGG (SEQ ID NO:2);
GGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGGG (SEQ ID NO:3);
GUGUGUGUGUGUUUUUUUUUUUUUUUUGUGUGUGUGUGU (SEQ ID NO:4);
GGUUGGUUGGUUUUUUUUUUUUUUUUUGGUUGGUUGGUU (SEQ ID NO:5);
GGGGGGGGGUUUGGGGGGGG (SEQ ID NO:6);
GGGGGGGGUUUUGGGGGGGG (SEQ ID NO:7);
GGGGGGGUUUUUUGGGGGGG (SEQ ID NO:8);
GGGGGGGUUUUUUUGGGGGG (SEQ ID NO:9);
GGGGGGUUUUUUUUGGGGGG (SEQ ID NO:10);
GGGGGGUUUUUUUUUGGGGG (SEQ ID NO:11);
GGGGGGUUUUUUUUUUGGGG (SEQ ID NO:12);
GGGGGUUUUUUUUUUUGGGG (SEQ ID NO:13);
GGGGGUUUUUUUUUUUUGGG (SEQ ID NO:14);
GGGGUUUUUUUUUUUUUGGG (SEQ ID NO:15);
GGGGUUUUUUUUUUUUUUGG (SEQ ID NO:16);
GGUUUUUUUUUUUUUUUUGG (SEQ ID NO:17);
GUUUUUUUUUUUUUUUUUUG (SEQ ID NO:18);
GGGGGGGGGGUUUGGGGGGGGG (SEQ ID NO:19);
GGGGGGGGGUUUUGGGGGGGGG (SEQ ID NO:20);
GGGGGGGGUUUUUUGGGGGGGG (SEQ ID NO:21);
GGGGGGGGUUUUUUUGGGGGGG (SEQ ID NO:22);
GGGGGGGUUUUUUUUGGGGGGG (SEQ ID NO:23);
GGGGGGGUUUUUUUUUGGGGGG (SEQ ID NO:24);
GGGGGGGUUUUUUUUUUGGGGG (SEQ ID NO:25);
GGGGGGUUUUUUUUUUUGGGGG (SEQ ID NO:26);
GGGGGGUUUUUUUUUUUUGGGG (SEQ ID NO:27);
GGGGGUUUUUUUUUUUUUGGGG (SEQ ID NO:28);
GGGGGUUUUUUUUUUUUUUGGG (SEQ ID NO:29);
GGGUUUUUUUUUUUUUUUUGGG (SEQ ID NO:30);
GGUUUUUUUUUUUUUUUUUUGG (SEQ ID NO:31);
GGGGGGGGGGGUUUGGGGGGGGGG (SEQ ID NO:32);
GGGGGGGGGGUUUUGGGGGGGGGG (SEQ ID NO:33);
GGGGGGGGGUUUUUUGGGGGGGGG (SEQ ID NO:34);
GGGGGGGGGUUUUUUUGGGGGGGG (SEQ ID NO:35);
GGGGGGGGUUUUUUUUGGGGGGGG (SEQ ID NO:36);
GGGGGGGGUUUUUUUUUGGGGGGG (SEQ ID NO:37);
GGGGGGGGUUUUUUUUUUGGGGGG (SEQ ID NO:38);
GGGGGGGUUUUUUUUUUUGGGGGG (SEQ ID NO:39);
GGGGGGGUUUUUUUUUUUUGGGGG (SEQ ID NO:40);
GGGGGGUUUUUUUUUUUUUGGGGG (SEQ ID NO:41);
GGGGGGUUUUUUUUUUUUUUGGGG (SEQ ID NO:42);
GGGGUUUUUUUUUUUUUUUUGGGG (SEQ ID NO:43);
GGGUUUUUUUUUUUUUUUUUUGGG (SEQ ID NO:44);
GUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUG (SEQ ID NO:45);
GGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGG (SEQ ID NO:46);
GGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGG (SEQ ID NO:47);
GGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGG (SEQ ID NO:48);
GGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGG(SEQ ID NO:49);
GGGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGGG(SEQ ID NO:50);
GGGGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGGGG(SEQ ID NO:51);
GGGGGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGGGGG(SEQ ID NO:52);
GGGGGGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGGGGGG (SEQ ID NO:53);
GGUUUGG (SEQ ID NO:54);
GGUUUUGG (SEQ ID NO:55);
GGUUUUUGG (SEQ ID NO:56);
GGUUUUUUGG (SEQ ID NO:57);
GGUUUUUUUGG (SEQ ID NO:58);
GGUUUUUUUUGG (SEQ ID NO:59);
GGUUUUUUUUUGG (SEQ ID NO:60);
GGUUUUUUUUUUGG (SEQ ID NO:61);
GGUUUUUUUUUUUGG (SEQ ID NO:62);
GGUUUUUUUUUUUUGG (SEQ ID NO:63);
GGUUUUUUUUUUUUUGG (SEQ ID NO:64);
GGUUUUUUUUUUUUUUGG (SEQ ID NO:65);
GGUUUUUUUUUUUUUUUGG (SEQ ID NO:66);
GGGUUUGGG (SEQ ID NO:67);
GGGUUUUGGG (SEQ ID NO:68);
GGGUUUUUGGG (SEQ ID NO:69);
GGGUUUUUUGGG (SEQ ID NO:70);
GGGUUUUUUUGGG (SEQ ID NO:71);
GGGUUUUUUUUGGG (SEQ ID NO:72);
GGGUUUUUUUUUGGG (SEQ ID NO:73);
GGGUUUUUUUUUUGGG (SEQ ID NO:74);
GGGUUUUUUUUUUUGGG (SEQ ID NO:75);
GGGUUUUUUUUUUUUGGG (SEQ ID NO:76);
GGGUUUUUUUUUUUUUGGG (SEQ ID NO:77);
GGGUUUUUUUUUUUUUUUGGGUUUUUUUUUUUUUUUGGGUUUUUUUUUU UUUUUGGG SEQ ID NO:78;
GGGUUUUUUUUUUUUUUUGGGGGGUUUUUUUUUUUUUUUGGG SEQ ID NO:79;
GGGUUUGGGUUUGGGUUUGGGUUUGGGUUUGGGUUUGGGUUUGGGUUUG GG SEQ ID NO:80.
Нуклеиновая кислота формулы (II), предлагаемая в изобретении, наиболее предпочтительно содержит по меньшей мере одну из следующих последовательностей, представленных в SEQ ID NO:81-83:
CCCUUUUUUUUUUUUUUUCCCUUUUUUUUUUUUUUUCCCUUUUUUUUUUU
UUUUCCC SEQ ID NO:81
CCCUUUCCCUUUCCCUUUCCCUUUCCCUUUCCCUUUCCCUUUCCCUUUCCC
SEQ ID NO:82
CCCUUUUUUUUUUUUUUUCCCCCCUUUUUUUUUUUUUUUCCC
SEQ ID NO:83
Нуклеотидные последовательности либо формулы (I), либо формулы (II), предлагаемые в изобретении, предпочтительно представляют собой не встречающиеся в естественных условиях или полученные синтетическим путем последовательности вирусного или бактериального происхождения.
Нуклеиновая кислота либо формулы (I), либо формулы (II), предлагаемая в изобретении, как правило, представляет собой «стабилизированный олигонуклеотид», т.е. олигорибонуклеотид или олигодезоксирибонуклеотид, устойчивый к расщеплению in vivo (например, экзо- или эндонуклеазой). Такую стабилизацию можно осуществлять, например, путем модификации фосфатного остова предлагаемой в изобретении нуклеиновой кислоты либо формулы (I), либо формулы (II). Нуклеотиды, которые предпочтительно используют для этих целей, содержат модифицированный фосфоротиоатом фосфатный остов, при этом предпочтительно по меньшей мере один из атомов кислорода фосфатов, входящих в фосфатный остов, заменен на атом серы. Другие стабилизированные олигонуклеотиды включают, например: неионнные аналоги, такие, например, как алкил- и арилфосфанаты, в которых заряженный кислород фосфаната заменен на алкильную или арильную группу, или сложные фосфодиэфиры и алкилфосфотриэфиры, в которых заряженный кислородный остаток присутствует в алкилированной форме.
Нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, также можно стабилизировать. Как отмечалось выше, любую нуклеиновую кислоту, например, ДНК или РНК, в принципе можно применять в качестве нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. Однако с точки зрения безопасности предпочтительным является применение РНК в качестве такой нуклеиновой кислоты. В частности, отсутствует риск того, что РНК будет стабильно интегрирована в геном трансфектированной клетки. Кроме того, РНК существенно легче расщепляется in vivo. Кроме того, к настоящему времени не обнаружены никакие антитела к РНК, вероятно вследствие относительно короткого времени полужизни РНК in vivo по сравнению с ДНК. По сравнению с ДНК РНК является существенно менее стабильной в растворе, главным образом благодаря расщепляющим РНК ферментам, так называемым РНКазам (рибонуклеазы). Наличие даже минимального загрязнения рибонуклеазами является достаточным для полного расщепления РНК в растворе. Указанные загрязнения РНКазами, как правило, можно удалять только специальной обработкой, в частности диэтилпирокарбонатом (DEPC). Таким образом, естественное расщепление мРНК в цитоплазме клеток очень точно регулируется. В этой связи в данной области известен целый ряд механизмов. Так, концевая структура, как правило, имеет решающее значение для мРНК in vivo. На 5'-конце встречающейся в естественных условиях мРНК обычно находится структура в виде так называемого «кэпа» (модифицированный гуанозиновый нуклеотид), а на 3'-конце находится последовательность, включающая вплоть до 200 аденозиновых нуклеотидов (так называемый поли-А хвост).
Таким образом, нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, если она представляет собой РНК, можно стабилизировать в отношении расщепления РНКазами путем добавления на 5'-конец так называемой «кэп-структуры» («5'-кэп-структура»). В этом плане наиболее предпочтительными в качестве «5'-кэп»-структуры являются m7G(5')ppp (5'(A,G(5')ppp(5')A или G(5')ppp(5')G. Однако такую модификацию интродуцируют только, если другая модификация, например липидная модификация, еще не интродуцирована на 5'-конец нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, или если модификация не влияет на иммуногенные свойства (немодифицированной или химически модифицированной) нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении.
В другом варианте 3'-конец нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, прежде всего, если она представляет собой РНК, можно модифицировать с помощью последовательности, содержащей по меньшей мере 50 аденозиновых рибонуклеотидов, предпочтительно по меньшей мере 70 аденозиновых рибонуклеотидов, более предпочтительно по меньшей мере 100 аденозиновых рибонуклеотидов, наиболее предпочтительно по меньшей мере 200 аденозиновых рибонуклеотидов (так называемый «поли-А хвост»). И в этом случае также такую модификацию можно интродуцировать только, если другая модификация, например липидная модификация, еще не интродуцирована на 3'-конец нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, или если модификация не влияет на иммуногенные свойства (немодифицированной или химически модифицированной) нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. Обе указанные выше модификации, если они представляют собой инсерцию «5'-кэп-структуры» или инсерцию «поли-А хвоста» на 3'-конец, предупреждают преждевременное расщепления нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, in vivo и, следовательно, стабилизируют нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, in vivo.
Таким образом, конкретным вариантом осуществления изобретения является нуклеиновая кислота либо формулы (I), GlXmGn, либо формулы (II), ClXmCn, предлагаемая в изобретении, которая может содержать липидную модификацию. Такая имеющая липидную модификацию нуклеиновая кислота, предлагаемая в изобретении, как правило, содержит нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, как она определена выше, по меньшей мере один линкер, ковалентно связанный с указанной нуклеиновой кислотой, предлагаемой в изобретении, и по меньшей мере один липид, ковалентно связанный с соответствующим линкером. Альтернативно этому, имеющая липидную модификацию нуклеиновая кислота, предлагаемая в изобретении, как правило, содержит (по меньшей мере одну) нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, как она определена выше, и по меньшей мере один (бифункциональный) липид, ковалентно связанный (без линкера) с нуклеиновой кислотой либо формулы (I), либо формулы (II), предлагаемой в изобретении. Согласно третьему альтернативному варианту имеющая липидную модификацию нуклеиновая кислота, предлагаемая в изобретении, содержит нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, как она определена выше, по меньшей мере один линкер, ковалентно связанный с указанной нуклеиновой кислотой, предлагаемой в изобретении, и по меньшей мере один липид, ковалентно связанный с соответствующим линкером, а также по меньшей мере один (бифункциональный) липид, ковалентно связанный (без линкера) с нуклеиновой кислотой, предлагаемой в изобретении.
Липид, входящий в имеющую липидную модификацию нуклеиновую кислоту, предлагаемую в изобретении, как правило, представляет собой липид или липофильный остаток, который предпочтительно сам обладает биологической активностью. Указанные липиды предпочтительно включают встречающиеся в естественных условиях субстанции или соединения, такие, например, как витамины, например, α-токоферол (витамин Е), в том числе RRR-α-токоферол (прежнее название D-α-токоферол), L-α-токоферол, рацемат D,L-α-токоферола, сукцинат витамина Е (VES), или витамин А и его производные, например, ретиноевую кислоту, ретинол, витамин D и его производные, например, витамин D, а также его эргостерольные предшественники, витамин Е и его производные, витамин К и его производные, например, витамин К и родственные хиноновые и фитольные производные, или стероиды, такие как желчные кислоты, например, холевая кислота, дезоксихолевая кислота, дегидрохолевая кислота, кортизон, дигоксигенин, тестостерон, холестерин или тиохолестерин. Другие липиды или липофильные остатки, подпадающие под объем настоящего изобретения, включают, но, не ограничиваясь только ими, полиалкиленгликоли (Oberhauser и др., Nucl. Acids Res., 20, 1992, с.533), алифатические группы, такие например, как С1-С20алканы, С1-С20алкены или C1-С20алканолы и т.д., такие, например, как додекандиольные, гексадеканольные или ундецильные остатки (Saison-Behmoaras и др., EMBO J, 10, 1991, с.111; Kabanov и др., FEBS Lett., 259, 1990, с.327; Svinarchuk и др., Biochimie, 75, 1993, с.49), фосфолипиды, такие, например, как фосфоатидилглицерин, диацетилфосфатидилглицерин, фосфатидилхолин, дипальмитоилфосфоатидилхолин, дистероилфосфоатидилхолин, фосфатидилсерин, фосфатидилэтаноламин, дигексадецил-рац-глицерин, сфинголипиды, цереброзиды, ганглиозиды или 1,2-ди-О-гексадецил-рац-глицеро-3-Н-фосфанат триэтиламмония (Manoharan и др., Tetrahedron Lett., 36, 1995, с.3651; Shea и др., Nucl. Acids Res., 18, 1990, с.83777), полиамины или полиалкиленгликоли, такие, например, как полиэтиленгликоль (ПЭГ) (Manoharan и др., Nucleosides & Nucleotides, 14, 1995, с.969), гексаэтиленгликоль (ГЭГ), пальмитиновые или пальмитильные остатки (Mishra и др., Biochim. Biophys. Acta, 1264, 1995, с.229), октадециламины или гексиламинокарбонилоксихолестериновые остатки (Crooke и др., J. Pharmacol. Exp. Ther., 277, 1996, с.923), а также воски, терпены, алициклические углеводороды, остатки насыщенных или моно- или полиненасыщенных жирных кислот и т.д.
Связь между липидом и нуклеиновой кислотой либо формулы (I), либо формулы (II), предлагаемой в изобретении, в принципе может присутствовать на любом нуклеотиде, основании или сахарном компоненте любого нуклеотида предлагаемой в изобретении нуклеиновой кислоты, на 3'- и/или 5'-конце, и/или на фосфатном остове нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. Согласно изобретению наиболее предпочтительной является концевая липидная модификация нуклеиновой кислоты, предлагаемой в изобретении, т.е. на ее 3'- и/или 5'-конце. Концевая модификация имеет несколько преимуществ по сравнению с модификацией внутри последовательности. С одной стороны, модификации внутри последовательности могут влиять на поведение при гибридизации, что может оказывать вредное воздействие в случае определяющих стеричность остатков. С другой стороны, в случае синтетических препаратов модифицированной липидом нуклеиновой кислоты, предлагаемой в изобретении, которая имеет только концевую модификацию, синтез нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, можно осуществлять с использованием поступающих в продажу мономеров, которые можно получать в больших количествах, и можно применять протоколы синтеза, известные в данной области техники.
Таким образом, согласно первому предпочтительному варианту осуществления изобретения связь между нуклеиновой кислотой, предлагаемой в изобретении, и по меньшей мере одним липидом, для присоединения которого применяют «линкер» (ковалентно связанный с нуклеиновой кислотой либо формулы (I), либо формулы (II), предлагаемой в изобретении). Согласно настоящему изобретению линкеры, как правило, имеют по меньшей мере 2 и необязательно 3, 4, 5, 6, 7, 8, 9, 10, 10-20, 20-30 или более реактивных групп, выбранных, например, из гидроксигруппы, аминогруппы, алкоксигруппы и т.д.. Одна реактивная группа предпочтительно служит для связи вышеуказанной нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, например, РНК олигонуклеотида. Эта реактивная группа может присутствовать в защищенной форме, например, в виде DMT-группы (диметокситритилхлорид), в виде Fmoc-группы, в виде ММТ (монометокситритил) группы, в виде ТФК-группы (трифторуксусная кислота) и т.д. Кроме того, сульфогруппы могут быть защищены дисульфидами, например, алкилтиолами, такими, например, как 3-тиопропанол, или активированными компонентами, такими как 2-тиопиридин. Согласно изобретению одна или несколько дополнительных реактивных групп служат для ковалентного связывания одного или нескольких липидов. Таким образом, согласно первому варианту осуществления изобретения нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, можно присоединять через ковалентно связанный линкер, предпочтительно, используя по меньшей мере один липид, например, 1, 2, 3, 4, 5, 5-10, 10-20, 20-30 или большее количество липидов, прежде всего предпочтительно по меньшей мере 3-8 или большее количество липидов на нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении. Таким образом, связанные липиды можно связывать по отдельности друг от друга в различных положениях нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, либо они могут присутствовать в форме комплекса в одном или нескольких положениях нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. Дополнительную реактивную группу линкера можно применять для непосредственного или опосредованного (расщепляемого) связывания с носителем, например, с твердой фазой. Предпочтительными линкерами согласно настоящему изобретению являются, например, гликоль, глицерин и производные глицерина, 2-аминобутил-1,3-пропандиол и 2-аминобутил-1,3-пропандиольные производные/соединения, имеющие 2-аминобутил-1,3-пропандиольный скелет, пирролидиновые линкеры или содержащие пирролидин органические молекулы (прежде всего для модификации на 3'-конце) и т.д. Глицерин или производные глицерина (C3-якорь) или 2-аминобутил-1,3-пропандиольные производные/соединения, имеющие 2-аминобутил-1,3-пропандиольный скелет, (С7-якорь) являются наиболее предпочтительными линкерами, применяемыми согласно изобретению. Производное глицерина (C3-якорь) представляет собой наиболее предпочтительный линкер, когда липидную модификацию можно интродуцировать через эфирную связь (связь, представляющую собой простой эфир). Если липидную модификацию следует интродуцировать через амиидную или уретановую связь, то предпочтительным является, например, 2-аминобутил-1,3-пропандиольный скелет (С7-якорь). Таким образом, природа связи между линкером и нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, предпочтительно является такой, чтобы удовлетворять условиям и химическим веществам, которые применяют в амидитной химии, это означает, что она предпочтительно не является лабильной ни в кислых, ни в щелочных условиях. Наиболее предпочтительными являются связи, которые легко можно получать синтетически и которые не гидролизуются при процедуре аммиачного расщепления в процессе синтеза нуклеиновых кислот. Приемлемыми в принципе являются все соответствующие связи, предпочтительно сложноэфирные связи, амидные связи, уретановые и эфирные связи. Помимо хорошего соответствия исходным продуктам (несколько стадий синтеза), наибольшее предпочтение отдается эфирной связи благодаря ее относительно высокой биологической стабильности в отношении ферментативного гидролиза.
Согласно второму предпочтительному варианту осуществления изобретения (по меньшей мере одну) нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, связывают непосредственно по меньшей мере с одним (бифункциональным) липидом, как описано выше. В этом случае (бифункциональный) липид, применяемый согласно изобретению, предпочтительно содержит по меньшей мере 2 реактивные группы или необязательно 3, 4, 5, 6, 7, 8, 9, 10 или большее количество реактивных групп, где первая реактивная группа служит для связывания липида непосредственно или опосредованно с носителем, указанным в настоящем описании, а по меньшей мере одна дополнительная реактивная группа служит для связывания нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. Согласно второму варианту осуществления изобретения нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, в результате можно предпочтительно связывать, используя по меньшей мере один липид (непосредственно без линкера), например 1, 2, 3, 4, 5, 5-10, 10-20, 20-30 или большее количество липидов, наиболее предпочтительно по меньшей мере 3-8 или большее количество липидов на нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении. Связанные липиды можно связывать по отдельности друг от друга в различных положениях нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, либо они могут присутствовать в форме комплекса в одном или нескольких положениях нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. В другом варианте по меньшей мере одну нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, например, необязательно 3, 4, 5, 6, 7, 8, 9, 10, 10-20, 20-30 или большее количество нуклеиновых кислот либо формулы (I), либо формулы (II), предлагаемых в изобретении, можно связывать согласно второму варианту осуществления изобретения с липидом, описанным выше, через их реактивные группы. Липиды, которые можно применять согласно второму варианту осуществления изобретения, наиболее предпочтительно представляют собой такие (бифункциональные) липиды, которые позволяют осуществлять сочетание (предпочтительно на их концах или необязательно внутри молекул), такие, например, как полиэтиленгликоль (ПЭГ) и его производные, гексаэтиленгликоль (ГЭГ) и его производные, алкандиолы, аминоалканы, тиоалканолы и т.д. Природа связи между (бифункциональным) липидом и нуклеиновой кислотой либо формулы (I), либо формулы (II), предлагаемой в изобретении, описанной выше, предпочтительно является такой же, которая описана в первом предпочтительном варианте осуществления изобретения.
Согласно третьему варианту осуществления изобретения связь между нуклеиновой кислотой либо формулы (I), либо формулы (II), предлагаемой в изобретении, и по меньшей мере одним описанным выше липидом осуществляют одновременно согласно описанным выше вариантам осуществления изобретения. Например, нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, можно связывать в одном положении нуклеиновой кислоты по меньшей мере с одним липидом через линкер (аналогично первому варианту осуществления изобретения), а в другом положении нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, непосредственно по меньшей мере с одним липидом без использования линкера (аналогично второму варианту осуществления изобретения). Например, на 3'-конце нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, по меньшей мере один липид, описанный выше, можно ковалентно связывать с нуклеиновой кислотой через линкер, а на 5'-конце нуклеиновой кислоты, предлагаемой в изобретении, описанный выше липид можно ковалентно связывать с нуклеиновой кислотой без линкера. В альтернативном варианте на 5'-конце нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, по меньшей мере один липид, описанный выше, можно ковалентно связывать с нуклеиновой кислотой через линкер, а на 3'-конце нуклеиновой кислоты, предлагаемой в изобретении, описанный выше липид можно ковалентно связывать с нуклеиновой кислотой без линкера. Ковалентная связь может также присутствовать не только на концах нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, но также внутри молекулы, как описано выше, например, на 3'-конце и внутри молекулы, на 5'-конце и внутри молекулы, на 3'- и на 5'-конце и внутри молекулы, только внутри молекулы и т.д.
Модифицированную липидом нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, предпочтительно можно получать различными методами. В принципе липидную модификацию, как указано выше, можно интродуцировать в любое положение в нуклеиновой кислоте либо формулы (I), либо формулы (П), предлагаемой в изобретении, например, на 3'- и/или 5'-конец фосфатного остова нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, и/или на любое основание или на любой сахарный фрагмент любого нуклеотида нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. Согласно изобретению предпочтение отдается концевым липидным модификациям на 3'- и/или 5'-конце нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. С помощью указанной концевой химической модификации согласно изобретению можно получать большое количество различным образом дериватизированных нуклеиновых кислот. Примеры вариантов, предлагаемых в изобретении, представлены на фиг.4. Метод получения таких модифицированных липидом нуклеиновых кислот либо формулы (I), либо формулы (II), предлагаемых в изобретении, предпочтительно выбирают в зависимости от положения липидной модификации.
Если, например, липидная модификация присутствует на 3'-конце нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, то липидную модификацию, как правило, осуществляют либо до, либо после получения нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. Получение нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, можно осуществлять посредством прямого синтеза нуклеиновых кислот или необязательно путем добавления легко синтезируемой нуклеиновой кислоты или нуклеиновой кислоты, выделенной из других источников.
Согласно первому варианту нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, синтезируют непосредственно перед интродукцией липида, как правило, с помощью процессов, известных в области синтеза нуклеиновых кислот. Для этой цели исходный нуклеозид предпочтительно связывают с твердой фазой, например, через обеспечивающую сочетание молекулу, например, сукцинильный остаток, и синтезируют нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, например, с помощью амидитной химии. Затем описанный выше линкер ковалентно связывают, предпочтительно через первую реактивную группу линкера, с 3'-концом нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. Описанный выше липид затем можно ковалентно связывать с линкером через вторую реактивную группу линкера. В альтернативном варианте линкер можно ковалентно связывать с липидом до его связывания с 3'-концом нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. В этом случае необходимо связывание только первой реактивной группы линкера с 3'-концом нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. После синтеза нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, или после связывания липида нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, можно отделять от твердой фазы и удалять защитные группы. Если синтез осуществляли в растворе, то можно осуществлять стадию отмывки и очистки для удаления непрореагировавших реактантов, а также растворителей и нежелательных вторичных продуктов после синтеза модифицированной липидом нуклеиновой кислоты, предлагаемой в изобретении (и необязательно перед отделением носителя).
Согласно другому варианту описанную выше модифицированную липидом на 3'-конце нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, синтезируют после интродукции липида на реактивную группу линкера или связывают с реактивной группой линкера в виде легко синтезируемой нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, или нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, выделенной из образцов (фиг.5). Для этой цели, например, первую реактивную группу описанного выше линкера можно подвергать взаимодействию с описанным выше липидом. Затем, предпочтительно на второй стадии, вторую реактивную группу ликера защищают с помощью стабильной в кислых условиях защитной группы, например, DMT, Fmoc и т.д., для обеспечения последующего связывания нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, с реактивной группой. Затем линкер можно связывать непосредственно или опосредованно с твердой фазой через третью реактивную группу линкера. Опосредованное связывание можно осуществлять, например, через (обеспечивающую сочетание) молекулу, которую можно связывать ковалентно и с линкером, и с твердой фазой. Такая (обеспечивающая сочетание) молекула представляет собой, например, сукцинильный остаток и т.д., которые описаны выше. Удаляют защитную группу на третьей реактивной группе линкера и затем осуществляют связывание или синтез нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, на реактивной группе, которая в результате стала доступной. И, наконец, модифицированную липидом нуклеиновую кислоту, предлагаемую в изобретении, как правило, отщепляют от носителя (и защитные группы на нуклеиновой кислоте необязательно удаляют). Кроме того, дополнительный липид необязательно можно сочетать с 3'-концом присоединенной нуклеиновой кислоты, предлагаемой в изобретении, предпочтительно с использованием одной из описанных выше стадий.
Согласно описанному выше альтернативному варианту линкер, указанный выше, можно связывать непосредственно или опосредованно с твердой фазой через первую реактивную группу. Стабильную в кислых условиях защитную группу затем сначала связывают со второй реактивной группой линкера. После связывания защитной группы со второй реактивной группой описанный выше липид можно сначала связывать с третьей реактивной группой линкера. Затем предпочтительно также осуществляют удаление защитной группы третьей реактивной группы линкера, связывают или синтезируют нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, на реактивной группе, которая в результате стала доступной, и модифицированную липидом нуклеиновую кислоту, предлагаемую в изобретении, отщепляют от носителя (и защитные группы на нуклеиновой кислоте необязательно удаляют).
Согласно наиболее предпочтительному варианту описанной выше модифицированной на 3'-конце липидом нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, такую модифицированную липидом нуклеиновую кислоту, предлагаемую в изобретении, можно синтезировать с использованием линкера, имеющего три реактивные группы (трехфункциональное соединение-якорь), на основе субстанции, являющейся производной глицерина (С3-якорь) и имеющей однофункциональной липид, такой, например, как пальмитильный остаток, холестерин или токоферол. При этом в качестве исходного продукта для синтеза линкера можно применять, например, альфа, бета-изопропилиденглицерин (глицерин, который содержит кетальную защитную группу), который предпочтительно сначала превращают в алкоголят с помощью гидрида натрия и подвергают взаимодействию с гексадецилбромидом и липидом в реакции синтеза Вильямсона с получением соответствующего простого эфира. В альтернативном варианте эфирную связь можно вводить на первой стадии с помощью другого метода, например, путем образования тозилата α,β-изопропилиденглицерина и взаимодействия тозилата с реактивной группой липида, например кислотным протоном, с получением соответствующего простого эфира. На второй стадии кетальную защитную группу можно удалять с помощью кислоты, например, уксусной кислоты, разбавленной соляной кислоты и т.д., а затем первичную гидроксигруппу диола можно избирательно защищать диметокситритилхлоридом (DMT-Cl). На последней стадии предпочтительно осуществляют взаимодействие продукта, полученного на предыдущей стадии, с янтарным ангидридом в присутствии DMAP в качестве катализатора. Такой линкер является особенно предпочтительным, например, для связывания пальмитильных остатков или токоферола в качестве липида (см., например, фиг.5).
Согласно другому варианту указанную выше липидную модификацию 3'-конца нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, осуществляют с помощью (бифункционального) липида, такого, например, как полиэтиленгликоль (ПЭГ) или гексаэтиленгликоль (ГЭГ), без использования линкера, как описано выше. Указанные бифункциональные липиды, как правило, имеют две указанные выше функциональные группы, при этом один конец бифункционального липида можно предпочтительно связывать с носителем через (обеспечивающую сочетание) молекулу, например, лабильный в щелочных условиях сукцинильный якорь, и т.д., которые описаны выше, и нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, можно синтезировать на другом конце бифункционального липида (Е. Bayer, М. Maier, K. Bleicher, H.-J. Gaus Z. Naturforsch. 50b, 1995, с.671). Благодаря отсутствию третьей функционализации и линкера соответственно, что описано выше, синтез такой модифицированной липидом нуклеиновой кислоты, предлагаемой в изобретении, упрощается (см., например, фиг.6). При таком получении в бифункциональном липиде, применяемом согласно изобретению, например, полиэтиленгликоле, как правило, сначала осуществляют одно замещение с помощью защитной группы, например, DMT. На второй стадии, как правило, осуществляют этерификацию до сложного эфира липида, защищенного на реактивной группой, с использованием янтарного ангидрида в присутствии DMAP в качестве катализатора с получением сукцината. Затем на третьей стадии бифункциональный липид можно сочетать с носителем и удалять защитные группы, после чего на четвертой стадии осуществлять синтез нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, согласно описанному выше процессу. Затем осуществляют удаление защитных групп нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, и необязательно отщепляют модифицированную липидом нуклеиновую кислоту от носителя.
Согласно другому предпочтительному варианту осуществления изобретения описанная выше липидная модификация нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, присутствует на 5'-конце нуклеиновой кислоты. При этом липидную модификацию, как правило, осуществляют либо после получения, либо после синтеза нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. Получение нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, можно осуществлять - как указано выше - путем непосредственного синтеза нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, либо путем добавления легко синтезируемой нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, либо нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, выделенной из образцов. Синтез нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, предпочтительно осуществляют аналогично описанному выше методу в соответствии с процессами синтеза нуклеиновых кислот, известных в данной области, более предпочтительно с помощью фосфорамидитного процесса (см., например, фиг.7).
Согласно наиболее предпочтительному варианту осуществления изобретения липидную модификацию нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, осуществляют на 5'-конце нуклеиновой кислоты, предлагаемой в изобретении, с помощью специальных модифицированных фосфорамидитов в соответствии с фосфорамидитным процессом синтеза нуклеиновых кислот. Указанные амидиты, которые можно получать с помощью относительно простого синтеза, удобно сочетать в качестве последнего мономера с поступающей в продажу или легко синтезируемой нуклеиновой кислотой. Эти реакции отличаются относительно быстрыми реакционными кинетическими характеристиками и очень высоким выходом реакции сочетания. Синтез модифицированных амидитов предпочтительно осуществляют путем взаимодействия фосфорамидита, например, бета-цианэтилмонохлорфосфорамидита (моно(2-цианэтиловый эфир) диизопропиламидхлорид ангибрида фосфорной кислоты), со спиртом, растворенном в приемлемом растворителе, например, в абсолютном дихлорметане, указанного выше липида, например, жирным спиртом токоферола, холестерина, гексадеканола, DMT-ПЭГ и т.д. Предпочтительно также в реакционный раствор добавляют DIPEA в качестве акцептора кислоты.
Указанные фосфорамидиты, применяемые для синтеза модифицированных липидом на 5'-конце нуклеиновых кислот, предлагаемых в изобретении, являются относительно устойчивыми к гидролизу и их можно (до синтеза) очищать хроматографически с помощью силикагеля. Для этой цели небольшое количество слабого основания, такого, например, как триэтиламин, как правило, добавляют к элюенту для того, чтобы избежать расщепления амидита. Важно вновь полностью удалять указанное основание из продукта для того, чтобы избегать низкого выхода реакции сочетания. Это можно осуществлять, например, с помощью простой сушки в вакууме, но предпочтительно путем очистки фосфорамидитов посредством их осаждения из простого метил-трет-бутилового эфира с использованием пентана. Если применяемые модифицированные липидом амидиты имеют очень высокую вязкость, например, присутствуют в виде вязкого масла, можно осуществлять также (экспресс) хроматографию на колонках, что позволяет распределять их с использованием триэтиламина в качестве основания. Однако такую очистку не осуществляют, как правило, в случае применения модифицированных ПЭГ амидитов, поскольку они содержат лабильную в кислотных условиях защитную группу DMT.
Для реакции сочетания модифицированных липидом фосфорамидитов с 5'-концом нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, предпочтительно применяют такие растворители, в которых применяемые амидиты достаточно растворимы. Например, с учетом высокой липофильности амидитов, применяемых согласно изобретению, их растворимость в ацетонитриле может быть ограниченной. Поэтому помимо ацетонитрила, являющегося наиболее широко применяемым растворителем, предпочтительно применять в реакция сочетания раствор хлорированных углеводородов, например, 0,1М раствор в (абсолютном) дихлорметане. Однако применение дихлорметана требует некоторых изменений в стандартном протоколе цикла синтеза. Например, для того, чтобы избежать осаждения амидита в трубах устройства для автоматического синтеза и на носителе, все клапаны и трубы, которые приходят в контакт с амидитами, промывают (абсолютным) дихлорметаном до и после фактической стадии сочетания и сушат продувкой.
Когда применяют модифицированные липидом амидиты, как правило, получают высокий выход реакции сочетания, сопоставимый с выходом реакции сочетания амидитов, которую принято использовать в данной области. Кинетика реакции с использованием модифицированных липидом амидитов обычно является более медленной. По этой причине, когда применяют модифицированные липидом амидиты, то время сочетания предпочтительно (существенно) удлиняют по сравнению со стандартными протоколами. Специалист в данной области легко может определять указанное время сочетания. Из-за того, что можно исключить стадию кэпирования после сочетания, при необходимости можно также осуществлять дополнительный цикл синтеза с использованием этого же модифицированного липидом амидита для увеличения общего выхода реакции. В этом случае стадию детритилирования обычно не проводят, например, в случае модифицированного липидами DMT, например, DMT-ПЭГ.
При синтезе модифицированных липидом на 5'-конце нуклеиновых кислот, предлагаемых в изобретении, фосфитный триэфир, через который липид связывают с нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, можно окислять с помощью сульфурирующего агента. Для этой цели предпочтительно применять сульфурирующий агент, при использовании которого достигается максимально полное окисление фосфотриэфира. В другом случае, реакция сульфурирования, например, из-за связанных со стеричностью причинам, может проходить столь неполно, что только небольшое количество продукта получают после аммиачного расщепления и удаление защитных групп MON, или вообще не получают продукт. Это явление зависит от типа модификации, применяемого сульфурирующего агента и условий сульфурирования. Таким образом, окисление предпочтительно осуществляют с использованием йода. В результате этого, хотя происходит интродукция фосфодиэфирной связи, не следует ожидать, что эта связь будет распознаваться в качестве субстрата нуклеазами из-за ее близости к липидному остатку.
При осуществлении липидной модификации линкеры или (бифункциональные) липиды, соединенные с нуклеиновой кислотой либо формулы (I), либо формулы (II), предлагаемой в изобретении, или необязательно саму нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, можно, как описано выше, сочетать непосредственно или опосредованно с носителем. Непосредственное сочетание осуществляют предпочтительно прямо с носителем, а опосредованное сочетание с носителем, как правило, осуществляют через дополнительную (обеспечивающую сочетание) молекулу. Связь, образовавшаяся с носителем, предпочтительно представляет собой (расщепляемую) ковалентную связь с линкером или бифункциональным липидом и/или (расщепляемую) ковалентную связь с твердой фазой. Соединения, пригодные в качестве (обеспечивающей сочетание) молекулы, представляют собой, например, дикарбоновые кислоты, например, сукцинильные остатки (сукцинильные якоря), оксалильные остатки (оксалильные якоря) и т.д. Линкеры, (бифункциональные) липиды или необязательно нуклеиновые кислоты либо формулы (I), либо формулы (II), предлагаемые в изобретении, которые подобно, например, аминоалкильным остаткам (например, аминопропильным или аминогексанильным остаткам), несущим свободную аминофункцию, можно связывать с носителем через фталимидный линкер. Тиолсодержащие линкеры, (бифункциональные) липиды или необязательно нуклеиновые кислоты либо формулы (I), либо формулы (II), предлагаемые в изобретении, можно связывать в дисульфидной форме с носителем. Приемлемыми согласно настоящему изобретению носителями являются прежде всего твердые фазы, такие как CPG (стекло с контролируемым размером пор), Tentagel®, функционализированный аминогруппой ПС(полистирол)-ПЭГ (Tentagel® S NH2) и т.д. предпочтительно Tentagel® или функционализированный аминогруппой ПС-ПЭГ (Tentagel® S NH2). Согласно конкретному варианту осуществления изобретения для сочетания с носителем, предназначенным для сочетания, можно применять, например, сукцинаты описанных выше линкеров или бифункциональных липидов, предпочтительно с использованием TBTU/NMM (тетрафторборат 1Н-бензотриазол-1-ил-1,1,3,3- тетраметиламмония/N-метилморфолин) в качестве реагента для сочетания с функционализированный аминогруппой ПС-ПЭГ (Tentagel® S NH2). В случае применения в качестве носителя ПС-ПЭГ в 1 мкмолярных количествах, что является традиционным, наилучшие результаты, как правило, получают при загрузке от 50 до 100 мкмолей/г (Е. Bayer, K. Bleicher, M. Maier Z., Naturforsch. 50b, 1995, с.1096). Однако, если согласно изобретению требуется крупномасштабный синтез нуклеотидов, то загрузка носителя предпочтительно является максимально возможной (≥100 мкмолей). Согласно изобретению такой процесс обеспечивает также хороший выход реакции сочетания (M. Gerster, M. Maier, N. Clausen, J. Schewitz, Е. Bayer Z. Naturforsch. 52b, 1997, с.110). Например, можно применять носители, такие, например, как смолы с загрузкой вплоть до 138 мкмоля/г или необязательно более высокой загрузкой, с получением хорошего выхода синтеза. Поскольку выход реакции сочетания с указанными выше линкерами или бифункциональными липидами составляет примерно 100%, загрузку носителя можно регулировать относительно точно, исходя из стехиометрии этих соединений. Загрузку предпочтительно оценивают путем количественного спектроскопического определения расщепленной защитной группы DMT (см. экспериментальную часть). Оставшуюся аминофункцию, которая еще сохраняется на носителе, можно кэпировать уксусным ангидридом. Указанное кэпирование, как правило, осуществляют после загрузки носителя, но его можно осуществлять также непосредственно при синтезе нуклеиновой кислоты, например в ДНК-синтезаторе. Для синтеза модифицированных липидами нуклеиновых кислот на дераватизированных ПС-ПЭГ носителях предпочтительно применяют циклы синтеза, разработанные специально для Tentagel®, в которых учтены характерные свойства этого материла (Е. Bayer, M. Maier, K. Bleicher, H.-J. Gaus Z. Naturforsch. 50b, 1995, с.671, Е. Bayer, K. Bleicher, M. Maier Z. Naturforsch. 50b, 1995, с.1096.). Предпочтительные изменения по сравнению со стандартным протоколом представляют собой:
- удлинение продолжительности реакции на стадиях сочетания, кэпирования и окисления;
- увеличение количества стадий детритилирования;
- удлинение стадий отмывки после каждой стадии;
- применение содержащего аскорбиновую кислоту промывочного раствора (0,1М в смеси диоксан/вода=9:1) после стадии окисления, которая обычно является необходимой (для окисления фосфитного триэфира) при амидитном процессе для удаления следовых количеств йода.
Следует отметить, что природа модификации может оказывать влияние на индивидуальные стадии цикла синтеза. Например, в случае применения дериватизированного ПЭГ1500 носителя, наблюдается заметно замедленная кинетика реакцией, что требует удлинения стадий детритилирования и кроме того удлинения времени сочетания. Такие изменения и адаптации известны обычному специалисту в данной области и в контексте настоящего описания их можно осуществлять в любой момент времени. При модификации указанным образом циклов реакции можно синтезировать и модифицированные липидом фосфодиэфиры и фосфоротиоаты. Выход реакции сочетания при применении согласно изобретению амидитов в сочетании с линкерами или бифункциональными липидами не зависит от липидных остатков и соответствует общепринятым значениям (97-99%). Возможность 5'-дериватизации и интродукции дополнительных модификаций, например, на основание, сахарный фрагмент или фосфатный остов, сохраняется, когда применяют указанные 3'-модификации.
Нуклеиновую кислоту либо формулы (I), либо формулы (II) в виде химически немодифицированной нуклеиновой кислоты или в виде (химически) модифицированной нуклеиновой кислоты, например, модифицированной липидом нуклеиновой кислоты либо формулы (I), либо формулы (II), можно также стабилизировать путем образования комплекса нуклеиновой кислоты либо формулы (I), либо формулы (II), например, но, не ограничиваясь только ими, с катионным полимером, катионными пептидами или полипептидами, предпочтительно с поликатионным полимером, таким как полилизин или полиаргинин, или в другом варианте с катионными липидами или липофектантами, с гистоном, нуклеолином, протамином, олигофектамином, спермином или спермидином и катионными полисахаридами, в частности хитозаном, TDM, MDP, мурамилом-дипептидом, плюрониками и/или одним из их производных. Гистоны и протамины представляют собой катионные белки, которые в естественных условиях тесно связаны с ДНК. Таким образом, они ответственны in vivo за конденсацию нетранскрибируемой ДНК и ДНК некоторых вирусов. В качестве гистонов, которые можно применять в контексте настоящего изобретения для образования комплекса с нуклеиновой кислотой либо формулы (I), либо формулы (II), в качестве наиболее предпочтительных следует отметить гистоны H1, Н2а, Н3 и Н4. Однако наиболее предпочтительными являются протамин (протамин Р1 или Р2) или катионные частичные последовательности протамина. В контексте настоящего изобретения наиболее предпочтительные соединения могут иметь пептидную последовательность, выведенную из протамина Р1 или Р2, и более конкретно соответствующую (катионной) последовательности (SRSRYYRQRQRSRRRRRR (SEQ ID NO:85) или RRRLHRIHRRQHRSCRRRKRR (SEQ ID NO:86). Другие соединения, которые можно использовать для образования комплекса с нуклеиновой кислотой либо формулы (I), либо формулы (II), предлагаемой в изобретении, можно выбирать из указанных выше адъювантов, но, не ограничиваясь только ими.
В этом контексте понятие «образовывать комплекс» должно означать, что нуклеиновую кислоту либо формулы (I), либо формулы (II) связывают со стабилизирующим соединением, указанным выше, например, катионным полимером, катионными пептидами или полипептидами и т.д., с образованием нековалентного комплекса между нуклеиновой кислотой и стабилизирующим соединением. В контексте настоящего описания понятие «нековалентный» означает, что образуется обратимая ассоциация нуклеиновой кислоты и стабилизирующего соединения в результате нековалентных взаимодействий этих молекул, где молекулы объединяются друг с другом с помощью определенного типа взаимодействия электронов, отличных от ковалентной связи, например, с помощью ван-дер-ваальсовой связи, т.е. слабого электростатического притяжения, возникающего благодаря неспецифическим силам притяжения обеих молекул. Ассоциация нуклеиновой кислотой либо формулы (I), либо формулы (II) и стабилизирующего соединения находится в равновесии с диссоциацией этого комплекса. Не вдаваясь в какую-либо теорию, можно ожидать, что равновесие внутри клетки сдвигается в сторону диссоциации нуклеиновой кислотой либо формулы (I), либо формулы (II) и стабилизирующего соединения.
Таким образом, согласно варианту осуществления изобретения нуклеиновая кислота либо формулы (I), либо формулы (II), предлагаемая в изобретении, может представлять собой иммуностимулятор, если ее вводить без какого-либо другого фармацевтически активного компонента, или ее можно применять в качестве адъюванта, если вводить в сочетании с фармацевтически активным компонентом, например, в виде композиции, содержащей как фармацевтически активный компонент, так и представляющий собой адъювант компонент (например, в виде композиции вакцины, содержащей специфический антиген и нуклеиновую кислоту либо формулы (I), либо формулы (II) в качестве адъюванта).
Нуклеиновая кислота либо формулы (I), либо формулы (II), предлагаемая в изобретении, в качестве «иммуностимулятора» предпочтительно обладает способностью инициировать не антиген специфический иммунный ответ (который связан с врожденной иммунной системой), предпочтительно путем имуностимуляции. Иммунный ответ, как правило, можно вызывать различными путями. Важным фактором получения приемлемого иммунного ответа является стимуляция различных Т-клеточных субпопуляций. Т-лимфоциты, как правило, в результате дифференцировки разделяются на две субпопуляции Т-клетки-хелперы 1 (Th1) и Т-клетки-хелперы (Th2), с помощью которых иммунная система может разрушать внутриклеточные (ТЫ) и внеклеточные (Th2) патогены (например антигены). Две Th-клеточные популяции отличаются схемой производства ими эффекторных белков (цитокины). Так, Th1-клетки обусловливают клеточный иммунный ответ путем активации макрофагов и цитотоксических Т-клеток. С другой стороны, Th2-клетки усиливают гуморальный иммунный ответ путем стимуляции конверсии В-клеток в плазменные клетки и путем образования антител (например против антигенов). Таким образом, соотношение Th1/Th2 имеет очень важное значение для иммунного ответа. В контексте настоящего изобретения соотношение Th1/Th2 - иммунного ответа предпочтительно смещается с помощью иммуностимулятора, а именно нуклеиновой кислотой либо формулы (I), либо формулы (II), предлагаемой в изобретении, в сторону клеточного ответа, т.е. Th1-ответа и главным образом клеточного иммунного ответа. Как указано выше, нуклеиновая кислота, предлагаемая в изобретении, сама усиливает неспецифический иммунный ответ, что позволяет применять нуклеиновую кислоту как таковую (без добавления другого фармацевтически активного компонента) в качестве иммуностимулятора. При ее введении с другим фармацевтически активным компонентом, предпочтительно специфическим иммуностимулирующим компонентом, нуклеиновая кислота, предлагаемая в изобретении, служит в качестве адъюванта, поддерживая специфический иммунный ответ, вызванный другим фармацевтически активным компонентом.
Настоящее изобретение относится также к фармацевтическим композициям, содержащим нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, или обе нуклеиновые кислоты и необязательно фармацевтически приемлемый носитель и/или дополнительные вспомогательные субстанции и добавки и/или адъюванты (первый вариант композиции, предлагаемой в изобретении). Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, или обе нуклеиновые кислоты, фармацевтически активный компонент и необязательно фармацевтически приемлемый носитель и/или дополнительные вспомогательные субстанции и добавки и/или адъюванты (второй вариант композиции, предлагаемой в изобретении).
Фармацевтические композиции, предлагаемые в настоящем изобретении, как правило, содержат описанную выше нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, или обе нуклеиновые кислоты в безопасном и эффективном количестве. В контексте настоящего описания понятие «безопасное и эффективное количество» означает, количество нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, или обеих нуклеиновых кислот, достаточное для индукции в значительной степени положительной модификации состояния, подлежащего лечению, например, опухоли, аутоиммунных заболеваний, аллергий или инфекционных болезней. Однако в то же время «безопасное и эффективное количество» является в достаточной степени невысоким, позволяющим избегать серьезных побочных действий, другими словами обеспечивает разумное соотношение между преимуществом и риском. Определение этих пределов, как правило, находится в компетенции соответствующих медицинских органов. Касательно нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, понятие «безопасное и эффективное количество» предпочтительно означает количество, пригодное для стимуляции иммунной системы таким образом, который не приводит к избыточным или вредным иммунным ответам, но также предпочтительно, к таким иммунным ответам, которые находятся ниже измеряемого уровня. «Безопасное и эффективное количество» нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, может варьироваться в зависимости от конкретного состояния, подлежащего лечению, а также возраста и физического состояния пациента, подлежащего лечению, серьезности состояния, продолжительности лечения, природы сопутствующей терапии, конкретного применяемого фармацевтически приемлемого носителя и аналогичных факторов, известных практикующему врачу. Фармацевтические композиции, предлагаемые в изобретении, можно применять согласно изобретению для лечения человека, а также в ветеринарной практике.
Таким образом, согласно первому варианту осуществления изобретения описанную выше нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, можно применять как таковую в качестве иммуностимулятора (без добавления любых других фармацевтически активных компонентов). Это реализуется, в частности, в случае, когда нуклеиновая кислота либо формулы (I), либо формулы (II), предлагаемая в изобретении, содержит липидную модификацию. Липид может усиливать иммуностимулирующие свойства предлагаемых в изобретении нуклеиновых кислот или может приводить также к образованию терапевтически активной молекулы, такой, например, как витамин или стероид, как описано выше, например, α-токоферол (витамин Е), D-альфа-токоферол, L-альфа-токоферол, D,L-альфа-токоферол, витамина Е сукцинат (VES), витамин А и их производные, витамин D и его производные, витамин K и его производные и т.д.
Фармацевтическая композиция согласно второму варианту осуществления изобретения содержит (помимо нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении) по меньшей мере один дополнительный фармацевтически активный компонент. В этом контексте фармацевтически активный компонент представляет собой соединение, которое оказывает терапевтическое действие на конкретное показание, предпочтительно раковые заболевания, аутоиммунное заболевание, аллергии или инфекционные болезни. К таким соединениям относятся, но, не ограничиваясь только ими, пептиды, белки, нуклеиновые кислоты, (терапевтически активные) низкомолекулярные органические или неорганические соединения (молекулярная масса менее 5000, предпочтительно менее 1000), сахара, антигены или антитела, терапевтические агенты, уже известные в данной области, антигенные клетки, антигенные клеточные фрагменты, клеточные фракции; модифицированные, ослабленные или деактивированные (например, химически или облучением) патогены (вирус, бактерии и т.д.) и т.д.
Согласно первой альтернативе второго варианта осуществления изобретения (композиции, предлагаемой в изобретении), фармацевтически активный компонент, входящий в фармацевтическую композицию, представляет собой иммуномодулирующий компонент, предпочтительно иммуностимулирующий компонент. Наиболее предпочтительно фармацевтически активный компонент представляет собой антиген или иммуноген. Под «антигеном» и «иммуногеном» подразумевают любую структуру, обладающую способностью вызывать образование антител и/или активацию клеточного иммунного ответа, т.е. специфического (и не связанного с адъювантом) иммунного ответа. Таким образом, согласно изобретению понятие «антиген» и «иммуноген» применяют как синонимы. Примерами антигенов являются пептиды, полипептиды, а также белки, клетки, клеточные экстракты, полисахариды, конъюгаты полисахаридов, липиды, гликолипиды и углеводы. Под понятие антиген подпадают, например, также опухолевые антигены, антигены вирусов, бактерий, грибов и простейших. Предпочтение отдается поверхностным антигенам опухолевых клеток и поверхностным антигенам, в частности секретируемым формам патогенов вирусов, бактерий, грибов или простейших. Антиген, естественно может присутствовать, например, в вакцине, предлагаемой в изобретении, также в виде гаптена, сшитого с приемлемым носителем. Можно применять также другие антигенные компоненты, например, деактивированные или ослабленные патогены (например, описанные выше).
Антигенные (поли)пептиды включают все известные антигенные пептиды, например, опухолевые антигены и т.д. Конкретными примерами опухолевых антигенов являются среди прочего специфические для опухоли поверхностные антигены (tumour-specific surface antigens (TSSA)), например, 5Т4, альфа5β1-интегрин, 707-АР, AFP, ART-4, B7H4, BAGE, β-катенин/m, Bcr-abl, антиген MN/C IX, СА125, CAMEL, CAP-1, CASP-8, бета-катенин/m, CD4, CD19, CD20, CD22, CD25, CDC27/m, CD 30, CD33, CD52, CD56, CD80, CDK4/m, CEA, CT, Cyp-B, DAM, EGFR, ErbB3, ELF2M, EMMPRIN, EpCam, ETV6-AML1, G250, GAGE, GnT-V, Gp100, HAGE, HER-2/new, HLA-A*0201-R170I, HPV-E7, HSP70-2M, HAST-2, hTERT (или hTRT), iCE, IGF-1R, IL-2R, IL-5, KIAA0205, LAGE, LDLR/FUT, MAGE. MART-1/мелан-А, MART-2/Ski, MC1R, моизин/m, MUC1, MUM-1, -2, -3, NA88-A, PAP, протеиназа-3, pl90 minor bcr-abl, Pml/RARα, FRAME, PSA, PSM, PSMA, RAGE, RU1 или RU2, SAGE, SART-1 или SART-3, сурвивин, TEL/AML1, TGFβ, TPI/m, TRP-1, TRP-2, TRP-2/INT2, VEGF и WT1, или антигены, полученные, например, из таких последовательностей как NY-Eso-1 или NY-Eso-B. Любой класс опухолевых антигенов можно применять для целей настоящего изобретения, например опухолевые антигены, в отношении которых известно, что они участвуют в неоваскуляризации, воздействуют на структуру внеклеточного матрикса и т.д. Опухолевые антигены можно применять в фармацевтической композиции в виде белкового антигена или в виде мРНК или ДНК, кодирующей опухолевые антигены, предпочтительно указанные выше опухолевые антигены.
Согласно второй альтернативе второго варианта осуществления изобретения (композиции, предлагаемой в изобретении, которая содержит предлагаемую в изобретении нуклеиновую кислоту (в качестве адъюванта) и дополнительный фармацевтически активный компонент) фармацевтически активный компонент представляет собой антитело. В этом контексте можно применять любое терапевтически приемлемое антитело. Наибольшее предпочтение согласно изобретению отдается антителу к антигенам, белкам или нуклеиновым кислотам, которые играют важную роль в раковых заболеваниях или инфекционных болезнях, например, к белкам клеточной поверхности, генам-суппрессорам опухоли или их ингибиторам, факторам роста и элонгации, связанным с апоптозом белкам, опухолевым антигенам или антигенам, описанным выше, и т.д.
Согласно третьей альтернативе второго варианта осуществления изобретения фармацевтически активный компонент, входящий в фармацевтическую композицию, предлагаемую в изобретении, представляют собой нуклеиновую кислоту. Указанная нуклеиновая кислота может быть одноцепочечной или двухцепочечной и может образовывать гомо- или гетеродуплекс, а также находиться в линейной или кольцевой форме. Нуклеиновая кислота, входящая в качестве фармацевтически активного компонента в фармацевтическую композицию, не ограничена ее длиной и может включать любую встречающуюся в естественных условиях нуклеотидную последовательность или ее комплемент или фрагмент. Кроме того, в этом контексте нуклеиновая кислота может быть частично или полностью синтетической по своей природе. Например, нуклеиновая кислота может включать нуклеиновую кислоту, которая кодирует (терапевтически значимый) белок и/или которая может вызывать иммунный ответ, например, антиген или нуклеиновую кислоту, кодирующую антиген. Антиген в этом контексте предпочтительно обозначает указанный выше антиген.
Предпочтительно нуклеиновая кислота, входящая в качестве фармацевтически активного компонента в фармацевтическую композицию, предлагаемую в изобретении, представляет собой мРНК. Указанную мРНК можно добавлять в ее «оголенной» форме в фармацевтическую композицию, предлагаемую в изобретении, или в стабилизированной форме с целью снижения или даже предотвращения расщепления нуклеиновой кислоты in vivo, например, экзо- и/или эндонуклеазами.
Например, мРНК, входящая в качестве фармацевтически активного компонента в фармацевтическую композицию, предлагаемую в изобретении, может быть стабилизирована с помощью указанного выше 5'-кэпа и/или поли-А хвоста на 3'-конце, состоящем по меньшей мере из 50 нуклеотидов, предпочтительно по меньшей мере 70 нуклеотидов, более предпочтительно по меньшей мере 100 нуклеотидов, наиболее предпочтительно по меньшей мере 200 нуклеотидов. Как уже отмечалось, концевая структура имеет решающее значение in vivo. Благодаря этим структурам РНК распознается как мРНК и ее расщепление регулируется. Однако кроме того, существуют другие процессы, которые стабилизируют или дестабилизируют РНК. Многие из этих процессов пока не известны, но при этом взаимодействие между РНК и белками часто, вероятно, имеет решающее значение. Например, недавно описана «система контроля мРНК» (Hellerin и Parker, Ann. Rev. Genet., 33, 1999, cc.229-260), с помощью которой распознаются неполные или несмысловые мРНК с использованием конкретных белковых взаимодействий с обратной связью в цитозоле и делаются чувствительными к расщеплению, большинство указанных процессов происходит с помощью экзонуклеаз.
Стабилизацию мРНК, входящей в качестве фармацевтически активного компонента в фармацевтическую композицию, предлагаемую в изобретении, можно осуществлять также путем ассоциации или образования комплекса или связывания мРНК с катионным соединением, прежде всего поликатионным соединением, например (поли)катионным пептидом или белком. В частности наиболее эффективным является применение протамина, нуклеолина, спермина или спермидина в качестве поликатионного, связывающегося с нуклеиновой кислотой белка. Кроме того, возможно также применение других катионных пептидов или белков, таких как поли-L-лизин или гистоны. Эта процедура стабилизации мРНК описана в ЕР-А-1083232, содержание которой полностью включено в настоящее описание в качестве ссылки. Кроме того, предпочтительные катионные субстанции, которые можно применять для стабилизации мРНК, присутствующей в качестве фармацевтически активного компонента, включают катионные полисахариды, например, хитозан, полибрен, полиэтиленимин (PEI) или поли-L-лизин (PLL) и т.д. Помимо активности модифицированной липидом нуклеиновой кислоты, предлагаемой в изобретении, в форме адъюванта в отношении повышения клеточной проницаемости, что уже является преимуществом, ассоциация или комплексообразование мРНК с катионными соединениями, например, катионными белками или катионными липидами, например, олигофектамином (в качестве комплексообразующего реагента на основе липида) предпочтительно повышает перенос мРНК, присутствующей в качестве фармацевтически активного компонента, в клетки, подлежащие обработке, или в организм, подлежащий обработке. Кроме того, упомянутое выше стабилизирующее действие в отношении нуклеиновой кислоты, предлагаемой в изобретении, путем комплексообразования, относится также к стабилизации мРНК.
Другой подход к стабилизации мРНК в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении, представляет собой целенаправленное изменение последовательности мРНК путем удаления так называемых элементов, дестабилизирующих последовательность (DSE). Сигнальные белки могут связываться с этими элементами, дестабилизирующими последовательность (DSE), которые встречаются, в частности, в мРНК эукариотических организмов, и регулируют ферментативное расщепление мРНК in vivo. Таким образом, для дополнительной стабилизации мРНК, присутствующей в качестве фармацевтически активного компонента, предпочтительно сделать одну или несколько замен по сравнению с соответствующей областью мРНК дикого типа, приводящих к тому, чтобы в ней больше не присутствовали элементы, дестабилизирующие последовательность. Естественно также предпочтительной согласно изобретению является элиминация из мРНК DSE, необязательно присутствующих в нетранслируемых областях (3'- и/или 5'-UTR). Примерами указанных выше DSE являются, например, последовательности, богатые AU («AURES»), которые встречаются в 3'-UTR-участках многочисленных нестабильных мРНК (Caput и др., Proc. Natl. Acad. Sci. USA, 83, 1986, cc.1670-1674). Таким образом, предпочтительно мРНК, применяемую в качестве фармацевтически активного компонента, модифицировать по сравнению с мРНК дикого типа, так, чтобы в ней больше не присутствовали какие-либо элементы, дестабилизирующие последовательность. Это относится также к таким мотивам последовательностей, которые, возможно, распознаются эндонуклеазами, например, к последовательности GAACAAG, которая входит в 3'-UTR-сегмент гена, кодирующего рецептор трансферина (Binder и др., EMBO J., 13, 1994, cc.1969-1980). Указанные мотивы последовательностей предпочтительно также элиминируют из модифицированной липидом нуклеиновой кислоты, предлагаемой в изобретении.
мРНК в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении, можно модифицировать также, например, для эффективной трансфекции, что может оказаться желательным, таким образом, чтобы имело место эффективное связывание рибосом с сайтом связывания рибосомы (последовательность Козака: GCCGCCACCAUGG (SEQ ID NO:84), где AUG представляет собой стартовый кодон). В этой связи следует отметить, что повышение содержания A/U в районе этого положения обеспечивает более эффективное связывание рибосом с мРНК.
Кроме того, можно интродуцировать один или несколько так называемых IRES («внутренний рибосомальный сайт проникновения») в мРНК, применяемую в качестве фармацевтически активного компонента. Таким образом, IRES может функционировать не только в качестве рибосомального сайта связывания, а служить также для получения мРНК, которая кодирует несколько пептидов или полипептидов, которые должны транслироваться рибосомами независимо друг от друга («мультицистронная мРНК»). Примерами последовательностей IRES, которые можно применять согласно изобретению, являются последовательности IRES пикорнавирусов (например, вирус ящура (FMDV)), вирусов чумы (CFFV), вирусов полиомы (PV), вирусов энцефаломиокардита (ECMV), вирусов ящура (FMDV), вирусов гепатита С (HCV), вирусов классической лихорадки свиней (CSFV), вирусов мышиного лейкоза (MLV), вирусов иммунодефицита обезьян (SIV) или вирусов паралича сверчков (CrPV)
мРНК, необязательно применяемая в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении, может содержать также стабилизирующие последовательности в 5'- и/или 3'-нетранслируемых областях, которые обладают способностью увеличивать времени полужизни мРНК в цитозоле. Такие стабилизирующие последовательности могут быть на 100% гомологичны встречающимся в естественных условиях последовательностям, которые присутствуют в вирусах, бактериях и эукариотических организмах, но могут быть также частично или полностью синтетическими. В качестве примера стабилизирующих последовательностей, которые можно применять в настоящем изобретении, можно отметить нетранслируемые последовательности (UTR) гена β-глобина, например. Homo sapiens или Xenopus laevis. Другим примером стабилизирующей последовательности является последовательность общей формулы (C/U)CCANxCCC(U/A)PyxUC(C/U)CC (SEQ ID NO:88), которая содержится в 3'-UTR обладающей очень высокой стабильностью мРНК, кодирующей α-глобин, (I)-коллаген, 15-липоксигеназу или тирозингидроксилазу (см. Holcik и др., Proc. Natl. Acad. Sci. USA, 94, 1997, cc.2410-2414). Естественно, такие стабилизирующие последовательности можно применять индивидуально или в сочетании друг с другом или также в сочетании с другими стабилизирующими последовательностями, известными специалисту в данной области. Для дальнейшего повышения конечной требуемой трансляции мРНК, применяемую в качестве фармацевтически активного компонента, можно подвергать дополнительным модификациям по сравнению с соответствующей мРНК дикого типа, при этом модификации могут присутствовать либо в качестве индивидуальных альтернативных вариантов, либо в сочетании друг с другом. С одной стороны, содержание G/C в модифицированной области мРНК, кодирующей пептид или полипептид, может быть выше по сравнению с содержанием G/C в кодирующей области соответствующей мРНК дикого типа, кодирующей пептид или полипептид, при этом аминокислотная последовательность, кодируемая модифицированной мРНК, остается неизмененной по сравнению с аминокислотной последовательностью, кодируемой мРНК дикого типа. Такая модификация основана на том факте, что для эффективной трансляции мРНК стабильность мРНК имеет решающее значение. В этом случае имеет важное значение состав и последовательность расположения различных нуклеотидов. В частности, последовательности, которые имеют высокое содержание G (гуанозин)/С (цитозин), являются более стабильными, чем последовательности, имеющие высокое содержание А (аденозин)/и (урацил). Таким образом, согласно изобретению целесообразно при сохранении транслируемой аминокислотной последовательности изменять кодоны по сравнению с мРНК дикого типа таким образом, чтобы они имели более высокое содержание G/C-нуклеотидов. Благодаря тому факту, что несколько кодонов кодируют одну и ту же аминокислоту (так называемая «вырожденность генетического кода»), оказывается возможным определять кодоны, которые предпочтительны с точки зрения стабильности (альтернативные наиболее часто встречающимся кодонам). В зависимости от аминокислотной последовательности, которая должна кодироваться модифицированной мРНК, существуют различные возможности для модификации последовательности мРНК по сравнению с последовательностью дикого типа. В том случае, когда аминокислоты кодируются кодонами, которые содержат только нуклеотиды G или С, не требуется осуществлять модификацию кодонов. Так, кодоны являются для Pro (CCC или CCG), Arg (CGC или CGG), Ala (GCC или GCG) и Gly (GGC или GGG) не требуется изменять, поскольку в них не присутствуют ни А, ни U. С другой стороны, кодоны, которые содержат нуклеотиды А и/или U, можно изменять путем замены на различные кодоны, которые кодируют ту же самую аминокислоту, но которые не содержат А и/или U. Такими примерами являются: кодоны для Pro можно изменять с CCU или ССА на CCC или CCG; кодоны для Arg можно изменять с CGU или CGA или AGA или AGG на CGC или CGG; кодоны для Ala можно изменять с GCU или GCA на GCC или GCG; кодоны для Gly можно изменять с GGU или GGA на GGC или GGG. В других случаях, хотя и нельзя исключать из кодонов нуклеотиды А и U, оказывается возможным уменьшать содержание А и U путем использования кодонов, которые имеют меньшее содержание нуклеотидов А и/или U. Такими примерами являются: кодоны для Phe можно изменять с UUU на UUC; кодоны для Leu можно изменять с UUA, UUG, CUU или CUA на CUC или CUG; кодоны для Ser можно изменять с UCU или UCA, или AGU на UCC, UCG или AGC; кодон для Tyr можно изменять с UAU на UAC; стоп-кодон UAA можно изменять на UAG или UGA; кодон для Cys можно изменять с UGU на UGC; кодон для His можно изменять с CAU на САС; кодон для Gln можно изменять с САА на CAG; кодоны для Не можно изменять с AUU или AUA на AUC; кодоны для Thr можно изменять с ACU или АСА на АСС или ACG; кодон для Asn можно изменять с AAU на ААС; кодон для Lys можно изменять с ААА на AAG; кодон для Val можно изменять с GUU или GUA на GUC или GUG; кодон для Asp можно изменять с GAU на GAC; кодон для Glu можно изменять с GAA на GAG. С другой стороны, в случае кодонов для Met (AUG) и Trp (UGG) невозможна модификация последовательности. Перечисленные выше замены, естественно, можно применять индивидуально, но также во всех возможных комбинациях для повышения содержания G/C в модифицированной мРНК по сравнению с исходной последовательностью. Так, например, все кодоны для Thr, встречающиеся в исходной (дикого типа) последовательности можно заменять на АСС (или ACG). Однако предпочтительно использовать следующие комбинации возможных замен: замену всех кодонов, кодирующих Thr в исходной последовательности, на АСС (или ACG) и замену всех кодонов, кодирующих Ser в исходной последовательности, на UCC (или UCG, или AGC); замену всех кодонов, кодирующих Ile в исходной последовательности, на AUC и замену всех кодонов, кодирующих Lys в исходной последовательности, на AAG и замену всех кодонов, кодирующих Tyr в исходной последовательности, на UAC; - замену всех кодонов, кодирующих Val в исходной последовательности, на (или GUG) и замену всех кодонов, кодирующих Glu в исходной последовательности, на GAG и замену всех кодонов, кодирующих Ala в исходной последовательности, на GCC (или GCG), и замену всех кодонов, кодирующих Arg в исходной последовательности, на CGC (или CGG); замену всех кодонов, кодирующих Val в исходной последовательности, на GUC (или GUG) и замену всех кодонов, кодирующих Glu в исходной последовательности, на GAG и замену всех кодонов, кодирующих Ala в исходной последовательности, на GCC (или GCG), и замену всех кодонов, кодирующих Gly в исходной последовательности, на GGC (или GGG), и замену всех кодонов, кодирующих Asn в исходной последовательности, на ААС; замену всех кодонов, кодирующих Val в исходной последовательности, на GUC (или GUG) и замену всех кодонов, кодирующих Phe в исходной последовательности, на UUC, и замену всех кодонов, кодирующих Cys в исходной последовательности, на UGC, и замену всех кодонов, кодирующих Leu в исходной последовательности, на CUG (или CUC) и замену всех кодонов, кодирующих Gln в исходной последовательности, на CAG, и замену всех кодонов, кодирующих Pro в исходной последовательности, на ССС (или CCG), и т.д. Предпочтительно содержание G/C в области (или в каждом другом сегменте, где они необязательно присутствуют) мРНК, которая кодирует пептид или полипептид, увеличивают по меньшей мере на 7%, более предпочтительно по меньшей мере на 15%, наиболее предпочтительно по меньшей мере на 20%, по сравнению с содержанием G/C кодирующей области мРНК дикого типа, которая кодирует соответствующий пептид или полипептид, и предпочтительно увеличение составляет по меньшей мере 50%, более предпочтительно по меньшей мере 70% и наиболее предпочтительно по меньшей мере 90%. В этой связи наиболее предпочтительным увеличением содержания G/C модифицированной таким образом мРНК по сравнению с последовательностью дикого типа является максимально возможное увеличение.
Еще одна предпочтительная модификация мРНК, применяемой в качестве фармацевтически активного компонента в фармацевтической композиции, основана на том факте, что эффективность трансляции определяется также различной частотой встречаемости тРНК в клетках. Если так называемые «редкие» кодоны присутствую в повышенных количествах в последовательности РНК, то соответствующая мРНК транслируется существенно хуже по сравнению с вариантом, когда присутствуют кодоны, кодирующие относительно «часто встречающиеся» тРНК. Таким образом, согласно изобретению кодирующую область мРНК, которую применяют в качестве фармацевтически активного компонента, модифицируют по сравнению с соответствующей областью мРНК дикого типа таким образом, чтобы по меньшей мере один кодон последовательности дикого типа, который кодирует относительно редкую тРНК в клетках, заменять на кодон, который кодирует относительно часто встречающуюся тРНК в клетках, которая несет такую же аминокислоту, что и относительно редкая тРНК. С помощью такой модификации последовательности РНК модифицируют так, что интродуцируют кодоны, доступные для часто встречающихся тРНК. тРНК, встречающиеся относительно часто в клетке, и наоборот, встречающиеся относительно редко, известны специалистам в данной области, см., например, Akashi, Curr. Opin. Genet. Dev., 11(6), 2001, cc.660-666. С помощью указанной модификации согласно изобретению можно заменять все кодоны последовательности дикого типа, которые кодируют относительно редкие тРНК в клетках, на кодон, который кодирует относительно часто встречающиеся тРНК в клетках, которая несет такую же аминокислоту, что и относительно редкая тРНК. Наиболее предпочтительным является объединение повышенного, предпочтительно до максимума, содержания в последовательности мРНК G/C, как описано выше, с «частыми» кодонами, без изменения аминокислотой последовательности антигенного пептид или полипептида (одного или нескольких), кодируемого кодирующей областью мРНК. Предпочтительные антигены, которые можно кодировать с использованием обогащенной G/C /оптимизированной мРНК, перечислены выше.
Согласно четвертой альтернативе второго варианта осуществления изобретения (для композиции, предлагаемой в настоящем изобретении) нуклеиновая кислота, входящая в качестве фармацевтически активного компонента в фармацевтическую композицию, предлагаемую в изобретении, представляет собой dsPHK, предпочтительно siPHK. DsPHK или siPHK представляет особый интерес в связи с явлением РНК-интерференции. При иммунологических исследованиях большое внимание уделяется явлению РНК-интерференции. В последние годы изучен защитный механизм, основанный на РНК, который имеет место как в царстве грибов и растений, так и в царстве животных и проявляется в виде «иммунной системы генома». Эта система первоначально описана в различных видах независимо друг от друга, сначала у С.elegans, до того, как удалось идентифицировать механизмы, лежащие в основе процесса: опосредуемая РНК устойчивость растений к вирусам, PTGS (пост-транскрипционное молчание гена) у растения и РНК-интерференция у эукариот, все они основаны на общем процессе. Методы in vitro РНК-интерференции (PHKi) основаны на применении двухцепочечных молекул РНК (dsPHK), которые инициируют специфическую для последовательности суппрессию экспрессии гена (Zamore, Nat. Struct. Biol. 9, 2001. cc.746-750; Sharp, Genes Dev. 5, 2001, cc.485-490: Hannon, Nature 41, 2002, cc.244-251). При трансфекции клеток млекопитающих длинной dsPHK активация протеинкиназы R и RnaseL вызывает неспецифические действия, такие, например, как ответ в виде выделения интерферонов (Stark и др., Annu. Rev. Biochem. 67, 1998. cc.227-264; Не и Katze, Viral Immunol. 15, 2002, cc.95-119). Эти неспецифические действия не возникают при применении более короткой, например, 21-23-мерной РНК, так называемой siPHK (малая интерферирующая РНК), поскольку неспецифические действия не инициируются siPHK, состоящей менее чем из 30 пар оснований (Elbashir и др., Nature 411, 2001, cc.494-498). В настоящее время молекулы dsPHK применяют также in vivo (McCaffrey и др., Nature 418, 2002, cc.38-39; Xia и др., Nature Biotech. 20, 2002, cc.1006-1010; Brummelkamp и др., Cancer Cell 2, 2002, cc.243-247).
Таким образом, двухцепочечная РНК (dsPHK), которую в конечном итоге применяют в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении, предпочтительно содержит последовательность, имеющую общую структуру 5'-(N17-29)-3', где N обозначает любое основание и представляет собой нуклеотиды. Общая структура состоит из двухцепочечной РНК, представляющую собой макромолекулу, которая состоит из рибонуклеотидов, рибонуклеотид состоит из пентозы (рибозы), органического основания и фосфата. Органические основания в РНК, предлагаемой в изобретении, представляют собой пуриновые основания аденозин (А) и гуанозин (G) и пиримидиновые основания цитозин (С) и урацил (U). DsPHK, которую в конечном итоге применяют в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении, содержит указанные нуклеотиды или нуклеотидные аналоги, имеющие ориентированную структуру. DsPHK, которые применяют в качестве фармацевтически активного компонента, предлагаемого в изобретении, предпочтительно имеет общую структура 5'-(N19-25)-3', более предпочтительно 5'-(N19-24)-3', еще более предпочтительно 5'-(N21-23)-3', где N обозначает любое основание. Предпочтительно по меньшей мере 90%, более предпочтительно 95% и наиболее предпочтительно 100% нуклеотидов dsPHK, которую применяют в качестве фармацевтически активного компонента, должны быть комплементарны сегменту последовательности (м)РНК (терапевтически значимого) белка или антигена, описанного (в качестве фармацевтически активного компонента) выше. 90%-ная комплементарность означает, что, если dsPHK, применяемая согласно изобретению, состоит, например из 20 нуклеотидов, то она содержит не более 2 нуклеотидов, имеющих несоответствующую комплементарность, относительно соответствующего сегмента (м)РНК. Однако последовательность двухцепочечной РНК, которую необязательно применяют в фармацевтической композиции, предлагаемой в изобретении, предпочтительно полностью комплементарна по своей общей структуре сегменту (м)РНК белка или антигена, описанного выше в качестве фармацевтически активного компонента.
В принципе, все сегменты длиной от 17 до 29, предпочтительно от 19 до 25, пар оснований, которые присутствуют в кодирующей области (м)РНК, могут служить в качестве последовательности-мишени для dsPHK, применяемой в конечном итого в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении. Равным образом, dsPHK, применяемые в качестве фармацевтически активного компонента, могут быть направлены против нуклеотидной последовательности (терапевтически значимого) белка или антигена, описанного (в качестве фармацевтически активного компонента) выше, который не расположен в кодирующей области, например, прежде всего расположен в 5'-некодирующей области (м)РНК, то есть направлен перед некодирующими областями (м)РНК, которые имеют регуляторную функцию. Таким образом, последовательность-мишень dsPHK, применяемой в качестве фармацевтически активного компонента белка или антигена, описанного выше, может находиться как в транслируемой, так и нетранслируемой области (м)РНК и/или находиться в области контролирующих элементов. Последовательность-мишень dsPHK, применяемой в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении, может находиться также в области, перекрывающей транслируемую и нетранслируемую последовательность; в частности последовательность-мишень может содержать по меньшей мере один нуклеотид, расположенный против хода транскрипции относительно исходного триплета кодирующей области (м)РНК.
Модифицированный нуклеотид может предпочтительно присутствовать в dsPHK, которую в конечном итоге применяют в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении. Понятие «модифицированный нуклеотид» в контексте изобретения означает, что представляющий интерес нуклеотид был химически модифицирован. Специалисту в данной области известно, что под понятием «химическая модификация» понимают, что модифицированный нуклеотид изменен по сравнению с встречающимися в естественных условиях нуклеотидами путем замены, добавления или удаления одного или нескольких атомов или групп атомов. По меньшей мере один модифицированный нуклеотид dsPHK, которую применяют согласно изобретению, служит, с одной стороны, для стабильности, а с другой стороны, для предупреждения диссоциации. Предпочтительно в dsPHK, применяемой согласно изобретению, модифицированными являются от 2 до 10 и более предпочтительно от 2 до 5 нуклеотидов. Предпочтительно по меньшей мере одна 2'-гидроксигруппа нуклеотидов dsPHK в двухцепочечной структуре заменена химической группой, предпочтительно 2'-аминогруппой или 2'-метильной группой. По меньшей мере один нуклеотид по меньшей мере в одной цепи двухцепочечной структуры может представлять собой так называемый «замкнутый нуклеотид», который имеет химически модифицированное сахарное кольцо, предпочтительно с помощью 2'-O,4'-С-метиленового мостика. Несколько нуклеотидов dsPHK, предлагаемой в изобретении, предпочтительно представляют собой замкнутые нуклеотиды. Кроме того, с помощью модификации остова dsPHK, применяемой в изобретении, можно предотвращать ее преждевременное расщепление. В этой связи наибольшее предпочтение следует отдавать dsPHK, модифицированной с образованием фосфоротиоата, 2'-O-метил-РНК, ЗНК (замкнутая нуклеиновая кислота), гапмеров ЗНК/ДНК и т.д., и поэтому имеющей более продолжительное время полужизни in vivo.
Концы двухцепочечной РНК (dsPHK), применяемой в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении, можно предпочтительно модифицировать для того, чтобы противостоять расщеплению в клетке или диссоциации в индивидуальных цепях, в частности для того, чтобы избежать преждевременного расщепления нуклеазами. Нежелательная в норме диссоциация индивидуальных цепей dsPHK имеет место, в частности, когда ее применяют в низких концентрациях или используют короткие цепи. Для наиболее эффективного ингибирования диссоциации, сцепление, осуществляемое парами нуклеотидов двухцепочечной структуры dsPHK, применяемой согласно изобретению, можно повышать с помощью по меньшей мере одной, предпочтительно более одной химической связи. DsPHK, которую применяют в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении, диссоциация которой понижена, обладает более высокой стабильностью в отношении ферментативного и химического расщепления в клетке или в организме (in vivo) или ех vivo и поэтому имеет более продолжительное время полужизни. Другая возможность предупреждения преждевременной диссоциации в клетке dsPHK, предлагаемой в изобретении, заключается в формировании петли(ель) шпильки на каждом конце цепей. Таким образом, в конкретном варианте осуществления изобретения dsPHK, которую применяют в фармацевтической композиции, предлагаемой в изобретении, имеет структуру типа шпильки, способствующую замедлению кинетики диссоциации. В такой структуре петля образуется предпочтительно на 5' и/или 3'-конце. Такая структура типа петли не содержит водородных связей и поэтому, как правило, между нуклеотидными основания отсутствует комплементарность. Как правило, такая петля состоит по меньшей мере из 5, предпочтительно по меньшей мере из 7 нуклеотидов и таким образом связывает две комплементарные индивидуальные цепи dsPHK, применяемой согласно изобретению. Для предупреждения диссоциации цепей нуклеотиды двух цепей dsPHK, которую применяют согласно изобретению, можно также предпочтительно модифицировать таким образом, чтобы создавать сильную водородную связь, например, путем повышения силы водородной связи между основаниями, необязательно модифицированных нуклеотидов. В результате стабильность взаимодействия между цепями повышается и dsPHK защищается от действия РНКаз.
Таким образом, согласно наиболее предпочтительному варианту осуществления изобретения dsPHK, которую применяют в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении, направлена против (м)РНК белка или антигена, указанного выше. DsPHK, предпочтительно применяемая для этой цели, подавляет трансляцию вышеуказанного белка или антигена в клетке по меньшей мере на 50%, более предпочтительно на 60%, еще более предпочтительно на 70% и наиболее предпочтительно по меньшей мере на 90%, т.е. клетка содержит предпочтительно не более половины встречающейся в естественных условиях (без обработки dsPHK, предлагаемой в изобретении) концентрации вышеуказанного белка или антигена в клетке. Подавление трансляции этих белков или антигенов в клетке после добавления молекул dsPHK, которые применяют согласно изобретению, основано на явлении РНК-интерференции, вызываемой указанными молекулами. DsPHK, применяемую согласно изобретении, в результате можно отнести к так называемой siPHK, которая инициирует явление РНК-интерференции и может связывать (м)РНК указанного выше белка или антигена. Оценку или демонстрацию подавления трансляции, инициируемой в клетках dsPHK, которую применяют согласно изобретению, можно осуществлять с помощью Нозерн-блоттинга, количественной ПЦР в реальном времени или на уровне белка с помощью специфических антител к указанному выше белку или антигену. DsPHK, которую в конечном итоге применяют в качестве фармацевтически активного компонента в фармацевтической композиции, предлагаемой в изобретении, и соответствующую siPHK можно получать методами, известными специалистам в данной области.
Фармацевтическая композиция (согласно первому или второму варианту осуществления изобретения), предлагаемая в изобретении, как правило, содержит фармацевтически приемлемый носитель. В контексте настоящего описания понятие «фармацевтически приемлемый носитель» предпочтительно включает жидкую или нежидкую основу композиции. Если композиция находится в жидкой форме, то носитель, как правило, представляет собой свободную от пирогенов воду; изотонический физиологический раствор или забуференные (водные) растворы, например, забуференные фосфатом, цитратом и т.д. растворы. Для инъекции фармацевтической композиции, предлагаемой в изобретении, можно прежде всего применять буфер, предпочтительно водный буфер, содержащий натриевую соль, предпочтительно натриевую соль в концентрации по меньшей мере 50 мМ, кальциевую соль, предпочтительно кальциевую соль в концентрации по меньшей мере 0,01 мМ, и необязательно калиевую соль, предпочтительно калиевую соль в концентрации по меньшей мере 3 мМ. Согласно предпочтительному варианту осуществления изобретения натриевые соли, кальциевые соли и необязательно калиевые соли, содержащиеся в инъекционном буфере, находятся в форме галогенидов, например хлоридов, йодидов или бромидов, в форме их гидроксидов, карбонатов, бикарбонатов или сульфатов. Примерами натриевых солей являются, но, не ограничиваясь только ими, например, NaCl, Nai, NaBr, Na2CO3, NaHCO3, Na2SO4, примерами необязательно присутствующих калиевых солей являются, но, не ограничиваясь только ими, например, KCl, KI, KBr, K2CO3, KHCO3, K2SO4, а примерами кальциевых солей являются, но, не ограничиваясь только ими, например, CaCl2, CaI2, CaBr2, СаСО3, CaSO4, Ca(OH)2. Кроме того, буфер может содержать также органические анионы указанных выше катионов. В более предпочтительном варианте осуществления изобретения пригодный для инъекции буфер, предлагаемый в изобретении, может содержать соли, выбранные из группы, включающей хлорид натрия (NaCl), хлорид кальция (CaCl2) и необязательно хлорид калия (KCl), причем в дополнение к хлоридам он может содержать также другие анионы. Указанные соли, как правило, присутствуют в инъекционном буфере в концентрации, составляющей по меньшей мере 50 мМ для хлорида натрия (NaCl), по меньшей мере 3 мМ для хлорида калия (KCl) и по меньшей мере 0,01 мМ для хлорида кальция (CaCl2).
Инъекционный буфер, предлагаемый в изобретении, может быть гипертоническим, изотоническим или гипотоническим по отношению к соответствующей среде, с которой производится сравнение (референс-среда), т.е. буфер может иметь более высокую, равную, или более низкую концентрацию солей по сравнению с конкретной референс-средой, причем предпочтительно следует применять такие концентрации указанных выше солей, которые не приводят к повреждению клеток, вызываемому осмосом или другими зависящими от концентрации явлениями. Референс-среды, представляют собой, например, жидкости, применяемые в методах in vivo, такие, например, как кровь, лимфатическая жидкость, цитозольные жидкости или другие жидкости, присутствующие в организме, или жидкости, которые можно использовать в качестве референс-сред в методах in vivo, такие как обычные буферы или жидкости. Такие обычные буферы и жидкости известны специалисту в данной области. В качестве жидкой основы наиболее предпочтительным является раствор Рингера лактата.
Однако можно применять также один или несколько совместимых твердых или жидких наполнителей или разбавителей или капсулирующих соединений, пригодных для введения индивидууму. В контексте настоящего описания понятие «совместимый» означает, что компоненты фармацевтической композиции способны смешиваться с обладающим фармацевтической активностью компонентом, с нуклеиновой кислотой, предлагаемой в изобретении, которую применяют в качестве иммуностимулятора или адъюванта, индивидуально, а также с другим компонентом таким образом, что не происходит никакого взаимодействия, которое может существенно снижать фармацевтическую эффективность композиции при ее применении в обычных условиях. Фармацевтически приемлемые носители должны, конечно, иметь достаточно высокую степень чистоты и обладать достаточно низкой токсичностью для того, чтобы их можно было вводить индивидууму, подлежащему лечению. Некоторыми примерами соединений, которые можно применять в качестве фармацевтически приемлемых носителей или их компонентов, являются сахара, такие, например, как лактоза, глюкоза и сахароза; крахмалы, такие, например, как кукурузный крахмал или картофельный крахмал; целлюлоза и ее производные, такие, например, как натриевая соль карбоксиметицеллюлозы, этилцеллюлоза, ацетат целлюлозы; порошкообразный трагакант; солод; желатин; жир; твердые вещества, улучшающие скольжение, такие, например, как стеариновая кислота, стеарат магния, сульфат кальция; растительные масла, такие, например, как арахисовое масло, хлопковое масло, кунжутное масло, оливковое масло, кукурузное масло и масло из теобромы крупноцветковой; полиолы, такие, например, как полипропиленгликоль, глицерин, сорбит, маннит и полиэтиленгликоль; альгиновая кислота.
В принципе выбор фармацевтически приемлемого носителя определяется методом введения фармацевтических композиций, предлагаемых в изобретении. Фармацевтические композиции, предлагаемые в изобретении, можно вводить, например, системно. Пути введения включают, например, трансдермальный, оральный, парентеральный, в том числе подкожные или внутривенные инъекции, местный и/или интраназальный пути. Пригодное количество фармацевтической композиции, которое следует вводить, можно определять с помощью стандартных экспериментов на моделях с использованием животных. Такие модели включают, но, не ограничиваясь только ими, модели с использованием кролика, овцы, мыши, крысы, собаки и приматов кроме человека. Предпочтительные формы для инъекции стандартной дозы включают стерильные водные растворы, физиологический раствор или их смеси. Значение рН таких растворов следует доводить примерно до 7,4. Пригодными для инъекции носителями являются гидрогели, устройства для контролируемого или замедленного высвобождения, полимолочная кислота и коллагеновые матрицы. Пригодными фармацевтически приемлемыми носителями для местного применения являются носители, которые можно использовать в лосьонах, кремах, гелях и т.п. Если соединение предназначено для перорального введения, то предпочтительными стандартными формами лекарственного средства являются таблетки, капсулы и т.п. Фармацевтически приемлемые носители для приготовления стандартных форм лекарственного средства, которые можно применять для орального введения, хорошо известны из существующего уровня техники. Их выбор должен зависеть от дополнительных обстоятельств, таких как вкус, стоимость и способность к хранению, которые не имеют решающего значения для целей настоящего изобретения, и специалист в данной области может без затруднений сделать этот выбор.
Для еще большего повышения иммуногенности в фармацевтическую композицию, предлагаемую в изобретении, можно дополнительно включать одну или несколько вспомогательных субстанций. При этом предпочтительно достигается синергетическое действие нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, и вспомогательной субстанции, которая необязательно дополнительно содержится в фармацевтической композиции (и, в конечном итоге, обладающего фармацевтической активностью компонента). В этом отношении, в зависимости от различных типов вспомогательных субстанций можно рассматривать различные механизмы действия. Например, к первому классу пригодных вспомогательных относятся субстанции, которые обеспечивают созревание дендритных клеток (DC), такие, например, как липополисахариды, TNF-α или лиганд CD40. Как правило, в качестве вспомогательного вещества можно применять любую субстанцию, которая оказывает влияние на иммунную систему в качестве «сигнала опасности» (LPS, GP96 и т.д.), или цитокины, такие как GM-CFS, которые позволяют усиливать иммунный ответ, продуцируемый иммуностимулирующим адъювантом, предлагаемым в изобретении, и/или направленно воздействовать на него. Наиболее предпочтительными вспомогательными субстанциями являются цитокины, такие как монокины, лимфокины, интерлейкины или хемокины, стимулирующие иммунный ответ, такие как IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12, IL-13, IL-14, IL-15, IL-16, IL-17, IL-18, IL-19, IL-20, IL-21, IL-22, IL-23, IL-24, IL-25, IL-26, IL-27, IL-28, IL-29, IL-30, IL-31. IL-32, IL-33, INF-α, IFN-β, INF-γ, GM-CSF, G-CSF, M-CSF, LT-β или TNF-α, факторы роста, такие как hGH.
Другими добавками, которые можно включать в композиции, предлагаемые в изобретении, являются эмульгаторы, такие, например, как Tween®; смачивающие вещества, такие, например, как лаурилсульфат натрия; красители; вещества, улучшающие вкус, фармацевтические носители; вещества для формирования таблеток; стабилизаторы; антиоксиданты; консерванты.
Фармацевтическая композиция, предлагаемая в изобретении (первый (без обладающего фармацевтической активностью компонента) и второй (с обладающим фармацевтической активностью компонентом) вариант осуществления изобретения) может дополнительно содержать также адъювант. Таким образом, нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении в качестве иммуностимулятора или адъюванта (согласно второму варианту фармацевтической композиции, предлагаемой в изобретении), можно объединять с другими иммуностимуляторами/адъювантами. Согласно настоящему изобретению пригодными для этих целей агентами/адъювантами являются прежде всего соединения, которые усиливают (с помощью одного или нескольких механизмов действия) биологическое свойство/свойства (модифицированной или немодифицированной) нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, т.е. прежде всего субстанции, которые усиливают иммуностимулирующее действие нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении. Примерами агентов/адъювантов, которые можно применять согласно изобретению, являются, но, не ограничиваясь только ими, стабилизирующие катионные пептиды или полипептиды, описанные выше, такие как протамин, нуклеолин, спермин или спермидин, и катионные полисахариды, прежде всего хитозан, TDM, MDP, мурамил-дипептид, плюроники, раствор квасцов, гидроксид алюминия, ADJUMER™ (полифосфазен); гель алюминия фосфата; глюканы из водорослей; алгаммулин; гель гидроксида алюминия (квасцы); гель гидроксида алюминия с высокой способностью адсорбировать белок; гель гидроксида алюминия с низкой вязкостью; AF или SPT (эмульсия сквалана (5%), Твин 80 (0,2%), плюроник L121 (1,25%), забуференный фосфатом физиологический раствор, рН 7,4); AVRIDINE™ (пропандиамин); BAY R1005™ ((N-(2-дезокси-2-L-лейциламино-b-D-глюкопиранозил)-N-октадецилдодеканоиламида гидроацетат); CALCITRIOL™ (1α,25-дигидрокси-витамин D3); гель фосфата кальция; САРТМ (наночастицы фосфата кальция); голотоксин холеры, слитый белок токсин холеры-A1-D-фрагмент белка-А, субъединица В токсина холеры; CRL 1005 (блок-сополимер Р1205); содержащие цитокин липосомы; DDA (диметилдиоктадециламмония бромид); DHEA (дегидроэпиандростерон); DMPC (димиристоилфосфатидилхолин); DMPG (димиристоилфосфатидилглицерин); комплекс DOC/квасцы (натриевая соль дезоксихолевой кислоты); полный адъювант Фрейнда; неполный адъювант Фрейнда; гамма-инулин; адъювант Gerbu (смесь: I) N-ацетилглюкозаминил-(Р1-4)-N-ацетилмурамил-L-аланил-D-глутамин (GMDP), II) диметилдиоктадециламмония хлорид (DDA), III) комплексная соль цинк-L-пролина (ZnPro-8); GM-CSF); GMDP (N-ацетилглюкозаминил-(b1-4)-N-ацетилмурамил-L-аланил-D-изоглутамин); имихимод (1-(2-метилпропил)-1Н-имидазо[4,5-с]хинолин-4-амин); ImmTher™ (N-ацетилглюкозаминил-N-ацетилмурамил-L-Ala-D-isoGlu-L-Ala-глицерина дипальмитат); DRV (иммунолипосомы, полученные путем дегидратации/регидратации везикул); интерферон-γ; интерлейкин-1β; интерлейкин-2; интерлейкин-7; интерлейкин-12; ISCOMS™ («стимулирующие иммунную систему комплексы» (Immune Stimulating Complexes)); ISCOPREP 7.0.3.™; липосомы; LOXORIBINE™ (7-аллил-8-оксогуанозин); оральный адъювант LT (лабильный энтеротоксин-протоксин E.coli); микросферы и микрочастицы любого состава; MF59™ (водная эмульсия сквалена); MONTANIDE ISA 51™ (очищенный неполный адъювант Фрейнда); MONTANIDE ISA 720™ (метаболизируемый масляный адъювант); MPL™ (3-Q-дезацил-4'-монофосфорил липид А); липосомы МТР-РЕ и МТР-РЕ ((N-ацетил-L-аланил-D-изоглутаминил-L-аланин-2-(1,2-дипальмитоил-sn-глицеро-3-(гидроксифосфорилокси))этиламид, однонатриевая соль); MURAMETIDE™ (Nac-Mur-L-Ala-D-Gln-OCH3); MURAPALMITINE™ и D-MURAPALMITINE™ (Nac-Mur-L-Thr-D-isoGIn-sn-глицериндипальмитоил); NAGO (нейраминидаза-галактозооксидаза); наносферы или наночастицы любого состава; NISV (везикулы из неионогенного поверхностно-активного вещества); PLEURAN™ (β-глюкан); PLGA, PGA и PLA (гомо- и сополимеры молочной кислоты и гликолевой кислоты; микро-/наносферы); ПЛЮРОНИК L121™; РММА (полиметилметакрилат); PODDS™ (протеноидные микросферы); полиэтиленкарбаматные производные; поли-rA:поли-rU (комплекс полиадениловая кислота-полиуридиновая кислота); полисорбат 80 (Твин 80); белковые кохлеаты (фирма Avanti Polar Lipids, Inc., Алабастер, шт.Алабама); STIMULON™ (QS-21); Quil-A (сапонин Quil-A); S-28463 (4-аминотекдиметил-2-этоксиметил-1Н-имидазо[4,5-с]хинолин-1-этанол); SAF-1™ («композиция адъювантов фирмы Syntex»); протеолипосомы вируса Сендаи и липидные матрицы, содержащие вирус Сендаи; Спан-85 (сорбитантриолеат); Specol (эмульсия Marcol 52, Спан 85 и Твин 85); сквален или Robane® (2,6,10,15,19,23-гексаметилтетракозан и 2,6,10,15,19,23-гексаметил-2,6,10,14,18,22-тетракозагексан); стеарилтирозин (октадецилтирозина гидрохлорид); Theramid (М-ацетилглюкозаминил-N-ацетилмурамил-L-Ala-D-isoGlu-L-Ala-дипальмитоксипропиламид); Theronyl-MDP (Termurtide™ или [thr 1]-MDP; N-ацетилмурамил-L-треонил-D-изоглутамин); Ту-частицы (Ту-ВПЧ или вирусоподобные частицы); липосомы Уолтера-Рида (липосомы, содержащие липид А, адсорбированный на гидроксиде алюминия) и т.п. Липопептиды, такие как Pam3Cys, также являются в достаточной степени пригодными для объединения с нуклеиновой кислотой либо формулы (I), либо формулы (II), предлагаемой в изобретении, когда она присутствует в качестве иммуностимулирующего адъюванта (см. Deres и др., Nature, 342, 1989, cc.561-564).
Адъюванты, как указано выше, можно разделить на несколько классов, включая адъюванты, пригодные для применения в качестве депо и для введения, для костимуляции, адъюванты, которые можно применять в качестве антагонистов и т.д. Предпочтительными адъювантами, пригодными для применения в качестве депо и для введения, могут служить, например, соли алюминия, такие как адъю-фос (Adju-phos), алгидрогель, регидрагель и т.д., эмульсии, такие как CFA, SAF, IFA, MF59, провакс, TiterMax, монтанид, ваксфектин и т.д., сополимеры, такие как оптивакс (Optivax, CRL1005), L121, полоксамер 4010) и т.д., липосомы, такие как «липосомы-невидимки (Stealth)» и т.д., кохлеаты, такие как BIORAL, и т.д., адъюванты растительного происхождения, такие как QS21, Quil A, Iscomatrix, ISCOM, и т.д. Предпочтительными адъювантами, пригодными для костимуляции, могут служить, например, томатин, биополимеры, такие как PLG, РММ, инулин и т.д., адъюванты микробного происхождения, такие как ромуртид, DETOX, MPL, CWS, манноза, CpG7909, ISS-1018, IC31, имидазохинолины, амплиген, Ribi529, IMOxine, IRIV, ВПЧ, токсин холеры, термолабильный токсин, Pam3Cys, флагеллин, GPI (гликозилфосфатидилинозитол)-якорь, LNFPIII/Lewis X, противомикробные пептиды, UC-1V150, слитый белок RSV, cdiGMP и т.д. Предпочтительными адъювантами, пригодными в качестве антагонистов, могут служить, например, нейропептид CGRP и т.д.
Любое соединение, в отношении которого известно, что оно обладает иммуностимулирующей активностью благодаря своей аффинности к связыванию (в качестве лигандов) с Толл-подобными рецепторами, такими как TLR1, TLR2, TLR3, TLR4, TLR5, TLR6, TLR7, TLR8, TLR9, TLR10, TLR11, TLR12 или TLR13, можно использовать в качестве еще одного компонента для дополнительной стимуляции иммунного ответа, индуцируемого нуклеиновыми кислотами, предлагаемыми в изобретении, в фармацевтических композициях, предлагаемых в изобретении.
К другому классу соединений, которые можно добавлять к фармацевтической композиции, предлагаемой в изобретении, относятся содержащие CpG нуклеиновые кислоты (CpG-нуклеиновые кислоты), прежде всего CpG-PHK или CpG-ДНК. CpG-PHK или CpG-ДНК может представлять собой одноцепочечную CpG-ДНК (ss CpG-ДНК), двухцепочечную CpG-ДНК (dsDNA), одноцепочечную CpG-PHK (ss CpG-PHK) или двухцепочечную CpG-PHK (ds CpG-PHK). Предпочтительно CpG-нуклеиновая кислота присутствует в форме CpG-PHK, более предпочтительно в форме одноцепочечной CpG-PHK (ss CpG-PHK). CpG-нуклеиновая кислота предпочтительно содержит по меньшей мере одну или большее количество (митогенных) цитозин/гуанин динуклеотидную(ых) последовательность(ей) (СрС-мотив(ы)). Согласно первому предпочтительному варианту осуществления изобретения в этих последовательностях содержится по меньшей мере один CpG-мотив, т.е. С (цитозин) и G (гуанин) в CpG-мотиве являются неметилированными. Все другие цитозиновые или гуаниновые остатки, необязательно присутствующие в этих последовательностях, могут быть либо метилированными, либо неметилированными. Однако согласно другому предпочтительному варианту осуществления изобретения С (цитозин) и G (гуанин) в CpG-мотиве могут присутствовать также в метилированной форме.
В наиболее предпочтительном варианте осуществления изобретения фармацевтическая композиция, предлагаемая в изобретении, может представлять собой также вакцину. Вакцины, предлагаемые в изобретении, как правило, представляют собой (соответствуют ей) фармацевтическую композицию предлагаемую в изобретении. Состав таких вакцин, предлагаемых в изобретении, характеризуется наличием определенного класса обладающих фармацевтической активностью компонентов, включенных в композицию вакцины. Как правило, обладающее фармацевтической активностью соединение должно представлять собой иммуностимулирующую субстанцию, которая вызывает специфический иммунный ответ против определенного(ых) антигена(ов). Вызываемый специфический иммунный ответ позволяет индивидууму вырабатывать иммунный ответ (проявляющийся в активной или пассивной форме), например, против конкретного патогена или конкретной опухоли.
Фармацевтические композиции и, прежде всего вакцины, предлагаемые в изобретении, характеризуются конкретным способом их введения. Как правило, фармацевтические композиции, предлагаемые в изобретении, прежде всего вакцины, предпочтительно вводят системным путем. Такие композиции/вакцины, как правило, можно вводить с помощью трансдермального, орального, парентерального, включая подкожные и внутривенные инъекции, местного и/или интраназального пути введения. В альтернативном варианте вакцины или фармацевтические композиции, предлагаемые в изобретении, можно вводить с помощью интрадермального, подкожного, внутримышечного пути введения. Таким образом, композиции/вакцины предпочтительно приготавливают в жидкой или твердой форме. В вакцины, предлагаемые в изобретении, предпочтительно следует включать другие вспомогательные субстанции (указанные выше), которые могут еще более повышать иммуногенность, прежде всего вакцины. Предпочтительно следует выбирать одну или несколько их указанных выше вспомогательных субстанций, в зависимости от иммуногенности и других свойств фармацевтически активного компонента, входящего в вакцину, предлагаемую в изобретении.
Согласно еще одному предпочтительному объекту настоящего изобретения фармацевтические композиции, предлагаемые в изобретении, наиболее предпочтительно вакцины, предлагаемые в изобретении, применяют для лечения показаний, указанных в качестве примера ниже. С помощью фармацевтических композиций, предлагаемых в изобретении, наиболее предпочтительно вакцин, предлагаемых в изобретении, можно лечить, например, болезни или состояния, ассоциированные с различным патологическим отсутствием иммунных ответов или которые в контексте терапии требуют иммунного ответа, предпочтительно повышенного иммунного ответа, например, специфические для опухоли или специфические для патогена заболевания, инфекционные болезни и т.д., или заболевания, которые можно лечить путем сдвига (избыточного) иммунного ответа в сторону преобладания ТН1-иммунного ответа и/или путем десенсибилизации пациента, страдающего избыточным иммунным ответом, как в случае аллергий или аутоиммунных заболеваний. Получение указанного иммунного ответа или повышение уже существующего иммунного ответа, но необязательно неадекватного иммунного ответа, с помощью фармацевтической композиции, предлагаемой в изобретении, основано главным образом на ее способности инициировать неантигенспецифический иммунный ответ. Важным фактором получения приемлемого иммунного ответа является стимуляция различных Т-клеточных субпопуляций. Т-лимфоциты, как правило, в результате дифференцировки разделяются на две субпопуляции Т-клетки-хелперы 1 (Th1) и Т-клетки-хелперы (Th2), с помощью которых иммунная система может разрушать внутриклеточные (Th1) и внеклеточные (Th2) патогены (например, антигены). Две Th-клеточные популяции отличаются схемой производства ими эффекторных белков (цитокины). Так, Th1-клетки обусловливают клеточный иммунный ответ путем активации макрофагов и цитотоксических Т-клеток. С другой стороны, Th2-клетки усиливают гуморальный иммунный ответ путем стимуляции конверсии В-клеток в плазменные клетки и путем образования антител (например, против антигенов). Таким образом, соотношение Th1/Th2 имеет очень важное значение для иммунного ответа. В контексте настоящего изобретения соотношении Th1/Th2 иммунного ответа предпочтительно смещают с помощью фармацевтической композиции, предлагаемой в изобретении, которая содержит нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, в сторону клеточного ответа, т.е. Th1-ответа, и при этом главным образом индуцируется клеточный иммунный ответ. Только с помощью указанного смещения и доминирующего или даже исключительного наличия ТН1-иммунного ответа возможно эффективное лечение вышеуказанных показаний. Таким образом, предпочтительно, фармацевтические композиции или вакцины, предлагаемые в настоящем изобретении, применяют для инициации специфических для опухоли или специфических для патогена иммунных ответов. Такие фармацевтические композиции или вакцины, предлагаемые в изобретении, наиболее предпочтительно следует применять для повышения иммунных ответов антигенпрезентирующих клеток (АПК).
Аналогичным образом наиболее предпочтительно следует применять фармацевтические композиции или вакцины, предлагаемые в изобретении, для лечения рака или опухолевых заболеваний, предпочтительно выбранных из группы, включающей карциномы ободочной кишки, меланомы, почечные карциномы, лимфомы, острый миелолейкоз (AML), острый лимфолейкоз (ALL), хронический миелолейкоз (CML), хронический лимфолейкоз (CLL), желудочно-кишечные опухоли, легочные карциномы, глиомы, опухоли щитовидной железы, карциномы молочной железы, опухоли предстательной железы, гепатомы, различные опухоли, индуцируемые вирусом, такие, например, как карцинома, вызываемая вирусом папилломы (например, карцинома шейки матки), аденокарциномы, опухоли, вызываемые вирусом герпеса (например, лимфома Беркитта, В-клеточная лимфома, индуцируемая вирусом Эпштейна-Барре (EBV)), опухоли, индуцируемые вирусом гепатита В (печеночно-клеточная карцинома), лимфомы, вызываемые HTLV-1 и HTLV-2, невромы/невриномы слухового нерва, рак шейки матки, рак легкого, рак глотки, анальные карциномы, глиобластомы, лимфомы, ректальные карциномы, астроцитомы, опухоли головного мозга, рак желудка, ретинобластомы, базалиомы, метастазы в головной мозг, медуллобластомы, рак матки, рак поджелудочной железы, рак яичек, меланомы, карциномы щитовидной железы, рак мочевого пузыря, синдром Хджкина, менингиомы, болезнь Шнеебергера, бронхиальные карциномы, опухоли гипофиза, грибовидный микоз, рак пищевода, рак молочной железы, карциноиды, невриномы, спиналиомы, лимфомы Беркитта, рак гортани, рак почек, тимомы, карцинома тела, рак костей, не-ходжкинские лимфомы, рак уретральный рак, CUP-синдром, опухоли головы/шеи, олигодендроглиомы, рак вульвы, рак кишечника, карциномы ободочной кишки, карциномы пищевода, патологическое перерождение бородавок, опухоли тонкого кишечника, краниофарингеомы, карциномы яичника, опухоли мягких тканей/саркомы, рак яичника, рак печени, карциномы поджелудочной железы, карциномы шейки матки, карциномы эндометрия, метастазы в печень, рак полового члена, рак языка, рак желчного пузыря, лейкоз, плазмоцитомы, рак матки, опухоль века, рак предстательной железы и т.д. Наиболее предпочтительным является вариант, в котором липид, применяемый в модифицированной липидом нуклеиновой кислоте или в качестве фармацевтически активного компонента в композиции, представляет собой α-токоферол (витамин Е), D-α-токоферол, L-α-токоферол, D,L-α-токоферол или витамина Е сукцинат (VES). α-токоферол (витамин Е) характеризуется не очень высокой токсичностью и обладает значительной противоопухолевой активностью (A. Bendich, L.J. Machlin, Am. J. dm. Nutr. 48, 1988, cc.612), что делает его очень привлекательным с точки зрения противораковой терапии. Известны два механизма, позволяющие объяснить ингибирование пролиферации опухолевых клеток или цитотоксическое действие на них: с одной стороны, витамин Е является сильным антиоксидантом и хорошим акцептором радикалов (С.Borek, Ann. NY Acad. Sci. 570, 1990, cc.417); с другой стороны, он обладает способностью предупреждать рост опухоли благодаря стимуляции иммунного ответа (G. Shklar, J. Schwartz, D.P. Trickier, S. Reid, J. Oral Pathol. Med., 19, 1990, с.60). Кроме того, в современных исследованиях была установлена связь между экспрессией гена-супрессора опухоли р53 в опухолевых клетках (плоскоклеточный рак рта) и лечением с использованием витамина Е сукцината (VES) (J. Schwartz, G. Shklar, D. Trickier, Oral Oncol. Europ.J. Cancer 29B, 1993, cc.313). Таким образом, можно наблюдать как стимуляцию производства р53 дикого типа, который действует в качестве супрессора опухоли, так и снижение уровня мутантного р53, который обладает онкогенной активностью. Интересно, что биологическая активность VES в отношении указанных опухолевых клеток зависит от дозы и проявляется по разному в двух диапазонах: в физиологических дозах (от 0,001 до 50 мкмолей/л) она вызывает обнаруживаемое усиление роста клеток; в фармакологических дозах (от 100 до 154 мкмолей/л) она ингибирует рост клеток. Это было продемонстрировано на культуре клеток (Т.М.А. Elattar, A.S. Virji, Anticancer Res. 19, 1999, с.365). С помощью обработки VES можно также индуцировать апоптоз в различных линях клеток рака молочной железы (W.Yu, K. Israel, Q.Y. Liao, С.М. Aldaz, B.G. Sanders, K. Kline, Cancer Res., 59, 1999 с.953). Индукция апоптоза инициируется в результате взаимодействия лиганда Fas и рецептора Fas. Это заслуживает особого внимания, поскольку до сих пор не удавалось выявить такой механизм в соответствующих линиях клеток. Существуют различные изомеры витамина Е, которые различаются количеством и положением метильных групп в ароматическом кольце. В процитированных публикациях применяли обладающую наибольшей биологической активностью форму встречающегося в естественных условиях витамина Е, а именно α-токоферол. Он, в свою очередь, встречается в виде различных стереоизомеров, поскольку молекула содержит три оптически активных центра. Встречающаяся в естественных условиях форма витамина Е представляет собой RRR-α-токоферол (прежнее название D-α-токоферол), однако в настоящее время в основном применяют рацемат (D,L-α-токоферол). Все вышеуказанные формы витамина Е, применяемые в качестве липидов, также подпадают под объем настоящего изобретения.
Наиболее предпочтительно также фармацевтические композиции, предлагаемые в изобретении, применяют для лечения инфекционных болезней. Такие инфекционные болезни предпочтительно выбирают, но не ограничиваясь только ими, из группы, включающей грипп, малярию, SARS (тяжелый острый респираторный синдром), желтую лихорадку, СПИД, боррелиоз Лайма, лейшманиоз, сибирскую язву, менингит, вирусные инфекционные заболевания, такие как СПИД, остроконечную кондилому (кондилома акумината), полые бородавки, лихорадку Денге, флеботомную лихорадку, вирус Эбола, простуду, весенне-летний менингоэнцефалит (FSME), грипп, опоясывающий лишай, герпес, герпес простой типа I, герпес простой типа II, опоясывающий герпес, грипп, японский энцефалит, лихорадку Ласса, вирус Марбурга, корь, ящур, мононуклеоз, эпидемический паротит, инфекцию, вызываемую вирусом Норвалк, железистую лихорадку Пфейффера, натуральную оспу, полиомиелит (детская хромота), ложный круп, инфекционную эритему, бешенство, warts, лихорадку Западного Нила, ветряная оспа, заболевание, вызываемое цитомегаловирусом (CMV), из группы бактериальных инфекционных заболеваний, включающей выкидыш (воспаление предстательной железы), сибирскую язву, аппендицит, боррелиоз, ботулизм, заболевание, вызываемое микроорганизмами Camphylobacter, хламидийная трахома (воспаление уретры, конъюнктивит), холеру, дифтерию, донованоз, эпиглоттит, сыпной тиф, газовую гангрену, гонорею, туляремию, заболевание, вызываемое Heliobacter pylori, коклюш, паховый лимфогранулематоз, остеомиелит, болезнь легионеров, проказу, листериоз, пневмонию, менингит, бактериальный менингит, сибирскую язву, воспаление среднего уха, заболевание, вызываемое Mycoplasma hominis, сепсис новорожденных (хориоамнионит), влажную гангрену, паратиф, чуму, синдром Рейтера, пятнистую лихорадку Скалистых гор, паратиф, вызываемый Salmonella, тиф, вызываемый Salmonella, скарлатину, сифилис, столбняк, триппер, японскую речную лихорадку, туберкулез, тиф, вагинит (кольпит), мягкий шанкр, и из группы инфекционных заболеваний, вызываемых паразитами, простейшими или грибами, включающих амебиаз, шистамоз, болезнь Шагаса, эпидермофию стопы, пятнистость, вызываемая дрожжевыми грибками, чесотку, малярию, онхоцеркоз (речная слепота), или грибных заболеваний, включая токсоплазмоз, трихомониаз, трипаносомоз (сонная болезнь), индийский висцеральный лейшманиоз, «подгузниковый»/«пеленочный» дерматит, шистосомоз, пищевое отравление рыбой (сигуатера), кандидоз, кожный лейшманиоз, лямблиоз (протозойная инфекция, вызываемая Lamblia intestinalis) или сонную болезнь, или инфекционными заболеваниями, вызываемыми Echinococcus, такими как дифиллоботриоз, птичий цепень, собачий солитер (эхинококк), педикулез, бычий цепень, свиной солитер, карликовый цепень.
Таким образом, нуклеиновую кислоту, предлагаемую в изобретении или обладающие фармацевтической активностью композиции, предлагаемые в изобретении, можно применять для приготовления лекарственного средства, предназначенного для лечения аллергических нарушений или заболеваний. Аллергия представляет собой состояние, которое, как правило, сопровождается аномальной приобретенной иммунологической гиперчувствительностью к определенным чужеродным антигенам или аллергенам. Аллергии обычно возникают в результате местного или системного воспалительного ответа на такие антигены или аллергены и приводят к иммунной реакции организма на эти аллергены. В контексте настоящего описания понятие «аллергены» включает, например, различные виды трав, пыльцу, плесень, лекарственные средства или многочисленные провоцирующие факторы окружающей среды и т.д. Не вдаваясь в теорию, можно предполагать, что в развитии аллергии участвуют несколько различных вызывающих заболевание механизмов. Согласно классификации, предложенной Р. Gell и R. Coombs, понятие «аллергия» было ограничено только гиперчувствительностями типа I, которые обусловлены классическим IgE-механизмом действия. Гиперчувствительность типа I характеризуется избыточной активацией под действием IgE тучных клеток и базофилов, приводящей к системному воспалительному ответу, который может вызывать симптомы от слабых, таких как ринорея, до угрожающего жизни анафилактического шока и смерти. К хорошо известным типам аллергии относятся, но не ограничиваясь только ими, аллергическая астма (приводящая к опуханию слизистой оболочки носа), аллергический конъюнктивит (приводящий к покраснению и зуду в конъюнктиве), аллергический ринит («сенная лихорадка»), анафилаксия, ангиодема, атонический дерматит (экзема), крапивница (сыпь), эозинофилия, респираторные аллергии на укусы насекомых, кожные аллергии (вызывающие и приводящие к различным видам сыпи, таким как экзема, сыпь (крапивница) и (контактный) дерматит), аллергии на пищу, аллергии на лекарственные средства и т.д. В настоящем изобретении предложена, например, фармацевтическая композиция, которая содержит аллерген (например, в кошачий аллерген, пылевой аллерген, антиген клещей домашней пыли, растительный антиген (например, антиген пыльцы березы) и т.д.) либо в качестве белка, либо мРНК (или ДНК), кодирующей белковый аллерген, в сочетании с нуклеиновой кислотой, предлагаемой в изобретении. Фармацевтические композиции, предлагаемые в настоящем изобретении, могут сдвигать (избыточный) иммунный ответ в сторону более сильного ТН1-ответа, тем самым подавляя или ослабляя нежелательный IgE-ответ.
В настоящем изобретении предложены также лекарственные средства, предназначенные для лечения аутоиммунных заболеваний. Аутоиммунные заболевания в целом можно разделить на системные и специфические в отношении определенного органа или локализованные аутоиммунные нарушения в зависимости от основных клинико-патологических признаков каждого заболевания. Аутоиммунные заболевания можно подразделять на категории системных синдромов, к которым относятся SLE (системная красная волчанка), синдром Шегрена, склеродерма и ревматоидный артрит и полимиозит, или локальных синдромов, которые могут быть эндокринологическими (DM (сахарный диабет) типа I, тироидит Хашимото, болезнь Аддисона и т.д.), дерматологическими (пузырчатка обыкновенная), гематологическими (аутоиммунная гемолитическая анемия), нервными (рассеянный склероз), или в которых может участвовать практически любая ограниченная масса ткани организма. Аутоиммунные заболевания, подлежащие лечению, можно выбирать из группы, включающей аутоиммунные заболевания типа I, аутоиммунные заболевания типа II, аутоиммунные заболевания типа III и аутоиммунные заболевания типа IV, такие, например, как рассеянный склероз (MS), ревматоидный артрит, диабеты, диабет типа I (сахарный диабет), системная красная волчанка (SLE), хронический полиартрит, базедова болезнь, аутоиммунные формы хронического гепатита, язвенный колит, аллергические заболевания типа I, аллергические заболевания типа II, аллергические заболевания типа III, аллергические заболевания типа IV, фибромиалгия, потеря волос, болезнь Бехтерева, болезнь Крона, тяжелая псевдопаралитическая миастения, нейродермит, ревматическая полимиалгия, прогрессирующий системный склероз (PSS), псориаз, синдром Рейтера, ревматический артрит, васкулит и т.д., или диабет типа II.
Хотя точный механизм, посредством которого иммунная система индуцирует иммунный ответ на аутоантигены, до сих пор не выяснен, имеется ряд данных касательно этиологии. Так, аутореакция может быть обусловлена отсутствием участия Т-клеток. В здоровой иммунной системе происходит активация В-клеток Т-клетками до того, как первые становятся способными продуцировать антитела в больших количествах. В редких случаях может отсутствовать необходимость в Т-клетках, например, в случае заражения организмами, продуцирующими суперантигены, обладающие способностью инициировать поликлональную активацию В-клеток или даже Т-клеток путем непосредственного связывания с β-субъединицей Т-клеточных рецепторов неспецифическим путем. Согласно другой версии аутоиммунные заболевания происходят вследствие молекулярной мимикрии. Экзогенный антиген может обладать структурным сходством с определенными антигенами организма-хозяина; таким образом, любое антитело, продуцируемое против этого антигена (который имитирует аутоантигены), теоретически может также связываться с антигенами организма-хозяина и усиливать иммунный ответ. Наиболее выраженная форма молекулярной мимикрии выявлена для бета-гемолитического стрептококка группы А, который обладает сходством с антигенами человеческого миокарда и ответствен за сердечные симптомы ревматической атаки. Таким образом, настоящее изобретение позволяет создавать фармацевтическую композицию, содержащую аутоантиген (в качестве белка, мРНК или ДНК, кодирующей белок аутоантигена) и нуклеиновую кислоту, предлагаемую в изобретении, которая, как правило, позволяет десенсибилизировать иммунную систему
Изобретение относится также к применению описанной выше нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, или их обеих, для приготовления фармацевтической композиции, предлагаемой в изобретении, или вакцины, предлагаемой в изобретении, которая предназначена для лечения описанных выше показаний, например, для лечения указанных опухолевых, аутоиммунных заболеваний, различных типов аллергии и инфекционных болезней. В альтернативном варианте изобретение относится к (терапевтическому) применению нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, или их обеих для лечения опухоли или инфекционных болезней, указанных выше.
Настоящее изобретение относится также к наборам содержащим нуклеиновую кислоту либо формулы (I), либо формулы (II), предлагаемую в изобретении, или обе нуклеиновые кислоты, и/или фармацевтическую композицию, предлагаемую в изобретении, и/или вакцину, предлагаемую в изобретении, а также необязательно технические инструкции по применению, содержащие информацию о введении и дозе нуклеиновой кислоты либо формулы (I), либо формулы (II), предлагаемой в изобретении, и/или фармацевтической композиции, предлагаемой в изобретении, и/или вакцины, предлагаемой в изобретении.
Изобретение относится также к способам лечения нарушения или заболевания, выбранного из группы, включающей раковые заболевания, инфекционные болезни, аутоиммунные заболевания и аллергии, заключающимся в том, что пациенту, нуждающемуся в этом, вводят в фармацевтически эффективном количестве нуклеиновую кислоту, предлагаемую в изобретении.
Описание чертежей:
На чертежах показано:
на фиг.1 - стимуляция мышиных BMDC (дендритные клетки костного мозга) нуклеиновыми кислотами, предлагаемыми в изобретении, последовательности которых представлены в SEQ ID NO:1 и 2. Стимуляция оказалась наиболее выраженной в случае SEQ ID NO:1. Высвобождение IL-6 и IL-12 (нг/мл) определяли в качестве меры иммунной стимуляции (см. пример 2). В качестве положительного контроля применяли обладающую иммуностимулирующим действием некэпированную мРНК бета-галактоз ид азы (lacZ) дикого типа, в виде комплекса с протамином;
на фиг.2 - стимуляция человеческих РВМС (hPBMC) олигонуклеотидами, предлагаемыми в изобретении, последовательности которых представлены в SEQ ID NO:1 и 2. Стимуляция оказалась наиболее выраженной в случае SEQ ID NO:1. Определяли высвобождение интерлейкина-6 (IL-6) и TNF-альфа (нг/мл) в качестве меры иммунной стимуляции (см. пример 2);
на фиг.3 - стимуляция человеческих РВМС (hPBMC) олигонуклеотидами, предлагаемыми в изобретении, последовательности которых представлены в SEQ ID NO:1, 2, 3, 4 и 5. Стимуляция оказалась наиболее выраженной в случае SEQ ID NO:1. Определяли высвобождение интерлейкина-6 (IL-6) (нг/мл) в качестве меры иммунной стимуляции (см. пример 2);
на фиг.4 - различные возможные варианты, предлагаемые в изобретении, концевой модификации нуклеиновых кислот либо формулы (I), либо формулы (II), предлагаемых в изобретении, с помощью липидов. В частности, представлены модифицированные липидом линкеры и бифункциональные пептиды соответственно, которые можно применять для сочетания или синтеза нуклеотидных последовательностей (сокращенно ODN-последовательность);
на фиг.5 - приведенный в качестве примера путь синтеза (трифункциональный) модифицированных липидом линкеров, с помощью которых, например, модификацию токоферолом, можно интродуцировать на 3'-конец нуклеиновой кислоты. Указанные соединения, приведенные в качестве примера, представляют собой промежуточные продукты при получении модифицированных липидами на 5'- или 3'-конце нуклеиновых кислот, предлагаемых в изобретении, и адъювантов, предлагаемых в изобретении;
На фиг.6 - приведенный в качестве примера бифункциональный липид с сукцинильным якорем, который применяли для 3'-модификации нуклеиновой кислоты бифункциональным липидом, например. ПЭГ;
На фиг.7 - схематическое изображение сочетания модифицированных липидом амидитов с 5'-концом нуклеиновых кислот;
На фиг.8 - стимуляция человеческих РВМС (hPBMC) олигонуклеотидами, предлагаемыми в изобретении, последовательности которых представлены в SEQ ID NO:78 и 79 (формула (I)). Стимуляция оказалась наиболее выраженной в случае SEQ ID NO:78, а также SEQ ID NO:79;
На фиг.9 - стимуляция человеческих РВМС (hPBMC) олигонуклеотидами, предлагаемыми в изобретении, последовательности которых представлены в SEQ ID NO:78 (формула (I)) и 81 (формула (II)), в сочетании с протамином и без него. Стимуляция оказалась наиболее выраженной при использовании протамина в случае SEQ ID NO:81. Неожиданно было установлено, что в случае SEQ ID NO:78 аналогичная стимуляция имеет место как при использовании протамина, так и без него (см. фиг.9);
На фиг.10 - стимуляция человеческих РВМС (hPBMC) олигонуклеотидами, предлагаемыми в изобретении, последовательности которых представлены в SEQ ID NO:81, 82 и 83 (формула (II)). Стимуляция оказалась очень высокой во всех изученных случаях, т.е. при использовании SEQ ID NO:81, 82 и 83;
на фиг.11 - высвобождение TNF-альфа после стимуляции человеческих РВМС (hPBMC) олигонуклеотидами формулы (I), предлагаемыми изобретении. Трансфекцию осуществляли с олигофектамином и без него. Последовательности, применяемые для трансфекции с олигофектамином, представляли собой а) SEQ ID NO:1 (Seq. 1), 6) SEQ ID NO:1, модифицированную на 3'-, 5'-конце токоферолом, в сравнении с в) RNA40 (SEQ ID NO:87); Последовательности, применяемые для трансфекции без олигофектамина, представляли собой г) среду (без последовательности), д) SEQ ID NO:1 (Seq. 1) и e) SEQ ID NO:1, модифицированную на 3'-,5'-конце токоферолом. Наилучшие результаты получали, когда для трансфекции применяли SEQ ID NO:I (Seq. 1), SEQ ID NO:1, модифицированную на 3'-,5'-конце токоферолом, в сочетании с олигофектамином. Таким образом, токоферол повышал трансфекцию РНК и тем самым повышал иммуностимуляцию;
на фиг.12 - системное высвобождение IL-12 после i.v. (внутривенной) инъекции олигонуклеотидов формулы (I), предлагаемых в изобретении. На графике представлены результаты трансфекции: а) буфером в качестве контроля, б) RNA40 (SEQ ID NO:87), в) RNA40 (SEQ ID NO:87) в виде комплекса с олигофектамином, г) SEQ ID NO:1 (Seq. 1) и д) SEQ ID NO:1, модифицированной на 3'-,5'-конце токоферолом. Как видно из графика, наилучшие и результаты и, как следствие, выраженная иммуностимуляция выявлена при использовании предлагаемой в изобретении SEQ ID NO:1, модифицированной на 3'-,5'-конце токоферолом;
на фиг.13 - результаты сравнительного эксперимента и воздействие различных модификаций предлагаемой в изобретении последовательности, представленной в SEQ ID NO:1, на иммунностимуляцию человеческих РВМС (hPBMC) и высвобождение TNF-альфа. В эксперименте применяли SEQ ID NO:1 (формулы (I)), модифицированную холестерином, SEQ ID NO:1 (формулы (I)), модифицированную токоферолом, и SEQ ID NO:1, модифицированную на 3'-,5'-конце токоферолом, при этом наилучшие результаты получены при использовании последней модификации. Однако модификации SEQ ID NO:1 либо холестерином, либо токоферолом, приводили также к существенной стимуляции hPBMC;.
На фиг.14 - воздействие вакцинации на размер опухоли при использовании a) IFA (неполный адъювант Фрейнда), б) IFA и куриный овальбумин, в) IFA и куриный овальбумин, и RNA40 (SEQ ID NO:87) и г) IFA и куриный овальбумин, и предлагаемая в изобретении последовательность SEQ ID NO:1 в виде комплекса с протамином. Наилучшие результаты, т.е. существенное снижение размера опухоли, получены при применении IFA и куриного овальбумина, и предлагаемой в изобретении последовательности SEQ ID NO:1 (г), что свидетельствует о том, что предлагаемые в изобретении последовательности обладают очень высокой активностью в качестве адъювантов;
На фиг.15 - воздействие внутриопухолевой инъекции на размер опухоли при использовании а) только ЗФР, б) предлагаемой в изобретении последовательности SEQ ID NO:1, модифицированной на 3'-, 5'-конце токоферолом, в ЗФР, в) предлагаемой в изобретении последовательности SEQ ID NO:1 в ЗФР и г) RNA40 (SEQ ID NO:87) в ЗФР. Наилучшие результаты, т.е. существенное снижение размера опухоли, получены при применении предлагаемой в изобретении последовательности SEQ ID NO:1, модифицированной на 3'-, 5'-конце токоферолом, далее по эффективности следовала немодифицированная предлагаемая в изобретении последовательность SEQ ID NO:1.
Примеры
1. Синтез приведенных в качестве примера нуклеиновых кислот формулы (I), предлагаемых в изобретении
Олигонуклеотиды РНК, в качестве примеров нуклеиновой кислоты общей формулы (I) GlXmGn (SEQ ID NO:1-80) и общей формулы (II) ClXmCn (SEQ ID NO:81-83), предлагаемой в изобретении, получали с помощью автоматического твердофазного синтеза с использованием фосфорамидитной химии. В каждом случае РНК-специфические 2'-гидроксильные группы нуклеотидов защищали с помощью защитных групп TBDMS. При синтезе фосфоротиоатов применяли реагент Бекаже для окисления. Расщепление носителя и лабильных в щелочной среде защитных групп осуществляли с помощью метиламина, а для отщепления защитной группы TBDMS применяли гидрохлорид триэтиламина.
Неочищенный продукт очищали с помощью ЖХВР либо с использованием ион-парной хроматографии, либо ионообменной хроматографии или комбинации обоих методов, обессоливали и сушили. Определяли чистоту продукта и правильный состав оснований с помощью масс-спектрометрии.
2. In vitro иммунностимуляции с помощью приведенных в качестве примера нуклеиновых кислот формулы (I), предлагаемых в изобретении
а) Для стимуляции мышиных BDMC (дендритные клетки костного мозга) 3 мкл олигофектамина смешивали с 30 мкл не содержащей FCS среды IMDM (фирма BioWhittaker, каталожный №. BE12-722F) и инкубировали при комнатной температуре в течение 5 мин. Смешивали 6 мкг нуклеиновых кислот формулы (I), предлагаемой в изобретении, последовательности которых представлены в SEQ ID NO:1-2, в форме РНК с 60 мкл не содержащей FCS среде IMDM и смешивали с олигофектамином/IMDM, и инкубировали в течение 20 мин при комнатной температуре. По 33 мкл указанной смеси сносили для культивирования в течение ночи в лунки 96-луночного культурального титрационного планшета, каждая из которых содержала по 200000 мышиных BDMC в 200 мкл не содержащей FCS среде IMDM. Через 4 ч добавляли 100 мкл среды IMDM, содержащей 20% FCS, и после 16-часовой совместной инкубации супернатант удаляли и оценивали содержание интерлейкина-6 (IL-6) и интерлейкина-12 (IL-12) с помощью ELISA для цитокинов. Осуществляли сравнительные анализы, аналогичные проводимым с использованием последовательностей SEQ ID NO:1 и 2, предлагаемых в изобретении, с применением обладающей иммуностимулирующим действием некэпированной мРНК бета-галактозидазы (lacZ) дикого типа в виде комплекса с протамином.
Было продемонстрировано, что нуклеиновые кислоты формулы (I), предлагаемые в изобретении, присутствующие в форме РНК, в частности, которые имеют последовательности, предлагаемые в изобретении, которые представлены в SEQ ID NO:1 и 2, обладают хорошими иммуностимулирующими свойствами. В частности, SEQ ID NO:1 обладала иммуностимулирующими свойствами, существенно превышающими иммуностимулирующую активность некэпированной мРНК бета-галактозидазы (lacZ) дикого типа, которую применяли для сравнения.
б) Человеческие РВМС получали с использованием разделения в градиенте плотности фиколла и культивировали в течение ночи в среде X-VIVO-15 (фирма BioWhittaker, каталожный № BE04-418Q), содержащей 1% глутамина и 1% пенициллина, в присутствии 10 мкг/мл нуклеиновых кислот формулы (I), предлагаемых в изобретении, а форме РНК, в частности, которые имеют последовательности, предлагаемые в изобретении, представленные в SEQ ID NO:1 и 2 (см. фиг.2) и SEQ ID NO:1-5 (см. фиг.3) соответственно.
Для стимуляции 3 мкл олигофектамина смешивали с 30 мкл среды X-VIVO-15 (фирма BioWhittaker, каталожный № BE04-418Q) и инкубировали при комнатной температуре в течение 5 мин. Смешивали 6 мкг нуклеиновых кислот формулы (I), предлагаемых в изобретении, а форме РНК, в частности, которые имеют последовательности, предлагаемые в изобретении, представленные в SEQ ID NO:1 и 2 (см. фиг.2) и SEQ ID NO:1-5 (см. фиг.3) соответственно, с 60 мкл среды X-VIVO-15 (фирма BioWhittaker, каталожный № BE04-418Q) и после смешения с олигофектамином/средой X-VIVO, инкубировали в течение 20 мин при комнатной температуре. Затем по 33 мкл этой смеси помещали для культивирования в течение ночи в лунки 96-луночного культурального титрационного микропланшета, каждая из которых содержала 200000 РВМС в 200 мкл среды X-VIVO-15 (фирма BioWhittaker, каталожный № BE04-418Q). После совместной инкубации в течение 16 ч супернатант удаляли и оценивали содержание интерлейкина-6 (IL-6) и интерлейкина-12 (IL-12) и TNFα с помощью ELISA для анализа цитокинов. Осуществляли сравнительные анализы, аналогичные проводимым с использованием последовательностей SEQ ID NO:1 и 2, предлагаемых в изобретении, с применением обладающего иммуностимулирующим действием олигонуклеотида RNA40 (5'-GCCCGUCUGUUGUGUGACUC-3', SEQ ID NO:87).
Удалось продемонстрировать, что нуклеиновые кислоты формулы (I), предлагаемые в изобретении, присутствующие в форме РНК, в частности, которые имеют последовательности, предлагаемые в изобретении, которые представлены в SEQ ID NO:1 и 2, обладают хорошими иммуностимулирующими свойствами. В частности, SEQ ID NO:1 обладала иммуностимулирующими свойствами, существенно превышающими иммуностимулирующую активность РНК-олигонуклеотида, применяемого для сравнения.
3. In vivo иммунностимуляции с помощью приведенных в качестве примера нуклеиновых кислот формулы (I), предлагаемых в изобретении, при их применении в качестве адъюванта
Мышам линии BALB/c (5 особей группу) инъецировали белок β-галактозидазу и адъювант (указанный в настоящем описании) в дни 0 и 10. Мышей умерщвляли в день 20 и сыворотку крови применяли для анализа антител к белку β-галактозидазы с помощью ELISA, а уровень IL-6, IL-12 и TNF-α определяли аналогично методу, описанному выше для анализа in vitro на культурах.
4. Синтез 1-(4,4'-диметокситритил)полиэтиленгликоля (DMT-ПЭГ1500)

Процедура: 21 г ПЭГ1500 (14 ммолей) растворяли дважды для сушки, используя каждый раз по 30 мл абсолютного пиридана, который затем отгоняли азеотропически. Высушенный исходный продукт растворяли в 35 мл абсолютного пиридина. К этому раствору добавляли по каплям 4,7 г 4,4'-диметокситритилхлорида (13,9 ммоля), растворенного в 35 мл абсолютного пиридина, в течение 30 мин. Перемешивание продолжали в течение 2 ч при КТ, при этом осуществляли мониторинг протекания реакции с ТСХ (тонкослойная хроматография). Помимо того, что выявляли DMT-группы с помощью УФ-лампы, пластины для ТСХ оценивали в две стадии: 1 - в насыщенной НС1 атмосфере для выявления DMT; 2 - в йодной камере для выявления ПЭГ; ПЭГ выявляли также с помощью реагента для опрыскивания Драгендорфа-Бюргера. После завершения реакции растворитель удаляли и продукт растворяли в 50 мл дихлорметана (ДХМ). Органическую фазу промывали дважды 25 мл 5%-ного раствора NaHCO3 и дважды 25 мл Н2О. Фазовое разделение водной и органической фазы происходило в течение длительного времени, поскольку ПЭГ является и гидрофобным, и гидрофильным по своей природе. После сушки над Na2SO4 растворитель удаляли и неочищенный продукт очищали колоночной хроматографией на силикагеле, используя смесь ДХМ/МеОН/ТЭА (триэтаноламин) в соотношении 18:2:0,5. Содержащие продукт фракции идентифицировали с помощью ТСХ, объединяли и концентрировали досуха. Получали масло желтоватого цвета, которое после сушки в глубоком вакууме превращалось в воскоподобное твердое вещество.
Выход: 18,3 г (72,5% от теоретического)
ТСХ (ДХМ/МеОН/ТЭА=18:2:0,5): значение Rf≈0,5 (распространение сигнала соответствует распределению молекулярной массы ПЭГ).
5. Синтез 1-(4,4'-диметокситритил)гексаэтиленгликоля (РМТ-ГЭГ)
Процедура; 10 г гексаэтиленгликоля (35 ммолей) сушили с помощью совместного упаривания с 2×30 мл абсолютного пиридина и затем растворяли в 20 мл абсолютного пиридина. Аналогично процедуре, описанной в примере 1, ГЭГ подвергали взаимодействию с 10 г DMT-C1 (29,5 ммоля), растворенного в 50 мл абсолютного пиридина. Очистку с помощью колоночной хроматографии осуществляли с помощью этилацетата/ТЭА=95:5. В качестве высушенного продукта получали вязкое масло желтого цвета.
Выход: 12,5 г (60,5% от теоретического)
ТСХ (ДХМ/МеОН=95:5): значение Rf=0,59
МС (FD): m/z 583,9 (М+)
1H-ЯМР (CDCl3): δ 3,21 (t, DMT-O-CH2-), 3,47-3,68 (m, -CH2-), 3,76 (s, -СН3), 6,77-7,46 (m, ароматическое соединение).
6. Синтез сукцината 1-(4,4'-диметокситритил)полиэтиленгликоля (DMT-ПЭГ-Suc)
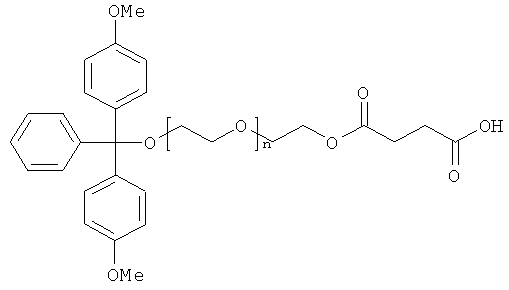
Описанную ниже процедуру можно применять для получения как DMT-ПЭГ1500, так DMT-ГЭГ.
Процедура: 5 г DMT-ПЭГ1500 (2,8 ммоля) растворяли в 25 мл смеси ДХМ/пиридин=5:1 и добавляли 420 мг янтарного ангидрида (4,2 ммоля, т.е. 1,5 экв.), растворенного в 7 мл пиридина, и 170 мг DMAP (1,4 ммоля, т.е. 0,5 экв.), растворенного в 3 мл пиридина. После перемешивания в течение 12 ч при КТ растворители удаляли в вакууме и остаток растворяли в ДХМ. Органическую фазу отмывали трижды раствором NaHCO3 (10% в Н2О) и дважды насыщенным водным раствором NaCl для удаления избытка янтарной кислоты. После сушки над Na2SO4 растворитель удаляют. После полной сушки в глубоком вакууме сукцинаты можно применять без дополнительной очистки для дальнейшей обработки с целью сочетания с модифицированными аминогруппой носителями.
TCX: DMT-ПЭГ1500-Suc (ДХМ/МеОН/ТЭА=18:2:0,5): значение Rf 0.41
DMT-ГЭГ-Suc (DCM/MeOH=9:2): значение Rf=0,70.
7. Синтез 1-тозил-2,3-изопропилиденглицерина

Процедура: 200 ммолей изопропилиденглицерина (26,4 г) растворяли в 200 мл ацетонитрила и добавляли 22,2 г триэтиламина (220 ммолей). Растворяли 220 ммолей хлорангидрида пара-толуолсульфоновой кислоты (41,9 г) в 250 мл ацетонитрила и добавляли по каплям к реакционной смеси при перемешивании в течение 2 ч. Перемешивание продолжали в течение еще 20 ч при КТ, при этом образовывался осадок белого цвета, который отфильтровывали после завершения реакции. Растворитель удаляли и неочищенный продукт очищали колоночной хроматографией на силикагеле с использованием смеси н-гексан/этилацетат=2:1. Содержащие продукт фракции идентифицировали с помощью TCX, объединяли и концентрировали досуха. Продукт сушили в глубоком вакууме. Получали масло желтоватого цвета (L.N. Markovskii и др., J. Org. Chem. UdSSR 26, 1990, с.2094.).
Выход: 31,3 г (54,7% от теоретического)
TCX (н-гексан/этилацетат=2:1): значение Rf=0,2
МС (FD): m/z 272.0 (M+-CH) (286,3 рассчитанное значение)
1Н-ЯМР (CDCl3): δ 1,31 (s); 1,34 (s); 2,45 (s); 3,74-3,79 (m); 3,90-4,08 (m); 4,23-4,32 (m); 7,36 (d); 7,77 (d)
13С-ЯМР: δ 21,7; 25,2; 26,7 (метил-С); 66,2; 69,5; 72,9 (глицерин-С); 110,1 (метилен-С); 128,0; 129,9; 132,7; 145,1 (ароматическое соединение-С)
8. Синтез 2,3-изопропилиден-1-D,L-α-токоферолглицерина (Toc1)

Процедура: 5,6 г порошкообразного гидроксида калия (100 ммолей) добавляли к 56 ммолям D,L-α-токоферола (24,06 г) в 280 мл ДМСО. После перемешивания в течение 2 ч при КТ, исключая доступ света, добавляли по каплям 56 ммолей 1-тозил-2,3-изопропилиденглицерина (16 г), растворенного в 20 мл ДМСО, и перемешивание продолжали в течение еще 12 ч при 60°C. Затем реакционную смесь гидролизовали 1 л ледяной воды и водную фазу экстрагировали 1,5 л толуола. После сушки над сульфатом натрия растворитель удаляли. Неочищенный продукт очищали колоночной хроматографией на силикагеле с использованием смеси н-гексан/этилацетат=1:1. Содержащие продукт фракции идентифицировали с помощью ТСХ, объединяли и концентрировали досуха. Продукт сушили в глубоком вакууме и хранили, исключая доступ света. Получали масло желтоватого цвета (D.W. Will, Т. Brown Tetrahedron Lett. 33, 1992, с.2729).
Выход: 24,2 г (79,2% от теоретического)
ТСХ (н-гексан/этилацетат=1:1): значение Rf=0,69
МС (FD): m/z 544,6 (M+)
9. Синтез 1-D,L-α-токоферилглицерина (Тос2)
Процедура: Растворяли 16,9 ммоля Toc1 (9,2 г) в 100 мл HCl(2М)/ТГФ (1:1) и перемешивали в течение 2 ч при КТ, исключая доступ света. Затем растворитель удаляли, к остатку добавляли 2×50 мл абсолютного этанола и смесь вновь концентрировали досуха. Неочищенный продукт очищали колоночной хроматографией на силикагеле с использованием смеси диэтиловый эфир/толуол=1:1. Содержащие продукт фракции идентифицировали с помощью ТСХ, объединяли и концентрировали досуха. Продукт сушили в глубоком вакууме и хранили, исключая доступ света. Получали масло желтого цвета.
Выход: 6,6 г (77,5% от теоретического)
ТСХ (диэтиловый эфир /толуол=1:1): значение Rf=0,22
МС (FD): m/z 504,4 (М+)
10. Синтез [1-(4,4'-диметокситритил)]-3-D,L-α-токоферилглицерина (Тос3)

Процедура: Для сушки дважды растворяли 6,6 г Тос2 (13 ммолей) в 15 мл абсолютного пиридина, который затем отгоняли азеотропически. Высушенный исходный продукт растворяли в 50 мл абсолютного пиридина и добавляли 5,34 г DMT-Cl (15,8 ммоля). После перемешивания в течение 12 ч при КТ, исключая доступ света, реакцию прекращали, добавляя 50 мл метанола, и реакционную смесь концентрировали досуха. Остаток растворяли в 500 мл дихлорметана и промывали дважды 150 мл водного насыщенного раствора NaCl, а затем однократно 150 мл воды. После сушки над Na2SO4 растворитель удаляли и остаток очищали колоночной хроматографией на силикагеле (н-гексан/диэтиловый эфир/триэтиламин=40:60:1). Содержащие продукт фракции идентифицировали с помощью ТСХ, объединяли и концентрировали досуха. Продукт сушили в глубоком вакууме. Получали масло желтоватого цвета, которое хранили, исключая доступ света.
Выход: 8,5 г (81% от теоретического)
ТСХ (н-гексан/диэтиловый эфир/ТЭА=40:60:1): значение Rf=0,43
МС (FD): m/z 807,2
11. Синтез of[1-(4,4'-диметокситритил)]-2-сукцинил-3-D,L-α-токоферилглицерина (Тос4)

Процедура: Растворяли дважды с целью сушки 1,45 г Тос3 (1,8 ммоля), используя каждый раз по 5 мл абсолютного пиридина, который вновь отгоняли азеотропически. После растворения исходного продукта в 8 мл абсолютного пиридина к нему добавляли 140 мг DMAP (1,08 ммоля) и 194,4 мг янтарного ангидрида (1,8 ммоля) в противотоке аргона. Затем смесь перемешивали в течение 18 ч при КТ, исключая доступ света. Для обработки к реакционному раствору добавляли 45 мл ДХМ и четырежды промывали, используя каждый раз по 50 мл воды. После сушки на сульфатом натрия растворитель удаляли и неочищенный продукт очищали колоночной хроматографией на силикагеле, используя смесь этилацетат/метанол/NH3 (25% в Н2О)=5:1:1. Содержащие продукт фракции идентифицировали с помощью ТСХ, объединяли и концентрировали досуха. После сушки в глубоком вакууме получали вязкое масло коричневатого цвета, которое хранили, исключая доступ света.
Выход: 1,35 г (82,8% от теоретического)
TLC (EtOAc/NH3/МеОН=5:1:1); значение Rf value=0,30
МС (FD): m/z 906,2 (M+), 604,2 (M+-DMT)
12. Синтез D,L-α-токоферил-β-цианэтил-N,N-диизопропил-фосфорамидита

Процедура: Добавляли 2×25 мл пиридина к 5 г D,L-α-токоферола (11,6 ммоля и смесь сушили путем азеотропического уноса. Исходный продукт растворяли в 40 мл ДХМабс. В противотоке аргона медленно по каплям добавляли 7,9 мл DIРЕАабс.(46,4 ммоля) и 2,5 мл 2-цианэтил-N,N-диизопропилфосфинхлорида (11 ммолей). После перемешивания реакционной смеси в течение 1 ч при КТ, исключая доступ света, ее разводили 100 мл смеси этилацетат/ТЭА (20:1) и промывали дважды 25 мл 10% NaHCO3 и дважды насыщенным раствором NaCl. Затем органическую фазу сушили над Na2SO4 и растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле, используя этилацетат.
Выход: 5,62 г (81% от теоретического)
ТСХ (EtOAc): значение Rf=0,75
MC(FD):m/z 630,1 (M+)
31Р-ЯМР: δ 152,24
13. Синтез [1-(4,4'-диметокситритил)]-(3-D,L-α-токоферил)глицерин-2-фосфорамидита

Процедура: Растворяли 1 г Тос3 (1,24 ммоля) с целью сушки, используя каждый раз по 10 мл абсолютного пиридина, который вновь отгоняли азеотропически. Затем исходный продукт растворяли 20 мл ДХМабс., и по каплям в противотоке аргона добавляли 0,84 мл DIРЕАабс. (4,96 ммоля). Затем медленно по каплям в противотоке аргона добавляли 0,27 мл 2-цианэтил-N,N-диизопропилфосфинхлорида (1,19 ммоля). Затем реакционную смесь перемешивали в течение 1 ч при КТ, после чего раствор разводили 50 мл смеси этилацетат/ТЭА (20:1). Органическую фазу промывали дважды 15 мл 10%-ного раствора NaHCO3 и дважды насыщенным раствором NaCl и сушили над Na2SO4. Растворитель удаляли и неочищенный продукт очищали колоночной хроматографией на силикагеле, используя смесь этилацетат/ТЭА (99:1). Содержащие продукт фракции идентифицировали с помощью ТСХ, объединяли и концентрировали досуха. После сушки в глубоком вакууме получали очень вязкое масло желтовато-коричневого цвета, которое охлаждали и хранили, исключая доступ света.
Выход: 0,76 г (63,4% от теоретического)
ТСХ (EtOAc, 1% ТЭА): значение Rf=0,68
МС (FD): m/z 1006.4 (M+), 953,6 (М+-цианэтил), 651,4 (M+-DMT,-цианэтил), 603,1 (M+-DMT,-диизопропиламин), 303,1 (DMT+)
31Р-ЯМР: δ 150,5
14. Синтез 1-гексадецил-2,3-изопропилиденглицерина (Pam1)

Процедура: Добавляли порциями в противотоке аргона 0,11 моля гидрида натрия (2,42 г) к 0,1 моля D,L-α,β-изопропилиденглицерина (12,4 мл) в 500 мл ТГФабс. После перемешивания в течение 12 ч при КТ к образовавшемуся алкоголяту добавляли по каплям 0,11 моля 1-бромгександекана (33,6 мл) в 80 мл ТГФабс. После добавления 0,5 ммоля йодида тетрабутиламмония в качестве катализатора смесь выдерживали при температуре кипения в течение 12 ч. После охлаждения реакционной смеси образовавшейся бромид натрия отфильтровывали и фильтрат концентрировали досуха. Остаток растворяли в диэтиловом эфире и эфирную фазу экстрагировали путем трехкратного встряхивания с H2O. После сушки органической фазы над Na2SO4 смесь концентрировали досуха и остаток очищали колоночной хроматографией на силикагеле (EtOAc/н-гексан=1:9) (S. Czernecki, С.Georgoulis, С.Provelenghiou Tetrahedron Lett. 39, 1976, с.3535).
Выход: 18,5 г (52% от теоретического)
ТСХ (EtOAc/н-гексан=1:9): значение Rf=0,47
МС (FD): m/z 357,6 (M++1)
1Н-ЯМР (CDCl3): δ 0.80 (t), 1,18 (s), 1,35 (s), 1,37 (s), 3,30-3,48 (m), 3,63-3,69 (dd), 3,96-4,01 (dd), 4,19 (q)
13С-ЯМР (CDCl3): δ 14,1; 22,7; 25,4; 26,1; 26,8; 29,4; 29,6; 29,7; 31,9 (алкильная цепь), 67,0 (алкил-С-O-), 71,8; 71,9; 74,7 (глицерин-С), 109,3 (кетил-С).
15. Синтез 1-гексадецилглицерина (Pam2)
Процедура: Перемешивали 18,5 г Pam1 (52 ммоля) в 300 мл уксусной кислоты (65%) в течение 24 ч при 40°С. Осадок белого цвета отфильтровывали и концентрировали досуха несколько раз с помощью н-гексана. Для очистки продукт суспендировали трижды в н-гексане и отфильтровывали. Исходный продукт, у которого в отличие от продукта не были удалены защитные группы, был растворим в н-гексане и поэтому его можно было отделять. Полученный остаток сушили в вакууме. Объединенные фильтраты также концентрировали досуха и вновь подвергали процедуре разделения (Н. Paulsen, E. Meinjohanns, F. Reck, I. Brockhausen Liebigs Ann. Chem, 1993, с.721).
Выход: 14,6 г (88% от теоретического)
ТСХ (EtOAc/н-гексан=1:1): значение Rf=0,2
MC(FD): m/z 317,4 (M++1)
1H-ЯМР (CDCl3): δ 0,86 (t), 1,23 (s), 3,30-3,48 (m), 3,41-3,49 (m), 3,55-3,75 (m), 3,78-3,9 (m)
13С-ЯМР (CDCl3): δ 14,1; 22,7; 25,4; 26,1; 29,4; 29,5; 29,6; 31,9 (алкильная цепь), 70,5 (алкил-С-O-), 64,2; 71,8; 72,4 (глицерин-С).
16. Синтез [1-(4,4'-диметокситритил)]-3-гексадецилглицерина (Pam3)

Процедура: Для сушки 10 ммолей Pam2 (3,16 г) растворяли в 15 мл пиридинаабс. и растворитель вновь удаляли. Указанную процедуру повторяли. Добавляли медленно по каплям 12 ммолей (4,06 г) диметокситритилхлорида (растворенного в 50 мл пиридина) к раствору диола в 100 мл пиридинаабс. и перемешивали в течение 24 при КТ. Затем реакцию прекращали с помощью 5 мл метанола и реакционную смесь концентрировали досуха. Оставшиеся следы пиридина удаляли путем азеотропического уноса с помощью толуола. Остаток растворяли в 300 мл ДХМ, промывали насыщенным водным раствором KCl и Н2О и сушили над Na2SO4. После удаления растворителя остаток хроматографировали на силикагеле (н-гексан/диэтиловый эфир/ТЭА=40:60:1) (R.A. Jones «Oligonucleotide Synthesis; A Practical Approach» под ред. M.J. Gait, изд-во IRL Press, 1984, с.23).
Выход: 4,9 г (79,3% от теоретического)
ТСХ (н-гексан/диэтиловый эфир/ТЭА=40:60:1): значение Rf=0,42
МС (FD): m/z 618,2 (М+), 303 (DMT+)
1Н-ЯМР (CDCl3): δ 0,80 (t), 1,20 (s), 1,96 (s), 2,36 (d), 2,72 (s), 3,27 (d), 3,32-3,48 (m), 3,75 (m), 6,61-6,76 (m), 7,08-7,37 (m)
17. Синтез [1-(4,4'-диметокситритил)]-2-сукцинил-3-гексадепилглицерина (Pam4)

Процедура: 1,26 ммоля Pam3 (0,78 г) сушили дважды с помощью пиридина. Добавляли 0,76 ммоля DMAP (92 мг) и 1,26 ммоля янтарного ангидрида (126 мг) к раствору спирта в 5 мл пиридинаабс. После перемешивания в течение 12 ч при КТ реакционный раствор растворяли в 30 мл ДХМ, промывали дважды 30 мл воды и сушили над Na2SO4. После удаления растворителя остаток хроматографировали на силикагеле (этилацетат/метанол/NH3 (25% в H2O)=5:1:1) (D.W. Will, T. Brown, Tetrahedron Lett. 33, 1992, 2729.).
Выход: 0,6 g (66,3%)
ТСХ (EtOAc/МеОН/NH3(H2O)=5:1:1): значение Rf=0,32
MC (FD): m/z 718 (M+), 303 (DMT+), 1020,7 (M+DMT)+
18. Стимуляция человеческих клеток адъювантом, предлагаемым в изобретении, в форме модифицированной липидом нуклеиновой кислоты
а) Для определения иммуногенной активности нуклеиновых кислот, предлагаемых в изобретении, в форме адъювантов модифицированные липидом нуклеиновые кислоты, которые содержат последовательности, представленные в SEQ ID NO:1, 2, 3, 4 или 5, совместно инкубировали с человеческими клетками. Для этой цели, например, человеческие РВМС-клетки совместно инкубировали в течение 16 ч в среде X-VIVO-15 (фирма BioWhittaker, каталожный номер ВЕ04-418Q), дополненной 2 мМ L-глутамином (фирма BioWhittaker), 10 ед./мл пенициллина (фирма BioWhittaker) и 10 мкг/мл стрептомицина, с 10 мкг/мл РНК (мРНК, которая кодирует β-галактозидазу, или нуклеиновая кислота формулы (I), предлагаемая в изобретении, модифицированная липидом) и необязательно с 10 мкг/мл протамина. Супернатанты удаляли и анализировали высвобождение IL-6 и TNFα с помощью ELISA.
б) В другом эксперименте высвобождение TNF-α из человеческих РВМС-клеток определяли после стимуляции РНК-олигонуклеотидами, применяемыми согласно изобретению (см. выше), а также адъювантами, предлагаемыми в изобретении.
Для этой цели человеческие РВМС-клетки совместно инкубировали в течение 16 ч с 10 мкг/мл РНК-олигонуклеотидов в среде X-VIVO 15 (фирма BioWhittaker), дополненной 2 мМ L-глутамином (фирма BioWhittaker), 10 ед./мл пенициллина (фирма BioWhittaker) и 10 мкг/мл стрептомицина. Супернатанты удаляли и анализировали с помощью ELISA.
19. Высвобождение TNF-альфа в hPBMC
Человеческие РВМС высевали в 96-луночный титрационный микропланшет (2×105 в объеме 200 мкл на лунку) в бессывороточной среде. Водные растворы РНК (либо в виде комплекса с протамином, либо свободные) добавляли в клетки (конечная концентрация РНК: 10 мкг/мл (в 33 мкл)) при интенсивном перемешивании и инкубировали при 37°С в течение 24 ч. Секрецию TNF-альфа оценивали в бесклеточных супернатантах клеток с помощью ELISA. Эксперименты осуществляли в трех повторностях.
В первом эксперименте оценивали стимуляцию человеческих РВМС (hPBMC) олигонуклеотидами, предлагаемыми в изобретении, последовательности которых представлены в SEQ ID NO:78 и 79 (формула (I)). Наиболее четко стимуляция обнаружена при применении SEQ ID NO:78, но также обнаружена и при использовании SEQ ID NO:79 (см. фиг.8).
Во втором (сравнительном) эксперименте оценивали стимуляцию человеческих РВМС (hPBMC) олигонуклеотидами, предлагаемыми в изобретении, последовательности которых представлены SEQ ID NO:78 (формула (II)) и 81 (формула (II)), с протамином или без него. Наиболее высокая стимуляция обнаружена при применении с протамином SEQ ID NO:81. При создании изобретения неожиданно было установлено, что в случае SEQ ID NO:78 близкий уровень стимуляции обнаружен как с протамином, так и без него (см. фиг.9).
В третьем эксперименте оценивали стимуляцию человеческих РВМС (hPBMC) олигонуклеотидами, предлагаемыми в изобретении, последовательности которых представлены в SEQ ID NO:81, 82 и 83 (формула (II)). Во всех случаях, т.е. при применении SEQ ID NO:81, 82 и 83, обнаружена выраженная стимуляция (см. фиг.10).
20. Высвобождение TNF-альфа в hPBMC - трансфекция комплексами с олигофектамином и без него
Человеческие РВМС высевали в 96-луночный титрационный микропланшет (2×105 в 200 мкл на лунку) в бессывороточной среде. Растворы РНК или растворы РНК в виде комплексов с олигофектамином добавляли в клетки (конечная концентрация РНК: 10 мкг/мл) при интенсивном перемешивании и инкубировали при 37°С в течение 24 ч. Секрецию TNF-альфа оценивали в бесклеточных супернатантах клеток с помощью ELISA. Последовательности, применяемые для трансфекции в виде комплексов с олигофектамином, представляли собой: a) SEQ ID NO:1 (Seq. 1), 6) SEQ ID NO:1, модифицированную на 3'-, 5'-конце токоферолом, в сравнении с в) RNA40 (SEQ ID NO:87); Последовательности, применяемые для трансфекции (без образования комплекса с олигофектамином), представляли собой: г) среду (без последовательности), д) SEQ ID NO:1 (Seq. 1) и е) SEQ ID NO:1, модифицированную на 3'-, 5'-конце токоферолом. Наилучшие результаты получали, когда для трансфекции применяли SEQ ID NO:1 (Seq. 1), SEQ ID NO:1, модифицированную на 3'-, 5'-конце токоферолом, в сочетании с олигофектамином. Эти эксперименты проводили в трех повторностях.
21. Системное высвобождение IL-12 в hPBMC после i.v. (внутривенной инъекции)
10 мкг РНК в 100 мкл раствор Рингера лактата вводили i.v. Через 4 ч после введения брали образцы крови у пациента и определяли уровни цитокинов в сыворотке. На фиг.12 показаны результаты трансфекции: а) буфером в качестве контроля, б) RNA40 (SEQ ID NO:87), в) RNA40 (SEQ ID NO:87) в виде комплекса с олигофектамином, г) SEQ ID NO:1 (Seq. 1) и д) SEQ ID NO:1, модифицированной на 3'-, 5'-конце токоферолом. Как видно из графика, наилучшие и результаты и, как следствие, выраженная иммуностимуляция выявлена при использовании предлагаемой в изобретении SEQ ID NO:1, модифицированной на 3'-, 5'-конце токоферолом.
22. Высвобождение TNF-альфа в hPBMC при применении различных модифицированных SEQ ID NO:1
И в этом эксперименте человеческие РВМС высевали в 96-луночный титрационный микропланшет (2×105 в объеме 200 мкл на лунку) в бессывороточной среде. 10 мкл растворов РНК (РНК в виде комплексов с олигофектамином или без олигофектамина) добавляли в клетки (конечная концентрация РНК: 10 мкг/мл) при интенсивном перемешивании и инкубировали при 37°С в течение 24 ч. Секрецию TNF-альфа оценивали в бесклеточных супернатантах клеток с помощью ELISA. В сравнительном эксперименте оценивали воздействие различных модификаций предлагаемой в изобретении последовательности, представленной в SEQ ID NO:1, на иммунностимуляцию человеческих РВМС (hPBMC) и высвобождение TNF-альфа. В эксперименте применяли SEQ ID NO:1 (формулы (I)), модифицированную холестерином, SEQ ID NO:1 (формулы (I)), модифицированную токоферолом, и SEQ ID NO:1, модифицированную на 3'-,5'-конце токоферолом, при этом наилучшие результаты получены при использовании последней модификации. Однако модификации SEQ ID NO:1 либо холестерином, либо токоферолом, приводили также к существенной стимуляции hPBMC (см. фиг.13).
23. Снижение размера опухолей после профилактической вакцинации овальбумином и предлагаемыми в изобретении олигонуклеотидами
Мышей линии С57/В16 возрастом 6-8 недель вакцинировали 10 мкл белка (куриный овальбумин) подкожно в бок. Белок (куриный овальбумин) и при необходимости олигонуклеотиды растворяли в IFA (неполный адъювант Фрейнда) и для каждой инъекции применяли общий объем 100 мкл. Растворы содержали: а) IFA, б) IFA и куриный овальбумин, в) IFA и куриный овальбумин, и RNA40 (SEQ ID NO:87) и г) IFA и куриный овальбумин, и предлагаемую в изобретении последовательность SEQ ID NO:1 в виде комплекса с протамином, вакцинацию осуществляли согласно следующей схеме:
Затем через 10 дней мышей реиммунизировали таким же смесями для вакцинации. Через 12 дней после реиммунизации осуществляли контрольное заражений мышей, используя по 1×106 опухолевых клеток (EG7.Ova), и размер опухолей оценивали каждые 2 недели.
Наилучшие результаты, т.е. существенное снижение размера опухоли, получены при применении IFA и куриного овальбумина, и предлагаемой в изобретении последовательности SEQ ID NO:1 (г), что свидетельствует о том, что предлагаемые в изобретении последовательности обладают очень высокой активностью в качестве адъювантов (см. фиг.14).
24. Снижение размера опухолей после внутриопухолевой инъекции
В день 0 (d0) мышам линии С57/В16 возрастом 6-8 недель имплантировали подкожно в бок 1×106 опухолевых клеток (EG7.Ova). В день 3 начинали обработку. 50 мкг РНК разводили в ЗФР и в целом в полость опухоли инъецировали объем, равный 100 мкл. Обработку и оценку размера опухолей осуществляли ежедневно. Когда объем опухоли достигал или превышал 160 мм3, мышей умерщвляли. Обрабатывали по 4 мыши на группу и результаты выражали в виде средних значений для 4 мышей. Для внутриопухолевой инъекции применяли: а) только ЗФР, б) предлагаемую в изобретении последовательность SEQ ID NO:1 (Seq. 1), модифицированную на 3'-,5'-конце токоферолом, в ЗФР, в) предлагаемую в изобретении последовательности SEQ ID NO:1 в ЗФР и г) RNA40 (SEQ ID NO:87) в ЗФР, используя следующий протокол:
Наилучшие результаты, т.е. существенное снижение размера опухоли, получены при применении предлагаемой в изобретении последовательности SEQ ID NO:1, модифицированной на 3'-, 5'-конце токоферолом, далее по эффективности следовала немодифицированная предлагаемая в изобретении последовательность SEQ ID NO:1 (см. фиг.15)
Преимущества изобретения:
Нуклеиновую кислоту общей формулы (I): GlXmGn или в виде нуклеиновой кислоты общей формулы (II): ClXmCn, предлагаемой в изобретении, можно применять в качестве иммуностимулятора так таковую. Предпочтительно нуклеиновая кислота, предлагаемая в изобретении, содержит липидную модификацию и ее можно применять как таковую (например, растворенную в фармацевтически активном носителе или наполнителе). При этом предлагаемые в изобретении нуклеиновые кислоты стимулируют врожденную иммунную систему и вызывают неспецифический иммунный ответ. Эту иммуностимулирующую активность можно значительно усиливать путем добавления других соединений, известных в данной области, в качестве активных стимуляторов врожденного иммунного ответа, к предлагаемым в изобретении нуклеиновым кислотам. Например, согласно изобретению предложен модифицированный 3'-холестерином фосфодиэфирный олигонуклеотид в качестве иммуностимулирующего адъюванта, который может индуцировать цитотоксическое действие в отношении специфических опухолевых клеток. Это неожиданно установленное при создании изобретения действие зависит главным образом от иммуностимулирующей активности применяемой необязательно модифицированной липидом нуклеиновой кислоты общей формулы (I) или (II), предлагаемой в изобретении, при этом 3'- или 5'-модификации (липидные модификации), играют важную роль. В целом, иммуностимулирующие свойства нуклеиновых кислот общей формула (I) или (II) делают эти молекулы эффективными в качестве терапевтических средств, предназначенных для индукции неспецифического связанного с иммунной стимуляцией ответа, либо при их индивидуальном применении (иммуностимулятор), либо - в качестве адъюванта - в сочетании с другими фармацевтически активными компонентами, которые, как правило, представляют собой иммуностимулирующие компоненты, активирующие специфический иммунный ответ на фармацевтически активные соединения.































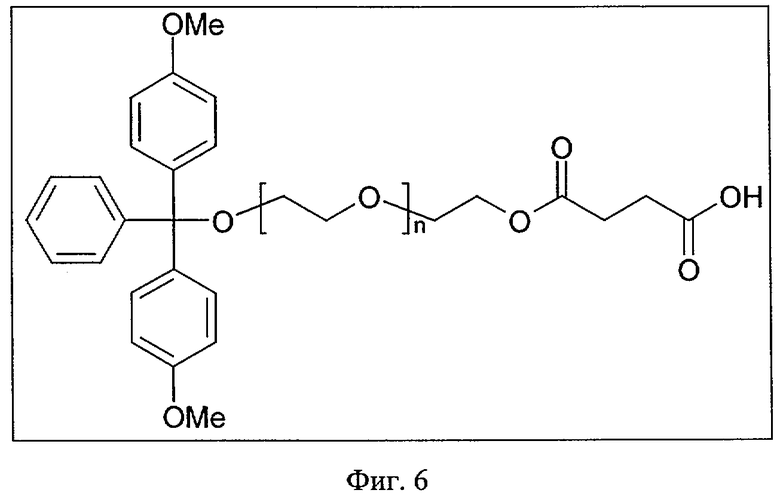
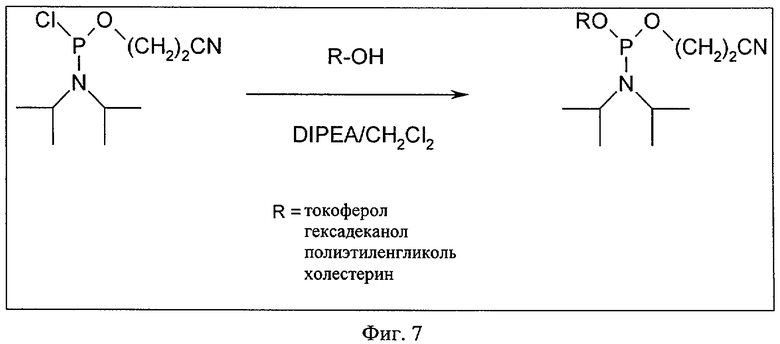

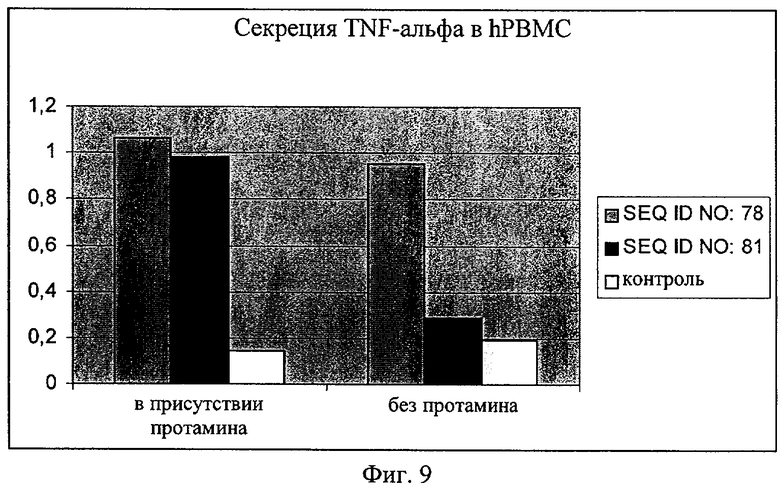

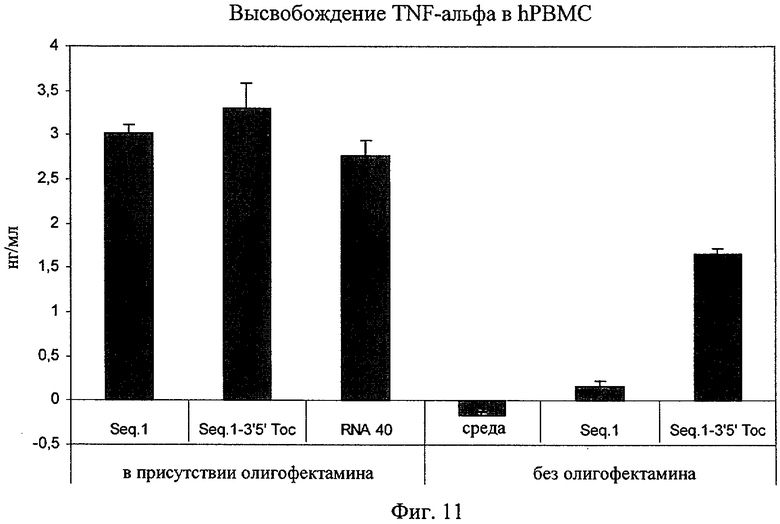

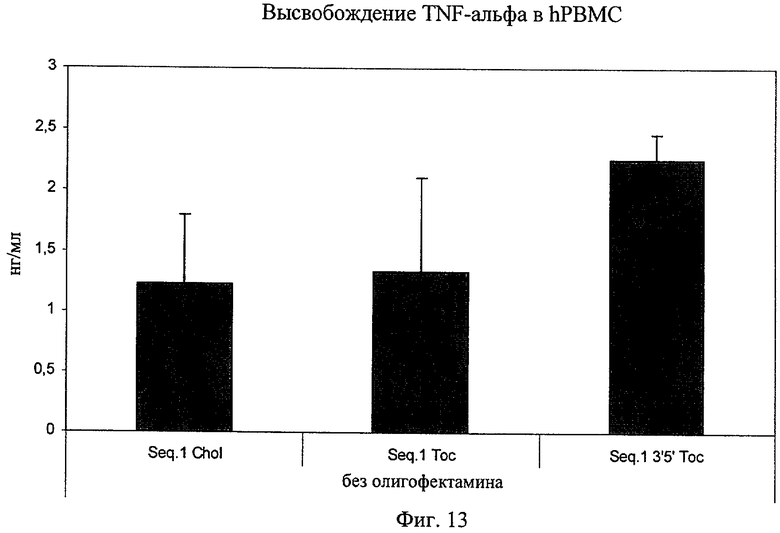

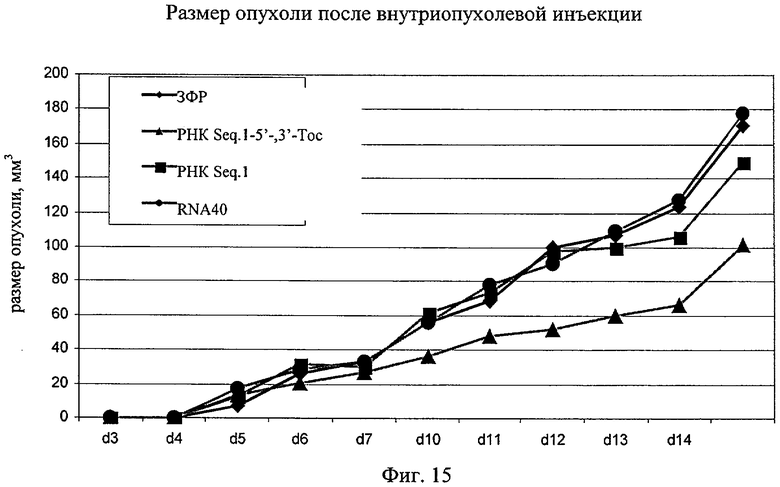
Изобретение относится к области биотехнологии. Описана однонитевая РНК общей формулы GlXmGn или ClXmCn, которая модифицирована липидом. Однонитевая РНК может действовать в качестве иммуностимулятора, индуцируя врожденный иммунный ответ. Описана также фармацевтическая композиция. Изобретение может быть использовано для лечения инфекционных болезней, аллергий или раковых заболеваний. 6 н. и 16 з.п. ф-лы, 15 ил., 24 пр., 2 табл.
1. Однонитевая РНК формулы (I), обладающая иммуностимулирующей активностью:
GlXmGn,
в которой G обозначает гуанозин, уридин или аналог гуанозина или уридина;
Х обозначает гуанозин, уридин, аденозин, тимидин, цитидин или аналог указанных выше нуклеотидов;
l обозначает целое число от 1 до 40,
где, если l обозначает 1, то G обозначает гуанозин или его аналог,
если l>1, то по меньшей мере 50% нуклеотидов представляют собой гуанозин или его аналог;
m обозначает целое число, и оно равно по меньшей мере 3;
где, если m равно 3, то Х обозначает уридин или его аналог,
если m>3, то встречаются по меньшей мере 3 последовательных остатка уридина или аналога уридина;
n обозначает целое число от 1 до 40,
где, если n равно 1, то G обозначает гуанозин или его аналог,
если n>1, то по меньшей мере 50% нуклеотидов представляют собой гуанозин или его аналог,
причем Xm имеет одинаковую урациловую последовательность длиной по меньшей мере 8 уридинов или аналогов уридинов;
причем эта РНК состоит из 15-60 нуклеотидов.
2. Однонитевая РНК по п.1, отличающаяся тем, что однонитевая РНК формулы (I) является линейной или кольцевой.
3. Однонитевая РНК по п.1, отличающаяся тем, что однонитевая РНК формулы (I) содержит по меньшей мере одну последовательность, выбранную из следующих последовательностей, представленных в SEQ ID NO:1-5, 10-18; 23-31; 36-53; 59-79:
GGUUUUUUUUUUUUUUUGGG (SEQ ID NO:1);
GGGGGUUUUUUUUUUGGGGG (SEQ ID NO:2);
GGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGGG (SEQ ID NO:3);
GUGUGUGUGUGUUUUUUUUUUUUUUUUGUGUGUGUGUGU (SEQ ID NO:4);
GGUUGGUUGGUUUUUUUUUUUUUUUUUGGUUGGUUGGUU (SEQ ID NO:5);
GGGGGGUUUUUUUUGGGGGG (SEQ ID NO:10);
GGGGGGUUUUUUUUUGGGGG (SEQ ID NO:11);
GGGGGGUUUUUUUUUUGGGG (SEQ ID NO:12);
GGGGGUUUUUUUUUUUGGGG (SEQ ID NO:13);
GGGGGUUUUUUUUUUUUGGG (SEQ ID NO:14);
GGGGUUUUUUUUUUUUUGGG (SEQ ID NO:15);
GGGGUUUUUUUUUUUUUUGG (SEQ ID NO:16);
GGUUUUUUUUUUUUUUUUGG (SEQ ID NO:17);
GUUUUUUUUUUUUUUUUUUG (SEQ ID NO:18);
GGGGGGGUUUUUUUUGGGGGGG (SEQ ID NO:23);
GGGGGGGUUUUUUUUUGGGGGG (SEQ ID NO:24);
GGGGGGGUUUUUUUUUUGGGGG (SEQ ID NO:25);
GGGGGGUUUUUUUUUUUGGGGG (SEQ ID NO:26);
GGGGGGUUUUUUUUUUUUGGGG (SEQ ID NO:27);
GGGGGUUUUUUUUUUUUUGGGG (SEQ ID NO:28);
GGGGGUUUUUUUUUUUUUUGGG (SEQ ID NO:29);
GGGUUUUUUUUUUUUUUUUGGG (SEQ ID NO:30);
GGUUUUUUUUUUUUUUUUUUGG (SEQ ID NO:31);
GGGGGGGGUUUUUUUUGGGGGGGG (SEQ ID NO:36);
GGGGGGGGUUUUUUUUUGGGGGGG (SEQ ID NO:37);
GGGGGGGGUUUUUUUUUUGGGGGG (SEQ ID NO:38);
GGGGGGGUUUUUUUUUUUGGGGGG (SEQ ID NO:39);
GGGGGGGUUUUUUUUUUUUGGGGG (SEQ ID NO:40);
GGGGGGUUUUUUUUUUUUUGGGGG (SEQ ID NO:41);
GGGGGGUUUUUUUUUUUUUUGGGG (SEQ ID NO:42);
GGGGUUUUUUUUUUUUUUUUGGGG (SEQ ID NO:43);
GGGUUUUUUUUUUUUUUUUUUGGG (SEQ ID NO:44);
GUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUG (SEQ ID NO:45);
GGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGG (SEQ ID NO:46);
GGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGG (SEQ ID NO:47);
GGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGG (SEQ ID NO:48);
GGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGG (SEQ ID NO:49);
GGGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGGG (SEQ ID NO:50);
GGGGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGGGG (SEQ ID NO:51);
GGGGGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGGGGG (SEQ ID NO:52);
GGGGGGGGGUUUUUUUUUUUUUUUUUUUUUUUUUUUUUUGGGGGGGG (SEQ ID NO:53);
GGUUUUUUUUGG (SEQ ID NO:59);
GGUUUUUUUUUGG (SEQ ID NO:60);
GGUUUUUUUUUUGG (SEQ ID NO:61);
GGUUUUUUUUUUUGG (SEQ ID NO:62);
GGUUUUUUUUUUUUGG (SEQ ID NO:63);
GGUUUUUUUUUUUUUGG (SEQ ID NO:64);
GGUUUUUUUUUUUUUUGG (SEQ ID NO:65);
GGUUUUUUUUUUUUUUUGG (SEQ ID NO:66);
GGGUUUGGG (SEQ ID NO:67);
GGGUUUUGGG (SEQ ID NO:68);
GGGUUUUUGGG (SEQ ID NO:69);
GGGUUUUUUGGG (SEQ ID NO:70);
GGGUUUUUUUGGG (SEQ ID NO:71);
GGGUUUUUUUUGGG (SEQ ID NO:72);
GGGUUUUUUUUUGGG (SEQ ID NO:73);
GGGUUUUUUUUUUGGG (SEQ ID NO:74);
GGGUUUUUUUUUUUGGG (SEQ ID NO:75);
GGGUUUUUUUUUUUUGGG (SEQ ID NO:76);
GGGUUUUUUUUUUUUUGGG (SEQ ID NO:77);
GGGUUUUUUUUUUUUUUUGGGUUUUUUUUUUUUUUUGGGUUUUUUUUUUUUUUUGGG (SEQ ID NO:78);
GGGUUUUUUUUUUUUUUUGGGGGGUUUUUUUUUUUUUUUGGG (SEQ ID NO:79).
4. Однонитевая РНК по п.1, отличающаяся тем, что по меньшей мере один нуклеотид нуклеиновой кислоты формулы (I) представляет собой не встречающийся в естественных условиях аналог встречающегося в естественных условиях нуклеотида.
5. Однонитевая РНК по п.1, отличающаяся тем, что однонитевая РНК формулы (I) присутствует дополнительно имеет на 5'-конце «кэп-структуру» и/или на 3'-конце имеет поли-А хвост.
6. Модифицированная липидом однонитевая РНК формулы (I) по пп.1-5, обладающая иммуностимулирующей активностью, где липид выбран из липидов или липофильных остатков, включая витамины, например α-токоферол (витамин Е), RRR-α-токоферол (D-α-токоферол), L-α-токоферол, рацемат D,L-α-токоферола, витамин А и его производные, включая ретиноевую кислоту, ретинол, витамин D и его производные, включая эргостерольные предшественники витамина D, витамин Е и его производные, включая сукцинат витамина Е (VES), витамин К и его производные, хиноновые и фитольные производные, стероиды, включая желчные кислоты, выбранные из холевой кислоты, дезоксихолевой кислоты, дегидрохолевой кислоты, или кортизон, дигоксигенин, тестостерон, холестерин или тиохолестерин, или полиалкиленгликоли, алифатические группы, включая С1-С20алканы, С1-С20алкены или C1-С20алканолы, додекандиольные, гексадеканольные или ундецильные остатки, или фосфолипиды, включая фосфатидилглицерин, диацетилфосфатидилглицерин, фосфатидилхолин, дипальмитоилфосфатидилхолин, дистероилфосфатидилхолин, фосфатидилсерин, фосфатидилэтаноламин или дигексадецил-рац-глицерин, сфинголипиды, цереброзиды, ганглиозиды, 1,2-ди-О-гексадецил-рац-глицеро-3-Н-фосфанат триэтиламмония, полиамины, полиалкиленгликоли, включая полиэтиленгликоль (ПЭГ), гексаэтиленгликоль (ГЭГ), или пальмитиновые или пальмитильные остатки, октадециламины или гексиламинокарбонилоксихолестериновые остатки, воски, терпены, алициклические углеводороды и остатки насыщенных или моно- или полиненасыщенных жирных кислот.
7. Модифицированная липидом однонитевая РНК формулы (I) по п.6, которая содержит по меньшей мере один линкер, ковалентно связанный с однонитевой РНК формулы (I) и липид, ковалентно связанный с соответствующим линкером.
8. Модифицированная липидом однонитевая РНК по п.7, отличающаяся тем, что содержит по меньшей мере один (бифункциональный) липид, ковалентно связанный по меньшей мере с одной однонитевой РНК формулы (I).
9. Модифицированная липидом однонитевая РНК по п.6, отличающаяся тем, что модифицированная липидом однонитевая РНК содержит один линкер, ковалентно связанный с по меньшей мере одной однонитевой РНК формулы (I), и по меньшей мере один липид, ковалентно связанный с соответствующим линкером, а также по меньшей мере один (бифункциональный) липид, ковалентно связанный с однонитевой РНК формулы (I).
10. Модифицированная липидом однонитевая РНК по п.6, отличающаяся тем, что модифицированная липидом однонитевая РНК содержит по меньшей мере от 3 до 8 липидов на нуклеиновую кислоту, где
а) все липиды ковалентно связаны с однонитевой РНК через линкер;
б) все липиды ковалентно связаны непосредственно с однонитевой РНК;
в) некоторые липиды ковалентно связаны с однонитевой РНК, а некоторые липиды ковалентно связаны непосредственно с однонитевой РНК.
11. Модифицированная липидом однонитевая РНК по п.6, отличающаяся тем, что связь между однонитевой РНК формулы (I) и липидом или между однонитевой РНК формулы (I) и линкером, связанным с липидом, находится на 3'- и/или 5'-конце однонитевой РНК формулы (I).
12. Фармацевтическая композиция для лечения раковых заболеваний, аллергий или инфекционных болезней, содержащая однонитевую РНК кислоту по любому из пп.1-11, фармацевтически приемлемый носитель и необязательно дополнительные вспомогательные субстанции, добавки и/или адъюванты.
13. Фармацевтическая композиция по п.12, дополнительно содержащая по меньшей мере один дополнительный фармацевтически активный компонент.
14. Фармацевтическая композиция по п.13, отличающаяся тем, что по меньшей мере один фармацевтически активный компонент выбран из группы, включающей пептиды, белки, нуклеиновые кислоты, (терапевтически активные) низкомолекулярные органические или неорганические соединения, имеющие молекулярную массу менее 5000, сахара, антигены, антитела, патогены, ослабленные патогены, деактивированные патогены, (человеческие) клетки, клеточные фрагменты или фракции и другие терапевтические агенты, которые предпочтительно адаптированы для усиления способности к трансфекции, например путем образования комплексов с липидами и/или поликатионными соединениями, например поликатионными пептидами.
15. Фармацевтическая композиция по п.12, отличающаяся тем, что композиция содержит по меньшей мере один дополнительный адъювант, который представляет собой иммуностимулятор, выбранный из группы, включающей катионные пептиды, в том числе такие полипептиды, как протамин, нуклеолин, спермин или спермидин, катионные полисахариды, включая хитозан, TDM, MDP, мурамил-дипептид, плюроники, раствор квасцов, гидроксид алюминия, ADJUMER™ (полифосфазен); гель алюминия фосфата; глюканы из водорослей; алгаммулин; гель гидроксида алюминия (квасцы); гель гидроксида алюминия с высокой способностью адсорбировать белок; гель гидроксида алюминия с низкой вязкостью; AF или SPT (эмульсия сквалана (5%), Твин 80 (0,2%), плюроник L121 (1,25%), забуференный фосфатом физиологический раствор, рН 7,4); AVRIDINE™ (пропандиамин); BAY R1005™ ((N-(2-дезокси-2-L-лейциламино-b-D-глюкопиранозил)-N-октадецилдодеканоиламида гидроацетат); CALCITRIOL™ (1α,25-дигидрокси-витамин D3); гель фосфата кальция; САРТМ (наночастицы фосфата кальция); голотоксин холеры, слитый белок токсин холеры-А1-D-фрагмент белка-А, субъединица В токсина холеры; CRL 1005 (блок-сополимер Р1205); содержащие цитокин липосомы; DDA (диметилдиоктадециламмония бромид); DHEA (дегидроэпиандростерон); DMPC (димиристоилфосфатидилхолин); DMPG (димиристоилфосфатидилглицерин); комплекс DOC/квасцы (натриевая соль дезоксихолевой кислоты); полный адъювант Фрейнда; неполный адъювант Фрейнда; гамма-инулин; адъювант Гербу (Gerbu) (смесь, содержащая I) N-ацетилглюкозаминил-(Р1-4)-N-ацетилмурамил-L-аланил-D-глутамин (GMDP), II) диметилдиоктадециламмония хлорид (DDA), III) комплексная соль цинк-L-пролина (ZnPro-8); GM-CSF); GMDP (N-ацетилглюкозаминил-(b1-4)-)N-ацетилмурамил-L-аланил-D-изоглутамин); имихимод (1-(2-метилпропил)-1Н-имидазо[4,5-с]хиполин-4-амин); ImmTher™ (N-ацетилглюкозаминил-N-ацетилмурамил-L-Ala-D-isoGlu-L-Ala-глицерина дипальмитат); DRV (иммунолипосомы, полученные путем дегидратации/регидратации везикул); интерферон-γ; интерлейкин-1β; интерлейкин-2; интерлейкин-7; интерлейкин-12; ISCOMS™ («иммунные стимулирующие комплексы»); ISCOPREP 7.0.3.™; липосомы; LOXORIBINE™ (7-аллил-8-оксогуанозин); оральный адъювант LT (лабильный энтеротоксин-протоксин E.coli); микросферы и микрочастицы любого состава; MF59™ (водная эмульсия сквалена); MONTANIDE ISA 51™ (очищенный неполный адъювант Фрейнда); MONTANIDE ISA 720™ (метаболизируемый масляный адъювант); MPL™ (3-Q-дезацил-4'-монофосфорил липид А); липосомы МТР-РЕ и МТР-РЕ ((N-ацетил-L-аланил-D-изоглутаминил-L-аланин-2-(1,2-дипальмитоил-sn-глицеро-3-(гидроксифосфорилокси))этиламид, однонатриевая соль); MURAMETIDE™ (Nac-Mur-L-Ala-D-Gln-OCH3); MURAPALMITINE™ и D-MURAPALMITINE™ (Nac-Mur-L-Thr-D-isoGIn-sn-глицериндипальмитоил); NAGO (нейраминидаза-галактозооксидаза); наносферы или наночастицы любого состава; NISV (везикулы из неионогенного поверхностно-активного вещества); PLEURAN™ (β-глюкан); PLGA, PGA и PLA (гомо- и сополимеры молочной кислоты и гликолевой кислоты; микро-/наносферы); плюроник L121™; РММА (полиметилметакрилат); PODDS™ (протеноидные микросферы); полиэтиленкарбаматные производные; поли-rA : поли-rU (комплекс полиадениловая кислота-полиуридиновая кислота); полисорбат 80 (Твин 80); белковые кохлеаты (фирма Avanti Polar Lipids, Inc., Алабастер. шт.Алабама); STIMULON™ (QS-21); Quil-A (сапонин Quil-A); S-28463 (4-аминоотекдиметил-2-этоксиметил-1Н-имидазо[4,5-с]хинолин-1-этанол);
SAF-1™ («композиция адъювантов фирмы Syntex»); протеолипосомы вируса Сендаи и липидные матрицы, содержащие вирус Сендаи; Спан-85 (сорбитантриолеат); Specol (эмульсия Marcol 52, Спан 85 и Твин 85); сквален или Robane® (2,6,10,15,19,23-гексаметилтетракозан и 2,6,10,15,19,23-гексаметил-2,6,10,14,18,22-тетракозагексан);
стеарилтирозин (октадецилтирозина гидрохлорид); Theramid® (N-ацетилглюкозаминил-N-ацетилмурамил-L-Ala-D-isoGlu-L-Ala-дипальмитоксипропиламид); Theronyl-MDP (TermurtideM или [thr 1]-MDP; N-ацетилмурамил-L-треонил-О-изоглутамин); Ту-частицы (Ту-ВПЧ или вирусоподобные частицы); липосомы Уолтера-Рида (липосомы, содержащие липид А, адсорбированный на гидроксиде алюминия), и липопептиды, включая Pam3Cys, прежде всего соли алюминия, такие как адъю-фос (Adju-phos), алгидрогель, регидрагель и; эмульсии, такие как CFA, SAF, IFA, MF59, провакс (Provax), TiterMax, монтанид (Montanide), ваксфектин (Vaxfectin); сополимеры, такие как оптивакс (Optivax, CRL1005), L121, полоксамер 4010); липосомы, такие как «липосомы-невидимки (Stealth)»; кохлеаты, такие как BIORAL; адъюванты растительного происхождения, такие как QS21, Quil A, Iscomatrix, ISCOM, предпочтительными адъювантами, пригодными для костимуляции, могут являться, например томатин, биополимеры, такие как PLG, РММ, инулин; адъюванты микробного происхождения, такие как ромуртид, DETOX, MPL, CWS, манноза, CpG7909, ISS-1018, IC31, имидазохинолины, амплиген, Ribi529, IMOxine, IRIV, ВПЧ, токсин холеры, термолабильный токсин, Pam3Cys, флагеллин, GPI (гликозилфосфатидилинозитол)-якорь, LNFPIII/Lewis X, противомикробные пептиды, UC-1V150, слитый белок RSV, cdiGMP; предпочтительными адъювантами, пригодными в качестве антагонистов, могут являться, например нейропептид CGRP.
16. Фармацевтическая композиция по п.13, отличающаяся тем, что фармацевтическая композиция представляет собой вакцину.
17. Применение однонитевой РНК по одному из пп.1-11 или композиции по одному из пп.12-16 для приготовления лекарственного средства, предназначенного для лечения раковых заболеваний, аллергий или инфекционных болезней.
18. Применение по п.17, отличающееся тем, что раковые заболевания выбраны из группы, включающей: карциномы ободочной кишки, меланомы, почечные карциномы, лимфомы, острый миелолейкоз (AML), острый лимфолейкоз (ALL), хронический миелолейкоз (CML), хронический лимфолейкоз (CLL), желудочно-кишечные опухоли, легочные карциномы, глиомы, опухоли щитовидной железы, карциномы молочной железы, опухоли предстательной железы, гепатомы, различные опухоли, индуцируемые вирусом, такие, например, как карцинома, вызываемая вирусом папилломы (например карцинома шейки матки), аденокарциномы, опухоли, вызываемые вирусом герпеса (например лимфома Беркитта, В-клеточная лимфома, индуцируемая EBV), опухоли, индуцируемые вирусом гепатита В (печеночно-клеточная карцинома), лимфомы, вызываемые HTLV-1 и HTLV-2, невромы/невриномы слухового нерва, рак шейки матки, рак легкого, рак глотки, анальные карциномы, глиобластомы, лимфомы, ректальные карциномы, астроцитомы, опухоли головного мозга, рак желудка, ретинобластомы, базалиомы, метастазы в головной мозг, медуллобластомы, рак матки, рак поджелудочной железы, рак яичек, меланомы, карциномы щитовидной железы, рак мочевого пузыря, синдром Ходжкина, менингиомы, болезнь Шнеебергера, бронхиальные карциномы, опухоли гипофиза, грибовидный микоз, рак пищевода, рак молочной железы, карциноиды, невриномы, спиналиомы, лимфомы Беркитта, рак гортани, рак почек, тимомы, карциному тела, рак костей, не-ходжкинские лимфомы, уретральный рак, CUP-синдром, опухоли головы/шеи, олигодендроглиомы, рак вульвы, рак кишечника, карциномы ободочной кишки, карциномы пищевода, патологическое перерождение бородавок, опухоли тонкого кишечника, краниофарингеомы, карциномы яичника, опухоли мягких тканей/саркомы, рак яичника, рак печени, карциномы поджелудочной железы, карциномы шейки матки, карциномы эндометрия, метастазы в печень, рак полового члена, рак языка, рак желчного пузыря, лейкоз, плазмоцитомы, рак матки, опухоль века, рак предстательной железы.
19. Применение по п.18, отличающееся тем, что инфекционные болезни выбраны из группы, включающей грипп, малярию, SARS (тяжелый острый респираторный синдром), желтую лихорадку, СПИД, боррелиоз Лайма, лейшманиоз, сибирскую язву, менингит, вирусные инфекционные заболевания, такие как СПИД, остроконечную кондилому, полые бородавки, лихорадку Денге, флеботомную лихорадку, вирус Эбола, простуду, весенне-летний менингоэнцефалит (FSME), грипп, опоясывающий лишай, герпес, герпес простой типа I, герпес простой типа II, опоясывающий герпес, грипп, японский энцефалит, лихорадку Ласса, вирус Марбурга, корь, ящур, мононуклеоз, эпидемический паротит, инфекцию, вызываемую вирусом Норвалк, железистую лихорадку Пфейффера, натуральную оспу, полиомиелит (детская хромота), ложный круп, инфекционную эритему, бешенство, бородавки, лихорадку Западного Нила, ветряную оспу, заболевание, вызываемое цитомегаловирусом (CMV), выбраны из группы бактериальных инфекционных болезней, включающей выкидыш (воспаление предстательной железы), сибирскую язву, аппендицит, боррелиоз, ботулизм, заболевание, вызываемое микроорганизмами Camphylobacter, хламидийную трахому (воспаление уретры, конъюнктивит), холеру, дифтерию, донованоз, эпиглоттит, сыпной тиф, газовую гангрену, гонорею, туляремию, заболевание, вызываемое Heliobacter pylori, коклюш, паховый лимфогранулематоз, остеомиелит, болезнь легионеров, проказу, листериоз, пневмонию, менингит, бактериальный менингит, сибирскую язву, воспаление среднего уха, заболевание, вызываемое Mycoplasma hominis, сепсис новорожденных (хориоамнионит), влажную гангрену, паратиф, чуму, синдром Рейтера, пятнистую лихорадку Скалистых гор, паратиф, вызываемый Salmonella, тиф, вызываемый Salmonella, скарлатину, сифилис, столбняк, триппер, японскую речную лихорадку, туберкулез, тиф, вагинит (кольпит), мягкий шанкр, и из группы инфекционных болезней, вызываемых паразитами, простейшими или грибами, включающих амебиаз, шистамоз, болезнь Шагаса, эпидермофию стопы, пятнистость, вызываемую дрожжевыми грибками, чесотку, малярию, онхоцеркоз (речная слепота), или грибных заболеваний, включая токсоплазмоз, трихомониаз, трипаносомоз (сонная болезнь), индийский висцеральный лейшманиоз, «подгузниковый»/«пеленочный» дерматит, шистосомоз, пищевое отравление рыбой (сигуатера), кандидоз, кожный лейшманиоз, лямблиоз (протозойная инфекция, вызываемая Lamblia intestinalis) или сонную болезнь, или выбраны из инфекционных болезней, вызываемых Echinococcus, таких как дифиллоботриоз, птичий цепень, собачий солитер (эхинококк), педикулез, бычий цепень, свиной солитер, карликовый цепень.
20. Применение по п.18, отличающееся тем, что аллергии выбраны из группы, включающей аллергическую астму (приводящую к опуханию слизистой оболочки носа), аллергический конъюнктивит (приводящий к покраснению и зуду в конъюнктиве), аллергический ринит («сенная лихорадка»), анафилаксию, ангиодему, атопический дерматит (экзема), крапивницу (сыпь), эозинофилию, респираторные аллергии на укусы насекомых, кожные аллергии (вызывающие и приводящие к различным видам сыпи, таким как экзема, сыпь (крапивница) и (контактный) дерматит), аллергии на пищу, аллергии на лекарственные средства.
21. Набор для лечения раковых заболеваний, аллергий или инфекционных болезней, содержащий однонитевую РНК по одному из пп.1-11 или фармацевтическую композицию по одному из пп.12-16, а также инструкции по применению с информацией о введении и дозе нуклеиновой кислоты или фармацевтической композиции.
22. Способ лечения нарушения или заболевания, выбранного из группы, включающей раковое заболевание, инфекционную болезнь и аллергию, заключающийся в том, что вводят пациенту, который нуждается в этом, в фармацевтически эффективном количестве однонитевую РНК по одному из пп.1-11 или фармацевтическую композицию по одному из пп.12-16.
| WO 2005097993 A2, 20.10.2005 | |||
| WO 2006029223 A2, 16.03.2006 | |||
| WO 03066649 A2, 14.08.2003. |

