Данная заявка является безусловной заявкой, притязающей на приоритет по предварительной заявке на патент США № 61/108616, поданной 27 октября 2008, озаглавленной “Oxidative monohalogenation of methane”, содержание которой включено путем ссылки в данную заявку, как если бы оно было полностью воспроизведено ниже в данной заявке.
Данное изобретение, в общем, относится к способу окислительного галогенирования (например, хлорирования, бромирования, иодирования или фторирования) метана с образованием метилгалогенида (например, метилхлорида) и к катализаторам, используемым для активации окислительного галогенирования метана. Данное изобретение, в частности, относится к такому способу, в котором метан, источник галогена и источник кислорода продуктивно контактируют с катализатором, который имеет большую селективность по метилгалогениду и монооксиду углерода, чем по метиленгалогениду, тригалогенметану или тетрагалогениду углерода.
Окислительное галогенирование представляет один путь конверсии метана (CH4) в галогенированный метан, предпочтительно в моногалогенированный метан (CH3X, где X представляет галоген) и более предпочтительно в монохлорированный метан (CH3Cl). Моногалогенированные метаны, такие как метилхлорид (CH3Cl), находят применение в производстве силиконов или в качестве интермедиатов в производстве различных товарных химикатов, таких как метанол, диметиловый эфир, легкие олефины (например, этилен, пропилен, бутен и более высокие углеводороды, включая газолины, которые имеют более пяти атомов углерода (C5 +)), газолин, винилхлорид и уксусная кислота. Хотя галогенированные метаны, которые имеют многочисленные (два, три или четыре) атома галогена (например, метиленхлорид) также находят некоторое применение, предпринимается ряд попыток улучшения селективности по моногалогенированным метанам, в особенности по метилхлориду, по сравнению с галогенированными метанами с двумя или более атомами галогена (например, метиленхлорид). Общее представление окисления показано ниже в формуле 1.
HX+½ O2+CH4→CH3X+H2O (Формула 1)
Несколько патентных публикаций описывают окислительное галогенирование метана с использованием разнообразных катализаторов. Ранние способы окислительного галогенирования, такие как раскрытые в патентах Соединенных Штатов Америки (США) 4769504 (Noceti с соавт.) и США 4795843 (Imai с соавт.), склонны производить большие фракции пергалогенированного продукта (например, тетрахлорида углерода), который типично имеет меньшую ценность, чем моногалогенированный продукт (например, CH3Cl). Такие способы также склонны производить неприемлемое количество продуктов глубокого окисления (номинально COx, причем монооксид углерода (CO) и диоксид углерода (CO2) служат конкретными примерами). Производство таких продуктов глубокого окисления загрязняет C1-углеводородное сырье (с одним атомом углерода), такое как CH4, и создает проблемы, такие как отделение продукта и утилизация побочных продуктов.
Патент США 6452058 (Schweizer с соавт.) раскрывает способ окислительного галогенирования, который включает приведение в контакт углеводорода-реагента, выбранного из CH4, галогенированного C1-углеводорода или их смеси, с источником галогена (например, хлоридом водорода) и, необязательно, с источником кислорода (например, молекулярным кислородом) совместно с катализатором в технологических условиях, достаточных для получения галогенированного C1-углеводорода с большим числом галогенных заместителей, чем содержится в углеводороде-реагенте. Катализатор представляет собой галогенид редкоземельного элемента или оксигалогенид редкоземельного элемента, который по существу не содержит железа или меди. Окислительное галогенирование, предпочтительно, по существу не дает пергалогенированного C1-углеводорода, и если и дает, то низкие содержания “нежелательных” продуктов окисления, таких как CO и CO2.
Патент США 6452058 (Schweizer с соавт.) также относится ко второму пути конверсии метана в галогенированный метан путем использования элементного галогена над нанесенным кислым катализатором или нанесенным катализатором на основе платинового металла и цитирует патент США 4523040 (Olah) и патент США 5354916 (Horvath с соавт.). Вариантные характеристики второго основного пути включают прямое галогенирование или электрофильное галогенирование. Представленная ниже формула 2 дает общее представление второго пути.
CH4+X2→CH3X+HCl (Формула 2)
Патентная заявка PCT (договор о патентной кооперации) WO 2006/118935 (Podkolzin с соавт.) описывает окислительное галогенирование углеводорода-реагента (например, CH4, галогенированного C1-углеводорода (например, CH3Cl или CH2Cl2) или их смеси) источником галогена и источником кислорода при молярном отношении углеводорода-реагента к источнику галогена в подаваемом в реактор материале при избытке 23:1 и/или при молярном отношении углеводорода-реагента к источнику кислорода при избытке 46:1 с использованием катализатора на основе галогенида редкоземельного элемента или оксигалогенида редкоземельного элемента.
WO 03/057318 (Weckhuysen с соавт.) описывает гидролитическое разрушение галогенированных углеводородов, таких как CCl4, над твердыми катализаторами на основе лантанидов совместно с паром при температуре в диапазоне от 200 градусов по Цельсию (°C) до 350°C.
Каждое из окислительного галогенирования и прямого или электрофильного галогенирования дает в некотором количестве метилгалогенид и каждое подает сырье для реакций дальнейшей переработки, которые дают, среди других желаемых продуктов, этилен. Каждое имеет положительные признаки и менее положительные признаки. Каждое имеет по меньшей мере один реагент и один побочный продукт, который отличается от другого. В случае окислительного галогенирования галогениды водорода (например, HCl) служат источником галогена, тогда как газообразный галоген (например, Cl2) обеспечивает галоген для прямого галогенирования. Окислительное галогенирование также требует источника кислорода, а прямое галогенирование не требует. Окислительное галогенирование дает воду в качестве побочного продукта, тогда как электрофильное галогенирование генерирует галогенид водорода как побочный продукт.
Конверсия метилгалогенида в этилен включает реакцию, представленную нижеприведенной формулой 3.
2 CH3X→ CH2=CH2 или этилен+HX (Формула 3)
Если основываться на реакции, показанной в формуле 3, ясно, что окислительное галогенирование является самодостаточным в том, что по меньшей мере при производстве более высокого углеводорода, такого как этилен, в качестве стадии дальнейшей переработки или второй стадии, следующей за получением метилгалогенида, галогенид водорода со второй стадии может быть использован при получении метилгалогенида. Предпочтительные результаты сопровождаются полной конверсией HX в CH3X, чтобы избежать образования влажного или водного HX. Электрофильное или прямое галогенирование, с другой стороны, требует воссоздания галогена в реакции, представленной ниже формулой 4. В каждом примере, упомянутом здесь, X представляет галоген, такой как хлор (Cl).
2 HX + ½ O2 → X2 + H2O (Формула 4)
В некоторых вариантах осуществления данное изобретение представляет собой способ окислительного галогенирования CH4, где способ включает приведение в контакт потока подаваемого материала, который включает CH4, источник галогена и источник кислорода, с первым катализатором и в условиях, достаточных, чтобы обеспечить поток продукта, который имеет большую селективность по метилгалогениду и CO, чем по метиленгалогениду, тригалогенметану или тетрагалогениду углерода, причем первый катализатор выбран из группы, состоящей из твердых суперкислот и твердых супероснований. Галоген представляет собой предпочтительно хлор, так что селективность по CH3Cl и CO превышает селективность по CH2Cl2, CHCl3 или CCl4.
В некоторых вариантах осуществления данного изобретения поток подаваемого материала также контактирует со вторым катализатором, который окислительно галогенирует по меньшей мере часть CH4, давая смесь, включающую по меньшей мере два члена группы, состоящей из метилгалогенида, метиленгалогенида, тригалогенметана, тетрагалогенида углерода, воды, галогенида водорода (например, HCl), непрореагировавшего галогена и непрореагировавшего кислорода. Второй катализатор предпочтительно выбран из группы, состоящей из галогенидов редкоземельных элементов и оксигалогенидов редкоземельных элементов.
В некоторых вариантах осуществления данного изобретения поток подаваемого материала контактирует со вторым катализатором перед тем, как он контактирует с первым катализатором, так что контакт со вторым катализатором дает смесь по меньшей мере двух членов группы, состоящей из метилгалогенида, метиленгалогенида, тригалогенметана, тетрагалогенида углерода, воды, галогенида водорода, непрореагировавшего галогена и непрореагировавшего кислорода, и контакт с первым катализатором превращает по меньшей мере часть метиленгалогенида, тригалогенметана и тетрагалогенида углерода в монооксид углерода, галогенид водорода и воду. Хотя первый и второй катализаторы могут быть пространственно разделены, чтобы обеспечить последовательный контакт, первый и второй катализаторы могут также включать каталитическую примесь.
В некоторых вариантах осуществления данного изобретения смесь продукта представляет собой эквимолярную смесь CO и CH3Cl, и эквимолярная смесь контактирует с катализатором карбонилирования в условиях, достаточных, чтобы превратить по меньшей мере часть эквимолярной смеси в по меньшей мере один из продуктов: ацетилхлорид и уксусную кислоту. В предпочтительной разновидности таких вариантов осуществления удаляют по меньшей мере части воды, произведенной посредством контакта с первым и вторым катализаторами, перед приведением эквимолярной смеси в контакт с катализатором карбонилирования.
При указании в настоящей заявке диапазонов, как в случае диапазона от 2 до 10, обе конечные точки диапазона (например, 2 и 10) и каждое численное значение, будь такое значение рациональным числом или иррациональным числом, включены в диапазон, если иное специально не указано.
Если не указано обратное, подразумевается из контекста или является общепринятым в данной области техники, что все части и проценты даны на массу. Выражения температуры могут быть даны в терминах либо градусов Фаренгейта (°F) вместе с эквивалентом в °C либо, типичнее, просто в °C.
“Конверсия” или “конв.” означает мольный процент CH4, который превращен во все продукты, включающие галогенированные метаны (например, CH3Cl, CH2Cl2, CHCl3 или CCl4) и окисленные побочные продукты (например, CO или CO2) в соответствии с различными вариантами осуществления данного изобретения.
“Селективность” или “сел.” означает мольный процент CH4, который превращен в конкретный продукт, такой как галогенированный C1-углеводородный продукт (например, CH3Cl) или окисленный побочный продукт (например, CO), поделенный на мольный процент всех произведенных продуктов. Например, определяют селективность для отдельного галогенированного метана (например, CH3Cl) относительно всех галогенированных метанов, присутствующих в качестве продуктов реакции (например, CH3Cl, CH2Cl2, CHCl3 и CCl4, где галоген является хлором), умножением на 100 частного, определенного с использованием моль отдельного галогенированного метана (например, CH3Cl) в качестве числителя и моль всех галогенированных метанов (например, CH3Cl, CH2Cl2, CHCl3 и CCl4) в качестве знаменателя.
В способах окислительного галогенирования различных вариантов осуществления данного изобретения галогенированный C1-углеводородный продукт, предпочтительно моногалогенированный C1-углеводородный продукт, более предпочтительно, моногалогенированный метановый продукт, и еще более предпочтительно, монохлорированный метановый продукт (например, CH3Cl) преобладает над другими галогенированными продуктами (например, CH2Cl2, CHCl3 и CCl4, где галоген представляет собой Cl).
Иллюстративные твердые суперкислоты включают вольфраматный оксид циркония или оксид вольфрама на носителе из оксида циркония (WO3/ZrO2), сульфатированный оксид циркония (SO4/ZrO2), сульфатированный оксид титана (SO4/TiO2), сульфатированный оксид титана-лантана (SO4/TiO2-La2O3), сульфатированный оксид олова (SO4/SnO2), сульфат церия на носителе из оксида циркония (Ce(SO4)2/ZrO2) и сульфат ванадия на носителе из оксида циркония (VSO4/ZrO2). В данной заявке размещение ZrO2 справа от диагональной черты (/) означает, что ZrO2 служит как каталитическим носителем каталитических материалов, показанных слева от диагональной черты, так и неотъемлемым компонентом самого катализатора. Твердая суперкислота представляет собой предпочтительно SO4/ZrO2.
Иллюстративные твердые супероснования включают фторид кальция на носителе из оксида циркония (CaF2/ZrO2), фторид бария на носителе из оксида циркония (BaF2/ZrO2); допированный калием оксид магния (K-допированный/MgO); и оксид натрия на носителе из оксида магния (Na2O/MgO). Твердое супероснование представляет собой предпочтительно CaF2/ZrO2.
Источником кислорода может быть любой кислородсодержащий газ или смесь таких газов. Иллюстративные кислородсодержащие газы включают по существу чистый или молекулярный кислород, воздух, обогащенный кислородом воздух или смесь кислорода с газом-разбавителем, который не мешает окислительному галогенированию. Газ-разбавитель представляет собой, предпочтительно, по меньшей мере (≥) один газ, выбранный из группы, состоящей из азота, аргона, гелия, монооксида углерода, диоксида углерода и метана. В случае смесей кислорода и газа-разбавителя газ-разбавитель присутствует в количестве ≥10 мольных процентов (мол.%) в расчете на сумму моль метана, источника галогена, источника кислорода и разбавителя. Количество газа-разбавителя, предпочтительно, не превышает (≤) 90 мол.% в расчете на сумму моль метана, источника галогена, источника кислорода и разбавителя.
Источник кислорода присутствует в количестве, которое удовлетворяет двум критериям. Во-первых, количество должно быть достаточным, чтобы дать желаемый метилгалогенид в качестве преобладающего галогенированного продукта, особенно относительно метиленгалогенида, и с желаемой степенью селективности по желаемому метилгалогениду относительно всех других продуктов реакции, включающих метиленгалогенид, тригалогенметан и тетрагалогенметан в качестве галогенированных продуктов реакции и CO и CO2 в качестве негалогенированных продуктов реакции. Во-вторых, количество является достаточным, чтобы обеспечить “богатую топливом” смесь источника кислорода и топлива, в данном случае метана, по соображениям безопасности и предпочтительно находится вне предела воспламеняемости при состоянии обогащения топливом для смеси метана и источника кислорода.
В некоторых вариантах осуществления данного изобретения условия, достаточные для того, чтобы обеспечить поток продукта, который имеет большую селективность по метилгалогениду и CO, чем к метиленгалогениду, тригалогенметану и тетрагалогениду метана, включают скорость потока подаваемого материала, достаточную, чтобы минимизировать конверсию метилгалогенида в CO и галогенид водорода. Предпочтительная скорость потока сырьевого материала представляет собой массовую часовую объемную скорость (WHSV) в диапазоне от 0,1 грамма (г) суммарной подачи метана, источника галогена, источника кислорода и необязательного разбавителя на г катализатора в час до менее чем (<) 100 г суммарной подачи метана, источника галогена, источника кислорода и, в случае присутствия, разбавителя на г катализатора в час.
Источник галогена представляет собой предпочтительно ≥ один галогенид водорода, выбранный из хлорида водорода (HCl), бромида водорода, фторида водорода и иодида водорода. Галоген представляет собой предпочтительно хлор, и источник галогена представляет собой предпочтительно хлорид водорода.
Галогенид, хотя наиболее предпочтительно хлорид, может также представлять собой бромид, иодид или фторид, как в случае бромида водорода, иодида водорода или фторида водорода. Если галогенид отличается от хлорида, иллюстративными моногалогенированными продуктами являются метилбромид, метилиодид и метилфторид. Смесь хлорида водорода и, например, бромида водорода предположительно дает смесь метилхлорида и метилбромида, если такая смесь является желательной.
Источник галогена предпочтительно присутствует в количестве, которое дает желаемый метилгалогенидный продукт в качестве преобладающего галогенированного продукта, независимо от того, рассматриваются ли галогенированные продукты одни или в сочетании с продуктами окисления углерода, конкретнее CO и CO2. Количество типично изменяется в зависимости от параметров процесса, таких как конкретная стехиометрия процесса, условия процесса (например, скорость потока реагентов и температура реакции), и выбора катализатора (например, твердая суперкислота или твердое супероснование).
Вышеупомянутые условия предпочтительно включают по меньшей мере одно из: температуру в диапазоне от 200°C до 600°C и давление в диапазоне от 95 килопаскаль (кПа) до 1100 кПа.
В некоторых вариантах осуществления данного изобретения условия являются достаточными, чтобы создать эквимолярную смесь CO и CH3Cl. Альтернативно, поток подаваемого материала может дополнительно включать количество CO, достаточное для того, чтобы обеспечить эквимолярную смесь CO и CH3Cl.
В некоторых вариантах осуществления данного изобретения эквимолярная смесь CO и CH3Cl контактирует с катализатором карбонилирования в условиях, достаточных для конверсии по меньшей мере части эквимолярной смеси в по меньшей мере один из продуктов: ацетилхлорид и уксусную кислоту. Катализатор карбонилирования представляет собой предпочтительно родиевый катализатор на угле. Удаление воды перед контактом с катализатором карбонилирования представляет предпочтительную разновидность таких вариантов осуществления.
Селективность по метилгалогениду предпочтительно находится в диапазоне от 35 мол.% до 100 мол.% в расчете на моли галогенированных продуктов, присутствующих в потоке продукта. Диапазон представляет собой предпочтительно от 50 мол.% до 99 мол.% в расчете на моли галогенированных продуктов, присутствующих в потоке продукта. Еще более предпочтительно диапазон представляет собой от 75 мол.% до 98 мол.% в расчете на моли галогенированных продуктов, присутствующих в потоке продукта.
Конверсия метана в метилгалогенид предпочтительно находится в диапазоне от 0,1 мол.% до 100 мол.% в расчете на моли метана, присутствующего перед конверсией. Более предпочтительно диапазон представляет собой от одного (1) мол.% до 75 мол.% в расчете на моли метана, присутствующего перед конверсией. Еще более предпочтительно диапазон представляет собой от пяти (5) мол.% до 50 мол.% в расчете на моли CH4, присутствующего перед конверсией.
В некоторых вариантах осуществления данного изобретения селективность по сочетанию метилгалогенида и CO предпочтительно находится в диапазоне от 50 мол.% до 100 мол.%. Более предпочтительно диапазон представляет собой от 75 мол.% до 97 мол.% и еще более предпочтительно от 90 мол.% до 95 мол.%.
В некоторых вариантах осуществления данного изобретения селективность по метилгалогениду относительно селективности по сочетанию метилгалогенида, метиленгалогенида, тригалогенметана и тетрагалогенида углерода предпочтительно находится в диапазоне от 85 мол.% до 100 мол.%. Более предпочтительно диапазон представляет собой от 90 мол.% до 100 мол.% и еще более предпочтительно от 95 мол.% до 100 мол.%.
Способ различных вариантов осуществления данного изобретения предпочтительно по существу не дает пергалогенированного продукта, такого как CCl4, если желаемым продуктом является CH3Cl. Использованное здесь выражение “по существу не дает пергалогенированного продукта” означает производство <5 мол.% пергалогенированного продукта, предпочтительно <2 мол.%, более предпочтительно ≤1 мол.% и еще более предпочтительно не более 0,1 мол.%, причем каждый мол.% рассчитан по сумме моль галогенированного продукта.
Окислительное галогенирование может быть проведено в реакторе любой традиционной конструкции, подходящей для газофазных процессов, включая реакторы периодического действия, реакторы с неподвижным слоем, реакторы с псевдоожиженным слоем, реакторы с транспортируемым слоем, проточные реакторы непрерывного и прерывистого действия и реакторы каталитической дистилляции. Условия способа (например, молярное отношение подаваемых компонентов, температура, давление, часовая объемная скорость газа (GHSV)) могут изменяться в широких пределах при условии, что они дают желаемый продукт галогенированного метана, предпочтительно CH3Cl. Типично, температура в способе выше (>) 200°C, предпочтительно >300°C и более предпочтительно >350°C. Типично, температура в способе составляет <600°C, предпочтительно <500°C и более предпочтительно <450°C. Обычно способ может быть осуществлен при атмосферном давлении, но при желании возможна работа при более высоких или более низких давлениях. Предпочтительно, давление ≥14 фунтов на квадратный дюйм, абсолютное (psia) (97 килопаскаль (кПа)), но <300 psia (2068 кПа). Типично, суммарная массовая часовая объемная скорость (WHSV) подаваемого материала (метана, источника галогена, источника кислорода и необязательного разбавителя) будет составлять >0,1 грамма суммарной подачи на г катализатора в час (ч-1) и предпочтительно >0,5 ч-1. Типично, суммарная WHSV подаваемого материала будет составлять <100 ч-1 и предпочтительно <20 ч-1.
Моногалогенированные и дигалогенированные углеводородные продукты, предпочтительно, моногалогенированные продукты, более предпочтительно CH3Cl или метилбромид, произведенные в способе окислительного галогенирования данного изобретения, могут быть использованы в качестве сырья в последующих способах переработки, которые производят высокоценные товарные химикаты, такие как метиловый спирт, диметиловый эфир, легкие олефины, включая этилен, пропилен и бутены; более высокие углеводороды, включая C5+-газолины; винилгалогенидный мономер и уксусная кислота. Гидролиз метилгалогенидов с образованием метилового спирта описан в данной области техники, и его репрезентативные публикации включают патент США 1086381 (Masland), патент США 4990696 (Stauffer), патент США 4523040 (Olah) и патент США 5969195 (Stabel с соавт.), а также G. Olah, Journal of the American Chemical Society, 1985, 107, 7097-7105 и I. Fells, Fuel Society Journal, 10 (1959), страницы 26-35. Гидролиз метилхлорида до метилового спирта может быть представлен следующей стехиометрической реакцией: CH3Cl + H2O → CH3OH + HCl.
Метилгалогенид, предпочтительно CH3Cl, полученный вышеупомянутым окислительным галогенированием CH4, может быть конденсирован с образованием легких олефинов, таких как этилен (CH2=CH2), пропилен, бутены и более высокие углеводороды, включающие C5+-газолины. Такая реакция конденсации может быть представлена следующим уравнением, показывающим конденсацию с образованием CH2=CH2 с хлоридом водорода (HCl) в качестве побочного продукта, который может быть рециркулирован для применения в окислительном галогенировании в качестве источника галогена
2 CH3Cl → CH2=CH2 + 2 HCl
Может быть использован любой катализатор, способный осуществлять конденсацию. Патент США 5397560 (Millar с соавт.), например, раскрывает применение алюмосиликатов, имеющих структурный код DCM-2, для конверсии метилгалогенидов в легкие олефины, преимущественно этилен и пропилен.
Примеры
Примеры (Пр.) настоящего изобретения обозначены арабскими цифрами, а сравнительные примеры (сравн. прим.) обозначены заглавными буквами алфавита. Если иное не указано, “комнатная температура” и “окружающая температура” составляют номинально 25°C.
Получение SO 4 /ZrO 2 , A
Нагревают два литра (2 л) деионизированной (DI) воды, pH которой регулируют до 10 гидроксидом аммония (NH4OH), до заданного значения температуры 50°C. Растворяют 65,1 г (0,202 моль) гидратированного оксихлорида циркония (ZrOCl2·8H2O) в нагретой и pH-регулированной DI-воде, затем добавляют достаточное количество DI-воды, чтобы увеличить объем раствора до 250 миллилитров (мл). Переносят полученный раствор в первую капельную воронку.
Разводят 140 г концентрированного NH4OH 500 мл DI-воды и переносят разведенный NH4OH во вторую капельную воронку.
В течение периода 15 минут прибавляют содержимое первой и второй капельных воронок в контейнер емкостью один (1) л, содержащий воду с pH, отрегулированным до 10 с помощью NH4OH, таким образом, чтобы по завершении периода 15 минут контейнер содержал в себе один объем содержимого первой капельной воронки на каждые два объема содержимого второй капельной воронки. Поддерживают pH содержимого контейнера при 10 добавлением концентрированного NH4OH по необходимости. Перемешивают содержимое контейнера в течение периода одного часа, затем позволяют осаждающимся твердым частицам (определены как оксигидрат циркония (ZrO(OH)2)) осесть на дно контейнера.
Извлекают осевшие твердые частицы четырьмя повторными фильтрованиями и повторным суспендированием твердых частиц в одном л DI-воды с последующим окончательным фильтрованием. Сушат твердые частицы в течение ночи в воздушной печи, работающей при заданной температуре 110°C. После сушки дробят твердые частицы до размера 14/30 меш (от 0,6 нанометра (нм) до 1,2 мм).
Сушат 23 г раздробленных твердых частиц в течение одного часа в воздушной печи, работающей при заданной температуре 140°C, чтобы удалить остаточную поверхностную воду. Прибавляют 25 г концентрированной (18 нормальной (н)) серной кислоты (H2SO4) к DI-воде, чтобы получить 100 мл 2,55 M раствора H2SO4. Пропитывают высушенные, раздробленные твердые частицы 7,49 г 2,55 M раствора H2SO4.
Прокаливают пропитанные твердые частицы в воздушной печи следующим образом: a) нагревают пропитанные твердые частицы от заданной температуры 25°C до заданной температуры 125°C в течение периода одного часа; b) удерживают температуру 125°C в течение периода двух часов; c) повышают заданную температуру до 600°C в течение периода четырех часов; d) удерживают температуру 600°C в течение четырех часов; e) охлаждают до заданной температуры 130°C в течение периода трех часов; f) выдерживают при температуре 130°C до удаления прокаленных твердых частиц из печи; и g) охлаждают прокаленные твердые частицы до окружающей температуры в эксикаторе. Прокаленные твердые частицы, извлеченные из эксикатора, имеют массу 20,5 г и имеют содержание сульфата шесть (6) мас.% в расчете на суммарную массу прокаленных твердых частиц и количество прибавленной серной кислоты.
Получение SO 4 /ZrO 2 , B
Повторяют получение SO4/ZrO2, A, с изменениями. Во-первых, прокаливают 23 г твердых частиц просеянного и высушенного ZrO(OH)2 в течение одного часа в воздухе в печи, работающей при заданной температуре 300°C, чтобы превратить ZrO(OH)2 в ZrO2. Во-вторых, добавляют 25 г концентрированной (приблизительно 18 н) серной кислоты (H2SO4) к DI-воде, чтобы приготовить 100 мл 2,55 M раствора H2SO4. В-третьих, пропитывают ZrO2 7,51 г раствора H2SO4. В-четвертых, прокаливают, как в получении A, получая 20,5 г номинальных шесть процентов по массе (6 мас.%) сульфатированного оксида циркония (SO4/ZrO2).
Пример 1 и сравнительный пример A
В первую трубку (пример 1) реактора с восходящим потоком, имеющую внутренний диаметр четыре (4) миллиметра (мм), сначала помещают слой 0,5 г оксихлорида лантана (LaOCl) (получен с использованием описания патента США 6452058 (Schweizer с соавт.)) вблизи середины трубки, затем помещают слой 0,5 г сульфатированного оксида циркония, полученного выше, поверх слоя LaOCl, так что текущие газы-реагенты сначала контактируют со слоем LaOCl, а затем контактируют со слоем сульфатированного оксида циркония. Во вторую трубку (сравнительный пример A) реактора с восходящим потоком помещают только слой 0,5 г LaOCl, как в примере 1.
Пропускают поток подаваемого материала (20 объемных процентов (об.%) CH4, 20 об.% HCl, 10 об.% кислорода (O2) и 50 об.% инертных газов (например, гелия (He), азота (N2) или их сочетания) через каждую из трубок при температурах (Темп.), показанных ниже в таблице 1, и определяют процент конверсии CH4 в CH3Cl при каждой из температур, причем значения конверсии также показаны ниже в таблице 1. Ниже в таблице 2 показаны данные по селективности по CH3Cl, CH2Cl2 и CHCl3 для примера 1.
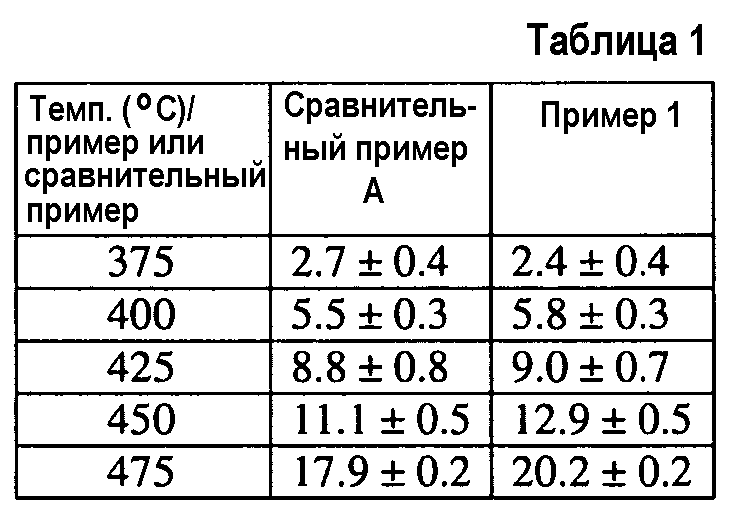
Данные в таблице 1 демонстрируют, что по меньшей мере при температурах 450°C и 475°C добавление слоя сульфатированного оксида циркония к слою LaOCl (пример 1) обеспечивает увеличение конверсии CH4 в CH3Cl относительно единственного слоя LaOCl. При температурах 375°C, 400°C и 425°C между примером 1 и сравнительным примером A не существует существенной разницы в конверсии CH4 в CH3Cl.
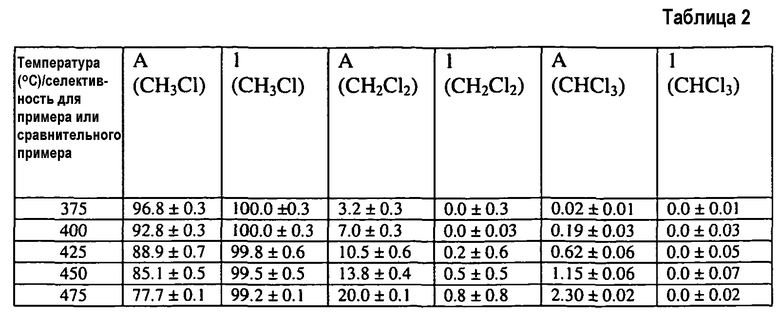
Данные, представленные в таблице 2, демонстрируют, что при всех температурах, показанных в таблице 2, добавление слоя сульфатированного оксида циркония к слою LaOCl (пример 1) эффективно повышает селективность по монохлорированному метану (CH3Cl) относительно полихлорированных метанов (CH2Cl2 и CHCl3) по сравнению с единственным слоем LaOCl (сравнительный пример A), даже если оно, как показано выше в таблице 1, не увеличивает конверсию метана в CH3Cl относительно конверсии для того же единственного слоя LaOCl при температурах 375°C, 400°C и 425°C.
Пример 2-4 и сравнительный пример B-C
Повторяют пример 1 при температуре 475°C и определяют селективность по CH3Cl, CH2Cl2 и CO в расчете на моли CH3Cl, CH2Cl2 и CO, а также относительную селективность по CH3Cl и CH2Cl2 в расчете на моли CH3Cl и CH2Cl2. Ниже в таблице 3 приведены результаты, причем селективность по CH3Cl, CH2Cl2 и CO показана слева от пустой колонки, а селективность по CH3Cl и CH2Cl2 справа от пустой колонки. В сравнительном примере B используют только 0,5 г сульфатированного оксида циркония. Пример 4 представляет менее предпочтительное расположение, в котором порядок слоев, показанный в вышеописанном примере 1, обращен. В примере 2 LaOCl и сульфатированный оксид циркония смешаны в единственный слой. В примере 3 используют такое же расположение катализатора, как в примере 1. В сравнительном примере C используют такое же расположение катализатора, как в сравнительном примере A.
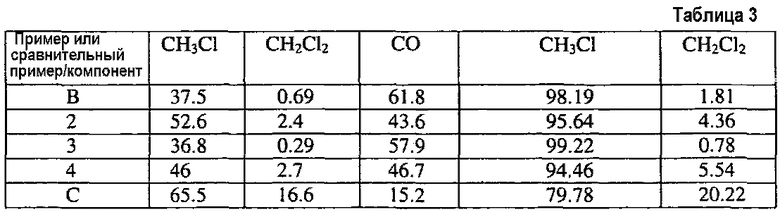
Данные, представленные в таблице 3, демонстрируют, что применение твердой суперкислоты (SO4/ZrO2) улучшает селективность по CH3Cl по сравнению с селективностью по CH2Cl2 относительно селективности по CH3Cl по сравнению с селективностью по CH2Cl2 при использовании одного LaOCl (сравнительный пример C). Фактически, твердая суперкислота сама по себе (сравнительный пример B) дает улучшение в селективности по сравнению с одним LaOCl (сравнительный пример C). Максимальное увеличение селективности по CH3Cl по сравнению с селективностью по CH2Cl2 происходит, когда поток подаваемого материала сначала контактирует со слоем LaOCl перед контактом со слоем суперкислоты (пример 3). Смесь твердой суперкислоты и LaOCl (пример 2), хотя не будучи такой же хорошей, как расположение (пример 3), все же дает лучшие результаты, чем один LaOCl (сравнительный пример C), как происходит и в случае примера 4 с обращением расположения слоев примера 3.
Пример 5 - пример 6 и сравнительный пример D
Используют реакторную систему, которая состоит из пяти реакторных трубок, погруженных в обычный нагреватель в виде песчаной бани, с 1,50 г LaOCl (получен с использованием описания патента США 6452058 (Schweizer с соавт.)) в одной трубке (сравнительный пример D), 1,51 г катализатора SO4/ZrO2, описанного в получении B, во второй трубке (пример 5) и 1,51 г катализатора SO4/ZrO2, описанного в получении A, в третьей трубке (пример 6). Четвертая и пятая трубка являются пустыми. Подают со скоростью 33 стандартных кубических сантиметра в минуту (sccm) смесь газов через контроллеры массового расхода в реакторную систему, чтобы получить газовую смесь 20 об.% CH4, 20 об.% HCl и 10 об.% O2, 5 мол.% азота (N2) и 45 об.% гелия, причем каждый об.% рассчитан на суммарный объем газа, присутствующего в газовой смеси. Использованный здесь в отношении газов об.% эквивалентен мольному проценту (мол.%). Анализ содержания в реакторных трубках на CH4, CH3Cl (MeCl), CH2Cl2 (MeCl2), CO и CO2 осуществляется газовой хроматографией с использованием Siemens MaxumTM Edition II Process Gas Chromatograph. Ниже в таблице 4 показаны аналитические результаты для содержания газов в реакторных трубках при температуре 430°C.
Пример 7 - пример 8 и сравнительный пример E
Повторяют пример 5, пример 6 и сравнительный пример D, соответственно в случае примера 7, примера 8 и сравнительного примера E, но изменяют температуру на 480°C. Аналитические результаты для содержания газов в реакторных трубках см. ниже в таблице 5.
Результаты в таблице 4 и таблице 5 подтверждают несколько наблюдений. Во-первых, каждый из катализаторов, использованных в примере 5 - примере 8, превращает по меньшей мере часть CH4 в CH3Cl с изменяющейся селективностью по CH3Cl, как это имеет место и в СП-D и СП-E. В каждом случае селективность по CH3Cl превышает селективность по CH2Cl2. Во-вторых, каждый из катализаторов (пример 5-8) дает некоторое количество оксидов углерода (CO и CO2), как это имеет место в случае СП-D и СП-E. Однако каждый из катализаторов (пример 5-8) показывает объединенную селективность CH3Cl+CO больше 84%, тогда как СП-D и СП-E показывают объединенную селективность CH3Cl+CO менее 84%. В-третьих, каждый из катализаторов (пример 5-8) показывает относительную селективность по CH3Cl более 95%, тогда как СП-D и СП-E показывают относительную селективность по CH3Cl менее 81%.
Хотя возможны многие варианты осуществления данного способа, пример 3 и сравнительный пример B из таблицы 3 показывают особенно высокую селективность (>96%) по моногалогенированному метану относительно метиленгалогенида. Данные в таблицах 4 и 5 показывают, что катализатор и конфигурации катализатора (пример 5 - пример 8) показывают высокую селективность по моногалогенированному метану (CH3Cl). Однако конверсия для равной массы катализатора хуже конверсии, наблюдаемой для катализатора в сравнительном примере D и E. Если выбирают максимальное увеличение селективности по моногалогенированному метану, то расположение (пример 3) (поток подаваемого материала контактирует со слоем LaOCl и затем поток подаваемого материала с прореагировавшими компонентами контактирует со слоем твердой суперкислоты) представляет предпочтительный вариант по сравнению с сравнительным примером B, сравнительным примером C или сравнительным примером D. Композиция катализатора и расположение примера 3 ведет как к более высокой активности, так и к более высокой селективности по моногалогенированному метану относительно применения каждого индивидуального катализатора отдельно (сравнительный пример B - сравнительный пример D).
Пример 9 - пример 14
Повторяют пример 5, но изменяют скорость газового потока (в sccm) и GHSV, как показано ниже в таблице 6. В примере 9-12 используют 1,51 г катализатор SO4/ZrO2 получения B. В примере 12 и примере 14 используют 3,02 г того же катализатора. В таблице 6 также показаны аналитические результаты определения содержания газов в реакторных трубках.
Пример 15 - пример 20
Повторяют примеры 9-14, но используют 1,51 г катализатор SO4/ZrO2 получения A для примера 15 - примера 18 и 2,99 г того же катализатора для примера 19 и примера 20. Ниже в таблице 7 показаны скорость потока газа, GHSV и аналитические результаты определения содержания газов в реакторных трубках.
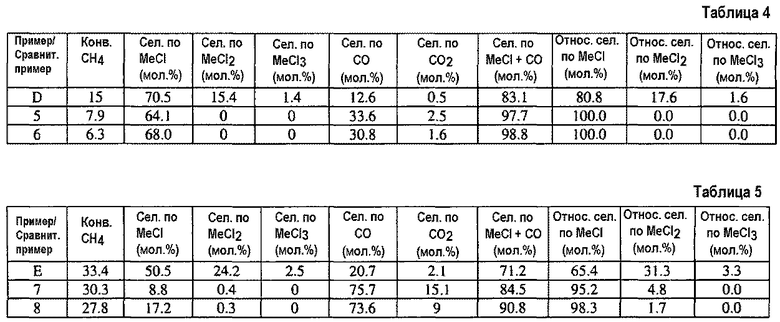
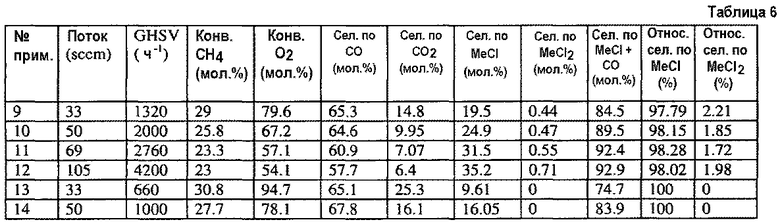
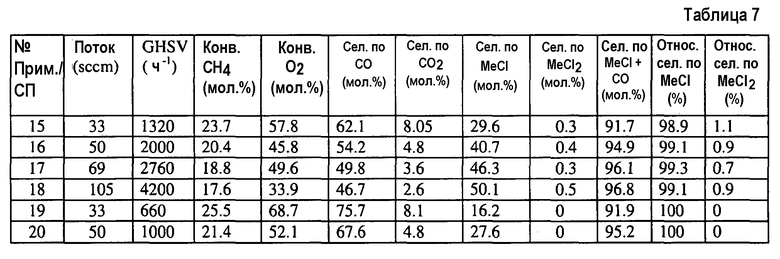
Результаты в таблице 6 и таблице 7 подтверждают несколько наблюдений. Во-первых, каждый из катализаторов, использованных в примере 8 - примере 19, превращает по меньшей мере часть CH4 в CH3Cl с изменяющейся селективностью по CH3Cl. В каждом случае селективность по CH3Cl превышает селективность по CH2Cl2. Во-вторых, каждый из катализаторов примера 8-19 дает некоторое количество оксидов углерода (CO и CO2). Однако каждый из катализаторов примера 8-19 показывает объединенную селективность CH3Cl+CO больше 74%. В-четвертых, каждый из катализаторов примера 8-19 показывает относительную селективность по CH3Cl более 97%.
Сходные результаты ожидаются для других сильнокислых материалов и технологических условий, все из которых раскрыты в данной заявке.
Получение CaF 2 /ZrO 2
Часть A:
Помещают два литра (2 л) деионизированной воды в стакан на 4 л. Регулируют pH деионизированной воды до 10 прибавлением гидроксид аммония (NH4OH), получая первый раствор. Нагревают первый раствор до температуры 40 градусов Цельсия (°C).
Растворяют 65,1 грамм (г) (0,202 молей (мол.)) гидратированного оксихлорида циркония (ZrOCl2·8H2O) в 100 миллилитрах (мл) деионизированной воды, затем добавляют достаточное количество деионизированной воды, чтобы получить 250 мл второго раствора. Помещают второй раствор в первую капельную воронку.
Разводят 140 г концентрированного (приблизительно 15 молярного (M)) NH4OH 500 мл деионизированной воды, получая 500 мл третьего раствора. Помещают третий раствор во вторую капельную воронку.
Прибавляют второй и третий растворы к первому раствору при быстром (более (>) 250 оборотов в минуту (об/мин)) перемешивании в течение периода 15 минут при скоростях, достаточных, чтобы прибавить один объем второго раствора на каждые два объема третьего раствора. Прибавляют дополнительное количество концентрированного NH4OH, чтобы поддержать pH содержимого стакана при 10. Продолжают перемешивание содержимого стакана в течение периода 30 минут, затем прекращают перемешивание и позволяют оседающим твердым частицам (оксигидроксид циркония ZrO(OH)2) осесть на дно стакана.
Часть B:
Растворяют 3,62 г хлорида кальция (CaCl2) в 100 мл деионизированной воды для образования пятого раствора и 3,98 г фторида калия (KF) в 100 мл деионизированной воды для образования шестого раствора.
Прибавляют пятый и шестой растворы к содержимому стакана при быстром перемешивании (>250 об/мин), затем повышают температуру содержимого стакана до 40°C и продолжают перемешивание в течение четырех с половиной часов перед тем, как позволить температуре вернуться к окружающей и осесть на дно стакана твердым компонентам содержимого стакана. Осаждение происходит в течение периода от примерно получаса до одного часа. Сохраняют осевшее содержимое в надосадочной жидкости до начала извлечения твердых компонентов.
Извлекают твердые компоненты из содержимого стакана фильтрованием и затем ресуспендируют твердые компоненты в 1 л деионизированной воды при перемешивании в течение 15 минут перед извлечением твердых частиц снова фильтрованием. Повторяют ресуспендирование и фильтрование четыре раза или пока анализ ресуспендированных твердых частиц с использованием нитрата серебра (AgNO3) не покажет отсутствие детектируемых ионов хлора (Cl¯), затем фильтруют еще один раз. Сушат твердые частицы, собранные фильтрованием, при температуре 110°C в течение двух часов, затем дробят твердые частицы и продолжают сушку в течение дополнительных 10 часов при температуре 115°C. Прокаливают высушенные твердые частицы в воздушной печи, нагретой до заданной температуры 800°C в течение пяти часов, затем выдерживают прокаленные твердые частицы в течение дополнительного периода четырех часов перед тем, как позволить прокаленным твердым частицам охладиться до окружающей температуры. Прокаленные твердые частицы (24,5 г) составляют фторид кальция на носителе из оксида циркония (CaF2/ZrO2).
Получение WO 3 /ZrO 2
Повторяют получение CaF2/ZrO2 вплоть до предоставления возможности осесть оседающим твердым частицам ZrO(OH)2 на дно стакана (вплоть по часть A). Сушат осевшие твердые частицы ZrO(OH)2 при температуре 110°C в течение по меньшей мере 12 часов. Дробят и просеивают высушенные твердые частицы ZrO(OH)2 до размера 14/30 меш (диапазон отверстия сита от 1,41 мм для 14 меш до 0,595 мм для 30 меш), что означает, что твердые частицы проходят через экран 14 меш, но остаются на экране 30 меш.
Растворяют 2,85 г метавольфрамата аммония ((NH4)6H2W12O40) в 7,5 г деионизированной (DI) воды с получением раствора метавольфрамата аммония. Прибавляют раствор метавольфрамата аммония к 17,5 г высушенных (непрокаленных) твердых частиц ZrO(OH)2 (собранных на экране 30 меш (отверстие сита 0,595 мм) путем пропитывания.
Прокаливают пропитанный материал следующим образом: нагревают твердые частицы в воздушной печи для прокаливания от окружающей температуры до заданной температуры 125°C в течение периода одного часа со скоростью нагрева 1,7°C в минуту; выдерживают твердые частицы при заданной температуре 125°C в течение периода двух часов; нагревают твердые частицы до заданной температуры 800°C в течение периода 10 часов со скоростью 1,1°C в минуту; выдерживают при 800°C в течение периода четырех часов; охлаждают до 130°C в течение периода трех часов со скоростью 3,7°C в минуту; выдерживают твердые частицы при 130°C в течение периода 4 часов; и удаляют твердые частицы из печи и помещают их в эксикатор для охлаждения до окружающей температуры. Охлажденные твердые частицы (16,4 г) составляют вольфрамат на носителе из оксида циркония (WO3/ZrO2).
Получение Ce(SO 4 ) 2 /ZrO 2
Повторяют получение WO3/ZrO2, но используют сульфат церия вместо метавольфрамата аммония. Растворяют 4,5 г гидратированного сульфата церия (Ce(SO4)2·nH2O) в 15,5 г DI-воды с получением раствора сульфата церия. Прибавляют 5,1 г раствора сульфата церия к 13,2 г высушенных (непрокаленных) твердых частиц ZrO(OH)2 путем пропитывания.
Прокаливают, как при получении CaF2/ZrO2, но изменяют заданную температуру 800°C до 600°C и сокращают период нагрева до 600°C до четырех часов. Охлажденные твердые частицы (11,4 г) составляют сульфат церия на носителе из оксида циркония (Ce(SO4)2/ZrO2).
Получение VO(SO 4 )/ZrO 2
Повторяют получение Ce(SO4)2/ZrO2, но используют 4,0 г раствора сульфата ванадия, полученного растворением 4,5 г гидратированного сульфата ванадия (VO(SO4)2·nH2O) в 15,6 г DI-воды вместо сульфата церия. Охлажденные твердые частицы (10,0 г) составляют сульфат ванадия на носителе из оксида циркония (VO(SO4)/ZrO2).
Получение ZrO(OH) 2
Повторяют получение CaF2/ZrO2 вплоть до предоставления возможности осесть оседающим твердым частицам ZrO(OH)2 на дно стакана (вплоть по часть A). Сушат осевшие твердые частицы ZrO(OH)2 при температуре 110°C в течение по меньшей мере 12 часов. Дробят и просеивают высушенные твердые частицы ZrO(OH)2 до размера 14/30 меш, как при получении WO3/ZrO2.
Примеры 21-25
Используют тот же реактор, который описан в случае примера 5, примера 6 и сравнительного D, и повторяют использованный в них способ, но изменяют скорость газового потока до 20 sccm и газовую смесь на 80 об.% CH4, 10 об.% HCl и 5 об.% O2 и 5 об.% N2, причем каждый об.% рассчитан на суммарный объем газа, присутствующего в газовой смеси. Помещают 0,5 г CaF2/ZrO2 в первую трубку (пример 21), 0,5 г WO3/ZrO2 во вторую трубку (пример 22), 0,5 г Ce(SO4)2/ZrO2 в третью трубку (пример 23), 0,5 г VO(SO4)/ZrO2 в четвертую трубку (пример 24) и 0,5 г SO4/ZrO2 в пятую трубку (пример 25). См. ниже таблицу 8 касательно аналитических результатов реакторных трубок при температурах, указанных для каждого примера, причем определения конверсии и селективности приведены выше.

Результаты в таблице 8 подтверждают несколько наблюдений. Во-первых, каждый из катализаторов, использованных в примере 21 - примере 25, превращает по меньшей мере часть CH4 в MeCl (CH3Cl) с изменяющейся селективностью по MeCl. В каждом случае селективность по MeCl превышает селективность по MeCl2. Хотя CaF2/ZrO2 (пример 21) и Ce(SO4)2/ZrO2 (пример 23) обеспечивают более высокую селективность по MeCl, чем катализаторы примера 22 (WO3/ZrO2), примера 23 (VO(SO4)/ZrO2) и примера 25 (SO4/ZrO2), катализаторы примера 22, примера 24 и примера 25 также являются подходящими. Во-вторых, каждый из катализаторов примера 21-25 дает некоторые количества оксидов углерода (CO и CO2), причем опять же в примере 21 и примере 23 получаются меньшие количества таких оксидов углерода. В-третьих, каждый из катализаторов примера 21-25 показывает объединенную селективность CH3Cl + CO больше 85%. В-четвертых, каждый из катализаторов примера 21-25 показывает относительную селективность по CH3Cl более 95%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОКИСЛИТЕЛЬНОГО ГАЛОГЕНИРОВАНИЯ C УГЛЕВОДОРОДОВ ДО ГАЛОГЕНИРОВАННЫХ C УГЛЕВОДОРОДОВ И СВЯЗАННЫЕ С НИМ ИНТЕГРИРОВАННЫЕ СПОСОБЫ | 2002 |
|
RU2286329C2 |
| СПОСОБ ОКИСЛИТЕЛЬНОГО ГАЛОГЕНИРОВАНИЯ И СПОСОБ ПОЛУЧЕНИЯ ГАЛОГЕНИРОВАННОГО C ПРОДУКТА | 2006 |
|
RU2409547C2 |
| СИНТЕЗ УГЛЕВОДОРОДОВ | 2004 |
|
RU2366642C2 |
| СПОСОБ ОКИСЛИТЕЛЬНОГО ГАЛОГЕНИРОВАНИЯ И НЕОБЯЗАТЕЛЬНОГО ДЕГИДРИРОВАНИЯ УГЛЕВОДОРОДОВ ОТ C ДО C (ВАРИАНТЫ) | 2002 |
|
RU2284984C2 |
| КОМПЛЕКСНЫЙ СПОСОБ КАТАЛИТИЧЕСКОЙ ПЕРЕРАБОТКИ ПРИРОДНОГО ГАЗА С ПОЛУЧЕНИЕМ НИЗШИХ ОЛЕФИНОВ | 2011 |
|
RU2451005C1 |
| ЭЛЕКТРОХИМИЧЕСКИЙ СПОСОБ ПОЛУЧЕНИЯ ГАЛОГЕНИРОВАННОГО СОЕДИНЕНИЯ, СОДЕРЖАЩЕГО КАРБОНИЛЬНУЮ ГРУППУ | 2007 |
|
RU2423553C2 |
| КАТАЛИЗАТОР ОКИСЛЕНИЯ МЕТАНА, ПРОЦЕСС ЕГО ПОЛУЧЕНИЯ И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2016 |
|
RU2715732C2 |
| КАТАЛИЗАТОР, СПОСОБ ПРЕВРАЩЕНИЯ УГЛЕВОДОРОДОВ И СПОСОБ ИЗОМЕРИЗАЦИИ ПАРАФИНОВОГО СЫРЬЯ | 2004 |
|
RU2342189C1 |
| КАТАЛИЗАТОР ИЗОМЕРИЗАЦИИ ЛЕГКИХ БЕНЗИНОВЫХ ФРАКЦИЙ И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2017 |
|
RU2633756C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОБАЛЬТОВОГО КАТАЛИЗАТОРА | 2012 |
|
RU2493914C1 |
Изобретение относится к способу окислительного галогенирования метана, включающему контактирование потока подаваемого материала, который содержит метан, источник галогена и источник кислорода, с первым катализатором и в условиях, достаточных для обеспечения потока продукта, где первый катализатор выбран из группы, состоящей из твердых суперкислот и твердых супероснований. Данный катализатор имеет большую селективность по метилгалогениду и монооксиду углерода, чем по метиленгалогениду, тригалогенметану или тетрагалогениду углерода. 9 з.п. ф-лы, 25 пр., 8 табл.
1. Способ окислительного галогенирования метана, включающий контактирование потока подаваемого материала, который содержит метан, источник галогена и источник кислорода, с первым катализатором и в условиях, достаточных для обеспечения потока продукта, который имеет большую селективность по метилгалогениду и монооксиду углерода, чем по метиленгалогениду, тригалогенметану или тетрагалогениду углерода, где первый катализатор выбран из группы, состоящей из твердых суперкислот и твердых супероснований.
2. Способ по п.1, в котором твердая суперкислота выбрана из группы, состоящей из вольфраматного оксида циркония, сульфатированного оксида циркония, сульфатированного оксида титана, сульфатированного оксида титана-лантана, сульфатированного оксида олова, сульфата церия на носителе из оксида циркония и сульфата ванадия на носителе из оксида циркония, и твердое супероснование выбрано из группы, состоящей из фторида кальция на носителе из оксида циркония, фторида бария на носителе из оксида циркония; допированного калием оксида магния (K-допированный/MgO); и оксида натрия на носителе из оксида магния.
3. Способ по п.1 или 2, в котором поток подаваемого материала также контактирует со вторым катализатором, который окислительно галогенирует по меньшей мере часть метана, с получением смеси, содержащей по меньшей мере два члена группы, состоящей из метилгалогенида, метиленгалогенида, тригалогенметана, тетрагалогенида углерода, воды, галогенида водорода, непрореагировавшего галогена и непрореагировавшего кислорода.
4. Способ по п.3, в котором второй катализатор выбран из группы, состоящей из галогенидов редкоземельных элементов и оксигалогенидов редкоземельных элементов.
5. Способ по п.3, в котором поток подаваемого материала контактирует со вторым катализатором перед тем, как он контактирует с первым катализатором, где контактирование со вторым катализатором дает смесь по меньшей мере двух членов группы, состоящей из метилгалогенида, метиленгалогенида, тригалогенметана, тетрагалогенида углерода, воды, галогенида водорода, непрореагировавшего галогена и непрореагировавшего кислорода, и где контактирование с первым катализатором превращает по меньшей мере часть метиленгалогенида, тригалогенметана и тетрагалогенида углерода в монооксид углерода, галогенид водорода и воду.
6. Способ по п.1, в котором источник галогена представляет собой по меньшей мере один галогенид водорода, выбранный из хлорида водорода, бромида водорода, фторида водорода и иодида водорода.
7. Способ по п.1, в котором условия являются достаточными для получения эквимолярной смеси монооксида углерода и метилхлорида.
8. Способ по п.1, в котором поток подаваемого материала дополнительно содержит количество монооксида углерода, достаточное для предоставления эквимолярной смеси монооксида углерода и метилхлорида.
9. Способ по п.7, в котором эквимолярная смесь монооксида углерода и метилхлорида контактирует с катализатором карбонилирования в условиях, достаточных для превращения по меньшей мере части эквимолярной смеси в по меньшей мере один из продуктов: ацетилхлорид и уксусную кислоту.
10. Способ по п.1, в котором а) селективность по метилгалогениду находится в диапазоне от 35 мол.% до 100 мол.%; b) конверсия метана в метилгалогенид находится в диапазоне от 0,1 мол.% до 100 мол.% в расчете на моли метана, присутствующего перед конверсией; с) селективность по сочетанию метилгалогенида и монооксида углерода находится в диапазоне от 50 мол.% до 100 мол.%; d) селективность по метилгалогениду относительно селективности по сочетанию метилгалогенида, метиленгалогенида, тригалогенметана и тетрагалогенида углерода находится в диапазоне от 85 мол.% до 100 мол.%; или е) имеет место сочетание двух или более из а)-d).
| WO 2006118935 A2, 09.11.2006 | |||
| BATAMACK PATRICE ET AL | |||
| Catalysis by solid superacids | |||
| Солесос | 1922 |
|
SU29A1 |
| Electrophilic chlorination of methane over superacidic sulfated zirconia, Catalysis Letters, Vol: 25, Nr: 1-2, Page (s): 11-19 | |||
| GEORGE A OLAH ET AL | |||
| Slective Monohalogenation of Methane over Supported Acid or Platinum Metal Catalysts and Hydrolysis of Methyl | |||

