Перекрестная ссылка
Данная заявка испрашивает приоритет по временной заявке США № 60/870009, зарегистрированной 14 декабря 2006 г., временной заявке США № 60/894835, зарегистрированной 14 марта 2007 г., и временной заявке США № 60/894829, зарегистрированной 14 марта 2007 г., временной заявке США № 60/894799, зарегистрированной 14 марта 2007 г., заявке США № 11/733642, зарегистрированной 10 апреля 2007 г., и заявке США № 11/749497, зарегистрированной 16 мая 2007 г., и временной заявке США № 60/979049, зарегистрированной 10 октября 2007 г., каждая из которых полностью включена в настоящий документ в качестве ссылки.
Уровень техники
(R)-прамипексол представляет собой энантиомер активного фармацевтического ингредиента разрешенного лекарственного средства для лечения болезни Паркинсона (PD) и синдрома усталых ног (RLS) Mirapex (прамипексол; (S)-прамипексол). Mirapex® представляет собой высокоаффинный (низкая, нМ, IC50) агонист рекомбинантных дофаминовых рецепторов D2 и D3 человека и грызунов, свойство, которое является фармакологической основой его эффективности при указанных расстройствах. Как было показано в доклинических исследованиях, как (R)-, так и (S)-энантиомеры обладают нейропротективными свойствами, которые не зависят от аффинности в отношении дофаминовых рецепторов.
Нейропротективные свойства (S)-прамипексола признаны потенциально полезными для лечения нейродегенеративных расстройств, но клинический опыт применения лекарственного средства для лечения расстройств, связанных с дефицитом дофамина, таких как PD, показал, что введение лекарственного средства ограничивается как временным фактором, необходимостью длительного титрования дозы, так и абсолютно в том, что касается максимально переносимой дозы (MTD), из-за побочных эффектов, связанных с действием агониста дофаминовых рецепторов. Указанные ограничения дозирования являются типичными для агонистов дофаминовых рецепторов указанного класса.
Предельно допустимая однократная начальная доза Mirapex® составляет 0,125 мг, которую вводят три раза в день (t.i.d.); а предельно допустимая доза Mirapex составляет 1,5 мг t.i.d., что дает максимальную суточную дозу 4,5 мг Mirapex® после 7-8 недель титрования.
В то время как указанные уровни доз Mirapex® являются пригодными для лечения признаков и симптомов PD и RLS, в нейропротективных исследованиях эффективность (S)-прамипексола в качестве нейропротектора приблизительно в 1000 раз ниже его эффективности в качестве агониста дофаминовых рецепторов. Это наводит на мысль о том, что при использовании данного энантиомера невозможно достичь терапевтически пригодных нейропротективных доз.
(R)-прамипексол обладает сходной нейропротективной эффективностью, но более низкой аффинностью в отношении дофаминовых рецепторов. Соответственно, его продвигали как соединение, потенциально более пригодное для лечения нейродегенеративных расстройств. Однако разница в аффинности в отношении дофаминовых рецепторов между (R)-прамипексолом и (S)-прамипексолом, о которой сообщалось ранее, все же должна будет накладывать клинически важные ограничения на дозы и все же потребует титрования дозы и ограничений дозы, чтобы избежать побочных эффектов, связанных с дофамином. В предыдущих исследованиях, касающихся применения (R)-прамипексола для лечения бокового амиотрофического склероза (ALS), быстро прогрессирующего смертельного нейродегенеративного расстройства, было предложено ограничить введение (R)-прамипексола и потребовать достоверного титрования дозы в экспериментах на животных. Принятое требование титрования дозы, конкретно, требование начинать введение с очень низких доз и повышать дозу до конечного, терапевтически эффективного уровня дозы в течение 7-8 недель, строго ограничивает пригодность нейропротективного потенциала энантиомера (R)-прамипексола. Кроме того, принятая MTD будет строго ограничивать регулярное использование нейропротективного потенциала энантиомера (R)-прамипексола как при острых, так и при хронических нейродегенеративных расстройствах.
Сущность изобретения
Настоящее изобретение раскрывает терапевтический потенциал (R)-прамипексола путем получения клинически очищенного (R)-прамипексола и определения фактической in vitro и in vivo связывающей аффинности и переносимости пациентом очищенного (R)-прамипексола. В соответствии с вариантами осуществления настоящего изобретения более высокие дозы (R)-прамипексола можно вводить пациенту, который в этом нуждается.
Настоящее изобретение дополнительно относится к способу лечения нейродегенеративного заболевания у пациента, который в этом нуждается, включающему введение пациенту суточной дозы в количестве приблизительно от 25 мг до 5000 мг (R)-прамипексола, более предпочтительно, приблизительно от 500 мг до 2100 мг, наиболее предпочтительно, более 500 мг и менее 2100 мг (R)-прамипексола ежедневно.
В некоторых вариантах осуществления настоящего изобретения заболевание, подлежащее лечению, является острым, а в других вариантах - хроническим. В некоторых вариантах осуществления настоящего изобретения хроническое нейродегенеративное заболевание выбрано из первичного нейродегенеративного заболевания, хореи Гентингтона, метаболически индуцированного неврологического повреждения, сенильной деменции типа Альцгеймера, когнитивной дисфункции, связанной с возрастом, сосудистой деменции, мультиинфарктной деменции, деменции с тельцами Леви, нейродегенеративной деменции, нейродегенеративного двигательного расстройства, атаксии, атаксии Фридрейха, рассеянного склероза, спинальной мышечной атрофии, первичного бокового склероза, расстройств, сопровождаемых припадками, расстройства или заболевания двигательных нейронов, воспалительного демиелинизирующего расстройства, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, печеночной энцефалопатии и хронического энцефалита. В некоторых вариантах осуществления настоящего изобретения хроническое нейродегенеративное заболевание представляет собой боковой амиотрофический склероз. В некоторых вариантах осуществления настоящего изобретения пациент является пациентом, который ранее не подвергался лечению.
Краткое описание рисунков
Фиг.1 отражает средние концентрации (R)-прамипексола в плазме после перорального введения натощак однократных доз 50 мг, 150 мг и 300 мг здоровым добровольцам.
Фиг.2 отражает средние концентрации (R)-прамипексола в плазме после перорального введения однократных доз 150 мг здоровым добровольцам натощак и после приема пищи.
Фиг.3 отражает средние концентрации (R)-прамипексола в плазме в дни 1 и 7 во время перорального введения 50 мг и 100 мг доз в день 1, каждые 12 часов в дни с 3 до 6 и однократной дозы в день 7 здоровым добровольцам натощак.
Фиг.4 отражает воздействие (AUC) в зависимости от дозы (мг/м2) для самцов и самок крыс и людей (обоих полов).
Фиг.5 отражает среднее воздействие (AUC) в зависимости от дозы (мг/м2) для самцов и самок карликовых свиней и людей (обоих полов).
Подробное описание
Настоящее изобретение предоставляет данные о том, что аффинность (R)-прамипексола в отношении дофаминовых рецепторов фактически является намного ниже, чем было известно ранее, что значительно увеличивает клиническую пригодность композиции. В настоящем документе показано также, что разница в функциональной аффинности между энантиомерами (S)-прамипексолом и (R)-прамипексолом (например, в 10000-20000 раз) намного больше, чем сообщалось ранее. Указанные данные показывают, что (R)-прамипексол можно использовать в дозах, которые могут более полно и неожиданно раскрыть менее эффективный нейропротективный потенциал соединения без теоретического ограничения, которое накладывается допущениями в отношении разделения аффинности дофаминовых рецепторов между энантиомерами. Указанное введение можно осуществлять без необходимости титрования доз. Указанные данные также показывают, что загрязнение композиции чистого (R)-прамипексола малыми количествами (S)-прамипексола приводит к резким сдвигам в направленности композиции. Данная заявка представляет способы применения хирально более чистых композиций (R)-прамипексола для лечения острых и хронических нейродегенеративных расстройств, которые, как полагали ранее, являются недоступными для указанного лекарственного средства в том, что касается немедленного проявления полной эффективности, и/или без титрования дозы.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, содержащей (R)-прамипексол в дозах, достаточных для получения нейропротективных, антиоксидантных, антиапоптотических или других благоприятных клеточных эффектов, без одновременного возникновения значительных побочных эффектов. Способность доставлять клинически эффективные дозы без ограничивающих дозы побочных эффектов стала возможной благодаря двум основным открытиям: (i) синтезу (R)-прамипексола, который является чистым в пределах обнаружения, обсуждаемых в настоящем документе; и (ii) открытию того факта, что (R)-прамипексол обладает значительно меньшей аффинностью в отношении рецепторов дофамина, чем сообщалось ранее. Фармацевтическая композиция по настоящему изобретению может зависеть, в некоторых вариантах осуществления настоящего изобретения, от оптической чистоты (R)-прамипексола, используемого в композиции, или от ограниченной дофаминергической активности хирально чистого (R)-прамипексола, используемого в композиции, или от того и другого.
Прежде чем описывать настоящие композиции и способы, следует понять, что настоящее изобретение не ограничивается описанными конкретными способами, композициями или методиками, поскольку они могут меняться. Следует также понимать, что терминология, используемая в описании, предназначена только для описания конкретных версий или вариантов осуществления настоящего изобретения и не предназначена для ограничения объема настоящего изобретения, которое будет ограничиваться только прилагаемой формулой изобретения. Все публикации, упомянутые в настоящем документе, полностью включены в настоящий документ в качестве ссылки, до той степени, в которой они поддерживают настоящее изобретение.
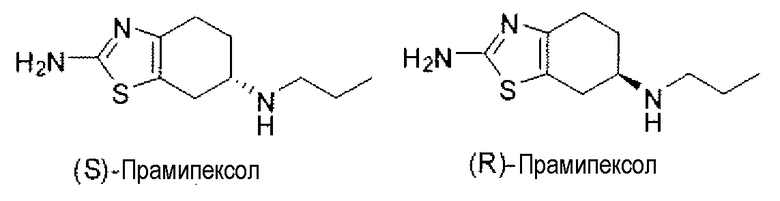
Соединение 2-амино-4,5,6,7-тетрагидро-6-(пропиламино)бензотиазол представляет собой синтетическое аминобензотиазольное производное, имеющее два энантиомера со структурами, показанными ниже. (S)-энантиомер представляет собой мощный агонист семейства дофаминовых рецепторов D2, с особой аффинностью в отношении подтипа рецепторов D3. В качестве агониста дофаминовых рецепторов (S)-прамипексол активирует дофаминовые рецепторы, имитируя, таким образом, эффекты нейротрансмиттера дофамина. Стереоизомер (S)-прамипексол представляет собой мощный агонист дофамина, лишь в очень малых необходимых суточных дозах и действительно переносимый пациентами. Оба энантиомера, как полагают, обладают нейропротективными свойствами за счет своей способности аккумулироваться в головном мозге, спинном мозге и митохондриях и, независимо от агонистической активности в отношении дофаминовых рецепторов, предположительно, посредством ингибирования перекисного окисления липидов, нормализации митохондриальной функции и/или детоксикации кислородных радикалов. Как таковые, указанные соединения могут быть пригодными в качестве ингибиторов каскадов клеточной гибели и утраты жизнеспособности, наблюдаемых при нейродегенеративных заболеваниях.
Степень, до которой введение молекулы имеет доказуемую фенотипическую активность, в основе которой лежит аффинность в отношении определенных рецепторов или других фармакологически эффективных белков, даже когда в основе активности лежат аффинности в отношении неизвестных мишеней, можно операционно определить с точки зрения того, вносит ли указанная активность положительный вклад (направленная активность) или отрицательный вклад (ненаправленная активность») в специфический и желательный терапевтический эффект. Для любой данной молекулы может быть теоретически идентифицирован ряд категорий ненаправленной активности, но направленная активность ограничена желательным терапевтическим эффектом. В той степени, которой указанные категории активности можно измерить и количественно охарактеризовать, или в которой можно осуществить сравнения с известными стандартами, можно вывести индекс активности для каждой из указанных категорий («эквивалент активности» или AE) и одно или более соотношений для сравнения направленной активности и ненаправленной активности, используемых для сравнения потенциальных соотношений риск-выгода у разных молекул.
В случае (R)-прамипексола две категории активности можно определить в данном контексте. Во-первых, активность, которая представляет собой агонистическую активность в отношении подгруппы человеческих дофаминовых рецепторов и результатом которой является поведенческий/токсикологический фенотип, представляет собой ненаправленную активность при большинстве нейродегенеративных расстройств. Указанная активность вызывает ограничивающие дозу побочные эффекты, возникающие в результате агонистической активности в отношении дофаминовых рецепторов, и для целей настоящего обсуждения может быть определена как эквивалент дофаминергической активности или DAE. В настоящей заявке термин «эквивалент дофаминергической активности» (DAE) будет относиться к средству измерения активности в отношении дофаминовых рецепторов, эквивалентной активности 1 мг (S)-прамипексола в отношении дофаминовых рецепторов. Например, доза (R)-прамипексола, имеющая DAE 0,01, будет обладать активностью в отношении дофаминовых рецепторов, которая эквивалентна активности 0,01 мг (S)-прамипексола. DAE также может относиться к ряду фармацевтических терминов, включая максимальную переносимую дозу (MTD), уровень, не вызывающий видимых неблагоприятных эффектов (NOAEL), и неэффективное дозовое количество, для ясности. Например, дозовое количество NOAEL для (S)-прамипексола, наиболее предпочтительно, составляет менее 0,05 мг. Это, в свою очередь, соответствует DAE менее 0,05. Дозовое количество (R)-прамипексола, имеющее DAE 0,01, будет, следовательно, менее DAE для наиболее предпочтительного дозового количества NOAEL (S)-прамипексола 0,05 мг. В некоторых вариантах осуществления настоящего изобретения DAE определяют измерением связывающей аффинности (IC50) или активности (EC50) для рецепторов D2 и/или D3 по отношению к тому же параметру для 1 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения DAE определяют по связывающей аффинности или активности в отношении рецепторов D2. В некоторых вариантах осуществления настоящего изобретения DAE определяют по связывающей аффинности или активности в отношении рецепторов D3. В некоторых вариантах осуществления настоящего изобретения DAE определяют по связывающей аффинности в отношении рецепторов D2. В некоторых вариантах осуществления настоящего изобретения DAE определяют по связывающей аффинности в отношении рецепторов D3. В некоторых вариантах осуществления настоящего изобретения DAE определяют с использованием подходящего анализа in vitro, такого как анализ связывающей аффинности IC50 для рецепторов D2 или D3, как описано, например, в публикации Schneider, C.S.; Mierau, J., “Dopamine Autoreceptor Agonists: Resolution and Pharmacological Activity of 2,6-Diaminotetrahydrobenzothiazole and an Aminothiazole Analogue of Apomorphine”, (1987). J. Med. Chem. 30:494-498; или Wong, S.K.-F.; Shrikhande, A.V., S.K.-F. Wong, “Activation of Extracellular Signal-Regulated Kinase by Dopamine D2 and D3 Receptors”, (2003). Society for Neuroscience Abstracts. Указанная ненаправленная активность для (R)-прамипексола при нейродегенеративных расстройствах (не являющихся болезнью Паркинсона) будет направленной активностью для его энантиомера (S)-прамипексола, используемого для лечения PD и синдрома усталых ног.
Исследования авторов предполагают, что DAE для (R)-прамипексола гораздо ниже, чем могли оценить ранее. Например, исследования авторов показали, что связывающая аффинность для (R)-прамипексола в отношении дофаминовых рецепторов D2 и D3 приблизительно в 290 и в 649 раз ниже, чем для (S)-прамипексола, соответственно, при использовании (R)-прамипексола высокой хиральной чистоты. Для сравнения, в литературе сообщается о том, что связывающая аффинность для (R)-прамипексола в отношении дофаминовых рецепторов D2 приблизительно в 9-21 раз ниже, чем для (S)-прамипексола, в то время как связывающая аффинность для (R)-прамипексола в отношении дофаминовых рецепторов D3 приблизительно в 50 раз ниже, чем для (S)-прамипексола.
Еще более поразительно, исследования авторов на собаках породы бигль показали, что соотношение доз MTD (R)-прамипексола и (S)-прамипексола составляет 10000, в то время как соотношение доз NOAEL (R)-прамипексола и (S)-прамипексола составляет 20000. В качестве биологического анализа, MTD и NOAEL у собак выявляют переносимость in vivo, ранее полностью непредсказуемую. Ввиду ограничений стандартного и количественного анализа, in vivo MTD и NOAEL на собаках могут действительно предположить, что даже малейшая примесь в 0,005% может, фактически, быть ответственной за побочные эффекты, связанные с дофаминовой агонистической активностью. Указанные сравнительные исследования наводят на мысль о том, что DAE для (R)-прамипексола намного ниже, чем оценивалось ранее.
Другой активностью (R)-прамипексола и (S)-прамипексола является нейропротекция. Нейропротекция представляет собой явление, независящее от механизма, и, следовательно, квалифицируется как категория активности. Данная направленная активность (R)-прамипексола для лечения нейродегенеративных расстройств поддается измерению и является приблизительно эквивалентной у обоих энантиомеров, и она может быть определена в относительных значениях как эквивалент нейропротективной активности или NAE. Эквивалент нейропротективной активности (NAE) относится к нейропротективной активности, присущей 1 мг (S)-прамипексола. В отличие от DAE, величина NAE, как было показано, является равной у обоих энантиомеров прамипексола в ряде тестов in vitro. В данном примере DAE рассматривается как стандартная мера потенциальных неблагоприятных эффектов, в то время как NAE рассматривается как стандартная мера потенциальной терапевтической пользы. Для данного примера величины NAE как для (R)-прамипексола, так и для Mirapex®, можно определить по концентрациям, необходимым для получения нейропротекции в анализах in vitro.
В некоторых вариантах осуществления настоящего изобретения NAE можно определить измерением нейропротективной активности в стандартном нейропротективном анализе in vitro по отношению к активности 1 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения нейропротективную активность определяют измерением клеточной гибели в присутствии МРР+ и/или ротенона на дофаминергических и/или недофаминергических клетках (в качестве неограничивающего примера см. анализ в M. Gu, Journal of Neurochemistry, 91:1075-1081 (2004)).
Предпочтительной целью настоящего изобретения является получение максимальных NAE, доставляемых пациенту, при минимизации количества эквивалентов активности, предполагающих неблагоприятные события, в данном случае, DAE.
(S)-прамипексол имеет высокое соотношение DAE/NAE благодаря высокой дофаминовой аффинности, в то время как соответствующее соотношение для (R)-прамипексола значительно ниже. С практической точки зрения варианты осуществления настоящего изобретения обеспечивают значительно более высокие уровни NAE и более высокие уровни NAE/DAE, чем полагали ранее, обеспечивая максимальную вероятность того, что терапевтически эффективное дозовое количество нейропротектора можно вводить нуждающемуся в этом пациенту. NAE и DAE могут быть полезными с точки зрения соотношения, особенно в качестве соотношения благоприятных и неблагоприятных эффектов, и пригодными для определения пределов, в которых можно вводить конкретную композицию.
Дозы (S)-прамипексола, однако, ограничены дофаминергической активностью (S)-энантиомера, которая может вызывать неблагоприятные побочные эффекты в дозах, превышающих «уровень, не вызывающий видимых неблагоприятных эффектов» (дозовое количество NOAEL). Доза NOAEL, используемая в настоящем документе, относится к количеству активного соединения или фармацевтического агента, которое не вызывает статистически или биологически значимых увеличений частоты или тяжести неблагоприятных эффектов при сравнении популяции, получающей лечение, и соответствующего контроля; некоторые эффекты могут возникать на данном уровне, но они не рассматриваются как неблагоприятные или как предшественники неблагоприятных эффектов. Популяцией, получающей лечение, может быть система, ткань, животное, индивидуум или человек, которым проводит лечение исследователь, ветеринар, врач или другой клиницист. Что касается (S)-прамипексола, примерами неблагоприятных событий являются головокружение, галлюцинации, тошнота, гипотензия, сонливость, запор, головная боль, тремор, боль в спине, постуральная гипотензия, гипертония, депрессия, боль в животе, тревожность, диспепсия, метеоризм, диарея, сыпь, атаксия, сухость во рту, экстрапирамидный синдром, судороги в ногах, подергивания мышц, фарингит, синусит, потливость, ринит, инфекция мочевыводящих путей, вазодилатация, гриппозный синдром, повышенное слюноотделение, заболевание зубов, одышка, кашель, нарушения походки, учащенное мочеиспускание, рвота, аллергическая реакция, гипертензия, зуд, гипокинезия, нервозность, нарушения сна, боль в груди, боль в шее, парестезия, тахикардия, вертиго, изменение голоса, конъюнктивит, паралич, шум в ушах, слезотечение, мидриаз и диплопия.
Например, доза 1,5 мг (S)-прамипексола, как было показано, вызывает сонливость у людей (публичное заявление по Mirapex®, внезапное засыпание, от европейского агентства по оценке медицинских продуктов; вкладыш для Mirapex®, продукта Boehringer Ingelheim, который указывает, что лекарственное средство вводят в виде трех доз в день). Кроме того, проведенные на собаках исследования, как представлено в настоящем документе (см. примеры и результаты, представленные в таблице 11), показывают, что доза NOAEL может составлять всего 0,00125 мг/кг, что эквивалентно дозе для человека 0,0007 мг/кг или 0,05 мг для индивидуума с массой тела 70 кг. Таким образом, что касается (S)-прамипексола, дозовое количество NOAEL может представлять собой количество менее 1,5 мг, менее 0,50 мг или, более предпочтительно, менее 0,05 мг. Что касается DAE, как определено в настоящем документе, доза NOAEL может иметь DAE менее 1,5, менее 0,5 или, более предпочтительно, менее 0,05.
Как правило, количество, превышающее неэффективное дозовое количество (S)-прамипексола, является необходимым для получения терапевтического эффекта при лечении заболеваний, облегчаемых дофаминовой агонистической активностью. Указанное количество, однако, может быть нежелательным, если добиваются нейропротективного эффекта, поскольку оно может вызывать описанные неблагоприятные побочные эффекты. Термин «неэффективное дозовое количество», используемый в настоящем документе, относится к количеству активного соединения или фармацевтического агента, которое вызывает биологический или лечебный ответ, сходный с биологическим или лечебным ответом на плацебо, наблюдаемый в ткани, системе, у животного, индивидуума или человека, которым проводит лечение исследователь, ветеринар, врач или другой клиницист. «Неэффективное дозовое количество» не может, таким образом, вызывать заметные отличия от плацебо по положительным эффектам, наблюдаемым в ткани, системе, у животного, индивидуума или человека, которым проводит лечение исследователь, ветеринар, врач или другой клиницист. Как таковое, «неэффективное дозовое количество» не предусмотрено для (1) профилактики заболевания; например, профилактики заболевания, состояния или расстройства у индивидуума, который может быть предрасположен к заболеванию, состоянию или расстройству, но до сих пор не ощущал и не демонстрировал патологии или симптоматики заболевания; (2) ингибирования заболевания; например, ингибирования заболевания, состояния или расстройства у индивидуума, который ощущает или демонстрирует патологию или симптоматику заболевания, состояния или расстройства (т.е. приостановки или замедления дальнейшего развития патологии и/или симптоматики) или (3) облегчения заболевания; например, облегчения заболевания, состояния или расстройства у индивидуума, который ощущает или демонстрирует патологию или симптоматику заболевания, состояния или расстройства (т.е. обратного развития или уменьшения патологии и/или симптоматики).
В качестве примера, у обезьян, которых лечили с использованием MPTP (1-метил-4-фенил-1,2,3,6-тетрагидропиридина), известного дофаминергического нейротоксина, (S)-прамипексол, как было показано, противостоит моторным дефицитам и паркинсоноподобным симптомам дозозависимым образом, при самой низкой эффективной пероральной дозе 0,053 мг/кг (см. научное обсуждение на http://www.emea.europa.eu/humandocs/PDFs/EPAR/Sifrol/059197EN6.pdf). Это должно быть эквивалентным дозе для человека 0,017 мг/кг, или 1,2 мг для индивидуума с массой тела 70 кг. В испытаниях на людях самая низкая эффективная пероральная доза (S)-прамипексола со значимым эффектом по сравнению с плацебо при лечении болезни Паркинсона составляла, как было показано, 1,1 мг/день. Отдельным пациентам могут потребоваться дозы, превышающие 1,1 мг/день, для получения эффектов, превышающих эффект плацебо (первоначальная научная дискуссия по разрешению использования Mirapex в европейском агентстве по оценке медицинских продуктов). В испытаниях на людях самая низкая эффективная пероральная доза со значимым эффектом по сравнению с плацебо при лечении синдрома усталых ног составляла, как было показано, 0,25 мг/день (вкладыш для Mirapex®, продукта Boehringer Ingelheim). Таким образом, по отношению к (S)-прамипексолу неэффективное дозовое количество может представлять собой количество менее 1,0 мг/день, менее 0,75 мг/день, менее 0,5 мг/день, менее 0,25 мг/день или, предпочтительно, менее 0,125 мг/день. Что касается DAE, неэффективное дозовое количество в день может иметь DAE в день менее 1,0, менее 0,75, менее 0,5, менее 0,25 или, предпочтительно, менее 0,125.
Другие ограничения для количества (S)-прамипексола, которое можно вводить пациенту, также включают максимальную рекомендованную терапевтическую дозу и максимальную переносимую дозу. «Максимальная рекомендованная терапевтическая доза» (MRTD) относится к дозам, приведенным центром FDA по оценке и исследованиям лекарственных средств, бюро фармацевтической науки, информатики и вычислительному анализу Staff безопасности максимальной рекомендованной терапевтической дозы, и описанным в Matthews, et al., “Assessment of the Health Effects of Chemicals in Humans: I. QSAR Estimation of the Maximum Recommended Therapeutic Dose (MRTD) and No Effect Level (NOEL) of Organic Chemicals Based on Clinical Trial Data”, Current Drug Discovery Technologies, 2004, 1:61-76). В базе данных FDA по MRTD приведены MRTD для (S)-прамипексола 0,1 мг/кг/день или 7,0 мг/день для индивидуума с массой тела 70 фунтов. Matthews, в свою очередь, доказывает, что NOEL (уровень отсутствия неблагоприятных эффектов) обычно составляет приблизительно одну десятую от MRTD, что соответствует 0,01 мг/кг или приблизительно 0,7 мг/день для индивидуума с массой тела 70 фунтов.
Ввиду своего неблагоприятного влияния на пациентов, не подвергавшихся ранее лечению, (S)-прамипексол следует титровать в течение нескольких недель, чтобы добиться указанных доз без ограничивающих дозу неблагоприятных эффектов (таких, которые описаны в документации во вкладыше для Mirapex®, продукта Boehringer Ingelheim). Например, при синдроме усталых ног рекомендованная начальная суточная доза Mirapex® составляет 0,125 мг один раз в день за 2-3 часа перед отходом ко сну. Для пациентов, требующих дополнительного облегчения симптомов, суточную дозу можно увеличить до 0,25 мг в течение периода 4-7 дней, а затем до 0,5 мг в течение еще одного периода в 4-7 дней. Для лечения болезни Паркинсона на вкладыше в упаковке рекомендована следующая схема титрования для Mirapex®:
Термин «максимальная переносимая доза» (MTD), используемый в настоящем документе, относится к количеству активного соединения или фармацевтического агента, которое вызывает значимую токсичность в ткани, системе, у животного, индивидуума или человека, которым проводит лечение исследователь, ветеринар, врач или другой клиницист. Токсичность однократной дозы (S)-прамипексола после перорального введения изучали на грызунах, собаках, обезьянах и людях. У грызунов смерть наступала при дозах 70-105 мг/кг и выше (первоначальная научная дискуссия по разрешению использования Mirapex в европейском агентстве по оценке медицинских продуктов). Это эквивалентно дозе для человека 7-12 мг/кг или приблизительно 500-850 мг для индивидуума с массой тела 70 кг. В случае испытаний на людях начальная суточная доза (S)-прамипексола, превышающая 0,20 мг, была непереносимой, когда ее вводили пациенту, ранее не подвергавшемуся лечению. У собак наблюдалась рвота при 0,0007 мг/кг и выше, в то время как у обезьян наблюдалось возбуждение при 3,5 мг/кг. Кроме того, вкладыш для продукта Mirapex® предписывает максимальную переносимую дозу для людей 4,5 мг/день, вводимую тремя однократными дозами по 1,5 мг. Однако дозу 4,5 мг/день не вводят пациенту, ранее не подвергавшемуся лечению, но, вместо этого, она достигается после схемы титрования (такой как описано в документации во вкладыше для продукта Mirapex®). В общем, начальная суточная доза для введения пациенту, ранее не подвергавшемуся лечению, составляет 0,125 мг, которую вводят три раза в день, и рекомендуется семинедельная схема титрования для достижения дозы 1,5 мг, которую вводят три раза ежедневно. У всех видов наблюдались признаки токсичности, связанные с повышенными фармакодинамическими ответами на (S)-прамипексол. Например, изменения поведения, включая гиперактивность, были широко распространены и приводили к ряду вторичных эффектов, таких как уменьшение массы тела и другие индуцированные стрессом симптомы. У минипигов и обезьян (S)-прамипексол умеренно влиял на сердечно-сосудистые параметры. У крыс мощный ингибирующий пролактин эффект прамипексола оказывал влияние на органы размножения (например, увеличенное corpora lutea, пиометра), и демонстрировал дозозивисимую дегенерацию сетчатки во время длительного воздействия (первоначальная научная дискуссия по разрешению использования Mirapex в европейском агентстве по оценке медицинских продуктов). Исследования на собаках показали, что количество MTD (S)-прамипексола может представлять собой количество менее 4,5 мг/день, предпочтительно менее 1,5 мг/день. Кроме того, количество MTD для человека может представлять собой количество менее 0,3 мг на дозу, на основании результатов исследований, описанных в настоящем документе, и, предпочтительно, менее 0,2 мг на дозу (см. таблицу 11). Что касается DAE, то количество MTD может иметь DAE ниже 1,5, ниже 0,3 или ниже 0,2.
При данных ограничениях количества (S)-прамипексола, которое можно вводить пациенту, применение вариантов осуществления настоящего изобретения представляет клинически важную альтернативу для разработки новых нейропротективных лекарственных средств. В литературе ранее сообщалось, что связывающая аффинность (R)-прамипексола в отношении рецепторов D2 приблизительно в 9-21 раз меньше таковой для (S)-прамипексола, в то время как связывающая аффинность (R)-прамипексола в отношении рецепторов D3 приблизительно в 50 раз меньше таковой для (S)-прамипексола (таблица 10). Указанные полученные из литературы сравнительные соотношения связывающей активности позволяют предположить, что (R)-прамипексол можно вводить только в несколько более высоких дозах по сравнению с (S)-прамипексолом. Указанное ограничение может иметь место в связи с тем, что сильная чувствительность тканей, систем, животных и человека к эффектам дофаминового агонизма будет препятствовать применению (R)-прамипексола в дозах, которые превышают переносимые дозы (S)-прамипексола на множитель, превышающий полученные из литературы сравнительные соотношения связывающей активности двух энантиомеров.
Кажущееся препятствие более высоких доз (R)-прамипексола можно продемонстрировать в отношении теоретической таблетки 50 мг. Предполагая 9-кратную разницу в величинах связывающей аффинности, таблетка 50 мг, которая является чистой на 99,95%, должна иметь DAE приблизительно 5,575 (5,55 DAE от (R)-прамипексола и 0,025 DAE от (S)-прамипексола). Аналогично, таблетка 25 мг, как ожидается, будет демонстрировать DAE 2,79 (2,78 от (R)-прамипексола и 0,0125 DAE от (S)-прамипексола). MTD (S)-прамипексола после семинедельной схемы титрования составляет 4,5 мг или 1,5 мг три раза в день, что эквивалентно 4,5 DAE в день или 1,5 DAE однократной дозой. Кроме того, дозовое количество NOAEL для (S)-прамипексола составляет менее 1,5 мг, предпочтительно, менее 0,50 мг или, более предпочтительно, менее 0,05 мг, которые эквивалентны 1,5 DAE, 0,5 DAE и 0,05 DAE, соответственно. Учитывая, что однократная доза MTD для (S)-прамипексола имеет 1,5 DAE, а NOAEL (S)-прамипексола имеет менее приблизительно 1,5 DAE, однократная доза 50 мг с DAE 5,55 и однократная доза 25 мг с DAE 2,79 должны быть устранены, если обращаться только к полученным из литературы сравнительным соотношениям связывающей аффинности. Кроме того, применение высокой хиральной чистоты 99,95%, которая используется в указанных теоретических дозах, должно приводить к неприемлемо высоким величинам DAE 5,55 и 2,79 свыше однократной дозы MTD DAE 1,5 мг, и намного выше предпочтительных величин NOAEL 0,5 DAE и 0,05 DAE.
Напротив, в некоторых вариантах осуществления, аспект настоящего изобретения включает неожиданно высокие величины хиральной чистоты, которые были получены. Указанные величины чистоты дают значения MTD или NOAEL для (R)-прамипексола, которые выше ранее оцененных величин, полученных на основании литературных сравнительных значений связывающей аффинности. В некоторых вариантах осуществления настоящее изобретение относится к фармацевтическим композициям, начальным дозам, способу лечения и наборам, включающим (R)-прамипексол высокой хиральной чистоты. В соответствии с приведенным выше обсуждением можно прогнозировать, что доза 25 мг со сходной хиральной чистотой 99,95% будет значительно превышать MTD или NOAEL для (S)-прамипексола и, следовательно, вызывать видимые неблагоприятные побочные эффекты. Исследования на собаках, однако, позволяют предположить, что (R)-прамипексол высокой хиральной чистоты обеспечивает дозовые количества NOAEL, неожиданно отличающиеся от оцененных ранее (таблица 10). Невероятно, но доза 25 мг/кг (R)-прамипексола, не содержащего обнаруживаемого количества (S)-прамипексола (0,05% граница обнаружения), не вызывала видимых эффектов у собак, что является неожиданным, если принимать во внимание литературные данные по связывающей аффинности.
Кроме того, исследования на собаках показали высокую (приближающуюся к абсолютной) хиральную чистоту композиций прамипексола для (R)-энантиомера. (R)-прамипексол вводят в высоких дозах в исследованиях, описанных в настоящем документе (эквивалентных дозам для человека от 1000 мг до 3000 мг; см. примеры), так что даже наименьшее количество (S)-прамипексола должно вносить свой вклад в наблюдаемые NOAEL и MTD. Например, что касается эквивалентных доз для человека, то на основании данных, полученных на собаках, MTD для (R)-энантиомера, как было показано, эквивалентна приблизительно 3000 мг для человека с массой тела 70 кг, в то время как эквивалентная MTD для (S)-энантиомера будет эквивалентна лишь 0,30 мг для указанного субъекта (таблица 11). Это представляет собой разницу в 10000 раз. Доза NOAEL для (R)-энантиомера в 20000 раз выше, чем для (S)-энантиомера (таблица 11). Таким образом, композиции (R)-прамипексола, использованные в настоящем исследовании, должны быть по меньшей мере на 99,99% чистыми, если допускается, что наблюдавшиеся побочные эффекты являются результатом лишь загрязнения (S)-энантиомером. С другой стороны, приведенные данные демонстрируют высокие уровни доз (R)-энантиомера прамипексола, которые можно вводить безопасно. Приведенные данные подчеркивают полезность высокой хиральной чистоты для (R)-энантиомера прамипексола в различных вариантах осуществления настоящего изобретения.
Настоящее изобретение относится также к фармацевтическим композициям, начальным дозам, способам и наборам, включающим (R)-прамипексол более высокой хиральной чистоты и в более высоких дозах. Как обсуждалось выше, в научной литературе ранее предполагали, что сравнительные соотношения связывающей аффинности в отношении рецепторов D2 и D3 составляют приблизительно от 9 до 21 и 50, соответственно (см. пример 1 и таблицу 10, ниже). Неожиданно было установлено, что сравнительные соотношения связывающей аффинности S:R в отношении рецепторов D2 и D3 составляют приблизительно 290 и 649, если используется (R)-прамипексол высокой хиральной чистоты.
Как обсуждается более подробно ниже, это предполагает, что сравнительные соотношения связывающей аффинности приблизительно в 13-32 раза превышают сравнительные соотношения связывающей аффинности, приведенные в литературе. Литература изобилует обсуждениями неблагоприятного влияния (S)-прамипексола. Несмотря на то, что данные in vitro, касающиеся связывающей аффинности, которые представлены в поддержку настоящего изобретения, являются убедительными, важность экономичного и эффективного синтеза становится очевидной после сравнения данных in vitro и in vivo, представленных в настоящем документе. In vivo клинические наблюдения с использованием собак породы бигль показывают, что соотношение дозы MTD (R)-прамипексола и (S)-прамипексола составляет 10000, в то время как соотношение дозы NOAEL (R)-прамипексола и (S)-прамипексола составляет 20000. Абсолютное соотношение дозы MTD может быть выше, поскольку величины хиральной чистоты, приведенные в настоящем документе, ограничены уровнем обнаружения (см. пример 2 и таблицу 11). На основании хиральной чистоты и in vitro сравнительных соотношений связывающей аффинности клинические соотношения дозы NOAEL и клинические соотношения дозы MTD (в настоящем документе - «сравнительные соотношения») в настоящее время позволяют предсказать DAE для данной дозы (R)-прамипексола. В таблице 1 приведены DAE для дозы 25 мг (R)-прамипексола в зависимости от сравнительного соотношения и хиральной чистоты. Приведенные данные показывают, что при использовании 25 мг лекарственной формы (R)-прамипексола может быть неожиданно получена намного более низкая величина DAE, чем оценивалось ранее, благодаря более низким сравнительным соотношениям, описанным в настоящем документе, по сравнению с приведенными в литературе сравнительными соотношениями.
Таблица 1
DAE для 25 мг дозы(R)-прамипексола в зависимости от % хиральной чистоты и сравнительного соотношения
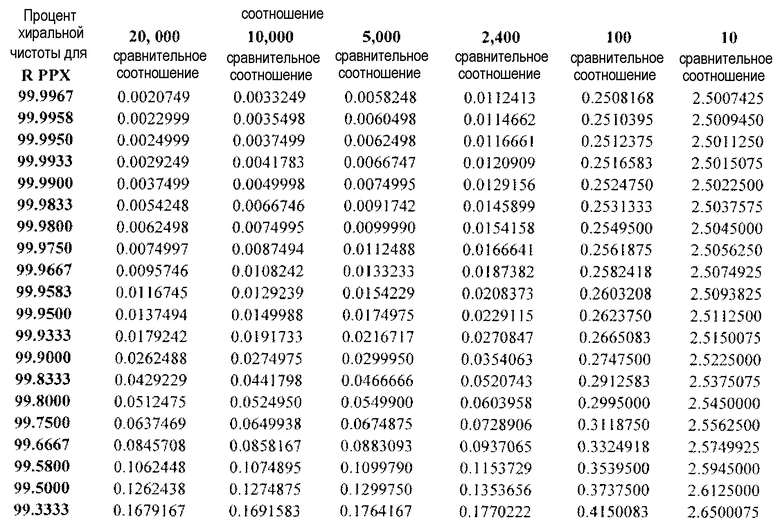

R PPX означает R-прамипексол
В таблице 1 предпринята попытка проиллюстрировать важность как чистоты, так и аффинности даже при использовании 25 мг однократной пероральной дозы. Допущения, касающиеся дофаминергической активности (R)-прамипексола в отношении дофаминовых рецепторов, должны, по-видимому, препятствовать даже высокой чистоте (даже 100% чистоте) 25 мг таблетки (R)-прамипексола. На основе описания настоящего изобретения можно непосредственно рассмотреть многочисленные таблетки для иллюстрации вопроса. Таблицы 1А и 1В, приведенные ниже, предназначены для иллюстрации важности чистоты для однократной пероральной лекарственной формы (R)-прамипексола путем иллюстрации влияния даже малейшего загрязнения композиции (S)-прамипексолом.
Дозы “NOAEL” композиций (R)-прамипексола (на основе DAE <0,05)
«Неэффективные» дозы композиций (R)-прамипексола (на основе DAE <0,125)
Никто не оценил или ясно сформулировал то, что синтетические методики необходимы для достижения степени чистоты, которая превышает пределы обычных способов обнаружения. Кроме того, никто не предложил то, что указанная однократная доза должна иметь чистоту 99,95% или более, чтобы соответствовать заявленной цели.
На основе сравнительных соотношений для связывающей аффинности, величин NOAEL и MTD, стало возможным, таким образом, предсказать количество (R)-прамипексола, которое можно вводить и которое было бы эквивалентным неэффективному дозовому количеству (S)-прамипексола. Таблица 2 показывает DAE в зависимости от дозы (R)-прамипексола (левый столбец) и сравнительного соотношения (верхний ряд). Ссылаясь на таблицу 2, можно выбрать стандартную дозу, которая предусматривает количество (R)-прамипексола, имеющее величину DAE, которая равна неэффективному количеству (S)-прамипексола. Действительно, если не требуется двойного эффекта DAE/NAE, то DAE в фармацевтической композиции следует избегать или минимизировать. Следовательно, не следует ожидать, что любая однократная доза, превышающая 25 миллиграммов, предотвратит ненаправленную активность, и опытный специалист категорически должен ее избегать. Это не соответствует действительности, если, как в настоящем изобретении, сравнительные соотношения превышают 200. Наилучшим образом данное обстоятельство иллюстрируется в таблице 2.
Таблица 2
DAE в зависимости от доз (R)-прамипексола и сравнительного соотношения (допуская 100% хиральную чистоту (R)-прамипексола)
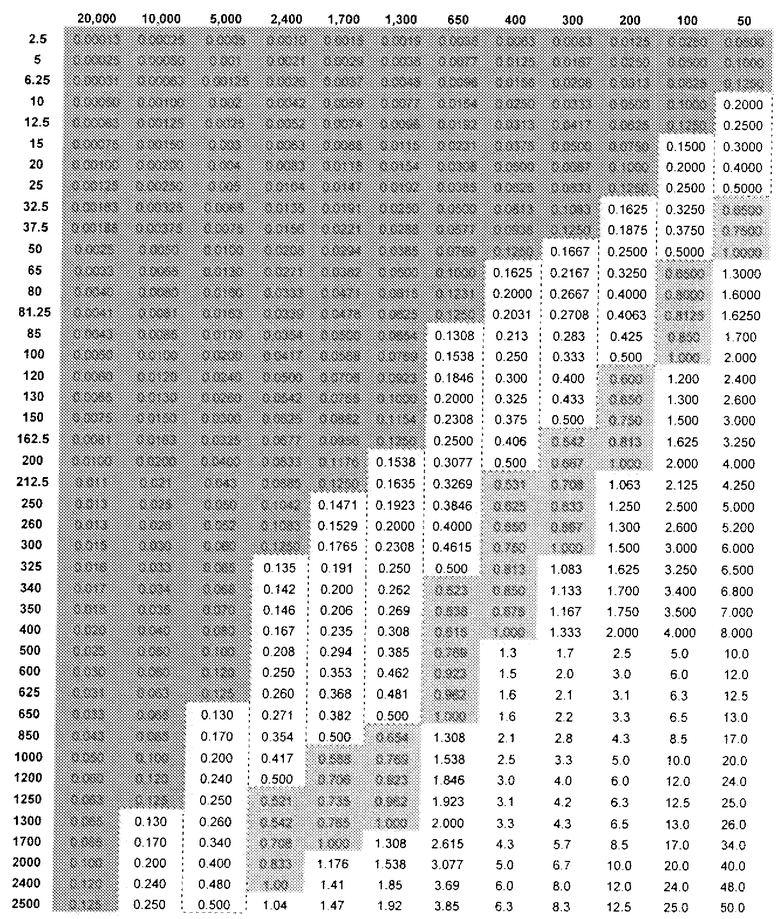
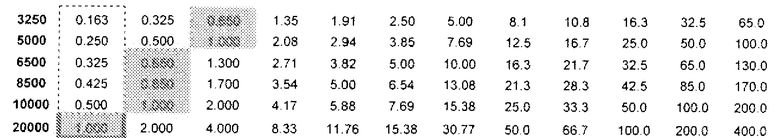
Величина DAE, эквивалентная предпочтительному неэффективному дозовому количеству (S)-прамипексола, может быть ниже 1,0 мг; более предпочтительно, ниже 0,5 мг и, более предпочтительно, ниже 0,125 мг.
Аналогично, можно установить количество (R)-прамипексола, которое можно вводить и которое было бы эквивалентным дозовому количеству (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов. Таблица 3 показывает DAE в зависимости от дозы (R)-прамипексола (левый столбец) и сравнительного соотношения (верхний ряд). Ссылаясь на таблицу 3, можно выбрать стандартную дозу, которая предусматривает количество (R)-прамипексола, имеющее величину DAE, равную дозовому количеству NOAEL (S)-прамипексола. В то время как использование 0,125 позволяет избежать нежелательных эффектов, при использовании менее 0,05 избегают NOAEL. Разница между сообщениями в литературе и фактическими результатами является еще более впечатляющей в таблице 3.
Таблица 3
DAE в зависимости от доз (R)-прамипексола и сравнительного соотношения (допуская 100% хиральную чистоту (R)-прамипексола)
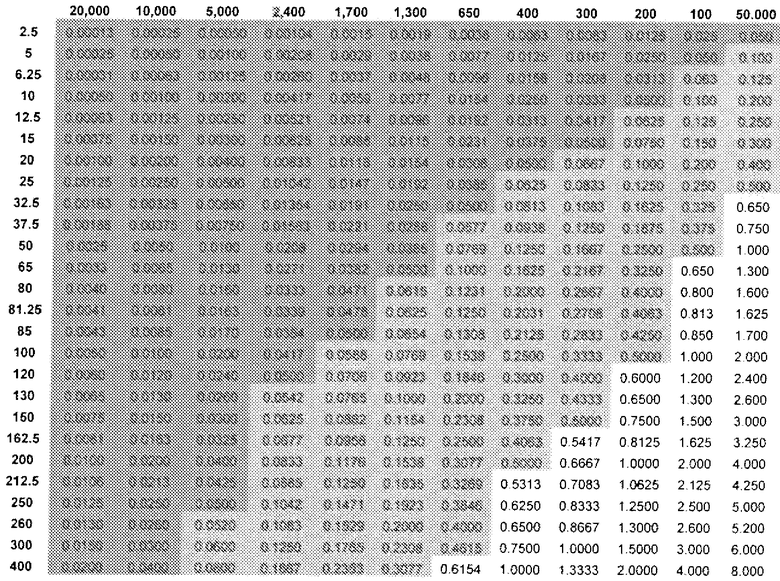
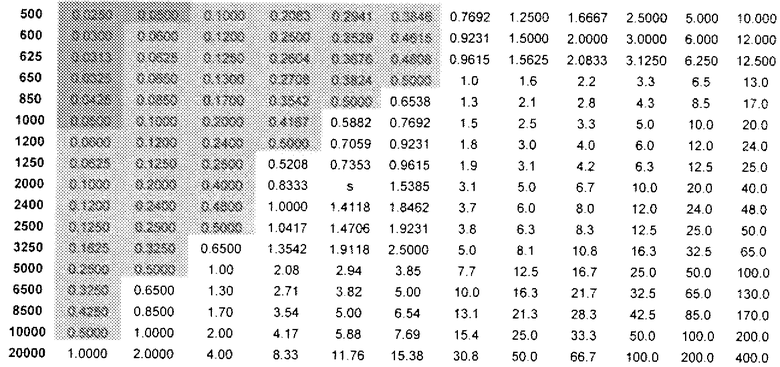
Величина DAE, эквивалентная предпочтительному дозовому количеству (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов (NOAEL), может быть ниже 0,5 мг, предпочтительно, ниже 0,05 мг.
Кроме того, таблица 4 показывает DAE в зависимости от дозы (R)-прамипексола (левый столбец) и сравнительного соотношения (верхний ряд). Ссылаясь на таблицу 4, можно выбрать стандартную дозу, которая предусматривает количество (R)-прамипексола, имеющее определенное значение DAE.
Таблица 4
DAE в зависимости от доз (R)-прамипексола и сравнительного соотношения (допуская 100% хиральную чистоту (R)-прамипексола)
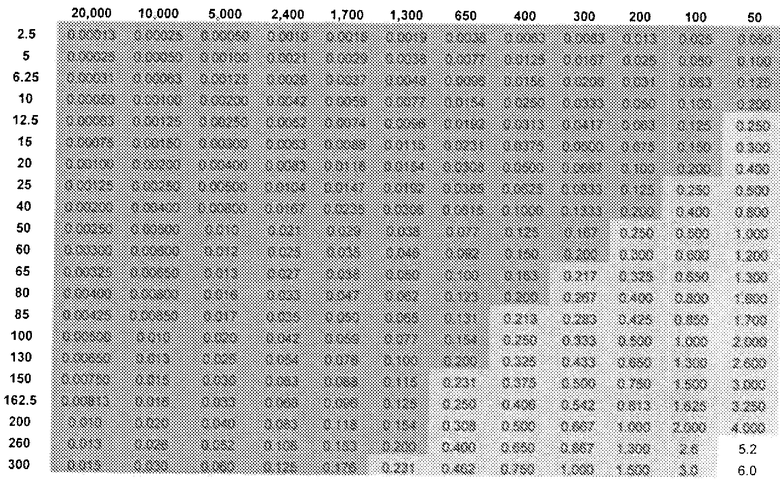
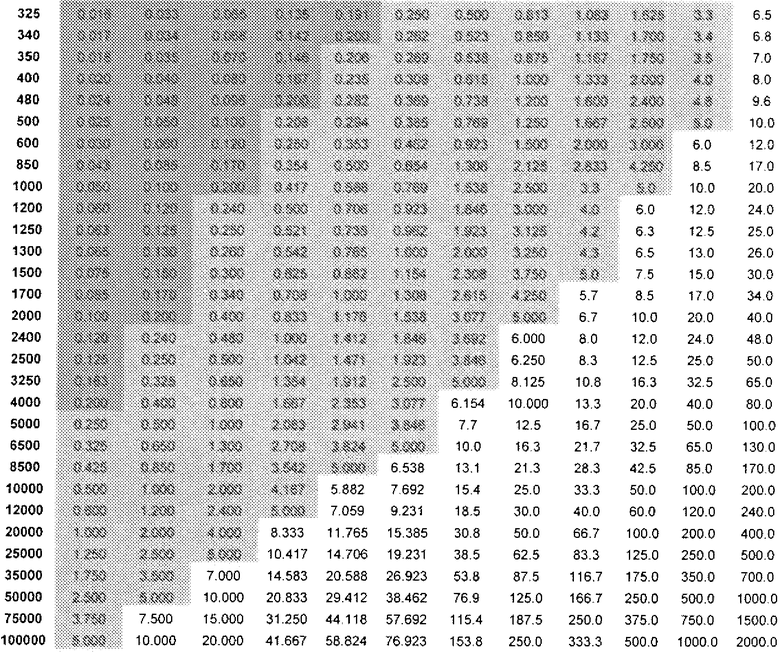
DAE ниже 0,2 или ниже 0,5 мг.
Более высокие сравнительные соотношения, описанные в настоящем документе, предполагают также, что данная доза (R)-прамипексола может содержать определенное количество примеси (S)-прамипексола, прежде чем превысит приемлемую величину DAE. Например, таблица 3 показывает, что 25 мг доза (R)-прамипексола обеспечивает 0,00125 DAE при сравнительном соотношении 20000, как предполагается соотношением NOAEL (R)-прамипексола и (S)-прамипексола в исследованиях на собаках, допуская 100% хиральную чистоту (R)-прамипексола. Теоретически, дополнительные 1,4 мг (S)-прамипексола могут быть добавлены без превышения DAE для однократной дозы MTD (S)-прамипексола, в то время как дополнительные 0,045 мг (S)-прамипексола могут быть добавлены без превышения предпочтительного дозового количества NOAEL (S)-прамипексола. Указанные композиции должны быть чистыми на 96% и на 99,8%. Напротив, 25 мг 100% чистого (R)-прамипексола должны приводить к 2,78 DAE, при использовании сравнительного соотношения связывающей аффинности 9, описанного в литературе, предполагая, что даже 100% чистота будет недостаточной для того, чтобы избежать неблагоприятных побочных эффектов. Следовательно, настоящее изобретение относится также к конкретным дозам (R)-прамипексола, которые неожиданно допускают наличие малых количеств примесей (S)-прамипексола.
Дополнительные определения
Следует отметить также, что используемые в настоящем документе и в прилагаемой формуле изобретения формы единственного числа включают в себя и формы множественного числа, если в контексте ясно не указано иное. Так, например, ссылка на «соль» представляет собой ссылку на один или более органических растворителей и их эквивалентов, известных специалистам, и т.п.
Используемый в настоящем документе термин «приблизительно» означает плюс или минус 10% численного значения, с которым он был употреблен. Таким образом, приблизительно 50% означает пределы 45%-55%. Если не указано иное, все технические и научные термины, используемые в настоящем документе, имеют общепринятое у специалистов значение.
Используемый в настоящем документе термин «сравнительное соотношение связывающей аффинности» относится к связывающей аффинности в отношении дофаминовых рецепторов D2 или D3 (величина IC50) (R)-прамипексола, разделенной на связывающую аффинность в отношении дофаминовых рецепторов D2 или D3 (величина IC50) (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения сравнительное соотношение связывающей аффинности относится к соотношению величин IC50 в отношении рецептора D2. В некоторых вариантах осуществления настоящего изобретения сравнительное соотношение связывающей аффинности относится к соотношению величин IC50 в отношении рецептора D3.
Используемый в настоящем документе термин «сравнительное соотношение» относится к следующему: 1) к соотношению величин IC50 в отношении рецепторов D2 или D3 для (R)-прамипексола:(S)-прамипексола; 2) к отношению количеств MTD для (R)-прамипексола:(S)-прамипексола; или 3) к отношению дозовых количеств NOAEL для (R)-прамипексола:(S)-прамипексола.
Используемый в настоящем документе термин «суточное дозовое количество» относится к количеству прамипексола в день, которое вводят или назначают пациенту. Указанное количество можно вводить в виде многократных стандартных доз или в виде однократной стандартной дозы, один раз в течение дня или многократно в течение дня.
Используемый в настоящем документе термин «эквивалент дофаминергической активности» (DAE) относится к мере активности в отношении дофаминовых рецепторов, которая является эквивалентной активности 1 мг (S)-прамипексола в отношении дофаминовых рецепторов.
«Дозовое количество», используемое в настоящем документе, обычно равно дозе активного ингредиента, которую можно вводить один раз в день, или можно вводить несколько раз в день (например, стандартная доза представляет собой часть желаемой суточной дозы). Например, неэффективное дозовое количество 0,5 мг/день (S)-прамипексола можно вводить в виде 1 дозы в 0,5 мг, в виде 2 доз по 0,25 мг каждая или в виде 4 доз по 0,125 мг. Термин «стандартная доза», используемый в настоящем документе, может использоваться для обозначения дискретного количества терапевтической композиции, которое включает заранее определенное количество активного соединения. Количество активного ингредиента обычно равно дозе активного ингредиента, которую можно вводить один раз в день, или можно вводить несколько раз в день (например, стандартная доза представляет собой часть желаемой суточной дозы). Стандартная доза может также использоваться для обозначения суммарной суточной дозы, которую можно вводить один раз в день или которую можно вводить в виде подходящей части указанной дозы (например, стандартная доза представляет собой суммарную суточную дозу, которую можно вводить частями, такими как, например, половина или треть дозы).
Используемые в настоящем документе термины «энантиомеры», «стереоизомеры» и «оптические изомеры» могут быть использованы взаимозаменяемо и относятся к молекулам, которые содержат асимметричный или хиральный центр и представляют собой не накладывающиеся друг на друга зеркальные изображения друг друга. Используемый в настоящем документе термин «хирально чистый» или «энантиомерно чистый» можно использовать для указания того факта, что соединение содержит по меньшей мере 99,95% единственного оптического изомера. Термин «энантиомерно обогащенный», если не указано число, можно использовать для указания того факта, что по меньшей мере 51% материала представляет собой единственный энантиомер. Термин «энантиомерное обогащение», используемый в настоящем документе, относится к увеличению количества одного энантиомера по сравнению с другим энантиомером. «Рацемическая смесь» представляет собой смесь равных количеств (R)- и (S)-энантиомеров хиральной молекулы.
Используемый в настоящем документе «набор» относится к одной или нескольким фармацевтическим композициям и инструкциям по введению или назначению одной или нескольких композиций. Инструкции могут состоять из вкладыша к продукту, инструкций на упаковке одной или нескольких фармацевтических композиций, или любой другой инструкции.
Используемый в настоящем документе термин «Mirapex®» относится к таблеткам, содержащим (S)-прамипексола дигидрохлорид, который имеет химическое наименование моногидрат дигидрохлорида (S)-2-амино-4,5,6,7-тетрагидро-6-(пропиламино)бензотиазола.
Используемый в настоящем документе термин «пациент, который ранее не подвергался лечению», относится к пациенту, которого ранее не лечили прамипексолом (или (R)-прамипексолом, или (S)-прамипексолом) или к которому ранее не применяли схему титрования прамипексола до получения начальной дозы прамипексола.
Используемый в настоящем документе термин «нейропротектор» относится к любому агенту, который может предотвратить или замедлить прогрессирование дегенерации нейронов и/или предотвратить гибель нейронов.
Термины «пациент» или «субъект» являются взаимозаменяемыми и могут использоваться для обозначения любого живого организма, который можно лечить соединениями по настоящему изобретению. Как таковые, термины «пациент» и «субъект» могут включать, без ограничения, любое млекопитающее, не являющееся человеком, примата или человека. В некоторых вариантах осуществления настоящего изобретения «пациент» или «субъект» представляет собой млекопитающее, такое как мышь, крысы, другие грызуны, кролики, собаки, кошки, свиньи, крупный рогатый скот, овцы, лошади, приматы или люди. В некоторых вариантах осуществления настоящего изобретения пациент или субъект представляет собой взрослого, ребенка или ребенка младшего возраста. В некоторых вариантах осуществления настоящего изобретения пациентом или субъектом является человек.
Используемый в настоящем документе термин «фармацевтически приемлемая соль» обозначает те соли, которые, в рамках здравого медицинского суждения, являются подходящими для применения в контакте с тканями человек и низших животных, без чрезмерной токсичности, раздражения, аллергической реакции и т.п., и соразмерными с разумным соотношением польза/риск. Фармацевтически приемлемые соли хорошо известны специалистам. Например, Berg et. al. (1977) J. Pharm Sciences, Vol. 6. 1-19, подробно описывает Фармацевтически приемлемые соли.
Термин «фармацевтическая композиция» будет означать композицию, содержащую по меньшей мере один активный ингредиент, в соответствии с чем композиция подлежит изучению на предмет специфического эффективного результата при действии на млекопитающего (например, без ограничения, человека). Специалисты поймут и примут во внимание методики, подходящие для того, чтобы определить, обеспечивает ли активный ингредиент желаемый эффективный результат в соответствии с потребностями исследователя.
Используемый в настоящем документе термин «(R)-прамипексол» относится к (R)-энантиомеру прамипексола или к его фармацевтически приемлемой соли, предпочтительно, к R(+)-энантиомеру прамипексола или к его фармацевтически приемлемой соли. «(R)-прамипексол» также может включать гидрат (R)-энантиомера прамипексола или его фармацевтически приемлемую соль. В некоторых вариантах осуществления настоящего изобретения (R)-прамипексол представляет собой моногидрат дигидрохлорида (R)-прамипексола.
Используемый в настоящем документе термин «(S)-прамипексол» относится к (S)-энантиомеру прамипексола или к его фармацевтически приемлемой соли, предпочтительно, к S(-)-энантиомеру прамипексола или к его фармацевтически приемлемой соли. «(S)-прамипексол» также может включать гидрат (S)-энантиомера прамипексола или его фармацевтически приемлемую соль.
Используемый в настоящем документе термин «соль» (R)-прамипексола представляет собой любую кислотно-аддитивную соль, предпочтительно, фармацевтически приемлемую кислотно-аддитивную соль, включая, без ограничения, соль галогеновой кислоты, такую как, например, соль бромистоводородной, хлористоводородной, фтористоводородной и йодистоводородной кислоты; соль неорганической кислоты, такую как, например, соль азотной, хлорной, серной и фосфорной кислоты; соль органической кислоты, такую как, например, соли сульфоновой кислоты (метансульфоновой, трифторметансульфоновой, этансульфоновой, бензолсульфоновой или п-толуолсульфоновой), соли уксусной, яблочной, фумаровой, янтарной, лимонной, бензойной, глюконовой, молочной, миндальной, муциновой, памоевой, пантотеновой, щавелевой и малеиновой кислоты; и соль аминокислоты, такую как, например, соли аспарагиновой или глутаминовой кислоты. Кислотно-аддитивная соль может представлять собой моно- или ди-кислотно-аддитивную соль, такую как соль ди-галогеноводородной, ди-серной, ди-фосфорной или ди-органической кислоты. Во всех случаях кислотно-аддитивную соль используют в качестве ахирального реагента, который не выбирают, основываясь на любом ожидаемом или известном предпочтении в отношении взаимодействия с конкретным оптическим изомером продуктов по настоящему изобретению или его осаждения (например, в противоположность специфическому использованию в предшествующем уровне техники D(+)-винной кислоты, которая может предпочтительно осаждать (R)-прамипексол).
Используемый в настоящем документе термин «начальное суточное дозовое количество» относится к количеству прамипексола в день, которое вводят или назначают в начале лечения прамипексолом пациенту, который ранее не подвергался лечению с использованием титрационной схемы прамипексола. Указанное количество можно вводить в виде многократных стандартных доз или в виде однократной стандартной дозы, один раз в течение дня или многократно в течение дня.
«Терапевтически эффективное количество», как используется в настоящем документе, относится к количеству активного соединения или фармацевтического агента, которое вызывает биологический или лечебный ответ в ткани, системе, у животного, индивидуума или человека, которым проводит лечение исследователь, ветеринар, врач или другой клиницист, которое включает одно или несколько из следующих мероприятий: 1) профилактика заболевания; например, профилактика заболевания, состояния или расстройства у индивидуума, который может быть предрасположен к заболеванию, состоянию или расстройству, но до сих пор не ощущал или не демонстрировал патологии или симптоматики заболевания; (2) ингибирование заболевания; например, ингибирование заболевания, состояния или расстройства у индивидуума, который ощущает или демонстрирует патологию или симптоматику заболевания, состояния или расстройства (т.е. приостановка или замедление дальнейшего развития патологии и/или симптоматики) и (3) облегчение заболевания; например, облегчение заболевания, состояния или расстройства у индивидуума, который ощущает или демонстрирует патологию или симптоматику заболевания, состояния или расстройства (т.е. обратное развитие или уменьшение патологии и/или симптоматики).
Термин «лечение» может означать профилактику конкретного расстройства, заболевания или состояния, облегчение симптомов, связанных с конкретным расстройством, заболеванием или состоянием, и/или профилактику симптомов, связанных с конкретным расстройством, заболеванием или состоянием. В некоторых вариантах осуществления настоящего изобретения указанный термин относится к замедлению прогрессирования расстройства, заболевания или состояния или к облегчению симптомов, связанных с конкретным расстройством, заболеванием или состоянием. В некоторых вариантах осуществления настоящего изобретения термин относится к замедлению прогрессирования расстройства, заболевания или состояния. В некоторых вариантах осуществления настоящего изобретения указанный термин относится к облегчению симптомов, связанных с конкретным расстройством, заболеванием или состоянием. В некоторых вариантах осуществления настоящего изобретения указанный термин относится к восстановлению функции, которая нарушена или утрачена в результате конкретного расстройства, заболевания или состояния.
Термин «порошкование» может использоваться для обозначения способа отверждения химического соединения. Порошкование включает перемешивание, взбивание или подобный способ до образования химическим соединением кристаллического твердого вещества или осадка. Указанное твердое вещество может действовать для затравки оставшегося химического соединения в растворе, вызывая его осаждение или кристаллизацию из раствора.
Несмотря на то, что любые способы и материалы, сходные или эквивалентные описанным в настоящем документе, можно использовать на практике или при испытании вариантов осуществления настоящего изобретения, далее будут описаны предпочтительные способы, устройства и материалы.
Фармацевтические композиции
Используемый в настоящем изобретении прамипексол высокой хиральной чистоты, (R)-прамипексол, позволяет терапевтическим композициям иметь широкие индивидуальные и суточные пределы доз. Что касается таковых, в первом аспекте настоящее изобретение относится к композиции, содержащей (R)-прамипексол. Композиция может дополнительно содержать фармацевтически приемлемый носитель.
В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 0,01 мг/кг/день до 10000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1000 мг/кг/день до 10000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 50 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения доза может составлять от 10 мг/день до 1500 мг/день, более предпочтительно, от 100 мг/день до 600 мг/день. Количество (R)-прамипексола в композициях может предпочтительно составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола в композициях может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз, предпочтительно, на две или три дозы в день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 50 мг до 5000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 100 мг до 3000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 300 мг до 1500 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму.
Композиция может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% или, более предпочтительно, по меньшей мере 99,99%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,99% или выше.
В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой капсулу. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой таблетку.
Варианты осуществления настоящего изобретения для количеств (R)-прамипексола в композиции, хиральной чистоты и лекарственной формы, которые описаны в настоящем документе, могут быть объединены в любой подходящей комбинации.
В другом аспекте настоящее изобретение относится к композициям, содержащим прамипексол, который представляет собой хирально чистый (R)-прамипексол. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 0,01 мг/кг/день до 10000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1000 мг/кг/день до 10000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 50 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения доза может составлять от 10 мг/день до 1500 мг/день, более предпочтительно, от 100 мг/день до 600 мг/день. В некоторых вариантах осуществления настоящего изобретения композиции вводят в дозах приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг или приблизительно от 500 мг до 1000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения композиции вводят в дозах приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день. Указанные дозы прамипексола предпочтительно, находятся в препаратах, которые имеют химическую чистоту 97% или выше и хиральную чистоту для (R)-прамипексола 99,6% или выше, 99,7% или выше, 99,8% или выше, 99,9% или выше, предпочтительно, 99,95% или выше и, более предпочтительно, 99,99% или выше. В предпочтительном варианте осуществления композиции, содержащие прамипексол, могут иметь хиральную чистоту для (R)-прамипексола 100%. Композиции могут дополнительно содержать носитель. Композиции по настоящему изобретению можно вводить перорально, предпочтительно, в виде твердой пероральной дозы и, более предпочтительно, в виде твердой пероральной дозы, которая может представлять собой капсулу или таблетку. В предпочтительных вариантах осуществления композиции по настоящему изобретению могут быть составлены в форме таблеток для перорального введения.
В другом аспекте настоящее изобретение дополнительно относится к композиции, содержащей терапевтически эффективное количество (R)-прамипексола. Композиция может дополнительно содержать фармацевтически приемлемый носитель.
В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 0,01 мг/кг/день до 10000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1000 мг/кг/день до 10000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 50 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения доза может составлять от 10 мг/день до 1500 мг/день, более предпочтительно, от 100 мг/день до 600 мг/день. Терапевтически эффективное количество (R)-прамипексола в композициях может предпочтительно составлять приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола в композициях может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола составляет приблизительно от 50 мг до 5000 мг. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола составляет приблизительно от 100 мг до 3000 мг. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола составляет приблизительно от 300 мг до 1500 мг. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола составляет приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму.
Композиция может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% или, более предпочтительно, по меньшей мере 99,99%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,99% или выше.
В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой капсулу. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой таблетку.
Варианты осуществления настоящего изобретения для количеств (R)-прамипексола в композиции, хиральной чистоты и лекарственной формы, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
В другом аспекте настоящее изобретение относится к композиции, состоящей по существу из терапевтически эффективного количества (R)-прамипексола, где хиральная чистота для (R)-прамипексола составляет 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,99% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%.
В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой капсулу. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой таблетку.
В еще одном аспекте настоящее изобретение дополнительно относится к композиции, содержащей терапевтически эффективное количество (R)-прамипексола и неэффективное дозовое количество (S)-прамипексола. Композиция может дополнительно содержать фармацевтически приемлемый носитель.
В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 0,01 мг/кг/день до 10000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1000 мг/кг/день до 10000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 50 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения доза может составлять от 10 мг/день до 1500 мг/день, более предпочтительно, от 100 мг/день до 600 мг/день. Количество (R)-прамипексола в композициях может предпочтительно составлять от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола в композициях может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 50 мг до 5000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 100 мг до 3000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 300 мг до 1500 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму.
Композиция может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% или, более предпочтительно, по меньшей мере 99,99%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,99% или выше.
В некоторых вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола представляет собой количество, которое не превышает приблизительно 1,0 мг. В более предпочтительных вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола представляет собой количество, которое не превышает приблизительно 0,75 мг, приблизительно 0,5 мг, приблизительно 0,25 мг или приблизительно 0,125 мг. В некоторых вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола составляет приблизительно менее 0,125 мг.
В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой капсулу. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой таблетку.
Варианты осуществления настоящего изобретения для количеств (R)-прамипексола в композиции, хиральной чистоты, неэффективного дозового количества и лекарственной формы, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество (R)-прамипексола и неэффективное дозовое количество (S)-прамипексола, которую вводят в виде стандартной лекарственной формы. Предпочтительные стандартные лекарственные формы включают формы, подходящие для перорального введения, включая, без ограничения, капсулы, таблетки и т.п. В таблице 5 показаны различные примеры вариантов осуществления настоящего изобретения. В каждом столбце таблицы 5 приводится количество (S)-прамипексола, которое можно совместно вводить в неэффективном дозовом количестве, в зависимости от хиральной чистоты композиции для (R)-энантиомера прамипексола. Терапевтически эффективное количество (R)-прамипексола может, предпочтительно, составлять приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день.
Неэффективное дозовое количество (S)-прамипексола может составлять предпочтительно менее 1,0 мг/день, менее 0,5 мг/день и менее 0,125 мг/день. Таким образом, в качестве неограничивающего примера, доза 500 мг/день, введенная пациенту в виде однократной стандартной дозы, может иметь хиральную чистоту для (R)-энантиомера прамипексола по меньшей мере приблизительно 99,80%, так что неэффективное дозовое количество (S)-прамипексола может оставаться менее 1,0 мг/день, более предпочтительно, приблизительно 99,90%, так что неэффективное дозовое количество (S)-прамипексола может оставаться менее 0,5 мг/день, и, более предпочтительно, приблизительно 99,975%, так что неэффективное дозовое количество (S)-прамипексола может оставаться менее 0,125 мг/день. Варианты осуществления настоящего изобретения для терапевтически эффективного количества (R)-прамипексола, неэффективного дозового количества (S)-прамипексола и хиральной чистоты, представленные в настоящем документе, могут быть объединены в любой подходящей комбинации. Ссылаясь на таблицу 5, можно использовать любую комбинацию хиральной чистоты и стандартной дозы, которая позволяет получить желаемую комбинацию терапевтически эффективного количества (R)-прамипексола и неэффективного дозового количества (S)-прамипексола, как заявлено в настоящем документе.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция является подходящей для перорального введения и содержит количество (R)-прамипексола, превышающее 100 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,125 мг. Другой предпочтительный вариант осуществления настоящего изобретения представляет собой подходящую для перорального введения фармацевтическую композицию, содержащую количество (R)-прамипексола, превышающее 250 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,125 мг. Еще один предпочтительный вариант осуществления настоящего изобретения представляет собой подходящую для перорального введения фармацевтическую композицию, содержащую количество (R)-прамипексола, превышающее 500 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,125 мг. Предпочтительные фармацевтические композиции для перорального введения включают таблетки, капсулы и т.п.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция составлена в форме таблетки, подходящей для перорального введения, и содержит количество (R)-прамипексола, превышающее 50 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,50 мг, предпочтительно, количество (R)-прамипексола, превышающее 100 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,50 мг, и, более предпочтительно, количество (R)-прамипексола, превышающее 250 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,50 мг. Другой предпочтительный вариант осуществления настоящего изобретения представляет собой фармацевтическую композицию, составленную в форме таблетки, подходящей для перорального введения, содержащую количество (R)-прамипексола, превышающее 500 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,50 мг.
Таблица 5
Предпочтительные неэффективные дозовые количества (S)-прамипексола на основе хиральной чистоты композиции для (R)-прамипексола
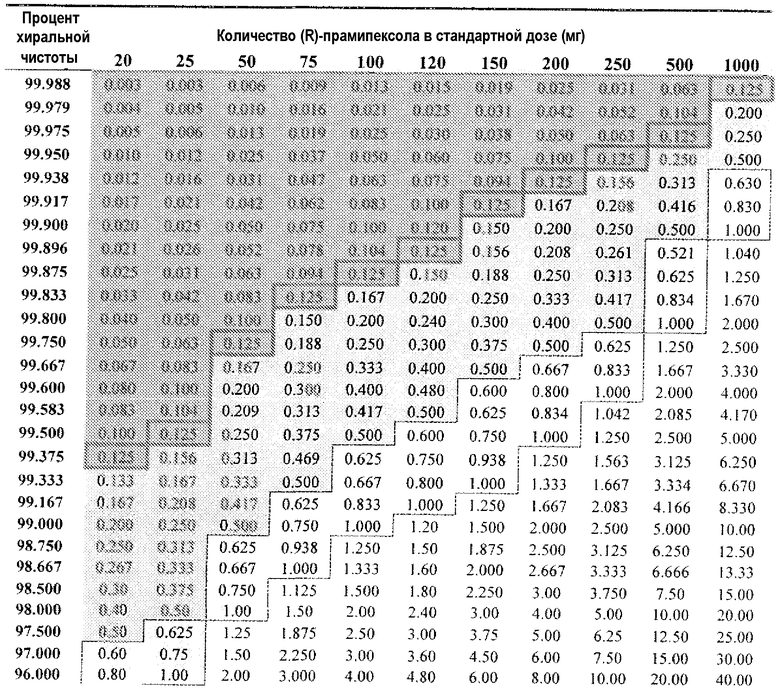
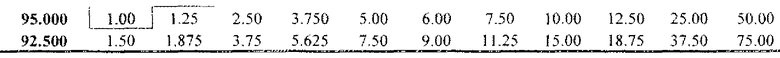
Предпочтительное неэффективное дозовое количество (S)-прамипексола может быть ниже 1,0 мг; более предпочтительно, ниже 0,5 мг и, более предпочтительно, ниже 0,125 мг.
Варианты осуществления настоящего изобретения для количеств (R)-прамипексола в композиции, хиральной чистоты, неэффективного дозового количества и лекарственной формы, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
Другой вариант осуществления настоящего изобретения представляет собой фармацевтическую композицию, составленную в форме таблетки, подходящей для перорального введения, содержащую количество (R)-прамипексола, превышающее 50 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,25 мг, предпочтительно, количество (R)-прамипексола, превышающее 100 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,25 мг, и, более предпочтительно, количество (R)-прамипексола, превышающее 250 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,25 мг. Другой предпочтительный вариант осуществления настоящего изобретения представляет собой фармацевтическую композицию, составленную в форме таблетки, подходящей для перорального введения, содержащую количество (R)-прамипексола, превышающее 500 мг, и неэффективное дозовое количество (S)-прамипексола, которое составляет приблизительно менее 0,25 мг.
В другом аспекте настоящее изобретение относится к композиции, содержащей терапевтически эффективное количество (R)-прамипексола и дозовое количество (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов (NOAEL). Терапевтическая композиция может дополнительно содержать фармацевтически приемлемый носитель.
В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 0,01 мг/кг/день до 10000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1000 мг/кг/день до 10000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 50 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения доза может составлять от 10 мг/день до 1500 мг/день, более предпочтительно, от 100 мг/день до 600 мг/день. Количество (R)-прамипексола в композициях может предпочтительно составлять приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола в композициях может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 50 мг до 5000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 100 мг до 3000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 300 мг до 1500 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму.
Композиция может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% или, более предпочтительно, по меньшей мере 99,99%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,99% или выше.
В некоторых вариантах осуществления настоящего изобретения дозовое количество (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов, составляет менее приблизительно 1,50 мг. В некоторых вариантах осуществления настоящего изобретения дозовое количество (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов, составляет менее приблизительно 0,5 мг. В некоторых вариантах осуществления настоящего изобретения дозовое количество (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов, составляет менее приблизительно 0,05 мг.
В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой капсулу. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой таблетку.
Варианты осуществления настоящего изобретения для количеств (R)-прамипексола в композиции, хиральной чистоты, дозового количества с уровнем, не вызывающим видимых неблагоприятных эффектов, и лекарственной формы, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество (R)-прамипексола и дозовое количество NOAEL (S)-прамипексола, которую вводят в виде стандартной лекарственной формы. Предпочтительные стандартные лекарственные формы включают формы, подходящие для перорального введения, включая, без ограничения, капсулы, таблетки и т.п. В таблице 6 показаны различные примеры вариантов осуществления настоящего изобретения. В каждом столбце таблицы 6 приводится количество (S)-прамипексола, которое можно совместно вводить в дозовом количестве NOAEL, в зависимости от хиральной чистоты композиции для R(+)-энантиомера прамипексола. Терапевтически эффективное количество (R)-прамипексола может предпочтительно составлять приблизительно от 50 мг до 5000 мг, предпочтительно, приблизительно от 100 мг до 3000 мг, предпочтительно, приблизительно от 300 мг до 1500 мг, более предпочтительно, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день.
Доза NOAEL (S)-прамипексола может составлять предпочтительно менее 1,5 мг, предпочтительно, менее 0,5 мг или, более предпочтительно, менее 0,05 мг. Таким образом, в качестве неограничивающего примера, вариант осуществления настоящего изобретения может представлять собой дозу 1500 мг/день, введенную пациенту в виде однократной стандартной дозы, которая может иметь хиральную чистоту для R(+)-энантиомера прамипексола, которая составляет по меньшей мере приблизительно 99,967%, так что доза (S)-прамипексола, не вызывающая видимых неблагоприятных эффектов, может оставаться менее 0,50 мг на дозу. Альтернативно, доза 1500 мг/день, введенная пациенту тремя отдельными дозами по 500 мг, может иметь хиральную чистоту для (R)-прамипексола, которая составляет по меньшей мере приблизительно 99,90%, так что доза (S)-прамипексола, не вызывающая видимых неблагоприятных эффектов, может оставаться менее 0,50 мг на дозу или 1,5 мг/день. Варианты осуществления настоящего изобретения для терапевтически эффективного количества (R)-прамипексола, дозового количества NOAEL (S)-прамипексола и хиральной чистоты, перечисленные в настоящем документе, могут быть объединены в любой подходящей комбинации. Ссылаясь на таблицу 6, можно использовать любую комбинацию хиральной чистоты и стандартной дозы, которая позволяет получить желаемую комбинацию терапевтически эффективного количества (R)-прамипексола и дозового количества (S)-прамипексола, не вызывающего видимых неблагоприятных эффектов, как заявлено в настоящем документе.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция составлена в готовую форму в виде таблетки, подходящей для перорального введения, и содержит количество (R)-прамипексола, превышающее 50 мг, и дозовое количество NOAEL (S)-прамипексола, которое составляет приблизительно менее 0,05 мг, предпочтительно, количество (R)-прамипексола, превышающее 100 мг, и дозовое количество NOAEL (S)-прамипексола, которое составляет приблизительно менее 0,05 мг, и, более предпочтительно, количество (R)-прамипексола, превышающее 250 мг, и дозовое количество NOAEL (S)-прамипексола, которое составляет приблизительно менее 0,05 мг. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция составлена в готовую форму в виде таблетки, подходящей для перорального введения, и содержит количество (R)-прамипексола, превышающее 500 мг, и дозовое количество NOAEL (S)-прамипексола, которое составляет приблизительно менее 0,05 мг.
Таблица 6
Предпочтительные дозы (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов, на основе хиральной чистоты композиции для (R)-прамипексола
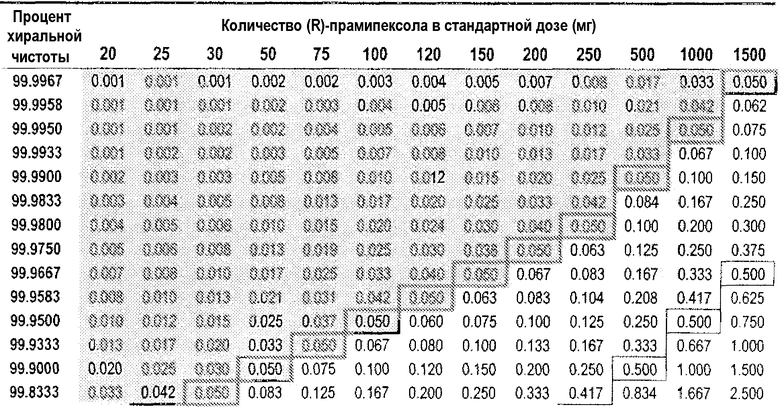
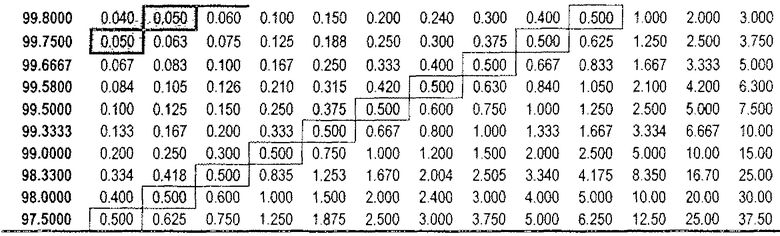
Предпочтительное дозовое количество (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов (NOAEL), может быть ниже 0,5 мг, предпочтительно, ниже 0,05 мг.
В некоторых вариантах осуществления настоящее изобретение относится к композиции для применения в качестве нейропротектора, содержащей терапевтически эффективное количество (R)-прамипексола и терапевтически эффективное количество (S)-прамипексола. Композиция может дополнительно содержать фармацевтически приемлемый носитель. Композиция может быть пригодной для лечения заболеваний, которые могут быть облегчены действием нейропротектора. Дополнительный вариант осуществления настоящего изобретения представляет собой терапевтическую композицию для применения в качестве нейропротектора, содержащую терапевтически эффективное количество (R)-прамипексола и терапевтически эффективное количество (S)-прамипексола. Композиция может дополнительно содержать фармацевтически приемлемый носитель. Терапевтическая композиция может быть пригодной для лечения заболеваний, связанных с дегенерацией нейронов или гибелью нейронов.
В одном варианте осуществления настоящего изобретения композиции (R)-прамипексола можно использовать для восстановления или улучшения функции нейронов, сетчатки и мускулатуры у взрослых и детей. Кроме того, композиции (R)-прамипексола можно использовать для лечения нейродегенеративных заболеваний или других заболеваний, связанных с митохондриальной дисфункцией или повышенным окислительным стрессом. В некоторых вариантах осуществления настоящего изобретения композиции (R)-прамипексола можно использовать для лечения нейродегенеративных деменций, нейродегенеративных двигательных расстройств и атаксий, расстройств, сопровождающихся припадками, расстройств или заболеваний двигательных нейронов и воспалительных демиелинизирующих расстройств у взрослых и детей. Композиции по настоящему изобретению также могут быть пригодными для лечения других расстройств, не перечисленных в настоящем документе, и любой перечень, приведенный в настоящем изобретении, служит лишь для примера и не является ограничивающим.
В некоторых вариантах осуществления настоящего изобретения композиции, которые содержат (R)-прамипексол, могут быть эффективными в качестве ингибиторов окислительного стресса, ингибиторов перекисного окисления липидов, для детоксикации кислородных радикалов и нормализации митохондриальной функции. Окислительный стресс может быть вызван увеличением количества кислородных и других свободных радикалов и связан со смертельным расстройством - боковым амиотрофическим склерозом (ALS). ALS представляет собой прогрессирующее нейродегенеративное расстройство, в которое вовлечены двигательные нейроны коры головного мозга, стволовой части головного мозга и спинного мозга. Приблизительно у 10% всех пациентов с ALS заболевание является наследственным, из которых 20% имеют мутации гена супероксиддисмутазы 1 (SOD-1). Фермент SOD-1 может играть главную роль в патогенезе и прогрессировании наследственного бокового амиотрофического склероза (FALS). Недавние исследования также связывают преждевременную гибель нейронов при ALS с мутировавшими митохондриальными генами, которые приводят к отклонениям в функционировании путем выработки энергии в митохондриях.
Композиции, которые содержат (R)-прамипексол, также могут быть эффективными для лечения возрастной дегенерации мышц. В качестве таковых, вариант осуществления настоящего изобретения может представлять собой композицию, содержащую (R)-прамипексол, подходящую для системного введения, окулярного введения или местного введения в глаза.
Таким образом, нейропротективный эффект композиций по настоящему изобретению может происходить, по меньшей мере частично, от способности (R)-энантиомера прамипексола предотвращать гибель нейронов посредством по меньшей мере одного из трех механизмов. Во-первых, (R)-энантиомер прамипексола может быть способен уменьшать образование реакционноспособных видов кислорода в клетках с нарушенной выработкой энергии в митохондриях. Во-вторых, (R)-энантиомер прамипексола может частично восстанавливать редуцированный митохондриальный мембранный потенциал, который коррелирует с болезнью Альцгеймера, болезнью Паркинсона и боковым амиотрофическим склерозом. В-третьих, (R)-энантиомер прамипексола может блокировать пути гибели клеток, которые производят фармакологические модели болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза и митохондриальных нарушений.
Композиции указанных нескольких вариантов осуществления настоящего изобретения, которые содержат (R)-прамипексол в качестве активного агента, могут быть эффективными в качестве ингибиторов окислительного стресса, ингибиторов перекисного окисления липидов, для детоксикации кислородных радикалов и нормализации митохондриальной функции. Кроме того, они могут быть эффективными для лечения нарушенной двигательной функции и дегенеративных заболеваний, которые могут поражать сердечную и поперечно-полосатые мышцы, а также ткани сетчатки. Как таковые, они могут быть эффективными для лечения нейродегенеративных заболеваний, таких как ALS, болезнь Паркинсона и болезнь Альцгеймера, а также дегенерация желтого пятна сетчатки.
Другой вариант осуществления настоящего изобретения представляет собой композицию, состоящую по существу из терапевтически эффективного количества (R)-прамипексола и неэффективного дозового количества (S)-прамипексола. Другой вариант осуществления настоящего изобретения представляет собой композицию, состоящую по существу из терапевтически эффективного количества (R)-прамипексола и дозового количества NOAEL (S)-прамипексола. Другой вариант осуществления настоящего изобретения представляет собой композицию, состоящую из терапевтически эффективного количества (R)-прамипексола и неэффективного дозового количества (S)-прамипексола. Такие композиции могут быть предпочтительно терапевтическими или фармацевтическими композициями. Другой вариант осуществления настоящего изобретения представляет собой композицию, состоящую из терапевтически эффективного количества (R)-прамипексола и дозового количества NOAEL (S)-прамипексола. Такие композиции могут быть предпочтительно терапевтическими или фармацевтическими композициями.
В другом аспекте настоящее изобретение относится к таблетке, содержащей по меньшей мере приблизительно 100 мг (R)-прамипексола и не более чем приблизительно 1,5 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 150 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 200 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 250 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 500 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 1000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит не более 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит не более 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит не более 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит не более 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит не более 0,125 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка дополнительно содержит фармацевтически приемлемый носитель.
В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 150 мг (R)-прамипексола и не более чем приблизительно 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 150 мг (R)-прамипексола и не более чем приблизительно 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 150 мг (R)-прамипексола и не более чем приблизительно 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 150 мг (R)-прамипексола и не более чем приблизительно 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 150 мг (R)-прамипексола и не более чем приблизительно 0,125 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 200 мг (R)-прамипексола и не более чем приблизительно 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 200 мг (R)-прамипексола и не более чем приблизительно 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 200 мг (R)-прамипексола и не более чем приблизительно 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 200 мг (R)-прамипексола и не более чем приблизительно 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 200 мг (R)-прамипексола и не более чем приблизительно 0,125 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 250 мг (R)-прамипексола и не более чем приблизительно 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 250 мг (R)-прамипексола и не более чем приблизительно 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 250 мг (R)-прамипексола и не более чем приблизительно 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 250 мг (R)-прамипексола и не более чем приблизительно 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 250 мг (R)-прамипексола и не более чем приблизительно 0,125 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 500 мг (R)-прамипексола и не более чем приблизительно 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 500 мг (R)-прамипексола и не более чем приблизительно 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 500 мг (R)-прамипексола и не более чем приблизительно 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 500 мг (R)-прамипексола и не более чем приблизительно 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 500 мг (R)-прамипексола и не более чем приблизительно 0,125 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 1000 мг (R)-прамипексола и не более чем приблизительно 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 1000 мг (R)-прамипексола и не более чем приблизительно 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 1000 мг (R)-прамипексола и не более чем приблизительно 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 1000 мг (R)-прамипексола и не более чем приблизительно 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения таблетка содержит приблизительно 1000 мг (R)-прамипексола и не более чем приблизительно 0,125 мг (S)-прамипексола.
Таблетка может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% или, более предпочтительно, по меньшей мере 99,99%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения таблетка имеет хиральную чистоту для (R)-прамипексола 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения таблетка имеет хиральную чистоту для (R)-прамипексола 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения таблетка имеет хиральную чистоту для (R)-прамипексола 99,99% или выше.
Варианты осуществления настоящего изобретения для количества (R)-прамипексола в таблетке, количества (S)-прамипексола в таблетке и хиральной чистоты, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
В другом аспекте настоящее изобретение относится к капсуле, содержащей по меньшей мере приблизительно 100 мг (R)-прамипексола и не более чем приблизительно 1,5 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 150 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 200 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 250 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 500 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 1000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит не более 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит не более 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит не более 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит не более 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит не более 0,125 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула дополнительно содержит фармацевтически приемлемый носитель.
В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 150 мг (R)-прамипексола и не более чем приблизительно 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 150 мг (R)-прамипексола и не более чем приблизительно 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 150 мг (R)-прамипексола и не более чем приблизительно 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 150 мг (R)-прамипексола и не более чем приблизительно 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 150 мг (R)-прамипексола и не более чем приблизительно 0,125 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 200 мг (R)-прамипексола и не более чем приблизительно 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 200 мг (R)-прамипексола и не более чем приблизительно 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 200 мг (R)-прамипексола и не более чем приблизительно 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 200 мг (R)-прамипексола и не более чем приблизительно 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 200 мг (R)-прамипексола и не более чем приблизительно 0,125 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 250 мг (R)-прамипексола и не более чем приблизительно 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 250 мг (R)-прамипексола и не более чем приблизительно 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 250 мг (R)-прамипексола и не более чем приблизительно 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 250 мг (R)-прамипексола и не более чем приблизительно 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 250 мг (R)-прамипексола и не более чем приблизительно 0,125 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 500 мг (R)-прамипексола и не более чем приблизительно 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 500 мг (R)-прамипексола и не более чем приблизительно 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 500 мг (R)-прамипексола и не более чем приблизительно 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 500 мг (R)-прамипексола и не более чем приблизительно 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 500 мг (R)-прамипексола и не более чем приблизительно 0,125 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 1000 мг (R)-прамипексола и не более чем приблизительно 1,0 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 1000 мг (R)-прамипексола и не более чем приблизительно 0,333 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 1000 мг (R)-прамипексола и не более чем приблизительно 0,3 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 1000 мг (R)-прамипексола и не более чем приблизительно 0,2 мг (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения капсула содержит приблизительно 1000 мг (R)-прамипексола и не более чем приблизительно 0,125 мг (S)-прамипексола.
Капсула может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% или, более предпочтительно, по меньшей мере 99,99%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения капсула имеет хиральную чистоту для (R)-прамипексола 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения капсула имеет хиральную чистоту для (R)-прамипексола 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения капсула имеет хиральную чистоту для (R)-прамипексола 99,99% или выше.
Варианты осуществления настоящего изобретения для количества (R)-прамипексола в капсуле, количества (S)-прамипексола в капсуле и хиральной чистоты, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
В еще одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей по меньшей мере приблизительно 25 мг (R)-прамипексола и менее чем приблизительно 1,5 эквивалента дофаминергической активности (“DAE”). В таблице 1 показаны DAE для 25 мг дозы (R)-прамипексола в зависимости от конкретной хиральной чистоты (R)-прамипексола в дозе и сравнительного соотношения связывающей аффинности.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция включает менее чем приблизительно 0,5 эквивалента дофаминергической активности (DAE). В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция включает менее чем приблизительно 0,05 эквивалента дофаминергической активности. Указанные величины DAE получены из дозовых количеств (R)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов, которые обсуждались в настоящем документе. В некоторых вариантах осуществления настоящего изобретения композиция имеет DAE, который меньше DAE, рассчитанного по количеству MTD или неэффективным дозовым количествам (S)-прамипексола. С учетом неэффективных дозовых количеств (S)-прамипексола в некоторых вариантах осуществления настоящего изобретения DAE не превышает приблизительно 1,0, не превышает приблизительно 0,75, не превышает приблизительно 0,5, не превышает приблизительно 0,25 или не превышает приблизительно 0,125. С учетом количества MTD композиция может иметь DAE менее 1,5, менее 0,3 или менее 0,2.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 50 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 75 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 125 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 150 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 200 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 250 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 300 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 400 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 500 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 600 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 750 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит по меньшей мере приблизительно 1000 мг (R)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой таблетку. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой капсулу.
Варианты осуществления настоящего изобретения для количества (R)-прамипексола в композиции, DAE, хиральной чистоты и лекарственной формы, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 0,01 мг/кг/день до 10000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1000 мг/кг/день до 10000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 50 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения доза может составлять от 10 мг/день до 1500 мг/день, более предпочтительно, от 100 мг/день до 600 мг/день. Количество (R)-прамипексола в композициях может предпочтительно составлять приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола в композициях может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 50 мг до 5000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 100 мг до 3000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 300 мг до 1500 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму.
Композиция может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% или, более предпочтительно, по меньшей мере 99,99%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,99% или выше.
Варианты осуществления настоящего изобретения для количества (R)-прамипексола в композиции, DAE, хиральной чистоты и лекарственной формы, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
В другом аспекте настоящее изобретение относится к начальной суточной дозе (R)-прамипексола по меньшей мере приблизительно 25 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 50 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 75 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 125 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 150 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 200 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 300 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 400 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 500 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 600 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 750 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит по меньшей мере приблизительно 1000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения начальная суточная доза содержит приблизительно от 600 мг до 900 мг (R)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола может составлять приблизительно от 0,01 мг/кг/день до 10000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1000 мг/кг/день до 10000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола может составлять приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 50 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество может составлять от 10 мг/день до 1500 мг/день, более предпочтительно, от 100 мг/день до 600 мг/день. Начальное суточное дозовое количество (R)-прамипексола в композициях может предпочтительно составлять приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола в композициях может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола составляет приблизительно от 50 мг до 5000 мг. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола составляет приблизительно от 100 мг до 3000 мг. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола составляет приблизительно от 300 мг до 1500 мг. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола составляет приблизительно от 500 мг до 1000 мг.
Композиция может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% или, более предпочтительно, по меньшей мере 99,99%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,99% или выше.
В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция представляет собой таблетку. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция представляет собой капсулу.
Варианты осуществления настоящего изобретения для начальной суточной дозы (R)-прамипексола в композиции, хиральной чистоты и лекарственной формы, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
В другом аспекте настоящее изобретение относится к готовой фармацевтической форме, содержащей микрокристаллическую целлюлозу в количестве приблизительно от 20% до 50% от массы указанной готовой формы; маннит в количестве приблизительно от 10% до 30% от массы указанной готовой формы; кросповидон в количестве приблизительно от 2% до 6% от массы указанной готовой формы; стеарат магния в количестве приблизительно от 0,01% до 2% от массы указанной композиции; и (R)-прамипексол. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит разбавитель в количестве приблизительно от 20% до 50% от массы указанной готовой формы; необязательно, второй разбавитель в количестве приблизительно от 10% до 30% от массы указанной готовой формы; необязательно, разрыхлитель в количестве приблизительно от 2% до 6% от массы указанной готовой формы; необязательно, смазывающий агент в количестве приблизительно от 0,01% до 2% от массы указанной композиции; и (R)-прамипексол. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит микрокристаллическую целлюлозу, маннит, натрий-кроскармеллозу, стеарат магния или их комбинации. В некоторых вариантах осуществления настоящего изобретения фармацевтически приемлемый носитель содержит микрокристаллическую целлюлозу, маннит или их комбинации, и дополнительно, необязательно, содержит натрий-кроскармеллозу или стеарат магния, или их комбинацию.
Готовая форма может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95%, или, более предпочтительно, по меньшей мере 99,99%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения готовая форма имеет хиральную чистоту для (R)-прамипексола 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения готовая форма имеет хиральную чистоту для (R)-прамипексола 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения готовая форма имеет хиральную чистоту для (R)-прамипексола 99,99% или выше.
Количество (R)-прамипексола в готовой форме может предпочтительно составлять приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола в готовой форме может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола в готовой форме составляет приблизительно от 600 мг до 900 мг.
В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции, содержащей микрокристаллическую целлюлозу в количестве приблизительно от 20% до 50% от массы указанной композиции; маннит в количестве приблизительно от 10% до 30% от массы указанной композиции; кросповидон в количестве приблизительно от 2% до 6% от массы указанной композиции; стеарат магния в количестве приблизительно от 0,01% до 2% от массы указанной композиции; и (R)-прамипексол. В некоторых вариантах осуществления настоящего изобретения композиция является подходящей для перорального введения. В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму.
Композиция может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% или, более предпочтительно, по меньшей мере 99,99%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения композиция имеет хиральную чистоту для (R)-прамипексола 99,99% или выше.
Количество (R)-прамипексола в композициях может предпочтительно составлять приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола в композициях может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. В некоторых вариантах осуществления настоящее изобретение дополнительно относится к фармацевтической композиции, содержащей (R)-прамипексол, имеющий приблизительно 25 эквивалентов нейропротективной активности и менее чем приблизительно 1,5 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет менее чем приблизительно 0,5 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет менее чем приблизительно 0,05 эквивалента дофаминергической активности.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет по меньшей мере приблизительно 50, по меньшей мере приблизительно 75, по меньшей мере приблизительно 125, по меньшей мере приблизительно 150, по меньшей мере приблизительно 200, по меньшей мере приблизительно 300, по меньшей мере приблизительно 400, по меньшей мере приблизительно 500, по меньшей мере приблизительно 700, по меньшей мере приблизительно 750 или по меньшей мере приблизительно 1000 эквивалентов нейропротективной активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 50 до 5000, приблизительно от 100 до 3000, приблизительно от 300 до 1500, приблизительно от 500 до 1000, приблизительно от 25 до 5000, приблизительно от 100 до 5000, приблизительно от 200 до 5000, приблизительно от 250 до 5000, приблизительно от 300 до 5000, приблизительно от 400 до 5000, приблизительно от 450 до 5000, приблизительно от 200 до 3000, приблизительно от 250 до 3000, приблизительно от 300 до 3000, приблизительно от 400 до 3000, приблизительно от 450 до 3000, приблизительно от 100 до 1000, приблизительно от 200 до 1000, приблизительно от 250 до 1000, приблизительно от 300 до 1000, приблизительно от 400 до 1000, приблизительно от 600 до 1000, приблизительно от 450 до 1000 или приблизительно от 600 до 900 эквивалентов нейропротективной активности.
В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 50 до 5000 эквивалентов нейропротективной активности и менее чем приблизительно 0,5 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 100 до 3000 эквивалентов нейропротективной активности и менее чем приблизительно 0,5 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 200 до 3000 эквивалентов нейропротективной активности и менее чем приблизительно 0,5 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 300 до 1500 эквивалентов нейропротективной активности и менее чем приблизительно 0,5 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 500 до 1000 эквивалентов нейропротективной активности и менее чем приблизительно 0,5 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 50 до 5000 эквивалентов нейропротективной активности и менее чем приблизительно 0,05 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 100 до 3000 эквивалентов нейропротективной активности и менее чем приблизительно 0,05 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 200 до 3000 эквивалентов нейропротективной активности и менее чем приблизительно 0,05 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 300 до 1500 эквивалентов нейропротективной активности и менее чем приблизительно 0,05 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция имеет приблизительно от 500 до 1000 эквивалентов нейропротективной активности и менее чем приблизительно 0,05 эквивалента дофаминергической активности.
В некоторых вариантах осуществления настоящего изобретения композиция представляет собой твердую пероральную лекарственную форму. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция представляет собой таблетку. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция представляет собой капсулу.
Варианты осуществления настоящего изобретения для эквивалентов нейропротективной активности, эквивалентов дофаминергической активности и лекарственных форм, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
Способы лечения, варианты применения и композиции и соединения для применения
В другом аспекте настоящее изобретение относится к способу лечения нейродегенеративного заболевания введением терапевтически эффективного количества (R)-прамипексола. В соответствии с указанным вариантом осуществления настоящего изобретения (R)-прамипексол может быть получен в виде фармацевтической или терапевтической композиции путем объединения одного или более фармацевтически приемлемых носителей. Варианты осуществления настоящего изобретения включают фармацевтические или терапевтические композиции, которые можно вводить перорально, предпочтительно, в виде твердой пероральной дозы и, более предпочтительно, в виде твердой пероральной дозы, которая может представлять собой капсулу или таблетку. В предпочтительном варианте осуществления настоящего изобретения фармацевтическую или терапевтическую композицию составляют в форме таблетки или капсулы для применения в путях перорального введения. Составы и количества неактивных ингредиентов в такой готовой форме могут зависеть от количества активного ингредиента и от размера и формы таблетки или капсулы. Указанные параметры может легко оценить и понять специалист в данной области. Терапевтически эффективное количество (R)-прамипексола может быть эффективным в качестве ингибитора окислительного стресса, ингибитора перекисного окисления липидов или для детоксикации кислородных радикалов.
В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 0,01 мг/кг/день до 10000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1000 мг/кг/день до 10000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 50 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения доза может составлять от 10 мг/день до 1500 мг/день, более предпочтительно, от 100 мг/день до 600 мг/день. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, предпочтительно, от 300 мг до 1500 мг или, более предпочтительно, от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола может составлять приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения терапевтически эффективное количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день.
Прамипексол может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% и, более предпочтительно, по меньшей мере 99,99%. В предпочтительном варианте осуществления настоящего изобретения хиральная чистота для R(+)-энатиометра прамипексола может составлять 100%.
В еще одном аспекте настоящее изобретение дополнительно относится к способу лечения острого нейродегенеративного заболевания у нуждающегося в этом пациента, включающему введение пациенту суточного дозового количества приблизительно от 25 мг до 5000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящее изобретение относится к использованию суточного дозового количества приблизительно от 25 мг до 5000 мг (R)-прамипексола для изготовления лекарственного средства для применения в способе лечения острого нейродегенеративного расстройства у пациента. В другом аспекте настоящее изобретение относится к суточному дозовому количеству приблизительно от 25 мг до 5000 мг (R)-прамипексола для применения в способе лечения острого нейродегенеративного расстройства у пациента.
В некоторых вариантах осуществления настоящего изобретения острое нейродегенеративное заболевание выбрано из инсульта, нейротравмы, острой метаболической дисфункции, последствий эпилептического припадка, эпилептического статуса и острого энцефалита.
В некоторых вариантах осуществления настоящего изобретения пациентом является пациент, который ранее не подвергался лечению.
В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола может составлять приблизительно от 0,01 мг/кг/день до 10000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1000 мг/кг/день до 10000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола может составлять приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 50 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество может составлять от 10 мг/день до 1500 мг/день, более предпочтительно, от 100 мг/день до 600 мг/день. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола составляет приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг или приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола составляет приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 500 мг до 1000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 50 мг до 5000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 100 мг до 3000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 200 мг до 3000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 300 мг до 1500 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 500 мг до 1000 мг (R)-прамипексола. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день.
В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,5% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,6% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,7% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,8% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,99% или выше.
В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество дополнительно включает количество (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов. В некоторых вариантах осуществления настоящего изобретения дозовое количество (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов, составляет менее 1,5 мг, менее 0,5 мг или менее 0,05 мг в день.
В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество дополнительно включает неэффективное дозовое количество (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола представляет собой количество, которое не превышает суммарную дозу 1,0 мг в день. В некоторых вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола представляет собой количество, которое не превышает суммарную дозу 0,75 мг/день, 0,5 мг/день, 0,25 мг/день или 0,125 мг/день. В некоторых вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола не превышает суммарную дозу 0,125 мг/день.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 100 мг до 3000 мг (R)-прамипексола, а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 200 мг до 3000 мг (R)-прамипексола, а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 300 мг до 1500 мг (R)-прамипексола, а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 500 мг до 1000 мг (R)-прамипексола, а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 100 мг до 3000 мг (R)-прамипексола и суточное дозовое количество дополнительно включает менее приблизительно 0,05 мг (S)-прамипексола.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 200 мг до 3000 мг (R)-прамипексола и суточное дозовое количество дополнительно включает менее приблизительно 0,05 мг (S)-прамипексола.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 300 мг до 1500 мг (R)-прамипексола и суточное дозовое количество дополнительно включает менее приблизительно 0,05 мг (S)-прамипексола.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 500 мг до 1000 мг (R)-прамипексола и суточное дозовое количество дополнительно включает менее приблизительно 0,05 мг (S)-прамипексола.
В другом варианте осуществления настоящего изобретения (R)-прамипексол в каждом из вариантов осуществления способов, описанных в настоящем документе, вводят в виде фармацевтической композиции. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция представляет собой таблетку. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция представляет собой капсулу. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит фармацевтически приемлемый носитель.
Варианты осуществления настоящего изобретения для болезненных состояний, типа пациента (не подвергавшегося лечению по сравнению с подвергавшимся лечению), суточных дозовых количеств, дозовых количеств с уровнем, не вызывающим видимых неблагоприятных эффектов, неэффективных дозовых количеств и степеней хиральной чистоты для способов по настоящему изобретению, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
Любой из вариантов осуществления настоящего изобретения, описанных в настоящем документе для способов, можно использовать для применения (R)-прамипексола для получения лекарственных средств для применения в способах лечения острого нейродегенеративного заболевания или в суточной дозе (R)-прамипексола для применения в способе лечения острого нейродегенеративного расстройства.
В еще одном аспекте настоящее изобретение дополнительно относится к способу лечения хронического нейродегенеративного заболевания у нуждающегося в этом пациента, включающему введение пациенту суточного дозового количества приблизительно от 25 мг до 5000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящее изобретение относится к применению суточного дозового количества приблизительно от 25 мг до 5000 мг (R)-прамипексола для получения лекарственного средства для применения в способе лечения хронического нейродегенеративного расстройства у пациента. В некоторых вариантах осуществления настоящее изобретение относится к суточному дозовому количеству приблизительно от 25 мг до 5000 мг (R)-прамипексола для применения в способе лечения хронического нейродегенеративного расстройства у пациента.
В некоторых вариантах осуществления настоящего изобретения хроническое нейродегенеративное заболевание выбрано из первичного нейродегенеративного заболевания, хореи Гентингтона, метаболически индуцированного неврологического повреждения, сенильной деменции типа Альцгеймера, когнитивной дисфункции, связанной с возрастом, сосудистой деменции, мультиинфарктной деменции, деменции с тельцами Леви, нейродегенеративной деменции, нейродегенеративного двигательного расстройства, атаксии, атаксии Фридрейха, рассеянного склероза, спинальной мышечной атрофии, первичного бокового склероза, расстройств, сопровождаемых припадками, расстройства или заболевания двигательных нейронов, воспалительного демиелинизирующего расстройства, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, печеночной энцефалопатии и хронического энцефалита.
В некоторых вариантах осуществления настоящего изобретения пациентом является пациент, который ранее не подвергался лечению.
В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола может составлять приблизительно от 0,01 мг/кг/день до 10000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1 мг/кг/день до 1000 мг/кг/день, приблизительно от 1000 мг/кг/день до 10000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола может составлять приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола может составлять приблизительно от 3 мг/кг/день до 50 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество может составлять от 10 мг/день до 1500 мг/день, более предпочтительно, от 100 мг/день до 600 мг/день. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола составляет приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, приблизительно от 300 мг до 1500 мг или приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола составляет приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 500 мг до 1000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 50 мг до 5000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 100 мг до 3000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 200 мг до 3000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 300 мг до 1500 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество составляет приблизительно от 500 мг до 1000 мг (R)-прамипексола. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день.
В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,5% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,6% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,7% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,8% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,99% или выше.
В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество дополнительно включает дозовое количество (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов. В некоторых вариантах осуществления настоящего изобретения дозовое количество (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов, составляет менее 1,5 мг, менее 0,5 мг или менее 0,05 мг в день.
В некоторых вариантах осуществления настоящего изобретения суточное дозовое количество дополнительно включает неэффективное дозовое количество (S)-прамипексола. В некоторых вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола представляет собой количество, которое не превышает суммарную дозу 1,0 мг в день. В некоторых вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола представляет собой количество, которое не превышает суммарную дозу 0,75 мг/день, 0,5 мг/день, 0,25 мг/день или 0,125 мг/день. В некоторых вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола не превышает суммарную дозу 0,125 мг в день.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 100 мг до 3000 мг (R)-прамипексола, а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 200 мг до 3000 мг (R)-прамипексола, а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 300 мг до 1500 мг (R)-прамипексола, а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 500 мг до 1000 мг (R)-прамипексола, а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 100 мг до 3000 мг (R)-прамипексола и суточное дозовое количество дополнительно включает менее приблизительно 0,05 мг (S)-прамипексола.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 200 мг до 3000 мг(R)-прамипексола и суточное дозовое количество дополнительно включает менее приблизительно 0,05 мг (S)-прамипексола.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 300 мг до 1500 мг (R)-прамипексола и суточное дозовое количество дополнительно включает менее приблизительно 0,05 мг (S)-прамипексола.
В некоторых вариантах осуществления способов по настоящему изобретению суточное дозовое количество составляет приблизительно от 500 мг до 1000 мг (R)-прамипексола и суточное дозовое количество дополнительно включает менее приблизительно 0,05 мг (S)-прамипексола.
В другом варианте осуществления настоящего изобретения (R)-прамипексол в каждом из вариантов осуществления способов, описанных в настоящем документе, вводят в виде фармацевтической композиции. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция представляет собой таблетку. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция представляет собой капсулу. В некоторых вариантах осуществления настоящего изобретения фармацевтическая композиция содержит фармацевтически приемлемый носитель.
Варианты осуществления настоящего изобретения для болезненных состояний, типа пациента (не подвергавшегося лечению по сравнению с подвергавшимся лечению), суточных дозовых количеств, дозовых количеств с уровнем, не вызывающим видимых неблагоприятных эффектов, неэффективных дозовых количеств и степеней хиральной чистоты для способов по настоящему изобретению, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
Любой из вариантов осуществления настоящего изобретения, описанных в настоящем документе для способов, можно использовать для применения (R)-прамипексола для получения лекарственных средств для применения в способах лечения хронического нейродегенеративного заболевания или в суточной дозе (R)-прамипексола для применения в способе лечения хронического нейродегенеративного расстройства.
В другом аспекте настоящее изобретение относится к набору, включающему одну или более фармацевтических композиций, содержащих (R)-прамипексол, и инструкции по введению или назначению одной или более фармацевтических композиций, включающие указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения начальной суточной дозы по меньшей мере приблизительно от 50 мг до 5000 мг (R)-прамипексола пациенту. Кроме того, в некоторых вариантах осуществления настоящее изобретение относится к набору, включающему одну или более фармацевтических композиций согласно любому из предыдущих вариантов осуществления композиций, описанных в настоящем документе, или любой их комбинации, и инструкции по введению или назначению одной или более фармацевтических композиций, включающие указания по введению или назначению одной или более фармацевтических композиций согласно вариантам осуществления способов, описанных в настоящем документе, или любой их комбинации.
Прамипексол для использования в наборах по настоящему изобретению может иметь хиральную чистоту для (R)-прамипексола по меньшей мере 99,5%, предпочтительно, по меньшей мере 99,6%, предпочтительно, по меньшей мере 99,7%, предпочтительно, по меньшей мере 99,8%, предпочтительно, по меньшей мере 99,9%, предпочтительно, по меньшей мере 99,95% и, более предпочтительно, по меньшей мере 99,99%. В предпочтительном варианте осуществления настоящего изобретения хиральная чистота для (R)(+)-энантиомера прамипексола составляет 100%. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,95% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота для (R)-прамипексола составляет 99,99% или выше.
В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения начальной суточной дозы (R)-прамипексола приблизительно от 0,1 мг/кг/день до 1000 мг/кг/день или приблизительно от 1 мг/кг/день до 100 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения начальной суточной дозы (R)-прамипексола приблизительно от 3 мг/кг/день до 70 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения начальной суточной дозы (R)-прамипексола приблизительно от 7 мг/кг/день до 40 мг/кг/день. В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения начальной суточной дозы (R)-прамипексола приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 3000 мг, предпочтительно, от 300 мг до 1500 мг или, более предпочтительно, приблизительно от 500 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения начальной суточной дозы (R)-прамипексола приблизительно от 25 мг до 5000 мг, приблизительно от 50 мг до 5000 мг, приблизительно от 100 мг до 5000 мг, приблизительно от 200 мг до 5000 мг, приблизительно от 250 мг до 5000 мг, приблизительно от 300 мг до 5000 мг, приблизительно от 400 мг до 5000 мг, приблизительно от 450 мг до 5000 мг, приблизительно от 200 мг до 3000 мг, приблизительно от 250 мг до 3000 мг, приблизительно от 300 мг до 3000 мг, приблизительно от 400 мг до 3000 мг, приблизительно от 450 мг до 3000 мг, приблизительно от 100 мг до 1000 мг, приблизительно от 200 мг до 1000 мг, приблизительно от 250 мг до 1000 мг, приблизительно от 300 мг до 1000 мг, приблизительно от 400 мг до 1000 мг, приблизительно от 600 мг до 1000 мг или приблизительно от 450 мг до 1000 мг. В некоторых вариантах осуществления настоящего изобретения начальное суточное дозовое количество (R)-прамипексола составляет приблизительно от 600 мг до 900 мг. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, вводимых в течение дня, например, на 1-5 доз в день, предпочтительно, на две или три дозы в день.
В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 100 мг до 3000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 200 мг до 3000 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 300 мг до 1500 мг (R)-прамипексола. В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 500 мг до 1000 мг (R)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения указания дополнительно касаются введения неэффективного дозового количества (S)-прамипексола. В вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола представляет собой количество, которое не превышает суммарную дозу 1,0 мг/день. В более предпочтительных вариантах осуществления настоящего изобретения неэффективное дозовое количество (S)-прамипексола представляет собой количество, которое не превышает суммарную дозу 0,75 мг/день, 0,5 мг/день, 0,25 мг/день и, предпочтительно, 0,125 мг/день. В некоторых вариантах осуществления настоящего изобретения неэффективное дозовое количество составляет менее чем приблизительно 0,125 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения указания дополнительно касаются введения дозового количества (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов (NOAEL). В некоторых вариантах осуществления настоящего изобретения дозовое количество (S)-прамипексола с уровнем, не вызывающим видимых неблагоприятных эффектов, может предпочтительно составлять менее 1,5 мг, предпочтительно, менее 0,5 мг или, более предпочтительно, менее 0,05 мг. В некоторых вариантах осуществления настоящего изобретения дозовое количество с уровнем, не вызывающим видимых неблагоприятных эффектов, составляет менее чем приблизительно 0,05 мг в день. В другом предпочтительном варианте осуществления настоящего изобретения дозовое количество (S)-прамипексола с NOAEL представляет собой количество, которое не превышает 0,0007 мг/кг на стандартную дозу. В некоторых вариантах осуществления настоящего изобретения указания дополнительно касаются введения менее приблизительно 1,5 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения указания дополнительно касаются введения менее приблизительно 0,5 эквивалента дофаминергической активности. В некоторых вариантах осуществления настоящего изобретения указания дополнительно касаются введения менее приблизительно 0,05 эквивалента дофаминергической активности.
В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 100 мг до 3000 мг (R)-прамипексола; а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 200 мг до 3000 мг (R)-прамипексола; а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 300 мг до 1500 мг (R)-прамипексола; а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 500 мг до 1000 мг (R)-прамипексола; а хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 100 мг до 3000 мг (R)-прамипексола и менее приблизительно 0,05 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 200 мг до 3000 мг (R)-прамипексола и менее приблизительно 0,05 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 300 мг до 1500 мг (R)-прамипексола и менее приблизительно 0,05 мг (S)-прамипексола.
В некоторых вариантах осуществления настоящего изобретения инструкции включают указания по введению или назначению одной или более фармацевтических композиций в количестве, достаточном для введения пациенту начальной суточной дозы приблизительно от 500 мг до 1000 мг (R)-прамипексола и менее приблизительно 0,05 мг (S)-прамипексола.
Варианты осуществления настоящего изобретения для суточных дозовых количеств, дозовых количеств с уровнем, не вызывающим видимых неблагоприятных эффектов, неэффективных дозовых количеств и степеней хиральной чистоты для наборов по настоящему изобретению, которые описаны в настоящем документе по отдельности ради краткости, могут быть объединены в любой подходящей комбинации.
Фармацевтические или терапевтические композиции по настоящему изобретению можно изготавливать, упаковывать, продавать в массе, в виде однократной стандартной дозы или в виде многократных стандартных доз. Композиции могут быть составлены в готовую форму для перорального, офтальмологического, внутривенного, внутримышечного, интраартериального, интрамедуллярного, интратекального, интравентрикулярного, чрескожного, подкожного, интраперитонеального, интравезикулярного, интраназального, энтерального, местного, сублингвального или ректального введения. Композиции по настоящему изобретению можно вводить перорально, предпочтительно, в виде твердой пероральной дозы и, более предпочтительно, в виде твердой пероральной дозы, которая может представлять собой капсулу или таблетку. В некоторых вариантах осуществления композиции по настоящему изобретению могут быть составлены в готовую форму в виде таблеток для перорального введения.
Соединения по настоящему изобретению можно вводить общепринятыми способами любым путем, где они являются активными. Введение может быть системным, местным или пероральным. Например, введение можно осуществлять, без ограничения, парентеральным, подкожным, внутривенным, внутримышечным, интраперитонеальным, чрескожным, пероральным, буккальным или окулярным путями, или интравагинально, интравезикулярно, ингаляцией, с использованием депо-инъекций или с использованием имплантатов. Таким образом, способы введения соединений по настоящему изобретению (как в отдельности, так и в комбинации с другими лекарственными средствами) могут представлять собой, без ограничения, сублингвальный, инъекционный (включая формы короткого действия, депо, имплантаты и гранулярные формы, которые инъецируют подкожно или внутримышечно) способы, или способы с использованием вагинальных кремов, суппозиториев, пессариев, вагинальных колец, ректальных суппозиториев, внутриматочных устройств и трансдермальных форм, таких как пластыри и кремы.
Дозы (R)-прамипексола, которые можно вводить нуждающемуся в этом пациенту, могут находиться в пределах приблизительно от 0,1 мг/кг в день до 1000 мг/кг в день. Указанную дозу можно вводить в виде однократной суточной дозы или ее можно разделить на несколько доз, которые вводят в течение дня, например, на 1-5 доз, предпочтительно, на две или три дозы в день. Путь введения может включать пероральный, сублингвальный, чрескожный, ректальный или любой доступный парентеральный путь. Специалист поймет и примет во внимание дозировки и распределение по времени введения доз пациенту, который в этом нуждается. Дозы и продолжительность лечения могут меняться и могут основываться на оценке специалиста, по результатам мониторинга и измеримых улучшений в нейрональных и не являющихся нейрональными тканях. Указанную оценку можно производить на основе внешних физикальных признаков улучшения, таких как улучшение контроля мускулатуры, или внутренних физиологических признаков или маркеров. Дозы могут зависеть также от состояния или заболевания, которое лечат, степени состояния или заболевания, которое лечат, а также от возраста и массы тела пациента.
Конкретные способы введения будут зависеть от показаний. Выбор конкретного пути введения и схемы введения может устанавливать или титровать клиницист, в соответствии со способами, известными клиницисту, с целью получения оптимального клинического ответа. Количество соединения, которое следует ввести, может представлять собой количество, которое является терапевтически эффективным. Доза, которую следует ввести, может зависеть от характеристик субъекта, которого лечат, например, конкретного животного или человека, возраста, массы тела, состояния здоровья, типов одновременного лечения, если оно проводится, и частоты проведения лечения, и может быть легко определена специалистом (например, клиницистом).
Предпочтительным путем введения композиций по настоящему изобретению может быть пероральный, при предпочтительном использовании таблеток, капсул, пастилок и т.п. В предпочтительных вариантах осуществления композиции по настоящему изобретению могут быть составлены в готовую форму в виде таблеток для перорального введения. Таблетку можно изготавливать прессованием или формованием, необязательно, с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки можно изготавливать прессованием в подходящей машине активного ингредиента в сыпучей форме, такой как порошок или гранулы, необязательно, в смеси со связующим агентом, смазывающим агентом, инертным разбавителем, смазывающим, поверхностно-активным или диспергирующим агентом. Формованные таблетки можно изготавливать формованием в подходящей машине порошкообразного соединения, увлажненного инертным жидким разбавителем.
Таблетки могут не иметь покрытия или на них может быть нанесено покрытие с использованием известных технологий, необязательно, для задержки дезинтеграции и всасывания в желудочно-кишечном тракте, и, таким образом, для обеспечения замедленного действия в течение более продолжительного периода времени. Покрытие может быть подобрано таким образом, чтобы высвобождать активное соединение по заранее определенному образцу (например, с целью получения композиции с контролируемым высвобождением), или оно может быть подобрано таким образом, чтобы не высвобождать активное соединение до тех пор, пока оно не пройдет через желудок (энтеросолюбильное покрытие). Покрытие может представлять собой сахарное покрытие, пленочное покрытие (например, на основе гидроксипропилметилцеллюлозы, метилцеллюлозы, метилгидроксиэтилцеллюлозы, гидроксипропилцеллюлозы, карбоксиметилцеллюлозы, акрилатных сополимеров, полиэтиленгликолей и/или поливинилпирролидона) или энтеросолюбильное покрытие (например, на основе сополимера метакриловой кислоты, ацетатфталата целлюлозы, фталата гидроксипропилметилцеллюлозы, ацетатсукцината гидроксипропилметилцеллюлозы, поливинилацетатфталата, шеллака и/или этилцеллюлозы). Кроме того, можно использовать материал, обеспечивающий задержку по времени, такой как, например, глицерилмоностеарат или глицерилдистеарат. Твердые таблеточные композиции могут включать покрытие, подобранное таким образом, чтобы защищать композицию от нежелательных химических изменений (например, химической деградации до высвобождения активного лекарственного вещества).
Фармацевтические композиции, содержащие соединения по настоящему изобретению и подходящий носитель, также могут представлять собой ряд твердых лекарственных форм, которые включают, без ограничения, таблетки, капсулы, облатки, мелкие гранулы, пилюли, порошки и гранулы; местные лекарственные формы, которые включают, без ограничения, растворы, порошки, жидкие эмульсии, жидкие суспензии, полутвердые формы, мази, пасты, кремы, гели и желе и пены; и парентеральные лекарственные формы, которые включают, без ограничения, растворы, суспензии, эмульсии и сухой порошок; содержащие эффективное количество полимера или сополимера по настоящему изобретению. Специалистам также известно, что активные ингредиенты могут содержаться в указанных готовых формах с фармацевтически приемлемыми разбавителями, наполнителями, разрыхлителями, связующими агентами, смазывающими агентами, поверхностно-активными веществами, гидрофобными носителями, водорастворимыми носителями, эмульгаторами, буферными агентами, гигроскопическим агентами, увлажняющими агентами, солюбилизаторами, консервантами и т.п. Средства и способы введения известны специалистам, и специалист может обратиться за информацией к различным фармакологическим источникам. Например, можно проконсультироваться в Modern Pharmaceutics, Banker & Rhodes, Marcel Dekker, Inc. (1979); и Goodman & Gilman's The Pharmaceutical Basis of Therapeutics, 6th Edition, MacMillan Publishing Co., New York (1980).
Соединения по настоящему изобретению могут быть составлены в готовую форму для парентерального введения путем инъекции, например, болюсной инъекции или непрерывной инфузии. Соединения можно вводить путем непрерывной инфузии в течение периода приблизительно от 15 минут до 24 часов. Композиции для инъекций могут быть представлены в виде стандартной лекарственной формы, например, в ампулах или в контейнерах, содержащих множество доз, с добавлением консерванта. Композициям можно придавать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, и они могут содержать агенты, способствующие изготовлению композиции, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты.
Для перорального введения соединения можно легко составить в готовую форму объединением указанных соединений с фармацевтически приемлемыми носителями, хорошо известными специалистам. Указанные носители способствуют составлению соединений по настоящему изобретению в готовую форму в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п. для перорального приема пациентом, подвергающимся лечению. Фармацевтические препараты для перорального применения можно получить добавлением твердого эксципиента, необязательно, размалыванием полученной смеси и обработкой смеси гранул, после добавления подходящих вспомогательных агентов, если это желательно, с получением ядер таблеток или драже. Подходящие эксципиенты включают, без ограничения, наполнители, такие как сахара, включая, без ограничения, лактозу, сахарозу, маннит и сорбит; препараты целлюлозы, такие как, без ограничения, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий-карбоксиметилцеллюлоза и поливинилпирролидон (PVP). Если желательно, можно добавлять разрыхлители, такие как, без ограничения, сшитый поливинилпирролидон, агар или альгиновая кислота, или ее соли, такие как альгинат натрия.
На ядра драже можно наносить подходящие покрытия. Для указанной цели можно использовать концентрированные растворы сахара, которые, необязательно, могут содержать аравийскую камедь, тальк, поливинилпирролидон, гель карбопол, полиэтиленгликоль и/или диоксид титана, растворы лаков и подходящие органические растворители или смеси растворителей. Красители или пигменты можно добавлять к покрытиям для таблеток или драже для идентификации или для обозначения различных комбинаций доз активного соединения.
Фармацевтические препараты, которые можно использовать перорально, включают, без ограничения, сборные капсулы, изготовленные из желатина, а также мягкие, герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Сборные капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как, например, лактоза, связующим агентом, таким как, например, крахмалы, и/или смазывающими агентами, такими как, например, тальк или стеарат магния, и, необязательно, стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, можно добавлять стабилизаторы. Все готовые формы для перорального введения должны быть в дозах, подходящих для указанного введения.
Для буккального или сублингвального введения композициям можно придавать форму таблеток, мгновенно тающих таблеток или пастилок, изготовленных общепринятыми способами.
Для введения путем ингаляции соединения для использования по настоящему изобретению удобно доставлять в форме аэрозоля, распыляемого из упаковок под давлением или небулайзера, с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением дозированную единицу можно определить установкой клапана для доставки отмеренного количества. Можно изготавливать капсулы и картриджи, например, из желатина для использования в ингаляторе или инсуффляторе, содержащие порошкообразную смесь соединения и подходящей порошкообразной основы, такой как лактоза или крахмал.
Соединения по настоящему изобретению можно также составить в готовую форму в виде ректальных композиций, таких как суппозитории, или удерживающих клизм, например, содержащих обычные основы для суппозиториев, такие как масло какао или другие глицериды.
Помимо готовых форм, описанных ранее, соединения по настоящему изобретению можно также составлять в форме депо-препарата. Такие готовые формы длительного действия можно вводить путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции.
Депо-инъекции можно вводить с интервалами приблизительно от 1 до 6 месяцев и более. Так, например, соединения могут быть составлены в готовые формы с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами, или в виде труднорастворимых производных, например, в виде труднорастворимой соли.
При чрескожном введении соединения по настоящему изобретению, например, можно наносить на пластырь или на чрескожные терапевтические системы, которыми впоследствии снабжают организм.
Фармацевтические и терапевтические композиции соединений могут также содержать подходящие твердофазные или гелевые носители или эксципиенты. Примеры таких носителей или эксципиентов включают, без ограничения, карбонат кальция, фосфат кальция, различные сахара, крахмалы, производные целлюлозы, желатин и полимеры, такие как, например, полиэтиленгликоли.
Соединения по настоящему изобретению можно также вводить в комбинации с другими активными ингредиентами, такими как, например, адъюванты, ингибиторы протеазы или другие совместимые лекарственные средства или соединения, когда указанная комбинация представляется желательной или выгодной для достижения желательных эффектов способов, описанных в настоящем документе.
Получение (R)- и (S)-прамипексола
Способы получения прамипексола описаны в патенте США № 4843086 и в патенте США № 4886812, выданном Griss et al., каждый из которых полностью включен в настоящий документ путем ссылки. (R)-прамипексол по настоящему изобретению может быть синтезирован и очищен способами, описанными во временной заявке США № 60/894829, озаглавленной «Способы синтеза и очистки (R)(+) и (S)-прамипексола», поданной 14 марта 2007 г., и во временной заявке США № 60/894814, озаглавленной «Способы энантиомерной очистки хиральных соединений», поданной 14 марта 2007 г., которые полностью включены в настоящий документ путем ссылки. Конкретно, препараты прамипексола, которые являются хирально чистыми для R(+)-энантиомера, могут быть получены с использованием реакции бимолекулярного нуклеофильного замещения (SN2). Диамин 2,6-диамино-4,5,6,7-тетрагидробензотиазол взаимодействует с пропилсульфонатом или пропилгалогенидом в полярных растворителях с образованием нерастворимой соли прамипексола по схеме синтеза в одном сосуде. Продукт реакции соль прамипексола имеет высокую химическую чистоту и повышенную оптическую чистоту по сравнению с реагентами, что может происходить благодаря ограниченной растворимости соли прамипексола в полярных растворителях реакционной смеси. Очистка конечного продукта синтеза прамипексола из реакционной смеси, таким образом, включает простое растирание и промывание осажденной соли прамипексола в летучем растворителе, таком как спирт или гептан, с последующей вакуумной сушкой.
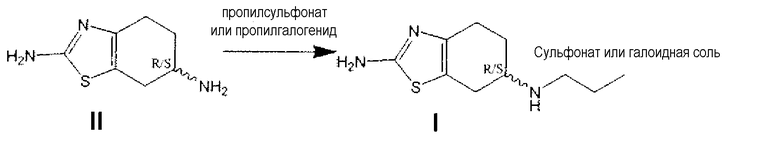
В некоторых вариантах осуществления настоящего изобретения (R)-прамипексол получают растворением диамина, 2,6-диамино-4,5,6,7-тетрагидробензотиазола, в органическом растворителе, взаимодействием диамина с пропилсульфонатом или пропилгалогенидом в условиях, достаточных для образования и осаждения соли прамипексола, и восстановлением соли прамипексола. В предпочтительном варианте осуществления настоящего изобретения пропилсульфонат может представлять собой пропилтозилат. В еще одном варианте осуществления настоящего изобретения пропилгалогенид может представлять собой пропилбромид. Соль прамипексола в виде продукта реакции имеет высокую химическую чистоту и повышенную оптическую чистоту по сравнению с реагентами. Не имея намерения связываться с какой бы то ни было теорией, можно предположить, что повышенная оптическая чистота может быть обусловлена ограниченной растворимостью продукта реакции, соли прамипексола, в полярных растворителях реакционной смеси. Очистка конечного продукта реакции прамипексола из реакционной смеси, таким образом, включает простое растирание и промывание осажденной соли прамипексола в летучем растворителе, таком как спирт или гептан, с последующей вакуумной сушкой.
В вариантах осуществления способа диамин может представлять собой R(+) диамин или смесь R(+) и S диамина. Химическая чистота конечной соли прамипексола может составлять по меньшей мере приблизительно 97% или выше, предпочтительно, 98% или выше, более предпочтительно, 99% или выше. R(+)-энантиомеры соли прамипексола, полученные с использованием указанного способа, получают из исходных диаминов, которые могут быть по меньшей мере на 55% оптически чистыми, предпочтительно, на 70% оптически чистыми и, более предпочтительно, более чем на 90% оптически чистыми. Конечный продукт прамипексол может быть обогащен до 99,6% оптической чистоты или выше, 99,7% оптической чистоты или выше, предпочтительно, 99,8% оптической чистоты или выше и, более предпочтительно, 99,9% оптической чистоты или выше, 99,95% оптической чистоты или выше, 99,99% оптической чистоты или выше. В некоторых вариантах осуществления настоящего изобретения оптическая чистота может составлять 100%.
В вариантах осуществления указанного способа органический растворитель может представлять собой полярный апротонный растворитель, такой как тетрагидрофуран, диметилформамид, диметилсульфоксид, диметилацетамид или гексаметилфосфорный триамид. Органический растворитель может представлять собой также низкомолекулярный спирт, такой как этанол, 1-пропанол или н-бутанол. Кроме того, органический растворитель может представлять собой любую комбинацию полярных апротонных растворителей и низкомолекулярных спиртов. Органический растворитель может иметь содержание воды приблизительно от 0 до 10 объемных процентов. Предпочтительно, растворители, используемые в практике настоящего изобретения, представляют собой стандартные растворители класса ACS. Кроме того, пропилсульфонат или пропилгалогенид можно добавлять в количестве приблизительно от 1,0 до 2,0 молярных эквивалентов диамина.
В дополнительных вариантах осуществления настоящего способа условия, достаточные для образования и осаждения соли прамипексола, могут включать нагревание растворенного диамина при повышенной температуре, добавление пропилсульфоната или пропилгалогенида, которые могут быть растворены в диизопропилэтиламине и органическом растворителе, с получением смеси, и перемешивание смеси приблизительно в течение 4 часов. Альтернативно, диизопропилэтиламин можно добавлять в реакционную смесь с диамином, а пропилсульфонат или пропилгалогенид можно растворять в органическом растворителе с получением смеси, которую можно добавлять в реакционную смесь при перемешивании в течение приблизительно 4 часов.
В указанном варианте осуществления настоящего изобретения повышенная температура реакционной смеси может быть ниже температуры кипения реакционной смеси, конкретно, ниже температуры кипения органического растворителя (растворителей) реакционной смеси. Повышенная температура может быть ниже приблизительно 125°C, предпочтительно, ниже приблизительно 100°C и, более предпочтительно, приблизительно 95°C или ниже. Время, необходимое для протекания реакции, может варьировать в зависимости от особенностей реагентов, системы растворителей и выбранной температуры.
В альтернативном варианте осуществления настоящего изобретения условия, достаточные для образования и осаждения соли прамипексола, могут включать использование диметилформамида в качестве органического растворителя, нагревание растворенного диамина при повышенной температуре, добавление пропилсульфоната или пропилгалогенида, которые могут быть растворены в диметилформамиде, с получением смеси, и перемешивание смеси приблизительно в течение 4 часов. Повышенная температура реакционной смеси может быть ниже температуры кипения реакционной смеси, конкретно, ниже температуры кипения органического растворителя (растворителей) реакционной смеси. Повышенная температура может быть ниже приблизительно 125°C, предпочтительно, ниже приблизительно 100°C и, более предпочтительно, приблизительно 75°C или ниже. Время, необходимое для протекания реакции, может варьировать в зависимости от особенностей реагентов, системы растворителей и выбранной температуры.
В предпочтительном альтернативном варианте осуществления настоящего изобретения условия, достаточные для образования и осаждения соли прамипексола, могут включать использование диметилформамида в качестве органического растворителя и нагревание растворенного диамина при повышенной температуре. Смесь пропилсульфоната или пропилгалогенида, предпочтительно, в количестве 1,25 молярных эквивалентов, растворенных в диметилформамиде, предпочтительно, в 10 объемах, и диизопропилэтиламина, предпочтительно, в количестве 1,25 молярных эквивалентов, медленно добавляют к нагретому диамину при перемешивании приблизительно в течение 4 часов. Альтернативно, диизопропилэтиламин можно добавлять в реакционную смесь с диамином, а пропилсульфонат или пропилгалогенид можно растворять в диметилформамиде с получением смеси, которую можно добавлять в реакционную смесь при перемешивании в течение приблизительно 4 часов. Повышенная температура реакционной смеси может быть ниже температуры кипения реакционной смеси, конкретно, ниже температуры кипения органического растворителя (растворителей) реакционной смеси. Повышенная температура может быть ниже приблизительно 125°C, предпочтительно, ниже приблизительно 100°C и, более предпочтительно, приблизительно 65°C или ниже. Время, необходимое для протекания реакции, может варьировать в зависимости от особенностей реагентов, системы растворителей и выбранной температуры.
Варианты осуществления способа дополнительно включают охлаждение реакционной смеси до температуры, приблизительно соответствующей комнатной температуре, приблизительно 25°C, и перемешивание реакционной смеси в течение приблизительно 2 часов. Способ может дополнительно включать фильтрование реакционной смеси для отделения твердого осадка, промывание осадка спиртом и сушку осадка в вакууме. Продукт реакции указанного способа, соль прамипексола, может иметь повышенную оптическую чистоту по сравнению с реагентами.
Альтернативно, сульфонат или галоидная соль прамипексола может взаимодействовать с концентрированной HCl в органическом растворителе, таком как спирт, при температуре приблизительно от 0 до 5°C. Можно добавлять органический растворитель, такой как метил-трет-бутиловый эфир (МТВЕ), и реакционную смесь можно перемешивать в течение еще одного часа. Продукт дигидрохлорид (R)-прамипексола можно выделить из реакционной смеси фильтрованием, промыванием спиртом и вакуумной сушкой.
В одном варианте осуществления способа, называемом условиями А в таблице 7 и в примерах, условия реакции, которые могут быть достаточными для образования продукта прамипексола, могут включать нагревание растворенного диамина формулы II при повышенной температуре и непрерывном перемешивании. Повышенная температура, предпочтительно, представляет собой температуру ниже температуры плавления выбранного органического растворителя, ниже приблизительно 125°C, предпочтительно, ниже приблизительно 100°C и, более предпочтительно, приблизительно 95°C. Раствор пропилсульфоната или пропилгалогенида, который растворен в диизопропилэтиламине и органическом растворителе с образованием смеси, медленно добавляют в течение нескольких часов. Указанную реакционную смесь можно затем перемешивать при выбранной температуре в течение дополнительного периода времени, например, в течение приблизительно 4 часов. Время, необходимое для протекания реакции, может варьировать в зависимости от особенностей реагентов и системы растворителей, а также от выбранной температуры, и будет понятно специалисту.
В альтернативном варианте осуществления настоящего изобретения диизопропилэтиламин можно добавлять в реакционную смесь с диамином, а пропилсульфонат или пропилгалогенид можно растворять в органическом растворителе с получением смеси, которую можно добавлять в реакционную смесь при перемешивании в течение нескольких часов. Затем реакционную смесь можно перемешивать при выбранной температуре в течение дополнительного периода времени, например, в течение по меньшей мере 4 часов. Время, необходимое для протекания реакции, может варьировать в зависимости от особенностей реагентов и системы растворителей, а также от выбранной температуры, и будет понятно специалисту.
В альтернативном варианте осуществления способа, называемом условиями В в таблице 7, условия реакции, которые могут быть достаточными для образования продукта прамипексола, могут включать использование диметилформамида в качестве органического растворителя и нагревание растворенного диамина формулы II при повышенной температуре и непрерывном перемешивании. Повышенная температура предпочтительно, представляет собой температуру ниже температуры плавления выбранного органического растворителя, ниже приблизительно 125°C, предпочтительно, ниже приблизительно 100°C и, более предпочтительно, приблизительно 75°C. Раствор пропилсульфоната или пропилгалогенида, который растворен в диметилформамиде, можно медленно добавлять в течение нескольких часов. Указанную реакционную смесь можно затем перемешивать при выбранной температуре в течение дополнительного периода времени, например, в течение приблизительно 4 часов. Время, необходимое для протекания реакции, может варьировать в зависимости от особенностей реагентов и системы растворителей, а также от выбранной температуры, и будет понятно специалисту.
В предпочтительном альтернативном варианте осуществления способа, называемом условиями С в таблице 7, реакционная смесь включает использование диметилформамида в качестве органического растворителя для растворения диамина. Диамин формулы II можно затем нагревать при повышенной температуре и непрерывном перемешивании. Повышенная температура предпочтительно, представляет собой температуру ниже температуры плавления выбранного органического растворителя, ниже приблизительно 125°C, предпочтительно, ниже приблизительно 100°C и, более предпочтительно, приблизительно 65°C. Раствор пропилсульфоната или пропилгалогенида, предпочтительно, в количестве приблизительно 1,25 молярных эквивалента, может быть растворен в диметилформамиде, предпочтительно, приблизительно в 10 объемах, и в диизопропилэтиламине, предпочтительно, в количестве 1,25 молярных эквивалентов, с образованием смеси. Указанную смесь затем медленно добавляют в течение нескольких часов к нагретому диамину. Указанную реакционную смесь можно затем перемешивать при выбранной температуре в течение дополнительного периода времени, например, в течение приблизительно 4 часов. Альтернативно, диизопропилэтиламин можно добавлять в реакционную смесь с диамином, а пропилсульфонат или пропилгалогенид можно растворять в диметилформамиде с получением смеси, которую можно добавлять в реакционную смесь при перемешивании в течение нескольких часов. Указанную реакционную смесь можно затем перемешивать при выбранной температуре в течение дополнительного периода времени, например, в течение приблизительно 4 часов. Время, необходимое для протекания реакции, может варьировать в зависимости от особенностей реагентов и системы растворителей, а также от выбранной температуры, и будет понятно специалисту.
Очистка конечного продукта прамипексола может включать охлаждение реакционной смеси до температуры приблизительно 25°C и перемешивание реакционной смеси в течение определенного периода времени, например, в течение приблизительно 2 часов. Очистка может дополнительно включать фильтрование реакционной смеси для выделения твердого осадка, промывание осадка спиртом и сушку осадка в вакууме. Конечные продукты реакции можно анализировать с использованием высокоэффективной жидкостной хроматографии (ВЭЖХ) на предмет химической и хиральной чистоты.
Кроме того, для подтверждения структуры продукта прамипексола можно использовать 1Н-ЯМР и 13С-ЯМР. Результаты примеров синтеза с использованием каждого из нескольких условий, которые представляют собой варианты осуществления настоящего описания, представлены в таблице 7. Несколько примеров синтеза прамипексола с использованием условий А и С настоящего описания подробно описаны в примерах 5-7.
Сульфонатные или галоидные соли прамипексола можно преобразовать в соль HCl с использованием концентрированного раствора HCl в этаноле. р-TSA соль прамипексола можно повторно растворить в спирте, таком как этанол, и смесь можно охладить до температуры приблизительно от 0 до 5°C при непрерывном перемешивании. Затем можно добавить концентрированную HCl, затем растворитель, такой как метил-трет-бутиловый эфир (МТВЕ), и смесь можно перемешивать в течение часа при температуре приблизительно от 0 до 5°C. Реакционную смесь затем можно фильтровать, промывать раствором МТВЕ/спирта и сушить в вакууме. Конечный продукт представляет собой дигидрохлорид прамипексола. Подробно пример указанного синтеза описан в примере 8.
Альтернативный способ преобразования сульфонатных или галоидных солей прамипексола в соль HCl включает использование концентрированного раствора HCl и изопропилацетата (IPAC). Сульфонатную или галоидную соль прамипексола можно поместить в IPAC и охладить до 15°C. HCl (газообразный) можно барботировать во взвесь в течение приблизительно 1 часа, после чего смесь можно фильтровать, промывать IPAC и сушить в вакууме при комнатной температуре с получением дигидрохлоридной соли прамипексола. Подробно пример указанного синтеза описан в примере 9.
Сульфонатные или галоидные соли прамипексола можно, альтернативно, преобразовать в форму свободного основания прамипексола. Соль р-TSA прамипексола можно растворить в дихлорметане (DCM) и воде. рН полученного раствора затем можно довести приблизительно до 11-12 с использованием NaOH. Можно получить две фазы, и водную фазу можно экстрагировать с использованием DCM, сушить над сульфатом магния (MgSO4), фильтровать через Celite® и концентрировать. Концентрированный остаток можно повторно растворить в МТВЕ и перемешивать в виде взвеси в течение нескольких часов. Твердые вещества затем можно отфильтровать, промыть с использованием МТВЕ и сушить в вакууме при температуре приблизительно 35°C. Конечный продукт представляет собой свободное основание прамипексола. Подробно пример указанного синтеза описан в примере 10.
Альтернативно, сульфонатные или галоидные соли прамипексола можно, альтернативно, преобразовать в форму свободного основания прамипексола вторым способом. В указанном втором способе р-TSA соль прамипексола растворяют в воде и охлаждают до температуры приблизительно 10°C. Указанную взвесь подщелачивают добавлением NaOH, разбавляют насыщенным раствором соли и несколько раз экстрагируют с использованием DCM. Объединенные органические фазы затем промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют досуха. Подробно пример указанного синтеза описан в примере 11.
Форму свободного основания прамипексола можно преобразовать в дигидрохлорид прамипексола барботированием газообразного HCl в охлажденный раствор свободного основания прамипексола в IPAC. Альтернативно, форму свободного основания прамипексола можно преобразовать в дигидрохлорид прамипексола смешиванием с концентрированной HCl при комнатной температуре в течение ночи. Подробные примеры указанных выше схем синтеза описаны в примерах 12 и 13, соответственно.
Альтернативно, форму свободного основания прамипексола можно преобразовать в фумарат прамипексола добавлением 2 молярных эквивалентов фумаровой кислоты.
Эксперименты по SN2 получению р-TSA соли прамипексола
Альтернативный способ получения энантиомерно чистого прамипексола из смеси (R)-прамипексола и (S)-прамипексола включает использование добавления кислоты и растирание (осаждение) энантиомерно чистого прамипексола на основе нерастворимости энантиомеров R(+) и S(-)) в полученном растворе ахиральной соли. В вариантах осуществления указанного способа энантиомерно чистый прамипексол осаждают из кислотно-аддитивного раствора, основываясь на нерастворимости энантиомеров в получаемых ахиральных солях. Указанный вариант осуществления способа получения энантиомерно чистого прамипексола включает растворение энантиомерно обогащенного прамипексола в органическом растворителе при повышенной температуре, добавление выбранной кислоты, охлаждение реакционной смеси до комнатной температуры, перемешивание охлажденной реакционной смеси при комнатной температуре в течение продолжительного периода времени и выделение энантиомерно чистого (R)-прамипексола. В предпочтительном варианте осуществления настоящего изобретения выбранную кислоту можно добавлять в количестве приблизительно от 1 молярного эквивалента до 2 молярных эквивалентов энантиомерно обогащенного прамипексола.
В одном варианте осуществления способа выбранная кислота представляет собой п-толуолсульфоновую кислоту (p-TSA), а органический растворитель представляет собой этанол. В другом варианте осуществления способа повышенная температура может составлять приблизительно от 65°C до 85°C, а охлаждение осуществляют со скоростью приблизительно 25°C в час. Повышенная температура также может быть температурой ниже 125°C, предпочтительно, ниже 100°C и, более предпочтительно, приблизительно 75°C. Время, необходимое для протекания реакции, может варьировать в зависимости от особенностей реагентов, системы растворителей и от выбранной температуры и будет понятно специалисту. В еще одном варианте осуществления способа выделение энантиомерно чистого прамипексола включает охлаждение реакционной смеси до температуры приблизительно 25°C и перемешивание реакционной смеси в течение по меньшей мере приблизительно 2 часов. Выделение может дополнительно включать фильтрование реакционной смеси для выделения твердого осадка, промывание осадка спиртом и сушку осадка в вакууме.
В различных вариантах осуществления способа органический растворитель может включать, без ограничения, ацетонитрил, ацетон, этанол, этилацетат, метил-трет-бутиловый эфир, метилэтилкетон, изопропилацетат и изопропиловый спирт. В предпочтительном варианте осуществления настоящего изобретения органический растворитель представляет собой этанол. Кислота может включать, без ограничения, галогеновые кислоты, такие как, например, бромистоводородная, хлористоводородная, фтористоводородная и йодистоводородная кислота; неорганические кислоты, такие как, например, азотная, хлорная, серная и фосфорная кислота; органические кислоты, такие как, например, сульфоновые кислоты (метансульфоновая, трифторметансульфоновая, этансульфоновая, бензолсульфоновая или п-толуолсульфоновая), уксусная, яблочная, фумаровая, янтарная, лимонная, бензойная, глюконовая, молочная, миндальная, муциновая, памоевая, пантотеновая, щавелевая и малеиновая кислота; и аминокислоты, такие как аспарагиновая или глутаминовая кислота. Кислота может представлять собой моно- или дикислоту, такую как дигидрогалогеновая, дисерная, дифосфорная или диорганическая кислота. Во всех случаях кислоту используют в качестве ахирального агента, которые не выбирают, основываясь на любом ожидаемом или известном преимуществе для взаимодействия с конкретным оптическим изомером продуктов указанного описания или для его осаждения. В предпочтительном варианте осуществления настоящего изобретения выбранная кислота представляет собой п-толуолсульфоновую кислоту.
В вариантах осуществления способа конечная хиральная чистота для R(+)-энантиомера соли прамипексола может составлять более 99%, когда исходная смесь содержит прамипексол, который является по меньшей мере на 55% оптически чистым для R(+)-энантиомера, предпочтительно, на 80% оптически чистым для R(+)-энантиомера, предпочтительно, на 85% оптически чистым для R(+)-энантиомера, более предпочтительно, на 90% оптически чистым для R(+)-энантиомера и, наиболее предпочтительно, на 95% оптически чистым для R(+)-энантиомера. Конечная хиральная чистота для S(-)-энантиомера соли прамипексола может составлять более 99%, когда исходная смесь содержит прамипексол, который является по меньшей мере на 55% оптически чистым для S(-)-энантиомера, предпочтительно, на 80% оптически чистым для S(-)-энантиомера, предпочтительно, на 85% оптически чистым для S(-)-энантиомера, более предпочтительно, на 90% оптически чистым для S(-)-энантиомера и, наиболее предпочтительно, на 95% оптически чистым для S(-)-энантиомера. Хиральная чистота конечной соли прамипексола может предпочтительно составлять 99,6% или выше, 99,7% или выше, предпочтительно, 99,8% или выше и, более предпочтительно, 99,9% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота конечной соли прамипексола может составлять 100%.
В некоторых вариантах осуществления настоящего изобретения, после растворения энантиомерно обогащенного прамипексола в органическом растворителе при повышенной температуре и добавления кислоты, реакционную смесь можно охладить до комнатной температуры со скоростью приблизительно 25°C в час. Затем энантиомерно чистый прамипексол можно выделить из реакционного раствора перемешиванием реакционной смеси в течение по меньшей мере приблизительно 2 часов, фильтрованием реакционной смеси для выделения твердого осадка, промыванием осадка спиртом и сушкой осадка в вакууме. Скорости охлаждения и время, необходимое для дополнительного перемешивания, могут варьировать в зависимости от выбранного органического растворителя и кислоты и будут понятны специалисту. Кроме того, объемы реакции могут диктовать степень оптической очистки и общий выход конечного продукта прамипексола. Указанные объемы будут понятны специалисту. Примеры конкретных периодов времени, температур и объемов, дающих возможность практического осуществления настоящего изобретения, представлены в примерах.
В вариантах осуществления способа хиральная чистота продукта соли прамипексола для R(+)-энантиомера может составлять более 99%, когда хиральная чистота исходной смеси прамипексола для R(+)-энантиомера превышает 55%, предпочтительно, превышает 70% или, более предпочтительно, превышает 90%. Хиральная чистота конечной соли прамипексола может составлять 99,6% или выше, 99,7% или выше, предпочтительно, 99,8% или выше и, более предпочтительно, 99,9% или выше, более предпочтительно, 99,95% или выше, еще более предпочтительно 99,99% или выше. В некоторых вариантах осуществления настоящего изобретения хиральная чистота конечной соли прамипексола может составлять 100%.
Хирально чистый прамипексол можно также получать с использованием способа осаждения единственного энантиомера прамипексола из смеси R(+) и (S)-прамипексола добавлением кислоты, на основе нерастворимости энантиомеров в полученном растворе ахиральной соли. Способ включает растворение энантиомерно обогащенного прамипексола в органическом растворителе при повышенной температуре, добавление приблизительно от 1,05 до 2,05 молярных эквивалентов выбранной кислоты, охлаждение реакционной смеси до комнатной температуры, перемешивание охлажденной реакционной смеси при комнатной температуре в течение продолжительного периода времени и выделение энантиомерно чистого прамипексола.
В вариантах осуществления настоящего изобретения повышенная температура реакционной смеси может быть ниже температуры кипения реакционной смеси, конкретно, ниже температуры кипения органического растворителя (растворителей) реакционной смеси. Повышенная температура может быть ниже приблизительно 125°C, предпочтительно, ниже приблизительно 100°C и, более предпочтительно, приблизительно 75°C. Время, необходимое для протекания реакции, может варьировать в зависимости от особенностей реагентов, системы растворителей и выбранной температуры и будет понятно специалисту.
В вариантах осуществления настоящего изобретения органический растворитель может включать, без ограничения, ацетонитрил, ацетон, этанол, этилацетат, метил-трет-бутиловый эфир, метилэтилкетон, изопропилацетат и изопропиловый спирт. В предпочтительном варианте осуществления настоящего изобретения органический растворитель представляет собой этанол. В указанном варианте осуществления настоящего изобретения кислота может включать, без ограничения, галогеновые кислоты, такие как, например, бромистоводородная, хлористоводородная, фтористоводородная и йодистоводородная кислота; неорганические кислоты, такие как, например, азотная, хлорная, серная и фосфорная кислота; органические кислоты, такие как, например, сульфоновые кислоты (метансульфоновая, трифторметансульфоновая, этансульфоновая, бензолсульфоновая или п-толуолсульфоновая), уксусная, яблочная, фумаровая, янтарная, лимонная, бензойная, глюконовая, молочная, миндальная, муциновая, памоевая, пантотеновая, щавелевая и малеиновая кислота; и аминокислоты, такие как аспарагиновая или глутаминовая кислота. Кислота может представлять собой моно- или дикислоту, такую как дигидрогалогеновая, дисерная, дифосфорная или диорганическая кислота. Во всех случаях кислоту используют в качестве ахирального агента, которые не выбирают, основываясь на любом ожидаемом или известном преимуществе для взаимодействия с конкретным оптическим изомером продуктов указанного описания или для его осаждения. В предпочтительном варианте осуществления настоящего изобретения выбранная кислота представляет собой п-толуолсульфоновую кислоту.
В дополнительных вариантах осуществления настоящего изобретения, после растворения энантиомерно обогащенного прамипексола в органическом растворителе при повышенной температуре и добавления кислоты, реакционную смесь можно охладить до комнатной температуры со скоростью приблизительно 25°C в час. Затем хирально чистый прамипексол можно выделить из реакционного раствора перемешиванием реакционной смеси в течение по меньшей мере приблизительно 2 часов, фильтрованием реакционной смеси для выделения твердого осадка, промыванием осадка спиртом и сушкой осадка в вакууме. Скорости охлаждения и время, необходимое для дополнительного перемешивания, могут варьировать в зависимости от выбранного органического растворителя и кислоты и будут понятны специалисту. Кроме того, объемы реакции могут диктовать степень оптической очистки и общий выход конечного продукта прамипексола. Указанные объемы будут понятны специалисту. Примеры конкретных периодов времени, температур и объемов, дающих возможность практического осуществления настоящего изобретения, представлены в примерах.
В вариантах осуществления способа хиральная чистота для R(+)-энантиомера выделенной соли прамипексола может составлять более 99%, когда исходный материал прамипексол имеет хиральную чистоту для R(+)-энантиомера выше 55%, предпочтительно, выше 70% или, более предпочтительно, выше 90%. Хиральная чистота конечной соли прамипексола для R(+)-энантиомера может составлять 99,6% или выше, 99,7% или выше, предпочтительно, 99,8% или выше и, более предпочтительно, 99,9% или выше, более предпочтительно, 99,95% или выше, еще более предпочтительно, 99,99% или выше. В наиболее предпочтительных вариантах осуществления настоящего изобретения хиральная чистота конечной соли прамипексола для R(+)-энантиомера может составлять 100%.
Способ может включать растворение энантиомерно обогащенного прамипексола в органическом растворителе при повышенной температуре, добавление приблизительно от 1,05 до 2,05 молярных эквивалентов выбранной кислоты, охлаждение реакционной смеси до комнатной температуры, перемешивание охлажденной реакционной смеси при комнатной температуре в течение продолжительного периода времени и выделение энантиомерно чистого прамипексола формулы I.
В одном варианте осуществления способа выбранная кислота представляет собой п-толуолсульфоновую кислоту (p-TSA), а органический растворитель представляет собой этанол. В другом варианте осуществления способа повышенная температура может составлять приблизительно от 65°C до 85°C, и охлаждение осуществляют со скоростью приблизительно 25°C в час. Повышенная температура также может быть температурой ниже 125°C, предпочтительно, ниже 100°C и, более предпочтительно, приблизительно 75°C. Время, необходимое для протекания реакции, может варьировать в зависимости от особенностей реагентов, системы растворителей и от выбранной температуры и будет понятно специалисту. В еще одном варианте осуществления способа выделение энантиомерно чистого прамипексола включает охлаждение реакционной смеси до температуры приблизительно 25°C и перемешивание реакционной смеси в течение по меньшей мере приблизительно 2 часов. Выделение может дополнительно включать фильтрование реакционной смеси для выделения твердого осадка, промывание осадка спиртом и сушку осадка в вакууме.
В различных вариантах осуществления способа органический растворитель может включать, без ограничения, ацетонитрил, ацетон, этанол, этилацетат, метил-трет-бутиловый эфир, метилэтилкетон, изопропилацетат и изопропиловый спирт. В предпочтительном варианте осуществления настоящего изобретения органический растворитель представляет собой этанол. Кислота может включать, без ограничения, галогеновые кислоты, такие как, например, бромистоводородная, хлористоводородная, фтористоводородная и йодистоводородная кислота; неорганические кислоты, такие как, например, азотная, хлорная, серная и фосфорная кислота; органические кислоты, такие как, например, сульфоновые кислоты (метансульфоновая, трифторметансульфоновая, этансульфоновая, бензолсульфоновая или п-толуолсульфоновая), уксусная, яблочная, фумаровая, янтарная, лимонная, бензойная, глюконовая, молочная, миндальная, муциновая, памоевая, пантотеновая, щавелевая и малеиновая кислота; и аминокислоты, такие как аспарагиновая или глутаминовая кислота. Кислота может представлять собой моно- или дикислоту, такую как дигидрогалогеновая, дисерная, дифосфорная или диорганическая кислота. Во всех случаях кислоту используют в качестве ахирального агента, которые не выбирают, основываясь на любом ожидаемом или известном преимуществе для взаимодействия с конкретным оптическим изомером продуктов указанного описания или для его осаждения. В предпочтительном варианте осуществления настоящего изобретения выбранная кислота представляет собой п-толуолсульфоновую кислоту.
В вариантах осуществления способа конечная хиральная чистота для R(+)-энантиомера соли прамипексола может составлять более 99%, когда исходная смесь содержит прамипексол, который является по меньшей мере на 55% оптически чистым для R(+)-энантиомера, предпочтительно, на 80% оптически чистым для R(+)-энантиомера, предпочтительно, на 85% оптически чистым для R(+)-энантиомера, более предпочтительно, на 90% оптически чистым для R(+)-энантиомера и, наиболее предпочтительно, на 95% оптически чистым для R(+)-энантиомера. Конечная хиральная чистота для S(-)-энантиомера соли прамипексола может составлять более 99%, когда исходная смесь содержит прамипексол, который является по меньшей мере на 55% оптически чистым для S(-)-энантиомера, предпочтительно, на 80% оптически чистым для S(-)-энантиомера, предпочтительно, на 85% оптически чистым для S(-)-энантиомера, более предпочтительно, на 90% оптически чистым для S(-)-энантиомера и, наиболее предпочтительно, на 95% оптически чистым для S(-)-энантиомера. Хиральная чистота конечной соли прамипексола может, предпочтительно, составлять 99,6% или выше, 99,7% или выше, предпочтительно, 99,8% или выше и, более предпочтительно, 99,9% или выше. В более предпочтительных вариантах осуществления настоящего изобретения хиральная чистота конечной соли прамипексола может составлять 100%.
Результаты примеров очистки с использованием каждого варианта нескольких условий, которые являются вариантами осуществления настоящего описания, представлены в таблице 8.
Эксперименты по получению R(+)-энантиомера прамипексола
Химическая и хиральная чистота препаратов (R)-прамипексола может быть подтверждена по меньшей мере данными ВЭЖХ, 13С-ЯМР, 1Н-ЯМР и FTIR. В предпочтительных вариантах осуществления настоящего изобретения (R)-прамипексол может быть синтезирован способом, описанным выше, который дает энантиомерно чистый продукт. Альтернативно, (R)-прамипексол может быть выделен из смесей R(+) и (S)-прамипексола с использованием схемы очистки, которая описана в находящейся одновременно на рассмотрении временной заявке США № 60/894829, озаглавленной «Способы синтеза и очистки (R)(+) и (S)-прамипексола», поданной 14 марта 2007 г., и во временной заявке США № 60/894814, озаглавленной «Способы энантиомерной очистки хиральных соединений», поданной 14 марта 2007 г., которые полностью включены в настоящий документ путем ссылки.
Для лучшего объяснения, не ограничиваясь теорией, растворимость (R)-прамипексола и (S)-прамипексола может быть одинаковой на стадии осаждения в процессах синтеза и очистки. В качестве примера, если процесс синтеза осуществляют с использованием 90 граммов R(+)-диамина и 10 граммов S(-)-диамина, а растворимость конечного продукта прамипексола составляет 10 граммов для каждого энантиомера, то 80 граммов продукта (R)-прамипексола и 0 граммов продукта (S)-прамипексола выпадут в осадок (допуская 100% химическое преобразование из диамина и отсутствие изменения молекулярной массы в процессе получения продукта прамипексола). Иными словами, можно ожидать, что 10 граммов каждого энантиомера прамипексола перейдут в раствор. Это приведет к получению продукта прамипексола 100% хиральной чистоты для R(+)-энантиомера. Противоположное соотношение исходных продуктов для процесса синтеза (90 граммов S(-)-диамина и 10 граммов R(+)-диамина) может дать продукт реакции из 90 граммов (S)-прамипексола и 10 граммов (R)-прамипексола. Можно ожидать, что из данной смеси продуктов реакции 80 граммов S(-)-энантиомера и 0 граммов R(+)-энантиомера прамипексола выпадут в осадок, что приведет к получению продукта прамипексола 100% хиральной чистоты для S(-)-энантиомера. С помощью данного мысленного эксперимента можно представить, что объемы, которые используются для реакции, могут оказывать большое потенциальное влияние на конечный выход и хиральную чистоту. Значит, слишком большой объем будет уменьшать выход, поскольку больше энантиомерных продуктов прамипексола будут переходить в раствор (но будет повышать хиральную чистоту), а слишком малый объем будет увеличивать выход, поскольку меньше энантиомерных продуктов прамипексола будут переходить в раствор (но будет понижать хиральную чистоту).
Для лучшего определения фактических ограничений объемов реакции и величины оптической чистоты, которые можно получить с использованием способов по настоящему описанию, были испытаны различные соотношения хиральной чистоты для исходного продукта диамина. Как показано в таблице 9, способ синтеза и очистки тестировали с использованием следующих соотношений исходных R(+) и S(-)-диаминов: 80:20, 20:80, 85:15, 15:85, 90:10, 10:90, 95:5 и 5:95. Дополнительно, тестировали три конкретных варианта условий реакции, в которых изменяли или объем реакции, или стадию выделения после реакции. Данные испытания показали, что энантиомеры прамипексола являются одинаково нерастворимыми (или растворимыми) в органических растворителях, которые использовали в различных вариантах осуществления способов синтеза, описанных в настоящем документе.
Эксперименты по SN2 получению чистых энантиомеров прамипексола
Условия Е: Реакцию осуществляли в 10 объемах ДМФА и 1,25 эквивалента пропилтозилата при 65-67°C. Реакционную смесь затем охлаждали до комнатной температуры без разбавления МТВЕ.
Данные, приведенные в таблице 9, показывают, что оба энантиомера прамипексола обладают сходной, если не одинаковой, растворимостью. Кроме того, данные показывают, что синтез является одинаково эффективным для каждого из энантиомеров прамипексола. Указанные данные также показывают, что энантиомеры ведут себя независимо друг от друга в том, что на растворимость одного энантиомера, как представляется, не влияет концентрация в растворе другого энантиомера. Например, различные реакции синтеза, осуществлявшиеся с использованием условий С, имели химический выход приблизительно 50%, независимо от процентной доли преобладающего энантиомера диамина в исходном продукте. В случае, когда объем используемого в реакции синтеза органического растворителя возрастает, химический выход уменьшается, но хиральный выход увеличивается. Это становится очевидным при сравнении реакции, осуществляемой при условиях С и D, когда соотношение диаминов R(+):S(-) 85:15 давало продукт прамипексол, имеющий 86,8% хиральной чистоты для R(+)-энантиомера, если в реакции использовали 10 объемов органического растворителя, и 99,8% хиральной чистоты для R(+)-энантиомера, если в реакции использовали 18 объемов органического растворителя. Следует заметить также, что химический выход уменьшался в реакции с использованием большего объема органического растворителя (43% выход при условиях С и 36% выход при условиях D).
В таблице 9 условия Е такие же, как и условия С, за исключением того, что стадия выделения не включает разбавление с использованием МТВЕ. МТВЕ, как было установлено, увеличивает выделение (выход) прамипексола из реакции синтеза, но может уменьшить общую хиральную чистоту. К данному выводу можно придти путем сравнения результатов испытаний, осуществляемых при соотношении диаминов R(+):S(-) 85:15, которое давало продукт прамипексол, имеющий 86,8% хиральной чистоты для R(+)-энантиомера, если реакционная смесь включала органический растворитель МТВЕ, и 99,9% хиральной чистоты для R(+)-энантиомера, если реакционная смесь не включала органический растворитель МТВЕ. Химический выход уменьшался за счет исключения разбавления с использованием МТВЕ на стадии выделения; 43% выход при условиях С по сравнению с 39% выходом при условиях Е.
Хирально чистый (R)-прамипексол, полученный любым из описанных выше способов, можно преобразовать в фармацевтически приемлемую соль (R)-прамипексола. Например, дигидрохлорид (R)-прамипексола представляет собой предпочтительную фармацевтическую соль благодаря своей высокой растворимости в воде. Дигидрохлорид (R)-прамипексола можно получить из других солей (R)-прамипексола одностадийным способом, включающим взаимодействие (R)-прамипексола или соли (R)-прамипексола с концентрированной HCl в органическом растворителе, таком как спирт, при пониженной температуре. В некоторых вариантах осуществления настоящего изобретения пониженная температура представляет собой температуру приблизительно от 0°C до 5°C. Можно добавить органический растворитель, такой как метил-трет-бутиловый эфир, и реакционную смесь можно перемешивать в течение еще одного часа. Продукт дигидрохлорид (R)-прамипексола можно выделить из реакционной смеси фильтрованием, промыванием спиртом и вакуумной сушкой.
Каждый из способов, описанных в настоящем документе, для получения и очистки (R)-прамипексола или его фармацевтически приемлемой соли может быть масштабирован, чтобы получить количества и выходы в промышленном масштабе, дающем продукты с высокой химической и хиральной чистотой. В некоторых вариантах осуществления настоящего изобретения энантиомерно чистый (R)-прамипексол можно получать большими партиями, которые могут потребоваться для крупномасштабного фармацевтического использования.
Различные аспекты настоящего изобретения далее будут проиллюстрированы с помощью следующих неограничивающих примеров.
ПРИМЕРЫ
ПРИМЕР 1 - Измерение величин аффинности R(+) и S(-)-энантиомеров прамипексола в отношении дофаминовых рецепторов.
S(-)-энантиомер прамипексола исторически характеризуется как высокоаффинный лиганд дофаминовых рецепторов D2 (как S-, так и L-изоформ), D3 и D4, хотя наибольшая аффинность наблюдается для подтипа рецепторов D3. Лигандная аффинность (S)-прамипексола и (R)-прамипексола в отношении дофаминовых рецепторов из журнальных публикаций была занесена в таблицу (данные представлены в таблице 10). Хотя условия, при которых осуществляли каждое исследование или эксперимент, слегка отличаются, и использовали различные радиолиганды, данные показывают сравнимые величины аффинности для различных дофаминовых рецепторов. Исследования, предпринятые заявителями, касающиеся величин аффинности S(-) и R(+)-энантиомеров прамипексола в отношении дофаминовых рецепторов, также показаны в таблице 10. Указанные данные демонстрируют неожиданно большую разницу в величинах аффинности двух энантиомеров прамипексола в отношении всех дофаминовых рецепторов. Таблица 10 показывает, что вместо ожидаемой 10-20-кратной разницы в связывающей аффинности в отношении рецепторов D2 и 50-кратной разницы в связывающей аффинности в отношении рецепторов D3, как следовало из литературных данных, величины, установленные заявителями, были обычно в 10 раз выше (в 290 и 649 раз, соответственно) (таблица 10).
Сравнительные данные по связывающей аффинности энантиомеров прамипексола
**Wong, S.K.-F.; Shrikhande, A.V., S.K.-F. Wong, "Activation of Extracellular Signal-Regulated Kinase by Dopamine D2 and D3 Receptors",. (2003) Society for Neuroscience Abstracts
***Данные из текущего исследования
(R)-прамипексол и (S)-прамипексол поставляли заявителям по контракту с партнером по исследованиям Cerep производителем AMRI. Растворы (R)-прамипексола и (S)-прамипексола готовили из маточных растворов в ДМСО. Восемь концентраций (R)-прамипексола или (S)-прамипексола (0,01 нМ, 0,1 нМ, 1 нМ, 10 нМ, 100 нМ, 1 мМ, 10 мМ и 100 мМ) использовали для вытеснения стандартных радиоактивно меченных агонистов дофамина. Указанные концентрации тестировали на клеточных линиях, экспрессирующих выбранные человеческие клонированные дофаминовые рецепторы (D2S, D3). Ранние работы, представленные в литературе, и исследования заявителей показали отсутствие значимого взаимодействия с дофаминовыми рецепторами D1 и D5. Результаты по группам для взаимодействия (R)-прамипексола или (S)-прамипексола с каждым видом рецепторов выражены в виде IC50 в таблице 10.
Указанные данные показывают, что величины IC50 (R)-прамипексола в отношении указанных рецепторов приблизительно в 290-649 раз превышают величины IC50 для (S)-прамипексола. Кроме того, указанные данные предполагают, что соотношение величин IC50 для (R)-прамипексола и (S)-прамипексола в отношении рецептора D2 приблизительно в 14-32 раза превышает соотношения, указанные в литературе, по меньшей мере, когда величины хиральной чистоты выходят за пределы обнаружения (LOD 0,05%) (хиральная чистота выше 99,95%). Аналогично, данные предполагают, что соотношение величин IC50 для (R)-прамипексола и (S)-прамипексола в отношении рецептора D3 приблизительно в 13 раз превышает соотношения, указанные в литературе, по меньшей мере, когда величины хиральной чистоты выходят за пределы обнаружения (хиральная чистота выше 99,95%). Указанные данные также предполагают, что если аффинность в отношении дофаминовых рецепторов является основным фактором, который вносит свой вклад в лимитирующую переносимость дозы S(-)-энантиомера, то чистые препараты R(+)-энантиомера должны иметь максимально переносимую дозу (MTD) и/или дозу с уровнем, не вызывающим видимых неблагоприятных эффектов (NOAEL), по меньшей мере в 290 раз выше, чем MTD и/или NOAEL для S(-)-энантиомера. Таким образом, даже небольшое загрязнение композиций (R)-прамипексола по настоящему изобретению S(-)-энантиомером на таких низких уровнях, как 0,5% или менее, может влиять на наблюдаемые MTD и NOAEL.
ПРИМЕР 2 - Исследования in vivo для определения MTD и NOAEL на собаках для 100% чистых препаратов R(+) и S(-)-энантиомеров прамипексола и смеси (R 99,5%/S 0,5%). Форма (R)-прамипексола представляла собой моногидрат дигидрохлорида (R)-прамипексола.
Следующие исследования in vivo на собаках породы бигль были предприняты для того, чтобы проверить гипотезу о том, что большая видимая разница в величинах связывающей аффинности в отношении рецепторов для R(+) и S(-)-энантиомеров прамипексола будет преобразовываться в большую видимую разницу в величинах максимально переносимой дозы (MTD) и/или уровня, не вызывающего видимых неблагоприятных эффектов (NOAEL) двух энантиомеров. Собакам вводили препараты каждого энантиомера, полученные в виде соединения высокой очистки (100% чистые препараты (в пределах аналитического обнаружения)), или препарат R(+)-энантиомера, загрязненный 0,5% S(-)-энантиомера прамипексола.
Три группы из четырех подвергавшихся ранее лечению собак породы бигль мужского пола использовали в исследовании. Каждой группе вводили различные дозы R(+) или S(-)-энантиомера, полученного в виде соединения высокой очистки, или препарат R(+)-энантиомера, загрязненный 0,5% S(-)-энантиомера прамипексола. Дозы вводили перорально с использованием желудочного зонда и осуществляли клинические наблюдения непрерывно после введения: каждый час в течение первых четырех часов, а затем дважды в день, вблизи клеток, в течение периода времени между введением доз или интервала после введения дозы. Наблюдали за клиническими признаками, смертностью, повреждениями и доступностью пищи и воды. Животные голодали в течение 24 часов перед введением лекарственного средства. Собаки в каждой группе получали только одно лекарственное средство, или комбинацию; каждую дозу вводили только один раз, с последующим введением дозы после периода восстановления в 4 дня. Данные суммированы в таблице 11.
Значение NOAEL было установлено на уровне 25 мг/кг для R(+)-энантиомера для собак, которые ранее подвергались лечению, в то время как уровень 75 мг/кг может рассматриваться как MTD для собак, которые ранее подвергались лечению. Для S(-)-энантиомера было установлено значение NOAEL 0,00125 мг/кг и MTD 0,0075 мг/кг для собак, которые ранее подвергались лечению. Для композиции, содержащей смесь двух энантиомеров (99,5% (R)-прамипексола и 0,5% (S)-прамипексола), NOAEL, как было установлено, составляет 0,25 мг/кг, что соответствует дозе 0,00125 мг/кг S(-)-энантиомера, в то время как MTD составляет 1,5 мг/кг, что соответствует дозе 0,0075 мг/кг S(-)-энантиомера. Указанные данные показывают, что NOAEL для R(+)-энантиомера прамипексола приблизительно в 20000 раз выше, чем для S(-)-энантиомера для собак, которые ранее подвергались лечению.
Клинические наблюдения собак породы бигль мужского пола, которым вводили композиции прамипексола
R(+) (День 1)
R(+) (День 4)
R(+) (День 8)
смесь (День 4)
** Смесь 99,5% (R)-прамипексола и 0,5% (S)-прамипексола
Данные, представленные в таблице 11, показывают, что идентифицированные величины аффинности в отношении рецепторов (см. таблицу 10) напрямую вносят свой вклад в видимые различия в дозах MTD и NOAEL для R(+) и S(-)-энантиомеров прамипексола. Указанные данные также показывают, что может потребоваться хиральная чистота R(+)-энантиомера прамипексола в вариантах осуществления композиций по настоящему изобретению (см. таблицы 5 и 6), превышающая 99,9%, в зависимости от конечной суммарной дозы, чтобы избежать неблагоприятных побочных эффектов (S)-прамипексола.
Кроме того, данные таблицы 11 показывают, что NOAEL и MTD для комбинированной композиции (99,5% (R)-прамипексола и 0,5% (S)-прамипексола) могут быть определены непосредственно дозой S(-)-энантиомера в композиции. Таким образом, малое (доли процента) загрязнение композиции (R)-прамипексола S(-)-энантиомером может уменьшить MTD и NOAEL композиции. Например, в указанных экспериментах MTD прамипексола была снижена с 75 мг/кг для R(+)-энантиомера до суммарной дозы 1,5 мг/кг смешанной композиции (в 50 раз), а NOAEL был снижен с 25 мг/кг до 0,25 мг/кг, соответственно (в 100 раз). Поскольку сдвиг в MTD и NOAEL может быть предсказан по дозе S(-)-энантиомера прамипексола в смеси, сдвиг для любой неизвестной смеси можно рассчитать на основе процентной доли загрязнения (R)-прамипексола S(-)-энантиомером, относительно MTD и NOAEL для (S)-прамипексола. Это показывает, что любое загрязнение дозирующего раствора (R)-прамипексола (S)-прамипексолом будет оказывать поддающееся измерению влияние на указанные показатели переносимости дозы.
ПРИМЕР 3.1 - Токсикологические исследования на крысах и минипигах и фаза I испытаний на здоровых взрослых добровольцах. Были завершены двухнедельные и трехмесячные токсикологические исследования (R)-прамипексола на крысах и минипигах. Были установлены дозовые уровни NOAEL 150 мг/кг в двухнедельных исследованиях и 100 мг/кг в трехмесячных исследованиях для крыс, и 75 мг/кг в двухнедельных исследованиях и 50 мг/кг в трехмесячных исследованиях для минипигов. Фаза I испытаний на здоровых взрослых добровольцах показала, что (R)-прамипексол в возрастающих однократных дозах до 300 мг и в многократных дозах до 200 мг в день в течение 4,5 дней являются безопасными и хорошо переносятся. Этикетка Mirapex® устанавливает начальную дозу 0,125 мг и максимальную суточную дозу 4,5 мг. Данные фазы I показывают, таким образом, что (R)-прамипексол можно безопасно вводить (1) в начальной дозе, которая по меньшей мере в 2400 раз выше начальной дозы Mirapex®, и (2) в установившемся режиме дозы, которая по меньшей мере в 44 раза выше высшей рекомендованной дозы Mirapex®. Форма (R)-прамипексола представляла собой моногидрат дигидрохлорида (R)-прамипексола.
Далее обсуждаются предварительные результаты клинических испытаний и токсикологических исследований. Воздействие в установившемся режиме на крысах, минипигах и людях является линейным для всех изученных доз. Через 3 месяца введения лекарственного средства подлинное дозовое количество с уровнем, не вызывающим видимых неблагоприятных эффектов (NOAEL) для крыс было определено на уровне 100 мг/кг; а подлинное значение NOAEL для минипигов было определено на уровне 50 мг/кг. Средняя AUC в установившемся режиме для крыс при дозе NOAEL 100 мг/кг составляла 61299 и 61484 ч*нг/мл для самцов и самок, соответственно, а для минипигов при дозе NOAEL 50 мг/кг составляла 91812 и 131731 ч*нг/мл для самцов и самок, соответственно. Средняя AUC в установившемся режиме для людей при дозе 100 мг каждые 12 часов (суммарная суточная доза 200 мг) составляла 2574 ч*нг/мл. Лекарственное средство было безопасным, хорошо переносилось и не вызывало клинически значимых неблагоприятных событий у взрослых здоровых субъектов при использовании однократных доз до 300 мг и многократных доз до 100 мг каждые 12 часов, и прогнозируемое воздействие на человека, связанное с суточной дозой 250 мг каждые 12 часов, как ожидается, будет более чем в 13 раз ниже, чем воздействия, наблюдаемые при NOAEL у минипигов-самцов, и приблизительно в 9 раз ниже, чем воздействия, наблюдаемые при NOAEL у самцов и самок крыс после 13 недель введения лекарственного средства.
3.2 - Клинические испытания. (R)-прамипексол изучали с использованием однократных суточных доз 50, 150 и 300 мг и двух доз в день 50 и 100 мг в течение 4,5 дней на здоровых взрослых добровольцах. Лекарственное средство было безопасным и хорошо переносилось в обоих исследованиях, и не наблюдалось серьезных неблагоприятных событий, прекращения участия в испытаниях из-за неблагоприятных событий или дозозависимых или клинически значимых неблагоприятных событий в обоих исследованиях. Наиболее частыми неблагоприятными событиями были головокружение и головная боль, которые имели степень тяжести от легкой до средней и разрешались без вмешательства.
3.2.1 - Краткое изложение (слепых) результатов по безопасности и фармакокинетике (R)-прамипексола (исследование с использованием возрастающих однократных доз). Три последовательные группы из 8 субъектов получали однократные дозы (R)-прамипексола (6 субъектов) или плацебо (2 субъекта) в возрастающих дозах 50, 150 и 300 мг. Наблюдения на предмет безопасности включали основные показатели жизненных функций, физикальное исследование, клинические лабораторные анализы, ЭКГ и сообщения о неблагоприятных событиях. Образцы крови и мочи отбирали перед введением дозы и в течение 72 часов после введения дозы для оценки фармакокинетики. Все 24 субъекта завершили свое участие в испытаниях, как планировалось. Серьезных неблагоприятных событий не наблюдалось; 46% всех субъектов сообщили по меньшей мере об одном нетяжелом неблагоприятном событии (АЕ). Большинство АЕ были легкими; наиболее частым АЕ было легкое головокружение у 21% субъектов. Клинически значимых событий, касающихся безопасности, не наблюдалось при использовании всех доз.
Фармакокинетические данные показали, что (R)-прамипексол быстро всасывается и достигает средних максимальных концентраций 125, 360 и 781 нг/мл приблизительно через 2 часа после введения лекарственного средства в группах, получавших дозы 50, 150 и 300 мг, соответственно (см. фиг.1 и таблицу 12, ниже). Среднее воздействие (AUC0-∝) составляло 1254, 3815 и 8623 ч*нг/мл для групп, получавших дозы 50, 150 и 300 мг, соответственно. Как Cmax, так и AUC возрастали пропорционально дозе по всем изучавшимся уровням доз. Выделение с мочой неизмененного лекарственного средства составляло приблизительно 70% элиминации лекарственного средства по всем уровням доз. Среднее Т1/2 составляло 6-7 часов и зависело от дозы. Сравнение средних концентраций в плазме (фиг.2) и средних фармацевтических параметров (таблица 12) после введения однократной дозы 150 мг после завтрака с высоким содержанием жиров и калорий и тех же параметров после введения дозы 150 мг натощак показывает практическое отсутствие влияние пищи на всасывание и элиминацию (R)-прамипексола.
Результаты данного исследования показывают, что однократные пероральные дозы 50, 150 и 300 мг (R)-прамипексола являются безопасными и хорошо переносятся. Лекарственное средство является биодоступным при пероральном введении, а фармакокинетика является линейной. На всасывание и элиминацию не влияет пища с высоким содержанием жиров и калорий.
Таблица 12
Краткое изложение фармакокинетических параметров для (R)-прамипексола после перорального введения здоровым добровольцам однократных доз 50 мг, 150 мг и 300 мг натощак и 150 мг после приема пищи
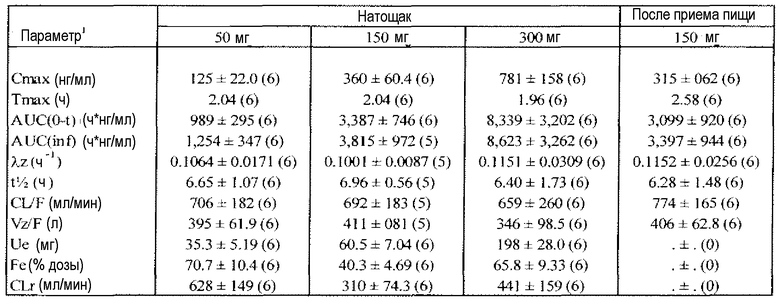
1 Среднее ± стандартное отклонение (N), кроме Tmax, для которого приводится медиана (N)
3.2.2 - Краткое изложение (слепых) результатов по безопасности и фармакокинетике (R)-прамипексола (исследование с использованием возрастающих многократных доз). Данное исследование в настоящее время продолжается и все еще является слепым в отношении распределения по видам лечения, и доступными на данный момент являются лишь клинические наблюдения и фармакокинетические данные по первым двум группам. К настоящему времени 2 последовательные группы по 8 субъектов каждая были зачислены в группы, получавшие многократные дозы (R)-прамипексола (6 субъектов) или плацебо (2 субъекта). Первая группа получала однократную дозу 50 мг, с последующими через 48 часов в течение 4,5 дней многократными (дважды в день) дозами по 50 мг каждые 12 часов. Вторая группа получала однократную дозу 100 мг, с последующими через 48 часов в течение 4,5 дней многократными (дважды в день) дозами по 100 мг каждые 12 часов. Наблюдения на предмет безопасности включали основные показатели жизненных функций, физикальное исследование, клинические лабораторные анализы, ЭКГ и сообщения о неблагоприятных событиях. Образцы крови отбирали перед введением дозы в день 1 и периодически в течение 48 часов после введения дозы для оценки фармакокинетики однократной дозы. Образцы крови отбирали перед введением дозы в дни 5, 6 и 7 для подтверждения достижения установившегося состояния и периодически в течение 72 часов после введения дозы в день 7 для оценки установившегося состояния фармакокинетики (R)-прамипексола. Образцы мочи отбирали в течение 12 часов после введения дозы в день 7 для оценки экскреции с мочой.
Все 16 субъектов, зачисленных для участия, к настоящему времени завершили исследование, как планировалось. Не наблюдалось смертей, сообщений о тяжелых неблагоприятных событиях или прекращений участия из-за неблагоприятных событий во время исследования. Оба уровня дозы хорошо переносились. В группе 1 все неблагоприятные события были легкими, за исключением умеренных головных болей, о которых сообщили 2 субъекта. В группе 2 все неблагоприятные события были легкими, за исключением умеренной «неподвижности в спине» и умеренной вазовагальной реакции, о которых сообщил 1 субъект. О бессимптомном легком повышении частоты сердечных сокращений при стоянии (без изменений кровяного давления) сообщил главный исследователь для 1 из 8 субъектов в группе 1 (получавшей 50 мг) и для 2 из 8 субъектов в группе 2 (получавшей 100 мг). Клинически значимых событий, касающихся безопасности, не наблюдалось при использовании всех доз.
Фармакокинетические данные показаны в таблице 13 и на фиг.3. Cmax и AUC(0-12) возрастали на 37% и 40%, соответственно, со дня 1 по день 7 для субъектов, получавших 50 мг каждые 12 часов, при практическом отсутствии изменений в Tmax. Среднее воздействие AUC(0-12) в день 7 составляло 1449 ч*нг/мл для группы, получавшей 50 мг каждые 12 часов. Cmax и AUC(0-12) возрастали на 24% и 38%, соответственно, со дня 1 по день 7 для субъектов, получавших 100 мг каждые 12 часов, при практическом отсутствии изменений в Tmax. Среднее воздействие AUC(0-12) в день 7 составляло 2465 ч*нг/мл для группы, получавшей 100 мг каждые 12 часов. Результаты данного исследования показывают, что многократные пероральные дозы 50 и 100 мг (R)-прамипексола, которые вводят два раза в день, являются безопасными и хорошо переносятся. Лекарственное средство является биодоступным при пероральном введении, а фармакокинетика является линейной в установившемся состоянии, без значимой аккумуляции.
Таблица 13
Краткое изложение фармакокинетических параметров для (R)-прамипексола после перорального введения натощак здоровым добровольцам доз 50 мг и 300 мг в день 1, каждые 12 часов в дни с 3 по 6 и однократной дозы в день 7
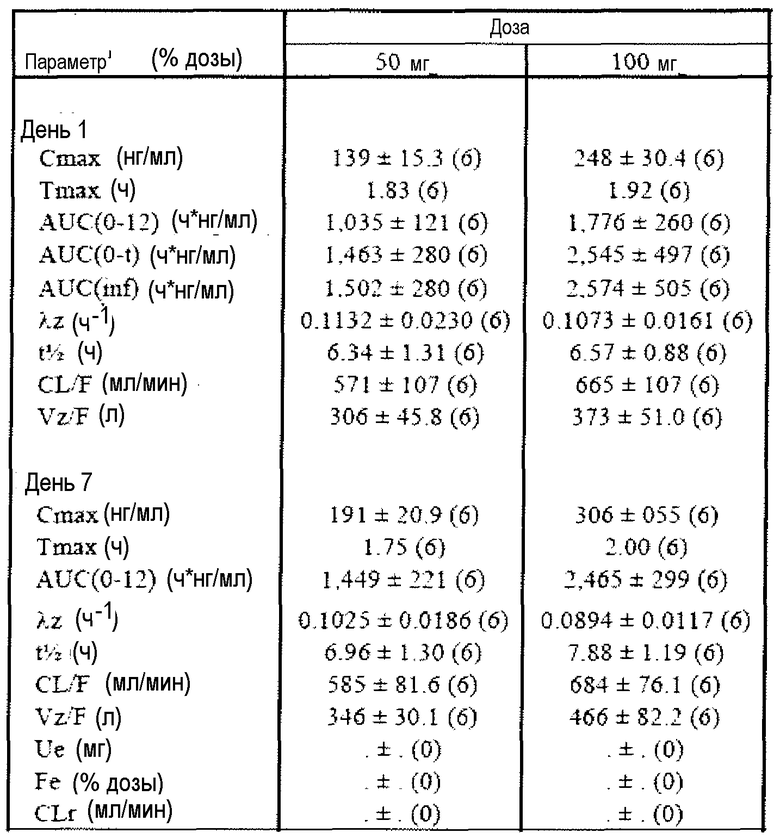
1Среднее ± стандартное отклонение (N), кроме Tmax, для которого приводится медиана (N)
3.3 - Токсикологические исследования. В токсикологических исследованиях, проводимых на крысах с использованием 2-недельных повторных доз, животные получали дозы (R)-прамипексола 50, 150 и 500 мг/кг в течение 14 дней. (R)-прамипексол вызывал смертность в высокой дозе 500 мг/кг, и статистически значимые изменения в прибавке массы тела и потреблении пищи для обоих полов наблюдались у животных, выживших до запланированного умерщвления. Не было идентифицировано токсичности в отношении органов-мишеней по результатам гистопатологического исследования при всех дозах. Было определено значение NOAEL для указанного 2-недельного исследования на крысах, составляющее 150 мг/кг. После указанного исследования 3- и 6-месячные токсикологические исследования с использованием повторных доз были проведены с дозами 30, 100 и 300 мг/кг. Результаты 3-месячного исследования показали некоторую токсичность в отношении органов-мишеней по результатам гистопатологического исследования при высшей дозе (300 мг/кг), при отсутствии смертей, связанных с испытуемым лекарственным средством и без значимых клинических наблюдений, кроме нескольких случаев судорог при использовании высокой дозы у крыс, которые продолжались приблизительно 2 минуты. Здоровье животных, как представляется, никак не пострадало вследствие произошедших судорог. Микроскопические изменения, связанные с испытуемым лекарственным средством, наблюдались в печени (минимальный холестаз, коррелирующий с повышением общего билирубина), в подвздошной кишке (минимальная минерализация) и в тимусе (минимальная лимфопения, коррелирующая с более низкими массами тимуса по сравнению с контролями). Было определено значение NOAEL для 3-месячного исследования на крысах, составляющее 100 мг/кг. Системное воздействие (AUC0-последний) на неделе 13 при дозе NOAEL 100 мг/кг составляло 61299 ч*нг/мл для самцов и 61484 ч*нг/мл для самок. Прижизненная фаза 6-месячного токсикологического исследования на крысах недавно была завершена, и результаты гистопатологических исследований ожидаются. Не наблюдалось смертности при всех уровнях доз среди животных, умерщвленных между 13 и 26 неделями.
В токсикологических исследованиях, проводимых на минипигах с использованием 2-недельных повторных доз, животные получали дозы (R)-прамипексола 7,5, 25 и 75 мг/кг в течение 14 дней. Не было идентифицировано токсичности в отношении органов-мишеней по результатам гистопатологического исследования при всех дозах. Клинические наблюдения включали слюнотечение, пониженную активность, рвоту и отсутствие аппетита; рвота наблюдалась чаще у самок, чем у самцов, и, главным образом, в группе, получавшей 75 мг/кг. Частота рвоты в группе, получавшей 75 мг/кг (по меньшей мере один эпизод у 5 из 8 животных, получавших дозу 75 мг/кг) наводит на мысль о том, что указанная доза является близкой к пределу переносимости для (R)-прамипексола при постоянном введении минипигам. Поскольку токсикологических изменений, связанных с испытуемым лекарственным средством, при высокой дозе не наблюдалось, значение NOAEL для 2-недельного исследования было расценено как превышающее или равное 75 мг/кг. На основе данного исследования 3-, 6- и 9-месячные токсикологические исследования с использованием повторных доз (R)-прамипексола были начаты с дозами 7,5, 25 и 75 мг/кг. На 2 месяц уровни доз были уменьшены до 7,5, 25 и 50 мг/кг из-за смертности при использовании доз 75 мг/кг. 3- и 6-месячные токсикологические исследования с использованием повторных доз в настоящее время завершены для доз 7,5, 25 и 50 мг/кг, а 9-месячное исследование с использованием повторных доз продолжается. Не было идентифицировано токсичности в отношении органов-мишеней по результатам гистопатологического исследования при всех дозах после умерщвления животных после 3 месяцев воздействия. Было определено значение NOAEL для 3-месячного исследования на минипигах, составляющее 50 мг/кг. Системное воздействие (AUC0-24) на неделе 13 при дозе NOAEL 50 мг/кг/день составляло 91812 ч*нг/мл для самцов и 131731 ч*нг/мл для самок. Прижизненная фаза 6-месячного токсикологического исследования на минипигах недавно была завершена, и результаты гистопатологических исследований ожидаются. Не наблюдалось смертности или клинически значимых наблюдений, связанных с испытуемым лекарственным средством, при всех уровнях доз среди животных, умерщвленных между 13 и 26 неделями. Продолжающиеся 9-месячные исследования на минипигах в настоящее время протекают 7-й месяц, и за указанное время не наблюдалось смертности или клинически значимых наблюдений, связанных с испытуемым лекарственным средством, при всех уровнях доз.
3.4. Дозы для людей. Разработка (R)-прамипексола в качестве лекарственного средства для лечения ALS основана на стратегии максимально переносимой дозы, полученной из данных по переносимости или данных по безопасности в исследованиях на людях, или по результатам токсикологических исследований на животных. К настоящему времени не наблюдается ограничивающей дозу переносимости у людей. Следовательно, с целью разработки дозирования для людей необходимо тщательно изучить воздействие, при котором наблюдается токсичность у крыс и минипигов. Полученные к настоящему времени фармакокинетические данные предполагают, что фармакокинетика у людей будет продолжать оставаться пропорциональной дозе при более высоких дозах, и что фактор аккумуляции будет постоянным. Результаты по безопасности и токсикокинетике 3-месячных токсикологических исследований на крысах и минипигах показывают отсутствие неблагоприятных эффектов при постоянном введении лекарственного средства в дозах до 100 мг/кг крысам и 50 мг/кг минипигам. Анализ пределов безопасности при воздействии (R)-прамипексола между NOAEL для минипигов и прогнозом воздействия на человека, следовательно, поддерживает увеличение суммарных суточных доз для людей до 500 мг. Прогнозируемое воздействие (R)-прамипексола при установившемся режиме с суммарной суточной дозой 500 мг, которую вводят в виде 250 мг каждые 12 часов, составляет приблизительно 7000 ч*нг/мл, что более чем в 13 раз ниже воздействий, наблюдающихся при NOAEL у самцов минипигов, и приблизительно в 9 раз ниже воздействий, наблюдающихся при NOAEL у самцов и самок крыс после 13 недель введения лекарственного средства.
Фиг.4 и 5 представляют собой график зависимости воздействия от дозы для крыс и минипигов, соответственно, по сравнению с людьми. Каждый график показывает взаимоотношения между воздействием, выраженным как AUC (ч*нг/мл), и дозой, выраженной площадью поверхности тела (мг/м2), для каждого уровня дозы, введенной каждому виду, как по 2-недельной, так и по 13-недельной оценке. Отдельные значения со столбцами ошибки представляют собой среднее ± СО. Пунктирная горизонтальная линия внизу обеих диаграмм иллюстрирует экстраполированную AUC при установившемся режиме (7000 ч*нг/мл) у людей при 250 мг каждые 12 часов. В таблице 17А и таблице 17В представлено объединенное изложение всех фармакокинетических оценок для людей, полученных в двух исследованиях фазы I.
Краткое изложение фармакокинетических оценок для людей, полученных в двух исследованиях фазы I с участием здоровых добровольцев
ОД = однократная доза
Краткое изложение фармакокинетических оценок для людей, полученных в двух исследованиях фазы I с участием здоровых добровольцев (продолжается)
±1,07(6)
±182(6)
±61,9(6)
±0,56(5)
±183(5)
±081(5)
4,69(6)
±1,73(6)
±260(6)
±98,5(6)
9,33(6)
±1,48(6)
±165(6)
±62,8(6)
±1,31(6)
±107(6)
±45,8(6)
±1,30(6)
±81,6(6)
±30,1(6)
±0,88(6)
±107(6)
±51,0(6)
±1,19(6)
±76,1(6)
±82,2(6)
ОД = однократная доза
Воздействие при установившемся режиме для крыс, минипигов и людей является линейным для всех изучавшихся доз. После 3 месяцев введения лекарственного средства было определено значение NOAEL для крыс 100 мг/кг, а для минипигов - 50 мг/кг. Средняя AUC для крыс при NOAEL составляла 61299 и 61484 ч*нг/мл для самцов и самок, соответственно, а для минипигов -91812 и 131731 ч*нг/мл для самцов и самок, соответственно. Средняя AUC для людей при установившемся режиме в дозе 100 мг каждые 12 часов (суммарная суточная доза 200 мг) составляла 2574 ч*нг/мл.
ПРИМЕР 4 - Изготовление капсул с (R)-прамипексолом. Моногидратом дигидрохлорида (R)-(+)-прамипексола наполняли твердые желатиновые капсулы без эксципиентов. Капсулы, используемые для лекарственного средства, представляли собой № 00 синие непрозрачные желатиновые капсулы от компании Hawkins Chemical Group. Изготавливали дозы 50 и 500 мг. Соответствующие капсулы плацебо наполняли микрокристаллической целлюлозой. Капсулы изготавливали с использованием взвешивания отдельных пустых капсул и регистрации веса (We). Конкретное количество активного лекарственного вещества взвешивали индивидуально и вручную помещали его в нижнюю часть капсулы, используя воронку для наполнения Torpac®. Фактор регулирования чистоты 1,0638 использовали для регулирования массы воды (моногидрата) в форме соли, т.е. 50 мг доза должна в конечном итоге наполняться 50×1,0638=53,16 мг. Верхние части капсул соединяли с заполненными нижними частями. Наполненные капсулы затем взвешивали и регистрировали вес (Wf). Рассчитанный вес лекарственного вещества в капсуле (Wf-We) регистрировали. Если рассчитанный вес находился в пределах ±5% от номинального веса, то капсулу чистили, полировали и помещали в должным образом маркированный контейнер. Если рассчитанный вес выходил за соответствующие пределы, то капсулу выбрасывали. Вес свободного основания на капсулу (вес свободного основания на мг содержимого капсулы, умноженный на вес содержимого капсулы) составлял от 90% до 100% от рассчитанной величины, заявленной на маркировке. Общее содержание примесей составляло ≤2%. Продукт представлял собой капсулу синего цвета, содержащую порошок от белого до грязно-белого цвета.
ПРИМЕР 4В - Изготовление таблеток с (R)-прамипексолом. Капсулы, содержащие дозу 125 мг, изготавливали с использованием композиции, представленной в таблице 17. Капсулы обычно изготавливали в условиях от 60 до 74°F и относительной влажности от 30 до 60%. Микрокристаллическую целлюлозу, маннит, кросповидон, стеарат магния и (R)-прамипексол (размолотый) взвешивали в количествах, указанных в столбце «Количество/Партия» в таблице 14. Микрокристаллическую целлюлозу, маннит, кросповидон и (R)-прамипексол затем вручную просеивали через сито из нержавеющей стали меш № 20 и переносили в V-блендер Maxiblend с корпусом емкостью 4 кварты. Материалы затем смешивали с использованием V-блендера Maxiblend в течение 10 минут. Стеарат магния затем просеивали вручную через сито из нержавеющей стали меш № 30 и переносили в блендер. Порошки затем смешивали в течение пяти минут. Затем конечную смесь помещали в маркированный двойной бак, покрытый изнутри РЕ, и регистрировали вес брутто, вес тары и вес нетто.
Таблетки изготавливали с использованием Minipress II B с 5 блоками 3/8” круглыми, стандартными, вогнутыми инструментами и гравитационным питателем. Конечную смесь помещали в загрузочную воронку и задавали параметры работы таблетировочного пресса, согласно описанию в таблице 15.
Композиции таблеток и партий
Параметры работы таблетировочного пресса
ПРИМЕР 5 - Получение p-TSA соли (R)-прамипексола: условия А: Все реагенты приобретали у CNH technologies, Fisher, Aldrich, G.J. Chemicals, Puritan, TCI and Spectrum и использовали в том виде, в котором они были предоставлены поставщиком. Спектры протонного ядерно-магнитного резонанса получали с использованием спектрометра Bruker AC 300 на 300 МГц. Анализ на хиральную чистоту методом ВЭЖХ осуществляли на колонке Chiralpak® IA (5 мкм, 250×4,6 мм) при 30°C с использованием в качестве подвижной фазы смеси гептан/этанол/диэтиламин (80:20:2 об./об./об.). Анализ на химическую чистоту методом ВЭЖХ осуществляли на колонке Sunfire® (3,5 мкм, 150×4,6 мм) при 30°C с использованием двух подвижных фаз: А - 0,5% ТФУК в воде; и В - 0,5% ТФУК в метаноле. Градиент от 5%В до 80%В использовали для разделения пиков диамина и прамипексола. Длину волны обнаружения 265 нм использовали для обоих анализов методом ВЭЖХ.
Каждый из способов, подробно описанных в примерах 5-14, может быть также масштабирован для производства в промышленных масштабах, как показано в примерах 15-17. Определенные примеры подробно описаны как в лабораторном, так и в промышленном масштабах, чтобы показать, что химические и хиральные выходы не зависят от масштабов синтеза.
Трехгорлую колбу емкостью 2,0 литра оборудовали расположенной сверху мешалкой, датчиком температуры, нагревательным кожухом, соединением Кляйзена, обратным холодильником и 500 мл дополнительной воронкой. В колбу помещали 45 граммов R(+)-2,6-диамино-4,5,6,7-тетрагидробензотиазола, а затем 750 мл н-пропанола. При непрерывном перемешивании смесь нагревали до температуры 95°C в течение 15 минут с получением прозрачного раствора. Дополнительную воронку загружали раствором 74 граммов пропилтозилата и 60 мл диизопропилэтиламина в 250 мл н-пропанола. Указанный раствор по каплям добавляли в 2-литровую колбу при непрерывном перемешивании в течение 4 часов. Реакция продолжалась при перемешивании в течение еще 8 часов при 95°C, после чего раствор охлаждали до комнатной температуры и перемешивание продолжали в течение еще 4 часов.
Выпавший в осадок продукт собирали фильтрованием и промывали три раза с использованием спирта реагентного сорта в количестве по 100 мл каждый раз. Промытый спиртом осадок затем промывали 100 мл гептана и сушили в высоком вакууме в течение 2 часов. Конечный вес сухого продукта составлял 53,2 грамма, что соответствовало 52,2% выходу. ВЭЖХ использовали для определения химической чистоты R(+)-2,6-диамино-4,5,6,7-тетрагидробензотиазола ((R)-прамипексола) как 98,2%, а хиральная чистота составляла более 99,5%. 1Н-ЯМР и 13С-ЯМР использовали для подтверждения структуры.
ПРИМЕР 6 - Получение p-TSA соли рацемического прамипексола: условия А: Трехгорлую колбу емкостью 250 мл оборудовали магнитной мешалкой, датчиком температуры, нагревательным кожухом, соединением Кляйзена, обратным холодильником и 100 мл дополнительной воронкой. В колбу помещали 5 граммов рацемического 2,6-диамино-4,5,6,7-тетрагидробензотиазола, а затем 80 мл н-пропанола. При непрерывном перемешивании смесь нагревали до температуры 95°C в течение 15 минут с получением прозрачного раствора. Дополнительную воронку загружали раствором 10,12 грамма пропилтозилата и 8,2 мл диизопропилэтиламина в 28 мл н-пропанола. Указанный раствор по каплям добавляли в 250 мл колбу при непрерывном перемешивании в течение 2 часов. Реакция продолжалась при перемешивании в течение еще 6 часов при 95°C, после чего раствор охлаждали до комнатной температуры и перемешивание продолжали в течение дополнительных 6 часов.
Выпавший в осадок продукт собирали фильтрованием и промывали два раза с использованием спирта реагентного сорта в количестве по 25 мл каждый раз. Промытый спиртом осадок затем промывали 25 мл гептана и сушили в высоком вакууме в течение 1 часа. Конечный вес сухого продукта составлял 5,12 грамма, что соответствовало 45% выходу. ВЭЖХ использовали для определения химической чистоты рацемического 2,6-диамино-4,5,6,7-тетрагидробензотиазола (рацемического прамипексола) как 97,12%, а хиральная чистота показала 1:1 смесь R(+) и (S)-прамипексола. 1Н-ЯМР использовали для подтверждения структуры.
ПРИМЕР 7 - Получение p-TSA соли (R)-прамипексола: условия С: Трехгорлую колбу емкостью 12 литров оборудовали расположенной сверху мешалкой, датчиком температуры, нагревательным кожухом, соединением Кляйзена, конденсатором и 500 мл дополнительной воронкой. В колбу помещали 250 граммов R(+)-2,6-диамино-4,5,6,7-тетрагидробензотиазола (R(+)-диамина), а затем 2 л диметилформамида (ДМФА). При непрерывном перемешивании смесь нагревали до температуры 65°C. Дополнительную воронку загружали раствором 386,6 грамма пропилтозилата (1,25 молярного эквивалента) и 322 мл диизопропилэтиламина (1,25 молярного эквивалента) в 500 мл ДМФА. Указанный раствор по каплям добавляли в 12 л колбу в течение 2,0 часов. Мониторинг реакции проводили с использованием анализа методом ВЭЖХ.
Реакция продолжалась при 65°C в течение еще 5 часов, после чего раствор постепенно охлаждали до комнатной температуры и перемешивали в течение ночи. Раствор разбавляли 2 л МТВЕ и перемешивали в течение еще 0,5 часа. Выпавший в осадок продукт собирали фильтрованием и промывали с помощью 500 мл МТВЕ, а затем промывали три раза спиртом реагентного сорта в количестве по 500 мл каждый раз. Промытый осадок сушили в высоком вакууме.
Конечный вес сухого продукта составлял 317,6 грамма, что соответствовало 56% выходу. ВЭЖХ использовали для определения химической чистоты R(+)-2,6-диамино-4,5,6,7-тетрагидробензотиазола ((S)-прамипексола) как 98,4%, а хиральная чистота составляла более 99,8%. 1Н-ЯМР и 13С-ЯМР использовали для подтверждения структуры: 1H-ЯМР (300 МГц, ДМСО-d6) δ 8,5 (шир.с, 2H), 7,5 (д, 2H), 71,2 (д, 1H), 6,8 (с, 2H), 3,4 (м, 1H), 2,95 (м, 3H), 2,6 (м, 2H, слившийся с пиком ДМСО), 2,3 (с, 3H), 2,15 (м, 1H), 1,8 (м, 1H), 1,55 (м, 2H), 0,9 (т, 3H); 13C-ЯМР (300 МГц, ДМСО-d6) δ 167,0, 145,5, 144,6, 138,4, 128,6, 125,8, 110,7, 53,9, 46,5, 25,8, 25,6, 24,5, 21,2, 19,6, 31,3.
ПРИМЕР 8 - Преобразование p-TSA соли (R)-прамипексола в дигидрохлорид (R)-прамипексола: p-TSA соль (R)-прамипексола (50 граммов, 0,13 моль) помещали в 150 мл абсолютного этанола и охлаждали до температуры 0-5°C при непрерывном перемешивании. Концентрированную HCl (33 мл) медленно добавляли к реакционной смеси, поддерживая температуру 0-5°C, и смесь перемешивали в течение еще 15 минут. МТВЕ (200 мл) добавляли к смеси и перемешивание продолжали в течение еще 1,5 часов при указанной температуре. Реакционную смесь затем фильтровали, два раза промывали раствором МТВЕ/этанол (2:1, объемы промывания 2×50 мл) и сушили в вакууме при 30°C в течение ночи. Конечный вес сухого продукта составлял 34 грамма дигидрохлорида (R)-прамипексола, что соответствовало 92% выходу, а 97,3% химическую чистоту определяли методом ВЭЖХ.
ПРИМЕР 9 - Преобразование p-TSA соли (R)-прамипексола в дигидрохлорид (R)-прамипексола: p-TSA соль (R)-прамипексола (10 граммов, 0,026 моль) растворяли в 200 мл IPAC и охлаждали до 15°C при непрерывном перемешивании. Газообразный HCl барботировали во взвесь в течение 1 часа. Смесь затем фильтровали, промывали с использованием IPAC и сушили в вакууме в течение ночи при комнатной температуре. Конечный вес продукта составлял 6,8 грамма дигидрохлорида (R)-прамипексола, что соответствовало 92% выходу, а 97% химическую чистоту определяли методом ВЭЖХ.
ПРИМЕР 10 - Преобразование p-TSA соли (R)-прамипексола в свободное основание (R)-прамипексола: p-TSA соль (R)-прамипексола (25 граммов, 0,065 моль) растворяли в 200 мл DCM и перемешивали с получением взвеси. Добавляли 10 мл воды и смесь подщелачивали 12 мл 6 н. NaOH до рН 11-12. Две фазы разделяли и водную фазу экстрагировали 200 мл DCM. Объединенные органические фазы сушили над MgSO4, фильтровали через Celite® и концентрировали. Остаток растворяли в 100 мл МТВЕ и суспендировали в течение нескольких часов. Твердые вещества затем отфильтровывали, промывали с использованием МТВЕ и сушили в вакууме при 35°C. Конечный вес продукта составлял 9,1 грамма дигидрохлорида (R)-прамипексола, что соответствовало 66% выходу, а 98% химическую чистоту определяли методом ВЭЖХ.
ПРИМЕР 11 - Преобразование p-TSA соли (R)-прамипексола в свободное основание (R)-прамипексола: Образование свободного основания осуществляли в масштабе 200 граммов. В трехгорлую круглодонную колбу емкостью 5 литров, оборудованную расположенной сверху мешалкой, термометром и дополнительной воронкой, помещали 200 г (0,522 моль) p-TSA соли (R)-прамипексола и 1 л воды. Смесь перемешивали и охлаждали до 10°C. Взвесь подщелачивали до рН приблизительно 11-12 медленным добавлением 200 мл 6 н. NaOH в течение 15 минут. Реакционную смесь разбавляли 500 мл насыщенного раствора соли (хлорид натрия, растворенный в воде) и экстрагировали 3×1 л дихлорметана. Объединенные органические фазы промывали 1,0 л насыщенного раствора соли, сушили над MgSO4, фильтровали и концентрировали досуха. Остаток растирали с 1 л 1:1 смеси IPAC:гептан, полученную взвесь перемешивали в течение 1 часа, фильтровали и осадок на фильтре промывали 2×250 мл 1:1 смеси IPAC:гептан. Осадок на фильтре собирали и сушили при 40°C в высоком вакууме в течение 24 часов, получая 94,1 грамма (R)-прамипексола (85,5%) в виде твердого вещества белого цвета. Химическая чистота составляла 100% AUC по результатам ВЭЖХ, а хиральная чистота составляла 100% AUC по результатам ВЭЖХ. 1Н-ЯМР и 13С-ЯМР использовали для подтверждения структуры: 1H-ЯМР (300 МГц, ДМСО-d6) δ 6,6 (с, 2H), 2,8 (м, 2H), 2,5 (м, 2H, слившийся с пиком ДМСО), 2,2 (м, 1H), 1,9 (м, 1H), 1,5-1,3 (м, 4H), 0,85 (т, 3H); 13C-ЯМР (300 МГц, ДМСО-d6) δ 166,2, 144,8, 113,6, 54,2, 49,1, 30,0, 29,6, 25,2, 23,5, 12,3.
ПРИМЕР 12 - Преобразование свободного основания (R)-прамипексола в дигидрохлорид (R)-прамипексола: Свободное основание (R)-прамипексола (4,8 грамма, 0,022 моль) растворяли в 200 мл IPAC и охлаждали до 15°C. Газообразный HCl барботировали во взвесь в течение 1 часа. Смесь затем фильтровали, промывали с использованием IPAC и сушили в вакууме в течение ночи при комнатной температуре. Конечный вес продукта составлял 6,4 грамма дигидрохлорида (R)-прамипексола, что соответствовало 100% выходу, а 97% химическую чистоту определяли методом ВЭЖХ.
ПРИМЕР 13 - Преобразование свободного основания (R)-прамипексола в дигидрохлорид (R)-прамипексола: Свободное основание (R)-прамипексола (50 граммов, 0,13 моль) растворяли в 500 мл IPAC. При непрерывном перемешивании в смесь медленно добавляли 78 мл концентрированной HCl при температуре 25°C. Смесь перемешивали в течение ночи при комнатной температуре (~25°C), фильтровали и сушили в вакууме при 40°C. Конечный вес продукта составлял 68 граммов дигидрохлорида (R)-прамипексола, что соответствовало 95% выходу.
ПРИМЕР 14 - Оптическая очистка (R)-прамипексола с использованием добавления ахиральной кислоты: Прамипексол, энантиомерно обогащенный R(+)-энантиомером (~300 мг), растворяли в 10 мл выбранного растворителя при 75°C (см. примеры в таблице 8; этанол или ацетонитрил). Полное растворение наблюдалось во всех образцах. Добавление кислоты осуществляли в количестве 1,05 молярного эквивалента для p-TSA (растворитель представлял собой этанол; 2,97 мл 0,5 М кислоты) и MSA (растворитель представлял собой ацетонитрил; 1,49 мл 1,0 М кислоты) и 2,05 молярного эквивалента для фумаровой (растворитель представлял собой ацетонитрил; 5,84 мл 0,5 М кислоты) и фосфорной (растворитель представлял собой ацетонитрил; 2,90 мл 1,0 М кислоты) кислоты. Реакционные смеси охлаждали до комнатной температуры со скоростью 25°C в час и перемешивали при комнатной температуре в течение еще 19 часов. Твердые вещества, полученные на указанной стадии осаждения, выделяли фильтрованием и сушили в высоком вакууме при комнатной температуре. Указанные продукты анализировали методом ВЭЖХ, 1Н-ЯМР, термического гравиметрического анализа, дифференциальной сканирующей калориметрии, рентгеновского порошкового дифракционного анализа (XPRD), инфракрасной спектроскопии на основе преобразования Фурье и сорбционного анализа moister. Паттерны XPRD показали, что солевые формы (R)-прамипексола с p-TSA, MSA и фумаровой кислотой являются кристаллическими, в то время как фосфатная солевая форма (R)-прамипексола является аморфной.
ПРИМЕР 15 - Разделение рацемического диамина в промышленном масштабе: В 72-литровый реактор без кожуха загружали рацемический 2,6-диамино-4,5,6,7-тетрагидробензотиазол (рац-диамин) (4,5 кг; 26,6 моль) и 58,5 л воды и нагревали в виде суспензии до температуры приблизительно 60-65°C. Разделения энантиомеров достигали добавлением одного эквивалента (D)-(-)-винной кислоты (3991 грамм; 26,6 моль) в 4,5 л воды, после чего полученный раствор нагревали до температуры приблизительно 70-75°C и поддерживали указанную температуру в течение приблизительно 1 часа. Смеси давали охладиться до температуры приблизительно 20-25°C и перемешивали в течение еще 15 часов, после чего смесь фильтровали и твердые вещества промывали три раза водой (по 6,3 л каждая промывка).
Влажные твердые вещества, которые содержали R(+)-энантиомер диамина, загружали в реактор, затем помещали в него 54 л воды и смесь нагревали до температуры приблизительно 70-75°C в течение 2 часов. Смеси давали охладиться до температуры приблизительно 20-25°C и перемешивали в течение 17 часов. Смесь затем фильтровали и твердые вещества промывали два раза водой (по 4,5 л каждая промывка). Влажные твердые вещества переносили в реактор с кожухом и помещали туда же 8,1 л воды. Смесь охлаждали до температуры приблизительно 0-5°C и осторожно добавляли 1,625 л концентрированной HCl, а затем 1,155 л 50% NaOH для достижения рН приблизительно 9-10. Во время добавления температуру поддерживали на уровне приблизительно 0-5°C и перемешивали в течение еще одного часа при указанной температуре. Полученную смесь затем фильтровали и твердые вещества промывали два раза холодной (0-5°C) водой (по 1,125 л каждая промывка). Твердые вещества переносили в реактор с кожухом и еще раз ресуспендировали с использованием 4,5 л воды при 0-5°C. Твердые вещества отфильтровывали и сушили теплым воздухом (40-45°C), получая 1940 граммов продукта (R(+)-диамина) в виде твердого вещества белого цвета, с 86% выходом для R(+)-энантиомера.
Маточные растворы с исходной стадии разделения, которые содержали S(-)-энантиомер диамина, концентрировали с получением диамина с 95,5% выходом для S(-)-энантиомера.
Эксперименты по разделению R(+)-энантиомера диамина в промышленном масштабе
ПРИМЕР 16 - Получение пропилтозилата в промышленном масштабе: В 110-литровый стеклянный реактор с кожухом загружали 1-пропанол (2,098 кг; 34,9 моль), триэтиламин (4,585 кг; 45,3 моль; 1,3 эквивалента) и DCM (20,1 л). Смесь охлаждали до температуры приблизительно 5-15°C и осторожно добавляли раствор п-толуолсульфонилхлорида (6 кг; 31,47 моль; 0,9 эквивалента) в DCM (10,5 л) в течение 30 минут. По завершении добавления смесь нагревали до температуры приблизительно 18-22°C и перемешивали в течение приблизительно 12 часов. Реакционную смесь анализировали с использованием 1Н-ЯМР (в CDCl3) и реакцию считали завершенной. Осторожно загружали HCl (6 н.; 2,98 л), поддерживая температуру ниже 25°C. Водную фазу удаляли, а органическую фазу промывали два раза водой (по 21 л каждая промывка), сушили над MgSO4 и фильтровали через Celite®. Отфильтрованные твердые вещества промывали с использованием DCM (4 л) и концентрировали с получением остатка. Остаток растворяли в гептане и снова концентрировали с получением конечного продукта пропилтозилата (6,385 кг; 95% выход).
Настоящее изобретение свидетельствует о том, что аффинность (R)-прамипексола в отношении дофаминовых рецепторов на самом деле гораздо ниже, чем считалось ранее. В исследовании на собаках породы бигль, представленном в настоящем документе, было показано, что разделение функций между энантиомерами (S)-прамипексолом и (R)-прамипексолом (в 10000-20000 раз) гораздо больше, чем считалось ранее. Указанные данные показывают также, что загрязнение композиции чистого (R)-прамипексола малыми, известными количествами (S)-прамипексола приводит к предсказуемому изменению MTD композиции. Указанные данные показывают, что (R)-прамипексол можно вводить в количествах, которые могут более полно и неожиданно задействовать менее мощный нейропротективный потенциал соединения без теоретического ограничения MTD, которое оценивалось ранее, и без необходимости титрования дозы. Заявка представляет способы применения чистых композиций (R)-прамипексола при острых и хронических нейродегенеративных расстройствах, которые ранее были недоступны для действия указанного лекарственного средства, и применения их непосредственно в полном объеме без титрования дозы и при более высоких теоретических MTD. Кроме того, данные, показывающие, что чистую композицию (R)-прамипексола можно смешивать с известным количеством (S)-прамипексола для получения агонистических эффектов в отношении дофаминовых рецепторов, определяемых только вкладом (S)-энантиомера, предусматривают применение композиций, содержащих смесь известных количеств чистых (R)- и (S)-энантиомеров, для лечения нейродегенеративных расстройств, поддающихся лечению агонистами обоих дофаминовых рецепторов и нейропротекции, таких как PD.
ПРИМЕР 17 - Получение p-TSA соли (R)-прамипексола в промышленном масштабе: В 72-литровый реактор без кожуха помещали 1,84 кг (10,87 моль) R(+)-2,6-диамино-4,5,6,7-тетрагидробензотиазола (R(+)-диамина), а затем 14,7 л диметилформамида (ДМФА). При непрерывном перемешивании смесь нагревали до температуры 65-68°C. Раствор 2926 граммов пропилтозилата и 1761 грамма диизопропилэтиламина в 3,455 л ДМФА медленно добавляли в течение 2 часов. Реакция продолжалась при 67°C в течение еще 4 часов, после чего раствор постепенно охлаждали до комнатной температуры (18-22°C) и перемешивали в течение еще 15 часов. Раствор разбавляли 14,72 л МТВЕ в течение 30 минут и перемешивали в течение еще 1 часа. Выпавший в осадок продукт собирали фильтрованием и промывали с использованием 7,32 л МТВЕ, затем промывали 3 раза с использованием 3,68 л этанола на каждую промывку, а затем промывали с использованием 9,2 л гептана. Промытый осадок сушили в высоком вакууме при температуре 30-35°C. Конечный вес сухого продукта составлял 2090 граммов, что соответствовало 50% выходу.
Несмотря на то, что настоящее изобретение было описано достаточно подробно со ссылкой на определенные предпочтительные варианты его осуществления, другие версии являются возможными. Таким образом, сущность и объем прилагаемой формулы изобретения не должны ограничиваться описанием и предпочтительными версиями, содержащимися в настоящем описании.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕЗ ХИРАЛЬНО ОЧИЩЕННЫХ ЗАМЕЩЕННЫХ БЕНЗОТИАЗОЛДИАМИНОВ | 2008 |
|
RU2454409C2 |
| БОЛЕУТОЛЯЮЩАЯ КОМПОЗИЦИЯ И СПОСОБ ДОСТИЖЕНИЯ БОЛЕУТОЛЯЮЩЕГО ЭФФЕКТА У ЧЕЛОВЕКА | 1993 |
|
RU2125873C1 |
| СПОСОБ УМЕНЬШЕНИЯ ОТЛОЖЕНИЯ АМИЛОИДА, АМИЛОИДНОЙ НЕЙРОТОКСИЧНОСТИ И МИКРОГЛИОЗА С ПОМОЩЬЮ (-)-ЭНАНТИОМЕРА НИЛВАДИПИНА | 2008 |
|
RU2490014C2 |
| НОВЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ СЕКСУАЛЬНЫХ РАССТРОЙСТВ | 2005 |
|
RU2445095C2 |
| КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2017 |
|
RU2764716C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2008 |
|
RU2470907C2 |
| АМИНОПИРАЗОЛЫ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2017 |
|
RU2757218C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2008 |
|
RU2470908C2 |
| ТАБЛЕТКА С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ, СОДЕРЖАЩАЯ РЕБОКСЕТИН | 2003 |
|
RU2292873C2 |
| ЛЕЧЕНИЕ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ С ИСПОЛЬЗОВАНИЕМ МОДУЛЯТОРОВ ИЗОФОРМ PIЗ-КИНАЗЫ | 2014 |
|
RU2705204C2 |
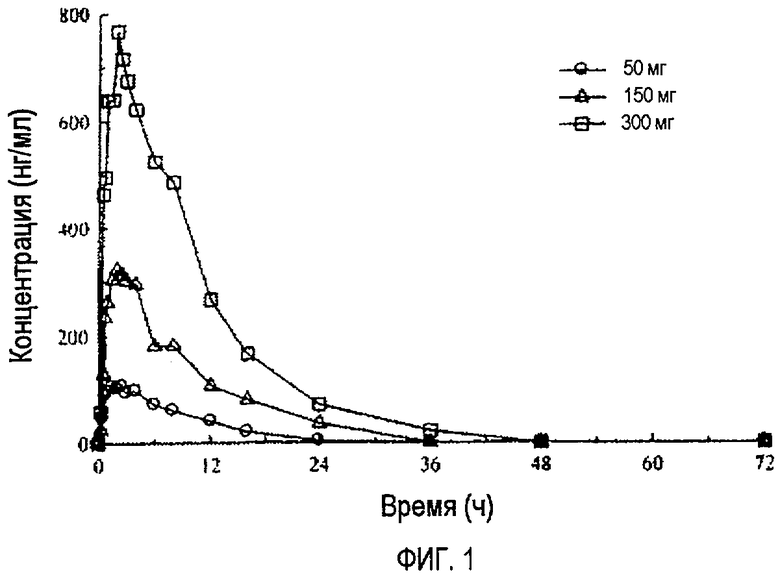
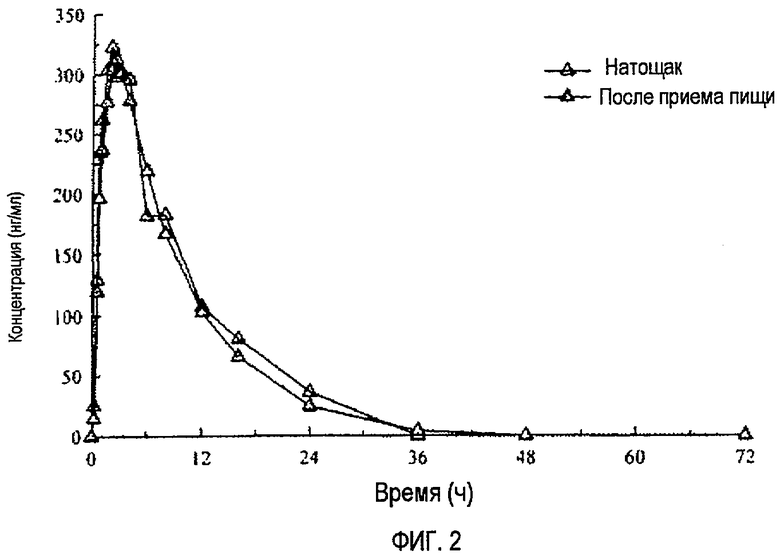
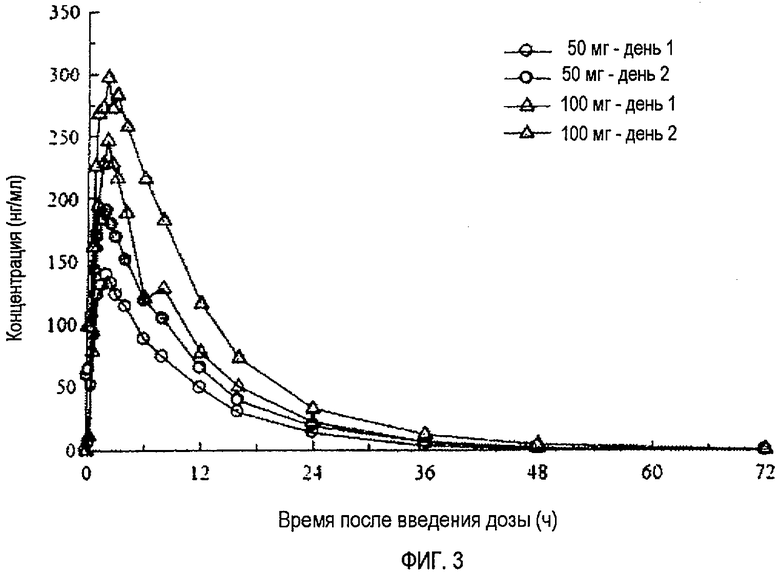
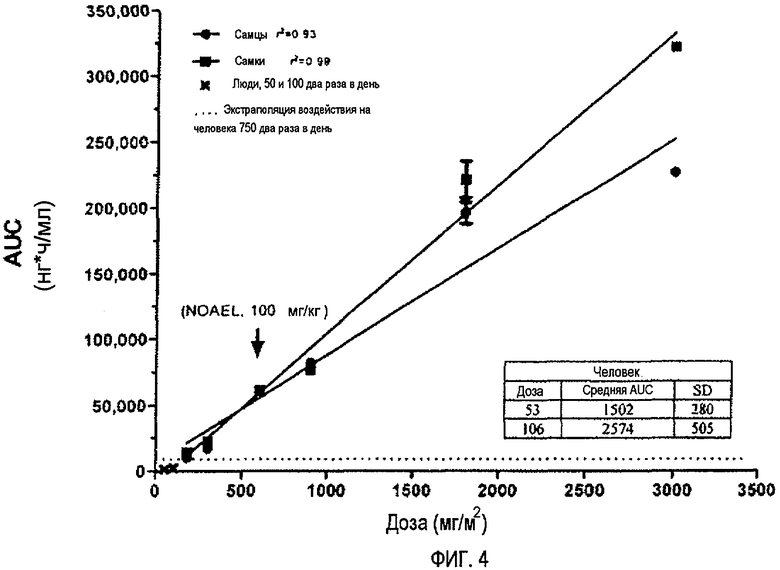
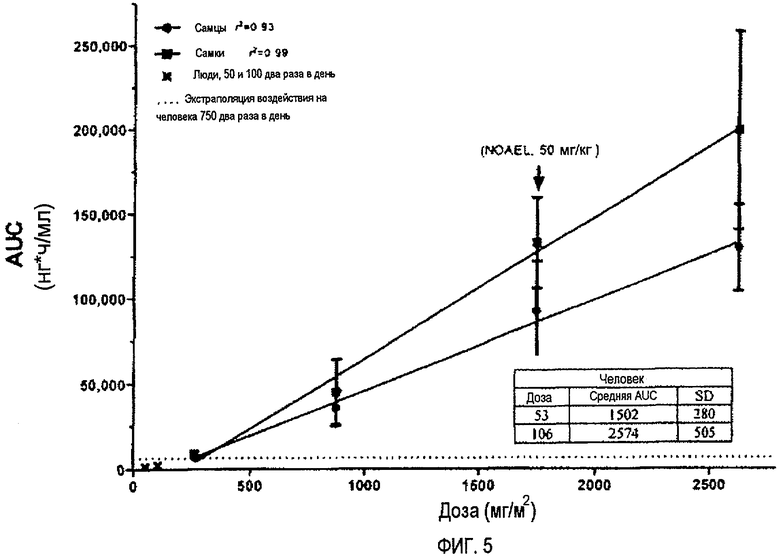
Группа изобретений относится к фармацевтическим композициям однократной дозы (R)-прамипексола высокой хиральной чистоты и способам применения указанных композиций для лечения нейродегенеративных заболеваний или заболеваний, связанных с дисфункцией митохондрий или повышенным окислительным стрессом. Группа изобретений позволяет приостановить или замедлить развития патологии и/или симптоматики путем введения однократной дозы композиции (R)-прамипексола. 3 н. и 26 з.п. ф-лы, 5 ил., 18 табл., 17 пр.
1. Композиция однократной дозы для лечения нейродегенеративного заболевания, содержащая от 150 до 5000 мг (R)-прамипексола, менее чем 1,5 мг (S)-прамипексола и фармацевтически приемлемый носитель.
2. Композиция однократной дозы по п.1, содержащая 150 мг (R)-прамипексола.
3. Композиция однократной дозы по п.1, содержащая 300 мг (R)-прамипексола.
4. Композиция однократной дозы по п.1, содержащая 600 мг (R)-прамипексола.
5. Композиция однократной дозы по п.1, которая представляет собой твердую пероральную лекарственную форму.
6. Композиция однократной дозы по п.1, которая представляет собой таблетку.
7. Композиция однократной дозы по п.1, которая представляет собой капсулу.
8. Фармацевтическая композиция однократной дозы для лечения нейродегенеративного заболевания, где фармацевтическая композиция однократной дозы содержит от 150 до 5000 мг (R)-прамипексола; указанная композиция имеет терапевтические нейропротективные эффекты и не имеет уровня неблагоприятных эффектов, присущих (R)-прамипексолу или любому (S)-прамипексолу.
9. Фармацевтическая композиция по п.8, в которой указанные уровни отсутствия неблагоприятных эффектов представляют собой уровень, не вызывающий видимых неблагоприятных эффектов у людей.
10. Фармацевтическая композиция по п.8, в которой хиральная чистота для (R)-прамипексола составляет 100% в пределах, поддающихся обнаружению.
11. Фармацевтическая композиция по п.8, содержащая 150 мг (R)-прамипексола.
12. Фармацевтическая композиция по п.8, содержащая 300 мг (R)-прамипексола.
13. Фармацевтическая композиция по п.8, содержащая 600 мг (R)-прамипексола.
14. Фармацевтическая композиция по п.8, которая представляет собой твердую пероральную лекарственную форму.
15. Фармацевтическая композиция по п.8, которая представляет собой таблетку.
16. Фармацевтическая композиция по п.8, которая представляет собой капсулу.
17. Способ лечения нейродегенеративного заболевания у пациента, который в этом нуждается, включающий введение начальной дозы от 150 до 5000 мг (R)-прамипексола в фармацевтической композиции.
18. Способ по п.17, в котором указанное нейродегенеративное заболевание представляет собой острое нейродегенеративное заболевание.
19. Способ по п.17, в котором указанное нейродегенеративное заболевание представляет собой хроническое нейродегенеративное заболевание.
20. Способ по п.17, в котором указанное хроническое нейродегенеративное заболевание выбрано из инсульта, нейротравмы, острой метаболической дисфункции, последствий церебрального припадка, эпилептического статуса, острого энцефалита, первичного нейродегенеративного заболевания, хореи Гентингтона, метаболически индуцированного неврологического повреждения, сенильной деменции типа Альцгеймера, когнитивной дисфункции, связанной с возрастом, сосудистой деменции, мультиинфарктной деменции, деменции с тельцами Леви, нейродегенеративной деменции, нейродегенеративного двигательного расстройства, атаксии, атаксии Фридрейха, рассеянного склероза, спинальной мышечной атрофии, первичного бокового склероза, расстройств, сопровождающихся припадками, расстройства или заболевания двигательных нейронов, воспалительного демиелинизирующего расстройства, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, печеночной энцефалопатии и хронического энцефалита.
21. Способ по п.17, в котором указанное нейродегенеративное заболевание представляет собой боковой амиотрофический склероз.
22. Способ по п.17, в котором указанный пациент представляет собой пациента, не подвергавшегося ранее лечению.
23. Способ по п.17, в котором указанная начальная доза составляет от 150 до 5000 мг (R)-прамипексола.
24. Способ по п.17, в котором указанная начальная доза составляет 150 миллиграммов (R)-прамипексола.
25. Способ по п.17, в котором указанная начальная доза составляет 300 мг (R)-прамипексола.
26. Способ по п.17, в котором указанная начальная доза составляет 600 мг (R)-прамипексола.
27. Способ по п.17, в котором хиральная чистота для (R)-прамипексола составляет 99,9% или выше.
28. Способ по п.17, в котором хиральная чистота для (R)-прамипексола составляет 99,95% или выше.
29. Способ по п.17, в котором хиральная чистота для (R)-прамипексола составляет 99,99% или выше.
| WO 03049705 A2, 19.06.2003 | |||
| US 6156777 A, 05.12.2000 | |||
| WO 2006012277 A2, 02.02.2006. |

